







- US6150383
- US6211205
- US6303640
- US6329404
- US8071130
- US7538125
- US7700128
- US7358366
Home » GENERICS
Category Archives: GENERICS
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
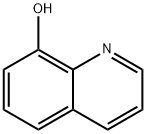
IODOQUINOL

IODOQUINOL
Diiodohydroxyquinoline
- Molecular FormulaC9H5I2NO
- Average mass396.951 Da
- NSC-8704
- SS-578
5,7-Diiodo-8-quinolinol
5,7-Diiodooxine
5,7-diiodoquinolin-8-ol
83-73-8[RN]
8-Hydroxy-5,7-diiodoquinoline
8-Quinolinol, 5,7-diiodo-
дийодогидроксихинолин[Russian][INN]
ثنائي إيودوهيدروكسيكينوليين[Arabic][INN]
双碘喹啉[Chinese][INN]
201-497-9[EINECS]
5,7-Diiodo-8-hydroxyquinoline
IodoquinolCAS Registry Number: 83-73-8
CAS Name: 5,7-Diiodo-8-quinolinol
Additional Names: diiodohydroxyquin; diiodo-oxyquinoline; 5,7-diiodo-8-hydroxyquinoline
Manufacturers’ Codes: SS-578
Trademarks: Diodoquin (Searle); Disoquin; Floraquin (Searle); Dyodin; Dinoleine; Searlequin; Diodoxylin; Rafamebin; Ioquin (Abbott); Direxiode (Delalande); Stanquinate; Yodoxin (Searle); Zoaquin; Enterosept; Embequin (M & B)
Molecular Formula: C9H5I2NO, Molecular Weight: 396.95
Percent Composition: C 27.23%, H 1.27%, I 63.94%, N 3.53%, O 4.03%
Literature References: Prepd by the action of iodine monochloride on 8-hydroxyquinoline: Papesch, Burtner, J. Am. Chem. Soc.58, 1314 (1936); by the action of KIO3 on 8-hydroxyquinoline: Zeifman, C.A.34, 3745. Electrolytic prepn: Brown, Berkowitz, Trans. Electrochem. Soc.75, 385 (1939). See also Claus, DE78880; Passek, DE411050; Matsumura, C.A.21, 1461 (1927); Pirrone, Cherubino, C.A.28, 3073 (1934).Properties: Crystals from xylene. The medicinal grade is a yellowish-brown powder. mp 200-215° (extensive decompn). Almost insol in water. Sparingly sol in alcohol, ether, and acetone; sol in hot pyridine and in hot dioxane.
Melting point: mp 200-215° (extensive decompn)
Therap-Cat: Antiamebic.
Keywords: Antiamebic.
The quinoline derivative diiodohydroxyquinoline (INN), or iodoquinol (USAN), can be used in the treatment of amoebiasis.[1]
It is poorly absorbed from the gastrointestinal tract and is used as a luminal amebicide. It acts by chelation of ferrous ions essential for metabolism.[2]
It was discovered by Adco Co. and introduced as diiodohydroxyquinoline.[3]
Susceptibility of Dientamoeba fragilis has been measured.[4]
Iodoquinol is an amebocide used against Entamoeba histolytica, and it is active against both cyst and trophozoites that are localized in the lumen of the intestine. It is considered the drug of choice for treating asymptomatic or moderate forms of amebiasis. The full mechanism of action is unknown. Iodoquinol is used for diseases caused by moderate intestinal amebiasis.
Diodoquin enhances zinc absorption in the zinc deficiency disorder Acrodermatitis enteropathica, probably because Diodoquin act as a zinc ionophore.[5]
5,7-Diiodo-8-quinolinol Chemical
Originator
Diiodohydroxyquinoline,Adco Co.
Uses
Antiamebic.
Uses
GABA prodrug
Uses
It acts as an amoebicidal and so used in the treatment of amoebiasis, balantidiasis (an infection caused by protozoa).
Indications
Iodoquinol (diiodohydroxyquin, Yodoxin, Moebiquin) is a halogenated 8-hydroxyquinoline derivative whose precise mechanism of action is not known but is thought to involve an inactivation of essential parasite enzymes. Iodoquinol kills the trophozoite forms of E. histolytica, B. coli, B. hominis, and Dientamoeba fragilis.
Iodoquinol is absorbed from the gastrointestinal tract and is excreted in the urine as glucuronide and sulfate conjugates. Most of an orally administered dose is excreted in the feces. Iodoquinol has a plasma half-life of about 12 hours.
Iodoquinol is the drug of choice in the treatment of asymptomatic amebiasis and D. fragilis infections. It is also used in combination with other drugs in the treatment of other forms of amebiasis and as an alternative to tetracycline in the treatment of balantidiasis.
Adverse reactions are related to the iodine content of the drug; the toxicity is often expressed as skin reactions, thyroid enlargement, and interference with thyroid function studies. Headache and diarrhea also occur. Chronic use of clioquinol, a closely related agent, has been linked to a myelitislike illness and to optic atrophy with permanent loss of vision.
Manufacturing Process
5,7-Diiodo-8-quinolinol widely used as an intestinal antiseptic, especially as an antiamebic agent. It is also used topically in other infections and may cause CNS and eye damage. It is known by very many similar trade names worldwide.
0.01 mol 8-oxychinoline and 0.01 mol salicylic acid were dissolved in 500 ml of water and then 0.05 mol potassium iodide was added. The mixture was heated to temperature 90°-100°C. After that 0.01 mol of KIO3 by little tiles was added. The next tile was added after a disappearence of discharging iodine. Then 10 ml 2 N HCl was added. The solid product was fallen, filtered off, washed with hot water and in 0.25 N NaOH dissolved. The solution was filtered and the clear filtrate precipitated with a very little excess of HCl. The product 5,7-diiodo-8-quinolinol was filtered, washed with hot water and dried. MP: 200°-250°C (with decomposition).
brand name
Quinadome (Bayer); Yodoxin (Glenwood).
Therapeutic Function
Antibacterial
Clinical Use
5,7-Diiodo-8-quinolinol, 5,7-diiodo-8-hydroxyquinoline,or diiodohydroxyquin (Yodoxin, Diodoquin, Diquinol) is ayellowish to tan microcrystalline, light-sensitive substancethat is insoluble in water. It is recommended for acute andchronic intestinal amebiasis but is not effective in extraintestinaldisease. Because a relatively high incidence of topicneuropathy has occurred with its use, iodoquinol should notbe used routinely for traveler’s diarrhea.
Safety Profile
Poison by ingestion and intravenous routes. Human systemic effects by ingestion: eye effects. Mutation data reported. When heated to decomposition it emits very toxic fumes of Iand Nox
Chemical Synthesis
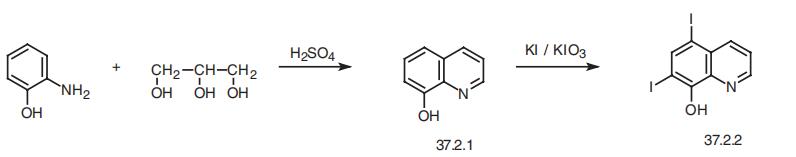
Iodoquinol, 5,7-diiodo-8-quinolinol (37.2.2), is made by iodination of 8-oxyquinoline (37.2.1) using a mixture of potassium iodide/potassium iodate. The initial 8-hydroxyquinolin (37.2.1) is made from 2-aminophenol and glycerol in the presence of sulfuric acid and nitrobenzene (Skraup synthesis).
Purification Methods
It crystallises from xylene and is dried at 70o in a vacuum. [Beilstein 21 II 58.]
5,7-Diiodo-8-quinolinol synthesis
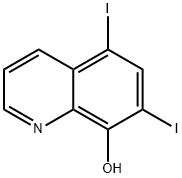
Synthesis of 5,7-Diiodo-8-quinolinol from 8-Hydroxyquinoline
SYN
DE 411050 DOI: 10.1021/ja01298a506

CLIP
Iodoquinol, 5,7-diiodo-8-quinolinol (37.2.2), is made by iodination of 8-oxyquinoline (37.2.1) using a mixture of potassium iodide/potassium iodate. The initial 8-hydroxyquinolin (37.2.1) is made from 2-aminophenol and glycerol in the presence of sulfuric acid and nitrobenzene (Skraup synthesis) [39,40]

Iodoquinol is an amebocide used against E. histolytica, and it is active against both cysts and trophozoites that are localized in the lumen of the intestine. It is considered the drug of choice for treating asymptomatic or moderate forms of amebiasis. The mechanism of action is unknown. Iodoquinol is used for diseases caused by moderate intestinal amebiasis. Synonyms of this drug are diquinol, iodoxin, diiodoquin, amebaquin, and others
39. F. Passek, Ger. Pat. 411.050 (1925). 40. V. Papesch, R.R. Burtner, J. Am. Chem. Soc., 58, 1314 (1936).

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Ghaskadbi S, Vaidya VG (March 1989). “In vivo antimutagenic effect of ascorbic acid against mutagenicity of the common antiamebic drug diiodohydroxyquinoline”. Mutat. Res. 222 (3): 219–22. doi:10.1016/0165-1218(89)90137-7. PMID 2493578.
- ^ Nagata, Noriyuki; Marriott, Deborah; Harkness, John; Ellis, John T.; Stark, Damien (2012). “Current treatment options for Dientamoeba fragilis infections”. International Journal for Parasitology: Drugs and Drug Resistance. 2: 204–215. doi:10.1016/j.ijpddr.2012.08.002. ISSN 2211-3207. PMC 3862407. PMID 24533282.
- ^ Publishing, William Andrew (2013-01-15). Pharmaceutical Manufacturing Encyclopedia (3rd ed.). Elsevier Science. p. 1312. ISBN 9780080947266.
- ^ Chan FT, Guan MX, Mackenzie AM, Diaz-Mitoma F (May 1994). “Susceptibility testing of Dientamoeba fragilis ATCC 30948 with iodoquinol, paromomycin, tetracycline, and metronidazole”. Antimicrob. Agents Chemother. 38 (5): 1157–60. doi:10.1128/aac.38.5.1157. PMC 188168. PMID 8067755.
- ^ Aggett, P.J.; Delves, H.T.; Harries, J.T.; Bangham, A.D. (March 1979). “The possible role of Diodoquin as a zinc ionophore in the treatment of acrodermatitis enteropathica”. Biochemical and Biophysical Research Communications. 87 (2): 513–517. doi:10.1016/0006-291X(79)91825-4. PMID 375935.
| Names | |
|---|---|
| Preferred IUPAC name5,7-Diiodoquinolin-8-ol | |
| Other namesDiquinol, iodoxin, diiodoquin, amebaquin | |
| Identifiers | |
| CAS Number | 83-73-8 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:5950 |
| ChEMBL | ChEMBL86754 |
| ChemSpider | 3597 |
| ECHA InfoCard | 100.001.362 |
| KEGG | D00581 |
| MeSH | Iodoquinol |
| PubChem CID | 3728 |
| UNII | 63W7IE88K8 |
| CompTox Dashboard (EPA) | DTXSID6023155 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C9H5I2NO |
| Molar mass | 396.951 |
| Pharmacology | |
| ATC code | G01AC01 (WHO) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
//////////////IODOQUINOL, Diiodohydroxyquinoline, NSC-8704, SS-578
OC1=C2N=CC=CC2=C(I)C=C1I

NEW DRUG APPROVALS
ONE TIME
$10.00
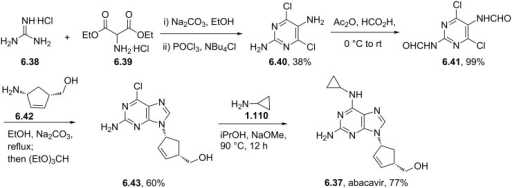
ABACAVIR
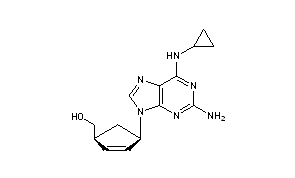
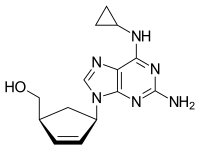
Abacavir
CAS Registry Number: 136470-78-5
CAS Name: (1S,4R)-4-[2-Amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
Additional Names: (-)-cis-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
Manufacturers’ Codes: 1592U89
Molecular Formula: C14H18N6O
Molecular Weight: 286.33
Percent Composition: C 58.73%, H 6.34%, N 29.35%, O 5.59%
Literature References: Nucleoside reverse transcriptase inhibitor (NRTI).
Prepn: S. M. Daluge, EP 349242 (1990 to Wellcome Found.); idem, US 5034394 (1991 to Burroughs Wellcome). Asymmetric synthesis: M. T. Crimmins, B. W. King, J. Org. Chem. 61,4192 (1996).
Pharmacology and biological profile: S. M. Daluge et al., Antimicrob. Agents Chemother. 41, 1082 (1997).
Review of antiviral activity and clinical evaluations: R. H. Foster, D. Faulds, Drugs 55, 729-736 (1998).
Clinical trial of triple nucleoside regimen in HIV patients: S. Staszewski et al., J. Am. Med. Assoc. 285, 1155 (2001).
Properties: White solid foam from acetonitrile, mp 165°. uv max (pH 1): 296, 255 nm (e 14000, 10700); uv max (pH 7): 284, 259 nm (e 15900, 9200); uv max (pH 13): 284, 259 nm (e 15800, 9100). [a]D20 -59.7°; [a]43620 -127.8°; [a]36520 -218.1° (c = 0.15 in methanol). Log P (1-octanol/0.1M sodium phosphate): 1.22 ±0.03 (pH 7.4). pKa 5.01. Soly in water (25°): >80 mM (pH 7).
Melting point: mp 165°
pKa: pKa 5.01
Optical Rotation: [a]D20 -59.7°; [a]43620 -127.8°; [a]36520 -218.1° (c = 0.15 in methanol)
Log P: Log P (1-octanol/0.1M sodium phosphate): 1.22 ±0.03 (pH 7.4)
Absorption maximum: uv max (pH 1): 296, 255 nm (e 14000, 10700); uv max (pH 7): 284, 259 nm (e 15900, 9200); uv max (pH 13): 284, 259 nm (e 15800, 9100)
Derivative Type: Sulfate
CAS Registry Number: 188062-50-2
Trademarks: Ziagen (GSK)
Molecular Formula: (C14H18N6O)2.H2SO4
Molecular Weight: 670.74
Percent Composition: C 50.14%, H 5.71%, N 25.06%, O 14.31%, S 4.78%
Therap-Cat: Antiviral.
Keywords: Reverse Transcriptase Inhibitor; Antiviral; Purines/Pyrimidinones.
Abacavir (ABC) is a medication used to prevent and treat HIV/AIDS.[1][2] Similar to other nucleoside analog reverse-transcriptase inhibitors (NRTIs), abacavir is used together with other HIV medications, and is not recommended by itself.[3] It is taken by mouth as a tablet or solution and may be used in children over the age of three months.[1][4]
Abacavir is generally well tolerated.[4] Common side effects include vomiting, trouble sleeping, fever, and feeling tired.[1] More severe side effects include hypersensitivity, liver damage, and lactic acidosis.[1] Genetic testing can indicate whether a person is at higher risk of developing hypersensitivity.[1] Symptoms of hypersensitivity include rash, vomiting, and shortness of breath.[4] Abacavir is in the NRTI class of medications, which work by blocking reverse transcriptase, an enzyme needed for HIV virus replication.[5] Within the NRTI class, abacavir is a carbocyclic nucleoside.[1]
Abacavir was patented in 1988 and approved for use in the United States in 1998.[6][7] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[8] It is available as a generic medication.[1] The wholesale cost in the developing world as of 2014 is between US$0.36 and US$0.83 per day.[9] As of 2016 the wholesale cost for a typical month of medication in the United States is US$70.50.[10] Commonly, abacavir is sold together with other HIV medications, such as abacavir/lamivudine/zidovudine, abacavir/dolutegravir/lamivudine, and abacavir/lamivudine.[4][5]
Medical uses
_300mg.jpg)
Two Abacavir 300mg tablets
Abacavir tablets and oral solution, in combination with other antiretroviral agents, are indicated for the treatment of HIV-1 infection.
Abacavir should always be used in combination with other antiretroviral agents. Abacavir should not be added as a single agent when antiretroviral regimens are changed due to loss of virologic response.
Side effects
Common adverse reactions include nausea, headache, fatigue, vomiting, diarrhea, loss of appetite and trouble sleeping. Rare but serious side effects include hypersensitivity reaction or rash, elevated AST and ALT, depression, anxiety, fever/chills, URI, lactic acidosis, hypertriglyceridemia, and lipodystrophy.[11]
People with liver disease should be cautious about using abacavir because it can aggravate the condition. Signs of liver problems include nausea and vomiting, abdominal pain, dark-colored urine and yellowing of the skin or whites of the eyes. The use of nucleosidedrugs such as abacavir can very rarely cause lactic acidosis. Signs of lactic acidosis include fast or irregular heartbeat, unusual muscle pain, fatigue, difficulty breathing and stomach pain with nausea and vomiting.[12] Abacavir can also lead to immune reconstitution inflammatory syndrome, a change in body fat as well as an increased risk of heart attack.
Resistance to abacavir has developed in laboratory versions of HIV which are also resistant to other HIV-specific antiretrovirals such as lamivudine, didanosine, and zalcitabine. HIV strains that are resistant to protease inhibitors are not likely to be resistant to abacavir.
Abacavir is contraindicated for use in infants under 3 months of age.
Little is known about the effects of Abacavir overdose. Overdose victims should be taken to a hospital emergency room for treatment.
Hypersensitivity syndrome
Hypersensitivity to abacavir is strongly associated with a specific allele at the human leukocyte antigen B locus namely HLA-B*5701.[13][14][15] There is an association between the prevalence of HLA-B*5701 and ancestry. The prevalence of the allele is estimated to be 3.4 to 5.8 percent on average in populations of European ancestry, 17.6 percent in Indian Americans, 3.0 percent in Hispanic Americans, and 1.2 percent in Chinese Americans.[16][17] There is significant variability in the prevalence of HLA-B*5701 among African populations. In African Americans, the prevalence is estimated to be 1.0 percent on average, 0 percent in the Yorubafrom Nigeria, 3.3 percent in the Luhya from Kenya, and 13.6 percent in the Masai from Kenya, although the average values are derived from highly variable frequencies within sample groups.[18]
Common symptoms of abacavir hypersensitivity syndrome include fever, malaise, nausea, and diarrhea. Some patients may also develop a skin rash.[19] Symptoms of AHS typically manifest within six weeks of treatment using abacavir, although they may be confused with symptoms of HIV, immune reconstitution syndrome, hypersensitivity syndromes associated with other drugs, or infection.[20] The U.S. Food and Drug Administration (FDA) released an alert concerning abacavir and abacavir-containing medications on July 24, 2008,[21] and the FDA-approved drug label for abacavir recommends pre-therapy screening for the HLA-B*5701 allele and the use of alternative therapy in subjects with this allele.[22] Additionally, both the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Group recommend use of an alternative therapy in individuals with the HLA-B*5701 allele.[23][24]
Skin-patch testing may also be used to determine whether an individual will experience a hypersensitivity reaction to abacavir, although some patients susceptible to developing AHS may not react to the patch test.[25]
The development of suspected hypersensitivity reactions to abacavir requires immediate and permanent discontinuation of abacavir therapy in all patients, including patients who do not possess the HLA-B*5701 allele. On March 1, 2011, the FDA informed the public about an ongoing safety review of abacavir and a possible increased risk of heart attack associated with the drug. A meta-analysis of 26 studies conducted by the FDA, however, did not find any association between abacavir use and heart attack [26][27]
Immunopathogenesis
The mechanism underlying abacavir hypersensitivity syndrome is related to the change in the HLA-B*5701 protein product. Abacavir binds with high specificity to the HLA-B*5701 protein, changing the shape and chemistry of the antigen-binding cleft. This results in a change in immunological tolerance and the subsequent activation of abacavir-specific cytotoxic T cells, which produce a systemic reaction known as abacavir hypersensitivity syndrome.[28]
Interaction
Abacavir, and in general NRTIs, do not undergo hepatic metabolism and therefore have very limited (to none) interaction with the CYP enzymes and drugs that effect these enzymes. That being said there are still few interactions that can affect the absorption or the availability of abacavir. Below are few of the common established drug and food interaction that can take place during abacavir co-administration:
- Protease inhibitors such as tipranavir or ritonovir may decrease the serum concentration of abacavir through induction of glucuronidation. Abacavir is metabolized by both alcohol dehydrogenase and glucuronidation.[29][30]
- Ethanol may result in increased levels of abacavir through the inhibition of alcohol dehydrogenase. Abacavir is metabolized by both alcohol dehydrogenase and glucuronidation.[29][31]
- Methadone may diminish the therapeutic effect of Abacavir. Abacavir may decrease the serum concentration of Methadone.[32][33]
- Orlistat may decrease the serum concentration of antiretroviral drugs. The mechanism of this interaction is not fully established but it is suspected that it is due to the decreased absorption of abacavir by orlistat.[34]
- Cabozantinib: Drugs from the MPR2 inhibitor (Multidrug resistance-associated protein 2 inhibitors) family such as abacavir could increase the serum concentration of Cabozantinib.[35]
Mechanism of action
Abacavir is a nucleoside reverse transcriptase inhibitor that inhibits viral replication. It is a guanosine analogue that is phosphorylated to carbovir triphosphate (CBV-TP). CBV-TP competes with the viral molecules and is incorporated into the viral DNA. Once CBV-TP is integrated into the viral DNA, transcription and HIV reverse transcriptase is inhibited.[36]
Pharmacokinetics
Abacavir is given orally and is rapidly absorbed with a high bioavailability of 83%. Solution and tablet have comparable concentrations and bioavailability. Abacavir can be taken with or without food.
Abacavir can cross the blood-brain barrier. Abacavir is metabolized primarily through the enzymes alcohol dehydrogenase and glucuronyl transferase to an inactive carboxylate and glucuronide metabolites. It has a half-life of approximately 1.5-2.0 hours. If a person has liver failure, abacavir’s half life is increased by 58%.
Abacavir is eliminated via excretion in the urine (83%) and feces (16%). It is unclear whether abacavir can be removed by hemodialysis or peritoneal dialysis.[36]
History
Robert Vince and Susan Daluge along with Mei Hua, a visiting scientist from China, developed the medication in the ’80s.[37][38][39]
Abacavir was approved by the Food and Drug Administration (FDA) on December 18, 1998, and is thus the fifteenth approved antiretroviral drug in the United States. Its patent expired in the United States on 2009-12-26.
Synthesis

Abacavir synthesis:[40]
References
From internet free resources


From internet free resources
From internet free resources



From internet free resources




ABOVE From internet free resources
SYN
- Crimmins, M. T.; King, B. W. (1996). “An Efficient Asymmetric Approach to Carbocyclic Nucleosides: Asymmetric Synthesis of 1592U89, a Potent Inhibitor of HIV Reverse Transcriptase”. The Journal of Organic Chemistry. 61 (13): 4192–4193. doi:10.1021/jo960708p. PMID 11667311.

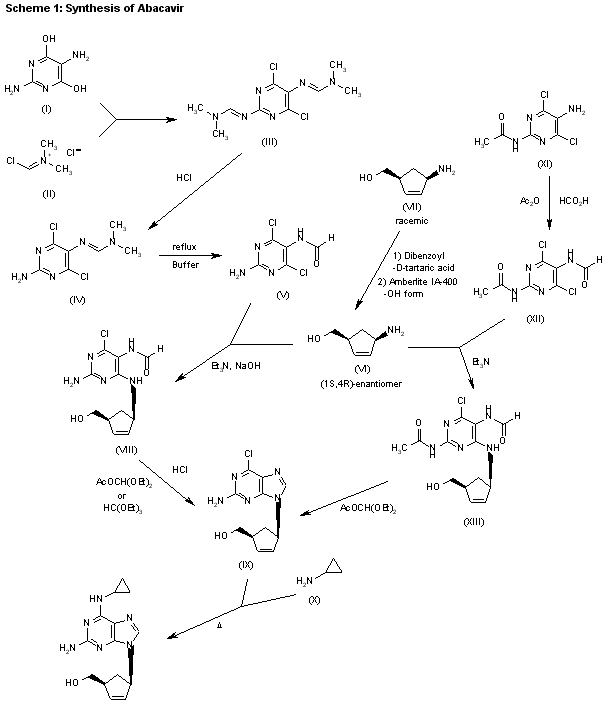
The reaction of 4,6-dihydroxypyrimidine-2,5-diamine (I) with (chloromethylene)dimethylammonium chloride (II) in refluxing chloroform gives 4,6-dichloro-2,5-bis(dimethylaminomethyleneamino)pyrimidine (III), which by reaction with aqueous HCl in hot ethanol yields monoamine (IV). The reaction of (IV) with a refluxing phosphate buffer (pH 3.2) affords N-(2-amino-4,6-dichloropyrimidin-5-yl)formamide (V). The condensation of (V) with (1S,4R)-4-amino-2-cyclopentene-1-methanol (VI) (which was obtained by optical resolution of the cis-racemate (VII) with D-dibenzoyltartaric acid, and elimination of the acid with ion exchange resin Amberlite IA-400, by means of triethylamine and NaOH in refluxing ethanol) gives N-[2-amino-4-chloro-6-[4(S)-(hydroxymethyl)-2-cyclopenten-1(R)-ylamino]pyrimidin-5-yl]formamide (VIII). The cyclization of (VIII) with refluxing diethoxymethyl acetate or triethyl orthoformate yields the corresponding purine derivative (IX), which is finally treated with cyclopropylamine (X) in refluxing n-butanol. 2) The formylation of N-(5-amino-4,6-dichloropyrimidin-2-yl)acetamide (XI) with 95% formic acid in acetic anhydride gives the expected formamide (XII), which is condensed with (1S,4R)-4-amino-2-cyclopentene-1-methanol (VI) by means of triethylamine in hot ethanol to yield the substituted pyrimidine (XIII). Finally, the cyclization of (XIII) with diethoxymethyl acetate as before affords the purine intermediate (IX).
| AU 8937025; EP 0349242; JP 1990045486; JP 1999139976; US 5034394; US 5089500
|
SYN 2
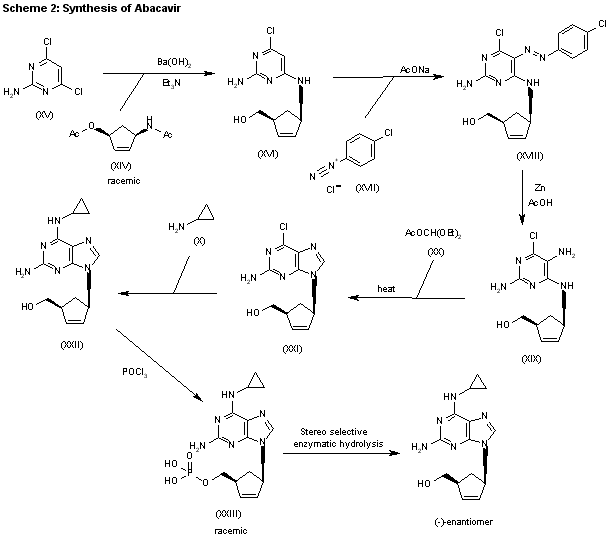
The condensation of (?-cis-4-acetamido-2-cyclopentenylmethyl acetate (XIV) with 2-amino-4,6-dichloropyrimidine (XV) by means of Ba(OH)2 and triethylamine in refluxing butanol gives the expected condensation product (XVI), which is treated with 4-chlorophenyldiazonium chloride (XVII) in water/acetic acid to yield the corresponding azo-compound (XVIII). The reduction of (XVIII) with Zn/acetic acid in ethanol affords the diamine (XIX), which is cyclized with refluxing diethoxymethyl acetate (XX) to afford the corresponding purine (XXI). The reaction of (XXI) with cyclopropylamine (X) in refluxing ethanol affords racemic abacavir (XXII), which is phosphorylated with POCl3 giving the racemic 4′-O-phosphate (XXIII). Finally, this compound is submitted to stereoselective enzymatic dephosphorylation using snake venom 5′-nucleotidase (EC 3.1.3.5) from Crotalus atrox yielding the (-)-enantiomer, abacavir.
SYN 3
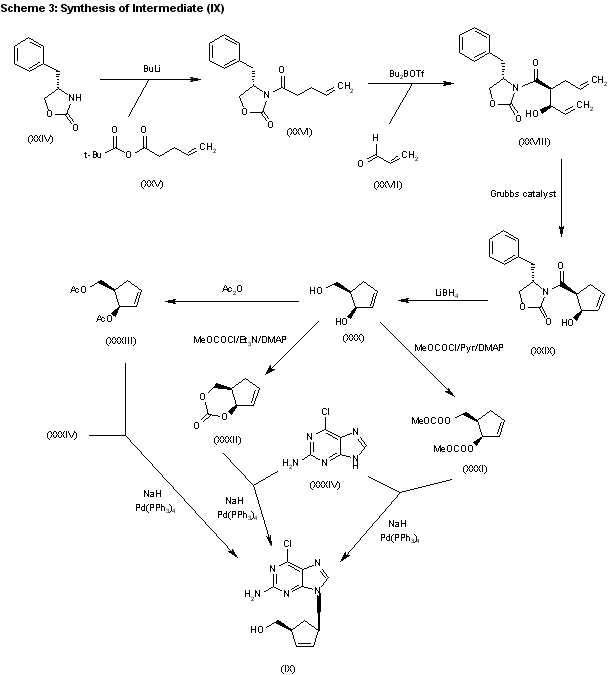
The acylation of 4(S)-benzyloxazolidin-2-one (XXIV) with 4-pentenoyl pivaloyl anhydride (XXV) by means of NaH in THF gives 4(S)-benzyl-3-(4-pentenoyl)oxazolidin-2-one (XXVI), which is submitted to a diastereoselective syn aldol condensation with acrolein (XXVII), using dibutylboron triflate as catalyst, affording the aldol (XXVIII). The cyclization of (XXVIII) by means of the Grubbs catalyst in dichloromethane yields the cyclopentenol (XXIX), which is reduced with LiBH4 in THF/methanol to give the key intermediate 5(R)-(hydroxymethyl)-2-cyclopenten-1(R)-ol (XXX). The reaction of (XXX) with methyl chloroformate/pyridine/DMAP or methyl chloroformate/triethylamine/DMAP or acetic anhydride gives the diols (XXXI), (XXXII) and (XXXIII), respectively, each of which coupled with 2-amino-6-chloropurine (XXXIV) in the presence of NaH and palladium tetrakis(triphenylphosphine) in THF/DMSO, affords the purine intermediate (IX) already reported.
SYN
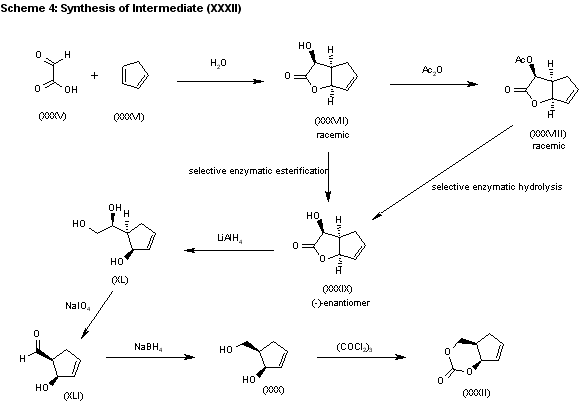
The water promoted condensation of glyoxylic acid (XXXV) with cyclopentadiene (XXXVI) gives the racemic cis-hydroxylactone (XXXVII), which is acetylated with acetic anhydride to the acetate (XXXVIII). The selective enzymatic hydrolysis of (XXXVIII) with Pseudomonas fluorescens lipase yields the pre (-)-enantiomer (XXXIX), which is reduced with LiAlH4 in refluxing THF, affording triol (XL). The oxidation of the vicinal glycol of (XL) with NaIO4 in ethyl ether/water yields the hydroxyaldehyde (XLI), which is reduced with NaBH4 in ethanol to give the key intermediate 5(R)-(hydroxymethyl)-2-cyclopenten-1(R)-ol (XXX). This compound, by reaction with triphosgene and triethylamine in dichloromethane, results in the cyclic carbonate intermediate (XXXII), already reported.
SYN
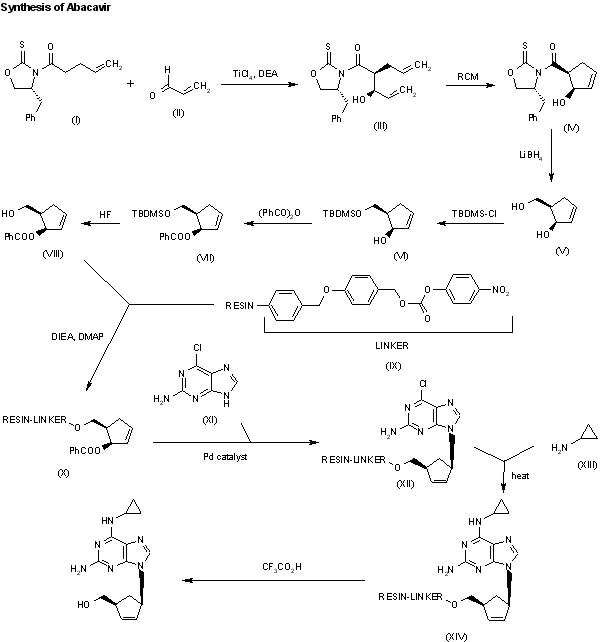
A new solid phase synthesis of abacavir has been reported: Condensation of the chiral 4(R)-benzyl-3-(4-pentenoyl)oxazolidin-2-thione (I) with acrolein (II) by means of TiCl4 and DIEA gives the adduct (III), which was transformed into the chiral cyclopentene (IV) by catalytic ring-closing metathesis. The reductive removal of the chiral auxiliary with LiBH4 affords the chiral diol (V), which is selectively silylated with TBDMSCl providing the primary silyl ether (VI). Acylation of the secondary alcohol of (VI) with benzoic anhydride gives the benzoate (VII), which is desilylated with HF in acetonitrile yielding the allylic benzoate (VIII). Benzoate (VIII) is condensed with a p-nitrophenyl Wang carbonate resin (IX) by means of DIEA and DMAP affording the solid phase resin (X) which is condensed with 2-amino-6-chloropurine (XI) by means of a Pd catalyst furnishing the adduct (XII). Thermal condensation of (XII) with cyclopropylamine (XIII) yields the diaminopurine resin (XIV) which, after cleavage from the resin by a treatment with TFA in dichloromethane, gives directly abacavir.
SYN
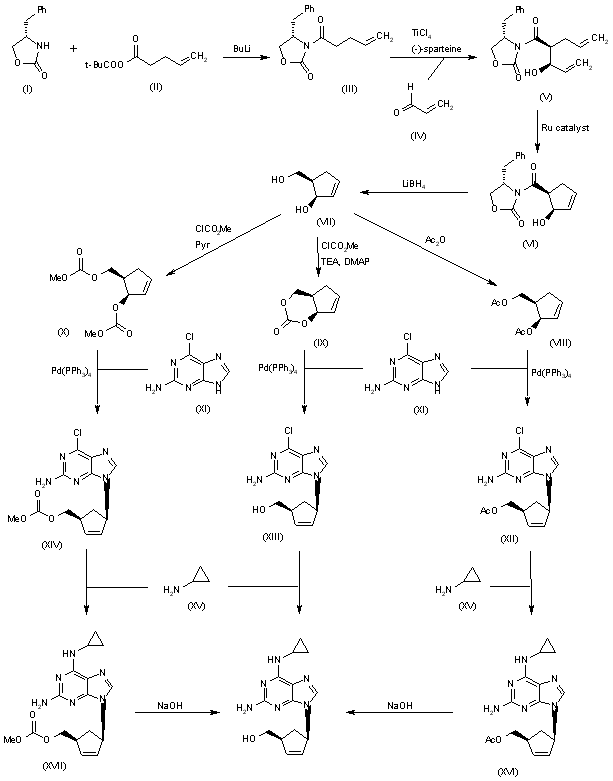
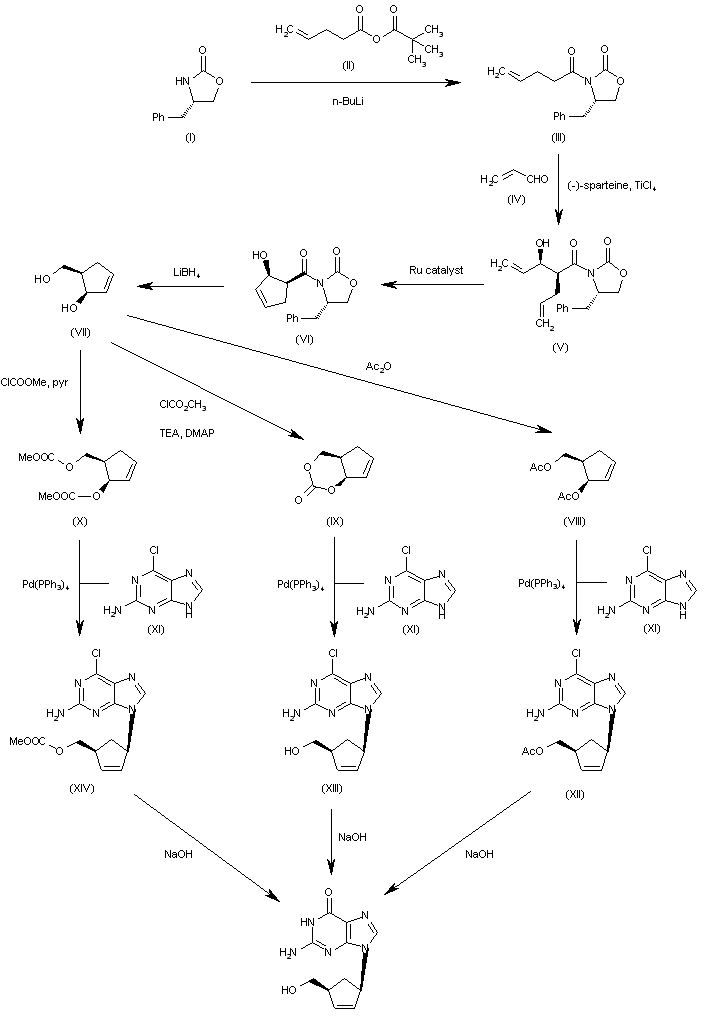 The condensation of the chiral oxazolidinone (I) with the pentenoic anhydride (II) by means of n-BuLi in THF gives the N-pentenoyloxazolidinone (III), which is condensed with acrolein (IV) catalyzed by TiCl4 and (-)-spartein in dichloromethane, yielding the chiral adduct (V). The ring-closing metathesis of (V) by means of a Ru catalyst in dichloromethane affords the chiral cyclopentenol derivative (VI), which is reduced to the (R,R)-5-(hydroxymethyl)-2-cyclopenten-1-ol (VII) by means of LiBH4 in THF. The reaction of diol (VII) with Ac2O; with methyl chloroformate, TEA and DMAP; or with ethyl chloroformate and pyridine gives the diacetate (VIII), the cyclic carbonate (IX) or the dicarbonate (X), respectively. The condensation of (VIII), (IX) or (X) with 2-amino-6-chloropurine (XI) by means of Pd(PPh3)4 yields the carbocyclic purines (XII), (XIII) or (XIV), respectively. Finally, these compounds are hydrolyzed with aqueous NaOH to the target carbocyclic guanine.
The condensation of the chiral oxazolidinone (I) with the pentenoic anhydride (II) by means of n-BuLi in THF gives the N-pentenoyloxazolidinone (III), which is condensed with acrolein (IV) catalyzed by TiCl4 and (-)-spartein in dichloromethane, yielding the chiral adduct (V). The ring-closing metathesis of (V) by means of a Ru catalyst in dichloromethane affords the chiral cyclopentenol derivative (VI), which is reduced to the (R,R)-5-(hydroxymethyl)-2-cyclopenten-1-ol (VII) by means of LiBH4 in THF. The reaction of diol (VII) with Ac2O; with methyl chloroformate, TEA and DMAP; or with ethyl chloroformate and pyridine gives the diacetate (VIII), the cyclic carbonate (IX) or the dicarbonate (X), respectively. The condensation of (VIII), (IX) or (X) with 2-amino-6-chloropurine (XI) by means of Pd(PPh3)4 yields the carbocyclic purines (XII), (XIII) or (XIV), respectively. Finally, these compounds are hydrolyzed with aqueous NaOH to the target carbocyclic guanine.
SYN
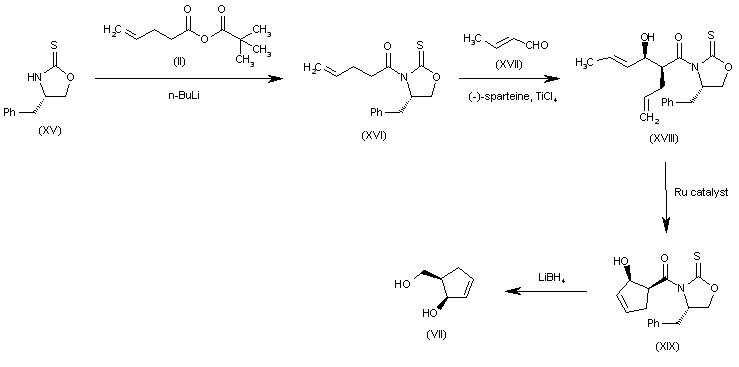
Alternatively, the (R,R)-5-(hydroxymethyl)-2-cyclopenten-1-ol (VII) can also be obtained as follows: The condensation of the chiral oxazolidinethione (XV) with the pentenoic anhydride (II) by means of n-BuLi in THF gives the N-pentenoyloxazolidinethione (XVI), which is condensed with crotonaldehyde (XVII) catalyzed by TiCl4 and (-)-spartein in dichloromethane, yielding the chiral adduct (XVIII). The ring-closing metathesis of (XVIII) by means of a Ru catalyst in dichloromethane affords the chiral cyclopentenol derivative (XIX), which is reduced to the target diol (VII) by means of LiBH4 in THF.
SYN
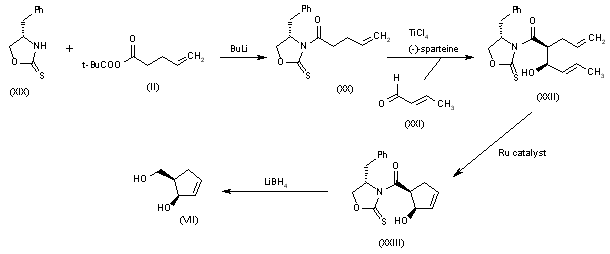
An efficient asymmetric synthesis of abacavir has been reported: Acylation of the chiral oxazolidinone (I) with the mixed anhydride (II) by means of BuLi in THF gives the N-pentenoyloxazolidinone (III), which by condensation with acrolein (IV) catalyzed by TiCl4 and (?-spartein in dichloromethane yields the chiral adduct (V). The ring-closing metathesis of adduct (V) by means of the ruthenium catalyst (Cy3P)Cl2Ru=CHPh in dichloromethane affords the chiral cyclopentenol (VI), which is reduced to 5(R)-(hydroxymethyl)-2-cyclopenten-1(R)-ol (VII) by means of LiBH4 in THF. Reaction of diol (VII) with a) Ac2O, TEA and DMAP, b) methyl chloroformate, TEA and DMAP or c) methyl chloroformate, pyridine and DMAP gives a) the diacetate (VIII), b) the cyclic carbonate (IX) or c) the dicarbonate (X), respectively. The condensation of diacetate (VIII), cyclic carbonate (IX) or dicarbonate (X) with 2-amino-6-chloropurine (XI) by means of Pd(PPh3)4 yields the carbocyclic purines (XII), (XIII) or (XIV), respectively. Treatment of these chloro-purines (XII), (XIII) and (XIV) with cyclopropylamine (XV) in hot DMSO provides the corresponding cyclopropylaminopurine carbonate (XVI), abacavir or cyclopropylaminopurine acetate (XVII), respectively. Finally, the protecting groups of purines (XVI) and (XVII) are hydrolyzed with aqueous NaOH.
SYN
Alternatively, 5(R)-(hydroxymethyl)-2-cyclopenten-1(R)-ol (VII) can also be obtained as follows: Acylation of the chiral oxazolidinethione (XIX) with the mixed anhydride (II) by means of BuLi in THF gives the N-pentenoyl-oxazolidinethione (XX), which by condensation with crotonaldehyde (XXI) catalyzed by TiCl4 and (?-spartein in dichloromethane yields the chiral adduct (XXII). The ring-closing metathesis of (XXII) by means of the ruthenium catalyst in dichloromethane affords the chiral cyclopentenol derivative (XXIII), which is reduced to the target diol (VII) by means of LiBH4 in THF.
SYN
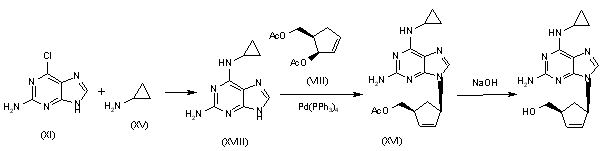
Alternatively, 2-amino-6-chloropurine (XI) is treated with cyclopropylamine (XV) in hot DMSO to give 2-amino-6-(cyclopropylamino)purine (XVIII), which is condensed with the chiral diacetate (VIII) by means of Pd(PPh3)4 to yield the carbocyclic purine acetate (XVI). Finally, purine (XVI) is deprotected by hydrolysis with aqueous NaOH.
CLIP

