
Home » rheumatoid arthritis
Category Archives: rheumatoid arthritis
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Iguratimod
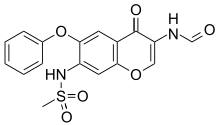
Iguratimod
- Molecular FormulaC17H14N2O6S
- Average mass374.368 Da
- UNII-4IHY34Y2NVигуратимодإيغوراتيمود艾拉莫德
123663-49-0[RN]
3-Formylamino-7-methylsulfonylamino-6-phenoxy-4H-1-benzopyran-4-one
4IHY34Y2NV8176IGU
Methanesulfonamide, N-[3-(formylamino)-4-oxo-6-phenoxy-4H-1-benzopyran-7-yl]-
N-(3-Formamido-4-oxo-6-phenoxy-4H-chromen-7-yl)methanesulfonamide
product patent US4954518
Research Code:T-614
Trade Name:Iremod® / Kolbet® / Careram®
MOA:Nuclear factor NF-κB activation inhibitor
Indication:Rheumatoid arthritis
Status:Approved
Company:Simcere (Originator) , Taisho Toyama,EisaiSales:ATC Code:
Approved Countries or Area
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-06-29 | Marketing approval | Careram | Rheumatoid arthritis | Tablet, Film coated | 25 mg | Eisai | |
| 2012-06-29 | Marketing approval | Kolbet | Rheumatoid arthritis | Tablet, Film coated | 25 mg | Toyama Chemical, Taisho Toyama |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-08-15 | Marketing approval | 艾得辛/Iremod | Rheumatoid arthritis | Tablet, Film coated | 25 mg | Simcere |
Iguratimod was first approved by China Food and Drug Administration (CFDA) on August 15, 2011, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on June 29, 2012. It was developed by Simcere and marketed as 艾得辛®/Iremod® by Simcere and as Kolbet® by Taisho Toyama and by Eisai in Japan.
Iguratimod is a nuclear factor NF-κB activation inhibitor used in the treatment of rheumatoid arthritis.
Iremod® is available as tablet for oral use, containing 25 mg of free Iguratimod, and the recommended dose is 25 mg once daily or 25 mg at a time, twice daily.
Iguratimod: (Iremod)-First approval: 2009
Iguratimod is a disease-modifying antirheumatic drug (DMARD) that was approved for use in rheumatoid arthritis (RA) patients in China and Japan in 2009
Toyama Chemical , Taisho Toyama Pharmaceutical , Eisai , Simcere and Tianjin Institute of Pharmaceutical Research have codeveloped and launched iguratimod, an inflammatory cytokine and IL-6 gene inhibiting compound that also inhibits immunoglobulin production in B cells. Iguratimod is indicated for the oral treatment of rheumatoid arthritis.
Iguratimod is an anti-inflammatory small molecule drug used for the treatment of rheumatoid arthritis, together with methotrexate in Japan and China.[1] As of 2015 the biological target was not known, but it prevents NF-κB activation and subsequently selectively inhibits COX-2 and several inflammatory cytokines.[1]
Adverse effects include elevated transaminases, nausea, vomiting, stomach pain; rashes, and itchiness.[1]
It is a derivative of 7-methanesulfonylamino-6-phenoxychromones and is a chromone with two amide groups; it was first published in 2000.[1][2] It was submitted for regulatory approval in Japan in 2003; the application was withdrawn in 2009, and it was resubmitted with additional data in 2011 and approved for marketing in Japan in 2012.[1] Eisai and Toyama Chemical market it in Japan.[3] Approval was obtained in China in 2011 by Simcere, independently of the Japanese originators.[1][4]
During discovery and development it was called T-614 and it is marketed under the names Careram and Kolbet.[5]
Syn
Indian Pat. Appl., 2014MU01507

SYN


Reference:1. US4954518.Route 2
Reference:1. Chem. Pharm. Bull. 2000, 48, 131-139.
2. Chin. J. New. Drugs. 2006, 15, 2042-2044.
3. Shanghai Chem. Ind. 2007, 32, 22-24.Route 3
Reference:1. CN1462748A.
2. Chem. Pharm. Bull. 2000, 48, 131-139.
SYN
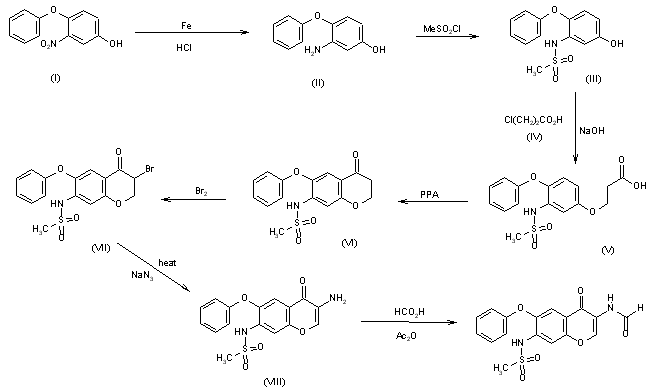
AU 8823489; CH 679397; FR 2621585; GB 2210879; JP 1995267943; US 4954518
The reduction of 3-nitro-4-phenoxyphenol (I) with Fe, aqueous HCl gives 3-amino-4-phenoxyphenol (II), which is acylated with methanesulfonyl chloride by means of pyridine in dichloromethane affording 3-(methylsulfonamido)-4-phenoxyphenol (III). The condensation of (III) with 3-chloropropionic acid (IV) by means of NaOH in water gives 3-[3-(methylsulfonamido)-4-phenoxyphenoxy]propionic acid (V), which is cyclized by means of polyphosphoric acid at 65-70 C yielding 7-(methylsulfonamido)-6-phenoxy-3,4-dihydro-2H-1-benzopyran-4-one (VI). The bromination of (VI) with Br2 in CHCl3 affords 3-bromo-7-(methylsulfonamido)-6-phenoxy-3,4-dihydro-2H-1-benzopyran-4-one (VII), which is treated with sodium azide in DMF at 70-75 C giving 3-amino-7-(methylsulfonamido)-6-phenoxy-2H-1-benzopyran-4-one (VIII). Finally, this compound is formylated with formic acid in acetic anhydride.
