
Home » cancer
Category Archives: cancer
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Foselutoclax
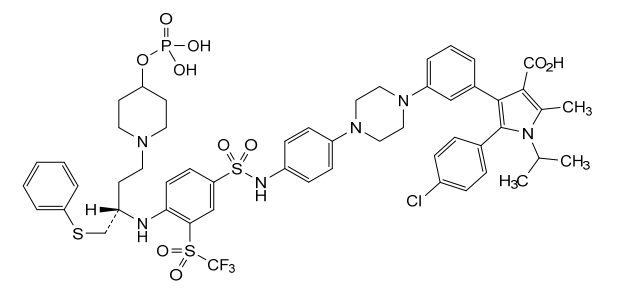
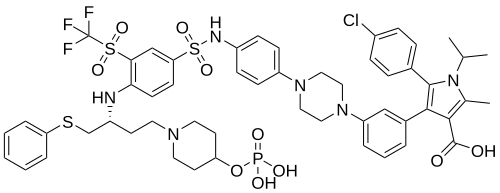
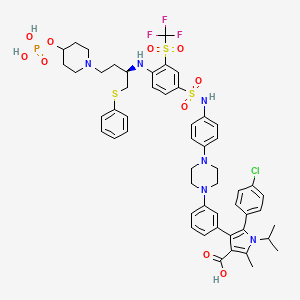
Foselutoclax
CAS 2271269-01-1
MF C53H59ClF3N6O10PS3 MW 1159.7 g/mol
(10R)-14-chloro-25-methyl-7,7-dioxo-10-[(phenylsulfanyl)methyl]-134-(phosphonooxy)-21-(propan-2-yl)-83-(trifluoromethanesulfonyl)-21H-7λ6-thia-6,9-diaza-4(1,4)-piperazina-13(1)-piperidina-2(2,3)-pyrrola-1(1),3(1,3),5,8(1,4)-tetrabenzenatridecaphane-24-carboxylic acid
5-(4-chlorophenyl)-2-methyl-4-[3-[4-[4-[[4-[[(2R)-1-phenylsulfanyl-4-(4-phosphonooxypiperidin-1-yl)butan-2-yl]amino]-3-(trifluoromethylsulfonyl)phenyl]sulfonylamino]phenyl]piperazin-1-yl]phenyl]-1-propan-2-ylpyrrole-3-carboxylic acid
B-cell lymphoma 2 (Bcl-2) inhibitor, antineoplastic, VT53CL5GES, UBX 1325
Foselutoclax is an investigational new drug that is being evaluated for the treatment of age-related eye diseases, particularly diabetic macular edema (DME) and wet age-related macular degeneration (AMD). Developed by Unity Biotechnology, this senolytic compound acts as a potent inhibitor of Bcl-xL, a protein that senescent cells rely on for survival.[1] Foselutoclax is designed to selectively eliminate senescent cells in the retina, potentially addressing the underlying causes of vision loss in these conditions.[2]
- Assess the Efficacy and Safety of Repeat Intravitreal Injections of Foselutoclax (UBX1325) in Patients With DME (ASPIRE)CTID: NCT06011798Phase: Phase 2Status: CompletedDate: 2025-08-05
- Safety, Tolerability and Evidence of Activity Study of UBX1325 in Patients With Diabetic Macular Edema (BEHOLD)CTID: NCT04857996Phase: Phase 2Status: CompletedDate: 2024-05-16
- Safety and Tolerability Study of UBX1325 in Patients With Diabetic Macular Edema or Neovascular Age-Related Macular DegenerationCTID: NCT04537884Phase: Phase 1Status: CompletedDate: 2022-03-10
REF
- Therapeutic targeting of cellular senescence in diabetic macular edema: preclinical and phase 1 trial resultsPublication Name: Nature MedicinePublication Date: 2024-02PMID: 38321220DOI: 10.1038/s41591-024-02802-4
- Senolytics in the treatment of diabetic retinopathyPublication Name: Frontiers in PharmacologyPublication Date: 2022-08-26PMCID: PMC9462063PMID: 36091769DOI: 10.3389/fphar.2022.896907
- Senolytic drugs: from discovery to translationPublication Name: Journal of Internal MedicinePublication Date: 2020-08-04PMCID: PMC7405395PMID: 32686219DOI: 10.1111/joim.13141
- bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell deathPublication Name: CellPublication Date: 1993-08-27PMID: 8358789DOI: 10.1016/0092-8674(93)90508-n
PAT
Treatment of Lung Diseases Using Pharmaceutical Agents that Eliminate Senescent Cells
Publication Number: US-2020354336-A9
Priority Date: 2017-08-11
- Senescent Cells and for Treating CancerPublication Number: US-2022017485-A1Priority Date: 2018-06-13
- Acyl sulfonamides that are bcl family antagonists for use in clinical management of conditions caused or mediated by senescent cells and for treating cancerPublication Number: EP-4335516-A2Priority Date: 2018-06-13
- Methods of Inhibiting Pathological AngiogenesisPublication Number: US-2020253991-A1Priority Date: 2017-10-31
- Methods of inhibiting pathological angiogenesisPublication Number: US-11129838-B2Priority Date: 2017-10-31Grant Date: 2021-09-28
- Treatment of Lung Diseases Using Pharmaceutical Agents that Eliminate Senescent CellsPublication Number: US-2020199103-A1Priority Date: 2017-08-11
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US279621490&_cid=P21-MGPXU3-15237-1
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US421382898&_cid=P21-MGPXWE-19244-1
A crystalline solid meglumine salt of of (R)-5-(4-chlorophenyl)-1-isopropyl-2-methyl-4-(3-(4-(4-((4-((1-(phenylthio)-4-(4-((phosphonooxy)methyl)piperidin-1-yl)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonamido)phenyl)piperazin-1-yl)phenyl)-1H-pyrrole-3-carboxylic acid, the compound of Formula I:
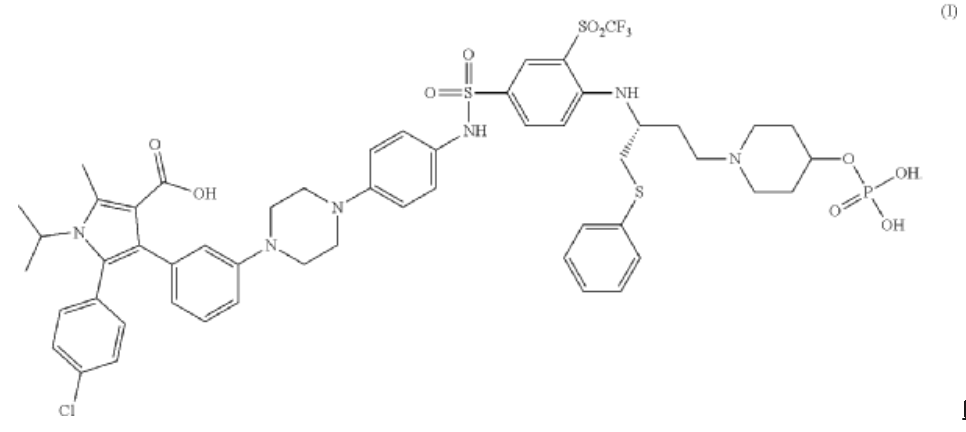
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US348024244&_cid=P21-MGPXWE-19244-1



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
| Clinical data | |
|---|---|
| Other names | UBX1325 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2271269-01-1 |
| PubChem CID | 147562879 |
| IUPHAR/BPS | 13366 |
| ChemSpider | 115277082 |
| UNII | VT53CL5GES |
| Chemical and physical data | |
| Formula | C53H59ClF3N6O10PS3 |
| Molar mass | 1159.69 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Crago SM (22 June 2023). “Design for Phase 2B ASPIRE Study of UBX1325 for DME announced by UNITY”. Modern Retina. Archived from the original on 13 August 2024.
- Macha N, Yu M, Sapieha P, Klier S, Ghosh A, White L, et al. (September 2024). “Multifocal Electroretinography Changes after UBX1325 (Foselutoclax) Treatment in Neovascular Age-Related Macular Degeneration”. Journal of Clinical Medicine. 13 (18): 5540. doi:10.3390/jcm13185540. PMC 11433175. PMID 39337030.
//////////foselutoclax, antineoplastic, VT53CL5GES, UBX 1325
Ezobresib
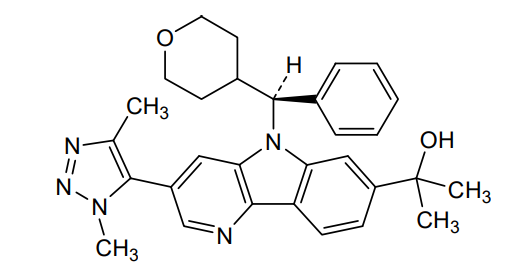
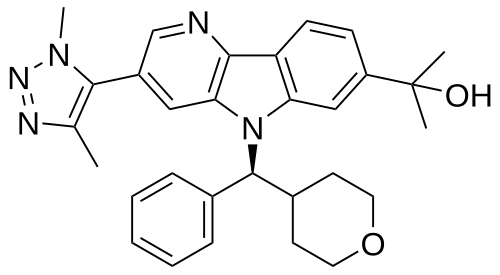
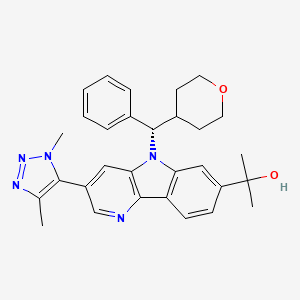
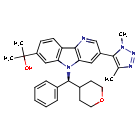
Ezobresib
CAS 1800340-40-2
MF C30H33N5O2 MW 495.6 g/mol
2-{3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-[(S)-(oxan-4-yl)(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl}propan-2-ol
bromodomain and extra-terminal motif (BET) inhibitor,
antineoplastic, BMS-986158, BMS 986158, Bristol Myers Squibb, antineoplastic, UNII-X8BW0MQ5PI
2-[3-(3,5-dimethyltriazol-4-yl)-5-[(S)-oxan-4-yl(phenyl)methyl]pyrido[3,2-b]indol-7-yl]propan-2-ol
Ezobresib is an investigational new drug that has been evaluated for the treatment of cancer. It inhibits Bromodomain and Extra-Terminal domain (BET) proteins, with potential antineoplastic activity.[1] Developed by Bristol Myers Squibb, this therapeutic agent has been studied for its efficacy in treating various cancers, including solid tumors and hematological malignancies.[2] Despite showing promise in early-phase clinical trials, recent developments suggest that Bristol Myers Squibb has decided to discontinue further development of ezobresib.[3]
BMS-986158 is under investigation in clinical trial NCT02419417 (Study of BMS-986158 in Subjects With Select Advanced Cancers).
Ezobresib is an inhibitor of the Bromodomain (BRD) and Extra-Terminal domain (BET) family of proteins, with potential antineoplastic activity. Upon administration, ezobresib binds to the acetyl-lysine binding site in the BRD of BET proteins, thereby preventing the interaction between BET proteins and acetylated histones. This disrupts chromatin remodeling and prevents the expression of certain growth-promoting genes, resulting in an inhibition of tumor cell growth. BET proteins (BRD2, BRD3, BRD4 and BRDT) are transcriptional regulators that bind to acetylated lysines on the tails of histones H3 and H4, and regulate chromatin structure and function; they play an important role in the modulation of gene expression during development and cellular growth
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US206490064&_cid=P21-MGLNPO-16484-1
Examples 54 & 55
2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
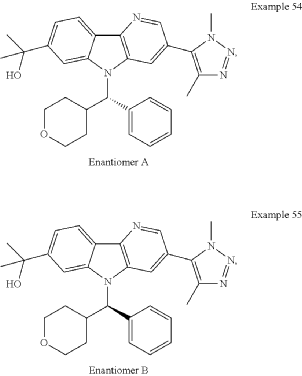
Step 1: 2-Chloro-5-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)pyridin-3-amine
Step 2: Methyl 3-((2-chloro-5-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)pyridin-3-yl)amino)benzoate
Step 3: Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5H-pyrido[3,2-b]indole-7-carboxylate
Alternate synthesis of Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 4: Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 5: 2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Alternate Synthesis of Examples 54
2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Step 1: 2-Chloro-5-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)pyridin-3-amine
Step 2: Methyl 3-((2-chloro-5-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)pyridin-3-yl)amino)benzoate
Step 3: Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5H-pyrido[3,2-b]indole-7-carboxylate
Alternate synthesis of Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 4: Methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 5: 2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Alternate Synthesis of Examples 54
2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Step 1: (S)-methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 2. (S)-2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
Step 1: (S)-methyl 3-(1,4-dimethyl-1H-1,2,3-triazol-5-yl)-5-(phenyl(tetrahydro-2H-pyran-4-yl)methyl)-5H-pyrido[3,2-b]indole-7-carboxylate
Step 2. (S)-2-[3-(Dimethyl-1H-1,2,3-triazol-5-yl)-5-[oxan-4-yl(phenyl)methyl]-5H-pyrido[3,2-b]indol-7-yl]propan-2-ol
PATENT
LIT
- BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor–resistant BRCA -mutant ovarian cancerPublication Name: Science Translational MedicinePublication Date: 2023-06-21PMCID: PMC10758289PMID: 37343085DOI: 10.1126/scitranslmed.add7872
- Recent updates on 1,2,3-triazole-containing hybrids with in vivo therapeutic potential against cancers: A mini-reviewPublication Name: European Journal of Medicinal ChemistryPublication Date: 2023-05-05PMID: 36893627DOI: 10.1016/j.ejmech.2023.115254
- Development of BET Inhibitors as Potential Treatments for Cancer: Optimization of Pharmacokinetic PropertiesPublication Name: ACS Medicinal Chemistry LettersPublication Date: 2022-07-05PMCID: PMC9290009PMID: 35859878DOI: 10.1021/acsmedchemlett.2c00219
- Synthesis of BMS-986158Publication Name: SynfactsPublication Date: 2021-11-17DOI: 10.1055/s-0041-1737090
- Development of BET inhibitors as potential treatments for cancer: A new carboline chemotypePublication Name: Bioorganic & Medicinal Chemistry LettersPublication Date: 2021-11-01PMID: 34560263DOI: 10.1016/j.bmcl.2021.128376
- Discovery and Preclinical Pharmacology of an Oral Bromodomain and Extra-Terminal (BET) Inhibitor Using Scaffold-Hopping and Structure-Guided Drug DesignPublication Name: Journal of Medicinal ChemistryPublication Date: 2021-09-20PMID: 34543572DOI: 10.1021/acs.jmedchem.1c00625
- Retrospective assessment of rat liver microsomal stability at NCATS: data and QSAR modelsPublication Name: Scientific ReportsPublication Date: 2020-11-26PMCID: PMC7693334PMID: 33244000DOI: 10.1038/s41598-020-77327-0
- High-Throughput Screening to Identify Inhibitors of the Type I Interferon–Major Histocompatibility Complex Class I Pathway in Skeletal MusclePublication Name: ACS Chemical BiologyPublication Date: 2020-05-27PMCID: PMC7859889PMID: 32459468DOI: 10.1021/acschembio.0c00343
- Predictive models of aqueous solubility of organic compounds built on A large dataset of high integrityPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2019-07-15PMCID: PMC8274818PMID: 31176566DOI: 10.1016/j.bmc.2019.05.037
- Highly predictive and interpretable models for PAMPA permeabilityPublication Name: Bioorganic & Medicinal ChemistryPublication Date: 2017-02-01PMCID: PMC5291813PMID: 28082071DOI: 10.1016/j.bmc.2016.12.049
- BET inhibitor resistance emerges from leukaemia stem cellsPublication Name: NaturePublication Date: 2015-09-14PMCID: PMC6069604PMID: 26367796DOI: 10.1038/nature14888
- Efficacy of BET Bromodomain Inhibition in Kras-Mutant Non–Small Cell Lung CancerPublication Name: Clinical cancer research : an official journal of the American Association for Cancer ResearchPublication Date: 2013-11-14PMCID: PMC3838895PMID: 24045185DOI: 10.1158/1078-0432.ccr-12-3904
- Discovery and Preclinical Evaluation of [4-[[1-(3-fluorophenyl)methyl]-1H-indazol-5-ylamino]-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yl]carbamic Acid, (3S)-3-Morpholinylmethyl Ester (BMS-599626), a Selective and Orally Efficacious Inhibitor of Human Epidermal Growth Factor Receptor 1 and 2 KinasesPublication Name: Journal of Medicinal ChemistryPublication Date: 2009-10-12PMID: 19821562DOI: 10.1021/jm9010065
- [Statistical analysis of cerebrospinal fluid acid-base equilibrium and cerebrospinal fluid lactate concentration in cases of brain tumors, cerebrocranial injuries and meningoencephalitis]Publication Name: Neurologia i neurochirurgia polskaPublication Date: 1976-07PMID: 8740
PAT
- Tricyclic compounds as anticancer agentsPublication Number: WO-2015100282-A1Priority Date: 2013-12-24
- Tricyclic compound as anticancer agentsPublication Number: EP-3466949-B1Priority Date: 2013-12-24Grant Date: 2020-12-23
- Novel tricyclic compounds as anticancer agentsPublication Number: TW-202028203-APriority Date: 2013-12-24
- Novel tricyclic compounds as anticancer agentsPublication Number: TW-I726544-BPriority Date: 2013-12-24Grant Date: 2021-05-01
- Tricyclic compounds as anticancer agentsPublication Number: CN-108558871-BPriority Date: 2013-12-24Grant Date: 2022-02-18
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Other names | BMS-986158 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1800340-40-2 |
| PubChem CID | 118196485 |
| DrugBank | DB15435 |
| ChemSpider | 58828664 |
| UNII | X8BW0MQ5PI |
| KEGG | D12710 |
| ChEMBL | ChEMBL4297458 |
| Chemical and physical data | |
| Formula | C30H33N5O2 |
| Molar mass | 495.627 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- Ma Z, Zhang C, Bolinger AA, Zhou J (October 2024). “An updated patent review of BRD4 degraders”. Expert Opinion on Therapeutic Patents. 34 (10): 929–951. doi:10.1080/13543776.2024.2400166. PMC 11427152. PMID 39219068.
- “Clinical Trials Using Ezobresib”. National Cancer Institute.
- Brown A. “Bristol backs out of BET inhibition”. ApexOnco.