https://www.sciencedirect.com/science/article/pii/S0960894X15007581
CLIP
CLIP
CLIP
Production of Abacavir
030-8 1.0g (0.0053mol), in the reaction flask was added cesium carbonate 1.75 g (0.0054 mol) and dry DMSO 50ml, stirred under N2 protection, the temperature was raised to 60 °C and stirred at this temperature for 2 h the mixture wascooled to room temperature, then add tetrakis (triphenylphosphine) combined palladium (TTP) [0.85 (0.00074mol)] and compound 030-5 [0.79g (0.0034 mol), DMSO (10 ml) solution was stirred and heated to 65 °C held 65 °C and stirred reaction 2.25h. The you can get the mixture containing compounds 030-9.
To the mixture was added methanol 100ml and K2CO3 is 2.10g, the mixture reaction was stirred for 45min at 40 °C, a solid precipitate which was filtered through a Celite layer and the filtrate was evaporated to a small volume under vacuum at 90 °C, and the remaining gum pounding mill was extracted with dichloromethane (100ml * 2) to give a brown solid residue was purified by silica gel (Merck 9385) column chromatography [eluent: dichloromethane / methanol (volume ratio 9:1)] to give a yellow foam was 030 0.26 g, yield 26.8.

CLIP
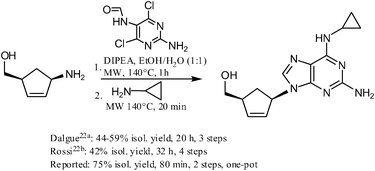
https://pubs.rsc.org/en/content/articlehtml/2012/ra/c2ra20842c
References
- ^ Jump up to:a b c d e f g h “Abacavir Sulfate”. The American Society of Health-System Pharmacists. Archived from the original on 8 September 2017. Retrieved 31 July 2015.
- ^ “Drug Name Abbreviations Adult and Adolescent ARV Guidelines”. AIDSinfo. Archived from the original on 2016-11-09. Retrieved 2016-11-08.
- ^ “What Not to Use Adult and Adolescent ARV Guidelines”. AIDSinfo. Archived from the original on 2016-11-09. Retrieved 2016-11-08.
- ^ Jump up to:a b c d Yuen, GJ; Weller, S; Pakes, GE (2008). “A review of the pharmacokinetics of abacavir”. Clinical Pharmacokinetics. 47 (6): 351–71. doi:10.2165/00003088-200847060-00001. PMID 18479171.
- ^ Jump up to:a b “Nucleoside reverse transcriptase inhibitors (NRTIs or ‘nukes’) – HIV/AIDS”. http://www.hiv.va.gov. Archived from the original on 2016-11-09. Retrieved 2016-11-08.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 505. ISBN 9783527607495. Archived from the original on 2017-09-08.
- ^ Kane, Brigid M. (2008). HIV/AIDS Treatment Drugs. Infobase Publishing. p. 56. ISBN 9781438102078. Archived from the original on 2017-09-08.
- ^ “WHO Model List of Essential Medicines (19th List)” (PDF). World Health Organization. April 2015. Archived (PDF) from the original on 13 December 2016. Retrieved 8 December 2016.
- ^ “International Drug Price Indicator Guide”. ERC. Retrieved 20 November 2016.
- ^ “NADAC as of 2016-12-07 | Data.Medicaid.gov”. Centers for Medicare and Medicaid Services. Archived from the original on 21 December 2016. Retrieved 12 December 2016.
- ^ “Abacavir Adverse Reactions”. Epocrates Online.
- ^ “Abacavir | Dosage, Side Effects | AIDSinfo”. AIDSinfo. Archived from the original on 2017-03-06. Retrieved 2016-11-08.
- ^ Mallal, S., Phillips, E., Carosi, G.; et al. (2008). “HLA-B*5701 screening for hypersensitivity to abacavir”. New England Journal of Medicine. 358 (6): 568–579. doi:10.1056/nejmoa0706135. PMID 18256392.
- ^ Rauch, A., Nolan, D., Martin, A.; et al. (2006). “Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study”. Clinical Infectious Diseases. 43 (1): 99–102. doi:10.1086/504874. PMID 16758424.
- ^ Dean, Laura (2012), Pratt, Victoria; McLeod, Howard; Rubinstein, Wendy; Dean, Laura (eds.), “Abacavir Therapy and HLA-B*57:01 Genotype”, Medical Genetics Summaries, National Center for Biotechnology Information (US), PMID 28520363, retrieved 2019-01-14
- ^ Heatherington; et al. (2002). “Genetic variations in HLA-B region and hypersensitivity reactions to abacavir”. Lancet. 359 (9312): 1121–1122. doi:10.1016/s0140-6736(02)08158-8. PMID 11943262.
- ^ Mallal; et al. (2002). “Association between presence of HLA*B5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir”. Lancet. 359(9308): 727–732. doi:10.1016/s0140-6736(02)07873-x. PMID 11888582.
- ^ Rotimi, C. N.; Jorde, L. B. (2010). “Ancestry and disease in the age of genomic medicine”. New England Journal of Medicine. 363 (16): 1551–1558. doi:10.1056/nejmra0911564. PMID 20942671.
- ^ Phillips, E., Mallal, S. (2009). “Successful translation of pharmacogenetics into the clinic”. Molecular Diagnosis & Therapy. 13: 1–9. doi:10.1007/bf03256308.
- ^ Phillips, E., Mallal S. (2007). “Drug hypersensitivity in HIV”. Current Opinion in Allergy and Clinical Immunology. 7 (4): 324–330. doi:10.1097/aci.0b013e32825ea68a. PMID 17620824.
- ^ “Postmarket Drug Safety Information for Patients and Providers – Information for Healthcare Professionals: Abacavir (marketed as Ziagen) and Abacavir-Containing Medications”. Center for Drug Evaluation and Research at the US FDA. Archived from the original on 2013-12-11. Retrieved 2013-11-29.
- ^ “Archived copy”. Archived from the original on 2014-08-08. Retrieved 2014-07-31.
- ^ Swen JJ, Nijenhuis M, de Boer A, et al. (May 2011). “Pharmacogenetics: from bench to byte–an update of guidelines”. Clin Pharmacol Ther. 89 (5): 662–73. doi:10.1038/clpt.2011.34. PMID 21412232.
- ^ Martin MA, Hoffman JM, Freimuth RR, et al. (May 2014). “Clinical Pharmacogenetics Implementation Consortium Guidelines for HLA-B Genotype and Abacavir Dosing: 2014 update”. Clin Pharmacol Ther. 95 (5): 499–500. doi:10.1038/clpt.2014.38. PMC 3994233. PMID 24561393.
- ^ Shear, N.H., Milpied, B., Bruynzeel, D. P.; et al. (2008). “A review of drug patch testing and implications for HIV clinicians”. AIDS. 22 (9): 999–1007. doi:10.1097/qad.0b013e3282f7cb60. PMID 18520343.
- ^ “FDA Alert: Abacavir – Ongoing Safety Review: Possible Increased Risk of Heart Attack”. Drugs.com. Archived from the original on 2013-12-10. Retrieved 2013-11-29.
- ^ Ding X, Andraca-Carrera E, Cooper C, et al. (December 2012). “No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis”. J Acquir Immune Defic Syndr. 61 (4): 441–7. doi:10.1097/QAI.0b013e31826f993c. PMID 22932321.
- ^ Illing, PT; et al. (2012). “Immune self-reactivity triggered by drug-modified HLA-peptide repertoire”. Nature. 486 (7404): 554–8. doi:10.1038/nature11147. PMID 22722860.
- ^ Jump up to:a b Prescribing information. Ziagen (abacavir). Research Triangle Park, NC: GlaxoSmithKline, July 2002
- ^ Vourvahis, M; Kashuba, AD (2007). “Mechanisms of Pharmacokinetic and Pharmacodynamic Drug Interactions Associated with Ritonavir-Enhanced Tipranavir”. Pharmacotherapy. 27 (6): 888–909. doi:10.1592/phco.27.6.888. PMID 17542771.
- ^ McDowell, JA; Chittick, GE; Stevens, CP; et al. (2000). ““, “Pharmacokinetic Interaction of Abacavir (1592U89) and Ethanol in Human Immunodeficiency Virus-Infected Adults”. Antimicrob Agents Chemother. 44 (6): 1686–90. doi:10.1128/aac.44.6.1686-1690.2000. PMC 89933. PMID 10817729.
- ^ Berenguer, J; Perez-Elias, MJ; Bellon, JM; et al. (2006). “Effectiveness and safety of abacavir, lamivudine, and zidovudine in antiretroviral therapy-naive HIV-infected patients: results from a large multicenter observational cohort”. J Acquir Immune Defic Syndr. 41 (2): 154–159. doi:10.1097/01.qai.0000194231.08207.8a. PMID 16394846.
- ^ Dolophine(methadone) [prescribing information]. Columbus, OH: Roxane Laboratories, Inc.; March 2015.
- ^ Gervasoni, C; Cattaneo, D; Di Cristo, V; et al. (2016). “Orlistat: weight lost at cost of HIV rebound”. J Antimicrob Chemother. 71 (6): 1739–1741. doi:10.1093/jac/dkw033. PMID 26945709.
- ^ Cometriq (cabozantinib) [prescribing information]. South San Francisco, CA: Exelixis, Inc.; May 2016.
- ^ Jump up to:a b Product Information: ZIAGEN(R) oral tablets, oral solution, abacavir sulfate oral tablets, oral solution. ViiV Healthcare (per Manufacturer), Research Triangle Park, NC, 2015.
- ^ “Dr. Robert Vince – 2010 Inductee”. Minnesota Inventors Hall of Fame. Minnesota Inventors Hall of Fame. Archived from the original on 15 February 2016. Retrieved 10 February 2016.
- ^ “Robert Vince, PhD (faculty listing)”. University of Minnesota. University of Minnesota. Archived from the original on 2016-02-17.
- ^ Daluge SM, Good SS, Faletto MB, Miller WH, St Clair MH, Boone LR, Tisdale M, Parry NR, Reardon JE, Dornsife RE, Averett DR (May 1997). “1592U89, a novel carbocyclic nucleoside analog with potent, selective anti-human immunodeficiency virus activity”. Antimicrobial Agents and Chemotherapy. 41 (5): 1082–1093. doi:10.1128/AAC.41.5.1082. PMC 163855. PMID 9145874.
- ^ Crimmins, M. T.; King, B. W. (1996). “An Efficient Asymmetric Approach to Carbocyclic Nucleosides: Asymmetric Synthesis of 1592U89, a Potent Inhibitor of HIV Reverse Transcriptase”. The Journal of Organic Chemistry. 61 (13): 4192–4193. doi:10.1021/jo960708p. PMID 11667311.
External links
- Full Prescribing Information
- Abacavir pathway on PharmGKB
- Abacavir dosing guidelines from the Clinical Pharmacogenetics Implementation Consortium (CPIC)
- Abacavir dosing guidelines from the Dutch Pharmacogenetics Working Group (DPWG)
References
-
- Crimmins, M.T. et al.: J. Org. Chem. (JOCEAH) 61 4192 (1996).
- b Olivo, H.F. et al.: J. Chem. Soc., Perkin Trans. 1 (JCPRB4) 1998, 391.
- US 5 089 500 (Burroughs Wellcome; 18.2.1992; GB-prior. 27.6.1988).
- a EP 434 450 (Wellcome Found.; 26.6.1991; appl. 21.12.1990; USA-prior. 22.12.1989).
- EP 1 857 458 (Solmag; appl. 5.5.2006).
- aa EP 424 064 (Enzymatix; appl. 24.4.1991; GB-prior. 16.10.1989).
- US 6 340 587 (SmithKline Beecham; 22.1.2002; appl. 20.8.1998; GB-prior. 22.8.1997).
- c US 5 034 394 (Welcome Found.; 23.7.1991; appl. 22.12.1989; GB-prior. 27.6.1988).
- d WO 9 924 431 (Glaxo; appl. 12.11.1998; WO-prior. 12.11.1997).
-
Alternative syntheses:
- EP 878 548 (Lonza; appl. 13.5.1998; CH-prior. 13.5.1997).
-
Preparation of chloropyrimidine intermediate V:
- US 6 448 403 (SmithKline Beecham; 10.9.2002; appl. 3.2.1995; GB-prior. 4.2.1994).
-
Condensation of pyrimidines with cyclopentylamine IV:
- Vince, R.; Hua, M.: J. Med. Chem. (JMCMAR) 33 (1), 17 (1990).
- Grumam, A. et al.: Tetrahedron Lett. (TELEAY) 36 (42), 7767 (1995).
- EP 349 242 (Wellcome Found.; appl. 26.6.1989; GB-prior. 27.6.1988).
- EP 366 385 (Wellcome Found.; appl. 23.10.1989; GB-prior. 24.10.1988).
- US 6 646 125 (SmithKline Beecham; 11.11.2003; appl. 14.10.1998; GB-prior. 14.10.1997).
- JP 1 022 853 (Asahi Glass Co.; appl. 17.7.1987).
-
Alternative preparation of 4-amino-2-cyclopentene-1-methanol:
- EP 926 131 (Lonza; appl. 24.11.1998; CH-prior. 27.11.1997).
- WO 9 745 529 (Lonza; appl. 30.5.1997; CH-prior. 30.5.1996).
- WO 9 910 519 (Glaxo; 4.3.1999; GB-prior. 20.8.1998).
- WO 9 824 741 (Glaxo; 11.6.1998; GB-prior. 7.12.1996).
- WO 2 001 017 952 (Chirotech; 15.3.2001; GB-prior. 9.9.1999).
-
Abacavir hemisulfate salt:
- US 6 294 540 (Glaxo Wellcome; 25.9.2001; appl. 14.5.1998; GB-prior. 17.5.1997).
-
Abacavir succinate as antiviral agent:
- WO 9 606 844 (Wellcome; 7.3.1996; appl. 25.8.1995; GB-prior. 26.8.1994).
-
Pharmaceutical formulations:
- US 6 641 843 (SmithKline Beecham; 4.11.2003; appl. 4.2.1999; GB-prior. 6.2.1998).
-
Synergistic combinations for treatment of HIV infection:
- WO 9 630 025 (Wellcome; 3.10.1996; appl. 28.3.1996; GB-prior. 30.3.1995).
 |
|

Chemical structure of abacavir
|
|
| Clinical data | |
|---|---|
| Pronunciation | /əˈbækəvɪər/ ( |
| Trade names | Ziagen, others[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a699012 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
By mouth (solution or tablets) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 83% |
| Metabolism | Liver |
| Elimination half-life | 1.54 ± 0.63 h |
| Excretion | Kidney (1.2% abacavir, 30% 5′-carboxylic acid metabolite, 36% 5′-glucuronide metabolite, 15% unidentified minor metabolites). Fecal (16%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| NIAID ChemDB | |
| ECHA InfoCard | 100.149.341 |
| Chemical and physical data | |
| Formula | C14H18N6O |
| Molar mass | 286.332 g/mol g·mol−1 |
| 3D model (JSmol) | |
| Melting point | 165 °C (329 °F) |
/////////
OLD POST
 |
|

Chemical structure of abacavir
|
{(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopent-2-en-1-yl}methanol
(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
(1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol
Abacavir
Abacavir (ABC) is a powerful nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. [Wikipedia] Chemically, it is a synthetic carbocyclic nucleoside and is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. In vivo, abacavir sulfate dissociates to its free base, abacavir.
Abacavir (ABC) ![]() i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
i/ʌ.bæk.ʌ.vɪər/ is a nucleoside analog reverse transcriptase inhibitor (NRTI) used to treat HIV and AIDS. It is available under the trade name Ziagen (ViiV Healthcare) and in the combination formulations Trizivir (abacavir, zidovudine andlamivudine) and Kivexa/Epzicom (abacavir and lamivudine). It has been well tolerated: the main side effect is hypersensitivity, which can be severe, and in rare cases, fatal. Genetic testing can indicate whether an individual will be hypersensitive; over 90% of patients can safely take abacavir. However, in a separate study, the risk of heart attack increased by nearly 90%.[1]
Viral strains that are resistant to zidovudine (AZT) or lamivudine (3TC) are generally sensitive to abacavir (ABC), whereas some strains that are resistant to AZT and 3TC are not as sensitive to abacavir.
It is on the World Health Organization’s List of Essential Medicines, a list of the most important medication needed in a basic health system.[2]
Abacavir is a nucleoside reverse transcriptase inhibitor (NRTI) with activity against Human Immunodeficiency Virus Type 1 (HIV-1). Abacavir is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis. The concentration of drug necessary to effect viral replication by 50 percent (EC50) ranged from 3.7 to 5.8 μM (1 μM = 0.28 mcg/mL) and 0.07 to 1.0 μM against HIV-1IIIB and HIV-1BaL, respectively, and was 0.26 ± 0.18 μM against 8 clinical isolates. Abacavir had synergistic activity in cell culture in combination with the nucleoside reverse transcriptase inhibitor (NRTI) zidovudine, the non-nucleoside reverse transcriptase inhibitor (NNRTI) nevirapine, and the protease inhibitor (PI) amprenavir; and additive activity in combination with the NRTIs didanosine, emtricitabine, lamivudine, stavudine, tenofovir, and zalcitabine.
Brief background information
| Salt | ATC | Formula | MM | CAS |
|---|---|---|---|---|
| – | J05AF06 | C 14 H 18 N 6 O | 286.34 g / mol | 136470-78-5 |
| succinate | J05AF06 | C 14 H 18 N 6 O · C 4 H 6 O | 356.43 g / mol | 168146-84-7 |
| sulfate | J05AF06 | C 14 H 18 N 6 O · 1 / 2H 2 SO 4 | 670.76 g / mol | 188062-50-2 |
| Systematic (IUPAC) name | |
|---|---|
| {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]cyclopent-2-en-1-yl}methanol | |
| Clinical data | |
| Trade names | Ziagen |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a699012 |
| Pregnancy cat. | B3 (AU) C (US) |
| Legal status | POM (UK) ℞-only (US) |
| Routes | Oral (solution or tablets) |
| Pharmacokinetic data | |
| Bioavailability | 83% |
| Metabolism | Hepatic |
| Half-life | 1.54 ± 0.63 h |
| Excretion | Renal (1.2% abacavir, 30% 5′-carboxylic acid metabolite, 36% 5′-glucuronide metabolite, 15% unidentified minor metabolites). Fecal (16%) |
| Identifiers | |
| CAS number | 136470-78-5 |
| ATC code | J05AF06 |
| PubChem | CID 441300 |
| DrugBank | DB01048 |
| ChemSpider | 390063 |
| UNII | WR2TIP26VS |
| KEGG | D07057 |
| ChEBI | CHEBI:421707 |
| ChEMBL | CHEMBL1380 |
| NIAID ChemDB | 028596 |
| Chemical data | |
| Formula | C14H18N6O |
| Mol. mass | 286.332 g/mol |
Abacavir is a carbocyclic synthetic nucleoside analogue and an antiviral agent. Intracellularly, abacavir is converted by cellular enzymes to the active metabolite carbovir triphosphate, an analogue of deoxyguanosine-5′-triphosphate (dGTP). Carbovir triphosphate inhibits the activity of HIV-1 reverse transcriptase (RT) both by competing with the natural substrate dGTP and by its incorporation into viral DNA. Viral DNA growth is terminated because the incorporated nucleotide lacks a 3′-OH group, which is needed to form the 5′ to 3′ phosphodiester linkage essential for DNA chain elongation.
Application
-
an antiviral agent, is used in the treatment of AIDS
-
ingibitor convertibility transkriptazы
Classes of substances
-
Adenine (6-aminopurines)
-
Aminoalcohols
-
Cyclopentenes and cyclopentadienes
-
Tsyklopropanы
-
-
-
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2289753 | 2007-01-23 | 2018-05-14 |
| Canada | 1340589 | 1999-06-08 | 2016-06-08 |
| Canada | 2216634 | 2004-07-20 | 2016-03-28 |
| United States | 6641843 | 2000-02-04 | 2020-02-04 |
| United States | 5089500 | 1992-12-26 | 2009-12-26 |
PATENT
US5034394
Synthesis pathway
Abacavir, (-) cis-[4-[2-amino-6-cyclopropylamino)-9H-purin-9-yl]-2-cyclopenten-yl]-1 – methanol, a carbocyclic nucleoside which possesses a 2,3-dehydrocyclopentene ring, is referred to in United States Patent 5,034,394 as a reverse transcriptase inhibitor. Recently, a general synthetic strategy for the preparation of this type of compound and intermediates was reported [Crimmins, et. al., J. Org. Chem., 61 , 4192-4193 (1996) and 65, 8499-8509-4193 (2000)].
-
-
Abacavir is the International Nonproprietary Name (INN) of {(1S,4R)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol and CAS No. 136470-78-5. Abacavir and therapeutically acceptable salts thereof, in particular the hemisulfate salt, are well-known as potent selective inhibitors of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
-
The structure of abacavir corresponds to formula (I):
-

-
EP 434450-A discloses certain 9-substituted-2-aminopurines including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
-
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring.
-
According to the teachings of EP 434450-A , the abacavir base is finally isolated by trituration using acetonitrile (ACN) or by chromatography, and subsequently it can be transformed to a salt of abacavir by reaction with the corresponding acid. Such isolation methods (trituration and chromatography) usually are limited to laboratory scale because they are not appropriate for industrial use. Furthermore, the isolation of the abacavir base by trituration using acetonitrile gives a gummy solid (Example 7) and the isolation by chromatography (eluted from methanol/ethyl acetate) yields a solid foam (Example 19 or 28).
-
Other documents also describe the isolation of abacavir by trituration or chromatography, but always a gummy solid or solid foam is obtained (cf. WO9921861 and EP741710 ), which would be difficult to operate on industrial scale.
-
WO9852949 describes the preparation of abacavir which is isolated from acetone. According to this document the manufacture of the abacavir free base produces an amorphous solid which traps solvents and is, therefore, unsuitable for large scale purification, or for formulation, without additional purification procedures (cf. page 1 of WO 9852949 ). In this document, it is proposed the use of a salt of abacavir, in particular the hemisulfate salt which shows improved physical properties regarding the abacavir base known in the art. Said properties allow the manufacture of the salt on industrial scale, and in particular its use for the preparation of pharmaceutical formulations.
-
However, the preparation of a salt of abacavir involves an extra processing step of preparing the salt, increasing the cost and the time to manufacture the compound. Generally, the abacavir free base is the precursor compound for the preparation of the salt. Thus, depending on the preparation process used for the preparation of the salt, the isolation step of the abacavir free base must also be done.
………………………………
http://www.google.co.in/patents/US5034394
EXAMPLE 21(-)-cis-4-[2-Amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol
The title compound of Example 7, (2.00 g, 6.50 mmol) was dissolved in 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone (Aldrich, 20 mL). Phosphoryl chloride (2.28 mL, 24.0 mmol) was added to the stirred, cooled (-10° C.) solution. After 3 minutes, cold water (80 mL) was added. The solution was extracted with chloroform (3×80 mL). The aqueous layer was diluted with ethanol (400 mL) and the pH adjusted to 6 with saturated aqueous NaOH. The precipitated inorganic salts were filtered off. The filtrate was further diluted with ethanol to a volume of 1 liter and the pH adjusted to 8 with additional NaOH. The resulting precipitate was filtered and dried to give the 5′-monophosphate of (±)-cis-4-[2-amino-6-(cyclopropylmethylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol as white powder (4.0 mmoles, 62% quantitated by UV absorbance); HPLC analysis as in Example 17 shows one peak. This racemic 5′ -monophosphate was dissolved in water (200 mL) and snake venom 5′-nucleotidase (EC 3.1.3.5) from Crotalus atrox (5,000 IU, Sigma) was added. After incubation at 37° C. for 10 days, HPLC analysis as in Example 17 showed that 50% of the starting nucleotide had been dephosphorylated to the nucleoside. These were separated on a 5×14 cm column of DEAE Sephadex A25 (Pharmacia) which had been preequilibrated with 50 mM ammonium bicarbonate. Title compound was eluted with 2 liters of 50 mM ammonium bicarbonate. Evaporation of water gave white powder which was dissolved in methanol, adsorbed on silica gel, and applied to a silica gel column. Title compound was eluted with methanol:chloroform/1:9 as a colorless glass. An acetonitrile solution was evaporated to give white solid foam, dried at 0.3 mm Hg over P2 O5 ; 649 mg (72% from racemate); 1 H-NMR in DMSO-d6 and mass spectrum identical with those of the racemate (title compound of Example 7); [α]20 D -48.0°, [α]20 436 -97.1°, [α]20 365 -149° (c=0.14, methanol).
Anal. Calcd. for C15 H20 N6 O.0.10CH3 CN: C, 59.96; H, 6.72; N, 28.06. Found: C, 59.93; H, 6.76; N, 28.03.
Continued elution of the Sephadex column with 2 liters of 100 mM ammonium bicarbonate and then with 2 liters of 200 mM ammonium bicarbonate gave 5′-monophosphate (see Example 22) which was stable to 5′-nucleotidase.
…………………………………………
| Синтез a) |
|---|
        |
| Синтез b) |
     |
| Preparation c) |
    |
| Synthesis d) |
 |

An enantiopure β-lactam with a suitably disposed electron withdrawing group on nitrogen, participated in a π-allylpalladium mediated reaction with 2,6-dichloropurine tetrabutylammonium salt to afford an advanced cis-1,4-substituted cyclopentenoid with both high regio- and stereoselectivity. This advanced intermediate was successfully manipulated to the total synthesis of (−)-Abacavir.

http://pubs.rsc.org/en/content/articlelanding/2012/ob/c2ob06775g#!divAbstract
………………………………….
http://www.google.com.ar/patents/EP2085397A1?cl=en
Example 1: Preparation of crystalline Form I of abacavir base using methanol as solvent
-
[0026]Abacavir (1.00 g, containing about 17% of dichloromethane) was dissolved in refluxing methanol (2.2 mL). The solution was slowly cooled to – 5 °C and, the resulting suspension, was kept at that temperature overnight under gentle stirring. The mixture was filtered off and dried under vacuum (7-10 mbar) at 40 °C for 4 hours to give a white solid (0.55 g, 66% yield, < 5000 ppm of methanol). The PXRD analysis gave the diffractogram shown in FIG. 1.
……………………………………..
http://www.google.com/patents/WO2008037760A1?cl=en
Abacavir, is the International Nonproprietary Name (INN) of {(1 S,4R)-4-[2- amino-6-(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol, and CAS No. 136470-78-5. Abacavir sulfate is a potent selective inhibitor of HIV-1 and HIV-2, and can be used in the treatment of human immunodeficiency virus (HIV) infection.
The structure of abacavir hemisulfate salt corresponds to formula (I):

(I)
EP 434450-A discloses certain 9-substituted-2-aminopuhnes including abacavir and its salts, methods for their preparation, and pharmaceutical compositions using these compounds.
Different preparation processes of abacavir are known in the art. In some of them abacavir is obtained starting from an appropriate pyrimidine compound, coupling it with a sugar analogue residue, followed by a cyclisation to form the imidazole ring and a final introduction of the cyclopropylamino group at the 6 position of the purine ring. Pyrimidine compounds which have been identified as being useful as intermediates of said preparation processes include N-2-acylated abacavir intermediates such as N-{6- (cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H-purin- 2-yl}acetamide or N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-
(hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide. The removal of the amino protective group of these compounds using acidic conditions is known in the art. According to Example 28 of EP 434450-A, the amino protective group of the N-{6-(cyclopropylamino)-9-[(1 R,4S)-4- (hydroxymethyl)cyclopent-2-enyl]-9H-purin-2-yl}isobutyramide is removed by stirring with 1 N hydrochloric acid for 2 days at room temperature. The abacavir base, after adjusting the pH to 7.0 and evaporation of the solvent, is finally isolated by trituration and chromatography. Then, it is transformed by reaction with an acid to the corresponding salt of abacavir. The main disadvantages of this method are: (i) the use of a strongly corrosive mineral acid to remove the amino protective group; (ii) the need of a high dilution rate; (iii) a long reaction time to complete the reaction; (iv) the need of isolating the free abacavir; and (v) a complicated chromatographic purification process.
Thus, despite the teaching of this prior art document, the research of new deprotection processes of a N-acylated {(1 S,4R)-4-[2-amino-6- (cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is still an active field, since the industrial exploitation of the known process is difficult, as it has pointed out above. Thus, the provision of a new process for the removal of the amino protective group of a N-acylated {(1 S,4R)-4-[2-amino-6-
(cyclopropylamino)-9H-purin-9-yl]-cyclopent-2-enyl}methanol is desirable.
Example 1 : Preparation of abacavir hemisulfate
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (6.56 g, 18.40 mmol) was slurried in a mixture of isopropanol (32.8 ml) and 10% solution of NaOH (36.1 ml, 92.0 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (32.8 ml) was added. The layers were separated and H2SO4 96% (0.61 ml, 11.03 mmol) was added dropwise to the organic layer. This mixture was cooled to 0-50C and the resulting slurry filtered off.
The solid was dried under vacuum at 40 0C. Abacavir hemisulfate (5.98 g, 97%) was obtained as a white powder.
Example 6: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.0 g, 2.80 mmol) was slurried in a mixture of isopropanol (2 ml) and 10% solution of NaOH (1.1 ml, 2.80 mmol). The mixture was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and tert-butyl methyl ether (2 ml) was added. The aqueous layer was discarded, the organic phase was cooled to 0-5 0C and the resulting slurry filtered off. The solid was dried under vacuum at 400C. Abacavir (0.62 g, 77%) was obtained as a white powder.
Example 7: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetone. Abacavir (0.47 g, 47%) was obtained as a white powder.
Example 8: Preparation of abacavir
N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent-2-enyl]-9H- purin-2-yl}isobutyramide (1.25 g, 3.51 mmol) was slurried in a mixture of isopropanol (2.5 ml) and 10% solution of NaOH (1.37 ml, 3.51 mmol). The mixture was refluxed for 1 h and concentrated to dryness. The residue was crystallized in acetonitrile. Abacavir (0.43 g, 43%) was obtained as a white powder.
Example 9: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in ethyl acetate (150 ml) to afford abacavir (7.2 g, 90%).
Example 10: Preparation of abacavir
A mixture of N-{6-(cyclopropylamino)-9-[(1 R,4S)-4-(hydroxymethyl)cyclopent- 2-enyl]-9H-purin-2-yl}isobutyramide (10 g, 28 mmol), isopropanol (100 ml) and 10% solution of NaOH (16.8 ml, 42 mmol) was refluxed for 1 h. The resulting solution was cooled to 20-25 0C and washed several times with 25% solution of NaOH (10 ml). The wet organic layer was neutralized to pH 7.0-7.5 with 17% hydrochloric acid and it was concentrated to dryness under vacuum. The residue was crystallized in acetone (300 ml) to afford abacavir (7.0 g, 88%).
…………………………………
http://www.google.com/patents/WO2004089952A1?cl=en
Abacavir of formula (1) :

or (1 S,4R)-4-[2-Amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1 – methanol and its salts are nucleoside reverse transcriptase inhibitors. Abacavir sulfate is a nucleoside reverse transcriptase inhibitor and used in the treatment of human immunodeficiency virus infection. Abacavir sulfate and related compounds and their therapeutic uses are disclosed in US 5,034,394.
Crystalline forms of abacavir sulfate have not been reported in the literature. Moreover, the processes described in the literature do not produce abacavir sulfate in a stable, well-defined and reproducible crystalline form. It has now been discovered that abacavir sulfate can be prepared in three stable, well-defined and consistently reproducible crystalline forms.
Example 1
Abacavir free base (3.0 gm, obtained by the process described in example 21 of US 5,034,394) is dissolved in ethyl acetate (15 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 3 hours at 20°C and filtered to give 3.0 gm of form I abacavir sulfate. Example 2 Abacavir free base (3.0 gm) is dissolved in acetone (20 ml) and cone, sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 6 hours at 25°C and filtered to give 2.8 gm of form I abacavir sulfate.
Example 3 Abacavir free base (3.0 gm) is dissolved in acetonitrile (15 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 2 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 4 Abacavir free base (3.0 gm) is dissolved in methyl tert-butyl ether (25 ml) and sulfuric acid (0.3 ml) is added to the solution. Then the contents are stirred for 1 hours at 25°C and the separated solid is filtered to give 3.0 gm of form II abacavir sulfate.
Example 5 Abacavir free base (3.0 gm) is dissolved in methanol (15 ml) and sulfuric acid (0.3 ml) is added to the solution. The contents then are cooled to 0°C and diisopropyl ether (15 ml) is added. The reaction mass is stirred for 2 hours at about 25°C and the separated solid is filtered to give 3.0 gm of form III abacavir sulfate
…………………………….
http://www.google.com.ar/patents/WO1999021861A1?cl=en
The present invention relates to a new process for the preparation of the chiral nucleoside analogue (1S, 4R)-4-[2-amino-6-(cyclopropylamino)-9H purin-9-yl]-2- cyclopentene-1 -methanol (compound of Formula (I)).
The compound of formula (I) is described as having potent activity against human immunodeficiency virus (HIV) and hepatitis B virus (HBV) in EPO34450.