SYN
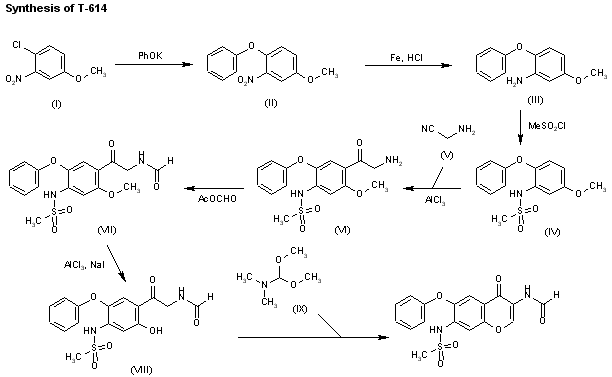
Chem Pharm Bull 2000,48(1),131
A preparative-scale synthetic route for T-614 has been reported: The reaction of 4-chloro-3-nitroanisole (I) with potassium phenolate in hot DMF gives 4-phenoxy-3-nitroanisole (II), which is reduced to the corresponding 3-amino compound (III) by treatment with Fe and HCl. The reaction of (III) with mesyl chloride in pyridine affords the sulfonamide (IV), which is acylated with 2-aminoacetonitrile (V) and AlCl3 in nitrobenzene/HCl giving the 2-aminoacetophenone (VI). Formylation of (VI) at the NH2 with acetic formic anhydride yields the formamide (VII), which is demethylated with AlCl3 and NaI in acetonitrile affording the phenol (VIII). Finally, this compound is cyclized with dimethylformamide dimethylacetal in DMF.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0968089614001230
Synthetic approaches to the 2012 new drugs
Hong X. Ding, … Christopher J. O’Donnell, in Bioorganic & Medicinal Chemistry, 2014

13 Iguratimod (Careram®, Iremod®)
Iguratimod, which was discovered by Toyama Pharmaceuticals and jointly co-developed with Eisai in Japan, was approved by the PMDA (Pharmaceuticals and Medical Devices Agency) of Japan on June 29, 2012 for the treatment of rheumatoid arthritis.83 This drug was also independently developed by Simcere Pharmaceutical Group and is marketed as Iremod® in China. The drug exhibited inhibitory effects on granuloma inflammation, and was shown to be efficacious for the prevention of joint destruction in adjuvant arthritis.84,85 While several synthesis of iguratimod have been published,86 the most likely scale synthesis, which does not require chromatographic purification, is described in Scheme 14.87
The synthesis began with commercially available 3-nitro-4-chloro anisole (78) which was reacted with potassium phenoxide (generated from phenol and potassium t-butoxide at 110 °C) to provide the corresponding nitrophenyl ether which was subsequently reduced and sulfonylated to furnish sulfonamide 79. Next, this diphenyl ether was submitted to a Friedel–Crafts reaction with aminoacetonitrile hydrochloride which gave rise to aminomethylacetophenone 80 in 90% yield. This aminoketone was then formylated with formic trimethylacetic anhydride 81 at room temperature to afford formamide 82 in 91% yield, and this material was immediately subjected to O-demethylation conditions with aluminum trichloride and sodium iodide in acetonitrile to give the phenol 83 in 95% yield. Finally, treatment of the aminomethyl acetophenone phenol 83 with N,N-dimethylformamide dimethylacetal in DMF at low temperatures furnished iguratimod (XII) in 87% yield
83. Eisai and Toyama Chemical Receive Approval to Market Anti-rheumatic Agent Iguratimod in Japan, 2012, http://www.eisai.com/news/news201239.html, [Access Date: 2012-July-29].
84. Tanaka, K.; Shimotori, T.; Makino, S.; Aikawa, Y.; Inaba, T.; Yoshida, C.; Takano, S. Arzneim.-Forsch. 1992, 42, 935.
85. Tanaka, K.; Makino, S.; Shimotori, T.; Aikawa, Y.; Inaba, T.; Yoshida, C. Arzneim.-Forsch. 1992, 42, 945.
86. Takano, S.; Yoshida, C.; Inaba, T.; Tanaka, K.; Takeno, R.; Nagaki, H.; Shimotori, T.; Makino, S. US Patent 4954518 A, 1990.
