
Home » FDA 2011
Category Archives: FDA 2011
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Abiraterone acetate, アビラテロン酢酸エステル

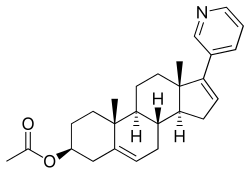
Abiraterone acetate
- Molecular FormulaC26H33NO2
- Average mass391.546 Da
Abiraterone, CB-7598, アビラテロン酢酸エステル
- (3β)-17-(pyridin-3-yl)androsta-5,16-dien-3-ol
- 17-(3-Pyridyl)androsta-5,16-dien-3beta-ol
Centocor Ortho Biotech
Abiraterone is a derivative of steroidal progesterone and is an innovative drug that offers clinical benefit to patients with hormone refractory prostate cancer. Abiraterone is administered as an acetate salt prodrug because it has a higher bioavailability and less susceptible to hydrolysis than abiraterone itself. FDA approved on April 28, 2011.
Used in combination with prednisone for the treatment of metastatic, castration-resistant prostate cancer.
- Originator The Institute of Cancer Research
- Developer All Ireland Cooperative Oncology Research Group; Cancer Research UK; Cougar Biotechnology; Janssen Research & Development; Johnson & Johnson; UNICANCER
- Class Androstenols; Antiandrogens; Antineoplastics; Small molecules
- Mechanism of Action CYP17A1 protein inhibitors
Highest Development Phases
- Marketed Prostate cancer
- Phase II Breast cancer; Ovarian cancer
- No development reported Congenital adrenal hyperplasia
Most Recent Events
- 06 Jun 2018 The National Institute for Health and Clinical Excellence does not recommend abiraterone for Prostate cancer (Combination therapy, First-line therapy, Hormone refractory, Metastatic disease)
- 06 Mar 2018 Janssen initiates a phase II trial for Prostate cancer (Combination therapy, Hormone refractory, Metastatic disease, Second-line therapy or greater) in USA (PO) (NCT03360721)
- 01 Mar 2018 Janssen plans the phase II OPTIMABI trial in Prostate cancer (Hormone refractory, Metastatic disease) in France (PO, Tablet) (NCT03458247)
- Abiraterone is associated with decreases in PSA levels, tumor shrinkage (as evaluated by RECIST criteria), radiographic regression of bone metastases and improvement in pain. Levels of adrenocorticotropic hormones increased up to 6-fold but this can be suppressed by dexamethasone.


FDA
NDA 202379, ZYTIGA (abiaterone acetate)
(3β)-17-(3-pyridinyl)androsta-5,16-dien-3-yl acetate
OND Division: NDA: Applicant: Stamp Date: PDUFA Goal Date: Established Name: Trade Name Dosage Form and Strength: Route of Administration: Indication: eCTD Reference for CMC Regulatory Filing Related IND Assessed by: Division of Drug Oncology Products 202-379 Centocor Ortho Biotech, Inc. 20 December, 2010 20 June, 2011 (Priority) Abiraterone Acetate ZYTIGA (proposed) Tablet – 250 mg Oral Indicated with prednisone for the treatment of metastatic (castrationresistant prostate cancer) in patients who have received prior chemotherapy containing a
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202379Orig1s000ChemR.pdf
Abiraterone acetate, the drug substance, is an acetyl ester of abiraterone. It is a pro-drug of the active metabolite abiraterone. Abiraterone acetate is converted in vivo to abiraterone which selectively inhibits the enzyme CYP17. Abiraterone acetate is designated chemically as (3β)-17- (3-pyridinyl)androsta-5,16-dien-3-yl acetate. It is a white to off-white, non-hygroscpic, crystalline powder. It is freely soluble in organic solvents like tetrahydrofuran and dichloromethane but practically insoluble in water. It shows some solubility in 0.1N HCl. It should be noted that abiraterone acetate contains a . The dissociation constant (pKa) of abiraterone acetate is 5.19. It indicates that most of the abiraterone acetate will be soluble in stomach pH and most of the drug will be absorbed in the unionized form in the intestine at higher pH. The partition coefficient (log P) value of abiraterone acetate is 5.12 indicating high lipophilicity. Based on low aqueous solubility and low permeability thru the cells in GI tract, the drug substance is considered BCS Class IV.
Abiraterone acetate, sold under the brand name Zytiga among others, is an antiandrogen medication which is used in the treatment of prostate cancer.[1] It is specifically indicated for use in conjunction with castration and prednisone for the treatment of metastaticcastration-resistant prostate cancer (mCRPC) and in the treatment of metastatic high-risk castration-sensitive prostate cancer (mCSPC).[1] It is taken by mouth once per day with food.[1]
Side effects of abiraterone acetate include fatigue, arthralgia, hypertension, nausea, edema, hypokalemia, hot flashes, diarrhea, vomiting, cough, headache, glucocorticoid deficiency, mineralocorticoid excess, and hepatotoxicity among others.[1] The drug is an androgen synthesis inhibitor – specifically, a CYP17A1 inhibitor – and thereby inhibits the production of androgens like testosterone and dihydrotestosterone in the body.[1] In doing so, it prevents the effects of these hormones in the prostate gland and elsewhere in the body.[1] Abiraterone acetate is a prodrug of abiraterone.[1]Abiraterone acetate, sold under the brand name Zytiga among others, is an antiandrogen medication which is used in the treatment of prostate cancer.[1] It is specifically indicated for use in conjunction with castration and prednisone for the treatment of metastaticcastration-resistant prostate cancer (mCRPC) and in the treatment of metastatic high-risk castration-sensitive prostate cancer (mCSPC).[1] It is taken by mouth once per day with food.[1]
Abiraterone acetate was first described in 1993 and was introduced for medical use in 2011.[5][6][7] It was approved for the treatment of mCRPC in 2011 and was subsequently approved for the treatment of mCSPC in 2018.[8] The medication is marketed widely throughout the world.[9] It is not available as a generic medication.[10]
Medical uses
Prostate cancer
Abiraterone acetate is indicated for use in combination with prednisone, a corticosteroid, as a treatment for mCRPC (previously called hormone-resistant or hormone-refractory prostate cancer).[11][12][13][14] This is a form of prostate cancer that is not responding to first-line androgen deprivation therapy or treatment with androgen receptor antagonists. Abiraterone acetate has received FDA (28 April 2011), EMA (23 September 2011), MHRA (5 September 2011) and TGA (1 March 2012) approval for this indication.[11][12][13][14] In Australia it is covered by the Pharmaceutical Benefits Scheme when being used to treat castration-resistant prostate cancer and given in combination with prednisone/prednisolone (subject to the conditions that the patient is not currently receiving chemotherapy, is either resistant or intolerant of docetaxel, has a WHO performance status of <2, and his disease has not since become progressive since treatment with PBS-subsidised abiraterone acetate has commenced).[15]
Clinical effectiveness
A phase III study in subjects previously treated with docetaxel started in 2008.[16] In September 2010, an independent panel found that the interim results in patients previously treated with docetaxel were so much better compared to those treated with placebo that it was unethical to keep half the study participants on placebo, and all patients began receiving the drug. Overall survival was increased by 3.9 months in to this study (14.8 months versus 10.9 months for placebo).[17]
A placebo-controlled double-blind randomized phase III study in patients with castration-refractory prostate cancer but who had not received chemotherapy opened to accrual in April 2009.[18][19] 1,088 men received either abiraterone acetate (1000 mg daily) plus prednisone (5 mg twice daily), or placebo plus prednisone. The median radiographic progression-free survival was 16.5 months with abiraterone acetate–prednisone and 8.3 months with prednisone alone (hazard ratio (HR) = 0.53; 95% confidence interval (CI), 0.45 to 0.62; P<0.001). After a median follow-up period of 22.2 months, overall survival was better with abiraterone acetate plus prednisone (median not reached) compared to placebo plus prednisone (27.2 months); HR = 0.75; 95% CI, 0.61 to 0.93; P=0.01).[20]
Available forms
Abiraterone acetate is available in the form of 250 mg and 500 mg film-coated oral tablets and 250 mg uncoated oral tablets.[1] It is used at a dosage of 1,000 mg orally once per day with food in conjunction with castration (via GnRH analogue therapy or orchiectomy) and in combination with 5 mg prednisone orally twice per day.[1]
Contraindications
Contraindications include hypersensitivity to abiraterone acetate. Although documents state that it should not be taken by women who are or who may become pregnant,[12][21] there is no medical reason that any woman should take it. Women who are pregnant should not even touch the pills unless they are wearing gloves.[21] Other cautions include severe baseline hepatic impairment, mineralocorticoid excess, cardiovascular disease including heart failure and hypertension, uncorrected hypokalemia, and adrenocorticoid insufficiency.[22]
Side effects
Side effects by frequency:[11][12][13][14][22]
Very common (>10% frequency):
Common (1-10% frequency):
- Hypertriglyceridaemia
- Sepsis
- Cardiac failure
- Angina pectoris
- Arrhythmia
- Atrial fibrillation
- Tachycardia
- Dyspepsia (indigestion)
- Rash
- Alanine aminotransferase increased
- Aspartate aminotransferase increased
- Fractures
- Hematuria
Uncommon (0.1-1% frequency):
Rare (<0.1% frequency):
- Allergic alveolitis
Overdose
Clinical experience with overdose of abiraterone acetate is limited.[1] There is no specific antidote for abiraterone acetate overdose, and treatment should consist of general supportive measures, including monitoring of cardiac and liver function.[1]
Interactions
Abiraterone acetate is a CYP3A4 substrate and hence should not be administered concurrently with strong CYP3A4 inhibitors such as ketoconazole, itraconazole, clarithromycin, atazanavir, nefazodone, saquinavir, telithromycin, ritonavir, indinavir, nelfinavir, voriconazole) or inducers such as phenytoin, carbamazepine, rifampin, rifabutin, rifapentine, phenobarbital.[22][21] It also inhibits CYP1A2, CYP2C9, and CYP3A4 and likewise should not be taken concurrently with substrates of any of these enzymes that have a narrow therapeutic index.[22][21]
Pharmacology
Pharmacodynamics

