
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Bimatoprost

BIMATOPROST
155206-00-1
Bimatoprost ophthalmic solution is a topical medication used for controlling the progression of glaucoma or ocular hypertension, by reducing intraocular pressure. It is a prostaglandin analogue that works by increasing the outflow of aqueous fluid from the eyes. It binds to the prostanoid FP receptor.
Allergan reported Lumigan® sales of US$625 million and Latisse® sales of US$100 million in 2013
| Systematic (IUPAC) name | |
|---|---|
| 7-[3,5-dihydroxy-2- (3-hydroxy-5-phenyl-pent-1-enyl)- cyclopentyl]-N-ethyl-hept-5-enamide | |
| Clinical data | |
| Trade names | Lumigan |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a602030 |
| Licence data | US Daily Med:link |
| Pregnancy cat. | C (US) |
| Legal status | ℞-only (US) |
| Routes | Topical (eye drops) |
| Identifiers | |
| CAS number | 155206-00-1 |
| ATC code | S01EE03 |
| PubChem | CID 5311027 |
| IUPHAR ligand | 1958 |
| DrugBank | DB00905 |
| ChemSpider | 4470565 |
| UNII | QXS94885MZ |
| Chemical data | |
| Formula | C25H37NO4 |
| Mol. mass | 415.566 g/mol |
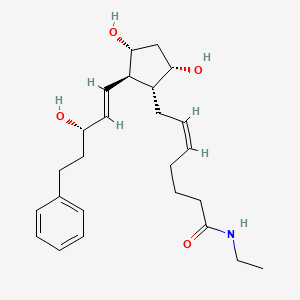
Bimatoprost (marketed in the U.S., Canada and Europe by Allergan, under the trade name Lumigan) is a prostaglandinanalog/prodrug used topically (as eye drops) to control the progression of glaucoma and in the management of ocular hypertension. It reduces intraocular pressure (IOP) by increasing the outflow of aqueous fluid from the eyes.[1] In December 2008, the indication to lengthen eyelashes was approved by the U.S. Food and Drug Administration (FDA); the cosmetic formulation of bimatoprost is sold as Latisse /ləˈtiːs/.[2] In 2008-2011, at least three case series suggested that bimatoprost has the ability to reduce adipose (fat) tissue.[3][4][5]
Allergen originally developed Bimatoprost and marketed it as Lumigan® for the treatment of elevated intraocular pressure (IOP), with open-angle glaucoma or ocular hypertension. Bimatoprost was later reformulated as a topical formulation and marketed as Latisse® for use in the treatment of hypotrichosis of the eyelashes.
Cosmetic use
In patients using ophthalmic prostaglandins such as travoprost and latanoprost, it has been anecdotally noted[by whom?] that there had been an increase in diameter, density and length of eyelashes. Allergan has initiatedclinical trials investigating the usage of Lumigan as a cosmetic drug.[6] On December 5, 2008, the FDA Dermatologic and Ophthalmic Drugs Advisory Committee voted to approve bimatoprost for the cosmetic use of darkening and lengthening eyelashes.The medical term for this is treatment of hypotrichosis, however, the FDA approval is for purely cosmetic purposes.[7]
For cosmetic purposes, it is administered once daily by applying the solution to the skin at the base of the eyelash using an applicator device “Application Guide”, where it has its effect upon the hair follicle.
Bimatoprost activates prostamide alpha F2 receptors found in the hair follicle to stimulate its growth rate. Research led by Professor Randall and the University of Bradford found that it may also offer a treatment for scalp hair regrowth in trials conducted on samples taken from men undergoing hair transplants.[8]
According to Allergan’s package labeling, users of its Latisse cosmetic product didn’t develop darker irises in clinical studies; however, “patients should be advised about the potential for increased brown iris pigmentation which is likely to be permanent.”[9]
Several cosmetics companies have released products based on prostaglandin analogs, as non-drug cosmetics.
- Age Intervention Eyelash by Jan Marini Skin Research
- RevitaLash by Athena Cosmetics Corp.
These companies have been sued by Allergan for patent infringement.[6] The FDA has seized Age Intervention Eyelash as an “unapproved and misbranded drug” because Jan Marini Skin Research promoted it as something that increases eyelash growth[10] and because it is “adulterated” with bimatoprost.[11]
Fat-reducing properties
Reductions in orbital fat (i.e., fat around the eye) have been observed in patients using bimatoprost as glaucoma therapy.[12] Of particular interest, the loss of orbital fat was unilateral in patients who used bimatoprost on only one eye.[13] The effect appears reversible upon cessation of bimatoprost use. The effect is likely to explain deepening of the lid sulcus described in a series of three patients on bimatoprost.[14] The mechanism for the apparent fat reduction remains unclear. However, bimatoprost is chemically analogous to prostaglandin F2α (PGF2α), a compound which is known to reduce fat by inhibition of adipocyte differentiation and survival.[15]
Formulations
Lumigan is a 0.03% solution of bimatoprost, and contains benzalkonium chloride as a preservative. Contact lenses should therefore be removed before use, and replaced no less than 15 minutes later;[1] other eye drops or ointments should be given no less than five minutes before or after bimatoprost.[1]
Efficacy
Studies have shown once-daily bimatoprost 0.03% ophthalmic solution to be more effective than timolol twice daily in reduction of intraocular pressure (IOP) and as effective as or more effective than the prostaglandin analogues latanoprost and travoprost in reducing IOP.[16]
Side effects
Possible side effects of this medication are:
- May cause blurred vision.
- May cause eyelid redness.
- May permanently darken eyelashes.
- May cause eye discomfort.
- May eventually cause permanent darkening of the iris to brown.
- May cause a temporary burning sensation during use.
- May cause thickening of the eyelashes.
- It may cause unexpected growth of hair if applied inappropriately, on the cheek, for example.
- It may cause infection if the one-time applicators which come with the genuine product are reused.
- Lashes may grow so long that they become ingrown and scratch the cornea.
- May cause darkening of the eyelid or of the area beneath the eye.[17]
On November 19, 2007, the FDA issued a warning during the seizure of a bimatoprost-containing cosmetic.[18] The warning stated that “the extra dose of bimatoprost may decrease the prescription drug’s effectiveness. Damage to the optic nerve may lead to decreased vision and possibly blindness.”
PATENTS
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| Canada | 2585691 | 2009-05-19 | 2026-03-14 |
| Canada | 2144967 | 2003-11-11 | 2013-09-09 |
| United States | 7351404 | 2004-05-25 | 2024-05-25 |
| United States | 6403649 | 1992-09-21 | 2012-09-21 |
Jiang Xing Chen, “Process for the production of intermediates for making prostaglandin derivatives such as latanaprost, travaprost, and bimatoprost.” U.S. Patent US20090287003, issued November 19, 2009.
US20090287003
LUMIGAN® 0.01% and 0.03% (bimatoprost ophthalmic solution) is a synthetic prostamide analog with ocular hypotensive activity. Its chemical name is (Z)-7-[(1R,2R,3R,5S)-3,5Dihydroxy-2-[(1E,3S)-3-hydroxy-5-phenyl-1-pentenyl]cyclopentyl]-5-N-ethylheptenamide, and Its molecular weight is 415.58. Its molecular formula is C24H37NO4. Its chemical structure is:
 |
Bimatoprost is a powder, which is very soluble in ethyl alcohol and methyl alcohol and slightly soluble in water. LUMIGAN® 0.01% and 0.03% is a clear, isotonic, colorless, sterile ophthalmic solution with an osmolality of approximately 290 mOsmol/kg.
LUMIGAN® 0.01% contains Active: bimatoprost 0.1 mg/mL; Preservative: benzalkonium chloride 0.2 mg/mL; Inactives: sodium chloride; sodium phosphate, dibasic; citric acid; and purified water. Sodium hydroxide and/or hydrochloric acid may be added to adjust pH. The pH during its shelf life ranges from 6.8-7.8.
LUMIGAN® 0.03% contains Active: bimatoprost 0.3 mg/mL; Preservative: benzalkonium chloride 0.05 mg/mL; Inactives: sodium chloride; sodium phosphate, dibasic; citric acid; and purified water. Sodium hydroxide and/or hydrochloric acid may be added to adjust pH. The pH during its shelf life ranges from 6.8-7.8.

SYNTHESIS


……………………………….
http://www.google.com/patents/EP2495235A1?cl=en

-
PGF2α prostaglandin synthetic routes so far reported may comprise the following two major steps:

-
The starting compound is generally protected on the secondary hydroxyl group:

where Y stands for p-phenylbenzoyl (PPB) (Ia) or benzoyl (Bz) (Ib) or an analogous substituted aryl group.
-
For example, EP0364417B1 (Kabi Pharmacia AB) describes the following synthetic sequence:

-
The product is obtained as a mixture of epimers where the 15-OH can be in α-position or β-position. The removal of unwanted β-isomer and of other impurities represents one of the main difficulties in the preparation of prostaglandins and many methods have been proposed to reduce the amount of the undesired diastereoisomers formed during the preparation.
-
In EP0544899B1 (Pharmacia AB) the reduction of the α,β-unsaturated ketone is performed with lithium-tri(sec-butyl)borohydride at ―130°C.
-
In this kind of processes the low selectivity in the reduction of the keto group leads to a tedious separation of the diastereomers and generally decreases the global yield of the synthesis.
-
A different method of stereoselective reduction of the ketone is proposed in US7674921 B1 (Cayman Chemical Co.) where the reaction with lithium aluminium hydride in the presence of (S)-binaphtol in tetrahydrofuran at -78°C is reported.
-
An alternative is presented in US6927300B2 and in US 7157590B2(Finetech Lab. Ltd.) where the critical step of the reduction of the α,β-unsaturated keto-group is performed on the compound reported below

where R1 is an aryl carbonyl group in the presence of (-)β-chlorodiisopinocampheylborane in tetrahydrofuran at ―25°C which allows to obtain a ratio of 95/5 of the two diastereoisomers (R)-(IV)/(S)-(IV).

