






( Angew. Chem., Int. Ed. 2015,54, 4945−4948).
Volume 54, Issue 16April 13, 2015 Pages 4945–4948
PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
Home » MANUFACTURING
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
If you have difficulty in viewing click,,,,,,,,,,http://www.allfordrugs.com/2016/08/07/pharmaceutical-manufacturing-encyclopedia-3rd-edition/
This industry standard encyclopedia on pharmaceutical manufacturing processes has been completely updated to include FDA drugs approved up to the summer of 2004. The encyclopedia gives details for the manufacture of 2226 pharmaceuticals that are being marketed as a trade-named product somewhere in the world. Each entry includes:
ò Therapeutic function
ò Chemical and common name
ò Structural Formula
ò Chemical Abstracts Registry no.
ò Trade name, manufacturer, country, and year introduced
ò Raw Materials
ò Manufacturing Process
In addition, references are also cited under each drug’s entry to major pharmaceutical works where additional information can be obtained on synthesis and the pharmacology of the individual products.

//////////
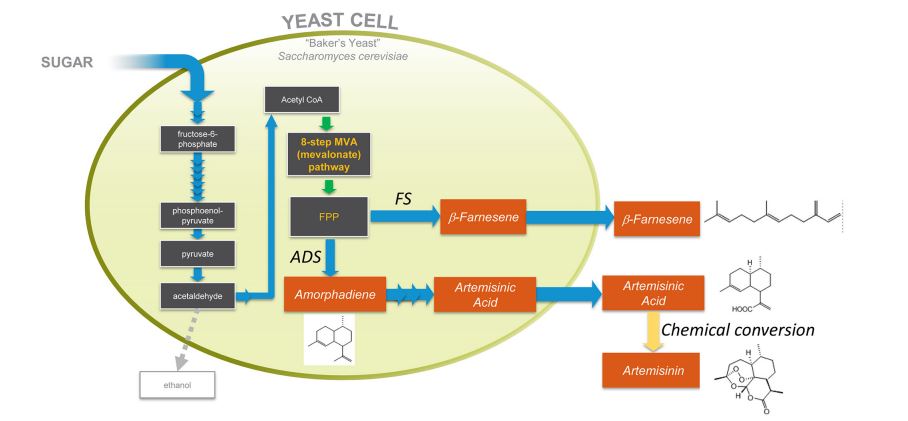
Figure 1. Production of artemisinic acid or β-farnesene by engineered yeast. The sesquiterpene alkenes β-farnesene and amorphadiene are both derived from FPP (farnesyl diphosphate) by the action of specific enzymes introduced from plants: amorphadiene synthase (ADS) generates amorphadiene and β-farnesene synthase (FS) generates β-farnesene. Production strains express either ADS or FS, not both. Oxidation of amorphadiene to artemisinic acid is accomplished by the action of five plant enzymes expressed in the engineered yeast.17 Conversion of purified artemisinic acid to artemisinin is accomplished by in vitro organic chemistry. Isoprenoid production strains make little ethanol.
The antimalarial drug artemisinin and the specialty chemical β-farnesene are examples of natural product isoprenoids that can help solve global challenges, but whose usage has previously been limited by supply and cost impediments. This review describes the path to commercial production of these compounds utilizing fermentation of engineered yeast. Development of commercially viable yeast strains was a substantial challenge that was addressed by creation and implementation of an industrial synthetic biology pipeline. Using the engineered strains, production of β-farnesene from Brazilian sugarcane offers several environmental advantages. Among the many commercial applications of β-farnesene, its use as a feedstock for making biodegradable lubricants is highlighted. This example, along with others, highlight a powerful new suite of technologies that will become increasingly important for production of chemicals, spanning from pharmaceuticals through commodity chemicals.
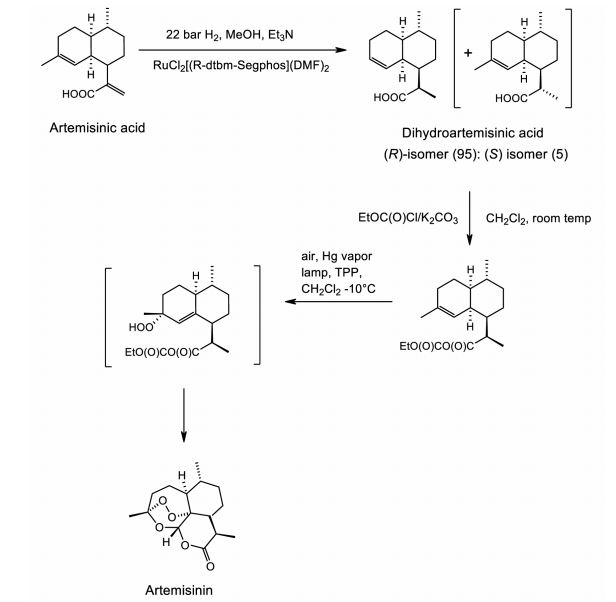
Figure 2. Sanofi industrial semi-synthesis of artemisinin. The process starts with a moderate pressure catalytic diastereoselective hydrogenation of artemisinic acid to produce a high (95:5) ratio of the desired (R)-isomer. To avoid formation of a lactone byproduct, dihydro-epi-deoxyarteannuin B, during the photooxidation, the carboxylic acid is protected as a mixed anhydride. The final step combines formation of the intermediate hydroperoxide via photoxidation using a Hg vapor lamp and commercially available tetraphenylporphyin (TPP) as sensitizer with a Hock cleavage and rearrangement catalyzed by trifluoroacetic acid to give, after workup, the best yield reported to date of pure isolated artemisinin (55%).
Synthetic Biology and the Development of Commercial β-Farnesene Production Strains Semi-synthetic artemisinin is a pharmaceutical with a price point comparable to plant-derived artemisinin,20 namely above $150 per kg. β-Farnesene, however, is a specialty chemical with multiple uses (more details below); most specialty and commodity chemicals have significantly lower price points, often below $10 per kg. For these product categories, it is of paramount importance that fermentative production be as efficient as possible, with high yields (namely, grams of product made per gram of feed substrate), productivities (grams of product/liter of culture/hour) and concentration (also known as titer; grams of product per liter of culture). Developing yeast strains capable of the yield, productivity and titer required for chemical production requires extensive development, and has been enabled over the last decade by the new discipline of synthetic biology. Synthetic biology seeks to extend approaches and concepts from engineering and computation to redesign biology for a chosen function;21recent advances in the application of design automation, i.e., the use of software, hardware and robotics22 have enabled the creation and screening of hundreds of thousands of strain variants (created by both design and random mutagenesis) for the properties required for commercial production of β-farnesene. Notable enabling technologies developed for routine usage include rapid and reliable assembly of large (i.e., multiple kilobase) deoxyribonucleic acid (DNA) constructs;23-25 high throughput, cost effective, verification of structural DNA assemblies by both initial restriction digest26 and by low-cost DNA sequencing;27 and whole genome sequencing of yeast strains.28 In addition, there is a need to effectively identify the best new strains (akin to panning for gold!) through high throughput, rapid, and accurate methods to screen thousands of strains. Further, the results of small-scale (< 1 milliliter) tests must correspond to the results of large-scale (> 50,000 liter) production. Development and implementation of these technologies required considerable investment by Amyris. The outcome is a robust pipeline for efficient, cost-effective strain generation allied with screening for the properties required for commercial production of β-farnesene by fermentation (i.e., at a price point required for its use as a specialty chemical).
As the world’s population and economies grow, the demand for a wide variety of specialty, commodity, and pharmaceutical chemicals will outpace the supply available from current sources. There is an urgent need to develop alternative, sustainable sources of many existing chemicals and to develop abundant sources of currently scarce chemicals with novel beneficial properties. Synthetic biology and industrial fermentation, combined with synthetic chemistry, will be an increasingly important source of chemicals in the decades ahead; artemisinin and β-farnesene provide good examples of this relatively new approach to chemical production. Brazil’s plentiful sugar cane feedstock and fermentation expertise make it an excellent location for this type of manufacturing, which can expand and diversify the nation’s industrial base and international importance.
J. Braz. Chem. Soc. 2016, 27(8), 1339-1345
Kirsten R. Benjamin; Iris R. Silva; João P. Cherubim; Derek McPhee; Chris J. Paddon
 Genes encoding the biosynthetic pathway for production of a valuable product (e.g., farnesene) in a native organism are expressed in a heterologous microbial host (e.g., yeast). The engineered yeast produces farnesene by commercial fermentation. Copyright © 2016 Amyris, inc. All rights reserved.
Genes encoding the biosynthetic pathway for production of a valuable product (e.g., farnesene) in a native organism are expressed in a heterologous microbial host (e.g., yeast). The engineered yeast produces farnesene by commercial fermentation. Copyright © 2016 Amyris, inc. All rights reserved.
http://jbcs.sbq.org.br/imagebank/pdf/v27n8a04.pdf
Kirsten R. Benjamin,a Iris R. Silva,b João P. Cherubim,c Derek McPheea and Chris J. Paddon*,a a Amyris, Inc., 5885 Hollis Street, Suite 100, CA 94608 Emeryville, USA b Amyris Brasil Ltda, Rua John Dalton 301-Bloco B-Edificio 3, Condominio Techno Plaza, 13069-330 Campinas-SP, Brazil c Amyris Brasil Ltda, Rodovia Brotas/Torrinha-km 7.5, 17380-000 Brotas-SP, Brazil

