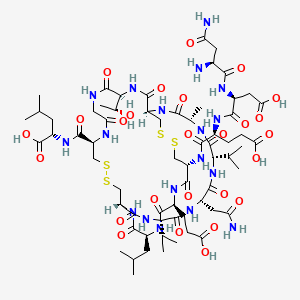

PLECANATIDE; UNII-7IK8Z952OK; (3-Glutamic acid(D>E))human uroguanylin (UGN); 467426-54-6;
IUPAC Condensed
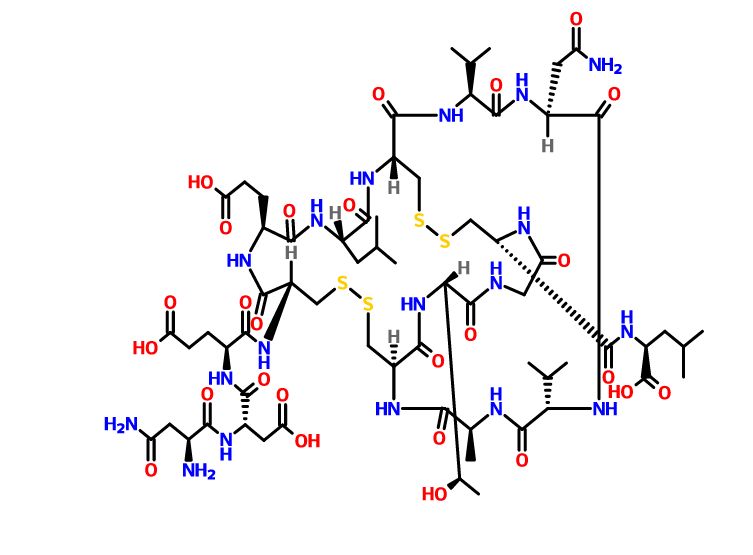
H-Asn-Asp-Glu-Cys(1)-Glu-Leu-Cys(2)-Val-Asn-Val-Ala-Cys(1)-Thr-Gly-Cys(2)-Leu-OH
(2S)-2-[[(1R,4S,7S,10S,13S,16R,21R,27S,34R,37S,40S)-10-(2-amino-2-oxoethyl)-34-[[(2S)-4-carboxy-2-[[(2S)-3-carboxy-2-[[(2S)-2,4-diamino-4-oxobutanoyl]amino]propanoyl]amino]butanoyl]amino]-37-(2-carboxyethyl)-27-[(1R)-1-hydroxyethyl]-4-methyl-40-(2-methylpropyl)-3,6,9,12,15,23,26,29,35,38,41-undecaoxo-7,13-di(propan-2-yl)-18,19,31,32-tetrathia-2,5,8,11,14,22,25,28,36,39,42-undecazabicyclo[14.13.13]dotetracontane-21-carbonyl]amino]-4-methylpentanoic acid
L-Asparaginyl-L-α-aspartyl-N-{(1R,4S,7S,10S,13S,16R,19S,22S,25R,32S,38R)-10-(2-amino-2-oxoethyl)-22-(2-carboxyethyl)-38-{[(1S)-1-carboxy-3-methylbutyl]carbamoyl}-32-[(1R)-1-hydroxyethyl]-19-isobut yl-7,13-diisopropyl-4-methyl-3,6,9,12,15,18,21,24,30,33,36-undecaoxo-27,28,40,41-tetrathia-2,5,8,11,14,17,20,23,31,34,37-undecaazabicyclo[14.13.13]dotetracont-25-yl}-L-α-glutamine
Plecanatide
RN: 467426-54-6
- Molecular FormulaC65H104N18O26S4
- Average mass1681.886 Da
-
- 105: PN: WO2012037380 SEQID: 105 claimed protein
- 1: PN: US20100152118 SEQID: 1 claimed protein
- 1: PN: WO2008151257 SEQID: 1 claimed protein
- 1: PN: WO2010065751 SEQID: 1 claimed protein
- 1: PN: WO2011069038 SEQID: 1 claimed sequence
- 1: PN: WO2012037380 SEQID: 1 claimed protein
- 1: PN: WO2014131024 SEQID: 1 claimed protein
- 1: PN: WO2015054649 SEQID: 1 claimed protein
- 20: PN: WO02078683 SEQID: 20 claimed protein
- 78: PN: WO2010065751 SEQID: 105 claimed protein
- Plecanatide
- SP 304


Plecanatide
- Molecular FormulaC65H104N18O26S4
- Average mass1681.886 Da
плеканатид [Russian] [INN]
بليكاناتيد [Arabic] [INN]
7IK8Z952OK
Guanilib
L-Asparaginyl-L-α-aspartyl-N-{(1R,4S,7S,10S,13S,16R,19S,22S,25R,32S,38R)-10-(2-amino-2-oxoethyl)-22-(2-carboxyethyl)-38-{[(1S)-1-carboxy-3-methylbutyl]carbamoyl}-32-[(1R)-1-hydroxyethyl]-19-isobut yl-7,13-diisopropyl-4-methyl-3,6,9,12,15,18,21,24,30,33,36-undecaoxo-27,28,40,41-tetrathia-2,5,8,11,14,17,20,23,31,34,37-undecaazabicyclo[14.13.13]dotetracont-25-yl}-L-α-glutamine
L-α-Glutamine, L-asparaginyl-L-α-aspartyl-N-[(1R,4S,7S,10S,13S,16R,19S,22S,25R,32S,38R)-10-(2-amino-2-oxoethyl)-22-(2-carboxyethyl)-38-[[[(1S)-1-carboxy-3-methylbutyl]amino]carbonyl]-32-[(1R)- 1-hydroxyethyl]-4-methyl-7,13-bis(1-methylethyl)-19-(2-methylpropyl)-3,6,9,12,15,18,21,24,30,33,36-undecaoxo-27,28,40,41-tetrathia-2,5,8,11,14,17,20,23,31,34,37-undecaazabicyclo[14.13.13]dotetracont-2 5-yl]-

CLIPS
April 19, 2016
Novel Chronic Idiopathic Constipation Drug Under FDA Review
 Plecanatide is a once-daily, oral, uroguanylin analog
Plecanatide is a once-daily, oral, uroguanylin analogSynergy Pharmaceuticals announced the Food and Drug Administration (FDA) has accepted for review the New Drug Application (NDA) for plecanatide for the treatment of chronic idiopathic constipation (CIC).
The NDA submission was based on data from two double-blind, placebo-controlled Phase 3 trials and one open-label long term safety study in over 3,500 patients with CIC.
RELATED: NDA Submitted for Chronic Idiopathic Constipation Drug Plecanatide
The FDA has set a Prescription Drug User Fee Act (PDUFA) target action date of January 29, 2017 to make a decision on the NDA.
Plecanatide is a once-daily, oral, uroguanylin analog currently under development for the treatment of CIC and irritable bowel syndrome with constipation (IBS-C). It is designed to replicate the function of uroguanylin, a naturally occurring GI peptide, by working locally in the upper GI tract to stimulate digestive fluid movement and support regular bowel function.
PATENT
CN 104628827
http://www.google.com/patents/CN104628827A?cl=en
Prica exenatide Synergy Pharmaceuticals developed by the United States for the GC-C receptor in development of drugs, administered orally Limited.Currently underway include chronic idiopathic constipation (CIC) and constipation irritable bowel syndrome (IBS-C), including the phase III clinical trials. It is expected to receive US FDA clearance to market in recent years. Prica that peptides CAS: 467426-54-6 English name plecanatide, structural formula is as follows:
Preparation Prica that peptides from Shenzhen Han Yu medicine was first reported (CN103694320A), using a solid-phase synthesis of linear peptides in solution and then the two-step method to get into the ring, respectively. Since the method to form a ring carved in solution twice, the solution of complex composition, separation and purification difficult, the method should be improved.
Example 1
Weigh the degree of substitution of 0. 51mmol / g of Fmoc-Leu- Wang resin 10g (5. Lmmol), added to the solid phase reactor, DMF washing 3 times, the swelling 3h. The volume ratio of 1: 4 piperidine: DMF was added to the reactor the reaction, after the reaction was washed with DCM and washed twice, DMF 4 times. Weigh Fmoc-Cys (Acm) -OH 6. 34g, H0Bt 2. 07g, DIC 2. 37mL was dissolved in DMF, added to the reactor uniformly mixed, the reaction at room temperature 2h. Ninhydrin color reaction control endpoint, the resin was colorless indicates the end of the reaction, the reaction is continued if the color to colorless. After completion of the reaction, DCM was washed twice, DMF and washed 4 times.