Abiraterone, the active metaboliteof abiraterone acetate.
Antiandrogenic activity
Abiraterone, the active metabolite of abiraterone acetate, inhibits CYP17A1, which manifests as two enzymes, 17α-hydroxylase (IC50 = 2.5 nM) and 17,20-lyase (IC50 = 15 nM) (approximately 6-fold more selective for inhibition of 17α-hydroxylase over 17,20-lyase)[23][24] that are expressed in testicular, adrenal, and prostatic tumor tissues. CYP17A1 catalyzes two sequential reactions: (a) the conversion of pregnenolone and progesterone to their 17α-hydroxy derivatives by its 17α-hydroxylase activity, and (b) the subsequent formation of dehydroepiandrosterone (DHEA) and androstenedione, respectively, by its 17,20-lyase activity.[25] DHEA and androstenedione are androgens and precursors of testosterone. Inhibition of CYP17A1 activity by abiraterone thus decreases circulating levels of androgens such as DHEA, testosterone, and dihydrotestosterone (DHT). Abiraterone acetate, via its metabolite abiraterone, has the capacity to lower circulating testosterone levels to less than 1 ng/dL (i.e., undetectable) when added to castration.[23][26] These concentrations are considerably lower than those achieved by castration alone (~20 ng/dL).[26] The addition of abiraterone acetate to castration was found to reduce levels of DHT by 85%, DHEA by 97 to 98%, and androstenedione by 77 to 78% relative to castration alone.[26] In accordance with its antiandrogenic action, abiraterone acetate decreases the weights of the prostate gland, seminal vesicles, and testes.[27]
Abiraterone also acts as a partial antagonist of the androgen receptor (AR), and as an inhibitor of the enzymes 3β-hydroxysteroid dehydrogenase (3β-HSD), CYP11B1 (steroid 11β-hydroxylase), CYP21A2 (Steroid 21-hydroxylase), and other CYP450s (e.g., CYP1A2, CYP2C9, and CYP3A4).[22][28][29][30] In addition to abiraterone itself, part of the activity of the drug has been found to be due to a more potent active metabolite, δ4-abiraterone (D4A), which is formed from abiraterone by 3β-HSD.[31] D4A is an inhibitor of CYP17A1, 3β-hydroxysteroid dehydrogenase/Δ5-4 isomerase, and 5α-reductase, and has also been found to act as a competitive antagonist of the AR reportedly comparable to the potent antagonist enzalutamide.[31] However, the initial 5α-reduced metabolite of D4A, 3-keto-5α-abiraterone, is an agonist of the AR, and promotes prostate cancer progression.[32] Its formation can be blocked by the coadministration of dutasteride, a potent and selective 5α-reductase inhibitor.[32]
Estrogenic activity
There has been interest in the use of abiraterone acetate for the treatment of breast cancer due to its ability to lower estrogen levels.[33] However, abiraterone has been found to act as a direct agonist of the estrogen receptor, and induces proliferation of human breast cancer cells in vitro.[33] If abiraterone acetate is used in the treatment of breast cancer, it should be combined with an estrogen receptor antagonist like fulvestrant.[33] In spite of its antiandrogenic and estrogenic properties, abiraterone acetate does not appear to produce gynecomastia as a side effect.[34]
Other activities
Due to inhibition of glucocorticoid biosynthesis, abiraterone acetate can cause glucocorticoid deficiency, mineralocorticoid excess, and associated adverse effects.[35] This is why the medication is combined with prednisone, a corticosteroid, which serves as a means of glucocorticoid replacement and prevents mineralocorticoid excess.[36]
Abiraterone acetate, along with galeterone, has been identified as an inhibitor of sulfotransferases (SULT2A1, SULT2B1b, SULT1E1), which are involved in the sulfation of DHEA and other endogenous steroids and compounds, with Ki values in the sub-micrmolar range.[37]
Pharmacokinetics
After oral administration, abiraterone acetate, the prodrug form in the commercial preparation, is converted into the active form, abiraterone. This conversion is likely to be esterase-mediated and not CYP-mediated. Administration with food increases absorption of the drug and thus has the potential to result in increased and highly variable exposures; the drug should be consumed on an empty stomach at least one hour before or two hours after food. The drug is highly protein bound (>99%), and is metabolised in the liver by CYP3A4 and SULT2A1 to inactive metabolites. The drug is excreted in feces (~88%) and urine (~5%), and has a terminal half-life of 12 ± 5 hours.[21]
Chemistry
Abiraterone acetate, also known as 17-(3-pyridinyl)androsta-5,16-dien-3β-ol acetate, is a synthetic androstane steroid and a derivative of androstadienol (androsta-5,16-dien-3β-ol), an endogenous androstane pheromone. It is specifically a derivative of androstadienol with a pyridine ring attached at the C17 position and an acetate ester attached to the C3β hydroxyl group. Abiraterone acetate is the C3β acetate ester of abiraterone.
History
In the early 1990s, Mike Jarman, Elaine Barrie, and Gerry Potter of the Cancer Research UK Centre for Cancer Therapeutics in the Institute of Cancer Research in London set out to develop drug treatments for prostate cancer. With the nonsteroidal androgen synthesis inhibitor ketoconazole as a model, they developed abiraterone, filing a patent in 1993 and publishing the first paper describing it the following year.[5][38] Rights for commercialization of the drug were assigned to BTG, a UK-based specialist healthcare company. BTG then licensed the product to Cougar Biotechnology, which began development of the commercial product.[39] In 2009, Cougar was acquired by Johnson & Johnson, which developed and sells the commercial product, and is conducting ongoing clinical trials to expand its clinical uses.[40]
Abiraterone acetate was approved by the United States Food and Drug Administration on April 28, 2011.[6][7] The FDA press release made reference to a phase III clinical trial in which abiraterone use was associated with a median survival of 14.8 months versus 10.9 months with placebo; the study was stopped early because of the successful outcome.[41]Abiraterone acetate was also licensed by the European Medicines Agency.[42] Until May 2012 the National Institute for Health and Clinical Excellence (NICE) did not recommend use of the drug within the NHS on cost-effectiveness grounds. This position was reversed when the manufacturer submitted revised costs.[43] The use is currently limited to men who have already received one docetaxel-containing chemotherapy regimen.[44][45]
Society and culture
Generic names
Abiraterone acetate is the generic name of the drug and its USAN, BANM, and JAN, while abiraterone is the INN and BAN of abiraterone, its deacetylated form.[9] Abiraterone acetate is also known by its developmental code names CB-7630 and JNJ-212082, while CB-7598 was the developmental code name of abiraterone.[9][46]
Brand names
Abiraterone acetate is marketed by Janssen Biotech (a subsidiary of Johnson & Johnson) under the brand name Zytiga.[9] In addition, Intas Pharmaceuticals markets the drug under the brand name Abiratas, Cadila Pharmaceuticals markets the drug as Abretone, and Glenmark Pharmaceuticals as Abirapro.[citation needed]
Availability
Abiraterone acetate is marketed widely throughout the world, including in the United States, Canada, the United Kingdom, Ireland, elsewhere in Europe, Australia, New Zealand, Latin America, Asia, and Israel.[9]
Research
Abiraterone acetate is under development for the treatment of breast cancer and ovarian cancer and as of March 2018 is in phase II clinical trials for these indications.[46] It was also under investigation for the treatment of congenital adrenal hyperplasia, but no further development has been reported for this potential use.[46] An oral ultramicrosize tablet formulation of abiraterone acetate (also known as abiraterone acetate fine particle (AAFP) or submicron abiraterone acetate) with improved bioavailability is in pre-registration in the United States for the treatment of prostate cancer as of April 2018 and has the tentative brand name Yonza.[47]
PAPER
https://pubs.acs.org/doi/abs/10.1021/op500044p
Improved Procedure for Preparation of Abiraterone Acetate

