
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Polymorph case study……….Duvelisib


Duvelisib
Infinity and AbbVie partner to develop and commercialise duvelisib for cancer
INK 1197; IPI 145; 8-Chloro-2-phenyl-3-[(1S)-1-(9H-purin-6-ylamino)ethyl]-1(2H)-isoquinolinone
1(2H)-Isoquinolinone, 8-chloro-2-phenyl-3-((1S)-1-(9H-purin-6-ylamino)ethyl)-
8-Chloro-2-phenyl-3-((1S)-1-(7H-purin-6-ylamino)ethyl)isoquinolin-1(2H)-one
(S)-3-(l-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-l(2H)-one
UNII-610V23S0JI; IPI-145; INK-1197;
Originator…….. Millennium Pharmaceuticals
| Molecular Formula | C22H17ClN6O | |
| Molecular Weight | 416.86 | |
| CAS Registry Number | 1201438-56-3 |

Infinity Pharmaceuticals has partnered with AbbVie to develop and commercialise its duvelisib (IPI-145), an oral inhibitor of phosphoinositide-3-kinase (PI3K)-delta and PI3K-gamma, to treat patients with cancer.

Duvelisib
The filing of patents claiming new crystalline forms, usually 4−6 years after the original product patent, is a typical strategy applied by such companies to extend patent protection. This patent protection approach by big pharma forces generic bulk producers to discover and file patents on new polymorphs if they want to market the drug after expiry of the product patents.
Polymorphism is of paramount importance due to its effect on some physical characteristics of powders such as melting point, flowability, vapour pressure, bulk density, chemical reactivity, apparent solubility and dissolution rate, and optical and electrical properties. In other words, polymorphism can affect drug stability, manipulation, and bioavailability
the principal aim of generic bulk producers was to generate a competitive market advantage by protecting their new crystal form.
Polymorphic forms of a compound of Formula (I):.US8809349

herein referred to as Form A, Form B, Form C, Form D, Form E, Form F, Form G, Form H, Form I, Form J, or an amorphous form of a compound of Formula (I), or a salt, solvate, or hydrate thereof; or a mixture of two or more thereof. In one embodiment, the polymorphic form of a compound of Formula (I) can be a crystalline form, a partially crystalline form, an amorphous form, or a mixture of crystalline form(s) and/or amorphous form(s).
(XRPD) peaks
“Enantiomerically pure”
As used herein, and unless otherwise specified, the term “enantiomerically pure” means a stereomerically pure composition of a compound having one or more chiral center(s).
As used herein, and unless otherwise specified, the terms “enantiomeric excess” and “diastereomeric excess” are used interchangeably herein. In some embodiments, compounds with a single stereocenter can be referred to as being present in “enantiomeric excess,” and those with at least two stereocenters can be referred to as being present in “diastereomeric excess.” For example, the term “enantiomeric excess” is well known in the art and is defined as:
eea=(conc.ofa-conc.ofbconc.ofa+conc.ofb)×100
Thus, the term “enantiomeric excess” is related to the term “optical purity” in that both are measures of the same phenomenon. The value of ee will be a number from 0 to 100, zero being racemic and 100 being enantiomerically pure. A compound which in the past might have been called 98% optically pure is now more precisely characterized by 96% ee. A 90% ee reflects the presence of 95% of one enantiomer and 5% of the other(s) in the material in question.
Some compositions described herein contain an enantiomeric excess of at least about 50%, 75%, 90%, 95%, or 99% of the S enantiomer. In other words, the compositions contain an enantiomeric excess of the S enantiomer over the R enantiomer. In other embodiments, some compositions described herein contain an enantiomeric excess of at least about 50%, 75%, 90%, 95%, or 99% of the R enantiomer. In other words, the compositions contain an enantiomeric excess of the R enantiomer over the S enantiomer.
GRAPHS
FIG. 1 shows an X-ray powder diffraction (XRPD) for Polymorph Form A.
FIG. 2 shows an XRPD for Polymorph Form B.
FIG. 3 shows an XRPD for Polymorph Form C.
FIG. 4 shows an XRPD for Polymorph Form D.
FIG. 5 shows an XRPD for Polymorph Form E.
FIG. 6 shows an XRPD for Polymorph Form F.
FIG. 7 shows an XRPD for Polymorph Form G.
FIG. 8 shows an XRPD for Polymorph Form H.
FIG. 9 shows an XRPD for Polymorph Form I.
FIG. 10 shows an XRPD for Polymorph Form J.
FIG. 11 shows an XRPD for amorphous compound of Formula (I).
FIG. 12 shows a differential scanning calorimetry (DSC) thermogram for Polymorph Form A.
FIG. 13 shows a DSC for Polymorph Form B.
FIG. 14 shows a DSC for Polymorph Form C.
FIG. 15 shows a DSC for Polymorph Form D.
FIG. 16 shows a DSC for Polymorph Form E.
FIG. 17 shows a DSC for Polymorph Form F.
FIG. 18 shows a DSC for Polymorph Form G.
FIG. 19 shows a DSC for Polymorph Form H.
FIG. 20 shows a DSC for Polymorph Form I.
FIG. 21 shows a DSC for Polymorph Form J.
FIG. 22 shows a DSC thermogram and a thermogravimetric analysis (TGA) for Polymorph Form A.
FIG. 23 shows two DSC thermograms for Polymorph Form C.
FIG. 24 shows a DSC and a TGA for Polymorph Form F.
FIG. 25 shows a panel of salts tested for formation of crystalline solids in various solvents.
FIG. 26 shows a single crystal X-ray structure of Polymorph Form G MTBE (t-butyl methyl ether) solvate of a compound of Formula (I).
FIG. 27 shows an FT-IR spectra of Polymorph Form C.
FIG. 28 shows a 1H-NMR spectra of Polymorph Form C.
FIG. 29 shows a 13C-NMR spectra of Polymorph Form C.
FIG. 30 shows a dynamic vapor sorption (DVS) analysis of Polymorph Form C.
FIG. 31 shows representative dissolution profiles of capsules containing Polymorph Form C.
DRAWINGS
FIG. 1 shows an X-ray powder diffraction (XRPD) for Polymorph Form A.

FIG. 1 shows a representative X-ray powder diffraction (XRPD) for polymorph Form A.
In one embodiment, polymorph Form A can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 1. In one embodiment, polymorph Form A can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 12.2° (±0.2°), and 18.3° (±0.2°). In one embodiment, polymorph Form A can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 12.2° (±0.2°), and 18.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=15.6° (±0.2°) and 19.2° (±0.2°). In another embodiment, polymorph Form A can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 12.2° (±0.2°), 15.6° (±0.2°), 18.3° (±0.2°), and 19.2° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.1° (±0.2°), 9.4° (±0.2°), 12.4° (±0.2°), 14.8° (±0.2°), 16.3° (±0.2°), 17.7° (±0.2°), 21.1° (±0.2°), 21.9° (±0.2°), 24.0° (±0.2°), and 26.9° (±0.2°). In one embodiment, polymorph Form A can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 1.
FIG. 2 shows an XRPD for Polymorph Form B.

FIG. 2 shows a representative XRPD for polymorph Form B.
In one embodiment, polymorph Form B can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 2. In one embodiment, polymorph Form B can be characterized as having at least one XRPD peak selected from 2θ=7.9° (±0.2°), 13.4° (±0.2°), and 23.4° (±0.2°). In one embodiment, polymorph Form B can be characterized as having at least one XRPD peak selected from 2θ=7.9° (±0.2°), 13.4° (±0.2°), and 23.4° (±0.2°) in combination with at least one XRPD peak selected from 2θ=14.0° (±0.2°) and 15.0° (±0.2°). In another embodiment, polymorph Form B can be characterized as having at least one XRPD peak selected from 2θ=7.9° (±0.2°), 13.4° (±0.2°), 14.0° (±0.2°), 15.0° (±0.2°), and 23.4° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.5° (±0.2°), 12.7° (±0.2°), 13.6° (±0.2°), 14.2° (±0.2°), 15.7° (±0.2°), 19.0° (±0.2°), 22.3° (±0.2°), 24.2° (±0.2°), 24.8° (±0.2°), and 26.9° (±0.2°). In one embodiment, polymorph Form B can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 2.
FIG. 3 shows an XRPD for Polymorph Form C.
In one embodiment, polymorph Form C can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 3. In one embodiment, Form C can be characterized by having at least one XRPD peak selected from 2θ=10.5° (±0.2°), 13.7° (±0.2°), and 24.5° (±0.2°). In another embodiment, Form C can be characterized by having at least one XRPD peak selected from 2θ=10.4° (±0.2°), 13.3° (±0.2°), and 24.3° (±0.2°). In one embodiment, polymorph Form C can be characterized as having at least one XRPD peak selected from 2θ=10.4° (±0.2°), 13.3° (±0.2°), and 24.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=6.6° (±0.2°) and 12.5° (±0.2°). In another embodiment, polymorph Form C can be characterized as having at least one XRPD peak selected from 2θ=6.6° (±0.2°), 10.4° (±0.2°), 12.5° (±0.2°), 13.3° (±0.2°), and 24.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=8.8° (±0.2°), 9.9° (±0.2°), 13.4° (±0.2°), 15.5° (±0.2°), 16.9° (±0.2°), 19.8° (±0.2°), 21.3° (±0.2°), 23.6° (±0.2°), 25.3° (±0.2°), and 27.9° (±0.2°). In one embodiment, polymorph Form C can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 3.

FIG. 4 shows an XRPD for Polymorph Form D.

In one embodiment, polymorph Form D can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 4. In one embodiment, polymorph Form D can be characterized as having at least one XRPD peak selected from 2θ=11.4° (±0.2°), 17.4° (±0.2°), and 22.9° (±0.2°). In one embodiment, polymorph Form D can be characterized as having at least one XRPD peak selected from 2θ=11.4° (±0.2°), 17.4° (±0.2°), and 22.9° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.2° (±0.2°) and 18.3° (±0.2°). In another embodiment, polymorph Form D can be characterized as having at least one XRPD peak selected from 2θ=9.2° (±0.2°), 11.4° (±0.2°), 17.4° (±0.2°), 18.3° (±0.2°), and 22.9° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.8° (±0.2°), 12.2° (±0.2°), 15.8° (±0.2°), 16.2° (±0.2°), 16.8° (±0.2°), 18.9° (±0.2°), 19.9° (±0.2°), 20.0° (±0.2°), 24.9° (±0.2°), and 29.3° (±0.2°). In one embodiment, polymorph Form D can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 4.
FIG. 5 shows an XRPD for Polymorph Form E. US8809349
In one embodiment, polymorph Form E can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 5. In one embodiment, polymorph Form E can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.3° (±0.2°), and 24.4° (±0.2°). In one embodiment, polymorph Form E can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.3° (±0.2°), and 24.4° (±0.2°) in combination with at least one XRPD peak selected from 2θ=12.7° (±0.2°) and 13.9° (±0.2°). In another embodiment, polymorph Form E can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.3° (±0.2°), 12.7° (±0.2°), 13.9° (±0.2°), and 24.4° (±0.2°) in combination with at least one XRPD peak selected from 2θ=12.4° (±0.2°), 13.3° (±0.2°), 14.3° (±0.2°), 15.5° (±0.2°), 17.4° (±0.2°), 18.5° (±0.2°), 22.0° (±0.2°), 23.9° (±0.2°), 24.1° (±0.2°), and 26.4° (±0.2°). In one embodiment, polymorph Form E can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 5.
FIG. 6 shows an XRPD for Polymorph Form F. US8809349
In one embodiment, polymorph Form F can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 6. In one embodiment, polymorph Form F can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 17.3° (±0.2°), and 24.6° (±0.2°). In one embodiment, polymorph Form F can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 17.3° (±0.2°), and 24.6° (±0.2°) in combination with at least one XRPD peak selected from 2θ=14.0° (±0.2°) and 19.2° (±0.2°). In another embodiment, polymorph Form F can be characterized as having at least one XRPD peak selected from 2θ=9.6° (±0.2°), 14.0° (±0.2°), 17.3° (±0.2°), 19.2° (±0.2°), and 24.6° (±0.2°) in combination with at least one XRPD peak selected from 2θ=12.4° (±0.2°), 16.1° (±0.2°), 16.6° (±0.2°), 17.1° (±0.2°), 20.8° (±0.2°), 21.5° (±0.2°), 22.0° (±0.2°), 24.3° (±0.2°), 25.2° (±0.2°), and 25.4° (±0.2°). In one embodiment, polymorph Form F can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 6.
FIG. 7 shows an XRPD for Polymorph Form G. US8809349
In one embodiment, polymorph Form G can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 7. In one embodiment, polymorph Form G can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.5° (±0.2°), and 19.0° (±0.2°). In one embodiment, polymorph Form G can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.5° (±0.2°), and 19.0° (±0.2°) in combination with at least one XRPD peak selected from 2θ=10.6° (±0.2°) and 19.6° (±0.2°). In another embodiment, polymorph Form G can be characterized as having at least one XRPD peak selected from 2θ=6.7° (±0.2°), 9.5° (±0.2°), 10.6° (±0.2°), 19.0° (±0.2°), and 19.6° (±0.2°) in combination with at least one XRPD peak selected from 2θ=13.4° (±0.2°), 15.0° (±0.2°), 15.8° (±0.2°), 17.8° (±0.2°), 20.7° (±0.2°), 21.2° (±0.2°), 22.8° (±0.2°), 23.8° (±0.2°), 24.3° (±0.2°), and 25.6° (±0.2°). In one embodiment, polymorph Form G can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 7.
FIG. 8 shows an XRPD for Polymorph Form H. US8809349
In one embodiment, polymorph Form H can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 8. In one embodiment, polymorph Form H can be characterized as having at least one XRPD peak selected from 2θ=8.9° (±0.2°), 9.2° (±0.2°), and 14.1° (±0.2°). In one embodiment, polymorph Form H can be characterized as having at least one XRPD peak selected from 2θ=8.9° (±0.2°), 9.2° (±0.2°), and 14.1° (±0.2°) in combination with at least one XRPD peak selected from 2θ=17.3° (±0.2°) and 18.5° (±0.2°). In another embodiment, polymorph Form H can be characterized as having at least one XRPD peak selected from 2θ=8.9° (±0.2°), 9.2° (±0.2°), 14.1° (±0.2°), 17.3° (±0.2°), and 18.5° (±0.2°) in combination with at least one XRPD peak selected from 2θ=7.1° (±0.2°), 10.6° (±0.2°), 11.3° (±0.2°), 11.6° (±0.2°), 16.2° (±0.2°), 18.3° (±0.2°), 18.8° (±0.2°), 20.3° (±0.2°), 21.7° (±0.2°), and 24.7° (±0.2°). In one embodiment, polymorph Form H can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 8.
FIG. 9 shows an XRPD for Polymorph Form I.
In one embodiment, polymorph Form I can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 9. In one embodiment, polymorph Form I can be characterized as having at least one XRPD peak selected from 2θ=9.7° (±0.2°), 19.3° (±0.2°), and 24.5° (±0.2°). In one embodiment, polymorph Form I can be characterized as having at least one XRPD peak selected from 2θ=9.7° (±0.2°), 19.3° (±0.2°), and 24.5° (±0.2°) in combination with at least one XRPD peak selected from 2θ=11.4° (±0.2°) and 14.2° (±0.2°). In another embodiment, polymorph Form I can be characterized as having at least one XRPD peak selected from 2θ=9.7° (±0.2°), 11.4° (±0.2°), 14.2° (±0.2°), 19.3° (±0.2°), and 24.5° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.2° (±0.2°), 14.7° (±0.2°), 15.5° (±0.2°), 16.7° (±0.2°), 17.3° (±0.2°), 18.4° (±0.2°), 21.4° (±0.2°), 22.9° (±0.2°), 29.1° (±0.2°), and 34.1° (±0.2°). In one embodiment, polymorph Form I can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 9.
FIG. 10 shows an XRPD for Polymorph Form J.
In one embodiment, polymorph Form J can be characterized by any one, two, three, four, five, six, seven, eight, nine, ten, or more of significant peak(s) of FIG. 10. In one embodiment, polymorph Form J can be characterized as having at least one XRPD peak selected from 2θ=9.1° (±0.2°), 17.3° (±0.2°), and 18.3° (±0.2°). In one embodiment, polymorph Form J can be characterized as having at least one XRPD peak selected from 2θ=9.1° (±0.2°), 17.3° (±0.2°), and 18.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=16.4° (±0.2°) and 17.9° (±0.2°). In another embodiment, polymorph Form J can be characterized as having at least one XRPD peak selected from 2θ=9.1° (±0.2°), 16.4° (±0.2°), 17.3° (±0.2°), 17.9° (±0.2°), and 18.3° (±0.2°) in combination with at least one XRPD peak selected from 2θ=9.4° (±0.2°), 10.1° (±0.2°), 10.7° (±0.2°), 14.0° (±0.2°), 14.3° (±0.2°), 15.5° (±0.2°), 16.9° (±0.2°), 19.9° (±0.2°), 24.0° (±0.2°), and 24.7° (±0.2°). In one embodiment, polymorph Form J can be characterized in that it has substantially all of the peaks in its XRPD pattern as shown in FIG. 10.
FIG. 11 shows an XRPD for amorphous compound of Formula (I).
FIG. 12 shows a differential scanning calorimetry (DSC) thermogram for Polymorph Form A.
FIG. 13 shows a DSC for Polymorph Form B.
FIG. 14 shows a DSC for Polymorph Form C.
FIG. 15 shows a DSC for Polymorph Form D.
FIG. 16 shows a DSC for Polymorph Form E.
FIG. 17 shows a DSC for Polymorph Form F.
FIG. 18 shows a DSC for Polymorph Form G.
FIG. 19 shows a DSC for Polymorph Form H.
FIG. 20 shows a DSC for Polymorph Form I.
FIG. 21 shows a DSC for Polymorph Form J.
FIG. 22 shows a DSC thermogram and a thermogravimetric analysis (TGA) for Polymorph Form A.
FIG. 23 shows two DSC thermograms for Polymorph Form C.
FIG. 24 shows a DSC and a TGA for Polymorph Form F.
FIG. 25 shows a panel of salts tested for formation of crystalline solids in various solvents.
FIG. 26 shows a single crystal X-ray structure of Polymorph Form G MTBE (t-butyl methyl ether) solvate of a compound of Formula (I).
FIG. 27 shows an FT-IR spectra of Polymorph Form C.