Repeat the above steps, in accordance with the order of the sequence, followed by deprotection, coupling Fmoc-Gly-OH, Fmoc-Thr (tBu) -OH, Fmoc-Cys- (Mmt) -OH, Fmoc-Ala-OH, Fmoc- Val-OH, Fmoc-Asn (Trt) -〇H, Fmoc-Val-OH, Fmoc-Cys (Acm) -OH, Fmoc-Leu-OH, Fmoc-Glu (OtBu) -OH, Fmoc-Cys (StBu) -OH, Fmoc-Glu (OtBu) -OH, Fmoc-Asp (OtBu) -OH, Boc-Asn (Trt) -〇H〇
To a prepared peptide resin reactor volume percentage of 15% DMF solution of mercapto ethanol, reaction 2h; then DCM was added a solution of 20-fold amount DTNP reaction lh; was added after washing 1% TFA containing TIS 5% of DCM solution reaction 20min.
Preparation of peptide resin obtained after sufficiently washed with DMF, DMF was added 10 times the amount in the reaction solution 12 lh. Full wash sash.
After the preparation of the peptide resin was added in a volume ratio of 95/2/2/1 TFA / TIS / EDT / H lysis reagent 20 is added in an amount 20mL / g, the reaction ice bath lh, stirring was continued at room temperature 5h, then filtration.After lysis reagent suction filtrate using a rotary evaporator until no overflow TFA, precipitated reagent was added standing; Pulika centrifugation the precipitated crude peptide was peptide to give 8. 67g〇
The preparation of the crude peptide was obtained Pulika peptide using preparative HPLC system, wavelength 214nm, C18 reversed-phase column packing for the separation, the mobile phase of water and acetonitrile were used, with a gradient elution method to collect the target polypeptide The absorption peak. Using rotary evaporation at 30 ° C to remove most of the acetonitrile, were freeze-dried to obtain a purified Prica exenatide refined products.
Example 2
Weigh the degree of substitution of 0. 2mmol / g of Fmoc-Leu- Wang resin 10g (2mmol), added to the solid phase reactor. DMF washing 3 times, the swelling 3h. The volume ratio of 1: 4 piperidine: DMF was added to the reactor the reaction, after the reaction was washed with DCM and washed twice, DMF 4 times. Weigh Fmoc-Cys (Acm) -OH1. 24g, HOBtO. 406g, DIC 0 • 465mL dissolved in DMF solution, after mixing into the reactor at room temperature the reaction 2h.Ninhydrin color reaction control endpoint, the resin was colorless indicates the end of the reaction, the reaction is continued if the color to colorless. After completion of the reaction, DCM was washed twice, DMF and washed 4 times.
Repeat the above steps, in accordance with the order of the sequence, followed by deprotection, coupling Fmoc-Gly-OH, Fmoc-Thr (tBu) -OH, Fmoc-Cys- (Mmt) -OH, Fmoc-Ala-OH, Fmoc- Val-OH, Fmoc-Asn (Trt) -〇H, Fmoc-Val-OH, Fmoc-Cys (Acm) -OH, Fmoc-Leu-OH, Fmoc-Glu (OtBu) -OH, Fmoc-Cys (StBu) -OH, Fmoc-Glu (OtBu) -OH, Fmoc-Asp (OtBu) -OH, Boc-Asn (Trt) -〇H〇
[0053] To illustrate the preparation of the present embodiment obtained peptide resin reactor volume percent of a DMF solution of 30% mercaptoethanol, reaction 4h; then 5-fold amount DTNP in DCM reaction lh; was added after washing 1% TFA containing TIS 5% in DCM reaction 20min.
Preparation of peptide resin obtained after sufficiently washed with DMF, 20 times the amount of DMF was added in the reaction solution 12 lh. Full wash sash.
Peptide Resin [0055] Preparation was added volume ratio of 82. 5/5/5/5/2. 5 TFA / thioanisole / H20 / phenol / EDT cleavage reagents, added in an amount 10mL / g, the reaction ice bath 0 After. 5h, stirring was continued at room temperature for lh, then suction filtered. After lysis reagent suction filtrate to the non-use of force blowing TFA overflow, adding precipitation reagent standing; centrifugation precipitated Prica exenatide crude peptide to give 1. 52g.
The preparation of the crude peptide was obtained Pulika peptide using preparative HPLC system, wavelength 214nm, C18 reversed-phase column packing for the separation, the mobile phase of water and acetonitrile were used, with a gradient elution method to collect the target polypeptide The absorption peak. Using rotary evaporation at 30 ° C to remove most of the acetonitrile, were freeze-dried to obtain a purified Prica exenatide refined products.
Example 3
Weigh the degree of substitution of 0. 6mmol / g of Fmoc-Leu- Wang resin 10g (6mmol), added to the solid phase reactor, DMF washing 3 times, the swelling 3h. The volume ratio of 1: 4 piperidine: DMF was added to the reactor the reaction, after the reaction was washed with DCM and washed twice, DMF 4 times. Weigh Fmoc-Cys (Acm) -OH 7. 46g, H0Bt2. 44g, DIC 2. 79mL was dissolved in DMF, added to the reactor uniformly mixed, the reaction at room temperature 2h.Ninhydrin color reaction control endpoint, the resin was colorless indicates the end of the reaction, the reaction is continued if the color to colorless. After completion of the reaction, DCM was washed twice, DMF and washed 4 times.
Repeat the above steps, in accordance with the order of the sequence, followed by deprotection, coupling Fmoc-Gly-OH, Fmoc-Thr (tBu) -OH, Fmoc-Cys- (Mmt) -OH, Fmoc-Ala-OH, Fmoc- Val-OH, Fmoc-Asn (Trt) -〇H, Fmoc-Val-OH, Fmoc-Cys (Acm) -OH, Fmoc-Leu-OH, Fmoc-Glu (OtBu) -OH, Fmoc-Cys (StBu) -OH, Fmoc-Glu (OtBu) -OH, Fmoc-Asp (OtBu) -OH, Boc-Asn (Trt) -〇H〇
To the prepared peptide resin reactor volume percentage of 25% DMF solution of mercapto ethanol, reaction 3h; then 10-fold amount DTNP in DCM reaction lh; was added 1% TFA washed containing TIS5% DCM solution Reaction 20min〇
Preparation of peptide resin obtained after sufficiently washed with DMF, 15 times the amount of DMF was added in the reaction solution 12 lh. Full wash sash.
Preparation of the peptide resin was added in a volume ratio of 90/5/3/2 TFA / thioanisole / anisole / EDT cleavage reagents, added in an amount 20mL / g, the ice bath was reacted 0.lh, stirring was continued at room temperature The reaction 10h, then filtration. After lysis reagent suction filtrate using a rotary evaporator until no overflow TFA, precipitated reagent was added standing; Pulika centrifugation the precipitated crude peptide was peptide to give 8. 46g.
The preparation of the crude peptide was obtained Pulika peptide using preparative HPLC system, wavelength 214nm, C18 reversed-phase column packing for the separation, the mobile phase of water and acetonitrile were used, with a gradient elution method to collect the target polypeptide The absorption peak. Using rotary evaporation at 30 ° C to remove most of the acetonitrile, were freeze-dried to obtain a purified Prica exenatide refined products.
Although the above has been described with general, specific embodiments and test, the present invention has been described in detail, but on the basis of the present invention, it may make some changes or improvements, which the skilled artisan It is obvious. Thus, the present invention without departing from the spirit on the basis of these modifications or improvements made, belong to the scope of the invention as claimed.
PATENT
CN 104211777
http://www.google.com/patents/CN104211777A?cl=en
The pickup exenatide (Plecanatide) is a synthetic analogue of guanylin urine (urine guanylin is a natriuretic hormone, can regulate gastrointestinal transport of ions and liquid), pickup exenatide enter After in vivo and guanylate gastrointestinal tract endothelial cells cyclase C binding and activation, activation of the cystic fibrosis transmembrane conductance regulator (CFTR), to promote chloride and water into the intestine, thereby promoting bowel motility, improve constipation symptoms.
Synergy company announced its pick in the research of new drugs that peptide (code: SP304) on October 6, 2010 the treatment of gastrointestinal disorders II a clinical experimental results. The study, conducted in patients with chronic constipation showed that the drugs can improve bowel function in patients, promote intestinal motility and reduce abdominal discomfort shape. In the experiment, there was no diarrhea and other adverse reactions, at the doses tested did not detect the pickup system that peptides are absorbed. The drug is expected for the treatment of chronic constipation (CC), constipation-predominant irritable bowel syndrome (IBS-C) and other gastrointestinal disorders. CC and IBS-C is a common gastrointestinal disease that can cause serious impact on the work and the quality of life of patients. Synergy will continue to conduct clinical trials of other pickups that peptide.