An improved procedure for the preparation of abiraterone acetate is described. The present process highlights reduced reaction time, isolation with acid–base treatment without involving column chromatography, multiple crystallization and is amenable to large-scale synthesis.
Abiraterone Acetate (1)
1 in 81% yield (1.8 kg). HPLC Purity: 99.72%, Assay: 98.8% (HPLC, w/w). MS: m/z = 392.7 [M + H]+. IR (KBr) (cm–1): 3047, 2936, 1735, 1244, 1035, 801, 714. 1H NMR (400 MHz, DMSO-d6): δ 8.58 (s, 1 H), 8.43–8.42 (d, 1 H), 7.76–7.74 (d, 1 H), 7.34–7.31 (dd, 1 H), 6.11 (s, 1 H), 5.38 (s, 1 H), 4.44 (m, 1H), 2.19–2.50 (m, 3H), 1.98–2.08 (m, 6H), 1.39–1.85 (m, 9H), 1.03–1.11 (m, 8H). 13C NMR (CDCl3): δ 170.4, 151.6, 147.9, 147.8, 140.0, 133.6, 132.9, 129.1, 122.9, 122.2, 73.8, 57.4, 50.2, 47.3, 38.1, 36.9, 36.7, 35.1, 31.7, 31.4, 30.3, 27.7, 21.4, 20.8, 19.2, 16.5.
https://pubs.acs.org/doi/suppl/10.1021/op500044p/suppl_file/op500044p_si_001.pdf




Abiraterone (2)
The abiraterone acetate was the ester of formula (Abiraterone acetate) structure.
[0004]

[0005] So far, the search route may abiraterone acetate ester (Abiraterone acetate) are two.
[0006] Patent W09509178, CN 102030798, WO 2006021777, 2006021777, WO2006021776, J.Med.Chem.38,2463-2471,1995, synthetic route reported in the literature like the following formula WO.
[0007]

[0008] The route is DHEA as raw material, with an acetyl group protecting the hydroxyl group, the product obtained is then reacted with trifluoromethanesulfonic anhydride to give triflate product was finally reagent under palladium catalysis, Suzuki coupling reaction with 3-pyridyl diethyl borane, to give an ester of abiraterone acetate.
[0009] Patent GB 2282377,0PPI, 29 (I), 123-134,1997 the reported another method of synthesis.
The synthetic procedure the following formula.
[0010]

[0011] The route is DHEA as raw material, the reaction with hydrazine hydrate, and then reacted with iodine to give the 17-iodo – androsta-5,16-diene–3beta- alcohol, and catalytic agent in the button with Li-yl-pyrazol-diethyl _3_ boron burning Suzuki coupling reaction to give abiraterone, and finally acetylated abiraterone acetate to give abiraterone acetate.
[0012] By comparing the two lines, a synthetic routes can be found with a reagent such as trifluoromethanesulfonic anhydride, 2,6-di-t-butyl-4-methylpyridine and the like are expensive, relatively high chemical costs. In comparison, two synthetic route mild reaction conditions, the reagents are cheap, and therefore have more industrialized prospects. However, according to the synthesis process reported in the literature, the route to industrial production, there are still some technical problems.
[0013] More specifically, to 17- iodo – androsta-5,16-diene–3beta_ when alcohol (2) Synthesis of abiraterone (3) as a raw material for the Suzuki coupling reaction, the solvent is tetrahydrofuran, the solvent high cost; shall reaction refluxed for 4 days, the reaction time is too long. More importantly, when the Suzuki coupling reaction, starting material 17- iodo – male left diene-5,16-ol _3beta_
(2) will react with the impurities abiraterone (3) 4, 4 impurities not removed by recrystallization, can only be purified by column chromatography. If the compound is not 4 Ex, abiraterone prepared by acetylation reaction of abiraterone acetate ester, the impurities will be converted to 4 5 impurities, the impurities by recrystallization 5 likewise not removed, only purified by column chromatography.
[0014]

[0015] The abiraterone acetate ester synthesis, synthesis is reported abiraterone 24h the reaction with acetic anhydride and pyridine at room temperature, the reaction time is too long. The mixture was then evaporated under reduced pressure to be excess acetic anhydride and pyridine, and then crystallized from diethyl ether again, to give the final acetate abiraterone acetate was purified by column chromatography.
[0016] In summary, two synthetic routes reported in the literature of the last two long reaction time and complicated operation, product purification difficult. All this has seriously hampered the industrialization prospects abiraterone acetate esters.It is essential to two synthetic routes to optimize the improvement, in order to achieve the industrial production of abiraterone acetate ester.

] Example 1
Preparation of [0031] 17- (3-pyrazol Li-yl) androsta-5,16-diene-_3beta_ alcohol (abiraterone) of
[0032] A 750ml NMP was added to a 3L three-necked flask, were added with stirring 50gl7_ iodo – androsta-5,16-diene–3beta- alcohol, 88 mg of bis (triphenylphosphine) palladium chloride and diethyl 19.74g yl – (3-pyridyl) borane, and finally adding 345ml 2mol / L Na2CO3 solution. Heating, holding temperature of about 70-80 ° C, TLC monitored the reaction was complete. The reaction was cooled to room temperature, the reaction solution was added 1500ml of water, stirred, filtered and washed with water. Blast drying, 26.3g abiraterone.
[0033] Example 2
[0034] Preparation and purification of abiraterone acetate ester
[0035] The abiraterone 26g 156ml dissolved in pyridine, 52 ml of acetic anhydride was added at room temperature, heating, holding temperature of about 70-80 ° C, the reaction for about 4 h, TLC monitoring of the reaction was complete. The reaction was cooled to room temperature, the ice bath, 560ml of ice water was added to the reaction mixture, the precipitated white solid was stirred 20min, filtered, washed with water. 55 ° C blast drying. The crude product was added to 26ml of ethanol was dissolved by heating to clarify. Water was added 26ml, stirred for lh. Cooled to room temperature and filtered. Blast drying. Abiraterone acetate to give the final acetate 22.lg, HPLC> 99.5%.
[0036] Example 3
Preparation of [0037] 17- (3-pyridyl) androsta-5,16-diene-_3beta_ alcohol (abiraterone) of
[0038] The IlOL NMP was added to a 3L three-necked flask, were added with stirring 7.5kgl7_ iodo – androsta-5,16-diene–3beta- alcohol, 132 g of bis (triphenylphosphine) palladium chloride and 29.6kg two ethyl – (3-pyridyl) borane and finally 500L2mol / L Na2CO3 solution. To maintain the internal temperature of about 70_80 ° C, TLC monitored the reaction was complete. The reaction was cooled to room temperature, 220L of water was added to the reaction mixture, stirred for 30min, filtered, washed with water. Blast drying, 39.2kg abiraterone.
[0039] Example 4
[0040] Preparation and purification of abiraterone acetate ester
[0041] The abiraterone 39kg dissolved in pyridine 230L, 78L of acetic anhydride was added at room temperature, heating, holding temperature of about 70-80 ° C, the reaction for about 4 h, TLC monitoring of the reaction was complete. The reaction was cooled to room temperature, the ice bath, ice water was added to the 840L reaction solution, stirred 30min, filtered, washed with water, 50_55 ° C blast drying. The crude product was added to 39L of ethanol was dissolved by heating to clarify. Water was added 39L, stirred for lh then cooled to room temperature and filtered. Blast drying. Abiraterone acetate to obtain the final ester 33.2kg, HPLC> 99.5% ο
Abiraterone acetate [17-(3-pyridyl)-5,16-androstadien-33-acetate] is a steroid compound which inhibits selectively and efficiently the enzyme 17-ohydroxylase-C17- 20-lyase, which catalyzes the conversion of dehydroepiandrosterone and androstenedione to testosterone. The inhibition of said enzyme causes a strong decrease of testosterone levels in the patient and therefore this drug is used in the treatment of certain hormone-dependent tumors resistant to chemotherapy such as prostate cancer. This compound has the followin chemical formula:

This product was disclosed for the first time in WO 93/20097, which also provides a synthetic process for its preparation including as last step the reaction of an enol triflate with a pyridine borate by Suzuki coupling (see scheme below). However, this process is not viable in practice, mainly because of the difficulty in preparing the enol trifluorosulfonate at the 17-position 2: this step, apart from proceeding with a poor conversion and low yield, gives place to the impurity tri-unsaturated 3 in a 10% yield, which only may be removed by column chromatography. Further, the product obtained after the subsequent Suzuki coupling must be also purified by column chromatography according to the examples provided therein.

Abiraterone-acetate
The above-mentioned impurity was prevented in later processes (EP 1 781 683 y EP 1 789 432) thanks to the use of alternative bases to that previously employed (i.e. 2,5-ditert-butyl-4-methylpyridine) such as DABCO, DBU or tryethylamine. However, in the sole example described in said documents, whilst the final product is achieved without using any column chromatography, it is obtained in a global yield of scarcely 21 % and shows a purity of only 96.4%.

EP 0 721 461 proposes the use of a vinyl iodide or bromide intermediate instead of the enol triflate, as depicted in the following scheme:

However, the iodo-enol is much less reactive than the triflate in the coupling with the pyridine borane, resulting in long reaction times (48 hours – 4 days) with a part of the starting material unreacted and wherein until a 5% of a dimeric impurity is obtained, which can only be removed by purification by means of reverse phase column chromatograp

