


| Patent ID | Title | Submitted Date | Granted Date |
|---|---|---|---|
| US8383596 | ANTIBACTERIAL AMINOGLYCOSIDE ANALOGS |
2010-04-22
|
|
| US8822424 | Antibacterial aminoglycoside analogs |
2013-01-04
|
2014-09-02
|
| US2012208781 | AMINOGLYCOSIDE DOSING REGIMENS |
2011-11-11
|
2012-08-16
|
| US2012214759 | TREATMENT OF KLEBSIELLA PNEUMONIAE INFECTIONS WITH ANTIBACTERIAL AMINOGLYCOSIDE COMPOUNDS |
2011-11-11
|
2012-08-23
|
| US2012214760 | TREATMENT OF URINARY TRACT INFECTIONS WITH ANTIBACTERIAL AMINOGLYCOSIDE COMPOUNDS |
2011-11-11
|
2012-08-23
|
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
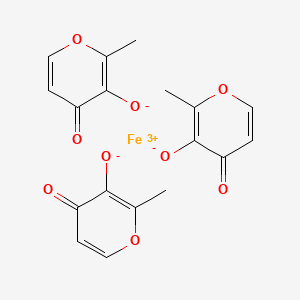
Ferric Maltol, マルトール第二鉄
Ferric Maltol
Iron, tris(3-hydroxy-2-methyl-4H-pyran-4-onato-O3,O4)-
| Molecular Formula: | C18H15FeO9 |
|---|---|
| Molecular Weight: | 431.154 g/mol |
iron(3+);2-methyl-4-oxopyran-3-olate
RN: 33725-54-1
UNII: MA10QYF1Z0
Feraccru
Ferric maltol; UNII-MA10QYF1Z0; MA10QYF1Z0; Ferric maltol (INN); Ferric maltol [INN]; 33725-54-1
Shield Therapeutics, under license from Vitra Pharmaceuticals
UPDATE
- Originator University of Cambridge; University of London
- Developer Shield Therapeutics
- Class Antianaemics; Ferric compounds; Pyrones; Small molecules
- Mechanism of Action Iron replacements
- Marketed Iron deficiency anaemia
Most Recent Events
- 26 Jul 2019 Registered for Iron deficiency anaemia (In adults) in USA (PO)
- 24 Apr 2019 Swissmedic approves a extension of the approved indication for ferric maltol to include treatment of all adults with iron deficiency (ID) with or without anaemia
- 14 Mar 2019 The European Patent Office decides in favour of Shield Therapeutics in relation to the group’s patent No. 2 668 175
The US Food and Drug Administration (FDA) has approved oral ferric maltol(Accrufer, Shield Therapeutics) AUG 2019 for the treatment of iron deficiency in adults.
The product is already approved in the European Union and Switzerland for the treatment of iron deficiency in adults, where it is sold as Feraccru.
OLD DATA NDA filing expected in US in 2H 2018, Ph 3 trial is planned in 2018/19 for treatment of iron deficiency anemia (IDA) in children., Expected dose form: Oral Capsule; 30 mg
Treatment of iron deficiency anemia (IDA) associated with inflammatory bowel disease (IBD) and Chronic Kidney disease.
Iron deficiency anaemia (IDA) occurs when iron levels are insufficient to support red blood cell production and is defined – according to the WHO – as haemoglobin levels below 13 g/dL in men over 15 years, below 12 g/dL in non-pregnant women over 15 years, and below 11 g/dL in pregnant women. Iron is absorbed at the apical surface of enterocytes to be transported by ferroportin, the only known iron exporter, across the basolateral surface of the enterocyte into circulation. Inflammation from IBD interferes with iron absorption by causing an increase in hepcidin, a peptide hormone synthesized in the liver that inhibits ferroportin activity. Anaemia is the most common extra-intestinal complication of inflammatory bowel disease (IBD) and although it often involves a combination of IDA and anaemia of chronic disease, IDA remains an important contributor in this condition due to chronic intestinal bleeding and decreased iron intake (from avoidance of foods that may exacerbate symptoms of IBD). In a variety of populations with IBD, the prevalence of iron deficiency anaemia ranges from 36%-76%. The serum markers of iron deficiency are low ferritin, low iron, raised total iron binding capacity, raised red cell protoporhyrin and increased transferrin binding receptor (sTfR). Serum ferritin is the most powerful test for iron deficiency. The cut-off level of ferritin which is diagnostic varies between 12-15 µg/L. Higher levels of serum ferritin do not exclude the possibility of iron deficiency, and a serum ferritin level of <100 μg/L may still be consistent with iron deficiency in patients with IBD. A transferrin saturation of <16% is indicative of iron deficiency, either absolute or functional. Other findings on a complete blood count panel that are suggestive of iron deficiency anaemia, but are not considered diagnostic, include microcytosis, hypochromia, and elevation of red cell distribution width.
A deficiency of iron can have a significant impact on a patient’s quality of life. Appropriate diagnosis and treatment of iron deficiency anaemia are important to improve or maintain the quality of life of patients. The goals of treatment are to treat the underlying cause, limit further blood loss or malabsorption, avoid blood transfusions in haemodynamically stable patients, relieve symptoms, and improve quality of life. More specifically, therapeutic goals of treatment include normalizing haemoglobin levels within 4 weeks (or achieving an increase of >2 g/dL) and replenishing iron stores (transferrin saturation >30%). Oral iron supplementation has been considered standard treatment because of an established safety profile, lower cost, and ease of administration. It has been shown to be effective in correcting anaemia and repleting iron stores. One concern with higher doses of daily oral iron is intolerance due to GI side effects. Symptoms include nausea, vomiting, diarrhea, abdominal pain, constipation, and melena-like stools. Guidelines on the Diagnosis and Management of Iron Deficiency and Anaemia in Inflammatory Bowel Diseases recommend IV iron therapy over oral iron supplementation in the treatment of iron deficiency anaemia in patients with IBD, citing faster and prolonged response to treatment, decreased irritation of existing GI inflammation, improved patient tolerance, and improved quality of life. Patients with severe anaemia (haemoglobin level of <10 g/dL), failure to respond or intolerance to oral iron therapy, severe intestinal disease or patients receiving concomitant erythropoietin are recommended indications for IV iron therapy. Other conditions where patients should be considered for first-line IV therapy over oral therapy include congestive heart failure, upper GI bleeding, and in situations where rapid correction of anaemia may be required.
Across EU there are several iron (Fe+2) oral preparations as ferrous fumarate, ferrous gluconate, ferrous sulphate and ferrous glycine sulfate, formulated as tablet, solution or gastroresistent capsules. All ferrous compounds are oxidised in the lumen of the gut or within the mucosa with release of activated hydroxyl radicals, which may attack the gut wall and can effect a range of gastrointestinal symptoms and discomfort. Ferric preparations also exist but with less bioavailability. Across EU there are also several IV products on the market: iron (III) hydroxide dextran complex, iron sucrose, ferric carboxymaltose, iron isomaltoside. IV iron therapy, however, is inconvenient, invasive and associated with the risk of rare but serious hypersensitivityreactions; it is used in those situations when oral preparations cannot be used or when there is a need to deliver iron rapidly. Feraccru is a trivalent iron, oral iron replacement preparation. The active substance of Feraccru is ferric maltol (also known as 3-hydroxy-2-methyl-4H-pyrane-4-one iron (III) complex, or ST10, or ferric trimaltol or ferric maltol) an oral ferric iron/maltol complex. It is presented as red hard gelatine capsules containing 30 mg iron (ferric iron). Maltol is a sugar derivative that strongly chelates iron in the ferric form (FeIII) rendering the iron stable and available for absorption. Upon dissociation of the ferric maltol complex, the maltol molecules are absorbed and glucuronidated in the intestinal wall, and within the liver during first pass metabolism, and subsequently eliminated from the body in the urine. The iron is absorbed via the endogenous dietary iron uptake system. The indication finally agreed with the CHMP was: Feraccru is indicated in adults for the treatment of iron deficiency anaemia (IDA) in patients with inflammatory bowel disease (IBD) (see section 5.1). The proposed dosage is one 30 mg capsule twice daily on an empty stomach, corresponding to 60 mg ferric iron per day. There was agreement in the paediatric investigation plan to grant a deferral and a waiver for iron as iron (III)-maltol complex (EMEA-001195-PIP01-11).The PDCO granted a waiver in infants under 6 months of age and a referral for the completion of the planned paediatric studies (ST10-021 PK-PED/ST10-01-102, an open label, randomised, multiple-dose, parallel PK study and ST10-01-303, a randomised, open label comparative safety and efficacy study of ST10 and oral ferrous sulphate as comparator) until the adult studies are completed.

SYN
Patent
https://patents.google.com/patent/WO2017167963A1/en
The sugar derivative maltol is a hydroxypyrone (IUPAC name: 3- hydroxy-2-methyl-4£f-pyran-4-one) and it strongly chelates iron and the resulting complex (ferric trimaltol) is well absorbed, unlike many other ferric iron therapies. Ferric trimaltol appears well tolerated even in populations highly susceptible to gastrointestinal side-effects, such as IBD patients (Harvey et al . , 1998), and as such it provides a valuable alternative to patients who are intolerant of oral ferrous iron products, notably in place of intravenous iron. Clinical trials using ferric trimaltol have been carried out, see for example, Gasche et al., 2015.
However, despite the evidence of bioavailability and tolerability for ferric trimaltol, its clinical development has been limited by the absence of adequate synthetic routes. In particular, most manufacturing processes require the use of organic solvents, which increase manufacturing costs, for example to deal with post-synthesis solvent removal, and require additional safety measures, for example to deal with flammability . Critically, solvent-based syntheses are not robust and often generate ferric hydroxide, described in the prior art to be an unwanted impurity of the synthesis.
WO 03/097627 (Vitra Pharmaceuticals Limited) describes the synthesis of ferric trimaltol from iron salts of carboxylic acids in aqueous solution at a pH greater than 7. In a first
synthesis, ferric citrate is added to a solution of sodium hydroxide at room temperature and maltol is added to a second solution of sodium hydroxide at pH 11.6. The ferric citrate solution is added to the maltol solution, leading to the
production of a deep red precipitate. This composition is then evaporated until dryness and the material is powdered and dried. Alternative syntheses are described using ferrous fumarate or ferrous gluconate as the iron carboxylate salt starting material, and by dissolving maltol in sodium carbonate solution in place of sodium hydroxide. However, despite the fact that this process is fully aqueous, several of the iron carboxylate salts employed are expensive, especially as they need to be pharmaceutical grade if the ferric trimaltol is to be suitable for human administration. More importantly, this process introduces high levels of
carboxylates (equimolar to iron or greater) to the synthesis that are not easily removed by filtration or centrifugation of the ferric trimaltol cake. Instead these water soluble contaminants must be washed off (e.g. water washed), but this would result in considerable losses of the product due to the amphipathic nature of ferric trimaltol.
WO 2012/101442 (Iron Therapeutic Holdings AG) describes the synthesis of ferric trimaltol by reacting maltol and a non- carboxylate iron salt in an aqueous solution at alkaline pH .
However, despite the lower cost of non-carboxylate iron salts, pharmaceutically appropriate grades are still required if the ferric trimaltol is to be suitable for human administration and hence are comparatively expensive starting materials.
Importantly, the use of non-carboxylate iron salts (e.g. ferric chloride) results in the addition of considerable levels of the respective counter-anion (e.g. three moles of chloride per every mole of iron) of which a significant part is retained in the filtration (or centrifugation) cake and thus must be washed off. As such, WO 2012/101442 does not address the problem of product losses in WO 03/097627. Furthermore, the addition of a non- carboxylate iron salt (e.g. ferric chloride) to a very alkaline solution, as described in WO 2012/101442, promotes the formation of stable iron oxides, which is an unwanted contaminant in ferric trimaltol . As a consequence, further costly and time-consuming processing of the material would be required for manufacturing .
Overall, the cost of the current aqueous syntheses is driven by regulatory demands for low levels of toxic heavy metals and residual reagents in the final pharmaceutical formulation, which force the use of highly purified, and thus expensive, iron salts as well as thorough washing of the final product (resulting in significant losses of product) . This will impact on the final price of ferric trimaltol and potentially limits patient access to this therapy. As such, there is a need for a process that can use lower iron grades and limited wash cycles, whilst producing ferric trimaltol of adequate purity.
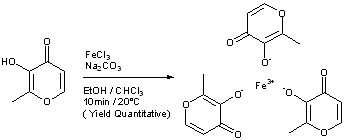
Ferric maltols are a class of compounds that include ferric trimaltol, a chemical complex formed between ferric iron (Fe3+) and the hydroxypyrone, maltol (IUPAC name: 3-Hydroxy-2-methyl-4£f- pyran-4-one) , in a molar ratio of ferric iron to maltol of 3:1. Maltol strongly chelates the ferric iron and the resulting complex (ferric trimaltol which may also be written as ferric tri-maltol) is well absorbed, in contrast to some other ferric iron supplements, fortificants and therapies. Maltol binds metal cations mainly in the form of a dioxobidentate ligand in a similar manner proposed for other 4 ( 1H) -pyranones :

