
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
1,2 Diaminocyclohexane from Synthesis with Catalysts Pvt Ltd

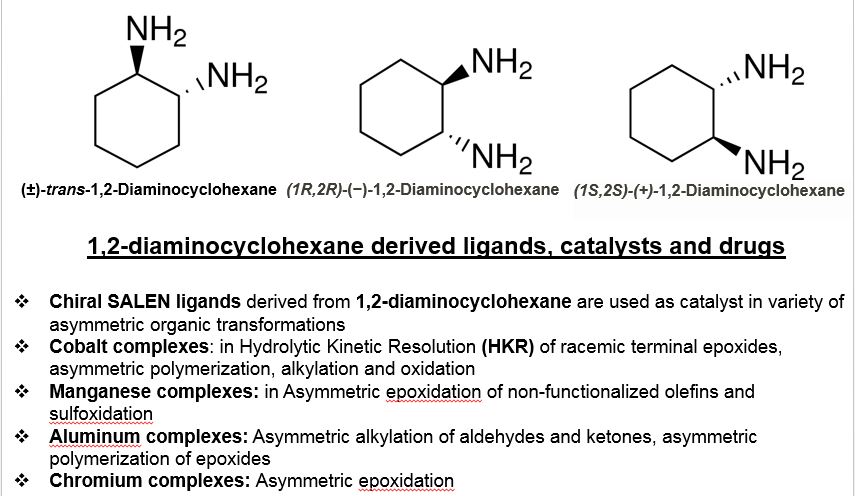
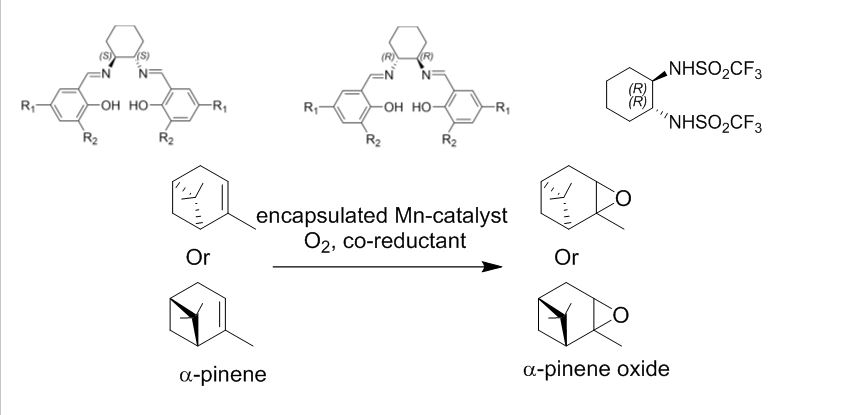
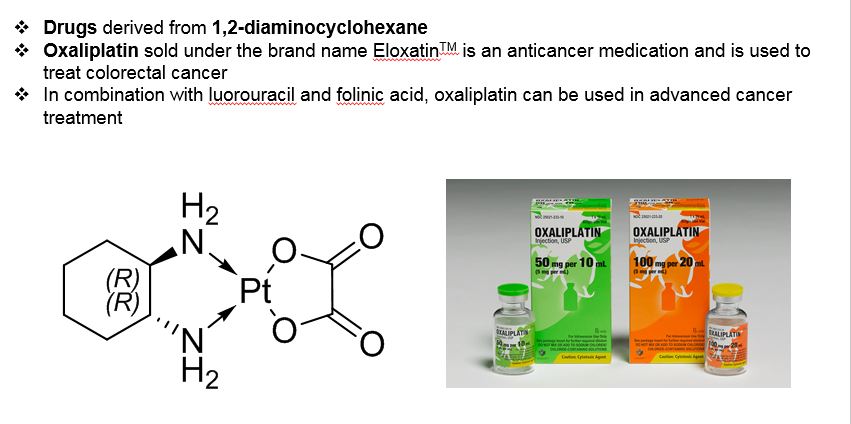
![]()
 +91- 999-997-2051
+91- 999-997-2051bpk@synthesiswithcatalysts.com

Axay Parmar

shr@synthesiswithcatalysts.com, ba@synthesiswithcatalysts.com
Synthesis with Catalysts Pvt Ltd was founded with an aim to help aromatic chemical, essential oil, pharmaceutical, API manufacturers to develop new products, increase productivity and improve production methodologies.
We have an advanced research and development centre where we innovate new chemical processes and improve the existing ones and help our customers implement the same. We support our research with pilot production of the products.
We are also developing precious metal complexes, Catalysts, Legends etc.
We are continuously working to reset standards of purity with our products.
The team at Synthesis with Catalysts Pvt Ltd has an vast experience and well renowned scientists of India have found it suitable to continue their Research in our facilities. The team with Synthesis with Catalysts Pvt Ltd has presented countless number of research papers all across the world.
We have a world class lab with all advance analytical testing machines.
RESEARCH & DEVELOPMENT
Synthesis with Catalysts Pvt Ltd.’s strength lies in its state-of- the-art infrastructure and R&D capabilities. Innovative process development is the foundation of SWC’s success. Our team of highly qualified R&D experts in process of research and technology development work 24 hours for inventing new processes and Optimizing product development capabilities. Our main focus is developing innovative processes, which could help our partners in reducing their costs and production time. Our Scientists constantly work for cost-effective ways of developing products, ensuring better service for our clients.

////////////1,2 Diaminocyclohexane, Synthesis with Catalysts Pvt Ltd
Roquinimex
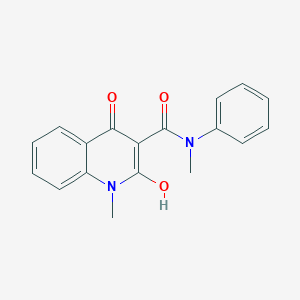

Roquinimex
- Molecular FormulaC18H16N2O3
- Average mass308.331 Da
LS-2616
PNU-212616
Roquinimex (Linomide) is a quinoline derivative immunostimulant which increases NK cell activity and macrophage cytotoxicity. It also inhibits angiogenesis and reduces the secretion of TNF alpha.
Investigated as a treatment for some cancers (including as adjuvant therapy after bone marrow transplantation in acute leukemia) and autoimmune diseases, such as multiple sclerosis and recent-onset type I diabetes.
Roquinimex has been investigated as a treatment for some cancers (including as adjuvant therapy after bone marrow transplantation in acute leukemia) and autoimmune diseases, such as multiple sclerosis and recent-onset type I diabetes. Several trials have been terminated due to serious cardiovascular toxicity.
Synthesis

Roquinimex synthesis:[1]
Ethyl 2-(methylamino)benzoate is condensed with ethyl malonate. Amine-ester ineterchange of that compound with N-methylanilineresults in formation of the amide roquinimex.
PAPER
Using DOE to Achieve Reliable Drug Administration: A Case Study

Design of experiments (DOE), a statistical tool, and mathematical modeling techniques are established and proven methodologies for process and product improvements in the pharmaceutical industry. This contribution presents a case study where an unsatisfactory dissolution capacity for the drug Roquinimex was overcome by investigating the process parameters with the help of an experimental design. By elucidating the detailed effects of temperature, dosing time, and dilution, conformity in the particle size distribution of the active pharmaceutical ingredient (API) from batch to batch in full-scale manufacturing could be ensured. As a direct result the manufactured drug met its specified dissolution capacity, which was a prerequisite for obtaining the desired bioavailability of the pharmaceutical oral formulation. This work demonstrates how the use of DOE in chemical process development adds value by allowing efficient and reliable improvements of a given synthetic step.
1 H NMR (4): δ 12.4 (broad s, 1H, OH), 8.1 (m, 1H, Ar), 7.5 (m, 1H, Ar), 7.1 (m, 7H, Ar), 3.5 (s, 3H, NCH3 ), 3.3 5 (s, 3H, NCH3 ).
SYN
BE 0904431; DE 3609052; GB 2172594; JP 1986221194; US 4672057
 By condensation of 4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acid ethyl ester (I) with N-methylaniline (II) by heating at 125 C and distillation of the ethanol formed.
By condensation of 4-hydroxy-1-methyl-2-oxo-1,2-dihydroquinoline-3-carboxylic acid ethyl ester (I) with N-methylaniline (II) by heating at 125 C and distillation of the ethanol formed.
CLIP
https://www.sciencedirect.com/science/article/abs/pii/S0731708597001076
 1H-NMR spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
1H-NMR spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
 13C-DEPT experiment of linomide in DMSO at 298 K recorded on a Bruker AC
13C-DEPT experiment of linomide in DMSO at 298 K recorded on a Bruker AC


A 2D 13C–1H COLOC experiment of linomide in DMSO at 298 K recorded on

 COSY 45° spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
COSY 45° spectrum of linomide in DMSO at 298 K recorded on a Bruker AC
PATENT
https://patents.google.com/patent/US5912349
U.S. Pat. No. 4,738,971 discloses roquinimex and a method to produce it. The disclosed method starts with N-methylisatoic anhydride (I) and requires three steps. The improved process of the present invention starts with the same N-methylisatoic anhydride (I) and requires fewer steps.
The process of the present invention is practiced according to EXAMPLE 2. It is preferred to perform the claimed process in an aprotic solvent. Suitable aprotic solvents include DMF, THF, glyme, dioxane and ether and mixtures thereof.
The roquinimex produced by the process of the invention (EXAMPLE 2) can be upgraded or purified by the process of EXAMPLE 3.
Roquinimex is known to be useful as a pharmaceutical agent, see U.S. Pat. No. 4,738,971. It is preferably used in treating multiple sclerosis, in particular the treatment of relapsing remitting and secondary progressive multiple sclerosis. In treating multiple sclerosis roquinimex is administered in an oral dose of from about 2.0 to about 5.0 mg/day.
Example 1
N-Methyl-N-Phenyl-α-Carbomethoxyacetamide (V)
Mono-methyl malonate potassium salt also known as potassium methyl malonate (73.32 g, 0.47 mol) and water (50 ml) are cooled to 5° with an ice bath, and concentrated hydrochloric acid (40 ml) is added over a 30 minute period while the temperature is maintained below 10°. The mixture is filtered with suction to remove potassium chloride, and the precipitate washed with methyl t-butylether (75 ml). The aqueous layer of the filtrate is separated and washed with methyl t-butyl ether (3×50 ml). The combined methyl t-butyl ether extracts are dried over anhydrous sodium sulfate; then the solvent was removed under reduced pressure at 45-50° to give carbomethoxy acetic acid. This product was checked by NMR for complete removal of the methyl t-butyl ether solvent.
Carbomethoxy acetic acid (100 g, 0.84 mol) is dissolved in methylene chloride (400 ml). Thionyl chloride (100 g, 0.84 mol) is added via a dropping funnel. It can be added rapidly as there is little, if any, exotherm produced during the addition. After addition, the reaction is refluxed at 40-45° for 1 hr. At the end of the reflux period, 50% of the methylene chloride is removed (200 ml) by distillation at atmospheric pressure and 40-45°. Fresh methylene chloride is added (200 ml) followed by distillation to again remove 50% of the total volume. This add-distillation procedure is repeated two times to give the carbomethoxy acetyl chloride.
The carbomethoxy acetyl chloride mixture is cooled in an ice-salt bath to -5 to 0° and N-methyl aniline (55.64 g, 0.52 mol) in methylene chloride (200 ml) is added at a rate so as to maintain the temperature of the reaction mixture between -5 to 0°. The addition is performed using an addition funnel and can normally be carried out over a 3-5 min time period to control the slight exotherm. Pyridine (66.36 g, 0.84 mol) in methylene chloride (200 ml) is then added to the above mixture. The addition rate is adjusted so as to keep the temperature of the reaction between -5 to 0° during the addition. The addition is performed using an addition funnel and can normally be carried out over a 3-5 min time period to control the slight exotherm. After the addition is complete (as measured by HPLC) the reaction is quenched by pouring the reaction mixture into water (500 ml) and stirring continued for 30 min. The reaction is equilibrated and the methylene chloride layer separated. Additional methylene chloride (400-500 ml) is added and the methylene chloride mixture is washed successively with hydrochloric acid (1N, 2×300 ml), saturated sodium bicarbonate solution (2×300 ml), saline (1×600 ml) and the methylene chloride mixture dried through anhydrous sodium sulfate. Concentration of the mixture under reduced pressure at 40-45° gives the title compound, HPLC (Nucleosil column; acetonitrile/water, 45/55, 1 ml/min, UV=229 nm; Retention times for N-methyl-N-phenyl-α-carbomethoxyacetamide˜6.0 min; N-methyl aniline˜11.0-12.0 min.
Example 2
Preparation of Roquinimex (IV) from N-Methylisotoic anhydride (I) and N-Methyl-α-carbomethoxyacetamide (V)
N-Methyl-N-phenyl-α-carbomethoxyacetamide (V, EXAMPLE 1, 139 g, 0.671 mole) and DMF (695 mL). The mixture is subject to reduced pressure and purged with nitrogen three times. While at room temperature (20-25°), potassium t-butoxide solution (1.714 M in THF, 367 mL, 0.630 mole) is added in one portion. A small exotherm and slight darkening of the mixture followed this addition. The mixture is heated to 80-90° and kept at this temperature for 1.5 hr.
A -78° cooling bath is placed on the receiving flask of the distillation assembly, the nitrogen flow is shut off and the mixture is subject to reduced pressure over 0.5 hr to remove the THF solvent. The pot temperature at the end of the distillation is 72-76°. The amount of distillate collected should be nearly identical to the amount of potassium tert-butoxide reagent used, (367 ml). The mixture is then heated to 80-85° and N-methylisotoic anhydride (I, 70.72 g, 0.400 mole) is added in one portion followed by a 5-10 mL DMF wash. Gas evolution with foaming followed the addition and subsequent wash. The equipment is modified at this point to include a reflux condenser with a vacuum port. With the temperature still at 80-85°, the mixture is placed under reduced pressure and the mixture refluxed for 30 min. After refluxing the temperature is 79°. The reduced pressure and heat source are removed, the system is repressurized with nitrogen and the temperature is allowed to drop to 30° (±2°). Hydrochloric acid (0.6 N, 2.295 L) is added slowly via an addition funnel attached to the claisen head over 2.5 hr, to pH=1.0-1.5, making sure the temperature does not exceed 32°. The temperature control is especially critical at the beginning of the addition when a mild exotherm occurs. The temperature at the end of the addition is nearly room temperature (24-25°). When the acid addition is complete, the resulting slurry is stirred for 30 min and then let stand overnight before filtration. The solids are washed with water (2 ×330 mL) and dried on a nitrogen press to give the title compound, HPLC (Nucleosil column; acetonitrile/water, 45/55, 1 ml/min, UV=229 nm; Retention times 2.29 min.
Example 3
Purification of Roquinimex (IV)
Roquinimex crude is taken up in water (1.5 L) and the slurry is stirred vigorously at 20-25°. The pH is adjusted to 7.5-7.7 with sodium hydroxyde (7%, about 170 mL). (The base can be added as fast as possible but requires longer pH equilibration near the end of the addition (about 1-2 hr total addition time). It is recommended that 85% of the base is initially added to a stable pH and the rest is added dropwise until the pH has stabilized and falls into the desired range of 7.5-7.7.) Nearly all solids should be dissolved (some may remain however). After the base is added and the pH is stabilized for more than 30 min, Darco (charcoal, 15.00 g) is added and the mixture is stirred for 30 min. The mixture is filtered through a 0.45 micron Millipore filter and the filter cake is washed with water (2×175 mL). The filtrate is transferred to a flask.
The mixture is stirred vigorously, heated to 28-32° and hydrochloric acid (6 N, about 120 mL) is added over 30 to 45 min to a pH of 0.5 to 1.0. After the addition is over, the mixture is stirred for 15 min then allowed to stand, without stirring, at the above temperature for 2 hr before filtration. The filter cake is washed with water (2×180 mL) and dried on a nitrogen press to give essentially pure title compound.
PATENT
https://patents.google.com/patent/US6605616
The present invention relates to novel substituted quinoline-3-carboxamide derivatives, to methods for their preparation, to compositions containing them, and to methods and use for clinical treatment of diseases resulting from autoimmunity, such as multiple sclerosis, insulin-dependent diabetes mellitus, systemic lupus erythematosus, rheumatoid arthritis, inflammatory bowel disease and psoriasis and, furthermore, diseases where pathologic inflammation plays a major role, such as asthma, atherosclerosis, stroke and Alzheimer’s disease. More particularly, the present invention relates to novel quinoline derivatives suitable for the treatment of, for example, multiple sclerosis and its manifestations.
BACKGROUND OF THE INVENTION
Autoimmune diseases, e.g., multiple sclerosis (MS), insulin-dependent diabetes mellitus (IDDM), systemic lupuis erythematosus (SLE), rheumatoid arthritis (RA), inflammatory bowel disease (IBD) and psoriasis represent assaults by the body’s immune system which may be systemic in nature, or else directed at individual organs in the body. They appear to be diseases in which the immune system makes mistakes and, instead of mediating protective functions, becomes the aggressor (1).
MS is the most common acquired neurologic disease of young adults in western Europe and North America. It accounts for more disability and financial loss, both in lost income and in medical care, than any other neurologic disease of this age group. There are approximately 250.000 cases of MS in the United States. Although the cause of MS is unknown, advances in brain imaging, immunology, and molecular biology have increased researchers’ understanding of this disease. Several therapies are currently being used to treat MS, but no single treatment has demonstrated dramatic treatment efficacy. Current treatment of MS falls into three categories: treatment of acute exacerbations, modulation of progressive disease, and therapy for specific symptoms.
MS affects the central nervous system and involves a demyelination process, i.e., the myelin sheaths are lost whereas the axons are preserved. Myelin provides the isolating material that enables rapid nerve impulse conduction. Evidently, in demyelination, this property is lost. Although the pathogenic mechanisms responsible for MS are not understood, several lines of evidence indicate that demyelination has an immunopathologic basis. The pathologic lesions, the plaques, are characterized by infiltration of immunologically active cells such as macrophages and activated T cells (2).
In U.S. Pat. No. 4,547,511 and in U.S. Pat. No. 4,738,971 and in EP 59,698 some derivatives of N-aryl-1,2-dihydro-4-substituted-1-alkyl-2-oxo-quinoline-3-carboxamide are claimed as enhancers of cell-mediated immunity. The compound

