PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
Triheptanoin is a source of heptanoate fatty acids, which can be metabolized without the enzymes of long chain fatty acid oxidation.4 In clinical trials, patients with long chain fatty acid oxidation disorders (lc-FAODs) treated with triheptanoin are less likely to develop hypoglycemia, cardiomyopathy, rhabdomyolysis, and hepatomegaly.1,2 Complications in lc-FAOD patients are reduced from approximately 60% to approximately 10% with the addition of triheptanoin.2
Triheptanoin was granted FDA approval on 30 June 2020.4
Triheptanoin, sold under the brand name Dojolvi, is a medication for the treatment of children and adults with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD).[1][2][3]
The most common adverse reactions include abdominal pain, diarrhea, vomiting, and nausea.[1][2][3]
Triheptanoin was approved for medical use in the United States in June 2020.[4][2][3]
Since triheptanoin is composed of odd-carbon fatty acids, it can produce ketone bodies with five carbon atoms, as opposed to even-carbon fatty acids which are metabolized to ketone bodies with four carbon atoms. The five-carbon ketones produced from triheptanoin are beta-ketopentanoate and beta-hydroxypentanoate. Each of these ketone bodies easily crosses the blood–brain barrier and enters the brain.
Medical uses
Dojolvi is indicated as a source of calories and fatty acids for the treatment of children and adults with molecularly confirmed long-chain fatty acid oxidation disorders (LC-FAOD).[1][2]
Triheptanoin was approved for medical use in the United States in June 2020.[4][2]
The FDA approved triheptanoin based on evidence from three clinical trials (Trial 1/NCT018863, Trial 2/NCT022141 and Trial 3/NCT01379625).[3] The trials enrolled children and adults with LC-FAOD.[3] Trials 1 and 2 were conducted at 11 sites in the United States and the United Kingdom, and Trial 3 was conducted at two sites in the United States.[3]
Trial 1 and Trial 2 were used to evaluate the side effects of triheptanoin.[3] Both trials enrolled children and adults diagnosed with LC-FAOD.[3] In Trial 1, participants received triheptanoin for 78 weeks.[3] Trial 2 enrolled participants from other trials who were already treated with triheptanoin (including those from Trial 1) as well as participants who were never treated with triheptanoin before.[3] Trial 2 is still ongoing and is planned to last up to five years.[3]
The benefit of triheptanoin was evaluated in Trial 3 which enrolled enrolled children and adults with LC-FAOD.[3] Half of the participants received triheptanoin and half received trioctanoin for four months.[3] Neither the participants nor the investigators knew which treatment was given until the end of the trial.[3] The benefit of triheptanoin in comparison to trioctanoin was assessed by measuring the changes in heart and muscle function.[3]
Names
Triheptanoin is the international nonproprietary name.[17]
^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3): 694. hdl:10665/330879. License: CC BY-NC-SA 3.0 IGO.
Further reading
de Almeida Rabello Oliveira M, da Rocha Ataíde T, de Oliveira SL, de Melo Lucena AL, de Lira CE, Soares AA, et al. (March 2008). “Effects of short-term and long-term treatment with medium- and long-chain triglycerides ketogenic diet on cortical spreading depression in young rats”. Neurosci. Lett. 434 (1): 66–70. doi:10.1016/j.neulet.2008.01.032. PMID18281154. S2CID7754768.
“Triheptanoin”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT01379625 for “Study of Triheptanoin for Treatment of Long-Chain Fatty Acid Oxidation Disorder (Triheptanoin)” at ClinicalTrials.gov
Prevention and Treatment of Thromboembolic Disorders
Milvexian, also known as BMS-986177, is a blood coagulation factor XIa inhibitor.Bristol-Myers Squibb , in collaboration with Janssen , is developing milvexian (BMS-986177, JNJ-70033093; JNJ-3093), an antithrombotic factor XIa (FXIa) inhibitor, for the oral prevention and treatment of thrombosis.
