
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
RADOTINIB

RADOTINIB
- Molecular FormulaC27H21F3N8O
- Average mass530.504 Da
4-Methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-{[4-(2-pyrazinyl)-2-pyrimidinyl]amino}benzamide
4-methyl-N-[3-(4-methylimidazole-l-yl)-5-trifluoromethyl-phenyl] –
3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzamide
9242
926037-48-1[RN]
| 926037-48-1 (Radotinib); 926037-85-6 (Radotinib 2HCl); |
Benzamide, 4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(2-pyrazinyl)-2-pyrimidinyl]amino]-
I284LJY110, IY5511
UNII-I284LJY110
радотиниб
رادوتينيب
雷度替尼
MOA:Bcr-Abl tyrosine kinase inhibitor
Indication:Chronic myeloid leukemia (CML )
Company:IL-Yang (Originator)
IY-5511; IY-5511A3001
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-01-05 | Marketing approval | Supect | Chronic myeloid leukemia (CML ) | Capsule | 100 mg/200 mg | IL-Yang |
Radotinib dihydrochloride was approved by Korea Food and Drug Administration (KFDA) on January 5, 2012. It was developed and marketed as Supect® by IL-Yang in KR.
Radotinib dihydrochloride is a second-generation tyrosine kinase inhibitor of Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR). It is indicated for the second-line treatment of patients with Philadelphia chromosome-positive (Ph+) CML that is refractory to Imatinib mesilate.
Supect® is available as capsule for oral use, containing 100 mg or 200 mg of free Radotinib. The recommended dose is 400 mg twice daily.
Radotinib (INN; trade name Supect), and sometimes referred to by its investigational name IY5511, is a drug for the treatment of different types of cancer, most notably Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML)[1] with resistance or intolerance of other Bcr-Abl tyrosine-kinase inhibitors, such as patients resistant or intolerant to imatinib.
Radotinib is being developed by Ilyang Pharmaceutical Co., Ltd of South Korea[2] and is co-marketed by Daewoong Pharmaceutical Co. Ltd, in South Korea.[3] Radotinib completed a multi-national Phase II clinical trial study in 2012[4] and in August 2011, Ilyang initiated a Phase III, multinational, multi-center, open-label, randomized study for first-line indication.[5] Its mechanism of action involves inhibition of the Bcr-Abl tyrosine kinase and of platelet-derived growth factor receptor (PDGFR).[6]
In January 2012, radotinib hydrochloride (marketed as Supect ®) obtained its approval from the KFDA (Korea Food and Drug Administration) for the treatment of patients with Philadelphia chromosomepositive chronic myeloid leukemia (CML) who have become resistant to existing drugs such as Gleevec, Tasigna and Sprycel. Originally developed by IL-YANG pharmaceuticals of South Korea as an orally second-generation tyrosine kinase inhibitor, the drug inhibits both Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR).
Chemical Synthesis
Because of the structural similarity of radotinib to that of nilotinib (Tasigna ®), the process-scale synthetic route (which is depicted in the scheme) is capable of furnishing both drugs.Claisen condensation of commerical 2-acetylpyrazine (142) with N,N-dimethylformamide dimethylacetal gave rise to the enamino ketone 143 in 81% yield. Under basic conditions, vinylogous amide 143 was coupled with commercial guanidine nitrate 144187 to produce aminopyridine 145. Subsequent condensation with commercial aniline (146) by means of potassium t-butoxide in THF constructed radotinib 147 in 85% yield as the free base, and this material could be converted to the radotinib dihydrochloride (XXII) upon exposure to concentrated hydrochloric acid in chilled acetone. 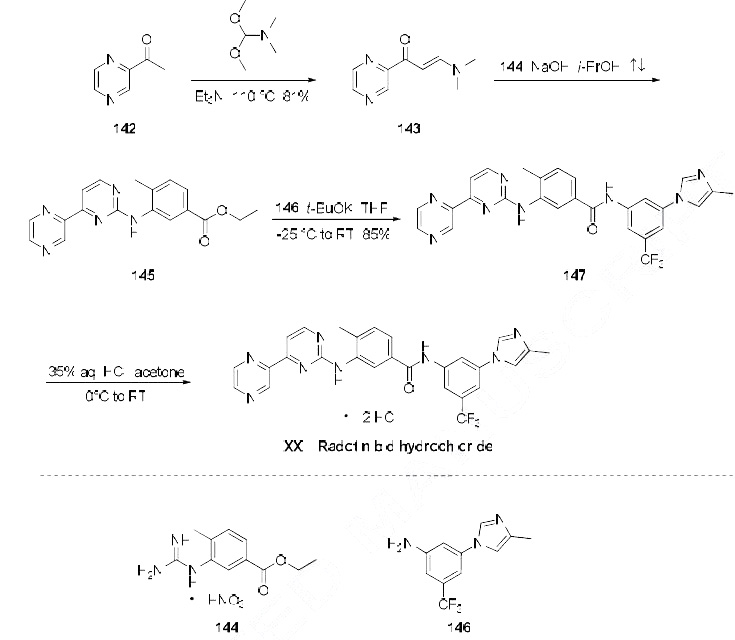
PATENT1.
WO2007018325A1 / US7501424B2.
https://patents.google.com/patent/WO2007018325A1/en
PATENT
WO2010018895A1 / CN101648946A.
https://patents.google.com/patent/WO2010018895A1/en
The compound represented by Formula 1 was disclosed in Korea Patent Registration
No. 10-0674813. A preferred compound according to Formula 1 includes 4-methyl-N- [3-(4-methylimidazole- 1 -yl)-5-trifluoromethyl-phenyl] -3-(4-pyrazine-2-yl -pyrimidine-2-yl amino)benzamide. It has been known that the compound represented by Formula 1 can inhibit at least one kind of tyrosine kinase, for example, c-Abl, Bcr- AbI, and receptor tyrosine kinases (PDGF-R, Flt3, VEGF-R, EGF-R and c-Kit). Accordingly, the compound represented by Formula 1 may be used for treatment of various kinds of cancers in a warm blooded animal, such as lung cancer, stomach cancer, colon cancer, pancreatic cancer, liver cancer, prostate cancer, breast cancer, chronic or acute leukemia, hematological malignancy, brain tumor, bladder cancer, rectal cancer, uterine cervical cancer, lymphoma, etc.
[7] According to a conventional method, the compound represented by Formula 1 is synthesized through hydrolysis of ethyl ester into carboxylic acid and then a reaction with aniline, and herein, diethyl cyano phosphonate is used as a coupling agent (see Reaction Scheme 1).
[8] [Reaction Scheme 1]
NsOIf


( 2 ) { s :

Diethyl cyano phosphate

( 1 )
[10] The above method requires a process of hydrolyzing ethyl ester (2) into carboxylic acid (3). In order to obtain the compound represented by Formula 3 as shown in Reaction Scheme 1, a preparation process and a purifying process require a long time. Also, in the condensation reaction, there have been problems such as high production cost due to a low yield (30 to 40%) of the compound represented by Formula 1. Especially, it is very difficult to treat carboxylic acid (3) after purification and reaction, due to its very low solubility in general organic solvent. Also, diethyl cyano phosphonate used for the condensation reaction is an expensive reagent, and an environmentally harmful and very toxic material, which has LD50 values of 25mg/Kg and 4mg/Kg in mice and rabbits (that is, rodents), respectively. Therefore, there is a requirement for an alternative method of conveniently, consistently, efficiently and rapidly preparing a high-purity compound (represented by Formula 1) with low production cost in high yield, which is not harmful for humans and the environment.


Example 2
[69] Synthesis of 4-methyl-N-[3-(4-methylimidazole-l-yl)-5-trifluoromethyl-phenyl] –
3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzamide
[70]
[71] Method A
[72] A pale yellow solid final compound (18.7g, yield 85%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4-methyl -3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)-benzoic acid ethyl ester in a similar manner as described in Method A of Example 1, except that 4-methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino) -benzoic acid ethyl ester (15.3g, 45.60mmol) was used, instead of 4-methyl-3-(4-thiazole-2-yl-pyrimidine-2-yl amino)benzoic acid ethyl ester.
[73] 1H-NMR(DMSOd , δ= 2.21(s,3H), 2.38(s,3H), 7.35(s,lH), 7.39(s,lH), 7.54(s,lH),
7.63(d,lH), 7.75(d,lH), 8.14(d,2H), 8.38(d,2H), 8.54(d,2H), 8.68(s,lH), 9.06(s,lH), 9.45(s, IH), 10.56(s,lH)
[74]
[75] Method B
[76] A pale yellow solid final compound (18.3g, yield 83%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4-methyl -3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)-benzoic acid methyl ester in a similar manner as described in Method A of Example 1, except that 4-methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino) -benzoic acid methyl ester (14.7g, 45.60mmol) was used, instead of 4-methyl-3-(4-thiazole-2-yl-pyrimidine-2-yl amino)benzoic acid ethyl ester.
[77]
[78] Method C
[79] A pale yellow solid final compound (17.2g, yield 78%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4- methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzoic acid methyl ester (14.7g, 45.60mmol) in a similar manner as described in Method A of Example 1, except that sodium tert-butoxide was used, instead of potassium tert-butoxide.
[80]
[81] Method D
[82] A pale yellow solid final compound (16. Ig, yield 73%) was obtained by reacting
3-(4-methyl-imidazole-l-yl)-5-trifluoromethyl-phenylamine (1Og, 41.46mmol) with 4- methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino)benzoic acid phenyl ester in a similar manner as described in Method A of Example 1, except that 4-methyl-3-(4-pyrazine-2-yl-pyrimidine-2-yl amino) -benzoic acid phenyl ester (17.5g, 45.60mmol) was used, instead of 4-methyl-3-(4-thiazole-2-yl-pyrimidine-2-yl amino)benzoic acid ethyl ester.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0968089614001230

Radotinib hydrochloride (Supect) In January 2012, radotinib hydrochloride (marketed as Supect) obtained its approval from the KFDA (Korea Food and Drug Administration) for the treatment of patients with Philadelphia chromosome-positive chronic myeloid leukemia (CML) who have become resistant to existing drugs such as Gleevec, Tasigna and Sprycel.181 Originally developed by IL-YANG pharmaceuticals of South Korea as an oral second-generation tyrosine kinase inhibitor, the drug inhibits both Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR).182 Because of the structural similarity of radotinib to that of nilotinib (Tasigna), the processscale synthetic route (which is depicted in Scheme 27) is capable of furnishing both drugs.183–185 Claisen condensation of commerical 2-acetylpyrazine (142) with N,N-dimethylformamide dimethylacetal gave rise to the enamino ketone 143 in 81% yield.186 Under basic conditions, vinylogous amide 143 was coupled with commercial guanidine nitrate 144187 to produce aminopyridine 145. 184 Subsequent condensation with commercial aniline (146) by means of potassium t-butoxide in THF constructed radotinib 147 in 85% yield as the free base, and this material could be converted to the radotinib dihydrochloride (XXII) upon exposure to concentrated hydrochloric acid in chilled acetone.185
181. Droppert, P. In Biotech Strategy Blog: http://biotechstrategyblog.com/2012/01/ radotinib-approved-in-south-korea-for-cml.html/, 2012.
182. Radotinib hydrochloride http://www.cancer.gov/drugdictionary?cdrid= 723999.
183. Davies, S.; Bolos, J.; Serradell, N.; Bayes, M. Drugs Future 2007, 32, 17.
184. Kim, D.-Y.; Cho, D.-J.; Lee, G.-Y.; Kim, H.-Y.; Woo, S.-H.; Kim, Y.-S.; Lee, S.-A.; Han, B.-C. WO Patent 2007/018325 A1, 2007.
185. Kim, D. Y.; Cho, D. J.; Lee, G. Y.; Kim, H. Y.; Woo, S. H. WO Patent 2010/018895 A1, 2010.
///////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Joanne Bronson; Amelia Black; T. G. Murali Dhar; Bruce A. Ellsworth; J. Robert Merritt (2013). “To Market, To Market – 2012”. Radotinib (Anticancer). Annual Reports in Medicinal Chemistry. Vol. 48. pp. 523–524. doi:10.1016/b978-0-12-417150-3.00028-4. ISBN 9780124171503.
- ^ “Il-Yang Pharmaceutical”.
- ^ http://www.dailypharm.com/Users/News/EnglishNews.html?NewsID=3108&nStart=1023&mode=&searchValue=[dead link]
- ^ Kim SH, Menon H, Jootar S, Saikia T, Kwak JY, Sohn SK, Park JS, Jeong SH, Kim HJ, Kim YK, Oh SJ, Kim H, Zang DY, Chung JS, Shin HJ, Do YR, Kim JA, Kim DY, Choi CW, Park S, Park HL, Lee GY, Cho DJ, Shin JS, Kim DW (2014). “Efficacy and safety of radotinib in chronic phase chronic myeloid leukemia patients with resistance or intolerance to BCR-ABL1 tyrosine kinase inhibitors”. Haematologica. 99 (7): 1191–6. doi:10.3324/haematol.2013.096776. PMC 4077080. PMID 24705186.
- ^ https://clinicaltrials.gov/ct2/show/NCT01511289?term=radotinib&rank=1
- ^ “Radotinib hydrochloride”. NCI Drug Dictionary. National Cancer Institute. 2011-02-02.
| Clinical data | |
|---|---|
| Trade names | Supect |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 926037-48-1 |
| PubChem CID | 16063245 |
| ChemSpider | 17222861 |
| UNII | I284LJY110 |
| CompTox Dashboard (EPA) | DTXSID90239069 |
| Chemical and physical data | |
| Formula | C27H21F3N8O |
| Molar mass | 530.515 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Patent
Publication numberPriority datePublication dateAssigneeTitle
WO2003066613A1 *2002-02-072003-08-14Novartis AgN-phenyl-2-pyrimidine-amine derivatives
WO2004005281A1 *2002-07-052004-01-15Novartis AgInhibitors of tyrosine kinases
KR100674813B1 *2005-08-052007-01-29일양약품주식회사N-phenyl-2-pyrimidine-amine derivatives and process for the preparation thereof
Publication numberPriority datePublication dateAssigneeTitle
US9132126B22011-04-192015-09-15Il-Yang Pharm. Co., Ltd.Phenyl-isoxazole derivatives and preparation process thereof
KR20180032784A *2016-09-232018-04-02재단법인 대구경북첨단의료산업진흥재단Novel imidazolyl pyrimidine derivatives, preparation method thereof, and pharmaceutical composition for use in preventing or treating cancer containing the same as an active ingredient
Family To Family Citations
KR101956586B1 *2012-03-272019-03-11일양약품주식회사Pharmaceutical composition and preparation method thereof
////////////////////RADOTINIB, UNII-I284LJY110, радотиниб , رادوتينيب , 雷度替尼 , IY5511, IY 5511, korea 2012, Chronic myeloid leukemia
Cyclobenzaprine


Cyclobenzaprine
- Molecular FormulaC20H21N
- Average mass275.387 Da
- MK-130
- TNX-102
1-(3-Dimethylaminopropylidene)-2,3:6,7-dibenzo-4-suberene
1-Propanamine, 3-(5H-dibenzo[a,d]cyclohepten-5-ylidene)-N,N-dimethyl-[ACD/Index Name]
206-145-8[EINECS]
3-(5H-Dibenzo[a,d]cyclohepten-5-ylidene)-N,N-dimethyl-1-propanamine
303-53-7[RN]
5-(3-Dimethylaminopropylidene)dibenzo[a,e]cycloheptatriene
циклобензаприн[Russian][INN]
سيكلوبنزابرين[Arabic][INN]
环苯扎林[Chinese][INN]
Cyclobenzaprine, CAS Registry Number: 303-53-7
CAS Name: 3-(5H-Dibenzo[a,d]cyclohepten-5-ylidene)-N,N-dimethyl-1-propanamine
Additional Names:N,N-dimethyl-5H-dibenzo[a,d]cyclohepten-D5,g-propylamine; 5-(3-dimethylaminopropylidene)dibenzo[a,e]cycloheptatriene; 1-(3-dimethylaminopropylidene)-2,3:6,7-dibenzo-4-suberene; proheptatriene
Manufacturers’ Codes: MK-130; Ro-4-1577; RP-9715
Molecular Formula: C20H21N, Molecular Weight: 275.39
Percent Composition: C 87.23%, H 7.69%, N 5.09%
Literature References: Prepn: GB858187 (1961 to Hoffmann-La Roche); Villani et al.,J. Med. Pharm. Chem.5, 373 (1962); Winthrop et al.,J. Org. Chem.27, 230 (1962). Pharmacology: C. D. Barnes, W. L. Adams, Neuropharmacology17, 445 (1978); N. N. Share, ibid. 721; and toxicology: J. Metysova et al.,Arch. Int. Pharmacodyn. Ther.144, 481 (1963). Metabolism: G. Belvedere et al.,Biomed. Mass Spectrom.1, 329 (1974); H. B. Hucker et al.,Drug Metab. Dispos.6, 184 (1978). Bioavailability: eidem,J. Clin. Pharmacol.17, 719 (1977). Clinical studies: J. V. Basmajian, Arch. Phys. Med. Rehabil.5, 58 (1978); B. R. Brown, J. Womble, J. Am. Med. Assoc.240, 1151 (1978). Comprehensive description: M. L. Cotton, G. R. B. Down, Anal. Profiles Drug Subs.17, 41-72 (1988).
Properties: bp1 175-180°. uv max: 224, 289 nm (log e 4.57, 4.02), (Villani et al.)
Boiling point: bp1 175-180°
Absorption maximum: uv max: 224, 289 nm (log e 4.57, 4.02), (Villani et al.)
Derivative Type: Hydrochloride
CAS Registry Number: 6202-23-9
Trademarks: Flexeril (Merck & Co.); Flexiban (Merck & Co.)
Molecular Formula: C20H21N.HCl, Molecular Weight: 311.85
Percent Composition: C 77.03%, H 7.11%, N 4.49%, Cl 11.37%
Literature References: Use as muscle relaxant: N. N. Share, FR2100873 (1972 to Frosst), C.A.78, 47801n (1973).
Properties: Crystals from isopropanol, mp 216-218°. Soly in water: >20 g/100 ml. Freely sol in water, methanol, ethanol; sparingly sol in isopropanol; slightly sol in chloroform, methylene chloride. Practically insol in hydrocarbons. uv max: 226, 295 nm (e 52300, 12000). LD50 in mice (mg/kg): 35 i.v., 250 orally (Metysova).
Melting point: mp 216-218°
Absorption maximum: uv max: 226, 295 nm (e 52300, 12000)
Toxicity data: LD50 in mice (mg/kg): 35 i.v., 250 orally (Metysova)
Therap-Cat: Muscle relaxant (skeletal).
Keywords: Muscle Relaxant (Skeletal).
Cyclobenzaprine, a centrally-acting muscle relaxant, was first synthesized in 196111 and has been available for human use since 1977.10 It was initially studied for use as antidepressant given its structural similarity to tricyclic antidepressants – it differs from Amitriptyline by only a single double bond.11,10 Since its approval, it has remained relatively popular as an adjunctive, short-term treatment for acute skeletal muscle spasms secondary to musculoskeletal injury.
Cyclobenzaprine (sold under the brand name Flexeril, among others) is a medication used for muscle spasms from musculoskeletal conditions of sudden onset.[6] It is not useful in cerebral palsy.[6] It is taken by mouth.[6] Use is not recommended for more than a few weeks.[6]
Common side effects include headache, feeling tired, dizziness, and dry mouth.[6] Serious side effects may include an irregular heartbeat.[6] There is no evidence of harm in pregnancy, but it has not been well studied in this population.[6] It should not be used with an MAO inhibitor.[6] How it works is unclear.[6]
Cyclobenzaprine was approved for medical use in the United States in 1977.[6] It is available as a generic medication.[6] In 2019, it was the 45th most commonly prescribed medication in the United States, with more than 15 million prescriptions.[7][8] It was not available in the United Kingdom as of 2012.[9]
Synthesis Reference
Villani, F.J.; US. Patent 3,409,640; November 5,1968; assigned to Schering Corporation.
Paper
By: Gowda, Narendra B.; Rao, Gopal Krishna; Ramakrishna, Ramesha A.
Tetrahedron Letters (2010), 51, (43), 5690-5693.
https://www.sciencedirect.com/science/article/abs/pii/S0040403910014668
A simple and convenient protocol for deoxygenation of aliphatic and aromatic N-oxides to the corresponding amines in good to excellent yield using sodium borohydride–Raney nickel in water is reported. Other functional moieties such as alkenes, halides, ethers, and amides are unaffected under the present reaction condition.
Graphical abstract

Cyclobenzaprine N-oxide, CAS RN: 6682-26-4
Dissolve (1 mmol) of cyclobenzaprine N-oxide in 2.5 mL of water at 60 °C. 2. Add Raney nickel (0.10 g, W6 grade) to the solution. 3. Stir the reaction mixture for 10 minutes. 4. Add (2 mmol) of sodium borohydride slowly in portions over 15-20 minutes to the reaction mixture. 5. Stir the reaction mixture at the same temperature for 2.5 hours (the completion of the reaction as monitored by TLC). 6. Once the reaction is completed, add chloroform (50 mL) to the reaction mixture. 7. Filter the resulted mixture to remove Raney nickel. 8. Dry the chloroform layer over anhydrous magnesium sulfate. 9. Filter the reaction mixture. 10. Evaporate the solvent under vacuum. 11. Purify the obtained residue through short path flash chromatography with silica gel and chloroform.
1H NMR (400 MHz, CDCl3) δ: 1.12 (s, 6H, N-CH3), 1.23- 1.34 (m, 4H, CH2), 4.58 (t, J= 4.0 Hz, 1H, CH), 5.82(d, J= 4.0 Hz, 2H, CH), 6.21- 6.33 (m, 8H, ArH).
13C NMR (100 MHz, CDCl3) δ: 27.89, 45.93, 60.12, 127.40, 127.55, 128.30, 128.59, 128.92, 129.33, 129.45, 129.67, 131.74, 131.96, 132.40, 134.63, 135.39, 137.97, 142.95, 143.30.
SYN
PATENT
https://patents.google.com/patent/WO2012098563A2/en
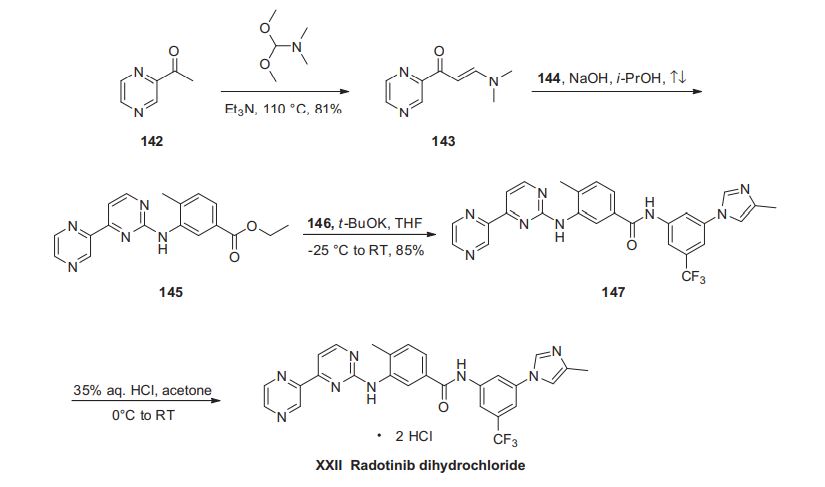
Cyclobenzaprine hydrochloride, chemically known as 5-(3-dimethylaminopropylidene)- dibenzo (a,e) cycloheptatriene hydrochloride (Formula I),

Formula I is a commonly prescribed tricyclic amine having muscle relaxant pharmaceutical activity. After sustaining an injury, muscle spasms may occur to stabilize the affected body part and prevent further damage. Cyclobenzaprine hydrochloride is used to treat such muscle spasm associated with acute, painful musculoskeletal conditions.
Few multistep processes for the preparation of this tricyclic amine are already available in the literature which involves isolation and purification of intermediate compounds. The conventional route of synthesis as reported in US3454643, ES8201950 includes preparation of Grignard reagent (GR) of 3-dimethylaminopropyl chloride in a first step, reacting with 5-dibenzosuberenone (Formulall) in a second step. The reaction mass was extracted with benzene, solid obtained was recrystallized from alcohol to produce 5- hydroxy intermediate (Formula III) and further dehydrated in third step using acetyl chloride or acetic anhydride in presence of chloroform as a solvent medium followed by purging HC1 gas to produce hydrochloride salt (Formula I). CH,
CI-(CH2)3 NS
CH,
Dimeth laminopropyl chloide

Di methy lam i nopropy I 5-dibenzosubrenone – y roxy compoun magnesium chloide
(Formula II) (Formula III)

Cyclobenzaprine base Cyclobenzaprine hydrochloride
(Formula IV) (Formula I)
The multistep synthesis is cumbersome and use of hazardous solvents and reagents like chloroform, benzene and acetyl chloride etc are not recommended for the preparation of pharmaceutical substances.
J. Org. Chem. Vol. 27, 230-240 (1961) also portrayed similar procedure for the synthesis of cyclobenzaprine hydrochloride, wherein 5-hydroxy compound of formula III was isolated and recrystallized before dehydration reaction.
Synthetic Comm. 11 (3), 241-246 (1981) described a process which involves isolation and purification of the intermediate at magnesium -complex stage. Hydrolysis of the isolated complex afforded desired tricyclic amine. GB858186 and GB858187 jointly described a process which comprises preparation of 5- hydroxy compound (Formula III) and subsequent conversion of the same to cyclobenzaprine hydrochloride. However the overall yield reported is significantly low.
In a different approach, a high temperature dehydrogenation of amitriptyline base resulting in formation of cyclobenzaprine hydrochloride is reported in Indian patent application 387/CHE/2005.