Structure of maltol (3-hydroxy-2-methyl-4 (H) -pyran-4-one) and dioxo-chelation to metal cations (M) such as iron. For ferric trimaltol three maltol groups surround one iron.
Examples
Example 1: Ferric trimaltol from L-lyslne coated ferric hydroxide
Synthesis of lysine-coated ferric hydroxide colloid
14.87g FeCl3. 6H20 was added to 25 mL UHP water and stirred until dissolved. 14.9g NaOH 5M was then added drop-wise to this solution with constant stirring, during which a ferric hydroxide colloid was gradually produced. This colloidal suspension was then added to a L-Lysine suspension (5.02g in 25mL ddH2<D) .
Ferric trimaltol synthesis
7 g NaOH pellets was added to 25 mL UHP water and stirred until dissolved. Next, 24.5g maltol was added and stirred until dissolved. Then, the suspension of lysine-coated ferric
hydroxide colloids was gradually added to the maltol with vigorous stirring, producing a dark red precipitate (with a significant brown hue) . This suspension was incubated overnight during which time it became lighter and the brown hue
disappeared. This precipitate was then recovered by
centrifugation (4500 rpm x 5min) and dried overnight (50°C) .
Example 2: Ferric trimaltol from L-lysine modified ferric hydroxide
Synthesis of lysine-modified ferric hydroxide gel
14.87g FeCl3.6H20 and 5.02g L-Lysine were added to 25 mL UHP water and stirred until dissolved. 32 mL NaOH 5M was then gradually added to this solution producing a ferric hydroxide gel .
Ferric trimaltol synthesis
7 g NaOH pellets was added to 25 mL UHP water and stirred until dissolved. Next, 24.5g maltol was added and stirred until dissolved. Next, the lysine-modified ferric hydroxide gel was gradually added to this solution with vigorous stirring. A 1.2 M HC1 solution was then used to drop the pH of the solution to 10, which was then incubated for 70 min. Finally, a dark red precipitate (i.e., ferric trimaltol) was recovered by
centrifugation (4500 rpmx5min) and dried overnight (45°C) .
Example 3: Absence of ferric hydroxide in ferric trimaltol
Ferric trimaltol is soluble in ethanol whereas ferric hydroxide (a potential contaminant) is not. As such ferric trimaltol powders produced as per Examples 1 and 2 were dissolved in ethanol. The material from Example 2 dissolved completely confirming the absence of iron hydroxides whereas the material from Example 1 did not. This supported the preference in the present invention for ligand modification, rather just surface coating, to ensure full conversion to ferric trimaltol .
Example 4: Ferric trimaltol from tartrate-modified ferric hydroxide
Synthesis of tartrate-modified ferric hydroxide gel
14.87g FeCl3.6H20 (0.055 mol) was added to 25 mL UHP water and stirred until dissolved. 4.12 g tartaric acid (0.0275 mol) was added to this solution and stirred until dissolved. 38 mL NaOH 5M was then gradually added to this solution producing a ferric hydroxide gel .
Ferric trimaltol synthesis
2 g NaOH pellets was added to 25 mL UHP water and stirred until dissolved. Next, 24.5g maltol was added and stirred. This produced a slurry in which most of the maltol remained
undissolved. Next, the tartrate-modified ferric hydroxide gel was gradually added to this solution with vigorous stirring during which the remainder of maltol dissolved. After 15 min a dark red precipitate (i.e. ferric trimaltol) had been formed and pH had stabilised at 8.5. The material was then washed by (1)
centrifuging, (2) disposing of the supernatant and (3)
resuspending in water back to its original volume. Finally, the material was recovered by centrifugation (4500 rpm x 5min) and dried overnight (50°C) . Previously disclosed synthetic processes for the production of ferric trimaltol under aqueous conditions require the addition of NaOH (or other suitable bases) for conversion of maltol from its protonated form to its deprotonated form prior to complexation of iron. However this results in the formation of unwanted sodium ions which must be washed off. In contrast, the use of ferric hydroxides according to the methods of the present invention reduces the requirements for base and associated counter cation (e.g. sodium), which is a favourable feature. Note that ferric hydroxides are represented above as Fe (OH) 3 for illustrative purposes only. Different iron hydroxides possess different structures and elemental compositions (see Cornell & Schwertmann, The Iron Oxides Structure, Properties, Reactions, Occurrence and Uses. 2nd edition, 1996, VCH Publishers, New York) . Example 5: Ferric trimaltol from tartrate-modified ferric hydroxide (with removal of contaminants from ferric hydroxide)
Material prepared as in Example 4, except excess reactants and reaction products (e.g. unbound tartaric acid, sodium chloride) were removed from the ferric hydroxide gel. This was achieved by centrifuging the ferric hydroxide gel after its synthesis and discarding the supernatant, which contained unwanted soluble species. Finally, the ferric hydroxide gel was re-suspended in water back to its original volume prior to being added to a maltol slurry.
Example 6: Ethanolic clean up for ferric trimaltol produced from ligand coated ferric hydroxide
Ferric trimaltol precipitate was purified as it contained an unwanted iron oxide fraction. Part of the wet pellet recovered by centrifugation (4.5 g) was dissolved in 1L ethanol. The iron oxide fraction (which remained undissolved) was then removed by filtration, producing a turbidity-free solution. Next, ethanol was evaporated (40°C in a rotavapor under vacuum) producing a concentrated ferric trimaltol slurry. This was then recovered and oven dried overnight at 50°C.
PATENT
https://patents.google.com/patent/WO2012101442A1/en
Comparative Example 1
Preparation of Iron Trimaltol from Pure Maltol Maltol was dissolved in an aqueous solution of ferric chloride and ferric trimaltol was precipitated upon the addition of sodium hydroxide.
An accurate mass of ferric chloride hexahydrate granules (330g) was dissolved in distilled water to yield a pH of 0.6. To this solution, an equimolar amount of maltol was added (490g in total, initially 250g) and allowed to dissolve with continuous stirring. The pH of this solution was found to be zero and the colour of this solution was deep- purple. Spectroscopy showed that the initial solution was mainly a 1 :1 Fe/maltol mixture with some 1 :2 component. The remaining maltol was added. After an hour of stirring, sodium hydroxide (147g NaOH in 750 ml water) was added dropwise to the solution until a pH of 8.3 was achieved. The solution and precipitate were red. The precipitate was collected using a Buchner funnel under vacuum. The precipitate was dried at 40°C under vacuum.
Maltol is only slightly soluble in an aqueous acidic reaction medium. After an hour of stirring, traces of undissolved maltol were visible on the surface of the ferric chloride/maltol solution, on the walls of the reaction vessel and on the stirrer. Upon addition of sodium hydroxide, there appeared to be lumps of a brownish-black substance on the walls of the reaction vessel and on the stirrer which seemed to add to the impurities in the desired product.
An attempt to heat the ferric chloride/maltol solution so as to assist the maltol to dissolve in the ferric chloride solution resulted in a burnt, off spec, colour iron maltol sample. This method also produces two by-products which consume expensive maltol namely Fe(OH)2 (Maltol) and Fe (OH) (Maltol)2.
The sodium hydroxide solution has to be added extremely slowly to prevent “gumming up” and formation of undesirable lumps at the bottom of the reaction vessel. A yield of about 78% ferric trimaltol was obtained using this method of preparation.
When maltol is added to a ferric chloride solution at a low pH, no ferric trimaltol is formed and ferric hydroxide is generated with ferric monomaltol and a small percentage of ferric dimaltol species. The charge neutralisation of these complexes is either the hydroxy! functional group or the chloride anion. This addition also results in the formation of black deposits and gums consisting of ferric chloride/ferric hydroxide polymers. These black deposits are also produced if the solutions are heated. Therefore it is not possible to obtain the correct stoichiometry for the formation of ferric trimaltol and manufacture a pharmaceutically acceptable product using this method.
The addition of maltol to an aqueous solution of ferric or ferrous chloride was deemed impractical for scale up and manufacturing purposes and Examples 2 to 4 investigate the addition of the iron chlorides to maltol in solution.
The problem of working in an aqueous environment
Ferric chloride as a hydrated ion in aqueous solution is a strong Lewis acid with a Ka of 7x 103 and ferrous chloride as a hydrated ion in aqueous solution is also a strong Lewis acid with a Ka of 5 x 10“9. Over the desired range for using iron chlorides as starting materials for the synthesis of ferric trimaltol, ferric chloride in aqueous solution has a pH value in the range of 1-3 and ferrous chloride has a pH in the range of 3-5. Furthermore, commercial solutions of iron chlorides have a pH circa 1 because they are stabilised by the addition of hydrochloric acid to prevent the precipitation of ferric hydroxide species.
The present invention recognises that maltol is virtually insoluble at these low pH values and has limited solubility when dissolved in water in the pH range 6-8. The maximum aqueous solubility is 1g/100m! at 20°C. However, the solubility of maltol can be increased to 10g/100ml by heating to near boiling temperatures. Maltol is stable in aqueous solution at these temperatures and this property has been employed in Example 4 to synthesise ferric trimaltol. At low pH values ferric trimaltol is not the preferred species due to disproportionation. In order to obtain significant amounts of ferric trimaltol using a stoichiometric ratio of iron salt to hydroxypyrone of 1 :3, the eventual pH of the solution must exceed 7 since below that pH ferric dimaltol and monomaltol species will exist. Therefore two methods of increasing the pH were researched 1) using sodium carbonate and 2) using sodium hydroxide. Other alkali hydroxides could be used such as potassium hydroxide. The sodium carbonate neutralisation was found to be less preferable due to C02 generation. This research lead to an improved synthesis of ferric trimaltol.
Example 2
Maltol was dissolved in an aqueous solution of sodium hydroxide and iron maltol was precipitated upon the addition of ferric chloride.
In view of some of the difficulties experienced in Example 1 , and the fact that maltol is very soluble in aqueous alkali hydroxide solutions, it was decided to change the manufacturing procedure.
The initial work using this method of preparation showed that a 90% yield was achieved. Various operating parameters were then optimised and the following procedure outlines the final method chosen. A yield of 95% was then achieved. An accurate mass of sodium hydroxide pellets (20g) was dissolved in distilled water to yield a pH of 13.50. An equimolar amount of maltol (63g) was added to this aqueous solution of NaOH to give a clear yellow coloured solution with a pH of 11.6. Almost immediately a stoichiometric amount of ferric chloride (45g) was added slowly to this solution to give a pH of 7.1 and a red precipitate formed, which was then collected using a Buchner funnel under vacuum. The precipitate was then dried at 40°C under vacuum.
Adding the maltol solution in sodium hydroxide to ferric chloride as in method 1 is not preferred since it gives an off spec product and gums and a black precipitate.
Maltol is very soluble in aqueous alkali hydroxide solutions giving a yellow solution. The concentration of the hydroxide solution preferably does not exceed 20%.
This method is advantageous since it has the potential to produce only one by-product viz, ferric hydroxide Fe(OH)3 which consumes some of the iron intended to complex with the maltol. This is not easily measurable in the presence of iron maltol and so the following method was used to measure the ferric hydroxide. Fe(OH)3 is insoluble in ethanol and so the iron maltol product was dissolved in ethanol. It was found that small amounts of Fe(OH)3 may be present in the batches of iron maltol synthesized according to Example 2.
Taking the extremes of the specification, in one embodiment, the amount of Fe(OH)3 present in the active material may not exceed 2 wt. % Fe(OH)3 based on the total weight of the composition. In view of its well known inert characteristics the level of this compound is adequately controlled and a final specification including controlled ferric hydroxide should be acceptable.
The mass balance for maltol and iron was closed at 99%.
A yield of 95% iron maltol was obtained using this method of preparation.
Example 3
Maltol was dissolved in an aqueous solution of Sodium Carbonate and Iron Maltol was precipitated upon the addition of Ferric Chloride.
An accurate mass of sodium carbonate (Na2C03) (53g) was dissolved in distilled water to give a solution having pH = 11.5. An equimolar amount of maltol (65g) was added to this aqueous alkali solution to give a murky creme coloured solution of pH = 9.9. A stoichiometric amount of a ferric chloride solution was added drop wise to this solution to a pH of 8.00. A further 15 grams of Na2C03 was added to this solution to increase the pH to 9.00. The remainder of the ferric chloride solution was then added to give a solution pH = 8.77 and a red coloured precipitate appeared.
The precipitate was collected using a Buchner funnel under vacuum. The precipitate was then dried at 40°C under vacuum. The release of C02 during the reaction tends to make this process less desirable due to foaming on the surface. The final product is a gellike solid when wet and the removal of moisture during drying can therefore be time consuming. The process may not be preferred but the ferric trimaltol produced could be acceptable.
Example 4 Maltol was dissolved in water and heated to a near boiling temperature and ferric or ferrous chloride was added to form a 1 :1/1:2 mixture of ferric maltol. The solution was allowed to cool and was added to maltol dissolved in sodium hydroxide. Stage 1
Depending on the batch size required, the ferric chloride was added slowly to a maltol solution in water at a pH of 6-7. The solubility of maltol is greatly enhanced up to 10g/100ml by heating to temperatures above 60°C. Addition of ferric chloride or ferrous chloride and monitoring the pH of the solution and maintaining the pH> 3 mainly produces ferric dimaltol species but very little ferric trimaltol. Above pH 3, no ferric hydroxide appeared to be generated. Ferric monomaltol and dimaltol species either with hydroxy or chloride giving the charge neutralisation are very soluble and a concentrated solution in excess of 30g/100ml can be generated. In order to obtain the correct stoichiometry for the formation of ferric trimaltol, further maltol is required and the pH needs to be corrected to values higher than 7.
As anhydrous ferrous or ferric chloride either 126g or 162g in 200ml of water can be added to a litre of water containing 120g of maltol. This ratio of iron to maltol does not provide sufficient maltol to produce any significant amounts of ferric trimaltol which does not precipitate at this stage.
Stage 2 Maltol in alkaline solution has been described as set out above. Conveniently, because maltol solutions up to 20% in sodium hydroxide have a pH circa 11.6, mixing of this solution with the ferric mono/dimaltol solutions from stage 1 yields a precipitate of ferric trimaltol with a deep characteristic burgundy red colour of high purity as determined by UV-vis spectroscopy. The filtrate yields product which is suitable for a GMP (good manufacturing process). The sodium chloride which is generated by this process is found in the supernatant since it has a much higher solubility at 35g/100ml than ferric trimaltol. The small amounts of sodium chloride in the ferric trimaltol can be reduced, if required, by washing in water. A further, surprising feature of the research resulted from work on ferrous chloride. Ferrous chloride may be substituted in stage 1 to form ferric dimaltol since the maltol was found to auto-oxidise the ferrous to ferric during the process of chelation. One aspect of this work which was considered to be potentially very useful if larger batch sizes were required arose from the finding that being a weaker Lewis acid than ferric chloride the pH of the starting solution was in excess of 3. Therefore the risk of generating ferric hydroxide was lower than with the use of ferric chloride at higher concentrations.
Ferrous and ferric chloride in solution or as a solid may be added to an alkaline solution of maltol in sodium hydroxide, combining stages 1 & 2. Providing a small excess of maltol up to about 10% is added then a precipitate of ferric trimaltol with a small amount of maltol is obtained. Such a preparation would be satisfactory as a GMP ferric trimaltol product.
PATENT
https://patents.google.com/patent/WO2017167963A1/en
AMPLE 1
Synthesis of ferric trimaltol using ferric citrate
NaOH (12g, 0.3 moles) is dissolved in water (50 ml) to form a sodium hydroxide solution. 20 ml of the sodium hydroxide solution is placed in a separate vessel.
Ferric citrate (30g, 0.11 moles) is slowly added to the sodium hydroxide solution in the separate vessel at room temperature with gentle stirring. Further portions of the sodium hydroxide solution are added to the solution of ferric citrate, as necessary, in order to ensure that all of the ferric citrate is dissolved.
Maltol (49g, 0.39 moles) is added to the remaining volume of sodium hydroxide solution and dissolved. The pH of the maltol solution is 11.6.
The ferric citrate solution is slowly added to the maltol solution with gentle stirring. A deep red precipitate forms; the supernatant is a deep red colour.
The solution is slowly evaporated to dryness at 60 to 80° C until the material is suitable for powdering. The material is powdered and the powder is then dried to a constant weight.
The yield of the final product is 87g. The final product comprises ferric trimaltol and sodium citrate. The product was assayed, using elemental analysis, for iron and sodium content. The iron content is 7.89% (theoretical 7.8%) and the sodium content is 13.45%.
The pH of a solution of the final product in water was measured. The pH of a 1% solution of the product by total weight of aqueous solution is 9.9 at 20°C.
EXAMPLE 2
Synthesis of ferric trimaltol using ferrous fumarate
NaOH (40g, 1 mole) is dissolved in water (100 ml) to form a sodium hydroxide solution. The pH of the solution is approximately 13.0.
Ferrous fumarate (170g, 1 mole) is slowly added to the sodium hydroxide solution at room temperature with gentle stirring.
Maltol (408g, 3.23 moles) is added to a separate volume of sodium hydroxide (40g, 1 mole) dissolved in water (100 ml) and dissolved. The pH of the solution is approximately 11.
The ferrous fumarate solution is slowly added to the maltol solution with gentle stirring. A deep red precipitate forms; the supernatant is a deep red colour.
The solution is slowly evaporated to dryness at 60 to 80° C until the material is suitable for powdering. The material is powdered and the powder is then dried to a constant weight. The yield of the final product is 615g.
The final product comprises ferric trimaltol and sodium fumarate.
EXAMPLE 3
Synthesis of ferric trimaltol using sodium carbonate to vary pH
Sodium carbonate (2.5g) is dissolved in 10ml of distilled water at room temperature. The pH of the solution is 11.6. Maltol (9.6g – three molar equivalents of sodium carbonate) is added to the sodium carbonate solution to give a cream coloured solution having a pH of 10.0.
A stoichiometric amount of ferric citrate (5g, allowing for a small excess of maltol) in an aqueous solution of sodium hydroxide (lg in 5ml of distilled water) is added slowly to the solution of maltol. The pH of the combined solutions is about 9. A red precipitate appears which is separated by decantation and dried at 80°C in an oven.
The red precipitate is ferric trimaltol, as confirmed by UV-Vis spectrometry.
EXAMPLE 4
Synthesis of ferric trimaltol using ferrous gluconate
Potassium hydroxide (5.5g) is dissolved in 50ml of distilled water at room temperature. To 25ml of this solution, maltol (16.5g, 0.13 moles) is added and gently heated to form a clear solution. To the other 25ml aliquot of the potassium hydroxide solution ferrous gluconate (22.5g) is added. This is gently heated to form a dark green saturated solution. The ferrous gluconate solution is added to the maltol solution and immediately a colour change to dark brown is noted.
On cooling, a deep brown precipitate forms (which is ferric trimaltol). The supernatant is a deep brown solution containing ferric trimaltol and potassium gluconate. The precipitate and the supernatant are dried separately at 80°C in an oven. The ferric trimaltol is a deep red brown powder with a characteristic caramel odour and UV-vis spectrum in aqueous solution.
EXAMPLE 5
Synthesis of ferric trimaltol using solid ferrous gluconate
Example 4 was repeated with the modification that the maltol is added to all of the 50 ml solution of potassium hydroxide and then solid ferrous gluconate is added directly to the maltol solution. This method gives similar end products to Example 4.
EXAMPLE 6
Synthesis of ferric trimaltol using sodium ferrous citrate
A 20% solution w/v of sodium ferrous citrate in distilled water is prepared from 7.5g of sodium ferrous citrate in 37.5ml of water. The solution of sodium ferrous citrate is dark green with an iron content of about 20%. A solution of maltol (containing 10g/50ml) in 20% sodium hydroxide is added to the solution of sodium ferrous citrate. A characteristic deep red/brown iron complex of ferric trimaltol is formed.
EXAMPLE 7
Synthesis of ferric trimaltol using solid sodium ferrous citrate
Example 6 was repeated using the same amounts and concentrations of components but the method is varied in that solid sodium ferrous citrate (7.5g) is added directly to the maltol solution (containing lOg of maltol in 50ml). Ferric trimaltol is formed using this alternative method.
EXAMPLE 8
Synthesis of ferric trimaltol using sodium ferric citrate
A 20% solution w/v of sodium ferric citrate in distilled water is prepared from 7.5g of sodium ferric citrate in 37.5ml of water. The solution of sodium ferric citrate is dark brown with an iron content of about 20%.
A solution of maltol (containing 10g/50ml) in 20% sodium hydroxide is added to the solution of sodium ferric citrate. A characteristic deep red/brown iron complex of ferric trimaltol is formed. EXAMPLE 9
Example 8 was repeated using the same amounts and concentrations of components but the method is varied in that solid sodium ferric citrate (7.5g) is added directly to the maltol solution (containing lOg of maltol in 50ml). Ferric trimaltol is formed using this alternative method.
If any of Examples 3 to 9 are repeated using maltol in a neutral or acidic aqueous medium, such as for example in buffered citric acid, brown/black impurities appear and insoluble fractions are formed (probably of ferric hydroxide) and the UN-vis spectra of the solutions are not correct. In particular, there is a peak shift towards 510nm indicating the formation of mono or dimaltol complexes or compounds.
PATENT
WO 2017167970
POLYMORPH
GB 2531742
PATENT
WO 2016066555
https://patents.google.com/patent/WO2016066555A1/en
An adequate supply of iron to the body is an essential requirement for tissue growth and the maintenance of good health in both man and animals. Moreover, in certain pathological conditions where there is an insidious blood loss, or where there is a mal-distribution of iron in the body, there may be a state of low iron stores in the body leading to an iron deficiency and a concomitant chronic anaemia. This is seen in inflammatory diseases of the gastrointestinal tract, such as gastric and peptic ulcers, reflux oesophagitis, ulcerative colitis and Crohn’s disease.
Anaemia can also follow operations that result in serious blood loss and can be associated with gastrointestinal infections, such as those caused by Helicobacter pylori.
Ferric maltol comprises a complex of one ferric iron and three maltol anions and has the following molecular formula: (C6H503)3Fe. Maltol is also known as 3-hydroxy-2-methyl-4- pyrone.
Polymorphic forms occur where the same composition of matter crystallises in a different lattice arrangement, resulting in different thermodynamic properties and stabilities specific to the particular polymorphic form. WO 03/097627 A1 discloses a method of forming iron hydroxypyrone compounds.
EP 0 159 917 A3 describes a pharmaceutical composition containing a hydroxypyrone-iron complex. WO 2012/101442 A1 discloses a method of forming iron hydroxypyrone compounds.
Schlindwein et al (Dalton Transactions, 2006, Vol. 10, pages 1313-1321) describes lipophilic 3-hydroxy-4-pyridinonate iron(lll) complexes. Ferric maltol has been known for about 100 years but no polymorphs have been identified or studied prior to this invention.
We have now found that it is possible to produce different polymorphs of ferric maltol, which crystalline forms may be referred to herein as the “compounds of the invention”. One polymorph form can be preferable in some circumstances when certain aspects, such as ease of preparation and stability, such as thermodynamic stability are required. In other situations, a different polymorph may be preferred for greater solubility and/or superior pharmacokinetics. The polymorphs of the invention can provide advantages in terms of improved or better bioavailability or improved or better stability or solubility.
The term “ferric maltol” as used herein refers to both ferric trimaltol and the designation INN ferric maltol. In one aspect of the invention there is provided a Form I polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising characteristic crystalline peaks expressed in degrees 2-theta at each of 15.6 and 22.5 ± 0.25 or 0.2 degrees, optionally wherein the Form I polymorph comprises greater than about 92 wt.% ferric maltol based on the weight of the polymorph, such as greater than about 95 wt.%, preferably greater than about 96 wt.%, or about 98 wt.%, or about 99 wt.% such as about 99.8 wt.%.
In a further aspect of the invention there is provided a Form II polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising a peak expressed in degrees 2-theta at 8.3 ± 0.25 degrees.
In a yet further aspect of the invention there is provided a Form III polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising a peak expressed in degrees 2-theta at 7.4 ± 0.25 degrees. In a still further aspect of the invention there is provided a Form IV polymorph of ferric maltol characterized by a powder X-ray diffraction pattern comprising peaks expressed in degrees 2-theta at 9.5 and 14.5 ± 0.2 degrees.
The measurements of degrees 2-theta generally refer to measurements at ambient temperature, such as from about 5 to about 40°C, preferably about 10 to about 30°C. The relative intensities of the peaks can vary, depending on the sample preparation technique, the sample mounting procedure, the particular instrument employed, and the morphology of the sample. Moreover, instrument variation and other factors can affect the 2-theta values. Therefore, XRPD peak assignments for the polymorphs of the invention, as defined herein in any embodiment, can vary by, for example, ± 0.2, such as ±0.1 or ±0.05. The term “about” in relation to XRPD peak values may include for example, ±0.25 or ± 0.2, such as ±0.1 or ±0.05. These ranges may apply to any of the peak values in degrees referred to herein.
In another embodiment of the invention, there is provided a process for the preparation of a ferric maltol polymorph, such as Form I or Form II polymorph, which comprises combining ferric citrate with maltol anions to form a mixture comprising ferric maltol and wherein the process comprises the use of a ferric maltol seed crystal. The seed crystal may comprise a Form I and/or Form II polymorph as described herein and these polymorphs may be prepared using the methods described herein.
In another aspect of the invention, there is provided a process for the preparation of Form I polymorph, which comprises combining ferric citrate with maltol anions to form a mixture comprising ferric maltol polymorph Form I wherein the process comprises the use of a ferric maltol seed crystal comprising Form I and/or Form II polymorph and preferably wherein the polymorph formed is washed (typically with water) prior to drying.
In a further aspect of the invention, there is provided a process for the preparation of Form II polymorph, which comprises combining ferric citrate with maltol anions in solution to form a mixture comprising ferric maltol polymorph Form II, wherein the process preferably comprises the use of a ferric maltol seed crystal comprising Form I and/or Form II polymorph and preferably wherein the polymorph formed is washed (typically with water) prior to drying.
The invention also provides a pharmaceutical composition comprising a polymorph according to the invention, or mixtures thereof, and a pharmaceutically acceptable adjuvant, diluent or carrier. In addition, the invention provides a composition comprising Form I and Form II polymorphs as defined herein.
In an aspect of the invention, the polymorph of the invention is for use in the prevention or treatment of iron deficiency with or without anaemia in a subject. The anaemia is preferably iron deficiency anaemia.
In a further aspect of the invention there is provided the use of a polymorph of the invention for the manufacture of a medicament for the prevention or treatment of iron deficiency with or without anaemia in a subject. The anaemia is preferably iron deficiency anaemia.
The invention further provides a method for the prevention or treatment of iron deficiency with or without anaemia which method comprises the administration of a polymorph according to the invention to a subject in need of such treatment. The anaemia is preferably iron deficiency anaemia.
Preferably the polymorphs of the invention are obtained in forms that are greater than about 90%, such as greater than about 95%, crystalline (e.g. greater than about 98% crystalline and, particularly, 100%, or nearly 100%, crystalline). By “substantially crystalline” we include greater than about 60%, preferably greater than about 75%, and more preferably greater than about 80% (such as about 90%) crystalline. The degree (%) of crystallinity may be determined by the skilled person using X-ray powder diffraction (XRPD). Other techniques, such as solid state NMR, FT-IR, Raman spectroscopy, differential scanning calorimetry (DSC) microcalorimetry and calculations of the true density may also be used.
The polymorphs of the invention may be characterised by an X-ray powder diffraction pattern comprising the following characteristic crystalline peaks with approximate 2-Theta values (in degrees) as well as an indication of the relative intensity of those peaks in brackets, where a percentage relative intensity of approximately 25- 00% is termed “vs” (very strong), approximately 10-25% is termed “s” (strong), approximately 3-10% is termed “m” (medium) and approximately 1-3% is termed “w” (weak).
Form I: The Form I polymorph preferably comprises characteristic crystalline peaks with 2-Theta values (in degrees) of around (i.e. at about or at approximately) 15.6 and 22.5 ± 0.25, or 0.2 degrees. The diffraction pattern typically does not comprise peaks at one or more, or all, or each of, about 6.9, 7.4, 8.3, 9.3, 10.5, or about 11.8 degrees, such as 8.3 or 11.8 ± 0.25, or ± 0.2, or ±0.1 such as about ±0.05 degrees.
Form II:
The form II polymorph preferably comprises a characteristic crystalline peak with 2-Theta value (in degrees) of around (i.e. at about or at approximately) 8.3 ± 0.25, or ± 0.2, or +0.1 such as about ±0.05 degrees. The diffraction pattern typically does not comprise peaks at one or more, or all, or each of, about 6.9, 7.4, 9.3, 9.5, 10.5, 11.4 or about 13.7 degrees, such as 11.4 or 13.8 ±0.25, or ±0.2, or ±0.1 such as about ±0.05 degrees.
The Form III polymorph preferably comprises a characteristic crystalline peak with 2-Theta value (in degrees) of around (i.e. at about or at approximately) 7.4 ±0.3, ±0.25, or 0.2, or ±0.1 such as about ±0.05 degrees. The diffraction pattern typically does not comprise peaks at one or more, or two or more, or three or more or each of, about 6.9, 8.3, 9.5, 11.3, 12.0, 12.5, 12.9, 14.5, or about 15.8 degrees, such as 6.9, 9.5, 11.3 ±0.25, or ±0.2, or ±0.1 such as about ±0.05 degrees.
The form IV polymorph preferably comprises a characteristic crystalline peaks with 2-Theta values (in degrees) of around (i.e. at about or at approximately) 9.5 and 14.5 +0.2, or ±0.1 such as about ±0.05 degrees. The diffraction pattern typically does not comprise peaks at one or more, or two or more, or three or more or each of, about 6.9, 8.3, 10.5, 11.7, 12.0, 12.2, 12.5, 13.0, 13.4, and about 15.8 degrees, such as 6.9, 8.3, 11.7 ±0.25, or ±0.2, or ±0.1 such as about ±0.05 degrees.
Example 1 : Form I 9.04 kg ferric citrate was combined with 29 litres of purified water. Separately, 12.2 kg of maltol was combined with 15.2 litres of sodium hydroxide solution (20 % w/w). The ferric citrate and sodium hydroxide were charged into a vessel with the addition of 4 litres of water and then stirred at 20 to 25°C. A seed was then added. The seed was 65g of ferric maltol polymorph in 12 litres of water. The seed crystal was prepared by the same process as described in Example 1 but without the use of a seed crystal. The seed was added to the vessel to aid a consistent crystallisation/precipitation. The mixture was held in the vessel, as a suspension, to allow crystal growth and then filtered and washed three times, each time with 13 litres of water. The resulting solid was dried at less than 80°C and produced 13.25 kg of dried ferric maltol.
The ferric maltol in Example 1 was produced on a scale of 12 to 15 kg in different batches. The analysis of the ferric maltol produced showed the % w/w of iron present was about 12.8 to 13.0 and the % w/w of maltol present was about 87.6 to 89.3.
Patent
Publication numberPriority datePublication dateAssigneeTitle
EP0159917A2 *1984-04-191985-10-30National Research Development CorporationPharmaceutical composition containing a hydroxypyrone-iron complex
WO2003097627A1 *2002-05-182003-11-27Vitra Pharmaceuticals LimitedMethod of forming iron hydroxypyrone compounds
Family To Family Citations
EP0107458B1 *1982-10-221987-07-29National Research Development CorporationPharmaceutical compositions
* Cited by examiner, † Cited by third party
Publication numberPriority datePublication dateAssigneeTitle
Family To Family Citations
WO2003097627A1 *2002-05-182003-11-27Vitra Pharmaceuticals LimitedMethod of forming iron hydroxypyrone compounds
US20080188555A1 *2007-02-062008-08-07Jonathan Joseph PowellLigand modified poly oxo-hydroxy metal ion materials, their uses and processes for their preparation
REFERENCES
Inorganica Chimica Acta (1990), 170(2), 241-3
Dalton Transactions (2006), (10), 1313-1321.
EP 0,107,458 [ 1984, to National Research Development Corporation]
Journal of Chemical Research, Synopses (1980), (9), 314.
Chemistry for Sustainable Development (2007) 15(4), PP- 448 – 458
US 5,028,411 [ 1991, to National Research Development Corporation]
Journal of Coordination Chemistry (1978), 8(1), 27-33
Polyhedron (1988), 7(19-20), 1973-9.
US 7,459,569 [2008, to Vitra Pharmaceuticals Limited].
Journal of Pharmaceutical Sciences (1972), 61(8), 1209-12
WO 2012 / 101,442 [ 2012, to Iron Therapeutics Holdings Ag]
Chemistry Letters (1975), (4), 339-42
//////////////Ferric Maltol, マルトール第二鉄 , Feraccru, FDA 2019
CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].[Fe+3]
CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].CC1=C(C(=O)C=CO1)[O-].[Fe+3]
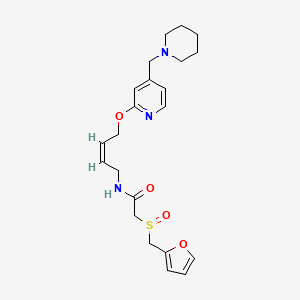
LAFUTIDINE, ラフチジン
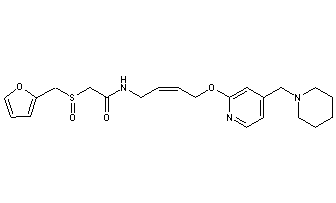