Example 1. Preparation of (9i?,135)-13-{4-[5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl]-6-oxo- 1 ,6-dihydropyrimidin- 1 -yl} -3-(difluoromethyl)-9-methyl-3,4,7, 15-tetraazatricyclo[ 12.3.1.02‘6] -8-one trifluoroacetate
1A. Preparation of l-(difluoromethyl)-4-nitro-lH-pyrazole
CS2CO3 (14.41 g, 44.2 mmol) was suspended in a solution of 4-nitro-lH-pyrazole (5.00 g, 44.2 mmol) and DMF (40 mL). After heating to 120 °C for 5 min, solid sodium 2-chloro-2,2-difluoroacetate (13.48 g, 88 mmol) was added in 10 equal portions over 20 min. The reaction was complete after 10 min of additional heating. The mixture was added to a separatory funnel containing 100 mL water and extracted with Et20 (2 x 50 mL). The combined organic layers were concentrated. Purification by normal-phase chromatography eluting with a gradient of hexanes/EtOAc yielded l-(difluoromethyl)-4-nitro-lH-pyrazole (6.99 g, 42.9 mmol, 97% yield) as a clear, colorless oil. 1H NMR (500MHz, CDCI3) δ 8.58 (s, 1H), 8.22 (s, 1H), 7.39 – 7.05 (t, J= 60 Hz, 1H).
IB. Preparation of (S)-tert-butyl (l-(4-(l-(difluoromethyl)-4-nitro-lH-pyrazol-5-yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate
To a N2 flushed, 500 mL RBF was added {S)-tert-bvXy\ (l-(4-chloropyridin-2-yl)but-3-en-l-yl)carbamate, prepared as described in Example 3, (10 g, 35.4 mmol), 1-(difluoromethyl)-4-nitro-lH-pyrazol (6.34 g, 38.9 mmol) and dioxane (100 mL). The solution was bubbled with N2 for 5 min. Then Pd(OAc)2 (0.40 g, 1.7 mmol),
di(adamantan-l-yl)(butyl)phosphine (1.27 g, 3.5 mmol), K2CO3 (14.7 g, 106 mmol) and PvOH (1.08 g, 10.61 mmol) were added. The reaction mixture was bubbled with N2 for 5 min then the reaction mixture was heated to 100 °C for 3 h. After this time, the solution was cooled to rt and water (200 mL) was added. The reaction mixture was then extracted with EtOAc (2 x 200 mL). The combined organic extracts were washed with water (200 mL), brine (200 mL), dried over Na2S04, filtered and concentrated in vacuo. Purification by normal phase chromatography eluting with a gradient of hexanes/EtOAc afforded (S)-tert-butyl ( 1 -(4-( 1 -(difluoromethyl)-4-nitro- lH-pyrazol-5 -yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate (12.91 g, 31.5 mmol, 89% yield) as a slightly yellow oil. MS(ESI) m/z: 410.4 [M+H]+. 1H NMR (400MHz, CDC13) δ 8.80 (dd, J=5.1, 0.7 Hz, 1H), 8.36 (s, 1H), 7.34 (s, 1H), 7.31 (dd, J=5.1, 1.5 Hz, 1H), 7.27 – 6.91 (t, J=58 Hz, 1H), 5.79 – 5.63 (m, 1H), 5.16 – 5.03 (m, 2H), 4.92 (d, J=5.9 Hz, 1H), 2.67 (t, J=6.4 Hz, 2H), 1.46 (br. s., 9H).
