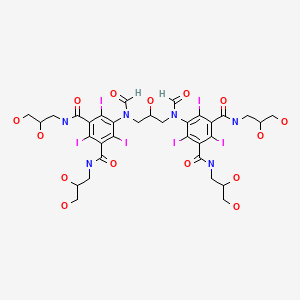
ioforminol
Ioforminol [INN], UNII-95FNF21CDN, 1095110-48-7, FEK-256-062
5-[formyl-[3-[formyl-[3,5-bis(2,3-dihydroxypropylcarbamoyl)-2,4,6- triiodophenyl]amino]-2-hydroxypropyl]amino]-N,N’-bis(2,3-dihydroxypropyl)-2,4,6-triiodobenzene- 1 ,3-dicarboxamide
1,3-Benzenedicarboxamide, 5,5′-[(2-hydroxy-1,3-
propanediyl)bis(formylimino)]bis[N1,N3-bis(2,3-dihydroxypropyl)-2,4,6-triiodo-
All-ambo-5,5′-[2-hydroxypropane-1,3-diylbis(formylazanediyl)]bis[N,N’-bis(2,3-
dihydroxypropyl)-2,4,6-triiodobenzene-1,3-dicarboxamide]
https://download.ama-assn.org/resources/doc/usan/x-pub/ioforminol.pdf
MOLECULAR FORMULA C33H40I6N6O15
MOLECULAR WEIGHT 1522.1
SPONSOR GE HealthCare Ltd
CODE DESIGNATION FEK-256-062
CAS REGISTRY NUMBER 1095110-48-7
WHO NUMBER 9245
Visualisation of anatomical structures of the body during computed tomography for diagnostic purposes
All diagnostic imaging is based on the achievement of different signal levels from different structures within the body. Thus, in X-ray imaging for example, for a given body structure to be visible in the image, the X-ray attenuation by that structure must differ from that of the surrounding tissues. The difference in signal between the body structure and its surroundings is frequently termed contrast and much effort has been devoted to means of enhancing contrast in diagnostic imaging since the greater the contrast between a body structure and its surroundings the higher the quality of the images and the greater their value to the physician performing the diagnosis. Moreover, the greater the contrast the smaller the body structures that may be visualized in the imaging procedures, i.e. increased contrast can lead to increased spatial resolution. The diagnostic quality of images is strongly dependent on the inherent noise level in the imaging procedure, and the ratio of the contrast level to the noise level can thus be seen to represent an effective diagnostic quality factor for diagnostic images.
For the last 50 years the field of X-ray contrast agents has been dominated by soluble iodine containing compounds. Commercial available contrast media containing iodinated contrast agents are usually classified as ionic monomers such as diatrizoate (Gastrografen™), ionic dimers such as ioxaglate (Hexabrix™), nonionic monomers such as iohexol (Omnipaque™), iopamidol (Isovue™), iomeprol (lomeron™) and the non-ionic dimer iodixanol (Visipaque™). The most widely used commercial non-ionic X-ray contrast agents such as those mentioned above are considered safe. Contrast media containing iodinated contrast agents are used in more than 20 million of X-ray examinations annually in the USA and the number of adverse reactions is considered acceptable. However, since a contrast enhanced X- ray examination will require up to about 200 ml contrast media administered in a total dose, there is a continuous drive to provide improved contrast media.
Achieving improvement in such a diagnostic quality factor has long been and still remains an important goal.
In techniques such as X-ray, one approach to improve the diagnostic quality factor has been to introduce contrast enhancing materials formulated as contrast media into the body region being imaged. Thus for X-ray, early examples of contrast agents were insoluble inorganic barium salts which enhanced X-ray attenuation in the body zones into which they distributed. For the last 50 years the field of X-ray contrast agents has been dominated by soluble iodine containing compounds.
Commercial available contrast media containing iodinated contrast agents are usually classified as ionic monomers such as diatrizoate (marketed e.g. under the trade mark Gastrografen™), ionic dimers such as ioxaglate (marketed e.g. under the trade mark Hexabrix™), nonionic monomers such as iohexol (marketed e.g. under the trade mark Omnipaque™), iopamidol (marketed e.g. under the trade mark Isovue™), iomeprol (marketed e.g. under the trade mark Iomeron™) and the non-ionic dimer iodixanol (marketed under the trade mark Visipaque™). The clinical safety of iodinated X-ray contrast media has continuously been improved over the recent decades through development of new agents; from ionic monomers (Isopaque™) to non-ionic monomers (e.g. Omnipaque™) and non-ionic dimers (e.g. Visipaque™).