. EXAMPLE:
In a reaction vessel, THF (1 10ml), magnesium turnings 20gm (0.823mole) were charged and the mixture was warmed to 45-55°C for 20 min. A solution of l OOgm (0.823mole) of 3-dimethylaminopropyl chloride prepared in 1 10ml THF was added dropwise to the reaction mixture by controlling the reflux generated due to reaction initiation and maintained for 2hrs. The formed Grignard reagent was then cooled to 0-5°C and a solution of lOOgm (0.485mole) 5-dibenzosuberenone prepared in 220ml THF was charged to the reaction mass at temperature below 10°C. The reaction mass was stirred for 45 min at temperature 10-15°C. The absence of 5-dibenzosuberenone was checked by TLC and 770ml of 20% aq. HC1 was charged to the reaction mass at a temperature below 10°C. The reaction mass was then heated to 70-80°C for 3 hrs. The acidic mass was neutralized by using aqueous Na2C03 solution and extracted with 900ml methylene dichloride. The solvent was removed completely under reduced pressure and oil thus formed was dissolved in 450ml IPA and acidified with 240 ml of 20% IPA .HC1 solution and stirred for 2 hrs at 0-5°C for complete precipitation. The precipitate is filtered, recrystallized from IPA (800 ml) and dried to obtain 1 18 gm (78%) white crystalline cyclobenzaprine hydrochloride with purity 99.93% by HPLC.

PATENT
PATENT
CN 111393305
CLIP
Muscle Relaxants
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Cyclobenzaprine
Cyclobenzaprine, N,N-dimethyl-3-(dibenzo[a,d]cyclohepten-5-ylidene) propylamine (15.3.9), is synthesized by reacting 5H-dibenzo[a,d]cyclohepten-5-one with 3-dimethylaminopropylmagnesium chloride and subsequent dehydration of the resulting carbinol (15.3.8) in acidic conditions into cyclobenzaprine (15.3.9) [30–32].

Cyclobenzaprine is structurally similar to tricyclic antidepressants. It acts at the brain stem level. It is used as an adjuvant agent for relieving muscle spasms associated with severe diseased conditions of the muscle. A synonym of this drug is flexeril.
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical use
Cyclobenzaprine is used, in conjunction with physical therapy, to treat muscle spasms that occur because of acute musculoskeletal conditions.[10] After sustaining an injury, muscle spasms to stabilize the affected body part occur, which may increase pain to prevent further damage. Cyclobenzaprine is used to treat such muscle spasms associated with acute, painful musculoskeletal conditions.[11] It decreases pain in the first two weeks,[12][13] peaking in the first few days, but has no proven benefit after two weeks.[12][14] Since no benefit is proven beyond that, therapy should not be continued long-term.[11] It is the best-studied muscle relaxer.[12] It is not useful for spasticity due to neurologic conditions such as cerebral palsy.[11][15]
A 2004 review found benefit for fibromyalgia symptoms, with a reported number needed to treat of 4.8 (meaning that 1 person out of every 4.8 benefits from treatment) for pain reduction, but no change in fatigue or tender points.[16] A 2009 Cochrane review found insufficient evidence to justify its use in myofascial pain syndrome.[17] It may also be used along with other treatments for tetanus.[18]
Side effects
Cyclobenzaprine results in increased rates of drowsiness (38%), dry mouth (24%), and dizziness (10%).[14] Drowsiness and dry mouth appear to intensify with increasing dose.[19] The sedative effects of cyclobenzaprine are likely due to its antagonistic effect on histamine, serotonin, and muscarinic receptors.[medical citation needed]
Agitation is a common side effect observed, especially in the elderly. Some experts[who?] believe that cyclobenzaprine should be avoided in elderly patients because it can cause confusion, delirium, and cognitive impairment.[20][21] In general, the National Committee for Quality Assurance recommends avoiding the use of cyclobenzaprine in the elderly because of the potential for more severe side effects.[22]
Dysphagia, a life-threatening side-effect, may rarely occur.[23] Treatment protocols and support should follow the same as for any structurally related tricyclic, such as tricyclic antidepressants.[24]
Overdose
The most common effects of overdose are drowsiness and tachycardia.[11] Rare but potentially critical complications are cardiac arrest, abnormal heart rhythms, severe low blood pressure, seizures, and neuroleptic malignant syndrome.[11] Life-threatening overdose is rare,[11] however, as the median lethal dose is about 338 milligrams/kilogram in mice and 425 mg/kg in rats.[11] The potential harm is increased when central nervous system depressants and antidepressants are also used; deliberate overdose often includes alcohol among other drugs.[11]
Interactions
Cyclobenzaprine has major contraindications with monoamine oxidase inhibitors (MAOIs). At least one study also found increased risk of serotonin syndrome when cyclobenzaprine was taken with the serotonergic drugs duloxetine or phenelzine.[25]
These substances may interact with cyclobenzaprine:
- Central nervous system depressants (e.g. alcohol, opioids, benzodiazepines, nonbenzodiazepines, phenothiazines, carbamates, barbiturates, major tranquilizers)
- Monoamine oxidase inhibitors taken within two weeks of cyclobenzaprine may result in serious, life-threatening side effects.[11]
Cyclobenzaprine may affect the medications used in surgical sedation and some surgeons request that patients temporarily discontinue its use prior to surgery.[26]
Pharmacology
Cyclobenzaprine is a centrally acting muscle relaxant.[27] Cyclobenzaprine is a 5-HT2 receptor antagonist; it relieves muscle spasm through action on the central nervous system at the brain stem, rather than targeting the peripheral nervous system or muscles themselves.[28]
Pharmacodynamics
| Site | CBP | NCBP | Action | Ref |
|---|---|---|---|---|
| 5-HT1A | 5.3 | 3.2 | Agonist | [29] |
| 5-HT2A | 5.2 | 13 | Antagonist | [29] |
| 5-HT2B | 100 | ??? | Antagonist | [29] |
| 5-HT2C | 5.2 | 43 | Antagonist | [29] |
| α1A | 5.6 | 34 | ND | [29] |
| α2A | 4.3 | 6.4 | Antagonist | [29] |
| α2B | 21 | 150 | ND | [29] |
| α2C | 21 | 48 | ND | [29] |
| H1 | 1.3 | 5.6 | ND | [29] |
| M1 | 7.9 | 30 | ND | [29] |
| Values are Ki (nM), unless otherwise noted. The smaller the value, the more strongly the drug binds to the site. |
Pharmacokinetics
Cyclobenzaprine has an oral bioavailability of about 55% and approximately 93% is bound to proteins in plasma. The half-life of the drug is 18 hours and it has a plasma clearance of 0.7 litres per minute.[27][30][31]
Comparison to other medications
Cyclobenzaprine has been found to be not inferior to tizanidine, orphenadrine, and carisoprodol in the treatment of acute lower back pain, although none have been proven to be effective for long-term use (beyond two weeks of treatment). No differences in pain or spasm scores were noted among these agents, nor when compared to benzodiazepines.[32] However, nonbenzodiazepine (including cyclobenzaprine) treatment was found to have a lower risk of medication abuse and continuation of use against medical advice.[medical citation needed] Side effects such as sedation and ataxia are also less pronounced with nonbenzodiazepine antispasmodics.[medical citation needed]
In a study on the treatment of musculoskeletal pain treatment with cyclobenzaprine alone or in combination with ibuprofen, no significant differences in pain scores were noted among the three treatment groups. Peak benefit was found to occur on day seven of the treatment for all groups.[33]
Formulations

Cyclobenzaprine 10mg tablets
By mouth, cyclobenzaprine is marketed as Apo-Cyclobenzaprin, Fexmid, Flexeril and Novo-Cycloprine. It is available in generic form. A once-a-day, extended-release formulation, Amrix, is available.[34] Cyclobenzaprine is also used by compounding pharmacies in topical creams.[citation needed]
References
- ^ Micromedex® 2010 – DRUGDEX Evaluations (Cyclobenzaprine Hydrochloride)
- ^ “Cyclobenzaprine Hydrochloride Tablets USP Revised: April 2005 Rx only”. nih.gov. Retrieved 1 October 2016.
- ^ Teva Pharmaceuticals USA, Inc (May 2016). “AMR40470 (Amrix) Prescribing Information” (PDF).
- ^ U.S. Food and Drug Administration. “NDA 17-821/S-045 Flexeril (Cyclobenzaprine HCl) Tablets” (PDF).
- ^ Teva Pharmaceuticals USA, Inc (May 2016). “AMR40470 (Amrix) Prescribing Information” (PDF).
- ^ Jump up to:a b c d e f g h i j k “Cyclobenzaprine Monograph for Professionals”. Drugs.com. AHFS. Retrieved 22 December 2018.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Cyclobenzaprine – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ “Fibromyalgia, psychiatric comorbidity, and the somatosensory cortex”. British Journal of Medical Practitioners. 5 (2): a522. 2012.
- ^ Yang YW, Macdonald JB, Nelson SA, Sekulic A (December 2017). “Treatment of vismodegib-associated muscle cramps with cyclobenzaprine: A retrospective review”. Journal of the American Academy of Dermatology. 77 (6): 1170–1172. doi:10.1016/j.jaad.2016.12.017. PMID 29132849. S2CID 8265576.
- ^ Jump up to:a b c d e f g h i “Cyclobenzaprine- cyclobenzaprine hydrochloride tablet, film coated”. DailyMed. 30 December 2019. Retrieved 26 September 2020.
- ^ Jump up to:a b c Chou R, Peterson K, Helfand M (August 2004). “Comparative efficacy and safety of skeletal muscle relaxants for spasticity and musculoskeletal conditions: a systematic review”. Journal of Pain and Symptom Management. 28 (2): 140–75. doi:10.1016/j.jpainsymman.2004.05.002. PMID 15276195.
- ^ van Tulder MW, Touray T, Furlan AD, Solway S, Bouter LM (2003). Van Tulder MW (ed.). “Muscle relaxants for non-specific low back pain”. The Cochrane Database of Systematic Reviews. 2 (2): CD004252. doi:10.1002/14651858.CD004252. PMC 6464310. PMID 12804507.
- ^ Jump up to:a b Browning R, Jackson JL, O’Malley PG (July 2001). “Cyclobenzaprine and back pain: a meta-analysis”. Archives of Internal Medicine. 161 (13): 1613–20. doi:10.1001/archinte.161.13.1613. PMID 11434793.
- ^ Ashby P, Burke D, Rao S, Jones RF (October 1972). “Assessment of cyclobenzaprine in the treatment of spasticity”. Journal of Neurology, Neurosurgery, and Psychiatry. 35 (5): 599–605. doi:10.1136/jnnp.35.5.599. PMC 494138. PMID 4563483.
- ^ Tofferi JK, Jackson JL, O’Malley PG (February 2004). “Treatment of fibromyalgia with cyclobenzaprine: A meta-analysis”. Arthritis and Rheumatism. 51 (1): 9–13. doi:10.1002/art.20076. PMID 14872449.
- ^ Leite FM, Atallah AN, El Dib R, Grossmann E, Januzzi E, Andriolo RB, da Silva EM (July 2009). “Cyclobenzaprine for the treatment of myofascial pain in adults”. The Cochrane Database of Systematic Reviews (3): CD006830. doi:10.1002/14651858.CD006830.pub3. PMC 6481902. PMID 19588406.
- ^ Smith BT (2014). Pharmacology for Nurses. Jones & Bartlett Publishers. p. 122. ISBN 9781449689407.
- ^ “Flexeril: Side effects”. RxList.com. Archived from the original on 12 September 2008. Retrieved 22 February 2010.
- ^ “Long-term Use of Cyclobenzaprine for Pain: A Review of the Clinical Effectiveness”. CADTH Rapid Response Reports. Ottawa, Ontario: Canadian Agency for Drugs and Technologies in Health. 23 February 2015. PMID 25763449.
- ^ Potentially inappropriate medications for the elderly according to the revised Beers criteria. 2012. Duke Clinical Research Institute website. [1]
- ^ “High risk medications” (PDF). National Committee for Quality Assurance. Archived from the original (PDF) on 1 February 2010. Retrieved 22 February 2010.
- ^ “MEDICATIONS AND DYSPHAGIA/ SWALLOWING RISKS” (PDF).
- ^ Chabria SB (July 2006). “Rhabdomyolysis: a manifestation of cyclobenzaprine toxicity”. Journal of Occupational Medicine and Toxicology. 1 (1): 16. doi:10.1186/1745-6673-1-16. PMC 1540431. PMID 16846511.
- ^ Keegan MT, Brown DR, Rabinstein AA (December 2006). “Serotonin syndrome from the interaction of cyclobenzaprine with other serotoninergic drugs”. Anesthesia and Analgesia. 103 (6): 1466–8. doi:10.1213/01.ane.0000247699.81580.eb. PMID 17122225.
- ^ Medical Practice of William H. Gorman, M.D. (18 February 2014). “Medications to Avoid, Continue, or Stop – Before & After Surgery”.
- ^ Jump up to:a b “Cyclobenzaprine”. http://www.drugbank.ca.
- ^ Kobayashi H, Hasegawa Y, Ono H (September 1996). “Cyclobenzaprine, a centrally acting muscle relaxant, acts on descending serotonergic systems”. European Journal of Pharmacology. 311 (1): 29–35. doi:10.1016/0014-2999(96)00402-5. PMID 8884233.
- ^ Jump up to:a b c d e f g h i j k “Cyclobenzaprine (CBP) and Its Major Metabolite Norcyclobenzaprine (nCBP) Are Potent Antagonists of Human Serotonin Receptor 2a (5HT2a), Histamine Receptor H-1 and á-Adrenergic Receptors: Mechanistic and Safety Implications for Treating Fibromyalgia Syndrome by Improving Sleep Quality”. ACR Meeting Abstracts. Retrieved 27 January 2022.
- ^ “Cyclobenzaprine”. pubchem.ncbi.nlm.nih.gov.
- ^ Winchell GA, King JD, Chavez-Eng CM, Constanzer ML, Korn SH (January 2002). “Cyclobenzaprine pharmacokinetics, including the effects of age, gender, and hepatic insufficiency”. Journal of Clinical Pharmacology. 42 (1): 61–9. doi:10.1177/0091270002042001007. PMID 11808825. S2CID 7749001.
- ^ “Medscape: Medscape Access”. medscape.com. Retrieved 1 October 2016.
- ^ Childers MK, Petri M, Laudadio C, Harrison D, Silber S, Bowen D (2004). “Comparison of cyclobenzaprine alone versus cyclobenzaprine plus ibuprofen in patients with acute musculoskeletal spasm and pain”. Annals of Emergency Medicine. 44 (4): S87–S88. doi:10.1016/j.annemergmed.2004.07.286.
- ^ “Patient Web site for Amrix (Cyclobenzaprine Hydrochloride Extended‐Release Capsules)”. amrix.com. Retrieved 1 October 2016.
External links
- “Cyclobenzaprine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Flexeril, Amrix, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a682514 |
| License data | US DailyMed: Cyclobenzaprine |
| Routes of administration | By mouth |
| ATC code | M03BX08 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 33–55%[1][2] |
| Protein binding | 93% |
| Metabolism | major: CYP3A4, CYP1A2; minor: CYP2D6, N-demethylation[5] |
| Metabolites | Norcyclobenzaprine |
| Elimination half-life | 32 hours (extended-release, range 8-37 hours),[3] 18 hours (immediate release, range 8–37 hours)[4] |
| Excretion | Kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 303-53-7 |
| PubChem CID | 2895 |
| IUPHAR/BPS | 7152 |
| DrugBank | DB00924 |
| ChemSpider | 2792 |
| UNII | 69O5WQQ5TI |
| KEGG | D07758 |
| ChEBI | CHEBI:3996 |
| ChEMBL | ChEMBL669 |
| CompTox Dashboard (EPA) | DTXSID0046933 |
| ECHA InfoCard | 100.005.588 |
| Chemical and physical data | |
| Formula | C20H21N |
| Molar mass | 275.395 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
///////////////cyclobenzaprine, циклобензаприн , سيكلوبنزابرين , 环苯扎林 , MK-130, TNX-102, Muscle Relaxant
CN(C)CCC=C1C2=CC=CC=C2C=CC2=CC=CC=C12

NEW DRUG APPROVALS
ONE TIME
$10.00
Pyritinol
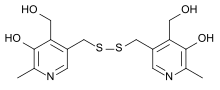
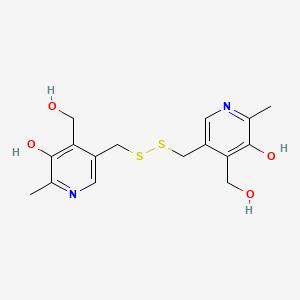
Pyritinol
- Molecular FormulaC16H20N2O4S2
- Average mass368.471 Da
1098-97-1[RN]
1308
214-150-1[EINECS]
233-178-5[EINECS]
3,3′-[Dithiobis(methylene)]bis[5-hydroxy-6-methyl-4-pyridinemethanol]
4-Pyridinemethanol, 3,3′-[dithiobis(methylene)]bis[5-hydroxy-6-methyl-
пиритинол[Russian][INN]
بيريتينول[Arabic][INN]
吡硫醇[Chinese][INN]
Pyritinol, CAS Registry Number: 1098-97-1
CAS Name: 3,3¢-[Dithiobis(methylene)]bis[5-hydroxy-6-methyl-4-pyridinemethanol]
Additional Names: bis(4-hydroxymethyl-5-hydroxy-6-methyl-3-pyridylmethyl) disulfide; bis[(3-hydroxy-4-hydroxymethyl-2-methyl-5-pyridyl)methyl] disulfide; dipyridoxolyldisulfide; pyridoxine-5-disulfide; pyrithioxin
Molecular Formula: C16H20N2O4S2, Molecular Weight: 368.47
Percent Composition: C 52.15%, H 5.47%, N 7.60%, O 17.37%, S 17.40%
Literature References: Prepn: Zima, Schorre, US3010966 (1961 to E. Merck); Iwanami et al.,Bitamin36, 122 (1967); J. Vitaminol.14, 321, 326 (1968). HPLC determn in urine: K. Kitao et al.,Chem. Pharm. Bull.25, 1335 (1977). Pharmacokinetics and metabolism: Darge et al.,Arzneim.-Forsch.19, 5, 9, (1969); Nowak, Schorre, ibid. 11. Clinical trial in dementia: S. Hoyer et al.,ibid.27, 671 (1977); A. J. Cooper, R. V. Magnus, Pharmacotherapeutica2, 317 (1980); in cerebrovascular disorders: Y. Tazaki et al.,J. Int. Med. Res.8, 118 (1980).
Properties: Crystals, mp 218-220°.
Melting point: mp 218-220°
Derivative Type: Dihydrochloride monohydrate
Trademarks: Biocefalin (Benvegna); Bonifen (Merck KGaA); Enbol (Chugai); Encephabol (Merck KGaA); Enerbol (Polfa); Epocan (Merck KGaA); Life (SIT)
Molecular Formula: C16H20N2O4S2.2HCl.H2O, Molecular Weight: 459.41
Percent Composition: C 41.83%, H 5.27%, N 6.10%, O 17.41%, S 13.96%, Cl 15.43%
Properties: mp 184°. Note: Has no vitamin B6 activity.
Melting point: mp 184°
Therap-Cat: Nootropic.
Keywords: Nootropic.
Derivatives
Dihydrochloride monohydrate
- Formula:C16H20N2O4S2 • 2HCl • H2O
- MW:459.42 g/mol
- CAS-RN:10049-83-9
- EINECS:233-178-5
- LD50:221 mg/kg (M, i.v.); 5786 mg/kg (M, p.o.);
300 mg/kg (R, i.v.); 6 g/kg (R, p.o.)
Pyritinol has been used in trials studying the treatment of Dementia, Depression, Schizophrenia, Anxiety Disorders, and Psychosomatic Disorders.
Pyritinol also called pyridoxine disulfide or pyrithioxine (European drug names Encephabol, Encefabol, Cerbon 6) is a semi-synthetic water-soluble analog of vitamin B6 (Pyridoxine HCl). It was produced in 1961 by Merck Laboratories by bonding 2 vitamin B6 compounds (pyridoxine) together with a disulfide bridge. Since the 1970s, it has been a prescription and OTC drug in several countries for cognitive disorders, rheumatoid arthritis,[1] and learning disorders in children. Since the early 1990s it has been sold as a nootropic dietary supplement in the United States.

SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 39984-49-1 | C8H10Br3NO | 3,4-bis(bromomethyl)-5-hydroxy-6-methylpyridine hydrobromide | 3-Pyridinol, 4,5-bis(bromomethyl)-2-methyl- |
| 92147-37-0 | C11H15NO3S2 | ethylxanthic acid [5-hydroxy-4-(hydroxymethyl)-6-methyl-3-pyridyl]methyl ester | Xanthic acid, ethyl-, [5-hydroxy-4-(hydroxymethyl)-6-methyl-3-pyridyl]methyl ester |
| 140-89-6 | C3H5KOS2 | potassium ethylxanthogenate | Carbonodithioic acid, O-ethyl ester, potassium salt |

PATENT
PATENT
https://patents.google.com/patent/CN103992268A/en
Pyritinol, it is the derivative of vitamin B6, for nootropic agents, can promote glucose and amino acid metabolism in brain, improve whole body assimilation, increase Flow of carotid artery, improve cerebral blood flow (CBF), be applicable to the dizzy distending pain, insomnia, hypomnesis of cerebral trauma sequela, encephalitis and meningitis sequela etc., the improvement of absent minded, emotional change; Also for cerebral arteriosclerosis, senile dementia mental symptom etc.
The pyritinol of applying clinically at present, it is pyritinol hydrochloride, be specially the monohydrate of hydrochloride, its chemical name is 3,3-(dithio methylene radical) two (5-hydroxyl-6-methyl-pyridine methane) dihydrochloride monohydrate, has recorded in < < Chinese Pharmacopoeia version > > in 2010.The preparation of this product listing has sheet, capsule and sterile powder injection, and its injection easily causes venous stimulation when clinical application, has greatly limited clinical application.The powder injection of pyritinol hydrochloride easy caking after standing storage, not soluble or dissolve and thoroughly cause liquid unclarity, particulate matter to exceed standard and easily cause the untoward reactions such as Microembolization during use.
CN101003509A discloses hydrobromate and the mesylate of pyritinol, record its stability having had, solvability and bland advantage, but in fact, Hydrogen bromide pyritinol, methylsulfonic acid pyritinol store easy moisture absorption under normal condition, in purification refine, be difficult to separate out with conventional crystallization method, need loaded down with trivial details aftertreatment technology, Hydrogen bromide and methylsulfonic acid have strong corrodibility in addition, comparatively difficult to its suitability for industrialized production.
CN101066266A discloses organic acid salt of pyritinol and preparation method thereof, wherein preferred pyritinol nicotinate.Yet, in nicotinic acid pyritinol water solvability a little less than, and nicotinic acid pyritinol preparation technology used dry-out benzene, toxicity is larger, and aftertreatment technology is complicated, is not suitable for suitability for industrialized production.
Yet, existing pyritinol or its salt, or pyritinol salt exists defect in the use, or the production technique that obtains this pyritinol salt is unsuitable for suitability for industrialized production.For this reason, need to provide a kind of safe, pyritinol salt and production method thereof of stablizing, meeting industrialization production requirements.
Embodiment 1: pyritinol maleate synthetic
Get 5.0g pyritinol powder, drop in reaction flask, add 100ml purified water, then under agitation add toxilic acid 3.8g, finish, be heated to 60-65 ℃ and stir 30min and all dissolve to solid, remove heating fluid, stirred crystallization under room temperature, separate out a large amount of white solids, use a small amount of cold water washing, 45 ℃ of vacuum-dryings, obtain white powder 5.97g, yield 72.9%.Purity: 99.5%; M.p.:134~137 ℃; Ultimate analysis (C16H20N2O4S22C4H4O4): C:47.9%, H:4.8%, N:4.6%, S:10.6%, O:32.1% (theory: C:48.0%, H:4.7%, N:4.7%, S:10.7%, O:32.0%); 1H-NMR (600MHz, DMSO) δ: 2.39 (6H, s), 3.93 (4H, s), 4.76 (4H, s), 6.18 (4H, s), 7.87 (2H, s).By the 1H-NMR (Fig. 2) of toxilic acid pyritinol and the 1H-NMR (Fig. 1) of pyritinol contrast, in a part toxilic acid pyritinol, contain 2 molecule toxilic acids.
Embodiment 2: pyritinol maleate synthetic
Get 5.0g pyritinol powder, drop in reaction flask, add 100ml ethanol, then under agitation add toxilic acid 3.0g, finish, be heated to return stirring 30min and all dissolve to solid, remove heating fluid, stirred crystallization under room temperature, separate out a large amount of white solids, use a small amount of cold water washing, 45 ℃ of vacuum-dryings, obtain white powder 5.50g, yield 67.5%.After measured, the toxilic acid pyritinol that structure makes with embodiment 1.
PATENT
https://patents.google.com/patent/CN105153021A/en
Embodiment 1
Toxilic acid 3.8g is dissolved in 100ml ethanol, be warming up to 60 DEG C clearly molten, add pyritinol 5.0g, stir clearly molten, react 1 hour, cooling crystallization, filter, solid is drying under reduced pressure at 50 DEG C, obtains white crystalline solid toxilic acid pyritinol crystal form A 4.9g.X-ray powder diffraction analysis, as Fig. 1, its 2 θ value is as following table.
Embodiment 2
Toxilic acid 3.8g is dissolved in 100ml acetone, be warming up to 45 DEG C clearly molten, add pyritinol 5.0g, stir clearly molten, react 1.5 hours, cooling crystallization, filter, solid is drying under reduced pressure at 50 DEG C, obtains white crystalline solid 5.2g.It is toxilic acid pyritinol crystal form A that dry product does X-ray powder diffraction.
Embodiment 3
Toxilic acid 3.8g is dissolved in and adds 100ml Virahol, be warming up to 60 DEG C clearly molten, add pyritinol 5.0g, stir clearly molten, react 2 hours, cooling crystallization, filter, solid is drying under reduced pressure at 50 DEG C, obtains white crystalline solid 5.1g.It is toxilic acid pyritinol crystal form A that dry product does X-ray powder diffraction.
PATENT
https://patents.google.com/patent/CN101066266A/en
Specific embodiment:
Embodiment 1: nicotinic acid pyritinol salt synthetic
Get nicotinic acid 24.6g, fully be dissolved in the 300ml anhydrous benzene, heated and stirred is to molten entirely, under complete molten state, add pyritinol 40.5g, reflux mixture 3 hours, TLC thin layer identification (developing solvent: ethyl acetate: ethanol: glacial acetic acid=5: 6: 0.6) fully, the cooling back adds the 200ml dehydrated alcohol slightly, mixture is put into refrigerator fully cool off, sucking filtration is separated out white crystals, with a small amount of cold absolute ether washing solid.65 ℃ of vacuum dryings get 62.1g nicotinic acid pyritinol salt, yield 89.7%.Determination of acid-basetitration nicotinic acid and pyritinol content are measured moisture with the karl Fischer method.The result is: nicotinic acid 37.2%, and pyritinol 62.0%, water 5.8%, approaching with theoretical value, contain 2 water of crystallization.Elementary analysis: theoretical value C52.8% H5.3% O25.2%N6.6% S10.1%; Measured value C52.4% H5.2% O25.1%N6.5% S10.0%.
Embodiment 2: fumaric acid pyritinol salt synthetic
Get fumaric acid 11.6g, fully be dissolved in the 300ml anhydrous benzene, heated and stirred is to molten entirely, under complete molten state, add pyritinol 40.5g, reflux mixture 3 hours, TLC thin layer identification (developing solvent: ethyl acetate: ethanol: glacial acetic acid=5: 4: 0.8) fully, the cooling back adds the 200ml dehydrated alcohol slightly, mixture is put into refrigerator fully cool off, sucking filtration is separated out white crystals, with a small amount of cold absolute ether washing solid.65 ℃ of vacuum dryings get 49.9g fumaric acid pyritinol salt, yield 88.9%.Determination of acid-basetitration fumaric acid and pyritinol content are measured moisture with the karl Fischer method.The result is: fumaric acid 20.8%, and pyritinol 72.7%, water 6.5%, approaching with theoretical value, contain 2 water of crystallization.Elementary analysis: theoretical value C49.6% H5.0%O26.4% N5.8% S13.2%; Measured value C49.4% H5.2% O26.5% N5.9%S13.1%.
PATENT
https://patents.google.com/patent/CN102516297A/en
Embodiment 1: the preparation of compd A
With Pyrithioxine hydrochloride 10g, be dissolved in the 20ml pyridine, slowly drip POCl3 solution 10ml under the room temperature; Drip and finish, stirring at room reaction 12 hours slowly adds the 100g frozen water and stirred hydrolysis reaction 2 hours; Toluene gradation extraction 30ml * 3, water layer evaporated under reduced pressure, Virahol dissolution residual substance; Filter, evaporate to dryness gets compd A 4.2g.
Embodiment 2: the preparation of compd B
With Pyrithioxine hydrochloride 10g, be dissolved in the 40ml THF, add 4gNaH, 30 ℃ were stirred 2 hours; Add the 20ml POCl3, stirring reaction 16 hours slowly adds the 100g frozen water and stirred hydrolysis reaction 2 hours; ETHYLE ACETATE gradation extraction 30ml * 3, the water layer evaporated under reduced pressure adds 80ml Virahol dissolution residual substance; Add 40ml water, freezing crystallization gets compd B 5.6g.
Embodiment 3: the preparation of Compound C
With Pyrithioxine hydrochloride 10g, be dissolved in the 40ml THF, add 4gNaH, 30 ℃ were stirred 2 hours; Add the 20ml chloroiodomethane, stirring reaction 16 hours, 60 ℃ of evaporated under reduced pressure add 20ml acetonitrile dissolution residual substance; As midbody, other gets triethylamine 9ml and is dissolved in the 10ml acetonitrile, drips 3.6ml phosphoric acid, after dropping finishes; Stir down and slowly splash into midbody, continued 60 ℃ of stirring reactions 12 hours, steaming desolventizes; Residue adds water 20ml dissolving, and water layer filters clarification, and freeze-drying promptly gets compd B 6.7g.
Embodiment 4: the preparation of Compound D
Serine 3 grams, ethylene bromohyrin 2.5g, N with the BOC protection; N-Dimethylamino pyridine 3g and NSC 57182 3g are dissolved in the THF; Stirring at room 10 hours, vacuum concentration is with the thick product of chromatography purification (with the ETHYLE ACETATE/normal hexane wash-out of normal hexane to 30%); Merging filtrate, evaporate to dryness gets intermediate A; Pyrithioxine hydrochloride 2g and intermediate A 2.5g are dissolved with THF 30ml, add triphenyl phosphorus 2g, slowly drip diethyl azodiformate solution 2ml, room temperature reaction 5 hours; Reaction is finished, and evaporated under reduced pressure adds ETHYLE ACETATE 50ml dissolving, filters insolubles; With the thick product of chromatography purification (with the ETHYLE ACETATE/normal hexane wash-out of normal hexane to 10%), merging filtrate, evaporate to dryness dissolves with methylene dichloride 20ml then; Feed hydrogen chloride gas to saturated, stirring reaction 5 hours filters; Get the hydrochloride of Compound D, transferring pH behind the use dissolved in distilled water is about 8, and the water layer lyophilize gets Compound C 0.27g.
Embodiment 5: the preparation of compd E
Get compd A 10g, be dissolved in the 30ml Virahol, add 25gBoc-Ser-OBZL in batches, 50 ℃ of stirring reactions; HPLC monitoring react to compd B less than 5%, add 0.1M hydrochloric acid soln 20ml, 60 ℃ of heating hydrolysis 5 hours are regulated pH to 7; Evaporated under reduced pressure adds anhydrous alcohol solution, removes by filter insolubles, evaporated under reduced pressure; Add the 5ml water dissolution, filtering, lyophilize get compd E 6.9g
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Availability
It is approved for “symptomatic treatment of chronically impaired brain function in dementia syndromes” and for “supportive treatment of sequelae of craniocerebral trauma” in various European countries, including Austria, Germany, France, Italy, Portugal, and Greece. In France it is also approved for rheumatoid arthritis as a disease modifying drug, on the basis of the results of clinical trials. In many countries it is available over the counter and is widely advertised on the internet as being for “memory disturbances.”
Effects
review refs needed
Adverse effects
Adverse effects include nausea, headache,[2] and rarely allergic reaction (mild skin reactions).[3] A 2004 survey of six case reports suggested a link between pyritinol and severe cholestatic hepatitis when on several drugs for certain diseases.[4]
Other rare side effects: acute pancreatitis[5] and photoallergic eruption.[6]
References
- ^ Lemmel EM (May 1993). “Comparison of pyritinol and auranofin in the treatment of rheumatoid arthritis. The European Multicentre Study Group”. British Journal of Rheumatology. 32 (5): 375–82. doi:10.1093/rheumatology/32.5.375. PMID 8495257.
- ^ Nachbar F, Korting HC, Vogl T (1993). “Erythema multiforme-like eruption in association with severe headache following pyritinol”. Dermatology. 187 (1): 42–6. doi:10.1159/000247196. PMID 8324277.
- ^ de Groot, Anton C.; Nater, Johan Pieter; Weyland, J. Willem. Unwanted Effects of Cosmetics and Drugs Used in Dermatology.[full citation needed][page needed]
- ^ Maria V, Albuquerque A, Loureiro A, Sousa A, Victorino R (March 2004). “Severe cholestatic hepatitis induced by pyritinol”. BMJ. 328 (7439): 572–4. doi:10.1136/bmj.328.7439.572. PMC 381054. PMID 15001508.
- ^ Straumann A, Bauer M, Pichler WJ, Pirovino M (August 1998). “Acute pancreatitis due to pyritinol: an immune-mediated phenomenon”. Gastroenterology. 115 (2): 452–4. doi:10.1016/S0016-5085(98)70212-4. PMID 9679051.
- ^ Tanaka M, Niizeki H, Shimizu S, Miyakawa S (October 1996). “Photoallergic drug eruption due to pyridoxine hydrochloride”. The Journal of Dermatology. 23 (10): 708–9. doi:10.1111/j.1346-8138.1996.tb02685.x. PMID 8973037. S2CID 28810619.
External links
- Media related to Pyritinol at Wikimedia Commons
| Clinical data | |
|---|---|
| ATC code | N06BX02 (WHO) |
| Pharmacokinetic data | |
| Elimination half-life | 2.5 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1098-97-1 |
| PubChem CID | 14190 |
| ChemSpider | 13561 |
| UNII | AK5Q5FZH2R |
| KEGG | D02160 |
| ChEMBL | ChEMBL488093 |
| CompTox Dashboard (EPA) | DTXSID3048362 |
| ECHA InfoCard | 100.012.864 |
| Chemical and physical data | |
| Formula | C16H20N2O4S2 |
| Molar mass | 368.473 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
//////////////Pyritinol, пиритинол , بيريتينول , 吡硫醇 , Nootropic,

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Liranaftate
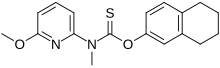
Liranaftate
リラナフタート
88678-31-3
(6-Methoxy-2-pyridinyl)methylcarbamothioic Acid O-(5,6,7,8-Tetrahydro-2-naphthalenyl) Ester
O-(5,6,7,8-Tetrahydronaphthalen-2-yl) (6-methoxypyridin-2-yl)methylcarbamothioate
Zefnart;Piritetrate;M-732
лиранафтат
ليرانافتات
利拉萘酯
| Formula | C18H20N2O2S |
|---|---|
| CAS | 88678-31-3 |
| Mol weight | 328.4286 |
| Efficacy | Antifungal, Ergosterol biosynthesis inhibitor |
|---|---|
| Comment | Thiocarbamate |
Liranaftate (trade name Zefnart) is a topical antifungal drug.[1] It is used as a 2% cream used to treat tinea pedis (athlete’s foot), tinea corporis (ringworm), and tinea cruris (jock itch).[2] It was approved for use in Japan in August 2000.[3][4]
Liranaftate works by inhibiting the fungal enzyme squalene epoxidase that is necessary for the fungus to synthesize sterols which are essential for cell membrane integrity.[5]
SYN
IN 2010MU02699
PAPER
Journal of Chemical and Pharmaceutical Research (2013), 5(11), 219-222,
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007010744
Conventionally, 0-aryl N- (6-alkoxy-2-pyridyl) -N-alkylthio-force rubamate has generally been produced by a method using thiophosgen. For example, in Patent Document 1, 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N- represented by the following reaction formula 0 or ii) A method for producing methylthiolbamate (4) is disclosed.
(Example 1)
1) Sodium 5, 6, 7, 8-Tetrahydro-2-naphthoside synthesis
[hua 6]
,She
To methanol (10 ml), 0.54 g (10.0 mmol) of sodium methoxide was added, and the mixture was stirred at room temperature. There, 1.50 g (10.0 mmol) of 5,6,7,8-tetrahydro-2-naphthol was added and he stirred for 1 hour at room temperature. The solvent was distilled off under reduced pressure to obtain 3.75 g ( q uant.) Of white powder. I left it overnight in a desiccator.
2) 2- [Ν- (1-imidazolithiocarbol) -Ν’-methyl] amino-6-methoxypyridin compound
[hua 7]
To ethyl acetate (30 ml), 2.07 g (15.0 mmol) of 6-methoxy-2-methylaminoviridin and 2.67 g (15.0 mmol) of 1,1, -thiocarboldiimidazole were added, and the mixture was heated under reflux for 2 hours. After allowing to cool, the solvent was distilled off under reduced pressure to obtain 3.70 g of brown oil. (Yield 99.3%). If necessary, further purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1) to obtain pale yellow crystals.
Melting point: 58.0~60.0°C
NMR(CDC1 3 ) δ ppm:3.86(3H,s), 3.87(3H,s), 6.38 (lH’dd, J=7.5Hz, 0.7Hz), 6.61 (1H
,dd, J=8.3Hz, 0.7Hz), 6.82 (lH,t, J=1.0Hz) , 7.03 (lH,t, J=1.0Hz) , 7.46 (lH’dd, J= 8.3Hz, 7.5Hz), 7.72 (lH,t, J=1.0Hz)
IR(KBr)cm_1: 1604, 1590, 1571, 1465, 1359, 1303, 1120, 1013, 986, 822, 798 MS m/z: 248(M+)
3) Synthesis of 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) -N- (6-methoxy-2-pyridyl) -N-methylthiocarbamate
Dissolve 2- [N- (1-imidazolithiocarbol) -N-methyl] amino-6-methoxypyridin 250 mg (1.0 mmol) in N, N-dimethylformamide (4 ml), and then dissolve. At room temperature, Natrium 5, 6, 7, 8-tetrahydro-2-naphthoside 360 mg (2.0 mmol) was added. -After stirring at room temperature, the reaction solution was extracted with ethyl acetate (10 mlx2), and the insoluble material was filtered off on the way. The organic layer was washed with saturated brine, dried over magnesium sulfate, filtered off magnesium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) gave the title compound 266.6 mg (yield 81.3%).
Melting point: 99~100°C
NMR(CDCl 3) δ ppm:1.77(4H,bs), 2.75(4H,bs), 3.75(3H,s), 3.93(3H,s), 6.65(lH,d, J
=8.0Hz), 6.78-7.08(4H,m), 7.64(lH,t,J=8.0Hz)
IR(KBr) cm_1 : 1603, 1460, 1413, 1369, 1325, 1262, 1175, 1035, 808, 785
MS m/z: 328(M+)
(Example 2)
0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N-methylthio force Rubamate synthesis
[Chemical 9]
1.34 g (33.6 mmol) of 60% sodium hydride was added to N, N-dimethylformamide (20 ml), followed by the addition of 5, 6, 7, 8-tetrahydro-2-naphthol 4.65 g (30.5 mmol). After gas generation is complete, add 2- [N- (1-imidazolthiocarbonyl) -N-methyl] amino-6-methoxypyridin 7.45 g (30.0 mmol) and zinc chloride 2.05 g (15.0 mmol). rice field. After heating and stirring at 60 ° C for 3 hours and allowing to cool, the reaction solution was extracted with ethyl acetate (150 mlx2), and the insoluble material was filtered off on the way. The organic layer is washed with saturated brine, dried over magnesium sulfate, and filtered through magnesium sulfate.
Separately, the solvent was distilled off under reduced pressure. The obtained crystals were purified by one of the following methods.
[0028] A) Purification was performed by silica gel column chromatography (eco-gel C 200, hexane: ethyl silicate = 10: 1) to obtain 9.80 g of the indicated compound (yield 99.5%).
B) Suspended in hexane (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 9.65 g of crystals. Further, the mixture was suspended in methanol (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 8.62 g (yield 87.5%) of the indicated compound.
The physics and physics data of the obtained compound were consistent with the compounds obtained in the examples.
(Example 3)
1) Synthesis of 2- [N- [1-2 (1H) -pyridonylthiocarbol] -N-methyl] amino-6-methoxypyridine
[Chemical 10]
OMe
Add 6-methoxy-2-methylaminoviridin 690 mg (5.0 mmol) and 1, 1, -thiocarbol-di-2 (1H) -pyridone 1.16 g (5.0 mmol) to ethyl acetate (15 ml). Heated and refluxed for 1 hour. After allowing to cool, the solvent was distilled off under reduced pressure, and purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1)! ヽ, 297.4 mg of brown oil was obtained. (Yield 21.6%).
NMR(CDC1 3 ) δ ppm:3.77(3H,s), 3.93(3H,s), 6.66 (lH’dd, J=8.0Hz, 0.7Hz), 7.07 ( lH,d, J=8.0Hz), 7.14 (lH,d, J=7.5Hz) , 7.25 (lH’dd, J=8.0Hz, 4.0Hz) , 7.62 (lH’dd , J=8.0Hz, 7.5Hz), 7.78 (lH’dd, J=2.0Hz, 0.7Hz) , 8.43 (lH’dd, J=4.0Hz, 0.7Hz)
MS m/z: 275(M+)
[0031] 2) Synthesis of 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N-methylthiocarbamate
[Chemical 11]
OMe
N, N-dimethylformamide (2 ml), 2- [N- [1-2 (1H) -pyridonylthiocarbol] –N-methyl] amino-6-methoxypyridin 297 mg (1.08 mmol) and sodium 5 , 6, 7, 8-Tetrahydro-2-naphthoside 390 mg (2.16 mmol) was added and stirred overnight at room temperature. The reaction mixture was extracted with ethyl acetate (50 mlx2), the organic layer was washed with saturated brine, dried over magnesium sulfate, magnesium sulfate was filtered off, and the solvent was distilled off under reduced pressure. The obtained crystals were purified by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) to obtain the title compound 288.2 mg (81.4%).
SYN
CN 104725302
| Liranafate is a new-generation antifungal drug, a squalene cyclooxygenase inhibitor and a cell wall synthesis inhibitor, with the chemical name of 6-methoxy-2-N-methyl-pyridylamino-thio Formic acid-(5,6,7,8-tetrahydro)-β-naphthyl ester. A new type of antifungal drug jointly developed by Tosoh Corporation of Japan and Zenyaku Kogyo Corporation was first listed in Japan by Torii Corporation in August 2000. The antifungal drug exerts antifungal activity by inhibiting the squalene epoxidation reaction of fungal cells and inhibiting the synthesis of ergosterol, a component of cell membranes. effect is particularly evident. Today, with the increasing concern of the world about environmental pollution, the development of new green and effective drug synthesis methods is an important task faced by the research of drug synthesis. In recent years, room temperature ionic liquids have been widely used in various organic synthesis reactions as a new type of environmentally friendly reaction media. Compared with traditional organic solvents, ionic liquids have many advantages, such as extremely low vapor pressure, non-flammability, good thermal stability and recyclability. |
| At present, the main synthetic route of liranaftate is as follows: |
| |
| Among the four synthetic routes, the pyridine derivative intermediates of routes C and D need to be prepared through multi-step reactions, the routes are long, the steps are cumbersome, the actual operation is cumbersome, the cost is high, and they are not suitable for industrialized large-scale production. Although route A has simple steps, the yield of pyridine derivatives is low. Each intermediate structure in route B is relatively simple and easy to prepare, but this route uses 6-methoxy-2-methylaminopyridine and 5,6,7,8-tetrahydro-2-naphthoxysulfuryl chloride as raw materials to synthesize the In the process of lanaphthalate, isopropanol-water is used as the reaction medium, and the experiment shows that with the progress of the reaction, the reaction solution becomes viscous, and the reaction is difficult to complete. |
| Example 1 |
| (1) Ionic liquid [bmim]BF 4 Synthesis |
| |
| Add N-methylimidazole (14.8g, 0.18mol) and trichloroethane (80mL) to a dry 250mL three-neck flask, stir to make the mixture uniform, add 20.4mL of freshly distilled n-bromine to the dropping funnel Butane (26.03g, 0.19mol) was added dropwise for about 30min, and the reaction was refluxed for 4-5h (the reflux temperature was about 78±1℃). With the progress of the reaction, the reaction solution changed from colorless and transparent to white turbidity, light yellow turbidity, and the color gradually became darker until brownish red. After the reaction is completed, the liquids are separated into layers, the upper layer is lighter in color, which is the trichloroethane layer, and the lower layer is darker in color (brown red), which is the ionic liquid [bmim]Br layer. The prepared ionic liquid [bmim]Br and trichloroethane were separated, and the ionic liquid [bmim]Br was washed twice with trichloroethane, and then the trichloroethane in the ionic liquid [bmim]Br was washed with a water pump. The alkane was pumped away until the ionic liquid [bmim]Br liquid was no longer turbid, and then dried in a vacuum drying oven at 90 °C for 10-12 h to obtain relatively pure ionic liquid [bmim]Br. |
| |
| Then prepare 0.03mol NaBF 4 of aqueous solution. Add 6.58g (about 0.03mol) ionic liquid [bmim]Br and 5-10mL water to a 100mL round-bottomed single diameter flask, stir, ice-water bath, and dropwise add NaBF 4 The solution (completed dropwise addition in about 5min), continue to stir for 10-20min, the solution is yellow and transparent, pour it into a separatory funnel, extract twice with dichloromethane, combine the dichloromethane layers, and wash the dichloromethane layer 2 with 50 mL of water times, and then the dichloromethane layer was washed with anhydrous MgSO 4 Dry, filter, evaporate the dichloromethane under normal pressure in a water bath (50-52°C), and dry the remaining dark yellow viscous liquid in a vacuum drying oven at 90°C for 10-12h to obtain the ionic liquid [bmim]BF 4 。 |
| |
| (2) Synthesis of 6-methoxy-2-chloropyridine 2 |
| 2,6-dichloropyridine (10g, 0.068mol) and sodium methoxide (24.5g, 0.136mol) were put into the reaction flask, heated under reflux for 4-5h, and the reaction was completed by TLC (ethyl acetate: petroleum ether=1 : 15), concentrated to remove methanol, added 100 mL of water, extracted with ethyl acetate, combined the organic phases, washed with saturated brine, dried, filtered, and the filtrate was concentrated to obtain 9 g of a crude colorless oily product with a yield of 92.5%. used for the next reaction. |
| (3) Synthesis of 6-methoxy-2-methylaminopyridine 3 |
| Take 6-methoxy-2-chloropyridine 2 (9g, 0.127mol), cuprous chloride (1.72g, 0.0017mol) and methylamine aqueous solution (29mL, mass concentration is 25%-30%) and add it to the autoclave , sealed and heated to 120 °C for 7 h, the reaction was stopped, ethyl acetate was added for extraction, the organic phases were combined, washed with saturated brine, dried, and the filtrate was concentrated to obtain 6.18 g of brown oil, the yield was 71.2%, and the HPLC purity was 98% . |
| (4) Synthesis of 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride 4 |
| Mix 50 mL of ethyl acetate, thiophosgene (4.25 mL, 0.056 mol) and 5,6,7,8-tetrahydro-2-naphthol (6.3 g, 0.0425 mol), and cool it in an ice-salt bath to below 0 °C. Add 10 mL of potassium carbonate (3 g, 0.022 mol) solution, continue to stir the reaction after the dropwise addition, and check by TLC (developing solvent: petroleum ether) that the reaction is complete, add 100 mL of water, extract with ethyl acetate, wash the organic phase with saturated brine, Dry, filter, and concentrate the filtrate to obtain 8.7 g of yellow oil with a yield of 90.4%, which can be directly used in the next reaction without purification. |
| (5) Synthesis of Liranaftate 1 |
| The prepared ionic liquid [bmim]BF 4 (100mL), 6-methoxy-2-methylaminopyridine 3 (5.7g, 0.0413mol) and potassium carbonate (5.7g, 0.0413mol) were mixed, cooled with ice water, and slowly added dropwise 5,6,7,8 -Tetrahydro-2-naphthyloxysulfuryl chloride 4 (8.7g, 0.0385mol) was added dropwise for 4h, slowly added 150mL of water under full stirring, continued to stir for 20min, filtered, washed with deionized water to obtain 12.2g of crude product, collected The yield was 96.81%, and acetone was recrystallized to obtain 11 g of white crystalline powder, the yield was 90%, and the HPLC purity was 99.7%. mp: 98.8-99.5°C, IR (2973cm -1 , 2930cm -1 , 2852cm -1 , 1416cm -1 , 1264cm -1 , 1037cm -1 ), 1 HNMR: 1.8 (m, 4H); 6.68(d, 1H) ;6.86(dd,1H);3.78(s,3H);3.98(s,3H);6.68(d,1H);6.86(dd,1H);7.05(d,1H);7.10(d.1H); 7.65 (dd, 1H), MS (m/z: 328, 181, 165, 108). |
| Example 2 |
| Under the same conditions, the ionic liquid 1-n-butyl-3-methylimidazolium tetrafluoroborate ([bmim]BF 4 ), N-ethylpyridine tetrafluoroborate ([EPy]BF 4 ), 1-n-butyl-3-methylimidazolium hexafluorophosphate ([bmim]PF 6 ), 1-hydroxyethyl-2,3-dimethylimidazolium chloride (LOH), 1-cyanopropyl-3-methylimidazolium chloride (LCN), 1-carboxyethyl-3-methylimidazole Chloride salt (LOOH), [Hnmp]HSO 4 The effects of and [bmim]OH on the synthesis of liranaftate are shown in Table 1. The results show that different ionic liquids have little effect on the yield of the synthesis and the yields are relatively high. |
| Table 1 Effects of different ionic liquids on the reaction yield |
| ionic liquidYield/%[bmim] BF 496.81[EPy]BF 496.83[bmim]PF 696.82LOH96.75LCN96.67LOOH96.05[Hnmp]HSO 496.06[bmim]OH95.98 |
| Example 3 |
| Whether the reaction medium used can be recovered and reused is an important content of “green chemistry”. This example specifically examines the reuse of ionic liquid for synthesizing liranaftate. After 5 times of use of ionic liquid, the product yield It just started to decrease, which shows that the ionic liquid can be recovered and reused effectively, and the reuse performance is good. It is a recyclable green solvent. |
SYN
| Comparative Example 1: |
| Put 10 g of 2,6-dichloropyridine, 100 ml of methanol, and 15 g of sodium methoxide into a reaction flask, heat under reflux for about 4 to 5 hours, concentrate to remove methanol, add 150 ml of water, extract with ethyl acetate, and concentrate under reduced pressure to remove ethyl acetate. 6-Methoxy2-chloropyridine was obtained as a colorless oil. |
| 9 g of 6-methoxy 2-chloropyridine, 1.72 g of cuprous chloride, and 29 ml of 30% methylamine aqueous solution were put into the reaction flask, heated and added with a mass fraction of 11.6 g of cuprous chloride, and the temperature was kept at 120 ° C for the reaction 8h, extracted three times with 150 ml of ethyl acetate, washed with saturated brine, concentrated under reduced pressure to remove the ethyl acetate to obtain 6.18 g of 6-methoxy-2-methylaminopyridine as a brown oily product. The two-step yield was 71.2%. |
| 50ml of carbon tetrachloride, 4.25g of thiophosgene, 6.3g of 5,6,7,8-tetrahydro-2-naphthol were added to the reaction flask, the ice-salt bath was lowered to below 0°C, and 10ml of 3g potassium carbonate aqueous solution was added dropwise. , Continue the reaction at 0°C after the dropwise addition, and detect by TLC (developing solvent: petroleum ether) after the reaction is completed, separate the organic phase, wash three times with saturated brine, and concentrate under reduced pressure to obtain red oily products 5, 6, 7 , 8.7g of 8-tetrahydro-2-naphthyloxysulfuryl chloride was directly used in the next reaction. |
| 100ml of acetone, 5.7g of 6-methoxy-2-methylaminopyridine and 5.7g of potassium carbonate were added to the reaction flask, cooled with ice water, and 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride was added dropwise 8.7g, continue to stir and react for 4h after dropping, add 150ml of water, continue to stir for 30min, and filter to obtain the crude product. The crude product was recrystallized with acetone to obtain 11 g of off-white crystalline powder. The weight yield was 174.6% based on 5,6,7,8-tetrahydro-2-naphthol. The maximum single impurity content determined by HPLC was 1.5%, which did not meet the requirements of the Pharmacopoeia. |
SYN
CN 106632018
| Example 1 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours; |
| Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, and the consumption of purified water was 25.0 g each time; |
| Drying: put the wet product into a drying box, control the temperature to 45 ℃ and dry for 4 hours, to obtain 24 g of the crude product of lira naphthate; |
| The synthesis yield is 81%; |
| (2) preparation of Liranaftate fine product: |
| Impurity removal: put 23g of Liranaftate crude product and 115g of absolute ethanol into the reaction flask, add 1.38g of medicinal charcoal, decolorize at 55°C under temperature control, remove impurities for 30 minutes, filter, transfer the filtrate to the reaction flask, control the temperature Crystallize at 55°C, centrifuge, dry, pulverize, and pack to obtain 22g of Lira naphthate fine product. |
| The purification yield was 92%. |
| Example 2 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 500g of absolute ethanol was added to the reaction flask, 25g of 2-methoxy-6-methylaminopyridine, 17.6g of anhydrous sodium carbonate and 62.6g of purified water were added to the reaction flask in turn, stirred for 30 minutes, and slowly added 2-(5,6,7,8-tetrahydronaphthyloxy) chlorothioformate 37.6g, added in 2.5 hours; |
| Reaction: control the temperature at 25°C for 2.5 hours, add 250 g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 50 g each time; |
| Drying: put the wet product into a drying box, control the temperature to 55 ℃ and dry for 4 hours to obtain 49 g of the crude product of lira naphthate; |
| The synthesis yield is 82%; |
| (2) preparation of Liranaftate fine product: |
| Impurity removal: put 49g of Liranaftate crude product and 245g of absolute ethanol into the reaction flask, add 2.9g of medicinal charcoal, decolorize at 55~65 ℃ of temperature, remove impurities for 30 minutes, filter, and transfer the filtrate to the reaction flask, The temperature was controlled at 65°C for crystallization, centrifugation, drying, pulverization, and packaging to obtain 45g of fine lanaftate. |
| The purification yield was 92%. |
| Example 3 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours; |
| Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 25.0 g each time; |
| Drying: put the wet product into a drying oven, control the temperature to 45~55 ℃ and dry for 4 hours, to obtain the crude product, 23.3 g of the crude liranaftate; |
| The synthesis yield is 82%; |
| (2) preparation of Liranaftate fine product: |
| Removal of impurities: 140g of absolute ethanol was added to the reaction flask, 23.3g of crude liranaftate was added, the temperature was controlled at 50°C and stirred for 30 minutes, 1.5g of medicinal charcoal was added, the temperature was controlled at 60°C for decolorization for 30 minutes, filtered, and the temperature was controlled Crystallize at 60°C, centrifuge, dry, pulverize, and package to obtain 23g of Lira naphthate fines. |
| The purification yield was 92%. |
SYN
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Koga H, Nanjoh Y, Makimura K, Tsuboi R (2009). “In vitro antifungal activities of luliconazole, a new topical imidazole”. Medical Mycology. 47 (6): 640–7. doi:10.1080/13693780802541518. PMID 19115136.
- ^ “Torii Pharmaceutical to Launch Antifungal Agent for External Use, “ZEFNART SOLUTION 2%”, in Japan” (Press release). Torii Pharmaceutical Co. Retrieved June 27, 2021.
- ^ “Liranaftate”. ncats.io. Retrieved June 27, 2021.
- ^ “Liranaftate”. Adis Insight. Retrieved June 27, 2021.
- ^ “Liranaftate”. targetmol.com. Retrieved June 27, 2021.
///////////////////Liranaftate , リラナフタート , Zefnart, Piritetrate, M-732, лиранафтат , ليرانافتات , 利拉萘酯 , ANTIFUNGAL, JAPAN 2000