known as roquinimex (Merck Index 12th Ed., No. 8418; Linomide®, LS2616, N-phenyl-N-methyl-1,2-dihydro-4-hydroxy-1-methyl-2-oxo-quinoline-3-carboxamide) belongs to this series of compounds. Roquinimex has been reported to have multiple immunomodulatory activities not accompanied with general immunosuppression (3-12). Furthermore, in U.S. Pat. No. 5,580,882 quinoline-3-carboxarnide derivatives are claimed to be useful in the treatment of conditions associated with MS. The particular preferred compound is roquinimex. In U.S. Pat. No. 5,594,005 quinoline-3-carboxamide derivatives are claimed to be useful in the treatment of type I diabetes. The particular preferred compound is roquinimex. In WO 95/24195 quinoline-3-carboxamide derivatives are claimed to be useful in the treatment of inflammatory bowel disease. Particularly preferred compounds are roquinimex or a salt thereof. In WO95/24196 quinoline-3-carboxamide derivatives are claimed to be useful in the treatment of psoriasis. Particularly preferred compounds are roquinimex or a salt thereof.
In clinical trials comparing roquinimex to placebo, roquinimex was reported to hold promise in the treatment of conditions associated with MS (13, 14). There are, however, some serious drawbacks connected to roquinimex. For example, it has been found to be teratogenic in the rat, and to induce dose-limiting side effects in man, e.g., a flu-like syndrome, which prevents from using the full clinical potential of the compound.
Further, in WO 92/18483 quinoline derivatives substituted in the 6-position with a RAS (O)n-group (RA=lower alkyl or aryl; n=0−2) are claimed, which possess an immunomodulating, anti-inflammatory and anti-cancer effect.
PAPER
Modified synthesis and antiangiogenic activity of linomide
https://www.sciencedirect.com/science/article/pii/S0960894X00006995?via%3Dihub

PAPER
https://pubs.acs.org/doi/full/10.1021/jm031044w
1H NMR (CDCl3) δ 3.28 (s, br, 3H, 1-NCH3), 3.50 (s, 3H, 12-NCH3), 7.1−7.3 (m, 7H, 6,8,2‘,3‘,4‘,5‘,6‘-aromatic CH), 7.56 (dt, JHCCH = 7.5 and 8.5 Hz, JHCCCH = 1.5 Hz, 1H, 7-aromatic CH), 8.09 (dd, JHCCH = 8.0 Hz, JHCCCH = 1.5 Hz, 1H, 5-aromatic CH), 12.3 (s, br, 1H, 4-OH). 13C NMR (CDCl3) δ 28.7 (1C, 1-NCH3), 38.3 (1C, br, 12-NCH3), 104.6 (1C, 3-C), 113.5 (1C, 8-CH), 115.3 (1C, 10-C), 121.4 (1C, 6-CH), 124.6 (1C, 5-CH), 125.5 (2C, 2‘,6‘-CH), 126.7 (1C, 4‘-CH), 128.5 (2C, 3‘,5‘-CH), 132.3 (1C, 7-CH), 140.1 (1C, 9-C), 143.8 (1C, 1‘-C), 158.8 (1C, 2-CO), 164.3 (1C, 4-C), 169.4 (1C, 11-CO). MS-ESI: m/z 309 [MH]+. Anal. (C18H16N2O3) C, H, N.
PATENT
1,2-dihydro-4-hydroxy-2-oxo-quinoline-3-carboxanilides have been described in the literature since the 1970s (refs 1-4). The most well-known compound in this class, roquinimex (Linomide), was first described by AB Leo as an immuno-stimulating agent (ref 4) but was later also found to have immuno-modulating effects, as well as anti-angiogenetic effects (refs 5a, b). Roquinimex has been claimed beneficial for the treatment of autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, systemic lupus erythematosus, inflammatory bowel disease, diabetes type 1, and psoriasis, as well as for the treatment of cancer (refs 6a-d, 9d and refs therein).


The compound laquinimod (a 5-Cl, N-Et carboxanilide derivative) has been reported by Active Biotech AB to convey a better therapeutic index compared with roquinimex (refs 7a, b) and is currently in phase III clinical studies for the treatment of multiple sclerosis. Laquinimod has also entered clical trials in Crohn’s disease and
SLE. Two other compounds in the same class under clinical evaluation are tasquinimod (prostate cancer) and paquinimod (systemic sclerosis). Recently, a molecular target for laquinimod was identified as S100A9 (ref 8).
Fujisawa has reported on similar compounds with inhibitory activity on nephritis and on B16 melanoma metastases (refs 9a-d). Also the closely related thieno-pyridone analogs have been described as immunomodulating compounds with anti-inflammatory properties (ref
10).
Another closely related compound class are the corresponding N-pyridyl-carboxamide derivatives, which have been reported to have antitubercular activity as well as anti-inflammatory properties (ref
11). However, according to litterature (ref 10) these derivatives are less active as immunomodulating agents.
The N-hydrogen 3-carboxanilides (“N-H derivatives”) and the N-alkyl 3-carboxanilides (“N-alkyl derivatives”), respectively, are described in the prior art documents relating to inflammation, immunomodulation, and cancer as a homogenous group of compounds in terms of biological effects. Prior art also teaches that the N-alkyl derivatives are the preferred compound derivatives.