-
A further improvement in the preparation of PGF2α is directed to the reduction of lactone to lactol in order to introduce the α-chain by subsequent Wittig reaction with 4-carboxybutyltriphenylphosphonium bromide.
-
The reaction was usually carried out using diisobutylaluminum hydride at very low temperatures, namely between -80°C and ―40°C. Unfortunately, in these conditions the partial removal of the protecting group PPB or benzoyl (Bz) is possible.
-
To avoid the inconvenient of working with mixtures of products, the p-phenylbenzoyl group or the benzoyl group R is removed by basic hydrolysis and a suitable protecting group is introduced at the two hydroxyl groups of the so obtained compound according to the following scheme:

-
In this way two additional steps of protection and deprotection are introduced possibly leading to a decrease of the global yield of the process at an advanced stage of the synthesis.
-
Moreover when P’ is a silylated group as reported in US7268239B2(Resolution Chemical Ltd.), a mixture of products is obtained because of the migration of the silyl group as shown in scheme 3

-
The same approach of hydrolysis of the aroyl group R followed by diprotection of the two hydroxyl groups with trialkylsilyl group or triaryl silyl group or tetrahydropiranyl group is disclosed in US7642370B2(Daichii Fine Chemical Co.). The protection of the two hydroxyl groups coming before the reduction of the lactone ring to lactol is described also in US7674921B1 (Cayman Chemical Co.) and in US6689901B2(Pharmacia and Upjohn Company).
-
It follows that one important issue in the synthesis of PGF2α is the involvement of intermediates with the most appropriate hydroxyl protecting group. As a consequence the use of the starting Corey lactone carrying a protection able to survive to the conditions of the subsequent reactions allows to form intermediates easy to isolate and to purify. Moreover the protection should be selectively removed in mild conditions.
-
In US5359095 (Pharmacia AB) the preparation of PGF2α prostaglandins is described starting from the commercially available (-)-Corey lactone (Ia) which is oxidized to the corresponding aldehyde (IIa)

-
The reaction is carried out in the presence of dicyclohexylcarbodiimide in dimethylsulfoxide and 1,2-dimethoxyethane; after quenching with orthophosphoric acid the aldehyde is obtained. In the same application it is reported that the crude aldehyde (II) is particularly unstable and must be used within a short period after preparation.
-
In US2010/0010239A1 (Sandoz AG) compound (I) is preferably oxidized in dichloromethane with oxalyl chloride and DMSO; the aldehyde (II) is not isolated and is processed directly in the solution where it is obtained or, when necessary, stored in solution at a temperature between ―20 and 0°C.
-
In US7268239B2 (Resolution Chem. Ltd.) the aldehyde (II)

where Z is (C6-C10)-aryl optionally substituted with one to three substituents independently selected from the group consisting of halo, C1 to C6 alkyl and unsubstituted C6 to C10 aryl, is formed by oxidation of the alcohol with sodium hypochlorite and 2,2,6,6-tetramethtyl-1-piperidinyloxy free radical (TEMPO); in order to avoid the risk of degradation, the aldehyde is directly used in the organic solution where it is synthesized.
-
According to US2009/0287003A1 (Eastar Chem. Corp.) five steps can be performed to prepare the aldehyde to be used as starting material for the synthesis of latanoprost. The five steps are reported in the scheme below:

-
In WO2010/097672 (Sifavitor) the starting material is the Corey lactone where the hydroxyl group is protected astert-butyldimethyl silyl derivative and during the synthesis a second protection is introduced according the scheme reported below referred to the preparation of Bimatoprost:

-
In case Bimatoprost is the end product, compound 9 with A=benzyl (in the following scheme, compound 9a) is directly converted by Wittig reaction to the acid 10 which is then converted in one step to ethylamide affording Bimatoprost, according to the following scheme:
-
In a preferred embodiment, the conversion of compound 10a to Bimatoprost is performed by reaction with ethylamine in an organic solvent, which is chosen among amides, ethers, ketones and chlorinated solvents, more preferably N,N-dimethylformamide, at a temperature between ―35 and 25°C, more preferably between -20°C and -10°C, in the presence of triethylamine and a suitable coupling reagent, which is preferably 1-(methylsulfonyloxy)benzotriazole. The molar ratio between compound 10a and ethylamine is comprised between 1 and 5, more preferably is 3.5, while the molar ratio between compound 10a and the coupling reagent is comprised between 1 and 2.5, more preferably is 1.8.
-
When Latanoprost is the desired product the double bond on the side chain of compound 9a is hydrogenated to form compound 11, then by Wittig reaction with 4-carboxybutyltriphenylphosphonium bromide compound 11 is converted into Latanoprost acid 12. By conversion of the carboxylic acid into isopropyl ester, the final product Latanoprost is obtained:

-
In the case of Travoprost, compound 9 with A=3-(trifluoromethyl)phenoxy (in the following scheme, compound 9b) is converted into 10b, which in turn is converted into Travoprost by esterification of the carboxylic acid by reaction with 2-iodopropane, according to scheme 10:

-














EXAMPLE 13
(Z)-7-((1R,2R,3R,5S)-3,5-dihydroxy-2-((S,E)-3-hydroxy-5-phenylpent-1-enyl)cyclopentyl)hept-5-enoic acid (Bimatoprost free acid)
-

-
4-Carboxybutyltriphenylphosphonium bromide 15 (65.4 g, 0.148 mol) was suspended in tetrahydrofuran (150.0 mL) at 0°C under nitrogen atmosphere. A solution of potassium tert-butoxide in tetrahydrofuran (296.0 mL, 0.296 mol) was added dropwise and the mixture turned into orange. After stirring for 45 minutes at 0°C the system was cooled to ―15°C. A solution of (3aR,4R,5R,6aS)-4-((S,E)-3-hydroxy-5-phenylpent-1-enyl)hexahydro-2H-cyclopenta[b]furan-2,5-diol (10.0 g, 0.033 mol) in tetrahydrofuran (46.0 mL) was added dropwise at a temperature lower than – 10°C. After three hours at -15°C no more starting material was visible on TLC and water (200 mL) was added. The mixture was extracted with diisopropyl ether (144 mL) and the layers were separated. The aqueous phase was treated with 0.6 N HCl to pH 6.0. Two extractions with ethyl acetate (2x 250 mL) were then performed and the combined organic layers were concentrated under vacuum at 40°C. An oil (26.80 g) was obtained which was purified on silica gel (eluent: dichloromethane:methanol from 97.5:2.5 to 85:15). The fractions of interest were combined and concentrated at 35°C under reduced pressure affording a colorless oil (11.7 g, 0.030 mol, 91%).
-
1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.29-7.25 (m, 2H, Ph), 7.19-7.15 (m, 3H, Ph), 5.60 (dd, J=7.2, 15.2 Hz, 1H, vinyl), 5.51-5.41 (m, 2H, H vinyl), 5.38-5.31 (m, 1H, vinyl), 4.5-3.8 (m, 7H), 2.67 (m, 2H, -CH 2-Ph), 2.37-1.43 (m, 14H).
-
13C-NMR {400 MHz, CDCl3, δ (ppm)}: 177.2 (C), 141.9 (C), 134.9 (CH), 133.0 (CH), 129.6 (CH), 129.1 (CH), 128.4 (2xCH, arom.), 128.3 (2xCH arom.),125.8 (CH), 77.4 (CH), 72.3 (CH), 72.2 (CH), 55.1 (CH), 50.5 (CH), 42.7 (CH2), 38.5 (CH2), 32.9 (CH2), 31.8 (CH2), 26.3 (CH2), 25.2 (CH2), 24.4 (CH2).
-
HPLC-MS (ESI): [M-H2O+1]+= 371; [M+Na]+ = 411; [2M+Na]+ = 799.

EXAMPLE 14
(5Z)-7-[(2R)-3,5-Dihydroxy-2-[(1E)-3-hydroxy-5-phenylpent-1-en-1-yl]cyclopentyl]-N-ethylhept-5-enamide (Bimatoprost)
-

-
Bimatoprost acid (11.50 g, 0.030 mol) was dissolved in dimethylformamide (92.0 mL), and stirred at -15°C. Triethylamine (7.25 mL, 5.26 g, 0.052 mol) was then added over 5 minutes followed by the portionwise addition of 1-(methylsulfonyloxy)benzotriazole 16 (prepared according to Bulletin of the Chemical Society of Japan, 1978, 51(11), 3320-3329) (11.27 g, 0.053 mol). The mixture was then stirred for one hour at -15°C and an aqueous solution of ethylamine (70% weight, 8.4 mL, 0.104 mol) was added dropwise over 5 minutes. The temperature was allowed to reach 0°C and the reaction was checked by TLC. The mixture was washed with water (172.0 mL) and extracted four times with ethyl acetate (4x 230.0 mL). The combined organic layers were washed with 5% sodium bisulfate solution (100 mL, 50 mL, 50 mL). The bisulfate aqueous phases were extracted with ethyl acetate (50.0 mL). The organic layers were concentrated at 40°C under reduced pressure affording the crude product as an oil (16.98 g). Treatment with dichloromethane and diisopropylether at 0°C for one hour followed by filtration afforded a solid which was then recrystallized from ethyl acetate (6.79 g, 0.016 mol, 55%).
-
1H-NMR {400 MHz, CDCl3, δ (ppm)}: 7.26 (m, 2H, Ph), 7.17 (m, 3H, Ph), 6.07 (t, J=5.6Hz, 1H, -NH-), 5.57 (dd, J=7.6, 15.2 Hz, 1H, H-14), 5.45 (dd, J=8.8, 15.2 Hz, 1H, H-13), 5.35 (m, 2H, H-5+H-6), 4.32 (d, J=4.8 Hz, 1H, OH-11), 4.08 (m, 2H, H-9+H-15), 3.90 (m, 1H, H-11), 3.73 (m, 2H, OH-9+OH-15), 3.20 (m, 2H, -N-CH 2-CH3), 2.64 (m, 2H, -CH2-17), 2.30 (m, 1H, H-12), 2.18 (m, 2H, H-7+H-10), 2.11 (m, 3H, CH2-2+H-7), 2.03 (m, 2H, H-4), 1.89 (m, 1H, H-16), 1.77 (m, 2H, H-10+H-16), 1.64 (m, 2H, H-3), 1.45 (m, 1H, H-8), 1.09 (t, J=6.8Hz, 3H, -N-CH2-CH3).
-
13C-NMR {400 MHz, CDCl3, δ (ppm)}: 173.4 (C), 142.0 (C), 135.0 (CH), 133.2 (CH), 129.6 (CH), 129.1 (CH), 128.4 (2xCH arom), 128.3 (2xCH arom), 125.7 (CH arom), 77.5 (CH), 72.22 (CH), 72.20 (CH), 55.4 (CH), 50.1 (CH), 42.8 (CH2), 38.7 (CH2), 35.8 (CH2), 34.3 (-N-CH2), 31.8 (CH2), 26.6 (CH2), 25.6 (CH2), 25.3 (CH2), 14.7 (CH3).
-
HPLC-MS (ESI): [M+Na]+ = 438, [(M-H2O) +H]+=398, [(M-2H2O) +H]+=380.

………………………………..
http://www.google.com.ar/patents/US20090287003
Bimatoprost refers to (Z)-7-[(1R,2R,3R,5S)-3,5-Dihydroxy-2-[1E,3S)-3-hydroxy-5-phenyl-1-pentenyl]cyclopentyl]-5-N-ethylheptenamide, and its molecular weight is 415.58. Its molecular formula is C25H37NO4. Its chemical structure is:


……………………………
http://www.google.com/patents/EP2454227A1?cl=en
Example 1
Synthesis of Bimatoprost

9a-iso

Bimatoprost
Scheme 3. Synthesis of Bimatoprost.
An exemplary synthesis of bimatoprost, a prostaglandin analog, is shown in Scheme 3. The synthesis is scalable, highly convergent and includes a conjugate addition between two chiral synthons, cyclopentenone derivative 6 and vinyl iodide 7a to form ketone 8a. 7a and similar vinyl halides can be prepared in a manner analogous to that shown for the corresponding THP- protected vinyl iodide in U.S. Patent No. 7,109,371 to Clissold et al., or by other methods known in the art. Ketone 8a is reduced to the corresponding isomeric alcohols 9a and 9a-iso, followed by esterification with 5-hexenoic acid to produce ester intermediate 10a. A single isomer of the alcohol can be produced, if desired, by using a stereoselective reducing agent, such as a SELECTRIDE™ (Sigma-Aldrich, St. Louis, Missouri, United States of America) reducing agent. Ring-closure metathesis (RCM) of 10a produced 10- membered ring lactone 11a, which was subsequently deprotected to form lactone 12a. Ring-opening of lactone 12a with ethylamine produced Bimatoprost. The overall yield of Bimatoprost starting from 6 and 7a was good, with each step having a yield of about 60% or greater.
Individual steps in the synthesis of Bimatoprost are described further hereinbelow in Examples 5-10. An alternative step for the synthesis of ketone 8a, using an alkyne reagent, is also shown hereinbelow in Example 13. Ring- opening of 11a prior to deprotection to form a hydroxy-protected Bimatoprost and its subsequent deprotection are described in Examples 11 and 12. As shown below in Schemes 4-6 of Examples 2-4, other exemplary prostanoids were prepared via analogous routes as that shown in Scheme 3. Details regarding individual steps in these syntheses are also shown hereinbelow in Examples 5-10.
Synthesis of Bimatoprost from 12a:
As shown in Scheme 3 in Example 1 , a 250 ml_ 3-necked round-bottom flask equipped with a magnetic bar, a temperature probe, rubber septa, and a nitrogen gas inlet was charged at room temperature with 4.1 g (11.1 mmol) of deprotected lactone 12a in 20 ml_ of THF, 22.2 mi_( 44.3 mmol) of 2 M trimethylaluminum in THF, and 67 ml_ (133 mmol) of 2 M ethylamine in THF. The reaction mixture was heated at 40 °C for 18 h and TLC analysis indicated complete reaction. The mixture was diluted with 50 mL of water and the pH was adjusted to 6 with 1 N HCI. The layers were separated and the aqueous layer was back extracted with 20 mL of ethyl acetate for two times. The combined organic layers were washed with 40 mL of brine, dried over sodium sulfate, filtered, and concentrated.
The crude product was triturated with 20 mL of MTBE at 35 0C for 3 h, cooled to room temperature, and filtered to obtain 3.1 g (67.3% yield) of Bimatoprost, confirmed by 1H NMR.
Example 11
Synthesis of Protected Bimatoprost