1C. Preparation of (l-(4-(4-amino-l -(difluoromethyl)- lH-pyrazol-5-yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate
To a 100 mL, 3-necked RBF was added a solution of (S)-tert-butyl (l-(4-(l-(difluoromethyl)-4-nitro-lH-pyrazol-5-yl)pyridin-2-yl)but-3-en-l-yl)carbamate (0.78 g, 1.90 mmol) in MeOH (12 mL) and a solution of NH4C1 (1.02 g, 19 mmol) in water (3 mL). To the solution was added Fe (0.53 g, 9.49 mmol). The reaction mixture was heated to 65 °C for 3 h. Water (50 mL) was added. After cooling to rt, the mixture was filtered through a CELITE® pad and rinsed with MeOH (200 mL). The filtrate was concentrated in vacuo. The residue was partitioned between EtOAC (100 mL) and water (100 mL). The organic phase was separated, washed with water (100 mL), brine (100 mL), dried over Na2S04, filtered and concentrated in vacuo. Purification by normal phase chromatography eluting with a gradient of DCM/MeOH yielded (S)-tert-butyl (l-(4-(4-amino- 1 -(difluoromethyl)- lH-pyrazol-5 -yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate (0.585 g, 1.54 mmol, 81% yield) as an oil. MS(ESI) m/z: 380.1 [M+H]+. 1H NMR (400MHz,
ID. Preparation of tert-butyl ((5)-l-(4-(l-(difiuoromethyl)-4-((i?)-2-methylbut-3-enamido)- lH-pyrazol-5-yl)pyridin-2-yl)but-3-en- 1 -yl)carbamate
To a N2 flushed, 3 -necked, 250 mL RBF was added a solution of {S)-tert-bvXy\ (1-(4-(4-amino-l-(difluoromethyl)-lH-pyrazol-5-yl)pyridin-2-yl)but-3-en-l-yl)carbamate (5 g, 13.18 mmol) and EtOAc (50 ml). The solution was cooled to -10 °C and (R)-2-methylbut-3-enoic acid, as prepared in Example 2, (1.72 g, 17.13 mmol), pyridine (4.26 ml, 52.7 mmol). and T3P® (23.54 ml, 39.5 mmol) were added. The cooling bath was removed and the solution was allowed to warm to rt and then stir over a period of 20 h. Water (30 mL) and EtOAc (30 mL) were added and the mixture was stirred for 30 min. The organic phase was separated and the aqueous layer was extracted with EtOAc (30 mL). The combined organic extracts were washed with brine (50 mL), dried over
Na2SC”4, filtered and concentrated in vacuo. Purification by normal phase
IE. Preparation of tert-butyl N-[(9i?,10E,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,10,14,16-hexaen-13-yl] carbamate
To a N2 flushed, 2 L, 3 -necked, RBF was added a solution of tert-butyl ((S)-l-(4-(1 -(difluoromethyl)-4-((i?)-2-methylbut-3 -enamido)- lH-pyrazol-5 -yl)pyridin-2-yl)but-3 -en-l-yl)carbamate (3 g, 6.50 mmol) in EtOAc (1300 ml). The solution was sparged with argon for 15 min. Grubbs II (1.38 g, 1.63 mmol) was added in one portion. The reaction mixture was heated to reflux for 24 h. After cooling to rt, the solvent was removed and the residue was purified by normal phase chromatography eluting with a gradient of DCM/MeOH to yield tert-butyl N-[(9R, 10E, 135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,10,14,16-hexaen-13-yl]carbamate (2.13 g, 4.91 mmol, 76% yield) as a tan solid. MS(ESI) m/z: 434.4 [M+H]+. 1H NMR (400MHz, CDC13) δ 8.71 (d, J=5.1 Hz, 1H), 7.78 (s, 1H), 7.44 – 7.40 (m, 1H), 7.36 (br. s., 1H), 7.27 (t, J=58 Hz, 1H), 6.87 (s, 1H), 6.49 – 6.39 (m, 1H), 5.78 (s, 1H), 4.80 (br. s., 2H), 3.18 – 3.08 (m, 1H), 3.08 – 2.98 (m, 1H), 2.06 – 1.93 (m, 1H), 1.51 (s, 9H), 1.19 (d, J=6.6 Hz, 3H).
