
Home » Priority review
Category Archives: Priority review
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Omaveloxolone



Omaveloxolone
CAS
1474034-05-3
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,2,3,4,4a,5,6,6a,6b,7,8,8a,9,10,12a,14,14a,14b-octadecahydropicen-4a-yl]-2,2-difluoropropanamide
N-[(4aS,6aR,6bS,8aR,12aS,14aR,14bS)-11-cyano-2,2,6a,6b,9,9,12a-heptamethyl-10,14-dioxo-1,3,4,5,6,7,8,8a,14a,14b-decahydropicen-4a-yl]-2,2-difluoropropanamide
FDA 2023, 2/28/2023, To treat Friedrich’s ataxia
Drug Trials Snapshot
WeightAverage: 554.723
Monoisotopic: 554.331999611
Chemical FormulaC33H44F2N2O3
- RTA 408
- RTA-408
- OriginatorDartmouth College; University of Texas M. D. Anderson Cancer Center
- DeveloperBiogen
- ClassAnalgesics; Anti-inflammatories; Antineoplastics; Eye disorder therapies; Neuroprotectants; Small molecules; Triterpenes
- Mechanism of ActionNF-E2-related factor 2 stimulants
- Orphan Drug StatusYes – Friedreich’s ataxia; Malignant melanoma
- MarketedFriedreich’s ataxia
- Phase IIMitochondrial disorders; Ocular inflammation; Ocular pain
- Phase I/IIMalignant melanoma
- PreclinicalBrain disorders
- DiscontinuedDuchenne muscular dystrophy; Non-small cell lung cancer; Radiation-induced skin damage
- 08 Apr 2025Biogen completes a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
- 17 Mar 2025Registered for Friedreich’s ataxia (In adolescents, In adults) in Canada (PO)
- 18 Oct 2024Biogen initiates enrolment in a phase I pharmacokinetics trial (In volunteers) in USA (PO) (NCT06612879)
Omaveloxolone, sold under the brand name Skyclarys, is a medication used for the treatment of Friedreich’s ataxia.[2][5] It is taken by mouth.[2]
The most common side effects include an increase in alanine transaminase and an increase of aspartate aminotransferase, which can be signs of liver damage, headache, nausea, abdominal pain, fatigue, diarrhea and musculoskeletal pain.[5]
Omaveloxolone was approved for medical use in the United States in February 2023,[2][5][6][7][8] and in the European Union in February 2024.[3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication.[9]
SYNTHESIS

PATENT
Sheikh, AY et al. (2018). Bardoxolonmethyl-2,2-difluoropropionamide derivatives, polymorphe forms and procedures for use thereof. DK/EP 2989114 T3. Danish Patent and Trademark Office. Available at https://patentimages.storage.googleapis.com/51/87/43/97d0fb3e69ee73/DK2989114T3.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP159939262&_cid=P21-MAKI10-93498-1

[0164] Reagents and conditions: (a) (PhO) 2PON 3 (DPPA), triethylamine, toluene, 0 °C for 5 minutes, then ambient temperature overnight, ∼94%; (b) benzene, 80 °C for 2 hours; (c) HCl, CH 3CN, ambient temperature for 1 hour; (d) CH 3CF 2CO 2H, dicyclohexylcarbodiimide, 4-(dimethylamino)pyridine, CH 2Cl 2, ambient temperature overnight, 73% from RTA 401 (4 steps).
[0165]Compound 1: RTA 401 (20.0 g, 40.6 mmol), triethylamine (17.0 mL, 122.0 mmol), and toluene (400 mL) were added into a reactor and cooled to 0 °C with stirring. Diphenyl phosphoryl azide (DPPA) (13.2 mL, 61.0 mmol) was added with stirring at 0 °C over 5 minutes, and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no RTA 401 left). The reaction mixture was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 5% ethyl acetate in CH 2Cl 2) to give compound 1 (19.7 g, ∼94%, partially converted into compound 2) as a white foam.
[0166]Compound 2: Compound 1 (19.7 g, ∼38.1 mmol) and benzene (250 mL) were added into a reactor and heated to 80 °C with stirring for 2 hours (HPLC-MS check shows no compound 1 left). The reaction mixture was concentrated at reduced pressure to afford crude compound 2 as a solid residue, which was used for the next step without purification.
[0167]Compound 3: Crude compound 2 (≤38.1 mmol) and CH 3CN (200 mL) were added into a reactor and cooled to 0 °C with stirring. HCl (12 N, 90 mL) was added at 0 °C over 1 minute, and the mixture was continually stirred at room temperature for 1 hour (HPLC-MS check shows no compound 2 left). The reaction mixture was cooled to 0 °C and 10% NaOH (∼500 mL) was added with stirring. Then, saturated NaHCO 3 (1 L) was added with stirring. The aqueous phase was extracted by ethyl acetate (2×500 mL). The combined organic phase was washed by H 2O (200 mL), saturated NaCl (200 mL), dried over Na 2SO 4, and concentrated to afford crude compound 3 (16.62 g) as a light yellow foam, which was used for the next step without purification.
[0168]RTA 408: Crude amine 3 (16.62 g, 35.9 mmol), CH 3CF 2CO 2H (4.7388 g, 43.1 mmol), and CH 2Cl 2 (360 mL) were added into a reactor with stirring at room temperature. Then, dicyclohexylcarbodiimide (DCC) (11.129 g, 53.9 mmol) and 4-(dimethylamino)pyridine (DMAP) (1.65 g, 13.64 mmol) were added and the mixture was continually stirred at room temperature overnight (HPLC-MS check shows no compound 3 left). The reaction mixture was filtered to remove solid by-products, and the filtrate was directly loaded on a silica gel column and purified by column chromatography (silica gel, 0% to 20% ethyl acetate in hexanes) twice to give compound RTA 408 (16.347 g, 73% from RTA 401 over 4 steps) as a white foam: 1H NMR (400 MHz, CD 3Cl) δ ppm 8.04 (s, 1H), 6.00 (s, 1H), 5.94 (s, br, 1H), 3.01 (d, 1H, J = 4.8 Hz), 2.75-2.82 (m, 1H), 1.92-2.18 (m, 4H), 1.69-1.85 (m, 7H), 1.53-1.64 (m, 1H), 1.60 (s, 3H), 1.50 (s, 3H), 1.42 (s, 3H), 1.11-1.38 (m, 3H), 1.27 (s, 3H), 1.18 (s, 3H), 1.06 (s, 3H), 1.04 (s, 3H), 0.92 (s, 3H); m/z 555 (M+1).
SYNTHESIS
J. Med. Chem. 2025, 68, 2147−2182
Omaveloxolone (Skyclarys). Omaveloxolone (6) was approved in February 2023 for the treatment of Friedreich’s Ataxia (FRDA), a genetic, neurodegenerative disease. Patients with FRDA have lowered activity of the frataxin gene (FXN), attributed to an expansion of a guanine-adenine-adenine (GAA)
triplet. The resulting decrease in frataxin limits the production of iron−sulfur clusters, leading to accumulation of iron in the mitochondria and oxidative stress which in turn leads to cell damageanddeath.49
Omaveloxoloneactivates the nuclear factor erythroid 2-related factor 2 (Nrf2), an important pathway in
oxidative stress. It acts by preventing ubiquitination and subsequent degradation of Nrf2, keeping levels high enough to counteract the oxidative stress associated with FRDA. 50
Omaveloxolone was developed by Reata Pharmaceuticals (which was acquired by Biogen in September 2023) and was granted orphan drug, fast track, priority review, and rare pediatric disease designations. 51Omaveloxolone (6) is a semisynthetic triterpenoid based on the oleanolic acid scaffold.52
advanced intermediate 6.1,The synthesis started from the53also known as CDDO orbardoxolone, which has individually been investigated fortherapeutic benefits from Nrf2 activation (Scheme 10).
Treatment of acid 6.1 with DPPA produced the azide, and subsequent heating in benzene generated isocyanate 6.2 via aCurtius rearrangement. Hydrolysis with aqueous acid generated amine 6.3, and an amidation with 2,2-difluoropropanoic acid produced omaveloxolone (6). A yield of 73% over the sequence was reported, and intermediates were used crude with no purification between steps.
(49) Ghanekar, S. D.; Miller, W. W.; Meyer, C. J.; Fenelon, K. J.;
Lacdao, A.; Zesiewicz, T. A. Orphan drugs in development for the
treatment of Friedreich’s ataxia: focus on omaveloxolone. Degener.
Neurol. Neuromuscular Dis. 2019, 9, 103−107.
(50) Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-inducer
prevents mitochondrial defects and oxidative stress in Friedreich’s
ataxia models. Front. Cell. Neurosci. 2018, 12, 188.
(51) Lee,A.Omaveloxolone:first approval. Drugs 2023, 83, 725−729.
(52) Anderson, E.; Decker, A.; Liu, X. Synthesis, pharmaceutical use,
and characterization of crystalline forms of 2,2-difluoropropionamide
derivatives of bardoxolone methyl. WO 2013163344, 2013.
(53) Honda, T.; Rounds, B. V.; Gribble, G. W.; Suh, N.; Wang, Y.;
Sporn, M. B. Design and synthesis of 2-cyano-3,12-dioxoolean-1,9
dien-28-oic acid, a novel and highly active inhibitor of nitric oxide
production in mouse macrophages. Bioorg. Med. Chem. Lett. 1998, 8,
2711−2714.

SYN
European Journal of Medicinal Chemistry 265 (2024) 116124
Omaveloxolone (Skyclarys)
Omaveloxolone was granted FDA approval on February 28, 2023, to treat Friedrich’s ataxia in individuals aged 16 and older [2]. Omaveloxolone possesses antioxidant and anti-inflammatory properties, making it a semi-synthetic triterpenoid compound. It has the ability to function as a stimulator of nuclear factor-erythroid 2 related factor 2(Nrf2), a transcription factor that reduces oxidative stress. In individuals
suffering from FA, a genetic disorder characterized by mitochondrial dysfunction, the Nrf2 pathway is compromised, leading to a decrease in Nrf2 activity. Hence, Omaveloxolone, an Nrf2 activator, can be
employed as a therapeutic option for the management of these in dividuals [23].The process route of Omaveloxolone is described below in Scheme 724]. The substitution reaction of carboxylic acid OMAV-001 with diphenylphosphoryl azide (DPPA) gave the acyl azide OMAV-002,which underwent Curtius-rearrangement under heating conditions to produce isocyanate OMAV-003. The amine OMAV-004 was obtained under acidic conditions. OMAV-004 was condensed with 2,2-difluoro propionic acid to obtain the final product Omaveloxolone.
[23] B.L. Probst, I. Trevino, L. McCauley, R. Bumeister, I. Dulubova, W.C. Wigley, D.
A. Ferguson, RTA 408, A novel synthetic triterpenoid with broad anticancer and
anti-inflammatory activity, PLoS One 10 (2015) e0122942.
[24] E. Anderson, A. Decker, X. Liu Synthesis, Pharmaceutical Use, and
Characterization of Crystalline Forms of 2,2-difluoropropionamide Derivatives of
Bardoxolone Methyl, 2013. WO2013163344.

.
Medical uses
Omaveloxolone is indicated for the treatment of Friedreich’s ataxia.[2][5]
Friedreich’s ataxia causes progressive damage to the spinal cord, peripheral nerves, and the brain, resulting in uncoordinated muscle movement, poor balance, difficulty walking, changes in speech and swallowing, and a shortened lifespan.[5] The condition can also cause heart disease.[5] This disease tends to develop in children and teenagers and gradually worsens over time.[5]
Although rare, Friedreich’s ataxia is the most common form of hereditary ataxia in the United States, affecting about one in every 50,000 people.[5]
Mechanism of action
The mechanism of action of omaveloxolone and its related compounds has been demonstrated to be through a combination of activation of the antioxidative transcription factor Nrf2 and inhibition of the pro-inflammatory transcription factor NF-κB.[10]
Nrf2 transcriptionally regulates multiple genes that play both direct and indirect roles in producing antioxidative potential and the production of cellular energy (i.e., adenosine triphosphate or ATP) within the mitochondria. Consequently, unlike exogenously administered antioxidants (e.g., vitamin E or Coenzyme Q10), which provide a specific and finite antioxidative potential, omaveloxolone, through Nrf2, broadly activates intracellular and mitochondrial antioxidative pathways, in addition to pathways that may directly increase mitochondrial biogenesis (such as PGC1α) and bioenergetics.[11]
History
Omaveloxolone is a second generation member of the synthetic oleanane triterpenoid compounds and in clinical development by Reata Pharmaceuticals. Preclinical studies have demonstrated that omaveloxolone possesses antioxidative and anti-inflammatory activities[10][12] and the ability to improve mitochondrial bioenergetics.[11] Omaveloxolone is under clinical investigation for a variety of indications, including Friedreich’s ataxia, mitochondrial myopathies, immunooncology, and prevention of corneal endothelial cell loss following cataract surgery.
The efficacy and safety of omaveloxolone was evaluated in a 48-week randomized, placebo-controlled, and double-blind study [Study 1 (NCT02255435)] and an open-label extension.[5] Study 1 enrolled 103 individuals with Friedreich’s ataxia who received placebo (52 individuals) or omaveloxolone 150 mg (51 individuals) for 48 weeks.[5] Of the research participants, 53% were male, 97% were white, and the mean age was 24 years at study entry.[5] Nine (18%) patients were younger than age 18.[5] The primary objective was to evaluate the change in the modified Friedreich’s Ataxia Rating Scale (mFARS) score compared to placebo at week 48.[5] The mFARS is a clinical assessment that measures disease progression, namely swallowing and speech (bulbar), upper limb coordination, lower limb coordination, and upright stability.[5] Individuals receiving omaveloxolone performed better on the mFARS than people receiving placebo.[5]
The US Food and Drug Administration (FDA) granted the application for omaveloxolone orphan drug, fast track, priority review, and rare pediatric disease designations.[5][9]
Society and culture
Legal status
Omaveloxolone was approved for medical use in the United States in February 2023.[2][5]
In December 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Skyclarys, intended for the treatment of Friedreich’s ataxia.[3] The applicant for this medicinal product is Reata Ireland Limited.[3] Omaveloxolone was approved for medical use in the European Union in February 2024.[3][4]
References
- ^ “Register of Innovative Drugs”. Health Canada. 3 November 2006. Retrieved 17 April 2025.
- ^ Jump up to:a b c d e f “Skyclarys- omaveloxolone capsule”. DailyMed. 12 May 2023. Archived from the original on 1 July 2023. Retrieved 16 December 2023.
- ^ Jump up to:a b c d e “Skyclarys EPAR”. European Medicines Agency (EMA). 14 December 2023. Archived from the original on 15 December 2023. Retrieved 16 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Skyclarys product information”. Union Register of medicinal products. 12 February 2024. Retrieved 19 February 2024.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “FDA approves first treatment for Friedreich’s ataxia”. U.S. Food and Drug Administration (FDA). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Reata Pharmaceuticals Announces FDA Approval of Skyclarys (Omavaloxolone), the First and Only Drug Indicated for Patients with Friedreich’s Ataxia”. Reata Pharmaceuticals Inc. (Press release). 28 February 2023. Archived from the original on 1 March 2023. Retrieved 28 February 2023.
- ^ Lee A (June 2023). “Omaveloxolone: First Approval”. Drugs. 83 (8): 725–729. doi:10.1007/s40265-023-01874-9. PMID 37155124. S2CID 258567442. Archived from the original on 9 December 2023. Retrieved 16 December 2023.
- ^ Subramony SH, Lynch DL (May 2023). “A Milestone in the Treatment of Ataxias: Approval of Omaveloxolone for Friedreich Ataxia”. Cerebellum. 23 (2): 775–777. doi:10.1007/s12311-023-01568-8. PMID 37219716. S2CID 258843532.
- ^ Jump up to:a b New Drug Therapy Approvals 2023 (PDF). U.S. Food and Drug Administration (FDA) (Report). January 2024. Archived from the original on 10 January 2024. Retrieved 9 January 2024.
- ^ Jump up to:a b Reisman SA, Lee CY, Meyer CJ, Proksch JW, Ward KW (July 2014). “Topical application of the synthetic triterpenoid RTA 408 activates Nrf2 and induces cytoprotective genes in rat skin”. Archives of Dermatological Research. 306 (5): 447–454. doi:10.1007/s00403-013-1433-7. PMID 24362512. S2CID 25733020.
- ^ Jump up to:a b Neymotin A, Calingasan NY, Wille E, Naseri N, Petri S, Damiano M, et al. (July 2011). “Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis”. Free Radical Biology & Medicine. 51 (1): 88–96. doi:10.1016/j.freeradbiomed.2011.03.027. PMC 3109235. PMID 21457778.
- ^ Reisman SA, Lee CY, Meyer CJ, Proksch JW, Sonis ST, Ward KW (May 2014). “Topical application of the synthetic triterpenoid RTA 408 protects mice from radiation-induced dermatitis”. Radiation Research. 181 (5): 512–520. Bibcode:2014RadR..181..512R. doi:10.1667/RR13578.1. PMID 24720753. S2CID 23906747.
External links
Clinical trial number NCT02255435 for “RTA 408 Capsules in Patients With Friedreich’s Ataxia – MOXIe” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Skyclarys |
| Other names | RTA 408 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Omaveloxolone |
| Routes of administration | By mouth |
| ATC code | N07XX25 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3][4] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1474034-05-3 |
| PubChem CID | 71811910 |
| IUPHAR/BPS | 7573 |
| DrugBank | DB12513 |
| ChemSpider | 34980948 |
| UNII | G69Z98951Q |
| KEGG | D10964 |
| ChEBI | CHEBI:229661 |
| CompTox Dashboard (EPA) | DTXSID101138251 |
| Chemical and physical data | |
| Formula | C33H44F2N2O3 |
| Molar mass | 554.723 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
- Zesiewicz TA, Hancock J, Ghanekar SD, Kuo SH, Dohse CA, Vega J: Emerging therapies in Friedreich’s Ataxia. Expert Rev Neurother. 2020 Dec;20(12):1215-1228. doi: 10.1080/14737175.2020.1821654. Epub 2020 Sep 21. [Article]
- Jiang Z, Qi G, Lu W, Wang H, Li D, Chen W, Ding L, Yang X, Yuan H, Zeng Q: Omaveloxolone inhibits IL-1beta-induced chondrocyte apoptosis through the Nrf2/ARE and NF-kappaB signalling pathways in vitro and attenuates osteoarthritis in vivo. Front Pharmacol. 2022 Sep 27;13:952950. doi: 10.3389/fphar.2022.952950. eCollection 2022. [Article]
- Shekh-Ahmad T, Eckel R, Dayalan Naidu S, Higgins M, Yamamoto M, Dinkova-Kostova AT, Kovac S, Abramov AY, Walker MC: KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain. 2018 May 1;141(5):1390-1403. doi: 10.1093/brain/awy071. [Article]
- Probst BL, Trevino I, McCauley L, Bumeister R, Dulubova I, Wigley WC, Ferguson DA: RTA 408, A Novel Synthetic Triterpenoid with Broad Anticancer and Anti-Inflammatory Activity. PLoS One. 2015 Apr 21;10(4):e0122942. doi: 10.1371/journal.pone.0122942. eCollection 2015. [Article]
- Lynch DR, Farmer J, Hauser L, Blair IA, Wang QQ, Mesaros C, Snyder N, Boesch S, Chin M, Delatycki MB, Giunti P, Goldsberry A, Hoyle C, McBride MG, Nachbauer W, O’Grady M, Perlman S, Subramony SH, Wilmot GR, Zesiewicz T, Meyer C: Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann Clin Transl Neurol. 2018 Nov 10;6(1):15-26. doi: 10.1002/acn3.660. eCollection 2019 Jan. [Article]
- Zighan M, Arkadir D, Douiev L, Keller G, Miller C, Saada A: Variable effects of omaveloxolone (RTA408) on primary fibroblasts with mitochondrial defects. Front Mol Biosci. 2022 Aug 12;9:890653. doi: 10.3389/fmolb.2022.890653. eCollection 2022. [Article]
- FDA Approved Drug Products: SKYCLARYS (omaveloxolone) capsules for oral use (February 2023) [Link]
- EMA Approved Drug Products: Skyclarys (omaveloxolone) Oral Capsules [Link]
- Health Canada Approved Drug Products: SKYCLARYS (Omaveloxolone) Capsules For Oral Use [Link]
///////////Omaveloxolone, Skyclarys, Friedrich’s ataxia, FDA 2023, APPROVALS 2023, RTA 408, RTA-408, omaveloxolona, RTA 408, 63415, PP415, orphan drug, fast track, priority review, rare pediatric disease



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
Suzetrigine



Suzetrigine
CAS
2649467-58-1 |
Average: 473.4
Monoisotopic: 473.137396951
Chemical Formula
C21H20F5N3O4
FDA 1/30/2025, Journavx
To treat moderate to severe acute pain
Press Release
- 2-Pyridinecarboxamide, 4-[[[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)tetrahydro-4,5-dimethyl-5-(trifluoromethyl)-2-furanyl]carbonyl]amino]-
- 4-[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)-4,5- dimethyl-5-(trifluoromethyl)oxolane-2- carboxamido]pyridine-2-carboxamide
- 4-[(2R,3S,4S,5R)-3-(3,4-difluoro-2-methoxyphenyl)-4,5-dimethyl-5-(trifluoromethyl)oxolane-2-amido]pyridine2-carboxamide
- 4-[[[(2R,3S,4S,5R)-3-(3,4-Difluoro-2-methoxyphenyl)tetrahydro-4,5-dimethyl-5-(trifluoromethyl)-2-furanyl]carbonyl]amino]-2-pyridinecarboxamide
- CS-0641183
- HY-148800
- VX 548
- VX-548
- VX548
- Management of
Acute, moderate pain
Suzetrigine, sold under the brand name Journavx, is a medication used for the management of pain.[1][2] It is a non-opioid, small-molecule analgesic that works as a selective inhibitor of Nav1.8-dependent pain-signaling pathways in the peripheral nervous system,[3][4] avoiding the addictive potential of opioids. Suzetrigine is taken by mouth.[1]
The most common adverse reactions include itching, muscle spasms, increased blood level of creatine kinase, and rash.[1][2]
It was developed by Vertex Pharmaceuticals,[5] and was approved for medical use in the United States in January 2025.[2][6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines.[2]
Medical uses
Suzetrigine is indicated for the treatment of moderate to severe acute pain in adults.[1][2]
FDA Approves Novel Non-Opioid Treatment for Moderate to Severe Acute Pain
First Drug Approved in New Class of Non-Opioid Pain Medicines; Agency Continues to Take Steps to Support New Approaches for Pain Management
For Immediate Release:January 30, 2025
Today, the U.S. Food and Drug Administration approved Journavx (suzetrigine) 50 milligram oral tablets, a first-in-class non-opioid analgesic, to treat moderate to severe acute pain in adults. Journavx reduces pain by targeting a pain-signaling pathway involving sodium channels in the peripheral nervous system, before pain signals reach the brain.
Journavx is the first drug to be approved in this new class of pain management medicines.
Pain is a common medical problem and relief of pain is an important therapeutic goal. Acute pain is short-term pain that is typically in response to some form of tissue injury, such as trauma or surgery. Acute pain is often treated with analgesics that may or may not contain opioids.
The FDA has long supported development of non-opioid pain treatment. As part of the FDA Overdose Prevention Framework, the agency has issued draft guidance aimed at encouraging development of non-opioid analgesics for acute pain and awarded cooperative grants to support the development and dissemination of clinical practice guidelines for the management of acute pain conditions.
“Today’s approval is an important public health milestone in acute pain management,” said Jacqueline Corrigan-Curay, J.D., M.D., acting director of the FDA’s Center for Drug Evaluation and Research. “A new non-opioid analgesic therapeutic class for acute pain offers an opportunity to mitigate certain risks associated with using an opioid for pain and provides patients with another treatment option. This action and the agency’s designations to expedite the drug’s development and review underscore FDA’s commitment to approving safe and effective alternatives to opioids for pain management.”
The efficacy of Journavx was evaluated in two randomized, double-blind, placebo- and active-controlled trials of acute surgical pain, one following abdominoplasty and the other following bunionectomy. In addition to receiving the randomized treatment, all participants in the trials with inadequate pain control were permitted to use ibuprofen as needed for “rescue” pain medication. Both trials demonstrated a statistically significant superior reduction in pain with Journavx compared to placebo.
The safety profile of Journavx is primarily based on data from the pooled, double-blind, placebo- and active-controlled trials in 874 participants with moderate to severe acute pain following abdominoplasty and bunionectomy, with supportive safety data from one single-arm, open-label study in 256 participants with moderate to severe acute pain in a range of acute pain conditions.
The most common adverse reactions in study participants who received Journavx were itching, muscle spasms, increased blood level of creatine phosphokinase, and rash. Journavx is contraindicated for concomitant use with strong CYP3A inhibitors. Additionally, patients should avoid food or drink containing grapefruit when taking Journavx.
The application received Breakthrough Therapy, Fast Track and Priority Review designations by the FDA.
The FDA granted approval of Journavx to Vertex Pharmaceuticals Incorporated.
PATENTS
https://patentimages.storage.googleapis.com/08/4f/6e/4f104b27a3772f/US11919887.pdf
https://patentscope.wipo.int/search/en/detail.jsf?docId=US407339565&_cid=P22-M90R90-47554-1