Protected Bimatoprost
Scheme 7. Synthesis of Protected Bimatoprost.
As shown in Scheme 7 above, a 250 mL 3-necked round-bottom flask, equipped with a magnetic stirring bar, a temperature probe, rubber septa, and nitrogen inlet, was charged at room temperature, under nitrogen, with 3.0 g (5.01 mmol) of compound 11a in 3O mL of THF, and 15mL ethylamine, 2.0 M in THF. The mixture heated at 40 °C for 24 h and then reflux for another 3 h. TLC analysis (hexanes/ethyl acetate, 10:1) indicated complete reaction. The mixture was cooled to room temperature and diluted with 30 mL of MTBE and 25 mL of water. The layers were separated and the aqueous layer was washed with 15 mL of MTBE. The combined organic extracts were washed with 25 mL of brine, dried over sodium sulfate, filtered, concentrated, and chromatographically purified to afford 2.90 g (90.0% yield) of bis-silylated bimatoprost, confirmed by 1H NMR.
Example 12
Deprotection of Protected Bimatoprost

(8)
Protected Bimatoprost
Bimatoprost Scheme 8. Deprotection of Protected Bimatoprost.
As shown in Scheme 8 above, a 250 ml_ 3-necked round-bottom flask, equipped with a magnetic stirring bar, a temperature probe, rubber septa, and nitrogen inlet, was charged at room temperature, under nitrogen, with 3.Og (3.11 mmol) of protected bimatoprost from Example 12, 30 ml. of THF1 and 0.9 g (90.8 mmol) of ammonium hydrogen difluoride. The reaction mixture was heated at 40 0C for 24 h and TLC analysis (hexanes/ethyl acetate, 1 :1) indicated complete reaction. The mixture was then diluted with 30 ml. of MTBE and 25 ml_ of water followed by layer separation. The aqueous layer was back extracted with 15 ml_ of MTBE. The combined organic extracts were washed with 25 mL of brine, dried over sodium sulfate, filtered, concentrated, and chromatographically purified to afford 1.02 g (80.0% yield) of bimatoprost confirmed by 1H NMR.
………………………………
http://www.google.com/patents/WO2013186550A1?cl=en

Example 7 – Experimental procedures for the synthesis of bimatoprost
A synthesis of bimatoprost is shown and described below.

bimatoprost (97) 7A. (±)-5-Phenyl-l-(trimethylsilyl)pent-l-yn-3-ol, 81

Following a modified procedure of Matsuda (Matsuda, F. Et al., C em. Eur. J. 5, 3252-3259 (1999)); n-butyllithium (1.6 M in hexanes, 8.8 ml, 14.1 mmol, 1.0 eq.) was added dropwise to a solution of ethynyltrimethylsilane (2.0 ml, 14.1 mmol, 1.0 eq.) in THF (6 ml) at -78 °C. After addition, the mixture was allowed to warm slowly to 0 °C and stirred for 1 h. The mixture was cooled to -78 °C and a solution of hydrocinnamaldehyde (2.2 ml, 17.0 mmol, 1.2 eq.) in THF (3 ml) was added dropwise. The mixture was then allowed to slowly warm to -10 °C and stirred for 1 h before being quenched by the addition of saturated NH4CI solution (10 ml) followed by EtOAc (10 ml). The aqueous layer was extracted with EtOAc (3 x 10 ml), the combined organic phases were washed with brine (15 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/EtOAc (100:5), giving the title product 81 as a clear, colourless oil (3.1 g, 94 %). Analytical data consistent with the literature (Matsuda, F. Et al., Chem. Eur. J. 5, 3252-3259 (1999)). max (filmVcm-1 3326 (broad), 2951, 2172, 1604, 1496, 1454, 1249, 1045, 838
*H NMR (400 MHz; CDCI3) δΗ = 0.23 (s, 9H, 3 x CH3), 2.07 (m, 3H, CH2, OH), 2.84 (t, J = 7.8 Hz, 2H, CH2), 4.40 (t, J = 6.6 Hz, 1H, CHO ), 7.25 (m, 3H, ArCH’s), 7.33 (app t, J = 7.3 Hz, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 0.0 (3 x CH3), 31.6 (CH2), 39.3 (CH2), 62.3 (CH), 90.0 (C), 106.7 (C), 126.1 (ArCH), 128.6 (2 x ArCH), 128.7 (2 x ArCH), 141.4 (ArC)
m/z (ESI+) 255.1 [MNa]+, 215.1
7B. Methyl 3-phenylpropanoate, 82

82 Hydrocinnamic acid (10.0 g, 66.6 mmol, 1 eq.) was dissolved in methanol (90 ml), cone. H2S04 (1 ml) added dropwise with stirring and the reaction mixture was stirred under reflux for 5 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. The residue was dissolved in water (100 ml) and extracted with EtOAc (3 x 50 ml). The combined organic phases were washed with 10 % NaHC03 aq. (2 x 50 ml), brine (50 ml) before being dried (MgS04), filtered and concentrated to give the title compound 82 (10.8 g, 99 %) as a clear, colourless oil. Analytical data consistent with the literature (Black, P. J. et al., Eur. J. Org. Chem. 4367-4378 (2006)). vmax (filmVcm-1 3028, 2952, 1734, 1436, 1194, 1160, 749, 697
*H NMR (400 MHz; CDCI3) δΗ = 2.68 (t, J = 7.8 Hz, 2H, CH2), 3.00 (t, J = 7.8 Hz, 2H,
CH2), 3.71 (s, 3H, CH3), 7.25 (m, 3H, ArCH’s), 7.33 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 31.1 (CH2), 35.8 (CH2), 51.7 (CH3), 126.4 (2 x ArCH),
128.4 (ArCH), 128.6 (2 x ArCH), 140.6 (ArC), 173.5 (C=0)
m/z (CI+) 165.1 [MH]+ (20%), 133.1 (100%), 105.1 (55%), 93.1 (51%), 85.0 (57%)
7C. /V-Methoxy-/V-methyl-3-phenylpropanamide, 83

Following a procedure of Trost (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754
(2006)); to a slurry of Λ/,Ο-dimethylhydroxylamine hydrochloride (4.91 g, 50.4 mmol, 2.1 eq.) in toluene (50 ml) at -10 °C was added AIMe3 (2 M in hexanes, 25.2 ml, 50.4 mmol, 2.1 eq.) dropwise. After addition, the mixture was allowed to warm to r.t. and stirred for 1 h. The mixture was cooled to -5 °C and a solution of methyl 3-phenylpropanoate 82 (3.94 g, 24.0 mmol, 1 eq.) in toluene (40 ml) was added dropwise. The reaction mixture was then allowed to warm slowly to r.t. and stirred for 3 h. The solution was cooled to 0 °C and quenched carefully by dropwise addition of HCI and the reaction mixture was extracted with EtOAc (4 x 70 ml). The combined organic phases were washed with brine (50 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/EtOAc (80:20 to 75:25), giving the Weinreb amide 83 as a clear, colourless oil (4.45 g, 97 %). Analytical data consistent with the literature (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754 (2006); Murphy, J. A. et al., Org. Lett. 7, 1427-1429 (2005)). max (film cm-1 3017, 2937, 1659, 1453, 1414, 1383, 1176, 988, 750
*H NMR (400 MHz; CDCI3) δΗ = 2.74 (t, J = 7.8 Hz, 2H, CH2), 2.97 (t, J = 7.8 Hz, 2H, CH2), 3.18 (s, 3H, CH3), 3.60 (s, 3H, CH30), 7.25 (m, 5H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 30.7 (CH3), 32.2 (CH2), 33.8 (CH2), 61.2 (CH30), 126.1
(ArCH), 128.4 (4 x ArCH), 141.3 (ArC), 173.7 (C=0)
m/z (CI+) 194.1 [MH]+ (100%), 164.1 (20%), 133.1 (12%)
7D. 5-Phenyl-l-(tri/sopropylsilyl)pent-l-yn-3-one, 84

Following a procedure of Trost (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754
(2006)); /7-Butyllithium (2.5 M in hexanes, 10.4 ml, 26 mmol, 1.7 eq.) was added dropwise to a solution of tri/sopropylsilyl acetylene (5.8 ml, 26 mmol, 1.7 eq.) in THF (53 ml) at -78 °C. After addition, the mixture was allowed to warm slowly to 0 °C and stirred for 1 h. The mixture was cooled to -78 °C and a solution of 83 (3.0 g, 15.3 mmol, 1 eq.) in THF (20 ml) was added dropwise. The mixture was then allowed to slowly warm to -10 °C and stirred for 1 h before being quenched by the addition of saturated aq. NH4CI (50 ml). The mixture was extracted with EtOAc (3 x 30 ml) and the combined organic phases were washed with brine (50 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (100:0 to 98:2), giving the title product 84 as a clear, colourless oil (4.61 g, 96 %). max (filmycnr1 2944, 2866, 1675, 1462, 1445, 1103, 881, 696
*H NMR (400 MHz; CDCI3) δΗ = 1.12 (m, 21 H, 6 x CH3, 3 x CH), 2.92 (m, 2H, CH2), 3.03 (m, 2H, CH2), 7.22 (m, 3H, ArCH’s), 7.31 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 11.1 (3 x CH), 18.6 (6 x CH3), 30.2 (CH2), 47.4 (CH2), 96.2 (C), 104.2 (C), 126.4 (ArCH), 128.4 (2 x ArCH), 128.7 (2 x ArCH), 140.4 (ArC), 186.7 (C=0) m/z (CI+) 315.3 [MH]+ (100%), 271.2 52%), 157.2 (27%)
HRMS (CI+) calcd for C20H3iOSi [MH]+ 315.2144, found 315.2139
7E. (S)-5-Phenyl-l-(tri/sopropylsilyl)pent-l-yn-3-ol, 85