Dr. Paddon has a PhD in Biochemistry from Imperial College, London, but now considers himself a synthetic biologist. After postdoctoral work at the National Institutes of Health in Bethesda, MD, he worked in the pharmaceutical industry (GlaxoSmithKline), and then for two Bay Area biopharmaceutical companies (Affymax and Xenoport) before joining Amyris, Inc. in 2005 as its sixth employee and first scientist. He was project leader for the semi-synthetic artemisinin project at Amyris, Inc. and has subsequently led a number of other projects and programs there.
//////////// Commercial Production, Semi-Synthetic , Artemisinin, farnesene, fermentation, natural product, lubricant

Sreeni Labs Private Limited, Hyderabad, India is ready to take up challenging synthesis projects from your preclinical and clinical development and supply from few grams to multi-kilo quantities. Sreeni Labs has proven route scouting ability to design and develop innovative, cost effective, scalable routes by using readily available and inexpensive starting materials. The selected route will be further developed into a robust process and demonstrate on kilo gram scale and produce 100’s of kilos of in a relatively short time.
Accelerate your early development at competitive price by taking your route selection, process development and material supply challenges (gram scale to kilogram scale) to Sreeni Labs…………
Sreeni Labs based in Hyderabad, India is working with various global customers and solving variety of challenging synthesis problems. Their customer base ranges from USA, Canada, India and Europe. Sreeni labs Managing Director, Dr. Sreenivasa Reddy Mundla has worked at Procter & Gamble Pharmaceuticals and Eli Lilly based in USA.
The main strength of Sreeni Labs is in the design, development of innovative and highly economical synthetic routes and development of a selected route into a robust process followed by production of quality product from 100 grams to 100s of kg scale. Sreeni Labs main motto is adding value in everything they do.
They have helped number of customers from virtual biotech, big pharma, specialty chemicals, catalog companies, and academic researchers and drug developers, solar energy researchers at universities and institutions by successfully developing highly economical and simple chemistry routes to number of products that were made either by very lengthy synthetic routes or by using highly dangerous reagents and Suzuki coupling steps. They are able to supply materials from gram scale to multi kilo scale in a relatively short time by developing very short and efficient synthetic routes to a number of advanced intermediates, specialty chemicals, APIs and reference compounds. They also helped customers by drastically reducing number of steps, telescoping few steps into a single pot. For some projects, Sreeni Labs was able to develop simple chemistry and avoided use of palladium & expensive ligands. They always begin the project with end in the mind and design simple chemistry and also use readily available or easy to prepare starting materials in their design of synthetic routes
Over the years, Sreeni labs has successfully made a variety of products ranging from few mg to several kilogram scale. Sreeni labs has plenty of experience in making small select libraries of compounds, carbocyclic compounds like complex terpenoids, retinal derivatives, alkaloids, and heterocyclic compounds like multi substituted beta carbolines, pyridines, quinolines, quinolones, imidazoles, aminoimidazoles, quinoxalines, indoles, benzimidazoles, thiazoles, oxazoles, isoxazoles, carbazoles, benzothiazoles, azapines, benzazpines, natural and unnatural aminoacids, tetrapeptides, substituted oligomers of thiophenes and fused thiophenes, RAFT reagents, isocyanates, variety of ligands, heteroaryl, biaryl, triaryl compounds, process impurities and metabolites.
Sreeni Labs is Looking for any potential opportunities where people need development of cost effective scalable routes followed by quick scale up to produce quality products in the pharmaceutical & specialty chemicals area. They can also take up custom synthesis and scale up of medchem analogues and building blocks. They have flexible business model that will be in sink with customers. One can test their abilities & capabilities by giving couple of PO based (fee for service) projects.
Some of the compounds prepared by Sreeni labs;