The structure of the peptide pickup that is:
H-Asn-Asp-Asp-Cys-Glu-Leu-Cys-Val-Asn-Val-Ala-Cys-Thr-Gly-C ys-Leu-〇H (4-12 disulfide, 7- 15)
Example 30:
H-Asn-Asp-Asp-Cys-Glu-Leu-Cys-Val-Asn-Val-Ala-Cys-Thr-Gly-C ys-Leu-〇H (4-12 disulfide, 7- 15) Preparation of
embodiments will be prepared by the method of Example 18 H-Asn (Trt) -Asp (OtBu) -Asp (OtBu) -Cys (mmt) -Glu (Ot Bu) -Leu-Cys (StBu) -Val-Asn ( Trt) -Val-Ala-Cys (mmt) -Thr (tBu) -Gly-Cys (StBu) -Leu-CT C resin (IOOmmol, 472. 88g) disposed cracking reactor to 10ml / g resin ratio Add lysis reagent (TFA: EDT: water = 95: 2 5:.. 2 5 (V / V)), stirred at room temperature 2h. The reaction was filtered with sand core funnel, and then added a small amount of TFA The resin was washed in the funnel, collecting the filtrate, the combined filtrate was concentrated. Frozen in dry diethyl ether was added (100ml / g peptide purpose tree months) and the solution was precipitated, centrifuged to remove the precipitate was washed with diethyl ether after dry ether three times, and dried in vacuo to give a white solid powder was approximately 180g, i.e., H-Asn-Asp-Asp -Cys-Glu-Leu-Cys (StBu) -Val-Asn-Val-Ala-Cys-Thr-Gly-Cy s (StBu) -Leu-OH. The solid was dissolved with water to lmg / ml solution. Was added an aqueous solution of 1% by volume of H2O2, the reaction was stirred at room temperature 30min, to prepare H-Asn-Asp-Asp-Cys-Glu-Leu-Cys (StBu) -Val-Asn-Val-Ala-Cys-Thr-Gl y-Cys (StBu) -Leu-OH (disulfide 4-12) was treated with a rotary evaporator after drying the compound containing 500ml 20% β- mercaptoethanol and 0. IM N- methylmorpholine were dissolved in water, followed by stirring After 12h the reaction, the reaction solution was diluted with water to 3mg / ml was about 60L, dissolved in ethanol was added with IL 300mmol I2 solution, the reaction was stirred at room temperature 2h. Adding an appropriate amount Vc remove excess I2, until the color of the reaction solution was transparent, i.e., to give H-Asn-Asp-Asp-Cys-Glu-Leu-Cys-Val-As n-Val-Ala-Cys-Thr-Gly-Cys-L eu_0H (disulfide bonds 4-12, 7-15).
PATENT
WO 2014197720
CN 103694320
WO 2012118972
WO 2012037380
WO 2011069038
US 20100152118
WO 2010065751
///Plecanatide, 普卡那肽 , ليكاناتيد , плеканатид, 467426-54-6, Chronic Idiopathic Constipation, NDA, SP 304, SYNERGY, PEPTIDE,
C[C@H]1C(=O)N[C@H]2CSSC[C@@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@H](NC(=O)CNC(=O)[C@@H](NC2=O)[C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)O)C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N1)C(C)C)CC(=O)N)C(C)C)CC(C)C)CCC(=O)O)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CC(=O)N)N
OR
O=C(N[C@@H](CC(=O)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H]1CSSC[C@@H]2NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CCC(=O)O)NC1=O)CC(C)C)CSSC[C@H](NC(=O)CNC(=O)[C@@H](NC2=O)[C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)O)C(C)C)C(C)C)[C@@H](N)CC(N)=O
updated
Plecanatide (brand name Trulance), is a drug approved by the FDA for the treatment of chronic idiopathic constipation (CIC)[1] and irritable bowel syndrome with constipation. Plecanatide is an agonist of guanylate cyclase-C. Plecanatide increases intestinal transit and fluid through a buildup of cGMP.[2][3]
Medical uses
As of January 2017, plecanatide is approved in the United States for the treatment of chronic idiopathic constipation in adults.[1] The presence of this condition is determined using the Rome III diagnostic criteria for chronic constipation which requires that the patient meet stool frequency, stool consistency, incomplete evacuation, and straining requirements in addition to not being a likely candidate for irritable bowel syndrome.[4] The symptoms should also have been present for at least three of the last six months to establish the chronic nature of the condition before treatment with plecanatide is indicated.[4]
Plecanatide has been shown to be safe and effective. It has shown to be at least equally as effective as its main competitor, linaclotide (brand name Linzess), but has been shown to have a lower rate of diarrhea as an adverse drug reaction.[5]
Contraindications
Plecanatide has not been shown to be safe or effective in persons 6 years to 18 years of age.[6] Use of plecanatide by persons under the age of 6 poses a serious dehydration risk and studies have demonstrated plecanatide can cause death in juvenile mice due to this dehydrating effect.[6]
Use of plecanatide is also contraindicated in persons who are suspected of having a mechanical gastrointestinal obstruction.[6]
Pharmacology
Structure and function
Plecanatide is a 16 amino acid peptide with the amino acid sequence:
H-Asn1-Asp2-Glu3-Cys4-Glu5-Leu6-Cys7-Val8-Asn9-Val10-Ala11-Cys12-Thr13-Gly14-Cys15-Leu16-OH
Is nearly structurally identical to human uroguanylin, apart from the substitution of Asp3 with Glu3.[7] Disulfide bonds exist between Cys4 and Cys12, as well as Cys7 and Cys15.[8]
Plecanatide has two important motifs. The first being the acidic residues Asp2 and Glu3 which modulate the affinity for its receptor in response to environmental pH.[6][7][9] Simulations predict the optimal activity of Plecanatide to occur at pH 5, making it suitable for targeting cells within the proximal intestine, which has a pH of between 5 and 6.[6] The second is the ACTGC motif (residues Ala11 to Cys15) which is the region responsible for its binding to the receptor, guanylate cyclase-C.[10]
Mechanism of action
Plecanatide works as a laxative by drawing water in to the gastrointestinal tract thereby softening stool and encouraging its natural passage.
Similar to its endogenous counterpart, plecanatide activates guanylate cyclase-C on endothelial cells within the gastrointestinal tract.[7] The activation of guanylate cyclase-C catalyses the production of the second messenger guanosine 3’,5’-cyclic monophosphate (cGMP) which leads to the protein kinase A (PKA) and protein kinase G II (PKGII)-mediated phosphorylation of the cystic fibrosis transmembrane conductance regulator (CFTR) protein.[11][12] CFTR is an anion channel and upon activation it will secrete negatively charged ions, particularly chloride (Cl−) and bicarbonate (HCO3−) in to the GI tract lumen.[13][14] This disruption to the electrochemical gradient is in part rectified by the passive secretion of positively charged sodium ions in to the lumen and water follows by osmosis.[13]
Plecanatide is also known to have an anti-nociceptive effect in animal models, however the exact mechanism of action is not yet fully elucidated.[6] It has been suggested that this may be in part to the anti-inflammatory action of guanylate cyclase-C by its inhibition of pro-inflammatory cytokines, or through the inhibition of associated sensory neurons.[15]
Pharmacokinetics and metabolism
As plecanatide acts on receptors present on the apical side of endothelial cells lining the gastrointestinal tract it is able to impart its effect without ever entering circulation.[7] As with most orally ingested peptides, plecanatide is degraded by intestinal enzymes, and so very little of the active drug enters systemic circulation.[6] Minimal amounts of the drug are expected to be transported in to the body, and concentrations of plecanatide and its metabolites are undetectable in plasma following the recommended dosage of 3 mg.[6][7] It has also been shown that dosages up to 48.6 mg produced no detectable concentration of plecanatide in human plasma at any time point after ingestion.[7]
Commercialization
Plecanatide, branded as Trulance, is manufactured by Synergy Pharmaceuticals.[16]
PATENT
WO-2020250102
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020250102&tab=PCTDESCRIPTION&_cid=P12-KJ0SK6-65801-1Plecanatide is an agonist of the guanylate cyclase type-C receptor (“GCC agonists”). Plecanatide is a 16 amino acid peptide with the following chemical name: L- Leucine, L-asparaginyl-L-a-aspartyl-L-a-glutamyl-L-cysteinyl-L-a-glutamyl-L-leucyl-L-cysteinyl-L-valyl-L-asparaginyl-L-valyl-L-alanyl-L-cysteinyl-L-threonylglycyl-Lcysteinyl-, cyclic (4 12),(7 15)-bis(disulfide). The amino acid sequence for Plecanatide is shown below:

Formula I
Plecanatide is approved in the United States under the trade name TRULANCE™ for treatment of Chronic Idiopathic Constipation (CIC) in adult patients.