Following a modified procedure of Trost (Trost, B. M. et al., J. Am. C em. Soc. 128, 6745- 6754 (2006)); Potassium hydroxide (8.5 mg, 0.15 mmol, 1.2 mol%) and RuCI(p- cymene)[(S,S)-Ts-DPEN] (80.7 mg, 0.127 mmol, 1 mol%) were added to /PrOH (110 ml) and the resultant mixture was stirred for 2 min at r.t.. 5-Phenyl-l-(tri/sopropylsilyl)pent-l-yn-3-one 84 (4.0 g, 12.7 mmol, 1 eq.) was added via syringe and the mixture was stirred at r.t. for 45 min. The mixture was concentrated in vacuo to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (20:1 to 10:1), giving the title product 85 as a clear, colourless oil (4.0 g, 99 %). The enantiomeric excess was determined to be 99 % via HPLC analysis of its derivative 87. Analytical data consistent with the literature (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754 (2006)). max (filmVcm-1 3322 (broad), 2942, 2864, 1462, 1045, 1011, 996, 882, 697, 675
*H NMR (300 MHz; CDCI3) δΗ = 1.07-1.11 (m, 21 H, 6 x CH3, 3 x CH), 1.83 (d, J = 5.5 Hz, 1H, OH), 2,03 (m, 2H, CH2), 2.82 (t, J = 7.9 Hz, 2H, CH2), 4.40 (dd, J = 6.4, 5.5 Hz, 1H, CHOH), 7.17-7.32 (m, 5H, ArCH’s)
13C NMR (75 MHz; CDCfe) 5C = 11.2 (3 x CH), 18.7 (6 x CH3), 31.6 (CH2), 39.7 (CH2), 62.4 (CH), 86.1 (C), 108.5 (C), 126.1 (ArCH), 128.6 (4 x ArCH), 141.5 (ArC)
m/z (ESI+) 339.2 [MNa]+, 299.2., 225.0
HRMS (ESI+) calcd for C20H32OSiNa [MNa]+ 339.2114, found 339.2127
[a]D 22 + 28.5 (c. 2.0, CHCI3) (lit., +27.17 (c. 2.14, CH2CI2)) 7F. (S)-5-Phenylpent-l-yn-3-ol, 86

Following a modified procedure of Trost (Trost, B. M. et al., J. Am. C em. Soc. 128, 6745- 6754 (2006)); Tetrabutylammonium fluoride (1.0 M in THF, 25 ml, 25 mmol, 2.5 eq.) was added to a solution of (S)-5-phenyl-l-(tri/sopropylsilyl)pent-l-yn-3-ol 85 (3.165 g, 10 mmol, 1 eq.) in THF (95 ml). The reaction mixture was stirred for 1 h at r.t. and then quenched by addition of saturated aq. NH4CI (50 ml). The mixture was extracted with Et20 (3 x 40 ml), the combined organic phases were washed with brine (50 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (10: 1 to 9:1), giving the title product 86 as a clear, colourless oil (1.6 g, 99 %). max (filmVcm-1 3289 (broad), 1603, 1496, 1454, 1300 (broad), 1040, 1010, 744
*H NMR (400 MHz; CDCI3) δΗ = 2.10 (m, 3H, CH2, OH), 2.55 (d, J = 2.2 Hz, 1H, C≡CH), 2.85 (t, J = 7.8 Hz, 2H, CH2), 4.40 (m, 1H, CHOH), 7.25 (m, 3H, ArCH’s), 7.33 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = 31.4 (CH2), 39.2 (CH2), 61.7 (CHOH), 73.5 (C≡CH), 84.8 (C≡CH), 126.2 (2 x ArCH), 128.6 (3 x ArCH), 141.2 (ArC)
m/z (CI+) 161.1 [MH]+ (15%), 155.1 (37%), 143.1 (88%), 119.1 (42%), 91.1 (100%) HRMS (CI+) calcd for CnH130 [MH]+161.0966, found 161.0972
[a]D 22 + 20.0 (c. 2.0, CHC )
7G. (S)-5-Phenyl-l-(trimethylsilyl)pent-l-yn-3-ol, 87
1) nBuLi, THF
-78 °C to 0 °C
OH 2) Me3SiCI OH
3) Citric acid
86 87 Following a procedure of Trost (Trost, B. M. et al., J. Am. C em. Soc. 128, 6745-6754 (2006)); /7-butyllithium (2.5 M in hexanes, 474 pL, 1.18 mmol, 3 eq.) was added dropwise to a solution of (S)-5-phenylpent-l-yn-3-ol 86 (63.3 mg, 0.39 mmol, 1 eq.) in THF (1 ml) at -78 °C. The mixture was allowed to warm to 0 °C and stirred 30 min before being cooled to -78 °C. TMSCI (148.2 pL, 1.18 mmol, 3 eq.) was added dropwise and the mixture was allowed to warm to r.t. and stirred for 2 h. Citric acid (65 mg) in methanol (0.7 ml) was added and the mixture stirred for 1 h. The mixture was poured into a mixture of brine (3 ml) and Et20 (5 ml). The aqueous layer was extracted with Et20 (3 x 10 ml) and the combined organic phases were washed with brine (10 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (9:1), giving the title product 87 as a clear, colourless oil (18.5 mg, 15.5 %). Analytical data consistent with the literature (Trost, B. M. et al., J. Am. Chem. Soc. 128, 6745-6754 (2006)). vmax(filmVcm-1 3344 (broad), 2956, 2172, 1496, 1454, 1249, 1046, 838
*H NMR (400 MHz; CDCI3) δΗ = 0.19 (s, 9H, 3 x CH3), 1.80 (d, J = 5.6 Hz, 1H, OH), 2.02 (m, 2H, CH2), 2.80 (t, J = 7.8 Hz, 2H, CH2), 4.36 (dd, J = 6.3, 5.6 Hz, 1H, CHOH), 7.20 (m, 3H, ArCH’s), 7.29 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = -0.1 (3 x CH3), 31.4 (CH2), 39.2 (CH2), 62.2 (CHOH), 89.9 (C), 106.4 (C), 125.9 (ArCH), 128.4 (2 x ArCH), 128.5 (2 x ArCH), 141.3 (ArC)
m/z (ESI+) 255.1 [MNa]+
HRMS (ESI+) calcd for Ci4H20OSiNa [MNa]+ 255.1175, found 255.1185
[a]D 22 + 28.6 (c. 0.735, CHCI3) (lit., +32.91 (c. 2.37, CH2CI2))
Chiral-HPLC data: ee = >99 % (Chiralcel OD column, 210 nm, hexane/2-propanol: 90/10, flow rate: 0.7 mlVmin, room temperature; ¾: minor 15.5 min, major 10.7 min)
7H. (S)-tert-Butyldimethyl(5-phenylpent-l-yn-3-yloxy)silane, 88

Following a procedure of Noyori (Suzuki, M. et al., J. Med. Chem. 41, 3084-3090 (1998)); Imidazole (919.1 mg, 13.5 mmol, 1.8 eq.) and t-butylchlorodimethylsilane (1.35 g, 9.0 mmol) were added to a solution of (S)-5-phenylpent-l-yn-3-ol 86 (1.2 g, 7.5 mmol, 1 eq.) in CH2CI2 (18 ml), cooled to 0 °C. The reaction mixture was then stirred at room temperature for 14 h before being poured into 1 M HCI (50 ml). The mixture was extracted with 40/60 petroleum ether (3 x 50 ml). The combined organic phases were washed with brine (50 ml) before being dried (MgS04), filtered and concentrated to give the crude product. This was purified by column chromatography on silica, eluting with petrol/Et20 (99:1), giving the title product 88 as a clear, colourless oil (1.77 g, 86 %). Analytical data consistent with the literature (Kiyotsuka, Y. et al., Org. Lett. 10, 1719-1722 (2008).; Sato, F. et al., EP 1211241 Al, Taisho Pharmaceutical co., LTD (2002)). max (filmVcm-1 3309, 2954, 2929, 2886, 2857, 1251, 1091, 834, 776
*H NMR (400 MHz; CDCI3) δΗ = 0.15 (s, 3H, CH3), 0.17 (s, 3H, CH3), 0.95 (s, 9H, 3 x CH3), 2.04 (m, 2H, CH2), 2.46 (d, J = 2.1 Hz, C≡CH), 2.81 (m, 2H, CH2), 4.41 (dt, J = 6.3, 2.1 Hz, CHOTBDMS), 7.24 (m, 3H, ArCH’s), 7.32 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = -4.9 (CH3), -4.4 (CH3), 18.3 (C), 25.9 (3 x CH3), 31.4
(CH2), 40.3 (CH2), 62.2 (CHOTBDMS), 72.5 (C≡CH), 85.4 (C≡CH), 125.9 (ArCH), 128.5 (2 x ArCH), 128.6 (2 x ArCH), 141.7 (ArC)
m/z (CI+) 275.2 [MH]+ (12%), 259.1 (37%), 217.1 (50%), 189.1 (38%), 143.1 (100%) HRMS (CI+) calcd for Ci7H27OSi [MH]+275.1831, found 275.1825
[a]D 22 -17.0 (c. 2.0, CHCI3)
71. (S,£)-teif-Butyl(l-iodo-5-phenylpent-l-en-3-yloxy)dimethylsila

A flame dried Schlenk flask, evacuated and purged with nitrogen, was charged with alkyne 88 (1.50 g, 5.46 mmol, 1 eq.). Anhydrous CH2CI2 (35 ml) was added and the reaction stirred at r.t. Zr(Cp)2HCI (2.82 g, 10.9 mmol, 2 eq.) was added as a solid, in portions. The yellow suspension was stirred at r.t. for 1 h. The resulting yellow solution was cooled to 0 °C and iodine (1.52 g, 6.01 mmol, 1.1 eq.) added as a solid, in one portion. The cooling bath was removed and the reaction mixture stirred at room temperature for 1 h. The reaction mixture was poured into water (100 ml) and extracted with 40/60 petroleum ether (4 x 50 ml). The combined organic phases were washed with water (100 ml), saturated Na2S203 solution (2 x 100 ml) and brine (100 ml) before being dried (MgS04), filtered, and concentrated to give the crude material. This was purified by flash chromatography, eluting with 40/60 petroleum ether. The fractions containing product were combined and washed with saturated Na2S203 solution (20 ml), dried (MgS04), filtered, and concentrated to give the title compound 89 (1.98 g, 90%) as a clear, colourless oil. Analytical data consistent with the literature (Sato, F. et al., EP 1211241 Al, Taisho Pharmaceutical co., LTD (2002)). max (filmVcm-1 2952, 2928, 2856, 1604, 1360, 1251, 1086, 942, 833, 774
*H NMR (400 MHz; CDCI3) δΗ = 0.04 (s, 3H, CH3), 0.06 (s, 3H, CH3), 0.92 (s, 9H, 3 x CH3), 1.82 (m, 2H, CH2), 2.65 (m, 2H, CH2), 4.15 (dq, J = 6.0, 1.2 Hz, 1H, C /-/OTBDMS), 6.25 (dd, J = 14.3, 1.2 Hz, 1H, CH=CHI), 6.57 (dd, J = 14.3, 6.0 Hz, 1H, CH=CHI), 7.18 (m, 3H, ArCH’s), 7.29 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = -4.9 (CH3), -4.5 (CH3), 18.2 (C), 25.8 (3 x CH3), 31.0 (CH2), 39.1 (CH2), 74.6 (CHOTBDMS), 76.1 (CH=CHI), 125.8 (ArCH), 128.3 (2 x ArCH), 128.4 (2 x ArCH), 141.8 (ArC), 148.8 (CH=CHI)
m/z (ESI+) 425.1 [MNa]+
HRMS (ESI+) calcd for Ci7H27IOSiNa [MNa]+ 425.0768, found 425.0754
[a]D 22 -4.5 (c. 2.0, CHCI3)
73. tert-Butyl((S,l£)-l-((3aR,4R,6aS)-2-methoxy-5- ((trimethylsilyloxy)methylene)hexahydro-2H-cyclopenta[d]furan-4-yl)-5- phenylpent-l-en-3-yloxy)dimethylsilane, 92