LAFUTIDINE
N-[4-[4-(Piperidin-1-ylmethyl)pyridin-2-yloxy]-(Z)-but-2-en-1-yl]-2-(furfurylsulfinyl)acetamide
-
- FRG-8813
- ATC:A02B
- Use:antisecretory, gastric H2-antagonist
- (+)-2-[(2-furanylmethyl)sulfinyl]-N-[(2Z)-4-[[4-(1-piperidinylmethyl)-2-pyridinyl]oxy]-2-butenyl]acetamide
- Formula:C22H29N3O4S
- MW:431.56 g/mol
- CAS-RN:118288-08-7
-
(±)-2-(Furfurylsulfinyl)-N-(4-(4-(piperidinomethyl)-2-pyridyl)oxy-(Z)-2-butenyl)acetamide
-
(Z)-2-((2-Furanylmethyl)sulfinyl)-N-(4-((4-(1-piperidinylmethyl)-2-pyridinyl)oxy)-2-butenyl)acetamide
-
118288-08-7
FRG‐8813、2‐(Furfurylsulfinyl)‐N‐[(Z)‐4‐[[4‐(piperidinomethyl)‐2‐pyridinyl]oxy]‐2‐butenyl]acetamide、ロクチジン、Loctidine、ラフチジン・・・
2-[(furan-2-ylmethyl)sulfinyl]-N-[(2Z)-4-{[4-(piperidin-1-ylmethyl)pyridin-2-yl]oxy}but-2-en-1-yl]acetamide
49S4O7ADLC
Lafutidine , also named N-[4-[4-(piperidin-1-ylmethyl)pyridin-2-yloxy]-(Z)-but-2-en-1-yl]-2-(furfurylsulfinyl)acetamide, is a histamine H2 receptor antagonist that was first produced in Japan by Taiho and UCB Japan for the oral treatment of peptic ulcers in 2000. In 2010 it was approved for the treatment of mild gastroesophageal reflux disease, and in 2012 it was approved to help improve symptoms of gastric mucosal lesions due to gastritis
Lafutidine (INN) is a second generation histamine H2 receptor antagonist having multimodal mechanism of action and used to treat gastrointestinal disorders. It is marketed in Japan and India.
Medical use
Lafutidine is used to treat gastric ulcers, duodenal ulcers, as well as wounds in the lining of the stomach associated with acute gastritis and acute exacerbation of chronic gastritis.[1][2]
Adverse effects
Adverse events observed during clinical trials included constipation, diarrhea, drug rash, nausea, vomiting and dizziness.[2]
Mechanism of action
Like other H2 receptor antagonists it prevents the secretion of gastric acid.[2] It also activates calcitonin gene-related peptide, resulting in the stimulation of nitric oxide (NO) and regulation of gastric mucosal blood flow, increases somatostatin levels also resulting in less gastric acid secretion, causes the stomach lining to generate more mucin, inhibits neutrophil activation thus preventing injury from inflammation, and blocks the attachment of Helicobacter pylori to gastric cells.[2]

Trade names
It is marketed in Japan as Stogar by UCB[1] and in India as Lafaxid by Zuventus Healthcare.[2]





N-[4-[4-(Piperidin-1-ylmethyl)pyridin-2-yloxy]-(Z)-but-2-en-1-yl]-2-(furfurylsulfinyl)acetamide 1 as a white solid (15.8 kg, 91.3%).(2,3)
1H NMR (600 MHz, CDCl3): δ 1.43 (m, 2H), 1.56–1.60 (m, 4H), 2.36 (m, 4H), 3.34 (d, 1H, J = 14.4 Hz), 3.40 (s, 2H), 3.59 (d, 1H, J = 14.4 Hz), 4.10 (t, 2H, J = 6.6 Hz), 4.17 (d, 1H, J = 13.8 Hz), 4.31 (d, 1H, J = 13.8 Hz), 4.93 (d, 2H, J = 6.6 Hz), 5.67–5.69 (m, 1H), 5.83–5.87 (m, 1H), 6.39 (dd, 1H, J = 1.8, 3.0 Hz), 6.47 (d, 1H, J = 3.0 Hz), 6.72 (s, 1H), 6.87 (d, 1H, J = 5.4 Hz), 7.19 (s, 1H), 7.43 (d, 1H, J = 1.8 Hz), 8.03 (d, 1H, J = 5.4 Hz).
13C NMR (150 MHz, CDCl3): δ 24.2, 26.0, 26.0, 37.2, 50.2, 53.4, 54.6, 54.6, 61.4, 62.4, 110.8, 111.3, 112.2, 117.7, 128.4, 128.9, 143.3, 143.9, 146.3, 151.5, 163.6, 163.6.
IR (KBr): 3325, 2935, 1638, 1613, 1041 cm–1.
ESI-MS: m/z 431.1.
Increasing the Purity of Lafutidine Using a “Suicide Substrate”
Chengjun Wu  , Zhen Li, Chunchao Wang, Yanan Zhou, and Tiemin Sun*
, Zhen Li, Chunchao Wang, Yanan Zhou, and Tiemin Sun*
Key Laboratory of Structure-Based Drug Design and Discovery, Shenyang Pharmaceutical University, Ministry of Education, Shenyang 110016, P. R. China
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00070/suppl_file/op8b00070_si_001.pdf
CLIP
http://www.drugfuture.com/synth/syndata.aspx?ID=145925

EP 0282077; JP 1988225371; JP 1989230556; JP 1989230576; US 4912101
1) The reaction of 2-bromo-4-(piperidin-1-ylmethyl)pyridine (I) with 4-amino-2(Z)-buten-1-ol (II) by means of NaH in THF gives 4-[4-(piperidin-1-ylmethyl)pyridin-2-yloxy]-2(Z)-buten-1-amine (III), which is then condensed with 2-(2-furylmethylsulfinyl)acetic acid (IV) by means of 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDCD) in dichloromethane.
 EP 0582304; JP 1994192195
EP 0582304; JP 1994192195
The condensation of 2-chloro-4-(piperidin-1-ylmethyl)pyridine (V) with 4-(tetrahydropyranyloxy)-2(Z)-buten-1-ol (VI) by means of NaH in THF gives 4-(piperidin-1-ylmethyl)-2-[4-(tetrahydropyranyloxy)-2(Z)-butenyloxy)pyridine (VII), which is deprotected with 4-methylbenzenesulfonic acid in methanol, yielding the free butenol (VIII). The acylation of (VIII) with methanesulfonyl chloride in toluene affords the corresponding mesylate (IX), which is finally condensed with 2-(2-furylmethylsulfonyl)acetamide (X) (obtained from the corresponding 4-nitrophenyl ester (XI) with ammonia) by means of potassium tert-butoxide in toluene.
 Chem Pharm Bull 1998,46(4),616
Chem Pharm Bull 1998,46(4),616
A new synthesis of lafutidine has been described: The condensation of 2-bromopyridine-4-carbaldehyde ethylene ketal (I) with 4-(tetrahydropyranyloxy)-2(Z)-buten-1-ol (II) by means of NaOH, K2CO3 and tetrabutylammonium bisulfate in refluxing toluene gives the corresponding substitution product (III), which by treatment with pyridinium p-toluenesulfonate (PPTS) in hot ethanol yields the 2(Z)-butenol (IV). The reaction of (IV) with SOCl2 and then with potassium phthalimide (V) affords the substituted phthalimide (VI), which by treatment with hydrazine hydrate in refluxing methanol gives the 2(Z)-butenamine (VII). The condensation of (VII) with 2-(2-furylmethylsulfinyl)acetic acid 4-nitrophenyl ester (VIII) in THF yields the expected amide (IX), which is treated with p-toluenesulfonic acid in refluxing acetone/water to eliminate the ethylene ketal protecting group yilding the aldehyde (X). Finally, this compound is reductocondensed with piperidine (XI) by means of NaBH4 in ethanol.
CLIP
Synthesis Path
Lafutidine
CAS Registry Number: 118288-08-7
CAS Name: 2-[(2-Furanylmethyl)sulfinyl]-N-[(2Z)4-[[4-(1-piperidinylmethyl)-2-pyridinyl]oxy]-2-butenyl]-acetamide
Additional Names: 2-(furfurylsulfinyl)-N-[(Z)-4-[[4-(piperidinomethyl)-2-pyridyl]oxy]-2-butenyl]acetamide
Manufacturers’ Codes: FRG-8813
Trademarks: Protecadin (Taiho); Stogar (Fujirebio)
Molecular Formula: C22H29N3O4S
Molecular Weight: 431.55
Percent Composition: C 61.23%, H 6.77%, N 9.74%, O 14.83%, S 7.43%
Literature References: Second generation histamine H2-receptor antagonist. Prepn of racemate: N. Hirakawa et al., EP 282077; eidem, US 4912101 (1988, 1990 both to Fujirebio); and pharmacology: eidem, Chem. Pharm. Bull. 46, 616 (1998). Pharmacology: S. Onodera et al., Jpn. J. Pharmacol. 68, 161 (1995). Mode of action study: M. Umeda et al., J. Gastroenterol. Hepatol. 14, 859 (1999). Gastroprotective effects in rats: H. Ajioka et al., Pharmacology 61, 83 (2000). Clinical pharmacokinetics: S. Haruki et al.,Yakuri to Chiryo 23, 3049 (1995). Toxicology study: A. Broadmeadow et al., Oyo Yakuri 50, 167 (1995).
Properties: Prepd as the (±) mixture, crystals from benzene-hexane, mp 92.7-94.9°. Slightly bitter taste. Freely sol in DMF, glacial acetic acid; sol in methanol; sparingly sol in dehydrated ethanol; very slightly sol in ether. Practically insol in water.
Melting point: mp 92.7-94.9°
Therap-Cat: Antiulcerative.
Keywords: Antiulcerative; Histamine H2-Receptor Antagonist.
References
References
- ^ Jump up to:a b UCB Japan Revised: April 2005 Stogar tablets
- ^ Jump up to:a b c d e Zuventus Healthcare Ltd. India Lafaxid tablets
-
- a EP 582 304 (Fujirebio; 5.8.1993; J-prior. 7.8.1992).
-
preparation of 2-benzenesulfonyl-4-methylpyridine:
- EP 931 790 (Kuraray; 26.1.1999; J-prior. 26.1.1998).
-
chlorination of 2-benzenesulfonyl-4-methylpyridine:
- JP 10 231 288 (Kuraray; 2.9.1998; J-prior. 21.2.1997).
- WO 9 626 188 (Sagami Res. Center; 21.2.1996; J-prior. 22.2.1995).
- b EP 282 077 (Fujirebio; 11.3.1988; J-prior. 13.3.1987).
- US 4 912 101 (Fujirebio; 27.3.1990; J-prior. 13.3.1987).
-
preparation of I:
- JP 10 231 288 (Kuraray; 2.9.1998; J-prior. 21.2.1997).
-
chlorination of 2-chloromethylpyridines forming 2-chloro-4-trichloromethylpyridine:
- EP 557 967 (Central Glass Co.; 1.9.1993; J-prior. 24.2.1993).
-
treatment of I with (Z)-4-(tetrahydro-2H-pyran-2-yloxy)-2-buten-1-ol:
- US 5 382 589 (Fujirebio; 17.1.1995; J-prior. 27.1.1992).
-
preparation of furfuryl acetate and derivatives:
- JP 8 198 844 (Fujirebio; 6.8.1996; J-prior. 23.1.1995).
- JP 8 198 843 (Fujirebio; 6.8.1996; J-prior. 23.1.1995).
- JP 07 010 860 (Central Glass Co.; 13.1.1995; J-prior. 25.6.1993).
- JP 07 010 864 (Central Glass Co.; 13.1.1995; J-prior. 25.6.1993).
-
2-(furfurylsulfinyl)acetic acid nitrophenyl ester:
- JP 07 010 862 (Central Glass Co.; 13.1.1995; J-prior. 25.6.1993).
-
4-(tetrahydro-2-pyranyloxy)-2(Z)-buten-1-ol from 2(Z)-butene-1,4-diol:
- Nishiguchi, T. et al.: J. Org. Chem. (JOCEAH) 63, 23, 8183 (1998).
- Davis, K. J. et al.: Synth. Commun. (SYNCAV) 29, 10, 1679 (1999).
- Nishiguchi, T. et al.: J. Chem. Soc., Perkin Trans. 1 (JCPRB4) 1995, 24, 2491.
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ECHA InfoCard | 100.118.935 |
| Chemical and physical data | |
| Formula | C22H29N3O4S |
| Molar mass | 431.54 g/mol |
| 3D model (JSmol) | |
/////////////////LAFUTIDINE, ラフチジン , FRG-8813, ATC:A02B
FRG‐8813
2‐(Furfurylsulfinyl)‐N‐[(Z)‐4‐[[4‐(piperidinomethyl)‐2‐pyridinyl]oxy]‐2‐butenyl]acetamide
ロクチジン
Loctidine
ラフチジン
Laftidine
2‐[[(2‐Furyl)methyl]sulfinyl]‐N‐[(Z)‐4‐[[4‐(piperidinomethyl)‐2‐pyridyl]oxy]‐2‐butenyl]acetamide
N‐[(Z)‐4‐[4‐(Piperidinomethyl)‐2‐pyridyloxy]‐2‐butenyl]‐2‐(furfurylsulfinyl)acetamide
(Z)‐2‐フルフリルスルフィニル‐N‐[4‐(4‐ピペリジノメチル‐2‐ピリジルオキシ)‐2‐ブテニル]アセトアミド
ストガー
Stogar
プロテカジン
Protecadin
(+)‐ラフチジン
(+)‐Laftidine
ラフルチジン
Laflutidine
(Z)‐2‐Furfurylsulfinyl‐N‐[4‐(4‐piperidinomethyl‐2‐pyridyloxy)‐2‐butenyl]acetamide
2‐[[(2‐フリル)メチル]スルフィニル]‐N‐[(Z)‐4‐[[4‐(ピペリジノメチル)‐2‐ピリジル]オキシ]‐2‐ブテニル]アセトアミド
N‐[(Z)‐4‐[4‐(ピペリジノメチル)‐2‐ピリジルオキシ]‐2‐ブテニル]‐2‐(フルフリルスルフィニル)アセトアミド
2‐(フルフリルスルフィニル)‐N‐[(Z)‐4‐[[4‐(ピペリジノメチル)‐2‐ピリジニル]オキシ]‐2‐ブテニル]アセトアミド
C1CCN(CC1)CC2=CC(=NC=C2)OCC=CCNC(=O)CS(=O)CC3=CC=CO3
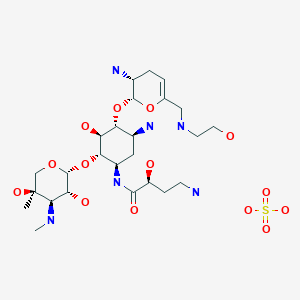
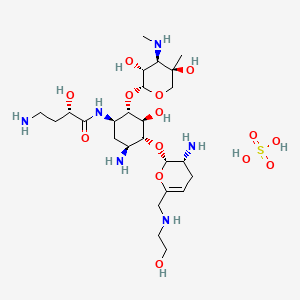
Plazomicin sulfate, プラゾマイシン硫酸塩 ,


Plazomicin
- Molecular FormulaC25H48N6O10
- Average mass592.683 Da
(2S)-4-Amino-N-[(1R,2S,3S,4R,5S)-5-amino-4-{[(2S,3R)-3-amino-6-{[(2-hydroxyéthyl)amino]méthyl}-3,4-dihydro-2H-pyran-2-yl]oxy}-2-{[3-désoxy-4-C-méthyl-3-(méthylamino)-β-L-arabinopyranosyl]oxy}-3-hyd roxycyclohexyl]-2-hydroxybutanamide [French][ACD/IUPAC Name]
1154757-24-0 [RN]
9522
ACHN-490
Plazomicin Sulfate
| Molecular Formula: | C25H50N6O14S |
|---|---|
| Molecular Weight: | 690.763 g/mol |
Plazomicin Sulfate; UNII-A78L6MT746; Plazomicin Sulfate [USAN]; A78L6MT746; 1380078-95-4; Plazomicin sulfate (USAN),
|
6′-(hydroxylethyl)-1-(haba)-sisomicin
Plazomicin is a neoglycoside antibiotic with activity against a broad range of Gram-positive and Gram-negive pathogens. Plazomicin showed potent in vitro activity against multidrug-resistant Klebsiella pneumoniae and Escherichia coli.
- Mechanism of ActionProtein synthesis inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
Highest Development Phases
- MarketedUrinary tract infections
- RegisteredPyelonephritis
- PreregistrationBacteraemia; Nosocomial pneumonia
- PreclinicalGram-negative infections
- No development reportedRespiratory tract infections; Tularaemia; Yersinia infections
Most Recent Events
- 27 Jun 2018Registered for Pyelonephritis (Treatment-resistant) in USA (IV)- First Global Approval
- 27 Jun 2018Registered for Urinary tract infections (Treatment-resistant) in USA (IV)- First Global Approval
- 26 Jun 2018Achaogen receives complete response letter from the FDA for Plazomicin in Bloodstream infection
| Synonyms: O-2-Amino-2,3,4,6-tetradeoxy-6-[(2-hydroxyethyl)amino]-α-D-glycero-hex-4-enopyranosyl-(1→4)-O-[3-deoxy-4-C-methyl-3-(methylamino)-β-L-arabinopyranosyl-(1→6)]-N1-[(2S)-4-amino-2-hydroxy-1-oxobutyl]-2-deoxy-D-streptamine; ACHN 490; |
| CAS Number: 1154757-24-0
Sulfate 1380078-95-4, プラゾマイシン硫酸塩; |
| Achaogen (USA)Phase II completed |
| Mol. Formula: C25H48N6O10 |
| Aminoglycosides, Broad-spectrum, |
| Mol. Weight: 592.68 |
FDA
Click to access 210303Orig1s000lbl.pdf
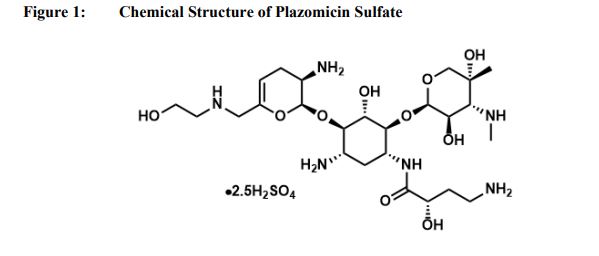
Developed by Achaogen biopharmaceuticals, plazomicin is a next-generation aminoglycoside synthetically derived from [DB12604]. The structure of plazomicin was established via appending hydroxylaminobutyric acid to [DB12604] at position 1 and 2-hydroxyethyl group at position 6′ [A33942]. It was designed to evade all clinically relevant aminoglycoside-modifying enzymes, which contribute to the main resistance mechanism for aminoglycoside therapy [A33942]. However, acquired resistance of aminoglycosides may arise through over expression of efflux pumps and ribosomal modification by bacteria, which results from amino acid or rRNA sequence mutations [A33942]. Like other aminoglycosides, plazomicin is ineffective against bacterial isolates that produce 16S rRNA methyltransferases [FDA Label]. Plazomicin mediates the antibacterial activity against pathogens including carbapenem-resistant (CRE) and extended-spectrum beta-lactamase (ESBL) producing _Enterobacteriaceae_. It mediates the antibacterial activity by binding to bacterial 30S ribosomal subunit and inhibiting protein synthesis [FDA Label]. On June 28th, 2018, plazomicin sulfate was approved by the FDA for use in adult patients for the treatment of complicated urinary tract infections (cUTI) including Pyelonephritis. It is marketed as Zemdri and is administered via once-daily intravenous infusion.
Developed by Achaogen biopharmaceuticals, plazomicin is a next-generation aminoglycoside synthetically derived from Sisomicin. The structure of plazomicin was established via appending hydroxylaminobutyric acid to Sisomicin at position 1 and 2-hydroxyethyl group at position 6′ [1]. It was designed to evade all clinically relevant aminoglycoside-modifying enzymes, which contribute to the main resistance mechanism for aminoglycoside therapy [1]. However, acquired resistance of aminoglycosides may arise through over expression of efflux pumps and ribosomal modification by bacteria, which results from amino acid or rRNA sequence mutations [1]. Like other aminoglycosides, plazomicin is ineffective against bacterial isolates that produce 16S rRNA methyltransferases [Label]. Plazomicin mediates the antibacterial activity against pathogens including carbapenem-resistant (CRE) and extended-spectrum beta-lactamase (ESBL) producing Enterobacteriaceae. It mediates the antibacterial activity by binding to bacterial 30S ribosomal subunit and inhibiting protein synthesis [Label]. On June 28th, 2018, plazomicin sulfate was approved by the FDA for use in adult patients for the treatment of complicated urinary tract infections (cUTI) including Pyelonephritis. It is marketed as Zemdri and is administered via once-daily intravenous infusion.
Plazomicin (INN,[1] ZEMDRI) is a next-generation aminoglycoside (“neoglycoside”) antibacterial derived from sisomicin by appending a hydroxy-aminobutyric acid (HABA) substituent at position 1 and a hydroxyethyl substituent at position 6′.[2][3]
Plazomicin has been reported to demonstrate in vitro synergistic activity when combined with daptomycin or ceftobiprole versus methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant S. aureus (VRSA) and against Pseudomonas aeruginosawhen combined with cefepime, doripenem, imipenem or piperacillin/tazobactam.[3] It also demonstrates potent in vitro activity versus carbapenem-resistant Acinetobacter baumannii.[4]
In 2012, U.S. Food and Drug Administration granted fast track designation for the development and regulatory review of plazomicin.[5]
It is being developed by Achaogen, Inc. to treat serious bacterial infections due to multidrug-resistant Enterobacteriaceae, including carbapenem-resistant Enterobacteriaceae (CRE)[6] and was in Phase III clinical trials as of April 7, 2016.[7]
In June 2018 the FDA approved plazomicin (ZEMDRI) for adults with complicated urinary tract infections (cUTI), including pyelonephritis, caused by Escherichia coli, Klebsiella pneumoniae, Proteus mirabilis, or Enterobacter cloacae, in patients who have limited or no alternative treatment options. Zemdri is an intravenous infusion, administered once daily.[8][9] The FDA declined approval for treating bloodstream infections due to lack of effectiveness.[10]
To continue the development of plazomicin, the company has received a contract option of US$ 60M from the Biomedical Advanced Research and Development Authority (BARDA) to support a global Phase III clinical study. The study will evaluate plazomicin in treating patients with serious Gram-negative bacterial infections due to carbapenem-resistant Enterobacteriaceae. The study is expected to start in the fourth quarter of 2013 [4].
PATENT
WO 2009067692
WO 2010132770
PAPER
Synthesis and spectrum of the neoglycoside ACHN-490
Antimicrobial Agents and Chemotherapy (2010), 54, (11), 4636-4642
https://aac.asm.org/content/54/11/4636



PAPER
Plazomicin Retains Antibiotic Activity against Most Aminoglycoside Modifying Enzymes
ACS Infectious Diseases (2018), 4, (6), 980-987.
https://pubs.acs.org/doi/abs/10.1021/acsinfecdis.8b00001


PAPER
Effects of the 1-N-(4-Amino-2S-hydroxybutyryl) and 6′-N-(2-Hydroxyethyl) Substituents on Ribosomal Selectivity, Cochleotoxicity, and Antibacterial Activity in the Sisomicin Class of Aminoglycoside Antibiotics
ACS Infectious Diseases (2018), 4, (7), 1114-1120.
https://pubs.acs.org/doi/abs/10.1021/acsinfecdis.8b00052

Syntheses of the 6′-N-(2-hydroxyethyl) and 1-N-(4-amino-2S-hydroxybutyryl) derivatives of the 4,6-aminoglycoside sisomicin and that of the doubly modified 1-N-(4-amino-2S-hydroxybutyryl)-6′-N-(2-hydroxyethyl) derivative known as plazomicin are reported together with their antibacterial and antiribosomal activities and selectivities. The 6′-N-(2-hydroxyethyl) modification results in a moderate increase in prokaryotic/eukaryotic ribosomal selectivity, whereas the 1-N-(4-amino-2S-hydroxybutyryl) modification has the opposite effect. When combined in plazomicin, the effects of the two groups on ribosomal selectivity cancel each other out, leading to the prediction that plazomicin will exhibit ototoxicity comparable to those of the parent and the current clinical aminoglycoside antibiotics gentamicin and tobramycin, as borne out by ex vivo studies with mouse cochlear explants. The 6′-N-(2-hydroxyethyl) modification restores antibacterial activity in the presence of the AAC(6′) aminoglycoside-modifying enzymes, while the 1-N-(4-amino-2S-hydroxybutyryl) modification overcomes resistance to the AAC(2′) class but is still affected to some extent by the AAC(3) class. Neither modification is able to circumvent the ArmA ribosomal methyltransferase-induced aminoglycoside resistance. The use of phenyltriazenyl protection for the secondary amino group of sisomicin facilitates the synthesis of each derivative and their characterization through the provision of sharp NMR spectra for all intermediates.
https://pubs.acs.org/doi/suppl/10.1021/acsinfecdis.8b00052/suppl_file/id8b00052_si_001.pdf