////////////Ezobresib, antineoplastic, BMS-986158, BMS 986158, Bristol Myers Squibb, antineoplastic, UNII-X8BW0MQ5PI
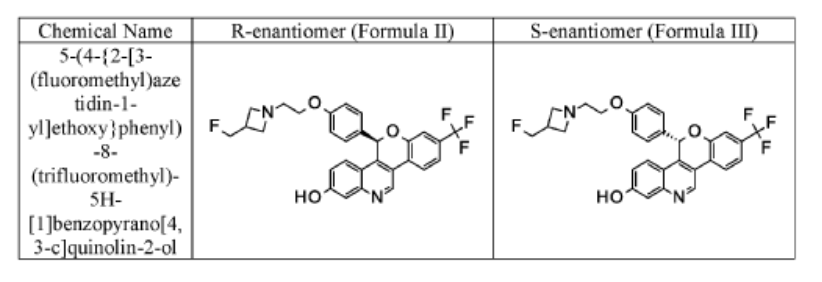
Imlunestrant
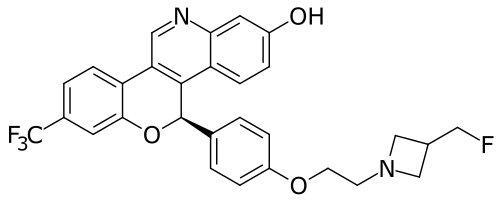
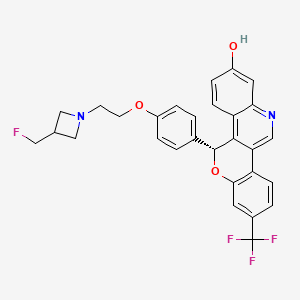

Imlunestrant
CAS 2408840-26-4
as tosylate: 2408840-41-3
(5R)-5-[4-[2-[3-(fluoromethyl)azetidin-1-yl]ethoxy]phenyl]-8-(trifluoromethyl)-5H-chromeno[4,3-c]quinolin-2-ol
- (5r)-5-(4-(2-(3-(fluoromethyl)azetidin-1-yl)ethoxy)phenyl)-8-(trifluoromethyl)-5h-(1)benzopyrano(4,3-c)quinolin-2-ol
- 5h-(1)benzopyrano(4,3-c)quinolin-2-ol, 5-(4-(2-(3-(fluoromethyl)-1-azetidinyl)ethoxy)phenyl)-8-(trifluoromethyl)-, (5r)-
MF C29H24F4N2O3 MW 524.516
FDA 9/25/2025, Inluriyo, LY3484356, LY-3484356, To treat estrogen receptor-positive, human epidermal growth factor receptor 2-negative, estrogen receptor-1-mutated advanced or metastatic breast cancer with disease progression following at least one line of endocrine therapy
Imlunestrant, sold under the brand name Inluriyo, is an anti-cancer medication used for the treatment of breast cancer.[1] It is an is an estrogen receptor antagonist.[1] It is used as the salt, imlunestrant tosylate.[2] It is taken by mouth.[1] It was developed by Eli Lilly and Company.[2]
The most common adverse events and laboratory abnormalities include decreased hemoglobin, musculoskeletal pain, decreased calcium, decreased neutrophils, increased AST, fatigue, diarrhea, increased ALT, increased triglycerides, nausea, decreased platelets, constipation, increased cholesterol, and abdominal pain.[2]
Imlunestrant was approved for medical use in the United States in September 2025.[2]
SYN
- Imlunestrant with or without Abemaciclib in Advanced Breast CancerPublication Name: The New England journal of medicinePublication Date: 2025-03-27PMID: 39660834DOI: 10.1056/nejmoa2410858
- Targeting the Estrogen Receptor for the Treatment of Breast Cancer: Recent Advances and ChallengesPublication Name: Journal of Medicinal ChemistryPublication Date: 2023-06-28PMID: 37377342DOI: 10.1021/acs.jmedchem.3c00136
- Novel endocrine therapies: What is next in estrogen receptor positive, HER2 negative breast cancer?Publication Name: Cancer Treatment ReviewsPublication Date: 2023-06PMID: 37146385DOI: 10.1016/j.ctrv.2023.102569
- Oral Selective Estrogen Receptor Degraders (SERDs) as a Novel Breast Cancer Therapy: Present and Future from a Clinical PerspectivePublication Name: International Journal of Molecular SciencesPublication Date: 2021-07-22PMCID: PMC8345926PMID: 34360578DOI: 10.3390/ijms22157812
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US281655517&_cid=P12-MG7DCV-14904-1
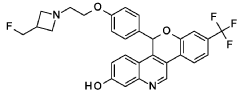
Example 1A
5-(4-{2-[3-(Fluoromethyl)azetidin-1-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[1]benzopyrano[4,3-c]quinolin-2-ol, Isomer 1Separate the two enantiomers of 5-(4-{2-[3-(fluoromethyl)azetidin-1-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[1]benzopyrano[4,3-c]quinolin-2-ol by chiral SFC with the following conditions: Column: LUX® Cellulose-1, 5×25 cm; eluting with a mobile phase of 30% iPrOH (with 0.5% DMEA) in CO 2; column temperature: 40° C.; flow rate: 300 g/minute; UV detection wavelength: 270 nm to give Example 1A as the first eluting enantiomer (Isomer 1). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 1 by chiral analytical SFC, >99% ee, t (R): 1.30 minutes; column: CHIRALCEL® OD-H, 4.6×150 mm; eluting with a mobile phase of 30% MeOH (0.2% IPA) in CO 2; column temperature: 40° C.; flow rate: 5 mL/minute; UV detection wavelength: 225 nm. Isolate the title compound of Example 1B to give the second eluting enantiomer (Isomer 2). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 2 by chiral analytical SFC, 98% ee, t (R): 2.03 minutes; column: CHIRALCEL® OD-H, 4.6×150 mm; eluting with a mobile phase of 30% MeOH (0.2% IPA) in CO 2; column temperature: 40° C.; flow rate: 5 mL/minute; UV detection wavelength: 225 nm.
Alternate Preparation Example 1B
Crystalline 5-(4-{2-[3-(Fluoromethyl)azetidin-1-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[1]benzopyrano[4,3-c]quinolin-2-ol, Isomer 2
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020014435&_cid=P12-MG7DHN-18354-1
EXAMPLE 1
Racemic 5-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [ 1 ]benzopyrano[4,3 -c]quinolin-2-ol
Cool a solution of (4-{2-[3-(fluoromethyl)azetidin-l-yl]ethoxy}phenyl){3-[2-fluoro-4-(trifluoromethyl)phenyl]-7-hydroxyquinolin-4-yl}methanone (5.27 g, 9.71 mmol) in 1,4-dioxane (100 mL) to 5 °C. Add lithium triethylborohydride (1 M in THF, 30.0 mL, 30.0 mmol). Remove the cooling bath and stir for 1.5 hours at room temperature. Quench the mixture with water. Add saturated NH4Cl solution and EtOAc. Separate the layers and extract the aqueous layer with EtOAc. Combine the organic extracts, dry over anhydrous MgS04, filter, and concentrate the filtrate. Dissolve the crude residue in THF (100 mL).
Add sodium hydride (60% in mineral oil, 1.94 g, 48.5 mmol). Reflux the solution for 1.5 hours. Add additional sodium hydride (60% in mineral oil, 1.94 g, 48.5 mmol), then reflux for an additional 30 minutes. Cool the solution to room temperature and quench with water. Add EtOAc and saturated NH4Cl solution. Separate the layers and extract the aqueous layer with EtOAc. Combine the organic extract, dry over anhydrous MgS04, filter, and concentrate the filtrate. Purify the residue by silica gel column chromatography eluting with a gradient of 5-7% MeOH in DCM to give the title compound (3.70 g, 72%) as a light yellow foam. ES/MS (m/z): 525.2 (M+H).
Prepare the following compounds in a manner essentially analogous to the method of Example 1, with the following variations in procedure. For the reduction, use 3 to 5 equivalents of lithium triethylborohydride with reaction times from 30 minutes to one hour and drying of the organic layers over magnesium sulfate or sodium sulfate. ETse the crude residue directly or purify by silica gel column chromatography eluting with a gradient of 0-5-7.5-10% MeOH in DCM before cyclization. Complete the cyclization by refluxing in THF for up to 16 hours, or in DMF, from 2 hours at room temperature for Ex 2, to 2 hours at 85 °C for Ex 8. Extract with DCM or EtOAc and dry organic layers over magnesium sulfate or sodium sulfate. Purify by silica gel column chromatography using up to 10% (MeOH or 7 M ammoniated MeOH) in DCM (Ex 2: gradient 0-10% MeOH in DCM; Ex 5: gradient 4-10% 7 M ammoniated MeOH in DCM; Ex 8: gradient 5-7.5% 7 M ammoniated MeOH in DCM) or by high pH reversed phase HPLC as noted.
EXAMPLE 1A
-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [l]benzopyrano[4,3-c]quinolin-2-ol, Isomer 1
and
EXAMPLE 1B
5-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [l]benzopyrano[4,3-c]quinolin-2-ol, Isomer 2
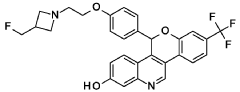
Separate the two enantiomers of 5-(4-{2-[3-(fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[l]benzopyrano[4,3-c]quinolin-2-ol by chiral SFC with the following conditions: Column: LUX® Cellulose-l, 5 x 25 cm; eluting with a mobile phase of 30% iPrOH (with 0.5% DMEA) in C02; column temperature: 40 °C; flow rate: 300 g/minute; UV detection wavelength: 270 nm to give Example 1 A as the first eluting enantiomer (Isomer 1). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 1 by chiral analytical SFC, >99% ee, /(R>: 1.30 minutes; column: CHFRALCEL® OD-H, 4.6 x 150 mm; eluting with a mobile phase of 30% MeOH (0.2% IP A) in C02; column temperature: 40 °C; flow rate: 5 mL/minute; UV detection wavelength: 225 nm. Isolate the title compound of Example 1B to give the second eluting enantiomer (Isomer 2). ES/MS (m/z): 525.2 (M+H). Confirm enantiomeric enrichment of Isomer 2 by chiral analytical SFC, 98% ee, /(R>: 2.03 minutes; column: CHIRALCEL® OD-H, 4.6 x 150 mm; eluting with a mobile phase of 30% MeOH (0.2% IP A) in C02; column temperature: 40 °C; flow rate: 5 mL/minute; UV detection wavelength: 225 nm.
Alternate Preparation EXAMPLE 1B
Crystalline 5-(4-{2-[3-(Fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H- [l]benzopyrano[4,3-c]quinolin-2-ol, Isomer 2
Stir 5-(4-{2-[3-(fluoromethyl)azetidin-l-yl]ethoxy}phenyl)-8-(trifluoromethyl)-5H-[l]benzopyrano[4,3-c]quinolin-2-ol, 4-methylbenzenesulfonic acid, Isomer 2 (23.8 g, 0.034 mol) in water (250 mL) at 1000 rpm. Add NaOH (76 pL) and stir the solution for 2 hours. Add DCM (600 mL). Separate the mixture, dry the DCM extract with magnesium sulfate, filter the material through a syringe filter (0.45 pm), and concentrate to dryness. Allow the material to sit under a N2 stream over a weekend. Add 1 : 1 EtOH/water (80 mL) and stir the mixture with sonication. Collect a tan solid by filtration on a nylon membrane to give the title compound (10.47 g, 0.02 mol, 59%).
PAT
- Selective estrogen receptor degradersPublication Number: US-2023234960-A1Priority Date: 2018-07-12
- Selective estrogen receptor degraderPublication Number: CN-112424205-BPriority Date: 2018-07-12Grant Date: 2023-10-31
- selective estrogen receptor degraderPublication Number: CN-117379428-APriority Date: 2018-07-12
- Selective estrogen receptor degradersPublication Number: US-11993608-B2Priority Date: 2018-07-12Grant Date: 2024-05-28
- Selective estrogen receptor degradersPublication Number: US-12128040-B2Priority Date: 2018-07-12Grant Date: 2024-10-29
PAT
https://patents.google.com/patent/US11926634B2/en
Selective estrogen receptor degraders (SERDs) bind to the estrogen receptor (ER) and downregulate ER-mediated transcriptional activity. The degradation and downregulation caused by SERDs can be useful in the treatment of various proliferative immune mediated disorders, cell proliferation disorders, including cancers such as breast cancer, ovarian cancer, endometrial cancer, prostate cancer, uterine cancer, gastric cancer, and lung cancer as well as mutations due to emerging resistance. Some small molecule examples of SERDs have been disclosed in the literature (see, e.g., WO2005073204, WO2014205136, and WO2016097071). Nonetheless, there is a need for new SERDs to treat ER-positive cancers, such as breast cancer, gastric cancer, and/or lung cancer.
As described in U.S. Pat. No. 10,654,866 (the ‘866 patent) a series of SERDs of the following formula have been discovered, along with pharmaceutically acceptable salts thereof:
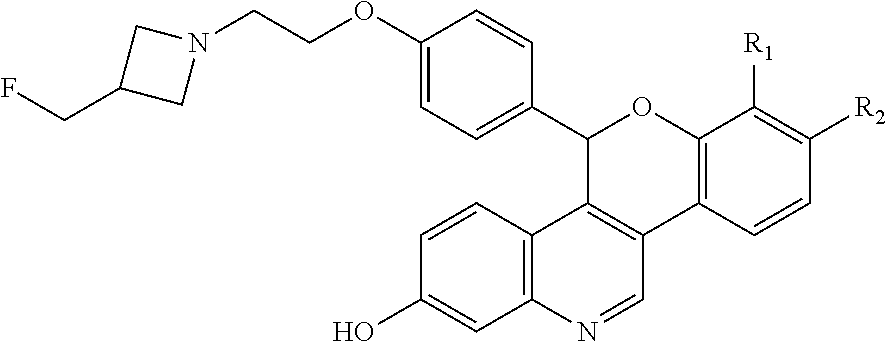
wherein one of R1 and R2 are independently Cl, F, —CF3, or —CH3, and the other is H.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
| Clinical data | |
|---|---|
| Trade names | Inluriyo |
| Other names | LY3484356, LY-3484356 |
| AHFS/Drugs.com | Inluriyo |
| License data | US DailyMed: Imlunestrant |
| Routes of administration | By mouth |
| Drug class | Estrogen receptor antagonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2408840-26-4as tosylate: 2408840-41-3 |
| PubChem CID | 146603228 |
| DrugBank | DB19043 |
| ChemSpider | 115010421 |
| UNII | 9CXQ3PF69Uas tosylate: F7UDT90EW5 |
| KEGG | D12216as tosylate: D12217 |
| ChEMBL | ChEMBL5095183 |
| Chemical and physical data | |
| Formula | C29H24F4N2O3 |
| Molar mass | 524.516 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
References
- https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/218881s000lbl.pdf
- “FDA approves imlunestrant for ER-positive, HER2-negative, ESR1-mutated advanced or metastatic breast cancer”. U.S. Food and Drug Administration (FDA). 25 September 2025. Retrieved 27 September 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - “U.S. FDA approves Inluriyo (imlunestrant) for adults with ER+, HER2-, ESR1-mutated advanced or metastatic breast cancer” (Press release). Eli Lilly. 25 September 2025. Retrieved 27 September 2025 – via PR Newswire.
- World Health Organization (2022). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 88”. WHO Drug Information. 36 (3). hdl:10665/363551.
Further reading
- Jhaveri, Komal L.; Jeselsohn, Rinath; Lim, Elgene; Hamilton, Erika P.; Yonemori, Kan; Beck, J. Thaddeus; et al. (June 2022). “A phase 1a/b trial of imlunestrant (LY3484356), an oral selective estrogen receptor degrader (SERD) in ER-positive (ER+) advanced breast cancer (aBC) and endometrial endometrioid cancer (EEC): Monotherapy results from EMBER”. Journal of Clinical Oncology. 40 (16_suppl): 1021. doi:10.1200/JCO.2022.40.16_suppl.1021. S2CID 249445691.
- Jhaveri, Komal; O’Shaughnessy, Joyce; Andre, Fabrice; Goetz, Matthew P.; Harbeck, Nadia; Martín, Miguel; et al. (March 2023). “Abstract OT1-01-02: EMBER-4: A phase 3 adjuvant trial of imlunestrant vs standard endocrine therapy (ET) in patients with ER+, HER2- early breast cancer (EBC) with an increased risk of recurrence who have previously received 2 to 5 years of adjuvant ET”. Cancer Research. 83 (5_Supplement): OT1–01–02-OT1-01–02. doi:10.1158/1538-7445.SABCS22-OT1-01-02.
- Neven, P.; Stahl, N.; Losada, M.J. Vidal; Jimenez, M. Martin; Kaufman, P.A.; Harbeck, N.; et al. (October 2023). “273P A preoperative window-of-opportunity (WOO) study of imlunestrant in ER+, HER2- early breast cancer (EBC): Final analysis from EMBER-2”. Annals of Oncology. 34: S292 – S293. doi:10.1016/j.annonc.2023.09.470. S2CID 264385454.
External links
- Clinical trial number NCT04975308 for “A Study of Imlunestrant, Investigator’s Choice of Endocrine Therapy, and Imlunestrant Plus Abemaciclib in Participants With ER+, HER2- Advanced Breast Cancer (EMBER-3)” at ClinicalTrials.gov
/////////Imlunestrant, FDA 2025, APPROVALS 2025, Inluriyo, CANCER, LY3484356, LY 3484356, 9CXQ3PF69U
Oritinib
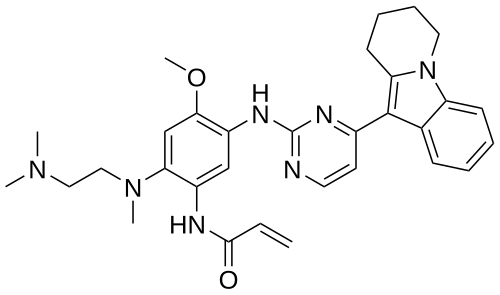
Oritinib
- CAS 2035089-28-0
- MESYLATE CAS 2180164-79-6
- SH-1028
- SK593H37SC
- N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide
- 539.7 g/mol, C31H37N7O2
- rilertinib
CHINA 2024, Nanjing Sanhome Pharmaceutical.
N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl]amino]phenyl]prop-2-enamide
Oritinib is an investigational new drug currently under investigation for its potential use in cancer treatment.[1][2] As a epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, oritinib targets specific enzymes involved in the signaling pathways that regulate cell division and survival, which are often dysregulated in cancer cells.[1]
Oritinib (SH-1028), an irreversible third-generation EGFR TKI, overcomes T790M-mediated resistance in non-small cell lung cancer. Oritinib (SH-1028), a mutant-selective inhibitor of EGFR kinase activity, inhibits EGFRWT, EGFRL858R, EGFRL861Q, EGFRL858R/T790M, EGFRd746-750 and EGFRd746-750/T790M kinases, with IC50s of 18, 0.7, 4, 0.1, 1.4 and 0.89 nM, respectively.
PAT
https://patents.google.com/patent/CN115974845B/en
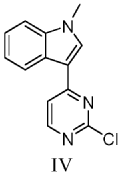
Reaction condition optimization experiment:
The experimental group numbered 1 referred to in table 1 below is the preparation of 1-methyl-3- (2-chloro-4-pyrimidinyl) indole, which was prepared as follows:
To a 10mL reaction tube, 2, 4-dichloropyrimidine (74.5 mg,0.05 mol), zinc triflate (67.3 mg,0.37 equiv), scandium triflate (7.4 mg,0.03 equiv) and 1-methylindole (78.6 mg,1.2 equiv) were added under inert gas atmosphere, and acetonitrile (2.5 mL) were heated to 80℃to react for 24 hours. The reaction was quenched with 30ml of ethyl acetate, the above mixture was added to a separating funnel, 50ml of saturated aqueous sodium carbonate and 50ml of saturated aqueous ammonium chloride were added thereto, and the mixture was shaken for 2 minutes, and the organic phase was taken after the liquid in the separating funnel had settled and separated. The aqueous phase was rinsed with 30ml of ethyl acetate under shaking for 2 times, the whole organic phase was collected, silica gel powder and anhydrous sodium sulfate were added thereto, and the mixture was dried under reduced pressure and packed into a silica gel column. Sequential gradient elution was performed using 250ml (PE: EA: triethylamine 16:4:1), 250ml (PE: EA: triethylamine 15:5:1), 250ml (PE: EA: triethylamine 40:20:3) as developing reagent. The eluent is collected and dried under reduced pressure to obtain pale yellow solid with the yield of 90 percent.
The nuclear magnetic resonance spectrum of 1-methyl-3- (2-chloro-4-pyrimidinyl) indole is as follows:
1H NMR(400MHz,DMSO-d6)δ8.51(d,J=5.9Hz,2H),8.40(dd,1H),7.82(d,J=5.4Hz,1H),7.56(dd,1H),7.28(pd,J=7.1,1.4Hz,2H),3.88(s,3H).
13C NMR(101MHz,DMSO)δ164.55,160.32,158.75,137.84,134.83,125.30,122.81,121.74,121.64,114.43,110.90,110.76,33.31.
