
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lodenafil Carbonate … an Erectile Dysfunction Drug in Phase III

Lodenafil carbonate
UNII-29X84F932D, CRIS-031
bis-(2-{4-[4-ethoxy-3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-benzenesulfonyl]piperazin-1-yl}-ethyl)carbonate
5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one. IS THE NAME OF MONOMER
398507-55-6 CAS
Cristalia (Originator)
| C47 H62 N12 O11 S2= MF | |
| Molecular Weight | 1035.199 |
Lodenafil is a drug belonging to a class of drugs called PDE5 inhibitor, which many other erectile dysfunction drugs such as sildenafil, tadalafil, and vardenafil also belong to. Like udenafil and avanafil it belongs to a new generation of PDE5 inhibitors.
- Sildenafil, tadalafil, vardenafil, and the newer udenafil and avanafil selectively inhibit PDE5, which is cGMP-specific and responsible for the degradation of cGMP in the corpus cavernosum. These phosphodiesterase inhibitors are used primarily as remedies for erectile dysfunction, as well as having some other medical applications such as treatment of pulmonary hypertension.
Lodenafil is formulated as a dimer, lodenafil carbonate, which breaks down in the body to form two molecules of the active drug lodenafil. This formulation has higher oral bioavailability than the parent drug.[1]
It is manufactured by Cristália Produtos Químicos e Farmacêuticos in Brazil and sold there under the brand-name Helleva.[2]

Helleva (Lodenafil Carbonate) is an oral PDE5 inhibitor prescribed to treat men suffering from erectile dysfunction. It operates by relaxing muscles and dilating blood vessels in the penis to increase circulation making it easier to attain and maintain an erection.
It has undergone Phase III clinical trials,[3][4][5] but is not yet approved for use in the United States by the U.S. Food and Drug Administration.

………..
SYNTHESIS
WO 2002012241 OR US7148350
MONOMER synthesis
 PIPERAZINE
PIPERAZINE
AND
 ETHYL CHLORO ACETATE
ETHYL CHLORO ACETATE
WILL GIVE
Ethyl 1-piperazinylacetate
SEE RXN 1 BELOW
Reaction 1:
Synthesis of Piperazine Ethyl Acetate
To a reaction blend containing 100 g (3 Eq, 0.515 mol, MW=194) of piperazine, 26.3 mL (1.1 Eq, 0.189 mol, MW=101, d=0.726) of triethylamine in 200 mL of isopropanol, add to a solution previously prepared of 18.4 mL (1 Eq., 0.172 mol, MW=122.55, d=1.15) of chloroacetate of ethyl in 140 mL of isopropanol under stirring, at room temperature. Keep the reaction medium under stirring, monitoring the reaction termination by means of a chromatography of the thin layer (about 2–3 hours). Add a solution of 40.6 g (0.344 mol) of succinic acid in 140 mL of isopropanol. Keep the system under stirring for about 30 minutes to assure total precipitation of the succinate salt of piperazine formed. Filter this salt and concentrate the filtrate containing the mono and dialkyled derivatives. We obtain a slightly yellowish oil, which is used in later phases without purification.
Mass obtained=33 g
GC/MS: Monoalkylated derivative 72%, and dialkylated 22%.
NEXT
Piperazine Ethyl Acetate
AND
![5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one Structure](https://i0.wp.com/www.chemicalbook.com/CAS/GIF/139756-22-2.gif)
5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one
WILL REACT TO GIVE… 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one AS IN RXN 4 BELOW
Reaction 4:
Synthesis of 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one.
Suspend 24.6 g (60 mmol, MW=410.9) of 5-(5-chlorosulfonyl-2-etoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one in 900 mL of ethanol absolute. Under stirring and at room temperature, add at only one time, a solution containing 31.0 g (3 Eq., 180 mmol MW=172) of N-piperazine ethyl acetate (Reaction 1) dissolved in 150 mL of ethanol absolute. In an interval of 2–10 minutes, all solid is consumed, forming a clean and homogeneous solution, and after that starts the precipitation of the expected product. At the end of the reaction, which lasts 2–3 hours (monitored by chromatography of thin layer), the product is vacuum filtered and the solid is washed with two portions of 50 mL of iced absolute ethanol. 29 g are obtained (yielding=89%) from the product as a white solid of MP=165.5–166.5° C.
Reaction 7:
Intermediate 1
5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one. IS MONOMER
please note during LAH redn …………. the PIP CH2-C=O-O CH2 CH3 BECOMES PIP-CH2CH2-OH
To a suspension of lithium aluminum hydride (0.74 g 2.2 Eq. MW=37.9) in 25 mL of THF, slowly add, under stirring and at room temperature, a suspension of 5.0 g (9.1 mmol, MW=546.6) of 5-{2-ethoxy-5-[(4-ethyl acetate 1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-di-hydro-7H-pyrazole[4,3-d]pyrimidin-7-one in 50 mL of THF. The system is maintained under stirring, monitoring the consumption of the product by chromatography of thin layer, until the complete consumption of the starting reagent (about 5–6 hours). Slowly add water to the reaction medium and, when there is no longer release of H2, add HCl 1M regulating pH for 7. Extract the product with 3 200 mL-portions of chloroform, dry with anhydrous sodium sulfate and vacuum concentrate the product. It is obtained 3.8 g of the product as a cream solid MP=183–187° C. yielding 83%. The same was crystallized from methanol and DMF yielding a slightly yellowish solid with melting point at 189–192° C.

note …………. the PIP CH2-C=O-O CH2 CH3 BECOMES PIP-CH2CH2-OH
HOMODIMER CARBONATE
EXAMPLE 1B
Homodimer Carbonate of Intermediate 1—Alternative Method
A phosgene solution (3.5 g, 35 mmol) dissolved in 20 mL of toluene was added dropwise to a solution of 2.02 g (4 mmol) of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, suspended in 44 mL of toluene. The reaction mixture resulting is stirred and followed by chromatography analysis of thin layer every hour until the reagent conversion in its chloroformate was completed. When the analysis indicates the complete consumption of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, the volatile compounds of the reaction are vacuum removed (solvents and phosgene), yielding the esther chloroformate raw derivative of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one.
The raw chloroformate obtained above (4.0 mmol, 2.27 g) is dissolved in about 30 mL of dichloromethane, to which is added 2.07 g (4.1 mmol) of 5-{2-ethoxy-5-[(4-hydroxyethyl-1-piperazinyl)sulfonyl]phenyl}-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one, followed by the addition of 4 mL of dichloromethane containing 450 mg of triethylamine. The reaction mixture is maintained under stirring, being followed by chromatography of thin layer every hour until this indicates the end of the reaction (disappearing of chloroformate derivative). The reaction mixture is then diluted with 60 mL of dichloromethane, washed with NaCl saturated solution, after with sodium bicarbonate saturated solution and again with NaCl saturated solution. Organic phase is separated and dry with anhydrous sodium sulfate. The solvent is then evaporated to dry, yielding the dimer carbonate as a slightly yellowish solid.
This compound is re-crystallized from ethanol:DMF, yielding a pale white solid. Yielding m=3.2 g (76%)
Microanalysis: Theoretical C, (54.53%); H, (6.04%); N, (16.24%);
Obtained C, (54.45%); H, (6.02%); N, (16.17%).
INFO ABOUT INTERMEDIATE
5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3n-propyl-1,6-dihydro-7H-pyrazole[4,3-d]pyrimidin-7-one
| CAS No. | 139756-22-2 |
| Chemical Name: | 5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one |
| Synonyms: | Sildenafil Chlorosulfone IMpurity;Sildenafil Chlorosulfonyl IMpurity;5-(5-CHLOROSULFONYL-2-ETHOXY PHENYL)-1-METHYL-3-N-PROPYL-1;3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1 H-pyrazolo-(4-3-d)-pyrimidine-5;5-(5-Chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one;3-(4,7-Dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-4-ethoxy-benzenesulfonyl Chloride;4-Ethoxy-3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyriMidin-5-yl)benzene-1-sulfonyl chloride |
| CBNumber: | CB11175931 |
| Molecular Formula: | C17H19ClN4O4S |
http://www.chemicalbook.com/ChemicalProductProperty_EN_CB11175931.htm
…………..
SYNTHESIS OF

http://www.google.co.in/patents/US6362178

22.27 g (250 mmol) of D,L-alanine and 55.66 g (550 mmol) of triethylamine are dissolved in 250 ml of dichloromethane, and the solution is cooled to 0° C. 59.75 g (550 mmol) of trimethylsilyl chloride are added dropwise, and the solution is stirred for 1 hour at room temperature and for 1 hour at 40° C. After cooling to −10° C., 26.64 g (250 mmol) of butyryl chloride are added dropwise, and the resulting mixture is stirred for 2 hours at −10° C. and for one hour at room temperature.
With ice-cooling, 125 ml of water are added dropwise and the reaction mixture is stirred at room temperature for 15 minutes. The aqueous phase is evaporated to dryness, the residue is titrated with acetone and the mother liquor is filtered off with suction. The solvent is removed and the residue is chromatographed. The resulting product is dissolved in 3N aqueous sodium hydroxide solution and the resulting solution is evaporated to dryness. The residue is taken up in conc. HCl and once more evaporated to dryness. The residue is stirred with acetone, precipitated solid is filtered off with suction and the solvent is removed under reduced pressure. This gives 28.2 g (71%) of a viscous oil which crystallizes after some time.
200 MHz 1H-NMR (DMSO-d6): 0.84, t, 3H; 1.22, d, 3H; 1.50, hex, 2H; 2.07, t, 2H; 4.20, quin., 1H; 8.09, d, 1H.

25 g (210 mmol) of 2-hydroxybenzonitrile are refluxed with 87 g of potassium carbonate and 34.3 g (314.8 mmol) of ethyl bromide in 500 ml of acetone overnight. The solid is filtered off, the solvent is removed under reduced pressure and the residue is distilled under reduced pressure. This gives 30.0 g (97%) of a colourless liquid.
200 MHz 1H-NMR (DMSO-d6): 1.48, t, 3H; 4.15, quart., 2H; 6.99, dt, 2H; 7.51, dt, 2H.

21.4 g (400 mmol) of ammonium chloride are suspended in 375 ml of toluene, and the suspension is cooled to 0° C. 200 ml of a 2M solution of trimethylaluminium in hexane are added dropwise, and the mixture is stirred at room temperature until the evolution of gas has ceased. After addition of 29.44 g (200 mmol) of 2-ethoxybenzonitrile, the reaction mixture is stirred at 80° C. (bath) overnight.
With ice-cooling, the cooled reaction mixture is added to a suspension of 100 g of silica gel and 950 ml of chloroform, and the mixture is stirred at room temperature for 30 minutes. The mixture is filtered off with suction, and the filter residue is washed with the same amount of methanol. The mother liquor is concentrated, the resulting residue is stirred with a mixture of dichloromethane and methanol (9:1), the solid is filtered off with suction and the mother liquor is concentrated. This gives 30.4 g (76%) of a colourless solid.
200 MHz 1H-NMR (DMSO-d6): 1.36, t, 3H; 4.12, quart., 2H; 7.10, t, 1H; 7.21, d, 1H; 7.52, m, 2H; 9.30, s, broad, 4H.
EXAMPLE 10A 2-(2-Ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one

7.16 g (45 mmol) of 2-butyrylamino-propionic acid and 10.67 g of pyridine are dissolved in 45 ml of THF and, after addition of a spatula tip of DMAP, heated to reflux. 12.29 g (90 mmol) of ethyl oxalyl chloride are slowly added dropwise, and the reaction mixture is refluxed for 3 hours. The mixture is poured into ice-water and extracted three times with ethyl acetate and the organic phase is dried over sodium sulphate and concentrated using a rotary evaporator. The residue is taken up in 15 ml of ethanol and refluxed with 2.15 g of sodium bicarbonate for 2.5 hours. The cooled solution is filtered.
With ice-cooling, 2.25 g (45 mmol) of hydrazine hydrate are added dropwise to a solution of 9.03 g (45 mmol) of 2-ethoxybenzamidine hydrochloride in 45 ml of ethanol, and the resulting suspension is stirred at room temperature for another 10 minutes. The ethanolic solution described above is added to this reaction mixture, and the mixture is stirred at a bath temperature of 70° C. for 4 hours. After filtration, the mixture is concentrated, the residue is partitioned between dichloromethane and water, the organic phase is dried over sodium sulphate and the solvent is removed under reduced pressure.
This residue is dissolved in 60 ml of 1,2-dichloroethane and, after addition of 7.5 ml of phosphorus oxychloride, refluxed for 2 hours. The mixture is diluted with dichloromethane and neutralized by addition of sodium bicarbonate solution and solid sodium bicarbonate. The organic phase is dried and the solvent is removed under reduced pressure. Chromatography using ethyl acetate and crystallization afford 4.00 g (28%) of a colourless solid, Rf=0.42 (dichloromethane/methanol=95:5)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.56, t, 3H; 1.89, hex, 2H; 2.67, s, 3H; 3.00, t, 2H; 4.26, quart., 2H; 7.05, m, 2H; 7.50, dt, 1H; 8.17, dd, 1H; 10.00, s, 1H.
EXAMPLE 15A 4-Ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride

At 0° C., 2.00 g (6.4 mmol) of 2-(2-ethoxy-phenyl)-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are slowly added to 3.83 ml of chlorosulphonic acid. At room temperature, the reaction mixture is stirred ovemight, and then poured into ice-water and extracted with dichloromethane. This gives 2.40 g (91%) of a colourless foam.
200 MHz 1H-NMR (CDCl3): 1.03, t, 3H; 1.61, t, 2H; 1.92, hex, 2H; 2.67, s, 3H; 3.10, t, 2H; 4.42, quart., 2H; 7.27, t, 1H; 8.20, dd, 1H; 8.67, d, 1H; 10.18, s, 1H.
Example 22 2-[2-Ethoxy-5-(4-hydroxyethyl-1-amino-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one
By the same method, starting with 0.04 g (0.097 mmol) of 4-ethoxy-3-(5-methyl-4-oxo-7-propyl-3,4-dihydro-imidazo[5,1-f][1,2,4]triazin-2-yl)-benzenesulphonyl chloride and 0.04 g (0.29 mmol) of 1-amino-4-hydroxyethylpiperazine, 46 mg (91%) of 2-[2-ethoxy-5-(4-hydroxyethyl-1-amino-piperazine-1-sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5,1-f][1,2,4]triazin-4-one are obtained.
Rf=0.08 (dichloromethane/methanol=19:1)
200 MHz 1H-NMR (CDCl3): 1.02, t, 3H; 1.59, t, 3H; 1.90, sex., 2H; 2.49, m, 6H; 2.62, s, 3H; 2.71, m, 4H; 3.00, t, 2H; 3.55, t, 2H; 4.31, quart., 2H; 7.14, d, 1H; 8.05, dd, 1H; 8.60, d, 1H.
…………..
Methods of analysis
The development of lodenafil carbonate was reported by Toque et al. (2008). They observed the effects of lodenafil carbonate on rabbit and human corpus cavernosum relaxation, activity of PDE5 in human platelets, stability and metabolic studies in comparison with sildenafil and lodenafil, as well as the pharmacological evaluation of lodenafil carbonate after intravenous and oral administration in male beagles.
The determination of PDE activity, stability of lodenafil carbonate in human, dog and rat plasma and the pharmacokinetic parameters after a single intravenous or oral dose was carried out by LC-MS/MS analysis
Codevilla et al. (2011a) developed a stability-indicating reversed-phase liquid chromatography method using ultraviolet (UV) detection for the quantitative determination of lodenafil carbonate in tablets. The method can be useful for routine quality control assay and stability studies.
Another study for the determination of lodenafil carbonate in tablets was developed by Codevilla et al. (2011b). As an alternative to the LC method the authors suggested a UV-spectrophotometric method for the analysis of lodenafil carbonate in pharmaceutical form. The UV method offers advantages over other analytical methods due to its rapidity, simplicity, and lower cost. Recently, Codevilla et al. (2012) developed and validated a capillary zone electrophoresis (CZE) method for determination of lodenafil carbonate in drug products. There are some advantages to use the CZE method, such as rapid analysis, small sample and reagent consumption, high separation efficiency (Furlanetto et al., 2001; Yang et al., 2010). The results obtained from the UV-spectrophotometric method and CZE method were compared statistically with the LC method (Codevilla et al., 2011a) and the results showed no significant difference between these methods.
References
- Toque HA, Teixeira CE, Lorenzetti R, Okuyama CE, Antunes E, De Nucci G (September 2008). “Pharmacological characterization of a novel phosphodiesterase type 5 (PDE5) inhibitor lodenafil carbonate on human and rabbit corpus cavernosum”. European Journal of Pharmacology 591 (1–3): 189–95. doi:10.1016/j.ejphar.2008.06.055. PMID 18593576.
- Cristália Product page. Retrieved on September 16, 2009.
- ukmedix Lodenafil article. Retrieved on September 16, 2009.
- Glina S, Toscano I, Gomatzky C, de Góes PM, Júnior AN, Claro JF, Pagani E (February 2009). “Efficacy and tolerability of lodenafil carbonate for oral therapy in erectile dysfunction: a phase II clinical trial”. The Journal of Sexual Medicine 6 (2): 553–7. doi:10.1111/j.1743-6109.2008.01079.x.PMID 19040623.
- Glina S, Fonseca GN, Bertero EB, Damião R, Rocha LC, Jardim CR, Cairoli CE, Teloken C, Torres LO, Faria GE, da Silva MB, Pagani E (February 2010). “Efficacy and Tolerability of Lodenafil Carbonate for Oral Therapy of Erectile Dysfunction: A Phase III Clinical Trial”. The Journal of Sexual Medicine 7 (5): 1928–1936. doi:10.1111/j.1743-6109.2010.01711.x. PMID 20214718.
- Toque H A et al., (2008) European Journal of Pharmacology, 591(1-3):189-95.
- Exploring the role of PDE5 inhibition in the treatment of muscular dystrophy
Drugs Fut 2011, 36(4): 321
VARDENAFIL

GSK’s diabetes drug Eperzan moves towards approval in Europe
GlaxoSmithKline has received a positive opinion from the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) for albiglutide, under the brand name Eperzan, for treatment of type 2 diabetes.
CLICK ON TITLE
GSK’s diabetes drug Eperzan moves towards approval in Europe
Mirodenafil 米罗那非 标准品 ………..An erectogenic agent.

862189-95-5 (free base)
SK Chemicals (Originator)
Treatment of Erectile Dysfunction , hypertention
Mirodenafil belongs to a class of drugs called PDE5 inhibitors, which many other erectile dysfunction drugs such as sildenafil, tadalafil, andvardenafil also belong to. It was developed by SK Chemicals Life Science and is marketed under the trade name of Mvix tab which comes in different doses (50 mg, 100 mg).
Mirodenafil is also available under the name of Mvix S ODF 50 mg as an orally dissolving film (ODF) which dissolves on the tongue without water. It is the first licensed medicine for the treatment of erectile dysfunction as a dosage form of film.
Mirodenafil is a newly developed oral phosphodiesterase type 5 inhibitor, currently under investigation as a treatment for erectile dysfunction (ED).
MIRODENAFIL米罗那非 标准品
Mirodenafil hydrochloride is a high selective PDE5 inhibitor commercialized by SK Chemicals which had been in early clinical development for the treatment of erectile dysfunction (ED). Early clinical studies had also been ongoing for the treatment of hypertension in patients taking amlodipine; however, no recent development has been reported for this research. The development of compound started in 1998 jointly by SK Chemicals and a bio-venture In2Gen.
Several clinical trials were conducted,[1][2][3] but mirodenafil has not been approved for use in the United States by the U.S. Food and Drug Administration.

Mirodenafil dihydrochloride
|
||||||||||||
Korean Patent No. 358083 discloses pyrrolopyrimidinone derivatives having good inhibition activity against PDE-5, a method of its preparation thereof, an intermediate compound used to prepare the same and their use for prevention and treatment of erectile dysfunction, pulmonary arterial hypertension, chronic obstructive pulmonary disease, benign prostatic hypertrophy and lower urinary tract diseases.
Of the pyrrolopyrimidinone derivatives disclosed in Korean Patent No. 358083, 5-ethyl-2-{5-[4- (2-hydroxyethyl)piperazin-1-ylsulfonyl]-2-n-propoxyphenyl}-7-n-propyl-l-3,5-dihydro-4 H-pyrrolo[3,2-d]pyrimidin-4-one (hereinafter, “SK-3530”) represented by the following formula (1 ) is an excellent selective inhibitor PDE-5 over other PDEs and is under clinical trial for the treatment of erectile dysfunction after passing through the preclinical stage.

The dihydrochloride salt (2HCI) of SK-3530 has been under investigation through the preclinical and clinical stages.
The SK-3530 dihydrochloride salt has good solubility and can be easily stabilized for pharmaceutical preparation. But, it has the following drawbacks.
First, because the SK-3530 dihydrochloride salt is hygroscopic, it easily absorbs moisture from the atmosphere and becomes discolored when the moisture content is high. And, due to the hygroscopic property, an anhydrous solvent condition and a dry air condition have to be provided to obtain a stable product. Second, the SK-3530 dihydrochloride salt should be kept at a temperature lower than room temperature because it does not show enough stability at room temperature. In particular, the SK-3530 dihydrochloride salt is labile to heat or light, and thus any prolonged exposure to heat or light results in various impurities.
Third, the SK-3530 dihydrochloride salt could corrode the punch during tablet ting due to its somewhat corrosive properties. This is because the SK-3530 dihydrochloride salt is a simple amorphous salt rather than being a stable crystalline acid addition salt or hydrate form. Thus, one of the two hydrochloric acid groups with a relatively weak ionic bond character may leave the molecule under severe conditions. As aforementioned, the SK-3530 dihydrochloride salt may be endowed with a sufficient stability for pharmaceutical preparation. But, some additional techniques and costs are needed due to the deficiency in intrinsic physicochemical property and stability of the compound.