In fact, very few studies (refs 4, 9d) of N-hydrogen derivatives, especially in vivo studies, have been reported. Furthermore, no fundamental biological differences between the N-alkyl derivatives and the N-hydrogen derivatives, respectively, have been described.
However, some chemical properties of the N-hydrogen and the N-alkyl derivatives are different (ref 12). N-Alkyl derivatives adopt a twisted 3D-structure, whereas the N-H derivatives are stabilized by intramolecular hydrogen bonds in a planar structure. The N-alkyl derivatives are more soluble in aqueous media, but also inherently unstable towards nucleophiles, such as amines and alcohols (refs 12, 13).
The N-alkyl derivatives roquinimex (N-Me) and laquinimod (N-Et) have been reported to be metabolized in human microsomes to give the corresponding N-hydrogen derivatives, via N-dealkylation catalyzed mainly by CYP3A4 (refs 14a, b).
bHLH-PAS (basic helix-loop-helix Per-Arnt-Sim) proteins constitute a recently descovered protein family functioning as transcripon factors as homo or hetero protein dimers (refs 15a, b). The N-terminal bHLH domain is responsible for DNA binding and contributes to dimerization with other family members. The PAS region (PAS-A and PAS-B) is also involved in protein-protein interactions determining the choice of dimerization partner and the PAS-B domain harbors a potential ligand binding pocket.
The aryl hydrocarbon receptor (AhR or dioxin receptor) and its dimerization partner ARNT (AhR nuclear translocator) were the first mammalian protein members to be identified. AhR is a cytosolic protein in its non-activated form, associated in a protein complex with Hsp90, p23, and XAP2. Upon ligand activation, typically by chlorinated aromatic hydrocarbons like TCDD, the Ahr enters the nucleus and dimerizes with ARNT. The AhR/ARNT dimer recognizes specific xenobiotic response elements (XREs) to regulate TCDD-responsive genes. The ligand binding domain of AhR (AhR-LBD) resides in the PAS-B domain.
Recently, it has been demonstrated that AhR is involved in Thl7 and Treg cell development and AhR has been proposed as a unique target for therapeutic immuno-modulation (refs 16a-c). The AhR ligand TCDD was shown to induce development of Treg (FoxP3+) cells, essential for controlling auto-immunity, and to suppress symptoms in the EAE model. In addition, activation of AhR has been shown essential for the generation of IL-10 producing regulatory Trl cells (ref 16d), and Ahr ligands have also been proven efficacious in other models of auto-immunity, e.g. diabetes type 1, IBD, and uveitis (refs 16e-h). Apart from controlling autoimmune disorders, AhR activation and Treg cell development have been implicated as a therapeutic strategy for other conditions with an immunological component, such as allergic lung inflammation, food allergy, transplant rejection, bone loss, and type 2 diabetes and other metabolic disorders (refs 17a-e).
Apart from its role as a transcription factor, AhR has been reported to function as a ligand-dependent E3 ubiguitin ligase (ref 18), and ligand-induced degradation of β-catenin has been demonstrated to suppress intestinal cancer in mice (ref 19). In addition, activation of AhR has been implicated to play a protective role in prostate cancer (ref 20).
Other members of the bHLH-PAS family are the HIF-α (hypoxia inducible factor alpha) proteins, which also hetero-dimerize with ARNT. In conditions with normal oxygen levels (normoxia), HIF-α proteins are rapidly degraded by the ubiquitin-proteasome system and they are also inactivated by asparagine hydroxylation. Under hypoxic conditions, however, the proteins are active and upregulate genes as a response to the hypoxic state, e.g. genes for erythropoietin and vascular endothelial growth factor (VEGF). VEGF is essential for blood vessel growth (angiogenesis) and is together with HIF-1α considered as interesting targets for anti-angiogenetic tumour theraphy (ref 21). HIF-α proteins can be negatively and indirectly regulated by AhR ligands, which upon binding with AhR reduce the level of the common dimerization partner ARNT. Anti-angiogenetic effects can possibly also be achieved directly by AhR activity via upregulation of thrombospondin-1 (ref 22).
 |
|
Title: Roquinimex
CAS Registry Number: 84088-42-6
CAS Name: 1,2-Dihydro-4-hydroxy-N,1-dimethyl-2-oxo-N-phenyl-3-quinolinecarboxamide
Additional Names: N-phenyl-N-methyl-1,2-dihydro-4-hydroxy-1-methyl-2-oxoquinoline-3-carboxamide; 1,2-dihydro-4-hydroxy-N,1-dimethyl-2-oxo-3-quinolinecarboxanilide
Manufacturers’ Codes: LS-2616
Trademarks: Linomide (Pfizer)
Molecular Formula: C18H16N2O3
Molecular Weight: 308.33
Percent Composition: C 70.12%, H 5.23%, N 9.09%, O 15.57%
Literature References: Biological response modifier. Prepn: E. Eriksoo et al., EP 59698; eidem, US 4738971 (1982, 1988 both to AB Leo). Immunopharmacology: A. Tarkowski et al., Immunology 59, 589 (1986). Mechanism of action study: E.-L. Larsson et al.,Int. J. Immunopharmacol. 9, 425 (1987). Clinical evaluation in cancer patients: J. C. S. Bergh et al., Cancer Invest. 15, 204 (1997).
Properties: Crystals from pyridine, mp 200-204°.
Melting point: mp 200-204°
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Immunomodulators.
|
 |
|
| Clinical data | |
|---|---|
| ATC code | |
| Pharmacokinetic data | |
| Biological half-life | 26-42 hours |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ECHA InfoCard | 100.163.758 |
| Chemical and physical data | |
| Formula | C18H16N2O3 |
| Molar mass | 308.331 g/mol |
| 3D model (JSmol) | |
| |
|
Roquinimex (Linomide) is a quinoline derivative immunostimulant which increases NK cell activity and macrophage cytotoxicity. It also inhibits angiogenesis and reduces the secretion of TNF alpha.
/////////////////Roquinimex, Linomide, FCF-89, LS-2616, PNU-212616
CN1C2=CC=CC=C2C(=O)C(=C1O)C(=O)N(C)C3=CC=CC=C3
ビガバトリン , Vigabatrin
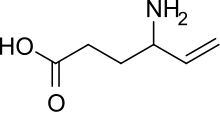
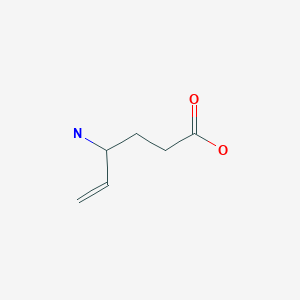
Vigabatrin
CAS: 60643-86-9
- Molecular FormulaC6H11NO2
- Average mass129.157 Da
Infantile spasms, Anticonvulsant, Antiepileptic
orphan drug designation
GVG
M071754
MDL-71754
ORP-001
RMI-71754
RMI-71890 ((+)-enantiomer)
Vigabatrin, brand name Sabril, is an antiepileptic drug that inhibits the breakdown of γ-aminobutyric acid (GABA) by acting as a suicide inhibitor of the enzyme GABA transaminase (GABA-T). It is also known as γ-vinyl-GABA, and is a structural analogue of GABA, but does not bind to GABA receptors.[1]
Medical uses
Epilepsy
In Canada, vigabatrin is approved for use as an adjunctive treatment (with other drugs) in treatment resistant epilepsy, complex partial seizures, secondary generalized seizures, and for monotherapy use in infantile spasms in West syndrome.[1]
As of 2003, vigabatrin is approved in Mexico for the treatment of epilepsy that is not satisfactorily controlled by conventional therapy (adjunctive or monotherapy) or in recently diagnosed patients who have not tried other agents (monotherapy).[2]
Vigabatrin is also indicated for monotherapy use in secondarily generalized tonic-clonic seizures, partial seizures, and in infantile spasms due to West syndrome.[2]
On August 21, 2009, Lundbeck announced that the U.S. Food and Drug Administration had granted two New Drug Application approvals for vigabatrin. The drug is indicated as monotherapy for pediatric patients one month to two years of age with infantile spasms for whom the potential benefits outweigh the potential risk of vision loss, and as adjunctive (add-on) therapy for adult patients with refractory complex partial seizures (CPS) who have inadequately responded to several alternative treatments and for whom the potential benefits outweigh the risk of vision loss.
In 1994, Feucht and Brantner-Inthaler reported that vigabatrin reduced seizures by 50-100% in 85% of children with Lennox-Gastaut syndrome who had poor results with sodium valproate.[3]
Others
Vigabatrin reduced cholecystokinin tetrapeptide-induced symptoms of panic disorder, in addition to elevated cortisol and ACTH levels, in healthy volunteers.[4]
Vigabatrin is also used to treat seizures in succinic semialdehyde dehydrogenase deficiency (SSADHD), which is an inborn GABA metabolism defect that causes intellectual disability, hypotonia, seizures, speech disturbance, and ataxia through the accumulation of γ-Hydroxybutyric acid (GHB). Vigabatrin helps lower GHB levels through GABA transaminase inhibition. However, this is in the brain only; it has no effect on peripheral GABA transaminase, so the GHB keeps building up and eventually reaches the brain.[5]
Adverse effects
Central nervous system
Sleepiness (12.5%), headache (3.8%), dizziness (3.8%), nervousness (2.7%), depression (2.5%), memory disturbances (2.3%), diplopia (2.2%), aggression (2.0%), ataxia (1.9%), vertigo (1.9%), hyperactivity (1.8%), vision loss (1.6%) (See below), confusion(1.4%), insomnia (1.3%), impaired concentration (1.2%), personality issues (1.1%).[1] Out of 299 children, 33 (11%) became hyperactive.[1]
Some patients develop psychosis during the course of vigabatrin therapy,[6] which is more common in adults than in children.[7] This can happen even in patients with no prior history of psychosis.[8] Other rare CNS side effects include anxiety, emotional lability, irritability, tremor, abnormal gait, and speech disorder.[1]
Gastrointestinal
Abdominal pain (1.6%), constipation (1.4%), vomiting (1.4%), and nausea (1.4%). Dyspepsia and increased appetite occurred in less than 1% of subjects in clinical trials.[1]
Body as a whole
Fatigue (9.2%), weight gain (5.0%), asthenia (1.1%).[1]
Teratogenicity
A teratology study conducted in rabbits found that a dose of 150 mg/kg/day caused cleft palate in 2% of pups and a dose of 200 mg/kg/day caused it in 9%.[1] This may be due to a decrease in methionine levels, according to a study published in March 2001.[9] In 2005, a study conducted at the University of Catania was published stating that rats whose mothers had consumed 250–1000 mg/kg/day had poorer performance in the water maze and open-field tasks, rats in the 750-mg group were underweight at birth and did not catch up to the control group, and rats in the 1000 mg group did not survive pregnancy.[10]
There is no controlled teratology data in humans to date.
Sensory
In 2003, vigabatrin was shown by Frisén and Malmgren to cause irreversible diffuse atrophy of the retinal nerve fiber layer in a retrospective study of 25 patients.[11] This has the most effect on the outer area (as opposed to the macular, or central area) of the retina.[12] Visual field defects had been reported as early as 1997 by Tom Eke and others, in the UK. Some authors, including Comaish et al. believe that visual field loss and electrophysiological changes may be demonstrable in up to 50% of Vigabatrin users.
The retinal toxicity of vigabatrin can be attributed to a taurine depletion.[13]
Interactions
A study published in 2002 found that vigabatrin causes a statistically significant increase in plasma clearance of carbamazepine.[14]
In 1984, Drs Rimmer and Richens at the University of Wales reported that administering vigabatrin with phenytoin lowered the serum phenytoin concentration in patients with treatment-resistant epilepsy.[15] Five years later, the same two scientists reported a fall in concentration of phenytoin of 23% within five weeks in a paper describing their failed attempt at elucidating the mechanism behind this interaction.[16]
Pharmacology
Vigabatrin is an irreversible mechanism-based inhibitor of gamma-aminobutyric acid aminotransferase (GABA-AT), the enzyme responsible for the catabolism of GABA, which increases the level of GABA in the brain.[1][17] Vigabatrin is a racemic compound, and its [S]-enantiomer is pharmacologically active.[18],[19]
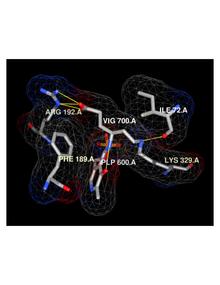
Crystal Structure (pdb:1OHW) showing vigabatrin binding to specific residues in the active site of GABA-AT, based off experiments by Storici et al.[20]
Pharmacokinetics
With most drugs, elimination half-life is a useful predictor of dosing schedules and the time needed to reach steady state concentrations. In the case of vigabatrin, however, it has been found that the half-life of biologic activity is far longer than the elimination half-life.[21]
For vigabatrin, there is no range of target concentrations because researchers found no difference between the serum concentration levels of responders and those of non-responders.[22] Instead, the duration of action is believed to be more a function of the GABA-T resynthesis rate; levels of GABA-T do not usually return to their normal state until six days after stopping the medication.[19]
History
Vigabatrin was developed in the 1980s with the specific goal of increasing GABA concentrations in the brain in order to stop an epileptic seizure. To do this, the drug was designed to irreversibly inhibit the GABA transaminase, which degrades the GABA substrate. Although the drug was approved for treatment in the United Kingdom in 1989, the authorized use of Vigabatrin by US Food and Drug Administration was delayed twice in the United States before 2009. It was delayed in 1983 because animal trials produced intramyelinic edema, however, the effects were not apparent in human trials so the drug design continued. In 1997, the trials were temporarily suspended because it was linked to peripheral visual field defects in humans.[23]
Society and culture
Brand Names
Vigabatrin is sold as Sabril in Canada,[24] Mexico,[2] and the United Kingdom.[25] The brand name in Denmark is Sabrilex. Sabril was approved in the United States on August 21, 2009 and is currently marketed in the U.S. by Lundbeck Inc., which acquired Ovation Pharmaceuticals, the U.S. sponsor in March 2009.
Synthesis
http://www.drugfuture.com/synth/syndata.aspx?ID=90252

This compound can be prepared in two different ways: 1) The reaction of 1,4-dichloro-2-butene (I) with diethyl malonate (II) by means of sodium ethoxide in refluxing ethanol gives 1,1-bis(ethoxycarbonyl)-2-vinylcyclopropane (III), which by reaction with ammonia gas in DMF at 120 C is converted into 3-carboxamido-5-vinyl-2-pyrrolidone (IV). Finally, this compound is treated with concentrated HCl in refluxing acetic acid. 2) The treatment of (IV) with sodium ethoxide in refluxing ethanol gives 3-carboxy-5-vinyl-2-pyrrolidone (V), which is decarboxylated by treatment with refluxing acetic acid to afford 5-vinyl-2-pyrrolidone (VI). The bromination of (VI) with Br2 in CCl4 yields 5-(1,2-dibromoethyl)-2-pyrrolidone (VII), which by treatment with Na in liquid NH3 in a pressure vessel at 25 C is converted into 4-aminohex-5-inoic acid (VIII). Finally, this compound is partially reduced with H2 over a suitable catalyst.

The synthesis of [14C]-labeled vigabatrin has been described: The reduction by known methods of pyroglutamic acid (I) to the alcohol (II) and its acylation with p-toluenesulfonyl chloride gives 5-(tosyloxymethyl)pyrrolidin-2-one (III), which by reaction with [14C]-labeled sodium cyanide in hot DMF yields 5-([14C]-cyanomethyl)pyrrolidin-2-one (IV). The reduction of (VI) with H2 over Pd/Al2O3 and treatment with dimethylamine affords 5-[2-(dimethylamino)ethyl]pyrrolidin-2-one (VI), which is oxidized with H2O2 in water to the N-oxide (VI). The treatment of (VI) with K2CO3 in refluxing xylene affords 5-([14C]-vinyl)pyrrolidin-2-one (VII), which is finally submitted to ring opening with hot 5 M aqueous HCl, followed by neutralization with triethylamine.

An efficient new synthesis for [14C]-labeled vigabatrin has been described: The reaction of triphenylphosphine (I) with [14C]-labeled methyl iodide (II) in benzene gives the corresponding phosphonium salt (III), which is submitted to a Wittig condensation with 1-(1-butenyl)-5-oxopiperidin-2-carbaldehyde (IV) to afford the vinylpyrrolidone (V). Finally, this compound is hydrolyzed with 6N HCl at 95 C.
 The enantiocontrolled addition of phthalimide (I) to 1,3-butadiene monoepoxide (II) with a chiral palladium catalyst and Na2CO3 in dichloromethane gives N-(2-hydroxy-1(S)-vinylethyl)phthalimide (III), which is treated with triflic anhydride and TEA in dichloromethane to yield the triflate (IV). The condensation of (IV) with dimethyl malonate (V) by means of NaH in THF affords the alkylated malonate (VI), which is finally decarboxylated and deprotected by a treatment with aqueous refluxing HCl. Note that the synthesis of the biologically active (S)-enantiomer simply requires a change in the chirality of the Pd catalyst used in the first step of the synthesis.
The enantiocontrolled addition of phthalimide (I) to 1,3-butadiene monoepoxide (II) with a chiral palladium catalyst and Na2CO3 in dichloromethane gives N-(2-hydroxy-1(S)-vinylethyl)phthalimide (III), which is treated with triflic anhydride and TEA in dichloromethane to yield the triflate (IV). The condensation of (IV) with dimethyl malonate (V) by means of NaH in THF affords the alkylated malonate (VI), which is finally decarboxylated and deprotected by a treatment with aqueous refluxing HCl. Note that the synthesis of the biologically active (S)-enantiomer simply requires a change in the chirality of the Pd catalyst used in the first step of the synthesis.

