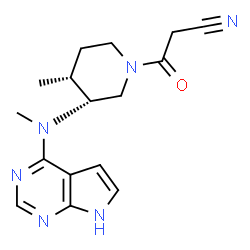

Tofacitinib Citrate, 的合成
托法替布, トファシチニブクエン酸塩, Тофацитиниба Цитрат
3-{(3R,4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile citrate salt
CAS : 540737-29-9
ROTATION +
Tofacitinib; Tasocitinib;
477600-75-2 base ; CP-690550;
3-((3R,4R)-4-methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile;
3-{(3R,4R)-4-methyl-3-rmethyl-(7H-pyrrolor2,3-dlpyrimidin-4-yl)-amino1- piperidin-1-yl}-3-oxo-propionitrile mono citrate salt
CP 690550 Tofacitinib; CP-690550; CP-690550-10; Xeljanz; Jakvinus; Tofacitinib citrate
Trademarks: Xeljanz; Jakvinus
MF: C16H20N6O
CAS : 477600-75-2 BASE ; 540737-29-9(citrate) 3-[(3R,4R)-4-methyl-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]piperidin-1-yl]-3-oxopropanenitrile
Molecular Weight: 312.369
SMILES: C[C@@H]1CCN(C[C@@H]1N(C)C2=NC=NC3=C2C=CN3)C(=O)CC#N
Activity: Treatment of Rheumatoid Arthritis; RA Treatment, JAK Inhibitor; Protein Kinase Inhibitor; JAK3 Inhibitor; Janus Kinase 3 Inhibitor; JAK-STAT Signaling Pathway; JAK1 Kinase Inhibitor; Selective Immunosuppressants
Status: Launched 2012
Originator: Pfizer

Pfizer Inc’s oral JAK inhibitor tofacitinib was approved on November 6, 2012 by US FDA for the treatment of rheumatoid arthritis.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Tofacitinib (trade names Xeljanz and Jakvinus, formerly tasocitinib,[1] CP-690550[2]) is a drug of the janus kinase (JAK) inhibitor class, discovered and developed by Pfizer. It is currently approved for the treatment of rheumatoid arthritis (RA) in the United States,Russia, Japan and many other countries, is being studied for treatment of psoriasis, inflammatory bowel disease, and other immunological diseases, as well as for the prevention of organ transplant rejection. 


Yogesh S. Patil, Nilesh L. Bonde, Ankush S. Kekan, Dhananjay G. Sathe*1, and Arijit Das
Publication Date (Web): November 17, 2014 (Article)
DOI: 10.1021/op500274j
MS m/z 313 (M+ + 1);
mp 201–202 °C;
|
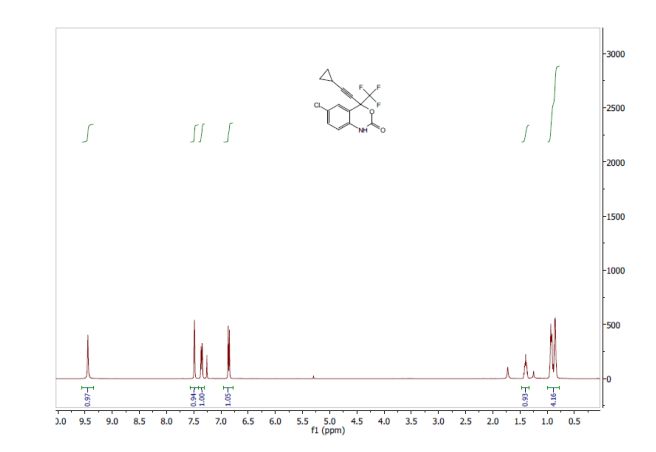
1H NMR (CDCl3) δ 8.34 (s, 1H), δ 7.38 (d, 1H, J = 2.4 Hz), δ 6.93 (d, 1H, J = 2.4 Hz), δ 4.97 (m, 1H), δ 3.93–4.03 (m, 4H), δ 3.66 (m, 1H), δ 3.50 (m, 4H), δ 2.91 (d, 2H, J = 15.6 Hz), δ 2.80 (t, 2H, J = 12.8 Hz), δ 2.55 (m, 1H), δ 1.99 (m, 1H), δ 1.77 (m, 1H), δ 1.13–1.18 (m, 3H).
|
TEAMWORK
Part of the Pfizer group responsible for Xeljanz: Front row, from left: Sally Gut Ruggeri, Chakrapani Subramanyam, Eileen Elliott Mueller, and Frank Busch. Second row, from left: Matthew Brown, Mark Flanagan, and Robert Dugger. Back row, from left: Elizabeth Kudlacz and Douglas Ball.
Credit: Pfizer
Mark Flanagan, who was on the team at Pfizer that discovered Xeljanz, (tofacitinib citrate), an oral treatment for rheumatoid arthritis, remembers testing the drug in a rat model and seeing the drug decrease the level of inflammation in the rats’ footpads. “What we look for is physical measurements of the size of the joint. In the control animals, there was quite a bit of inflammation in the joints, whereas animals treated with different doses of the drug showed a dose-dependent decrease in the size of the joint. “Tofacitinib showed robust efficacy in the first such study run. I can remember the excitement that this data generated on the team,” he says.
Tofacitinib, chemically known as (3R,4R)-4-methyl-3-(methyl-7H-pyrrolo [2,3- d]pyrimidin-4-ylamino)-B-oxo-l -piperidinepi panenitrile, is represented Formula I. Tofacitinib citrate, a janus kinase inhibitor, is approved as XELJANZ® tablets for treatment .of rheumatoid arthritis.
Various intermediates and processes for preparation of tofacitinib are disclosed in patents like US7301 023 and US8232394.