The utility of the contrast media is governed largely by its toxicity, by its diagnostic efficacy, by adverse effects it may have on the subject to which the contrast medium is administered, but also by the ease of production, storage and administration. The toxicity and adverse biological effects of a contrast medium are contributed to by the components of the formulation medium, i.e. of the diagnostic composition, e.g. the solvent or carrier as well as the contrast agent itself and its components such as ions for the ionic contrast agents and also by its metabolites.
The manufacture of non-ionic X-ray contrast media involves the production of the active
pharmaceutical ingredient (API), i.e. the contrast agent prepared in the primary production, followed by the formulation into the drug product, herein denoted the X-ray composition, prepared in the secondary production. In the preparation of an X-ray composition, the contrast agent is admixed with additives, such as salts, optionally after dispersion in a physiologically tolerable carrier. The contrast agent has to be completely solved in the carrier when additives are included and the composition is prepared. A well-known process for preparing X-ray compositions includes heating the contrast agent in the carrier, such as water for injection, to ensure complete dissolution. For instance, for the contrast media Visipaque™ the secondary production process includes dissolution of the contrast agent iodixanol in water for injection and heating to about 98 °C. Heating at this temperature for an adequate period of time ensures that the contrast agent is completely dissolved.
However, different X-ray contrast agents have different solubility. For instance WO 2009/008734 of GE Healthcare AS discloses a new class of compounds and their use as X-ray contrast agents. The compounds are dimers containing two linked iodinated phenyl groups. Compound I, now called
Ioforminol, falling within the formula I of WO2009/008734, has been found by the applicant to have particularly favourable properties. Ioforminol is supersaturated at the relevant storage conditions.
Compound I, Ioforminol:
5-[formyl-[3-[formyl-[3,5-bis(2,3-dihydroxypropylcarbamoyl)-2,4,6- triiodophenyl]amino]-2-hydroxypropyl]amino]-N,N’-bis(2,3-dihydroxypropyl)-2,4,6-triiodobenzene- 1 ,3-dicarboxamide.
A solution in which the concentration of the solute (API) exceeds the equilibrium solute concentration at a given temperature is said to be supersaturated. This is possible because the solute does not precipitate immediately when the solution is cooled below the saturation temperature. Such solutions are denoted supersaturated.
As the solubility of Ioforminol decreases with decreasing temperature, the supersaturation increases. At room temperature the solubility of Ioforminol is limited. To achieve solutions with a concentration higher than the thermodynamic equilibrium concentration, at room temperature, Ioforminol is dissolved at a temperature above room temperature. When a clear solution has been achieved the solution is cooled and enters a state defined as supersaturated.
Supersaturated solutions are thermodynamically unstable and prone to nucleate and therefore to precipitate on storage. Among several factors, the onset of the precipitation depends on the degree of supersaturation, presence of the crystals of the solute and foreign particles such as dust or other impurities, i.e. purity, and storage temperature of the solution.
The injection solution of Ioforminol, i.e. the administrable X-ray composition, is highly supersaturated. The nucleation (precipitation) in the injection solution at storage conditions is strongly undesirable. The physical stability of the solution, i.e. prevention of the nucleation for a certain time at storage conditions, may be improved substantially by heat treatment of the solution well above its saturation temperature for a sufficiently long period of time.
WO2011/117236 of the applicant is directed to a process involving hea treatment at low pH to avoid degradation and precipitation of an X-ray contrast agent composition. However, a high heat load is needed to obtain a seed- free solution. This heat load causes a greater degradation of the product and a lower pH in the final product resulting in liberation of iodine. This sets a restriction to the total heat load that may be given to the formulated solution.
………………..
WO2014052091A1
X-ray contrast media containing a chemical compound as the active pharmaceutical ingredient(s) having two triiodinated phenyl groups linked by a linking group are usually referred to as dimeric contrast agents or dimers. During the years a wide variety of iodinated dimers have been proposed. Currently, one contrast medium having an iodinated non-ionic dimer as the active pharmaceutical ingredient is on the market^ the product Visipaque™ containing the compound iodixanol. In WO2009/008734 of the applicant a novel dimeric contrast agent named loforminol is disclosed.