FIG. 28 shows a 1H-NMR spectra of Polymorph Form C.

FIG. 29 shows a 13C-NMR spectra of Polymorph Form C.

FIG. 30 shows a dynamic vapor sorption (DVS) analysis of Polymorph Form C.
FIG. 31 shows representative dissolution profiles of capsules containing Polymorph Form C.
Enantiomers
Enantiomers can be isolated from racemic mixtures by any method known to those skilled in the art, including chiral high pressure liquid chromatography (HPLC), the formation and crystallization of chiral salts, or prepared by asymmetric syntheses. See, for example, Enantiomers, Racemates and Resolutions (Jacques, Ed., Wiley Interscience, New York, 1981); Wilen et al., Tetrahedron 33:2725 (1977); Stereochemistry of Carbon Compounds (E. L. Eliel, Ed., McGraw-Hill, NY, 1962); and Tables of Resolving Agents and Optical Resolutions p. 268 (E. L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, Ind. 1972).
“Tautomer”
The term “tautomer” is a type of isomer that includes two or more interconvertable compounds resulting from at least one formal migration of a hydrogen atom and at least one change in valency (e.g., a single bond to a double bond, a triple bond to a single bond, or vice versa). “Tautomerization” includes prototropic or proton-shift tautomerization, which is considered a subset of acid-base chemistry. “Prototropic tautomerization” or “proton-shift tautomerization” involves the migration of a proton accompanied by changes in bond order. The exact ratio of the tautomers depends on several factors, including temperature, solvent, and pH. Where tautomerization is possible (e.g., in solution), a chemical equilibrium of tautomers can be reached. Tautomerizations (i.e., the reaction providing a tautomeric pair) can be catalyzed by acid or base, or can occur without the action or presence of an external agent. Exemplary tautomerizations include, but are not limited to, keto-to-enol; amide-to-imide; lactam-to-lactim; enamine-to-imine; and enamine-to-(a different) enamine tautomerizations. An example of keto-enol tautomerization is the interconversion of pentane-2,4-dione and 4-hydroxypent-3-en-2-one tautomers. Another example of tautomerization is phenol-keto tautomerization. Another example of phenol-keto tautomerization is the interconversion of pyridin-4-ol and pyridin-4(1H)-one tautomers.
As defined herein, the term “Formula (I)” includes (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one in its imide tautomer shown below as (1-1) and in its lactim tautomer shown below as (1-2):