PAT
CN109705118
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN242181067&_cid=P20-MEGI3F-20821-1
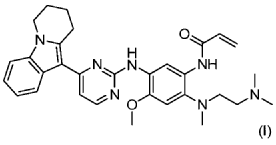
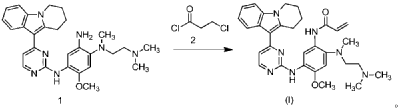
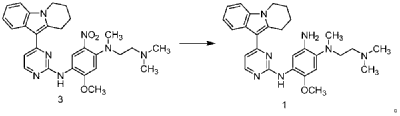
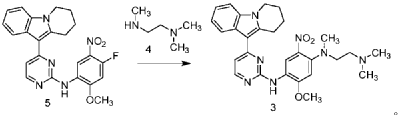
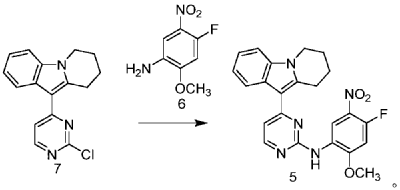
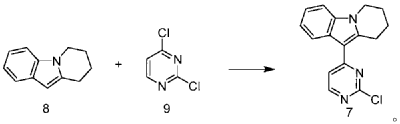
| Step 1: Synthesis of 10-(2-chloropyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole |
| |
| In a 100L vertical jacketed glass reactor, add ethylene glycol dimethyl ether (39.15kg) and 2,4-dichloropyrimidine (3.915kg). Cool the solid-liquid mixture to below 10°C, then add anhydrous aluminum chloride (3.855kg) in batches, controlling the addition rate to keep the temperature below 30°C. After the addition is complete, stir at 25±5°C for 30 minutes, then add 6,7,8,9-tetrahydropyrido[1,2-a]indole (4.500kg). Raise the temperature to 60±5°C and react for 3 hours. Monitor by HPLC until the 6,7,8,9-tetrahydropyrido[1,2-a]indole content does not exceed 1.0%, confirming the reaction is complete. The reaction solution was cooled to below 25° C., purified water (90.0 kg) was added, stirred, and filtered. The filter cake was added to acetonitrile (17.8 kg), slurried, filtered, and dried to obtain a yellow powdery solid, a total of 6.652 kg, with a yield of 89.2%. |
| Step 2: Synthesis of N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-amine |
| |
| To a 500L glass-lined reactor, sec-butyl alcohol (80.82kg), 10-(2-chloropyrimidin-4-yl)-6,7,8,9-tetrahydropyrido[1,2-a]indole (6.652kg), 4-fluoro-2-methoxy-5-nitroaniline (4.363kg), and p-toluenesulfonic acid monohydrate (4.816kg) were added to obtain a solid-liquid mixture. The reaction mixture was heated to reflux, and the solid gradually dissolved. As the reaction proceeded, a yellow solid precipitated. After reflux for 7.5 hours, the reaction was monitored by HPLC to confirm completion. Heating was stopped, the reaction mixture was cooled to below 15°C, stirred for 1 hour, and the solid was centrifuged and filtered. Acetonitrile (31.5kg) was added to the filter cake, and the mixture was slurried at 25±5°C for 1.5 hours. The mixture was centrifuged and dried to obtain the title compound, a total of 9.548kg, with a yield of 94.0%. |
| Step 3: Synthesis of N 1 -(2-dimethylaminoethyl)-5-methoxy-N 1 -methyl-2-nitro-N 4 -(4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)phenyl-1,4-diamine |
| |
| To a 100 L vertical jacketed glass reactor, add N,N-dimethylacetamide (44.7 kg), N-(4-fluoro-2-methoxy-5-nitrophenyl)-4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-amine (9.548 kg), N,N,N’-trimethylethylenediamine (3.380 kg), and N,N-diisopropylethylamine (4.841 kg). Under nitrogen, the reaction mixture was reacted at 85±5°C for 2 hours and monitored by HPLC until the reaction was complete. The reaction solution was cooled to below 70°C, purified water (95.5 kg) was added, filtered, and dried to obtain the title compound, a total of 8.206 kg, with a yield of 72.2%. |
| Step 4: Synthesis of N 1 -(2-(dimethylamino)ethyl)-5-methoxy-N 1 -methyl-N 4 -(4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)benzene-1,2,4-triamine |
| |
| A 100 L vertical jacketed reactor was charged with anhydrous ethanol (32.39 kg), purified water (14.32 kg), N 1 -(2-dimethylaminoethyl)-5-methoxy-N 1 -methyl-2-nitro-N 4 -(4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)phenyl-1,4-diamine (4.103 kg), reduced iron powder (2.224 kg), and ammonium chloride (2.129 kg). The reaction mixture was refluxed for 1.5 hours and monitored by HPLC until the reaction was complete. The reaction mixture was cooled to below 50°C and filtered through diatomaceous earth to remove the solid. The filtrate was concentrated, and tetrahydrofuran (3.45 kg) and purified water (34.71 kg) were added to the residue. The mixture was slurried, filtered, and dried to obtain 3.244 kg of the title compound in an 84.0% yield. |
| Step 5: Synthesis of N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide |
| |
| Add N,N-dimethylacetamide (48.6 kg) to a 100 L vertical jacketed glass reactor. Raise the temperature to 40°C, then add N₁- ( 2-(dimethylamino)ethyl)-5-methoxy- N₁ -methyl- N₄- (4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)benzene-1,2,4-triamine (6.487 kg). Then, begin the dropwise addition of 3-chloropropionyl chloride (1.777 kg). Control the addition rate to no more than 60°C. After the addition is complete, cool the reaction mixture and stir at 40±5°C for 1 hour. Sample the mixture and monitor the reaction by HPLC until complete. Add purified water (0.253 kg) and stir for 30 minutes. |
| The reaction mixture was heated at 80±5°C, triethylamine (13.52 kg) was added, and the temperature was raised to 95±5°C. After reacting for 2 hours, the reaction was complete as determined by HPLC. The temperature was then lowered, and methanol (83.0 kg) was added. The mixture was then cooled and crystallized, filtered, and dried to obtain 4.953 kg of the title compound, with a yield of 68.6% and a purity of 97.37%. |
| Step 6: Purification of N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide |
| Anhydrous ethanol (31.25 kg) was added to a 100 L reactor and heated to above 70°C. The crude N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide prepared in step 5 was added. The reaction mixture was heated and stirred under nitrogen until dissolved. The reaction mixture was cooled to below 10°C, the precipitated solid was centrifuged and dried under vacuum at 60±5°C for more than 12 hours to obtain 4.559 kg of the title compound with a yield of 92.1% and a purity of 98.73%. 1 H NMR (300 MHz, DMSO-d 6 )δ10.20(s,1H),8.65(s,1H),8.34(d,1H),8.11(s,1H),8.06(d,1H),7.43(d, 1H),7.19-7.03(m,3H),6.98(s,1H),6.57-6.41(m,1H),6.28-6.15(m,1H),5.8 2-5.71(m,1H),4.09(t,2H),3.84(s,3H),3.18(t,2H),3.06-2.92(m,2H),2.66 (s,3H),2.47-2.40(m,2H),2.27(s,6H),2.08-1.96(m,2H),1.87-1.74(m,2H). ESI-Ms m/z: 540.3 [M+H] + . |
| Example 2: Synthesis of N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide |
| |
| The preparation method is the same as that in step 5 of Example 1, except that N,N-dimethylacetamide is replaced by N,N-dimethylformamide. The purity of the obtained title compound is 69%. |
| The N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(6,7,8,9-tetrahydropyrido[1,2-a]indol-10-yl)pyrimidin-2-yl)amino)phenyl)allylamide of the present invention prepared according to the above method has a high yield and purity, mild reaction conditions, easy purification, stable process, easy operation, environmental friendliness, and can meet the requirements of industrial-scale production and application. |
Syn
European Journal of Medicinal Chemistry 291 (2025) 117643
Oritinib represents a third-generation EGFR TKI engineered by Nanjing Sanhome Pharmaceutical. This agent specifically targets both EGFR-sensitizing mutations and the T790 M resistance mutation,
thereby addressing resistance mechanisms linked to prior-generation EGFR-TKIs. In 2024, the NMPA granted approval for Oritinib to treat adult patients with locally advanced or metastatic NSCLC who have experienced disease progression during or following EGFR-TKI therapy and possess confirmed EGFR T790 M mutation-positive status. The mechanism of action of Oritinib involves irreversible binding to mutant EGFR, including the T790 M variant, which in turn suppresses down stream signaling pathways responsible for tumor cell proliferation and survival [28]. The mechanism of Oritinib effectively inhibits tumor growth in patients harboring T790M-mediated resistance to first- and second-generation EGFR-TKIs. Clinical efficacy was established in a Phase II trial (NCT03823807) enrolling patients with EGFR T790 Mmutation-positive NSCLC who had experienced disease progression following prior EGFR-TKI therapy. This study documented an ORR of 60.5 % and a median PFS of 9.6 months, highlighting substantial anti
tumor efficacy in this specific patient cohort. In terms of safety, Oritinib exhibited favorable tolerability. The predominant treatment-related adverse events were rash, diarrhea, and elevated liver enzymes, pri
marily of mild (Grade 1) or moderate (Grade 2) severity. No dose-limiting toxicities were encountered, and the overall safety profile aligned with those observed for other third-generation EGFR-TKIs [29].
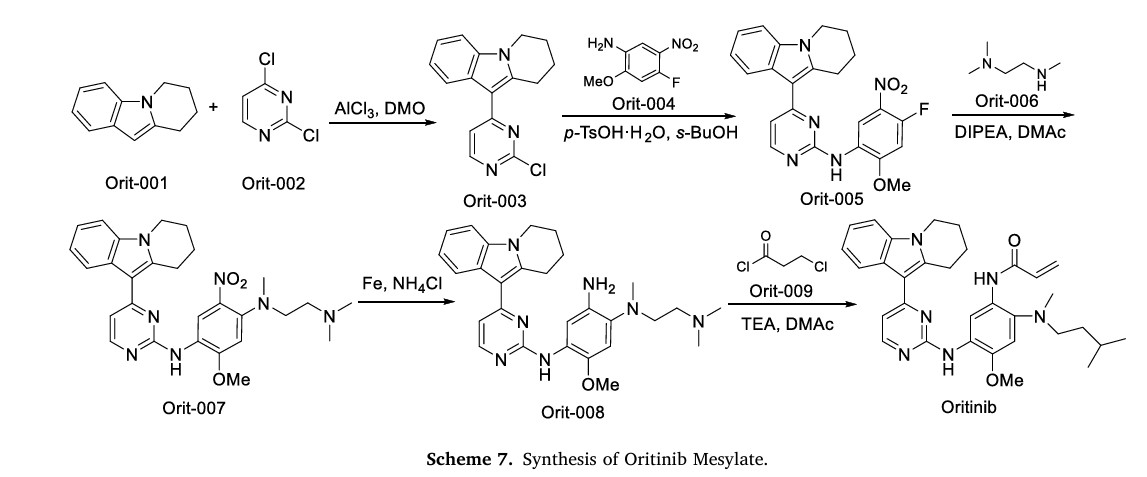
The synthetic route of Oritinib Mesylate, shown in Scheme 7, begins with nucleophilic substitution reaction between Orit-001 and Orit-002 to yield Orit-003, which further reacts with Orit-004 via nucleophilic substitution to produce Orit-005 [30]. Orit-005 subsequently undergoes another nucleophilic substitution with Orit-006 to generate Orit-007. Following this, Orit-007 is reduced to form Orit-008. Finally, an amidation reaction between Orit-008 and Orit-009 affords Oritinib.
[28] C. Zhou, A. Xiong, L. Miao, J. Chen, K. Li, H. Liu, Z. Ma, H. Wang, Z. Lu, J. Shen,
P51.03 oritinib (SH-1028), a third-generation EGFR-TKI in advanced NSCLC
patients with positive EGFR T790M: results of a single-arm phase Ib trial,
J. Thorac. Oncol. 16 (2021) S1119–S1120.
[29] C. Zhou, A. Xiong, J. Zhao, W. Li, M. Bi, J. Chen, K. Li, L. Miao, Y. Mao, D. Wang,
7MO oritinib (SH-1028) a third-generation EGFR tyrosine kinase inhibitor in
locally advanced or metastatic NSCLC patients with positive EGFR T790M: results
of a single-arm phase II trial, Ann. Oncol. 33 (2022) S31.
[30] L. Zhao, W. Fu, W. Wu, J. Liu, J. Jin, Method for Preparing Tricyclic Compound as
EGFR Kinase Inhibitor, 2019. CN109705118A.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Xiong A, Ren S, Liu H, Miao L, Wang L, Chen J, et al. (October 2022). “Efficacy and Safety of SH-1028 in Patients With EGFR T790M-Positive NSCLC: A Multicenter, Single-Arm, Open-Label, Phase 2 Trial”. Journal of Thoracic Oncology. 17 (10): 1216–1226. doi:10.1016/j.jtho.2022.06.013. PMID 35798241.
- “Rilertinib – Nanjing Sanhome Pharmaceutical”. AdisInsight. Springer Nature Switzerland AG.
| Clinical data | |
|---|---|
| Other names | SH-1028 |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2035089-28-0 |
| PubChem CID | 122666966 |
| ChemSpider | 115007246 |
| UNII | SK593H37SC |
| Chemical and physical data | |
| Formula | C31H37N7O2 |
| Molar mass | 539.684 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- New drugs approved by the NMPA in 2024: Synthesis and clinical applicationsPublication Name: European Journal of Medicinal ChemistryPublication Date: 2025-07-05PMID: 40262297DOI: 10.1016/j.ejmech.2025.117643
- Safety, efficacy, and pharmacokinetics of SH‐1028 in EGFR T790M‐positive advanced non–small cell lung cancer patients: A dose‐escalation phase 1 studyPublication Name: CancerPublication Date: 2023-02-22PMID: 36813747DOI: 10.1002/cncr.34697
- SH-1028, An Irreversible Third-Generation EGFR TKI, Overcomes T790M-Mediated Resistance in Non-Small Cell Lung CancerPublication Name: Frontiers in PharmacologyPublication Date: 2021-04-27PMCID: PMC8111447PMID: 33986687DOI: 10.3389/fphar.2021.665253
- [1]. Luwei Han, et al. SH-1028, An Irreversible Third-Generation EGFR TKI, Overcomes T790M-Mediated Resistance in Non-Small Cell Lung Cancer. Front Pharmacol. 2021 Apr 27;12:665253. [Content Brief]
/////////Oritinib, CHINA 2024, APPROVALS 2024, 2035089-28-0, SH 1028, SK593H37SC, rilertinib, Oritinib mesylate, Nanjing Sanhome Pharmaceutical,
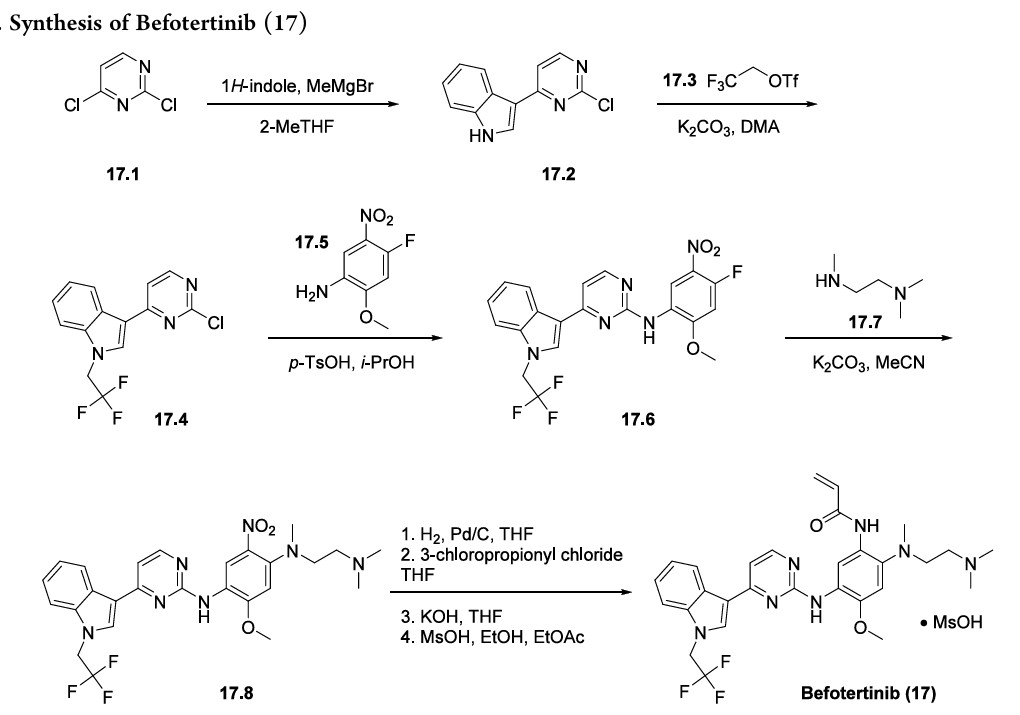
Befotertinib
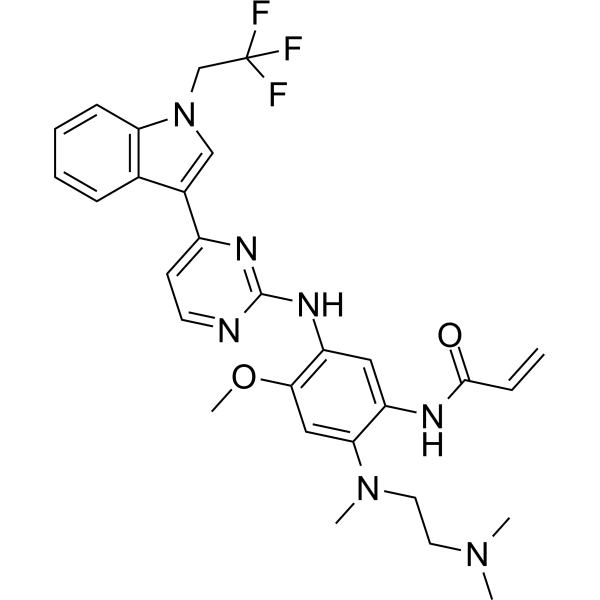
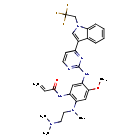
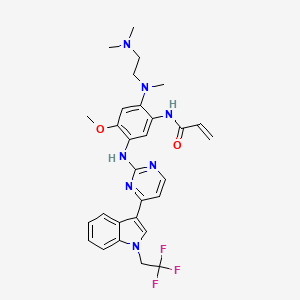
Befotertinib
D-0316, 0XT2CPR891
CAS No. : 1835667-63-4, MESYLATE CAS No. 2226167-02-6
- 2-propenamide, n-(2-((2-(dimethylamino)ethyl)methylamino)-4-methoxy-5-((4-(1-(2,2,2-trifluoroethyl)-1h-indol-3-yl)-2-pyrimidinyl)amino)phenyl)-
- N-(2-((2-(dimethylamino)ethyl)(methyl)amino)-4-methoxy-5-((4-(1-(2,2,2-trifluoroethyl)-1h-indol-3-yl)pyrimidin-2-yl)amino)phenyl)prop-2-enamide
- N-[2-[2-(dimethylamino)ethyl-methylamino]-4-methoxy-5-[[4-[1-(2,2,2-trifluoroethyl)indol-3-yl]pyrimidin-2-yl]amino]phenyl]prop-2-enamide
| Molecular Weight | 567.61 |
|---|---|
| Formula | C29H32F3N7O2 |
Befotertinib (D-0316) is an orally active EGFR tyrosine kinase inhibitor. Befotertinib can inhibit the proliferation of tumor cells. Befotertinib can be used in the research of EGFR T790M-positive non-small cell lung cancer (NSCLC).
Befotertinib is an orally available inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. Upon administration, befotertinib specifically binds to and inhibits EGFR T790M, a secondarily acquired resistance mutation, which prevents EGFR-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. Compared to some other EGFR inhibitors, befotertinib may have therapeutic benefits in tumors with T790M-mediated drug resistance. EGFR, a receptor tyrosine kinase that is mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization.
PAPER
J. Med. Chem. 2017, 60, 6480−6515.
PATENT
WO 2019218987
https://patentscope.wipo.int/search/en/WO2019218987
[0054]
U.S. Publication No. 2017/0355696 A1 describes a method of preparing Compound 4 and various pharmaceutically acceptable salts thereof. The exemplified synthetic process in U. S. Publication No. 2017/0355696 A1 includes a two-step conversion from the aniline compound, corresponding to Compound 1 of this disclosure, into the bismesylate of Compound 4, which has a low yield.
[0055]
As shown herein, representative methods of preparation of Compound 4, or a pharmaceutically acceptable salt, (or alternatively referred to as synthetic methods) , can provide the desired Compound 4, or a pharmaceutically acceptable salt, in improved yield and high purity and can be adapted for large-scale manufacture.
[0056]
In various embodiments, the present invention provides a novel method of preparing Compound 4, or a pharmaceutically acceptable salt thereof. The method typically includes converting a compound of Formula III, or a salt thereof, into compound 4, typically under an elimination reaction condition:
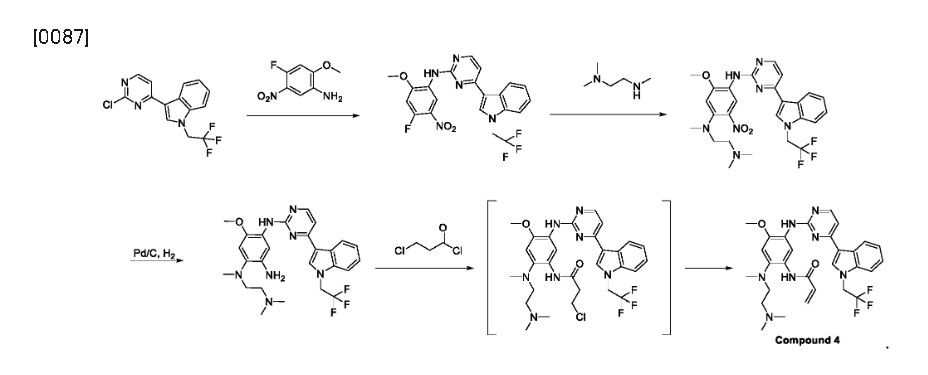
Syn
https://doi.org/10.1021/acs.jmedchem.4c02079
J. Med. Chem. 2025, 68, 2147−2182
Befotertinib (Surmana). Befotertinib (17), an oral, highly selective, third generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) developed by Betta Pharmaceuticals and InventisBio, was approved in China in May 2023 for the second-line treatment of patients
with locally advanced or metastatic nonsmall cell lung cancer (NSCLC) with positive EGFR T790 M mutation who have disease progression on previous EGFR TKI therapy. 140 139 NSCLC
has a high incidence and disease burden in China, which has spurred the development of multiple EGFR TKIs by Chinese companies.