Plecanatide is described in US patent No. 7,041,786. The US‘786 patent discloses that the peptide including plecanatide are synthesized and purified (>95% purity) using a published procedure of Klodt, et al., J. Peptide Res. 50:222-230 (1997). The article discloses general solid-phase synthesis, deprotection of Fmoc groups with 20% piperidine in NMP, deprotection of dry peptidyl resins by using a mixture of TFA/EDT/H2O, formation of disulfide bond by air oxidation and with iodine, acidification with TFA, purification using preparative C1S-HPLC column (buffer A: 0.1% TFA, buffer B: 0.1% TFA in MeCN/water, 80:20).
US Patent No. US 9,580,471 describes a process for preparation of plecanatide using combination of solid and solution phase synthesis. Further, US ‘471 describes purification process of peptide on RP-HPLC column followed by desalination by eluting column with aq. alcohol, concentration of obtained fractions and then precipitation with diethyl ether or MTBE to obtain Plecanatide.
The inventors of the present invention developed an improved process for the preparation of pure Plecanatide, which is simple, cost-effective, and avoids or reduces content of impurities and makes the process robust.
EXAMPLES
EXAMPLE 1: Synthesis of H-Gly-Cys(Acm)-Leu-Otbu (Fragment B)
Step -I: Synthesis of H-Cys(Acm)-Lue-Otbu.
H-Leu-(Otbu).HCl (356.27gm,l.leq), HOBT(222.97gm,1.0eq) were added to a solution of Fmoc-Cys(Acm)-OH (600.0 g, l.Oeq) in DMF (3.0L) and then cooled to 5-10°C. HBTU (603.9gm, 1.1 eq) and DIPEA(883.4 ml, 3.5eq) were added to the reaction mass. After completion of the reaction, the product was extracted with ethyl acetate (6.0L) and then washed with HC1, 5% aq sodium bicarbonate soln, 10%NaCl solution and water. The organic layer was collected, filtered and the filtrate was concentrated to obtain Fmoc-protected dipeptide. The obtained product was proceeded for next step without any further purification. Yield: 945 g Fmoc-Cys (Acm)-Leu-Otbu (945 g, leq) was taken in flask containing DMF (1.89 L) and cooled the solution to 5-10°C. Tertiary butyl amine (255 ml 1.5eq) was added slowly to the solution and stirred for 15min. After completion of reaction, the reaction mass was cooled to 5-10°C. Water and IN HC1 were added to the
solution and the obtained aqueous solution was washed with hexane. The pH of product was adjusted to 8 to 8.5 with saturated sodium bicarbonate solution and the product was extracted with ethyl acetate. The ethyl acetate layer was washed with 10% NaCl solution and water and then filtered. The obtained filtrate was evaporated completely to obtained light yellow thick residue H-Cys(Acm)-Leu-Otbu. Yield: 450 g.
Step -II: Synthesis of H-Gly-Cys (Acm)-Lue-Otbu.
H-Cys (Acm)-Leu-Otbu (450.0 g, l.leq) was taken in flask containing DMF (1.68 L). Fmoc-Gly-OH (336.45g, l.Oeq), HOBT (174.2g, l.Oeq) were added to the solution and then cooled to 5-10°C. HBTU (472g, l.leq) and DIPEA (414.15ml, 2.1eq) were added and stirred. After completion of the reaction, ethyl acetate was added to the above reaction mass and washed with pre-cooled 0.5N HC1, 5% aqueous NaHCC solution (pre-cooled), 10%NaCl solution and water. The resultant organic layer was evaporated completely to give Fmoc-Gly-Cys (Acm)-Leu-Otbu. Yield: 825 g.
Fmoc-Gly-Cys(Acm)-Leu-Otbu (825g, leq) was taken in flask containing THF (1650ml) and cooled to 5-10° C. Tertiary butyl amine (338 ml 2.5eq) was added slowly to the solution. After completion of reaction, the solution was cooled to 5-10°C and then water (2.48L) was added to the reaction mass. The aqueous reaction mass was washed with hexane, 50% ethyl acetate in hexane, then the product was extracted with dichlorome thane. The dichloromethane layer was washed with 10%NaCl solution and water. The (collected) organic layer was dried with anhydrous sodium sulphate and then filtered. The obtained filtrate was evaporated completely to obtain H-Gly-Cys (Acm)-Leu-Otbu as light yellow semi solid. Yield: 392 g; HPLC purity: -92%.
EXAMPLE 2: Fmoc-Cvs(Acm)-Val-Asn(Trt)-Val-Ala-Cvs(Trt)-Thr(Otbu)-2CTC. (Fragment-A)
Step A: CTC resin (750gm) was taken in a SPPS reactor, 7.5L of dry dichloromethane (DCM) was added and allowed it to swell for 20min and drained.
Step B: A solution of Fmoc-Thr(otbu)-OH (954gm, 2eq) and DIEA (627.6ml, 3eq) in dry dichloromethane (3.75L) were added to the resin at step A and stirred for at room temperature and drained.
The resin was then capped with DIEA (1%) solution in DCM: methanol (1: 1)) and then drained. Thereafter, washed the resin with one bed volume of DMF (2 times), DCM (2 times) and MTBE (2 times), isolated and dried. Yield: 1150gm
The above resin was deblocked with 20% piperidine in DMF and washed with DMF (2times), IPA (2 times) and DMF (2 times).
Step C: To a solution of Fmoc-Cys (Trt)-OH (808g, 1.5eq.) and HOBT (212.5g, 1.5eq) in DMF, DIC (323ml, 2.25eq) was added. The obtained reaction mixture was added to the resin in Step B and stirred. After completion of the reaction the resin was drained and washed with DMF (2 times) and then resin was deblocked with 20% piperidine in DMF and then washed with DMF, IPA and DMF.
Step-D: Followed by sequential coupling of Fmoc-Ala-OH, Fmoc-Val-OH, Fmoc-Asn (Trt)-OH, Fmoc-Val-OH similar to the procedure described in step-C.
Step E: To a solution of Fmoc-Cys (acm)-OH (572g, 1.5eq.) and HOBT (212.5g, 1.5eq) in DMF, DIC (323ml, 2.25eq) was added. The obtained reaction mixture was added to the resin in Step D and stirred. After completion of the reaction, the resin was drained and washed with DMF (2 times) DCM (2 times) and MTBE (2 times). It was isolated and dried to give FMOC-Cys (Acm)-Val-Asn (Trt)-Val-Ala-Cys (Trt)-Thr(otbu)-2CTC Resin. Yield: 2.17 kg.
Step F: Selective cleavage of 2-chloro trityl resin from the Fmoc-Cys(acm)-Val-Asn(Trt)-Val-Ala-Cys(Trt)-Thr(otbu)-2CTC Resin was performed with a mixture 1%TFA in dichloromethane and then above peptidyl resin was taken in SPPS reactor and treated with a solution of 1% TFA in DCM and drained. The filtrate was immediately neutralized with precooled saturated NaHC03 Solution to precipitate the product. The same process was repeated 3 more times and dried to obtain off-white solid. The obtained solid was further purified by treating with MTBE (2ml/g of product) to give Fmoc-Cys(Acm)-Val-Asn(Trt)-Val-Ala-Cys(Trt)-Thr(tbu)-OH. Yield: 1133gm; Purity: 94.29%.
EXAMPLE-3: Synthesis of Boc-Asn(Xan)-Asp(Otbu)-Glu(Otbu)-Cys(Trt)-Glu(Otbu)-Leu-2CTC (Fragment D)
Step A: CTC resin (445gm) was taken in a SPPS reactor, 4.45L of dry dichloromethane was added and allowed it to swell for 20min and drained.
Step B: A solution of Fmoc-Leu-OH (525gm, 2eq) and DIEA (388.6ml, 3eq) in dry dichloromethane (2.22L) was added to the resin at step A and stirred at room temperature and drained.
The resin was then capped with DIEA (1%) solution in DCM: methanol (1:1)). Thereafter, washed the resin with one bed volume of DMF (2 times), DCM (2 times) and MTBE (2 times), isolated and dried. Yield: 700gm.
The above resin was deblocked with 20% piperidine in DMF for 10 and 15 minutes and washed with of DMF (2times), IPA (2 times) and DMF (2 times).
Step C: To a solution of Fmoc-Glu(Otbu)-OH (553gm, 2.0eq.) and HOBT (200.5gm, 2.0eq) in DMF, DIC (305ml, 3.0 eq) was added. It was added to the resin in Step B. After completion of the reaction, the resin was drained and washed with DMF.
