
Home » INDIA 2020
Category Archives: INDIA 2020
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Azelnidipine
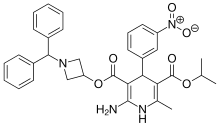
Azelnidipine
C33H34N4O6, 582.6 g/mol
CAS 123524-52-7
3-(1-Benzhydrylazetidin-3-yl) 5-isopropyl 2-amino-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate
CS-905, RS-9054
Approved India cdsco 2020
SYN REF https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4245158/
MP 95-98 °C AND NMR WO 2004058745 . EP 266922
Azelnidipine is a dihydropyridine calcium channel blocker. It is marketed by Daiichi-Sankyo pharmaceuticals, Inc. in Japan. It has a gradual onset of action and produces a long-lasting decrease in blood pressure, with only a small increase in heart rate, unlike some other calcium channel blockers. It is currently being studied for post-ischemic stroke management.
Azelnidipine (INN; marketed under the brand name CalBlock — カルブロック) is a dihydropyridine calcium channel blocker. Azelnidipine is L and T calcium channel blocker. It is sold in Japan by Daiichi-Sankyo pharmaceuticals, Inc. Unlike nicardipine, it has a gradual onset and has a long-lasting hypotensive effect, with little increase in heart rate. Drug Controller General Of India (DCGI) has approved the use of azelnipine in India. It is launched under the brand name Azusa (ajanta pharma ltd.)[1] In 2020.
Chemical Synthesis
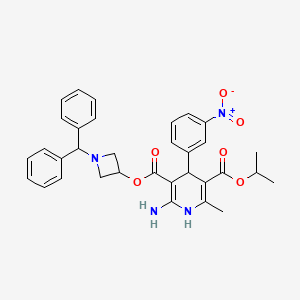
A solution of benzhydrylamine (46) and epichlorohydrin (47) was mixed without adding solvent to give azetidinol 48 in 57% yield. DCC coupling between cyanoacetic acid (49) and azetidinol 48 in hot THF gave ester 50 in 93% yield. Cyanoester 50 was treated with ethanol and HCl gas in chloroform to give imidate HCl salt 51, which was treated with ammonia gas in chloroform and ammonium acetate in acetonitrile to give the corresponding amidinoacetate 52. A modified Hantzsch reaction was employed to construct the 2-amino-1,4- dihydropyridine core structure. Compound 52 was condensed with 2-(3-nitrobenzylidene)acetic acid isopropyl ester (55) in the presence of NaOMe in refluxing isopropanol to give the cyclized product, azelnidipine (V) in 74% yield. Benzylideneacetoacetate 55 was obtained through the Knoevenagel reaction employing 3-nitrobenzaldehyde (53) and isopropyl acetoacetate (54) in isopropanol containing a catalytic amount of piperidinium acetate at 45-55oC in 65% yield.
PATENT
EP 266922
IN 201621044802
CN 106279109
CN 107188885
CN 105461691
CN 103509003
CN 103183663
CN 102382104
JP 2012020970 A
PAPER
Bioanalysis (2019), 11(4), 251-266.
PAPER
Asian Journal of Chemistry (2014), 26(15), 4675-4678.
PAPER
http://www.asianjournalofchemistry.co.in/User/ViewFreeArticle.aspx?ArticleID=26_16_30

Azelnidipine is designated chemically as 3-(1-benzhydrylazetidin-3-yl)-5-isopropyl-2-amino-6-methyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate. Its literature synthesis (Scheme-I) involves 3-nitrobenzaldehyde 5 with isopropyl acetoacetate 6. The product of (Z)-isopropyl 2-(3- nitrobenzylidene)-3-oxobutanoate (7a, b, c), on treatment with piperidine and acetic acid, coupling of (7) and 1-benzhydrylazetidin-3-yl 3-amino-3-iminopropanoate acetate (8) gave azelnidipine (1).
PAPER
International Research Journal of Pharmacy (2012), 3(8), 191-192.
Chemical & Pharmaceutical Bulletin (1995), 43(5), 797-817.
PATENT
https://patents.google.com/patent/WO2014139410A1/en
The invention belongs to the technical field of medicine and provides an important intermediate of dihydropyridine calcium antagonist adipine, 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidine The synthesis process of ester acetate. Background technique
Azelnidipine is a new type of dihydropyridine calcium channel blocker developed by Sankyo and Ube Industries of Japan. It was approved for sale in Japan in late May 2003 under the trade name Calblock. Adipine has a selective blockade of calcium channels in arterial smooth muscle cells, it can dilate blood vessels, reduce peripheral vascular resistance and arterial pressure, and is widely used clinically for mild or moderate essential hypertension, renal disorders with hypertension And treatment of severe hypertension. Compared with nicardipine and nifedipine dihydropyridine calcium channel blockers, adipine is superior in selectivity, long-lasting and long-lasting, and has little effect on the heart.

阿折地平的结构式
A flat floor structure
At present, references to the preparation of agdipine include: European patents EP0266922; Chinese patent CN201010516967.7; Chinese Journal of Medicinal Chemistry, 2010, 20 (3): 192-194; Chinese Journal of Pharmaceutical Industry, 2008, 39 (3): 163-165; Chemical Industry and Engineering, 2009, 26 ( 1 ): 15-18; Qilu Pharmacy, 2005, 24 (6): 365-366. The preparation method of adipine in these literatures is based on the reaction of epichlorohydrin and diphenylamine with N-alkylation, cyclization, esterification, Pinner synthesis, neutralization, and oxime reaction. The intermediate 3-amino-3-iminopropionic acid-1-(diphenylfluorenyl)-3-azetidinyl acetate is prepared first, followed by 2-(3-nitrobenzylidene)acetyl Acepinedipine was obtained by the Hantzsch condensation of isopropyl acetate.
The control of the solvent and reaction conditions in the esterification, Pinner synthesis and neutralization three-step reaction in this route is critical. Using the preparation methods provided by these documents, we found that the operation was cumbersome and the yield and purity were not satisfactory.
In the esterification reaction, according to the method specifically reported in the above literature, the highest yield of the obtained product is only 85%, and the purity is poor, it is difficult to purify, and it is difficult to obtain a solid product.

副产物 (7 )和(8 )结构式 发明内容 We have found that 3-amino-3-iminopropionic acid-1- (3) is prepared by a three-step reaction from cyanoacetate-1-diphenylhydrazin-3-azetidinyl ester (3) according to the method specifically reported in the above literature. Diphenylhydrazino)-3-azetidinyl acetate (6), the reaction operation is cumbersome, and it is easy to produce by-products of hydrolysis of ester bonds and hydrolysis of imid bonds (7) and (8), three-step reaction. The total yield is only 20~30%, and the purification of the product is difficult, which seriously affects the quality of the final product and greatly increases the production cost.
Byproducts (7) and (8) structural formula Summary of the invention
It is an object of the present invention to provide a process for the preparation of the key intermediate of adipine, 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidinyl acetate. The adipine intermediate of the present invention 3-amino-3-iminopropionic acid-1-(diphenylhydrazinyl)-3-azetidinyl acetate acetate has the following structural formula:

The preparation method of 3-amino-3-iminopropionic acid-1-(diphenylindenyl)-3-azetidinyl acetate of the present invention comprises the following steps: 1) Esterification: 1-diphenylhydrazin-3-azetidinol (2), cyanoacetic acid (1) and N,N-dicyclohexylcarbodiimide (DCC) in organic solvent at 0~ Reacting at 80 ° C, to obtain 7-diphenylindolyl-3-azetidinyl cyanoacetate (3);
2) Pinner reaction: Add intermediate (3), absolute ethanol to dichlorosilane, stir and cool
To -20~25 °C, dry hydrogen chloride gas is passed, and then the reaction solution is kept sealed at -20~25 °C to obtain 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl) -3-azetidinyl ester hydrochloride (4);
3) Neutralization reaction: The intermediate (4) is dissolved in dichloromethane, and the base is added at -5 to 25 ° C to obtain 3-imino-3-ethoxypropionic acid-1-(diphenylhydrazine). Benzyl-3-azetidinyl ester (5);
4) Formation reaction: The intermediate (5) is dissolved in acetonitrile, ammonium acetate is added, and the temperature is raised to 40 to 60 ° C to obtain 3-amino-3-iminopropionic acid-1-(diphenylfluorenyl)-3. – azetidinium acetate compound (6). detailed description
Example
1. Preparation of cyanoacetic acid-1-diphenylhydrazine-3-azetidine (esterification)

Method 1: Add 1-diphenylhydrazin-3-azetidinol (2, 235 g, 0.983 mol) and cyanoacetic acid (1, 100 g, 1.18 mol) to 1.5 mL of dichloromethane, and stir until fully dissolved. Ν, Ν-dicyclohexylcarbodiimide (DCC, 243 g, 1.18 mol) was added at 0-10 ° C and allowed to react at room temperature for 3 h. After the completion of the reaction, the reaction mixture was cooled to 0 to 5 ° C, and filtered, filtered, washed with a small portion of dichloromethane. The organic solvent was evaporated to dryness under reduced pressure and dried to give 275 g of white solid.
Method 2: chloroform was used as the reaction solvent, and the operation was the same as above, and the reaction was carried out at 55 ° C for 5 hours, the HPLC purity was 98.7%, and the product yield was 95.3%.
Method 3: Ethyl acetate was used as the reaction solvent, and the operation was the same as above, and the reaction was carried out at 55 ° C for 2 h, the HPLC purity was 98.9%, and the product yield was 96.1%.

Method 4: Using hydrazine as the reaction solvent, the operation was the same as above, and the reaction was carried out at 55 ° C for 7 h, the HPLC purity was 98.5%, and the product yield was 94.7%. 2. Preparation of 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl)-3-azetidinyl ester hydrochloride (Pinner reaction)
Intermediate 3 (270 g, 0.882 mol), absolute ethanol (61.8 mL, 1.06 mol) was added to 1.5 L of dry dichloromethane, cooled to -5 to 0 ° C in a water salt bath, and dried. HC1 gas for 2.5 h, after the completion of the aeration, the reaction solution was kept under stirring at 0 ° C for 6 h.
Allow to stand overnight at 0-4 °C. After completion of the reaction, the solvent was evaporated under reduced pressure to give an oily viscous intermediate 4 .
3. Preparation of 3-imino-3-ethoxypropionic acid-1-(diphenylfluorenyl)-3-azetidinyl ester

Method 1: Add 1.4 L of dichloromethane to Intermediate 4, cool to 0-5 ° C, add dry diethylamine (182 mL, 1.76 mol) to the solution, adjust pH 7-8, continue to stir after the dropwise addition. 2h. The mixture was suction filtered, and the filtrate was evaporated to dryness vacuo.
Method 2: Diamine is used for neutralization, and the operation is the same as above.
Method 3: Triethylamine is used for neutralization, and the operation is the same as above.
Method 4: Ethylenediamine is used for neutralization, and the operation is the same as above.
Method 5: Add 1.4 L of dichloromethane to Intermediate 4, cool to 0-5 ° C, add potassium carbonate (242.88 g, 1.76 mol) to the solution in portions, adjust pH 7-8, continue stirring for 2 h. . The mixture was suction filtered, and the filtrate was evaporated to dryness vacuo. Method 6: Neutralize with sodium carbonate, and operate as above.
Method 7: Neutralize with sodium hydroxide, and operate as above.