IF. Preparation of tert-butyl N-[(9i?,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-13-yl]carbamate
Pd/C (0.60 g, 0.570 mmol) was added to a 250 mL Parr hydrogenation flask containing a solution of tert-butyl N-[(9i?,10E,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,10,14,16-hexaen-13-yljcarbamate (2.46 g, 5.68 mmol) in EtOH (100 mL). The flask was purged with N2 and pressurized to 55 psi of H2 allowed to stir for 18 h. The reaction was filtered through CELITE® and concentrated to yield tert-butyl N-[(9i?,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-13-yl]carbamate (2.17 g, 88% yield) as a tan solid. MS(ESI) m/z: 436.3 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 9.32 (s, 1H), 8.71 (d, J=5.0 Hz, 1H), 7.96 (t, J=58 Hz, 1H), 7.43 (s, 1H), 7.32 (d, J=4.8 Hz, 1H), 7.22 (d, J=7.3 Hz, 1H), 4.66 (d, J=8.3 Hz, 1H), 2.62 (br. s., 1H), 1.88 (d, J=12.8 Hz, 1H), 1.77 – 1.59 (m, 2H), 1.42 – 1.28 (m, 9H), 1.15 (d, J=18.2 Hz, 2H), 0.83 (d, J=7.0 Hz, 3H).
I G. Preparation of (9R, 13S)-l 3-amino-3-(difiuoromethyl)-9-methyl-3,4,7, 15-tetraazatricyclo[ 12.3.1.02‘6]octadeca- 1(18),2(6),4, 14,16-pentaen-8-one
4 N HC1 in dioxane (3.88 mL, 15.5 mmol) was added to a solution of tert-butyl N-[(9R, 13S)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7, 15-tetraazatricyclo[12.3.1.02‘6] octadeca-l(18),2(6),4,14,16-pentaen-13-yl]carbamate (2.25 g, 5.2 mmol) in MeOH (10 mL). The reaction was allowed to stir at rt for 2 h. The reaction was cooled in an ice bath, and 7 N NH3 in MeOH (13.3 mL, 93.0 mmol) was added. After 5 min, the reaction was diluted with CH2C12 (80 mL) and the solid that formed was filtered. The filtrate was concentrated to yield (9i?,135)-13-amino-3-(difluoromethyl)-9-methyl-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-8-one (1.3 g, 3.88 mmol, 75% yield). MS(ESI) m/z: 336.3 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 9.33 (s, 1H), 8.71 (d, J=5.0 Hz, 1H), 7.94 (t, J=58 Hz, 1H), 7.85 (s, 1H), 7.40 (s, 1H), 7.32 (d, J=5.0 Hz, 1H), 4.01 (dd, J=10.2, 5.1 Hz, 1H), 2.63 – 2.53 (m, 1H), 1.90 – 1.69 (m, 2H), 1.53 -1.36 (m, 2H), 1.16 – 1.00 (m, 1H), 0.85 (d, J=7.0 Hz, 3H).
