PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Literature References: Derivative of artemisinin, q.v. Prepn: Y. Li et al.,K’o Hsueh T’ung Pao24, 667 (1979), C.A.91, 211376u (1979); eidem,Acta Pharm. Sin.16, 429 (1981). Absolute configuration: X.-D. Luo et al.,Helv. Chim. Acta67, 1515 (1984). NMR spectral study: F. S. El-Feraly et al.,Spectrosc. Lett.18, 843 (1985). Inhibition of protein synthesis: H. M. Gu et al.,Biochem. Pharmacol.32, 2463 (1983). Antimalarial activity: S. Thaithong, G. H. Beale, Bull. WHO63, 617 (1985). Series of articles on chemistry, pharmacology and antimalarial efficacy: China Cooperative Research Group on Qinghaosu, J. Tradit. Chin. Med.2, 3-50 (1982). Toxicity data: eidem,ibid. 31. Clinical trial in cerebral malaria in children: M. B. van Hensbroek et al.,N. Engl. J. Med.335, 69 (1996). Review: R. N. Price, Expert Opin. Invest. Drugs9, 1815-1827 (2000).
Properties: Crystals, mp 86-88°. [a]D19.5 +171° (c = 2.59 in CHCl3). LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu).
Melting point: mp 86-88°
Optical Rotation: [a]D19.5 +171° (c = 2.59 in CHCl3)
Toxicity data: LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu)
Therap-Cat: Antimalarial.
Keywords: Antimalarial.
Artemether is an antimalarial agent used in combination with lumefantrine for the treatment of acute uncomplicated malaria caused by Plasmodium falciparum.
Artemether is an antimalarial agent used to treat acute uncomplicated malaria. It is administered in combination with lumefantrine for improved efficacy. This combination therapy exerts its effects against the erythrocytic stages of Plasmodium spp. and may be used to treat infections caused by P. falciparum and unidentified Plasmodium species, including infections acquired in chloroquine-resistant areas.
Artemether is a natural product which effectively kills both malarial parasites P. falciparum and P. vivax. Artemether is usually used in combination with Lumefantrine for the treatment of malaria. Arthemether also kills trematodes of the species Schistosoma, providing protection against schistosomiasis. Sesquiterpene lactones like artemether, artesunate, and artemisinin have potential applications in certain types of cancer and inflammatory conditions.
Artemether causes relatively few side effects.[5] An irregular heartbeat may rarely occur.[5] While there is evidence that use during pregnancy may be harmful in animals, there is no evidence of concern in humans.[5] The World Health Organization (WHO) therefore recommends its use during pregnancy.[5] It is in the artemisinin class of medication.[5]
Haynes RK, Vonwiller SC: Extraction of artemisinin and artemisinic acid: preparation of artemether and new analogues. Trans R Soc Trop Med Hyg. 1994 Jun;88 Suppl 1:S23-6. Pubmed.
Malaria is a serious parasitic disease caused by Plasmodium parasites in the human body. Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malaria and Plasmodium knowlesi are the parasites that live in humans, of which P. vivax and P. falciparum are the most common.
Traditional anti-malarial drugs mainly include quinine, chloroquine, primaquine, and pyrimethamine. In 1972, the antimalarial active ingredient artemisinin extracted from the Compositae plant Artemisia annuaL by Chinese scientists is the most popular antimalarial effect after chloroquine, pyrimethamine, primary amine and sulfonamide. Drugs, especially for the treatment of cerebral malaria and anti-chloroquine malaria.
At present, a large number of artemisinin derivatives have been synthesized and screened for antimalarial activity. Artemether is a compound with excellent curative effect. In addition to the advantages of artemisinin’s quick effect and low toxicity, its solubility in oil is also higher than that of artemisinin. Artemisinin is large, which is especially beneficial for the preparation of preparations. Since artemether has two products, α and β epimers, and the antimalarial activity of artemether is mainly isomer β, so the industrial automation and intelligent production of β-artemether and the improvement of the process are realized. , reducing the impurities produced by the reaction, improving the quality of the product, and improving the purity of the product are the problems that need to be solved in today’s scientific research.
Patent CN104557965B discloses a preparation process of β-artemether, which mainly includes adding dihydroartemisinin and etherification reagent to alcohol to form a reaction system, and then adding acid to the reaction system for reaction. Water or non-alkaline aqueous solution is added to the reaction system to crystallize, namely β-artemether. The preparation process claims to effectively inhibit the production of isomer α-artemether in the reaction, and can make the etherification reaction proceed mildly, with simple post-treatment and high purity; although the purity of the product has been improved, the yield and Purity needs to be further improved.
Patent CN102731523B discloses a method for preparing β-artemether, which mainly includes the reaction of artemisinin under the action of a reducing agent to generate dihydroartemisinin, and the reaction of dihydroartemisinin with p-toluenesulfonic acid to generate β-artemisinin. The crude artemether is crystallized with methanol, ethanol, ethylene glycol or isopropanol, filtered, washed and dried. The method for preparing B-artemether of the invention has mild conditions, is environmentally friendly, is suitable for industrial production, and has a product yield of over 90 percent and a purity of 99.2 percent. The crystallization step of the invention adopts organic reagents, which adversely affects the quality control of subsequent products.
Patent CN103180325B discloses a method for preparing β-artemether, which uses dihydroartemisinin as a raw material and undergoes etherification reaction with trimethyl orthoformate in organic solvents including esters and alkanes to obtain β-artemether. The method of the invention is easy to control in process operation, high in yield, low in cost and high in product quality, and is suitable for industrial production. The method requires vacuum distillation, the obtained crude product needs to be redissolved with methanol, decolorized with activated carbon, etc., new impurities are easily introduced, the operation is not simple enough, and the efficiency is low.
Patent CN107793428A discloses a preparation method of artemether, hydrogenating artemisinin to obtain dihydroartemisinin, adding trimethyl orthoformate, reacting with boron trifluoride ether solution, slowly adding saturated sodium bicarbonate solution dropwise, The system was adjusted to neutrality, the liquids were separated, the aqueous phase was extracted with dichloromethane, the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure to obtain a solid; the obtained solid was dissolved in methanol, and an appropriate amount of activated carbon was added to obtain a solid. Reflux and decolorize, filter, add pure water dropwise to the filtrate, crystallize, wash with water, and dry to obtain artemether. However, this method requires steps such as extraction with an organic reagent dichloromethane and decolorization with activated carbon, which is cumbersome to handle.
Therefore, the following problems generally exist in the process of preparing β-artemether at present:
(1) when preparing β-artemether, the reaction time is longer, the impurities are large, and the purity and yield of the product are not high enough;
(2) The use of organic reagents in the subsequent purification process has a certain impact on the quality control of the product;
(3) The batch production equipment is adopted, the subsequent process steps are many, the degree of industrialization is low, the production efficiency is low, and it does not meet the requirements of GMP.
Example 1
This embodiment includes the following steps:
(1) at room temperature, add methanol 2400L in the 3000L stirred tank (1), then add 600kg of dihydroartemisinin through the solid feed pump, and circulate and disperse evenly;
(2) add etherification agent trimethyl orthoformate and acid catalyst acetyl chloride through three-way automatic feeding mixing reactor again, the volume ratio is 500:100:3, the mixing reactor control temperature is 5 ℃, and the flow rate of control feeding is 5L /min;
(3) in the continuous flow pipeline, enter the second mixer and add 5% sodium bicarbonate solution to neutralize, and the adding speed is 1.0L/min, and is filtered through the fine filter;
(4) Then directly enter the 2000L crystallization reaction kettle 11 with 300L of water added in advance and keep the temperature at 10°C. At the same time, purified water was added to the reaction kettle at a rate of 12L/min, and the crystallization was continued for 1.5h; the jacket of the crystallization kettle was fed with -10°C chilled water for 30min, and the temperature of the system was controlled to 5°C.
(5) centrifugal washing, obtaining crude artemether 704.5kg, drying to obtain artemether fine product 608.6kg, β-artemether purity 99.83%, α-artemether impurity 0.12%, and other single impurities less than 0.1%, The content is 99.8%, the mass yield is 96.1%, and the molar yield is 91.42%.
Example 2
This embodiment includes the following steps:
(1) at room temperature, 2400L of methanol was pumped into the 3000L reactor 1, and then 800kg of dihydroartemisinin was added by the solid feed pump, and the circulation was uniformly dispersed;
(2) add etherifying agent dimethyl phosphate and acid catalyst boron trifluoride ether through the three-way automatic feeding mixing reactor again, the volume ratio is 500:105:3.5, the mixing reactor control temperature is 3 ℃, and the control feeding flow rate is 3L/min;
(3) in the continuous flow pipeline, enter the second mixer and add 3% sodium bicarbonate solution to neutralize, and the speed of addition is 1.8L/min, through the fine filter;
(4) Directly enter the 2000L crystallization reaction kettles 11 and 12 with 300L of water added in advance and the temperature kept at 10°C. At the same time, purified water was added to the reaction kettle at 9 L/min, and the crystallization was continued for 2.5 hours; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 10 °C
(5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 96.2%, and the molar yield is 91.6%.
Example 3
This embodiment includes the following steps:
(1) 2400L of methanol was pumped into the 3000L reactor F1 at room temperature, and then 400kg of dihydroartemisinin was added through the solid feed pump, and the circulation was uniformly dispersed;
(2) Add etherification agent dimethyl phosphate and acid catalyst trimethylchlorosilane through the three-way automatic feeding mixing reactor, the volume ratio is 500:95:2.5, the mixing reactor is controlled at a temperature of 8 °C, and the feeding liquid is controlled to be added. The flow rate is 7L/min, and the reaction time is;
(3) in the continuous flow pipeline, enter the second mixer and add 8% sodium bicarbonate solution for neutralization, and the rate of addition is 0.6L/min, passing through the fine filter;
(4) Directly enter into the 2000L crystallization reactor J2 with 300L water added in advance and keeping the temperature at 10°C. At the same time, purified water was added to the reaction kettle at 15 L/min, and the crystallization was continued for 1 hour; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 0 °C
(5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 95.5%, and the molar yield is 90.9%.
Comparative Example 1
The difference between this embodiment and Example 1 is that hydrochloric acid is used instead of the acidic catalyst. Finally, 633.6kg of crude artemether was obtained, and 550.3kg of fine artemether was obtained by drying. The purity of β-artemether was 94.20%, and the impurities of α-artemether were 3.66%. %, and the molar yield was 80.6%.
Comparative Example 2
The difference between this embodiment and Example 1 is that the step of adding water in advance in the crystallization kettle is removed. Finally, 645.1kg of crude artemether was obtained, and 562.2kg of fine artemether was obtained by drying. The purity of β-artemether was 99.68%, the impurity of α-artemether was 0.22%, and the average of single and impurity was less than 0.1%. The mass yield was 88.7%. %, and the molar yield was 84.4%.
In Comparative Example 2, the step of adding water in advance in the crystallization was removed, the purity of β-artemether was 99.68%, and the yield was 88.7%. The yield dropped by 7.6%.
The above detailed description is a specific description of one of the feasible embodiments of the present invention, and this embodiment is not intended to limit the patent scope of the present invention. Any equivalent implementation or modification that does not depart from the present invention shall be included in the present invention. within the scope of the technical solution.
SYN1
Synthetic Reference
Continuous synthesis of artemisinin-derived medicines; Gilmore, Kerry; Kopetzki, Daniel; Lee, Ju Weon; Horvath, Zoltan; McQuade, D. Tyler; Seidel-Morgenstern, Andreas; Seeberger, Peter H. Chemical Communications (Cambridge, United Kingdom); Volume 50; Issue 84; Pages 12652-12655; Journal; 2014
SYN2
Synthetic Reference
An Improved Manufacturing Process for the Antimalaria Drug Coartem. Part I; Boehm, Matthias; Fuenfschilling, Peter C.; Krieger, Matthias; Kuesters, Ernst; Struber, Fritz; Organic Process Research & Development; Volume 11; Issue 3; Pages 336-340; Journal; 2007
SYN3
Synthetic Reference
Some transition metal complexes bearing artemisinin derivatives and (N-N-O) tridentate chromium (III) complexes ligated by 2-benzolmidazo-yl-6-acetyl-pyridines for catalytic behaviour towards ethylene; Obaleye, Joshua Ayoola; Amolegbe, Saliu Alao; Adewuyi, Sheriff; Sun, Wenhua; Oshodi, Margaret Damilola; Journal of Chemistry and Chemical Engineering; Volume 4; Issue 12; Pages 23-32; Journal; 2010
SYN4
Synthetic Reference
Method and apparatus for the synthesis of dihydroartemisinin and artemisinin derivatives; Kopetzki, Daniel; McQuade, David Tyler; Seeberger, Peter H.; Gilmore, Kerry; Assignee Max-Planck-Gesellschaft zur Foerderung der Wissenschaften e.V., Germany; 2015; Patent Information; Jan 21, 2015; EP 2826779 A1
An efficient one pot green synthesis of β-artemether/arteether from artemisinin has been developed using a sodium borohydride-cellulose sulfuric acid (CellSA) catalyst system. The green methodology is high yielding and the catalyst has good recyclability.
Fig. 2 Conventional approaches for synthesis of artemether from artemisinin.
Scheme 1 One-pot conversion of β-artemisinin to artemether.
Experimental section
Representative procedure for catalyst preparation
Preparation of cellulose sulfuric acid.To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 °C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder.17
General procedure for the arteether from artemisinin in one-pot
To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 ml) and trimethyl orthoacetate (0.5 ml) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 °C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40 ± 5 °C in water. The reaction mass was seeded with pure β-arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried.
General procedure for the artemether from artemisinin in one-pot
Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.
Preparation of cellulose sulfuric acid. To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 0 C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder. K General procedure for the arteether from artemisinin in one-pot. To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 mL) and trimethyl orthoacetate (0.5 mL) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 0 C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40± 5 0 C in water. The reaction mass was seeded with pure β–arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried. General procedure for the artemether from artemisinin in one-pot. Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.
Approximately, out of the 4 billion people suffering from malaria, 1-3 million, mostly children die every year worldwide. The rapidly spreading multidrug resistant parasite to standard quinoline based antimalarial drugs such as chloroquine and mefloquine based antimalarial complicate chemotherapy treatment of malaria patients.
Artemether is a methyl ether derivative of dihydroartemisinin. Dihydroartemisinin is derived from arternisinin, a novel sesquiterpene endoperoxide isolated from the plant Artemisia annua. Artemisinin and its derivative artemether, arteether, artelinate and artesunate a novel class of antimalarials derived from Artemisia annua are now proving their promising activity and being used for the treatment; of uncomplicated severe complicated/cerebral and multi drug resistant malaria.
Artemether, developed in France and China has undergone extensive preclinical, animal, toxicological studies as well as clinical studies. Artemether is more potential as compared to artemisinin and an antimalarial drug especially for treating multi drug resistant and complicated strains of Plasmodium falciparum.
Artemether shows rapid shizonticidal action with quicker parasite clearance rate, short half life less side effect and low recrudence rate. Brossi, et al (Brossi, A; Venugopalan, B, Domingueg, G L; Yeh, H. J. C; Flippend-Anderson, J. L.; Buchs, P; Luo, X. D.; Milhous,W and peters, W; J. Med. Chem. 31, 646-649, 1988) reported the preparation of arteether, the ethyl ether derivative of dihydroartemisinin in two steps: First artemisinin was reduced with an excess of sodium borohydride in methanol at 0 to −5 degree C. in 3 hours to dihydroartemisinin in 79% yield. In the second step arteether is prepared by dissolving the dihydroartemisinin in the solvent mixture of benzene and ethanol at 45 degree C. followed by addition of BF3 etherate and refluxing the reaction mixture at 70 degree C. for one hour. After completion of the reaction it was worked up, dried over anhydrous sodium sulphate with removal of the solvent dichloromethane. The reaction yielded arteether along with some impurities. Column chromatography of the reaction mixture over silica gel, 1:20 ratio yielded pure alpha and beta arteether in nearly qualitative yield.
EL-Feraly etal. (E L Feraly, F. S; Al-Yahya M A; Orabi, K. Y; Mc-Phail D R and Me Phail A. T. J.Nat.Prod. 55, 878-883 1992) reported the preparation of arteether by a process in which anhydrodihydroartemisinin, prepared from artemisinin was dissolved in absolute alcohol. The reaction mixture was stirred in the presence of p-toluene sulphonic acid used as a catalyst. On workup it yielded a mixture of beta arteether and C-11 epimer in the ratio of 3:1. In this process only beta arteether, is obtained and separation of C-11 epimer is difficult and preparation of anhydrodihydroartemisinin is a tedious process. The reaction took 22 hours to complete. The lewis acid catalyst used in this reaction is required in large amount (60 mg. acid catalyst by 100 mg. anhydrodihydroartemisinin).
In another method Bhakuni etal (Bhakuni, R. S.; Jain D. C and Sharma R. P. Indian. J. Chemistry, 34B, 529-30, 1995) arteether, artemether and other ether derivatives were prepared from dihydroartemisinin in different alcohol and benzene in the presence of chlorotrimethylsilane catalyst in 2-4 hours at room temperature. After workup of the reaction mixture and removal of the solvent, the impure reaction products were purified over silica gel column to obtained the pure mixture of alpha, beta ethers.
Another method is reported by Lin et al. (Lin, A. J. and Miller, R. E, J.Med Chero. 38,764-770, 1995) In this method the new ether derivatives were prepared by dissolving dihydroarternisinin in anhydrous ether and appropriate alcohol followed by BF3-etherate. The reaction mixture was stirred at room temperature for 24 hours. The yield of the purified products ranged from 40-90%. Purification was achieved by the use of silica gel chromatography.
Another method described by Jain et al (Jain D. C, Bhakuni R. S, Saxena S, kumar, S and Vishwakarma, R. A.) the preparation of arteether from artemisinin comprises: Reduction of artemisinin into dihydroartemisinin. Isolation of dihydroartemisinin. Acylation of dihydroartemisinin by dissolving it in alcohol and adding trialkylorthoformate in the reaction mixture, which produce ethers in quantitative yield in 10 hours at 40 degree C.
The above mentioned methods carry some disadvantages being less cost effective and more time consuming as compared to the present invention. Moreover, benzene, a carcinogenic solvent, used in the previous methods is not acceptable according to the health standard. Further, all the above methods require at least two separate steps to convert artemisinin into ethers i.e. reduction of the artemisinin into dihydroartemisinin in the first pot followed by isolation of dihydroartemisinin and then comes the second step of conversion of dihydroartemisinin into different ethers in the second pot. However, the present invention provide an efficient method for conversion of artemisinin into artemether
EXAMPLE 1
Artemisinin (3 g.) was dissolved in dry methanol (40 ml) at room temperature. It was cooled to −5 degree C. Now sodium borohydride (700 mg) was added slowly for 30 minutes and the reaction mixture was stirred for about 1.5 hours. The reaction was monitored by TLC to check completion of the reduction step. Now cation exchange resin (8 g) was added slowly at cooling temperature and the reaction mixture was further stirred at room temperature for about 2 hours. Cooled water was added to the reaction mixture and the resin was filtered.
The filtrate was neutralized with 5% sodium bicarbonate solution followed by extracting with dichloromethane (3×50 ml). The dichloromethane extract was dried over anhydrous sodium sulphate and evaporation of the solvent yielded 3.21 g, of artemether along with some impurities. The impure artemether was purified over silica gel column (1:5 ratio) in hexane:ethyl acetate (96:4) furnished pure alpha and beta artemether 2.43 g (81% w/w). Small portion of artemether was separated by prep TLC into alpha and beta isomers and characterized by the analysis of their IR, Mass and 1H NMR data.
EXAMPLE 2
The experiment was carried out following example 1 except in place of solid acid catalyst in the second reaction. Liquid acid catalyst chlorotrimethylsilane was added at cooling temperature for methylation reaction. The overall yield of pure alpha, beta artemether after column chromatography was 2.46 gm (82% w/w).
EXAMPLE 3
Artemisinin (100 g.) was dissolved in dry methanol (3 ml). Added sodium borohydride (30 mg.) at −5° C. The reaction mixture was stirred for 2 hours. After completion of the reaction, trifluroacetic acid (0.5 ml) was added and the reaction mixture was stirred for 5 hours. The methylation was incompleted and after workup the artemether was purified by prep TLC to yield 46 mg (46%) pure alpha, beta artemether.
EXAMPLE 4
The experiment was carried following example 1 except before column chromatography, the beta isomer (40%) was recrystallized in hexane from impure artemether and remaining mother liquor was purified over silica gel column in 1:5 ratio to yield alpha and beta artemether in 80% w/w.
The earlier developed flow protocol for stoichiometric reduction of an important biologically derived pharmaceutical precursor, artemisinin, to dihydroartemisinin was extended to a sequential reaction to produce one of the final APIs, artemether. A highly active heterogeneous catalyst was found for the etherification reaction. The use of QuadraSil catalyst allows to eliminate one step of reaction workup. A comparative Life Cycle Assessment of both reactions has shown advantages of the flow process over the optimized literature batch protocols. Results of LCA highlight the significance of solvents in pharmaceuticals manufacture and the advantage of flow technology, enabling small solvent inventories to be used.
In a previous episode chemical company Sanofi was granted exclusive access to certain yeast cells that produce a precursor to anti-malarial drug artemisinin. One of the charities making this all possible is the Bill and Melinda Gates Foundation. Another charity that has apparently entered into the drug business is the Clinton Health Access Initiative. Bill together with Rodger Stringham and David Teager report on an improved process for the conversion of artemisinin to artemether in Organic Process Research & Development (DOI). Does the Clinton Health Access Initiative have a pilot-plant facility or even an organic lab? Unless it is all cramped in suite 400 on Dorchester Avenue in Boston, the article is not very explicit. The acknowledgements mention Mangalam Drugs and Organics. Case at hand: artemether has the carbonyl group replaced by a methoxy group in a two-step reduction – methylation. So far so good. The point is that principal supplier Novartis reports up to 68% overall yields but that many Indian and Chinese suppliers working with the procedure generously supplied by same Novartis, report considerably lower figures (58-62%). But Why? And how can the process be improved? Any organic chemist knows reported yields in the literature should be considered with caution. Chemists tend to be over-optimistic / self-delusionional but this scenario was not considered. No bottlenecks were encountered in step 1, the reduction with sodium borohydride. Only the beta form was isolated due to its poor solubility in the quench. Drying the product without heat prevented formation of one byproduct. Moving on to step two, the methylation with HCl in methanol was more troublesome. The byproducts lurking around the corner are the anomer and the elimination product. Co-solvent (co-reagent?) trimethyl orthoformate made all the difference. The critical element in the workup was first adding more methanol before adding the base quench otherwise you end up with a nasty gum. The new record yield for the improved synthesis is 72%. But what have all these suppliers been doing wrong with the existing Novartis procedure? The answer to that question, remains unclear. The Novartis yield for step two with co-solvent methylacetate (not the formate) was confirmed so no surprise there.
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Artemether is an antimalarial drug for uncomplicated malaria caused by P. falciparum (and chloroquine-resistant P. falciparum) or chloroquine-resistant P. vivax parasites.[8] Artemether can also be used to treat severe malaria.[2]
Artemether can also be used in treating and preventing trematode infections of schistosomiasis when used in combination with praziquantel.[11]
Artemether is rated category C by the FDA based on animal studies where artemisinin derivatives have shown an association with fetal loss and deformity. Some studies, however, do not show evidence of harm.[12][13]
Side effects
Possible side effects include cardiac effects such as bradycardia and QT interval prolongation.[14] Also, possible central nervous system toxicity has been shown in animal studies.[15][16]
Drug interactions
Plasma artemether level was found to be lower when the combination product was used with lopinavir/ritonavir.[16] There is also decreased drug exposure associated with concurrent use with efavirenz or nevirapine.[17][18]
Artemether/lumefantrine should not be used with drugs that inhibit CYP3A4.[19]
Hormonal contraceptives may not be as efficacious when used with artemether/lumefantrine.[19]
Artemether interact with ferriprotoporphyrin IX (heme) or ferrous ions in the acidic parasite food vacuole, and generates cytotoxic radical species
The accepted mode of action of the peroxide containing drug involve its interaction with heme (byproduct of hemoglobin degradation), derived from proteolysis of haemoglobin. This interaction results in the formation of toxic oxygen and carbon centered radicals.