87. Inaba, T.; Tanaka, K.; Takeno, R.; Nagaki, H.; Yoshida, C.; Takano, S. Chem. Pharm. Bull. 2000, 48, 131.
SYN

https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/slct.202003553A Convenient Synthesis of Iguratimod‐Amine Precursor via NHC‐Catalyzed Aldehyde‐Nitrile Cross Coupling ReactionNithya MurugeshProf. Ramasamy KarvembuDr. Seenuvasan VedachalamFirst published: 24 November 2020https://doi.org/10.1002/slct.202003553
A protocol for the synthesis of iguratimod‐amine precursor has been developed using N‐heterocyclic carbene (NHC)‐catalyzed aldehyde‐nitrile cross coupling reaction with overall atom efficiency of 71 %. The first step involves a nucleophilic aromatic substitution (SNAr) of 1‐chloro‐4‐methoxy‐2‐nitrobenzene (1) with phenol to produce 4‐methoxy‐2‐nitro‐1‐phenoxybenzene (2) which further undergoes nitro reduction followed by mesylation to produce N‐(5‐methoxy‐2‐phenoxyphenyl)methanesulfonamide (4). Furthermore, it was subjected to Vilsmeier‐Haack formylation and demethylation (using BBr3) to produce N‐(4‐formyl‐5‐hydroxy‐2‐phenoxyphenyl)methanesulfonamide (6). Subsequently, O‐alkylation followed by NHC‐catalyzed aldehyde‐nitrile cross coupling yields the amine precursor of iguratimod (8).
N-(3-Amino-4-oxo-6-phenoxy-4H-chromen-7-yl)methanesulfonamide (8):4=4. S. Vedachalam, J. Zeng, B. K. Gorityala, M. Antonio, X.-W. Liu, Org. Lett. 2010, 12, 352–355.
Compound 7 (290 mg, 0.83 mmol) and triazolium carbene catalyst (34 mg, 0.1245 mmol) were dissolved in dry CH2Cl2 under N2 atmosphere. To this, DBU (24.7 µL, 0.16 mmol) was added at room temperature, and the mixture was stirred for 12 h. After the completion of reaction, the reaction mixture was dried, and the residue was purified by column chromatography to yield compound 8. Yield: 200 mg, 70 %; m. p. 162 ℃; 1H NMR (500 MHz, DMSO-d6): δ 8.25 (s, 1H), 7.96 (s, 1H), 7.78 (s, 1H), 7.45 (t, J = 8.0 Hz, 2H), 7.25–7.21 (m, 3H), 7.16 (s, 1H), 4.91 (s, 2H), 3.23 (s, 3H); 13C NMR (125 MHz, DMSO-d6): δ 171.2, 155.0, 152.1, 150.4, 137.9, 133.0, 132.8, 131.0 (2C), 125.9, 122.4, 121.1 (2C), 117.2, 110.0, 39.0; FTIR (KBr): v 3423, 3347, 1620, 1592, 1487, 1342, 1210, 1155, 970, 757 cm-1 ; HR-MS (ESI): m/z calcd. for C16H15N2O5S 347.0702, found 347.0714 [M+H]+ …..https://chemistry-europe.onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fslct.202003553&file=slct202003553-sup-0001-misc_information.pdf
Patent
WO 2021020481
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021020481&tab=FULLTEXT&_cid=P11-KKXGLE-06477-1The following formula (1)
[Chemical 1]
Iguratimod (chemical name: N- [3- (formylamino) -4-oxo-6-phenoxy-4H-chromen-7-yl] methanesulfonamide), which is indicated by, has excellent anti-inflammatory and antipyretic and analgesic effects. It is a very useful compound as a therapeutic agent exhibiting anti-arthritis and anti-allergic effects (see Patent Document 1).
A plurality of synthetic routes are known for the method for producing iguratimod and its derivatives (hereinafter, may be referred to as iguratimod derivatives as a term meaning both iguratimod and its derivatives), and all of them use intermediates of iguratimod derivatives. This is a stepwise manufacturing method. The present inventors have studied the stepwise production of intermediates of iguratimod derivatives of formulas (II) to (XI) by the following synthetic route to synthesize the iguratimod derivatives represented by formula (XII). doing.
[Chemical formula 2]R 1 = hydroxyl protecting group, R 2 = amino protecting group, Ar = aromatic ring group which may have a substituent, X 2 = halogen atom.[Chemical 3]
Ms = methylsulfonyl group[Chemical 4]
Production Example 1
(Production of igratimodo: Patent Documents 3 and 4)
N, N-dimethylformamide 150 mL, N, N-dimethylformamide dimethyl acetal 40.9 g (N, N-dimethylformamide dimethyl acetal ) in a 1000 mL four-necked flask equipped with two stirring blades having a diameter of 10 cm ( 343 mmol) was added, and the mixture was cooled to 10 ° C. with stirring. 8.2 g (137 mmol) of glacial acetic acid and 50.0 g (137 mmol) of formylaminomethyl (2-hydroxy-4-methylsulfonylamino-5-phenoxyphenyl) ketone were sequentially added thereto, and the temperature was raised to 20 ° C. The reaction was carried out at the same temperature for 5 hours. 250 mL of methylene chloride was added to the reaction suspension, and 500 mL of water was added dropwise to the obtained solution. After adjusting the pH to 5 with a 10% aqueous hydrochloric acid solution, the mixture was stirred at 20 ° C. for 1 hour. The obtained precipitated crystals were separated, washed successively with 50 mL of methylene chloride, 50 mL of water and 50 mL of ethanol, and then dried at 50 ° C. for 12 hours. Next, the obtained crystals were dissolved in a mixed solvent of 7.7 g (137 mmol) of potassium hydroxide, 750 mL of water and 750 mL of acetone, and then neutralized with 2N hydrochloric acid water, and the obtained precipitated crystals were separated. Then, after washing with 50 mL of water, it was dried at 50 ° C. for 12 hours to obtain 42.8 g of iguratimod (iguratimod purity: 99.72%, N-methyl compound: 0.23%).