Results presented at the 34th Interscience Conference on Antimicrobial Agents and Chemotherapy (October 4-7, 1994) demonstrate that the compound of formula I has significant activity against HIV comparable to, and if not better than, some current anti HIV drugs, such as zidovudine and didanosine.
Currently the compound of Formula (I) is undergoing clinical investigation to determine its safety and efficacy in humans. Therefore, there exists at the present time a need to supply large quantities of this compound for use in clinical trials.
Current routes of synthesising the compound of formula (I) involve multiple steps and are relatively expensive. It will be noted that the compound has two centres of asymmetry and it is essential that any route produces the compound of formula (I) substantially free of the corresponding enantiomer, preferably the compound of formula (I) is greater than 95% w/w free of the corresponding enantiomer.
Processes proposed for the preparation of the compound of formula (I) generally start from a pyrimidine compound, coupling with a 4-amino-2-cyclopentene-1- methanol analogue, cyciisation to form the imidazole ring and then introduction of the cyclopropylamine group into the 6 position of the purine, such routes include those suggested in EPO434450 and WO9521161. Essentially both routes disclosed in the two prior patent applications involve the following steps:-
(i) coupling (1S, 4R)-4-amino-2-cyclopentene-1 -methanol to N-(4,6-dichloro-5- formamido-2-pyrimidinyl) acetamide or a similar analogue thereof, for example N- (2-amino-4,6-dichloro-5-pyrimidinyl) formamide;
(ii) ring closure of the resultant compound to form the intermediate (1 S, 4R)-4- (2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1 -methanol;
(iii) substituting the halo group by a cyclopropylamino group on the 6 position of the purine ring.
The above routes are multi-step processes. By reducing the number of processing steps significant cost savings can be achieved due to the length of time to manufacture the compound being shortened and the waste streams minimised.
An alternative process suggested in the prior art involves the direct coupling of carbocyclic ribose analogues to the N atom on the 9 position of 2-amino-6-chloro purine. For example WO91/15490 discloses a single step process for the formation of the (1S, 4R)- 4-(2-amino-6-chloro-9H-purin-9-yl)-2-cyclopentene-1- methanol intermediate by reacting (1S, 4R)-4-hydroxy-2-cyclopentene-1 -methanol, in which the allylic hydroxyl group has been activated as an ester or carbonate and the other hydroxyl group has a blocking group attached (for example 1 ,4- bis- methylcarbonate) with 2-amino-6-chloropurine.
However we have found that when synthesising (1S, 4R)-4-(2-amino-6-chloro-9H- purin-9-yl)-2-cyclopentene-1- methanol by this route a significant amount of an N- 7 isomer is formed (i.e. coupling has occurred to the nitrogen at the 7- position of the purine ring) compared to the N-9 isomer desired. Further steps are therefore required to convert the N-7 product to the N-9 product, or alternatively removing the N-7 product, adding significantly to the cost. We have found that by using a transition metal catalysed process for the direct coupling of a compound of formula (II) or (III),

Example 1 (1 S. 4R)-4-[2-Amino-6-(cvclopropylamino)-9H purin-9-vπ-2-cvclopentene-1 – methanol
Triphenylphosphine (14mg) was added, under nitrogen, to a mixture of (1S.4R)- 4-hydroxy-2-cyclopentene -1 -methanol bis(methylcarbonate) (91 mg), 2-amino-6- (cyclopropylamino) purine (90mg), tris(dibenzylideneacetone)dipalladium (12mg) and dry DMF (2ml) and the resulting solution stirred at room temperature for 40 min.
The DMF was removed at 60° in vacuo and the residue partitioned between ethyl acetate (25ml.) and 20% sodium chloride solution (10ml.). The ethyl acetate solution was washed with 20% sodium chloride (2x12ml.) and with saturated sodium chloride solution, then dried (MgSO4) and the solvent removed in vacuo.
The residue was dissolved in methanol (10ml.), potassium carbonate (17mg) added and the mixture stirred under nitrogen for 15h.
The solvent was removed in vacuo and the residue chromatographed on silica gel
(Merck 9385), eluting with dichloromethane-methanol [(95:5) increasing to (90:10)] to give the title compound (53mg) as a cream foam.
δ(DMSO-d6): 7.60 (s.1 H); 7.27 (s,1 H); 6.10 (dt,1 H); 5.86 (dt, 1 H); 5.81 (s,2H); 5.39 (m,1H); 4.75 (t,1H); 3.44 (t,2H); 3.03 (m, 1H): 2.86 (m,1H);2.60 (m,1H); 1.58 (dt, 1 H); 0.65 (m, 2H); 0.57 (m,2H).
TLC SiO2/CHCI3-MeOH (4:1 ) Rf 0.38; det. UN., KMnO4
Trade Names
| Page | Trade name | Manufacturer |
|---|---|---|
| Germany | Kiveksa | GlaxoSmithKline |
| Trizivir | -»- | |
| Ziagen | -»- | |
| France | Kiveksa | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| United Kingdom | Kiveksa | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| Italy | Trizivir | -»- |
| Ziagen | -»- | |
| Japan | Épzikom | -»- |
| Ziagen | -»- | |
| USA | Épzikom | -»- |
| Trizivir | -»- | |
| Ziagen | -»- | |
| Ukraine | Virol | Ranbaksi Laboratories Limited, India |
| Ziagen | GlaksoSmitKlyayn Inc.., Canada | |
| Abamun | Tsipla Ltd, India | |
| Abacavir sulfate | Aurobindo Pharma Limited, India |
Formulations
-
Oral solution 20 mg / ml;
-
Tablets of 300 mg (as the sulfate);
-
Trizivir tablets 300 mg – abacavir in fixed combination with 150 mg of lamivudine and 300 mg zidovudine
ZIAGEN is the brand name for abacavir sulfate, a synthetic carbocyclic nucleoside analogue with inhibitory activity against HIV-1. The chemical name of abacavir sulfate is (1S,cis)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol sulfate (salt) (2:1). Abacavir sulfate is the enantiomer with 1S, 4R absolute configuration on the cyclopentene ring. It has a molecular formula of (C14H18N6O)2•H2SO4 and a molecular weight of 670.76 daltons. It has the following structural formula:
 |
Abacavir sulfate is a white to off-white solid with a solubility of approximately 77 mg/mL in distilled water at 25°C. It has an octanol/water (pH 7.1 to 7.3) partition coefficient (log P) of approximately 1.20 at 25°C.
ZIAGEN Tablets are for oral administration. Each tablet contains abacavir sulfate equivalent to 300 mg of abacavir as active ingredient and the following inactive ingredients: colloidal silicon dioxide, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate. The tablets are coated with a film that is made of hypromellose, polysorbate 80, synthetic yellow iron oxide, titanium dioxide, and triacetin.
ZIAGEN Oral Solution is for oral administration. Each milliliter (1 mL) of ZIAGEN Oral Solution contains abacavir sulfate equivalent to 20 mg of abacavir (i.e., 20 mg/mL) as active ingredient and the following inactive ingredients: artificial strawberry and banana flavors, citric acid (anhydrous), methylparaben and propylparaben (added as preservatives), propylene glycol, saccharin sodium, sodium citrate (dihydrate), sorbitol solution, and water.
In vivo, abacavir sulfate dissociates to its free base, abacavir. All dosages for ZIAGEN are expressed in terms of abacavir.
History
Abacavir was approved by the Food and Drug Administration (FDA) on December 18, 1998 and is thus the fifteenth approved antiretroviral drug in the United States. Its patent expired in the United States on 2009-12-26.
Links
-
US 5 089 500 (Burroughs Wellcome; 18.2.1992; GB-prior. 27.6.1988).
-
Synthesis a)
-
EP 434 450 (Wellcome Found .; 26.6.1991; appl. 21.12.1990; prior-USA. 22.12.1989).
-
Crimmins, MT et al .: J. Org. Chem. (JOCEAH) 61 4192 (1996).
-
EP 1 857 458 (Solmag; appl. 5.5.2006).
-
EP 424 064 (Enzymatix; appl. 24.4.1991; GB -prior. 16.10.1989).
-
U.S. 6 340 587 (Beecham SMITHKLINE; 22.1.2002; appl. 20.8.1998; GB -prior. 22.8.1997).
-
-
Синтез b)
-
Olivo, HF et al .: J. Chem. Soc., Perkin Trans. 1 (JCPRB4) 1998, 391.
-
-
Preparation c)
-
U.S. 5 034 394 (Wellcome Found .; 23.7.1991; appl. 22.12.1989; GB -prior. 27.6.1988).
-
-
Synthesis d)
-
WO 9 924 431 (Glaxo; appl. 12.11.1998; WO-prior. 12.11.1997).
-
| WO2008037760A1 * | Sep 27, 2007 | Apr 3, 2008 | Esteve Quimica Sa | Process for the preparation of abacavir |
| EP1905772A1 * | Sep 28, 2006 | Apr 2, 2008 | Esteve Quimica, S.A. | Process for the preparation of abacavir |
| US8183370 | Sep 27, 2007 | May 22, 2012 | Esteve Quimica, Sa | Process for the preparation of abacavir |
| EP0434450A2 | 21 Dec 1990 | 26 Jun 1991 | The Wellcome Foundation Limited | Therapeutic nucleosides |
| EP0741710A1 | 3 Feb 1995 | 13 Nov 1996 | The Wellcome Foundation Limited | Chloropyrimide intermediates |
| WO1998052949A1 | 14 May 1998 | 26 Nov 1998 | Glaxo Group Ltd | Carbocyclic nucleoside hemisulfate and its use in treating viral infections |
| WO1999021861A1 | 24 Oct 1997 | 6 May 1999 | Glaxo Group Ltd | Process for preparing a chiral nucleoside analogue |
| WO1999039691A2 * | 4 Feb 1999 | 12 Aug 1999 | Brooks Nikki Thoennes | Pharmaceutical compositions |
| WO2008037760A1 * | 27 Sep 2007 | 3 Apr 2008 | Esteve Quimica Sa | Process for the preparation of abacavir |
References
- Jump up^ SFGate.com
- Jump up^ “WHO Model List of EssentialMedicines”. World Health Organization. October 2013. Retrieved 22 April 2014.
- Jump up^ https://online.epocrates.com/noFrame/showPage.do?method=drugs&MonographId=2043&ActiveSectionId=5
- Jump up^ Mallal, S., Phillips, E., Carosi, G. et al. (2008). “HLA-B*5701 screening for hypersensitivity to abacavir”. New England Journal of Medicine 358: 568–579.doi:10.1056/nejmoa0706135.
- Jump up^ Rauch, A., Nolan, D., Martin, A. et al. (2006). “Prospective genetic screening decreases the incidence of abacavir hypersensitivity reactions in the Western Australian HIV cohort study”. Clinical Infectious Diseases 43: 99–102. doi:10.1086/504874.
- Jump up^ Heatherington et al. (2002). “Genetic variations in HLA-B region and hypersensitivity reactions to abacavir”. Lancet 359: 1121–1122.
- Jump up^ Mallal et al. (2002). “Association between presence of HLA*B5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir”. Lancet359: 727–732. doi:10.1016/s0140-6736(02)07873-x.
- Jump up^ Rotimi, C.N.; Jorde, L.B. (2010). “Ancestry and disease in the age of genomic medicine”. New England Journal of Medicine 363: 1551–1558.
- Jump up^ Phillips, E., Mallal, S. (2009). “Successful translation of pharmacogenetics into the clinic”. Molecular Diagnosis & Therapy 13: 1–9. doi:10.1007/bf03256308.
- Jump up^ Phillips, E., Mallal S. (2007). “Drug hypersensitivity in HIV”. Current Opinion in Allergy and Clinical Immunology 7: 324–330. doi:10.1097/aci.0b013e32825ea68a.
- Jump up^http://www.fda.gov/drugs/drugsafety/postmarketdrugsafetyinformationforpatientsandproviders/ucm123927.htmAccessed November 29, 2013.
- Jump up^ http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=ca73b519-015a-436d-aa3c-af53492825a1
- Jump up^ Martin MA, Hoffman JM, Freimuth RR et al. (May 2014). “Clinical Pharmacogenetics Implementation Consortium Guidelines for HLA-B Genotype and Abacavir Dosing: 2014 update”. Clin Pharmacol Ther. 95 (5): 499–500. doi:10.1038/clpt.2014.38.PMC 3994233. PMID 24561393.
- Jump up^ Swen JJ, Nijenhuis M, de Boer A et al. (May 2011). “Pharmacogenetics: from bench to byte–an update of guidelines”. Clin Pharmacol Ther. 89 (5): 662–73.doi:10.1038/clpt.2011.34. PMID 21412232.
- Jump up^ Shear, N.H., Milpied, B., Bruynzeel, D.P. et al. (2008). “A review of drug patch testing and implications for HIV clinicians”. AIDS 22: 999–1007.doi:10.1097/qad.0b013e3282f7cb60.
- Jump up^ http://www.drugs.com/fda/abacavir-ongoing-safety-review-possible-increased-risk-heart-attack-12914.html Accessed November 29, 2013.
- Jump up^ Ding X, Andraca-Carrera E, Cooper C et al. (December 2012). “No association of abacavir use with myocardial infarction: findings of an FDA meta-analysis”. J Acquir Immune Defic Syndr. 61 (4): 441–7. doi:10.1097/QAI.0b013e31826f993c.PMID 22932321.
- Illing PT et al. 2012, Nature, doi:10.1038/nature11147
External links
- Full Prescribing Information
- Abacavir pathway on PharmGKB
- Abacavir dosing guidelines from the Clinical Pharmacogenetics Implementation Consortium (CPIC)
- Abacavir dosing guidelines from the Dutch Pharmacogenetics Working Group (DPWG)
EXTRA INFO
How to obtain carbocyclic nucleosides?
Carbocyclic nucleosides are synthetically the most challenging class of nucleosides, requiring multi-step and often elaborate synthetic pathways to introduce the necessary stereochemistry. There are two main strategies for the preparation of carbocyclic nucleosides. In the linear approach a cyclopentylamine is used as starting material and the heterocycle is built in a stepwise manner (see Scheme 1).
Scheme 1: Linear approach for the synthesis of abacavir.[5]

The more flexible strategy is a convergent approach: a functionalized carbocyclic moiety is condensed with a heterocycle rapidly leading to a variety of carbocyclic nucleosides. Initially, we started our syntheses from cyclopentadiene 1 that is deprotonated and alkylated with benzyloxymethyl chloride to give the diene 2. This material is converted by a hydroboration into cyclopentenol 3 or isomerized into two thermodynamically more stable cyclopentadienes 4a,b. With the protection and another hydroboration step to 5 we gain access to an enantiomerically pure precursor for the synthesis of a variety of carbocyclic 2’-deoxynucleosides e.g.:carba-dT, carba-dA or carba-BVDU.[6] The isomeric dienes 4a,b were hydroborated to the racemic carbocyclic moiety 6.

Scheme 2: Convergent approach for the synthesis of carba-dT.
The asymmetric synthesis route and the racemic route above are short and efficient ways to diverse carbocyclic D- or L-nucleosides (Scheme 2). Different heterocycles can be condensed to these precursors leading to carbocyclic purine- and pyrimidine-nucleosides. Beside α- and β-nucleosides, carbocyclic epi– andiso-nucleosides in the 2’-deoxyxylose form were accessable.[7]
What else is possible? The racemic cyclopentenol 6 can be coupled by a modified Mitsunobu-reaction.Moreover, this strategy offers the possibility of synthesizing new carbocyclic nucleosides by functionalizing the double bond before or after introduction of the nucleobase (scheme 3).[8]

Scheme 3: Functionalized carbocyclic nucleosides based on cyclopentenol 6.
Other interesting carbocyclic precursors like cyclopentenol 7 can be used to synthesize several classes of carbocyclic nucleoside analogues, e.g.: 2’,3’-dideoxy-2’,3’-didehydro nucleosides (d4-nucleosides), 2’,3’-dideoxynucleosides (ddNs), ribonucleosides, bicyclic nucleosides or even 2’-fluoro-nucleosides.

Scheme 4: Functionalized carbocyclic thymidine analogues based on cyclopentenol 7.
[1] V. E. Marquez, T. Ben-Kasus, J. J. Barchi, K. M. Green, M .C. Nicklaus, R. Agbaria, J. Am. Chem. Soc.2004,126, 543.
[2] A. D. Borthwick, K. Biggadike, Tetrahedron 1992, 48, 571.
[3] H. Bricaud, P. Herdewijn, E. De Clercq, Biochem. Pharmacol. 1983, 3583.
[4] P. L. Boyer, B. C. Vu, Z. Ambrose, J. G. Julias, S. Warnecke, C. Liao, C. Meier, V. E. Marquez, S. H. Hughes, J. Med. Chem. 2009, 52, 5356.
[5] S. M. Daluge, M. T. Martin, B. R. Sickles, D. A. Livingston, Nucleosides, Nucleotides Nucleic Acids 2000,19, 297.
[6] O. R. Ludek, C. Meier, Synthesis 2003, 2101.
[7] O. R. Ludek, T. Kraemer, J. Balzarini, C. Meier, Synthesis 2006, 1313.
[8] M. Mahler, B. Reichardt, P. Hartjen, J. van Lunzen, C. Meier, Chem. Eur. J. 2012, 18, 11046-11062.
Dipivefrine, дипивефрин , ديبيفيفرين , 地匹福林 , ジピベフリン
Dipivefrine
- Molecular FormulaC19H29NO5
- Average mass351.437 Da
4-[1-hydroxy-2-(methylamino)ethyl]benzene-1,2-diyl bis(2,2-dimethylpropanoate)
52365-63-6 [RN]
(±)-3,4-Dihydroxy-a-[(methylamino)methyl]benzyl Alcohol 3,4-Dipivalate
1-(3′,4′-Dipivaloyloxyphenyl)-2-methylamino-1-ethanol
2,2-Dimethylpropanoic acid 4-[1-hydroxy-2-(methylamino)ethyl]-1,2-phenylene ester
дипивефрин [Russian] [INN]
ديبيفيفرين [Arabic] [INN]
地匹福林 [Chinese] [INN]
ジピベフリン
4-[1-hydroxy-2-(methylamino)ethyl]-o-phenylene divavalate
D Epifrin [Trade name]
Diopine [Trade name]
MFCD00673243 [MDL number]
Pivalephrine [Trade name]
Pro-Epinephrine
Propine [Trade name]
Thilodrin [Trade name]
ATC:S01EA02
Use:antiglaucoma
Dipivefrine (INN) or dipivefrin (USAN), trade name Propine among others, is a prodrug of epinephrine, and is used to treat open-angle glaucoma.[1][2] It is available as a 0.1% ophthalmic solution. It is no longer available in the United States.[3]
Dipivefrin is a prodrug with little or no pharmacologically activity until it is hydrolyzed into epinephrine inside the human eye. The liberated epinephrine, an adrenergic agonist, appears to exert its action by stimulating α -and/or β2-adrenergic receptors, leading to a decrease in aqueous production and an enhancement of outflow facility. The dipivefrin prodrug delivery system is a more efficient way of delivering the therapeutic effects of epinephrine, with fewer side effects than are associated with conventional epinephrine therapy. Dipivefrin is used as initial therapy for the control of intraocular pressure in chronic open-angle glaucoma.

Contraindications
Use in narrow-angle glaucoma may be dangerous because it could make the eye susceptible to an attack of angle closure,[2] causing an increase in pressure and pain, and possibly loss of vision.
Side effects
The most common side effects of dipivefrine are burning, stinging and other irritations of the eye. Possible, but uncommon, side effects are those of epinephrine: tachycardia (fast heartbeat), hypertension (high blood pressure) and arrhythmias (irregular heartbeat).[2]
Pharmacology
Dipivefrine penetrates the cornea and is then hydrolysed to epinephrine by esterase enzymes. It increases outflow of the aqueous humour and also reduces its formation (mediated by its action on α1 and α2 receptors), thus reducing pressure inside the eye. It also increases the conductivity of trabecular filtering cells (a β2 receptor mediated action). It is preferred to epinephrine because it is longer acting, more consistent in its action and better tolerated.[1]
Patent
https://patents.google.com/patent/CN102153485A/en

Example 1 [0023] Embodiment
[0024] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 5 ° C, was added 666g (6. 58mol) of triethylamine, and then was added dropwise 784g (6. 5mol) pivaloyl chloride was added dropwise and stirring was continued after the pool. Filtered off with suction, the filtrate by rotary evaporation; to give 990g yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 96.2%. [0025] The 35mol) N- methyl amine section, 370g (3. 66mol) of triethylamine, 25g (0. 15mol) KI, 3L DMF was added 4-neck flask of the IOL. Cooled to 0 ° C, was added dropwise 990g (2. 8mol) 4- (2- chloroacetyl) -I, DMF solution tank 2-phenyl pivalate ester. At room temperature was stirred for 4h.
[0026] suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 923. Og. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 96.5%.
[0027] Take 625g (1. 422mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 65g (1.71mol) of sodium borohydride. At room temperature was stirred for 4h. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 552. 5g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.2%.
[0028] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 13g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0029] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0030] After the solution was washed with methanol hydrochloride salt to give an off-white solid 119. 9g, dipivefrin i.e., the content of 98.9%.
[0031] m.p. 161 ~162 ° C;
[0032] 1H NMR (CDCl3) δ: 1. 35 (s, 18Η), 2 68 (s, 3Η), 3 07-3 13 (m, 2Η), 5 36-5 39 (m….. , 1H),
[0033] 7. 06-7. 30 (m, 3H), 8. 61 (s, 1H), 9. 48 (s, 1H)
Dipivefrin prepared: Example 2 [0034] Embodiment
[0035] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 10 ° C, was added 666g (6. 58mol) of triethylamine, and then dropwise 78½ (6. 5mol) pivaloyl chloride was added dropwise and stirring was continued after the pool. Filtered off with suction, the filtrate by rotary evaporation; 978. 2g to give yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 96. 2% o
[0036] The 35mol) N- methyl amine section, 370g (3. 66mol) of triethylamine, 25g (0. 15mol) KI, 3L DMF was added 4-neck flask of the IOL. Cooled to O0C, dropwise 978. 2g (2. 77mol) 4- (2- chloroacetyl) of DMF solution tank Laid-1,2-phenyl valerate. At room temperature was stirred for 4h.
[0037] suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 910. 2g. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 96.3%.
[0038] Take 625g (1. 422mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 97g (l. SOmol) potassium borohydride. Stirred cell at room temperature. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 532. 7g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.0%.
[0039] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 15g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0040] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0041] After the solution was washed with methanol hydrochloride salt to give an off-white solid was 112. 8g, i.e., dipivefrin, content 98.6%.
3 [0042] Example 2: Preparation of dipivefrin
[0043] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 5 ° C, was added 897g (6. 5mol) of potassium carbonate, and then drops was added 784g (6. 5mol) pivaloyl chloride addition was completed stirring was continued Syndrome. Filtered off with suction, the filtrate by rotary evaporation; to give 900g yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 95.6%.
[0044] A 526g (4. 35mol) N_ methylbenzylamine, 414g (3. Omol) of potassium carbonate, 25g (0. 15mol) KI, 3L DMF force Λ IOL of four port flask. Cooled to O0C, was added dropwise 900g (2. 55mol) 4- (2- chloroacetyl) of DMF solution of 1,2-Shan Laid phenyl valerate. It was stirred at room temperature Mi.
[0045] The suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 820g. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 95.6%.
[0046] Take 625g (1. 42mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 65g (1.71mol) of sodium borohydride. Stirred cell at room temperature. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 512. 5g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.0%.
[0047] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 16g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0048] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0049] After the solution was washed with methanol hydrochloride salt to give an off-white solid was 109. 8g, i.e., dipivefrin, content 98.5%.
SYN
Dipivefrin
CAS Registry Number: 52365-63-6
CAS Name: 2,2-Dimethylpropanoic acid 4-[1-hydroxy-2-(methylamino)ethyl]-1,2-phenylene ester
Additional Names: (±)-3,4-dihydroxy-a-[(methylamino)methyl]benzyl alcohol 3,4-dipivalate; 1-(3¢,4¢-dipivaloyloxyphenyl)-2-methylamino-1-ethanol; dipivalyl epinephrine; DPE
Molecular Formula: C19H29NO5
Molecular Weight: 351.44
Percent Composition: C 64.93%, H 8.32%, N 3.99%, O 22.76%
Literature References: Dipivalyl ester of epinephrine, q.v. Prepn: D. Henschler et al., DE 2152058; eidem, US 4085270 (1973, 1978 both to Klinge); A. Hussain, J. E. Truelove, DE 2343657; eidem, US 3809714 and US 3839584 (all 1974 to Interx). In vitrostudy: A. H. Neufeld, E. D. Page, Invest. Ophthalmol. Visual Sci. 16, 1118 (1977). Pharmacology: B. C. Wang et al., J. Pharmacol. Exp. Ther. 203, 442 (1977). Effects on intraocular pressure in dogs: R. M. Gwin et al., Am. J. Vet. Res. 39, 83 (1978). Metabolism: I. Abramovsky, J. S. Mindel, Arch. Ophthalmol. 97, 1937 (1979). Clinical study: M. A. Kass et al., ibid. 1865. General pharmacology, toxicology and clinical experience in glaucoma: D. A. McClure, ACS Symp. Ser. 14, 224-235 (1975). Comprehensive description: G. M. Wall, T. Y. Fan, Anal. Profiles Drug Subs. Excip. 22, 229-262 (1993).
Properties: Crystals from ether, mp 146-147°.
Melting point: mp 146-147°
Derivative Type: Hydrochloride
CAS Registry Number: 64019-93-8
Trademarks: Diopine (Allergan); d Epifrin (Allergan); Diphemin (Alcon); Pivalephrine (Santen); Propine (Allergan)
Molecular Formula: C19H29NO5.HCl
Molecular Weight: 387.90
Percent Composition: C 58.83%, H 7.80%, N 3.61%, O 20.62%, Cl 9.14%
Properties: Crystals from ethyl acetate, mp 158-159°. Sol in water and ethanol. pKa 8.40.
Melting point: mp 158-159°
pKa: pKa 8.40
Therap-Cat: Adrenergic (ophthalmic); antiglaucoma.
Keywords: a-Adrenergic Agonist; Antiglaucoma.
SYN
2-chloro-3′,4′-dihydroxyacetophenone, 99-40-1
3′,4′-dihydroxy-2-methylaminoacetophenone, 99-45-6
2,2-dimethylpropanoic acid 4-[(methylamino)acetyl]-1,2-phenylene ester, 52245-00-8
Pivaloyl chloride, 3282-30-2
Trimethylacetyl chloride, 3282-30-2

1-(3,4-dipivaloyloxyphenyl)-2-(benzylmethylamino)ethan-1-one, 42146-03-2
SPECTROSCOPY

infrared spectral assignments for dipiveh hydrochloride
Wavelength (cm-1) Assignment
3255,2804,2475, 2397 RflHz+-NH stretch
2974-2875 sp3 C-H stretch
1273, 1258-1163 C-0-C stretch
3600-3400 0-H stretch
phenyl ester C=O stretch 1761
aromatic C-C stretch 1614, 1595, 1562, 1504
sp3 C-H bending and scissoring 1481, 1461, 1441, 1397
tert-butyl C-H bending1368, 1332
secondary alcohol C-0 stretch 1 124- 1028
out-of-plane bending for 1,substituted benzene ring 3,4 891,842

Ultraviolet absorption of dipivefrin hydrochloride
E (176, 1 cm)
Solvent 210 nm 264 Nn 270 nm
Acetonitrile 267.3 14.8 13.4
Ethanol 246.8 14.5 13.1
pH 3 Buffer 266.7 12.4 10.4
pH 7 Buffer 257.6 10.8 8.9
Water 278.0 18.0 16.2




References
- ^ Jump up to:a b KD Tripari. Essentials of Medical Pharmacology (5 ed.). Jaypee Brothers Medical Publishers(P) Ltd. p. 88. ISBN 81-8061-187-6.
- ^ Jump up to:a b c Dipivefrin FDA Professional Drug Information.
- ^ Zhang L, Weizer JS, Musch DC (2017). “Perioperative medications for preventing temporarily increased intraocular pressure after laser trabeculoplasty”. Cochrane Database Syst Rev. 2: CD010746. doi:10.1002/14651858.CD010746.pub2. PMC 5477062. PMID 28231380.
-
- Hussain, A.; Truelove, J.E.: J. Pharm. Sci. (JPMSAE) 65, 1510 (1976).
- US 3 839 584.
- a DOS 2 343 657 (Interx Res. Corp.; appl. 30.8.1973; USA-prior. 31.8.1972).
- US 3 809 714 (Interx; 7.5.1974; prior. 31.8.1972) also racemate resolution.
- b DOS 2 152 058 (Klinge; appl. 19.10.1971).
 |
|
| Clinical data | |
|---|---|
| Trade names | Propine, Pivalephrine |
| Synonyms | Dipivefrin |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686005 |
| Pregnancy category |
|
| Routes of administration |
Eye drops |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C19H29NO5 |
| Molar mass | 351.437 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////дипивефрин , ديبيفيفرين , 地匹福林 , Dipivefrine, antiglaucoma, GENERIC, ジピベフリン
DIOSMIN, диосмин , ديوسمين , 地奥司明 ,

DIOSMIN
- Molecular FormulaC28H32O15
- Average mass608.545 Da
5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-7-{[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-({[(2R,3R,4R,5R,6S)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxy}methyl)oxan-2-yl]oxy}-4H-chromen-4-one
3′,5,7-Trihydroxy-4′-methoxyflavone-7-rutinoside
4H-1-Benzopyran-4-one, 7-((6-O-(6-deoxy-α-L-mannopyranosyl)-β- D-glucopyranosyl)oxy)-5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-
4H-1-Benzopyran-4-one, 7-((6-O-(6-deoxy-α-L-mannopyranosyl)-β-D-glucopyranosyl)oxy)-5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-
4H-1-Benzopyran-4-one, 7-[[6-O-(6-deoxy-α-L-mannopyranosyl)-β-D-glucopyranosyl]oxy]-5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-
520-27-4 [RN]
5-Hydroxy-2-(3-hydroxy-4-methoxyphenyl)-4-oxo-4H-chromen-7-yl 6-O-(6-deoxy-α-L-mannopyranosyl)-β-D-glucopyranoside
диосмин [Russian] [INN]
ديوسمين [Arabic] [INN]
地奥司明 [Chinese] [INN]
Barosmin / Dalfon (Servier) / Detralex / Diosven / Dioven / Diovenor / Flebosmil / Flebosten / Hemerven / Insuven / Litosmil / Varinon / Ven-detrex / Venosmine, Barosmin, Diosmine, Venosmine, Diosmil,
Diosmin is a bioflavonoid that strengthens vascular walls.
Diosmin is a semisynthetic drug indicated for the treatment of venous disease. Diosmin is a flavone that can be found in the plant Teucrium gnaphalodes. Diosmin is available as a prescription medicine in several European countries, and is available as a nutritional supplement in the United States and the rest of Europe. It should be noted that clinical studies have been inconclusive and no articles have been published pertaining to its use in the treatment of vascular disease. When used in rats, diosmin has been effective at mitigating hyperglycaemia, and may also have antineurodegenerative properties.
Diosmin is a flavone, a member of the flavonoid family. Diosmin aglycone is diosmetin. It can be found in Teucrium gnaphalodes, a plant endemic to the Iberian Peninsula.[1]


IR KBR





SYNTHESIS

Drug
Diosmin is a semisynthetic flavonoid molecule derived from citrus d (modified hesperidin). It is an oral phlebotropic drug used in the treatment of venous disease, i.e., chronic venous insufficiency (CVI) including spider and varicose veins, leg swelling (edema), stasis dermatitis and venous ulcers. It is also used as a stand-alone or surgical adjunctive therapy in hemorrhoidal disease (HD).
There are extensive clinical trials that show diosmin improves all stages of venous disease including venous ulcers and improves quality of life.[2] There are no prospective studies in arterial disease.
Diosmin is currently a prescription medication in some European countries (under the Dio-PP, Venotec, Daflon etc. tradenames), and is sold as a nutritional supplement in the United States.
Diosmin has been found to be effective in mitigating hyperglycemia in diabetic rats.[3] It is also speculated that diosmin might have potential in the treatment of neurodegenerative diseases,[4] such as Alzheimer’s disease.
Mechanisms
Diosmin improves lymphatic drainage by increasing the frequency and intensity of lymphatic contractions, and by increasing the total number of functional lymphatic capillaries. Furthermore, diosmin with hesperidine decreases the diameter of lymphatic capillaries and the intralymphatic pressure.Diosmin prolongs the vasoconstrictor effect of norepinephrine on the vein wall, increasing venous tone, and therefore reducing venous capacitance, distensibility, and stasis. This increases the venous return and reduces venous hyperpressure present in patients suffering from CVI.
At the microcirculation level, diosmin reduces capillary hyperpermeability and increases capillary resistance by protecting the microcirculation from damaging processes.
Diosmin reduces the expression of endothelial adhesion molecules (ICAM1, VCAM1), and inhibits the adhesion, migration, and activation of leukocytes at the capillary level. This leads to a reduction in the release of inflammatory mediators, principally oxygen free radicals and prostaglandins (PGE2, PGF2a).
Society and culture
Diosmin is distributed in the U.S. as a dietary supplement.[5][6]
Diosmin was first reported by O. A. Osterle and G. Wander in HeIv. Chim. Acta. 8, 519 – 536, 1925 and is a naturally occurring flavonoid glycoside that can be isolated from various plant sources, i.e from the peel of the citrus fruit or hesperidin. Diosmin is a protecting agent and is used for the treatment of chronic venous insufficiency, lymphedema, hemorrhoids and varicose veins. It has been also used for other therapeutic purposes such as cancer, premenstrual syndrome, colitis, and diabetes.
The several references are reported in the prior art for conversion of hesperidin to diosmin.
Zemplen and Bogner, in Ber. 76, 452, 1943 reported monobromination of acetylated flavanones by liquid bromine in chloroform solution in presence of ultraviolet radiation to obtain flavone derivative by following loss of hydrogen bromide and deacetylation with alcoholic alkali. The conversion of hesperidin to diosmin reported is 37%.
In the journal reference, J. Org. Chem., 16, 930 – 933, 1951, by N. B. Lorette et. al. N-bromosuccinimide was used for the bromination of acetylated hesperidin in chloroform and benzoyl peroxide was used as a catalyst. Diosmin yield was 44%.
Studies in Organic Chemistry (Amsterdam) (1982), Volume Date 1981, 11, 115-119 describes conversion of Hesperidin, neohesperidin and naringin to diosmin, neodiosmin, and rhoifolin respectively by dehydrogenation with iodine in pyridine. Tianran Chanwu Yabjiu Yu Kaif (2006), 18(6), 896-899 describes separation and purification of diosmin by macroporous resins, and reported 95% pure diosmin.
ES459076 describes the preparation of diosmin by bromination and debromination of hesperidin acetate in tetrahydrofuran with 2-carboxy ethyl triphenyl phosphonium bromide followed by saponification with potassium tertiary butoxide.
ES465156 describes diosmin preparation by reaction of hesperidin with aqueous sodium hydroxide, iodine and pyridine with 66% yield.
DE2740950 describes iodination-dehydroiodination of hesperidin in the presence of pyridine and iodine resulting 89% of diosmin.
EP52086 claims a process for the preparation of diosmin comprising of total acetylation of hesperidin or related flavone by heating it in acetic anhydride and pyridine followed by selective dehydrogenation or oxidation by means of SeO2 in isoamyl alcohol and then deprotection by means of alkaline hydrolysis with inorganic bases under hot condition. The isolated diosmin is purified by base acid treatment with overall reported yield is of 60%.
US4078137 describes a process for diosmin comprising of acetylation of hesperidin, thereby brominating it and the brominated product is hydrolysed to isolate diosmin with bromine content less then 0.1% with over all 65% yield.
In BE 904614, diosmin was prepared by iodination of hesperidin followed by elimination of HI. In the process, iodine in dimethylformamide and pyridine were successively added to hesperidin and the resulting mixture was heated at 100°C to give 96% pure diosmin.
EP 860443 describes the process that involves reaction of hesperidin with iodine in presence of pyridine at reflux temperature for 5 hours. The reaction mixture is cooled to 5°C and the isolated diosmin is purified using base acid treatment to get the quality of diosmin above 90% with 75 % yield.
FR2760015 provides industrial dehydrogenation of hesperidin with potassium iodide in DMSO in presence of cone. H2SO4 resulted in diosmin with 73% yield and pharmacopoeial quality.
WO2000011009 describes reaction of hesperidin with iodine in presence of pyridine and anhydrous alkaline earth metal base. The process involves purifying the reaction mass using morpholine followed by base acid treatment which resulted in diosmin with 80% yield and purity of diosmin meets with pharmacopoeial norms.
EP 1086953 discloses the process for purification of diosmin by reacting with pulverized zinc in aqueous solution followed by filtration and acidification.
Diosmin which is produced by many of the prior art processes is often found to contain impurities and is contaminated with various byproducts, for instance hesperidin, Isorhoifin, acetyl lisovanilone, 6-Iododiosmin, linarin, diosmetin and other organic volatile impurities. Some of the major impurities are resulted from hesperidin during extraction. The impurities of hesperidin have a major effect on the final assay of diosmin. The impurities vary depending upon the source of hesperidin. It is worthy to note that direct crystallization of crude diosmin with aqueous base acid solution does not necessarily improve the assay / purity of diosmin.
The process described in Studies in Organic Chemistry (Amsterdam) (1982), Volume Date 1981, 11, 115-119 is different from the present inventors process.
Although ES459076 teaches the preparation of diosmin by bromination and debromination of hesperidin acetate in tetrahydrofuran with 2-carboxy ethyl triphenyl phosphonium bromide, it does not teach about the final purity of diosmin with pharmaceutical quality as required.
ES465156 and DE2740950 although disclose method of preparation, does not teach a process that gives yields as are taught by present invention.
EP52086 and US4078137 uses acetic anhydride for acetylation of hesperidine with yields around 60%, which are phenomenally less as compared with yields of process described by present invention.
Sequence of addition of reactants in the process as taught by BE 904614 is different than the teachings of the present invention.
FR2760015 teaches use of different reactants under conditions that are different from the teachings of the present invention.
Invention disclosed in present application does not use morpholine as disclosed in WO2000011009.
Though there are reported several processes for preparation of diosmin in the prior art, present invention describes a novel systematic process for the preparation of diosmin by converting hesperidin to diosmin at optimum level i.e. % conversion, and keeping the impurities at minimum level which results in consistently pure diosmin with good yield and the desired quality. The process allows recovery and recycle of major contributing chemicals and solvents such as methanol, pyridine and iodine, without impact on quality, purity or yield of the process making the process more economical and ecofriendly. It is surprisingly found that quality diosmin output obtained is independent of hesperidin quality used