The reaction of 3-aminotetrahydrofuran-2-one (I) with benzyloxycarbonyl chloride (II) and TEA in chloroform gives the carbamate (III), which is reduced to the lactol (IV) by means of DIBAL in toluene. It has been observed that lactol (IV) is in equilibrium with its tautomeric open chain aldehydic form.(V). The reaction of (IV)??(V) with phosphonium bromide (VI) by means of Bu-Li in THF yields 3-amino-4-penten-1-ol (VII), which is reprotected with benzyloxycarbonyl chloride (II) and TEA to afford the carbamate (VIII). The reaction of (VIII) with CBr4 and PPh3 in dichloromethane provides the pentenyl bromide (IX), which is treated with LiCN in THF to give 4-(benzyloxycarbonylamino)-5-hexenenitrile (X). Finally this compound is hydrolyzed with conc. HCl to yield the target 4-amino-5-hexenoic acid.
References
- ^ Jump up to:a b c d e f g h i Long, Phillip W. “Vigabatrin.” Archived April 23, 2006, at the Wayback Machine. Internet Mental Health. 1995–2003.
- ^ Jump up to:a b c DEF Mexico: Sabril Archived September 14, 2005, at the Wayback Machine. Diccionario de Especialdades Farmaceuticas. Edicion 49, 2003.
- Jump up^ Feucht M, Brantner-Inthaler S (1994). “Gamma-vinyl-GABA (vigabatrin) in the therapy of Lennox-Gastaut syndrome: an open study” (PDF). Epilepsia. 35 (5): 993–8. doi:10.1111/j.1528-1157.1994.tb02544.x. PMID 7925171. Retrieved 2006-05-25.
- Jump up^ Zwanzger P, Baghai TC, Schuele C, Strohle A, Padberg F, Kathmann N, Schwarz M, Moller HJ, Rupprecht R (2001). “Vigabatrin decreases cholecystokinin-tetrapeptide (CCK-4) induced panic in healthy volunteers”. Neuropsychopharmacology. 25 (5): 699–703. doi:10.1016/S0893-133X(01)00266-4. PMID 11682253.
- Jump up^ Pearl, Phillip L; Robbins, Emily; Capp, Philip K; Gasior, Maciej; Gibson, K Michael (May 5, 2004). “Succinic Semialdehyde Dehydrogenase Deficiency”. GeneReviews. Seattle, Washington: University of Washington. Retrieved September 6, 2010.
- Jump up^ Sander JW, Hart YM (1990). “Vigabatrin and behaviour disturbance”. Lancet. 335 (8680): 57. doi:10.1016/0140-6736(90)90190-G. PMID 1967367.
- Jump up^ Chiaretti A, Castorina M, Tortorolo L, Piastra M, Polidori G (1994). “[Acute psychosis and vigabatrin in childhood]”. La Pediatria Medica e Chirurgica : Medical and surgical pediatrics. 16 (5): 489–90. [Article in Italian] PMID 7885961
- Jump up^ Sander JW, Hart YM, Trimble MR, Shorvon SD (1991). “Vigabatrin and psychosis”. Journal of Neurology, Neurosurgery, and Psychiatry. 54 (5): 435–9. doi:10.1136/jnnp.54.5.435. PMC 488544
 . PMID 1865207.
. PMID 1865207. - Jump up^ Abdulrazzaq YM, Padmanabhan R, Bastaki SM, Ibrahim A, Bener A (2001). “Placental transfer of vigabatrin (gamma-vinyl GABA) and its effect on concentration of amino acids in the embryo of TO mice”. Teratology. 63 (3): 127–33. doi:10.1002/tera.1023. PMID 11283969.
- Jump up^ Lombardo SA, Leanza G, Meli C, Lombardo ME, Mazzone L, Vincenti I, Cioni M (2005). “Maternal exposure to the antiepileptic drug vigabatrin affects postnatal development in the rat”. Neurological Sciences. 26 (2): 89–94. doi:10.1007/s10072-005-0441-6. PMID 15995825.
- Jump up^ Frisén L, Malmgren K (2003). “Characterization of vigabatrin-associated optic atrophy”. Acta Ophthalmologica Scandinavica. 81 (5): 466–73. doi:10.1034/j.1600-0420.2003.00125.x. PMID 14510793.
- Jump up^ Buncic JR, Westall CA, Panton CM, Munn JR, MacKeen LD, Logan WJ (2004). “Characteristic retinal atrophy with secondary “inverse” optic atrophy identifies vigabatrin toxicity in children”. Ophthalmology. 111 (10): 1935–42. doi:10.1016/j.ophtha.2004.03.036. PMC 3880364 . PMID 15465561.
- Jump up^ Gaucher D; Arnault E; Husson Z; et al. (November 2012). “Taurine deficiency damages retinal neurones: cone photoreceptors and retinal ganglion cells”. Amino Acids. 43 (5): 1979–1993. doi:10.1007/s00726-012-1273-3. PMC 3472058 . PMID 22476345.
- Jump up^ Sanchez-Alcaraz, Agustín; Quintana MB; Lopez E; Rodriguez I; Llopis P (2002). “Effect of vigabatrin on the pharmacokinetics of carbamazepine”. Journal of Clinical Pharmacology and Therapeutics. 27 (6): 427–30. doi:10.1046/j.1365-2710.2002.00441.x. PMID 12472982.
- Jump up^ Rimmer EM, Richens A (1984). “Double-blind study of gamma-vinyl GABA in patients with refractory epilepsy”. Lancet. 1 (8370): 189–90. doi:10.1016/S0140-6736(84)92112-3. PMID 6141335.
- Jump up^ Rimmer EM, Richens A (1989). “Interaction between vigabatrin and phenytoin”. British Journal of Clinical Pharmacology. 27 (Suppl 1): 27S–33S. doi:10.1111/j.1365-2125.1989.tb03458.x. PMC 1379676 . PMID 2757906.
- Jump up^ Rogawski MA, Löscher W (2004). “The neurobiology of antiepileptic drugs”. Nat Rev Neurosci. 5 (7): 553–564. doi:10.1038/nrn1430. PMID 15208697.
- Jump up^ Sheean, G.; Schramm T; Anderson DS; Eadie MJ. (1992). “Vigabatrin–plasma enantiomer concentrations and clinical effects”. Clinical and Experimental Neurology. 29: 107–16. PMID 1343855.
- ^ Jump up to:a b Gram L, Larsson OM, Johnsen A, Schousboe A (1989). “Experimental studies of the influence of vigabatrin on the GABA system”. British Journal of Clinical Pharmacology. 27(Suppl 1): 13S–17S. doi:10.1111/j.1365-2125.1989.tb03455.x. PMC 1379673 . PMID 2757904.
- Jump up^ Storici Paola; De Biase D; Bossa F; Bruno S; Mozzarelli A; Peneff C; Silverman R; Schirmer T. (2003). “Structures of γ-Aminobutyric Acid (GABA) Aminotransferase, a Pyridoxal 5′-Phosphate, and [2Fe-2S] Cluster-containing Enzyme, Complexed with γ-Ethynyl-GABA and with the Antiepilepsy Drug Vigabatrin”. The Journal of Biochemistry. 279(1): 363–73. doi:10.1074/jbc.M305884200. PMID 14534310.
- Jump up^ Browne TR (1998). “Pharmacokinetics of antiepileptic drugs”. Neurology. 51 (5 suppl 4): S2–7. doi:10.1212/wnl.51.5_suppl_4.s2. PMID 9818917.
- Jump up^ Lindberger M, Luhr O, Johannessen SI, Larsson S, Tomson T (2003). “Serum concentrations and effects of gabapentin and vigabatrin: observations from a dose titration study”. Therapeutic Drug Monitoring. 25 (4): 457–62. doi:10.1097/00007691-200308000-00007. PMID 12883229.
- Jump up^ Ben-Menachem E. (2011). “Mechanism of Action of vigabatrin: correcting misperceptions”. Acta Neurologica Scandinavica. 124: 5. doi:10.1111/j.1600-0404.2011.01596.x.
- Jump up^ drugs.com Vigabatrin Drug Information
- Jump up^ Treatments for Epilepsy – Vigabatrin Norfolk and Waveney Mental Health Partnership NHS Trust
///////////Vigabatrin, ビガバトリン , MDL-71754; RMI-71754, orphan drug designation
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Sabril |
| Synonyms | γ-Vinyl-GABA |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a610016 |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 80–90% |
| Protein binding | 0% |
| Metabolism | not metabolized |
| Biological half-life | 5–8 hours in young adults, 12–13 hours in the elderly. |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.165.122 |
| Chemical and physical data | |
| Formula | C6H11NO2 |
| Molar mass | 129.157 g/mol |
| 3D model (JSmol) | |
| Melting point | 171 to 177 °C (340 to 351 °F) |
ANTHONY M CRASTO GETS PHARMA EXCELLENCE AWARD AT THE GOLDEN GLOBE TIGER AWARDS 2018

Pic…. Shobha crasto, Lionel and Aishal collecting my award named The Golden Globe Tigers Awards 2018, for Excellence in Pharma, at kuala lumpur, Malaysia, 23 april 2018 at Pullman city centre hotel, Kuala lumpur, Malaysia. Dr Anthony could not travel as he is wheelchair bound and 90 percent paralysed

DR ANTHONY M CRASTO
The Golden Globe Tigers Award 2018 is the highest recognition amongst individual and organizations who have achieved the highest levels of standards and benchmark in numerous areas such as CSR, Pharma, Social Media & Digital Marketing, Education Leadership Award and so on. The award ceremony took place in Pullman Kuala Lumpur City Centre Hotel & Residences on the 23rd of April, where a number of respectable attendees were present not only from Malaysia but from many different parts of the world such as Iceland, Saudi Arabia, India, China, South Africa and such!
The Golden Globe Tigers Award not only aims to increase awareness on CSR practices but also continuously innovate practices towards sustainable development. It is organized by the founder of the World CSR Day, World Sustainability Congress and World Women Leadership Congress.








Dr. Anthony Melvin Crasto, graduated from Mumbai University, Completed his Ph.D from ICT, 1991, Mumbai, India, in the field of Organic Chemistry, Currently he is working with GLENMARK PHARMACEUTICALS LTD, Research Centre as a Principal Scientist, in Process Research at Mahape, Navi Mumbai, India, for the last 10 years, His total Industry experience is 30 +yrs with major Multinationals companies.
Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable Scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri , Dr T.V. Radhakrishnan and Dr B. K. Kulkarni, etc, He did Custom Synthesis for various multinationals in his career like BASF, Novartis, Sanofi, Pfizer etc., He has worked in Drug Discovery, Natural products, Bulk Drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, Pharma Plant, API plant etc, he is now helping millions, His friends call him worlddrugtracker.
His New Drug Approvals, All about drugs, Eurekamoments, Organic spectroscopy international, etc in Organic/ Medchem are some most read blogs. He has hands on experience in initiation and developing Novel routes for Drug molecules and implementing them on commercial scale over a 30 year tenure till date Feb’ 2018, Around 30 plus commercial products in his career. He has good knowledge of IPM, GMP, QbD, Regulatory aspects, Technology transfer, Manufacturing, Formulations, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc. He has several International patents published worldwide.
He suffered a one in a million disease in the form of a paralytic stroke/ Acute Transverse Mylitis in Dec’ 2007 and is 90 % paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, he has several million hits on Google, 60 Lakh plus views on dozen plus blogs, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto
He is a prolific presenter and is invited to major conferences in Mumbai, where he can travel easily. He speaks at universities on topics of Drug discovery, Patents, Qbd, GMP, Tech transfer, polymorphism, Literature search tools, Computer programs and Topics of interest to Pharma Students.
His extraordinary skill on the Computers give him the edge to write/present his thoughts. He demonstrates them to students and professionals alike.
Notably he has 20 lakh plus views on New Drug Approvals Blog in 216 countries, This blog has 3.5 lakh viewers in USA alone.
AWARDS
“100 Most Impactful Health care Leaders Global listing”, conferred at Taj lands end, Mumbai, India on 14 Feb 2014 by World Health Wellness congress and awards
“Best Worlddrugtracker “ Award for lifetime acheivement in Pharma… 7 th July 2017 , The venue…Taj land ends, Bandra, Mumbai India
“Lifetime achievement award” WORLD HEALTH CONGRESS 2017 in Hyderabad, 22 aug 2017 at JNTUH KUKATPALLY. HYDERABAD, TELANGANA, INDIA,
” Lifetime Achievement Award”, at the The Middle East Healthcare Leadership Awards – 12th October, 2017 -The Address, Dubai Mall, Dubai…UAE……….Mohammed Bin Rashid Boulevard, Downtown Dubai – Dubai – United Arab Emirates
International award for Outstanding contribution in Pharma at World Health and wellness Congress award, 14th Feb, 2018, at Taj Lands ends, Bandra, Mumbai, India
Conferred very prestigious IDMA award for contribution to society in Pharma at INDIAN DRUGS ANNUAL DAY 2018 VMCC IITBombay Powai, Mumbai India 22 Feb 2018,I was Guest of honor at and was felicitated by president,Indian Drug manufacturers association (IDMA)
The Golden Globe Tigers Awards 2018, for Excellence in Pharma, at kuala lumpur, Malaysia, 23 april 2018 at Pullman hotel.
Biography
READ MY BIOGRAPHY……….http://scijourno.com/2017/01/09/dr-anthony-crasto/ Written by Mr. Amrit B. Karmarkar
Director, InClinition
///////////////
The Green ChemisTREE: 20 years after taking root with the 12 principles

