
Home » EU 2022
Category Archives: EU 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Nirsevimab
(Heavy chain)
QVQLVQSGAE VKKPGSSVMV SCQASGGLLE DYIINWVRQA PGQGPEWMGG IIPVLGTVHY
GPKFQGRVTI TADESTDTAY MELSSLRSED TAMYYCATET ALVVSETYLP HYFDNWGQGT
LVTVSSASTK GPSVFPLAPS SKSTSGGTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP
AVLQSSGLYS LSSVVTVPSS SLGTQTYICN VNHKPSNTKV DKRVEPKSCD KTHTCPPCPA
PELLGGPSVF LFPPKPKDTL YITREPEVTC VVVDVSHEDP EVKFNWYVDG VEVHNAKTKP
REEQYNSTYR VVSVLTVLHQ DWLNGKEYKC KVSNKALPAP IEKTISKAKG QPREPQVYTL
PPSREEMTKN QVSLTCLVKG FYPSDIAVEW ESNGQPENNY KTTPPVLDSD GSFFLYSKLT
VDKSRWQQGN VFSCSVMHEA LHNHYTQKSL SLSPGK
(Light chain)
DIQMTQSPSS LSAAVGDRVT ITCQASQDIV NYLNWYQQKP GKAPKLLIYV ASNLETGVPS
RFSGSGSGTD FSLTISSLQP EDVATYYCQQ YDNLPLTFGG GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H153-H209, H229-L214, H235-H’235, H238-H’238, H270-H330, H376-H434, H’22-H’96, H’153-H’209, H’229-L’214, H’270-H’330, H’376-H’434, L23-L88, L’23-L’88, L134-L194, L’134-L’194)
>Heavy_chain QVQLVQSGAEVKKPGSSVMVSCQASGGLLEDYIINWVRQAPGQGPEWMGGIIPVLGTVHY GPKFQGRVTITADESTDTAYMELSSLRSEDTAMYYCATETALVVSETYLPHYFDNWGQGT LVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFP AVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKRVEPKSCDKTHTCPPCPA PELLGGPSVFLFPPKPKDTLYITREPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKP REEQYNSTYRVVSVLTVLHQDWLEGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTL PPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLT VDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
>Light_chain DIQMTQSPSSLSAAVGDRVTITCQASQDIVNYLNWYQQKPGKAPKLLIYVASNLETGVPS RFSGSGSGTDFSLTISSLQPEDVATYYCQQYDNLPLTFGGGTKVEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
Nirsevimab
EMS APPROVED 2022/10/31, Beyfortus, AstraZeneca AB
| Formula | C6494H10060N1708O2050S46 |
|---|---|
| CAS | 1989556-22-0 |
| Mol weight | 146334.5658 |
Monoclonal antibody
Prevention of respiratory syncytial virus infection
- Immunoglobulin g1-kappa, anti-(human respiratory syncytial virus fusion glycoprotein f0 (protein f))human monoclonal antibody.gamma.1 heavy chain (1-456) (human vh (homo sapiens ighv1-69*01(ighd)-ighj4*01 (90.1%)) (8.8.19) (1-126) -homo sapiens ighg1*03
- Immunoglobulin g1, anti-(human respiratory syncytial virus fusion protein)(human monoclonal med18897 .gamma.1-chain), disulfide with monoclonal med18897 .kappa.-chain, dimer
Synthesis Reference
Khan, AA et al. (2020) Dosage regimens for and compositions including anti-rsv antibodies. (U.S. Patent No. 2020/0347120 A1). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/6b/d2/10/a841b66e0c90cf/US20200347120A1.pdf
Nirsevimab, sold under the brand name Beyfortus, is a human recombinant monoclonal antibody with activity against respiratory syncytial virus, or RSV for infants.[2][3] It is under development by AstraZeneca and Sanofi.[2][3] Nirsevimab is designed to bind to the fusion protein on the surface of the RSV virus.[4][5]
The most common side effects reported for nirsevimab are rash, pyrexia (fever) and injection site reactions (such as redness, swelling and pain where the injection is given).[6]
Nirsevimab was approved for medical use in the European Union in November 2022.[1][7]
Nirsevimab (MEDI8897) is a recombinant human immunoglobulin G1 kappa (IgG1ĸ) monoclonal antibody used to prevent respiratory syncytial virus (RSV) lower respiratory tract disease in neonates and infants.6 It binds to the prefusion conformation of the RSV F protein, a glycoprotein involved in the membrane fusion step of the viral entry process, and neutralizes several RSV A and B strains.6,1 Compared to palivizumab, another anti-RSV antibody, nirsevimab shows greater potency at reducing pulmonary viral loads in animal models. In addition, nirsevimab was developed as a single-dose treatment for all infants experiencing their first RSV season, whereas palivizumab requires five monthly doses to cover an RSV season.5 This is due to a modification in the Fc region of nirsevimab that grants it a longer half-time compared to typical monoclonal antibodies.1,6
On November 2022, nirsevimab was approved by the EMA for the prevention of RSV lower respiratory tract disease in newborns and infants during their first RSV season.6
////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | F protein of RSV |
| Clinical data | |
| Trade names | Beyfortus |
| Other names | MED-18897, MEDI8897 |
| Routes of administration | Intramuscular |
| ATC code | None |
| Legal status | |
| Legal status | EU: Rx-only [1] |
| Identifiers | |
| CAS Number | 1989556-22-0 |
| PubChem SID | 384585358 |
| DrugBank | DB16258 |
| UNII | VRN8S9CW5V |
| KEGG | D11380 |
| ChEMBL | ChEMBL4297575 |
| Chemical and physical data | |
| Formula | C6494H10060N1708O2050S46 |
| Molar mass | 146336.58 g·mol−1 |
Adverse effects
No major hypersensitivity reactions have been reported, and adverse events of grade 3 or higher were only reported in 8% (77 of 968) of participants in clinical trial NCT02878330.[8][4]
Pharmacology
Mechanism of action
Nirsevimab binds to the prefusion conformation of the RSV fusion protein, i.e. it binds to the site at which the virus would attach to a cell; effectively rendering it useless. It has a modified Fc region, extending the half-life of the drug in order for it to last the whole RSV season.[4]
History
The opinion by the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) is based on data from two randomized, double-blind, placebo-controlled multicenter clinical trials that investigated the efficacy and safety of nirsevimab in healthy preterm (premature) and full-term infants entering their first respiratory syncytial virus (RSV) season.[6] These studies demonstrated that nirsevimab prevents lower respiratory tract infection caused by RSV requiring medical attention (such as bronchiolitis and pneumonia) in term and preterm infants during their first RSV season.[6]
The safety of nirsevimab was also evaluated in a phase II/III, randomized, double‑blind, multicenter trial in infants who were born five or more weeks prematurely (less than 35 weeks gestation) at higher risk for severe RSV disease and infants with chronic lung disease of prematurity (i.e. long-term respiratory problems faced by babies born prematurely) or congenital heart disease.[6] The results of this study showed that nirsevimab had a similar safety profile compared to palivizumab (Synagis).[6]
Society and culture
Legal status
On 15 September 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Beyfortus, intended for the prevention of respiratory syncytial virus (RSV) lower respiratory tract disease in newborns and infants.[9][6] Beyfortus was reviewed under EMA’s accelerated assessment program.[9] The applicant for this medicinal product is AstraZeneca AB.[9] Nirsevimab was approved for medical use in the European Union in November 2022.[1][7]
Research
Nirsevimab is being investigated as an experimental vaccine against respiratory syncytial virus, RSV, in the general infant population.[2][3] The MELODY study is an ongoing, randomized, double-blind, placebo-controlled to evaluate the safety and efficacy of nirsevimab in late preterm and term infants. Initial results have been promising, with nirsevimab reducing LRTI (lower respiratory tract infections) by 74.5% compared to placebo in infants born at term or late preterm.[5][10][11]
Ongoing trials for nirsevimab are:
- “Evaluate the Safety and Efficacy of Nirsevimab in Healthy Preterm and Term Infants in China (CHIMES)”.
- “A Study to Evaluate the Safety and Efficacy of MEDI8897 for the Prevention of Medically Attended Lower Respiratory Tract Infection Due to Respiratory Syncytial Virus in Healthy Late Preterm and Term Infants (MELODY)”.
- “Evaluate the Safety and Tolerability, for Nirsevimab in Immunocompromised Children (MUSIC)”.
References
- ^ Jump up to:a b c “Beyfortus”. Union Register of medicinal products. 3 November 2022. Retrieved 6 November 2022.
- ^ Jump up to:a b c “Nirsevimab demonstrated protection against respiratory syncytial virus disease in healthy infants in Phase 3 trial” (Press release). Sanofi. 26 April 2021. Archived from the original on 27 December 2021. Retrieved 27 December 2021.
- ^ Jump up to:a b c “Nirsevimab MELODY Phase III trial met primary endpoint of reducing RSV lower respiratory tract infections in healthy infants” (Press release). AstraZeneca. 26 April 2021. Archived from the original on 26 December 2021. Retrieved 27 December 2021.
- ^ Jump up to:a b c Griffin MP, Yuan Y, Takas T, Domachowske JB, Madhi SA, Manzoni P, et al. (Nirsevimab Study Group) (July 2020). “Single-Dose Nirsevimab for Prevention of RSV in Preterm Infants”. The New England Journal of Medicine. 383 (5): 415–425. doi:10.1056/NEJMoa1913556. PMID 32726528. S2CID 220876651.
- ^ Jump up to:a b Hammitt LL, Dagan R, Yuan Y, Baca Cots M, Bosheva M, Madhi SA, et al. (March 2022). “Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants”. The New England Journal of Medicine. 386 (9): 837–846. doi:10.1056/NEJMoa2110275. PMID 35235726. S2CID 247220023.
- ^ Jump up to:a b c d e f “New medicine to protect babies and infants from respiratory syncytial virus (RSV) infection”. European Medicines Agency (EMA) (Press release). 16 September 2022. Archived from the original on 19 September 2022. Retrieved 18 September 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Beyfortus approved in the EU for the prevention of RSV lower respiratory tract disease in infants”. AstraZeneca (Press release). 4 November 2022. Retrieved 6 November 2022.
- ^ Clinical trial number NCT02878330 at ClinicalTrials.gov
- ^ Jump up to:a b c “Beyfortus: Pending EC decision”. European Medicines Agency (EMA). 15 September 2022. Archived from the original on 19 September 2022. Retrieved 18 September 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Zacks Equity Research (25 March 2022). “Pfizer’s (PFE) RSV Jab Gets Another Breakthrough Therapy Tag”. Nasdaq. Archived from the original on 8 April 2022. Retrieved 8 April 2022.
- ^ “Nirsevimab significantly protected infants against RSV disease in Phase III MELODY trial”. AstraZeneca (Press release). 3 March 2022. Retrieved 6 November 2022.
////////////Nirsevimab, EU 2022, APPROVALS 2022, PEPTIDE, Monoclonal antibody, respiratory syncytial virus infection, ANTIVIRAL, 1989556-22-0, MED-18897, MEDI8897, AstraZeneca AB

NEW DRUG APPROVALS
ONE TIME
$10.00
Lutetium (177Lu) chloride

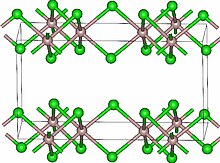

Lutetium (177Lu) chloride
塩化ルテチウム (177Lu)
| Formula | Lu. 3Cl |
|---|---|
| CAS | 16434-14-3 |
| Mol weight | 281.326 |
2022/9/15 EMA 2022, Illuzyce
EndolucinBeta
(177Lu)lutetium(3+) trichloride
| Diagnostic aid, Radioactive agent |
Lutetium 177 is an isotope of a rare-earth lanthanide metal lutetium. Radioactive decay of Lu 177 produces electrons with low energies making the isotope suitable for treatment of metastatic disease. A complex of Lu177 and somatostatin analog DOTA-TATE was approved by the FDA for the treatment of somatostatin receptor-positive gastroenteropancreatic neuroendocrine tumors, including foregut, midgut, and hindgut neuroendocrine tumors in adults. It is marketed under a tradename Lutathera. Lutetium in the complex with other carriers – phosphonates and monoclonal antibodies – was investigated in clinical trials as radiotherapy to prostate, ovarian, renal and other types of cancer.Lutetium (177Lu) chloride is a radioactive compound used for the radiolabeling of pharmaceutical molecules, aimed either as an anti-cancer therapy or for scintigraphy (medical imaging).[5][6] It is an isotopomer of lutetium(III) chloride containing the radioactive isotope 177Lu, which undergoes beta decay with a half-life of 6.65 days.
Medical uses
Lutetium (177Lu) chloride is a radiopharmaceutical precursor and is not intended for direct use in patients.[5] It is used for the radiolabeling of carrier molecules specifically developed for reaching certain target tissues or organs in the body. The molecules labeled in this way are used as cancer therapeutics or for scintigraphy, a form of medical imaging.[5] 177Lu has been used with both small molecule therapeutic agents (such as 177Lu-DOTATATE) and antibodies for targeted cancer therapy[8][9]
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Trade names | Lumark, EndolucinBeta, Illuzyce |
| AHFS/Drugs.com | Lumark UK Drug Information EndolucinBeta UK Drug Information |
| License data | EU EMA: by INN |
| Pregnancy category | AU: X (High risk)[1][2] |
| ATC code | None |
| Legal status | |
| Legal status | AU: Unscheduled [3][4]EU: Rx-only [5][6][7]In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 16434-14-3 |
| PubChem CID | 71587001 |
| DrugBank | DBSALT002634 |
| ChemSpider | 32700269 |
| UNII | 1U477369SN |
| KEGG | D10828 |
| CompTox Dashboard (EPA) | DTXSID20167745 |
| Chemical and physical data | |
| Formula | Cl3Lu |
| Molar mass | 281.32 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILES[Cl-].[Cl-].[Cl-].[177Lu+3] |
Contraindications
Medicines radiolabeled with lutetium (177Lu) chloride must not be used in women unless pregnancy has been ruled out.[5]
Adverse effects
The most common side effects are anaemia (low red blood cell counts), thrombocytopenia (low blood platelet counts), leucopenia (low white blood cell counts), lymphopenia (low levels of lymphocytes, a particular type of white blood cell), nausea (feeling sick), vomiting and mild and temporary hair loss.[5]
Society and culture
Legal status
Lutetium (177Lu) chloride (Lumark) was approved for use in the European Union in June 2015.[5] Lutetium (177Lu) chloride (EndolucinBeta) was approved for use in the European Union in July 2016.[6]
On 21 July 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Illuzyce, a radiopharmaceutical precursor.[10] Illuzyce is not intended for direct use in patients and must be used only for the radiolabelling of carrier medicines that have been specifically developed and authorized for radiolabelling with lutetium (177Lu) chloride.[10] The applicant for this medicinal product is Billev Pharma ApS.[10] Illuzyce was approved for medical use in the European Union in September 2022.[7]
References
- ^ “Lutetium (177Lu) Chloride”. Therapeutic Goods Administration (TGA). 21 January 2022. Archived from the original on 5 February 2022. Retrieved 5 February 2022.
- ^ “Updates to the Prescribing Medicines in Pregnancy database”. Therapeutic Goods Administration (TGA). 12 May 2022. Archived from the original on 3 April 2022. Retrieved 13 May 2022.
- ^ “TGA eBS – Product and Consumer Medicine Information Licence”. Archived from the original on 5 February 2022. Retrieved 5 February 2022.
- ^ http://www.ebs.tga.gov.au/servlet/xmlmillr6?dbid=ebs/PublicHTML/pdfStore.nsf&docid=1C7A40803A3A3F94CA2587D4003CE48A&agid=(PrintDetailsPublic)&actionid=1 Archived 30 July 2022 at the Wayback Machine[bare URL PDF]
- ^ Jump up to:a b c d e f g “Lumark EPAR”. European Medicines Agency (EMA). Archived from the original on 25 October 2020. Retrieved 7 May 2020. Text was copied from this source under the copyright of the European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c “EndolucinBeta EPAR”. European Medicines Agency (EMA). Archived from the original on 28 October 2020. Retrieved 7 May 2020. Text was copied from this source under the copyright of the European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b “Illuzyce EPAR”. European Medicines Agency (EMA). 18 July 2022. Archived from the original on 22 September 2022. Retrieved 21 September 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Lundsten S, Spiegelberg D, Stenerlöw B, Nestor M (December 2019). “The HSP90 inhibitor onalespib potentiates 177Lu‑DOTATATE therapy in neuroendocrine tumor cells”. International Journal of Oncology. 55 (6): 1287–1295. doi:10.3892/ijo.2019.4888. PMC 6831206. PMID 31638190.
- ^ Michel RB, Andrews PM, Rosario AV, Goldenberg DM, Mattes MJ (April 2005). “177Lu-antibody conjugates for single-cell kill of B-lymphoma cells in vitro and for therapy of micrometastases in vivo”. Nuclear Medicine and Biology. 32 (3): 269–78. doi:10.1016/j.nucmedbio.2005.01.003. PMID 15820762.
- ^ Jump up to:a b c “Illuzyce: Pending EC decision”. European Medicines Agency. 21 July 2022. Archived from the original on 30 July 2022. Retrieved 30 July 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Lutetium chloride Lu-177”. Drug Information Portal. U.S. National Library of Medicine.
.///////////Lutetium (177Lu) chloride, EMA 2022, EU 2022, APPROVALS 2022, Illuzyce, EndolucinBeta, 塩化ルテチウム (177Lu),

NEW DRUG APPROVALS
ONE TIME
$10.00
Lenacapavir sodium
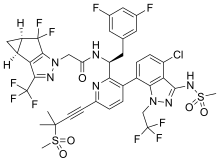
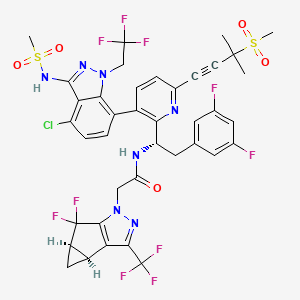
Lenacapavir sodium
レナカパビルナトリウム
| Formula |
C39H31ClF10N7O5S2. Na
C39H32ClF10N7O5S2 FREE FORM
|
|---|---|
| CAS |
2283356-12-5
2189684-44-2 FEE FORM
|
| Mol weight |
990.2641
968.28 FREE FORM
|
2022/8/17 EMA APPROVED, SUNLECA
N-[(1S)-1-[3-[4-chloro-3-(methanesulfonamido)-1-(2,2,2-trifluoroethyl)indazol-7-yl]-6-(3-methyl-3-methylsulfonylbut-1-ynyl)pyridin-2-yl]-2-(3,5-difluorophenyl)ethyl]-2-[(2S,4R)-5,5-difluoro-9-(trifluoromethyl)-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetamide
|
Treatment of HIV-1 infection
|
PF-3540074, to GS-CA1, GS-6207, GS-HIV, GS-CA1, GS-CA2
Lenacapavir, sold under the brand name Sunlenca, is a medication used to treat HIV/AIDS.[1] It is taken by mouth or by subcutaneous injection.[1]
The most common side effects include reactions at the injection site and nausea.[1]
Lenacapavir was approved for medical use in the European Union in August 2022.[1]
HIV/AIDS remains an area of concern despite the introduction of numerous successful therapies, mainly due to the emergence of multidrug resistance and patient difficulty in adhering to treatment regimens.1,2 Lenacapavir is a first-in-class capsid inhibitor that demonstrates picomolar HIV-1 inhibition as a monotherapy in vitro, little to no cross-resistance with existing antiretroviral agents, and extended pharmacokinetics with subcutaneous dosing.1,2,3,5
Lenacapavir was first globally approved by the European Commission to treat adults with multi-drug resistant HIV infection.7 It is currently being investigated in clinical trials in the US.
U.S. Patent Application No. 15/680,041 discloses novel compounds useful for treating a Retroviridae viral infection, including an infection caused by the HIV virus. One specific compound identified therein is a compound of formula I:
PATENTS
- WO 2018/035359 A1
- Different formulations and salts: WO 2019/035904 A1; WO 2019/035973 A1
PATENT
WO 2019/161280 A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019161280
I. Synthesis of Starting Materials and Intermediates
Example la: Preparation of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan- 1-amine (VIII-02), or a co-crystal, solvate, salt, or combination thereof, and starting materials and/or intermediates therein
wherein R4 and R5 are each independently hydrogen, methyl, phenyl, benzyl, 4-nitrobenzyl, 4-chlorobenzyl, 4-brornobenzylamine, or 4-methoxybenzyl
Synthesis of 3,6-dibromopicolinaldehyde (1a)
[00553] A dry reaction flask with magnetic stir-bar was charged with 2,5-dibromopyridine (1.0 g). The flask was inerted under nitrogen, THF (4.2 mL) was added, and the thin slurry agitated. Separately, a dry glass reactor was charged with 2,2,6,6-tetramethylpiperidinylmagnesium chloride, lithium chloride complex (TMPMgCl●LiCl) (5.8 mL, 6.3 mmol). The TMPMgCl●LiCl solution was agitated and cooled to about -20 °C. The 2,5-dibromopyridine solution was added to the TMPMgCl●LiCl solution over about 30 min, maintaining a temperature below about -18 °C. Upon completing the addition, the flask was rinsed forward to the reactor with three additional portions of THF (1 mL x 2), and aged at about -20 for about 1 hour. A solution of N,N-dimethylformamide (1.6 mL, 20 mmol) in THF (1.6 mL) was added to the reactor over about 15 min. The reaction mixture was aged for a further 15 min. and quenched by the addition of a solution of acetic acid (1.9 mL, 34 mmol) in water (10 mL) over about 20 minutes, maintaining a temperature of no more than about 0 °C. To the reactor was added isopropyl acetate (10 mL) and the reaction mixture was warmed to about 20 °C. After aging for 30 min, the mixture was filtered through diatomaceous earth and the reactor rinsed with a mixture of isopropyl acetate (10 mL), saturated aqueous ammonium chloride (10 mL) and 0.2 M aqueous hydrochloric acid (10 mL). The reactor rinse was filtered and the pH of the combined reaction mixture was adjusted to about 8-9 by the addition of a 10% aqueous sodium hydroxide solution (about 6 mL). The mixture was filtered a second time to remove magnesium salts and transferred to a separatory funnel. The phases were separated and the aqueous phase was extracted with isopropyl acetate (3 x 10 mL). The combined organic extracts were washed with 50% saturated aqueous sodium chloride (20 mL), dried over anhydrous sodium sulfate, and filtered. The solution was concentrated to dryness by rotary evaporation and purified by chromatography (eluting with 0-100% ethyl acetate in heptane) to afford 3,6-dibromopicolinaldehyde (1a) as a solid. 1H NMR (400 MHz, DMSO-d6) δ 9.94 (q, J = 0.6 Hz, 1H), 8.19 (dq, J = 8.4, 0.6 Hz, 1H), 7.82 (dt, J = 8.4, 0.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 189.33, 148.59, 145.66, 140.17, 133.19, 120.27.
Synthesis of 3,6-dibromopicolinaldehyde (1a)
[00554] A solution of 2,5-dibromo-6-methylpyridine (8.03 g) in THF (81 mL) was cooled to about 0 °C. To this solution was charged tert-butyl nitrite (4.33 g), followed by a dropwise addition of potassium tert-butoxide (28 mL, 1.5 equiv, 20 wt% solution in THF). The reaction mixture was agitated at about 0 °C until the reaction was complete. The reaction mixture was diluted with THF (24 mL), and quenched with ammonium chloride (6.38 g, 119 mmol) in water (43 mL). The reaction mixture was distilled under vacuum to approximately 55 mL to afford a slurry, which was filtered and washed twice with water (2x 24 mL) to afford 1h. 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.67 (s, 1H), 7.61 (d, J = 8.5 Hz, 1H).