90 92 Vinyl iodide 89 (1.45 g, 3.6 mmol, 1.2 eq.) was added via syringe to a flame dried Schlenk flask (evacuated and purged with nitrogen several times and allowed to cool). Anhydrous Et20 (14.5 ml) was added via syringe and the resulting solution cooled to -78 °C. t-BuLi (1.6 M, 4.5 ml, 7.2 mmol, 2.4 eq.) was added dropwise and the reaction mixture stirred at -78 °C for 2 h and -40 °C for 2 h before being cooled back to -78 °C. Meanwhile, thiophene (303 mg, 288 μΙ, 3.6 mmol, 1.2 eq.) was added via syringe to a flame dried Schlenk flask (evacuated and purged with nitrogen several times and allowed to cool). Anhydrous THF (14.5 ml) was added via syringe and the resulting solution cooled to -30 °C. n-BuLi (1.6 M, 2.25 ml, 3.6 mmol, 1.2 eq.) was added dropwise and the solution stirred at -30 °C for 30 min. The solution was then cooled to -78 °C and CuCN (322.4 mg, 3.6 mmol, 1.2 eq.) added as a solid, in one portion. The cooling bath was removed and the suspension allowed to warm to r.t. The resulting tan/brown solution of cuprate was added dropwise via syringe to the Schlenk flask containing the vinyl lithium and anhydrous THF (14.5 ml) added. The mixture was stirred at -20 °C for 1 h to allow formation of mixed cuprate 90. This was cooled to -78 °C and a solution of enal 24 (504.6 mg, 3.0 mmol, 1.0 eq.) in anhydrous THF (14.5 ml) was added dropwise. The mixture was stirred at -78 °C for 1 h and then allowed to warm slowly to -20 °C. TMSCI (2.2 ml) was added via syringe followed by NEt3 (2.8 ml). The reaction was quenched by the addition of saturated NH4CI solution (80 ml) and extracted with Et20 (3 x 80 ml). The combined organic phases were washed with saturated NH4CI solution (40 ml) before being dried (MgS04), filtered, and concentrated to give the crude material as a yellow oil. This was used directly in the next step.
7K. (3aR 4R,5R,6aS)-4-((S,£)-3-(tert-Butyldimethylsilyloxy)-5-phenylpent-l- enyl)-2-methoxyhexahydro-2H-cyclopenta[d]furan-5-ol, 93

92 93
The crude material from the conjugate addition / trapping experiment, containing 92, was dissolved in C^Cb/MeOH (3:1) (32 ml) and cooled to -78 °C. A stream of ozone was passed through the stirred solution. The reaction was monitored periodically by TLC in order to judge completion of the ozonolysis (judged by consumption of silyl enol ether). At this point, the flask was purged with a stream of nitrogen during 10 min and NaBH4 (204 mg, 5.4 mmol) was added in one portion. The reaction mixture was stirred at -78 °C for 2 h before the cooling bath was removed and the reaction allowed to warm to r.t.. The reaction was stirred at r.t. for 1 h. The reaction mixture was poured into saturated NaCI solution (20 ml) and extracted with EtOAc (3 x 40 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the crude product as a pale yellow oil. This was purified by column chromatography on silica, eluting with petrol/EtOAc (9:1 to 8:2), giving the alcohol 93 (as a mixture of diastereoisomers) as a clear, colourless oil (731.0 mg, 56.0% (2 steps from enal 24)). max (filmVcm-1 3434 (broad), 2928, 1496, 1471, 1454, 1360, 1250, 1098, 1044, 1003, 970, 834, 774
*H NMR (400 MHz; CDCI3)
δΗ = (mixture of 2 diastereoisomers, signals of minor diastereoisomer indicated by *) 0.05 (s, 3H, CH3), 0.06* (s, 3H, CH3), 0.07 (s, 3H, CH3), 0.08* (s, 3H, CH3), 0.93 (s, 9H, 3 x CH3), 0.94* (s, 9H, 3 x CH3), 1.74-2.52* (m, 8H, 3 x CH2, 2 x CH), 1.74-2.52 (m, 7H, 3 x CH2, CH), 2.56-2.76* (m, 2H, CH2), 2.56-2.76 (m, 3H, CH2, CH), 3.35 (s, 3H, OCH3), 3.39* (s, 3H, OCH3), 3.81* (m, 1H, CHOH), 3.94 (m, 1H, CHOH), 4.15* (m, 1H, C/-OTBDMS), 4.15 (m, 1H, CHOTBDMS), 4.53 (app td, J = 6.6, 3.2 Hz, 1H, CH), 4.63* (app td, J = 7.5, 4.6 Hz, 1H, CH), 5.09* (app d, J = 5.6 Hz, 1H, CH), 5.14 (app d, J = 4.4 Hz, 1H, CH), 5.48* (m, 1H, =CH), 5.48 (m, 1H, =CH), 5.60* (m, 1H, =CH), 5.60 (m, 1H, =CH), 7.20* (m, 3H, ArCH’s), 7.20 (m, 3H ArCH’s), 7.30* (m, 2H, ArCH’s), 7.30 (m, 2H, ArCH’s)
13C NMR (100 MHz; CDCI3) 5C = (mixture of 2 diastereoisomers, signals of minor diastereoisomer indicated by *) -4.6 (CH3), -4.6* (CH3), -4.0* (CH3), -3.9 (CH3), 18.4 (C), 18.4* (C), 26.0 (3 x CH3), 26.0* (3 x CH3), 31.8 (CH2), 31.9 (CH2), 38.0 (CH2), 39.3 (CH2), 39.7 (CH2), 40.2 (CH2), 40.3 (CH2), 42.5 (CH2), 45.8 (CH), 46.1* (CH), 54.6* (CH30), 55.0 (CH30), 56.7* (CH), 57.6 (CH), 72.8 (CHOTBDMS), 72.9* (CHOTBDMS), 77.9* (CHOH), 78.9 (CHOH), 81.2 (CH), 83.3* (CH), 106.6 (CH), 107.7* (CH), 125.7 (ArCH), 125.8 (ArCH), 126.0 (ArCH), 128.4 (ArCH), 128.5 (ArCH), 128.6 (ArCH), 130.3* (=CH), 131.1 (=CH), 135.1* (=CH), 135.2 (=CH), 142.2* (ArC), 142.5 (ArC)
m/z (ESI+) 455.1 [MNa]+
HRMS (ESI+) calcd for C25H4o04SiNa [MNa]+ 455.2588, found 455.2587 7L. (3a ?,4 ?,5 ?,6aS)-4-((S,E)-3-Hydroxy-5-phenylpent-l-enyl)hexahyd cyclopenta[b]furan-2,5-diol, 94

93 94
Alcohol 93 (210 mg, 0.485 mmol) was stirred with 1.5% aqueous HCI / THF (3:2) (10 ml) at r.t. for 16 h. The mixture was extracted with CH2CI2 (5 x 15 ml) and the combined organic phases were dried (MgS04), filtered, and concentrated to give the triol 94 and silanol byproduct as a clear, colourless oil. This material was taken forward for the subsequent transformation without purification.
7M. 5-Bromo-/V-ethylpentanamide, 95
1) SOCI2, DMF cat.
Toluene, 55 °C

95
Dimethylformamide (116 μΙ_, 1.5 mmol, 0.1 eq.) and thionyl chloride (1.63 ml, 22.5 mmol, 1.5 eq.) were added to a solution of 5-bromovaleric acid (2.71 g, 15 mmol, 1 eq.) in toluene (20 ml) and the reaction mixture was stirred at 50 °C for 4 h. The volatiles were removed under high vacuum, THF (130 ml) was added and the mixture was cooled to 0 °C. N,N- Diisopropylethylamine (4.2 ml, 24 mmol, 1.6 eq.) and ethylamine (2 M solution in THF, 9.0 ml, 18 mmol, 1.2 eq.) were added dropwise. The reaction mixture was stirred for 1 h at 0 °C before being quenched by the addition of saturated aq. NH4CI (100 ml). The reaction mixture was extracted with Et20 (4 x 75 ml) and the combined organic phases were washed with brine (100 ml) before being dried (MgS04), filtered, and concentrated to give the crude material. This was purified by flash chromatography, eluting with petrol/EtOAc (4:6), giving the title product 95 as a clear, colourless oil (2.75 g, 88 %). Analytical data consistent with the literature (Halazy, S. et al., WO 9612713, Pierre Fabre Medicament (1996)). max (filmVcm-1 3275 (broad), 2970, 2933, 1640, 1543, 1439, 1276, 1150, 643
*H NMR (400 MHz; CDCI3) δΗ = 1.13 (t, J = 7.2 Hz, 3H, CH3), 1.78 (m, 2H, CH2), 1.89 (m, 2H, CH2), 2.19 (t, J = 7.3 Hz, CH2CO), 3.27 (dt, J = 7.2, 5.5 Hz, 2H, NHCH2), 3.41 (t, J = 6.5 Hz, 2H, BrCH2), 5.79 (broad s, 1H, NH)
13C NMR (100 MHz; CDCI3) 5C = 15.0 (CH3), 24.4 (CH2), 32.2 (CH2Br), 33.4 (CH2), 34.4 (NHCH2), 35.7 (CH2C=0), 172.3 (C=0)
m/z (CI+) 210.0 [M81BrH]+ (80%), 208.0 [M79BrH]+ (80%), 128.1 (100%), 107.1 (60%) HRMS (CI+) calcd for C7Hi5 81BrNO [M81BrH]+ 210.0317, found 210.0320; calcd for
C7H15 79BrNO [M79BrH]+ 208.0337, found 208.0341
(5-(Ethylamino)-5-oxopentyl)triphenylphosphonium bromide, 96

Triphenylphosphine (2.88 g, 11 mmol, 1.1 eq.) was added to a solution of 5-bromo-A/- ethylpentanamide 95 (2.08 g, 10 mmol, 1 eq.) in MeCN (5 ml) and the mixture was stirred for 14 h at 80 °C. The mixture was allowed to cool to r.t. and concentrated under vacuum. The residue was added dropwise into Et20 (100 ml) and stirred vigourously for 10 min. The resulting solid was filtered, washed with Et20 (2 x 10 ml) and dissolved in CH2CI2 (15 ml). This solution was then added dropwise into Et20 (200 ml) and stirred for 10 min. Solids were filtered, washed with Et20 (2 x 10 ml) and dried under high vacuum to give the title product 96 as a white powder (3.87 g, 82 %).
Mp 177-179 °C
max (filmVcm“1 3275 (broad), 2970, 2933, 1640, 1543, 1439, 1276, 1150, 643
*H NMR (400 MHz; CDCI3) δΗ = 1.06 (t, J = 7.3 Hz, 3H, CH3), 1.63 (m, 2H, CH2), 1.91 (quin., J = 6.5 Hz, CH2), 2.57 (t, J = 6.5 Hz, 2H, CH2C=0), 3.15 (q, J = 7.3, 5.6 Hz, 2H, NHCH2), 3.66 (m, 2H, CH2P+), 7.66 (m, 6H, ArCH’s), 7.75-7.82 (m, 9H, ArCH’s), 8.26 (t, J = 5.6 Hz, 1H, NH)
13C NMR (100 MHz; CDCI3) 5C = 14.8 (s, CH3), 21.1 (d, J = 4.6 Hz, CH2), 22.4 (d, J = 49.9 Hz, CH2P+), 26.1 (d, J = 17.7 Hz, CH2), 34.1 (s, CH2), 34.2 (s, CH2NH), 118.2 (d, J = 86.1 Hz, 3 x ArC), 130.6 (d, J = 12.3 Hz, 6 x ArCH), 133.8 (d, J = 10.0 Hz, 6 x ArCH), 135.1 (d, J = 3.1 Hz, 3 x ArCH), 172.7 (C=0) m/z (ESI+) 390.2 [M-Br]+
HRMS (ESI+) calcd for C25H29NOP [M-Br]+ 390.1981, found 390.1971
70. (Z)-7-((lR,2R,3R,5S)-3,5-Dihydroxy-2-((S,£)-3-hydroxy-5-phenylpent-l- enyl)cyclopentyl)-/V-ethylhept-5-enamide, bimatoprost, 97