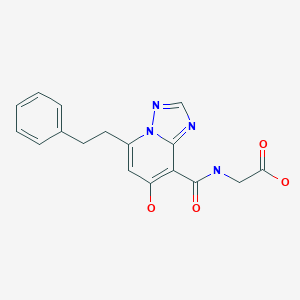
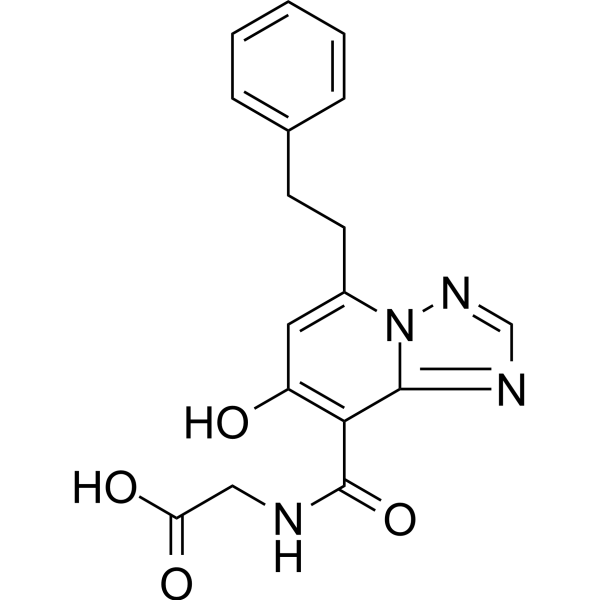
To a 100 mL flask containing a white suspension of 6-(5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl)pyrimidin-4-ol (0.83 g, 2.7 mmol), as prepared in Example 4 in ACN (36 mL) was added HATU (1.12 g, 3.0 mmol) and DBU (0.53 mL, 3.5 mmol). The resulting clear, yellow solution was stirred at rt. After 5 min, (9i?,135)-13-amino-3-(difluoromethyl)-9-methyl-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-8-one (0.9 g, 2.68 mmol) was added and the resulting suspension was stirred at rt for 3 h. The reaction was then concentrated and purified by normal phase silica gel chromatography, eluting with a gradient of 0% to 100% EtOAc in hexanes to yield (9i?,135)-13-{4-[5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl]-6-oxo- 1 ,6-dihydropyrimidin- 1 -yl} -3-(difluoromethyl)-9-methyl-3 ,4,7, 15-tetraazatricyclo [12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-8-one (0.87 g, 50% yield) as a white solid. MS(ESI) m/z: 626.2 [M+H]+. 1H NMR (500MHz, CD3OD) δ 8.91 – 8.83 (m, 1H), 8.78 – 8.71 (m, 1H), 8.33 (s, 1H), 7.88 (d, J=2.5 Hz, 1H), 7.74 (s, 2H), 7.69 – 7.67 (m, 1H), 7.65 (s, 1H), 7.63 (t, J=58 Hz, 1H), 7.52 – 7.50 (m, 1H), 6.36 (d, J=0.8 Hz, 1H),
Cetuximab Sarotalocan Sodium is an antibody-drug-conjugate (molecular weight: 156,000-158,000) consisting of tetrasodium salt of Sarotalocan (6-({[3-({(OC-6-13)-bis({3-[bis(3-sulfopropyl)(3-sulfonatopropyl)azaniumyl]propyl}dimethylsilanolato-κO,κO‘)[(phtalocyaninato(2-)κN29,κN30,κN31,κN32)-1-yl]silicon}oxy)propoxy]carbonyl}amino)hexanoyl (C70H96N11O24S6Si3; molecular weight: 1,752.22)) attached to an average of 2-3 Lys residues of Cetuximab.
Originator Japan Tobacco Developer Japan Tobacco; JW Pharmaceutical Class Acetic acids; Amides; Antianaemics; Pyridones; Small molecules; Triazoles Mechanism of Action Hypoxia-inducible factor-proline dioxygenase inhibitors
Preregistration Anaemia
27 Dec 2019 Japan Tobacco and SalubrisBio enter into a development and marketing agreement for enarodustat (JTZ 951) in China, Hong Kong, Macau and Taiwan for Anaemia 29 Nov 2019 Preregistration for Anaemia in Japan (PO) 31 Oct 2019 Phase I development in Anaemia is ongoing in USA
Enarodustat is a potent and orally active factor prolyl hydroxylase inhibitor, with an EC50 of 0.22 μM. Enarodustat has the potential for renal anemia treatment
Inhibition of hypoxia inducible factor prolyl hydroxylase (PHD) represents a promising strategy for the discovery of a next generation treatment for renal anemia. We identified several 5,6-fused ring systems as novel scaffolds of the PHD inhibitor on the basis of pharmacophore analysis. In particular, triazolopyridine derivatives showed potent PHD2 inhibitory activities. Examination of the predominance of the triazolopyridines in potency by electrostatic calculations suggested favorable π–π stacking interactions with Tyr310. Lead optimization to improve the efficacy of erythropoietin release in cells and in vivo by improving cell permeability led to the discovery of JTZ-951 (compound 14), with a 5-phenethyl substituent on the triazolopyridine group, which increased hemoglobin levels with daily oral dosing in rats. Compound 14 was rapidly absorbed after oral administration and disappeared shortly thereafter, which could be advantageous in terms of safety. Compound 14 was selected as a clinical candidate.