Therefore, there is still a need of developing new processes for obtaining 17-(3- pyridyl)-5,16-androstadien-33-ol and related compounds, some of which are of therapeutic interest (e.g. abiraterone acetate) which overcome all or part of the drawbacks associated to the known processes belonging to the state of the art.
The chemical name of abiraterone acetate ⑽) -17- (3- pyridyl) – androsta-5,16-diene-3-acetate, by the oral US Centocor Ortho developed CYP17 inhibitor . As anti-cancer drugs on the market April 28, 2011 by the US Food and Drug Administration (FDA) approval, in combination with prednisone therapy with castration-resistant prostate cancer. Trade name Zyitga. Abiraterone acetate is an oral androgen synthesis inhibitors, capable of inhibiting 17a- hydroxylase / C17,20-lyase (CYP17). Clinical results show that abiraterone acetate can significantly prolong patients with advanced prostate cancer include the use of one or both docetaxel-containing chemotherapy but her condition is still deteriorating lives of patients, the risk of death by 35%, and the side effects of drugs is very small, good safety.
[0003] Currently, the synthesis of abiraterone acetate routes are mainly three: (1) dehydroepiandrosterone acetic acid as a starting material, first-butyl-4-methylpyridine 2,6_ di ( esterified by trifluoromethanesulfonic anhydride, then with diethyl under DTBMP) under catalytic bis-triphenylphosphine palladium chloride – acetate proceeded abiraterone acetate Suzuki coupling (2-pyridyl) borane the total yield of the reaction is 48.7%; short reaction step of the process, but after the first-stage reaction a lot of by-products, to be purified by column chromatography, and the double bonds can not be divided by-products, and therefore remains a need for second-stage reaction column chromatography Analysis and purified by recrystallization complicated operation; (2) DHEA as a starting material, a condensation reaction of a hydrazone with hydrazine hydrate, and the presence (TMG) in 1,1,3,3-tetramethylguanidine ene reaction with iodine to generate iodine compound 17- iodo – male left -5,16_ diene -30- alcohol, then in the catalytic bis-triphenylphosphine palladium dichloride and diethyl – (3-pyridyl ) borane was prepared by Suzuki coupling abiraterone abiraterone acetate to give finally acetylated hydroxyl prepared. The total yield of the reaction is 41.5%. This synthesis step is longer, lower yields, and since the active iodides easy to generate high polymer impurities that can not be removed by recrystallization or column chromatography, can only be purified by preparative chromatography to give A in the Suzuki coupling process Long bit acetate pure, can not meet the requirements of industrial production; (3) in the method (1) was treated with triethylamine instead of DTBMP, reduces the formation of byproducts double bond, then after the reaction the remaining starting material was recrystallized removed. This reaction increases the process steps and the purity of the final product was only 96.4%, the drug does not meet the quality standards.
Example 1
[0021] One method of synthesis of abiraterone acetate, comprising the steps of:
[0022] A, in a 100ml round bottom flask were sequentially added 0 • 95g (5mmol) of p-toluenesulfonyl chloride, 15 mL of toluene, sufficiently stirred to dissolve, to obtain X-solution, (solution cooled to 15 ° C X) at 15 ° C under a slow was added dropwise by molar ratio of 1: 2 was added 1.5mL (lOmmol) 80% hydrazine hydrate to the solution X, dropwise within 5min; 3〇min reaction was continued, white precipitate appears in the flask. TLC analysis of the reaction end point is determined. After completion of the reaction, cold water was added 3〇mllO ° C., Stirred, filtered off with suction, the filter cake was then washed with purified water 3-5 times, and dried to obtain a white crystalline p-toluene sulfonyl hydrazide billion • 82g, 86.3% yield ( literature values: yield 92%).
[0023] B, DHEA -17- Synthesis of p-toluenesulfonyl hydrazone
[0024] In a 100ml round bottom flask were sequentially added in square • 75g CMmo 1) dehydroepiandrosterone (DHEA), 25 mL of methanol, 0.81 g sufficiently stirred to dissolve the p-toluene sulfonyl hydrazide, rt (15_25 ° C), was added O.lmL 0.2 mol .L-1 sulfuric acid, 60 ° C in an oil bath at reflux for 2h, TLC analysis of the reaction end point is determined. After completion of the reaction, the solvent was largely removed by rotary evaporation, a heavy white precipitate appeared in the flask. Was added 30mL of water, filtered off with suction, the filter cake was then washed with purified water 3-5 times to remove water at one thousand bake 50 ° C, to give a white solid 1.27g i.e. -17- Dehydroepiandrosterone p-toluenesulfonyl hydrazone, yield 81.4%.
[0025] C, 17- (3- pyridyl) androsta-5,16-diene–30- _ Synthesis of alcohol
[0026] In a 100ml round bottom flask was added 0.91g (2mmol) -17- Dehydroepiandrosterone p-toluenesulfonyl hydrazone, 25mLl, 4- dioxane, 〇.27g (3 mmol of) lithium tert-butoxide, 0_012g (0.013mmol) Pd2 (dba) 3,0.023g stirred for five minutes to fully dissolve the (0_005mmol) Xphos, at room temperature, wherein Pd2 (dba) 3 were added under nitrogen, and then quickly added 0.39g (2.5mmol) 3- bromo pyridine, 95 ° C oil bath reactor 12h, TLC analysis of the reaction end point is determined. After the reaction, ice water was added 30mL0 ° C, thoroughly shaken, ethyl acetate was added 20mL of acetic acid, liquid separation, was extracted with ethyl acetate (1 〇ml each, extracted three times) and the combined organic phase was dried over anhydrous sodium sulfate (by lg / ml was added over anhydrous sodium ratio) sulfate, filtered, and the filtrate rotary evaporated to give a pale yellow solid which was recrystallized from n-hexane (20ml) to give a white solid that is 17- (3-pyridyl) – male stay -5, 16- -3P- diene alcohols, a yield of 42.6%.
[0027] D, Synthesis of abiraterone acetate
[0028] In 0.39gl7- successively added 100mL round bottom flask (3-pyridyl) – androsta-5,16-diene-fir -3 – ol, 3〇111 dagger diethyl ether, 0.15mL (0.25mmol) triethylamine amine, are hook stirring, was slowly added dropwise 0.3 mL (2mm〇l) of acetyl chloride, the reaction stirred at room temperature Jiao 3h, TLC analysis of the reaction end point is determined. The mixture was then suction filtered, the filtrate was decolorized with charcoal, rotary evaporation, to give a white solid, i.e. abiraterone acetate product yield of 80.6%.
[0029] Example 2
[0030] A, in a 100ml round bottom flask were added 1. (^ (5.311111101) p-toluenesulfonyl chloride, 15 mL of toluene, sufficiently stirred to dissolve to give the solution X, at 15 ° C was slowly added dropwise 1 • 7mL (12mmol) X 80% hydrazine hydrate to the solution in dropwise within 5min; reaction was continued for 30min, a white precipitate appeared .TLC analysis to determine the end of the reaction after the completion of the reaction flask was added 30ml 10 ° C water with stirring, suction filtered, then the filter cake. washed 3-5 times purified water, was dry, i.e., p-toluenesulfonyl hydrazide to give white crystals 0.93 g, 86.7% yield (literature: yield 92%).
[0031] B, DHEA -17- Synthesis of p-toluenesulfonyl hydrazone
[0032] successively added 0 • 97g (3mmo 1) dehydroepiandrosterone (DHEA) in a 100ml round bottom flask, 25 mL of methanol, 1 • 08g p-toluene sulfonyl hydrazide, fully dissolved with stirring at room temperature, was added O.lmL 0.2mol • L_1 sulfuric acid, 60 ° C in an oil bath at reflux for 2h, TLC analysis of the reaction end point is determined. After completion of the reaction, the solvent was largely removed by rotary evaporation, a heavy white precipitate appeared in the flask. Was added 30mL of water, filtered off with suction, the filter cake was then washed with purified water 3-5 times, 5 (TC drying under removal of water, to give a white solid 1.43g i.e. Dehydroepiandrosterone p-toluenesulfonyl hydrazone -17- yield 80.9%.
[0033] C, 17- (3- pyridyl) -30- _ male left diene -5,16-ol Synthesis
[0034] In a 100ml round bottom flask was added in 1.32g (3 mmol of) -17- Dehydroepiandrosterone p-toluenesulfonyl hydrazone, 25mLl, 4- dioxane, 0.35g (4mmol) of lithium t-butoxide, 0.012g (0.013_ol) Pd2 (dba) 3,0.023g stirring for five minutes under fully dissolved (0.005mmol) Xphos, at room temperature, wherein Pd2 (dba) 3 were added under nitrogen, and then quickly added 0.48g (3mmol) 3- bromo pyridine, 80 ° C oil bath and the reaction 19h, TLC analysis of the reaction end point is determined. After the reaction, 30mL of ice water was added, shaken well, 2〇mL ethyl acetate was added, liquid separation, was extracted with 30ml ethyl acetate (10ml each, extracted three times) and the combined organic phases were scaled lg / ml was added anhydrous dried over sodium sulfate, filtered, and the filtrate was rotary evaporated to give a pale yellow solid which was recrystallized from 20ml of n-hexane to give a white solid that is 17- (3-pyridyl) – androsta-5,16-diene–3P- alcohol, 42 • 6% yield.
[0035] D, Synthesis of abiraterone acetate
[0036] In 0.41gl7- successively added 100mL round bottom flask (3-pyridyl) -! -33- androst-5,16-diene-ol, ^ 3〇 diethyl ether, 0.2mL (0 • 3 round 〇1 ) of triethylamine, stir, slowly added dropwise 0.3mL (2mmol) of acetyl chloride, the reaction was stirred at room temperature for 3h, TLC analysis of the reaction end point is determined. The mixture was then suction filtered, the filtrate was decolorized with charcoal, rotary evaporation, to give a white solid, i.e. abiraterone acetate product yield of 81 • 2%.
[0037] Example 3
[0038] A, were added in a 100ml round-bottomed flask 1.08g (5.7mmo 1) p-toluenesulfonyl chloride, 15 mL of toluene, sufficiently stirred to dissolve to give the solution X, at 15 ° C was slowly added dropwise 1 • 8mL (13mmo 1 ) of 80% hydrazine hydrate to the solution X, dropwise within 5min; reaction was continued for 30min, a white precipitate appeared in the flask. TLC analysis of the reaction end point is determined. After completion of the reaction, water 30ml 10 ° C with stirring, filtered off with suction, the filter cake was then washed with purified water 3-5 times, and dried to obtain a white crystalline p-toluene sulfonyl hydrazide 0.94g, 85.6% (Yield literature values: yield 92%).
[0039] B, DHEA -17- Synthesis of p-toluenesulfonyl hydrazone
[0040] 1 • 18g were added in a 100ml round bottom flask (3.3 mmol) Dehydroepiandrosterone, 25 mL of methanol, 1.28 g of p-toluene sulfonyl hydrazide, fully dissolved with stirring at room temperature, was added O.lmL 0.2mol • I / a sulfate, an oil bath at reflux for 2h, TLC analysis of the reaction end point is determined. After completion of the reaction, the solvent was largely removed by rotary evaporation, a heavy white precipitate appeared in the flask. Was added 30mL of water, filtered off with suction, the filter cake was then washed with purified water 3-5 times, 5 (TC drying under removal of water, to give a white solid 1.51g i.e. Dehydroepiandrosterone p-toluenesulfonyl hydrazone -17- yield 80.9%.
[0041] C, 17- (3- pyridyl) – androst _5,16- diene synthetic alcohols -3P-
[0042] Add l_45g (3.2mmol) -17- Dehydroepiandrosterone p-toluenesulfonyl hydrazone, 25mLl, 4- dioxane, 0 • 35g in 100ml round bottom flask (4mmol) of lithium tert-butoxide, 0.012 g (0.013mmol) Pd2 (dba) 3,0.023g (0.005ramol) Xphos, fully dissolved after stirred at room temperature for five minutes, wherein Pd2 (dba) 3 were added under nitrogen, then added rapidly 0 • 60g (4mmol) 3 – bromopyridine, 120 ° C oil bath and the reaction 9h, TLC analysis of the reaction end point is determined. After the reaction, 30mL of ice water was added, shaken well, was added 20mL of ethyl acetate, separated, extracted with 30ml ethyl acetate (10ml each, extracted three times) and the combined organic phases were scaled lg / ml was added over anhydrous sodium to intervene sulfate, filtered, and the filtrate rotary evaporated to give a pale yellow solid which was recrystallized from burning 2〇ml n-hexyl, i.e. 17_ to give a white solid (3-Jie ratio piperidyl) – androst -5,16_ diene -30- alcohols, a yield of 43.1%.
[0043] D, Synthesis of abiraterone acetate
[0044] successively added in a round bottom flask i〇〇mL 0.52gl7- (3- pyridyl) -! -30- androst-5,16-diene-ol, ^ 3〇 diethyl ether, 0.25mL (0.36mmol) triethylamine, stir, slowly added dropwise 0.35 mL (2.2 mmol) of acetyl chloride, the reaction was stirred at room temperature for 3h, TLC analysis of the reaction end point is determined. The mixture was then suction filtered, the filtrate was decolorized with charcoal, rotary evaporation, to give a white solid, i.e. abiraterone acetate product yield of 81.8%.
[0045] In each of the above embodiments, the improved synthetic route abiraterone acetate to DHEA as raw materials by the condensation of p-toluenesulfonyl hydrazide, and then reacted with 3-bromopyridine coupling occurs, acetylation, etc. 3 target product was synthesized from abiraterone acetate, 43.4% overall yield.Route and the mild reaction conditions, readily available and inexpensive raw materials, low production cost.
PAPER
Four-Step Synthesis of Abiraterone Acetate from Dehydroepiandrosterone
https://link.springer.com/article/10.1007/s11094-016-1459-1
Balaev, A.N., Gromyko, A.V. & Fedorov, V.E. Pharm Chem J (2016) 50: 404. https://doi.org/10.1007/s11094-016-1459-1
Syn 1
J Med Chem 1995,38(13),2463