Formula I or isomers or a mixture of isomers thereof by following any method provided in the prior art, for example, by following Example 14 of U.S. Patent No. RE41,783 or by following Example 6 of U.S. Patent No. 7,301,023. Tofacitinib of Formula I or isomers of tofacitinib or a mixture of isomers thereof may be converted into a salt by following any method provided in the prior art, for example, by following Example 1 of U.S. Patent No. 6,965,027 or by following Example 1 or Example 8 of PCT Publication No. WO 2012/135338. The potential significance of JAK3 inhibition was first discovered in the laboratory of John O’Shea, an immunologist at the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH).[5] In 1994, Pfizer was approached by the NIH to form a public-private partnership in order to evaluate and bring to market experimental compounds based on this research.[5] Pfizer initially declined the partnership but agreed in 1996, after the elimination of an NIH policy dictating that the market price of a product resulting from such a partnership would need to be commensurate with the investment of public taxpayer revenue and the “health and safety needs of the public.”[5] The drug discovery, preclinical development, and clinical development of tofacitinib took place exclusively at Pfizer.[6] In November 2012, the U.S. Food and Drug Administration (FDA) approved tofacitinib for treatment of rheumatoid arthritis. Once on the market, rheumatologists complained that the $2,055 a month wholesale price was too expensive, though the price is 7% less than related treatments.[6] A 2014 study showed that tofacitinib treatment was able to convert white fat tissues into more metabolically active brown fat, suggesting it may have potential applications in the treatment of obesity.[7] It is an inhibitor of the enzyme janus kinase 1 (JAK1) and janus kinase 3 (JAK 3) , which means that it interferes with the JAK-STAT signaling pathway, which transmits extracellular information into the cell nucleus, influencing DNA transcription.[3] Recently it has been shown in a murine model of established arthritis that tofacitinib rapidly improved disease by inhibiting the production of inflammatory mediators and suppressing STAT1-dependent genes in joint tissue. This efficacy in this disease model correlated with the inhibition of both JAK1 and 3 signaling pathways, suggesting that tofacitinib may exert therapeutic benefit via pathways that are not exclusive to inhibition of JAK3.[4]
Preparation of 3-{(3R,4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile citrate salt (Tofacitinib citrate, Xeljanz, CP-690550-10)
To a round-bottomed flask fitted with a temperature probe, condenser, nitrogen source, and heating mantle, methyl-[(3R,4R)-4-methyl-piperidin-3-yl]-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (5.0 g, 20.4 mmol) was added followed by 1-butanol (15 mL), ethyl cyanoacetate (4.6 g, 40.8 mmol), and DBU (1.6 g, 10.2 mmol). The resulting amber solution was stirred at 40 °C for 20 h. Upon reaction completion, citric acid monohydrate (8.57 g, 40.8 mmol) was added followed by water (7.5 mL) and 1-butanol (39.5 mL). The mixture was heated to 81 °C and held at that temperature for 30 min. The mixture was then cooled slowly to 22 ºC and stirred for 2 h. The slurry was filtered and washed with 1-butanol (20 mL). The filter cake was dried in a vacuum oven at 80 °C to afford 9.6 g (93%) of tofacitinib citrate as an off-white solid.
1H NMR (500 MHz, d6-DMSO): δ 8.14 (s, 1H), 7.11 (d, J=3.6 Hz, 1H), 6.57 (d, J=3.6 Hz, 1H), 4.96 (q, J=6.0 Hz, 1H), 4.00-3.90 (m, 2H), 3.80 (m, 2H), 3.51 (m, 1H), 3.32 (s, 3H), 2.80 (Abq, J=15.6 Hz, 2H), 2.71 (Abq, J=15.6 Hz, 2H), 2.52-2.50 (m, 1H), 2.45-2.41 (m, 1H), 1.81 (m, 1H), 1.69-1.65 (m, 1H), 1.04 (d, J=6.9 Hz, 3H).
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
PAPER
3-((3R,4R)-4-Methyl-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)piperidin-1-yl)-3-oxopropanenitrile (1) Monocitrate
J. Med. Chem., 2010, 53 (24), pp 8468–8484
DOI: 10.1021/jm1004286
1monocitrate as a white crystalline solid (mp = 201 dec).
LRMS: m/z 313.2 (MH+).
1H NMR (400 MHz) (D2O) δ HOD: 0.92 (2 H, d, J = 7.2 Hz), 0.96 (1 H, d, J = 7.6 Hz), 1.66 (1 H, m), 1.80 (1 H, m), 2.37 (1 H, m), 2.58 (2 H, 1/2 ABq, J = 15.4 Hz), 2.70 (2 H, 1/2 ABq, J = 15.4 Hz), 3.23 (2 H, s), 3.25 (1 H, s), 3.33 (1 H, m), 3.46 (1 H, m), 3.81 (4 H, m), 4.55 (1 H, m), 6.65 (1 H, d, J = 3.2 Hz), 7.20 (1 H, t, J = 3.2 Hz), 8.09 (1 H, m).
Anal. Calcd for C22H28N6O8: C, 52.38; H, 5.59; N, 16.66. Found: C, 52.32; H, 5.83; N, 16.30. For additional characterization of the monocitrate salt of 1 see WO 03/048162.
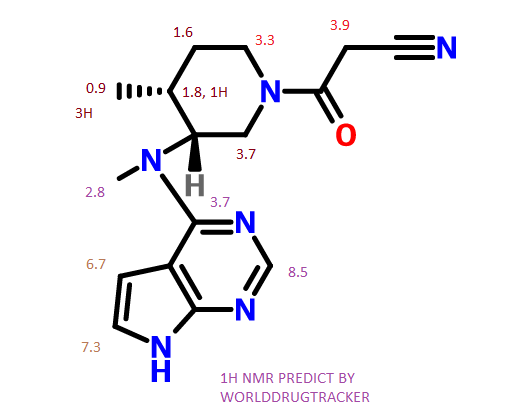
NMR PREDICT
References:
Weiling Cai, James L. Colony,Heather Frost, James P. Hudspeth, Peter M. Kendall, Ashwin M. Krishnan,Teresa Makowski, Duane J. Mazur, James Phillips, David H. Brown Ripin, Sally Gut Ruggeri, Jay F. Stearns, and Timothy D. White; Investigation of Practical Routes for the Kilogram-Scale Production of cis-3-Methylamino-4-methylpiperidines; Organic Process Research & Development 2005, 9, 51−56
Ripin, D. H.B.; 3-amino-piperidine derivatives and methods of manufacture, US patent application publication, US 2004/0102627 A1
Ruggeri, Sally, Gut;Hawkins, Joel, Michael; Makowski, Teresa, Margaret; Rutherford, Jennifer, Lea; Urban,Frank,John;Pyrrolo[2,3-d]pyrimidine derivatives: their intermediates and synthesis, PCT pub. No. WO 2007/012953 A 2, US20120259115 A1, United States Patent US8232393. Patent Issue Date: July 31, 2012
Kristin E. Price, Claude Larrive´e-Aboussafy, Brett M. Lillie, Robert W. McLaughlin, Jason Mustakis, Kevin W. Hettenbach, Joel M. Hawkins, and Rajappa Vaidyanathan; Mild and Efficient DBU-Catalyzed Amidation of Cyanoacetates, Organic Letters, 2009, vol.11, No.9, 2003-2006
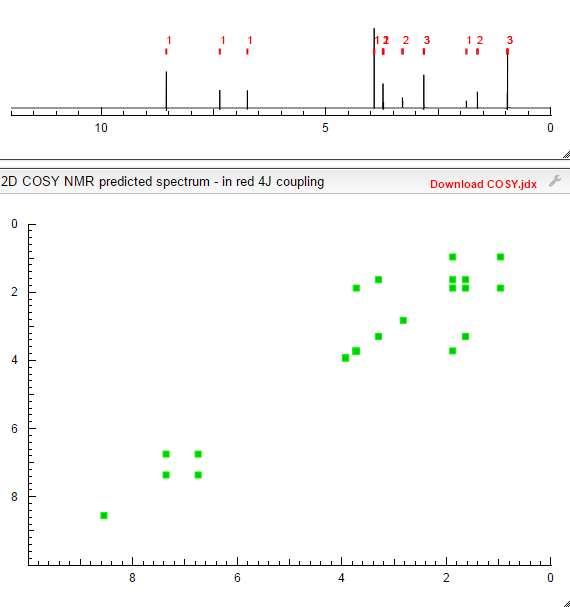
MORE NMR PREDICT