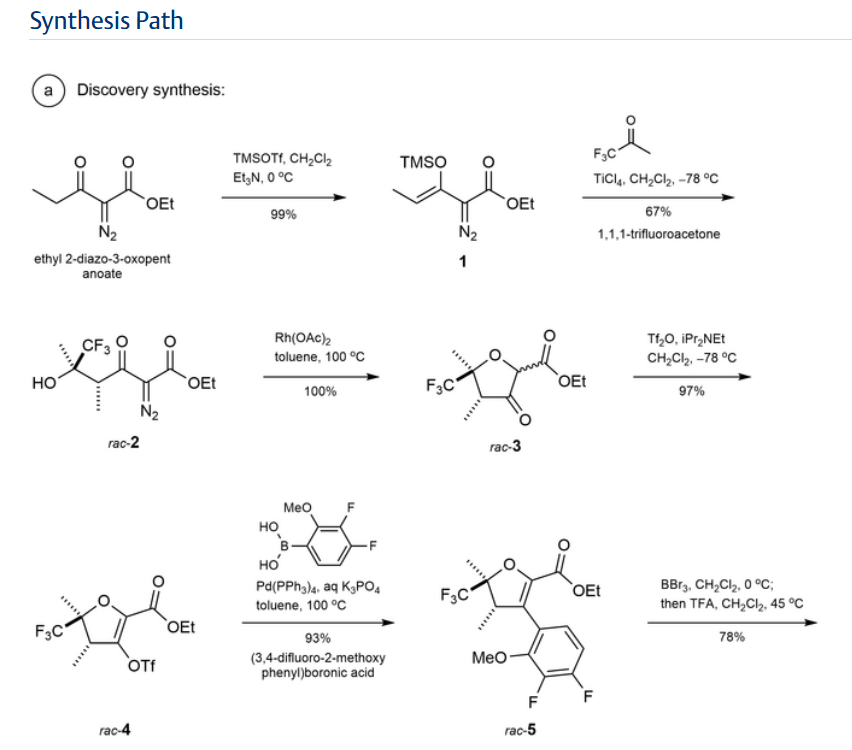
Step 1:
NEt₂ (7.7 mL, 55.2 mmol) was added to a solution of
ethyl 2-diazo-3-oxo-pentanoate (6.69 g, 39.3 mmol) in
DCM (80 mL) with stirring at 0° C. under nitrogen. Trimethylsilyl trifluoromethanesulfonate (8.5 mL, 47.0 mmol)
was added dropwise over 5 mins and the mixture was stirred
for a further 30 mins at 0° C. The reaction mixture was
diluted with pentane (100 mL), the layers separated and the
organic phase washed with dilute aqueous sodium bicarbonate (100 mL) and brine (100 mL). The organic layer was
dried (MgSO4), and concentrated in vacuo to give ethyl
(Z)-2-diazo-3-trimethylsilyloxy-pent-3-enoate (9.4 g, 99%)
as a red oil. H NMR (500 MHz, Chloroform-d) 8 5.33 (q,
J=7.0 Hz, 1H), 4.25 (q, J=7.1 Hz, 2H), 1.67 (d, J=7.0 Hz,
3H), 1.29 (t, J=7.1 Hz, 3H), 0.22 (s, 9H) ppm.
Step 2:
To a solution of 1,1,1-trifluoropropan-2-one (8 mL, 89.4
mmol) in DCM (80 mL) stirring at -78° C. was added TiCl
(70 mL of 1 M in DCM, 70.00 mmol) via cannula. To the
resulting solution, a solution of ethyl (Z)-2-diazo-3-trimethylsilyloxy-pent-3-enoate (36.1 g of 31.3% w/w, 46.6 mmol)
in 40 mL of DCM was added dropwise over 15 mins. After
100 mins the reaction was carefully quenched with water,
allowing the temperature to rise slowly, and then extracted
with DCM. The combined organic layers were dried
(MgSO), filtered, and concentrated in vacuo. Purification
by flash chromatography (330 g SiO₂, 0 to 20% EtOAc in
heptane) gave ethyl 2-diazo-6,6,6-trifluoro-5-hydroxy-4,5-
dimethyl-3-oxo-hexanoate (8.82 g, 67%), which was stored
as a solution in toluene. H NMR (500 MHz, Chloroform-d)
8 4.33 (q, J=7.1 Hz, 2H), 4.14 (q, J=7.0 Hz, 1H), 3.98 (s,
1H), 1.43 (q, J=1.2 Hz, 3H), 1.35 (t, J=7.1 Hz, 3H), 1.31 (dq.
J=7.0, 1.4 Hz, 3H) ppm. ESI-MS m/z calc. 282.08273, found
283.1 (M+1)*; 281.0 (M-1)-.
Step 3:
A solution of rhodium tetraacetate (245 mg, 0.55 mmol)
in benzene (32 mL) was heated at reflux for 10 min before
a solution of ethyl 2-diazo-6,6,6-trifluoro-5-hydroxy-4,5-
dimethyl-3-oxo-hexanoate (10 g, 35.4 mmol) in benzene (13
mL) was added slowly via addition funnel while refluxing
for 60 mins. The mixture was then concentrated in vacuo to
give ethyl rac-(4R, 5R)-4,5-dimethyl-3-oxo-5-(trifluoromethyl)tetrahydrofuran-2-carboxylate (9.0 g, 100%) as a
green coloured residue containing residual catalyst, and as a
mixture of epimers at the position next to the ester. This
material was used without further purification. H NMR
(500 MHz, Chloroform-d) 8 4.83-4.57 (m, 1H), 4.38-4.16
(m, 2H), 2.60 (dddd, J=9.3, 8.2, 5.6, 1.4 Hz, 1H), 1.73-1.63
(m, 3H), 1.30 (t, J=7.1 Hz, 3H), 1.24 (ddq, J=6.4, 4.1, 1.9
Hz, 3H) ppm.
Step 4:
To a stirred solution of ethyl rac-(4R,5R)-4,5-dimethyl- 5
3-oxo-5-(trifluoromethyl)tetrahydrofuran-2-carboxylate (48
g, 188.83 mmol) in DCM (400 mL) stirring at -78° C. was
added DIPEA (29.680 g, 40 mL, 229.64 mmol). A solution
of trifluoromethylsulfonyl trifluoromethanesulfonate
(53.440 g, 32 mL, 189.41 mmol) in DCM (200 mL) was 10
added to the reaction mixture at the same temperature over
1 h. The reaction mixture was stirred for 30 mins at 0° С.
before being quenched with 100 mL saturated aqueous
NaHCO3 solution. The organic layer was separated and
aqueous layer extracted with DCM (160 mL). The combined 15
organic layers were dried (MgSO) and concentrated in
vacuo to give ethyl rac-(4R,5R)-2,3-dimethyl-2-(trifluoromethyl)-4-(trifluoromethylsulfonyloxy)-3H-furan-5-carboxylate (71 g, 97%). H NMR (400 MHz, Chloroform-d) 8
4.38-4.32 (m, 2H), 3.29-3.23 (m, 1H), 1.64 (s, 3H), 1.37- 20
1.33 (m, 6H) ppm.
STEP 5
To stirred a solution of ethyl rac-(4R,5R)-2,3-dimethyl2-(trifluoromethyl)-4-(trifluoromethylsulfonyloxy)-3Hfuran-5-carboxylate (26 g, 67.311 mmol) in toluene (130.00
mL) was added (3,4-difluoro-2-methoxy-phenyl)boronic
acid (14 g, 74.5 mmol) followed by K3PO4 (100 mL of 2 M,
200.00 mmol) under an argon atmosphere. The reaction was
degassed before tetrakis(triphenylphosphine)palladium(0)
(4 g, 3.46 mmol) was added. After further degassing, the
reaction was heated at 100° C. for 2 hours. The reaction was
diluted in water and the aqueous layer extracted with EtOAc
(2×100 mL). The combined organic layers were concentrated in vacuo. Purification by flash chromatography (SiO.
0 to 10% EtOAc in heptane) gave ethyl 4-(3,4-difluoro-2- 35
methoxy-pheny1)-2,3-dimethyl-2-(trifluoromethyl)-3Hfuran-5-carboxylate (24.4 g, 93%) as a 6:1 diastereomeric
mixture, with the major isomer believed to be ethyl rac-(4R,
5R)-4-(3,4-difluoro-2-methoxy-phenyl)-2,3-dimethyl-2-
(trifluoromethyl)-3H-furan-5-carboxylate. Major isomer: H 40
NMR (400 MHz, Chloroform-d) 8 6.88-6.79 (m, 2H), 4.17-
4.09 (m, 2H), 3.90 (s, 3H), 3.46 (q, J=7.4 Hz, 1H), 1.67 (s,
3H), 1.12 (t, J=7.4 Hz, 3H), 1.06 (dd, J=5.4, 2.7 Hz, 3Н)
ppm. Minor isomer ¹H NMR (400 MHz, Chloroform-d) 8
6.88-6.79 (m, 2H), 4.17-4.09 (m, 2H), 3.88 (s, 3H), 3.76- 45
3.71 (m, 1H), 1.51 (s, 3H), 1.12 (t, J=7.4 Hz, 3H), 0.99 (dd,
J=5.4, 2.7 Hz, 3H) ppm. ESI-MS m/z calc. 380.1047, found
381.02 (M+1)+.
Step 6:
To an ice-cooled solution of ethyl 4-(3,4-difluoro-2- 50
methoxy-phenyl)-2,3-dimethyl-2-(trifluoromethyl)-3Hfuran-5-carboxylate (110 g, 243.0 mmol) in DCM (360 mL)
was added BBr, (370 mL of 1 M, 370.0 mmol) dropwise.
Upon completion the mixture was quenched by addition of
water and aqueous sodium bicarbonate solution, the aqueous 55
layer extracted with DCM and the combined organic layers
dried (MgSO) and concentrated in vacuo. The residue was
dissolved in DCM (430 mL) at ambient temperature and
TFA (40 mL, 519.2 mmol) was added, then the reaction was
heated to 45° C. Upon completion, the mixture was
quenched by addition of aqueous sodium bicarbonate solution and the aqueous layer extracted with DCM, dried
(MgSO) and concentrated in vacuo to give the desired
product in a 5:1 mixture of diastereomers. Recrystallization
was carried out by solubilizing the crude in the smallest
possible amount of DCM and adding a layer of heptane on
top of this solution (liquid-liquid diffusion). After approx. 1



https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021113627&_cid=P22-M90RUB-70989-1

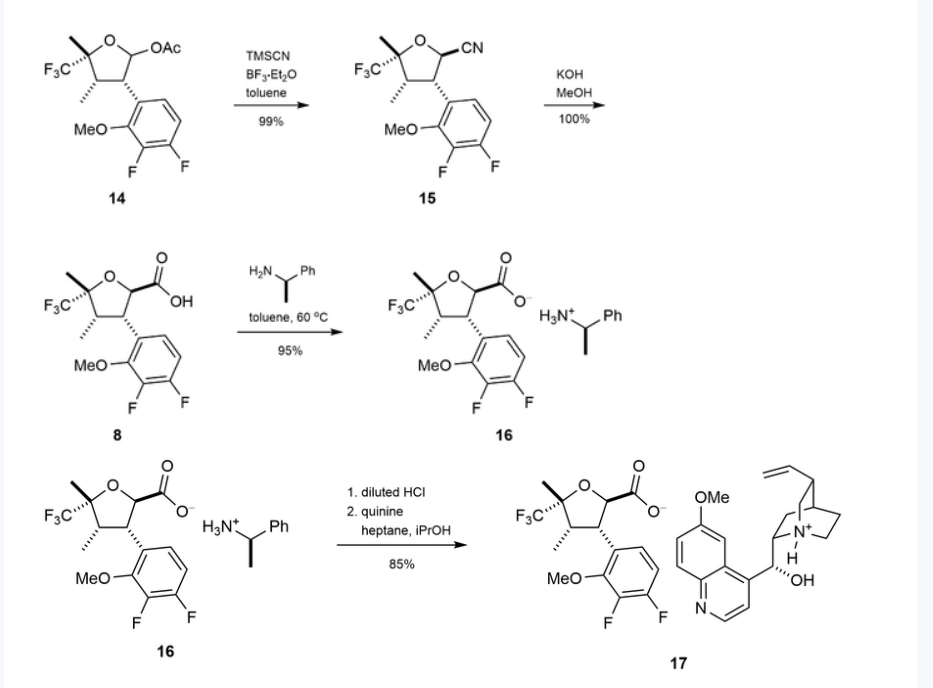
Example 6
rel-(2S,3R,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (20), (2S,3R,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)- 5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (21), rel- (2R,3S,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2- carbonyl]amino]pyridine-2-carboxamide (22), and (2R,3S,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy- phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (23)
[00676] Step 7:
[00677] (4-[[3-(3-Chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (420 mg, 0.8827 mmol) was separated by chiral SFC [(R,R)-Whelk-O1 column, 5 µm particle size, 25 cm x 21.2 mm from Regis Technologies, MeOH, 20 mM NH3], followed by further purification of one or more of the fractions by chiral SFC using a Chiralpak IC column, 5 µm particle size, 25 cm x 20 mm from Daicel or a Chiralpak ID column, 5 µum particle size, 25 cm x 20 mm from Daicel to give:
[00678] First Eluting Isomer: rel-(2S,3R,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (20, 30 mg, 7.1%) (further purified by chiral SFC using Chiralpak IC column). 1H NMR (500 MHz, Chloroform-d) δ 8.92 (s, 1H), 8.47 (d, J = 5.5 Hz, 1H), 8.21 (dd, J = 5.6, 2.1 Hz, 1H), 8.09 (d, J = 2.2 Hz, 1H), 7.87 (d, J = 4.1 Hz, 1H), 7.26 (dd, J = 8.8, 5.8 Hz, 1H), 7.03 (t, J = 8.4 Hz, 1H), 5.87 – 5.82 (m, 1H), 4.77 (d, J = 10.6 Hz, 1H), 3.98 (td, J = 11.2, 8.3 Hz, 1H), 3.88 (s, 3H), 2.51 (dd, J = 13.2, 11.7 Hz, 1H), 2.42 (dd, J = 13.2, 8.3 Hz, 1H), 1.69 (s, 3H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00679] Second Eluting Isomer: (2S,3R,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (21, 29 mg, 6.7%) (further purified by chiral SFC using Chiralpak ID column). 1H NMR (500 MHz, Chloroform-d) δ 8.56 (s, 1H), 8.48 (d, J = 5.5 Hz, 1H), 8.08 (dd, J = 5.5, 2.2 Hz, 1H), 7.98 (d, J = 2.1 Hz, 1H), 7.86 (d, J = 4.4 Hz, 1H), 7.23 (dd, J = 8.8, 5.8 Hz, 1H), 7.01 (t, J = 8.4 Hz, 1H), 5.86 (d, J = 4.2 Hz, 1H), 4.80 (d, J = 9.7 Hz, 1H), 4.10 – 4.00 (m, 1H), 3.93 (s, 3H), 3.52 – 3.48 (m, 1H), 2.86 (dd, J = 13.9, 8.4 Hz, 1H), 2.16 -2.07 (m, 1H), 1.64 (s, 2H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00680] Third Eluting Isomer: rel-(2R,3S,5R)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (22, 42 mg, 9.5%).
1H NMR (500 MHz, Chloroform-d) δ 8.87 (s, 1H), 8.33 (d, J = 5.6 Hz, 1H), 8.08 (dd, J = 5.6, 2.2 Hz, 1H), 7.98 (d, J = 2.2 Hz, 1H), 7.74 (d, J = 4.5 Hz, 1H), 7.12 (dd, J = 8.8, 5.8 Hz, 1H), 6.89 (t, J = 8.4 Hz, 1H), 5.79 (d, J = 4.5 Hz, 1H), 4.63 (d, J = 10.7 Hz, 1H), 3.85 (td, J = 11.2, 8.4 Hz, 1H), 3.74 (s, 3H), 2.37 (dd, J = 13.2, 11.7 Hz, 1H), 2.28 (dd, J = 13.1, 8.4 Hz, 1H), 1.55 (s, 3H) ppm. ESI-MS m/z calc.
475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00681] Fourth Eluting Isomer: (2R,3S,5S)-4-[[3-(3-chloro-4-fluoro-2-methoxy-phenyl)-5-methyl-5-(trifluoromethyl)tetrahydrofuran-2-carbonyl]amino]pyridine-2-carboxamide (23, 40 mg, 8.8%).
1H NMR (500 MHz, Chloroform-d) δ 8.43 (s, 1H), 8.35 (d, J = 5.5 Hz, 1H), 7.95 (dd, J = 5.5, 2.2 Hz, 1H), 7.85 (d, J = 2.2 Hz, 1H), 7.73 (d, J = 4.3 Hz, 1H), 7.10 (dd, J = 8.8, 5.9 Hz, 1H), 6.87 (t, J = 8.4 Hz, 1H), 5.76 – 5.71 (m, 1H), 4.67 (d, J = 9.7 Hz, 1H), 3.97 – 3.87 (m, 1H), 3.80 (s, 3H), 2.73 (dd, J = 13.9, 8.4 Hz, 1H), 1.98 (dd, J = 13.9, 11.6 Hz, 1H), 1.51 (s, 3H) ppm. ESI-MS m/z calc.475.0922, found 476.4 (M+1)+; 474.4 (M-1)-.
[00682] Compound 22 – Solid Form A
Efficacy
When people used suzetrigine in clinical studies conducted through 2024, there was a reduction in pain typically from seven to four on the standard numerical scale used to rate pain.[7][8] Suzetrigine provided pain relief equal to a combination of hydrocodone and paracetamol (acetaminophen) (5 mg of hydrocodone bitartrate and 325 mg of acetaminophen).[8][9]
Suzetrigine suppresses pain at the same level as an opioid, but without the risks of addiction, sedation, or overdose.[10] An alternative to opioids, it is the first pain medication to be approved by the Food and Drug Administration in two decades.[10]
The efficacy of suzetrigine was evaluated in two randomized, double-blind, placebo- and active-controlled trials of acute surgical pain, one following abdominoplasty and the other following bunionectomy.[2] Both trials found that suzetrigine reduced pain more effectively than a placebo.[2]
Contraindications
Concomitant use of suzetrigine with strong CYP3A inhibitors is contraindicated.[1][2]
Adverse effects
Common adverse effects of suzetrigine may include itching, rash, muscle spasms, and increased levels of creatine kinase.[2] Mild side effects may include nausea, constipation, headache, and dizziness.[7][8] As of 2024, long-term safety and side effects remain undetermined.[8]
In preliminary research, suzetrigine had no serious neurological, behavioral, or cardiovascular effects.[3]
Interactions
Consuming grapefruit while using suzetrigine may cause an adverse grapefruit–drug interaction.[1][2]
Mechanism of action
Suzetrigine operates on peripheral nerves, avoiding the addictive potential of opioids which affect the central nervous system.[3][4][7] Unlike opioid medications, which reduce pain signals in the brain, suzetrigine works by closing sodium channels in peripheral nerves, inhibiting pain-signaling nerves from transmitting painful sensations to the brain.[3][4][7]
In pharmacological studies, suzetrigine selectively inhibited Nav1.8 channels, but not other voltage-gated sodium channels, and bound to a unique site on these sodium channels with a novel allosteric mechanism, by binding to the channel’s second voltage sensing domain, thereby stabilizing the closed state, causing tonic inhibition. It exerts its action on dorsal root ganglion.[3]
History
Vertex Pharmaceuticals announced in January 2024 that suzetrigine had successfully met several endpoints in its Phase III clinical trials.[5] The company announced in July 2024 that the FDA had accepted a new drug application for suzetrigine.[11] The FDA granted the application for suzetrigine priority review, fast track, and breakthrough therapy designations.[2][11] In January 2025, the FDA granted approval of Journavx to Vertex Pharmaceuticals.[2]
Society and culture
Legal status
Suzetrigine was approved for medical use in the United States in January 2025.[2]
Names
Suzetrigine is the international nonproprietary name.[12]
Suzetrigine is sold under the brand name Journavx.[1][2]





References
a) WO2021113627A1 (Vertex, 10.06.2021; USA-prior. 06.12.2019).
US11834441B2 (Vertex, 05.12.2023; USA-prior. 06.12.2019).
b) WO2022256660A1 (Vertex, 08.12.2022; USA-prior. 04.06.2021).
WO2024123815A1 (Vertex, 13.06.2024; USA-prior. 06.12.2022).
WO2022256708A1 (Vertex, 08.12.2022; USA-prior. 04.06.2021, 02.12.2021).
Source:
Suzetrigine, in Kleemann A., Kutscher B., Reichert D., Bossart M., Pharmaceutical Substances, Thieme. https://pharmaceutical-substances.thieme.com/lexicon/KD-19-0151, accessed: 05-29-2025
| Clinical data | |
|---|---|
| Pronunciation | /suˈzɛtrɪdʒiːn/ soo-ZE-tri-jeen |
| Trade names | Journavx |
| Other names | VX-548 |
| AHFS/Drugs.com | Journavx |
| License data | US DailyMed: Suzetrigine |
| Routes of administration | By mouth |
| Drug class | Nav1.8 sodium channel blocker; Analgesic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2649467-58-1 |
| PubChem CID | 156445116 |
| DrugBank | DB18927 |
| ChemSpider | 128942439 |
| UNII | LOG73M21H5 |
| KEGG | D12860 |
| ChEMBL | ChEMBL5314487 |
| Chemical and physical data | |
| Formula | C21H20F5N3O4 |
| Molar mass | 473.400 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
References
- ^ Jump up to:a b c d e f g h “Journavx- suzetrigine tablet, film coated”. DailyMed. 6 February 2025. Retrieved 2 April 2025.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves Novel Non-Opioid Treatment for Moderate to Severe Acute Pain” (Press release). U.S. Food and Drug Administration (FDA). 30 January 2025. Archived from the original on 7 February 2025. Retrieved 30 January 2025.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e Osteen, Jeremiah D.; Immani, Swapna; Tapley, Tim L.; Indersmitten, Tim; Hurst, Nicole W.; Healey, Tiffany; et al. (January 2025). “Pharmacology and Mechanism of Action of Suzetrigine, a Potent and Selective NaV1.8 Pain Signal Inhibitor for the Treatment of Moderate to Severe Pain”. Pain and Therapy. doi:10.1007/s40122-024-00697-0. PMID 39775738.
- ^ Jump up to:a b c Jones, Jim; Correll, Darin J.; Lechner, Sandra M; Jazic, Ina; Miao, Xiaopeng; Shaw, David; et al. (August 2023). “Selective Inhibition of NaV1.8 with VX-548 for Acute Pain”. The New England Journal of Medicine. 389 (5): 393–405. doi:10.1056/NEJMoa2209870. PMID 37530822. S2CID 260377748.
- ^ Jump up to:a b “Vertex Announces Positive Results From the VX-548 Phase 3 Program for the Treatment of Moderate-to-Severe Acute Pain” (Press release). Vertex. 30 January 2024. Archived from the original on 25 December 2024. Retrieved 31 January 2025 – via Business Wire.
- ^ “Novel Drug Approvals for 2025”. U.S. Food and Drug Administration (FDA). 21 February 2025. Retrieved 9 March 2025.
- ^ Jump up to:a b c d Broadfoot, Marla (20 August 2024). “New Painkiller Could Bring Relief to Millions — without Addiction Risk”. Scientific American. Archived from the original on 30 December 2024. Retrieved 31 January 2025.
- ^ Jump up to:a b c d Hang Kong, Aaron Yik; Tan, Hon Sen; Habib, Ashraf S. (September 2024). “VX-548 in the Treatment of Acute Pain”. Pain Management. 14 (9): 477–486. doi:10.1080/17581869.2024.2421749. PMC 11721852. PMID 39552600.
- ^ Kingwell, Katie (December 2024). “NaV1.8 inhibitor poised to provide opioid-free pain relief”. Nature Reviews. Drug Discovery. 24 (1): 3–5. doi:10.1038/d41573-024-00203-3. PMID 39668193.
- ^ Jump up to:a b Dolgin, Elie (January 2025). “US drug agency approves potent painkiller – the first non-opioid in decades”. Nature. 638 (8050): 304–305. doi:10.1038/d41586-025-00274-1. PMID 39885357.
- ^ Jump up to:a b “Vertex Announces FDA Acceptance of New Drug Application for Suzetrigine for the Treatment of Moderate-to-Severe Acute Pain” (Press release). Vertex. 30 July 2024. Retrieved 31 January 2025 – via Business Wire.
- ^ World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 90”. WHO Drug Information. 37 (3). hdl:10665/373341.
Further reading
- Oliver, Brian; Devitt, Catherine; Park, Grace; Razak, Alina; Liu, Sun Mei; Bergese, Sergio D. (2025). “Drugs in Development to Manage Acute Pain”. Drugs. 85 (1): 11–19. doi:10.1007/s40265-024-02118-0. PMID 39560856.
External links
- “Suzetrigine (Code C199115)”. NCI Thesaurus.
- Clinical trial number NCT05661734 for “A Single-arm Study to Evaluate Safety and Effectiveness of VX-548 for Acute Pain” at ClinicalTrials.gov
- Clinical trial number NCT05558410 for “Evaluation of Efficacy and Safety of VX-548 for Acute Pain After an Abdominoplasty” at ClinicalTrials.gov
//////////Suzetrigine, Journavx, FDA 2025, APPROVALS 2025, CS-0641183, HY-148800, VX 548, VX-548, VX548, Breakthrough Therapy, Fast Track, Priority Review
Maribavir

Maribavir
- Molecular FormulaC15H19Cl2N3O4
- Average mass376.235 Da
FDA APROVED 11/23/2021, Livtencity1263 W94, 1263W94
176161-24-3[RN]
1H-Benzimidazol-2-amine, 5,6-dichloro-N-(1-methylethyl)-1-β-L-ribofuranosyl-
UNII-PTB4X93HE1, марибавир , ماريبافير ,马立巴韦 , BW-1263W94
Camvia, D04859, G1263, GW257406X
1263W94; BW-1263W94; GW-1263; GW-257406X; SHP-620; VP-41263
Company:GlaxoSmithKline (Originator) , Shire
MOA:UL97 kinase inhibitorIndication:CMV prophylaxis
To treat post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV
Press Release
Reference:1. WO9601833A1.
Syn
US 6204249
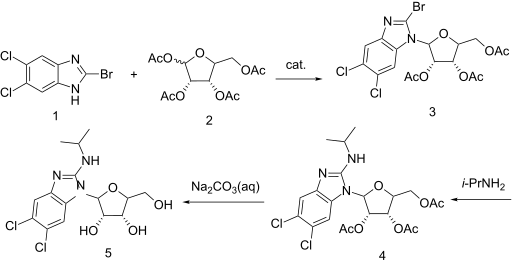

https://patents.google.com/patent/WO2001077083A1/enExample 7: 5,6-Dichloro-2-(isoproylamino)-1-(β-L-ribofuranosyl)-1 H-benzimidazolesoprylamino (10 mL) and 2-bromo-5,6-dichloro-1-(2,3,5-tri-0-acetyl-β-L- ribofuranosyl)-1 H-benzimidazole (1.0 g, 1.9 mmol) were combined with absolute ethanol (20 mL) and stirred at 75°C for 48 h. The reaction mixture was concentrated and purified on a silica gel column (2.5 vm x 16 cm, 230-400 mesh) with 1 :20 methanol: dichloromethane to give product contaminated with a small amount of higher Rf material. This was repurified on a chromatotron, fitted with a 2 mm silica gel rotor, with 1 :25 methanol.dichloromethane to give a white solid (0.43 g, 1.15 mmol, 60o/o); [a]20D=(-)22.4 (c=0.5 DMF); UVλ™* (E): pH 7.0:304 nm (95,00), 275 (1 ,800) 260 (8,300); 0.1 NaOH: 304 nm (9,900), 275 (19,00), 260 (8,100); MS (Cl): m/z (re/, intensity) 376 (100, M+1); ‘H NMR (DMSO-de) d 7.59 (s, 1 H, Ar-H), 7.35 (s, 1 H, Ar- H), 6.90 (d, 1 H, NH, J=7.8 Hz), 5.73 (d, 1 H, H-1′, J=6.5 Hz), 5.62 (t, 1 H, OH, J=4.2 Hz), 5.27-5.23 (m, 2H, OH), 4.27 (apparent dd, 1 H, J=13.4 Hz, J=7.6 Hz), 4.11 -3.99 (m, 2H), 3.97 (br. s, 1 H), 3.72-3.61 (m, 2H, H-5’), 1.18 (d, 6H, CH(CH3)2, J=6.6 Hz).Anal. Calcd. for