…………………………

The invention relates to a series of pyrrolopyrimidinone derivatives of the formula (1):

R1 ETHYL
R2=H
R3= PROPYL
R4 = PROPYL
R5=R5=SO2NR6R7, NR6R7 is 4-(3-hydroxypropyl)piperazinyl) IS MIRODENAFIL
ANALOGOUS METHOD
BELOW IS CUT PASTE OF R1 METHYL ANALOGUE ……………..R1 =METHYL AND NOT ETHYL ….CAUTION
Example 39 Preparation of
5-(5-(4-(2-hydroxyethyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one hydrochloride (a compound of the formula (1) wherein R5=SO2NR6R7, R1=CH3, R2=H, R3=CH2CH2CH3, R4=CH2CH2CH3; NR6R7 is 4-(2-hydroxyethyl)piperazinyl)
The titled compound was prepared as described in Example 2 by using 5-(5-(4-(2-hydroxyethyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one in place of 5-(2-ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one.
yield: 99%
mp 66.5° C. dec;
IR (neat) 3332 (NH and OH), 1676 (C═O), 1166 (SO2) cm−1;
1H NMR (DMSO-d6) δ 0.92 (t, J=7.2 Hz, 3H, CH2CH2CH3), 0.96 (t, J=7.2 Hz, 3H, OCH2CH2CH3), 1.56-1.80 (m, 4H, 2 CH2CH2CH3), 2.59 (t, J=7.5 Hz, 2H, CH2CH2CH3), 2.91 (br t, J=11.7 Hz, 2H, 2 SO2NCHax), 3.12-3.27 (m, 4H, NCH2CH2 and 2 SO2NCHeq), 3.58 (br d, J=11.7 Hz, 2H, 2 +HNCHax), 3.68-3.85 (m, 4H, CH2CH2OH and 2 +HNCHeq), 4.00 (s, 3H, NCH3), 4.15 (t, J=6.3 Hz, 2H, OCH2CH2CH3), 4.66 (br s, 1H, OH), 7.28 (s, 1H, H-2), 7.44 (d, J=9.0 Hz, 1H, H-3′), 7.89 (dd, J=9.0 Hz, 2.4 Hz, 1H, H-4′), 8.01 (d, J=2.4 Hz, 1H, H-6′), 10.85 (br s, 1H, NH+), 12.01 (br s, 1H, NH).
Example 42 Preparation of
5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one (a compound of the formula (1) wherein R5=SO2NR6R7, R1=CH3, R2=H, R3=CH2CH2CH3, R4=CH2CH2CH3; NR6R7 is 4-(3-hydroxypropyl)piperazinyl)
The titled compound was prepared as described in Example 1 by using 5-(5-chlorosulfonyl-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one and 1-(3-hydroxypropyl)piperazine in place of 5-(5-chlorosulfonyl-2-ethoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one and 1-methylpiperazine.
yield: 94%
mp 162.5° C. dec (EtOAc/hexanes);
IR (neat) 3484, 3302 (NH and OH), 1669 (C═O), 1170 (SO2) cm−1;
1H NMR (CDCl3/TMS) δ 1.00 (t, J=7.5 Hz, 3H, CH2CH2CH3), 1.20 (t, J=7.5 Hz, 3H, OCH2CH2CH3), 1.64-1.80 (m, 4H, CH2CH2CH2OH and CH2CH2CH3), 1.99-2.11 (m, 2H, OCH2CH2CH3), 2.58-2.64 (m, 6H, NCH2CH2 and 2 NCH2), 2.71 (t, J=7.5 Hz, 2H, CH2CH2CH3), 3.08 (br s, 4H, 2 SO2NCH2), 3.71 (t, J=5.4 Hz, 2H, CH2CH2OH), 4.08 (s, 3H, NCH3), 4.26 (t, J=6.3 Hz, 2H, OCH2CH2CH3), 4.28 (br s, 1H, OH), 6.88 (s, 1H, H-2), 7.14 (d, J=8.7 Hz, 1H, H-3′), 7.77 (dd, J=8.7 Hz, 2.7 Hz, 1H, H-4′), 8.87 (d, J=2.7 Hz, 1H, H-6′), 10.69 (br s, 1H, NH); MS (FAB) m/z 532 (MH+).
Example 43 Preparation of
5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one hydrochloride (a compound of the formula (1) wherein R5=SO2NR6R7, R1=CH3, R2=H, R3=CH2CH2CH3, R4=CH2CH2CH3; NR6R7 is 4-(3-hydroxypropyl)piperazinyl)
The titled compound was prepared as described in Example 2 by using 5-(5-(4-(3-hydroxypropyl)piperazinylsulfonyl)-2-n-propoxyphenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one in place of 5-(2-ethoxy-5-(4-methylpiperazinylsulfonyl)phenyl)-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrrolo[4,3-d]pyrimidin-7-one.
yield: 99%
mp 62.5° C. dec;
IR (neat) 3347, 3321 (NH and OH), 1689 (C═O), 1168 (SO2) cm−1;
1H NMR (DMSO-d6) δ 0.93 (t, J=7.5 Hz, 3H, CH2CH2CH3), 0.96 (t, J=7.5 Hz, 3H, OCH2CH2CH3), 1.57-1.87 (m, 6H, CH2CH2CH2OH and 2 CH2CH2CH3), 2.59 (t, J=7.5 Hz, 2H, CH2CH2CH3), 2.89 (br t, J=11.7 Hz, 2H, 2 SO2NCHax), 3.01-3.19 (m, 4H, NCH2CH2 and 2 SO2NCHeq), 3.44 (t, J=6.0 Hz, 2H, CH2CH2OH), 3.52 (br d, J=11.7 Hz, 2H, 2 +HNCHax), 3.79 (br d, J=11.7 Hz, 2H, 2 +HNCHeq), 4.00 (s, 3H, NCH3), 4.15 (t, J=6.6 Hz, 2H, OCH2CH2CH3), 4.71 (br s, 1H, OH), 7.29 (s, 1H, H-2), 7.44 (d, J=8.7 Hz, 1H, H-3′), 7.89 (dd, J=8.7 Hz, 2.4 Hz, 1H, H-4′), 8.02 (d, J=2.4 Hz, 1H, H-6′), 11.13 (br s, 1H, NH+), 12.05 (br s, 1H, NH).
……………………………
Synthesis from patent and some construction by me
you can synthesize as follows, A CHEMIST CAN PICK THIS UP, this is not available clearly anywhere
Chlorosulfonation of the methyl salicylate with ClSO3H in SOCl2 affords the Methyl 3-Chlorosulfonyl-6-hydroxybenzoate described below
THESE INTERMEDIATES FROM PATENT MAY HELP YOU
 methyl salicylate
methyl salicylate
 X=CL, R8=ME
X=CL, R8=ME
- Methyl 3-Chlorosulfonyl-6-hydroxybenzoate
Example 1 EP1362858A1
-
To a cooled solution of SOCl2 (156 g, 1. 31 mol) and ClSO3H (460 g, 3.94 mol) at 0°C was added slowly methyl salicylate (200 g, 1.31 mol) for 30 minutes, and the mixture was stirred at room temperature for 20 hours. The reaction mixture was poured slowly into the ice (2 kg) and H2O (3 L) mixture, and the resulting white precipitates were collected by filtration. The filtered solid was washed with H2O (3 L), air-dried for 2 days and then dried under vacuum at 40°C for 2 days to afford the titled product (232 g, 93%) as a white solid.
mp 76.5-77.5 °C (toluene/hexanes);
IR (neat) 1699 (C=O) cm-1;
1H NMR (CDCl3/TMS) δ 3. 90 (s, 3 H, OCH3), 6. 93 (d, J= 8. 7 Hz, 1 H, H-3), 7. 70 (dd, J= 8. 7 Hz, 2. 4 Hz, 1 H, H-4), 8. 03 (d, J= 2. 4 Hz, 1 H, H-6).
- Methyl 3-Chlorosulfonyl-6-hydroxybenzoate
Example 2 EP1362858A1
-
 1-(2-hydroxyethyl)piperazine
1-(2-hydroxyethyl)piperazine  R8=ME, W=N, n=2
R8=ME, W=N, n=2-
- Methyl 2-Hydroxy-5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]benzoate
-
To a mixture of 1-(2-hydroxyethyl)piperazine (27 mg, 0. 21 mmol) and K2CO3 (33 mg, 0. 24 mmol) in DMF (5 mL) was added methyl 3-chlorosulfonyl-6-hydroxybenzoate (50 mg, 0. 20 mmol), and the mixture was stirred at room temperature for 1 hour. The reaction mixture was washed with H2O (10 mL), and the aqueous layer was further extracted with 5% MeOH in CH2Cl2 (20 mL). The combined organic layer was dried (MgSO4), filtered, and the filtrate was evaporated to dryness under reduced pressure. The crude residue was purified by MPLC on silica gel (5% MeOH in CH2Cl2) to afford the titled compound (59 mg, 86%) as white solid.
mp 152 °C (dec) (CH2Cl2/ether);
IR (neat) 1685 (C=O) cm-1;
1H NMR (CDCl3/TMS) δ 2. 30 (br s, 1 H, CH2OH), 2. 63 (t, J = 5. 4 Hz, 2 H, NCH 2CH2O), 2. 70 (m, 4 H, 2 NCH2), 3. 12 (m, 4 H, 2 SO2NCH2), 3. 64 (t, J= 5. 4 Hz, 2 H, NCH2CH 2O), 4. 01 (s, 3 H, OCH3), 7. 12 (d, J= 8. 7 Hz, 1 H, H-3), 7. 81 (dd, J= 8. 7 Hz, 2. 4 Hz, 1 H, H-4), 8. 26 (d, J = 2. 4 Hz, 1 H, H-6), 11. 26 (br s, 1 H, OH);
MS (FAB) m/z 345 (MH+).
- Methyl 2-Hydroxy-5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]benzoate
Example 3 EP1362858A1
Methyl 3-[4-(2-Hydroxyethyl)piperazin-1-ylsulfonyl]-6-n-propoxybenzoate
-
To a mixture of methyl 2-hydroxy-5-(4-(2-hydroxyethyl)piperazin-1-ylsulfonyl)benzoate (800 mg, 2. 32 mmol) and K2CO3 (482 mg, 3. 49 mmol) in DMF (5 mL) was added 1-bromopropane (253 µL, 2.79 mmol), and the mixture was stirred at 60°C overnight. The reaction mixture was evaporated to dryness under reduced pressure, washed with H2O (10 mL), and the aqueous layer was further extracted with CH2Cl2 (50 mL x 2). The combined organic layer was dried (MgSO4), filtered, and the filtrate was evaporated to dryness under reduced pressure. The crude residue was purified by MPLC on silica gel (3% MeOH in CHCl3) to afford the titled compound (309 mg, 80%) as a white solid.
mp 88-89 °C (EtOAc/hexanes);
IR (neat) 3242 (OH), 1741 (C=O) cm-1;
1H NMR (CDCl3/TMS) δ 1. 09 (t, J = 7. 5 Hz, 3 H, OCH2CH2CH 3), 1. 84-1. 95 (m, 2 H, OCH2CH 2CH3), 2. 23 (br s, 1 H, CH2OH), 2. 54 (t, J= 5. 4 Hz, 2 H, NCH 2CH2O), 2. 60 (m, 4 H, 2 NCH2), 3. 04 (m, 4 H, 2 SO2NCH2), 3. 58 (t,J = 5. 4 Hz, 2 H, NCH2CH 2O), 3. 91 (s, 3 H, OCH3), 4. 08 (t, J= 6. 6 Hz, 2 H, OCH 2CH2CH3), 7. 07 (d, J = 9. 0 Hz, 1 H, H-3), 7. 82 (dd, J = 9. 0 Hz, 2. 4 Hz, 1 H, H-4), 8. 15 (d, J = 2. 4 Hz, 1 H, H-6);
MS (FAB) m/z 387 (MH+).
- FURTHER INFO OTHER THAN ABOVE PATENT
- HYDROLYSE Methyl 3-[4-(2-Hydroxyethyl)piperazin-1-ylsulfonyl]-6-n-propoxybenzoate TO -COOLi SALT using LiOH
- CONDENSE WITH 3-amino-1-ethyl-4-propyl-1H-pyrrole-2-carboxamide USING HOBt AND DMAP/ PYRIDINE

9……….Methyl 3-[4-(2-Hydroxyethyl)piperazin-1-ylsulfonyl]-6-n-propoxybenzoate R8= ME, R4=PROPYL, W=N, n=2
10……….3-amino-1-ethyl-4-propyl-1H-pyrrole-2-carboxamide R1=ETHYL, R2=H, R3=PROPYL, IN ABOVE
YOU WILL GET A COMPD

R1 ETHYL
R2=H
R3= PROPYL
R4 = PROPYL
W=N
n=2
IS MIRODENAFIL precursor ie n-1 compund
- CYCLIZE THIS WITH BuOK/tBuOH AND USE ACID TO GET FINAL PRODUCT MIRODENAFIL
- A cyclization reaction is generally carried out by heating at an elevated temperature, for example 50-150° C., in the presence of an acid or a base in a suitable solvent such as an aqueous C1-C4 alkanol, water, a halogenated hydrocarbon, or acetonitrile. Thus, for example, the cyclization may be affected by treatment of a compound with an inorganic or organic base such as sodium hydroxide, potassium carbonate or potassium tert-butoxide, in an alcoholic aqueous medium, preferably potassium tert-butoxide in tert-butanol at 60° C. to reflux temperature.
SYNTHESIS OF 1-(2-hydroxyethyl)piperazine needed for MIRODENAFIL SYNTHESIS
Compounds of the formula (29) can be prepared from the compounds of the formula (30):

wherein X and P are as previously defined.
note X=N ATOM, n = 2
…………………………….
 MIRODENAFIL
MIRODENAFIL
Two methods were published for the determination of mirodenafil in biological fluids. Choi et al. (2009) describe an isocratic reversed-phase liquid chromatographic method for simultaneous analysis of mirodenafil and its two main metabolites, SK3541 and SK3544, in rat plasma, urine, and tissue homogenates. The authors used a simple deproteinization procedure for sample preparation, and the compounds were separated on a C18 column (250 mm x 4.6 mm, i.d.; 5 µm particle size; Shiseido, Tokyo, Japan). The mobile phase was constituted with 0.02 M ammonium acetate buffer (pH 6):acetonitrile (52:48, v/v) at a flow rate of 1.4 mL/min. UV detection was at 254 nm.
Lee et al. (2009) developed a study with the proposed method to determine sildenafil and mirodenafil in the plasma and corpus cavernosum tissue of rats using LC–MS/MS. A CapcellPak phenyl column (2.1mm x 150 mm, 5µm) maintained constant at 40 ºC was used for the separation. The mobile phase consisted of 90% acetonitrile in 5 mM ammonium formate (pH 6.0). A gradient program was used for the LC separation with a flow rate of 0.2 mL/min.
References
- Paick JS, Ahn TY, Choi HK, Chung WS, Kim JJ, Kim SC, Kim SW, Lee SW, Min KS, Moon KH, Park JK, Park K, Park NC, Suh JK, Yang DY, Jung HG (November 2008). “Efficacy and safety of mirodenafil, a new oral phosphodiesterase type 5 inhibitor, for treatment of erectile dysfunction”. The Journal of Sexual Medicine 5 (11): 2672–80. doi:10.1111/j.1743-6109.2008.00945.x. PMID 18638004.
- Kim BH, Yi S, Kim J, Lim KS, Kim KP, Lee B, Shin SG, Jang IJ, Yu KS (June 2009). “Influence of alcohol on the hemodynamic effects and pharmacokinetic properties of mirodenafil: a single-dose, randomized-sequence, open-label, crossover study in healthy male volunteers in Korea”.Clinical Therapeutics 31 (6): 1234–43. doi:10.1016/j.clinthera.2009.06.008. PMID 19695390.
- Shin KH, Kim BH, Kim TE, Kim JW, Yi S, Yoon SH, Cho JY, Shin SG, Jang IJ, Yu KS (December 2009). “The effects of ketoconazole and rifampicin on the pharmacokinetics of mirodenafil in healthy Korean male volunteers: an open-label, one-sequence, three-period, three-treatment crossover study”.Clinical Therapeutics 31 (12): 3009–20. doi:10.1016/j.clinthera.2009.12.012. PMID 20110038.
- Synthesis of 5-ethyl-2-[5-[4-(2-hydroxyethyl)piperazin-1-ylsulfonyl]-2-n-propoxyphenyl]-7-n-propyl-3,5-dihydro-4H-pyrrolo[3,2-d]-[2-14C]pyrimidin-4-one·2 HCl (14C-SK3530·2 HCl)J Label Compd Radiopharm 2006, 49(13): 1141
- More information about mirodenafil can be found at Paick J S et al., (2008) The Journal of Sexual Medicine, 5 (11): 2672-80.
- PDE-5 inhibitor that came into the market recently (Choi et al., 2009; Lee et al., 2009).not currently approved for use in the United States but clinical trials are being conducted.
- Crystal forms of SK-3530.
Song HO, Sohn YT.Arch Pharm Res. 2010 Dec;33(12):2033-6. doi: 10.1007/s12272-010-1220-3. Epub 2010 Dec 30. - Looking to the future for erectile dysfunction therapies.Hatzimouratidis K, Hatzichristou DG.Drugs. 2008;68(2):231-50. Review.
-
- Paick JS, Ahn TY, Choi HK, Chung WS, Kim JJ, Kim SC, Kim SW, Lee SW, Min KS, Moon KH, Park JK, Park K, Park NC, Suh JK, Yang DY, Jung HG (November 2008). “Efficacy and safety of mirodenafil, a new oral phosphodiesterase type 5 inhibitor, for treatment of erectile dysfunction”. The Journal of Sexual Medicine 5 (11): 2672–80. doi:10.1111/j.1743-6109.2008.00945.x. PMID 18638004.
- Kim BH, Yi S, Kim J, Lim KS, Kim KP, Lee B, Shin SG, Jang IJ, Yu KS (June 2009). “Influence of alcohol on the hemodynamic effects and pharmacokinetic properties of mirodenafil: a single-dose, randomized-sequence, open-label, crossover study in healthy male volunteers in Korea”. Clinical Therapeutics 31 (6): 1234–43. doi:10.1016/j.clinthera.2009.06.008. PMID 19695390.
- Shin KH, Kim BH, Kim TE, Kim JW, Yi S, Yoon SH, Cho JY, Shin SG, Jang IJ, Yu KS (December 2009). “The effects of ketoconazole and rifampicin on the pharmacokinetics of mirodenafil in healthy Korean male volunteers: an open-label, one-sequence, three-period, three-treatment crossover study”. Clinical Therapeutics 31 (12): 3009–20. doi:10.1016/j.clinthera.2009.12.012. PMID 20110038.
- Matheny, C., et al., Drug Metab. Dispos., 32, 1008 (2004)
Gupta, M., et al., J. Clin. Pharmacol., 45, 987 (2005)
Ek, M., et al., Biochem. Pharmacol., 74, 496 (2007)
Lee, H., et al., Xenobiotica, 38, 21 (2008)

| WO2006018088A1 * | Jul 15, 2005 | Feb 23, 2006 | Switch Biotech Ag | Use of a pde 5 inhibitor for treating and preventing hypopigmentary disorders |
| KR20010083637A * | Title not available |
| US6962911 * | Feb 15, 2001 | Nov 8, 2005 | Sk Chemicals Co., Ltd. | Pyrrolopyrimidinone derivatives, process of preparation and use |
| US20100069632 * | Jul 3, 2007 | Mar 18, 2010 | Sk Chemicals Co., Ltd | Salts of pyrrolopyrimidinone derivatives and process for preparing the same |
| EP2038282A1 * | Jul 3, 2007 | Mar 25, 2009 | SK Chemicals, Co., Ltd. | Salts of pyrrolopyrimidinone derivatives and process for preparing the same |


DASANTAFIL
 Dasantafil
Dasantafil

569351-91-3 CAS NO
405214-79-1 (racemate)
THERAPEUTIC CLAIM treatment of erectile dysfunction (phosphodiesterase (PDE) 5 isoenzyme inhibitor)
read all at
ALL ABOUT DRUGS
CLICK BELOW
http://www.allfordrugs.com/2014/01/29/dasantafil-for-treatment-of-erectile-dysfunction/
Gisadenafil for erectile dysfunction
GISEDENAFIL
GISEDENAFIL BESYLATE
334826-98-1 free form
334827-98-4 (as besylate)
- UK 369003
- UK-369,003
- UK0369,003
- UNII-S6G4R7DI1C
THERAPEUTIC CLAIM Treatment of lower urinary tract
symptoms associated with BPH
LEARN SPECTROSCOPY USING GISADENAFIL INTERMEDIATES
CHEMICAL NAMES FREE FORM
1. ……..7H-Pyrazolo[4,3-d]pyrimidin-7-one, 5-[2-ethoxy-5-[(4-ethyl-1-
piperazinyl)sulfonyl]-3-pyridinyl]-3-ethyl-2,6-dihydro-2-(2-methoxyethyl)-
2. …….5-{2-ethoxy-5-[(4-ethylpiperazin-1-yl)sulfonyl]pyridin-3-yl}-3-ethyl-2-(2-
methoxyethyl)-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one
3………1-(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine
MOLECULAR FORMULA C23H33N7O5S
MOLECULAR WEIGHT 519.6
CODE DESIGNATION UK-369,003
CAS REGISTRY NUMBER 334826-98-1
5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulfonyl)pyridin-3-yl]-3-ethyl-2-(2-methoxyethyl)-6,7-dihydro-2H-pyrazolo[4,3-d]pyrimidin-7-one
Phosphodiesterase PDE5A Inhibitors , Treatment of Erectile Dysfunction
Pfizer (Originator)
UK-369003 is a phosphodiesterase V (PDE V) inhibitor which had been under development for the treatment of erectile dysfunction, pulmonary hypertension and for the treatment of lower urinary tract symptoms, but no recent development has been reported for these indications. Trials for the treatment of benign prostatic hyperplasia were discontinued.
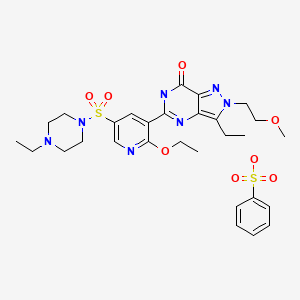
D09622, 334827-98-4
M.Wt:677.79
5-(2-ethoxy-5-(4-ethylpiperazin-1-ylsulfonyl)pyridin-3-yl)-3-ethyl-2-(2-methoxyethyl)-2H-pyrazolo[4,3-d]pyrimidin-7(6H)-one benzenesulfonate
1-[[6-Ethoxy-5-[3-ethyl-4,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridinyl]sulfonyl]-4-ethylpiperazine Monobenzenesulfonate
Formula:C23H33N7O5S.C6H6O3S
| Certificate of Analysis |
|
| Biological Activity:Potent and selective PDE5 inhibitor (IC50: 1.23 nM) with improved selectivity over PDE6(PDE5/6 selectivity value 117 and >3000-fold selectivity over other PDEs).Gisadenafil has the potential for oral bioavailability and dose-proportional pharmacokinetics. Close analogue of Sildenafil (Viagra; Axon 2046) |
Gisadenafil besylate is a PDE5 inhibitor. Inhibition of PDE5 prevents the breakdown of cyclic phosphodiester secondary messenger molecules. This has the effect of prolonging and enhancing signal transduction.
CLINICAL TRIALS
http://clinicaltrials.gov/search/intervention=UK-369,003
………………………….
PAPERS
Bioorganic and Medicinal Chemistry, 2012 , vol. 20, 1 p. 498 – 509
http://www.sciencedirect.com/science/article/pii/S0968089611008303


Scheme 1.
Reagents and conditions: (i) 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, hydroxybenzotriazole, di-isopropylethylamine, THF, 20 °C, 20 h; (ii) caesium carbonate, alkyl mesylate or alkyl chloride, DMF, 20 °C, 20 h; (iii) KHMDS, R1OH, 120 °C, 20 h.

Scheme 2.
Reagents and conditions: (i) KHMDS, nBuOH, 120–130 °C, pressure vessel (ii) TFA, CH2Cl2; (iii) methanesulphonyl chloride, NEt3, CH2Cl2; (iv) HOAc, NaCNBH3, CH2O (v) KHMDS, nBuOH, reflux.

Scheme 3.
Reagents and conditions: (i) caesium carbonate, RCl, DMF; (ii) 50 psi H2, 10% Pd/C (iii) 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride, HOBT, di-isopropylethylamine, THF, 20 °C, 20 h; (iv) KHMDS, ethanol, 120 °C, pressure vessel; (v) TFA, CH2Cl2; (vi) CH2O, HOAc, NaCNBH3; (vii) R1OH, KHMDS, 120 °C.

Scheme 4.
Reagents and conditions: (i) NaNO2, HCl, H2O; (ii) TFAA, Et2O; (iii) ethyl propynoate, xylene, reflux, 2 h; (iv) NaOH, H2O, dioxan; (v) HNO3/H2SO4, 40–55 °C; (vi) (COCl)2, CH2Cl2, DMF; (vii) NH3, THF; (viii) 10% Pd/C, EtOH, 60 psi H2, 20 °C, 14 h; (ix) acid chloride of 3, NEt3, CH2Cl2; (x) KHMDS, EtOH, 130 °C, 14 h, pressure vessel; (xi) methoxyethanol, KHMDS, reflux, 14 h.
……………………………
PAPERS
http://pubs.acs.org/doi/abs/10.1021/op0300241

………………………….