References
- ^ Jump up to:a b c d e f Tanaka K, Yamaguchi T, Hara M (May 2015). “Iguratimod for the treatment of rheumatoid arthritis in Japan”. Expert Review of Clinical Immunology. 11 (5): 565–73. doi:10.1586/1744666X.2015.1027151. PMID 25797025. S2CID 25134255.
- ^ Inaba T, Tanaka K, Takeno R, Nagaki H, Yoshida C, Takano S (January 2000). “Synthesis and antiinflammatory activity of 7-methanesulfonylamino-6-phenoxychromones. Antiarthritic effect of the 3-formylamino compound (T-614) in chronic inflammatory disease models”. Chemical & Pharmaceutical Bulletin. 48 (1): 131–9. doi:10.1248/cpb.48.131. PMID 10705489.
- ^ Bronson J, Dhar M, Ewing W, Lonberg N (2012). “Chapter Thirty-One – To Market, To Market—2011”. Annual Reports in Medicinal Chemistry. 47: 499–569. doi:10.1016/B978-0-12-396492-2.00031-X.
- ^ “Iguratimod – Simcere”. AdisInsight. Retrieved 27 May 2018.
- ^ “Iguratimod – Toyama Chemical”. AdisInsight. Retrieved 27 May 2018.
| Clinical data | |
|---|---|
| Trade names | Careram; Kolbet |
| Other names | T-614 |
| ATC code | None |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 123663-49-0 |
| PubChem CID | 124246 |
| ChemSpider | 110694 |
| UNII | 4IHY34Y2NV |
| ChEMBL | ChEMBL2107455 |
| CompTox Dashboard (EPA) | DTXSID0048971 |
| ECHA InfoCard | 100.236.037 |
| Chemical and physical data | |
| Formula | C17H14N2O6S |
| Molar mass | 374.37 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=S(=O)(Nc3c(Oc1ccccc1)cc2c(O/C=C(\C2=O)NC=O)c3)C | |
| InChI[hide]InChI=1S/C17H14N2O6S/c1-26(22,23)19-13-8-15-12(17(21)14(9-24-15)18-10-20)7-16(13)25-11-5-3-2-4-6-11/h2-10,19H,1H3,(H,18,20)Key:ANMATWQYLIFGOK-UHFFFAOYSA-N |
////////////IGURATIMOD, UNII-4IHY34Y2NV , игуратимод , إيغوراتيمود , 艾拉莫德 , T-614, T 614, Kolbet, Careram, Rheumatoid arthritis, JAPAN 2012, CHINA 2011
CS(=O)(=O)NC1=C(C=C2C(=C1)OC=C(C2=O)NC=O)OC3=CC=CC=C3
WXFL-10203614
![(7R)-7-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carbonitrile.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=134405105&t=l)
WXFL-10203614
CAS 2054932-34-0 R isomer, (S isomer 2054932-33-9 )
C15 H15 N7, 293.33
(7R)-7-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine-2-carbonitrile
- (7R)-5,6,7,8-Tetrahydro-7-(methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)imidazo[1,2-a]pyridine-2-carbonitrile
-
Imidazo[1,2-a]pyridine-2-carbonitrile, 5,6,7,8-tetrahydro-7-(methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)-, (7R)-
Wuxi Fortune Pharmaceutical Co Ltd
Jak1 tyrosine kinase inhibitor
Wuxi Fuxin Pharmaceutical Research and Development , in collaboration with Wuxi Apptec , is investigating a tablet formulation of WXFL-10203614 , a JAK1 tyrosine kinase inhibitor, for the oral treatment of rheumatoid arthritis. In January 2019, a phase I trial was planned.
- Imidazo[1,2-a]pyridine-2-carbonitrile, 5,6,7,8-tetrahydro-7-(methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)-, (7R)-, 4-methylbenzenesulfonate, hydrate (1:1:1)
- cas 2226936-85-0
- Imidazo[1,2-a]pyridine-2-carbonitrile, 5,6,7,8-tetrahydro-7-(methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)-, (7R)-, 2,2,2-trifluoroacetate (1:1)
- cas 2226936-87-2
syn

PATENT
WO2018095345 claiming novel crystalline salt forms of similar compound
PATENT
WO 2016192563
PATENT
US-20190218231
Novel crystalline forms of 7h-pyrrolo[2,3-D]pyrimidine compounds (designated as forms A to E) useful as JAK1 and JAK2 inhibitors for treating arthritis, inflammation and autoimmune diseases.