“polymorph” can be used herein to describe a crystalline material, e.g., a crystalline form. In certain embodiments, “polymorph” as used herein are also meant to include all crystalline and amorphous forms of a compound or a salt thereof, including, for example, crystalline forms, polymorphs, pseudopolymorphs, solvates, hydrates, co-crystals, unsolvated polymorphs (including anhydrates), conformational polymorphs, tautomeric forms, disordered crystalline forms, and amorphous forms, as well as mixtures thereof, unless a particular crystalline or amorphous form is referred to. Compounds of the present disclosure include crystalline and amorphous forms of those compounds, including, for example, crystalline forms, polymorphs, pseudopolymorphs, solvates, hydrates, co-crystals, unsolvated polymorphs (including anhydrates), conformational polymorphs, tautomeric forms, disordered crystalline forms, and amorphous forms of the compounds or a salt thereof, as well as mixtures thereof.
As used herein, and unless otherwise specified, a particular form of a compound of Formula (I) described herein (e.g., Form A, B, C, D, E, F, G, H, I, J, or amorphous form of a compound of Formula (I), or mixtures thereof) is meant to encompass a solid form of a compound of Formula (I), or a salt, solvate, or hydrate thereof, among others.
The polymorphs made according to the methods provided herein can be characterized by any methodology known in the art. For example, the polymorphs made according to the methods provided herein can be characterized by X-ray powder diffraction (XRPD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), dynamic vapor sorption (DVS), hot-stage microscopy, optical microscopy, Karl Fischer analysis, melting point, spectroscopy (e.g., Raman, solid state nuclear magnetic resonance (ssNMR), liquid state nuclear magnetic resonance (1H- and 13C-NMR), and FT-IR), thermal stability, grinding stability, and solubility, among others.
“Solid form”
The terms “solid form” and related terms herein refer to a physical form comprising a compound provided herein or a salt or solvate or hydrate thereof, which is not in a liquid or a gaseous state. Solid forms can be crystalline, amorphous, disordered crystalline, partially crystalline, and/or partially amorphous.
“Crystalline,”
The term “crystalline,” when used to describe a substance, component, or product, means that the substance, component, or product is substantially crystalline as determined, for example, by X-ray diffraction. See, e.g., Remington: The Science and Practice of Pharmacy, Lippincott Williams & Wilkins, 21st ed. (2005).
As used herein, and unless otherwise specified, the term “crystalline form,” “crystal form,” and related terms herein refer to the various crystalline material comprising a given substance, including single-component crystal forms and multiple-component crystal forms, and including, but not limited to, polymorphs, solvates, hydrates, co-crystals and other molecular complexes, as well as salts, solvates of salts, hydrates of salts, other molecular complexes of salts, and polymorphs thereof. In certain embodiments, a crystal form of a substance can be substantially free of amorphous forms and/or other crystal forms. In other embodiments, a crystal form of a substance can contain about 1%, about 2%, about 3%, about 4%, about 5%, about 10%, about 15%, about 20%, about 25%, about 30%, about 35%, about 40%, about 45% or about 50% of one or more amorphous form(s) and/or other crystal form(s) on a weight and/or molar basis.
Certain crystal forms of a substance can be obtained by a number of methods, such as, without limitation, melt recrystallization, melt cooling, solvent recrystallization, recrystallization in confined spaces, such as, e.g., in nanopores or capillaries, recrystallization on surfaces or templates, such as, e.g., on polymers, recrystallization in the presence of additives, such as, e.g., co-crystal counter-molecules, desolvation, dehydration, rapid evaporation, rapid cooling, slow cooling, vapor diffusion, sublimation, grinding, solvent-drop grinding, microwave-induced precipitation, sonication-induced precipitation, laser-induced precipitation, and/or precipitation from a supercritical fluid. As used herein, and unless otherwise specified, the term “isolating” also encompasses purifying.
Characterizing crystal forms and amorphous forms
Techniques for characterizing crystal forms and amorphous forms can include, but are not limited to, thermal gravimetric analysis (TGA), differential scanning calorimetry (DSC), X-ray powder diffractometry (XRPD), single crystal X-ray diffractometry, vibrational spectroscopy, e.g., infrared (IR) and Raman spectroscopy, solid-state nuclear magnetic resonance (NMR) spectroscopy, optical microscopy, hot stage optical microscopy, scanning electron microscopy (SEM), electron crystallography and quantitative analysis, particle size analysis (PSA), surface area analysis, solubility studies, and dissolution studies.
PEAK
As used herein, and unless otherwise specified, the term “peak,” when used in connection with the spectra or data presented in graphical form (e.g., XRPD, IR, Raman, and NMR spectra), refers to a peak or other special feature that one skilled in the art would recognize as not attributable to background noise. The term “significant peak” refers to peaks at least the median size (e.g., height) of other peaks in the spectrum or data, or at least 1.5, 2, or 2.5 times the background level in the spectrum or data.
“Pharmaceutically acceptable carrier”
“pharmaceutically acceptable carrier” or “pharmaceutically acceptable excipient” includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like. The use of such media and agents for pharmaceutically active substances is known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in the therapeutic compositions of the present disclosure is contemplated. Supplementary active ingredients can also be incorporated into the compositions.
“Substantially pure”
the term “substantially pure” when used to describe a polymorph, a crystal form, or a solid form of a compound or complex described herein means a solid form of the compound or complex that comprises a particular polymorph and is substantially free of other polymorphic and/or amorphous forms of the compound. A representative substantially pure polymorph comprises greater than about 80% by weight of one polymorphic form of the compound and less than about 20% by weight of other polymorphic and/or amorphous forms of the compound; greater than about 90% by weight of one polymorphic form of the compound and less than about 10% by weight of other polymorphic and/or amorphous forms of the compound; greater than about 95% by weight of one polymorphic form of the compound and less than about 5% by weight of other polymorphic and/or amorphous forms of the compound; greater than about 97% by weight of one polymorphic form of the compound and less than about 3% by weight of other polymorphic and/or amorphous forms of the compound; or greater than about 99% by weight of one polymorphic form of the compound and less than about 1% by weight of other polymorphic and/or amorphous forms of the compound.
“Stable”
The term “stable” refers to a compound or composition that does not readily decompose or change in chemical makeup or physical state. A stable composition or formulation provided herein does not significantly decompose under normal manufacturing or storage conditions. In some embodiments, the term “stable,” when used in connection with a formulation or a dosage form, means that the active ingredient of the formulation or dosage form remains unchanged in chemical makeup or physical state for a specified amount of time and does not significantly degrade or aggregate or become otherwise modified (e.g., as determined, for example, by HPLC, FTIR, or XRPD). In some embodiments, about 70 percent or greater, about 80 percent or greater, about 90 percent or greater, about 95 percent or greater, about 98 percent or greater, or about 99 percent or greater of the compound remains unchanged after the specified period. In one embodiment, a polymorph provided herein is stable upon long-term storage (e.g., no significant change in polymorph form after about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 18, 24, 30, 36, 42, 48, 54, 60, or greater than about 60 months).
Amorphous form
In one embodiment, an amorphous form of a compound of Formula (I), or a pharmaceutically acceptable salt, solvate, or hydrate thereof, can be made by dissolution of a crystalline form followed by removal of solvent under conditions in which stable crystals are not formed. For example, solidification can occur by rapid removal of solvent, by rapid addition of an anti-solvent (causing the amorphous form to precipitate out of solution), or by physical interruption of the crystallization process. Grinding processes can also be used. In other embodiments, an amorphous form of a compound of Formula (I), or a pharmaceutically acceptable salt, solvate, or hydrate thereof, can be made using a process or procedure described herein elsewhere.
In certain embodiments, an amorphous form can be obtained by fast cooling from a single solvent system, such as, e.g., ethanol, isopropyl alcohol, t-amyl alcohol, n-butanol, methanol, acetone, ethyl acetate, or acetic acid. In certain embodiments, an amorphous form can be obtained by slow cooling from a single solvent system, such as, e.g., ethanol, isopropyl alcohol, t-amyl alcohol, or ethyl acetate.
In certain embodiments, an amorphous form can be obtained by fast cooling from a binary solvent system, for example, with acetone or DME as the primary solvent. In certain embodiments, an amorphous form can be obtained by slow cooling from a binary solvent system, for example, with ethanol, isopropyl alcohol, THF, acetone, or methanol as the primary solvent. In some embodiments, an amorphous form can be obtained by dissolution of a compound of Formula (I) in t-butanol and water at elevated temperature, followed by cooling procedures to afford an amorphous solid form.
Salt Forms
In certain embodiments, a compound of Formula (I) provided herein is a pharmaceutically acceptable salt, or a solvate or hydrate thereof. In one embodiment, pharmaceutically acceptable acid addition salts of a compound provided herein can be formed with inorganic acids and organic acids. Inorganic acids from which salts can be derived include, but are not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like. Organic acids from which salts can be derived include, but are not limited to, acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid, and the like. In other embodiments, if applicable, pharmaceutically acceptable base addition salts of a compound provided herein can be formed with inorganic and organic bases. Inorganic bases from which salts can be derived include, but are not limited to, sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum, and the like. Organic bases from which salts can be derived include, but are not limited to, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like. Exemplary bases include, but are not limited to, isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, and ethanolamine. In some embodiments, a pharmaceutically acceptable base addition salt is ammonium, potassium, sodium, calcium, or magnesium salt. In one embodiment, bis salts (i.e., two counterions) and higher salts (e.g., three or more counterions) are encompassed within the meaning of pharmaceutically acceptable salts.
In certain embodiments, salts of a compound of Formula (I) can be formed with, e.g., L-tartaric acid, p-toluenesulfonic acid, D-glucaronic acid, ethane-1,2-disulfonic acid (EDSA), 2-naphthalenesulfonic acid (NSA), hydrochloric acid (HCl) (mono and bis), hydrobromic acid (HBr), citric acid, naphthalene-1,5-disulfonic acid (NDSA), DL-mandelic acid, fumaric acid, sulfuric acid, maleic acid, methanesulfonic acid (MSA), benzenesulfonic acid (BSA), ethanesulfonic acid (ESA), L-malic acid, phosphoric acid, and aminoethanesulfonic acid (taurine).
(R)- and (S)-isomers
In some embodiments, the (R)- and (S)-isomers of the non-limiting exemplary compounds, if present, can be resolved by methods known to those skilled in the art, for example by formation of diastereoisomeric salts or complexes which can be separated, for example, by crystallization; via formation of diastereoisomeric derivatives which can be separated, for example, by crystallization, gas-liquid or liquid chromatography; selective reaction of one enantiomer with an enantiomer-specific reagent, for example enzymatic oxidation or reduction, followed by separation of the modified and unmodified enantiomers; or gas-liquid or liquid chromatography in a chiral environment, for example on a chiral support, such as silica with a bound chiral ligand or in the presence of a chiral solvent. Alternatively, a specific enantiomer can be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
XRPD
Compounds and polymorphs provided herein can be characterized by X-ray powder diffraction patterns (XRPD). The relative intensities of XRPD peaks can vary depending upon the sample preparation technique, the sample mounting procedure and the particular instrument employed, among other parameters. Moreover, instrument variation and other factors can affect the 2θ peak values. Therefore, in certain embodiments, the XRPD peak assignments can vary by plus or minus about 0.2 degrees theta or more, herein referred to as “(±0.2°)”.
XRPD patterns for each of Forms A-J and amorphous form of the compound of Formula (I) were collected with a PANalytical CubiX XPert PRO MPD diffractometer using an incident beam of CU radiation produced using an Optix long, fine-focus source. An elliptically graded multilayer mirror was used to focus Cu Kα X-rays through the specimen and onto the detector. Samples were placed on Si zero-return ultra-micro sample holders. Analysis was performed using a 10 mm irradiated width and the following parameters were set within the hardware/software:
| X-ray tube: | Cu Kα, 45 kV, 40 mA | ||
| Detector: | X′Celerator | ||
| Slits: | ASS Primary Slit: Fixed 1° | ||
| Divergence Slit (Prog): | Automatic – 5 mm irradiated length | ||
| Soller Slits: | 0.02 radian | ||
| Scatter Slit (PASS): | Automatic – 5 mm observed length | ||
| Scanning | |||
| Scan Range: | 3.0-45.0° | ||
| Scan Mode: | Continuous | ||
| Step Size: | 0.03° | ||
| Time per Step: | 10 s | ||
| Active Length: | 2.54° | ||
DSC
Compounds and polymorphs provided herein can be characterized by a characteristic differential scanning calorimeter (DSC) thermogram. For DSC, it is known in the art that the peak temperatures observed will depend upon the rate of temperature change, the sample preparation technique, and the particular instrument employed, among other parameters. Thus, the peak values in the DSC thermograms reported herein can vary by plus or minus about 2° C., plus or minus about 3° C., plus or minus about 4° C., plus or minus about 5° C., plus or minus about 6° C., to plus or minus about 7° C., or more. For some polymorph Forms, DSC analysis was performed on more than one sample which illustrates the known variability in peak position, for example, due to the factors mentioned above. The observed peak positional differences are in keeping with expectation by those skilled in the art as indicative of different samples of a single polymorph Form of a compound of Formula (I).
Impurities in a sample can also affect the peaks observed in any given DSC thermogram. In some embodiments, one or more chemical entities that are not the polymorph of a compound of Formula (I) in a sample being analyzed by DSC can result in one or more peaks at lower temperature than peak(s) associated with the transition temperature of a given polymorph as disclosed herein.
DSC analyses were performed using a Mettler 822e differential scanning calorimeter. Samples were weighed in an aluminum pan, covered with a pierced lid, and then crimped. General analysis conditions were about 30° C. to about 300° C.-about 350° C. ramped at about 10° C./min. Several additional ramp rates were utilized as part of the investigation into the high melt Form B, including about 2° C./min, about 5° C./min, and about 20° C./min. Samples were analyzed at multiple ramp rates to measure thermal and kinetic transitions observed.
Isothermal holding experiments were also performed utilizing the DSC. Samples were ramped at about 10° C./min to temperature (about 100° C. to about 250° C.) and held for about five minutes at temperature before rapid cooling to room temperature. In these cases, samples were then analyzed by XRPD or reanalyzed by DSC analysis.
TGA
A polymorphic form provided herein can give rise to thermal behavior different from that of an amorphous material or another polymorphic form. Thermal behavior can be measured in the laboratory by thermogravimetric analysis (TGA) which can be used to distinguish some polymorphic forms from others. In one embodiment, a polymorph as disclosed herein can be characterized by thermogravimetric analysis.
TGA analyses were performed using a Mettler 851e SDTA/TGA thermal gravimetric analyzer. Samples were weighed in an alumina crucible and analyzed from about 30° C. to about 230° C. and at a ramp rate of about 10° C./min.
DVS
Compounds and polymorphs provided herein can be characterized by moisture sorption analysis. This analysis was performed using a Hiden IGAsorp Moisture Sorption instrument. Moisture sorption experiments were carried out at about 25° C. by performing an adsorption scan from about 40% to about 90% RH in steps of about 10% RH and a desorption scan from about 85% to about 0% RH in steps of about −10% RH. A second adsorption scan from about 10% to about 40% RH was performed to determine the moisture uptake from a drying state to the starting humidity. Samples were allowed to equilibrate for about four hours at each point or until an asymptotic weight was reached. After the isothermal sorption scan, samples were dried for about one hour at elevated temperature (about 60° C.) to obtain the dry weight. XRPD analysis on the material following moisture sorption was performed to determine the solid form.
Optical Microscopy
Compounds and polymorphs provided herein can be characterized by microscopy, such as optical microscopy. Optical microscopy analysis was performed using a Leica DMRB Polarized Microscope. Samples were examined with a polarized light microscope combined with a digital camera (1600×1200 resolution). Small amounts of samples were dispersed in mineral oil on a glass slide with cover slips and viewed with 100× magnification.
Karl Fischer Analysis
Compounds and polymorphs provided herein can be characterized by Karl Fischer analysis to determine water content. Karl Fischer analysis was performed using a Metrohm 756 KF Coulometer. Karl Fisher titration was performed by adding sufficient material to obtain 50 μg of water, about 10 to about 50 mg of sample, to AD coulomat.
Raman Spectroscopy
Compounds and polymorphs provided herein can be characterized by Raman spectroscopy. Raman spectroscopy analysis was performed using a Kaiser RamanRXN1 instrument with the samples in a glass well. Raman spectra were collected using a PhAT macroscope at about 785 nm irradiation frequency and about 1.2 mm spot size. Samples were analyzed using 12 to 16 accumulations with about 0.5 to about 12 second exposure time and utilized cosmic ray filtering. The data was processed by background subtraction of an empty well collected with the same conditions. A baseline correction and smoothing was performed to obtain interpretable data when necessary.
FT-IR
Compounds and polymorphs provided herein can be characterized by FT-IR spectroscopy. FT-IR spectroscopy was performed using either a Nicolet Nexus 470 or Avatar 370 Infrared Spectrometer and the OMNIC software. Samples were analyzed using a diamond Attenuated Total Reflection (ATR) accessory. A compound sample was applied to the diamond crystal surface and the ATR knob was turned to apply the appropriate pressure. The spectrum was then acquired and analyzed using the OMNIC software. Alternative sample preparations include solution cells, mulls, thin films, and pressed discs, such as those made of KBr, as known in the art.
NMR
Compounds and polymorphs provided herein can be characterized by nuclear magnetic resonance (NMR). NMR spectra were obtained using a 500 MHz Bruker AVANCE with 5-mm BBO probe instrument. Samples (approximately 2 to approximately 10 mg) were dissolved in DMSO-d6 with 0.05% tetramethylsilane (TMS) for internal reference. 1H-NMR spectra were acquired at 500 MHz using 5 mm broadband observe (1H-X) Z gradient probe. A 30 degree pulse with 20 ppm spectral width, 1.0 s repetition rate, and 32-64 transients were utilized in acquiring the spectra.
High-Performance Liquid Chromatography
Compounds and polymorphs provided herein can be analyzed by high-performance liquid chromatography using an Agilent 1100 instrument. The instrument parameters for achiral HPLC are as follows:
| Column: | Sunfire C18 4.6 × 150 mm | ||
| Column Temperature: | Ambient | ||
| Auto-sampler Temperature: | Ambient | ||
| Detection: | UV at 250 nm | ||
| Mobile Phase A: | 0.05% trifluoroacetic acid in water | ||
| Mobile Phase B: | 0.05% trifluoroacetic acid in MeCN | ||
| Flow Rate: | 1.0 mL/minute | ||
| Injection Volume: | 10 μL | ||
| Data Collection time: | 20 minutes | ||
| Re-equilibration Time: | 5 minutes | ||
| Diluent & Needle Wash: | MeOH | ||
Gradient Conditions:
| Time (minutes) | % A | % B |
| 0.0 | 90 | 10 |
| 3.5 | 90 | 10 |
| 10.0 | 10 | 90 |
| 15.0 | 10 | 90 |
| 18.0 | 90 | 10 |
| 20.0 | 90 | 10 |
Compounds and polymorphs provided herein can be analyzed by high-performance liquid chromatography using a chiral HPLC column to determine % ee values:
| Column: | Chiralpak IC, 4.6 mm × 250 mm, 5 μm. |
| Column Temperature: | Room Temperature |
| Sample Temperature: | Room Temperature |
| Detection: | UV at 254 nm |
| Mobile Phase A: | 60% Hexane 40% (IPA: EtOH = 2:3) with 0.2% |
| Acetic Acid and 0.1% DEA | |
| Isocratic: | 100% A |
| Flow Rate: | 1 mL/min |
| Diluent: | Methanol |
| Injection Volume: | 10 μL |
| Analysis Time: | 25 min |
Example 8
Analytical Data of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one
Provided herein are analytical data of various purified samples of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one, the compound of Formula (I). Confirmation of the structure of the compound of Formula (I) was obtained via single crystal X-ray diffraction, FT-IR, 1H-NMR and 13C-NMR spectra.
A single crystal structure of a tert-butyl methyl ether solvate of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one (e.g., polymorph Form G) was generated and single crystal X-ray data was collected. The structure is shown in FIG. 26, which further confirmed the absolute stereochemistry as the S-enantiomer.
FT-IR spectra of Form C of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one was obtained, and shown in FIG. 27.
1H-NMR and 13C-NMR spectra of a sample of Form C of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one were obtained, and are provided in FIG. 28 and FIG. 29, respectively.
Example 9
General Methods for the Preparation of Polymorphs Form A, B, C, D, E, F, G, H, I, J of the Compound of Formula (I)
General Method A: Single Solvent Crystallization with Fast Cooling or Slow Cooling
A sample of a compound of Formula (I) (e.g., Form A or Form C) is placed into a vial equipped with stir bar and dissolved with a minimal amount of solvent (such as about 0.2 mL to about 0.3 mL) at an elevated temperature. The resulting solution is polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the vial is placed in a refrigerator (e.g., about 4° C.) overnight in a fast cooling procedure, or cooled to ambient temperature at a rate of about 20° C./h and allowed to equilibrate without stiffing at ambient temperature overnight in a slow cooling procedure. Optionally, a sample without solids can be scratched with an implement known in the art (e.g., a spatula) to initiate crystallization. The solution can be allowed to equilibrate for a period of time, such as approximately 8 hours. For a slow cooling sample, if scratching does not provide solids after about 8 hours, then a stir bar can be added and the sample then stirred overnight. A sample without precipitation can be evaporated to dryness under a gentle gas stream, such as argon, nitrogen, ambient air, etc. The precipitated solids can be recovered by vacuum filtration, centrifuge filtration, or decanted as appropriate to afford the Form as indicated below.
General Method B: Multi-Solvent Crystallization with Fast Cooling or Slow Cooling
Multi-solvent (e.g., binary) solvent crystallizations can be performed. Primary solvents include, but are not limited to, ethanol, isopropyl alcohol, methanol, tetrahydrofuran, acetone, methyl ethyl ketone, dioxane, NMP, DME, and DMF. Anti-solvents include, but are not limited to, MTBE, DCM, toluene, heptane, and water.
A sample of a compound of Formula (I) (e.g., Form A or Form C) is placed into a vial equipped with stir bar and dissolved with a minimal amount of solvent (such as about 0.2 mL to about 0.3 mL) at an elevated temperature. The resulting solution is polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the anti-solvent is added until turbidity is observed. After hot filtration, the vial is placed in a refrigerator (e.g., about 4° C.) overnight in a fast cooling procedure, or cooled to ambient temperature at a rate of about 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight in a slow cooling procedure. Optionally, a sample without solids can be scratched with an implement known in the art (e.g., a spatula) to initiate crystallization. The solution can be allowed to equilibrate for a period of time, such as approximately 8 hours. For a slow cooling sample, if scratching does not provide solids after about 8 hours, then a stir bar can be added and the sample then stirred overnight. A sample without precipitation can be evaporated to dryness under a gentle gas stream, such as argon, nitrogen, ambient air, etc. The precipitated solids can be recovered by vacuum filtration, centrifuge filtration, or decanted as appropriate to afford the Form as indicated below.
General Method C: Slurry Procedures to Afford Formula (I) Polymorph Forms
A mixture of one or more Forms (e.g., Form A or Form C) of the compound of Formula (I) are placed in a vial equipped with a stir bar. A minimal amount of solvent (e.g., a single solvent or a mixture/solution of two or more solvents) is added to the vial to form a heterogeneous slurry. Optionally, the vial can be sealed to prevent evaporation. The slurry is stirred for a period of time ranging from less than about an hour, to about 6 hours, to about 12 hours, to about 24 hours, to about 2 days, to about 4 days, to about 1 week, to about 1.5 weeks, to about 2 weeks or longer. Aliquots can be taken during the stirring period to assess the Form of the solids using, for example, XRPD analysis. Optionally, additional solvent(s) can be added during the stirring period. Optionally, seeds of a given polymorph Form of the compound of Formula (I) can be added. In some cases, the slurry is then stirred for a further period of time, ranging as recited above. The recovered solids can be recovered by vacuum filtration, centrifuge filtration, or decanted as appropriate to afford the Form as indicated below.
Example 10
Preparation of Polymorphs Form A, B, C, D, E, F, G, H, I, J of the Compound of Formula (I)
Form A
Single Solvent Crystallizations to Afford Formula (I) Form A
1. Fast Cooling Procedure From MeCN: Approximately 23 mg of Formula (I) Form A was placed into a 20-mL glass vial equipped with a stir bar. To the vial was added a minimal amount of acetonitrile (7.4 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the vial was placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by decanting off the liquid and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
2. Slow Cooling Procedure From MeCN: Approximately 24 mg of Formula (I) Form A was placed into a 20-mL glass vial equipped with a stir bar. To the vial was added a minimal amount of acetonitrile (8 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by decanting off the liquids and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
3. Slow Cooling Procedure From n-Butanol: Approximately 23 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of n-butanol (0.6 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, the vials were cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. To further induce crystallization, a stir bar was added to the vial and the contents stirred overnight. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
Binary Solvent Crystallizations to Afford Formula (I) Form A
1. Fast Cooling Procedure From Acetone/DCM: Approximately 23.5 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of acetone (2.6 ml) to just dissolve the solids at 50° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (5.0 ml) was added portion-wise. After the anti-solvent addition, the vials were placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
2. Fast Cooling Procedure From MEK/DCM: Approximately 23 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of MEK (2.2 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (5.0 ml) was added portion-wise. After the anti-solvent addition, the vial was placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
3. Fast Cooling Procedure From DMF/DCM: Approximately 24 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of DCM (0.2 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (7.0 ml) was added portion-wise. After the anti-solvent addition, the vial was placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
4. Fast Cooling Procedure From Dioxane/DCM: Approximately 24.4 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of dioxane (0.8 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (7.0 ml) was added portion-wise. After the anti-solvent addition, the vial was placed in a refrigerator (4° C.) overnight. Once at 4° C., the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
5. Slow Cooling Procedure From Acetone/DCM: Approximately 22 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of acetone (2.5 ml) to just dissolve the solids at 50° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (5.0 ml) was added portion-wise. After the anti-solvent addition, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. To further induce crystallization, a stir bar was added to the vial and the contents stirred overnight. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
6. Slow Cooling Procedure From MEK/DCM: Approximately 23.4 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of MEK (2.2 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (5.0 ml) was added portion-wise. After the anti-solvent addition, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
7. Slow Cooling Procedure From Dioxane/DCM: Approximately 24 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of dioxane (0.8 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (7.0 ml) was added portion-wise. After the anti-solvent addition, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. To further induce crystallization, a stir bar was added to the vial and the contents stirred overnight. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
8. Slow Cooling Procedure From DMF/DCM: Approximately 23.5 mg of Formula (I) Form A was placed into a 2-dram glass vial equipped with a stir bar. To the vial was added a minimal amount of DMF (0.2 ml) to just dissolve the solids at 70° C. The resulting solution was polish filtered through a 0.45 μm syringe filter into a clean preheated vial. After hot filtration, DCM (7.0 ml) was added portion-wise. After the anti-solvent addition, the vial was cooled to ambient temperature at a rate of 20° C./h and allowed to equilibrate without stirring at ambient temperature overnight. After the equilibration hold at ambient temperature, the contents of the vial were periodically scratched with a spatula to induce crystallization, and then allowed to equilibrate for approximately 8 hours. To further induce crystallization, a stir bar was added to the vial and the contents stirred overnight. To further induce crystallization, the contents of the vial were concentrated under a gentle stream of nitrogen to near dryness. The resulting crystals were collected by filtration and dried under vacuum (30 inches Hg) at ambient temperature overnight. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
Slurry Procedure to Afford Formula (I) Form A
1. Procedure from CH2Cl2 and from IPA: Form C (1 g) was slurried in five volumes of dichloromethane. After holding for 15 hours, filtration, and drying, Form A was isolated in 82% yield. Scale-up was performed on a 20 g scale with a water-wet cake of Form C to yield Form A in 92% yield. Drying at 70° C. for six days indicated no degradation in chemical or chiral purity. Slurrying dry Form C in isopropyl alcohol using a similar method also yielded Form A.
2. Procedure for Competitive Slurry Experiment (using forms A, B and C): Competitive slurries were performed by charging approximately a 50/50 mixture of Forms A and C (11.2 mg of Form A and 11.7 mg Form C) to a 1-dram glass vial equipped with a glass stir bar. To the vial was added 600 μL of MeCN. The vial cap was wrapped with parafilm to prevent evaporation. The slurry was stirred for 1 day and an aliquot was taken. The contents of the vial were allowed to stir for an additional week and another aliquot was taken. Both aliquots were centrifuge filtered for five minutes at 8000 RPM. XRPD analysis was performed on the solids from each aliquot to show that the Formula (I) had converted to Form A at both time points. After the one week aliquot was taken, an additional 300 μL of acetonitrile was added to the remaining slurry and allowed to equilibrate for one day. The slurry was then seeded with approximately 3.2 mg of Form B and allowed to equilibrate for an additional three days. The solids were isolated by centrifuge filtration (5 minutes at 8000 RPM) and dried over night under vacuum. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
3. Procedure for Competitive Slurry Experiment (using forms A, C, D, and E): Competitive slurries were performed by charging an approximately equal mixture of each form (7.8 mg of Form A, 7.7 mg Form C, 7.7 mg of Form D, and 8.2 mg of Form E) to a 1-dram glass vial equipped with a glass stir bar. To the vial was added 1 ml of 2-propanol. The vial cap was wrapped with parafilm to prevent evaporation. The slurry was mixed for 1 day and an aliquot was taken. The contents of the vial were allowed to stir for an additional week and another aliquot was taken. Both aliquots were centrifuge filtered for five minutes at 8000 RPM. XRPD analysis was performed on the solids from each aliquot to show that the Formula (I) had converted to Form A at both time points. After the one week aliquot was taken, the remaining solids were isolated by centrifuge filtration (5 minutes at 8000 RPM) and dried over night under vacuum. The dried solids were evaluated for crystallinity and form by XRPD which indicated the crystalline material was polymorph Form A.
Using the General Method B of Example 9, the following experiments detailed in Tables 4 and 5 were performed to afford Formula (I) Form C. Table 4 experiments were conducted using the fast cooling procedure, while Table 5 experiments were conducted using the slow cooling procedure.
Table 4. Fast Cooling Procedure

Table 5. Slow Cooling Procedure

Using General Method C of Example 9, the following experiments detailed in Table 6 were performed to afford the polymorph Form of the compound of Formula (I) as indicated.
Table 6:

Example 12
XRPD Studies
[00653] Using the XRPD instrument and parameters described above, the following XRPD peaks were observed for Formula (I) Polymorph Forms A, B, C, D. E, F, G, H, I, and J. The XRPD traces for these ten polymorph forms are given in Figures 1-10, respectively. In Table 7, peak position units are °2Θ. In one embodiment, a given polymorph Form can be characterized as having at least one of the five XRPD peaks given in Set 1 in Table 7. In another embodiment, the given Form can be characterized as having at least one of the five XRPD peaks given in Set 1 in combination with at least one of the XRPD peaks given in Set 2 in Table 7. In some embodiments, one or more peak position values can be defined as being modified by the term “about” as described herein. In other embodiments, any given peak position is with ±0.2 2Θ (e.g., 9.6+0.2 2Θ).
Table 7.

Example 13
Differential Scanning Calorimetry (DSC) Studies
[00654] Using the DSC instrument and parameters described above, the following DSC peaks were observed for the compound of Formula (I) polymorph Forms A, B, C, D. E, F, G, H, I, and J. The DSC thermograms for these nine polymorph forms are given in FIGS. 12-24, respectively, and peak positions are given in Table 8. Further DSC data for Polymorph Forms A, B, C, D. E, F, G, H, I, and J is given in Table 9 below. Unless marked with a Λ that indicates an exothermic peak, all peaks are endothermic.
Table 8.