Tofacitinib 

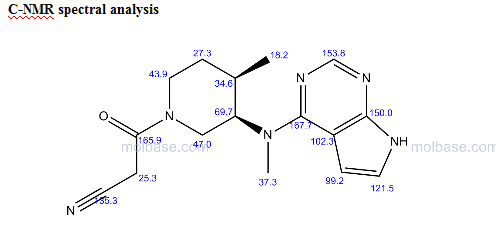
13C NMR PREDICT 
SEE…….https://newdrugapprovals.org/2015/07/24/tofacitinib-%E7%9A%84%E5%90%88%E6%88%90-spectral-visit/
COSY PREDICT  सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
SEE………http://orgspectroscopyint.blogspot.in/2014/12/tofacitinib-citrate.html
NMR PICTURE FROM THE NET

PAPER
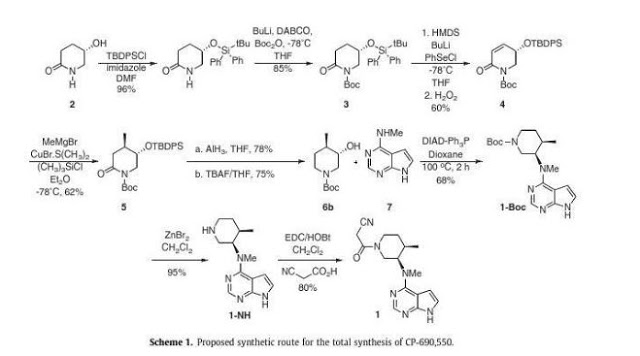
Asymmetric total synthesis of Tofacitinib
- a Laboratory of Asymmetric Synthesis, Chemistry Institute of Natural Resources, University of Talca, P.O. Box 747, Talca, Chile
- b Laboratory of Natural Products, Department of Chemistry, University of Antofagasta, P.O. Box 170, Antofagasta, Chile
http://dx.doi.org/10.1016/j.tetlet.2013.07.042
Abstract
A novel stereoselective synthesis of Tofacitinib (CP-690,550), a Janus tyrosine kinase (JAK3) specific inhibitor, has been achieved starting from (5S)-5-hydroxypiperidin-2-one in 10 steps from 2 with a 9.5% overall yield. The potentiality of this synthetic route is the obtention of tert-butyl-(3S,4R)-3-hydroxy-4-methylpiperidine-1-carboxylate (6b) as a new chiral precursor involved in the synthesis of CP690,550, in a three-step reaction, without epimerizations, rather than the 5 or more steps used in described reactions to achieve this compound from analogues of 6b.
…………………. Tofacitinib synthesis: US2001053782A1
Tofacitinib synthesis: WO2002096909A1
Tofacitinib synthesis: Org Process Res Dev 2014, 18(12), 1714-1720 (also from a chinese publication, same procedure just slight changes in reagents/conditions)
References:
1. Blumenkopf, T. A.; et. al. Pyrrolo[2,3-d]pyrimidine compounds. US2001053782A1
2. Flanagan, M. E.; et. al. Optical resolution of (1-benzyl-4-methylpiperidin-3-yl) -methylamine and the use thereof for the preparation of pyrrolo 2,3-pyrimidine derivatives as protein kinases inhibitors. WO2002096909A1
3. Das, A.; et. al. An Improved and Efficient Process for the Preparation of Tofacitinib Citrate. Org Process Res Dev2014, 18(12), 1714-1720.
Synthesis of Tofacitinib Citrate
Author(s): ZHAO Fanglu,
,CHEN Guohua,
,YAO Shilun,
,ZHANG Zhenxia Journal: Chinese Journal of Pharmaceuticals, Year 2014 , Issue 3 , Page 201-204,253 Keyword: tofacitinib citrate%;
anti-rheumatoid arthritis drug%;
synthesis%;
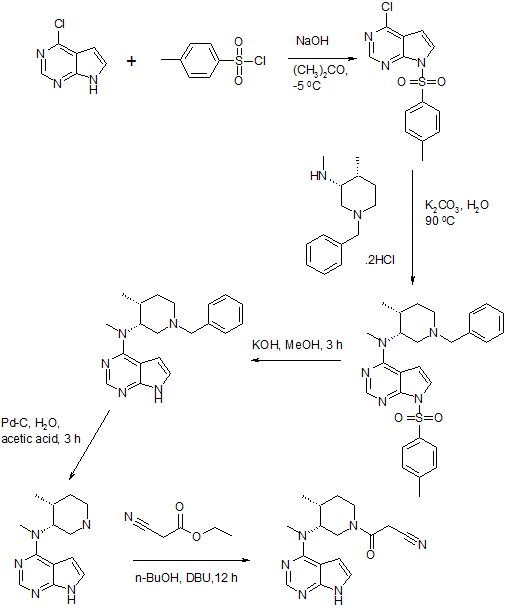
PATENT https://www.google.co.in/patents/WO2003048162A1?cl=en The crystalline form of the compound of this invention 3-{4-methyl-3-[methyl- (7H-pyrrolot2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile mono citrate salt is prepared as described below. Scheme 1
Scheme 2