4. Preparation of 3-amino-3-iminopropionic acid-1-(diphenylindenyl)-3-azetidinyl acetate (formed into 脒)
To the intermediate 5, 1.2 L of acetonitrile was added, and after dissolution, ammonium acetate (68.0 g, 0.882 mol) was added, and the mixture was heated to 55 ° C for 6 h. After the reaction, it was naturally cooled, crystallization, suction filtration, acetonitrile washing cake, and dried to give 236 g of a white solid. The total yield of the three-step reaction was 69.9 73.1%.
PAPER
https://pubs.rsc.org/en/content/articlelanding/2015/cc/c4cc09337b#!divAbstract
Abstract
A protocol for the coupling of 3-iodoazetidines with Grignard reagents in the presence of an iron catalyst has been developed. A variety of aryl, heteroaryl, vinyl and alkyl Grignards were shown to participate in the coupling process to give the products in good to excellent yields. Furthermore, a short formal synthesis towards a pharmacologically active molecule was shown.

http://www.rsc.org/suppdata/cc/c4/c4cc09337b/c4cc09337b1.pdfPATENThttps://patents.google.com/patent/CN103509003A/zhAzelnidipine, whose chemical name is 3-(1-diphenylmethylazetidin-3-yl) 5-isopropyl 2-amino-1,4-dihydro-6-methyl 4-(3-nitrophenyl)-3,5-pyridinedicarboxylate, developed by Japan Sankyo Co., Ltd. and approved to be marketed in Japan in late May 2003. The existing synthesis method of azedipine is cumbersome, and the preparation of intermediate (VI) adopts column chromatography method, and the purification of product (I) also uses column chromatography method, which is not suitable for industrial production.
A method for preparing azeldipine, which is characterized in that it is prepared by the following steps.
[0006]

Description of the drawings:
Figure 1 is a flow chart of the synthesis process of azeldipine.
[0025] Example 12-Preparation of (3-nitrobenzylidene) isopropyl acetoacetate (III)
[0026] Add 2.1kg of 3-nitrobenzaldehyde and 5L of isopropanol to the reaction kettle, start stirring, add 3kg of isopropyl acetoacetate, and stir. Add 43ml of anhydrous piperidine and 12ml of glacial acetic acid, and continue to stir until the solid is completely dissolved. Heat the temperature to 45°C and keep the reaction for 6h, then lower the temperature, stir and crystallize for 16h. Filter and collect the resulting filter cake. Put the obtained filter cake and 16L ethanol (industrial) into the reaction kettle, start stirring, beating, filtering, and collecting the filter cake. Put the filter cake in the baking tray, put it in the oven, and dry at 70-80°C. Collect the product 2-(3-nitrobenzylidene) isopropyl acetoacetate (III), about 2.7 kg.
[0027] Example 21-Preparation of benzhydryl-3-hydroxyazetidine (Intermediate V)
[0028] 9.6L of methanol, 5.4kg of benzhydrylamine (IV) and 3.33kg of epichlorohydrin were added to the reaction kettle, stirred at room temperature for 48 hours, the reaction was completed, the temperature was raised to 68°C, and the reaction was refluxed for 72h. Cool to room temperature. Concentrate under reduced pressure to remove methanol, and collect the filter cake by filtration. The filter cake was put into the reaction kettle, 19.2L of ether and 13.75L of 3mol/L NaOH solution were added, stirred, and the water layer was released after standing still. The ether layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, filtered, and the filtrate was collected. The ether was recovered under reduced pressure to dryness to obtain about 3.05 kg of 1-benzyl-3-hydroxyazetidine (Intermediate V).
[0029] Example 3 Preparation of cyanoacetic acid (1-diphenylmethylazetidin-3-yl) ester (Intermediate VI)
[0030] Put about 3.05g of intermediate (V), 27L of tetrahydrofuran and 1.7kg of cyanoacetic acid into the reactor, start stirring, turn on the chilled water of the reactor to cool down, and slowly add 3.1kgN, N’-dicyclohexyl to the reactor Diimine, control the temperature at IO0C -15°C, after the addition, close the chilled water in the reactor. Turn on the heating system, slowly increase the temperature to 55-60°C, and react for 10 hours. The material liquid was cooled to room temperature, filtered, and the filtrate was concentrated to dryness. Put 16.8L of ethyl acetate into the reaction kettle, stir to dissolve, then wash with water, dry with anhydrous sodium sulfate, filter, and collect the filtrate. Ethyl acetate was recovered under reduced pressure, petroleum ether was added to the solid residue, stirred, and filtered to obtain cyanoacetic acid (1-diphenylmethylazetidin-3-yl) ester (Intermediate VI), about 3.19 kg.
[0031] Example 4 Preparation of amidinoacetic acid (1-diphenylmethylazetidin-3-yl) ester acetate (VII)
[0032] Put 25L of dichloromethane, about 3.19kg of intermediate (VI), and 430g of ethanol into the reactor, start stirring, cool to below 0°C, and pass in hydrogen chloride gas until the temperature stabilizes below 0°C, at 0°C Let stand for 14 hours at °C. Concentrate under reduced pressure to remove most of the hydrogen chloride gas and recover the solvent dichloromethane. Add 25L of dichloromethane to the residue of the reaction kettle, stir, cool to below 0°C, and pass in ammonia until the temperature stabilizes below 0°C, and filter . The filtrate was poured into the reactor, concentrated under reduced pressure to recover the solvent to obtain a colorless liquid, added 22.8L of acetonitrile and 905g of amine acetate, heated to 55-60°C for 1.5 hours, stopped the reaction, filtered while hot, and recovered the filtrate under reduced pressure Solvent to dryness, add 3L of ether to the residue to crystallize, filter, and dry to obtain amidinoacetic acid (1-diphenylmethylazetidin-3-yl) ester acetate (Intermediate VII) about 3.2kg .
[0033] Example 5. Add about 3.2kg of Intermediate (VII), about 2.7kg of Intermediate (III), 21L of isopropanol and 585g of sodium methoxide to the reaction kettle, start stirring, heat to reflux and react for 4 hours, and cool to Below 10°C, filter, the filtrate is decompressed to recover the solvent to dryness, add 35L ethyl acetate to the residue to dissolve, wash with 6.5LX3 water, release the water layer, add anhydrous sodium sulfate to the ethyl acetate layer to dry, filter , Collect the filtrate, recover ethyl acetate under reduced pressure, add 4.2L of toluene to the residue,
3.4L of n-hexane was heated to dissolve, filtered, the filtrate was stirred to room temperature to crystallize, filtered and collected and dried, and the product was placed in an oven at 45-55°C to dry to obtain the crude azedipine (I), about 2.3kg.
[0034] Example 6, Refining
[0035] Put 8.8L ethyl acetate and 8.8L n-hexane into the reaction kettle, turn on the stirring, put about 2.3kg of the crude azeldipine into the reaction kettle, slowly heat up until the material is dissolved, add 180g of activated carbon and stir for 0.5h, while it is hot Filter, hydraulically filter the material to the crystallization dad, wash the filter cake with 5.5L ethyl acetate and 4.5L n-hexane solution, combine with the filtrate, cool to 0~5°C to crystallize, filter, collect the product, and place it in a hot air circulating oven After drying at 45-55°C, 2.2 g of azeldipine is obtained. The purity is 99.6% as measured by high performance liquid chromatography. The refined yield is 96.0%.
[0036] Example 7 Azedipin Refining
[0037] The mixed solvent was prepared according to the volume ratio of ethyl acetate and n-hexane of 2:1, 22L of the mixed solvent was put into the reactor, about 2.3kg of azedipine crude product was put into the reactor, and the temperature was slowly heated until the material was dissolved, Add 180g of activated carbon and stir for 0.5h, filter while hot, filter the material hydraulically into a crystallization kettle, wash the filter cake with a mixed solvent, combine the washing liquid with the filtrate, cool to 0~5°C for crystallization, filter, collect the product, and circulate the hot air Dry in an oven at a temperature of 45-55°C to obtain 2.2 g of azeldipine fine product, with a purity of 99.7% measured by high performance liquid chromatography.
[0038] Example 8 prepared a mixed solvent at a volume ratio of ethyl acetate and n-hexane of 1.5:1, put 22L of the mixed solvent into the reactor, put about 2.3kg of crude azeldipine into the reactor, and slowly heated to Dissolve the material, add 180g of activated carbon and stir for 0.5h, filter while it is hot, filter the material hydraulically into a crystallization kettle, wash the filter cake with a mixed solvent, combine the washing liquid and the filtrate, cool to 0~5°C to crystallize, filter, and collect the product. Dry in a hot air circulating oven at a temperature of 45-55°C to obtain
2.2g azeldipine is a fine product with a purity of 99.6% measured by high performance liquid chromatography.
PATENT
https://patents.google.com/patent/CN103183663B/zh
Azelnidipine (Azelnidipine) is a new type of dihydropyridine calcium channel blocker jointly developed by Sankyo Co., Ltd. and Ube Industries Co., Ltd., which inhibits the entry of calcium ions into excitable tissues and causes peripheral blood vessels And coronary artery vasodilation plays a role in lowering blood pressure. Clinically, it is widely used in patients with mild or moderate symptoms of primary hypertension, hypertension with renal dysfunction, and severe hypertension. Compared with similar antihypertensive drugs, azeldipine has a slow and long-lasting antihypertensive effect.
[0004] The chemical structure of azeldipine is similar to that of nifedipine:

[0006] The Chinese patent CN87107150.9 reported the compound earlier and gave a detailed introduction to its synthesis; afterwards, most of the synthesis of azeldipine adopts this route:

[0008] The reaction takes o-nitrobenzaldehyde and isopropyl acetoacetate as raw materials to prepare intermediate compound 5; takes benzhydrylamine and epichlorohydrin as raw materials to prepare compound 2, compound 2 and cyanoacetic acid act in DCC Compound 3 is prepared by the next reaction. Compound 3 is added with ethanol under the action of hydrogen chloride gas, ammonia gas ammonolysis, and acetate anion exchange to obtain compound 4. Compound 4 and compound 5 are under the action of sodium methoxide to obtain compound 1, namely azeldipine.
[0009] Wherein: Compound 3 can be purchased as an industrial product, or can be prepared according to the traditional method reported in the literature; Compound 5 is prepared according to the traditional method reported in the literature.
[0010] In the process of preparing amidine 4 in the traditional reaction route, hydrogen chloride gas and ammonia gas need to be passed in successively. Therefore, the reaction requires anhydrous reagents. According to literature reports, the reaction yield is about 70%. From the perspective of industrial synthesis, The application of anhydrous reagents will undoubtedly increase the cost, while the use of gas will increase the difficulty of operation and require the use of high-pressure equipment. At the same time, post-reaction processing is difficult and industrial production is difficult. Therefore, this step of the reaction requires further improvement.
With acetonitrile as a solvent, the crude product of reaction 2) was stirred until dissolved, ammonium acetate was added, and acetate anion exchange was performed to obtain the amidine compound 4;

[0018] The second step: use toluene as a solvent, compound 4 and compound 5 in the use of sodium amide to obtain compound 1, namely azedipine

[0020] The preferred technical solution of the present invention is characterized in that the temperature of reaction 1) is controlled below _5°C
Example 1: Preparation of azeldipine
[0030] Add 50 g of compound 3, 1500 mL of dichloromethane, and 16.64 mL of absolute ethanol to a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5 °C to saturation, and after saturation, keep the reaction at -5 °C for 24 hours. Protect from light and nitrogen, slowly add the above reaction system to 1665ml of ammonia water with a concentration of 2.5-3.0% under the control of 0-5°C. After the addition, stir for 0.5h, stand for 0.5h, and separate the liquids. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C -60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 57.55 g of amidine 4, the yield was 91.2%, the HPLC purity was 99.63%, and the melting point was 130-132.3°C.
[0031] 50g amidine 4, 43.5g compound 5, 1000mL toluene, and 7.7g sodium amide were added into a 1000mL three-necked flask, mechanically stirred, heated to reflux, and reacted for 4 hours. TLC detects that the reaction is complete and cools to room temperature to crystallize. Filter, put the solid directly into the mixed solution of toluene and n-hexane (1:1.2-1.5) without drying, heat up to reflux to clear, cool to 56°C naturally, add seed crystals, stop stirring, and cool to 25° C, filter. The solid was purified once more according to the above method, and dried under reduced pressure at 40°C for 48 hours to obtain 66.87g of α-crystal form of Azedipine, yield 88.2%, melting point: 121-123°C.
[0032] Example 2; Preparation of Azeldipine
[0033] Add 50g of compound 3, 1500mL of dichloromethane, 16·64mL of absolute ethanol into a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5°C to saturation, and after saturation, -6°C to -8°C Incubate the reaction for 24h. Under the control of 0-5 °C, slowly add the above reaction system to ammonia water with a concentration of 2.5-3.0%, adjust the pH to 7.8-8.5, after adding, stir for 0.5h, stand for 0.5h, and separate. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C-60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 59.0 lg of amidine 4 with a yield of 93.5%, an HPLC purity of 99.52%, and a melting point of 130.1-132.0°C.
[0034] 50g amidine 4, 43.5g compound 5, 1000mL toluene and 7.7g sodium amide were added to a 1000mL three-necked flask, mechanically stirred, heated to reflux, and reacted for 4 hours. TLC detects that the reaction is complete and cools to room temperature to crystallize. Filter, put the solid directly into the mixed solution of toluene and n-hexane (1:1.2-1.5) without drying, heat up to reflux to clear, cool to 56°C naturally, add seed crystals, stop stirring, and cool to 25° C, filter. The solid was refined once more according to the above method, and dried under reduced pressure at 40°C for 48 hours to obtain 68.31 g of α-crystal azedipine, yield 90.01%, melting point: 121 -123 °C.
[0035] Example 3: Preparation of Amidine 4
[0036] Add 50g of compound 3, 1500mL of dichloromethane, 16·64mL of absolute ethanol into a 5L three-necked flask, and under mechanical stirring, pass HC1 gas below -5°C to saturation, and after saturation, -7°C to -9°C Incubate the reaction for 24h. Under the control of 0-5 °C, slowly add the above reaction system to the ammonia water with a concentration of 2.5-3.0%, adjust the pH to 8.5-9.5, after adding, stir for 0.5h, stand for 0.5h, and separate. The dichloromethane layer was washed once with 2000 mL of saturated brine, left standing for 1.0 h, separated, and the dichloromethane layer was drained under reduced pressure to obtain a white solid. Without drying, it was directly added to 2000 mL of acetonitrile, and the temperature was slowly heated to dissolve. Add 11.7g of ammonium acetate, control the temperature at 55°C-60°C, and react for 2h under mechanical stirring. After cooling, the solid precipitated, filtered, and dried to obtain 59.5 g of amidine 4, HPLC purity 99.78%, melting point: 130.7-132·2°C.