One of the proposed mechanisms is that through inhibiting anti-oxidant and metabolic enzymes, artemisinin derivatives inflict oxidative and metabolic stress on the cell. Some pathways affected may concern glutathione and glucose metabolism. As a consequence, lesions and reduced growth of the parasite may result.[20]
Another possible mechanism of action suggests that arteristinin drugs exert their cidal action through inhibiting PfATP6. Since PfATP6 is an enzyme regulating cellular calcium concentration, its malfunctioning will lead to intracellular calcium accumulation, which in turns causes cell death.[21]
Pharmacokinetics
Absorption of artemether is improved 2- to 3-fold with food. It is highly bound to protein (95.4%). Peak concentrations of artemether are seen 2 hours after administration.[4]
Artemether is metabolized in the human body to the active metabolite, dihydroartemisinin, primarily by hepatic enzymes CYP3A4/5.[4] Both the parent drug and active metabolite are eliminated with a half-life of about 2 hours.[4]
Chemistry
Artemether is a methylether derivative of artemisinin, which is a peroxide-containing lactone isolated from the antimalarial plant Artemisia annua. It is also known as dihydroartemisinin methyl ether, but its correct chemical nomenclature is (+)-(3-alpha,5a-beta,6-beta,8a-beta, 9-alpha,12-beta,12aR)-decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano(4,3-j)-1,2-benzodioxepin. It is a relatively lipophilic and unstable drug,[22] which acts by creating reactive free radicals in addition to affecting the membrane transport system of the plasmodium organism.[14]
^“Artemether and Lumefantrine”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 28 November 2016.
^World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. 2019. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Piola P, Nabasumba C, Turyakira E, et al. (2010). “Efficacy and safety of artemether—lumefantrine compared with quinine in pregnant women with uncomplicated Plasmodium falciparum malaria: an open-label, randomised, non-inferiority trial”. Lancet Infect Dis. 10 (11): 762–769. doi:10.1016/S1473-3099(10)70202-4. hdl:10144/116337. PMID20932805.
^ Kiang, Tony K. L.; Wilby, Kyle J.; Ensom, Mary H. H. (2013-10-26). “Clinical Pharmacokinetic Drug Interactions Associated with Artemisinin Derivatives and HIV-Antivirals”. Clinical Pharmacokinetics. 53 (2): 141–153. doi:10.1007/s40262-013-0110-5. ISSN0312-5963. PMID24158666. S2CID1281113.
^ Saeed, ME; Krishna, S; Greten, HJ; Kremsner, PG; Efferth, T (August 2016). “Antischistosomal activity of artemisinin derivatives in vivo and in patients”. Pharmacological Research. 110: 216–26. doi:10.1016/j.phrs.2016.02.017. PMID26902577.
The invention relates to triaminopyrimidine compd. of formula I, pharmaceutically acceptable salts thereof, hydrates, solvates, polymorphs, optically active forms thereof, in solid state forms useful for preventing or treating malaria. The invention also relates to a process for prepn. of triaminopyrimidine compd. and intermediates thereof. Compd. I was prepd. by condensation of 5-bromouracil with tert-Bu (R)-2-methylpiperazine-1-carboxylate to give tert-Bu (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine-1-carboxylate, which underwent chlorination followed by condensation with 1,5-dimethyl-1H-pyrazol-3-amine followed by condensation with 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride to give (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine, which underwent Boc-deprotection followed by methylation to give I.
Malaria is caused by protozoan parasites of the genus Plasmodium that infect and destroy red blood cells, leading to fever, severe anemia, cerebral malaria and, if untreated, death.
International (PCT) Publication No. WO 2015/165660 (the WO ‘660) discloses triaminopyrimidine compounds, intermediates, pharmaceutical compositions and methods for use for preventing or treating malaria. The WO ‘660 discloses a process for preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine (compound 5) as depicted in scheme-1.
Scheme 1
WO ‘660 discloses a process for preparation of triaminopyrimidine compounds depicted in scheme-2.
WO ‘660 discloses the preparation of compounds 8 and 4 by using microwave technique using Biotage microwave vial. WO ‘660 in example- 13, discloses the isolation of compound 1 by concentration of reaction mixture to obtain crude product, which was purified through reverse phase HPLC GILSON instrument to obtain pure solid compound 1 in 40.8% yield, without providing the purity of the solid compound 1. The process disclosed in WO ‘660 is not industrially advantageous as it requires microwave conditions as well as chromatographic purification and provides compound 1 with lower yields. The compound 1 prepared may not be suitable for pharmaceutical preparations based on various regulatory requirements.
Polymorphism, the occurrence of different crystalline forms, is a property of some molecules. A single molecule can exist in different crystalline forms having distinct physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – TGA, or different scanning calorimetry – DSC, Powder x-ray diffraction pattern – PXRD, infrared absorption – IR). One or more these techniques may be used to distinguish different polymorphic forms of a compound.
Different salts and solid states (e.g. solvates, hydrates) of an active pharmaceutical ingredient may possess different physio-chemical properties. Such variation in the properties of different salts and solid states forms may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (both chemical and polymorph) and shelf-life. These variations in the properties of different salts and solid states forms may offer improvements to the final dosage form for example, to improve bioavailability. Different salts and solid state forms of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms or amorphous form, which may in turn provide additional opportunities to assess variations in the properties and characteristics of an active pharmaceutical ingredient.
In view of the above, the present invention provides a process for the preparation of triaminopyrimidine compound 1 or pharmaceutically acceptable salts thereof or hydrates or solvates or polymorphs or optically active forms thereof, which is industrially scalable, environment friendly and efficient so as to obtain compounds of the invention in higher yields and purity.
The process for the preparation of triaminopyrimidine compound 1 or intermediates thereof of the present invention, takes the advantage by using appropriate solvent systems and isolation techniques as well as purification techniques, thereby to overcome problems of lower yields, chromatography purifications and microwave reactions of the prior art.
SUMMARY OF THE INVENTION
The present invention provides solid state forms of triaminopyrimidine compound
1,
1
Examples: Preparation of Intermediates
Example-1: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a 250 mL 4N round bottom flask, process water (30 ml) and cyclopropanecarboxylic acid (14.19 g, 164.88 mmol) were added at 25 to 35°C and started stirring. Sulphuric acid (4.4 ml, 82.44 mmol) was charged to the reaction mixture. Silver nitrate (4.18 g, 24.73 mmol), 6-Chloro-3-fluoro-2-methylpyridine (6 g, 41.22 mmol) were charged to the reaction mixture. Aqueous solution of ammonium persulphate (65.85 g, 288.54 mmol in 90 mL water) was added to the reaction mixture in 30 to 60 min at temperature NMT 60 °C. After the completion of the reaction as monitored by HPLC, toluene (30 ml) was added to the reaction mixture and stirred for 15 min. The reaction mixture filtered, separated layers from filtrate and extracted aqueous layer using toluene (30 mL). The organic layer was washed with aqueous sodium carbonate solution (30 mL) and water. The organic layer was distilled completely under vacuum at 60 °C to obtain 3.37 g syrupy mass as titled compound.
Example-2: Preparation of 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine
In a suitable glass assembly, process water (7.5 L) and cyclopropanecarboxylic acid (3.55 Kg, 41.24 mol) were added at 25 to 35 °C and stirred. Sulphuric acid (2.02 Kg, 20.59 mol), silver nitrate (1.05 Kg, 6.21 mol), 6-chloro-3-fluoro-2-methylpyridine (1.5 Kg, 10.3 mol) were added to the reaction mixture. Aqueous solution of ammonium persulphate (16.46 g, 72.13 mmol in 22.5 L water) was added to the reaction mixture at 55 to 60 °C and maintained. After the completion of the reaction as monitored by HPLC, toluene (7.5 L) was added to the reaction mixture and stirred for 15 min. The reaction mixture was filtered, organic layer was separated and aqueous layer was extracted using toluene (6 L), filtered the reaction mixture and washed the solid with toluene (1.5 L). The combined organic layer was washed with 20% sodium carbonate solution (9 L) and water. The organic layer was concentrated completely under vacuum at 60 °C to obtain 880 g (86.50%) syrupy mass of titled compound.
Example-3: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a 100 mL 3N round bottom flask, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (2.69 g, 14.48 mmol) and toluene (30 mL) were added at 25 to 35 °C. Diphenylmethanimine (3.15 g, 17.38 mmol) was charged to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (270 mg, 0.43 mmol) and palladium acetate (98 mg, 0.43 mmol) were added to the reaction mixture. Sodium-ie/ -butoxide (2.78 g, 28.96 mmol) was added to the reaction mixture and heated to 100 to 110° C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C and filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-4: Preparation of N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenyl-methanimine
In a suitable assembly, 6-chloro-4-cyclopropyl-3-fluoro-2-methylpyridine (880) and toluene (7.5 L) were added at 25 to 35 °C. Diphenylmethanimine (787 g, 4.34 mmol) and BOC anhydride (237 g, 1.086 mol) was added to the reaction mixture and stirred for 5-10 min under nitrogen purging. Racemic BINAP (67.6 g, 0.108 mmol) and palladium acetate (24.4 g, 0.108 mol) were added to the reaction mixture. S odium- ieri-butoxide (870 g, 9.05 mol) was added to the reaction mixture and heated to 100 to 110 °C under nitrogen. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C, water (6 L) was added. The reaction mixture was filtered over hyflo bed and washed with toluene. The filtrate containing titled compound was preserved for next stage of reaction.
Example-5: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a 100 mL 3N round bottom flask, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-3 was added water (25 mL) at 25 to 35° C. The cone. HCl (3 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride, charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (9 mL) and ethyl acetate (9 mL) was added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 1.62 g title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity.
Example-6: Preparation of 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride monohydrate
In a suitable glass assembly, N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-l,l-diphenylmethanimine in toluene as obtained in example-4 was added water (6 L) at 25 to 35° C. The cone. HCl (750 mL) was charged to the reaction mixture and heated to 40 to 50 °C. After the completion of the reaction as monitored by HPLC, the reaction mixture was cooled to 25 to 35 °C. Layers were separated. The aqueous layer was treated with methylene dichloride (3 L) and pH was adjusted to 7.5 to 8.5 using sodium carbonate solution, stirred for 15 min and layers were separated. Aqueous layer was extracted with methylene dichloride (3 L), charcoaled and acidified to pH 3 to 4 with aqueous HCl. The solvent was distilled completely and acetonitrile (1.5 L) and ethyl acetate (1.5 L) were added. The reaction mixture was stirred for 1 hour at 25 to 35° C. The product was filtered and washed with ethyl acetate. The product was dried at 50° C for 4 hours under vacuum to obtain 489 g (96.80%) title compound as monohydrate yellow crystalline solid having 99.51% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.5), Differential scanning calorimetry (FIG.6) and Thermogravimetric analysis (FIG.7).
Example 7: Preparation of 2,3-dibromobutanenitrile
In a 2 L round bottom flask, dichloromethane (550 mL) and 2-butenenitrile 110 g
(1.64 mol) were cooled to 20 to 25 °C. A solution of bromine 275 g (1.72 mol) in dichloromethane (220 mL) was dropwise added at 20 to 25 °C. Hydrobromic acid 1.43 ml (0.0082 mol) in acetic acid (33%) solution was added into the reaction mixture and stirred for 4 hours. After the completion of reaction, Na2S203 (550 mL) 4% aqueous solution was added and the reaction mixture was stirred for 15 min. The separated organic layer was distilled under vacuum completely to obtain 364.2 g (97.9%) of title compound as an oil.
Example 8: Preparation of l,5-dimethyl-lH-pyrazol-3-amine
In a 5 L round bottom flask, water (1. 36 L), sodium hydroxide 340 g (8.99 mol) were added and the reaction mixture was cooled to 0 to 5°C. A solution of methyl hydrazine sulphate 237.8 g (1.65 mol) in 680 mL water was added dropwise to the reaction mixture and stirred below 10 °C. 2,3-dibromobutanenitrile 340 g (1.5 mol) prepared in example-7 was added and the reaction mixture was stirred below 10 °C for 2 hours. After the completion of reaction, toluene (630 mL) was added and the reaction mixture was stirred for 15 min. The aqueous layer was separated and the organic layer was removed. The aqueous layer was extracted with dichloromethane (5.1 L). The combined organic layer was distilled completely under vacuum to obtain residue. Diisopropyl ether (680 mL) was added and the reaction mixture was stirred at 0 to 5 °C for 1 hour. The reaction mixture was filtered, washed with diisopropyl ether and dried to obtained 121.5 g (72.93%) of title compound having 95.63% purity.
Examples: Preparation of triaminopyrimidine compounds
Example-9: Preparation of tert-butyl (R)-4-(2,4-dioxo-l,2,3,4-tetrahydro- pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 2 L four neck round bottom flask, 1.25 Kg (6.545 mol) 5-bromouracil, 1.87 Kg (9.360 mol) tert-butyl (R)-2-methylpiperazine-l-carboxylate and 5L pyridine were added at 25 to 35° C. The reaction mass was stirred for 15 hours at 115 to 120°C. After completion, the reaction mass was cooled to 25 to 35°C. 12.5 L water was added and stirred for 1 hour. The reaction mass was filtered, washed with 2.5 L water and dried to obtain 1.37 Kg (67.4%) of title compound.
Example-10: Preparation of tert-butyl (R)-4-(2,4-dichloropyrimidin-5-yl)-2-methylpiperazine- 1 -carboxylate
In 20 L four neck round bottom flask, 1.36 Kg (4.382 mmol) tert-butyl (R)-4-(2,4-dioxo-1, 2,3, 4-tetrahydropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate and 6.8 L phosphorus oxychloride were added at 25 to 35° C. 26.5 mL pyridine (0.329 mol) was added and the reaction mass was heated to 105 to 110 °C and stirred for 4 hours. After the completion of the reaction, phosphorus oxychloride was distilled completely at atmospheric pressure. 2.72 L acetone was added and the reaction mixture was quenched into 4.08 L water. Acetone was removed by distillation under vacuum. 20% sodium carbonate solution was added to adjust pH 7.5-8.5 of the reaction mixture. 1.14 Kg (5.258 mol) di-tert-butyl dicarbonate and 9.52 L ethyl acetate were added and stirred for 2 hours at 25 to 35 °C. After the completion of the reaction, the organic layer was separated and aqueous layer was extracted with 6.8 L ethyl acetate. The combined ethyl layers were distilled to remove ethyl acetate completely under vacuum to obtain residue. 1.36 L isopropyl alcohol was added to the residue and isopropyl alcohol was removed completely. 4.08 L isopropyl alcohol and 6.8 L water were added to the residue and stirred for 1 hour. The reaction mass was filtered, washed with water and dried to obtain 1.25 Kg of title compound.
Example-11: Preparation of tert-butyl (R)-4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 640 g (1.843 mol) tert-butyl (R)-4-(2, 4-dichloropyrimidin-5-yl)-2-methylpiperazine-l -carboxylate, 225.3 g (2.027 g) 1,5-dimethyl-lH-pyrazol-3-amine and 9.6L toluene were added at 25 to 35°C. 1.2 Kg (3.686 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 12.41 g (0.0553 mol) palladium acetate and 34.43 g (0.0553 mol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added and the reaction mass was maintained for 16 hours at 110 to 115 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed the bed with 1.28 L toluene. Toluene was distilled completely and 2.56 L dichlromethane was added. The compound was adsorbed by 1.92 Kg silica gel (60-120 mesh). The dichloromethane was distilled completely under vacuum and 12.8 L mixture of ethyl acetate and hexane was added to the residue and stirred for 2 hours. The silica gel was filtered and the filtrate was distilled completely under vacuum to obtain 595 g title compound.
Example-12: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 20 L round bottom flask, 595 g (1.40 mol) tert-butyl (R)- 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 305 g (1.38 mol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride and 11.5 L toluene were added at 25 to 35°C. 1.08 Kg (3.32 mol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. 17.21 g (27.6 mmol) palladium acetate and 6.21 g (27.6 mmol) racemic 2,2′-bis(diphenylphosphino)-l, -binaphthyl were added. The reaction mass was stirred for 6 hours at 110 tol l5 °C under nitrogen. After the completion of the reaction, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
Example-13: Preparation of tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate
In 500 mL four neck round bottom flask, 7.5 g (17.77 mmol) (R)-tert-butyl 4-(2-chloro-4-[(l,5-dimethyl-lH-pyrazol-3-yl)amino)pyrimidin-5-yl]-2-methylpiperazine-l-carboxylate, 3.92 g (17.77 mmol) 4-cyclopropyl-5-fluoro-6-methylpyridin-2-amine hydrochloride compound and 150 mL toluene were added at 25 to 35 °C. 20 g (61.3 mmol) cesium carbonate was added. The reaction mixture was purged for 15 min under nitrogen. Then, 130 mg (0.58 mmol) palladium acetate and 360 mg (0.58 mmol) racemic 2,2′-bis(diphenylphosphino)-l,l’-binaphthyl were added. The reaction mass was stirred for 18 hours at 110 to 115° C under nitrogen. After completion, the reaction mixture was filtered through a celite bed and washed with toluene. The filtrate was used as such in the next step without further treatment.
In 50 L glass assembly, the filtrate containing tert-butyl (R)-4-(2-((4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)amino)-4-((l,5-dimethyl-lH-pyrazol-3-yl)amino) pyrimidin-5-yl)-2-methylpiperazine-l-carboxylate from example 13 was taken. 11.5 L water and 1.28 L Cone. HC1 were added at 25 to 35 °C. The reaction mass was stirred for 2 hours at 50 to 55 °C. After the completion of the reaction, reaction mixture was cooled to room temperature and filtered over celite bed and washed with water. The separated the aqueous layer from filtrate was basified by using 20% sodium carbonate solution and extracted with 12.8 L methylene dichloride. The organic layer was distilled completely under vacuum to obtain residue. 9.6 L acetonitrile was added to the residue and heated to reflux for 30 min. The reaction mixture was cooled and stirred at 25 to 35 °C for 1 hour. The reaction mixture was filtered, washed with 640 mL acetonitrile and dried to obtain 360 g titled compound.
In 250 mL four neck round bottom flask, 4.7 g (10.4 mmol) (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine was dissolved in 56 mL ethanol. 1.89 g (23.32 mmol) formaldehyde and 1.44 g (22.90 mmol) sodium cyanoborohydride were added. Adjusted pH 5-6 using acetic acid and stirred the reaction mass at 25 to 35 °C for 2 hours. After completion, ethanol was distilled completely under vacuum. 47 mL water was added to the residue. The reaction mass was basified by 20% sodium carbonate solution and extracted with methylene dichloride. Both the organic layers were combined and distilled completely under vacuum. 94 mL acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mass was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 5 mL acetonitrile and dried to obtain 3.7 g title compound as crystalline solid, having HPLC purity of about 99.61%.
In 20 L round bottom flask, 725 g (1.60 mol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazine-l-yl)pyrimidine-2,4-diamine was dissolved in 6.52 L dichloromethane. 261.5 g (3.2 mol) formaldehyde and 510.4 g (2.4 mol) sodium triacetoxyborohydride were added and stirred the reaction mixture at 25 to 35 °C for 2 hours. After the completion of the reaction, 3.63 L water was added into the reaction mixture. The reaction mixture was basified by 20% sodium carbonate solution and the organic layer was separated. The aqueous layer was extracted with 1.45 L methylene dichloride. The combined organic layers were distilled completely under vacuum. 14.5 L acetonitrile was added to the residue and heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35° C and stirred for 1 hour. The reaction mass was filtered, washed with 1.45 L acetonitrile and dried to obtain 632 g of title compound as crystalline solid having 99.01% HPLC purity. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.l) and Differential Scanning Calorimetry (FIG.2).
2 4
Example-17: Preparation of (R)-N -(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N -(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine In a 10 mL round bottom flask, 300 mg (0.644 mmol) (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(l,5-dimethyl-lH-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-l-yl)pyrimidine-2,4-diamine, 2.7 mL acetonitrile and 0.3 mL water were added and the reaction mixture was heated to reflux for 15 min. The reaction mixture was cooled to 25 to 35 °C and stirred for 1 hour. The reaction mass was filtered, washed with acetonitrile and dried to obtain 201 mg (67%) title compound as crystalline solid. The crystalline compound is characterized by Powder x-ray diffraction pattern (FIG.3) and Differential Scanning Calorimetry (FIG.4).
In a 50 mL round-bottomed flask (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (190 mg, 0.42 mmol, Example 2) was taken in DCM (2 mL) to give a yellow suspension. To this Hunig’s Base (0.184 mL, 1.05 mmol) was added and the suspension turned clear. After 10 minutes, it turned into a white suspension. After another 10 minutes, the mixture was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 mL) and formaldehyde (0.042 mL, 0.63 mmol) was added and stirred for 10 minutes. White suspension slowly cleared to yellow solution. To this clear solution sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get white suspension. After 30 minutes LCMS showed completion of reaction. The reaction mixture was concentrated and the crude was purified through reverse phase HPLC GILSON instrument to get the pure solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8 %).1H NMR (300
Hameed P., S., Solapure, S., Patil, V. et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun6, 6715 (2015). https://doi.org/10.1038/ncomms7715
The widespread emergence of Plasmodium falciparum (Pf) strains resistant to frontline agents has fuelled the search for fast-acting agents with novel mechanism of action. Here, we report the discovery and optimization of novel antimalarial compounds, the triaminopyrimidines (TAPs), which emerged from a phenotypic screen against the blood stages of Pf. The clinical candidate (compound 12) is efficacious in a mouse model of Pf malaria with an ED99 <30 mg kg−1 and displays good in vivo safety margins in guinea pigs and rats. With a predicted half-life of 36 h in humans, a single dose of 260 mg might be sufficient to maintain therapeutic blood concentration for 4–5 days. Whole-genome sequencing of resistant mutants implicates the vacuolar ATP synthase as a genetic determinant of resistance to TAPs. Our studies highlight the potential of TAPs for single-dose treatment of Pf malaria in combination with other agents in clinical development.
(A) Pyridine, microwave, 150 °C, 45 min. (B) (i) POCl3, reflux, 6 h (ii) sodium carbonate, di-tert-butyl dicarbonate, room temperature, 16 h. (C) N,N-Diisopropylethylamine (DIPEA), ethanol, microwave, 110 °C, 1 h. (D) (i) Potassium tert-butoxide, 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP), pd2(dba)3, toluene, reflux, 12 h. (E) HCl (4 N) in dioxane, 15–30 min. (F) Compound 9, DIPEA, dichloromethane, formaldehyde (HCHO), sodium cyanoborohydride, 15 min.