Example 1 3-{(3R,4R)-4-methyl-3-rmethyl-(7H-pyrrolor2,3-dlpyrimidin-4-yl)-amino1- piperidin-1-yl}-3-oxo-propionitrile mono citrate salt Ethanol (13 liters), (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H-pyrrolo[2,3- d]pyrimidin-4-yl)-amine (1.3 kg), cyano-acetic acid 2,5-dioxo-pyrrolidin-1-yl ester (1.5 kg), and triethylamine (1.5 liters) were combined and stirred at ambient temperature. Upon reaction completion (determined by High Pressure Liquid Chromotography (HPLC) analysis, approximately 30 minutes), the solution was filtered, concentrated and azeotroped with 15 liters of methylene chloride. The reaction mixture was washed sequentially with 12 liters of 0.5 N sodium hydroxide solution, 12 liters of brine and 12 liters of water. The organic layer was concentrated and azeotroped with 3 liters of acetone (final pot temperature was 42°C). The resulting solution was cooled to 20°C to 25°C followed by addition of 10 liters of acetone. This solution was filtered and then aqueous citric acid (0.8 kg in 4 liters of water) added via in-line filter. The reaction mixture was allowed to granulate. The slurry was cooled before collecting the solids by filtration. The solids were dried to yield 1.9 kg (71 %) (3R, 4R)- 3-{4-Methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile mono citrate. This material was then combined with 15 liters of a 1:1 ratio of ethanol/water and the slurry was agitated overnight. The solids were filtered and dried to afford 1.7 kg (63% from (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine) of the title compound as a white crystalline solid. 1H NMR (400 MH2)(D20) δ HOD: 0.92 (2H, d, J = 7.2 Hz), 0.96 (1H, d, J = 7.6 Hz), 1.66 (1H, m), 1.80 (1H, m), 2.37 (1H, m), 2.58 (2H, 1/2 ABq, J = 15.4 Hz), 2.70 (2H, 3 ABq, J = 154 Hz), 3.23 (2H, s), 3.25 (1H, s), 3.33 (1H, m), 3.46 (1H, m), 3.81 (4H, m), 4.55 (1 H, m), 6.65 (1 H, d, J = 3.2 Hz), 7.20 (1 H, t, J = 3.2 Hz), 8.09 (1 H, m).
Patent
http://www.google.co.in/patents/EP1913000A2?cl=en Example 10 Preparation of methyl-[(3R, 4R)-4-methyl-piperidin-3-yl]-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine:
KEY INTERMEDIATE
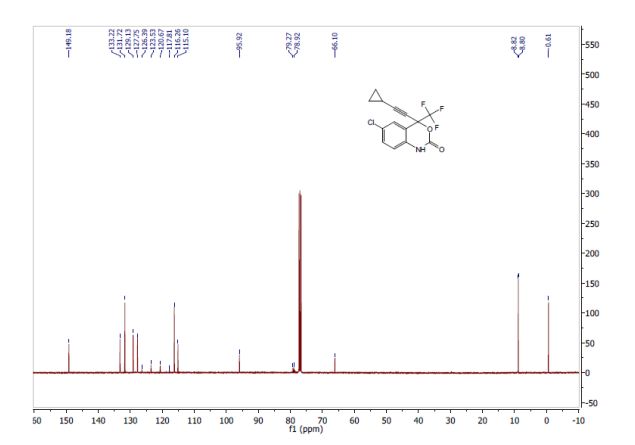
To a clean, dry, nitrogen-purged 2 L hydrogenation reactor were charged 20 wt% Pd(OH)2/C (24.0 g, 50% water wet), water (160 ml), isopropanol (640 ml), (1-benzyl-4-methyl-piperidin-3-yI)-methyi- (7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (160.0 g, 0.48 mol), and acetic acid (28.65 g, 0.48 mol). The reactor was purged with three times at 50 psi with nitrogen and three times at 50 psi with hydrogen. Once purging was complete, the reactor was heated to 45-55°C and pressurized to 50 psi with hydrogen through a continuous feed. The hydrogen uptake was monitored until no hydrogen was consumed for 1 hour. The reactor was cooled to 20-300C and purged three times at 50 psi with nitrogen. The reaction mixture was filtered through wet Celite and the filtrate was sent to a clean, dry, nitrogen-purged vessel. A solution of sodium hydroxide (39.33 g) in water (290 ml) was charged and the mixture was stirred for a minimum of 1 hour then heated to 75-900C. The isopropanol was removed by distillation. The reaction mixture was cooled to 20-30°C and 2-methyltetrahydrofuran (1.6 L) was added. The aqueous layer was drained off and the 2-methyltetrahydrofuran was displaced with toluene (1.6 L). The distillation was continued until the final volume was 800 ml. The slurry was cooled to 20-30°C and held for a minimum of 7 hours. The resulting solids were isolated by filtration and washed with toluene (480 ml). After drying under vacuum between 40-50DC for a minimum of 24 hours with a slight nitrogen bleed 102.3 g (87.3%) of the title compound were isolated. Mp 158.6-159.8°C. 1H NMR (400 MHz, CDCI3): δ 11.38 (bs, 1H), 8.30 (s, 1H), 7.05 (d, J=3.5 Hz, 1H), 6.54 (d, J=3.5 Hz, 1H), 4.89-4.87 (m, 1H), 3.39 (s, 3H), 3.27 (dd, J=12.0, 9.3 Hz, 1 H), 3.04 (dd, J=12.0, 3.9 Hz, 1H), 2.94 (td, J=12.6, 3.1 Hz, 1H0, 2.84 (dt, J=12.6, 4.3 Hz, 1H), 2.51-2.48 (m, 1H), 2.12 (bs, 2H), 1.89 (ddt, J=13.7, 10.6, 4 Hz, 1 H), 1.62 (dq, J=13.7, 4Hz, 1 H), 1.07 (d, J=7.3 Hz, 3H). 13C NMR (400 MHz, CDCI3): δ 157.9, 152.0, 151.0, 120.0, 103.0, 102.5, 56.3, 46.2, 42.4, 34.7, 33.4, 32.4, 14.3.  KEY INT