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Fabomotizole

Fabomotizole
Afobazole
- Molecular FormulaC15H21N3O2S
- Average mass307.411 Da
0F8K1X115C
173352-21-1[RN], 173352-21-1 (free base) 173352-39-1 (HCl) 189638-30-0 (2HCl)
1H-Benzimidazole, 6-ethoxy-2-[[2-(4-morpholinyl)ethyl]thio]-
Obenoxazine, Afobazol, Afobazole, Aphobazole, Fabomotizole dihydrochloride, CM-346, CM346, CM 346,
фабомотизол[Russian][INN]
فابوموتيزول[Arabic][INN]
法莫替唑[Chinese][INN]
Fabomotizole dihydrochloride
CAS#: 189638-30-0 (2HCl)
Chemical Formula: C15H23Cl2N3O2S
Molecular Weight: 380.33
Fabomotizole (also known as Afobazole) is a selective non-benzodiazepine anxiolytic which was developed in Russia and launched in 2006. The drug is used for the treatment of wide range of diseases: generalized anxious disorders, neurasthenia, adaptation disorders, sleep disorders, for alleviation of withdrawal syndrome. According to the drug label (in Russian), its action is related to the interaction with sigma-1 receptors.
Fabomotizole (INN;[1] brand name Afobazole) is an anxiolytic drug launched in Russia in the early 2000s. It produces anxiolytic and neuroprotective effects without any sedative or muscle relaxant actions.[citation needed] Its mechanism of action remains poorly defined however, with GABAergic, NGF– and BDNF-release-promoting, MT1 receptor agonism, MT3 receptor antagonism, and sigma agonism suggested as potential mechanisms. Fabomotizole was shown to inhibit MAO-A reversibly and there might be also some involvement with serotonin receptors.[2][3][4][5][6] Clinical trials have shown fabomotizole to be well tolerated and reasonably effective for the treatment of anxiety.[7]
Experiments of mice have shown antimutagenic and antiteratogenic properties.[8]
Fabomotizole has found little clinical use outside Russia and has not been evaluated by the FDA.
PATENT
WO 9534304
https://patents.google.com/patent/WO1995034304A1/en

PAPER
European Journal of Medicinal Chemistry (2021), 211, 113110
https://www.sciencedirect.com/science/article/abs/pii/S0223523420310825?
A ligand-based virtual screening study to search for giardicidal compounds on a 6551 ChEMBL drugs database was carried out using molecular similarity. Three fingerprints implemented in MayaChemTools with different design and validated by ROC curves, were used. Twelve compounds were retrieved from this screening, from which, four representative compounds were selected to carry out biological assays. Whereas two compounds were commercially available, the additional two compounds were synthesized during the development of this work. The biological assays revealed that the compounds possess in vitro activity against five strains of Giardia intestinalis, each with different susceptibility/resistance rates to metronidazole, albendazole and nitazoxanide. Particularly, tenatoprazole showed the best effect against the WB and IMSS strains. Furthermore, fabomotizole, tenatoprazole and ipriflavone showed a higher activity against resistant strains than the reference drugs: metronidazole, albendazole and nitazoxanide.
Graphical abstract


///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Afobazole |
| Other names | Fabomotizole |
| Routes of administration | Oral |
| ATC code | N05BX04 (WHO) |
| Legal status | |
| Legal status | US: Unscheduled Not FDA approved |
| Pharmacokinetic data | |
| Bioavailability | 43.64%, pronounced first-pass effect |
| Metabolism | extensive hepatic |
| Onset of action | 0.85±0.13 hours |
| Elimination half-life | 0.82±0,54 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 173352-39-1 |
| PubChem CID | 9862937 |
| ChemSpider | 8038633 |
| UNII | HDO6HX6NZU |
| CompTox Dashboard (EPA) | DTXSID00169606 |
| Chemical and physical data | |
| Formula | C15H21N3O2S |
| Molar mass | 307.41 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
References
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). WHO Drug Information. 26 (1): 63. 2012. Retrieved 21 March 2015.
- ^ Neznamov, GG; Siuniakov, SA; Chumakov, DV; Bochkarev, VK; Seredenin, SB (2001). “Clinical study of the selective anxiolytic agent afobazol”. Eksperimental’naia i Klinicheskaia Farmakologiia. 64 (2): 15–9. PMID 11548440.
- ^ Silkina, IV; Gan’shina, TC; Seredin, SB; Mirzoian, RS (2005). “Gabaergic mechanism of cerebrovascular and neuroprotective effects of afobazole and picamilon”. Eksperimental’naia i Klinicheskaia Farmakologiia. 68 (1): 20–4. PMID 15786959.
- ^ Seredin, SB; Melkumian, DS; Val’dman, EA; Iarkova, MA; Seredina, TC; Voronin, MV; Lapitskaia, AS (2006). “Effects of afobazole on the BDNF content in brain structures of inbred mice with different phenotypes of emotional stress reaction”. Eksperimental’naia i Klinicheskaia Farmakologiia. 69 (3): 3–6. PMID 16878488.
- ^ Antipova, TA; Sapozhnikova, DS; Bakhtina, LIu; Seredenin, SB (2009). “Selective anxiolytic afobazole increases the content of BDNF and NGF in cultured hippocampal HT-22 line neurons”. Eksperimental’naia i Klinicheskaia Farmakologiia. 72 (1): 12–4. PMID 19334503.
- ^ Seredenin, SB; Antipova, TA; Voronin, MV; Kurchashova, SY; Kuimov, AN (2009). “Interaction of afobazole with sigma1-receptors”. Bulletin of Experimental Biology and Medicine. 148 (1): 42–4. doi:10.1007/s10517-009-0624-x. PMID 19902093. S2CID 37411324.
- ^ Medvedev, VE; Trosnova, AP; Dobrovol’skiĭ, AV (2007). “Psychopharmacotherapy of anxiety disorders in patients with cardio-vascular diseases: the use of aphobazole”. Zh Nevrol Psikhiatr Im S S Korsakova. 107 (7): 25–9. PMID 18379478.
- ^ Durnev AD, Zhanataev AK, Shreder OV, Seredenin SB (Jan–Feb 2009). “Antimutagenic and antiteratogenic properties of afobazole”. Eksp Klin Farmakol. 72 (1): 46–51. PMID 19334511.
//////////////Fabomotizole, Afobazole, фабомотизол , فابوموتيزول , 法莫替唑 , Obenoxazine, Afobazol, Afobazole, Aphobazole, Fabomotizole dihydrochloride, CM-346, CM346, CM 346,
CCOc1ccc2c(c1)[nH]c(n2)SCCN3CCOCC3.Cl.Cl

NEW DRUG APPROVALS
ONE TIMETO MAINTAIN THIS BLOG
$10.00
Sutimlimab-jome
(Heavy chain)
EVQLVESGGG LVKPGGSLRL SCAASGFTFS NYAMSWVRQA PGKGLEWVAT ISSGGSHTYY
LDSVKGRFTI SRDNSKNTLY LQMNSLRAED TALYYCARLF TGYAMDYWGQ GTLVTVSSAS
TKGPSVFPLA PCSRSTSEST AALGCLVKDY FPEPVTVSWN SGALTSGVHT FPAVLQSSGL
YSLSSVVTVP SSSLGTKTYT CNVDHKPSNT KVDKRVESKY GPPCPPCPAP EFEGGPSVFL
FPPKPKDTLM ISRTPEVTCV VVDVSQEDPE VQFNWYVDGV EVHNAKTKPR EEQFNSTYRV
VSVLTVLHQD WLNGKEYKCK VSNKGLPSSI EKTISKAKGQ PREPQVYTLP PSQEEMTKNQ
VSLTCLVKGF YPSDIAVEWE SNGQPENNYK TTPPVLDSDG SFFLYSRLTV DKSRWQEGNV
FSCSVMHEAL HNHYTQKSLS LSLGK
(Light chain)
QIVLTQSPAT LSLSPGERAT MSCTASSSVS SSYLHWYQQK PGKAPKLWIY STSNLASGVP
SRFSGSGSGT DYTLTISSLQ PEDFATYYCH QYYRLPPITF GQGTKLEIKR TVAAPSVFIF
PPSDEQLKSG TASVVCLLNN FYPREAKVQW KVDNALQSGN SQESVTEQDS KDSTYSLSST
LTLSKADYEK HKVYACEVTH QGLSSPVTKS FNRGEC
(Disulfide bridge: H22-H96, H132-L216, H145-H201, H224-H’224, H227-H’227, H259-H319, H365-H423, H’22-H’96, H’132-L’216, H’145-H’201, H’259-H’319, H’365-H’423, L23-L89, L136-L196, L’23-L’89, L’136-L’196)
Sutimlimab-jome
スチムリマブ (遺伝子組換え)
| Formula | C6436H9912N1700O2016S46 |
|---|---|
| CAS | 2049079-64-1 |
| Mol weight | 144832.7369 |
- BIVV009
- Sutimlimab
- Sutimlimab [INN]
- Sutimlimab [WHO-DD]
- TNT009
- UNII-GNWE7KJ995
- WHO 10757
| Efficacy | Anti-anemic, Anti-complement C1s antibody |
|---|---|
| Comment | Monoclonal antibody |
FDA APPROVED 2/4/2022, To decrease the need for red blood cell transfusion due to hemolysis in cold agglutinin disease, Enjaymo
A Humanized Antibody for the Specific Inhibition of the Classical Complement Pathway.