PAPERS

UK-369,003 was nominated for development as the lead candidate for treatment of benign prostatic hyperplasia (BPH). The free base was found to be moderately crystalline with a melting point of 168 °C. Solubility of the free base at physiological pH was found to be poor hence necessitating a comprehensive screen for a suitable salt form of the API. Benzenesulfonic acid was found to form the most suitable counterion for the API with a melting point of 248 °C and satisfied all our requirements for primary and secondary processing. The process for the formation of the benzenesulfonic acid salt involved the use of water/methyl ethyl ketone (4% water by volume) as the reaction medium. The water level at 4% ensured an optimum balance between product quality (purging of impurities) and the reaction yield. The cyclisation reaction (step 2/Scheme 01) involves the use of ethanol as the reaction media. Any residual amount of ethanol in the isolated step 2 product was therefore considered to be a considerable risk factor in the potential formation of ethyl besylate during the final step processing (step 3/Scheme 01).

aCDI = carbonyl diimidazole; MEK = methyl ethyl ketone; EtOAc = ethyl acetate; KOtBu = potassium tertiary butoxide; EtOH = ethanol.
……………………
SYNTHESIS
Compound 1E is also known as 5-[2-Ethoxy-5-(4-ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one, or alternatively as 1-{6-ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-pyridyl sulphonyl}-4-ethylpiperazine (the compound of Example 103 of WO 01/27113 and exemplified hereinafter as Example 1).
Preparation 1
2,2-dimethoxybutane:
Methyl ethyl ketone (672 mL) was charged to a 2 L round bottomed flask and stirred at room temperature before being treated with, trimethylorthoformate (763 mL) and para-toluenesulphonic acid (6.65 g, 0.5 mol %). Over a 15 min period the internal temperature rose to 46° C., so the reaction was cooled to 0° C. for 30 min. The reaction was then stirred at room temperature for 2 h. The reaction was then neutralised by pouring onto sodium carbonate (ca. 750 g) with constant stirring. The resultant slurry was filtered under vacuum and the resultant filtrate was distilled at atmospheric pressure. The fraction boiling in the range 118° C.-124° C. was collected as a colourless liquid, 582 g, 70%.
1H NMR (CDCl3): δ=0.88 (3H, t), 1.24 (3H, s), 1.61 (2H, q), 3.17 (6H, s).
Example 1 N-[3-Carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl]-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide
(a) Ethyl 3-ethyl-1H-pyrazole-5-carboxylate (IIA) from (IlI) and (V)

To a stirred solution of 2,2-dimethoxybutane (10 g, 84.7 mMol) in CH2Cl2 (50 mL) under a nitrogen atmosphere at 0° C. was added pyridine (13.7 mL, 169.5 mMol). The reaction mixture was maintained at 0° C. and a solution of trichloroacetyl chloride (18.9 mL, 169.5 mMol) in CH2Cl2 (35 mL) was added over 1 hour with constant stirring. The yellow-orange solution begins to precipitate a white solid as the reaction progresses. The reaction mixture is allowed to warm to room temperature over 20 h. The reaction mixture was diluted with ethanol (150 mL) and re-cooled to 0° C. before treatment with hydrazine hydrate (8.2 mL, 169.5 mMol) as a solution in ethanol (35 mL) over 30 min. The reaction was heated to 50° C. and solvent was distilled at atmospheric pressure. The temperature was increased until the head temperature reached 78° C. Reflux was maintained for a further 2 h, before cooling to room temperature. The reaction mixture was diluted with water (250 mL) and ethanol was removed by evaporation at reduced pressure. The resultant mixture was extracted with CH2Cl2 (3×200 mL). The combined organics were dried (MgSO4), filtered and evaporated at reduced pressure to afford the title compound as a brown oil, 12.05 g, 85%.
1H NMR (300 MHz, CDCl3): δ=1.20 (3H, t), 1.28 (3H, t), 2.67 (2H, q), 4.29 (2H, q), 6.55 (1H, s), 12.56 (1H, s).
LRMS m/z=167.1 [M-H]+, C8H12N2O2 requires 168.2.
(b) Ethyl 3-ethyl-1H-pyrazole-5-carboxylic acid (IIA) from (IIA) via route 1

Aqueous sodium hydroxide solution (10M; 100 ml, 1.0 mol) was added dropwise to a stirred suspension of the title compound of Example (a) (66.0 g, 0.39 mol) in methanol and the resulting solution heated under reflux for 4 hours. The cool reaction mixture was concentrated under reduced pressure to ca. 200 ml, diluted with water (200 ml) and this mixture washed with toluene (3×100 ml). The resulting aqueous phase was acidified with concentrated hydrochloric acid to pH 4 and the white precipitate collected and dried by suction to provide the title compound (34.1 g). δ (DMSOd6): 1.13 (3H,t), 2.56 (2H,q), 6.42 (1H,s).
(c) 4-Nitro-3-n-propyl-1H-pyrazole-5-carboxylic acid
Fuming sulphuric acid (17.8 ml) was added dropwise to stirred, ice-cooled fuming nitric acid (16.0 ml), the resulting solution heated to 50° C., then 3-n-propyl-1H-pyrazole-5-carboxylic acid (Chem. Pharm. Bull., 1984, 32,1568; 16.4 g, 0.106 mol) added portionwise over 30 minutes whilst maintaining the reaction temperature below 60° C. The resulting solution was heated for 18 hours at 60° C., allowed to cool, then poured onto ice. The white precipitate was collected, washed with water and dried by suction to yield the title compound (15.4 g), m.p. 170-172° C. Found: C, 42.35; H, 4.56; N, 21.07. C7H9N3O4requires C, 42.21; H, 4.55; N, 21.10%. δ (DMSOd6): 0.90 (3H,t), 1.64 (2H,m), 2.83 (2H,m), 14.00 (1 H,s).
(d) 3-Ethyl-4-nitro-1H-pyrazole-5-carboxylic acid (IIA) to (AA) via route 2

Obtained from the title compound of Example (b), by analogy with the process of Example (c), as a brown solid (64%). δ (DMSOd6): 1.18 (3H,t), 2.84 (2H,m), 13.72 (1 H,s).
(e) 4-Nitro-3-n-propyl-1H-pyrazole-5-carboxamide
A solution of the title compound of Example (c) (15.4 g, 0.077 mol) in thionyl chloride (75 ml) was heated under reflux for 3 hours and then the cool reaction mixture evaporated under reduced pressure. The residue was azeotroped with tetrahydrofuran (2×50 ml) and subsequently suspended in tetrahydrofuran (50 ml), then the stirred suspension ice-cooled and treated with gaseous ammonia for 1 hour. Water (50 ml) was added and the resulting mixture evaporated under reduced pressure to give a solid which, after trituration with water and drying by suction, furnished the title compound (14.3 g).
m.p. 197-199° C. Found: C, 42.35; H, 5.07; N, 28.38. C7H10N4O3 requires C, 42.42; H, 5.09; N, 28.27%. δ (DMSOd6): 0.90 (3H,t), 1.68 (2H,m), 2.86 (2H,t), 7.68 (1 H,s), 8.00 (1 H,s).
(f) 3-Ethyl-4-nitro-1H-pyrazole-5-carboxamide BA from AA via route 3

Obtained from the title compound of Example (d), by analogy with Example (e), as a white solid (90%). δ (DMSOd6): 1.17 (3H,t), 2.87 (2H,m), 7.40 (1H,s), 7.60 (1H,s), 7.90 (1H,s). LRMS: m/z 185 (M+l)+.
(g)(i) 5-Ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide CA from BA via route 4

A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (2.5 kg, 13.6 Mol), sodium carbonate (1.8 Kg, 17.0 Mol) and 2-bromoethyl methyl ether (1.98 kg, 14.2 Mol) in THF (22.5 L) and water (2.5 L) was heated under reflux and stirred for 20 hours. The mixture was cooled to ambient temperature and CH2Cl2 (67.5 L) and water (22.5 L) were added. The resultant organic and aqueous layers were separated. The aqueous phase was extracted with CH2Cl2 (22.5 L) and the combined organic solution was distilled under atmospheric pressure and replaced with ethyl acetate (33 L) to a final volume of 17 L. The cooled mixture was granulated at ambient temperature for 2 hours, filtered and washed with ethyl acetate (2.5 L). This afforded 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a white crystalline solid, 2.10 kg, 57%. m.p.=140° C. Found: C, 44.46; H, 5.79; N, 23.01. C9H14N4O4 requires C, 44.63; H, 5.79; N, 23.14%.
δ (CDCl3): 1.18 (3H, t), 2.98 (2H, q), 3.22 (3H, s), 3.77 (2H, t), 4.28 (2H, q), 6.03 (1H, s), 7.36 (1H, s).
LRMS: m/z=243 (M+1)+
(g)(ii) 5-Ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide.
A mixture of 3-ethyl-4-nitro-1H-pyrazole-5-carboxamide (25 g, 0.136 Mol), sodium carbonate (18 g, 0.17 Mol) and sodium iodide (20.4 g, 0.136 Mol) were suspended in ethyl methyl ketone (125 mL) at room temperature. 2-bromoethyl methyl ether (12.8 mL, 0.142 Mol) was added and the mixture was heated to reflux and stirred for 70 hours. The mixture was cooled to ambient temperature and water (250 mL) was added. The resultant slurry was warmed to reflux and held at that temperature for 30 min before cooling to room temperature. The resultant precipitate was granulated at room temperature for 3 h, filtered and vacuum dried to afford 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide as a yellow crystalline solid 24.3 g, 74%. Data as reported for Example (g)(i).
(h) 4-Amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide (IA) from CA via route 5

A mixture of 5-ethyl-1-(2-methoxyethyl)-4-nitro-1H-pyrazole-3-carboxamide (20 g, 82.6 mMol) and 5% Pd/C (1 g) in methanol (200 mL) was pressurised at 50psi/25° C. in a sealed vessel and stirred for 15 hours. At the end of the reaction the mixture was filtered through arbocel and the filter cake was washed with methanol. The methanolic solution was distilled at atmospheric pressure and replaced with ethyl acetate to a final volume of 100 mL. The cooled mixture was granulated at ambient temperature for 2 h filtered and washed with ethyl acetate (20 mL) to afford 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide as a white crystalline solid, 15 g, 88%. m.p.=131° C. Found: C, 50.75; H, 7.62; N, 26.38. C9H16N4O2 requires C, 50.94; H, 7.55; N, 26.42%. δ (CDCl3): 1.20 (3H, t), 2.63 (2H, q), 3.32 (3H, s), 3.74 (2H, t), 3.95 (2H, s), 4.15 (2H, t), 5.27 (1H, s), 6.59 (1H, s).
LRMS: m/z=213 (M+1)+
(i) N-[3-Carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl]-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide.

2-ethoxy-5-(4-ethyl-1-piperazinylsulfonyl)nicotinic acid (2.31 kg, 6.73 Mol) was suspended in ethyl acetate (16.2 L) and 1,1-carbonyldimidazole (1.09 kg, 6.73 Mol) was added at room temperature. The reaction mixture was heated at 45° C. for 40 minutes and then the reaction was stirred for a further 40 minutes at reflux. After cooling to ambient temperature 4-amino-5-ethyl-1-(2-methoxyethyl)-1H-pyrazole-3-carboxamide (1.5 kg, 7.06 Mol) was added to the cooled mixture, and the reaction stirred for a further 15 hours under reflux. The mixture was cooled filtered and the filter cake was washed with 90% water/10% ethyl acetate, (2 mL /g) to afford N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide as an off white crystalline solid, 3.16 kg, 88%. m.p.=156° C. Found: C, 51.33; H, 6.56; N, 18.36. C23H35N7O6S requires C, 51.40; H, 6.53; N, 18.25%.
δ (CDCl3): 1.04 (3H, t), 1.22 (3H, t), 1.60 (3H, t), 2.44 (2H, q), 2.54 (4H, m), 2.96 (2H, q), 3.12 (4H, m), 3.36 (3H, s), 3.81 (2H, t), 4.27 (2H, t), 4.80(2H, q), 5.35(1H, s), 6.68 (1H, s), 8.66 (1H, d), 8.86 (1H, d), 10.51 (1H, s).
LRMS: m/z=539 (M+1)+
(i) 1-(6-Ethoxy-5-[3-ethyll-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine•ethyl acetate solvate.

GISADENAFIL
A mixture of N-[3-carbamoyl-5-ethyl-1-(2-methoxyethyl)-1H-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1-piperazinyl sulfonyl) nicotinamide (1.18 kg, 2.2 Mol), potassium tert-butoxide (500 g, 4.4 moles) and ethyl acetate (193 g) in ethanol (11.8 L) was heated at 120° C. for 20 hours. The reaction mixture was then concentrated under reduced pressure, in total approx. 10 L of solvent were distilled. To the residue water (2.9 L) was added and the mixture stirred at room temperature while aqueous HCl was added until pH 7.5 was obtained. Ethyl acetate (7.5 L) was added and the two phase mixture was warmed to 55° C. The organic phase was separated and the aqueous phase was extracted with further ethyl acetate (3.0 L). The combined organic phases were distilled at atmospheric pressure to a final volume of 4 L. The precipitated solids were granulated at 5° C. for 1 h, filtered and washed with ethyl acetate (1.2 L) and dried under vacuum. This afforded 1-(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazole[4,3-d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine as a light yellow crystalline solid, 877 g, 78%. m.p.=157° C. Found: C, 52.65; H, 6.46; N, 17.76. C23H33N705S. 0.2 C2H5CO2CH3 requires C, 53.21; H, 6.49; N, 18.25%.
δ (CDCl3): 1.07 (3H, t), 1.42 (3H, t), 1.61 (3H, t), 2.44 (2H, q), 2.57 (4H, m), 3.08 (2H, q), 3.15 (4H, m), 3.32 (3H, s), 3.92 (2H, q), 4.48 (2H, q), 4.77 (2H, q), 8.65 (1H, d), 9.06 (1H, d). The spectrum also has signals that correspond to a solvate with ethyl acetate.
LRMS: m/z=520 (M+1)+
……………..
Example 102
1-(6-Ethoxy-5-f3-ethyll-6,7-dihvdro-2-(2-methoxyethvn-7-oxo-2r7-pyrazoler4.3- cf1pyrimidin-5-vn-3-pyridylsulfonyl)-4-ethylpiperazine»ethyl acetate solvate.

To prepare the compound of Example 8 a mixture of Λ/-[3-carbamoyl-5-ethyl- 1 -(2-methoxyethyl)-1 /-/-pyrazol-4-yl}-2-ethoxy-5-(4-ethyl-1 -piperazinyl sulfonyl) nicotinamide (1.18 kg, 2.2 Mol), potassium tert-butoxide (500 g, 4.4 moles) and ethyl acetate (193 g) in ethanol (11.8 L) was heated at 120°C for 20 hours. The reaction mixture was then concentrated under reduced pressure, in total approx. 10 L of solvent were distilled. To the residue water (2.9 L) was added and the mixture stirred at room temperature while aqueous HCl was added until pH 7.5 was obtained. Ethyl acetate (7.5 L) was added and the two phase mixture was warmed to 55°C. The organic phase was separated and the aqueous phase was extracted with further ethyl acetate (3.0 L). The combined organic phases were distilled at atmospheric pressure to a final volume of 4L. The precipitated solids were granulated at 5°C for 1 h, filtered and washed with ethyl acetate (1.2 L) and dried under vacuum. This afforded 1 -(6-Ethoxy-5-[3-ethyl]-6,7-dihydro-2-(2-methoxyethyl)-7-oxo- 2H-pyrazole[4,3-o pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine as a light yellow crystalline solid, 877 g, 78%. m.p. = 157°C. Found: C, 52.65; H, 6.46; N, 17.76. C23H33N705S. 0.2 C2H5C02CH3 requires C, 53.21 ; H, 6.49; N, 18.25%.
δ(CDCI3): 1.07 (3H, t), 1.42 (3H, t), 1.61 (3H, t), 2.44 (2H, q), 2.57 (4H, m), 3.08 (2H, q), 3.15 (4H, m), 3.32 (3H, s), 3.92 (2H, q), 4.48 (2H, q), 4.77 (2H, q), 8.65 (1 H, d), 9.06 (1 H, d). The spectrum also has signals that correspond to a solvate with ethyl acetate.
LRMS: m/z = 520 (M+1)+
Example 103
1-(6-ethoxy-5-r3-ethyl-6.7-dihvdro-2-(2-methoxyethvn-7-oxo-2H-pyrazolor4.3- dlpyrimidin-5-vn-3-pyridylsulfonyl)-4-ethylpiperazine

GISADENAFIL
10g (0.019 mol) of the compound of Example 8 and Example 102, 1-{6- ethoxy-5-[3-ethyl-6,7-dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3- d]pyrimidin-5-yl]-3-pyridylsulfonyl}-4-ethylpiperazine ethyl acetate solvate, was charged followed by 12ml/g (120mls) of 16% water in ethyl alcohol. The slurry was heated to reflux to yield a solution and 6ml/g (60mls) distilled off at atmospheric pressure. The solution was then cooled to room temperature with crystallisation occurring at 40°C. The slurry was then cooled to 5-10°C and granulated for 30 minutes following which it was filtered and washed with 2ml/g ethyl alcohol (20 mis). The damp solid was dried in vacuo overnight at 55-60 °C to yield a white crystalline solid. (Yield 7.6g, 76%). Melting Point 162- 165°C.
δ (CDCI3): 1.05 (3H,t), 1.42 (3H,t), 1.58 (3H,t), 2.43 (2H,q), 2.57 (4H,t), 3.09 (2H, t), 3.15 (4H,t), 3.30 (3H,s), 3.93 (2H,t), 4.48 (2H,t), 4.90 (2H,q), 8.65 (1 H,d), 9.05 (1 H,d), 10.65 (1 H,s).
In the process of Example 103, water and pharmaceutically acceptable alcohols such as methanol, ethanol, propanol, butanol and mixtures thereof can be used to prepare the compound of Examples 8 and 102.
BESYLATE SALT
Example 104 1-(6-ethoxy-5-r3-ethyl-6,7-dihvdro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolor4.3- d]pyrimidin-5-yl]-3-pyridylsulfonyl)-4-ethylpiperazine benzene-sulfonate salt.

170g (0.33 mol) of the compound of Example 103, 1-{6-ethoxy-5-[3-ethyl-6,7- dihydro-2-(2-methoxyethyl)-7-oxo-2H-pyrazolo[4,3- d]pyrimidin-5-yl]-3- pyridylsulfonyl}-4-ethylpiperazine, was charged followed by a water/ 2- butanone (4% v/v) at 10 ml/g (1.7 litres) and warmed to reflux. 53g (0.33 mol) of benzene sulfonic acid dissolved in water (23mls, resulting in 70 % w/w solution) was added to the refluxing solution over 30 minutes.5.3ml/g (0.9 litres) of 2-butanone were striped and replaced and the slurry cooled. The slurry was cooled to 5-10°C and granulated for 2 hours after which it was filtered and washed with 2ml/g (0.3 litres) of 2-butanone. The salt was dried overnight in vacuo at 55-60°C to yield a white crystalline solid. Yield 215g, 96.4%. Mpt 242-244°C. δ (DMSO): 1.17 (3H, t), 1.28 (3H, t), 1.35 (3H, t), 2.73 (2H, q), 2.97 (2H, q), 3.2 (3H, s), 3.58 (2H, t), 3.78 (3H, t), 3.81 (2H, t), 4.49 (2H, t) 4.51 (2H, q), 7.29-7.33 (3H, m), 7.57-7.60 (2H, m), 8.28 (1 H, d), 8.73 (1 H, d), 9.13 (1 H,s), 11.90(1 H,s).
The powder X-ray diffraction (PXRD) pattern for this salt, having Mpt 242- 244°C, was determined using a Siemens D5000 powder X-ray diffractometer fitted with a theta-theta goniometer, automatic beam divergence slits, a secondary monochromator and a scintillation counter. The specimen was rotated whilst being irradiated with copper K-alpha1 X-rays (Wavelength = 1.5046 Angstroms) filtered with a graphite monochromator (λ = 0.15405nm) with the X-ray tube operated at 40 kV/mA. The main peaks (in degrees θ) of the PXRD pattern are illustrated in Table I.
Table


The same besylate salt, as defined by the XRD pattern described in Table 1 , when made via alternative routes can have a melting point in the range of from 235-246°C (measured using a Perkin Elmer DSC7 at a heating rate of 20°C/minute).

References
1 The discovery of UK-369003, a novel PDE5 inhibitor with the potential for oral bioavailability and dose-proportional pharmacokinetics
Bioorg Med Chem 2012, 20(1): 498………….MP 161 – 162 °C
2. Hajikarimian, Y.; Yeo, S.; Ryan, R.W.; Levett, P.; Stoneley, C.; Singh, P.
Investigation into the formation of the genotoxic impurity ethyl besylate in the final step manufacturing process of UK-369,003-26, a novel PDE5 inhibitor
Org Process Res Dev 2010, 14(4): 1027
3. Bentham; Dawson; Dunn; Papadopoulos; Taylor; Mitchell; Snowden; Taylor
Organic Process Research and Development, 2004 , vol. 8, 4 PG. 674 – 679 ………….AS ENTRY B
- Bloch, W., et al.: Prostate, 33, 1 (1997)
- • Glowienke, S., et al.: Mutat. Res., 581, 23 (1997)
- • Chapple, C., et al.: Eur. Urol., 54, 563 (1997)
- • Elder, D., et al.: J. Pharm. Pharmacol., 61, 269 (1997)
PATENTS
1. WO 2010062366
2. WO 2007072156
3 WO 2007072156
4.US2002/22732 A1,
5.US2002/28799 A1,
6.
| WO1998049166A1 * | Apr 10, 1998 | Nov 5, 1998 | Mark Edward Bunnage | PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3′,5′-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION |
| WO1999054333A1 * | Mar 25, 1999 | Oct 28, 1999 | Mark Edward Bunnage | Pyrazolopyrimidinone cgmp pde5 inhibitors for the treatment of sexual dysfunction |
| US4666921 * | 15 окт 1985 | 19 май 1987 | Ludwig Heumann & Co. Gmbh | Pyrazole derivatives, processes for their preparation and pharmaceutical preparations containing these compounds |
| US5808092 * | 15 окт 1997 | 15 сен 1998 | Ube Industries, Ltd. | Process for preparing-1-ethyl-5-hydroxypyrazole |
| US6015911 * | 24 мар 1998 | 18 янв 2000 | Dow Agrosciences Llc | Process for preparing 1-alkyl-4-(2-chloro-3-alkoxy-4-alkylsulfonylbenzoyl)-5-hydroxypyrazole and related compounds |
| EP0463756A1 | 7 июн 1991 | 2 янв 1992 | Pfizer Limited | Pyrazolopyrimidinone antianginal agents |
| EP0812845A1 | 4 июн 1997 | 17 дек 1997 | Pfizer Limited | Process for preparing sildenafil |
| EP0994115A2 | 11 окт 1999 | 19 апр 2000 | Pfizer Limited | Process for preparation of pyrazolo-(4,3-d)pyrimidin-7-ones and intermediates thereof |
| EP0995750A1 | 15 окт 1999 | 26 апр 2000 | Pfizer Inc. | Pyrazolopyrimidinone cGMP PDE5 inhibitors for the treatment of sexual dysfunction |
| WO1998049166A1 | 10 апр 1998 | 5 ноя 1998 | Mark Edward Bunnage | PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3′,5′-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION |
| WO1999054333A1 | 25 мар 1999 | 28 окт 1999 | Mark Edward Bunnage | Pyrazolopyrimidinone cgmp pde5 inhibitors for the treatment of sexual dysfunction |
| WO2001027112A1 | 4 окт 2000 | 19 апр 2001 | Charlotte Moira Norfo Allerton | 5-(2-substituted-5-heterocyclylsulphonylpyrid-3-yl)-dihydropyrazolo[4,3-d]pyrimidin-7-ones as phosphodiesterase inhibitors |
| WO2001027113A2 | 11 окт 2000 | 19 апр 2001 | Mark Edward Bunnage | PYRAZOLO `4,3-d! PYRIMIDINE DERIVATIVES |
PDE5 inhibitors mirodenafil

sildenafil

tadalafil

udenafil 3-(l-methyl-7-oxo-3-propyl-4H-pyrazolo[5,4-e]pyrimidin-5-yl)-N- [2-(l -methylpyrrolidin-2-yl)ethyl] -4-propoxybenzenesulfonamide

vardenafil 4-[2-ethoxy-5-(4-ethylpiperazin-l-yl)sulfonyl-phenyl]-9-methyl-7- propyl- 3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one

avanafil 4-[(3-chloro-4-methoxy-phenyl)methylamino]-2-[(2S)-2- (hydroxymethyl)pyrrolidin- 1 -yl] -N-(pyrimidin-2- ylmethyl)pyrimidine-5-carboxamide

dasantafil 7-[(3-bromo-4-methoxyphenyl)methyl]-l-ethyl-8-[[(lR,2R)-2- hydroxycyclopentyl]amino]-3-(2-hydroxyethyl)purine-2,6-dione
NM 702 (Nissan Chemical Industries)

SLX 101 (Surface Logix) – Structure Not Available
UK 369003 (Pfizer) – Gisadenafil besylate


334827-98-4 (as besylate)
- UK 369003
- UK-369,003
- UK0369,003
- UNII-S6G4R7DI1C
symptoms associated with BPH

 1…………..
1…………..