JAK belongs to the family of tyrosine kinases involved in inflammation, autoimmune diseases, proliferative diseases, transplant rejection, impaired cartilage turnover-related diseases, congenital cartilage malformations, and/or diseases associated with excessive secretion of IL6. The present invention also provides a method for preparing the compound or a pharmaceutical composition comprising the compound, and a method for preventing and/or treating inflammation, autoimmune diseases, proliferative diseases, transplant rejection, impaired cartilage turnover-related diseases, congenital cartilage malformations, and/or diseases associated with excessive secretion of IL6 by administrating the compound of the present invention.
Janus kinase (JAK) is a cytoplasmic tyrosine kinase that transduces a cytokine signal from a membrane receptor to an STAT transcription factor. The prior art has described four members of the JAK family: JAK1, JAK2, JAK3 and TYK2. When cytokines bind to their receptors, JAK family members are auto-phosphorylated and/or trans-phosphorylated from each other, followed by STATs phosphorylation, and then are migrated into the cell nucleus to regulate the transcription. JAK-STAT intracellular signal transduction is suitable for interferons, most interleukins, as well as various cytokines and endocrine factors, such as EPO, TPO, GH, OSM, LIF, CNTF, GM-CSF and PRL (Vainchenker W. et al. (2008)).
A combinatorial study of a genetic model and a small molecule JAK inhibitor has revealed the therapeutic potential of several JAKs. It has been confirmed by mouse and human genetics that JAK3 is an immunosuppressive target (O’Shea J. et al. (2004)). A JAK3 inhibitor has been successfully used in clinical development. At first, it was used in organ transplant rejection, and later also used in other immunoinflammatory indications such as rheumatoid arthritis (RA), psoriasis and Crohn’s disease (http://clinicaltrials.gov/). It has been confirmed by human genetics and mouse knockout studies that TYK2 is a potential target for immunoinflammatory diseases (Levy D. and Loomis C. (2007)). JAK1 is a new target in the field of immunoinflammatory diseases. The heterodimerization of JAK1 and other JAKs arouses a transduction of cytokine-driven pro-inflammatory signaling. Thus, it is expected that inhibition of JAK1 and/or other JAKs has a therapeutic benefit for a series of inflammatory diseases and other diseases driven by JAK-mediated signal transduction.
transduction.
Example 1: Preparation of Compound 1
Step 1: 2-chloro-4-nitro-1-oxo-pyridin-1-ium (40.0 g, 229.2 mmol) and (4-methoxyphenyl)methylamine (63 g, 458.4 mmol) were dissolved in EtOH (400 mL), and the resulting solution was stirred at reflux for 5 hours. TLC (PE:EA=2:1) showed that the reaction was complete. The EtOH was concentrated to half of its volume and was cooled in an ice bath for 2-3 hours. The resulting cold mixture was filtered, and the isolated solid was washed with PE (60 mL*3) and ice water (60 mL*3), respectively. Drying in vacuum given an orange solid, N-[(4-methoxyphenyl)methyl]-4-nitro-1-oxo-pyridin-1-ium-2-amine (2) (38.6 g, 140.2 mmol, with a yield of 61.2%). MS (ESI) calcd. For r C 13H 13N 3O 4 [M+H] + 275, found 276.
Step 2: to a solution of N-[(4-methoxyphenyl)methyl]-4-nitro-1-oxo-pyridin-1-ium-2-amine (5.0 g, 18.16 mmol) in CHCI 3 (50 mL) was dropwise added PCI 3 (8.4 g, 60.8 mmol) at 0° C. After the addition, the reaction mixture was heated to 25° C. and stirred vigorously for 16 hours. TLC (PE:EA=1:1) showed that the reaction was complete. The reaction mixture was filtered, and the resulting solid was washed with PE (30 mL*3) to give a yellow solid compound, N-[(4-methoxyphenyl)methyl]-4-nitro-pyridin-2-amine (3) (4.2 g, a crude product) which was directly used in the next step without further purification. MS (ESI) calcd. For C 15H 18N 6 [M+H] +259, found 260.
Step 3: to a solution of N-[(4-methoxyphenyl)methyl]-4-nitro-pyridin-2-amine (4.2 g, 16.2 mmol) in toluene (10 mL) was dropwise added TFA (5.0 mL) at atmospheric temperature. Then, the mixture was stirred at 80° C. for 2 hours. TLC (PE:EA=1:1) showed that the reaction was complete. The mixture was concentrated under reduced pressure to remove the solvent. The residue was diluted with H 2O (50 mL), and its pH was adjusted to be neutral with solid NaHCO 3. The aqueous phase was extracted with EA (50 mLE*3). The combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography (silica, petroleum ether/ethyl acetate=1/0-1:1) to obtain an orange solid compound, 4-nitropyridine-2-amine (4) (700 mg, 5.0 mmol, with a yield of 31.1%). MS (ESI) calcd. For C 5H 5N 3O 2 [M+H] + 139, found 140.
Step 4: to a solution of 4-nitropyridine-2-amine (200 mg, 1.4 mmol) in DME (5 mL) was added 3-bromo-2-oxo-propanoate (280 mg, 1.4 mmol) at atmospheric temperature. The resulting mixture was stirred at 25° C. for 1 hour, and then was concentrated under reduced pressure to remove the solvent. The residue was dissolved with EtOH (10 mL); and then was refluxed for 3 hours. TLC showed that the reaction was complete. The reaction solution was cooled to room temperature, and the solvent was concentrated under reduced pressure. The residue was basified with saturated NaHCO 3 aqueous solution (25 mL). The aqueous phase was extracted with DCM (15 mL*3); and the combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (EA:PE=10-60%) to obtain a light yellow solid compound, ethyl 7-nitroimidazo[1,2-]pyridin-2-carboxylate (5) (302 mg, with a yield of 88.9%). MS (ESI) calcd. For C 10H 9N 3O 4 [M+H] + 235, found 236.