Step 2: Using toluene as a solvent, compound 4 and compound 5 under the action of sodium amide to obtain compound 1, namely azeldipine

PATENThttps://patents.google.com/patent/CN102453023A/zh
detailed description
[0007] In the synthesis workshop, benzhydrylamine is used as a raw material to be synthesized by addition, cyclization, esterification, acidification, ammoniation, condensation and other reactions. The crude azeodipine is refined, dried, mixed and packaged in a clean area. Fold the ground. The specific response is as follows:
[0008] 1. Addition and cyclization reaction
[0009] Methanol, benzhydrylamine, and epichlorohydrin were added to the reaction kettle, stirred at room temperature for 24hr, the reaction was completed, the reaction was heated to reflux for 24hr, cooled, filtered to collect the precipitated solid, and then the mother liquor was concentrated to recover the raw materials, and the heating was continued to reflux 18 After hours, collect the product, add dichloromethane and H2O to the obtained solid, adjust the pH to 10-11 with 40% NaOH while stirring in an ice bath, stand still, separate the organic layer, dry with anhydrous magnesium sulfate, and recover the dichloromethane under reduced pressure To dryness, a colorless solid compound III (1-benzyl-3-hydroxyazetidine) is obtained. After improvement, the raw materials are fully reacted, and the reaction yield of this step is improved. The mass yield is 75%. % Mentioned 85%.
[0010]

[0011] 2. Esterification reaction
[0012] Add THF, compound (III), and cyanoacetic acid to the reaction kettle, stir evenly, add DCC in batches under ice bath stirring, control the temperature at 10°C~15°C, after the addition, remove the ice water bath, and slowly heat up React at 55°C~60°C for 18h. After the reaction is complete, cool, filter to remove insoluble materials, concentrate the filtrate to dryness, add ethyl acetate to the residue to dissolve, wash with water, dry with anhydrous magnesium sulfate, and recover ethyl acetate under reduced pressure. The residue was added with petroleum ether and stirred for crystallization, and the solid was collected by filtration to obtain compound IV (1-diphenylmethyl-3-azetidinyl cyanoacetate).
[0013]

[0014] 3. Acidification and amination reaction
[0015] Dichloromethane, ethanol and intermediate (IV) were added to the reaction kettle respectively, mixed and stirred, cooled to about _5 ° C in an ice salt bath, and dried hydrogen chloride gas was introduced until saturation (about 1.5 hours) after . Let stand overnight at about -5°C, recover the solvent under reduced pressure at room temperature, add dichloromethane to the residue and stir, cool to about _5°C in an ice-salt bath, pass in the dried ammonia gas until saturation (about 3 hours) , Filtration to remove the insoluble matter, and the filtrate was decompressed to recover solvent at room temperature. Acetonitrile and ammonium acetate were added to the residue respectively, and the temperature was raised to 55~60°C for 2 hours with stirring. After the reaction was completed, it was cooled and filtered. 3-Azacyclobutanylamidinoacetate acetate), the reaction in this step is controlled at about _5°C, and the transesterification
The side reaction is reduced, and the reaction yield is improved.
[0016]

[0017] 4. Condensation reaction
[0018] Add isopropanol, intermediate (III’), sodium methoxide and compound V to the reaction kettle, mix and stir, heat to reflux and react for 5 hours. After the reaction is complete, cool and filter, and the filtrate is decompressed to recover the solvent to dryness, leaving residue Add ethyl acetate to dissolve, wash with water, dry with anhydrous magnesium sulfate, recover ethyl acetate under reduced pressure to 1/4 of the total volume, add n-hexane, and stir at 50°C for 30 min. After cooling and crystallization, the solid was collected by filtration, and air-dried at 45°C to obtain the crude azedipine (I). After the crude product was dissolved in ethyl acetate-n-hexane mixed solvent, activated carbon was added for decolorization and impurity removal to achieve the purpose of purification.

[0020] The refined product is dissolved in dioxane, refluxed with n-hexane, cooled and crystallized, and dried to obtain a solid that is boiled in cyclohexane, cooled and filtered, and dried to obtain α-crystalline form Azedipine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCN102453023A *2010-10-212012-05-16大丰市天生药业有限公司Process for producing azelnidipineCN103130700A *2013-03-142013-06-05沈阳中海药业有限公司Preparation method of azelnidipine intermediateCN103509003A *2012-06-272014-01-15威海威太医药技术开发有限公司Preparation method of azelnidipine
JP3491506B2 *1997-10-142004-01-26宇部興産株式会社Method for producing dihydropyridine derivativeCN101475521B *2008-11-132010-11-10青岛黄海制药有限责任公司Method for synthesizing acetate of 1-benzhydryl-3-azetidine amidino acetic ester
TitleLIU, JIAN-FENG ET AL.: “Improved Synthesis of Azelnidipine”, CHINESE JOURNAL OF MEDICINAL CHEMISTRY, vol. 20, no. 3, 30 June 2010 (2010-06-30), pages 192 – 194 *ZHANG, KAI ET AL.: “Synthesis of Azelnidipine”, CHINESE JOURNAL OF PHARMACEUTICALS, vol. 39, no. 3, 31 March 2008 (2008-03-31), pages 163 – 165, XP025959789, DOI: doi:10.1016/j.ejphar.2008.12.041 *
CN103130700B *2013-03-142015-04-29沈阳中海药业有限公司Preparation method of azelnidipine intermediateCN104860855B *2014-12-082017-06-16宁夏紫光天化蛋氨酸有限责任公司A kind of preparation method of the methylmercapto butyric acid ester of 2 hydroxyl of the D of high-purity, L 4CN105949102A *2016-06-202016-09-21许昌豪丰化学科技有限公司Production method of azelnidipine intermediatePublication numberPriority datePublication dateAssigneeTitleWO2014139410A1 *2013-03-142014-09-18Shenyang Zhonghai Pharmaceutical Co., Ltd.A kind of preparation method of azeldipine intermediateCN105461691A *2015-12-312016-04-06Weihai Disu Pharmaceutical Co., Ltd.A kind of preparation method of azeldipineCN106279109A *2016-08-182017-01-04Weihai Disu Pharmaceutical Co., Ltd.A kind of preparation method of azeldipineCN106543061A *2016-10-202017-03-29Weihai Disu Pharmaceutical Co., Ltd.Preparation method of N-diphenylmethylcyclobutane-3-alcohol
References
- ^ Oizumi K, Nishino H, Koike H, Sada T, Miyamoto M, Kimura T (September 1989). “Antihypertensive effects of CS-905, a novel dihydropyridine Ca++ channel blocker”. Jpn. J. Pharmacol. 51 (1): 57–64. doi:10.1254/jjp.51.57. PMID 2810942.
| Clinical data | |
|---|---|
| Trade names | CalBlock,AZUSA,Azovas |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral |
| ATC code | none |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 123524-52-7 |
| PubChem CID | 65948 |
| ChemSpider | 59352 |
| UNII | PV23P19YUG |
| KEGG | D01145 |
| ChEMBL | ChEMBL1275868 |
| CompTox Dashboard (EPA) | DTXSID3020120 |
| ECHA InfoCard | 100.162.151 |
| Chemical and physical data | |
| Formula | C33H34N4O6 |
| Molar mass | 582.657 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| hideSMILES[O-][N+](=O)c1cccc(c1)C5C(/C(=O)OC(C)C)=C(\NC(\N)=C5\C(=O)OC4CN(C(c2ccccc2)c3ccccc3)C4)C | |
| hideInChIInChI=1S/C33H34N4O6/c1-20(2)42-32(38)27-21(3)35-31(34)29(28(27)24-15-10-16-25(17-24)37(40)41)33(39)43-26-18-36(19-26)30(22-11-6-4-7-12-22)23-13-8-5-9-14-23/h4-17,20,26,28,30,35H,18-19,34H2,1-3H3 Key:ZKFQEACEUNWPMT-UHFFFAOYSA-N |
/////////Azelnidipine, CS-905, RS-9054, INDIA 2020, APPROVALS 2020
#Azelnidipine, #CS-905, #RS-9054, #INDIA 2020, #APPROVALS 2020
CC1=C(C(C(=C(N1)N)C(=O)OC2CN(C2)C(C3=CC=CC=C3)C4=CC=CC=C4)C5=CC(=CC=C5)[N+](=O)[O-])C(=O)OC(C)C
Centhaquine
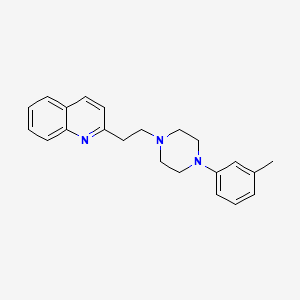
Centhaquine
PMZ-2010
CAS 57961-90-7
2-[2-[4-(3-methylphenyl)piperazin-1-yl]ethyl]quinoline
INDIA 2020, 14.05.2020, Centhaquine citrate bulk and Centhaquine citrate injection 1.0mg/vial, Add on resuscitative agent for hypovolemic shock
- OriginatorMidwestern University; Pharmazz
- DeveloperPharmazz
- ClassAnalgesics; Antihaemorrhagics; Antihypertensives; Cardiovascular therapies; Piperazines; Quinolines; Small molecules
- Mechanism of ActionAlpha 1 adrenergic receptor antagonists; Alpha 2 adrenergic receptor agonists
- RegisteredHaemorrhagic shock
- Phase IHeart arrest; Postoperative pain
- 20 Jul 2020Pharmazz plans to launch centhaquin for Haemorrhagic shock (Adjuvant therapy) in India by the middle of September 2020
- 20 Jul 2020Efficacy data from a phase III trial in Haemorrhagic shock released by Pharmazz
- 02 Jun 2020Centhaquine is still in phase I trials for Postoperative pain in USA (Pharmazz pipeline, June 2020)
SYNCenthaquin is a compound that produces hypotension and bradycardia in higher doses and resuscitation in lower doses. It is water insoluble, and is unsuitable for intravenous use. We prepared the citrate salt of centhaquin and evaluated its cardiovascular efficacy vs. centhaquin. Centhaquin citrate was prepared and characterized; its purity was determined by HPLC. Mean arterial pressure (MAP), heart rate (HR), pulse pressure (PP), cardiac output (CO), stroke volume (SV) and stroke work (SW) following intravenous administration of centhaquin and the citrate (0.05, 0.15 and 0.45 mg.kg(-1)) were determined in anaesthetized male Sprague-Dawley rats. Centhaquin citrate was 99.8% pure and water soluble. Centhaquin (0.05, 0.15 and 0.45 mg.kg(-1)) produced a maximal decrease in MAP of 15.6, 25.2 and 28.1%, respectively; while centhaquin citrate produced a greater (p<0.001) decrease of 35.7, 47.1 and 54.3%, respectively. The decrease in PP and HR produced by the citrate was greater than centhaquin (p<0.001). At 0.45 mg.kg(-1) centhaquin produced a maximal decrease of 20.9% (p<0.01) in CO, while centhaquin citrate produced a decrease of 42.1% (p<0.001). Reduction in SV (p<0.01) and SW (p<0.001) produced by centhaquin citrate were greater than centhaquin. Centhaquin citrate has greater cardiovascular activity compared to centhaquin.https://www.semanticscholar.org/paper/Synthesis-and-characterization-of-centhaquin-and-a-Reniguntala-Lavhale/6ca3975b114b0f23753e7a47710eff2467bc2dae