Sutimlimab, sold under the brand name Enjaymo, is a monoclonal antibody that is used to treat adults with cold agglutinin disease (CAD).[1][2][3] It is given by intravenous infusion.[1]
The most common side effects include respiratory tract infection, viral infection, diarrhea, dyspepsia (indigestion), cough, arthralgia (joint stiffness), arthritis, and swelling in the lower legs and hands.[2]
Sutimlimab prevents complement-enhanced activation of autoimmune human B cells in vitro.[4]
This drug is being developed by Bioverativ, a Sanofi company.[5] Sutimlimab was approved for medical use in the United States in February 2022.[2][6]
Sutimlimab-jome, a classical complement inhibitor, is a humanized monoclonal antibody expressed by recombinant in Chinese hamster ovary (CHO) cells and produced in vitro using standard mammalian cell culture methods. Sutimlimab-jome is composed of two heterodimers. Each heterodimer is composed of a heavy and a light polypeptide chain. Each heavy chain (H-chain) is composed of 445 amino acids and each light chain (L-chain) contains 216 amino acids. Sutimlimab-jome has a molecular weight of approximately 147 kDa.
ENJAYMO (sutimlimab-jome) injection is a sterile, clear to slightly opalescent, colorless to slightly yellow, preservative-free solution for intravenous use. Each single-dose vial contains 1,100 mg sutimlimab-jome at a concentration of 50 mg/mL with a pH of 6.1. Each mL contains 50 mg of sutimlimab-jome and also contains polysorbate 80 (0.2 mg), sodium chloride (8.18 mg), sodium phosphate dibasic heptahydrate (0.48 mg), sodium phosphate monobasic monohydrate (1.13 mg), and Water for Injection, USP. https://www.rxlist.com/enjaymo-drug.htm#clinpharm
Medical uses
Sutimlimab is indicated to decrease the need for red blood cell transfusion due to hemolysis (red blood cell destruction) in adults with cold agglutinin disease (CAD).[1][2]
History
The effectiveness of sutimlimab was assessed in a study of 24 adults with cold agglutinin disease who had a blood transfusion within the past six months.[2] All participants received sutimlimab for up to six months and could choose to continue therapy in a second part of the trial.[2] Based on body weight, participants received either a 6.5g or 7.5g infusion of sutimlimab into their vein on day 0, day 7, and every 14 days through week 25.[2]
In total, 54% of participants responded to sutimlimab.[2] The response was defined in the study as an increase in hemoglobin (an indirect measurement of the amount of red blood cells that are not destroyed) of 2 g/dL or greater (or to 12 g/dL or greater), and no red blood cell transfusions after the first five weeks of treatment; and no other therapies for cold agglutinin disease as defined in the study.[2]
The application for sutimlimab received orphan drug,[2][7] breakthrough therapy,[2] and priority review designations.[2]
Society and culture
Names
Sutimlimab is the International nonproprietary name (INN).[8]
//////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
https://www.sanofi.com/en/media-room/press-releases/2022/2022-02-04-23-00-00-2379517
FDA approves Enjaymo™ (sutimlimab-jome), first treatment for use in patients with cold agglutinin disease
- Enjaymo is the only approved treatment to decrease the need for red blood cell transfusion due to hemolysis, the destruction of red blood cells, in adults with cold agglutinin disease (CAD)
- Enjaymo addresses a serious and chronic unmet medical need for adults living with CAD, a rare blood disorder
Paris, February 4, 2022. The U.S. Food and Drug Administration (FDA) has approved Enjaymo™ (sutimlimab-jome) to decrease the need for red blood cell transfusion due to hemolysis in adults with cold agglutinin disease (CAD). Enjaymo is the first and only approved treatment for people with CAD and works by inhibiting the destruction of red blood cells (hemolysis).
Bill Sibold
Executive Vice President, Head of Specialty Care
“Until now, people living with cold agglutinin disease haven’t had an approved treatment option to manage the constant destruction of red blood cells. Without healthy, viable red blood cells, a chain reaction of debilitating signs and symptoms can be triggered, starting with severe anemia. Enjaymo is the only approved treatment to inhibit red blood cell destruction in CAD and help stop the chain reaction from the start.”
CAD, a rare autoimmune hemolytic anemia, is caused by antibodies called cold agglutinins binding to the surface of red blood cells, which starts a process that causes the body’s immune system to mistakenly attack healthy red blood cells and cause their rupture (hemolysis). As red blood cells have the vital job of carrying oxygen throughout the body, patients with CAD may experience severe anemia, which can result in fatigue, weakness, shortness of breath, light-headedness, chest pain, irregular heartbeat, and other potential complications. CAD is a chronic and rare blood disorder that impacts the lives of an estimated 5,000 people in the U.S.
Enjaymo, targeting C1s in the classical complement pathway
Enjaymo is a humanized monoclonal antibody that is designed to selectively target and inhibit C1s in the classical complement pathway, which is part of the innate immune system. By blocking C1s, Enjaymo inhibits the activation of the complement cascade in the immune system and inhibits C1-activated hemolysis in CAD to prevent the abnormal destruction of healthy red blood cells. Enjaymo does not inhibit the lectin and alternative pathways.
Enjaymo Phase 3 pivotal CARDINAL study results supporting approval
The approval of Enjaymo in the U.S. is based on positive results from the 26-week open label, single arm pivotal Phase 3 study in patients with CAD (n=24) who have a recent history of blood transfusion, also known as the CARDINAL study.
Catherine Broome, MD
Associate professor of medicine at Georgetown University Lombardi Comprehensive Cancer Center, and a principal investigator in the CARDINAL study
“For people living with cold agglutinin disease, it is as if their body’s immune system is waging a war on itself. The relentless destruction of healthy red blood cells is a daily, silent reality for people with CAD. For the first time, we have a treatment that targets complement-mediated hemolysis, which is the underlying cause of the red blood cell destruction in many CAD patients. In the pivotal study, patients treated with sutimlimab had an improvement in anemia as measured by hemoglobin and bilirubin levels during the 26-week study.”
In the study, Enjaymo met its primary efficacy endpoint, which was a composite endpoint defined as the proportion of patients who achieved normalization of hemoglobin (Hgb) level ≥12 g/dL or demonstrated an increase from baseline in Hgb level ≥2 g/dL at the treatment assessment time point (mean value from weeks 23, 25, and 26) and no blood transfusion from weeks 5 through 26 or medications prohibited per the protocol from weeks 5 through 26. Secondary endpoints were also met, including improvements in hemoglobin and normalization of bilirubin.
- The majority of patients (54%; n=13) met the composite primary endpoint criteria with 63% (n=15) of patients achieving a hemoglobin ≥ 12 g/dL or an increase of at least 2 g/dL; 71% (n=17) of patients remaining transfusion-free after week five; and 92% (n=22) of patients did not use other CAD-related treatments.
- For the secondary measures on disease process, patients enrolled experienced a mean increase in hemoglobin level of 2.29 g/dL (SE: 0.308) at week 3 and 3.18 g/dL (SE: 0.476) at the 26-week treatment assessment timepoint from the mean baseline level of 8.6 g/dL. The mean reduction in bilirubin levels (n=14) was by -2.23 mg/dL (95% CI: -2.49 to -1.98) from a mean baseline level of 3.23 mg/dL (2.7-fold ULN).
In the CARDINAL study, the most common adverse reactions occurring in 10 percent or more of patients were respiratory tract infection, viral infection, diarrhea, dyspepsia, cough, arthralgia, arthritis, and peripheral edema. Serious adverse reactions were reported in 13 percent (3/24) of patients who received Enjaymo. These serious adverse reactions were streptococcal sepsis and staphylococcal wound infection (n=1), arthralgia (n=1), and respiratory tract infection (n=1). None of the adverse reactions led to discontinuation of Enjaymo in the study. Dosage interruptions due to an adverse reaction occurred in 17 percent (4/24) of patients who received Enjaymo.
Following the completion of the 26-week treatment period of CARDINAL (Part A), eligible patients continued to receive Enjaymo in an extension study.
The recommended dose of Enjaymo is based on body weight (6,500 mg for people 39-75 kg and 7,500 mg for people >75 kg). Enjaymo is administered intravenously weekly for the first two weeks with administration every two weeks thereafter.
Enjaymo is expected to be available in the U.S. in the coming weeks. The U.S. list price, or wholesale acquisition cost, of Enjaymo is $1,800 per vial. Actual costs to patients are generally anticipated to be lower as the list price does not reflect insurance coverage, co-pay support, or financial assistance from patient support programs. As part of our commitment to ensure treatment access and affordability for innovative therapies, Enjaymo Patient Solutions provides disease education, financial and co-pay assistance programs and other support services to eligible patients. For more information, please call 1-833-223-2428.
Enjaymo received FDA Breakthrough Therapy and Orphan Drug designation, and priority review, which is reserved for medicines that, if approved, would represent significant improvements in safety or efficacy in treating serious conditions. Outside of the U.S., sutimlimab has been submitted to regulatory authorities in Europe and Japan and reviews are ongoing.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people’s lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761164s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA approves treatment for adults with rare type of anemia”. U.S. Food and Drug Administration. 4 February 2022. Retrieved 6 February 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Tvedt TH, Steien E, Øvrebø B, Haaverstad R, Hobbs W, Wardęcki M, et al. (February 2022). “Sutimlimab, an investigational C1s inhibitor, effectively prevents exacerbation of hemolytic anemia in a patient with cold agglutinin disease undergoing major surgery”. American Journal of Hematology. 97 (2): E51–E54. doi:10.1002/ajh.26409. PMID 34778998. S2CID 244116614.
- ^ Nikitin PA, Rose EL, Byun TS, Parry GC, Panicker S (February 2019). “C1s Inhibition by BIVV009 (Sutimlimab) Prevents Complement-Enhanced Activation of Autoimmune Human B Cells In Vitro”. Journal of Immunology. 202 (4): 1200–1209. doi:10.4049/jimmunol.1800998. PMC 6360260. PMID 30635392.
- ^ “Sutimlimab FDA Approval Status”. FDA. 19 May 2020.
- ^ “FDA approves Enjaymo (sutimlimab-jome), first treatment for use in patients with cold agglutinin disease”. Sanofi (Press release). 4 February 2022. Retrieved 6 February 2022.
- ^ “Sutimlimab Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 27 July 2016. Retrieved 6 February 2022.
- ^ World Health Organization (2018). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 80”. WHO Drug Information. 32 (3). hdl:10665/330907.
External links
- “Sutimlimab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03347396 for “A Study to Assess the Efficacy and Safety of BIVV009 (Sutimlimab) in Participants With Primary Cold Agglutinin Disease Who Have a Recent History of Blood Transfusion (Cardinal Study)” at ClinicalTrials.gov
//////////////Sutimlimab-jome, Enjaymo, FDA 2022, APPROVALS 2022, agglutinin disease, BIVV009, TNT009, UNII-GNWE7KJ995, WHO 10757, PEPTIDE, MONOCLONAL ANTIBODY, スチムリマブ (遺伝子組換え),

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG SUBSCRIPTIONS
$10.00
ARTEMETHER
ARTEMETHER
- Molecular FormulaC16H26O5
- Average mass298.375 Da
(3R,5aS,6R,8aS,9R,10S,12R,12aR)-10-methoxy-3,6,9-trimethyldecahydro-3,12-epoxy[1,2]dioxepino[4,3-i]isochromene
(4S,5R,8S,9R,10S,12R,13R)-10-Methoxy-1,5,9-trimethyl-11,14,15,16-tetraoxatetracyclo[10.3.1.04,13.08,13]hexadecane[
3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, decahydro-10-methoxy-3,6,9-trimethyl-, (3R,5aS,6R,8aS,9R,10S,12R,12aR)-
3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, decahydro-10-methoxy-3,6,9-trimethyl-, (5aS,6R,8aS,9R,10S,12R,12aR)-
71963-77-4[RN]
dihydroartemisinin methyl ether
Dihydroqinghaosu Methyl Ether
KD4165000
PALUTHER
- SM 224
- SM-224
- 3,12-Epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin, decahydro-10-methoxy-3,6,9-trimethyl-, [3R-(3α,5aβ,6β,8aβ,9α,10α,12β,12aR*)]-
- (3R,5aS,6R,8aS,9R,10S,12R,12aR)-Decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin
- (+)-Artemether
Artemether
CAS Registry Number: 71963-77-4
CAS Name: (3R,5aS,6R,8aS,9R,10S,12R,12aR)-Decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano[4,3-j]-1,2-benzodioxepin
Additional Names: dihydroartemisinin methyl ether; dihydroqinghaosu methyl ether; o-methyldihydroartemisinin
Manufacturers’ Codes: SM-224
Trademarks: Paluther (RPR)
Molecular Formula: C16H26O5, Molecular Weight: 298.37,
Percent Composition: C 64.41%, H 8.78%, O 26.81%
Literature References: Derivative of artemisinin, q.v. Prepn: Y. Li et al.,K’o Hsueh T’ung Pao24, 667 (1979), C.A.91, 211376u (1979); eidem,Acta Pharm. Sin.16, 429 (1981). Absolute configuration: X.-D. Luo et al.,Helv. Chim. Acta67, 1515 (1984). NMR spectral study: F. S. El-Feraly et al.,Spectrosc. Lett.18, 843 (1985). Inhibition of protein synthesis: H. M. Gu et al.,Biochem. Pharmacol.32, 2463 (1983). Antimalarial activity: S. Thaithong, G. H. Beale, Bull. WHO63, 617 (1985). Series of articles on chemistry, pharmacology and antimalarial efficacy: China Cooperative Research Group on Qinghaosu, J. Tradit. Chin. Med.2, 3-50 (1982). Toxicity data: eidem,ibid. 31. Clinical trial in cerebral malaria in children: M. B. van Hensbroek et al.,N. Engl. J. Med.335, 69 (1996). Review: R. N. Price, Expert Opin. Invest. Drugs9, 1815-1827 (2000).
Properties: Crystals, mp 86-88°. [a]D19.5 +171° (c = 2.59 in CHCl3). LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu).
Melting point: mp 86-88°
Optical Rotation: [a]D19.5 +171° (c = 2.59 in CHCl3)
Toxicity data: LD50 i.m. in mice: 263 mg/kg (China Cooperative Research Group on Qinghaosu)
Therap-Cat: Antimalarial.
Keywords: Antimalarial.
Artemether is an antimalarial agent used in combination with lumefantrine for the treatment of acute uncomplicated malaria caused by Plasmodium falciparum.
Artemether is an antimalarial agent used to treat acute uncomplicated malaria. It is administered in combination with lumefantrine for improved efficacy. This combination therapy exerts its effects against the erythrocytic stages of Plasmodium spp. and may be used to treat infections caused by P. falciparum and unidentified Plasmodium species, including infections acquired in chloroquine-resistant areas.
Artemether is a natural product which effectively kills both malarial parasites P. falciparum and P. vivax. Artemether is usually used in combination with Lumefantrine for the treatment of malaria. Arthemether also kills trematodes of the species Schistosoma, providing protection against schistosomiasis. Sesquiterpene lactones like artemether, artesunate, and artemisinin have potential applications in certain types of cancer and inflammatory conditions.
Artemether is a medication used for the treatment of malaria.[2] The injectable form is specifically used for severe malaria rather than quinine.[2] In adults, it may not be as effective as artesunate.[2] It is given by injection in a muscle.[2] It is also available by mouth in combination with lumefantrine, known as artemether/lumefantrine.[3][4]
Artemether causes relatively few side effects.[5] An irregular heartbeat may rarely occur.[5] While there is evidence that use during pregnancy may be harmful in animals, there is no evidence of concern in humans.[5] The World Health Organization (WHO) therefore recommends its use during pregnancy.[5] It is in the artemisinin class of medication.[5]
Artemether has been studied since at least 1981, and been in medical use since 1987.[6] It is on the World Health Organization’s List of Essential Medicines.[7]
Synthesis Reference
Haynes RK, Vonwiller SC: Extraction of artemisinin and artemisinic acid: preparation of artemether and new analogues. Trans R Soc Trop Med Hyg. 1994 Jun;88 Suppl 1:S23-6. Pubmed.
REF
ChemMedChem (2007), 2, (10), 1448-1463
PAT
| Malaria is a serious parasitic disease caused by Plasmodium parasites in the human body. Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium malaria and Plasmodium knowlesi are the parasites that live in humans, of which P. vivax and P. falciparum are the most common. |
| Traditional anti-malarial drugs mainly include quinine, chloroquine, primaquine, and pyrimethamine. In 1972, the antimalarial active ingredient artemisinin extracted from the Compositae plant Artemisia annuaL by Chinese scientists is the most popular antimalarial effect after chloroquine, pyrimethamine, primary amine and sulfonamide. Drugs, especially for the treatment of cerebral malaria and anti-chloroquine malaria. |
| At present, a large number of artemisinin derivatives have been synthesized and screened for antimalarial activity. Artemether is a compound with excellent curative effect. In addition to the advantages of artemisinin’s quick effect and low toxicity, its solubility in oil is also higher than that of artemisinin. Artemisinin is large, which is especially beneficial for the preparation of preparations. Since artemether has two products, α and β epimers, and the antimalarial activity of artemether is mainly isomer β, so the industrial automation and intelligent production of β-artemether and the improvement of the process are realized. , reducing the impurities produced by the reaction, improving the quality of the product, and improving the purity of the product are the problems that need to be solved in today’s scientific research. |
| Patent CN104557965B discloses a preparation process of β-artemether, which mainly includes adding dihydroartemisinin and etherification reagent to alcohol to form a reaction system, and then adding acid to the reaction system for reaction. Water or non-alkaline aqueous solution is added to the reaction system to crystallize, namely β-artemether. The preparation process claims to effectively inhibit the production of isomer α-artemether in the reaction, and can make the etherification reaction proceed mildly, with simple post-treatment and high purity; although the purity of the product has been improved, the yield and Purity needs to be further improved. |
| Patent CN102731523B discloses a method for preparing β-artemether, which mainly includes the reaction of artemisinin under the action of a reducing agent to generate dihydroartemisinin, and the reaction of dihydroartemisinin with p-toluenesulfonic acid to generate β-artemisinin. The crude artemether is crystallized with methanol, ethanol, ethylene glycol or isopropanol, filtered, washed and dried. The method for preparing B-artemether of the invention has mild conditions, is environmentally friendly, is suitable for industrial production, and has a product yield of over 90 percent and a purity of 99.2 percent. The crystallization step of the invention adopts organic reagents, which adversely affects the quality control of subsequent products. |
| Patent CN103180325B discloses a method for preparing β-artemether, which uses dihydroartemisinin as a raw material and undergoes etherification reaction with trimethyl orthoformate in organic solvents including esters and alkanes to obtain β-artemether. The method of the invention is easy to control in process operation, high in yield, low in cost and high in product quality, and is suitable for industrial production. The method requires vacuum distillation, the obtained crude product needs to be redissolved with methanol, decolorized with activated carbon, etc., new impurities are easily introduced, the operation is not simple enough, and the efficiency is low. |
| Patent CN107793428A discloses a preparation method of artemether, hydrogenating artemisinin to obtain dihydroartemisinin, adding trimethyl orthoformate, reacting with boron trifluoride ether solution, slowly adding saturated sodium bicarbonate solution dropwise, The system was adjusted to neutrality, the liquids were separated, the aqueous phase was extracted with dichloromethane, the organic phases were combined, washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure to obtain a solid; the obtained solid was dissolved in methanol, and an appropriate amount of activated carbon was added to obtain a solid. Reflux and decolorize, filter, add pure water dropwise to the filtrate, crystallize, wash with water, and dry to obtain artemether. However, this method requires steps such as extraction with an organic reagent dichloromethane and decolorization with activated carbon, which is cumbersome to handle. |
| Therefore, the following problems generally exist in the process of preparing β-artemether at present: |
| (1) when preparing β-artemether, the reaction time is longer, the impurities are large, and the purity and yield of the product are not high enough; |
| (2) The use of organic reagents in the subsequent purification process has a certain impact on the quality control of the product; |
| (3) The batch production equipment is adopted, the subsequent process steps are many, the degree of industrialization is low, the production efficiency is low, and it does not meet the requirements of GMP. |
| Example 1 |
| This embodiment includes the following steps: |
| (1) at room temperature, add methanol 2400L in the 3000L stirred tank (1), then add 600kg of dihydroartemisinin through the solid feed pump, and circulate and disperse evenly; |
| (2) add etherification agent trimethyl orthoformate and acid catalyst acetyl chloride through three-way automatic feeding mixing reactor again, the volume ratio is 500:100:3, the mixing reactor control temperature is 5 ℃, and the flow rate of control feeding is 5L /min; |
| (3) in the continuous flow pipeline, enter the second mixer and add 5% sodium bicarbonate solution to neutralize, and the adding speed is 1.0L/min, and is filtered through the fine filter; |
| (4) Then directly enter the 2000L crystallization reaction kettle 11 with 300L of water added in advance and keep the temperature at 10°C. At the same time, purified water was added to the reaction kettle at a rate of 12L/min, and the crystallization was continued for 1.5h; the jacket of the crystallization kettle was fed with -10°C chilled water for 30min, and the temperature of the system was controlled to 5°C. |
| (5) centrifugal washing, obtaining crude artemether 704.5kg, drying to obtain artemether fine product 608.6kg, β-artemether purity 99.83%, α-artemether impurity 0.12%, and other single impurities less than 0.1%, The content is 99.8%, the mass yield is 96.1%, and the molar yield is 91.42%. |
| Example 2 |
| This embodiment includes the following steps: |
| (1) at room temperature, 2400L of methanol was pumped into the 3000L reactor 1, and then 800kg of dihydroartemisinin was added by the solid feed pump, and the circulation was uniformly dispersed; |
| (2) add etherifying agent dimethyl phosphate and acid catalyst boron trifluoride ether through the three-way automatic feeding mixing reactor again, the volume ratio is 500:105:3.5, the mixing reactor control temperature is 3 ℃, and the control feeding flow rate is 3L/min; |
| (3) in the continuous flow pipeline, enter the second mixer and add 3% sodium bicarbonate solution to neutralize, and the speed of addition is 1.8L/min, through the fine filter; |
| (4) Directly enter the 2000L crystallization reaction kettles 11 and 12 with 300L of water added in advance and the temperature kept at 10°C. At the same time, purified water was added to the reaction kettle at 9 L/min, and the crystallization was continued for 2.5 hours; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 10 °C |
| (5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 96.2%, and the molar yield is 91.6%. |
| Example 3 |
| This embodiment includes the following steps: |
| (1) 2400L of methanol was pumped into the 3000L reactor F1 at room temperature, and then 400kg of dihydroartemisinin was added through the solid feed pump, and the circulation was uniformly dispersed; |
| (2) Add etherification agent dimethyl phosphate and acid catalyst trimethylchlorosilane through the three-way automatic feeding mixing reactor, the volume ratio is 500:95:2.5, the mixing reactor is controlled at a temperature of 8 °C, and the feeding liquid is controlled to be added. The flow rate is 7L/min, and the reaction time is; |
| (3) in the continuous flow pipeline, enter the second mixer and add 8% sodium bicarbonate solution for neutralization, and the rate of addition is 0.6L/min, passing through the fine filter; |
| (4) Directly enter into the 2000L crystallization reactor J2 with 300L water added in advance and keeping the temperature at 10°C. At the same time, purified water was added to the reaction kettle at 15 L/min, and the crystallization was continued for 1 hour; the jacket of the crystallization kettle was fed with -10 °C chilled water for 30 minutes, and the temperature of the system was controlled to 0 °C |
| (5) centrifugal washing, obtain crude artemether 939.3kg, oven dry to obtain artemether fine product 809.7kg, β-artemether purity 99.81%, α-artemether impurity 0.11%, other single impurities are less than 0.1%, The content is 99.8%, the mass yield is 95.5%, and the molar yield is 90.9%. |
| Comparative Example 1 |
| The difference between this embodiment and Example 1 is that hydrochloric acid is used instead of the acidic catalyst. Finally, 633.6kg of crude artemether was obtained, and 550.3kg of fine artemether was obtained by drying. The purity of β-artemether was 94.20%, and the impurities of α-artemether were 3.66%. %, and the molar yield was 80.6%. |
| Comparative Example 2 |
| The difference between this embodiment and Example 1 is that the step of adding water in advance in the crystallization kettle is removed. Finally, 645.1kg of crude artemether was obtained, and 562.2kg of fine artemether was obtained by drying. The purity of β-artemether was 99.68%, the impurity of α-artemether was 0.22%, and the average of single and impurity was less than 0.1%. The mass yield was 88.7%. %, and the molar yield was 84.4%. |
| In Comparative Example 2, the step of adding water in advance in the crystallization was removed, the purity of β-artemether was 99.68%, and the yield was 88.7%. The yield dropped by 7.6%. |
| The above detailed description is a specific description of one of the feasible embodiments of the present invention, and this embodiment is not intended to limit the patent scope of the present invention. Any equivalent implementation or modification that does not depart from the present invention shall be included in the present invention. within the scope of the technical solution. |
SYN1
Synthetic Reference
Continuous synthesis of artemisinin-derived medicines; Gilmore, Kerry; Kopetzki, Daniel; Lee, Ju Weon; Horvath, Zoltan; McQuade, D. Tyler; Seidel-Morgenstern, Andreas; Seeberger, Peter H. Chemical Communications (Cambridge, United Kingdom); Volume 50; Issue 84; Pages 12652-12655; Journal; 2014
SYN2
Synthetic Reference
An Improved Manufacturing Process for the Antimalaria Drug Coartem. Part I; Boehm, Matthias; Fuenfschilling, Peter C.; Krieger, Matthias; Kuesters, Ernst; Struber, Fritz; Organic Process Research & Development; Volume 11; Issue 3; Pages 336-340; Journal; 2007
SYN3
Synthetic Reference
Some transition metal complexes bearing artemisinin derivatives and (N-N-O) tridentate chromium (III) complexes ligated by 2-benzolmidazo-yl-6-acetyl-pyridines for catalytic behaviour towards ethylene; Obaleye, Joshua Ayoola; Amolegbe, Saliu Alao; Adewuyi, Sheriff; Sun, Wenhua; Oshodi, Margaret Damilola; Journal of Chemistry and Chemical Engineering; Volume 4; Issue 12; Pages 23-32; Journal; 2010
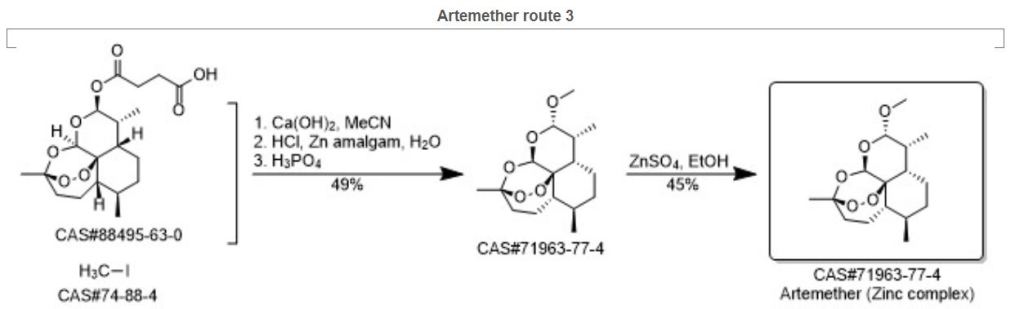
SYN4
Synthetic Reference
Method and apparatus for the synthesis of dihydroartemisinin and artemisinin derivatives; Kopetzki, Daniel; McQuade, David Tyler; Seeberger, Peter H.; Gilmore, Kerry; Assignee Max-Planck-Gesellschaft zur Foerderung der Wissenschaften e.V., Germany; 2015; Patent Information; Jan 21, 2015; EP 2826779 A1
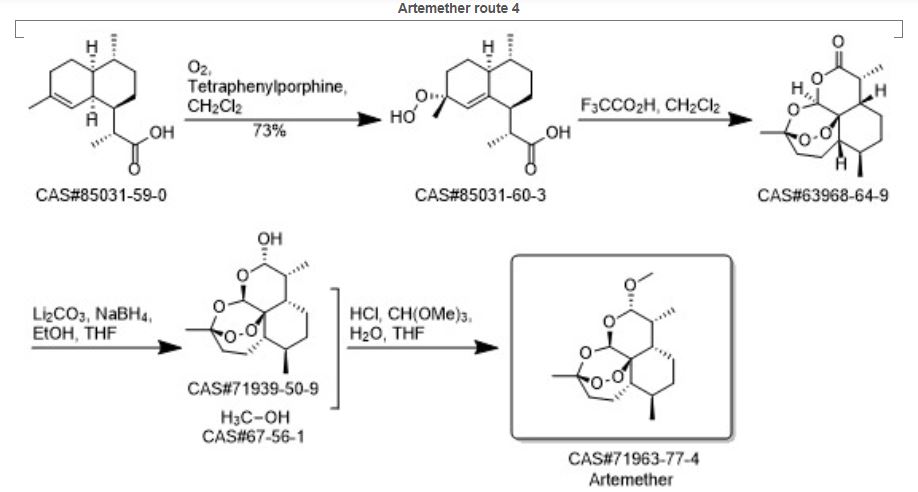
PAPER
https://pubs.rsc.org/en/content/articlehtml/2014/ra/c4ra05531d
An efficient one pot green synthesis of β-artemether/arteether from artemisinin has been developed using a sodium borohydride-cellulose sulfuric acid (CellSA) catalyst system. The green methodology is high yielding and the catalyst has good recyclability.
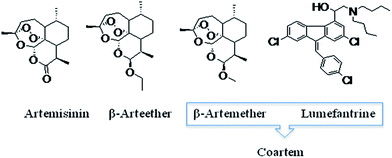
Experimental section
Representative procedure for catalyst preparation
Preparation of cellulose sulfuric acid.To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 °C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder.17
General procedure for the arteether from artemisinin in one-pot
To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 ml) and trimethyl orthoacetate (0.5 ml) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 °C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40 ± 5 °C in water. The reaction mass was seeded with pure β-arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried.
General procedure for the artemether from artemisinin in one-pot
Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at −5 to 0 °C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.
Preparation of cellulose sulfuric acid. To a magnetically stirred mixture of 5.00 g of cellulose (DEAE for column chromatography, Merck) in 20 ml of n-hexane, 1.0 g of chlorosulfonic acid (9 mmol) was added dropwise at 0 0 C over 2 h. HCl gas was removed from the reaction vessel immediately. After the addition was complete, the mixture was stirred for 2 h. Then, the mixture was filtered, washed with 30 ml of acetonitrile, and dried at room temperature to obtain 5.47 g cellulose sulfuric acid as a white powder. K General procedure for the arteether from artemisinin in one-pot. To a solution of artemisinin (200 mg, 0.71 mmol) in ethanol (15 mL) and trimethyl orthoacetate (0.5 mL) was added NaBH4 (67 mg, 1.77 mmol, 2.5 equ.) and cellulose sulfuric acid (0.015 g). Reaction mixture was was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. Then we added a solution of sodium bicarbonate to quenched the reaction. The slurry was stirred in an below 20 0 C for 1 h and allowed to settle for 30 min. Solid crude arteether was collected by filtration, and the cake was washed with of ethanol. The reaction mass was heated to 40± 5 0 C in water. The reaction mass was seeded with pure β–arteether. Then it was filtered, washed with chilled 50% solution of ethanol in water and dried. General procedure for the artemether from artemisinin in one-pot. Artemisinin (200 mg, 0.71 mmol) in methanol (15 ml) and trimethylorthoformate (0.5 ml), cellulose sulfuric acid (0.015 g), was carried out at -5 to 0°C for 60 min, and then stirred at room temperature for 1.5 h. The reaction was monitored by TLC and HPLC to check completion of the reaction. The cellulose sulfuric acid was removed by filtration, the filtrate was concentrated. Then we added a solution of sodium bicarbonate to terminate the reaction. Then, follow above recrystallization method.
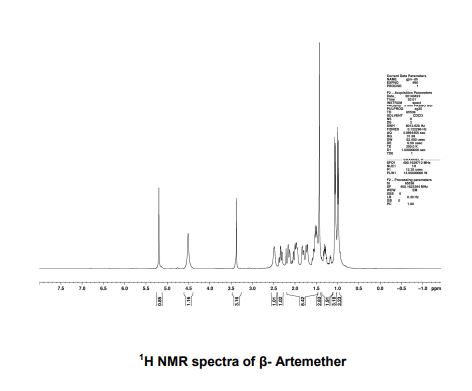
PATENT
https://patents.google.com/patent/US6683193B2/en
Approximately, out of the 4 billion people suffering from malaria, 1-3 million, mostly children die every year worldwide. The rapidly spreading multidrug resistant parasite to standard quinoline based antimalarial drugs such as chloroquine and mefloquine based antimalarial complicate chemotherapy treatment of malaria patients.
Artemether is a methyl ether derivative of dihydroartemisinin. Dihydroartemisinin is derived from arternisinin, a novel sesquiterpene endoperoxide isolated from the plant Artemisia annua. Artemisinin and its derivative artemether, arteether, artelinate and artesunate a novel class of antimalarials derived from Artemisia annua are now proving their promising activity and being used for the treatment; of uncomplicated severe complicated/cerebral and multi drug resistant malaria.
Artemether, developed in France and China has undergone extensive preclinical, animal, toxicological studies as well as clinical studies. Artemether is more potential as compared to artemisinin and an antimalarial drug especially for treating multi drug resistant and complicated strains of Plasmodium falciparum.
Artemether shows rapid shizonticidal action with quicker parasite clearance rate, short half life less side effect and low recrudence rate. Brossi, et al (Brossi, A; Venugopalan, B, Domingueg, G L; Yeh, H. J. C; Flippend-Anderson, J. L.; Buchs, P; Luo, X. D.; Milhous,W and peters, W; J. Med. Chem. 31, 646-649, 1988) reported the preparation of arteether, the ethyl ether derivative of dihydroartemisinin in two steps: First artemisinin was reduced with an excess of sodium borohydride in methanol at 0 to −5 degree C. in 3 hours to dihydroartemisinin in 79% yield. In the second step arteether is prepared by dissolving the dihydroartemisinin in the solvent mixture of benzene and ethanol at 45 degree C. followed by addition of BF3 etherate and refluxing the reaction mixture at 70 degree C. for one hour. After completion of the reaction it was worked up, dried over anhydrous sodium sulphate with removal of the solvent dichloromethane. The reaction yielded arteether along with some impurities. Column chromatography of the reaction mixture over silica gel, 1:20 ratio yielded pure alpha and beta arteether in nearly qualitative yield.
EL-Feraly etal. (E L Feraly, F. S; Al-Yahya M A; Orabi, K. Y; Mc-Phail D R and Me Phail A. T. J.Nat.Prod. 55, 878-883 1992) reported the preparation of arteether by a process in which anhydrodihydroartemisinin, prepared from artemisinin was dissolved in absolute alcohol. The reaction mixture was stirred in the presence of p-toluene sulphonic acid used as a catalyst. On workup it yielded a mixture of beta arteether and C-11 epimer in the ratio of 3:1. In this process only beta arteether, is obtained and separation of C-11 epimer is difficult and preparation of anhydrodihydroartemisinin is a tedious process. The reaction took 22 hours to complete. The lewis acid catalyst used in this reaction is required in large amount (60 mg. acid catalyst by 100 mg. anhydrodihydroartemisinin).
In another method Bhakuni etal (Bhakuni, R. S.; Jain D. C and Sharma R. P. Indian. J. Chemistry, 34B, 529-30, 1995) arteether, artemether and other ether derivatives were prepared from dihydroartemisinin in different alcohol and benzene in the presence of chlorotrimethylsilane catalyst in 2-4 hours at room temperature. After workup of the reaction mixture and removal of the solvent, the impure reaction products were purified over silica gel column to obtained the pure mixture of alpha, beta ethers.
Another method is reported by Lin et al. (Lin, A. J. and Miller, R. E, J.Med Chero. 38,764-770, 1995) In this method the new ether derivatives were prepared by dissolving dihydroarternisinin in anhydrous ether and appropriate alcohol followed by BF3-etherate. The reaction mixture was stirred at room temperature for 24 hours. The yield of the purified products ranged from 40-90%. Purification was achieved by the use of silica gel chromatography.
Another method described by Jain et al (Jain D. C, Bhakuni R. S, Saxena S, kumar, S and Vishwakarma, R. A.) the preparation of arteether from artemisinin comprises: Reduction of artemisinin into dihydroartemisinin. Isolation of dihydroartemisinin. Acylation of dihydroartemisinin by dissolving it in alcohol and adding trialkylorthoformate in the reaction mixture, which produce ethers in quantitative yield in 10 hours at 40 degree C.
The above mentioned methods carry some disadvantages being less cost effective and more time consuming as compared to the present invention. Moreover, benzene, a carcinogenic solvent, used in the previous methods is not acceptable according to the health standard. Further, all the above methods require at least two separate steps to convert artemisinin into ethers i.e. reduction of the artemisinin into dihydroartemisinin in the first pot followed by isolation of dihydroartemisinin and then comes the second step of conversion of dihydroartemisinin into different ethers in the second pot. However, the present invention provide an efficient method for conversion of artemisinin into artemether
EXAMPLE 1
Artemisinin (3 g.) was dissolved in dry methanol (40 ml) at room temperature. It was cooled to −5 degree C. Now sodium borohydride (700 mg) was added slowly for 30 minutes and the reaction mixture was stirred for about 1.5 hours. The reaction was monitored by TLC to check completion of the reduction step. Now cation exchange resin (8 g) was added slowly at cooling temperature and the reaction mixture was further stirred at room temperature for about 2 hours. Cooled water was added to the reaction mixture and the resin was filtered.
The filtrate was neutralized with 5% sodium bicarbonate solution followed by extracting with dichloromethane (3×50 ml). The dichloromethane extract was dried over anhydrous sodium sulphate and evaporation of the solvent yielded 3.21 g, of artemether along with some impurities. The impure artemether was purified over silica gel column (1:5 ratio) in hexane:ethyl acetate (96:4) furnished pure alpha and beta artemether 2.43 g (81% w/w). Small portion of artemether was separated by prep TLC into alpha and beta isomers and characterized by the analysis of their IR, Mass and 1H NMR data.
EXAMPLE 2
The experiment was carried out following example 1 except in place of solid acid catalyst in the second reaction. Liquid acid catalyst chlorotrimethylsilane was added at cooling temperature for methylation reaction. The overall yield of pure alpha, beta artemether after column chromatography was 2.46 gm (82% w/w).
EXAMPLE 3
Artemisinin (100 g.) was dissolved in dry methanol (3 ml). Added sodium borohydride (30 mg.) at −5° C. The reaction mixture was stirred for 2 hours. After completion of the reaction, trifluroacetic acid (0.5 ml) was added and the reaction mixture was stirred for 5 hours. The methylation was incompleted and after workup the artemether was purified by prep TLC to yield 46 mg (46%) pure alpha, beta artemether.
EXAMPLE 4
The experiment was carried following example 1 except before column chromatography, the beta isomer (40%) was recrystallized in hexane from impure artemether and remaining mother liquor was purified over silica gel column in 1:5 ratio to yield alpha and beta artemether in 80% w/w.