The properties of this is described in more detail in the publications Chai et al. “Predicting cardiotoxicity propensity of the novel iodinated contrast medium GE-145: ventricular fibrillation during left coronary arteriography in pigs”, Acta Radiol, 2010, and in Wistrand, L.G., et al “GE-145, a new low-osmolar dimeric radiographic contrast medium”, Acta Radiol, 2010. loforminol (GE-145) is named Compound 1 herein and has the following structure:
Compound 1 :
5,5′-(2-Hydroxypropane-1 ,3-diyl)bis(formylazanediyl)bis(N1 ,N3-bis(2,3- dihydroxypropyl)-2,4,6-triiodoisophthalamide)
The manufacture of non-ionic X-ray contrast media involves the production of the chemical drug, the active pharmaceutical ingredient (API), i.e. the contrast agent, followed by the formulation into the drug product, herein denoted the X-ray composition. WO2009/008734 of the applicant provides a synthetic route for preparing the API loforminol.
loforminol can e.g., as provided by the general preparation description and Example 1 of WO2009/008734, be synthesized from 5- amino-N,N’-bis-(2,3-dihydroxy-propyl)-2,4,6-triiodo-isophthalamide (compound (4)), which is commercially available. The preparation of this compound is known from the synthesis of both iohexol and iodixanol and can also be prepared from 5- nitroisophthalic acid for instance as described in WO2006/016815, including hydrogenation and subsequent iodination e.g. by iodine chloride, I CI. Alternatively,
5-amino-2,4,6-triiodoisophthalic acid may be used, which is commercially available precursor, e.g. from Sigma-Aldrich. The free amino group of the isophthalamide compound (compound (4)) is then acylated and the hydroxyl groups in the substituents may also be protected by acylation. The protecting groups may be removed for example by hydrolysis to give N1 ,N3-bis(2,3-dihydroxypropyl)-5- formylamino-2,4,6-triiodoisophthalamide.
In a dimerization step this is reacted e.g. with epichlorohydrin to provide the loforminol contrast agent compound. The state of the art synthesis of loforminol, as disclosed in examples 1 and 2 of WO2009/008734, is shown in Scheme 1 below.
Scheme 1 .
As described in WO2009/008734 compound 3 is a mixture comprising 1 – formylamino-3,5-bis(2,3-bis(formyloxy)propan-1 -ylcarbamoyl)-2,4,6-trioodobenzene, and X is then a formyl group. In each synthetic step it is important to optimize the yield and minimize the production of impurities. The problem to be solved by the present invention may be regarded as the provision of optimizing the process for preparation of compound mixture (3) of scheme 1 , i.e. a mixture comprising 1 -formylamino-3,5-bis(2,3- bis(formyloxy)propan-1 -ylcarbamoyl)-2,4,6-trioodobenzene.
The process is hence directed to the preparation of compound mixture (3) by the formylation of the amino function of 5-amino-N1 ,N3-bis(2,3-dihydroxypropyl)-2,4,6-triiodoisophthalamide (4), including a work-up procedure.
Examples
Example 1 : Preparation of compound mixture (3) comprising 1-formylamino- 3,5-bis(2,3-bis(formyloxy)propan-1-ylcarbamoyl)-2,4,6-trioodobenzene
5-amino-N1 ,N3-bis(2,3-dihydroxypropyl)-2,4,6-triiodoisophthalamide (compound (4)) (7.5 kg, 10.6 moles) was dissolved in formic acid (4.9 I) and heated to 45 until a clear solution was obtained (~4 hours), then the thick amber solution was cooled to 10 °C.
Formic acid (9.4 I) was charged into a different reactor and cooled to 10 <€, after reaching the target temperature acetic anhydride was added at such a rate that the temperature did not exceeded 15 <€.
After 2.5 hours all acetic anhydride was added to the formic acid and the mixed anhydride solution was added drop wise to the compound (4) solution. The rate of addition was adjusted so that the temperature never exceeded 20 °C. After 2 hours all mixed anhydride had been added and the reaction was left stirring at 15 °C for additional 1 hour. Isopropanol (4.9 I) was added carefully and the suspension became noticeable thicker and was left stirring at ambient temperature. After 16 hours the reaction slurry was filtered on a vacuum nutch and washed with isopropanol (3 * 1 .5 I) to give compound mixture (3) comprising 1 -formylamino-3,5- bis(2,3-bis(formyloxy)propan-1 -ylcarbamoyl)-2,4,6-trioodobenzene as a dense white powder (7.98kg). The quantitative yield with regards to N-formylation was > 99 %.
…………….
