
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Acetaminosalol
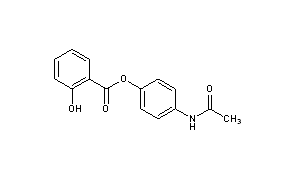

Acetaminosalol
- Molecular FormulaC15H13NO4
- Average mass271.268 Da
- ацетаминосалол [Russian] [INN], أسيتامينوسالول [Arabic] [INN], 醋氨沙洛 [Chinese] [INN]
(1E)-N-{4-[(2-Hydroxybenzoyl)oxy]phenyl}ethanimidic acid118-57-0[RN]
204-261-3[EINECS]
CAS Registry Number: 118-57-0
CAS Name: 2-Hydroxybenzoic acid 4-(acetylamino)phenyl ester
Additional Names:p-acetamidophenyl salicylate; acetylaminophenyl salicylate; acetyl-p-aminosalol; p-acetylaminophenol salicylic acid ester; phenetsal
Trademarks: Salophen (Bayer); Phenosal
Molecular Formula: C15H13NO4
Molecular Weight: 271.27
Percent Composition: C 66.41%, H 4.83%, N 5.16%, O 23.59%
Literature References: Prepn: Brewster, J. Am. Chem. Soc.40, 1136 (1918).
Properties: Crystals from hot ethanol, mp 187°. Practically insol in petr ether, cold water, more sol in warm water. Sol in alcohol, ether, benzene. Incompatible with alkalies and alkaline solns which dissolve it with decompn. The alkaline soln gradually becomes blue when boiled, the blue color being discharged upon continued boiling and again produced upon cooling and exposure to air.
Melting point: mp 187°
Therap-Cat: Analgesic; antipyretic; anti-inflammatory.
Therap-Cat-Vet: Analgesic; antipyretic.
Keywords: Analgesic (Non-Narcotic); Anti-inflammatory (Nonsteroidal); Salicylic Acid Derivatives; Antipyretic.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Acetaminosalol is an organic compound with the chemical formula C15H13NO4.
It is an esterification product of salicylic acid and paracetamol. It was marketed by Bayer under the brand name Salophen as an analgesic in the late 19th and early 20th centuries.
Action and uses
In a warm alkaline solution acetaminosalol is broken up into salicylic acid and paracetamol. It is decomposed in the intestines, even when given as an injection. It was used as a substitute for salicylic acid in acute rheumatism, and as an intestinal antiseptic. It was similarly effective and much safer than salol, another intestinal antiseptic commonly used at the time. The fact that it is tasteless renders it easy to administer.Advertisement for early 20th century Bayer products, including Salophen
SYNJournal of Organic Chemistry, 86(5), 4254-4261; 2021

| Names | |
|---|---|
| Preferred IUPAC name4-Acetamidophenyl 2-hydroxybenzoate | |
| Identifiers | |
| CAS Number | 118-57-0 |
| 3D model (JSmol) | Interactive imageInteractive image |
| ChEBI | CHEBI:250620 |
| ChEMBL | ChEMBL92590 |
| ChemSpider | 1907 |
| ECHA InfoCard | 100.003.875 |
| EC Number | 204-261-3 |
| MeSH | Salophen |
| PubChem CID | 1984 |
| UNII | O3J7H54KMD |
| CompTox Dashboard (EPA) | DTXSID7045865 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C15H13NO4 |
| Molar mass | 271.272 g·mol−1 |
| Density | 1.327 g cm−3 |
| log P | 2.562 |
| Acidity (pKa) | 7.874 |
| Basicity (pKb) | 6.123 |
| Hazards | |
| Flash point | 241.9 °C (467.4 °F; 515.0 K) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
///////////////Acetaminosalol, nalgesic , Anti-inflammatory, Salicylic Acid Derivatives, Antipyretic, ацетаминосалол , أسيتامينوسالول , 醋氨沙洛 ,

NEW DRUG APPROVALS
ONE TIME
$10.00
LINZAGOLIX
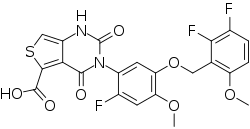
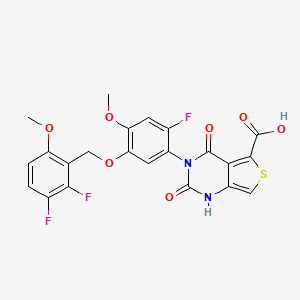
LINZAGOLIX
CAS 935283-04-8
C22H15F3N2O7S
- Hormone Antagonists
3-[5-[(2,3-difluoro-6-methoxyphenyl)methoxy]-2-fluoro-4-methoxyphenyl]-2,4-dioxo-1H-thieno[3,4-d]pyrimidine-5-carboxylic acid
- WHO 10711
- Treatment of Endometriosis Pain and Uterine Myoma-Associated Heavy Menstrual Bleeding
- OriginatorKissei Pharmaceutical
- DeveloperKissei Pharmaceutical; ObsEva
- Class2 ring heterocyclic compounds; Antihormones; Antineoplastics; Carboxylic acids; Fluorinated hydrocarbons; Ketones; Pyrimidines; Small molecules; Thiophenes
- Mechanism of ActionLHRH receptor antagonists
- PreregistrationUterine leiomyoma
- Phase IIIEndometriosis
- Phase IIAdenomyosis
- 22 Nov 2021FDA assigns PDUFA action date of (13/09/2022) for linzagolix for Uterine leiomyoma
- 22 Nov 2021The US FDA accepts NDA for linzagolix for Uterine leiomyoma for review
- 20 Oct 2021Efficacy and adverse events data from a phase II trial in Adenomyosis presented at the American Society for Reproductive Medicine (ASRM) 2021 Scientific Congress & Expo


Linzagolix choline
CAS#: 1321816-57-2 (choline)
Chemical Formula: C27H28F3N3O8S
Exact Mass: 611.1549
Molecular Weight: 611.58
Linzagolix is an orally bioavailable gonadotropin-releasing hormone (GnRH or LHRH) receptor antagonist, with potential hormone production inhibitory activity. Upon oral administration of linzagolix, this agent competes with GnRH for receptor binding and inhibits GnRH receptor signaling in the anterior pituitary gland, thereby inhibiting the secretion and release of luteinizing hormone (LH) and follicle stimulating hormone (FSH). In males, the inhibition of LH secretion prevents the release of testosterone. As a result, this may relieve symptoms associated with hormonally dependent disease states such as hormone-dependent prostate cancer. In women, this prevents the production of estrogen by the ovaries and may relieve symptoms from sex-hormone dependent diseases, such as pain associated with endometriosis, heavy menstrual bleeding or uterine fibroids.
Linzagolix (INN; developmental code names KLH-2109, OBE-2109; tentative brand name Yselty) is a small-molecule, non-peptide, orally active gonadotropin-releasing hormone antagonist (GnRH antagonist) which is under development by Kissei Pharmaceutical and ObsEva for the treatment of uterine fibroids, endometriosis, and adenomyosis.[1][3][2] As of December 2020, it is under review for approval for uterine fibroids, is in phase III clinical trials for endometriosis, and is in phase II clinical studies for adenomyosis.[1]
Estrogen-dependent disorders represent a challenging class of diseases that have a high incidence in the general population and are often associated with particularly severe symptomology. Uterine fibroids, for example, also referred to as leiomyomata, are among the most common benign tumors in women. Symptoms associated with uterine fibroids commonly include heavy or prolonged menstrual bleeding, pelvic pressure and pelvic organ compression, back pain, and adverse reproductive outcomes. Heavy menstrual bleeding may lead to iron deficiency anemia, a key symptom of uterine fibroids and the leading cause of surgical interventions that may include hysterectomy. Endometriosis is another estrogen-dependent gynecological condition, characterized by the presence of endometrial-like tissue outside the uterus.
Additional examples of estrogen-dependent diseases include adenomyosis and rectovaginal endometriosis, which are particularly severe endometrial growth disorders characterized by the invasion of endometrial tissue into the uterine myometrium and rectovaginal zones, respectively. The term adenomyosis or uterine adenomyosis is used to describe the presence of both endometrial glands and stroma deep within the myometrium. This condition is associated with hypertrophy and hyperplasia of the subjacent muscle cells, which may ultimately result in an altered size and globulous morphology of the uterus. Due to the severity of this disorder, one of the key symptoms is strong menstrual and even non-menstrual pelvic pain with abnormal uterine bleeding. Like adenomyosis, rectovaginal endometriosis patients present with a variety of pain symptoms including dysmenorrhea, dyspareunia, chronic pelvic pain, dysuria, and dyschezia. Treatment options for rectovaginal endometriosis are limited. Since medical therapies are either ineffective or have considerable side effects, rectovaginal endometriosis patients often undergo surgical procedures to reduce the endometrial node, and may even be subject to resection of the bowel if the node infiltrates the rectal or sigmoidal wall.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Obseva Announces U.S. FDA Acceptance of New Drug Application for Linzagolix
November 22, 2021 01:05 ET | Source: ObsEva SA………. https://www.globenewswire.com/news-release/2021/11/22/2338610/0/en/Obseva-Announces-U-S-FDA-Acceptance-of-New-Drug-Application-for-Linzagolix.html
FDA Accepts NDA for Linzagolix for the Management of Heavy Menstrual Bleeding Associated with Uterine Fibroids
GENEVA, Switzerland November 22, 2021 – Obseva SA (NASDAQ: OBSV; SIX: OBSN), a biopharmaceutical company developing and commercializing novel therapies to improve women’s reproductive health, today announced that the New Drug Application (NDA) for linzagolix for the management of heavy menstrual bleeding associated with uterine fibroids in premenopausal women has been accepted for review by the United States Food and Drug Administration (FDA). The submission is based on data from the two Phase 3 PRIMROSE trials. Linzagolix has a differentiated profile and if approved, would be the first and only GnRH receptor antagonist with flexible dosing options for uterine fibroids, including a low dose option to address the needs of women who cannot or do not want to take hormones.1,4 The FDA set a target action date of September 13, 2022 for this NDA under the Prescription Drug User Fee Act (PDUFA).
“Today marks an important milestone not only in the linzagolix clinical development process, but for Obseva as a company, and most importantly, the millions of women living with uterine fibroids throughout the US. Linzagolix is a significant innovation in the field of women’s health – an area that is consistently underinvested in – and we are incredibly excited about the potential of bringing this important treatment to market” said Brian O’Callaghan, CEO of Obseva. “We are encouraged by our positive Phase 3 PRIMROSE results. If approved, we believe linzagolix will address a significant unmet need in offering a more individualized treatment option for a broader range of women.”
The Phase 3 PRIMROSE trials of linzagolix (PRIMROSE 1: US; n=574 and PRIMROSE 2: Europe and US; n=535) investigated the efficacy and safety of two dosing regimens, 100mg once daily and 200mg once daily, alone or in combination with hormonal ABT (1 mg estradiol and 0.5 mg norethisterone acetate) for the treatment of heavy menstrual bleeding associated with uterine fibroids. The NDA submission comprises positive 24-week treatment results from both studies, as well as supportive results from Week 52 and the 76-week post-treatment follow-up.
“Uterine fibroids can have a devastating impact on women’s day-to-day life. With its unique dosing options, linzagolix has the potential to significantly advance medical options for women,” stated Elizabeth Garner, MD, MPH, Chief Medical Officer of Obseva. “A dosing option without hormonal ABT would be welcomed by the significant number of women who either have contraindications to or a personal preference to avoid the use of estrogen-based therapies, while also providing a dosing option for women in whom hormonal ABT is indicated.”
The linzagolix marketing authorization application (MAA) was validated by the European Medicine Agency (EMA) with an approval recommendation from the Committee for Medicinal Products for Human Use (CHMP) expected in Q4 2021. Obseva announced previously that the company has entered into a partnership with Syneos Health to support commercialization of linzagolix in the US and EU.
About Linzagolix
Linzagolix is a novel, once daily, oral GnRH receptor antagonist with a potentially best-in-class profile1,2,3. Linzagolix is the subject of submitted marketing authorization applications for the treatment of heavy menstrual bleeding associated with uterine fibroids and is currently in late-stage clinical development for the treatment of pain associated with endometriosis. Obseva licensed linzagolix from Kissei in late 2015 and retains worldwide commercial rights, excluding Asia, for the product. Linzagolix is not currently approved anywhere in the world.
About the Phase 3 PRIMROSE Program in Uterine Fibroids
PRIMROSE 1 & 2 were prospective, randomized, parallel group, double-blind, placebo-controlled Phase 3 studies that investigated the efficacy and safety of two dosing regimens of linzagolix, 100 mg and 200 mg once daily, alone and in combination with hormonal ABT (1 mg estradiol and 0.5 mg norethisterone acetate) for the treatment of heavy menstrual bleeding associated with uterine fibroids. PRIMROSE 1 was conducted in the United States and enrolled 574 women. PRIMROSE 2 was conducted in Europe and the United States and enrolled 535 women. Both trials comprised a 52-week treatment period followed by a 6-month post treatment follow-up period. Additional information can be found here.
About Uterine Fibroids
Uterine fibroids are common benign tumors of the muscular tissue of the uterus which affect women of childbearing age and can vary in size from undetectable to large bulky masses. Few long-term medical treatments are available, and as a result, approximately 300,000 hysterectomies are performed for uterine fibroids every year in the US.
The symptoms of uterine fibroids are wide-ranging and include heavy menstrual bleeding, anemia, pelvic pressure and bloating, urinary frequency and pain that can be extremely debilitating with a significant impact on quality of life. These symptoms can also have an impact on mental health, creating the additional burden of anxiety and distress.
About Obseva
Obseva is a biopharmaceutical company built to address some of the most challenging unmet needs in women’s health – an under-researched, under-invested field of medicine. With deep expertise in clinical development, Obseva is passionate about the pursuit of advances that benefit women and their health and the importance of delivering truly meaningful innovation in this space. Through strategic in-licensing and disciplined drug development, Obseva has established a late-stage clinical pipeline with development programs focused on new therapies for the treatment of uterine fibroids, endometriosis, and preterm labor. Obseva is listed on the Nasdaq Global Select Market and is traded under the ticker symbol “OBSV” and on the SIX Swiss Exchange where it is traded under the ticker symbol “OBSN”. For more information, please visit http://www.ObsEva.com.
About Kissei
Kissei is a Japanese pharmaceutical company with approximately 70 years of history, specialized in the field of urology, kidney-dialysis and unmet medical needs. Silodosin is a Kissei product for the treatment of the signs and symptoms of benign prostatic hyperplasia which is sold worldwide through its licensees. KLH-2109/OBE2109 is a new chemical entity discovered by Kissei R&D.

……………………………

PATENT
WO 2007046392
https://patents.google.com/patent/WO2007046392A1/en
PATENT
WO 2014042176
https://patents.google.com/patent/WO2014042176A1/en