Step 5: a solution of ethyl 7-nitroimidazo[1,2-a]pyridin-2-carboxylate (150 mg, 637.8 mmol) in ethanol (20 mL) was added HCl (7 mg, 0.2 mmol) and PtO 2 (15 mg, 0.6 mmol) at atmospheric temperature. The reaction system was repeatedly vacuumed and filled with N 2 for three times, then filled with H 2(50 psi), and was stirred at 50° C. for 16 hours. TLC (PE:EA=1:1) showed that the reaction was complete. The reaction mixture was concentrated to half of its volume, and filtered to obtain a white solid compound, ethyl 7-amino-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate hydrochloride (6) (120 mg, a crude product). MS (ESI) calcd. For C 10H 15N 3O 2 [M+H] + 209, found 210.
Step 6: ethyl 7-amino-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate hydrochloride (100 mg, 0.4 mmol) and 4-chloro-7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidine (137 mg, 0.4 mmol) were dissolved in n-BuOH (5 mL), and DIEA (158 mg, 1.2 mmol) were added to the above solution. The resulting mixture was stirred under reflux for 16 hours. LC-MS showed that the reaction was complete. The reaction mixture was concentrated under reduced pressure, and the resulting residue was diluted with H 2O (10 mL). The aqueous phase was extracted with EA (20 mL*3); and the combined organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting residue was purified by preparative TLC (PE:EA=0:1) to obtain a light yellow solid compound, ethyl 7-[[7-(p-toluenesulfonyl) pyrrolo[2,3-d]pyrimidin-4-yl] amino]-5,6,7,8-tetrahydroimidazo[1,2-α]pyridin-2-carboxylate (7) (55 mg, 0.11 mmol, with a yield of 28.1%). MS (ESI) calcd. For C 23H 24N 6O 4S [M+H] + 480, found 481.
Step 7: to a solution of ethyl 7-[[7-(p-toluenesulfonyl) pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-α]pyridin-2-carboxylate (3.0 g, 6.2 mmol) in THF (150 mL) was added NaH (499 mg, 12.5 mmol) in portions under N 2 atmosphere at 0° C. The mixture was stirred at that temperature for 1 hour, and then was dropwise added MeI (7.1 g, 50.2 mmol). After the addition, the mixture was stirred at atmospheric temperature for 1 hour. TLC showed that the reaction was complete. The reaction was quenched by the addition of saturated NH 4Cl (10 mL), and then was diluted by the addition of ice water (50 mL). The aqueous phase was extracted with a mixed solvent of DCM/MeOH (3:1, 50 mL*3). The combined organic phase was dried over sodium sulfate, filtered, and concentrated under reduced pressure. The resulting crude product was purified by flash column chromatography (DCM:MeOH=10:1) to obtain a light yellow solid, ethyl 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate (8) (1.5 g, with a yield of 45%). MS (ESI) calcd. For C 24H 26N 6O 4S [M+H] + 494, found 495.
Step 8: to a solution of 7-[methyl-[7-(p-toluenesulfonyl) pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxylate (4.0 g, 8.1 mmol) in THF (40 mL) and H 2O (8 mL) was added LiOH.H 2O (509 mg, 12.1 mmol), and the mixture was stirred at 20° C. for 10 hours. TLC showed that the reactants were completely consumed. THF in the reaction mixture was removed under reduced pressure; and the pH of the residue was adjusted to 2-3 with 2M HCl (4 mL) to form a white solid. The solid was filtered out, and was concentrated under reduced pressure to obtain 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1, 2-a]pyridin-2-carboxylic acid (9) as a white solid (3.6 g, with a yield of 95.4%). MS (ESI) calcd. For C 22H 22N 6O 4S [M+H] + 466, found 467.
Step 9: to a solution of 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1, 2-a]pyridin-2-carboxylic acid (1.8 g, 3.9 mmol) in DMF (20 mL) was added CDI (751 mg, 4.6 mmol) at 0° C. The reaction solution was heated to 25° C. and stirred for 2 hours, and after that, solid ammonium chloride (2.1 g, 38.6 mmol) was added, and then the reaction was kept overnight at atmospheric temperature. LC-MS showed that the reactants were completely consumed. The reaction mixture was poured into ice water (50 mL), and a white solid was precipitated. The solid was filtered out, washed with water (20 mL), and was dried under reduced pressure in a rotating manner to obtain 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxamide (10) as a white solid (2.5 g, a crude product) which product was directly used in the next step. MS (ESI) calcd. For C 22H 23N 7O 3S [M+H] + 465, found 466.