See presentation below
LINK ON SLIDESHARE
Managing Director at Sreeni Labs Private Limited\
QUOTE………….
One virtual biotech company customer from USA, through a common friend approached Sreeni Labs and told that they are buying a tetrapeptide from Bachem on mg scale at a very high price and requested us to see if we can make 5g. We accepted the challenge and developed solution phase chemistry and delivered 6g and also the process procedures in 10 weeks time. The customer told that they are using same procedures with very minor modifications and produced the tetrapeptide ip to 100kg scale as the molecule is in Phase III.
One East coast customer in our first meeting told that they are working with 4 CROs of which two are in India and two are in China and politely asked why they should work with Sreeni Labs. We told that give us a project where your CROs failed to deliver and we will give a quote and work on it. You pay us only if we deliver and you satisfy with the data. They immediately gave us a project to make 1.5g and we delivered 2g product in 9 weeks. After receiving product and the data, the customer was extremely happy as their previous CRO couldn’t deliver even a milligram in four months with 3 FTEs.
One Midwest biotech company was struggling to remove palladium from final API as they were doing a Suzuki coupling with a very expensive aryl pinacol borane and bromo pyridine derivative with an expensive ligand and relatively large amount of palldium acetate. The cost of final step catalyst, ligand and the palladium scavenging resin were making the project not viable even though the product is generating excellent data in the clinic. At this point we signed an FTE agreement with them and in four months time, we were able to design and develop a non suzuki route based on acid base chemistry and made 15g of API and compared the analytical data and purity with the Suzuki route API. This solved all three problems and the customer was very pleased with the outcome.
One big pharma customer from east coast, wrote a structure of chemical intermediate on a paper napkin in our first meeting and asked us to see if we can make it. We told that we can make it and in less than 3 weeks time we made a gram sample and shared the analytical data. The customer was very pleased and asked us to make 500g. We delivered in 4 weeks and in the next three months we supplied 25kg of the same product.
Through a common friend reference, a European customer from a an academic institute, sent us an email requesting us to quote for 20mg of a compound with compound number mentioned in J. med. chem. paper. It is a polycyclic compound with four contiguous stereogenic centers. We gave a quote and delivered 35 mg of product with full analytical data which was more pure than the published in literature. Later on we made 8g and 6g of the same product.
One West coast customer approached us through a common friend’s reference and told that they need to improve the chemistry of an advanced intermediate for their next campaign. At that time they are planning to make 15kg of that intermediate and purchased 50kg of starting raw material for $250,000. They also put five FTEs at a CRO for 5 months to optimize the remaining 5 steps wherein they are using LAH, Sodium azide, palladium catalyst and a column chromatography. We requested the customer not to purchase the 50kg raw material, and offered that we will make the 15kg for the price of raw material through a new route in less than three months time. You pay us only after we deliver 15 kg material. The customer didn’t want to take a chance with their timeline as they didn’t work with us before but requested us to develop the chemistry. In 7 weeks time, we developed a very simple four step route for their advanced intermediate and made 50g. We used very inexpensive and readily available starting material. Our route gave three solid intermediates and completely eliminated chromatographic purifications.
One of my former colleague introduced an academic group in midwest and brought us a medchem project requiring synthesis of 65 challenging polyene compounds on 100mg scale. We designed synthetic routes and successfully prepared 60 compounds in a 15 month time.
UNQUOTE…………
The man behind Seeni labs is Dr. Sreenivasa Reddy Mundla

Managing Director at Sreeni Labs Private Limited
Sreeni Labs Private Limited
Road No:12, Plot No:24,25,26
Links
LINKEDIN https://in.linkedin.com/in/sreenivasa-reddy-10b5876
FACEBOOK https://www.facebook.com/sreenivasa.mundla
RESEARCHGATE https://www.researchgate.net/profile/Sreenivasa_Mundla/info
EMAIL mundlasr@hotmail.com, Info@sreenilabs.com, Sreeni@sreenilabs.com
Dr. Sreenivasa Reddy Mundla

Dr. M. Sreenivasa Reddy obtained Ph.D from University of Hyderabad under the direction Prof Professor Goverdhan Mehta in 1992. From 1992-1994, he was a post doctoral fellow at University of Wisconsin in Professor Jame Cook’s lab. From 1994 to 2000, worked at Chemical process R&D at Procter & Gamble Pharmaceuticals (P&G). From 2001 to 2007 worked at Global Chemical Process R&D at Eli Lilly and Company in Indianapolis.
In 2007 resigned to his job and founded Sreeni Labs based in Hyderabad, Telangana, India and started working with various global customers and solving various challenging synthesis problems.
The main strength of Sreeni Labs is in the design, development of a novel chemical route and its development into a robust process followed by production of quality product from 100 grams to 100’s of kg scale.
They have helped number of customers by successfully developing highly economical simple chemistry routes to number of products that were made by Suzuki coupling. they are able to shorten the route by drastically reducing number of steps, avoiding use of palladium & expensive ligands. they always use readily available or easy to prepare starting materials in their design of synthetic routes.
Sreeni Labs is Looking for any potential opportunities where people need development of cost effective scalable routes followed by quick scale up to produce quality products in the pharmaceutical & specialty chemicals area. They have flexible business model that will be in sink with customers. One can test their abilities & capabilities by giving PO based projects
August 2007 – Present (8 years 11 months)
Sreeni Labs Profile

March 2001 – August 2007 (6 years 6 months)

July 1994 – February 2001 (6 years 8 months)


With Sreenivasa Mundla, Narahara sastry, Ram Kishan Rao, Jagadeesh Bharatam, Jagadish Gunjur and Jagadish Bharatham.
PUBLICATIONS
Jianye Zhang · Zhiqian Dong · Sreenivasa Reddy Mundla · X Eric Hu · William Seibel ·Ruben Papoian · Krzysztof Palczewski · Marcin Golczak
Article: ChemInform Abstract: Regioselective Synthesis of 4Halo ortho-Dinitrobenzene Derivative
Aug 2010 · ChemInform
Hong-yu Li · William T. McMillen · Charles R. Heap · Denis J. McCann · Lei Yan · Robert M. Campbell · Sreenivasa R. Mundla · Chi-Hsin R. King · Elizabeth A. Dierks · Bryan D. Anderson · Karen S. Britt · Karen L. Huss
Apr 2008 · Journal of Medicinal Chemistry
Hong-yu Li · Yan Wang · William T. McMillen · Arindam Chatterjee · John E. Toth ·Sreenivasa R. Mundla · Matthew Voss · Robert D. Boyer · J. Scott Sawyer
Feb 2008 · ChemInform
Hong-yu Li · Yan Wang · William T. McMillen · Arindam Chatterjee · John E. Toth ·Sreenivasa R. Mundla · Matthew Voss · Robert D. Boyer · J. Scott Sawyer
Nov 2007 · Tetrahedron
Hong-yu Li · Yan Wang · Charles R Heap · Chi-Hsin R King · Sreenivasa R Mundla · Matthew Voss · David K Clawson · Lei Yan · Robert M Campbell · Bryan D Anderson · Jill R Wagner ·Karen Britt · Ku X Lu · William T McMillen · Jonathan M Yingling
Apr 2006 · Journal of Medicinal Chemistry

Hui Cao · Sreenivasa R. Mundla · James M. Cook
Aug 2003 · Tetrahedron Letters
Article: ChemInform Abstract: A New Method for the Synthesis of 2,6-Dinitro and 2Halo6-nitrostyrenes
Nov 2000 · ChemInform
Article: ChemInform Abstract: A Novel Method for the Efficient Synthesis of 2-Arylamino-2-imidazolines


TGF-β inhibitors
The present invention provides 2-(6-methyl-pyridin-2-yl)-3-[6-amido-quinolin-4-yl) -5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole monohydrate, i.e., Formula I.