(Process 1)
Compound (D) can be produced by reacting compound (B) or a salt thereof with compound (C) in the presence of a base in a solvent. Examples of the solvent include halogen solvents such as dichloromethane, cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran, and tetrahydropyran, amide solvents such as N, N-dimethylformamide, aromatic hydrocarbon solvents such as toluene, A nitrile solvent such as acetonitrile, an ester solvent such as ethyl acetate, or a mixed solvent thereof and a mixed solvent thereof and water are preferable, and a mixed solvent of tetrahydrofuran and water is preferable. Examples of the base include organic bases such as triethylamine and pyridine, and inorganic bases such as sodium hydrogen carbonate, potassium hydrogen carbonate, cesium carbonate, sodium carbonate, and potassium carbonate, preferably triethylamine, sodium hydrogen carbonate, or potassium carbonate Is mentioned. The equivalent of the base may be an equivalent amount capable of neutralizing the salt and neutralizing the acid generated by the reaction. The equivalent of (C) can be used in an amount of 0.8 to 1.1 equivalents relative to (B), preferably 1.0 equivalent. The reaction temperature is usually 0 to 30 ° C., and the reaction time is usually 0.5 to 3 hours, although it varies depending on the raw material used, the solvent, the reaction temperature and the like. Examples of the salt of the compound (B) include a salt with an inorganic acid, a salt with an organic acid, a salt with an acidic amino acid, and the like. Examples of the salt with an inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like. Examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, p-toluene And salts with sulfonic acid and the like. Examples of salts with acidic amino acids include salts with aspartic acid, glutamic acid and the like. Among these salts, salts with hydrochloric acid and methanesulfonic acid are preferable. Compound (C) used in Scheme 1 may be a commercially available product, or can be produced according to a known method or a method analogous thereto. Compound (D) may be isolated before the next step, but it can also be used in the next step without isolation.(Process 2)
Compound (F) can be produced by reacting compound (D) with compound (E) or a salt thereof in a solvent in the presence or absence of a base. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran, tetrahydropyran, amide solvents such as N, N-dimethylformamide, aromatic hydrocarbon solvents such as toluene, nitrile solvents such as acetonitrile, An ester solvent such as ethyl acetate or a mixed solvent thereof and a mixed solvent thereof with water, and the like are preferable, and a mixed solvent of tetrahydrofuran and water is preferable. Examples of the base include organic bases such as N, N-dimethylaminopyridine, triethylamine, N-methylpyrrolidine, N-methylmorpholine, diisopropylethylamine, and preferably N, N-dimethylaminopyridine, triethylamine and the like. . The equivalent of the base can be used in an amount of 0.1 to 2.0 equivalents relative to the compound (E), preferably 0.1 to 0.5 equivalents (provided that when a salt of the compound (E) is used, Further base necessary for neutralization is required). The reaction temperature is from room temperature to 60 ° C., and the reaction time is usually from 1 to 24 hours, although it varies depending on the raw material used, the solvent, the reaction temperature, and the like. Examples of the salt of compound (E) include a salt with an inorganic acid, a salt with an organic acid, a salt with an acidic amino acid, and the like. Examples of the salt with an inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like. Examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, p-toluene And salts with sulfonic acid and the like. Examples of salts with acidic amino acids include salts with aspartic acid, glutamic acid and the like. Compound (F) may be isolated before the next step, but it can also be used in the next step without isolation.(Process 3)
The intramolecular cyclization and hydrolysis reaction in this step can be performed simultaneously or separately.
(Step 3-1)
Compound (A) can be produced by subjecting compound (F) to intramolecular cyclization and hydrolysis in the presence of a base in a solvent. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran and tetrahydropyran, lower alcohols such as methanol, ethanol and 2-propanol, amide solvents such as N, N-dimethylformamide, and nitriles such as acetonitrile. Examples thereof include a solvent and the like or a mixed solvent of a mixed solvent thereof and water, and a mixed solvent of tetrahydrofuran / methanol / water is preferable. Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide and sodium hydride, and metal alkoxides such as sodium methoxide and potassium tert-butoxide, preferably lithium hydroxide and sodium And methoxide. The base can be used in an amount of 3.0 to 6.0 equivalents, preferably 4.0 to 4.5 equivalents, relative to compound (F). The reaction temperature is usually from 0 to 20 ° C., and the reaction time is usually from 1 to 10 hours, although it varies depending on the raw material used, solvent, reaction temperature and the like.
(Step 3-2)
When isolating compound (G), compound (G) can be produced by subjecting compound (F) to an intramolecular cyclization reaction in a solvent in the presence of a base. Examples of the solvent include cyclic ethers such as tetrahydrofuran, 2-methyltetrahydrofuran and tetrahydropyran, lower alcohols such as methanol, ethanol and 2-propanol, amide solvents such as N, N-dimethylformamide, and nitriles such as acetonitrile. Examples thereof include a solvent and the like or a mixed solvent thereof, and a mixed solvent of tetrahydrofuran / methanol is preferable. Examples of the base include inorganic bases such as sodium hydroxide, potassium hydroxide, lithium hydroxide or sodium hydride, metal alkoxides such as sodium methoxide and potassium tert-butoxide, and lithium hydroxide, sodium methoxide and the like. preferable. The base can be used in an amount of 0.1 to 1.5 equivalents, preferably 1.0 to 1.1 equivalents, relative to compound (F). The reaction temperature is usually from 0 to 20 ° C., and the reaction time is usually from 1 to 10 hours, although it varies depending on the raw material used, solvent, reaction temperature and the like.
(Step 3-3)
The hydrolysis reaction in this step can be performed by the same method as in step 3-1 or a method analogous thereto.(Process 4)
Compound (A) can be converted to a salt thereof by a conventional method. Examples of such salts include inorganic salts such as sodium salt, potassium salt, calcium salt, magnesium salt, triethylamine, diisopropylamine, N, N′-dibenzylethylenediamine, ethanolamine, (2-hydroxyethyl) trimethylammonium. (Hereinafter referred to as choline), addition salts with organic bases such as N-methylglucamine, arginine, lysine and the like, and choline salts are preferred. Examples of the reagent used for conversion to the choline salt include choline hydroxide, choline bicarbonate, choline chloride and choline acetate.Here, the compound (B) and the salt thereof used in the above-mentioned scheme 1 are commercially available, or manufactured by the method described in a) to c), the method described in the reference examples, or a method analogous thereto. Can do.
a) JP-A 64-29373
b) Synthetic Communications, 32, 2565 (2002)
c) Synthesis, 200 (1977)Further, the compound (E) or a salt thereof used in the scheme 1 can be produced by the method described in Patent Document 1, the method described in Reference Examples, or a method analogous thereto.The compound obtained in the production process in the present specification includes hydrates or solvates thereof, and any of them can be used. Furthermore, the compound obtained in the production process in the present specification may have tautomers and / or geometric isomers, any of which can be used, and also a mixture thereof. be able to.By the production method of the present invention, the compound (A) useful as a pharmaceutical product or a salt thereof can be obtained in high yield and high purity through the compound (D) which is a production intermediate.The content of the present invention will be described in more detail by the following examples, but the present invention is not limited to the content.Reference example 1
Dimethyl 4-oxothiolane-2,3-dicarboxylate methylthioglycolate (15.0 g), tetrahydrofuran (45 g), piperidine (0.361 g) in a reaction mixture at room temperature with dimethyl maleate (21.4 g) in tetrahydrofuran (30 g) The solution was added. To the reaction mixture was added 20% sodium methoxide in methanol (43 g) at 55 ° C. under a nitrogen atmosphere. The reaction mixture was stirred at reflux for 3 hours. Diisopropyl ether (105 g) and acetic acid (0.85 g) were added to the reaction mixture at 45-50 ° C., and then cooled. The suspension was filtered to obtain wet crystals (43.3 g) of sodium salt of dimethyl 4-oxothiolane-2,3-dicarboxylate. The wet crystals were added to a mixture of 85% phosphoric acid (9.8 g), water (20 g) and ethyl acetate (150 g) at room temperature, and the aqueous layer was removed. The obtained organic layer was washed with 10% brine and then dried over anhydrous magnesium sulfate. The drying agent was removed by filtration, and the filtrate was concentrated under reduced pressure to obtain the title compound (22.7 g).Reference example 2
Dimethyl 4- (hydroxyimino) thiolane-2,3-dicarboxylate Dimethyl 4-oxothiolane-2,3-dicarboxylate (10.0 g), pyridine (5.44 g), hydroxylamine hydrochloride (3.34 g) Was stirred at 50 ° C. for 1 hour. Ethyl acetate and 7% aqueous phosphoric acid solution were added to the reaction mixture at room temperature, and the aqueous layer was removed. The obtained organic layer was washed with 5% sodium bicarbonate water and 10% brine. The organic layer was dried over anhydrous sodium sulfate. After removing the desiccant by filtration, the filtrate was concentrated under reduced pressure to obtain the title compound (10.4 g).Reference example 3
4-Aminothiophene-2,3-dicarboxylic acid dimethyl hydrochloride 4- (hydroxyimino) thiolane-2,3-dicarboxylate (10.4 g) in acetic acid (32 g) solution in 4N-hydrogen chloride / ethyl acetate solution ( 120 g) was added at room temperature. The reaction mixture was stirred at room temperature for 8 hours. After filtering the suspension, the obtained solid was dried to obtain the title compound (9.42 g).Reference example 4
4-Aminothiophene-2,3-dicarboxylic acid dimethyl methanesulfonate To a solution of methanesulfonic acid (80.0 g) in ethyl acetate (900 g), dimethyl 4- (hydroxyimino) thiolane-2,3-dicarboxylate (97. 1 g) of ethyl acetate (500 g) was added at 65-75 ° C. The reaction mixture was stirred at the same temperature for 2 hours. Methyl isobutyl ketone (100 g) was added at 45-50 ° C. and cooled to room temperature. After filtering the suspension, the obtained solid was dried to obtain the title compound (102 g).Reference Example 5
1,2-difluoro-3-[(4-fluoro-2-methoxyphenoxy) methyl] -4-methoxybenzene sodium borohydride in a solution of 2,3-difluoro-6-methoxybenzaldehyde (150 g) in toluene (900 g) (13.2 g) of 0.1N sodium hydroxide aqueous solution (180 g) was added at 35 to 39 ° C. The reaction mixture was stirred at the same temperature for 5 hours. After cooling the reaction mixture to room temperature, the aqueous layer was removed. The obtained organic layer was washed with 20% brine to obtain a toluene solution of 2,3-difluoro-6-methoxybenzyl alcohol. To this solution was added concentrated hydrochloric acid (610 g) at room temperature. The reaction mixture was stirred at 38-43 ° C. for 5 hours. After cooling the reaction mixture to room temperature, the aqueous layer was removed. The obtained organic layer was washed with water and 20% brine to obtain a toluene solution of 3- (chloromethyl) -1,2-difluoro-4-methoxybenzene. To this solution, 4-fluoro-2-methoxyphenol (125 g) and tetrabutylammonium bromide (56.2 g) were added at room temperature. A 25% aqueous sodium hydroxide solution (170 g) was added to the reaction mixture at 60 to 63 ° C., and the mixture was stirred at the same temperature for 4 hours. Water was added to the reaction mixture and the aqueous layer was removed. The obtained organic layer was washed with water and concentrated under reduced pressure. The residue was dissolved in 2-propanol and water was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (232 g).Reference Example 6
1,2-difluoro-3-[(4-fluoro-2-methoxy-5-nitrophenoxy) methyl] -4-methoxybenzene 1,2-difluoro-3-[(4-fluoro-2-methoxyphenoxy) methyl ] To a solution of 4-methoxybenzene (158 g) in acetic acid (1200 g) was added 60% nitric acid (72.2 g) at 59-62 ° C., and the mixture was stirred at the same temperature for 2 hours. Water (1200 g) was added to the suspension at 15 to 19 ° C., and the mixture was stirred at the same temperature for 1 hour. After filtering the suspension, the obtained solid was washed with water to obtain wet crystals of the title compound (190 g, Net amount 168 g).Reference Example 7
2-Fluoro-5-[(2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline Raney nickel (2.5 g), ethyl acetate (180 g), 1,2-difluoro-3-[(4 -Fluoro-2-methoxy-5-nitrophenoxy) methyl] -4-methoxybenzene wet crystal (10.9 g, Net amount 10.0 g) was stirred at room temperature under a hydrogen atmosphere for 4 hours. The catalyst was removed by filtration, and the filtrate was concentrated under reduced pressure. The residue was dissolved with methanol and water was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (7.97 g).Example 1
4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylic acid dimethyl potassium carbonate (17.1 g), water (90 g), tetrahydrofuran (150 g) and 4-aminothiophene-2,3-dicarboxylic acid dimethyl hydrochloride (30 0.06) was added phenyl chloroformate (18.6 g) at 6-13 ° C. The reaction mixture was stirred at 12-13 ° C. for 30 minutes, and then the aqueous layer was removed. To the obtained organic layer, tert-butyl methyl ether was added and washed with 20% brine. The obtained organic layer was concentrated under reduced pressure. The residue was dissolved with diisopropyl ether and n-hexane was added. After filtering the suspension, the obtained solid was dried to obtain the title compound (37.0 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.82 (3H, s), 3.82 (3H, s), 7.13-7.30 (3H, m), 7.40-7.46 (2H, m), 7.80 (1H, s ), 10.24 (1H, s)Example 2
4- {3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} dimethyl thiophene-2,3-dicarboxylate 2-fluoro-5-[( 2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline (7.70 g), dimethyl 4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylate (8.65 g), triethylamine (0. 37 g) and tetrahydrofuran (80 mL) were stirred at room temperature for 24 hours. The reaction mixture was concentrated under reduced pressure. Ethyl acetate and methanol were added to the residue. After filtering the suspension, the obtained solid was dried to obtain the title compound (12.0 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.71 (3H, s), 3.82 (3H, s), 3.83 (3H, s), 3.89 (3H, s), 5.00 (2H, d, J = 1.6 Hz), 6.87-6.93 (1H, m), 7.00 (1H, d, J = 12.8Hz), 7.41-7.50 (1H, m), 7.75 (1H, d, J = 8.0Hz), 7.94 (1H, s ), 8.82 (1H, s), 8.95 (1H, s)Example 3
3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] -2,4-dioxo-1,2,3,4-tetrahydrothieno [3,4 d] methyl pyrimidine-5-carboxylate 4- {3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} thiophene-2,3-dicarboxylic acid A methanol solution (3.48 g) of 28% sodium methoxide was added to a suspension of dimethyl (10.0 g) in tetrahydrofuran (40 g), stirred at room temperature for 3 hours, and acetic acid (1.30 g) was added. The reaction mixture was concentrated under reduced pressure. Methanol was added to the residue, and water was further added. After filtering the suspension, the obtained solid was dried to obtain the title compound (8.58 g).
1 H-NMR (DMSO-d 6 ) δ ppm: 3.79 (3H, s), 3.81 (3H, s), 3.84 (3H, s), 4.95 (2H, s), 6.88-6.94 (1H, m), 7.08 (1H, d, J = 11.6Hz), 7.19-7.23 (2H, m), 7.44-7.53 (1H, m), 11.62 (1H, s)Example 4
4- (phenoxycarbonylamino) thiophene-2,3-dicarboxylate potassium carbonate (9.38 kg), water (49 kg), tetrahydrofuran (82 kg), dimethyl 4-aminothiophene-2,3-dicarboxylate hydrochloride (16 4 kg) of the reaction mixture was stirred for 40 minutes, and then phenyl chloroformate (10.1 kg) was added at 11-21 ° C. The reaction mixture was stirred for 30 minutes, and then the aqueous layer was removed to obtain a tetrahydrofuran solution of the title compound.Example 5
4- {3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} dimethyl thiophene-2,3-dicarboxylate 4-obtained in Example 4 To a tetrahydrofuran solution of dimethyl (phenoxycarbonylamino) thiophene-2,3-dicarboxylate, 2-fluoro-5-[(2,3-difluoro-6-methoxyphenyl) methoxy] -4-methoxyaniline (17.0 kg), Tetrahydrofuran (8.5 kg) and triethylamine (1.1 kg) were added, and the mixture was stirred at 50 ° C. for 3.5 hours to obtain a tetrahydrofuran solution of the title compound.Example 6
3- [2-Fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] -2,4-dioxo-1,2,3,4-tetrahydrothieno [3,4 d] pyrimidine-5-carboxylic acid tetrahydrofuranate 4- {3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) -4-methoxyphenyl] ureido} obtained in Example 5 Methanol (41 kg) and water (47 kg) are added to a tetrahydrofuran solution of dimethyl thiophene-2,3-dicarboxylate, a 7.3% lithium hydroxide aqueous solution (80.1 kg) is added at 11 to 13 ° C., and 90 ° C. at 11 ° C. Stir for minutes. Acetic acid (11.4 kg) was added to the reaction mixture at 9 to 16 ° C., and acetic acid (13.0 kg) was further added at 29 to 31 ° C. Seed crystals were added to the reaction mixture, and the mixture was stirred at the same temperature for 30 minutes. Water (34 kg) was added to the suspension and stirred at 30 ° C. for 40 minutes. The suspension was stirred at 4-9 ° C. for 90 minutes. After the suspension was filtered, the obtained solid was washed with a mixed solution of methanol (54 kg) and water (68 kg) to give wet crystals of the title compound (31.64 kg, Net amount (compound (A) free form equivalent)) 26 0.7 kg) was obtained.
A part of the wet crystals of the title compound was dried under reduced pressure at an external temperature of 60 ° C., and 1 H-NMR, HPLC and powder X-ray diffraction were measured on the obtained dried crystals of the title compound.
1 H-NMR (DMSO-d 6 ) δ ppm: 1.68-1.82 (3H, m), 3.53-3.65 (3H, m), 3.80 (3H, s), 3.81 (3H, s), 4.94-4.98 (2H , m), 6.87-6.94 (1H, m), 7.13 (1H, d, J = 11.2Hz), 7.25 (1H, d, J = 7.2Hz), 7.39 (1H, s), 7.43-7.52 (1H, m), 11,99 (1H, s), 14.53 (1H, s)
PATENT
WO 2020089190
https://patents.google.com/patent/WO2020089190A2/enFor example, the GnRH antagonist may be 3-[2-fluoro-5-(2,3-difluoro-6-methoxybenzyloxy)4- methoxyphenyl]-2,4-dioxo-1 ,2,3,4- tetrahydrothieno [3,4d]pyrimidine-5-carboxylic acid, or a pharmaceutically acceptable salt thereof. The salt may be, for instance, the choline salt thereof, represented by formula (Via), below.
Compound (VI) and pharmaceutically acceptable salts thereof, such as the choline salt thereof (compound (Via)), can be synthesized, for example, using the methodology described in WO 2014/042176, the disclosure of which is incorporated herein by reference in its entirety. An exemplary synthetic scheme that may be used for the preparation of compound (VI) and the choline salt thereof is shown in Scheme 1 , below.Scheme 1 . Exemplary preparation of compound (VI) and the choline salt thereof




wherein Ri and R are each independently C alkoxy groups; LG is a nucleofugal leaving group, such as chlorine or bromine, among others; R represents an optional substituent, such as halogen, acyl group, C alkyl group, or a nitro substituent; DMAP denotes A/-dimethylaminopyridine; and TEA denotes trimethylamine.Crystalline compound (Via) has been characterized spectroscopically, for instance, in US Patent No. 9,169,266, the disclosure of which is incorporated herein by reference in its entirety. The foregoing crystalline form has been shown to exhibit characteristic X-ray powder diffraction peaks at about 7.10 2Q, about 11 .5° 2Q, about 19.4° 2Q, about 21 .5° 2Q, about 22.0° 2Q, about 22.6° 2Q, about 23.5° 2Q, and about 26.2° 2Q. Additionally, this crystalline form exhibits 13C solid-state nuclear magnetic resonance (NMR) peaks centered at about 55.5 ppm, about 57.1 ppm, about 58.7 ppm, about 69.8 ppm, about 98.1 ppm, about 110.3 ppm, about 1 1 1 .6 ppm, about 113.7 ppm, about 1 18.0 ppm, about 145.3 ppm, about 149.8 ppm, and about 155.8 ppm. This crystalline form further exhibits 19F solid-state NMR peaks centered at about -151.8 ppm, -145.2 ppm, and -131 .6 ppm.Compound (VI), as well as pharmaceutically acceptable salts thereof, such as the choline salt thereof, exhibit a high affinity for human GnRH receptor (27.4 nM). Using the compositions and methods described herein, a patient that is presenting with or has been diagnosed as having, adenomyosis or rectovaginal endometriosis may be administered a compound of formula (VI), or a pharmaceutically acceptable salt thereof, such as the choline salt thereof, to treat the disease or ameliorate one or more symptoms of the disease. Exemplary doses of compound (VI) and pharmaceutically acceptable salts thereof, such as the choline salt thereof, include doses of from 25 mg to 500 mg daily, such as doses of 100 mg per day and 200 mg per day. Additional dosing information is provided below.3-Aminoalkyl pyrimidine-2, 4(1 H,3H)-dionesAdditional GnRH antagonists that may be used in conjunction with the compositions and methods described herein include optionally substituted 3-aminoalkyl pyrimidine-2, 4(1 H,3H)-dione derivatives, such as compounds represented by formula (VII)

PATENTWO 2021023876https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021023876&_cid=P11-KWFRM2-91270-1
In some embodiments, the compound is the choline salt of the compound represented by formula (VI), choline 3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) 4-methoxyphenyI] -2,4- dioxo-1,2,3,4-
tetrahydrothieno [3,4d] pyrimidine-5-carboxylate. It is to be understood that references herein to a compound represented by formula (VI) specifically include the choline salt of compound (VI), which is represented by formula (VIa), below.
In some embodiments, the choline 3- [2-fluoro-5- (2,3-difluoro-6-methoxybenzyloxy) 4-methoxyphenyI] -2,4-dioxo-1,2,3,4- tetrahydrothieno [3,4d ] pyrimidine-5-carboxylate is in a crystalline state.
PATENT
WO 2021023877
References
- ^ Jump up to:a b c “Linzagolix – Kissei Pharmaceutical/ObsEva – AdisInsight”.
- ^ Jump up to:a b Ezzati M, Carr BR (2015). “Elagolix, a novel, orally bioavailable GnRH antagonist under investigation for the treatment of endometriosis-related pain”. Womens Health (Lond). 11 (1): 19–28. doi:10.2217/whe.14.68. PMID 25581052.
- ^ Chodankar, Rohan; Allison, Jennifer (2018). “New Horizons in Fibroid Management”. Current Obstetrics and Gynecology Reports. 7 (2): 106–115. doi:10.1007/s13669-018-0242-6. ISSN 2161-3303.
External links
| Clinical data | |
|---|---|
| Trade names | Yselty |
| Other names | KLH-2109; OBE-2109 |
| Routes of administration | By mouth[1][2] |
| Drug class | GnRH modulator; GnRH antagonist; Antigonadotropin |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 935283-04-8 |
| PubChem CID | 16656889 |
| ChemSpider | 17590169 |
| UNII | 7CDW97HUEX |
| KEGG | D11608 |
| ChEMBL | ChEMBL3668014 |
| Chemical and physical data | |
| Formula | C22H15F3N2O7S |
| Molar mass | 508.42 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////LINZAGOLIX, Hormone Antagonists, WHO 10711, KLH-2109, KLH 2109, OBE-2109, OBE 2109

NEW DRUG APPROVALS
ONE TIME
$10.00
Ropeginterferon alfa-2b
PCDLPQTHSL GSRRTLMLLA QMRRISLFSC LKDRHDFGFP QEEFGNQFQK AETIPVLHEM
IQQIFNLFST KDSSAAWDET LLDKFYTELY QQLNDLEACV IQGVGVTETP LMKEDSILAV
RKYFQRITLY LKEKKYSPCA WEVVRAEIMR SFSLSTNLQE SLRSKE
(Disulfide bridge: 2-99, 30-139)
Ropeginterferon alfa-2b
- AOP2014
CAS 1335098-50-4
UNII981TME683S
FDA APPROVED, 2021/11/12, BESREMI
PEPTIDE, Antineoplastic, Antiviral
Polycythemia vera (PV) is the most common Philadelphia chromosome-negative myeloproliferative neoplasm (MPN), characterized by increased hematocrit and platelet/leukocyte counts, an increased risk for hemorrhage and thromboembolic events, and a long-term propensity for myelofibrosis and leukemia.1,2 Interferon alfa-2b has been used for decades to treat PV but requires frequent dosing and is not tolerated by all patients.2 Ropeginterferon alfa-2b is a next-generation mono-pegylated type I interferon produced from proline-IFN-α-2b in Escherichia coli that has high tolerability and a long half-life.4,6 Ropeginterferon alfa-2b has shown efficacy in PV in in vitro and in vivo models and clinical trials.3,4
Ropeginterferon alfa-2b was approved by the FDA on November 12, 2021, and is currently marketed under the trademark BESREMi by PharmaEssentia Corporation.6
Ropeginterferon alfa-2b, sold under the brand name Besremi, is a medication used to treat polycythemia vera.[1][2][3][4] It is an interferon.[1][3] It is given by injection.[1][3]
The most common side effects include low levels of white blood cells and platelets (blood components that help the blood to clot), muscle and joint pain, tiredness, flu-like symptoms and increased blood levels of gamma-glutamyl transferase (a sign of liver problems).[3] Ropeginterferon alfa-2b can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness.[2] Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).[2]
It was approved for medical use in the European Union in February 2019,[3] and in the United States in November 2021.[2][5] Ropeginterferon alfa-2b is the first medication approved by the U.S. Food and Drug Administration (FDA) to treat polycythemia vera that people can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.[2]
https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-rare-blood-disease#:~:text=FDA%20NEWS%20RELEASE-,FDA%20Approves%20Treatment%20for%20Rare%20Blood%20Disease,FDA%2DApproved%20Option%20Patients%20Can%20Take%20Regardless%20of%20Previous%20Therapies,-ShareFor Immediate Release:November 12, 2021
Today, the U.S. Food and Drug Administration approved Besremi (ropeginterferon alfa-2b-njft) injection to treat adults with polycythemia vera, a blood disease that causes the overproduction of red blood cells. The excess cells thicken the blood, slowing blood flow and increasing the chance of blood clots.
“Over 7,000 rare diseases affect more than 30 million people in the United States. Polycythemia vera affects approximately 6,200 Americans each year,” said Ann Farrell, M.D., director of the Division of Non-Malignant Hematology in the FDA’s Center for Drug Evaluation and Research. “This action highlights the FDA’s commitment to helping make new treatments available to patients with rare diseases.”
Besremi is the first FDA-approved medication for polycythemia vera that patients can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.
Treatment for polycythemia vera includes phlebotomies (a procedure that removes excess blood cells though a needle in a vein) as well as medicines to reduce the number of blood cells; Besremi is one of these medicines. Besremi is believed to work by attaching to certain receptors in the body, setting off a chain reaction that makes the bone marrow reduce blood cell production. Besremi is a long-acting drug that patients take by injection under the skin once every two weeks. If Besremi can reduce excess blood cells and maintain normal levels for at least one year, then dosing frequency may be reduced to once every four weeks.
The effectiveness and safety of Besremi were evaluated in a multicenter, single-arm trial that lasted 7.5 years. In this trial, 51 adults with polycythemia vera received Besremi for an average of about five years. Besremi’s effectiveness was assessed by looking at how many patients achieved complete hematological response, which meant that patients had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots. Overall, 61% of patients had a complete hematological response.
Besremi can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness. Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).
Interferon alfa products like Besremi may cause or worsen neuropsychiatric, autoimmune, ischemic (not enough blood flow to a part of the body) and infectious diseases, which could lead to life-threatening or fatal complications. Patients who must not take Besremi include those who are allergic to the drug, those with a severe psychiatric disorder or a history of a severe psychiatric disorder, immunosuppressed transplant recipients, certain patients with autoimmune disease or a history of autoimmune disease, and patients with liver disease.
People who could be pregnant should be tested for pregnancy before using Besremi due to the risk of fetal harm.
Besremi received orphan drug designation for this indication. Orphan drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted the approval of Besremi to PharmaEssentia Corporation.
Medical uses
In the European Union, ropeginterferon alfa-2b is indicated as monotherapy in adults for the treatment of polycythemia vera without symptomatic splenomegaly.[3] In the United States it is indicated for the treatment of polycythemia vera.[1][2][5]
History
The effectiveness and safety of ropeginterferon alfa-2b were evaluated in a multicenter, single-arm trial that lasted 7.5 years.[2] In this trial, 51 adults with polycythemia vera received ropeginterferon alfa-2b for an average of about five years.[2] The effectiveness of ropeginterferon alfa-2b was assessed by looking at how many participants achieved complete hematological response, which meant that participants had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots.[2] Overall, 61% of participants had a complete hematological response.[2] The U.S. Food and Drug Administration (FDA) granted the application for Ropeginterferon_alfa-2b orphan drug designation and granted the approval of Besremi to PharmaEssentia Corporation[2]
REF
- Bartalucci N, Guglielmelli P, Vannucchi AM: Polycythemia vera: the current status of preclinical models and therapeutic targets. Expert Opin Ther Targets. 2020 Jul;24(7):615-628. doi: 10.1080/14728222.2020.1762176. Epub 2020 May 18. [Article]
- How J, Hobbs G: Use of Interferon Alfa in the Treatment of Myeloproliferative Neoplasms: Perspectives and Review of the Literature. Cancers (Basel). 2020 Jul 18;12(7). pii: cancers12071954. doi: 10.3390/cancers12071954. [Article]
- Verger E, Soret-Dulphy J, Maslah N, Roy L, Rey J, Ghrieb Z, Kralovics R, Gisslinger H, Grohmann-Izay B, Klade C, Chomienne C, Giraudier S, Cassinat B, Kiladjian JJ: Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer J. 2018 Oct 4;8(10):94. doi: 10.1038/s41408-018-0133-0. [Article]
- Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, Wolf D, Kralovics R, Gisslinger B, Strecker K, Egle A, Melchardt T, Burgstaller S, Willenbacher E, Schalling M, Them NC, Kadlecova P, Klade C, Greil R: Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015 Oct 8;126(15):1762-9. doi: 10.1182/blood-2015-04-637280. Epub 2015 Aug 10. [Article]
- EMA Approved Products: Besremi (ropeginterferon alfa-2b ) solution for injection [Link]
- FDA Approved Drug Products: BESREMi (ropeginterferon alfa-2b-njft) injection [Link]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d e https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761166s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA Approves Treatment for Rare Blood Disease”. U.S. Food and Drug Administration (FDA) (Press release). 12 November 2021. Retrieved 12 November 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f g “Besremi EPAR”. European Medicines Agency (EMA). Retrieved 14 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Wagner SM, Melchardt T, Greil R (March 2020). “Ropeginterferon alfa-2b for the treatment of patients with polycythemia vera”. Drugs of Today. Barcelona, Spain. 56 (3): 195–202. doi:10.1358/dot.2020.56.3.3107706. PMID 32282866. S2CID 215758794.
- ^ Jump up to:a b “U.S. FDA Approves Besremi (ropeginterferon alfa-2b-njft) as the Only Interferon for Adults With Polycythemia Vera” (Press release). PharmaEssentia. 12 November 2021. Retrieved 14 November 2021 – via Business Wire.
External links
- “Ropeginterferon alfa-2b”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01193699 for “Safety Study of Pegylated Interferon Alpha 2b to Treat Polycythemia Vera (PEGINVERA)” at ClinicalTrials.gov
- Clinical trial number NCT02218047 for “AOP2014 vs. BAT in Patients With Polycythemia Vera Who Previously Participated in the PROUD-PV Study. (CONTI-PV)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Besremi |
| Other names | AOP2014, ropeginterferon alfa-2b-njft |
| License data | EU EMA: by INNUS DailyMed: Ropeginterferon_alfa |
| Pregnancy category | Contraindicated |
| Routes of administration | Subcutaneous |
| Drug class | Interferon |
| ATC code | L03AB15 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2]EU: Rx-only [3] |
| Identifiers | |
| CAS Number | 1335098-50-4 |
| DrugBank | DB15119 |
| UNII | 981TME683S |
| KEGG | D11027 |
/////////Ropeginterferon alfa-2b, FDA 2021, APPROVALS 2021, BESREMI, PEPTIDE, Antineoplastic, Antiviral, AOP 2014, PharmaEssentia