[00555] A solution of glyoxylic acid (407 L, 50 wt% in water) was heated to about 80 °C and in portions was charged with 1h (40.69 kg, 145.4 mol) . Reaction mixture was held at this temperature until the reaction was complete. The reaction mixture was cooled to about 20 °C, filtered, and the filter cake was washed with water until the filtrate had a pH ≥ 5, to afford 1a. 1H NMR (400 MHz, DMSO-d6) δ 9.95 (s, 1H), 8.22 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 8.4 Hz, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00556] Compound 1a (5.0 g, 18.0 mmol) in toluene (20 mL) was heated to about 50 °C and benzhydrylamine (3.47 g, 18.9 mmol) was charged in one portion and agitated at this temperature until the reaction was deemed complete. Methanol (61 mL) was charged and the reaction mixture was distilled to a volume of approximately 25 mL. Methanol (40 mL) was charged and the reaction mixture was distilled to a volume of approximately 30 mL. The resulting slurry was filtered and rinsed with two portions of methanol (15 mL each) and dried under vacuum to afford 1b-02.
[00557] Alternatively, compound 1a (10.0 g, 37.8 mmol) in 2-methyltetrahydrofuran (50 mL) was heated to about 50 °C and benzhydrylamine (7.28 g, 39.7 mmol) was charged dropwise. The reaction was agitated at this temperature until it was deemed complete. The reaction mixture was distilled to a volume of approximately 30 mL. To the reaction mixture was charged heptane (100 mL) and 1b-02 seed (59.3 mg, 0.138 mmol). The resulting slurry was filtered, rinsed with two portions of heptane (2x 20 mL), and dried under vacuum to afford 1b-02. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.44 – 7.40 (m,
4H), 7.38 – 7.32 (m, 4H), 7.28 – 7.22 (m, 2H), 5.88 (s, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00558] 1a (2.00 g) was combined with isopropanol (7.6 mL) and agitated at ambient temperature. To this mixture was added potassium metabisulfite (0.96 g) in water (3.8 mL), dropwise. This mixture was agitated for at least 90 minutes and the resulting slurry was filtered. The filter cake was rinsed twice with isopropanol (6 mL then 12 mL) to afford 1i-1. 1H NMR (400 MHz, DMSO-d6) δ 7.92 (d, J = 8.3 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 5.48 – 5.38 (m, 2H).
[00559] li-1 (1.00 g) was combined with 2-methyltetrahydrofuran (3.5 mL) and agitated at ambient temperature. To this slurry was charged potassium hydroxide (443.8 mg, 7.91 mmol) in water (4 mL) and the biphasic mixture was agitated for 2 hours. The layers were separated and the aqueous layer was extracted with an additional portion of 2-methyltetrahydrofuran (3.5 mL). To the combined organics was charged benzhydrylamine (0.47 mL, 2.7 mmol). The reaction mixture was concentrated in vacuo (-300 mbar, 45 °C bath) to a volume of approximately 3 mL. Heptane (7 mL) was charged and the mixture was agitated. The resulting slurry was filtered to afford 1b-02 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.44 – 7.40 (m, 4H), 7.38 – 7.32 (m, 4H), 7.28 – 7.22 (m, 2H), 5.88 (s, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00560] Compound 1a (1.0 g) was added to a reactor, and toluene (6.0 mL) was added to the reactor. The mixture was agitated. Aminodiphenylmethane (0.73 g, 1.05 equiv.) was added to the reaction mixture. The jacket was heated to about 60 °C, and the mixture was allowed to age for about 1 hour. After about one hour, the mixture was carried forward to the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.4 Hz, 4H), 7.40 – 7.34 (m, 7H), 7.29 (td, J = 6.9, 6.5, 1.7 Hz, 5H), 7.22 – 7.16 (m, 3H), 5.81 (s, 1H).
Synthesis of N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-1,1-diphenylmethanimine (1d-02)
[00561] A solution of1b-02 in toluene (1.0 g in 3.8 mL) was stirred in a reactor at about 60 °C. Tetrabutylammonium bromide (0. 08 g, 0.10 equiv.) was added, 3,5-difluorobenzylbromide (0.60 g, 1.20 equiv.) was added, and potassium hydroxide (50% in water, 1.3 g, 5 equiv.) was added. The mixture was aged for about 4 hours and sampled for conversion. When the reaction was complete, the aqueous phase was removed, and water (3.1 mL) was added to the reactor. Contents were agitated and phases were allowed to settle. The aqueous phase was removed, and the toluene solution of1d-02 was carried forward to the next step. 1H NMR (400 MHz, Chloroform-d) δ 7.78 (dd, J = 8.6, 1.0 Hz, 1H), 7.64 – 7.60 (m, 2H), 7.59 – 7.53 (m, 1H), 7.49 (d, J = 8.3 Hz, 1H), 7.47 (s, 0H), 7.45 (s, 0H), 7.43 (d, J = 0.7 Hz, 0H), 7.41 – 7.34 (m, 3H), 7.33 (t, J = 1.4 Hz, 1H), 7.28 (t, J = 7.3 Hz, 2H), 7.22 (s, 0H), 7.18 (d, J = 8.3 Hz, 1H), 6.87 (dd, J = 7.7, 1.7 Hz, 2H), 6.55 (dt, J = 9.0, 2.3 Hz, 1H), 6.50 (dd, J = 7.0, 4.9 Hz, 3H), 5.26 (s, 0H), 5.16 (t, J = 6.9 Hz, 1H), 3.32 (dd, J = 13.2, 6.6 Hz, 1H), 3.16 (dd, J = 13.1, 7.2 Hz, 1H).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) from N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-1,1-diphenylmethanimine (1d-02)
[00562] A solution of 1d-02 in toluene (1.0 g in 3.0 mL) was stirred in a reactor at about 60 °C. Sulfuric acid (0.93 g, 5 equiv.) was diluted into water (3.5 mL), and added to the reactor. The mixture was aged for about 4 hours. When the reaction was complete, the aqueous phase was removed. The aqueous phase was recharged to the reactor, and heptane (2.5 mL) was added. The mixture was agitated and agitation stopped and layers allowed to settle. The aqueous phase was removed, and heptane was discharged to waste. Toluene (5.0 mL) and potassium hydroxide (50% in water, 2.1 g, 10 equiv.) was added to the reactor. The aqueous acidic solution was added to the reactor. The mixture was agitated for about 10 minutes, and agitation stopped and phases allowed to settle. The aqueous phase was discharged to waste. Water (2.5 mL) was added to the reactor, and the mixture was agitated for about 5 minutes, and agitation was stopped and the phases were allowed to settle. The aqueous phase was discharged to waste. The toluene solution of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) was carried forward to the next step. 1H NMR (400 MHz, Chloroform-d) δ 7.60 (d, J = 8.3 Hz, 1H), 7.21 (d, J = 8.3 Hz, 1H), 6.74 – 6.67 (m, 2H), 6.66 – 6.58 (m, 1H), 4.57 – 4.45 (m, 1H), 3.02 (dd, J = 13.5, 5.2 Hz, 1H), 2.72 (dd, J = 13.5, 8.6 Hz, 1H), 1.77 (s, 3H).
Synthesis of (S)-1-(3.6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-2-hydroxy-2-phenyl acetate (VIII-03)
[00563] A solution of X in toluene (1.0 g in 7.1 mL) was stirred in a reactor at about 60 °C. The mixture was distilled to minimum volumes (2.9 mL), and methyl tert-butyl ether was added (7.1 mL). (R)-(-)-Mandelic acid (0.41 g, 1 equiv.) was added, and the mixture was cooled to about 0 °C. The newly formed slurry was filtered, providing (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-2-hydroxy-2-phenylacetate (VIII-03). 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 8.4 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.34 (d, J = 7.3 Hz, 2H), 7.28 – 7.14 (m, 4H), 7.01 (tt, J = 9.4, 2.3 Hz, 1H), 6.79 (d, J = 7.4 Hz, 3H), 4.77 (s, 1H), 4.55 (d, J = 6.6 Hz, 1H), 3.02 (s, 1H), 2.92 (d, J = 6.7 Hz, 2H), 1.05 (s, 2H).
Synthesis of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine N-acetyl-D- Leucine (VIII-04)
[00564] A reactor was charged with X (15.0 g), N-acetyl-D-leucine (8.28 g) and zinc oxide (0.311 g). Toluene (375 mL) was charged to the reactor followed by 2-pyridinecarboxaldehyde (183 μL). The mixture was aged at about 55 °C for about 6 hrs. and then held at about 35 °C for about 4 days. The mixture was cooled to about 0 °C and held for about 17 hrs. The product was isolated by filtration and the filter cake was washed with cold toluene (2 x 75 mL). The filter cake was re-charged to the reactor. Ethanol (150 mL) was added and the mixture distilled to remove residual toluene. Once the toluene was removed, the reactor volume was adjusted with ethanol to about 90 mL and the mixture was cooled to about 25 °C. Water (210 mL) was added over approximately 10 min. and the mixture aged for approximately 12 hrs. The slurry was filtered and the solids were dried to afford VIII-04. 1H NMR (400 MHz, DMSO-d6) δ 8.03 (d, J = 8.0 Hz, 1H). 7.95 (d, J = 8.3 Hz, 1H), 7.49 (d, 7 8.3 Hz, 1H), 7.03 (tt, J = 9.5, 2.4 Hz, 1H),
6.87 (dtd, J = 8.4, 6.2, 2.2 Hz, 2H), 5.49 (s, 3H), 4.42 (dd, J = 7.9, 5.9 Hz, 1H), 4.18 (q, J = 7.8 Hz, 1H), 2.93 (dd, J = 13.3, 5.9 Hz, 1H), 2.85 (dd, J = 13.2, 8.0 Hz, 1H), 1.83 (s, 3H), 1.71 -1.54 (m, 1H), 1.47 (dd, J = 8.4, 6.2 Hz, 2H), 0.88 (d, J = 6.6 Hz, 3H), 0.83 (d, J = 6.5 Hz, 3H).
13C NMR (101 MHz, DMSO-d6) δ 174.72, 169.03, 162.07 (dd, J = 245.5, 13.3 Hz), 161.79, 143.51, 142.82 (t, J = 9.4 Hz), 139.72, 128.39, 119.30, 113.36 – 111.39 (m), 101.73 (t, J = 25.7 Hz), 55.19, 50.69, 41.74 (d, J = 2.3 Hz), 40.51, 24.36, 22.91, 22.44, 21.46.
Example 1b: Preparation of alternative starting materials and intermediates for use in the formation of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difliiorophenyl)ethan-1-amine (VIII), or a co-crystal, solvate, salt, or combination thereof
Synthesis of (R)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-ol (XII)
[00565] A stainless steel autoclave equipped with a glass inner tube was charged with compound XI (1.00 g) and (A)-RuCY-XylBINAP (16 mg, 0.05 equiv.). EtOH (1.0 mL) and IPA (1.0 mL) followed by tert-BuOK (1.0 M solution in THE, 0.51 mL, 0.2 equiv.) were added to the autoclave. After being purged by H2, the autoclave was charged with 3 MPa
of H2. The mixture was stirred at about 20 °C for about 10 h. To the mixture, cone. HCl aqueous solution was added and pH was adjusted to 2. 1H NMR (400 MHz, CDCl3): δ 7.72 ( d, J = 8.2 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 6.80 -6.72 (m, 2H), 6.68 (tt, J = 9.2, 2.4 Hz, 1H), 5.16 (dd, J = 8.2, 3.4 Hz, 1H), 3.60 (br, 1H), 3.12 (dd, J = 13.8, 3.4 Hz, 1H), 2.81 (dd, J = 13.8, 8.2 Hz,
1H). 13C NMR (100 MHz, CDC13): d 162.8 (dd, J= 246.4, 12.9 Hz), 160.1, 143.0, 141.3 (t, j = 9.1 Hz), 139.8, 128.7 (t, J= 35.7 Hz), 117.9, 112.3 (m), 102.1 (t, J= 25.0 Hz), 72.0, 43.0. 19F NMR (376 MHz, CDCl3): δ -112.1 (m).
Synthesis of N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-15-chloranimine (X-02)
[00566] Compound XIII (.0 g) was dissolved in THF (4.2 mL) and was cooled over an ice bath. Diphenylphosphoryl azide (0.66 mL, 1.2 equiv.) was added followed by DBU (0.46 mL, 1.2 equiv.) over about 25 min at below about 4 °C. The dark mixture was aged about 1 hour, and the cooling bath was removed. After about 2.5 hours age at RT, some starting material was still present so more diphenylphosphoryl azide (0.15 equiv.) and DBU (0.15 equiv.) were added after cooling over an ice bath. After about 2 hours, more diphenylphosphoryl azide (0.08 equiv.) and DBU (0.08 equiv.) were added. The reaction mixture was allowed to age overnight for about 16 h to allow the conversion to azide intermediate complete. The reaction mixture was cooled over an ice bath and triphenylphosphine (1.0 g, 1.5 equiv.) was added over about 15 min at about 6 °C). The cooling bath was removed after about 10 min and the reaction mixture was agitated for additional about 2.5 hours. To this reaction mixture was added water (0.18 mL, 4 equivalents) and the resulting mixture was aged for about 15 hours at room temperature. The mixture was diluted with EtOAc (5.0 mL) and was washed with water (4.2 mL + 2.0 mL). The aqueous layer was back extracted with EtOAc (4.0 mL) and the EtOAc layer was washed with water (1.0 mL). The organic layers were combined, concentrated via rotary evaporation and evaporated with EtOAc (4 x 4.0 mL) to dry. The residue was dissolved to a 50 ml solution in EtOAc, and cooled over an ice bath to become slurry. To the cold slurry 4N HCl/dioxane (0.76 mL, 1.2 equiv.) was added and the slurry was aged about 2 hours at room temperature. The solid product was filtered and the filter cake was rinsed with EtOAc and dried at about 35 to 50 °C under vacuum to give X-02.
[00567] Recrystallization: A portion of the above obtained X-02 (1.0 g) was mixed with EtOAc (10 mL) and was heated to 65 °C to afford thick slurry. The slurry was aged at about 65 °C for about 2 hours, and overnight at room temperature. The solids were filtered with recycling the mother liquor to help transfer the solids. The filter cake was rinsed with EtOAc, and dried overnight at about 50 °C vacuum to afford X-02. 1H NMR (300 MHz, DMSO-d) δ 8.78 (br s, 3 H), 8.06-8.02 (m, 1 H), 7.64-7.61 (m, 1 H), 7.15-7.08 (m, 1 H), 6.83-6.78 (m, 2 H), 4.87-4.82 (m, 1 H), 3.35-3.25 (m, 1 H), 3.17-3.05 (m, 1 H). 19F NMR (282.2 MHz, Chloroform-d) δ – 109.9-110.1 (m).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl methanesulfonate (XIII-A)
[00568] Compound XIII (1.0 g) and DMAP (0.1 equiv.) were dissolved in THF (4.5 mL) and cooled over an ice bath. Triethylamine (Et3N) (0.39 mL, 1.1 equiv.) was added followed by methanesulfonyl chloride (218 μL, 1.1 equiv.). The cooling bath was removed, and the mixture was aged about 1.5 hours at room temperature. The reaction mixture was cooled over an ice bath and quenched with water (10 mL). The mixture was diluted with EtOAc and the phases were separated. The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (Na2SO4) and was passed through silica gel with EtOAc. The filtrate was concentrated to afford the mesylate (XIII-A). 1H NMR (300 MHz, Chloroform-d) δ 7.72-7.66 (m, 1 H), 7.38-7.32 (m, 1 H), 6.78-6.63 (m, 3 H), 6.17-6.13 (m, 1 H), 3.40-3.25 (m, 2 H), 2.87 (s, 3 H). 19F NMR (282.2 MHz, Chloroform-d) δ -109.3—109.5 (m).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) from 1-(3,6- dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl methanesulfonate (XIII-A)
[00569] A glass pressure bottle was charged with the mesylate (XIII-A) (1.0 g), 28-30% ammonium hydroxide (19 mL) and MeOH (4.7 mL). The mixture was sealed and heated at about 70 °C for about 16 hours, and extracted with 2-MeTHF/ EtOAc. The organic layer was dried (Na2SO4) and purified by silica gel chromatography (10-60% EtOAc/hexanes) to afford racemic amine X. 1H NMR (300 MHz, Chloroform-d) δ 7.70-7.60 (m, 1 H), 7.30-7.20 (m, 1 H), 6.78-6.60 (m, 3 H), 4.46-4.58 (m, 1 H), 3.00-3.16 (m, 1 H), 2.70-2.80 (m, 1 H). 19F NMR (282.2 MHz, Chloroform-d) δ -110.3 – 110.4 (m).
Synthesis of (Z)-N-(1-(3,6-dibrornopyridin-2-yl)-2-(3,5-difluorophenyl)vinyl)acetamide (1f)
[00570] A glass reactor was charged with XI (1.0 g). Ethanol (5.0 mL) was added, and the slurry was agitated while hydroxylamine hydrochloride (0.88 g) was charged. Pyridine (1.0 mL) was added and the mixture heated at about 55-65 °C for about two hours. The mixture was cooled to about 20 °C, transferred to a flask, and concentrated to approximately 75 mL by rotary evaporation. The concentrate was returned to the reactor, rinsing through with isopropyl acetate (5.0 mL). Residue remaining in the flask was carefully (gas evolution) rinsed into the reactor with saturated aqueous sodium bicarbonate (5.0 mL). The bi-phasic mixture was agitated, the phases separated, and the organic extract washed with water (3.2 mL) and saturated sodium chloride (3.2 mL). The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated to dryness by rotary evaporation to yield 1e which was used without further purification.
[00571] A glass reactor was charged with iron powder (<10 micron, 0.30 g mmol) followed by acetic acid (1.6 mL) and acetic anhydride (0.72 mL). The slurry was de-gassed by holding the reactor contents under vacuum until bubbling was observed, and back-filled with nitrogen (3 cycles). The mixture was heated at 115-120 °C for 2 hours and cooled to 40 °C. Compound le from the previous step in isopropyl acetate (2.0 mL) was added over 30 min. Upon completing the addition, the temperature was raised to 45-65 °C and the mixture aged for about 2 hours. A slurry of diatomaceous earth (1.0 g) in isopropyl acetate (2.0 mL) was added, followed by toluene (2.0 mL). The slurry was filtered, hot, through a Buchner funnel and the reactor and filter cake were washed with warm isopropyl acetate (3 x 1.8 mL). The filtrate was transferred to a reactor and the solution washed with 0.5% aqueous sodium chloride (4.2 mL). Water (3.1 mL) was added to the reactor and the mixture was cooled to about 5 °C. The pH was adjusted to 7-9 with the addition of 50 wt% aqueous sodium hydroxide; following separation, the organic extract was warmed to room temperature and washed with aqueous 1% (w/w) sodium chloride NaCl (3.6 mL). The organic extract was discharged to a flask and dried over anhydrous sodium sulfate (ca. 0.8 g), filtered through diatomaceous earth, and concentrated to approximately 4 mL at 100 mmHg and 45 °C water bath. The warm solution was returned to the reactor, rinsing forward with isopropyl acetate to a produce a total volume of approximately 5.2 mL. This solution was heated further to 50 °C with agitation, cooled to about 35 °C, and seeded with pure 1f (0.006 g). Heptane (9.6 mL) was added over a period of about 4 hours, the solution was cooled to about 10 °C, and the product was isolated by filtration. The filter cake was washed with 33.3% iPAc in heptane (4.0 mL) and dried in a vacuum oven at 40 °C with nitrogen sweep for approximately 24 hours. Compound 1f, a mixture of geometric isomers (approximately 94:6 ratio) was isolated. Major isomer: 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.66 (d, J= 8.4 Hz, 1H), 7.05 (s, 1H), 6.97 (tt, J = 9.2, 2.2 Hz, 1H), 6.40 – 6.31 (m,
2H), 1.97 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.37, 162.04 (dd, J = 245.1, 13.9 Hz), 154.47, 143.63, 139.45, 139.40 – 139.18 (m), 135.99, 129.44, 120.66, 113.80, 111.23 – 109.68 (m), 101.77 (t, J = 26.0 Hz), 23.49.
Synthesis of (S)-N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)acetamide (1g)
[00572] Preparation of catalyst solution: A flask was charged with [IrCl(cod)((S)-segphos)] (110 mg) and the internal atmosphere was replaced with N2. EtOAc (200 mL) was added to the flask and the mixture was stirred until the catalyst solid was dissolved.
[00573] A stainless steel autoclave was charged with compound 1f (1.0 mg). EtOAc (16 mL) and followed by the catalyst solution prepared above (4.0 mL, 0.001 equiv.) were added to the autoclave. After being purged by H2, the autoclave was charged with 3 MPa of H2.The mixture was stirred at about 130 °C for about 6 hours and cooled to room temperature and H2 was vented out. The reaction mixture was purified by silica gel column chromatography (EtOAc/Hexane = 1/4 to 1/1) to afford 1g. 1H NMR (400 MHz, CD2Cl2): d 7.70 ( d, J = 8.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 6.68 (tt, J = 9.2, 2.4 Hz, 1H), 6.64 -6.58 (m, 2H), 6.49 (brd, j = 8.0 Hz, 1H), 5.74 (ddt, J = 8.0, 7.2, 6.4 Hz, 1H), 3.10 (dd, J = 13.6, 6.4 Hz, 1H), 2.99 (dd, J = 13.6, 7.2 Hz), 1.95 (s, 3H). 13C NMR (100 MHz, CD2Cl2): δ 169.5, 163.3 (dd, J = 246.0, 12.9 Hz), 159.1, 143.6, 141.4 (t, J = 9.1 Hz), 140.7, 129.1, 119.9, 112.9 (m), 102.6 (t, J= 25.1 Hz), 53.0, 41.3, 23.6. 19F NMR (376 MHz, CD2Cl2): δ -111.3 (m).
Synthesis of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VIII) from 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-one (XI), Method 1
[00574] A glass-lined reactor was charged with isopropylamine (about 18 g) and triethanolamine (3.8 g). Water (231 mL) was added and the pH was adjusted to about 7.5 by the addition of concentrated hydrochloric acid. A portion of the buffer solution (23 mL) was removed. The transaminase enzyme (2.5 g) was added to the reactor as a suspension in buffer solution (12 mL), followed by addition of pyridoxal phosphate monohydrate (50 mg) as a solution in buffer solution (12 mL). A solution of XI (1.0 g) in dim ethyl sulfoxide (23 mL) was added to the reactor and the mixture was heated at about 35 °C for about 48 hours with constant nitrogen sparging of the solution. The reaction mixture was cooled to about 20 °C the unpurified amine was removed by filtration. The filter cake was washed with water (3 x 7.7 mL) and the product was dried at about 60 °C under vacuum with nitrogen sweep to afford VIII.