4 (19 mg, 40%). [α]D 25 = +46.5 (c = 0.01, H2O);
1 H NMR (600 MHz, D2O): δ 5.51 ( s, 1H, H-1ʹ), 5.16 (t, J = 3.5 Hz, H, H-4ʹ), 4.99 (d , J = 4.0 Hz, 1H, H-1ʹʹ), 4.11 (dd , J =9.4 Hz, 3.9 Hz, 1H, CH(OH)CH2CH2), 4.00 (d , J = 12.8 Hz, 1H, H-5ʹʹ), 3.99-3.93 (m, 1H, H-1), 3.84 (dd, J = 11.0 Hz, 4.0 Hz, 1H, H-2ʹʹ), 3.81 (t, J = 9.9 Hz, 1H, H-4), 3.77 (t, J = 5.3 Hz, 1H, H-2ʹ), 3.71 (t, J = 5.1 Hz, 2H, NHCH2CH2O), 3.69 – 3.65 (m, 2H, H-6, H-6ʹ), 3.64 – 3.44 (m , 2H, H-5, H-6ʹ), 3.35 – 3.26 (m , 1H, H-3), 3.24 (d, J = 12.8 Hz, 1H, H-5ʹʹ), 3.15 (d, J = 11.0 Hz, 1H, H-3ʹʹ), 3.09 – 3.06 (m, 2H, NHCH2CH2O), 3.01 (t, J = 7.2 Hz, 2H, CH(OH)CH2CH2), 2.74 (s, 3H, NCH3), 2.58 – 2.52 (m, 1H, H-3ʹ), 2.29 – 2.24 (m, 1H, H-3ʹ), 2.07 (dt, J = 13.2 Hz, 4.4 Hz, 1H, H-2), 2.04 – 1.98 (m, 1H, CH(OH)CH2CH2), 1.84 – 1.79 (m, 1H, CH(OH)CH2CH2), 1.64 (q, 1H, J = 12.5 Hz, H-2), 1.17 (s, 3H, 4ʹʹ-CH3);
13C NMR (151 MHz, D2O): δ 181.2 (s, CH3COOH), 175.4 (s, NHCO), 141.7 (s, C-5ʹ), 102.5 (s, C-4ʹ), 98.0 (s, C-1ʹʹ), 96.9 (s, C-1ʹ), 79.8 (s, C-4), 78.8 (s, C-6), 73.8 (s, C-5), 69.8 (s, C-4ʹʹ), 69.4 (s, CH(OH)CH2CH2), 66.8 (s, C-5ʹʹ), 65.9 (s, C-2ʹʹ), 64.2 (s, C-3ʹʹ), 56.4 (s, NHCH2CH2O), 48.8 (s, C-1), 48.31 (s, NHCH2CH2O), 48.26 (s, C-3), 47.9 (s, C-6ʹ), 45.9 (s, C2ʹ), 36.8 (s, CH(OH)CH2CH2), 34.9 (s, NCH3), 30.7 (s, CH(OH)CH2CH2), 30.4 (s, C-2), 23.1 (s, CH3COOH), 23.0 (s, C-3ʹ), 20.8 (s, 4ʹʹ-CH3).
ESI-HRMS: m/z calcd. for C25H49N6O10 [M+H]+ 593.3510, found: 593.3481.
PATENT
http://www.google.com/patents/US20100099661
Common Intermediates Sisomicin

Amberlite IRA-400 (OH form) (200 g) was washed with MeOH (3×200 m1). To a stirring suspension of the washed resin in MeOH (150 mL) was added sisomicin sulfate (20.0 g, 0.029 mol) and the mixture was stirred overnight. The resin was then filtered and washed with MeOH (100 mL) and the combined organic layers were concentrated to dryness to yield the desired sisomicin (11.57 g, 0.026 mol, 89.6% yield): MS m/e [M+H]+ calcd 448.3, found 448.1.
Example 1 6′-(2-Hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin

6′-(2-tert-Butyldimethylsililoxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin
2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (0.10 g, 0.105 mmol) was treated with tert-butyldimethylsilyloxy acetaldehyde following Procedure 1-Method A to yield the desired 6′-(2-tert-butyldimethylsilyloxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (MS m/e [M+H]+ calcd 1107.6, found 1107.4), which was carried through to the next step without further purification.

6′-(2-Hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin
6′ -(2-tert-butyldimethylsililoxy-ethyl)-2′,3,3″-triBoc-1-(N-Boc-4-amino-2(S)-hydroxy-butyryl)-sisomicin (0.105 mmol) was submitted to Procedure 3-Method B for Boc removal to yield a crude, which was purified by RP HPLC Method 1-Column A to yield 6′-(2-hydroxy-ethyl)-1-(4-amino-2(S)-hydroxy-butyryl)-sisomicin: MS m/e [M+H]+ calcd 593.3, found 593.2, [M+Na]+615.3 ; CLND 97.5% purity.
- Achaogen. Study for the treatment of complicated urinary tract infection and acute pyelonephritis.Available online: http://www.clinicaltrials.gov/ct2/show/NCT01096849 (accessed on 11 April 2013).
- Zhanel, G.G.; Lawson, C.D.; Zelenitsky, S.; Findlay, B.; Schweizer, F.; Adam, H.; Walkty, A.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; et al. Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin. Expert Rev. Anti-Infect. Ther. 2012, 10, 459–473, doi:10.1586/eri.12.25.
- Endimiani, A.; Hujer, K.M.; Hujer, A.M.; Armstrong, E.S.; Choudhary, Y.; Aggen, J.B.; Bonomo, R.A. ACHN-490, a neoglycoside with potent in vitro activity against multidrug-resistant Klebsiella pneumoniae isolates. Antimicrob. Agents Chemother. 2009, 53, 4504–4507.
- Achaogen. Achaogen pipeline. Available online: http://www.achaogen.com (accessed on 30 August 2012).
- Achaogen. Achaogen Awarded $60M Contract Option by BARDA for the Clinical Development of Plazomicin. Available online: http://www.achaogen.com/news/151/15 (accessed on 19 June 2013).
- Achaogen. Achaogen announces all objectives met in Phase 2 Plazomicin complicated urinary tract infections study and start of first-in-human study with ACHN-975. Available online: http://www.achaogen.com/uploads/news/id148/Achaogen_PressRelease_2012–05–15.pdf (accessed on 10 April 2013).
- Achaogen. Achaogen Announces Agreement with FDA on a Special Protocol Assessment for a Phase 3 Clinical Trial of Plazomicin to Treat Infections Caused by Carbapenem-Resistant Enterobacteriaceae (CRE); Achaogen: San Francisco, CA, USA, 2013.
- Comparison of the next-generation aminoglycoside plazomicin to gentamicin, tobramycin and amikacin
-
4-23-2010ANTIBACTERIAL AMINOGLYCOSIDE ANALOGS

| US8318685 | Nov 14, 2011 | Nov 27, 2012 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8367625 | Apr 7, 2011 | Feb 5, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8372813 | Apr 7, 2011 | Feb 12, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8377896 | Mar 9, 2011 | Feb 19, 2013 | Isis Pharmaceuticals, Inc | Antibacterial 4,6-substituted 6′, 6″ and 1 modified aminoglycoside analogs |
| US8399419 | Mar 9, 2011 | Mar 19, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8481502 | Apr 6, 2012 | Jul 9, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8492354 | Nov 14, 2011 | Jul 23, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8524675 | Nov 14, 2011 | Sep 3, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8524689 | Nov 14, 2011 | Sep 3, 2013 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8569264 | Jan 5, 2012 | Oct 29, 2013 | Isis Pharmaceuticals, Inc. | Antibacterial 4,5-substituted aminoglycoside analogs having multiple substituents |
| US8653041 | Oct 15, 2012 | Feb 18, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8653042 | Nov 14, 2011 | Feb 18, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
| US8658606 | Nov 14, 2011 | Feb 25, 2014 | Achaogen, Inc. | Antibacterial aminoglycoside analogs |
References
- Jump up^ “WHO Drug Information, Vol. 26, No. 3, 2012. International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 68”(PDF). World Health Organization. p. 314. Retrieved 27 April 2016.
- Jump up^ Aggen, JB; Armstrong, ES; Goldblum, AA; Dozzo, P; Linsell, MS; Gliedt, MJ; Hildebrandt, DJ; Feeney, LA; Kubo, A; Matias, RD; Lopez, S; Gomez, M; Wlasichuk, KB; Diokno, R; Miller, GH; Moser, HE (30 August 2010). “Synthesis and Spectrum of the Neoglycoside ACHN-490” (PDF). Antimicrobial Agents and Chemotherapy. 54 (11): 4636–4642. doi:10.1128/AAC.00572-10. PMC 2976124
 . PMID 20805391. Retrieved 27 April2016.
. PMID 20805391. Retrieved 27 April2016. - ^ Jump up to:a b Zhanel, GG; Lawson, CD; Zelenitsky, S; Findlay, B; Schweizer, F; Adam, H; Walkty, A; Rubinstein, E; Gin, AS; Hoban, DJ; Lynch, JP; Karlowsky, JA (10 January 2014). “Comparison of the Next-Generation Aminoglycoside Plazomicin to Gentamicin, Tobramycin and Amikacin”. Expert Review of Anti-infective Therapy. 10 (4): 459–73. doi:10.1586/eri.12.25. PMID 22512755.
- Jump up^ García-Salguero, C; Rodríguez-Avial, I; Picazo, JJ; Culebras, E (October 2015). “Can Plazomicin Alone or in Combination Be a Therapeutic Option against Carbapenem-Resistant Acinetobacter baumannii?” (PDF). Antimicrobial Agents and Chemotherapy. 59 (10): 5959–66. doi:10.1128/AAC.00873-15. PMC 4576036 . Retrieved 27 April 2016.
- Jump up^ “Achaogen Announces Plazomicin Granted QIDP Designation by FDA”. GlobeNewswire, Inc. Retrieved 27 April 2016.
- Jump up^ “Achaogen — Plazomicin”. Achaogen, Inc. Retrieved 27 April2016.
- Jump up^ “Plazomicin — AdisInsight”. Springer International Publishing AG. Retrieved 27 April 2016.
- Jump up^ “Medscape Log In”. http://www.medscape.com. Retrieved 2018-07-03.
- Jump up^ “BioCentury – FDA approves plazomicin for cUTI, but not blood infections”. http://www.biocentury.com. Retrieved 2018-06-28.
- Jump up^ “Drugs@FDA: FDA Approved Drug Products”. http://www.accessdata.fda.gov. Retrieved 2018-06-28.
 |
|
| Names | |
|---|---|
| IUPAC name
(2S)-4-Amino-N-[(1R,2S,3S,4R,5S)-5-amino-4-[[(2S,3R)-3-amino-6-[(2-hydroxyethylamino)methyl]-3,4-dihydro-2H-pyran-2-yl]oxy]-2-[(2R,3R,4R,5R)-3,5-dihydroxy-5-methyl-4-(methylamino)oxan-2-yl]oxy-3-hydroxycyclohexyl]-2-hydroxybutanamide
|
|
| Other names
6′-(hydroxylethyl)-1-(HABA)-sisomicin
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEMBL | |
| ChemSpider | |
| KEGG | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C25H48N6O | |
| Molar mass | 592.683 g/mol |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F],
|
|
Achaogen is a clinical-stage biopharmaceutical company passionately committed to the discovery, development, and commercialization of novel antibacterials to treat multi-drug resistant, or MDR, gram-negative infections.

Achaogen (a-KAY-o-jen) is developing plazomicin, its lead product candidate, for the treatment of serious bacterial infections due to MDR Enterobacteriaceae, including carbapenem-resistant Enterobacteriaceae, or CRE. In 2013, the Centers for Disease Control and Prevention identified CRE as a “nightmare bacteria” and an immediate public health threat that requires “urgent and aggressive action.” We expect to initiate a Phase 3 superiority trial of plazomicin in the first quarter of 2014.
CRE are one of many types of MDR gram-negative pathogens threatening patients. Bacteria such as Pseudomonas aeruginosa, Acinetobacter baumannii, and extended-spectrum beta-lactamase producing Enterobacteriaceae each pose “serious” resistance threats, according to the CDC, and also drive a great need for new, safe, and effective antibiotics. We have assembled the chemistry and microbiology expertise and capabilities required to develop new agents for the treatment of gram-negative infections. Plazomicin was the first clinical candidate from our gram-negative antibiotic discovery engine. In addition, our research and development pipeline includes two antipseudomonal programs targeting P. aeruginosa—a program to discover and develop small molecule inhibitors of LpxC, which is an enzyme essential for the synthesis of the outer membrane of gram-negative bacteria, and a therapeutic antibody program. We are also pursuing small molecule research programs targeting other essential gram-negative enzymes.
Achaogen has built an exceptional research and development team with deep expertise in the discovery and development of new drugs from research through commercialization. Our executive team has over 60 years of combined industry experience, and a proven track record of leadership, global registration, and lifecycle management for over 20 products. Our facility is located on the shores of the San Francisco Bay, ten minutes from the San Francisco International Airport, and only fifteen minutes from downtown San Francisco.

ZEMDRITM (plazomicin) Approved by FDA for the Treatment of Adults with Complicated Urinary Tract Infections (cUTI)
― ZEMDRI is a new treatment for patients with cUTI, including pyelonephritis, due to certain Enterobacteriaceae ―
― ZEMDRI is the only once-daily aminoglycoside therapy approved for use in cUTI ―
― ZEMDRI has microbiological activity against pathogens designated by the CDC as urgent and serious public health threats, including carbapenem-resistant (CRE) and extended spectrum beta-lactamase (ESBL)- producing Enterobacteriaceae ―
SOUTH SAN FRANCISCO, Calif., June 26, 2018 (GLOBE NEWSWIRE) — Achaogen, Inc. (NASDAQ:AKAO), a biopharmaceutical company developing and commercializing innovative antibacterial agents to address multidrug resistant (MDR) gram-negative infections, today announced that the U.S. Food and Drug Administration (FDA) has approved ZEMDRI™ (plazomicin) for adults with complicated urinary tract infections (cUTI), including pyelonephritis, caused by certain Enterobacteriaceae in patients who have limited or no alternative treatment options. ZEMDRI is an intravenous infusion, administered once daily.
“The approval of ZEMDRI marks a significant milestone for Achaogen and we are excited to offer healthcare practitioners a new treatment option for patients with certain serious bacterial infections. ZEMDRI is designed to retain its potent activity in the face of certain difficult-to-treat MDR infections, including CRE and ESBL- producing Enterobacteriaceae,” said Blake Wise, Achaogen’s Chief Executive Officer. “Today’s milestone was made possible by our employees, by patients and investigators involved in our clinical trials, and by BARDA, who contributed significant funding for the development of ZEMDRI. This marks an important step in our commitment to fighting MDR bacteria and we are excited to launch ZEMDRI, a much needed once-daily antibiotic.”
“Bacteria continue to circumvent existing antibiotics, making certain infections notoriously hard to treat and putting some patients at high risk for mortality,” said James A. McKinnell, Assistant Professor of Medicine at the David Geffen School of Medicine and LA Biomed at Harbor-UCLA. “Aminoglycosides are a familiar and very effective class of antibiotics. I look forward to adding plazomicin to my short list of available treatment options and to its potential impact on patient outcomes.”
Regarding the potential indication for plazomicin for the treatment of bloodstream infection (BSI), the FDA issued a Complete Response Letter (CRL) stating that the CARE study does not provide substantial evidence of effectiveness of plazomicin for the treatment of BSI. The Company intends to meet with the FDA to determine whether there is a feasible resolution to address the CRL.
Achaogen will work with hospitals, providers, and insurers to ensure patients are able to receive this treatment. Patients, physicians, pharmacists, or other healthcare professionals with questions about ZEMDRI should contact 1.833.252.6400 or visit www.ZEMDRI.com.
ZEMDRI Phase 3 Clinical Results
The approval of ZEMDRI is supported in part by data from the EPIC (Evaluating Plazomicin In cUTI) clinical trial, which was the first randomized controlled study of once-daily aminoglycoside therapy for the treatment of cUTI, including pyelonephritis.
In the Phase 3 EPIC cUTI trial, ZEMDRI demonstrated non-inferiority to meropenem for the co-primary efficacy endpoints of composite cure (clinical cure and microbiological eradication) in the microbiological modified intent-to-treat (mMITT; N=388) population at Day 5 and test-of-cure (TOC) visit (Day 17 + 2). Composite cure rates at Day 5 were 88.0% (168/191) for ZEMDRI vs 91.4% (180/197) for meropenem (difference -3.4%, 95% CI, -10.0 to 3.1). Composite cure rates at TOC were 81.7% (156/191) for ZEMDRI vs 70.1% (138/197) for meropenem (difference 11.6%, 95% CI, 2.7 to 20.3). Composite cure at the TOC visit in patients with concomitant bacteremia at baseline was achieved in 72.0% (18/25) of patients in the ZEMDRI group and 56.5% (13/23) of patients in the meropenem group. The most common side effects (≥1% of patients treated with ZEMDRI) were decreased kidney function, diarrhea, hypertension, headache, nausea, vomiting, and hypotension.1
The FDA approved a breakpoint of <= 2 mcg/mL; greater than 99% of Escherichia coli, Klebsiella pneumoniae and Enterobacter cloacae in U.S. surveillance are susceptible to Zemdri when applying this breakpoint.2
About cUTI
cUTI is defined as a UTI occurring in a patient with an underlying complicating factor of the genitourinary tract, such as a structural or functional abnormality.3 Patients with pyelonephritis, regardless of underlying abnormalities of the urinary tract, are considered a subset of patients with cUTI.4 An estimated 3 million cases of cUTI are treated in the hospital setting in the US each year.5 Enterobacteriaceae are the most common pathogens causing cUTIs6, and resistance within this family is a global concern. High rates of resistance to previous mainstays of therapy necessitate alternative treatment options. Ineffectively managed cUTI can lead to increased treatment failure rates, recurrence of infection, increased re-hospitalization, and increased morbidity and mortality. cUTI infections place an economic burden on hospitals and payers.6,7
About ZEMDRI
ZEMDRI is an aminoglycoside with once-daily dosing that has activity against certain Enterobacteriaceae, including CRE and ESBL- producing Enterobacteriaceae. Achaogen’s EPIC clinical trial successfully evaluated the safety and efficacy of ZEMDRI in adult patients with cUTI, including pyelonephritis. ZEMDRI was engineered to overcome aminoglycoside-modifying enzymes, the most common aminoglycoside-resistance mechanism in Enterobacteriaceae, and has in vitro activity against ESBL- producing, aminoglycoside- resistant, and carbapenem- resistant isolates. The Centers for Disease Control and Prevention (CDC) has characterized ESBL- producing Enterobacteriaceae as a “serious threat” and CRE as “nightmare bacteria”, which is an immediate public health threat that requires urgent and aggressive action.
Working in the Lab

Working in the Lab
Achaogen, Inc.
Blake Wise, Chief Executive Officer at Achaogen

Blake Wise, Chief Executive Officer at Achaogen
Achaogen, Inc.
High-Resolution Achaogen company logo