EXAMPLE 1 Preparation of 2-(6-methyl-pyridin-2-yl)-3-[6-amido-quinolin-4-yl-5,6-dihydro-4H -pyrrolo[1,2-b]pyrazole monohydrate

Galunisertib
1H NMR (CDCl3): δ=9.0 ppm (d, 4.4 Hz, 1H); 8.23-8.19 ppm (m, 2H); 8.315 ppm (dd, 1.9 Hz, 8.9 Hz, 1H); 7.455 ppm (d, 4.4 Hz, 1H); 7.364 ppm (t, 7.7 Hz, 1H); 7.086 ppm (d, 8.0 Hz, 1H); 6.969 ppm (d, 7.7 Hz, 1H); 6.022 ppm (m, 1H); 5.497 ppm (m, 1H); 4.419 ppm (t, 7.3 Hz, 2H); 2.999 ppm (m, 2H); 2.770 ppm (p, 7.2 Hz, 7.4 Hz, 2H); 2.306 ppm (s, 3H); 1.817 ppm (m, 2H). MS ES+: 370.2; Exact: 369.16
ABOVE MOLECULE IS
https://newdrugapprovals.org/2016/05/04/galunisertib/
Galunisertib
Phase III
A TGF-beta receptor type-1 inhibitor potentially for the treatment of myelodysplastic syndrome (MDS) and solid tumours.
LY-2157299
CAS No.700874-72-2
READ MY PRESENTATION ON
KEYWORDS Sreenivasa Mundla Reddy, Managing Director, Sreeni Labs Private Limited, Hyderabad, Telangana, India, new, economical, scalable routes, early clinical drug development stages, Custom synthesis, custom manufacturing, drug discovery, PHASE 1, PHASE 2, PHASE 3, API, drugs, medicines

Continuous production is a flow production method used to manufacture, produce, or process materials without interruption. Continuous production is called a continuous process or a continuous flow process because the materials, either dry bulk or fluids that are being processed are continuously in motion, undergoing chemical reactions or subject to mechanical or heat treatment. Continuous processing is contrasted with batch production.
Continuous usually means operating 24 hours per day, seven days per week with infrequent maintenance shutdowns, such as semi-annual or annual. Some chemical plants can operate for more than one or two years without a shutdown. Blast furnaces can run four to ten years without stopping.[1]
Production workers in continuous production commonly work in rotating shifts.
Processes are operated continuously for practical as well as economic reasons. Most of these industries are very capital intensive and the management is therefore very concerned about lost operating time.
Shutting down and starting up many continuous processes typically results in off quality product that must be reprocessed or disposed of. Many tanks, vessels and pipes cannot be left full of materials because of unwanted chemical reactions, settling of suspended materials or crystallization or hardening of materials. Also, cycling temperatures and pressures from starting up and shutting down certain processes (line kilns, boilers, blast furnaces, pressure vessels, etc.) may cause metal fatigue or other wear from pressure or thermal cycling.

In the more complex operations there are sequential shut down and start up procedures that must be carefully followed in order to protect personnel and equipment. Typically a start up or shut down will take several hours.
Continuous processes use process control to automate and control operational variables such as flow rates, tank levels, pressures, temperatures and machine speeds.[2]
Many processes such as assembly lines and light manufacturing that can be easily shut down and restarted are today considered semi-continuous. These can be operated for one or two shifts if necessary.
The oldest continuous flow processes is the blast furnace for producing pig iron. The blast furnace is intermittently charged with ore, fuel and flux and intermittently tapped for molten pig iron and slag; however, the chemical reaction of reducing the iron and silicon and later oxidizing the silicon is continuous.
Semi-continuous processes, such as machine manufacturing of cigarettes, were called “continuous” when they appeared.
Many truly continuous processes of today were originally batch operations.
The Fourdrinier paper machine, patented in 1799, was one of the earliest of the industrial revolution era continuous manufacturing processes. It produced a continuous web of paper that was formed, pressed, dried and reeled up in a roll. Previously paper had been made in individual sheets.
Another early continuous processes was Oliver Evans‘es flour mill (ca. 1785), which was fully automated.
Early chemical production and oil refining was done in batches until process control was sufficiently developed to allow remote control and automation for continuous processing. Processes began to operate continuously during the 19th century. By the early 20th century continuous processes were common.
In addition to performing maintenance, shut downs are also when process modifications are performed. These include installing new equipment in the main process flow or tying-in or making provisions to tie-in sub-processes or equipment that can be installed while the process is operating.
Shut-downs of complicated processes may take weeks or months of planning. Typically a series of meetings takes place for co-ordination and planning. These typically involve the various departments such as maintenance, power, engineering, safety and operating units.
All work is done according to a carefully sequenced schedule that incorporates the various trades involved, such as pipe-fitters, millwrights, mechanics, laborers, etc., and the necessary equipment (cranes, mobile equipment, air compressors, welding machines, scaffolding, etc.) and all supplies (spare parts, steel, pipe, wiring, nuts and bolts) and provisions for power in case power will also be off as part of the outage. Often one or more outside contractors perform some of the work, especially if new equipment is installed.
Safety meetings are typically held before and during shutdowns. Other safety measures include providing adequate ventilation to hot areas or areas where oxygen may become depleted or toxic gases may be present and checking vessels and other enclosed areas for adequate levels of oxygen and insure absence of toxic or explosive gases. Any machines that are going to be worked on must be electrically disconnected, usually through the motor starter, so that it cannot operate. It is common practice to put a padlock on the motor starter, which can only be unlocked by the person or persons who is or are endangered by performing the work. Other disconnect means include removing couplings between the motor and the equipment or by using mechanical means to keep the equipment from moving. Valves on pipes connected to vessels that workers will enter are chained and locked closed, unless some other means is taken to insure that nothing will come through the pipes.
Continuous Production can be supplemented using a Continuous Processor. Continuous Processors are designed to mix viscous products on a continuous basis by utilizing a combination of mixing and conveying action. The Paddles within the mixing chamber (barrel) are mounted on two co-rotating shafts that are responsible for mixing the material. The barrels and paddles are contoured in such a way that the paddles create a self-wiping action between themselves minimizing buildup of product except for the normal operating clearances of the moving parts. Barrels may also be heated or cooled to optimize the mixing cycle. Unlike an extruder, the Continuous Processor void volume mixing area is consistent the entire length of the barrel ensuring better mixing and little to no pressure build up. The Continuous Processor works by metering powders, granules, liquids, etc. into the mixing chamber of the machine. Several variables allow the Continuous Processor to be versatile for a wide variety of mixing operations:[3]
Continuous Processors are used in the following processes:
The Continuous Processor has an unlimited material mixing capabilities but, it has proven its ability to mix:

In the development of a new route to bendamustine hydrochloride, the API in Treanda, the key benzimidazole intermediate 5 was generated via catalytic heterogeneous hydrogenation of an aromatic nitro compound using a batch reactor. Because of safety concerns and a site limitation on hydrogenation at scale, a continuous flow hydrogenation for the reaction was investigated at lab scale using the commercially available H-Cube. The process was then scaled successfully, generating kilogram quantities on the H-Cube Midi. This flow process eliminated the safety concerns about the use of hydrogen gas and pyrophoric catalysts and also showed 1200-fold increase in space–time yield versus the batch processing.



Volume 54, Issue 16April 13, 2015 Pages 4945–4948


The synthesis of 2-(2′-hydroxy-5′-methylphenyl)benzotriazole from 2-nitro-2′-hydroxy-5′-methylazobenzene over Pd/γ-Al2O3 in a fixed-bed reactor was investigated. Pd/γ-Al2O3 catalysts were prepared by two methods and characterized by XRD, TEM, H2-TPR, and N2 adsorption–desorption. Employed in the above reaction, the palladium catalyst impregnated in hydrochloric acid exhibited much better catalytic performance than that impregnated in ammonia–water, which was possibly attributed to the better dispersion of palladium crystals on γ-Al2O3. This result demonstrated that the preparation process of the catalyst was very important. Furthermore, the reaction parameters were optimized. Under the optimized conditions (toluene, NAB/triethylamine molar ratio 1:2, 60 °C, 2.5 MPa hydrogen pressure, 0.23 h–1 liquid hourly space velocity), about 90% yield of 2-(2′-hydroxy-5′-methylphenyl)benzotriazole was obtained. Finally, the time on stream performance of the catalyst was evaluated, and the reaction could proceed effectively over 200 h without deactivation of the catalyst.
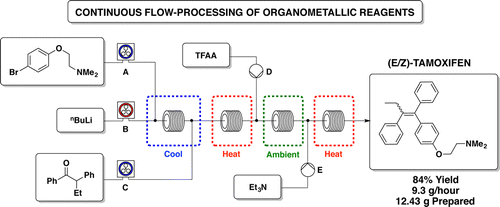
A new enabling technology for the pumping of organometallic reagents such as n-butyllithium, Grignard reagents, and DIBAL-H is reported, which utilises a newly developed, chemically resistant, peristaltic pumping system. Several representative examples of its use in common transformations using these reagents, including metal–halogen exchange, addition, addition–elimination, conjugate addition, and partial reduction, are reported along with examples of telescoping of the anionic reaction products. This platform allows for truly continuous pumping of these highly reactive substances (and examples are demonstrated over periods of several hours) to generate multigram quantities of products. This work culminates in an approach to the telescoped synthesis of (E/Z)-tamoxifen using continuous-flow organometallic reagent-mediated transformations.

A versatile multi-step continuous flow synthesis for the preparation of substituted pyrazoles is presented.
The automated synthesis utilises a metal-free ascorbic acid mediated reduction of diazonium salts prepared from aniline starting materials followed by hydrolysis of the intermediate hydazide and cyclo-condensation with various 1,3-dicarbonyl equivalents to afford good yields of isolated functionalised pyrazole products.
The synthesis of the COX-2 selective NSAID was demonstrated using this approach.

Continuous flow methodologyhas been used to enhance several steps in the synthesis of a precursor to Sacubitril.
In particular, a key carboethoxyallylation benefited from a reducedprocessing time and improved reproducibility, the latter attributable toavoiding the use of a slurry as in the batch procedure. Moreover, in batchexothermic formation of the organozinc species resulted in the formation ofside products, whereas this could be avoided in flow because heat dissipationfrom a narrow packed column of zinc was more efficient
Cyclohexaneperoxycarboxylic acid (6, has been developed as a safe, inexpensive oxidant, with demonstrated utility in a Baeyer−Villiger rearrangement.34 Solutions of cyclohexanecarboxylic acid in hexane and 50% aqueous H2O2 were continuously added to 45% H2SO4 at 50−70 °C and slightly reduced pressure. The byproduct H2O was removed azeotropically, and the residence time in the reactor was 3 h. Processing was adjusted to maintain a concentration of 6 at 17−19%, below the detonable level, and the product was kept as a stable solution in hexane. These operations enhanced the safety margin in preparing 6.

Scheme . Generation of cyclohexaneperoxycarboxylic acid

The conversion of a batch process to continuous (flow) operation has been investigated. The manufacture of 4,d-erythronolactone at kilogram scale was used as an example. Fully continuousprocessing was found to be impracticable with the available plant because of the difficulty in carrying out a multiphase isolation step continuously, so hybrid batch–continuous options were explored. It was found that very little additional laboratory or process safety work other than that required for the batch process was required to develop the hybrid process. A hybrid process was chosen because of the difficulty caused by the precipitation of solid byproduct during the isolation stage. While the project was a technical success, the performance benefits of the hybrid process over the batch were not seen as commercially significant for this system.





The FDA gave Johnson & Johnson’s ($JNJ) Janssen drug unit the thumbs up last week for the continuous manufacturing process that it has been working on for 5 years. The agency approved a switchover from batch to the new technology for production of HIV drug Prezista on a line at its plant in Gurabo, Puerto Rico……http://www.fiercepharma.com/manufacturing/fda-urges-companies-to-get-on-board-continuous-manufacturing
 |
|
 |
SEE……http://www.en-cphi.cn/news/show-29367.html
Just after opening a refurbished manufacturing facility in Cape Town, South Africa earlier this year, pharma giant Johnson & Johnson ($JNJ) recently opened the doors to its Global Public Health Africa Operations office there.
The company has invested $21 million (300 million rand) in the facilities. The global public health facility will focus on HIV, tuberculosis and maternal, newborn and child health, South Africa – The Good News reported.
“This (investment) tells us that South Africa has the capability to provide a facility for world-class manufacturing,” Rob Davies, minister of the Department of Trade and Industry told the publication.
Johnson & Johnson, which has operated in South Africa for more than 86 years, planned to close the Cape Town manufacturing plant by the end of 2008 but was persuaded to keep the facility open for local manufacturing to serve sub-Saharan business. By 2015, the plant was cited by J&J as the most-improved in cost competitiveness from 30 company plants worldwide.
Earlier this month, the FDA gave J&J’s Janssen drug unit the go-ahead to proceed with the continuous manufacturing process it’s been working on for 5 years. The agency approved a switchover from batch to the new technology for production of HIV drug Prezista, Darunavir on a line at its plant in Gurabo, Puerto Rico.