Treatment of dehydroepiandrosterone 3-acetate (I) with triflic anhydride and 2,6-di-tert-butyl-4-methylpyridine provided the desired enol triflate (III) along with some 3,5-diene (II), which were separated by column chromatography. Subsequent coupling of triflate (III) with pyridylborane (IV) using bis(triphenylphosphine)- palladium(II) chloride as the catalyst afforded the (3-pyridyl)androstadiene (V), which after hydrolysis of the acetate ester with NaOH provided the target compound.
Abiraterone
-
- Synonyms:CB-7598
- ATC:L02BX03
- Use:androgen biosynthesis inhibitor for treating prostate cancer
- Chemical name:(3β)-17-(3-pyridinyl)androsta-5,16-dien-3-ol
- Formula:C24H31NO
- MW:349.51 g/mol
- CAS-RN:154229-19-3
Substance Classes
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 853-23-6 | C21H30O3 | dehydroepiandrosterone-3-acetate | Androst-5-en-17-one, 3-(acetyloxy)-, (3β)- |
| 89878-14-8 | C9H14BN | diethyl (3-pyridyl)borane | Pyridine, 3-(diethylboryl)- |
| C26H33NO2 | (3β)-acetoxy-17-(3-pyridyl)androsta-5,16-diene |
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| EU | Zytiga | Janssen Cilag, 2011 | |
| USA | Zytiga | Johnson & Johnson, 2011 |
Formulations
- tabs. 250 mg
References
-
- Potter, G. A. et al., J. Med. Chem., (1995) 38, 2463.
- US 5 604 213 (British Technology Group; 18.2.1997; appl. 30.9.1994; GB-prior. 31.3.1992).
- EP 633 893 (Brit. Technology Group; 18.1.1995; appl. 15.3.1993; GB-prior. 31.3.1992).
- WO 9 320 097 (Brit. Technology Group; 14.10.1993; appl. 15.3.1993; GB-prior. 31.3.1992).
-
large scale synthesis of acetate:
- Potter, G. A. et al., Org. Prep. Proced. Int., (1997) 29, 123.
CLIP
Abiraterone acetate, the active ingredient of ZYTIGA is the acetyl ester of abiraterone. Abiraterone is an inhibitor of CYP17 (17α-hydroxylase/C17,20-lyase). Each ZYTIGA tablet contains either 250 mg or 500 mg of abiraterone acetate. Abiraterone acetate is designated chemically as (3β)-17-(3-pyridinyl) androsta-5,16-dien-3-yl acetate and its structure is:
 |
Abiraterone acetate is a white to off-white, non-hygroscopic, crystalline powder. Its molecular formula is C26H33NO2 and it has a molecular weight of 391.55. Abiraterone acetate is a lipophilic compound with an octanol-water partition coefficient of 5.12 (Log P) and is practically insoluble in water. The pKa of the aromatic nitrogen is 5.19.
ZYTIGA tablets are available in 500 mg film-coated tablets, 250 mg film-coated tablets and 250 mg uncoated tablets with the following inactive ingredients:
- 500 mg film-coated tablets: colloidal silicon dioxide, croscarmellose sodium, hypromellose, lactose monohydrate, magnesium stearate, silicified microcrystalline cellulose, and sodium lauryl sulfate. The coating, Opadry® II Purple, contains iron oxide black, iron oxide red, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
- 250 mg film-coated tablets: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate. The coating, Opadry® II Beige, contains iron oxide red, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
- 250 mg uncoated tablets: colloidal silicon dioxide, croscarmellose sodium, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and sodium lauryl sulfate.
PAPER
A CONVENIENT, LARGE-SCALE SYNTHESIS OF ABIRATERONE ACETATE [3β-ACETOXY-17-(3-PYRIDYL)ANDROSTA-5,16-DIENE], A POTENTIAL NEW DRUG FOR THE TREATMENT OF PROSTATE CANCER
Organic Preparations and Procedures International , The New Journal for Organic Synthesis , Volume 29, 1997 – Issue 1
https://www.tandfonline.com/doi/abs/10.1080/00304949709355175
- 10.1080/00304949709355175
- 3P-Acetoxy- 17-(3-pyridyl)androsta-5,16-diene (abiraterone acetate, 5) is a pro-drug for 17- (3-pyridyl)androsta-5,16-dien-3P-ol (abiraterone, 4), a potent inhibitor of human cytochrome P450,,, (steroidal 17a-hydroxylase-C,,,,,-lyase).’ This enzyme is a potential target for a drug designed to treat hormone-dependent prostatic carcinoma, and 5 has been approved for clinical trial in patients with this cancer. In connection with this trial, a method amenable to the synthesis of 5 on a large scale (ca. 100 g or more) was required. The present paper reports such a synthesis.