Certain glycopyrronium salts and related compounds, as well as processes for making and methods of using these glycopyrronium salts and related compounds, are known. See, for example, US Patent No. 8,558,008, which issued to assignee Dermira, Inc. See also, for example, US Patent No. 2,956,062, which issued to assignee Robins Co Inc. A H. See also, for example, International Patent Application Publication Nos. WO 98/00132 Al and WO 2009/00109A1, both of which list applicant Sepracor, Inc., as well as US Patent Nos. 6,063,808 and 6,204,285, both of which issued to assignee Sepracor, Inc. Certain methods of treating hyperhidrosis using glycopyrronium salts and related compounds are known. See, for example GB 1,080,960. Certain forms of applying glycopyrrolate compounds to a subject are known. See, for example US Patent Nos. 6,433,003 and 8,618,160, both of which issued to assignee Rose U; also US Patent Nos. 7,060,289; 8,252,316; and 8,679,524, which issued to PurePharm, Inc.
[0004] One glycopyrronium salt which is useful in certain medical applications is the following compound:
[0005] As illustrated above, the absolute configuration at the three asymmetric chiral positions is 2R3’R1’RS. This means that the carbon indicated with the number, 2, has the stereochemical R configuration. The carbon indicated with the number, 3′, also has the stereochemical R configuration. The quatemary ammonium nitrogen atom, indicated with a positive charge, may have either the R or the S stereochemical configuration. As drawn, the compound above is a mixture of two diastereoisomers.
[0006] Certain processes for making glycopyrronium salts are known. However, these processes are not as safe, efficient, stereospecific, or stereoselective as the new processes disclosed herein, for example with respect to large-scale manufacturing processes. Certain publications show that higher anticholinergic activity is attributed to the 2R3’R configuration. However, to date, processes for making the 2R3’R isomers, as well as the 2R3’R1’R isomers are low yielding, involve too many reaction steps to be economically feasible, use toxic materials, and/or are not sufficiently stereospecific or stereoselective with respect to the products formed.
EXAMPLE 2
[0179] The below synthetic description refers to the numbered compounds illustrated in FIG. 2. Numbers which refer to these compounds in FIG. 2 are bolded and underlined in this Example.
[0180] Synthesis of R(-)-Cyclopentylmandelic acid (4)
[0181] R(-)-cyclopentylmandelic acid (compound 4) can be synthesized starting with
R(-)-mandelic acid (compound 1) according to Example 1.
[0182] Step 1 : Making Compound 2.
[0183] R(-)-mandelic acid (1) was suspended in hexane and mixed with pivaldehyde and a catalytic amount of trifluoromethanesulfonic acid at room temperature to form a mixture. The mixture was warmed to 36 °C and then allowed to react for about 5 hours. The mixture was then cooled to room temperature and treated with 8% aqueous sodium bicarbonate. The aqueous layer was removed and the organic layer dried over anhydrous sodium sulfate. After filtration and removal of the solvent under vacuum, the crude product was recrystallized to give (5R)-2-(tert-butyl)-5-phenyl-l,3-dioxolan-4-one (compound 2) in 88% yield (per S-enantiomer yield).
[0184] Step 2: Making Compound 3.
[0185] Compound 2 was reacted with lithium hexamethyl disilazide (LiHMDS) in hexane at -78 °C under stirring for one hour. Next, cyclopentyl bromide was added to the reaction mixture including compound 2 and LiHMDS . The reaction was kept cool for about four (4) hours and then slowly warmed to room temperature and allowed to react for at least twelve (12) more hours. The resulting mixture was then treated with 10% aqueous ammonium chloride. The aqueous layer was discarded and the organic layer dried over anhydrous sodium sulfate. The solvent was removed under vacuum and the residue recrystallized from hexane to give pure product (5R)-2-(tert-butyl)-5-cyclopentyl-5-phenyl- l,3-dioxolan-4-one (3) in 63% yield (per S-enantiomer yield).