Title: Diosmin
CAS Registry Number: 520-27-4
CAS Name: 7-[[6-O-(6-Deoxy-a-L-mannopyranosyl)-b-D-glucopyranosyl]oxy]-5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-4H-1-benzopyran-4-one
Additional Names: 3¢,5,7-trihydroxy-4¢-methoxyflavone-7-rutinoside; 5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-7-(O6-a-L-rhamnopyranosyl-b-D-glucopyranosyloxy)chromen-4-one; 5-hydroxy-2-(3-hydroxy-4-methoxyphenyl)-7-b-rutinosyloxy-4H-chromen-4-one; diosmetin 7-b-rutinoside; barosmin; buchu resin
Trademarks: Diosmil (Bellon); Diosven (CT); Diovenor (Innotha); Flebosmil (Bouchara); Flebosten (Bonomelli); Hemerven (Interdelta); Insuven (Berenguer); Litosmil (Evans); Tovene (Kali-Chemie); Varinon (Exa); Ven-Detrex (Zyma); Venosmine (Geymonat)
Molecular Formula: C28H32O15
Molecular Weight: 608.54
Percent Composition: C 55.26%, H 5.30%, O 39.44%
Literature References: Naturally occurring flavonic glycoside; rhamnoglycoside of diosmetin, q.v. Isolation from various plant sources: O. A. Oesterle, G. Wander, Helv. Chim. Acta 8, 519 (1925). Elucidation of structure: G. Zemplén, R. Bognár, Ber. 76, 452 (1943). Prepn from hesperidin, q.v.: eidem, ibid.; N. B. Lorette et al., J. Org. Chem. 16, 930 (1951). Isoln from lemon peel (Citrus limon Linn. Rutaceae): R. M. Horowitz, J. Org. Chem. 21, 1184 (1956); from Zanthoxylum avicennae, Rutaceae: H. R. Arthur et al.,J. Chem. Soc. 1956, 632; H. R. Arthur et al., ibid. 1959, 4007; from flowers of Sophora microphylla Ait. Leguminosae: L. H. Briggs et al., ibid. 1960, 1955. Toxicology studies: H. Heusser, W. Osswald, Arch. Farmacol. Toxicol. 3, 33 (1977). NMR spectrum: J. L. Nieto, A. M. Gutierrez, Spectrosc. Lett. 19, 427 (1986). Mechanism of action: C. Boudet, L. Peyrin, Arch. Int. Pharmacodyn. 283,312 (1986). Pharmacology: J. R. Caseley-Smith, J. R. Caseley-Smith, Agents Actions 17, 1 (1985); M. Damon et al., Arzneim.-Forsch. 37, 1149 (1987). HPLC determn in biological fluids: D. Baylocq et al., Ann. Pharm. Fr. 41, 115 (1983). Clinical study in post-phlebitic ulcers: M. C. Nguyen, K. Morere, Gaz. Med. 92, 71 (1985); in acute hemorrhoids: A. Tajana et al., Minerva Med. 79,387 (1988). Clinical trial in chronic venous insufficiency: R. Laurent et al., Int. Angiol. 7, Suppl. 2, 39 (1988).
Derivative Type: Monohydrate
Molecular Formula: C28H32O15.H2O
Molecular Weight: 626.56
Percent Composition: C 53.67%, H 5.47%, O 40.86%
Properties: mp 275-277° (dec) (Zemplén). Also reported as fine needles from aq pyridine or aq DMF, mp 283° (dec) (Briggs). uv max (ethanol): 255, 268, 345 nm (log e 4.28, 4.25, 4.30). Practically insol in water, alcohol.
Melting point: mp 275-277° (dec) (Zemplén); mp 283° (dec) (Briggs)
Absorption maximum: uv max (ethanol): 255, 268, 345 nm (log e 4.28, 4.25, 4.30)
Derivative Type: Flavonoid extract
Trademarks: Daflon (Servier); Flebopex (Profarma); Flebotropin (Bago)
Therap-Cat: Capillary protectant.
Keywords: Vasoprotectant.
PAPER
Lee, Sanghyun; Natural Product Sciences 2002, VOL 8(4), P127-128
Siciliano, Tiziana; Journal of Agricultural and Food Chemistry 2004, VOL 52(21), P6510-6515
Yin, Feng; Zhongguo Tianran Yaowu 2004, VOL 2(3), P149-151
Markovic, D.; Farm. Glasnik 1949, VOL 5(No. 7;No. 8), P135-48;153-62
Nakaoki, Tahitiro; Yakugaku Zasshi 1938, VOL 58, P639-47(in German 197-201)
Wander, G.; Pharmaceutical Journal 1925, VOL 115, P520
Morita, Naokata; Yakugaku Zasshi 1967, VOL 87(3), P319-20
“PhysProp” data were obtained from Syracuse Research Corporation of Syracuse, New York (US)
Narasimhachari, N.; Proceedings – Indian Academy of Sciences, Section A 1949, VOL 30A, P151-62
Horowitz, Robert M.; Journal of Organic Chemistry 1956, VOL 21, P1184-5
paper
Spectroscopy Letters , An International Journal for Rapid Communication , Volume 19, 1986 – Issue 5, 1H NMR Spectra at 360 MHz of Diosmin and Hesperidin in DMSO Solution
Pages 427-434 | Received 03 Jan 1986, Accepted 03 Jan 1986, Published online: 06 Dec 2006
PAPER
Journal of Natural Products, 2013, vol. 76, 1, pg. 8 – 12
https://pubs.acs.org/doi/suppl/10.1021/np300460a/suppl_file/np300460a_si_001.pdf
1 H NMR, 13C NMR, HMQC and HMBC spectra of diosmin (5) ……………….S2



PAPER
Journal of Molecular Liquids, 2014, vol. 199, pg. 35 – 41

PATENT
https://patents.google.com/patent/CN102653549A/en
BRIEF DESCRIPTION
[0012] BRIEF I: iodine purification process Figure 2: Synthesis of diosmin roadmap
detailed description
[0013] Main reaction: The 80Kg Hesperidin, 40Kg soda ash, 400kg90% ethanol, 80L pyridine, 24kg iodine successively into reactor closed good pot opening, with stirring and heated to 110 ° c with a microwave, heat stirring, until the orange leather glycosides completely dissolved, about ten minutes. Hesperidin is completely dissolved, the solvent was slowly added to 80L of pyridine, combined with sodium iodide 8Kg, heated to 110 ° C, the reaction was stirred for 3-4 hr incubation, the sample is then detected by HPLC detection method, when a peak area less than hesperidin the reaction was terminated when 5% diosmin peak area, heat recovery of the solvent pyridine.
[0014] The filter press: End recovered 25Kg pyridine was added a paste of methanol, was press iodine recycling of waste, the recovery is completed, washed with purified water of 62 ° C, colorless and transparent until the washing water to the water 3 t, remove the filter cake to afford crude diosmin, 125. 4Kg.
[0015] Purification: 16Kg sodium hydroxide into dissolving tank, add purified water 500Kg, dissolved under stirring, and after dissolution the crude into the tank, and the water plus t I stir crystals were filtered into a stainless steel frame filter kettle, adding 42Kg hydrochloric acid, adjusted to PH 6.7, 25Kg of methanol was added, after stirring for 30min, the precipitate was allowed to stand, the I h.
[0016] Washing: The crystalline material tank into the filter press, ere washed with purified water, the washing water to colorless far, four tons of water, remove the filter cake to afford fine diosmin, 118. 5Kg. [0017] The dried, pulverized, mixed: semi-finished products into the oven dried 11.2 hours, 82 ° C temperature conditions, the dried material was crushed with a grinder, then put double cone blender and mixed overall speed 15r / min, each of the positive and negative inversion 20min, diosmin have finished 72Kg, a yield of 90.0%.
[0018] Packaging: for medical packaging with double polyethylene bags, into the drum after passing inspection, into finished products.
[0019] The recovery of iodine: iodine-containing filtrate generated pressure filtration step was slowly added sulfuric acid to adjust the PH 4, left for 5 hours, vacuum distillation, collecting high-boiling fraction, 20Kg hydrogen peroxide was slowly added, stand for 2 hours, filtered, the recovered iodine cloth, can be re-purified to obtain purified iodine! .
[0020] Processing pyridine in water: pyridine pyridine recovered after 400Kg containing moisture added to the kettle, 35Kg of potassium hydroxide was added, heated to 105 ± 5 ° C, collecting it pyridine (105 ° C before the liquid front , is defective, back again into the reaction vessel, then 105 ° C out is a good product), Hugh moisture meter by Karl Fischer detected, less than 2%.
PATENT
https://patents.google.com/patent/CN102875621A/en
diosmin chemical name is 3 ‘, 5,7-trihydroxy-4’ – methoxy flavone, i.e. (7 – {[6-0- (6-deoxy-mannose -a -L- sugar) _β -D- glucopyranosyl] oxy} -5_ hydroxy-2- (3-hydroxy-_4_ methoxyphenoxy) -4H-L–benzopyran-4-one), the following structure Figure:
[0003]

[0004] Diosmin has a comprehensive effect on vascular transfusion system to the venous system, micro-circulatory system and the lymphatic system has a powerful effect. Diosmin can be significantly reduced in addition to the adhesion of leukocytes to vascular endothelial cells, migration, inhibition of leukocyte disintegration and release of inflammatory mediators such as histamine, bradykinin, complement, leukotrienes, prostaglandins, free radical scavenging and the like, It may also reduce blood viscosity, to enhance flow of red blood cells, thus reducing the microcirculation stasis, mainly used in clinical treatment of chronic venous insufficiency.
[0005] diosmin content in natural plant is very low, direct extraction of high cost, so it is through the oxygen
Hesperidin is prepared by chemical synthesis; hesperidin formula below:
[0006]

[0007] diosmin synthesis process generally as follows:
[0008] hesperidin and an oxidant, and a solvent after mixing an alkaline reagent can be synthesized by heating the reaction Diosmin; wherein said oxidizing agent is iodine, mainly basic agent mainly inorganic bases, typically hydrogen sodium hydroxide, potassium hydroxide, sodium carbonate or potassium carbonate, an alkaline substance, etc., the solvent is pyridine or dimethylformamide.
[0009] Preparation of diosmin conventional synthesis method will inevitably pyridine or dimethylformamide as the reaction solvent, in particular in the main pyridine; as pyridine, dimethylformamide as a reaction solvent after the treatment process is not easy divisible, thus resulting in higher residual solvent in the product; the same time, since the two types of the organic solvent is pyridine, a large irritating odor on the human body have a greater toxic effects, and therefore in the production process and on the environment endanger personnel more apparent.
Example 8
[0081] In addition hesperidin 1000L reaction vessel 100 g, 47 g of sodium hydroxide and 12 g of iodine, and finally adding morpholine: water (60: 40) mixed solvent O. 8 liters, stir until completely dissolved. after heating to 85-90 ° C. was stirred incubated for 9 hours. the reaction liquid becomes viscous liquid was added 3 g of sodium thiosulfate, recovered at 85-90 ° C under vacuum conditions to a 70-80% morpholinyl, after complete recovery morpholine, O. 8 liters of water was added, stirred uniformly filtered to collect the waste. washed with water to give diosmin crude product. the crude product diosmin O. 8-liter and water was added 30 g of sodium hydroxide. stirred to dissolve completely after high-speed centrifuge filtration. into the crystallizer, water was added to the filtrate I. 5 liters of sulfuric acid was added slowly acidified to PH 2-3. standing, filtered and washed with water. diosmin give crude crystals. The crude crystals add water O. 8-liter and 30 g of sodium hydroxide and stirred to dissolve completely, placed in a crystallizer tank, to force saliva I. 5 liters of sulfuric acid was added slowly acidified to PH 2-3. Standing, was filtered, washed with water the drying, grinding to give the finished Diosmin 80. I g. Product purity by HPLC 95.26%, the yield was 80.1%, iodine residual, residual solvent, associated impurities, the content of all standards
PATENT
https://patents.google.com/patent/WO2010092592A2/en
Example – 1
100 gm of hesperidin , 700 ml of pyridine, 9.8 gm of sodium hydroxide and 45.6 gm of iodine were charged in 2 liter clean glass assembly, The resulting solution was heated to 95-1050C for 9 – 10 hours. Reaction was monitored by HPLC to get hesperidin less than 1 %. The pyridine was recovered completely by distillation. Charged methanol to the resulting solid, the reaction mass was heated to reflux and filtered at room temperature. Iodine was recovered from mother liquor, solid obtained was treated with sodium thiosulfate solution and 900 ml, 5% aqueous NaOH solution. pH 2-4 was adjusted with cone, sulfuric acid. Reaction mass was filtered to obtain crude diosmin. Yield : 80 – 86 gm. Recovery of iodine from above methanol mother liquor: Distilled methanol and pyridine mixture. The obtained residue was acidified with sulfuric acid. The resulting pH was less than 1. The brown precipitate formed was filtered. The resulting filtrate was oxidized with hydrogen peroxide at 0-10°C and filtered to obtain crude iodine having assay 50 – 60 %, which was steam distilled to obtain pure iodine with assay 95 %.
Example – 2
100 gm of crude diosmin as prepared in example 1 and 1800 ml of dimethylformamide was charged in 3 liter clean glass assembly. The resulting mass was heated to 90-950C to obtain clear solution. 200 ml of water was added at 90- 950C and maintained for 30 min. The reaction mass was cooled and filtered. The wet solid was collected.
Charged wet solid obtained in 3 liter clean glass assembly and charged 900 ml of water, 900 ml of 5% aqueous NaOH solution. Distilled out approximately 900 ml of water under vacuum below 5O0C. Charged 1000 ml of water and the resulting reaction mass was treated with charcoal and filtered through hyflow. pH 1.8-2.2 was adjusted using sulfuric acid. Stirred the mass for 30 min, filtered and washed it with water, hot water. Solid was dried. Yield : 80 – 85 gm. Assay : 99.9 %.
Example – 3
100 gm of crude diosmin as prepared in example 1, 1800 ml of dimethylformamide and 1800 ml of water was charged in 5 liter clean glass assembly. The resulting solution was heated to 90-950C to obtain slurry and maintained for 30 min. Cooled the reaction mass and filtered, washed with water and hot water. The obtained solid was dried. Yield: 90 – 95 gm. Assay : 97 %. Example – 4
100 gm of crude diosmin as prepared in example 1 and 1800 ml of dimethylformamide was charged in 3 liter clean glass assembly. The resulting solution was heated to 90-950C to obtain clear solution.charged 360 ml of water at 90-950C and maintained for 30 min. The reaction mass was filtered, washed with water and with hot water. Solid obtained was dried. Yield: 90 – 95 gm. Assay : 99.5 %.
Example – 5
100 gm of crude diosmin as prepared in example 1 and 1800 ml of dimethylformamide was charged in 3 liter clean glass assembly. The resulting solution was heated to 90-950C to obtain clear solution, charged 900 ml of water at 90-950C and maintained for 30 min. The reaction mass was filtered, washed with water and with hot water. Solid obtained was dried. Solid obtained was 90 — 95 gm. Assay obtained was 98.8 %.
Example – 6
100 gm of hesperidin , 700 ml of recovered pyridine, 9.8 gm of sodium hydroxide and 45.6 gm of iodine were charged in 2 liter clean glass assembly . The resulting solution was heated to 95-1050C for 9 – 10 hrs. Reaction was monitored by HPLC. Pyridine was recovered completely by distillation. Charged methanol to the resulting solid, the reaction mass was heated to reflux and filtered at room temperature. The solid obtained was treated with sodium thiosulfate solution and 900 ml, 5% aqueous NaOH solution. pH 2-4 was adjusted with cone, sulfuric acid. Reaction mass was filtered to obtain crude diosmin. Crude diosmin obtained was 80 – 86 gm. Purity was 98.6 %.
Example – 7
100 gm of hesperidin , 700 ml of recovered Pyridine, 9.8 gm of sodium hydroxide and 48 gm (assay 95 %) of recovered iodine were charged in 2 liter clean glass assembly.. The resulting solution was heated to 95-1050C for 9 – 10 hours. Reaction was monitored by HPLC to get hesperidin less than 1 %. The pyridine was recovered by distillation. Charged methanol to the resulting solid, the reaction mass was heated to reflux and filtered at room temperature. The solid obtained was treated with sodium thiosulfate solution and 900 ml, 5% aqueous NaOH solution. pH 2-4 was adjusted with cone, sulfuric acid. Reaction mass was filtered to obtain crude diosmin. Yield:80 – 86 gm. Purity : 95.3 %.
PATENT
Method for preparing diosminum comprising the steps of mixing amide solvent, hesperidin, alkaline reagent and iodine, and heating the reaction to obtain diosmin . Diosmin is a naturally occurring flavonoid glycoside that can be obtained from various plant sources. It is used in therapy due to its pharmacological activity as phlebotonic and vascular protecting agent, and useful for treating chronic venous insufficiency.
0047]
Example 1
1.1 Oxidation reaction: Open the vacuum pump, vacuum inhale 1500L of dimethylformamide into the reaction tank, and add sodium hydroxide to adjust the pH of the solvent between 6 and 7. Add 250.00kg of hesperidin and stir it while feeding. The material and the solvent are in full contact; 125 kg of iodine is added into the reaction tank at a constant rate for reaction; the temperature of the reaction tank is controlled between 70° C. and 100° C. for 14 hours.
1.2 Solvent recovery: After the reaction is complete, open the valve of the turnover tank and dehydration tank and close the return valve. Control the temperature of the material to start depressurizing the solvent at 90°C to 110°C. During the recovery process, attention should be paid to observe the recovery temperature and recovery conditions. When the material is dilute, stop the solvent recovery and enter the next process.
1.3 Crude product crystallization, filtration, washing: open the reaction tank vacuum valve, pump 1500L of purified water from the upper part of the reaction tank, start the mixer, stir for 10-20min, then add 1500L of purified water and stir for 1h; after the mixing is accepted, when the temperature of the crystallization liquid drops After 35°C, the crystallization liquid is pumped into the plate and frame filter press for filtration, and the filtrate is temporarily stored in the storage tank; after the filtered plate nozzle no liquid flows out, the 18000L purified water pump is pumped into the frame filter press for washing; The water wash is discharged into a waterless collection tank for sewage treatment, and the water is washed until the pH of the effluent is measured with a pH test paper of 6 to 7. The beaker sample is observed to be colorless and transparent; after the washing is completed, the water inlet valve is closed and the air pressure is turned on. The valve is air-pressed, the air pressure is controlled at 0.07~0.09 MPa, and the time is maintained for 3 hours. The filter cake is collected and put into the turnover barrel for marking.
[0051]
1.4 Secondary Dissolution and Filtration: Open the dissolving tank and stir. In the dissolving tank, add 75 kg of alkali A to 1300 L of purified water. After the solution is completely dissolved, add the filter cake in the circulating drum to the tank; after the filter cake is added and stirring is continued for 1 h, Add 1300L of purified water into the tank, stir for 30 minutes, and stand for 3 hours. Filter the filtrate with a frame filter press. The filtered solution is finely filtered by a fine filter and then pumped into a clean area crystallizer.
[0052]
1.5 secondary crystallization: open the stirrer, slowly put 160 ~ 230kg 36% hydrochloric acid into the clean area crystallization tank, the measured pH of the solution is between 5.0 ~ 6.0, after stirring 20min measured the pH of the solution should be stable and qualified , Stir 1h, make the original record of the process.
1.6 Fine filtration and washing: The crystallization liquid is vacuum-inhaled into the box type filter press for filtration; after the filtration is completed, the washed water is washed with 18,000 to 20,000 L of purified water. After the washing is completed, it is checked that the washing liquid should be colorless and transparent, and the filtrate should be filtrated. Discharge into the sewage treatment system.
1.7 Fine Drying: Open the hot air circulation oven, control the temperature at 100 °C ~ 130 °C, drying time is maintained at 10 ~ 16h; when dried 10h, timely sampling, with a quick moisture meter to determine the moisture, when the sample moisture is less than 5%, Stop drying; after passing the drying, close the oven and transport the material to the next process.
1.8 Fine-grinding: The dried product is crushed with a crusher. The crushing sieve is 80 mesh to obtain Diosmin.
The quality standards of all raw materials in Example 1 are shown in Table 1.
Table 1 Raw material quality standards
[Table 0001]
| Original accessories name | specification | Quality Standard |
| Hesperidin | EC | In line with “Hesperidin Quality Standard” |
| New solvent | Industrial grade | Meet the “new solvent quality standards” |
| Alkali A | Industrial grade | In accordance with the “Alkaline A Quality Standard” |
| iodine | Pharmaceutical grade | In line with “Iodine Quality Standards” |
| hydrochloric acid | Analytical purity | In line with the “Hydrochloric Acid Quality Standard” |
Diosmin Yield Calculation Method:
Diosmin product yield = (Diosmin Fine Quality) / (Hesperidin weight) × 100%
References
- Jump up^ Flavonoid Aglycones and Glycosides from Teucrium gnaphalodes. F. A. T. Barberán, M. I. Gil, F. Tomás, F. Ferreres and A. Arques, J. Nat. Prod., 1985, 48 (5), pages 859–860, doi:10.1021/np50041a040
- Jump up^ Jantet, G. (2002-06-01). “Chronic venous insufficiency: worldwide results of the RELIEF study. Reflux assEssment and quaLity of lIfe improvEment with micronized Flavonoids”. Angiology. 53(3): 245–256. ISSN 0003-3197. PMID 12025911.
- Jump up^ Leelavinothan Pari, Subramani Srinivasan, Antihyperglycemic effect of diosmin on hepatic key enzymes of carbohydrate metabolism in streptozotocin-nicotinamide-induced diabetic rats, Biomedicine & Pharmacotherapy, Volume 64, Issue 7, September 2010, Pages 477-481.
- Jump up^ Sirlak, Mustafa; Akar, A. Ruchan; Eryilmaz, Sadik; Cetinkanat, Elif Kuzgun; Ozcinar, Evren; Kaya, Bulent; Elhan, Atilla Halil; Ozyurda, Umit (2010-01-01). “Micronized purified flavonoid fraction in pretreating CABG patients”. Texas Heart Institute Journal. 37 (2): 172–177. ISSN 1526-6702. PMC 2851420
 . PMID 20401289.
. PMID 20401289. - Jump up^ “Nutratech dietary supplement notification” (PDF). FDA. November 3, 2000. Archived from the original (PDF) on December 9, 2006.
- Jump up^ “Stragen Pharma dietary supplement notification” (PDF). FDA. September 6, 2005.
Patent
Publication numberPriority datePublication dateAssigneeTitle
EP0860443A1 *1997-02-211998-08-26Innokem, SARLIndustrial process for the production of diosmine starting from hesperidine
WO2000011009A2 *1998-08-192000-03-02Innokem, S.A.R.L.Method for industrial production of diosmin from hesperidin by reaction with iodine and pyridine
Publication numberPriority datePublication dateAssigneeTitle
CN102653549A *2011-12-282012-09-05长沙富能生物技术有限公司Synthesis method of diosmin raw medicine meeting EP7 version quality standards
RU2481353C1 *2011-12-222013-05-10Закрытое акционерное общество “Активный Компонент”Commercial method for preparing officinal diosmin and crystalline form thereof (versions)
CN103772336A *2014-02-232014-05-07闻永举Semi-synthesis method of phenolic hydroxyl flavonoid compounds and iodine recycling method
Publication numberPriority datePublication dateAssigneeTitle
WO2010092592A2 *2009-02-112010-08-19Elder Pharmaceuticals Ltd.Process for the preparation of diosmin
CN102653549A *2011-12-282012-09-05长沙富能生物技术有限公司Synthesis method of diosmin raw medicine meeting EP7 version quality standards
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
oral |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.007.537 |
| Chemical and physical data | |
| Formula | C28H32O15 |
| Molar mass | 608.545 g/mol |
| 3D model (JSmol) | |
//////////DIOSMIN, диосмин , ديوسمين , 地奥司明 , Barosmin, Diosmine, Venosmine, Diosmil,
COC1=C(O)C=C(C=C1)C1=CC(=O)C2=C(O)C=C(O[C@@H]3O[C@H](CO[C@@H]4O[C@@H](C)[C@H](O)[C@@H](O)[C@H]4O)[C@@H](O)[C@H](O)[C@H]3O)C=C2O1
Entecavir, энтекавир , إينتيكافير , 恩替卡韦 , エンテカビル
Entecavir
- Molecular FormulaC12H15N5O3
- Average mass277.279 Da
NNU2O4609D
QA-0464
SQ 34,676
SQ34676
Teviral
UNII:NNU2O4609D
Entecavir; 142217-69-4; Baraclude; BMS 200475; Anhydrous entecavir; UNII-NNU2O4609D
энтекавир [Russian] [INN]
إينتيكافير [Arabic] [INN]
恩替卡韦 [Chinese] [INN]
エンテカビル JAPANESE
2-amino-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylidenecyclopentyl]-9H-purin-6-ol
6H-Purin-6-one, 2-amino-1,9-dihydro-9-((1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl)-
6H-Purin-6-one, 2-amino-1,9-dihydro-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-
9H-purin-6-ol, 2-amino-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-
Baraclude (Entecavir) Film Coated Tablets & Oral Solution
Company: Bristol-Myers Squibb Pharmaceutical Co.
Application No.: 021797 & 021798
Approval Date: 03/29/2005
Chemistry Review(s) (PDF)
BARACLUDE® is the tradename for entecavir, a guanosine nucleoside analogue with selective activity against HBV. The chemical name for entecavir is 2-amino-1,9-dihydro-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purin-6-one, monohydrate. Its molecular formula is C12H15N5O3•H2O, which corresponds to a molecular weight of 295.3. Entecavir has the following structural formula:
 |
Entecavir is a white to off-white powder. It is slightly soluble in water (2.4 mg/mL), and the pH of the saturated solution in water is 7.9 at 25° C ± 0.5° C.
BARACLUDE film-coated tablets are available for oral administration in strengths of 0.5 mg and 1 mg of entecavir. BARACLUDE 0.5 mg and 1 mg film-coated tablets contain the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, crospovidone, povidone, and magnesium stearate. The tablet coating contains titanium dioxide, hypromellose, polyethylene glycol 400, polysorbate 80 (0.5 mg tablet only), and iron oxide red (1 mg tablet only). BARACLUDE Oral Solution is available for oral administration as a ready-to-use solution containing 0.05 mg of entecavir per milliliter. BARACLUDE Oral Solution contains the following inactive ingredients: maltitol, sodium citrate, citric acid, methylparaben, propylparaben, and orange flavor.

Title: Entecavir
CAS Registry Number: 142217-69-4
CAS Name: 2-Amino-1,9-dihydro-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purin-6-one
Molecular Formula: C12H15N5O3
Molecular Weight: 277.28
Percent Composition: C 51.98%, H 5.45%, N 25.26%, O 17.31%
Literature References: Deoxyguanine nucleoside analog; inhibits hepatitis B virus (HBV) DNA polymerase. Prepn: R. Zahler, W. A. Slusarchyk, EP481754; eidem,US5206244 (1992, 1993 both to Squibb); G. S. Bisacchi et al.,Bioorg. Med. Chem. Lett.7, 127 (1997). In vitro antiviral activity: S. F. Innaimo et al,Antimicrob. Agents Chemother.41, 1444 (1997). Review of pharmacology and clinical experience: P. Honkoop, R. A. de Man, Expert Opin. Invest. Drugs12, 683-688 (2003); T. Shaw, S. Locarnini, Expert Rev. Anti Infect. Ther.2, 853-871 (2004). Clinical comparisons with lamivudine in chronic hepatitis B: T.-T. Chang et al., N. Engl. J. Med.354, 1001 (2006); C.-L. Lai et al., ibid. 1011.
Derivative Type: Monohydrate
CAS Registry Number: 209216-23-9
Manufacturers’ Codes: BMS-200475; SQ-200475
Trademarks: Baraclude (BMS)
Molecular Formula: C12H15N5O3.H2O
Molecular Weight: 295.29
Percent Composition: C 48.81%, H 5.80%, N 23.72%, O 21.67%
Properties: White to off-white powder, mp >220°. [a]D +35.0° (c = 0.38 in water). Soly in water: 2.4 mg/ml. pH of saturated soln in water is 7.9 at 25°±0.5°.
Melting point: mp >220°
Optical Rotation: [a]D +35.0° (c = 0.38 in water)
Therap-Cat: Antiviral.
Keywords: Antiviral; Purines/Pyrimidinones.

Antiviral agents used against HBV
Entecavir is an oral antiviral drug used in the treatment of hepatitis B infection. It is marketed under the trade name Baraclude (BMS).
Entecavir is a guanine analogue that inhibits all three steps in the viral replication process, and the manufacturer claims that it is more efficacious than previous agents used to treat hepatitis B (lamivudine and adefovir). It was approved by the U.S. Food and Drug Administration (FDA) in March 2005.
For the treatment of chronic hepatitis B virus infection in adults with evidence of active viral replication and either evidence of persistent elevations in serum aminotransferases (ALT or AST) or histologically active disease.
Entecavir (ETV), sold under the brand name Baraclude, is an antiviral medication used in the treatment of hepatitis B virus (HBV) infection.[1] In those with both HIV/AIDS and HBV antiretroviral medication should also be used.[1] Entecavir is taken by mouth as a tablet or solution.[1]
Common side effects include headache, nausea, high blood sugar, and decreased kidney function.[1] Severe side effects include enlargement of the liver, high blood lactate levels, and liver inflammation if the medication is stopped.[1] While there appears to be no harm from use during pregnancy, this use has not been well studied.[4] Entecavir is in the nucleoside reverse transcriptase inhibitors(NRTIs) family of medications.[1] It prevents the hepatitis B virus from multiplying by blocking reverse transcriptase.[1]
Entecavir was approved for medical use in 2005.[1] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[5] In the United States as of 2015 it is not available as a generic medication.[6]The wholesale price is about 392 USD for a typical month supply as of 2016 in the United States.[7]
Medical uses
Entecavir is mainly used to treat chronic hepatitis B infection in adults and children 2 years and older with active viral replication and evidence of active disease with elevations in liver enzymes.[2] It is also used to prevent HBV reinfection after liver transplant[8] and to treat HIV patients infected with HBV. Entecavir is weakly active against HIV, but is not recommended for use in HIV-HBV co-infected patients without a fully suppressive anti-HIV regimen[9] as it may select for resistance to lamivudine and emtricitabine in HIV.[10]
The efficacy of entecavir has been studied in several randomized, double-blind, multicentre trials. Entecavir by mouth is effective and generally well tolerated treatment.[11]
Pregnancy and breastfeeding
It is considered pregnancy category C in the United States, and currently no adequate and well-controlled studies exist in pregnant women.[12]
Side effects
The majority of people who use entecavir have little to no side effects.[13] The most common side effects include headache, fatigue, dizziness, and nausea.[2] Less common effects include trouble sleeping and gastrointestinal symptoms such as sour stomach, diarrhea, and vomiting.[14]
Serious side effects from entecavir include lactic acidosis, liver problems, liver enlargement, and fat in the liver.[15]
Laboratory tests may show an increase in alanine transaminase (ALT), hematuria, glycosuria, and an increase in lipase.[16] Periodic monitoring of hepatic function and hematology are recommended.[2]
Mechanism of action
Entecavir is a nucleoside analog,[17] or more specifically, a deoxyguanosine analogue that belongs to a class of carbocyclic nucleosidesand inhibits reverse transcription, DNA replication and transcription in the viral replication process. Other nucleoside and nucleotide analogues include lamivudine, telbivudine, adefovir dipivoxil, and tenofovir.
Entecavir reduces the amount of HBV in the blood by reducing its ability to multiply and infect new cells.[18]
Administration
Entecavir is take by mouth as a tablet or solution. Doses are based on a person’s weight.[15] The solution is recommended for children more than 2 years old who weigh up to 30 kg. Entecavir is recommended on an empty stomach at least 2 hours before or after a meal, generally at the same time every day. It is not used in children less than 2 years old. Dose adjustments are also recommended for people with decreased kidney function.[15]
History
- 1992: SQ-34676 at Squibb as part of anti-herpes virus program[19]
- 1997: BMS 200475 developed at BMS pharmaceutical research institute as antiviral nucleoside analogue à Activity demonstrated against HBV, HSV-1, HCMV, VZV in cell lines & no or little activity against HIV or influenza[20]
- Superior activity observed against HBV pushed research towards BMS 200475, its base analogues and its enantiomer against HBV in HepG2.2.15 cell line[20]
- Comparison to other NAs, proven more selective potent inhibitor of HBV by virtue of being Guanine NA[21]
- 1998: Inhibition of hepadnaviral polymerases was demonstrated in vitro in comparison to a number of NAs-TP[22]
- Metabolic studies showed more efficient phosphorylation to triphosphate active form[23]
- 3-year treatment of woodchuck model of CHB à sustained antiviral efficacy and prolonged life spans without detectable emergence of resistance[24]
- Efficacy # LVD resistant HBV replication in vitro[25]
- Superior activity compared to LVD in vivo for both HBeAg+ & HBeAg− patients[26][27]
- Efficacy in LVD refractory CHB patients[28]
- Entecavir was approved by the U.S. FDA in March 2005.
Patent information
Bristol-Myers Squibb was the original patent holder for Baraclude, the brand name of entecavir in the US and Canada. The drug patent expiration for Baraclude was in 2015.[29][30]On August 26, 2014, Teva Pharmaceuticals USA gained FDA approval for generic equivalents of Baraclude 0.5 mg and 1 mg tablets;[31] Hetero Labs received such approval on August 21, 2015;[32] and Aurobindo Pharma on August 26, 2015.[33]
Chronic hepatitis B virus infection is one of the most severe liver diseases in morbidity and death rate in the worldwide range. At present, pharmaceuticals for treating chronic hepatitis B (CHB) virus infection are classified to interferon α and nucleoside/nucleotide analogue, i.e. Lamivudine and Adefovir. However, these pharmaceuticals can not meet needs for doctors and patients in treating chronic hepatitis B virus infection because of their respective limitation. Entecavir (ETV) is referred to as 2′-cyclopentyl deoxyguanosine (BMS2000475) which belongs to analogues of Guanine nucleotide and is phosphorylated to form an active triple phosphate in vivo. The triple phosphate of entecavir inhibits HBV polymerase by competition with 2′-deoxyguanosine-5′-triphosphate as a nature substrate of HBV polymerase, so as to achieve the purpose of effectively treating chronic hepatitis B virus infection and have strong anti-HBV effects. Entecavir, [1S-(1α,3α,4β)]-2-amino-1,9-dihydro-9-[4-hydroxy-3-hydroxymethyl]-2-methylenecyclopentyl]-6H-purin-6-one, monohydrate, and has the molecular formula of C12H15N5O3.H2O and the molecular weight of 295.3. Its structural formula is as follows:

Entecavir was successfully developed by Bristol-Myers Squibb Co. of USA first and the trademark of the product formulation is Baraclude™, including two types of formulations of tablet and oral solution having 0.5 mg and 1 mg of dosage. Chinese publication No. CN1310999 made by COLONNO, Richard, J. et al discloses a low amount of entecavir and uses of the composition containing entecavir in combination with other pharmaceutically active substances for treating hepatitis B virus infection, however, the entecavir is non-crystal. In addition, its oral formulations such as tablet and capsule are made by a boiling granulating process. The process is too complicated to control quality of products during humidity heat treatment even though ensuring uniform distribution of the active ingredients.
Entecavir, [1-S-(1α,3α,4β)]-2-amino-1,9-dihydro-9-[4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purin-6-one, is currently used for treating hepatitis B virus infection, whose structure is composed of a cyclopentane ring having purine, exomethylene, hydroxymethyl, and hydroxy substituents at the 1S-, 2-, 3R-, and 4S-positions, respectively. There have been conducted a number of studies to develop methods for preparing entecavir.
For example, U.S. Pat. No. 5,206,244 and WO 98/09964 disclose a method for preparing entecavir shown in Reaction Scheme 1:
The above method, however, has difficulties in that: i) the cyclopentadiene monomer must be maintained at a temperature lower than -30 °C in order to prevent its conversion to dicyclopentadiene; ii) residual sodium after the reaction as well as the sensitivity of the reaction toward moisture cause problems; iii) the process to obtain the intermediate of formula a) must be carried out at an extremely low temperature of below -70 °C in order to prevent the generation of isomers; iv) a decantation method is required when (-)-Ipc2BH (diisopinocampheylborane) is used for hydroboration; v) the process of the intermediate of formula a) does not proceed smoothly; and, vi) separation by column chromatography using CHP-20P resin is required to purify entecavir.
WO 2004/52310 and U.S. Pat. Publication No. 2005/0272932 disclose a method for preparing entecavir using the intermediate of formula (66), which is prepared as shown in Reaction Scheme 2:

The above preparation method of the intermediate of formula (66) must be carried out at an extremely low temperature of -70 °C or less, and the yield of the desired product in the optical resolution step is less than 50%.
PATENT
https://patents.google.com/patent/EP2382217B1

(3-4) Preparation of [1-S-(1α,3α,4β)]-2-amino-1,9-dihydro-9-[4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purine-6-one (a compound of formula (1))
34 mg (0.115 mmol) of 4-(2-amino-6-chloro-purine-9-yl)-2-hydroxymethyl-3-methylene-cyclopentanol (a compound of formula (5)) obtained in (3-3) was added to 0.7 ml of 2N aqueous sodium hydroxide, and the resulting mixture was stirred. The solution thus obtained was heated to 72 °C and stirred for 3.5 hrs. After completion of the reaction, the resulting mixture was cooled to 0 °C, controlled to pH 6.3 by adding 2N aqueous hydrochloric acid and 1N aqueous hydrochloric acid, and condensed to obtain 24 mg of the title compound (yield: 70 %, purity: 99 %).
NMR(300MHz, DMSO-d6): δ 10.58 (s, 1H), 7.67 (s, 1H), 6.42 (s, 2H), 5.36 (t, 1H), 5.11 (s, 1H), 4.86 (d, 1H), 4.83 (t, 1H), 4.57 (s, 1H), 4.24 (s, 1H), 3.54 (t, 2H), 2.53(s, 1H), 2.27-2.18 (m, 1H), 2.08-2.01(m, 1H).
PAPER
https://www.sciencedirect.com/science/article/pii/S0040403911020144



PAPER
https://www.sciencedirect.com/science/article/pii/S0040402017313029


PAPER


Total Synthesis of Entecavir: A Robust Route for Pilot Production
Launch-Pharma Technologies, Ltd., 188 Kaiyuan Boulevard, Building D, Fifth Floor, The Science Park of Guangzhou, Guangzhou 510530, China
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.8b00007
Publication Date (Web): February 12, 2018
Copyright © 2018 American Chemical Society
*E-mail: jinyehua@launch-pharma.com., *E-mail: qiufayang@launch-pharma.com.