Synthesis of (S)-1-(3.6-dibromopyridin-2-yl)-2-(3.5-difluorophenyl)ethan-1-amine (VIII) from 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-one (XI), Method 2
[00575] A stainless steel reactor was charged with XI (1.0 g) and p-toluenesulfonic acid (0.49 g). Ammonia (7 M in methanol, 3.7 mL) was added and the vessel was sealed and heated at about 60 °C for about 18 hours. The mixture was cooled to about 20 °C and sparged for about 30 min to remove excess ammonia. A solution of diacetato[(R)-5,5′-bis(diphenylphosphino)-4,4′-bi-1,3-benzodioxole]ruthenium(II) (0.10 g) in methanol (0.5 mL) was added to the reactor, which was sealed and heated at about 60 °C under a hydrogen atmosphere (400 psi) for a further about 6-10 hours. Upon cooling to about 20 °C the mixture was filtered through a plug of silica, rinsing with additional methanol (5.0 mL). Concentration of the filtrate by rotary evaporation affords VIII.
Example 1c: Preparation of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyI)ethan-1-amine (X) by racemization of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VIII)
[00576] A vial was charged with zinc acetate (25 mol %), enantioenriched VIII (1.0 g, 92:8 enantiomer ratio), toluene (10 mL), and 2-formylpyridine (5 mol %). The vial was wanned to about 60 °C and stirred for about 4 h.
Example 2: Preparation of (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI)
[00577] A glass-lined reactor was charged with (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-mandelic acid salt (VIII-03) (1.0 g), 3-methyl-3-(methylsulfonyl)but-1-yne (IX) (about 0.3 g), and dichlorobis(triphenylphosphine)palladium(II) (about 0.39 g). The reactor was evacuated and purged with nitrogen to inert. To this reactor was added 2-methyltetrahydrofuran (6.4 kg) and triethylamine (0.92 kg 5.0 equiv.). The reaction mixture was agitated at about 65-75 °C until the reaction was deemed complete by HPLC analysis. Upon cooling to about 30-40 °C the reaction mixture was discharged to another reactor and the parent reactor was rinsed with 2-methyltetrahydrofuran (4.6 g) and the resulting solution transferred to the receiving reactor. To the reactor was added water (5.0 g) and the biphasic mixture agitated at about 30-40 °C for about 30 min. Agitation was ceased and the mixture was allowed to layer for 30 min. The lower aqueous layer was discharged and the remaining organic solution held for about 15 hours. A solution of A-acetyl-L-cysteine (196 g) and sodium hydroxide (0.80 g) in water (11.8 g) was prepared. To the reactor was added approximately half of the N-acetyl-L-cysteine solution (6.7 g). The mixture was agitated at about 55-65 °C for about 30 min. The temperature was adjusted to about 30-40 °C and agitation was ceased. After about 30 min had elapsed, the lower aqueous phase was discharged. The remaining alkaline N-acetyl-L-cysteine solution (5.4 kg) was added and the mixture was heated, with agitation, to about 55-65 °C and held for about 30 min. The temperature was adjusted to about 30-40 °C and agitation was ceased. After about 30 min had elapsed, the lower aqueous phase was discharged. To the reactor was added a solution of sodium chloride (0.26 g) in water (4.9 g) and the mixture agitated at about 30-40 °C for about 30 min. Agitation was ceased and the biphasic mixture allowed to layer for about 30 min. The lower aqueous layer was discharged and the contents cooled to about 15-25 °C and held for about 16 hours. The mixture was concentrated at about 55-65 °C. The concentrated solution was cooled to about 30-40 °C and heptane (3.4 kg) was added over about 2 hours. The resulting slurry was cooled to about 20 °C and aged for about 20 h, and filtered. The filter cake was washed with 2-methyltetrahydrofuran/heptane (1:1 v/v,2 mL) and the solids dried in a vacuum oven at about 40 °C to yield (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI)). 1H NMR (400 MHz, DMSO-d6) δ 8.05 (d, J = 8.2 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.01 (tt, J = 9.5, 2.4 Hz, 1H), 6.97 – 6.84 (m, 2H), 4.41 (dd, J = 8.5, 5.2 Hz, 1H), 3.20 (s, 3H), 2.93 (dd, J = 13.3, 5.2 Hz, 1H), 2.79 (dd, J = 13.3, 8.5 Hz, 1H), 1.99 (s, 2H), 1.68 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 162.25, 162.00 (dd, J = 245.2, 13.4 Hz), 143.88 (t, J= 9.4 Hz), 141.09, 139.72, 127.51, 120.08, 112.58 – 112.12 (m), 101.45 (t, J= 25.7 Hz), 87.94, 84.25, 57.24, 55.90, 42.57, 34.99, 22.19.
Example 2a: Preparation of 3-methyl-3-(methylsulfonyl)but-1-yne (IX)
[00578] Sodium methansulfmate (418.1 g), copper (II) acetate (26.6 g), N,N,N’,N’- Tetramethylethylenediamine (TMEDA, 34.0 g), and isopropyl acetate (2100 mL) were added to a reactor and the suspension was agitated at 20 – 25 °C. 3-Chloro-3-methylbut-1-yne (3-CMB,
300 g) was added slowly to maintain a constant temperature of about 20 – 25 °C. The reaction mixture was then heated to about 30 °C until the reaction was complete. The mixture was cooled to about 20 °C and washed twice with 5% aqueous sulfuric acid (600 mL). The combined
aqueous layers were then extracted with isopropyl acetate (600 mL). The combined organic layers were then washed with water (600 mL). The product was then isolated by crystallization from isopropyl acetate (900 mL) and n-heptane (1.8 kg) at about 0 °C. The wet cake was then washed with cold n-heptane to afford IX. 1H NMR (400 MHz, DMSO-d6) δ 3.61 (s, 1H), 3.07 (s, 3H), 1.55 (s, 6H); 13C NMR (10Q MHz, DMSO) d 82.59, 77.76, 56.95, 34.95, 22.77.
Example 3a: Preparation of (3bS,4aR)-3-(trifluoromethyI)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from lithium (Z)-2,2,2-trifluoro-1-(3-oxobicyclo[3.1.0]hexan-2-ylidene)ethan-1-olate (3a)
Synthesis of 3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazole (3b)
[00579] A reactor was charged with 3a (1.0 g) and AcOH (4.2 ml) and the resulting solution was adjusted to about 20 °C. Hydrazine hydrate (0.29 g, 1.4 equiv.) was added over about 60 min at about 17-25 °C and the reaction mixture was stirred for about 2 hours at about 20-25 °C, warmed up to about 45 to 50 °C over about 30 min, and aged at about 50 °C overnight. Water was slowly (5 mL) added at about 50 °C and product started to crystallize after addition of 5 mL of water. Another 5 mL of water was added at about 50 °C, and the slurry was cooled down to about 20 °C in about one hour and held overnight at about 20 °C. The solids were filtered, washed with water (4X 3 mL), and dried under vacuum at about 30 °C to yield 3b. 1H NMR (400 MHz, Chloroform-d) δ 2.99 (dd, J = 17.0, 6.1 Hz, 1H), 2.89 – 2.78 (m, 1H), 2.14 (dddd, J = 9.1, 7.9, 3.6, 2.5 Hz, 2H), 1.13 (td, J = 7.8, 5.1 Hz, 1H), 0.36 – 0.26 (m, 1H).
Isolation of (3bS,4aS)-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazole (3c)
[00580] Chiral purification of 3b (1.0 g) was achieved using a 8×50 mm simulated moving bed (SMB) chromatography system and Chiralpak IG (20 μ particle size) stationary phase using acetonitrile as a mobile phase to afford 3c. 1H NMR (400 MHz, Chloroform-d) δ 3.00 (dd, J = 17.0, 5.7 Hz, 1H), 2.90 – 2.77 (m, 1H), 2.21 – 2.05 (m, 2H), 1.13 (td, J = 7.8, 5.1 Hz, 1H), 0.35 – 0.27 (m, 1H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV)
[00581] A reactor was charged with water (7 mL) and CuCl2 ● 2H2O (0.09 g, 0.1 equiv). To the reactor was added pyridine (0.42 g, 1 equiv.) and 3c. tert-Butylhydroperoxide (70% in water, 5.5 g, 8 equiv.) was added over about 0.5 hour. The reaction mixture was stirred at about 20 °C for about 2.5 days and quenched with aqueous sodium metabisulfite solution (0.73 g in 2.5 mL water). The quenched reaction mixture was extracted with isopropyl acetate (20 mL), and the aqueous layer was back extracted with isopropyl acetate (2.0 ml). The organic layers were combined and washed with aqueous ethylenediaminetetraacetic acid (EDTA) solution 0.16 g EDTA 10 ml in water), the aqueous layer was dropped, and the organic layer was further washed with aqueous EDTA solution (0.015 g EDTA in 20 ml water). The washed organic layer was concentrated to dryness. To the residue was added isopropyl acetate (2.0 ml) and heptane (2.0 mL). The solution was seeded and stirred overnight at about 20 °C, further diluted with heptane (2.0 mL), and the mixture was concentrated to dryness. The residue was suspended in heptane (4.0 mL) at about 40 °C. The solid was filtered and the filter cake was washed with heptane (1.0 mL) and dried at about 40 °C to yield XV. 1H NMR (400 MHz, Chloroform-d) δ 2.84 (dt, J = 6.8, 4.2 Hz, 1H), 2.71 – 2.64 (m, 1H), 1.79 – 1.67 (m, 2H).
Example 3b: Preparation of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from lithium (Z)-1-((1S,5R)-4,4- dimethoxy-3-oxobicyclo[3.1.0]hexan-2-ylidene)-2,2,2-trifluoroethan-1-olate (3d-02)
[00582] Hydrazine sulfate (0.45 g, 0.95 equiv.) and ketal lithium salt 3d-02 (1.0 g) were dissolved in ethylene glycol (9.5 mL), and the solution was heated to about 40 °C for about 16 hours. Reaction was cooled to room temperature and water (9.0 mL) was added. Reaction was polish filtered andThe filtrate was collected and to this receiving flask was added water (10 mL, 2x). Slurry was cooled in ice water bath for about five hours, and filtered. Solids were washed with ice water (10 mL, 2x), deliquored, and dried to afford XV. 1H NMR (400 MHz, CDCl3) δ 11.83 (bs, 1H), 2.93 – 2.77 (m, 1H), 2.77 – 2.58 (m, 1H), 1.86 – 1.57 (m, 2H). 19F NMR (376 MHz, CDCl3) δ -61.69. 13C NMR (101 MHz, CDCl3) δ 188.56, 144.08, 142.92, 121.82, 119.15, 36.28, 31.87, 14.15.
Example 3c: Preparation of (3bS,4aR)-3-(trifiuoromethyl)-1,3b,4,4a-tetrahydro-5H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from (1S,2S)-2-iodo-N-methoxy-N- methylcyclopropane-1-carboxamide (3f) and 1-(4-methoxybenzyl)-4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)-1H-pyrazole (3i) and preparation of starting materials and/or intermediates therein
Synthesis of (1S,2S)-2-iodo-N-methoxy-N-methylcyclopropane-1-carboxamide (3f)
[00583] Starting material iodoacid 3e is a mixture of 3e and cyclopropane carboxylic acid (des-iodo 3e) with mole ratio of 3e to des-iodo 3e of 2:1 by NMR. A mixture of 3e (1.0 g),
N,O-dimethyl hydroxyl amine-HCl (0.46 g) and carbonyl diimidazole (1.72 g) in THF was stirred overnight at room temperature. The reaction mixture was diluted with water, extracted with CH2Cl2, and concentrated to afford unpurified 3f (1.8 g). The unpurified 3f was purified by column chromatography to afford 3f which was a mixture of Wei nr eb amide 3f and des-iodo-3f (about 80:20 by HPLC).
Synthesis of 1-(4-methoxybenzyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3- (trifluoromethyl)-1H-pyrazole (3i)
[00584] To a suspension of NaH (60%, 0.31 g, 1.1 equiv.) in DMF (7.5 mL), a solution of 3g (1.0 g) in DMF (7.5 mL) was added dropwise over about 15 min at about 3 to 7 °C. The reaction mixture was stirred at room temperature for about 1 h and a solution of PMBCl (1.2 g, 1.05 equiv.) in DMF (4.2 mL) was added dropwise in about 25 min at room temperature. The reaction mixture was stirred at room temperature overnight, poured into water (17 mL), and extracted with diethyl ether (3×17 mL). The ether layers were combined and washed with water (2 x 17 mL) and brine (17 mL), dried over Na2SO4, and concentrated in vacuo to give unpurified 3h. Unpurified 3h was absorbed in silica gel (4.3 g) and purified by silica gel chromatography (eluting with 5-25% EtOAc in hexanes) to give 3h (1.5 g).
[00585] To solution of iodopyrazole 3h (1.0 g) in THF (8 mL) i-PrMgCl (2M in ether, 1.8 mL, 1.1 equiv.) was added dropwise over about 10 min at below about 5 °C. The resulting solution was stirred at about 0 °C for about 70 min and 2-methoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (970 mg, 1.81 equiv.) was added at below about 6 °C. The reaction mixture was warmed up to room temperature, quenched by addition of saturated NH4Cl (20 mL), and
extracted with EtOAc (2 x 20 mL). The combined organic layer was washed with saturated NH4Cl (10 mL) and concentrated to unpurified oil, which was combined with the unpurified oil from a previous batch (prepared using 1.1 g of 3h), absorbed on silica gel (6 g), and purified via silica gel chromatography (eluting with 5-40% EtOAc/Hexanes,). Boronate 3i was obtained. 1H NMR (300 MHz, Chloroform-d) δ 7.60 (s, 1 H), 7.23-7.19 (m, 2 H), 6.90-6.85 (m, 2 H), 5.25
(s, 2 H), 3.81 (m, 3 H), 1.29 (s, 12 H).
Synthesis of (1R,2S)-N-methoxy-2-(1-(4-methoxybenzyl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)-N-methylcyclopropane-1-carboxamide (3j)
[00586] A mixture of unpurified iodide 3f (1.0 g), boronate 3i (about 2.2 g), CsF (4.5 equiv.), Pd(OAc)2 (0.1 equiv.), and PPh3 (0.5 equiv.) in DMF (58 mL) was degassed by bubbling N2 and heated at about 87 °C for about 15 hours. The reaction mixture was diluted with water,
extracted with MTBE, concentrated and the unpurified product was purified by column chromatography to give 3j. 1H NMR (300 MHz, Chloroform-d) δ 7.18-7. 14 (m, 3 H), 6.86-6.82 (m, 2 H), 5.24-5.08 (m, 2 H), 3.77 (s, 3 H), 3.63 (s, 3 H), 3.05 (s, 3 H), 2.37-2.32 (m, 1 H), 1.50-1.42 (m, 1 H), 1.32-1.21 (m, 2 H).
Synthesis of (3bS,4aR)-1-(4-methoxybenzyl)-3-ftrifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta91,2-c]pyrazol-5-one (3k)
[00587] Compound 3j (1.0 g) was treated with freshly prepared LDA (3.3 eq then 0.7 equiv.) at about -67 °C for about 2.5 hours. The reaction mixture was quenched with saturated NH4Cl (12.5 mL) and diluted with MTBE (63 mL). The organic layer was washed with brine, concentrated, and purified by column chromatography to give 3k. 1H NMR (300 MHz, Chloroform-d) δ 7.36-7.33 (m, 2 H), 6.86-6.83 (m, 2 H), 5.28 (s, 2 H), 3.78 (s, 3 H), 2.73-2.65
(m, 1 H), 2.60-2.53 (1 H), 1.70-1.61 (m, 2 H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1.2-c]pyrazol-5-one (XV)
[00588] A mixture of 3k (1.0 g) and TFA (5 mL) was heated at about 75 °C for about 3 hours and concentrated. The residue was dissolved in DCM (50 mL), washed with saturated NaHCO3 and brine, concentrated, and purified by column chromatography to give XV. 1H NMR (300 MHz, Chloroform-d) δ 2.86-2.80 (m, 1 H), 2.68-2.63 (m, 1 H), 1.77-1.65 (m, 2 H).
Example 3d: Resolution of 2-(2,2,2-trifluoroacetyl)bicyclo[3.1.0]hexan-3-one (3I) with quinine
[00589] A flask was charged with 3I (1.0 g), acetone (2.5 ml), and quinine (1.7 g, 0.65 equiv). The mixture was stirred at about 15 to 25 °C for about 18 hours and the solids were isolated by filtration and washed with acetone to provide the quinine salt 3n.
Example 4a: Preparation of ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV) from (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV)
[00590] Acetonitrile (5 vol.) was added to a reactor containing XV (1.0 g). N,N-Diisopropylethylamine (0.80 g, 1.25equiv.) was added at about 0 °C. Ethyl bromoacetate (0.91 g, 1.1 equiv.) was added over about 1 hour at about 0 °C. The reaction was stirred at about 5 °C for about 30 minutes and warmed to about 10 °C. The reaction was stirred until complete as determined by HPLC, warmed to about 20 °C, and extracted with MTBE (2 vol.) and saturated NaCl (6 vol.). The aqueous layer was removed and the organic phase was concentrated and diluted with EtOH (3 vol.). The reaction was crystallized by the addition of H2O (7.8 vol.) at about 20 °C. The mixture was cooled to about 5 °C over about 2 hours and maintained at about 5 °C for about 0.5 hour. The mixture was filtered at about 5 °C and washed with cold water (4 vol). The product was dried at about 40 °C under vacuum to give XIV. 1H NMR (400 MHz, Chloroform-d) δ 4.97 (s, 2H), 4.31 – 4.17 (m, 2H), 2.77 (dddd, J= 6.4, 5.2, 2.9, 2.3Hz, 1H), 2.65 – 2.55 (m, 1H), 1.74 – 1.64 (m, 2H), 1.34 – 1.19 (m, 5H), 0.94 – 0.84 (m, 1H).
Example 4b: Preparation of ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV) from (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
Synthesis of (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-ol (4b-02)
[00591] Potassium hydroxide (KOH) (2.2 g, 3.50 equiv.) and anhydrous methanol (13 mL) were added to a reactor and the reaction mixture was warmed to about 55 °C and agitated until
KOH solids were dissolved completely. The mixture was adjusted to about 0 to 6 °C and compound 4a (1.0 g) was slowly added while maintaining the internal temperature at NMT 6 °C. The reaction mixture was agitated for about 45 min at about 0 to 6 °C. Diacetoxy iodobenzene (PhI(OAc)2, 5.0 g, 1.5 equiv.) was added over about 2 hours while maintaining the internal temperature at NMT 6 °C. The reaction mixture was agitated for NLT 1 hour at about 0 to 6 °C. Water (10 g) and heptane (10 mL) were added to the reaction mixture and the biphasic was agitated for NLT 30 min at about 19 to 25 °C The aqueous layer was separated and washed with heptane (10 mL). The combined organic layer was extracted twice with aqueous solution of methanol (MeOH, 10 mL) and water (5 g). The combined aqueous layer was concentrated under vacuum. The aqueous layer was extracted twice with DCM (15 mL and 5 mL). The combined organic layer was concentrated and dried under vacuum. The unpurified compound 4b-02 was obtained. 1H NMR (600 MHz, CDCl3): d 3.98 (d, 1H), 3.45 (s, 3H), 3.25 (s, 3H),
2.40 (s, 1H), 2.21 (m, 1H), 1.78 (d, 1H), 1.48 (m, 1H), 1.38 (m, 1H), 0.83 (q, 1H), 0.58 (m, 1H).
13C NMR (150 MHz, CDCl3): δ 110.91, 72.19, 51.18, 49.02, 34.08, 21.66, 14.75, 8.37.
Synthesis of (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-one (4c-02)
[00592] Oxalyl chloride (0.96 g, 1.20 equiv.) and dichloromethane (10 mL) were added to a reactor and the mixture was cooled to about -78 °C. Dimethyl sulfoxide (DMSO, 1.2 g, 2.4 equiv.) was added over about 30 min while maintaining the internal temperature below about -60 °C. After agitation for about 5 min, the solution of compound 4b-02 (1.0 g) in dichloromethane (6 mL) was added over about 30 min while maintaining the internal temperature below about -60 °C and the reaction mixture was agitated for about 20 min at about -60 °C. Triethylamine (TEA, 3.1 g, 4.8 equiv.) was added over about 40 min at about -60 °C, and the reaction mixture was warmed to about 10 to 20 °C. Water (15 g) was added and the biphasic was agitated about 30 min at about 10 to 20 °C. After phase separation, the aqueous layer was back-extracted with dichloromethane (10 mL). Combined organic layer was concentrated until no distillate was observed. To the residue was added MTBE (1 mL), filtered and evaporated to afford unpurified compound 4c-02. 1H NMR (600 MHz, CDCl3): d 3.45 (s,
3H), 3.27 (s, 3H), 2.79 (ddd, 1H), 2.30 (d, 1H), 1.73 (td, 1H), 1.63 (m, 1H), 0.96 (m, 1H), 0.25 (td, 1H). 13C NMR (150 MHz, CDCl3): δ 207.75, 102.13, 50.93, 50.50, 38.87, 19.15, 9.30, 8.56.
Synthesis of lithium (Z)-1-((1S,5R)-4,4-dimethoxy-3-oxobicyclo[3.1.0]hexan-2-ylidene)-2,2,2-trifluoroethan-1-olate (3d-02)
[00593] A reactor was charged with compound 4c-02 (1.0 g), ethyl trifluoroacetate (CF3COOEt, 0.91 g, 1.0 equiv.) and tetrahydrofuran (THF, 0.5 mL) and the reaction mixture was cooled to about -10 to 0 °C. The 1M solution of lithium bis(trimethylsilyl)amide (LiHMDS, 7.0 mL, 1.10 equiv.) was added over about 40 min while maintaining the internal temperature below about 0 °C. The reaction mixture was agitated for about 2 hours at about -10 to 0 °C until the reaction was complete. After then, the reaction mixture was wanned to about 20 °C followed by charging tert-butyl methyl ether (MTBE, 10 mL) and water (10 g). After agitating for about 30 min, the organic layer was separated and the aqueous layer was back-extracted twice with mixture of MTBE (6 mL) and THF (4 mL). The combi ned organic layer was concentrated until no distillate was observed. To the unpurified solids, THF (3 mL) and heptane (15 mL) were added at about 20 °C, and the reaction mixture was cooled to about 0 °C and agitated about 1 hour. The resulting slurry was filtered and wet cake was washed with heptane (7 g) and dried under vacuum at about 40 °C to afford compound 3d-02. 1H NMR (600
MHz, DMSO-d6): d 3.31 (s, 3H), 3.27 (s, 3H) 2.01 (m, 1H), 1.42 (td, 1H), 0.96 (m, 1H), 0.08 (q, 1H). (600 MHz, CDCl3 with THF) δ 3.44 (s, 3H), 3.24 (s, 3H), 2.26 (m, 1H), 1.48 (m, 1H), 1.04 (q, 1H), 0.25 (m, 1H). 13C NMR (150 MHz, DMSO-d6): 193.20, 120.78, 118.86, 105.53,
104.04, 50.66, 49.86, 17.34, 16.20, 13.78.
Synthesis of ethyl 2-((3bS.4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00594] Compound 3d-02 (1.0 g), ethyl hydrazinoacetate hydrochloride (EHA-HCl, 0.60 g,
1.0 equiv.) and absolute ethanol (EtOH, 15 mL) were added to a reactor and the reaction mixture was cooled to about 0 – 5 °C. Sulfuric acid (H2SO4, 0.19 g, 0.50 equiv.) was added while maintaining the internal temperature below about 5 °C. Triethyl orthoformate (0.86 g, 1.50 equiv.) was added and the reaction mixture was agitated at about 0 to 5 °C for about 15 hours. The reaction mixture was warmed to about 20 to 25 °C and water (30 g) was added over about 15 minutes. The content was cooled to about 0 to 5 °C and agitated for about 1 hour. The slurry was filtered and wet cake was washed with water (5 g) and dried under vacuum at about 45 °C to afford XIV 1H NMR (600 MHz, CDCl3): d 4.97 (s, 1H), 4.23 (qd, 2H), 2.77 (quint. 1H), 2.60 (quint, 1H), 1.69 (m, 2H), 1.28 (t, 3H). 13C NMR (150 MHz, CDCl3): d 187.14, 165.98, 143.35, 143.12, 121.37, 119.59, 62.34, 51.83, 35.35, 31.72, 14.00, 13.73.