[0186] Step 3: Making Compound 4.
[0187] R(-)-cyclopentylmandelic acid (compound 4) was prepared by providing compound 3 in aqueous methanolic potassium hydroxide at 65 °C for four hours. After cooling this mixture to room temperature and removing the methanol under vacuum, the aqueous solution was acidified with aqueous hydrochloric acid. The aqueous solution was then extracted twice with ethyl acetate and the organic phase dried with anhydrous sodium sulfate. After removing the solvent and performing a recrystallization, pure R(-)- cyclopentylmandelic acid (compound 4) was obtained in 62% yield (based on S-enantiomer yield).
[0188] Next, a racemic mixture of l -methyl-3-pyrridinol (20) was provided:
[0189] Synthesis of 2R3 ‘R-glycopyrrolate base (8)
[0190] Step 4: Making Compound 8.
[0191] Enantiomerically pure R(-)-cyclopentylmandelic acid (4) was coupled to racemic l-methyl-3-pyrridinol (20) using 1, 1 -carbonyldiimideazole (CDI) activated esterification to make an enantiomerically pure mixture of the following erythro- and threo- glycopyrrolate bases (compounds 8 and 21, respectively):
[0192] The 2R3’R-glycopyrrolate base (compound 8) was then resolved using the 5- nitroisophthalate salt procedure in Finnish Patent 49713, to provide enantiomerically pure 2R3 Έ. {erythro) as well as pure 2R3 ‘S {threo). In this example, the 2R3 ‘S {threo) was discarded. The 2R3 Έ. {erythro) was separated as stereomerically pure compound 8.
[0193] Step 6: Making Compound 9.
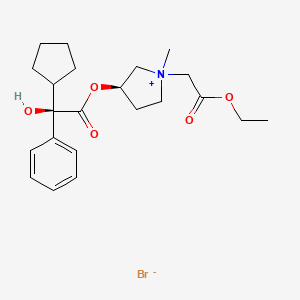
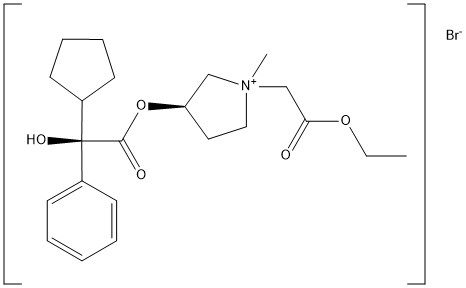
[0194] The glycopyrrolate base, compound 8, was treated in dry acetonitrile with methyl bromoacetate at room temperature under stirring for three (3) hours. The crude product was dissolved in a small volume of methylene chloride and poured into dry ethyl ether to obtain a precipitate. This procedure was repeated three times to provide (3R)-3-((R)- 2-cyclopentyl-2-hydroxy-2-phenylacetoxy)-l -(2-ethoxy-2-oxoethyl)-l-methylpyrrolidin-l – ium bromide, also known as 3′(R)-[R-Cyclopentylphenylhydroxyacetoy]- -ethyl- l ‘methoxycarbonylpyrrolidinium bromide (compound 9) in 89% yield. Compound 9 included the following stereoisomers:

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....

















.png)


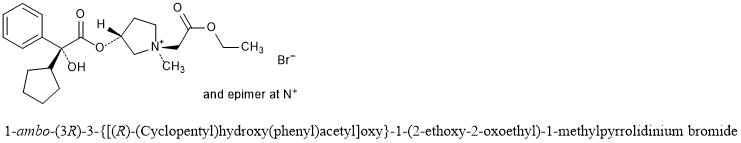







{kind=link}
{kind=link}