7…………………



1.07 (3H, t), METHYL OF -N CH2-CH3 ON PIPERAZINE RING
1.42 (3H, t), METHYL OF -CH2-CH3 ON PYRAZOLE SIDE CHAIN
1.61 (3H, t), METHYL OF -O-CH2-CH3 ON PYRIMIDINE RING
2.44 (2H, q), CH2 OF -N CH2-CH3 ON PIPERAZINE RING
2.57 (4H, m),4H OF –NCH2 ON PIPERAZINE RING BOTH SIDE OF N ATOM
3.08 (2H, q), CH2 OF –CH2-CH3 ON PYRAZOLE SIDE CHAIN
3.15 (4H, m),4H OF –NCH2 ON PIPERAZINE RING BOTH SIDE OF N ATOM CLOSE TO SO2 GP
3.32 (3H, s),METHYL OF -OCH3 ON PYRAZOLE SIDE CHAIN
3.92 (2H, q), CH2 OF NCH2-CH2-O-CH3 ON PYRAZOLE SIDECHAIN
4.48 (2H, q), CH2 OF NCH2 –CH2-O-CH3 ON PYRAZOLE SIDECHAIN
4.77 (2H, q), CH2 OF O-CH2 CH3 ON PYRIMIDINE RING
8.65 (1H, d), PYRIMIDINE AROM H …..AWAY/PARA TO C=O-NH -PYRAZOLE GP
9.06 (1H, d). PYRIMIDINE AROM H …..CLOSER/ORTHO TO C=O-NH -PYRAZOLE GP, reason this signal will shift from 8,86 delta to 9.06 after cyclization in this step ie formation of GISADENAFIL
The spectrum also has signals that correspond to a solvate with ethyl acetate.
Orphan Drugs: Global Regulatory Events
This Blog Post discusses recent global non-US regulatory events for orphan drugs.
I – Europe
At a January meeting, the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) gives the following opinions for three orphan drugs :
• Positive recommendation for Bayer’s Adempas (Riociguat) for the treatment of Chronic Thromboembolic Pulmonary Hypertension (CTEPH) and Pulmonary Arterial Hypertension (PAH)
• Negative opinion for AB Science’s Masiviera (Masitinib) which is intended for the treatment of non resectable locally advanced or metastatic pancreatic cancer
• Negative opinion for PTC Therapeutics’ Translarna (Ataluren) which is intended for the treatment of Duchenne Muscular Dystrophy.
Also, EMA’s Committee for Orphan Medicinal Products (COMP) issues 15 positive opinions for ODD at their January meeting (Reference Blog Post). These ODDs are to be presented to the European Commission (EC) for final approval. If the EC approves these ODDs, the drugs receive ODD in the…
View original post 230 more words
UDENAFIL …The Eastern Viagra (like)

UDENAFIL
An oral phosphodiesterase 5 inhibitor used for the treatment of erectile dysfunction.
268203-93-6 CAS NO
LAUNCHED 2005 MEZZION DA-8159 ME-3113 Udzire Zydena MEZZION …INNOVATOR
POWERPOINT PRESENTATION BY INNOVATOR.. CLICK HERE
| Synonyms: | Zydena;Udenafi;Da-8159;Da 8159;Udenafil;Udenafil(DA 8159,Zydena);5-(2-Propyloxy-5-(1-methyl-2-pyrollidinylethylamidosulfonyl)phenyl)-1-methyl-3-propyl-1,6-dihydro-7H-pyrazolo(4,3-D)pyrimidine-7-one;5-[2-propyloxy-5-[2-(1-Methyl-2-pyrrolidinyl)ethylaMinosulfonyl]phenyl]-1-Methyl-3-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyriMidine-7-one;5-[2-propyloxy-5-(2-(1-Methylpyrrolidin-2-yl)ethylaMinosulphonyl)phenyl]-1-Methyl-3-propyl-6,7-dihydro-1H-pyrazolo(4,3-d)pyriMidin-7-one;3-(6,7-Dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methyl-2-pyrrolidinyl)ethyl]-4-propoxybenzenesulfonamide |
| Molecular Formula: | C25H36N6O3S2 |
| Formula Weight: | 516.66 |
3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide
(5- [2-propyloxy-5- (1- methyl-2-pyrolidinylethylamidosulfonyl) phenyl] -1-methyl- propyl-1, β-dihydro-7H-pyrazolo (4 , 3-d) pyrimidin-7-one)
A pyrazolo-pyrimidinone similar to sildenafil; phosphodiesterase type 5 inhibitor. Udenafil is a new phosphodiesterase type 5 (PDE5) inhibitor used to treat erectile dysfunction (ED). It has been approved in South Korea and will be marketed under the brand name Zydena.
It is not yet approved for use in the U.S., E.U., or Canada. Udenafil (Zydena®) is also a potent and selective PDE5i developed by Dong-A Pharmaceutical Company in Korea (Kim et al., 2008; Han et al., 2010).
It has not yet been approved by FDA or the European Medicines Agency (EMEA) and was only approved by the Korean Food and Drug Administration (KFDA), being currently used in Korea and Russia (Alwaal et al., 2011; Cho et al., 2012).
- DA 8159
- DA-8159
- Udenafil
- UNII-L5IB4XLY36
- Zydena
Udenafil is a drug used in urology to treat erectile dysfunction. It belongs to a class of drugs called PDE5 inhibitor, which many other erectile dysfunction drugs such as sildenafil, tadalafil, and vardenafil also belong to. It was developed by Dong-A Pharmaceutical Co., Ltd. and is marketed under the trade name Zydena™.[2] With a T max of 1.0-1.5 h and a T 1/2 of 11-13 h (a relatively rapid onset and a long duration of action), both on-demand and once-daily use of udenafil have been reported.[3] Typical doses are 100 and 200 mg. It is not approved for use in the United States by theU.S. Food and Drug Administration. Udenafil (DA-8159), a pyrazolopyramidinone derivative that acts as a phosphodiesterase 5 (PDE5) inhibitor, was launched by Dong-A Pharmtech (currently Mezzion Pharma) in late 2005 in Korea for the oral treatment of erectile dysfunction (ED). The company is currently conducting phase III clinical trials in the U.S. for this indication.
Dong-A Pharmatech is conducting phase III clinical trials for the treatment of patients with portal hypertension resulting from liver disease and for the treatment of benign prostatic hyperplasia (BPH). Phase II/III clinical studies at Dong-A Pharmatech for the treatment of secondary Raynaud phenomenon have been completed. Meiji Seika Pharma is developing the compound in phase I clinical trials for the treatment of BPH in Japan.
Phosphodiesterases regulate the tissue concentration of cyclic guanosine monophosphate (cGMP), which in turn triggers smooth muscle relaxation, allowing blood to flow into the penis and resulting in erection. PDE5 is the most abundant phosphodiesterase in the human corpus cavernosum, and as such its inhibition by DA-8159 enhances erectile function by increasing the concentration of cGMP. Results from phase I studies indicate that udenafil has a unique pharmacokinetic profile with a relatively rapid onset and sufficiently long duration to make it effective for up to 24 hours. In 2009, the compound was licensed to Warner Chilcott (acquired by Actavis in 2013) by Dong-A Pharmatech for development and marketing in the U.S. for the oral treatment of erectile dysfunction.
In 2011, udenafil was licensed to Meiji Seika Pharma by Dong-A ST in Japan for the treatment of benign prostatic hyperplasia. Udenafil is a potent novel phosphodiesterase-5 inhibitor approved for use in Korea. Udenafil has unique properties, with a T max of 1.0–1.5 h and a T 1/2 of 11–13 h (a relatively rapid onset and a long duration of action). Therefore, both on-demand and once-daily use of udenafil have been reported. Udenafil’s efficacy and tolerability have been evaluated in several studies, and recent and continuing studies have demonstrated udenafil’s promise in both dosing regimens. Presently, tadalafil is the only FDA-approved drug for daily dosing, but udenafil can be used as a once-daily dose for erectile dysfunction patients who cannot tolerate tadalafil due to phosphodiesterase subtype selectivity. Udenafil as an on-demand or once-daily dose is effective and tolerable, but more studies are needed in patients of other ethnicities and with comorbid conditions such as diabetes mellitus, hypertension, and benign prostate hyperplasia.
Erectile dysfunction (ED) is defined as the inability to achieve and maintain a sufficient erection to permit satisfactory intercourse [Montorsi et al. 2010]. Numerous strategies have been used to overcome ED. Therapies for ED include intracavernosal injection, vacuum erection devices, intraurethral suppositories, penile prosthesis surgery and oral phosphodiesterase-5 (PDE5) inhibitors [Dinsmore and Evans, 1999]. Oral PDE5-inhibitor medications have revolutionized the treatment of ED. Men prefer oral medications as the first-line therapeutic option in the absence of a specific contraindication to their use [Ding et al. 2012].
There are currently four PDE5 inhibitors (sildenafil, vardenafil, tadalafil, and avanafil) approved worldwide for the treatment of male erectile dysfunction, with two other agents (udenafil and mirodenafil) currently approved only in Korea [Bell and Palmer, 2011]. The choice of PDE5 inhibitor for each patient should be determined after physician and patient discuss the characteristics of different drugs and the individual patient’s sexual habits, preferences, and expectations [Hatzimouratidis et al. 2010]. There are two types of treatment usage of PDE5 inhibitors according to their pharmacological characteristics. On-demand treatment of ED with PDE5 inhibitors allows the patient to have intercourse within 1 hour, but can remove spontaneity from sexual activity and be burdensome to patients and their partners [Hanson-Divers et al. 1998]. Once-daily dosing of a PDE5 inhibitor is an alternative for couples that prefer spontaneous sexual activities.
A new oral selective PDE5 inhibitor, udenafil (Zydena, Dong-A, Seoul, Korea), has recently been developed for the treatment of ED. Udenafil is a novel pyrazolopyrimidinone compound developed by Dong-A Pharmaceutical Co., Ltd (Seoul, Korea) for the treatment of ED which has the same mechanism of action as sildenafil [Kim et al. 2008]. Udenafil is rapidly absorbed, reaching peak plasma concentrations at 0.8–1.3 h, then declining monoexponentially with a terminal half-life (T 1/2) between 7.3 and 12.1 hours, giving it the unique pharmacokinetics of both relatively rapid onset and long duration [Salem et al. 2006]. Thus, both on-demand treatment and once-daily dosing have been reported in the literature. The purpose of this review is to evaluate the efficacy and tolerability of udenafil for patients with ED according to the currently available literature.
Udenafil” refers to the chemical compound, 3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide and has the following chemical formula:

More information about udenafil can be found at Kouvelas D. et al., (2009) Curr Pharm Des, 15(30):3464-75. Udenafil is marketed under the trade name Zydena® but not approved for use in the United States. TRADE NAME IN INDIA – UDEZIRE Erectile dysfunction (ED) is an inability to achieve or sustain an erection suitablefor sexual intercourse.
Sexologists say at least 10% men in India may have to use these drugs at some point. Studies have shown that 40% of men up to the age of 40 years have ED andit goesup 70% by 60 years. The commonly prescribed drugs for the disorder in India are sildenafil(Viagra) and tadalafil,which belong to a category called phosphodiesterasetype5 drugs.
Now, Zydus, a pharmaceutical company, has got exclusive permission to sell udenafil. It’s not always that the release of a drug is celebrated by many, particularly men. A drug that was released in India last week is the recent in the list of drugs that has a cure for erectile dysfunction. The manufacturers say udenafil, which will be marketed under the brand name Udezire, will be long-acting, but with minimal side effects. Erectile dysfunction (ED) is an inability to achieve or sustain an erection suitable for sexual intercourse. Sexologists say at least 10% men in India may have to use these drugs at some point. Studies have shown that 40% of men up to the age of 40 years have ED and it goes up 70% by 60 years

UDENAFIL
……………………
INTRODUCTION
Udenafil (Zydena®) is a therapeutic agent hypothesized to improve erectile function endpoints through interaction with the phosphodiesterase type 5 (PDE5) enzyme. As such, udenafil belongs to the class of such agents that includes tadalafil (Clalis®), sildenafil (Viagra®), and vardenafil (Levitra®). These agents are purported to promote erectile response through inhibition of PDE5, the predominant PDE within the penis, which leads to higher intracellular levels of cyclic guanylate cyclase (cGMP). cGMP is a second messenger for the smooth-muscle relaxing effects of nitric oxide within the penis. The various agents differ in pharmacology primarily based on 1) onset and duration of action and 2) selectivity profiles vs. other PDEs. All three marketed agents have proven remarkably safe. These agents should not be taken by patients with unstable cardiovascular disease. Udenafil has been shown to exhibit greater selectivity against the known PDE homologues, than other PDE5 inhibitors. Udenafil is comparable to tadalafil in many respects, such as duration of action and high selectivity for PDE6, but udenafil has greater selectivity for PDE11 than tadalafil.
Tadalafil, with a half life of 17.5 hours, has a much longer duration of action and improved exercise tolerance than either sildenfail or vardenafil, which have half lives of 4-5 hours. Consequently, tadalafil is associated with less planning or pressure to have sexual intercourse after dosing. Dissociation of the sexual activity from the time of dosing is associated with higher rates of patient and partner satisfaction. In prospective, randomized crossover clinical studies, patients preferred tadalafil over sildenafil by margins ranging from 7:3 to 9:1. Sildenafil and vardenafil both modulate PDE6 at higher rate than tadalafil. PDE6 modulation has been associated with chromatopsia. The side effects of chromatopsia, such as sensitivity to light and blurred vision, are therefore higher in patients taking sildenafil or vardenafil, about 2-3%, than patients taking tadalafil, about <0.1%. Tadalafil is less selective than sildenafil and vardenafil for PDE5 and for PDE11a. Activity at PDE11a is suspected to have a causal relationship with myalgia and testicular toxicity. The selectivity profile for udenafil is similar to sildenafil, which should impart greater safety for this agent.

The benefits and shortcomings of these drugs have been reviewed. Some of these shortcomings can be traced to metabolism-related phenomena. Udenafil is converted in vivo by oxidative and conjugative degradation to multiple metabolites. Phase I metabolism leads to demethylation of the pyrazole, hydroxylation of the pyrazole propyl group, and dealkylation alpha to the sulfonamide nitrogen to afford an active metabolite. Because udenafil is metabolized primarily by cytochrome P450 subtype 3A4 (CYP3A4), exposure to udenafil can influence polypharmacy. For example, CYP3A4 inhibitors such as HIV protease inhibitors, azole antifungals, and erythromycin can lead to higher than otherwise expected blood levels of udenafil. Conversely, co-administration of CYP3A4 inducers such as rifampin can decrease the otherwise expected blood levels of udenafil. Thus, the polypharmacy of udenafil is necessarily complex and has potential for adverse events. In addition, there may be increased inter-patient variability in response to polypharmacy.
Analogs of udenafil as described herein have the potential to alleviate the problems associated with the commercially available PDE5 inhibitors while maintaining or improving efficacy. It is believed that the reduction in CYP3A4 clearance of udenafil analogs will be expected to increase the proportion of clearance via mechanisms less susceptible to polypharmaceutical complications. In addition, analogs of udenafil having an attenuated rate of oxidative metabolism will have an increased half-life, further augmenting their advantages vs. tadalafil, sildenafil and vardenafil. Potentially, a single dose of an udalafil analog, described herein, having an increased half-life may provide therapeutic coverage for an entire weekend or beyond while increasing safety parameters by reducing the likelihood of drug-drug interactions and by increasing safety as a result of the increased selectivity.

The compounds of formula 1 may contain asymmetric centers and thus they can exist as enantiomers. The present invention includes both mixtures and separate individual isomers . Male erectile dysfunction is one of the most common sexual dysfunctions in men. Although erectile dysfunction can be primarily psychogenic in origin, it often accompanies chronic illnesses, such as diabetes mellitus, heart disease, hypertension, and a variety of neurological diseases. Its prevalence is strongly related to age, with a estimated prevalence of 2% at age 40 years rising to 25-30% by age of 65. Although no data are available on the prevalence of erectile dysfunction in men aged over 75, it is probably over 50%. Various treatment options for erectile dysfunction are available, such as counseling, hormonal therapy, self-injection or transurethral application of vasodilator agents, vacuum devices, prosthesis implantation, and venous/arterial surgery. However, these therapeutic options have several limitations such as side effects, high-cost and low efficacy.
Therefore it has called for research efforts to develop new, high effective and simple to use treatment methods, potentially oral medication. Recently, sildenafil has been developed as a therapeutic agent for male erectile dysfunction by oral administration. Sildenafil is the first in a new class of drugs known as inhibiting phosphodiesterase-5 enzyme distributed specifically in corpus cavernosal tissues and induces relaxation of the corpus cavernosal smooth muscle cells, so that blood flow to the penis is enhanced, leading to an erection.
Sildenafil has shown a response rate of around 80% in men with erectile dysfunction of organic cause. On the other hand, USP 3,939,161 discloses that 1 , 3 -dimethyl -lH-pyrazolopyrimidinone derivatives exhibit anticonvulsant and sedative activiity, and also exhibit anti-inflammatory activity and gastric antisecretory activity; EP 201,188 discloses that 5-substituted pyrazolopyrimidinone derivatives have effects of antagonizing adenosine receptor and of inhibiting phosphodiesterase enzymes and can be used for the treatment of cardiovascular disorders such as heart failure or cardiac insufficiency; EP 463,756, EP 526,004, WO 93/6,104 and WO 93/7,149 disclose that pyrazolopyrimidinone derivatives which inhibit c-GMP phosphodiesterase more selectively than c-AMP phosphodiesterase have efficacy on cardiovascular disorders such as angina pectoris, hypertension, heart failure, atherosclerosis, chronic asthma, etc.; and WO 94/28,902, WO 96/16,644, WO 94/16,657 and WO 98/49,166 disclose that the known inhibitors of c-GMP phosphodiesterase including the pyrazolopyrimidinone derivatives of the above mentioned patents can be used for the treatment of male erectile dysfunction Since sildenafil has been developed, various compounds for inhibiting phosphodiesterase-5 have been reported.
Among them, pyrazolopyrimidinone compounds of formula 1 (KR Pat. No. 99-49384) were reported having better potency than that of sildenafil, based on the mechanism of inhibiting phosphodiesterase-5 and having better selectivity over phosphodiesterase-6 distributed in retina and phosphodiesterase-3 distributed in heart to reduce the side effects. Further, the pyrazolopyrimidinone compounds of formula 1 were said to be improved the solubility and the metabolism in the liver, which are very important factor affecting the rate of the absorption when administered orally.
The KR patent No. 99-49384 also disclosed a process for preparing the pyrazolopyrimidinone compounds of formula , comprising the steps of: a) reacting chlorosulfonated alkoxy bonzoic acid with a primary amine to obtain sulfonamide-substituted benzoic acid; b) reacting the obtained sulfonamide-substituted benzoic acid with pyrazolamine in the presence of activating reagent of carboxylic group or coupling agent of carboxylic group with amine group to obtain corresponding amide compound; and, c) performing an intramolecular cyclization of the obtained amide compound to obtain the pyrazolopyrimidinone compound of formula 1. This reaction is represented in scheme 1 Scheme 1

…………………..
SYNTHESIS
The present invention provides an agent comprising a pyrazolopyrimidinone compound (5- [2-propyloxy-5- (1- methyl-2-pyrolidinylethylamidosulfonyl) phenyl] -1-methyl- propyl-1, 6-dihydro-7H-pyrazolo (4, 3-d) pyrimidin-7-one) expressed as formula 1 as an effective ingredient for preventing and treating benign prostatic hyperplasia (BPH) . Formula 1

The pyrazolopyrimidinone compound represented as formula 1 is one of the PDE-5 inhibitors and has characteristics in that it has a strong inhibitive activity and an excellent selectivity for PDE-5; it is readily absorbed as its solubility is improved; it has a good bioavailability and a large volume of distribution; and it has an in vivo half-life longer three times than sildenafil or vardenafil, a drug of the same mechanism. Physicochemical properties of the pyrazolopyrimidinone compound of formula 1 are as follows: it is hardly dissolved in water; however, it is readily dissolved in acetic acid, methanol, chloroform and the like; and it is a white or pale yellow powder, not a hydrate or a solvate, having a melting point of 158 to 161 “Q and having pKal and pKa2 of about 6.5 and 12.5, respectively. The pyrazolopyrimidinone compound represented as formula 1 is prepared via a synthetic process consisting of roughly three steps. The inventors of the present invention have disclosed a method for preparing the same in WO2000/027847 (Corresponding Korean Patent No.0353014), which will now be described roughly as follows. First, in the first step, 4- [2-propyloxy-5- (chlorosulfonyl) benzamido] -l-methyl-3-propyl-5-carbamoyl pyrazole is prepared.
For such preparation, a specified amount of 4- [2-propyloxybenzamido] -l-methyl-3-propyl-5- carbamoyl pyrazole is added to a specified amount of chlorosulfonic acid cooled to 0 °Q then, the resultant mixture is stirred, filtered, washed and dried to obtain 4- [2-propyloxy-5- (chlorosulfonyl) benzamido] -l-methyl-3- propyl-5-carbomoyl pyrazole. In the second step, from the pyrazole compound prepared in the first step, 4- [2-propyloxy-5- ( l-methyl-2- pyrolidinylethylamidosulfonyl) benzamido] -l-methyl-3- propyl-5-carbomoyl pyrazole is prepared. For such preparation, a specified amount of 2- (2-aminoethyl) -1- methyl pyrolidine is added in dichloromethane solution of the specified amount of 4- [2-propyloxy-5- (chlorosulfonyl) benzamido] -l-methyl-3-propyl-5-carbamoyl pyrazole prepared in the first step to be stirred. Then, the reactant solution is diluted with dichloromethane. The organic layer is washed, dried, concentrated and filtered to obtain 4- [2-propyloxy-5- (l-methyl-2- pyrolidinylethylamidosulfonyl) benzamido] -l-methyl-3- propyl-5-carbomoyl pyrazole is obtained.
Last, in the third step, the pyrazolopyrimidinone compound of the present invention (5- [2-propyloxy-5- (1- methyl-2-pyrolidinylethylamidosulfonyl) phenyl] -1-methyl- propyl-1, β-dihydro-7H-pyrazolo (4 , 3-d) pyrimidin-7-one) is prepared from the compound obtained in the second step. For such preparation, the specified amount of pyrazole compound prepared in the second step is dissolved in t- butanol . A specified amount of potassium t-butoxide is added in the resultant solution and, then, reflux-stirred for a predetermined time. After the resultant solution is cooled, diluted, washed and dried, distillation under reduced pressure, solvolysis and silica gel column chromatography are carried out, thus obtaining a specified amount of pure pyrazolopyrimidinone compound of the present invention
. …………………………..
SYNTHESIS WO2000027848A1
REACTION SCHEME 2

The process for preparation according to the present invention comprises the steps of : 1) reacting the chlorosulfonated compound of formula ( 2 ) and primary amine (3_) under the condition of suitable temperature and suitable solvent to give sulfonamide (4.) (step 1) ; 2) reacting the carboxylic acid (4.) prepared in step 1 and pyrazoleamine (5) to give an amide (6.) by the known method preparing amide from carboxylic acid and amine (step 2) ; and 3) cyclizing the amide (6.) prepared in step 2 to give the desired compound of formula 1 by the known cyclization method used for preparation of pyrimidinone (step 3) .
In step 1, a little excess of 2 equivalents of amine may be used, or a little excess of 1 equivalent of amine and 1 equivalent of acid scavenger such as tertiary amine are may be used together. The reaction temperature is preferred below 20 °C. The known method preparing amide from carboxylic acid and amine in step 2 is the process, for example, in which carboxyl group is transformed into activated acid chloride or acid anhydride by using thionyl chloride, pivaloyl chloride, trichlorobenzoyl chloride, carbonyldiimidazole, diphenylphosphinic chloride, etc. and followed by reacting with amine group, or the process using coupling agents such as DCC (1,3-dicyclo hexylcarbodiimide) or EEDQ (N-ethoxycarbonyl -2 -ethoxy- 1, 3-dihydroquinoline) .
The cyclization process in step 3 may be carried out in the presence of a suitable base and a suitable solvent. Preferred bases which are employed in step 3 are metal alkoxides; metal salts of ammonia; amine; hydrides of alkali metal or alkaline earth metal; hydroxides; carbonates; bicarbonates ; and bicyclic amidines such as DBU (1 , 8-diazabicyclo [5.4.0] undec -7-ene) and DBΝ (1 , 5-diazabicyclo [4.3.0] non-5-ene) . Preferred solvents which are employed in step 3 are alcohols such as methanol, ethanol, isopropanol, t-butanol, etc.; ethers such as tetrahydrofuran, dimethoxyethane, dioxane, etc.; aromatic – hydrocarbons such as benzene, toluene, xylene, chlorobenzene, etc.; acetonitrile; dimethylsulfoxide; dimethylformamide; N-methylpyrrolidin-2 -one ; and pyridine.
SEE ENTRY no 68
5- [2-propyloxy-5- ( 1-methyl-2-pyrrolidinylethyl amidosulfonyl) phenyl] -l-methyl-3 -propyl-1 , 6-dihydro-7 H-pyrazolo (4 , 3-d) yrimidin-7-one (compound of example68) 
ACCORDING TO ME ENTRY IS 68 ANY ERROR, amcrasto@gmail.com
Synthesis WO2001098304A1
The present invention relates to a process for preparing pyrazolopyrimidinone derivatives of formula 1 and pharmaceutically acceptable salts thereof which have an efficacy on impotence, comprising the steps of chlorosulfonation of pyrazolamide compounds of formula 2, followed by amination with a primary amine and intramolecular cyclization. Formula 1

Formula 2

The compounds of formula 1 may exist in tautomeric equilibrium as shown below.