Example 4c: Kinetic resolution of ethyl 2-(5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XVII) to form ethyl 2- ((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00595] Compound XVII (1.0 g), (R)-2-methyl-CBS-oxazaborolidine (0.0.05 g, 0.05 equiv.), and tetrahydrofuran (11.9 g) were combined and cooled to about 0 to 5 °C. A solution of borane dimethyl sulfide complex (0.14 g, 0.55 equiv.) in tetrahydrofuran (0.67 g) was added to the mixture, and the mixture was agitated at about 0 to 5 °C until the reaction was deemed complete. Methanol (1 mL) was added to the mixture at about 0 to 5 °C over about 1 h, and the mixture was adjusted to about 15 to 25 °C. The mixture was concentrated under vacuum and combined with tetrahydrofuran (2.7 g). The mixture was combined with 4-dimethylaminopyridine (0.18, 0.44 equiv.) and succinic anhydride (0.30 g, 0.87 equiv.) and agitated at about 15 to 25 °C until the reaction was deemed complete. The mixture was combined with tert-butyl methyl ether (5.2 g) and washed with 1 M aqueous HCl (6.7 g), twice with 5 wt % aqueous potassium carbonate (6.7 g each), and 5 wt % aq. sodium chloride (6.7 g). The organics were concentrated under reduced pressure to an oil which was dissolved in dichloromethane (0.1 g) and purified by flash column chromatography (2.0 g silica gel, 20:80 to 80:20 gradient of ethyl acetate:hexanes). The combined fractions were concentrated under vacuum to give XIV.
Example 4d: Preparation of (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
[00596] 4-Tosyloxycyclohexanone (50 mg), (8α,9S)-6′-methoxycinchonan-9-amine trihydrochloride (16 mg), trifluoroacetic acid (28 μL), lithium acetate (49 mg), water (3.4 μL), and 2-methyltetrahydrofuran (0.75 mL) were combined in a vial. The mixture was agitated at about 20 °C until the reaction was complete. 4a was isolated by vacuum distillation. 1H NMR (400 MHz, CDCl3) δ2.05 (m, 5H), 1.74 (m, 1H), 1.18 (m, 1H), 0.91 (m, 1H).
Example 5: Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a- dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane]-1(3bH)- yl)acetate (5h) from (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-ol (4b-02)
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan1-3-ol (5d)
[00597] A mixture of ketal alcohol 4b-02 (1.0 g), ethanedi thiol (0.91 g), MeCN (7.5 ml) and BiCl3 (0.30 g) was agitated at r.t. overnight. The solids were removed by filtration and the filtrate was concentrated and the residue was further purified by flash column on silica gel to obtain the two isomers. Major product: 1H NMR (400 MHz, Chloroform-d) δ 3.82 (ddt, J = 6.1, 1.3, 0.6 Hz, 1H), 3.41 – 3.32 (m, 2H), 3.31 -3.23 (m, 1H), 3.14 – 3.06 (m, 1H), 2.71 (s, 1H),
2.33 (dddd, J = 14.0, 6.2, 4.8, 1.4 Hz, 1H), 2.00 (d, J = 13.9 Hz, 1H), 1.79 – 1.72 (m, 1H), 1.54 -1.46 (m, 1H), 1.04 (dt, J = 5.1, 3.9 Hz, 1H), 0.63 – 0.54 (m, 1H). Minor product: 1H NMR (400 MHz, Chloroform-d) δ 3.83 (q, J = 9.1 Hz, 1H), 3.43 – 3.34 (m, 2H), 3.33 – 3.25 (m, 2H), 2.35 (d, J= 11.2 Hz, 1H), 2.18 (ddd, J = 12.7, 6.7, 0.4 Hz, 1H), 1.84 (ddd, J= 8.1, 6.3, 3.7 Hz, 1H),
1.60 – 1.51 (m, 1H), 1.43 – 1.35 (m, 1H), 0.65 (tdt, J= 8.1, 5.9, 0.8 Hz, 1H), 0.57 (dddd, J= 5.9, 4.2, 3.7, 0.6 Hz, 1H).
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan1-3-one (5e)
[00598] To a dried flask was sequentially added dithiolane alcohol 5d (1.0 g), CH2Cl2 (25 ml), anhydrous DMSO (8.5 ml), and tri ethylamine (3.5 ml) and the resulting mixture was aged at room temperature for about 21 hours. The reaction mixture was transferred to a separatory funnel, diluted with CH2Cl2 (30 ml), washed with 1 M HCl (25 ml), and water (25 ml). The CH2Cl2 layer was concentrated to a solid and further purify by flash column chromatography on silica gel eluted with gradient EtOAc/n-heptane (0-20%) to obtain 5e. 1H NMR (400 MHz, Chloroform-d) δ 3.57 (dddd, J = 10.5, 5.6, 4.3, 0.5 Hz, 1H), 3.49 – 3.41 (m, 1H), 3.39 – 3.28 (m, 2H), 3.10 (ddd, J = 18.3, 5.6, 2.2 Hz, 1H), 2.29 (d, J = 18.3 Hz, 1H), 1.89 (ddd, J = 8.0, 7.0, 3.9
Hz, 1H), 1.63 (tdd, J= 7.3, 5.6, 4.1 Hz, 1H), 1.05 (tdd, J = 8.0, 6.3, 2.2 Hz, 1H), 0.21 (dt J = 6.4, 4.0 Hz, 1H).
Synthesis of lithium (Z)-2,2,2-trifluoro-1-((1R,5S)-3-oxospiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-4-ylidene)ethan-1-olate (5f)
[00599] To a flask with dithiolane ketone 5e (1.0 g) under N2 was added anhydrous THF (8.8 ml), and the mixture was cooled to about -78 °C and followed by addition of LiHMDS (1 M in THF, 7.4 ml) over about 5 min. The resulting mixture was agitated at about -78 °C for about 0.5 hours, and ethyl trifluoroacetate (0.88 ml) was added. The resulting mixture was agitated at about -78 °C for about 10 minutes, at about 0 °C for about 1 hour, and at room temperature overnight. THF was removed under reduced pressure and the residue was crystallized in n-heptane (about 18 ml). The solid product was isolated by filtration, and the filter cake was rinsed with n-heptane (4.1 ml), and dried at about 50 °C under vacuum to provide 5f. 1H NMR (400 MHz, Acetonitrile-d3) δ 6.98 (s, 0H), 5.20 (s, 0H), 3.60 – 3.50 (m, 2H), 3.46 – 3.36 (m, 2H), 2.28 – 2.20 (m, 1H), 1.80 (ddd, J = 8.3, 7.2, 4.1 Hz, 1H), 1.39 (s, 1H), 1.03 (ddd, J = 8.3, 6.7, 4.8 Hz, 1H), 0.17 (ddd, J = 4.7, 4.2, 3.6 Hz, 1H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydrospiro[cvciopropa[3.4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane] (5g)
[00600] To flask containing the dithiolane lithium salt 5f (1.0 g) was added water (10 ml), hydrazine hydrate (0.88 ml) and acetic acid (10 ml). The reaction mixture was heated at about 35 °C for about 2 hours, and at about 55 °C for about 2 hours. Water was removed under reduced pressure and the residue was diluted with acetic acid (20 ml) and heated at about 55 °C for about 0.5 hour and held at room temperature overnight. The reaction mixture was further heated at about 65 °C for about 20 hours, and cooled down and concentrated to remove volatile components by rotavap. The residue was triturated with water (50 ml) at about 0 °C and the solid residue was isolated and further washed with ice-cold water (2×10 ml). The solids were further dried to afford unpurified 5g. 1H NMR (400 MHz, Chloroform-d) δ 3.65 – 3.46 (m, 4H), 2.60 (dddd, J = 8.3, 5.6, 4.2, 0.7 Hz, 1H), 2.47 – 2.38 (m, 1H), 1.33 (dddd, J= 8.2, 7.4, 5.7, 0.7 Hz, 1H), 0.66 (dddd, J = 5.7, 4.3, 3.6, 0.7 Hz, 1H)
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5.2′-[1,3]dithiolane]-1(3bH)-yl)acetate
(5h) from (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane] (5g)
[00601] A reactor was charged with dithiolane pyrazole 5g (1.0 g) and THF (15 ml). The contents were adjusted to about 0 to -5 °C and followed by addition of ethyl bromoacetate (0.44 ml, 1.1 equiv.). To the resulting mixture NaHMDS (2 M, 2.0 ml, 1.1 equiv.) was added over about 10 min via syringe pump at about -2.5 to 0 °C and the mixture was held for about 3 hours, a second portion of ethyl bromoacetate (0.050 ml, 0.12 equiv.) was added, and the mixture was aged for about 1 hour. The reaction mixture was quenched by excess water (2 ml) to form 5h.
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolanel-1(3bH)-yl)acetate
(5h) from lithium (Z)-2,2,2-trifluoro-1-((1R,5S)-3-oxospiro[bicyclo[3.1.0]hexane-2.2′- [1,3]dithiolanl-4-ylidene)ethan-1-olate (5f)
[00602] A 100 ml flask was charged with ethanol (5 ml). The contents were cooled to about 0 °C and acetyl chloride (1.1 g, 4.0 equiv.) was added over about 10 min. The mixture was agitated at about 0 °C for about 20 minutes and at room temperature for about 20 minutes. To the freshly prepared HCl ethanol solution was added EHA.HCl (0.68 g, 1.2 equiv.) and dithiolane lithium salt 5f (1.0 g). The reaction mixture was heated at about 40 °C for about 22 hours. Ethanol was removed under reduced pressure, and the residue was partitioned between ethyl acetate (5 ml) and water (5 ml). The aqueous layer was discarded, and the organic layer was sequentially washed with aqueous NaHCO3 (5%, 5 ml) and brine (5%, 5 ml) and 5h was
obtained in the EtOAc layer. 1H NMR (400 MHz, DMSO-d6) d 5.14 – 4.97 (m, 2H), 4.14 (qd, J = 7.1, 1.0 Hz, 2H), 3.67 – 3.35 (m, 4H), 2.69 (ddd, J= 8.2, 5.6, 4.2 Hz, 1H), 2.44 (ddd, J= 7.2,
5.5, 3.5 Hz, 1H), 1.37 – 1.29 (m, 1H), 1.21 – 1.14 (m, 3H), 0.44 (ddd, J = 5.3, 4.2, 3.6 Hz, 1H).
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolanel-1 (3bH)-yl)acetate (5h) from (1R,5R)-spiro[bicyclo[3.1.0]hexane-2.2′-[1,3]dithiolanl-3-one (5e)
[00603] 5e (756 mg) was charged to a vessel and dissolved in 2-methyltetrahydrofuran (7.6 mL). To this solution was charged ethyl trifluoroacetate (0.57 g) and the resulting solution was cooled to about 0 °C. Lithium hexamethyldisilazide (1.0 M solution in THF, 4.5 g) was charged over about 60 minutes and reaction was agitated until complete. A solution of sulfuric acid (2.0 g) in water (5.6 mL) was charged, then the reaction was warmed to about 20 °C and agitated for about 20 minutes. Layers were separated and aqueous layer was extracted twice with 2-methyltetrahydrofuran (5.3 mL). Combined organic layer was concentrated to about 0.4 mL and N,N-diisopropylamine (0.5 g) was charged. The product was crystallized by the addition of heptane (11 ml). The slurry was filtered and the filter cake was washed with heptane, then deliquored thoroughly, and dried to afford 5f-01. 1H NMR (400 MHz, Acetonitrile-d3) δ 7.84 (m, 2H), 3.58 (d, J = 8.7 Hz, 2H), 3.47 – 3.27 (m, 4H), 2.20 (s, 1H), 1.81 – 1.68 (m, 1H), 1.24 (dd, J = 6.5, 0.6 Hz, 12H), 0.99 (q, J = 6.5 Hz, 1H), 0.13 (s, 1H).
[00604] Acetyl chloride (1.02 g) was charged to a cooled reaction vessel containing ethanol (5.0 mL) at about 0 °C, then warmed to about 20 °C and agitated for about 30 minutes. In a separate vessel, 5f-01 (1.00 g), ethyl hydrazinoacetate hydrochloride (0.48 g), and lithium chloride (0.39 g) were combined, and the acetyl chloride/ethanol solution was charged to this mixture, followed by tri ethyl orthoformate (1.16 g). The mixture was heated to about 45 °C and agitated until reaction was complete. The reaction was then concentrated to 2 volumes and dichlorom ethane (5.0 mL) was added followed by water (5.0 mL). Layers were separated and organic layer was washed with 5 wt % aqueous sodium bicarbonate followed by 10 wt % aqueous sodium chloride to afford a solution of 5h in dichloromethane that was carried forward into the subsequent step. 1H NMR (400 MHz, DMSO-d6) δ 5.27 – 4.79 (m, 2H), 4.14 (qd, J =
7.1, 1.1 Hz, 2H), 3.70 – 3.42 (m, 4H), 2.68 (dtd, J = 8.0, 6.4, 5.9, 4.4 Hz, 1H), 2.44 (ddd, J = 7.2, 5.5, 3.6 Hz, 1H), 1.32 (ddd, J = 8.2, 7.2, 5.4 Hz, 1H), 1.18 (t, J = 7.1 Hz, 3H), 0.44 (dt, J = 5.4, 3.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 167.14, 148.36, 133.80 (q, J = 38.3 Hz), 128.77 (m), 121.54 (q, J = 268.4 Hz), 65.33, 61.79, 51.14, 41.30, 40.98, 40.49, 23.57, 15.52, 14.33; 19F NMR (376 MHz, DMSO-d6) δ -60.31.
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-3-one (5e) from (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-3-one (5e) from (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
[00605] Tert-butyl nitrite (1.31 g) was charged to a vessel containing 4a (1.00 g, 1.0 equiv) and tetrahydrofuran (5.0 mL) at about 20 °C. Potassium tert-butoxide (6.1 g, 1.7M in tetrahydrofuran) was charged over not less than 30 minutes. The mixture was then agitated until the reaction was complete. The reaction was quenched with aqueous citric acid (2.00 g in 10.00 g water) and extracted with dichloromethane (10.0 mL, 3x). This solution was partially concentrated and the product was isolated by the addition of heptane (6.0 mL). The slurry was filtered and the filter cake was washed with heptane (2.0 mL), then deliquored thoroughly to afford 4d 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 2.73 (d, J = 18.5 Hz, 1H), 2.63 (ddd, J = 18.6, 5.3, 2.0 Hz, 1H), 2.17 – 2.01 (m, 2H), 1.34 (dddd, J= 9.2, 7.1, 4.9, 2.0 Hz, 1H), 0.77 (td, J= 4.6, 3.4 Hz, 1H).
[00606] 1,2-Ethanedithiol (0.41 g) was charged to a vessel containing a solution of 4d (0.50 g, 4.0 mmol) in glacial acetic acid (2.5 mL) at about 20 °C. para-toluenesulfonic acid monohydrate (0.15 g) was added and the mixture was agitated until the reaction was complete. The product was isolated by the addition of water (2 mL). The slurry was filtered and the filter cake was washed with water, then deliquored thoroughly to afford 5i. 1H NMR (400 MHz,
DMSO-d6) δ 10.93 (s, 1H), 3.63 – 3.51 (m, 2H), 3.51 – 3.42 (m, 1H), 3.39 – 3.31 (m, 1H), 2.83 (d, J= 17.4 Hz, 1 H), 2.59 – 2.52 (m, 1H), 1.87 (ddd, J = 8.0, 6.2, 3.7 Hz, 1H), 1.65 (dddd, J=
7.7, 6.2, 5.2, 3.9 Hz, 1H), 0.93 (tdd, J = 7.6, 5.5, 1.7 Hz, 1H), 0.02 (dt, J= 5.5, 3.8 Hz, 1H).
[00607] Para-toluenesulfonic acid (0.90 g) was charged to a vessel containing a suspension of 5i (0.50 g, 2.5 mmol) in methyl ethyl ketone (2.5 mL) and water (2.5 mL). The mixture was agitated at about 85 °C until the reaction was complete. The product was isolated from the reaction mixture by cooling to about 20 °C, adding water (2.50 mL), and cooling to about 0 °C. The slurry was filtered and the filter cake was washed with water, then deliquored thoroughly to afford 5e. 1H NMR (400 MHz, DMSO-d6) δ 3.55 – 3.37 (m, 3H), 3.28 – 3.13 (m, 1H), 3.03 (ddd, J = 18.5, 5.6, 2.2 Hz, 1H), 2.20 (d, J = 18.5 Hz, 1H), 1.84 (ddd, J = 8.0, 7.0, 3.8 Hz, 1H), 1.66 (tdd, J = 7.2, 5.6, 4.1 Hz, 1H), 1.03 (tdd, J = 7.9, 5.9, 2.1 Hz, 1H), 0.06 (dt, J = 6.0, 4.0 Hz, 1H).
Example 6: Preparation of 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetic acid (VII) from ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane]-1(3bH)-yl)acetate (5h) from ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00608] Dichloromethane (27 g) was added to a reactor containing XIV (1.0 g) and cooled to about 10 °C. To this was added 1,2-ethanedithiol (0.18 g, 1.2 equiv.). To this was added boron trifluoride acetic acid complex (3.3 g, 2.5 equivalents) over about 25 minutes, and the reaction mixture was agitated at about 20 °C until complete. A solution of calcium chloride dihydrate (0.80g, 0.78 equiv) in 0.10 N hydrochloric acid (16 g) was added over about 1 hour at about 10 °C, and the mixture was agitated for about 90 minutes at about 20 °C. The organic layer was washed successively with water (8 g) and sodium bicarbonate solution (5 wt/wt%). The organic layer was concentrated to afford 5h. 1H NMR (400 MHz, DMSO-d6) δ 5.27 – 4.79 (m, 2H),
4.14 (qd, J = 7.1, 1.1 Hz, 2H), 3.70 – 3.42 (m, 4H), 2.68 (dtd, J = 8.0, 6.4, 5.9, 4.4 Hz, 1H), 2.44 (ddd, J = 7.2, 5.5, 3.6 Hz, 1H), 1.32 (ddd, J = 8.2, 7.2, 5.4 Hz, 1H), 1.18 (t, J= 7.1 Hz, 3H), 0.44 (dt, J = 5.4, 3.9 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 167. 14, 148.36, 133.80 (q, J= 38.3 Hz), 128.77 (m), 121.54 (q, J= 268.4 Hz), 65.33, 61.79, 51.14, 41.30, 40.98, 40.49, 23.57,
15.52, 14.33. 19F NMR (376 MHz, DMSO-d6) δ -60.31.
Synthesis of ethyl 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (VII-A)
[00609] Dichloromethane (26 g) was added to a reactor containing 1,3-dibromo-5,5-dimethylhydantoin (DBDMH, 2.4 g, 3.1 equiv.) and cooled to about -10 °C. To this was added 70% hydrofluoric acid/pyridine complex (1.3 g, 17 equiv.), followed by a solution of 5h (1.0 g) in dichloromethane (3 g). The reaction was agitated at about 0 °C until complete. A solution of potassium hydroxide (3.7 g, 25 equivalents) and potassium sulfite (1 .9 g, 4 equiv.) in water (24 g) was added, maintaining an internal temperature of about 5 °C, and agitated for about 30 minutes at about 20 °C. Layers were separated and organic layer was washed with hydrochloric acid (1.1 g, 4 equiv.) in water (9.6 g). The organic layer was concentrated to afford VII-A. 1H NMR (400 MHz, DMSC-d6) δ 5.31 – 5.04 (m, 2H), 4.17 (q, J = 7.1 Hz, 2H), 2.78 – 2.57 (m,
2H), 1.47 (dddd, J = 8.5, 7.1, 5.5, 1.4 Hz, 1H), 1.19 (t, J = 7.1 Hz, 3H), 1.04 (tdt, J= 5.3, 4.0,
1.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 166.79, 143.15 (t, J= 29.4 Hz), 134.65 (q, J=
39.0 Hz), 132.99, 121.05 (q, J= 268.4 Hz), 120.52 (t, J= 243.3 Hz), 62.09, 52.49, 27.95 (dd, J = 34.7, 29.0 Hz), 23.82 (d, J = 2.6 Hz), 14.25, 12.14 (t, J = 3.1 Hz). 19F NMR (376 MHz, DMSO-d6) δ -60.47, -79.68 (dd, J= 253.5, 13.2 Hz), -103.09 (dd, J = 253.3, 9.8 Hz).
Synthesis of 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetic acid (VII)
[00610] A reactor was charged with a solution of VII-A (1.0 g) in dichloromethane (18 g) and cooled to about 5 °C. To this was added ethanol (1.5 g), followed by potassium hydroxide (45 wt/wt%, 0.74 g, 2.0 equiv.). The reaction mixture was agitated at about 20 °C until complete. Water (3.7 g) was added and the reaction mixture was agitated for about 30 minutes. Organic layer was removed and reaction was cooled to about 10 °C. Dichloromethane (18 g) was added, followed by 2N hydrochloric acid (3.3 g, 2,2 equiv.). Reaction was warmed to about 20 °C and agitated for 10 minutes. Layers were separated and aqueous phase was washed with dichloromethane (18 g). Organic layers were combined and concentrated on rotary evaporator to afford VII. 1H NMR (400 MHz, DMSO-d6) δ 13.50 (s, 1H), 5.14 – 4.81 (m, 2H), 2.82 – 2.56 (m, 2H), 1.46 (dddd, J = 8.5, 7.1, 5.5, 1.4 Hz, 1H), 1.08 – 1.00 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.16, 143.05 (t, J = 29.4 Hz), 134.40 (q, J = 38.9 Hz), 132.80, 121.11 (q, J = 268.4 Hz), 120.55 (t, J = 243.3 Hz), 52.54, 27.97 (dd, J = 34.7, 29.0 Hz), 23.81 (d, J = 2.5 Hz), 12.13 (t, J = 3.1 Hz). 19F NMR (376 MHz, DMSO-d6) δ -60.39 (d, J = 1.4 Hz), -79.83 (dd, J = 253.2, 13.1 Hz), -102.97 (dd, J= 253.2, 9.8 Hz).