The Green ChemisTREE: 20 years after taking root with the 12 principles
View original post 314 more words
Cadexomer Iodine
Cadexomer Iodine
Cadex, Declat, Decrat, Dextrinomer iodine, Iodoflex, Iodosorb, NI-009
CAS 94820-09-4
Listed in 1984 (Perstorp, Finland). For the treatment of exudative and infectious wounds, such as venous ulcers. This product is in contact with wound exudate to form a non-adhesive protective layer and release antibacterial iodine
Product of reaction of dextrin with epichlorohydrin coupled with ion-exchange groups and iodine
Cadexomer iodine is an iodophor that is produced by the reaction of dextrin with epichlorhydrin coupled with ion-exchange groups and iodine. It is a water-soluble modified starch polymer containing 0.9% iodine, calculated on a weight-weight basis, within a helical matrix.[1]
The Central Drugs Standard Control Organization (CDSCO) is the Central Drug Authority for discharging functions assigned to the Central Government under the Drugs and Cosmetics Act. One of the major functions of CDSCO is approval of new drugs in the country. During the month of March 2018, CDSCO has approved the following drugs classifying them as New Drug Approvals
Cadexomer Iodine Bulk & Powder 100 % w/w (contain 0.9 % w/v Iodine) or Cadexomer Iodine Ointment 500 mg (contains 0.9% w/v iodine)
For the treatment of chronic exuding wounds such as leg ulcers, pressure ulcers and diabetes ulcers infected traumatic and surgical wounds.
Cadexomer iodine is an iodophor that is produced by the reaction of dextrin with epichlorhydrin coupled with ion-exchange groups and iodine. It is a water-soluble modified starch polymer containing 0.9% iodine, calculated on a weight-weight basis, within a helical matrix.
In India, M/s Virchow Biotech Private Limited presented their proposal for grant of license to manufacture and market this product in India. The firm presented the Phase III Clinical trial report titled ‘Safety and efficacy of Dexadine (Cadexomer Iodine) in the treatment of chronic wounds’ before the CDSCO’s Subject Expert Committee on Antibiotics & Antivirals. After detailed deliberation, the committee recommended the manufacturing and marketing of the products (Cadexomer Iodine Ointment & Cadexomer Iodine Powder), as topical preparations for the treatment of chronic exuding wounds
History
Cadexomer iodine was developed in the early 1980s in Sweden by Perstorp AB, and given the name Iodosorb. The product was shown to be effective in the treatment of venous ulcers,.[2][3] More recently, it has been shown in studies in animals and humans that, unlike the iodophor povidone-iodine, Iodosorb causes an acceleration of the healing process in chronic human wounds. This is due to an increase in epidermal regeneration and epithelialization in both partial-thickness and full-thickness wounds.[4] In this way cadexomer iodine acts as a cicatrizant.
Properties
When formulated as a topical wound dressing Iodosorb adsorbs exudate and particulate matter from the surface of granulating wounds and, as the dressing becomes moist, iodine is released. The product thus has the dual effect of cleansing the wound and exerting a bactericidal action.
Uses
In addition to other manufacturers, Smith & Nephew distributes cadexomer iodine as Iodosorb and Iodoflex in many countries of the world for the treatment and healing of various types of wounds. The dosage forms are a paste dressing, an ointment and a gel, all of which contain 0.9% iodine.
PATENT
WO2001070242
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001070242
PATENT
WO 2008117300
https://patents.google.com/patent/WO2008117300A2/und
Improved Process for the Preparation Of
Cadexomer Iodine
The present invention describes an improved method for the preparation of cadexomer iodine. Cadexomer iodine is a hydrophilic modified starch polymer containing 0.9%w/w iodine within the helical matrix. It is used for its absorbent and antiseptic properties in the management of chronic wounds such as venous leg ulcers, pressure sores, etc. It is applied as a powder or as an ointment over the wound.
Background of the invention
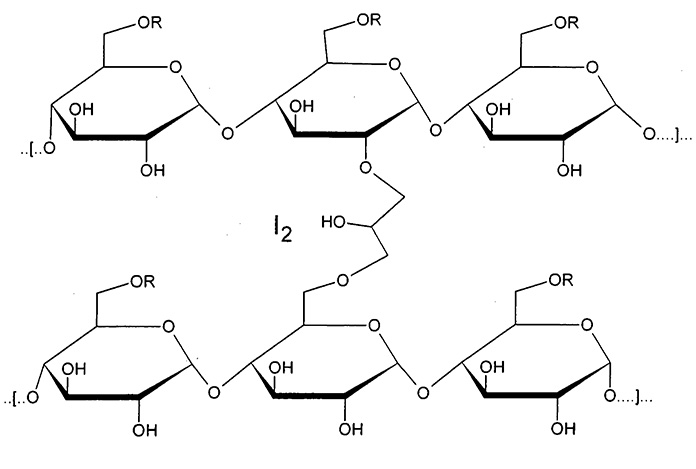
Cadexomer iodine is an iodophor that releases iodine. It contains 0.9%w/w iodine in hydrophilic modified starch carrier. It is used for its absorbent and antiseptic properties, in the management of venous leg ulcers and pressure sores, burn wounds etc. It is applied as a powder of microbeads or ointment containing iodine 0.9%w/w. When applied to the wound it absorbs fluids, removing exudates, pus and debris. As they swell, iodine is released which kills bacteria. When the color of the gel changes it indicates that the dressing should be changed. It is structurally represented as shown figure 1 , and chemically is known as2-hydroxy methylene cross-linked (1-4) α-D-glucan wther containing iodine.

R=H, CH2COOH
Figure: ! Structural representation of cadexomer iodine
The method of preparation of cadexomer iodine and it applications in clinical use is described in the US patent 4,010,259(1977). The process basically consists of two steps. The step one involves preparation of water insoluble, gel forming, and water swell able organic hydrophilic carrier. The next stage involves complexation of iodine with the above organic polymeric carrier.
The carrier is prepared by a polymerization /cross-linking reaction of a polyhydroxylic organic substance by means of a bifunctional organic cross-linking agent of the type Y-R-Z, wherein Y and Z each represent epoxy groups or halogen atoms and R is an organic residue. In this polymerization/cross linking reaction each of the functional groups Y and Z react with a hydroxy group of the polyhydroxylic organic material to form ether bonds. The linking has to proceed to the extent that the formed polymer becomes insoluble in water, but is capable of absorbing water.
The polyhydroxylic starting material used is dextrin or carboxy methyl dextrin and the cross linking agent used for the polymerization reaction is a bifunctional glycerol derivative such as epichlorohydrin, which is capable of forming ether bridges. The reaction between polyhdyroxy starting material and cross-linking agent epichlorohydrin is carried out by emulsion/suspension of polymerization reaction. This type reaction requires specially designed reactors with efficient stirring and an agent to disperse/ stabilize the reaction mass.
The reaction conditions mentioned in the patent uses toluene/water emulsion system, and which is stabilized/dispersed using toluene solution of a mixture of mono and di-esters of ortho phosphoric acid. This process has the following disadvantages:
Disadvantages of the prior art process
1. During cross-linking, the reaction mixture gets dried-up and sticks to the reaction vessel.
2. Efficient stirring is not possible due to formation of lumps.
3. Particles size of the cross-linked carrier is not uniform.
4. Iodine incorporation to carrier is not efficient; hence large excess has to be used.
5. The color of the product obtained by this process is dark brown, whereas product is expected to be golden yellow in color.
6. Results are not reproducible and batch-to-batch variations observed.
7. The stabilizer solution referred in the patent (US 4,010,259) is a solution of a mixture of mono and di-esters of ortho phosphoric acid, which is not available commercially..
Essentially similar procedures are described in Fr, Demande 2,320,1 12 (1977),
Australian 506,419 (1980), Finn 59,014(1981 ), Dan Dk 150,781 ( 1989). However the chemical nature and details of composition of stabilizer solution are not disclosed in these patents also.
An improved method for the preparation of cadexomer iodine is now developed free of these problems and which can easily scaled up to manufacturing level.
ADVANTAGES OF PRESENT INVENTION
1. The particle size of cadexomer iodine by the present process is fine and uniform, which is highly suitable for powder and ointment formulations.
2. Iodine incorporation to the cross-linked dextrin is efficient and consistent and swelling is appropriate
3. The color of cadexomer iodine obtained is golden yellow which is consistent and as per the expected color of the product.
4. The process is simple and economical and can be carried out in regular reactor with out any extra investment on the specialized equipment
5. Present process uses the dispersing agents, which are available commercially.
The details of the invention are described in examples given below which are provided to illustrate the invention only and therefore should not be construed to limit the scope of the present invention.
Example 1
Commercial dextrin (5Og) is dissolved in sodium hydroxide (50ml of 3.1N) containing sodium borohydride (0.75g), to this dispersing agent; sorbitan monooleate (Span 80, 3.75g) dissolved in toluene (125ml) is added. Then of epichlorohydrin (10 g) is added and reaction mixture is heated at 700C for 5h. After completion of 5h, water (600ml) is added to the reaction mixture, and then neutralized to a pH of 6.5 with hydrochloric acid (2N). The product is filtered washed with acetone (500ml). The product is again washed with water (1000ml) and finally with acetone (300ml). The wet product is treated with a solution of iodine (7.8g) in acetone (196ml) and stirred at 250C for 20 hours, then at O0C for 2 hours. The product is filtered in a sintered funnel under nitrogen atmosphere, washed with chilled acetone (150ml) and dried at 250C for 24h in a vacuum
desiccator.
Yield: 33g
Iodine content: .0.91 % w/w
Swelling capacity: 5.0ml/g
Example 2
Commercial dextrin (1Og) is dissolved in sodium hydroxide (10ml of 3.1N) containing sodium borohydride (0.15g); to this dispersing agent; cetrimide (0.25g) dissolved in toluene (25ml) is added. Then of epichlorohydrin (2.Og) is added and reaction mixture is heated at 700C for 5h. After completion of 5h, water (150 ml) is added, and then the reaction mixture was neutralized to a pH of 6.5 with hydrochloric acid (2N). The separated product was filtered and washed with acetone (100ml). Again the product washed with water (200ml) and finally with acetone (60ml). The wet product (carrier) is treated with a solution of iodine ( 1 ,6g) in acetone (40 ml) and stirred at 250C for 20 hours, then at O0C for 2 hours. The product is filtered in a sintered funnel under nitrogen atmosphere, washed with chilled acetone (40ml) and dried at 250C for 24h in a vacuum desiccator.
Yield: 4.2g
Iodine content: 0.91% w/w
Swelling capacity: 6.0ml/g
Example 3
Commercial dextrin (1Og) is dissolved in sodium hydroxide (10ml of 3.1N) containing sodium borohydride (0.15g), to this dispersing agent; glyceryl monostearate (0.25g) dissolved in toluene (25ml) is added. Then of epichlorohydrin (2.Og) is added and reaction mixture is heated at 700C for 5h. After the completion of 5h, water (150 ml) is added, and then the reaction mixture is neutralized to a pH of 6.5 with hydrochloric acid (2N). The separated product is filtered and washed with acetone (100ml). Again the product is washed with water (200ml) and finally with acetone (60ml). The wet product (carrier) is treated with a solution of iodine (1.6g) in acetone (40 ml) and stirred at 250C for 20 hours, then at O0C for 2 hours. The product is filtered in a sintered funnel under nitrogen atmosphere, washed with chilled acetone (40ml) and dried at 250C for 24h in a vacuum desiccator.
Yield: 3.3g
Iodine content: 0.9% w/w
Swelling capacity: 6.2ml/g
Example 4
Commercial carboxymethyl dextrin (20g) was dissolved in sodium hydroxide (20ml of 3.1N) containing sodium borohydride (0.3g), to this dispersing agent; glyceryl monostearate (1.Og) dissolved in toluene (75ml) is added. Then of epichlorohydrin (6.Og) is added and reaction mixture is heated at 700C for 5h After completion of 5h, water (280 ml) is added, then the reaction mixture is neutralized to a pH of 6.5 with hydrochloric acid (2N). The separated product is filtered and washed with acetone (250ml). Again the product is washed with water (500ml) and finally with acetone (150ml). The wet product (carrier) is treated with a solution of iodine (3.Ig) in acetone (60 ml) and stirred at 250C for 20 hours, then at O0C for 2 hours. The product is filtered in a sintered funnel under nitrogen atmosphere, washed with chilled acetone (60ml) and dried at 250C for 24h in a vacuum desiccator.
Yield: 16gms
Iodine content: 0.92 % w/w.
Swelling capacity: 5.8 ml per gram.
References
- Jump up^ Merck Index, 14th Edition, p262 Merck & Co. Inc.
- Jump up^ Skog, E. et al. (1983). A randomized trial comparing cadexomer iodine and standard treatment in the out-patient management of chronic venous ulcers. British Journal of Dermatology 109, 77. PMID 6344906
- Jump up^ Ormiston, M.C., Seymour, M.T., Venn, G.E., Cohen, R.I. and Fox, J.A. (1985). Controlled trial of Iodosorb in chronic venous ulcers. British Medical Journal (Clinical Research Edition) 291, 308-310. PMID 3962169
- Jump up^ Drosou Anna, Falabella Anna, and Kirsner Robert S. (2003) Antiseptics on Wounds: An area of controversy. Wounds 159(5) 149-166. http://cme.medscape.com/viewarticle/456300_2Retrieved 02/03/2009
Tang, M.B.; Tan, E.S.
Hailey-Hailey disease: Effective treatment with topical cadexomer iodine
J Derm Treat 2011, 22(5): 304
Early diagnosis and early corticosteroid administration improves healing of peristomal pyoderma gangrenosum in inflammatory bowel disease
Dis Colon Rectum 2009, 52(2): 311
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATC code | |
| Identifiers | |
| CAS Number | |
| ChemSpider |
|
//////////////Cadexomer Iodine, ind 2018, Cadex, Declat, Decrat, Dextrinomer iodine, Iodoflex, Iodosorb, NI-009,
Clofarabine
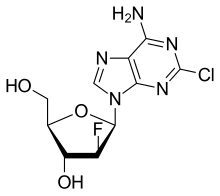
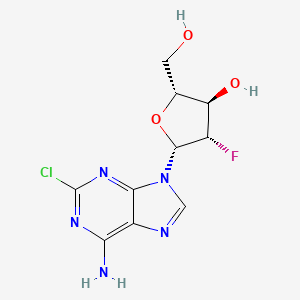
Clofarabine
- Molecular FormulaC10H11ClFN5O3
- Average mass303.677 Da