The key step in the previously reported’ synthesis of 5 was the palladium-catalysed crosscoupling reaction between diethyl(3-pyridy1)borane and the 17-en01 triflate derived from the 3-acetate of dehydroepiandrosterone 1. The procedure has potential drawbacks as a method for large-scale synthesis. Aside from the use of the expensive and noxious triflic anhydride, the formation of the enol triflate requires the expensive hindered base 2,6-di-tert-b~tyl-4-methylpyridine.~ Further, it was accompanied by some elimination of acetic acid to give androsta-3,5,16-trien- 17-yl triflate which required chromatographic separation from the desired product, and contributed to reducing its isolated yield from the acetate of 1 to a moderate 58%. These problems prompted consideration of an altemative steroidal precursor suitable for the cross-coupling reaction. It occurred to us that the vinyl iodide 3 might provide a viable alternative to an enol triflate in the palladium catalyzed cross-coupling step. Such steroidal vinyl iodides are easily and cheaply obtained via the corresponding 17-h~drazones.’-~ The synthesis of 3 iself from the hydrazone3 2 by oxidation with iodine in the presence of a hindered guanidine base has been optimi~ed~.~ to obtain the product on a small scale (0.13 g) in 95% yield. We were able to repeat this reaction on a large scale and obtain a similar yield of 3. The palladium catalysed cross-coupling reaction of 3 with diethyl(3-pyridy1)borane proceeded without the need to protect the 3-hydroxyl function to give 4, whereas the use of an enol triflate in the coupling reaction does not conveniently allow this option. However, coupling with the iodide was much slower, requiring 4 days at 80″ as compared with the 1 hr required when an enol triflate precursor was used.’ RO A ‘ OR Recrystallization of the crude 4 obtained by the foregoing procedure gave a product with melting point lower than that previously reported,’ and TLC revealed a less mobile contaminant that was not removed by further recrystallization. The crude product was therefore acetylated to give the crude target compound 5 contaminated with a by-product. This by-product was 6, formed from a precursor 7 present as a contaminant in crude 4. The prolonged reaction time required for the cross-coupling reaction using the vinyl iodide 3 had enabled a Heck-type reaction7 to occur between the initial product 4 and the bis(tripheny1phosphine)- palladium derivative of 3 to form 7. The very recently reported8 palladium-catalysed dimerisation of 17-i0dod’~-steroids to give 16,17′-coupled products provides a precedent for this side-reaction. Whereas column chromatography on silica-gel of crude 5 afforded pure 6, which was eluted first, compound 6 contaminated later fractions and could not be completely removed from 5 by recrystallization. However subsequent reverse phase chromatography allowed the complete separation of the now faster eluting 5 from 6, and recovery of >lo0 g of pure 5 by batchwise chromatography of the crude product. The by-product 6 was deacetylated to give 7, the contaminant present in 4 prepared by the present route. Neither of the new compounds 6 and 7 was appreciably inhibitory towards the human cytochrome P450,,, (S. E. Barrie, personal communication). The availability of pure 7 affords the option of exploring the purification of 4 prior to acetylation. However, for chromatographic purification, the greater solubilities of 5 and 6 in suitable organic solvents compared with their non-acetylated counterparts favor the present choice of purification after acetylation.
3P-Acetoxy-17-(3-pyridyl)androsta-5,16-diene ( 5) and 3~-acetoxy-16-(3~-acetoxyandrosta-5,16- dien-17-yl)-17-(3-pyridyl)androsta-5,16-diene (6).- To a stirred suspension of the product from the foregoing reaction (36.5 g, 0.104 mol) in dry pyridine (200 mL) in a 500 mL round-bottomed flask was added acetic anhydride (75 mL) and the mixture stirred at room temperature for 24 hrs. The pyridine and excess acetic anhydride was removed on a rotary evaporator, initially at water pump pressure with the water bath at 70 “, and finally under high vacuum at 80″ for 30 min. The resulting oil was dissolved in Et,O (500 mL), washed with saturated aqueous NaHCO, (2 x 200 mL), dried (N%CO,), and concentrated to an oil which crystallised on standing. The crude 5 was partly purified by preparative flash chromatography on silica gel using a 9 cm diameter column, eluting with dichloromethane. A by-product (6) eluted first and was followed by fractions variously enriched in 5. The foregoing reaction and purification procedure was carried out a total of four times, thus using a total of 146 g (0.41 8 mol) of crude 4. The dichloromethane fractions containing the least by-product were combined and concentrated. Recrystallisation from hexane afforded product (1 08 g) consisting of 5 containing 6.8% w/wof 6 as impurity as determined by analytical HPLC. The more contaminated fractions similarly afforded product (25 g) containing 21 3% w/w of 6 (we thank Dr C. P. Quarterman, Aston Molecules Ltd, Birmingham U.K. for these analyses). A pure sample of 6 (4 g) was isolated from the combined initial fractions as pale yellow crystals, mp. 269-270” (from hexane); IR 1732 cm-‘ (GO str); ‘H-NMR: 6 0.85 (s, 3, H-18′), 1.02, 1.04 (2s, 6, H-19,19′), 1.06 (s, 3, H-18), 2.034, 2.039 (2s, 6, CH,CO), 4.59 (2m, 2, H-3,3’), 5.13 (s, 1, H-16), 5.39 (dd, 2, H-6,6), 7.62 (dd, 1, Js,4 = 8.0 Hz, Js,6 = Anal. Calcd for C,,H,,NO,: C, 80.18; H, 8.73; N, 1.99. Found: C, 80.19; H, 8.78; N, 1.95
The crude 5 was purified by reverse-phase column chromatography. A solution of material from the 108 g batch (10 g) in a hot mixture of acetonitrile (200 mL) and methanol (40 mL) was allowed to cool and filtered. The filtrate was applied to a 10 cm diameter column (500 g) of LiChroprep@ RP-8 reverse-phase C, packing Art. No. 9324. The column was eluted with acetonitrile-0.05 M ammonium acetate (20: 1) with a flow rate of 25 amin and 500 mL fractions were collected and analysed by analytical HPLC (see below). Fractions 4-10 contained pure 5. After a further two fractions, the eluant was changed to acetonitrile-acetic acid (20:l) and pure 6 was completely eluted in fractions 16-19. In 3 subsequent runs using the same column, 25 g portions of the same batch of crude 5 were each dissolved in a mixture of hot acetonitrile (350 mL) and methanol (100 mL) and processed as before. Fractions 2-7 contained pure 5 and, following the change of solvent, fractions 8-12 contained 6. The four eluates containing 5 were combined and recrystallised from acetonitrile (1200 mL) to give pure 5 (57.5 g), mp. 146-148″, lit.’ mp. 144-145′, in which 6 was not detected by analytical HPLC (for procedure, see below) at the limit of detection (<0,05% w/w 6). Anal. Calcd for C,,H,,NO,: C, 79.75; H, 8.50; N, 3.58. Found: C, 79.73; H, 8.48; N, 3.62
Further material (14 g) from the 108 g batch was combined with a portion (22 g) of the more impure 25 g batch and the total of 36 g was chromatographed in one batch as above, again giving complete separation of 5 from 6. Recrystallisation from acetonitrile (600 mL) gave a further 28.5 g of 5 of purity equal to the foregoing crop of 57.5 g 5. Concentration of the combined mother liquors from these crops followed by addition of water (MeCN:&O, 12:l v/v) gave further pure 5 (17.5 g). The total recovery of pure 5 was therefore 103.5 g (36% based on 3). The spectroscopic data (NMR, IR, and MS) of the final products from this procedure were identical with those reported for the product obtained by the route previously described.’
Procedure for Analysis of Purity of Batches of 5 Using Analytical HPLC.- The eluant was acetonitrile-0.05M ammonium acetate and the flow rate 1.5 mumin. Components were monitored either by fluorescence detection (excitation wavelength hex 262 nm, emission wavelength hem 353 nm) or by UV detection (254 nm). Typical retention times were: for 5,225 sec; for 6, 1162 sec. For analysis of crystalline products, a solution (5 mg/mL) in acetonitrile was diluted 50 fold to 100 pg/mL and 100 pl of this solution was injected onto the column.
1 G. A. Potter, S. E. Barrie, M. Jarman and M. G. Rowlands, J. Med. Chem., 38,2463 (1995).
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q Janssen Biotech, Inc. “ZYTIGA Prescribing Information”(PDF). Horsham, PA: U.S. Food and Drug Administration.
- ^ Jump up to:a b c d “Meeting Library – Meeting Library”. meetinglibrary.asco.org.
- ^ Jump up to:a b c d Benoist GE, Hendriks RJ, Mulders PF, Gerritsen WR, Somford DM, Schalken JA, van Oort IM, Burger DM, van Erp NP (November 2016). “Pharmacokinetic Aspects of the Two Novel Oral Drugs Used for Metastatic Castration-Resistant Prostate Cancer: Abiraterone Acetate and Enzalutamide”. Clin Pharmacokinet. 55 (11): 1369–1380. doi:10.1007/s40262-016-0403-6. PMC 5069300
 . PMID 27106175.
. PMID 27106175. - Jump up^ Potter et al. J. Med. Chem., 1995, 38 (13), pp 2463–2471
- ^ Jump up to:a b Scowcroft H (2011-09-21). “Where did abiraterone come from?”. Cancer Research UK. Retrieved 2011-09-28.
- ^ Jump up to:a b “Drugs@FDA – FDA Approved Drug Products – Zytiga”. http://www.fda.gov. United States Food and Drug Administration. Retrieved 4 March 2016.