The above resin was deblocked with 20% piperidine in DMF for 10 and 15 minutes and washed with of DMF (2times), IPA (2 times) and DMF (2 times).
Step D: Sequential coupling of Fmoc-Cys(Trt)-OH, Fmoc-Glu(Otbu)-OH, Fmoc-Asp(Otbu)-OH, Fmoc-Asp(Otbu)-OH similar to the procedure of step-C.
Step-E: To a solution of Boc-Asn(xan)-OH (537gm, 2.0eq.) and HOBT (200.5gm, 2.0eq) in DMF, DIC (305ml, 3.0eq) were added and stirred. It was added to the resin in Step F and stirred. After completion of the reaction, the resin was drained and washed with DMF (2 times), DCM (2 times) and MTBE (2 times). It was isolated and dried to give Boc-Asn(Xan)-Asp(Otbu)-Glu(Otbu)-Cys(Trt)-Glu(Otbu)-Leu-2CTC resin. Yield: 1.37kg.
Step H: Selective cleavage of 2-chloro trityl resin from the Boc-Asn(Xan)-Asp(Otbu)-Glu(Otbu)-Cys(Trt)-Glu(Otbu)-Leu-2CTC resin was performed with a mixture 1%TFA in dichloromethane.
The above peptidyl resin was taken in SPPS reactor and treated with a solution of 1% TFA in DCM. The filtrate was neutralized with precooled saturated NaHC03
solution. The same process was repeated 3 more times, the collected organic solution was washed with water, dried with sodium sulphate and evaporated to obtain off-white solid. The obtained solid was further purified by treating with MTBE (2ml/g of product) to give Boc-Asn(Xan)-Asp(Otbu)-Glu(Otbu)-Cys(Trt)-Glu(Otbu)-Leu-OH. Yield: 720g; Purity: 90.74%.
EXAMPLE 4: Synthesis of H-Cys(Acm)-Val-Asn(Trt)-Val-Ala-Cys(Trt)-Thr(tbu) -Gly- Cys( Acm) -Leu-Otbu .
Fmoc-Cys(Acm)-Val-Asn(Trt)-Val-Ala-Cys(Trt)-Thr(tbu)-OH (Fragment- A) (500.0 g, l.Oeq) was taken in a round bottom flask containing DMF (2.5F). H-Gly-Cys (Acm)-Feu-Otbu (Fragment B) (209g, 1.5eq) was added to the reaction mass and cooled to -10 to -15°C. HOAT (50g, l.leq) and HATU (151.75g , 1.2 eq) were added to reaction mass and then DIPEA (116ml, 2eq) was added drop wise while stirring at -10 to-15°C. After completion of the reaction, methanol (15. OF) was added to the above reaction mass (solid formation was observed), then pH of the reaction mixture was adjusted with IN HC1 up to pH 3, stirred for one hour, filtered and dried to give dec apep tide. Yield: 700g.
The above obtained decapeptide (700g, FOeq) was taken in a round bottom flask containing DMF (4.2 F) and cooled the solution to 5-10°C. Tertiary butyl amine (77.30ml 2.0eq) was added and stirred the reaction mass for 15min at 5-10°C and then stirred at R.T. After completion of the reaction, methanol (25.20F) was added to precipitate the product. The obtained solid was filtered and dried to give NH2-Cys (Acm)-Val-Asn(Trt)-Val-Ala-Cys(Trt)-Thr(otbu)- Gly-Cys (Acm)-Fue-Otbu. Yield: 437g; HPFC purity: -97%
EXAMPLE 5: Synthesis of Boc-Asn(Xan)-Asp(Otbu)-Glu(Otbu)-Cys(Trt)- Glu(Otbu)-Leu-Cys(Acm)-Val-Asn(Trt)-Val-Ala-Cys(Trt)-Thr(otbu)-Gly- Cys(Acm)-Leu-Otbu.
NH2-Cys(Acm)-Val-Asn(Trt)-Val-Ala-Cys(Trt)-Thr(otbu)-Gly-Cys-(Acm)-Lue-Otbu, (435.0g,F0eq) and Boc-Asn(Xan)-Asp(Otbu)-Glu(Otbu)-Cys(Trt)-Glu(Otbu)-Leu-OH (Fragment D), (365.0 g, 0.9eq) were taken in a round bottom
flask containing DMF (2.61 L). The solution obtained was cooled to -10 to -15°C and then HO AT (42.0g, 1.2eq) and HATU (118g, 1.2eq) were added. DIPEA (90 ml, 2.0eq) was added to the reaction mass and stirred at -10 to-15°C. Methanol (13.0L) was added to the above reaction mass to precipitate the product. The obtained solid was filtered and dried to give Boc-Asn(Xan)-Asp(Otbu)-Glu(Otbu)-Cys(Trt)-Glu(Otbu)-Leu-Cys(Acm)-Val-Asn(Trt)-Val-Ala-Cys(Trt)-Thr(otbu)-Gly-Cys(Acm)-Leu-Otbu as off-white solid. Yield: 750g.
EXAMPLE 6: Synthesis of H-Asn-Asp-Glu-Cys-Glu-Leu-Cys(Acm)-Val-Asn-Val-Ala-Cys-Thr-Gly-Cys(Acm)-Leu-OH.
Protected peptide (500.0 g) obtained in example 5 was treated with a pre-cooled solution of 84% TFA (4200 ml), 5% TIPS (250 ml), 5% H20 (250 ml), 5% DTT (250 gm), 1%DMS (50ml) for 2 hrs at R.T. The product was precipitated by the addition of reaction mass to the pre-cooled MTBE, filtered the product under nitrogen and washed with MTBE and dried. Yield: 310.0g; Purity: 73 %.
EXAMPLE 7: PREPARATION OF CRUDE PLECANATIDE
Linear 1-16 peptide obtained from example 6 was dissolved in degassed 0.015 M ammonium hydroxide solution at a concentration of lg /0.75 L, pH was adjusted between 8 .5to 9.0 by using ammonia solution. After dissolution of compound, H2O2 (200 pi / g) was added. After completion of oxidation, the pH was adjusted between 3 to 4 by IN HC1 to obtain mono cyclized 1-16 peptide solution and then solution was treated with 5% iodine in acetonitrile, till the yellow color persist. The reaction mixture with excess of iodine was quenched with 0.1M aqueous ascorbic acid solution and the pH was adjusted between 6.5 and 7 by using ammonia solution. The solution was filtered through 2.4 micron. The obtained filtrate was used as such for next stage purification.
EXAMPLE 8: PURIFICATION OF PLECANATIDE
Stage 1: Crude Plecanatide solution obtained from example 7 was purified on preparative HPLC, column was packed with reverse phase C18 hybrid silica using Tris HC1 pH 7(as buffer A) and 100% acetonitrile (as buffer B).The fractions were collected and purity of fractions were monitored by analytical HPLC. Fractions containing > 94% pure Plecanatide were pooled as main pool; and fractions not meeting the pooling criteria were re-processed in a similar manner.
Stage 2: The main pool obtained from stage 1 purification were diluted with equal amount of purified water and re-purified on preparative HPLC, column was packed with reverse phase C18 hybrid silica using Tris HC1 pH 7(as buffer A) and 100% acetonitrile (as buffer B).The fractions were collected and purity of fractions were monitored by analytical HPLC. Fractions containing > 98.5% pure Plecanatide were pooled as main pool for desalting. (OR)
The main pool obtained from stage- 1 purification were diluted with equal amount of purified water and re-purified on preparative HPLC, column was packed with reverse phase C18 hybrid silica using TEAP (as buffer A) and acetonitrile (as buffer B).The fractions were collected and purity of fractions were monitored by analytical HPLC. Fractions containing > 98.5% pure Plecanatide were pooled as main pool for desalting.
EXAMPLE 9: DE SALTING AND LYOPHILIZATION
The main pool obtained from the purification were diluted with equal amount of purified water and loaded on preparative HPLC, column was packed with reverse phase C18 hybrid silica.
De-salting was done by passing 5 void volume of 0.1% acetic acid in purified water fallowed by elution of product from the column by using 30% acetonitrile (HPLC grade) in purified water containing 0.1% acetic acid. The fractions were collected and purity of fractions were monitored by analytical HPLC.
The fractions containing pure Plecanatide (>98.5%) were pooled; the organic modifier was removed under reduced pressure and filtered through 0.2 micron filter. The resulting peptide solution was freeze-dried to isolate Plecanatide.