PATENT
https://patents.google.com/patent/WO2014035446A1/en
Shock due to severe hemorrhage accounts for a large proportion of posttraumatic deaths, particularly during early stages of injury (Wu, Dai et al. 2009). A majority of deaths due to hemorrhage occur within the first six hours after trauma (Shackford, Mackersie et al. 1993), but many of these deaths can be prevented (Acosta, Yang et al. 1998).
[0003] Shock is accompanied by circulatory failure which is the primary cause of mortality and morbidity. Presently, the recommended fluid therapy uses large volumes of Lactated Ringer’s solution (LR), which is effective in restoring hemodynamic parameters, but presents logistic and physiologic limitations (Vincenzi, Cepeda et al. 2009). For example, resuscitation using a large volume of crystalloids, like LR, has been associated with secondary abdominal compartment syndrome, pulmonary edema, cardiac dysfunction, and paralytic ileus (Balogh, McKinley et al. 2003). Therefore, a need exists in the art for a resuscitation agent that improves survival time, and can be used with a small volume of resuscitation fluid, for resuscitation in hypovolemic shock.
[0004] Centhaquin (2-[2-(4-(3-methyphenyl)-l-piperazinyl) ethyl-quinoline) is a centrally acting antihypertensive drug. The structure of centhaquin was determined (Bajpai et al., 2000) and the conformation of centhaquin was confirmed by X-ray diffraction (Carpy and Saxena, 1991).

Structure of centhaquin (2-[2-(4-(3-methyphenyl)- 1 -piperazinyl) ethyl] -quinoline) (as free base)
[0005] Centhaquin is an active cardiovascular agent that produces a positive inotropic effect and increases ventricular contractions of isolated perfused rabbit heart (Bhatnagar, Pande et al. 1985). Centhaquin does not affect spontaneous contractions of the guinea pig right auricle, but significantly potentiates positive inotropic effect of norepinephrine (NE) (Srimal, Mason et al. 1990). The direct or indirect positive inotropic effect of centhaquin can lead to an increase in cardiac output (CO). Centhaquin produces a decrease in mean arterial pressure (MAP) and heart rate (HR) in anesthetized rats and conscious freely moving cats and rats (Srimal, Gulati et al. 1990) due to its central sympatholytic activity (Murti, Bhandari et al. 1989; Srimal, Gulati et al. 1990; Gulati, Hussain et al. 1991). When administered locally into a dog femoral artery, centhaquin (10 and 20 μg) increased blood flow, which was similar to that observed with acetylcholine and papaverine. However, the vasodilator effect of centhaquin could not be blocked by atropine or dibenamine (Srimal, Mason et al. 1990). The direct vasodilator or central sympatholytic effect of centhaquin is likely to decrease systemic vascular resistance (SVR).
[0006] It was found that centhaquin enhances the resuscitative effect of hypertonic saline (HS) (Gulati, Lavhale et al. 2012). Centhaquin significantly decreased blood lactate and increases MAP, stroke volume, and CO compared to hypertonic saline alone. It is theorized, but not relied upon, that the cardiovascular actions of hypertonic saline and centhaquin are mediated through the ventrolateral medulla in the brain (Gulati, Hussain et al. 1991 ; Cavun and Millington 2001) and centhaquin may be augmenting the effect of hypertonic saline.
[0007] A large volume of LR (i.e., about three times the volume of blood loss) is the most commonly used resuscitation fluid therapy (Chappell, Jacob et al. 2008), in part because LR does not exhibit the centrally mediated cardiovascular effects of hypertonic saline. Large volume resuscitation has been used by emergency medical personnel and surgeons to reverse hemorrhagic shock and to restore end-organ perfusion and tissue oxygenation. However, there has been a vigorous debate with respect to the optimal methods of resuscitation (Santry

ased on the molecular weight of centhaquin (free base) (MW-332) and centhaquin citrate (MW-523), for identical doses of centhaquin (as free base) and centhaquin citrate, centhaquin citrate provides only 63.5% of centhaquin free base compared to the dose of centhaquin free base, e.g., a 0.05 mg dose of centhaquin citrate contains a 0.0318 mg of centhaquin (as free base). Similarly, a dose of centhaquin citrate dihydrate (MW-559) provides 59.4% centhaquin (free base) of the same dose as centhaquin (as free base), i.e., a 0.0005 mg dose of centhaquin citrate dihydrate contains 0.030 mg of centhaquin (as free base). Surprisingly, and as demonstrated below, at the same mg/kg dose centhaquin citrate and centhaquin citrate dihydrate provides greater cardiovascular effects than centhaquin free base.

Synthesis of Centhaquin

[0061] The synthesis of centhaquin was reported by Murthi and coworkers (Murthi et al U.S. Patent No. 3,983,121 ; Murti, Bhandari et al. 1989). In one procedure, reactants 1 and 2 were stirred at reflux for 15 hours. The resulting product was purified by evaporating the solvents to obtain an oil, which was heated in vacuo (100°C, 1 mm Hg). The remaining residue was recrystallized from ether-petroleum ether to obtain the final centhaquin product 3. The melting point reported for centhaquin was 76-77°C. In a subsequent publication (Murti, Bhandari et al. 1989), the reaction mixture was concentrated following 24 hours of reflux, diluted with water, and basified with aqueous NaOH. The basic mixture was extracted with ethyl acetate, and the ethyl acetate extracts were dried over anhydrous sodium sulfate and evaporated in vacuo to give centhaquin which was crystallized from hexane. The melting point of centhaquin (free base) obtained in this procedure was 82°C. The product obtained using either purification method is light tan in color, which is indicative of small amounts of impurities that were not completely removed using previously reported purification methods.
[0062] In accordance with the present invention, an improved purification method was found. According to the improved method, reactants 1 and 2 were stirred at reflux for 24 hours. The solvents were evaporated in vacuo and the resulting mixture was diluted with water and basified (10% NaOH). The basic mixture was extracted with ethyl acetate and the combined ethyl acetate extracts are dried over anhydrous sodium sulfate and evaporated in vacuo to obtain a residue, which was further purified with column chromatography (Si02, ethyl acetate). The resulting product can be decolorized using activated charcoal or directly crystallized from hot hexane to yield pure centhaquin. The resulting product is an off-white crystalline solid having a melting point of 94-95°C (free base). The product was
characterized using proton NMR, mass spectral, and elemental analysis and indicated high purity and superior quality.
[0063] Synthesis and characterization of centhaquin (free base): A mixture of 2- vinylquinoline (1) (5.0 g, 32.2 mmol, 98.5%) and 1 -(3-methylphenyl)piperazine (2) (5.68 g, 32.2 mmol, 99.0%) in absolute ethyl alcohol (150 ml) and glacial acetic acid (3.5 ml) was stirred at reflux for 24 hours in a round bottom flask. The reaction mixture was concentrated in vacuo, diluted with water (150 ml) and treated with 10% aqueous NaOH (150 ml). The residue was extracted with ethyl acetate (4 x 125 ml), dried with anhydrous Na2S04, and concentrated under reduced pressure to yield a crude product which was purified by column chromatography using silica gel (100-200 mesh) with ethyl acetate as an eluent. The resulting compound was recrystallized from hot hexane and filtered, to yield centhaquin as an off- white crystalline solid (7.75 g, 23.4 mmol, 73% yield); mp. 94-95°C; i? 0.30 (100% ethyl acetate); 1H NMR (300 MHz, CDC13): δ 8.07 (t, J= 7.5 Hz, 2 H), 7.78 (d, J= 7.8 Hz, 1 H), 7.70 (t, J= 7.8 Hz, 1 H), 7.50 (t, J= 7.5 Hz, 1 H), 7.36 (d, J= 8.4 Hz, 1 H), 7.16 (t, J = 7.5 Hz, 1 H), 6.77 – 6.74 (m, 2 H), 6.69 (d, J= 7.2 Hz, 1 H), 3.26- 3.21 (m, 6 H), 2.97 – 2.92 (m, 2 H), 2.76 – 2.73 (m, 4 H), 2.32 (s, 3 H); HRMS (ESI) m/z 332.2121 [M+l]+ (calcd for C22H26N3 332.2122); Anal. (C22H25N3) C, H, N.
[0064] Preparation of centhaquin citrate: Centhaquin (free base) (5.62 g, 16.98 mmol) was treated with citric acid (3.26 g, 16.98 mmol) in a suitable solvent and converted to the citrate salt obtained as an off-white solid (7.96 g, 15.2 mmol, 90%); m.p. 94-96°C ; Anal.
10065] Figs. 1(a) and 1(b) are high resolution mass spectral analyses of centhaquin free base (Fig 1(a)) and centhaquin citrate (Fig. 1(b)). Compound samples were analyzed following ionization using electrospray ionization (ESI).
[0066J For centhaquin free base in Fig 1(a), a base peak [M+l]+ was observed at m z 332.2141 (theory: 332.2121) consistent with the elemental composition of protonated centhaquin (C22H26N3).
[0067] For centhaquin citrate in Fig 1(b), the mass spectrum was identical to the mass spectrum obtained for the free base. An [M+l]+base peak was observed at m z 332.2141 (theory: 332.2121), which corresponds to the elemental composition of protonated centhaquin (C22H26N3). This result is typical of salts of organic bases to yield the [M+l]+ of the free base as observed here with centhaquin citrate.
[0068] Mass spectrometry is one of the most sensitive analytical methods, and examination of the mass spectra of Fig. 1 indicate that the samples are devoid of any extraneous peaks and are of homogeneous purity (>99.5).
PATENT
https://patents.google.com/patent/WO2014035446A1/en
////////////Centhaquine, PMZ-2010, PMZ 2010, INDIA 2020, 2020 APPROVALS
DOFETILIDE