- ^ Jump up to:a b “FDA approves Zytiga for late-stage prostate cancer” (Press release). United States Food and Drug Administration. 2011-04-28.
- Jump up^ NCI Staff. “Abiraterone Approved for Earlier Use in Men with Metastatic Prostate Cancer”. U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute.
- ^ Jump up to:a b c d e “Abiraterone”. Drugs.com.
- Jump up^ “Generic Zytiga Availability”. Drugs.com.
- ^ Jump up to:a b c “Zytiga (abiraterone acetate) tablet [Janssen Biotech, Inc.]”. DailyMed. Janssen Biotech, Inc. September 2013. Retrieved 24 January 2014.
- ^ Jump up to:a b c d “Zytiga: EPAR – Product Information” (PDF). European Medicines Agency. Janssen-Cilag International N.V. 29 October 2013. Retrieved 24 January 2014.
- ^ Jump up to:a b c “Zytiga 250 mg tablets – Summary of Product Characteristics”. electronic Medicines Compendium. Janssen-Cilag Ltd. 21 January 2014. Retrieved 24 January 2014.
- ^ Jump up to:a b c “Zytiga abiraterone acetate product information” (PDF). TGA eBusiness Services. Janssen-Cilag Pty Ltd. 1 March 2012. Retrieved 24 January 2014.
- Jump up^ “Pharmaceutical Benefits Scheme – Abiraterone”. Pharmaceutical Benefits Scheme. Retrieved 24 January 2014.
- Jump up^ Clinical trial number NCT00638690 for “Abiraterone Acetate in Castration-Resistant Prostate Cancer Previously Treated With Docetaxel-Based Chemotherapy” at ClinicalTrials.gov
- Jump up^ de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Fléchon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI (May 2011). “Abiraterone and increased survival in metastatic prostate cancer”. The New England Journal of Medicine. 364 (21): 1995–2005. doi:10.1056/NEJMoa1014618. PMC 3471149 . PMID 21612468.
- Jump up^ Clinical trial number NCT00887198 for “Abiraterone Acetate in Asymptomatic or Mildly Symptomatic Patients With Metastatic Castration-Resistant Prostate Cancer” at ClinicalTrials.gov
- Jump up^ “BTG and Ortho Biotech’s Prostate Cancer Trial Unblinded”. Genetic Engineering & Biotechnology News. 2010-09-10. Retrieved 2011-05-26.
- Jump up^ Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, Carles J, Mulders PF, Basch E, Small EJ, Saad F, Schrijvers D, Van Poppel H, Mukherjee SD, Suttmann H, Gerritsen WR, Flaig TW, George DJ, Yu EY, Efstathiou E, Pantuck A, Winquist E, Higano CS, Taplin ME, Park Y, Kheoh T, Griffin T, Scher HI, Rathkopf DE (January 2013). “Abiraterone in metastatic prostate cancer without previous chemotherapy”. The New England Journal of Medicine. 368 (2): 138–48. doi:10.1056/NEJMoa1209096. PMC 3683570 . PMID 23228172.
- ^ Jump up to:a b c d e “Zytiga prescribing information” (pdf). Janssen Biotech. May 2012. Retrieved 4 March 2016.
- ^ Jump up to:a b c d e “Zytiga (abiraterone) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 24 January 2014.
- ^ Jump up to:a b Neidle S (30 September 2013). Cancer Drug Design and Discovery. Academic Press. pp. 341–342. ISBN 978-0-12-397228-6.
- Jump up^ Fernández-Cancio, Mónica; Camats, Núria; Flück, Christa E.; Zalewski, Adam; Dick, Bernhard; Frey, Brigitte M.; Monné, Raquel; Torán, Núria; Audí, Laura (2018-04-29). “Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency”. Pharmaceuticals. 11 (2): 37. doi:10.3390/ph11020037.
- Jump up^ Attard G, Belldegrun AS, de Bono JS (December 2005). “Selective blockade of androgenic steroid synthesis by novel lyase inhibitors as a therapeutic strategy for treating metastatic prostate cancer”. BJU International. 96 (9): 1241–6. doi:10.1111/j.1464-410X.2005.05821.x. PMID 16287438.
- ^ Jump up to:a b c Small EJ (November 2014). “Can targeting the androgen receptor in localized prostate cancer provide insights into why men with metastatic castration-resistant prostate cancer die?”. Journal of Clinical Oncology. 32 (33): 3689–91. doi:10.1200/JCO.2014.57.8534. PMID 25311216.
Abiraterone acetate is a prodrug for abiraterone, a CYP17 inhibitor, which has the capacity to lower serum testosterone levels to less than 1 ng/dL (compared with levels closer to 20 ng/dL that are achieved with conventional ADT).19 […] Relative to LHRHa alone, the addition of abiraterone resulted in an 85% decline in dihydrotestosterone (DHT) levels, a 97% to 98% decline in dehydroepiandrosterone (DHEA) levels, and a 77% to 78% decline in androstenedione levels.
- Jump up^ Tindall DJ, James M (20 April 2009). Androgen Action in Prostate Cancer. Springer Science & Business Media. pp. 748–. ISBN 978-0-387-69179-4.
- Jump up^ Yin L, Hu Q (January 2014). “CYP17 inhibitors–abiraterone, C17,20-lyase inhibitors and multi-targeting agents”. Nature Reviews. Urology. 11 (1): 32–42. doi:10.1038/nrurol.2013.274. PMID 24276076.
- Jump up^ Malikova, Jana; Brixius-Anderko, Simone; Udhane, Sameer S.; Parween, Shaheena; Dick, Bernhard; Bernhardt, Rita; Pandey, Amit V. (2017). “CYP17A1 inhibitor abiraterone, an anti-prostate cancer drug, also inhibits the 21-hydroxylase activity of CYP21A2”. The Journal of Steroid Biochemistry and Molecular Biology. 174: 192–200. doi:10.1016/j.jsbmb.2017.09.007. ISSN 1879-1220. PMID 28893623.
- Jump up^ Udhane, Sameer S.; Dick, Bernhard; Hu, Qingzhong; Hartmann, Rolf W.; Pandey, Amit V. (2016). “Specificity of anti-prostate cancer CYP17A1 inhibitors on androgen biosynthesis”. Biochemical and Biophysical Research Communications. 477 (4): 1005–1010. doi:10.1016/j.bbrc.2016.07.019. ISSN 1090-2104. PMID 27395338.
- ^ Jump up to:a b Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N (July 2015). “Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer”. Nature. 523 (7560): 347–51. doi:10.1038/nature14406. PMC 4506215 . PMID 26030522.
- ^ Jump up to:a b Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, Balk SP, Taplin ME, Auchus RJ, Sharifi N (May 2016). “Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy”. Nature. 533 (7604): 547–51. doi:10.1038/nature17954. PMID 27225130.
- ^ Jump up to:a b c Capper CP, Larios JM, Sikora MJ, Johnson MD, Rae JM (May 2016). “The CYP17A1 inhibitor abiraterone exhibits estrogen receptor agonist activity in breast cancer”. Breast Cancer Research and Treatment. 157 (1): 23–30. doi:10.1007/s10549-016-3774-3. PMID 27083183.
- Jump up^ Alesini D, Iacovelli R, Palazzo A, Altavilla A, Risi E, Urbano F, Manai C, Passaro A, Magri V, Cortesi E (2013). “Multimodality treatment of gynecomastia in patients receiving antiandrogen therapy for prostate cancer in the era of abiraterone acetate and new antiandrogen molecules”. Oncology. 84 (2): 92–9. doi:10.1159/000343821. PMID 23128186.
- Jump up^ Figg WD, Chau CH, Small EJ (14 September 2010). Drug Management of Prostate Cancer. Springer Science & Business Media. p. 97. ISBN 978-1-60327-829-4.
- Jump up^ Rosenthal L, Burchum J (17 February 2017). Lehne’s Pharmacotherapeutics for Advanced Practice Providers – E-Book. Elsevier Health Sciences. p. 936. ISBN 978-0-323-44779-9.
- Jump up^ Yip CK, Bansal S, Wong SY, Lau AJ (April 2018). “Identification of Galeterone and Abiraterone as Inhibitors of Dehydroepiandrosterone Sulfonation Catalyzed by Human Hepatic Cytosol, SULT2A1, SULT2B1b, and SULT1E1”. Drug Metabolism and Disposition. 46 (4): 470–482. doi:10.1124/dmd.117.078980. PMID 29436390.
- Jump up^ “A new way to treat prostate cancer: The story of abiraterone”. The Institute of Cancer Research. 2012-09-10. Retrieved 2012-11-12.
- Jump up^ “Abiraterone Acetate (CB7630)”. Cougar Biotechnology. Archived from the original on 7 September 2008. Retrieved 2008-08-20.
- Jump up^ “Johnson & Johnson Announces Definitive Agreement to Acquire Cougar Biotechnology, Inc” (Press release). Cougar Biotechnology. 2009-05-11. Archived from the original on 29 May 2009. Retrieved 2009-06-03.
- Jump up^ “FDA Approval for Abiraterone Acetate”. U.S. Department of Health and Human Services, National Institutes of Health, National Cancer Institute.
- Jump up^ “EMA assessment of Zytiga (abiraterone)”. European Medicines Agency.
- Jump up^ “Prostate cancer (metastatic, castration resistant) – abiraterone (following cytoxic therapy): final appraisal determination guidance” (PDF). NICE guidance. 15 May 2012. Archived from the original (PDF) on February 19, 2013.
- Jump up^ “NICE technology appraisal guidance [TA259]”. NICE guidance. June 2012.
- Jump up^ “NICE appraisal of earlier treatment with abiraterone for prostate cancer”. NICE press release. 14 August 2014.
- ^ Jump up to:a b c “Abiraterone acetate – Johnson & Johnson”. Adis Insight.
- Jump up^ “Abiraterone acetate – Churchill Pharmaceuticals”. Adis Insight.
External links
 |
|
| Clinical data | |
|---|---|
| Trade names | Zytiga, others |
| Synonyms | CB-7630; JNJ-212082; 17-(3-Pyridinyl)androsta-5,16-dien-3β-ol acetate |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a611046 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth (tablets)[1] |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Unknown, but may be 50% at most on empty stomach[3] |
| Protein binding | Abiraterone: ~99.8% (to albumin and α1-AGp)[3][1][2] |
| Metabolism | Esterases, CYP3A4, SULT2A1[2] |
| Metabolites | Abiraterone, others[1][3] |
| Elimination half-life | Abiraterone: 12–24 hours[1][3] |
| Excretion | Feces: 88%[1][2] Urine: 5%[1][2] |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C26H33NO2 |
| Molar mass | 391.555 g/mol |
| 3D model (JSmol) | |
| Melting point | 144 to 145 °C (291 to 293 °F) [4] |