| CENTRAL DRUGS STANDARD CONTROL ORGANIZATION |
| DIRECTOR GENERAL OF HEALTH SERVICES, |
| MINISTRY OF HEALTH AND FAMILY WELFARE, |
| GOVERNMENT OF INDIA |
approved
|
Clofarabine Bulk & Injection 20 mg/20ml vial
|
For the treatment of patients 1 to 21 years old with relapsed or refractory acute lymphoblastic leukemia after at least two prior regimens. This indication is based upon response rate
|
16.01.2018 |
Clofarabine is a purine nucleoside antimetabolite marketed in the US and Canada as Clolar. In Europe and Australia/New Zealand the product is marketed under the name Evoltra. It is FDA-approved for treating relapsed or refractory acute lymphoblastic leukaemia(ALL) in children after at least two other types of treatment have failed. It is not known if it extends life expectancy. Some investigations of effectiveness in cases of acute myeloid leukaemia (AML) and juvenile myelomonocytic leukaemia (JMML) have been carried out. Ongoing trials are assessing its efficacy, if any, for managing other cancers.
Clofarabine is a purine nucleoside antimetabolite that is being studied in the treatment of cancer. It is marketed in the U.S. and Canada as Clolar. In Europe and Australia/New Zealand the product is marketed under the name Evoltra.
Clofarabine is used in paediatrics to treat a type of leukaemia called relapsed or refractory acute lymphoblastic leukaemia (ALL), only after at least two other types of treatment have failed. It is not known if the drug extends life expectancy. Some investigations of effectiveness in cases of acute myeloid leukaemia (AML) and juvenile myelomonocytic leukaemia (JMML) have been carried out.
For the treatment of pediatric patients 1 to 21 years old with relapsed or refractory acute lymphocytic (lymphoblastic) leukemia after at least two prior regimens. It is designated as an orphan drug by the FDA for this use.
Approval
Clolar was Food and Drug Administration (FDA) approved 28 December 2004. (Under accelerated approval regulations requiring further clinical studies.)

Side effects
- Tumor lysis syndrome (TLS). Clofarabine quickly kills leukaemia cells in the blood. The body may react to this. Signs include hyperkalemia, hyperuricemia, and hyperphosphatemia. TLS is very serious and can lead to death if it is not treated right away.
- Systemic inflammatory response syndrome (SIRS): symptoms include fast breathing, fast heartbeat, low blood pressure, and fluid in the lungs.
- Bone marrow problems (suppression). Clofarabine can stop the bone marrow from making enough red blood cells, white blood cells, and platelets. Serious side effects that can happen because of bone marrow suppression include severe infection (sepsis), bleeding, and anemia.
- Effects on pregnancy and breastfeeding. Girls and women should not become pregnant or breastfeed during treatment which may harm the baby.
- Dehydration and low blood pressure. Clofarabine can cause vomiting and diarrhea which may lead to low body fluid (dehydration). Signs and symptoms of dehydration include dizziness, lightheadedness, fainting spells, or decreased urination.
- Other side effects. The most common side effects are stomach problems (including vomiting, diarrhea, and nausea), and effects on blood cells (including low red blood cells count, low white blood cell count, low platelet count, fever, and infection. Clofarabine can also cause tachycardia and can affect the liver and kidneys.
Contraindications
- pregnancy or planned pregnancy
- breast-feeding
- liver problems
- kidney problems
Drug interactions
- nephrotoxic drugs
- hepatotoxic drugs
Delivery
- By intravenous infusion.
- Dosage is a 2-hour infusion (52 mg/m²) every day for five days. The cycle is repeated every 2 to 6 weeks.
- Regular blood tests to monitor his or her blood cells, kidney function, and liver function.
Biology
Clofarabine is a second-generation purine nucleoside analog designed to overcome biological limitations observed with ara-A and fludarabine. A 2´(S)-fluorine in clofarabine significantly increased the stability of the glycosidic bond in acidic solution and toward phosphorolytic cleavage as compared to fludarabine.[1] A chlorine substitution at the 2-position of the adenine base avoids production of a 2-fluoroadenine analog, a precursor to the toxic 2-fluoro-adenosine-5´-triphosphate and prevents deamination of the base as compared to ara-A.[2]
Clofarabine can be administered intravenously or given orally. Clofarabine enters cells via hENT1, hENT2, and hCNT2, where upon it is phosphorylated by deoxycytidine kinase to generate clofarabine-5´-monophosphate. The rate-limiting step in clofarabine metabolism is clofarabine-5´-diphosphosphate. Clofarabine-5´-triphosphate is the active-metabolite, and it inhibits ribonucleotide reductase, resulting in a decrease cellular dNTP concentrations, which promotes greater incorporation of clofarabine-5´-triphosphate during DNA synthesis. Embedded clofarabine-5´-monophosphate in the DNA promotes polymerase arrest at the replication fork, triggering DNA repair mechanisms that without repair lead to DNA strand breaks in vitro and cytochrome c-mediated apoptosis in vitro. Studies using cell lines have shown that clofarabine-5´-triphosphate can also be incorporated into RNA.[3]
Mechanisms of resistance and turnover have been reported. Clofarabine-resistance arises from decreased deoxycytidine kinase activity in vitro.[4] ABC transporter ABCG2 promotes export of clofarabine-5´-monophosphate and thus limits the cytotoxic effects of this analog in vivo.[5] Biochemically, clofarabine-5’-triphosphate was shown to be substrate for SAMHD1, thus potentially limiting the amount of active compound in cells.[6]

Synthesis
Production of Clofarabine
The reaction flask was added 2-chloro-9-(2-deoxy-2-fluoro-3,5-di-O-benzoyl-beta-D arabinose yl) adenine 1.5g (3mmol) and methanol 40ml,mixed with stirring. Then it was added sodium methoxide, 0.05g (content> 50%), the reaction was stirred for 40min. Then the mixture was cooled to room temperature, adjusted to pH 7 with acetic acid, filtered, and the filter cake was washed with an ice-methanol 10ml, added to the methanol 40ml, and heated to 63 °C, and then cooled to -10 o C. Still 1h, filtered, and the filter cake was washed with an ice-methanol 10ml, drained, dried under reduced pressure to give an off-white powdery solid clofarabine 0.48g. The yield is 54%.

CLIP

http://pubs.rsc.org/en/content/articlehtml/2017/ra/c6ra27790j
CLIP

SYN 1
JP 1993502014; US 5034518; WO 9014352

Reaction of 1,2:5,6-di-O-isopropylidene-3-O-tosyl-a-D-allofuranose (I) with KF in acetamide at 210 oC gives 3-deoxy-3-fluoro-1,2:5,6-di-O-isopropylidene-a-D-glucofuranose (II), which is treated with a 1:1 mixture of metha-nol and 0.7% aqueous H2SO4 to yield 3-deoxy-3-fluoro-1,2-isopropylidene-a-D-glucofuranose (III). Selective acylation of the sugar (III) with benzoyl chloride in pyridine affords the 6-O-benzoyl derivative (IV), which is treated with Amberlite IR-100 (H+) ion-exchange resin in hot dioxane to provide 6-O-benzoyl-3-deoxy-3-fluoro-D-glucofuranose (V). The oxidative cleavage of glucofuranose (V) by means of KIO4 in water results in rearrangement to give 5-O-benzoyl-2-deoxy-2-fluoro-3-O-formyl-D- arabinofuranose (VI), which is deformylated by means of NaOMe in methanol to provide 5-O-benzoyl-2-deoxy-2-fluoro-D-arabinofuranose (VII). Acylation of the arabinofuranose (VII) with acetic anhydride in pyridine affords the 1,3-di-O-acetyl derivative (VIII), which is treated with HBr in AcOH/CH2Cl2 to yield 3-O-acetyl-5-O-benzoyl-2-deoxy-2-fluoro-D-arabinofuranosyl bromide (IX). Condensation of compound (IX) with 2-chloroadenine (X) by means of potassium tert-butoxide in different solvents gives the acylated 2-chloroadenosine derivative (XI), which is finally deacylated by means of NaOMe in methanol
Carbohydr Res 1975,42(2),233
Drugs Fut 2004,29(2),112
| J Med Chem 1992,35(2),397 |
US 2003114663; WO 0311877
CA 2400470; EP 1261350; WO 0160383
References
- Jump up^ Parker WB, Allan PW, Hassan AE, Secrist JA 3rd, Sorscher EJ, Waud WR (Jan 2003). “Antitumor activity of 2-fluoror-2’deoxyadenosine against tumors that express Escherichia coli purine nucleoside phosphorylase”. Cancer Gene Ther. 10 (1): 23–29. doi:10.1038/sj.cgt.7700520. PMID 12489025.
- Jump up^ Bonate PL, Arthaud L, Cantrell WR Jr, Stephenson K, Secrist JA 3rd, Weitman S (Feb 2014). “Discovery and development of clofarabine: a nucleoside analogue for treating cancer”. nat Rev Drug Discov. 5 (10): 855–63. doi:10.1038/nrd2055. PMID 17016426.
- Jump up^ Shelton J, Lu X, Hollenbaugh JA, Cho JH, Amblard F, Schinazi RF (Dec 2016). “Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs”. Chem Rev. 116 (23): 14379–14455. doi:10.1021/acs.chemrev.6b00209. PMID 27960273.
- Jump up^ Lotfi K, Månsson E, Spasokoukotskaja T, Pettersson B, Liliemark J, Peterson C, Eriksson S, Albertioni F (1999). “Biochemical pharmacology and resistance to 2-chloro-2′-arabino-fluoro-2’deoxyadenosine, a novel analogue of cladribine in human leukemic cells”. Clin Cancer Res. 5 (9): 2438–44. PMID 10499616.
- Jump up^ Nagai S, Takenaka K, Nachagari D, Rose C, Domoney K, Sun D, Sparreboom A, Schuetz JD (Mar 2011). “Deoxycytidine kinase modulates the impact of the ABC transporter ABCG2 on clofarabine cytotoxicity”. Cancer Res. 75 (1): 1781–91. doi:10.1158/0008-5472.CAN-10-1919. PMC 3531552 . PMID 21245102.
- Jump up^ Arnold LH, Kunzelmann S, Webb MR, Taylor IA (Jan 2015). “A continuous enzyme-coupled assay for triphosphohydrolase activity of HIV-1 restriction factor SAMHD1”. Antimicrob Agents Chemother. 59 (1): 186–92. doi:10.1128/AAC.03903-14. PMC 4291348 . PMID 25331707.
External links
 |
|
| Clinical data | |
|---|---|
| Trade names | Clolar, Evoltra |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a607012 |
| License data | |
| Routes of administration |
Intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.159.663 |
| Chemical and physical data | |
| Formula | C10H11ClFN5O3 |
| Molar mass | 303.677 g/mol |
| 3D model (JSmol) | |
//////////////////ind 2018, Clofarabine, Nucleotides
C1=NC2=C(N1C3C(C(C(O3)CO)O)F)N=C(N=C2N)Cl
FDA approves first therapy Crysvita (burosumab) for rare inherited form of rickets, x-linked hypophosphatemia

FDA approves first therapy for rare inherited form of rickets, x-linked hypophosphatemia
The U.S. Food and Drug Administration today approved Crysvita (burosumab), the first drug approved to treat adults and children ages 1 year and older with x-linked hypophosphatemia (XLH), a rare, inherited form of rickets. XLH causes low levels of phosphorus in the blood. It leads to impaired bone growth and development in children and adolescents and problems with bone mineralization throughout a patient’s life.
April 17, 2018
Release
The U.S. Food and Drug Administration today approved Crysvita (burosumab), the first drug approved to treat adults and children ages 1 year and older with x-linked hypophosphatemia (XLH), a rare, inherited form of rickets. XLH causes low levels of phosphorus in the blood. It leads to impaired bone growth and development in children and adolescents and problems with bone mineralization throughout a patient’s life.
“XLH differs from other forms of rickets in that vitamin D therapy is not effective,” stated Julie Beitz, M.D., director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “This is the first FDA-approved medication for the treatment of XLH and a real breakthrough for those living with this serious disease.”
XLH is a serious disease affecting approximately 3,000 children and 12,000 adults in the United States. Most children with XLH experience bowed or bent legs, short stature, bone pain and severe dental pain. Some adults with XLH experience persistent discomfort or complications, such as joint pain, impaired mobility, tooth abscesses and hearing loss.
The safety and efficacy of Crysvita were studied in four clinical trials. In the placebo-controlled trial, 94 percent of adults receiving Crysvita once a month achieved normal phosphorus levels compared to 8 percent of those receiving placebo. In children, 94 to 100 percent of patients treated with Crysvita every two weeks achieved normal phosphorus levels. In both children and adults, X-ray findings associated with XLH improved with Crysvita therapy. Comparison of the results to a natural history cohort also provided support for the effectiveness of Crysvita.
The most common adverse reactions in adults taking Crysvita were back pain, headache, restless leg syndrome, decreased vitamin D, dizziness and constipation. The most common adverse reactions in children were headache, injection site reaction, vomiting, decreased vitamin D and pyrexia (fever).
Crysvita was granted Breakthrough Therapy designation, under which the FDA provides intensive guidance to the company on efficient drug development, and expedites its review of drugs that are intended to treat serious conditions where clinical evidence shows the drug may represent a substantial improvement over other available therapies. Crysvita also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The sponsor is receiving a Rare Pediatric Disease Priority Review Voucher under a program intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. A voucher can be redeemed at a later date to receive Priority Review of a subsequent marketing application for a different product. This is the 14th Rare Pediatric Disease Priority Review Voucher issued by the FDA since the program began.
The FDA granted approval of Crysvita to Ultragenyx Pharmaceutical Inc.
////////////fda 2018, Crysvita, burosumab, Breakthrough Therapy, priority review. Ultragenyx Pharmaceutical Inc
WO 2018066004, NEW PATENT, INDOCO REMEDIES LIMITED, DORZOLAMIDE
![]()
(WO2018066004) PROCESS FOR THE PREPARATION OF DORAOLZMIDE HYDROCHLORIDE
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018066004&redirectedID=true
| Applicants: | INDOCO REMEDIES LIMITED [IN/IN]; Indoco House, 166 C.S.T. Road, Santacruz (East) Mumbai, Maharashtra 400098 (IN) |
| Inventors: | SHETH, Nilima; (IN). KUDUVA, Srinivasan Subramanian; (IN). RAMESAN, Palangat Vayalileveetil; (IN). PANANDIKAR, Aditi Milind; (IN) |