Dofetilide
115256-11-6[RN]
6756
b-((p-Methanesulfonamidophenethyl)methylamino)methanesulfono-p-phenetidide
Methanesulfonamide, N-[4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]-
MFCD00869707 [MDL number]
- Molecular FormulaC19H27N3O5S2
- Average mass441.565 Da
- UK68798UNII:R4Z9X1N2NDUNII-R4Z9X1N2NDXelideβ-((p-Methanesulfonamidophenethyl)methylamino)methanesulfono-p-phenetidideдофетилидدوفيتيليد多非利特
N-[4-[2-[methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]-methanesulfonamideNCGC00164549-01PB0478000SMR000466333Tikosyn (TN)Research Code:UK-68798Trade Name:Tikosyn®MOA:Atrial potassium channel blockerIndication:Atrial flutter; Atrial fibrillationStatus:ApprovedCompany:Pfizer (Originator)Sales:ATC Code:C01BD04
INDIA 31/7/2020 APPROVED CDSCO
Dofetilide was first approved by the U.S. Food and Drug Administration (FDA) on Oct 1, 1999, then approved by European Medicine Agency (EMA) on Nov 29, 1999. It was developed and marketed as Tikosyn® by Pfizer.
Dofetilide is a selective blocker of delayed rectifier outward potassium current (IKr). It is indicated for the maintenance of normal sinus rhythm (delay in time to recurrence of atrial fibrillation/atrial flutter [AF/AFl]) in patients with atrial fibrillation/atrial flutter of greater than one week duration who have been converted to normal sinus rhythm.
Tikosyn® is available capsule for oral use, containing 0.125, 0.25 or 0.5 mg of free Dofetilide. The recommended dose is 500 µg orally twice daily.
Dofetilide is a class III antiarrhythmic agent.[1] It is marketed under the trade name Tikosyn by Pfizer, and is available in the United States in capsules containing 125, 250, and 500 µg of dofetilide. It is not available in Europe[2] or Australia.[3] In the United States it is only available by mail order or through specially trained local pharmacies.[4]
Medical uses
Dofetilide is used for the maintenance of sinus rhythm in individuals prone to the occurrence of atrial fibrillation and flutter arrhythmias, and for chemical cardioversion to sinus rhythm from atrial fibrillation and flutter.[5][6]
Based on the results of the Danish Investigations of Arrhythmias and Mortality on Dofetilide (“DIAMOND”) study,[7] dofetilide does not affect mortality in the treatment of patients post-myocardial infarction with left ventricular dysfunction, however it was shown to decrease all-cause readmissions as well as CHF-related readmissions.[8] Because of the results of the DIAMOND study, some physicians use dofetilide in the suppression of atrial fibrillation in individuals with LV dysfunction, however use appears limited: After initially receiving marketing approval in Europe in 1999, Pfizer voluntarily withdrew this approval in 2004 for commercial reasons[2] and it is not registered in other first world countries.
It has clinical advantages over other class III antiarrhythmics in chemical cardioversion of atrial fibrillation, and maintenance of sinus rhythm, and does not have the pulmonary or hepatotoxicity of amiodarone, however atrial fibrillation is not generally considered life-threatening, and dofetilide causes an increased rate of potentially life-threatening arrhythmias in comparison to other therapies.[9]
Contraindications
Prior to administration of the first dose, the corrected QT (QTc) must be determined. If the QTc is greater than 440 msec (or 500 msec in patients with ventricular conduction abnormalities), dofetilide is contraindicated. If heart rate is less than 60 bpm, the uncorrected QT interval should be used. After each subsequent dose of dofetilide, QTc should be determined and dosing should be adjusted. If at any time after the second dose of dofetilide the QTc is greater than 500 msec (550 msec in patients with ventricular conduction abnormalities), dofetilide should be discontinued. [4]
Adverse effects
Torsades de pointes is the most serious side effect of dofetilide therapy. The incidence of torsades de pointes is 0.3-10.5% and is dose-related, with increased incidence associated with higher doses. The majority of episodes of torsades de pointes have occurred within the first three days of initial dosing. Patients should be hospitalized and monitored for the first three days after starting dofetilide.[10]
The risk of inducing torsades de pointes can be decreased by taking precautions when initiating therapy, such as hospitalizing individuals for a minimum of three days for serial creatinine measurement, continuous telemetry monitoring and availability of cardiac resuscitation.
Pharmacology
Mechanism of action
Dofetilide works by selectively blocking the rapid component of the delayed rectifier outward potassium current (IKr).[11]
This causes the refractory period of atrial tissue to increase, hence its effectiveness in the treatment of atrial fibrillation and atrial flutter.
Dofetilide does not affect dV/dTmax (the slope of the upstroke of phase 0 depolarization), conduction velocity, or the resting membrane potential.

Dofetilide synthesis
There is a dose-dependent increase in the QT interval and the corrected QT interval (QTc). Because of this, many practitioners will initiate dofetilide therapy only on individuals under telemetry monitoring or if serial EKG measurements of QT and QTc can be performed.
Pharmacokinetics
Peak plasma concentrations are seen two to three hours after oral dosing when fasting. Dofetilide is well absorbed in its oral form, with a bioavailability of >90%. Intravenous administration of dofetilide is not available in the United States. [12]
The elimination half-life of dofetilide is roughly 10 hours; however, this varies based on many physiologic factors (most significantly creatinine clearance), and ranges from 4.8 to 13.5 hours. Due to the significant level of renal elimination (80% unchanged, 20% metabolites), the dose of dofetilide must be adjusted to prevent toxicity due to impaired renal function.[13]
Dofetilide is metabolized predominantly by CYP3A4 enzymes predominantly in the liver and GI tract. This means that it is likely to interact with drugs that inhibit CYP3A4, such as erythromycin, clarithromycin, or ketoconazole, resulting in higher and potentially toxic levels of dofetilide. [14]
Metabolism
A steady-state plasma level of dofetilide is achieved in 2–3 days.
80% of dofetilide is excreted by the kidneys, so the dose of dofetilide should be adjusted in individuals with chronic kidney disease, based on creatinine clearance.
In the kidneys, dofetilide is eliminated via cation exchange (secretion). Agents that interfere with the renal cation exchange system, such as verapamil, cimetidine, hydrochlorothiazide, itraconazole, ketoconazole, prochlorperazine, and trimethoprim should not be administered to individuals taking dofetilide.
About 20 percent of dofetilide is metabolized in the liver via the CYP3A4 isoenzyme of the cytochrome P450 enzyme system. Drugs that interfere with the activity of the CYP3A4 isoenzyme can increase serum dofetilide levels. If the renal cation exchange system is interfered with (as with the medications listed above), a larger percentage of dofetilide is cleared via the CYP3A4 isoenzyme system.
History
After its initial US FDA approval, due to the pro-arrhythmic potential it was only made available to hospitals and prescribers that had received education and undergone specific training in the risks of treatment with dofetilide; however, this restriction was subsequently removed in 2016. [15
SYN