NEW DRUG APPROVALS
ONE TIME
$10.00
SULCONAZOLE

SULCONAZOLEсульконазол , سولكونازول , 硫康唑
- Molecular FormulaC18H15Cl3N2S
- Average mass397.749 Da
1H-Imidazole, 1-[2-[[(4-chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]- [ACD/Index Name]
4332
5D9HAA5Q5S
61318-90-9[RN]
(±)-1-[2,4-Dichloro-b-[(p-chlorobenzyl)thio]phenethyl]imidazole
1-[2-[[(4-Chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole: SulconazoleCAS Registry Number: 61318-90-9
CAS Name: 1-[2-[[(4-Chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole
Additional Names: (±)-1-[2,4-dichloro-b-[(p-chlorobenzyl)thio]phenethyl]imidazole
Molecular Formula: C18H15Cl3N2S
Molecular Weight: 397.75
Percent Composition: C 54.35%, H 3.80%, Cl 26.74%, N 7.04%, S 8.06%
Literature References: Prepn: K. A. M. Walker, DE2541833; idem,US4055652 (1976, 1977 both to Syntex). HPLC determn in plasma: M. Fass et al.,J. Pharm. Sci.70, 1338 (1981). Mechanism of action study: W. H. Beggs, Biochem. Arch.10, 117 (1994). Clinical trial in tinea pedis: W. A. Akers et al.,J. Am. Acad. Dermatol.21, 686 (1989). Review of pharmacology and clinical efficacy: P. Benfield, S. P. Clissold, Drugs35, 143-153 (1988).
Derivative Type: Nitrate
CAS Registry Number: 61318-91-0
Manufacturers’ Codes: RS-44872
Trademarks: Exelderm (Syntex); Myk (Cassenne); Sulcosyn (Syntex)
Molecular Formula: C18H15Cl3N2S.HNO3
Molecular Weight: 460.76
Percent Composition: C 46.92%, H 3.50%, Cl 23.08%, N 9.12%, S 6.96%, O 10.42%
Properties: Colorless crystals from acetone, mp 130.5-132°.
Melting point: mp 130.5-132°
Therap-Cat: Antifungal.
Keywords: Antifungal (Synthetic); Imidazoles.
Sulconazole (trade name Exelderm) is an antifungal medication of the imidazole class. It is available as a cream or solution to treat skin infections such as athlete’s foot, ringworm, jock itch, and sun fungus.[1][2] Although not used commercially for insect control, sulconazole nitrate exhibits a strong anti-feeding effect on the keratin-digesting Australian carpet beetle larvae Anthrenocerus australis.[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

DE 2541833 US 4038409
(Read example 5 and 9 in US patent.)
https://patents.google.com/patent/US4038409A/en
EXAMPLE 5Alternative Route to 1-[β-(R-carbonylthio)phenethyl]imidazolesA. Preparation of 1-[2,4-dichloro-β-(methylcarbonylthio)-phenethyl]imidazole, oxalate.1-(β,2,4-Trichlorophenethylimidazole (1.19g) in 5 ml of dry tetrahydrofuran was added to preformed sodium thioacetate, generated in situ from 720 mg thioacetic acid and sodium hydride (480 mg 57% dispersion in mineral oil) in 20 ml. tetrahydrofuran and the mixture stirred and refluxed under nitrogen for 18 hours. The solvent was removed under reduced pressure, water (20 ml) added and the product extracted with ether. The extracts were washed with water, dried (MgSO4), evaporated and the residue chromatographed on silica gel eluting with 10-20% acetone in dichloromethane. The pure product in ether was treated dropwise with ethereal oxalic acid until precipitation was complete, and the thus obtained oxalate salt of 1-[2,4-dichloro-β-(methylcarbonylthio)phenethyl]imidazole recrystallized from acetone/ethyl acetate with mpBy substituting other available sodium thioacids for sodium thioacetate, other compounds of this invention may be prepared.
EXAMPLE 9A. Preparation of 1-[2,4-dichloro-β-(4-chlorobenzylthio)-phenethyl]imidazoleTo a stirred solution of 330 mg sodium hydroxide in 30 ml methanol under nitrogen is added 810 mg of 1-[2,4-dichloro-β-(methylcarbonylthio)phenethyl]imidazole oxalate and the mixture is stirred at room temperature for ca. 30 minutes (until thin layer chromatography shows the disappearance of the ester). α,p-dichlorotoluene (350 mg) is then added, the solution stirred a further 15 minutes and the solvent removed under reduced pressure. Ether and water are then added to the residue and the ether extract washed with water, dried (MgSO4) and concentrated. Dropwise addition of nitric acid (d = 1.42) until precipitation is complete gives the nitrate salt of 1-[2,4-dichloro-β-(4-chlorobenzylthio)phenethyl]imidazole, recrystallized from acetone, mp 130.5°-132° C.B. By using other compounds of this invention exemplified by those set forth in Examples 2 and 4 and other suitable (substituted) hydrocarbyl halides (or mesylates, tosylates), other compounds may be prepared.
SYN
https://www.sciencedirect.com/science/article/pii/S2095177917301405
SYN
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 6258-66-8 | C7H7ClS | 4-chlorobenzyl mercaptan | Benzenemethanethiol, 4-chloro- |
| 24155-42-8 | C11H10Cl2N2O | 1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethanol | 1H-Imidazole-1-ethanol, α-(2,4-dichlorophenyl)- |
References
- ^ Drugs.com: sulconazole topical
- ^ Fromtling RA (April 1988). “Overview of medically important antifungal azole derivatives”. Clinical Microbiology Reviews. 1 (2): 187–217. doi:10.1128/CMR.1.2.187. PMC 358042. PMID 3069196.
- ^ Sunderland MR, Cruickshank RH, Leighs SJ (2014). “The efficacy of antifungal azole and antiprotozoal compounds in protection of wool from keratin-digesting insect larvae”. Textile Research Journal. 84 (9): 924–931. doi:10.1177/0040517513515312.
| Clinical data | |
|---|---|
| Trade names | Exelderm |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a698018 |
| Routes of administration | Topical |
| ATC code | D01AC09 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 61318-90-9 |
| PubChem CID | 5318 |
| ChemSpider | 5127 |
| UNII | 5D9HAA5Q5S |
| KEGG | D08535 |
| ChEBI | CHEBI:9325 |
| ChEMBL | ChEMBL1221 |
| CompTox Dashboard (EPA) | DTXSID8044129 |
| Chemical and physical data | |
| Formula | C18H15Cl3N2S |
| Molar mass | 397.74 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////SULCONAZOLE, сульконазол , سولكونازول , 硫康唑 , Antifungal,