High-Resolution Achaogen company logo
Achaogen, Inc.
/////////Plazomicin, ZEMDRI, FDA 2018, fast track designation, Plazomicin SULFATE, ACHN 490 sulfate, cUTI, Achaogen
CC1(COC(C(C1NC)O)OC2C(CC(C(C2O)OC3C(CC=C(O3)CNCCO)N)N)NC(=O)C(CCN)O)O
CN[C@@H]1[C@@H](O)[C@@H](O[C@H]2[C@@H](C[C@H](N)[C@@H](O[C@H]3OC(CNCCO)=CC[C@H]3N)[C@@H]2O)NC(=O)[C@@H](O)CCN)OC[C@]1(C)O
Inotersen sodium, イノテルセンナトリウム
Inotersen sodium,
UNII: 950736UC77
Data Source for DNA, d(P-thio)((2′-O-(2-methoxyethyl))m5rU-(2′-O-(2-methoxyethyl))m5rC-(2′-O-(2-methoxyethyl))m5rU-(2′-O-(2-methoxyethyl))m5rU-(2′-O-(2-methoxyethyl))rG-G-T-T-A-m5C-A-T-G-A-A-(2′-O-(2-methoxyethyl))rA-(2′-O-(2-methoxyethyl))m5rU-(2′-O-(2-methoxyethyl))m5rC-(2′-O-(2-methoxyethyl))m5rC-(2′-O-(2-methoxyethyl))m5rC)
STR https://chem.nlm.nih.gov/chemidplus/rn/1432726-13-0
IUPAC Condensed
Thy-MeOEt(-2)Ribf-sP-m5Cyt-MeOEt(-2)Ribf-sP-Thy-MeOEt(-2)Ribf-sP-Thy-MeOEt(-2)Ribf-sP-Gua-MeOEt(-2)Ribf-sP-dGuo-sP-dThd-sP-dThd-sP-dAdo-sP-m5Cyt-dRibf-sP-dAdo-sP-dThd-sP-dGuo-sP-dAdo-sP-dAdo-sP-Ade-MeOEt(-2)Ribf-sP-Thy-MeOEt(-2)Ribf-sP-m5Cyt-MeOEt(-2)Ribf-sP-m5Cyt-MeOEt(-2)Ribf-sP-m5Cyt-MeOEt(-2)Ribf.19Na+
IUPAC
O2′-(2-methoxyethyl)-5-methyl-P-thio-uridylyl-(3′->5′)-O2′-(2-methoxyethyl)-5-methyl-P-thio-cytidylyl-(3′->5′)-O2′-(2-methoxyethyl)-5-methyl-P-thio-uridylyl-(3′->5′)-O2′-(2-methoxyethyl)-5-methyl-P-thio-uridylyl-(3′->5′)-O2′-(2-methoxyethyl)-P-thio-guanylyl-(3′->5′)-2′-deoxy-P-thio-guanylyl-(3′->5′)-P-thio-thymidylyl-(3′->5′)-P-thio-thymidylyl-(3′->5′)-2′-deoxy-P-thio-adenylyl-(3′->5′)-2′-deoxy-5-methyl-P-thio-cytidylyl-(3′->5′)-2′-deoxy-P-thio-adenylyl-(3′->5′)-P-thio-thymidylyl-(3′->5′)-2′-deoxy-P-thio-guanylyl-(3′->5′)-2′-deoxy-P-thio-adenylyl-(3′->5′)-2′-deoxy-P-thio-adenylyl-(3′->5′)-O2′-(2-methoxyethyl)-P-thio-adenylyl-(3′->5′)-O2′-(2-methoxyethyl)-5-methyl-P-thio-uridylyl-(3′->5′)-O2′-(2-methoxyethyl)-5-methyl-P-thio-cytidylyl-(3′->5′)-O2′-(2-methoxyethyl)-5-methyl-P-thio-cytidylyl-(3′->5′)-O2′-(2-methoxyethyl)-5-methyl-cytidine sodium salt
|
イノテルセンナトリウム
|
| Formula |
C230H299N69O121P19S19. 19Na
|
|---|---|
| Cas |
1432726-13-0
Antisense oligonucleotide; TTR mRNA
|
| Mol weight |
7600.7669
|
- ClassAntisense oligonucleotides; Neuroprotectants
- Mechanism of ActionPrealbumin expression inhibitors
- Orphan Drug StatusYes – Amyloid polyneuropathy
- New Molecular Entity Yes
Highest Development Phases
- RegisteredAmyloid polyneuropathy
- Phase IIAmyloidosis; Cardiomyopathies
Most Recent Events
- 07 Aug 2018PTC Therapeutics announces intention to submit regulatory application in Latin America
- 06 Aug 2018Akcea Therapeutics intends to launch inotersen in Germany after Summer 2018
- 02 Aug 2018Inotersen licensed to PTC Therapeutics in Latin America
UNII-950736UC77; 950736UC77; Inotersen sodium; Inotersen sodium [USAN]; ISIS 420915 salt; 1432726-13-0
////////////////Inotersen sodium, eu 2018, イノテルセンナトリウム ,
SMILES
[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].COCCO[C@@H]1[C@H](O)[C@@H](COP(=O)([S-])O[C@@H]2[C@@H](COP(=O)([S-])O[C@@H]3[C@@H](COP(=O)([S-])O[C@@H]4[C@@H](COP(=O)([S-])O[C@@H]5[C@@H](COP(=O)([S-])O[C@H]6C[C@@H](O[C@@H]6COP(=O)([S-])O[C@H]7C[C@@H](O[C@@H]7COP(=O)([S-])O[C@H]8C[C@@H](O[C@@H]8COP(=O)([S-])O[C@H]9C[C@@H](O[C@@H]9COP(=O)([S-])O[C@H]%10C[C@@H](O[C@@H]%10COP(=O)([S-])O[C@H]%11C[C@@H](O[C@@H]%11COP(=O)([S-])O[C@H]%12C[C@@H](O[C@@H]%12COP(=O)([S-])O[C@H]%13C[C@@H](O[C@@H]%13COP(=O)([S-])O[C@H]%14C[C@@H](O[C@@H]%14COP(=O)([S-])O[C@H]%15C[C@@H](O[C@@H]%15COP(=O)([S-])O[C@@H]%16[C@@H](COP(=O)([S-])O[C@@H]%17[C@@H](COP(=O)([S-])O[C@@H]%18[C@@H](COP(=O)([S-])O[C@@H]%19[C@@H](COP(=O)([S-])O[C@@H]%20[C@@H](CO)O[C@H]([C@@H]%20OCCOC)N%21C=C(C)C(=O)NC%21=O)O[C@H]([C@@H]%19OCCOC)N%22C=C(C)C(=NC%22=O)N)O[C@H]([C@@H]%18OCCOC)N%23C=C(C)C(=O)NC%23=O)O[C@H]([C@@H]%17OCCOC)N%24C=C(C)C(=O)NC%24=O)O[C@H]([C@@H]%16OCCOC)n%25cnc%26C(=O)NC(=Nc%25%26)N)n%27cnc%28C(=O)NC(=Nc%27%28)N)N%29C=C(C)C(=O)NC%29=O)N%30C=C(C)C(=O)NC%30=O)n%31cnc%32c%31ncnc%32N)N%33C=C(C)C(=NC%33=O)N)n%34cnc%35c%34ncnc%35N)N%36C=C(C)C(=O)NC%36=O)n%37cnc%38C(=O)NC(=Nc%37%38)N)n%39cnc%40c%39ncnc%40N)n%41cnc%42c%41ncnc%42N)O[C@H]([C@@H]5OCCOC)n%43cnc%44c%43ncnc%44N)O[C@H]([C@@H]4OCCOC)N%45C=C(C)C(=O)NC%45=O)O[C@H]([C@@H]3OCCOC)N%46C=C(C)C(=NC%46=O)N)O[C@H]([C@@H]2OCCOC)N%47C=C(C)C(=NC%47=O)N)O[C@H]1N%48C=C(C)C(=NC%48=O)N
CC1=CN(C(=O)NC1=O)C2CC(C(O2)COP(=S)([O-])OC3CC(OC3COP(=S)([O-])OC4CC(OC4COP(=S)([O-])OC5C(OC(C5OCCOC)N6C=NC7=C6N=C(NC7=O)N)COP(=S)([O-])OC8C(OC(C8OCCOC)N9C=C(C(=O)NC9=O)C)COP(=S)([O-])OC1C(OC(C1OCCOC)N1C=C(C(=O)NC1=O)C)COP(=O)(OC1C(OC(C1OCCOC)N1C=C(C(=NC1=O)N)C)COP(=S)([O-])OC1C(OC(C1OCCOC)N1C=C(C(=O)NC1=O)C)CO)[S-])N1C=NC2=C1N=C(NC2=O)N)N1C=C(C(=O)NC1=O)C)OP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=CN=C2N)OP(=S)([O-])OCC1C(CC(O1)N1C=C(C(=NC1=O)N)C)OP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=CN=C2N)OP(=S)([O-])OCC1C(CC(O1)N1C=C(C(=O)NC1=O)C)OP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=C(NC2=O)N)OP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=CN=C2N)OP(=S)([O-])OCC1C(CC(O1)N1C=NC2=C1N=CN=C2N)OP(=S)([O-])OCC1C(C(C(O1)N1C=NC2=C1N=CN=C2N)OCCOC)OP(=S)([O-])OCC1C(C(C(O1)N1C=C(C(=O)NC1=O)C)OCCOC)OP(=S)([O-])OCC1C(C(C(O1)N1C=C(C(=NC1=O)N)C)OCCOC)OP(=S)([O-])OCC1C(C(C(O1)N1C=C(C(=NC1=O)N)C)OCCOC)OP(=S)([O-])OCC1C(C(C(O1)N1C=C(C(=NC1=O)N)C)OCCOC)O.[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+]
Amfonelic acid, амфонеловая кислота , حمض أمفونيليك , 安福萘酸 , アンホネル酸
Amfonelic acid
- Molecular FormulaC18H16N2O3
- Average mass308.331 Da
1,8-Naphthyridine-3-carboxylic acid, 1-ethyl-1,4-dihydro-4-oxo-7-(phenylmethyl)-
15180-02-6 [RN]
1-Ethyl-1,4-dihydro-4-oxo-7-(phenylmethyl)-1,8-naphthyridine-3-carboxylic Acid
2324
RR302AR19Y
NSC 100638
амфонеловая кислота [Russian] [INN]
حمض أمفونيليك [Arabic] [INN]
安福萘酸 [Chinese] [INN]
Lopac0_000416
MFCD00055095 [MDL number]
NCA
NSC-100638
UNII:RR302AR19Y
UNII-RR302AR19Y
Win 25,978
Amfonelic acid (AFA; WIN 25,978) is a research chemical and dopaminergic stimulant with antibiotic properties.[1]
History
The stimulant properties of AFA were discovered serendipitously at Sterling-Winthrop in the midst of research on the antibiotic nalidixic acid.[1] In addition to behaving as antibiotics, it was found that many derivatives of nalidixic acid have either stimulant or depressant effects on the central nervous system.[2] Researchers at Sterling-Winthrop found that AFA had a higher potency and therapeutic indexthan cocaine or amphetamine and so it was singled out for further study.[1][3] A small number of clinical trials were held in the 1970s, but when it was found that AFA exacerbated psychotic symptoms in schizophrenic patients and produced undesirable stimulant properties in geriatric depressives clinical evaluation of AFA was discontinued.[1] AFA remains a widely used pharmacological tool for study of the brain’s reward system, dopamine pathways, and the dopamine transporter.[1] Since 2013 AFA has been sold on the gray market and there are numerous anecdotal reports detailing its non-medical use.[1]
Pharmacology
In studies it proved to be a potent and highly selective dopamine reuptake inhibitor (DRI) in rat brain preparations.[4][5] A study found a moderately long half-life of approximately 12 hours and a dopaminergic potency approximately 50 fold that of methylphenidate in rat brain preparations.[6] Despite lack of direct serotonin activity, rats treated with subchronic doses of amfonelic acid display subsequent decreases in 5HT and 5HIAA.[7] Amfonelic acid displays no activity in the norepinephrine system.[8]
Despite its different mechanism of action, amfonelic acid displays discriminatory substitution with 150% the stimulant potency of dextroamphetamine.[9] Amfonelic acid has been shown to be neuroprotective against methamphetamine damage to dopamine neurons.[10] It also increases the effects of the antipsychotic drugs haloperidol, trifluoperazine and spiperone.[11] Rats are shown to self-administer amfonelic acid in a dose-dependent manner.[12]
Though AFA was discovered in the course of antibiotic research, there is very little data available on the drug’s antimicrobial activity. In 1988 the biologist G.C. Crumplin wrote, “[AFA] is less active against bacteria than are many other 4-quinolones, but studies in our laboratory on selected mammalian cell lines have shown it to be markedly more toxic to these cells than are the 4-quinolones that are more active antibacterial agents. Furthermore, it can be shown that sublethal doses induced marked changes in the pattern of proteins produced by the cell, thus suggesting a possible effect of 4-quinolones on gene transcription in mammalian cells.”[13] When evaluated via broth microdilution the MIC of AFA for Escherichia coli is 125 μg/mL, a concentration thirty times higher than the MIC for nalidixic acid in the same E. coli strain.[1]
References
- ^ Jump up to:a b c d e f g Morris, Hamilton (October 2015). “Sad Pink Monkey Blues”. Harper’s Magazine. Retrieved 2015-09-19.
- Jump up^ US patent 3590036, “Naphthyridine-3-carboxylic Acids, Their Derivatives and Preparation Thereof”
- Jump up^ Aceto, M.A. (1970). “Pharmacologic properties and mechanism of action of amfonelic acid”. European Journal of Pharmacology. 10: 344–354. doi:10.1016/0014-2999(70)90206-2. PMID 4393073.
- Jump up^ Fuller, R. W.; Perry, K. W.; Bymaster, F. P.; Wong, D. T. (1978). “Comparative effects of pemoline, amfonelic acid and amphetamine on dopamine uptake and release in vitro and on brain 3,4-dihydroxyphenylacetic acid concentration in spiperone-treated rats”. Journal of Pharmacy and Pharmacology. 30 (3): 197–198. doi:10.1111/j.2042-7158.1978.tb13201.x. PMID 24701.
- Jump up^ McMillen, B. A.; Shore, P. A. (1978). “Amfonelic acid, a non-amphetamine stimulant, has marked effects on brain dopamine metabolism but not noradrenaline metabolism: Association with differences in neuronal storage systems”. Journal of Pharmacy and Pharmacology. 30 (7): 464–466. doi:10.1111/j.2042-7158.1978.tb13293.x. PMID 27622.
- Jump up^ Izenwasser, S.; Werling, L. L.; Cox, B. M. (1990). “Comparison of the effects of cocaine and other inhibitors of dopamine uptake in rat striatum, nucleus accumbens, olfactory tubercle, and medial prefrontal cortex”. Brain Research. 520 (1–2): 303–309. doi:10.1016/0006-8993(90)91719-W. PMID 2145054.
- Jump up^ McMillen, BA; Scott, SM; Williams, HL (1991). “Effects of subchronic amphetamine or amfonelic acid on rat brain dopaminergic and serotonergic function”. Journal of neural transmission. General section. 83 (1–2): 55–66. doi:10.1007/BF01244452. PMID 2018630.
- Jump up^ Agmo, A; Belzung, C; Rodríguez, C (1997). “A rat model of distractibility: Effects of drugs modifying dopaminergic, noradrenergic and GABAergic neurotransmission”. Journal of neural transmission (Vienna, Austria : 1996). 104 (1): 11–29. doi:10.1007/BF01271291. PMID 9085190.
- Jump up^ Aceto, MD; Rosecrans, JA; Young, R; Glennon, RA (1984). “Similarity between (+)-amphetamine and amfonelic acid”. Pharmacology Biochemistry and Behavior. 20 (4): 635–7. doi:10.1016/0091-3057(84)90316-2. PMID 6728880.
- Jump up^ Pu, C; Fisher, JE; Cappon, GD; Vorhees, CV (1994). “The effects of amfonelic acid, a dopamine uptake inhibitor, on methamphetamine-induced dopaminergic terminal degeneration and astrocytic response in rat striatum”. Brain Research. 649 (1–2): 217–24. doi:10.1016/0006-8993(94)91067-7. PMID 7953636.
- Jump up^ Waldmeier, PC; Huber, H; Heinrich, M; Stoecklin, K (1985). “Discrimination of neuroleptics by means of their interaction with amfonelic acid: An attempt to characterize the test”. Biochemical Pharmacology. 34 (1): 39–44. doi:10.1016/0006-2952(85)90097-8. PMID 2857083.
- Jump up^ Porrino, LJ; Goodman, NL; Sharpe, LG (1988). “Intravenous self-administration of the indirect dopaminergic agonist amfonelic acid by rats”. Pharmacology Biochemistry and Behavior. 31 (3): 623–6. doi:10.1016/0091-3057(88)90240-7. PMID 2908003.
- Jump up^ Crumplin, G.C. (1988). “Aspects of Chemistry in the Development of the 4-Quinolone Antibacterial Agents”. Reviews of Infectious Diseases. 10 Suppl 1 (10): S2–S9. doi:10.1093/clinids/10.Supplement_1.S2. PMID 3279494.
External links
 |
|
| Clinical data | |
|---|---|
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C18H16N2O3 |
| Molar mass | 308.3329 g/mol |
| 3D model (JSmol) | |
////////////Amfonelic acid, RR302AR19Y, амфонеловая кислота , حمض أمفونيليك , 安福萘酸 , アンホネル酸
CCN1C=C(C(=O)C2=C1N=C(C=C2)CC3=CC=CC=C3)C(=O)O
BINIMETINIB, биниметиниб , بينيميتينيب , 美替尼 , ビニメチニブ

Binimetinib
MEK-162
биниметиниб [Russian] [INN]
بينيميتينيب [Arabic] [INN]
贝美替尼 [Chinese] [INN]
ビニメチニブ
5-[(4-bromo-2-fluorophenyl)amino]-4-fluoro-N-(2-hydroxyethoxy)-1-methyl-1H-benzimidazole-6-carboxamide
5-(4-Bromo-2-fluorophenylamino)-4-fluoro-1-methyl-1H-benzimidazole-6-carbohydroxamic acid 2-hydroxyethyl ester
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide
606143-89-9 CAS
C17H15BrF2N4O3, 441.227
UNII-181R97MR71
181R97MR71
1H-Benzimidazole-6-carboxamide, 5-[(4-bromo-2-fluorophenyl)amino]-4-fluoro-N-(2-hydroxyethoxy)-1-methyl-
tyrosine kinase inhibitor, antineoplastic
Array BioPharma Inc;PHASE 3 Cancer, ovary (serous)
Novartis PHASE 3 Melanoma
CAS 606143-89-9 [RN]
9764
ARRY-162
ARRY-438162, NVP-MEK162
MEK-1 protein kinase inhibitor; MEK-2 protein kinase inhibitor
Liver injury; Melanoma; Noonan syndrome; Ovary tumor; Solid tumor
On June 27, 2018, the Food and Drug Administration approved encorafenib and binimetinib in combination patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test
Binimetinib, also known as Mektovi and ARRY-162, is an anti-cancer small molecule that was developed by Array Biopharma to treat various cancers.[1] Binimetinib is a selective inhibitor of MEK, a central kinase in the tumor-promoting MAPK pathway.[2] Inappropriate activation of the pathway has been shown to occur in many cancers.[2] In June 2018 it was approved by the FDA in combination with encorafenib for the treatment of patients with unresectable or metastatic BRAF V600E or V600K mutation-positive melanoma.[3]
Binimetinib, also known as Mektovi, is a potent is a potent and selective oral mitogen-activated protein kinase 1/2 (MEK 1/2) inhibitor which is combined with Encorafenib [4],[8].
On June 27, 2018, the Food and Drug Administration approved the combination of Encorafeniband binimetinib (BRAFTOVI and MEKTOVI, from Array BioPharma Inc.) in combination for patients with unresectable or metastatic melanoma with the BRAF V600E or V600K mutations, as detected by an FDA-approved test [8].
Binimetinib was originally developed by Array BioPharma, then licensed to Novartis for worldwide development in 2010. But Array Biopharma regained full worldwide rights of the product in 2015. And in 2015, Pierre Fabre acquired exclusive rights to commercialize the product.
Mechanism of action
Binimetinib is an orally available inhibitor of mitogen-activated protein kinase kinase (MEK), or more specifically, a MAP2K inhibitor.[4]MEK is part of the RAS pathway, which is involved in cell proliferation and survival. MEK is upregulated in many forms of cancer.[5]Binimetinib, uncompetitive with ATP, binds to and inhibits the activity of MEK1/2 kinase, which has been shown to regulate several key cellular activities including proliferation, survival, and angiogenesis.[6] MEK1/2 are dual-specificity threonine/tyrosine kinases that play key roles in the activation of the RAS/RAF/MEK/ERK pathway and are often upregulated in a variety of tumor cell types.[7] Inhibition of MEK1/2 prevents the activation of MEK1/2 dependent effector proteins and transcription factors, which may result in the inhibition of growth factor-mediated cell signaling.[8] As demonstrated in preclinical studies, this may eventually lead to an inhibition of tumor cell proliferation and an inhibition in production of various inflammatory cytokines including interleukin-1, -6 and tumor necrosis factor.[8]
Development
In 2015, it was in phase III clinical trials for ovarian cancer,[9] BRAF mutant melanoma,[10] and NRAS Q61 mutant melanoma.[11]
In December 2015, the company announced that the mutant-NRAS melanoma trial was successful.[12] In the trial, those receiving binimetinib had a median progression-free survival of 2.8 months versus 1.5 months for those on the standard dacarbazinetreatment.[13] NDA submitted Jun 2016,[14] and the FDA should decide by 30 June 2017.[15]
In April 2016, it was reported that the phase III trial for low-grade ovarian cancer was terminated due to lack of efficacy.[16]
Binimetinib was studied for treatment of rheumatoid arthritis, but a phase II trial did not show benefit.
In 2017, the FDA informed Array Biopharma that the phase III trial data was not sufficient and the New Drug Application was withdrawn.[17]
In June 2018 it was approved for the treatment of certain melanomas by the FDA in combination with encorafenib.[3]
Growth factor-mediated proliferative signals are transmitted from the extracellular environment to the nucleus through several pathways, including the RAS/RAF/ MEK pathway. The RAS/RAF/MEK kinase signal transduction pathway is activated through initial extracellular binding and stimulation of tyrosine receptor kinases (RTKs) by their respective cognate ligands. Upon autophosphorylation of specific tyrosine residues in the cytosolic domain of RTKs, the Grb2-Sos complex translocates to the plasma membrane, and converts the inactive RAS’GDP to active RAS’GTP. The interaction between the Grb2 docking protein and the activated kinases or the phosphorylated receptor associated proteins is mediated by the Src Homology (SH2) domain of the signaling protein that recognizes specific phosphotyrosine sequences. RAS undergoes a conformational change upon guanosine 5 ‘-triphosphate (GTP) binding and causes the recruitment of RAF- 1 to the cytoplasmic membrane where it is phosphorylated by several kinases and simultaneous disphosphorylated at key residues by protein phosphatase-2B. Activated RAF phosphorylates the mitogen- activated protein kinase kinase (MEK) on two serine residues in the activation loop, which results in the activation of this protein kinase. MEK then phosphorylates and activates extracellular signal-regulated kinase (ERK), allowing its translocation to the nucleus where it phosphorylates transcriptional factors permitting the expression of a variety of genes.
The RAS/RAF/MEK signal transduction pathway is deregulated, often through mutations that result in ectopic protein activation, in roughly 1/3 of human cancers. This deregulation in turn results in a wide array of cellular changes that are integral to the etiology and maintenance of a cancerous phenotype including, but not limited to, the promotion of proliferation and evasion of apoptosis (Dhillon et al., Oncogene, 2007, 26: 3279-3290).
Accordingly, the development of small molecule inhibitors of key members of the RAS/ RAF/ MEK signal transduction pathway has been the subject of intense effort within the pharmaceutical industry and oncology community.
MEK is a major protein in the RAS/ RAF/ MEK pathway, which signals toward cell proliferation and survival, and frequently activated in tumors that have mutations in the RAS or RAF oncogenes or in growth receptor tyrosine kinases. MEK is a key player in the RAS/RAF/MEK pathway as it is downstream of RAS and RAF. Despite being only rarely mutated in cancer (Murugan et al., Cell Cycle, 2009, 8: 2122-2124; Sasaki et al., J. Thorac. Oncol., 2010, 5: 597-600), inhibitors of the MEK1 and MEK2 proteins have also been targeted for small molecule inhibition owing to their central position within the RAS/ RAF/ MEK signal transduction pathway signaling cascade (Fremin and Meloche, J. Hematol.
Oncol., 2010, 3:8). Recently a potent MEK inhibitor failed to demonstrate efficacy in clinical trials in patients with advanced non-small cell lung cancer (Haura et al., Clin. Cancer Res., 2010, 16: 2450-2457). The reason for failure in this trial is not clear.
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (hereinafter, “Compound A”) is a benzimidazole compound that is a known potent and selective inhibitor of the MEK1 and MEK2 proteins, and useful in the treatment of hyperproliferative diseases, particularly cancer, in mammals. For example, in a recently published Phase I study of 28 patients suffering from unresectable, locally advanced or metastatic biliary cancer and who had received < 1 prior systemic therapy, oral Compound A treatment (60 mg twice daily) resulted in 1 complete regression, 1 partial regression and 11 stable disease diagnoses after at least 6 weeks of treatment (Finn et al., J. Clin. Oncol. 30, 2012 (Supplement 4, 2012 Gastrointestinal Cancers Symposium, Abstract No. 220). Compound A has also been demonstrated to be effective in the treatment of patients with either BRAFV600 or NRAS-mutant melanoma (Ascierto et al., J. Clin. Oncol. 30, 2012 (Supplement, 2012 ASCO Annual Meeting, Abstract No. 8511).
The compound, as well as a process for its preparation, is disclosed in PCT Pub. No. WO 03/077914
MEK-162, a potent, orally active MEK1/2 inhibitor, is in phase III clinical trials at Array BioPharma and licensee Novartis for the treatment of metastatic or unresectable cutaneous melanoma with NRAS mutations and in combination with LGX-818 in adult patients with BRAF V600. Phase III studies are also under way at Array BioPharma for the treatment of low grade serous carcinomas of the ovary, fallopian tube or primary peritoneum following at least one prior platinum-based chemotherapy regimen and no more than three lines of prior chemotherapy regimens. Novartis and Array BioPharma are also conducting phase II clinical studies for the treatment of locally advanced and unresectable or metastatic malignant cutaneous melanoma, harboring BRAFV600E mutations; in BRAF mutated melanoma in combination with AMG-479 and for the treatment of Noonan’s syndrome, and in non-small cell lung cancer harboring KRAS or EGFR mutation and in combination with erlotinib. MEK-162 is being evaluated in phase I/II as first line treatment of advanced biliary tract carcinoma and for the treatment of adult patients with mutant or wild-type RAS metastatic colorectal cancer. The product is in early clinical trials at Array Biopharma for the treatment of biliary cancer.
According to Array, MEK-162 may also provide broad therapeutic benefits in the treatment of chronic degenerative diseases. However, a phase II trial for the treatment of stable rheumatoid arthritis (RA) did not meet its primary endpoint. Based on these data, the company focused development of MEK-162 solely in oncology.
In 2010, MEK-162 was licensed to Novartis by Array BioPharma for worldwide development. In 2013, orphan drug designation was assigned in Japan for the treatment of malignant melanoma with NRAS or BRAF V600 mutation.
WO-2014063024 DEALS WITH Preparation, crystalline forms, and formulations comprising binimetinib. Binimetinib is a MEK-1/2 inhibitor originally claimed in WO03077914, which Array and Novartis are developing for the treatment of cancer, including melanoma, low-grade serous ovarian cancer, and other solid tumors, as well as Noonan syndrome hypertrophic cardiomyopathy and hepatic impairment. See also WO2014018725 for the most recent filing on the agent
SYNTHESIS

PATENT
WO 03/077914
http://www.google.com/patents/WO2003077914A1?cl=en
Schemes 1-4.
Scheme 1