Achromatography-free process route to befotertinib (17) has been reported in the patent literature by researchers at InventisBio (Scheme 29), although details about scale and yields were not provided.
141 142 The reaction sequence closely follows that of osimertinib, a third generation EGFR inhibitor
that was first approved in 2015 and was covered in our previous review.
Osimertinib and befotertinib share a common backbone, differing only in N-substitution on the indole ring.
Friedel−Crafts arylation of 1H-indole with 2,4-dichloropyrimidine (17.1) gave the 3-pyrimidinyl indole 17.2. The trifluoroethyl moiety in indole 17.4 was introduced via Nalkylation of 17.2 with triflate 17.3. This was followed by an SAr reaction with nitroaniline 17.5 to provide amino pyrimidine 17.6. Next, N,N,N′-trimethylethylenediamine (17.7) displaced the electrophilic aryl fluoride in an SNArreaction to generate intermediate 17.8. The acrylamide moiety was installed using a three-step sequence: hydrogenolytic
reduction of the nitro group to the corresponding aniline, acylation with 3-chloropropanoyl chloride, and immediate elimination to the acrylamide. Mesylate salt formation and crystallization furnished befotertinib mesylate (17) in eight steps from 17.1.
(139) Blair, H. A. Befotertinib: first approval. Drugs 2023, 83, 1433−
1437.
(140) Lau, S. C. M.; Ou, S.-H. I. And still they come over troubled
waters: can Asia’s third-generation EGFR tyrosine kinase inhibitors
(Furmonertinib, Aumolertinib, Rezivertinib, Limertinib, Befotertinib,
SH-1028, and Lazertinib) affect global treatment of EGFR+ NSCLC. J.
Thorac. Oncol. 2022, 17, 1144−1154.
(141) Dai, X.; Jiang, Y. Preparation of pyrimidine derivative and its
pharmaceutical salt as EGFR inhibitors for the treatment of cancer and
other diseases. WO 2019218987, 2019.
(142) Flick, A. C.; Ding, H. X.; Leverett, C. A.; Kyne, R. E.; Liu, K. K.
C.; Fink, S. J.; O’Donnell, C. J. Synthetic approaches to the new drugs
approved during 2015. J. Med. Chem. 2017, 60, 6480−6515.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. Nagasaka M, et, al. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors For Advanced EGFR+ NSCLC. J Thorac Oncol. 2021 May;16(5):740-763. [Content Brief][2]. Blair HA. Befotertinib: First Approval. Drugs. 2023 Oct;83(15):1433-1437. [Content Brief]
/////////Befotertinib, APPROVALS 2023, CHINA 2023, Betta Pharmaceuticals, InventisBio, CANCER, D-0316, D 0316, 0XT2CPR891
Zongertinib
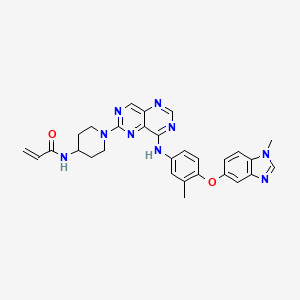
Zongertinib
CAS No. : 2728667-27-2,
BI-1810631, BI1810631
| Molecular Weight | 535.60 |
|---|---|
| Formula | C29H29N9O2 |
FDA 8/8/2025, Hernexeos, To treat adults with unresectable or metastatic non-squamous non-small cell lung cancer whose tumors have HER2 tyrosine kinase domain activating mutations, as detected by an FDA-approved test, and who have received prior systemic therapy
- N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide
- N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo(d)imidazol-5-yl)oxy)phenyl)amino)pyrimido(5,4-d)pyrimidin-2-yl)piperidin-4-yl)acrylamide
- 884-819-6
Zongertinib is an orally bioavailable inhibitor of the receptor tyrosine kinase human epidermal growth factor receptor 2 (HER2; ErbB2; HER-2), with potential antineoplastic activity. Upon oral administration, zongertinib covalently binds to and inhibits the activity of both wild-type and HER2 mutants, including HER2 mutants with exon 20 insertion (ex20ins) mutations. This prevents HER2-mediated signaling and may lead to cell death in HER2-expressing tumor cells. HER2, a receptor tyrosine kinase overexpressed on a variety of tumor cell types, plays an important role in tumor cell proliferation and tumor vascularization.
REF
Synthesis of zongertinib (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-
548 yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide)
Methods
Synthesis of Zongertinib (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acrylamide)
An overview of the synthetic routes to zongertinib and BI-3999 is shown in Supplementary Fig. S1, and graphical NMR spectra are shown in Supplementary Fig. S2.
3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)aniline (500 mg, 1.97 mmol) and 8-chloro-2-(methylthio)pyrimido[5,4-d]pyrimidine hydrochloride (492 mg, 1.97 mmol) were suspended in isopropanol, and the resulting reaction mixture stirred at 50°C for 3 hours, at which time high-performance liquid chromatography–mass spectrometry (HPLC-MS) indicated full conversion. The reaction mixture was concentrated under reduced pressure, and the crude product was redissolved in dichloromethane and washed with aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–15% methanol in dichloromethane) to afford the product (840 mg).
N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)-6-(methylthio)pyrimido[5,4-d]pyrimidin-4-amine (860 mg, 90%, 1.80 mmol) was suspended in dichloromethane (30 mL), and the resulting mixture was cooled to 0°C to 5°C. mCPBA (3-chloroperbenzoic acid, 444 mg, 77%, 1.98 mmol) was added portionwise over 1 hour, and the resulting reaction mixture was stirred at room temperature overnight, at which time HPLC-MS indicated full conversion. The reaction mixture was diluted with dichloromethane and washed with aqueous NaHCO3. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product which was used directly in the next step (767 mg, crude).
N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)-6-(methylsulfinyl)pyrimido[5,4-d]pyrimidin-4-amine (5.42 g, 80%, 9.73 mmol) was dissolved in N,N-dimethyl formamide (DMF, 50 mL) and diisopropylethylamine (2.8 mL, 16 mmol). 4-Boc-amino-1-piperidine (2.39 g, 11.9 mmol) was added, and the reaction was stirred at 60°C overnight. Then, the reaction mixture was concentrated, and the crude product was used directly in the next step (5.66 g, crude).
Tert-butyl (1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)carbamate (5.66 g, 9.73 mmol) was dissolved in dichloromethane (100 mL) and methanol (30 mL). Four mol/L HCl in dioxane (11 mL, 44 mmol) was added, and the resulting reaction mixture was heated to 45°C for 7 hours. HPLC-MS indicated some remaining starting material; therefore, the reaction mixture was stirred at room temperature overnight. Four mol/L HCl in dioxane (1 mL, 0.40 mmol) was added, and the reaction mixture was reheated to 45°C for 4 hours, at which time HPLC-MS indicated full conversion. The reaction mixture was concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–20% methanol in dichloromethane) to afford the product (4.5 g, 70% purity).
1-[8-({3-methyl-4-[(1-methyl-1H-1,3-benzodiazol-5-yl)oxy]phenyl}amino)-[1,3]diazino[5,4-d]pyrimidin-2-yl]piperidin-4-amine (4.5 g, 70%, 6.9 mmol) was suspended in dichloromethane (150 mL) and triethyl amine (4 mL, 28 mmol), and dimethylaminopyridine (115 mg, 0.941 mmol) was added. Then, acroyloyl anhydride (1.36 g, 95%, 10.3 mmol) was added, and the resulting reaction mixture was stirred at room temperature for 1 hour, at which time HPLC-MS indicated full conversion. The reaction mixture was diluted with dichloromethane (50 mL) and washed with aqueous NaHCO3 and brine. The organic layer was dried over Na2SO4 and concentrated, and the resulting crude product was purified by column chromatography (SiO2, gradient of 0%–20% methanol in dichloromethane) to afford the product (2.49 g).
1H NMR (DMSO-d6, 500 MHz) δ 9.58 (s, 1H), 9.08 (s, 1H), 8.39 (s, 1H), 8.19 (s, 1H), 8.10 (d, 1H, J = 7.6 Hz), 7.84 (d, 1H, J = 2.2 Hz), 7.77 (dd, 1H, J = 8.8 Hz, J = 2.2 Hz), 7.57 (d, 1H, J = 8.8 Hz), 7.09 (d, 1H, J = 2.2 Hz), 7.00 (dd, 1H, J = 2.2, 8.5 Hz), 6.89 (d, 1H, J = 8.8 Hz), 6.20 (dd, 1H, J = 10.1, 17.0 Hz), 6.10 (dd, 1H, J = 2.2, 17.0 Hz), 5.6 (dd, 1H, J = 2.2, 9.8 Hz), 4.86 (m, 2H), 3.99 (m, 1H), 3.84 (s, 3H), 3.25 (m, 2H), 2.26 (s, 3H), 1.92 (m, 2H), and 1.43 (m, 2H).
Synthesis of BI-3999 (N-(1-(8-((3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)amino)pyrimido[5,4-d]pyrimidin-2-yl)piperidin-4-yl)acetamide)
6-(4-aminopiperidin-1-yl)-N-(3-methyl-4-((1-methyl-1H-benzo[d]imidazol-5-yl)oxy)phenyl)pyrimido[5,4-d]pyrimidin-4-amine (100 mg, 208 mmol) and 4-dimethylaminopyridine (2.5 mg, 0.02 mmol) were suspended in 5 mL dichloromethane. Acetic anhydride (25 μL, 0.23 mmol) was added, and the resulting reaction mixture was stirred at room temperature for one hour. Then, the reaction mixture was diluted with dichloromethane and washed with aqueous NaHCO3 and brine. Then, the layers were separated, and the organic layer was dried over MgSO4 and concentrated. The crude product was purified by column chromatography (SiO2, gradient of 0%–10% methanol in dichloromethane) to afford the product (75 mg).
1H NMR (DMSO-d6, 400 MHz) δ 9.58 (s, 1H), 9.07 (s, 1H), 8.39 (s, 1H), 8.17 (s, 1H), 7.88 (d, 1H, J = 7.9 Hz), 7.84 (d, 1H, J = 2.5 Hz), 7.77 (dd, 1H, J = 2.7, 8.7 Hz), 7.57 (d, 1H, J = 8.9 Hz), 7.09 (d, 1H, J = 2.3 Hz), 7.00 (dd, 1H, J = 2.3, 8.6 Hz), 6.89 (d, 1H, J = 8.6 Hz), 4.85 (m, 2H), 3.90 (m, 1H), 3.84 (s, 3H), 3.23 (m, 2H), 2.26 (s, 3H), 1.88 (m, 2H), 1.82 (s, 3H), and 1.38 (m, 2H).
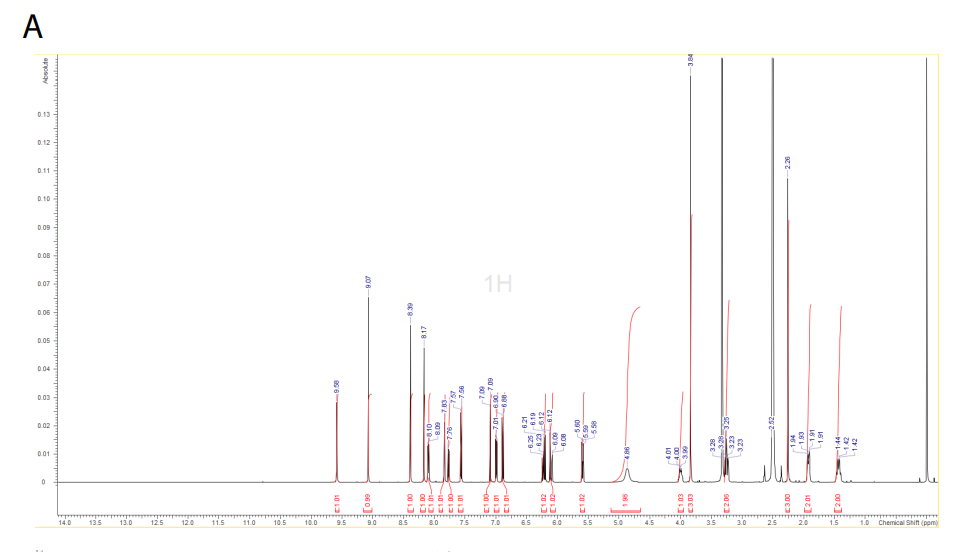
A) 1H NMR spectrum of zongertinib
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021213800&_cid=P10-ME52KD-62836-1
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
- [1]. WHO Drug Informat ion – World Health Organization (WHO).[2]. Wilding Birgit, et al. Synthesis of diazino-pyrimidines as anticancer agents: World Intellectual Property Organization, WO2021213800. 2021-10-28.[3]. Li S, et al. Emerging Targeted Therapies in Advanced Non-Small-Cell Lung Cancer. Cancers (Basel). 2023 May 24;15(11):2899. [Content Brief]
////////////Zongertinib, Hernexeos, APPROVALS 2025, FDA 2025, lung cancer, BI-1810631, BI1810631, DRH7R67UVL
Olverembatinib

Olverembatinib
1257628-77-5- 3-((1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl)-4-methyl-N-(4-((4-methylpiperazin-1-yl)methyl)-3-(trifluoromethyl)phenyl)benzamide
- HQP1351
- 4-methyl-N-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[2-(1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl]benzamide
- HQP1351 is under investigation in clinical trial NCT03883100 (A Pivotal Study of HQP1351 in Patients of Chronic Myeloid Leukemia in Accelerated Phase With T315I Mutation).
- 4-methyl-N-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[2-(1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl]benzamide
- D-824
- GZD824
WeightAverage: 532.571
Monoisotopic: 532.219844002, Chemical FormulaC29H27F3N6O
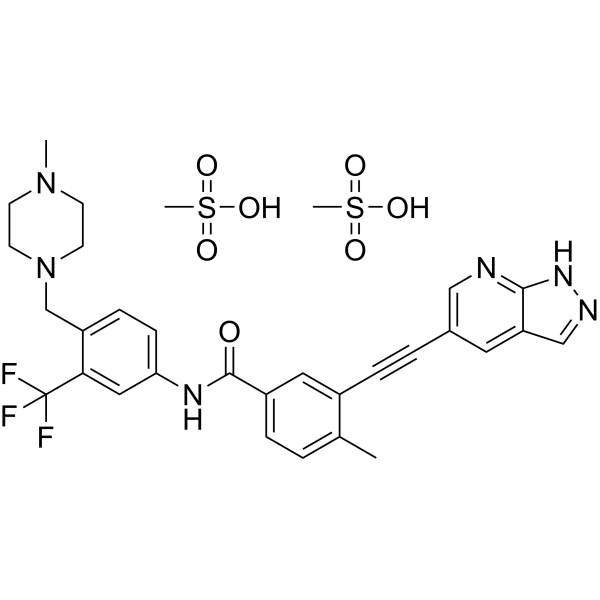
| Molecular Weight | 724.77 |
|---|---|
| Formula | C31H35F3N6O7S2 |
Olverembatinib (GZD824) dimesylate is a potent and orally active pan-Bcr-Abl inhibitor. Olverembatinib dimesylate potently inhibits a broad spectrum of Bcr-Abl mutants. Olverembatinib dimesylate strongly inhibits native Bcr-Abl and Bcr-AblT315I with IC50s of 0.34 nM and 0.68 nM, respectively. Olverembatinib dimesylate has antitumor activity. Olverembatinib (dimesylate) is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Olverembatinib is a BCR-ABL1 tyrosine kinase inhibitor developed by Ascentage Pharma. In 2021, it was approved in China “for the treatment of adult patients with TKI-resistant chronic-phase CML (CML-CP) or accelerated-phase CML (CML-AP) harbouring the T315I mutation”.[1][2][3]
SYN
Ren, Xiaomei;Pan, Xiaofen;Zhang, Zhang;Wang, Deping;Lu, Xiaoyun;Li, Yupeng;Wen, Donghai;Long, Huoyou;Luo, Jinfeng;Feng, Yubing;Zhuang, Xiaoxi;Zhang, Fengxiang;Liu, Jianqi;Leng, Fang;Lang, Xingfen;Bai, Yang;She, Miaoqin;Tu, Zhengchao;Pan, Jingxuan;Ding, Ke [Journal of Medicinal Chemistry,2013,vol. 56,# 3,p. 879 – 894]
https://pubs.acs.org/doi/10.1021/jm301581y
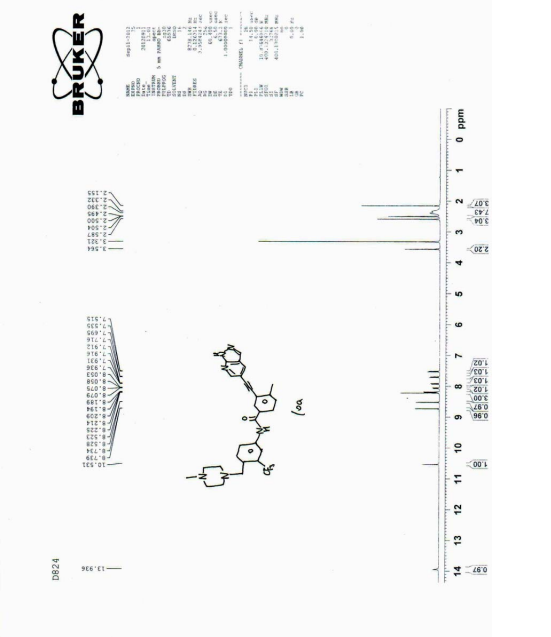
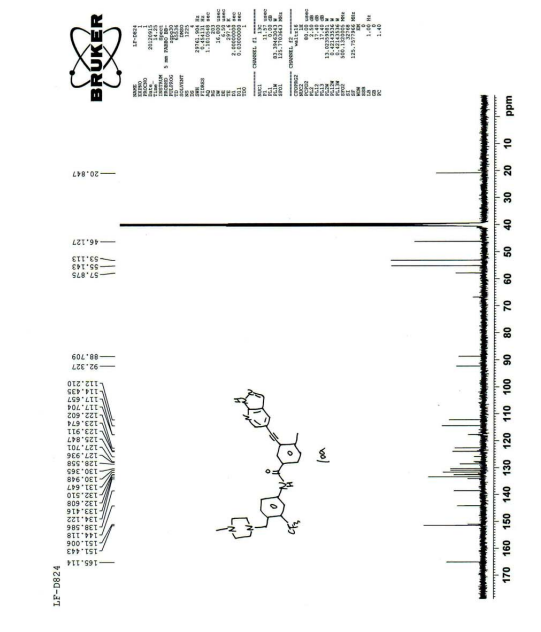
PATENT
CN 114163434