Table 9 summarizes non-limiting exemplary preparation techniques for Formula (I) Polymorph Forms A-J and representative analytical data as described below and elsewhere.
Table 9.


http://www.google.com/patents/US8809349?cl=en
Extras…….
SEE NMR ……….http://www.medkoo.com/Product-Data/IPI-145/IPI-145-QC-SSC20130422Web.pdf
http://www.chemietek.com/Files/Line2/CHEMIETEK,%20IPI-145%20(01),%20NMR.pdf

New Parathyroid Disease Drug Etelcalcetide Seeks FDA Approval
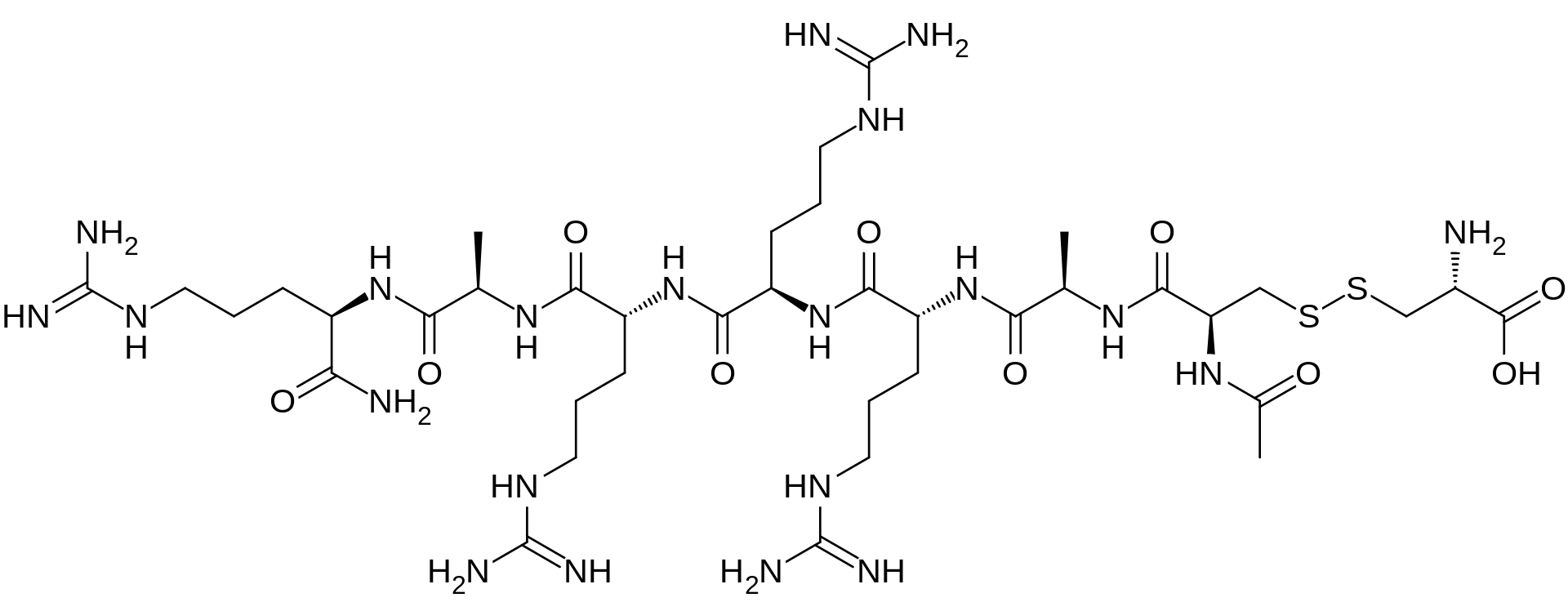
Etelcalcetide, AMG 416
AMG-416; Etelcalcetide hydrochloride; KAI-4169; KAI-4169-HCl; ONO-5163; Telcalcetide; Velcalcetide; Velcalcetide hydrochloride
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine,
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine
Secondary hyperparathyroidism
- Originator KAI Pharmaceuticals…Kai Pharmaceuticals, Inc.
- Developer Amgen; KAI Pharmaceuticals; Ono Pharmaceutical
- ClassDisulfides; Peptides
- Mechanism of ActionCalcium-sensing receptor agonists
New Parathyroid Disease Drug Seeks FDA Approval
Amgen is seeking FDA approval for etelcalcetide (AMG 461), the first calcimimetic agent administered intravenously after dialysis to treat secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD).
SHPT is a common and serious condition that is often progressive among CKD patients. It usually manifests as high amounts of parathyroid hormone (PTH) associated with abnormal calcium and phosphorus levels in the body.
– See more at: http://www.pharmacytimes.com/product-news/new-parathyroid-disease-drug-seeks-fda-approval
Etelcalcetide is a D-amino peptide calcimimetic undergoing clinical evaluation for the treatment of secondary hyperparathyroidismfor patients with chronic kidney disease (CKD) on hemodialysis. Etelcalcetide is administered intravenously at the end of each dialysis session.[1][2] It exerts a pharmacological effect by binding to and activating the calcium-sensing receptor (CaSR) in theparathyroid gland, resulting in parathyroid hormone (PTH) reduction and suppression.[1] Elevated PTH is often observe in patients with CKD.[3]
On August 25, 2015 Amgen Inc. announced its submission of a New Drug Application to the Food and Drug Administration for etelcalcetide.[1]
| CAS Registry Number | 1262780-97-1 |
|---|---|
| Synonyms | Velcalcetide |
| Chemical data | |
| Formula | C38H73N21O10S2 |
| Molecular mass | 1,048.26 g·mol−1 |
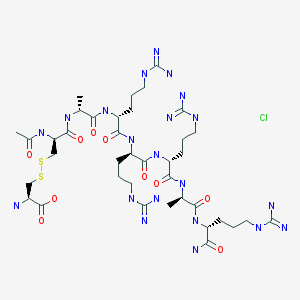
Etelcalcetide hydrochloride
RN: 1334237-71-6
UNII: 72PT5993DU
The term “AMG 416” refers to the compound having the chemical name: JV-acetyl-D- cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-arginamide disulfide with L- cysteine, which may be represented as:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The terms “AMG 416 hydrochloride” or “AMG 416 HQ” are interchangeable and refer to the compound having the chemical name: N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl- D-arginyl-D-alanyl-D-arginamide disulfide with L-cysteine hydrochloride, which may be represented as:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · xHCl
D-Argininamide, N-acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-, disulfide with L-cysteine, hydrochloride (1:?)
N-Acetyl-D-cysteinyl-D-alanyl-D-arginyl-D-arginyl-D-arginyl-D-alanyl-D-argininamide disulfide with L-cysteine hydrochloride
Amgen today announced the submission of a New Drug Application (NDA) with the United States Food and Drug Administration (FDA) for etelcalcetide (formerly AMG 416) for the treatment of secondary hyperparathyroidism (SHPT) in patients with chronic kidney disease (CKD) on hemodialysis. If approved, etelcalcetide will be the first calcimimetic agent that can be administered intravenously at the end of the dialysis session.
“Secondary hyperparathyroidism is a serious, progressive disease that can lead to significant clinical consequences and is also associated with a high pill burden for patients,” said Sean E. Harper, M.D., executive vice president of Research and Development at Amgen. “We look forward to working with regulatory authorities during the review process to bring this important treatment to market, helping to fill an unmet need for the many patients impacted by this disease.”
Etelcalcetide is a novel calcimimetic agent that suppresses the secretion of parathyroid hormone and is in clinical development for the treatment of SHPT in patients with CKD on hemodialysis. Etelcalcetide is administered intravenously three times per week at the end of each dialysis session. It acts by binding to and activating the calcium-sensing receptor on the parathyroid gland, thereby causing decreases in parathyroid hormone (PTH). Sustained elevations in PTH are known to be associated with significant clinical consequences for patients with CKD.
The submission includes data from three Phase 3 studies, all of which met the primary endpoints, including two pooled placebo-controlled trials in more than 1,000 patients and a head-to-head study evaluating etelcalcetide compared with cinacalcet.
About Secondary Hyperparathyroidism
SHPT is a common and serious condition that is often progressive among patients with CKD, and it affects many of the approximately two million people throughout the world who are receiving dialysis, including 450,000 people in the U.S. The disorder develops early in the course of CKD and usually manifests as increased levels of PTH as a result of increased production from the parathyroid glands (four small glands in the neck). Patients with end stage renal disease who require maintenance dialysis often have substantial elevations of PTH that are commonly associated with abnormal calcium and phosphorus levels and an increased risk of significant clinical consequences.
About Etelcalcetide (AMG 416)
Etelcalcetide is a novel calcimimetic agent in clinical development for the treatment of SHPT in CKD patients on hemodialysis that is administered intravenously at the end of the dialysis session. Etelcalcetide binds to and activates the calcium-sensing receptor on the parathyroid gland, thereby decreasing PTH levels.
About Sensipar® (cinacalcet)
Sensipar® (cinacalcet) is the first oral calcimimetic agent approved by the FDA for the treatment of SHPT in adult patients with CKD on dialysis. Sensipar is not indicated for use in adult patients with CKD who are not on dialysis because of an increased risk of hypocalcemia. The therapy is also approved in the U.S. for treatment of hypercalcemia in adult patients with parathyroid carcinoma and hypercalcemia in adult patients with primary HPT for whom parathyroidectomy would be indicated on the basis of serum calcium levels, but who are unable to undergo parathyroidectomy. Sensipar binds to the calcium-sensing receptor, resulting in a drop in PTH levels by inhibiting PTH synthesis and secretion. In addition, the reductions in PTH lower serum calcium and phosphorus levels.
…………………
WO 2011014707
http://www.google.com/patents/WO2011014707A2?cl=en
……………………..
WO 2014210489
http://www.google.com/patents/WO2014210489A1?cl=en
A variety of compounds having activity for lowering parathyroid hormone levels have been described. See International Publication No. WO 2011/014707. In one embodiment, the compound may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2
The main chain has 7 amino acids, all in the D-configuration and the side-chain cysteine residue is in the L-configuration. The amino terminal is acetylated and the carboxyl-terminal is amidated. This compound (“AMG-416”) has utility for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis patients. A liquid formulation comprising AMG-416 may be administered to a subject intravenously. The hydrochloride salt of AMG-416 may be represented as follows:
H-L-Cys-OH
S— S
Ac-D-Cys-D-Ala-D-Arg-D-Arg-D-Arg-D-Ala-D-Arg-NH2 · x(HCl)
Therapeutic peptides pose a number of challenges with respect to their formulation. Peptides in general, and particularly those that contain a disulfide bond, typically have only moderate or poor stability in aqueous solution. Peptides are prone to amide bond hydrolysis at both high and low pH. Disulfide bonds can be unstable even under quite mild conditions (close to neutral pH). In addition, disulfide containing peptides that are not cyclic are particularly prone to dimer formation. Accordingly, therapeutic peptides are often provided in lyophilized form, as a dry powder or cake, for later reconstitution. A lyophilized formulation of a therapeutic peptide has the advantage of providing stability for long periods of time, but is less convenient to use as it requires the addition of one or more diluents and there is the potential risk for errors due to the use of an improper type or amount of diluent, as well as risk of contamination. In addition, the lyophilization process is time consuming and costly.
Accordingly, there is a need for an aqueous liquid formulation comprising a peptide agonist of the calcium sensing receptor, such as AMG 416. It would be desirable for the liquid formulation to remain stable over a relevant period of time under suitable storage conditions and to be suitable for administration by intravenous or other parenteral routes.
…………………………………
Milestones
- 25 Aug 2015Preregistration for Secondary hyperparathyroidism in USA (IV)
- 29 May 2015Pooled analysis efficacy and adverse events data from two phase III trials in secondary hyperparathyroidism released by Amgen
- 21 Apr 2015Amgen plans to submit Biological License Application to USFDA and Marketing Authorisation Application to EMA for Secondary hyperparathyroidism
References
- “Amgen Submits New Drug Application For Novel Intravenous Calcimimetic Etelcalcetide (AMG 416)”
- “Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects
- “Evidence for Chronic Kidney Disease-Mineral and Bone Disorder Associated With Metabolic Pathway Changes”
KAI-4169, a novel calcium sensing receptor agonist, decreases serum iPTH, FGF-23 and improves serum bone markers in a phase 2 study in hemodialysis subjects with chronic kidney disease-mineral and bone disorder
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO054
KAI-4169, a novel peptide agonist of the calcium sensing receptor, attenuates PTH and soft tissue calcification and restores parathyroid gland VDR levels in uremic rats
49th Congr Eur Renal Assoc – Eur Dialysis Transpl Assoc (May 24-27, Paris) 2012, Abst SAO014
Long term safety and efficacy of velcalcetide (AMG 416), a calcium-sensing receptor (CaSR) agonist, for the treatment of secondary hyperparathyroidism (SHPT) in hemodialysis (HD) patients
Kidney Week (November 5-10, Atlanta, GA) 2013, Abst SA-PO575
Preclinical PK and PD relationship for KAI-4169, a novel calcimimetic
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P1-198
KAI-4169, a novel calcimimetic for the treatment of secondary hyperparathyroidism
93rd Annu Meet Endo Soc (June 4-7, Boston) 2011, Abst P2-98
Characterization of KAI-4169, a novel peptide for the treatment of chronic kidney disease – Mineral and bone disorder, in a phase I study in healthy males
44th Annu Meet Am Soc Nephrol (ASN) (November 8-13, Philadelphia) 2011, Abst FR-PO1238
| WO2011014707A2 | Jul 29, 2010 | Feb 3, 2011 | Kai Pharmaceuticals, Inc. | Therapeutic agents for reducing parathyroid hormone levels |
////Etelcalcetide, Parathyroid Disease, Amgen Inc, AMG 416, KAI-4169; KAI-4169-HCl, ONO-5163, Telcalcetide, Velcalcetide, Velcalcetide hydrochloride
FDA approves Repatha to treat certain patients with high cholesterol
August 27, 2015
Release
The U.S. Food and Drug Administration today approved Repatha (evolocumab) injection for some patients who are unable to get their low-density lipoprotein (LDL) cholesterol under control with current treatment options.
Repatha, the second drug approved in a new class of drugs known as PCSK9 inhibitors, is approved for use in addition to diet and maximally-tolerated statin therapy in adult patients with heterozygous familial hypercholesterolemia (HeFH), homozygous familial hypercholesterolemia (HoFH), or clinical atherosclerotic cardiovascular disease, such as heart attacks or strokes, who require additional lowering of LDL cholesterol.
Familial hypercholesterolemia (encompassing both HeFH and HoFH) is an inherited condition that causes high levels of LDL cholesterol. A high level of LDL cholesterol in the blood is linked to cardiovascular or heart disease. Heart disease is the number one cause of death for Americans, both men and women. According to the Centers for Disease Control and Prevention, about 610,000 people die of heart disease in the United States every year– that equals one in every four deaths.
“Repatha provides another treatment option in this new class of drugs for patients with familial hypercholesterolemia or with known cardiovascular disease who have not been able to lower their LDL cholesterol enough with statins,” said John Jenkins, M.D., director of the Office of New Drugs, Center for Drug Evaluation and Research. “Cardiovascular disease is a serious threat to the health of Americans, and the FDA is committed to facilitating the development and approval of effective and safe drugs to address this important public health problem.”
Repatha is an antibody that targets a specific protein, called PCSK9. PCSK9 reduces the number of receptors on the liver that remove LDL cholesterol from the blood. By blocking PCSK9’s ability to work, more receptors are available to get rid of LDL cholesterol from the blood and, as a result, lower LDL cholesterol levels.
The efficacy and safety of Repatha were evaluated in one 52-week placebo-controlled trial and eight 12-week placebo-controlled trials in participants with primary hyperlipidemia, including two that specifically enrolled participants with HeFH and one that enrolled participants with HoFH. In one of the 12-week studies, 329 participants with HeFH, who required additional lowering of LDL cholesterol despite statins with or without other lipid-lowering therapies, were randomized to receive Repatha or placebo for 12 weeks. Participants taking Repatha had an average reduction in LDL cholesterol of approximately 60 percent, compared to placebo.
The most common side effects of Repatha include nasopharyngitis, upper respiratory tract infection, flu, back pain, and reactions such as redness, pain, or bruising where the injection is given. Allergic reactions, such as rash and hives, have been reported with the use of Repatha. Patients should stop using Repatha and get medical help if they experience symptoms of a serious allergic reaction.
Multiple clinical trials have demonstrated that statins lower the risk of having a heart attack or stroke. A trial evaluating the effect of adding Repatha to statins for reducing cardiovascular risk is ongoing.
Repatha is marketed by Amgen Inc., of Thousand Oaks, Calif.
Vintafolide