NEW DRUG APPROVALS
ONE TIME
$10.00
TNO 155
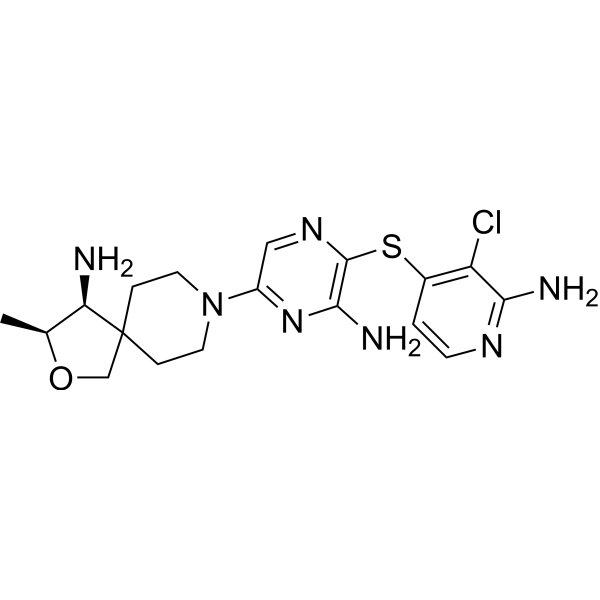
TNO 155
2-Oxa-8-azaspiro[4.5]decan-4-amine, 8-[6-amino-5-[(2-amino-3-chloro-4-pyridinyl)thio]-2-pyrazinyl]-3-methyl-, (3S,4S)-
- (3S,4S)-8-[6-Amino-5-[(2-amino-3-chloro-4-pyridinyl)thio]-2-pyrazinyl]-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine
- (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine
| Molecular Weight |
421.95 |
|---|---|
| Formula |
C₁₈H₂₄ClN₇OS |
| CAS No. |
- PTPN11 inhibitor TNO155
- SHP2 inhibitor TNO155
- TNO-155
- TNO155
- UNII-FPJWORQEGI
TNO155 is a potent selective and orally active allosteric inhibitor of wild-type SHP2 (IC50=0.011 µM). TNO155 has the potential for the study of RTK-dependent malignancies, especially advanced solid tumors.
- Originator Novartis
- Developer Mirati Therapeutics; Novartis
- Class Antineoplastics
- Mechanism of ActionProtein tyrosine phosphatase non receptor antagonists
- Phase I/IISolid tumours
- Phase IColorectal cancer
- 11 Jul 2021Phase I trial in Solid tumours is still ongoing in USA, Canada, Japan, South Korea, Netherlands, Singapore, Spain, Taiwan (NCT03114319)
- 04 Jun 2021Efficacy, safety and pharmacokinetics data from phase I trial in Solid tumours presented at 57th Annual Meeting of the American Society of Clinical Oncology (ASCO-2021)
- 08 Jan 2021Novartis plans a phase Ib/II trial for Solid tumours (Combination therapy, Inoperable/Unresectable, Late-stage disease, Metastatic disease, Second-line therapy or greater) in February 2021 (NCT04699188)
CLIP
Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling
Results: In EGFR-mutant lung cancer models, combination benefit of TNO155 and the EGFRi nazartinib was observed, coincident with sustained ERK inhibition. In BRAFV600E colorectal cancer models, TNO155 synergized with BRAF plus MEK inhibitors by blocking ERK feedback activation by different RTKs. In KRASG12C cancer cells, TNO155 effectively blocked the feedback activation of wild-type KRAS or other RAS isoforms induced by KRASG12Ci and greatly enhanced efficacy. In addition, TNO155 and the CDK4/6 inhibitor ribociclib showed combination benefit in a large panel of lung and colorectal cancer patient–derived xenografts, including those with KRAS mutations. Finally, TNO155 effectively inhibited RAS activation by colony-stimulating factor 1 receptor, which is critical for the maturation of immunosuppressive tumor-associated macrophages, and showed combination activity with anti–PD-1 antibody.
Conclusions: Our findings suggest TNO155 is an effective agent for blocking both tumor-promoting and immune-suppressive RTK signaling in RTK- and MAPK-driven cancers and their tumor microenvironment. Our data provide the rationale for evaluating these combinations clinically.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENT
WO 2015107495
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015107495
PATENT
WO 2020065453
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020065453
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine, which has the formula I,
WO/2015/107495 A1 describes a method for the manufacture of the compound of the formula I which can be characterized by the following reaction scheme 1:
[0008] The last compound resulting from step g above was then reacted as in the following scheme 2:
Scheme 2:
[0009] Thus the compound of formula I is obtained (last compound in the scheme 2, above). The synthesis requires at least the 9 steps shown and is rather appropriate for synthesis in laboratory amounts.
Scheme 1A:
[0016] Therefore, the process, though readily feasible on a laboratory scale, is not ideal for manufacture at a large scale.
[0017] The compound added in reaction b in Scheme 2 is obtained in WO
2015/107495 A1 as “Intermediate 10” follows:
Scheme 3:
[0018] An issue here is the relatively low yield of the amine resulting from reaction a in
Scheme 3.
[0019] In addition, while WO 2015/107495 A1 generically mentions that pharmaceutically acceptable salts of the compound of the formula I may be obtainable, no concrete reason for obtaining such salts and no specific examples of salts are described.
[0020] In addition, given the many potentially salt forming groups in formula I, it is not clear whether any salts with a clear stoichiometry can be formed at all.
Example 1
Method of synthesis of the compound of the formula I ((3S,4S)-8-(6-amino-5-((2-amino-3- chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine):
The overall synthesis can be described by the following Reaction Scheme A:
Scheme A:
Step a
[00293] To a solution of A1 (10.4 kg, 100 mol, 1.0 Eq) in CH2Cl2 (50 L) was added imidazole (8.16 kg, 120 mol, 1.2eq) and TBSCl (18 kg, 120 mol, 1.2 Eq) at 0 °C. After addition, the mixture was stirred at 0°C for 4 h . GC showed the reaction was finished. (A1/ (A1 + A2) < 1%). The reaction mixture was quenched with saturated NaHCO3 (14L) at 0-5°C. Phases were separated. The organic phase was washed with brine (14L). The organic layer was dried over Na2SO4, concentrated under vacuum at 40-45°C to afford A2 (23.3 kg, assay 88%, yield 94%) which was used for the next step directly. 1H NMR (400 MHz, CDC13) δ = 4.35 (d, J= 8.8 Hz, 1H), 3.74 (s, 3H), 2.48 (s, J= 8.8
Hz, 3H), 0.93 (s, 9H), 0.09 (s, 6H).
Step b
[00294] To a solution of A2 (7.5 kg, 34.3 mol, 1.0 Eq) and N,O-dimethylhydroxylamine hydrochloride (6.69 kg, 68.6mol, 2.0 Eq) in THF (20 L) was added drop-wise a solution
of chloro(isopropyl)magnesium (2 M, 51.45 L, 3.5 Eq) at 0 °C under N2 over 5-6 h. After addition, the reaction mixture was stirred at 0 °C for 1h, GC showed the reaction was finished (A2/(A2+A3) < 2 %). The mixture was quenched with NH4Cl (25 L) slowly by keeping the temperature at 0-5°C. After addition, the reaction mixture was stirred for 30min. Phase was separated. The aqueous layer was extracted with EA(2 x 20 L). The combined organic phase was washed with brine (25L), dried over Na2SO4, concentrated to give A3(9.4 kg, assay 86%, yield 95%) which was used for the next step directly. 1HNMR (400 MHz, CDCl3) δ = 4.67 (m, J= 6.6 Hz, 1H), 3.70 (s, 3H), 3.21 (s, 3H), 3.17 (d, 3H)2.48 (s , J= 6.6 Hz, 3H), 0.90 (s, 9H), 0.10 (s, 3H), 0.08 (s, 3H).
[00295] To a solution of A3 (7.1 kg, assay 86%, 24.65 mol, 1.0 Eq) in DCM (30 L) was added dropwise a solution of LiAlH4 (2.4 M, 11.3 L, 1.1 Eq) at -70 °C under N2. Then the reaction mixture was stirred at -70 °C for 3h, and TLC showed the reaction was finished (PSC-1). The mixture was warmed to 0 °C, and then quenched with sat. potassium sodium tartrate (35 L) at 0 °C. After addition, DCM (20L) was added and stirred for 2h at 20-25°C. Phases were separated. The aqueous layer was extracted with DCM (25 L). The combined organic phase was charged with sat. citric acid (45L) and stirred at 0°C for 8h. Phase was separated. The organic phase was washed with NaHCO3 (25L), brine (25 L), dried over Na2SO4, and the solvent was removed under vacuum at 25-30°C. n-Heptane (10 L) was added to the residue and concentrated under vacuum at 30-35°C. n-Heptane (10 L) was added to the residue again and concentrated under vacuum at 30-35°C to give A4 (4.2 kg, assay
60%, yield 54%) which was used for the next step directly.
Step d
[00296] To a solution of diisopropylamine (3.06 kg, 30.3 mol, 1.5 eq) in THF (20 L) cooled to approximately -10°C was added 2.5 M n-BuLi (12.12 L, 30.3 mol, 1.5 eq) under N2. The resulting mixture was stirred at approximately -10 °C for 30min, then a solution of A5 (5.2 kg, 20.20 mol, 1.0eq) in THF (10 L) was added slowly. After addition, the reaction mixture was stirred at -10°C for 30 min, and then cooled to -50°C. A4 (4.18 kg, 22.22 mol, 1.1eq) was added dropwise. After addition, the reaction mixture was stirred at -50°C for 30 min. The mixture was quenched with saturated aqueous NH4Cl (30L) and water (10L) at -50°C. The reaction mixture was warmed to 20-25°C. Phase was separated. The aqueous phase was extracted with EA (3 x 20 L). All organic phases were combined and washed with brine(20L), then concentrated to a yellow oil which was purified by column (silica gel, 100-200 mesh, eluted with n-heptane:EA from 50:1 to 10:1) to give A6 (5.5 kg, assay 90 %, yield 55%) as pale yellow oil. 1H NMR (400 MHz, CDCl3) δ = 4.35-4.15 (m, 2H), 3.95-3.74 (m, 3H), 3.52 (m, 2H), 2.67(m, 2H), 2.12-1.98 (m, 2H), 1.75-1.52 (m, 4H), 1.49 (s, 9H), 1.35-1.10 (m, 6H), 0.98 (s,
9H), 0.02 (s, 6H).
Step e
[00297] To a solution of A6 (11.4 kg, 25.58 mol, 1.0eq) in THF (60 L) was added LiBH4
(836 g, 38.37 mol, 1.5eq) in portions at 5-10 °C, and the reaction mixture was stirred at 20-25 °C for 18 h. HPLC showed the reaction was finished (A6/(A6+A7)<2%). The mixture was cooled to l0°C and slowly quenched with saturated NaHCO3 solution (15 L) and water (25L) with vigorously stirring. After gas formation stopped, vacuum filtration was applied to remove solids. The solid was washed with EA (2 x 15 L). Phase was separated; the aqueous phase was extracted with EA (3 x15L). All organic phases were combined and washed with brine (15L), and concentrated to obtain crude A7 (13.8 kg, assay 58%, yield 77%) which was used for the next step directly.
[00298] To a solution of A7 (8 kg, 19.82 mol, 1.0 eq) in THF (40 L) under nitrogen atmosphere was added TsCl (5.28 kg, 27.75 mol, 1.4 eq) at 10-15°C. After addition, the mixture was cooled to 0 °C, and 1M LiHMDS (29.7 L, 29.73 mol, 1.5 eq) was added dropwise during 2h. After addition, the mixture was stirred at 0°C for 3h. HPLC showed the reaction was finished (PSC-1 A7/ (A7+A8)<7%). TBAF (20.72 kg, 65.67 mol, 3.3 eq) was added into the mixture at 0 °C and the reaction mixture was stirred at 25-30 °C for 48h. HPLC showed the reaction was finished ( PSC-2, A9-intermedaite/(A9-intermediate+A9) < 2%). The mixture was quenched with saturated aqueous sodium bicarbonate solution (32L) and stirred for 30min at 0 °C. Phase was separated, and the aqueous phase was extracted with EA (3 x 20 L). The combined organic phase was washed with brine(20 L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 10:1 to 1:1) to give A9 (4.42 kg, assay 90%, yield 74 %) as pale yellow solid.
Step g
[00299] To a solution of A9 (4.0 kg, 14.74 mol, 1.0 eq) in DCM (40 L) cooled on an ice-bath was added DMP (9.36 kg, 23.58mol, 1.6eq) in portions, and it resulted in a suspension. After addition, the mixture stirred for 4 hours at 20-25°C. HPLC showed the reaction was finished (A9/(A9+A10)<2%). DCM (30L) was added at 0°C. After addition, the mixture was quenched with saturated aqueous Na2SO3 (20 L). The mixture was stirred for 30min at 0 °C, filtered and the white solid was washed with DCM (2 x15L). Phase was separated, and the organic phase was cooled to 0°C, to which was added saturated aqueous NaHCO3 (20L) and stirred for 1h. Phase was separated, and the organic phase was washed with brine(25L), dried over Na2SO4, and concentrated to a yellow oil which was purified by column (eluted with n-heptane:EA from 50:1 to 10:1) to give A10 (3.70 kg, assay 88%, ee value 95.3%, yield 82%) as white solid. 1H NMR (400 MHz, DMSO-d6) δ = 4.20 (d, J = 8.0 Hz,
1H), 3.98-3.67 (m, 4H), 3.08-2.90 (m, 2H), 1.54-1.39(m, 13H), 1.18 (d, J = 8.0 Hz, 3H).
Step h
[00300] To a solution of A10 (4.60 kg, 17.08 mol, 1.0 eq) in THF (40 L) was added
Ti(OEt)4 (15.58 kg, 68.32 mol, 4.0 eq) and (R)-t-Butyl sulfmamide (4.14 kg, 34.16 mol, 2.0 eq) at 25 °C. After addition, the mixture was heated to 70°C and stirred for 20h. HPLC showed the reaction was finished (PSC-l, A10/(A10+A12)<4%). The mixture was cooled to -30— 40°C, and MeOH (4 L) was added dropwise within 30 min and stirred for 1 h. 2M L1BH4 (8.1 L) solution was added dropwise to the reaction mixture at -40- -50°C and stirred for 1h. HPLC indicated all of imine was consumed (PSC-2, A12/(A12+A13)<1%). The mixture was warmed to -30 °C and stirred for 1h, then warmed to 0 °C within 2 h and stirred for 1h, then warmed to 20-25 °C and stirred for 30min. IP AC ( 25L) was added to above mixture, NaHCO3(5L) was added dropwise in about 1h at 25 °C and stirred for 30 min. The mixture was filtered under vacuum and the cake was washed with IP AC (8 x15L). The combined organic phase was washed with brine (25L), then evaporated under vacuum to get a solution of A13
(about 28kg) which was used for next step.
[00301] To a mixture of A13 in IPAC (about 28 kg, 17.08 mol, 1.0 eq) was added dropwise
4M HCl/IPA (8.54 L, 34.16 mol, 2.0 eq) at -5 °C and stirred for 5h at -5 °C. HPLC showed that A13 was consumed completely (A13/(A14+A13)<1%). MTBE (25 L) was added to above mixture within
30 min and stirred for 30 min at -5 °C .The solid was collected by vacuum filtration. The cake was washed with MTBE (2 x 2.5 L). The wet cake was used for next step directly.
Step j
[00302] The wet solid A14 (from 9.2 kg A10) was stirred in MTBE(76 L) at 25°C, then the
16% NaOH (9.84 kg) solution was added dropwise to the MTBE suspension while maintaining IT<10ºC. After addition, the mixture was stirred for 15 min and all solids were dissolved at 0°C. The organic phase was separated, and the aqueous phase was extracted with MTBE (2 x 20L). The combined organic phase was washed with brine (10 L) and evaporated under vacuum to remove all MTBE. ACN (24 L) was added to above residue, and the mixture was evaporated under vacuum to remove the organic solvents and yielded a crude A15 (5.42 kg, qnmr 90%, 18.04 mol, 1.0 eq). ACN (34.68 kg) was added to above residue and stirred for 10 min at 65°C. A solution of (-)-O-acetyl-D-mandelic acid (3.15kg,16.2 mol, 0.9 eq) in ACN(11.6 kg) was added drop-wise to the mixture (firstly added 1/3, stirred for 0.5 h, then added the others) over 3h. The mixture was stirred for 1 h at 65°C, then cooled to 25°C over 4h and stirred for l2h at 25°C . The solid was collected by vacuum filtration, and the cake was washed with pre-cooled ACN (2 x15kg) (PSC-1) and dried under vacuum to give
A16 (7.36 kg, yield 46% from A10 to A16). 1H NMR (400 MHz, DMSO-d6) δ = 7.43-7.29 (m, 5H),
5.58 (s, 2H), 4.12-4.07 (m, 1H), 3.75-3.65 (m, 3H), 3.51-3.49 (m, 1H), 3.18-3.17 (m, 1H), 2.84 (bs,
2H), 2.05 (s, 3H), 1.60-1.40 (m, 13H), 1.14-1.12 (d, J= 8.0 Hz, 3H).
Step k
[00303] To a solution of A16 (15 g) in MeOH (90 mL) was added dropwise 5N HC1/IPA
(45 mL) at room temperature within 15 minutes. After the addition, the mixture was stirred for 6 hours.
IP AC (180 mL) was added dropwise to above mixture within 1h at room temperature. The resulting mixture was stirred for another 30 minutes before it was cooled to 0-5 °C. The mixture was stirred at 0- 5 °C for another 2h and the precipitants were collected by filtration. The cake was washed with (45*2 mL) IP AC, dried under vacuum at 60 °C overnight to afford the product as a white solid. 1H NMR (400
MHz, DMSO-d6) δ = 9.37 (br s, 1H), 9.25 (br s, 1H), 8.42 (br s, 3H), 4.26 – 4.17 (m, 1H), 3.72 (ABq, J
= 9.1 Hz, 2H), 3.50 – 3.41 (m, 1H), 3.28 – 3.18 (m, 1H), 3.18 – 3.09 (m, 1H), 2.99 – 2.74 (m, 2H), 2.07 – 1.63 (m, 4H), 1.22 (d, J= 6.5 Hz, 3H).
Step l
[00304] To a mixture of A17 (10 g) and Z17a (9.5 g) in DMAC (60 mL) was added K2CO3
(22.5 g) and H2O (40 mL) at room temperature. The mixture was degassed with nitrogen and stirred at
90 °C overnight. The mixture was cooled to room temperature, diluted with Me-THF (500 mL) and
H2O (280 mL). The organic phase was separated and the aqueous phase was extracted with Me-THF
(300 mL*2). The combined organic phases were washed with brine (200 mL*3), concentrated under
vacuum to remove most of the solvent. The residue was diluted with IPA (60 mL) and H2O (20 mL), stirred at 50 °C for 1h, cooled to 5 °C within 3h, stirred at this temperature for 1h. The solid was collected by vacuum filtration, dried under vacuum to afford the product as a yellow solid (l2g,
87.4%). 1H NMR (400 MHz, DMSO-d6)δ = 7.64 (d, J= 6.2 Hz, 1H), 7.62 (s, 1H), 6.26 (s, 2H), 6.13 (s, 2H), 5.74 (d, J= 5.3 Hz, 1H), 4.12 – 4.02 (m, 1H), 3.90 – 3.78 (m, 2H), 3.67 (d, J= 8.4 Hz, 1H), 3.49 (d, J= 8.4 Hz, 1H), 3.33 (s, 2H), 2.91 (d, J= 5.1 Hz, 1H), 1.78 – 1.68 (m, 1H), 1.67 – 1.57 (m, 1H), 1.56 – 1.41 (m, 2H), 1.08 (d, J= 6.5 Hz, 3H).
Example 2
Formation of the succinate salt of the compound of the formula I:
[00305] The reaction is summarized by the following Reaction Scheme:
[00306] To a mixture of A18 (10 g) in MeOH (76 g) and H2O (24 g) was added succinic acid (2.94 g) at room temperature. The mixture was heated to 50 °C and stirred for 30 minutes to dissolve all solid. The solution was added to IPA (190 mL) at 60-65 °C. The resulting mixture was stirred at 60 °C >5 hours, cooled to -15 °C within 5 hours and stirred at this temperature >4 hours. The solid was collected by vacuum filtration, dried under vacuum to afford the product as an off-white solid(l0.8 g, 82.8%). 1H NMR (400 MHz, DMSO-d6)δ = 7.64 (d, J= 6.2 Hz, 1H), 7.63 (s, 1H), 6.26 (s, 2H), 6.16 (s, 2H), 5.74 (d, J= 5.3 Hz, 1H), 4.12 – 4.02 (m, 1H), 3.90 – 3.78 (m, 2H), 3.67 (d, J= 8.4 Hz, 1H), 3.49 (d, J= 8.4 Hz, 1H), 3.33 (s, 2H), 2.91 (d, J= 5.1 Hz, 1H), 2.34 (s, 4H), 1.71 – 1.60 (m, 4H), 1.13 (d, J = 6.5 Hz, 3H).
[00307] In a special variant, the reaction follows the following Reaction Scheme, also including an optional milling to yield the final product:
Example 3
Formation of the intermediate Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2- amine). Variant 1:
[00308] The compound Z17a was obtained by reaction according to the following Reaction
Scheme:
[00309] In detail, the synthesis of Compound Z17a was carried out as follows:
Step a
[00310] Under nitrogen atmosphere, n-BuLi (2.5M, 7.6 L) was added dropwise to a solution of 3-chloro-2-fluoropyridine (2 kg) in THF (15 L) at -78°C. Then the resultant mixture was stirred for 1h. Then a solution of I2 (4.82 kg) in THF (6 L) was added dropwise. After addition, the reaction mixture was stirred for 30 min, and then quenched with sat. Na2SO3 (10 L), and warmed to 20- 25°C. Phase was separated. The aqueous phase was extracted with EA (2 x 10 L). The combined organic phase was washed with sat.Na2SO3 (2 x 8 L), brine (8 L), and dried over Na2SO4. The organic phase was concentrated under vacuum. The residue was slurried in MeOH (4 L), filtered, and dried to offer 3-chloro-2-fluoro-4-iodopyridine 1c (2.2 kg, yield 68%).
Step b
[00311] Into a solution of Compound 1c (8 kg) in DMSO (48 L) was passed through NH3
(gas) at 80 °C overnight. TLC showed the reaction was finished. The reaction mixture was cooled to RT. The reaction mixture was added to water (140 L). The solid was collected and washed with water (25 L), dried to afford Z17b (6.91 kg, yield 87%). 1H NMR (400 MHz, CDC13) δ = 7.61 (d, J= 6.8 Hz,
1H), 7.14 (s , J= 6.8 Hz, 1H), 5.09 (bs, 2H).
Step c
[00312] A solution of 2-amino-6-chloro-pyrazine la (1 kg, 7.69 mol) in DCM (15 L) was heated to reflux, to which was charged NBS (4l7g) in portions during 1 h. The reaction was cooled to room temperature. The reaction mixture was washed with water (3 L) and brine (3 L). The organic phase was evaporated, and the residue was purified by column chromatography to give product Z17f
(3-bromo-6-chloropyrazin-2-amine) (180 g, 11% yield).
[00313] To a solution of 3-bromo-6-chloropyrazin-2-amine Z17f (6.0 kg, 28.78 mol) in 1,4- Dioxane (40 L) was added Pd(OAc)2 (64.56 g, 287.6 mmol), Xantphos (333 g, 575.6 mmol), and DIPEA (7.44 kg, 57.56 mol) at room temperature under nitrogen. After another 30 minutes purging with nitrogen, methyl 3-mercaptopropanoate (3.81 kg, 31.70 mol) was added, resulting in darkening of the orange mixture. The mixture was heated to 90°C. HPLC showed complete conversion of the starting material. The mixture was allowed to cool to about room temperature, then diluted with EtOAc (40L). After aging for 30 min with stirring, the entire mixture was filtered and solids were washed with EtOAc (3 x 15L). The combined orange filtrate was concentrated to dryness and the solid residue was suspended in DCM (45 L). The mixture was heated to 35-40 °C and stirred for 1h until all solids were dissolved. Then n-heptane (45L) was added dropwise. Upon complete addition, the mixture was cooled to 15-20 °C with stirring for 1h. The solids were collected by vacuum filtration and solids were washed with cold 1:1 DCM/heptane (25 L), then heptane (25 L) (PSC-2). The solids were dried over the weekend to give Z17d (5.32 kg, yield 75%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs,
2H), 3.73 (s, 3H), 3.47 (t, J= 9.2 Hz, 2H), 2.79 (t, J= 9.2 Hz, 2H).
Step e
[00314] To a solution of Z17d (8.0 kg, assay 95%, 30.68 mol) in THF (70 L) was added
EtONa (prepared from 776 g Na and 13.6 L EtOH) at room temperature and the mixture was stirred at
ambient temperature for 1 hour. The mixture was then concentrated to a wet yellow solid by rotary evaporation and the residue was suspended in DCM (40L). The mixture stirred under N2 for l6h. The solids were collected by vacuum filtration and the cake was washed with DCM (about 15 L) until the filtrate was colorless (PSC-2). The solids were then dried under vacuum to give Z17c (6.93 kg, qNMR
72 %, yield 88%). 1H NMR (400 MHz, D2O) δ = 7.37 (s, 1H).
Step f
[00315] To a mixture of Z17c (6.95 kg, assay 72%, 27.23 mol) in l,4-dioxane (72 L) was added Xantphos (233 g, 411 mmol, 0.015 eq), Pd2(dba)3 (186 g, 206 mmol, 0.0075 eq), Z17b (7.13 kg, 28.02 mol) and DIPEA (7.02 kg, 54.46 mol). The system was vacuated and purged with nitrogen gas three times. The mixture was stirred at 65 °C for 16 h under N2. The mixture was cooled to RT and water (50 L) was added, filtered. The cake was washed with EA (25 L). The filtrate was extracted with EA (4 x 20 L). The organic phase was concentrated in vacuum to offer the crude product which was combined with the cake. Then DCM (60 L) was added to the crude product and stirred at 25-30°C for l8h and then filtered. The filter cake was slurried with CH2Cl2 (30 L) for 4 hrs and filtered. The filter cake was slurred in CH2Cl2 (30 L) for 16 hrs and filtered. Then the filter cake was dried in vacuum to give Z17a (3-((2-amino-3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine; 9.1 kg, 84 %) as light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.89 (s, 1H), 7.7 (d, J= 7.6 Hz, 1H), 7.18 (bs, 2H), 6.40 (bs, 2H), 5.97 (d, J= 7.6 Hz, 1H).
Example 4
Alternative formation of the intermediate Z17a (here also named Y7a)
[00316] By way of alternative and according to a preferred reaction method, the compound of the formula Z17a was obtained according to the following Reaction Scheme:
In detail, the synthesis of the compound of the formula Y7a = Z17a was carried out as follows:
Step a
[00317] 2, 3, 5-trichloropyrazine (70.50 g, 384.36 mmol, 1 equiv) and ammonia solution
(25% wt, 364.00 g, 400 mL, 2.68 mol, 6.14 equiv) were added to a 1-L sealed reactor. The mixture was heated to 80 °C and stirred for 24 h, and the reaction was completed. The reaction mixture was cooled to 30 °C and filtered to give a brown filter cake. The brown filter cake was dissolved in acetone
(50 mL), and filtered. To the filtrate was added petroleum ether (300 mL). The suspension was stirred for 4 h, and filtered to give the crude product. The crude product was slurried in combined solvents of petroleum ether and acetone (10/1, 200 mL) and filtered to give the product Y7d (51.00 g, 307.91 mmol, 80% yield) as a light yellow solid. 1H NMR (400 MHz, DMSO-d6) δ = 7.63 (s, 1H).
[00318] To a 200 mL round bottom flask was added Na2S (10.816 g, 44wt% containing crystalline water, 60.978mmol) and toluene (100 mL). The mixture was heated to reflux, and water was removed with a Dean-Stark trap (about 5~6 mL water was distilled out). After cooling, the mixture was concentrated to dryness.
[00319] To above round bottom flask was added Y7d (5.000 g, 30.489mmol) and 2-methylbutan-2-ol (50 mL), the reaction was heated to reflux and stirred for 36 h. After cooling to 25 °C, the mixture was filtered. The solvent of the filtrate was exchanged with n-heptane (5 V, 3 times, based on Y7d), and finally concentrated to IV residue. THF (25 mL) was charged to the residue at 25 °C and stirred. The suspension was filtered and washed with THF/n-heptane (5 mL/5 mL) to give a brown solid (6.200 g).
[00320] To another 200 mL round bottom flask was added above brown solid (6.200 g),
10% brine (25 mL), Me-THF (30 mL) and n-Bu4NBr (9.829 g, 30.489 mmol). The mixture was stirred for 0.5 h at room temperature, and the phases were separated. The organic phase was washed with 20% brine (25 mL), and exchanged the solvent with iso-propanol (5 V *3 times, based on Y7d) to give the iso-propanol solution of Y7c (27.000g, 99.2% purity by HPLC area, 58.08% assay yield). 1H NMR (400 MHz, DMSO-d6) δ = 6.88 (s, 1H), 2.97 – 2.92 (m, 14H), 1.38 – 1.31 (m, 14H), 1.13 – 1.04 (m,
14H), 0.73 – 0.69 (t, 21H).
Step c
[00321] To a 25-mL round-bottom flask was added Y7c (4.7g, 23.27wt%, IPA solution from Step b, 2.723 mmol, 1.0 equiv), Y7b (1.052 g, 4.085 mmol, 1.5 equiv), l,lO-Phenanthroline (0.05 g, 0.272 mmol) and water (8 mL). The mixture was purged with nitrogen gas three times, and Cul (0.026 g, 0.136 mmol) was added under nitrogen atmosphere. The mixture was heated up to 65 °C and stirred for 3 h, and the reaction was completed. The reaction was cooled to room temperature and filtered, and the filter cake was washed with water (4 mL*3). The filter cake was slurried in MTBE (6 mL) for 30 min and filtered. The filter cake was washed with MTBE (6 mL) and dried to afford Y7a which is Z17a (565 mg, 72% yield).
[00322] Z17b is synthesized as described in Example 3 Step a and Step b.
Example 5
Alternative Synthesis of the intermediate Z17a:
[00323] According to another preferred method, the compound of the formula Z17a was obtained in accordance with the following Reaction Scheme:
[00324] The reactions were carried out as follows:
Step a
Y7d was synthesised as described in Example 4 step a.
[00325] To a three-necked round-bottle flask was added Y7d (200 mg, 1.22 mmol, 1 equiv), dioxane (4 mL). The solution was vacuated and purged with nitrogen gas three times. Xantphos (14mg, 0.024 mmol, 0.02 equiv), PdCl2(dppf) (8.9 mg, 0.012 mmol, 0.1 equiv), and DIPEA (0.32 g, 2.44 mmol, 2.0 equiv) were added under nitrogen atmosphere. The solution was heated to 85 °C for overnight. The reaction was cooled and evaporated. The residue was purified by column chromatography (eluent/ethyl acetate/heptane = 1/1) to give Z17d (259 mg, 0.99 mmol, 81%). 1H NMR (400 MHz, CDCl3) δ = 7.83 (s, 1H), 4.88 (bs, 2H), 3.73 (s, 3H), 3.47 (t, J= 9.2 Hz, 2H), 2.79 (t, J= 9.2 Hz, 2H).
[00326] The remaining steps were carried out as described in Example 4, Steps e and f, to yield Z17a. Z17b was synthesized as described in Example 3 Step a and Step b.
Example 6
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8- azaspiro[4.5]decan-4-amine. succinate (1:1) hemihydrate. modification (form) HA:Variant a)
[00327] 50 ml ethanol and 2.5 ml water were added to a 100ml flask containing 3.0 g of free base of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (obtained as A18 for example as described in Example 1) and 848.0 mg of succinic acid. The mixture was heated to 50°C to generate a clear solution. The temperature was lowered to 15°C during a period of 3 hours. The solution was kept stirring at 15°C overnight.
Precipitated solid was separated via suction filtration and 50 ml of acetone was added to produce a suspension. The suspension was stirred at 50°C for 3 hours. The solid was separated with suction filtration and dried at room temperature under vacuum for 3 hours. Yield was about 60%.
[00328] The succinate appeared as a highly crystalline solid, with a melting point onset of
94.4°C and an accompanying enthalpy of 96 J/g. The succinate salt crystals showed aggregates of broken drusy tabular particles.
[00329] Variant b)
[00330] 14.34 g of 3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)- 3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine free form (obtained as A18 for example as described in Example 1) and 4.053 g of succinic acid were equilibrated in 100 mL 95% EtOH at 50°C. Add 5 mL of water into the system and heat to 70-75 °C. Add 95 mL of pure EtOH and heat for 30 min more. Stir over night at 25 oC. Filter the mixture wash with EtOH and dry under vacuum in an oven at room temperature. Yield is 87.5%.
PATENT
WO 2020065452
PATENT
PHARMACEUTICAL COMBINATION COMPRISING TNO155 AND NAZARTINIB
PAPER
Journal of Medicinal Chemistry (2020), 63(22), 13578-13594.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c01170
SHP2 is a nonreceptor protein tyrosine phosphatase encoded by the PTPN11 gene and is involved in cell growth and differentiation via the MAPK signaling pathway. SHP2 also plays an important role in the programed cell death pathway (PD-1/PD-L1). As an oncoprotein as well as a potential immunomodulator, controlling SHP2 activity is of high therapeutic interest. As part of our comprehensive program targeting SHP2, we identified multiple allosteric binding modes of inhibition and optimized numerous chemical scaffolds in parallel. In this drug annotation report, we detail the identification and optimization of the pyrazine class of allosteric SHP2 inhibitors. Structure and property based drug design enabled the identification of protein–ligand interactions, potent cellular inhibition, control of physicochemical, pharmaceutical and selectivity properties, and potent in vivo antitumor activity. These studies culminated in the discovery of TNO155, (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (1), a highly potent, selective, orally efficacious, and first-in-class SHP2 inhibitor currently in clinical trials for cancer.

file:///C:/Users/Inspiron/Downloads/jm0c01170_si_001.pdf
(3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8- azaspiro[4.5]decan-4-amine (1):