Step 10: 7-[methyl-[7-(p-toluenesulfonyl)pyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1, 2-a]pyridin-2-carboxamide (2.5 g, 5.4 mmol) was dissolved in a mixture of THF (20 mL), MeOH (10 mL) and H 2O (6 mL), and NaOH (429.6 mg, 10.7 mmol) was added. The mixture was heated to 60° C. and stirred for 30 minutes. LC-MS showed that the reactants were completely consumed. The reaction mixture was concentrated under reduced pressure to obtain 7-[methyl-[heptahydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-α]pyridin-2-carboxamide (11) as a white solid (2.5 g, a crude product) which was directly used in the next step. MS (ESI) calcd. For C 15H 17N 7O [M+H] + 311, found 312.
Step 11: to a solution of 7-[methyl-[heptahydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-carboxamide (2.0 g, 6.4 mmol) and triethylamine (3.9 g, 38.5 mmol) in THF (20 mL) was dropwise added TFAA (4.1 g, 19.3 mmol) at 0° C. After the addition, the reaction solution was stirred at atmospheric temperature for 30 minutes. LC-MS showed the starting materials were completely consumed. The reaction mixture was poured into ice water (20 mL), and extracted with DCM/MeOH (5:1, 100 mL*2). The combined organic layer was washed with saturated saline (20 mL), dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to obtain a residue. The residue was purified by column chromatography (DCM/MeOH=40/1 to 20:1) to obtain 7-[methyl-[7-hydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-nitrile (12,378 mg, with a yield of 19.8%). MS (ESI) calcd. For C 15H 15N 7 [M+H] + 293, found 294. 1H NMR (400 MHz, DMSO-d6) 11.44-11.71 (m, 1H), 7.99-8.17 (m, 2H), 7.11-7.20 (m, 1H), 6.63 (dd, J=1.76, 3.26 Hz, 1H), 5.33 (br. s., 1H), 4.21-4.21-4.31 (m, 1H), 4.13 (dt, J=4.14, 12.49 Hz, 1H), 3.27 (s, 3H), 2.91-3.11 (m, 2H), 2.31-2.44 (m, 1H), 2.07 (d, J=11.54 Hz, 1H).
Step 12: racemic 7-[methyl-[7-hydropyrrolo[2,3-d]pyrimidin-4-yl]amino]-5,6,7,8-tetrahydroimidazo[1,2-a]pyridin-2-nitrile (30 mg, 102.3 umol) was separated by a chiral column to obtain the compound 1 (10 mg, with a yield of 32.8%).
Compound 1: retention time 6.407 min; MS (ESI) calcd. For C 15H 15N 7 [293, found 294 M+H]+. Purity 98.8%, e.e. was 98.9%; [α] D 20=+78.4° (c=0.6, DMSO). MS ESI calcd. For C 15H 15N 7 [M+H] + 294, found 294. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.02-2.15 (m, 1H) 2.39 (qd, J=12.42, 5.90 Hz, 1H) 2.92-3.12 (m, 2H) 3.28 (s, 3H) 4.05-4.36 (m, 2H) 5.20-5.45 (m, 1H) 6.64 (dd, J=3.39, 1.88 Hz, 1H) 7.17 (dd, J=3.26, 2.51 Hz, 1H) 8.02-8.17 (m, 2H) 11.69 (br s, 1H).
//////////WXFL-10203614, WXFL 10203614 , WXFL10203614, Wuxi Fuxin, arthritis, inflammation, autoimmune diseases, Wuxi Apptec, JAK1, JAK2 inhibitors
N#Cc1cn2CC[C@H](Cc2n1)N(C)c4ncnc3nccc34
Piclidenoson, иклиденозон , بيكليدينوسون , 匹利诺生 ,
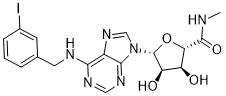

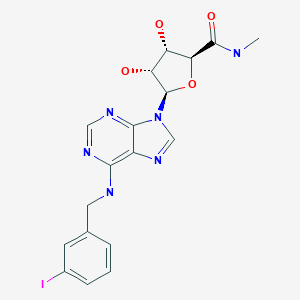
CF 101, Piclidenoson
ALB-7208
CAS 152918-18-8
Chemical Formula: C18H19IN6O4
Molecular Weight: 510.28
(2S,3S,4R,5R)-3,4-Dihydroxy-5-{6-[(3-iodobenzyl)amino]-9H-purin-9-yl}-N-methyltetrahydro-2-furancarboxamide
N6-(3-Iodobenzyl)adenosine-5′-N-methyluronamide
β-D-Ribofuranuronamide, 1-deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-
1-Deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-β-D-ribofuranuronamide
CF 101 (known generically as IB-MECA) is an anti-inflammatory drug for rheumatoid arthritis patients. Its novel mechanism of action relies on antagonism of adenoside A3 receptors. CF101 is supplied as an oral drug and has an excellent safety profile. It is also being considered for the treatment of other autoimmune-inflammatory disorders, such as Crohn’s disease, psorasis and dry eye syndrome.