H2O: C, 45.70; H, 5.37; N, 10.66. Found: C, 45.75; H, 4.98; N, 10.50.
Maribavir was in phase II clinical trials for the treatment of cytomegalovirus (CMV) infection. It was granted orphan drug designation by the FDA for the indication.
The drug was originally developed by the University of Michigan and was licensed to GlaxoSmithKline. ViroPharma (now subsidiary of Shire) acquired worldwide rights to the drug from GlaxoSmithKline in 2003.
Maribavir, sold under the brand name Livtencity, is an antiviral medication that is used to treat post-transplant cytomegalovirus (CMV).[1][2]
The most common side effects include taste disturbance, nausea, diarrhea, vomiting and fatigue.[2]
Maribavir is a cytomegalovirus pUL97 kinase inhibitor that works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.[2]
Maribavir was approved for medical use in the United States in November 2021.[2][3]
Medical uses
Maribavir is indicated to treat people twelve years of age and older and weighing at least 35 kilograms (77 lb) with post-transplant cytomegalovirus infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for cytomegalovirus.[2]
Contraindications
Maribavir may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these medications is not recommended.[2]
History
Maribavir is licensed by ViroPharma from GlaxoSmithKline in 2003, for the prevention and treatment of human cytomegalovirus (HCMV) disease in hematopoietic stem cell/bone marrow transplant patients. The mechanism by which maribavir inhibits HCMV replication is by inhibition of an HCMV encoded protein kinase enzyme called UL97 or pUL97.[4] Maribavir showed promise in Phase II clinical trials and was granted fast track status, but failed to meet study goals in a Phase III trial.[5] However, the dosage used in the Phase III trial may have been too low to be efficacious.[6]
A Phase II study with maribavir demonstrated that prophylaxis with maribavir displayed strong antiviral activity, as measured by statistically significant reduction in the rate of reactivation of CMV in recipients of hematopoietic stem cell/bone marrow transplants.[7] In an intent-to-treat analysis of the first 100 days after the transplant, the number of subjects who required pre-emptive anti-CMV therapy was statistically significantly reduced with maribavir compared to placebo.
ViroPharma conducted a Phase III clinical study to evaluate the prophylactic use for the prevention of cytomegalovirus disease in recipients of allogeneic stem cell transplant patients. In February 2009, ViroPharma announced that the Phase III study failed to achieve its goal, showing no significant difference between maribavir and a placebo at reducing the rate at which CMV DNA levels were detected in patients.[8]
The safety and efficacy of maribavir were evaluated in a Phase III, multicenter, open-label, active-controlled trial that compared maribavir with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat cytomegalovirus: ganciclovir, valganciclovir, foscarnet, or cidofovir.[2] In the study, 352 transplant recipients with cytomegalovirus infections who did not respond (with or without resistance) to treatment randomly received maribavir or treatment assigned by a researcher for up to eight weeks.[2] The study compared the two groups’ plasma cytomegalovirus DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable.[2] Of the 235 participants who received maribavir, 56% had levels of cytomegalovirus DNA below what was measurable versus 24% of the 117 participants who received an investigator-assigned treatment.[2]
The U.S. Food and Drug Administration (FDA) granted the application for maribavir orphan drug, breakthrough therapy and priority review designations.[2][3][9][10] The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.[2][3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs
Approval is for Cytomegalovirus, a Type of Herpes Virus
https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-common-type-post-transplant-infection-resistant-other-drugsFor Immediate Release:November 23, 2021
Today, the U.S. Food and Drug Administration approved Livtencity (maribavir) as the first drug for treating adults and pediatric patients (12 years of age and older and weighing at least 35 kilograms) with post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV. Livtencity works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.
“Transplant recipients are at a much greater risk for complications and death when faced with a cytomegalovirus infection,” said John Farley, M.D., M.P.H., director of the Office of Infectious Diseases in the FDA’s Center for Drug Evaluation and Research. “Cytomegalovirus infections that are resistant or do not respond to available drugs are of even greater concern. Today’s approval helps meet a significant unmet medical need by providing a treatment option for this patient population.”
CMV is a type of herpes virus that commonly causes infection in patients after a stem cell or organ transplant. CMV infection can lead to CMV disease and have a major negative impact on transplant recipients, including loss of the transplanted organ and death.
Livtencity’s safety and efficacy were evaluated in a Phase 3, multicenter, open-label, active-controlled trial that compared Livtencity with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat CMV: ganciclovir, valganciclovir, foscarnet or cidofovir. In the study, 352 transplant recipients with CMV infections who did not respond (with or without resistance) to treatment randomly received Livtencity or treatment assigned by a researcher for up to eight weeks.
The study compared the two groups’ plasma CMV DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable. Of the 235 patients who received Livtencity, 56% had levels of CMV DNA below what was measurable versus 24% of the 117 patients who received an investigator-assigned treatment.
The most common side effects of Livtencity include taste disturbance, nausea, diarrhea, vomiting and fatigue. Livtencity may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these drugs is not recommended. Virologic failure due to resistance can occur during and after treatment with Livtencity, therefore CMV DNA levels should be monitored and Livtencity resistance should be checked if the patient is not responding to treatment or relapses.
Livtencity received Breakthrough Therapy and Priority Review designations for this indication. Breakthrough Therapy designation is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s). Priority Review designation directs overall attention and resources to the evaluation of applications for drugs that, if approved, would be significant improvements in the safety or effectiveness of the treatment, diagnosis or prevention of serious conditions when compared to standard applications.
The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.
Related Information
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215596lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs”. U.S. Food and Drug Administration (FDA) (Press release). 23 November 2021. Retrieved 23 November 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “Takeda’s Livtencity (maribavir) Approved by U.S. FDA as the First and Only Treatment for People Ages 12 and Older with Post-Transplant Cytomegalovirus (CMV), Refractory (With or Without Genotypic Resistance) to Conventional Antiviral Therapies”. Takeda (Press release). 23 November 2021. Retrieved 26 November 2021.
- ^ Biron KK, Harvey RJ, Chamberlain SC, Good SS, Smith AA, Davis MG, et al. (August 2002). “Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action”. Antimicrobial Agents and Chemotherapy. 46 (8): 2365–72. doi:10.1128/aac.46.8.2365-2372.2002. PMC 127361. PMID 12121906.
- ^ Marty FM, Ljungman P, Papanicolaou GA, Winston DJ, Chemaly RF, Strasfeld L, et al. (April 2011). “Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial”. The Lancet. Infectious Diseases. 11 (4): 284–92. doi:10.1016/S1473-3099(11)70024-X. PMID 21414843.
- ^ Snydman DR (April 2011). “Why did maribavir fail in stem-cell transplants?”. The Lancet. Infectious Diseases. 11 (4): 255–7. doi:10.1016/S1473-3099(11)70033-0. PMID 21414844.
- ^ Phase 2 Data Shows Maribavir Markedly Reduced Rate Of Cytomegalovirus Infection And Disease In Bone Marrow Transplant Patients, Medical News Today, Jun 2, 2008
- ^ ViroPharma:Maribavir Phase III Study Missed Goal;Shares Plunge, CNN Money, February 09, 2009
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 1 February 2007. Retrieved 26 November 2021.
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 7 June 2011. Retrieved 26 November 2021.
External links
- “Maribavir”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02931539 for “Efficacy and Safety Study of Maribavir Treatment Compared to Investigator-assigned Treatment in Transplant Recipients With Cytomegalovirus (CMV) Infections That Are Refractory or Resistant to Treatment With Ganciclovir, Valganciclovir, Foscarnet, or Cidofovir” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Livtencity |
| Other names | 1263W94 |
| License data | USDailyMed: Maribavir |
| Routes of administration | By mouth |
| ATC code | J05AX10 (WHO) |
| Legal status | |
| Legal status | US:℞-only[1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 176161-24-3 |
| PubChemCID | 471161 |
| DrugBank | DB06234 |
| ChemSpider | 413807 |
| UNII | PTB4X93HE1 |
| ChEMBL | ChEMBL515408 |
| NIAID ChemDB | 070966 |
| CompTox Dashboard (EPA) | DTXSID60170091 |
| Chemical and physical data | |
| Formula | C15H19Cl2N3O4 |
| Molar mass | 376.23 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////Maribavir, APPROVALS 2021, FDA 2021, Livtencity, Takeda, Breakthrough Therapy, Priority Review , ORPHAN, UNII-PTB4X93HE1, марибавир , ماريبافير ,马立巴韦 , BW-1263W94, Camvia, D04859, G1263, GW257406X, 1263W94, BW-1263W94, GW-1263, GW-257406X, SHP-620, VP-41263,

NEW DRUG APPROVALS
ONE TIME
$10.00
Pafolacianine

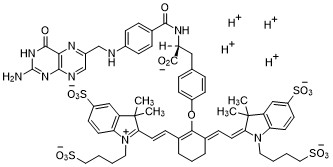
Pafolacianine
OTL-38
- Molecular FormulaC61H67N9O17S4
- Average mass1326.495 Da
FDA APPROVED NOV 2021
2-{(E)-2-[(3E)-2-(4-{2-[(4-{[(2-Amino-4-oxo-3,4-dihydro-6-pteridinyl)methyl]amino}benzoyl)amino]-2-carboxyethyl}phenoxy)-3-{(2E)-2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene ]ethylidene}-1-cyclohexen-1-yl]vinyl}-3,3-dimethyl-1-(4-sulfobutyl)-3H-indolium-5-sulfonate OTL-38Tyrosine, N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-O-[(6E)-6-[(2E)-2-[1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene]ethylidene]-2-[(E)-2-[3,3-dimethy l-5-sulfo-1-(4-sulfobutyl)-3H-indolium-2-yl]ethenyl]-1-cyclohexen-1-yl]-, inner salt
2-(2-(2-(4-((2S)-2-(4-(((2-amino-4-oxo-3,4-dihydropteridin-6-yl)methyl)amino)benzamido)-2-carboxyethyl)phenoxy)-3-(2-(3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene)ethylidene)cyclohex-1-en-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-3H-indolium inner salt,sodium salt (1:4)
- 3H-Indolium, 2-(2-(2-(4-((2S)-2-((4-(((2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl)amino)benzoyl)amino)-2-carboxyethyl)phenoxy)-3-(2-(1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene)ethylidene)-1-cyclohexen-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1 (4-sulfobutyl)-, inner salt,sodium salt (1:4)
1628423-76-6 [RN]
Pafolacianine sodium [USAN]
RN: 1628858-03-6
UNII: 4HUF3V875C
C61H68N9Na4O17S4+5
- Intraoperative Imaging and Detection of Folate Receptor Positive Malignant Lesions
Pafolacianine, sold under the brand name Cytalux, is an optical imaging agent.[1][2]
The most common side effects of pafolacianine include infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity.[2]
It was approved for medical use in the United States in November 2021.[2][3]
Pafolacianine is a fluorescent drug that targets folate receptor (FR).[1]
Medical uses
Pafolacianine is indicated as an adjunct for intraoperative identification of malignant lesions in people with ovarian cancer.[1][2]
History
The safety and effectiveness of pafolacianine was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.[2] Of the 134 women (ages 33 to 81 years) who received a dose of pafolacianine and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pafolacianine orphan drug, priority review, and fast track designations.[2][4] The FDA granted the approval of Cytalux to On Target Laboratories, LLC.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

WO 2014149073
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014149073
In another aspect of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0029] In a fourth embodiment of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0030]
[0032] wherein C is any carbon isotope. In this embodiment, the amino acid linker is selected from a group consisting of methyl 2-di-tert-butyl dicarbonate-amino-3-(4-phenyl)propanoate, 3-(4-hydroxyphenyl)-2-(di-tert-butyl-dicarbonate methylamino)propanoic acid, 2-amino-4-(4-hydroxyphenyl)butanoic acid, and Tert-butyl (2-di-tert-butyl dicarbonate- amino)-3-(4-hydroxyphenyl)propanoate . In a particular embodiment, the aqueous base is potassium hydroxide (KOH). The method of this embodiment may also further include purifying the compound by preparatory HPLC.
EXAMPLE 1 : General synthesis of Pte – L Tyrosine – S0456 (OTL-0038)
[0088] Scheme:
C33H37CIF3N
Reactants for Step I:
[0089] A 500 mL round bottom flask was charged with a stirring bar, pteroic acid
(12.0 g, 29.40 mmol, 1 equiv), (L)-Tyr(-OfBu)-OfBu- HCI (1 1 .63 g, 35.28 mmol, 1 .2
equiv) and HATU (13.45 g, 35.28 mmol, 1 .2 equiv) then DMF (147 mL) was added to give a brown suspension [suspension A]. DIPEA (20.48 mL, 1 17.62 mmol, 4.0 equiv) was added slowly to suspension A at 23 °C, over 5 minutes. The suspension turned in to a clear brown solution within 10 minutes of addition of DIPEA. The reaction was stirred at 23 °C for 2.5 h. Reaction was essentially complete in 30 minutes as judged by LC/MS but was stirred further for 2.5 h. The formation of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI (Figure 12) was confirmed by LC/MS showing m/z 409→m/z 684. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column . The reaction mixture was cannulated as a steady stream to a stirred solution of aq. HCI (2.0 L, 0.28 M) over the period of 30 minutes to give light yellow precipitate of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The precipitated Pte_N 10(TFA)_L_Tyr(- OfBu)-OfBu HCI was filtered using sintered funnel under aspirator vacuum, washed with water (8 * 300 mL) until the pH of the filtrate is between 3 and 4. The wet solid was allowed to dry under high vacuum for 12 hours on the sintered funnel. In a separate batch, where this wet solid (3) was dried under vacuum for 48 hours and then this solid was stored at -20 0 C for 48 h. However, this brief storage led to partial decomposition of 3. The wet cake (58 g) was transferred to a 500 mL round bottom flask and was submitted to the next step without further drying or purification.
Reactants for Step II:
The wet solid (58 g) was assumed to contain 29.40 mmol of the desired compound (3) (i. e. quantitative yield for the step I ).
[0090] A 500 mL round bottom flask was charged with a stirring bar, Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI as a wet cake (58 g, 29.40 mmol, 1 equiv). A solution of TFA:TIPS:H20 (95:2.5:2.5, 200 mL) was added at once to give a light brown suspension. The reaction content was stirred at 23°C for 1 .5 hours and was monitored by LC/MS. The suspension became clear dull brown solution after stirring for 5 minutes. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column. The formation of Pte_TFA_L_Tyr (Figure 12) was confirmed by showing m/z 684→m/z 572. Reaction time varies from 30 min to 1 .5 hours depending on the water content of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The reaction mixture was cannulated as a steady stream to a stirred MTBE (1 .8 L) at 23 °C or 100 °C to give light yellow precipitate of Pte_TFA_L_Tyr. The precipitated Pte_TFA_L_Tyr was filtered using sintered funnel under aspirator vacuum, washed with MTBE (6 * 300 mL) and dried under high vacuum for 8 hours to obtain Pte_TFA_L_Tyr (14.98 g, 83.98% over two steps) as a pale yellow solid. The MTBE washing was tested for absence of residual TFA utilizing wet pH paper (pH between 3-4). The yield of the reaction was between 80-85% in different batches. The deacylated side product was detected in 3.6% as judged by LC/MS. For the different batches this impurity was never more than 5%.
Reactants for Step III:
[0091] A 200 mL round bottom flask was charged with a stirring bar and Pte_TFA_L_Tyr (13.85 g, 22.78 mmol, 1 equiv), then water (95 mL) was added to give a yellow suspension [suspension B]. A freshly prepared solution of aqueous 3.75 M NaOH (26.12 mL, 97.96 mmol, 4.30 equiv), or an equivalent base at a corresponding temperature using dimethylsulfoxide (DMSO) as a solvent (as shown in Table 1 ), was added dropwise to suspension B at 23 °C, giving a clear dull yellow solution over 15 minutes [solution B]. The equivalence of NaOH varied from 3.3 to 5.0 depending on the source of 4 (solid or liquid phase synthesis) and the residual TFA. Trianion 5 (Figure 12) formation was confirmed by LC/MS showing m/z 572→m/z 476 while the solution pH was 9-10 utilizing wet pH paper. The pH of the reaction mixture was in the range of 9-10. This pH is crucial for the overall reaction completion. Notably, pH more than 10 leads to hydrolysis of S0456. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. The presence of hydrolysis by product can be visibly detected by the persistent opaque purple/blue to red/brown color.
TABLE 1 : Separate TFA deprotection via trianion formation; S0456
[0092] The precipitated OTL-0038 product could also be crashed out by adding the reaction solution steady dropwise to acetone, acetonitrile, isopropanol or ethyl acetate/acetone mixture. Acetone yields optimal results. However, viscous reactions could be slower due to partial insolubility and/or crashing out of S0456. In this reaction, the equivalence of the aqueous base is significant. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. This solution phase synthesis provides Pte_N10(TFA)_Tyr-OH »HCI salt and desires approximately 4.1 to approximately 4.8 equiv base as a source to hydrolyze the product. Particularly, precipitation of Pte_Tyr_S0456 was best achieved when 1 mL of reaction mixture is added dropwise to the stirred acetone (20 mL). Filtration of the precipitate and washing with acetone (3 x10 mL) gave the highest purity as judged from LC/MS chromatogram.
[0093] During experimentation of this solution-phase synthesis of Pte – L Tyrosine -S0456 (OTL-0038) at different stages, some optimized conditions were observed:
Mode of addition: Separate TFA deprotection via trianion formation; S0456 @ 23 °C; reflux.
Stability data of Pte – L Tyrosine – S0456 (OTL-0038):
Liquid analysis: At 40 °C the liquid lost 8.6% at 270 nm and 1 % at 774 nm. At room temperature the liquid lost about 1 .4% at 270 nm and .5% at 774 nm. At 5 °C the
270 nm seems stable and the 774 nm reasonably stable with a small degradation purity.
Source Purity Linker S0456 Base Solvent Duration % Conversion
4.3-4.6
Solution 0.95
95% 1 equiv equiv H20 15 min 100% phase equiv
K2C03
PATENT
US 20140271482
FDA approves pafolacianine for identifying malignant ovarian cancer lesions
On November 29, 2021, the Food and Drug Administration approved pafolacianine (Cytalux, On Target Laboratories, LLC), an optical imaging agent, for adult patients with ovarian cancer as an adjunct for interoperative identification of malignant lesions. Pafolacianine is a fluorescent drug that targets folate receptor which may be overexpressed in ovarian cancer. It is used with a Near-Infrared (NIR) fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
Efficacy was evaluated in a single arm, multicenter, open-label study (NCT03180307) of 178 women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer scheduled to undergo primary surgical cytoreduction, interval debulking, or recurrent ovarian cancer surgery. All patients received pafolacianine. One hundred and thirty-four patients received fluorescence imaging evaluation in addition to standard of care evaluation which includes pre-surgical imaging, intraoperative palpation and normal light evaluation of lesions. Among these patients, 36 (26.9%) had at least one evaluable ovarian cancer lesion detected with pafolacianine that was not observed by standard visual or tactile inspection. The patient-level false positive rate of pafolacianine with NIR fluorescent light with respect to the detection of ovarian cancer lesions confirmed by central pathology was 20.2% (95% CI 13.7%, 28.0%).
The most common adverse reactions (≥1%) occurring in patients were nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, pruritus, and hypersensitivity.
The recommended pafolacianine dose is 0.025 mg/kg administered intravenously over 60 minutes, 1 to 9 hours before surgery. The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of pafolacianine.
View full prescribing information for Cytalux.
This application was granted priority review, fast track designation, and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
USFDA approves new drug to help identify cancer lesions
This drug is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery.By The Health Master -December 2, 2021
The U.S. Food and Drug Administration (USFDA) has approved Cytalux (pafolacianine), an imaging drug intended to assist surgeons in identifying ovarian cancer lesions. The drug is designed to improve the ability to locate additional ovarian cancerous tissue that is normally difficult to detect during surgery.
Cytalux is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery. The drug is a diagnostic agent that is administered in the form of an intravenous injection prior to surgery.
Alex Gorovets, M.D., deputy director of the Office of Specialty Medicine in the FDA’s Center for Drug Evaluation and Research said, “The FDA’s approval of Cytalux can help enhance the ability of surgeons to identify deadly ovarian tumors that may otherwise go undetected.
By supplementing current methods of detecting ovarian cancer during surgery, Cytalux offers health care professionals an additional imaging approach for patients with ovarian cancer.”
The American Cancer Society estimates there will be more than 21,000 new cases of ovarian cancer and more than 13,000 deaths from this disease in 2021, making it the deadliest of all female reproductive system cancers.
Conventional treatment for ovarian cancer includes surgery to remove as many of the tumors as possible, chemotherapy to stop the growth of malignant cells or other targeted therapy to identify and attack specific cancer cells.
Ovarian cancer often causes the body to overproduce a specific protein in cell membranes called a folate receptor. Following administration via injection, Cytalux binds to these proteins and illuminates under fluorescent light, boosting surgeons’ ability to identify the cancerous tissue.
Currently, surgeons rely on preoperative imaging, visual inspection of tumors under normal light or examination by touch to identify cancer lesions. Cytalux is used with a Near-Infrared fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
The safety and effectiveness of Cytalux was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.
Of the 134 women (ages 33 to 81 years) who received a dose of Cytalux and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.
The most common side effects of Cytalux were infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity. Cytalux may cause fetal harm when administered to a pregnant woman.
The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of Cytalux. There is a risk of image interpretation errors with the use of Cytalux to detect ovarian cancer during surgery, including false negatives and false positives.
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/214907s000lbl.pdf
- ^ Jump up to:a b c d e f g h i “FDA Approves New Imaging Drug to Help Identify Ovarian Cancer Lesions”. U.S. Food and Drug Administration (FDA) (Press release). 29 November 2021. Retrieved 30 November 2021. This article incorporates text from this source, which is in the public domain.
- ^ “On Target Laboratories Announces FDA Approval of Cytalux (pafolacianine) injection for Identification of Ovarian Cancer During Surgery”. On Target Laboratories. 29 November 2021. Retrieved 30 November 2021 – via PR Newswire.
- ^ “Pafolacianine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 23 December 2014. Retrieved 30 November 2021.
External links
- “Pafolacianine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Cytalux |
| Other names | OTL-0038 |
| License data | US DailyMed: Pafolacianine |
| Pregnancy category | Not recommended |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1628423-76-6 |
| PubChem CID | 135565623 |
| DrugBank | DB15413 |
| ChemSpider | 64880249 |
| UNII | F7BD3Z4X8L |
| ChEMBL | ChEMBL4297412 |
| Chemical and physical data | |
| Formula | C61H67N9O17S4 |
| Molar mass | 1326.49 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Pafolacianine, FDA 2021, APPROVALS 2021, Cytalux, OVARIAN CANCER, OTL 38,
[Na+].[Na+].[Na+].[Na+].CC1(C)\C(=C/C=C/2\CCCC(=C2Oc3ccc(C[C@H](NC(=O)c4ccc(NCc5cnc6N=C(N)NC(=O)c6n5)cc4)C(=O)O)cc3)\C=C\C7=[N](CCCCS(=O)(=O)O)c8ccc(cc8C7(C)C)S(=O)(=O)O)\N(CCCCS(=O)(=O)O)c9ccc(cc19)S(=O)(=O)O

NEW DRUG APPROVALS
ONE TIME
$10.00
Avalglucosidase alfa
QQGASRPGPR DAQAHPGRPR AVPTQCDVPP NSRFDCAPDK AITQEQCEAR GCCYIPAKQG
LQGAQMGQPW CFFPPSYPSY KLENLSSSEM GYTATLTRTT PTFFPKDILT LRLDVMMETE
NRLHFTIKDP ANRRYEVPLE TPRVHSRAPS PLYSVEFSEE PFGVIVHRQL DGRVLLNTTV
APLFFADQFL QLSTSLPSQY ITGLAEHLSP LMLSTSWTRI TLWNRDLAPT PGANLYGSHP
FYLALEDGGS AHGVFLLNSN AMDVVLQPSP ALSWRSTGGI LDVYIFLGPE PKSVVQQYLD
VVGYPFMPPY WGLGFHLCRW GYSSTAITRQ VVENMTRAHF PLDVQWNDLD YMDSRRDFTF
NKDGFRDFPA MVQELHQGGR RYMMIVDPAI SSSGPAGSYR PYDEGLRRGV FITNETGQPL
IGKVWPGSTA FPDFTNPTAL AWWEDMVAEF HDQVPFDGMW IDMNEPSNFI RGSEDGCPNN
ELENPPYVPG VVGGTLQAAT ICASSHQFLS THYNLHNLYG LTEAIASHRA LVKARGTRPF
VISRSTFAGH GRYAGHWTGD VWSSWEQLAS SVPEILQFNL LGVPLVGADV CGFLGNTSEE
LCVRWTQLGA FYPFMRNHNS LLSLPQEPYS FSEPAQQAMR KALTLRYALL PHLYTLFHQA
HVAGETVARP LFLEFPKDSS TWTVDHQLLW GEALLITPVL QAGKAEVTGY FPLGTWYDLQ
TVPIEALGSL PPPPAAPREP AIHSEGQWVT LPAPLDTINV HLRAGYIIPL QGPGLTTTES
RQQPMALAVA LTKGGEARGE LFWDDGESLE VLERGAYTQV IFLARNNTIV NELVRVTSEG
AGLQLQKVTV LGVATAPQQV LSNGVPVSNF TYSPDTKVLD ICVSLLMGEQ FLVSWC
(Disulfide bridge:26-53, 36-52, 47-71, 477-502, 591-602, 882-896)
Avalglucosidase alfa
アバルグルコシダーゼアルファ (遺伝子組換え)
Avalglucosidase alfa (USAN/INN);
Avalglucosidase alfa (genetical recombination) (JAN);
Avalglucosidase alfa-ngpt
To treat late-onset Pompe disease
| Formula | C4490H6818N1197O1299S32 |
|---|---|
| CAS | 1802558-87-7 |
| Mol weight | 99375.4984 |
FDA APPROVED Nexviazyme, 2021/8/6, Enzyme replacement therapy product
Treatment of Pompe disease
Biologic License Application (BLA): 761194
Company: GENZYME CORP
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-pompe-diseaseFor Immediate Release:August 06, 2021
Today, the U.S. Food and Drug Administration approved Nexviazyme (avalglucosidase alfa-ngpt) for intravenous infusion to treat patients 1 year of age and older with late-onset Pompe disease.
Patients with Pompe disease have an enzyme deficiency that leads to the accumulation of a complex sugar, called glycogen, in skeletal and heart muscles, which cause muscle weakness and premature death from respiratory or heart failure. Normally, glycogen—the stored form of glucose—breaks down to release glucose into the bloodstream to be used as fuel for the cells.
“Pompe disease is a rare genetic disease that causes premature death and has a debilitating effect on people’s lives,” said Janet Maynard, M.D., deputy director of the Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine in the FDA’s Center for Drug Evaluation and Research. “Today’s approval brings patients with Pompe disease another enzyme replacement therapy option for this rare disease. The FDA will continue to work with stakeholders to advance the development of additional new, effective and safe therapies for rare diseases, including Pompe disease.”
Nexviazyme, an enzyme replacement therapy, is an intravenous medication that helps reduce glycogen accumulation. The effectiveness of Nexviazyme for the treatment of Pompe disease was demonstrated in a study of 100 patients who were randomized to take Nexviazyme or another FDA-approved enzyme replacement therapy for Pompe disease. Treatment with Nexviazyme improved lung function similar to the improvement seen with the other therapy.
The most common side effects included headache, fatigue, diarrhea, nausea, joint pain (arthralgia), dizziness, muscle pain (myalgia), itching (pruritus), vomiting, difficulty breathing (dyspnea), skin redness (erythema), feeling of “pins and needles” (paresthesia) and skin welts (urticaria). Serious reactions included hypersensitivity reactions like anaphylaxis and infusion-associated reactions, including respiratory distress, chills and raised body temperature (pyrexia). Patients susceptible to fluid volume overload or with compromised cardiac or respiratory function may be at risk for serious acute cardiorespiratory failure.
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Nexviazyme also received an orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Nexviazyme to Genzyme Corporation.
###
NEW DRUG APPROVALS
one time
$10.00
FDA grants priority review for avalglucosidase alfa, a potential new therapy for Pompe disease
- The FDA decision date for avalglucosidase alfa, an investigational enzyme replacement therapy, is set for May 18, 2021
- Regulatory submission based on positive data from two trials in patients with late-onset and infantile-onset Pompe disease, respectively
- Avalglucosidase alfa received FDA Breakthrough Therapy and Fast Track designations for the treatment of people with Pompe Disease
- Pompe disease, a rare degenerative muscle disorder, affects approximately 3,500 people in the U.S.
- Milestone reinforces 20+year commitment to Pompe disease community
PARIS – November 18, 2020 – The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application (BLA) for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease (acid α-glucosidase deficiency). The target action date for the FDA decision is May 18, 2021.
Avalglucosidase alfa is an investigational enzyme replacement therapy designed to improve the delivery of acid alpha-glucosidase (GAA) enzyme to muscle cells, and if approved, would offer a potential new standard of care for patients with Pompe disease.
In October, the European Medicines Agency accepted for review the Marketing Authorization Application for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease. The Medicines and Healthcare Products Regulatory Agency in the UK has granted Promising Innovative Medicine designation for avalglucosidase alfa.
“The hallmarks of Pompe disease are the relentless and debilitating deterioration of the muscles, which causes decreased respiratory function and mobility,” said Karin Knobe, Head of Development for Rare Diseases and Rare Blood Disorders at Sanofi. “Avalglucosidase alfa is specifically designed to deliver more GAA enzyme into the lysosomes of the muscle cells. We have been greatly encouraged by positive clinical trial results in patients with late-onset and infantile-onset Pompe disease.”
Pompe disease is a rare, degenerative muscle disorder that can impact an individual’s ability to move and breathe. It affects an estimated 3,500 people in the U.S. and can manifest at any age from infancy to late adulthood.i
The BLA is based on positive data from two trials:
- Pivotal Phase 3, double-blind, global comparator-controlled trial (COMET), which evaluated the safety and efficacy of avalglucosidase alfa compared to alglucosidase alfa (standard of care) in patients with late-onset Pompe disease. Results from this trial were presented during a Sanofi-hosted virtual scientific session in June 2020 and in October 2020 at World Muscle Society and the American Association of Neuromuscular and Electrodiagnostic Medicine.
- The Phase 2 (mini-COMET) trial evaluated the safety and exploratory efficacy of avalglucosidase alfa in patients with infantile-onset Pompe disease previously treated with alglucosidase alfa. Results from this trial were presented at the WORLDSymposium, in February 2020.
Delivery of GAA to Clear Glycogen
Pompe disease is caused by a genetic deficiency or dysfunction of the lysosomal enzyme GAA, which results in build-up of complex sugars (glycogen) in muscle cells throughout the body. The accumulation of glycogen leads to irreversible damage to the muscles, including respiratory muscles and the diaphragm muscle supporting lung function, and other skeletal muscles that affect mobility.
To reduce the glycogen accumulation caused by Pompe disease, the GAA enzyme must be delivered into the lysosomes within muscle cells. Research led by Sanofi has focused on ways to enhance the delivery of GAA into the lysosomes of muscle cells by targeting the mannose-6-phosphate (M6P) receptor that plays a key role in the transport of GAA.
Avalglucosidase alfa is designed with approximately 15-fold increase in M6P content, compared to standard of care alglucosidase alfa, and aims to help improve cellular enzyme uptake and enhance glycogen clearance in target tissues.ii The clinical relevance of this difference has not been confirmed.
Avalglucosidase alfa is currently under clinical investigation and its safety and efficacy have not been evaluated by any regulatory authority worldwide.
| About Sanofi Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe. Sanofi, Empowering Life |
/////////Avalglucosidase alfa, FDA 2021, Nexviazyme, APPROVALS 2021, PEPTIDE, Enzyme replacement therapy , Pompe disease, アバルグルコシダーゼアルファ (遺伝子組換え), Fast Track, Priority Review, Breakthrough Therapy, orphan drug designation, genzyme, sanofi
BELUMOSUDIL