Synthesis of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3, 4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (12). (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1,5-dimethyl-1H-pyrazol-3-yl)-5-(3-methylpiperazin-1-yl)pyrimidine-2,4-diamine hydrochloride (compound 9, 190 mg, 0.42 mmol) was taken in dichloromethane (2 ml) to give a yellow suspension. To this Hunig’s Base (0.184 ml, 1.05 mmol) was added and the suspension turned clear. After 10 min of stirring, reaction mixture turned into a white suspension and then it was concentrated to dryness. Resultant residue was dissolved in ethanol (absolute, 99.5%) (3 ml), and formaldehyde (0.042 ml, 0.63 mmol) was added and stirred for 10 min. To this clear solution, sodium cyanoborohydride (26.4 mg, 0.42 mmol) was added in one portion to get a white suspension. The reaction mixture was concentrated and the crude product was purified through reverse-phase chromatography to get the pure off-white solid of (R)-N2-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-N4-(1, 5-dimethyl-1H-pyrazol-3-yl)-5-(3,4-dimethylpiperazin-1-yl)pyrimidine-2,4-diamine (80 mg, 40.8%). Yield: 40.8%, purity: >95% by HPLC (ultraviolet at 220 and 254 nm). 1H NMR (300 MHz, DMSO-d6) δ 9.26 (s,1H), 8.03 (s, 1H) 8.00 (s, 1H) 7.67 (d, J=5.1 Hz, 1H) 6.83 (s, 1H) 3.33 (s, 3H) 2.96–2.73 (m, 4H) 2.75–2.50 (m, 1H) 2.38–2.30 (m, 4H) 2.23 (s, 7H) 2.10–1.96 (m, 1H),1.08–1.02 (m, 2H) 1.00 (d, J=6.2 Hz, 3H) 0.78–0.67 (m, 2H). 13C-NMR (126 MHz, DMO-d6) δ 155.30, 154.67, 152.10, 150.93, 148.98, 146.81. 145.29, 141.95, 140.31, 138.81, 124.91, 106.20, 97.07, 58.78, 51.87, 42.16, 35.28, 17.23. 10.99 and 8.77, HRMS (ESI): m/z calculated for C24H32FN9+H [M+H]: 466.2765. Found: 466. 2838. Traces of LC-MS, HRMS, 1H NMR and 13C-NMR of compound 12 are shown in Supplementary Figs 1–3.
Product vision
Uncomplicated malaria treatment and resistance management
MoA
Unknown
Key features
Predicted human dose 900mg for a 9-log parasite killing
Low resistance potential from in vitro studies
Challenges
Synthesis and cost of goods
Status
First-in-human study started in February 2019
Next milestone
Initiate phase IIb study of ZY19489 with FQ
Previously
Discovery partnership between MMV and AstraZeneca, Bangalore
Name AZ13721412; full reference name is MMV674253
Zydus receives Orphan Drug Designation from USFDA for ZY-19489, a novel compound to treat malaria;
ZY19489 is a novel antimalarial compound active against all current clinical strains of P. falciparum and P. vivax, including drug-resistant strains.
December 16, 2021 11:38 IST | India Infoline News Service
Zydus Cadila listed as Cadila Healthcare Limited announced that its antimalarial compound ZY19489 (MMV253), currently in development together with Medicines for Malaria Venture (MMV), a leading product development partnership (PDP) in antimalarial drug research, has received Orphan Drug Designation from the USFDA.
Orphan drug designation provides eligibility for certain development incentives, including tax credits for qualified clinical testing, prescription drug user fee exemptions, and seven-year marketing exclusivity upon FDA approval.
The company said that the Phase I study of ZY19489 has demonstrated a long half-life and potential for a single-dose cure for malaria. In a separate malaria challenge trial, potent antimalarial activity has been demonstrated following single-dose oral administration of ZY19489.
“As a global community facing threats from rapidly mutating malaria strains and the rise in artemisinin resistance cases, we have to be prepared with novel therapeutic drugs. ZY-19489 is a potential single dose radical cure for P. falciparum and P. vivax malaria which is a major global health risk today,” Pankaj R. Patel, Chairman, Zydus Group, said.
“ZY19489 is a potent, first in class molecule, originally discovered and elaborated in India” said Dr. Timothy Wells, Chief Scientific Officer, MMV. “It has tremendous potential as part of a new generation of treatments and is fully active against drug resistant strains of malaria which are increasingly a concern.”
Artemisinin resistance is seen as a mounting challenge to the global fight against malaria. ZY19489 is being developed to provide an effective alternative to the current front-line antimalarial drugs for the treatment of P. falciparum and P. vivax malaria, as artemisinin-based combination therapies (ACTs) are under threat of resistance.
As per the World Malaria Report 2021, there were an estimated 241 million cases of malaria worldwide and the estimated number of malaria deaths stood at 627,000 in 2020. A major health concern, it is estimated that a child dies from malaria every minute. About 96% of malaria deaths globally were in 29 countries. India accounted for about 82% of all malaria deaths in the WHO South-East Asia Region.
Graphical depiction of circumsporozoite (CSP) and RTS,S structures. CSP comprises an N-terminal region containing a signal peptide sequence and Region I that binds heparin sulfate proteoglycans and has embedded within it a conserved five amino acid (KLKQP) proteolytic cleavage site sequence; a central region containing four-amino acid (NANP/NVDP) repeats; and a C-terminal region containing Region II [a thrombospondin (TSP)-like domain] and a canonical glycosylphosphatidylinositol (GPI) anchor addition sequence. The region of the CSP included in the RTS,S vaccine includes the last 18 NANP repeats and C-terminus exclusive of the GPI anchor addition sequence. Hepatitis B virus surface antigen (HBsAg) monomers self-assemble into virus-like particles and approximately 25% of the HBsAg monomers in RTS,S are genetically fused to the truncated CSP and serve as protein carriers. The CSP fragment in RTS,S contains three known T-cell epitopes: a highly variable CD4 + T-cell epitope before the TSP-like domain (TH2R), a highly variable CD8 + T-cell epitope within the TSP-like domain (TH3R), and a conserved “universal” CD4 + T cell epitope (CS.T3) at the C-terminus. (Figure courtesy of a recent publication16 and open access, PATENTWO 2009080715
Example 1Recipe for component for a single pediatric dose of RTS, S malaria vaccine (2 vial formulation)Component AmountRTS,S 25μgNaCl 2.25mgPhosphate buffer (NaZK2) 1OmMMonothioglycerol 125μgWater for Injection Make volume to 250 μLThe above is prepared by adding RTS, S antigen to a mix of Water for Injection, NaCl 150OmM, phosphate buffer (NaZK2) 50OmM (pH 6.8 when diluted x 50) and an aqueous solution of monothioglycerol at 10%. Finally pH is adjusted to 7.0 ± 0.1.This may be provided as a vial together with a separate vial of adjuvant, for example a liposomal formulation of MPL and QS21Component Amount l,2-di-oleoyl-5/?-glycero-3-phosphocholine (DOPC) 500 μgCholesterol 125 μgMPL 25 μgQS21 25 μgNaCl 2.25mg Phosphate buffer (NaZK2) 1 OmMWater for Injection Make volume to250 μLFor administration the adjuvant formulation is added to the component formulation, for example using a syringe, and then shaken. Then the dose is administered in the usual way. The pH of the final liquid formulation is about 6.6 +/- 0.1.Example IAA final pediatric liquid formulation (1 vial) according to the invention may be prepared according to the following recipe.Component AmountRTS,S 25μgNaCl 4.5mgPhosphate buffer (NaZK2) 1OmMMonothioglycerol 125μg1 ,2-di-oleoyl-5/?-glycero-3-phosphocholine (DOPC) 500 μgCholesterol 125 μgMPL 25 μgQS21 25 μgWater for Injection Make volume to500 μLThe pH of the above liquid formulation is either adjusted to 7.0 +/- 0.1 (which is favorable for antigen stability, but not favorable at all for the MPL stability), or to 6.1 +/- 0.1 (which is favorable for MPL stability, but not favorable at all for RT S, S stability). Therefore this formulation is intended for rapid use after preparation.The above is prepared by adding RTS, S antigen to a mix of Water for Injection, NaCl 150OmM, phosphate buffer (NaZK2) 50OmM (pH 6.8 when diluted x 50) and an aqueous solution of monothioglycerol at 10%. Then a premix of liposomes containing MPL with QS21 is added, and finally pH is adjusted. Example IBA final adult dose (1 vial formulation) for the RTS, S according to the invention may be prepared as follows:Component AmountRTS,S 50μgNaCl 4.5mgPhosphate buffer (NaZK2) 1OmMMonothioglycerol 250μg1 ,2-di-oleoyl-5/?-glycero-3-phosphocholine (DOPC) 1000 μgCholesterol 250 μgMPL 50 μgQS21 50 μgWater for Injection Make volume to500 μLExample 1CExample 1C may prepared by putting Example 1, IA or IB in an amber vial, for example flushed with nitrogen before filing.
The World Health Organization (WHO) is recommending widespread use of the RTS,S/AS01 (RTS,S) malaria vaccine among children in sub-Saharan Africa and in other regions with moderate to high P. falciparum malaria transmission. The recommendation is based on results from an ongoing pilot programme in Ghana, Kenya and Malawi that has reached more than 800 000 children since 2019.
“This is a historic moment. The long-awaited malaria vaccine for children is a breakthrough for science, child health and malaria control,” said WHO Director-General Dr Tedros Adhanom Ghebreyesus. “Using this vaccine on top of existing tools to prevent malaria could save tens of thousands of young lives each year.”
Malaria remains a primary cause of childhood illness and death in sub-Saharan Africa. More than 260 000 African children under the age of five die from malaria annually.
In recent years, WHO and its partners have been reporting a stagnation in progress against the deadly disease.
“For centuries, malaria has stalked sub-Saharan Africa, causing immense personal suffering,” said Dr Matshidiso Moeti, WHO Regional Director for Africa. “We have long hoped for an effective malaria vaccine and now for the first time ever, we have such a vaccine recommended for widespread use. Today’s recommendation offers a glimmer of hope for the continent which shoulders the heaviest burden of the disease and we expect many more African children to be protected from malaria and grow into healthy adults.”
WHO recommendation for the RTS,S malaria vaccine
Based on the advice of two WHO global advisory bodies, one for immunization and the other for malaria, the Organization recommends that:
WHO recommends that in the context of comprehensive malaria control the RTS,S/AS01 malaria vaccine be used for the prevention of P. falciparum malaria in children living in regions with moderate to high transmission as defined by WHO. RTS,S/AS01 malaria vaccine should be provided in a schedule of 4 doses in children from 5 months of age for the reduction of malaria disease and burden.
Summary of key findings of the malaria vaccine pilots
Key findings of the pilots informed the recommendation based on data and insights generated from two years of vaccination in child health clinics in the three pilot countries, implemented under the leadership of the Ministries of Health of Ghana, Kenya and Malawi. Findings include:
Feasible to deliver: Vaccine introduction is feasible, improves health and saves lives, with good and equitable coverage of RTS,S seen through routine immunization systems. This occurred even in the context of the COVID-19 pandemic.
Reaching the unreached: RTS,S increases equity in access to malaria prevention.
Data from the pilot programme showed that more than two-thirds of children in the 3 countries who are not sleeping under a bednet are benefitting from the RTS,S vaccine.
Layering the tools results in over 90% of children benefitting from at least one preventive intervention (insecticide treated bednets or the malaria vaccine).
Strong safety profile: To date, more than 2.3 million doses of the vaccine have been administered in 3 African countries – the vaccine has a favorable safety profile.
No negative impact on uptake of bednets, other childhood vaccinations, or health seeking behavior for febrile illness. In areas where the vaccine has been introduced, there has been no decrease in the use of insecticide-treated nets, uptake of other childhood vaccinations or health seeking behavior for febrile illness.
High impact in real-life childhood vaccination settings: Significant reduction (30%) in deadly severe malaria, even when introduced in areas where insecticide-treated nets are widely used and there is good access to diagnosis and treatment.
Highly cost-effective: Modelling estimates that the vaccine is cost effective in areas of moderate to high malaria transmission.
Next steps for the WHO-recommended malaria vaccine will include funding decisions from the global health community for broader rollout, and country decision-making on whether to adopt the vaccine as part of national malaria control strategies.
Financial support
Financing for the pilot programme has been mobilized through an unprecedented collaboration among three key global health funding bodies: Gavi, the Vaccine Alliance; the Global Fund to Fight AIDS, Tuberculosis and Malaria; and Unitaid.
Note to editors:
The malaria vaccine, RTS,S, acts against P. falciparum, the most deadly malaria parasite globally, and the most prevalent in Africa.
The Malaria Vaccine Implementation Programme is generating evidence and experience on the feasibility, impact and safety of the RTS,S malaria vaccine in real-life, routine settings in selected areas of Ghana, Kenya and Malawi.
Pilot malaria vaccine introductions are led by the Ministries of Health of Ghana, Kenya and Malawi.
The pilot programme will continue in the 3 pilot countries to understand the added value of the 4th vaccine dose, and to measure longer-term impact on child deaths.
The Malaria Vaccine Implementation Programme is coordinated by WHO and supported by in-country and international partners, including PATH, UNICEF and GSK, which is donating up to 10 million doses of the vaccine for the pilot.
The RTS,S malaria vaccine is the result of 30 years of research and development by GSK and through a partnership with PATH, with support from a network of African research centres.
The Bill & Melinda Gates Foundation provided catalytic funding for late-stage development of RTS,S between 2001 and 2015.
RTS,S/AS01 (trade name Mosquirix) is a recombinant protein-based malaria vaccine. In October 2021, the vaccine was endorsed by the World Health Organization (WHO) for “broad use” in children, making it the first malaria vaccine candidate, and first vaccine to address parasitic infection, to receive this recommendation.[3][4][5]
Potential malaria vaccines have been an intense area of research since the 1960s.[12] SPf66 was tested extensively in endemic areas in the 1990s, but clinical trials showed it to be insufficiently effective.[13] Other vaccine candidates, targeting the blood-stage of the malaria parasite’s life cycle, have also been insufficient on their own.[14] Among several potential vaccines under development that target the pre-erythrocytic stage of the disease, RTS,S has shown the most promising results so far.[15]
Approval history
The EMA approved the RTS,S vaccine in July 2015, with a recommendation that it be used in Africa for babies at risk of getting malaria. RTS,S was the world’s first malaria vaccine to get approval for this use.[16][8] Preliminary research suggests that delayed fractional dosing could increase the vaccine’s efficacy up to 86%.[17][18]
On 17 November 2016, WHO announced that the RTS,S vaccine would be rolled out in pilot projects in three countries in sub-Saharan Africa. The pilot program, coordinated by WHO, will assess the extent to which the vaccine’s protective effect shown in advanced clinical trials can be replicated in real-life settings. Specifically, the programme will evaluate the feasibility of delivering the required four doses of the vaccine; the impact of the vaccine on lives saved; and the safety of the vaccine in the context of routine use.[19]
Vaccinations by the ministries of health of Malawi, Ghana, and Kenya began in April and September 2019 and target 360,000 children per year in areas where vaccination would have the highest impact. The results are planned to be used by the World Health Organization to advise about a possible future deployment of the vaccine.[10][11][20] In 2021 it was reported that the vaccine together with other anti-malaria medication when given at the most vulnerable season could reduce deaths and illness from the disease by 70%.[21][22]
Funding
RTS,S has been funded, most recently, by the non-profit PATH Malaria Vaccine Initiative (MVI) and GlaxoSmithKline with funding from the Bill and Melinda Gates Foundation.[23] The RTS,S-based vaccine formulation had previously been demonstrated to be safe, well tolerated, immunogenic, and to potentially confer partial efficacy in both malaria-naive and malaria-experienced adults as well as children.[24]
Components and mechanism
The RTS,S vaccine is based on a protein construct first developed by GlaxoSmithKline in 1986. It was named RTS because it was engineered using genes from the repeat (‘R’) and T-cell epitope (‘T’) of the pre-erythrocytic circumsporozoite protein (CSP) of the Plasmodium falciparum malaria parasite together with a viral surface antigen (‘S’) of the hepatitis B virus (HBsAg).[7] This protein was then mixed with additional HBsAg to improve purification, hence the extra “S”.[7] Together, these two protein components assemble into soluble virus-like particles similar to the outer shell of a hepatitis B virus.[25]
A chemical adjuvant (AS01, specifically AS01E) was added to increase the immune system response.[26] Infection is prevented by inducing humoral and cellular immunity, with high antibody titers, that block the parasite from infecting the liver.[27]
Wilby KJ, Lau TT, Gilchrist SE, Ensom MH (March 2012). “Mosquirix (RTS,S): a novel vaccine for the prevention of Plasmodium falciparum malaria”. The Annals of Pharmacotherapy. 46 (3): 384–93. doi:10.1345/aph.1Q634. PMID22408046.
Asante KP, Abdulla S, Agnandji S, Lyimo J, Vekemans J, Soulanoudjingar S, et al. (October 2011). “Safety and efficacy of the RTS,S/AS01E candidate malaria vaccine given with expanded-programme-on-immunisation vaccines: 19 month follow-up of a randomised, open-label, phase 2 trial”. The Lancet. Infectious Diseases. 11 (10): 741–9. doi:10.1016/S1473-3099(11)70100-1. PMID21782519.
A poster advertising trials of the RTS,S vaccine[2]
A malaria vaccine is a vaccine that is used to prevent malaria. The only approved vaccine as of 2021, is RTS,S, known by the brand name Mosquirix.[1] It requires four injections.[1]
Research continues with other malaria vaccines. The most effective malaria vaccine is R21/Matrix-M, with a 77% efficacy rate shown in initial trials, and significantly higher antibody levels than with the RTS,S vaccine.[2] It is the first vaccine that meets the World Health Organization‘s (WHO) goal of a malaria vaccine with at least 75% efficacy.[3][2]
RTS,S (developed by PATH Malaria Vaccine Initiative (MVI) and GlaxoSmithKline (GSK) with support from the Bill and Melinda Gates Foundation) is the most recently developed recombinant vaccine. It consists of the P. falciparumcircumsporozoite protein (CSP) from the pre-erythrocytic stage. The CSP antigen causes the production of antibodies capable of preventing the invasion of hepatocytes and additionally elicits a cellular response enabling the destruction of infected hepatocytes. The CSP vaccine presented problems in the trial stage, due to its poor immunogenicity. RTS,S attempted to avoid these by fusing the protein with a surface antigen from hepatitis B, hence creating a more potent and immunogenic vaccine. When tested in trials an emulsion of oil in water and the added adjuvants of monophosphoryl A and QS21 (SBAS2), the vaccine gave protective immunity to 7 out of 8 volunteers when challenged with P. falciparum.[4]
RTS,S/AS01 (commercial name Mosquirix),[5] was engineered using genes from the outer protein of P. falciparum malaria parasite and a portion of a hepatitis B virus plus a chemical adjuvant to boost the immune response. Infection is prevented by inducing high antibody titers that block the parasite from infecting the liver.[6] In November 2012, a Phase III trial of RTS,S found that it provided modest protection against both clinical and severe malaria in young infants.[7]
As of October 2013, preliminary results of a Phase III clinical trial indicated that RTS,S/AS01 reduced the number of cases among young children by almost 50 percent and among infants by around 25 percent. The study ended in 2014. The effects of a booster dose were positive, even though overall efficacy seems to wane with time. After four years reductions were 36 percent for children who received three shots and a booster dose. Missing the booster dose reduced the efficacy against severe malaria to a negligible effect. The vaccine was shown to be less effective for infants. Three doses of vaccine plus a booster reduced the risk of clinical episodes by 26 percent over three years, but offered no significant protection against severe malaria.[8]
In a bid to accommodate a larger group and guarantee a sustained availability for the general public, GSK applied for a marketing license with the European Medicines Agency (EMA) in July 2014.[9] GSK treated the project as a non-profit initiative, with most funding coming from the Gates Foundation, a major contributor to malaria eradication.[10]
On 24 July 2015, Mosquirix received a positive opinion from the European Medicines Agency (EMA) on the proposal for the vaccine to be used to vaccinate children aged 6 weeks to 17 months outside the European Union.[11][12][1] A pilot project for vaccination was launched on 23 April 2019, in Malawi, on 30 April 2019, in Ghana, and on 13 September 2019, in Kenya.[13][14]
In October 2021, the vaccine was endorsed by the World Health Organization for “broad use” in children, making it the first malaria vaccine to receive this recommendation.[15][16][17]
Agents under development
A completely effective vaccine is not available for malaria, although several vaccines are under development. Multiple vaccine candidates targeting the blood-stage of the parasite’s life cycle have been insufficient on their own.[18] Several potential vaccines targeting the pre-erythrocytic stage are being developed, with RTS,S the only approved option so far.[19][7]
R21/Matrix-M
The most effective malaria vaccine is R21/Matrix-M, with 77% efficacy shown in initial trials. It is the first vaccine that meets the World Health Organization’s goal of a malaria vaccine with at least 75% efficacy.[3] It was developed through a collaboration involving the University of Oxford, the Kenya Medical Research Institute, the London School of Hygiene & Tropical Medicine, Novavax, the Serum Institute of India, and the Institut de Recherche en Sciences de la Santé in Nanoro, Burkina Faso. The R21 vaccine uses a circumsporozoite protein (CSP) antigen, at a higher proportion than the RTS,S vaccine. It includes the Matrix-M adjuvant that is also utilized in the Novavax COVID-19 vaccine.[20]
A Phase II trial was reported in April 2021, with a vaccine efficacy of 77% and antibody levels significantly higher than with the RTS,S vaccine. A Phase III trial is planned with 4,800 children across four African countries. If the vaccine is approved, over 200 million doses can be manufactured annually by the Serum Institute of India.[2]
Nanoparticle enhancement of RTS,S
In 2015, researchers used a repetitive antigen display technology to engineer a nanoparticle that displayed malaria specific B cell and T cell epitopes. The particle exhibited icosahedral symmetry and carried on its surface up to 60 copies of the RTS,S protein. The researchers claimed that the density of the protein was much higher than the 14% of the GSK vaccine.[21][22]
The PfSPZ vaccine is a candidate malaria vaccine developed by Sanaria using radiation-attenuated sporozoites to elicit an immune response. Clinical trials have been promising, with trials taking place in Africa, Europe, and the US protecting over 80% of volunteers.[23] It has been subject to some criticism regarding the ultimate feasibility of large-scale production and delivery in Africa, since it must be stored in liquid nitrogen.
In April 2019, a phase 3 trial in Bioko was announced, scheduled to start in early 2020.[25]
saRNA vaccine against PMIF
A patent was published in February 2021 for a Self-amplifying RNA (saRNA) vaccine that targets the protein PMIF, which is produced by the plasmodium parasite to inhibit the body’s T-cell response. The vaccine has been tested in mice and is described as, “probably the highest level of protection that has been seen in a mouse model” according to Richard Bucala, co-inventor of the vaccine. There are plans for phase one tests in humans later in 2021.[26]
Other developments
SPf66 is a synthetic peptide based vaccine developed by Manuel Elkin Patarroyo team in Colombia, and was tested extensively in endemic areas in the 1990s. Clinical trials showed it to be insufficiently effective, with 28% efficacy in South America and minimal or no efficacy in Africa.[27]
The CSP (Circum-Sporozoite Protein) was a vaccine developed that initially appeared promising enough to undergo trials. It is also based on the circumsporozoite protein, but additionally has the recombinant (Asn-Ala-Pro15Asn-Val-Asp-Pro)2-Leu-Arg(R32LR) protein covalently bound to a purified Pseudomonas aeruginosa toxin (A9). However at an early stage a complete lack of protective immunity was demonstrated in those inoculated. The study group used in Kenya had an 82% incidence of parasitaemia whilst the control group only had an 89% incidence. The vaccine intended to cause an increased T-lymphocyte response in those exposed, this was also not observed.[citation needed]
The NYVAC-Pf7 multi-stage vaccine attempted to use different technology, incorporating seven P.falciparum antigenic genes. These came from a variety of stages during the life cycle. CSP and sporozoite surface protein 2 (called PfSSP2) were derived from the sporozoite phase. The liver stage antigen 1 (LSA1), three from the erythrocytic stage (merozoite surface protein 1, serine repeat antigen and AMA-1) and one sexual stage antigen (the 25-kDa Pfs25) were included. This was first investigated using Rhesus monkeys and produced encouraging results: 4 out of the 7 antigens produced specific antibody responses (CSP, PfSSP2, MSP1 and PFs25). Later trials in humans, despite demonstrating cellular immune responses in over 90% of the subjects, had very poor antibody responses. Despite this following administration of the vaccine some candidates had complete protection when challenged with P.falciparum. This result has warranted ongoing trials.[citation needed]
In 1995 a field trial involving [NANP]19-5.1 proved to be very successful. Out of 194 children vaccinated none developed symptomatic malaria in the 12-week follow up period and only 8 failed to have higher levels of antibody present. The vaccine consists of the schizont export protein (5.1) and 19 repeats of the sporozoite surface protein [NANP]. Limitations of the technology exist as it contains only 20% peptide and has low levels of immunogenicity. It also does not contain any immunodominant T-cell epitopes.[28]
A chemical compound undergoing trials for treatment of tuberculosis and cancer—the JmJc inhibitor ML324 and the antitubercular clinical candidate SQ109—is potentially a new line of drugs to treat malaria and kill the parasite in its infectious stage. More tests still need to be carried out before the compounds would be approved as a viable treatment.[29]
Considerations
The task of developing a preventive vaccine for malaria is a complex process. There are a number of considerations to be made concerning what strategy a potential vaccine should adopt.