- Originator Can-Fite BioPharma
- Class Amides; Anti-inflammatories; Antineoplastics; Antipsoriatics; Antirheumatics; Eye disorder therapies; Iodobenzenes; Neuroprotectants; Purine nucleosides; Ribonucleosides; Small molecules
- Mechanism of Action Adenosine A3 receptor agonists; Immunosuppressants; Interleukin 23 inhibitors; Interleukin-17 inhibitors
- Phase III Plaque psoriasis; Rheumatoid arthritis
- Phase II Glaucoma; Ocular hypertension
- Phase I Uveitis
- Preclinical Osteoarthritis
- Discontinued Colorectal cancer; Dry eyes; Solid tumours
- 05 Feb 2019 Can-Fite BioPharma receives patent allowance for A3 adenosine receptor (A3AR) agonists in USA
- 05 Feb 2019 Can-Fite BioPharma receives patent allowance for A3 adenosine receptor (A3AR) agonists in North America, South America, Europe and Asia
- 21 Aug 2018 Phase-III clinical trials in Plaque psoriasis (Monotherapy) in Israel (PO)
Piclidenoson, also known as CF101, is a specific agonist to the A3 adenosine receptor, which inhibits the development of colon carcinoma growth in cell cultures and xenograft murine models. CF101 has been shown to downregulate PKB/Akt and NF-κB protein expression level. CF101 potentiates the cytotoxic effect of 5-FU, thus preventing drug resistance. The myeloprotective effect of CF101 suggests its development as an add-on treatment to 5-FU.
Piclidenoson is known to be a TNF-α synthesis inhibitor and a neuroprotectant. use as an A3 adenosine receptor agonist, useful for treating rheumatoid arthritis (RA), psoriasis, osteoarthritis and glaucoma.
Can-Fite BioPharma , under license from the National Institutes of Health (NIH), is developing a tablet formulation of CF-101, an adenosine A3 receptor-targeting, TNF alpha-suppressing low molecular weight molecule for the potential treatment of psoriasis, RA and liver cancer. The company is also investigating a capsule formulation of apoptosis-inducing namodenoson, the lead from a program of adenosine A3 receptor agonist, for treating liver diseases, including hepatocellular carcinoma (HCC). In January 2019, preclinical data for the treatment of obesity were reported. Also, see WO2019105217 , WO2019105359 and WO2019105082 , published alongside.
PATENT
WO-2019105388
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019105388&tab=FULLTEXT&maxRec=1000
Novel crystalline forms of CF-101 (also known as piclidenoson; designated as Forms CS1, CS2 and CS3), processes for their preparation, compositions comprising them and their use as an A3 adenosine receptor agonist for treating rheumatoid arthritis, psoriasis, osteoarthritis and glaucoma are claimed
PAPER
Journal of medicinal chemistry (1994), 37(5), 636-46
https://pubs.acs.org/doi/pdf/10.1021/jm00031a014
PAPER
Journal of medicinal chemistry (1998), 41(10), 1708-15
https://pubs.acs.org/doi/abs/10.1021/jm9707737
PAPER
Bioorganic & Medicinal Chemistry (2006), 14(5), 1618-1629
PATENT
WO 2015009008
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015009008


PATENT
WO 2008111082
REFERENCES
1: Avni I, Garzozi HJ, Barequet IS, Segev F, Varssano D, Sartani G, Chetrit N, Bakshi E, Zadok D, Tomkins O, Litvin G, Jacobson KA, Fishman S, Harpaz Z, Farbstein M, Yehuda SB, Silverman MH, Kerns WD, Bristol DR, Cohn I, Fishman P. Treatment of Dry Eye Syndrome with Orally Administered CF101 Data from a Phase 2 Clinical Trial. Ophthalmology. 2010 Mar 19. [Epub ahead of print] PubMed PMID: 20304499.
2: Bar-Yehuda S, Rath-Wolfson L, Del Valle L, Ochaion A, Cohen S, Patoka R, Zozulya G, Barer F, Atar E, Piña-Oviedo S, Perez-Liz G, Castel D, Fishman P. Induction of an antiinflammatory effect and prevention of cartilage damage in rat knee osteoarthritis by CF101 treatment. Arthritis Rheum. 2009 Oct;60(10):3061-71. PubMed PMID: 19790055.
3: Borea PA, Gessi S, Bar-Yehuda S, Fishman P. A3 adenosine receptor: pharmacology and role in disease. Handb Exp Pharmacol. 2009;(193):297-327. Review. PubMed PMID: 19639286.
4: Moral MA, Tomillero A. Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2008 Mar;30(2):149-71. PubMed PMID: 18560631.
5: Silverman MH, Strand V, Markovits D, Nahir M, Reitblat T, Molad Y, Rosner I, Rozenbaum M, Mader R, Adawi M, Caspi D, Tishler M, Langevitz P, Rubinow A, Friedman J, Green L, Tanay A, Ochaion A, Cohen S, Kerns WD, Cohn I, Fishman-Furman S, Farbstein M, Yehuda SB, Fishman P. Clinical evidence for utilization of the A3 adenosine receptor as a target to treat rheumatoid arthritis: data from a phase II clinical trial. J Rheumatol. 2008 Jan;35(1):41-8. Epub 2007 Nov 15. PubMed PMID: 18050382
/////////////CF 101, Piclidenoson, CF101, CF-101, CF 101, ALB-7208, ALB 7208, ALB7208, IB MECA, Phase III, Plaque psoriasis, Rheumatoid arthritis, UNII-30679UMI0N, Пиклиденозон , بيكليدينوسون , 匹利诺生 , Can-Fite BioPharma
CNC(=O)[C@H]1O[C@H]([C@H](O)[C@@H]1O)N1C=NC2=C(NCC3=CC(I)=CC=C3)N=CN=C12

{kind=link}
{kind=link}
{kind=link}
{kind=link}