BELUMOSUDIL
MW 452.5
911417-87-3, SLx-2119, KD-025, KD 025, WHO 11343
2-[3-[4-(1H-indazol-5-ylamino)quinazolin-2-yl]phenoxy]-N-propan-2-ylacetamide
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
Belumosudil mesylate
KD025 mesylate
2109704-99-4
UPDATE FDA APPROVED 7/16/2021 To treat chronic graft-versus-host disease after failure of at least two prior lines of systemic therapy, Rezurock
New Drug Application (NDA): 214783
Company: KADMON PHARMA LLC
200 MG TABLET
FDA approves belumosudil for chronic graft-versus-host disease
On July 16, 2021, the Food and Drug Administration approved belumosudil (Rezurock, Kadmon Pharmaceuticals, LLC), a kinase inhibitor, for adult and pediatric patients 12 years and older with chronic graft-versus-host disease (chronic GVHD) after failure of at least two prior lines of systemic therapy.
Efficacy was evaluated in KD025-213 (NCT03640481), a randomized, open-label, multicenter dose-ranging trial that included 65 patients with chronic GVHD who were treated with belumosudil 200 mg taken orally once daily.
The main efficacy outcome measure was overall response rate (ORR) through Cycle 7 Day 1 where overall response included complete response (CR) or partial response (PR) according to the 2014 criteria of the NIH Consensus Development Project on Clinical Trials in Chronic Graft-versus-Host Disease. The ORR was 75% (95% CI: 63, 85); 6% of patients achieved a CR, and 69% achieved a PR. The median time to first response was 1.8 months (95% CI: 1.0, 1.9). The median duration of response, calculated from first response to progression, death, or new systemic therapies for chronic GVHD, was 1.9 months (95% CI: 1.2, 2.9). In patients who achieved response, no death or new systemic therapy initiation occurred in 62% (95% CI: 46, 74) of patients for at least 12 months since response.
The most common adverse reactions (≥ 20%), including laboratory abnormalities, were infections, asthenia, nausea, diarrhea, dyspnea, cough, edema, hemorrhage, abdominal pain, musculoskeletal pain, headache, phosphate decreased, gamma glutamyl transferase increased, lymphocytes decreased, and hypertension.
The recommended dosage of belumosudil is 200 mg taken orally once daily with food.
View full prescribing information for Rezurock.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Australia’s Therapeutic Goods Administration, Health Canada, Switzerland’s Swissmedic, and the United Kingdom’s Medicines and Healthcare products Regulatory Agency.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application 6 weeks ahead of the FDA goal date.
This application was granted priority review and breakthrough therapy designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Belumosudil mesylate is an orally available rho kinase 2 (ROCK 2) inhibitor being developed at Kadmon. In 2020, the drug candidate was submitted for a new drug application (NDA) in the U.S., under a real-time oncology review pilot program, for the treatment of chronic graft-versus-host disease (cGVHD). The compound is also in phase II clinical development for the treatment of idiopathic pulmonary fibrosis and diffuse cutaneous systemic sclerosis. Formerly, the company had also been conducting clinical research for the treatment of psoriasis and non-alcoholic steatohepatitis (NASH); however, no further development has been reported for these indications. Originally developed by Nano Terra, the product was licensed to Kadmon on an exclusive global basis in 2011. In 2019, Kadmon entered into a strategic partnership with BioNova Pharmaceuticals and established a joint venture, BK Pharmaceuticals, to exclusively develop and commercialize KD-025 for the treatment of graft-versus-host disease in China. The compound has been granted breakthrough therapy designation in the U.S. for the treatment of cGVHD and orphan drug designations for cGVHD and systemic sclerosis. In the E.U. belumosudil was also granted orphan drug status in the E.U. for the treatment of cGVHD.
Kadmon , under license from NT Life Sciences , is developing belumosudil as mesylate salt, a ROCK-2 inhibitor, for treating IPF, chronic graft-versus-host disease, hepatic impairment and scleroderma. In July 2021, belumosudil was reported to be in pre-registration phase.
Belumosudil (formerly KD025 and SLx-2119) is an experimental drug being explored for the treatment of chronic graft versus host disease (cGvHD), idiopathic pulmonary fibrosis (IPF), and moderate to severe psoriasis. It is an inhibitor of Rho-associated coiled-coil kinase 2 (ROCK2; ROCK-II).[1] Belumosudil binds to and inhibits the serine/threonine kinase activity of ROCK2. This inhibits ROCK2-mediated signaling pathways which play major roles in pro- and anti-inflammatory immune cell responses. A genomic study in human primary cells demonstrated that the drug also has effects on oxidative phosphorylation, WNT signaling, angiogenesis, and KRAS signaling.[2] Originally developed by Surface Logix, Inc,[1] Belumosudil was later acquired by Kadmon Corporation. As of July 2020 the drug was in completed or ongoing Phase II clinical studies for cGvHD, IPF and psoriasis.[3]
cGvHD is a complication that can follow stem cell or hematopoietic stem cell transplantation where the transplanted cells (graft) attack healthy cells (host). This causes inflammation and fibrosis in multiple tissues. Two cytokines controlled by the ROCK2 signaling pathway, IL-17 and IL-21, have a major role in the cGvHD response. In a 2016 report using both mouse models and a limited human clinical trial ROCK2 inhibition with belumosudil targeted both the immunologic and fibrotic components of cGvHD and reversed the symptoms of the disease.[4] In October 2017 KD025 was granted orphan drug status in the United States for treatment of patients with cGvHD.[5]
IPF is a progressive fibrotic disease where the lining of the lungs become thickened and scarred.[6] Increased ROCK activity has been found in the lungs of humans and animals with IPF. Treatment with belumosudil reduced lung fibrosis in a bleomycin mouse model study.[7] Belumosudil may have a therapeutic benefit in IPF by targeting the fibrotic processes mediated by the ROCK signaling pathway.
Psoriasis is an inflammatory skin condition where patients experiences eruptions and remissions of thickened, erythematous, and scaly patches of skin. Down-regulation of pro-inflammatory responses was observed with KD025 treatment in Phase 2 clinical studies in patients with moderate to severe psoriasis.[8]
“Substance Name:Substance Name: Belumosudil [USAN]”.
PATENT
| WO2012040499 |
https://patents.google.com/patent/WO2012040499A2/en
PATENT
| CN106916145 |
https://patents.google.com/patent/CN106916145A/en

WO 2014055996, WO 2015157556



Patent
WO-2021129589
Novel crystalline polymorphic forms (N1, N2 and N15) of KD-025 (also known as belumosudil ), useful as a Rho A kinase 2 (ROCK-2) inhibitor for treating multiple sclerosis, psoriasis, rheumatoid arthritis, idiopathic pulmonary fibrosis (IPF), atherosclerosis, non-alcoholic fatty liver and systemic sclerosis. Represents the first filing from Sunshine Lake Pharma or its parent HEC Pharm that focuses on belumosudil.KD-025 is a selective ROCK2 (Rho-associated protein kinase 2, Rho-related protein kinase 2) inhibitor. It has multiple clinical indications such as the treatment of multiple sclerosis, psoriasis, rheumatoid arthritis, and Primary pulmonary fibrosis, atherosclerosis, non-alcoholic fatty liver, etc., among which many indications are in clinical phase I, and psoriasis and systemic sclerosis are in clinical phase II.
The structure of KD-025 is shown in the following formula (1).
Example 1 Preparation method of crystal form N1 of KD-025[0222]300mg of KD-025 solid was suspended and stirred in 10mL methanol at room temperature. After 22h, it was filtered, suction filtered and placed in a drying oven at 50°C under vacuum overnight to obtain 262mg of powder. The obtained crystal was detected by XPRD and confirmed to be KD-025 crystal form N1; its X-ray powder diffraction pattern was basically the same as that of Fig. 1, its DSC pattern was basically the same as that of Fig. 2, and the TGA pattern was basically the same as that of Fig. 3.
PATENT
WO2006105081 ,
Belumosudil product pat,
protection in the EU states until March 2026, expires in the US in May 2029 with US154 extension.
Example 82
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
[0257] A suspension of 2-(3-(4-(lH-indazol-5-ylamino)qumazolin-2-yl)ρhenoxy)acetic acid (70 mg, 0.14 mmol), PyBOP® (40 mg, 0.077 mmol), DlEA (24 μL, 0.14 mmol) in dry CH2Cl2 : DMF (2 : 0.1 mL) was stirred at RT for 15 minutes. To this solution of activated acid was added propan-2-amine (5.4 mg, 0.091 mmol). After 30 minutes, 1.0 equivalent of DIEA and 0.55 equivalents of PyBOP® were added. After stirring the solution for 15 minutes, 0.65 equivalents of propan-2-aminewere added and the mixture was stirred for an additional 30 minutes. The solvent was removed in vacuo and the crude product was purified using prep HPLC (25-50 90 rnins) to afford 2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide. (40 mg, 0.086 mmol, 61 %).
References
- ^ Jump up to:a b Boerma M, Fu Q, Wang J, Loose DS, Bartolozzi A, Ellis JL, et al. (October 2008). “Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin”. Blood Coagulation & Fibrinolysis. 19 (7): 709–18. doi:10.1097/MBC.0b013e32830b2891. PMC 2713681. PMID 18832915.
- ^ Park J, Chun KH (5 May 2020). “Identification of novel functions of the ROCK2-specific inhibitor KD025 by bioinformatics analysis”. Gene. 737: 144474. doi:10.1016/j.gene.2020.144474. PMID 32057928.
- ^ “KD025 – Clinical Trials”. ClinicalTrials.gov. Retrieved 25 July 2020.
- ^ Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. (April 2016). “Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism”. Blood. 127 (17): 2144–54. doi:10.1182/blood-2015-10-678706. PMC 4850869. PMID 26983850.
- ^ Shanley M (October 6, 2017). “Therapy to Treat Transplant Complications Gets Orphan Drug Designation”. RareDiseaseReport. Retrieved 25 July 2018.
- ^ “Pulmonary Fibrosis”. The Mayo Clinic. Retrieved July 25, 2018.
- ^ Semedo D (June 5, 2016). “Phase 2 Study of Molecule Inhibitor for Idiopathic Pulmonary Fibrosis Begins”. Lung Disease News. BioNews Services, LLC. Retrieved 25 July 2018.
- ^ Zanin-Zhorov A, Weiss JM, Trzeciak A, Chen W, Zhang J, Nyuydzefe MS, et al. (May 2017). “Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10”. Journal of Immunology. 198 (10): 3809–3814. doi:10.4049/jimmunol.1602142. PMC 5421306. PMID 28389592.
| Clinical data | |
|---|---|
| Routes of administration |
Oral administration (tablets or capsules) |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 911417-87-3 |
| PubChem CID | 11950170 |
| UNII | 834YJF89WO |
| CompTox Dashboard (EPA) | DTXSID80238425 |
| Chemical and physical data | |
| Formula | C26H24N6O2 |
| Molar mass | 452.518 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////BELUMOSUDIL, SLx-2119, KD-025, KD 025, WHO 11343, PHASE 2, cGvHD, IPF, psoriasis, Breakthrough Therapy, Orphan Drug Designation
CC(C)NC(=O)COC1=CC=CC(=C1)C2=NC3=CC=CC=C3C(=N2)NC4=CC5=C(C=C4)NN=C5
NEW DRUG APPROVALS
ONE TIME
$10.00
Pegcetacoplan
Sequence:
1ICVWQDWGAH RCTXK
Sequence:
1ICVWQDWGAH RCTXK
Sequence Modifications
| Type | Location | Description |
|---|---|---|
| terminal mod. | Lys-15 | C-terminal amide |
| terminal mod. | Lys-15′ | C-terminal amide |
| bridge | Cys-2 – Cys-12 | disulfide bridge, dimer |
| bridge | Lys-15 – Lys-15′ | covalent bridge, dimer |
| bridge | Cys-2′ – Cys-12′ | disulfide bridge, dimer |
| uncommon | Oaa-14 | – |
| uncommon | Oaa-14′ | – |
Pegcetacoplan
ペグセタコプラン;
FDA APPROVED Empaveli, 2021/5/14
Protein Sequence
Sequence Length: 30, 15, 15multichain; modifiedPoly(oxy-1,2-ethanediyl), α-hydro-ω-hydroxy-, 15,15′-diester with N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-α-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-[2-(2-aminoethoxy)ethoxy]acetyl-N6-carboxy-L-lysinamide cyclic (2→12)-(disulfide)Polymer
Poly(oxy-1,2-ethanediyl), alpha-hydro-omega-hydroxy-, 15,15′-diester with N-acetyl-Lisoleucyl-L-cysteinyl-L-valyl-1-methyl-L-tryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-N6-carboxy-L-lysinamide cyclic (2�->12)-(disulfide)
O,O’-bis((S2,S12-cyclo(N-acetyl-L-isoleucyl-L-cysteinyl-L-valyl-1-methyl-Ltryptophyl-L-glutaminyl-L-alpha-aspartyl-L-tryptophylglycyl-L-alanyl-L-histidyl-L-arginyl-L-cysteinyl-L-threonyl-2-(2-(2-aminoethoxy)ethoxy)acetyl-L-lysinamide))-N6.15-carbonyl)polyethylene glycol(n = 800-1100)
- APL-2
- WHO 10743
| Formula | C170H248N50O47S4. (C2H4O)n3872.40 g·mol−1 |
|---|---|
| EfficacyDisease | Complement inhibitorParoxysmal nocturnal hemoglobinuria |
| CAS | 2019171-69-6 |
| Comment | Treatment of paroxysmal nocturnal hemoglobinuria (PNH), complement-mediated nephropathies, and age-related macular degeneration (AMD) |
- OriginatorApellis Pharmaceuticals
- ClassAnti-inflammatories; Anti-ischaemics; Antianaemics; Cyclic peptides; Eye disorder therapies; Polyethylene glycols; Urologics
- Mechanism of ActionComplement C3 inhibitors
- Orphan Drug StatusYes – Paroxysmal nocturnal haemoglobinuria; Autoimmune haemolytic anaemia; Glomerulonephritis
- RegisteredParoxysmal nocturnal haemoglobinuria
- Phase IIIAge-related macular degeneration
- Phase IIAmyotrophic lateral sclerosis; Autoimmune haemolytic anaemia; Glomerulonephritis; IgA nephropathy; Lupus nephritis; Membranous glomerulonephritis
- Phase I/IIWet age-related macular degeneration
- DiscontinuedIschaemia
- 02 Jun 2021Apellis Pharmaceuticals plans a phase III trial for Glomerulonephritis in the second half of 2021
- 25 May 2021Top-line efficacy and safety results from the phase III PRINCE trial for Paroxysmal nocturnal haemoglobinuria released by Apellis Pharmaceuticals
- 18 May 2021Registered for Paroxysmal nocturnal haemoglobinuria in USA (SC) – First global approval
Pegcetacoplan, sold under the brand name Empaveli, is a medication used to treat paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
The most common side effects include injection-site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.[2]
Paroxysmal nocturnal hemoglobinuria is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells).[1]
Pegcetacoplan is the first treatment for paroxysmal nocturnal hemoglobinuria that binds to complement protein C3.[1] Pegcetacoplan was approved for medical use in the United States in May 2021.[1][3]
Pegcetacoplan is a complement inhibitor indicated in the treatment of paroxysmal nocturnal hemoglobinuria (PNH).5,7 Prior to its FDA approval, patients with PNH were typically treated with the C5 inhibiting monoclonal antibody eculizumab.5 Patients given eculizumab experienced less hemolysis caused by the membrane attack complex, but were still somewhat susceptible to hemolysis caused by C3b opsonization.5,6 Pegcetacoplan was developed out of a need for an inhibitor of complement mediated hemolysis further upstream of C5.5,6 Pegcetacoplan is a pegylated C3 inhibitor that can disrupt the processes leading to both forms of hemolysis that threaten patients with PNH.5
Pegcetacoplan was granted FDA approval on 14 May 2021.7
Medical uses
Pegcetacoplan is indicated to treat adults with paroxysmal nocturnal hemoglobinuria (PNH).[1][2]
EMPAVELI contains pegcetacoplan, a complement inhibitor. Pegcetacoplan is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to the ends of a linear 40-kiloDalton (kDa) PEG molecule. The peptide portions of pegcetacoplan contain 1-methyl-L-tryptophan (Trp(Me)) in position 4 and amino(ethoxyethoxy)acetic acid (AEEA) in position 14.
The molecular weight of pegcetacoplan is approximately 43.5 kDa. The molecular formula is C1970H3848N50O947S4. The structure of pegcetacoplan is shown below.
 |
EMPAVELI injection is a sterile, clear, colorless to slightly yellowish aqueous solution for subcutaneous use and is supplied in a 20-mL single-dose vial. Each 1 mL of solution contains 54 mg of pegcetacoplan, 41 mg of sorbitol, 0.384 mg of glacial acetic acid, 0.490 mg of sodium acetate trihydrate, and Water for Injection USP. EMPAVELI may also contain sodium hydroxide and/or additional glacial acetic acid for adjustment to a target pH of 5.0.
FDA approves new treatment for adults with serious rare blood disease..
FDA has approved Empaveli (pegcetacoplan) injection to treat adults with paroxysmal nocturnal hemoglobinuria (PNH), a rare, life-threatening blood disease. Empaveli is the first PNH treatment that binds to compliment protein C3.
PNH is characterized by red blood cell destruction, anemia (red blood cells unable to carry enough oxygen to tissues), blood clots, and impaired bone marrow function (not making enough blood cells). The disease affects 1-1.5 people per million. Individuals are typically diagnosed around ages 35 to 40. PNH can be serious, with median survival of 10 years after diagnosis. However, some patients live for decades with only minor symptoms.
PNH is caused by gene mutations that affect red blood cells. Red blood cells in people with these mutations are defective and can be destroyed by the immune system, which causes anemia.
The effectiveness of Empaveli was evaluated in a study enrolling 80 patients with PNH and anemia who had been taking eculizumab, a treatment previously approved for PNH. Patients first completed a four-week period during which they received Empaveli 1,080 mg twice weekly in addition to eculizumab at their previous dose. After the first four weeks, patients were randomly assigned to receive either Empaveli or their current dose of eculizumab for 16 weeks.
After 16 weeks, the severity of anemia was compared in the two treatment groups on the basis of hemoglobin concentration (a laboratory measure of anemia). In both treatment groups, the average hemoglobin was 8.7 g/dL at baseline, indicating severe anemia. (Normal hemoglobin values in adult men are 14 g/dL or above; normal values in adult women are 12 g/dL or above.) During the 16 weeks of treatment, patients in the Empaveli group had an average increase in their hemoglobin of 2.4 g/dL. Meanwhile, patients in the eculizumab group had an average decrease in their hemoglobin of 1.5 g/dL.
Empaveli is available only through a restricted program under a risk evaluation and mitigation strategy. Meningococcal (a type of bacteria) infections can occur in patients taking Empaveli and can become life-threatening or fatal if not treated early. Empaveli may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria. Patients should be monitored for infusion-related reactions. Empaveli can interfere with certain laboratory tests. The most common side effects are injection site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.
Empaveli received priority review, fast track and orphan drug designations for this indication.
FDA granted the approval of Empaveli to Apellis Pharmaceuticals.
Adverse effects
Meningococcal (a type of bacteria) infections can occur in people taking pegcetacoplan and can become life-threatening or fatal if not treated early.[1] Pegcetacoplan may also predispose individuals to serious infections, especially infections caused by encapsulated bacteria.[1]
History
The effectiveness of pegcetacoplan was evaluated in a study enrolling 80 participants with paroxysmal nocturnal hemoglobinuria and anemia who had been taking eculizumab, a treatment previously approved for paroxysmal nocturnal hemoglobinuria.[1]
References
- ^ Jump up to:a b c d e f g h i “FDA approves new treatment for adults with serious rare blood disease”. U.S. Food and Drug Administration (FDA). 14 May 2021. Retrieved 14 May 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d https://pi.apellis.com/files/PI_Empaveli.pdf
- ^ “Apellis Announces U.S. Food and Drug Administration (FDA) Approval of Empaveli (pegcetacoplan) for Adults with Paroxysmal Nocturnal Hemoglobinuria (PNH)” (Press release). Apellis Pharmaceuticals. 14 May 2021. Retrieved 14 May 2021 – via GlobeNewswire.
External links
- “Pegcetacoplan”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03500549 for “Study to Evaluate the Efficacy and Safety of APL-2 in Patients With Paroxysmal Nocturnal Hemoglobinuria (PNH)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Empaveli |
| Other names | APL-2 |
| License data | US DailyMed: Pegcetacoplan |
| Routes of administration | Subcutaneous infusion |
| Drug class | Complement inhibitor |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 2019171-69-6 |
| UNII | TO3JYR3BOU |
| KEGG | D11613 |
| ChEMBL | ChEMBL4298211 |
| Chemical and physical data | |
| Formula | C170H248N50O47S4 |
| Molar mass | 3872.40 g·mol−1 |
/////////Pegcetacoplan, ペグセタコプラン , FDA 2021, APPROVALS 2021, APL-2, WHO 10743, Apellis Pharmaceuticals, Empaveli, priority review, fast track, orphan drug
https://www.sec.gov/Archives/edgar/data/1492422/000156459020007350/apls-10k_20191231.htm
NEW DRUG APPROVALS
ONE TIME
$10.00
Sotorasib

![4-((S)-4-Acryloyl-2-methylpiperazin-1-yl)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=137278711&t=l)
Sotorasib
6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-propan-2-ylpyridin-3-yl)-4-[(2S)-2-methyl-4-prop-2-enoylpiperazin-1-yl]pyrido[2,3-d]pyrimidin-2-one
AMG 510
AMG-510
AMG510
| Formula | C30H30F2N6O3 |
|---|---|
| CAS | 2296729-00-3 |
| Mol weight | 560.5944 |
FDA APPROVED, 2021/5/28 Lumakras
Antineoplastic, Non-small cell lung cancer (KRAS G12C-mutated)
ソトラシブ (JAN);
Sotorasib
(1M)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one
C30H30F2N6O3 : 560.59
[2296729-00-3]
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
 |
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras™, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
View full prescribing information for Lumakras.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
This application was granted priority review, fast-track, breakthrough therapy and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Sotorasib, sold under the brand name Lumakras is an anti-cancer medication used to treat non-small-cell lung cancer (NSCLC).[1][2] It targets a specific mutation, G12C, in the protein KRAS which is responsible for various forms of cancer.[3][4]
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
The U.S. Food and Drug Administration (FDA) granted the application for sotorasib orphan drug, fast track, priority review, and breakthrough therapy designations.[2] The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA).[2] The application reviews are ongoing at the other regulatory agencies.[2]
The FDA granted approval of Lumakras to Amgen Inc.[2]
Society and culture
Economics
Sotorasib costs US$17,900 per month.[5]
Names
Sotorasib is the recommended international nonproprietary name (INN).[14]
PAPER
Nature (London, United Kingdom) (2019), 575(7781), 217-223
https://www.nature.com/articles/s41586-019-1694-1
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310540

KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180

KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
AMG 510 [(R)-38]. (1R)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one
………… concentrated in vacuo. Chromatographic purification of the residue (silica gel; 0–100% 3:1 EtOAc–EtOH/heptane) followed by chiral supercritical fluid chromatography (Chiralpak IC, 30 mm × 250 mm, 5 μm, 55% MeOH/CO2, 120 mL/min, 102 bar) provided (1R)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510; (R)-38; 2.25 g, 43% yield) as the first-eluting peak. 1H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ −115.6 (d, J = 5.2 Hz, 1 F), −128.6 (br s, 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M + H]+ calcd for C30H30F2N6O3 561.24202. Found 561.24150.