Vintafolide, EC-145 , MK-8109
mw 1917.041, cas 742092-03-1, mf C86 H109 N21 O26 S2
(2S)-2-[(4-{[(2-amino-4-oxo-3H-pteridin-6-yl)methyl]amino}phenyl)formamido]-4-{[(1S)-1-{[(1S)-4-carbamimidamido-1-{[(1S)-2-carboxy-1-{[(1S)-2-carboxy-1-{[(1R)-1-carboxy-2-({2-[({[(1R,9R,10S,11R,12R,19R)-12-ethyl-4-[(13S,15R,17S)-17-ethyl-17-hydroxy-13-(methoxycarbonyl)-1,11-diazatetracyclo[13.3.1.04,12.05,10]nonadeca-4(12),5,7,9-tetraen-13-yl]-10,11-dihydroxy-5-methoxy-8-methyl-8,16-diazapentacyclo[10.6.1.01,9.02,7.016,19]nonadeca-2,4,6,13-tetraen-10-yl]formohydrazido}carbonyl)oxy]ethyl}disulfanyl)ethyl]carbamoyl}ethyl]carbamoyl}ethyl]carbamoyl}butyl]carbamoyl}-2-carboxyethyl]carbamoyl}butanoic acid
Vincaleukoblastin-23-oic acid, O4-deacetyl-, 2-[(2-mercaptoethoxy)carbonyl]hydrazide, disulfide with N-[4-[[(2-amino-1,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine
Endocyte innovator
Vintafolide is an investigational targeted cancer therapeutic currently under development by Endocyte and Merck & Co.[1] It is a small molecule drug conjugate consisting of a small molecule targeting the folate receptor, which is overexpressed on certain cancers, such as ovarian cancer, and a potent chemotherapy drug, vinblastine.[2] It is being developed with a companion imaging agent, etarfolatide, that identifies patients that express the folate receptor and thus would likely respond to the treatment with vintafolide.[3] A Phase 3 study evaluating vintafolide for the treatment of platinum-resistant ovarian cancer (PROCEED trial) and a Phase 2b study(TARGET trial) in non-small-cell lung carcinoma (NSCLC) are ongoing.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
A Marketing Authorization Application (MAA) filing for vintafolide and etarfolatide for the treatment of patients withfolate receptor-positive platinum-resistant ovarian cancer in combination with doxorubicin, pegylated liposomal doxorubicin (PLD), has been accepted by the European Medicines Agency.[6] The drug received an orphan drug status in Europe in March 2012.[1] Merck & Co. acquired the development and marketing rights to this experimental cancer drug from Endocyte in April 2012.[1] The drug received orphan drug status in Europe in March 2012.[3]Endocyte remains responsible for the development and commercialization of etarfolatide, a non-invasive companion imaging agent used to identify patients expressing the folate receptor that will likely respond to treatment with vintafolide.[4] Vintafolide is designed to deliver the toxic vinblastine drug selectively to cells expressing the folate receptor using folate targeting.[5]
In 2014 Merck and Endocyte stopped a late-stage study of vintafolide in treating ovarian cancer on the recommendation of a data safety monitoring board, saying that the drug failed to improve progression-free survival.[7]
Vintafolide is folate-conjugated with DAVBLH, which is a derivative of the vinca alkaloid vinblastine.Vinblastine is a microtubule-destabilizing agent that binds tubulin and causes M phase-specific cell cycle arrest and apoptosis of mitotically active cells. Vinblastine is an extremely potent chemotherapeutic agent but has significant toxicities including bone marrow suppression, neurotoxicity, gastrointestinal toxicity and vesicant injury.
Endocyte’s desacetylvinblastinehydrazide/folate conjugate (EC-145) is a folate-targeted cytotoxic anticancer drug in early development for the treatment of non-small cell lung cancer (NSCLC) and breast cancer. The compound had been pre-registered in the E.U. by Merck for the treatment of ovarian cancer, but the application was withdrawn due to lack of efficacy.
In 2012, the product was licensed to Merck & Co. by Endocyte for worldwide exclusive development and commercialization. In 2014, however, this license agreement was terminated and Endocyte regained all rights.
Folates can serve as one-carbon donors in reactions that are critical in the de novo biosynthesis of purines and thymidylate, amino acid metabolism and methylation reactions. Folate can enter a cell by two routes: RFC or by membrane-bound FRs. RFC is a bidirectional anion transporter that is the normal entry method for reduced folates in most cells. By contrast, FRs are expressed in a limited distribution in normal tissues but are overexpressed in multiple cancers including ovarian, lung, breast and colorectal cancer. FRs bind folate derivatives with high affinity and mediate their internalization by endocytosis. Given that FRs are not typically expressed on the luminal surface of epithelial cells, making them inaccessible to normal circulation, they are attractive therapeutic targets with limited toxicity. In addition to the therapeutic agent vintafolide, a radiodiagnostic agent (99mTc-etarfolatide [EC20]) has been developed to allow single-photon emission computed tomography (SPECT) imaging to identify FR-expressing tissues (tumors).
In 2012, orphan drug designations were assigned in the E.U. for the treatment of ovarian cancer and to be used with folic acid for the diagnosis of positive folate-receptor status in ovarian cancer. In 2013, orphan drug designation was assigned in the U.S. for the treatment of ovarian cancer.
Vintafolide is a water-soluble derivative of folic acid and the vinca alkaloid DAVLBH. The molecules are connected through a hydrophilic L-peptide spacer and a disulfide linker (Figure 1). The disulfide linker serves as a cleavable bond that is necessary for drug release following receptor mediated endocytosis. The disulfide bond is reduced in the acidic environment of the endosome, leading to efficient release of vinblastine.

Vintafolide.
DAVBLH: Desacetylvinblastine hydrazide

Structure of vintafolide and mechanism of release of the payload in the endosome.

Mechanism of action
Folate is required for cell division, and rapidly dividing cancer cells often express folate receptors in order to capture enough folate to support rapid cell growth. Elevated expression of the folate receptor occurs in many diseases, including other aggressively growing cancers and inflammatory disorders.[8] Vintafolide binds to the folate receptor and is subsequently taken up by the cell through a natural internalization process called endocytosis. Once inside the cell, vintafolide’s linker releases the chemotherapy drug which kills the cell.[3]
……………
Bioorganic & Medicinal Chemistry Letters (2006), 16(19), 5093-5096
http://www.sciencedirect.com/science/article/pii/S0960894X06008079
An efficient synthesis of the folate receptor (FR) targeting conjugate EC145 is described. EC145 is a water soluble derivative of the vitamin folic acid and the potent cytotoxic agent, desacetylvinblastine monohydrazide. Both molecules are connected in regioselective manner via a hydrophilic peptide spacer and a reductively labile disulfide linker.

………approach for the design and regioselective synthesis of a FA-vinca alkaloid conjugate 1 (EC145,BELOW). As indicated in the retrosynthetic scheme, 1 can be assembled by tethering a FA-Spacer unit 2 to the highly potent cytotoxic molecule, desacetylvinblastine monohydrazide 3, via a linker containing a reducible disulfide bond. The latter is important for drug delivery applications since real-time imaging using a fluorescence resonance energy transfer technique has recently demonstrated that reduction-mediated release of the drug cargo from a disulfide linked FA-conjugate efficiently occurs within the endosomes of cancer cells.

-
Scheme 1.
Reagents and conditions: (i) a—Fmoc-Asp(OtBu)-OH, PyBOP, DIPEA, RT, 1 h; b—20% piperidine/DMF, rt, 10 min; (ii) a—Fmoc-Arg(Pbf)-OH, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iii) a—Fmoc-Glu-OtBu, PyBOP, DIPEA, rt, 1 h; b—20% piperidine/DMF, rt, 10 min; (iv) N10-TFA-pteroic acid, PyBOP, DIPEA, rt, 1.5 h; (v) TFA/H2O/TIPS/EDT (92.5:2.5:2.5:2.5), rt, 1 h; (vi) aq NH4OH, pH 9.3, rt, 1 h.
- Selected 1H NMR data for 2 (D2O, 300 MHz): δ 8.68 (s, 1H, FA H-7), 7.57 (d, 2H,J = 8.4 Hz, FA H-12 & 16), 6.67 (d, 2H, J = 9 Hz, FA H-13 & 15), 4.40–4.75 (series of m, 5H), 4.35 (m, 2H), 4.16 (m, 1H), 3.02 (m, 2H), 2.55–2.95 (series of m, 8H), 2.42 (m, 2H), 2.00–2.30 (m, 2H), 1.55–1.90 (m, 2H), 1.48 (m, 2H).
- 1H NMR for compound 6 (DMSO-d6, 300 MHz): δ 8.38 (m, 1H), 8.16 (dt, 1H, J = 8 Hz, 1 Hz), 8.02 (dt, 1H, J = 8 Hz, 1 Hz), 7.88 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz), 7.7 (m, 2H), 7.63 (ddd, 1H, J = 8 Hz, 7 Hz, 1 Hz,), 7.4–7.2 (br, 1H), 7.2 (m, 1H), 4.72 (t, 2H,J = 6 Hz), 3.36 (t, 2H, J = 6 Hz).
- Selected 1H NMR data for
EC145 (D2O, 300 MHz): δ 8.67 (s, 1H, FA H-7), 7.50 (br s, 1H, VLB H-11′), 7.30–7.40 (br s, 1H, VLB H-14′), 7.35 (d, 2H, J = 7.8 Hz, FA H-12 & 16), 7.25 (m, 1H, VLB H-13′), 7.05 (br s, 1H, VLB H-12′), 6.51 (d, 2H, J = 8.7 Hz, FA H-13 & 15), 6.4 (s, 2H, VLB H-14 & 17), 5.65 (m, 1H, VLB H-7), 5.5 (m, 1H, VLB H-6), 4.15 (m,1H, VLB H-8′), 3.82 (s, 3H, VLB C18′ –CO2CH3), 3.69 (s, 3H, VLB C16 –OCH3), 2.8 (s, 3H, VLB N–CH3), 1.35 (br s, 1H, VLB H-3′), 1.15 (m, 1H, VLB H-2′), 0.9 (t, 3H, J = 7 Hz, VLB H-21′), 0.55 (t, 3H, J = 6.9 Hz, VLB H-21).
 CLICK ON IMAGE FOR CLEAR VIEW
CLICK ON IMAGE FOR CLEAR VIEW
…………
WO 2004069159
http://www.google.com/patents/WO2004069159A2?cl=en
EXAMPLE 16b

The compounds of Examples 16a and 16b were prepared from the peptidyl fragment Pte-Glu-Asp-Arg-Asp-Asp-Cys-OH , prepared according to the general procedure described in Scheme 12. The Michael addition of this peptidyl fragment to the maleimido derivative of seco-CBI-bis-indole resulted in the folate conjugates Example 16a. The peptidyl fragment also reacted with either the thiosulfonate or pyridyldithio-activated vinblastine to form Example 16b. The maleimido derivative of seco-CBI-bis-indole, and the thiosulfonate and pyridyldithio- activated vinblastine intermediates were prepared using the procedures described herein for other examples.
……………..
https://www.google.com/patents/WO2012142281A1?cl=en
Folate-targeted drugs have been developed and are being tested in clinical trials as cancer therapeutics. EC145, also known as vintafolide, comprises a highly potent vinca alkaloid cytotoxic compound, desacetylvinblastine hydrazide (DAVLBH), conjugated to folate. The EC 145 molecule targets the folate receptor found at high levels on the surface of epithelial tumors, including non-small cell lung carcinomas (NSCLC), ovarian, endometrial and renal cancers, and others, including fallopian tube and primary peritoneal carcinomas. It is believed that EC 145 binds to tumors that express the folate receptor delivering the vinca moiety directly to cancer cells while avoiding normal tissue. Thus, upon binding, EC 145 enters the cancer cell via endocytosis, releases DAVLBH and causes cell death or inhibits cell function. EC 145 has the following formula

EC145
and has been accorded the Chemical Abstracts Registry Number 742092-03-1. As used herein, according to the context, the term EC 145 means the compound, or a pharmaceutically acceptable salt thereof; and the compound may be present in a solid, solution or suspension in an ionized form, including a protonated form. EC145 is disclosed in U.S. Patent No. 7,601,332; and particular uses and an aqueous liquid pH 7.4, phosphate-buffered formulation for intravenous administration are disclosed in WO 2011/014821. As described in WO 2011/014821, it is necessary to store the aqueous liquid formulation in the frozen state to ensure its stability. To avoid this necessity, a formulation is needed which has adequate stability at ambient temperature.
As one aspect of the invention described herein, there is provided a pharmaceutical composition of EC145 which is a lyophilized solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
In another aspect of the invention, there is provided a pharmaceutical composition of EC 145 which is an X-ray amorphous solid which has adequate stability for storage at ambient temperature and which is capable of redissolving in an aqueous diluent prior to administration.
| Systematic (IUPAC) name | |
|---|---|
|
N-(4-{[(2-Amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}benzoyl)-L-γ-glutamyl-L-α-aspartyl-L-arginyl-L-α-aspartyl-L-α-aspartyl-L-cysteine disulfide with methyl (5S,7R,9S)-5-ethyl-9-[(3aR,4R,5S,5aR,10bR,13aR)-3a-ethyl-4,5-dihydroxy-8-methoxy-6-methyl-5-({2-[(2-sulfanylethoxy)carbonyl]hydrazinyl}carbonyl)-3a,4,5,5a,6,11,12,13a-octahydro-1H-indolizino[8,1-cd]carbazol-9-yl]-5-hydroxy-1,4,5,6,7,8,9,10-octahydro-2H-3,7-methanoazacycloundecino[5,4-b]indol-9-carboxylate
|
|
| Clinical data | |
| Legal status |
|
| Identifiers | |
| CAS Registry Number | 742092-03-1 |
| ATC code | L01CA06 |
| ChemSpider | 27444385 |
| Synonyms | EC-145 |
| Chemical data | |
| Formula | C86H109N21O26S2 |
| Molecular mass | 1917 g/mol |
References
- Sridharan, Balaji (Apr 16, 2012). “Endocyte soars on cancer drug deal with Merck”. Reuters.
- Statement on a nonproprietary name adopted by the USAN Council, United States Adopted Names (USAN) Council, 6 April 2012
- Kuo, Phillip H. (February 2013). “Companion Imaging Diagnostics for Targeted Therapies”. Radiology Today 14 (2): 32.
- “Merck, Endocyte in Development Deal”. Drug Development & Discovery magazine. 2012-04-25.
- Dosio, F.; Milla, P.; Cattel, L. (2010). “EC-145, a folate-targeted Vinca alkaloid conjugate for the potential treatment of folate receptor-expressing cancers”. Current opinion in investigational drugs (London, England : 2000) 11 (12): 1424–1433. PMID 21154124.
- “EMA Accepts For Review MAA Filings For Vintafolide And Etarfolatide”. rttnews.com. 2012-11-27.
- Garde, Damian (2014-05-02). “Merck halts study of the billion-dollar cancer drug vintafolide”. Fierce Biotech. Retrieved 21 April 2015.
- “Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay” 338 (2). March 2005. pp. 284–93. doi:10.1016/j.ab.2004.12.026.PMID 15745749.
-
WO2008098970A1 * Feb 13, 2008 Aug 21, 2008 Pf Medicament Anhydrous crystalline vinflunine salts, method of preparation and use thereof as a drug and means of vinflunine purification WO2010150100A1 * Jun 23, 2010 Dec 29, 2010 Entarco Sa The use of spinosyns and spinosyn compositions against diseases caused by protozoans, viral infections and cancer WO2011014821A1 * Jul 30, 2010 Feb 3, 2011 Endocyte, Inc. Folate-targeted diagnostics and treatment US20100247669 * Sep 30, 2010 Cerulean Pharma Inc. Polymer-agent conjugates, particles, compositions, and related methods of use
////////Vintafolide, BMS-753493, DAVBLH, Desacetylvinblastine hydrazide, EC-145 , MK-8109 , phase 2
Sandoz, Pfizer closing Mumbai API plant

Sandoz closing Mumbai API plant but says it remains committed to India

Sandoz will shutter an Indian API facility in 2016 as part of a manufacturing refocus in the region.
From online sources
Drug major Sandoz will discontinue operations at its Turbhe site (Maharashtra) by end December 2016, as part of global plans to optimise its manufacturing network.

The Turbhe sites employs 170 people and manufactures antibiotics and active pharmaceutical ingredients (API), a note from the company said. Sandoz is the generic drugs arm of pharmaceutical company Novartis.
“Sandoz will refocus its manufacturing set up in India as part of its strategy to optimise its global manufacturing network, while continuing to serve patients in India,” the company said. As part of the plan, Sandoz will focus its manufacturing at other sites which employ over 1,300 employees and produce over three billion tablets and 180 tonnes of API annually, it added. The company has two manufacturing facilities at Kalwe and Mahad.
“We made the announcement today to ensure our associates are informed as soon as possible about our decisions and to ensure a transparent process,” Vivek Devaraj, Sandoz Country Head in India, said in the statement. “We are committed to managing the process with care, sensitivity and respect for all impacted associates at Turbhe, to supporting our customers through the transition and to meeting patient needs for access to important medicines,” he added.
In 2012, the company had shut down its formulations and API development centres, respectively. Drug companies have in the past shut down plants in India as a fallout of global strategies, mergers and acquisitions. At present, Pfizer’s plant in Thane faces an uncertain future.

……………………
ABOUT PFIZER
 There has been practically no production at Pfizer’s Thane plant (pic above) since 2013.
There has been practically no production at Pfizer’s Thane plant (pic above) since 2013.

Sandoz India’s Turbhe plant to down shutters by December 2016
Less than a week after Sandoz, the generic division of Novartis, announced that it would discontinue operations at its Turbhe site by end December 2016, another MNC, Pfizer India announced the closure of its manufacturing facility at Thane, two months from today, from September 16, 2015.
According to Pfizer India spokesperson, the Thane plant was commissioned in the 1960s, manufacturing medicines for both domestic and international markets but there has been ‘practically been no production activity at this plant since 2013′. Hence closure of the site would not impact supply of Pfizer India’s medicines.
Both plant closures are a consolidation of manufacturing facilities, with the shutting down of older facilities and re-direction to more modern facilities, with Pfizer India’s statement attributing the decision to ‘an assessment of its long term viability and its ability to achieve the needed production.’
132 of the 212 Pfizer India workmen at the Thane plant had already taken up the voluntary retirement scheme (VRS) offered by the company and the statement indicated that the remaining 80 workmen who continued to receive full wages despite plant inactivity, would also receive requisite compensation as mandated by law.
While the close down process is in the final stages at Pfizer India’s Thane facility, Sandoz’ July 10 announcement is the beginning of the process at its Turbhe plant, which employs 170 associates and manufactures antibiotics and APIs.
“We made the announcement (on July 10) to ensure our associates are informed as soon as possible about our decisions and to ensure a transparent process,” said Vivek Devaraj, Sandoz Country Head in India. He said the company was “committed to managing the process with the utmost care, sensitivity and respect for all impacted associates at Turbhe, to supporting our customers through the transition and to meeting patient needs for access to important medicines.” Manufacturing would now focus at its other sites which employ over 1,300 associates and produce over three billion tablets and 180 tonnes of API annually.”
/////////Sandoz, shutter, Indian API facility, Pfizer
Mahendra Chemicals gets FDA Warning Letter with Focus on “Data Integrity”
DRUG REGULATORY AFFAIRS INTERNATIONAL

A Warning Letter issued by the US Food & Drug Administration (FDA) to an Indian API manufacturer on 13 July 2015 shows again a clear focus on the missing integrity of data. Specifically, the following issues are addressed:
1. Activities were not recorded at the time they were carried out and original data were deleted:
Entries in the manufacturing protocols were made only days after the relevant activities had been conducted. Further, batches were released before all results were available.
In particular the use of “rough notes” was criticised as these original data were completely destroyed after transfer in the batch records.
2. Due to unauthorised access to data systems, data could be modified or deleted:
Specifically HPLC, GC, and Karl Fischer Titrators were concerned. For instance, for the GC instrument multiple copies of raw data were found in the waste. And there was no password regulation for the data systems…
View original post 100 more words
FDA approves Praluent for the treatment of high LDL cholesterol
26 August 2015
Sanofi and Regeneron have announced that the US Food and Drug Administration (FDA) has approved Praluent® (alirocumab) Injection.