REF
Reference:1. US5079248A / US4959366A.
2. J. Med. Chem. 1990, 33, 1151-1155.
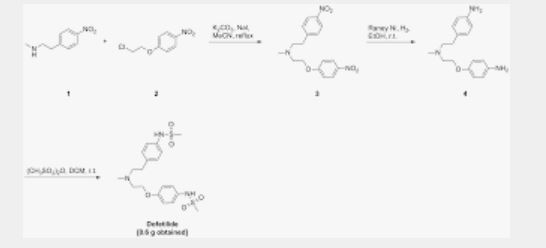
SYN
SYN
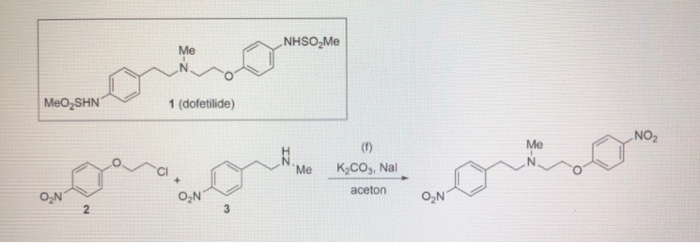
SYN
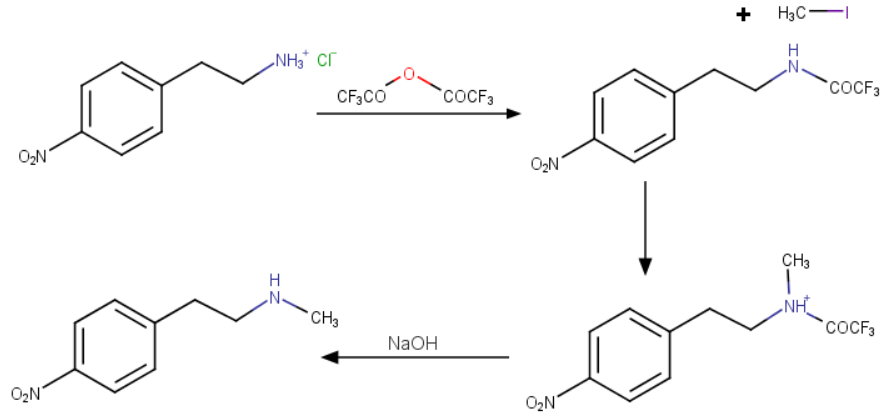
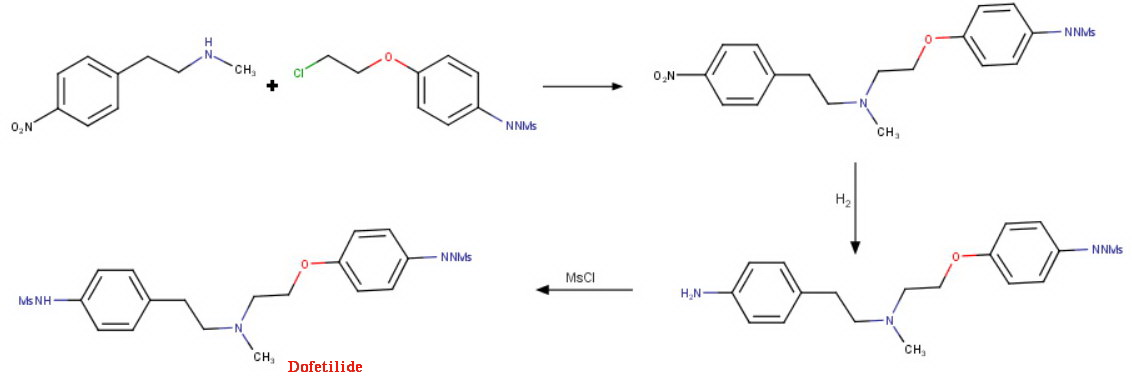
SYN
EP 0245997; JP 1987267250; US 4959366; US 5079248
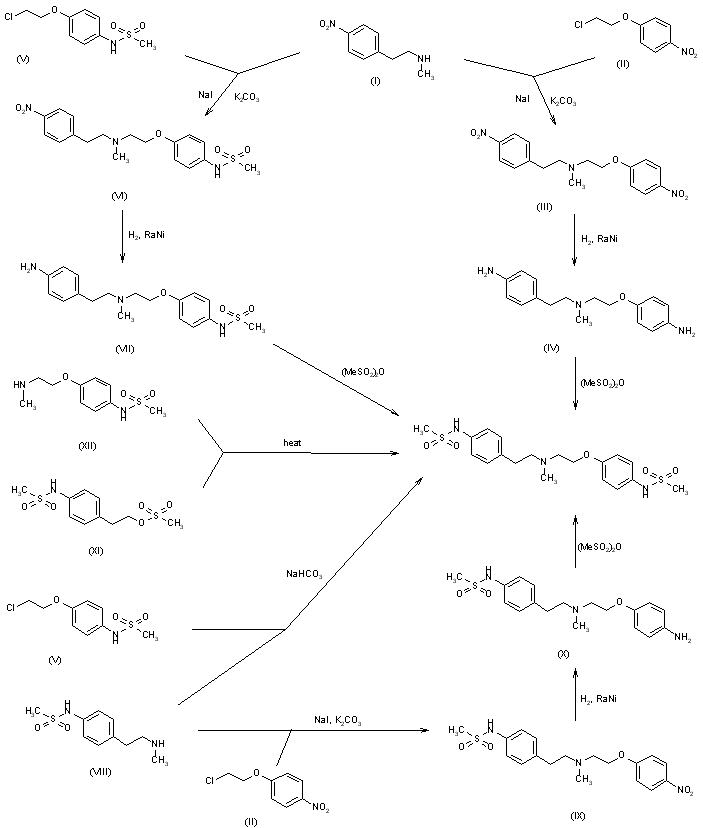
This compound can be prepared by several related ways: 1) The condensation of N-methyl-2-(4-nitrophenyl)ethylamine (I) with 4-(2-chloroethoxy)nitrobenzene (II) by means of NaI and K2CO3 in refluxing acetonitrile gives 1-(4-nitrophenoxy)-5-(4-nitrophenyl)-3-methyl-3-azapentane (III), which is reduced with H2 over Pd/C in ethanol, yielding the corresponding diamino derivative (IV). Finally, this compound is acylated with methanesulfonyl anhydride in dichloromethane. 2) The condensation of (I) with N-[4-(2-chloroethoxy)phenyl]methanesulfonamide (V) with NaI and K2CO3 as before gives 1-[4-(methanesulfonamide)phenoxy]-3-methyl-5-(4-nitrophenyl)-3-azapentane (VI), which is reduced with H2 over Pd/C as before, yielding the corresponding amino derivative (VII). Finally, this compound is acylated with methanesulfonyl anhydride as usual. 3) The condensation of (II) with N-[4-[2-(methylamino)ethyl]phenyl]methanesulfonamide (VIII) with NaI and K2CO3 as usual gives 1-[4-(methanesulfonamido)phenyl]-3-methyl-5-(4-nitrophenoxy)-3-azapentane (IX), which is reduced with H2 and RaNi to the corresponding amino derivative (X). Finally, this compound is acylated with methanesulfonyl chloride and pyridine. 4) By condensation of N-[4-[2-(methanesulfonyloxy)ethyl]phenyl]methanesulfonamide (XI) with N-[4-[2-(methylamino)ethoxy]phenyl]methanesulfonamide (XII) in refluxing ethanol. 5) By condensation of (V) with (VIII) by means of NaHCO3.
References
- ^ Lenz TL; Hilleman DE (July 2000). “Dofetilide, a new class III antiarrhythmic agent”. Pharmacotherapy. 20 (7): 776–86. doi:10.1592/phco.20.9.776.35208. PMID 10907968.
- ^ Jump up to:a b Wathion, Noel (2004-04-13). “Public Statement on Tikosyn (dofetilide): Voluntary Withdrawal of the Marketing Authorisation in the European Union” (PDF). European Agency for the Evaluation of Medicinal Products.
- ^ Australian Medicines Handbook 2014
- ^ Jump up to:a b TIKOSYN® (dofetilide). Pfizer. <http://www.tikosyn.com/>.
- ^ Banchs JE; Wolbrette DL; Samii SM; et al. (November 2008). “Efficacy and safety of dofetilide in patients with atrial fibrillation and atrial flutter”. J Interv Card Electrophysiol. 23(2): 111–5. doi:10.1007/s10840-008-9290-6. PMID 18688699. S2CID 25162347.
- ^ Lenz TL; Hilleman DE (November 2000). “Dofetilide: A new antiarrhythmic agent approved for conversion and/or maintenance of atrial fibrillation/atrial flutter”. Drugs Today. 36 (11): 759–71. doi:10.1358/dot.2000.36.11.601530. PMID 12845335.
- ^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. The New England Journal of Medicine. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID 10486417.
- ^ Torp-Pedersen C; ller M; Mø Bloch-Thomsen PE; et al. (September 1999). “Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group”. N. Engl. J. Med. 341 (12): 857–65. doi:10.1056/NEJM199909163411201. PMID 10486417.
- ^ Micromedex Drugdex drug evaluations micromedex.com
- ^ Torp-Pedersen C, Møller M, Bloch-Thomsen PE, et al. Dofetilide in patients with congestive heart failure and left ventricular dysfunction. Danish Investigations of Arrhythmia and Mortality on Dofetilide Study Group. N Engl J Med 1999; 341:857.
- ^ Roukoz H; Saliba W (January 2007). “Dofetilide: a new class III antiarrhythmic agent”. Expert Rev Cardiovasc Ther. 5 (1): 9–19. doi:10.1586/14779072.5.1.9. PMID 17187453. S2CID 11255636.
- ^ 1Rasmussen HS, Allen MJ, Blackburn KJ, et al. Dofetilide, a novel class III antiarrhythmic agent. J Cardiovasc Pharmacol 1992; 20 Suppl 2:S96.
- ^ “Dofetilide.” Lexicomp. Wulters Kluwer Health, n.d. Web. <online.lexi.com>.
- ^ Walker DK, Alabaster CT, Congrave GS, et al. Significance of metabolism in the disposition and action of the antidysrhythmic drug, dofetilide. In vitro studies and correlation with in vivo data. Drug Metab Dispos 1996; 24:447.
- ^ “Information for Tikosyn (dofetilide)”. US Food and Drug Administration. 2016-03-09.
DofetilideCAS Registry Number: 115256-11-6CAS Name:N-[4-[2-[Methyl[2-[4-[(methylsulfonyl)amino]phenoxy]ethyl]amino]ethyl]phenyl]methanesulfonamideAdditional Names: 1-(4-methanesulfonamidophenoxy)-2-[N-(4-methanesulfonamidophenethyl)-N-methylamino]ethaneManufacturers’ Codes: UK-68798Trademarks: Tikosyn (Pfizer)Molecular Formula: C19H27N3O5S2Molecular Weight: 441.56Percent Composition: C 51.68%, H 6.16%, N 9.52%, O 18.12%, S 14.52%Literature References: Potassium channel blocker. Prepn: J. E. Arrowsmith et al.,EP245997; P. E. Cross et al.,US4959366 (1987, 1990 both to Pfizer); idemet al.,J. Med. Chem.33, 1151 (1990). HPLC determn in urine: D. K. Walker et al.,J. Chromatogr.568, 475 (1991). Mechanism of action study: D. Carmeliet, J. Pharmacol. Exp. Ther.262, 809 (1992). Review of pharmacology and pharmacokinetics: H. S. Rasmussen et al.,ibid.20, Suppl. 2, S96-S105 (1992). Clinical trial in atrial fibrillation and flutter: B. L. Norgaard et al.,Am. Heart J.137, 1062 (1999); in congestive heart failure: C. Torp-Pedersen et al.,N. Engl. J. Med.341, 857 (1999).Properties: Crystals from ethyl acetate/methanol (10:1), mp 147-149° (Cross); from hexane/ethyl acetate, mp 151-152° (Arrowsmith). Also reported as white crystalline solid, mp 161° (Rasmussen). pKa 7.0, 9.0, 9.6. Distribution coefficient (pH 7.4): 0.96. Sol in 0.1M NaOH, acetone, 0.1M HCl; very slightly sol in water, propan-2-ol.Melting point: mp 147-149° (Cross); mp 151-152° (Arrowsmith); mp 161° (Rasmussen)pKa: pKa 7.0, 9.0, 9.6Therap-Cat: Antiarrhythmic (class III).Keywords: Antiarrhythmic; Potassium Channel Blocker.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a601235 |
| ATC code | C01BD04 (WHO) |
| Pharmacokinetic data | |
| Bioavailability | 96% (oral) |
| Protein binding | 60% -70% |
| Elimination half-life | 10 hours |
| Identifiers | |
| IUPAC name[show] | |
| CAS Number | 115256-11-6 |
| PubChem CID | 71329 |
| IUPHAR/BPS | 2604 |
| DrugBank | DB00204 |
| ChemSpider | 64435 |
| UNII | R4Z9X1N2ND |
| KEGG | D00647 |
| ChEBI | CHEBI:4681 |
| ChEMBL | ChEMBL473 |
| CompTox Dashboard (EPA) | DTXSID5046433 |
| ECHA InfoCard | 100.166.441 |
| Chemical and physical data | |
| Formula | C19H27N3O5S2 |
| Molar mass | 441.56 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES[hide]O=S(=O)(Nc1ccc(cc1)CCN(CCOc2ccc(cc2)NS(=O)(=O)C)C)C | |
| InChI[hide]InChI=1S/C19H27N3O5S2/c1-22(13-12-16-4-6-17(7-5-16)20-28(2,23)24)14-15-27-19-10-8-18(9-11-19)21-29(3,25)26/h4-11,20-21H,12-15H2,1-3H3 Key:IXTMWRCNAAVVAI-UHFFFAOYSA-N |
////////////DOFETILIDE, 2020 APPROVALS, INDIA 2020, UK 68798, UNII:R4Z9X1N2ND, дофетилид , دوفيتيليد ,多非利特 , TIKOSYN
Pretomanid, プレトマニド;
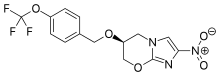
Pretomanid
プレトマニド;
| Formula |
C14H12F3N3O5
|
|---|---|
| CAS |
187235-37-6
|
| Mol weight |
359.2574
|
- (S)-PA 824
2019/8/14 FDA 2109 APPROVED
Antibacterial (tuberculostatic),
MP 149-150 °C, Li, Xiaojin; Bioorganic & Medicinal Chemistry Letters 2008, Vol 18(7), Pg 2256-2262 and Orita, Akihiro; Advanced Synthesis & Catalysis 2007, Vol 349(13), Pg 2136-2144
150-151 °C Marsini, Maurice A.; Journal of Organic Chemistry 2010, Vol 75(21), Pg 7479-7482
Pretomanid is an antibiotic used for the treatment of multi-drug-resistant tuberculosis affecting the lungs.[1] It is generally used together with bedaquiline and linezolid.[1] It is taken by mouth.[1]
The most common side effects include nerve damage, acne, vomiting, headache, low blood sugar, diarrhea, and liver inflammation.[1] It is in the nitroimidazole class of medications.[2]
Pretomanid was approved for medical use in the United States in 2019.[3][1] Pretomanid was developed by TB Alliance,[4] a not-for-profitproduct development partnership dedicated to the discovery and development of new, faster-acting and affordable medicines for tuberculosis (TB).[5]
Global Alliance for the treatment of tuberculosis (TB).
The compound was originally developed by PathoGenesis (acquired by Chiron in 2000). In 2002, a co-development agreement took place between Chiron (acquired by Novartis in 2005) and the TB Alliance for the development of the compound. The compound was licensed to Fosunpharma by TB Alliance in China.
History
Pretomanid is the generic, nonproprietary name for the novel anti-bacterial drug compound formerly called PA-824.[6] Pretomanid is referred to as “Pa” in regimen abbreviations, such as BPaL. The “preto” prefix of the compound’s name honors Pretoria, South Africa, the home of a TB Alliance clinical development office where much of the drug’s development took place. The “manid” suffix is used to group compounds with similar chemical structures. This class of drug is variously referred to as nitroimidazoles, nitroimidazooxazines or nitroimidazopyrans. Development of this compound was initiated because of the urgent need for new antibacterial drugs effective against resistant strains of tuberculosis. Also, current anti-TB drugs are mainly effective against replicating and metabolically active bacteria, creating a need for drugs effective against persisting or latent bacterial infections as often occur in patients with tuberculosis.[7]
Discovery and pre-clinical development
Pretomanid was first identified in a series of 100 nitroimidazopyran derivatives synthesized and tested for antitubercular activity. Importantly, pretomanid has activity against static M. tuberculosis isolates that survive under anaerobic conditions, with bactericidal activity comparable to that of the existing drug metronidazole. Pretomanid requires metabolic activation by Mycobacterium for antibacterial activity. Pretomanid was not the most potent compound in the series against cultures of M. tuberculosis, but it was the most active in infected mice after oral administration. Oral pretomanid was active against tuberculosis in mice and guinea pigs at safely tolerated dosages for up to 28 days.[7]
Limited FDA approval
FDA approved pretomanid only in combination with bedaquiline and linezolid for treatment of a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug resistant pulmonary tuberculosis. Pretomanid was approved under the Limited Population Pathway (LPAD pathway) for antibacterial and antifungal drugs. The LPAD Pathway was established by Congress under the 21st Century Cures Act to expedite development and approval of antibacterial and antifungal drugs to treat serious or life-threatening infections in a limited population of patients with unmet need. Pretomanid is only the third tuberculosis drug to receive FDA approval in more than 40 years.[3][8]
PATENT
IN 201641030408
HETERO RESEARCH FOUNDATION
http://ipindiaservices.gov.in/PatentSearch/PatentSearch/ViewPDF
- By Reddy, Bandi Parthasaradhi; Reddy, Kura Rathnakar; Reddy, Adulla Venkat Narsimha; Krishna, Bandi Vamsi
- From Indian Pat. Appl. (2018), IN 201641030408
The nitroimidazooxazine Formula I (PA-824) is a new class of bioreductive drug for tuberculosis. The recent introduction of the nitroimidazooxazine Formula I (PA-824) to clinical trial by the Global Alliance for TB Drug Development is thus of potential significance, since this compound shows good in vitro and in vivo activity against Mycobacterium tuberculosis in both its active and persistent forms. Tuberculosis (TBa) remains a leading infectious cause of death worldwide, but very few new drugs have been approved for TB treatment ifi the past 35 years, the current drug therapy for TB is long and complex, involving multidrug combinations.
The mechanism of actiém of Pretomanid is thoughrto involve reduction of the nitro group, in a‘ process dependent on the Bacterial ‘ m E Nfilw‘fieéFPEOEPEa‘e fillyeifiaasnfi (F8189); $943“; 20mm; “q Mcyarecent Swiss on mutant strains showed that a 151-amino acid (17.37 kDa) protein of unknown function, Rv3547, also, appears to be critical for this activation. Equivalent genes are present in M. boVis and MaVium.
Pretomanid and its pharmace’utically acceptable salts Were generically disclosed in US 5,668,127 A and Specifically disclosed in US 6,087,358 A. US ‘358 patent discloses a process for the preparation of Pretomanid, which is as shown below in scheme 1:

CN 104177372 A discloses a process for the preparation of Pretomanid, which is as shown below in scheme II: 
Bioorganic & Medicinal Chemistry Letters 2008, Volume: 18, Issue: 7, Pages: 2256-2262 discloses a process for the preparation of Pretomanid, which is as shown below in scheme Ill: 
US 7,!15,736 B2-discloses_a process fdr the preparation of 3S-Hydroxy-6-nitrQ-2H-3, 4— dihydro-[2-1b]-imidazopyran which is a key intermediate of Pretomanid, which is as shown below in scheme IV:

Journal Medicinal Chemistry 2009, Volume: 52, Pages: 637 — 645 discloses a process for the preparation of ‘Pretomanid, which is as shown below in scheme V:

Joumal Organic Chemistry 2010; Volume: 75 (2]), Pages: 7479—82 discloses a process for. the preparation of Pretomanid, which is as shown below in scheme VI:


Example 3: Preparation of Pretomanid (S) 1- -(3 (tert- -Butyldomethylsilyloxy)- -2- -(-4 -(trifluoromethoxy)-71benzyloxy2‘- propyl)- 2- -methylP AT E N4Tnitro- fi-Eimigazole (Efgm Awlas (3315;501:1691 gin! %etra%1y7drofuraen (18(150 ml) at room temperature and stirred for 5 to 10 minutes then TBAF (9516 ml) was added to the reaction mixture and stirred for 2 hours, at room temperature, afler completion of the reaction removed solvent through vacuum to obtained residue, dissolved the residue in MDC (1800 ml) and water (1800 ml) stirred, separated the layers and the organic layer washed with 10% ‘ sodium bicarbonate the obtained organic solution was concentrated under atmospheric pressure to obtained residue added MeOH (1730 ml) at room temperature and the reaction mixture was cooled to 0°C to 5°C, added KOH (24.5 gm) at the same temperaturé then cooled to room temperature and stirred for 24 hours. After completion of reaction DM Water added drop wise over 30 minutes at 10°C to 15° C and stirred for 1 hour to 1 hour 30 minutes at room’lemperature, filtrated the compound and washed with DM wa‘er (133 ml) and dried under vacuum for 10 hours at 50° C. Yield: 53 gm , Chromatographic purity: 97.69% (by HPLC):
Example 4: Purification of Pretomanid Pretomanid (53 gm) was dissolved in MDC (795 ml) at room temperatur’e and stirred for 10 to 15 minutes, added charcoal (10 gm) and stirred for 30-35 minutes, remove the charcoal and concentrated to obtained residue: Dissolved the obtained residue in IPA (795 ml) and the reaction mixture was heated to 80°C maintained for 10-15 minutes, added cyclohexane (1600ml) for 30 minutes at 80° C, then cooled to room temperature and stirred the reaction mass for overnight, filtered the solid and washed with cyclohexane (265 ml), and dried under vacuum for 10 hours at 50° C. Yield: 48 gm (Percentage of Yield: 90%) Chromatographic purity: 99.97% by HPLC).
CLIP

ReferencE
CN104177372A.
WO9701562A1.
IN 201641030408
IN 201621026053
CN 107915747
CN 106632393
CN 106565744
CN 104177372
WO 9701562
US 6087358
PAPER
Science (Washington, DC, United States) (2008), 322(5906), 1392-1395.
Paper
PAPER
Huagong Shikan (2010), 24(4), 32-34, 51.
Xiaojin; Bioorganic & Medicinal Chemistry Letters 2008, Vol 18(7), Pg 2256-2262
PAPER
Orita, Akihiro; Advanced Synthesis & Catalysis 2007, Vol 349(13), Pg 2136-2144
https://onlinelibrary.wiley.com/doi/abs/10.1002/adsc.200700119
https://application.wiley-vch.de/contents/jc_2258/2007/f700119_s.pdf



Marsini, Maurice A.; Journal of Organic Chemistry 2010, Vol 75(21), Pg 7479-7482

Scheme 1

aDHP = 3,4-dihydropyran; p-TsOH = p-toluenesulfonic acid; MsOH = methanesulfonic acid.
Scheme 3

aCl3CCN = trichloroacetonitrile; TBME = tert-butylmethyl ether; TfOH = trifluoromethanesulfonic acid.



PAPER
Journal of Medicinal Chemistry (2010), 53(1), 282-294.
Journal of Medicinal Chemistry (2009), 52(3), 637-645.
PATENT

References
- ^ Jump up to:a b c d e “FDA approves new drug for treatment-resistant forms of tuberculosis that affects the lungs”. FDA. 14 August 2019. Retrieved 28 August 2019.
- ^ “Compounds | TB Alliance”. http://www.tballiance.org. Retrieved 2019-04-18.
- ^ Jump up to:a b Abutaleb Y (14 August 2019). “New antibiotic approved for drug-resistant tuberculosis”. Washington Post.
- ^ “TB Medicine Pretomanid Enters Regulatory Review Process in the United States | TB Alliance”. http://www.tballiance.org. Retrieved 2019-04-18.
- ^ “About TB Alliance”. TB Alliance. Retrieved 2019-04-18.
- ^ “PA-824 has a New Generic Name: Pretomanid”. TB Alliance. Retrieved 2019-04-18.
- ^ Jump up to:a b Lenaerts AJ, Gruppo V, Marietta KS, Johnson CM, Driscoll DK, Tompkins NM, Rose JD, Reynolds RC, Orme IM (June 2005). “Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models”. Antimicrobial Agents and Chemotherapy. 49 (6): 2294–301. doi:10.1128/AAC.49.6.2294-2301.2005. PMC 1140539. PMID 15917524.
- ^ FDA News Release. FDA approves new drug for treatment-resistant forms of tuberculosis that affects the lungs.
 |
|
| Legal status | |
|---|---|
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| Chemical and physical data | |
| Formula | C14H12F3N3O5 |
| Molar mass | 359.261 g·mol−1 |
| 3D model (JSmol) | |
//////////////Pretomanid, FDA 2109, プレトマニド , Antibacterial, tuberculostatic, PA-824, ANTI tuberculostatic
Pixantrone
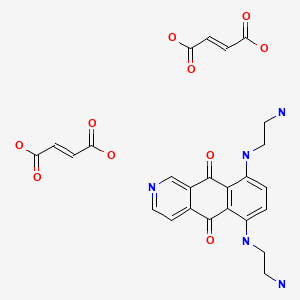

Pixantrone
BBR-2778 , CTK0H5262
- Pixolti
- Pixuvri
- UNII-P0R64C4CR9
An immunosuppressant.
144510-96-3 [RN]
5,8-Bis((2-aminoethyl)amino)-2-aza-anthracene-9,10-dione
6,9-Bis((2-aminoethyl)amino)benz(g)isoquinoline-5,10-dione
5,8-Bis((2-aminoethyl)amino)-2-aza-anthracene-9,10-dione
6,9-Bis((2-aminoethyl)amino)benz(g)isoquinoline-5,10-dione
CTI BioPharma receives Israeli approval for aggressive B-cell non-Hodgkin’s lymphoma therapy
CTI BioPharma has obtained Israeli Ministry of Health’s approval for Pixuvri (pixantrone), as a monotherapy to treat adult patients with multiply relapsed or refractory aggressive B-cell non-Hodgkin’s lymphoma who have received up to three previous courses of treatment.
The company also announced that the Dutch Healthcare Authority and the College voor zorgverzekeringen of the Netherlands have approved funding for Pixuvri as an add-on drug for patients who need a third or fourth-line treatment option for aggressive B-cell lymphoma.
Tel Aviv University faculty of medicine Dr Abraham Avigdor said: “The approval of PIXUVRI in Israel provides patients with aggressive B-cell NHL who have failed second or third-line therapy a new approved option, where none existed before, that can effectively treat their disease with manageable side-effects.
read at
| Pixantrone | |
|---|---|
 |
|
| Identifiers | |
| CAS number | 144510-96-3 |
| PubChem | 134019 |
| ChemSpider | 118174 |
| KEGG | D05522 |
| ChEMBL | CHEMBL167731 |
| ATC code | L01 |
| Jmol-3D images | Image 1 |
| Properties | |
| Molecular formula | C17H19N5O2 |
| Molar mass | 325.365 g/mol |
| Appearance | Blue solid |
| Pharmacology | |
| Routes of administration |
Intravenous |
| Elimination half-life |
9.5–17.5 hours |
| Excretion | Fecal (main route of excretion) and renal (4–9%) |
| Except where noted otherwise, data are given for materials in their standard state (at 25 °C (77 °F), 100 kPa) | |

Pixantrone dimaleate [USAN]
CAS 144675-97-8
Molecular Formula
-
C17-H19-N5-O2.2C4-H4-O4
Molecular Weight
- 441.4417
-
Benz(g)isoquinoline-5,10-dione, 6,9-bis((2-aminoethyl)amino)-, (2Z)-2-butenedioate (1:2)
On May 10, 2012, the European Commission issued a conditional marketing authorization valid throughout the European Union for pixantrone for the treatment of adult patients with multiply relapsed or refractory aggressive non-Hodgkin’s B-cell lymphoma (NHL). Pixantrone is a cytotoxic aza-anthracenedione that directly alkylates DNA-forming stable DNA adducts and cross-strand breaks. The recommended dose of pixantrone is 50 mg/m2 administered on days 1, 8, and 15 of each 28-day cycle for up to 6 cycles. In the main study submitted for this application, a significant difference in response rate (proportion of complete responses and unconfirmed complete responses) was observed in favor of pixantrone (20.0% vs. 5.7% for pixantrone and physician’s best choice, respectively), supported by the results of secondary endpoints of median progression-free and overall survival times (increase of 2.7 and 2.6 months, respectively). The most common side effects with pixantrone were bone marrow suppression (particularly of the neutrophil lineage) nausea, vomiting, and asthenia. This article summarizes the scientific review of the application leading to approval in the European Union. The detailed scientific assessment report and product information, including the summary of product characteristics, are available on the European Medicines Agency website (http://www.ema.europa.eu).
Pixantrone (rINN; trade name Pixuvri) is an experimental antineoplastic (anti-cancer) drug, an analogue of mitoxantrone with fewertoxic effects on cardiac tissue.[1] It acts as a topoisomerase II poison and intercalating agent.[2][3] The code name BBR 2778 refers topixantrone dimaleate, the actual substance commonly used in clinical trials.[4]
History
Anthracyclines are important chemotherapy agents. However, their use is associated with irreversible and cumulative heart damage. Investigators have attempted to design related drugs that maintain the biological activity, but do not possess the cardiotoxicity of the anthracyclines.[5] Pixantrone was developed to reduce heart damage related to treatment while retaining efficacy.[1]
Random screening at the US National Cancer Institute of a vast number of compounds provided by the Allied Chemical Company led to the discovery of ametantrone as having significant anti-tumor activity. Further investigation regarding the rational development of analogs of ametantrone led to the synthesis of mitoxantrone, which also exhibited marked anti-tumor activity[5] Mitoxantrone was considered as an analog of doxorubicin with less structural complexity but with a similar mode of action. In clinical studies, mitoxantrone was shown to be effective against numerous types of tumors with less toxic side effects than those resulting from doxorubicin therapy. However, mitoxantrone was not totally free of cardiotoxicity. A number of structurally modified analogs of mitoxantrone were synthesized and structure-activity relationship studies made.[5] BBR 2778 was originally synthesized by University of Vermont researchers Miles P. Hacker and Paul A. Krapcho[5] and initially characterized in vitro for tumor cell cytotoxicity and mechanism of action by studies at the Boehringer Mannheim Italia Research Center, Monza, and University of Vermont, Burlington.[4]Other studies have been completed at the University of Texas M. D. Anderson Cancer Center, Houston, the Istituto Nazionale Tumori,Milan, and the University of Padua.[2][6][4] In the search for novel heteroanalogs of anthracenediones, it was selected as the most promising compound. Toxicological studies indicated that BBR 2778 was not cardiotoxic, and US patents are held by the University of Vermont. An additional US patent application was completed in June 1995 by Boehringer Mannheim, Italy.[5]
Novuspharma, an Italian company, was established in 1998 following the merger of Boehringer Mannheim and Hoffmann-La Roche, and BBR 2778 was developed as Novuspharma’s leading anti-cancer drug, pixantrone.[7] A patent application for the injectable preparation was filed in May 2003.[8]
In 2003, Cell Therapeutics, a Seattle biotechnology company, acquired pixantrone through a merger with Novuspharma.[9]
Clinical trials
Pixantrone is a substance that is being studied in the treatment of cancer. It belongs to the family of drugs called antitumor antibiotics.[10] phase III clinical trials of pixantrone have been completed.[11][12] Pixantrone is being studied as an antineoplastic for different kinds of cancer, including solid tumors and hematological malignancies such as non-Hodgkin lymphomas.
Animal studies demonstrated that pixantrone does not worsen pre-existing heart muscle damage, suggesting that pixantrone may be useful in patients pretreated with anthracyclines. While only minimal cardiac changes are observed in mice given repeated cycles of pixantrone, 2 cycles of traditional anthracyclines doxorubicin or mitoxantrone result in marked or severe heart muscle degeneragion.[1]
Clinical trials substituting pixantrone for doxorubicin in standard first-line treatment of patients with aggressive non-Hodgkin’s lymphoma, had a reduction in severe side effects when compared to patients treated with standard doxorubicin-based therapy. Despite pixantrone patients receiving more treatment cycles, a three-fold reduction in the incidence of severe heart damage was seen as well as clinically significant reductions in infections and thrombocytopenia, and a significant reduction in febrile neutropenia. These findings could have major implications for treating patients with breast cancer, lymphoma, and leukemia, where debilitating cardiac damage from doxorubicin might be prevented.[13]Previous treatment options for multiply relapsed aggressive non-Hodgkin lymphoma had disappointing response rates.[14]
The completed phase II RAPID trial compared the CHOP-R regimen of Cyclophosphamide, Doxorubicin, Vincristine, Prednisone, and Rituximab to the same regimen, but substituting Doxorubicin with Pixantrone. The objective was to show that Pixantrone was not inferior to Doxorubicin and less toxic to the heart.[15]
Pixantrone was shown to have potentially reduced cardiotoxicity and demonstrated promising clinical activity in these phase II studies in heavily pretreated non-Hodgkin lymphomapatients.[14]
The pivotal phase III EXTEND (PIX301) randomized clinical trial studied pixantrone to see how well it works compared to other chemotherapy drugs in treating patients with relapsed non-Hodgkin’s lymphoma.[16] The complete response rate in patients treated with pixantrone has been significantly higher than in those receiving other chemotherapeutic agents for treatment of relapsed/refractory aggressive non-Hodgkin lymphoma.[14]
Administration
It can be administered through a peripheral vein rather than a central implanted catheter as required for other similar drugs.[8][14]
Regulatory approval
U.S. Food and Drug Administration
The FDA granted fast track designation for pixantrone in patients who had previously been treated two or more times for relapsed or refractory aggressive NHL. Study sponsor Cell Therapeutics announced that Pixantrone achieved the primary efficacy endpoint. The minutes of the Oncologic Drugs Advisory Committee meeting of March 22, 2010[17]show that this had not in fact been achieved with statistical significance and this combined with major safety concerns lead to the conclusion that the trial was not sufficient to support approval. In April 2010 the FDA asked for an additional trial.[18]
European Medicines Agency
On May 5, 2009, Pixantrone became available in Europe on a Named-Patient Basis. A named-patient program is a compassionate use drug supply program under which physicians can legally supply investigational drugs to qualifying patients. Under a named-patient program, investigational drugs can be administered to patients who are suffering from serious illnesses prior to the drug being approved by the European Medicines Evaluation Agency. “Named-patient” distribution refers to the distribution or sale of a product to a specific healthcare professional for the treatment of an individual patient. In Europe, under the named-patient program the drug is most often purchased through the national health system.[19] In 2012 pixantrone received conditional marketing authorization in the European Union as Monotherapy to Treat Adult Patients with Multiply Relapsed or Refractory Aggressive Non-Hodgkin B-Cell Lymphomas.
Research
Pixantrone is as potent as mitoxantrone in animal models of multiple sclerosis.[20] Pixantrone has a similar mechanism of action as mitoxantrone on the effector function of lymphomonocyte B and T cells in experimental allergic encephalomyelitis but with lower cardiotoxicity. Pixantrone inhibits antigen specific and mitogen induced lymphomononuclear cell proliferation, as well as IFN-gamma production.[21] Clinical trials are currently ongoing in Europe.
Pixantrone also reduces the severity of experimental autoimmune myasthenia gravis in Lewis rats,[22] and in vitro cell viability experiments indicated that Pixantrone significantly reduces amyloid beta (A beta(1-42)) neurotoxicity, a mechanism implicated in Alzheimer’s disease.[23]