d (1R)-6-Fluoro7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1- piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one ((R)-38; AMG 510; 2.25 g, 43% yield) as the first-eluting peak.1 H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ –115.6 (d, J = 5.2 Hz, 1 F), –128.6 (br. s., 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M+H]+ Calcd for C30H30F2N6O3 561.24202; Found 561.24150. Atropisomer configuration (R vs. S) assigned crystallographically.The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180.
PATENT
WO 2021097212
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
PATENT
WO 2020102730
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020102730
PATENT
US 20180334454
References
- ^ Jump up to:a b c d e “Lumakras- sotorasib tablet, coated”. DailyMed. Retrieved 6 June 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves First Targeted Therapy for Lung Cancer Mutation Previously Considered Resistant to Drug Therapy”. U.S. Food and Drug Administration (FDA). 28 May 2021. Retrieved 28 May 2021. This article incorporates text from this source, which is in the public domain.
- ^ “KRAS mutant-targeting AMG 510”. NCI Drug Dictionary. National Cancer Institute. 2 February 2011. Retrieved 16 November2019.
- ^ Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (November 2019). “The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity”. Nature. 575 (7781): 217–23. Bibcode:2019Natur.575..217C. doi:10.1038/s41586-019-1694-1. PMID 31666701.
- ^ Jump up to:a b “FDA approves Amgen drug for lung cancer with specific mutation”. CNBC. 28 May 2021. Retrieved 28 May 2021.
- ^ Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (2020). “KRASG12C inhibition with sotorasib in advanced solid tumors”. N Engl J Med. doi:10.1056/NEJMoa1917239. PMC 7571518.
- ^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
- ^ “The Discovery Of Amgen’s Novel Investigational KRAS(G12C) Inhibitor AMG 510 Published In Nature” (Press release). Amgen. 30 October 2019. Retrieved 16 November 2019.
- ^ Irving M (24 December 2019). “Drug targeting common cancer cause enters phase 2 clinical trials”. New Atlas. Retrieved 24 December 2019.
- ^ Jump up to:a b c d Halford B (3 April 2019). “Amgen unveils its KRas inhibitor in human clinical trials: AMG 510 shuts down a mutant version of the cancer target via covalent interaction”. Chemical & Engineering News. 97 (4). Retrieved 16 November 2019.
- ^ Al Idrus A (9 September 2019). “Amgen’s KRAS drug continues to deliver but faces ‘curse’ of high expectations”. fiercebiotech.com. Retrieved 16 November 2019.
- ^ Kaiser J (30 October 2019). “Two new drugs finally hit ‘undruggable’ cancer target, providing hope for treatments”. Science Magazine. AAAS. Retrieved 16 November 2019.
- ^ Astor L (9 September 2019). “FDA Grants AMG 510 Fast Track Designation for KRAS G12C+ NSCLC”. targetedonc.com. Retrieved 16 November 2019.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85” (PDF). WHO Drug Information. 35 (1).
Further reading
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (September 2020). “KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors”. N Engl J Med. 383 (13): 1207–17. doi:10.1056/NEJMoa1917239. PMC 7571518. PMID 32955176.
- Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. (January 2020). “Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors”. J Med Chem. 63 (1): 52–65. doi:10.1021/acs.jmedchem.9b01180. PMID 31820981.
External links
- “Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lumakras |
| Other names | AMG 510 |
| License data | US DailyMed: Sotorasib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2252403-56-6 |
| PubChem CID | 137278711 |
| DrugBank | DB15569 |
| ChemSpider | 72380148 |
| UNII | 2B2VM6UC8G |
| KEGG | D12055 |
| Chemical and physical data | |
| Formula | C30H30F2N6O3 |
| Molar mass | 560.606 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////Sotorasib, ソトラシブ , FDA 2021, APPROVALS 2021, Lumakras, CANCER, ANTINEOPLASTIC, AMG 510, AMG-510, AMG510, AMGEN, priority review, fast-track, breakthrough therapy, orphan drug
CC1CN(CCN1C2=NC(=O)N(C3=NC(=C(C=C32)F)C4=C(C=CC=C4F)O)C5=C(C=CN=C5C(C)C)C)C(=O)C=C
Sotorasib
6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-propan-2-ylpyridin-3-yl)-4-[(2S)-2-methyl-4-prop-2-enoylpiperazin-1-yl]pyrido[2,3-d]pyrimidin-2-one
AMG 510
AMG-510
AMG510
| Formula | C30H30F2N6O3 |
|---|---|
| CAS | 2296729-00-3 |
| Mol weight | 560.5944 |
FDA APPROVED, 2021/5/28 Lumakras
Antineoplastic, Non-small cell lung cancer (KRAS G12C-mutated)
ソトラシブ (JAN);
Sotorasib
(1M)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one
C30H30F2N6O3 : 560.59
[2296729-00-3]
Sotorasib is an inhibitor of the RAS GTPase family. The molecular formula is C30H30F2N6O3, and the molecular weight is 560.6 g/mol. The chemical name of sotorasib is 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2enoyl) piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one. The chemical structure of sotorasib is shown below:
|
Sotorasib has pKa values of 8.06 and 4.56. The solubility of sotorasib in the aqueous media decreases over the range pH 1.2 to 6.8 from 1.3 mg/mL to 0.03 mg/mL.
LUMAKRAS is supplied as film-coated tablets for oral use containing 120 mg of sotorasib. Inactive ingredients in the tablet core are microcrystalline cellulose, lactose monohydrate, croscarmellose sodium, and magnesium stearate. The film coating material consists of polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, and iron oxide yellow.
FDA grants accelerated approval to sotorasib for KRAS G12C mutated NSCLC
On May 28, 2021, the Food and Drug Administration granted accelerated approval to sotorasib (Lumakras™, Amgen, Inc.), a RAS GTPase family inhibitor, for adult patients with KRAS G12C ‑mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA ‑approved test, who have received at least one prior systemic therapy.
FDA also approved the QIAGEN therascreen® KRAS RGQ PCR kit (tissue) and the Guardant360® CDx (plasma) as companion diagnostics for Lumakras. If no mutation is detected in a plasma specimen, the tumor tissue should be tested.
Approval was based on CodeBreaK 100, a multicenter, single-arm, open label clinical trial (NCT03600883) which included patients with locally advanced or metastatic NSCLC with KRAS G12C mutations. Efficacy was evaluated in 124 patients whose disease had progressed on or after at least one prior systemic therapy. Patients received sotorasib 960 mg orally daily until disease progression or unacceptable toxicity.
The main efficacy outcome measures were objective response rate (ORR) according to RECIST 1.1, as evaluated by blinded independent central review and response duration. The ORR was 36% (95% CI: 28%, 45%) with a median response duration of 10 months (range 1.3+, 11.1).
The most common adverse reactions (≥ 20%) were diarrhea, musculoskeletal pain, nausea, fatigue, hepatotoxicity, and cough. The most common laboratory abnormalities (≥ 25%) were decreased lymphocytes, decreased hemoglobin, increased aspartate aminotransferase, increased alanine aminotransferase, decreased calcium, increased alkaline phosphatase, increased urine protein, and decreased sodium.
The recommended sotorasib dose is 960 mg orally once daily with or without food.
The approved 960 mg dose is based on available clinical data, as well as pharmacokinetic and pharmacodynamic modeling that support the approved dose. As part of the evaluation for this accelerated approval, FDA is requiring a postmarketing trial to investigate whether a lower dose will have a similar clinical effect.
View full prescribing information for Lumakras.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada, and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, the Assessment Aid, and the Product Quality Assessment Aid (PQAA), voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 10 weeks ahead of the FDA goal date.
This application was granted priority review, fast-track, breakthrough therapy and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Sotorasib, sold under the brand name Lumakras is an anti-cancer medication used to treat non-small-cell lung cancer (NSCLC).[1][2] It targets a specific mutation, G12C, in the protein KRAS which is responsible for various forms of cancer.[3][4]
The most common side effects include diarrhea, musculoskeletal pain, nausea, fatigue, liver damage and cough.[1][2]
Sotorasib is an inhibitor of the RAS GTPase family.[1]
Sotorasib is the first approved targeted therapy for tumors with any KRAS mutation, which accounts for approximately 25% of mutations in non-small cell lung cancers.[2] KRAS G12C mutations represent about 13% of mutations in non-small cell lung cancers.[2] Sotorasib was approved for medical use in the United States in May 2021.[2][5]
Sotorasib is an experimental KRAS inhibitor being investigated for the treatment of KRAS G12C mutant non small cell lung cancer, colorectal cancer, and appendix cancer.
Sotorasib, also known as AMG-510, is an acrylamide derived KRAS inhibitor developed by Amgen.1,3 It is indicated in the treatment of adult patients with KRAS G12C mutant non small cell lung cancer.6 This mutation makes up >50% of all KRAS mutations.2 Mutant KRAS discovered in 1982 but was not considered a druggable target until the mid-2010s.5 It is the first experimental KRAS inhibitor.1
The drug MRTX849 is also currently being developed and has the same target.1
Sotorasib was granted FDA approval on 28 May 2021.6
Medical uses
Sotorasib is indicated for the treatment of adults with KRAS G12C-mutated locally advanced or metastatic non-small cell lung cancer (NSCLC), as determined by an FDA-approved test, who have received at least one prior systemic therapy.[1][2]
Clinical development
Sotorasib is being developed by Amgen. Phase I clinical trials were completed in 2020.[6][7][8] In December 2019, it was approved to begin Phase II clinical trials.[9]
Because the G12C KRAS mutation is relatively common in some cancer types, 14% of non-small-cell lung cancer adenocarcinoma patients and 5% of colorectal cancer patients,[10] and sotorasib is the first drug candidate to target this mutation, there have been high expectations for the drug.[10][11][12] The Food and Drug Administration has granted a fast track designation to sotorasib for the treatment of metastatic non-small-cell lung carcinoma with the G12C KRAS mutation.[13]
Chemistry and pharmacology
Sotorasib can exist in either of two atropisomeric forms and one is more active than the other.[10] It selectively forms an irreversible covalent bond to the sulfur atom in the cysteine residue that is present in the mutated form of KRAS, but not in the normal form.[10]
History
Researchers evaluated the efficacy of sotorasib in a study of 124 participants with locally advanced or metastatic KRAS G12C-mutated non-small cell lung cancer with disease progression after receiving an immune checkpoint inhibitor and/or platinum-based chemotherapy.[2] The major outcomes measured were objective response rate (proportion of participants whose tumor is destroyed or reduced) and duration of response.[2] The objective response rate was 36% and 58% of those participants had a duration of response of six months or longer.[2]
The U.S. Food and Drug Administration (FDA) granted the application for sotorasib orphan drug, fast track, priority review, and breakthrough therapy designations.[2] The FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), Health Canada and the United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA).[2] The application reviews are ongoing at the other regulatory agencies.[2]
The FDA granted approval of Lumakras to Amgen Inc.[2]
Society and culture
Economics
Sotorasib costs US$17,900 per month.[5]
Names
Sotorasib is the recommended international nonproprietary name (INN).[14]
PAPER
Nature (London, United Kingdom) (2019), 575(7781), 217-223
https://www.nature.com/articles/s41586-019-1694-1
KRAS is the most frequently mutated oncogene in cancer and encodes a key signalling protein in tumours1,2. The KRAS(G12C) mutant has a cysteine residue that has been exploited to design covalent inhibitors that have promising preclinical activity3,4,5. Here we optimized a series of inhibitors, using novel binding interactions to markedly enhance their potency and selectivity. Our efforts have led to the discovery of AMG 510, which is, to our knowledge, the first KRAS(G12C) inhibitor in clinical development. In preclinical analyses, treatment with AMG 510 led to the regression of KRASG12C tumours and improved the anti-tumour efficacy of chemotherapy and targeted agents. In immune-competent mice, treatment with AMG 510 resulted in a pro-inflammatory tumour microenvironment and produced durable cures alone as well as in combination with immune-checkpoint inhibitors. Cured mice rejected the growth of isogenic KRASG12D tumours, which suggests adaptive immunity against shared antigens. Furthermore, in clinical trials, AMG 510 demonstrated anti-tumour activity in the first dosing cohorts and represents a potentially transformative therapy for patients for whom effective treatments are lacking.
Paper
Scientific Reports (2020), 10(1), 11992
PAPER
European journal of medicinal chemistry (2021), 213, 113082.
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310540
KRAS is the most commonly altered oncogene of the RAS family, especially the G12C mutant (KRASG12C), which has been a promising drug target for many cancers. On the basis of the bicyclic pyridopyrimidinone framework of the first-in-class clinical KRASG12C inhibitor AMG510, a scaffold hopping strategy was conducted including a F–OH cyclization approach and a pyridinyl N-atom working approach leading to new tetracyclic and bicyclic analogues. Compound 26a was identified possessing binding potency of 1.87 μM against KRASG12C and cell growth inhibition of 0.79 μM in MIA PaCa-2 pancreatic cancer cells. Treatment of 26a with NCI–H358 cells resulted in down-regulation of KRAS-GTP levels and reduction of phosphorylation of downstream ERK and AKT dose-dependently. Molecular docking suggested that the fluorophenol moiety of 26a occupies a hydrophobic pocket region thus forming hydrogen bonding to Arg68. These results will be useful to guide further structural modification.
PAPER
Journal of Medicinal Chemistry (2020), 63(1), 52-65.
https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180
KRASG12C has emerged as a promising target in the treatment of solid tumors. Covalent inhibitors targeting the mutant cysteine-12 residue have been shown to disrupt signaling by this long-“undruggable” target; however clinically viable inhibitors have yet to be identified. Here, we report efforts to exploit a cryptic pocket (H95/Y96/Q99) we identified in KRASG12C to identify inhibitors suitable for clinical development. Structure-based design efforts leading to the identification of a novel quinazolinone scaffold are described, along with optimization efforts that overcame a configurational stability issue arising from restricted rotation about an axially chiral biaryl bond. Biopharmaceutical optimization of the resulting leads culminated in the identification of AMG 510, a highly potent, selective, and well-tolerated KRASG12C inhibitor currently in phase I clinical trials (NCT03600883).
AMG 510 [(R)-38]. (1R)-6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one
………… concentrated in vacuo. Chromatographic purification of the residue (silica gel; 0–100% 3:1 EtOAc–EtOH/heptane) followed by chiral supercritical fluid chromatography (Chiralpak IC, 30 mm × 250 mm, 5 μm, 55% MeOH/CO2, 120 mL/min, 102 bar) provided (1R)-6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1-piperazinyl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510; (R)-38; 2.25 g, 43% yield) as the first-eluting peak. 1H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ −115.6 (d, J = 5.2 Hz, 1 F), −128.6 (br s, 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M + H]+ calcd for C30H30F2N6O3 561.24202. Found 561.24150.
d (1R)-6-Fluoro7-(2-fluoro-6-hydroxyphenyl)-1-[4-methyl-2-(1-methylethyl)-3-pyridinyl]-4-[(2S)-2-methyl-4-(1-oxo-2-propen-1-yl)-1- piperazinyl]-pyrido[2,3-d]pyrimidin-2(1H)-one ((R)-38; AMG 510; 2.25 g, 43% yield) as the first-eluting peak.1 H NMR (600 MHz, DMSO-d6) δ ppm 10.20 (s, 1H), 8.39 (d, J = 4.9 Hz, 1H), 8.30 (d, J = 8.9 Hz, 0.5H), 8.27 (d, J = 8.7 Hz, 0.5H), 7.27 (q, J = 8.4 Hz, 1H), 7.18 (d, J = 4.9 Hz, 1H), 6.87 (dd, J = 16.2, 10.8 Hz, 0.5H), 6.84 (dd, J = 16.2, 10.7 Hz, 0.5H), 6.74 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 8.4 Hz, 1H), 6.21 (d, J = 16.2 Hz, 0.5H), 6.20 (d, J = 16.2 Hz, 0.5H), 5.76 (d, J = 10.8 Hz, 0.5H), 5.76 (d, J = 10.7 Hz, 0.5H), 4.91 (m, 1H), 4.41 (d, J = 12.2 Hz, 0.5H), 4.33 (d, J = 12.2 Hz, 1H), 4.28 (d, J = 12.2 Hz, 0.5H), 4.14 (d, J = 12.2 Hz, 0.5H), 4.02 (d, J = 13.6 Hz, 0.5H), 3.69 (m, 1H), 3.65 (d, J = 13.6 Hz, 0.5H), 3.52 (t, J = 12.2 Hz, 0.5H), 3.27 (d, J = 12.2 Hz, 0.5H), 3.15 (t, J = 12.2 Hz, 0.5H), 2.72 (m, 1H), 1.90 (s, 3H), 1.35 (d, J = 6.7 Hz, 3H), 1.08 (d, J = 6.7 Hz, 3H), 0.94 (d, J = 6.7 Hz, 3H).
19F NMR (376 MHz, DMSO-d6) δ –115.6 (d, J = 5.2 Hz, 1 F), –128.6 (br. s., 1 F).
13C NMR (151 MHz, DMSO-d6) δ ppm 165.0 (1C), 163.4 (1C), 162.5 (1C), 160.1 (1C), 156.8 (1C), 153.7 (1C), 151.9 (1C), 149.5 (1C), 148.3 (1C), 145.2 (1C), 144.3 (1C), 131.6 (1C), 130.8 (1C), 127.9 (0.5C), 127.9 (0.5C), 127.8 (0.5C), 127.7 (0.5C), 123.2 (1C), 122.8 (1C), 111.7 (1C), 109.7 (1C), 105.7 (1C), 105.3 (1C), 51.4 (0.5C), 51.0 (0.5C), 48.9 (0.5C), 45.4 (0.5C), 44.6 (0.5C), 43.7 (0.5C), 43.5 (0.5C), 41.6 (0.5C), 29.8 (1C), 21.9 (1C), 21.7 (1C), 17.0 (1C), 15.5 (0.5C), 14.8 (0.5C).
FTMS (ESI) m/z: [M+H]+ Calcd for C30H30F2N6O3 561.24202; Found 561.24150. Atropisomer configuration (R vs. S) assigned crystallographically.The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b01180.
PATENT
WO 2021097212
The present disclosure relates to an improved, efficient, scalable process to prepare intermediate compounds, such as compound of Formula 6A, having the structure,
useful for the synthesis of compounds for the treatment of KRAS G12C mutated cancers.
BACKGROUND
[0003] KRAS gene mutations are common in pancreatic cancer, lung adenocarcinoma, colorectal cancer, gall bladder cancer, thyroid cancer, and bile duct cancer. KRAS mutations are also observed in about 25% of patients with NSCLC, and some studies have indicated that KRAS mutations are a negative prognostic factor in patients with NSCLC. Recently, V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations have been found to confer resistance to epidermal growth factor receptor (EGFR) targeted therapies in colorectal cancer; accordingly, the mutational status of KRAS can provide important information prior to the prescription of TKI therapy. Taken together, there is a need for new medical treatments for patients with pancreatic cancer, lung adenocarcinoma, or colorectal cancer, especially those who have been diagnosed to have such cancers characterized by a KRAS mutation, and including those who have progressed after chemotherapy.
Related Synthetic Processes
[0126] The following intermediate compounds of 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one are representative examples of the disclosure and are not intended to be construed as limiting the scope of the present invention.
[0127] A synthesis of Compound 9 and the relevant intermediates is described in U.S. Serial No.15/984,855, filed May 21, 2018 (U.S. Publication No.2018/0334454, November 22, 2018) which claims priority to and the benefit claims the benefit of U.S. Provisional Application No.62/509,629, filed on May 22, 2017, both of which are incorporated herein by reference in their entireties for all purposes. 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one was prepared using the following process, in which the isomers of the final product were isolated via chiral chromatography.
[0128] Step 1: 2,6-Dichloro-5-fluoronicotinamide (Intermediate S). To a mixture of 2,6-dichloro-5-fluoro-nicotinic acid (4.0 g, 19.1 mmol, AstaTech Inc., Bristol, PA) in dichloromethane (48 mL) was added oxalyl chloride (2M solution in DCM, 11.9 mL, 23.8 mmol), followed by a catalytic amount of DMF (0.05 mL). The reaction was stirred at room temperature overnight and then was concentrated. The residue was dissolved in 1,4-dioxane (48 mL) and cooled to 0 °C. Ammonium hydroxide solution (28.0-30% NH3 basis, 3.6 mL, 28.6 mmol) was added slowly via syringe. The resulting mixture was stirred at 0 °C for 30 min and then was concentrated. The residue was diluted with a 1:1 mixture of EtOAc/Heptane and agitated for 5 min, then was filtered. The filtered solids were discarded, and the remaining mother liquor was partially concentrated to half volume and filtered. The filtered solids were washed with heptane and dried in a reduced-pressure oven (45 °C) overnight to provide 2,6-dichloro-5-fluoronicotinamide. 1H NMR (400 MHz, DMSO-d6) δ ppm 8.23 (d, J = 7.9 Hz, 1 H) 8.09 (br s, 1 H) 7.93 (br s, 1 H). m/z (ESI, +ve ion): 210.9 (M+H)+.
[0129] Step 2: 2,6-Dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. To an ice-cooled slurry of 2,6-dichloro-5-fluoronicotinamide (Intermediate S, 5.0 g, 23.9 mmol) in THF (20 mL) was added oxalyl chloride (2 M solution in DCM, 14.4 mL, 28.8 mmol) slowly via syringe. The resulting mixture was heated at 75 °C for 1 h, then heating was stopped, and the reaction was concentrated to half volume. After cooling to 0 °C, THF (20 mL) was added, followed by a solution of 2-isopropyl-4-methylpyridin-3-amine (Intermediate R, 3.59 g, 23.92 mmol) in THF (10 mL), dropwise via cannula. The resulting mixture was stirred at 0 °C for 1 h and then was quenched with a 1:1 mixture of brine and saturated aqueous ammonium chloride. The mixture was extracted with EtOAc (3x) and the combined organic layers were dried over anhydrous sodium sulfate and concentrated to provide 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide. This material was used without further purification in the following step. m/z (ESI, +ve ion): 385.1(M+H)+.
[0130] Step 3: 7-Chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione. To an ice-cooled solution of 2,6-dichloro-5-fluoro-N-((2-isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (9.2 g, 24.0 mmol) in THF (40 mL) was added KHMDS (1 M solution in THF, 50.2 mL, 50.2 mmol) slowly via syringe. The ice bath was removed and the resulting mixture was stirred for 40 min at room temperature. The reaction was quenched with saturated aqueous ammonium chloride and extracted with EtOAc (3x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% 3:1 EtOAc-EtOH/heptane) to provide 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione.1H NMR (400 MHz, DMSO-d6) δ ppm 12.27 (br s, 1H), 8.48-8.55 (m, 2 H), 7.29 (d, J = 4.8 Hz, 1 H), 2.87 (quin, J = 6.6 Hz, 1 H), 1.99-2.06 (m, 3 H), 1.09 (d, J = 6.6 Hz, 3 H), 1.01 (d, J = 6.6 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -126.90 (s, 1 F). m/z (ESI, +ve ion): 349.1 (M+H)+.
[0131] Step 4: 4,7-Dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. To a solution of 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (4.7 g, 13.5 mmol) and DIPEA (3.5 mL, 20.2 mmol) in acetonitrile (20 mL) was added phosphorus oxychloride (1.63 mL, 17.5 mmol), dropwise via syringe. The resulting mixture was heated at 80 °C for 1 h, and then was cooled to room temperature and concentrated to provide 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one. This material was used without further purification in the following step. m/z (ESI, +ve ion): 367.1 (M+H)+.
[0132] Step 5: (S)-tert-Butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. To an ice-cooled solution of 4,7-dichloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidin-2(1H)-one (13.5 mmol) in acetonitrile (20 mL) was added DIPEA (7.1 mL, 40.3 mmol), followed by (S)-4-N-Boc-2-methyl piperazine (3.23 g, 16.1 mmol, Combi-Blocks, Inc., San Diego, CA, USA). The resulting mixture was warmed to room temperature and stirred for 1 h, then was diluted with cold saturated aqueous sodium bicarbonate solution (200 mL) and EtOAc (300 mL). The mixture was stirred for an additional 5 min, the layers were separated, and the aqueous layer was extracted with more EtOAc (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-50% EtOAc/heptane) to provide (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. m/z (ESI, +ve ion): 531.2 (M+H)+.
[0133] Step 6: (3S)-tert-Butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate. A mixture of (S)-tert-butyl 4-(7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (4.3 g, 8.1 mmol), potassium trifluoro(2-fluoro-6-hydroxyphenyl)borate (Intermediate Q, 2.9 g, 10.5 mmol), potassium acetate (3.2 g, 32.4 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II), complex with dichloromethane (661 mg, 0.81 mmol) in 1,4-dioxane (80 mL) was degassed with nitrogen for 1 min. De-oxygenated water (14 mL) was added, and the resulting mixture was heated at 90 °C for 1 h. The reaction was allowed to cool to room temperature, quenched with half-saturated aqueous sodium bicarbonate, and extracted with EtOAc (2x) and DCM (1x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-60% 3:1 EtOAc-EtOH/heptane) to provide (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate.1H NMR (400 MHz, DMSO-d6) δ ppm 10.19 (br s, 1 H), 8.38 (d, J = 5.0 Hz, 1 H), 8.26 (dd, J = 12.5, 9.2 Hz, 1 H), 7.23-7.28 (m, 1 H), 7.18 (d, J = 5.0 Hz, 1 H), 6.72 (d, J = 8.0 Hz, 1 H), 6.68 (t, J = 8.9 Hz, 1 H), 4.77-4.98 (m, 1 H), 4.24 (br t, J = 14.2 Hz, 1 H), 3.93-4.08 (m, 1 H), 3.84 (br d, J=12.9 Hz, 1 H), 3.52-3.75 (m, 1 H), 3.07-3.28 (m, 1 H), 2.62-2.74 (m, 1 H), 1.86-1.93 (m, 3 H), 1.43-1.48 (m, 9 H), 1.35 (dd, J = 10.8, 6.8 Hz, 3 H), 1.26-1.32 (m, 1 H), 1.07 (dd, J = 6.6, 1.7 Hz, 3 H), 0.93 (dd, J = 6.6, 2.1 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ: -115.65 (s, 1 F), -128.62 (s, 1 F). m/z (ESI, +ve ion): 607.3 (M+H)+.
[0134] Step 7: 6-Fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one. Trifluoroacetic acid (25 mL, 324 mmol) was added to a solution of (3S)-tert-butyl 4-(6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(2-isopropyl-4-methylpyridin-3-yl)-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl)-3-methylpiperazine-1-carboxylate (6.3 g, 10.4 mmol) in DCM (30 mL). The resulting mixture was stirred at room temperature for 1 h and then was concentrated. The residue was dissolved in DCM (30 mL), cooled to 0 °C, and sequentially treated with DIPEA (7.3 mL, 41.7 mmol) and a solution of acryloyl chloride (0.849 mL, 10.4 mmol) in DCM (3 mL; added dropwise via syringe). The reaction was stirred at 0 °C for 10 min, then was quenched with half-saturated aqueous sodium bicarbonate and extracted with DCM (2x). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The residue was purified by silica gel chromatography (eluent: 0-100% 3:1 EtOAc-EtOH/heptane) to provide 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-1-(4-methyl-2-(2-propanyl)-3-pyridinyl)-4-((2S)-2-methyl-4-(2-propenoyl)-1-piperazinyl)pyrido[2,3-d]pyrimidin-2(1H)-one.1H NMR (400 MHz, DMSO-d6) δ ppm 10.20 (s, 1 H), 8.39 (d, J = 4.8 Hz, 1 H), 8.24-8.34 (m, 1 H), 7.23-7.32 (m, 1 H), 7.19 (d, J = 5.0 Hz, 1 H), 6.87 (td, J = 16.3, 11.0 Hz, 1 H), 6.74 (d, J = 8.6 Hz, 1 H), 6.69 (t, J = 8.6 Hz, 1 H), 6.21 (br d, J = 16.2 Hz, 1 H), 5.74-5.80 (m, 1 H), 4.91 (br s, 1 H), 4.23-4.45 (m, 2 H), 3.97-4.21 (m, 1 H), 3.44-3.79 (m, 2 H), 3.11-3.31 (m, 1 H), 2.67-2.77 (m, 1 H), 1.91 (s, 3 H), 1.35 (d, J = 6.8 Hz, 3 H), 1.08 (d, J = 6.6 Hz, 3 H), 0.94 (d, J = 6.8 Hz, 3 H).19F NMR (376 MHz, DMSO-d6) δ ppm -115.64 (s, 1 F), -128.63 (s, 1 F). m/z (ESI, +ve ion): 561.2 (M+H)+.
[0135] Another synthesis of Compound 9 and the relevant intermediates was described in a U.S. provisional patent application filed November 16, 2018, which is incorporated herein by reference in its entirety for all purposes.
Representative Synthetic Processes
[0136] The present disclosure comprises the following steps wherein the synthesis and utilization of the boroxine intermediate is a novel and inventive step in the manufacture of AMG 510 (Compound 9):
Raw Materials
Step la
[0137] To a solution of 2,6-dichloro-5-fluoro-3-pyridinecarboxylic acid (25kg; 119. lmol) in dichloromethane (167kg) and DMF (592g) was added Oxalyl chloride (18.9kg; 148.9mol) while maintaining an internal temp between 15-20 °C. Additional dichloromethane (33kg) was added as a rinse and the reaction mixture stirred for 2h. The reaction mixture is cooled then quenched with ammonium hydroxide (40.2L; 595.5mol) while maintaining internal temperature 0 ± 10°C. The resulting slurry was stirred for 90min then the product collected by filtration. The filtered solids were washed with DI water (3X 87L) and dried to provide 2,6-dichloro-5-fluoronicotinamide (Compound 1).
Step 1b
[0138] In reactor A, a solution of 2,6-dichloro-5-fluoronicotinamide (Compound 1) (16.27kg; 77.8mol) in dichloromethane (359.5kg) was added oxalyl chloride (11.9kg;
93.8mol) while maintaining temp ≤ 25°C for 75min. The resulting solution was then headed to 40°C ± 3°C and aged for 3h. Using vacuum, the solution was distilled to remove dichloromethane until the solution was below the agitator. Dichloromethane (300 kg) was then added and the mixture cooled to 0 ± 5°C. To a clean, dry reactor (reactor B) was added,2-isopropyl-4-methylpyridin-3-amine (ANILINE Compound 2A) (12.9kg; 85.9mol) followed by dichloromethane (102.6 kg). The ANILINE solution was azeodried via vacuum distillation while maintaining an internal temperature between 20-25 °), replacing with additional dichloromethane until the solution was dry by KF analysis (limit ≤ 0.05%). The solution volume was adjusted to approx. 23L volume with dichloromethane. The dried ANILINE solution was then added to reactor A while maintaining an internal temperature of 0 ± 5°C throughout the addition. The mixture was then heated to 23 °C and aged for 1h. the solution was polish filtered into a clean reactor to afford 2,6-dichloro-5-fluoro-N-((2- isopropyl-4-methylpyridin-3-yl)carbamoyl)nicotinamide (Compound 3) as a solution in DCM and used directly in the next step.
Step 2
[0139] A dichloromethane solution of 2,6-dichloro-5-fluoro-N-{[4-methyl-2-(propan-2- yl)pyridin-3-yl]carbamoyl}pyridine-3-carboxamide (UREA (Compound 3)) (15kg contained; 38.9mol) was solvent exchanged into 2-MeTHF using vacuum distillation while maintaining internal temperature of 20-25 °C. The reactor volume was adjusted to 40L and then
additional 2-MeTHF was charged (105.4 kg). Sodium t-butoxide was added (9.4 kg;
97.8mol) while maintaining 5-10 °C. The contents where warmed to 23 °C and stirred for 3h. The contents where then cooled to 0-5C and ammonium chloride added (23.0kg; 430mol) as a solution in 60L of DI water. The mixture was warmed to 20 C and DI water added (15L) and further aged for 30min. Agitation was stopped and the layers separated. The aqueous layer was removed and to the organic layer was added DI water(81.7L). A mixture of conc HCl (1.5kg) and water (9L) was prepared then added to the reactor slowly until pH measured between 4-5. The layers were separated, and the aqueous layer back extracted using 2-MeTHF (42.2kg). The two organic layers combined and washed with a 10% citric acid solution (75kg) followed by a mixture of water (81.7L) and saturated NaCl (19.8 kg). The organic layer was then washed with saturated sodium bicarbonate (75kg) repeating if necessary to achieve a target pH of ≥ 7.0 of the aqueous. The organic layer was washed again with brine (54.7kg) and then dried over magnesium sulfate (5kg). The mixture was filtered to remove magnesium sulfate rinsing the filtered bed with 2-MeTHF (49.2 kg). The combined filtrate and washes where distilled using vacuum to 40L volume. The concentrated solution was heated to 55 °C and heptane (10-12kg) slowly added until cloud point. The solution was cooled to 23 °C over 2h then heptane (27.3 kg) was added over 2h. The product slurry was aged for 3h at 20-25 °C then filtered and washed with a mixture of 2-MeTHF (2.8kg) and heptane (9kg). The product was dried using nitrogen and vacuum to afford solid 7-chloro-6-fluoro-1-(2-isopropyl-4-methylpyridin-3-yl)pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (rac-DIONE (Compound 4)).
Step 3
[0140] To a vessel, an agitated suspension of Compound 4, (1.0 eq.) in 2- methylterahydrofuran (7.0 L/kg) was added (+)-2,3-dibenzoyl-D-tartaric acid (2.0 eq.) under an atmosphere of nitrogen. 2-MeTHF is chiral, but it is used as a racemic mixture. The different enantiomers of 2-MeTHF are incorporated randomly into the co-crystal. The resulting suspension was warmed to 75°C and aged at 75°C until full dissolution was observed (< 30 mins.). The resulting solution was polish filtered at 75°C into a secondary vessel. To the polish filtered solution was charged n-Heptane (2.0 L/kg) at a rate that maintained the internal temperature above 65°C. The solution was then cooled to 60°C, seeded with crystals (0.01 kg/kg) and allowed to age for 30 minutes. The resulting suspension was cooled to 20°C over 4 hours and then sampled for chiral purity analysis by HPLC. To the suspension, n-Heptane (3.0 L/kg) was charged and then aged for 4 hours at 20°C under an atmosphere of nitrogen. The suspension was filtered, and the isolated solids were washed two times with (2:1) n-Heptane:2-methyltetrahydrofuran (3.0 L/kg). The material was dried with nitrogen and vacuum to afford M-Dione:DBTA: Me-THF complex (Compound 4a).
Step 4
[0141] To vessel A, a suspension of disodium hydrogen phosphate (21.1 kg, 2.0 equiv) in DI water (296.8 L, 6.3 L/kg) was agitated until dissolution was observed (≥ 30 min.). To vessel B, a suspension of the M-Dione:DBTA: Me-THF complex (Composition 4a)[46.9 kg (25.9 kg corrected for M-dione, 1.0 equiv.)] in methyl tert-butyl ether (517.8 L, 11.0 L/kg) was agitated for 15 to 30 minutes. The resulting solution from vessel A was added to vessel B, and then the mixture was agitated for more than 3 hours. The agitation was stopped, and the biphasic mixture was left to separate for more than 30 minutes. The lower aqueous phase was removed and then back extracted with methyl tert-butyl ether (77.7 L, 1.7 L/kg). The organic phases were combined in vessel B and dried with magnesium sulfate (24.8 kg, 0.529 kg/kg). The resulting suspension from vessel B was agitated for more than three hours and then filtered into vessel C. To vessel B, a methyl tert-butyl ether (46.9 L, 1.0 L/kg) rinse was charged and then filtered into vessel C. The contents of vessel C were cooled to 10 °C and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until 320-350 kg (6.8-7.5 kg/kg) of methyl tert-butyl ether was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (278.7 L, 5.9 L/kg) was charged a second time over one hour and then distilled under vacuum while slowly being warmed to 35°C. Distillation was continued until a 190-200 kg (4.1-4.3 kg/kg) mixture of methyl tert-butyl ether and n-Heptane was collected. After cooling the contents of vessel C to 20°C, n-Heptane (195.9 L, 4.2 L/kg) was charged a third time over one hour and then sampled for solvent composition by GC analysis. The vessel C suspension continued to agitate for more than one hour. The suspension was filtered, and then washed with a n-Heptane (68.6 L, 1.5 L/kg) rinse from vessel C. The isolated solids were dried at 50°C, and a sample was submitted for stock suitability. Afforded 7-chloro-6-fluoro-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidine-2,4(1H,3H)-dione (M-DIONE) Compound 5M.
[0142] The first-generation process highlighted above has been successfully scaled on 200+ kg of rac-dione starting material (Compound 4). In this process, seeding the crystallization with the thermodynamically-stable rac-dione crystal form (which exhibits low solubility) would cause a batch failure. Based on our subsequent studies, we found that increasing the DBTA equivalents and lowering the seed temperature by adjusting heptane
charge schedule improves robustness of the process. The improved process is resistant to the presence of the thermodynamically-stable rac-dione crystal form and promotes successful separation of atropisomers. Subsequent batches will incorporate the improved process for large scale manufacture.
Step 5
Note: All L/kg amounts are relative to M-Dione input; All equiv. amounts are relative to M-Dione input after adjusted by potency.
[0143] M-Dione (Compound 5M, 1.0 equiv.) and Toluene-1 (10.0 L/kg) was charged to Vessel A. The resulting solution was dried by azeotropic distillation under vacuum at 45 °C until 5.0 L/kg of solvents has been removed. The contents of Vessel A were then cooled to 20 °C.
[0144] Vessel C was charged with Toluene-3 (4.5 L/kg), Phosphoryl chloride (1.5 equiv.) and N,N-Diisopropylethylamine-1 (2.0 equiv.) while maintaining the internal temperature below 20 ± 5 °C.
Upon finishing charging, Vessel C was warmed to 30 ± 5 °C. The contents of Vessel A were then transferred to Vessel C over 4 hours while maintaining the internal temperature at 30 ± 5°C. Vessel A was rinsed with Toluene-2 (0.5 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated at 30°C for an additional 3 hours. The contents of Vessel C were cooled to 20 ± 5 °C. A solution of (s)-1-boc-3-methylpiperazine (1.2 equiv.), N,N-Diisopropylethylamine-2 (1.2 equiv.) in isopropyl acetate-1 (1.0 L/kg) was prepared in Vessel D. The solution of Vessel D was charged to vessel C while maintaining a batch temperature of 20 ± 5 °C (Note: Exotherm is observed). Upon the end of transfer, Vessel D was rinsed with additional dichloromethane (1.0 L/kg) and transferred to Vessel C. The contents of Vessel C were agitated for an additional 60 minutes at 20 °C. A solution of sodium bicarbonate [water-1 (15.0 L/kg + Sodium bicarbonate (4.5 equiv.)] was then charged into Vessel C over an hour while maintaining an internal temperature at 20 ± 5 °C throughout the addition. The contents of Vessel C were agitated for at least 12 hours at which point the Pipazoline (Compound 6) product was isolated by filtration in an agitated filter dryer. The cake was washed with water-2 and -3 (5.0 L/kg x 2 times, agitating each wash for 15 minutes) and isopropyl acetate-2 and 3 (5.0 L/kg x 2 times, agitating each wash for 15 min). The cake as dried under nitrogen for 12 hours.
Acetone Re-slurry (Optional):
[0145] Pipazoline (Compound 6) and acetone (10.0 L/kg) were charged to Vessel E. The suspension was heated to 50 °C for 2 hours. Water-4 (10.0 L/kg) was charged into Vessel E over 1 hour. Upon completion of water addition, the mixture was cooled to 20 °C over 1 hour. The contents of Vessel E were filtered to isolate the product, washing the cake with 1:1 acetone/water mixture (5.0 L/kg). The cake was dried under nitrogen for 12 hours.
Step 6
General Note: All equivalents and volumes are reported in reference to Pipazoline input
Note: All L/kg and kg/kg amounts are relative to Pipazoline input
[0146] Reactor A is charged with Pipazoline (Compound 6, 1.0 equiv), degassed 2- MeTHF (9.0 L/kg) and a solution of potassium acetate (2.0 equiv) in degassed water (6.5 L/kg). The resulting mixture is warmed to 75 ± 5 °C and then, charge a slurry of
Pd(dpePhos)Cl2 (0.003 equiv) in 2-MeTHF (0.5 L/kg). Within 2 h of catalyst charge, a solution of freshly prepared Boroxine (Compound 6A, 0.5 equiv) in wet degassed 2-MeTHF (4.0 L/kg, KF > 4.0%) is charged over the course of >1 hour, but < 2 hours, rinsing with an additional portion of wet 2-MeTHF (0.5 L/kg) after addition is complete. After reaction completion ( <0.15 area % Pipazoline remaining, typically <1 h after boroxine addition is complete), 0.2 wt% (0.002 kg/kg) of Biaryl seed is added as a slurry in 0.02 L/kg wet 2- MeTHF, and the resulting seed bed is aged for > 60 min. Heptane (5.0 L/kg) is added over 2 hours at 75 ± 5 °C. The batch is then cooled to 20 ± 5 °C over 2 hours and aged for an additional 2 h. The slurry is then filtered and cake washed with 1 x 5.0L/kg water, 1 x 5.0L/kg 1:1 iPrOH:water followed by 1 x 5.0 L/kg 1:1 iPrOH:heptane (resuspension wash: the cake is resuspended by agitator and allow to set before filtering) . The cake (Biaryl, Compound 7) is then dried under vacuum with a nitrogen sweep.
Note: If the reaction stalls, an additional charge of catalyst and boroxine is required
Step 7 Charcoal Filtration for Pd removal
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to crude Biaryl input
[0147] In a clean Vessel A, charge crude Biaryl (1 equiv) and charge DCM (10 L/kg). Agitate content for > 60 minutes at 22 ± 5 °C, observing dissolution. Pass crude Biaryl from Vessel A, through a bag filter and carbon filters at a flux ≤ 3 L2/min/m and collect filtrate in clean Vessel B. Charge DCM rinse (1 L/kg) to Vessel A, and through carbon filters to collect in vessel B.
[0148] From filtrate in Vessel B, pull a solution sample for IPC Pd content. Sample is concentrated to solid and analyzed by ICP-MS. IPC: Pd ≤ 25 ppm with respect to Biaryl. a. If Pd content is greater than 25 ppm with respect to Biaryl on first or second IPC sample, pass solution through carbon filter a second time at ≤ 3 L2/min/m2, rinsing with 1 L/kg DCM; sample filtrate for IPC.
b. If Pd content remains greater than 25 ppm after third IPC, install and condition fresh carbon discs. Pass Biaryl filtrate through refreshed carbon filter, washing with 1 L/kg DCM. Sample for IPC.
[0149] Distill and refill to appropriate concentration. Prepare for distillation of recovered filtrate by concentrating to ≤ 4 L/kg DCM, and recharge to reach 5.25 ± 0.25 L/kg DCM prior to moving into Step 7 Boc-deprotection reaction.
Step 7
General Note: All equivalents and volumes are reported in reference to crude Biaryl input
Note: All L/kg and kg/kg amounts are relative to Biaryl input
[0150] To Reactor A was added: tert-butyl (3S)-4-{6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-2-oxo-1,2-dihydropyrido[2,3-d]pyrimidin-4-yl}-3-methylpiperazine-1-carboxylate (Biaryl) (1.0 equiv), dichloromethane (5.0 L/kg), and the TFA (15.0 equiv, 1.9 L/kg) is charged slowly to maintain the internal temperature at 20 ± 5 °C. The reaction was stirred for 4 h at 20 ± 5 °C.
[0151] To Reactor B was added: potassium carbonate (18.0 equiv), water (20.0 L/kg), and NMP (1.0) to form a homogenous solution. While agitating at the maximum acceptable rate for the equipment, the reaction mixture in A was transferred into the potassium carbonate solution in B over 30 minutes (~ 0.24 L/kg/min rate). The mixture was stirred at 20 ± 5 °C for an additional 12 h.
[0152] The resulting slurry was filtered and rinsed with water (2 x 10 L/kg). The wet cake was dried for 24 h to give 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-4-[(2S)-2-methylpiperazin- 1-yl]-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Des- Boc, Compound 8).
Step 8
Note: All L/kg and kg/kg amounts are relative to Des-Boc input
[0153] Des-Boc (Compound 8, 1.0 equiv) and NMP (4.2 L/kg) are charged to Vessel A under nitrogen, charge the TFA (1.0 equiv.) slowly to maintain the Tr <25 °C. The mixture is aged at 25 °C until full dissolution is observed (about 0.5 hour). The solution is then polish filtered through a 0.45 micron filter into Vessel B, washing with a NMP (0.8 L/kg). The filtrate and wash are combined, and then cooled to 0 °C. To the resulting solution, Acryloyl Chloride (1.3 equiv.) is added while maintaining temperature < 10 C. The reaction mixture is then aged at 5 ±5°C until completed by IPC (ca.1.5 hrs).
Preparation of Aqueous Disodium Phosphate Quench:
[0154] Disodium Phosphate (3.0 equiv) and Water (15.0 L/kg) are charged to Vessel C. The mixture is aged at 25 °C until full dissolution is observed. The solution is warmed to 45 ±5°C. A seed slurry of AMG 510 (0.005 equiv.) in Water (0.4 L/kg) is prepared and added to Vessel C while maintaining temperature at 45 ±5°C.
[0155] The reaction mixture in Vessel B is transferred to Vessel C (quench solution) while maintaining temperature at 45 ±5°C (ca.1 hrs). Vessel B is washed with a portion of NMP (0.5 L/kg). The product slurry is aged for 2 hrs at 45 ±5°C, cooled to 20 °C over 3 hrs, aged at 20 °C for a minimum of 12 hrs, filtered and washed with Water (2 x 10.0 L/kg). The product is dried using nitrogen and vacuum to afford Crude AMG 510 (Compound 9A).
Step 9
General Note: All equivalents and volumes are reported in reference to crude AMG 510 input
Note: All L/kg and kg/kg amounts are relative to Crude AMG 510 input
[0156] Reactor A was charged with 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4- methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1- yl]pyrido[2,3-d]pyrimidin-2(1H)-one (Crude AMG 510) (1.0 equiv), ethanol (7.5 L/kg), and water (1.9 L/kg). The mixture heated to 75 °C and polish filtered into a clean Reactor B. The solution was cool to 45 °C and seeded with authentic milled AMG 510 seed (0.015 േ 0.005
1 Seed performs best when reduced in particle size via milling or with other type of mechanical grinding if mill is not available (mortar/ pestle). Actual seed utilized will be based on seed availability. 1.0- 2.0% is seed is target amount.
kg/kg); the resulting slurry was aged for 30 min. Water (15.0 L/kg) was added over 5h while maintaining an internal temperature > 40 °C; the mixture was aged for an additional 2h.
[0157] The mixture was cooled to 20 °C over 3 hours and aged for 8h, after which the solid was collected by filtration and washed using a mixture of ethanol (2.5 L/kg) and water (5.0 L/kg). The solid was dried using vacuum and nitrogen to obtain 6-fluoro-7-(2-fluoro-6-hydroxyphenyl)-(1M)-1-[4-methyl-2-(propan-2-yl)pyridin-3-yl]-4-[(2S)-2-methyl-4-(prop-2-enoyl)piperazin-1-yl]pyrido[2,3-d]pyrimidin-2(1H)-one (AMG 510, Compound 9).
Compound 6A Boroxine Synthesis:
Lithiation/borylation
[0158] Reactor A was charged with THF (6 vol), a secondary amine base, Diisopropylamine (1.4 equiv), and a catalyst, such as triethylamine hydrochloride (0.01 equiv.). The resulting solution was cooled to -70 °C and a first base, n-BuLi (2.5 M in hexane, 1.5 equiv) was slowly added. After addition is complete, a solution of 3-fluoroanisole (1.0 equiv) in THF (6 vol) was added slowly and kept at -70 °C for 5 min. Concurrently or subsequently, a reagent, B(EtO)3 (2.0 equiv), was added slowly and kept at -70 °C for 10 min. The reaction mixture was quenched with an acid, 2N HCl. The quenched reaction mixture was extracted with MTBE (3 x 4 vol). The combined organic phases were concentrated to 1.5-3 total volumes. Heptane (7-9 vol) was added drop-wise and the mixture was cooled to 0-10 °C and stirred for 3 h. The mixture was filtrated and rinsed with heptane (1.5 vol). The solid was dried under nitrogen at < 30 °C to afford (2-fluoro-6-methoxyphenyl)boronic acid.
Demethylation:
Note: All L/kg and kg/kg amounts are relative to (2-fluoro-6-methoxyphenyl)boronic acid input
[0159] To a reactor, charge dichloromethane (solvent, 4.0 L/kg) and an acid, BBr3 (1.2 equiv), and cool to -20 °C. To this solution, a suspension of (2-fluoro-6-methoxyphenyl)boronic acid (1.0 equiv) in dichloromethane (4.0 L/kg) was added into the BBr3/DCM mixture while keeping temperature -15 to -25 °C. The reaction was allowed to proceed for approximately 2 hours while monitored by HPLC [≤1% (2-fluoro-6-methoxyphenyl)boronic acid] before reverse quenching into water (3.0 L/kg). The precipitated solid was then isolated by filtration and slurried with water (3.0 L/kg) on the filter prior to deliquoring. The filtrates were adjusted to pH 4-6 by the addition of sodium bicarbonate. The bottom organic phase was separated and the resulting aqueous layer was washed with dichloromethane (solvent, 5.0 Vol) and adjusted to pH = 1 by addition of concentrated hydrochloric acid. The resulting solids were isolated by filtration, washing the cake with water (2 x 5.0 L/kg)
Purification via Reslurry (required)
[0160] The combined crude solids were charged into a reactor and slurried with 5% EtOH/water (5.0 L/kg) at 20 °C for >1 h. The purified product was then isolated by filtration and rinsed with water (2 x 3 L/kg) before drying on the filter at < 30 °C to with nitrogen/vacuum to afford 2,2′,2”-(1,3,5,2,4,6-trioxatriborinane-2,4,6-triyl)tris(3-fluorophenol) (Boroxine, Compound 6A).
PATENT
WO 2020102730
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2020102730
PATENT
US 20180334454
References
- ^ Jump up to:a b c d e “Lumakras- sotorasib tablet, coated”. DailyMed. Retrieved 6 June 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “FDA Approves First Targeted Therapy for Lung Cancer Mutation Previously Considered Resistant to Drug Therapy”. U.S. Food and Drug Administration (FDA). 28 May 2021. Retrieved 28 May 2021. This article incorporates text from this source, which is in the public domain.
- ^ “KRAS mutant-targeting AMG 510”. NCI Drug Dictionary. National Cancer Institute. 2 February 2011. Retrieved 16 November2019.
- ^ Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. (November 2019). “The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity”. Nature. 575 (7781): 217–23. Bibcode:2019Natur.575..217C. doi:10.1038/s41586-019-1694-1. PMID 31666701.
- ^ Jump up to:a b “FDA approves Amgen drug for lung cancer with specific mutation”. CNBC. 28 May 2021. Retrieved 28 May 2021.
- ^ Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (2020). “KRASG12C inhibition with sotorasib in advanced solid tumors”. N Engl J Med. doi:10.1056/NEJMoa1917239. PMC 7571518.
- ^ Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation ” at ClinicalTrials.gov
- ^ “The Discovery Of Amgen’s Novel Investigational KRAS(G12C) Inhibitor AMG 510 Published In Nature” (Press release). Amgen. 30 October 2019. Retrieved 16 November 2019.
- ^ Irving M (24 December 2019). “Drug targeting common cancer cause enters phase 2 clinical trials”. New Atlas. Retrieved 24 December 2019.
- ^ Jump up to:a b c d Halford B (3 April 2019). “Amgen unveils its KRas inhibitor in human clinical trials: AMG 510 shuts down a mutant version of the cancer target via covalent interaction”. Chemical & Engineering News. 97 (4). Retrieved 16 November 2019.
- ^ Al Idrus A (9 September 2019). “Amgen’s KRAS drug continues to deliver but faces ‘curse’ of high expectations”. fiercebiotech.com. Retrieved 16 November 2019.
- ^ Kaiser J (30 October 2019). “Two new drugs finally hit ‘undruggable’ cancer target, providing hope for treatments”. Science Magazine. AAAS. Retrieved 16 November 2019.
- ^ Astor L (9 September 2019). “FDA Grants AMG 510 Fast Track Designation for KRAS G12C+ NSCLC”. targetedonc.com. Retrieved 16 November 2019.
- ^ World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 85” (PDF). WHO Drug Information. 35 (1).
Further reading
- Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, et al. (September 2020). “KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors”. N Engl J Med. 383 (13): 1207–17. doi:10.1056/NEJMoa1917239. PMC 7571518. PMID 32955176.
- Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. (January 2020). “Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors”. J Med Chem. 63 (1): 52–65. doi:10.1021/acs.jmedchem.9b01180. PMID 31820981.
External links
- “Sotorasib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03600883 for “A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Lumakras |
| Other names | AMG 510 |
| License data | US DailyMed: Sotorasib |
| Routes of administration | By mouth |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2252403-56-6 |
| PubChem CID | 137278711 |
| DrugBank | DB15569 |
| ChemSpider | 72380148 |
| UNII | 2B2VM6UC8G |
| KEGG | D12055 |
| Chemical and physical data | |
| Formula | C30H30F2N6O3 |
| Molar mass | 560.606 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////Sotorasib, ソトラシブ , FDA 2021, APPROVALS 2021, Lumakras, CANCER, ANTINEOPLASTIC, AMG 510, AMG-510, AMG510, AMGEN, priority review, fast-track, breakthrough therapy, orphan drug
CC1CN(CCN1C2=NC(=O)N(C3=NC(=C(C=C32)F)C4=C(C=CC=C4F)O)C5=C(C=CN=C5C(C)C)C)C(=O)C=C
NEW DRUG APPROVALS
ONE TIME
$10.00
Loncastuximab tesirine