Example 7: Preparation of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1- (2,2,2-trifluoroethyl)-1H-indazol-3-amine (V-02) and its mesylated derivatives
Synthesis of 4-chloro-7-bromo-1-(2,2,2-trifluoroethyl)-1H-indazol-3-amine (V-A)
[00611] To a reactor was added tetrahydrofuran (THF, 275 kg) and diisopropyl amine (DIPA, 30 kg) and the mixture was cooled to about -35 °C. nButyl lithium (2.5 mol/L in hexanes, 74 kg) was charged slowly keeping the reaction temperature less than -30 °C. The mixture was agitated at-35 °C until the reaction was complete. 1-bromo-4-chloro-2-fluorobenzene (52 kg) was charged keeping reaction temperature less than 30 °C and the mixture was agitated at -35°C until reaction was complete. N,N-dimethylformamide (DMF, 36 kg) was charged keeping reaction temperature less than -30 °C and the mixture was agitated at about -35 °C until reaction was complete. Hydrochloric acid (HCl, 18 mass% in water, 147 kg) was charged keeping reaction temperature less than -5 °C. The reaction was warmed to about 0 °C, water (312 kg) was added, and the reaction was extracted with methyl tert-butyl ether (MTBE, 770 kg). The organic was warmed to about 20 °C and washed with brine (NaCl, 23.5 mass% in water, 1404 kg). The mixture was distilled to about 3-4 volumes and heptane was charged (354 kg). The product was isolated by distillation to 3-4 volumes. The slurry was filtered and washed with heptane (141 kg) and dried to afford 6a. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (d, J = 1.2 Hz, 1H), 8.00 (dd, J = 8.7, 1.4 Hz, 1H), 7.44 (dd, J = 8.7, 1.4 Hz, 1H).
[00612] 6a (98.5 kg) was charged to a reactor containing acetic anhydride (105 kg) and acetic acid (621 kg) at 20 °C. The mixture was heated to about 45 °C and hydroxyl amine hydrochloride (31.5 kg) was charged. The reaction was heated to about 75 °C and agitated until the reaction was complete. The product was isolated from the reaction mixture by adding water (788 kg) at about 45 °C. The mixture was cooled to about 25 °C and then the slurry was filtered. The filtered cake was washed with water (197 kg,). The cake was dried to afford 6b. 1H NMR (400 MHz, DMSO-d6) δ 8.11 (dd, J= 8.8, 1.4 Hz, 1H), 7.58 (dd, J = 8.8, 1.4 Hz, 1H).
[00613] To a reactor was charged 6b (84 kg), isopropanol (318 kg), and water (285 kg).
Hydrazine hydrate (20 wt% in water, 178 kg) was charged and the mixture was heated to about 80 °C until the reaction was complete. The product was isolated by cooling the reaction to about 25 °C. The slurry was filtered and the filtered cake was washed with a mixture of isopropanol (127 kg) and water (168 kg). The wet solids were recharged to the reactor and water (838 g) was added. The mixture was agitated at about 25 °C and then filtered and washed with water
(168 g, 2 rel). The cake was dried to afford 6c 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.41 (d, J= 7.9 Hz, 1H), 6.84 (d, J= 7.9 Hz, 1H), 5.31 (s, 2H).
[00614] 6c (75 kg) was charged to a reactor containing N,N-dimethylformamide (75 kg). Potassium phosphate (99.8 kg) was charged to the reactor at about 25 °C and the mixture was agitated. 2,2,2-trifluoroethyl trifluoromethanesulfonate (74.3 kg) was charged at about 25 °C and the mixture was agitated until the reaction was complete. Water (375 kg) was charged and the mixture was agitated at about 20 °C. The slurry was filtered and washed with water (150 kg). N,N-dimethylformamide (424 kg) and the wet solid were charged to a reactor at about 20 °C.
The mixture was agitated at about 45 °C. 5 % hydrochloric acid (450 kg) was charged drop-wise to the mixture at about 45 °C. The mixture was cooled to about 25 °C. The slurry was filtered and washed with water (375 g). Water (375 kg) and the filtered solid were charged to a reactor at about 20 °C. The mixture was agitated for about 1 hour at about 20 °C. The slurry was filtered and washed with water (375 kg). The cake was dried to afford V-A. 1H NMR (400 MHz, DMSO-d6) δ 7.57 (d, J= 8.1 Hz, 1H), 6.98 (d, J = 8.1 Hz, 1H), 5.70 (s, 2H), 5.32 (q, J = 8.6 Hz,
2H).
Synthesis of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2-trifluoroethyl)- 1 H-indazol-3-amine (V-02)
[00615] A reactor containing tetrahydrofuran (27 g) and V-A (1.0 g) was cooled to about 0 °C. Chlorotrimethylsilane (7.6 g, 2.3 equiv) was added, followed by the slow addition of lithium bis(trimethylsilyl)amide (5.7 g, 1 M in THF, 2.1 equiv.). The mixture was stirred at about 0 °C until bistrimethylsilane protection was complete. The solution was washed with ammonium chloride in water (10 wt%, 52 g), toluene (44 g) was added, and the biphasic mixture was filtered through celite. The organic and aqueous phases were separated and the aqueous phase was washed with toluene (44 g). The organics were combined, washed with brine (58 g), and azeotropically distilled . The solution was cooled to about 0 °C, isopropylmagnesium chloride lithium chloride complex (2.7 g, 1.3 M in THF, 1.2 equiv.) was added and the reaction was stirred at about 0 °C until lithium halogen exchange was complete. Isopropoxyboronic acid pinacol ester (6.8 g, 1.2 equiv.) was added and the reaction was stirred at about 0°C until botylation was complete. At about 0 °C, The reaction was quenched with aqueous hydrochloric acid (52 g, 1 M), acetonitrile (16 g) was added, and the mixture was stirred until trimethylsilane deprotection was complete. The solution was extracted with ethyl acetate (45 g) and the organic was washed twice with brine (2 x 58 g). The solution was concentrated to low volumes (26 g), dim ethylformami de (47 g) was added, and the solution was concentrated again (51 g). The product was crystallized by the addition of water (50 g). The slurry was filtered and filter cake was washed with heptane (14 g). The solids were dried to afford V-02. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J = 7.6, 1.0 Hz, 1H), 7.07 (dd, J = 7.6, 1.0 Hz, 1H), 5.58 (s, 2H), 5.46 (q, J = 9.1Hz, 2H), 1.32 (s, 12H).
Synthesis of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2-trifiuoroethyl)- 1 H-indazol-3-amine (V-02)
[00616] To a reactor was charged V-A (30 kg), bis(pinacolato)diboron (27.9 kg), bis(triphenylphosphine)palladium (II) dichloride (0.9 kg, 1.5 mol%), N,N-dimethylformamide (56 kg, 2 rel. vol.) and toluene (157 kg, 6 rel vol.). The mixture was heated to about 105 °C until the reaction was complete. The mixture was cooled to about 25 °C, filtered through celite (15 kg, 0.5 rel. wt.) and rinsed forward with ethyl acetate (270 kg, 10 rel vol.). PSA-17 palladium scavenger (3 kg, 10 wt%) was added and the mixture was stirred at about 45 °C. The mixture was filtered and the cake was washed with ethyl acetate (54 kg, 2 rel. vol.). The mixture was washed twice with lithium chloride (180 kg, 6 rel. vol.) and once with brine (NaCl, 23.5 mass% in water, 180 kg, 6 rel. vol.). The mixture was then concentrated to about 5-6 rel. vol. under vacuum, heated to about 45 °C then cooled to about 25 °C. Heptane (102 kg, 5 rel. vol.) was charged and the mixture was concentrated to about 4-5 rel. vol. The product was isolated by charging heptane (41 kg, 2 rel. vol.) and cooling the mixture to about 0 °C. The slurry was filtered and washed with heptane (41 kg, 2 rel. vol.). The wet solids were recharged to the reactor with ethyl acetate (27 kg, 1 rel. vol.) and heptane (82 kg, 4 rel. vol.), heated to about 65 °C, and then cooled to about 5 °C. The slurry was filtered and washed with heptane (41 kg, 2 rel. vol.). The cake was dried to afford V-02. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J =
7.6, 1.0 Hz, 1H), 7.07 (dd, J = 7.6, 1.0 Hz, 1H), 5.58 (s, 2H), 5.46 (q, J = 9.1Hz, 2H), 1.32 (s, 12h).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)-N-(methylsulfonyl)methanesulfonamide (V-04)
[00617] To a 100 mL reactor was added V-02 (5.00 g), 2-methyltetrahydrofuran (50 mL), and triethylamine (11.1 mL). The mixture was cooled to about 10 °C and methanesulfonyl chloride (2.58 mL, 33.3 mmol) was added to the mixture. The mixture was agitated at about 10 °C until reaction was complete. The mixture was concentrated to dryness and the residue was purified by column chromatography to afford V-04. 1H NMR (400 MHz, DMSO-d6) δ 7.96 (d, J = 7.7 Hz, 1H), 7.50 (d, J = 7.6 Hz, 1H), 5.95 (q, J = 8.8 Hz, 2H), 3.66 (s, 6H), 1.37 (s, 12H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2)-1-(2,2,2,- trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-03)
[00618] To a 100 mL reactor was added V-02 (5.00 g), 2-methyltetrahydrofuran (50 mL), and triethylamine (11.1 mL, 79.6 mmol). The mixture was cooled to about 10 °C and methanesulfonyl chloride (2.58 mL) was added to the mixture. The mixture was agitated at about 10 °C until reaction was complete. To the mixture was added 2-methyltetrahydrofuran (21.5 g) and sodium hydroxide (0.43 g) and the mixture was agitated at about 25 °C until the reaction was complete. To the resulting solution was added 2-methyltetrahydrofuran (21.5 g), water (25 g) and acetic acid to achieve a pH of less than 7. The lower aqueous layer was then removed and the organic layer was washed with brine (5 wt%, 7.8g). The organic layer was then concentrated to dryness and the residue was purified by column chromatography to afford V-03. 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.34 (d, J = 7.6 Hz, 1H), 5.80 (q, J = 8.9 Hz, 2H), 3.22 (s, 3H), 1.36 (s, 12H).
Synthesis of N-(7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)-N- (methylsulfonyl)methanesulfonamide (V-06)
[00619] To a reactor was added V-A (3 g), 2-methyltetrahydrofuran (25.8 g), and triethylamine (7.6 mL). The mixture was cooled to about 10 °C, methanesulfonyl chloride (1.8 mL) was added, and the mixture was stirred until reaction was complete. The reaction mixture was washed with aqueous sodium chloride (30 mL) and the organic layer was evaporated to dryness. The residue was purified by column chromatography to afford V-06. 1H NMR (400 MHz, DMSO-d6) δ 7.83 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 5.79 (q, J = 8.5 Hz, 2H), 3.62 (s, 6H).
Synthesis of N-(7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-05)
[00620] To a reactor was added V-02 (3 g), 2-methyltetrahydrofuran (30 mL), and triethylamine (7.6 mL). The mixture was cooled to about 10 °C, methanesulfonyl chloride (1.8 mL) was added, and the mixture was stirred until reaction was complete. The reaction mixture was washed with aqueous sodium chloride (30 mL) and the organic portion was concentrated to dryness.
[00621] To the resulting mixture (2.7g) was added 2-methyltetrahydrofuran (15 mL) and sodium hydroxide (1M in water, 15 mL). The mixture was stirred at about 20 °C until the reaction was complete. The aqueous layer was removed and the organic was washed with acetic acid (0.7M in water, 10 mL) and sodium chloride (5 wt% in water, 10 mL).The organic layer was then concentrated to dryness and the residue was purified by column chromatography to afford V-05. 1H NMR (400 MHz, DMSO-D6) δ 10.03 (s, 1H), 7.71 (dd, J = 8.0, 1.6 Hz, 1H), 7.20 (dd, J = 8.1, 1.6 Hz, 1H), 5.64 (q, J = 8.7 Hz, 3H), 3.19 (2, 3H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2,-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)-N-(methylsulfonyl)methanesulfonamide (V-04)
[00622] To a reactor was charged V-06 (148 mg), bis(pinacolato)diboron (93 mg), potassium acetate (90 mg) and bis(triphenylphosphine)palladium (II) chloride (4.3 mg, 1.5 mol%). N,N- dimethylformamide (0.2 mL) and toluene (0.6 mL) were added and the reaction was heated to about 105 °C until completion. V-04 was formed. 1H NMR (400 MHz, DMSO-D6) δ 7.96 (d, J = 7.7 Hz, 1H), 7.50 (d, J= 7.6 Hz, 1H), 5.95 (q, J= 8.8 Hz, 2H), 3.66 (s, 6H), 1.37 (s, 12H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-03)
[00623] To a reactor was charged V-05 (124 mg), bis(pinacolato)diboron (93 mg), potassium acetate (90 mg) and bis(triphenylphosphine)palladium (II) chloride (4.3 mg, 1.5 mol%). N,N- dimethylform amide (0.2 mL.) and toluene (0.6 mL, 6 rel. vol.) were added and the reaction was heated to about 105 °C until completion. V-03 was formed. 1H NMR (400 MHz, DMSO-d6) δ
9.96 (s, 1 H), 7.86 (d, J= 7.6 Hz, 1H), 7.34 (d, J= 7.6 Hz, 1H), 5.80 (q, J = 8.9 Hz, 2H), 3.22 (s,
II. Synthesis of the Compound of Formula I
Example 8: Preparation of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1- yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
Synthesis of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2- (3,5-difluorophenyl)ethyl)-2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV) from (S)-1-(3-bromo-6-(3- methyl-3-(methylsulfbnyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3.5-difluorophenyl)ethan-1-amine (VI) Method 1
[00624] n-Propyl phosphonic anhydride (T3P, 3.1 g, 1.5 equiv.) was slowly added to a reactor containing amine VI (1.5 g), acid VII (1.0 g, 1.1 equiv.), triethylamine (Et3N, 0.5 g, 1.5 equiv.), and acetonitrile (MeCN, 8.0 g). The mixture was agitated at about 20 °C until the reaction was complete. The product was crystallized from the reaction mixture with DMF (0.63 g), and water (15 g). The slurry was filtered and the filter cake was washed with a mixture of acetonitrile and water (2 x 2.5 g). The cake was dried to afford IV. 1H NMR (400 MHz, DMSO-d6) δ9.19 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.07 (tt, J = 9.4, 2.4 Hz, 1H),
6.96 – 6.87 (m, 2H), 5.52 (td), J = 8.8, 5.3 Hz, 1 H), 4.93 – 4.73 (m, 2H), 3.22 (s, 3H), 3.11 -2.90 (m, 2H), 2.66 – 2.52 (m, 2H), 1.69 (s, 6H), 1.45 – 1.36 (m, 1H), 1.02 – 0.93 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.42, 163.62, 163.49, 161.17, 161.04, 158.19, 142.92, 142.20, 142.10, 142.01, 141.63, 140.23, 134.11, 133.73, 132.14, 128.66, 122.23, 120.49, 119.56, 112.49, 112.25, 104.75, 102.25, 88.62, 84.20, 57.44, 53.85, 53.03, 35.21, 23.41, 22.46, 22.40, 11.79.
Synthesis of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV) from (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI) Method 2
[00625] N-methylmorpholine (NMM, 0.51 g, 2.3 equiv.) was added to a vessel containing amine VI (1.0 g), acid VII (1.0 g), 1-hydroxybenzotriazole hydrate (HOBt ● H2O, 0.17 g, 0.5 equiv.), N-(3-dimethylaminopropyi)-N’-ethylcarbodiimide (EDCI ● HCl, 0.52 g, 1.25 equiv.), and acetonitrile (MeCN, 7.8 g). The mixture was agitated at about 20 °C until the reaction was complete. The product was crystallized from the reaction mixture with DMF (2.8 g), and water (10 g). The slurry was filtered and the filter cake was washed with a mixture of acetonitrile and water. The cake was dried to afford IV. 1H NMR (400 MHz, DMSO-d6) δ9.19 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.07 (tt, J = 9.4, 2.4 Hz, 1H), 6.96 – 6.87 (m, 2H), 5.52 (td), J = 8.8, 5.3 Hz, 1 H), 4.93 – 4.73 (m, 2H), 3.22 (s, 3H), 3.11 – 2.90 (m, 2H), 2.66 – 2.52 (m, 2H), 1.69 (s, 6H), 1.45 – 1.36 (m, 1H), 1.02 – 0.93 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.42, 163.62, 163.49, 161.17, 161.04, 158.19, 142.92, 142.20, 142.10, 142.01, 141.63, 140.23, 134.11, 133.73, 132.14, 128.66, 122.23, 120.49, 119.56, 112.49, 112.25, 104.75, 102.25, 88.62, 84.20, 57.44, 53.85, 53.03, 35.21, 23.41, 22.46, 22.40, 11.79.
Example 9: Preparation of N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H- indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5- difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
Synthesis of compound III-03
[00626] To a reactor was added IV (1 .0 g), potassium bicarbonate (0.43 g, 1.3 equiv), dichlorobis(tricyclohexylphosphine)palladium(II) (28 mg, 2.5mol%), V-02 (0.67 g), butyl acetate (7.3 g) and water (2.1 g). The reactor was inerted and the mixture was agitated at about 85 °C (75-90 °C) until the reaction was complete. The mixture was cooled to about 40 °C and passed through celite (0.52 g). The celite cake was rinsed with butyl acetate (1.8 g). The filtrate and rinse were combined and this solution was washed twice with a mixture of N-acetyl-L-
cysteine (0.31 g) dissolved in water (5.2 g) and sodium hydroxide in water (5 wt%, 5.4 g). The organics were washed twice with sodium chloride in water (5 wt%, 11 g). The solution was azeotropically distilled into 1-propanol (3.3 g). To the propanol solution at about 50 °C was added methanesulfonic acid (0.31 g, 2.25 equiv.) and the product was crystallized using dibutyl ether (5.1 g). The slurry was cooled to about 10 °C, filtered, and the filter cake was washed with a 5:1 mixture of propanol in dibutyl ether (1.6 g). The solids were dried to afford III-03 1H NMR (400 MHz, DMSO-d6) δ 9.19 (d, J = 8.3 Hz, 2H), 7.84 – 7.69 (m, 4H), 7.11 (d, J = 7.7 Hz, 2H), 7.07 – 6.95 (m, 3H), 6.82 (d, J = 7.7 Hz, 2H), 6.54 – 6.40 (m, 4H), 4.90 (d, J = 16.4 Hz, 2H), 4.76 – 4.60 (m, 4H), 4.15 (dq, J = 16.6, 8.4 Hz, 2H), 3.75 (dt, J = 16.3, 8.7 Hz, 2H), 3.25 (s, 7H), 2.99 – 2.86 (m, 4H), 2.63 – 2.50 (m, 3H), 2.41 (s, 14H), 1.73 (d, J = 2.1 Hz, 13H), 0.93 (dd, J = 6.1, 3.9 Hz, 2H).
Synthesis of N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
[00627] Aqueous sodium hydroxide (0.2 M; 2.2 equivalents; 9.2 g) was added to a reactor containing III-03 (1.0 g) in MeTHF (8.3 g) at about 20 °C. The biphasic mixture was agitated for about 15 min, and the aqueous layer was removed. The organic layer was washed four times with 2.0 wt% aqueous sodium chloride (9.8 g) and was distilled. The solution containing III was used directly in the II process below. A sample was concentrated to dryness for analysis. 1H NMR (400 MHz, CDCl3): δ 7.44 ( m, 1H), 7.39 (br, 1H), 7.18 (m, 1H), 6.90 (m, 1H), 6.65 (m 1H), 4.10 (m, 2H), 3.72 (m, 4H), 2.78 (m 2H), 2.56 (br, 4H), 1.31 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 176.88, 158.95, 141,06, 129.55, 112.79, 109.56, 106.83, 66.66, 65.73, 57.45,
Example 10: Preparation of N-((S)-1-(3-(4-chloro-3-(N- (methylsulfonyl)methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (II)
[00628] Methanesulfonyl chloride (0.32 g, 2.5 equivalents) was added to a reactor containing III (1.0 g), triethylamine (0.69 g, 6.0 equivalents), and MeTHF (11 g) at about 10 °C. The mixture was agitated at about 10 °C until the reaction was complete. The reaction mixture was washed with water (6.4 g) for about 15 minutes, and warmed to about 20 °C. The layers were separated and the organic layer was washed for about 15 minutes with 10 wt% aqueous sodium chloride (6.9 g). The layers were separated and the organic layer was used directly in the next step. An aliquot was concentrated to dryness for analysis. 1H NMR (400 MHz, δ6-DMSO; 9: 1 mixture of atropi somers): δ 9.20 (d, J = 7.9 Hz 1 H), 8.99* (d, J = 8.6 Hz, 1 H), 7.96* (d, J = 7.9 Hz, 1 H), 7.83 (d, J = 8.0 Hz, 1 H), 7.80* (d, J = 7,9 Hz, 1 H), 7.76 (d, J – 8.0 Hz, 1 H), 7.45 (d, J = 7.7 Hz, 1 H), 7.41* (d, J = 7.8 Hz, 1 H), 7.31* (d, J = 7.8 Hz, 1 H), 7.02 (tt, J = 9.4, 2.1 Hz,
1 H), 6.92* (s, 1 H), 6.91 (d, J = 7.7 Hz, 1 H), 6.48 (m, 2 H), 4.92* (s, 1 H), 4.88 (d, J = 16.4 Hz, 1 H), 4.79* (d, J = 16.8 Hz, 1 H), 4.73* (d, J = 16.4 Hz, 1 H), 4.71* (m, 1 H), 4.69 (m, 1 H), 4.62* (s, 1 H), 4.60 (m, 1 H), 4.38* (dq, J = 16.4, 8.2 Hz, 1 H), 4.12 (dq, J = 16.7, 8.4 Hz, 1 H), 3.68* (s, 3 H), 3.66* (s, 3 H), 3.63 (s, 3 H), 3.58 (s, 3 H), 3.26 (s, 3 H), 3.12* (dd, 7 = 13.8, 10.5 Hz, 1 H), 3.05 (dd, J = 13.5, 5.8 Hz, 1 H), 2.97 (dd, J = 13.5, 8.5 Hz, 1 H), 2.78* (dd, J = 13.7, 3.9 Hz, 1 H), 2.59 (m, 1 H), 2.53 (m, 1 H), 1.75 (s), 1.75 (s, 6 H), 1 .39 (m, 1 H), 0.98 (m, 1 H).