Step a: A mixture of (3S,4S)-tert-butyl 4-((R)-1,1-dimethylethylsulfinamido)-3-methyl-2-oxa-8- azaspiro[4.5]decane-8-carboxylate (51 mg, 0.136 mmol) and HCl (4 M in dioxane, 340 L, 1.362 mmol) in MeOH (5 mL) was stirred for 1 h at 40 °C. After cooling to RT, the volatiles were removed under reduced pressure to give (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine which was used in next step without further purification. MS m/z 171.1 (M+H)+. Step b: A mixture of (3S,4S)-3-methyl-2-oxa-8-azaspiro[4.5]decane-4-amine crude, 3-((2-amino3-chloropyridin-4-yl)thio)-6-chloropyrazin-2-amine (35.5 mg, 0.123 mmol), and DIPEA (193 L, 1.11 mmol) in DMSO (600 L) was stirred for 16 h at 100 °C. After cooling to RT, the volatiles were removed under reduced pressure and the resulting residue was purified by HPLC (gradient elution 15-40% acetonitrile in water, 5 mM NH4OH modifier) to give (3S,4S)-8-(6-amino-5-((2-amino-3-chloropyridin-4-yl)thio)pyrazin-2-yl)-3-methyl-2-oxa-8-azaspiro[4.5]decan-4-amine (11 mg, 0.026 mmol). 1 H NMR (400 MHz, METHANOL-d4) δ ppm 7.67-7.47 (m, 2 H), 5.91 (d, J=5.5 Hz, 1 H), 4.22 (qd, J=6.4, 4.8 Hz, 1 H), 4.03 (ddt, J=13.5, 8.9, 4.7 Hz, 2 H), 3.86 (d, J=8.7 Hz, 1 H), 3.71 (d, J=8.7 Hz, 1 H), 3.37 (td, J=9.9, 4.9 Hz, 1 H), 3.29-3.23 (m, 1 H), 3.00 (d, J=5.0 Hz, 1H) 1.91-1.56 (m, 4 H), 1.21 (d, J=6.4 Hz, 3 H). HRMS calcd for C18H25ClN7OS (M+H)+ 422.1530, found 422.1514.
//////////TNO 155, CANCER
CILENGITIDE
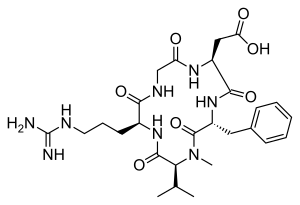
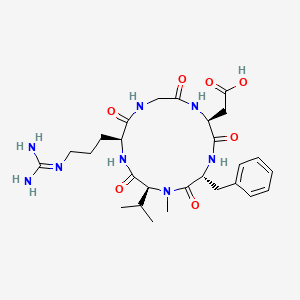
| IUPAC Condensed | cyclo[Arg-Gly-Asp-D-Phe-N(Me)Val] |
|---|---|
| HELM | PEPTIDE1{R.G.D.[dF].[meV]}$PEPTIDE1,PEPTIDE1,5:R2-1:R1$$$ |
| IUPAC | cyclo[L-arginyl-glycyl-L-alpha-aspartyl-D-phenylalanyl-N-methyl-L-valyl] |
CILENGITIDE
- Molecular FormulaC27H40N8O7
- Average mass588.656 Da
2-[(2S,5R,8S,11S)-5-benzyl-11-[3-(diaminomethylideneamino)propyl]-7-methyl-3,6,9,12,15-pentaoxo-8-propan-2-yl-1,4,7,10,13-pentazacyclopentadec-2-yl]acetic acid188968-51-6[RN]
4EDF46E4GI
7823
циленгитид
سيلانجيتيد
西仑吉肽
EMD 121974, EMD-121974, UNII-4EDF46E4GI
Cilengitide has been in phase III clinical trials by Merck Serono and NCI for the treatment of glioblastoma multiforme. However, this research has been discontinued.
Cilengitide was originally developed by Merck KGaA in collaboration with the Technical University of Munich, then received orphan drug designation from FDA for the treatment of glioma in 2005.
Cilengitide (EMD 121974) is a molecule designed and synthesized at the Technical University Munich in collaboration with Merck KGaA in Darmstadt. It is based on the cyclic peptide cyclo(-RGDfV-), which is selective for αv integrins, which are important in angiogenesis (forming new blood vessels), and other aspects of tumor biology. Hence, it is under investigation for the treatment of glioblastoma, where it may act by inhibiting angiogenesis, and influencing tumor invasion and proliferation.[1][2]
The European Medicines Agency has granted cilengitide orphan drug status.[3]
Cilengitide seems to function by inhibiting the FAK/src/AKT pathway and inducing apoptosis in endothelial cells.[4] Preclinical studies in mice of cilengitide were able to demonstrate efficacious tumor regression.[4]
In a rat xenograft model, cilengitide was able to potentiate the cytotoxic effects of radiation when cilengitide was administered prior to radiation therapy.[5] When combined with radiation, inhibition of integrin expression by cilengitide synergistically improves the cytotoxic effects of ionizing radiation for glioblastoma.[5]
Clinical trials
Phase II studies were able to demonstrate that cilengitide as a potential monotherapy in patients with recurrent glioblastoma[6] with high intratumor drug levels when 2000 mg of cilengitide is given twice weekly.[7]
Cilengitide is well tolerated, in combination with radiation and temozolomide, at a dose of 2000 mg in patients with newly diagnosed glioblastoma, regardless of MGMT promoter status.[8] In a phase I/IIa study, the addition of cilengitide to the standard of care for newly diagnosed glioblastoma (surgical resection followed by temozolomide and radiation therapy) improves progression-free survival and overall survival in patients with MGMT promoter methylation.[9]
However, in a subsequent study, cilengitide does not seem to alter the pattern of glioblastoma progression,[10]
and in an EORTC phase III randomized, controlled, multicenter clinical trial, consisting of over 500 patients in 23 countries, the addition of cilengitide to the standard of care did not improve overall survival in patients with newly diagnosed glioblastoma and methylated MGMT promoter status [11] A phase II study, the CORE trial, is currently being conducted in patients with newly diagnosed glioblastoma and unmethylated MGMT promoter status.[12]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
Angewandte Chemie, International Edition, 55(4), 1540-1543; 2016

SYN
Chemistry – A European Journal, 16(18), 5385-5390, S5385/1-S5385/36; 2010
Reference:1. WO0047228A1 / US7115261B1.
2. US6001961A.Route 2
Reference:1. CN102731627A.PATENTWO/2021/224234ANTIVIRAL USE OF CILENGITIDEhttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021224234&_cid=P20-KW0M52-85135-1
PATENThttps://patents.google.com/patent/CN102731627A/enEMD121974 (Cilengitide), the Chinese another name: ring (L-arginyl glycyl-L-aspartoyl-D-phenylalanyl-N-methyl-L-valyl) is an a kind of new classification cancer therapy drug of synthetic.Merkel company discovers that EMD121974 amalgamation radiotherapy (merging to reach assists TM to add radiotherapy) possibly prolong lifetime; Simultaneously integrate plain supressor antitumor drug as first; Got into the III clinical trial phase, its important mechanism is to grow targeting that the blood supply structure of nutrition, the growth of promotion cancer cell is provided in tumour and for tumour through line artery.The EMD121974 molecular formula is: C 27H 40N 8O 7, have following structure:
The preparation method of cyclic peptide mainly contains liquid phase synthesis process, solid phase synthesis precursor peptide cyclization process, process for solid phase synthesis in liquid phase at present; Wherein preceding two kinds of synthesis techniques all are the cyclisation in liquid phase of synthetic precursor peptide, and this method needs reactant in extremely rare solvent, to react (10 -3~10 -4Mol/L), and intermolecular be prone to react generation line style or cyclic polymer, greatly reduced the cyclisation yield, bring trouble for follow-up purifying, and in large-scale production, produce a large amount of waste liquids, be unfavorable for suitability for industrialized production.In conjunction with the structure of EMD121974, utilize the false rare principle of benefit of solid phase, developed a kind of efficient cyclization reaction, the cyclisation time shortens to 20%~30% of liquid phase cyclisation, and the 2%-8% of solvent as liquid phase used in reaction.Embodiment 1The preparation of Fmoc-L-Asp (OtBu)-Wang ResinThe Wang Resin that takes by weighing the 10g substitution degree and be 0.5mmol/g joins in the reactor drum, adds an amount of DCM, and swelling 30min takes out DCM; 6.17g Fmoc-L-Asp-OtBu, DIC 2.40ml, HOBT2.1g are dissolved among the 30ml DMF; At 0-5 ℃ of activation 15min, activation solution is joined in the reactor drum that contains Wang Resin, behind the reaction 10min; Add DMAP 0.18g again, at 0~30 ℃ of reaction 1~5h.After reaction finishes, add sealing Wang Resin unreacted hydroxylation reagent diacetyl oxide 1ml and pyridine 0.5ml, behind the capping 1h, DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min.Through detecting, obtain the Fmoc-L-Asp that substitution degree is 0.47mmol/g (OtBu)-Wang Resin.Embodiment 2The EMD121974 precursor:The preparation of A-Wang Resin (Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin)Fmoc-L-Asp (OtBu)-Wang Resin is joined in the reactor drum, behind DMF swelling 30min, take out solvent, the piperidines-DMF that adds 80ml 25% reacts 5min, and 80ml DMF washs 1 time (3min), and the piperidines-DMF that adds 80ml 25% reacts 15min; DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min; With 4.45g Fmoc-Gly-OH, 5.68g HBTU, 2.03g HOBt, be dissolved among the DMF of 30ml, dissolve the back and added DIEA 2.45ml; 0~5 ℃ of activation 15min; Activation solution is joined in the above-mentioned reactor drum, and behind reaction 1-3h under 0~30 ℃, reaction end detects with ninhydrin method.Adopt aforesaid method coupling Fmoc-L-Arg (Mtr)-OH, Fmoc-N-Me-L-Val, Fmoc-D-Phe-OH successively, finally obtain Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin.Embodiment 3EMD121974 precursor peptide: the preparation of B-Wang Resin (D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp-Wang Resin)With volume ratio is that piperidines-DMF of 25% is the Fmoc deprotection agent of Fmoc-D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin; Add piperidines-DMF 80ml of 25% first time; Reaction 5min, 80ml DMF washs 1 time (3min), adds piperidines-DMF 80ml of 25% for the second time; Behind the reaction 15min, DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min gets D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp (OtBu)-Wang Resin after washing finishes.80% the PhOH-DCM solution that adds volume ratio and be 100ml takes off OtBu with the TFA of catalytic amount, reacts 8h; DMF, DCM, the CH of 80ml used in washing successively 3OH, DMF washing 2,1,1,2 times, each 1min gets D-Phe-N-Me-L-Val-L-Arg (Mtr)-Gly-L-Asp-Wang Resin.Embodiment 4The preparation of EMD121974-Wang Resin (Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp)-Wang Rsin)In above-mentioned reactor drum, add cyclization reagent 3.9g DPPA, 2.5ml DIEA (reactant cyclization reagent amount of substance ratio is 1: 3), at 10~40 ℃ of reaction 3h, the multiple cyclization reagent reaction 3~5h (reaction end detects with ninhydrin method) that throws once above-mentioned equivalent; DMF, DCM, the CH of 80ml used in washing successively 3OH washing 2,1,3 times, each 3min gets Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp)-Wang Rsin.Embodiment 5The preparation of EMD121974 (Cyclo (D-Phe-N-Me-L-Val-L-Arg-Gly-L-Asp))In above-mentioned reactor drum, add the TFA/H of lytic reagent 120ml again 2Behind O/TlS (volume ratio is 95: 2.5: 2.5) the reaction 3h, suction filtration is removed resin, and filtrating slowly joins in the no water-ice ether; Static 2-5h, high speed centrifugation obtain thick peptide, prepare through high-pressure liquid phase; Lyophilize gets smart EMD121974; Its purity>99.5%, single impurity<0.2%, total recovery reaches 63%.Choosing substitution degree in the present embodiment is the Wang Resin of 0.5mmol/g, and can also choose substitution degree is the arbitrary Wang Resin and Fmoc-L-Asp-OtBu prepared in reaction Fmoc-L-Asp (the OtBu)-Wang Resin of 0.4~0.9mmol/g scope.All can realize technical scheme of the present invention, and obtain technique effect of the present invention.Above content is an EMD121974 and become one of best preferred version of route; And to further explain that the present invention did; But can not assert that practical implementation of the present invention is only limited to these explanations; Under the prerequisite that does not break away from the present invention’s design, can also make some simple deductions and replacement, all should be regarded as protection domain of the present invention.
CLIPhttps://www.eurekaselect.net/article/2607Cilengitide, a cyclic RGD pentapeptide, is currently in clinical phase III for treatment of glioblastomas and in phase II for several other tumors. This drug is the first anti-angiogenic small molecule targeting the integrins αvβ3, αvβ5 and α5β1. It was developed by us in the early 90s by a novel procedure, the spatial screening. This strategy resulted in c(RGDfV), the first superactive αvβ3 inhibitor (100 to 1000 times increased activity over the linear reference peptides), which in addition exhibited high selectivity against the platelet receptor αIIbβ3. This cyclic peptide was later modified by N-methylation of one peptide bond to yield an even greater antagonistic activity in c(RGDf(NMe)V). This peptide was then dubbed Cilengitide and is currently developed as drug by the company Merck-Serono (Germany). This article describes the chemical development of Cilengitide, the biochemical background of its activity and a short review about the present clinical trials. The positive anti-angiogenic effects in cancer treatment can be further increased by combination with “classical” anti-cancer therapies. Several clinical trials in this direction are under investigation.
CLIPJournal of Protein Chemistry

Schematic of the one-step chemoenzymatic synthesis of cilengitide using wild-type Mcy TE. (1) The chemically synthesised (SPPS, solid-phase peptide synthesis) mimetic substrate was condensed with benzyl mercaptane to produce pentapeptide thioester (pentapeptide-BMT). (2) Models of the substrate-O-TE acyl enzyme intermediate are marked with brackets (protein data bank, 1JMK). (3) Mechanism of TE domain catalysis: a pentapeptide -O-TE acyl-enzyme intermediate is formed by transfer of the peptidyl chain from the phosphopantethiene of the terminal peptidyl carrier protein (PCP), which was substituted by benzyl mercaptane, to the active site serine of the TE domain. For hydrolyzing TE domains, the intermediate is captured by water, generating the linear peptide; for cyclizing TE domains, an intramolecular nucleophile captures the intermediate, resulting in “cilengitide”
PATENTWO 9745447
WO 9745137
DE 19534177
WO 2000053627
WO 2000047228
US 20040063790
WO 2009124754
WO 2011079015
WO 2011069629
WO 2011144756WO 2016059622
PATENTWO 2012062777https://patents.google.com/patent/WO2012062777A1/enSynthesis of cyclic peptidesCyclo[-Arg-Gly-Asp- 6 or 7 -Phe-Val-Ala-] (1 and 2). Resin loading. 2- chlorotrityl chloride-resin ( 1 50 m g , 1 .5m m ol/g ) was p laced i n a 20 m l polypropylene syringe fitted with a polyethylene filter disk. The resin was then washed with CH2CI2 (5 χ 0.5 min), and a solution of Fmoc-L-Gly-OH (334 mg, 1 .125 mmol, 5 equiv) and DIEA (239 μΙ_, 6.25 equiv) in CH2CI2 (2.5 ml_) was added. The mixture was then stirred for 15 min. Extra DIEA (239 μΙ_, total 12.5 mmol) was added, and the mixture was stirred for an additional 45 min. The reaction was stopped by adding 3 χ DCM/ MeOH/ DIEA (85: 10:5) and stirring for 1 0 m in. The Fmoc-L-Gly-O-resin product was subjected to the following washings/treatments with CH2CI2 (3 χ 0.5 min), DMF (3 χ 0.5 min), piperidine and DMF (5 χ 0.5 min). The loading was 0.50 mmol/g, as calculated by Fmoc determination.Peptide coupling. Fmoc-L-Arg(Pbf)-OH (243 mg, 0.375 mmol, 5 equiv), Fmoc- L-Ala-OH (1 17 mg, 0.375 mmol, 5 equiv), Fmoc-L-Val-OH ( 127 mg, 0.375 mmol, 5 equiv) and Fmoc- L-Phe-OH ( 145 mg, 0.375 mmol, 5 equiv) were added sequentially to the above obtained H-L-Gly-O-resin using HCTU (155 mg, 0.375 mmol, 5 equiv), HOBt (50 mg, 0.375 mmol, 5 equiv) and DIEA (127 μΙ_, 0.75 mmol, 10 equiv) in DMF (2.5 ml_). In all cases, after 90 min of coupling, the ninhydrin test was negative. Removal of Fmoc group and washings were performed as described in general procedures. /V-Alloc-thiazole 6 or 7 (92 mg, 0.375 mmol, 5 equiv) was coupled with HATU (143 mg, 0.375 mmol, 5 equiv), HOAt (51 mg, 0.375 mmol, 5 equiv) and DIEA (127 μΙ_, 0.75 mmol, 10 equiv) for 90 min. This coupling was repeated twice in the same conditions. The Alloc group of the peptide resin was removed with Pd (PPh3)4 (9 mg, 0.0075 mmol, 0.1 equiv) in the presence of PhSiH3 (92.5 μΙ_, 0.75 mmol, 10 equiv) in DCM for 20 min. This deprotection was repeated three times in the same conditions. After washing, the resin was treated with dry THF (2ml_) for 15 min. Meanwhile, Fmoc-L-Asp(tBu)-OH (154 mg, 0.375 mmol, 5 equiv) was added to a 68 mM solution of triphosgene in dry THF (1 .15 equiv). Sym-collidine (99.5 μΙ_, 0.75 mmol, 10 equiv) was added to the clear solution, upon which a precipitate of collidinium chloride was formed. DIEA (102 μΙ_, 0.6 mmol, 8 equiv) was added to the resin, immediately followed by addition of the suspension. This coupling was repeated four times in the same conditions. The reaction mixture was stirred at 50 °C during 48 h.Peptide cleavage. Following Fmoc deprotection, the peptidyl-resin was treated with TFA-CH2CI2 (1 :99) (5 χ 30 s). The filtrate was collected on H20 (4 ml_) and the H20 was partially removed under reduced pressure. MeCN was then added to dissolve solid that formed during the removal of H20, and the solution was lyophilized to give 12 mg and 10 mg of the linear compounds 28 and 29 respectively with a purity of > 91 % as checked by HPLC (Column A, Rt 7.43 min and Rt 7.38 min respectively, linear gradient 35%-40% ACN in 15 min.)], which was used without further purification. MALDI-TOF-MS calculated for C50H71 N11 O13S2 1098.29; found mlz 1099.29 [M + H]+, 1 121 .28 [M + Na]+, 1 137.39 [M + K]+.Synthesis in solution. Cyclization. The protected linear peptides 28 and 29 were dissolved in DMF (1 L, 10“4 M), and HOAt (9.6 mg, 0.07 mmol, 5 equiv), DIPEA (24 μΙ_, 0.14 mmol, 10 equiv), and PyAOP (36.6 mg, 0.07 mmol, 5 equiv) were added. The mixture was stirred for 24 h at room temperature, and the course of the cyclization step was then checked by HPLC (Column A, Rt 1 1 -67 min and Rt 10.70 min respectively, linear gradient 45%-55% ACN in 15 min.). The solvent was removed by evaporation under reduced pressure and the protected cycle 30 and 31 were used in the next step without further purification. MALDI-TOF-MS calculated for C50H69N11 O12S2 1080.28; found mlz 1081 .28 [M + H]+, 1 103.27 [M + Na]+, 1 1 19.38 [M + K]+.Side chain deprotection. The protected cyclopeptides 30 and 31 (14.7 mg, 19.04 pmol) were treated with TFA-H20 (95: 5) during 1 h. The solvent was removed by evaporation under reduced pressure.Peptide purification. The crude product was purified by HPLC (Symmetry C8 5 μη-Ί, 30 mm x 100 mm), gradient of MeCN (30% to 75% in 15 min) MeCN (+0.05% TFA) in water (+0.05% TFA), 20 mL/min, detection at 220 nm, to give the cyclopeptides 1 and 2 (4.5 mg, 5.8 pmol and 6.5 mg, 8.37 pmol, 7.7% and 12% yield respectively). The products were characterized by HPLC (Rt 8.99 min, and Rt 8.02 min Column A, respectively, linear gradient 0%-100% ACN in 1 5 min. ) and by MALDI-TOF-MS: calculated for C33H45N11 O9S 771 .84; found mlz 772.84 [M + H]+, 794.83 [M + Na]+, 810.94 [M + K]+.Cyc/o-[Arg-Gly-Asp-Thz1X-] (3). General procedure for cyclopeptide synthesis. Solid phase synthesis: The synthesis of the linear peptide H- Asp(tBu)-XX-Arg(Pbf)-Gly-OH was performed using Fmoc-based solid phase peptide synthesis with 2-chlorotrityl chloride resin (2.0 g, 3.2 mmol).Resin loading: Fmoc-Gly-OH (594 mg, 2.0 mmol) was attached to the resin with DIPEA in DCM at room temperature for 1 .5 h. The remaining trityl groups were capped adding 0.5 mL of MeOH for 30 min. After that, the resin was filtered and washed with DCM (2x), DMF (2x). The loading of the resin was determined by titration of the Fmoc group (Chan WC and White PD. Fmoc Solid Phase Peptide Synthesis. Oxford University Press: New York, 2000). The final loading was 2.0 mmol/g. The Fmoc group was eliminated by treatment with 20% piperidine in DMF (2X10 min). The resin was washed with DMF (3x), DCM (3x). Peptide coupling: Fmoc-Arg(Pbf)-OH (5.19 g, 8.0 mmol), DIPCDI (1.23 mL, 8.0 mmol) and HOBt (1.08 g, 8.0 mmol) were dissolved in DMF and added to the resin for 1 .5 h. The end of the coupling was monitored by ninhydrin test (free amine group) (Kaiser E et al. Anal Biochem 1970, 34:595-598). The resin was filtered and washed with DMF (3X) and DCM (3X). The Fmoc group was eliminated with 20 % piperidine in DMF (2X10 min).The coupling of the thiazole module was carried out with 8 (1 .14 g, 3.0 mmol), PyAOP (1 .56 g, 3.0 mmol) and DIPEA (1 .02 mL, 6.0 mmol) in DMF for 1 .5 h. The completion of the reaction was checked with the ninhydrin test. Finally the deprotection of the amine and coupling of the Fmoc-Asp(‘Bu)-OH were carried out under the same conditions of the second amino acid.Peptide cleavage: The resin bound peptide was treated with 2% TFA in DCM (6 x 30 sec.) The resin was washed with DCM and the combined solution was evaporated under vacuum with Et20 several times, furnishing the linear peptide 32 as a white solid. The peptide was used for the next step without purification.H PLC (gradient 20 to 80% of CH3CN in 1 5 m in): tR= 8.33 min. HPLC-MS (ES(+)): m/z 795.3.Synthesis in solution. Cyclization: The product 32 (200 mg, 0.251 mmol) was dissolved in anhydrous DMF (50 mL, 5 mM), PyAOP (262 mg, 0.503 mmol) and DIPEA (213 μί, 1 .255 mmol) were added. The reaction was monitored by HPLC. Once the reaction was finished, the DMF was evaporated under vacuum. The crude was dissolved in AcOEt and the solution was washed with NH4CISat and Na2CO3 sat. The organic layer was collected, dried over Na2SO4, filtered and concentrated under vacuum. The peptide was purified by flash chromatography (CHCIs/MeOH 8:2) furnishing the protected cyclic peptide 33 as a white solid (1 56 mg, XX%). HPLC (gradient 40 to 90% of CH3CN in 1 5 min): tR= 8.86 min. HPLC-MS (ES(+)): m/z 778.2Side chain deprotection: The protected peptide 33 (125 mg, XX mmol), was treated with 25 mL of a solution of TFA H2O (95:5). After 3 h, the solvent was evaporated under vacuum and the residue was precipitated with Et2O (4X). The Et2O solution was discarded and the white solid was lyophilized to afford 3 55 mg (XX%).
Peptide purification. The end product 3 was dissolved in 5 ml MilliQ water and it was filtered through a 0.2 pm filter. The cyclic peptide was purified by semipreparative RP-HPLC using acetronitrile (0.05% TFA)/water (0.1 % TFA). The HPLC sample was vacuum concentred and transformed into the hydrochloride salt lyophilized with water with 0.05% HCI.1H-NMR (500 MHz, H20:D20-d2 9: 1 , 278 K): δ = 9.29 (t, NH Gly), 9.20 (d, J = 7.24 Hz, NH Asp), 8.90 (t, J = 5.89/5.89 Hz, NH Thz), 8.46 (d, J = 8.93 Hz, NH Arg), 7.79 (s, CH Thz), 7.22 (t, J = 5.39/5.39 Hz, ΝΗε Arg), 4.75 (m, CHa Arg), 4.63 (m, CHa Asp), 4.04 (dd, J = 3.35/14.90 Hz, CHa Gly), 3.82 (dd, J = 6.69/14.96 Hz, CHa Gly), 3.17 (m, CH25 Arg), 2.89 (m, CH2p Asp), 1 .92 (m, CH p Arg), 1 .82 (m, CHP Arg), 1 .63 (m, CH2 Arg). HPLC (gradient 0 to 20% of CH3CN in 15 min): tR= 10.52 m in. HRMS (E IS) m/z calculated 468.1540