Scheme la

Scheme 2

Scheme 3

17 18
Scheme 4

25
Scheme 5


General synthetic methods which may be referred to for preparing some of the compounds of the present invention are provided in PCT published application number WO 00/42022 (published July 20, 2000). The foregoing patent application is incorporated herein by reference in its entirety.
similar ie chloro instead of fluoro
Example 52

6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy)-amide (lOcc) Step A: 3-Chloro-2,4-difluoro-5-nitro-benzoic acid 2a
3-Chloro-2,4-difluoro-benzoic acid la (3.00 g, 15.6 mmol) is added to a stirred solution of concentrated H2SO4 (16 mL) and fuming nitric acid (0.85 mL, 20.3 mmol). After 3 hours a precipitate forms. The yellow slurry is poured onto ice water (100 mL). The aqueous mixture is extracted with diethyl ether (3x). The organic extracts are dried (Na2SO4) and concentrated under reduced pressure to give 3.50 g (95%) of clean desired product as a pale yellow solid.
Step B: 4-Amino-3-chloro-2-fluoro-5-nitro-benzoic acid 3a
Ammonium hydroxide solution (6.88 g, -30% in water, 58.9 mmol) is added to a solution of 3-chloro-2,4-difluoro-5-nitro-benzoic acid 2a (3.5 g, 14.7 mmol) in water (16 mL) at 0 °C with stirring. Upon completion of the ammonium hydroxide addition the reaction mixture is warmed to room temperature. After 5 hours the reaction mixture is cooled to 0 °C and concentrated HCl is carefully added until the pH of the reaction mixture is near zero. The solid is collected by filtration and washed with water and diethyl ether. The solids are transferred to a round bottom flask as a solution in MeOH and EtOAc and concentrated under reduced pressure to give 2.96 g of a yellow solid. The filtrate is partitioned between diethyl ether and water and the organic layer is washed with brine. The combined organic extracts are dried (Na2SO ) and concentrated under reduced pressure to give 0.65 g of product. Recovered a total of 3.61 g (104%) of pure desired product, that is carried forward without further purification.
Step C: 4~Amino-3-chloro-2-fluoro-5-nitro-benzoic acid methyl ester 4a
To a stirred solution of 4-amino-3-chloro-2-fluoro-5-nitro-benzoic acid 3a (3.61 g, 15.4 mmol) in THF (30 mL) and MeOH (10 mL), TMS diazomethane (9.23 mL, 2.0 M solution in hexanes, 18.5 mmol) is added. After completion of reaction, the reaction mixture is concentrated via rotary evaporation with acetic acid in the trap. The recovered oily solid is triturated with diethyl ether to provide 1.51 g of a yellow solid. The filtrate is concentrated and triturated with diethyl ether to give an additional 0.69 g of yellow solid. A total of 2.20 g (57%) of pure desired product is recovered.
Step D: 4-Amino-3-chloro-5-nitro-2-phenylamino-benzoic acid methyl ester 5c
4-Amino-3-chloro-2-fluoro-5-nitro-benzoic acid methyl ester 4a (2.20 g, 8.84 mmol) is suspended in MeOH (9.4 mL) and aniline (3.22 mL, 35.4 mmol) is added. The reaction mixture is heated to reflux with stirring under a nitrogen atmosphere. After 19 hours, the reaction is complete. Distilled water (3.22 mL) is added to the reaction mixture and refluxing is continued for one hour. The reaction mixture is cooled to 0 °C in an ice bath for 20 minutes. The reaction mixture is filtered and washed with 3:10 distilled water/MeOH (65 mL total) and then with MeOH. The solid is dissolved with CH2C12 and concentrated under reduced pressure to give 2.40 g (84%) of pure desired product. MS APCI (-) m/z 320.3 (M-l) detected.
Step E: 4, 5-Diamino-3-chloro-2-phenylamino-benzoic acid methyl ester 6b
4-Amino-3-chloro-5-nitro-2-phenylamino-benzoic acid methyl ester 5c (0.50 g, 1.55 mmol) is dissolved into 2:1 EtOH/MeOH (15.5 mL). Saturated aqueous NH4C1 (15 mL), Zn powder (1.02 g, 15.6 mmol), and THF (10 mL) are added. After stirring for 20 hours, the reaction mixture is diluted with CH C12/THF and water. The organic layer is washed with water (3x). The combined organic extracts are dried (Na2SO4) and concentrated under reduced pressure. The solids are triturated with ether to give 0.32 g (70%) clean desired product. Step F: 7-Chloro-6-phenylamino-3H-benzoimidazole-5-carboxylic acid methyl ester 7c
4,5-Diamino-3-chloro-2-phenylamino-benzoic acid methyl ester 6b (0.32 g, 1.09 mmol) and formamidine acetate (72 mg, 1.64 mmol) in EtOH (36 mL) are heated, with stirring, to 80 °C. After 44 hours, the reaction mixture is cooled to room temperature and diluted with EtOAc and washed with water (3x), saturated NaHCO3, and brine. The combined organic extracts are dried (Na2SO4) and concentrated under reduced pressure to give 0.33 g (99%) clean desired product as a solid. MS APCI (+) m/z 302.3 (M+l) detected.
Step G: 6-(4-Bromo-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester 8g
7-Chloro-6-phenylamino-3H-benzoimidazole-5-carboxylic acid methyl ester 7c (0.327 g, 1.08 mmol) is dissolved into DMF (16 mL) and NBS (0.193 g, 1.08 mmol) is added. After one hour, the reaction mixture is quenched by the addition of saturated aqueous NaHSO3. The reaction mixture is then partitioned between EtOAc/THF and water. The organic layer is washed with water and brine. The combined organic extracts are dried (Na2SO ) and concentrated under reduced pressure. The recovered solid is triturated with ether to give 0.225 g (54%) pure desired product. MS ESI (+) m/z 382, 384 (M+, Br pattern) detected.
Step H: 6-(4-Bromo-2-chloro-phenylamino)- 7 -chloro-3H-benzoimidazole-5 -carboxylic acid methyl ester lOdd 6-(4-Bromo-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester 8g (0.225 g, 0.591 mmol) is dissolved in DMF (2 mL) and NCS (79 mg, 0.591 mmol) is added. After the NCS is in solution concentrated HCl (0.005 mL, 0.059 mmol) is added. After 2 hours, sodium bicarbonate, water and NaHSO3 are added to the reaction mixture. Solids are filtered and washed with water and ether to give 0.141 g (57%) of clean desired product as a tan solid. MS APCI (-) m/z 414, 416 (M-, Br pattern) detected.
Step I: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester lOee
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3H-benzoimidazole-5-carboxylic acid methyl ester lOdd (0.141 g, 0.34 mmol), potassium carbonate (0.141 g, 1.02 mmol), and iodomethane (0.063 mL, 1.02 mmol) are dissolved in dimethylformamide (3 mL). After 20 hours, the reaction mixture is diluted with EtOAc and washed with water (3x), potassium carbonate, and brine. The organic layer is dried (Na2SO4) and concentrated to a brown oil. The N3 and Nl alkylated regioisomers are separated by flash chromatography (EtOAc). The recovery of the N3 alkylated regioisomer is 20.4 mg (28%). MS ESI (+) m/z 428, 430 (M+, Br pattern) detected.
Step J: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid 10 ff
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid methyl ester lOee (21 mg, 0.048 mmol) is dissolved into 2:1 THF/water (1.2 mL) and NaOH (0.190 mL, 1.0 M aqueous solution, 0.190 mmol) is added. After stirring for 4 hours the reaction is diluted with water and acidified to pH 2 by addition of 1.0 M HCl. The mixture is then extracted with 3:1 EtOAc/THF (3x), dried (Na2SO ) and concentrated to give quantitative yield of desired prodcut as a white solid. MS APCI (+) m/z 414, 416 (M+, Br pattern) detected.
Step K: 6-(4-Bromo-2’chloro-phenylamino)- 7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-vinyloxy-ethoxy) -amide lOgg
6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid lOff (32 mg, 0.077 mmol), O-(2-vinyloxy-ethyl)-hydroxylamine (0.010 mL, 0.092 mmol), HOBt (13 mg, 0.093 mmol), triethylamine (0.011 mL, 0.077 mmol), and EDCI (19 mg, 0.10 mmol) are dissolved into dimethylformamide (1.0 mL) and allowed to stir under a nitrogen atmosphere at room temperature for 24 hours. The reaction mixture is diluted with EtOAc, washed with water (3x), 10% potassium carbonate (2x), saturated ammonium chloride, brine, dried (Na2SO4), and concentrated under reduced pressure to give 39 mg of 85% pure material. MS APCI (-) m/z 497, 501 (M-, Br pattern) detected.
Step L: 6-(4-Bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H-benzoimidazole-5- carboxylic acid (2-hydroxy-ethoxy)-amide lOcc
Hydrochloric acid (0.78 mL, 1.0 M aqueous solution, 0.78 mmol) is added to a suspension of 6-(4-bromo-2-chloro-phenylamino)-7-chloro-3-methyl-3H- benzoimidazole-5-carboxylic acid lOgg (2-vinyloxy-ethoxy)-amide (39 mg, 0.078 mmol) in MeOH (1 mL). After one hour, the reaction mixture is neutralized to pH 7 and concentrated under reduced pressure. The solids are dissolved in EtOAc, washed with brine, dried (Na SO4), and concentrated under reduced pressure. Flash chromatography (20:1 CH2Cl2/MeOH) provides 9 mg (23%) of pure product: MS APCI (+) m/z 473, 475 (M+, Br pattern) detected; 1H NMR (400 MHz, CDC13) δ 8.30 (s, IH), 8.08 (s, IH), 7.57
(d, IH), 7.15 (dd, IH), 6.21 (d, IH), 3.97 (s, 3H) 3.86 (m, 2H), 3.57 (m, 2H).
actual is below
Example 18
The following compounds are prepared by methods similar to those described in
Example 10 by using methyl ester 8d and the appropriate alkylating agent (Step A) and
the appropriate hydroxylamine (Step C):

PATENT
 COMPD A
COMPD A
Example 1. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-

In an inertized (N2) reaction vessel at internal temperature 20°C and under exclusion of humidity and air, Compound 1 (1.0 eq.) and Compound 2 (1.2 eq.) are reacted in the presence of cesium carbonate (2.4 eq.), tris(dibenzylidenaceton) dipalladium(O) (0.035 eq.) and Xantphos (0.07 eq.) in a mixture of toluene and 1 ,4-dioxane at internal temperature of 99°C. After 8 hours, the mixture is cooled to internal temperature of 60°C.
Subsequently, dimethylformamide (DMF), filter aid (CEFOK) and activated charcoal (EKNS) are added, and the mixture is stirred and cooled to internal temperature of 35 °C. The solids are filtered off and washed with a mixture of dimethylformamide and toluene. To the filtrate, which contains the product Compound 3, is introduced at internal temperature of
25 °C hydrogen chloride gas (CLC) whereupon the HQ salt of Compound 3 crystallizes. The palladium residue mainly remains in solution. After warming to 60 °C and cooling to 0°C, the solids are filtered using a centrifuge and are washed with a mixture of toluene and dimethylformamide.
The damp Compound 3 HC1 salt is charged to a reactor (equipped with pH probe) together with dimethylformamide and is heated to 60°C. By adding a 4 wt% of aqueous tripotassium phosphate solution, the pH is adjusted to a pH range of 6.8-7.6 (with a target of pH 7.2) while Compound 3 crystallizes as free base. After cooling to 22°C and stirring, the solids are filtered using a centrifuge and are washed with drinking water. The moist solids are dried at 50 °C under vacuum to give dry, crude Compound 3.
In order to remove residual palladium, dry, crude Compound 3 is dissolved in dimethylformamide at internal temperature of 60°C and stirred together with Smopex-234 (commercially available from Johnson Matthey) and activated charcoal for 90 minutes. The solids are filtered off at internal temperature of 60°C and are washed with
dimethylformamide. To the filtrate are added drinking water and Compound 3 seed crystals. More drinking water is added while Compound 3 crystallizes. After cooling to internal temperature of 20 °C, the solids are filtered using a centrifuge and are washed with a mixture of deionized water and dimethylformamide and with deionized water. The moist solids are dried at 50°C under vacuum, providing 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester (Compound 3).
Example 2. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide
A. “One-pot” Synthesis

In an inertized reaction vessel at internal temperature 20-25 °C under nitrogen, 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid methyl ester (Compound 3, 1.0 eq.) is added to a mixture of DMF and THF. To this slurry, a solution of potassium trimethylsilanolate (1.05 eq.) in THF is added to the mixture at internal temperature of 25 °C over a period of about 40 minutes, and the resulting mixture is stirred for about 1 hour, providing a potassium salt solution of Intermediate 1. A THF/methanol mixture is then sequentially distilled off from the mixture at 85-120°C during about 2 hours.
The potassium salt solution is then added to a suspension of CDI (1.25 eq.) and imidazole hydrochloride (1.40 eq.) in THF at internal temperature of 25 °C over a period of about 1 hour. The resulting mixture is then stirred for approximately 1 hour at 50°C, and the following imidazolide intermediate
The imidazolide intermediate is not further isolated.
Subsequently, 1.2 eq. of 0-(2-tert-butoxyethyl)hydroxylamine (Compound 4, CAS No. 1023742-13-3, available from suppliers such as Huhu Technology, Inc.®) is added over a period of about 30 minutes at 50°C and stirred for 1.5 hours. Demineralized water is then added at 50°C, producing a precipitate. After cooling to 20°C and stirring for about 3-16 hours, the slurry is filtered off, washed with THF/ demineralized water (1 :2) in 2 portions and with demineralized water in three portions, and dried at 50°C / <70 mbar for about 17 hours, providing 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) as monohydrate.
B. A synthesis method with isolation of the intermediate of step a) from the reaction mixture of step a) prior to the reaction of step b)
Alternatively, 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5 -carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) can be made by the synthesis method as shown below. Compound 3, which is a methyl ester, is first converted to a carboxylic acid, which is then isolated by a crystallization to form Compound
6. Compound 6 is then coupled with Compound 4 to form Compound 5 as monohydrate.
The crystallization step in this method removes starting materials such as Compound 1, process impurities, and the dba ligand from the prior catalyst before the coupling reaction with Compound 4, and at the same time maintains the overall yield of the synthesis.


6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-memy acid In an inertized (N2) reaction vessel at internal temperature of 60°C, Compound 3 (1.0 eq.) is dissolved in DMF and stirred with a fiber, which is sold under the trademark
SMOPEX 234, and activated charcoal for the removal of palladium to not more than 100 ppm. The fiber and activated charcoal are removed by filtration at 60°C and washed with DMF.
The filtrate (containing Compound 3) is transferred to a second inertized (N2) reaction vessel and cooled to an internal temperature of 30°C. A thin suspension can form at this point of time. 30% sodium hydroxide (1.1 eq.) and water (for rinsing) are added, and the resulting reaction mixture is vigorously stirred for 3 hours at an internal temperature of 30 °C. The methyl ester is saponified. Conversion is checked by an IPC (HPLC). As soon as the IPC criterion is met, a filter aid, which is sold under the trademark HYFLO, is added. The mixture is stirred for 15 minutes and then filtered at 30°C via a plate filter and polish filter to a third reaction inertized (N2) vessel.
An aqueous HC1 solution 7.5 % is added to the clear filtrate in the third vessel at an internal temperature of 30 °C until a pH value of 8 is reached. Then the solution is seeded at an internal temperature of 30°C with Compound 6, and an aqueous HC1 solution 7.5 % is added under vigorous stirring until a pH value of pH 2.8 is reached. The product gradually crystalizes. The suspension is cooled over 60 min to an internal temperature of 25 °C and
water is added. The suspension is stirred for at least 4 hours at an internal temperature of 25°C.
The resulting solid is collected by centrifugation or filtration. The filter cake is first washed with DMF/water 1 :1 (w/w) and then with water, discharged and dried in a vacuum at 50°C. The water content is controlled by IPC. The crystalline product Compound 6 is discharged as soon as the IPC criterion is met.
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid- (2-tert-butoxyethoxy) – amide
An inertized (N2) reaction vessel is charged with Compound 6 (1.0 eq.), DMF, and
THF at room temperature. The suspension is heated to 25 °C under stirring with flow of nitrogen. After CDI (1.13 eq.) is added, the suspension can get thinner and slight evolution of gases can be observed. After the suspension finally becomes a solution, it is then monitored by IPC (HPLC).
As soon as the IPC (HPLC) criterion is met, the reaction mixture is heated to 50°C over 20 minutes and imidazole hydrochloride (0.3 eq.) is added, forming a solution of
Intermediate 2.
To the solution of Intermediate 2, Compound 4 (1.3 eq.) is added over 60 minutes at internal temperature of 50°C under stirring at a speed of 300 rpm with flow of nitrogen. As soon as the IPC (HPLC) criterion is met, the mixture is cooled to 20-25 °C over 30 minutes. The mixture is then stored at ambient temperature overnight under nitrogen without stirring. DMF is added to the mixture followed by heating it to 50 °C over 30 minutes. Complete conversion of Intermediate 2 to Compound 5 is confirmed by IPC (HPLC).
Water is added to the mixture at internal temperature of 50 °C over 20 minutes. Then the solution is seeded with Compound 5. After stirring at 50 °C for 60 minutes, more water is added to the suspension at 50 °C over 90 minutes. After vigorous stirring, the suspension is cooled to 20 °C over 2 hours and filtered. The filter cake is washed twice with THF/water (v/v: 1 :2) at 20 °C, and twice with water at 20 °C. Finally, the filter cake is dried at 50 °C under vacuum to provide 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) as monohydrate.
Example 3. Preparation of 6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A)

Compound 5 Compound A
6-(4-Bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid-(2-tert-butoxyethoxy)-amide (Compound 5) monohydrate is added in 3 portions to a premixed solution of Acetonitrile and excess Phosphoric acid (85 % aqueous solution) at internal temperature 20-25 °C. After stirring for about 15 minutes, the suspension is heated to internal temperature 50-53 °C. The suspension is maintained at this temperature for 6 hours, cooled to internal temperature 20-25 °C. The mixture is then heated to internal temperature 35-37°C and diluted with Ethanol- Water (3 :1 v/v). EKNS and CEFOK are added, the reaction mixture is stirred approximately 15 minutes and filtered over a funnel coated with CEFOK. The filtrate is cooled to approximately 30°C. 3 N aqueous potassium hydroxide (ΚΟΗ) is added to the cooled filtrate over a period of 90 minutes until a pH- value of about 8.1 is reached. The suspension is heated to internal temperature 60-63 °C, stirred at this temperature for a period of about 2 hours, cooled to 20-23 °C over a period of about 45 minutes, filtered over a funnel, and dried at 50°C pressure <100 mbar over a period of about 17 hours, providing 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A) as a white powder.
Example 4. Preparation of Crystallized 6-(4-bromo-2-fluorophenylamino)-7-fluoro-3-methyl-3H-benzoimidazole-5-carboxylic acid (2-hydroxyethyoxy)-amide (Compound A) In a dry vessel at room temperature, Compound A is added to a premixed solvent solution of methanol/THF/water (35/35/30 w/w). The suspension is heated to internal temperature 53-55°C, and the resulting solution is hot filtered by deep and membrane filtration (via a paper filter and PTFE membrane) at internal temperature 53-56°C. The clear solution is stirred and cooled to 47-48°C, and the seed crystals suspension (i.e., seed crystals of crystallized Compound A in water, 10% m/m) is added (0.2 to 0.5% of crystallized Compound A expected yield mass). After about 20 minutes, water is slowly added within 25 hours (33.3% within 15 hours and 66.6% within 10 hours with at least 10 minute stirring after addition of water) to obtain a final ratio of methanol THF/water (20/20/60 w/w). After the water is added, the suspension is cooled down to internal temperature 3-5 °C within 10 hours and stirred for 0.5 hours. The white suspension is filtered over a sinter glass nutsche (75 ml, diameter = 6 cm, pore 3) suction filter and washed once with ice cold methanol/THF/water (15/15/70 w/w at 2-4 °C), and two times with ice cold water (2-4 °C). Drying takes place in a vacuum oven dryer at 20°C for 10 hours, and then at 40°C for 10 hours, and then at 60°C for at least 12 hours with pressure < lOmbar, providing crystallized Compound A.
CLIP

http://blog.sina.com.cn/s/blog_de171b9b0101dvov.html
CLIP
https://www.pharmacodia.com/yaodu/html/v1/chemicals/675f9820626f5bc0afb47b57890b466e.html
References
- Jump up^ “Binimetinib”. Array Biopharma.
- ^ Jump up to:a b Koelblinger P, Dornbierer J, Dummer R (August 2017). “A review of binimetinib for the treatment of mutant cutaneous melanoma”. Future Oncology. 13 (20): 1755–1766. doi:10.2217/fon-2017-0170. PMID 28587477.
- ^ Jump up to:a b Research, Center for Drug Evaluation and. “Approved Drugs – FDA approves encorafenib and binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations”. http://www.fda.gov. Retrieved 2018-07-17.
- Jump up^ Wu PK, Park JI (December 2015). “MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms”. Seminars in Oncology. 42 (6): 849–62. doi:10.1053/j.seminoncol.2015.09.023. PMC 4663016 . PMID 26615130.
- Jump up^ “Binimetinib”. PubChem.
- Jump up^ Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A, Niazi F, Wandel S, Peters M, Zubel A, Dummer R (March 2013). “MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study”. The Lancet. Oncology. 14(3): 249–56. doi:10.1016/S1470-2045(13)70024-X. PMID 23414587.
- Jump up^ Mehdizadeh A, Somi MH, Darabi M, Jabbarpour-Bonyadi M (February 2016). “Extracellular signal-regulated kinase 1 and 2 in cancer therapy: a focus on hepatocellular carcinoma”. Molecular Biology Reports. 43 (2): 107–16. doi:10.1007/s11033-016-3943-9. PMID 26767647.
- ^ Jump up to:a b Woodfield SE, Zhang L, Scorsone KA, Liu Y, Zage PE (March 2016). “Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression”. BMC Cancer. 16: 172. doi:10.1186/s12885-016-2199-z. PMC 4772351 . PMID 26925841.
- Jump up^ Clinical trial number NCT01849874 for “A Study of MEK162 vs. Physician’s Choice Chemotherapy in Patients With Low-grade Serous Ovarian, Fallopian Tube or Peritoneal Cancer” at ClinicalTrials.gov
- Jump up^ Clinical trial number NCT01909453 for “Study Comparing Combination of LGX818 Plus MEK162 Versus Vemurafenib and LGX818 Monotherapy in BRAF Mutant Melanoma (COLUMBUS)” at ClinicalTrials.gov
- Jump up^ Clinical trial number NCT01763164 for “Study Comparing the Efficacy of MEK162 Versus Dacarbazine in Unresectable or Metastatic NRAS Mutation-positive Melanoma” at ClinicalTrials.gov
- Jump up^ Hufford A (December 2015). “Array BioPharma Has Successful Trial for Cancer Drug Binimetinib”. Wall Street Journal.
- Jump up^ “Array BioPharma announces Phase 3 binimetinib trial meets primary endpoint for NRAS-mutant melanoma”. Metro Denver. December 2015.
- Jump up^ Array Bio submits marketing application in U.S. for lead product candidate in certain type of melanoma. June 2016
- Jump up^ House DW (1 September 2016). “FDA accepts Array Bio’s NDA for binimetinib, action date June 30”. Seeking Alpha.
- Jump up^ House DW (1 April 2016). “Array bags Phase 3 study of binimetinib in ovarian cancer; shares down 4%”. Seeking Alpha.
- Jump up^ Adams B (20 March 2017). “Losing Nemo: Array pulls skin cancer NDA for binimetinib”. Fierce Biotech.
 |
|
| Clinical data | |
|---|---|
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C17H15BrF2N4O3 |
| Molar mass | 441.23 g/mol |
| 3D model (JSmol) | |
- Koelblinger P, Dornbierer J, Dummer R: A review of binimetinib for the treatment of mutant cutaneous melanoma. Future Oncol. 2017 Aug;13(20):1755-1766. doi: 10.2217/fon-2017-0170. Epub 2017 Jun 7. [PubMed:28587477]
- Queirolo P, Spagnolo F: Binimetinib for the treatment of NRAS-mutant melanoma. Expert Rev Anticancer Ther. 2017 Nov;17(11):985-990. doi: 10.1080/14737140.2017.1374177. Epub 2017 Sep 8. [PubMed:28851243]
- Dummer R, Schadendorf D, Ascierto PA, Arance A, Dutriaux C, Di Giacomo AM, Rutkowski P, Del Vecchio M, Gutzmer R, Mandala M, Thomas L, Demidov L, Garbe C, Hogg D, Liszkay G, Queirolo P, Wasserman E, Ford J, Weill M, Sirulnik LA, Jehl V, Bozon V, Long GV, Flaherty K: Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017 Apr;18(4):435-445. doi: 10.1016/S1470-2045(17)30180-8. Epub 2017 Mar 9. [PubMed:28284557]
- Bendell JC, Javle M, Bekaii-Saab TS, Finn RS, Wainberg ZA, Laheru DA, Weekes CD, Tan BR, Khan GN, Zalupski MM, Infante JR, Jones S, Papadopoulos KP, Tolcher AW, Chavira RE, Christy-Bittel JL, Barrett E, Patnaik A: A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br J Cancer. 2017 Feb 28;116(5):575-583. doi: 10.1038/bjc.2017.10. Epub 2017 Feb 2. [PubMed:28152546]
- Gardner AM, Vaillancourt RR, Lange-Carter CA, Johnson GL: MEK-1 phosphorylation by MEK kinase, Raf, and mitogen-activated protein kinase: analysis of phosphopeptides and regulation of activity. Mol Biol Cell. 1994 Feb;5(2):193-201. [PubMed:8019005]
- Wang ZQ, Wu DC, Huang FP, Yang GY: Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res. 2004 Jan 16;996(1):55-66. [PubMed:14670631]
- Cancer.gov link [Link]
- FDA approves encorafenib and binimetinib in combination for unresectable or metastatic melanoma with BRAF mutations [Link]
- A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor [Link]
- Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression [Link]
- Binimetinib [File]
- EMA assessment [File]
/////////////BINIMETINIB, FDA 2018, MEK-162, биниметиниб , بينيميتينيب , 美替尼 , ビニメチニブ , 606143-89-9 , 9764, ARRY-162, ARRY-438162, NVP-MEK162
CN1C=NC2=C(F)C(NC3=CC=C(Br)C=C3F)=C(C=C12)C(=O)NOCCO
https://cen.acs.org/articles/95/i23/Array-licenses-cancer-compounds-Ono.html