13C NMR (100 MHz, DMSO-d6, 9:1 mixture of atropi somers): δ 164.5, 163.6*, 162.1 (dd, ,7 = 246.3, 13.4 Hz), 162.0* (dd, J = 246.1, 13.3 Hz), 158.7, 158.4*, 142.7 (t, J = 29.3 Hz), 142.3, 142.0*, 141.8 (t, J= 9.4 Hz), 140.6*, 139.9, 139.7*, 139.3, 135.8*, 135.0, 133.8 (q, J = 39.0 Hz), 132.2*, 132.1 (m), 131.6, 129.6, 129.4*, 126.7, 125.3, 125.2*, 124.1*, 123.4, 122.8*, 122.7 (q, J= 280.9 Hz), 120.7 (q, J = 268.3 Hz), 119.9 (t, J = 243.7 Hz), 119.8, 119.5*, 119.0*, 118.9, 112.0, 102.2 (t, J= 225.7 Hz), 101.8*, 88.4, 84.5, 57.3, 52.93, 52.86, 52.7, 52.5*, 50.7 (q, J = 33.8 Hz), 50.3*, 42.6*, 42.4, 42.3*, 42.2, 39.51, 39.5, 38.9*, 35.1, 27.5 (dd, J = 35.0, 28.6 Hz), 23.1, 22.4, 22.3, 11.5. (* signals arising from minor atropisomer)
Example 11: Preparation of N-((S)-1-(3-(4-chIoro-3-(methylsuIfonamido)-1-(2,2,2- trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)- 2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5- tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I)
Synthesis of sodium (4-chloro-7-(2-((S)-1-(2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamido)-2-(3,5- difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide (1-02)
[00629] Sodium hydroxide (1 M, 2.9 g, 3.0 equiv.) was added to a reactor containing II (1.0 g) and 2-methyltetrahydrofuran (8.4 g) at about 35 °C. The mixture was agitated until the reaction was deemed complete. The reaction mixture was adjusted to between about 20 and 40 °C and the bottom layer was removed. The organic layer was washed with water (2.9 g) for about 15 minutes, and the bottom layer was removed. The organic solvent was swapped for ethanol and the solution was concentrated to about 5 volumes and the temperature was adjusted to about 35 °C. n-Heptane (3.4 g) was slowly added, and the mixture was aged for about 12 hours. The solids were collected by filtration, and the filter cake was washed with ethanol/n- heptane (1:1). The resultant wet cake was dried under vacuum to afford 1-02. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (d, J = 8.0 Hz, 1H), 8.93* (d, J = 8.5 Hz), 7.80 – 7.72* (m), 7.71 (s, 2H), 6.99 (tt, J = 9.5, 2.4 Hz, 1H), 6.94 (d, J = 7.6 Hz, 1H), 6.90* (d, J = 6.3 Hz), 6.69 (d, J = 7.6 Hz, 1H), 6.57 – 6.51* (m), 6.48 – 6.40 (m, 2H), 4.90 (d, J = 16.5 Hz, 1H), 4.77 (d, J = 16.4
Hz, 1H), 4.70 (td, J = 8.3, 5.2 Hz, 1H), 4.63* (d, J = 16.5 Hz), 4.22 (dq, J= 16.7, 8.4 Hz, 1H), 3.90 – 3.75 (m, 1H), 3.26 (s, 3H), 2.92 (td, J = 13.8, 8.5 Hz, 2H), 2.83* (s), 2.80 (s, 3H), 2.64 – 2.51 (m, 2H), 1.74 (d, J = 2,2 Hz, 6H), 1.44 – 1.34 (m, 1H), 0.94 (dq, J = 6.0, 3.7 Hz, 1H); 13C NMR (100 MHz, dmso) δ 164.39, 163.43, 163.39, 163.25, 160.94, 160.91, 160.81, 158.93,
158.22, 152.64, 151.94, 142.92, 142.72, 142.63, 142.43, 142.34, 142.19, 142.10, 142.00, 141.43,
141.14, 139.55, 139.36, 133.95, 133.56, 133.17, 132.12, 131.93, 131.68, 129.66, 129.56, 128.17,
127.91, 126.86, 126.76, 125.02, 122.35, 122.21, 122.08, 122.05, 119.93, 119.88, 119.38, 118.88,
118.18, 117.54, 117.21, 117.04, 112.18, 112.02, 111.95, 111.84, 111.78, 102.28, 102.03, 101.81,
88.14, 88.00, 84.69, 84.65, 57.33, 53.22, 52.96, 52.76, 52.44, 40.15, 39.94, 39.73, 39.52, 39.31, 39.10, 38.97, 38.89, 38.65, 35.10, 35.08, 27.86, 27.56, 27.52, 27.23, 23.19, 22.42, 22.41, 22.30, 22.28, 11.63. * Signals arising from minor atropisomer. 13C NMR data is reported for the mixture of atropisomers.
Synthesis of N-((S)-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I) from sodium (4-chioro-7-(2-((S)-1-(2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-l-yl)acetamido)-2-(3.5-difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide (I-02)
[00630] Compound I-02 (1.0 g) and glacial acetic acid (2.1 g) were combined at about 20 °C and were agitated until dissolved. The resultant solution was transferred to a reactor containing water (15 g) over about 1 hour. The resultant slurry was further agitated for about one hour, and was filtered. The wet cake was washed with water (2 x 5 g), deliquored, and dried at about 60 °C under vacuum to provide I. 1H NMR (400 MHz, δ6-DMSO; 5:1 mixture of atropi somers) δ 10.11* (s), 10.00 (s, 1 H), 9.25 (d, J= 8.0 Hz, 1 H), 8.92* (d, J = 8.4 Hz), 7.90* (d, J = 7.6 Hz), 7.81 (d, J = 8.0 Hz, 1 H), 7.76 (d, J= 8.0 Hz, 1 H), 7.32 (d, J = 7.6 Hz, 1 H), 7.23* (d, J = 8.0 Hz), 7.19* (d, J = 8.0 Hz), 7.02 (tt, J = 9.4, 2,4 Hz, 1 H), 6.94* (m), 6.86 (d, J = 7.6 Hz, 1 H), 6.54* (m), 6.48 (m, 2 H), 4.92 (d, J = 16.4 Hz, 1 H), 4.77* (d, J = 16.4 Hz), 4.71 (d, J = 16.4 Hz, 1 H), 4.68* (m), 4.51 (dq, J = 16.4, 8.3 Hz, 1 H), 4.19* (dq, J = 16.4, 8.2 Hz), 3.96 (dq, J = 16.8,
8.4 Hz, 1 H), 3.27 (s, 3 H), 3.24* (s), 3.17 (s, 3 H), 3.11* (dd, J = 13.0, 3.4 Hz), 3.02 (dd, J = 13.6, 5.6 Hz, 1 H), 2.95 (dd, J = 13.8, 8.6 Hz, 1 H), 2.92* (m), 2.60 (m, 1 H), 2.55 (m, 1 H), 1.74 (s, 6 H), 1.40 (m, 1 H), 0.96 (m, 1 H); 13C NMR (100 MHz, δ6-DMSO; 5:1 mixture of atropisomers) δ 164.5, 163.4*, 162.1 (dd, 7 = 246.0, 13.4 Hz), 162.0* (dd, 7 = 246.1, 13.4 Hz), 158.8, 158.1 *, 142.7 (t, 7 = 29.3 Hz), 142.3, 142.1* (m), 141.9 (t, J= 9.5 Hz), 141.7*, 140.2*, 140.0*, 139.8*, 139.5, 139.3, 139.2, 133.8 (q, J= 38.7 Hz), 132.0 (m), 131.7*, 131.1, 130.3*, 130.0, 126.8, 126.4, 126.2*, 123.0* (m), 122.9 (q, J = 281.7 Hz), 122.7*, 122.1, 120.7 (q, J = 268.3 Hz), 119.9 (t, J= 243.4 Hz), 119.0, 118.7*, 117.5*, 117.4, H2.0 (m), 102.1 (t, J= 25.6 Hz), 101.9* (m), 88.5*, 88.4, 84.5, 57.3, 52.8, 52.7, 52.4*, 50.2 (q, J= 33.3 Hz), 50.0 (m),
41.4*, 41.2, 39.8, 38.7, 35.1, 27.5 (dd, J= 35.1, 29.0 Hz), 23.2, 22.4, 22.3, 22.2*, 11.6. * Signals arising from the minor atropisomer.
[00631] Alternatively, a premixed solution of acetic acid (1.5 g), ethanol (12 g), and water (0.3 g) were combined with Compound I-02 at 20 °C and were agitated until dissolved. The resultant solution was transferred to a reactor containing water (100 g) over about 30 minutes. The resultant slurry was further agitated for about one hour, and was filtered. The wet cake was washed with water (2 x 25 g), deliquored, and dried at about 60 °C under vacuum to provide I.
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,44a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide(I) from N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
[00632] A reactor was charged with III (1.0 g) followed by cyclopentyl methyl ether (2.0 mL). The contents were adjusted to about 80 °C. In a separate reactor, methanesulfonic acid anhydride (0.3g, 1.5 equiv.) was dissolved in cyclopentyl methyl ether (6 mL). The solution was added to the first reactor via a syringe pump over 5 h. Following addition, the reaction mixture was aged for 16 h. The reaction mixture was quenched with water (10 mL). UPLC analysis of the organic phase showed I with 94.8% purity.
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I) from N-((S)-1-(3-bromo-6-(3- methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
[00633] To a 40 mL vial was added IV (1 .00 g), potassium bicarbonate (420 mg), palladium(II) chloride (4.9 mg, 2.0 mol%), cyclohexyl diphenylphosphine (13.4 mg, 3.6 mol%), V-03 (849 mg), 2-methyltetrahydrofuran (8.0 mL) and water (2.0 mL). The vial was inerted and the mixture was agitated at about 68 °C (65-73 °C) until the reaction was complete. The mixture was cooled to about 40 °C and the aqueous layer was removed. The organic layer was washed with aqueous acetic acid (5% w/v, 5.1 g). The organic was then concentrated to dryness and the residue was purified by column chromatography to afford I. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 0.2H), 10.00 (s, 1H), 9.25 (d, J = 8.2 Hz, 1H), 8.92 (d, J = 8.6 Hz, 0H),
7.90 (d, J = 7.9 Hz, 0.1H), 7.85 – 7.71 (m, 2H), 7.52-7.50 (m, 0.1H), 7.32 (d, J = 7.7 Hz, 1H),
7.21 (q, J= 9.6 Hz, 0.4H), 7.11 – 6.97 (m, 1H), 6.94-6.89 (m, 0.2H), 6.86 (d, J = 7.7 Hz, 1H),
6.55 (d, J = 7.4 Hz, 0.4H), 6.52 – 6.43 (m, 2H), 4.92 (d, J = 16.4 Hz, 1H), 4.81-4.66 (m, 1.5H),
4.64-4.45 (m, 2.4H), 4.28-4.13 (m, 0.2H), 4.08-3.92 (m, 1.6H), 3.32 (s, 0.7H), 3.30-3.22 (m, 4.4H), 3.17 (s, 3H), 3.08-2.89 (m, 2.2H), 2.69 – 2.53 (m, 2.2H), 2.12 (s, 0.2H), 1.99 (s, 1H), 1.91 (s, 0.3H), 1.80 – 1.70 (m, 6H), 1.48-1.36 (m, 1.2H), 1.23 – 1.12 (m, 1.3H), 0.96 (s, 1.2H).
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3.5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cvclopenta[1,2-c]pyrazol-1-yl)acetamide(I) from N-((S)-1-(3-bromo-6-(3- methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
[00634] To a 40 mL vial was added IV (1.00 g), potassium bicarbonate (420 mg), palladium(II) chloride (4.9 mg, 2.0 mol%), cyclohexyl diphenylphosphine (13.4 mg, 3.6 mol%), V-04 (923 mg), 2-methyltetrahydrofuran (8.0 mL) and water (2.0 mL). The vial was inerted and the mixture was agitated at about 68 °C (65-73 °C) until the reaction was complete. The mixture was cooled to about 40 °C and the aqueous layer was removed. The organic was stirred with aqueous sodium hydroxide (5 % w/w, 6.3 g) at 40 °C until reaction was complete. The organic was washed with aqueous acetic acid (5% w/v, 5.1 g). The organic was then concentrated to dryness and the residue was purified by column chromatography to afford I. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 0.2H), 10.00 (s, 1H), 9.25 (d, J = 8.2 Hz, 1H), 8.92 (d, J = 8.6 Hz, 0H), 7.90 (d, J = 7.9 Hz, 0.1H), 7.85 – 7.71 (m, 2H), 7.52-7.50 (m, 0.1H), 7.32 (d, J = 7.7 Hz, 1H), 7.21 (q, J = 9.6 Hz, 0.4H), 7.11 – 6.97 (m, 1H), 6.94-6.89 (m, 0.2H), 6.86 (d, J =
7.7 Hz, 1H), 6.55 (d, J = 7.4 Hz, 0.4H), 6.52 – 6.43 (m, 2H), 4.92 (d, J = 16.4 Hz, 1H), 4.81- 4.66 (m, 1.5H), 4.64-4.45 (m, 2.4H), 4.28-4.13 (m, 0.2H), 4.08-3.92 (m, 1.6H), 3.32 (s, 0.7H), 3.30-3.22 (m, 4.4H), 3.17 (s, 3H), 3.08-2.89 (m, 2.2H), 2.69 – 2.53 (m, 2.2H), 2.12 (s, 0.2H), 1.99 (s, 1H), 1.91 (s, 0.3H), 1.80 – 1.70 (m, 6H), 1.48-1.36 (m, 1.2H), 1.23 – 1.12 (m, 1.3H), 0.96 (s, 1.2H).
SYN
Luíza Cruz
https://drughunter.com/lenacapavir-synthesis-highlights/


see below at end
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
 |
|
| Clinical data | |
|---|---|
| Trade names | Sunlenca |
| Other names | GS-CA1, GS-6207 |
| Routes of administration |
By mouth, subcutaneous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C39H32ClF10N7O5S2 |
| Molar mass | 968.28 g·mol−1 |
| 3D model (JSmol) | |
History
Lenacapavir is being developed by Gilead Sciences.[2]
As of 2021, it is in phase II/III clinical trials.[3] It is being investigated as a treatment for HIV patients infected with multidrug-resistant virus and as a twice-yearly injectable for pre-exposure prophylaxis (PrEP).[3][4]
Society and culture
Legal status
On 23 June 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Sunlenca, intended for the treatment of adults with multidrug‑resistant human immunodeficiency virus type 1 (HIV‑1) infection.[5] The applicant for this medicinal product is Gilead Sciences Ireland UC.[5] Lenacapavir was approved for medical use in the European Union in August 2022.[1]
References
- ^ Jump up to:a b c d e f “Sunlenca EPAR”. European Medicines Agency (EMA). 22 June 2022. Archived from the original on 26 August 2022. Retrieved 25 August 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Link JO, Rhee MS, Tse WC, Zheng J, Somoza JR, Rowe W, et al. (August 2020). “Clinical targeting of HIV capsid protein with a long-acting small molecule”. Nature. 584 (7822): 614–618. Bibcode:2020Natur.584..614L. doi:10.1038/s41586-020-2443-1. PMC 8188729. PMID 32612233. S2CID 220293679.
- ^ Jump up to:a b Boerner H (11 March 2021). “Lenacapavir Effective in Multidrug Resistant HIV”. Medscape. Archived from the original on 16 March 2021. Retrieved 15 March 2021.
- ^ Highleyman L (15 March 2021). “Lenacapavir Shows Promise for Long-Acting HIV Treatment and Prevention”. POZ. Archived from the original on 19 July 2021. Retrieved 15 March 2021.
- ^ Jump up to:a b “Sunlenca: Pending EC decision”. European Medicines Agency. 23 June 2022. Archived from the original on 26 June 2022. Retrieved 26 June 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Lenacapavir”. Drug Information Portal. U.S. National Library of Medicine.
- “Lenacapavir sodium”. Drug Information Portal. U.S. National Library of Medicine.
- “Lenacapavir”. Clinical Info. National Institutes of Health.
////////////Lenacapavir sodium, approvals 2022, ema 2022, レナカパビルナトリウム , HIV, SUNLECA, GS-6207, GS-HIV, GS-CA1, GS-CA2, PF-3540074, GS-CA1, eu 2022
[H][C@]12C[C@@]1([H])C(F)(F)C1=C2C(=NN1CC(=O)N[C@@H](CC1=CC(F)=CC(F)=C1)C1=NC(=CC=C1C1=CC=C(Cl)C2=C1N(CC(F)(F)F)N=C2NS(C)(=O)=O)C#CC(C)(C)S(C)(=O)=O)C(F)(F)F
Syn
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
Lenacapavir (Sunlenca). Lenacapavir was first approved in the EU in 2022 for use in combination with
other antiretroviral(s) in adults with multidrug resistant HIV infection and for whom it is otherwise not possible to construct a suppressive antiviral regimen.1314Later in the year the drug also received approval in Canada and the US for similar indications.
Lenacapavir received first-in-class designation as 15,16 the first approved drug known to inhibit the capsid of HIV-1, aprotein shell that encompasses the genetic material of the virus and is known to be involved in multiple stages of the HIV life cycle.
Lenacapavir is available as both an oral and injectable therapy. As a slow-release, long lasting treatment
for HIV-1, dosing may be required as infrequently as every 6months when taken in combination with other antiretrovirals, with the goal of improving patient adherence and medication
compliance. Lenacapavir was developed by Gilead Sciences, Inc. and is currently being evaluated in a number of other clinical trials for both HIV-1 treatment and prevention. 13 The synthesis of lenacapavir is achieved by joining four advancedintermediates:17boronateester2.5,18 cyclopropane carboxylic acid 2.13a,17 chiral amino pyridine 2.19,17 and alkynyl sulfone2.20(Schemes2, 3, 4, and5, respectively).18 Thesynthesisof thesekeyintermediateshavebeendescribed inthepatent literatureemployingavarietyof routes17−22and is also exemplified on scales ranging fromsingle gram to multikilogramscale, albeit without reporting isolated yields. For thepurposeof this review, the largest reportedroute to each intermediate will be described. First, synthesis of boronateester2.5beganwitharylcyanofluorideintermediate 2.1 and reliedon a condensation/intramolecular cyclization processwithhydrazinehydrate in i-PrOH/water togenerate
aminopyrazole2.2onmultikilogramscaleafter coolingand precipitation of the product from the reaction mixture (Scheme2).18Next, nucleophilicdisplacementof triflate2.3, facilitated by potassium phosphate in DMF, generated ultimately providing 2.13a as a single isomer to carry forward to lenacapavir. Chiral amine intermediate 2.19 was accessed from 3,6 dibromopicolinaldehyde (2.14), using (S)-2-ethylpropane-2 sulfinamide (2.15) as a chiral auxiliary for installation of the chiral amine (Scheme 4). 17
Condensation of aldehyde 2.14 and sulfinamide 2.15 in the presence of Cs2CO3 and NMP
furnished chiral sulfinimine 2.16, which was precipitated from the reaction mixture by addition of water. The resulting solid was then treated with (3,5-difluorobenzyl)zinc bromide using a controlled addition protocol before HCl-mediated cleavage of the chiral auxiliary. Precipitation from the reaction mixture
yielded the HCl salt of 2.18 as a single isomer, which was treated with Boc2O under aqueous basic conditions to yield the Boc-protected chiral amine 2.19. 17 It is important to note that while this chiral auxiliary-based method constitutes the largest scale route to 2.13a published to date, patent literature
by Gilead has recently emerged, describing synthesis of similar analogs on a small scale using chiral salt-based resolution methods.
The final steps in the synthesis of lenacapavir began with the Sonogashira cross coupling of advanced pyridyl bromide intermediate 2.19 and 3-methyl-3-(methylsulfonyl)but-1-yne (2.20) (Scheme 5). 17
After precipitation from the reaction mixture, the resulting product, 2.21, was subjected to Suzuki
coupling with boronate ester 2.5, furnishing 2.22. Bis sulfonylation of amine 2.22 with MsCl in TEA and subsequent Boc group cleavage with TFA allowed for generation of 2.23 following a hexane wash and neutralization process. Finally, incorporation of the chiral cyclopropane fragment was made
possible by HATU-mediated coupling of amine 2.23 and cyclopropane-carboxylic acid intermediate 2.13a in DMF and DIPEA to give a bis-substituted methylsulfonamide. Selective cleavage of one methanesulfonyl group was performed by addition of 2 N NaOH to the crude reaction mixture,
completing the synthesis of lenacapavir (2).17
(13) Paik, J. Lenacapavir: First approval. Drugs 2022, 82, 1499−
1504.
(14) Tuan, J.; Ogbuagu, O. Lenacapavir: a twice-yearly treatment for
adults with multidrug-resistant HIV infection and limited treatment
options. Expert Rev. Anti Infect. Ther. 2023, 21, 565−570.
(15) Mullard, A. FDA approves first-in-class HIV capsid inhibitor.
Nat. Rev. Drug Discovery 2023, 22, 90.
(16) Dvory-Sobol, H.; Shaik, N.; Callebaut, C.; Rhee, M. S.
Lenacapavir: A first-in-class HIV-1 capsid inhibitor. Curr. Opin. HIV
AIDS 2022, 17, 15−21.
(17) Graupe, M.; Henry, S. J.; Link, J. O.; Rowe, C. W.; Saito, R. D.;
Schroeder, S. D.; Stefanidis, D.; Tse, W. C.; Zhang, J. R. Preparation
of cyclopropacyclopentapyrazolylacetamide compounds useful for the
prophylactic or therapeutic treatment of HIV virus infection. WO
2018035359, 2018.
(18) Allan, K. M.; Batten, A. L.; Brizgys, G.; Dhar, S.; Doxsee, I. J.;
Goldberg, A.; Heumann, L. V.; Huang, Z.; Kadunce, N. T.; Kazerani,
S.; et al. Methods and intermediates for preparation of antiretroviral
pyridine derivative useful for treatment of HIV-1 infections. WO
2019161280, 2019.
.
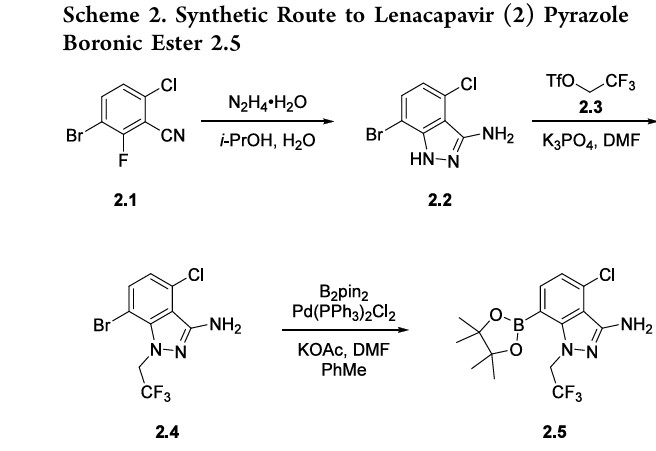



.
RISPERIDONE

Risperidone
EU APPROVED 2022/2/14, Okedi
- R-64,766
- R-64766
- RCN-3028
- RCN3028
Risperidone, R-64766, Risperdal M-Tab, Risperdal Consta, Rispolept, Belivon, Risperdal
| Formula | C23H27FN4O2 |
|---|---|
| CAS | 106266-06-2 |
| Mol weight | 410.4845 |
3-{2-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]ethyl}-2-methyl-4H,6H,7H,8H,9H-pyrido[1,2-a]pyrimidin-4-one
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Risperidone tartrate | 0S6B72E3LK | 666179-92-6 | KSWIOGDSXUFKOC-LREBCSMRSA-N |
Risperidone
CAS Registry Number: 106266-06-2
CAS Name: 3-[2-[4-(6-Fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]ethyl]-6,7,8,9-tetrahydro-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one
Manufacturers’ Codes: R-64766
Trademarks: Belivon (Organon); Risperdal (J & J)
Molecular Formula: C23H27FN4O2, Molecular Weight: 410.48
Percent Composition: C 67.30%, H 6.63%, F 4.63%, N 13.65%, O 7.80%
Literature References: Combined serotonin (5-HT2) and dopamine (D2) receptor antagonist. Prepn: L. E. J. Kennis, J. Vandenberk, EP196132; eidem,US4804663 (1986, 1989 both to Janssen). Pharmacology: P. A. J. Janssen et al.,J. Pharmacol. Exp. Ther.244, 685 (1988). Receptor binding studies: J. E. Leysen et al.,ibid.247, 661 (1988). HPLC determn in plasma: A. Avenoso et al.,J. Chromatogr. B746, 173 (2000). Clinical study in psychoses: Y. G. Gelders et al.,Pharmacopsychiatry23, 206 (1990); in autism: L. Scahill et al., N. Engl. J. Med.347, 314 (2002). Brief review: M. G. Livingston, Lancet343, 457-460 (1994). Review of pharmacology and therapeutic potential: S. Grant, A. Fitton, Drugs48, 253-273 (1994); B. Green, Curr. Med. Res. Opin.16, 57-65 (2000); of clinical experience in schizophrenia: H.-J. Möller, Expert Opin. Pharmacother.6, 803-818 (2005),
Properties: Crystals from DMF + 2-propanol, mp 170.0°. LD50 in male, female mice, rats, dogs (mg/kg): 29.7, 26.9, 34.3, 35.4, 14.1, 18.3 i.v.; 82.1, 63.1, 113, 56.6, 18.3, 18.3 orally (Janssen, 1988).