Praluent is indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease (ASCVD), who require additional lowering of low-density lipoprotein (LDL) cholesterol. The effect of Praluent on cardiovascular morbidity and mortality has not been determined.
////////Sanofi, Regeneron, US Food and Drug Administration, FDA, approved, Praluent® , alirocumab
Non compliance at Parabolic drugs
DRUG REGULATORY AFFAIRS INTERNATIONAL

Statement “non compliance GMP”. Officina Farmaceutica: Parabolic Drugs Limited – INDIA (30/07/2015)
Following the inspection, conducted by the inspectorate Italian, under the program of inspections of the EDQM, at the Indian site in question, the same was not “in compliance” with the GMP.
It calls on companies to verify, with urgency, if the medicines containing the following active substances / intermediate production Dicloxacillin SODIUM, amoxicillin trihydrate, PIVAMPICILLIN, Flucloxacillin SODIUM, SODIUM cloxacillin, AMPICILLIN trihydrate, AMPICILLIN ANHYDROUS, Bacampicillin HYDROCHLORIDE authorized for the Italian market and / or products for export, showing this as a possible supplier of active / intermediate Officina Farmaceutica: PARABOLIC DRUGS LIMITED, PDL-2 – Plot No. 45, Industrial Area, Phase II, Panchkula District of Haryana, 134113 , INDIA .
The communication must be sent only by all companies Holders of marketing authorizations or Officine pharmaceutical manufacturers of medicines containing these materials pharmacologically active / production intermediates produced at…
View original post 194 more words
Pevonedistat

Millennium Pharmaceuticals, Inc. INNOVATOR
Millennium Pharmaceuticals, Inc., a subsidiary of Takeda Pharmaceutical Company Limited,
MLN4924, MLN 4924-003, TAK-924
905579-51-3 BASE
1160295-21-5 HcL
A potent and selective inhibitor of NAE. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. The ubiquitin-proteasome pathway mediates the destruction of unwanted proteins.
(((1S,2S,4R)-4-{4-[(S)-2,3-Dihydro-1H-inden-1-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate hydrochloride) (pevonedistat), a novel NEDD8-activating enzyme (NAE) inhibitor, has demonstrated in vitro cytotoxic activity against a variety of human malignancies and is currently being developed by Takeda Pharmaceuticals Company Limited as a clinical candidate for the treatment of cancer
In 2011, orphan drug designation was assigned to MLN-4924 for the treatment of MDS and for the treatment of acute myelogenous leukemia.
PHASE 1…….CANCER SOLID TUMOR

………………….
PATENT
http://www.google.com/patents/US20120330013

preparing a compound represented by the following formula 1 by reacting the compound of formula 11 with TFA (step 9):


The retrosynthetic analysis of MLN4924 (1), as the final desired nucleoside, is shown in the following.

MLN 4924 (1) can be synthesized by condensing cyclic sulfate 3 as the glycosyl donor with a purine base. The glycosyl donor 3 can be produced from diol 4, which in turn can be obtained from cyclopentanone 5 via a stereoselective reduction and a regioselective cleavage of the isopropylidene moiety. The cyclopentanone 5 can be synthesized from cyclopentenone 6 by stereoselective reduction. The intermediate cyclopentenone 6 can be easily derived from D-ribose according to our previously published procedure (Jeong, L. S. et al., J. Org. Chem. 2004, 69, 2634-2636).
The synthetic route for the glycosyl donor 3 is shown in the following scheme 1.

Example 1 Preparation of MLN4924 Step 1: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-one (Compound 5)

To a suspension of the compound 6 (20.0 g, 47.1 mmol) in methanol (400 ml) was added 10% palladium on activated carbon (1.0 g), and the mixture was stirred at room temperature overnight under H2 atmosphere. After filtration of the reaction mixture, the solvent was removed and the residue was dissolved in methylene chloride and then filtered through short pad silica gel. Then, the solvent was evaporated to give the compound 5 (20.1 g, 100%) as a colorless syrup.
[α]20 D −28.32 (c 1.49, MeOH); HR-MS (ESI): m/z calcd for C25H32NaO4Si [M+Na]+ 447.1968, Found 447.1956; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.40 (m, 6H), 4.84 (t, J=4.4 Hz, 1H), 4.22 (dd, J=1.2, 4.8 Hz, 1H), 3.96 (dd, J=8.0, 10.0 Hz, 1H), 3.82 (dd, J=6.8, 10.0 Hz, 1H), 2.37 (m, 1H), 2.30 (ddd, J=1.2, 8.4, and 18.4 Hz, 1H), 2.20 (ddd, J=1.2, 12.0, and 18.4 Hz, 1H), 1.37 (s, 3H), 1.35 (s, 3H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 112.6, 80.5, 77.6, 77.2, 76.9, 63.6, 38.1, 36.9, 27.1, 27.02, 27.01, 25.3, 19.5; Anal. Calcd for C25H32O4Si: C, 70.72; H, 7.60. Found: C, 70.79; H, 7.75.
Step 2: Preparation of 6-(tert-butyl-diphenyl-silanyloxymethyl)-2,2-dimethyl-tetrahydro-cyclopenta[1,3]dioxol-4-ol (Compound 7)

To a suspension of the compound 5 (20.1 g, 47.1 mmol) in methanol (500 ml) were added sodium borohydride (2.17 g, 57.4 mmol) and cerium (III) chloride heptahydrate (21.3 g, 57.2 mmol) at 0° C., and the mixture was stirred at room temperature for 30 min. After the solvent was removed, the residue was partitioned between ethyl acetate and water. The organic layer was then washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=5/1) to give the compound 7 (20.86 g, 98%) as a colorless syrup.
[α]20 D +34.55 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C25H34NaO4Si [M+Na]+: 449.2124; Found: 449.2110; 1H NMR (400 MHz, CDCl3) δ 7.69 (m, 4H), 7.39 (m, 6H), 4.62 (t, J=5.6 Hz, 1H), 4.44 (t, J=5.6 Hz, 1H), 3.89 (dd, J=6.0, 7.6 Hz, 1H), 3.84 (m, 1H), 3.68 (dd, J=6.4, 10.0 Hz, 1H), 1.91 (m, 2H), 1.26 (m, 1H), 1.42 (s, 3H), 1.33 (s, 3H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 135.8, 134.2, 134.1, 129.8, 129.7, 127.8, 127.7, 110.6, 79.4, 78.9, 77.6, 77.2, 76.9, 72.5, 62.9, 41.6, 33.4, 27.0, 25.9, 27.0, 25.9, 24.4, 19.5; Anal. Calcd for C25H34O4Si: C, 70.38; H, 8.03. Found: C, 70.41; H, 8.08.
Step 3: Preparation of 3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentane-1,2-diol (Compound 4)

To a solution of the compound 7 (20.86 g, 47.12 mmol) in methylene chloride was added trimethylaluminum (2.0 M in toluene, 132.1 ml) at 0° C., and the mixture was stirred at room temperature for 2 days. The mixture was cooled to 0° C., slowly quenched with an aqueous saturated ammonium chloride solution, filtered, and evaporated. The residue was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 4 (13.42 g, 62%) as a colorless syrup.
[α]20 D +3.30 (c 0.55, MeOH); HR-MS (ESI): m/z calcd for C26H38NaO4Si [M+Na]+: 465.2437; Found: 465.2423; 1H NMR (400 MHz, CDCl3) δ 7.70 (m, 4H), 7.41 (m, 6H), 4.05 (dd, J=4.4, 7.2 Hz, 1H), 3.93 (m, 1H), 3.72 (m, 2H), 3.59 (dd, J=3.6, 12.0 Hz, 2H), 2.70 (d, J=20.8 Hz, 1H), 2.10 (m, 2H), 1.60 (m, 1H), 1.20 (s, 9H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.9, 133.5, 130.0, 129.9, 127.9, 127.9, 77.6, 77.2, 76.9, 74.9, 73.8, 72.7, 72.1, 63.3, 42.1, 34.0, 28.5, 27.0, 19.4; Anal. Calcd for C26H38O4Si: C, 70.55; H, 8.65. Found: C, 70.61; H, 8.70.
Step 4: Preparation of (4-tert-butoxy-2,2-dioxo-tetrahydro-2-yl-6-cyclopenta[1,3,2]-dioxathiol-5-ylmethoxy)-tert-butyl-diphenyl-silane (Compound 3)

To a solution of the compound 4 (13.42 g, 30.3 mmol) in methylene chloride were added triethyl amine (14.5 ml, 101.0 mmol) and thionyl chloride (3.7 ml, 47.4 mmol) at 0° C., and the reaction mixture was stirred at 0° C. for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the cyclic sulfite (14.37 g, 97%) as a white foam.
[α]20 D +20.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO5SSi [M+Na]+: 511.1950; Found: 511.1929; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.40 (m, 6H), 5.23 (m, 1H), 5.04 (dd, J=4.4, 6.0 Hz, 1H), 4.01 (t, J=4.8 Hz, 1H), 3.68 (dd, J=3.6, 10.4 Hz, 1H), 3.56 (dd, J=8.0, 10.4 Hz, 1H), 2.07 (m, 2H), 1.96 (m, 1H), 1.14 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.8, 135.7, 133.9, 133.8, 129.9, 129.9, 127.9, 127.8, 85.7, 83.2, 77.6, 77.2, 76.9, 75.0, 71.1, 62.7, 44.7, 31.4, 28.5, 27.1, 19.4; Anal. Calcd for C26H36O5SSi: C, 63.90; H, 7.42; S, 6.56. Found: C, 63.94; H, 7.45; S, 6.61.
To a solution of the cyclic sulfite obtained above (14.37 g, 29.4 mmol) in the mixture of carbon tetrachloride, acetonitrile and water (1:1:1.5, 210 ml) were added sodium metaperiodate (18.56 g, 56.4 mmol) and ruthenium chloride (1.72 g, 8.25 mmol), and the reaction mixture was stirred at room temperature for 10 minutes. The reaction mixture was partitioned between methylene chloride and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=4/1) to give the compound 3 (13.36 g, 90%) as a white solid.
mp 101-104° C.; [α]20 D −80.00 (c 0.05, MeOH); HR-MS (ESI): m/z calcd for C26H36NaO6SSi [M+Na]+: 527.1900; Found: 527.1881; 1H NMR (400 MHz, CDCl3) δ 7.64 (m, 4H), 7.41 (m, 6H), 5.13 (m, 1H), 4.83 (dd, J=4.4, 6.8 Hz, 1H), 4.13 (t, J=4.0 Hz, 1H), 3.92 (dd, J=6.4, 10.4 Hz, 1H), 3.69 (dd, J=5.2, 10.4 Hz, 1H), 2.11 (m, 2H), 2.02 (m, 1H), 1.15 (s, 9H), 1.05 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 135.7, 135.0, 133.8, 133.7, 130.0, 128.0, 127.9, 83.5, 82.2, 77.6, 77.2, 76.9, 75.4, 70.4, 70.4, 62.2, 43.9, 31.3, 28.2, 27.1, 26.8, 19.4; Anal. Calcd for C26H36O6SSi: C, 61.87; H, 7.19; S, 6.35. Found: C, 61.91; H, 7.14; S, 6.30.
Step 5: Preparation of 2-tert-butoxy-3-(tert-butyl-diphenyl-silanyloxymethyl)-5-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 8)

A suspension of N6-indanyl-7-deazaadenine (8.80 g, 35.2 mmol), sodium hydride (1.38 g, 45.7 mmol) and 18-crown-6 (9.11 g, 45.7 mmol) in THF (200 ml) was stirred at 80° C. To the reaction mixture was added a solution for the compound 3 (13.36 g, 26.5 mmol) in THF (150 ml), and the stirring was continued at 80° C. overnight. The reaction mixture was cooled down to 0° C., and conc. HCl was added slowly until pH reaches 1-2. Then the reaction mixture was further stirred at 80° C. for 2 hours. After neutralized with saturated aqueous NaHCO3 solution, the reaction mixture was partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. The residue was purified by silica gel column chromatography (hexane/ethyl acetate=2/1) to give the compound 8 (11.62 g, 65%) as a white foam.
UV (CH2Cl2) λmax 272.5 nm; [α]20 D −8.89 (c 0.45, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O3Si [M+H]+: 675.3730; Found: 675.3717; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.70 (m, 4H), 7.41 (m, 6H), 6.92 (d, J=3.6 Hz, 1H), 6.29 (d, J=3.2 Hz, 1H), 5.91 (dd, J=7.6, 14.8 Hz, 1H), 5.14 (br d, J=6.8 Hz, 1H), 4.77 (m, 1H), 4.36 (t, J=6.0 Hz, 1H), 4.22 (dd, J=5.2, 10.8 Hz, 1H), 3.84 (dd, J=5.6, 10.4 Hz, 1H), 3.73 (dd, J=8.4, 10.4 Hz, 1H), 3.37 (d, J=5.6 Hz, 1H), 3.06 (m, 1H), 2.95 (m, 1H), 2.75 (m, 1H), 2.75 (m, 1H), 2.58 (m, 1H), 2.38 (m, 1H), 2.15 (m, 1H), 1.98 (m, 1H), 1.65 (s, 1H), 1.55 (s, 1H), 1.16 (s, 9H), 1.07 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.8, 150.3, 144.1, 143.8, 135.9, 134.0, 129.9, 128.2, 127.9, 127.9, 127.0, 125.1, 124.4, 123.3, 103.8, 97.4, 77.8, 77.6, 77.2, 76.9, 74.9, 72.4, 63.5, 62.1, 56.3, 43.9, 34.9, 30.5, 30.5, 28.5, 27.2, 19.5; Anal. Calcd for C41H50N4O3Si: C, 72.96; H, 7.47; N, 8.30. Found: C, 73.01; H, 7.45; N, 8.36.
Step 6: Preparation of {7-[3-tert-butoxy-4-(tert-butyl-diphenyl-silanyloxymethyl)-cyclopentyl]-7H-pyrrolo[2,3-d]pyrimidin-4-yl}-indan-1-yl-amine (Compound 9)

To a solution of the compound 8 (11.62 g, 17.2 mmol) in methylene chloride (300 ml) were added N,N-dimethylaminopyridine (5.64 g, 51.6 mmol) and phenyl chlorothionocarbonate (4.3 ml, 34.4 mmol), and the reaction mixture was stirred at room temperature overnight. After the solvent was removed, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=6/1) to give the thiocarbonate (13.82 g, 99%) as a white foam.
UV (MeOH) λmax 271.50 nm; [α]20 D +10.00 (c 0.15, MeOH); HR-MS (ESI): m/z calcd for C48H55N4O4SSi [M+H]+: 811.3713; Found: 811.3687; 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1H), 7.61 (dd, J=1.6, 7.6 Hz, 4H), 7.34 (m, 5H), 7.26 (m, 4H), 7.18 (m, 6H), 6.86 (s, 1H), 6.25 (d, J=3.2 Hz, 1H), 6.00 (dd, J=3.2, 8.4 Hz, 1H), 5.83 (d, J=6.8 Hz, 1H), 5.19 (m, 1H), 5.07 (br s, 1H), 4.48 (t, J=3.6 Hz, 1H), 3.82 (dd, J=7.2, 10.4 Hz, 1H), 3.52 (dd, J=7.2, 10.0 Hz, 1H), 2.99 (m, 1H), 2.88 (m, 2H), 2.69 (m, 2H), 2.18 (dd, J=11.2, 13.6 Hz, 1H), 1.94 (m, 2H), 1.12 (s, 9H), 0.98 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 194.9, 153.5, 152.1, 143.9, 135.9, 135.8, 134.1, 129.9, 129.6, 128.3, 127.9, 127.0, 126.7, 125.1, 124.6, 123.2, 122.0, 87.9, 77.6, 77.2, 76.9, 74.6, 70.4, 63.5, 57.3, 42.8, 35.0, 30.7, 30.5, 29.9, 28.7, 27.1, 19.4; Anal. Calcd for C48H54N4O4SSi: C, 71.08; H, 6.71; N, 6.91; S, 3.95. Found: C, 71.14; H, 6.75; N, 6.95; S, 4.01.
To a solution of the thiocarbonate obtained above (13.82 g, 17.0 mmol) in toluene (200 ml) were added tri-n-butyltinhydride (9.4 ml, 34.1 mmol) and 2,2′-azo-bis-isobutyronitrile (4.32 g, 26.3 mmol), and the reaction mixture was stirred at 110° C. for 1 hour. After the mixture was cooled down, the solvent was removed. The resulting residue was purified by silica gel column chromatography (hexane/ethyl acetate=3/1) to give the compound 9 (9.21 g, 82%) as a white foam.
UV (MeOH) λmax 272.50 nm; [α]20 D −10.00 (c 0.20, MeOH); HR-MS (ESI): m/z calcd for C41H51N4O2Si [M+H]+: 659.3781; Found: 659.3757; 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.69 (m, 4H), 7.41 (m, 6H), 7.29 (m, 2H), 7.23 (m, 2H), 6.92 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.6 Hz, 1H), 5.90 (dd, J=7.2, 14.8 Hz, 1H), 5.38 (m, 1H), 5.15 (br s, 1H), 4.33 (dd, J=5.2, 8.4 Hz, 1H), 3.88 (dd, J=6.4, 10.0 Hz, 1H), 3.68 (dd, J=7.2, 10.4 Hz, 1H), 3.05 (m, 1H), 2.96 (dd, J=7.6, 15.6 Hz, 1H), 2.76 (m, 1H), 2.45 (d, J=5.2 Hz, 1H), 2.29 (m, 2H), 2.06 (m, 1H), 1.95 (m, 2H), 1.55 (s, 1H), 1.13 (s, 9H), 1.06 (s, 9H);13C NMR (100 MHz, CDCl3) δ 156.3, 151.9, 144.1, 143.9, 135.9, 135.8, 134.3, 129.8, 128.2, 127.8, 127.0, 125.1, 124.6, 121.8, 77.6, 77.2, 76.7, 73.5, 72.2, 63.6, 56.4, 52.8, 46.8, 42.8, 34.9, 34.5, 30.5, 28.6, 27.2, 28.7, 19.4; Anal. Calcd for C41H50N4O2Si: C, 74.73; H, 7.65; N, 8.30. Found: C, 74.79; H, 7.61; N, 8.25.
Step 7: Preparation of 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentanol (Compound 10)