A practical synthetic route for pilot production of entecavir is described. It is safe, robust, and scalable to kilogram scale. Starting from (S)-(+)-carvone, this synthetic route consists of a series of highly efficient reactions including a Favorskii rearrangement-elimination-epimerization sequence to establish the cyclopentene skeleton, the Baeyer–Villiger oxidation/rearrangement to afford the correct configuration of the secondary alcohol, and a directed homoallylic epoxidation followed by epoxide ring-opening to introduce the hydroxyl group suitable for the Mitsunobu reaction. In addition, the synthesis contains only four brief chromatographic purifications.
1: white crystalline solid; HRMS (m/z) calcd for C12H16N5O3 [M + H]+ 278.1253, found 278.1255; [α]D +27.2° [c 1.07, DMF/H2O (1:1)];
1H NMR (500 MHz, DMSO) δ 10.55 (s, 1H), 7.65 (s, 1H), 6.40 (s, 2H), 5.36 (dd, J = 10.3, 8.0 Hz, 1H), 5.10 (s, 1H), 4.85 (d, J = 3.1 Hz, 1H), 4.81 (t, J = 5.3 Hz, 1H), 4.56 (s, 1H), 4.23 (s, 1H), 3.54 (t, J = 6.1 Hz, 2H), 2.55–2.50 (m, 1H), 2.26–2.17 (m, 1H), 2.04 (dd, J = 12.5, 7.8 Hz, 1H);
13C NMR (126 MHz, DMSO) δ 156.8, 153.4, 151.4, 151.3, 135.9, 116.2, 109.2, 70.4, 63.0, 55.1, 54.1, 39.2.
Clips
EP 0481754; JP 1992282373; US 5206244, WO 9809964

The regioselective reaction of cyclopentadiene (I) and sodium or commercial sodium cyclopentadienide (II) with benzyl chloromethyl ether (III) by means of the chiral catalyst (-)-diisopinocampheylborane in THF, followed by hydroxylation with H2O2/NaOH, gives (1S-trans)-2-(benzyloxymethyl)-3-cyclopenten-1-ol (IV), which is regioselectively epoxidized with tert-butyl hydroperoxide and vanadyl acetylacetonate in 2,2,4-trimethylpentane, yielding [1S-(1alpha,2alpha,3beta,5alpha)-2-(benzyloxymethyl)-6-oxabicyclo[3.1.0]hexan-3-ol (V). The protection of (V) with benzyl bromide and NaH affords the corresponding ether (VI), which is condensed with 6-O-benzylguanine (VII) by means of LiH in DMF to give the guanine derivative (VIII). The protection of the amino group of (VIII) with 4-methoxyphenyl(diphenyl)chloromethane (IX), TEA and DMAP in dichloromethane gives intermediate (X), which is oxidized at the free hydroxyl group with methylphosphonic acid, DCC and oxalic acid in DMSO or Dess Martin periodinane in dichloromethane, yielding the cyclopentanone derivative (XI). The reaction of (XI) with (i) Zn/TiCl4/CH2Br2 complex in THF/CH2Cl2, (ii) activated Zn/PbCl2/CH2I2/TiCl4 in THF/CH2Cl2 (2), (iii) Nysted reagent/TiCl4 in THF/CH2Cl2 or (iv) Tebbe reagent in toluene affords the corresponding methylene derivative (XII), which is partially deprotected with 3N HCl in hot THF, providing the dibenzylated compound (XI). Finally, this compound is treated with BCl3 in dichloromethane
PAPER
Bioorg Med Chem Lett 1997,7(2),127
BMS-200475, a novel carbocyclic 2′-deoxyguanosine analog with potent and selective anti-hepatitis B virus activity in vitro
BMS-200475, a novel carbocyclic analog of 2′-deoxyguanosine, is a potent inhibitor of hepatitis B virus in vitro (ED50 = 3 nM) with relatively low cytotoxicity (CC50 = 21–120 μM). A practical 10-step asymmetric synthesis was developed affording BMS-200475 in 18% overall chemical yield and >99% optical purity. The enantiomer of BMS-200475 as well as the adenine, thymine, and iodouracil analogs are much less active.
BMS-200475, a novel carbocyclic analog of 2′-deoxyguanosine, is a potent inhibitor of hepatitis B virus in vitro (ED50 = 3nM) with relatively low cytotoxicity (CC50 = 21–120 μM).

PATENT
https://patents.google.com/patent/US20140220120
Fourier transform infrared (FTIR) spectrogram: The range of wave numbers is measured by using the Nicolet NEXUS 670 FT-IR spectrometer with KBr pellet method, and the range of wave numbers is about 400 to 4000 cm−1. FIG. 3 is a Fourier transform infrared spectrogram of the sample. The infrared spectrogram shows that there are groups in the molecular structure of the sample, such as NH, NH2, HN—C═O, C═C, OH.

PAPER
Total Synthesis of Entecavir
† Departament de Química Orgànica and Institut de Biomedicina de la Universitat de Barcelona (IBUB), Facultat de Química, Universitat de Barcelona, Martí i Franquès 1, 08028-Barcelona, Spain
‡ R&D Department, Esteve Química S.A., Caracas 17-19, 08030-Barcelona, Spain
§ CIBER Fisiopatología de la Obesidad y la Nutrición (CIBERobn), Instituto de Salud Carlos III, Madrid, Spain
J. Org. Chem., 2013, 78 (11), pp 5482–5491
DOI: 10.1021/jo400607v

Entecavir (BMS-200475) was synthesized from 4-trimethylsilyl-3-butyn-2-one and acrolein. The key features of its preparation are: (i) a stereoselective boron–aldol reaction to afford the acyclic carbon skeleton of the methylenecylopentane moiety; (ii) its cyclization by a Cp2TiCl-catalyzed intramolecular radical addition of an epoxide to an alkyne; and (iii) the coupling with a purine derivative by a Mitsunobu reaction.
2-Amino-9-((1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl)-1H-purin-6(9H)-one Monohydrate (1)
1 (2.102 g, 64% overall yield, 99.47% HPLC purity) with a 6.7% water content (as determined by Karl Fischer titration). Mp 248 °C. [α]D25 +35.0 (c 0.4, H2O). IR (ATR): 3445, 3361, 3296, 3175, 3113, 2951, 2858, 2626, 1709 cm–1.
1H NMR (DMSO-d6, 400 MHz) δ: 10.59 (s, 1H), 7.66 (s, 1H), 6.42 (bs, 2H), 5.36 (ddt, J = 10.6, 7.8, 2.7 Hz, 1H), 5.10 (dd, J = 2.7, 2.2 Hz, 1H), 4.87 (d, J = 3.1 Hz, 1H), 4.84 (t, J = 5.3 Hz, 1H), 4.56 (t, J = 2.4 Hz, 1H), 4.23 (m, 1H), 3.53 (m, 2H), 2.52 (m, 1H), 2.22 (ddd, J = 12.6, 10.8, 4.6 Hz, 1H), 2.04 (ddt, J = 12.6, 7.7, 1.9 Hz, 1H).
13C NMR (DMSO-d6, 101 MHz) δ: 156.9, 153.5, 151.5, 151.3, 136.0, 116.2, 109.3, 70.4, 63.1, 55.2, 54.1, 39.2. HRMS (ESI): m/z calcd for C12H16N5O3+ [M + H]+ 278.1253; found 278.1262.
PATENTS
JP5788398B2 *2009-10-122015-09-30ハンミ・サイエンス・カンパニー・リミテッドNovel production methods and intermediates used to entecavir
US8481728B22010-02-162013-07-09Scinopharm Taiwan, Ltd.Process for preparing entecavir and its intermediates
CA2705953A1 *2010-05-312011-11-30Alphora Research Inc.Carbanucleoside synthesis and intermediate compounds useful therein
CN106928227A2010-07-152017-07-07浙江奥翔药业股份有限公司Synthetic method of entecavir and intermediate compound thereof
EP2474548A12010-12-232012-07-11Esteve Química, S.A.Preparation process of an antiviral drug and intermediates thereof
EP2597096A12011-11-242013-05-29Esteve Química, S.A.Process for preparing entecavir and intermediates thereof
WO2014175974A32013-03-132015-04-09Elitech Holding B.V.Artificial nucleic acids derived from 2-((nucleobase)methyl)butane-1,3-diol
WO2015051900A12013-10-082015-04-16Pharmathen S.A.Process for the preparation of entecavir through novel intermediates
WO2015051903A1 *2013-10-082015-04-16Pharmathen S.A.A novel process for the preparation of chiral cyclopentanone intermediates
CN103675185B *2013-12-102015-10-07上海景峰制药股份有限公司The method of one kind TU entecavir tablet all-trans isomer by high performance liquid chromatography
KR101647061B12014-04-022016-08-10서강대학교산학협력단Path Generation Method and apparatus for Unmanned Autonomous Vehicle
CN105037363B *2015-07-132016-08-24山东罗欣药业集团股份有限公司En one kind of new synthetic method of compound entecavir
* Cited by examiner, † Cited by third party, ‡ Family to family citation
PublicationPublication DateTitle
EP0391652A11990-10-10Interphenylène 7-oxabicycloheptyl substituted heterocyclic amide prostaglandin analogs useful in the treatment of thrombotic and vasopastic disease
EP1626045A12006-02-15Processes for producing 3-substituted 2-chloro-5-fluoropyridine or salt thereof
US20060211855A12006-09-21Method for the production of oh protected{4-(2.6-diamino-9h-purine-9-yl)-1.3-dioxolane-2-yl] methanol derivatives
EP2186815A12010-05-19Method for producing pyripyropene derivative and production intermediate thereof
Oikawa et al.1986Highly stereoselective synthesis of methynolide, the aglycone of the 12-membered ring macrolide methymycin, from D-glucose
Just et al.1980C-Nucleosides and related compounds. XV. The synthesis of d, l-2′-epi-showdomycin and d, l-showdomycin
EP0277599A21988-08-10Fluorine containing cyclopentane derivatives and processes for their production
Tulshian et al.1984Out-of-ring Claisen rearrangements are highly stereoselective in pyranoses: routes to gem-dialkylated sugars
Corey et al.1984Methods for the interconversion of protective groups. Transformation of mem ethers into isopropylthiomethyl ethers or cyanomethyl ethers
WO2009125841A12009-10-15Process for production of ethynylthymidine compound using 5-methyluridine as starting raw material
ApplicationPriority dateFiling dateTitle
PCT/KR2009/0077862008-12-262009-12-24Novel intermediate and process for preparing entecavir using same
References
- “Entecavir”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 28 November 2016.
- “Baraclude (entecavir) Tablets for Oral Use & Oral Solution. U.S. Full Prescribing Information. Archived 2014-02-22 at the Wayback Machine.” Bristol-Myers Squibb Company, 2005. Revised December 2013.
- The Merck Index (14th ed.). 2006. p. 613. ISBN 978-0-911910-00-1.
- “Entecavir (Baraclude) Use During Pregnancy”. http://www.drugs.com. Archived from the original on 7 November 2016. Retrieved 6 December 2016.
- “WHO Model List of Essential Medicines (19th List)” (PDF). World Health Organization. April 2015. Archived (PDF) from the original on 13 December 2016. Retrieved 8 December 2016.
- Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 76. ISBN 9781284057560.
- “NADAC as of 2016-11-30 | Data.Medicaid.gov”. Centers for Medicare and Medicaid Services. Archived from the original on 30 November 2016. Retrieved 6 December 2016.
- Fung, J; Cheung, C; Chan, SC; et al. (2011). “Entecavir Monotherapy is Effective in Suppressing Hepatitis B Virus After Liver Transplantation”. Gastroenterology. 141 (4): 1212–9. doi:10.1053/j.gastro.2011.06.083. PMID 21762659.
- “Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents” (PDF). Panel on Antiretroviral Guidelines for Adults and Adolescents. Archived (PDF) from the original on 1 November 2016. Retrieved 15 March 2015.
- McMahon, Moira (21 June 2007). “The Anti-Hepatitis B Drug Entecavir Inhibits HIV-1 Replication and Can Select HIV-1 Variants Resistant to Antiretroviral Drugs”. N Engl J Med. 356 (25): 2614–2621. doi:10.1056/NEJMoa067710. PMC 3069686 . PMID 17582071. Archived from the original on 2 April 2015. Retrieved 15 March 2015.
- Scott, LJ; Keating, GM (2009). “Entecavir”. Drugs. 69 (8): 1003–1033. doi:10.2165/00003495-200969080-00005. Archived from the original on 2011-10-08.
- Jump up^ “Entecavir (Baraclude) Use During Pregnancy”. http://www.drugs.com. Archived from the original on 2016-11-07. Retrieved 2016-11-07.
- Jump up^ “Entecavir: Indications, Side Effects, Warnings – Drugs.com”. http://www.drugs.com. Archived from the original on 2016-11-07. Retrieved 2016-11-10.
- Jump up^ “Entecavir Side Effects in Detail – Drugs.com”. http://www.drugs.com. Archived from the original on 2016-11-10. Retrieved 2016-11-10.
- ^ Jump up to:a b c “DailyMed – BARACLUDE- entecavir tablet, film coated BARACLUDE- entecavir solution”. dailymed.nlm.nih.gov. Archived from the original on 2016-11-08. Retrieved 2016-11-09.
- Jump up^ “DailyMed – BARACLUDE- entecavir tablet, film coated BARACLUDE- entecavir solution”. dailymed.nlm.nih.gov. Archived from the original on 2016-11-09. Retrieved 2016-11-10.
- Jump up^ Sims KA, Woodland AM (December 2006). “Entecavir: a new nucleoside analog for the treatment of chronic hepatitis B infection”. Pharmacotherapy. 26 (12): 1745–57. doi:10.1592/phco.26.12.1745. PMID 17125436.[permanent dead link]

- Jump up^ “Entecavir: Indications, Side Effects, Warnings – Drugs.com”. http://www.drugs.com. Archived from the original on 2016-11-07. Retrieved 2016-11-07.
- Jump up^ Slusarchyk, W. A., A. K. Field, J. A. Greytok, P. Taunk, A. V. Tooumari, M. G. Young, and R. Zahler. 4-Hydroxy-3-(hydroxymethyl)-2-methylcyclopentyl purines and pyrimidines, a novel class of anti-herpesvirus agents. Abstract from the Fifth International Conference on Antiviral Research. Antivir Res 1992.17(Suppl. 1):98
- ^ Jump up to:a b Bisacchi, G. S.; Chao, S. T.; Bachard, C.; Daris, J. P.; Innaimo, S. F.; Jacobs, J. A.; Kocy, O.; Lapointe, P.; Martel, A.; Merchant, Z.; Slusarchyk, W. A.; Sundeen, J. E.; Young, M. G.; Colonno, R.; Zahler, R. (1997). “BMS-200475, a novel carbocyclic 29-deoxyguanosine analog with potent and selective antihepatitis B virus activity in vitro”. Bioorg. Med. Chem. Lett. 7: 127–132. doi:10.1016/s0960-894x(96)00594-x.
- Jump up^ Innaimo, S F; Seifer, M; Bisacchi, G S; Standring, D N; Zahler, R; Colonno, R J (1997). “Identification of BMS-200475 as a Potent and Selective Inhibitor of Hepatitis B Virus. Antimicrob”. Agents Chemother. 41 (7): 1444–1448.
- Jump up^ Seifer, M.; Hamatake, R. K.; Colonno, R. J.; Standring, D. N. (1998). “In vitro inhibition of hepadnavirus polymerases by the triphosphates of BMS-200475 and lobucavir. Antimicrob”. Agents Chemother. 42: 3200–3208.
- Jump up^ Yamanaka, G.; Wilson, T.; Innaimo, S.; Bisacchi, G. S.; Egli, P.; Rinehart, J. K.; Zahler, R.; Colonno, R. J. (1999). “Metabolic studies on BMS-200475, a new antiviral compound active against hepatitis B virus. Antimicrob”. Agents Chemother. 43: 190–193.
- Jump up^ Colonno, R. J.; Genovesi, E. V.; Medina, I.; Lamb, L.; Durham, S. K.; Huang, M. L.; Corey, L.; Littlejohn, M.; Locarnini, S.; Tennant, B. C.; Rose, B.; Clark, J. M. (2001). “Long-term entecavir treatment results in sustained antiviral efficacy and prolonged life span in the woodchuck model of chronic hepatitis infection”. J. Infect. Dis. 184: 1236–1245. doi:10.1086/324003.
- Jump up^ Levine, S.; Hernandez, D.; Yamanaka, G.; Zhang, S.; Rose, R.; Weinheimer, S.; Colonno, R. J. (2002). “Efficacies of entecavir against lamivudine-resistant hepatitis B virus replication and recombinant polymerases in vitro. Antimicrob”. Agents Chemother. 46: 2525–2532. doi:10.1128/aac.46.8.2525-2532.2002.
- Jump up^ Chang, T. T. (2006). “A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B”. N. Engl. J. Med. 354: 1001–1010. doi:10.1056/nejmoa051285.
- Jump up^ Lai CL, Shouval D, Lok AS, Chang TT, Cheinquer H, Goodman Z, DeHertogh D, Wilber R, Zink RC, Cross A, Colonno R, Fernandes L (9 March 2006). “Entecavir versus Lamivudine for Patients with HBeAg-Negative Chronic Hepatitis B”. The New England Journal of Medicine. 354 (10): 1011–20. doi:10.1056/NEJMoa051287. PMID 16525138.
- Jump up^ Sherman, M.; Yurdaydin, C.; Sollano, J.; Silva, M.; Liaw, Y. F.; Cianciara, J.; Boron-Kaczmarska, A.; Martin, P.; Goodman, Z.; Colonno, R. J.; Cross, A.; Denisky, G.; Kreter, B.; Hindes, R. (2006). “Entecavir for the treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B”. Gastroenterology. 130: 2039–2049. doi:10.1053/j.gastro.2006.04.007.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. http://www.accessdata.fda.gov. Archived from the original on 4 March 2016. Retrieved 2015-08-29.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. Orange Book. Patent and Exclusivity for: N021798. Archived from the original on 15 November 2016. Retrieved 14 November 2016.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. http://www.accessdata.fda.gov. Search results from the “OB_Rx” table for query on “202122.”. Archived from the original on 22 December 2015. Retrieved 2015-08-29.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. http://www.accessdata.fda.gov. Search results from the “OB_Rx” table for query on “205740.”. Archived from the original on 4 March 2016. Retrieved 2015-08-29.
- Jump up^ “Orange Book: Approved Drug Products with Therapeutic Equivalence Evaluations”. http://www.accessdata.fda.gov. Search results from the “OB_Rx” table for query on “206217.”. Archived from the original on 4 March 2016. Retrieved 2015-08-29.
External links
 |
|
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ɛnˈtɛkəvɪər/ en-TEK-a-vir or en-TE-ka-veer |
| Trade names | Baraclude[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a605028 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
by mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | n/a (≥70)[2] |
| Protein binding | 13% (in vitro) |
| Metabolism | negligible/nil |
| Biological half-life | 128–149 hours |
| Excretion | Renal 62–73% |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.111.234 |
| Chemical and physical data | |
| Formula | C12H15N5O3 |
| Molar mass | 277.279 g/mol |
| 3D model (JSmol) | |
| Melting point | 220 °C (428 °F) value applies to entecavir monohydrate and is a minimum value[3] |
///////////////Entecavir, энтекавир , إينتيكافير , 恩替卡韦 , BMS-200475, SQ-200475, エンテカビル,
NC1=NC(=O)C2=C(N1)N(C=N2)[C@H]1C[C@H](O)[C@@H](CO)C1=C
NMR PREDICT
1H NMR AND 13C NMR
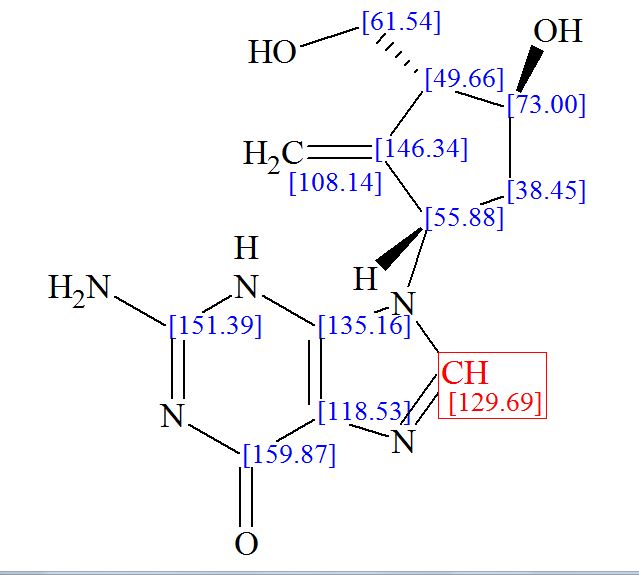
13C PREDICT VALUES
GLIMEPIRIDE
Glimepiride
- Molecular FormulaC24H34N4O5S
- Average mass490.616 Da
-
HOE 490UNII:6KY687524K
3-Ethyl-N-{2-[4-({(E)-hydroxy[(trans-4-methylcyclohexyl)imino]methyl}sulfamoyl)phenyl]ethyl}-4-methyl-2-oxo-2,5-dihydro-1H-pyrrole-1-carboximidic acid
93479-97-1 [RN]
1-{[4-(2-{[(3-ethyl-4-methyl-2-oxo-2,5-dihydro-1H-pyrrol-1-yl)carbonyl]amino}ethyl)phenyl]sulfonyl}-3-(trans-4-methylcyclohexyl)urea
3-Ethyl-4-methyl-N-[2-(4-{[(trans-4-methylcyclohexyl)carbamoyl]sulfamoyl}phenyl)ethyl]-2-oxo-2,5-dihydro-1H-pyrrole-1-carboxamide
1H-Pyrrole-1-carboxamide, 3-ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methylcyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-
Amarel [Trade name]
Amaryl [Trade name]
Endial [Trade name]
Glimepiride (original trade name Amaryl) is an orally available medium-to-long-acting sulfonylurea antidiabetic drug. It is sometimes classified as either the first third-generation sulfonylurea,[1] or as second-generation.[2]
Glimepiride is a Sulfonylurea. The chemical classification of glimepiride is Sulfonylurea Compounds.
Glimepiride is a long-acting, third-generation sulfonylurea with hypoglycemic activity. Compared to other generations of sulfonylurea compounds, glimepiride is very potent and has a longer duration of action. This agent is metabolized by CYP2C9 and shows peroxisome proliferator-activated receptor gamma (PPARgamma) agonistic activity.
Glimepiride is only found in individuals that have used or taken this drug. It is the first III generation sulphonyl urea it is a very potent sulphonyl urea with long duration of action. The mechanism of action of glimepiride in lowering blood glucose appears to be dependent on stimulating the release of insulin from functioning pancreatic beta cells, and increasing sensitivity of peripheral tissues to insulin. Glimepiride likely binds to ATP-sensitive potassium channel receptors on the pancreatic cell surface, reducing potassium conductance and causing depolarization of the membrane. Membrane depolarization stimulates calcium ion influx through voltage-sensitive calcium channels. This increase in intracellular calcium ion concentration induces the secretion of insulin.
Indications
Glimepiride is indicated to treat type 2 diabetes mellitus; its mode of action is to increase insulin production by the pancreas. It is not used for type 1 diabetes because in type 1 diabetes the pancreas is not able to produce insulin.[3]
Contraindications
Its use is contraindicated in patients with hypersensitivity to glimepiride or other sulfonylureas.
Adverse effects
Side effects from taking glimepiride include gastrointestinal tract (GI) disturbances, occasional allergic reactions, and rarely blood production disorders including thrombocytopenia, leukopenia, and hemolytic anemia. In the initial weeks of treatment, the risk of hypoglycemia may be increased. Alcohol consumption and exposure to sunlight should be restricted because they can worsen side effects.[3]
Pharmacokinetics

Two generic oral tablets of glimepiride, 2 mg each
Gastrointestinal absorption is complete, with no interference from meals. Significant absorption can occur within one hour, and distribution is throughout the body, 99.5% bound to plasma protein. Metabolism is by oxidative biotransformation, it is hepatic and complete. First, the medication is metabolized to M1 metabolite by CYP2C9. M1possesses about 1⁄3 of pharmacological activity of glimepiride, yet it is unknown if this results in clinically meaningful effect on blood glucose. M1 is further metabolized to M2metabolite by cytosolic enzymes. M2 is pharmacologically inactive. Excretion in the urine is about 65%, and the remainder is excreted in the feces.
Mechanism of action
Like all sulfonylureas, glimepiride acts as an insulin secretagogue.[4] It lowers blood sugar by stimulating the release of insulin by pancreatic beta cells and by inducing increased activity of intracellular insulin receptors.
Not all secondary sufonylureas have the same risks of hypoglycemia. Glibenclamide (glyburide) is associated with an incidence of hypoglycemia of up to 20–30%, compared to as low as 2% to 4% with glimepiride. Glibenclamide also interferes with the normal homeostatic suppression of insulin secretion in reaction to hypoglycemia, whereas glimepiride does not. Also, glibenclamide diminishes glucagon secretion in reaction to hypoglycemia, whereas glimepiride does not.[5]

Interactions
Nonsteroidal anti-inflammatory drugs (such as salicylates), sulfonamides, chloramphenicol, coumadin and probenecid may potentiate the hypoglycemic action of glimepiride. Thiazides, other diuretics, phothiazides, thyroid products, oral contraceptives, and phenytoin tend to produce hyperglycemia.
SYNTHESIS
EP 0031058; US 4379785, Arzneim-Forsch Drug Res 1988,38(8),1079

The condensation of 3-ethyl-4-methyl-3-pyrrolin-2-one (I) with 2-phenylethyl isocyanate (II) at 150 C gives 3-ethyl-4-methyl-2-oxo-N-(2-phenylethyl)-3-pyrrolin-1-carboxamide (III), which is sulfonated with chlorosulfonic acid at 40 C to yield the corresponding benzenesulfonyl chloride (IV). The reaction of (IV) with concentrated NH4OH affords the sulfonamide (V), which is finally condensed with 4-methylcyclohexyl isocyanate (VI) in acetone.
clip

CLIP

Following is one of the synthesis routes: 3-Ethyl-4-methyl-3-pyrrolin-2-one could be condensed (I) with 2-phenylethyl isocyanate (II) at 150 C to produce 3-ethyl-4-methyl-2-oxo-N-(2-phenylethyl)-3-pyrrolin-1-carboxamide (III), which is sulfonated with chlorosulfonic acid at 40 C to yield the corresponding benzenesulfonyl chloride (IV). The reaction of (IV) with concentrated NH4OH affords the sulfonamide (V), which is finally condensed with 4-methylcyclohexyl isocyanate (VI) in acetone.
CLIP
http://science24.com/paper/6906

PAPER
https://www.sciencedirect.com/science/article/pii/S073170850500378X

PATENT
https://www.google.com/patents/US20070082943
-
Glimepiride, according to U.S. Pat. No. 4,379,785 (EP 031058) issued to Hoechst is prepared via reaction of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV) with trans-4-methylcyclohexyl isocyanate (VIII). U.S. Pat. No. 4,379,785 (EP 031058) (hereafter referred to as the ‘785 patent) discloses heterocyclic substituted sulfonyl ureas, particularly 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1H-pyrrole-1-carboxamide i.e. Glimepiride (I). The ‘785 patent teaches the preparation of Glimepiride starting from 3-Ethyl-4-methyl-3-pyrolidine-2-one (II) and 2-phenyl ethyl isocyanate to give [2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene (III). The [2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene is converted to the 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV), by reacting with chlorosulphonic acid, followed by treatment with ammonia solution. This intermediate compound (IV) is then finally reacted with trans-4-methylcyclohexyl isocyanate (VIII) prepared from trans-4-methyl cyclohexylamine HCl (VII) to form Glimepiride.
-
[0004]Glimepiride can also be synthesized by reaction of N-[[4-[2-(3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido)-ethyl]phenyl]sulphonyl]methylurethane (IX) with trans-4-methyl cyclohexyl amine (VII) as reported by R. Weyer, V. Hitzel in Arzneimittel Forsch 38, 1079 (1988).
-
[0005]trans-4Methylcyclohexyl isocyanate (VIII) is prepared from trans-4-methyl cyclohexyl amine HCl (VII), by phosgenation.
-
[0006]H. Ueda et. al., S.T.P Pharma Sciences, 13(4) 281-286, 2003 discloses a novel polymorph of Glimepiride, Form II obtained by recrystallisation from a solvent mixture of ethanol and water. It also discloses that earlier known form is Form I. Reported solvents for obtaining Form I are methanol, acetonitrile, chloroform, butyl acetate, benzene and toluene.
-
[0007]An alternative route is disclosed in WO03057131(Sun Pharmaceutical), where 3-ethyl-4-methyl-2,5-dihydro-N-(4-nitrophenyloxycarbonyl)-pyrrole-2-one is treated with 4-(2-aminoethyl)-benzene sulphonamide to obtain 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV) which was then converted to Glimepiride (I). However, nonavailability of raw material and the yield being poor, the process as described in U.S. Pat. No. 4,379,785 is preferred.
-
[0008]To obtain Glimepiride of highest purity, following intermediates should be of highest quality:
-
[0009]a) 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV) with lowest possible content of ortho and meta isomers.
-
[0010]b) Trans-4-methyl cyclohexyl amine (VII) and its respective isocyanate (VIII) should have lowest content of the cis isomer.
-
[0011]The preparation of the 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide is well disclosed in the patent U.S. Pat. No. 4,379,785. It is prepared by condensation of 3-ethyl-4-methyl-3-pyrrolidine-2-one of Formula (II) with 2-phenyl ethyl isocyanate. The condensed product is then chlorosulphonated with chlorosulphonic acid followed by ammonolysis with liq. ammonia to give compound of Formula (IV). The purity is not well documented in the patents, and by following the patented process, ˜85 to 88% of desired para isomer is obtained. This is evident as the chlorosulphonation is ortho-para directing.
-
[0012]Hence, there is a need to develop purification process to maintain undesired ortho and meta isomers below 0.1%.
-
[0013]The other key intermediate trans-4-methylcyclohexyl amine HCl (VII) should preferably have lowest possible content of the cis isomer. The commonly used procedure is reduction of 4-methyl cyclohexanone oxime (V) with sodium in alcohol, preferably ethanol.
-
[0014]T. P. Johnston, et. al., J. Med. Chem., 14, 600-614 (1971); H. Booth, et. al., J. Chem. Soc (B) 1971, 1047-1050 and K. Ramalingam et. al., Indian Journal of Chem Vol. 40, 366-369 (April 1972) all report the abovementioned reduction. The amine obtained via this process typically contains between 8 to 10% of the cis isomer. However, use of high excess sodium metal (25 eqv.) for reduction makes process commercially and environmentally unviable. Also, the purification of trans amine from the mixture via the distillation is very difficult as the boiling points differ only by about 2° C. Also there is an inherent drawback of said free amine as, it immediately forms carbonate salt. Further purification of the amine to reduce the cis content via crystallization of its salt is not sufficiently documented. Prior art describes purification of crude trans-4-methylcyclohexylamine HCl by crystallization of its hydrochloride but the yield and purity are not sufficiently discussed. A description of such purification is provided in J. Med. Chem, 14, 600-614 (1971), wherein trans-4-methylcyclohexylamine HCl is obtained by triple crystallization in acetonitrile of the crude hydrochloride (m.p. 260° C.) in 27% yield.
-
[0015]WO 2004073585 (Zentiva) describes a process for preparation of trans-4-methylcyclohexylamine HCl wherein the highlights of the invention are the use of sodium metal and purification via the pivalic acid salt. However drawbacks of the process are use of sodium metal, which is hazardous and pivalic acid which is expensive. The overall yield is ˜40%.
-
[0016]Thus considering the current stringent pharmacopieal requirements for cis content, there is a need for obtaining Glimepiride having cis impurity content well below 0.15% by a cost effective process.
-
[0017]Key factors in the production of Glimepiride are:
-
[0018]a) Substantial purity of trans-4-methyl cyclohexyl amine HCl (VII) with the lowest possible content of the cis isomer.
-
[0019]b) Substantial purity of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV) with the lowest possible content of the ortho and meta isomer.
-
[0020]The purity of intermediate compound of Formula (IV) when prepared by the process disclosed in ‘785 patent, was found to be 82 to 85% by HPLC.
-
-
schemes I to III.



-
[0037]The purification of trans-4-methyl cyclohexylamine HCl (VII) is accomplished by using an appropriate solvent combination. The mixture of cis/ trans stereoisomers (i.e. 50:50) were dissolved in diluted methanol and the desired trans isomer is coprecipitated by adding acetone to it. The process is repeated with different proportions of the solvent mixture to get the trans-4-Methyl cyclohexylamine HCl (VII) >99.5% with cis isomer less than 0.15%. The overall yield from 4-methyl cyclohexanone is ˜30%. The purification has been achieved using a solvent mixture of alcohol and ketone. A preferred alcohol for dissolution is an aliphatic one wherein carbon chain may be preferably C1-C4. Preferably methanol is used to dissolve the crude trans-4-Methyl cyclohexylamine HCl. The ratio of substrate:methanol:acetone is fixed at 1:1.5:6 for achieving the desired purity. The cosolvent used for precipitation is an aliphatic ketone. The preferred ketone is acetone. The precipitation is carried out at a temperature between 20 to 50° C., preferably between 30 to 50° C. and most preferably at about 40° C. The addition of acetone is carried out over a period of 2 to 6 hrs, more preferably for about 2 to 4 hrs and most preferably in about 3 hrs. The compound thus obtained has a purity >95% by gas chromatography.
-
[0038]The enriched trans-4-Methyl cyclohexylamine HCl (VII) (>95%) is further purified using different proportions of the same solvent mixture. The enriched trans isomer is dissolved in alcohol and reprecipitated using an aliphatic ketone. The ratio of substrate:methanol:acetone ratio is fixed at 1:1.5:13.6 for obtaining purity greater than 99.8%.
-

-
- EXAMPLE 1trans-4-Methyl cyclohexylamine HCl (VII)
-
[0053]1.5 Kg of crude 4-Methyl cyclohexyloxime (V) was dissolved in 8.33 L Methanol. To this 0.15 Kg Raney nickel was added. Then the mixture was hydrogenated at 4-5 Kg/cm2 pressure at 50 to 55° C. After the absorption of H2 ceases, the reaction mass is cooled down and filtered. From resulting reaction mixture, methanol was distilled completely. Crude concentrated oil obtained is cooled to 15 to 20° C. to which methanolic hydrochloric acid (12 to 13%) is added slowly, when the product i.e. 4-Methylcyclohexylamine HCl precipitates out. The yield obtained 1.5 Kg of crude 4-methyl cyclohexylamine HCl (85%) with ˜50% content of trans isomer. The crude 4-Methyl cyclohexylamine HCl 1.5 Kg (wet) was further purified in methanol/acetone mixture. The crude 4-methyl cyclohexylamine HCl (1.5 Kg) was dissolved in 2.25 L of methanol at 25 to 30° C. Slowly started addition of 13.5 L of acetone over a period of 3 hrs. The trans-4-methyl cyclohexylamine HCl precipitated out. Yield 0.6 Kg. The purity achieved of trans isomer is >95%. The cis isomer at this stage is ˜2 to 3%.
-
[0054]The trans-4-methyl cyclohexylamine HCl (0.6 Kg) thus obtained is again taken in 0.9 L of methanol and is dissolved completely at 25 to 30° C. 8.1 L acetone is added slowly over a period of 3 hrs when pure trans isomer precipitates out completely. The purity achieved at this stage is >99.8% and cis isomer well below 0.15%. The yield thus obtained after the second purification is 0.48 Kg of trans-4-Methyl cyclohexylamine HCl (27.2% yield calculated on the starting oxime). Purity of the desired trans isomer is greater than 99.8% by G.C.
-
[0055]Melting point of the trans-4-methyl cyclohexylamine HCl thus obtained is 262° C. to 263° C.
-
EXAMPLE 2Preparation of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (IV)
-
-
[0056]3-Ethyl-4-methyl-2,5-dihydro-1H-pyrrole-2-one (II) (1.0 Kg) and β-phenylethyl isocyanate (1.488 Kg) were mixed in anhydrous toluene (4.0 L) and refluxed for 4 hrs. The toluene was distilled off and hexane (8.0 L) was added to the reaction mixture at 50° C. The product precipitated is cooled to 0 to 5° C. to obtain the solid compound viz. 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene (2.17 Kg). It was filtered & washed with 2.0 L of hexane.
-
[0057]To a cooled (15 to 25° C.) solution of chlorosulfonic acid (2.8 L), 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene (2.0 Kg) was added in small portions over a period of 2 to 3 hrs. Further it was stirred for 30 min at this temperature and then temperature was gradually raised to 30 to 35° C. The reaction mass is stirred further for 2 hrs. The reaction mixture was then quenched into ice-water and stirred for 1 hr and filtered to obtain the product 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonyl chloride (2.0 kg). To a cooled (15 to 20° C.) solution of diluted ammonia (1.4 L) 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonyl chloride was added in small portion over 1 to 2 hrs. The reaction mixture was then heated to 70° C. for 2 hrs when ammonolysis is complete. The product converted is then stirred for 1 hr at R.T. and filtered and dried at 90 to 100° C. to obtain crude 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (2.2 Kg) having HPLC purity in the range of 82 to 88%. The crude compound 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (2.2 Kg) is then purified from mixture of organic solvents chosen from Methanol, Acetone & toluene.
-
EXAMPLE 3APurification of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (IV)
-
-
[0058]1st Purification
-
[0059]In a reaction vessel containing Toluene (12.0 L), 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide (2.0 Kg) was charged at 25 to 30° C. Slowly the temperature was raised to 60 to 65° C. and methanol (5.0 L) was added via the dosing tank slowly when the product dissolved completely. Refluxed it for 0.5 hr. Charcoalised and filtered the product in another reaction vessel. Distill off toluene/methanol mixture till total recovery about 65% under vacuum. White crystalline product precipitated out. After the recovery, cool the reaction mass to 15 to 20° C. The resulting crystallized solid product was filtered and washed two times with chilled acetone (about 2 L) each. The resulting product was dried at 90 to 100° C. in air oven till constant weight to obtain about 1.4 Kg of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide with greater than 95% HPLC purity.
-
EXAMPLE 3BPurification of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (IV)
-
-
[0060]2nd Purification
-
[0061]In a reaction vessel containing Acetone (8.4 L), (1.4 Kg) of 1st purified 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide was charged at 25 to 30° C. slowly and the temperature was raised to 55 to 60° C. Methanol was added (5.6 L) via the dose tank at this reflux temperature to dissolve it completely. Refluxed it for further 30 min. Distilled off acetone/ methanol mixture till total recovery about 65 to 70%. White crystalline product precipitated out. After the recovery slowly cooled the product to 15 to 20° C. The resultant solid product was filtered, washed two times with chilled acetone (1.4 L) each. The 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido) ethyl] benzene sulfonamide was dried at 90 to 100° C. in air oven till constant weight to obtain about 1.12 Kg of 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (IV) with greater than 99.5% purity with other isomers i.e. ortho and meta well below 0.2% respectively.
-
EXAMPLE 4Preparation of 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1 H-pyrrole-1-carboxamide (I).
-
-
[0062]In a reaction vessel containing (24.2 L) Acetone, 4-[2-(3-Ethyl-4-methyl-2-carbonyl pyrrolidine amido)ethyl] benzene sulfonamide (1.0 Kg) and potassium carbonate (0.46 Kg) was added and refluxed at about 55 to 60° C. for 1 hr. trans-4-Methyl-cyclohexyl isocyanate was obtained by method known in art from trans-4-methyl-cyclohexylamine. A solution of trans-4-methyl-cyclohexyl isocyanate (0.515 Kg) in toluene (5 L) was prepared and added to the above reaction mixture. This reaction mixture is refluxed for 12 hrs, then cooled. To this cooled reaction mass charge 27 L of water. The reaction mass was filtered and the pH was adjusted to 5.5 to 6.0 by adding acetic acid at about 20 to 25° C. The solid obtained was filtered and washed with water. The 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1H-pyrrole-1-carboxamide (I) obtained is then dried at 90 to 100° C. till constant weight. Yield of the product is 86.3%.
-
EXAMPLE 5Purification of 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methyl cyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1H-pyrrole-1-carboxamide (I)
-
[0063]In a reaction vessel containing 6.0 L methanol and 1.0 Kg crude Glimepiride, dry ammonia gas was purged at 20 to 25° C. till all Glimepiride dissolves and a clear solution is obtained. This homogeneous mass was then charcoalised, filtered and finally neutralized with Glacial acetic acid to pH 5.5 to 6.0, till the entire product precipitates out. The pure Glimepiride was then filtered and dried at 65° C. to 70° C. till constant weight. Yield obtained was ˜90%.
CLIP
- Journal of Pharmaceutical Sciences 100(11):4700-9
- DOI
- 10.1002/jps.22662

Magnified 1H NMR spectra of (a) glimepiride and its solid dispersions with hyperbranched polymers containing the (b) hydroxyl and (c) the tertiary amino functional groups.