KEY INT
Example 11 Preparation of 3-{(3R, 4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3- oxo-propionitrile….TOFACITINIB BASE
To a clean, dry, nitrogen-purged 1.0 L reactor were charged methyl-(4-methyl-piperidin-3-yI)-(7H- pyrroIo[2,3-d]pyrimidin-4-yl)-amine (32.0 g, 0.130 mol), toluene (160 ml), ethyl cyanoacetate (88.53 g, 0.783 mol) and triethyl amine (26.4 g, 0.261 mol). The reaction was heated to 1000C and held for 24 hours. The reaction was washed with water (160 ml). The organic layer concentrated to a volume of 10 ml and water (20 ml) was added. The residual toluene was removed by distillation and the mixture was cooled to room temperature. Acetone (224 ml) was added followed by citric acid (27.57 g, 0.144 mol) in water (76 ml). The resulting slurry was stirred for 7 hours. The solids were isolate by filtration, washed with acetone (96 ml), and dried under vacuum to afford 42.85 g (65.3%) of the title compound. Example 13 Preparation of 3-{(3R, 4R)~4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile citrate salt:…………..TOFACITINIB CITRATE To a clean, dry, nitrogen-purged 500 ml reactor were charged methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine (25.0 g, 0.102 mol) and methylene chloride (250 ml). The mixture was stirred at room temperature for a minimum of 2.5 hours. To a clean, dry, nitrogen-purged 1 L reactor were charged cyanoacetic acid (18.2 g, 0.214 mol), methylene chloride (375 ml), and triethyl amine (30.1 ml, 0.214 mol). The mixture was cooled to -15.0— 5.00C over one hour and trimethylacetyl chloride (25.6 ml, 0.204 mol) was added at a rate to maintain the temperature below O0C. The reaction was held for a minimum of 2.5 hours, then the solution of the amine was added at a rate that maintained the temperature below O0C. After stirring for 1 hour, the mixture was warmed to room temperature and 1 M sodium hydroxide (125 ml) was added. The organic layer was washed with water (125 ml) The methylene chloride solution.was displaced with acetone until a volume of 500 ml and a temperature of 55-650C had been achieved. Water (75 ml) was charged to the mixture while maintaining the temperature at 55-65°C. A solution of citric acid (20.76 g, 0.107 mol) in water (25.0) was charged and the mixture was cooled to room temperature. The reactor was stirred for a minimum of 5 hours and then the resulting solids were isolated by filtration and washed with acetone (2×75 ml), which was sent to the filter. The salt was charged into a clean, dry, nitrogen-purged 1L reactor with 2B ethanol (190 ml) and water (190 ml). The slurry was heated to 75-850C for a minimum of 4 hours. The mixture was cooled to 20-300C and stirred for an additional 4 hours. The solids were isolated by filtration and washed with 2B ethanol (190 ml). After drying in a vacuum oven at 500C with a slight nitrogen bleed, 34.6 g (67.3%) of the title compound were isolated. 1H NMR (500 MHz, CZ6-DMSO): δ 8.14 (s, 1 H), 7.11 (d, J=3.6 Hz, 1 H), 6.57 (d, J=3.6 Hz, 1 H), 4.96 (q, J=6.0 Hz, 1 H), 4.00-3.90 (m, 2H), 3.80 (m, 2H), 3.51 (m, 1 H), 3.32 (s, 3H), 2.80 (Abq, J=15.6 Hz, 2H), 2.71 (Abq, J=15.6 Hz, 2H), 2.52-2.50 (m, 1 H), 2.45-2.41 (m, 1 H), 1.81 (m, 1 H), 1.69-1.65 (m, 1 H), 1.04 (d, J=6.9 Hz, 3H)
PAPER
Org. Lett., 2009, 11 (9), pp 2003–2006
DOI: 10.1021/ol900435t
http://pubs.acs.org/doi/full/10.1021/ol900435t 
PATENT
http://www.omicsonline.org/open-access/advances-in-the-inhibitors-of-janus-kinase-2161-0444.1000540.php?aid=29799  ……………..
…………….. 

 सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Clinical trials
Rheumatoid arthritis
Phase II clinical trials tested the drug in rheumatoid arthritis patients that had not responded to DMARD therapy. In a tofacitinib monotherapy study, the ACR score improved by at least 20% (ACR-20) in 67% of patients versus 25% who received placebo; and a study that combined the drug with methotrexate achieved ACR-20 in 59% of patients versus 35% who received methotrexate alone. In a psoriasis study, the PASI score improved by at least 75% in between 25 and 67% of patients, depending on the dose, versus 2% in the placebo group.[8] The most important side effects in Phase II studies were increased blood cholesterol levels (12 to 25 mg/dl LDL and 8 to 10 mg/dl HDL at medium dosage levels) andneutropenia.[8] Phase III trials testing the drug in rheumatoid arthritis started in 2007 and are scheduled to run until January 2015.[9] In April 2011, four patients died after beginning clinical trials with tofacitinib. According to Pfizer, only one of the four deaths was related to tofacitinib.[10] By April 2011, three phase III trials for RA had reported positive results.[11] In November 2012, the U.S. FDA approved tofacitinib “to treat adults with moderately to severely active rheumatoid arthritis who have had an inadequate response to, or who are intolerant of, methotrexate.”[12]
Psoriasis
As of April 2011 a phase III trial for psoriasis is under way.[11]
Alopecia
In June 2014, scientists at Yale successfully treated a male patient afflicted with alopecia universalis. The patient was able to grow a full head of hair, eyebrows, eyelashes, facial, armpit, genitalia and other hair. No side effects were reported in the study.[13]
Ulcerative colitis
The OCTAVE study of Tofacitinib in Ulcerative Colitis started in 2012. It is currently enrolling patients, though the NIH trials page states that they expect the trial to close in June 2015.[14]
Vitiligo
In a June 2015 study, a 53-year-old woman with vitiligo showed noticeable improvement after taking tofacitinib for five months.[15]
Development of Safe, Robust, Environmentally Responsible Processes for New Chemical Entities
– Dr. V. Rajappa, Director & Head-Process R&D, Bristol-Myers Squibb, India
A PRESENTATION
 The presentation will load below
The presentation will load below