Melting point: mp 170.0°
Toxicity data: LD50 in male, female mice, rats, dogs (mg/kg): 29.7, 26.9, 34.3, 35.4, 14.1, 18.3 i.v.; 82.1, 63.1, 113, 56.6, 18.3, 18.3 orally (Janssen, 1988)
Therap-Cat: Antipsychotic.
Keywords: Antipsychotic; Benzisoxazoles; Serotonin-Dopamine Antagonist.
Risperidone, sold under the brand name Risperdal among others, is an atypical antipsychotic[2] used to treat schizophrenia and bipolar disorder.[2] It is taken either by mouth or by injection (subcutaneous or intramuscular).[2] The injectable versions are long-acting and last for 2-4 weeks.[6]
Common side effects include movement problems, sleepiness, dizziness, trouble seeing, constipation, and increased weight.[2][7] Serious side effects may include the potentially permanent movement disorder tardive dyskinesia, as well as neuroleptic malignant syndrome, an increased risk of suicide, and high blood sugar levels.[2][6] In older people with psychosis as a result of dementia, it may increase the risk of death.[2] It is unknown if it is safe for use in pregnancy.[2] Its mechanism of action is not entirely clear, but is believed to be related to its action as a dopamine and serotonin antagonist.[2]
Study of risperidone began in the late 1980s and it was approved for sale in the United States in 1993.[2][8][4] It is on the World Health Organization’s List of Essential Medicines.[9] It is available as a generic medication.[6] In 2019, it was the 149th most commonly prescribed medication in the United States, with more than 4 million prescriptions.[10][11]
Synthesis ReferenceUS4804663
SYN
| EP 0196132; ES 8705881; JP 1986221186; US 4804663 |
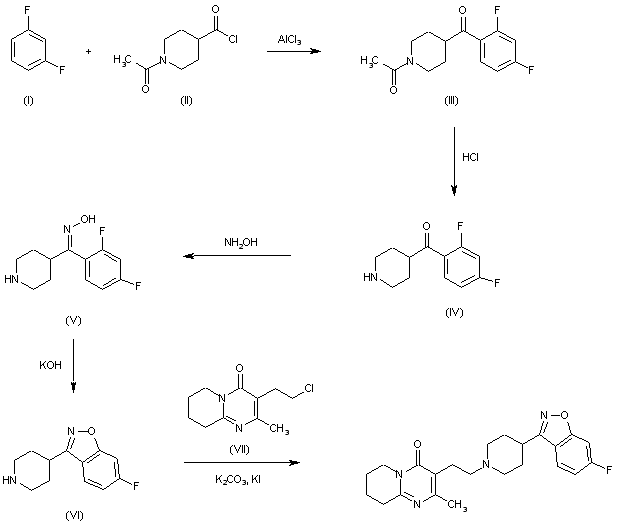
The Friedel-Crafts condensation of 1,3-difluorobenzene (I) with 1-acetylpiperidine-4-carbonyl chloride (II) by means of AlCl3 in dichloromethane gives 1-acetyl-4-(2,4-difluorobenzoyl)piperidine (III), which is hydrotyzed with refluxing 6N HCl to yield 4-(2,4-difluorobenzoyl)piperidine (IV). The reaction of (IV) with hydroxylamine in refluxing ethanol affords the corresponding oxime (V), which is cyclized by means of KOH in boiling water giving 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole (VI). Finally, this compound is condensed with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (VII) by means of K2CO3 and Kl in a variety of solvents.
SYN
ES 2050069
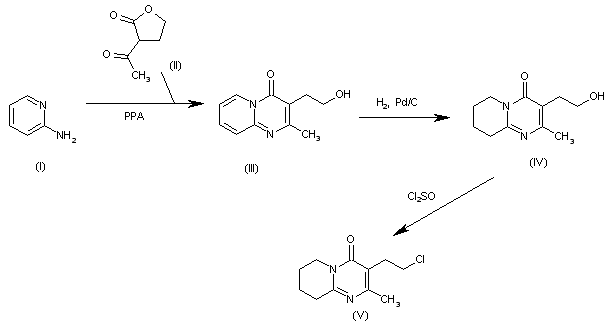
The intermediate 3-(2-chloroethyl)-2-methyl-6, 7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (V) has been obtained as follows: The cyclization of 2-aminopyridine (I) with 3-acetyltetrahydrofuran-2-one (II) by means of polyphosphoric acid (PPA) at 160 C gives 3-(2-hydroxyethyl)-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one (III), which is hydrogenated with H2 over Pd/C in ethanol/water to yield the tetrahydro derivative (IV). Finally, the OH group of (IV) is treated with SOCl2 in dichloromethane to afford the target 2-chloroethyl intermediate (V).
SYN
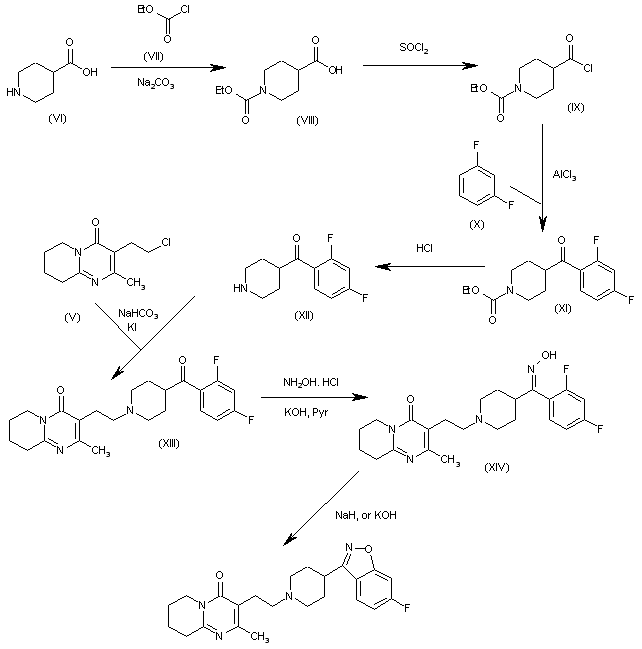
The condensation of piperidine-4-carboxylic acid (VI) with ethyl chloroformate (VII) by means of Na2CO3 in toluene/water gives 1-(ethoxycarbonyl)piperidine-4-carboxylic acid (VIII), which is treated with SOCl2 to yield the corresponding acyl chloride (IX). The Friedel-Crafts condensation of (IX) with refluxing 1,3-difluorobenzene (X) by means of AlCl3 gives 4-(2,4-difluorobenzoyl)piperidine-1-carboxylic acid ethyl ester (XI), which is treated with concentrated HCl at 100 C to yield 4-(2,4-difluorobenzoyl)piperidine (XII). The condensation of piperidine (XII) with the 2-chloroethyl intermediate (V) by means of KI and NaHCO3 in refluxing acetonitrile affords the adduct (XIII), which is treated with hydroxylamine hydrochloride and KOH in refluxing pyridine/ethanol to provide the corresponding oxime (XIV). Finally, this compound is cyclized by means of KOH in refluxing water or with NaH in refluxing THF to afford in both cases the target 1,2-benzisoxazole.
SYN
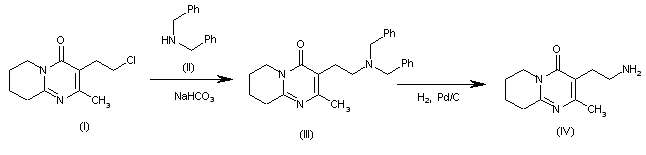
The intermediate 3-(2-aminoethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (IV) has been obtained as follows: The condensation of 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (I) with dibenzylamine (II) by means of NaHCO3 in refluxing acetonitrile gives the tertiary amine (III), which is debenzylated by hydrogenation with H2 over Pd/C in warm ethanol to afford the target intermediate (IV).
SYN
The condensation of tetrahydropyran-4-carbonyl chloride (V) with refluxing 1,3-difluorobenzene (VI) by means of AlCl3 gives 1-(2,4-difluorophenyl)-1-(tetrahydropyran-4-yl)methanone (VII), which is treated with hydroxylamine hydrochloride and sodium acetate in refluxing ethanol/water to yield the corresponding oxime (VIII). The cyclization of (VIII) by means of KOH in refluxing methanol affords 6-fluoro-3-(tetrahydropyran-4-yl)-1,2-benzisoxazole (IX), which is treated with NaI and Ac-Cl and then with K2CO3 in refluxing acetonitrile to provide the 5-iodopentanol derivative (X). The reaction of the OH group of (X) with Ms-Cl and TEA in dichloromethane gives the corresponding mesylate (XI), which is finally cyclized with the intermediate amine (IV) by means of NaHCO3 in refluxing acetonitrile to yield the target piperidine.
SYN

SYN
Eur. Pat. Appl. 196132

SYN
- Production Route of Risperidone
- (CAS NO.: ), with other name of 4H-Pyrido(1,2-a)pyrimidin-4-one, 6,7,8,9-tetrahydro-3-(2-(4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl)ethyl)-2-methyl-, could be produced through many synthetic methods.Following is one of the synthesis routes:
The Friedel-Crafts condensation of 1,3-di (I) with 1-acetylpiperidine-4-carbonyl chloride (II) by means of AlCl3 in dichloromethane gives 1-acetyl-4-(2,4-difluorobenzoyl)piperidine (III), which is hydrotyzed with refluxing 6N HCl to yield 4-(2,4-difluorobenzoyl)piperidine (IV). The reaction of (IV) with hydroxylamine in refluxing ethanol affords the corresponding oxime (V), which is cyclized by means of KOH in boiling water giving 6-fluoro-3-(4-piperidinyl)-1,2-benzisoxazole (VI). Finally, this compound is condensed with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (VII) by means of K2CO3 and Kl in a variety of solvents.
- SYN
Piperidine-Based Nonfused Biheterocycles With C–N and C–C Coupling
Ruben Vardanyan, in Piperidine-Based Drug Discovery, 2017
Risperidone (15970)
Risperidone (7.2.1) (Risperdal) is the first second-generation antipsychotic that was specifically designed as a combined D2 and serotonin 5-HT(2A) receptor antagonist, thus following the pharmacological mechanism thought to be responsible for the antipsychotic effects. After its advent in the 1990s as the first novel second-generation antipsychotic, risperidone has achieved worldwide acceptance. It was initially approved for use in schizophrenia, mania of bipolar disorder, and irritability and aggression of autism. But it is also effectively used in other instances of psychosis, including schizoaffective disorder, depression with psychotic features, and psychosis secondary to general medical conditions. Risperidone may be effective in other conditions such as major depression, various anxiety disorders, delirium, dementia, for Alzheimer’s dementia, which occurs in 6–8% of persons older than 65 and increases to 30% among those 85 years or older, and substance abuse disorders [84–113].
Risperidone is proposed for inclusion in the WHO Model List of Essential Medications for treatment of schizophrenia, mania, and autism.
Risperidone (7.2.1) was synthesized starting from 1-acetyl-4-piperidine-carbonyl chloride (7.2.4), which was used to acylate 1,3-difluorobenzene (7.2.5) in dichloromethane using aluminum chloride as Lewis acid. The reaction gave 1-(4-(2,4-difluorobenzoyl)piperidin-1-yl)ethan-1-one (7.2.6). The protecting acetyl group of the last was removed off by hydrolysis in 6 N hydrochloric acid on reflux, which gave (2,4-difluorophenyl)(piperidin-4-yl)methanone (7.2.7). The obtained product was converted further to corresponding oxime (7.2.8) on reaction with hydroxylamine hydrochloride in ethanol in the presence of N,N-diethylenethanamine. Synthesized oxime (7.2.8) was cyclized to 6-fluoro-3-(piperidin-4-yl)benzo[d]isoxazole (7.2.9) on reflux with 50% potassium hydroxide solution in water. At the final stage the obtained product (7.2.9) was alkylated with 3-(2-chloroethyl)-2-methyl-6,7,8,9-tetrahydro-4H-pyrido[1,2-a]pyrimidin-4-one (7.2.10) on heating at 85–90°C in dimethylformamide in the presence of sodium carbonate and potassium iodide, which gave the desired product, risperidone (7.2.1) [114,115]. Later, another method of (7.2.7) → (7.2.1) transformation was proposed, which involved the reductive alkylation of (2,4-difluorophenyl)(piperidin-4-yl)methanone (7.2.7) with aldehyde (7.2.11) and sodium cyanoborohydride, which gave compound (7.2.12), coherently converted to oxime (7.2.13) and further to the desired compound, risperidone (7.2.1) [116] (Scheme 7.7).

///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Risperidone is mainly used for the treatment of schizophrenia, bipolar disorder, and irritability associated with autism.[12]
Schizophrenia
Risperidone is effective in treating psychogenic polydipsia and the acute exacerbations of schizophrenia.[13][14]
Studies evaluating the utility of risperidone by mouth for maintenance therapy have reached varying conclusions. A 2012 systematic review concluded that evidence is strong that risperidone is more effective than all first-generation antipsychotics other than haloperidol, but that evidence directly supporting its superiority to placebo is equivocal.[15] A 2011 review concluded that risperidone is more effective in relapse prevention than other first- and second-generation antipsychotics with the exception of olanzapine and clozapine.[16] A 2016 Cochrane review suggests that risperidone reduces the overall symptoms of schizophrenia, but firm conclusions are difficult to make due to very low-quality evidence. Data and information are scarce, poorly reported, and probably biased in favour of risperidone, with about half of the included trials developed by drug companies. The article raises concerns regarding the serious side effects of risperidone, such as parkinsonism.[17] A 2011 Cochrane review compared risperidone with other atypical antipsychotics such as olanzapine for schizophrenia:[18]
| Summary |
|---|
| Risperidone seems to produce somewhat more extrapyramidal side effects and clearly more prolactin increase than most other atypical antipsychotics. It may also differ from other compounds in the occurrence of other adverse effects such as weight gain, metabolic problems, cardiac effects, sedation, and seizures. Nevertheless, the large proportion of participants leaving studies early and incomplete reporting of outcomes makes drawing firm conclusions difficult.[18] |
| showOutcomeFindings in wordsFindings in numbersQuality of evidence |
Long-acting injectable formulations of antipsychotic drugs provide improved compliance with therapy and reduce relapse rates relative to oral formulations.[19][20] The efficacy of risperidone long-acting injection appears to be similar to that of long acting injectable forms of first generation antipsychotics.[21]
Bipolar disorder
Second-generation antipsychotics, including risperidone, are effective in the treatment of manic symptoms in acute manic or mixed exacerbations of bipolar disorder.[22][23][24] In children and adolescents, risperidone may be more effective than lithium or divalproex, but has more metabolic side effects.[25] As maintenance therapy, long-acting injectable risperidone is effective for the prevention of manic episodes but not depressive episodes.[26] The long-acting injectable form of risperidone may be advantageous over long acting first generation antipsychotics, as it is better tolerated (fewer extrapyramidal effects) and because long acting injectable formulations of first generation antipsychotics may increase the risk of depression.[27]
Autism
Compared to placebo, risperidone treatment reduces certain problematic behaviors in autistic children, including aggression toward others, self-injury, challenging behaviour, and rapid mood changes.[28] The evidence for its efficacy appears to be greater than that for alternative pharmacological treatments.[29] Weight gain is an important adverse effect.[4][30] Some authors recommend limiting the use of risperidone and aripiprazole to those with the most challenging behavioral disturbances in order to minimize the risk of drug-induced adverse effects.[31] Evidence for the efficacy of risperidone in autistic adolescents and young adults is less persuasive.[32]
Other uses
Risperidone has shown promise in treating therapy-resistant obsessive–compulsive disorder, when serotonin reuptake inhibitors alone are not sufficient.[33]
Risperidone has not demonstrated a benefit in the treatment of eating disorders or personality disorders, except for limited evidence in schizotypal personality disorder.[34]
While antipsychotic medications such as risperidone have a slight benefit in people with dementia, they have been linked to higher incidence of death and stroke.[34] Because of this increased risk of death, treatment of dementia-related psychosis with risperidone is not FDA approved and carries a black box warning.[4]
Forms
Available forms of risperidone include tablet, oral dissolving tablet, oral solution, and powder and solvent for suspension for injection.[35]
Adverse effects
See also: List of adverse effects of risperidone
Common side effects include movement problems, sleepiness, dizziness, trouble seeing, constipation, and increased weight.[2][7] About 9 to 20% of people gained more than 7% of the baseline weight depending on the dose.[2] Serious side effects may include the potentially permanent movement disorder tardive dyskinesia, as well as neuroleptic malignant syndrome, an increased risk of suicide, and high blood sugar levels.[2][6] In older people with psychosis as a result of dementia, it may increase the risk of death.[2]
While atypical antipsychotics appear to have a lower rate of movement problems as compared to typical antipsychotics, risperidone has a high risk of movement problems among the atypicals.[36][37] Atypical antipsychotics however are associated with a greater amount of weight gain.[37]
Drug interactions
- Carbamazepine and other enzyme inducers may reduce plasma levels of risperidone.[4] If a person is taking both carbamazepine and risperidone, the dose of risperidone will likely need to be increased. The new dose should not be more than twice the patient’s original dose.[4]
- CYP2D6 inhibitors, such as SSRI medications, may increase plasma levels of risperidone and those medications.[4]
- Since risperidone can cause hypotension, its use should be monitored closely when a patient is also taking antihypertensive medicines to avoid severe low blood pressure.[4]
- Risperidone and its metabolite paliperidone are reduced in efficacy by P-glycoprotein inducers such as St John’s wort[38][39]
Discontinuation
The British National Formulary recommends a gradual withdrawal when discontinuing antipsychotic treatment to avoid acute withdrawal syndrome or rapid relapse.[40] Some have argued the additional somatic and psychiatric symptoms associated with dopaminergic super-sensitivity, including dyskinesia and acute psychosis, are common features of withdrawal in individuals treated with neuroleptics.[41][42][43][44] This has led some to suggest the withdrawal process might itself be schizomimetic, producing schizophrenia-like symptoms even in previously healthy patients, indicating a possible pharmacological origin of mental illness in a yet unknown percentage of patients currently and previously treated with antipsychotics. This question is unresolved, and remains a highly controversial issue among professionals in the medical and mental health communities, as well as the public.[45]
Dementia
Older people with dementia-related psychosis are at a higher risk of death if they take risperidone compared to those who do not. Most deaths are related to heart problems or infections.[4]
Pharmacology
Pharmacodynamics
See also: Atypical antipsychotic § Pharmacodynamics, and Antipsychotic § Comparison of medications
| Site | Ki (nM) | Action |
|---|---|---|
| 5-HT1A | 423 | Antagonist |
| 5-HT1B | 14.9 | Antagonist |
| 5-HT1D | 84.6 | Antagonist |
| 5-HT2A | 0.17 | Inverse agonist |
| 5-HT2B | 61.9 | Inverse agonist |
| 5-HT2C | 12.0 | Inverse agonist |
| 5-HT5A | 206 | Antagonist |
| 5-HT6 | 2,060 | Antagonist |
| 5-HT7 | 6.60 | Irreversible antagonist[47] |
| α1A | 5.0 | Antagonist |
| α1B | 9.0 | Antagonist |
| α2A | 16.5 | Antagonist |
| α2B | 108 | Antagonist |
| α2C | 1.30 | Antagonist |
| D1 | 244 | Antagonist |
| D2 | 3.57 | Antagonist |
| D2S | 4.73 | Antagonist |
| D2L | 4.16 | Antagonist |
| D3 | 3.6 | Inverse agonist |
| D4 | 4.66 | Antagonist |
| D5 | 290 | Antagonist |
| H1 | 20.1 | Inverse agonist |
| H2 | 120 | Inverse agonist |
| mACh | >10,000 | Negligible |
Risperidone pharmacodynamics excluding D-amino acid oxidase inhibition
Risperidone has been classified as a “qualitatively atypical” antipsychotic agent with a relatively low incidence of extrapyramidal side effects (when given at low doses) that has more pronounced serotonin antagonism than dopamine antagonism. Risperidone contains the functional groups of benzisoxazole and piperidine as part of its molecular structure. Although not a butyrophenone, it was developed with the structures of benperidol and ketanserin as a basis. It has actions at several 5-HT (serotonin) receptor subtypes. These are 5-HT2C, linked to weight gain, 5-HT2A, linked to its antipsychotic action and relief of some of the extrapyramidal side effects experienced with the typical neuroleptics.[48]
It has been found that D-amino acid oxidase, the enzyme that catalyses the breakdown of D-amino acids (e.g. D-alanine and D-serine — the neurotransmitters) is inhibited by risperidone.[49]
Risperidone acts on the following receptors:
Dopamine receptors: This drug is an antagonist of the D1 (D1, and D5) as well as the D2 family (D2, D3 and D4) receptors, with 70-fold selectivity for the D2 family. This drug has “tight binding” properties, which means it has a long half-life and like other antipsychotics, risperidone blocks the mesolimbic pathway, the prefrontal cortex limbic pathway, and the tuberoinfundibular pathway in the central nervous system. Risperidone may induce extrapyramidal side effects, akathisia and tremors, associated with diminished dopaminergic activity in the striatum. It can also cause sexual side effects, galactorrhoea, infertility, gynecomastia and, with chronic use reduced bone mineral density leading to breaks, all of which are associated with increased prolactin secretion.[48]
Serotonin receptors: Its action at these receptors may be responsible for its lower extrapyramidal side effect liability (via the 5-HT2A/2C receptors) and improved negative symptom control compared to typical antipsychotics such as haloperidol for instance. Its antagonistic actions at the 5-HT2C receptor may account, in part, for its weight gain liability.[medical citation needed]
Alpha α1 adrenergic receptors: This action accounts for its orthostatic hypotensive effects and perhaps some of the sedating effects of risperidone.[48]
Alpha α2 adrenergic receptors: Perhaps greater positive, negative, affective and cognitive symptom control.[50]
Histamine H1 receptors: effects on these receptors account for its sedation and reduction in vigilance. This may also lead to drowsiness and weight gain.[48]
Voltage-gated sodium channels: Because it accumulates in synaptic vesicles, Risperidone inhibits voltage-gated sodium channels at clinically used concentrations.[51]
Though this medication possesses similar effects to other typical and atypical antipsychotics, it does not possess an affinity for the muscarinic acetylcholine receptors. In many respects, this medication can be useful as an “acetylcholine release-promoter” similar to gastrointestinal drugs such as metoclopramide and cisapride.[medical citation needed]
Pharmacokinetics
Risperidone undergoes hepatic metabolism and renal excretion. Lower doses are recommended for patients with severe liver and kidney disease.[4] The active metabolite of risperidone, paliperidone, is also used as an antipsychotic.[52]
Society and culture

Risperdal (risperidone) 4 mg tablets (UK)
Legal status
Risperidone was approved by the United States Food and Drug Administration (FDA) in 1993 for the treatment of schizophrenia.[63] In 2003, the FDA approved risperidone for the short-term treatment of the mixed and manic states associated with bipolar disorder. In 2006, the FDA approved risperidone for the treatment of irritability in autistic children and adolescents.[64] The FDA’s decision was based in part on a study of autistic people with severe and enduring problems of violent meltdowns, aggression, and self-injury; risperidone is not recommended for autistic people with mild aggression and explosive behavior without an enduring pattern.[65] On 22 August 2007, risperidone was approved as the only drug agent available for treatment of schizophrenia in youths, ages 13–17; it was also approved that same day for treatment of bipolar disorder in youths and children, ages 10–17, joining lithium.