(5-(Ethylamino)-5-oxopentyl)triphenylphosphonium bromide 96 (1.37 g, 2.91 mmol, 6 eq.) was added to a flame dried Schlenk flask, under N2, and anhydrous THF (10 ml) added. The resulting suspension was cooled to 0 °C. KOt-Bu (653.0 mg, 5.82 mmol, 12 eq.) was added in one portion and the resulting orange mixture stirred at 0 °C for 40 min. A solution of crude triol 94 (0.485 mmol, 1 eq.) in anhydrous THF (2.5 ml) was added dropwise via syringe. After complete addition the mixture was stirred at r.t. for 1 h. The reaction was quenched with saturated aq. NH4CI (10 ml) and extracted with EtOAc (5 x 10 ml). The combined organic phases were dried (MgS04), filtered, and concentrated to give the crude material as solids. These were triturated with EtOAc (10 ml) and the solids filtered and washed with EtOAc (4 x 10 ml). The filtrate was concentrated under vacuum and purified by column chromatography on silica, eluting with EtOAc/MeOH (97.5:2.5 to 95:5) to give 97 (99.2 mg) as a yellowish oil which was further purified by preparative TLC (EtOAc/MeOH 5%) to give BIMATOPROST97 (82.6 mg, 41% over 2 steps) as a clear, colourless oil. Analytical data consistent with the literature (Zanoni, G. et al., Tetrahedron 66, 7472-7478 (2010); Gutman, A. et al., US 20090163596 (2009)).
max (filmVcm-1 3300 (broad), 2930, 1643, 1550, 1453, 1332, 1293, 1048, 1029, 968, 729, 698
*H NMR (400 MHz; CDCI3) δΗ = 1.09 (t, J = 7.1 Hz, 3H, CH3), 1.42-2.40 (m, 14H, 6 x CH2, 2 x CH), 2.67 (m, 2H, CH2), 3.22 (dq, J = 7.1, 6.3 Hz, 2H, CH2NH), 3.41 (broad s, 3H, 3 x OH), 3.80-4.30 (broad m, 3H, 3 x CHOH), 5.37 (m, 2H, 2 x =CH), 5.47 (dd, J = 15.2, 7.9 Hz, 1H, =CH), 5.59 (dd, J = 15.2, 7.9 Hz, 1H, =CH), 5.90 (broad s, 1H, NH), 7.17 (m, 3H, ArCH’s), 7.26 (m, 2H, ArCH’s)
13C NMR ( 100 MHz; CDCI3) 5C = 14.8 (CH3), 25.4 (CH2), 25.6 (CH2), 26.7 (CH2), 31.9 (CH2), 34.4 (CH2NH), 35.8 (CH2C=0), 38.8 (CH2), 42.9 (CH2), 50.2 (CH), 55.5 (CH), 72.3 (CHOH), 72.4 (CHOH), 77.7 (CHOH), 125.8 (ArCH), 128.4 (2 x ArCH), 128.5 (2 x ArCH), 129.1 (=CH), 129.7 (=CH), 133.7 (=CH), 135.1 (=CH), 142.0 (ArC), 173.4 (C=0)
m/z (ESI+) 438.2 [MNa]+
HRMS (ESI+) calcd for Q^IV^Na [MNa]+ 438.2614, found 438.2615
[a]D 22 +41.1 (c. 0.35, CH2CI2) (lit. – Zanoni, G. et al., Tetrahedron 66, 7472-7478 (2010), +32.7 (c. 0.33, CH2CI2)) (lit. – Gutman, A. et al., US 20090163596 (2009), +36 (c. 1, MeOH))
References
- “Bimatoprost Ophthalmic”. MedlinePlus. January 1, 2003. Archived from the original on 2007-10-05. Retrieved 2007-11-19.
- “Allergan gets FDA approval for eyelash treatment”. BusinessWeek. Associated Press. December 26, 2008. Retrieved December 26, 2008.
- Park J, Cho HK, Moon JI (2011). “Changes to upper eyelid orbital fat from use of topical bimatoprost, travoprost, and latanoprost.”. Japanese Ophthalmological Society 55 (1): 22–27.doi:10.1007/s10384-010-0904-z. PMID 21331688.
- Jayaprakasam A, Ghazi-Nouri S. (2010). “Periorbital fat atrophy – an unfamiliar side effect of prostaglandin analogues.”. Orbit 29 (6): 357–359. doi:10.3109/01676830.2010.527028.PMID 21158579.
- Filippopoulos T, Paula JS, Torun N, Hatton MP, Pasquale LR, Grosskreutz CL. (2008). “Periorbital changes associated with topical bimatoprost.”. Ophthalmology Plastic and Reconstructive Surgery 24 (4): 302–307. doi:10.1097/IOP.0b013e31817d81df. PMID 18645437.
- Rundle, Rhonda L. (2007-11-19). “Drug That Lengthens Eyelashes Sets Off Flutter”. The Wall Street Journal. Retrieved 2007-11-19.
- The Pink Sheet: [1] Lauren Smith December 15, 2008; Volume 70, Number 050,Page[verification needed]
- Federation of American Societies for Experimental Biology Journal “The prostamide-related glaucoma therapy, bimatoprost, offers a novel approach for treating scalp alopecias” Randall et all. October 26, 2012.
- Latisse prescribing information: “Important Safety Information”
- MSNBC: FDA Seizes $2 Million Of Potentially Harmful SJ Eye Product KNTV-TV November 17, 2007[dead link]
- Reuters: “U.S. seizes discontinued eyelash product”. Jim Wolf. November 16, 2007.
- Tappeiner C, Perren B, Iliev ME, Frueh BE, Goldblum D (May 2008). “Orbitale Fettgewebsatrophie bei lokaler Bimatoprost-Therapie – Kann Bimatoprost einen Enophthalmus verursachen?” [Orbital fat atrophy in glaucoma patients treated with topical bimatoprost–can bimatoprost cause enophthalmos?]. Klinische Monatsblätter für Augenheilkunde (in German)225 (5): 443–5. doi:10.1055/s-2008-1027362. PMID 18454393.
- Filippopoulos T, Paula JS, Torun N, Hatton MP, Pasquale LR, Grosskreutz CL (2008). “Periorbital changes associated with topical bimatoprost”. Ophthalmic Plastic and Reconstructive Surgery 24 (4): 302–7. doi:10.1097/IOP.0b013e31817d81df. PMID 18645437.
- Peplinski LS, Albiani Smith K (August 2004). “Deepening of lid sulcus from topical bimatoprost therapy”. Optometry and Vision Science 81 (8): 574–7.doi:10.1097/01.opx.0000141791.16683.4a. PMID 15300114.
- Serrero G, Lepak NM (April 1997). “Prostaglandin F2alpha receptor (FP receptor) agonists are potent adipose differentiation inhibitors for primary culture of adipocyte precursors in defined medium”. Biochemical and Biophysical Research Communications 233 (1): 200–2. doi:10.1006/bbrc.1997.6433. PMID 9144422.
- Curran MP (2009). “Bimatoprost: a review of its use in open-angle glaucoma and ocular hypertension”. Drugs Aging 26 (12): 1049–71. doi:10.2165/11203210-000000000-00000.PMID 19929032.
- “Long Lashes Without Prescription, but With Risks”. Catherine Saint Louis. The New York Times. May 1, 2010
- “Potentially Harmful “Cosmetic” Eye Product Seized” (Press release). U.S. Food and Drug Administration (FDA). November 19, 2007. Retrieved 2007-12-05.
Citations
- Chen M, Cheng C, Chen Y, Chou C, Hsu W (2006). “Effects of bimatoprost 0.03% on ocular hemodynamics in normal tension glaucoma.”. J Ocul Pharmacol Ther 22 (3): 188–93. doi:10.1089/jop.2006.22.188. PMID 16808680.
- Kruse P, Rieck P, Sherif Z, Liekfeld A (2006). “Cystoid macular edema in a pseudophakic patient after several glaucoma procedures. Is local therapy with bimatoprost the reason?”. Klinische Monatsblätter für Augenheilkunde 223 (6): 534–7. doi:10.1055/s-2005-858992. PMID 16804825.
- Steinhäuser S (2006). “Decreased high-density lipoprotein serum levels associated with topical bimatoprost therapy.”. Optometry 77 (4): 177–9.doi:10.1016/j.optm.2006.02.001. PMID 16567279.
- Park J, Cho HK, Moon JI (2011). “Changes to upper eyelid orbital fat from use of topical bimatoprost, travoprost, and latanoprost.”. Japanese Ophthalmological Society 55 (1): 22–27. doi:10.1007/s10384-010-0904-z. PMID 21331688.
- Jayaprakasam A, Ghazi-Nouri S. (2010). “Periorbital fat atrophy – an unfamiliar side effect of prostaglandin analogues.”. Orbit 29 (6): 357–359.doi:10.3109/01676830.2010.527028. PMID 21158579.
- Filippopoulos T, Paula JS, Torun N, Hatton MP, Pasquale LR, Grosskreutz CL. (2008). “Periorbital changes associated with topical bimatoprost.”. Ophthalmology Plastic and Reconstructive Surgery 24 (4): 302–307. doi:10.1097/IOP.0b013e31817d81df. PMID 18645437.
External links
- Medical News Today: FDA Seizes $2 Million Of Cosmetic Eye Product Which Contains Drug Ingredient And Makes Unapproved Drug Claims. Christian Nordqvist. 18 November 2007
- Wired Science: FDA Seizes Cosmetic That Can Blind. Brandon Keim. November 19, 2007
- Eye Drops: [The generic name of the Latisse eye drop is Bimatoprost Ophthalmic Solution 0.03%]. Crazzy Paul. Aug 01, 2013
|
1-25-2012
|
Method of enhancing hair growth
|
|
|
1-25-2012
|
NITRIC OXIDE DONATING PROSTAMIDES
|
|
|
11-25-2011
|
BIMATOPROST CRYSTALLINE FORM I
|
|
|
10-19-2011
|
Method of Enhancing Hair Growth
|
|
|
7-22-2011
|
IMPROVED PROCESS FOR THE PRODUCTION OF BIMATOPROST
|
|
|
6-3-2011
|
Process for the Preparation of Prostaglandin Analogues and Intermediates Thereof
|
|
|
5-27-2011
|
COMPOSITIONS AND METHODS FOR STIMULATING HAIR GROWTH
|
|
|
5-27-2011
|
ENHANCED BIMATOPROST OPHTHALMIC SOLUTION
|
|
|
5-25-2011
|
Bimatoprost crystalline form I
|
|
|
5-13-2011
|
COMPOSITIONS FOR ENHANCING HAIR GROWTH
|
|
3-2-2011
|
Process for the Preparation of Prostaglandin Analogues and Intermediates Thereof
|
|
|
12-24-2010
|
METHOD FOR THE PURIFICATION OF PROSTAGLANDINS
|
|
|
12-15-2010
|
Enhanced bimatoprost ophthalmic solution
|
|
|
9-17-2010
|
Compositions and Methods for Reducing Body Fat
|
|
|
5-28-2010
|
COMPLEXES OF PROSTAGLANDIN DERIVATIVES AND MONOSUBSTITUTED, CHARGED BETA-CYCLODEXTRINS
|
|
|
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
|
|
|
4-30-2010
|
AMINO ACID SALTS OF PROSTAGLANDINS
|
|
|
2-24-2010
|
Compositions and methods for reducing body fat
|
|
|
1-27-2010
|
10-HYDROXY-11-DIHYDROPROSTAGLANDIN ANALOGS AS SELECTIVE EP4 AGONISTS
|
|
|
1-15-2010
|
Process for the Production of Prostaglandins and Prostaglandin Analogs
|
|
11-20-2009
|
Process for the production of intermediates for making prostaglandin derivatives such as latanaprost, travaprost, and bimatoprost
|
|
|
11-4-2009
|
Enzymatic transformation of a prostaglandin (bimatoprost) intermediate
|
|
|
10-16-2009
|
Method for preparing prostaglandin F analogue
|
|
|
8-14-2009
|
METHOD OF ENHANCING HAIR GROWTH
|
|
|
6-12-2009
|
Enhanced Bimatoprost Ophthalmic Solution
|
|
|
4-3-2009
|
METHOD FOR SCREENING OF PROSTAGLANDIN COMPOUNDS COMPRISING AN OPTIMAL FORMULATION FOR THE ENHANCEMENT OF HAIR GROWTH AND THE STIMULATION OF FOLLICULAR ANAGEN AND FORMULATIONS RESULTING THEREFROM
|
|
|
10-15-2008
|
5-Thiopiperdinyl prostaglandin e analogs
|
|
|
2-20-2008
|
COMPOSITIONS AND METHODS COMPRISING PROSTAGLANDIN-RELATED COMPOUNDS AND TREFOIL FACTOR FAMILY PEPTIDES FOR THE TREATMENT OF GLAUCOMA WITH REDUCED HYPEREMIA
|
|
|
2-15-2008
|
Novel Prostamides For The Treatment Of Glaucoma And Related Diseases
|
|
|
11-23-2007
|
COMPOSITIONS AND METHODS COMPRISING PROSTAGLANDIN-RELATED COMPOUNDS AND TREFOIL FACTOR FAMILY PEPTIDES FOR THE TREATMENT OF GLAUCOMA WITH REDUCED HYPEREMIA
|
|
7-18-2007
|
Compositions and methods comprising prostaglandin related compounds and trefoil factor family peptides for the treatment of glaucoma with reduced hyperemia
|
|
|
5-18-2007
|
NOVEL PROSTAMIDES FOR THE TREATMENT OF GLAUCOMA AND RELATED DISEASES
|
|
|
4-27-2007
|
Compositions comprising benzo (g) quinoline derivatives and prostaglandin derivatives
|
|
|
3-7-2007
|
Prostamides for the treatment of glaucoma and related diseases
|
|
|
1-31-2007
|
10-Hydroxy-11-dihydroprostaglandin analogs as selective EP4 agonists
|
|
|
1-24-2007
|
Process for the preparation of prostaglandin derivatives
|
|
|
1-10-2007
|
Protected diols for prostaglandin synthesis
|
|
|
1-3-2007
|
Process for the preparation of 17-phenyl-18,19,20-thinor-pgf 2a and its derivatives
|
|
|
11-29-2006
|
Protected and unprotected triols for prostaglandin synthesis
|
|
|
9-20-2006
|
Prostaglandin synthesis
|
|
9-6-2006
|
Cyclopentane heptan(ENE)OIC acid, 2-heteroarylalkenyl derivatives as therapeutic agents
|
|
|
7-5-2006
|
Method for imparting artificial tan to human skin
|
|
|
8-24-2005
|
Inhibition of irritating side effects associated with use of a topical ophthalmic medication
|
|
|
3-18-2005
|
Methods for the treatment of gray hair using cyclopentane(ene) heptan(en)oic acid amides
|
|
|
3-9-2005
|
9,11-cycloendoperoxide pro-drugs of prostaglandin analogues for treatment of ocular hypertension and glaucoma
|
|
|
2-11-2005
|
Compositions for delivery of therapeutics into the eyes and methods for making and using same
|
|
|
5-29-2002
|
Ocular hypotensive lipids
|
| EP0364417A1 | Sep 6, 1989 | Apr 18, 1990 | Pharmacia AB | Prostaglandin derivatives for the treatment of glaucoma or ocular hypertension |
| EP0364417B1 | Sep 6, 1989 | Feb 9, 1994 | Pharmacia AB | Prostaglandin derivatives for the treatment of glaucoma or ocular hypertension |
| EP0544899B1 | Jun 19, 1992 | Sep 6, 1995 | CHINOIN Gyogyszer és Vegyészeti Termékek Gyára RT. | Prostaglandins |
| US5359095 | Feb 8, 1994 | Oct 25, 1994 | Pharmacia Ab | Hydrogenation of doulbe bond in intermediate compound without deoxygenatio of allytic alcohol |
| US6689901 | Jun 25, 2002 | Feb 10, 2004 | Pharmacia & Upjohn Company | Process and intermediates to prepare latanoprost |
| US6927300 | Jan 26, 2001 | Aug 9, 2005 | Finetech Laboratories Ltd | Process for the preparation of Latanoprost |
| US7157590 | May 3, 2002 | Jan 2, 2007 | Finetech Laboratories Ltd. | Chemical intermediate for vision defect drug |
| US7268239 | Jul 27, 2005 | Sep 11, 2007 | Resolution Chemicals Limited | Process for the preparation of prostaglandins and analogues thereof |
| US7642370 | Mar 20, 2007 | Jan 5, 2010 | Daiichi Fine Chemical Co., Ltd. | Method for preparing prostaglandin derivative |
| US7674921 | Feb 25, 2008 | Mar 9, 2010 | Cayman Chemical Company, Inc. | cloprostenol or latanoprost; topical use; highly crystalline structures that are easy to formulate into ophthalmic solutions; hydrolysis of these analogs releases only the active PGF2 alpha analog free acid, without the production of toxic and irritant small aliphatic alcohol coproducts |
| US20090259066 * | Jul 3, 2008 | Oct 15, 2009 | Everlight Usa, Inc. | Method for preparing prostaglandin F analogue |
| US20090287003 | Sep 29, 2006 | Nov 19, 2009 | Jiang Xing Chen | Process for the production of intermediates for making prostaglandin derivatives such as latanaprost, travaprost, and bimatoprost |
| US20100010239 | Jul 10, 2009 | Jan 14, 2010 | Sandoz Ag | Process for the Production of Prostaglandins and Prostaglandin Analogs |
| WO2010097672A1 | Feb 18, 2010 | Sep 2, 2010 | Sifavitor S.R.L. | Process for the preparation of prostaglandin derivatives |
Organic spectroscopy should be brushed up and you get confidence
read my blog
ORGANIC SPECTROSCOPY INTERNATIONAL is the blog
Organic chemists from Industry and academics to interact on Spectroscopy techniques for Organic compounds ie NMR, MASS, IR, UV Etc. email me ……….. amcrasto@gmail.com
http://orgspectroscopyint.blogspot.in/ is the link
 amcrasto@gmail.com
amcrasto@gmail.com
Sage Kills 51% of Melanoma Cells in Vitro and Reduces Risk in Humans