The compounds of formula 1 may also contain asymmetric centers and thus they can exist as enantiomers. The present invention includes both racemic mixture and separate individual enantiomers. Scheme 2

……………………………….
SYNTHESIS WO2010013925A2
INTERMEDIATES
4-[2-propyloxy benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole
CHLOROSULPHONIC ACID
4-[2-propyloxy-5-(chlorosulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole
2-(2-aminoethyl)-l-methylpyrrolidine 4-[2-propyloxy-5-(l-methyl-2-pyrrolidinylethyl amido- sulfonyl)benzamido]-l-methyl-3-propyl-5-carbamoyl pyrazole
potassium t-butoxide
3, 5-[2-propyloxy-5-(l-methyl-2-pyrrolidinylethyl amido- sulfonyl)phenyl]-l-methyl-3-propyl-l,6-dihydro-7H-pyrazolo(4,3-d)pyrimidin-7-one UDENAFIL
The present invention provides a pharmacological compound containing 5- [2-propyloxy-5-( 1 -methyl-2-pyrolidinylethylamidosulphonyl)phenyl] – 1 -methyl-prop yl- 1 ,6-dihydro-7H-pyrazolo(4,3-d)pyrimidin-7-one, a pyrazolopyrimidinone compound, represented by the following Chemical Formula 1 or pharmaceutically acceptable salts thereof, as an active ingredient for prevention and treatment of respiratory diseases. [14] [Chemical Formula 1]

………………
SYNTHESIS
EXAMPLE 2 3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methyl-pyrrolidin-2-yl)-ethyl]-4-propoxy-benzenesulfonamide

Step 1

2,4-Dioxo-heptanoic acid methyl ester: Sodium (25.3 g, 1.1 mol) was proportionally added to ethanol (350 mL) at ambient temperature with vigorous stirring, and the solution was cooled to 0° C. Pentan-2-one (86 g, 1.0 mol) and diethyl oxalate (146 g, 1.0 mol) were added sequentially at 0° C., and stirring was continued for 1 hour at 0° C., and overnight at ambient temperature. The solvent was removed under reduced pressure, diethyl ether (200 mL) and cold dilute hydrochloric acid (500 mL) were added. Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound (141 g, 76%). 1H-NMR (300 MHz, CDCl3) δ 14.51 (broad s, 1H), 6.37 (s, 1H), 4.35 (q, 2H, J=6.6 Hz), 2.47 (t, 2H, J=7.2 Hz), 1.76-1.66 (m, 2H), 1.38 (t, 3H, J=7.2 Hz), 0.97 (t, 3H, J=7.5 Hz); GC-MS: 186 (M)+, 113 (M-73)+
Step 2

5-Propyl-2H-pyrazole-3-carboxylic acid ethyl ester: Hydrazine hydrate (41.4 g, 827 mmol) was slowly added to a solution of 2,4-dioxo-heptanoic acid methyl ester (140 g, 753 mmol) in 280 mL of acetic acid at 0° C. The mixture was heated to reflux for 8 hours and cooled. The solvent was removed under reduced pressure; the residue was diluted with diethyl ether (300 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound as a white solid (131 g, 96%). 1H NMR (300 MHz, CDCl3) δ 9.27 (broad s, 1H), 6.61 (s, 1H), 4.37 (q, 2H, J=7.2 Hz), 2.68 (t, 2H, J=7.5 Hz), 1.75-1.62 (m, 2H), 1.37 (t, 3H, J=6.6 Hz), 0.96 (t, 3H, J=7.2 Hz); LC-MS: m/z=183 (MH)+;
Step 3

2-Methyl-5-propyl-2H-pyrazole-3-carboxylic acid ethyl ester: A mixture of 5-propyl-2H-pyrazole-3-carboxylic acid ethyl ester (32.8 g, 180 mmol) and dimethyl sulfate (24.9 g, 198 mmol) was heated at 90° C. for 3 hours. The reaction was cooled and diluted with dichloromethane (200 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield a crude residue which was purified by flash chromatography on silica gel to give the title compound as a colorless oil (23 g, 65%). 1H NMR (300 MHz, CDCl3) δ 6.59 (s, 1H), 4.37 (q, 2H, J=7.2 Hz), 2.58 (t, 2H, J=7.2 Hz), 1.76-1.64 (m, 2H), 1.40 (t, 3H, J=6.6 Hz), 1.01 (t, 3H, J=7.2 Hz), 4.40 (q, 2H), 3.89 (s, 3H), 2.59 (t, 2H), 1.69 (2H), 1.37 (t, 3H), 1.01 (t, 3H); LC-MS: m/z=197 (MH)+.
Step 4

2-Methyl-5-propyl-2H-pyrazole-3-carboxylic acid: 2-methyl-5-propyl-2H-pyrazole-3-carboxylic acid ethyl ester (29.4 g, 150 mmol) was suspended in 6N sodium hydroxide (120 mL, 720 mmol) and heated to 80° C. for 2 hours, cooled, diluted with water (100 mL) and acidified with 5N hydrochloric acid (200 mL) to give a precipitate which was filtered off and dried to give the title compound as a white solid (24.2 g, 96%). 1H NMR (300 MHz, CDCl3) δ 6.76 (s, 1H), 4.17 (s, 3H), 2.63 (t, 2H, J=7.2 Hz), 1.70-1.68 (m, 2H), 0.98 (t, 3H, J=7.2 Hz); LC-MS: m/z=169 (M+H)+;
Step 5

2-Methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxylic acid: A solution of 2-methyl-5-propyl-2H-pyrazole-3-carboxylic acid (22 g, 131 mmol) in concentrated sulfuric acid (98%, 85 mL) was heated to 50° C. and treated with a mixture of fuming nitric acid (95%, 7.7 mL) and concentrated sulfuric acid (98%, 18 mL), while keeping the reaction temperature between 50 and 55° C. The reaction mixture was kept for 8 hours at 50° C., cooled to ambient temperature, and slowly added to cold water (600 mL, 4° C.), keeping the temperature below 25° C. The precipitate was collected by filtration, and dried below 80° C. to give the title compound as a white solid (25 g, 90%). 1H NMR (300 MHz, CDCl3) δ 4.25 (s, 3H), 2.92 (t, 2H, J=7.5 Hz), 1.77-1.70 (m, 2H), 1.03 (t, 3H, J=7.2 Hz); LC-MS: m/z=214 (M+H)+
Step 6

2-Methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxamide: To a suspension of 2-methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxylic acid (17.0 g, 79.8 mmol) in dry toluene (85 mL) was added a catalytic quantity of dimethylformamide (0.6 mL). The mixture was heated to 50° C. and thionyl chloride (17.1 g, 143.7 mmol) was added over 30 minutes. The reaction was stirred and heated at 55-60° C. for 6 hours. The solvent was removed, dry toluene (80 mL) was added and the mixture was cooled to 20° C. and cold (5° C.) concentrated ammonium hydroxide (100 mL) was added. The precipitate was filtered, washed with water and dried to give the title compound as an off-white solid (14.8 g, 87%). LC-MS: m/z=213 (M+H)+, 235 (M+Na)+.
Step 7

4-Amino-2-methyl-5-propyl-2H-pyrazole-3-carboxamide: To a suspension of 2-methyl-4-nitro-5-propyl-2H-pyrazole-3-carboxamide (14.7 g, 69.3 mmol) in ethyl acetate (130 mL), was added 10% palladium on carbon (3.3 g). The mixture was reacted at 50° C. and 4 atm hydrogen pressure overnight. The reaction mixture was cooled, and the catalyst was filtered off and washed with ethyl acetate and dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure to give the title compound (13.8 g, 98%). 1H NMR (300 MHz, CDCl3) δ 4.12 (s, 3H), 2.84 (s, 2H), 2.55 (t, 2H, J=7.2 Hz), 1.71-1.61 (m, 2H), 0.99 (t, 3H, J=7.2 Hz); LC-MS: m/z=183 (MH)+
Step 8

2-Methyl-4-(2-propoxybenzoylamino)-5-propyl-2H-pyrazole-3-carboxamide: A solution of 2-propoxybenzoic acid (13.7 g, 76.1 mmol) and thionyl chloride (36.2 g, 304.4 mmol) in dry dichloromethane (80 mL) was heated for 3 hours at reflux. The solvent and excess thionyl chloride were distilled off under reduced pressure. The residue was taken up in dry dichloromethane (60 mL) and reacted with a solution of 4-amino-2-methyl-5-propyl-2H-pyrazole-3-carboxamide (12.6 g, 69.2 mmol), dry triethylamine (7 g, 69.2 mmol) and 4-(N,N-dimethylamino)pyridine (84.5 mg, 0.7 mmol) in dry dichloromethane (200 mL) at 0° C. Stirring was maintained for 1 hour, and the reaction mixture was successively washed with water (150 mL), saturated aqueous sodium carbonate solution (200 mL) and saturated brine (200 mL). The organic layer was dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated to about 60 mL, and then hexane (150 mL) was added to give precipitate product as a white solid (22 g, 92%). 1H NMR (300 MHz, CDCl3) δ 9.47 (s, 1H), 8.28 (d, 1H, J=7.8 Hz), 7.87 (br.s, 1H), 7.57-7.52 (m, 1H), 7.16-7.05 (m, 2H), 5.53 (s, 1H), 4.20 (t, 2H, J=6.6 Hz), 4.09 (s, 3H), 2.54 (t, 2H, J=7.5 Hz), 1.97-1.85 (m, 2H), 1.69-1.26 (m, 2H), 1.07 (t, 3H, J=7.2 Hz), 0.95 (t, 3H, J=7.5 Hz). LC-MS: m/z=345 (M+H)+
Step 9

3-(5-Carbamoyl-1-methyl-3-propyl-1H-pyrazol-4-ylcarbamoyl)-4-propxy-benzenesulfonyl chloride: 2-Methyl-4-(2-propoxybenzoylamino)-5-propyl-2H-pyrazole-3-carboxamide (20 g, 58.1 mmol) was added to chlorosulfonic acid (81.3 g, 698 mmol) at 0° C. and the reaction was warmed to ambient temperature and stirred for 2 hours. The reaction mixture was poured into ice water (800 g) and mechanically stirred for 1 hour to give a white solid, which was filtered and washed with water. Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound (8 g, 31%). 1H NMR (300 MHz, CDCl3) δ 9.19 (s, 1H), 8.97 (s, 1H), 8.19 (t, 1H, J=8.9 Hz), 7.56 (br. s, 1H), 4.35 (t, 2H, J=6.6 Hz), 4.07 (s, 3H), 2.53 (t, 2H, J=7.5 Hz), 2.06-1.94 (m, 2H), 1.78-1.60 (m, 2H), 1.18 (t, 3H, J=7.5 Hz), 0.95 (t, 3H, J=7.2 Hz); LC-MS: m/z=443.1 (M+H)+
Step 10

2-Methyl-4-{5-[2-(1-methyl-pyrrolidin-2-yl)-ethylsulfamoyl]-2-propoxy-benzoylamino}-5-propyl-2H-pyrazole-3-carboxamide: To a solution of 3-(5-carbamoyl-1-methyl-3-propyl-1H-pyrazol-4-ylcarbamoyl)-4-propoxy-benzenesulfonyl chloride (2.12 g, 4.8 mmol) and dry triethylamine (0.5 g, 4.8 mmol) in dichloromethane (20 mL), was added 2-(2-aminoethyl)-1-methylpyrrolidine (0.6 g, 4.8 mmol) at 0° C. The reaction was warmed to ambient temperature, stirred for 1 hour at ambient temperature, and diluted with dichloromethane (40 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield the title compound (2.2 g) which was used directly in the next step. LC-MS: m/z=535 (M+H)+
Step 11

3-(1-Methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methyl-pyrrolidin-2-yl)-ethyl]-4-propoxy-benzenesulfonamide: Potassium tert-butoxide (0.9 g, 8.0 mmol) was added to a solution of crude 2-methyl-4-{5-[2-(1-methyl-pyrrolidin-2-yl)-ethylsulfamoyl]-2-propoxy-benzoylamino}-5-propyl-2H-pyrazole-3-carboxamide (2.14 g, 4.0 mmol) in dry tert-butanol (50 mL), and the mixture was heated to reflux for 8 hours. The reaction mixture was cooled to ambient temperature and diluted with ethyl acetate (300 mL). Following standard extractive work up, the solvent was evaporated under reduced pressure to yield a crude residue which was purified by flash chromatography to give the title compound (1.1 g, 53%).
1H NMR (300 MHz, CDCl3) δ 10.90 (broad s, 1H), 8.93 (s, 1H), 7.96 (d, 1H, J=8.7 Hz), 7.15 (d, 1H, J=8.7 Hz), 4.28-4.24 (m, 3H), 4.24 (s, 2H), 3.13 (t, 3H, J=6.9 Hz), 2.93 (t, 3H, J=7.8 Hz), 2.56 (s, 1H), 2.40 (s, 3H), 2.26-2.24 (m, 1H), 2.10-1.99 (m, 2H), 1.89-1.80 (m, 4H), 1.67 (s, 3H, J=7.2 Hz), 1.56-1.52 (m, 1H), 1.22 (t, 3H, J=7.5 Hz), 1.03 (t, 3H, J=7.2 Hz);
LC-MS: m/z=517 (MH)+
…………………….
References
- Udenafil Information
- Zydena (udenafil) product information page. Dong-A Pharmaceutical. Retrieved on April 13, 2009.
- Udenafil: efficacy and tolerability in the management of erectile dysfunction.
- http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3607490/
- British Journal of Pharmacology, 2008 , vol. 153, 7 PG. 1568 – 1578
- Arzneimittel-Forschung/Drug Research, 2009 , vol. 59, 12 pg. 641 – 646
- Chemical and Pharmaceutical Bulletin, 2011 , vol. 59, 9 PG. 1083 – 1088
- WO2010/13925 A2, …
- US2010/173915 A1
- WO2010/95849 A2,
- WO2007/114534 A1, …..
- Life Sciences, 2004 , vol. 75, 9 pg. 1075 – 1083 …………..mp 162 – 164 °C
- US2008/194529 A1,
-
WO2008100886A1 * Feb 12, 2008 Aug 21, 2008 Auspex Pharmaceuticals Inc Preparation and use of deuterated udenafil analogues as highly selective pde5 modulators for the treatment of erectile dysfunction -
US6333330 * Oct 22, 1999 Dec 25, 2001 Pfizer Inc. Pyrazolopyrimidinone CGMP PDE5 inhibitors for the treatment of sexual dysfunction US20040029891 * Sep 2, 2003 Feb 12, 2004 Pfizer Inc. Use of PDE5 inhibitors in the treatment of polycystic ovary syndrome -
WO1993006104A1 * Sep 4, 1992 Apr 1, 1993 Pfizer Pyrazolopyrimidinone antianginal agents WO1994028902A1 * May 13, 1994 Dec 22, 1994 Peter Ellis Pyrazolopyrimidinones for the treatment of impotence WO1996016657A1 * Oct 16, 1995 Jun 6, 1996 Simon Fraser Campbell Bicyclic heterocyclic compounds for the treatment of impotence WO1998049166A1 * Apr 10, 1998 Nov 5, 1998 Mark Edward Bunnage PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3′,5′-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION EP0463756A1 * Jun 7, 1991 Jan 2, 1992 Pfizer Limited Pyrazolopyrimidinone antianginal agents -
WO1993006104A1 * Sep 4, 1992 Apr 1, 1993 Pfizer Pyrazolopyrimidinone antianginal agents WO1998049166A1 * Apr 10, 1998 Nov 5, 1998 Mark Edward Bunnage PYRAZOLOPYRIMIDINONES WHICH INHIBIT TYPE 5 CYCLIC GUANOSINE 3′,5′-MONOPHOSPHATE PHOSPHODIESTERASE (cGMP PDE5) FOR THE TREATMENT OF SEXUAL DYSFUNCTION WO2000027848A1 * Nov 10, 1999 May 18, 2000 Byoung Ok Ahn Pyrazolopyrimidinone derivatives for the treatment of impotence EP0463756A1 * Jun 7, 1991 Jan 2, 1992 Pfizer Limited Pyrazolopyrimidinone antianginal agents -
WO2004108726A1 May 14, 2004 Dec 16, 2004 Tianjin North Pharma Sci Tech 2-SUBSTITUTED PHENYL-5,7-DIALKYL-3,7-DIHYDROPYRROLE [2,3-d] PYRIMIDINE-4-ONE DERIVATIVES, THE PREPARATION AND THE PHARMACEUTICAL USE THEREOF US7741483 Mar 6, 2008 Jun 22, 2010 Yangtze River Pharmaceutical (Group) Co., Ltd. Process for making substituted pyrrolo[2,3-d]pyrimidine derivatives as inhibitors of phosphodiesterase 5

Dolutegravir approved by the EU Commission (synthesis included in this post)

Dolutegravir
2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide
(4R,12aS)-N-(2,4-difluorobenzyl)-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Trade Name:Tivicay
Synonym:GSK1349572, S-349572, GSK572
Date of Approval: August 12, 2013 (US)
Indication:HIV infection
Drug class: Integrase strand transfer inhibitor
Company: ViiV Healthcare,GlaxoSmithKline
INNOVATOR …ViiV Healthcare
CAS number: 1051375-16-6
MF:C20H19F2N3O5
MW:419.4
Chemical Name: (4R,12aS)-N-[(2,4-difluorophenyl)methyl]-7-hydroxy-4-methyl-6,8-dioxo-3,4,6,8,12,12a- hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide
Patent: US8129385
Patent expiration date: Oct 5, 2027
PCT patent application: W02006116764
Dolutegravir (DTG, GSK1349572) is an integrase inhibitor being developed for the treatment of human immunodeficiency virus (HIV)-1 infection by GlaxoSmithKline (GSK) on behalf of Shionogi-ViiV Healthcare LLC. DTG is metabolized primarily by uridine diphosphate glucuronyltransferase (UGT)1A1, with a minor role of cytochrome P450 (CYP)3A, and with renal elimination of unchanged drug being extremely low (< 1% of the dose).
The European Commission has on 21 January 2014 Dolutegravir (Tivicay, ViiV) permit as part of combination therapy for the treatment of HIV-infected persons over the age of 12 years.Dolutegravir (Tivicay, ViiV) is an integrase inhibitor, in combination with other antiretroviral drugs in adults and adolescents can be used from 12 years for the treatment of HIV infection.
Source: Communication from the European Commission
Dolutegravir[1] is a FDA-approved drug[2] for the treatment of HIV infection. Dolutegravir is an integrase inhibitor. Known as S/GSK1349572 or just “572” the drug is marketed as Tivicay[3] by GlaxoSmithKline (GSK). In February, 2013 the Food and Drug Administration announced that it would fast track dolutegravir’s approval process.[4] On August 13, 2013, dolutegravir was approved by the FDA. On November 4, 2013, dolutegravir was approved by Health Canada.[5]
The oral HIV integrase inhibitor S-349572 was originated by Shionogi-GlaxoSmithKline and Shionogi-ViiV Healthcare. In 2013, the product was approved and launched in the U.S. for the treatment of HIV-1 in adults and children aged 12 years and older, in combination with other antiretroviral agents. A positive opinion was received in the E.U for this indication and, in 2014, approval was attained in Europe for this indication. Registration is pending in Japan.
In 2013, orphan drug designation in Japan was assigned to the compound.
Dolutegravir is approved for use in a broad population of HIV-infected patients. It can be used to treat HIV-infected adults who have never taken HIV therapy (treatment-naïve) and HIV-infected adults who have previously taken HIV therapy (treatment-experienced), including those who have been treated with other integrase strand transfer inhibitors. Tivicay is also approved for children ages 12 years and older weighing at least 40 kilograms (kg) who are treatment-naïve or treatment-experienced but have not previously taken other integrase strand transfer inhibitors.[6]
Dolutegravir has also been compared head-to-head with a preferred regimen from the DHHS guidelines in each of the three classes (i.e. 1.) nuc + non-nuc, 2.) nuc + boosted PI, and 3.) nuc + integrase inhibitor).
SPRING-2 compared dolutegravir to another integrase inhibitor, raltegravir, with both coformulated with a choice of TDF/FTC orABC/3TC. After 48 weeks of treatment 88% of those on dolutegravir had less than 50 copies of HIV per mL compared to 85% in the raltegravir group, thus demonstrating non-inferiority.[9]
The FLAMINGO study has been presented at scientific meetings but as of early 2014 has not yet been published. It is an open-label trial of dolutegravir versus darunavir boosted with ritonavir. In this trial 90% of those on dolutegravir based regimens had viral loads < 50 at 48 weeks compared to 83% in the darunavir/r.[10] This 7% difference was statistically significant for superiority of the dolutegravir based regimens.
Another trial comparing dolutegravir to efavirenz, SINGLE, was the first trial to show statistical superiority to an efavirenz/FTC/TDF coformulated regimen for treatment naive patients.[11] After 48 weeks of treatment, 88% of the dolutegravir group had HIV RNA levels < 50 copies / mL versus 81% of the efavirenz group. This has led one commentator to predict that it may replace efavirenz as the first line choice for initial therapy as it can also be formulated in one pill, once-a-day regimens.[12]
Doultegravir has also been studied in patients who have been on previous antiretroviral medications. The VIKING trial looked at patients who had known resistance to the first generation integrase inhibitor raltegravir. After 24 weeks 41% of patients on 50mg dolutegravir once daily and 75% of patients on 50mg twice daily (both along with an optimized background regimen) achieved an HIV RNA viral load of < 50 copies per mL. This demonstrated that there was little clinical cross-resistance between the two integrase inhibitors. [13]
Dolutegravir (also known as S/GSK1349572), a second-generation integrase inhibitor under development by GlaxoSmithKline and its Japanese partner Shionogi for the treatment of HIV infection, was given priority review status from the US Food and Drug Administration (FDA) in February, 2013.
GlaxoSmithKline marketed the first HIV drug Retrovir in 1987 before losing out to Gilead Sciences Inc. (GILD) as the world’s biggest maker of AIDS medicines. The virus became resistant to Retrovir when given on its own, leading to the development of therapeutic cocktails.
The new once-daily drug Dolutegravir, which belongs to a novel class known as integrase inhibitors that block the virus causing AIDS from entering cells, is owned by ViiV Healthcare, a joint venture focused on HIV in which GSK is the largest shareholder.
Raltegravir (brand name Isentress) received approval by the U.S. Food and Drug Administration (FDA) on 12 October 2007, the first of a new class of HIV drugs, the integrase inhibitors, to receive such approval. it is a potent and well tolerated antiviral agent. However, it has the limitations of twice-daily dosing and a relatively modest genetic barrier to the development of resistance, prompting the search for agents with once-daily dosing.
Elvitegravir, approved by the FDA on August 27, 2012 as part of theelvitegravir/cobicistat/tenofovir disoproxil fumarate/emtricitabine fixed-dose combination pill (Quad pill, brand name Stribild) has the benefit of being part of a one-pill, once-daily regimen, but suffers from extensive cross-resistance with raltegravir.
Gilead’s Atripla (Emtricitabine/Tenofovir/efavirenz), approved in 2006 with loss of patent protection in 20121, is the top-selling HIV treatment. The $3.2 billion medicine combines three drugs in one pill, two compounds that make up Gilead’s Truvada (Emtricitabine/Tenofovir) and Bristol- Myers Squibb Co.’s Sustiva (Efavirenz).
A three-drug combination containing dolutegravir and ViiV’s older two-in-one treatment Epzicom(Abacavir/Lamivudine, marketed outside US as Kivexa) proved better than Gilead’s market-leading Atripla in a clinical trial released in July, 2012 (See the Full Conference Report Here), suggesting it may supplant the world’s best-selling AIDS medicine as the preferred front-line therapy. In the latest Phase III study, after 48 weeks of treatment, 88% of patients taking the dolutegravir-based regimen had reduced viral levels to the goal compared with 81% of patients taking Atripla. More patients taking Atripla dropped out of the study because of adverse events compared with those taking dolutegravir — 10% versus just 2% — which was the main driver of the difference in efficacy. The result was the second positive final-stage clinical read-out for dolutegravir, following encouraging results against U.S. company Merck & Co’s rival Isentress in April, 2012 (See the Conference Abstract Here)..
Dolutegravir is viewed by analysts as a potential multibillion-dollar-a-year seller, as its once-daily dosing is likely to be attractive to patients. The FDA is scheduled to issue a decision on the drug’s approval by August 17。
TIVICAY contains dolutegravir, as dolutegravir sodium, an HIV INSTI. The chemical name of dolutegravir sodium is sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazin-7-olate. The empirical formula is C20H18F2N3NaO5 and the molecular weight is 441.36 g/mol. It has the following structural formula:
 |
Dolutegravir sodium is a white to light yellow powder and is slightly soluble in water.
Each film-coated tablet of TIVICAY for oral administration contains 52.6 mg of dolutegravir sodium, which is equivalent to 50 mg dolutegravir free acid, and the following inactive ingredients: D-mannitol, microcrystalline cellulose, povidone K29/32, sodium starch glycolate, and sodium stearyl fumarate. The tablet film-coating contains the inactive ingredients iron oxide yellow, macrogol/PEG, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
……………………………………
INTRODUCTION
Among viruses, human immunodeficiency virus (HIV), a kind of retrovirus, is known to cause acquired immunodeficiency syndrome (AIDS). The therapeutic agent for AIDS is mainly selected from a group of reverse transcriptase inhibitors (e.g., AZT, 3TC) and protease inhibitors (e.g., Indinavir), but they are proved to be accompanied by side effects such as nephropathy and the emergence of resistant viruses. Thus, the development of anti-HIV agents having the other mechanism of action has been desired.
On the other hand, a combination therapy is reported to be efficient in treatment for AIDS because of the frequent emergence of the resistant mutant. Reverse transcriptase inhibitors and protease inhibitors are clinically used as an anti-HIV agent, however agents having the same mechanism of action often exhibit cross-resistance or only an additional activity. Therefore, anti-HIV agents having the other mechanism of action are desired.
Under the circumstances above, an HIV integrase inhibitor has been focused on as an anti-HIV agent having a novel mechanism of action (Ref: Patent Documents 1 and 2). As an anti-HIV agent having such a mechanism of action, known are carbamoyl-substituted hydroxypyrimidinone derivative (Ref: Patent Documents 3 and 4) and carbamoyl-substituted hydroxypyrrolidione derivative (Ref: Patent Document 5). Further, a patent application concerning carbamoyl-substituted hydroxypyridone derivative has been filed (Ref: Patent Document 6, Example 8).
Other known carbamoylpyridone derivatives include 5-alkoxypyridine-3-carboxamide derivatives and γ-pyrone-3-carboxamide derivatives, which are a plant growth inhibitor or herbicide (Ref: Patent Documents 7-9).
Other HIV integrase inhibitors include N-containing condensed cyclic compounds (Ref: Patent Document 10).
- [Patent Document 1] WO03/0166275
- [Patent Document 2] WO2004/024693
- [Patent Document 3] WO03/035076
- [Patent Document 4] WO03/035076
- [Patent Document 5] WO2004/004657
- [Patent Document 6] JP Patent Application 2003-32772
- [Patent Document 7] JP Patent Publication 1990-108668
- [Patent Document 8] JP Patent Publication 1990-108683
- [Patent Document 9] JP Patent Publication 1990-96506
- [Patent Document 10] WO2005/016927
-
Patent Document 1 describes compounds (I) and (II), which are useful as anti-HIV drugs and shown by formulae:

-
This document describes the following reaction formula as a method of producing compound (I).


-
Furthermore, Patent Documents 2 to 6 describe the following reaction formula as an improved method of producing compound (I).


-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
- [PATENT DOCUMENTS]
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-



…………………………………………
Dolutegravir synthesis (EP2602260, 2013). LiHMDS as the non-nucleophilic strong base pulling compound 1 carbonyl group proton alpha position with an acid chloride after 2 and ring closure reaction to obtain 3 , 3 via primary amine 4 ring opening ring closure to obtain 5 , NBS the bromine under acidic conditions to obtain aldehyde acetal becomes 6 , 6 of the aldehyde and amino alcohols 7 and turn off the condensation reaction obtained by the ring 8 , alkaline hydrolysis 8 of bromine into a hydroxyl group and hydrolyzable ester obtained 9 after the 10 occurred acid condensation Dolutegravir.
………………………………………………………
Synthesis of Dolutegravir (S/GSK1349572, GSK1349572)

………………………
SYNTHESIS

2H-Pyrido[1′,2′:4,5]pyrazino[2,1-b][1,3]oxazine-9-carboxamide, N-[(2,4-difluorophenyl)methyl]-3,4,6,8,12,12a-hexahydro-7-hydroxy-4-methyl-6,8-dioxo-, (4R,12aS) ………..dolutegravir
PATENT US8129385

Desired isomer
Example Z-1
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

a)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide. To a solution of 16a (409 mg, 0.87 mmol) in dichloroethane (20 mL) was added (2R)-2-amino-1-propanol (0.14 mL, 1.74 mmol) and 10 drops of glacial acetic acid. The resultant solution was heated at reflux for 2 h. Upon cooling, Celite was added to the mixture and the solvents removed in vacuo and the material was purified via silica gel chromatography (2% CH3OH/CH2Cl2 gradient elution) to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 92%) as a glass. 1H NMR (CDCl3) δ 10.38 (m, 1H), 8.42 (s, 1H), 7.54-7.53 (m, 2H), 7.37-7.24 (m, 4H), 6.83-6.76 (m, 2H), 5.40 (d, J=10.0 Hz, 1H), 5.22 (d, J=10.0 Hz, 1H), 5.16 (dd, J=9.6, 6.0 Hz, 1H), 4.62 (m, 2H), 4.41 (m, 1H), 4.33-4.30 (m, 2H), 3.84 (dd, J=12.0, 10.0 Hz, 1H), 3.63 (dd, J=8.4, 7.2 Hz, 1H), 1.37 (d, J=6.0 Hz, 3H); ES+MS: 496 (M+1).
b)
(3R,11aS)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt. To a solution of (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (396 mg, 0.80 mmol) in methanol (30 mL) was added 10% Pd/C (25 mg). Hydrogen was bubbled through the reaction mixture via a balloon for 2 h. The resultant mixture was filtered through Celite with methanol and dichloromethane.
The filtrate was concentrated in vacuo to give (3R,11aS)—N-[(2,4-difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide , DOLUTEGRAVIR as a pink tinted white solid (278 mg, 86%).
1H NMR (CDCl3) δ 11.47 (m, 1H), 10.29 (m, 1H), 8.32 (s, 1H), 7.36 (m, 1H), 6.82 (m, 2H), 5.31 (dd, J=9.6, 3.6 Hz, 1H), 4.65 (m, 2H), 4.47-4.38 (m, 3H), 3.93 (dd, J=12.0, 10.0 Hz, 1H), 3.75 (m, 1H), 1.49 (d, J=5.6 Hz, 3H); ES+ MS: 406 (M+1).
DOLUTEGRAVIR NA SALT
The above material (278 mg, 0.66 mmol) was taken up in ethanol (10 mL) and treated with 1 N sodium hydroxide (aq) (0.66 ml, 0.66 mmol). The resulting suspension was stirred at room temperature for 30 min. Ether was added and the liquids were collected to provide the sodium salt of the title compound as a white powder (291 mg, 99%). 1H NMR (DMSO-d6) δ 10.68 (m, 1H), 7.90 (s, 1H), 7.35 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.20 (m, 1H), 4.58 (m, 1H), 4.49 (m, 2H), 4.22 (m, 2H), 3.74 (dd, J=11.2, 10.4 Hz, 1H), 3.58 (m, 1H), 1.25 (d, J=4.4 Hz, 3H).
UNDESIRED ISOMER
Example Z-9
(3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide sodium salt

The title compound was made in two steps using a similar process to that described in example Z-1. 16a (510 mg, 1.08 mmol) and (25)-2-amino-1-propanol (0.17 mL, 2.17 mmol) were reacted in 1,2-dichloroethane (20 mL) with acetic acid to give (3S,11aR)—N-[(2,4-difluorophenyl)methyl]-3-methyl-5,7-dioxo-6-[(phenylmethyl)oxy]-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (500 mg, 93%). This material was hydrogenated in a second step as described in example Z-1 to give (3S,11aR)—N-[(2,4-Difluorophenyl)methyl]-6-hydroxy-3-methyl-5,7-dioxo-2,3,5,7,11,11a-hexahydro[1,3]oxazolo[3,2-a]pyrido[1,2-d]pyrazine-8-carboxamide (386 mg, 94%) as a tinted white solid. 1H NMR (CDCl3) δ 11.46 (m, 1H), 10.28 (m, 1H), 8.32 (s, 1H), 7.35 (m, 1H), 6.80 (m, 2H), 5.30 (dd, J=10.0, 4.0 Hz, 1H), 4.63 (m, 2H), 4.48-4.37 (m, 3H), 3.91 (dd, J=12.0, 10.0 Hz, 1H), 3.73 (m, 1H), 1.48 (d, J=6.0 Hz, 3H); ES+ MS: 406 (M+1). This material (385 mg, 0.95 mmol) was treated with sodium hydroxide (0.95 mL, 1.0 M, 0.95 mmol) in ethanol (15 mL) as described in example Z-1 to provide its corresponding sodium salt (381 mg, 94%) as a white solid. 1H NMR (DMSO-d6) δ 10.66 (m, 1H), 7.93 (s, 1H), 7.33 (m, 1H), 7.20 (m, 1H), 7.01 (m, 1H), 5.19 (m, 1H), 4.59 (m, 1H), 4.48 (m, 2H), 4.22 (m, 2H), 3.75 (m, 1 H), 3.57 (m, 1H), 1.24 (d, J=5.6 Hz, 3H).
SYNTHESIS OF INTERMEDIATES

IN ABOVE SCHEME SYNTHESIS UPTO COMPD 9 MAY BE USEFUL IN SYNTHESIS BUT READERS DISCRETION IS SOUGHT IN THIS ?????????????????
1) Maltol 1 (189 g, 1.5 mol) was dissolved in dimethylformamide (1890 ml), and benzyl bromide (184 ml, 1.5 mol) was added. After the solution was stirred at 80° C. for 15 minutes, potassium carbonate (228 g, 1.65 mol) was added, and the mixture was stirred for 1 hour. After the reaction solution was cooled to room temperature, an inorganic salt was filtered, and the filtrate was distilled off under reduced pressure. To the again precipitated inorganic salt was added tetrahydrofuran (1000 ml), this was filtered, and the filtrate was distilled off under reduced pressure to obtain the crude product (329 g, >100%) of 3-benzyloxy-2-methyl-pyran-4-one 2 as a brown oil.
NMR (CDCl3) δ: 2.09 (3H, s), 5.15 (2H, s), 6.36 (1H, d, J=5.6 Hz), 7.29-7.41 (5H, m), 7.60 (1H, d, J=5.6 Hz).
2) The compound 2 (162.2 g, 750 mmol) was dissolved in ethanol (487 ml), and aqueous ammonia (28%, 974 ml) and a 6N aqueous sodium hydroxide solution (150 ml, 900 mmol) were added. After the reaction solution was stirred at 90° C. for 1 hour, this was cooled to under ice-cooling, and ammonium chloride (58 g, 1080 mmol) was added. To the reaction solution was added chloroform, this was extracted, and the organic layer was washed with an aqueous saturated sodium bicarbonate solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure, isopropyl alcohol and diethyl ether were added to the residue, and precipitated crystals were filtered to obtain 3-benzyloxy-2-methyl-1H-pyridine-4-one 3 (69.1 g, 43%) as a pale yellow crystal.
NMR (DMSO-d6) δ: 2.05 (3H, s), 5.04 (2H, s), 6.14 (1H, d, J=7.0 Hz), 7.31-7.42 (5H, m), 7.46 (1H, d, J=7.2 Hz), 11.29 (1H, brs).
3) The above compound 3 (129 g, 699 mmol) was suspended in acetonitrile (1300 ml), and N-bromosuccinic acid imide (117 g, 659 mmol) was added, followed by stirring at room temperature for 90 minutes. Precipitated crystals were filtered, and washed with acetonitrile and diethyl ether to obtain 3-benzyloxy-5-bromo-2-methyl-pyridine-4-ol 4 (154 g, 88%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 5.04 (2H, s), 7.32-7.42 (5H, m), 8.03 (1H, d, J=5.5 Hz), 11.82 (1H, brs).
4) To a solution of the compound 4 (88 g, 300 mmol), palladium acetate (13.4 g, 60 mmol) and 1,3-bis(diphenylphosphino)propane (30.8 g, 516 mmol) in dimethylformamide (660 ml) were added methanol (264 ml) and triethylamine (210 ml, 1.5 mol) at room temperature. The interior of a reaction vessel was replaced with carbon monoxide, and the material was stirred at room temperature for 30 minutes, and stirred at 80 degree for 18 hours. A vessel to which ethyl acetate (1500 ml), an aqueous saturated ammonium chloride solution (1500 ml) and water (1500 ml) had been added was stirred under ice-cooling, and the reaction solution was added thereto. Precipitates were filtered, and washed with water (300 ml), ethyl acetate (300 ml) and diethyl ether (300 ml) to obtain 5-benzyloxy-4-hydroxy-6-methyl-nicotinic acid methyl ester 5 (44.9 g, 55%) as a colorless crystal.
NMR (DMSO-d6) δ: 2.06 (3H, s), 3.72 (3H, s), 5.02 (2H, s), 7.33-7.42 (5H, m), 8.07 (1H, s).
5) After a solution of the compound 5 (19.1 g, 70 mmol) in acetic anhydride (134 ml) was stirred at 130° C. for 40 minutes, the solvent was distilled off under reduced pressure to obtain 4-acetoxy-5-benzyloxy-6-methyl-nicotinic acid methyl ester 6 (19.9 g, 90%) as a flesh colored crystal.
NMR (CDCl3) δ: 2.29 (3H, s), 2.52 (3H, s), 3.89 (3H, s), 4.98 (2H, s), 7.36-7.41 (5H, m), 8.85 (1H, s).
6) To a solution of the compound 6 (46.2 g, 147 mmol) in chloroform (370 ml) was added metachloroperbenzoic acid (65%) (42.8 g, 161 mmol) in portions under ice-cooling, and this was stirred at room temperature for 90 minutes. To the reaction solution was added a 10% aqueous potassium carbonate solution, and this was stirred for 10 minutes, followed by extraction with chloroform. The organic layer was washed with successively with a 10% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under induced pressure, and the residue was washed with diisopropyl ether to obtain 4-acetoxy-5-benzyloxy-6-methyl-1-oxy-nicotinic acid methyl ester 7 (42.6 g, 87%) as a colorless crystal.
NMR (CDCl3) δ: 2.30 (3H, s), 2.41 (3H, s), 3.90 (3H, s), 5.02 (2H, s), 7.37-7.39 (5H, m), 8.70 (1H, s).
7) To acetic anhydride (500 ml) which had been heated to stir at 130° C. was added the compound 7 (42.6 g, 129 mmol) over 2 minutes, and this was stirred for 20 minutes. The solvent was distilled off under reduced pressure to obtain 4-acetoxy-6-acetoxymethyl-5-benzyloxy-nicotinic acid methyl ester 8 (49.6 g, >100%) as a black oil.
NMR (CDCl3) δ: 2.10 (3H, s), 2.28 (3H, s), 3.91 (3H, s), 5.07 (2H, s), 5.20 (2H, s), 7.35-7.41 (5H, m), 8.94 (1H, s).
8) To a solution of the compound 8 (46.8 g, 125 mmol) in methanol (140 ml) was added a 2N aqueous sodium hydroxide solution (376 ml) under ice-cooling, and this was stirred at 50° C. for 40 minutes. To the reaction solution were added diethyl ether and 2N hydrochloric acid under ice-cooling, and precipitated crystals were filtered. Resulting crystals were washed with water and diethyl ether to obtain 5-benzyloxy-4-hydroxy-6-hydroxymethyl-nicotinic acid 9 (23.3 g, 68%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.49 (2H, s), 5.19 (2H, s), 5.85 (1H, brs), 7.14-7.20 (2H, m), 7.33-7.43 (7H, m), 8.30 (1H, s), 10.73 (1H, t, J=5.8 Hz), 11.96 (1H, brs).
9) To a solution of the compound 9 (131 g, 475 mmol), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (219 g, 1140 mmol) and 1-hydroxybenzotriazole (128 g, 950 mmol) in dimethylformamide (1300 ml) was added 4-fluorobenzylamine (109 ml, 950 mmol), and this was stirred at 80° C. for 1.5 hours. After the reaction solution was cooled to room temperature, hydrochloric acid was added, followed by extraction with ethyl acetate. The extract was washed with a 5% aqueous potassium carbonate solution, an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. The solvent was distilled off under reduced pressure to obtain a mixture (175 g) of 10 and 11. the resulting mixture was dissolved in acetic acid (1050 ml) and water (1050 ml), and zinc (31.1 g, 475 mmol) was added, followed by heating to reflux for 1 hour. After the reaction solution was cooled to room temperature, a 10% aqueous potassium carbonate solution was added, followed by extraction with ethyl acetate. The extract was washed with an aqueous saturated ammonium chloride solution, and an aqueous saturated sodium chloride solution, and dried with anhydrous sodium sulfate. After the solvent was distilled off under reduced pressure, this was washed with diethyl ether to obtain 5-benzyloxy-N-(4-fluoro-benzyl)-4-hydroxy-6-hydroxymethyl-nicotinic acid amide 10 (107 g, 59%) as a colorless crystal.
NMR (DMSO-d6) δ: 4.45 (2H, d, J=4.3 Hz), 4.52 (2H, d, J=5.8 Hz), 5.09 (2H, s), 6.01 (1H, brs), 7.36-7.43 (5H, m), 8.31 (1H, s), 12.63 (1H, brs).
………………..
SYNTHESIS
- Example 3

3H IS DOLUTEGRAVIR
Step 1
-
N,N-dimethylformamide dimethyl acetal (4.9 ml, 36.5 mmol) was added dropwise to compound 3A (5.0 g, 30.4 mmol) under cooling at 0°C. After stirring at 0°C for 1 hour, 100 ml of ethyl acetate was added to the reaction solution, and the organic layer was washed with a 0.5 N aqueous hydrochloric acid solution (50 ml). The aqueous layer was separated, followed by extraction with ethyl acetate (50 ml). The organic layers were combined, washed with a saturated aqueous solution of sodium bicarbonate and saturated saline in this order, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 1:1 (v/v) → ethyl acetate) to obtain 4.49 g (yield: 67%) of compound 3B as an oil.
1H-NMR (CDCl3)δ:1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Step 2
-
Lithium hexamethyldisilazide (1.0 M solution in toluene, 49 ml, 49.0 mmol) was diluted with tetrahydrofuran (44 ml). A tetrahydrofuran (10 ml) solution of compound 3B (4.49 g, 20.4 mmol) was added dropwise thereto under cooling at -78°C, and a tetrahydrofuran (10 ml) solution of ethyl oxalyl chloride (3.35 g, 24.5 mmol) was then added dropwise to the mixture. The mixture was stirred at -78°C for 2 hours and then heated to 0°C. 2 N hydrochloric acid was added to the reaction solution, and the mixture was stirred for 20 minutes, followed by extraction with ethyl acetate (200 ml x 2). The organic layer was washed with a saturated aqueous solution of sodium bicarbonate and saturated saline and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (n-hexane-ethyl acetate: 7:3 → 5:5 → 0:10 (v/v)) to obtain 1.77 g (yield: 31%) of compound 3C as a white solid.
1H-NMR (CDCl3)δ:1.36-1.46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Step 3
-
Aminoacetaldehyde dimethyl acetal (0.13 ml, 1.20 mmol) was added to an ethanol (6 ml) solution of compound 3C (300 mg, 1.09 mmol) at 0°C, and the mixture was stirred at 0°C for 1.5 hours, then at room temperature for 18 hours, and at 60°C for 4 hours. The solvent in the reaction solution was distilled off under reduced pressure, and the obtained residue was then purified by silica gel column chromatography (n-hexane-ethyl acetate: 5:5 → 0:10 (v/v)) to obtain 252 mg (yield: 64%) of compound 3D as an oil.
1H-NMR (CDCl3)δ:1.36-1.47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Step 4
-
62% H2SO4 (892 mg, 5.64 mmol) was added to a formic acid (10 ml) solution of compound 3D (1.02 g, 2.82 mmol), and the mixture was stirred at room temperature for 16 hours. The formic acid was distilled off under reduced pressure. To the residue, methylene chloride was added, and the mixture was pH-adjusted to 6.6 by the addition of a saturated aqueous solution of sodium bicarbonate. The methylene chloride layer was separated, while the aqueous layer was subjected to extraction with methylene chloride. The methylene chloride layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 531.8 mg of compound 3E as a yellow oil.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Step 5
-
Methanol (0.20 ml, 5.0 mmol), (R)-3-amino-butan-1-ol (179 mg, 2.0 mmol), and acetic acid (0.096 ml, 1.70 mmol) were added to a toluene (5 ml) solution of compound 3E (531 mg, 1.68 mmol), and the mixture was heated to reflux for 4 hours. The reaction solution was cooled to room temperature, then diluted with chloroform, and then washed with a saturated aqueous solution of sodium bicarbonate. The aqueous layer was subjected to extraction with chloroform. The chloroform layers were combined, washed with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by silica gel column chromatography (chloroform-methanol: 100:0 → 90:10) to obtain 309.4 mg of compound 3F as a brown oil.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
Step 6
-
Potassium trimethylsilanolate (333 mg, 2.34 mmol) was added to a 1,2-dimethoxyethane (2 ml) solution of compound 3F (159 mg, 0.47 mmol), and the mixture was stirred at room temperature for 7 hours. 1 N hydrochloric acid and saturated saline were added to the reaction solution, followed by extraction with chloroform. The chloroform layers were combined and dried over anhydrous sodium sulfate. The solvent was distilled off to obtain 34.4 mg (yield: 25%) of compound 3G as an orange powder.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H,m), 4.06-4.10 (2H, m), 4.31 (1H, dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz), 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
Step 7
-
Compound 3G (16 mg, 0.054 mmol) and 2,4-difluorobenzylamine (17 mg, 0.12 mmol) were dissolved in N,N-dimethylformamide (1 ml). To the solution, N,N,N’,N’-tetramethyl-O-(7-aza-benzotriazol-1-yl)uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol) and N-methylmorpholine (0.031 ml, 0.28 mmol) were added, and the mixture was stirred at room temperature for 16 hours. 2,4-difluorobenzylamine (17 mg, 0.12 mmol), HATU (64 mg, 0.17 mmol), and N-methylmorpholine (0.037 ml, 0.34 mmol) were further added thereto, and the mixture was stirred at room temperature for additional 16 hours. 0.5 N hydrochloric acid was added to the reaction solution, followed by extraction with ethyl acetate. The ethyl acetate layers were combined, washed with 0.5 N hydrochloric acid and then with saturated saline, and then dried over anhydrous sodium sulfate. The solvent was distilled off, and the obtained residue was purified by preparative high-performance liquid chromatography to obtain 12.5 mg (yield: 55%) of compound 3H as an orange solid.
- DOLUTEGRAVIR
-
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s).
ISOMERS OF DOLUTEGRAVIR
- Reference Example 1

Step 1
-
Acetic acid (180 mg, 3.00 mmol) was added to a toluene (90 ml) solution of compound A-1 (4.39 g, 9.33 mmol) and (R)-3-aminobutan-1-ol (998 mg, 11.2 mmol), and the mixture was stirred at 50°C for 90 minutes. The reaction solution was allowed to cool to room temperature and then poured to a saturated aqueous solution of sodium bicarbonate. The organic layer was separated, while the aqueous layer was subjected to extraction three times with ethyl acetate. The combined extracts were washed with saturated saline and then dried over sodium sulfate. The solvent was distilled off to obtain 4.29 g of crude product A-2.
Step 2
-
The crude product A-2 obtained in the preceding step was dissolved in ethanol (40 ml). To the solution, a 2 N aqueous sodium hydroxide solution (20 ml) was added at room temperature, and the mixture was stirred at the same temperature for 2 hours. The reaction solution was neutralized to pH 7 using a 2 N aqueous hydrochloric acid solution. The solvent was directly distilled off. The obtained crude product A-3 was subjected to azeotropy with toluene (100 ml) and used in the next step without being purified.
Step 3
-
HOBt (1.65 g, 12.2 mmol) and WSC HCl (2.34 g, 12.2 mmol) were added at room temperature to a DMF (100 ml) solution of the crude product A-3 obtained in the preceding step, and the mixture was stirred at the same temperature for 15 hours. Water was added to the reaction solution, followed by extraction three times with ethyl acetate. The combined extracts were washed with water three times and then dried over sodium sulfate. The solvent was distilled off, and the obtained oil was subjected to silica gel column chromatography for purification. Elution was performed first with n-hexane-ethyl acetate (3:7, v/v) and then with only ethyl acetate. The fraction of interest was concentrated, and the obtained oil was then dissolved in ethyl acetate. The solution was crystallized with diisopropyl ether as a poor solvent. The obtained crystals were collected by filtration and dissolved again in ethyl acetate. The solution was recrystallized to obtain 1.84 g of compound A-4.
1HNMR (CDCl3) δ: 1.49 (3H, d, J = 6.6 Hz), 1.88-1.96 (1H, m), 2.13-2.26 (1H, m), 3.90-4.17 (4H, m), 4.42-4.47 (1H, m), 4.63 (2H, d, J = 6.0 Hz), 5.12-5.17 (1H, m), 5.17 (1H, d, J = 9.9 Hz), 5.33 (1H, d, J = 9.9 Hz), 6.77-6.87 (2H, m), 7.27-7.42 (4H, m), 7.59-7.62 (2H, m), 8.35 (1H, s), 10.41 (1H, t, J = 5.7 Hz).
Step 4
-
The compound A-4 was subjected to the hydroxy deprotection reaction described in Step F of the paragraph [0088] to obtain compound A-5.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).
- Reference Example 2
-

-
Compound A-1 was reacted with (S)-3-aminobutan-1-ol in Step 1. Compound B-5 was obtained in the same way as in Reference Example 1.
1HNMR (DMSO-d6) δ:1.41 (3H, d, J = 6.3 Hz), 1.85-1.92 (1H, m), 1.50-1.75 (1H, m), 4.02-4.09 (3H, m), 4.28-4.34 (1H, m), 4.53 (2H, d, J = 5.7 Hz), 4.64 (1H, dd, J = 3.9 Hz, 12.6 Hz), 5.45 (1H, dd, J = 3.6 Hz, 9.3 Hz), 7.06 (1H, ddd, J = 2.7 Hz, 8.4 Hz, 8.4 Hz), 7.20-7.28 (1H, m), 7.35-7.42 (1H, m), 8.43 (1H, s),10.37 (1H, t, J = 6.0 Hz),12.37 (1H, brs).