WO2009008734A2
Preparation of intermediates (when not commercially available)
The precursors to the compounds of formulas (IVa) and (IVb), the tri-iodinated phenyl groups having a free amino group are commercially available or can be produced following procedures described or referred to e.g. in WO95/35122 and WO98/52911. 5-amino-2,4,6-triiodo-isophtalic acid for example is available e.g. from Aldrich and 5-amino-2,4,6-triiodo-N,N’-bis(2,3-dihydroxypropyl)-isophtalamide is commercially available e.g. from Fuji Chemical Industries, Ltd.
Examples of commercial available precursors of the compounds of formulas (IVa) and (IVb), either commercially available or previously described in the literature include:
5-Amino-N,N’-bis-(2,3-dihydroxy-propyl)-2,4,6-triiodo-isophthalamide
5-Amino-N-(2,3-dihydroxy-propyl)-N’-(2-hydroxy-1-hydroxymethyl-ethyl)- 2,4,6-triiodo-isophthalamide (WO2002044125)
5-Amino-N,N’-bis-(2,3-dihydroxy-propyl)-2,4,6-triiodo-N,N’-dimethyl- isophthalamide
5-Amino-N-(2,3-dihydroxy-propyl)-N’-(2-hydroxy-ethyl)-2,4,6-triiodo-is ophthalamide (WO 8700757)
The compounds of formulas (IVa) and (IVb), may be prepared by acylation of the corresponding compounds having free amino groups. In this reaction, hydroxyl groups in the substituents R may also be protected by acylation.
Acylation may be effected by any convenient method, e.g. by use of activated formic acid such as mixed anhydrides which can prepared by a variety of methods described in the literature.
A convenient method of preparing mixed anhydrides is to add a carboxylic acid anhydride to an excess of formic acid under controlled temperature. It is also possible to make mixed anhydrides by addition of a carboxylic acid chloride to a solution of a formic acid salt. Formyl-mixed anhydrides may include acetyl, isobutyryl, pivaloyl, benzoyl etc.
In the present implementation acetic-formic mixed anhydride is employed. To an excess of cooled pre-prepared acetic-formic mixed anhydride is added a 5-amino- monomer and the mixture is stirred overnight. The mixture is concentrated in vacuo and may be used directly in the alkylation step as described in the experimental section (procedure B) or alternatively the O-acylated groups may be hydrolysed prior to alkylation as described in the experimental section (procedure A). Hydrolysis is conveniently performed in aqueous basic media as exemplified in the experimental section or may alternatively be effected by alcoholysis e.g. as described in WO1997000240.
It is also possible to dissolve the 5-aminomonomer in formic acid and subsequently add the carboxylic acid anhydride but in order to reduce unwanted acylation it is preferred to prepare the mixed anhydride separately and subsequently mix this with the 5-aminomonomer as described above.
Experimental
Example 1
5,5′-(2-hvdroxypropane-1 ,3-diyl)bis(formylazanediyl)bis(N1,N3-bis(2,3- dihvdroxypropyl)-2.4,6-triiodoisophthalamide)
Procedure A:
1 a) N,N’-Bis-(213-dihvdroxy-propyl)-5-formylamino-2,4,6-triiodo-isophthalamide Formic acid (300 ml) was charged in a dry 1000 ml flask fitted with a dropping funnel, stir bar, thermometer and a gas inlet. The acid was cooled on an ice bath under a nitrogen blanket and acetic anhydride (144.8 g, 1.418 mol) was added drop wise at a rate so that the temperature did not exceed 2.5 C. After complete addition, the ice bath was removed and the temperature was allowed to reach 10 °C. The mixture was again ice cooled and 5-amino-N,N’-bis(2,3-dihydroxypropyl)-2,4,6- triiodo-isophthalamide (100 g, 141.8 mmol) was added over 5 minutes and the mixture was left stirring over night while attaining ambient temperature. The mixture was evaporated to dryness and methanol (300 ml) and water (300 ml) was added. 2 M potassium hydroxide was added until all material was in solution and a stable pH 12.5 was attained. The methanol was removed in vacuo. The mixture was neutralized with 4 M HCI and a slow precipitation started. 300 ml water was added and the product was precipitated over night. The precipitate was collected and rinsed with a small amount of water and dried on filter to a moist cake and further dried in vacuo to yield 84.8 g ( 81.5 %) of N,N’-bis-
(2,3-dihydroxy-propyl)-5-formylamino-2,4,6-triiodo-isophthalamide.