After completion of lyophilization cycle, the compound was unloaded and dissolved in purified water at a concentration Of 70 g /L, filtered through 0.2 micron filter. The resulting peptide solution was freeze-dried to obtain white solid lyophilized powder as Plecanatide. Purity: 99.1 %
EXAMPLE 10: PREPARATION OF MONO CYCLIZED PLECANATIDE
Linear 1-16 peptide obtained from example 6 was dissolved in degassed 0.015 M ammonium hydroxide solution at a concentration of lg /0.75 L, the pH was adjusted between 8 .5to 9.0 by using ammonia solution. After dissolution of compound, H2O2 (200pl / g) was added and stirred for 30 minutes. The progress of oxidation was monitored by analytical reverse phase HPLC & Ellman’s test. After completion of oxidation, the pH was adjusted between 6.5 and 7 by using IN HC1 to obtain mono cyclized 1-16 peptide solution.
EXAMPLE 11: PURIFICATION OF MONO CYCLIZED PLECANATIDE
Mono cyclized solution obtained from example 10 was purified on preparative HPLC, column packed with reverse phase C18 hybrid silica using Tris HC1 pH 7(as buffer A) and 100% acetonitrile (as buffer Bj.Thc fractions were collected and purity were monitored by analytical HPLC. Lractions containing > 95% pure Plecanatide were pooled as main pool; and fractions not meeting the pooling criteria were re-processed in a similar manner.
EXAMPLE 12: PREPARATION OF CRUDE PLECANATIDE
The resultant purified solution was diluted with equal amount of water, adjusted pH 3.5 with IN HC1 and treated with 5% iodine in acetonitrile, till the yellow color persist and the reaction mass was stirred for two hours.
The completion of oxidation was monitored by analytical reverse phase HPLC; quenched the excess iodine with 0.1 M aqueous ascorbic acid solution and then pH was adjusted between 6.5 and 7 by using ammonia solution. The solution was filtered through 2.4 micron filter and used as such for next stage purification.
EXAMPLE 13: PURIFICATION OF PLECANATIDE
The main pool obtained from stage 1 purification were purified on preparative HPLC, column was packed with reverse phase C18 hybrid silica using Tris HC1 pH 7 (buffer A) and 100% acetonitrile (buffer B). The fractions were collected and purity of fractions were monitored by analytical HPLC. Fractions containing > 98.5% pure Plecanatide were pooled as main pool for desalting. (OR)
The main pool obtained from stage 1 purification further purified on preparative HPLC, the column was packed with reverse phase C18 hybrid silica using TEAP (as buffer A) and acetonitrile (as buffer B). Fractions containing > 98.5% pure Plecanatide were pooled as main pool for desalting.
EXAMPLE 14: DE SALTING AND LYOPHILIZATION
The main pool obtained from the purification were diluted with equal amount of purified water and loaded on preparative HPLC, column packed with reverse phase C18 hybrid silica.
De-salting was done by passing 5 void volume of 0.1% acetic acid in purified water fallowed by elution of product from the column by using 30% acetonitrile (HPLC grade) in purified water containing 0.1% acetic acid. The fractions were collected and purity of fractions were monitored by analytical HPLC. The fractions containing pure Plecanatide (>98.5%) were pooled; the organic modifier was removed under reduced pressure and filtered through 0.2 micron filter. The resulting peptide solution was freeze-dried to isolate Plecanatide.
After completion of lyophilization cycle, unload the compound and dissolve in purified water at a concentration Of 70 g /L, filtered through 0.2 micron filter. The resulting peptide solution was freeze-dried to obtain white solid lyophilized powder as Plecanatide.
Purity: 99% by HPLC
CLIPS
April 19, 2016
Novel Chronic Idiopathic Constipation Drug Under FDA Review
Plecanatide is a once-daily, oral, uroguanylin analog
Synergy Pharmaceuticals announced the Food and Drug Administration (FDA) has accepted for review the New Drug Application (NDA) for plecanatide for the treatment of chronic idiopathic constipation (CIC).
The NDA submission was based on data from two double-blind, placebo-controlled Phase 3 trials and one open-label long term safety study in over 3,500 patients with CIC.
RELATED: NDA Submitted for Chronic Idiopathic Constipation Drug Plecanatide
The FDA has set a Prescription Drug User Fee Act (PDUFA) target action date of January 29, 2017 to make a decision on the NDA.
Plecanatide is a once-daily, oral, uroguanylin analog currently under development for the treatment of CIC and irritable bowel syndrome with constipation (IBS-C). It is designed to replicate the function of uroguanylin, a naturally occurring GI peptide, by working locally in the upper GI tract to stimulate digestive fluid movement and support regular bowel function.
PATENT
CN 104628827
http://www.google.com/patents/CN104628827A?cl=en
Prica exenatide Synergy Pharmaceuticals developed by the United States for the GC-C receptor in development of drugs, administered orally Limited.Currently underway include chronic idiopathic constipation (CIC) and constipation irritable bowel syndrome (IBS-C), including the phase III clinical trials. It is expected to receive US FDA clearance to market in recent years. Prica that peptides CAS: 467426-54-6 English name plecanatide, structural formula is as follows:
Preparation Prica that peptides from Shenzhen Han Yu medicine was first reported (CN103694320A), using a solid-phase synthesis of linear peptides in solution and then the two-step method to get into the ring, respectively. Since the method to form a ring carved in solution twice, the solution of complex composition, separation and purification difficult, the method should be improved.
Example 1
Weigh the degree of substitution of 0. 51mmol / g of Fmoc-Leu- Wang resin 10g (5. Lmmol), added to the solid phase reactor, DMF washing 3 times, the swelling 3h. The volume ratio of 1: 4 piperidine: DMF was added to the reactor the reaction, after the reaction was washed with DCM and washed twice, DMF 4 times. Weigh Fmoc-Cys (Acm) -OH 6. 34g, H0Bt 2. 07g, DIC 2. 37mL was dissolved in DMF, added to the reactor uniformly mixed, the reaction at room temperature 2h. Ninhydrin color reaction control endpoint, the resin was colorless indicates the end of the reaction, the reaction is continued if the color to colorless. After completion of the reaction, DCM was washed twice, DMF and washed 4 times.
Repeat the above steps, in accordance with the order of the sequence, followed by deprotection, coupling Fmoc-Gly-OH, Fmoc-Thr (tBu) -OH, Fmoc-Cys- (Mmt) -OH, Fmoc-Ala-OH, Fmoc- Val-OH, Fmoc-Asn (Trt) -〇H, Fmoc-Val-OH, Fmoc-Cys (Acm) -OH, Fmoc-Leu-OH, Fmoc-Glu (OtBu) -OH, Fmoc-Cys (StBu) -OH, Fmoc-Glu (OtBu) -OH, Fmoc-Asp (OtBu) -OH, Boc-Asn (Trt) -〇H〇
To a prepared peptide resin reactor volume percentage of 15% DMF solution of mercapto ethanol, reaction 2h; then DCM was added a solution of 20-fold amount DTNP reaction lh; was added after washing 1% TFA containing TIS 5% of DCM solution reaction 20min.
Preparation of peptide resin obtained after sufficiently washed with DMF, DMF was added 10 times the amount in the reaction solution 12 lh. Full wash sash.
After the preparation of the peptide resin was added in a volume ratio of 95/2/2/1 TFA / TIS / EDT / H lysis reagent 20 is added in an amount 20mL / g, the reaction ice bath lh, stirring was continued at room temperature 5h, then filtration.After lysis reagent suction filtrate using a rotary evaporator until no overflow TFA, precipitated reagent was added standing; Pulika centrifugation the precipitated crude peptide was peptide to give 8. 67g〇
The preparation of the crude peptide was obtained Pulika peptide using preparative HPLC system, wavelength 214nm, C18 reversed-phase column packing for the separation, the mobile phase of water and acetonitrile were used, with a gradient elution method to collect the target polypeptide The absorption peak. Using rotary evaporation at 30 ° C to remove most of the acetonitrile, were freeze-dried to obtain a purified Prica exenatide refined products.
Example 2
Weigh the degree of substitution of 0. 2mmol / g of Fmoc-Leu- Wang resin 10g (2mmol), added to the solid phase reactor. DMF washing 3 times, the swelling 3h. The volume ratio of 1: 4 piperidine: DMF was added to the reactor the reaction, after the reaction was washed with DCM and washed twice, DMF 4 times. Weigh Fmoc-Cys (Acm) -OH1. 24g, HOBtO. 406g, DIC 0 • 465mL dissolved in DMF solution, after mixing into the reactor at room temperature the reaction 2h.Ninhydrin color reaction control endpoint, the resin was colorless indicates the end of the reaction, the reaction is continued if the color to colorless. After completion of the reaction, DCM was washed twice, DMF and washed 4 times.