Magnified 13C NMR spectra of (a) glimepiride and its solid dispersions with hyperbranched polymers containing (b) the hydroxyl and (c) the tertiary amino functional groups.

The difference spectra of the solid dispersions of glimepiride and the hyperbranched polymer containing (a) the hydroxyl groups and (b) the tertiary amino groups. The difference spectra were obtained by subtraction of the spectra of the pure hyperbranched polymers from the spectra of the solid dispersions. The ATR spectra of the pure hyperbranched polymers were recorded on samples that were prepared under the same conditions as solid dispersions, only without the presence of the glimepiride drug.
Patents
FDA Orange Book Patents
| FDA Orange Book Patents: 1 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7358366 |
| Expiration | Apr 19, 2020. 7358366*PED expiration date: Oct 19, 2020 |
| Applicant | SB PHARMCO |
| Drug Application | |
from FDA Orange Book
| FDA Orange Book Patents: 2 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 8071130 |
| Expiration | Jun 8, 2028 |
| Applicant | TAKEDA PHARMS USA |
| Drug Application |
|
from FDA Orange Book
| FDA Orange Book Patents: 3 of 3 (FDA Orange Book Patent ID) | |
|---|---|
| Patent | 7700128 |
| Expiration | Jan 30, 2027 |
| Applicant | TAKEDA PHARMS USA |
| Drug Application |
|
from FDA Orange Book
CLIP
Journal of China Pharmaceutical University 1999 , 30(3):163 ~ 165
Ethyl acetoacetate (2) Preparation of literature more, such as with ethyl iodide or ethyl bromide as ethyl reagents will produce a double ethylation or oxyethylation, it is difficult to separate. We use dimethylamine and ethyl acetoacetate reaction enamine, then diethyl sulfate as ethylating agent, you can reduce the side reactions, product purity, the yield up to 80%. Preparation of cyanohydrin (3) Hydrochloric acid anhydrous literature, toxicity, difficult to operate, we use solid sodium cyanide and sodium bisulfite in the aqueous phase reaction, get 3, easy to operate. In the literature 1-acetyl-3- Ethyl-4-methyl-3-pyrrolin-2-one (4) was purified by high vacuum distillation and then hydrolyzed to give 3-ethyl- In the distillation of the product easy to loss, after the change to the crude hydrolysis, two-step yield of 33%. Reported in the literature 5 and phenethyl isocyanate (6) without solvent direct reaction of 3-ethyl-4-methyl-2-oxo-3-pyrroline-1 – N- (2 – phenethyl) A Amide (7), the experiment found that the reaction heat when the heating easy to red material, we add toluene as a solvent, the reaction is smooth, easy to post-treatment. 6 preparation, the general method is to use phosgene, but phosgene often Temperature of gas, highly toxic, difficult to operate, we use triphosgene instead of triphosgene as a yellow solid, easy to transport, weighing, laboratory convenience. We refer to the process of domestic glyburide, 7 chlorosulfonated, ammoniated sulfonamide (9), two-step yield of 77%. The last 9 reacts with trans-4-methylcyclohexylisocyanate to form glimepiride (1). Ethyl acetoacetate as the starting material, eight-step total yield of 11.5%.
Journal of China Pharmaceutical University 1999 , 30(3):163 ~ 165
Glimepi ride (1) trade name Amary l, chemical name 1- [4- [2- (3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido ) Ethyl] phenylsulfonyl] -3- (trans-4-methylcyclohexyl) urea, a new sulfonylurea hypoglycemic agent developed by Hoechst AG in Germany and listed in the Netherlands and Switzerland in 1995, In 1996 the United States FDA approval
- 1 Campbell RK.Glimepiride :Role of a new sulfonylurea in the t reatment of type 2 diabetes mellitus .An n Pharmacother , 1998 , 32 :1044
- 2 Hans P , Joachim K .Synt hesis of oxyopsopy rrolecarboxyli c acid
and further investigations in the pyrrolone series.Ann Chem ,
1964 , 680 :60
- 3 Glimepiride.Dr ugs F ut , 1992 , 17(9):774
- 4 Corson BB, Dodge RA , Harris SA , et al .M andeli c acid.Org Syn , 1941 , Coll Vol 1 :329
- 5 M aurice WG , Roy VD, Brian I , et a l .A new synt hesis of iso- cyanates .J Chem Soc , Perkin Ⅰ , 1976 :141
- 6 天津医药工业研究所.糖尿病药物-优降糖的新合成法.医药工业, 1974 , 4 :11
- 7 Weyer R, Gei sen K , Hitzel V , et al .Heterocyclic subst ituted sul- f onyl ureas and their use .Ger O f fen , 1979 :2951135 A 1
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4379785 * | Dec 17, 1980 | Apr 12, 1983 | Hoechst Aktiengesellschaft | Heterocyclic substituted sulfonyl ureas, and their use |
References
- Jump up^ Hamaguchi T, Hirose T, Asakawa H, et al. (December 2004). “Efficacy of glimepiride in type 2 diabetic patients treated with glibenclamide”. Diabetes Res. Clin. Pract. 66 Suppl 1: S129–32. doi:10.1016/j.diabres.2003.12.012. PMID 15563963.
- Jump up^ Davis SN (2004). “The role of glimepiride in the effective management of Type 2 diabetes”. J. Diabetes Complicat. 18 (6): 367–76. doi:10.1016/j.jdiacomp.2004.07.001. PMID 15531188.
- ^ Jump up to:a b “Glimepiride: MedlinePlus Drug Information”. nih.gov.
- Jump up^ Nissen SE, Nicholls SJ, Wolski K, et al. (April 2008). “Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial”. JAMA. 299 (13): 1561–73. doi:10.1001/jama.299.13.1561. PMID 18378631.
- Jump up^ Davis, Stephen N. (2005). “60. Insulin, oral hypoglycemic agents, and the pharmacology of the endocrine pancreas”. In Brunton, Laurence L.; Lazo, John S.; Parker, Keith L. (eds.). Goodman & Gilman’s The Pharmacological Basis of Therapeutics. New York: McGraw-Hill. p. 1636. ISBN 0-07-142280-3.
External links
- http://www.theodora.com/drugs/amaryl_tablets_sanofi_aventis.html
- http://www.rxlist.com/cgi/generic/glimepiride.htm
Title: Glimepiride
CAS Registry Number: 93479-97-1
CAS Name: 3-Ethyl-2,5-dihydro-4-methyl-N-[2-[4-[[[[(trans-4-methylcyclohexyl)amino]carbonyl]amino]sulfonyl]phenyl]ethyl]-2-oxo-1H-pyrrole-1-carboxamide
Additional Names: N-[4-[2-(3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido)-ethyl]-benzenesulfonyl]-N¢-4-methylcyclohexylurea; 1-[4-[2-(3-ethyl-4-methyl-2-oxo-3-pyrroline-1-carboxamido)ethyl]phenylsulfonyl]-3-(4-methylcyclohexyl)urea
Manufacturers’ Codes: HOE-490
Trademarks: Amaryl (Aventis)
Molecular Formula: C24H34N4O5S
Molecular Weight: 490.62
Percent Composition: C 58.75%, H 6.99%, N 11.42%, O 16.31%, S 6.54%
Literature References: Sulfonylurea. Prepn: R. Weyer et al., DE 2951135; eidem, US 4379785 (1981, 1983 both to Hoechst). Synthesis: R. Weyer, V. Hitzel, Arzneim.-Forsch. 38, 1079 (1988). Pharmacology: K. Geisen, ibid., 1120. Effects on insulin and glucagon secretion: V. Leclercq-Meyer et al., Biochem. Pharmacol. 42, 1634 (1991). HPLC determn in biological fluids: K. H. Lehr, P. Damm, J. Chromatogr. 526, 497 (1990). Clinical pharmacokinetics: K. Ratheiser et al., Arzneim.-Forsch. 43, 856 (1993). Toxicity study: U. Schollmeier et al., ibid. 1038. Series of articles on pharmacology and clinical efficacy: Diabetes Res. Clin. Pract. 28Suppl., S115-S149 (1995).
Properties: mp 207°.
Melting point: mp 207°
Therap-Cat: Antidiabetic.
Keywords: Antidiabetic; Sulfonylurea Derivatives.
 |
|
| Clinical data | |
|---|---|
| Trade names | Amaryl |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a696016 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral (tablets) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 100% |
| Protein binding | >99.5% |
| Metabolism | Complete hepatic (1st stage through CYP2C9) |
| Biological half-life | 5–8 hours |
| Excretion | Urine (~60%), feces (~40%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.170.771 |
| Chemical and physical data | |
| Formula | C24H34N4O5S |
| Molar mass | 490.617 g/mol |
| 3D model (JSmol) | |
///////////Amaryl, glimepiride, glymepiride, HOE 490
CCC1=C(CN(C1=O)C(=O)NCCC2=CC=C(C=C2)S(=O)(=O)NC(=O)NC3CCC(CC3)C)C
OXCARBAZEPINE

Oxcarbazepine
- Molecular FormulaC15H12N2O2
- Average mass252.268 Da
-
10-Oxo-10,11-dihydro-dibenzo[b,f]azepine-5-carboxylic acid amide28721-07-5 [RN]5H-Dibenz[b,f]azepine-5-carboxamide, 10,11-dihydro-10-oxo-

UV Spectroscopy The UV absorption spectrum of carbamazepine in methanol shown in Fig. 1 was recorded using Shimadzu UV–vis Spectrometer 1601 PC. The compound exhibited maxima at 288 and 259 nm. Clarke reported the following: methanol—237 and 285 nm (A 1%, 1 cm¼490) [1].
1 A.C. Moffat (Ed.), Clarke’s Isolation and Identification of Drugs, second ed.,
The Pharmaceutical Press, London, 1986, p. 428.


Vibrational Spectroscopy The FT-infrared absorption spectrum of carbamazepine was obtained in a KBr pellet using a Perkin-Elmer FT-infrared spectrophotometer. FTinfrared spectrum is shown in Fig. 2, where the principal peaks are observed at 3465, 3157, 1675, 1604, 1594, 1488, 1381, 1307, 870, 800, 762, and 724 cm1 .

1 H NMR Spectra The proton nuclear resonance (1 H NMR) spectra of carbamazepine were obtained using a Bruker instrument operating at 500 MHz. Standard Bruker software was used to execute the recording of the 1D and 2D spectra. The sample was dissolved in DMSO-d6 and all resonance bands were referencedto tetramethylsilane (TMS) as internal standard. The entire proton spectra are shown in Figs. 3 and 4. A singlet resonates at δ 5.54 representing the two protons of the amino group. An additional singlet which resonates at δ 6.99 ppm is assigned to the olefinic protons at positions 10 and 11. The two multiplets which resonate at δ 7.30–7.34 and δ 7.41–7.43 ppm are assigned to the aromatic protons of the two phenyl rings.

13C NMR Spectra A noise-modulated, broadband decoupling 13C NMR spectrum (Fig. 5) showed 11 carbon absorptions in accordance with what is anticipated for the structure of carbamazepine. Carbon resonance bands at δ 127.1, 129.0, 129.2, 129.3, 129.8, 130.3, 131.0, and 134.8 ppm account for the CH functions. A carbon band at δ 140.6 ppm represents the ethylene carbons. The carbonyl carbon resonates at δ 156.3 ppm. A DEPT experiment (Fig. 6) permitted the identification and confirmation of the methyl and methine carbons. Another confirmation was obtained through the HSQC experiment (Fig. 7).



SYN 1
http://www.drugfuture.com/synth/syndata.aspx?ID=117845
DD 153835; EP 0028028; JP 1045366; JP 1045367; JP 1045368; US 4452738; US 4540514; US 4559174; US 4579683

The nitration of 5-cyano 5H-dibenz[f,b]azepine (IV) with NaNO2 in acetic anhydride – acetic acid gives 5-cyano 10-nitro-5H-dibenz[b, f]azepine (V), which is then treated with BrF3 and powdered Fe in hot acetic acid.
SYN2
DE 2011087

The reaction of 10-methoxy-5H-dibenz[b,f]azepine (I) with phosgene in hot toluene gives 10-methoxy-5H-dibenz[b,f]azepine-5-carbonyl chloride (II), which is treated with NH3 in refluxing ethanol to afford 10-methoxy-5H-dibenz[b,f]azepine-5-carboxamide (III). Finally, this compound is hydrolyzed with refluxing 2N HCl.
SYN 3
WO 9621649

This compound has been obtained by two related ways: 1. The hydrolysis of 10-methoxy-5H-dibenzo[b,f]azepine (I) with refluxing 2N HCl gives 10,11-dihydro-5H-dibenzo[b,f]azepin-5-one (II), which is then treated with chlorosulfonyl isocyanate in chloroform to yield the target carboxamide. 2. The reaction of 10-methoxy-5H-dibenzo[b,f]azepine (I) with potassium cyanate in hot sulfuric acid also gives the target carboxamide. In this reaction sodium cyanate can also be used instead of the potassium salt. Other strong acids such as trichloroacetic acid or anhydrous HCl in acetic acid can be used instead of sulfuric acid.
SYN 4
The reaction of 10-methoxy-5H-dibenz[b,f]azepine (I) with phosgene in hot toluene gives 10-methoxy-5H-dibenz[b,f]azepine-5-carbonyl chloride (II), which is treated with NH3 in refluxing ethanol to afford 10-methoxy-5H-dibenz[b,f]azepine-5-carboxamide (III). Finally, this compound is hydrolyzed with refluxing 2N HCl.
SYN 5
WO 0156992

A new process for the preparation of oxcarbamazepine has been reported: Reaction of 1-phenyl-2,3-dihydro-1H-indol-2-one (I) with NaOH in refluxing THF gives 2-[2-(phenylamino)phenyl]acetic acid (II), which is condensed with dimethyl carbonate (III) by means of butyl lithium in the same solvent to yield 2-[2-[N-(methoxycarbonyl)-N-phenylamino]phenyl]acetic acid (IV). Cyclization of compound (IV) by means of polyphosphoric acid (PPA) at 100 C, followed by treatment of the reaction mixture with hot methanol (65 C) affords 10-methoxy-5H-dibenzo[b,f]azepine-5-carboxylic acid methyl ester (V), which is treated with NaOH in polyethyleneglycol at 100 C to provide 10-methoxy-5H-dibenzo[b,f]azepine (VI). Reaction of (VI) with sodium
SYN 6
Tetrahedron Lett 2001,42(3),385

Syntheses of intermediate (V), 5-benzyl-10,11-dihydro-5H-dibenz[b,f]azepin-10-one: Cyclization of either 2-[N-benzyl-N-(2-methylphenyl)amino]-N,N-dimethylbenzamide (I), 2-[N-benzyl-N-(2-methylphenyl)amino]-N,N-diethylbenzamide (II), 2-[N-benzyl-N-(2-methylphenyl)amino]-N,N-diisopropylbenzamide (III) or the morpholine derivative (IV) by means of LDA and TMEDA in THF

| Syntheses of intermediate (VIII), 5-(4-methoxybenzyl)-10,11-dihydro-5H-dibenz[b,f]azepin-10-one: Cyclization of 2-[N-(4-methoxybenzyl)-N-(2-methylphenyl)amino]-N,N-dimethylbenzamide (VI) or 2-[N-(4-methoxybenzyl)-N-(2-methylphenyl)amino]-N,N-diethylbenzamide (VII)) by means of LDA and TMEDA in THF. |

Syntheses of intermediate (XI), 5-allyl-10,11-dihydro-5H-dibenz[b,f]azepin-10-one: Cyclization of 2-[N-allyl-N-(2-methylphenyl)amino]-N,N-dimethylbenzamide (IX) or 2-[N-allyl-N-(2-methylphenyl)amino]-N,N-diethylbenzamide (X) by means of LDA and TMEDA in THF.

Finally, deprotection of either intermediate (V) with TMS-Cl and NaI, intermediate (VIII) with TiCl4 or intermediate (XI) with Rh(PPh3)3Cl give, in all cases, 10,11-dihydro-5H-dibenz[b,f]azepin-10-one (XII), which is finally treated with chlorosulfonyl isocyanate to afford oxcarbazepine.
PATENT

https://www.google.com/patents/US6670472
Oxcarbazepine is an anticonvulsant drug (as described in U.S. Pat. No. 3,642,775), and has been proposed for use as an anti-epileptical agent in the treatment of AIDS-related neural disorders (as described in PCT patent specification no. WO 94/20110); and for the treatment of Parkinson’s disease and/or Parkinsonian syndromes (as described in U.S. Pat. No. 5,658,900 and European patent specification no. 678 026).
Various processes for preparing oxcarbazepine have been described in the prior art. For example, U.S. Pat. No. 3,642,775 describes the preparation of oxcarbazepine from 10-methoxyiminostilbene (Scheme-1), which is first phosgenated in toluene, followed by amidation (ethanol and ammonia) and hydrolysis in an acidic medium to furnish the desired product. The main drawback of this process is the use of phosgene (COCl2), a toxic and hazardous substance.
Canadian patent specification no. 1 112 241 describes an alternative preparation of oxcarbazepine from the catalysed re-arrangement of 10,11-epoxycarbamazepine, which itself may be prepared from carbamazepine by reaction with m-chloroperbenzoic acid (CPBA) (Scheme-2). However, the drawbacks of this process are: use of carbamazepine, an expensive raw material; and converting this into its corresponding epoxide in poor yields and quality.


Another process, disclosed in European patent specification no. 028 028, starts from 5-cyanoiminostilbene through nitration, reduction and hydrolysis stages (Scheme-3). However, the drawback of the process is in the preparation of the 5-cyanoiminostilbene itself, which can be made from iminostilbene and cyanogen chloride. The latter is also toxic, hazardous and difficult to handle.

Another alternative is described in Swiss patent specification no. 642 950 and comprises hydrolysis, using concentrated sulphuric acid, of the corresponding chloride (10-chloro-5H-dibenz[b,f]azepin-5carboxamide) to form the oxcarbazepine.
More recently, a process has been described in PCT patent specification no. WO 96/21649 (Scheme-4), which starts with 10-methoxyiminostilbene and treats it with an alkali or alkaline earth metal cyanate and acid to produce 10-methoxycarbamazepine which, on acid hydrolysis, furnishes oxcarbazepine. Alternatively, 10-methoxyiminostilbene is first hydrolysed to produce 10-oxo-iminodibenzyl (10-keto-iminodibenzyl) which, upon condensation with chlorosulphonyl isocyanate followed by hydrolysis, furnishes oxcarbazepine. Chlorosulphonyl isocyanate is a very costly, highly moisture-sensitive and toxic reagents which is the main drawback of this latter process.
The biggest problem with the former process is that 10-methoxyiminostilbene undergoes two kinds of competitive reactions when an alkali metal cyanate and an acid are added. The enol-ether moiety of the compound undergoes hydrolysis to give the corresponding ketone (“oxo” compound), which does not undergo a carboxamidation reaction with HOCN, whereas the imino function of the intact 10-methoxyiminostilbene does undergo a carboxamidation reaction. Therefore, the end result is that a mixture of oxcarbazepine, oxo-iminodibenzyl and impurities are obtained, after hydrolysis, making the subsequent crystallization process highly tedious and uneconomical.

The acids that are used in this reaction (Scheme-4), according to the Examples of WO 96/21649, include acetic acid, mono-, di- and tri-chloroacetic acids, dry HCl and concentrated sulphuric acid etc. The general description teaches that concentrated mineral acids are to be used, optionally in solution in the organic acids. Nevertheless, all these acids produce substantial quantities of side products, ie oxo-iminodibenzyl and impurities formed therefrom. Due to this, although the conversion is high, the selectivity leading to the carboxamidation reaction is poor.
Furthermore, international patent specification no. WO 01/56992 describes the use of acetic acid in the absence of an additional solvent in this process, which is stated to result in an improved yield. Nothing about the purity of the end-product (oxcarbazepine) is mentioned, however, and the specific example given shows that the yield thereof is less than or equal to 78% after hydrolysis with water and sulphuric acid in the absence of a solvent such as toluene.
All the known methods therefore suffer from disadvantages, in particular, the requirement to use “environmentally unfriendly” reactants, and/or result in poor yields due to side reactions as mentioned above. In particular, the method described in WO 01/56992 precludes the use of a solvent, which imposes unfavourable limitations on the subsequent processing of the intermediate in the preparation of the end-product.
We have surprisingly found that reaction of 10-methoxyiminostilbene with cyanic acid (HOCN) in the presence of a mild acidic reagent, especially an aromatic acid, enables the disadvantages of the prior art preparation of 10-methoxycarbamazepine to be overcome. In particular, it allows for the use of a solvent in the subsequent reaction steps, which has advantages as will be further described hereinbelow
Accordingly, the present invention provides a process for the preparation of 10-methoxycarbamazepine, which process comprises reacting 10-methoxyiminostilbene with HOCN in a solvent therefor in the presence of a mild acidic reagent. It is important that the mild acidic reagent be chosen so that the enol-ether function is not rapidly hydrolysed. Accordingly, this reagent is preferably a weak acid, such as an aromatic acid. Preferred aromatic acids include weak, non-aliphatic organic acids, such as benzoic acid and substituted benzoic acids; suitable substituents being halo, especially chloro eg para-chlorobenzoic acid. Suitably, the acid has a pKa value in the range of from about 10−4 to 10−5.
Furthermore, the mild acidic reagent is preferably relatively insoluble in the solvent, especially at room temperature but also preferably at the temperature of the reaction, compared to other acids, such as acetic acid. Suitably, the mild acidic reagent has a solubility in the solvent of less than 75%, preferably less than 50% and more preferably less than 25% in the solvent. Especially preferred is when the mild acidic reagent has a solubility of less than about 10-12%, even at elevated temperatures, such as at the temperature of the reaction, and particularly preferred is when the mild acidic reagent has a solubility of less than about 1% at room/ambient Temperature. In this context, it is to be understood that ‘room temperature’ is less than 35° C. and more usually about 20-25° C., such as 21-22° C. Of all the aromatic acids, benzoic acid is the most suitable acid in terms of selectivity (by ‘selectivity’ in this context is meant preference for the carboxamidation reaction over the enol-ether hydrolysis).
Excess molar quantity of the weak acid is preferably used in comparison to the 10-methoxyiminostilbene starting material; for example, in the range of from 2 to 10 molar excess, more preferably about 5 to 8 times, eg 6-7 times, benzoic acid is most preferably employed in the reaction. Most of the acidic reagent can be easily recovered and re-used, such as up to 90-95% can be re-cycled. Such acids less readily hydrolyse the enol-ether moiety present in the 10-methoxyiminostilbene, while nevertheless being able readily to catalyse the reaction between the 10-methoxyiminostilbene and the HOCN.
In another aspect, the present invention provides a process for the preparation of 10-methoxycarbamazepine, which process comprises reacting 10-methoxyiminostilbene with HOCN in the absence of a strong acid. In particular, the present invention provides a process for the preparation of 10-methoxycarbamazepine, which process comprises reacting 10-methoxyiminostilbene with HOCN in the absence of an acid having a high solubility in the solvent. In this context, a strong acid is one that would rapidly hydrolyse the enol-ether function of the starting material, such as aliphatic organic acids (including acetic acid, which also has a high solubility in solvents such as toluene) and mineral acids. For example, when aliphatic acids, such as acetic acid, monochloro-acetic acid, ethylhexanoic acid and phenylacetic acid etc, were used in the reaction, the percentage formation of 10-methoxycarbamazepine was very poor, varying from 26% to 51%. Worse still, when mineral acids, such as hydrochloric acid and sulphuric acid, were tried, the percentage formation of 10-methoxycarbamazepine was even more poor (˜1%). In all the above reactions (ie when aliphatic acids or mineral acids were used), a significant percentage of 10-oxo-iminostilbene and impurities were formed. Table 1 below shows the results, using sodium cyanate in all reactions and 10 volumes of toluene per part of 10-methoxyiminostilbene.
| TABLE 1 | ||||||
| HPLC Analysis | ||||||
| % of 10- | % of | |||||
| Reflux | Conversion | methoxycarba- | Oxo- | Total | % of Unreacted | |
| Acid used | (hours) | (%) | mazepine | IDB | Impurity | 10-methoxy ISB |
| Hydrochloric acid | 4 | 89.63 | 0.24 | 70.19 | 19.19 | 10.37 |
| Sulphuric acid | 4 | 99.48 | 1.12 | 93.67 | 4.69 | 0.52 |
| Acetic acid | 12 | 59.05 | 26.22 | 12.97 | 19.86 | 40.95 |
| Monochloro-acetic acid | 12 | 96.32 | 51.5 | 24.00 | 20.82 | 3.68 |
| Ethylhexanoic acid | 22 | 44.14 | 22.86 | 12.93 | 8.35 | 55.86 |
| Benzoic acid | 12 | 98.00 | 75.50 | 9.10 | 13.40 | 2.00 |
| p-Chlorobenzoic acid | 12 | 99.66 | 56.44 | 20.00 | 23.22 | 0.34 |
| o-Chlorobenzoic acid | 12 | 98.13 | 31.25 | 54.77 | 12.11 | 1.87 |
| 2,4-Dichlorobenzoic acid | 6 | 98.48 | 55.45 | 30.04 | 12.99 | 1.52 |
| Phenylacetic acid | 6 | 72.88 | 34.38 | 18.36 | 20.14 | 27.12 |
On the contrary, when the aromatic acids such as mentioned above are used, the selectivity of the main reaction (ie the carboxamidation reaction as compared to hydrolysis of the enol-ether moiety) can increase to more than 75%. This results in improved efficiency and eventually in simpler methods of purification of the end product oxcarbazepine, resulting in easier commercialization of the process.
The carboxamidation of the 10-methoxyiminostilbene according to the present invention is preferably carried out in an organic medium, most preferably under reflux conditions. The organic medium is suitably an aromatic hydrocarbon solvent or an aliphatic chlorinated solvent, such as benzene, toluene, xylene, dichloromethane, chloroform and dichloroethane etc, including others described in relation to the Scheme-4 synthesis mentioned above and in WO 96/21649. The solvent(s) used in the carboxamidation reaction also play an important role in the selectivity and completion of reaction. We have found that toluene is the best solvent both in terms of selectivity and completion of reaction. It is important that the solvent is chosen such that the starting material and the HOCN are both soluble therein. Furthermore, as indicated above, it is important that the weak acid is relatively insoluble therein.
The HOCN reacts with the imino function to produce desired intermediate, 10-methoxycarbamazepine, which can afford the pharmacologically active end-product, ie oxcarbazepine, after hydrolysis.
The HOCN may be generated in situ by reaction of an alkali metal cyanate with the mild acidic reagent. Suitable cyanates include sodium and potassium, preferably sodium, cyanates. However, other methods of generating the HOCN, such as from cyanuric acid (as described in the Merck Index or by Linhard in Anorg Allgem Chem 236 200 (1938)) or other means may be used. Nevertheless, we have found that the method using sodium cyanate and an aromatic organic acid, especially benzoic acid, is commercially the most viable. In the preferred method of this invention, therefore, the mild acidic reagent is also capable of reacting with an alkali metal cyanate to produce cyanic acid (HOCN).
Accordingly, the present invention in a preferred aspect provides a process for the preparation of 10-methoxycarbamazepine, which process comprises reacting 10-methoxyiminostilbene with an alkali metal cyanate and a mild acidic reagent, as defined above.
Accordingly, the present invention further provides an improved method for preparing oxcarbazepine from 10-methoxystilbene, wherein the improvement comprises preparing the intermediate 10-methoxycarbamazepine according to the method described above.
The intermediate 10-methoxycarbamazepine is then preferably hydrolysed with an acid, more preferably a dilute mineral acid, such as hydrochloric and sulphuric acids, especially hydrochloric acid (HCI) to furnish oxcarbazepine. Finally, the oxcarbazepine thus obtained may be purified in a mixture of solvent systems selected from both a protic solvent with either an aromatic hydrocarbon solvent or a halogenated aliphatic solvent and an aromatic hydrocarbon solvent with a halogenated aliphatic solvent. Preferably, the mixed solvent system is one wherein the oxcarbazepine is soluble at elevated temperatures, suitably in the range of from 45 to 75° C., but crystallizes therefrom upon cooling. The oxcarbazepine may not be appreciably soluble in any of these solvents individually, but may be soluble in the mixture at elevated temperature. Examples of suitable mixtures include those such as methanol:toluene; dichloromethane:toluene; dichloroethane:toluene; dichloromethane:methanol; and dichloroethane:methanol.
Hydrolysis of the methoxycarbamazepine is preferably carried out in a biphasic system chosen such that the oxcarbazepine is substantially insoluble in both phases, whereas the by-products or impurities are soluble in at least one of the phases. The biphasic system comprises an organic phase and an aqueous phase in which the organic phase preferably comprises the solvent used in the carboxylation reaction eg toluene. Preferably, an excess of this solvent, compared with the amount of impurity or by-product to be produced, is used in the process of this invention. The preferred aqueous phase comprises an aqueous solution of the acid for the hydrolysis step and is therefore most preferably dilute hydrochloric acid. The advantage of this biphasic system is that oxcarbazepine formed in the reaction is thrown out from both the solvents, whereas the impurities remain soluble in the toluene.
Accordingly, the present invention further provides an improved method of hydrolyzing 10-methoxycarbazepine, which improvement comprises carrying out the hydrolysis in a biphasic system as described above.
Especially preferred is when both improved processes of the invention are used, consecutively. The improved processes of the invention enable the oxcarbazepine thereby produced to be purified in a single step.
An especially preferred method according to this invention comprises reaction of 10-methoxy-5H-dibenz[b,f]azepine with benzoic acid and sodium cyanate in toluene at reflux temperature to give 10-methoxy-5H-dibenz[b,f]azepine carboxamide as a major product (such as about 75%), along with 10-oxo-iminodibenzyl and other impurities. The reaction mixture is thereafter filtered and washed with water, and the toluene layer taken as such for hydrolysis in a biphasic system (aqueous hydrochloric acid/toluene) to furnish oxcarbazepine, which is purified just once (whereas twice at least is needed when the prior art process is carried out) in a mixture of methanol and dichloromethane (Scheme-5).