Sage Kills 51% of Melanoma Cells in Vitro and Reduces Risk in Humans: The herb sage is rich in the powerful anti-cancer compound thujone, which in this study was shown to kill 51% of human melanoma cells in vitro.
But does sage reduce melanoma risk in humans? Yes, according to a study out of the Italy, where this herb is often consumed as part of the traditional diet: people eating sage at least once weekly had 32% less risk of melanoma. Apart from thujone, this super-herb contains several other compounds with proven health benefits, including cineole, carnosol, caffeic acid and chlorogenic acid.
Sage has been used in traditional medicine for centuries to treat a variety of conditions, and it’s many health benefits are now being confirmed by some remarkable new clinical trials. One trial showed that 333 mg of sage extract daily significantly improved memory performance in older adults while in another clinical trial, 60 drops daily of liquid extract significantly enhanced cognitive performance in patients with Alzheimer’s.
Sage is also such a powerful natural antibacterial that one clinical trial (out of Switzerland) showed that a spray of sage + echinacea performed as well as chlorhexidine + lidocaine in treating sore throats! Sage can be consumed as supplements, prepared as a tea, or used generously as a herb in a variety of dishes.
http://www.ncbi.nlm.nih.gov/pubmed/21647317


Sage – Nature Wonder Herb
(Salvia officinalis) is cultivated as a spice and medicinal herb. This plant is known from parts of Europe, especially the Balkans, where is used for obtaining essential oils.
The Latin name of the whole genus Salvia comes from the Latin word “salvare“, which means “rescue saving curing …” because the Romans 2000 years ago appreciated and used sage for healing.

The effectiveness of the leaves is due primarily to the presence of etheric oil (1.5 – 2.5 percent), which has antimicrobial and anti-inflammatory action.
It has been proven that etheric oil destroys bacteria Eshericia colli, Schigela sonei, Salmonela, and has a slightly weaker activity against bacteria of a group of staphylococci and streptococci.
It is effective in destroying certain types of fungi as: cadida albicas, cadida krusei, cadidapseudotropicalis, torulopsis glabrata and cyptococcus eoformas.

Etheric sage oil is colorless or yellow-green liquid with an aromatic and bitter taste.
Because of this effect, sage is a useful herb for treating inflammation and infections of the mucous membranes in the mouth, gums and throat. For these diseases gargle from sage tea is recommended.
 -Put 2 tablespoons of crushed dried sage leaves in 2 cups of boiling water water. Cover it and let it stay like that for 20 or 30 minutes, then drain it.
-Put 2 tablespoons of crushed dried sage leaves in 2 cups of boiling water water. Cover it and let it stay like that for 20 or 30 minutes, then drain it.
For treatment to be successful, you must do gargle regularly , every 3 hours .
Sage softens the mucous secretions from the inflamed mucosa of the respiratory organs. Because of that you can drink sage tea against bronchitis.
-The tea is prepared with 1 tablespoon dried leaves and 2 cups of boiling water. Drink it three times a day.
You should avoid long term drinking tea because etheric oil from sage contains toxic substance thujone.
Dry leaves are used as a spice because it improves the taste and scent of food, and helps the digestion too.
Tea can be used for improving the functions of bile and liver, because bitter substances and etheric oil increase the secretion of digestive juices in the body.
Sage tea has a diuretic effect, which is poorly expressed and due to the presence of flavonoids . It can help with chronic disease of urinary tract .

From a long time ago is known that sage tea is very effective cure for sweating. This power of sage it’s explained by its effect on the nervous center which regulates the glands that secrete sweat.
Commonly is recommended against perspiration of neuromuscular origin or the heat waves that occur during menopause.
Applied on the skin , tea tightens skin and calms inflammation. Especially good for oily skin with open pores and irritated skin.

Sage is a remarkable tool for whitening teeth, strengthening gums and aid in periodontitis. A small spoon of sage leaves is mixed with 1 drop of peppermint etheric oil and abit of baking soda. With this mixture rub the teeth and gums twice a week.
Lumiracoxib…Selective cyclooxygenase-2-(COX-2) inhibitor. Anti-inflammatory.