Loncastuximab tesirine
ZYNLONTA FDA APPROVED 2021/4/23
| Formula | C6544H10048N1718O2064S52 |
|---|---|
| Exact mass | 147387.9585 |
| CAS | 1879918-31-6 |
| Efficacy | Antineoplasitc, Anti-CD19 antibody |
| Disease | Diffuse large B-cell lymphoma not otherwise specified [DS:H02434] |
| Comment | Antibody-drug conjugate Treatment of hematological cancers |
ロンカスツキシマブテシリン; ADCT-402, ADCX 19
Immunoglobulin G1, anti-(human CD19 antigen) (human-Mus musculus monoclonal RB4v1.2 γ1-chain), disulfide with human-Mus musculus monoclonal RB4v1.2 κ-chain, dimer, bis(thioether) with N-[31-(3-mercapt-2,5-dioxo-1-pyrrolidinyl)-1,29-dioxo-4,7,10,13,16,19,22,25-octaoxa-28-azahentriacont-1-yl]-L-valyl-N-[4-[[[[(11S,11aS)-8-[[5-[[(11aS)-5,11a-dihydro-7-methoxy-2-methyl-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-8-yl]oxy]pentyl]oxy]-11,11a-dihydro-11-hydroxy-7-methoxy-2-methyl-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-10(5H)-yl]carbonyl]oxy]methyl]phenyl]-L-alaninamide
NEW DRUG APPROVALS
ONETIME
$10.00
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | CD19 |
| Clinical data | |
| Trade names | Zynlonta |
| Other names | ADCT-402, loncastuximab tesirine-lpyl |
| License data | US DailyMed: Loncastuximab_tesirine |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1879918-31-6 |
| DrugBank | DB16222 |
| ChemSpider | none |
| UNII | 7K5O7P6QIU |
| KEGG | D11338 |
| Chemical and physical data | |
| Formula | C6544H10048N1718O2064S52 |
| Molar mass | 147481.45 g·mol−1 |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Zynlonta | Injection, powder, lyophilized, for solution | 5 mg/1mL | Intravenous | ADC Therapeutics America, Inc. | 2021-04-30 | Not applicable |  |
Loncastuximab tesirine-lpyl is a CD19-directed antibody and alkylating agent conjugate, consisting of a humanized IgG1 kappa monoclonal antibody conjugated to SG3199, a pyrrolobenzodiazepine (PBD) dimer cytotoxic alkylating agent, through a protease-cleavable valine–alanine linker. SG3199 attached to the linker is designated as SG3249, also known as tesirine.
|
Loncastuximab tesirine-lpyl has an approximate molecular weight of 151 kDa. An average of 2.3 molecules of SG3249 are attached to each antibody molecule. Loncastuximab tesirine-lpyl is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
ZYNLONTA (loncastuximab tesirine-lpyl) for injection is supplied as a sterile, white to off-white, preservative-free, lyophilized powder, which has a cake-like appearance, for intravenous infusion after reconstitution and dilution. Each single-dose vial delivers 10 mg of loncastuximab tesirine-lpyl, L-histidine (2.8 mg), L-histidine monohydrochloride (4.6 mg), polysorbate 20 (0.4 mg), and sucrose (119.8 mg). After reconstitution with 2.2 mL Sterile Water for Injection, USP, the final concentration is 5 mg/mL with a pH of approximately 6.0.
Loncastuximab tesirine , sold under the brand name Zynlonta, is used for the treatment of large B-cell lymphoma. It is an antibody-drug conjugate (ADC) composed of a humanized antibody targeting the protein CD19, which is expressed in a wide range of B cell hematological tumors.[2] The experimental drug, developed by ADC Therapeutics is being tested in clinical trials for the treatment of B-cell non-Hodgkin lymphoma (NHL) and B-cell acute lymphoblastic leukemia (ALL).
On April 23, 2021, the Food and Drug Administration granted accelerated approval to loncastuximab tesirine-lpyl (Zynlonta, ADC Therapeutics SA), a CD19-directed antibody and alkylating agent conjugate, for adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma.
Approval was based on LOTIS-2 (NCT03589469), an open-label, single-arm trial in 145 adult patients with relapsed or refractory DLBCL or high-grade B-cell lymphoma after at least two prior systemic regimens. Patients received loncastuximab tesirine-lpyl 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles. Patients received treatment until progressive disease or unacceptable toxicity.
The main efficacy outcome measure was overall response rate (ORR), as assessed by an independent review committee using Lugano 2014 criteria. The ORR was 48.3% (95% CI: 39.9, 56.7) with a complete response rate of 24.1% (95% CI: 17.4, 31.9). After a median follow-up of 7.3 months, median response duration was 10.3 months (95% CI: 6.9, NE). Of the 70 patients who achieved objective responses, 36% were censored for response duration prior to 3 months.
Most common (≥20%) adverse reactions in patients receiving loncastuximab tesirine-lpyl, including laboratory abnormalities, are thrombocytopenia, increased gamma-glutamyltransferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal pain.
The prescribing information provides warnings and precautions for adverse reactions including edema and effusions, myelosuppression, infections, and cutaneous reactions.
The recommended loncastuximab tesirine-lpyl dosage is 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles, by intravenous infusion over 30 minutes on day 1 of each cycle (every 3 weeks). Patients should be premedicated with dexamethasone 4 mg orally or intravenously twice daily for 3 days beginning the day before loncastuximab tesirine-lpyl.
Technology
The humanized monoclonal antibody is stochastically conjugated via a valine-alanine cleavable, maleimide linker to a cytotoxic (anticancer) pyrrolobenzodiazepine (PBD) dimer. The antibody binds to CD19, a protein which is highly expressed on the surface of B-cell hematological tumors[3] including certain forms of lymphomas and leukemias. After binding to the tumor cells the antibody is internalized, the cytotoxic drug PBD is released and the cancer cells are killed. PBD dimers are generated out of PBD monomers, a class of natural products produced by various actinomycetes. PBD dimers work by crosslinking specific sites of the DNA, blocking the cancer cells’ division that cause the cells to die. As a class of DNA-crosslinking agents they are significantly more potent than systemic chemotherapeutic drugs.[4]
Clinical trials
Two phase I trials are evaluating the drug in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma and relapsed or refractory B-cell acute lymphoblastic leukemia.[5] At the 14th International Conference on Malignant Lymphoma interim results from a Phase I, open-label, dose-escalating study designed to evaluate the treatment of loncastuximab tesirine in relapsed or refractory non-Hodgkin’s lymphoma were presented.[6] Among the patients enrolled at the time of the data cutoff the overall response rate was 61% in the total patient population (42% complete response and 19% partial response) and in patients with relapsing or refractory diffuse large B-cell lymphoma (DLBCL) the overall response rate was 57% (43% complete response and 14% partial response).[7][8]
Orphan drug designation
Loncastuximab tesirine was granted Orphan Drug Designation by the U.S. Food and Drug Administration (FDA) for the treatment of diffuse large B-cell lymphoma and mantle cell lymphoma.[9]
References
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf
- ^ WHO Drug Information: International Nonproprietary Names for Pharmaceutical Substances
- ^ Wang K, Wei G, Liu D (November 2012). “CD19: a biomarker for B cell development, lymphoma diagnosis and therapy”. Experimental Hematology & Oncology. 1 (1): 36. doi:10.1186/2162-3619-1-36. PMC 3520838. PMID 23210908.
- ^ “Pyrrolobenzodiazepine”. ADC Review.
- ^ Clinical trial number NCT02669017 for “ADCT-402 in B-NHL” at ClinicalTrials.gov
- ^ Kahl B, Hamadani M, Caimi PF, Reid EG, Havenith K, He S, Feingold JM, O’Connor O (June 2017). “First clinical results of ADCT‐402, a novel pyrrolobenzodiazepine-based antibody drug conjugate (ADC), in relapsed/refractory B‐cell linage NHL” (PDF). Hematol Oncol. 35 (S2): 49–51. doi:10.1002/hon.2437_33.
- ^ “First clinical results of ADCT-402”. ADC Review.
- ^ Bainbridge K. “Grandfather fighting deadly cancer reveals scans of tumors after testing new drug”. Mirror.
- ^ “ADCT-402 Orphan Drug Designation” (PDF). ADC Therapeutics press release.
External links
- “Loncastuximab tesirine”. Drug Information Portal. U.S. National Library of Medicine.
/////////Loncastuximab tesirine, FDA 2021, APPROVALS 2021, ZYNLONTA, ロンカスツキシマブテシリン, ORPHAN DRUG, ADCT-402, priority review, ADCX 19
Fosdenopterin hydrobromide