Repeat the above steps, in accordance with the order of the sequence, followed by deprotection, coupling Fmoc-Gly-OH, Fmoc-Thr (tBu) -OH, Fmoc-Cys- (Mmt) -OH, Fmoc-Ala-OH, Fmoc- Val-OH, Fmoc-Asn (Trt) -〇H, Fmoc-Val-OH, Fmoc-Cys (Acm) -OH, Fmoc-Leu-OH, Fmoc-Glu (OtBu) -OH, Fmoc-Cys (StBu) -OH, Fmoc-Glu (OtBu) -OH, Fmoc-Asp (OtBu) -OH, Boc-Asn (Trt) -〇H〇
[0053] To illustrate the preparation of the present embodiment obtained peptide resin reactor volume percent of a DMF solution of 30% mercaptoethanol, reaction 4h; then 5-fold amount DTNP in DCM reaction lh; was added after washing 1% TFA containing TIS 5% in DCM reaction 20min.
Preparation of peptide resin obtained after sufficiently washed with DMF, 20 times the amount of DMF was added in the reaction solution 12 lh. Full wash sash.
Peptide Resin [0055] Preparation was added volume ratio of 82. 5/5/5/5/2. 5 TFA / thioanisole / H20 / phenol / EDT cleavage reagents, added in an amount 10mL / g, the reaction ice bath 0 After. 5h, stirring was continued at room temperature for lh, then suction filtered. After lysis reagent suction filtrate to the non-use of force blowing TFA overflow, adding precipitation reagent standing; centrifugation precipitated Prica exenatide crude peptide to give 1. 52g.
The preparation of the crude peptide was obtained Pulika peptide using preparative HPLC system, wavelength 214nm, C18 reversed-phase column packing for the separation, the mobile phase of water and acetonitrile were used, with a gradient elution method to collect the target polypeptide The absorption peak. Using rotary evaporation at 30 ° C to remove most of the acetonitrile, were freeze-dried to obtain a purified Prica exenatide refined products.
Example 3
Weigh the degree of substitution of 0. 6mmol / g of Fmoc-Leu- Wang resin 10g (6mmol), added to the solid phase reactor, DMF washing 3 times, the swelling 3h. The volume ratio of 1: 4 piperidine: DMF was added to the reactor the reaction, after the reaction was washed with DCM and washed twice, DMF 4 times. Weigh Fmoc-Cys (Acm) -OH 7. 46g, H0Bt2. 44g, DIC 2. 79mL was dissolved in DMF, added to the reactor uniformly mixed, the reaction at room temperature 2h.Ninhydrin color reaction control endpoint, the resin was colorless indicates the end of the reaction, the reaction is continued if the color to colorless. After completion of the reaction, DCM was washed twice, DMF and washed 4 times.
Repeat the above steps, in accordance with the order of the sequence, followed by deprotection, coupling Fmoc-Gly-OH, Fmoc-Thr (tBu) -OH, Fmoc-Cys- (Mmt) -OH, Fmoc-Ala-OH, Fmoc- Val-OH, Fmoc-Asn (Trt) -〇H, Fmoc-Val-OH, Fmoc-Cys (Acm) -OH, Fmoc-Leu-OH, Fmoc-Glu (OtBu) -OH, Fmoc-Cys (StBu) -OH, Fmoc-Glu (OtBu) -OH, Fmoc-Asp (OtBu) -OH, Boc-Asn (Trt) -〇H〇
To the prepared peptide resin reactor volume percentage of 25% DMF solution of mercapto ethanol, reaction 3h; then 10-fold amount DTNP in DCM reaction lh; was added 1% TFA washed containing TIS5% DCM solution Reaction 20min〇
Preparation of peptide resin obtained after sufficiently washed with DMF, 15 times the amount of DMF was added in the reaction solution 12 lh. Full wash sash.
Preparation of the peptide resin was added in a volume ratio of 90/5/3/2 TFA / thioanisole / anisole / EDT cleavage reagents, added in an amount 20mL / g, the ice bath was reacted 0.lh, stirring was continued at room temperature The reaction 10h, then filtration. After lysis reagent suction filtrate using a rotary evaporator until no overflow TFA, precipitated reagent was added standing; Pulika centrifugation the precipitated crude peptide was peptide to give 8. 46g.
The preparation of the crude peptide was obtained Pulika peptide using preparative HPLC system, wavelength 214nm, C18 reversed-phase column packing for the separation, the mobile phase of water and acetonitrile were used, with a gradient elution method to collect the target polypeptide The absorption peak. Using rotary evaporation at 30 ° C to remove most of the acetonitrile, were freeze-dried to obtain a purified Prica exenatide refined products.
Although the above has been described with general, specific embodiments and test, the present invention has been described in detail, but on the basis of the present invention, it may make some changes or improvements, which the skilled artisan It is obvious. Thus, the present invention without departing from the spirit on the basis of these modifications or improvements made, belong to the scope of the invention as claimed.
PATENT
CN 104211777
http://www.google.com/patents/CN104211777A?cl=en
The pickup exenatide (Plecanatide) is a synthetic analogue of guanylin urine (urine guanylin is a natriuretic hormone, can regulate gastrointestinal transport of ions and liquid), pickup exenatide enter After in vivo and guanylate gastrointestinal tract endothelial cells cyclase C binding and activation, activation of the cystic fibrosis transmembrane conductance regulator (CFTR), to promote chloride and water into the intestine, thereby promoting bowel motility, improve constipation symptoms.
Synergy company announced its pick in the research of new drugs that peptide (code: SP304) on October 6, 2010 the treatment of gastrointestinal disorders II a clinical experimental results. The study, conducted in patients with chronic constipation showed that the drugs can improve bowel function in patients, promote intestinal motility and reduce abdominal discomfort shape. In the experiment, there was no diarrhea and other adverse reactions, at the doses tested did not detect the pickup system that peptides are absorbed. The drug is expected for the treatment of chronic constipation (CC), constipation-predominant irritable bowel syndrome (IBS-C) and other gastrointestinal disorders. CC and IBS-C is a common gastrointestinal disease that can cause serious impact on the work and the quality of life of patients. Synergy will continue to conduct clinical trials of other pickups that peptide.
The structure of the peptide pickup that is:
H-Asn-Asp-Asp-Cys-Glu-Leu-Cys-Val-Asn-Val-Ala-Cys-Thr-Gly-C ys-Leu-〇H (4-12 disulfide, 7- 15)
Example 30:
H-Asn-Asp-Asp-Cys-Glu-Leu-Cys-Val-Asn-Val-Ala-Cys-Thr-Gly-C ys-Leu-〇H (4-12 disulfide, 7- 15) Preparation of
embodiments will be prepared by the method of Example 18 H-Asn (Trt) -Asp (OtBu) -Asp (OtBu) -Cys (mmt) -Glu (Ot Bu) -Leu-Cys (StBu) -Val-Asn ( Trt) -Val-Ala-Cys (mmt) -Thr (tBu) -Gly-Cys (StBu) -Leu-CT C resin (IOOmmol, 472. 88g) disposed cracking reactor to 10ml / g resin ratio Add lysis reagent (TFA: EDT: water = 95: 2 5:.. 2 5 (V / V)), stirred at room temperature 2h. The reaction was filtered with sand core funnel, and then added a small amount of TFA The resin was washed in the funnel, collecting the filtrate, the combined filtrate was concentrated. Frozen in dry diethyl ether was added (100ml / g peptide purpose tree months) and the solution was precipitated, centrifuged to remove the precipitate was washed with diethyl ether after dry ether three times, and dried in vacuo to give a white solid powder was approximately 180g, i.e., H-Asn-Asp-Asp -Cys-Glu-Leu-Cys (StBu) -Val-Asn-Val-Ala-Cys-Thr-Gly-Cy s (StBu) -Leu-OH. The solid was dissolved with water to lmg / ml solution. Was added an aqueous solution of 1% by volume of H2O2, the reaction was stirred at room temperature 30min, to prepare H-Asn-Asp-Asp-Cys-Glu-Leu-Cys (StBu) -Val-Asn-Val-Ala-Cys-Thr-Gl y-Cys (StBu) -Leu-OH (disulfide 4-12) was treated with a rotary evaporator after drying the compound containing 500ml 20% β- mercaptoethanol and 0. IM N- methylmorpholine were dissolved in water, followed by stirring After 12h the reaction, the reaction solution was diluted with water to 3mg / ml was about 60L, dissolved in ethanol was added with IL 300mmol I2 solution, the reaction was stirred at room temperature 2h. Adding an appropriate amount Vc remove excess I2, until the color of the reaction solution was transparent, i.e., to give H-Asn-Asp-Asp-Cys-Glu-Leu-Cys-Val-As n-Val-Ala-Cys-Thr-Gly-Cys-L eu_0H (disulfide bonds 4-12, 7-15).