SHETH, Nilima
Aditi Kare Panandikar, Managing Director, Indoco Remedies
Process for preparing dorzolamide hydrochloride is claimed. It is disclosed that dorzolamide hydrochloride is a carbonic anhydrase inhibitor.
Trusopt is an ophthalmic solution containing the carbonic anhydrase inhibitor dorzolamide hydrochloride for treating intraocular pressure in patients with ocular hypertension or open-angle glaucoma, which was developed and launched by Merck & Co , and is also now marketed by Santen Pharmaceuticals and Mundipharma International .
In April 2018, Newport Premium™ reported that Indoco Remedies was capable of producing commercial quantities of dorzolamide hydrochloride and holds an active US DMF since 2010
Dorzolamide hydrochloride is a carbonic anhydrase (CA) inhibitor. It is chemically represented by (4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride; and is structurally represented

Formula I
It acts as an anti -glaucoma agent, in open-angle glaucoma and ocular hypertension. It is used in ophthalmic solutions to lower intraocular pressure (IOP).
The compound dorzolamide hydrochloride has been in the market for very long time. It is administered as a topical ophthalmic in the form of a solution and marketed under the brand name T rusopt.
Dorzolamide hydrochloride and process for its preparation are first described in the patent, US 4,797,413 (US “413 Patent) and its corresponding European patent, E P 0296879. The process described in US “413 patent involves reacting thiophene-2-thiol with but-2-enoic acid and further proceeds with formation of racemic 4- ( ethyl ami no) – 6- methyl -5, 6- di hydro-4H -thi eno[2, 3- b] thi opy ran-2-sul f onami de 7, 7-di oxide (dorzolamide base).
A number of further processes for the preparation of dorzolamide hydrochloride have been devised and in many of these, as well as in the above US “413 Patent, the last step of the process involves the removal of diastereomeric impurity from the racemic mixture of dorzolamide base. To obtain pure dorzolamide hydrochloride devoid of the diastereomeric impurity of cis-isomer from the racemic compound, as per the patent US ~413, racemic mixture of dorzolamide base is subjected to column chromatography and then resolution is carried out using resolving agent di-p-tol uoyl-L -tartaric acid monohydrate in n-propanol. The salt formed is treated with base to get dorzolamide free base, which is reacted with ethanolic hydrochloric acid to get dorzolamide hydrochloride. The compound is further recrystallised from mixture of solvents viz., methanol and isopropanol to get pure dorzolamide hydrochloride.
US 5,688,968 describes a process for preparation of dorzolamide hydrochloride, wherein chiral hydroxyl sulfone compound having fixed chirality, proceeds via Ritter reaction to obtain dorzolamide base having mixture of cis- and trans-isomer. The compound dorzolamide base is reacted with maleic acid to isolate maleate salt of dorzolamide. The salt is again converted to free base and then reacted with hydrochloric acid in ethyl acetate to get required pure trans-isomer of dorzolamide hydrochloride.
The PCT patent publication W 02006038222 discloses the preparation of dorzolamide hydrochloride, wherein the cis- and trans-isomer of racemic dorzolamide base is separated using resolution via chiral salt formation with di benzoyl -L -tartaric acid monohydrate or di-p-tol uoyl-L -tartaric acid monohydrate in methanol which on neutralization results in dorzolamide base. The base is then reacted with hydrochloric acid in isopropanol to give
dorzol amide hydrochloride which is recrystalised in isopropanol to obtain pure dorzol amide hydrochloride.
Another US patent US 7,109,353 discloses the process for preparation of dorzolamide hydrochloride, wherein racemic 4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide is treated with mineral acid to form the corresponding salt, which is then converted to racemic trans-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide and resolved with di-p-toluoyl-D -tartaric acid followed by neutralization of the chiral salt to isolate trans-(4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide. The compound trans-(4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide on reaction with hydrochloric acid in ethanol results in required trans-dorzolamide hydrochloride.
PCT patent publication WO2007122130 discloses the process for preparation of (4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide, wherein racemic 4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide having trans:cis diastereomeric mixture of 80:20 is treated with maleic acid in acetone to isolate racemic trans-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide maleate salt having trans:cis diastereomeric mixture of 95:5. The isolated maleate salt is then treated with base and reacted with (1 S)-(+)-10-camphorsulfonic acid to get corresponding (4S,6S)-4-(ethylamino)-6-methyl-5, 6-di hydro-4H -thi eno[2, 3- b] thi opy ran-2-sul f onami de 7, 7-di ox i de ( 1 S ) -( + )- 10-camphorsulfonate salt, which is neutralized to give pure (4S,6S)-4-(ethylamino)-6- methyl -5, 6-di hydro-4H -thi eno[2,3- b] thi opyran-2-sulf onami de 7, 7-di oxi de.
PCT patent publication W 02008135770 discloses the process for the preparation of dorzolamide hydrochloride, wherein the racemic 4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide is treated with
carboxylic acid selected from the group consisting of fumaric acid, benzoic acid, acetic acid, salicylic acid and p-hydroxybenzoic acid, which selectively forms an acid addition salt with the trans- isomer and removes the undesirable c is- isomer from the mixture of cis and trans- isomers. The trans-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide acid addition salt is converted to trans-(e)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide by conventional methods. The compound trans-(e)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide is resolved with di-p-toluoyl-L -tartaric acid followed by neutralization of the chiral salt yields the compound (4S,6S)4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide, which on reaction with hydrochloric acid in isopropanol results in the required (4S,6S)4-( ethyl ami no) – 6- methyl -5, 6- di hydro-4H -thi eno[2, 3- b] thi opy ran-2-sul f onami de 7, 7-di oxide hydrochloride.
PCT patent publication WO2010061398 discloses the process for the preparation of dorzolamide hydrochloride, wherein the racemic 4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide is treated with maleic acid in water to get trans-dorzolamide maleate salt. The maleate salt is further neutralized and then resolution with di-p-toluoyl-L -tartaric acid followed by neutralization of the chiral salt yields (4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide. The compound (4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide on reaction with hydrochloric acid in isopropanol results in the required pure trans-dorzolamide hydrochloride.
PCT patent publication WO2011101704 and corresponding Indian Patent application 426/C H E/2010 describes the process for the preparation of trans-dorzolamide hydrochloride by forming the maleate salt of racemic 4-( ethyl ami no) – 6- methyl -5, 6- di hydro-4H -thi eno[2, 3- b] thi opy ran-2-sul f onami de 7, 7-di oxide. The maleate salt is further neutralized and then resolution with di-p-
toluoyl-L -tartaric acid followed by neutralization of the chiral salt yields trans-(4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide. The compound trans-(4S,6S)-4-(ethylamino)-6-methyl- 5.6- dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide on reaction with hydrochloric acid in isopropanol results in the required trans-(S,S)-dorzol amide hydrochloride.
Indian Patent application 3431 /M U M/2012 discloses the process wherein racemic 4-( ethyl ami no) -6- methyl – 5, 6- di hydro-4H -thi eno[2, 3- b] thi opy ran-2-sul f onami de
7.7- dioxide is resolved using di benzoyl- L -tartaric acid monohydrate or di-p-toluoyl-L -tartaric acid monohydrate in methanol followed by neutralization of the chiral salt and then the (4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide thus obtained is treated with hydrochloric acid in isopropanol to result in the required trans-(S,S)-dorzolamide hydrochloride. The compound is further recrystallised in isopropanol to isolate pure dorzol amide hydrochloride.
The prior art processes disclosed as above have several drawbacks in the preparation of pure trans-dorzolamide hydrochloride viz.,
1. involves column chromatography for separation of the desired diastereomer;
2. involves preparation of corresponding diastereomeric salts and converting again to base before preparation and isolation of pure trans-dorzolamide hydrochloride;
3. involves additional step of reacting the racemic dorzolamide base with mineral acid to isolate corresponding dorzolamide salt which is again converted to dorzolamide base and further resolved using resolving agent to form the corresponding salt, neutralization and isolation of the chiral dorzolamide base before reacting with hydrochloric acid to obtain dorzolamide hydrochloride; and
4. involves an additional step of reacting the racemic dorzolamide base with carboxylic acid to isolate corresponding dorzolamide salt which is again converted to dorzolamide base and resolved using resolving agent to form the corresponding salt, neutralization and isolation of the chiral dorzolamide base before reacting with hydrochloric acid to obtain dorzolamide hydrochloride.
As is evident from the cursory review of the prior arts that the preparation of pure dorzolamide hydrochloride involves either column chromatography for isolation of trans- isomer followed by use of resolving agent or involves repeated preparations of chiral or diastereomeric salts, use of resolving agent followed by converting into dorzolamide base and then isolating pure dorzolamide hydrochloride devoid of the diastereomeric impurity of cis-isomer.
Therefore, there remains a need in the art to develop a simple and cost effective process for the preparation of dorzolamide hydrochloride which ameliorates the above drawbacks of the prior arts and makes the process industrially viable and economically advantageous. The present invention therefore seeks to address these issues by providing an improved and cost-effective process that can easily be scaled for industrial production of dorzolamide hydrochloride.
The present inventors have developed an alternative process for isolating pure dorzolamide hydrochloride substantially free from the cis-isomer without using the time consuming column chromatography technique, repeated preparation of chiral salts, diastereomeric salts and converting into base before hydrochloride salt formation to isolate pure trans-(S,S)-dorzolamide hydrochloride.
E xamples:
E xample 1 : Preparation of (6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H -thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride:
In a dry flask charged (6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide (50.0 gm) in acetone (700 ml) under stirring and cooled to OeC. Maintaining the temperature at OeC to 5eC purged hydrochloric acid gas to adjust the pH to acidic between the range of 1-2. After attaining desired pH, maintained the reaction mass for two hours at OeC to 5eC under stirring. Filtered the precipitated compound (6S)-4-(ethylamino)-6-methyl-5, 6-di hydro-4H -thi eno[2,3- b] thi opyran-2-sulf onami de 7,7-di oxi de hydrochl ori de and washed the solid mass with chilled acetone (50 ml). Dried the compound at 60-65eC till constant weight.
Dry weight: 50 g
H PL C purity: 77.62% [cis-isomer: 22.11 % ]
E xample 2: Preparation of trans-(4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H -thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride [C rude dorzolamide hydrochloride]:
In a dry flask charged (6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride (19.0 g) and methanol (190 ml) at temperature of 25eC to 30eC. Under stirring raised the temperature of the reaction mass to reflux and maintained at reflux temperature for a period of two hours. After maintaining cooled the reaction mass gradually to 10eC to 15eC. Filtered the compound trans-(4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride and washed the solid mass with chilled methanol. Dried the compound at 60-65eC till constant weight.
Dry weight: 12.8 g
H PL C purity: 99.33% [cis-isomer: 0.5% ]
E xample 3: Purification of trans-(4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H -thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride: In a dry flask charged trans-(4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride (11.0 g), acetone (11 ml) and purified water (5.5 ml) at the temperature of 25eC to 30eC. Raised the temperature of the reaction slurry to reflux and maintained at reflux for one hour. Diluted the reaction mass with fresh acetone (44 ml) maintaining the temperature at reflux and continued maintaining at reflux temperature further for one hour. Cooled the reaction mass gradually to 10eC to 15eC and maintained. Filtered the compound pure (4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride solid mass and washed the pure compound with chilled acetone (11 ml). Dried at 55eC to 60eC till constant weight.
Dry weight: 8.8 g
H PL C purity: 99.89% [cis-isomer: not detected]
[T otal impurities: 0.11 % ]
E xample 4: Preparation of trans-(4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H -thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride [C rude dorzolamide hydrochloride]:
Charged (6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride (5.0 g), methanol (22.5 ml) and 2.5 ml purified water at temperature of 25eC to 30eC. Under stirring raised the temperature of the reaction mass to reflux and maintained at reflux temperature for a period of two hours. After maintaining cooled the reaction mass gradually to 30eC to 35eC. Filtered the solid compound trans-(4S,6S)-4-(ethylamino)-6-methyl-5,6-dihydro-4H-thieno[2,3-b]thiopyran-2-sulfonamide 7,7-dioxide hydrochloride and washed with methanol (10 ml). Dried the compound at 60eC to 65eC till constant weight.
Dry weight: 3.2 g
H PL C purity: 99.47% [cis-isomer: 0.43% ]
////////WO 2018066004, NEW PATENT, INDOCO REMEDIES LIMITED, DORZOLAMIDE
WO 2018067805, NEW PATENT, SOTAGLIFLOZIN, TEVA
![]()
WO-2018067805
(WO2018067805) SOLID STATE FORMS OF SOTAGLIFLOZIN
TEVA PHARMACEUTICAL INDUSTRIES LTD.
GIAFFREDA, Stefano Luca; (IT).
MODENA, Enrico; (IT).
IANNI, Cristina; (IT).
MUTHUSAMY, Anantha Rajmohan; (IN).
KANNIAH, Sundara Lakshmi; (IN)