PAPER
https://www.sciencedirect.com/science/article/abs/pii/S0920586114003307
The earlier developed flow protocol for stoichiometric reduction of an important biologically derived pharmaceutical precursor, artemisinin, to dihydroartemisinin was extended to a sequential reaction to produce one of the final APIs, artemether. A highly active heterogeneous catalyst was found for the etherification reaction. The use of QuadraSil catalyst allows to eliminate one step of reaction workup. A comparative Life Cycle Assessment of both reactions has shown advantages of the flow process over the optimized literature batch protocols. Results of LCA highlight the significance of solvents in pharmaceuticals manufacture and the advantage of flow technology, enabling small solvent inventories to be used.
Graphical abstract

PAPER
http://chem.vander-lingen.nl/articles/Target:_Artemether/id/126/itemid/663
In a previous episode chemical company Sanofi was granted exclusive access to certain yeast cells that produce a precursor to anti-malarial drug artemisinin. One of the charities making this all possible is the Bill and Melinda Gates Foundation. Another charity that has apparently entered into the drug business is the Clinton Health Access Initiative. Bill together with Rodger Stringham and David Teager report on an improved process for the conversion of artemisinin to artemether in Organic Process Research & Development (DOI).
Does the Clinton Health Access Initiative have a pilot-plant facility or even an organic lab? Unless it is all cramped in suite 400 on Dorchester Avenue in Boston, the article is not very explicit. The acknowledgements mention Mangalam Drugs and Organics.
Case at hand: artemether has the carbonyl group replaced by a methoxy group in a two-step reduction – methylation. So far so good. The point is that principal supplier Novartis reports up to 68% overall yields but that many Indian and Chinese suppliers working with the procedure generously supplied by same Novartis, report considerably lower figures (58-62%). But Why? And how can the process be improved?
Any organic chemist knows reported yields in the literature should be considered with caution. Chemists tend to be over-optimistic / self-delusionional but this scenario was not considered. No bottlenecks were encountered in step 1, the reduction with sodium borohydride. Only the beta form was isolated due to its poor solubility in the quench. Drying the product without heat prevented formation of one byproduct. Moving on to step two, the methylation with HCl in methanol was more troublesome. The byproducts lurking around the corner are the anomer and the elimination product. Co-solvent (co-reagent?) trimethyl orthoformate made all the difference. The critical element in the workup was first adding more methanol before adding the base quench otherwise you end up with a nasty gum. The new record yield for the improved synthesis is 72%.
But what have all these suppliers been doing wrong with the existing Novartis procedure? The answer to that question, remains unclear. The Novartis yield for step two with co-solvent methylacetate (not the formate) was confirmed so no surprise there.
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Artemether is an antimalarial drug for uncomplicated malaria caused by P. falciparum (and chloroquine-resistant P. falciparum) or chloroquine-resistant P. vivax parasites.[8] Artemether can also be used to treat severe malaria.[2]
The World Health Organization (WHO) recommends the treatment of uncomplicated P. falciparum with artemisinin-based combination therapy.[9] Given in combination with lumefantrine, it may be followed by a 14-day regimen of primaquine to prevent relapse of P. vivax or P. ovale malarial parasites and provide a complete cure.[10]
Artemether can also be used in treating and preventing trematode infections of schistosomiasis when used in combination with praziquantel.[11]
Artemether is rated category C by the FDA based on animal studies where artemisinin derivatives have shown an association with fetal loss and deformity. Some studies, however, do not show evidence of harm.[12][13]
Side effects
Possible side effects include cardiac effects such as bradycardia and QT interval prolongation.[14] Also, possible central nervous system toxicity has been shown in animal studies.[15][16]
Drug interactions
Plasma artemether level was found to be lower when the combination product was used with lopinavir/ritonavir.[16] There is also decreased drug exposure associated with concurrent use with efavirenz or nevirapine.[17][18]
Artemether/lumefantrine should not be used with drugs that inhibit CYP3A4.[19]
Hormonal contraceptives may not be as efficacious when used with artemether/lumefantrine.[19]
Pharmacology
Mechanism of action
Artemether is an artemisinin derivative and the mechanism of action for artemisinins is.[medical citation needed]
Artemether interact with ferriprotoporphyrin IX (heme) or ferrous ions in the acidic parasite food vacuole, and generates cytotoxic radical species
The accepted mode of action of the peroxide containing drug involve its interaction with heme (byproduct of hemoglobin degradation), derived from proteolysis of haemoglobin. This interaction results in the formation of toxic oxygen and carbon centered radicals.
One of the proposed mechanisms is that through inhibiting anti-oxidant and metabolic enzymes, artemisinin derivatives inflict oxidative and metabolic stress on the cell. Some pathways affected may concern glutathione and glucose metabolism. As a consequence, lesions and reduced growth of the parasite may result.[20]
Another possible mechanism of action suggests that arteristinin drugs exert their cidal action through inhibiting PfATP6. Since PfATP6 is an enzyme regulating cellular calcium concentration, its malfunctioning will lead to intracellular calcium accumulation, which in turns causes cell death.[21]
Pharmacokinetics
Absorption of artemether is improved 2- to 3-fold with food. It is highly bound to protein (95.4%). Peak concentrations of artemether are seen 2 hours after administration.[4]
Artemether is metabolized in the human body to the active metabolite, dihydroartemisinin, primarily by hepatic enzymes CYP3A4/5.[4] Both the parent drug and active metabolite are eliminated with a half-life of about 2 hours.[4]
Chemistry
Artemether is a methyl ether derivative of artemisinin, which is a peroxide-containing lactone isolated from the antimalarial plant Artemisia annua. It is also known as dihydroartemisinin methyl ether, but its correct chemical nomenclature is (+)-(3-alpha,5a-beta,6-beta,8a-beta, 9-alpha,12-beta,12aR)-decahydro-10-methoxy-3,6,9-trimethyl-3,12-epoxy-12H-pyrano(4,3-j)-1,2-benzodioxepin. It is a relatively lipophilic and unstable drug,[22] which acts by creating reactive free radicals in addition to affecting the membrane transport system of the plasmodium organism.[14]
References
- ^ “Artemether – Drugs.com”. http://www.drugs.com. Archived from the original on 20 December 2016. Retrieved 7 December 2016.
- ^ Jump up to:a b c d e f Esu, Ekpereonne B.; Effa, Emmanuel E.; Opie, Oko N.; Meremikwu, Martin M. (18 June 2019). “Artemether for severe malaria”. The Cochrane Database of Systematic Reviews. 6: CD010678. doi:10.1002/14651858.CD010678.pub3. ISSN 1469-493X. PMC 6580442. PMID 31210357.
- ^ “Artemether and Lumefantrine”. The American Society of Health-System Pharmacists. Archived from the original on 20 December 2016. Retrieved 28 November 2016.
- ^ Jump up to:a b c d “Coartem- artemether and lumefantrine tablet”. DailyMed. 5 August 2019. Retrieved 26 April 2020.
- ^ Jump up to:a b c d e Kovacs, SD; Rijken, MJ; Stergachis, A (February 2015). “Treating severe malaria in pregnancy: a review of the evidence”. Drug Safety. 38 (2): 165–81. doi:10.1007/s40264-014-0261-9. PMC 4328128. PMID 25556421.
- ^ Rao, Yi; Zhang, Daqing; Li, Runhong (2016). Tu Youyou and the Discovery of Artemisinin: 2015 Nobel Laureate in Physiology or Medicine. World Scientific. p. 162. ISBN 9789813109919. Archived from the original on 2017-09-10.
- ^ World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. 2019. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Makanga, Michael; Krudsood, Srivicha (2009-10-12). “The clinical efficacy of artemether/lumefantrine (Coartem)”. Malaria Journal. 8 (Suppl 1): S5. doi:10.1186/1475-2875-8-S1-S5. ISSN 1475-2875. PMC 2760240. PMID 19818172.
- ^ Treatment of Uncomplicated Plasmodium falciparum Malaria. World Health Organization. 2015-01-01. Archived from the original on 2017-09-10.
- ^ Treatment Of Uncomplicated Malaria Caused By P. vivax, P. ovale, P. malariae or P. knowlesi. World Health Organization. 2015-01-01. Archived from the original on 2017-09-10.
- ^ Pérez del Villar, Luis; Burguillo, Francisco J.; López-Abán, Julio; Muro, Antonio (2012-01-01). “Systematic review and meta-analysis of artemisinin based therapies for the treatment and prevention of schistosomiasis”. PLOS ONE. 7 (9): e45867. Bibcode:2012PLoSO…745867P. doi:10.1371/journal.pone.0045867. ISSN 1932-6203. PMC 3448694. PMID 23029285.
- ^ Dellicour S, Hall S, Chandramohan D, Greenwood B (2007). “The safety of artemisinins during pregnancy: a pressing question”. Malaria Journal. 6: 15. doi:10.1186/1475-2875-6-15. PMC 1802871. PMID 17300719.
- ^ Piola P, Nabasumba C, Turyakira E, et al. (2010). “Efficacy and safety of artemether—lumefantrine compared with quinine in pregnant women with uncomplicated Plasmodium falciparum malaria: an open-label, randomised, non-inferiority trial”. Lancet Infect Dis. 10 (11): 762–769. doi:10.1016/S1473-3099(10)70202-4. hdl:10144/116337. PMID 20932805.
- ^ Jump up to:a b “Artemether”. http://www.antimicrobe.org. Archived from the original on 2017-02-23. Retrieved 2016-11-09.
- ^ “WHO Model Prescribing Information: Drugs Used in Parasitic Diseases – Second Edition: Protozoa: Malaria: Artemether”. apps.who.int. Archived from the original on 2016-11-10. Retrieved 2016-11-09.
- ^ Jump up to:a b Askling, Helena H.; Bruneel, Fabrice; Burchard, Gerd; Castelli, Francesco; Chiodini, Peter L.; Grobusch, Martin P.; Lopez-Vélez, Rogelio; Paul, Margaret; Petersen, Eskild (2012-01-01). “Management of imported malaria in Europe”. Malaria Journal. 11: 328. doi:10.1186/1475-2875-11-328. ISSN 1475-2875. PMC 3489857. PMID 22985344.
- ^ van Geertruyden, J.-P. (2014). “Interactions between malaria and human immunodeficiency virus anno 2014”. Clinical Microbiology and Infection. 20 (4): 278–285. doi:10.1111/1469-0691.12597. PMC 4368411. PMID 24528518.
- ^ Kiang, Tony K. L.; Wilby, Kyle J.; Ensom, Mary H. H. (2013-10-26). “Clinical Pharmacokinetic Drug Interactions Associated with Artemisinin Derivatives and HIV-Antivirals”. Clinical Pharmacokinetics. 53 (2): 141–153. doi:10.1007/s40262-013-0110-5. ISSN 0312-5963. PMID 24158666. S2CID 1281113.
- ^ Jump up to:a b Stover, Kayla R.; King, S. Travis; Robinson, Jessica (2012-04-01). “Artemether-Lumefantrine: An Option for Malaria”. Annals of Pharmacotherapy. 46 (4): 567–577. doi:10.1345/aph.1Q539. ISSN 1060-0280. PMID 22496476. S2CID 7678606.
- ^ Saeed, ME; Krishna, S; Greten, HJ; Kremsner, PG; Efferth, T (August 2016). “Antischistosomal activity of artemisinin derivatives in vivo and in patients”. Pharmacological Research. 110: 216–26. doi:10.1016/j.phrs.2016.02.017. PMID 26902577.
- ^ Guo, Zongru (2016-03-01). “Artemisinin anti-malarial drugs in China”. Acta Pharmaceutica Sinica B. 6 (2): 115–124. doi:10.1016/j.apsb.2016.01.008. PMC 4788711. PMID 27006895.
- ^ De Spiegeleer, B.M.J.; D’Hondt, M.; Vangheluwe, E.; Vandercruyssen, K.; De Spiegeleer, B.G.I.; Jansen, H.; Koijen, I.; Van Gompel, J. (2012). “Relative response factor determination of artemether degradants with a dry heat stress approach”. Journal of Pharmaceutical and Biomedical Analysis. 70: 111–116. doi:10.1016/j.jpba.2012.06.002. hdl:1854/LU-2938963. PMID 22770733.
| Clinical data | |
|---|---|
| Trade names | Many[1] |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Intramuscular[2] |
| ATC code | P01BE02 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 71963-77-4 |
| PubChem CID | 68911 |
| DrugBank | DB06697 |
| ChemSpider | 62138 |
| UNII | C7D6T3H22J |
| KEGG | D02483 |
| ChEBI | CHEBI:195280 |
| ChEMBL | ChEMBL1237051 |
| PDB ligand | D8Z (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID7040651 |
| ECHA InfoCard | 100.189.847 |
| Chemical and physical data | |
| Formula | C16H26O5 |
| Molar mass | 298.379 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 86 to 88 °C (187 to 190 °F) |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////ARTEMETHER, ANTIMALARIAL, SM 224, SM-224
[H][C@@]12CC[C@@H](C)[C@]3([H])CC[C@@]4(C)OO[C@@]13[C@]([H])(O[C@H](OC)[C@@H]2C)O4