To a solution of the compound 9 (9.21 g, 13.97 mmol) in the mixture of THF and pyridine (1:1, 160 ml) was added dropwise pyridine hydrofluoride (18.42 ml, 190.0 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 1 hour. The mixture was neutralized with saturated aqueous NaHCO3 solution and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried with anhydrous MgSO4, filtered, and evaporated. Then, the residue was purified by silica gel column chromatography (hexane/ethyl acetate=1/3) to give the compound 10 (5.63 g, 99%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −6.36 (c 1.10, MeOH); HR-MS (ESI): m/z calcd for C25H33N4O2 [M+H]+: 421.2604; Found: 421.2599; 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1H), 7.30 (d, J=7.6 Hz, 1H), 7.22 (d, J=7.2 Hz, 2H), 7.15 (t, J=6.8 Hz, 1H), 6.88 (d, J=3.2 Hz, 1H), 6.23 (d, J=3.6 Hz, 1H), 5.83 (dd, J=7.2, 15.2 Hz, 1H), 5.28 (m, 1H), 5.06 (m, 1H), 4.47 (dd, J=5.6, 10.4 Hz, 1H), 3.78 (m, 1H), 3.70 (m, 1H), 3.24 (t, J=5.2 Hz, 1H), 2.98 (m, 1H), 2.87 (m, 1H), 2.68 (m, 1H), 2.46 (m, 1H), 2.37 (m, 2H), 1.93 (m, 2H), 1.18 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 156.2, 151.8, 147.9, 143.9, 143.9, 128.3, 126.9, 125.1, 124.5, 121.9, 97.7, 77.6, 77.2, 76.9, 75.5, 74.9, 63.4, 56.4, 53.8, 44.2, 42.2, 34.9, 33.2, 30.5, 28.6; Anal. Calcd for C25H32N4O2: C, 71.40; H, 7.67; N, 13.32. Found: C, 71.46; H, 7.60; N, 13.35.
Step 8: Preparation of sulfamic acid 2-tert-butoxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 11)

Preparation of 2.0 M solution of chlorosulfonamide in acetonitrile: Formic acid (14.15 ml, 166.0 mmol) was added dropwise to chlorosulfonyl isocyanate (32.0 ml, 162.5 mmol) under nitrogen atmosphere at 0° C. When the addition was completed, the mixture was solidified. To the mixture was added acetonitrile (61.3 ml), and the resulting solution was left to stand under nitrogen source at room temperature overnight.
To a solution of the compound 10 (5.63 g, 13.83 mmol) and triethyl amine (9.7 ml, 0.74 mmol) in acetonitrile (278 ml) was added 2.0 M solution of chlorosulfonamide in acetonitrile (13.83 ml, 27.76 mmol) at 0° C., and the reaction mixture was stirred at room temperature for 45 minutes. Additional 2.0 M chlorosulfonamide solution in acetonitrile (13.83 ml, 27.76 mmol) was added and the mixture was stirred at room temperature for 15 minutes. The reaction was quenched with methanol, and the solvent was removed. The residue was purified by silica gel column chromatography (methylene chloride/methanol=20/1) to give the compound 11 (6.37 g, 92%) as a white foam.
UV (MeOH) λmax 273.00 nm; [α]20 D −18.00 (c 0.50, MeOH); HR-MS (ESI): m/z calcd for C25H34N5O4S [M+H]+: 500.2332; Found: 500.2331; 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.36 (d, J=7.2 Hz, 1H), 7.29 (d, J=7.2 Hz, 1H), 7.22 (m, 2H), 6.95 (d, J=3.6 Hz, 1H), 6.31 (d, J=3.2 Hz, 1H), 5.89 (d, J=6.4 Hz, 1H), 5.10 (s, 2H), 4.41 (m, 2H), 4.26 (m, 1H), 3.05 (m, 1H), 2.94 (m, 1H), 2.76 (m, 2H), 2.27 (m, 3H), 2.06 (m, 1H), 1.97 (m, 1H), 1.76 (br s, 1H); 13C NMR (100 MHz, CDCl3) δ 156.4, 151.9, 149.9, 143.9, 143.8, 128.3, 126.9, 125.1, 124.5, 121.9, 121.9, 103.5, 97.9, 77.4, 77.2, 76.9, 74.3, 71.9, 71.3, 56.4, 53.1, 49.0, 42.3, 34.9, 34.3, 30.5, 28.6; Anal. Calcd for C25H33N5O4S: C, 60.10; H, 6.66; N, 14.02; S, 6.42. Found: C, 60.15; H, 6.71; N, 13.98; S, 6.39.
Step 9: Preparation of sulfamic acid 2-hydroxy-4-[4-(indan-1-ylamino)-pyrrolo[2,3-d]pyrimidin-7-yl]-cyclopentylmethyl ester (Compound 1)

A solution of the compound 11 (6.37 g, 12.72 mmol) in 70% trifluoroacetic acid (149.24 ml) was stirred at room temperature for 2 hours. The solvent was removed and the residue was purified by silica gel column chromatography (hexane/ethylene acetate=1/2) to give the compound 1 (5.08 g, 90%) as a white foam. BASE
UV (MeOH) λmax 279.50 nm; [α]20 D −6.41 (c 2.34, MeOH);
HR-MS (ESI): m/z calcd for C21H26N5O4S [M+H]+: 444.1705; Found: 444.1706;
1H NMR (400 MHz, CD3OD) δ 8.17 (d, J=1.6 Hz, 1H), 7.25 (m, 2H), 7.18 (m, 2H), 6.64 (d, J=3.6 Hz, 1H), 5.86 (t, J=7.6 Hz, 1H), 5.46 (m, 1H), 4.49 (d, J=2.8 Hz, 1H), 3.07 (m, 1H), 2.92 (m, 1H), 2.80 (m, 1H), 2.64 (m, 1H), 2.35 (m, 1H), 2.25 (m, 2H), 2.03 (m, 2H);
13C NMR (100 MHz, CD3OD) δ 152.1, 145.3, 144.6, 128.8, 127.6, 125.7, 125.2, 122.6, 100.5, 73.1, 70.9, 56.9, 54.0, 44.8, 43.6, 34.9, 34.6, 31.1;
Anal. Calcd for C21H25N5O4S: C, 56.87; H, 5.68; N, 15.79; S, 7.23. Found: C, 56.91; H, 5.73; N, 15.82; S, 7.26.
…………………….
http://www.google.com/patents/WO2010132110A1?cl=en
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (//) is described in Intl. App. Pub. No. WO 07/092213, U.S. App. Pub. No. 2007/0191293, and U.S. App. Pub. No. 2009/0036678. The potassium salt of ((lS,2S,4R)-4-{4-[( 1 S)-2,3-dihydro- 1 H-inden- 1 -ylamino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl } -2-hydroxycyclopentyl)methyl sulfamate is disclosed in Intl. App. Pub. No. WO 07/092213 and U.S. App. Pub. No. 2007/0191293.

(H)
((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH- inden-l-ylamino]-7H-pyπOlo[2,3-d]pyrimidin-7-yl}-2-hydroxycyclopentyl)methyl sulfamate (/):

Step 3: Synthesis of ((lS,2S.4R)-4-(4-r(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolor2.3-dlpyrimidin-7-yl}-2-hvdroxycvclopentyl)methyl sulfamate hydrochloride Form 1
[0158] A reactor was charged with ((lS,2S,4R)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2,3-d]pyrimidin-7-yl }-2-hydroxycyclopentyl)methyl sulfamate (13.4 Kg, 30.2 mol) and 200-proof ethanol (106.2 Kg). The mixture was heated to reflux to afford a clear solution. The mixture was cooled to 50 ± 5 0C and passed through a cartridge filter. 200 proof ethanol (8.9 Kg) was used to rinse the filter. 1.27M hydrogen chloride in ethanol (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then seeded with Form 1 (67 g). Further 1.27M HCl (10.2 Kg) was added via a cartridge filter at a rate to maintain a temperature of 50 ± 5 0C. The mixture was then stirred at 50 ± 5 0C for about 3 hours. The mixture was then cooled to 20 ± 5 0C over about 3 hours and then stirred for about 2.5 hours. The solid product was then isolated by filtration and washed with 200-proof ethanol (I x 20.4 Kg and 1 x 21.2 Kg). The solids were dried by aspiration on the filter until no supernatant was seen to be collected, and then further dried under reduced pressure at <30 0C to afford the title compound (12.2 Kg) as a white solid determined to be Form 1 by XRPD. IH NMR (300MHz, DMSO, δ): 9.83 (s, IH), 8.34 (s, IH), 7.62 (s, IH), 7.44 (s, 2H), 7.30 (m, 3H), 7.22 (t, IH), 7.07 (s, IH), 5.86 (dd, IH), 5.42 (m, IH), 4.32 (m, IH), 4.21 (dd, IH), 4.02 (dd, IH), 3.04 (m, IH), 2.88 (m, IH), 2.67 (m, 2H), 2.15 (m, 2H), 2.08 (m, 2H), 1.94 (m, IH). XRPD data for Form 1 is shown in FIGURE 1 and Table 1; DSC data is shown in FIGURE 2, and TGA data for Form 1 is shown in FIGURE 3.
…………..
http://www.google.com/patents/WO2007092213A2?cl=en
Example 70: Diastereoisomeric mixture of (lS/2R/4R)-4-{4-[(lS)-2/3-dihydro-lH-inden-l- ylaimnol-ZH-pyrrolop^-dlpyxirnidin-Z-ylJ^-hydroxycyclopentyl s ulf amate and (lRf2S/4S)-4-{4-[(lS)-2,3-dihydro-lH-inden-l-ylaminol-7H-pyrrolo[2,3d]- pyrimidin-7-yl}-2-hydroxycyclopentyl sulfamate (Compounds 1-77 and 1-78)

Step a: Cyclopent-3-en-l-yl methanesulfonate
[0335] 3-Cydopentene-l-ol (0.500 g, 5.94 mmol) was stirred in DCM (95 mL).
Pyridine (2.40 mL), N,N-dimethylaminopyridine (0.10 g, 1.00 mmol) and methanesulfonyl chloride (0.690 mL, 8.92 mmol) were added, and the reaction mixture was stirred at 350C for 4 h. N,N-Dimethylarrιinopyridirιe (0.14 g, 1.2 mmol) and methanesulfonyl chloride (0.69 mL, 8.92 mmol) were added, and the reaction was stirred overnight. TLC indicated complete conversion. The reaction mixture was cooled and concentrated. The residue was purified by silica gel chromatography, eluting with DCM, to afford the title compound as a clear oil (0.660 g, 68%).
Step b: 7-Cyclopent-3-en-l-yl-N-r(lSV2,3-dihydro-lH-inden-l-yn-7H-pyrrolor2,3-rfl- pyrmτidin-4-arnine
[0336] N-[(lS)-2,3-DihydrcHlH-mden-l-yl]-7H-pyrrolo[2/3-d]p3αimidin-4-amine (1.32 g, 5.29 mmol) was azeotroped with toluene and placed under high vacuum for 30 min. N,N-Dimethylformamide (17.7 mL) was added, followed by cesium carbonate (1.99 g, 6.10 mmol). The mixture was stirred at 700C for 10 min. Cyclopent-3-en-l-yl methanesulfonate (0.660 g, 4.07 mmol) in N,N-dimethylformarnide (12.6 mL) was added dropwise. The reaction mixture was heated to 1100C for 1 h. The reaction mixture was cooled, quenched with brine and diluted with H2O. The aqueous layer was extracted with EtOAc (3x), washed with H2O and brine, dried (Na2SO4), filtered, and concentrated. The residue -was purified by via silica gel chromatography, eluting with a gradient of 0 to 5% MeOH in DCM followed by 25 to 50% EtOAc in hexanes, to afford the title compound as a pale brown solid (0.684 g, 53%). LC/MS: R1 = 1.38 min, ES+ 317 (FA standard). Step c: (lR,2S,45)-4-{4-r(lS)-2,3-dihydro-lH-inden-l-ylaininol-7H-pyrrolof2.3- rf1pyrimidin-7-yl}cyclopentane-l,2-diol
[0337] 7-Cyclopent-3-en-l-yl-N-[(lS)-2^-dihyrdo-lH-inden-l-yl]-7H-pyrrolo[2,3- d]pyτimidin-4-amine (0.312 g, 0.986 mmol) was stirred in tert-butyl alcohol (4.9 mL) and H2O (4.9 mL). AD-mix-α (Sigma- Aldrich, 1.4 g) was added, and the suspension was stirred at rt overnight. TLC indicated complete conversion. The reaction was quenched with sodium sulfite (1.48 g, 11.7 mmol), and the mixture was stirred for 5 h. The reaction mixture was diluted with EtOAc and H2O, and the aqueous layer was extracted with EtOAc (2x). The organic layer was dried (Na2SO4), filtered, and concentrated. The residue was purified via silica gel chromatography, eluting with EtOAc, to afford the title compound as a white solid (0.190 g, 55%).
Step d: Diastereoisomeric mixture of (lS,2R,4R)-4-{4-r(15)-23-dihydro-lH-inden-l- ylarninoi^jH-pyrrolofΣ^dlpyrirnidin-y-yll-l-hydroxycyclopentyl sulfamate and (lR,2S,4S)-4-{4-iαSV2,3-dihydro-lH-inden-l-ylarninol-7H-pyrrolor2,3- rf1pyrimidm-7-yl)-2-hydroxycyclopenryl sulfamate (Compounds 1-77 and 1-78)
[0338] (lR,2S,4S)-4-{4-[(lS)-2,3-Dihydro-lH-inden-l-ylarnino]-7H-pyrrolo[2/3- d]pyrimidin-7-ylJcyclopentane-l,2-diol (0.080 g, 0.23 mmol) was azeotroped with toluene and then was dissolved in anhydrous acetonitrile (2.3 mL). Pyridine (0.0369 mL, 0.458 mmol) was added. The reaction mixture was cooled to 00C, and a 2N solution of chlorosulfonamide in acetonitrile (0.144 mL) was added dropwise. The reaction was stirred for 1 h, and then additional 2N chlorosulfonamide in acetonitrile (0.028 mL) was added. After 30 min, additional 2N chlorosulfonamide in acetonitrile (0.0342 mL) was added, and the reaction mixture was stirred for 2 h. The reaction was quenched with methanol, and the mixture was concentrated in vacuo. The residue was purified by preparative thin layer chromatography using DCM:AcCN:MeOH (50:45:5). The relevant band was cut, washed with acetone, filtered, and concentrated to give a mixture of diastereomers as a white solid. (11 mg, 11%). 1H NMR (CDCl3, 400 NMR, δ): 8.36-8.27 (m, IH); 7.38-7.09 (m, 5H); 6.90-6.80 (m, IH); 6.36- 6.20 (m, IH); 5.95-5.76 (m, IH); 5.51-5.22 (m, 2H); 4.83-4.68 (m, IH); 3.87-3.72 (m, IH); 3.12- 2.83 (m, 2H); 2.75-2.53 (m, IH); 2.50-2.14 (m, 2H); 2.08-1.79 (m, 2H) ppm. LC/MS: R, = 1.16 min, ES+ 430 (FA standard).
…………
WO 2012061551
http://www.google.im/patents/WO2012061551A1?cl=en
The compound ((lS,2S,4R)-4-(4-((lS)-2,3-dihydro-lH-inden-l-ylamino)-7H-pyrrolo[2,3-d]- pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl sulfamate:

also known as MLN4924, is an inhibitor of NEDD8-activating enzyme (NAE). Inhibition of NAE has been shown to induce cancer cell death and inhibit the growth of tumors in xenograft models. See, e.g., T.A. Soucy et al., Nature, 2009, 458, 732-737; T.A. Soucy ei al., Clin. Cancer Res., 2009, 15 (12), 3912-3916; and J.E. Brownell et al., Mol. Cell., 2010, 37 (1), 102-111, each of which is hereby incorporated by reference herein in its entirety. MLN4924, pharmaceutical compositions of MLN4924, processes for its synthesis, and polymorphic forms have been described previously. See, e.g., US Patent Appl. Nos. 11/700,614 (Publ. No. 2007/0191293), 12/221,399 (Publ. No. 2009/0036678) and 12/779,331 (Publ. No. 2011/0021544),
……………

A practical synthesis of a novel NEDD8-activating enzyme (NAE) inhibitor pevonedistat (MLN4924) is described. Key steps include an enantioselective synthesis of an amino-diol cyclopentane intermediate containing three chiral centers and a novel, regioselective sulfamoylation using N-(tert-butoxycarbonyl)-N-[(triethylenediammonium)sulfonyl]azanide. The linear process, involving six solid isolations, has been carried out in multiple cGMP productions on 15–30 kg scale to produce pevonedistat in 98% (a/a) chemical purity and 25% overall yield.