……………..

ENTRY 68
………………………….
WO 2010068262
…………………………
WO 2010068253
…………………………………
WO 2011119566
…………………………..
Synthesis
Example 3

I was under cooling added dropwise at 0 ℃ (4.9 ml, 36.5 mmol) and N, N-dimethylformamide dimethyl acetal (5.0 g, 30.4 mmol) in the first step compound 3A. After stirring for 1 hour at 0 ℃, ethyl acetate was added to 100ml, the reaction mixture was washed with 0.5N aqueous hydrochloric acid (50 ml). Was extracted with ethyl acetate (50ml) and solution was separated and the aqueous layer. The organic layers were combined, washed successively with saturated aqueous sodium bicarbonate solution and saturated brine, and then dried over anhydrous sodium sulfate. After the solvent was distilled off, silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) → ethyl acetate 1:1) to an oil (67% yield) of Compound 3B 4.49 g I got a thing.
1 H-NMR (CDCl 3) δ: 1.32 (3H, t, J = 7.1 Hz), 2.90 (3H, br s), 3.29 (3H, br s), 4.23 (2H, q, J = 7.1 Hz), 4.54 (2H, s), 7.81 (1H, s).
Diluted with tetrahydrofuran (44 ml) (1.0M toluene solution, 49 ml, 49.0 mmol) the second step lithium hexamethyldisilazide, under cooling at -78 ℃, compound 3B (4.49 g, 20.4 mmol) in this After dropwise tetrahydrofuran (10 ml) was added dropwise tetrahydrofuran (3.35 g, 24.5 mmol) of ethyl oxalyl chloride and (10 ml) solution. After stirring for 2 hours at -78 ℃, I was warmed to 0 ℃. After washing (200 ml x 2), saturated aqueous sodium bicarbonate solution and the organic layer with saturated brine After stirring for 20 minutes, extracted with ethyl acetate by adding 2N hydrochloric acid, the reaction solution was dried over anhydrous sodium sulfate. After removal of the solvent, silica gel column chromatography and the residue obtained – was purified (n-hexane (v / v) ethyl acetate 7:3 → 5:5 → 0:10), compound 3C 1.77 g (yield I as a white solid 31%).
1 H-NMR (CDCl 3) δ :1.36-1 .46 (6H, m), 4.35-4.52 (8H, m), 8.53 (1H, s).
Was added at 0 ℃ (0.13 ml, 1.20 mmol) the aminoacetaldehyde dimethyl acetal ethanol (300 mg, 1.09 mmol) of the third step compound 3C to (6 ml) solution, 1 hour and 30 minutes at 0 ℃, 18 hours at room temperature , then I was stirred for 4 hours at 60 ℃. After the solvent was evaporated under reduced pressure and the reaction mixture by silica gel column chromatography and the residue obtained was – and purified by (n-hexane (v / v) ethyl acetate 5:5 → 0:10), compound 3D 252 mg (yield: I got as an oil 64%) rate.
1 H-NMR (CDCl 3) δ :1.36-1 .47 (6H, m), 3.42 (6H, s), 3.90 (2H, d, J = 5.2 Hz), 4.37 (3H, q, J = 7.2 Hz), 4.50 (2H, q, J = 7.2 Hz), 8.16 (1H, s).
Was added (892 mg, 5.64 mmol) and 2 SO 4 62-H% formic acid (1.02 g, 2.82 mmol) in a fourth step the compound for 3D (10 ml) solution was stirred at room temperature for 16 hours. Methylene chloride was added to the residue Shi distilled off under reduced pressure and formic acid was adjusted to pH = 6.6 by addition of saturated aqueous sodium bicarbonate. The solution was separated methylene chloride layer was extracted with methylene chloride and the aqueous layer. I was dried over anhydrous sodium sulfate combined methylene chloride layers. The solvent was then distilled off and was obtained as a yellow oil 531.8 mg compound 3E.
1H-NMR (CDCl3) δ: 1.28-1.49 (6H, m), 4.27-4.56 (4H, m), 4.84 (2H, s), 8.10 (1H, s), 9.72 (1H, s).
Amino – – butane – 1 – ol (179 mg, 2.0 mmol), methanol (0.20 ml, 5.0 mmol), (R) -3 toluene (531 mg, 1.68 mmol) in the fifth step to compound 3E (5 ml) solution was added (0.096 ml, 1.70 mmol) acetic acid was heated under reflux for 4 hours. After dilution with chloroform, cooled to room temperature, the reaction mixture was washed with a saturated aqueous sodium bicarbonate solution, and the aqueous layer was extracted with chloroform. After washing with saturated brine combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was then distilled off, silica gel column chromatography and the residue obtained – and (chloroform methanol 100:0 → 90:10), was obtained as a brown oil 309.4 mg compound 3F.
1H-NMR (CDCl3) δ: 1.40 (3H, t, J = 7.1 Hz), 1.40 (3H, d, J = 7.1 Hz), 1.55-1.61 (1H, m), 2.19-2.27 (1H, m), 4.00 (1H, d, J = 1.5 Hz), 4.03 (1H, d, J = 2.5 Hz), 4.10 (1H, dd, J = 13.2, 6.3 Hz), 4.26 (1H, dd, J = 13.2, 3.8 Hz ), 4.38 (2H, q, J = 7.1 Hz), 5.00-5.05 (1H, m), 5.31 (1H, dd, J = 6.4, 3.9 Hz), 8.10 (1H, s).
1,2 (159 mg, 0.47 mmol) in the sixth step compound 3F – was added (333 mg, 2.34 mmol) and potassium trimethylsilanolate dimethoxyethane (2 ml) solution was stirred for 7 hours at room temperature. Brine was added to the 1N-hydrochloric acid to the reaction mixture, followed by extraction with chloroform. The combined chloroform layer was dried over anhydrous sodium sulfate. The solvent was removed by distillation, and I as an orange powder (25% yield) of compound 3G 34.4 mg.
1H-NMR (CDCl3) δ: 1.46 (3H, d, J = 3.5 Hz), 1.58-1.65 (1H, m), 2.26-2.30 (1H, m), 4.06-4.10 (2H, m), 4.31 (1H , dd, J = 13.8, 5.6 Hz), 4.48 (1H, dd, J = 13.6, 3.9 Hz), 5.03 (1H, t, J = 6.4 Hz), 5.36 (1H, dd, J = 5.5, 4.0 Hz) , 8.44 (1H, s), 12.80 (1H, s), 14.90 (1H, s).
2,4 (16 mg, 0.054 mmol) and the seventh step compound 3G – was dissolved in N, N-dimethylformamide (1 ml) (17 mg, 0.12 mmol) difluorobenzyl amine, N, N, N ‘, N was added (0.031 ml, 0.28 mmol) and N-methylmorpholine uronium hexafluorophosphate (HATU) (53 mg, 0.14 mmol), and ‘- tetramethyl-O-(yl 7 – aza – – benzo triazolopyrimidine -1) I was stirred at room temperature for 16 h. 2,4 – was added (0.037 ml, 0.34 mmol) and N-methylmorpholine (64 mg, 0.17 mmol) and (17 mg, 0.12 mmol), HATU difluorobenzylamine, and the mixture was stirred for 16 hours at room temperature. I was extracted with ethyl acetate addition of 0.5N-hydrochloric acid to the reaction mixture. 0.5N-hydrochloric acid and then was washed with saturated brine, and dried over anhydrous sodium sulfate and combined ethyl acetate layer. The solvent was then distilled off, and purified by preparative high performance liquid chromatography residue was obtained as an orange solid (55% yield) of compound 3H 12.5 mg.
1H-NMR (DMSO-d6) δ: 1.36 (3H, d, J = 6.9 Hz), 1.55-1.60 (1H, m), 2.01-2.05 (1H, m), 3.92-3.94 (1H, m), 4.04 (1H, t, J = 12.6 Hz), 4.38-4.41 (1H, m), 4.57-4.60 (1H, m), 4.81-4.83 (1H, m), 5.46-5.49 (1H, m), 7.08-7.11 (1H, m), 7.25-7.30 (1H, m), 7.41 (1H, dd, J = 15.3, 8.7 Hz), 8.53 (1H, s), 10.38 (1H, s), 12.53 (1H, s)
References
- [1] American Medical Association (AMA), STATEMENT ON A NONPROPRIETARY NAME ADOPTED BY THE USAN COUNCIL (Dolutegravir) Accessed 3 December 2011.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- “U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay”. Reuters. 12 August 2013. Retrieved 13 February 2013.
- “GSK wins priority status for new HIV drug in U.S”. Reuters. 16 February 2013. Retrieved 18 February 2013.
- “ViiV Healthcare receives approval for Tivicay™ (dolutegravir) in Canada for the treatment of HIV”. Retrieved 11 November 2013.
- FDA approves new drug to treat HIV infection http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm364744.htm Aug. 12, 2013
- U.S. FDA approves GlaxoSmithKline’s HIV drug Tivicay http://www.reuters.com/article/2013/08/12/us-glaxosmithkline-hivdrug-idUSBRE97B0WU20130812 Mon Aug 12, 2013 6:40pm EDT
- “Dolutegravir Prescribing Information”. Retrieved 3 January 2014.
- Raffi, F; Jaeger, H; Quiros-Roldan, E; Albrecht, H; Belonosova, E; Gatell, JM; Baril, JG; Domingo, P; Brennan, C; Almond, S; Min, S; extended SPRING-2 Study, Group (Nov 2013). “Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial.”. The Lancet infectious diseases 13 (11): 927–35. PMID 24074642.
- http://www.natap.org/2013/ICAAC/ICAAC_24.htm
- Walmsley, Sharon L.; Antela, Antonio; Clumeck, Nathan; Duiculescu, Dan; Eberhard, Andrea; Gutiérrez, Felix; Hocqueloux, Laurent; Maggiolo, Franco; Sandkovsky, Uriel; Granier, Catherine; Pappa, Keith; Wynne, Brian; Min, Sherene; Nichols, Garrett (7 November 2013). “Dolutegravir plus Abacavir–Lamivudine for the Treatment of HIV-1 Infection”. New England Journal of Medicine 369 (19): 1807–1818. doi:10.1056/NEJMoa1215541.
- Sax, Paul. “SINGLE Study Underscores Waning of the Efavirenz Era — But Probably Just in the USA – See more at:http://blogs.jwatch.org/hiv-id-observations/index.php/single-study-underscores-waning-of-the-efavirenz-era-but-probably-just-in-the-usa/2013/11/06/#sthash.A39SderN.dpuf”. Retrieved 19 December 2013.
- Eron, JJ; Clotet, B; Durant, J; Katlama, C; Kumar, P; Lazzarin, A; Poizot-Martin, I; Richmond, G; Soriano, V; Ait-Khaled, M; Fujiwara, T; Huang, J; Min, S; Vavro, C; Yeo, J; VIKING Study, Group (2013 Mar 1). “Safety and efficacy of dolutegravir in treatment-experienced subjects with raltegravir-resistant HIV type 1 infection: 24-week results of the VIKING Study.”. The Journal of infectious diseases 207 (5): 740–8. PMID 23225901.
-
WO2010011812A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds WO2010011819A1 * Jul 23, 2009 Jan 28, 2010 Smithkline Beecham Corporation Chemical compounds -
-
- [Patent Document 1] International publication No.2006/116764 pamphlet
- [Patent Document 2] International publication No.2010/011812 pamphlet
- [Patent Document 3] International publication No.2010/011819 pamphlet
- [Patent Document 4] International publication No.2010/068262 pamphlet
- [Patent Document 5] International publication No.2010/067176 pamphlet
- [Patent Document 6] International publication No.2010/068253 pamphlet
- [Patent Document 7] US Patent 4769380A
- [Patent Document 8] International applicationPCT/JP2010/055316
[NON-PATENT DOCUMENTS]
-
- [Non-Patent Document 1] Journal of Organic Chemistry, 1991, 56(16), 4963-4967
- [Non-Patent Document 2] Science of Synthesis, 2005, 15, 285-387
- [Non-Patent Document 3] Journal of Chemical Society Parkin Transaction. 1, 1997, Issue. 2, 163-169
-
…………………
Sources:
Johns, Brian Alvin; Kawasuji, Takashi; Taishi, Teruhiko; Taoda, Yoshiyuki ; Polycyclic carbamoylpyridone derivative having HIV integrase inhibitory activity and their preparation; PCT Int. Appl., WO2006116764, 02 Nov 2006
Johns, Brian Alvin; Weatherhead, Jason Gordon;Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection; PCT Int. Appl., WO2010011812, 28 Jan 2010
Johns, Brian Alvin; Weatherhead, Jason Gordon; Tricyclic heterocyclic compounds as antiviral agents and their preparation and use in the treatment of HIV infection;PCT Int. Appl., WO2010011819, 28 Jan 2010
Yoshida, Hiroshi; Taoda, Yoshiyuki; Johns, Brian Alvin; Synthesis of fused tricyclic carbamoylpyridone HIV integrase inhibitors and intermediates;PCT Int. Appl.,WO2010068253, 17 Jun 2010
Johns, Brian Alvin; Duan, Maosheng; Hakogi, Toshikazu;Processes and intermediates for fused tricyclic carbamoylpyridone HIV integrase inhibitors;PCT Int. Appl., WO2010068262, 17 Jun 2010
Sumino, Yukihito; Okamoto, Kazuya; Masui, Moriyasu; Yamada, Daisuke; Ikarashi, Fumiya;Preparation of compounds having HIV integrase inhibitory activity; PCT Int. Appl.,WO2012018065, 09 Feb 2012
Kawasuji, Takashi; Johns, Brian A.;Discovery of dolutegravir and S/GSK1265744: Carbamoyl pyridone HIV-1 integrase inhibitors;Abstracts, 64th Southeast Regional Meeting of the American Chemical Society, Raleigh, NC, United States, November 14-17 (2012), SERM-176.
Kawasuji, Takashi; Johns, Brian A.; Yoshida, Hiroshi; Weatherhead, Jason G.; Akiyama, Toshiyuki; Taishi, Teruhiko; Taoda, Yoshiyuki; Mikamiyama-Iwata, Minako; Murai, Hitoshi; Kiyama, Ryuichi; Fuji, Masahiro; Tanimoto, Norihiko; Yoshinaga, Tomokazu; Seki, Takahiro; Kobayashi, Masanori; Sato, Akihiko; Garvey, Edward P.; Fujiwara, Tamio; Carbamoyl Pyridone HIV-1 Integrase Inhibitors. 2. Bi- and Tricyclic Derivatives Result in Superior Antiviral and Pharmacokinetic Profiles;Journal of Medicinal Chemistry (2013), 56(3), 1124-1135
Walmsley S et al. Dolutegravir (DTG; S/GSK1349572) + abacavir/lamivudine once daily statistically superior to tenofovir/emtricitabine/efavirenz: 48-week results – SINGLE (ING114467). 52nd ICAAC, 9-12 September 2012, San Francisco. Abstract H-556b.
http://www.abstractsonline.com/Plan/ViewAbstract.aspx?sKey=e1c18d5b-830f-4b4e-8671-35bcfb20eed5&cKey=af219b7d-2171-46b2-91ef-b8049552c9e5&mKey=%7b6B114A1D-85A4-4054-A83B-04D8B9B8749F%7d
http://www.natap.org/2012/ICAAC/ICAAC_06.htm
http://i-base.info/htb/20381
Raffi F et al. Once-daily dolutegravir (DTG; S/GSK1349572) is non-inferior to raltegravir (RAL) in antiretroviral-naive adults: 48 week results from SPRING-2 (ING113086). 19th International AIDS Conference. 22-27 July 2012, Washington. Late breaker oral presentation THLBB04.
http://pag.aids2012.org/abstracts.aspx?aid=20990
National Institutes of Health (U.S.). A trial comparing GSK1349572 50 mg plus abacavir/lamivudine once daily to Atripla (also called the SINGLE trial). Available from:http://clinicaltrials.gov/ct2/show/NCT01263015.
Stellbrink HJ, Reynes J, Lazzarin A, et al. Dolutegravir in combination therapy exhibits rapid and sustained antiviral response in ARV-naïve adults: 96-week results from SPRING-1 (ING112276) (Abstract 102LB). Paper presented at: 19th Conference on Retroviruses and Opportunistic Infections; 2012 March 5–8; Seattle, WA. Available from:http://www.retroconference.org/2012b/Abstracts/45432.html
AVANAFIL …..A PDE5 inhibitor.

AVANAFIL
A phosphodiesterase (PDE5) inhibitor, used to treat erectile dysfunction.

Avanafil is a new phosphodiesterase-5 inhibitor that is faster acting and more selective than other drugs belonging to the same class. Chemically, it is a derivative of pyrimidine and is only available as the S-enantiomer. FDA approved on April 27, 2012.
CAS RN: 330784-47-9
4-{[(3-chloro-4-methoxyphenyl)methyl]amino}-2-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide
| (S)-2-(2-Hydroxymethyl-1-pyrrolidinyl)-4-(3-chloro-4-methoxybenzylamino)-5-[(2-pyrimidinylmethyl)carbamoyl]pyrimidine |
| 4-[[(3-Chloro-4-methoxyphenyl)methyl]amino]-2-[(2S)-2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide |
| TA 1790 |
Molecular Formular: C23H26ClN7O3
Molecular Mass: 483.95064
- Stendra
- TA 1790
- TA-1790
- UNII-DR5S136IVO
- NDA 202276
INNOVATOR — VIVUS
APPROVED FDA 27/4/2-12
| Patent No | Patent Expiry | patent use code |
|---|---|---|
| 6656935 | Sep 13, 2020 | U-155 |
| 7501409 | May 5, 2023 |
U 155… TREATMENT OF ERECTILE DYSFUNCTION
| Exclusivity Code | Exclusivity_Date |
|---|---|
| NCE | Apr 27, 2017 |
Stendra (avanafil) was given the green light by the US Food and Drug Administration 27/4/2012, but there has been no launch yet as Vivus has been seeking a partner. The latest data should be attractive to potential suitors and could help Stendra take on other phosphodiesterase type 5 (PDE5) inhibitors, notably Pfizer’s Viagra (sildenafil) but also Eli Lilly’s Cialis (tadalafil) and Bayer’s Levitra (vardenafil).
read all at
http://www.pharmatimes.com/Article/13-06-20/Vivus_ED_drug_gets_to_work_in_less_than_15_mins.aspx
STENDRA (avanafil) is a selective inhibitor of cGMP-specific PDE5.
。

Avanafil is designated chemically as (S)-4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2pyrimidinylmethyl)-5-pyrimidinecarboxamide and has the following structural formula:
 |
Avanafil occurs as white crystalline powder, molecular formula C23H26ClN7O3 and molecular weight of 483.95 and is slightly soluble in ethanol, practically insoluble in water, soluble in 0.1 mol/L hydrochloric acid. STENDRA, for oral administration, is supplied as oval, pale yellow tablets containing 50 mg, 100 mg, or 200 mg avanafil debossed with dosage strengths. In addition to the active ingredient, avanafil, each tablet contains the following inactive ingredients: mannitol, fumaric acid, hydroxypropylcellulose, low substituted hydroxypropylcellulose, calcium carbonate, magnesium stearate, and ferric oxide yellow.
AVANAFIL

Avanafil is a PDE5 inhibitor approved for erectile dysfunction by FDA on April 27, 2012 [1] and by EMA on June 21, 2013.[2] Avanafil is known by the trademark names Stendra and Spedra and was developed by Vivus Inc. In July 2013 Vivus announced partnership with Menarini Group, which will commercialise and promote Spedra in over 40 European countries plus Australia and New Zealand.[3] Avanafil acts by inhibiting a specificphosphodiesterase type 5 enzyme which is found in various body tissues, but primarily in the corpus cavernosum penis, as well as the retina. Other similar drugs are sildenafil, tadalafil and vardenafil. The advantage of avanafil is that it has very fast onset of action compared with other PDE5 inhibitors. It is absorbed quickly, reaching a maximum concentration in about 30–45 minutes.[4] About two-thirds of the participants were able to engage in sexual activity within 15 minutes.[4]
Avanafil is a highly selective PDE5 inhibitor that is a competitive antagonist of cyclic guanosine monophosphate. Specifically, avanafil has a high ratio of inhibiting PDE5 as compared with other PDE subtypes allowing for the drug to be used for ED while minimizing adverse effects. Absorption occurs quickly following oral administration with a median Tmax of 30 to 45 minutes and a terminal elimination half-life of 5 hours. Additionally, it is predominantly metabolized by cytochrome P450 3A4. As such, avanafil should not be co-administered with strong cytochrome P450 3A4 inhibitors. Dosage adjustments are not warranted based on renal function, hepatic function, age or gender. Five clinical trials suggest that avanafil 100 and 200 mg doses are effective in improving the Sexual Encounter Profile and the Erectile Function Domain scores among men as part of the International Index of Erectile Function. A network meta-analysis comparing the PDE5 inhibitors revealed avanafil was less effective on Global Assessment Questionnaire question 1 while safety data indicated no major differences among the different PDE5 inhibitors. The most common adverse effects reported from the clinical trials associated with avanafil were headache, flushing, nasal congestion, nasopharyngitis, sinusitis, and dyspepsia.
A “phosphodiesterase type 5 inhibitor” or “PDE5 inhibitor” refers to an agent that blocks the degradative action of phosphodiesterase type 5 on cyclic GMP in the arterial wall smooth muscle within the lungs and in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. PDE5 inhibitors are used for the treatment of pulmonary hypertension and in the treatment of erectile dysfunction. Examples of PDE5 inhibitors include, without limitation, tadalafil, avanafil, lodenafil, mirodenafil, sildenafil citrate, vardenafil and udenafil and pharmaceutically acceptable salts thereof.
“Avanafil” refers to the chemical compound 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide, and its pharmaceutically acceptable salts. Avanafil is described in Limin M. et al., (2010) Expert Opin Investig Drugs, 19(11):1427-37. Avanafil has the following chemical formula:

Avanafil is being developed for erectile dysfunction. Avanafil currently has no trademarked term associated with it but it is being developed by Vivus Inc.
…………………………………
DESCRIPTION IN A PATENT
EXAMPLE 92-145
The corresponding starting compounds are treated in a similar manner to give the compounds as listed in the following Table 7.
| TABLE 7 |
 |
 |
 |
Amorphous MS(m/z): 484(MH+) |
ENTRY 98 IS AVANAFIL
…………………………………………………….
The invention discloses a preparation method of Avanafil (Avanafil, I), which comprises the following steps: carrying out a substitution reaction on 6-amino-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (XII) and 3-chloro-4-methoxy benzyl chloride (XIII) so as to obtain 6-(3-chloro-4-methoxy benzyl amino)-1, 2-dihydro pyrimidine-2-keto-5-carboxylic acid ethyl ester (IXV); carrying out condensation on the compound (IXV) and S-hydroxymethyl pyrrolidine (II) so as to generate 4-[(3-chloro-4-methoxy benzyl) amino]-2-[2-(hydroxymethyl)-1-pyrrole alkyl] pyrimidine-5-carboxylic acid ethyl ester (XI); and carrying out hydrolysis on the compound (XI) and then carrying out an acylation reaction on the compound (XI) and the compound (XI) so as to obtain Avanafil (I). The preparation method is simple in process, economic and environmental-friendly, suitable for the requirements of industrialization amplification.
……………………………………………………
The invention discloses a method for preparing avanafil (Avanafil, I). The method comprises the steps of taking cytosine as an initial material; and orderly carrying out replacement, halogen addition and condensation reaction on a side chain 3-chlorine-4-methoxy benzyl halide (III), N-(2-methylpyrimidine) formamide (IV) and S-hydroxymethyl pyrrolidine (II), so as to obtain a target product avanafil (I). The preparation method is available in material, concise in technology, economic and environment-friendly, and suitable for the demands of industrial amplification.
…………………………………………………….
SYNTHESIS
Avanafil can be synthesized from a benzylamine derivative and a pyrimidine derivative REF 5:Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709

- ………………………………………………………
- SYNTHESIS
- A cutting that phenanthrene by a methylthio urea ( a ) and ethoxy methylene malonate ( 2 ) cyclization of 3 , chloride, phosphorus oxychloride get 4 , 4 with benzyl amine 5 occurred SNAr the reaction product after oxidation with mCPBA 6 . In pyrimidine, if the 2 – and 4 – positions are active simultaneously the same leaving group in the case, SNAr reaction occurs preferentially at 4 – position, but does not guarantee the 2 – side reaction does not occur. Here is an activity of the poor leaving group sulfide spans 2 – bit, and a good leaving group active chlorine occupy four – position, thus ensuring a high regioselectivity of the reaction. 4 – position after completion of the reaction, then the 2 – position of the group activation, where sulfide sulfoxide better than the leaving group. Amino alcohols 7 and 6 recurrence SNAr reaction 8 , 8 after alkaline hydrolysis and acid alpha amidation get that phenanthrene.
AVANAFIL
- …………………………….