NEW DRUG APPROVALS
ONE TIME
$10.00
BIFONAZOLE
BIFONAZOLE
- Molecular FormulaC22H18N2
- Average mass310.392 Da
(±)-1-(p,a-Diphenylbenzyl)imidazole
(±)-Bifonazole
1-([1,1′-Biphenyl]-4-ylphenylmethyl)-1H-imidazole
1-(p,α-Diphenylbenzyl)imidazole
262-336-6[EINECS]
4887
60628-96-8[RN]
бифоназол
بيفونازول
联苯苄唑
- BAY H 4502
- BAY-H-4502
Bifonazole
CAS Registry Number: 60628-96-8
CAS Name: 1-([1,1¢-Biphenyl]-4-ylphenylmethyl)-1H-imidazole
Additional Names: (±)-1-(p,a-diphenylbenzyl)imidazole
Manufacturers’ Codes: Bay h 4502
Trademarks: Amycor (Lipha); Azolmen (Menarini); Bedriol (Andromaco); Mycospor (Bayer); Mycosporan (Bayer)
Molecular Formula: C22H18N2, Molecular Weight: 310.39
Percent Composition: C 85.13%, H 5.85%, N 9.03%
Literature References: Antimycotic deriv of imidazole. Prepn: E. Regel et al.,DE2461406; eidem,US4118487 (1976, 1978 both to Bayer). Series of articles on in vitro and in vivo antimycotic efficacy, microscopic studies, pharmacokinetics, efficacy in dermatomycoses and comparison with clotrimazole and miconazole, q.q.v.:Arzneim.-Forsch.33, 517-551, 745-754 (1983). Toxicology: G. Schlüter, ibid. 739.
Properties: Crystals from acetonitrile, mp 142°. Very lipophilic. Sol in alcohols, DMF, DMSO. Soly in water at pH 6: <0.1 mg/100 ml. Stable in aq soln at pH 1-12. LD50 in male mice, rats (mg/kg): 2629, 2854 orally (Schlüter).
Melting point: mp 142°
Toxicity data: LD50 in male mice, rats (mg/kg): 2629, 2854 orally (Schlüter)
Therap-Cat: Antifungal.
Keywords: Antifungal (Synthetic); Imidazoles.
BrandsAmycor (Merck) / Azolmen (Menarini) / Bayclear Plus (Bayer) / Bifonol (Mayado Seiyaku) / Canespor (Bayer) / Canesten (Bayer) / Mycospor (Bayer)
Bifonazole (trade name Canespor among others[1]) is an imidazole antifungal drug used in form of ointments.
It was patented in 1974 and approved for medical use in 1983.[2] There are also combinations with carbamide for the treatment of onychomycosis.
Bifonazole is an azole antifungal drug used to treat fungal skin infections, such as dermatomycosis.
- Synonyms:Bifonazolum
- ATC:D01AC10
- MW:310.40 g/mol
- CAS-RN:60628-96-8
- InChI Key:OCAPBUJLXMYKEJ-UHFFFAOYSA-N
- InChI:InChI=1S/C22H18N2/c1-3-7-18(8-4-1)19-11-13-21(14-12-19)22(24-16-15-23-17-24)20-9-5-2-6-10-20/h1-17,22H
- EINECS:262-336-6
- LD50:57 mg/kg (M, i.v.); 2629 mg/kg (M, p.o.);
63 mg/kg (R, i.v.); 1463 mg/kg (R, p.o.);
>500 mg/kg (dog, p.o.)
Derivatives
Monohydrochloride
- Formula:C22H18N2 • HCl
- MW:346.86 g/mol
- CAS-RN:60629-09-6
Sulfate
- Formula:C22H18N2 • xH2O4S
- MW:unspecified
- CAS-RN:60629-08-5
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 98-88-4 | C7H5ClO | benzoyl chloride | Benzoyl chloride |
| 92-52-4 | C12H10 | biphenyl | 1,1′-Biphenyl |
| 7515-73-3 | C19H15Cl | (±)-4-(chlorophenylmethyl)biphenyl | 1,1′-Biphenyl, 4-(chlorophenylmethyl)- |
| 288-32-4 | C3H4N2 | imidazole | 1H-Imidazole |
SYN
Synthesis Reference
Regal, E., Draber, W., Buchel, K.H.and Plempel, M.; U.S. Patent 4,118,487; October 3,1978; assigned to Bayer A.G.
SYN

SYN
(CAS NO.: ), with its systematic name of , 1-(alpha-(4-biphenylyl)benzyl)-, could be produced through many synthetic methods.
Following is one of the synthesis routes: (I) could be reduced with NaBH4 in ethanol to produce 4-phenylbenzhydrol (II), and the yielding product is then condensed with imidazole (III) in the presence of SOCl2 in acetonitrile.

PAT
https://patents.google.com/patent/DE10332684B3/en
- The The present invention relates to a process for the preparation of Bifonazole (1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole) by reacting 1-biphenyl-4-yl (phenyl) methanol with a chlorinating reagent in cyclohexane and subsequent coupling with imidazole.
- [0002]The compound bifonazole (1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole) is off DE-A 2 461 406 known and corresponds to the formula (I). Due to its antifungal activity, it can be used as an agent for the treatment of fungal diseases.
- [0003]Various methods for preparing this compound are known. So describes DE-A 2 461 406 the synthesis (process 1) of bifonazole (Example 1) starting from biphenyl-4-yl (phenyl) methanol by reaction with imidazole and thionyl chloride in acetonitrile with a yield of only 56% of theory. An alternative synthesis described therein (process 2) starting from 4- [chloro (phenyl) methyl] biphenyl, which is prepared from biphenyl-4-yl (phenyl) methanol by reaction with thionyl chloride in toluene, by reaction with trimethylsilylimidazole bifonazole provides only in a yield of 52% of theory.
- [0004]ES-A 2 024 363 describes also starting from 4- [chloro (phenyl) methyl] biphenyl, which is prepared from biphenyl-4-yl (phenyl) methanol by reaction with hydrogen chloride in acetonitrile, by reaction with imidazole in acetonitrile using a phase transfer catalyst, the synthesis (method 3) of bifonazole.
- [0005]AT-B 396 931 describes the preparation (method 4) of bifonazole by means of reductive amination of biphenyl-4-yl (phenyl) methanone with imidazole and formic acid. However, this requires high reaction temperatures (220 ° C.) and long reaction times. DE-A 3 538 873 describes a comparable process (process 5) with the additional use of p-toluenesulfonic acid, wherein the reaction temperature is 180 ° C.
- [0006]This in ES 539 345 described method (method 6) for the preparation of bifonazole involves a Gringard reaction between 4-biphenylmagnesium bromide and benzoylated imidazole. Finally, it is tosylated and reduced to bifonazole.
- [0007]ES 549 793 describes the synthesis (method 7) of bifonazole starting from a cyclocondensation between biphenyl-4-yl (phenyl) methylamine, 2-chloro-1-aminoethane and ethyl orthoacetate. The final dehydrogenation is carried out by reaction with 2,3-dichloro-5,6-dicyano-p-benzoquinone in benzene.
- [0008]All known processes have various disadvantages which are particularly unfavorable in the preparation of the compound of the formula (I) on an industrial scale. The solvents used in processes 1 and 2 acetonitrile and toluene are of concern to health. Their use should be avoided in the manufacture of active ingredients used in medicines. By using toluene in process 2, chlorination to give 4- [chloro (phenyl) methyl] biphenyl also produces a toluene-specific, undesired by-product which can only be removed incompletely and thus deteriorates the product quality. The yield is unsatisfactory in both processes. A significant disadvantage of method 3 is, in addition to the use of acetonitrile as solvent, the use of a phase transfer catalyst, which is difficult to separate from the product during work-up. Methods 4 and 5 both operate at very high temperatures and are therefore disadvantageous in a technical use due to the energy consumption and the potential hazard. In method 6, the use of the Gringard reagent is disadvantageous, since this must be produced under considerable safety expense and difficult to handle on an industrial scale. Disadvantage in process 7 is the use of the very toxic compounds 2,3-dichloro-5,6-dicyano-p-benzoquinone and benzene. Their use should be avoided especially in the production of active ingredients used in pharmaceuticals
- Embodiment:
- Synthesis of bifonazole (1- [Biphenyl-4-yl (phenyl) methyl] -1H-imidazole)
- 1st step: 4- [chloro (phenyl) methyl] biphenyl (III)
- [0038]140 g (0.54 mol) dry (water content <0.3%) biphenyl-4-yl (phenyl) methanol (II) are suspended in 1550 ml of cyclohexane and treated with 90 g (0.76 mol) thionyl chloride at a temperature of 50 to 55 ° C added. The reaction mixture is stirred for 0.5 h at a temperature of 50 to 55 ° C stirred. Subsequently, in the Vacuum (<100 mbar) Distilled off thionyl chloride and cyclohexane. A distillation bottoms containing 4- [chloro (phenyl) methyl] biphenyl remains.
- 2nd step: 1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole (Bifonazole)
- [0039]162 g (2.4 mol) of imidazole are suspended in 1350 ml of acetone and dissolved at 50 ° C. This solution is added to the distillation bottoms from step 1 containing 4- [chloro (phenyl) methyl] biphenyl (III). The reaction mixture is heated at reflux for 3 h. After cooling, the reaction solution is mixed with 2 g of activated carbon and 2 g of bleaching earth at a temperature of 50 to 55 ° C, stirred for 0.5 h and filtered. The filtrate is cooled to about 0 ° C. The title compound crystallizes by addition of seed crystals, is filtered off and washed with a mixture of acetone / water (1: 1). For recrystallization, the product is dissolved in 1250 ml of isopropanol, treated with 0.5 g of activated charcoal and 0.5 g of bleaching earth, heated to reflux and filtered hot. The filtrate is cooled to 10 ° C. The title compound crystallizes out by addition of seed crystals, is filtered off, washed with isopropanol and dried. The yield is 101 g (61.9% of theory). The purity of the product is 98.68% by weight.
Melting point: 142 ° C - Comparative method:
- [0040]In the comparative method, instead of cyclohexane, toluene is used as solvent in step 1 as in DE-A 2 461 406 described. Step 2 is performed as described above. 1- [biphenyl-4-yl (phenyl) methyl] -1H-imidazole (bifonazole) is obtained in a purity of 97.66% by weight.
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Adverse effects
The most common side effect is a burning sensation at the application site. Other reactions, such as itching, eczema or skin dryness, are rare.[3] Bifonazole is a potent aromatase inhibitor in vitro.[4][5]
Pharmacology
Mechanism of action
Bifonazole has a dual mode of action. It inhibits fungal ergosterol biosynthesis at two points, via transformation of 24-methylendihydrolanosterol to desmethylsterol, together with inhibition of HMG-CoA. This enables fungicidal properties against dermatophytes and distinguishes bifonazole from other antifungal drugs.[3][6]
Pharmacokinetics
Six hours after application, bifonazole concentrations range from 1000 µg/cm³ in the stratum corneum to 5 µg/cm³ in the papillary dermis.[3]
References
- ^ International Drug Names: Bifonazole.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 502. ISBN 9783527607495.
- ^ Jump up to:a b c Haberfeld H, ed. (2015). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Canesten Bifonazol-Creme.
- ^ Trösken ER, Fischer K, Völkel W, Lutz WK (February 2006). “Inhibition of human CYP19 by azoles used as antifungal agents and aromatase inhibitors, using a new LC-MS/MS method for the analysis of estradiol product formation”. Toxicology. 219 (1–3): 33–40. doi:10.1016/j.tox.2005.10.020. PMID 16330141.
- ^ Egbuta C, Lo J, Ghosh D (December 2014). “Mechanism of inhibition of estrogen biosynthesis by azole fungicides”. Endocrinology. 155 (12): 4622–8. doi:10.1210/en.2014-1561. PMC 4239419. PMID 25243857.
- ^ Berg D, Regel E, Harenberg HE, Plempel M (1984). “Bifonazole and clotrimazole. Their mode of action and the possible reason for the fungicidal behaviour of bifonazole”. Arzneimittel-Forschung. 34 (2): 139–46. PMID 6372801.
Further reading
- Lackner TE, Clissold SP (August 1989). “Bifonazole. A review of its antimicrobial activity and therapeutic use in superficial mycoses”. Drugs. 38 (2): 204–25. doi:10.2165/00003495-198938020-00004. PMID 2670516.
| Clinical data | |
|---|---|
| Trade names | Canespor, many others |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Topical |
| ATC code | D01AC10 (WHO) |
| Legal status | |
| Legal status | In general: Over-the-counter (OTC) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 60628-96-8 |
| PubChem CID | 2378 |
| DrugBank | DB04794 |
| ChemSpider | 2287 |
| UNII | QYJ305Z91O |
| KEGG | D01775 |
| ChEBI | CHEBI:31286 |
| ChEMBL | ChEMBL277535 |
| CompTox Dashboard (EPA) | DTXSID9045631 |
| ECHA InfoCard | 100.056.651 |
| Chemical and physical data | |
| Formula | C22H18N2 |
| Molar mass | 310.400 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////BIFONAZOLE, бифоназол , بيفونازول , 联苯苄唑 , BAY H 4502, BAY-H-4502
C1=CN(C=N1)C(C1=CC=CC=C1)C1=CC=C(C=C1)C1=CC=CC=C1

NEW DRUG APPROVALS
ONE TIME
$10.00
Melitracen

Melitracen
- Molecular FormulaC21H25N
- Average mass291.430 Da
10563-70-9[RN]
1568
1-Propanamine, 3-(10,10-dimethyl-9(10H)-anthracenylidene)-N,N-dimethyl-
225-858-5[EINECS], 234-150-5[EINECS]
3-(10,10-Dimethyl-9(10H)-anthracenyliden)-N,N-dimethyl-1-propanamine
Q7T0Y1109Z
Thymeol
мелитрацен[Russian][INN]
ميليتراسان[Arabic][INN]
美利曲辛[Chinese][INN]
Melitracen
CAS Registry Number: 5118-29-6
CAS Name: 3-(10,10-Dimethyl-9(10H)-anthracenylidene)-N,N-dimethyl-1-propanamine
Additional Names:N,N,10,10-tetramethyl-D9(10H),g-anthracenepropylamine; 9,10-dihydro-10,10-dimethyl-9-(3-dimethylaminopropylidene)anthracene; 9-[3-(dimethylamino)propylidene]-10,10-dimethyl-9,10-dihydroanthracene; N,N-dimethyl-3-(10,10-dimethyl-9(10H)-anthrylidene)propylamine
Molecular Formula: C21H25N, Molecular Weight: 291.43
Percent Composition: C 86.55%, H 8.65%, N 4.81%
Literature References: Prepn of the hydrochloride: Holm, Acta Chem. Scand.17, 2437 (1963); idem,GB939856 corresp to US3177209 (1963, 1965, both to Kefalas A/S). Crystal structure: J. Lopez de Lerma et al.,Acta Crystallogr.B35, 1739 (1979). Toxicity data: P. V. Petersen et al.,Acta Pharmacol. Toxicol.24, 121 (1966).
Derivative Type: Hydrochloride
CAS Registry Number: 10563-70-9
Manufacturers’ Codes: U-24973A
Trademarks: Melixeran (Lusofarmaco); Trausabun (Promonta); Dixeran (Lundbeck)
Molecular Formula: C21H25N.HCl, Molecular Weight: 327.89
Percent Composition: C 76.92%, H 7.99%, N 4.27%, Cl 10.81%
Properties: Crystals from acetone, mp 245-248°. LD50 i.v. in mice: 52 mg/kg (Petersen).
Melting point: mp 245-248°
Toxicity data: LD50 i.v. in mice: 52 mg/kg (Petersen)
Therap-Cat: Antidepressant.
Keywords: Antidepressant; Tricyclics.
Melitracen (brand names Melixeran) is a tricyclic antidepressant (TCA), for the treatment of depression and anxiety.[1][2][3][4] In addition to single drug preparations, it is also available as Deanxit, marketed by Lundbeck, a combination product containing both melitracen and flupentixol.[5][6][7][8]
The pharmacology of melitracen has not been properly investigated and is largely unknown, but it is likely to act in a similar manner to other TCAs. Indeed, melitracen is reported to have imipramine and amitriptyline-like effects and efficacy against depression and anxiety, though with improved tolerability and a somewhat faster onset of action.[9][10]
- ATC:N06AA14
- MW:291.44 g/mol
- CAS-RN:5118-29-6
- InChI Key:GWWLWDURRGNSRS-UHFFFAOYSA-N
- InChI:InChI=1S/C21H25N/c1-21(2)19-13-7-5-10-17(19)16(12-9-15-22(3)4)18-11-6-8-14-20(18)21/h5-8,10-14H,9,15H2,1-4H3
- EINECS:225-858-5
- LD50:52 mg/kg (M, i.v.); 315 mg/kg (M, p.o.);
170 mg/kg (R, p.o.)
Derivatives
hydrochloride
- Formula:C21H25N • HCl
- MW:327.90 g/mol
- CAS-RN:10563-70-9
- EINECS:234-150-5
- LD50:52 mg/kg (M, i.v.); 315 mg/kg (M, p.o.);
170 mg/kg (R, p.o.)
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 90-44-8 | C14H10O | anthrone | 9(10H)-Anthracenone |
| 85118-29-2 | C21H27NO | 9-[3-(dimethylamino)propyl]-9,10-dihydro-10,10-dimethyl-9-anthracenol | 9-Anthracenol, 9-[3-(dimethylamino)propyl]-9,10-dihydro-10,10-dimethyl- |
| 19070-16-7 | C5H12ClMgN | 3-dimethylaminopropylmagnesium chloride | Magnesium, chloro[3-(dimethylamino)propyl]- |
| 5447-86-9 | C16H14O | 10,10-dimethylanthrone | 9(10H)-Anthracenone, 10,10-dimethyl- |
SYN

English: DOI number: 10.3891/acta.chem.scand.17-2437 GB 939856 corresp to US 3177209 (1963, 1965, both to Kefalas A/S).
SYN
https://pubs.rsc.org/en/content/articlehtml/2020/re/d0re00087f
| Fig. 10 Synthesis of melitracen HCl-(36) by Kiil and co-workers making use of a one-flow system. Adapted with permission from Org. Process Res. Dev., 2018, 22, 228–235. Copyright 2018 American Chemical Society.35 |
Grignard reactions are commonly used for the construction of carbon–carbon bonds and show exothermic behaviour which can be dangerous in large-scale batch processes. The use of Grignard reagents in flow can be beneficial because of the high control of reaction conditions, facile heat transport and small effective reaction volume.6,34 A recent example was published by Kiil and co-workers, who synthesised melitracen (36) in a one-flow system.35 Kiil hypothesised that the seven unit operations required in batch could be decreased by combining a hydrolysis and dehydration step, and removing a phase separation (Fig. 10).
The investigation commenced with finding a suitable solvent for the Grignard reaction in which starting materials 34, 35 and intermediate products would dissolve. After having identified THF as the most suitable option, the next challenge was to find an acid that could induce both hydrolysis and dehydration in a single step. Hydrochloric acid was able to perform both transformations, however, precipitation was observed. Thus, hydrochloric acid molarities ranging from 1–12 M were tested. However, while even at the lowest molarity precipitation was observed, it also appeared that below 6 M the dehydration reaction did not proceed. Since the precipitation could not be prevented, a molarity of 12 M was eventually used. The individually optimised transformations were then combined in a one-flow continuous system. Most troublesome was that addition of HCl to the reaction mixture led to an exothermic reaction and boiling of the solvent. Therefore, a back-pressure regulator was employed so that melitracen (36) could be successfully synthesised as its HCl-salt in approximately 85% yield.
SYN
https://pubs.acs.org/doi/pdf/10.1021/acs.oprd.7b00368
A Grignard-based batch process, for the preparation of Melitracen HCl, has been redesigned to fit a continuous reactor system. The Grignard addition is carried out at room temperature, with subsequent hydrolysis of the magnesium alkoxide intermediate followed by dehydration of the resulting alcohol. The product undergoes further workup by simple gravimetric phase separation and then crystallization with 2 M HCl in diethyl ether to afford pure Melitracen HCl. All steps in the laboratory setup were concatenated, and the setup was proven capable of producing a significant portion of the commercial quantities of Melitracen HCl. The flow setup profits from a reduced footprint, lower energy consumption, fewer synthetic steps, and reduced raw material usage compared to the batch process.

As illustrated in Scheme 1, four synthetic steps are involved in the manufacturing of Melitracen HCl (6). The four steps are a classic Grignard addition to a ketone, a hydrolysis of a magnesium alkoxide, a dehydration of an alcohol and a salt precipitation to isolate the API. The Grignard addition is between 10,10-dimethylanthrone (10,10-DMA (1)) and 3-(N,N-dimethylamino)propylmagnesium chloride (DMPC-MgCl (2)), resulting in formation of the magnesium alkoxide 3. The magnesium alkoxide 3 is then hydrolyzed to the alcohol 4 and dehydrated to form product 5. The last step is a crystallization of the API as a salt, where HCl is added to obtain the Melitracen HCl (6)
Scheme 1: Syntheses of magnesium alkoxide 3, alcohol 4 and dehydrated product 5 in the manufacturing process of Melitracen HCl 6, from ketone 1 and Grignard reagent 2.