Fosdenopterin hydrobromide
FDA APPR 2021/2/26, NULIBRY
BBP-870/ORGN001
a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.
| ホスデノプテリン臭化水素酸塩水和物; |
| Formula | C10H14N5O8P. 2H2O. HBr |
|---|---|
| CAS | 2301083-34-9DIHYDRATE |
| Mol weight | 480.1631 |
2301083-34-9
(1R,10R,12S,17R)-5-amino-11,11,14-trihydroxy-14-oxo-13,15,18-trioxa-2,4,6,9-tetraza-14λ5-phosphatetracyclo[8.8.0.03,8.012,17]octadeca-3(8),4-dien-7-one;dihydrate;hydrobromide
1,3,2-DIOXAPHOSPHORINO(4′,5′:5,6)PYRANO(3,2-G)PTERIDIN-10(4H)-ONE, 8-AMINO-4A,5A,6,9,11,11A,12,12A-OCTAHYDRO-2,12,12-TRIHYDROXY-, 2-OXIDE, HYDROBROMIDE, HYDRATE (1:1:2), (4AR,5AR,11AR,12AS)-
| CYCLIC PYRANOPTERIN MONOPHOSPHATE MONOHYDROBROMIDE DIHYDRATE |
(4aR,5aR,11aR,12aS)-8-Amino-2,12,12-trihydroxy-4a,5a,6,7,11,11a,12,12aoctahydro-2H-2lambda5-(1,3,2)dioxaphosphinino(4′,5′:5,6)pyrano(3,2-g)pteridine-2,10(4H)-dione, hydrobromide (1:1:2)
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide, hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
1,3,2-Dioxaphosphorino(4′,5′:5,6)pyrano(3,2-g)pteridin-10(4H)-one, 8-amino-4a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-, 2-oxide,hydrobromide, hydrate (1:1:2), (4aR,5aR,11aR,12aS)-
ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780



C10H14N5O8P, Average: 363.223
150829-29-1
- ALXN-1101
- WHO 11150
- Synthesis ReferenceClinch K, Watt DK, Dixon RA, Baars SM, Gainsford GJ, Tiwari A, Schwarz G, Saotome Y, Storek M, Belaidi AA, Santamaria-Araujo JA: Synthesis of cyclic pyranopterin monophosphate, a biosynthetic intermediate in the molybdenum cofactor pathway. J Med Chem. 2013 Feb 28;56(4):1730-8. doi: 10.1021/jm301855r. Epub 2013 Feb 19.
Fosdenopterin (or cyclic pyranopterin monophosphate, cPMP), sold under the brand name Nulibry, is a medication used to reduce the risk of death due to a rare genetic disease known as molybdenum cofactor deficiency type A (MoCD-A).[1]
Adverse effects
The most common side effects include complications related to the intravenous line, fever, respiratory infections, vomiting, gastroenteritis, and diarrhea.[1]
Mechanism of action
People with MoCD-A cannot produce cyclic pyranopterin monophosphate (cPMP) in their body.[1] Fosdenopterin is an intravenous medication that replaces the missing cPMP.[1][2] cPMP is a precursor to molybdopterin, which is required for the enzyme activity of sulfite oxidase, xanthine dehydrogenase/oxidase and aldehyde oxidase.[3]
History
Fosdenopterin was developed by José Santamaría-Araujo and Guenter Schwarz at the German universities TU Braunschweig and the University of Cologne.[4][5]
The effectiveness of fosdenopterin for the treatment of MoCD-A was demonstrated in thirteen treated participants compared to eighteen matched, untreated participants.[1][6] The participants treated with fosdenopterin had a survival rate of 84% at three years, compared to 55% for the untreated participants.[1]
The U.S. Food and Drug Administration (FDA) granted the application for fosdenopterin priority review, breakthrough therapy, and orphan drug designations along with a rare pediatric disease priority review voucher.[1] The FDA granted the approval of Nulibry to Origin Biosciences, Inc., in February 2021.[1] It is the first medication approved for the treatment of MoCD-A.[1]
References
- ^ Jump up to:a b c d e f g h i j “FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A”. U.S. Food and Drug Administration (FDA) (Press release). 26 February 2021. Retrieved 26 February 2021. This article incorporates text from this source, which is in the public domain.
- ^ DrugBank DB16628 . Accessed 2021-03-05.
- ^ Santamaria-Araujo JA, Fischer B, Otte T, Nimtz M, Mendel RR, Wray V, Schwarz G (April 2004). “The tetrahydropyranopterin structure of the sulfur-free and metal-free molybdenum cofactor precursor”. The Journal of Biological Chemistry. 279 (16): 15994–9. doi:10.1074/jbc.M311815200. PMID 14761975.
- ^ Schwarz G, Santamaria-Araujo JA, Wolf S, Lee HJ, Adham IM, Gröne HJ, et al. (June 2004). “Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli”. Human Molecular Genetics. 13 (12): 1249–55. doi:10.1093/hmg/ddh136. PMID 15115759.
- ^ Tedmanson S (5 November 2009). “Doctors risk untried drug to stop baby’s brain dissolving”. TimesOnline.
- ^ Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, et al. (November 2015). “Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study”. Lancet. 386 (10007): 1955–63. doi:10.1016/S0140-6736(15)00124-5. PMID 26343839. S2CID 21954888.
External links
- “Fosdenopterin”. Drug Information Portal. U.S. National Library of Medicine.
Molybdenum cofactor deficiency (MoCD) is an exceptionally rare autosomal recessive disorder resulting in a deficiency of three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde oxidase.1 Signs and symptoms begin shortly after birth and are caused by a build-up of toxic sulfites resulting from a lack of SOX activity.1,5 Patients with MoCD may present with metabolic acidosis, intracranial hemorrhage, feeding difficulties, and significant neurological symptoms such as muscle hyper- and hypotonia, intractable seizures, spastic paraplegia, myoclonus, and opisthotonus. In addition, patients with MoCD are often born with morphologic evidence of the disorder such as microcephaly, cerebral atrophy/hypodensity, dilated ventricles, and ocular abnormalities.1 MoCD is incurable and median survival in untreated patients is approximately 36 months1 – treatment, then, is focused on improving survival and maintaining neurological function.
The most common subtype of MoCD, type A, involves mutations in MOCS1 wherein the first step of molybdenum cofactor synthesis – the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP) – is interrupted.1,3 In the past, management strategies for this disorder involved symptomatic and supportive treatment,5 though efforts were made to develop a suitable exogenous replacement for the missing cPMP. In 2009 a recombinant, E. coli-produced cPMP was granted orphan drug designation by the FDA, becoming the first therapeutic option for patients with MoCD type A.1
Fosdenopterin was approved by the FDA on Februrary 26, 2021, for the reduction of mortality in patients with MoCD type A,5 becoming the first and only therapy approved for the treatment of MoCD. By improving the three-year survival rate from 55% to 84%,7 and considering the lack of alternative therapies available, fosdenopterin appears poised to become a standard of therapy in the management of this debilitating disorder.
Fosdenopterin replaces an intermediate substrate in the synthesis of molybdenum cofactor, a compound necessary for the activation of several molybdenum-dependent enzymes including sulfite oxidase (SOX).1 Given that SOX is responsible for detoxifying sulfur-containing acids and sulfites such as S-sulfocysteine (SSC), urinary levels of SSC can be used as a surrogate marker of efficacy for fosdenopterin.7 Long-term therapy with fosdenopterin has been shown to result in a sustained reduction in urinary SSC normalized to creatinine.7
Animal studies have identified a potential risk of phototoxicity in patients receiving fosdenopterin – these patients should avoid or minimize exposure to sunlight and/or artificial UV light.7 If sun exposure is necessary, use protective clothing, hats, and sunglasses,7 in addition to seeking shade whenever practical. Consider the use of a broad-spectrum sunscreen in patients 6 months of age or older.8
Molybdenum cofactor deficiency (MoCD) is a rare autosomal-recessive disorder in which patients are deficient in three molybdenum-dependent enzymes: sulfite oxidase (SOX), xanthine dehydrogenase, and aldehyde dehydrogenase.1 The loss of SOX activity appears to be the main driver of MoCD morbidity and mortality, as the build-up of neurotoxic sulfites typically processed by SOX results in rapid and progressive neurological damage. In MoCD type A, the disorder results from a mutation in the MOCS1 gene leading to deficient production of MOCS1A/B,7 a protein that is responsible for the first step in the synthesis of molybdenum cofactor: the conversion of guanosine triphosphate into cyclic pyranopterin monophosphate (cPMP).1,4
Fosdenopterin is an exogenous form of cPMP, replacing endogenous production and allowing for the synthesis of molybdenum cofactor to proceed.7
- Mechler K, Mountford WK, Hoffmann GF, Ries M: Ultra-orphan diseases: a quantitative analysis of the natural history of molybdenum cofactor deficiency. Genet Med. 2015 Dec;17(12):965-70. doi: 10.1038/gim.2015.12. Epub 2015 Mar 12. [PubMed:25764214]
- Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG, Hennermann JB, Jameson E, Konig K, McGregor TL, Font-Montgomery E, Santamaria-Araujo JA, Santra S, Vaidya M, Vierzig A, Wassmer E, Weis I, Wong FY, Veldman A, Schwarz G: Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study. Lancet. 2015 Nov 14;386(10007):1955-63. doi: 10.1016/S0140-6736(15)00124-5. Epub 2015 Sep 3. [PubMed:26343839]
- Iobbi-Nivol C, Leimkuhler S: Molybdenum enzymes, their maturation and molybdenum cofactor biosynthesis in Escherichia coli. Biochim Biophys Acta. 2013 Aug-Sep;1827(8-9):1086-101. doi: 10.1016/j.bbabio.2012.11.007. Epub 2012 Nov 29. [PubMed:23201473]
- Mendel RR: The molybdenum cofactor. J Biol Chem. 2013 May 10;288(19):13165-72. doi: 10.1074/jbc.R113.455311. Epub 2013 Mar 28. [PubMed:23539623]
- FDA News Release: FDA Approves First Treatment for Molybdenum Cofactor Deficiency Type A [Link]
- OMIM: MOLYBDENUM COFACTOR DEFICIENCY, COMPLEMENTATION GROUP A (# 252150) [Link]
- FDA Approved Drug Products: Nulibry (fosdenopterin) for intravenous injection [Link]
- Health Canada: Sun safety tips for parents [Link]
SYN
Journal of Biological Chemistry (1995), 270(3), 1082-7.
https://linkinghub.elsevier.com/retrieve/pii/S0021925818829696
PATENT
WO 2005073387
PATENT
WO 2012112922
PAPER

Journal of Medicinal Chemistry (2013), 56(4), 1730-1738
https://pubs.acs.org/doi/10.1021/jm301855r

Cyclic pyranopterin monophosphate (1), isolated from bacterial culture, has previously been shown to be effective in restoring normal function of molybdenum enzymes in molybdenum cofactor (MoCo)-deficient mice and human patients. Described here is a synthesis of 1 hydrobromide (1·HBr) employing in the key step a Viscontini reaction between 2,5,6-triamino-3,4-dihydropyrimidin-4-one dihydrochloride and d-galactose phenylhydrazone to give the pyranopterin (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one (10) and establishing all four stereocenters found in 1. Compound 10, characterized spectroscopically and by X-ray crystallography, was transformed through a selectively protected tri-tert-butoxycarbonylamino intermediate into a highly crystalline tetracyclic phosphate ester (15). The latter underwent a Swern oxidation and then deprotection to give 1·HBr. Synthesized 1·HBr had in vitro efficacy comparable to that of 1 of bacterial origin as demonstrated by its enzymatic conversion into mature MoCo and subsequent reconstitution of MoCo-free human sulfite oxidase–molybdenum domain yielding a fully active enzyme. The described synthesis has the potential for scale up.
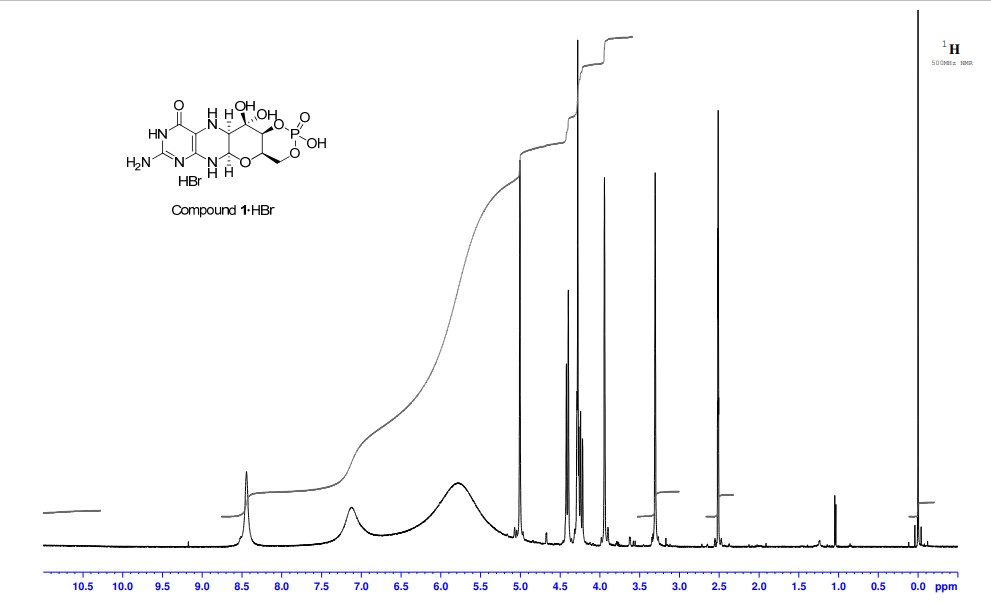
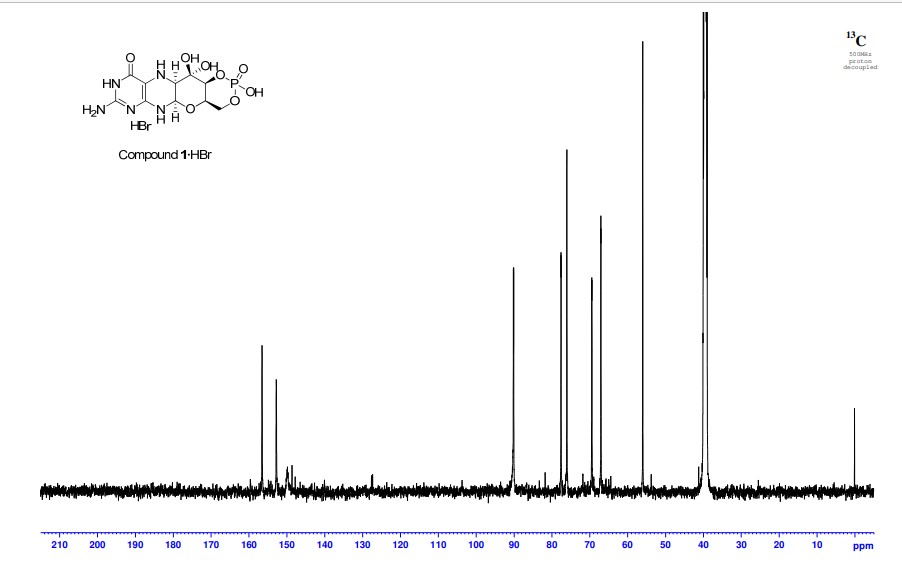





PAPER
European Journal of Organic Chemistry (2014), 2014(11), 2231-2241.
https://chemistry-europe.onlinelibrary.wiley.com/doi/abs/10.1002/ejoc.201301784
Abstract
The first synthesis of an oxygen‐stable analogue of the natural product cyclic pyranopterin monophosphate (cPMP) is reported. In this approach, the hydropyranone ring is annelated to pyrazine by a sequence comprising ortho‐lithiation/acylation of a 2‐halopyrazine, followed by nucleophilic aromatic substitution. The tetrose substructure is introduced from the chiral pool, from D‐galactose or D‐arabitol.

Abstract
Molybdenum cofactor (Moco) deficiency is a lethal hereditary metabolic disease. A recently developed therapy requires continuous intravenous supplementation of the biosynthetic Moco precursor cyclic pyranopterin monophosphate (cPMP). The limited stability of the latter natural product, mostly due to oxidative degradation, is problematic for oral administration. Therefore, the synthesis of more stable cPMP analogues is of great interest. In this context and for the first time, the synthesis of a cPMP analogue, in which the oxidation‐labile reduced pterin unit is replaced by a pyrazine moiety, was achieved starting from the chiral pool materials D‐galactose or D‐arabitol. Our synthesis, 13 steps in total, includes the following key transformations: i) pyrazine lithiation, followed by acylation; ii) closure of the pyrane ring by nucleophilic aromatic substitution; and iii) introduction of phosphate.
Patent
https://patents.google.com/patent/US9260462B2/en
Molybdenum cofactor (Moco) deficiency is a pleiotropic genetic disorder. Moco consists of molybdenum covalently bound to one or two dithiolates attached to a unique tricyclic pterin moiety commonly referred to as molybdopterin (MPT). Moco is synthesized by a biosynthetic pathway that can be divided into four steps, according to the biosynthetic intermediates precursor Z (cyclic pyranopterin monophosphate; cPMP), MPT, and adenylated MPT. Mutations in the Moco biosynthetase genes result in the loss of production of the molybdenum dependent enzymes sulfite-oxidase, xanthine oxidoreductase, and aldehyde oxidase. Whereas the activities of all three of these cofactor-containing enzymes are impaired by cofactor deficiency, the devastating consequences of the disease can be traced to the loss of sulfite oxidase activity. Human Moco deficiency is a rare but severe disorder accompanied by serious neurological symptoms including attenuated growth of the brain, untreatable seizures, dislocated ocular lenses, and mental retardation. Until recently, no effective therapy was available and afflicted patients suffering from Moco deficiency died in early infancy.
It has been found that administration of the molybdopterin derivative precursor Z, a relatively stable intermediate in the Moco biosynthetic pathway, is an effective means of therapy for human Moco deficiency and associated diseases related to altered Moco synthesis (see U.S. Pat. No. 7,504,095). As with most replacement therapies for illnesses, however, the treatment is limited by the availability of the therapeutic active agent.
Scheme 3.

Scheme 4.