http://www.chemdrug.com/databases/8_0_mhpyqlxgrykqdwig.html
3,4-Pyridinedicarboxylic acid (I) was converted to the cyclic anhydride (II) upon heating with acetic anhydride. Friedel-Crafts condensation of anhydride (II) with p-difluorobenzene (III) in the presence of AlCl3 gave rise to a mixture of two regioisomeric keto acids, (IV) and (V). Cyclization of this mixture in fuming sulfuric acid at 140 C generated the benzoisoquinoline (VI) (1,2). Subsequent displacement of the fluorine atoms of (VI) with ethylenediamine ( VII) in pyridine provided the target bis (2-aminoethylamino) derivative, which was finally converted to the stable dimaleate salt. Alternatively, ethylenediamine (VII) was protected as the mono-N-Boc derivative (VIII) by treatment with Boc2O. Condensation of the difluoro compound (VI) with the protected ethylenediamine (VIII) furnished (IX). The Boc groups of (IX) were then removed by treatment with trifluoroacetic acid. After adjustment of the pH to 4.2 with KOH, treatment with maleic acid provided BBR-2778.
J Med Chem1994,37, (6): 828
SEE MORE
http://www.chemdrug.com/databases/8_0_mhpyqlxgrykqdwig.html
References
- Cavalletti E, Crippa L, Mainardi P, Oggioni N, Cavagnoli R, Bellini O, Sala F. (2007). “Pixantrone (BBR 2778) has reduced cardiotoxic potential in mice pretreated with doxorubicin: comparative studies against doxorubicin and mitoxantrone”. Invest New Drugs. 25 (3): 187–95. doi:10.1007/s10637-007-9037-8. PMID 17285358.
- De Isabella P, Palumbo M, Sissi C, Capranico G, Carenini N, Menta E, Oliva A, Spinelli S, Krapcho AP, Giuliani FC, Zunino F. (1995). “Topoisomerase II DNA cleavage stimulation, DNA binding activity, cytotoxicity, and physico-chemical properties of 2-aza- and 2-aza-oxide-anthracenedione derivatives”. Mol Pharmacol. 48 (1): 30–8.PMID 7623772.
- Evison BJ, Mansour OC, Menta E, Phillips DR, Cutts SM (2007). “Pixantrone can be activated by formaldehyde to generate a potent DNA adduct forming agent”. Nucleic Acids Res. 35 (11): 3581–9. doi:10.1093/nar/gkm285. PMC 1920253.PMID 17483512.
- Krapcho AP, Petry ME, Getahun Z, Landi JJ Jr, Stallman J, Polsenberg JF, Gallagher CE, Maresch MJ, Hacker MP, Giuliani FC, Beggiolin G, Pezzoni G, Menta E, Manzotti C, Oliva A, Spinelli S, Tognella S (1994). “6,9-Bis[(aminoalkyl)amino]benzo[g]isoquinoline-5,10-diones. A novel class of chromophore-modified antitumor anthracene-9,10-diones: synthesis and antitumor evaluations”. J Med Chem. 37 (6): 828–37. doi:10.1021/jm00032a018. PMID 8145234.
- US patent 5587382, Krapcho AP, Hacker MP, Cavalletti E, Giuliani FC, “6,9-bis[(2-aminoethyl) amino]benzo [g]isoquinoline-5,10- dione dimaleate; an aza-anthracenedione with reduced cardiotoxicity”, issued 1996-12-24, assigned to Boehringer Mannheim Italia, SpA
- Zwelling LA, Mayes J, Altschuler E, Satitpunwaycha P, Tritton TR, Hacker MP. (1993). “Activity of two novel anthracene-9,10-diones against human leukemia cells containing intercalator-sensitive or -resistant forms of topoisomerase II”. Biochem Pharmacol. 46 (2): 265–71. doi:10.1016/0006-2952(93)90413-Q. PMID 8394077.
- Borchmann P, Reiser M (May 2003). “Pixantrone (Novuspharma)”. IDrugs 6 (5): 486–90. PMID 12789604.
- EP patent 1503797, Bernareggi A, Livi V, “Injectable Pharmaceutical Compositions of an Anthracenedione Derivative with Anti-Tumoral Activity”, published 2003-11-27, issued 2008-09-29, assigned to Cell Therapeutics Europe S.R.L.
- Pollack, Andrew (2003-06-17). “Company News; Cell Therapeutics Announces Plan To Buy Novuspharma”. The New York Times. Retrieved 2010-05-22.
- Jump up^ Mosby’s Medical Dictionary, 8th edition. © 2009, Elsevier. “definition of antineoplastic antibiotic”. Free Online Medical Dictionary, Thesaurus and Encyclopedia. Retrieved 2012-01-31.
- Jump up^ “NCT00088530”. BBR 2778 for Relapsed, Aggressive Non-Hodgkin’s Lymphoma (NHL). ClinicalTrials.gov. Retrieved 2012-01-31.
- “NCT00551239”. Fludarabine and Rituximab With or Without Pixantrone in Treating Patients With Relapsed or Refractory Indolent Non-Hodgkin Lymphoma. ClinicalTrials.gov. 2012-01-31. Retrieved 2012-01-31.
- “Pixantrone Combination Therapy for First-line Treatment of Aggressive Non-Hodgkin’s Lymphoma Results in Reduction in Severe Toxicities Including Heart Damage When Compared to Doxorubicin-based Therapy”. Press Release. Retrieved 2012-01-31.
- ^ Jump up to:a b c d Engert A, Herbrecht R, Santoro A, Zinzani PL, Gorbatchevsky I (September 2006). “EXTEND PIX301: a phase III randomized trial of pixantrone versus other chemotherapeutic agents as third-line monotherapy in patients with relapsed, aggressive non-Hodgkin’s lymphoma”. Clin Lymphoma Myeloma 7 (2): 152–4.doi:10.3816/CLM.2006.n.055. PMID 17026830.
- Jump up^ “NCT00268853”. A Trial in Patients With Diffuse Large-B-cell Lymphoma Comparing Pixantrone Against Doxorubicin. ClinicalTrials.gov. Retrieved 2012-01-31.
- Jump up^ “NCT00101049”. BBR 2778 for Relapsed, Aggressive Non-Hodgkin’s Lymphoma (NHL). ClinicalTrials.gov. Retrieved 2012-01-31.
- Jump up^ Vesely N, Eckhardt SG (2010-03-22). “NDA 022-481 PIXUVRI (pixantrone dimaleate) injection” (pdf). Summary Minutes of the Oncologic Drugs Advisory Committee. United States Food and Drug Administration. Retrieved 2012-01-31.
- Jump up^ “Cell Therapeutics Formally Appeals FDA’s Nonapprovable Ruling for Pixantrone”. GEN News. 2010-12-03.
- Jump up^ “Pixantrone Now Available in Europe on a Named-Patient Basis”. Retrieved 2012-01-31.
- Jump up^ Gonsette RE, Dubois B (August 2004). “Pixantrone (BBR2778): a new immunosuppressant in multiple sclerosis with a low cardiotoxicity”. J. Neurol. Sci. 223(1): 81–6. doi:10.1016/j.jns.2004.04.024. PMID 15261566.
- Jump up^ Mazzanti B, Biagioli T, Aldinucci A, Cavaletti G, Cavalletti E, Oggioni N, Frigo M, Rota S, Tagliabue E, Ballerini C, Massacesi L, Riccio P, Lolli F (November 2005). “Effects of pixantrone on immune-cell function in the course of acute rat experimental allergic encephalomyelitis”. J. Neuroimmunol. 168 (1-2): 111–7.doi:10.1016/j.jneuroim.2005.07.010. PMID 16120465.
- Jump up^ Ubiali F, Nava S, Nessi V, Longhi R, Pezzoni G, Capobianco R, Mantegazza R, Antozzi C, Baggi F (February 2008). “Pixantrone (BBR2778) reduces the severity of experimental autoimmune myasthenia gravis in Lewis rats”. J. Immunol. 180 (4): 2696–703. PMID 18250482.
- Jump up^ Colombo R, Carotti A, Catto M, Racchi M, Lanni C, Verga L, Caccialanza G, De Lorenzi E (April 2009). “CE can identify small molecules that selectively target soluble oligomers of amyloid beta protein and display antifibrillogenic activity”. Electrophoresis 30(8): 1418–29. doi:10.1002/elps.200800377. PMID 19306269.

{kind=link}
{kind=link}
{kind=link}