 Sundara Lakshmi Kanniah
Sundara Lakshmi KanniahSotagliflozin has the chemical name (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(methylthio)tetrahydro-2H- pyran-3,4,5-triol. Sotagliflozin has the following chemical structure:

[0003] Sotagliflozin is an orally available L-xyloside based molecule that apparently inhibits both sodium-glucose transporter type 1 (SGLT1) and type 2 (SGLT2). SGLT1 is primarily responsible for glucose and galactose absorption in the gastrointestinal tract, and SGLT2 is responsible for most of the glucose reabsorption performed by the kidney.
[0004] Sotagliflozin is known from WO 2008/109591. Amorphous forms and crystalline forms (i.e. Form 1 and Form 2) of Sotagliflozin are disclosed in WO2010/009197.
[0005] Polymorphism, the occurrence of different crystal forms, is a property of some molecules and molecular complexes. A single compound, like Sotagliflozin, may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, thermal behaviors (e.g. measured by thermogravimetric analysis – “TGA”, or differential scanning calorimetry – “DSC”), powder X-ray diffraction (PXRD) pattern, infrared absorption fingerprint, Raman absorption fingerprint, and solid state (13C-) NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
[0006] Different salts and solid state forms (including solvated forms) of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms and solvates may provide a basis for improving
formulation, for example, by facilitating better processing or handling characteristics, improving the dissolution profile, or improving stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also provide improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts and solid state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to use variations in the properties and characteristics of a solid active pharmaceutical ingredient for providing an improved product.
[0007] Discovering new salts, solid state forms and solvates of a pharmaceutical product can provide materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other salts or polymorphic forms. New salts, polymorphic forms and solvates of a pharmaceutically useful compound can also provide an opportunity to improve the performance characteristics of a pharmaceutical product (dissolution profile, bioavailability, etc.). It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, e.g., a different crystal habit, higher crystallinity or polymorphic stability which may offer better processing or handling characteristics, improved dissolution profile, or improved shelf-life. For at least these reasons, there is a need for additional salts and solid state forms (including solvated forms) of Sotagliflozin.
EXAMPLES
Sotagliflozin Form-2 may be prepared according to WO2010/009197. Sotaglifiozin Form-2 may also be prepared according to Example 16 below.
Working examples:
Example- 1 : Preparation of Sotagliflozin (Amorphous Form)
[0080] 2 g of Sotagliflozin (Form-2) was taken in 250ml round bottom flask and applied vacuum (approx. 50 mbar) with continuous rotation of the flask. The flask was externally heated by hot air flow maintained at few centimetres from the rotating flask wall for few minutes until the compound melts at around 130°C and the melt was quenched to room temperature (25°C) with water bath. The amorphous solid (1.8 g) was scratched from the walls of the flask.
Example-2: Preparation of Sotagliflozin Form A
[0081] 100 mg of Sotagliflozin (Amorphous form, prepared according to example 1) was suspended in 2 ml of water, was left at variable temperature as follows: heating from 10°C to 50°C at the rate of 20°C/hr, held at 50°C for 3 hrs; cooling from 50°C to 10°C at the rate of 20°C/hr, held at 10°C for 3hrs; again heating from 10°C to 50°C at the rate of 10°C/hr, held at 50°C for 3hrs; again cooling from 50°C to 10°C at the rate of 10°C/hr, held at 10°Cfor 3hrs; further heating from 10°C to 50°C at the rate of 5°C/hr, held at 50°C for 3hrs; further cooling from 50°C to 10°C at the rate of 5°C/hr, held at 10°C for 3hrs; followed by raising the temperature from 10°C to 25°C at the rate of 10°C/hr, held at 25°C for 24hrs. The suspension was filtered under vacuum and was dried at room temperature by vacuum suction. Sotagliflozin Form A has been confirmed by PXRD as presented in figure 1.
Example-3 : Preparation of Sotagliflozin Form B
[0082] 100 mg of Sotagliflozin (Amorphous form, prepared according to example 1) was suspended in 2 ml of Toluene at room temperature (20-25 °C). The suspension was stirred for 15days which was filtered under vacuum and was dried at room temperature by vacuum suction. Sotagliflozin Form B has been confirmed by PXRD as presented in figure 2.
Example-4: Preparation of Sotagliflozin Form B
[0083] 100 mg of Sotagliflozin (Amorphous form, prepared according to example 1) was suspended in 2 ml of Heptane at room temperature (20-25 °C). The suspension was stirred for 15days which was filtered under vacuum and was dried at room temperature by vacuum suction. Sotagliflozin Form B has been confirmed by PXRD.
Example-5 : Preparation of Sotagliflozin Form B
[0084] 100 mg of Sotagliflozin (Amorphous form, prepared according to example 1) was suspended in 2 ml of Mesitylene at room temperature (20-25°C). The suspension was stirred for 15days which was filtered under vacuum and was dried at room temperature by vacuum suction. Sotagliflozin Form B has been confirmed by PXRD.
Example-6: Preparation of Sotagliflozin Form B
[0085] 100 mg of Sotagliflozin (Amorphous form, prepared according to example 1) was suspended in 2 ml of p-Xylene at room temperature (20-25 °C). The suspension was stirred for 15days which was filtered under vacuum and was dried at room temperature by vacuum suction. Sotagliflozin Form B has been confirmed by PXRD.
Example-7: Preparation of Sotagliflozin Form C
[0086] 100 mg of Sotagliflozin (Amorphous form, prepared according to example 1) was suspended in 2 ml of Water at 50°C. The suspension was stirred for 72hrs which was filtered under vacuum and was dried at room temperature by vacuum suction. Sotagliflozin Form C has been confirmed by PXRD as presented in figure 3.
Example-8: Preparation of Sotagliflozin Form D
[0087] 30 mg of Sotagliflozin (Form-2) was dissolved in 3ml of ethanol. The solution was stirred at 25°C for lhr (for dissolution) and then filtered. The solution was kept in a 20 ml vial and left open to allow evaporation of the solvent (25°C/1 atm). Solid was observed after 3 days; it was collected and analyzed by PXRD. Sotagliflozin Form D has been confirmed by PXRD as presented in figure 4.
Example-9: Preparation of Sotagliflozin Form E
[0088] 30 mg of Sotagliflozin (Form-2) was dissolved in 3ml of isopropyl acetate. The solution was stirred at 25°C for lhr (for dissolution) and then filtered. The solution was kept in a 20ml vial and left opened in the refrigerator (4-7°C/l atm) to allow evaporation of the solvent. The crystals were observed after 9 days; it was collected and analyzed by PXRD. Sotagliflozin Form E has been confirmed by PXRD as presented in figure 5.
Example-9: Preparation of Sotagliflozin Form F
[0089] 30 mg of Sotagliflozin (Form-2) was dissolved in 3ml of 2-propanol. The solution was stirred at 25°C for lhr (for dissolution) and then filtered. The solution was kept in a 20ml vial and left opened to allow evaporation of the solvent (4-7°C/l atm). The crystals were observed after 13 days; it was collected and analyzed by PXRD. Sotagliflozin Form F has been confirmed by PXRD as presented in figure 6.
Example- 10: Preparation of Sotagliflozin Form G
[0090] 30 mg of Sotagliflozin (Form-2) was dissolved in 3ml of 1 -propanol. The solution was stirred at 25°C for lhr (for dissolution) and then filtered. The solution was kept in a 20ml vial and left opened in the refrigerator (4-7°C/l atm) to allow evaporation of the solvent. Solid was observed after 24 days; it was collected and analyzed by PXRD.
Sotagliflozin Form G has been confirmed by PXRD as presented in figure 7.
Example- 11 : Preparation of Sotagliflozin Form H
[0091] 15 mg of Sotagliflozin (Form- A) was kept in DVS (dynamic vapor sorption) instrument. The kinetic sorption measurement was performed at 25 °C in two full cycle of sorption and desorption as follows, from 40%RH to 90%RH, 90%RH to 0%RH then again from 0% to 90%RH, 90%RH to 0%RH. After completion of experiment, the powder was collected and analyzed by PXRD. Sotagliflozin Form H has been confirmed by PXRD as presented in figure 8.
Example- 12: Preparation of Sotagliflozin Form I
[0092] Procedure to prepare saturated solution: 1500 mg of Sotagliflozin were dissolved in 1ml of 2-Methoxyethanol and the solution was stirred overnight at 25°C; the solution was then filtered. Taken ΙΟΟμί from above saturated stock solution, 500μί of Diisopropylether was added drop by drop, no solid was observed left the solution overnight under stirring, added again 500μί of Diisopropylether into the entire solution. The solid was precipitated, stirred for 30min and filtered under vacuum. Sotagliflozin Form I has been confirmed by PXRD as presented in figure 9.
Example-13: Preparation of Sotagliflozin Form K
[0093] 10-20mg of Sotagliflozin (Form D) was kept for drying in a natural air convection oven (MPM instruments modelM40-VN) at 60°C for lh. Sotagliflozin Form K has been confirmed by PXRD as presented in figure 10.
Example-14: Preparation of Sotagliflozin Form E:
[0094] Sotagliflozin (2g) and ethyl acetate (6ml) were heated to reflux temperature (71-74°C). Heptane (6ml) was added at reflux, reaction mass was stirred for additional 15 minutes and then cooled to room temperature. Solid was precipitated out during cooling at about 57°C. A mixture of ethyl acetate and heptane (1 : 1 v/v, 24 ml) was added and the reaction mixture was heated to reflux temperature (71-74°C) to obtain a clear solution which was maintained for 15 minutes. Reaction mass was cooled to room temperature (25-30°C) and stirred for 3 hours. The slurry was filtered, washed with a mixture of ethyl acetate and heptane (l : lv/v, 8ml) and dried under vacuum at 50°C for 2Hrs. The obtained solid (1.8g) was analyzed by PXRD-Form E.
Example-15: Preparation of Sotagliflozin Form D:
[0095] 2g of sotagliflozin Form F was kept in glass petri-dish and exposed to 80%RH for 60hrs at room temperature. Solid was collected (2g) and analyzed for PXRD-Form D.
Example-16: Preparation of Sotagliflozin Form 2:
[0096] 50 gm of (2S,3S,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(Methylthio) tetrahydro-2H-pyran-3,4,5-triyl triacetate (SOT-1) and 500ml of methanol were charged in round bottom flask, the slurry was cooled to 20°C then added sodium methoxide solution prepared in methanol (2.45gm of sodium methoxide in 50ml of methanol) at 20°C
over the period of 10 min and stirred the mass for 2hr at 20°C.The reaction completion was ensured by HPLC. Once the reaction is completed added 2.5gm Norit carbon to the reaction mass at 23°C and stirred for 30min. Filtered the reaction mass through Hyflo bed and washed with 20ml of methanol. Taken the filtrate into the flask and concentrated under vacuum at 45°C up to 3 volumes with respect to SOT-1 then cooled to 21°C over the period of 60 min, added 560ml of Water at 21°C over the period of 30min and stirred for 30min at 21 °C, the reaction mass left overnight (without stirring) and stirred for lhr. The obtained slurry was filtered under vacuum and washed with 55ml*3times of water then kept for suction at 20-30°C for 30min. The material was dried at 50-60°C for 9hrs under vacuum to obtain 35gm of Sotagliflozin. 5.7gm of Sotagliflozin (5.7gr, Sotagliflozin) and 28.5ml of Methyl ethyl ketone (28.5ml) were charged in round bottom flask, the slurry was stirred at 22-25°C for 5-10min gradually raised the temperature to 78°C then added 114 ml of n-Heptane (114ml) at 78°C over the period of 55min. Once the addition of n-heptane was completed, seeds of Form-2 (20 mg) were added, the slurry was gradually cooled down to 25-27°C over the period of 60 min. The obtained slurry was stirred for 2-3hrs at 25-27°C and the mass was kept overnight (without stirring) at 25-27°C then stirred for 3hr at 23 °C. The mass was filtered under vacuum and washed with 10ml of n-Heptane then kept for suction for 30min at 25-30°C. The material was dried at 50°C for 2hrs under vacuum to obtain Form-2 of Sotagliflozin.
Preparation of Form 2- Seeds
[0097] Sotagliflozin (2gr, amorphous) was dissolved in methyl ethyl ketone (10ml) The slurry was heated to 80°C, then n-Heptane (40ml) was added over 60mins. The hazy solution was cooled to 20-30° over 60mins and stirred for 3hr. The slurry was kept overnight at 20-30°C (without stirring). The obtained slurry was filtered under vacuum and washed with n-Heptane (10ml) . The material was dried at 50 °C for 4hrs under vacuum to obtain the 1.9gm of Sotagliflozin Form -2 as confirmed by PXRD.
///////////WO 2018067805, NEW PATENT, SOTAGLIFLOZIN, TEVA