(I).
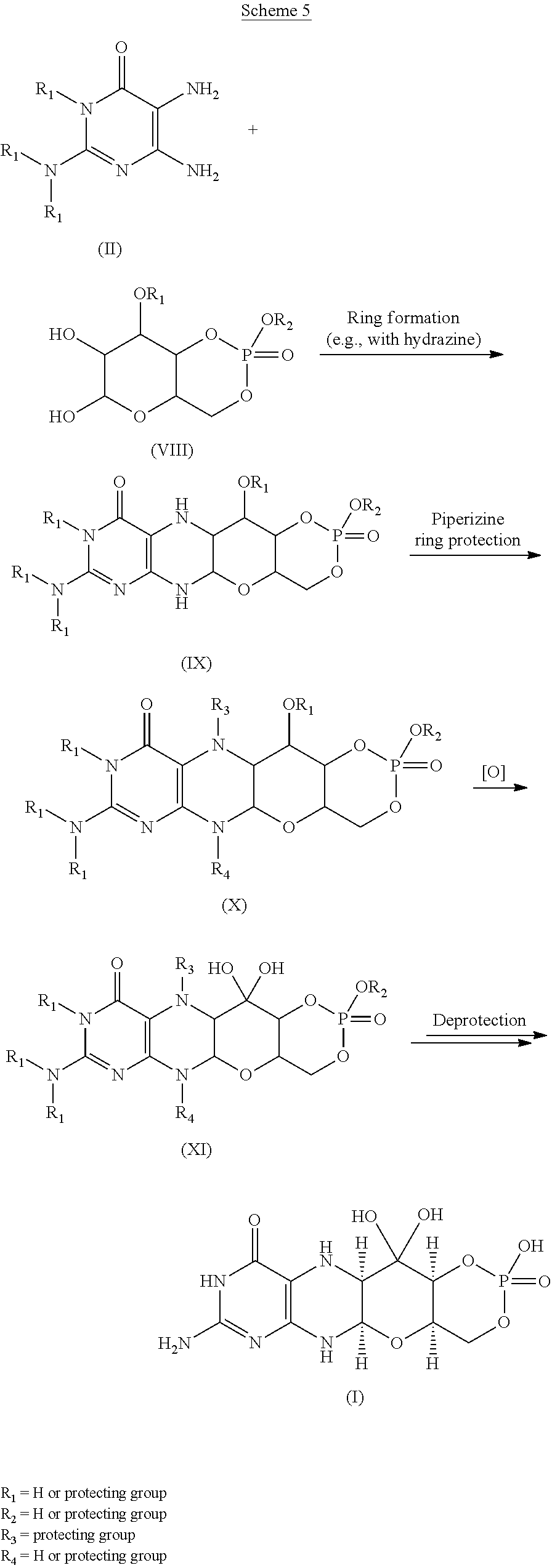
Scheme 6.

(I).

Scheme 8.

(I).

Scheme 10.

EXAMPLESExample 1Preparation of Precursor Z (cPMP)


Experimental
Air sensitive reactions were performed under argon. Organic solutions were dried over anhydrous MgSO4 and the solvents were evaporated under reduced pressure. Anhydrous and chromatography solvents were obtained commercially (anhydrous grade solvent from Sigma-Aldrich Fine Chemicals) and used without any further purification. Thin layer chromatography (t.l.c.) was performed on glass or aluminum sheets coated with 60 F254 silica gel. Organic compounds were visualized under UV light or with use of a dip of ammonium molybdate (5 wt %) and cerium(IV) sulfate 4H2O (0.2 wt %) in aq. H2SO4 (2M), one of I2 (0.2%) and KI (7%) in H2SO4 (1M), or 0.1% ninhydrin in EtOH. Chromatography (flash column) was performed on silica gel (40-63 μm) or on an automated system with continuous gradient facility. Optical rotations were recorded at a path length of 1 dm and are in units of 10−1 deg cm2 g−1; concentrations are in g/100 mL. 1H NMR spectra were measured in CDCl3, CD3OD (internal Me4Si, δ 0 ppm) or D2O(HOD, δ 4.79 ppm), and 13C NMR spectra in CDCl3 (center line, δ 77.0 ppm), CD3OD (center line, δ 49.0 ppm) or DMSO d6 (center line δ 39.7 ppm), D2O (no internal reference or internal CH3CN, δ 1.47 ppm where stated). Assignments of 1H and 13C resonances were based on 2D (1H—1H DQF-COSY, 1H—13C HSQC, HMBC) and DEPT experiments. 31P NMR were run at 202.3 MHz and are reported without reference. High resolution electrospray mass spectra (ESI-HRMS) were recorded on a Q-TOF Tandem Mass
Spectrometer. Microanalyses were performed by the Campbell Microanalytical Department, University of Otago, Dunedin, New Zealand.
A. Preparation of (5aS,6R,7R,8R,9aR)-2-amino-6,7-dihydroxy-8-(hydroxymethyl)-3H,4H,5H,5aH,6H,7H,8H,9aH,10H-pyrano[3,2-g]pteridin-4-one mono hydrate (1)
2,5,6-Triamino-3,4-dihydropyrimidin-4-one dihydrochloride (Pfleiderer, W.; Chem. Ber. 1957, 90, 2272; Org. Synth. 1952, 32, 45; Org. Synth. 1963, Coll. Vol. 4, 245, 10.0 g, 46.7 mmol), D-galactose phenylhydrazone (Goswami, S.; Adak, A. K. Tetrahedron Lett. 2005, 46, 221-224, 15.78 g, 58.4 mmol) and 2-mercaptoethanol (1 mL) were stirred and heated to reflux (bath temp 110° C.) in a 1:1 mixture of MeOH—H2O (400 mL) for 2 h. After cooling to ambient temperature, diethyl ether (500 mL) was added, the flask was shaken and the diethyl ether layer decanted off and discarded. The process was repeated with two further portions of diethyl ether (500 mL) and then the remaining volatiles were evaporated. Methanol (40 mL), H2O (40 mL) and triethylamine (39.4 mL, 280 mmol) were successively added and the mixture seeded with a few milligrams of 1. After 5 min a yellow solid was filtered off, washed with a little MeOH and dried to give 1 as a monohydrate (5.05 g, 36%) of suitable purity for further use. An analytical portion was recrystallized from DMSO-EtOH or boiling H2O. MPt 226 dec. [α]D 20 +135.6 (c1.13, DMSO). 1H NMR (DMSO d6): δ 10.19 (bs, exchanged D2O, 1H), 7.29 (d, J=5.0 Hz, slowly exchanged D2O, 1H), 5.90 (s, exchanged D2O, 2H), 5.33 (d, J=5.4 Hz, exchanged D2O, 1H), 4.66 (ddd, J˜5.0, ˜1.3, ˜1.3 Hz, 1H), 4.59 (t, J=5.6 Hz, exchanged D2O, 1H), 4.39 (d, J=10.3 Hz, exchanged D2O, 1H), 3.80 (bt, J˜1.8 Hz, exchanged D2O, 1H), 3.70 (m, 1H), 3.58 (dd, J=10.3, 3.0 Hz, 1H), 3.53 (dt, J=10.7, 6.4 Hz, 1H), 3.43 (ddd, J=11.2, 5.9, 5.9 Hz, 1H), 3.35 (t, J=6.4 Hz, 1H), 3.04 (br m, 1H). 13C NMR (DMSO d6 center line 6 39.7): δ 156.3 (C), 150.4 (C), 148.4 (C), 99.0 (C), 79.4 (CH), 76.5 (CH), 68.9 (CH), 68.6 (CH), 60.6 (CH2), 53.9 (CH). Anal. calcd. for C10H15N5O5H2O 39.60; C, 5.65; H, 23.09; N. found 39.64; C, 5.71; H, 22.83; N.
B. Preparation of Compounds 2 (a or b) and 3 (a, b or c)
Di-tert-butyl dicarbonate (10.33 g, 47.3 mmol) and DMAP (0.321 g, 2.63 mmol) were added to a stirred suspension of 1 (1.5 g, 5.26 mmol) in anhydrous THF (90 mL) at 50° C. under Ar. After 20 h a clear solution resulted. The solvent was evaporated and the residue chromatographed on silica gel (gradient of 0 to 40% EtOAc in hexanes) to give two product fractions. The first product to elute was a yellow foam (1.46 g). The product was observed to be a mixture of two compounds by 1H NMR containing mainly a product with seven Boc groups (2a or 2b). A sample was crystallized from EtOAc-hexanes to give 2a or 2b as a fine crystalline solid. MPt 189-191° C. [α]D 20 −43.6 (c 0.99, MeOH). 1H NMR (500 MHz, CDCl3): δ 5.71 (t, J=1.7 Hz, 1H), 5.15 (dt, J=3.5, ˜1.0, 1H), 4.97 (t, J=3.8, 1H), 4.35 (br t, J=˜1.7, 1H), 4.09-3.97 (m, 3H), 3.91 (m, 1H), 1.55, 1.52, 1.51, 1.50, 1.45 (5s, 45H), 1.40 (s, 18H). 13C NMR (125.7 MHz, CDCl3): δ 152.84 (C), 152.78 (C), 151.5 (C), 150.9 (C), 150.7 (2×C), 150.3 (C), 149.1 (C), 144.8 (C), 144.7 (C), 118.0 (C), 84.6 (C), 83.6 (C), 83.5 (C), 82.7 (3×C), 82.6 (C), 76.3 (CH), 73.0 (CH), 71.4 (CH), 67.2 (CH), 64.0 (CH2), 51.4 (CH), 28.1 (CH3), 27.8 (2×CH3), 27.7 (CH3), 27.6 (3×CH3). MS-ESI+ for C45H72N5O19 +, (M+H)+, Calcd. 986.4817. found 986.4818. Anal. calcd. for C45H71N5O19H2O 54.39; C, 7.39; H, 6.34; N. found 54.66; C, 7.17; H, 7.05; N. A second fraction was obtained as a yellow foam (2.68 g) which by 1H NMR was a product with six Boc groups present (3a, 3b or 3c). A small amount was crystallized from EtOAc-hexanes to give colorless crystals. [α]D 2O −47.6 (c, 1.17, CHCl3). 1H NMR (500 MHz, CDCl3): δ 11.10 (br s, exchanged D2O, 1H), 5.58 (t, J=1.8 Hz, 1H), 5.17 (d, J=3.4 Hz, 1H), 4.97 (t, J=3.9 Hz, 1H), 4.62 (s, exchanged D2O, 1H), 4.16 (dd, J=11.3, 5.9 Hz, 1H), 4.12 (dd, J=11.3, 6.4 Hz, 1H), 3.95 (dt, J=6.1, 1.1 Hz, 1H), 3.76 (m, 1H), 1.51, 1.50, 1.49, 1.48, 1.46 (5s, 54H). 13C NMR (125.7 MHz, CDCl3): δ 156.6 (C), 153.0 (C), 152.9 (C), 151.9 (C), 150.6 (C), 149.4 (2×C), 136.2 (C), 131.8 (C), 116.9 (C), 85.0 (2×C), 83.3 (C), 82.8 (C), 82.49 (C), 82.46 (C), 73.3 (CH), 71.5 (CH), 67.2 (CH), 64.5 (CH2), 51.3 (CH), 28.0, 27.72, 27.68, 27.6 (4×CH3). MS-ESI+ for C40H64N5O17 +, (M+H)+calcd. 886.4287. found 886.4289.
C. Preparation of Compound 4a, 4b or 4c
Step 1—The first fraction from B above containing mainly compounds 2a or 2b (1.46 g, 1.481 mmol) was dissolved in MeOH (29 mL) and sodium methoxide in MeOH (1M, 8.14 mL, 8.14 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
Step 2—The second fraction from B above containing mainly 3a, 3b or 3c (2.68 g, 3.02 mmol) was dissolved in MeOH (54 mL) and sodium methoxide in MeOH (1M, 12.10 mL, 12.10 mmol) added. After leaving at ambient temperature for 20 h the solution was neutralized with Dowex 50WX8 (H+) resin then the solids filtered off and the solvent evaporated.
The products from step 1 and step 2 above were combined and chromatographed on silica gel (gradient of 0 to 15% MeOH in CHCl3) to give 4a, 4b or 4c as a cream colored solid (1.97 g). 1H NMR (500 MHz, DMSO d6): δ 12.67 (br s, exchanged D2O, 1H), 5.48 (d, J=5.2 Hz, exchanged D2O, 1H), 5.43 (t, J=˜1.9 Hz, after D2O exchange became a d, J=1.9 Hz, 1H), 5.00 (br s, exchanged D2O, 1H), 4.62 (d, J=5.7 Hz, exchanged D2O, 1H), 4.27 (d, J=6.0 Hz, exchanged D2O, 1H), 3.89 (dt, J=5.2, 3.8 Hz, after D2O became a t, J=3.9 Hz, 1H), 3.62 (dd, J=6.0, 3.7 Hz, after D2O exchange became a d, J=3.7 Hz, 1H), 3.52-3.39 (m, 4H), 1.42 (s, 9H), 1.41 (s, 18H). 13C NMR (125.7 MHz, DMSO d6): δ 157.9 (C), 151.1, (C), 149.8 (2×C), 134.6 (C), 131.4 (C), 118.8 (C), 83.5 (2×C), 81.3 (C), 78.2 (CH), 76.5 (CH), 68.1 (CH), 66.8 (CH), 60.6 (CH2), 54.4 (CH), 27.9 (CH3), 27.6 (2×CH3). MS-ESI+ for C25H40N5O11 +, (M+H)+ calcd. 586.2719. found 586.2717.
D. Preparation of Compound 5a, 5b or 5c
Compound 4a, 4b or 4c (992 mg, 1.69 mmol) was dissolved in anhydrous pyridine and concentrated. The residue was dissolved in anhydrous CH2Cl2 (10 mL) and pyridine (5 mL) under a nitrogen atmosphere and the solution was cooled to −42° C. in an acetonitrile/dry ice bath. Methyl dichlorophosphate (187 μL, 1.86 mmol) was added dropwise and the mixture was stirred for 2 h 20 min. Water (10 mL) was added to the cold solution which was then removed from the cold bath and diluted with ethyl acetate (50 mL) and saturated NaCl solution (30 mL). The organic portion was separated and washed with saturated NaCl solution. The combined aqueous portions were extracted twice further with ethyl acetate and the combined organic portions were dried over MgSO4 and concentrated. Purification by silica gel flash column chromatography (eluting with 2-20% methanol in ethyl acetate) gave the cyclic methyl phosphate 5a, 5b or 5c (731 mg, 65%). 1H NMR (500 MHz, CDCl3,): δ 11.72 (bs, exchanged D2O, 1H), 5.63 (t, J=1.8 Hz, 1H), 5.41 (s, exchanged D2O, 1H), 4.95 (d, J=3.2 Hz, 1H), 4.70 (dt, J=12.4, 1.8 Hz, 1H), 4.42 (dd, J=22.1, 12.1 Hz, 1H). 4.15 (q, J=3.7 Hz, 1H), 3.82 (s, 1H), 3.75 (s, 1H), 3.58 (d, J=11.7 Hz, 3H), 2.10 (bs, exchanged D20, 1H+H2O), 1.50 (s, 9H), 1.46 (s, 18H). 13C NMR (125.7 MHz, CDCl3, centre line δ 77.0): δ 157.5 (C), 151.2 (C), 149.6 (2×C), 134.5 (C), 132.3 (C), 117.6 (C), 84.7 (2×C), 82.8 (C), 77.3 (CH), 74.8 (d, J=4.1 Hz, CH), 69.7 (CH2), 68.8 (d, J=4.1 Hz, CH), 68.6 (d, J=5.9 Hz, CH), 56.0 (d, J=7.4 Hz, CH3), 51.8 (CH), 28.1 (CH3), 27.8 (CH3). MS-ESI+ for C26H40N5NaO13P+ (M+Na)+, calcd. 684.2252. found 684.2251.
E. Preparation of Compound 6a, 6b or 6c
Compound 5a, 5b or 5c (223 mg, 0.34 mmol) was dissolved in anhydrous CH2Cl2 (7 mL) under a nitrogen atmosphere. Anhydrous DMSO (104 μL, 1.46 mmol) was added and the solution was cooled to −78° C. Trifluoroacetic anhydride (104 μL, 0.74 mmol) was added dropwise and the mixture was stirred for 40 min. N,N-diisopropylethylamine (513 μL, 2.94 mmol) was added and the stirring was continued for 50 min at −78° C. Saturated NaCl solution (20 mL) was added and the mixture removed from the cold bath and diluted with CH2Cl2 (30 mL). Glacial acetic acid (170 μL, 8.75 mmol) was added and the mixture was stirred for 10 min. The layers were separated and the aqueous phase was washed with CH2Cl2 (10 mL). The combined organic phases were washed with 5% aqueous HCl, 3:1 saturated NaCl solution:10% NaHCO3 solution and saturated NaCl solution successively, dried over MgSO4, and concentrated to give compound 6a, 6b or 6c (228 mg, quant.) of suitable purity for further use. 1H NMR (500 MHz, CDCl3): δ 5.86 (m, 1 H), 5.07 (m, 1 H), 4.70-4.64 (m, 2 H), 4.49-4.40 (m, 1 H), 4.27 (m, 1 H), 3.56, m, 4 H), 1.49 (s, 9 H), 1.46 (s, 18 H) ppm. 13C NMR (500 MHz, CDCl3): δ 157.5 (C), 151.1 (C), 150.6 (2 C), 134.6 (C), 132.7 (C), 116.6 (C), 92.0 (C), 84.6 (2 C), 83.6 (C), 78.0 (CH), 76.0 (CH), 70.4 (CH2), 67.9 (CH), 56.2 (CH3) δ6.0 (CH), 28.2 (3CH3), 26.8 (6 CH3) ppm. 31P NMR (500 MHz, CDCl3): δ−6.3 ppm.
F. Preparation of compound 7: (4aR,5aR,11aR,12aS)-1,3,2-Dioxaphosphorino[4′,5′:5,6]pyrano[3,2-g]pteridin-10(4H)-one,8-amino-4-a,5a,6,9,11,11a,12,12a-octahydro-2,12,12-trihydroxy-2-oxide
Compound 6a, 6b or 6c (10 mg, 14.8 μmol was dissolved in dry acetonitrile (0.2 mL) and cooled to 0° C. Bromotrimethylsilane (19.2 μL, 148 μmol) was added dropwise and the mixture was allowed to warm to ambient temperature and stirred for 5 h during which time a precipitate formed. HCl(aq) (10 μl, 37%) was added and the mixture was stirred for a further 15 min. The mixture was centrifuged for 15 min (3000 g) and the resulting precipitate collected. Acetonitrile (0.5 mL) was added and the mixture was centrifuged for a further 15 min. The acetonitrile wash and centrifugation was repeated a further two times and the resulting solid was dried under high vacuum to give compound 7 (4 mg, 75%). 1H NMR (500 MHz, D2O): δ 5.22 (d, J=1.6 Hz, 1H), 4.34 (dt, J=13, 1.6 Hz, 1H), 4.29-4.27 (m, 1H), 4.24-4.18 (m, 1H), 3.94 (br m, 1H), 3.44 (t, J=1.4 Hz, 1H). 31P NMR (500 MHz, D2O): δ −4.8 MS-ESI+ for C10H15N5O8P+, (M+H)+calcd. 364.0653. found 364.0652.
Example 2Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. Coli in the In vitro Synthesis of Moco
In vitro synthesis of Moco was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. Moco synthesis also involved the use of the purified components E. coli MPT synthase, gephyrin, molybdate, ATP, and apo-sulfite oxidase. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. The assay is based on the conversion of cPMP into MPT, the subsequent molybdate insertion using recombinant gephyrin and ATP, and finally the reconstitution of human apo-sulfite oxidase.
As shown in FIG. 1, Moco synthesis from synthetic cPMP was confirmed, and no differences in Moco conversion were found in comparison to E. coli purified cPMP.
Example 3Comparison of Precursor Z (cPMP) Prepared Synthetically to that Prepared from E. coli in the In vitro Synthesis of MPT
In vitro synthesis of MPT was compared using samples of synthetic precursor Z (cPMP) and cPMP purified from E. coli. MPT synthesis also involved the use of in vitro assembled MPT synthase from E. coli. See U.S. Pat. No. 7,504,095 and “Biosynthesis and molecular biology of the molybdenum cofactor (Moco)” in Metal Ions in Biological Systems, Mendel, Ralf R. and Schwarz, Gunter, Informa Plc, 2002, Vol. 39, pages 317-68. Three repetitions of each experiment were performed and are shown in FIGS. 2 and 3.
As shown in FIGS. 2 and 3, MPT synthesis from synthetic cPMP confirmed, and no apparent differences in MPT conversion were found when compared to E. coli purified cPMP. A linear conversion of cPMP into MPT is seen in all samples confirming the identity of synthetic cPMP (see FIG. 2). Slight differences between the repetitions are believed to be due to an inaccurate concentration determination of synthetic cPMP given the presence of interfering chromophores.
Example 4Preparation of Precursor Z (cPMP)
A. Preparation of Starting Materials

B. Introduction of the protected Phosphate

The formation of the cyclic phosphate using intermediate [10] (630 mg) gave the desired product [11] as a 1:1 mixture of diastereoisomers (494 mg, 69%).

C. Oxidation and Overall Deprotection of the Molecule
Oxidation of the secondary alcohol to the gem-diol did prove successful on intermediate [12], but the oxidized product [13] did show significant instability and could not be purified. For this reason, deprotection of the phosphate was attempted before the oxidation. However, the reaction of intermediate [11] with TMSBr led to complete deprotection of the molecule giving intermediate [14]. An attempt to oxidize the alcohol to the gem-diol using Dess-Martin periodinane gave the aromatized pteridine [15].
Oxidation of intermediate [11] with Dess-Martin periodinane gave a mixture of starting material, oxidized product and several by-products. Finally, intermediate [11] was oxidized using the method described Example 1. Upon treatment, only partial oxidation was observed, leaving a 2:1 mixture of [11]/[16]. The crude mixture was submitted to the final deprotection. An off white solid was obtained and analyzed by 1H-NMR and HPLC-MS. These analyses suggest that cPMP has been produced along with the deprotected precursor [11].
Because the analytical HPLC conditions gave a good separation of cPMP from the major impurities, this method will be repeated on a prep-HPLC in order to isolate the final material.
CLIP
BridgeBio Pharma And Affiliate Origin Biosciences Announces FDA Acceptance Of Its New Drug Application For Fosdenopterin For The Treatment Of MoCD Type A
Application accepted under Priority Review designation with Breakthrough Therapy Designation and Rare Pediatric Disease Designation previously grantedThere are currently no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury for infants and children.This is BridgeBio’s first NDA acceptanceSAN FRANCISCO, September 29, 2020 – BridgeBio Pharma, Inc. (Nasdaq: BBIO) and affiliate Origin Biosciences today announced the US Food and Drug Administration (FDA) has accepted its New Drug Application (NDA) for fosdenopterin (previously BBP-870/ORGN001), a cyclic pyranopterin monophosphate (cPMP) substrate replacement therapy, for the treatment of patients with molybdenum cofactor deficiency (MoCD) Type A.The NDA has been granted Priority Review designation. Fosdenopterin has previously been granted Breakthrough Therapy Designation and Rare Pediatric Disease Designation in the US and may be eligible for a priority review voucher if approved. It received Orphan Drug Designation in the US and Europe. This is BridgeBio’s first NDA acceptance.“We want to thank the patients, families, scientists, physicians and all others involved who helped us reach this critical milestone,” said BridgeBio CEO and founder Neil Kumar, Ph.D. “MoCD Type A is a devastating disease with a median survival of less than four years and we are eager for our investigational therapy to be available to patients, who currently have no approved treatment options. BridgeBio exists to help as many patients as possible afflicted with genetic diseases, no matter how rare. We are grateful that the FDA has accepted our first NDA for priority review and we look forward to submitting our second NDA later this year for infigratinib for second line treatment of cholangiocarcinoma.”About Fosdenopterin
Fosdenopterin is being developed for the treatment of patients with MoCD Type A. Currently, there are no approved therapies for the treatment of MoCD Type A, which results in severe and irreversible neurological injury with a median survival between 3 to 4 years. Fosdenopterin is a first-in-class cPMP hydrobromide dihydrate and is designed to treat MoCD Type A by replacing cPMP and permitting the two remaining MoCo synthesis steps to proceed, with activation of MoCo-dependent enzymes and elimination of sulfites.About Molybdenum Cofactor Deficiency (MoCD) Type A
MoCD Type A is an ultra-rare, autosomal recessive, inborn error of metabolism caused by disruption in molybdenum cofactor (MoCo) synthesis which is vital to prevent buildup of s-sulfocysteine, a neurotoxic metabolite of sulfite. Patients are often infants with severe encephalopathy and intractable seizures. Disease progression is rapid with a high infant mortality rate.Those who survive beyond the first few month’s experience profuse developmental delays and suffer the effects of irreversible neurological damage, including brain atrophy with white matter necrosis, dysmorphic facial features, and spastic paraplegia. Clinical presentation that can be similar to hypoxic-ischemic encephalopathy (HIE) or other neonatal seizure disorders may lead to misdiagnosis and underdiagnosis. Immediate testing for elevated sulfite levels and S-sulfocysteine in the urine and very low serum uric acid may help with suspicion of MoCD.About Origin Biosciences
Origin Biosciences, an affiliate of BridgeBio Pharma, is a biotechnology company focused on developing and commercializing a treatment for Molybdenum Cofactor Deficiency (MoCD) Type A. Origin is led by a team of veteran biotechnology executives. Together with patients and physicians, the company aims to bring a safe, effective treatment for MoCD Type A to market as quickly as possible. For more information on Origin Biosciences, please visit the company’s website at www.origintx.com.
About BridgeBio Pharma
BridgeBio is a team of experienced drug discoverers, developers and innovators working to create life-altering medicines that target well-characterized genetic diseases at their source. BridgeBio was founded in 2015 to identify and advance transformative medicines to treat patients who suffer from Mendelian diseases, which are diseases that arise from defects in a single gene, and cancers with clear genetic drivers. BridgeBio’s pipeline of over 20 development programs includes product candidates ranging from early discovery to late-stage development. For more information visit bridgebio.com.
| Clinical data | |
|---|---|
| Trade names | Nulibry |
| Other names | Precursor Z, ALXN1101 |
| License data | US DailyMed: Fosdenopterin |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 150829-29-1 |
| PubChem CID | 135894389 |
| DrugBank | DB16628 |
| ChemSpider | 17221217 |
| UNII | 4X7K2681Y7 |
| KEGG | D11779 |
| ChEMBL | ChEMBL2338675 |
| CompTox Dashboard (EPA) | DTXSID90934067 |
| Chemical and physical data | |
| Formula | C10H14N5O8P |
| Molar mass | 363.223 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILESNC1=NC(=O)C2=C(N[C@@H]3O[C@@H]4COP(=O)(O)O[C@@H]4C(O)(O)[C@@H]3N2)N1 | |
| hideInChIInChI=1S/C10H14N5O8P/c11-9-14-6-3(7(16)15-9)12-4-8(13-6)22-2-1-21-24(19,20)23-5(2)10(4,17)18/h2,4-5,8,12,17-18H,1H2,(H,19,20)(H4,11,13,14,15,16)/t2-,4-,5+,8-/m1/s1Key:CZAKJJUNKNPTTO-AJFJRRQVSA-N |
//////////Fosdenopterin hydrobromide, ホスデノプテリン臭化水素酸塩水和物 , ALXN1101 HBr, UNII-X41B5W735T, X41B5W735T, D11780, BBP-870/ORGN001, Priority Review designation, Breakthrough Therapy Designation, Rare Pediatric Disease Designation, Orphan Drug Designation, molybdenum cofactor deficiency, ALXN-1101, WHO 11150, FDA 2021, APPROVALS 2021
#Fosdenopterin hydrobromide, #ホスデノプテリン臭化水素酸塩水和物 , #ALXN1101 HBr, #UNII-X41B5W735T, X41B5W735T, #D11780, #BBP-870/ORGN001, #Priority Review designation, #Breakthrough Therapy Designation, #Rare Pediatric Disease Designation, #Orphan Drug Designation, #molybdenum cofactor deficiency, #ALXN-1101, #WHO 11150, #FDA 2021, #APPROVALS 2021
C1C2C(C(C3C(O2)NC4=C(N3)C(=O)NC(=N4)N)(O)O)OP(=O)(O1)O.O.O.Br

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}