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN355399053&_cid=P10-MDPKRT-75688-1
| Example |
| The following examples further illustrate but do not limit the present invention. It should be noted that those skilled in the art can make various modifications and improvements without departing from the inventive concept of the present invention, all of which are included in the scope of protection of the present invention. |
| The specific conditions not disclosed in the experimental methods of the following examples can be selected according to conventional methods and conditions, or according to the product instructions. |
| Unless otherwise specified, “room temperature” in the following examples refers to 20°C to 25°C. The term “h” used herein refers to hours. |
| Example 1 |
| Step 1: |
| |
| Under nitrogen, N-methylpyrrolidone (137.6 g) was heated to 30-35°C to obtain the compound of Formula 1 (14.4 g, 1.3 eq) and the compound of Formula 2 (19.14 g, 1 eq). Bis(triphenylphosphate)palladium dichloride (0.46 g, 0.01 eq) and cuprous iodide (0.113 g, 0.01 eq) were added sequentially. Triethylamine (9.45 g, 1.5 eq) was then added under nitrogen. The reaction mixture was heated to 65-75°C and maintained at this temperature for 2 hours. The reaction process was monitored by liquid chromatography-mass spectrometry. The reaction was terminated when the content of the compound of Formula 2 was ≤0.1%. After completion of the reaction, the reaction solution was cooled to 35-45°C and N-acetyl-L-cysteine (1 g, 0.1 eq) was added directly. The reaction was stirred for 4-5 hours. The resulting product was cooled to room temperature, precipitated with water, centrifuged, and washed with pure water to obtain a crude filter cake. The crude filter cake was vacuum-dried and then slurried with a mixture of ethyl acetate and n-heptane (5 mL of the mixed solvent, wherein the volume ratio of ethyl acetate to n-heptane was 1:1) at a rate of 5 mL per gram of crude filter cake. The resulting slurry was vacuum-dried to yield the compound of Formula 3 with a yield of 85.97% and a purity of 98.2%. |
| The NMR data for the compound of Formula 3 are as follows : 1 H NMR (400 MHz, d-DMSO): δ ppm: 8.93 (1H, d, J = 2.0 Hz); 8.63 (1H, d, J = 2.0 Hz); 8.49 (1H, s); 8.11 (1H, d, J = 2.0 Hz); 7.92 (1H, dd, J = 1.6 Hz; J = 8.0 Hz); 7.52 (1H, d, J = 8.0 Hz); 3.88 (3H, s); 2.59 (3H, s); 1.65 (9H, s). |
| Step 2: |
| |
| Under nitrogen, methanol (160 g) and water (50 g) were sequentially added to the compound of formula 3 (20 g, 1.0 eq). The reaction system was stirred at reflux for 18 hours with process control. The resulting product was cooled to room temperature and filtered to obtain a filter cake (no drying required). Recrystallization was performed by adding 10 times the mass of the filter cake in methanol. The resulting mixture was stirred at 60-70°C for 8-10 hours, then cooled to 40-50°C and subjected to a gradient cooling process at a cooling rate of 5°C per 1 to 1.5 hours to slowly form a solid precipitate. The resulting mixture was filtered, the filter cake was washed with methanol, and vacuum dried to obtain the compound of formula 4 in a 91% yield and 99.7% purity. |
| The NMR data for the compound of Formula 4 are as follows : 1 H NMR (400 MHz, d-DMSO): δ ppm: 8.73 (1H, d, J = 2.0 Hz); 8.52 (1H, t, J = 2.0 Hz); 8.21 (1H, d, J = 2.0 Hz); 8.06 (1H, s); 7.86 (1H, dd, J1 = 2.0 Hz; J2 = 8.0 Hz); 7.49 (1H, dd, J1 = 1.6 Hz; J2 = 7.6 Hz); 3.86 (3H, s); 2.56 (3H, s). |
| Step 3: |
| |
| Under nitrogen, THF (448 mL), compound of formula 4 (29.1 g, 1 eq), and compound of formula 5 (24.6 g, 0.9 eq) were added, stirred, and cooled to -65°C to -60°C. At this temperature, potassium tert-butoxide (19 g x 3) was added in batches every 0.5 h. The reaction process was controlled by liquid phase detection. After 2 hours, the reaction temperature was raised to -5 to 0°C. The reaction solution was washed with purified water, stirred for 0.5-1 hour, washed with brine, and separated to obtain an organic phase. N-acetyl-L-cysteine (11.41 g, 0.7 eq) was added to the organic phase, stirred, washed with brine, neutralized, and concentrated under reduced pressure. The resulting filter cake was washed with purified water and made into a slurry. The resulting product was washed again with purified water and dried under vacuum to obtain compound of formula 6 with a yield of 88.2% and a purity of 98.6%. |
| The NMR data for the compound of formula 6 are as follows : 1 H NMR (400 MHz, d-DMSO): δ ppm: 10.53 (1H, s); 8.75 (d, J = 2.0); 8.53 (d, J = 2.4); 8.24 (1H, s); 8.23 (d, J = 2.4); 8.21 (d, J = 1.6); 8.09 (dd, J1 = 1.6; J2 = 8.4); 7.94 (dd, J1 = 2.0; J2 = 8.0); 7.71 (d, J = 8.8); 7.53 (d, J = 8.0); 3.56 (2H, s); 2.59 (3H, s); 2.34-2.35 (8H, m), 2.16 (3H, s). |
| Its carbon spectrum data are 13 C NMR (100 MHz, d-DMSO): δ ppm: 20.38, 45.65, 52.64, 54.67, 57.41, 88.26, 91.86, 111.76, 113.98, 117.19, 122.14, 123.43, 127.35 (q), 124.30 (q), 128.10, 129.89, 130.49, 131.15, 132.02, 132.13, 132.93, 133.66, 138.15, 143.65, 150.55, 164.64. |
PATENT
CN 101885722
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN84081329&_cid=P10-MDPKML-68458-1
| Example 23 |
| 3-((1H-pyrazolo[3,4-b]pyridine-5-substituted)ethynyl)-4-methyl-N-(4-((4-methylpiperazine-1-substituted)methyl)3-(trifluoromethyl)phenyl)benzamide (D824) |
| (3-((1H-pyrazolo[3,4-b]pyridin-5-yl)ethynyl)-4-methyl-N-(4-((4-methylpiperazin-1-yl)m ethyl)-3-(trifluoromethyl)phenyl)benzamide) |

| The synthesis method is the same as in Example 1. |
| 1 HNMR (400MHz, d-DMSO), δ13.92 (s, 1H), 10.55 (s, 1H), 8.72 (d, J=2.0Hz, 1H), 8.52 (d, J=2.0Hz, 1H), 8.17 (m, 3H), 8.10 (d, J=8.0Hz, 1H), 7.92 (dd, J=8.0, 2.0Hz, 1H), 7.70 (d, J=8.8Hz, 1H), 7.53 (d, J=8.0Hz, 1H), 3.80 (s, 2H), 3.10 (brs, 8H), 2.71 (s, 3H), 2.57 (s, 3H). |
| MS(ESI), m/z: 533, (M + +H + ). |
SYN
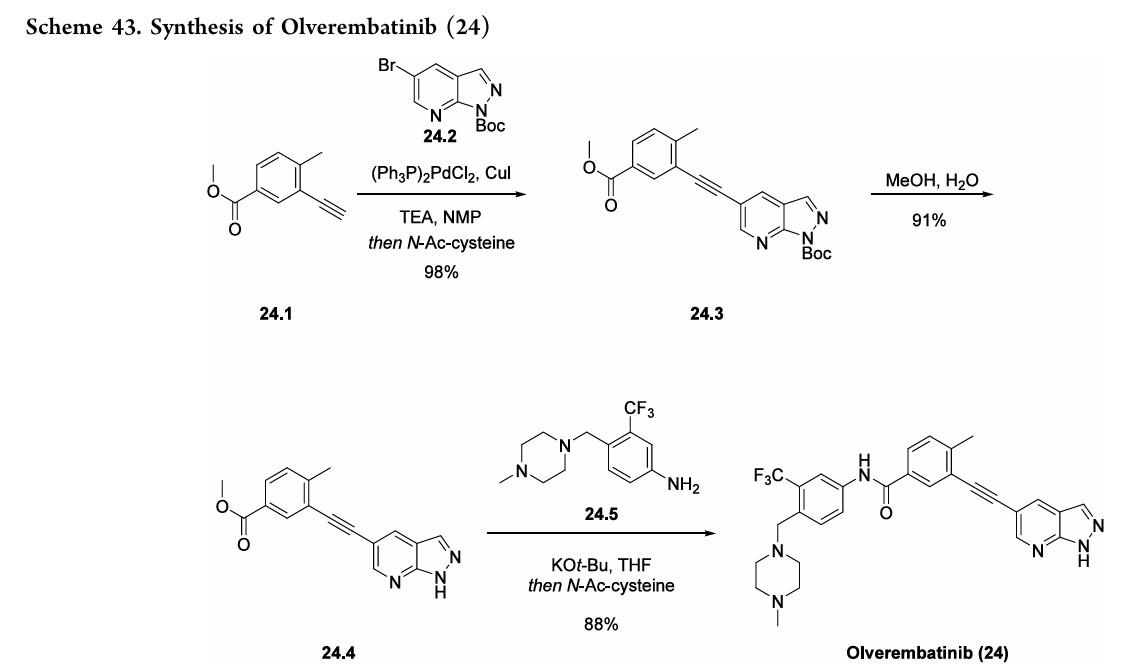
Olverembatinib(24) wasdeveloped by Ascentage Pharma as anorally available, third-generation
tyrosinekinase inhibitor (TKI) for the treatment of chronic myeloid leukemia (CML), acute myeloid leukemia, acute lymphoblastic leukemia (ALL), and solid tumors.167 It received its first approval inChina inNovember 2021 and was approved for use in adults with TKI-resistant CML chronicphaseandCML-acceleratephaseharboringtheT315I “gatekeeper” mutation.168 The current mainstay of CML
treatmentiscenteredaroundTKIs;however,resistancetoTKItherapy, often through BCR-ABL1 kinase domain point mutations, remains a challenge for early generation therapies.169Olverembatinibretainsitsefficacybyfunctioningasan ATP-bindingsiteinhibitorofwild-typeBCR-ABL1kinaseand broadly relatedmutants including T315I, which otherwise confers resistance against all first and second generation TKIs.168
Thesynthesisofolverembatinibhasbeenreportedinseveral patents,170−172 aswell as a journal article173 that details the divergentapproachtorelatedanalogues. Inarecentpatent,170 the synthesis of olverembatinib began with a Sonogashira coupling of commercially available alkyne 24.1 with
bromopyridine24.2toaffordester24.3in98%yield(Scheme43). Cleavage of the N-Boc group was accomplished by refluxingcarbamate24.3inaMeOHandwatermixturetogive pyrazole24.4 in91%yield. AfinalKOtBumediatedamide formation with aniline 24.5 resulted in the isolation of
olverembatinib(24) in88%yield.
(167) Dhillon, S. Olverembatinib: First approval. Drugs 2022, 82,
469−475.
(168) Braun, T. P.; Eide, C. A.; Druker, B. J. Response and resistance
to BCR-ABL1-targeted therapies. Cancer Cell 2020, 37, 530−542.
(169) Shoukier, M.; Kubiak, M.; Cortes, J. Review of new-generation
tyrosine kinase inhibitors for chronic myeloid leukemia. Curr. Oncol.
Rep. 2021, 23, 91.
(170) Wen, J.; Feng, J.; Wu, T.; Cai, M.; Teng, S. Preparation
method of alkynyl containing compound and its intermediate. China
Patent CN 114163434, 2022.
(171) Guo, M.; Wen, J.; Teng, S.; Wu, T.; Feng, J. Preparation of
(trifluoromethylphenyl)(pyrazolo[3,4-b]pyridinylethynyl)benzamide
derivative. China Patent CN 113292556, 2021.
(172) Ding, K.; Wang, D.; Pei, D.; Zhang, Z.; Shen, M.; Luo, K.;
Feng, Y. Heterocyclic alkynylbenzene derivatives as cancer cell line
inhibitors and their preparation, pharmaceutical compositions and use
in the treatment of cancer. China Patent CN 101885722, 2010.
(173) Ren, X.; Pan, X.; Zhang, Z.; Wang, D.; Lu, X.; Li, Y.; Wen, D.;
Long, H.; Luo, J.; Feng, Y.; et al. Identification of GZD824 as an
orally bioavailable inhibitor that targets phosphorylated and non
phosphorylated breakpoint cluster region−abelson (Bcr-Abl) kinase
and overcomes clinically acquired mutation-induced resistance against
imatinib. J. Med. Chem. 2013, 56, 879−894.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Dhillon, Sohita (March 2022). “Olverembatinib: First Approval”. Drugs. 82 (4): 469–475. doi:10.1007/s40265-022-01680-9. PMID 35195876. S2CID 247027755.
- Jiang, Qian; Li, Zongru; Qin, Yazhen; Li, Weiming; Xu, Na; Liu, Bingcheng; Zhang, Yanli; Meng, Li; Zhu, Huanling; Du, Xin; Chen, Suning; Liang, Yang; Hu, Yu; Liu, Xiaoli; Song, Yongping; Men, Lichuang; Chen, Zi; Niu, Qian; Wang, Hengbang; Lu, Ming; Yang, Dajun; Zhai, Yifan; Huang, Xiaojun (18 August 2022). “Olverembatinib (HQP1351), a well-tolerated and effective tyrosine kinase inhibitor for patients with T315I-mutated chronic myeloid leukemia: results of an open-label, multicenter phase 1/2 trial”. Journal of Hematology & Oncology. 15 (1): 113. doi:10.1186/s13045-022-01334-z. PMC 9389804. PMID 35982483.
- Jiang, Qian; Huang, Xiaojun; Chen, Zi; Niu, Qian; Shi, Dayu; Li, Zongru; Hou, Yue; Hu, Yu; Li, Weiming; Liu, Xiaoli; Xu, Na; Song, Yongping; Zhang, Yanli; Meng, Li; Hong, Zhenya; Liu, Bingcheng; Zeng, Shan; Men, Lichuang; Li, Yan; Chen, Suning; Xue, Mengxing; Zhu, Huanling; Li, He; Du, Xin; Lou, Jin; Zhang, Xiaohan; Liang, Yang; Dai, Yujun; Lu, Ming; Wang, Hengbang; Ji, Jiao; Yue, Changai; Yang, Dajun; Zhai, Yifan (5 November 2020). “Novel BCR-ABL1 Tyrosine Kinase Inhibitor (TKI) HQP1351 (Olverembatinib) Is Efficacious and Well Tolerated in Patients with T315I-Mutated Chronic Myeloid Leukemia (CML): Results of Pivotal (Phase II) Trials”. Blood. 136 (Supplement 1): 50–51. doi:10.1182/blood-2020-142142. S2CID 228875477.
| Clinical data | |
|---|---|
| Other names | GZD-824; GZD824 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1257628-77-5 |
| PubChem CID | 51038269 |
| IUPHAR/BPS | 10630 |
| DrugBank | DB16185 |
| ChemSpider | 29395146 |
| UNII | KV1M7Q3CBP |
| ChEMBL | ChEMBL2316582 |
| CompTox Dashboard (EPA) | DTXSID301352011 |
| Chemical and physical data | |
| Formula | C29H27F3N6O |
| Molar mass | 532.571 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
[1]. Ren X, Pan X, Zhang Z, Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated breakpoint cluster region-Abelson (Bcr-Abl) kinase and overcomes clinically acquired mutation-induced resistance against imatinib. J Med Chem. 2013 Feb 14;56(3):879-94. [Content Brief]
//////////Olverembatinib, approvals 2021, china 2021, Ascentage Pharma, cancer, HQP1351, HQP 1351, D-824, D 824, KV1M7Q3CBP, GZD824
Sontigidomide
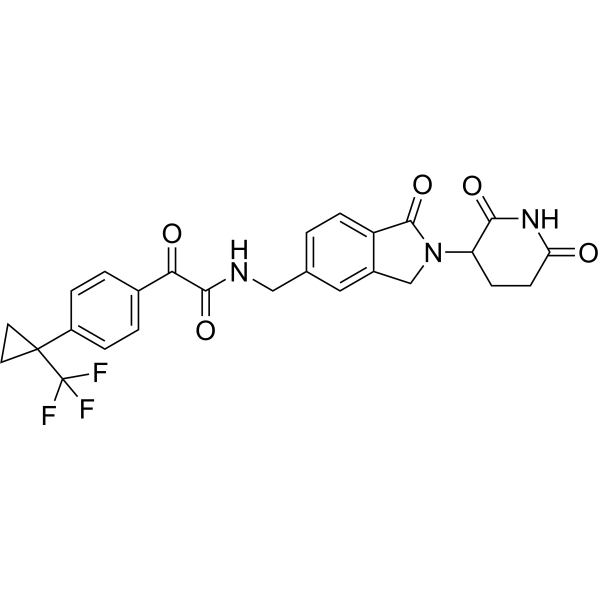
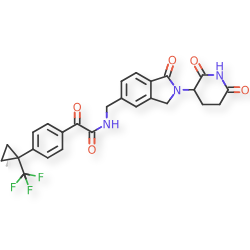
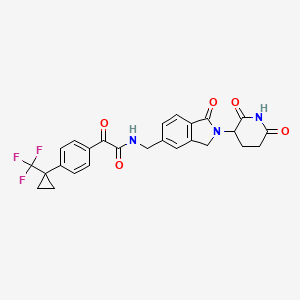
Sontigidomide
CAS 2560577-69-5
| Molecular Weight | 513.47 |
|---|---|
| Formula | C26H22F3N3O5 |
N-[[2-(2,6-Dioxo-3-piperidinyl)-2,3-dihydro-1-oxo-1H-isoindol-5-yl]methyl]-α-oxo-4-[1-(trifluoromethyl)cyclopropyl]benzeneacetamide
enzeneacetamide, N-[[2-(2,6-dioxo-3-piperidinyl)-2,3-dihydro-1-oxo-1H-isoindol-5-yl]methyl]-α-oxo-4-[1-(trifluoromethyl)cyclopropyl]-
FDD2NVW84X, Sontigidomida
Sontigidomide (Compound 5) is an antineoplastic compound. Sontigidomide inhibits MOLM-13 cell proliferation more than 80% at 1 μM (3 days).
SCHEME
COUPLER………….
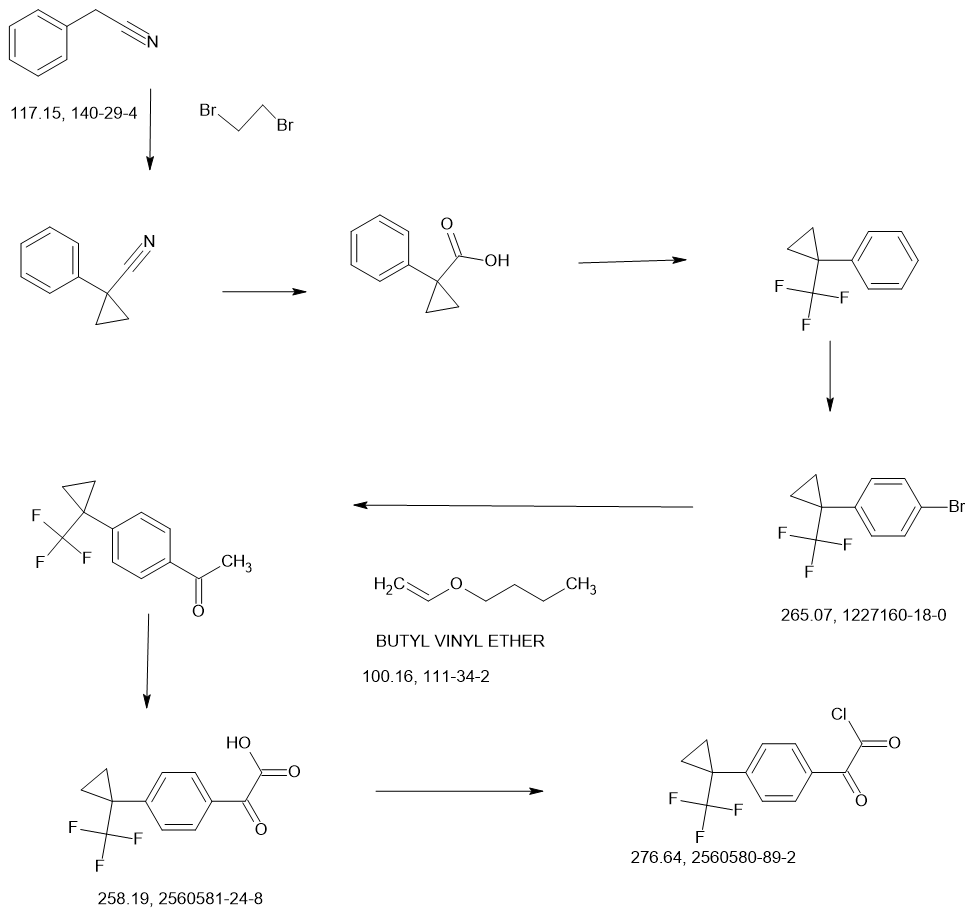
MAIN……….