AN EXAMPLE NOT RELATED TO DARUNAVIR
![]()
![]()
May 20-21, 2014 (Link to 2016 Meeting Website)
https://iscmp.mit.edu/white-papers/white-paper-4
READ
https://iscmp.mit.edu/white-papers/white-paper-1
//////
//////FDA, HIV drug, Prezista, Darunavir, Gurabo, Puerto Rico


Phosgene
Phosgene is the chemical compound with the formula COCl2. This colorless gas gained infamy as a chemical weapon during World War I. It is also a valued industrial reagent and building block in synthesis of pharmaceuticals and other organic compounds. In low concentrations, its odor resembles freshly cut hay or grass.[3] In addition to its industrial production, small amounts occur naturally from the breakdown and the combustion oforganochlorine compounds, such as those used in refrigeration systems.[4] The chemical was named by combining the Greek words ‘phos’ (meaning light) and genesis (birth); it does not mean it contains any phosphorus (cf. phosphine).
 TRIPHOSGENE
TRIPHOSGENE
Triphosgene (bis(trichloromethyl) carbonate (BTC), C3Cl6O3) is a chemical compound that is used as a safer substitute for phosgene, because at room temperature it is a solid crystal, as opposed to phosgene which is a gas.Triphosgene crystals decompose above 200 °C READ …….http://www.buss-ct.com/e/company/publications/reaction_technology/eckert_reprint_CO6_2011-hr2.pdf
This compound is commercially available. It is prepared by exhaustive free radical chlorination of dimethyl carbonate:
Triphosgene can be easily recrystallized from boiling hexanes to yield pure white crystals.
Phosgene is a planar molecule as predicted by VSEPR theory. The C=O distance is 1.18 Å, the C—Cl distance is 1.74 Å and the Cl—C—Cl angle is 111.8°.[5] It is one of the simplest acid chlorides, being formally derived from carbonic acid.
The reaction is exothermic, therefore the reactor must be cooled. Typically, the reaction is conducted between 50 and 150 °C. Above 200 °C, phosgene reverts to carbon monoxide and chlorine, Keq (300K) = 0.05. World production of this compound was estimated to be 2.74 million tonnes in 1989.[4]
Because of safety issues, phosgene is often produced and consumed within the same plant, and extraordinary measures are made to contain this toxic gas. It is listed on schedule 3 of the Chemical Weapons Convention: All production sites manufacturing more than 30 tonnes per year must be declared to the OPCW.[6] Although less dangerous than many other chemical weapons, such as sarin, phosgene is still regarded as a viablechemical warfare agent because it is so easy to manufacture when compared to the production requirements of more technically advanced chemical weapons such as the first-generation nerve agent tabun.[7]

Upon ultraviolet (UV) radiation in the presence of oxygen, chloroform slowly converts into phosgene by a radical reaction. To suppress thisphotodegradation, chloroform is often stored in brown-tinted glass containers. Chlorinated compounds used to remove oil from metals, such as automotive brake cleaners, are converted to phosgene by the UV rays of arc welding processes.[8]
Phosgene may also be produced during testing for leaks of older-style refrigerant gases. Chloromethanes (R12, R22 and others) were formerly leak-tested in situ by employing a small gas torch (propane, butane or propylene gas) with a sniffer tube and a copper reaction plate in the flame nozzle of the torch. If any refrigerant gas was leaking from a pipe or joint, the gas would be sucked into the flame via the sniffer tube and would cause a colour change of the gas flame to a bright greenish blue. In the process, phosgene gas would be created due to the thermal reaction. No valid statistics are available, but anecdotal reports suggest that numerous refrigeration technicians suffered the effects of phosgene poisoning due to their ignorance of the toxicity of phosgene, produced during such leak testing.[citation needed] Electronic sensing of refrigerant gases phased out the use of flame testing for leaks in the 1980s. Similarly, phosgene poisoning is a consideration for people fighting fires that are occurring in the vicinity of freon refrigeration equipment, smoking in the vicinity of a freon leak, or fighting fires using halon or halotron.
The great majority of phosgene is used in the production of isocyanates, the most important being toluene diisocyanate (TDI) and methylene diphenyl diisocyanate (MDI). These two isocyanates are precursors to polyurethanes.
Significant amounts are also used in the production of polycarbonates by its reaction with bisphenol A.[4] Polycarbonates are an important class of engineering thermoplastic found, for example, in lenses in eye glasses. Diols react with phosgene to give either linear or cyclic carbonates (R = H, alkyl, aryl):
The synthesis of isocyanates from amines illustrates the electrophilic character of this reagent and its use in introducing the equivalent of “CO2+“:[9]
Such reactions are conducted in the presence of a base such as pyridine that absorbs the hydrogen chloride.
In the research laboratory phosgene still finds limited use in organic synthesis. A variety of substitutes have been developed, notably trichloromethyl chloroformate (“diphosgene“), a liquid at room temperature, and bis(trichloromethyl) carbonate (“triphosgene“), a crystalline substance.[10] Aside from the above reactions that are widely practiced industrially, phosgene is also used to produceacid chlorides and carbon dioxide from carboxylic acids:
Such acid chlorides react with amines and alcohols to give, respectively, amides and esters, which are commonly used intermediates. Thionyl chloride is more commonly and more safely employed for this application. A specific application for phosgene is the production of chloroformic esters:
Analogously, with ammonia, one obtains urea:
Halide exchange with nitrogen trifluoride and aluminium tribromide gives COF2 and COBr2, respectively.[4]
Phosgene was synthesized by the British chemist John Davy (1790–1868) in 1812 by exposing a mixture of carbon monoxide and chlorine to sunlight. He named it “phosgene” in reference of the use of light to promote the reaction; from Greek, phos (light) and gene (born).[11] It gradually became important in the chemical industry as the 19th century progressed, particularly in dye manufacturing.
Following the extensive use of phosgene gas in combat during World War I, it was stockpiled by various countries as part of their secret chemical weapons programs.[12][13][14]
In May 1928, eleven tons of phosgene escaped from a war surplus store in central Hamburg.[15] 300 people were poisoned of whom 10 died.[15]
.