Scroll with mouse to view 76 pages


- Herper, Matthew (2 March 2011). “Why Pfizer’s Biggest Experimental Drug Got A Name Change”. Forbes. Retrieved 3 March 2011.
- Kremer, J. M.; Bloom, B. J.; Breedveld, F. C.; Coombs, J. H.; Fletcher, M. P.; Gruben, D.; Krishnaswami, S.; Burgos-Vargas, R. N.; Wilkinson, B.; Zerbini, C. A. F.; Zwillich, S. H. (2009). “The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo”. Arthritis & Rheumatism 60 (7): 1895–1905. doi:10.1002/art.24567. PMID 19565475. edit
- “Tasocitinib”. Drugs in R&D 10 (4): 271–284. 2010. doi:10.2165/11588080-000000000-00000. PMC 3585773. PMID 21171673. edit
- Ghoreschi, K.; Jesson, M. I.; Li, X.; Lee, J. L.; Ghosh, S.; Alsup, J. W.; Warner, J. D.; Tanaka, M.; Steward-Tharp, S. M.; Gadina, M.; Thomas, C. J.; Minnerly, J. C.; Storer, C. E.; Labranche, T. P.; Radi, Z. A.; Dowty, M. E.; Head, R. D.; Meyer, D. M.; Kishore, N.; O’Shea, J. J. (2011). “Modulation of Innate and Adaptive Immune Responses by Tofacitinib (CP-690,550)”. J Immunol. 186 (7): 4234–4243. doi:10.4049/jimmunol.1003668. PMC 3108067. PMID 21383241. edit
- ^ Jump up to:a b c “Seeking Profit for Taxpayers in Potential of New Drug”, Jonathan Weisman, New York Times, March 18, 2013
- Ken Garber (9 January 2013). “Pfizer’s first-in-class JAK inhibitor pricey for rheumatoid arthritis market”. Nature Biotechnology 31 (1): 3–4. doi:10.1038/nbt0113-3. PMID 23302910.
- Jump up^ Moisan A, et al. White-to-brown metabolic conversion of human adipocytes by JAK inhibition. Nature Cell Biology, 8 December 2014. DOI 10.1038/ncb3075
- “EULAR: JAK Inhibitor Effective in RA But Safety Worries Remain”. MedPage Today. June 2009. Retrieved 9 February 2011.
- Clinical trial number NCT00413699 for “Long-Term Effectiveness And Safety Of CP-690,550 For The Treatment Of Rheumatoid Arthritis” at ClinicalTrials.gov
- Matthew Herper. “Pfizer’s Key Drug Walks A Tightrope”. Forbes.
- “Two Phase III Studies Confirm Benefits of Pfizer’s Tofacitinib Against Active RA”. 28 Apr 2011.
- “FDA approves Xeljanz for rheumatoid arthritis”. 6 Nov 2012.
- “Hairless man grows full head of hair in yale arthritis drug trial”. 19 Jun 2014.
- https://clinicaltrials.gov/ct2/show/NCT01465763?term=A3921094&rank=1
- “This Drug Brought Pigment Back for Woman with Vitiligo”. TIME. June 27, 2015. Retrieved June 29, 2015.
- Nordqvist, Christian (27 April 2013). “Pfizer’s Arthritis Drug Xeljanz (tofacitinib) Receives A Negative Opinion In Europe”. Medical News Today. Retrieved 2 August 2013.
- “”XALEJANZ PRESCRIBING INFORMATION @ Labeling.Pfizer.com””.
SEE………http://orgspectroscopyint.blogspot.in/2014/12/tofacitinib-citrate.html
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.

सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये।
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on Linkedin

Join me on Facebook  FACEBOOK
FACEBOOK
Join me on twitter

 amcrasto@gmail.com
amcrasto@gmail.com

LIONEL MY SON
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
//////

































 ,
, 





















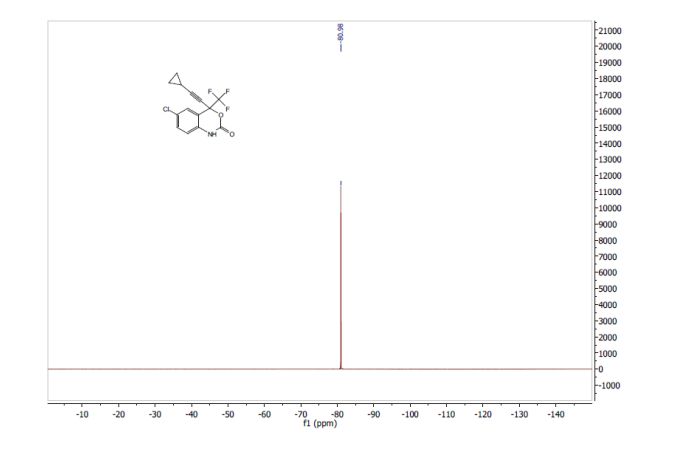


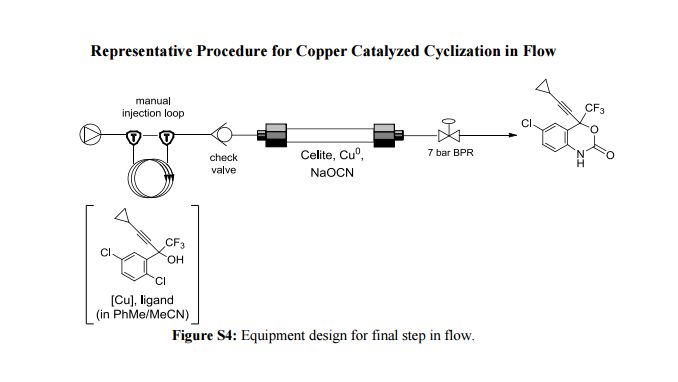


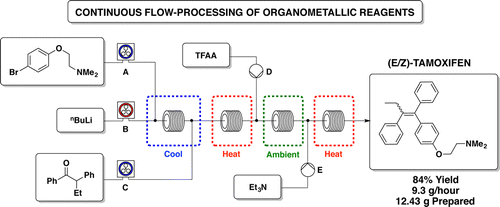



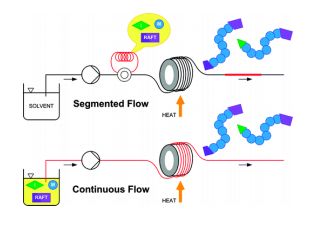









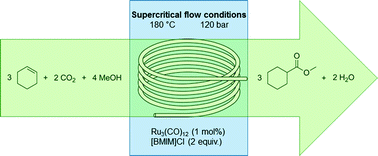







































nonanoic acid methyl ester")

 Instructions
Instructions