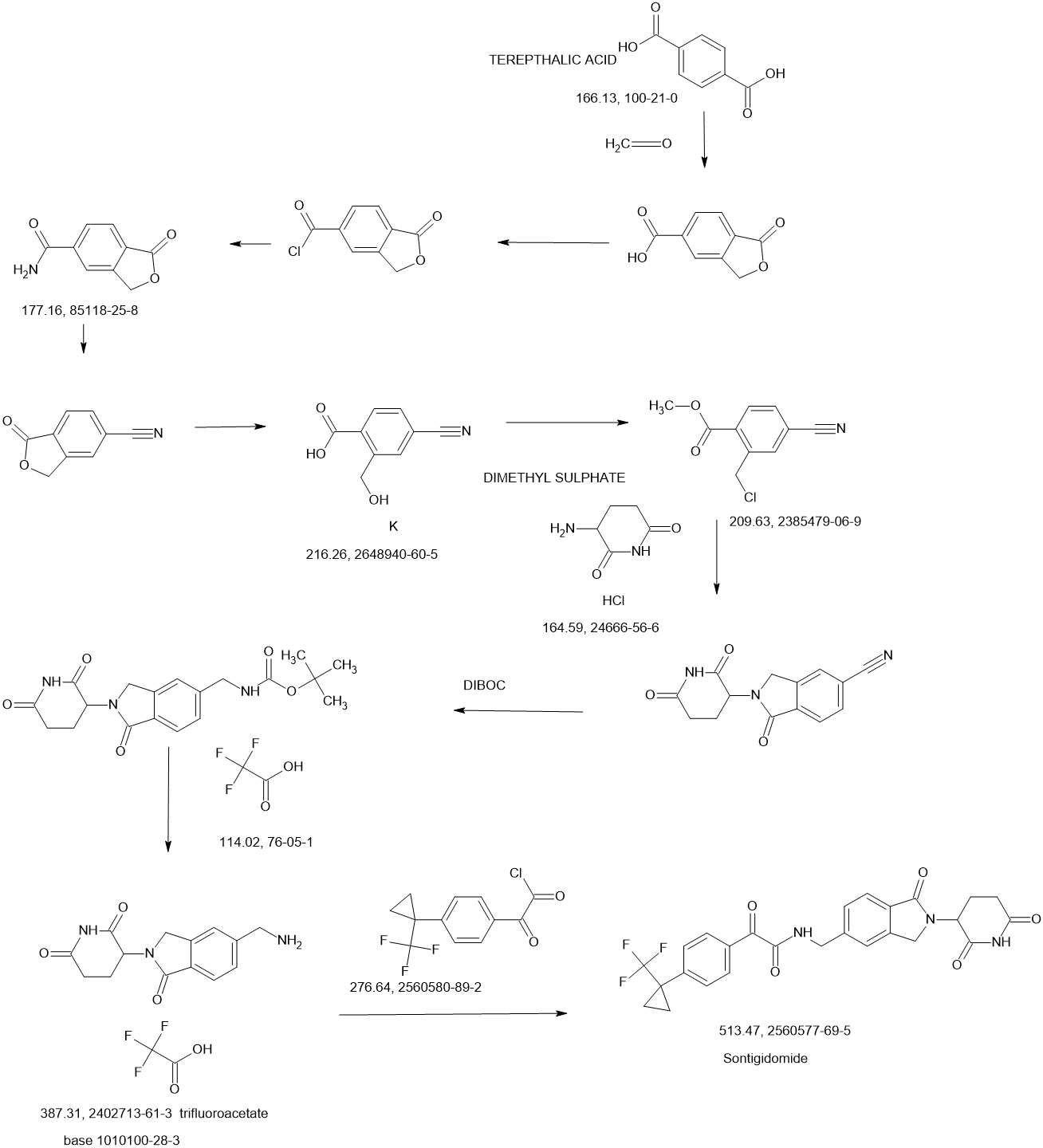
PATENTS
WO2023070120 BioTheryX, Inc.
PATENT
US20200369679
https://patentscope.wipo.int/search/en/detail.jsf?docId=US311579044&_cid=P20-MD87Y5-18242-1
Example 5
Compound I-5: N-((2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-5-yl)methyl)-2-oxo-2-(4-(1-(trifluoromethyl)cyclopropyl)phenyl)acetamide
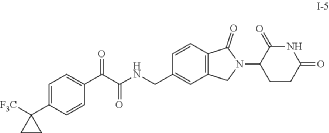
Compound I-5 was synthesized as shown in Scheme 5.
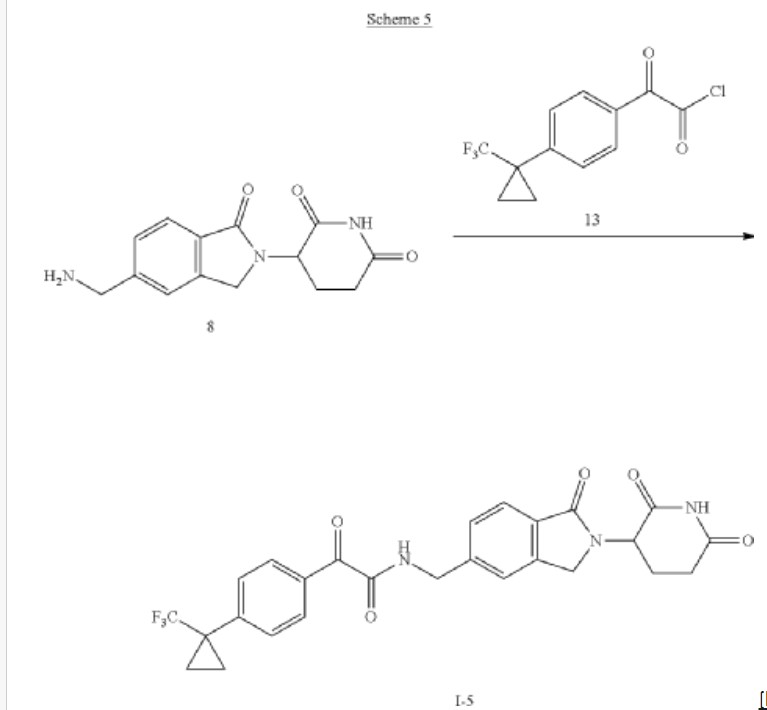
To a solution of 3-(5-(aminomethyl)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 8 (80 mg, 0.258 mmol) in DCM (4 mL) at 0° C. was added TEA (52.2 mg, 0.516 mmol). After stirring for 2 min, 2-oxo-2-(4-(1-(trifluoromethyl)cyclopropyl)phenyl)acetyl chloride 13 (71.3 mg, 0.258 mmol) was added and the mixture was stirred at RT for 2 h. After concentration, the residue was purified using prep-HPLC eluting with ACN/H 2O (0.1% TFA) from 10% to 95% to afford compound I-5 (16.1 mg) in 12% yield. MS (ESI) m/z: 514.0 [M+H] +; 1H NMR (400 MHz, DMSO-d 6) δ 10.98 (s, 1H), 9.57 (t, J=6.0 Hz, 1H), 8.03-8.01 (m, 2H), 7.74-7.48 (m, 7H), 5.13-5.09 (m, 1H), 4.59-4.57 (m, 2H), 4.49-4.31 (m, 2H), 2.95-2.87 (m, 1H), 2.63-2.58 (m, 1H), 2.45-2.38 (m, 1H), 2.03-1.99 (m, 1H), 1.43-1.40 (m, 2H), 1.24-1.21 (m, 2H).
- Ketoamides for treating malignancyPublication Number: WO-2023070120-A1Priority Date: 2021-10-22
- Protein-targeting compounds and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: US-2020369679-A1Priority Date: 2019-05-24
- Compounds targeting proteins and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: WO-2020242960-A1Priority Date: 2019-05-24
- Compounds targeting proteins and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: AU-2020283744-A1Priority Date: 2019-05-24
- Targeted protein compound, its pharmaceutical composition and therapeutic applicationPublication Number: CN-114502543-APriority Date: 2019-05-24
- Compounds targeting proteins and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: EP-3976623-A1Priority Date: 2019-05-24
- Protein targeting compounds, pharmaceutical compositions thereof and therapeutic applications thereofPublication Number: KR-20220023343-APriority Date: 2019-05-24
- Protein-targeting compounds and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: US-11345712-B2Priority Date: 2019-05-24Grant Date: 2022-05-31
- Protein-targeting compounds and pharmaceutical compositions thereof, and their therapeutic applicationsPublication Number: US-2022298172-A1Priority Date: 2019-05-24
////////Sontigidomide, FDD2NVW84X, CANCER, Sontigidomida
..
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Alflutinib, Furmonertinib, Firmonertinib



FIRMOMERTINIB, Furmonertinib, Alflutinib
CAS 1869057-83-9
, AST 2818, UNII-A49A7A5YN4
N-[2-[[2-(Dimethylamino)ethyl]methylamino]-5-[[4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]-6-(2,2,2-trifluoroethoxy)-3-pyridinyl]-2-propenamide
N-[2-[2-(dimethylamino)ethyl-methylamino]-5-[[4-(1-methylindol-3-yl)pyrimidin-2-yl]amino]-6-(2,2,2-trifluoroethoxy)pyridin-3-yl]prop-2-enamide
C28H31F3N8O2 568.6 g/mol
2-Propenamide, N-[2-[[2-(dimethylamino)ethyl]methylamino]-5-[[4-(1-methyl-1H-indol-3-yl)-2-pyrimidinyl]amino]-6-(2,2,2-trifluoroethoxy)-3-pyridinyl]-
Alflutinib is under investigation in clinical trial NCT03452592 (Efficacy and Safety of Alflutinib in Locally Advanced or Metastatic Non-small Cell Lung Cancer Patients With T790M).
Firmonertinib is an orally available selective inhibitor of the epidermal growth factor receptor (EGFR) mutant form T790M, with potential antineoplastic activity. Upon administration, firmonertinib specifically binds to and inhibits the tyrosine kinase activity of EGFR T790M, a secondarily acquired resistance mutation. This prevents EGFR T790M-mediated signaling and leads to cell death in EGFR T790M-expressing tumor cells. EGFR, a receptor tyrosine kinase that is mutated in many tumor cell types, plays a key role in tumor cell proliferation and tumor vascularization. Compared to some other EGFR inhibitors, alflutinib may have therapeutic benefits in tumors with T790M-mediated drug resistance.
FIRMONERTINIB is a small molecule drug with a maximum clinical trial phase of III (across all indications) and has 4 investigational indications.
SCHEME

CONTD……..

REF
https://patentscope.wipo.int/search/en/detail.jsf?docId=US201062358&_cid=P22-MBFXFH-62339-1
Example 3: N-{2-{[2-(dimethylamino)ethyl](methyl)amino}-6-(2,2,2-trifluoroethoxyl)-5-{[4-(1-methyl-H-indol-3-yl)pyrimidin-2-yl]amino}pyridin-3-yl}acrylamide
Step 1: Synthesis of N2-methyl-N2-[2-(dimethylamino)ethyl]-6-(2,2,2-trifluoroethoxyl)-N5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]-3-nitropyridin-2,5-diamine
| The compound was synthesized in the same manner as those in Step 1 of Example 1 with a yield of 86%. MS m/z: 545 [M+1]. |
Step 2: Synthesis of N2-methyl-N2-[2-(dimethylamino)ethyl]-6-(2,2,2-trifluoroethoxyl)-N5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]pyridin-2,3,5-triamine
| The compound was synthesized in the same manner as those in Step 2 of Example 2 with a yield of 56%. MS m/z: 515 [M+1]. |
Step 3: Synthesis of N-{2-{[2-(dimethylamino)ethyl](methyl)amino}-6-(2,2,2-trifluoroethoxyl)-5-{[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-yl]amino}pyridin-3-yl}acrylamide
| The compound was synthesized in the same manner as those in Step 3 of Example 1 with a yield of 23%. MS m/z: 569 [M+1]. |
PATENT
CN110606842
https://patentscope.wipo.int/search/en/detail.jsf?docId=CN280196686&_cid=P22-MBFXJY-67679-1
Patent application CN105315259A protects the compound of formula I and discloses its preparation method as follows:



| Example 1: Preparation of 6-chloro-3-nitro-2-(2,2,2-trifluoroethoxy)pyridine (XI-1) |
| Add toluene (24.0L) to the reactor, then add 2,6-dichloro-3-nitropyridine (3000g, 15.54mol), adjust the internal temperature between -20℃ and -10℃, and add sodium hydrogen (933g, 23.33mol) in batches. Add 2,2,2-trifluoroethanol (1586g, 16.00mol) toluene (6.0L) solution dropwise. React for 2h, and monitor the reaction end point by TLC and HPLC. After the reaction is completed, add 10% ammonium chloride solution (6.0L) dropwise. Let stand and separate. Wash the organic phase with water (6.0L) and concentrate under reduced pressure. Add ethyl acetate (0.3L), heat to 40-50℃, add n-heptane (2.7L) dropwise, cool to -15 to -5℃ after dripping, and continue crystallization for 3 hours, and filter with suction. Obtain 3017g of product solid, with a yield of 75.65%. |
| 1H NMR(500MHz,DMSO-d6)δ8.60(d,J=8.0Hz,1H),7.50(d,J=8.5Hz,lH),5.13(q,J=9.0Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ153.20,151.09,139.34,132.67,123.38(q,J=277.2Hz),119.14,63.34(q,J=36Hz); |
| MS m/z:256.99[M+1]。 |
| Example 2: Preparation of 6-chloro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (X-1) |
| At room temperature, add acetonitrile (21.0L) and water (21.0L) to the reactor, start stirring, add 6-chloro-3-nitro-2-(2,2,2-trifluoroethoxy)pyridine (3017.0g, 11.76mol) obtained in Example 1, and add hydrosulfite (15.1Kg, 70.54mol). Control the temperature at 27-33°C to react for 2 hours. Add 36% concentrated hydrochloric acid (11.9Kg, 117.60mol) dropwise, and continue to react for 1.5 hours. Add solid sodium bicarbonate (12.8Kg, 12.96mol). Filter, separate the mother liquor, wash the organic phase with saturated brine (21.0L), and concentrate under reduced pressure to obtain an oily substance. Theoretically calculated for the next step reaction. |
| 1H NMR(500MHz,DMSO-d6)δ7.03(d,J=8.0Hz,1H),6.90(d,J=8.0Hz,1H),5.21(s,2H),4.93(q,J=9.0Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ148.16,131.72,130.55,123.93(q,J=278.5Hz),121.02,118.42,61.72(q,J=34.0Hz); |
| MS m/z:227.01[M+1]。 |
| Example 3: Preparation of 6-chloro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (IX-1) |
| At room temperature, dichloromethane (10.4 L) was added to the reaction kettle, stirring was started, 6-chloro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (2664 g, 11.76 mol) obtained in Example 2 was added, diisopropylethylamine (2279 g, 17.64 mol) was added, the temperature was controlled at -15 to -10°C, a dichloromethane (5.2 L) solution of trifluoroacetic anhydride (2963 g, 14.11 mol) was added dropwise, and stirring was continued for 20 minutes after the addition was completed. Water (13.0 L) was added dropwise, the layers were separated, the organic phase was concentrated under reduced pressure, and the next step reaction was theoretically calculated. |
| 1 H NMR(400MHz,DMSO-d6)δ11.23(s,7H),7.95(d,J8.0Hz,1H),7.34(d,J8.0Hz,1H),5.03(q,J8.9Hz,2H) |
| 13C NMR(101MHz,DMSO-d6)δ155.74(q,J=46.6Hz),155.60,145.37,140.24,124.01(q,J=278.8Hz),119.07,118.30,116.19(q,J=289.9Hz),62.99(q,J=35.4Hz); |
| MS m/z.322.99[M+1]。 |
| Example 4: Preparation of 6-chloro-5-nitro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (VIII-1) |
| At room temperature, concentrated sulfuric acid (11.7 L) was added to the reaction kettle, stirring was started, 6-chloro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (3.9 Kg, 11.76 mol) obtained in Example 3 was added, and potassium nitrate solid (1783.4 g, 17.64 mol) was added in batches. After the addition, stirring was continued for about 40 minutes. After monitoring the reaction, the temperature was lowered to control the internal temperature at 10-25°C, and dichloromethane (27.3 L) was added dropwise. Stirring was continued, stirring was continued for 45 minutes, and the layers were separated. The organic phase was taken and washed once with water (11.7 L). The organic phase was concentrated under reduced pressure and theoretically calculated for the next step reaction. |
| 1H NMR(500MHz,DMSO-d6)δ11.58(s,1H),8.78(s,1H),5.17(q,J=8.7Hz,2H); |
| 13C NMR(126MHz,DMSO-d6)δ155.89,155.43(q,J=37.8Hz),138.84,138.57,135.05,123.22(q,J=273.4Hz),118.47,115.51(q,J=278.5Hz),63.65(q,J=35.3Hz); |
| MS m/z:367.98[M+1]。 |
| Example 5: Preparation of 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (VII-1) |
| At room temperature, methanol (13.0 L) was added to the reactor, 6-chloro-5-nitro-3-(2,2,2-trifluoroacetamido)-2-(2,2,2-trifluoroethoxy)pyridine (4322 g, 11.76 mol) obtained in Example 4 was added, p-toluenesulfonic acid monohydrate (3355 g, 17.64 mol) was added, the temperature was controlled at 60-65°C for 15 hours, and the methanol was removed under reduced pressure. Methyl tert-butyl ether (13.0 L) and water (6.5 L) were added, and the pH was adjusted to 7-8 with potassium carbonate. Layering was performed, the organic phase was washed once with water (8.6 L), separated, and concentrated under reduced pressure. n-heptane (21.5 L) was added, the temperature was controlled at 60-65°C and stirred for 1 hour, cooled to room temperature, filtered, and the filter cake was dried with air at 50°C for 18 hours to obtain 1475 g of the product. |
| The total yield of the five-step reaction from Example 1 to Example 5 is 34.9%. |
| 1H NMR(500 MHz,DMSO-d6)δ7.62(s,1H),5.92(s,2H),5.05(q,J=8.9Hz,2H). |
| 13C NMR(126MHz,DMSO-d6)δ149.30,139.53,132.84,123.46,123.44(q,J=278.5Hz),116.25,62.52(q,J=35.3Hz); |
| MS m/z:272.00[M+1]。 |
| Example 6: Preparation of 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (V-1) |
| Toluene (50 mL) was added to a 100 mL reaction bottle, and the compound of formula VII-1, 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (5.0 g, 18.4 mmol), the compound of formula VI, 3-(2-chloropyrimidin-4-yl)-1-methyl-1H-indole (5.8 g, 23.8 mmol), p-toluenesulfonic acid monohydrate (1.8 g, 9.2 mmol) were added in sequence, and the reaction mixture was heated to 110-115°C and reacted for 24 hours. The temperature was lowered to 22°C, filtered by suction, and the filter cake was dried at 50°C for 20 hours to obtain the compound of formula V-1, 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (10.4 g, 74.7 HPLC area% purity). According to the HPLC purity conversion, the next step reaction was carried out. |
| 1H NMR(400MHz,DMSO-d6)δ9.43(s,1H),8.76(s,1H),8.46-8.45(d,J=5.4Hz,1H),8.39(s,1H),8.38-8.36(d,J=7.8Hz,1H),7.57-7.55(d,J=8.2Hz,1H),7.41-7.40(d,J=5.4Hz,1H),7.31-7.27(t,J=7.5Hz,1H),7.20-7.16(t,J=7.5Hz,1H),5.23-5.16(q,J=8.8Hz,2H),3.90(s,3H); |
| MS m/z:479.08[M+1]。 |
| Example 7: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Add N,N-dimethylformamide (30 mL) to a 250 mL reaction bottle, add the compound of formula V-1 obtained in Example 6, 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (10.4 g, 16.22 mmol), stir, add potassium carbonate (4.48 g, 32.44 mol), N,N,N’-trimethylethylenediamine (2.48 g, 24.33 mol) in sequence, heat the reaction mixture to 77-82°C, keep warm for 1-1.5 hours. Add water (60 mL), and cool to room temperature after addition. Filter by suction, transfer the filter cake to a 50 L reactor, add acetonitrile (40 mL), and heat to reflux for 2 hours. The mixture was cooled to room temperature and filtered with suction. The filter cake was dried at 50°C for 18 hours to give a compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (6.7 g). The total yield of the two-step reaction with Example 6 was 66.8%. |
| 1H NMR(500MHz,DMSO-d6)δ8.62(s,1H),8.41(s,1H),8.26(s,2H),8.24(s,1H),7.48(d,J=8.2Hz,1H),7.21(t,J=7.6Hz,1H),7.16(d,J=5.3Hz,1H),7.05(t,J=7.3Hz,1H),5.04(q,J=8.9Hz,2H),3.84(s,3H),3.69(t,J=6.9Hz,2H),2.89(s,3H),2.55(t,J=6.9Hz,2H),2.17(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ162.15,160.55,156.99,154.98,148.42,137.53,132.83,132.68,125.50,123.58(q,J=279.7Hz),124.38,122.11,122.06,120.67,113.38,112.27,110.30,107.11,62.14(q,J=35.3Hz),56.10,49.51,45.34,45.33,39.35,32.98。 |
| MS m/z.:545.22[M+1]。 |
| Example 8: Preparation of 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (V-1′) |
| Toluene (7.43 L) was added to a 20 L reactor, and compound VII-1 6-chloro-5-nitro-3-amino-2-(2,2,2-trifluoroethoxy)pyridine (743.0 g, 2.74 mol), compound VI 3-(2-chloropyrimidin-4-yl)-1-methyl-1H-indole (866.7 g, 3.56 mol), p-toluenesulfonic acid monohydrate (780.7 g, 4.10 mol) were added in sequence, stirred, and the reaction mixture was heated to 110-115°C and reacted for 36 hours. The temperature was controlled at 15-30°C, tetrahydrofuran (3.72 L) was added and stirred for 30 minutes. Filtered by suction, the filter cake was transferred to a 50 L reactor, tetrahydrofuran (4.46 L) was added, and heated to reflux for 3 hours. The temperature was lowered to 15-25°C, filtered, and the filter cake was dried at 50°C for 17 hours to obtain 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1719 g, 85.96 HPLC area% purity). The purity was calculated according to HPLC and used for the next step reaction. |
| Melting point: 216.0-218.3℃ |
| 1H NMR(500MHz,DMSO-d6)δ9.70(s,1H),9.21(s,1H),8.62(s,1H),8.40(d,J=6.2Hz,1H),8.24(d,J=7.8Hz,1H),7.59(d,J=8.3Hz,1H),7.50(d,J=6.5Hz,1H),7.49(d,J=8.3Hz,2H),7.32(t,J=7.6Hz,1H),7.18(t,J=7.5Hz,1H),7.12(d,J=7.9Hz,2H),5.17(q,J=8.8Hz,2H),3.91(s,3H),2.29(d,J=5.2Hz,3H); |
| 13C NMR(126MHz,DMSO-d6)δ166.66,157.35,155.72,147.40,140.87,139.90,139.72,138.59,135.83,130.09,129.99,129.98,129.97,127.39,127.38,127.37,127.15,125.22(q,J=278.5Hz),124.97,123.85,123.69,113.63,112.97,110.27,63.58(q,J=35.3Hz),35.57,22.81。 |
| Example 9: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Add N,N-dimethylformamide (5.14L) to a 50L reactor, add 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1714.0g, 2.261mol) obtained in Example 8, stir, add potassium carbonate (624.7g, 4.52mol), N,N,N’-trimethylethylenediamine (346.2g, 3.39mol) in sequence, heat the reaction mixture to 77-82°C, keep warm for 1-1.5 hours. Add water (10.28L), and cool to room temperature after adding. Filter by suction, transfer the filter cake to a 50L reactor, add acetonitrile (6.86L), and heat to reflux for 2 hours. The temperature was lowered to 15-25°C, filtered with suction, and the filter cake was dried at 50°C for 18 hours to obtain the compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1142 g). The total yield of the two-step reaction with Example 8 was 76.54%. |
| 1H NMR(500MHz,DMSO-d6)δ8.62(s,1H),8.41(s,1H),8.26(s,2H),8.24(s,1H),7.48(d,J=8.2Hz,1H),7.21(t,J=7.6Hz,1H),7.16(d,J=5.3Hz,1H),7.05(t,J=7.3Hz,1H),5.04(q,J=8.9Hz,2H),3.84(s,3H),3.69(t,J=6.9Hz,2H),2.89(s,3H),2.55(t,J=6.9Hz,2H),2.17(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ162.15,160.55,156.99,154.98,148.42,137.53,132.83,132.68,125.50,123.58(q,J=279.7Hz),124.38,122.11,122.06,120.67,113.38,112.27,110.30,107.11,62.14(q,J=35.3Hz),56.10,49.51,45.34,45.33,39.35,32.98。 |
| MS m/z:545.22[M+1]。 |
| Example 10: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (IV) |
| Acetonitrile (10 mL) was added to a 50 L reactor, and 2-chloro-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine p-toluenesulfonate (1.0 g, 1.5 mmol) obtained in Example 8 was added, and stirred. Potassium carbonate (577 mg, 3 mmol) and N,N,N’-trimethylethylenediamine (320 mg, 2.25 mmol) were added in sequence. The reaction mixture was heated to 77-82°C and kept for 1-2 hours. Water (10 mL) was added and the temperature was cooled to room temperature after the addition. The product was filtered to give a compound of formula IV, 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (629 mg) with a purity of 95.94%. The total yield of the two-step reaction with Example 8 was 77%. |
| Example 11: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| Add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0 g, 7.34 mmol) to a 100 mL reaction bottle at room temperature, add tetrahydrofuran (27 mL) and water (13 mL), and stir for 10 to 20 minutes. Add hydrosulfite (9.6 g, 44.1 mmol) to the reactor in batches. After addition, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 30 to 35 ° C for reaction. The purity of the product compound of formula III’ was 64.68% after sampling the liquid phase after 2 hours of reaction. The reaction was continued until 17 hours after the reaction. 40 mL of water was added to the reaction solution, and the layers were separated by standing. The tetrahydrofuran phase was taken, and the aqueous phase was extracted twice with 100 mL of dichloromethane. The organic phases were combined, washed with saturated brine, separated by standing, and concentrated under reduced pressure to obtain 3.2 g of solid with a purity of 62.32%. |
| 1H NMR(500MHz,DMSO)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,2H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| MS m/z:515.24[M+1]。 |
| Example 12: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| In a 100mL single-mouth bottle, there is 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (2.0g, 3.67mmol), palladium carbon (200mg), ethanol (20mL), hydrogen balloon replacement twice, hydrogen gas, magnetic stirring, room temperature overnight (17 hours). After the reaction is completed, suction filtration, the filtrate is taken, and it is concentrated to dryness under reduced pressure to obtain 2.1g of product with a purity of 56.93%. |
| Example 13: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1317.0 g, 2.42 mol) to a 50 L reactor, add tetrahydrofuran (8.8 L) and water (4.3 L), and stir for 10 to 20 minutes. Add hydrosulfite (2970.0 g, 14.52 mol) to the reactor in batches. After adding, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 40-45 ° C and react for 2 hours. Add concentrated hydrochloric acid (5882.2 g, 58.08 mol) to the reactor. After the addition is complete, heat to 42 to 47 ° C and react for 15 hours. Add 30% sodium hydroxide (2323.2g, 58.08mol) aqueous solution dropwise, and then add solid sodium bicarbonate (1219.7g, 14.52mol) in batches to adjust the pH value to 6-8. After stirring for 20 minutes, filter with suction, let the filtrate stand and separate. The organic phase is concentrated under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine, with a purity of 97.1%. Calculated based on the theoretical yield of 100%, it is directly used in the next step reaction. |
| 1H NMR(500MHz,DMSO)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,2H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| MS m/z:515.24[M+1]。 |
| Example 14: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (III-1) |