US Army phosgene identification poster from World War II
Phosgene was then only frequently used by the Imperial Japanese Army against the Chinese during the Second Sino-Japanese War.[16] Gas weapons, such as phosgene, were produced by Unit 731 and authorized by specific orders given by Hirohito (Emperor Showa) himself, transmitted by the chief of staff of the army. For example, the Emperor authorized the use of toxic gas on 375 separate occasions during the battle of Wuhan from August to October 1938.[17]
Phosgene is an insidious poison as the odor may not be noticed and symptoms may be slow to appear.[18] The odor detection threshold for phosgene is 0.4 ppm, four times the threshold limit value. Its high toxicity arises from the action of the phosgene on the proteins in the pulmonary alveoli, the site of gas exchange: their damage disrupts the blood-air barrier, causing suffocation. It reacts with the amines of the proteins, causing crosslinking by formation of urea-like linkages, in accord with the reactions discussed above. Phosgene detection badges are worn by those at risk of exposure.[4]
Sodium bicarbonate may be used to neutralise liquid spills of phosgene. Gaseous spills may be mitigated with ammonia.[19]
.
TRIPHOSGENE HANDLING
.

Left, reaction vessel with amino acid and triphosgene dissolved in THF; middle, appearance of the reaction mixture after addition of 2,4,6-collidine; and right, appearance of the reaction mixture after microwave irradiation.

Typical glassware standard equipment for the safety phosgenation with phosgene supply from triphosgene: (A) phosgene generator (V = 1 L, T = 85 °C) loaded with 600 g of triphosgene; (B) refluxer (water cooled, T = 15 °C); (C) phosgene line (Viton hose); (D) phosgenation reactor (V = 10 L, T = 110 °C); (E) refluxer (cryostat cooled, T = −30 °C); (F) off-gas line (Viton hose) from the top of the refluxer (E); (G) cooling trap (dry ice cooled, T = −60 °C); (H) off-gas line; (I) cryostat. The assembly of the equipment is somewhat reduced to effect more clarity of the ensemble.
.

.
Phosgene is quantitatively formed from solid triphosgene in a solvent-free and safe process without any reaction heat, catalyzed by planar N-heterocycles with deactivated imino functions.
The rate of phosgene generation is adjustable to the rate of phosgene consumption in the subsequent phosgenation reaction by thermal control, catalyst concentration, and in some cases, specific properties of selected metal phthalocyanines. A thermal runaway reaction of this process is impossible.
.
Use a safer process for generating phosgene.

Decomposition of triphosgene (1a) into carbon tetrachloride, carbon dioxide, and 1 equiv of phosgene (3)
Phosgene (COCl2) is useful in organic synthesis for chlorination, chlorocarbonylation, carbonylation, and dehydration; but its high toxicity discourages its use. Until now, the best substitute for COCl2 has been triphosgene [(CCl3O)2CO], a stable solid that has low vapor pressure. Although (CCl3O)2CO can be used in phosgenation reactions, removing the unreacted reagent from reaction mixtures is difficult because of its high boiling point. In contrast, COCl2 is easily removed by evaporating it.
(CCl3O)2CO reacts with silica gel, metal salts, or Lewis acids to generate 1 equiv of phosgene by an electrocyclic reaction. H. Eckert* and J. Auerweck at the University of Technology, Munich (Germany) report that pyridine and phthalocyanine derivatives catalyze the decomposition of (CCl3O)2CO to generate 3 equiv of COCl2.
The catalysts, phenanthridine , poly(2-vinylpyridine) , and phthalocyanines , convert liquid (CCl3O)2CO to the desired COCl2. The size and structure of the catalysts allow (CCl3O)2CO to react by the mechanism shown. The reaction was run at the 100-g scale to generate 22 L of gaseous COCl2 with an oil bath or an IR heater as the heat source. Because the catalysts are not soluble in (CCl3O)2CO, the process is considered to be heterogeneous catalysis.
.

Controlled transformation of triphosgene (1) into 3 equiv of phosgene (3) catalyzed by 4
Compounds 1 and 4a−4 h are commercially available products from Sigma-Aldrich, with the following purities: 1, 98% (IR νC═O 1820 cm−1, 13C NMR δ 108.0, 140.9); 4a, 98%; 4c, n.a.; 4d, 99%;4e, 97%; 4f, 97%; 4g, 90%; 4h, 85%.
Because the reaction is controlled by temperature, turning off the heat source causes the liquid (CCl3O)2CO to crystallize and stops the reaction, making the process safe. The reaction can be used to generate COCl2 externally or to produce it in situ. According to the authors, this method fulfills the goal of “safety phosgenation on demand of consumer”.
ORGANIC PROCESS RESEARCH AND DEVELOPMENT
A FRET approach towards potential detection of phosgene is presented, which is based on a selective chemical reaction between phosgene (or triphosgene as a simulant) and donor and acceptor fluorophores.

FRET has been applied in an experimental method for the detection of phosgene. In it, phosgene or rather triphosgene as a safe substitute serves as a linker between an acceptor and a donor coumarine (forming urea groups).[3] The presence of phosgene is detected at 5×10-5M with a typical FRET emission at 464 nm.


EXAMPLES OF USE OF TRIPHOSGENE
Chlorination of Aliphatic Primary Alcohols via Triphosgene-Triethylamine Activation
Caitlan E. Ayala, Andres Villalpando, Alex L. Nguyen, Gregory T. McCandless and Rendy Kartika*
*Department of Chemistry, 232 Choppin Hall, Louisiana State University, Baton Rouge, Louisiana 70803, United States, Email: rkartika lsu.edu
lsu.edu
C. E. Ayala, A. Villalpando, A. L. Nguyen, G. T. McCandless, R. Kartika, Org. Lett., 2012, 14, 3676-3679.
DOI: 10.1021/ol301520d (free Supporting Information)

Activation of primary aliphatic alcohols with triphosgene and triethylamine mixtures afforded either alkyl chloride or diethylcarbamate products, and the switch in selectivity appeared to be driven by sterics. The reaction conditions to achieve this highly useful transformation were unexceptionally mild and readily tolerated by a wide range of sensitive functionalities.

…………………………
.