((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate (1)
((1S,2S,4R)-4-(4-(((S)-2,3-Dihydro-1H-inden-1-yl)amino)-7H-pyrrolo[2,3-d]pyrimidin-7-yl)-2-hydroxycyclopentyl)methyl Sulfamate·Hydrochloride (Pevonedistat)

MLN4924 (1), which is in clinical trials as an anticancer agent, was stereoselectively synthesized from d-ribose via a route involving stereoselective reduction, regioselective cleavage of an isopropylidene moiety, and selective displacement of a cyclic sulfate moiety as key steps.
Sulfamic Acid 2-Hydroxy-4-[4-(indan-1-ylamino)pyrrolo[2,3-d]pyrimidin-7-yl]cyclopentylmethyl Ester (1) BASE



| WO2012061551A1 * | Nov 3, 2011 | May 10, 2012 | Millennium Pharmaceuticals, Inc. | Administration of nedd8-activating enzyme inhibitor |
| WO2013028832A2 * | Aug 23, 2012 | Feb 28, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| WO2013028832A3 * | Aug 23, 2012 | May 2, 2013 | Millennium Pharmaceuticals, Inc. | Inhibitors of nedd8-activating enzyme |
| US8809356 | Aug 23, 2012 | Aug 19, 2014 | Millennium Pharmaceuticals, Inc. | Inhibitors of NEDD8-activating enzyme |
| Reference | ||
|---|---|---|
| 1 | * | Lee, et. al., Journal of Organic Chemistry (2011), 76(9), 3557-3561. |
1H NMR PREDICT

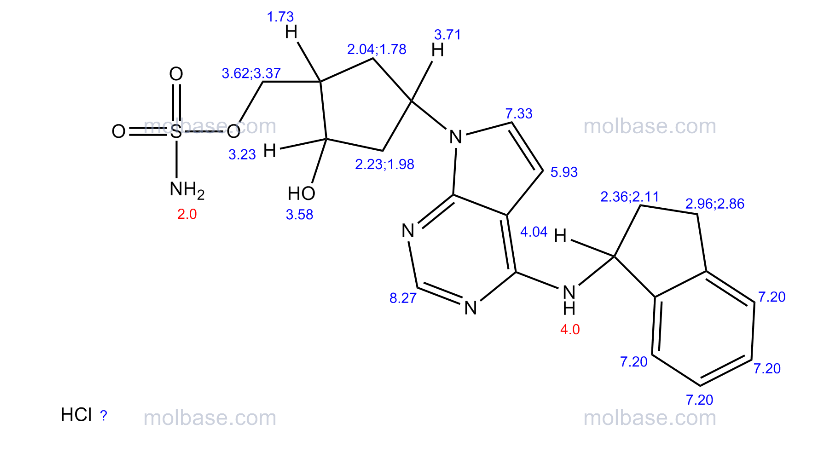
13 C NMR


//////////Pevonedistat, MLN4924, Millennium Pharmaceuticals, TAKEDA, TAK-924 , PHASE 1, orphan drug designation
AN IMPROVED PROCESS FOR THE PREPARATION OF DOLUTEGRAVIR

 Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
Aurobindo Pharma MD and CEO N. Govindarajan at a company research centre. “It [the transition] is purely driven by the need to get more into areas where there is scope for better profit margins,
Dolutegravir (I) is chemically known as (4/?,12aS)-N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-2//-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide. Dolutegravir is a human immunodeficiency virus type 1 (HIV-1) integrase strand transfer inhibitor (INSTI) indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection. Dolutegravir is being marketed under the trade name Tivicay®. US 8,129,385 disclosed Dolutegravir or its pharmaceutically acceptable salts thereof. US ‘385 also discloses a process for the preparation of Dolutegravir (I). The process involves the condensation of 5-benzyloxy-4-hydroxy-6-hydroxymethyl nicotinic acid (II) with 2,4-difluorobenzylamine (III) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-4-hydroxy-6-hydroxymethyl nicotinic acid amide (IV), which is further under goes oxidation using manganese dioxide (Mn02) to produce 5-benzyloxy-N-(2,4-difluorobenzyl)-6-formyl-4-hydroxy-nicotinic acid amide (V). This amide compound (V) is reacted with sodium chlorite (NaClCh) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4- hydroxy-pyridine-2-carboxylic acid (VI), which is further treated with methanol (MeOH) to produce 3-benzyloxy-5-(2,4-difluorobenzyl)-4-hydroxy-pyridine-2-carboxylic acid methyl ester (VII).

The methyl ester compound (VII) is reacted with 3-bromopropene to produce l-allyl-3-benzyloxy-5-(2,4-difluorobenzyl)-4-oxo-l,4-dihydro-pyridine-2- carboxylic acid methyl ester (VIII), which is further reacted with potassium osmate dihydrate (K2OSO4.2H2O) to produce 3-benzyloxy-5-(2,4-difluorobenzylcarbamoyl)-4-oxo-l-(2-oxo-ethyl)-l,4-dihydropyridine-2-carboxylic acid methyl ester (IX). The compound (IX) is reacted with (R)-3-amino-l-butanol (X) to produce benzyloxy Dolutegravir (XI), which is deprotected by treating with TFA to produce Dolutegravir (I). The process is as shown in scheme-I below:
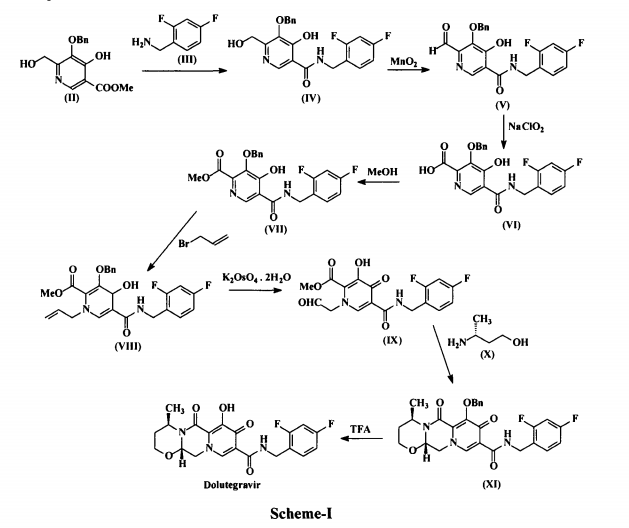
The major disadvantage with the above prior-art process is that it involves large no of steps and tedious work-up procedures to isolate the required product. This results a longer period of time cycle is required to produce Dolutegravir (I), which in turn renders the process more costly and less eco friendly. Further the above processes are low yielding and with less purity. US 8,217,034 discloses variant process for the preparation of Dolutegravir.
This process involves the reaction of methyl l-(2,2-dihydroxyethyl)-4-oxo-3-[(phenylmethyl)oxy]-l,4-dihydro-2-pyridine carboxylate (XII) with (R)-3-amino-l-butanol (X) to produce (4R, 12o5)-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2//-pyrido[ 1 \2′,4,5] pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIII), which is further undergoes bromination using NBS to produce (4R,12aS)-9-bromo-4-methyl-7-[(phenylmethyl)oxy]-3,4,12,12a-tetrahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-6,8-dione (XIV). The bromo Compound (XIV) is condensed with 2,4-difluorobenzylamine (III) in the presence of Tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) to produce benzyloxy Dolutegravir (XI), which is hydrogenated in the presence of Pd/C to produce Dolutegravir (I). The process is as shown in Scheme-II below:

The major disadvantage with the above prior art process of preparing Dolutegravir is the use of expensive reagent tetrakis(triphenylphosphine)palladium (Pd(PPh3)4> in coupling step. Use of this reagent on industrial scale is not preferred, which makes the process more expensive. WO 2011/119566 discloses another variant process for the preparation of Dolutegravir.
This process involves the reaction of l-(2,2-dimethoxyethyl)-5-methoxy-6-(methoxycarbonyl)-4-oxo-l,4-dihydropyridine-3-carboxylic acid (XV) with acetic acid in presence of methane sulfonic acid to produce 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), which is further condensed with (R)-3-amino-l-butanol (X) to produce (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[ 1 ‘,2’:4,5]pyrazino[2,1 -b] [ 1,3]-oxazine-9-carboxylic acid (XVII). This acid Compound XVII is acylated with 2,4-difluorobenzylamine (III) in the presence of carbonyldiimidazole (CDI) to produce methoxy Dolutegravir (XVIII), which is demethylated in the presence of lithium bromide (LiBr) to produce Dolutegravir (I).
The process is as shown in Scheme-3 below:
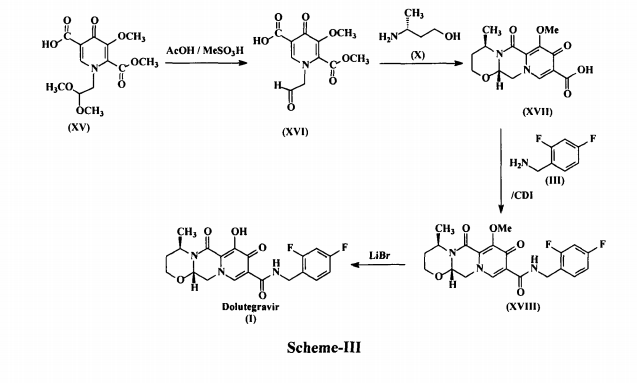
The major disadvantage of the above prior art process of preparing Dolutegravir is the use of expensive and highly moisture sensitive reagent, 1,1-carbonyldiimidazole (CDI), during acylation. Use of this reagent on industrial scale is not preferred due to anhydrous conditions required in the process. However, there is always a need for alternative preparative routes, which for example, involve fewer steps, use reagents that are less expensive and/or easier to handle, consume smaller amounts of reagents, provide a higher yield of product, have smaller and/or more eco-friendly waste products, and/or provide a product of higher purity. Hence, there is a need to develop cost effective and commercially viable process for the preparation of Dolutegravir of formula (I). The present invention is related to a process for the preparation of pure Dolutegravir of formula (I), wherein optically active acid addition salt of (R)-3-amino-l-butanol (X) is directly condensed with 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI) instead of condensing with free base of (R)-3-amino-1-butanol (X). The present invention is also related to a process for the preparation of pure Dolutegravir of formula (I), wherein, inexpensive and easily handling condensing reagents in the condensation of (4R, 12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2//-pyrido[l’,2′:4,5]pyrazino [2,l-b][l,3]oxazine-9-carboxylic acid (XVII) with 2,4-difluorobenzylamine (III).


AN IMPROVED PROCESS FOR THE PREPARATION OF DOLUTEGRAVIR
| APPLICATION NUMBER | 1361/CHE/2013 |
| APPLICANT NAME | AUROBINDO PHARMA LTD |
| DATE OF FILING | 27/03/2013 |
| PUBLICATION DATE (U/S 11A) | 16/01/2015 |
In another embodiment, 5-methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4- dihydropyridine-3-carboxylic acid (XVI) used in the present invention is prepared by reacting 4-methoxyacetoacetate (XIX) with N,N-dimethyl-l,l- bis(methyloxy)methanamine (DMF-DMA) (XX) to produce methyl-2- (dimethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-(dimethylamino)-2 [(methyloxy)acetyl]-2-propenoate) (XXI), which is reacted with aminoacetaldehyde dimethyl acetal (XXII) to produce methyl-2-(2,2-dimethoxyethylaminomethylene)-4-methoxy-3-oxo-butanoate(methyl-3-{[2,2-bis(methyloxy)ethyl]amino}-2-[(methyloxy) acetyl]-2-propenoate) (XXIII).
The compound (XXIII) is contacted with dimethyl ethanedioate in presence of alkali metal alkoxide to produce dimethyl-1-(2,2-dimethoxyethyl)-3-methoxy-4-oxo-l ,4-dihydropyridine-2,5-dicarboxylate (XXIV), which is selectively hydrolyzed with a base to produce l-[2,2-bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l ,4-dihydro-3-pyridinecarboxylic acid (XV). The compound (XV) is treated with a catalytic amount of a strong protic acid in the presence of acetic acid in an organic solvent to produce a reaction mixture containing 5- methoxy-6-(methoxycarbonyl)-4-oxo-l-(2-oxoethyl)-l,4-dihydropyridine-3-carboxylic acid (XVI), The process is as shown in Scheme-IV below:

The following examples illustrate the nature of the invention and are provided for illustrative purposes only and should not be construed to limit the scope of the invention.
Example-1:
EXAMPLES: Example-1: Process for the preparation of Dolutegravir
Step-i: Preparation of (/?)-3-amino-l-butanol tartarate salt: D-(+) Tartaric acid (12.7 g, 0.085 mol) was added in to a solution of (i?,5)-3-amino-l-butnaol (7.5 g, 0.084 mol) in methanol (100 ml) at 40 °C. The reaction mixture was stirred for about 1 hour at 35-40 °C and the reaction mass was cooled to 0-5°C and maintained for 30-40 minutes. The obtained solid was filtered and washed with chilled methanol (10 ml) at 0-5 °C. The solid was dried to get (i?)-3-amino-l-butanol tartarate salt (8.0 g, 40%).
Step-ii: Preparation of (4rt,12a£)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[l’,2′;4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxylic acid (XVII): l-[2,2-Bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyloxy)carbonyl]-4-oxo-l,4-dihydro-3-pyridinecarboxylic acid (XV) (lOOg; 0.3175 moles) was suspended in acetonitrile (800 ml) and heated to 80-82°C. A mixture of acetic acid (95.25 g), methanesulfonic acid (9.14 g; 0.09525 moles) and acetonitrile (200 ml) were added to the slurry at 80-82°C. The reaction mass was continued at 80-82°C to complete the reaction. After completion of the reaction, anhydrous sodium acetate (65 g) and (/?)-3-amino-l-butanol tartrate salt (79.68g; 0.3334 moles) were added at 20-25°C and stirred at 60-65°C to complete the reaction. The reaction mass was concentrated and acidified with IN aqueous hydrochloric acid (750 ml) and extracted with methylene chloride (1500 ml) at ice cold temperature. The organic layer was separated, concentrated, treated with hot methanol (350 ml) for 2 h, filtered, washed with methanol and dried to yield (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxylic acid (XVII) (72 g; HPLC purity: 99.07%).
Step-iii: Process for the preparation of Dolutegravir (I). Method A: Triethylamine (3.61 g; 0.0357 moles) was added to the suspension of (4R,12aS)-7- methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 – b][l,3]oxazine-9-carboxylic acid (XVII) (10 g; 0.0325 moles) in methylene chloride (50 ml), and cooled to 10-15°C. Pivaloyl chloride (4.3 g; 0.0357 moles) was added to the reaction mass, and stirred at 10-15°C for 1 h. Thereafter, 2,4-difiuorobenzylamine (5.58 g; 0.0389 moles) was added at 10-15°C and then warmed to 20-25°C to complete the reaction. After completion of the reaction, IN aqueous hydrochloric acid (20 ml) was added, organic layer was separated, washed with 5% w/w aqueous sodium bicarbonate solution (10 ml) followed by 15% w/w aqueous sodium chloride solution (10 ml) and concentrated. To the concentrated mass, acetonitrile (100 ml) and Lithium bromide (5.08 g; 0.0584 moles) were added and heated to 65-70°C for 3 h to complete the reaction. After completion of the reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml), concentrated to about 50 ml and DM water was added to crystallize the product at 20-25°C. The slurry was stirred for 2 h, filtered, washed with DM water and dried to yield (4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino[2,1 -b] [ 1,3]oxazine-9-carboxamide (I) (11.5 g, HPLC purity: 99.63%).
Method B: Isobutyl chloroformate (4.65 gm, 0.03404 moles) in methylene chloride (10 ml) was added to the solution of N-methylmorpholine (3.45 gm, 0.03410 moles) and (4R,12aS)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[ 1′ ,2′ :4,5]pyrazino-[2,1 -b][l,3]oxazine-9-carboxy!ic acid (XVII) (10.0 gm, 0.03245 moles) in methylene chloride (60 ml) at -10 to 0°C in about 1 h. 2,4-Difloro benzyl amine (4.88 gm, 0.03409 moles) in methylene chloride (10 ml) was added to the cold reaction mass, and stirred at 20-30°C for completion of reaction. After completion of reaction, the reaction mass was washed with 5%w/w aqueous sodium bicarbonate solution (20 ml), IN hydrochloric acid (20 ml), DM water (20 ml) and concentrated. Acetonitrile (120 ml) and lithium bromide (4.8 gm, 0.05516 moles) were added to the concentrated mass, and stirred at 70-80°C for 3 h to complete the reaction. After completion of reaction, the reaction mass was acidified with 5N aqueous hydrochloric acid (40 ml) and concentrated to about 50 ml. DM Water (100 ml) was added to the concentrated reaction mass and stirred for 2 h at 25-30°C to crystallize the product. The product was filtered, washed with DM Water (50 ml) and dried to yield Dolutegravir (I) (10.7 gm, HPLC purity: 99.60%).
Example-2: Process for the preparation of Dolutegravir (I) (4R, 12aS)-N-(2,4-difluorobenzyl)-7-methoxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a,-hexahydro-2H-pyrido[r,2′:4,5]pyrazino[2,l-b][l,3]oxazine-9-carboxamide (XVIII) (2 g, 0.0046 moles) was suspended in isopropyl alcohol (20 ml) and lithium bromide (0.8 g, 0.00924 moles) was added and stirred at 70-80°C for 15 h to complete the reaction. After completion of reaction the reaction mass was acidified with 5N aqueous hydrochloric acid (5 ml) and concentrated. DM Water (20 ml) was added to the concentrated mass and stirred at 25-30°C to crystallize the product. The product was filtered, washed with DM Water and dried to yield Dolutegravir (I) (1.5 g, HPLC purity: 97.93%).


/////