Parasite diversity
P. falciparum has demonstrated the capability, through the development of multiple drug-resistant parasites, for evolutionary change. The Plasmodium species has a very high rate of replication, much higher than that actually needed to ensure transmission in the parasite’s life cycle. This enables pharmaceutical treatments that are effective at reducing the reproduction rate, but not halting it, to exert a high selection pressure, thus favoring the development of resistance. The process of evolutionary change is one of the key considerations necessary when considering potential vaccine candidates. The development of resistance could cause a significant reduction in efficacy of any potential vaccine thus rendering useless a carefully developed and effective treatment.[30]
Choosing to address the symptom or the source
The parasite induces two main response types from the human immune system. These are anti-parasitic immunity and anti-toxic immunity.
“Anti-parasitic immunity” addresses the source; it consists of an antibody response (humoral immunity) and a cell-mediated immune response. Ideally a vaccine would enable the development of anti-plasmodial antibodies in addition to generating an elevated cell-mediated response. Potential antigens against which a vaccine could be targeted will be discussed in greater depth later. Antibodies are part of the specific immune response. They exert their effect by activating the complement cascade, stimulating phagocytic cells into endocytosis through adhesion to an external surface of the antigenic substances, thus ‘marking’ it as offensive. Humoral or cell-mediated immunity consists of many interlinking mechanisms that essentially aim to prevent infection entering the body (through external barriers or hostile internal environments) and then kill any micro-organisms or foreign particles that succeed in penetration. The cell-mediated component consists of many white blood cells (such as monocytes, neutrophils, macrophages, lymphocytes, basophils, mast cells, natural killer cells, and eosinophils) that target foreign bodies by a variety of different mechanisms. In the case of malaria both systems would be targeted to attempt to increase the potential response generated, thus ensuring the maximum chance of preventing disease.[citation needed]
“Anti-toxic immunity” addresses the symptoms; it refers to the suppression of the immune response associated with the production of factors that either induce symptoms or reduce the effect that any toxic by-products (of micro-organism presence) have on the development of disease. For example, it has been shown that Tumor necrosis factor-alpha has a central role in generating the symptoms experienced in severe P. falciparum malaria. Thus a therapeutic vaccine could target the production of TNF-a, preventing respiratory distress and cerebral symptoms. This approach has serious limitations as it would not reduce the parasitic load; rather it only reduces the associated pathology. As a result, there are substantial difficulties in evaluating efficacy in human trials.
Taking this information into consideration an ideal vaccine candidate would attempt to generate a more substantial cell-mediated and antibody response on parasite presentation. This would have the benefit of increasing the rate of parasite clearance, thus reducing the experienced symptoms and providing a level of consistent future immunity against the parasite.
Anti-host erythrocyte, antibodies blocking invasion; anti receptor ligand, anti-soluble toxin
Gametocytes
Anti-gametocyte. Anti-host erythrocyte, antibodies blocking fertilisation, antibodies blocking egress from the mosquito midgut.
By their very nature, protozoa are more complex organisms than bacteria and viruses, with more complicated structures and life cycles. This presents problems in vaccine development but also increases the number of potential targets for a vaccine. These have been summarised into the life cycle stage and the antibodies that could potentially elicit an immune response.
The epidemiology of malaria varies enormously across the globe, and has led to the belief that it may be necessary to adopt very different vaccine development strategies to target the different populations. A Type 1 vaccine is suggested for those exposed mostly to P. falciparum malaria in sub-Saharan Africa, with the primary objective to reduce the number of severe malaria cases and deaths in infants and children exposed to high transmission rates. The Type 2 vaccine could be thought of as a ‘travellers’ vaccine’, aiming to prevent all cases of clinical symptoms in individuals with no previous exposure. This is another major public health problem, with malaria presenting as one of the most substantial threats to travellers’ health. Problems with the available pharmaceutical therapies include costs, availability, adverse effects and contraindications, inconvenience and compliance, many of which would be reduced or eliminated entirely if an effective (greater than 85–90%) vaccine was developed.[citation needed]
The life cycle of the malaria parasite is particularly complex, presenting initial developmental problems. Despite the huge number of vaccines available, there are none that target parasitic infections. The distinct developmental stages involved in the life cycle present numerous opportunities for targeting antigens, thus potentially eliciting an immune response. Theoretically, each developmental stage could have a vaccine developed specifically to target the parasite. Moreover, any vaccine produced would ideally have the ability to be of therapeutic value as well as preventing further transmission and is likely to consist of a combination of antigens from different phases of the parasite’s development. More than 30 of these antigens are being researched[when?] by teams all over the world in the hope of identifying a combination that can elicit immunity in the inoculated individual. Some of the approaches involve surface expression of the antigen, inhibitory effects of specific antibodies on the life cycle and the protective effects through immunization or passive transfer of antibodies between an immune and a non-immune host. The majority of research into malarial vaccines has focused on the Plasmodium falciparum strain due to the high mortality caused by the parasite and the ease of a carrying out in vitro/in vivo studies. The earliest vaccines attempted to use the parasitic circumsporozoite protein (CSP). This is the most dominant surface antigen of the initial pre-erythrocytic phase. However, problems were encountered due to low efficacy, reactogenicity and low immunogenicity.[citation needed]
The initial stage in the life cycle, following inoculation, is a relatively short “pre-erythrocytic” or “hepatic” phase. A vaccine at this stage must have the ability to protect against sporozoites invading and possibly inhibiting the development of parasites in the hepatocytes (through inducing cytotoxicT-lymphocytes that can destroy the infected liver cells). However, if any sporozoites evaded the immune system they would then have the potential to be symptomatic and cause the clinical disease.
The second phase of the life cycle is the “erythrocytic” or blood phase. A vaccine here could prevent merozoite multiplication or the invasion of red blood cells. This approach is complicated by the lack of MHC molecule expression on the surface of erythrocytes. Instead, malarial antigens are expressed, and it is this towards which the antibodies could potentially be directed. Another approach would be to attempt to block the process of erythrocyte adherence to blood vessel walls. It is thought that this process is accountable for much of the clinical syndrome associated with malarial infection; therefore a vaccine given during this stage would be therapeutic and hence administered during clinical episodes to prevent further deterioration.
The last phase of the life cycle that has the potential to be targeted by a vaccine is the “sexual stage”. This would not give any protective benefits to the individual inoculated but would prevent further transmission of the parasite by preventing the gametocytes from producing multiple sporozoites in the gut wall of the mosquito. It therefore would be used as part of a policy directed at eliminating the parasite from areas of low prevalence or to prevent the development and spread of vaccine-resistant parasites. This type of transmission-blocking vaccine is potentially very important. The evolution of resistance in the malaria parasite occurs very quickly, potentially making any vaccine redundant within a few generations. This approach to the prevention of spread is therefore essential.
Another approach is to target the protein kinases, which are present during the entire lifecycle of the malaria parasite. Research is underway on this, yet production of an actual vaccine targeting these protein kinases may still take a long time.[31]
Report of a vaccine candidate capable to neutralize all tested strains of Plasmodium falciparum, the most deadly form of the parasite causing malaria, was published in Nature Communications by a team of scientists from the University of Oxford in 2011.[32] The viral vector vaccine, targeting a full-length P. falciparum reticulocyte-binding protein homologue 5 (PfRH5) was found to induce an antibody response in an animal model. The results of this new vaccine confirmed the utility of a key discovery reported from scientists at the Wellcome Trust Sanger Institute, published in Nature.[33] The earlier publication reported P. falciparum relies on a red blood cell surface receptor, known as ‘basigin’, to invade the cells by binding a protein PfRH5 to the receptor.[33] Unlike other antigens of the malaria parasite which are often genetically diverse, the PfRH5 antigen appears to have little genetic diversity. It was found to induce very low antibody response in people naturally exposed to the parasite.[32] The high susceptibility of PfRH5 to the cross-strain neutralizing vaccine-induced antibody demonstrated a significant promise for preventing malaria in the long and often difficult road of vaccine development. According to Professor Adrian Hill, a Wellcome Trust Senior Investigator at the University of Oxford, the next step would be the safety tests of this vaccine. At the time (2011) it was projected that if these proved successful, the clinical trials in patients could begin within two to three years.[34]
PfEMP1, one of the proteins known as variant surface antigens (VSAs) produced by Plasmodium falciparum, was found to be a key target of the immune system’s response against the parasite. Studies of blood samples from 296 mostly Kenyan children by researchers of Burnet Institute and their cooperators showed that antibodies against PfEMP1 provide protective immunity, while antibodies developed against other surface antigens do not. Their results demonstrated that PfEMP1 could be a target to develop an effective vaccine which will reduce risk of developing malaria.[35][36]
Plasmodium vivax is the common malaria species found in India, Southeast Asia and South America. It is able to stay dormant in the liver and reemerge years later to elicit new infections. Two key proteins involved in the invasion of the red blood cells (RBC) by P. vivax are potential targets for drug or vaccine development. When the Duffy binding protein (DBP) of P. vivax binds the Duffy antigen (DARC) on the surface of RBC, process for the parasite to enter the RBC is initiated. Structures of the core region of DARC and the receptor binding pocket of DBP have been mapped by scientists at the Washington University in St. Louis. The researchers found that the binding is a two-step process which involves two copies of the parasite protein acting together like a pair of tongs which “clamp” two copies of DARC. Antibodies that interfere with the binding, by either targeting the key region of the DARC or the DBP will prevent the infection.[37][38]
Antibodies against the Schizont Egress Antigen-1 (PfSEA-1) were found to disable the parasite ability to rupture from the infected red blood cells (RBCs) thus prevent it from continuing with its life cycle. Researchers from Rhode Island Hospital identified Plasmodium falciparum PfSEA-1, a 244 kd malaria antigen expressed in the schizont-infected RBCs. Mice vaccinated with the recombinant PfSEA-1 produced antibodies which interrupted the schizont rupture from the RBCs and decreased the parasite replication. The vaccine protected the mice from lethal challenge of the parasite. Tanzanian and Kenyan children who have antibodies to PfSEA-1 were found to have fewer parasites in their blood stream and milder case of malaria. By blocking the schizont outlet, the PfSEA-1 vaccine may work synergistically with vaccines targeting the other stages of the malaria life cycle such as hepatocyte and RBC invasion.[39][40]
Mix of antigenic components
Increasing the potential immunity generated against Plasmodia can be achieved by attempting to target multiple phases in the life cycle. This is additionally beneficial in reducing the possibility of resistant parasites developing. The use of multiple-parasite antigens can therefore have a synergistic or additive effect.
One of the most successful vaccine candidates in clinical trials[which?][when?] consists of recombinant antigenic proteins to the circumsporozoite protein.[41] (This is discussed in more detail below.)[where?]
Delivery system
The selection of an appropriate system is fundamental in all vaccine development, but especially so in the case of malaria. A vaccine targeting several antigens may require delivery to different areas and by different means in order to elicit an effective response. Some adjuvants can direct the vaccine to the specifically targeted cell type—e.g. the use of Hepatitis B virus in the RTS,S vaccine to target infected hepatocytes—but in other cases, particularly when using combined antigenic vaccines, this approach is very complex. Some methods that have been attempted include the use of two vaccines, one directed at generating a blood response and the other a liver-stage response. These two vaccines could then be injected into two different sites, thus enabling the use of a more specific and potentially efficacious delivery system.
To increase, accelerate or modify the development of an immune response to a vaccine candidate it is often necessary to combine the antigenic substance to be delivered with an adjuvant or specialised delivery system. These terms are often used interchangeably in relation to vaccine development; however in most cases a distinction can be made. An adjuvant is typically thought of as a substance used in combination with the antigen to produce a more substantial and robust immune response than that elicited by the antigen alone. This is achieved through three mechanisms: by affecting the antigen delivery and presentation, by inducing the production of immunomodulatory cytokines, and by affecting the antigen presenting cells (APC). Adjuvants can consist of many different materials, from cell microparticles to other particulated delivery systems (e.g. liposomes).
Adjuvants are crucial in affecting the specificity and isotype of the necessary antibodies. They are thought to be able to potentiate the link between the innate and adaptive immune responses. Due to the diverse nature of substances that can potentially have this effect on the immune system, it is difficult to classify adjuvants into specific groups. In most circumstances they consist of easily identifiable components of micro-organisms that are recognised by the innate immune system cells. The role of delivery systems is primarily to direct the chosen adjuvant and antigen into target cells to attempt to increase the efficacy of the vaccine further, therefore acting synergistically with the adjuvant.
There is increasing concern that the use of very potent adjuvants could precipitate autoimmune responses, making it imperative that the vaccine is focused on the target cells only. Specific delivery systems can reduce this risk by limiting the potential toxicity and systemic distribution of newly developed adjuvants.
Studies into the efficacy of malaria vaccines developed to date[when?] have illustrated that the presence of an adjuvant is key in determining any protection gained against malaria. A large number of natural and synthetic adjuvants have been identified throughout the history of vaccine development. Options identified thus far for use combined with a malaria vaccine include mycobacterial cell walls, liposomes, monophosphoryl lipid A and squalene.
History
Individuals who are exposed to the parasite in endemic countries develop acquired immunity against disease and death. Such immunity does not however prevent malarial infection; immune individuals often harbour asymptomatic parasites in their blood. This does, however, imply that it is possible to create an immune response that protects against the harmful effects of the parasite.
Research shows that if immunoglobulin is taken from immune adults, purified and then given to individuals who have no protective immunity, some protection can be gained.[42]
Irradiated mosquitoes
In 1967, it was reported that a level of immunity to the Plasmodium berghei parasite could be given to mice by exposing them to sporozoites that had been irradiated by x-rays.[43] Subsequent human studies in the 1970s showed that humans could be immunized against Plasmodium vivax and Plasmodium falciparum by exposing them to the bites of significant numbers of irradiated mosquitos.[44]
^ Clyde DF (May 1975). “Immunization of man against falciparum and vivax malaria by use of attenuated sporozoites”. The American Journal of Tropical Medicine and Hygiene. 24 (3): 397–401. doi:10.4269/ajtmh.1975.24.397. PMID808142.
Good MF, Levine MA, Kaper JB, Rappuoli R, Liu MA (2004). New Generation Vaccines. New York, N.Y: Marcel Dekker. ISBN978-0-8247-4071-9.
Hoffman SL, Doolan DL, Richie TL (January 2004). “Malaria: a complex disease that may require a complex vaccine.”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). New Generation Vaccines (3rd ed.). CRC Press. pp. 1763–1790. ISBN978-0-429-15186-6.
Good M, Kemp D. “Overview of Vaccine Strategies for Malaria”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). ibid (3rd ed.). CRC Press. ISBN978-0-429-15186-6.
Saul A. “Malaria Transmission-Blocking Vaccines”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). New Generation Vaccines (3rd ed.). CRC Press. ISBN978-0-429-15186-6.
Heppner DG, Cummings JF, Ockenhouse CF, Kester KE, Cohen J, Ballou WR (2004). “Adjuvanted RTS, S and other protein-based pre-erythrocytic stage malaria vaccines.”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). New generation vaccines (3rd ed.). CRC Press. pp. 851–60. ISBN978-0-429-15186-6.
Stanisic DI, Martin LB, Good MF, Anders RF. “Plasmodium falciparum Asexual Blood Stage Vaccine Candidates: Current Status.”. In Levine MM, Kaper JB, Rappuoli R, Liu MA, Good MR (eds.). New Generation Vaccines (3rd ed.). CRC Press. ISBN978-0-429-15186-6.
Aponte JJ, Aide P, Renom M, Mandomando I, Bassat Q, Sacarlal J, et al. (November 2007). “Safety of the RTS,S/AS02D candidate malaria vaccine in infants living in a highly endemic area of Mozambique: a double blind randomised controlled phase I/IIb trial”. Lancet. 370 (9598): 1543–51. doi:10.1016/S0140-6736(07)61542-6. PMID17949807. S2CID19372191.
Percent Composition: C 67.59%, H 8.19%, Cl 11.08%, N 13.14%
Literature References: Prepd by the condensation of 4,7-dichloroquinoline with 1-diethylamino-4-aminopentane: DE683692 (1939); H. Andersag et al.,US2233970 (1941 to Winthrop); Surrey, Hammer, J. Am. Chem. Soc.68, 113 (1946). Review: Hahn in Antibioticsvol. 3, J. W. Corcoran, F. E. Hahn, Eds. (Springer-Verlag, New York, 1975) pp 58-78. Comprehensive description: D. D. Hong, Anal. Profiles Drug Subs.5, 61-85 (1976). Comparative clinical trial with dapsone in rheumatoid arthritis: P. D. Fowler et al.,Ann. Rheum. Dis.43, 200 (1984); with penicillamine: T. Gibson et al.,Br. J. Rheumatol.26, 279 (1987).
Percent Composition: C 41.91%, H 6.25%, Cl 6.87%, N 8.15%, P 12.01%, O 24.81%
Properties: Bitter, colorless crystals. Dimorphic. One modification, mp 193-195°; the other, mp 215-218°. Freely sol in water; pH of 1% soln about 4.5; less sol at neutral and alkaline pH. Stable to heat in solns of pH 4.0 to 6.5. Practically insol in alcohol, benzene, chloroform, ether.
Chloroquine is a medication used primarily to prevent and to treat malaria in areas where that parasitic disease is known to remain sensitive to its effects.[1] A benefit of its use in therapy, when situations allow, is that it can be taken by mouth (versus by injection).[1] Controlled studies of cases involving human pregnancy are lacking, but the drug may be safe for use for such patients.[verification needed][1][2] However, the agent is not without the possibility of serious side effects at standard doses,[1][3] and complicated cases, including infections of certain types or caused by resistant strains, typically require different or additional medication.[1] Chloroquine is also used as a medication for rheumatoid arthritis, lupus erythematosus, and other parasitic infections (e.g., amebiasis occurring outside of the intestines).[1] Beginning in 2020, studies have proceeded on its use as a coronavirusantiviral, in possible treatment of COVID-19.[4]
Chloroquine, otherwise known as chloroquine phosphate, is in the 4-aminoquinoline class of drugs.[1] As an antimalarial, it works against the asexual form of the malaria parasite in the stage of its life cycle within the red blood cell.[1] In its use against rheumatoid arthritis and lupus erythematosus, its activity as a mild immunosuppressive underlies its mechanism.[1] Antiviral activities, established and putative, are attributed to chloroquines inhibition of glycosylation pathways (of host receptor sialylation or virus protein post-translational modification), or to inhibition of virus endocytosis (e.g., via alkalisation of endosomes), or other possible mechanisms.[5] Common side effects resulting from these therapeutic uses, at common doses, include muscle problems,[clarification needed] loss of appetite, diarrhea, and skin rash.[clarification needed][1] Serious side effects include problems with vision (retinopathy), muscle damage, seizures, and certain anemias.[1][6]
Distribution of malaria in the world:[11]
♦ Elevated occurrence of chloroquine- or multi-resistant malaria
♦ Occurrence of chloroquine-resistant malaria
♦ No Plasmodium falciparum or chloroquine-resistance
♦ No malaria
Chloroquine has been extensively used in mass drug administrations, which may have contributed to the emergence and spread of resistance. It is recommended to check if chloroquine is still effective in the region prior to using it.[14] In areas where resistance is present, other antimalarials, such as mefloquine or atovaquone, may be used instead. The Centers for Disease Control and Prevention recommend against treatment of malaria with chloroquine alone due to more effective combinations.[15]
Amebiasis
In treatment of amoebic liver abscess, chloroquine may be used instead of or in addition to other medications in the event of failure of improvement with metronidazole or another nitroimidazole within 5 days or intolerance to metronidazole or a nitroimidazole.[16]
Mental/mood changes (such as confusion, personality changes, unusual thoughts/behavior, depression, feeling being watched, hallucinating)[17][18]
Signs of serious infection (such as high fever, severe chills, persistent sore throat)[17]
Skin itchiness, skin color changes, hair loss, and skin rashes.[18][19]
Chloroquine-induced itching is very common among black Africans (70%), but much less common in other races. It increases with age, and is so severe as to stop compliance with drug therapy. It is increased during malaria fever; its severity is correlated to the malaria parasite load in blood. Some evidence indicates it has a genetic basis and is related to chloroquine action with opiate receptors centrally or peripherally.[20]
Unpleasant metallic taste
This could be avoided by “taste-masked and controlled release” formulations such as multiple emulsions.[21]
This manifests itself as either conduction disturbances (bundle-branch block, atrioventricular block) or Cardiomyopathy – often with hypertrophy, restrictive physiology, and congestive heart failure. The changes may be irreversible. Only two cases have been reported requiring heart transplantation, suggesting this particular risk is very low. Electron microscopy of cardiac biopsies show pathognomonic cytoplasmic inclusion bodies.
Chloroquine has not been shown to have any harmful effects on the fetus when used for malarial prophylaxis.[24] Small amounts of chloroquine are excreted in the breast milk of lactating women. However, this drug can be safely prescribed to infants, the effects are not harmful. Studies with mice show that radioactively tagged chloroquine passed through the placenta rapidly and accumulated in the fetal eyes which remained present five months after the drug was cleared from the rest of the body.[23][25] Women who are pregnant or planning on getting pregnant are still advised against traveling to malaria-risk regions.[24]
Elderly
There is not enough evidence to determine whether chloroquine is safe to be given to people aged 65 and older. Since it is cleared by the kidneys, toxicity should be monitored carefully in people with poor kidney functions.[23]
Chloroquine is very dangerous in overdose. It is rapidly absorbed from the gut. In 1961, a published compilation of case reports contained accounts of three children who took overdoses and died within 2.5 hours of taking the drug. While the amount of the overdose was not stated, the therapeutic index for chloroquine is known to be small.[26] One of the children died after taking 0.75 or 1 gram, or twice a single therapeutic amount for children. Symptoms of overdose include headache, drowsiness, visual disturbances, nausea and vomiting, cardiovascular collapse, seizures, and sudden respiratory and cardiac arrest.[23]
An analog of chloroquine – hydroxychloroquine – has a long half-life (32–56 days) in blood and a large volume of distribution (580–815 L/kg).[27] The therapeutic, toxic and lethal ranges are usually considered to be 0.03 to 15 mg/l, 3.0 to 26 mg/l and 20 to 104 mg/l, respectively. However, nontoxic cases have been reported up to 39 mg/l, suggesting individual tolerance to this agent may be more variable than previously recognised.[27]
Pharmacology
Chloroquine’s absorption of the drug is rapid. It is widely distributed in body tissues. It’s protein binding is 55%.[ It’s metabolism is partially hepatic, giving rise to its main metabolite, desethylchloroquine. It’s excretion os ≥50% as unchanged drug in urine, where acidification of urine increases its elimination It has a very high volume of distribution, as it diffuses into the body’s adipose tissue.