1H-NMR 500 MHz (solvent: D2O, ref. H2O=4.8 ppm, 25 0C): 8.35 and 8.05 ppm (2s,
1 H), 3.94 ppm (m, 2H), 3.67 ppm (m, 2H), 3.55 ppm (m, 2H), 3.45 ppm (m, 2H),
3.34 ppm (m, 2H).
LC-MS (column Agilent Zorbax SB-Aq 3.5 μm 3.0 x 100 mm, solvents: A = water/ 0.1 % formic acid and B = acetonitrile/ 0.1% formic acid; gradient 0-30 % B over 20 min; flow 0.3 ml/ min, UV detection at 214 and 254 nm, ESI-MS) gave two peaks centred at 5.5 minutes with m/z (M + H+) 733.828, m/z (M + NH4+) 750.855, m/z (M + Na+) 755.817 corresponding to the structure.
1 b) 5,5′-(2-hvdroxypropane-1 ,3-diyl)bis(formylazanediyl)bis(N1,N3-bis(2,3- dihvdroxypropyl)-2,4,6-triiodoisophthalamide)
Potassium hydroxide (1.07 g) was dissolved in water (6.9 ml) and methanol (3.4 ml) in a 50 ml round bottomed flask fitted with a magnetic stir bar. Boric acid (0.41 g, 6.6 mmol) and N,N’-bis-(2,3-dihydroxy-propyl)-5-formylamino-2,4,6-triiodo- isophthalamide (7.0 g, 9.56 mmol) was added to the stirred solution.. Epichlorohydrin (260 ul, 3.32 mmol) was added to the solution and a pH electrode was fitted in the flask and the pH was maintained at pH 12.7 by drop wise addition of 4 M potassium hydroxide for 4 h. At this point, the mixture was left stirring over night. The pH was adjusted with 4 M hydrochloric acid to pH 4 and the methanol was removed in vacuo. The remaining aqueous solution was diluted with water (75 ml) and treated with ion exchangers (AMB200C and IRA67) to zero conductivity. The ion exchangers were removed by filtration and rinsed with water and the combined aqueous filtrates were freeze dried. The crude product was purified by preparative HPLC (column Phenomenex Luna C18 10 μm solvents: A = water and B = acetonitrile; gradient 05-20 % B over 60 min. After freeze drying 3.80 g of 5,5′- (2-hydroxypropane-1 ,3-diyl)bis(formylazanediyl)bis(N1,N3-bis(2,3-dihydroxypropyl)- 2,4,6-triiodoisophthalamide) (74.8 % yield) was obtained.
1H-NMR 500 MHz (solvent: D2O, ref. H2O=4.8 ppm, 25 0C): 8.34 and 8.08 ppm (m, 2 H), 2.80-4.80 ppm (m 26 H). LC-MS TOF; 1522.68 m/z (M + H+), 1544.66 m/z (M + Na+).
…………
New patent
WO-2014052091
Process for the preparation of 1-formylamino-3,5-bis(2,3-bis(formyloxy)propan-1-ylcarbamoyl)-2,4,6-trioodobenzene, used as a key intermediate in the preparation of ioforminol. Also claims a process for the preparation of ioforminol, useful in X-ray imaging. GE Healthcare is developing ioforminol (GE-145; AN-113111) as an iv contrast agent (Phase 2). See WO2013104690 claiming X-ray imaging contrast media with low iodine concentration and X-ray imaging process. Also see concurrently published WO2014052092 claiming preparation of ioforminol. Appears to be the first filing from Medi-Physics on this compound.
……………
The most preferred iodinated agents are;
Diatrizoic acid
loxaglinic acid
loversol
lodixanol
lomeprol
lobitriol
The most preferred chelates are:
Gadopentetate
Ňadoversetamide
Gadoxetinic acid

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....





















 GADOBUTROL
GADOBUTROL




 WORLDCUP FOOTBALL WEEK 2014 BRAZIL
WORLDCUP FOOTBALL WEEK 2014 BRAZIL










































 NAV4694 is designed to aid visual detection and quantification of cerebral beta amyloid in diagnosing Alzheimer’s disease (AD). One hallmark of AD is the accumulation of beta amyloid plaques between nerve cells in the brain.
NAV4694 is designed to aid visual detection and quantification of cerebral beta amyloid in diagnosing Alzheimer’s disease (AD). One hallmark of AD is the accumulation of beta amyloid plaques between nerve cells in the brain.
