10-methoxyiminostilbene, the key starting material in the following Examples, maybe prepared according to the process disclosed in Belgian patent specification no. 597 793 and Swiss patent specification no. 392 515.
EXAMPLE A Using Monochloro-Acetic Acid and Sodium CyanateA mixture of 100 gms of 10-methoxyiminostilbene in 1000 mL of toluene containing 106 gms of monochloro-acetic acid and 73 gms of sodium cyanate were heated to 40° C. under stirring and maintained for 4 hours. After completion of the reaction (monitored by HPLC and/or TLC), the mixture was cooled to room temperature, filtered and washed with 5% sodium carbonate solution followed by water. The toluene layer was then added to 1000 mL of 2N hydrochloric acid, and the mixture was heated to 75-80° C. and maintained for 2 hours under good agitation. It was then cooled to 0-5° C. and maintained for 2 hours, and the product oxcarbazepine was separated by filtration. This was then purified twice in toluene:methanol followed by methanol:dichloromethane solvent mixture to furnish 28 gms of pure oxcarbazepine.
EXAMPLE 1 Using benzoic acid and sodium cyanateA mixture of 100 gms of 10-methoxyiminostilbene in 2000 mL of toluene containing 274 gms of benzoic acid and 370 gms of sodium cyanate were heated to reflux temperature under stirring and maintained for 12 hours. The reaction mixture was then cooled to room temperature and filtered. The clear toluene filtrate was washed with 5% sodium carbonate solution followed by water. The toluene layer was then added to 1000 mL of 2N hydrochloric acid and the mixture was heated at 75-90° C. for a period of 2 hours under good agitation. It was then cooled to 0-5° C., maintained for 2 hours and the product oxcarbazepine was separated by filtration. This was then purified once in a dichloromethane:methanol mixture to furnish 46 gms of pure oxcarbazepine. Purity was determined by HPLC to be 99.45%.
EXAMPLE 2 Using para-chlorobenzoic acid and sodium cyanateA mixture of 100 gms of 10-methoxyiminostilbene in 1000 mL of toluene containing 351 gms of para-chlorobenzoic acid and 370 gms of sodium cyanate were heated to reflux and refluxed for 12 hours. The reaction mixture was then cooled to room temperature and filtered. The clear toluene filtrate was then washed with 5% sodium carbonate solution followed by water. The toluene layer was then added to 1000 mL of 2N hydrochloric acid and the mixture was heated at 75-80° C. for a period of 2 hours under good agitation. It was then cooled to 0-5° C., maintained for 2 hours and the product oxcarbazepine was separated by filtration. This was then purified once in a dichloromethane methanol mixture to furnish 44 gms of pure oxcarbazepine.
EXAMPLE 3 Alternative Use of benzoic acid and sodium cyanateThe method of Example 1 was repeated, but using 1000 ml toluene; 164 g benzoic acid and 44 g of sodium cyanate, which were heated to 85-90° C. for 14 hours with the 10-methoxyiminostilbene to result in 55 gms of pure oxcarbazepine, found to be 99.45% pure by HPLC.
EXAMPLE 4 Using 2,4-dichloro benzoic acid and sodium cyanateA mixture of 100 gms of 10-methoxyiminostilbene in 1000 mL of toluene containing 430 gms of 2,4-dichlorobenzoic acid and 370 gms of sodium cyanate were heated to reflux and refluxed for 6 hours. The reaction mixture was then cooled to room temperature and filtered. The clear toluene filtrate was then washed with 5% sodium carbonate solution followed by water. The toluene layer was then added to 1000 mL of 2N hydrochloric acid and the mixture was heated at 75-80° C. for a period of 2 hours under good agitation. It was then cooled to 0-5° C., maintained for 2 hours and the product oxcarbazepine was separated by filtration. This was then purified once in a dichloromethane:methanol mixture to furnish 40 gms of pure oxcarbazepine.
EXAMPLE 5 Using benzoic acid and potassium cyanateThe method was carried out according to that described in Example 1, but replacing sodium cyanate with potassium cyanate (461.5 gm) and reflux maintained for 24 hrs to complete consumption of starting material. Following the similar process for hydrolysis and purification produced 32.00 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found 98.80%.
EXAMPLE 6The method was carried out according to that described in Example 1, but replacing 2N hydrochloric with 2N sulphuric acid (1000 mL). Following a similar process of carboxamidation and purification produced 25.00 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found 98.50%.
EXAMPLE 7 Hydrolysis Step Using 2N monochloro-acetic acidThe method was carried out according to that described in Example 1, but replacing 2N hydrochloric acid with 2N monochloro-acetic acid (1000 mL). The reaction mixture was heated to 75° C. to 80° C. and maintained for 24 hrs (after which 20% of unreacted methoxy ISB was found to be present). Under similar conditions for the carboxamidation reaction and purification step, this comparative Example produced 20.00 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found to be 98.00%.
EXAMPLE 8 Purification Using toluene:methanol solvent systemThe method was carried out according to that described in Example 1, but replacing dichloromethane with toluene. Following a similar process of carboxamidation and hydrolysis produced 47.0 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found 98.50%.
EXAMPLE 9 Purification Using toluene:dichloromethane solvent systemThe method was carried out according to that described in Example 1, but replacing methanol with toluene. Following a similar process of carboxamidation and hydrolysis produced 45.00 gm of pure oxcarbazepine. Purity was determined according to Example 1 and found to be 98.00%.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO1996021649A1 | Jan 3, 1996 | Jul 18, 1996 | Trifarma, S.R.L. | A PROCESS FOR THE PREPARATION OF 10-OXO-10,11-DIHYDRO-5H-DIBENZ(b,f)AZEPIN-5-CARBOXAMIDE |
| WO2001056992A2 | Feb 7, 2001 | Aug 9, 2001 | Novartis Ag | Dibenzo (b,f) azepine intermediates |
| Citing Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US7091339 | Jun 13, 2003 | Aug 15, 2006 | Taro Pharmaceuticals Usa, Inc. | Method of preparing a 5H-dibenz(b,f)azepine-5-carboxamide |
| US7125987 | Jun 16, 2005 | Oct 24, 2006 | Apotex Pharmachem Inc. | Process for the preparation of oxcarbazepine and related intermediates |
| US7183272 | Feb 12, 2002 | Feb 27, 2007 | Teva Pharmaceutical Industries Ltd. | Crystal forms of oxcarbazepine and processes for their preparation |
| US7459553 | Mar 11, 2005 | Dec 2, 2008 | Glenmark Generics Ltd. | Process for the preparation of carboxamide compounds |
| US7722898 | Apr 13, 2007 | May 25, 2010 | Supernus Pharmaceuticals, Inc. | Modified-release preparations containing oxcarbazepine and derivatives thereof |
| US7723514 | Jun 29, 2006 | May 25, 2010 | Taro Pharmaceuticals U.S.A., Inc. | Method of preparing a 5H-dibenz(b,f)azepine-5-carboxamide |
| US8530647 | May 6, 2009 | Sep 10, 2013 | Mylan Laboratories Limited | Process for the preparation of oxcarbazepine |
| US20030004154 * | Feb 12, 2002 | Jan 2, 2003 | Judith Aronhime | New crystal forms of oxcarbazepine and processes for their preparation |
| US20040044200 * | Jun 13, 2003 | Mar 4, 2004 | Daniella Gutman | Method of preparing a 5H-dibenz(b,f)azepine-5-carboxamide |
| US20050203297 * | Mar 11, 2005 | Sep 15, 2005 | Sivakumar Bobba V. | Process for the preparation of carboxamide compounds |
| US20050282797 * | Jun 16, 2005 | Dec 22, 2005 | Apotex Pharmachem Inc. | Process for the preparation of oxcarbazepine and related intermediates |
| US20060241292 * | Jun 29, 2006 | Oct 26, 2006 | Taro Pharmaceuticals Usa, Inc | Method of preparing a 5H-dibenz(b,f)azepine-5-carboxamide |
| US20070254033 * | Apr 13, 2007 | Nov 1, 2007 | Supernus Pharmaceuticals, Inc. | Modified-release preparations containing oxcarbazepine and derivatives thereof |
| US20110065917 * | May 6, 2009 | Mar 17, 2011 | Matrix Laboratories Ltd | process for the preparation of oxcarbazepine |
| WO2009139001A2 * | May 6, 2009 | Nov 19, 2009 | Matrix Laboratories Ltd | An improved process for the preparation of oxcarbazepine |
| WO2009139001A3 * | May 6, 2009 | Jan 27, 2011 | Matrix Laboratories Ltd | An improved process for the preparation of oxcarbazepine |
| WO2014049550A1 | Sep 26, 2013 | Apr 3, 2014 | Ranbaxy Laboratories Limited | Process for the preparation of oxcarbazepine and its use as intermediate in the preparation of eslicarbazepine acetate |
Title: Oxcarbazepine
CAS Registry Number: 28721-07-5
CAS Name: 10,11-Dihydro-10-oxo-5H-dibenz[b,f]azepine-5-carboxamide
Additional Names: oxacarbazepine
Manufacturers’ Codes: GP-47680
Trademarks: Trileptal (Novartis)
Molecular Formula: C15H12N2O2
Molecular Weight: 252.27
Percent Composition: C 71.42%, H 4.79%, N 11.10%, O 12.68%
Literature References:
Ketoderivative of carbamazepine, q.v. Prepn: W. Schindler, DE 2011087 (1970 to Geigy); idem, US3642775 (1972 to Ciba-Geigy).
Improved prepn: D. Kaufmann et al., Tetrahedron Lett. 45, 5275 (2004). Metabolism: H. Schütz et al., Xenobiotica 16, 769 (1986).
Hyponatremic effects: O. A. Nielsen et al., Epilepsy Res. 2, 269 (1988). Determn of oxcarbazepine and main metabolites by GC in plasma: G. E. Von Unruh, W. D. Paar, J. Chromatogr. 345, 67 (1985); by HPLC: A. A. Elyas, V. D. Goldberg, ibid. 528, 473 (1990). Clinical evaluation in treatment of epilepsy: M. Dam et al., Epilepsy Res. 3, 70 (1989); in management of trigeminal neuralgia: J. M. Zakrzewska, P. N. Patsalos, J. Neurol. Neurosurg. Psychiatry 52, 472 (1989). Review of pharmacology and therapeutic efficacy: A. Beydoun, E. Kutluay, Expert Opin. Pharmacother. 3, 59-71 (2001).
Properties: Crystals from ethanol, mp 215-216°.
Melting point: mp 215-216°
Therap-Cat: Anticonvulsant.
Keywords: Anticonvulsant.
////////////
TRIENTINE HYDROCHLORIDE, 塩酸トリエンチン , 曲恩汀
TRIENTINE
- Molecular Formula C6H18N4
- Average mass 146.234 Da
112-24-3 CAS
曲恩汀, KD-034, MK-0681, MK-681, TECZA, TETA, TJA-250
1,2-Ethanediamine, N1,N2-bis(2-aminoethyl)-
1,8-diamino-3,6-diazaoctane

TRIENTINE HYDROCHLORIDE
- Molecular Formula C6H19ClN4
- Average mass 182.695 Da
38260-01-4 CAS
Launched – 1986 VALEANT, WILSONS DISEASE

塩酸トリエンチン
Trientine Hydrochloride

C6H18N4 2HCl : 219.16
2HCl : 219.16
[38260-01-4]
UPDATE CDSCO INDIA Trientine 08.06.2021 APPROVED
Trientine Tetrahydrochloride bulk and
Trientine Tetrahydrochloride capsules 333 mg
(Each capsule contains Trientine
tetrahydrochloride 333mg equivalent to
Trientine 167mg base)
For the treatment of Wilson’s disease
(hepatolenticular degeneration) in patients
intolerant to Penicillamine. It should be
used when continued treatment with
Penicillamine is no longer possible because
of intolerable or life endangering side
effects.
Aton Pharma, a subsidiary of Valeant Pharmaceuticals, has developed and launched Syprine, a capsule formulation of trientine hydrochloride, for treating Wilson disease.

Triethylenetetramine, abbreviated TETA and trien and also called trientine (INN), is an organic compound with the formula [CH2NHCH2CH2NH2]2. This oily liquid is colorless but, like many amines, assumes a yellowish color due to impurities resulting from air-oxidation. It is soluble in polar solvents. The branched isomer tris(2-aminoethyl)amine and piperazine derivatives may also be present in commercial samples of TETA.[1]
Trientine hydrochloride is a metal antagonist that was first launched by Merck, Sharp & Dohme in the U.S. in 1986 under the brand name Syprine for the oral treatment of Wilson’s disease.
Orphan drug designation has also been assigned in the U.S. for the treatment of patients with Wilson’s disease who are intolerant or inadequately responsive to penicillamine and in the E.U. by Univar for the treatment of Wilson’s disease

By condensation of ethylenediamine (I) with 1,2-dichloroethane (II)
Trientine hydrochloride is N,N’-bis (2-aminoethyl)-1,2-ethanediamine dihydrochloride. It is a white to pale yellow crystalline hygroscopic powder. It is freely soluble in water, soluble in methanol, slightly soluble in ethanol, and insoluble in chloroform and ether.
The empirical formula is C6H18N4·2HCI with a molecular weight of 219.2. The structural formula is:
NH2(CH2)2NH(CH2)2NH(CH2)2NH2•2HCI
Trientine hydrochloride is a chelating compound for removal of excess copper from the body. SYPRINE (Trientine Hydrochloride) is available as 250 mg capsules for oral administration. Capsules SYPRINE contain gelatin, iron oxides, stearic acid, and titanium dioxide as inactive ingredients.

Production
TETA is prepared by heating ethylenediamine or ethanolamine/ammonia mixtures over an oxide catalyst. This process gives a variety of amines, which are separated by distillation and sublimation.[2]
Uses
The reactivity and uses of TETA are similar to those for the related polyamines ethylenediamine and diethylenetriamine. It was primarily used as a crosslinker (“hardener”) in epoxy curing.[2]
The hydrochloride salt of TETA, referred to as trientine hydrochloride, is a chelating agent that is used to bind and remove copper in the body to treat Wilson’s disease, particularly in those who are intolerant to penicillamine. Some recommend trientine as first-line treatment, but experience with penicillamine is more extensive.[3]
Coordination chemistry
TETA is a tetradentate ligand in coordination chemistry, where it is referred to as trien.[4] Octahedral complexes of the type M(trien)Cl3 can adopt several diastereomeric structures, most of which are chiral.[5]
Trientine, chemically known as triethylenetetramine or N,N’-bis(2-aminoethyl)-l,2-ethanediamine belongs to the class of polyethylene polyamines. Trientine dihydrochloride is a chelating agent which is used to bind and remove copper in the body in the treatment of Wilson’s disease.
Trientine dihydrochloride (1)
Trientine dihydrochloride formulation, developed by Aton with the proprietary name SYPRINE, was approved by USFDA on November 8, 1985 for the treatment of patients with Wilson’s disease, who are intolerant to penicillamine. Trientine dihydrochloride, due to its activity on copper homeostasis, is being studied for various potential applications in the treatment of internal organs damage in diabetics, Alzheimer’s disease and cancer.
Various synthetic methods for preparation of triethylenetetramine (TETA) and the corresponding dihydrochloride salt have been disclosed in the prior art.
U.S. 4,806,517 discloses the synthesis of triethylenetetramine from ethylenediamine and monoethanolamine using Titania supported phosphorous catalyst while U.S. 4,550,209 and U.S. 5,225,599 disclose catalytic condensation of ethylenediamine and ethylene glycol for the synthesis of linear triethylenetetramine using catalysts like zirconium trimethylene diphosphonate, or metatungstate composites of titanium dioxide and zirconium dioxide.
U.S. 4,503,253 discloses the preparation of triethylenetetramine by reaction of an alkanolamine compound with ammonia and an alkyleneamine having two primary amino groups in the presence of a catalyst, such as supported phosphoric acid wherein the support is comprised of silica, alumina or carbon.
The methods described above for preparation of triethylenetetramine require high temperatures and pressure. Further, due to the various possible side reactions and consequent associated impurities, it is difficult to control the purity of the desired amine.
CN 102924289 discloses a process for trientine dihydrochloride comprising reduction of Ν,Ν’-dibenzyl-,N,N’-bis[2-(l,3-dioxo-2H-isoindolyl)ethyl]ethanediamine using hydrazine hydrate to give N,N’-dibenzyl-,N,N’-bis(2-aminoethyl)ethanediamine, which, upon condensation with benzyl chloroformate gave N,N’-dibenzyl-,N,N’-bis[2-(Cbz-amino)ethyl]ethanediamine, and further reductive deprotection to give the desired compound.
CS 197,093 discloses a process comprising reaction of triethylenetetramine with concentrated hydrochloric acid to obtain the crystalline tetrahydrochlonde salt. Further reaction of the salt with sodium ethoxide in solvent ethanol, filtration of the solid sodium chloride which is generated in the process, followed by slow cooling and crystallization of the filtrate provided the dihydrochloride salt. Optionally, aqueous solution of the tetrahydrochloride salt was passed through a column of an anion exchanger and the eluate containing free base was treated with a calculated amount of the tetrahydrochloride, evaporated, and the residue was crystallized from aqueous ethanol to yield the dihydrochloride salt.
The process is quite circuitous and cumbersome, requiring use of strong bases, filtration of sodium chloride and results in yields as low as 60%.
US 8,394,992 discloses a method for preparation of triethylenetetramine dihydrochloride wherein tertiary butoxycarbonyl (boc) protected triethylenetetramine is first converted to its tetrahydrochloride salt using large excess of hydrochloric acid in solvent isopropanol, followed by treatment of the resulting tetrahydrochloride salt with a strong base like sodium alkoxide to produce the amine free base (TETA) and sodium chloride salt in anhydrous conditions. The free amine is extracted with tertiary butyl methyl ether (TBME), followed by removal of sodium chloride salt and finally the amine free base TETA is treated with hydrochloric acid in solvent ethanol to give trientine hydrochloride salt.
PATENT
WO-2017046695
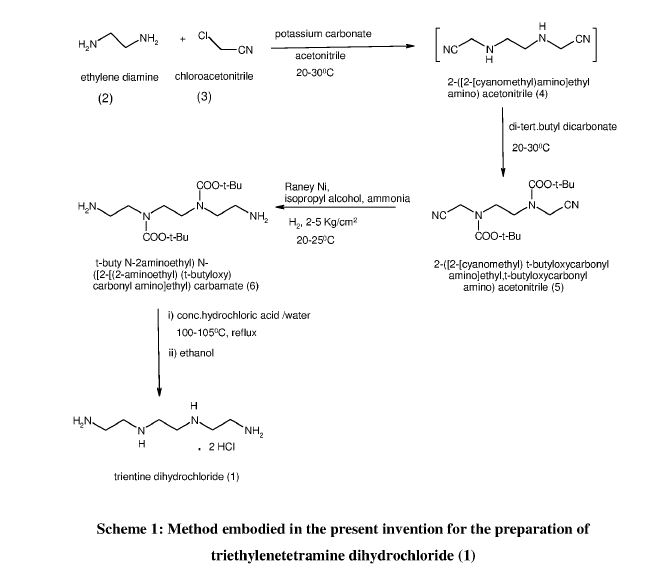
EXAMPLES
Example 1: Preparation of 2-([2-[cyanomethyl]-t-butyloxycarbonylamino]ethyl- 1-butyloxy carbonylamino)acetonitrile (5)
Potassium carbonate (481.9 g) was added to a stirred mixture of ethylenediamine (100.0 g) in acetonitrile (800 ml) and cooled to around 10°C. Chloroacetonitrile (263.8 g) was gradually added at same temperature and stirred at 25-30°C, till completion of the reaction, as monitored by HPLC. The mixture was cooled to 5-15°C and Boc-anhydride (762. lg) was added to it, followed by stirring at the same temperature. The temperature was raised to 25-30°C and the mass was stirred till completion of the reaction, as monitored by HPLC.
The reaction mass was filtered and the filtrate was concentrated. Toluene was added to the residue, and the mixture was heated to around 70°C followed by cooling and filtration to give 2-([2-[cyanomethyl)-t-butyloxycarbonylamino]ethyl-t-butyloxycarbonylamino) acetonitrile (5).
Yield: 506.8 g
% Yield: 89.9 %
Example 2: Preparation of t-butyl( N-2-aminoethyl)N-([2-[(2-aminoethyl)t-butyloxy)carbonylamino] ethyl) carbamate (6)
Raney nickel (120.0 g) in isopropanol (100 ml) was charged into an autoclave, followed by a mixture of Compound 5 (200 g) in isopropanol (400 ml). Cooled ammonia solution prepared by purging ammonia gas in 1400 ml isopropanol, equivalent to 125 g ammonia was gradually charged to the autoclave and the reaction was carried out around 15-25°C under hydrogen pressure of 2-5 Kg/cm2.
After completion of the reaction, as monitored by HPLC, the mass was filtered, concentrated, and methyl tertiary butyl ether was added to the residue. The mixture was heated to around 50°C, followed by cooling of the mass, stirring, optional seeding with compound 6 and filtration to give tertiary butyl-(N-2-aminoethyl)N-([2-[(2-aminoethyl)-(tert-butyloxy) carbonylamino] ethyl) carbamate.
Yield: 174 g
%Yield: 85 %
Example 3: Preparation of triethylenetetramine dihydrochloride (1)
Concentrated hydrochloric acid (121.5 g) was gradually added to a stirred mixture of tertiary-butyl-N-(2-aminoethyl)-N-2-[(2-aminoethyl)-(tert-butoxy) carbonyl] amino] ethyl} carbamate (Compound 6, 200.0 g) and water (1400 ml) at 20-30°C. The reaction mixture was heated in the temperature range of 100-105°C till completion of the reaction, as monitored by HPLC, with optionally distilling out water, if so required.
The reaction mass was concentrated and ethanol (600 ml) was added to the residue, followed by heating till a clear solution was obtained. The reaction mixture was gradually cooled with stirring, filtered and dried to provide triethylenetetramine dihydrochloride (1).
Yield: 88.9 g, (70 %)
Purity : > 99%
Patent
https://www.google.com/patents/US8394992
Trientine was said to be used in the synthesis of benzylidene-(2-{3-[2-(benzylidene-amino)-ethyl]-2-phenyl-imidazolidin-1-yl}-ethyl)-amine in French Patent No. FR2810035 to Guilard et al. Cetinkaya, E., et al., “Synthesis and characterization of unusual tetraminoalkenes,” J. Chem. Soc. 5:561-7 (1992), is said to be directed to synthesis of benzylidene-(2-{3-[2-(benzylidene-amino)-ethyl]-2-phenyl-imidazolidin-1-yl}-ethyl)-amine from trientine, as is Araki T., et al., “Site-selective derivatization of oligoethyleneimines using five-membered-ring protection method,” Macromol., 21:1995-2001 (1988). Triethylenetetramine may reportedly also be used in the synthesis of N-methylated triethylenetetramine, as reported in U.S. Pat. No. 2,390,766, to Zellhoefer et al.
Synthesis of polyethylenepolyamines, including triethylenetetramines, from ethylenediamine and monoethanolamine using pelleted group IVb metal oxide-phosphate type catalysts was reported by Vanderpool et al. in U.S. Pat. No. 4,806,517. Synthesis of triethylenetetramine from ethylenediamine and ethanolamine was also proposed in U.S. Pat. No. 4,550,209, to Unvert et al. U.S. Pat. No. 5,225,599, to King et al. is said to be directed to the synthesis of linear triethylene tetramine by condensation of ethylenediamine and ethylene glycol in the presence of a catalyst. Joint production of triethylenetetramine and 1-(2-aminoethyl)-aminoethyl-piperazine was proposed by Borisenko et al. in U.S.S.R. Patent No. SU1541204. U.S. Pat. No. 4,766,247 and European Patent No. EP262562, both to Ford et al., reported the preparation of triethylenetetramine by reaction of an alkanolamine compound, an alkaline amine and optionally either a primary or secondary amine in the presence of a phosphorous containing catalyst, for example phosphoric acid on silica-alumina or Group IIIB metal acid phosphate, at a temperature from about 175° C. to 400° C. under pressure. These patents indicate that the synthetic method used therein was as set forth in U.S. Pat. No. 4,463,193, to Johnson. The Ford et al. ‘247 patent is also said to be directed to color reduction of polyamines by reaction at elevated temperature and pressure in the presence of a hydrogenation catalyst and a hydrogen atmosphere. European Patent No. EP450709 to King et al. is said to be directed to a process for the preparation of triethylenetetramine and N-(2-aminoethyl)ethanolamine by condensation of an alkylenamine and an alkylene glycol in the presence of a condensation catalyst and a catalyst promoter at a temperature in excess of 260° C.
Russian Patent No. RU2186761, to Zagidullin, proposed synthesis of diethylenetriamine by reaction of dichloroethane with ethylenediamine. Ethylenediamine has previously been said to have been used in the synthesis of N-carboxylic acid esters as reported in U.S. Pat. No. 1,527,868, to Hartmann et al.
Japanese Patent No. 06065161 to Hara et al. is said to be directed to the synthesis of polyethylenepolyamines by reacting ethylenediamine with ethanolamine in the presence of silica-treated Nb205 supported on a carrier. Japanese Patent No. JP03047154 to Watanabe et al., is said to be directed to production of noncyclic polyethylenepolyamines by reaction of ammonia with monoethanolamine and ethylenediamine. Production of non-cyclic polyethylenepolyamines by reaction of ethylenediamine and monoethanolamine in the presence of hydrogen or a phosphorous-containing substance was said to be reported in Japanese Patent No. JP03048644. Regenerative preparation of linear polyethylenepolyamines using a phosphorous-bonded catalyst was proposed in European Patent No. EP115,138, to Larkin et al.
A process for preparation of alkyleneamines in the presence of a niobium catalyst was said to be provided in European Patent No. 256,516, to Tsutsumi et al. U.S. Pat. No. 4,584,405, to Vanderpool, reported the continuous synthesis of essentially noncyclic polyethylenepolyamines by reaction of monoethanolamine with ethylenediamine in the presence of an activated carbon catalyst under a pressure between about 500 to about 3000 psig., and at a temperature of between about 200° C. to about 400° C. Templeton, et al., reported on the preparation of linear polyethylenepolyamides asserted to result from reactions employing silica-alumina catalysts in European Patent No. EP150,558.
Production of triethylenetetramine dihydrochloride was said to have been reported in Kuhr et al., Czech Patent No. 197,093, via conversion of triethylenetetramine to crystalline tetrahydrochloride and subsequently to triethylenetetramine dihydrochloride. “A study of efficient preparation of triethylenetetramine dihydrochloride for the treatment of Wilson’s disease and hygroscopicity of its capsule,” Fujito, et al., Yakuzaigaku, 50:402-8 (1990), is also said to be directed to production of triethylenetetramine.
Preparation of triethylenetetramine salts used for the treatment of Wilson’s disease was said to be reported in “Treatment of Wilson’s Disease with Triethylene Tetramine Hydrochloride (Trientine),” Dubois, et al., J. Pediatric Gastro. & Nutrition, 10:77-81 (1990); “Preparation of Triethylenetetramine Dihydrochloride for the Treatment of Wilson’s Disease,” Dixon, et al., Lancet, 1(1775):853 (1972); “Determination of Triethylenetetramine in Plasma of Patients by High-Performance Liquid Chromatography,” Miyazaki, et al., Chem. Pharm. Bull., 38(4):1035-1038 (1990); “Preparation of and Clinical Experiences with Trien for the Treatment of Wilson’s Disease in Absolute Intolerance of D-penicillamine,” Harders, et al., Proc. Roy. Soc. Med., 70:10-12 (1977); “Tetramine cupruretic agents: A comparison in dogs,” Allen, et al., Am. J. Vet. Res., 48(1):28-30 (1987); and “Potentiometric and Spectroscopic Study of the Equilibria in the Aqueous Copper(II)-3,6-Diazaoctane-1,8-diamine System,” Laurie, et al., J.C.S. Dalton, 1882 (1976).
Preparation of Triethylenetetramine Salts by Reaction of Alcohol Solutions of Amines and acids was said to be reported in Polish Patent No. 105793, to Witek. Preparation of triethylenetetramine salts was also asserted in “Polycondensation of polyethylene polyamines with aliphatic dicarboxylic acids,” Witek, et al., Polimery, 20(3):118-119 (1975).
Baganz, H., and Peissker, H., Chem. Ber., 1957; 90:2944-2949; Haydock, D. B., and Mulholland, T. P. C., J. Chem. Soc., 1971; 2389-2395; and Rehse, K., et al., Arch. Pharm., 1994; 393-398, report on Strecker syntheses. Use of Boc and other protecting groups has been described. See, for example, Spicer, J. A. et al., Bioorganic & Medicinal Chemistry, 2002; 10: 19-29; Klenke, B. and Gilbert, I. H., J. Org. Chem., 2001; 66: 2480-2483.
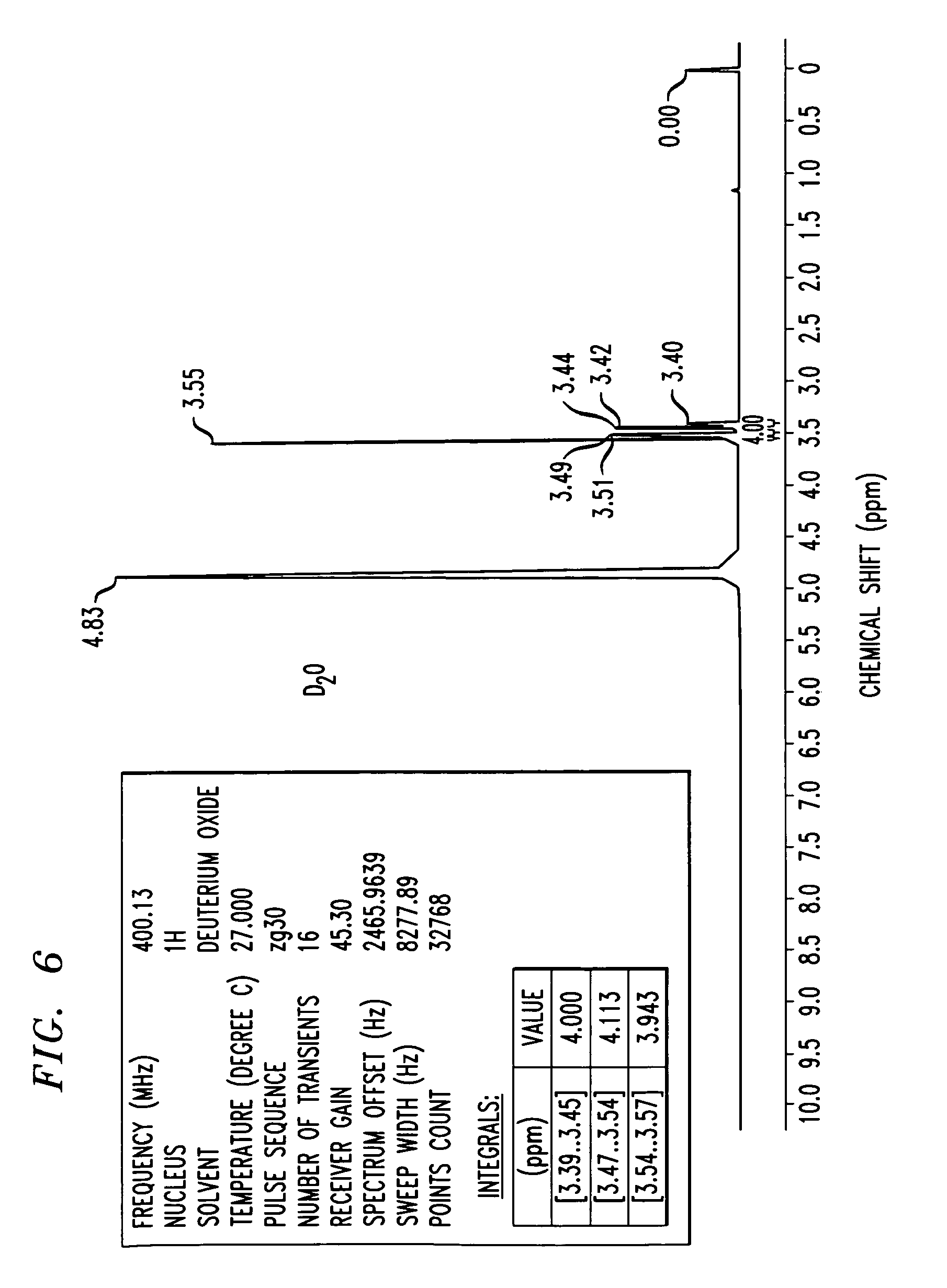
FIG. 6 shows an 1H-NMR spectrum of a triethylenetetramine hydrochloride salt in D2O, as synthesized in Example 3. NMR values include a frequency of 400.13 Mhz, a 1H nucleus, number of transients is 16, points count of 32768, pulse sequence of zg30, and sweep width of 8278.15 H
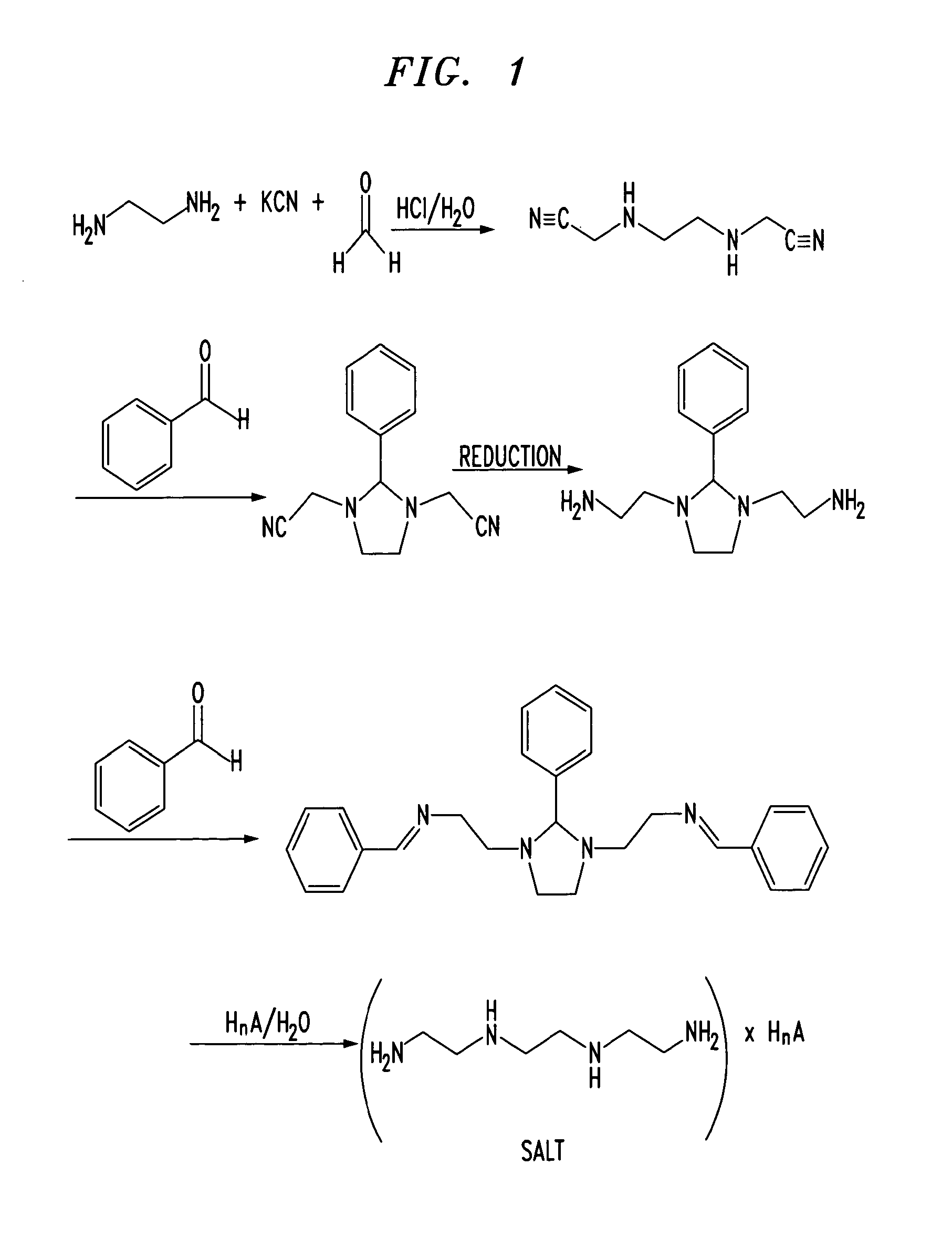
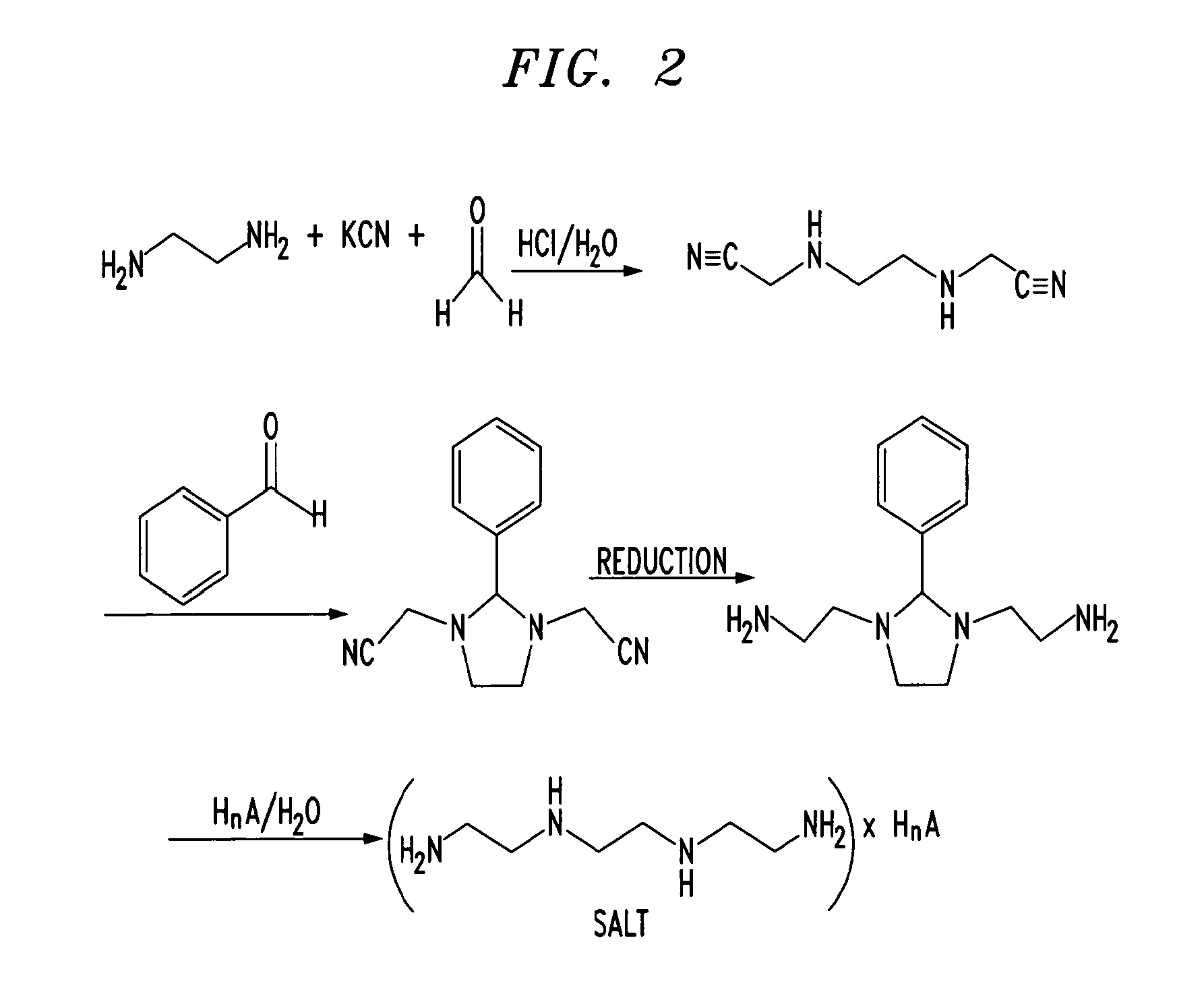

CLIP
Method of purification: Dissolve Trientine Hydrochloride in water while warming, and recrystallize by addition of ethanol (99.5). Or dissolve Trientine Hydrochloride in water while warming, allow to stand after addition of activated charcoal in a cool and dark place for one night, and filter. To the filtrate add ethanol (99.5), allow to stand in a cool and dark place, and recrystallize. Dry the crystals under reduced pressure not exceeding 0.67 kPa at 409C until ethanol odor disappears.
References
- “Ethyleneamines” (PDF). Huntsman. 2007.
- ^ Jump up to:a b Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. (2005). “Amines, Aliphatic”. Ullmann’s Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a02_001.
- Jump up^ Roberts, E. A.; Schilsky, M. L. (2003). “A practice guideline on Wilson disease” (pdf). Hepatology. 37 (6): 1475–1492. doi:10.1053/jhep.2003.50252. PMID 12774027.
- Jump up^ von Zelewsky, A. (1995). Stereochemistry of Coordination Compounds. Chichester: John Wiley. ISBN 047195599X.
- Utsuno, S.; Sakai, Y.; Yoshikawa, Y.; Yamatera, H. (1985). “Three Isomers of the Trans-Diammine-[N,N′-bis(2-Aminoethyl)-1,2-Ethanediamine]-Cobalt(III) Complex Cation”. Inorganic Syntheses. 23: 79–82. doi:10.1002/9780470132548.ch16.
 |
|
 |
|
 |
|
| Names | |
|---|---|
| Other names
N,N’-Bis(2-aminoethyl)ethane-1,2-diamine; TETA; Trien; Trientine (INN); Syprine (brand name)
|
|
| Identifiers | |
|
3D model (Jmol)
|
|
| 605448 | |
| ChEBI | |
| ChemSpider | |
| ECHA InfoCard | 100.003.591 |
| EC Number | 203-950-6 |
| 27008 | |
| KEGG | |
| MeSH | Trientine |
|
PubChem CID
|
|
| RTECS number | YE6650000 |
| UNII | |
| UN number | 2259 |
| Properties | |
| C6H18N4 | |
| Molar mass | 146.24 g·mol−1 |
| Appearance | Colorless liquid |
| Odor | Fishy, ammoniacal |
| Density | 982 mg mL−1 |
| Melting point | −34.6 °C; −30.4 °F; 238.5 K |
| Boiling point | 266.6 °C; 511.8 °F; 539.7 K |
| Miscible | |
| log P | 1.985 |
| Vapor pressure | <1 Pa (at 20 °C) |
|
Refractive index (nD)
|
1.496 |
| Thermochemistry | |
| 376 J K−1 mol−1 (at 60 °C) | |
| Pharmacology | |
| A16AX12 (WHO) | |
| Hazards | |
| GHS pictograms |   |
| GHS signal word | DANGER |
| H312, H314, H317, H412 | |
| P273, P280, P305+351+338, P310 | |
|
EU classification (DSD)
|
|
| R-phrases | R21, R34, R43, R52/53 |
| S-phrases | (S1/2), S26, S36/37/39, S45 |
| Flash point | 129 °C (264 °F; 402 K) |
| Lethal dose or concentration (LD, LC): | |
|
LD50 (median dose)
|
|
| Related compounds | |
|
Related amines
|
|
|
Related compounds
|
|
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
///////////////TRIENTINE, 112-24-3, 曲恩汀 , KD-034 , MK-0681, MK-681, TECZA, TETA, TJA-250, Orphan drug
NCCNCCNCCN
Calcifediol, カルシフェジオール