Accumulation of the drug may result in deposits that can lead to blurred vision and blindness. It and related quinines have been associated with cases of retinal toxicity, particularly when provided at higher doses for longer times. With long-term doses, routine visits to an ophthalmologist are recommended.
Chloroquine is also a lysosomotropic agent, meaning it accumulates preferentially in the lysosomes of cells in the body. The pKa for the quinoline nitrogen of chloroquine is 8.5, meaning—in simplified terms, considering only this basic site—it is about 10% deprotonated at physiological pH (per the Henderson-Hasselbalch equation) This decreases to about 0.2% at a lysosomal pH of 4.6.Because the deprotonated form is more membrane-permeable than the protonated form, a quantitative “trapping” of the compound in lysosomes results.
Mechanism of action
Medical quinolines
Malaria
Hemozoin formation in P. falciparum: many antimalarials are strong inhibitors of hemozoin crystal growth.
The lysosomotropic character of chloroquine is believed to account for much of its antimalarial activity; the drug concentrates in the acidic food vacuole of the parasite and interferes with essential processes. Its lysosomotropic properties further allow for its use for in vitro experiments pertaining to intracellular lipid related diseases,[28][29] autophagy, and apoptosis.[30]
Inside red blood cells, the malarial parasite, which is then in its asexual lifecycle stage, must degrade hemoglobin to acquire essential amino acids, which the parasite requires to construct its own protein and for energy metabolism. Digestion is carried out in a vacuole of the parasitic cell.[citation needed]
Hemoglobin is composed of a protein unit (digested by the parasite) and a heme unit (not used by the parasite). During this process, the parasite releases the toxic and soluble molecule heme. The heme moiety consists of a porphyrin ring called Fe(II)-protoporphyrin IX (FP). To avoid destruction by this molecule, the parasite biocrystallizes heme to form hemozoin, a nontoxic molecule. Hemozoin collects in the digestive vacuole as insoluble crystals.[citation needed]
Chloroquine enters the red blood cell by simple diffusion, inhibiting the parasite cell and digestive vacuole. Chloroquine then becomes protonated (to CQ2+), as the digestive vacuole is known to be acidic (pH 4.7); chloroquine then cannot leave by diffusion. Chloroquine caps hemozoin molecules to prevent further biocrystallization of heme, thus leading to heme buildup. Chloroquine binds to heme (or FP) to form the FP-chloroquine complex; this complex is highly toxic to the cell and disrupts membrane function. Action of the toxic FP-chloroquine and FP results in cell lysis and ultimately parasite cell autodigestion. [31] Parasites that do not form hemozoin are therefore resistant to chloroquine.[32]
Since the first documentation of P. falciparum chloroquine resistance in the 1950s, resistant strains have appeared throughout East and West Africa, Southeast Asia, and South America. The effectiveness of chloroquine against P. falciparum has declined as resistant strains of the parasite evolved. They effectively neutralize the drug via a mechanism that drains chloroquine away from the digestive vacuole. Chloroquine-resistant cells efflux chloroquine at 40 times the rate of chloroquine-sensitive cells; the related mutations trace back to transmembrane proteins of the digestive vacuole, including sets of critical mutations in the P. falciparum chloroquine resistance transporter (PfCRT) gene. The mutated protein, but not the wild-type transporter, transports chloroquine when expressed in Xenopus oocytes (frog’s eggs) and is thought to mediate chloroquine leak from its site of action in the digestive vacuole.[33] Resistant parasites also frequently have mutated products of the ABC transporterP. falciparum multidrug resistance (PfMDR1) gene, although these mutations are thought to be of secondary importance compared to Pfcrt. Verapamil, a Ca2+ channel blocker, has been found to restore both the chloroquine concentration ability and sensitivity to this drug. Recently, an altered chloroquine-transporter protein CG2 of the parasite has been related to chloroquine resistance, but other mechanisms of resistance also appear to be involved.[34] Research on the mechanism of chloroquine and how the parasite has acquired chloroquine resistance is still ongoing, as other mechanisms of resistance are likely.[citation needed]
Chloroquine has antiviral effects.[36] It increases late endosomal or lysosomal pH, resulting in impaired release of the virus from the endosome or lysosome – release requires a low pH. The virus is therefore unable to release its genetic material into the cell and replicate.[37][38]
Chloroquine also seems to act as a zinc ionophore, that allows extracellular zinc to enter the cell and inhibit viral RNA-dependent RNA polymerase.[39][40]
Other
Chloroquine inhibits thiamine uptake.[41] It acts specifically on the transporter SLC19A3.
In Peru the indigenous people extracted the bark of the Cinchona plant[42] trees and used the extract (Chinchona officinalis) to fight chills and fever in the seventeenth century. In 1633 this herbal medicine was introduced in Europe, where it was given the same use and also began to be used against malaria.[43] The quinoline antimalarial drug quinine was isolated from the extract in 1820, and chloroquine is an analogue of this.
Chloroquine was discovered in 1934, by Hans Andersag and coworkers at the Bayer laboratories, who named it “Resochin”.[44] It was ignored for a decade, because it was considered too toxic for human use. During World War II, United States government-sponsored clinical trials for antimalarial drug development showed unequivocally that chloroquine has a significant therapeutic value as an antimalarial drug. It was introduced into clinical practice in 1947 for the prophylactic treatment of malaria.[45]
Society and culture
Resochin tablet package
Formulations
Chloroquine comes in tablet form as the phosphate, sulfate, and hydrochloride salts. Chloroquine is usually dispensed as the phosphate.[46]
Names
Brand names include Chloroquine FNA, Resochin, Dawaquin, and Lariago.[47]
Chloroquine has been recommended by Chinese, South Korean and Italian health authorities for the treatment of COVID-19.[52][53] These agencies noted contraindications for people with heart disease or diabetes.[54] Both chloroquine and hydroxychloroquine were shown to inhibit SARS-CoV-2 in vitro, but a further study concluded that hydroxychloroquine was more potent than chloroquine, with a more tolerable safety profile.[55] Preliminary results from a trial suggested that chloroquine is effective and safe in COVID-19 pneumonia, “improving lung imaging findings, promoting a virus-negative conversion, and shortening the disease course.”[56] Self-medication with chloroquine has caused one known fatality.[57]
On 24 March 2020, NBC News reported[58] a fatality due to misuse of a chloroquine product used to control fish parasites.[59]
Other viruses
In October 2004, a group of researchers at the Rega Institute for Medical Research published a report on chloroquine, stating that chloroquine acts as an effective inhibitor of the replication of the severe acute respiratory syndrome coronavirus (SARS-CoV) in vitro.[60]
Chloroquine was being considered in 2003, in pre-clinical models as a potential agent against chikungunya fever.[61]
^Mittra, Robert A.; Mieler, William F. (1 January 2013). Ryan, Stephen J.; Sadda, SriniVas R.; Hinton, David R.; Schachat, Andrew P.; Sadda, SriniVas R.; Wilkinson, C. P.; Wiedemann, Peter; Schachat, Andrew P. (eds.). Retina (Fifth Edition). W.B. Saunders. pp. 1532–1554 – via ScienceDirect.
^Cortegiani A, Ingoglia G, Ippolito M, Giarratano A, Einav S (March 2020). “A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19”. Journal of Critical Care. doi:10.1016/j.jcrc.2020.03.005. PMID32173110.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^“Chloroquine (Base)”. International Drug Price Indicator Guide. Archived from the original on 27 August 2018. Retrieved 4 December 2015.
^CDC. Health information for international travel 2001–2002. Atlanta, Georgia: U.S. Department of Health and Human Services, Public Health Service, 2001.
^Ajayi AA (September 2000). “Mechanisms of chloroquine-induced pruritus”. Clinical Pharmacology and Therapeutics. 68 (3): 336. PMID11014416.
^Vaziri A, Warburton B (1994). “Slow release of chloroquine phosphate from multiple taste-masked W/O/W multiple emulsions”. Journal of Microencapsulation. 11 (6): 641–8. doi:10.3109/02652049409051114. PMID7884629.
^Tönnesmann E, Kandolf R, Lewalter T (June 2013). “Chloroquine cardiomyopathy – a review of the literature”. Immunopharmacology and Immunotoxicology. 35 (3): 434–42. doi:10.3109/08923973.2013.780078. PMID23635029.
^Hempelmann E (March 2007). “Hemozoin biocrystallization in Plasmodium falciparum and the antimalarial activity of crystallization inhibitors”. Parasitology Research. 100 (4): 671–6. doi:10.1007/s00436-006-0313-x. PMID17111179.
^Martin RE, Marchetti RV, Cowan AI, Howitt SM, Bröer S, Kirk K (September 2009). “Chloroquine transport via the malaria parasite’s chloroquine resistance transporter”. Science. 325 (5948): 1680–2. Bibcode:2009Sci…325.1680M. doi:10.1126/science.1175667. PMID19779197.
^Essentials of medical pharmacology fifth edition 2003, reprint 2004, published by-Jaypee Brothers Medical Publisher Ltd, 2003, KD Tripathi, pages 739,740.
^Alcantara LM, Kim J, Moraes CB, Franco CH, Franzoi KD, Lee S, et al. (June 2013). “Chemosensitization potential of P-glycoprotein inhibitors in malaria parasites”. Experimental Parasitology. 134 (2): 235–43. doi:10.1016/j.exppara.2013.03.022. PMID23541983.
^Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R (November 2003). “Effects of chloroquine on viral infections: an old drug against today’s diseases?”. The Lancet. Infectious Diseases. 3(11): 722–7. doi:10.1016/s1473-3099(03)00806-5. PMID14592603.
^Krafts K, Hempelmann E, Skórska-Stania A (July 2012). “From methylene blue to chloroquine: a brief review of the development of an antimalarial therapy”. Parasitology Research. 111 (1): 1–6. doi:10.1007/s00436-012-2886-x. PMID22411634.
^Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M (October 2004). “In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine”. Biochemical and Biophysical Research Communications. 323 (1): 264–8. doi:10.1016/j.bbrc.2004.08.085. PMID15351731.
^Devaux CA, Rolain JM, Colson P, Raoult D. New insights on the antiviral effects of chloroquine against coronavirus: what to expect for COVID-19? Int J Antimicrob Agents. 2020 Mar 11:105938. doi:10.1016/j.ijantimicag.2020.105938PMID32171740
^Yao X, Ye F, Zhang M, Cui C, Huang B, Niu P, et al. (March 2020). “In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)”. Clinical Infectious Diseases. doi:10.1093/cid/ciaa237. PMID32150618.
^Keyaerts E, Vijgen L, Maes P, Neyts J, Van Ranst M (October 2004). “In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine”. Biochemical and Biophysical Research Communications. 323 (1): 264–8. doi:10.1016/j.bbrc.2004.08.085. PMID15351731.
^Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R (November 2003). “Effects of chloroquine on viral infections: an old drug against today’s diseases?”. The Lancet. Infectious Diseases. 3(11): 722–7. doi:10.1016/S1473-3099(03)00806-5. PMID14592603.
^Savarino A, Lucia MB, Giordano F, Cauda R (October 2006). “Risks and benefits of chloroquine use in anticancer strategies”. The Lancet. Oncology. 7 (10): 792–3. doi:10.1016/S1470-2045(06)70875-0. PMID17012039.
^Sotelo J, Briceño E, López-González MA (March 2006). “Adding chloroquine to conventional treatment for glioblastoma multiforme: a randomized, double-blind, placebo-controlled trial”. Annals of Internal Medicine. 144 (5): 337–43. doi:10.7326/0003-4819-144-5-200603070-00008. PMID16520474.
“Summaries for patients. Adding chloroquine to conventional chemotherapy and radiotherapy for glioblastoma multiforme”. Annals of Internal Medicine. 144 (5): I31. March 2006. doi:10.7326/0003-4819-144-5-200603070-00004. PMID16520470.
External links
“Chloroquine”. Drug Information Portal. U.S. National Library of Medicine.
Hydroxychloroquine (HCQ), sold under the brand name Plaquenil among others, is a medication used for the prevention and treatment of certain types of malaria.[2] Specifically it is used for chloroquine-sensitive malaria.[3] Other uses include treatment of rheumatoid arthritis, lupus, and porphyria cutanea tarda.[2] It is taken by mouth.[2] It is also being used as an experimental treatment for coronavirus disease 2019 (COVID-19).[4]
Hydroxychloroquine was approved for medical use in the United States in 1955.[2] It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[6] The wholesale cost in the developing world is about US$4.65 per month as of 2015, when used for rheumatoid arthritis or lupus.[7] In the United States the wholesale cost of a month of treatment is about US$25 as of 2020.[8] In the United Kingdom this dose costs the NHS about £ 5.15.[9] In 2017, it was the 128th most prescribed medication in the United States with more than five million prescriptions.[10]
In 2014, its efficacy to treat Sjögren syndrome was questioned in a double-blind study involving 120 patients over a 48-week period.[11]
Hydroxychloroquine is widely used in the treatment of post-Lymearthritis. It may have both an anti-spirochaete activity and an anti-inflammatory activity, similar to the treatment of rheumatoid arthritis.[12]
Contraindications
The drug label advises that hydroxychloroquine should not be prescribed to individuals with known hypersensitivity to 4-Aminoquinoline compounds.[13] There are a range of other contraindications[14][15] and caution is required if patients have certain heart conditions, diabetes, psoriasis etc.
Side effects[
The most common adverse effects are a mild nausea and occasional stomach cramps with mild diarrhea. The most serious adverse effects affect the eye, with dose-related retinopathy as a concern even after hydroxychloroquine use is discontinued.[2] For short-term treatment of acute malaria, adverse effects can include abdominal cramps, diarrhea, heart problems, reduced appetite, headache, nausea and vomiting.[2]
For prolonged treatment of lupus or rheumatoid arthritis, adverse effects include the acute symptoms, plus altered eye pigmentation, acne, anemia, bleaching of hair, blisters in mouth and eyes, blood disorders, convulsions, vision difficulties, diminished reflexes, emotional changes, excessive coloring of the skin, hearing loss, hives, itching, liver problems or liver failure, loss of hair, muscle paralysis, weakness or atrophy, nightmares, psoriasis, reading difficulties, tinnitus, skin inflammation and scaling, skin rash, vertigo, weight loss, and occasionally urinary incontinence.[2] Hydroxychloroquine can worsen existing cases of both psoriasis and porphyria.[2]
Children may be especially vulnerable to developing adverse effects from hydroxychloroquine.[2]
One of the most serious side effects is retinopathy (generally with chronic use).[2][16] People taking 400 mg of hydroxychloroquine or less per day generally have a negligible risk of macular toxicity, whereas the risk begins to go up when a person takes the medication over 5 years or has a cumulative dose of more than 1000 grams. The daily safe maximum dose for eye toxicity can be computed from one’s height and weight using this calculator. Cumulative doses can also be calculated from this calculator. Macular toxicity is related to the total cumulative dose rather than the daily dose. Regular eye screening, even in the absence of visual symptoms, is recommended to begin when either of these risk factors occurs.[17]
Toxicity from hydroxychloroquine may be seen in two distinct areas of the eye: the cornea and the macula. The cornea may become affected (relatively commonly) by an innocuous cornea verticillata or vortex keratopathy and is characterized by whorl-like corneal epithelial deposits. These changes bear no relationship to dosage and are usually reversible on cessation of hydroxychloroquine.
The macular changes are potentially serious. Advanced retinopathy is characterized by reduction of visual acuity and a “bull’s eye” macular lesion which is absent in early involvement.
Overdose
Due to rapid absorption, symptoms of overdose can occur within a half an hour after ingestion. Overdose symptoms include convulsions, drowsiness, headache, heart problems or heart failure, difficulty breathing and vision problems.
Hydroxychloroquine overdoses are rarely reported, with 7 previous cases found in the English medical literature. In one such case, a 16-year-old girl who had ingested a handful of hydroxychloroquine 200mg presented with tachycardia (heart rate 110 beats/min), hypotension (systolic blood pressure 63 mm Hg), central nervous system depression, conduction defects (ORS = 0.14 msec), and hypokalemia (K = 2.1 meq/L). Treatment consisted of fluid boluses and dopamine, oxygen, and potassium supplementation. The presence of hydroxychloroquine was confirmed through toxicologic tests. The patient’s hypotension resolved within 4.5 hours, serum potassium stabilized in 24 hours, and tachycardia gradually decreased over 3 days.[18]
Specifically, the FDA drug label for hydroxychloroquine lists the following drug interactions [13]:
Digoxin (wherein it may result in increased serum digoxin levels)
Insulin or antidiabetic drugs (wherein it may enhance the effects of a hypoglycemic treatment)
Drugs that prolong QT interval and other arrhythmogenic drugs (as Hydroxychloroquine prolongs the QT interval and may increase the risk of inducing ventricular arrhythmias if used concurrently)
Mefloquine and other drugs known to lower the convulsive threshold (co-administration with other antimalarials known to lower the convulsion threshold may increase risk of convulsions)
Antiepileptics (concurrent use may impair the antiepileptic activity)
Methotrexate (combined use is unstudied and may increase the frequency of side effects)
Cyclosporin (wherein an increased plasma cylcosporin level was reported when used together).
Pharmacology[
Pharmacokinetics
Hydroxychloroquine has similar pharmacokinetics to chloroquine, with rapid gastrointestinal absorption and elimination by the kidneys. Cytochrome P450 enzymes (CYP2D6, 2C8, 3A4 and 3A5) metabolize hydroxychloroquine to N-desethylhydroxychloroquine.[21]
Pharmacodynamics
Antimalarials are lipophilic weak bases and easily pass plasma membranes. The free base form accumulates in lysosomes (acidic cytoplasmic vesicles) and is then protonated,[22] resulting in concentrations within lysosomes up to 1000 times higher than in culture media. This increases the pH of the lysosome from 4 to 6.[23] Alteration in pH causes inhibition of lysosomal acidic proteases causing a diminished proteolysis effect.[24] Higher pH within lysosomes causes decreased intracellular processing, glycosylation and secretion of proteins with many immunologic and nonimmunologic consequences.[25] These effects are believed to be the cause of a decreased immune cell functioning such as chemotaxis, phagocytosis and superoxide production by neutrophils.[26] HCQ is a weak diprotic base that can pass through the lipid cell membrane and preferentially concentrate in acidic cytoplasmic vesicles. The higher pH of these vesicles in macrophages or other antigen-presenting cells limits the association of autoantigenic (any) peptides with class II MHC molecules in the compartment for peptide loading and/or the subsequent processing and transport of the peptide-MHC complex to the cell membrane.[27]
In 2003, a novel mechanism was described wherein hydroxychloroquine inhibits stimulation of the toll-like receptor (TLR) 9 family receptors. TLRs are cellular receptors for microbial products that induce inflammatory responses through activation of the innate immune system.[29]
As with other quinoline antimalarial drugs, the mechanism of action of quinine has not been fully resolved. The most accepted model is based on hydrochloroquinine and involves the inhibition of hemozoinbiocrystallization, which facilitates the aggregation of cytotoxic heme. Free cytotoxic heme accumulates in the parasites, causing their deaths.[citation needed]
Brand names
It is frequently sold as a sulfate salt known as hydroxychloroquine sulfate.[2] 200 mg of the sulfate salt is equal to 155 mg of the base.[2]
Brand names of hydroxychloroquine include Plaquenil, Hydroquin, Axemal (in India), Dolquine, Quensyl, Quinoric.[30]
Hydroxychloroquine and chloroquine have been recommended by Chinese and South Korean health authorities for the experimental treatment of COVID-19.[31][32]In vitro studies in cell cultures demonstrated that hydroxychloroquine was more potent than chloroquine against SARS-CoV-2.[33]
On 17 March 2020, the AIFA Scientific Technical Commission of the Italian Medicines Agency expressed a favorable opinion on including the off-label use of chloroquine and hydroxychloroquine for the treatment of SARS-CoV-2 infection.[34]
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^“NADAC as of 2019-08-07”. Centers for Medicare and Medicaid Services. Retrieved 19 March 2020. Typical dose is 600mg per day. Costs 0.28157 per dose. Month has about 30 days.
^British national formulary: BNF 69 (69 ed.). British Medical Association. 2015. p. 730. ISBN9780857111562.
^Effects of Hydroxychloroquine on Symptomatic Improvement in Primary Sjögren Syndrome, Gottenberg, et al. (2014) “Archived copy”. Archived from the original on 11 July 2015. Retrieved 10 July 2015.
^Steere, AC; Angelis, SM (October 2006). “Therapy for Lyme Arthritis: Strategies for the Treatment of Antibiotic-refractory Arthritis”. Arthritis and Rheumatism. 54 (10): 3079–86. doi:10.1002/art.22131. PMID17009226.
^Mohammad, Samya; Clowse, Megan E. B.; Eudy, Amanda M.; Criscione-Schreiber, Lisa G. (March 2018). “Examination of Hydroxychloroquine Use and Hemolytic Anemia in G6PDH-Deficient Patients”. Arthritis Care & Research. 70 (3): 481–485. doi:10.1002/acr.23296. ISSN2151-4658. PMID28556555.
^Hurst, NP; French, JK; Gorjatschko, L; Betts, WH (1988). “Chloroquine and Hydroxychloroquine Inhibit Multiple Sites in Metabolic Pathways Leading to Neutrophil Superoxide Release”. The Journal of Rheumatology. 15 (1): 23–27. PMID2832600.
^Fox, R (1996). “Anti-malarial Drugs: Possible Mechanisms of Action in Autoimmune Disease and Prospects for Drug Development”. Lupus. 5: S4–10. doi:10.1177/096120339600500103. PMID8803903.
^Waller; et al. Medical Pharmacology and Therapeutics (2nd ed.). p. 370.
UNII F38R0JR742
CAS number 82186-77-4
Weight Average: 528.94
Monoisotopic: 527.154947772
Chemical Formula C30H32Cl3NO
Lumefantrine (or benflumetol) is an antimalarial drug. It is only used in combination with artemether. The term “co-artemether” is sometimes used to describe this combination.[1] Lumefantrine has a much longer half-life compared to artemether and so is therefore thought to clear any residual parasites that remain after combination treatment.[2]
Lumefantrine, along with pyronaridine and naphtoquine, were synthesized in course of the Project 523 antimalaria drug research initiated in 1967; these compounds are all used in combination antimalaria therapies.[3][4][5]
Lumefantrine is an antimalarial drug chemically known as 2-(dibutylamino)-1-[(9Z)-2, 7-dichloro-9-(4- chlorobenzylidene)-9H-floren-4-yl] ethanol, which is used in the prevention and treatment of Malaria in worm blooded animals. Lumefantrine is using the combination of β-Artemether in the treatment of Malaria
SYN
Synthetic Reference
Beutler, Ulrich; Fuenfschilling, Peter C.; Steinkemper, Andreas. An Improved Manufacturing Process for the Antimalaria Drug Coartem. Part II. Organic Process Research & Development. Volume 11. Issue 3. Pages 341-345. 2007.
SYN 2
Synthetic Reference
Rao, Dharmaraj Ramachandra; Kankan, Rajendra Narayanrao; Phull, Manjinder Singh. Process for preparation of lumefantrine as antimalarial agent with improved method. Assignee Cipla Co., Ltd., India. CN 1865227. (2006).