Array BioPharma has licensed Japan’s Ono Pharmaceutical the right to develop two late-stage oncology compounds, binimetinib and encorafenib, in Japan and South Korea. Array will get $32 million up front and up to $156 million in milestone payments. The compounds are in Phase III studies of patients with BRAF-mutant cancers. Array recently struck a deal to assess binimetinib with two Bristol-Myers Squibb immuno-oncology agents.
Cenegermin

Active Substance General information The active substance in Oxervate, cenegermin, is a recombinant human Nerve Growth factor (rhNGF) produced in E. coli strain HMS174. The molecule is identical to human Nerve Growth factor (NGF), a naturally occurring human protein. In humans, NGF is naturally produced as pre-pro-peptide, secreted into the endoplasmic reticulum and cleaved by furin protease. The pro-sequence is further cleaved during the production process by enzymatic hydrolysis. Therefore these two amino acid changes have no influence on the final active ingredient (rhNGF), which is identical to the naturally secreted human protein. The 3D structure of rhNGF is a non-covalent dimer with three intra-molecular disulphide bridges. Cenegermin contains 118 amino acids and has a relative molecular mass of 13,266 Daltons and the following molecular formula: C583H908N166O173S8. Figure 1 shows the protein sequence of recombinant human ProNGFrh ProNGF (Figure 1A), and a map of the disulphide bridges (Figure IB):

| Cenegermin sequence: | |
| SSSHPIFHRGEFSVCDSVSVWVGDKTTATDIKGKEVMVLGEVNIN | |
| NSVFKQYFFETKCRDPNPVDSGCRGIDSKHWNSYCTTTHTFVKAL | |
| TMDGKQAAWRFIRIDTACVCVLSRKAVR |
CAS 1772578-74-1
rhNGF, Nerve growth factor – Anabasis/Dompe; Oxervate; Sentinel
UNII B6E7K36KT8
- OriginatorAnabasis Pharma
- DeveloperDompe Farmaceutici; Ospedale San Raffaele
- ClassEye disorder therapies; Nerve growth factors; Neuroprotectants; Proteins
- Mechanism of ActionNerve growth factor receptor agonists; Neuron stimulants
- Orphan Drug StatusYes – Keratitis; Retinitis pigmentosa
- Highest Development Phases
- RegisteredKeratitis
- Phase II Dry eyes; Glaucoma; Retinitis pigmentosa
- APPROVED FDA AUG 2018
Most Recent Events
- 28 Jul 2018No recent reports of development identified for phase-I development in Glaucoma in Italy (Ophthalmic, Drops)
- 29 May 2018Phase-II clinical trials in Glaucoma (Ophthalmic) (http://www.dompe.com/RnD-Pipeline/)
- 01 May 2018Dompé Farmaceutici completes a phase I trial in Glaucoma in USA (Ophthalmic) (NCT02855450)

Cenegermin (planned brand names Oxervate, Sentinel), also known as recombinant human nerve growth factor (rhNGF), is a recombinant form of human nerve growth factor (NGF). It was approved in the European Union as an eye drop formulation for the treatment of moderate or severe neurotrophic keratitis in adults on 6 July 2017.[2][3][1] As a recombinant form of NGF, cenegermin is a peripherally selective agonist of the TrkA and LNGFR (p75NTR) which must be administered parenterally.[3] In addition to neurotrophic keratitis, cenegermin is also under development for the treatment of dry eyes, retinitis pigmentosa, and glaucoma.[3] It was developed by Anabasis Pharma, Dompé Farmaceutici, and Ospedale San Raffaele.[3]
Cenegermin is a human beta-nerve growth factor (beta-ngf)-(1-118)- peptide (non-covalent dimer) produced in escherichia coli. It received European Union Approval in July, 2017 for the treatment of moderate to severe neurotrophic keratitis.
In 2013, orphan drug designations in the E.U. and in the U.S. were assigned to the candidate for the treatment of retinitis pigmentosa. The product was granted additional orphan drug designation for the treatment of neurotrophic keratitis in the U.S. and the E.U. in 2014 and 2015, respectively.
Cenegermin, a recombinant human nerve growth factor developed by Dompé was first approved in July 2017 in the E.U. for the treatment of moderate to severe neurotrophic keratitis (NK) in adults
Clip
The U.S. Food and Drug Administration today approved the first drug, Oxervate (cenegermin), for the treatment of neurotrophic keratitis, a rare disease affecting the cornea (the clear layer that covers the colored portion of the front of the eye).
“While the prevalence of neurotrophic keratitis is low, the impact of this serious condition on an individual patient can be devastating,” said Wiley Chambers, M.D., an ophthalmologist in the FDA’s Center for Drug Evaluation and Research. “In the past, it has often been necessary to turn to surgical interventions; these treatments are usually only palliative in this disease. Today’s approval provides a novel topical treatment and a major advance that offers complete corneal healing for many of these patients.”
August 22, 2018
Release
The U.S. Food and Drug Administration today approved the first drug, Oxervate (cenegermin), for the treatment of neurotrophic keratitis, a rare disease affecting the cornea (the clear layer that covers the colored portion of the front of the eye).
“While the prevalence of neurotrophic keratitis is low, the impact of this serious condition on an individual patient can be devastating,” said Wiley Chambers, M.D., an ophthalmologist in the FDA’s Center for Drug Evaluation and Research. “In the past, it has often been necessary to turn to surgical interventions; these treatments are usually only palliative in this disease. Today’s approval provides a novel topical treatment and a major advance that offers complete corneal healing for many of these patients.”
Neurotrophic keratitis is a degenerative disease resulting from a loss of corneal sensation. The loss of corneal sensation impairs corneal health causing progressive damage to the top layer of the cornea, including corneal thinning, ulceration, and perforation in severe cases. The prevalence of neurotrophic keratitis has been estimated to be less than five in 10,000 individuals.
The safety and efficacy of Oxervate, a topical eye drop containing cenegermin, was studied in a total of 151 patients with neurotrophic keratitis in two, eight-week, randomized controlled multi-center, double-masked studies. In the first study, patients were randomized into three different groups. One group received Oxervate, a second group received an eye drop with a different concentration of cenegermin, and the third group received an eye drop without cenegermin. In the second study, patients were randomized into two groups. One group was treated with Oxervate eye drops and the other group was treated with an eye drop without cenegermin. All eye drops in both studies were given six times daily in the affected eye(s) for eight weeks. In the first study, only patients with the disease in one eye were enrolled, while in the second study, patients with the disease in both eyes were treated in both eyes (bilaterally). Across both studies, complete corneal healing in eight weeks was demonstrated in 70 percent of patients treated with Oxervate compared to 28 percent of patients treated without cenegermin (the active ingredient in Oxervate).
The most common adverse reactions in patients taking Oxervate are eye pain, ocular hyperemia (enlarged blood vessels in the white of the eyes), eye inflammation and increased lacrimation (watery eyes).
Oxervate was granted Priority Review designation, under which the FDA’s goal is to take action on an application within six months of application filing where the agency determines that the drug, if approved, would provide a significant improvement in the safety or effectiveness of the treatment, diagnosis or prevention of a serious condition. Oxervate also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted approval of Oxervate to Dompé farmaceutici SpA.
| Clinical data | |
|---|---|
| Trade names | Oxervate, Sentinel |
| Synonyms | Recombinant human nerve growth factor; rhNGF; human beta-nerve growth factor (beta-NGF)-(1-118) peptide (non-covalent dimer) produced in Escherichia coli[1] |
| Routes of administration |
Eye drops |
| ATC code | |
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C583H908N166O173S8 |
| Molar mass | 13266.94 g/mol |
References
- ^ Jump up to:a bhttp://www.who.int/medicines/publications/druginformation/issues/77_INN_Recommended_List.pdf
- Jump up^ “Authorisation details”. European Medicines Agency. Retrieved 19 February 2018.
- ^ Jump up to:a b c d http://adisinsight.springer.com/drugs/800035751
External links
////////////fda 2018, Oxervate, cenegermin, orphan drug, priority review, EU 2017, DOMPE, neurotrophic keratitis
FDA approves first drug Oxervate (cenegermin) for neurotrophic keratitis, a rare eye disease
The U.S. Food and Drug Administration today approved the first drug, Oxervate (cenegermin), for the treatment of neurotrophic keratitis, a rare disease affecting the cornea (the clear layer that covers the colored portion of the front of the eye).
“While the prevalence of neurotrophic keratitis is low, the impact of this serious condition on an individual patient can be devastating,” said Wiley Chambers, M.D., an ophthalmologist in the FDA’s Center for Drug Evaluation and Research. “In the past, it has often been necessary to turn to surgical interventions; these treatments are usually only palliative in this disease. Today’s approval provides a novel topical treatment and a major advance that offers complete corneal healing for many of these patients.”
August 22, 2018
Release
The U.S. Food and Drug Administration today approved the first drug, Oxervate (cenegermin), for the treatment of neurotrophic keratitis, a rare disease affecting the cornea (the clear layer that covers the colored portion of the front of the eye).
“While the prevalence of neurotrophic keratitis is low, the impact of this serious condition on an individual patient can be devastating,” said Wiley Chambers, M.D., an ophthalmologist in the FDA’s Center for Drug Evaluation and Research. “In the past, it has often been necessary to turn to surgical interventions; these treatments are usually only palliative in this disease. Today’s approval provides a novel topical treatment and a major advance that offers complete corneal healing for many of these patients.”
Neurotrophic keratitis is a degenerative disease resulting from a loss of corneal sensation. The loss of corneal sensation impairs corneal health causing progressive damage to the top layer of the cornea, including corneal thinning, ulceration, and perforation in severe cases. The prevalence of neurotrophic keratitis has been estimated to be less than five in 10,000 individuals.
The safety and efficacy of Oxervate, a topical eye drop containing cenegermin, was studied in a total of 151 patients with neurotrophic keratitis in two, eight-week, randomized controlled multi-center, double-masked studies. In the first study, patients were randomized into three different groups. One group received Oxervate, a second group received an eye drop with a different concentration of cenegermin, and the third group received an eye drop without cenegermin. In the second study, patients were randomized into two groups. One group was treated with Oxervate eye drops and the other group was treated with an eye drop without cenegermin. All eye drops in both studies were given six times daily in the affected eye(s) for eight weeks. In the first study, only patients with the disease in one eye were enrolled, while in the second study, patients with the disease in both eyes were treated in both eyes (bilaterally). Across both studies, complete corneal healing in eight weeks was demonstrated in 70 percent of patients treated with Oxervate compared to 28 percent of patients treated without cenegermin (the active ingredient in Oxervate).
The most common adverse reactions in patients taking Oxervate are eye pain, ocular hyperemia (enlarged blood vessels in the white of the eyes), eye inflammation and increased lacrimation (watery eyes).
Oxervate was granted Priority Review designation, under which the FDA’s goal is to take action on an application within six months of application filing where the agency determines that the drug, if approved, would provide a significant improvement in the safety or effectiveness of the treatment, diagnosis or prevention of a serious condition. Oxervate also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted approval of Oxervate to Dompé farmaceutici SpA.
/////////////fda 2018, Oxervate, cenegermin, orphan drug, priority review
USFDA has released GUIDANCE for Quality Attributes of *CHEWABLE TABLETS
DRUG REGULATORY AFFAIRS INTERNATIONAL

*CQAs of CHEWABLE TABLETS (CT)*
USFDA has released GUIDANCE for Quality Attributes of *CHEWABLE TABLETS*
According to this latest guideline, FDA has recommended sponsor/applicant should also incorporate following CQAs:
*1. PATIENT ACCEPTABILITY*
Acceptable Taste, Mouthfeel & Aftertaste With-
*2. HARDNESS / BREAKING FORCE / CRUSHING STRENGTH*
Hardness of CTshould be kept low (i.e. <12 kp).
A higher hardness value (e.g., >12 kp) may be considered if justified. An example of such justification could be demonstrating significant disintegration and/or reduction in hardness of such tablets following brief i.e. 30 seconds in-vitro exposure to simulated saliva (1 mL) before chewing to ensure patient compliance without GI obstruction (choking in throat / blocking bowel movement) in the case if patient swallow tablet without chewing due to high hardness
*3. CHEWING DIFFICULTY INDEX*
CDI is a value derived from the relationship between two methods used for measuring tablet strength: diametral compression (diametrical tensile strength)…
View original post 109 more words
Ambrisentan, أمبريسنتان , 安立生坦 ,アンブリセンタン

Ambrisentan
BSF-208075; LU-208075
(+)-(2S)-2-[(4,6-dimethylpyrimidin-2-yl)oxy]-3-methoxy-3,3-diphenylpropanoic acid
- Molecular FormulaC22H22N2O4
- Average mass378.421 Da
(2S)-2-[(4,6-dimethylpyrimidin-2-yl)oxy]-3-methoxy-3,3-diphenylpropanoic acid
177036-94-1 [RN]
8128
HW6NV07QEC
أمبريسنتان [Arabic] [INN]
安立生坦 [Chinese] [INN]
QA-7701
UNII:HW6NV07QEC
BSF208075
Letairis
Letairis®
LU208075
Trade Name:Letairis® / Volibris®
MOA:Type A endothelin receptor (ETA) antagonist
Indication:Pulmonary arterial hypertension
Company:Abbott (Originator) , Gilead,GlaxoSmithKline
| アンブリセンタン Ambrisentan  C22H22N2O4 : 378.42 [177036-94-1 |
Ambrisentan (U.S. trade name Letairis; E.U. trade name Volibris; India trade name Pulmonext by MSN labs) is a drug indicated for use in the treatment of pulmonary hypertension.
The peptide endothelin constricts muscles in blood vessels, increasing blood pressure. Ambrisentan, which relaxes those muscles, is an endothelin receptor antagonist, and is selective for the type A endothelin receptor (ETA).[1] Ambrisentan significantly improved exercise capacity (6-minute walk distance) compared with placebo in two double-blind, multicenter trials (ARIES-1 and ARIES-2).[2]
Ambrisentan was approved by the U.S. Food and Drug Administration (FDA) and European Medicines Agency, and designated an orphan drug, for the treatment of pulmonary hypertension.[3][4][5][6][7]
Ambrisentan is an endothelin receptor antagonist used in the therapy of pulmonary arterial hypertension (PAH). Ambrisentan has been associated with a low rate of serum enzyme elevations during therapy, but has yet to be implicated in cases of clinically apparent acute liver injury.
Ambrisentan was first approved by the U.S. Food and Drug Administration (FDA) on Jun 15, 2007, then approved by the European Medicines Agency (EMA) on Apr 21, 2008 and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jul 23, 2010. In 2000, Abbott, originator of ambrisentan, granted Myogen (acquired by Gilead in 2006) a license to the compound for the treatment of PAH. In 2006, GlaxoSmithKline obtained worldwide rights to market the compound for PAH worldwide, with the exception of the U.S. It is marketed as Letairis® by Gilead in US.
Ambrisentan is an endothelin receptor antagonist, and is selective for the type A endothelin receptor (ETA). It is indicated for the treatment of pulmonary arterial hypertension (PAH) (WHO Group 1) to improve exercise ability and delay clinical worsening. Studies establishing effectiveness included predominantly patients with WHO Functional Class II-III symptoms and etiologies of idiopathic or heritable PAH (64%) or PAH associated with connective tissue diseases (32%).
Letairis® is available as film-coated tablet for oral use, containing 5 or 10 mg of free Ambrisentan. The recommended starting dose is 5 mg once daily with or without food, and increase the dose to 10 mg once daily if 5 mg is tolerated.
Recent Developments and Publications
| Last Updated 9/2/2015 | |
|---|---|
| 8/15/2015Reprod. Toxicol. | Endothelin receptor activation mediates strong pulmonary vasoconstriction and positive inotropic effect on the heart. These physiologic effects are vital for the development of the fetal cardiopulmonary system. As such, endothelin receptor antagonists such as Ambrisentan are teratogenic.[8] |
| 8/27/2015NEJM | Ambrisentan when used in combination therapy with Tadalafil was found to be more efficacious in treating treatment naive patients with WHO class II or III Pulmonary Arterial Hypertension than monotherapy using either drug.[9] |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2007-06-15 | Marketing approval | Letairis | Pulmonary arterial hypertension | Tablet, Film coated | 5 mg/10 mg | Gilead | Priority; Orphan |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2008-04-21 | Marketing approval | Volibris | Pulmonary arterial hypertension | Tablet, Film coated | 5 mg/10 mg | GlaxoSmithKline | Orphan |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-07-23 | Marketing approval | Volibris | Pulmonary arterial hypertension | Tablet, Film coated | 2.5 mg | GlaxoSmithKline |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-10-19 | Marketing approval | 凡瑞克/Volibris | Pulmonary arterial hypertension | Tablet | 5 mg | GlaxoSmithKline | |
| 2010-10-19 | Marketing approval | 凡瑞克/Volibris | Pulmonary arterial hypertension | Tablet | 10 mg | GlaxoSmithKline |
Clinical uses
Ambrisentan is indicated for the treatment of pulmonary arterial hypertension (WHO Group 1) in patients with WHO class II or III symptoms to improve exercise capacity and delay clinical worsening.

Birth defects
Endothelin receptor activation mediates strong pulmonary vasoconstriction and positive inotropic effect on the heart. These physiologic effects are vital for the development of the fetal cardiopulmonary system. In addition to this, endothelin receptors are also known to play a role in neural crest cell migration, growth, and differentiation. As such, endothelin receptor antagonists such as Ambrisentan are known to be teratogenic.
Ambrisentan has a high risk of liver damage, and of birth defects if a woman becomes pregnant while taking it. In the U.S., doctors who prescribe it, and patients who take it, must enroll in a special program, the LETAIRIS Education and Access Program (LEAP), to learn about those risks. Ambrisentan is available only through specialty pharmacies.
External links
- Letairis website run by Gilead Sciences
- Prescribing information
- Information on the LETAIRIS Education and Access Program (LEAP)
PATENT
WO9611914A1 / US7109205B2.
WO2010070658A2 / US2011263854A1.
WO2011004402A2 / US2012184573A1.
WO2013030410A2 / US2014011992A1.
CN103709106A.
CN103420811A.