-
- FDA approves Stendra for erectile dysfunction” (Press release). Food and Drug Administration (FDA). April 27, 2012.
- http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002581/human_med_001661.jsp&mid=WC0b01ac058001d124
- http://ir.vivus.com/releasedetail.cfm?releaseid=775706
- Kyle, Jeffery; Brown, Dana (2013). “Avanafil for Erectile Dysfunction”. Annals of Pharmacotherapy (Sage Publishing). doi:10.1177/1060028013501989. Retrieved 28 September 2013.
- Yamada, K.; Matsuki, K.; Omori, K.; Kikkawa, K.; 2004, U.S. Patent 6,797,709
-
- Peterson CA. Hemodynamic effect of avanafil and glyceryl trinitrate coadministration. , Drugs Context , Volume 2013 , 2013 Feb 26
- Gur S. The Effect of Intracavernosal Avanafil, a Newer Phosphodiesterase-5 Inhibitor, on Neonatal Type 2 Diabetic Rats With Erectile Dysfunction. , Urology , 2013 Dec 9
- Hill JK. Avanafil for erectile dysfunction. , Ann Pharmacother , Volume 47 , Issue 10 , 2013 Oct
- Sanford M. Avanafil: a review of its use in patients with erectile dysfunction. , Drugs Aging , Volume 30 , Issue 10 , 2013 Oct
- Hellstrom WJ. PDE5 inhibitors: considerations for preference and long-term adherence. , Int J Clin Pract , Volume 67 , Issue 8 , 2013 Aug
- Aversa A. An update on pharmacological treatment of erectile dysfunction with phosphodiesterase type 5 inhibitors. , Expert Opin Pharmacother , Volume 14 , Issue 10 , 2013 Jul
- Oelke M. Phosphodiesterase inhibitors in clinical urology. , Expert Rev Clin Pharmacol , Volume 6 , Issue 3 , 2013 May
- Kukreja RC. Sildenafil and cardioprotection. , Curr Pharm Des , Volume 19 , Issue 39 , 2013
- Day WW. An open-label, long-term evaluation of the safety, efficacy and tolerability of avanafil in male patients with mild to severe erectile dysfunction. , Int J Clin Pract, Volume 67 , Issue 4 , 2013 Apr
- Tang J. Comparative effectiveness and safety of oral phosphodiesterase type 5 inhibitors for erectile dysfunction: a systematic review and network meta-analysis. , Eur Urol , Volume 63 , Issue 5 , 2013 May
United States APPROVED 6656935 2012-04-27 EXPIRY 2020-09-13 United States 7501409 2012-04-27 2023-05-05 - Faster-Working Erectile Dysfunction Drug?. CBS News. November 24, 2009.
- Vivus says men taking avanafil were more likely to be ready for sex within 15 minutes. The Gaea Times. January 11, 2010.
- “Avanafil is the New Player in The Erectile Dysfunction Field”. June 28, 2011.
-
- • Hatzimouratidis, K., et al.: Drugs, 68, 231 (2008)
-
4-20-2011Tablets quickly disintegrated in oral cavity7-16-2010Combination treatment for diabetes mellitus8-28-2009Roflumilast for the Treatment of Pulmonary Hypertension1-32-2008Cyclic compounds
US5242391 Oct 30, 1991 Sep 7, 1993 ALZA Corporation Urethral insert for treatment of erectile dysfunction US5474535 Jul 19, 1993 Dec 12, 1995 Vivus, Inc. Dosage and inserter for treatment of erectile dysfunction US5773020 Oct 28, 1997 Jun 30, 1998 Vivus, Inc. Treatment of erectile dysfunction US6656935 Aug 10, 2001 Dec 2, 2003 Tanabe Seiyaku Co., Ltd. Aromatic nitrogen-containing 6-membered cyclic compounds Update nov 2015
NEW PATENT WO 2015177807

WO 2015177807
Suryakant Shivaji Pol; Nitin Sharadchandra Pradhan; Shashikant Balu Padwal; Vihar Raghunath Telange; Nitn Shankar Bondre
Wanbury ltd

The present invention relates to a novel compound of Formula (II), and its use in preparation of Avanafil, [Formula should be inserted here] wherein R is -OH, -CI or -OR1 and R1 is C1 to C3 alkyl group
It having been developed and launched by VIVUS and JW Pharmaceutical, under license from Mitsubishi Tanabe Pharma, and Auxilium Pharmaceuticals, for treating ED.
A process for preparation of Avanafil was first disclosed in US 6,797,709 (depicted in Scheme I), wherein 4-chloro-5-ethoxycarbonyl-2-methylthio-pyrimidine is coupled with 3-chloro-4-methoxybenzylamine in presence of triethylamine to provide compound of Formula (A), which on oxidization provides a sulfonyl compound of Formula (B). Said compound of Formula (B) is reacted with L-prolinol and exert compound of Formula (C). The resulting compound of Formula (C) undergoes column chromatographic purification and crystallization, while further subjected to hydrolysis to obtain compound of Formula (D). The compound of Formula (D) is coupled with 2-aminomethylpyrimidine to obtain Avanafil of Formula (I). The final product obtained is purified by column chromatography. The need to purify the intermediate compound of Formula (C) and final product, by column chromatography makes this process cumbersome, time consuming and unviable for large scale production thereby contributing to main disadvantages of the process.
Scheme I

Formula (A)m-CPBA/chloroform

Formula (C) Formula (B)
NaOH/DMSO

Formula (D) Formula (I)CN 103254179, discloses a process for preparation of Avanafi, wherein 3-chloro-4-methoxybenzylhalide is coupled with cytosine to result compound of Formula (E), later on condensation with L-prolinol yields 4-[(3-chloro-4-methoxy benzyl)amino-2-(2-hydroxymethyl)-l -pyrrolinyl]pyrimidine of Formula (F). The compound of Formula (F) is then condensed with N-(2-pyrimidylmethyl)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme II
Scheme II

Formula (F) Formula (I)
CN 103254180 describes an alternate process for preparation of Avanafil of Formula (I), wherein a substitution reaction on 6-amino-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester and 3-chloro-4-methoxybenzylchloride provides 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid, ethyl ester of Formula (G) which on condensation with L-prolinoI generates 6-(3-chloro-4-methoxybenzylamino)-l ,2-dihydropyrimidine-2-keto-5-carboxylic acid ethyl ester of Formula (H). The compound of Formula (H) is then hydrolysed and coupled with N-(2-pyrimidylmethyI)formamide to obtain Avanafil of Formula (I). Process is depicted in Scheme III
Scheme III

Formula (H) Formula (Γ)
In all the prior art discussed above, chiral compound L-prolinol is coupled in molecule in earlier steps of synthesis. This approach seems to be less feasible for large scale production; the insertion of L-prolinol in early stage may need to exert number of purifications for intermediates. Further the main shortcoming in such process is that the chirality of molecule is disturbed by inserting L-prolinol in early stages because there are number of operations in line in process to obtain the target compound.
CN 103483323, discloses a synthetic method for preparation of avanafil, wherein amidation of pyrimidine-5-carbonyl chlorides with 2-(aminomethyl)pyrimidine at temperature ranging from -10 to 5°C resulted an amide (intermediates A); which underwent condensation with 3-chloro-4-methoxybenzylamine at the temperature ranging from 0 -3°C to give 4-[(3-chloro-4-methoxybenzyl)amino]-5-
pyrimidinecarboxamides (intermediates B), which further on condensation with L-prolinol gave avanafil. The disadvantage of this process is the need to maintain the reaction temperature in range of – 10 to 5°C which adds up to cost of process and makes the process complicated. The process is depicted in Scheme IV.
Scheme IV

Intermediate (A)

wherein, R’ & R2 are independently, hydrogen, halogen, alkoxy, alkoxyalkyl, cyno group, amino group
Hence, to overcome shortcomings of prior art the inventors of present invention have skillfully designed a process with novel intermediate which concomitantly result Avanafil compound of Formula (I), substantially free from impurities. Further this invention encompass L-proline in last stage of molecule in order to avoid the number of purifications of intermediate which relent the economic significances by taking into account yield of each stage.
Object of the invention
1. The main object of the invention is to provide a novel compound of Formula
(ID-
2. Another object of present invention is to provide a process for preparation of a novel compound of Formula (II).
3. Yet another object of present invention is to provide a process for preparation of Avanafil of Formula (I), in high yield and purity using a novel compound of Formula (II).
4. Yet another object of the present invention to provide simple, economic and industrially scalable process for the preparation of Avanafil o Formula (I).
Summary of the invention
According to an aspect of present invention, there is provided a novel compound of Formula (II).

Formula (II)
wherein R is -OH, -CI or -OR and R is Q to C3 alkyl group
The invention will be specifically described below with reference to Examples but it should not be construed that the scope of the invention is limited thereto. Since the starting compound was produced by a modified method from that described in prior art, it will be described as Referential Example 1 to 3. Here synthesis routes of Referential Example 1 to 3 and Example 1 to 10 are illustrated below in Scheme (V).
Scheme (V)

Formula (I) Referential Examples
Referential Example 1 – Preparation of ethyl 4-[(3-chloro-4-methoxybenzyl)amino]-2-(methyl sulfanyl)pyrimidine-5-carboxylate
To 600ml of methylene dichloride was added l OOg of ethyl 4-chloro-2-(methylsulfanyl) pyrimidine-5-carboxylate and 91.2g of 3-chloro-4-methoxybenzylamine. The reaction mixture was stirred and 500ml of water, 48g of sodium carbonate and Ig of tetra-butylammonium bromide were added to it. The reaction mixture was then maintained overnight at 25-30°C. After completion of reaction, methylene dichloride layer was separated, washed with water and evaporated to obtain 145g of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate having 95% of HPLC purity.
Above reaction can also be carried out using ammonia or triethylamine in same reaction conditions and parameters, in place of sodium carbonate.
Referential Example 2 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid
To 600ml of methanol was added l OOg of ethyl 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylate (Referential Example 1) and an aqueous solution of sodium hydroxide (15g of NaOH in 140ml of water). The reaction mixture was heated to reflux temperature. After completion of reaction, the pH of mixture was adjusted to 1 -2 using concentrated hydrochloric acid followed by stirring the mixture for 1 hour at 10-15°C. The solid product obtained was filtered, washed sequentially with water and methanol, and dried overnight at 70-75°C to get 87g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid.
Referential Example 3 – Preparation of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III)
To a mixture of 400ml of toluene and 0.5ml of dimethyl formamide was added 50g of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfanyl) pyrimidine-5-carboxylic acid (Referential Example 2) and 70g of thionyl chloride, and the reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain yellow solid mass. The solid mass thus obtained, was cooled to 15-20°C followed by addition of 1 75ml of methylene dichloride, 36. l g of 2-amino methyl pyrimidine mesylate and 35.55g of triaethylamine. The reaction mixture was stirred overnight at 25-30°C. After completion of reaction, methylene dichloride was distilled out to get residue. The residue was washed sequentially with 2.5% sodium carbonate solution and water. The residue was then treated with methanol to obtain 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) having HPLC purity of more than 95% (yield: 80%)
Referential Example 4 – Preparation of 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[(2S)-2-(hydroxymethyl)-l -pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide (Avanafil)
Step i)
To 200ml of dichloromethane was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyI)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting dichloromethane layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the dichloromethane layer obtained in step i), was added 2.57g of triethylamine followed by slow addition of 125ml solution of L-prolinol in dichloromethane (2.46g of L-prolinol in 125ml of dichlromethane). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water followed by evaporation of dichloromethane to obtain an oily mass. The oily mass thus obtained was treated with methanol to yield 8g of Avanafil.
Examples
Example 1 : Preparation of Compound of Formula (II) (wherein R is -OH)
Step i)
To 200ml of methylene dichloride was added lOg of 4-[(3-chloro-4-methoxybenzyl) amino]-2-(methyl sulfinyl)-N-(pyrimidin-2-ylmethyl) pyrimidine-5-carboxiamide of Formula (III) and 6.5g of m-chloro per benzoic acid and the mixture was stirred for 1 hour at 25-30°C. After completion of reaction, the reaction mixture was washed with aqueous solution of sodium carbonate and water. The resulting methylene dichloride layer comprising compound of Formula (IV) was taken to next step.
Step ii)
To the methylene dichloride layer comprising compound of Formula (IV) obtained in step i), was added 5g of triethylamine followed by slow addition of 125ml solution of L-proline in methylene dichloride (2.8g of L-proline in 125ml of methylene dichloride). The reaction mixture was maintained overnight. After completion of reaction, the reaction mixture was washed with water and 5% sodium carbonate solution, followed by evaporation of methylene dichloride to obtain an oily mass. The oily mass obtained was stripped with 50ml acetone to yield 9g of compound of Formula (II) having HPLC purity 98%.
Example 2: Preparation of Compound of Formula (II) (wherein R is -OC2H5)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filtrate containing product. The ethanol in filtrate is completely distilled out to isolate 10.45g of esterified compound of Formula (II).
Example 3 : Preparation of Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of toluene and 0.5ml of dimethylformamide was added 50g of compound of Formula (II) obtained in example 1 , and 70g of thionyl chloride. The reaction mixture was refluxed for 2.5 hours. After completion of reaction, solvent was distilled under vacuum and the residue was stripped with toluene to obtain 50.5g of oily carboxylic acid chloride compound of Formula (II).
Example 4: Preparation of Avanafil of Formula (I)
In an inert atmosphere, a solution of 30g of compound of Formula (II) obtained in example 1 or 2, in 150 ml of tetrahydrofuran was dropwise added to 180ml of suspension of 1.0M lithium aluminium hydride solution in tetrahydrofuran, The reaction mixture was refluxed for 5 hours. After completion of reaction, the mixture was cooled in ice-bath and saturated aqueous solution of sodium sulfate was added to decompose excess of lithium aluminium hydride. The mixture was then diluted with 200ml of methylene dichloride and thus formed organic layer was separated. The organic layer was washed with water (3 χ 100 ml), dried over MgS04 and concentrated to collect crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 22.8g of Avanafil of Formula (I) having HPLC purity of 99.20%.
Example 5 : Preparation of Avanafil of Formula (I)
To a mixture of 1.3g sodium borohydride, 1 ml methanesulfonic acid and 50ml ethanol was added l Og of compound of Formula (II) obtained in example 1 or 2, and the mixture was stirred at 25-30°C for 5 hours. After completion of reaction, 100ml water was added and the mixture was extracted with 1 00ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with ethyl acetate at 45- 50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 6 to Example 8
The procedure is carried out as in example 5 except for instead of methanesulfonic acid other reducing agents are used in combination with sodium borohydride. The results are given in Table I
Table I

Example 9: Preparation of Avanafil of Formula (I)
To 100ml of ethanol was added 0.5ml of sulphuric acid and l Og of compound of Formula (II) obtained in example 1 , and the reaction mixture was maintained at reflux temperature till completion of reaction. The reaction mixture was then cooled to 25-30°C and the pH of reaction mixture was adjusted to 7-8 using sodium carbonate. Filter the reaction mixture and collect filterate containing product. To the fi Iterate was added 1.2g of sodium borohydride and 2.6g of lithium bromide, and the mixture was stirred for 5 hours. After complete conversion of ester to final product, l OOml water was added and the mixture was extracted with 100ml methylene dichloride (50ml X 2). The methylene dichloride layer obtained was evaporated under reduced pressure to get an oily mass. The oily mass was stripped with 25ml ethyl acetate at 45-50°C. To the oily residue formed was added 50ml of ethyl acetate and the mixture was cooled to 0-5°C. The solid obtained was filtered, washed with ethyl acetate and dried to yield crude Avanafil of Formula (I) which was subjected to purification using methanol as solvent to yield 7.5g of Avanafil of Formula (I) having HPLC purity of 99%.
Example 10: Preparation of Avanafil of Formula (I) from Compound of Formula (II) (wherein R is -CI)
To a mixture of 400ml of tetrahydrofuran and 50g of carboxylic acid chloride compound of Formula (II) obtained in example 3, was added 12g sodium borohydride at 0-5°C. After completion of reaction, water was added to reaction mixture to decompose excess of sodium borohydride present. The reaction mixture was then concentrated and a solution of 30g of potassium hydroxide in 200 ml of water was added. The mixture was heated to 60-70°C and maintained for 15-18 hours. The mixture was then cooled to 25-30°C and 500 ml of methylene dichloride was added. The organic layer thus formed, was separated and evaporated to yield crude Avanafil
of Formula (I) which was then subjected to purification using methanol as solvent to obtain 40g of Avanafil of Formula (I) having HPLC purity of 99.01%.

Mr. K. Chandran Wholetime Director & Vice Chairman EXTRAS
A “phosphodiesterase type 5 inhibitor” or “PDE5 inhibitor” refers to an agent that blocks the degradative action of phosphodiesterase type 5 on cyclic GMP in the arterial wall smooth muscle within the lungs and in the smooth muscle cells lining the blood vessels supplying the corpus cavernosum of the penis. PDE5 inhibitors are used for the treatment of pulmonary hypertension and in the treatment of erectile dysfunction. Examples of PDE5 inhibitors include, without limitation, tadalafil, avanafil, lodenafil, mirodenafil, sildenafil citrate, vardenafil and udenafil and pharmaceutically acceptable salts thereof. In one aspect, the PDE5 inhibitor is tadalafil.
“Tadalafil” or “TAD” is described in U.S. Pat. Nos. 5,859,006 and 6,821,975. It refers to the chemical compound, (6R-trans)-6-(1,3-benzodioxol-5-yl)-2,3,6,7,12,12a-hexahydro-2-methyl-pyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-dione and has the following chemical formula:

Tadalafil is currently marketed in pill form for treating erectile dysfunction (ED) under the trade name Cialis® and under the trade name Adcirca® for the treatment of PAH.
“Avanafil” refers to the chemical compound 4-[(3-Chloro-4-methoxybenzyl)amino]-2-[2-(hydroxymethyl)-1-pyrrolidinyl]-N-(2-pyrimidinylmethyl)-5-pyrimidinecarboxamide, and its pharmaceutically acceptable salts. Avanafil is described in Limin M. et al., (2010) Expert Opin Investig Drugs, 19(11):1427-37. Avanafil has the following chemical formula:

Avanafil is being developed for erectile dysfunction. Avanafil currently has no trademarked term associated with it but it is being developed by Vivus Inc.
“Lodenafil” refers to the chemical compound, bis-(2-{4-[4-ethoxy-3-(1-methyl-7-oxo-3-propyl-6,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-benzenesulfonyl]piperazin-1-yl}-ethyl)carbonate and has the following chemical formula:

More information about lodenafil is available at Toque H A et al., (2008) European Journal of Pharmacology, 591(1-3):189-95. Lodenafil is manufactured by Cristália Produtos Químicose Farmacêuticos in Brazil and sold there under the brand-name Helleva®. It has undergone Phase III clinical trials, but is not yet approved for use in the United States by the U.S. FDA.
“Mirodenafil” refers to the chemical compound, 5-Ethyl-3,5-dihydro-2-[5-([4-(2-hydroxyethyl)-1-piperazinyl]sulfonyl)-2-propoxyphenyl]-7-propyl-4H-pyrrolo[3,2-d]pyrimidin-4-one and has the following chemical formula:

More information about mirodenafil can be found at Paick J S et al., (2008) The Journal of Sexual Medicine, 5 (11): 2672-80. Mirodenafil is not currently approved for use in the United States but clinical trials are being conducted.
“Sildenafil citrate,” marketed under the name Viagra®, is described in U.S. Pat. No. 5,250,534. It refers to 1-[4-ethoxy-3-(6,7-dihydro-1-methyl-7-oxo-3-propyl-1H-pyrazolo[4,3-d]pyrimidin-5-yl)phenylsulfonyl]-4-methylpiperazine and has the following chemical formula:

Sildenafil citrate, sold as Viagra®, Revatio® and under various other trade names, is indicated to treat erectile dysfunction and PAH.
“Vardenafil” refers to the chemical compound, 4-[2-Ethoxy-5-(4-ethylpiperazin-1-yl)sulfonyl-phenyl]-9-methyl-7-propyl-3,5,6,8-tetrazabicyclo[4.3.0]nona-3,7,9-trien-2-one and has the following chemical formula:

Vardenafil is described in U.S. Pat. Nos. 6,362,178 and 7,696,206. Vardenafil is marketed under the trade name Levitra® for treating erectile dysfunction.
“Udenafil” refers to the chemical compound, 3-(1-methyl-7-oxo-3-propyl-4,7-dihydro-1H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1-methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide and has the following chemical formula:

More information about udenafil can be found at Kouvelas D. et al., (2009) Curr Pharm Des, 15(30):3464-75. Udenafil is marketed under the trade name Zydena® but not approved for use in the United States.



THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D GLENMARK SCIENTIST , NAVIMUMBAI, INDIA
did you feel happy, a head to toe paralysed man’s soul in action for you round the clock need help, email or call me
MOBILE-+91 9323115463
web link
I was paralysed in dec2007, Posts dedicated to my family, my organisation Glenmark, Your readership keeps me going and brings smiles to my family