| To the 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine obtained in Example 13, THF (5.3 L) and ethanol (4.0 L) were added, the temperature was raised to 50-70°C, and concentrated hydrochloric acid (617.8 g, 6.1 mol) was added dropwise. After the addition was completed, the mixture was cooled to room temperature and stirred for 12 hours. Filtered by suction, the filter cake was dried by air at 50°C to obtain 1507.4 g of a crude product. Methanol (6.0 L) and ethanol (4.5 L) were added to a 20 L reaction bottle, and the above crude product was added, the temperature was raised to 55-60 ° C, hot slurry was added for 1-2 hours, the temperature was lowered to room temperature, and suction was filtered to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (1335.6 g), the liquid phase purity was 99.80%, and the total yield of the two-step reaction with Example 13 was 94.0%. Melting point: 236.6-240.8 ° C. |
| 1H NMR(500MHz,DMSO-d6)δ10.67(s,1H),10.36(s,1H),8.82(s,1H),8.18(s,1H),8.01(s,1H),7.59(d,J=8.2Hz,1H),7.45(d,J=6.8Hz,1H),7.32(t,J=7.5Hz,1H),7.24(s,1H),4.97(q,J=8.7Hz,1H),3.93(s,3H),3.75(s,2H),3.41(s,2H),3.10(s,3H),2.78(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ166.81,153.27,152.17,150.76,138.61,138.16,138.15,125.46,124.94,123.83(q.J=278.5Hz),123.42,123.41,122.60,122.59,120.52,111.34,111.17,106.29,62.14(q,J=35.3Hz),53.53,46.28,42.27,42.26,40.92,33.67。 |
| Example 15: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (III-1) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1136.0 g, 2.09 mol) to a 50 L reactor, add acetonitrile (7.95 L) and water (7.95 L), and stir for 10 to 20 minutes. Add hydrosulfite (2563.9 g, 12.50 mol) to the reactor in batches. After adding, continue stirring for 10 to 20 minutes. Control the temperature of the reactor to 35 to 40 ° C and react for 3 hours. Add concentrated hydrochloric acid (2505.3 g, 25.08 mol) to the reactor. After the addition is complete, heat to 35 to 45 ° C and react for 18 hours. 30% sodium hydroxide (1003.2 g, 25.08 mol) aqueous solution was added dropwise to adjust the pH value to 6-8. Solid sodium bicarbonate (1053.5 g, 12.54 mol) was added to adjust the pH value to 7-8. After stirring for 40 minutes, the mixture was filtered, the filtrate was allowed to stand, the layers were separated, and the organic phase was concentrated under reduced pressure. The purity of the liquid phase was detected to be 97.60%. |
| Add ethanol (5.68 L) to the product of the previous step, raise the temperature to 50-70°C, and drop concentrated hydrochloric acid (522 g, 5.23 mol). After the dropwise addition is completed, cool to room temperature and stir for 15 hours. Filter by suction, and air dry the filter cake at 50°C to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (780 g), with a liquid phase purity of 98.74%. |
| Example 16: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (II-1) |
| 2-[2-(Dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine dihydrochloride (1543.5 g, 2.63 mol) was added to a 50 L reactor, and dichloromethane (13.1 L) and triethylamine (532.2 g, 5.26 mol) were added. The mixture was stirred and cooled to -10 to -5 °C, and a solution of 3-chloropropionyl chloride (501.5 g, 3.95 mol) in dichloromethane (10.0 L) was added dropwise. After the addition is completed, keep warm and stir for 10 to 20 minutes, filter with suction, and the filter cake is formula II-12-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride wet product (2683.5g), which is calculated based on the theoretical yield of 100% and is directly used in the next reaction. |
| Melting point: 233.2-238.7℃ |
| 1H NMR(500MHz,DMSO-d6)δ10.18(s,1H),8.57(s,1H),8.42(s,1H),8.27(t,J=6.6Hz,2H),8.17(s,1H),7.51(d,J=8.1Hz,1H),7.26-7.22(m,1H),7.22-7.17(m,2H),4.99(q,J=9.1Hz,2H),3.91(d,J=6.3Hz,2H),3.89(s,3H),3.55(s,2H),3.13(s,2H),3.02(t,J=6.1Hz,2H),2.85(s,3H),2.64(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ168.41,161.88,160.22,157.34,148.05,146.73,137.62,133.25,130.86,125.43,124.09(q,J=279.2Hz),122.04,121.74,120.88,118.51,116.60,112.33,110.40,107.09,61.65(q,J=35.3Hz),54.90,40.96,40.95,40.60,38.71,32.96,32.95,32.94。 |
| Example 17: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| The wet product (2683.5 g) of Formula II 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride obtained in Example 16 was added to a 20L reactor, and acetonitrile (16.8L) and triethylamine (1329.3g, 13.15mol) were added, stirred, and heated to reflux for 4 hours. Cooled to room temperature, purified water (4.20L) was added, stirred at room temperature for 3-4 hours, and filtered. The filter cake was transferred to a 50L reactor, dichloromethane (17L) was added, and the pH value was adjusted to 7-8 with saturated sodium bicarbonate aqueous solution (17L). Liquid separation, the organic phase was transferred to a 20L reactor, activated carbon (84.3g) was added, refluxed for 1 hour, cooled to 20-30°C, and filtered. The filtrate was concentrated to dryness under reduced pressure to obtain the compound of formula I 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1390g), with a total yield of 92.9% and a purity of 99.21% for the two-step reaction with Example 16. |
| 1H NMR(500MHz,DMSO-d6)δ9.96(s,1H),8.71(s,1H),8.44(s,1H),8.29(d,J=5.3Hz,1H),8.26(d,J=7.7Hz,1H),8.13(s,1H),7.51(d,J=8.2Hz,1H),7.24(t,J=7.2Hz,1H),7.20(d,J=5.3Hz,1H),7.15(t,J=7.2Hz,1H),6.51(dd,J=17.0,10.2Hz,1H),6.28(dd,J=17.0,1.8Hz,1H),5.78(dd,J=10.2,1.8Hz,1H),5.00(q,J=9.1Hz,2H),3.89(s,3H),3.18(t,J=6.5Hz,2H),2.87(s,3H),2.48(t,J=6.5Hz,2H),2.22(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ163.40,161.84,160.26,157.35,148.07,147.15,137.60,133.23,131.61,130.07,126.67,125.41,124.03(q,J=278.5Hz),122.00,121.68,120.80,118.39,116.13,112.36,110.37,107.02,61.29(q,J=35.3Hz),56.57,52.44,45.60,45.59,38.54,32.93; |
| MS m/z:569.25[M+1]。 |
| Example 18: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (III’) |
| At room temperature, add the compound of formula IV 2-[2-(dimethylaminoethyl)methylamino]-3-nitro-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (20.0 g, 36.73 mmol) to a 1L reaction bottle, add tetrahydrofuran (134 mL) and water (66 mL), and stir for 10 to 20 minutes. Add hydrosulfite (47.9 g, 220.38 mmol) to the reaction bottle in batches. After addition, continue stirring for 10 to 20 minutes. Control the internal temperature to 35-40°C and react for 3 hours. Add concentrated hydrochloric acid (89.3 g, 881.52 mmol) to the reaction bottle. After the addition is complete, heat to 42 to 47°C and react for 17 hours. 30% sodium hydroxide (35.26 g, 881.52 mmol) aqueous solution was added dropwise, and solid sodium bicarbonate (18.5 g, 220.38 mmol) was added in batches to adjust the pH value to 6-8. After stirring for 30 minutes, the mixture was filtered, and the filtrate was allowed to stand and separated. The organic phase was concentrated to dryness under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (19.2 g) with a purity of 95.8% and a yield of 97.12%. |
| Example 19: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (II-1) |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5 g, 9.72 mmol) to a 250 mL reaction bottle, add dichloromethane (42 mL), stir, protect with argon, cool to -5 to 0°C, and add 3-chloropropionyl chloride (1.851 g) and dichloromethane (33 mL) dropwise. After the addition is complete, the mixture is stirred for 10-20 minutes at a temperature maintained at room temperature. After the reaction is complete, the mixture is concentrated under reduced pressure to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride (7.0 g) with a purity of 85.67%. Melting point: 233.5-238.9°C. |
| 1H NMR(500MHz,DMSO-d6)δ10.18(s,1H),8.57(s,1H),8.42(s,1H),8.27(t,J=6.6Hz,2H),8.17(s,1H),7.51(d,J=8.1Hz,1H),7.26-7.22(m,1H),7.22-7.17(m,2H),4.99(q,J=9.1Hz,2H),3.91(d,J=6.3Hz,2H),3.89(s,3H),3.55(s,2H),3.13(s,2H),3.02(t,J=6.1Hz,2H),2.85(s,3H),2.64(s,6H); |
| 13C NMR(126MHz,DMSO-d6)δ168.41,161.88,160.22,157.34,148.05,146.73,137.62,133.25,130.86,125.43,124.09(q,J=279.2Hz),122.04,121.74,120.88,118.51,116.60,112.33,110.40,107.09,61.65(q,J=35.3Hz),54.90,40.96,40.95,40.60,38.71,32.96,32.95,32.94。 |
| Example 20: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| The 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine hydrochloride obtained in Example 19 was added to a 250 mL reaction bottle, and acetonitrile (45 mL) and triethylamine (4.9 g) were added. The mixture was stirred magnetically and protected by argon. The temperature was raised to reflux in an oil bath. The reaction was allowed to react for 6 h. Water (23 mL) was added dropwise, and the mixture was naturally cooled to room temperature in an oil bath. The mixture was filtered with suction, and the filter cake was transferred to a 500 mL reaction bottle. Dichloromethane (100 mL) was added, and the pH value was adjusted to 7-8 with saturated aqueous sodium bicarbonate solution (100 mL). The liquids were separated and the organic phase was concentrated under reduced pressure. The solid was dried in an oven at 50°C to give 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.1 g) with a purity of 97.7%. The total yield of the two-step reaction with Example 19 was 74.17%. |
| Comparative Example 1: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1 g, 1.94 mmol) was added to a 50 mL multi-necked flask, tetrahydrofuran (10 mL) was used as the solvent, argon was replaced three times, and stirring was maintained at 0-5°C under argon protection, and 3-chloropropionyl chloride (0.37 g, 2.92 mmol), the addition was completed in 15 minutes, and the mixture was stirred at 0-5°C for 1 hour. Sodium hydroxide (0.31 g, 7.77 mmol) and water (1 mL) were added to the reaction solution, and the temperature was raised to 65°C and stirred for 15 hours. Saturated ammonium chloride solution (10 mL) was added, and the liquids were separated. The organic phase was washed with saturated sodium bicarbonate solution (10 mL). The liquids were separated and the organic phase was concentrated to dryness to obtain 1.04 g of a yellow solid with a yield of 94.9% and a purity of 87.35%. |
| Comparative Example 2: Preparation of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I, crude product) |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-amino-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g) to a 250 mL reaction bottle, add acetone (50 mL) and potassium carbonate (940 mg), stir, protect with argon, cool to -50°C, and add 3-chloropropionyl chloride (1.481 g) dropwise. After the addition is completed, the temperature is raised to -20°C and stirred for 30 minutes. A solution of sodium hydroxide (350 mg) and water (60 ml) is added dropwise over 10 minutes. The mixture is stirred at room temperature for 3 to 4 hours. The mixture is filtered and the filter cake is dried in an oven at 50°C to obtain a compound of formula II-1′, 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamide)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.28 g) with a purity of 64.18%. |
| 1H NMR(400MHz,DMSO-d 6 )δ10.32(s,1H),10.21(s,1H),8.54(s,1H),8.43(s,1H)8.29-8.28(d,J=5.1Hz,1H),8.28-8.26(d,J=6.2Hz,1H),8.19(s,1H),7.54-7.52(d,J=8.0Hz,1H),7.27-7.18(m,3H),5.77(s,2H),5.00(q,J=9.1Hz,1H),3.92(t,J=6.2Hz,1H),3.63(t,J=5.7Hz,2H),3.28(t,J=5.7Hz,2H),3.06-3.03(t,J=6.2Hz,2H),2.85(s,3H),2.74(s,6H). |
| MS m/z:605.23[M+1]。 |
| Add 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.28 g) to a 250 mL reaction bottle, add acetonitrile (45 ml) and triethylamine (3.606 g), stir magnetically, protect with argon, heat in an oil bath to reflux, and react for 6 h. Water (23 ml) was added dropwise, the temperature was naturally lowered in an oil bath and stirring was continued overnight (16 h), filtered with suction, and the solid was dried to obtain 2-[2-(dimethylaminoethyl)methylamino]-3-(3-chloro-propionamido)-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.3 g) with a purity of 95.13% and a two-step yield of 59.42%. |
| Example 21: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| The crude product (1390 g) of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine was transferred to a 50L reactor, acetone (25.0L) was added, argon was replaced 3 times, the temperature was raised to 45-50°C, all the solids were dissolved, and purified water (6.95L) was added dropwise. After the addition was completed, the mixture was cooled to 20-25°C and stirred for 2 hours. The mixture was filtered and the filter cake was vacuum dried at 50°C for 24 hours to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (895g). The reaction yield is 66.7% and the purity is 99.89%. |
| Example 22: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add ethyl acetate 100mL, heat to 70-75°C in an oil bath to dissolve all the solids, then cool naturally to 25°C in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.1g) with a purity of 99.73% and a yield of 62.0%. |
| Example 23: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add ethyl acetate 100mL, heat to 70-75°C in an oil bath, dissolve all the solids, continue to stir for 30min, and drop 150mL of n-heptane. After the drop is complete, cool to 25°C in an oil bath, filter by suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0g), with a purity of 99.32% and a yield of 80%. |
| Example 24: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add acetonitrile 75mL, heat in an oil bath to 77-82°C, dissolve all the solids, and drop 25mL of water. After dripping, naturally cool to 25°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.3g), with a purity of 99.64% and a yield of 86%. |
| Example 25: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add acetonitrile 75mL, heat in an oil bath to 77-82°C, dissolve all the solids, and continue to stir for 30min. Cool naturally to 25°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.0g), with a purity of 99.45% and a yield of 80%. |
| Example 26: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g), add 20mL of tetrahydrofuran, heat in an oil bath to 45-50°C to dissolve all the solids, continue to stir and maintain the temperature for 30 minutes, and add 40mL of n-heptane dropwise. After the addition was completed, the mixture was naturally cooled to 25°C in an oil bath, filtered and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.23 g) with a purity of 99.51% and a yield of 84.6%. |
| Example 27: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 100mL of isopropanol, heat to 50°C in an oil bath, dissolve all the solids, and continue to stir for 30 minutes. Cool naturally to 22°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (4.25g), with a purity of 99.51% and a yield of 85%. |
| Example 28: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 75mL of methanol, and heat to 55-60°C in an oil bath to dissolve all the solids. Cool naturally to 17°C in the oil bath, stir overnight, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.55g), with a purity of 99.63% and a yield of 71%. |
| Example 29: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0 g), add 50mL of dichloromethane, heat in an oil bath to 40°C to dissolve all the solids, continue to stir and maintain the temperature for 30 minutes, and add 100mL of n-heptane dropwise. The mixture was naturally cooled to 15°C in an oil bath, stirred overnight, filtered and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.78 g) with a purity of 99.56% and a yield of 75.6%. |
| Example 30: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (5.0g), add 100mL of toluene, heat to 65°C in an oil bath, dissolve all the solids, and continue to stir for 30 minutes. Cool naturally to 20°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.27g), with a purity of 99.57% and a yield of 65.4%. |
| Example 31: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add DMF50mL, heat to 80°C in an oil bath, dissolve all the solids, continue to stir for 30min, and drop 25mL of water. Naturally cool to 20°C in the oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.84g), with a purity of 99.77% and a yield of 76.8%. |
| Example 32: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 25 mL single-necked bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add tetrahydrofuran (6 mL), protect with argon, heat in an oil bath to 40-45°C until all the solution is dissolved, continue to stir and keep warm for 30 min, cool naturally to 22°C in an oil bath, filter and obtain a solid. The solid was transferred to a crystallization dish and dried to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (622 mg) with a purity of 99.83% and a yield of 62.2%. |
| Example 33: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add acetone (15 mL), protect with argon, heat in an oil bath to 45-50° C. until all the solution is dissolved, and then continue to stir and keep warm for 30 min, cool naturally to 22° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (537 mg) with a purity of 99.83% and a yield of 53.7%. |
| Example 34: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add tetrahydrofuran (8 mL), protect with argon, heat in an oil bath to 40-45° C. until all the solution is dissolved, and continue to stir and keep warm for 30 min. Add water (16 mL) dropwise, cool naturally to 21° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (880 mg) with a purity of 99.68% and a yield of 88.0%. |
| Example 35: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 100 mL single-mouth bottle, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1.0 g), add ethanol (35 mL), protect with argon, heat in an oil bath to 75-80° C. until all the solution is dissolved, add water (10 mL) dropwise over 10 min, cool naturally to 20° C. in an oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (915 mg), with a yield of 91.5% and a purity of 99.49%. |
| Example 36: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 50 mL single-mouth bottle, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (1.0 g), add xylene (20 mL), protect with argon, heat in an oil bath to 80° C. until all the solution is dissolved, cool naturally to 20° C. in an oil bath, filter with suction, and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (798 mg), with a yield of 79.8% and a purity of 99.48%. |
| Example 37: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| In a 250mL three-necked flask, add 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine crude product (5.0g), add ethanol 125mL, heat in an oil bath to 75-80°C to dissolve all the solids, continue to stir and keep warm for 30min, then cool naturally to 25°C in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (3.7g) with a purity of 99.66% and a yield of 74%. |
| Example 38: Purification of 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamido-5-[4-(1-methyl-1H-indol-3-yl)pyrimidin-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (I) |
| To a 100 mL single-mouth bottle, add crude 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (1.0 g), add methanol (35 mL), protect with argon, heat in an oil bath to 80° C. until all the solution is dissolved, add water (10 mL) dropwise over 10 min, cool naturally to 20° C. in an oil bath, filter and dry to obtain purified 2-[2-(dimethylaminoethyl)methylamino]-3-acrylamide-5-[4-(1-methyl-1H-indol-3-yl)pyrimidine-2-amino]-6-(2,2,2-trifluoroethoxy)pyridine (912 mg), with a yield of 91.2% and a purity of 99.53%. |
PATENT
CN110606842
WO2019238103
PAPER
https://www.nature.com/articles/s41401-020-0389-3
| NCT Number | Sponsor | Condition | Start Date | Phase |
|---|---|---|---|---|
| NCT02973763 | Allist Pharmaceuticals, Inc. | NSCLC | December 30, 2016 | Phase 1 |
| NCT03787992 | Allist Pharmaceuticals, Inc. | Locally Advanced or Metastatic EGFR Sensitising Mutation Positive Non-small Cell Lung Cancer | May 30, 2019 | Phase 3 |
| NCT03452592 | Allist Pharmaceuticals, Inc. | Advanced NSCLC Patients With T790M | April 30, 2018 | Phase 2 |
- [1]. Y. Shi, et al. P2.03-028 Third Generation EGFR Inhibitor AST2818 (Alflutinib) in NSCLC Patients with EGFR T790M Mutation: A phase1/2 Multi-Center Clinical Trial.[2]. Alexander I. Spira, et al. FURVENT: Phase 3 trial of furmonertinib vs chemotherapy as first-line treatment for advanced NSCLC with EGFR exon 20 insertion mutations (FURMO-004). Journal of Clinical Oncology. Volume 42, Number 16_suppl.