Calcifediol
カルシフェジオール
Ro 8-8892
U 32070E
(3b,5Z,7E)-9,10-Secocholesta-5,7,10(19)-triene-3,25-diol
(3S,5Z,7E,20R)-9,10-Secocholesta-5,7,10-trien-3,25-diol [German] [ACD/IUPAC Name]
(3S,5Z,7E,20R)-9,10-Secocholesta-5,7,10-triene-3,25-diol [ACD/IUPAC Name]
(3S,5Z,7E,20R)-9,10-Sécocholesta-5,7,10-triène-3,25-diol [French] [ACD/IUPAC Name]
19356-17-3 [RN]
1H-indene-1-pentanol, octahydro-4-[(2Z)-2-[(5S)-5-hydroxy-2-methylenecyclohexylidene]ethylidene]-a,a,e,7a-tetramethyl-, (eR,1R,3aS,4E,7aR)-
25(OH)D3
25-(OH)Vitamin D3
25-hydroxy Vitamin D3
25-HYDROXYCHOLECALCIFEROL-D6
25-hydroxycholecalciferolmonohydrate
25-hydroxyvitamin D
3-{2-[1-(5-Hydroxy-1,5-dimethyl-hexyl)-7a-methyl-octahydro-inden-4-ylidene]-ethylidene}-4-methylene-cyclohexanol
4-[(2Z)-2-[(5S)-5-hydroxy-2-methylenecyclohexylidene]ethylidene]octahydro-?,?,?,7a-tetramethyl-(?R,1R,3aS,4E,7aR)-1H-indene-1-pentanol
| Molecular form.: | C₂₇H₄₄O₂ |
| Appearance: | White to Off-White Solid |
| Melting Point: | 75-93ºC |
| Mol. Weight: | 400.64 |
Calcifediol (INN), also known as calcidiol, 25-hydroxycholecalciferol, or 25-hydroxyvitamin D (abbreviated 25(OH)D),[1] is a prehormone that is produced in the liver by hydroxylation of vitamin D3 (cholecalciferol) by the enzyme cholecalciferol 25-hydroxylase which was isolated by Michael F. Holick. Physicians worldwide measure this metabolite to determine a patient’s vitamin D status.[2] At a typical daily intake of vitamin D3, its full conversion to calcifediol takes approximately 7 days.[3]
Calcifediol is then converted in the kidneys (by the enzyme 25(OH)D-1α-hydroxylase) into calcitriol (1,25-(OH)2D3), a secosteroid hormone that is the active form of vitamin D. It can also be converted into 24-hydroxycalcidiol in the kidneys via 24-hydroxylation.[4][5]
Blood test
In medicine, a 25-hydroxy vitamin D (calcifediol) blood test is used to determine how much vitamin D is in the body.[6] The blood concentration of calcifediol is considered the best indicator of vitamin D status.[7]
This test can be used to diagnose vitamin D deficiency, and it is indicated in patients with high risk for vitamin D deficiency and when the results of the test would be used as supporting evidence for beginning aggressive therapies.[8] Patients with osteoporosis, chronic kidney disease, malabsorption, obesity, and some other infections may be high risk and thus have greater indication for this test.[8] Although vitamin D deficiency is common in some populations including those living at higher latitudes or with limited sun exposure, the 25(OH)D test is not indicated for entire populations.[8] Physicians may advise low risk patients to take over-the-counter vitamin D in place of having screening.[8]
It is the most sensitive measure,[9] though experts have called for improved standardization and reproducibility across different laboratories.[7] According to MedlinePlus, the normal range of calcifediol is 30.0 to 74.0 ng/mL.[6] The normal range varies widely depending on several factors, including age and geographic location. A broad reference range of 20–150 nmol/L (8-60 ng/ml) has also been suggested,[10] while other studies have defined levels below 80 nmol/L (32 ng/ml) as indicative of vitamin D deficiency.[11]
US labs generally report 25(OH)D levels as ng/mL. Other countries often use nmol/L. Multiply ng/mL by 2.5 to convert to nmol/L.
Clinical significance
Increasing calcifediol levels are associated with increasing fractional absorption of calcium from the gut up to levels of 80 nmol/L (32 ng/mL).[citation needed]Urinary calcium excretion balances intestinal calcium absorption and does not increase with calcifediol levels up to ~400 nmol/L (160 ng/mL).[12]
A study by Cedric F. Garland and Frank C. Garland of the University of California, San Diego analyzed the blood from 25,000 volunteers from Washington County, Maryland, finding that those with the highest levels of calcifediol had a risk of colon cancer that was one-fifth of typical rates.[13] However, randomized controlled trials failed to find a significant correlation between vitamin D supplementation and the risk of colon cancer.[14]
A 2012 registry study of the population of Copenhagen, Denmark, found a correlation between both low and high serum levels and increased mortality, with a level of 50–60 nmol/L being associated with the lowest mortality. The study did not show causation.[15][16]
Nmr
Regioselective Hydroxylation in the Production of 25-Hydroxyvitamin D by Coprinopsis cinerea Peroxygenase
ChemCatChem (2015), 7, (2), 283-290
1H NMR 500 MHz, CDCl3: δ= 0.55 (3 H, s, 18-H), 0.94 (1H, d, J= 6.5 Hz, 21-H), 1.06 (1H, m, 22-H), 1.22 (3 H, s, 26-H), 1.22 (3 H, s, 27-H), 1.23 (1H, m, 23-H), 1.27 (1H, m, 16-H), 1.28 (1H, m, 14-H), 1.29 (1H, m, 12-H), 1.37 (1H, m, 22-H), 1.38 (1H, m, 20-H), 1.39 (1H, m, 24-H), 1.42 (1H, m, 23-H), 1.44 (1H, m, 24-H), 1.47 (2 H, m, 11-H), 1.53 (1H, m, 15-H), 1.66 (1H, m, 15-H), 1.67 (1H, m, 2-H), 1.67 (1H, m, 9-H), 1.87 (1H, m, 16-H), 1.92 (1H, m, 2-H), 1.98 (1H, m, 17-H), 2.06 (1H, m, 12-H), 2.17 (1H, m, 1-H), 2.40 (1H, m, 1-H), 2.57 (1H, dd, J= 3.7, 13.1Hz, 4-H), 2.82 (1H, m, 9-H), 3.95 (1H, bm, 3-H), 4.82 (1H, m, 19-H), 5.05 (1H, m, 19-H), 6.03 (1H, d, J=11.2 Hz, 7-H), 6.23 ppm (1H, d, J= 11.2 Hz, 6-H).
13 C NMR 500 MHz, CDCl3: δ = 12.2 (C-18), 19.0 (C-21), 21.0 (C-23), 22.4 (C-11), 23.7 (C-15), 27.8 (C-16), 29.2 (C-9), 29.4 (C-27), 29.5 (C-26), 32.1 (C-1), 35.3 (C-2), 36.3 (C-20), 36.6 (C-22), 40.7 (C-12), 44.6 (C-24), 46.0 (C-13), 46.1 (C-4), 56.5 (C-17), 56.7 (C-14), 69.4 (C-3), 71.3 (C-25), 112.6 (C-19), 117.7 (C-7), 122.2 (C-6), 135.2 (C-5), 142.4 (C-8), 145.3 ppm (C-10).
PAPER
From Organic & Biomolecular Chemistry, 10(27), 5205-5211; 2012
http://pubs.rsc.org/en/content/articlelanding/2012/ob/c2ob25511a#!divAbstract
An efficient, two-stage, continuous-flow synthesis of 1α,25-(OH)2-vitamin D3 (activated vitamin D3) and its analogues was achieved. The developed method afforded the desired products in satisfactory yields using a high-intensity and economical light source, i.e., a high-pressure mercury lamp. In addition, our method required neither intermediate purification nor high-dilution conditions.

1H NMR(400 MHz, CDCl3): δ 8.13 (m, 2H), 7.68 (m, 2H), 6.64 (d, J = 8.3 Hz, 1H), 6.25 (d, J = 8.3 Hz, 1H), 5.19 (m, 2H), 3.93 (dd, J = 12.7, 8.2, 1H), 3.88 (dd, J = 14.6, 4.9 Hz, 1H), 3.58 (m, 1H), 1.02 (s, 3H), 1.02 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H), 0.86 (s, 9H), 0.80-0.84 (m, 9H), 0.09 (s, 3H), 0.00 (s, 3H)
13C NMR (100 MHz, CDCl3): δ 161.8, 159.6, 138.5, 135.3, 132.6, 132.5, 132.1, 130.6, 130.2, 128.7, 127.0, 126.5, 77.2, 68.5, 67.4, 67.1, 56.5, 50.6, 49.0, 44.2, 42.7, 40.4, 39.9, 39.3, 35.6, 34.7, 33.0, 30.5, 28.2, 25.9, 24.5, 21.9, 20.8, 19.9, 19.7, 18.5, 18.0, 17.4, 13.3, -4.4, -4.9
| IR (neat): 2957, 2872, 1653, 1603, 1462, 1311, 1093, 837, 762 cm-1 |
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles. [§ 1]
[[File:
|{{{bSize}}}px|alt=Vitamin D Synthesis Pathway]
Vitamin D Synthesis Pathway edit
- Jump up^ The interactive pathway map can be edited at WikiPathways: “VitaminDSynthesis_WP1531”.
References
- Jump up^ “Nomenclature of Vitamin D. Recommendations 1981. IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN)” reproduced at the Queen Mary, University of London website. Retrieved 21 March 2010.
- Jump up^ Holick, MF; Deluca, HF; Avioli, LV (1972). “Isolation and identification of 25-hydroxycholecalciferol from human plasma”. Archives of Internal Medicine. 129 (1): 56–61. doi:10.1001/archinte.1972.00320010060005. PMID 4332591.
- Jump up^ Am J Clin Nutr 2008;87:1738–42 PMID 18541563
- Jump up^ Bender, David A.; Mayes, Peter A (2006). “Micronutrients: Vitamins & Minerals”. In Victor W. Rodwell; Murray, Robert F.; Harper, Harold W.; Granner, Darryl K.; Mayes, Peter A. Harper’s Illustrated Biochemistry. New York: Lange/McGraw-Hill. pp. 492–3. ISBN 0-07-146197-3. Retrieved December 10, 2008 through Google Book Search.
- Jump up^ Institute of Medicine (1997). “Vitamin D”. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, D.C: National Academy Press. p. 254. ISBN 0-309-06403-1.
- ^ Jump up to:a b “25-hydroxy vitamin D test: Medline Plus”. Retrieved 21 March 2010.
- ^ Jump up to:a b Heaney, Robert P (Dec 2004). “Functional indices of vitamin D status and ramifications of vitamin D deficiency”. American Journal of Clinical Nutrition. 80 (6): 1706S–9S. PMID 15585791.
- ^ Jump up to:a b c d American Society for Clinical Pathology, “Five Things Physicians and Patients Should Question”, Choosing Wisely: an initiative of the ABIM Foundation, American Society for Clinical Pathology, retrieved August 1, 2013, which cites
-
- Sattar, N.; Welsh, P.; Panarelli, M.; Forouhi, N. G. (2012). “Increasing requests for vitamin D measurement: Costly, confusing, and without credibility”. The Lancet. 379 (9811): 95–96. doi:10.1016/S0140-6736(11)61816-3. PMID 22243814.
- Bilinski, K. L.; Boyages, S. C. (2012). “The rising cost of vitamin D testing in Australia: Time to establish guidelines for testing”. The Medical Journal of Australia. 197 (2): 90. doi:10.5694/mja12.10561. PMID 22794049.
- Lu, Chuanyi M. (May 2012). “Pathology consultation on vitamin D testing: Clinical indications for 25(OH) vitamin D measurement [Letter to the editor]”. American Journal Clinical Pathology. American Society for Clinical Pathology (137): 831–832., which cites
- Arya, S. C.; Agarwal, N. (2012). “Pathology Consultation on Vitamin D Testing: Clinical Indications for 25(OH) Vitamin D Measurement”. American Journal of Clinical Pathology. 137 (5): 832. doi:10.1309/AJCP2GP0GHKQRCOE. PMID 22523224.
- Holick, M. F.; Binkley, N. C.; Bischoff-Ferrari, H. A.; Gordon, C. M.; Hanley, D. A.; Heaney, R. P.; Murad, M. H.; Weaver, C. M. (2011). “Evaluation, Treatment, and Prevention of Vitamin D Deficiency: An Endocrine Society Clinical Practice Guideline”. Journal of Clinical Endocrinology & Metabolism. 96 (7): 1911–1930. doi:10.1210/jc.2011-0385. PMID 21646368.
-
- Jump up^ Institute of Medicine (1997), p. 259
- Jump up^ Bender, David A. (2003). “Vitamin D”. Nutritional biochemistry of the vitamins. Cambridge: Cambridge University Press. ISBN 0-521-80388-8. Retrieved December 10, 2008 through Google Book Search.
- Jump up^ Hollis BW (February 2005). “Circulating 25-hydroxyvitamin D levels indicative of vitamin D sufficiency: implications for establishing a new effective dietary intake recommendation for vitamin D”. J Nutr. 135 (2): 317–22. PMID 15671234.
- Jump up^ Kimball; et al. (2004). “Safety of vitamin D3 in adults with multiple sclerosis”. J Clin Endocrinol Metab. 86 (3): 645–51. PMID 17823429.
- Jump up^ Maugh II, Thomas H. “Frank C. Garland dies at 60; epidemiologist helped show importance of vitamin D: Garland and his brother Cedric were the first to demonstrate that vitamin D deficiencies play a role in cancer and other diseases.”, Los Angeles Times, August 31, 2010. Accessed September 4, 2010.
- Jump up^ Wactawski-Wende, J; Kotchen, JM, Women’s Health Initiative Investigators (Mar 9, 2006). “Calcium plus vitamin D supplementation and the risk of colorectal cancer.”. N Engl J Med. 354 (7): 684–96. doi:10.1056/NEJMoa055222. PMID 16481636. Retrieved December 28, 2013.
- Jump up^ “Too much vitamin D can be as unhealthy as too little” (Press release). University of Copenhagen. May 29, 2012. Retrieved 2015-05-27.
- Jump up^ Durup, D.; Jørgensen, H. L.; Christensen, J.; Schwarz, P.; Heegaard, A. M.; Lind, B. (May 9, 2012). “A Reverse J-Shaped Association of All-Cause Mortality with Serum 25-Hydroxyvitamin D in General Practice: The CopD Study”. The Journal of Clinical Endocrinology & Metabolism. Endocrine Society. 97 (8): 2644–2652. doi:10.1210/jc.2012-1176. Retrieved 2015-05-27.
 |
|
 |
|
| Names | |
|---|---|
| IUPAC names
(6R)-6-[(1R,3aR,4E,7aR)-4-[(2Z)-2-[(5S)-5-
Hydroxy-2-methylidene-cyclohexylidene] ethylidene]-7a-methyl-2,3,3a,5,6,7-hexahydro- 1H-inden-1-yl]-2-methyl-heptan-2-ol |
|
| Other names
25-Hydroxyvitamin D3
25-Hydroxycholecalciferol Calcidiol |
|
| Identifiers | |
| 19356-17-3 |
|
| 3D model (Jmol) | Interactive image |
| ChEBI | CHEBI:17933 |
| ChEMBL | ChEMBL1222 |
| ChemSpider | 4446820 |
| DrugBank | DB00146 |
| ECHA InfoCard | 100.039.067 |
| 6921 | |
| MeSH | Calcifediol |
| PubChem | 5283731 |
| UNII | T0WXW8F54E |
| Properties | |
| C27H44O2 | |
| Molar mass | 400.64 g/mol |
| Pharmacology | |
| A11CC06 (WHO) | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
Title: Calcifediol
CAS Registry Number: 19356-17-3
CAS Name: (3b,5Z,7E)-9,10-Secocholesta-5,7,10(19)-triene-3,25-diol
Additional Names: 25-hydroxyvitamin D3; 25-hydroxycholecalciferol; 25-HCC
Manufacturers’ Codes: U-32070E
Trademarks: Dedrogyl (DESMA); Didrogyl (Bruno); Hidroferol (FAES)
Molecular Formula: C27H44O2
Molecular Weight: 400.64
Percent Composition: C 80.94%, H 11.07%, O 7.99%
Literature References: The principal circulating form of vitamin D3, formed in the liver by hydroxylation at C-25: Ponchon, DeLuca, J. Clin. Invest. 48, 1273 (1969). It is the intermediate in the formation of 1a,25-dihydroxycholecalciferol, q.v., the biologically active form of vitamin D3 in the intestine. Identification in rat as an active metabolite of vitamin D3: Lund, DeLuca, J. Lipid Res. 7, 739 (1966); Morii et al., Arch. Biochem. Biophys. 120, 513 (1967). Evaluation of biological activity in comparison with vitamin D3: Blunt et al., Proc. Natl. Acad. Sci. USA 61, 717 (1968); ibid. 1503. Isoln from porcine plasma and establishment of structure: Blunt et al., Biochemistry 7, 3317 (1968). Synthesis: Blunt, DeLuca, ibid. 8, 671 (1969). Review of isoln, identification and synthesis: DeLuca, Am. J. Clin. Nutr. 22, 412 (1969). Review of bioassays: J. G. Haddad Jr., Basic Clin. Nutr. 2, 579-597 (1980).
Properties: uv max (ethanol): 265 nm (e 18000) (Blunt, DeLuca).
Absorption maximum: uv max (ethanol): 265 nm (e 18000) (Blunt, DeLuca)
Therap-Cat: Calcium regulator.
Keywords: Calcium Regulator.
/////////Calcifediol, カルシフェジオール
CC(CCCC(C)(C)O)C1CCC2C1(CCCC2=CC=C3CC(CCC3=C)O)C
Balsalazide

Balsalazide
| 80573-04-2; Colazal; Balsalazide Disodium; AC1NSFNR; P80AL8J7ZP; | |
| Molecular Formula: | C17H15N3O6 |
|---|---|
| Molecular Weight: | 357.322 g/mol |
(3E)-3-[[4-(2-carboxyethylcarbamoyl)phenyl]hydrazinylidene]-6-oxocyclohexa-1,4-diene-1-carboxylic acid
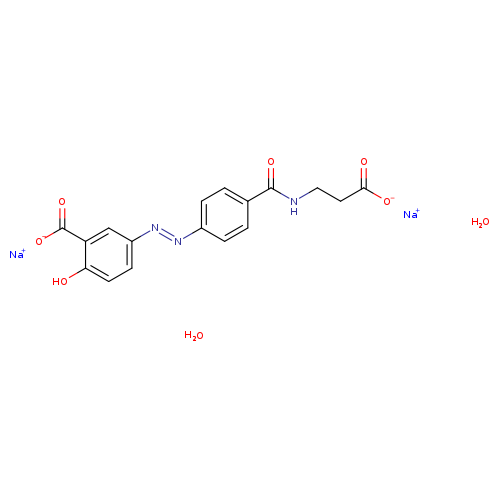 DISODIUMDIHYDRATE
DISODIUMDIHYDRATE
| CAS Number | 150399-21-6 |
|---|---|
| Weight | Average: 437.316 Monoisotopic: 437.08110308 |
| Chemical Formula | C17H17N3Na2O8 |
Balsalazide is an anti-inflammatory drug used in the treatment of inflammatory bowel disease. It is sold under the brand names Giazo, Colazal in the US and Colazide in the UK. It is also sold in generic form in the US by several generic manufacturers.
It is usually administered as the disodium salt. Balsalazide releases mesalazine, also known as 5-aminosalicylic acid, or 5-ASA,[1] in the large intestine. Its advantage over that drug in the treatment of ulcerative colitis is believed to be the delivery of the active agent past the small intestine to the large intestine, the active site of ulcerative colitis.
Balsalazide is an anti-inflammatory drug used in the treatment of Inflammatory Bowel Disease. It is sold under the name “Colazal” in the US and “Colazide” in the UK. The chemical name is (E)-5-[[-4-(2-carboxyethyl) aminocarbonyl] phenyl]azo] –2-hydroxybenzoic acid. It is usually administered as the disodium salt. Balsalazide releases mesalazine, also known as 5-aminosalicylic acid, or 5-ASA, in the large intestine. Its advantage over that drug in the treatment of Ulcerative colitis is believed to be the delivery of the active agent past the small intestine to the large intestine, the active site of ulcerative colitis.
Balsalazide disodium and its complete synthesis was first disclosed by Chan[18] in 1983, assigned to Biorex Laboratories Limited, England, claiming product ‘Balsalazide’ and process of its preparation. The synthesis involves converting 4-nitrobenzoyl chloride (6) to 4- nitrobenzoyl-β-alanine (7), hydrogenating with Pd/C (5%) in ethanol and isolating by adding diethyl ether to produce 4-aminobenzoyl-β-alanine (8). Thereafter, 4-aminobenzoyl-β-alanine (8) was treated with hydrochloric acid and sodium nitrite to generate N-(4-diazoniumbenzoyl)- β-alanine hydrochloride salt (9) which was reacted at low temperature with disodium salicylate to furnish Balsalazide disodium insitu which was added to dilute hydrochloric acid at low temperature to produce Balsalazide (1) (Scheme-1.1). Thus obtained Balsalazide was recrystallized with hot ethanol and converted to pharmaceutically acceptable salt (disodium salt).
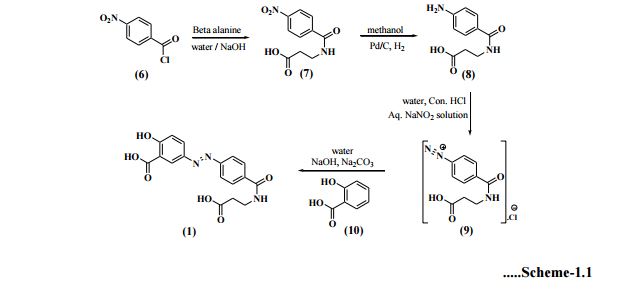
Optimization of this diazonium salt based process was performed by Huijun et al[19] and reported the preparation of the title compound in 64.6% overall yield. Zhenhau et al[20] have synthesized 1 from 4-nitrobenzoic acid (12) via chlorination, condensation, hydrogenation, diazotization, coupling and salt formation with overall yield 73%. Li et al[21] have given product in 73.9% total yield starting from 4-nitrobenzoyl chloride (6), where as Yuzhu et al[22] confirmed chemical structure of Balsalazide disodium by elemental analysis, UV, IR, 1H-NMR and ESI-MS etc. Shaojie et al[23] have also followed same process for its preparation. Yujie et al[24] synthesized 1 in this way; preparation of 4-nitrobenzoyl-β-alanine (7) under microwave irradiation of 420 W at 52oC for 10sec., reduction in ethyl acetate in the presence of Pd/C catalyst then diazotization, coupling and salt formation. Eckardt et al[25] have developed a process for the preparation of Balsalazide which comprises, conversion of 4-aminobenzoyl-β-alanine (8) to 4-ammoniumbenzoyl-β-alanine sulfonate salt using a sulfonic acid in water. This was treated with aq. sodium nitrite solution at low temperature to generate 4-diazoniumbenzoyl-β-alanine sulfonate salt (11) which was quenched with aq. disodium salicylate to furnish Balsalazide disodium solution. This was further acidified to allow isolation of 1 and then conversion to disodium salt (Scheme-1.2) in 76% yield.
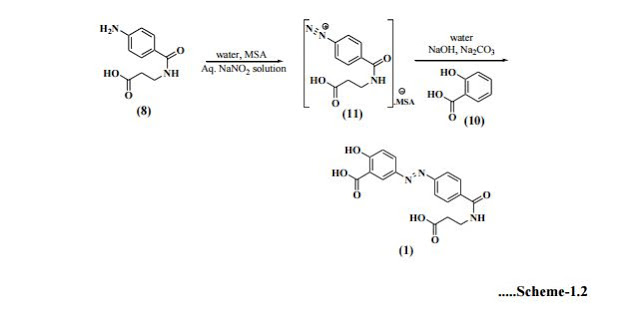
http://shodhganga.inflibnet.ac.in/bitstream/10603/101297/10/10_chapter%201.pdf
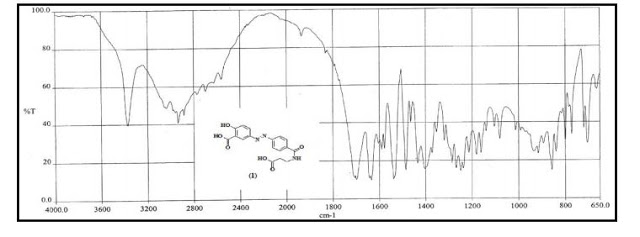



IR (KBr, cm-1 ): 3371 and 3039 (OH and NH), 1705 and 1699 (C=O), 1634 (C=O amide), 1590 and 1538 (C=C aromatic), 1464 and 1404 (aliphatic C-H), 1229 (C-N), 1073 (C-O), 773 and 738 (Ar-H out of plane bend). 1H NMR (DMSO-d6, 300 MHz, δ ppm): 2.54 (t, 2H), 3.50 (m, 2H), 6.95 (d, J = 8.8 Hz, 1H), 7.87 (d, J = 8.5 Hz, 2H), 8.02 (d, J = 8.5 Hz, 2H), 7.95 (dd, J = 8.8 Hz and 2.5 Hz, 1H), 8.34 (d, J = 2.5 Hz, 1H), 8.68 (t, J = 5.5 Hz, 1H), 12.12 (brs, 1H). MS m/z (ESI): 356 [(M-H)- ], Calculated; m/z 357.
Synthesis

Balsalazide synthesis: Biorex Laboratories, GB 2080796 (1986).
- Starting material is 4-aminohippuric acid, obtained by coupling para-aminobenzoic acid and glycine.
- That product is then treated with nitrous acid to give the diazonium salt.
- Reaction of this species with salicylic acid proceeds at the position para to the phenol to give balsalazide.
Sodium balsalazide (Balsalazide sodium)
Brief background information
Application
-
resolvent
Classes substance
-
β-alanine (3-aminopropionic acid)
-
m-aminobenzoic acid and esters and amides thereof
-
p-aminobenzoic acid and esters and amides thereof
-
azobenzene
-
salicylic acid
-
-
-
-
Synthesis Way
| Synthesis of a) |
|---|
   |

Trade names
| A country | Tradename | Manufacturer |
|---|---|---|
| United Kingdom | Kolazid | Shire |
| Italy | Balzid | Menarini |
| USA | Kolazal | Salix |
| Ukraine | no | no |
Formulations
-
capsules in 750 mg (as disodium salt)
PATENT
https://www.google.com/patents/US7271253
Balsalazide disodium (1) represents an effective gastrointestinal anti-inflammatory compound useful as a medicament for the treatment of diseases such as ulcerative colitis. It is delivered intact to the colon where it is cleaved by bacterial azoreduction thereby generating 5-aminosalicylic acid as the medicinally active component.

To date, relatively few patents or literature articles have dealt with the preparation of Balsalazide or the disodium salt. For instance, U.S. Pat. No. 4,412,992 (Biorex, 1983) is the first patent that we uncovered that claims the compound Balsalazide and a strategy of how to prepare it which strategy is depicted in Scheme 1.

Optimization of this diazonium-based process is detailed in Shan et al., Zhongguo Yaowu Huaxue Zazhi, 11, 110 (2001) and Shi et al., Zhongguo Yiyao Gongye Zazhi, 34, 537 (2003).
Problems arise with the above strategy and the optimization process.
It is well-documented in the literature, for instance in Thermochimica Acta, 225, 201-211 (1993), that diazonium salts can be involved in serious accidents in their use. A possible cause of some of the diazonium salt related accidents is that, for one reason or another, an intermediate material appeared in crystalline form in the vessel of the reaction. As a result, a potentially severe drawback of the above processes occurs. Since the intermediate hydrochloride salt of 4-aminobenzoyl-β-alanine has poor solubility in water, it may pose a safety-risk in the subsequent diazotation reaction.
Also, it is well-known that certain diazonium salts possess high mechanical and heat sensitivity and that their decomposition goes through the liberation of non-condensable nitrogen gas which results in the possibility of runaway reactions and explosions. Obviously this safety consideration becomes more pertinent upon further scale-up.
Therefore, for commercial production of Balsalazide disodium, there was a need to develop a scalable and intrinsically better process
Example 1 Batch Process
N-(4-Aminobenzoyl)-β-alanine (100 g) was suspended in water (1300 mL) and methanesulfonic acid (115.4 g) was added to this mixture. The mixture was cooled to 10° C. and a solution of sodium nitrite (34.46 g) in water (200 mL) was added at a rate such that the temperature stayed below 12° C. The mixture was stirred for 30 min and added to an ice-cold solution of salicylic acid (69.65 g), sodium hydroxide (40.35 g) and sodium carbonate (106.9 g) in 1 L water at 7-12° C. After 3 hours at 10° C., the mixture was heated to 60-65° C. and acidified to pH 4.0-4.5 by the addition of hydrochloric acid. After a further 3 hours at 60-65° C., the mixture was cooled to ambient temperature, filtered, washed with water and dried in vacuo to yield Balsalazide. Yield ca. 90%. Balsalazide was transformed into its disodium salt in ca. 85% yield by treatment with aqueous NaOH solution followed by crystallization from n-propanol/methanol.
1H-NMR (400 MHz; D2O): δ=8.04 ppm (s); 7.67 ppm (d; J=8.2 Hz); 7.62 ppm (d, J=9.2 Hz); 7.53 ppm (d; J=8.2 Hz); 6.84 ppm (d; J=8.9 Hz); 3.57 ppm (t, J=7.1 Hz); 2.53 ppm (t; J=7.2 Hz).
Example 2 Continuous Process
For the continuous operation, a conventional dual-head metering pump (Ratiomatic by FMI) was used to deliver the mesylate solution and the aqueous sodium nitrite solution. The schematic diagram shown in FIG. 4 represents a set-up used for the continuous process. The first pump-head was set at 13.9 g/min whereas the second was set at 2.1 g/min. These flow rates offered a residence time of 9.4 min. The yield of the coupled intermediate from this residence time was 93%. The working solutions were prepared as follow:
The mesylate solution was prepared by the addition into a 2 L 3-necked round bottom flask, of N-(4-aminobenzoyl) β-alanine (120 g) followed by of DI water (1560 g) and methanesulfonic acid (177.5 g) (Batch appearance: clear solution). The first pump-head was primed with this solution and the flow rate was adjusted to 13.9 g/min.
The sodium nitrite solution was prepared by dissolving of sodium nitrite (41.8 g) in of DI water (240 g) (Batch appearance: clear solution). The second pump-head was primed with this solution and the flow rate adjusted to 2.1 g/min.
The quenching solution (sodium salicylate) was made by adding salicylic acid (139.3 g) to DI water (900 g) followed by of sodium carbonate (106.9 g) and 50% aqueous sodium hydroxide (80 g).
The diazotation reaction was performed in a 500 ml jacketed flow reactor with a bottom drain valve. The drain valve was set at 16 g/min. For reactor start-up, the flow reactor was charged with 150 mL of DI water as a working volume and cooled to the reactions initial temperature of 0-5° C. Concomitantly, the additions of the mesylate and sodium nitrite solutions were started and the bottom valve of the flow reactor was opened. During the diazotization, the flow rate of both solutions remained fixed and the temperature was kept below 12° C. and at the end of additions the pumps were stopped while the remaining contents in the flow reactor were drained into the quenching salicylic acid solution. Analysis of the contents in the quenching reactor indicated no signs of uncoupled starting material (diazonium compound). The reactor contents were heated to 60-65° C. for 2-3 hrs before adjusting the pH to precipitate the coupling product. This provided 191.5 g of product.
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| US4412992 | Jul 8, 1981 | Nov 1, 1983 | Biorex Laboratories Limited | 2-Hydroxy-5-phenylazobenzoic acid derivatives and method of treating ulcerative colitis therewith |
| US6458776 * | Aug 29, 2001 | Oct 1, 2002 | Nobex Corporation | 5-ASA derivatives having anti-inflammatory and antibiotic activity and methods of treating diseases therewith |
| Reference | ||
|---|---|---|
| 1 | Chai, et al., Huaxi Yaoxue Zazhi, Jiangsu Institute of Materia Medica, Nanjing, China, 2004, 19(6), 431-433. | |
| 2 | Shan, et al., Zhongguo Yaowu Huaxue Zazhi, Institute of Materia Medica, Peking Union Medical College, Beijing China, 2001, 11(2), 110-111. | |
| 3 | Shi, et al., Zhongguo Yiyao Gongya Zazhi, Shanghai Institute of Pharmaceutical Industry, Shanghai, China, 2003, 34(11), 537-538. | |
| 4 | Su, et al., Huaxue Gongye Yu Gongcheng (Tianjin, China), College of Chemistry and Chemical Eng., Donghua Univ., Shanghai, China, 2005, 22(4), 313-315. | |
| 5 | Ullrich, et al., Decomposition of aromataic diazonium compounds, Thermochimica Acta, 1993, 225, 201-211. | |
References
-
Prakash, A; Spencer, CM: Drugs (DRUGAY) 1998 56 83- 89.
-
DE 3128819 (Biorex the Lab .; appl 07/21/1981;. GB -prior 07/21/1980, 07.07.1981.).
References
- Jump up^ Kruis, W.; Schreiber, I.; Theuer, D.; Brandes, J. W.; Schütz, E.; Howaldt, S.; Krakamp, B.; Hämling, J.; Mönnikes, H.; Koop, I.; Stolte, M.; Pallant, D.; Ewald, U. (2001). “Low dose balsalazide (1.5 g twice daily) and mesalazine (0.5 g three times daily) maintained remission of ulcerative colitis but high dose balsalazide (3.0 g twice daily) was superior in preventing relapses”. Gut. 49 (6): 783–789. doi:10.1136/gut.49.6.783. PMC 1728533. PMID 11709512.
| Patent ID | Patent Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8232265 | Multi-functional ionic liquid compositions for overcoming polymorphism and imparting improved properties for active pharmaceutical, biological, nutritional, and energetic ingredients | 2007-04-26 | 2012-07-31 |
| US2011319267 | AROMATIC CARBOXYLIC ACID DERIVATIVES FOR TREATMENT AND PROPHYLAXIS OF GASTROINTESTINAL DISEASES INCLUDING COLON CANCERS | 2011-12-29 | |
| US2007213304 | Use of Aminosalicylates in Diarrhoea-Predominent Irritable Bowel Syndrome | 2007-09-13 | |
| US7119079 | Bioadhesive pharmaceutical compositions | 2004-07-22 | 2006-10-10 |
| US6699848 | Bioadhesive anti-inflammatory pharmaceutical compositions | 2004-03-02 |
 |
|
| Clinical data | |
|---|---|
| Trade names | Colazal, Giazo |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a699052 |
| Pregnancy category |
|
| ATC code | A07EC04 (WHO) |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | <1% |
| Protein binding | ≥99% |
| Biological half-life | 12hr |
| Identifiers | |
| CAS Number | 80573-04-2 |
| PubChem (CID) | 5362070 |
| DrugBank | DB01014 |
| ChemSpider | 10662422 |
| UNII | P80AL8J7ZP |
| ChEBI | CHEBI:267413 |
| ChEMBL | CHEMBL1201346 |
| ECHA InfoCard | 100.117.186 |
| Chemical and physical data | |
| Formula | C17H15N3O6 |
| Molar mass | 357.318 g/mol |
| 3D model (Jmol) | Interactive image |
 CLICK ON IMAGE
CLICK ON IMAGE
Title: Balsalazide
CAS Registry Number: 80573-04-2
CAS Name: 5-[(1E)-[4-[[(2-Carboxyethyl)amino]carbonyl]phenyl]azo]-2-hydroxybenzoic acid
Additional Names: (E)-5-[[p-[(2-carboxyethyl)carbamoyl]phenyl]azo]-2-salicylic acid
Molecular Formula: C17H15N3O6
Molecular Weight: 357.32
Percent Composition: C 57.14%, H 4.23%, N 11.76%, O 26.87%
Literature References: Analog of sulfasalazine, q.v. Prodrug of 5-aminosalicylic acid where carrier molecule is 4-aminobenzoyl-b-alanine. Prepn: R. P. K. Chan, GB 2080796; idem, US 4412992 (1982, 1983 both to Biorex). Toxicology study and clinical metabolism: idem et al., Dig. Dis. Sci. 28, 609 (1983). Review of pharmacology and clinical efficacy in ulcerative colitis: A. Prakash, C. M. Spencer, Drugs 56, 83 (1998).
Properties: Crystals from hot ethanol, mp 254-255°.
Melting point: mp 254-255°
Derivative Type: Disodium salt dihydrate
CAS Registry Number: 150399-21-6; 82101-18-6 (anhydrous)
Manufacturers’ Codes: BX-661A
Trademarks: Colazal (Salix); Colazide (Shire)
Molecular Formula: C17H13N3Na2O6.2H2O
Molecular Weight: 437.31
Percent Composition: C 46.69%, H 3.92%, N 9.61%, Na 10.51%, O 29.27%
Properties: Orange to yellow microcrystalline powder, mp >350°. Nonhygroscopic. Freely sol in water, isotonic saline; sparingly sol in methanol, ethanol. Practically insol in organic solvents.
Melting point: mp >350°
Therap-Cat: Anti-inflammatory (gastrointestinal).
Keywords: Anti-inflammatory (Gastrointestinal); Anti-inflammatory (Nonsteroidal); Salicylic Acid Derivatives.
//////
O=C(O)c1cc(ccc1O)/N=N/c2ccc(cc2)C(=O)NCCC(O)=O
|
O.O.[Na+].[Na+].OC1=CC=C(C=C1C([O-])=O)\N=N\C1=CC=C(C=C1)C(=O)NCCC([O-])=O
|
P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.


{kind=link}
{kind=link}
{kind=link}