SYN 3
Synthetic Reference
Sethi, Madhuresh Kumar; Gonuguntla, Anantavena Rani; Arikatla, Siva Lakshmi Devi; Mulukutla, Suryanarayana; Yerramalla, Rajakrishna; Bontalakoti, Jaganmohanarao; Vemula, Lakshminarayana; Thirunavukarasu, Jayaprakash. Synthesis and characterization of novel related substances of Lumefantrine, an anti-malarial drug. Pharma Chemica. Volume 8. Issue 3. Pages 91-100. 2016.
SYN4
Synthetic Reference
Mathur, Prafull; Mathur, Suvigya; Vishwanath, Kannan; Mishra, Anand Kumar. Preparation of lumefantrine. Assignee Aanjaneya Lifecare Limited, India. IN 2013MU00611. (2015).
SYN 5
Synthetic Reference
Wu, Guang-liang; Dai, Ying-jie; Kang, Cong-min; Zi, Yan. A new synthetic technology of anti-malarial drug lumefantrine. Zhongguo Xinyao Zazhi. Volume 21. Issue 24. Pages 2944-2947. 2012.
SYN 6
Synthetic Reference
Krishna, Bettadapura Gundappa; Verma, Sudhakar; Krishna, Sujatha; Naik, Gajanan; Arulmoli, Thangavel. A process for preparation of lumefantrine. Assignee SeQuent Scientific Limited, India. IN 2012CH00470. (2012).
SYN 7
Synthetic Reference
Bansi, Lal; Genbhau, Gund Vitthal; Prabhakar, Bapat Chintamani; Popat, Bochiya Pravin; Banshi, Punde Dnyanadeo; Venkata, Reddy Prabhakar Gorla. Improved one pot process for the synthesis of lumefantrine. Assignee Calyx Chemicals and Pharmaceuticals Ltd., India. IN 2009MU01437. (2010).
SYN 8
Synthetic Reference
Shailesh, Singh; Dhaval, Vashi; Vinod, Gaikwad; Sanjay, Chowkekar; Sanjay, Bute. Process for the preparation of lumefantrine. Assignee Ajanta Pharma Ltd., India. IN 2008MU01677. (2010).
SYN 9
Synthetic Reference
Rawalnath, Sakhardande Rajiv; Kanji, Khatri Navin; Nilkanth, Firake Pandharinath; Vasant, Panchal Rajesh; Nagesh, Babrekar Chandan; Madhukar, Mohite Dhanaji. Preparation of lumefantrine. Assignee Saxena, Alok, India. IN 2006MU00260. (2007).
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Embodiment 1:The synthesis of (the chloro- 2- of 2- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) ethyoxyl) trimethyl silane
2,7- dichloro fluorenes -4- oxirane (100g, 0.368mol), zinc oxide are sequentially added in 1000ml there-necked flasks (2.94g, 0.036mol) and 500ml dichloromethane, TMSCl (43g, 0.4mol) are added drop-wise in above-mentioned reaction solution, room temperature reaction 2h.Filtering, a small amount of dichloromethane washing of filter cake, filtrate washing, organic phase anhydrous sodium sulfate drying, filtering, filtrate are rotated to analysis After going out solid, stop revolving, stand and separate out a large amount of yellow solids, filtering, drain to obtain (the chloro- 2- of 2- (2,7- bis- chloro- 9H- fluorenes- 4- yls) ethyoxyl) trimethyl silane yellow solid 102g, yield 73%.δ(1HNMR,CDCl3):7.78-7.80(m,1H), 7.56(s,1H),7.48(s,1H),7.41(s,1H),7.32-7.35(m,1H),5.64-5.67(m,1H),4.03-4.11(m, 2H),3.84(m,2H),0.63(s,9H),ppm。
Embodiment 2:N- butyl-N- (1- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) -2- (trimethylsiloxy group) ethyl) butyl – The synthesis of 1- amine:
(the chloro- 2- of 2- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) ethyoxyl) trimethyl silicane is sequentially added in 1000ml there-necked flasks Alkane (100g, 0.26mol), di-n-butylamine (67g, 0.52mol), potassium carbonate (71.65g, 0.52mol) and 800ml acetonitriles.Put It is changed to nitrogen system, back flow reaction 16h.After being down to room temperature, filtering, a small amount of acetonitrile of filter cake washs, and filtrate is evaporated to obtain fourth containing N- The crude yellow oil of base-N- (1- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) -2- (trimethylsiloxy group) ethyl) butyl -1- amine. Crude product is directly used in be synthesized in next step, without purifying.MS:479(MH+)
Embodiment 3:The synthesis of 2- dibutyl aminos -2- [the chloro- 9H- fluorenes -4- bases of 2,7- bis-] ethanol
By-the N- of butyl containing N- obtained in the previous step (1- (the chloro- 9H- fluorenes -4- bases of 2,7- bis-) -2- (trimethylsiloxy group) second Base) crude yellow oil of butyl -1- amine is dissolved in 500ml tetrahydrofurans, concentrated hydrochloric acid (1ml), nitrogen protection, heating is added dropwise To 50 DEG C of reactions, TLC tracing detections to raw material have reacted completely, cool, and separate out solid, filtering, the washing of filter cake tetrahydrofuran, take out Dry, filtrate is abandoned, and filter cake is transferred in beaker, adds 200ml dichloromethane, and saturated aqueous sodium carbonate is adjusted pH=8, separated Machine phase, water layer are extracted once with 200ml dichloromethane again, merge organic phase, are washed, and are dried, are concentrated to give yellow oil 68g, two step yields 64%.δ(1HNMR,CDCl3):7.80(s,1H),7.56(s,1H),7.38(m,2H),7.25(s,1H), 4.7(brs,1H),4.03-4.11(m,2H),3.99(m,1H),3.56(m,2H),2.42-2.85(m,4H),1.44-1.48 (m,4H),1.23-1.28(m,4H),0.86-0.91(m,6H).ppm。
Embodiment 4:(RS, Z) -2- dibutyl aminos -2- [bis- chloro- 9- of 2,7- (4- chlorobenzenes methylene) -9H- fluorenes -4- bases] second The synthesis of alcohol
2- dibutyl aminos -2- [the chloro- 9H- fluorenes -4- bases of 2,7- bis-] ethanol (68g, 0.167mol) is dissolved in 200ml ethanol In, 4-chloro-benzaldehyde (28.2g, 0.2mol) and sodium hydroxide (2.68g, 0.067mol) are added, nitrogen protection, is heated to reflux anti- Answer 1h.Room temperature is down to, watery hydrochloric acid, which is added dropwise, makes product be filtered into salting out, and the washing of filter cake ethanol, drains to obtain yellow solid, shifts Into beaker, 200ml dichloromethane is added, adds saturated aqueous sodium carbonate to dissociate, be layered, aqueous phase uses 200ml dichloromethane again Extraction once, merges organic phase, washing, and anhydrous sodium sulfate drying 6h is filtered, and filtrate is concentrated to give (RS, Z) -2- dibutyl aminos -2- [2,7- bis- chloro- 9- (4- chlorobenzenes methylene) -9H- fluorenes -4- bases] ethanol be yellow foam shape solid 74g, yield 83%, 0-5 DEG C Preserve.δ(1HNMR,CDCl3):7.75(brs,1H),7.66(s,1H),7.55(s,1H),7.44-7.48(m,5H),7.28- 7.29(m,1H),7.30-7.31(m,1H),4.7(brs,2H),3.88-3.89(m,2H),2.71-2.72(m,2H),2.60- 2.63(m,2H),1.44-1.48(m,4H),1.23-1.31(m,4H),0.86-0.90(m,6H).ppm。
Process for high purity lumefantrine Dr KrishnaSarma Pathy,Ramesh.D,P atchutaramaiah,Ch Sivasubramanyam Abstract. The present invention relates to Process for the preparation of lumefantrine Lumefantrine is a dichlorobenzylidine derivative effective for the treatment of various types of malaria. Chemically lumefantrine is 2-Dibutylamino-1-[ 2, 7-dichloro-9-(4- chlorobenzylidene)-9H-fluoren-4-yl]-Ethanol (racemate) The antimalarial agent is active against multi-drug resistant strains of Plasmodium falciparum. In combination with artemether, the drug is also used for the treatment of uncomplicated falciparum malaria. It has primary action as blood schizontocidal and secondary action as inhibition of nucleic acid and protein synthesis within the malarial parasite thus having a longer duration of anti malarial action. Thus, today lumefantrine is a drug of choice in antimalarial treatment against P. faliciparum. Therefore, development of an appropriate analytical procedure for the quantitative analysis of lumefantrine is of considerable importance to pharmaceutical industry. The spectroscopic techniques were used to confirm the identity of lumefantrine. The IR spectra, showed strong absorption band at 3404.67 cm-1(OH), 2953.28 cm-1(aliphatic and aromatic CH), 1757.31 cm-1(-C=C-), 933 cm-1(alkanes) and 696.37-373.22 cm-1 (Cl). Thus, IR spectra confirmed the presence of these functional groups in the structure of lumefantrine. The mass spectrum showed a sharp molecular ion peak at 528.0 m/z in Q1 MS (m/z, parent ion) parameter at negative polarity confirming the molecular weight of lumefantrine. The NMR spectra observed triplet at 0.943-0.989 (methyl protons of alkyl chain); a multiplet at 1.372-1.498 (methylene protons of alkyl chains); a multiplet at 2.449-2.909 (methylene protons of alkyl chain); broad singlet at 4.573 (OH proton); and multiplet at 7.314-7.733 (aromatic proton), thus confirming identity of lumefantrine. Summary of the process Stage1 Cl Cl 2,7-dichloro-9H-fluorene ClCl O Cl 2-chloro-1-(2,7-dichloro-9H-fluoren-3-yl)ethanone AlCl3 Chloroacetyl chloride In a 1 liter 3-necked flask, equipped with stirrer, thermometer and reflux condenser450.0 ml MDC cool to 0C.Start addition of 59.4 gm chloro acetyl chloride at 0-5C and stir for 15 min.Start addition of aluminium chloride at 0-5C. Stir for 30.0 min at same temp.Start addition of 2,7-dichloro fluorene soln. preprepared in 300.0 ml MDC d 150.0 ml saturated brine for washing and stir for 15-20 min. at 50-55 C.Allow to settle the layers at 50- 55C for 30.0 min.Separate the ORGANIC LAYER. at 50-55C.Again repeat Abovestep. for one time.Collect ORGANIC LAYER. and distill out dibutyl amine under vac. At 60- 90C.Disconnect vac. And cool reaction mass to 60C add 400.0 ml methanol and reflux to 30.0 min. After maintaining the reaction mass cool reaction mass to 50C.Seed the reaction mass by adding 500 mg stage 2 material at . 50C.Cool reaction mass to 30- 35C.Stir reaction mass further at same temp. fot 2 hrs.Filter reaction mass at 30- 35C.Suck dry and wash with 50.0 ml methanol at 30-35C.Suck dry and unload. Dry at 50-55C. Wt. of wet cake : 139.0 gm Wt. of dry material : 123.0 gm . . . a b c . . . a b c M.P: 72-76C Stage3: ClCl N OH + Cl COH ClCl N OH Cl 2,7-dichloro-alpha-[(dibutyl amino) p-chloro benzaldehyde methyl]-9H-fluoren-4-yl]oxirane Mol. Wt. 406 Lumefantrine Mol. Wt. 140.5Mol. Wt. 528.5 Under Nitrogen In a 2 liter 3-necked flask, equipped with stirrer, thermometer and reflux.Charge 840.0 ml ethanol at 30-35C.Chrge 120.0gm stage2 material at 30-35C.Charge 27.96 gm sodium methoxide in 30 min. at 30C exothermobserved temp. rise to 45C.Charge 41.52 gm p- Chloro benzaldehyde .Maintain reaction mass temp. 30-35C for 24 hrs. Check TLC a : Stage 2 b : Co spot c : reaction mass Mobile phase Hexane : Ethyl acetate 9 : 1 If TLC complies 1) 2) 3) Filter reaction mass at 30-35C. Suck dry and wash with 100.0 ml methanol. Suck dry and unload wet cake. Dry material at 50-55 C . . . a bc Wt. of wet cake : 184.0 gm Wt. Of dry material : 141.0 gm M.P : 122-126C Purification : In a 1 liter 3-necked flask, equipped with stirrer, thermometer and reflux .Charge 700.0 ml ethyl acetate at 30-35C.Chrge crude 141.0 gm material at 30-35C.Heat reaction mass to refluxand main for 30.0 min. to dissolve the material.Filter reaction mass at 75-80C.Collect filtrate cool to reaction mass to 0- 5C.Maintain reaction mass at same temp. for 30.0 min.Filter the reaction mass at 0- 5C.Suck dry and wash with 100.0 ml ethyl acetate at 0-5C.Suck dry and unload. Dry material at 50-55 C Wt. of wet cake : 100.0 gm Wt. Of dry material : 70-80 gm (19) (PDF) Process for high purity lumefantrine. Available from: https://www.researchgate.net/publication/221933417_Process_for_high_purity_lumefantrine [accessed Feb 06 2022].
M.P: 128-132C RESULTS:= Lumefantrine 99.15% (by HPLC) 128 – 130 Purity Melting Pointo C Liquid chromatography HPLC-UV investigation of the impurity profiles was performed on a HPLC-PDA apparatus consisting of a Waters Alliance 2695 separation module and a Waters 2998 photodiode array detector with Empower 2 software for data acquisition . For PDA detection, the UV spectrum was recorded at 190-400 nm. Quantification was performed at 266 nm. The positive ion ESI and the collision-induced dissociation (CID) mass spectra were obtained from the LC-UV/MS apparatus consisting of a Spectra System SN4000 interface, a Spectra System SCM1000 degasser, a Spectra System P1000XR pump, a Spectra System AS3000 autosampler, and a Finnigan LCQ Classic ion trap mass spectrometer in positive ion mode mass to charge range m/z 100 to m/z 2000 at unit resolution and with a peak width of 0.25 daltons/z, equipped with a Waters 2487 dual wavelength UV detector and Xcalibur 2.0 software (Thermo) for data acquisition. ESI was conducted using a needle voltage of 4.5 kV. Nitrogen was used as sheath and auxiliary gas with the heated capillary set at 250C. UV-detection was used for quantification (at 266 nm), while ESI-ion trap MS detection was used for identification. LC determination of impurities in lumefantrine samples was performed using a Purospher STAR RP-18 endcapped (150 4.6 mm, 5 m particle size) column (Merck, Darmstadt, Germany) with guard column at 30C under isocratic conditions with a mobile phase consisting of ammonium acetate (pH 4.9; 0.1 M) and acetonitrile (10:90, v/v). The flow rate was set at 2.0 mL/min (minimal run time: 30 min.). The injection volume was 10 l. Under these conditions, lumefantrine elutes at approximately 22 min. System suitability tests (SSTs) were established as the plate number on lumefantrine (N 8.2 103) and the oxidative stress degradation product (N 2.4 103), the signal-to-noise ratio of 0.5% l.c. lumefantrine solution (S/N 30), the peak area ratio of the 0.5% l.c. versus 100% l.c. (between 0.4 and 0.6) and the relative position of the in-situ prepared N-oxide by H2O2 treatment (RRTbetween 0.12 and 0.22). The LC method was validated for the determination of lumefantrine and its related impurities. The selectivity of the developed chromatographic method was established by the separation of lumefantrine and its impurities. A correlation coefficient (r2) of 0.9998 for lumefantrine (0.0006 to 0.01 mg/ml) and 1.0 for impurity A and DBK (0.001 to 0.1 mg/ml) demonstrated that the HPLC method is linear in the lower range. LOD/LOQ values for lumefantrine, DBK and impurity A were calculated (S/N = 3 resp. 10): 0.004 mg/mland 0.026 mg/ml for lumefantrine (0.004% respectively 0.026% l.c.), 0.011 mg/ml and 0.040 mg/ml for DBK (0.012% respectively 0.042% l.c.) and 0.110 mg/ml and 0.393 mg/ml for impurity A (0.115% respectively 0.409% l.c.). The analytical stability of lumefantrine, impurity A and DBK was confirmed over a storage period of 24 hours at 5C, i.e. the sample compartment temperature. Accuracy and precision were evaluated by repeated analysis (n = 6), with 102.6% l.c. recovery and 2.1%, respectively 2.86%, for repeatability, respectively intermediate precision. Structural information for the observed and/or reported lumefantrine related impurities # Compound [formula, mono-isotopic mass]Structure Origin # Compound [formula, mono-isotopic mass]Structure Origin Alkaline stress Oxidative stress Oxidative stress Metabolite 1Desbenzylketo N-oxide [C23H27NO3Cl2, MW 435.14] 2 Lumefantrine (mono-)desbutyl derivative [C26H24NOCl3, MW 473.09] 3Lumefantrine N-oxide [C30H32NO2Cl3, MW 543.15] Oxidative stress Degradation 4 2,7-dichloro-4-[2-(di-n-butylamino)-1-hydroxyethyl]- 9H-fluoren-9-one; Desbenzylketo derivative (DBK) [C23H27NO2Cl2, MW 419.14] Oxidative stress Acidic stress Degradation 5 2-(di-n-butylamino)-1-[2,7-dichloro-9H-fluoren-4- yl]ethanol; Desbenzyl derivative [C23H29NOCl2,405.16] Synthesis 6 Synthesis impurity found in lumefantrine API; Lumefantrine oxide [C30H32NO2Cl3, MW 543.14] Synthesis 7 Synthesis impurity found in lumefantrine API; Lumefantrine oxide [C30H32NO2Cl3, MW 543.14] Synthesis 8 (RS,Z)-2-(Dibutylamino)-2-(2,7-dichloro-9-(4- chloro-benzylidene)-9H-fluoren-4-yl)ethanol (isomeric compound); Impurity A (Ph. Int./USP Salmous) [C30H32NOCl3, MW 527.15] Synthesis 9 Synthesis (19) (PDF) Process for high purity lumefantrine. Available from: https://www.researchgate.net/publication/221933417_Process_for_high_purity_lumefantrine [accessed Feb 06 2022].
mpurity found in lumefantrine API; Lumefantrine oxide [C30H32NO2Cl3, MW 543.14] Synthesis 10 (1S,3R,5R)-1,3-bis((EZ)-2,7-Dichloro-9-(4- chlorobenzyl-idene)-9H-fluoren-4-yl)-2,6- dioxabicyclo[3.1.0]hexane; Impurity BA(USP Salmous) [C44H24Cl6O2, 797.39] 2-((EZ)-2,6-Dichloro-9- (4-chlorobenzylidene)-9H- fluoren-4-yl)-3′-((EZ)-2,7-dichloro-9-(4- chlorobenzylidene)-9H-fluoren-4-yl)-2,2′-bioxirane; Impurity BB (USP Salmous) [C44H24Cl6O2, 797.39] Percentage maximum actual levels of lumefantrine related impuritiesobserved(1) Synthesis 11 Synthesis # CompoundAPIFPP Unstressed Stressed Release Accelerated Stressed 1.39 0.560.11 21.32 1 Desbenzylketo N-oxide 2 Monodesbutyl derivative 3 Lumefantrine N-oxide 4 Desbenzylketo derivative 5 Desbenzyl derivative 6Lumefantrine oxide (RRT ~ 0.49) 7Lumefantrine oxide (RRT ~ 0.52) 8 Impurity A 9Lumefantrine oxide (RRT ~ 0.59) (1) RT: reporting threshold = 0.10% 0.60 0.12 0.34 0.86 4.26 HPLC characteristics of lumefantrine related impurities # CompoundRT(1)RRT(2) Most abundant m/z observed 436.14 474.00 544.08 420.13 406.09 544.12 544.12 528.10 544.12 528.10 1 Desbenzylketo N-oxide 2 Monodesbutyl derivative 3.25 3 Lumefantrine N-oxide 4 Desbenzylketo derivative 7.41 5 Desbenzyl derivative 6 Lumefantrine oxide 7 Lumefantrine oxide 8 Impurity A 9 Lumefantrine oxide L Lumefantrine (1) Retention time (min.) (2) Relative retention time reference 1.790.08 0.15 0.17 0.33 0.34 0.49 0.52 0.58 0.59 1.00 3.96 7.69 10.96 11.45 12.70 12.97 22.28 1. De Spiegeleer B, Vergote V, Pezeshki A, Peremans K, Burvenich C. Impurity profiling quality control testing of synthetic peptides using liquid chromatography-photodiode array-fluorescence and liquid chromatography-electrospray ionization-mass spectrometry: The obestatin case. Anal Biochem. 2008;376:229234. doi: 10.1016/j.ab.2008.02.014.[ 2. Nicolas EC, Scholz TH. Active drug substance impurity profiling – Part I. LC/UV diode array spectral matching. J Pharm Biomed Anal. 1998;16:813824. doi: 10.1016/S0731-7085(97)00131- 3Roy J. Pharmaceutical impurities – A mini review. AAPS PharmSciTech. 2002;3:article 6. doi: 10.1208/pt030206. ICH guidelines – International Conference on Harmonization, Q3A(R2) Impurities in new drug substances CPMP/ICH/2737/99. (October 2006) http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC50000 2675.pdf [Accessed on 5 November 2010 at 11:06] 3. Authorized Lumefantrine USP Salmous Standard (Februari 2009) http://www.usp.org/pdf/EN/nonUSStandards/lumefantrine.pdf [Accessed on 24 October 2010 at 17:14] 4. Lumefantrine: Document QAS/06.186/FINAL (WHO Ph. Int. – July 2008) http://www.who.int/medicines/publications/pharmacopoeia/Lumef_monoFINALQAS06_186_July 08.pdf [Accessed on 24 October 2010 at 17:37] 5. Lee H. Pharmaceutical applications of liquid chromatography coupled with mass spectrometry (LC/MS) J Liq Chromatogr Relat Technol. 2005;28:11611202. doi: 10.1081/JLC-200053022.] 6. Cesar ID, Nogueira FHA, Pianetti GA. Simultaneous determination of artemether and lumefantrine in fixed dose combination tablets by HPLC with UV detection. J Pharm Biomed Anal. 2008;48:951954. doi: 10.1016/j.jpba.2008.05.022 7. Cesar ID, Nogueira FHA, Pianetti GA. Comparison of HPLC, UV spectrophotometry and potentiometric titration methods for the determination of lumefantrine in pharmaceutical products. J Pharm Biomed Anal. 2008;48:223226. doi: 10.1016/j.jpba.2008.05.006. 8. Patil KR, Rane VP, Sangshetti JN, Shinde DB. A Stability-Indicating LC Method for Lumefantrine. Chromatographia. 2009;69:375379. doi: 10.1365/s10337-008-0894-x. [Cross Ref] 9. Munjal V, Paliwal N, Chaursia BK, Varshney B, Ahmed T, Paliwal J. LC-tandem mass spectrometry method for quantification of lumefantrine in human plasma and its application to bioequivalence study. Chromatographia. 2010;71:505510. doi: 10.1365/s10337-009-1446-8. [Cross Ref] 10. ICH guidelines – International Conference on Harmonization, Q1A(R2) Stability testing of new drug substances and products CPMP/ICH/2736/99. (August 2003) http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC5 (19) (PDF) Process for high purity lumefantrine. Available from: https://www.researchgate.net/publication/221933417_Process_for_high_purity_lumefantrine [accessed Feb 06 2022].
[1] Ulrich Beutler, C Peter.; Fuenfschilling.; and Andreas, Steinkemper.; Novartis Pharma AG; Chemical and Analytical Development: CH-4002 Basel, Switzerland, Organic Process Research & Development 2007, 11, 341- 345.
[2] Boehm, M. Fuenfschilling.; Krieger, P. C.; Kuesters, E. M.; Struber, F.; Org. Process Res. DeV. 2007, 11, 336- 340.