found 469.16099 (M+H)+.Cyc/o-[Arg-Gly-Asp-Thz2X-] (4). The cyclopeptide 4 was prepared according to the process followed for 3 and using bithiazole 9 (XX mg, YY mmol) instead of 8. The linear peptide 34: HPLC (gradient 0 to 100% CH3CN in 15 min.): tR = 10.34 min, HPLC-MS (ES(+)): m/z 877.81 . The protected peptide 35: HPLC (gradient 0 to 100% CH3CN in 15 min.): tR = 13.91 min, HPLC-MS (ES(+)): m/z 860.54. The final peptide 4: 1H-NMR (500 MHz, H20:D20-d2 9: 1 , 298 K): δ = 8.93 (sbroad, NH Gly), 8.82 (d, J = 7.62 Hz, NH Asp), 8.75 (t, J = 5.69/5.69 Hz, NH Thz), 8.51 (d, J = 7.62 Hz, NH Arg), 8.05 (s, CH Thz1), 7.50 (s, CH Thz2), 7.19 (t, J = 5.38/5.38 Hz, ΝΗε Arg), 4.13 (dd, J = 5.82/14.24 Hz, CH Gly), 3.87 (dd, J = 5.96/15.69 Hz, CH Gly), 3.21 (m , CH25 Arg), 2.94 (m, CH2p Asp), 1 .95 (m , CHP Arg), 1 .87 (m , CHP Arg), 1 .68 (m , CH2y Arg). HPLC (gradient 1 0 to 25% of CH3CN in 1 5 m in): tR = 8.73 min. HRMS (EIS) m/z calculated 551 .1369 (C2oH25N906S2) found 552.14392 (2M+2H)+.Cyc/o-[Arg-Gly-Asp-Thz3X-] (5). The cyclopeptide 5 was prepared according to the process for 3 and using trithiazole 10 (XX mg, YY mmol) instead of 8. The linear peptide 36: HPLC (gradient 20 to 80% of CH3CN in 15 min.): tR = 7.60 min, HPLC-MS (ES(+)): m/z 961 .23. The protected peptide 37: HPLC (gradient 20 to 80% of CH3CN in 15 m in. ): tR = 1 3.13 min, HPLC-MS (ES(+)): m/z 944.3. The final peptide 5: HPLC (gradient 10 to 30% CH3CN in 15 m in): tR = 8.26 m in. HRMS (E IS) m/z calculated 634.1 1 99 (C23H26N10O6S3) found 635.12683 (2M+2H)+. 1H-NMR (500 MHz, DMSO-d6 298 K): δ = 9.21 (t, J = 5.4, NH Gly), 8.72 (m, NH Asp + NH Thz), 8.37 (s, CH Thz1), 7.96 (d, J = 9.2, NHa Arg), 7.77 (s, CH Thz2), 7.68 (t, J = 6.0, ΝΗε Arg), 7.23 (s, CH Thz3), 4.83 (dd, J = 14.3, 8.5, CHa Arg), 4.72 (dd, J = 16.3, 6.6, CH Thz), 4.59 (m, CH Thz + CHa Asp), 3.89 (d, J = 1 1 .5, CH Gly), 3.59 (d, J = 9.7, CH Gly), 3.13 (dd, J = 12.6, 6.3, CH25 Arg), 2.81 (dd, J = 16.3, 4.3, CHP Asp), 2.58 (dd, J = 16.5, 8.7, CHP Asp), 1 .82 (m, CHP Arg), 1 .71 (m, CHP Arg), 1 .49 (m, CH2y Arg).Cilengitide. The cilengitide was prepared according to the method described in Dechantsreiter MA et al. (J Med Chem 1999, 42:3033-3040). 1H- NMR (500 MHz, H20:D20-d2 9: 1 , 298 K): δ = 8.55 (d, J = 8.06 Hz, NH Asp), 8.37 (d, J = 7.28 Hz, NH Arg), 8.13 ( d, J = 9.19 Hz, NH Phe), 7.97 (m, NH Gly), 7.34 (m, 2H, C6H5 Phe), 7.26 (m, 3H, C6H5 Phe), 7.22 (t, J = 5.53/5.53 Hz, ΝΗε Arg), 5.19 (dd, J = 8.58/16.02 Hz, CHa Phe), 4.56 (dd, J = 7.45/- Hz, CHa Asp), 4.34 (d, J = 10.89 Hz, CHa MeVal), 4.12 (dd, J = 7.80/14.63 Hz, CH Gly), 3.95 (dd, J = 6.84/15.33 Hz, CHa Arg), 3.54 (dd, J = 3.37/14.60 Hz, CH Gly), 3.20 (m , CH25 Arg), 3.02 (m, CH2p Phe), 2.88 (s, CH3 MeVal), 2.84 (dd, J = 7.26/16.68 Hz, CHP Asp), 2.63 (dd, J = 7.60/16.54 Hz, CHP Asp), 2.06 (m, CHP Val), 1 .91 (m, CH2p Arg), 1 .57 (m, CH2 Asp), 0.88 (d, J = 6.55 Hz, CH3 Val1), 0.56 (d, J = 6.49 Hz, CH3 Val2).
PAPERJournal of medicinal chemistry (1999), 42(16), 3033-40.Peptide Science (2001), Volume Date2000, 37th, 249-250. Current opinion in investigational drugs (London, England : 2000) (2003), 4(6), 741-5. Journal of medicinal chemistry (2005), 48(24), 7675-87.Peptide Science (2006), 43rd, 215-216Angewandte Chemie, International Edition (2010), 49(15), 2732-2737, S2732/1-S2732/53.Accounts of Chemical Research (2017), 50(7), 1541-1556.
References
- ^ Burke PA, DeNardo SJ, Miers LA, Lamborn KR, Matzku S, DeNardo GL (August 2002). “Cilengitide targeting of alpha(v)beta(3) integrin receptor synergizes with radioimmunotherapy to increase efficacy and apoptosis in breast cancer xenografts”. Cancer Research. 62 (15): 4263–72. PMID 12154028.
- ^ Goodman SL, Hölzemann G, Sulyok GA, Kessler H (February 2002). “Nanomolar small molecule inhibitors for alphav(beta)6, alphav(beta)5, and alphav(beta)3 integrins”. Journal of Medicinal Chemistry. 45 (5): 1045–51. doi:10.1021/jm0102598. PMID 11855984.
- ^ Spreitzer H (October 27, 2008). “Neue Wirkstoffe – Cilengitide”. Österreichische Apothekerzeitung (in German) (22/2008): 1136–7.
- ^ Jump up to:a b Yamada S, Bu XY, Khankaldyyan V, Gonzales-Gomez I, McComb JG, Laug WE (December 2006). “Effect of the angiogenesis inhibitor Cilengitide (EMD 121974) on glioblastoma growth in nude mice”. Neurosurgery. 59 (6): 1304–12, discussion 1312. doi:10.1227/01.NEU.0000245622.70344.BE. PMID 17277694. S2CID 19861713.
- ^ Jump up to:a b Mikkelsen T, Brodie C, Finniss S, Berens ME, Rennert JL, Nelson K, Lemke N, Brown SL, Hahn D, Neuteboom B, Goodman SL (June 2009). “Radiation sensitization of glioblastoma by cilengitide has unanticipated schedule-dependency”. International Journal of Cancer. 124 (11): 2719–27. doi:10.1002/ijc.24240. PMID 19199360.
- ^ Reardon DA, Fink KL, Mikkelsen T, Cloughesy TF, O’Neill A, Plotkin S, et al. (December 2008). “Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme”. Journal of Clinical Oncology. 26 (34): 5610–7. CiteSeerX 10.1.1.688.8987. doi:10.1200/JCO.2008.16.7510. PMID 18981465.
- ^ Gilbert MR, Kuhn J, Lamborn KR, Lieberman F, Wen PY, Mehta M, Cloughesy T, Lassman AB, Deangelis LM, Chang S, Prados M (January 2012). “Cilengitide in patients with recurrent glioblastoma: the results of NABTC 03-02, a phase II trial with measures of treatment delivery”. Journal of Neuro-Oncology. 106 (1): 147–53. doi:10.1007/s11060-011-0650-1. PMC 4351869. PMID 21739168.
- ^ Nabors LB, Mikkelsen T, Hegi ME, Ye X, Batchelor T, Lesser G, Peereboom D, Rosenfeld MR, Olsen J, Brem S, Fisher JD, Grossman SA (November 2012). “A safety run-in and randomized phase 2 study of cilengitide combined with chemoradiation for newly diagnosed glioblastoma (NABTT 0306)”. Cancer. 118 (22): 5601–7. doi:10.1002/cncr.27585. PMC 3423527. PMID 22517399.
- ^ Stupp R, Hegi ME, Neyns B, Goldbrunner R, Schlegel U, Clement PM, et al. (June 2010). “Phase I/IIa study of cilengitide and temozolomide with concomitant radiotherapy followed by cilengitide and temozolomide maintenance therapy in patients with newly diagnosed glioblastoma” (PDF). Journal of Clinical Oncology. 28(16): 2712–8. doi:10.1200/JCO.2009.26.6650. PMID 20439646.
- ^ Eisele G, Wick A, Eisele AC, Clément PM, Tonn J, Tabatabai G, et al. (March 2014). “Cilengitide treatment of newly diagnosed glioblastoma patients does not alter patterns of progression”(PDF). Journal of Neuro-Oncology. 117 (1): 141–5. doi:10.1007/s11060-014-1365-x. PMID 24442484. S2CID 21636884.
- ^ Merck Group. “Phase III Trial of Cilengitide Did Not Meet Primary Endpoint in Patients With Newly Diagnosed Glioblastoma, Date accessed: 3/24/2014.”
- ^ ASCO Meeting Library. [1] “Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma and methylated O6-methylguanine-DNA methyltransferase (MGMT) gene promoter: Key results of the multicenter, randomized, open-label, controlled, phase III CENTRIC study, Date accessed: 3/24/2014
| Names | |
|---|---|
| IUPAC name2-[(2S,5R,8S,11S)-5-benzyl-11-{3-[(diaminomethylidene)amino]propyl}-7-methyl-3,6,9,12,15-pentaoxo-8-(propan-2-yl)-1,4,7,10,13-pentaazacyclopentadecan-2-yl]acetic acid | |
| Identifiers | |
| CAS Number | 188968-51-6 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL429876 |
| ChemSpider | 154046 |
| IUPHAR/BPS | 6597 |
| KEGG | D03497 |
| MeSH | Cilengitide |
| PubChem CID | 176873 |
| UNII | 4EDF46E4GI |
| CompTox Dashboard (EPA) | DTXSID9044035 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C27H40N8O7 |
| Molar mass | 588.656 g/mol |
| Density | 1.417 g/mL |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
/////////CILENGITIDE, циленгитид , سيلانجيتيد ,西仑吉肽 , PHASE 3, EMD 121974, EMD-121974, UNII-4EDF46E4GI, orphan drug , MERCK, glioblastoma,
CC(C)C1C(=O)NC(C(=O)NCC(=O)NC(C(=O)NC(C(=O)N1C)CC2=CC=CC=C2)CC(=O)O)CCCN=C(N)N

NEW DRUG APPROVALS
ONE TIME
$10.00
Methotripremazine
Methotripremazine
- CL 36467
- CL 39743
- N05AA02
- RP 7044
- RP-7044
- SK&F 5116
- XP-03
- XP03
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Methotrimeprazine hydrochloride | 42BB1Y2586 | 1236-99-3 | ODLGFPIWRAEFAN-PFEQFJNWSA-N |
| Methotrimeprazine maleate | 5KN5Y9V01K | 7104-38-3 | IFLZPECPTYCEBR-VIEYUMQNSA-N |
Methotrimeprazine
CAS Registry Number: 60-99-1
CAS Name: (bR)-2-Methoxy-N,N,b-trimethyl-10H-phenothiazine-10-propanamine
Additional Names: (-)-10-(3-dimethylamino-2-methylpropyl)-2-methoxyphenothiazine; levomepromazine; 2-methoxytrimeprazine; levomeprazine
Manufacturers’ Codes: RP-7044
Trademarks: Sinogan-Debil; Tisercin (EGYT); Neozine (Rh>e-Poulenc); Nirvan; Nozinan (Rh>e-Poulenc); Levoprome (Lederle)
Molecular Formula: C19H24N2OS
Molecular Weight: 328.47
Percent Composition: C 69.47%, H 7.36%, N 8.53%, O 4.87%, S 9.76%
Literature References: Prepn: Courvoisier et al.,C.R. Seances Soc. Biol. Ses Fil.151, 1378 (1957); Jacob, Robert, US2837518 (1958 to Rhône-Poulenc).Optical Rotatory Power, -17, Conc: 5 g/100mL; Solv: chloroform; Wavlen: 589.3 nm; Temp: 20 °C
Derivative Type: Maleate
CAS Registry Number: 7104-38-3
Trademarks: Minozinan; Milezin (Spofa); Neuractil; Neurocil (Bayer); Sofmin (Dainippon); Veractil
Molecular Formula: C19H24N2OS.C4H4O4
Molecular Weight: 444.54
Percent Composition: C 62.14%, H 6.35%, N 6.30%, O 18.00%, S 7.21%
Properties: Crystals, darkened by light. Dec about 190°. Sparingly sol in water (0.3% at 20°) and in ethanol (0.4%). pH of a 0.3% aq soln is 4.3. The free base is levorotatory: [a]D20 -17° (c = 5 in chloroform).
Optical Rotation: [a]D20 -17° (c = 5 in chloroform)
Therap-Cat: Analgesic.
Keywords: Analgesic (Non-Narcotic).
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Methotrimeprazine is a phenothiazine used in the management of psychosis, particular those of schizophrenia, and manic phases of bipolar disorder.
A phenothiazine with pharmacological activity similar to that of both chlorpromazine and promethazine. It has the histamine-antagonist properties of the antihistamines together with central nervous system effects resembling those of chlorpromazine. (From Martindale, The Extra Pharmacopoeia, 30th ed, p604)
Levomepromazine, also known as methotrimeprazine, is a phenothiazine neuroleptic drug. Brand names include Nozinan, Levoprome, Detenler, Hirnamin, Levotomin and Neurocil. It is a low-potency antipsychotic (approximately half as potent as chlorpromazine) with strong analgesic, hypnotic and antiemetic properties that are primarily used in palliative care.[1][2]
Serious side effects include tardive dyskinesia, akathisia, abnormalities in the electrical cycle of the heart, low blood pressure and the potentially fatal neuroleptic malignant syndrome.[1][2]
As is typical of phenothiazine antipsychotics, levomepromazine is a “dirty drug“, that is, it exerts its effects by blocking a variety of receptors, including adrenergic receptors, dopamine receptors, histamine receptors, muscarinic acetylcholine receptors and serotonin receptors.[1][2]
Medical uses
It can be used as an analgesic for moderate to severe pain in non-ambulant patients (the latter being because of its strong sedative effects).[3]
Levomepromazine is also used at lower doses for the treatment of nausea and insomnia.[1]
Levomepromazine is frequently prescribed and valued worldwide in palliative care medicine for its multimodal action, to treat intractable nausea or vomiting, and for severe delirium/agitation in the last days of life. Palliative care physicians will commonly prescribe it orally or via subcutaneous syringe drivers in combination with opioid analgesics such as hydromorphone.[1][2]
Levomepromazine is used for the treatment of psychosis, particularly those of schizophrenia, and manic phases of bipolar disorder. It should only be used with caution in the treatment of agitated depressions, as it can cause akathisia as a side effect, which could worsen the agitation.[1][2] A 2010 systematic review compared the efficacy of levomepromazine with atypical antipsychotic drugs:
Adverse effects
The most common side effect is akathisia.[2] Levomepromazine has prominent sedative and anticholinergic/sympatholytic effects (dry mouth, hypotension, sinus tachycardia, night sweats) and may cause weight gain.[2] These side effects normally preclude prescribing the drug in doses needed for full remission of schizophrenia, so it has to be combined with a more potent antipsychotic.[2] In any case, blood pressure and EKG should be monitored regularly.[2]
A rare but life-threatening side effect is neuroleptic malignant syndrome (NMS).[2] The symptoms of NMS include muscle stiffness, convulsions and fever.[2]
PAPER
Bulletin de la Societe de Pharmacie de Bordeaux (1964), 103(4), 224-30.
The authors define an extn. equil. const., pKe. When a basic mol., A, in an org. solvent (immiscible with water) is shaken with an aq. acid, part of A passes into the aq. phase in the equil. A + H+ .rdblhar. AH+, and Ke and pKe are defined by the equations Ke = [A]org[H+]H2O/[AH+]H2O and pKe = pKa -log ([A]org/[A]H2O), resp. Values of pKe are reported for levomepromazine, properidiazine, thioridazine, chlorpromazine, alimenazine, propiomazine, promethazine, and aminopromazine. Where 2 C atoms sep. the 2 N chain atoms, pKe is of the order of 5, and if 3, the value is near 4.3.
PATENT
JP 40009030
A soln. of 10.5 g. l-3-dimethylamino-2-methylpropanol in xylene is added a suspension of 2.5 g. Na in xylene and a soln. of 18 g. p-tosyl chloride in xylene is dropped in to give l-3-dimethylamino-2-methylpropanol tosylate (I), hydrochloride m. 98-100%. I is treated with 18 g. 2-methoxyphenothiazine and NaNH2 (prepd. from 1.85 g. Na) to give 80% l-3-(2-methoxy-10-phenothiazinyl)-2-methyl-1-dimethylaminopropane, m. 125-6° (hexane). Similarly are prepd. l-3-(3-ethyl-10-phenothiazinyl)-2-methyl-1-dimethylaminopropane (maleate m. 136°) and l-3-(10-phenothiazinyl)-2-methyl-1-dimethylaminopropane (maleate m. 174-5°). The products are tranquilizers.
PATENT
HU 152208
HU 157158
PL 66636
PAPER
Bulletin de la Societe Chimique de France (1968), (8), 3220-2.
Folia medica (1970), 12(1), 88-9
Journal of pharmaceutical sciences (1987), 76(7), 541-4.
SYN
| IN201203390 |
Deprotonation of 2-methoxyphenothiazine by means of KOH in refluxing touene/DMSO, followed by condensation of resulting pottasium salt with N-(3-chloro-2-methylpropyl)-N,N-dimethylamine in refluxing toluene leads to racemic levomepromazine , which upon finally resolution using (-)-dibenzoyl-L-tartaric acid in acetone or using di-p-toluoyl-L-tartaric acid and, optionally, HCOOH in EtOH at 60 °C affords the target levomepromazine

SYN

References
- ^ Jump up to:a b c d e f Brayfield A, ed. (13 December 2013). “Levomepromazine”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 12 May 2014.
- ^ Jump up to:a b c d e f g h i j k Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ “Levomepromazine”. Farmacotherapeutisch Kompas (in Dutch). Retrieved 5 October 2016.
- ^ Jump up to:a b Sivaraman P, Rattehalli RD, Jayaram MB (October 2010). “Levomepromazine for schizophrenia”. The Cochrane Database of Systematic Reviews. 10 (10): CD007779. doi:10.1002/14651858.CD007779.pub2. PMC 3283151. PMID 20927765.
External links
- “Levomepromazine”. PubChem. National Center for Biotechnology Information.
- NOZINAN – Lévomépromazine Doctissimo Guides des Medicaments
- “Levomepromazine” (PDF). Grampians Palliative Care Team Publication. Victoria, Australia. May 2010. Archived from the original (PDF) on 2011-02-26.
- “Levomepromazine in Palliative Care” (PDF). Scotland, UK. August 2013. Archived from the original (PDF) on 2013-05-22.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| Pregnancy category | Only if clearly needed |
| Routes of administration | Oral, seldom IM |
| Drug class | Typical antipsychotic |
| ATC code | N05AA02 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | ~50–60% |
| Metabolism | Hepatic |
| Elimination half-life | ~20 hours |
| Excretion | In feces and urine (metabolites), unchanged drug only 1% |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 60-99-1 7104-38-3 (maleate), 1236-99-3 HCl) |
| PubChem CID | 72287 |
| IUPHAR/BPS | 7603 |
| DrugBank | DB01403 |
| ChemSpider | 65239 |
| UNII | 9G0LAW7ATQ |
| KEGG | D00403 |
| ChEBI | CHEBI:6838 |
| ChEMBL | ChEMBL1764 |
| CompTox Dashboard (EPA) | DTXSID1023289 |
| ECHA InfoCard | 100.000.450 |
| Chemical and physical data | |
| Formula | C19H24N2OS |
| Molar mass | 328.47 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////methotripremazine, L 36467, CL 39743, N05AA02, RP 7044, RP-7044, SK&F 5116, XP-03, XP03
O(c2cc1N(c3c(Sc1cc2)cccc3)C[C@H](C)CN(C)C)C