Current Batch Synthesis The current batch synthesis involves individual synthetic steps, as illustrated in Figure 1. DMPC-MgCl 2 is made in-house before it is used, due to its limited storage shelf life, in a toluene-THF solvent mixture. THF is present in trace amounts in order to stabilize the magnesium in the Grignard reagents.45 A solution of 10,10-DMA 1 is prepared in toluene and is slowly transferred to the DMPC-MgCl 2, maintaining a temperature of 50°C. DMPC-MgCl 2 is used in an equivalence of 1.6 compared to 10,10-DMA 1. The formed magnesium alkoxide 3 is hydrolyzed with water and acetic acid (80%). The aqueous phase is discarded and concentrated hydrochloric acid (37%) is used to dehydrate alcohol 4 to form dehydrated product 5. Toluene is replaced with ethanol by a solvent swap. Crystallization of the dehydrated product 5 from the ethanol phase is done with HCl gas to obtain the final Melitracen HCl (6), which is subsequently isolated by filtration.
Precipitation of Melitracen HCl from THF The dehydrated product 5 was crystallized as the final HCl salt in the THF in a batch experiment, in order to remove a solvent swap to ethanol. The crystallization was carried out with 2 M HCl in Et2O, as this was considered more suited for a later flow process and more easily implemented in the laboratory setup. An equivalence of 1.1 HCl was used and the requirement was an achievement of pH<2. The mixture was kept stirred during the crystallization and carried out at ambient temperature. After 10 minutes, fine white solids started to form, followed by a massive precipitation of Melitracen HCl 6. The Melitracen HCl 6 was filtered with a Büchner funnel and washed with THF. The isolated yield was 80% and within the specifications for the in-house analysis methods used in the routine production (CHN, TGA, UV-vis, HPLC, melting point). Figure 3 is a microscope picture of the isolated Melitracen HCl 6. For full-scale production, the HCl gas would still be more desirable for the crystallization and the 2 M HCl in Et2O merely serves as a proof of concept for the laboratory flow setup.
CLIP
PATENT
https://patents.google.com/patent/CN105418436B/en
Melitracen (Melitracen), is a kind of tricyclics, entitled 10, the 10- dimethyl -9- γ-two of chemistry Methylamino acrylic -9,10- dihydro-anthraquinone, Clinical practice is its hydrochloride.Melitracen can suppress in presynaptic membrane To the effect of the reuptake of norepinephrine and serotonin, and therefore improve containing for monoamine transmitterses in synaptic cleft Amount.
On the preparation method of melitracen, document report both domestic and external is seldom, existing as described below:
US3177209, GB939856, DK97400, are the compound patents of Lundbeck drugmaker of Denmark, it is mentioned that Synthetic method is that, with 10,10- dimethylanthracene -9- ketone and N, TMSDMA N dimethylamine base propyl group magnesium chloride is generated in the middle of melitracen Body, then by intermediate be dissolved under chloroform, reflux state lead to hydrogen chloride prepare melitracen crude product, then crystallized again with acetone Melitracen is obtained, this method needs to be passed through hydrogen chloride at reflux, there is substantial amounts of smog to produce, and reaction condition is not yet It is easy to control, it there is larger safety factor.
CN103877088A is Lundbeck drugmaker of Denmark in a kind of safe melitracen group disclosed in 2014 Compound, wherein the purity to melitracen in drug regimen proposes more strict requirements, especially to that may make in clinic Cause the impurity (formula I, formula II) of the adverse reactions such as anxiety, irritated and excitement in, even more propose:Formula I<0.1%, formula II< 0.1, I+formula of formula II<0.1% rigors.The melitracen of patent US3177209, GB939856, DK97400 method synthesis Impurity is more, and primary purification can not obtain satisfactory active pharmaceutical ingredient (API).
It is also mentioned that the preparation method of melitracen hydrochloride, this method is with 10,10- diformazans in patent CN103877088A The γ of base-9-dimethylaminopropyl-9- anthrols are raw material, add dichloromethane and hydrochloric acid, are heated to reflux, reaction system alkaline hydrolysis from The free alkali obtained afterwards, is re-dissolved in acetone and leads to hydrogen chloride into salt, obtain melitracen crude product, then isolated and purified with column chromatography Obtain the melitracen of high-purity.The melitracen yield that it is prepared into is low, and purifies and separates process needs column chromatography, it is impossible to meet The need for large-scale production.
Embodiment 1
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
10,10- dimethylanthracene -9- ketone carry out grignard reaction with 3- dimethylaminos-n-propyl chloride in the presence of initiator, obtain To melitracen intermediate, detailed process is as follows:
340g magnesium rods and 17.5L absolute ethers are added in 20L glass reaction kettles, stirring is warming up to 30~35 DEG C, addition 1.75kg 3- dimethylaminos-n-propyl chloride, finish insulated and stirred, add 1g iodine and 2mL 1,2- Bromofume as initiator, 9h is stirred at reflux, magnesium rod disappears completely, reaction system is cooled into 10~20 DEG C, 1.5kg 10,10- dimethyl is slowly added to Anthracene -9- ketone, then it is warming up to 30~35 DEG C, back flow reaction 1 hour;TLC monitoring reactions are complete, and reaction system is cooled into 10~20 DEG C, then add 5.5L water, ether layer is separated, anhydrous sodium sulfate is added and is concentrated under reduced pressure drying, obtain melitracen intermediate 2.03kg, receive The ﹪ of rate 97.2, purity 98.5%.
TLC monitoring methods:Add water and be quenched after sampling, take organic layer point plate;Solvent is petroleum ether:Ethyl acetate=2:1 (volume ratio);The Rf of 10,10- dimethylanthracene -9- ketone is 0.6, and the Rf of melitracen intermediate is 0.1.
(2) melitracen crude product is prepared
2kg melitracens intermediate, 10L chloroforms and 2.4L concentrated hydrochloric acids are put into 20L glass reaction kettles, stirred molten Solution, obtains pale yellow solution, and 60 DEG C of heating stirring reaction 2 hours, TLC monitoring reactions are complete, and separate aqueous layer, organic phase is concentrated under reduced pressure Dry, it is melitracen crude product 2.03kg, yield 95.7%, purity 99.41%, containing Formulas I to obtain white solid:0.20%, formula II:0.13%;Formulas I, II1HNMR spectrograms, melitracen crude product liquid phase spectrogram are shown in accompanying drawing 1,2,3 respectively;
TLC monitoring methods:Organic phase point plate is extracted reaction solution, solvent is dichloromethane:Methanol:Acetic acid=150:10:2 (volume ratio).
Formulas I:1H NMR(400MHz,DMSO)δ7.78-7.82(m,2H),δ7.50-7.53(m,2H),δ7.28-7.35 (m, 4H), δ 2.11 (S, 6H), δ 2.08 (d, J=6.8Hz, 2H), δ 1.96 (t, J=6.4Hz, 2H), δ 1.72 (s, 3H), δ 1.61(s,3H),δ1.26(brs,1H),δ1.02-1.09(m,2H)
Formula II:1H NMR(400MHz,DMSO)δ8.95(s,2H),δ7.47-7.63(m,4H),δ7.27-7.37(m, 4H), δ 6.06 (t, J=7.2Hz, 1H), δ 3.09 (t, J=7.2Hz, 2H), δ 2.91 (m, 2H), δ 2.54 (s, 3H), δ 1.53 (s,6H)
(3) purifying of melitracen crude product
Take 2.03kg melitracens crude product (purity 99.41%, Formulas I:0.20%, Formula II:0.13%) 4 times of amount (W/, are added V isopropanol), 20~25 DEG C of stirring 4h (mashing), is filtered, and is dried, is obtained product 2.0kg, yield is 98.5%, and purity is 99.61%, containing Formulas I:0.054%, without Formula II;Melitracen crude product is shown in accompanying drawing 4 through isopropanol mashing sample liquid chromatography(LC figure;
Product after 2kg is beaten is added in 30L glass reaction kettle, adds 16kg isopropanols, and backflow is dissolved, then Cool to 10 DEG C and stir crystallization and stay overnight, suction filtration is dried under reduced pressure, and obtains melitracen 1.89kg, and yield 94.5%, purity 99.98% contains Formulas I:0.0026%, without Formula II;See accompanying drawing 5 through isopropanol recrystallization liquid phase spectrogram.
Embodiment 2
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 1;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 1;
(3) purifying of melitracen crude product
Take 10g melitracens crude product (purity 99.41%, Formulas I:0.20%, Formula II:0.13%) 4 times of amounts (W/V), are added Ethanol, 20~25 DEG C stirring 4h (mashing), filtering, drying, obtain product 9.79g, yield is 97.9%, purity 99.69% contains Formula I 0.047%, containing formula II 0.005%;Melitracen crude product is shown in accompanying drawing 6 through ethanol mashing sample liquid chromatography(LC figure;
Product after 9.0g ethanol is beaten is added in 250mL round-bottomed flask, adds the dissolving of 230mL alcohol refluxs, Then 10 DEG C are cooled to stir crystallization and stay overnight, suction filtration is dried under reduced pressure, obtain melitracen 8.4g, yield 93.3%, purity 99.98%, Containing Formulas I:0.0041%, without Formula II;See accompanying drawing 7 through ethanol recrystallization liquid phase spectrogram.
Embodiment 3
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 1;
(2) melitracen crude product is prepared
100g melitracens intermediate, 500mL chloroforms and 120mL concentrated hydrochloric acids are put into 1L three-necked bottles, stirred molten Solution, obtains pale yellow solution, and 60 DEG C of heating stirring reaction 2 hours, TLC monitoring reactions are complete, and separate aqueous layer, organic phase is concentrated under reduced pressure Dry, it is melitracen crude product 104g, yield 98.3%, purity 99.38%, containing Formulas I to obtain white solid:0.22%, Formula II: 0.15%;Melitracen crude product liquid phase spectrogram is shown in accompanying drawing 8;
TLC monitoring methods:Organic phase point plate is extracted reaction solution, solvent is dichloromethane:Methanol:Acetic acid=150:10:2 (volume ratio).
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the methanol of 4 times of amounts (W/V) is added, 20~25 DEG C of stirring 4h (mashing) obtain product Weight is 18.48g, and yield is 92.4%, and purity is 99.66%, containing Formulas I:0.05%, Formula II:0.008%, see accompanying drawing 9.
Embodiment 4
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the n-butanol of 4 times of amounts (W/V) is added, 20~25 DEG C of stirring 5h (mashing) are produced Thing weight is 19.6g, and yield is 98%, and purity is 99.54%, containing Formulas I:0.05%, Formula II:0.009%, see accompanying drawing 10.
Embodiment 5
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the isopropanol of 4 times of amounts (W/V) is added, 30~35 DEG C of stirring 5h (mashing) are produced Thing weight is 18.06g, and yield is 90.3%.
Embodiment 6
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the isopropanol of 4 times of amounts (W/V) is added, 50 DEG C of stirring 3h (mashing) obtain product weight Measure as 14.2g, yield is 71%.
Embodiment 7
A kind of preparation method of melitracen hydrochloride, comprises the following steps:
(1) melitracen intermediate is prepared
This step is identical with the step (1) in embodiment 3;
(2) melitracen crude product is prepared
This step is identical with the step (2) in embodiment 3;
(3) purifying of melitracen crude product
Above-mentioned melitracen crude product is taken, the isopropanol of 4 times of amounts (W/V) is added, 5-10 DEG C of stirring 5h (mashing) obtains product Weight is 19.7g, and yield is 98.5%, and purity is 99.53%, containing Formulas I:0.054%, Formula II:0.014%, see accompanying drawing 11.
Embodiment 8
With reference to CN103877088A, crystallized using acetone, that is, take 10g melitracens intermediate and 24mL dichloromethane, 6.7mL concentrated hydrochloric acids are heated to reflux 2h and are cooled to room temperature, and pH is to 8-9 for regulation, then are extracted with dichloromethane and product, are concentrated to give free Alkali cpd, acetone is dissolved in by the free alkali compound, concentrated hydrochloric acid is added dropwise to pH=0.1, stirring, cooling separate out solid 7.1g, This solid crystallizes to obtain sample 6.4g with acetone again, and total recovery is 60.9%, and purity is 99.64%, containing Formulas I:0.09%, Formula II: 0.04%.Melitracen is shown in accompanying drawing 12 only with acetone crystallization liquid chromatography(LC figure.
Repeat literature method crystallized only with acetone obtained by product in impurity Formulas I, Formula II impurity summation be 0.13%, The adverse reactions such as anxiety, irritated and excitement may be caused in Clinical practice.
In summary, the effect of mashing is to make melitracen crude product rapid dispersion, and the effect of methanol mashing is similar with ethanol, But it is good without isopropanol effect, but methanol mashing yield is decreased obviously trend;N-butanol mashing needs the extension time to reach To the effect same with ethanol, but be not as good as isopropanol effect, and because the viscosity of n-butanol is slightly larger, melitracen crude product is at it In disperse slightly worse, invention has the granular solids that not readily dissolve after filtering, and the removal effect to other impurities is also poor;Isopropanol Temperature is raised during mashing, yield is decreased obviously, and reduces temperature, yield has no raising, though to the removal effect of impurity Formula II It can so control in the range of conforming to quality requirements, but compared to being decreased obviously in embodiment 1.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Swiss Pharmaceutical Society (2000). Index Nominum 2000: International Drug Directory (Book with CD-ROM). Boca Raton: Medpharm Scientific Publishers. ISBN 3-88763-075-0.
- ^ Hall, Chapman and; Chemical Abstracts Service, American Chemical Society; Rhodes, P. H (1996). Dictionary of organic compounds. London: Chapman & Hall. ISBN 0-412-54090-8.
- ^ O’Neil, Maryadele J. (2001). The Merck index: an encyclopedia of chemicals, drugs, and biologicals. Rahway, NJ: Merck Research Laboratories. ISBN 0-911910-13-1.
- ^ José Miguel Vela; Helmut Buschmann; Jörg Holenz; Antonio Párraga; Antoni Torrens (2007). Antidepressants, Antipsychotics, Anxiolytics: From Chemistry and Pharmacology to Clinical Application. Weinheim: Wiley-VCH. ISBN 978-3-527-31058-6.
- ^ Muller, Niels F; Dessing, Rudolf P; Pharmacy, European Society of Clinical (1998). European Drug Index, 4th Edition. Boca Raton: CRC Press. ISBN 3-7692-2114-1.
- ^ Van Moffaert M, Dierick M, De Meulemeester F, Vereecken A (1983). “Treatment of depressive anxiety states associated with psychosomatic symptoms. A double-blind multicentre clinical study: mianserin versus melitracen-flupentixol”. Acta Psychiatrica Belgica. 83 (5): 525–39. PMID 6670581.
- ^ Bin Yaacob H (April 1985). “Flupenthixol and Melitracen in the management of trigeminal neuralgia”. Dental Journal of Malaysia. 8 (2): 37–8. PMID 3917005.
- ^ Hashash JG, Abdul-Baki H, Azar C, et al. (June 2008). “Clinical trial: a randomized controlled cross-over study of flupenthixol + melitracen in functional dyspepsia”. Alimentary Pharmacology & Therapeutics. 27 (11): 1148–55. doi:10.1111/j.1365-2036.2008.03677.x. PMID 18331614. S2CID 40714136.
- ^ Aronson, Jeffrey Kenneth (2008). Meyler’s Side Effects of Psychiatric Drugs (Meylers Side Effects). Amsterdam: Elsevier Science. ISBN 978-0-444-53266-4.
- ^ Author Unknown (1970). Ann Reports Medicinal Chem V5 (v. 5). Boston: Academic Press. ISBN 0-12-040505-9.
{{cite book}}:|author=has generic name (help)
| Clinical data | |
|---|---|
| Trade names | Adaptol, Dixeran, Melixeran, Thymeol, Trausabun |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral, intramuscular injection |
| ATC code | N06AA14 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 5118-29-6 |
| PubChem CID | 25382 |
| ChemSpider | 23697 |
| UNII | Q7T0Y1109Z |
| KEGG | D08171 |
| ChEMBL | ChEMBL110094 |
| CompTox Dashboard (EPA) | DTXSID4048274 |
| ECHA InfoCard | 100.023.507 |
| Chemical and physical data | |
| Formula | C21H25N |
| Molar mass | 291.438 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
//////////Melitracen, Q7T0Y1109Z, Thymeol, мелитрацен , ميليتراسان , 美利曲辛 , U 24973A, Antidepressant, Tricyclics,

NEW DRUG APPROVALS
ONE TIME
$10.00
IODOQUINOL

IODOQUINOL
Diiodohydroxyquinoline
- Molecular FormulaC9H5I2NO
- Average mass396.951 Da
- NSC-8704
- SS-578
5,7-Diiodo-8-quinolinol
5,7-Diiodooxine
5,7-diiodoquinolin-8-ol
83-73-8[RN]
8-Hydroxy-5,7-diiodoquinoline
8-Quinolinol, 5,7-diiodo-
дийодогидроксихинолин[Russian][INN]
ثنائي إيودوهيدروكسيكينوليين[Arabic][INN]
双碘喹啉[Chinese][INN]
201-497-9[EINECS]
5,7-Diiodo-8-hydroxyquinoline
IodoquinolCAS Registry Number: 83-73-8
CAS Name: 5,7-Diiodo-8-quinolinol
Additional Names: diiodohydroxyquin; diiodo-oxyquinoline; 5,7-diiodo-8-hydroxyquinoline
Manufacturers’ Codes: SS-578
Trademarks: Diodoquin (Searle); Disoquin; Floraquin (Searle); Dyodin; Dinoleine; Searlequin; Diodoxylin; Rafamebin; Ioquin (Abbott); Direxiode (Delalande); Stanquinate; Yodoxin (Searle); Zoaquin; Enterosept; Embequin (M & B)
Molecular Formula: C9H5I2NO, Molecular Weight: 396.95
Percent Composition: C 27.23%, H 1.27%, I 63.94%, N 3.53%, O 4.03%
Literature References: Prepd by the action of iodine monochloride on 8-hydroxyquinoline: Papesch, Burtner, J. Am. Chem. Soc.58, 1314 (1936); by the action of KIO3 on 8-hydroxyquinoline: Zeifman, C.A.34, 3745. Electrolytic prepn: Brown, Berkowitz, Trans. Electrochem. Soc.75, 385 (1939). See also Claus, DE78880; Passek, DE411050; Matsumura, C.A.21, 1461 (1927); Pirrone, Cherubino, C.A.28, 3073 (1934).Properties: Crystals from xylene. The medicinal grade is a yellowish-brown powder. mp 200-215° (extensive decompn). Almost insol in water. Sparingly sol in alcohol, ether, and acetone; sol in hot pyridine and in hot dioxane.
Melting point: mp 200-215° (extensive decompn)
Therap-Cat: Antiamebic.
Keywords: Antiamebic.
The quinoline derivative diiodohydroxyquinoline (INN), or iodoquinol (USAN), can be used in the treatment of amoebiasis.[1]
It is poorly absorbed from the gastrointestinal tract and is used as a luminal amebicide. It acts by chelation of ferrous ions essential for metabolism.[2]
It was discovered by Adco Co. and introduced as diiodohydroxyquinoline.[3]
Susceptibility of Dientamoeba fragilis has been measured.[4]
Iodoquinol is an amebocide used against Entamoeba histolytica, and it is active against both cyst and trophozoites that are localized in the lumen of the intestine. It is considered the drug of choice for treating asymptomatic or moderate forms of amebiasis. The full mechanism of action is unknown. Iodoquinol is used for diseases caused by moderate intestinal amebiasis.
Diodoquin enhances zinc absorption in the zinc deficiency disorder Acrodermatitis enteropathica, probably because Diodoquin act as a zinc ionophore.[5]
5,7-Diiodo-8-quinolinol Chemical
Originator
Diiodohydroxyquinoline,Adco Co.
Uses
Antiamebic.
Uses
GABA prodrug
Uses
It acts as an amoebicidal and so used in the treatment of amoebiasis, balantidiasis (an infection caused by protozoa).
Indications
Iodoquinol (diiodohydroxyquin, Yodoxin, Moebiquin) is a halogenated 8-hydroxyquinoline derivative whose precise mechanism of action is not known but is thought to involve an inactivation of essential parasite enzymes. Iodoquinol kills the trophozoite forms of E. histolytica, B. coli, B. hominis, and Dientamoeba fragilis.
Iodoquinol is absorbed from the gastrointestinal tract and is excreted in the urine as glucuronide and sulfate conjugates. Most of an orally administered dose is excreted in the feces. Iodoquinol has a plasma half-life of about 12 hours.
Iodoquinol is the drug of choice in the treatment of asymptomatic amebiasis and D. fragilis infections. It is also used in combination with other drugs in the treatment of other forms of amebiasis and as an alternative to tetracycline in the treatment of balantidiasis.
Adverse reactions are related to the iodine content of the drug; the toxicity is often expressed as skin reactions, thyroid enlargement, and interference with thyroid function studies. Headache and diarrhea also occur. Chronic use of clioquinol, a closely related agent, has been linked to a myelitislike illness and to optic atrophy with permanent loss of vision.
Manufacturing Process
5,7-Diiodo-8-quinolinol widely used as an intestinal antiseptic, especially as an antiamebic agent. It is also used topically in other infections and may cause CNS and eye damage. It is known by very many similar trade names worldwide.
0.01 mol 8-oxychinoline and 0.01 mol salicylic acid were dissolved in 500 ml of water and then 0.05 mol potassium iodide was added. The mixture was heated to temperature 90°-100°C. After that 0.01 mol of KIO3 by little tiles was added. The next tile was added after a disappearence of discharging iodine. Then 10 ml 2 N HCl was added. The solid product was fallen, filtered off, washed with hot water and in 0.25 N NaOH dissolved. The solution was filtered and the clear filtrate precipitated with a very little excess of HCl. The product 5,7-diiodo-8-quinolinol was filtered, washed with hot water and dried. MP: 200°-250°C (with decomposition).
brand name
Quinadome (Bayer); Yodoxin (Glenwood).
Therapeutic Function
Antibacterial
Clinical Use
5,7-Diiodo-8-quinolinol, 5,7-diiodo-8-hydroxyquinoline,or diiodohydroxyquin (Yodoxin, Diodoquin, Diquinol) is ayellowish to tan microcrystalline, light-sensitive substancethat is insoluble in water. It is recommended for acute andchronic intestinal amebiasis but is not effective in extraintestinaldisease. Because a relatively high incidence of topicneuropathy has occurred with its use, iodoquinol should notbe used routinely for traveler’s diarrhea.
Safety Profile
Poison by ingestion and intravenous routes. Human systemic effects by ingestion: eye effects. Mutation data reported. When heated to decomposition it emits very toxic fumes of Iand Nox
Chemical Synthesis
Iodoquinol, 5,7-diiodo-8-quinolinol (37.2.2), is made by iodination of 8-oxyquinoline (37.2.1) using a mixture of potassium iodide/potassium iodate. The initial 8-hydroxyquinolin (37.2.1) is made from 2-aminophenol and glycerol in the presence of sulfuric acid and nitrobenzene (Skraup synthesis).
Purification Methods
It crystallises from xylene and is dried at 70o in a vacuum. [Beilstein 21 II 58.]
5,7-Diiodo-8-quinolinol synthesis
Synthesis of 5,7-Diiodo-8-quinolinol from 8-Hydroxyquinoline
SYN
DE 411050 DOI: 10.1021/ja01298a506

CLIP
Iodoquinol, 5,7-diiodo-8-quinolinol (37.2.2), is made by iodination of 8-oxyquinoline (37.2.1) using a mixture of potassium iodide/potassium iodate. The initial 8-hydroxyquinolin (37.2.1) is made from 2-aminophenol and glycerol in the presence of sulfuric acid and nitrobenzene (Skraup synthesis) [39,40]

Iodoquinol is an amebocide used against E. histolytica, and it is active against both cysts and trophozoites that are localized in the lumen of the intestine. It is considered the drug of choice for treating asymptomatic or moderate forms of amebiasis. The mechanism of action is unknown. Iodoquinol is used for diseases caused by moderate intestinal amebiasis. Synonyms of this drug are diquinol, iodoxin, diiodoquin, amebaquin, and others
39. F. Passek, Ger. Pat. 411.050 (1925). 40. V. Papesch, R.R. Burtner, J. Am. Chem. Soc., 58, 1314 (1936).
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Ghaskadbi S, Vaidya VG (March 1989). “In vivo antimutagenic effect of ascorbic acid against mutagenicity of the common antiamebic drug diiodohydroxyquinoline”. Mutat. Res. 222 (3): 219–22. doi:10.1016/0165-1218(89)90137-7. PMID 2493578.
- ^ Nagata, Noriyuki; Marriott, Deborah; Harkness, John; Ellis, John T.; Stark, Damien (2012). “Current treatment options for Dientamoeba fragilis infections”. International Journal for Parasitology: Drugs and Drug Resistance. 2: 204–215. doi:10.1016/j.ijpddr.2012.08.002. ISSN 2211-3207. PMC 3862407. PMID 24533282.
- ^ Publishing, William Andrew (2013-01-15). Pharmaceutical Manufacturing Encyclopedia (3rd ed.). Elsevier Science. p. 1312. ISBN 9780080947266.
- ^ Chan FT, Guan MX, Mackenzie AM, Diaz-Mitoma F (May 1994). “Susceptibility testing of Dientamoeba fragilis ATCC 30948 with iodoquinol, paromomycin, tetracycline, and metronidazole”. Antimicrob. Agents Chemother. 38 (5): 1157–60. doi:10.1128/aac.38.5.1157. PMC 188168. PMID 8067755.
- ^ Aggett, P.J.; Delves, H.T.; Harries, J.T.; Bangham, A.D. (March 1979). “The possible role of Diodoquin as a zinc ionophore in the treatment of acrodermatitis enteropathica”. Biochemical and Biophysical Research Communications. 87 (2): 513–517. doi:10.1016/0006-291X(79)91825-4. PMID 375935.
| Names | |
|---|---|
| Preferred IUPAC name5,7-Diiodoquinolin-8-ol | |
| Other namesDiquinol, iodoxin, diiodoquin, amebaquin | |
| Identifiers | |
| CAS Number | 83-73-8 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:5950 |
| ChEMBL | ChEMBL86754 |
| ChemSpider | 3597 |
| ECHA InfoCard | 100.001.362 |
| KEGG | D00581 |
| MeSH | Iodoquinol |
| PubChem CID | 3728 |
| UNII | 63W7IE88K8 |
| CompTox Dashboard (EPA) | DTXSID6023155 |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C9H5I2NO |
| Molar mass | 396.951 |
| Pharmacology | |
| ATC code | G01AC01 (WHO) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
//////////////IODOQUINOL, Diiodohydroxyquinoline, NSC-8704, SS-578
OC1=C2N=CC=CC2=C(I)C=C1I

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}