[3] (a) Rao, D. R.; Kankan, R. N.; Phull, M. S.; Patent Application CN 1009-3724 20060424, 2005. (b) Deng, R.; Zhong, J.; Zhao, D.; Wang, J.; Yaoxue, X. 2000, 35 (1), 22. (c) Allmendinger, Th.; Wernsdorfer, W. H. PCT WO 99/67197.
[4] Perrumattam, J.; Shao, Ch.; Confer, W. L. Synthesis 1994, 1181.
[5] Fuenfschilling, P. C.; Hoehn P.; Mutz J.-P. Organic Process Res. Dev. 2007, 11, 13.
[6] Di Nunno, L.; Scilimati, A. Tetrahedron 1988, 44, 3639.
Preparation of 2-(dibutylamino)-1-[(9Z)-2, 7-dichloro-9-(4-chlorobenzylidene)-9H-floren-4-yl] ethanol (Lumefantrine) 1.
To a stirred solution of NaOH (1.97 g 0.0492 mol) in methanol (100 ml) there was added 1-(2, 7- dichloro-9 H-fluren-4-yl)-2-(dibutyl amino) ethanol (10 g, 0.0246 mol) and para chloro benzaldehyde (5.24 g 0.0372). The suspension obtained was stirred at reflux temperature till the absence of starting material by TLC. After confirming the product formation reaction mixture was cooled to room temperature and further stirred at same temperature for overnight. The precipitated solids were filtered and washed with methanol and dried under vacuum at 50°C to get desired compound. (Purity by HPLC: 99%).
[2–4] Thus, today lumefantrine is a drug of choice in antimalarial treatment against P. …. The NMRspectra observed triplet at 0.943-0.989 (methyl protons of alkyl …
The spectroscopic techniques were used to confirm the identity of lumefantrine. The IR spectra, showed strong absorption band at 3404.67 cm-1 (OH), 2953.28 cm-1 (aliphatic and aromatic CH), 1757.31 cm-1 (-C=C-), 933 cm-1 (alkanes) and 696.37-373.22 cm-1 (Cl). Thus, IR spectra confirmed the presence of these functional groups in the structure of lumefantrine.
The mass spectrum showed a sharp molecular ion peak at 528.0 m/z in Q1 MS (m/z, parent ion) parameter at negative polarity confirming the molecular weight of lumefantrine.
The NMR spectra observed triplet at 0.943-0.989 (methyl protons of alkyl chain); a multiplet at 1.372-1.498 (methylene protons of alkyl chains); a multiplet at 2.449-2.909 (methylene protons of alkyl chain); broad singlet at 4.573 (OH proton); and multiplet at 7.314-7.733 (aromatic proton), thus confirming identity of lumefantrine.
IH NMR PREDICT
13C NMR PREDICT
References
Jump up^Toovey S, Jamieson A, Nettleton G (2003). “Successful co-artemether (artemether-lumefantrine) clearance of falciparum malaria in a patient with severe cholera in Mozambique”. Travel medicine and infectious disease. 1 (3): 177–9. doi:10.1016/j.tmaid.2003.09.002. PMID17291911.
Jump up^White, Nicholas J.; van Vugt, Michele; Ezzet, Farkad (1999). “Clinical Pharmacokinetics and Pharmacodynamics of Artemether-Lumefantrine”. Clinical Pharmacokinetics. 37 (2): 105–125. doi:10.2165/00003088-199937020-00002. ISSN0312-5963.
Percent Composition: C 68.12%, H 6.10%, Cl 20.11%, N 2.65%, O 3.02%
Literature References: Racemic aryl alcohol originally synthesized in the 1970’s by the Academy of Military Medical Sciences in Beijing, China. Inhibits hemozoin formation. Prepn: R. Deng et al.,CN1042535 (1990 to Acad. Military Med. Sci., Microbiol. & Epidemic Dis. Instit.); C.A.114, 6046 (1991). LC determn in plasma: A. Annerberg et al.,J. Chromatogr. B822, 330 (2005). In vitro activity against Plasmodium falciparum: B. Pradines et al.,Antimicrob. Agents Chemother.43, 418 (1999).
Properties: Odorless, yellow powder. Poorly sol in water, oil, and most organic solvents. Sol in unsaturated fatty acids.
Derivative Type: Co-artemether
CAS Registry Number: 141204-94-6
Manufacturers’ Codes: CPG-56697
Trademarks: Coartem (Novartis); Riamet (Novartis)
Literature References: Fixed 6:1 mixture with artemether, q.v. Clinical pharmacokinetics and bioavailability: F. Ezzet et al.,Br. J. Clin. Pharmacol.46, 553 (1998). Clinical trial in children against P. falciparum malaria: C. Hatz et al.,Trop. Med. Int. Health3, 498 (1998); in adults: S. Looareesuwan et al.,Am. J. Trop. Med. Hyg.60, 238 (1999). Review of comparative clinical trials in malaria: A. A. Omari et al., Trop. Med. Int. Health9, 192-199 (2004).
Figure 1. Production of artemisinic acid or β-farnesene by engineered yeast. The sesquiterpene alkenes β-farnesene and amorphadiene are both derived from FPP (farnesyl diphosphate) by the action of specific enzymes introduced from plants: amorphadiene synthase (ADS) generates amorphadiene and β-farnesene synthase (FS) generates β-farnesene. Production strains express either ADS or FS, not both. Oxidation of amorphadiene to artemisinic acid is accomplished by the action of five plant enzymes expressed in the engineered yeast.17 Conversion of purified artemisinic acid to artemisinin is accomplished by in vitro organic chemistry. Isoprenoid production strains make little ethanol.
The antimalarial drug artemisinin and the specialty chemical β-farnesene are examples of natural product isoprenoids that can help solve global challenges, but whose usage has previously been limited by supply and cost impediments. This review describes the path to commercial production of these compounds utilizing fermentation of engineered yeast. Development of commercially viable yeast strains was a substantial challenge that was addressed by creation and implementation of an industrial synthetic biology pipeline. Using the engineered strains, production of β-farnesene from Brazilian sugarcane offers several environmental advantages. Among the many commercial applications of β-farnesene, its use as a feedstock for making biodegradable lubricants is highlighted. This example, along with others, highlight a powerful new suite of technologies that will become increasingly important for production of chemicals, spanning from pharmaceuticals through commodity chemicals.
Figure 2. Sanofi industrial semi-synthesis of artemisinin. The process starts with a moderate pressure catalytic diastereoselective hydrogenation of artemisinic acid to produce a high (95:5) ratio of the desired (R)-isomer. To avoid formation of a lactone byproduct, dihydro-epi-deoxyarteannuin B, during the photooxidation, the carboxylic acid is protected as a mixed anhydride. The final step combines formation of the intermediate hydroperoxide via photoxidation using a Hg vapor lamp and commercially available tetraphenylporphyin (TPP) as sensitizer with a Hock cleavage and rearrangement catalyzed by trifluoroacetic acid to give, after workup, the best yield reported to date of pure isolated artemisinin (55%).
Synthetic Biology and the Development of Commercial β-Farnesene Production Strains Semi-synthetic artemisinin is a pharmaceutical with a price point comparable to plant-derived artemisinin,20 namely above $150 per kg. β-Farnesene, however, is a specialty chemical with multiple uses (more details below); most specialty and commodity chemicals have significantly lower price points, often below $10 per kg. For these product categories, it is of paramount importance that fermentative production be as efficient as possible, with high yields (namely, grams of product made per gram of feed substrate), productivities (grams of product/liter of culture/hour) and concentration (also known as titer; grams of product per liter of culture). Developing yeast strains capable of the yield, productivity and titer required for chemical production requires extensive development, and has been enabled over the last decade by the new discipline of synthetic biology. Synthetic biology seeks to extend approaches and concepts from engineering and computation to redesign biology for a chosen function;21recent advances in the application of design automation, i.e., the use of software, hardware and robotics22 have enabled the creation and screening of hundreds of thousands of strain variants (created by both design and random mutagenesis) for the properties required for commercial production of β-farnesene. Notable enabling technologies developed for routine usage include rapid and reliable assembly of large (i.e., multiple kilobase) deoxyribonucleic acid (DNA) constructs;23-25 high throughput, cost effective, verification of structural DNA assemblies by both initial restriction digest26 and by low-cost DNA sequencing;27 and whole genome sequencing of yeast strains.28 In addition, there is a need to effectively identify the best new strains (akin to panning for gold!) through high throughput, rapid, and accurate methods to screen thousands of strains. Further, the results of small-scale (< 1 milliliter) tests must correspond to the results of large-scale (> 50,000 liter) production. Development and implementation of these technologies required considerable investment by Amyris. The outcome is a robust pipeline for efficient, cost-effective strain generation allied with screening for the properties required for commercial production of β-farnesene by fermentation (i.e., at a price point required for its use as a specialty chemical).
As the world’s population and economies grow, the demand for a wide variety of specialty, commodity, and pharmaceutical chemicals will outpace the supply available from current sources. There is an urgent need to develop alternative, sustainable sources of many existing chemicals and to develop abundant sources of currently scarce chemicals with novel beneficial properties. Synthetic biology and industrial fermentation, combined with synthetic chemistry, will be an increasingly important source of chemicals in the decades ahead; artemisinin and β-farnesene provide good examples of this relatively new approach to chemical production. Brazil’s plentiful sugar cane feedstock and fermentation expertise make it an excellent location for this type of manufacturing, which can expand and diversify the nation’s industrial base and international importance.
Benjamin KR, Silva IR, Cherubim JP, Mcphee D, Paddon CJ. Developing Commercial Production of Semi-Synthetic Artemisinin, and of β-Farnesene, an Isoprenoid Produced by Fermentation of Brazilian Sugar. J. Braz. Chem. Soc. 2016;27(8):1339-1345
Kirsten R. Benjamin,a Iris R. Silva,b João P. Cherubim,c Derek McPheea and Chris J. Paddon*,a a Amyris, Inc., 5885 Hollis Street, Suite 100, CA 94608 Emeryville, USA b Amyris Brasil Ltda, Rua John Dalton 301-Bloco B-Edificio 3, Condominio Techno Plaza, 13069-330 Campinas-SP, Brazil c Amyris Brasil Ltda, Rodovia Brotas/Torrinha-km 7.5, 17380-000 Brotas-SP, Brazil
Dr. Paddon has a PhD in Biochemistry from Imperial College, London, but now considers himself a synthetic biologist. After postdoctoral work at the National Institutes of Health in Bethesda, MD, he worked in the pharmaceutical industry (GlaxoSmithKline), and then for two Bay Area biopharmaceutical companies (Affymax and Xenoport) before joining Amyris, Inc. in 2005 as its sixth employee and first scientist. He was project leader for the semi-synthetic artemisinin project at Amyris, Inc. and has subsequently led a number of other projects and programs there.
Chris Paddon is a Principal Scientist at Amyris, Inc. in Emeryville, CA. He was project leader for the Semi-Synthetic Artemisinin project, and subsequently led a number of projects at Amyris using synthetic biology for the production of natural products. He received his Bachelor’s degree in Microbiology from The University of Surrey (UK), and doctorate in Biochemistry from Imperial College (London, UK). Following postdoctoral work at The National Institutes for Health (Bethesda, MD) he joined the pharmaceutical industry, working for GSK (London, UK). He subsequently worked for Affymax (Palo Alto, CA) and Xenoport (Santa Clara, CA) before joining Amyris.
UCT’s H3D is a center of excellence for research and innovation with an already strong track record in malaria drug discovery. The vision of H3D is to be the leading organization for integrated drug discovery and development on the African continent.
ABOUT H3D
H3D is Africa’s first integrated drug discovery and development centre. The Centre was founded at the University of Cape Town in April 2011 and pioneers world-class drug discovery in Africa.
Our Vision
To be the leading organisation for integrated drug discovery and development from Africa, addressing global unmet medical needs.
Our Mission
To discover and develop innovative medicines for unmet medical needs on the African continent and beyond, by performing state-of-the-art research and development and bridging the gap between basic science and clinical studies.
We embrace partnerships with local and international governments, pharmaceutical companies, academia, and the private sector, as well as not-for-profit and philanthropic organisations, while training scientists to be world experts in the field.
The H3D collaboration with the Medicines for Malaria Venture (MMV) focuses on delivering potential agents against malaria that will be affordable and safe to use. In line with the global aim to eradicate malaria, projects are pursued that not only eliminates blood-stage Plasmodium falciparum and Plasmodium vivax infection, but also acts against liver stages and blocks transmission of the infection. The projects embrace multidisciplinary activities to optimise hit compounds from screening libraries through the drug discovery pipeline and deliver clinical candidates.
Merck Serono Announces Recipients of the Second Annual €1 Million Grant for Multiple Sclerosis Innovation
Darmstadt, Germany, September 12, 2014 – Merck Serono, the biopharmaceutical division of Merck, today announced the recipients of the second annual Grant for Multiple Sclerosis Innovation (GMSI) at MS Boston 2014, the joint meeting of the Americas Committee for Treatment and Research in MS (ACTRIMS) and European Committee for Treatment and Research in MS (ECTRIMS), taking place September 10-13 in Boston, U.S.A.
Merck signed a research agreement with the University of Cape Town (UCT), South Africa, to co-develop a new R&D platform. It aims at identifying new lead programs for potential treatments against malaria, with the potential to expand it to other tropical diseases. It combines Merck’s R&D expertise and the drug discovery capabilities of the UCT Drug Discovery and Development Centre, H3D.
UCT’s H3D is a center of excellence for research and innovation with an already strong track record in malaria drug discovery. The vision of H3D is to be the leading organization for integrated drug discovery and development on the African continent. They say that working with partners like Merck is critical to build up a comprehensive pipeline to tackle malaria and related infectious diseases.
Journal Publications:
Aminopyrazolo[1,5-a]pyrimidines as potential inhibitors of Mycobacterium tuberculosis: Structure activity relationships and ADME characterization C. Soares de Melo, T-S. Feng, R. van der Westhuyzen, R.K. Gessner, L. Street, G. Morgans, D. Warner, A. Moosa, K. Naran, N. Lawrence, H. Boshoff, C. Barry, C. Harris, R. Gordon, K. Chibale. Biorg. Med. Chem. 2015, 23, 7240-7250.
A Novel Pyrazolopyridine with in Vivo Activity in Plasmodium berghei- and Plasmodium falciparum- Infected Mouse Models from Structure−Activity Relationship Studies around the Core of Recently Identified Antimalarial Imidazopyridazines. C. Le Manach, T. Paquet, C. Brunschwig, M. Njoroge, Z. Han, D. Gonzàlez Cabrera, S. Bashyam, R. Dhinakaran, D. Taylor, J. Reader, M. Botha, A. Churchyard, S. Lauterbach, T. Coetzer, L-M. Birkholtz, S. Meister, E. Winzeler, D. Waterson, M. Witty, S. Wittlin, M-B. Jiménez-Díaz, M. Santos Martínez, S. Ferrer, I. Angulo-Barturen, L. Street, and K. Chibale, J. Med. Chem. 2015, XX, XXXX
Structure−Activity Relationship Studies of Orally Active Antimalarial 2,4-Diamino-thienopyrimidines. D. Gonzàlez Cabrera, F. Douelle, C. Le Manach, Z. Han, T. Paquet, D. Taylor, M. Njoroge, N. Lawrence, L. Wiesner, D. Waterson, M. Witty, S. Wittlin, L. Street and K. Chibale. J Med Chem. 2015, 58, 7572-7579.
Medicinal Chemistry Optimization of Antiplasmodial Imidazopyridazine Hits from High Throughput Screening of a SoftFocus Kinase Library: Part 2. Le Manach, T. Paquet, D. Gonzalez Cabrera, Y. Younis, D. Taylor, L. Wiesner, N. Lawrence, S. Schwager, D. Waterson, M.J. Witty, S. Wittlin, L. Street, and K. Chibale. J. Med. Chem. 2014, 57, 8839−8848.
Medicinal Chemistry Optimization of Antiplasmodial Imidazopyridazine Hits from High Throughput Screening of a SoftFocus Kinase Library: Part 1. Le Manach, D. González Cabrera, F. Douelle, A.T. Nchinda, Y. Younis, D. Taylor, L. Wiesner, K. White, E. Ryan, C. March, S. Duffy, V. Avery, D. Waterson, M. J. Witty, S. Wittlin; S. Charman, L. Street, and K. Chibale. J. Med. Chem. 2014, 57, 2789-2798.
2,4-Diamino-thienopyrimidines as Orally Active Antimalarial Agents. D. González Cabrera, C. Le Manach, F. Douelle, Y. Younis, T.-S. Feng, T. Paquet, A.T. Nchinda, L.J. Street, D. Taylor, C. de Kock, L. Wiesner, S. Duffy, K.L. White, K.M. Zabiulla, Y. Sambandan, S. Bashyam, D. Waterson, M.J. Witty, A. Charman, V.M. Avery, S. Wittlin, and K. Chibale. J. Med. Chem. 2014,57, 1014-1022.
Effects of a domain-selective ACE inhibitor in a mouse model of chronic angiotensin II-dependent hypertension. Burger, T.L. Reudelhuber, A. Mahajan, K. Chibale,E.D. Sturrock, R.M. Touyz. Clin. Sci. (Lond). 2014, 127(1), 57-63.
Pharmacokinetic evaluation of lisinopril-tryptophan, a novel C-domain ACE inhibitor. Denti, S.K. Sharp, W.L. Kröger, S.L. Schwager, A. Mahajan, M. Njoroge, L. Gibhard, I. Smit, K. Chibale, L. Wiesner, E.D. Sturrock, N.H. Davies. Eur. J. Pharm. Sci.2014, 56, 113-119.
Fragment-based design for the development of N-domain-selective angiotensin-1-converting enzyme inhibitors. R.G. Douglas, R.K. Sharma, G. Masuyer, L. Lubbe, I. Zamora, K.R. Acharya, K. Chibale, E.D. Sturrock. Sci. (Lond). 2014, 126(4),305-313.
Fast in vitro methods to determine the speed of action and the stage-specificity of anti-malarials in Plasmodium falciparum. Le Manach, C. Scheurer, S. Sax, S. Schleiferböck, D. González Cabrera, Y. Younis, T. Paquet, L. Street, P.J. Smith, X. Ding, D. Waterson, M.J. Witty, D. Leroy, K. Chibale and S. Wittlin*. Malaria Journal, 2013, 12, 424.
Structure-Activity-Relationship Studies Around the 2-Amino Group and Pyridine Core of Antimalarial 3,5-Diarylaminopyridines Lead to a Novel Series of Pyrazine Analogues with Oral in vivo Activity. Y. Younis, F. Douelle, González Cabrera, C. Le Manach, A.T. Nchinda, T. Paquet, L.J. Street, K.L. White, K. M. Zabiulla, J.T. Joseph, S. Bashyam, D. Waterson, M.J. Witty, S. Wittlin, S.A. Charman, and K. Chibale* J. Med. Chem. 2013, 56, 8860−8871.
Cell-based Medicinal Chemistry Optimization of High Throughput Screening (HTS) Hits for Orally Active Antimalarials-Part 2: Hits from SoftFocus Kinase and other Libraries. Y. Younis, L. J. Street, D. Waterson, M.J. Witty, and K. Chibale. J. Med. Chem. 2013, 56, 7750−7754.
Structure-Activity Relationship Studies of Orally active Antimalarial 3,5-Substituted 2-Aminopyridines. D. González Cabrera, F. Douelle, Y. Younis, T.-S. Feng, C. Le Manach, A.T. Nchinda, L.J. Street, C. Scheurer, J. Kamber, K. White, O. Montagnat, E. Ryan, K. Katneni, K.M. Zabiulla, J. Joseph, S. Bashyam, D. Waterson, M.J. Witty, S. Charman, S. Wittlin, and K. Chibale* J. Med. Chem. 2012, 55, 11022– 11030.
3,5-Diaryl-2-aminopyridines as a Novel Class of Orally Active Antimalarials Demonstrating Single Dose Cure in Mice and Clinical Candidate Potential. Y. Younis, F. Douelle, T.-S. Feng, D. González Cabrera, C. Le Manach, A.T. Nchinda, S. Duffy, K.L. White, M. Shackleford, J. Morizzi, J. Mannila, K. Katneni, R. Bhamidipati, K. M. Zabiulla, J.T. Joseph, S. Bashyam, D. Waterson, M.J. Witty, D. Hardick, S. Wittlin, V. Avery, S.A. Charman, and K. Chibale*. J. Med. Chem. 2012, 55, 3479−3487.
Novel Orally Active Antimalarial Thiazoles. D. González Cabrera, F. Douelle, T.-S Feng, A.T. Nchinda, Y. Younis, K.L. White, Wu,E. Ryan, J.N. Burrows,D. Waterson, M.J. Witty,S. Wittlin,S.A. Charman and K. Chibale. J. Med. Chem. 2011, 54, 7713–7719.
Synthesis and molecular modeling of a lisinopril-tryptophan analogue inhibitor of angiotensin I-converting enzyme. A.T. Nchinda, K. Chibale, P. Redelinghuys and E.D. Sturrock. Med. Chem. Lett. 2006, 16(17), 4616-4619.
Patents
Anti-Malarial Agents. Y. Younis, K. Chibale, M.J. Witty, D. Waterson. (2016) US9266842 B2.
New Anti-Malarial Agents. D. Waterson, M.J. Witty, K. Chibale, L. Street, D. González Cabrera, T. Paquet. EP patent application (2015), No. 15 176 514.6.
Preparation of aminopyrazine compounds as antimalarial agents for treatment of malaria. Y. Younis, K. Chibale, M.J. Witty, D. Waterson. PCT Int Appl. (2013), WO 2013121387 A1 20130822.
Preparation of peptides as angiotensin I-converting enzyme (ACE) inhibitors. E.D. Sturrock, A.T. Nchinda, K. Chibale. PCT Int. ppl. (2006), WO 2006126087 A2 20061130.
Preparation of peptides as angiotensin I-converting enzyme (ACE) inhibitors, E.D. Sturrock, A.T. Nchinda, K. Chibale. PCT Int. ppl. (2006), WO 2006126086 A2 20061130.
Head Office, Medicinal Chemistry Unit
Physical Address:
Department of Chemistry
7.32 H3D Lab Suite, PD Hahn Building, Level 7
North Lane off Ring Road
Upper Campus, University of Cape Town
Rondebosch, 7700, South Africa
T | 021 650 5495
F | 021 650 5195
Postal Address:
University of Cape Town
Private Bag X3
Rondebosch 7701
South Africa
P. D. Hahn Bldg, Rondebosch, Cape Town,
P. D. Hahn Bldg, Rondebosch, Cape Town, 7700, South Africa
//////H3D, Africa, integrated drug discovery and development centre, University of Cape Town
Artemisinin, a secondary metabolite of sweet wormwood, is the basis for the production of the most effective antimalarial drugs. Since the amount of artemisinin currently produced from plants is not sufficient to treat the worldwide malaria cases, an effective semisynthetic method was developed recently that is capable of producing artemisinin from dihydroartemisinic acid (DHAA). DHAA is a byproduct obtained during the extraction of artemisinin from plant leaves. The photocatalytic reaction to convert DHAA to artemisinin can be performed continuously in a tubular reactor using toluene as a solvent. The reactor effluent contains besides artemisinin the photocatalyst (dicyanoanthracene) and several compounds that are structurally similar to artemisinin, including unreacted DHAA starting material. To isolate artemisinin from the reaction mixture, two separation techniques were applied, crystallization and chromatography. The solid obtained by seeded cooling crystallization was highly enriched in artemisinin but contained also traces of the photocatalyst. In contrast, using a variant of continuously operated multicolumn simulated moving bed (SMB) chromatography, which splits the feed into three fractions, we were able to recover efficiently the photocatalyst in the raffinate stream. The extract stream provided already almost pure artemisinin, which could be finally further purified in a simple crystallization step.