/////////FIRMOMERTINIB, Furmonertinib, Alflutinib, AST 2818, UNII-A49A7A5YN4, PHASE 2, CANCER
Tibremciclib


Tibremciclib
cas 2397678-18-9, GTPL12881
CRB7BT5JDQ
518.6 g/mol, C28H32F2N8
N-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-[(1R)-6-fluoro-1-methyl-1,2,3,4-tetrahydropyrido[1,2-a]benzimidazol-8-yl]pyrimidin-2-amine
Tibremciclib is a CDK4 inhibitor with antineoplastic activity[1].
- Originator Betta Pharmaceuticals Co Ltd
- Class Antineoplastics; Small molecules
- Mechanism of Action Cyclin-dependent kinase 4 inhibitors; Cyclin-dependent kinase 6 inhibitors
- Phase III Breast cancer; Solid tumours
13 Sep 2024 Efficacy and adverse event data from a phase III trial in Breast cancer presented at the 49th European Society for Medical Oncology Congress 2024 (ESMO-2024)
- 30 Jun 2023Phase-III clinical trials in Breast cancer (Metastatic disease, Late-stage disease, Combination therapy, Second-line therapy or greater) in China (PO) (NCT05433480)
- 02 Jun 2023Efficacy, adverse events and PK data from a phase I trial in Solid tumours presented at the 59th Annual Meeting of the American Society of Clinical Oncology (ASCO-2023)
Cyclin-dependent kinases (CDKs) are a class of serine / threonine protein kinases that participate in the regulation of the cell cycle, transcription initiation, and control of certain specific metabolic cascades. Different CDKs and cyclins form CDK-cyclin complexes. If the CDK activity is dysregulated, it will directly or indirectly cause uncontrolled cell proliferation, genomic instability (increased DNA mutation, chromosome deletion, etc.) and chromosomal instability (change in chromosome number). )Wait.
The CDKs family has identified more than 20 subtypes. CDK1, CDK2, CDK4, and CDK6 are involved in cell cycle regulation; CDK7, CDK8, CDK9, and CDK11 are involved in transcription regulation; and other kinases include CDK3 and CDK5. Among them, CDK4 / 6 (cyclin-dependent kinases 4 and 6) is a key factor in regulating the cell cycle. Cancer-related cell cycle mutations mainly exist in the G1 and G1 / S phase transformation. CDK4 / 6 binds to CyclinD A complex with kinase activity is formed and phosphorylation of the tumor suppressor gene Rb product pRb releases the bound transcription factor E2F to initiate transcription of genes related to the S phase, prompting cells to pass the checkpoint and transfer from the G1 phase to the S phase. The specific activation of CDK4 / 6 is closely related to the proliferation of some tumors. About 80% of human tumors have abnormalities in the cyclin D-CDK4 / 6-INK4-Rb pathway. CDK4 / 6 inhibitors block the cell cycle in the G1 phase, thereby inhibiting tumor proliferation.
The development of drugs targeting CDK4 / 6 kinases is a significant area. The advantages of anti-tumor targets are: (1) Most proliferating cells rely on CDK2 or CDK4 / 6 to proliferate, but CDK4 / 6 inhibitors do not show Cytotoxicity of “pan-CDK inhibitors”, such as bone marrow suppression and intestinal response; (2) Preclinical experiments show that if the level of cyclin D or the inactivation of P16INK4a can increase the sensitivity of cells to drugs, due to tumors Compared with normal cells, cells have the above phenomenon, so the targeting of drugs is increased to a certain extent.
PCT International Application PCT / CN2017 / 117950 describes a class of benzimidazole derivatives that are used as CDK4 / 6 protein kinase inhibitors, and most of these compounds effectively inhibit CDK4 and CDK6. Because there are still unmet needs in the treatment options for kinase-mediated diseases, here we further screen the salt forms and crystal forms of benzimidazole derivatives to meet the medical needs of patients.
SCHEME
SIDE CHAIN

SIDE CHAIN

MAIN

Patent
Betta Pharmaceuticals Co., Ltd., WO2019242719
https://patents.google.com/patent/WO2019242719A1/en


Synthesis of 1-A1-01 (Step 1)
In a 50L reactor, add 20L of dichloromethane (DCM), 1-A1-S1 (300g), and triethylamine (390g). While stirring, lower the temperature to below -5 ° C, and add benzyl chloroformate / Cbz- Cl (570 g) was added dropwise for 5 hours, and the temperature was naturally raised to room temperature. TLC (ethyl acetate: n-hexane = 1: 3) was monitored until the reaction was completed. Water (1.5 L) was added, and concentrated hydrochloric acid (80 mL) was slowly added dropwise to control the pH to 1-2. The solution was allowed to stand and separate. The organic phase was washed with 15 L of water, dried over anhydrous sodium sulfate for 0.5 hours, filtered to remove the desiccant, and collected the filtrate. And concentrated to obtain 730 g of light yellow oily liquid, which is crude 1-A1-01, yield 95.4%
Synthesis of 1-A1-02 (Step 2)
720mL of DCM, N, N-dimethylsulfoxide (90g) was added to a 20L reaction flask, protected by nitrogen, and the temperature was lowered below -65 ° C under stirring, and oxalyl chloride (106g) was added dropwise. The addition was completed in 2 hours. Stir for 20 minutes under heat preservation; add 1-A1-01’s dichloromethane solution (143g / 500mL DCM) dropwise. After 40 minutes, the addition is complete and the reaction is held for 15 minutes. Controlled at this temperature, TEA was added dropwise. After the addition was completed for 2 hours, the temperature was naturally raised to -20 ° C. 250 L of water was added to the system. The pH of the system was adjusted to 1-2 with hydrochloric acid. × 2) Washed, dried over anhydrous sodium sulfate, filtered to remove the desiccant, collected the filtrate and concentrated to obtain 432 g of a yellow oily liquid, which is the crude product 1-A1-02, which was directly used in the next reaction.
Synthesis of 1-A1-03 (Step 3)
In a stirred state, 400 mL of tetrahydrofuran (THF) and potassium tert-butoxide (215 g) were sequentially added to a 1 L reaction kettle, the temperature was lowered to 5-15 ° C., and triethyl phosphoryl acetate (430 g) was added dropwise. The dropwise addition was completed in 50 minutes. At a controlled temperature of 15 ° C, a tetrahydrofuran solution of 1-A1-02 (431 g / 100 mL of THF) was added dropwise. After the dropwise addition was completed for 1 hour, TLC (ethyl acetate: n-hexane = 1: 3) was monitored to complete the reaction, and the system was added. Saturated aqueous sodium chloride solution (1.5L), allowed to stand and separate, and collected the tetrahydrofuran phase; the aqueous phase was extracted with dichloromethane (2L), and the organic phases were combined and dried over anhydrous sodium sulfate for 0.5 hours, and the drying agent was removed by filtration. The filtrate was collected and concentrated, and the concentrate was purified by column chromatography to obtain 390 g of a pale yellow oily liquid, which was 1-A1-03 product.
Synthesis of 1-A1-041 (step 4)
In a 5L reactor, an aqueous solution of sodium hydroxide (301 g / 1.5 L of water) was added to a tetrahydrofuran (601 g / 2.3 L of THF) solution of 1-A1-03, and the mixture was heated to reflux for 3-4 hours to stop the reaction. The temperature was lowered to 40-50 ° C, and the layers were left to stand. The organic phase (THF) was collected and concentrated to a large amount of solids; the solids were dissolved by adding water (20L), and the aqueous phase was sequentially treated with methyl tert-butyl ether (2L) and ethyl acetate. Ester (2L), methyl tert-butyl ether (2L) washing; the aqueous phase was adjusted to pH 1-2 with concentrated hydrochloric acid, extracted twice with ethyl acetate (1.5L, 3L), the organic phases were combined, and anhydrous sulfuric acid was used Sodium was dried for 0.5 hours; the desiccant was removed by filtration, and the filtrate was collected and concentrated to a large amount of solids. The solids were added with isopropyl ether (3L) and slurried for 2 hours. The solids were collected by filtration and the solids were rinsed with isopropyl ether (1L). The solid was air-dried at 50 ° C for 3-4 hours to obtain 331 g of a pale yellow solid, which is a 1-A1-041 product with a yield of 52.7%.
Synthesis of 1-051 (step 5)
In a stirred state, 1-A1-041 (600g), methanol (25L), and concentrated sulfuric acid were added to a 50L reactor, and the reaction was heated under reflux for 3-4 hours. After the reaction was completed, the temperature was reduced to room temperature. Dichloromethane (15L) was added to the concentrate, and the pH was adjusted to 9-10 with an aqueous solution of potassium carbonate. The organic phase was collected by stirring, standing, and separating. The organic phase was dried over anhydrous sodium sulfate for 0.5 hours. The desiccant was removed by filtration and the filtrate was collected. And concentrated to obtain 6.37 kg of off-white solid, which is 1-A1-051 product, with a yield of 97.3%.
Synthesis of 1-A1 (step 6)
In a 2L hydrogenation kettle, add 1-A1-051 (500g), methanol (1.8L), and palladium on carbon. The system replaces nitrogen 3 times and hydrogen 3 times in sequence. The system maintains a hydrogen atmosphere, and the temperature is increased to 85 ° C and the pressure is 3.0. The reaction was carried out at Mpa for 3 hours, and the reaction was completed. The temperature was lowered to room temperature, the palladium on carbon was removed by filtration, and the organic phase was collected and concentrated until a large amount of light yellow solid appeared. Isopropyl ether (3L) was added to freeze (-20 ° C) for crystallization, and the solid product was collected by filtration. Ether (500 mL) was rinsed to obtain 234 g of a pale yellow solid, which was a 1-A1 product with a yield of 90.5%.
Synthesis of 1-A2 (Step 7)
In a stirred state, 1-A1 (200g), 4-bromo-2,6-difluoroaniline (410g), and toluene (1.2L) were added to a 50L reactor, and phosphorus oxychloride (413g) was added dropwise to the system. The addition was completed in 1 hour. Triethylamine was added dropwise under an ice bath, and the addition was completed in 1 hour. The temperature was raised to 110 ° C, and the reaction was performed for 1 hour. Reduce the temperature of the system to 2-10 ° C, add 1L of water, adjust the pH = 9-10 with saturated potassium carbonate aqueous solution, extract twice with ethyl acetate (1.5L, 1L), and combine the organic phases with 2L saturated sodium chloride aqueous solution. Wash, dry with anhydrous sodium sulfate for 0.5 hours, remove the desiccant by filtration, collect the filtrate and concentrate to the appearance of a solid product, add isopropyl ether (1L) to beat the solid for 10 minutes, filter, and collect 460 g of a yellow solid as a 1-A2 product.
Synthesis of 1-A3 (step eight)
Under stirring, 1-A2 (450g), N, N-dimethylformamide (2L), and cesium carbonate (700g) were added to the reaction kettle, and the reaction was heated to 110 ° C for 24 hours, and the reaction was detected by TLC. Ethyl acetate (3L) was added to the system, and solid impurities were removed by filtration. The filtrate was washed with a saturated sodium chloride aqueous solution (1L × 5), and the organic phase was dried over anhydrous sodium sulfate for 0.5 hours, concentrated to the appearance of a large amount of solid, Butyl ether (1L × 2) was beaten for 30 minutes, and 382 g of pale yellow solid product was obtained by filtration, that is, 1-A3, and the yield was 90.10%.
Synthesis of 1-01 (step 9)
With stirring, 1-A3 (380 g), pinacol diborate (400 g), potassium acetate (340 g), palladium acetate (6 g), tricyclohexyl phosphorus (7 g), and 1,4-dioxane were sequentially added. The ring was added to the reaction kettle, protected by nitrogen, and heated to 90 ° C for 2 hours. TLC was monitored until the reaction was complete. The temperature was reduced to room temperature, and the filtrate was concentrated to remove a large amount of 1,4-dioxane. The concentrate was purified by n-hexane and dichloromethane column chromatography, and n-hexane (1.2 L) was slurried for 1 hour to obtain 334 g of a gray solid. That is 1-01, and the yield is 70.10%.
Synthesis of 1-02 (step 10)
Under stirring, take 1-01 (128g), 1,4-dioxane (1L), 1-S3 (85g), potassium carbonate (110g), and purified water and add them to a 2L three-necked flask in sequence. [1,1′-Bis (diphenylphosphine) ferrocene] palladium dichloromethane complex (Pd (dppf) Cl 2 .DCM) was added. The temperature was raised to 60 ° C. After 4 hours of reaction, the reaction was complete. The reaction solution was cooled to room temperature, and concentrated under reduced pressure to remove most of 1,4-dioxane. Dichloromethane (1.5 L) and purified water (1.1 L) were added, stirred, and allowed to stand and separate. The layers were separated, and water was added. The phases were extracted with dichloromethane (10 L), the organic phases were combined, washed with 0.5% dilute hydrochloric acid (1 L x 2), saturated aqueous sodium chloride solution (1 L), and the layers were separated. The organic phase was dried over anhydrous sodium sulfate (500 g), filtered to remove the drying agent, and the filtrate was concentrated under reduced pressure. Ethyl acetate (0.5 L) was added to the concentrate and the mixture was stirred for 30 minutes to precipitate a solid. After filtration, the obtained solid was rinsed with ethyl acetate (0.5 L) and dried under vacuum at 45 ° C for 3 hours to obtain 120 g of a yellow solid.
Synthesis of 1-03 (step 11)
Under stirring, take 1-02 (100g), 1,4-dioxane (1L), 1-C2 (80g), and cesium carbonate (163g) into a 2L three-necked bottle in sequence, protected by nitrogen, and add palladium acetate ( 2g) and 4,5-bisdiphenylphosphine-9,9-dimethylxanthracene (Xantphos) (4g), heated to 85 ° C. until the reaction was complete. The reaction solution was cooled to room temperature and filtered to obtain a solid product. The solid was rinsed with ethyl acetate, and then added to a mixed system of dichloromethane (1.5 L) and purified water (1.1 L), stirred, allowed to stand, and separated into layers. The aqueous phase was extracted with dichloromethane (700 mL). The organic phases were combined and washed with purified water (700 mL x 2). The organic phase was dried by adding anhydrous sodium sulfate (700 g), filtered to remove the desiccant, and the filtrate was concentrated. Methanol (0.5 L) was added, heated to 55-65 ° C. and stirred for 0.5 hours, lowered to room temperature, and filtered. The solid product was filtered and rinsed with 500 mL of ethyl acetate. The solid was dried under vacuum at 45 ° C for 8 hours to obtain 111.79 g of a pale yellow solid 1-03.
Synthesis of compound II (step twelve)
Under stirring, take 1-03 (500g) and anhydrous methanol (3.8L), add them to a 10L reactor in sequence, and heat to 65 ° C. After the reaction system is clarified for 0.5 hours, add L-tartaric acid in methanol (150.89) dropwise. g of tartaric acid is dissolved in 500mL of anhydrous methanol), and the dropping time is controlled to be 45 to 60 minutes. After the addition is complete, the reaction is kept at 65 ° C for 4 hours. ), Control the dropwise addition time to 30 to 45 minutes. After the dropwise addition is complete, hold the reaction at 65 ° C for 1 hour. Continue to dropwise add L-tartaric acid in methanol (36.55g of tartaric acid dissolved in 250mL of anhydrous methanol) and control the dropwise addition time to 30. To 45 minutes, the dropwise addition was completed. The temperature was kept at 65 ° C for 1.5 hours, and the heating was stopped. The temperature was naturally lowered to 20-30 ° C, filtered, the filter cake was rinsed with methanol (400mL × 2), and dried at 45 ° C under vacuum for 36 hours. 530.64 g of crystalline powder was Compound II, which was identified by X-ray powder diffraction, and showed that the crystal form was Form A of Compound II.
WO2022199656
WO2023131179
///Tibremciclib, GTPL12881, BETTA, PHASE 3, CANCER

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}