.
ABACAVIR SULPHATE

VESTIPITANT
The following synthetic route was reported by Giuseppe Guercio et al from GlaxoSmithKline:
The initial chemical development synthetic route, derived from the one used by medicinal chemistry, involved several hazardous reagents, gave low yields and produced high levels of waste. Through a targeted process of research and development, application of novel techniques and extensive route scouting, a new synthetic route for GW597599 was developed. This paper reports the optimisation work of the third and last stage in the chemical synthesis of GW597599 and the development of a pilot-plant-suitable process for the manufacturing of optically pure arylpiperazine derivative 1. In particular, the process eliminated the use of triphosgene in the synthesis of an intermediate carbamoyl chloride, substantially enhancing safety, overall yield, and throughput.

source:
Org. Process Res. Dev., 2009, 13 (6), pp 1100–1110.
Org. Process Res. Dev., 2009, 13 (3), pp 489–493.
Org. Process Res. Dev., 2008, 12 (6), pp 1188–1194.
.
TIVOZANIB

VIAGRA

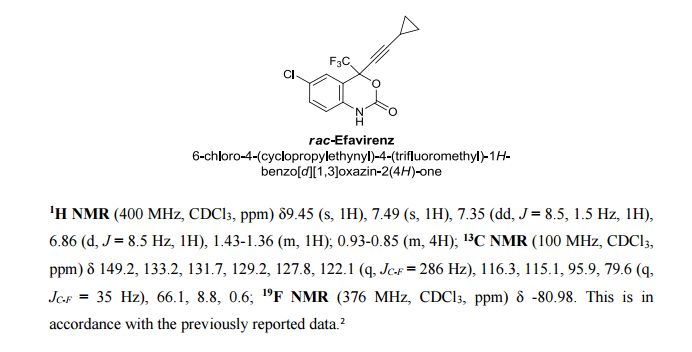
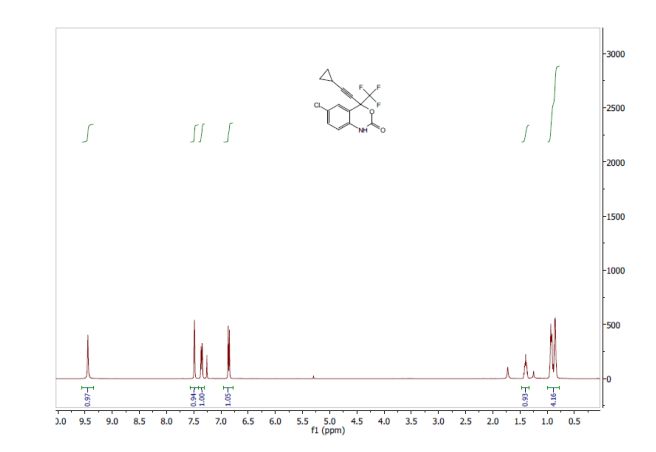
EFAVIRENZ ………EP2454244A1


Enantiomerically pure hydantoins are prepared from optically pure α-amino amides utilizing triphosgene. A mechanism for the racemization observed with 1,1′-carbonyldiimidazole (CDI) for this type of reaction is proposed.
D. Zhang, X. Xing, G. D. Cuny, J. Org. Chem., 2006, 71, 1750-1753.

(a) Cotarca, L. and Eckert, H. Phosgenations − A Handbook; Wiley-VCH: Weinheim, 2003.
(b) Cotarca, L. and Eckert, H. Phosgenations − A Handbook; Wiley-VCH:Weinheim, 2003; pp 20− 21.
(c) Cotarca, L. and Eckert, H. Phosgenations − A Handbook;Wiley-VCH: Weinheim, 2003; pp 44− 520.
(e) Cotarca, L. and Eckert, H.Phosgenations − A Handbook; Wiley-VCH: Weinheim, 2003; pp 14− 16, 613− 615.
Recent online information: www.ch.tum.de/oc1/HEckert/research.htm.
(b) Pasquato, L.; Modena, G.; Cotarca, L.; Delogu, P.; Mantovani, S. J. Org. Chem. 2000, 65,8224– 8228
(e) Nielsen, D. H.; Burke, T. G.; Woltz, P. J. H.; Jones, E. A. J. Chem. Phys. 1952, 20, 596– 604
(f) Gordon, E. P.;Enakaeva, V. G.; Korotchenko, A. V.; Mitrokhin, A. M. Russian Patent RU 2299852, 2007.
(a) Eckert, H.; Forster, B. Angew. Chem. 1987, 99, 922– 923 ; Angew. Chem., Int. Ed.,1987, 26, 894–895
(d) Triphosgene; Ubichem: U.K., 1999; CD-ROM.
Eckert, H.; Dirsch, N.; Gruber, B. (former Dr. Eckert GmbH, now Buss Chem Tech AG) German Offen. DE 19740577, 1999 (Sep. 15, 1997), Chem. Abstr. 1999, 130, 211406.;
WO 9914159, 1999; Eur. Pat. EP 1017623, 2002; U.S. Patent US 6399822, 2002; Japanese Patent JP 2001516692, 2001.
Mole percent 4 referring to 3 phosgene equivalents of 1 .
(b) Lever, A. B. P. Adv. Inorg. Chem. Radiochem. 1965, 7, 28– 114
(c) Ebert, N. A.; Gottlich, H. B. J. Am. Chem. Soc. 1952, 74, 2806
 ], [CAS]
], [CAS]The weighing error of this procedure mainly comes from icy condensed humidity at the cool glassware of the cooling trap and is less than 0.5 g, determined by a series of weighings under the same conditions, the same equipment, temperature (T = −78 °C), and handling time <10 s, but without 3. Under these conditions evaporation of 3 (bp 8 °C) hardly ever happens and can be ignored.
Monitox plus gas monitor (COCl2) and phosgene badges from Compurhttp://www.compur.com/gasmessgeraete/front_content.php?idcat=7&changelang=3.

chief executive and vice chairman Arun Kumar –Strides arcolab

Headquartered in India, Strides Arcolab is a pharmaceutical company with a key focus on the development and manufacture of IP-led niche products, particularly sterile injectables. It is among the world’s largest manufacturers of soft gelatin capsules. With 14 world-class manufacturing facilities, an innovative R&D hub and a marketing network in 70 countries, Strides is well positioned to meet the demands of the global pharmaceutical industry and has partnered with several of the world’s leading pharmaceutical companies.
2 August 2013
Strides Arcolab’s HIV drug, which is generic version of Gilead Sciences’ Truvada, gets tentative approval from the US FDA.
Tentative approval implies that the drug has met all standards but cannot be marketed in the US due to existing patent protections Good news for HIV AIDS patients
FDA has granted tentative approval to India-based Strides Arcolab’s HIV drug emtricitabine and tenofovir disoproxil fumarate tablets, 200 mg/300 mg. The drug is a generic version of Truvada, 200 mg/300 mg tablets, which is manufactured by Gilead Sciences.


The product is indicated for use in combination with other antiretroviral agents for the treatment of human immunodeficiency virus (HIV)-1


………………
………….
…………..
EMTRICITABINE

credit-chemdrug