NEW DRUG APPROVALS
one time
$10.00
Methiomeprazine
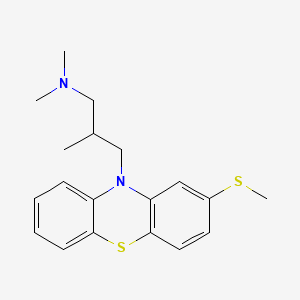
Methiomeprazine
N,N,2-trimethyl-3-(2-methylsulfanylphenothiazin-10-yl)propan-1-amine
CAS 7009-43-0
Molecular Formula, C19-H24-N2-S2, Molecular Weight, 344.5446,
- 10H-Phenothiazine-10-propanamine, N,N,β-trimethyl-2-(methylthio)-, (±)-
- Phenothiazine, 10-[3-(dimethylamino)-2-methylpropyl]-2-(methylthio)-, (±)- (8CI)
- N,N,β-Trimethyl-2-(methylthio)-10H-phenothiazine-10-propanamine
- (±)-10-(3-Dimethylamino-2-methylpropyl)-2-(methylthio)phenothiazine
- 10584-RP
- 2-Methylthio-10-(2-methyl-3-dimethylaminopropyl)phenothiazine
- Methiomeprazine
- SKF 6270
- (+-)-10-(3-Dimethylamino-2-methylpropyl)-2-(methylthio)phenothiazine
- Phenothiazine, 10-(3-(dimethylamino)-2-methylpropyl)-2-(methylthio)-, (+-)-
- 10584 RP
- EINECS 230-285-9
- Methiomeprazinum
- Methiomeprazinum [INN-Latin]
- Metiomeprazina
- Metiomeprazina [INN-Spanish]
- RP 10584
- SKF 6270
- UNII-X2R9QTF0OL
Methiomeprazine hydrochloride
14056-64-5
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
///////////////////////////////////////////////////////////////////////////////////////////////////// Methiomeprazine is an antiemetic drug.
PATENTFR 2705 M 19640831.The title compd. and its derivs. are prepd. and can be used in the prepn. of antiemetic compns. A soln. of 2.280 g. 3-methylthio-10-(3-dimethylamino-2-methylpropyl)phenothiazine (I) in 12 l. EtOH is heated to 70° and added to a soln. (60°) of 969 g. d-tartaric acid in 27 l. EtOH, the soln. kept overnight and filtered, and the mother liquors from the 1st and 2nd crystns. combined and evapd. The residue (2.352 g.) is dissolved in H2O, the soln. made alk. with 700 ml. NaOH (d. 1.33) and extd. with 4 l. CH2Cl2, the org. phase sepd., the aq. phase extd. with 1 l. CH2Cl2, and the exts. combined and evapd. at ∼20 mm. The residue (1.183 g.) is taken up in 7 l. EtOH at 60°, the soln. added to 370 g. maleic acid in 1.7 l. EtOH (60°), and the mixt. kept overnight to give 1.192 g. I acid maleate (II), m. 176-7° (EtOH), [α]24D -21.2° ± 1.5° (c 2, CHCl3). II (300 g.) is added to a mixt. of 1 l. H2O and 2 l. CH2Cl2, 150 ml. NaOH (d. 1.33) added, and the org. phase sepd. and distd. to give 185 g. (-)-3-methylthio-10-(3-dimethylamino-2-methylpropyl)phenothiazine (III), m. 84-5° (iso-PrOH), [α]23D -45° ± 3° (c 2.5, C6H6).
PAPERJournal of Organic Chemistry (1960), 25, 944-7.https://pubs.acs.org/doi/abs/10.1021/jo01076a019cf. CA 54, 15391b. The prepn. of various 10-aminoalkyl derivs. of the following phenothiazines was described: 2-hydroxyphenothiazine (I), 2-methylthiophenothiazine (II), 2-methylsulfonylphenothiazine (III), 2-trifluoromethylsulfonylphenothiazine (IV), 2-trifluoromethylthiophenothiazine (V), 2-azaphenothiazine (VI), and 8-chloro-2-azaphenothiazine (VII). The direct alkylation of I was not attempted. Instead, 2-benzoyloxyphenothiazine was alkylated with NaNH2 in xylene and the ester group removed by basic hydrolysis during the workup. The alkylation of IV with 3-(4-methylpiperazinyl)propyl chloride required 48 hrs. VI (15 g.), 6.8 g. NaNH2, and 500 ml. PhMe refluxed 45 min. under N, treated with 21 g. 3-chloro-1-(1-formyl-4-piperazinyl)propane-HCl and 300 ml. PhMe, the mixt. cooled, 150 ml. H2O added, the PhMe layer extd. with dil. HCl, the acid exts. made alk., extd. with C6H6, and the solvent evapd. gave 21 g. oil. The oil dissolved in 250 ml. alc., 60 ml. H2O and 7 ml. 40% NaOH, the mixt. refluxed 2 hrs., the solvents removed, the residual oil dissolved in C6H6, the soln. extd. with HCl, made alk., extd. with C6H6, and the whole distd. gave 11 g. 10-[3-(1-piperazinyl)propyl]-2-azaphenothiazine. The distd. material was dissolved in 250 ml. MeOH and refluxed 1.5 hrs. with 1.8 g. ethylene oxide, the solvent evapd., the residue dissolved in 250 ml. C6H6, the soln. azeotropically distd. during 1 hr., cooled, and refluxed 1 hr. with 6.5 g. AcCl, the solvents evapd., the gum treated with 10% NaOH, and the C6H6 evapd. gave 4.3 g. 4-[3-(2-azaphenothiazin-10-yl)propyl]-1-piperazineëthanol; acetate dimaleate m. 147-8° (decompn.) (EtOAc). 1-Piperazinepropanol (57.6 g.) refluxed 1 hr. with 48 g. HCO2Me, the excess HCO2Me removed, and the residue distd. gave 65.3 g. oil, b1.1 174.5-7.0°, n24D 1.5072. This oil (42.8 g.) in 300 cc. CHCl3 treated with excess HCl, then 19 g. SOCl2, the mixt. refluxed 0.5 hr., 3 g. SOCl2 added, refluxing continued 2.5 hrs., and the solvents removed gave a cryst. HCl salt. Conversion of this to the free base gave 60% 1-formyl-4-(3-chloropropyl)piperazine, yellow oil, b0.4 144.5-8.5°, n25D 1.5053. By starting with I-VII the following 2,10-disubstituted phenothiazines were obtained (substituents at 2, 10, b.p./mm., and % yield given); SMe, (CH2)3NMe2, 220-3°/0.7 (HCl salt m. 149-50°), 88; SMe, CH2CHMeCH2NMe2, 218-21°/0.1 (HCl salt m. 173-4°), 93; SMe, (CH2)3N.(CH2)2.NMe.CH2.CH2, 239-42°/0.1 (di-HCl salt m. 224-5°), 92; SMe, CH2CHMeCH2N.(CH2)2. NMe.CH2.CH2, 200-20°/0.03 (dimaleate m. 174-5°), 44; SMe, (CH2)3N.(CH2)2.N[(CH2)2OAc].CH2.CH2 – (dimaleate m. 165-6°), 33; SO2Me, (CH2)3NMe2, 115-16° (HCl salt m. 112-15°), 62; SO2Me, CH2CHMeCH2NMe2, 255-60°/0.2 (HCl salt m. 234-5°), 60; SCF3, (CH2)3NMe2, 153-7°/0.1, 64; SCF3, CH2CHMeCH2NMe2, 153-7°/0.1 (picrate m. 158.5-9.5°), 54; SCF3, I (CH2)3N.(CH2)2.NMe.CH2.CH2, 220-3°/0.3 (dimaleate m. 182-3°), 63; SO2CF3, (CH2)3NMe2, 235-40°/0.04 (HCl salt m. 174-5°), 15; SO2CF3, CH2CHMeCH2NMe2, 182-4°/0.2 (picrate m. 203-4°), 19; SO2CF3, (CH2)3N.(CH2)2.NMe.CH2.CH2, – [di-HCl salt m. 249.5° (decompn.)], 16; OH, (CH2)3NMe2, 220-5°/0.05, m. 90-1° (dimaleate m. 132-3°), 49. The following 8,10-substituted 2-azaphenothiazines were similarly prepd. (8,10 substituents, m.p. or b.p., % yield given): H, (CH2)3NMe2, 165-70°/0.007 [di-HCl salt m. 240.5-4.5° (decompn.)], 63; H, CH2CHMeCH2NMe2, 190-5°/0.6 (di-HCl salt m. 234-5°), 82; H, (CH2)3N.(CH2)2.N[(CH2)2OAc].CH2.CH2, – (dimaleate m. 147-8° (decompn.), 9; Cl, (CH2)3NMe2, 215-20°/1 (di-HCl salt m. 249-50°), 66.
PATENTGB 802725N-Aminoalkyl derivs. of I, where the alkyl is a straight or branched 2-5 C atom chain and the amino may be mono- or dialkylated or may be substituted by a pyrrolidino, piperidino, morpholino, or 4-alkyl-1-piperazinyl group, are prepd. by condensing I with the appropriate halo amine or by decompg. a phenothiazine-10-carboxylate of the appropriate amino alcohol. I (4.9 g.) was heated in 50 cc. boiling anhyd. xylene with 0.88 g. sodamide 1 hr., 2.71 g. 3-dimethylamino-1-chloropropane added, the soln. boiled 6 hrs., treated with H2O, then with dil. HCl, made alk. with NaOH, extd. with ether, and the solvent was evapd. in vacuo to give 4.5 g. 3-methylthio-10-(3-dimethylaminopropyl)phenothiazine (III), b0.2 206-18°; III.2HCl m. 160° (acetone-ether); picrate m. 135° (acetone). 3-Methylthio-10-(3-dimethylamino-2-methylpropyl)phenothiazine, m. 88-9°, was prepd. from I and 3-dimethylamino-2-methyl-1-chloropropane; picrate m. 145° (EtOH). The following were similarly prepd.: 3-methylthio-10-[3-(4-methyl-1-piperazinyl)propyl]phenothiazine, b0.1 250-6° [dihydrochloride m. 220° (decompn.) (acetone-ether); dipicrate m. 252-3° (acetone-iso-PrOH); 3-methylthio-10 – (2 – dimethylaminopropyl)phenothiazine, b0.2 202-6° (hydrochloride m. 205-6°; picrate m. 190°); 3-methylthio-10- (3-pyrrolidinopropyl)phenothiazine, b0.9 261° (hydrochloride m. 161°). I was phosgenated in toluene in the presence of pyridine to the 3-methylthiophenothiazine-10-carbonyl chloride (IV), m. 125°; IV heated in toluene with 3-(4- methyl-1-piperazinyl)-2-methylpropanol gave 3-(4-methyl-1- piperazinyl)-2-methylpropyl 3-methylthiophenothiazine-10- carboxylate (V) (dihydrochloride m. 225°). A soln. of 13 7 g. V in 60 cc. ο-Cl2C6H4 was boiled for 5 hrs. till CO2 evolution ceased, the soln. cooled, 60 cc. ether added and the mixt. H2O-washed, extd. with 10% HCl, made alk. with NaOH, and extd. with ether. The ether soln. was dried over anhyd. Na2SO4 and distd. in vacuo to yield 11.25 g. crude base which gave, with an EtOH soln. of maleic acid, 12.7 g. 3-methylthio-10-[3-(4-methyl-1-piperazinyl)-2-methyl-propyl]phenothiazinecarboxylic acid dimaleate, m. 199°. 3-Methylthio-10- [2,3-bis(dimethylamino)propyl] phenothiazine neutral fumarate, m. 198°, was similarly obtained by decarboxylating 1,3-bis(dimethylamino)-2-propyl 3-methylthiophenothiazine-10-carboxylate and treating with fumaric acid. 3-Methylthio-10-(3-diethylaminopropyl)phenothiazine-HCl, m. 172°, was prepd. from 3-methylthio-10-[3-(p-toluenesulfonyloxy)propyl]phenothiazine (VI) and Et2NH; 3-methylthio-10-(3-methylaminopropyl)phenothiazine (H oxalate m. 186°), from VI and MeNH2. VI heated with excess NH3 in toluene gave 3-methylthio-10-(3-aminopropyl)phenothiazine (VII) (oxalate m. 198°). VII in dioxane was neutralized with N HCl and treated with 30% aq. HCHO and PtO2 to give III. These compds. are antiemetics and potentiators of general anasthetics or neuroleptics.
SYN

///////////Methiomeprazine , antiemetic, Metiomeprazina, RP 10584, RP-10584, RP10584, RP 10584, SKF 6270
Systematic name (3):
- 10-[3-(ジメチルアミノ)-2-メチルプロピル]-2-(メチルチオ)-10H-フェノチアジン
- N,N,β-トリメチル-2-(メチルチオ)-10H-フェノチアジン-10-プロパン-1-アミン
- N,N,β-トリメチル-2-メチルチオ-10H-フェノチアジン-10-プロパン-1-アミン
Other name (6):
- メチオメプラジン
- Methiomeprazine
- 10-[3-(Dimethylamino)-2-methylpropyl]-2-(methylthio)-10H-phenothiazine
- SKF-6270
- N,N,β-Trimethyl-2-(methylthio)-10H-phenothiazine-10-propan-1-amine
CSc1ccc2Sc3ccccc3N(CC(C)CN(C)C)c2c1

NEW DRUG APPROVALS
ONE TIME
$10.00
Dichlorquinazine

CORRECT STR OF Dichlorquinazine
7-chloro-N-[1-[4-[2-[(7-chloroquinolin-4-yl)amino]propyl]piperazin-1-yl]propan-2-yl]quinolin-4-amine;methanesulfonic acid
- 1,4-Piperazinediethanamine, N,N’-bis(7-chloro-4-quinolinyl)-α,α’-dimethyl- (9CI)
- Quinoline, 4,4-[1,4-piperazinediylbis[(1-methylethylene)imino]]bis[7-chloro- (7CI)
- Quinoline, 4,4′-[1,4-piperazinediylbis[(1-methylethylene)imino]]bis[7-chloro- (8CI)
- N1,N4-Bis(7-chloro-4-quinolinyl)-α1,α4-dimethyl-1,4-piperazinediethanamine
- 1,4-Bis[2-(7-chloro-4-quinolylamino)propyl]piperazine
- Bis[(chloro-7”-quinolyl-4”)amino-2′-propyl]-1,4-piperazine
- Dichlorquinazine
- N,N’-Bis(7-chloro-4-quinolyl)-α,α’-dimethylpiperazine-1,4-diethylamine
- NSC 129790
- RP 12278
- WR 3863
WRONG STRUCTURE
WRONG STRUCTURE
Dichlorquinazine
- BRN 0867697
- Dichlorquinazine
- EINECS 234-130-6
- NSC 129790
- RP 12278
- UNII-HT3GAD2SCM
- WR 3863
cas 10547-40-7
C28H32Cl2N6, mw
| 523.5 |
7-chloro-N-[2-[4-[2-[(7-chloroquinolin-4-yl)amino]propan-2-yl]piperazin-1-yl]propan-2-yl]quinolin-4-amine
VARIANT
RN: 23256-65-7
Molecular Formula, C28-H32-Cl2-N6.C-H4-O3-S, Molecular Weight, 619.6144
- RP-12278 mesylate
- WR-3863 mesylate
- Quinoline, 4,4′-(1,4-piperazinediylbis((1-methylethylene)imino))bis(7-chloro-, tetramethanesulfonate bis((7-chloro-4”-quinolyl)-2′-aminopropyl)-1,4-piperazine methanesulfonate
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
PATENTS
BE 626239
4-(Chloro or alkoxy)quinolines are treated with a 1,4-bis(aminoalkyl)piperazine to give the title compds. which can be used as antiinflammatory agents and as amebicides. Thus, a mixt. of 16.3 g. 4-chloroquinoline, 10 g. 1,4-bis(3-aminopropyl)piperazine, 55 g. PhOH, and 0.2 g. NH4Cl is heated 5 hrs. at 175°, poured into a mixt. of 500 ml. H2O and 100 ml. NaOH (d. 1.33), filtered, the ppt. is treated with a mixt. of 80 ml. H2O and 20 ml. NaOH, the mixt. filtered, and the ppt. washed with 500 ml. H2O and dried to give 15.9 g. 1,4-bis[3-(4-quinolyl)aminopropyl]piperazine, m. 210°(MeOH-H2O). Similarly prepd. are the following I: n, R, R1, R2, X, Y, m.p.; 2, H, H, H, MeO, H, 245° (HCONMe2); 2, H, H, H, H, SO2NMe2, 271° (HCONMe2); 2, H, H, H, H, CF3, 293° (HCONMe2); 3, Me, H, H, H, H, ∼100°; 3, Me, Ac, H, H, H, -(1); 3, Me, H, H, MeOH, 180° and 190°; 3, Me, Ac, H, MeO, H, -(2); 1, Me, H, Me, H, Cl, 264°; 2, H, H, H, Cl, H, 264° (BuOH); 1, Me, H, H, H, CF3, 240° (MeCOEt); 2, H, H, H, H, MeO, 200° (EtOH); 2, H, H, Me, H, MeO, 216° (EtOH); 3, Me, H, H, H, MeO, 218° (CH2Cl2); (1) bis(acid maleate) m. 155° (iso-PrOH), (2) bis(acid maleate) m. 155° The following II were also prepd.: n, R, R1, R2, m.p.; 1, Me, A(R = R1 = X = Y = H,Z =Cl), A(R = R1 = X = Z = H,Y = Cl), 208-10° (HCONMe2); 1, Me, A(R = R1 = X = Y= H, Z = Cl), A(R = R1 = X = Y = H,Z = MeO), 206-8° (HCONMe2); 1, Me, A(R1 = X = Y = H, R = 4-ClC6H4, Z = Cl), A(R = R1 = X = Y = H,Z = Cl, 230-2° (HCONMe2) The following III were prepd.: n, R, m, R1, R2, m.p.; 3, Me, 1, H, A(R = R1 = X = Y = H, Z= Cl), 190-1° and 213-15°; 2, H, 2, H, A(R = X = Y = H, R1 = Me, Z =Cl), 198° (PrOH); 3, Me, 2, H, A(R = R1 = X = Y = H,Z = Cl), 160-2°; 1, Me, 1, H, A(R = R1 = X = Y = H,Z = Cl), 178°; 1, Me, 1, Me, A(R1 = X = Z = H,R = Me, Y =AcNH), 330° (decompn.) (EtOH); 2, H, 2, H, A(R1 = X = Y = H,R = 4-ClC6H4,Z = Cl), 320-1° (HCONMe2); 2, H, 2, H, A(R = Y = Z = H, R1 = Me, X = Cl) 96° (iso-PrOH); 1, Me, 1, Me, A(R = R1 = X = Z = H, Y = Cl), 220° and 246-8°; 1, Me, 1, Me, A(R1 = X = Z = H, R = Me, Y = NH2), 305° (EtOH-H2O); 1, Me, 1, Me, A(R1 = X = Z = H, R = Me, Y = MeO, 244° (EtOH) Also prepd. were (m.p. given): 1,4-bis[2-(7-chloro-4-quinolylamino)propyl]hexahydro-1,4-diazepine, 169°; 1-[5-(7-chloro-4-quinolylamino)-2-pentyl]-4-[2-(7-chloro-4-quinolylamino)propyl] piperazine, 210-12°(HCONMe2); 1,4-bis[3-(7-chloro- 4-quinolylamino)propyl] hexahydro-1,4-diazepine, 186° (HCONMe2). The following were prepd. (m.p. and optical rotation given):L(+)-1,4-bis[2-(7-chloro-4-quinolylamino)propyl]piperazine, 250-1°, [α]23.5D 382° ± 1° (c 4, 50:50 MeOH-H2O); D(-)-1,4- bis[2-(7-chloro-4-quinolylamino)propyl] piperazine, 250-1°, [α]25D -382.5° ± 1° (c 4, 50:50 MeOH-H2O); DL-1,4-bis[2-(7-chloro-4-quinolylamino)propyl]piperazine (IV), 266-8°, -; meso-1,4-bis [2-(7-chloro-4-quinolylamino)propyl] piperazine (V), 270-1° (HCONMe2), -; equimol. mixt. of IV and V, 250-2°, -; 1,4-bis[2-(6-chloro-4-quinolylamino)propyl]piperazine-form A (VI-form A), 227° -; VI-form B, 110° and 245°, -. Also prepd. are the following intermediates of the general formula VII (R = H) (X, Y, Z, and m.p. given): OH, H, SO2NMe2, ∼288°; Cl, H,SO2NMe2, 170°; HO(CH2)3CHMeNH, H, H, 158° (EtOH); AcO(CH2)3CHMeNAc, H, H, -; HO(CH2)3CHMeNAc, H, H, -; MeSO3(CH2)3CHMeNAc, H, H, -; N-(5-piperazino-2-pentyl)acetamido, H, H, -; HO(CH2)3CHMeNH, MeO, H, -; AcO(CH2)3CHMeNAc, MeO, H, -; HO(CH2)3CHMeNAc, MeO, H, -; MeSO3(CH2)3CHMeNAc, MeO, H, -; N-(5-piperazino-2-pentyl)acetamido, MeO, H, -; Me(HOCH2)CH, H, Cl, 210°; Me(ClCH2)CH, H, Cl, 148-50°; Me(HOCH2)CH, Cl, H, 192°; Me(ClCH2)CH, Cl, H, 142°; Me(HOCH2)CH, H, MeO, 170°; Me(ClCH2)CH, H, MeO, 160°. Also prepd. were (m.p. given): VII (R = CO2Et, X = OH, Y = H, Z = SO2NMe2), ∼335°; VII (R = CO2H, X = OH, Y = H, Z = SO2HMe2), 310° (decompn.); 1,4-bis(2-oxopropyl)hexahydro-1,4-diazepine, -; 1,4-bis(2-oximinopropyl)hexahydro-1,4-diazepine, 180-1°; 1,4-bis(2-aminopropyl)hexahydro-1,4-diazepine, -; 1,4-bis(2-cyanoethyl)-hexahydro-1,4-diazepine, -. The following were prepd. (m.p. and optical rotation given): L(+)-4-(3-hydroxy-2-propylamino)-7-chloroquinoline, 223-4°, [α]24D 28.5° ± 2° (c 1, EtOH); L(+)-4-(3-chloro-2-propylamino)-7-chloroquinoline, 146-7°, [α]24D 103 ± 1° (c 2, EtOH); L(+)-4-(3-piperazino-2-propylamino)-7-chloroquinoline, 128-30°, [α]23D 139 ± 1° (c 2, EtOH); D(-)-4-(3-hydroxy-2-propylamino)-7-chloroquinoline, 223-4°, [α]25D – 31 ± 2° (c 1, EtOH); D(-)-4-(3-chloro-2-propylamino)-7-chloroquinoline, 147-8°, [α]24D -101 ± 1° (c 2, EtOH); D(-)-4-(3-piperazino-2-propylamino)-7-chloroquinoline, 131-2°, [α]23D -137 ± 1° (c 2, EtOH)
PATENT
FR CAM42 19631007.
Piperazines (I) are antiinflammatory and anthelmintic agents. A mixt. of 8.25 g. MeCH(NH2)CH2OH, 19.8 g. 4,6-dichloroquinoline, and 55 g. PhOH is heated to give 16.0 g. 6-chloro-4-[(3-hydroxy-2-propyl)-amino]quinoline (II), m. 192°. II (14.0 g.) is treated with a soln. of 10.6 g. SOCl2 in 40 ml. CHCl3 to give 12.5 g. 6-chloro-4-[(3-chloro-2-propyl)amino]quinoline (III), m. 142°. A mixt. of 13.2 g. 1-[2-(7-chloro-4-quinolylamino)propyl]piperazine, 11.0 g. III, 6.4 g. NaI, 2.3 g. anhyd. Et3N, and 200 ml. AcEt is refluxed 18 hrs., the solvent is distd. in vacuo, and the residue is taken up in 100 ml. MeOH. The mixt. is made alk. with 110 ml. NaOH (d. 1.33), poured into 1000 ml. H2O, and the ppt. that forms is filtered off, washed with H2O, and recrystd. in HCONMe2 to give 11.0 g. 1-[2-(7-chloro-4-quinolylamino)propyl]-4-[2-(6-chloro-4-quinolylamino)propyl]piperazine, m. 208-10°. Similarly prepd. are the following I (R, m, R1, n, R2, R3, R4, and m.p. given): H, 2, H, 2, H, MeO, H, 245°; H, 2, H, 2, H, H, SO2NMe2, 271°; H, 2, H, 2, H, H, CF3, 293°; Me, 3, Me, 3, H, MeO, H, 180° and 190°; Me, 3, H, 1, H, H, Cl, 190-1° and 213-15°; H, 2, H, 2, H, Cl, H, 264°; Me, 1, Me, 1, H, H, CF3, 240°; H, 2, H, 2, H, H, MeO, 200°; Me, 3, H, 2, H, H, Cl, 160-2°; Me, 1, H, 1, H, H, Cl, 178°; Me, 1, Me, 1, Me, AcNH, H, 330°; H, 2, H, 2, p-ClC6H4, H, Cl, 320-1°; Me, 1, Me, 1, H, Cl, H, 227° (form A); Me, 1, Me, 1, H, Cl, H, 110° and 245° (form B); H, 3, H, 3, H, H, Cl, 239-41°; Me, 1, Me, 1, Me, NH2, H, 305°; Me, 1, Me, 1, Me, MeO, H, 244°; Me, 3, Me, 3, Me, 3, H, H, MeO, 218°; H, 3, H, 3, H, H, Cl, 240-2°. Also prepd. are (m.p. given): 1,4-bis[2-(7-chloro-4-quinolylamino)propyl]hexahydrodiazepine, 169°; 2,5-dimethyl-1,4-bis[2-(7-chloro-4-quinolylamino)propyl)piperazine, 264°; 1-[5-(7-chloro-4-quinolylamino [-2-pentyl]-4-[2-(7-chloro -4-quinolylamino)propyl]piperazine, 210-12°; 2,5-dimethyl-1,4-bis[3-(7-methoxy-4-quinolylamino)propyl]piperazine, 216°; 1,4-bis[3-(3-methyl-7-chloro-4-quinolylamino)propyl] piperazine, 198°; 1,4-bis[3-(7-chloro-4-quinolylamino)propyl]hexahydrodiazepine, 186°; 1-[2(7-chloro-4-quinolylamino)propyl]-4-[2-(7-methoxy-4-quinolylamino)propyl]piperazine, 206-8°; 1,4-bis[3-(3-methyl-5-chloro-4- quinolylamino)propyl]piperazine, 96°; 1 – [2 -[2 -(p – chlorophenyl)- 7- chloro- 4- quinolylamino]propyl] -4 – [2 – (7 – chloro – 4-quinolylamino)propyl]piperazine, 230-2°; L(+) 1,4-bis[2-(7-chloro-4-quinolylamino)propyl]piperazine, 250-1°, [α]23.5D + 382° ± 1° (c 4, 50/50 MeOH-H2O); L(+)-7-chloro-4-(3-hydroxy-2-propylamino)quinoline, 223-4°, [α]24D 28.5° ± 2° (c 1, EtOH); L(+)-7-chloro-4-(3-chloro-2-propylamino)quinoline, 146-7°, [α]24D 103° + 1° (c 2, EtOH); L(+)-7-chloro-4-(3-piperazino-2-propylamino)quinoline 128-30°, [α]23D 139° ± 1° (c 2, EtOH); D(–)-1,4-bis[2-(7-chloro-4-quinolylamino)propyl]piperazine, 250-1°, [α]25D -382° ± 1° (c 4, 50:50 MeOH-H2O); meso- 1,4 – bis [2 – (7 – chloro – 4 – quinolylamino)propyl] piperazine, 270-1°.
Patent Information
BE 612207

| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2016045487-A1 | Compositions and methods for treating neuropathy | 2013-03-27 | |
| WO-2014160811-A1 | Compositions and methods for treating neuropathy | 2013-03-27 | |
| AU-2014234258-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| AU-2014234258-B2 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | 2019-02-14 |
| CA-2907628-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| EP-2976069-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| EP-2976069-B1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | 2020-05-06 |
| US-2014322296-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| US-2016045447-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| US-9668979-B2 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | 2017-06-06 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2014147242-A1 | Piperaquine microcapsules and compositions containing them | 2013-03-22 | |
| AU-2009215107-A1 | Treatments for neuropathy | 2008-02-12 | |
| AU-2009215107-B2 | Treatments for neuropathy | 2008-02-12 | 2013-05-09 |
| AU-2013203934-A1 | Treatments for neuropathy | 2008-02-12 | |
| CA-2714676-A1 | Treatments for neuropathy | 2008-02-12 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| CA-2714676-C | Treatments for neuropathy | 2008-02-12 | 2015-04-14 |
| EP-2240177-A2 | Treatments for neuropathy | 2008-02-12 | |
| US-2009203735-A1 | Treatments for neuropathy | 2008-02-12 | |
| US-2011086878-A1 | Treatments for Neuropathy | 2008-02-12 | |
| US-2016058749-A1 | Treatments for neuropathy | 2008-02-12 |
////////////////Dichlorquinazine, BRN 0867697, Dichlorquinazine, EINECS 234-130-6, NSC 129790, RP 12278, UNII-HT3GAD2SCM, WR 3863
CC(C)(NC1=C2C=CC(=CC2=NC=C1)Cl)N3CCN(CC3)C(C)(C)NC4=C5C=CC(=CC5=NC=C4)Cl
WRONG
CC(CN1CCN(CC(C)Nc2ccnc3cc(Cl)ccc23)CC1)Nc4ccnc5cc(Cl)ccc45.CS(=O)(=O)O
AND
Clc1ccc2c(c1)nccc2NC(C)CN1CCN(CC(C)Nc2ccnc3cc(Cl)ccc32)CC1
CORRECT

NEW DRUG APPROVALS
ONETIME
$10.00
RP 12146


RP 12146, EX-A7910
cas 2732869-64-4
4-[[5-[(3R)-3-hydroxy-3-methyl-2-oxoindol-1-yl]pyridin-3-yl]methyl]-2H-phthalazin-1-one
| M.Wt | 398.422 | |
| Formula | C23H18N4O3 | |
RP12146 (RP12146) is a novel, selective, and potent small molecule inhibitor of PARP1/2 with IC50 of 0.6/0.5 nM, with several fold selectivity over other isoforms.
SCHEME
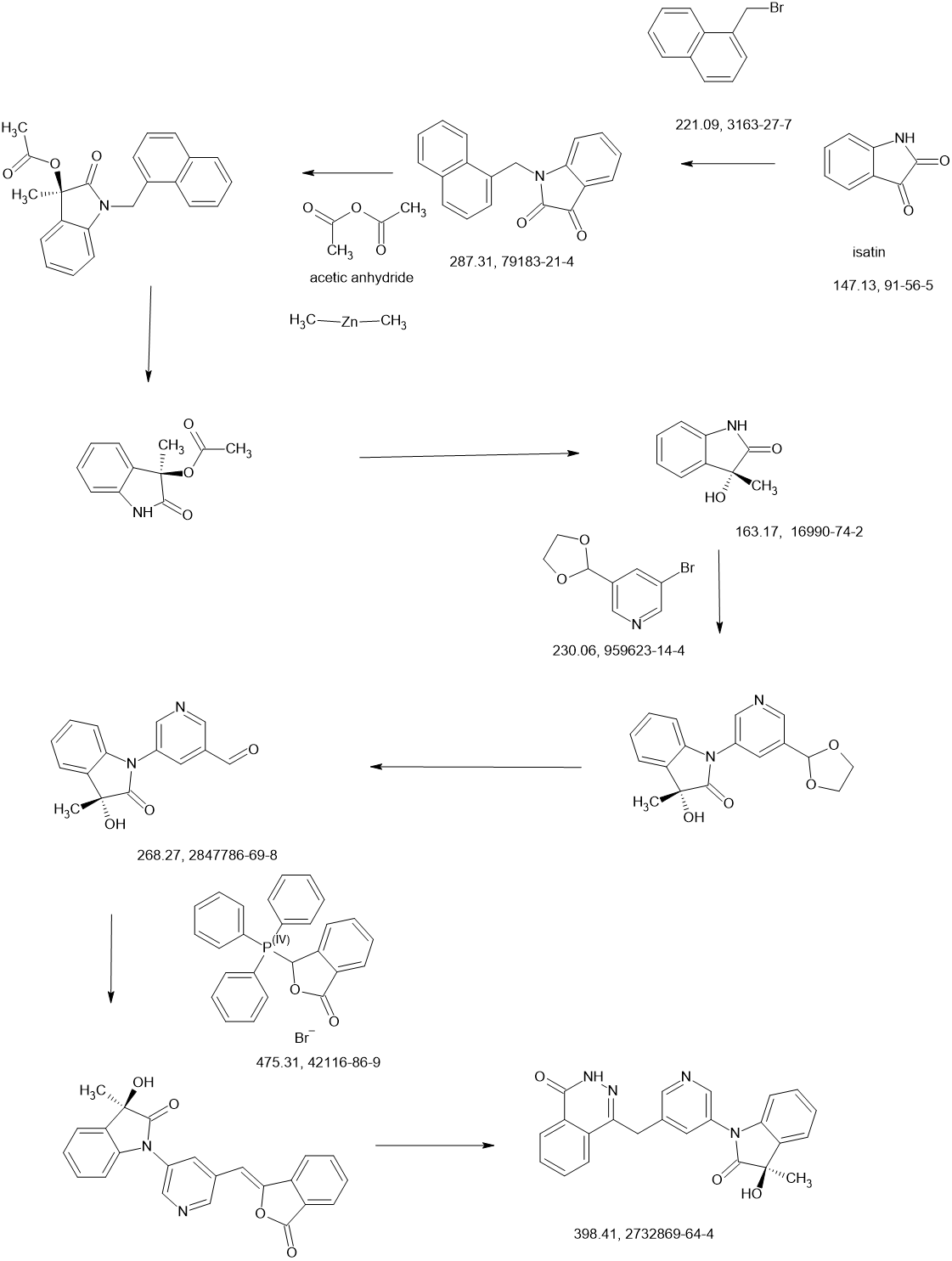
Ref
WO2021220120
Rhizen Pharmaceuticals AG
WO2022215034
Rhizen Pharmaceuticals AG; Incozen Therapeutics Pvt. Ltd.
RP-12146 is an oral poly (ADP-ribose) polymerase (PARP) inhibitor in phase I clinical development at Rhizen Pharmaceuticals for the treatment of adult patients with locally advanced or metastatic solid tumors.
Solid TumorExtensive-stage Small-cell Lung CancerLocally Advanced Breast CancerMetastatic Breast CancerPlatinum-sensitive Ovarian CancerPlatinum-Sensitive Fallopian Tube CarcinomaPlatinum-Sensitive Peritoneal Cancer
Poly(ADP-ribose) polymerase (PARP) defines a family of 17 enzymes that cleaves NAD+ to nicotinamide and ADP-ribose to form long and branched (ADP-ribose) polymers on glutamic acid residues of a number of target proteins, including PARP itself. The addition of negatively charged polymers profoundly alters the properties and functions of the acceptor proteins. Poly(ADP-ribosyl)ation is involved in the regulation of many cellular processes, such as DNA repair, gene transcription, cell cycle progression, cell death, chromatin functions and genomic stability. These functions have been mainly attributed to PARP-1 that is regarded as the best characterized member of the PARP family. However, the identification of novel genes encoding PARPs, together with the characterization of their structure and subcellular localization, have disclosed different roles for poly(ADP-ribosyl)ation in cells, including telomere replication and cellular transport.
Recently, poly(ADP-ribose) binding sites have been identified in many DNA damage checkpoint proteins, such as tumor suppressor p53, cyclin-dependent kinase inhibitor p21Cip1/waf1, DNA damage recognition factors (i.e., the nucleotide excision repair xeroderma pigmentosum group A complementing protein and the mismatch repair protein MSH6), base excision repair (BER) proteins (i.e. DNA ligase III, X-ray repair cross-complementing 1, and XRCC1), DNA-dependent protein kinase (DNA-PK), cell death and survival regulators (i.e.,
NF-kB, inducible nitric oxide synthase, and telomerase). These findings suggest that the different components of the PARP family might be involved in the DNA damage signal network, thus regulating protein-protein and protein-DNA interactions and, consequently, different types of cellular responses to genotoxic stress. In addition to its involvement in BER and single strand breaks (SSB) repair, PARP-1 appears to aid in the non-homologous end-joining (NHEJ) and homologous recombination (HR) pathways of double strand breaks (DSB) repair. See Lucio Tentori et al., Pharmacological Research, Vol. 45, No. 2, 2002, page 73-85.
PARP inhibition might be a useful therapeutic strategy not only for the treatment of BRCA mutations but also for the treatment of a wider range of tumors bearing a variety of deficiencies in the HR pathway. Further, the existing clinical data (e.g., Csaba Szabo et al., British Journal of Pharmacology (2018) 175: 192-222) also indicate that stroke, traumatic brain injury, circulatory shock and acute myocardial infarction are some of the indications where PARP activation has been demonstrated to contribute to tissue necrosis and inflammatory responses.
As of now, four PARP inhibitors, namely olaparib, talazoparib, niraparib, and rucaparib have been approved for human use by regulatory authorities around the world.
Patent literature related to PARP inhibitors includes International Publication Nos. WO 2000/42040, WO 2001/016136, WO 2002/036576, WO 2002/090334, WO2003/093261, WO 2003/106430, WO 2004/080976, WO 2004/087713, WO 2005/012305, WO 2005/012524, WO 2005/012305, WO 2005/012524, WO 2005/053662, W02006/033003, W02006/033007, WO 2006/033006, WO 2006/021801, WO 2006/067472, WO 2007/144637, WO 2007/144639, WO 2007/144652, WO 2008/047082, WO 2008/114114, WO 2009/050469, WO 2011/098971, WO 2015/108986, WO 2016/028689, WO 2016/165650, WO 2017/153958, WO 2017/191562, WO 2017/123156, WO 2017/140283, WO 2018/197463, WO 2018/038680 and WO 2018/108152, each of which is incorporated herein by reference in its entirety for all purposes.
There still remains an unmet need for new PARP inhibitors for the treatment of various diseases and disorders associated with cell proliferation, such as cancer.
PATENT
Illustration 1
CLIP
https://cancerres.aacrjournals.org/content/81/13_Supplement/1233
Abstract 1233: Preclinical profile of RP12146, a novel, selective, and potent small molecule inhibitor of PARP1/2
Srikant Viswanadha, Satyanarayana Eleswarapu, Kondababu Rasamsetti, Debnath Bhuniya, Gayatriswaroop Merikapudi, Sridhar Veeraraghavan and Swaroop VakkalankaProceedings: AACR Annual Meeting 2021; April 10-15, 2021 and May 17-21, 2021; Philadelphia, PA
Abstract
Background: Poly (ADP-ribose) polymerase (PARP) activity involves synthesis of Poly-ADP ribose (PAR) polymers that recruit host DNA repair proteins leading to correction of DNA damage and maintenance of cell viability. Upon combining with DNA damaging cytotoxic agents, PARP inhibitors have been reported to demonstrate chemo- and radio-potentiation albeit with incidences of myelosuppression. A need therefore exists for the development selective PARP1/2 inhibitors with a high therapeutic window to fully exploit their potential as a single agent or in combination with established therapy across various tumor types. Additionally, with the emerging concept of ‘synthetic lethality’, the applicability PARP inhibitors can be expanded to cancers beyond the well-defined BRCA defects. Herein, we describe the preclinical profile of RP12146, a novel and selective small molecule inhibitor of PARP1 and PARP2.
Methods: Enzymatic potency was evaluated using a PARP Chemiluminescent Activity Assay Kit (BPS biosciences). Cell growth was determined following incubation with RP12146 in BRCA1 mutant and wild-type cell lines across indications. Apoptosis was evaluated following incubation of cell lines with compound for 120 h, subsequent staining with Annexin-V-PE and 7-AAD, and analysis by flow cytometry. For cell cycle, cells were incubated with compound for 72 h, and stained with Propidium Iodide prior to analysis by flow cytometry. Expression of downstream PAR, PARP-trapping, phospho-γH2AX and cleaved PARP expression were determined in UWB1.289 (BRCA1 null) cells by Western blotting. Anti-tumor potential of RP12146 was tested in OVCAR-3 Xenograft model. Pharmacokinetic properties of the molecule were also evaluated. Results: RP12146 demonstrated equipotent inhibition of PARP1 (0.6 nM) and PARP2 (0.5 nM) with several fold selectivity over the other members of the PARP family. Compound caused a dose-dependent growth inhibition of both BRCA mutant and non-mutant cancer cell lines with GI50 in the range of 0.04 µM to 9.6 µM. Incubation of UWB1.289 cells with RP12146 caused a G2/M arrest with a corresponding dose-dependent increase in the percent of apoptotic cells. Expression of PAR was inhibited by 86% at 10 nM with a 2.3-fold increase in PARP-trapping observed at 100 nM in presence of RP12146. A four-fold increase in phospho-γH2AX and > 2-fold increase in cleaved PARP expression was observed at 3 µM of the compound. RP12146 exhibited anti-tumor potential with TGI of 28% as a single agent in OVCAR-3 xenograft model. Efficay was superior compared to Olaparib tested at an equivalent dose. Pharmacokinetic studies in rodents indicated high bioavailability with favorable plasma concentrations relevant for efficacy
Conclusions: Data demonstrate the therapeutic potential of RP12146 in BRCA mutant tumors. Testing in patients is planned in H1 2021.
Citation Format: Srikant Viswanadha, Satyanarayana Eleswarapu, Kondababu Rasamsetti, Debnath Bhuniya, Gayatriswaroop Merikapudi, Sridhar Veeraraghavan, Swaroop Vakkalanka. Preclinical profile of RP12146, a novel, selective, and potent small molecule inhibitor of PARP1/2 [abstract]. In: Proceedings of the American Association for Cancer Research Annual Meeting 2021; 2021 Apr 10-15 and May 17-21. Philadelphia (PA): AACR; Cancer Res 2021;81(13_Suppl):Abstract nr 1233.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
CLIP
Rhizen Pharmaceuticals AG Announces First Patient Dosing in a Phase I/Ib Study of Its Novel PARP Inhibitor (RP12146) in Patients With Advanced Solid Tumors
RHIZEN’S PARP INHIBITOR EFFORTS ARE PART OF A LARGER DDR PLATFORM THAT ALSO INCLUDES AN EARLY STAGE POLθ-DIRECTED PROGRAM; PLATFORM ENABLES PROPRIETARY IN-HOUSE COMBINATIONS
- Rhizen Pharma commences dosing in a phase I/Ib trial to evaluate its novel PARP inhibitor (RP12146) in patients with advanced cancers.
- Rhizen indicated that RP12146 has comparable preclinical activity vis-à-vis approved PARP inhibitors and shows improved preclinical safety that it expects will translate in the clinic.
- The two-part multi-center phase I/Ib study is being conducted in Europe and is designed to initially determine safety, tolerability and MTD/RP2D of RP12146 and to subsequently assess its anti-tumor activity in expansion cohorts with HRR mutation-enriched ES-SCLC, ovarian and breast cancer patients.
- RP12146 is part of a larger DDR platform at Rhizen that includes a preclinical-stage Polθ inhibitor program; the DDR platform enables novel, proprietary, in-house combinations
November 01, 2021 07:24 AM Eastern Daylight Time
BASEL, Switzerland–(BUSINESS WIRE)–Rhizen Pharmaceuticals AG (Rhizen), a Switzerland-based privately held, clinical-stage oncology & inflammation-focused biopharmaceutical company, announced today that it has commenced dosing in a multi-center, phase I/Ib trial to evaluate its novel poly (ADP-ribose) polymerase (PARP) inhibitor (RP12146) in patients with advanced solid tumors. This two-part multi-center phase I/Ib study is being conducted in Europe and has been designed to initially determine safety, tolerability, maximum tolerated dose (MTD), and/or recommended phase II dose (RP2D) of RP12146 and to subsequently assess its anti-tumor activity in expansion cohorts with HRR mutation-enriched ES-SCLC, ovarian and breast cancer patients.
“Our PARP program is foundational for our DDR platform efforts and will be the backbone for several novel proprietary combinations that we hope to bring into development going forward.”
Rhizen indicated that RP12146 has shown preclinical activity and efficacy comparable to the approved PARP inhibitor Olaparib, and shows improved safety as seen in the preclinical IND-enabling toxicology studies; an advantage that Rhizen hopes will translate in the clinical studies. Rhizen also announced that its PARP program is part of a larger DNA Damage Response (DDR) platform effort, which includes a preclinical-stage polymerase theta (Polθ) inhibitor program. Rhizen expects the platform to enable novel proprietary combinations of its PARP and Polθ assets given the mechanistic synergy and opportunity across PARP resistant/refractory settings.
“PARP inhibitors are a great success story in the DNA damage response area, but they are not without safety concerns that have limited realization of their full potential. Although our novel PARP inhibitor is competing in a crowded space, we expect its superior preclinical safety to translate into the clinic which will differentiate our program and allow us to extend its application beyond the current landscape of approved indications and combinations”, said Swaroop Vakkalanka, Founder & CEO of Rhizen Pharma. Swaroop also added that “Our PARP program is foundational for our DDR platform efforts and will be the backbone for several novel proprietary combinations that we hope to bring into development going forward.”
About Rhizen Pharmaceuticals AG.:
Rhizen Pharmaceuticals is an innovative, clinical-stage biopharmaceutical company focused on the discovery and development of novel oncology & inflammation therapeutics. Since its establishment in 2008, Rhizen has created a diverse pipeline of proprietary drug candidates targeting several cancers and immune associated cellular pathways.
Rhizen has proven expertise in the PI3K modulator space with the discovery of our first PI3Kδ & CK1ε asset Umbralisib, that has been successfully developed & commercialized in MZL & FL by our licensing partner TG Therapeutics (TGTX) in USA. Beyond this, Rhizen has a deep oncology & inflammation pipeline spanning discovery to phase II clinical development stages.
Rhizen is headquartered in Basel, Switzerland.
REF
Safety, Pharmacokinetics and Anti-tumor Activity of RP12146, a PARP Inhibitor, in Patients With Locally Advanced or Metastatic Solid Tumors….https://clinicaltrials.gov/ct2/show/NCT05002868
//////////RP 12146, oral poly (ADP-ribose) polymerase (PARP) inhibitor, phase I, clinical development, INCOZEN, Rhizen Pharmaceuticals, adult patients, locally advanced, metastatic solid tumors, PARP, CANCER, EX-A7910, EX A7910

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}