Properties: Crystalline stable material, mp 124°. Highly lipophilic, not sol in water. Stable in soln except in the presence of strong bases or strong reducing agents.
Melting point: mp 124°
Therap-Cat: Antimalarial
The oxidation of (5R)-(-)-carvone (I) with 3-chloroperbenzoic acid (3-CPB) in dichloromethane gives 5(R)-acetyl-2-methyl-2-cyclohexen-1-one (II), which is condensed with ethyltriphenylphosphonium bromide (III) by means of butyllithium in THF yielding 2-methyl-5(Z)-(1-methyl-1-propenyl)-2-cyclohexen-1-one (IV). The photochemical oxidation of (IV) in acetonitrile catalyzed by methylene blue affords (1R,4RS,5R,8S)-4,8-dimethyl-4-vinyl-2,3-dioxabicyclo[3.3.1]nonan-7-one (V), which is ozonolyzed with O3 in methanol to the corresponding aldehyde as a mixture of enantiomers, which is submitted to crystallization giving the (1S,4R,5R,8S)-isomer (VI). Finally, this compound is submitted to a Wittig condensation with 2,4-bis(trifluoromethyl)benzyltriphenylphosphonium bromide (VII) by means of sodium bis(trimethylsilyl)amide (NaBTSA) in dichloromethane.
……………………….
Literature References:
Synthetic sesquiterpene peroxide; structurally derived from the natural peroxides artemisinin, q.v. and yingzhaosu. Prepn: W. Hofheinz et al.,EP311955; eidem,US4977184 (1989, 1990 both to Hoffmann-La Roche).
Series of articles on prepn, biological activities, pharmacokinetics and clinical evaluations: Trop. Med. Parasitol.45, 261-291 (1994).

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











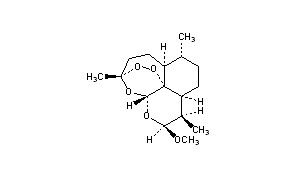
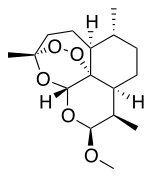
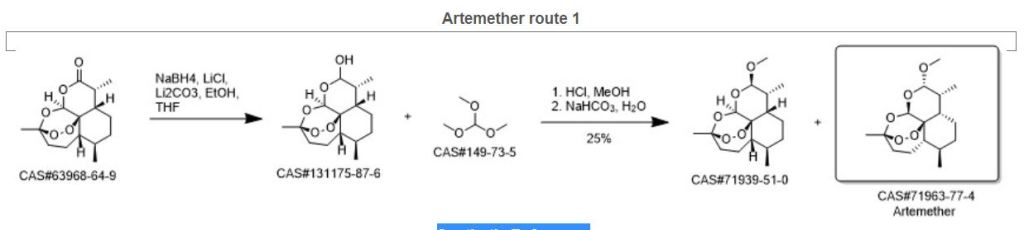
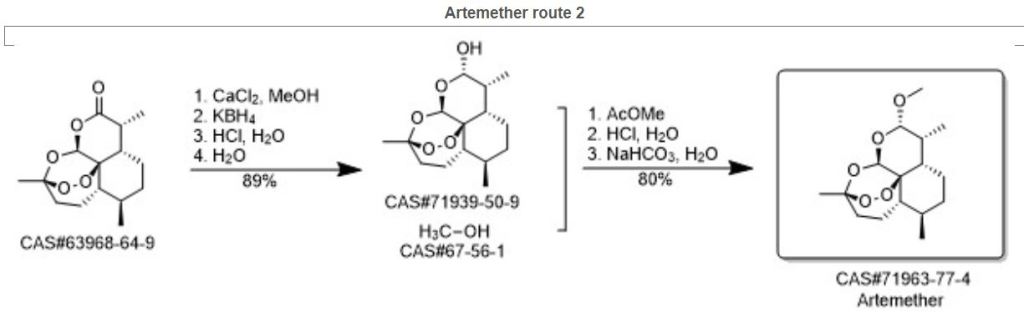
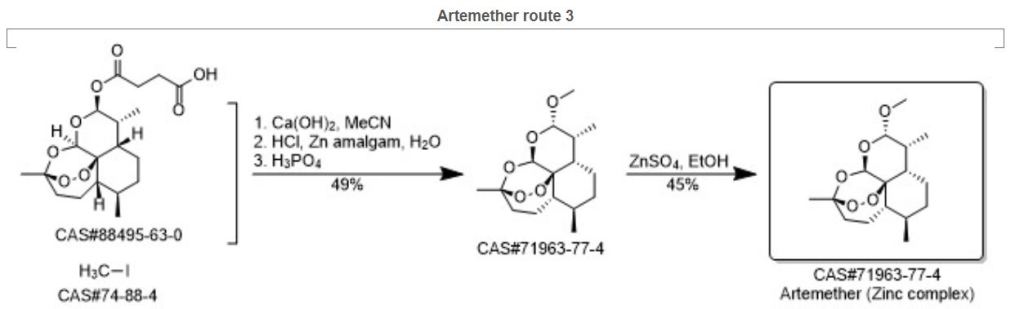
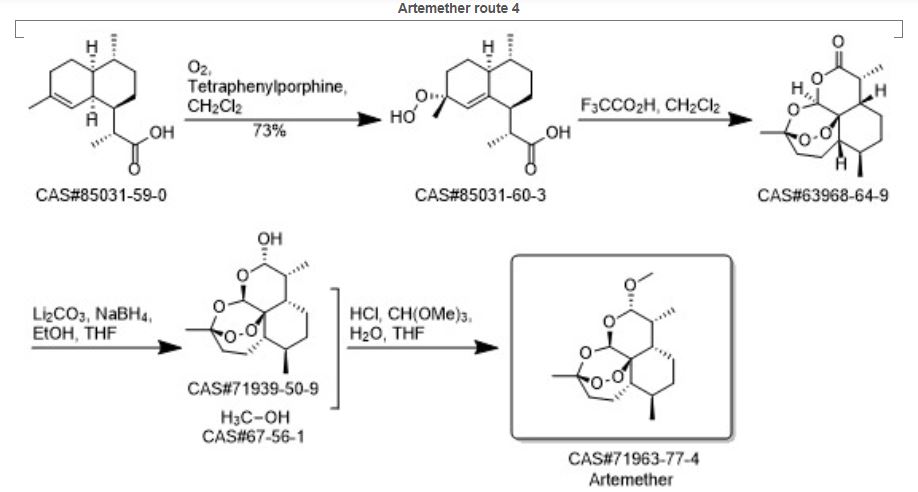
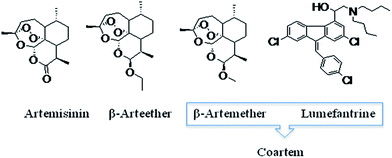
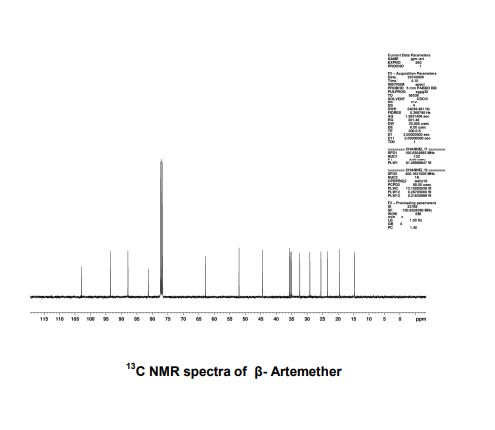






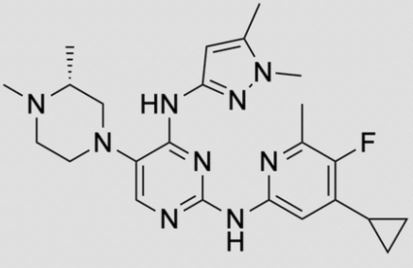

![2-N-(4-cyclopropyl-5-fluoro-6-methylpyridin-2-yl)-5-[(3R)-3,4-dimethylpiperazin-1-yl]-4-N-(1,5-dimethylpyrazol-3-yl)pyrimidine-2,4-diamine.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=92045019&t=l)









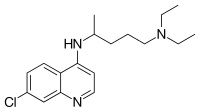
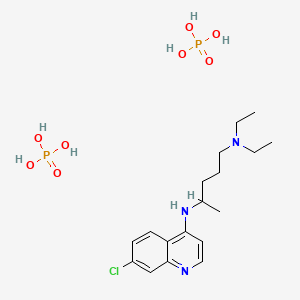



.jpg)


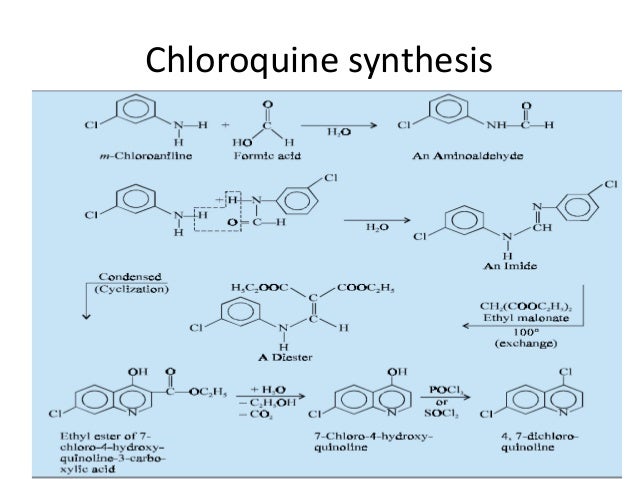
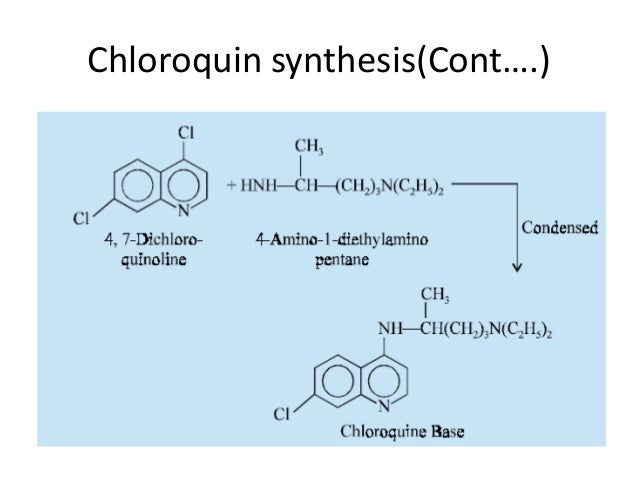

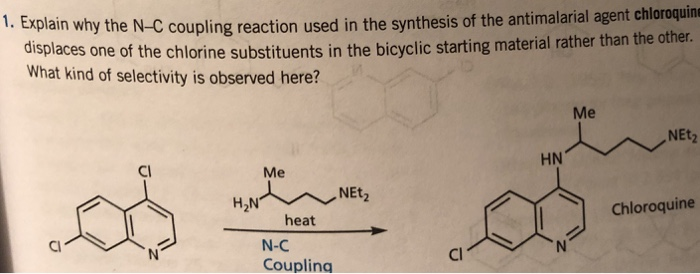




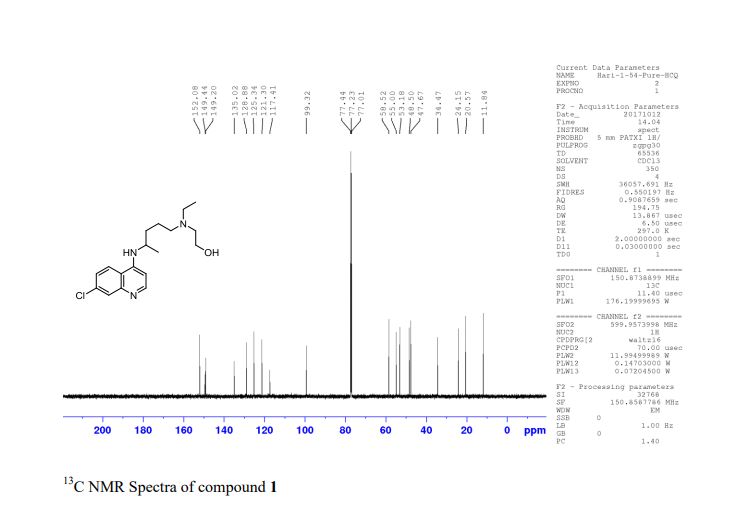
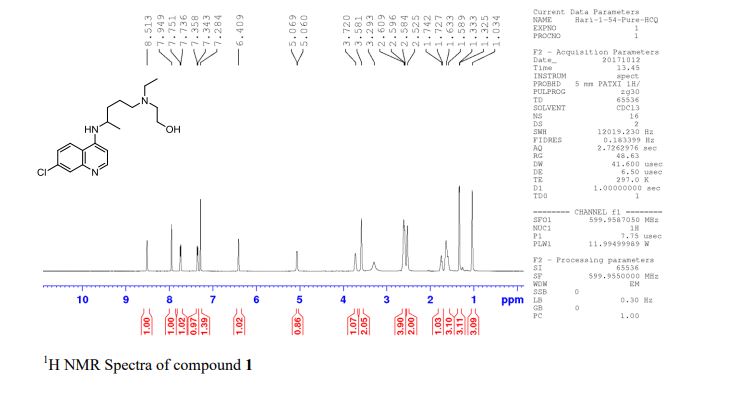












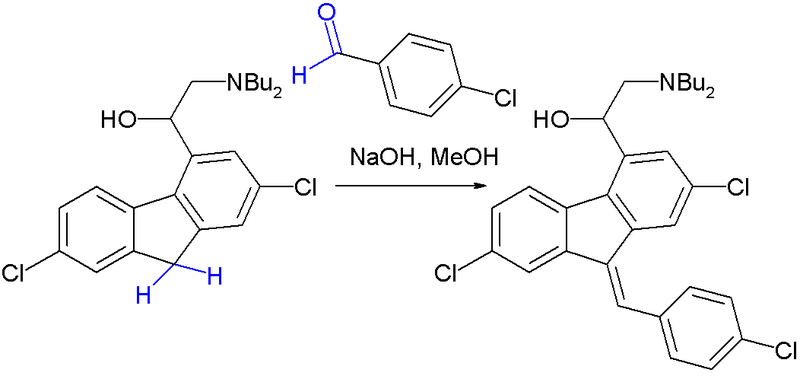








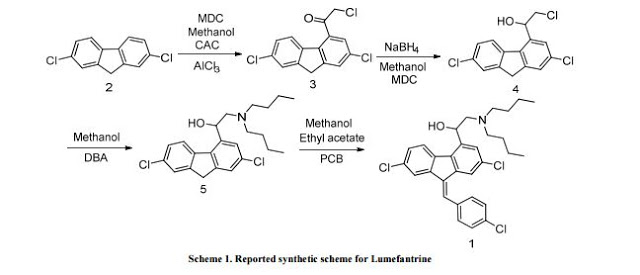




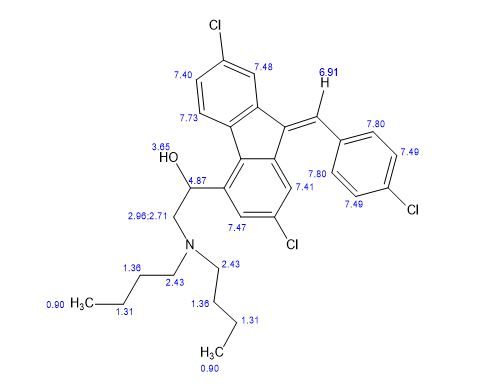
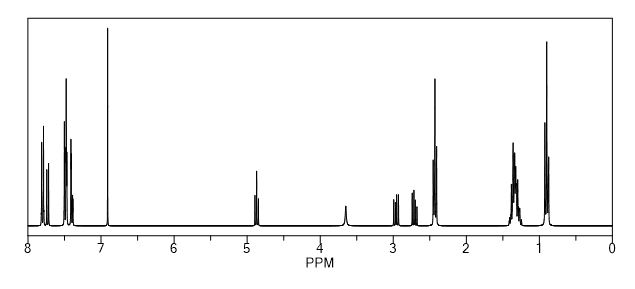
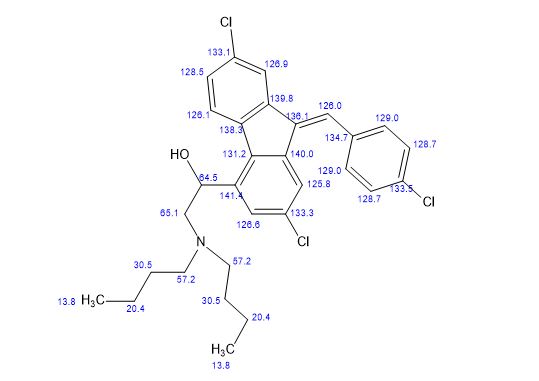
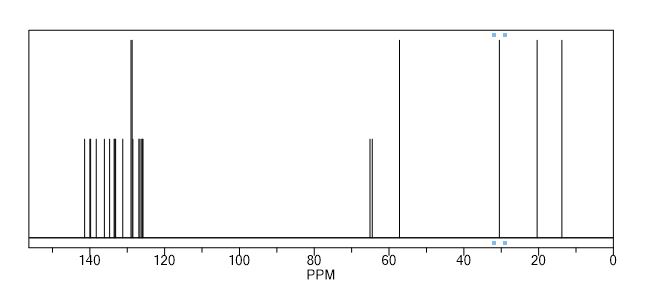



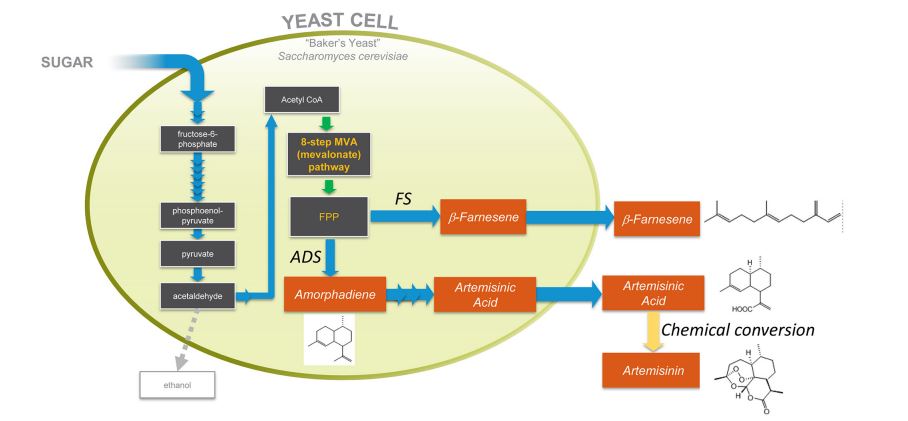
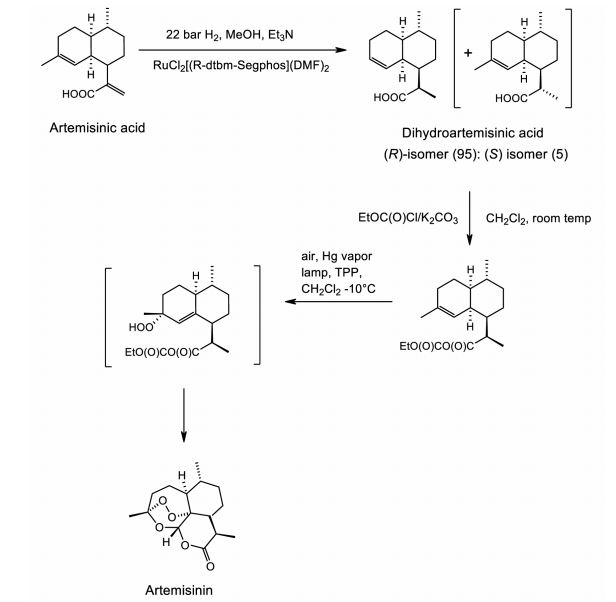
 Genes encoding the biosynthetic pathway for production of a valuable product (e.g., farnesene) in a native organism are expressed in a heterologous microbial host (e.g., yeast). The engineered yeast produces farnesene by commercial fermentation. Copyright © 2016 Amyris, inc. All rights reserved.
Genes encoding the biosynthetic pathway for production of a valuable product (e.g., farnesene) in a native organism are expressed in a heterologous microbial host (e.g., yeast). The engineered yeast produces farnesene by commercial fermentation. Copyright © 2016 Amyris, inc. All rights reserved.

















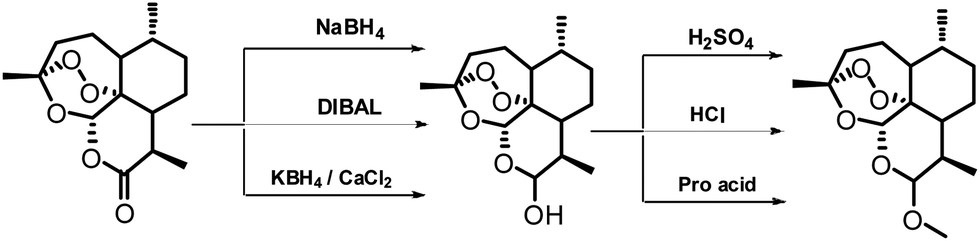{kind=link}
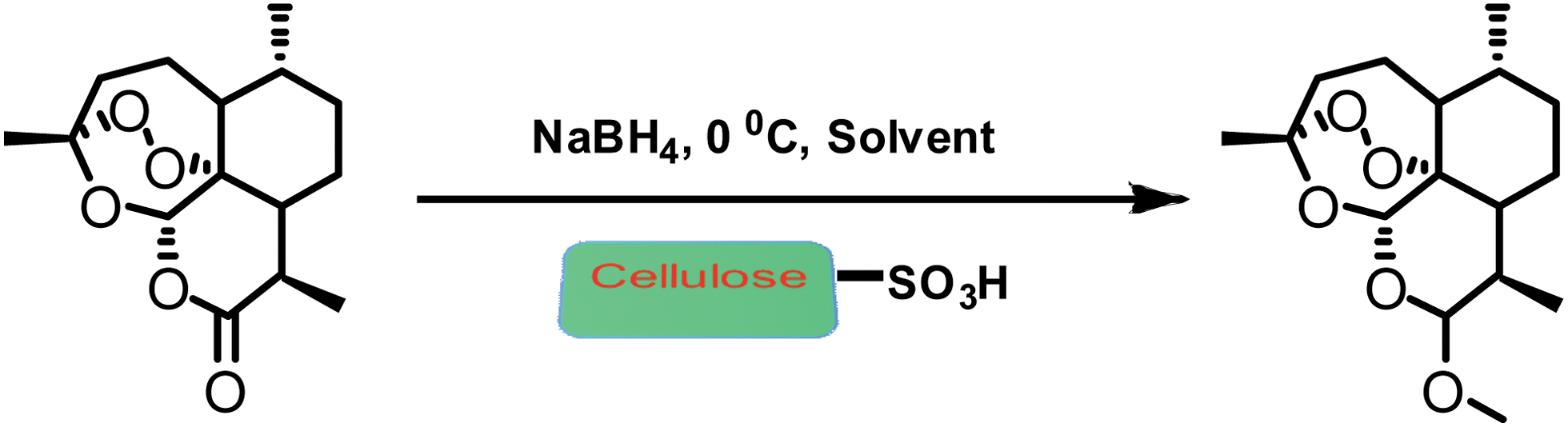{kind=link}
{kind=link}
{kind=link}
{kind=link}