On 16 December 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Okedi, intended for the treatment of schizophrenia in adults for whom tolerability and effectiveness has been established with oral risperidone.[66] The applicant for this medicinal product is Laboratorios Farmacéuticos Rovi, S.A.[66]
Availability
Janssen’s patent on risperidone expired on 29 December 2003, opening the market for cheaper generic versions from other companies, and Janssen’s exclusive marketing rights expired on 29 June 2004 (the result of a pediatric extension). It is available under many brand names worldwide.[1]
Risperidone is available as a tablet, an oral solution, and an ampule, which is a depot injection.[1]
Lawsuits
On 11 April 2012, Johnson & Johnson (J&J) and its subsidiary Janssen Pharmaceuticals Inc. were fined $1.2 billion by Judge Timothy Davis Fox of the Sixth Division of the Sixth Judicial Circuit of the U.S. state of Arkansas.[67] The jury found the companies had downplayed multiple risks associated with risperidone (Risperdal). The verdict was later reversed by the Arkansas State Supreme court.[68]
In August 2012, Johnson & Johnson agreed to pay $181 million to 36 U.S. states in order to settle claims that it had promoted risperidone and paliperidone for off-label uses including for dementia, anger management, and anxiety.[69]
In November 2013, J&J was fined $2.2 billion for illegally marketing risperidone for use in people with dementia.[70]
In 2015, Steven Brill posted a 15-part investigative journalism piece on J&J in The Huffington Post, called “America’s most admired lawbreaker”, which was focused on J&J’s marketing of risperidone.[71][72]
J&J has faced numerous civil lawsuits on behalf of children who were prescribed risperidone who grew breasts (a condition called gynecomastia); as of July 2016 there were about 1,500 cases in Pennsylvania state court in Philadelphia, and there had been a February 2015 verdict against J&J with $2.5 million awarded to a man from Alabama, a $1.75M verdict against J&J that November, and in 2016 a $70 million verdict against J&J.[73] In October 2019, a jury awarded a Pennsylvania man $8 billion in a verdict against J&J.[74]
Names
Brand names include Risperdal, Risperdal Consta, Risperdal M-Tab, Risperdal Quicklets, Risperlet, Okedi, and Perseris.[75]
References
- ^ Jump up to:a b c Drugs.com International trade names for risperidone Archived 18 March 2016 at the Wayback Machine Page accessed 15 March 2016
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “Risperidone”. The American Society of Health-System Pharmacists. Archived from the original on 2 December 2015. Retrieved 1 December 2015.
- ^ “Risperdal Consta 25 mg powder and solvent for prolonged-release suspension for injection – Summary of Product Characteristics (SmPC)”. (emc). 6 December 2018. Retrieved 29 January 2022.
- ^ Jump up to:a b c d e f g h i j “Risperdal- risperidone tablet Risperdal M-Tab- risperidone tablet, orally disintegrating Risperdal- risperidone solution”. DailyMed. Retrieved 31 December 2019.
- ^ “Okedi EPAR”. European Medicines Agency (EMA). 15 December 2021. Retrieved 2 March 2022.
- ^ Jump up to:a b c d Hamilton R (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. pp. 434–435. ISBN 9781284057560.
- ^ Jump up to:a b Hasnain M, Vieweg WV, Hollett B (July 2012). “Weight gain and glucose dysregulation with second-generation antipsychotics and antidepressants: a review for primary care physicians”. Postgraduate Medicine. 124 (4): 154–67. doi:10.3810/pgm.2012.07.2577. PMID 22913904. S2CID 39697130.
- ^ Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing textbook of psychopharmacology (4th ed.). Washington, D.C.: American Psychiatric Pub. p. 627. ISBN 9781585623099.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “The Top 300 of 2019”. ClinCalc. Retrieved 16 October 2021.
- ^ “Risperidone – Drug Usage Statistics”. ClinCalc. Retrieved 16 October 2021.
- ^ “Respiridone”. The American Society of Health-System Pharmacists. Archived from the original on 13 April 2011. Retrieved 3 April 2011.
- ^ Leucht S, Cipriani A, Spineli L, Mavridis D, Orey D, Richter F, et al. (September 2013). “Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis”. Lancet. 382 (9896): 951–62. doi:10.1016/S0140-6736(13)60733-3. PMID 23810019. S2CID 32085212.
- ^ Osser DN, Roudsari MJ, Manschreck T (2013). “The psychopharmacology algorithm project at the Harvard South Shore Program: an update on schizophrenia”. Harvard Review of Psychiatry. 21 (1): 18–40. doi:10.1097/HRP.0b013e31827fd915. PMID 23656760. S2CID 22523977.
- ^ Barry SJ, Gaughan TM, Hunter R (June 2012). “Schizophrenia”. BMJ Clinical Evidence. 2012. PMC 3385413. PMID 23870705.
- ^ Glick ID, Correll CU, Altamura AC, Marder SR, Csernansky JG, Weiden PJ, et al. (December 2011). “Mid-term and long-term efficacy and effectiveness of antipsychotic medications for schizophrenia: a data-driven, personalized clinical approach”. The Journal of Clinical Psychiatry. 72 (12): 1616–27. doi:10.4088/JCP.11r06927. PMID 22244023.
- ^ Rattehalli RD, Zhao S, Li BG, Jayaram MB, Xia J, Sampson S (December 2016). “Risperidone versus placebo for schizophrenia” (PDF). The Cochrane Database of Systematic Reviews. 2016 (12): CD006918. doi:10.1002/14651858.CD006918.pub3. PMC 6463908. PMID 27977041.
- ^ Jump up to:a b Komossa K, Rummel-Kluge C, Schwarz S, Schmid F, Hunger H, Kissling W, Leucht S (January 2011). “Risperidone versus other atypical antipsychotics for schizophrenia”. The Cochrane Database of Systematic Reviews (1): CD006626. doi:10.1002/14651858.CD006626.pub2. PMC 4167865. PMID 21249678.
- ^ Leucht C, Heres S, Kane JM, Kissling W, Davis JM, Leucht S (April 2011). “Oral versus depot antipsychotic drugs for schizophrenia–a critical systematic review and meta-analysis of randomised long-term trials”. Schizophrenia Research. 127 (13): 83–92. doi:10.1016/j.schres.2010.11.020. PMID 21257294. S2CID 2386150.
- ^ Lafeuille MH, Dean J, Carter V, Duh MS, Fastenau J, Dirani R, Lefebvre P (August 2014). “Systematic review of long-acting injectables versus oral atypical antipsychotics on hospitalization in schizophrenia”. Current Medical Research and Opinion. 30 (8): 1643–55. doi:10.1185/03007995.2014.915211. PMID 24730586. S2CID 24814527.
- ^ Nielsen J, Jensen SO, Friis RB, Valentin JB, Correll CU (May 2015). “Comparative effectiveness of risperidone long-acting injectable vs first-generation antipsychotic long-acting injectables in schizophrenia: results from a nationwide, retrospective inception cohort study”. Schizophrenia Bulletin. 41 (3): 627–36. doi:10.1093/schbul/sbu128. PMC 4393684. PMID 25180312.
- ^ Muralidharan K, Ali M, Silveira LE, Bond DJ, Fountoulakis KN, Lam RW, Yatham LN (September 2013). “Efficacy of second generation antipsychotics in treating acute mixed episodes in bipolar disorder: a meta-analysis of placebo-controlled trials”. Journal of Affective Disorders. 150 (2): 408–14. doi:10.1016/j.jad.2013.04.032. PMID 23735211.
- ^ Nivoli AM, Murru A, Goikolea JM, Crespo JM, Montes JM, González-Pinto A, et al. (October 2012). “New treatment guidelines for acute bipolar mania: a critical review”. Journal of Affective Disorders. 140 (2): 125–41. doi:10.1016/j.jad.2011.10.015. PMID 22100133.
- ^ Yildiz A, Vieta E, Leucht S, Baldessarini RJ (January 2011). “Efficacy of antimanic treatments: meta-analysis of randomized, controlled trials”. Neuropsychopharmacology. 36 (2): 375–89. doi:10.1038/npp.2010.192. PMC 3055677. PMID 20980991.
- ^ Peruzzolo TL, Tramontina S, Rohde LA, Zeni CP (2013). “Pharmacotherapy of bipolar disorder in children and adolescents: an update”. Revista Brasileira de Psiquiatria. 35 (4): 393–405. doi:10.1590/1516-4446-2012-0999. PMID 24402215.
- ^ Gitlin M, Frye MA (May 2012). “Maintenance therapies in bipolar disorders”. Bipolar Disorders. 14 Suppl 2: 51–65. doi:10.1111/j.1399-5618.2012.00992.x. PMID 22510036. S2CID 21101054.
- ^ Gigante AD, Lafer B, Yatham LN (May 2012). “Long-acting injectable antipsychotics for the maintenance treatment of bipolar disorder”. CNS Drugs. 26 (5): 403–20. doi:10.2165/11631310-000000000-00000. PMID 22494448. S2CID 2786921.
- ^ Jesner OS, Aref-Adib M, Coren E (January 2007). “Risperidone for autism spectrum disorder”. The Cochrane Database of Systematic Reviews (1): CD005040. doi:10.1002/14651858.CD005040.pub2. PMID 17253538.
- ^ Kirino E (2014). “Efficacy and tolerability of pharmacotherapy options for the treatment of irritability in autistic children”. Clinical Medicine Insights. Pediatrics. 8: 17–30. doi:10.4137/CMPed.S8304. PMC 4051788. PMID 24932108.
- ^ Sharma A, Shaw SR (2012). “Efficacy of risperidone in managing maladaptive behaviors for children with autistic spectrum disorder: a meta-analysis”. Journal of Pediatric Health Care. 26 (4): 291–9. doi:10.1016/j.pedhc.2011.02.008. PMID 22726714.
- ^ McPheeters ML, Warren Z, Sathe N, Bruzek JL, Krishnaswami S, Jerome RN, Veenstra-Vanderweele J (May 2011). “A systematic review of medical treatments for children with autism spectrum disorders”. Pediatrics. 127 (5): e1312–21. doi:10.1542/peds.2011-0427. PMID 21464191. S2CID 2903864.
- ^ Dove D, Warren Z, McPheeters ML, Taylor JL, Sathe NA, Veenstra-VanderWeele J (October 2012). “Medications for adolescents and young adults with autism spectrum disorders: a systematic review”. Pediatrics. 130 (4): 717–26. doi:10.1542/peds.2012-0683. PMC 4074627. PMID 23008452.
- ^ Dold M, Aigner M, Lanzenberger R, Kasper S (April 2013). “Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a meta-analysis of double-blind, randomized, placebo-controlled trials”. The International Journal of Neuropsychopharmacology. 16 (3): 557–74. doi:10.1017/S1461145712000740. PMID 22932229.
- ^ Jump up to:a b Maher AR, Theodore G (June 2012). “Summary of the comparative effectiveness review on off-label use of atypical antipsychotics”. Journal of Managed Care Pharmacy. 18 (5 Suppl B): S1–20. doi:10.18553/jmcp.2012.18.s5-b.1. PMID 22784311.
- ^ Joint Formulary Committee. British National Formulary (online) London: BMJ Group and Pharmaceutical Press http://www.medicinescomplete.com [Accessed on 2 February 2020]
- ^ Divac N, Prostran M, Jakovcevski I, Cerovac N (2014). “Second-generation antipsychotics and extrapyramidal adverse effects”. BioMed Research International. 2014: 656370. doi:10.1155/2014/656370. PMC 4065707. PMID 24995318.
- ^ Jump up to:a b Pillay J, Boylan K, Carrey N, Newton A, Vandermeer B, Nuspl M, MacGregor T, Jafri SH, Featherstone R, Hartling L (March 2017). “First- and Second-Generation Antipsychotics in Children and Young Adults: Systematic Review Update”. Comparative Effectiveness Reviews (184): ES–24. PMID 28749632. Report 17-EHC001-EF. Bookshelf ID: NBK442352.
Compared with FGAs, SGAs may decrease the risk for experiencing any extrapyramidal symptom (EPS). FGAs probably cause lower gains in weight and BMI.
- ^ Wang, J. S.; Ruan, Y.; Taylor, R. M.; Donovan, J. L.; Markowitz, J. S.; Devane, C. L. (2004). “The Brain Entry of Risperidone and 9-hydroxyrisperidone Is Greatly Limited by P-glycoprotein”. The International Journal of Neuropsychopharmacology. 7 (4): 415–9. doi:10.1017/S1461145704004390. PMID 15683552.
- ^ Gurley BJ, Swain A, Williams DK, Barone G, Battu SK (July 2008). “Gauging the clinical significance of P-glycoprotein-mediated herb-drug interactions: comparative effects of St. John’s wort, Echinacea, clarithromycin, and rifampin on digoxin pharmacokinetics”. Molecular Nutrition & Food Research. 52 (7): 772–9. doi:10.1002/mnfr.200700081. PMC 2562898. PMID 18214850.
- ^ BMJ Group, ed. (March 2009). “4.2.1”. British National Formulary (57 ed.). United Kingdom: Royal Pharmaceutical Society of Great Britain. p. 192. ISSN 0260-535X.
Withdrawal of antipsychotic drugs after long-term therapy should always be gradual and closely monitored to avoid the risk of acute withdrawal syndromes or rapid relapse.
- ^ Chouinard G, Jones BD (January 1980). “Neuroleptic-induced supersensitivity psychosis: clinical and pharmacologic characteristics”. The American Journal of Psychiatry. 137 (1): 16–21. doi:10.1176/ajp.137.1.16. PMID 6101522.
- ^ Miller R, Chouinard G (November 1993). “Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias, neuroleptic-induced supersensitivity psychosis and refractory schizophrenia”. Biological Psychiatry. 34 (10): 713–38. doi:10.1016/0006-3223(93)90044-E. PMID 7904833. S2CID 2405709.
- ^ Chouinard G, Jones BD, Annable L (November 1978). “Neuroleptic-induced supersensitivity psychosis”. The American Journal of Psychiatry. 135 (11): 1409–10. doi:10.1176/ajp.135.11.1409. PMID 30291.
- ^ Seeman P, Weinshenker D, Quirion R, Srivastava LK, Bhardwaj SK, Grandy DK, et al. (March 2005). “Dopamine supersensitivity correlates with D2High states, implying many paths to psychosis”. Proceedings of the National Academy of Sciences of the United States of America. 102 (9): 3513–8. Bibcode:2005PNAS..102.3513S. doi:10.1073/pnas.0409766102. PMC 548961. PMID 15716360.
- ^ Moncrieff J (July 2006). “Does antipsychotic withdrawal provoke psychosis? Review of the literature on rapid onset psychosis (supersensitivity psychosis) and withdrawal-related relapse”. Acta Psychiatrica Scandinavica. 114 (1): 3–13. doi:10.1111/j.1600-0447.2006.00787.x. PMID 16774655. S2CID 6267180.
- ^ National Institute of Mental Health. PDSD Ki Database (Internet) [cited 2013 Aug 10]. ChapelHill (NC): University of North Carolina. 1998-2013. Available from: “Archived copy”. Archived from the original on 8 November 2013. Retrieved 16 May 2016.
- ^ Smith C, Rahman T, Toohey N, Mazurkiewicz J, Herrick-Davis K, Teitler M (October 2006). “Risperidone irreversibly binds to and inactivates the h5-HT7 serotonin receptor”. Molecular Pharmacology. 70 (4): 1264–70. doi:10.1124/mol.106.024612. PMID 16870886. S2CID 1678887.
- ^ Jump up to:a b c d Brunton L, Chabner B, Knollman B. Goodman and Gilman’s The Pharmacological Basis of Therapeutics, Twelfth Edition. McGraw Hill Professional; 2010.
- ^ Abou El-Magd RM, Park HK, Kawazoe T, Iwana S, Ono K, Chung SP, et al. (July 2010). “The effect of risperidone on D-amino acid oxidase activity as a hypothesis for a novel mechanism of action in the treatment of schizophrenia”. Journal of Psychopharmacology. 24 (7): 1055–67. doi:10.1177/0269881109102644. PMID 19329549. S2CID 39050369.
- ^ Hecht EM, Landy DC (February 2012). “Alpha-2 receptor antagonist add-on therapy in the treatment of schizophrenia; a meta-analysis”. Schizophrenia Research. 134 (2–3): 202–6. doi:10.1016/j.schres.2011.11.030. PMID 22169246. S2CID 36119981.
- ^ Brauner, Jan M.; Hessler, Sabine; Groemer, Teja W.; Alzheimer, Christian; Huth, Tobias (2014). “Risperidone inhibits voltage-gated sodium channels”. European Journal of Pharmacology. 728: 100–106. doi:10.1016/j.ejphar.2014.01.062. PMID 24508524.
- ^ “The DrugBank database”. Archived from the original on 17 November 2011.
- ^ Parent M, Toussaint C, Gilson H (1983). “Long-term treatment of chronic psychotics with bromperidol decanoate: clinical and pharmacokinetic evaluation”. Current Therapeutic Research. 34 (1): 1–6.
- ^ Jump up to:a b Jørgensen A, Overø KF (1980). “Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels”. Acta Psychiatrica Scandinavica. Supplementum. 279: 41–54. doi:10.1111/j.1600-0447.1980.tb07082.x. PMID 6931472.
- ^ Jump up to:a b Reynolds JE (1993). “Anxiolytic sedatives, hypnotics and neuroleptics.”. Martindale: The Extra Pharmacopoeia (30th ed.). London: Pharmaceutical Press. pp. 364–623.
- ^ Ereshefsky L, Saklad SR, Jann MW, Davis CM, Richards A, Seidel DR (May 1984). “Future of depot neuroleptic therapy: pharmacokinetic and pharmacodynamic approaches”. The Journal of Clinical Psychiatry. 45 (5 Pt 2): 50–9. PMID 6143748.
- ^ Jump up to:a b Curry SH, Whelpton R, de Schepper PJ, Vranckx S, Schiff AA (April 1979). “Kinetics of fluphenazine after fluphenazine dihydrochloride, enanthate and decanoate administration to man”. British Journal of Clinical Pharmacology. 7 (4): 325–31. doi:10.1111/j.1365-2125.1979.tb00941.x. PMC 1429660. PMID 444352.
- ^ Young D, Ereshefsky L, Saklad SR, Jann MW, Garcia N (1984). Explaining the pharmacokinetics of fluphenazine through computer simulations. (Abstract.). 19th Annual Midyear Clinical Meeting of the American Society of Hospital Pharmacists. Dallas, Texas.
- ^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, van Nueten JM, et al. (November 1970). “The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug”. Arzneimittel-Forschung. 20 (11): 1689–98. PMID 4992598.
- ^ Beresford R, Ward A (January 1987). “Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis”. Drugs. 33 (1): 31–49. doi:10.2165/00003495-198733010-00002. PMID 3545764.
- ^ Reyntigens AJ, Heykants JJ, Woestenborghs RJ, Gelders YG, Aerts TJ (1982). “Pharmacokinetics of haloperidol decanoate. A 2-year follow-up”. International Pharmacopsychiatry. 17 (4): 238–46. doi:10.1159/000468580. PMID 7185768.
- ^ Larsson M, Axelsson R, Forsman A (1984). “On the pharmacokinetics of perphenazine: a clinical study of perphenazine enanthate and decanoate”. Current Therapeutic Research. 36 (6): 1071–88.
- ^ “Electronic Orange Book”. Food and Drug Administration. April 2007. Archived from the original on 19 August 2007. Retrieved 24 May 2007.
- ^ “FDA approves the first drug to treat irritability associated with autism, Risperdal” (Press release). FDA. 6 October 2006. Archived from the original on 28 August 2009. Retrieved 14 August 2009.
- ^ Scahill L (July 2008). “How do I decide whether or not to use medication for my child with autism? Should I try behavior therapy first?”. Journal of Autism and Developmental Disorders. 38 (6): 1197–8. doi:10.1007/s10803-008-0573-7. PMID 18463973. S2CID 20767044.
- ^ Jump up to:a b “Okedi: Pending EC decision”. European Medicines Agency. 15 December 2021. Retrieved 18 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Companies belittled risks of Risperdal, slapped with huge fine” Archived 12 April 2012 at the Wayback Machine, Los Angeles Times, 11 April 2012.
- ^ Thomas K (20 March 2014). “Arkansas Court Reverses $1.2 Billion Judgment Against Johnson & Johnson”. The New York Times. Archived from the original on 5 November 2015.
- ^ “NY AG: Janssen pays $181M over drug marketing”. The Seattle Times. 30 August 2012. Archived from the original on 7 April 2016.
- ^ “Johnson & Johnson to Pay More Than $2.2 Billion to Resolve Criminal and Civil Investigations”. Department of Justice, Office of Public Affairs. 4 November 2013. Archived from the original on 5 March 2015. Retrieved 23 December 2020.
- ^ Ashbrook T (22 September 2015). “Johnson & Johnson And The Big Lies Of Big Pharma”. On Point. Archived from the original on 22 November 2016.
- ^ Brill S (September 2015). “America’s Most Admired Lawbreaker”. The Huffington Post. Archived from the original on 2 October 2015.
- ^ Feeley J (1 July 2016). “J&J Hit With $70 Million Risperdal Verdict Over Male Breasts”. Bloomberg News. Archived from the original on 7 May 2017.
- ^ “Jury says J&J must pay $8 billion in case over male breast growth linked to Risperdal”. Reuters. 9 October 2019. Retrieved 9 October 2019.
- ^ “Risperidone: MedlinePlus Drug Information”. medlineplus.gov. Retrieved 28 September 2020.
Further reading
- Dean L (2017). “Risperidone Therapy and CYP2D6 Genotype”. In Pratt VM, McLeod HL, Rubinstein WS, et al. (eds.). Medical Genetics Summaries. National Center for Biotechnology Information (NCBI). PMID 28520384. Bookshelf ID: NBK425795.
“Risperidone”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Risperdal, others[1] |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a694015 |
| License data | US DailyMed: Risperidone |
| Pregnancy category | AU: C |
| Routes of administration | By mouth, intramuscular, subcutaneous |
| Drug class | Atypical antipsychotic[2] |
| ATC code | N05AX08 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)CA: ℞-onlyUK: POM (Prescription only) [3]US: ℞-only [4]EU: Rx-only [5] |
| Pharmacokinetic data | |
| Bioavailability | 70% (by mouth)[2] |
| Metabolism | Liver (CYP2D6 mediated to 9-hydroxyrisperidone)[2] |
| Elimination half-life | 20 hours (by mouth), 3–6 days (IM)[2] |
| Excretion | Urinary (70%) feces (14%)[2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 106266-06-2 |
| PubChem CID | 5073 |
| PubChem SID | 475100 |
| IUPHAR/BPS | 96 |
| DrugBank | DB00734 |
| ChemSpider | 4895 |
| UNII | L6UH7ZF8HC |
| KEGG | D00426 |
| ChEBI | CHEBI:8871 |
| ChEMBL | ChEMBL85 |
| PDB ligand | 8NU (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID8045193 |
| ECHA InfoCard | 100.114.705 |
| Chemical and physical data | |
| Formula | C23H27FN4O2 |
| Molar mass | 410.493 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////////Risperidone, R-64,766, R-64766, RCN-3028, RCN3028

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}