PATENT
WO 2014197720
CN 103694320
WO 2012118972
WO 2012037380
WO 2011069038
US 20100152118
WO 2010065751
///Plecanatide, 普卡那肽 , ليكاناتيد , плеканатид, 467426-54-6, Chronic Idiopathic Constipation, NDA, SP 304, SYNERGY, PEPTIDE,
C[C@H]1C(=O)N[C@H]2CSSC[C@@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@H](NC(=O)CNC(=O)[C@@H](NC2=O)[C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)O)C(=O)N[C@H](C(=O)N[C@H](C(=O)N[C@H](C(=O)N1)C(C)C)CC(=O)N)C(C)C)CC(C)C)CCC(=O)O)NC(=O)[C@H](CCC(=O)O)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](CC(=O)N)N
OR
O=C(N[C@@H](CC(=O)O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@H]1CSSC[C@@H]2NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CCC(=O)O)NC1=O)CC(C)C)CSSC[C@H](NC(=O)CNC(=O)[C@@H](NC2=O)[C@@H](C)O)C(=O)N[C@@H](CC(C)C)C(=O)O)C(C)C)C(C)C)[C@@H](N)CC(N)=O
References
- ^ Jump up to:a b “FDA approves Trulance for Chronic Idiopathic Constipation”. FDA.gov. U.S. Food and Drug Administration. Retrieved 20 January 2017.
- ^ “TRULANCE package insert” (PDF). Trulance website. Synergy Pharmaceuticals Inc. 420 Lexington Avenue, Suite 2012 New York, New York 10170. Retrieved 20 January 2017.
- ^ Thomas RH, Luthin DR (June 2015). “Current and emerging treatments for irritable bowel syndrome with constipation and chronic idiopathic constipation: focus on prosecretory agents”. Pharmacotherapy. 35 (6): 613–30. doi:10.1002/phar.1594. PMID 26016701.
- ^ Jump up to:a b Rome III : the functional gastrointestinal disorders. Drossman, Douglas A. (3rd ed.). McLean, Va.: Degnon Associates. 2006. ISBN 9780965683753. OCLC 79476570.
- ^ “Trulance – FDA prescribing information, side effects and uses”. Drugs.com. Retrieved 2017-10-27.
- ^ Jump up to:a b c d e f g h Al-Salama ZT, Syed YY (April 2017). “Plecanatide: First Global Approval”. Drugs. 77 (5): 593–598. doi:10.1007/s40265-017-0718-0. PMID 28255961.
- ^ Jump up to:a b c d e f Shailubhai K, Comiskey S, Foss JA, Feng R, Barrow L, Comer GM, Jacob GS (September 2013). “Plecanatide, an oral guanylate cyclase C agonist acting locally in the gastrointestinal tract, is safe and well-tolerated in single doses”. Digestive Diseases and Sciences. 58 (9): 2580–6. doi:10.1007/s10620-013-2684-z. PMID 23625291.
- ^ Chang WL, Masih S, Thadi A, Patwa V, Joshi A, Cooper HS, et al. (February 2017). “+/Min-FCCC mice”. World Journal of Gastrointestinal Pharmacology and Therapeutics. 8(1): 47–59. doi:10.4292/wjgpt.v8.i1.47. PMC 5292606. PMID 28217374.
- ^ Hamra FK, Eber SL, Chin DT, Currie MG, Forte LR (March 1997). “Regulation of intestinal uroguanylin/guanylin receptor-mediated responses by mucosal acidity”. Proceedings of the National Academy of Sciences of the United States of America. 94 (6): 2705–10. Bibcode:1997PNAS…94.2705H. doi:10.1073/pnas.94.6.2705. PMC 20153. PMID 9122260.
- ^ Forte LR (November 2004). “Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics”. Pharmacology & Therapeutics. 104 (2): 137–62. doi:10.1016/j.pharmthera.2004.08.007. PMID 15518884.
- ^ Hamra FK, Forte LR, Eber SL, Pidhorodeckyj NV, Krause WJ, Freeman RH, et al. (November 1993). “Uroguanylin: structure and activity of a second endogenous peptide that stimulates intestinal guanylate cyclase”. Proceedings of the National Academy of Sciences of the United States of America. 90 (22): 10464–8. Bibcode:1993PNAS…9010464H. doi:10.1073/pnas.90.22.10464. PMC 47797. PMID 7902563.
- ^ Bijvelds MJ, Loos M, Bronsveld I, Hellemans A, Bongartz JP, Ver Donck L, et al. (December 2015). “Inhibition of Heat-Stable Toxin-Induced Intestinal Salt and Water Secretion by a Novel Class of Guanylyl Cyclase C Inhibitors”. The Journal of Infectious Diseases. 212 (11): 1806–15. doi:10.1093/infdis/jiv300. PMID 25999056.
- ^ Jump up to:a b Gadsby DC, Vergani P, Csanády L (March 2006). “The ABC protein turned chloride channel whose failure causes cystic fibrosis”. Nature. 440 (7083): 477–83. Bibcode:2006Natur.440..477G. doi:10.1038/nature04712. PMC 2720541. PMID 16554808.
- ^ Park HW, Nam JH, Kim JY, Namkung W, Yoon JS, Lee JS, et al. (August 2010). “Dynamic regulation of CFTR bicarbonate permeability by [Cl-]i and its role in pancreatic bicarbonate secretion”. Gastroenterology. 139 (2): 620–31. doi:10.1053/j.gastro.2010.04.004. PMID 20398666.
- ^ Eutamene H, Bradesi S, Larauche M, Theodorou V, Beaufrand C, Ohning G, et al. (March 2010). “Guanylate cyclase C-mediated antinociceptive effects of linaclotide in rodent models of visceral pain”. Neurogastroenterology and Motility. 22 (3): 312–e84. doi:10.1111/j.1365-2982.2009.01385.x. PMID 19706070.
- ^ “Plecanatide – brand name list from Drugs.com”. Drugs.com.
Plecanatide
|
| Clinical data |
| Trade names |
Trulance |
| Other names |
SP-304 |
| License data |
|
Routes of
administration |
By mouth |
| ATC code |
|
| Legal status |
| Legal status |
|
| Identifiers |
| CAS Number |
|
| PubChem CID |
|
| IUPHAR/BPS |
|
| DrugBank |
|
| ChemSpider |
|
| UNII |
|
| KEGG |
|
| ChEMBL |
|
| CompTox Dashboard (EPA) |
|
| Chemical and physical data |
| Formula |
C65H104N18O26S4 |
| Molar mass |
1681.89 g·mol−1 |
| 3D model (JSmol) |
|
-
CC1C(=O)NC2CSSCC(C(=O)NC(C(=O)NC(C(=O)NC(CSSCC(NC(=O)CNC(=O)C(NC2=O)C(C)O)C(=O)NC(CC(C)C)C(=O)O)C(=O)NC(C(=O)NC(C(=O)NC(C(=O)N1)C(C)C)CC(=O)N)C(C)C)CC(C)C)CCC(=O)O)NC(=O)C(CCC(=O)O)NC(=O)C(CC(=O)O)NC(=O)C(CC(=O)N)N
|
-
InChI=1S/C65H104N18O26S4/c1-25(2)15-34-55(98)80-41-24-113-110-21-38(58(101)77-37(65(108)109)16-26(3)4)71-44(87)20-69-62(105)50(30(10)84)83-61(104)40(78-51(94)29(9)70-63(106)48(27(5)6)81-57(100)35(18-43(68)86)76-64(107)49(28(7)8)82-60(41)103)23-112-111-22-39(59(102)73-32(53(96)75-34)11-13-45(88)89)79-54(97)33(12-14-46(90)91)72-56(99)36(19-47(92)93)74-52(95)31(66)17-42(67)85/h25-41,48-50,84H,11-24,66H2,1-10H3,(H2,67,85)(H2,68,86)(H,69,105)(H,70,106)(H,71,87)(H,72,99)(H,73,102)(H,74,95)(H,75,96)(H,76,107)(H,77,101)(H,78,94)(H,79,97)(H,80,98)(H,81,100)(H,82,103)(H,83,104)(H,88,89)(H,90,91)(H,92,93)(H,108,109)/t29-,30+,31-,32-,33-,34-,35-,36-,37-,38-,39-,40-,41-,48-,49-,50-/m0/s1 
-
Key:NSPHQWLKCGGCQR-DLJDZFDSSA-N
|
////////////PLECATANIDE, плеканатид , بليكاناتيد , 普卡那肽 , 7IK8Z952OK, Guanilib

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

























.bmp)
.bmp)


























 D
D