PATENT
https://patents.google.com/patent/WO2012167406A1/en
Ambrisentan and darusentan first reported in U. Med. Chem. 1996, 39, 2123-2128), as a selective antagonist of endothelin receptor A, followed by their pharmacological properties have been studied further, published in J. Med. Chem. 1996, 39, 2123-2128), US patent US 5932730, WO 2009/017777 A2 in. The formula (I), when R is methyl, Chinese name is (+) – ambrisentan, Chinese chemical name is (+) – (2S) -2 – [(4, 6- dimethyl-pyrimidine 2-yl) – oxy] -3-methoxy-3,3-diphenyl-propionic acid; English name is (+) – ambrisentan, English name: (S) -2- (4,6-dimethylpyrimidin -2-yloxy) -3-methoxy-3,3-diphenylpropanoic acid; when R is methoxy, Chinese as (+) – darusentan, Chinese chemical name is (+) – (2S) -2- [ (4,6-dimethoxypyrimidin-2-yl) – oxy] -3-methoxy-3,3-diphenyl-propionic acid; English name is (+ darusentan, English name: (S) – . 2- (4,6-dimethoxypyrimidin-2-yloxy) -3-methoxy-3,3-diphenylpropanoic acid ambrisentan now been approved by the FDA in the United States, the trade name Letairis, for the oral treatment of pulmonary hypertension; up Lu bosentan new drugs may be resistant hypertension (Resistant hypertension) of.
Existing ambrisentan or darusentan synthetic techniques include benzophenone Darzens reaction of an epoxy compound and a racemic methyl chloroacetate, the racemic epoxide opening catalyst in a solution of boron trifluoride diethyl ether ring to give the chiral alcohol latent after substitution reaction and then after hydrolysis reaction ambrisentan or darusentan. Existing obtained optically pure (+) – ambrisentan or (+) – darusentan methods rely mainly on resolution techniques. For example, the split is by a latent chiral alcohol or R- L- proline methyl phenethylamine, see WO 2010/070658 A2, WO 2011/004402 A2. It is well known as chiral utilization of raw materials is not high, resulting in increased costs, limiting industrial-scale applications.
Example 1, (+) – ambrisentan ((2S) -2 – [(4,6- dimethyl-pyrimidin-2-yl) – oxy] -3-methoxy-3,3-diphenyl propionic acid) of
Preparation of 3,3-diphenyl-2,3-epoxy-propionate (1) (2S)

As indicated above Formula Scheme, wherein, Ph is phenyl; Ac is acetyl;
To a 50 L reactor equipped with a mechanical stirrer was added 3.0 L of acetonitrile was dissolved in 3,3-diphenyl acrylate (0.536 mol, 135.0 g), was dissolved in 1.5 L of acetonitrile to give a concentration of 0.12 M 4 M ethylenediamine of formula (IV) shown fructose derived chiral ketones and tetra-n-butylammonium hydrogen sulfate (36 mmol, 12.2 g), was then added containing 3.0 L Ι χ ΙΟ “an aqueous solution of disodium ; cooling liquid into the reaction vessel dissection, the kettle temperature adjusted to -5 ° C- + 5 ° C; was added in batches with stirring pulverized with the pulverizer medicine through a 1.85 kg potassium hydrogen sulfate complex salt mixture (Oxone®), and 0.78 kg NaHCO 3 (9.29mol), and takes about 4.5 hours complete addition of the above mixture, after the addition the reaction mixture was continued stirring the reaction under this condition (in the system, 3,3- diphenyl acrylate, over a potassium bisulfate salts and complexes of formula molar ratio of fructose derived chiral ketone (IV) is shown in h 5: 0.34), and the timing detection reactions by gas chromatography; the end of the reaction after 5 hours , 5.0 L of water was added to dilute the reaction solution, and extracted with 5.0 L of ethyl acetate; the aqueous phase was added 2.5 L of acetic Extracted with ethyl; organic phases were combined and concentrated to remove the solvent to give homogeneous Qing 162.56 g (2S) -3,3- diphenyl-2,3-epoxy-propionate, crude yield greater than 99%, No purification processing the next reaction, nuclear magnetic conversion was 92%, measured by HPLC enantiomeric excess of 86.9%, Analytical conditions: column model Chiralcel OD-H, n has a volume ratio of the embankment and isopropanol 98: 2 analysis of wavelength 210 nm, the mobile phase flow rate of 1 mL / min, t! = 9.5 min, t 2 = 13.01 min, 86.9% ee.
IR (fi lm) 1760, 1731 cm- 1; ¾ NMR [400 MHz, CDC1 3] δ 7.46-7.44 (m, 2H), 7.36-7.31 (m, 8H), 3.99 (m, 3H), 0.96 (t , J = 7.2Hz, 3H); 13 C NMR [100 MHz, CDC1 3] δ 166.99, 138.98, 135.62, 128.67, 128.53, 128.36, 128.13, 127.04, 66.57, 62.16, 61.43, 13.96.
(2) (2S) -2-phenyl-3,3-hydroxy-3-methoxy propionate

The step (1) 162.56 g obtained in unpurified (2S) – 3,3-diphenyl acrylate epoxy crude compound was dissolved in 100 mL of methanol, 1 mL of boron trifluoride etherate ((2S ) – mole fraction of ethylene-3,3-diphenyl acrylate and boron trifluoride diethyl ether ratio of 1: 0.013) for the epoxy ring opening reaction; after controlling the reaction temperature is 20 ° C, reacted for 8 hours , the reaction solution was concentrated, ethyl acetate and aqueous extraction of the reaction solution after the ethyl acetate was concentrated to give 166.0 g of intermediate (2S) -2- hydroxy-3-methoxy-3,3-diphenyl acetic acid ester, crude yield of 92%, measured by high performance liquid enantiomeric excess of 86.9%, Analytical conditions: column model Chiralcel OD-H, n-and isopropyl alcohol embankment has a volume ratio of 98: 2, the wavelength analysis 210 nm, mobile phase flow rate of 1 mL / min,
min, t 2 = 14.51 min, 85.8% ee .;
IR (film) 1769, 1758 cm “1; 1H NMR [400 MHz, CDC1 3] δ 7.50-7.28 (m, 10H), 5.18 (s, 1H), 4.10 (t, 2H), 3.20 (s, J = 7.2Hz, 3H), 3.03 (s , 1H), 1.17 (t, J = 7.2Hz, 3H); 13 C NMR [100 MHz, CDC1 3] δ 172.48, 141.13, 140.32, 128.97, 128.73, 128.99, 127.81, 127.76, 127.62, 85.01, 77.42, 61.76, 52.62, 14.07.
-3-methoxy-3,3-diphenyl propionate (3) (2S) -2- [- oxo – dimethyl-pyrimidin-2-yl)]

Step (2) obtained in 166.0 g of intermediate (2S) -2- hydroxy-3-methoxy-3,3-diphenyl-propionate were added N, N- dimethylformamide 750 mL , potassium carbonate 45.54 g, was added 4,6-dimethyl after stirring for about half an hour 2-methanesulfonyl-pyrimidin nucleophilic substitution reaction at 80 ° C in an oil bath, the system, (2S) -2- hydroxy -3-methoxy-3,3-diphenyl-ethyl, 4,6-dimethyl-2 molar fraction ratio methylsulfonylpyrimidine and potassium carbonate is 1: 1.2: 0.6; nuclear magnetic after complete consumption of starting material was monitored after about 3 hours, water was added and the reaction solution was extracted with ethyl acetate, the ethyl acetate layer was concentrated to give 237.70 g of intermediate (2S) -2 – [(4,6- dimethyl-pyrimidin-2-yl ) – oxy] -3-methoxy-3,3-diphenyl propionate, crude yield greater than 99%, measured by HPLC enantiomeric excess of 85.9%, Analytical conditions: column Chiralcel OD model volume -H, isopropanol and n has embankment ratio of 98: 2, analysis wavelength was 210 nm, the mobile phase flow rate of 1 mL / min, t ^ lO.15 min, t 2 = 11.87 min, 85.9% ee .
IR (film) 1750cm “VH NMR [400 MHz, CDC1 3] δ 7.45 (d, J = 7.2 Hz, 2H), 7.39 (d, J = 7.2 Hz, 2H), 7.33-7.19 (m, 7H), 6.70 (s, 1H), 6.12 ( s, 1H), 4.01-3.85 (m, 2H), 3.50 (s, 3H) 2.38 (s, 6H), 0.93 (t, J = 6.8 Hz, 3H); 13 C NMR [100 MHz, CDC1 3] δ 169.51, 168.70, 163.86, 142.50, 141.29, 128.54, 128.03, 127.97, 127.94, 127.47, 127.40, 115.03, 83.76, 79.23, 77.43, 60.66, 53.92, 23.99, 13.93;. Anal Calcd For C 24 H 26 N 2 O 4 : C, 70.92; H, 6.45; N, 6.89 Found:. C, 70.72; H, 6.47; N, 6.83.
(4) (28) -2 – [(4,6-dimethyl-2-yl) – oxy] -3-methoxy-3,3-diphenyl-propionic acid ((+) – Abe Students Tanzania) preparation
To step (3) 237.7 g of the intermediate obtained (2S) -2 – [(4,6- dimethyl-pyrimidin-2-yl) – oxy] -3-methoxy-3,3-diphenyl propionate was dissolved in 1.2 L of organic solvent is 1,4-dioxane was added 600 mL of an aqueous solution containing 92.3 g of sodium hydroxide (wherein, (2S) -2 – [(4,6- dimethyl pyrimidin-2-yl) – oxy] -3-methoxy-3,3-diphenyl propionate and sodium hydroxide molar fraction ratio of 1: 4), the reaction temperature was 80 ° C, the reaction after 8 hours, the reaction solution was concentrated, using (1 L, 0.5 L, 0.5 L) and extracted with ether to remove organic impurities, the aqueous phase was extracted after addition of hydrochloric acid to adjust pH 3, large amount of solid appears; then the aqueous phase was added 1.0 L ethyl acetate, filtered to remove insolubles (insolubles which was found after analysis racemic ambrisentan, 23.37 g), the organic layer was concentrated, i.e., optically pure can be obtained 103.9 g (+) – ambrisentan, from 3 , 3-diphenyl acrylate departure, the optically pure (+) – ambrisentan, a yield of 52.3%. A small amount of the obtained reaction with ambrisentan diazo embankment derived (2S) -2 – [(4,6- dimethyl-pyrimidin-2-yl) – oxy] -3-methoxy-3,3 methyl diphenyl measured enantiomeric excess ambrisentan. (2S) -2 – [(4,6- dimethyl-pyrimidin-2-yl) – oxy] -3-methoxy-3,3-diphenyl-propionic acid methyl ester: HPLC measured enantiomer excess of 99.1%, Analytical conditions: column model Chiralcel OD-H, n has a volume ratio of isopropanol embankment 98:! 2, analysis wavelength was 210 nm, the mobile phase flow rate of 1 mL / min, t = 11.61 min, t 2 = 14.05 min , 99.1% ee.
[a] D 25 = + 174.2 (c = 0.5, MeOH); mp> 150 ° C turns yellow,> 180 ° C into a black, 182 ° C melt; 1H NMR [400 MHz, CDC1 3] δ 7.43 ( d, J = Hz, 2H) , 7.29-7.19 (m, 8H), 6.63 (s, 1H), 6.30 (s, 1H), 3.26 (s, 3H) 2.31 (s, 6H); 13 C NMR [100 MHz, CDC1 3] δ 178.98,170.54, 169.70, 163.48, 139.91, 138.91, 128.77, 128.67, 128.22, 128.08, 115.34, 84.67, 77.55, 53.49, 23.93; 1H NMR [400 MHz, DMSO] δ 12.53 (s, 1H ), 7.34-7.20 (m, 10H) , 6.95 (s, 1H), 6.14 (s, 1H), 3.37 (s, 3H) 2.34 (s, 6H); 13 C NMR [100 MHz, DMSO] δ 169.01, 163.14, 142.59, 141.41, 127.80, 127.68, 127.64, 127.19, 126.95, 114.72, 83.12, 77.55, 52.99, 23.30.
CLIP
SEE https://www.pharmacodia.com/yaodu/html/v1/chemicals/a01610228fe998f515a72dd730294d87.html
CLIP
http://www.orgsyn.org/demo.aspx?prep=v89p0350#ref68

Shi and coworkers recently obtained 120 g of virtually enantiopure (+)-ambrisentan (97) without the need for column chromatography (Scheme 23).68 (+)-Ambrisentan, an endothelin-1 receptor antagonist, is currently used to treat hypertension. Ketone 2-catalyzed epoxidation afforded 96 in 90% conversion and 85% ee. Compound 97 was further enriched via precipitation and filtration of the racemate.
- Peng, X.; Li, P.; Shi, Y. J. Org. Chem. 2012, 77, 701-703.
Clip
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00184
Process Research for (+)-Ambrisentan, an Endothelin-A Receptor Antagonist
† Collaborative Innovation Center of Yangze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology, 18 Chaowang Road, Hangzhou 310014, China
‡ Department of Pharmaceutial Engineering, China Pharmaceutical University, 24 Tongjiaxiang, Nanjing 210009, China
§ Shanghai Institute of Pharmaceutical Industry, China State Institute of Pharmacetical Industry, 285 Gebaini Road, Pudong, Shanghai 201203, China
Org. Process Res. Dev., Article ASAP
DOI: 10.1021/acs.oprd.8b00184
Publication Date (Web): August 6, 2018
Copyright © 2018 American Chemical Society
*E-mail: zhangfuli1@sinopharm.com.

An efficient and robust synthetic route to (+)-ambrisentan ((+)-AMB) was designed by recycling the unwanted isomer from the resolution mother liquors. The racemization of AMB in the absence of either acid or base in the given solvents was reported. The recovery process was developed to produce racemates with purities over 99.5%. The mechanism of the formation of the process-related impurities of (+)-AMB is also discussed in detail. (+)-AMB was obtained in 47% overall yield with >99.5% purity and 99.8% e.e. by chiral resolution with only one recycling of the mother liquors on a 100-g scale without column purification.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00184/suppl_file/op8b00184_si_001.pdf




PaPER
https://pdfs.semanticscholar.org/3801/d5a98a526a4386c431e25d3ac99a328bfae2.pdf
CHEMICAL ENGINEERING TRANSACTIONS VOL. 46, 2015 A publication of The Italian Association of Chemical Engineering Online at http://www.aidic.it/cet Guest Editors: Peiyu Ren, Yancang Li, Huiping Song Copyright © 2015, AIDIC Servizi S.r.l., ISBN 978-88-95608-37-2; ISSN 2283-9216
Improved Synthesis Process of Ambrisentan and Darusentan Jian Lia , Lei Tian*b, c a School of Environmental Science, Nanjing Xiaozhuang University, 3601 Hongjing Road, Nanjing, Jiangsu, 211171, China b School of Petroluem Engineer, Yangtze University, Wuhan, Hubei, 430100, P. R. China c Key Laboratory of Exploration Technologies for Oil and Gas Resources (Yangtze University), Ministry of Education tianlei4665@163.com
2-hydroxy-3-phenoxy-3, 3-diphenylpropinate (5) was prepared from benzophenone via Darzens, methanolysis and hydrolysis reaction. The compound (5) was salified with (S)-dehydroabietylamine (7) and diasterotropic resolution was carried out to provide the key intermediate (S)-2-hydroxy-3-methoxy-3, 3-diphenylpropionic acid (6). Compound (6) was condensed with 2-methylsulfonyl-4, 6-dimethylpyrimidine and 2-methoxysulfonyl4, 6-dimethylpyrimidine to afford ambrisentan (1) and darusentan (7), respectively. Two products were with excellent charity and chemical purity. The total yield of the synthesis was 30.1% and 29.6%, respectively.


Synthesis of methyl 3, 3-diphenyloxirane-2-carboxylate (3) To a solution of sodium methanolate (4.3 g, 79.6 mmol) in dry THF (25 mL) was added the solution of benzophenone (7.2 g, 39.5 mmol) and methyl chloroacetate (6.6 g, 60.8 mmol) in dry THF (15 mL) and stirred at -10 °C for 2 h. The mixture was quenched with water (50 mL). The solution was extracted with diethyl ether (80 mL×3). The organic phases were combined and washed with saturated NaCl. The solution was dried over Na2SO4, filtered, and evaporated under reduced pressure to afford a light yellow oil. The residue (3) can apply in next step without further purification (8.24 g, 82.1%). 1H-NMR (CDCl3): δ 3.52 (s, 3H), 3.99 (s, 1H), 7.32-7.45 (m, 10H).
Synthesis of 2-hydroxy-3-methoxy-3, 3-diphenylpropanoic acid (5)
To a solution of compound (3) (8.2 g, 31.6 mmol) in methanol (40 mL) was added p-toluene sulfonic acid (0.5 g) and stirred at for 0.5 h to afford the solution containing compound (4). Aqueous solution of NaOH (10% wt.) (60 mL) was added to the solution of compound (4) and the mixture was stirred at refluxed for 1h (ester disappeared by TLC). The solution was evaporated in order to remove a lot of methanol. The residue was acidified to pH 2 by conc. HCl. The solution was stirred for overnight and white solid stayed at the aqueous layer. The precipitate was filtered and deeply dried under vacuum to afford (5). (7.34 g, 85.3%). 1H-NMR (CDCl3): δ 3.22 (s, 3H), 5.14 (br, 1H), 5.20 (d, 1H), 7.18-7.37 (m, 10H), 12.30 (1H, br). Synthesis of (S)-2-hydroxy-3-methoxy-3, 3-diphenylpropanoate (6) The solution of compound (5) (14 g, 51.4 mmol) in methyltertiarybutylether (140 mL) was stirred and refluxed for 0.5 h. Dehydroabietylamine (7) (14.7 g, 51.4 mmol) in methyltertiarybutylether (50 mL) was added dropwise in 10 min. After addition, the reaction mixture was stirred for 1 h under reflux temperature. The reaction mixture was cooled to 0 °C and continued to stir for 2 h. The solid ((R, S)-diastereoisomers) was precipitated from the solution, filtered, washed with acetonitrile. The filtrate was diluted with water (100 mL) and acidified to pH 2 by conc. HCl. The aqueous solution was extracted with methylteriarybutylether (50 mL×4). The organic phases were combined and washed with water (80 mL). The organic phase was separated, dried over anhydrous Na2SO4 and evaporated under reduced pressure to afford white residue. The residue was recystallized from toluene to afford (6) as a white solid. (5.53 g, 39.5%). 1H-NMR (CDCl3): δ 3.22 (s, 3H), 5.14 (br, 1H), 5.20 (d, 1H), 7.18-7.37 (m, 10H), 12.30 (1H, br). [α] 20 D =12.3°(c=1.8% in ethanol).
Synthesis of (+)-ambrisentan (1) To a solution of compound (6) (3.6 g, 13.1 mmol) and NaNH2 (1.0 g, 25.6 mmol) in DMF (20 mL) was added 4, 6-dimethyl-2-(methylsulfonyl) pyrimidine (3.63 g, 19.6 mmol) in DMF (10 mL) slowly. After addition, the reaction was stirred for 5 h at room temperature. The solution was quenched with water (20 mL) and acidified to pH 2 by 10% H2SO4 aqueous solution. The mixture was extracted with ethyl acetate (50 mL ×4). The combined organic layers were washed with water (30 mL) and saturated NaCl solution (30 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The resulting residue was recrystallized from iso-propyl alcohol (30 mL) and water (40 mL), and precipitate formed was filtered off. The cake was deeply dried under vacuum to afford (1) as a white solid. (4.27 g, 86.1%). 1H-NMR (CDCl3): δ 2.39 (s, 6H), 3.32 (s, 3H), 6.43 (s, 1H), 6.70 (s, 1H), 7.28-7.40 (m, 8H), 7.53-7.56 (d, 2H). MS-EI (m/z): 377(M-H). HPLC (XDB-C18, CH3OH/10mmol/L NaH2PO4 + 0.1% H3PO4 = 70/30, 1.0 mL/min): tR 5.2 min (>99.0%); ee= 99.0%.
Patent EP2547663A1



Title: Ambrisentan
CAS Registry Number: 177036-94-1
CAS Name: (aS)-a-[(4,6-Dimethyl-2-pyrimidinyl)oxy]-b-methoxy-b-phenylbenzenepropanoic acid
Manufacturers’ Codes: BSF-208075; LU-208075
Molecular Formula: C22H22N2O4
Molecular Weight: 378.42
Percent Composition: C 69.83%, H 5.86%, N 7.40%, O 16.91%
Literature References: Nonpeptide endothelin ETA receptor antagonist. Prepn: H. Riechers et al., WO 9611914; eidem, US5932730 (1996, 1998 both to BASF); H. Riechers et al., J. Med. Chem. 39, 2123 (1996). Pharmacology: H. Vatter et al., Clin. Neuropharmacol. 26, 73 (2003). Clinical evaluation in pulmonary arterial hypertension: N. Galié et al., J. Am. Coll. Cardiol. 46, 529 (2005). Review of development and therapeutic potential: G. E. Billman, Curr. Opin. Invest. Drugs 3, 1483-1486 (2002).
Derivative Type: (±)-Form
CAS Registry Number: 713516-99-5
Properties: Crystals from diethylether, mp 190-191°.
Melting point: mp 190-191°
Therap-Cat: Antihypertensive.
Keywords: Antihypertensive; Endothelin Receptor Antagonist.
References
- Jump up^ Vatter H, Seifert V (2006). “Ambrisentan, a non-peptide endothelin receptor antagonist”. Cardiovasc Drug Rev. 24 (1): 63–76. doi:10.1111/j.1527-3466.2006.00063.x. PMID 16939634.
- Jump up^ Frampton JE (2011). “Ambrisentan”. American Journal of Cardiovascular Drugs. 11 (4): 215–26. doi:10.2165/11207340-000000000-00000. PMID 21623643.
- Jump up^ Pollack, Andrew (2007-06-16). “Gilead’s Drug Is Approved to Treat a Rare Disease”. The New York Times. Archived from the original on May 24, 2013. Retrieved 2007-05-25.
- Jump up^ “U.S. Food and Drug Administration Approves Gilead’s Letairis Treatment of Pulmonary Arterial Hypertension” (Press release). Gilead Sciences. 2007-06-15. Archived from the original on 2007-09-27. Retrieved 2007-06-16.
- Jump up^ “FDA Approves New Orphan Drug for Treatment of Pulmonary Arterial Hypertension” (Press release). Food and Drug Administration. 2007-06-15. Archived from the original on 23 June 2007. Retrieved 2007-06-22.
- Jump up^ “GlaxoSmithKline’s Volibris (ambrisentan) receives authorisation from the European Commission for the treatment of Functional Class II and III Pulmonary Arterial Hypertension” (Press release). GlaxoSmithKline. 2008-04-25. Archived from the original on 30 April 2008. Retrieved 2008-04-29.
- Jump up^ Waknine, Yael (2005-05-09). “International Approvals: Ambrisentan, Oral-lyn, Risperdal”. Medscape. Retrieved 2007-06-16.
- Jump up^ de Raaf MA, Beekhuijzen M, Guignabert C, Vonk Noordegraaf A, Bogaard HJ (2015). “Endothelin-1 receptor antagonists in fetal development and pulmonary arterial hypertension”. Reproductive Toxicology. 56: 45–51. doi:10.1016/j.reprotox.2015.06.048. PMID 26111581.
- Jump up^ Galiè, Nazzareno; Barberà, Joan A.; Frost, Adaani E.; Ghofrani, Hossein-Ardeschir; Hoeper, Marius M.; McLaughlin, Vallerie V.; Peacock, Andrew J.; Simonneau, Gérald; Vachiery, Jean-Luc; Grünig, Ekkehard; Oudiz, Ronald J.; Vonk-Noordegraaf, Anton; White, R. James; Blair, Christiana; Gillies, Hunter; Miller, Karen L.; Harris, Julia H.N.; Langley, Jonathan; Rubin, Lewis J. (2015). “Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension”. New England Journal of Medicine. 373 (9): 834–44. doi:10.1056/NEJMoa1413687.
- US5703017
- CA2201785
- US5840722
- US7601730
- US8377933
- US7109205
- USRE42462
- US8349843
- US9474752
- US9549926
Patent
Publication numberPriority datePublication dateAssigneeTitle
WO2003066614A1 *2002-02-042003-08-14Colorado State University Research FoundationAsymmetric epoxidation of electron deficient olefins
WO2010070658A2 *2008-11-052010-06-24Msn Laboratories LimitedImproved process for the preparation of endothelin receptor antagonists
CN102276536A *2011-06-102011-12-14中国科学院化学研究所An optically pure (+) – ambrisentan and optically pure (+) – darusentan preparation
Family To Family Citations
US6030975A *1997-03-142000-02-29Basf AktiengesellschaftCarboxylic acid derivatives, their preparation and use in treating cancer
JP2010535210A2007-07-312010-11-18アボット ゲーエムベーハー ウント コンパニー カーゲーMetabolites and derivatives of ambrisentan
WO2011004402A32009-07-102011-03-10Cadila Healthcare LimitedImproved process for the preparation of ambrisentan and novel intermediates thereof
Non-Patent Citation
Title
WANG, BIN ET AL.: ‘A Diacetate Ketone-Catalyzed Asymmetric Epoxidation of Olefins’ J. ORG. CHEM. vol. 74, 23 April 2009, pages 3986 – 3989 *
Publication numberPriority datePublication dateAssigneeTitle
Family To Family Citations
CN102276536B *2011-06-102015-04-29中国科学院化学研究所Preparation method of optically pure (+)-ambrisentan and optically pure (+)-darusentan
CN103420811B *2012-05-182015-04-15上海医药工业研究院Intermediate compound used for preparing Ambrisentan, preparation method thereof, and preparation of Ambrisentan
CN103524425A *2012-07-042014-01-22天津药物研究院Crystal form V of ambrisentan as well as preparation method and application thereof
CN103755569A *2013-12-262014-04-30上海皓骏医药科技有限公司Preparation method for ambrisentan intermediate compound
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Monograph |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Undetermined |
| Protein binding | 99% |
| Elimination half-life | 15 hours (terminal) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| ECHA InfoCard | 100.184.855 |
| Chemical and physical data | |
| Formula | C22H22N2O4 |
| Molar mass | 378.421 g/mol |
| 3D model (JSmol) | |
/////////////Ambrisentan, أمبريسنتان , 安立生坦 , BSF-208075, LU-208075, アンブリセンタン
CC1=CC(=NC(=N1)OC(C(=O)O)C(C2=CC=CC=C2)(C3=CC=CC=C3)OC)C