Lumiracoxib
2-[(2-Chloro-6-fluorophenyl)amino]-5-methylbenzeneacetic Acid;
2-[2-(2-Chloro-6-fluorophenylamino)-5-methylphenyl]acetic Acid;
CGS 35189; COX 189; Prexige;
Applications: Selective cyclooxygenase-2-(COX-2) inhibitor. Anti-inflammatory.
| Systematic (IUPAC) name | |
|---|---|
| {2-[(2-chloro-6-fluorophenyl)amino]-5-methylphenyl} acetic acid |
|
| Clinical data | |
| Trade names | Prexige |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy cat. | C (AU) |
| Legal status | ℞-only Withdrawn (Australia, New Zealand, Canada, UK, Germany,Austria, Belgium, Cyprus, Brazil) |
| Routes | Oral |
| Pharmacokinetic data | |
| Bioavailability | 74-90%[1] |
| Protein binding | >98%[1] |
| Metabolism | Predominantly in the liver viaoxidation and hydroxylation(CYP2C9)[1] |
| Half-life | 5-8 hours[1] |
| Excretion | Urine (54%) and faeces (43%)[1] |
| Identifiers | |
| CAS number | 220991-20-8 |
| ATC code | M01AH06 |
| PubChem | CID 151166 |
| DrugBank | DB01283 |
| ChemSpider | 133236 |
| UNII | |
| PDB ligand ID | LUR (PDBe, RCSB PDB) |
| Chemical data | |
| Formula | C15H13ClFNO2 |
| Mol. mass | 293.72 g/mol |
Lumiracoxib (rINN) is a carboxylic acid COX-2 selective inhibitor non-steroidal anti-inflammatory drug, manufactured by Novartis and still sold in few countries, including Mexico, Ecuador and the Dominican Republic, under the trade name Prexige (sometimes misquoted as “Prestige” by the media).[1]
Lumiracoxib has several distinctive features. Its structure is different from that of other COX-2 inhibitors, such as celecoxib: lumiracoxib is an analogue of diclofenac (one chlorine substituted by fluorine, the phenylacetic acid has another methyl group in meta position), making it a member of the arylalkanoic acid class of NSAIDs; it binds to a different site on the COX-2 enzyme than do other COX-2 inhibitors; it is the only acidic coxib and has the highest COX-2 selectivity of any NSAID.[2]
Since its original approval, lumiracoxib has been withdrawn from the market in several countries, mostly due to its potential for causing liver failure (sometimes requiring liver transplantation). It has never been approved for use in the United States.[1]
History
The TARGET study (Therapeutic Arthritis Research and Gastrointestinal Event Trial) was conducted with more than 18,000 patients to test its gastrointestinal and cardiovascular safety against naproxen and ibuprofen and also study its efficacy against these two NSAIDs.
In November 2006, Prexige received marketing approval for all European Union countries through a common procedure called MRP. However, in August 2007, Prexige was withdrawn from the market in Australia following 8 serious liver adverse events, including 2 deaths and 2 liver transplants.[3] On September 27, 2007, the US Food and Drug Administration issued a not approvable letter for lumiracoxib, requiring additional safety data.[4] Canada withdrew Prexige (approved at 100 mg dose only) in October 2007.[5] Several European Union countries followed suit in November 2007.[6]
The FDA rejected Prexige as a trade name for lumiracoxib in 2003. Prexede was suggested as an alternative, but the FDA Division of Medication Errors and Technical Support (DMETS) subsequently recommended against it as well.[7]
Withdrawal from market
On August 11, 2007, Australia’s Therapeutic Goods Administration (TGA, the national agency responsible for regulation of pharmaceuticals) cancelled the registration of lumiracoxib in Australia due to concerns that it may cause liver failure.[8]
According to the TGA’s Principal Medical Adviser, Dr Rohan Hammett, as of 10 August 2007 the TGA had received 8 reports of serious adverse liver reactions to the drug, including two deaths and two liver transplants.
“The TGA and its expert advisory committee, the Adverse Drug Reactions Advisory Committee (ADRAC), have urgently investigated these reports. ADRAC has today recommended the cancellation of the registration of Lumiracoxib due to the severity of the reported side effects associated with this drug,” Dr Hammett said.
“The TGA has taken this advice to cancel the registration of Lumiracoxib in order to prevent further cases of severe liver damage.
“It seems that the longer people are on the medicine, the greater the chance of liver injury. The TGA is, therefore, advising people to stop taking the Lumiracoxib immediately and to discuss alternative treatments with their doctor,” Dr Hammett said.[9]
New Zealand has followed suit with Australia in recalling Prexige.[10]
On October 3, 2007, Health Canada requested sales of Prexige to stop. Novartis has agreed to the request and has taken steps to do so.[11] On December 13, 2007, the European Medicines Agency recommended the withdrawal for Prexige from all EU markets.[12]
On January 17, 2008, the Philippines Department of Health ordered Novartis Healthcare Phils. Inc. (Novartis) to remove (recall) all lumiracoxib from local drug stores in 2 weeks due to the harmful effects of the drug (potential serious liver-related side effects, hepatotoxicity or malfunction of the lungs).[13]
On July 22, 2008, The Brazilian National Health Surveillance Agency ordered the withdrawal of 100 mg formulations of lumiracoxib and suspended marketing of the 400 mg formulation for 90 days,[14] after a three-year safety review found a marked increase in adverse event reports; 35% of lumiracoxib-associated adverse events reported worldwide between July 2005 and April 2008 were found to have occurred in Brazil.[15] Lumiracoxib was definitively withdrawn from the Brazilian market on October 3, 2008.[16]
On November 12, 2008, INVIMA, the Colombian National Institute for Food and Drug Surveillance ordered the withdrawal of all presentations of lumiracoxib (Prexige), due to the international reports on hepatotoxicity.
MECHANISM

Synthesis




References
- Shi, S; Klotz, U (March 2008). “Clinical use and pharmacological properties of selective COX-2 inhibitors.”. European Journal of Clinical Pharmacology 64 (3): 233–52.doi:10.1007/s00228-007-0400-7. PMID 17999057.
- Tacconelli S, Capone ML, Patrignani P (2004). “Clinical pharmacology of novel selective COX-2 inhibitors”. Curr Pharm Des 10 (6): 589–601. doi:10.2174/1381612043453108.PMID 14965322.
- Urgent medicine recall – Lumiracoxib (PREXIGE)
- http://hugin.info/134323/R/1156327/223186.pdf
- Withdrawal of Market Authorization for Prexige
- Media releases
- http://www.fda.gov/ohrms/dockets/ac/05/briefing/2005-4090B1_33_GG-FDA-Tab-U.pdf
- Medicines Regulator cancels registration of anti inflammatory drug, Lumiracoxib, Therapeutic Goods Administration, 11 August 2007. Retrieved on 2007-08-11
- http://www.tga.gov.au/media/2007/070811-lumiracoxib.htm
- “NZ regulators ban arthritis drug”. The New Zealand Herald. 21 August 2007. Retrieved 12 September 2011.
- http://www.novartis.ca/downloads/en/letters/prexige_fact_20071003_e.pdf
- Press release: European Medicines Agency recommends withdrawal of the marketing authorisations for lumiracoxib-containing medicines, 13 December 2007
- Abs-Cbn Interactive, DOH recalls lumiracoxib, sets two-week deadline
- “Anvisa cancela registro do Prexige; consumidor deve substituir medicamento”. Folha de S. Paulo (in Portuguese). July 22, 2008. Retrieved 2008-07-22.
- “Anvisa cancela registro do antiinflamatório Prexige” (Press release) (in Portuguese). Anvisa. July 22, 2008. Retrieved 2008-07-22.
- “Anvisa suspende venda e uso de 2 antiinflamatórios” (in Portuguese). Terra. October 3, 2008. Retrieved 2008-10-03.
External links
- Prexige
- Forbes
- FDA request more information September 23, 2003
- NPS RADAR
- FDA – Background Document for Lumiracoxib 1/13/2005
- http://www.nzherald.co.nz/category/story.cfm?c_id=278&objectid=10459030

ROFECOXIB

ROFECOXIB
MK-966, MK-0966, Vioxx
162011-90-7
Rofecoxib /ˌrɒfɨˈkɒksɪb/ is a nonsteroidal anti-inflammatory drug (NSAID) that has now been withdrawn over safety concerns. It was marketed by Merck & Co. to treat osteoarthritis, acute pain conditions, and dysmenorrhoea. Rofecoxib was approved by the Food and Drug Administration (FDA) on May 20, 1999, and was marketed under the brand names Vioxx, Ceoxx, and Ceeoxx.
 |
|
 |
|
| Systematic (IUPAC) name | |
|---|---|
| 4-(4-methylsulfonylphenyl)-3-phenyl-5H-furan-2-one | |
| Clinical data | |
| Pregnancy cat. | C (AU) |
| Legal status | Prescription Only (S4) (AU)withdrawn |
| Routes | oral |
| Pharmacokinetic data | |
| Bioavailability | 93% |
| Protein binding | 87% |
| Metabolism | hepatic |
| Half-life | 17 hours |
| Excretion | biliary/renal |
| Identifiers | |
| CAS number | 162011-90-7 |
| ATC code | M01AH02 |
| PubChem | CID 5090 |
| DrugBank | DB00533 |
| ChemSpider | 4911 |
| UNII | 0QTW8Z7MCR |
| Chemical data | |
| Formula | C17H14O4S |
| Mol. mass | 314.357 g/mol |
Rofecoxib gained widespread acceptance among physicians treating patients with arthritis and other conditions causing chronic or acute pain. Worldwide, over 80 million people were prescribed rofecoxib at some time.[1]
On September 30, 2004, Merck withdrew rofecoxib from the market because of concerns about increased risk of heart attack and stroke associated with long-term, high-dosage use. Merck withdrew the drug after disclosures that it withheld information about rofecoxib’s risks from doctors and patients for over five years, resulting in between 88,000 and 140,000 cases of serious heart disease.[2] Rofecoxib was one of the most widely used drugs ever to be withdrawn from the market. In the year before withdrawal, Merck had sales revenue of US$2.5 billion from Vioxx.[3] Merck reserved $970 million to pay for its Vioxx-related legal expenses through 2007, and have set aside $4.85bn for legal claims from US citizens.
Rofecoxib was available on prescription in both tablet-form and as an oral suspension. It was available by injection for hospital use.
Mode of action
Cyclooxygenase (COX) has two well-studied isoforms, called COX-1 and COX-2. COX-1 mediates the synthesis of prostaglandinsresponsible for protection of the stomach lining, while COX-2 mediates the synthesis of prostaglandins responsible for pain and inflammation. By creating “selective” NSAIDs that inhibit COX-2, but not COX-1, the same pain relief as traditional NSAIDs is offered, but with greatly reduced risk of fatal or debilitating peptic ulcers. Rofecoxib is a selective COX-2 inhibitor, or “coxib”.
Others include Merck’s etoricoxib (Arcoxia), Pfizer’s celecoxib (Celebrex) and valdecoxib (Bextra). Interestingly, at the time of its withdrawal, rofecoxib was the only coxib with clinical evidence of its superior gastrointestinal adverse effect profile over conventional NSAIDs. This was largely based on the VIGOR (Vioxx GI Outcomes Research) study, which compared the efficacy and adverse effect profiles of rofecoxib and naproxen.[4]
Pharmacokinetics
The therapeutic recommended dosages were 12.5, 25, and 50 mg with an approximate bioavailability of 93%.[5][6][7] Rofecoxib crossed the placenta and blood–brain barrier,[5][6][8]and took 1–3 hours to reach peak plasma concentration with an effective half-life (based on steady-state levels) of approximately 17 hours.[5][7][9] The metabolic products are cis-dihydro and trans-dihydro derivatives of rofecoxib[5][9] which are primarily excreted through urine.
Fabricated efficacy studies
On March 11, 2009, Scott S. Reuben, former chief of acute pain at Baystate Medical Center, Springfield, Mass., revealed that data for 21 studies he had authored for the efficacy of the drug (along with others such as celecoxib) had been fabricated in order to augment the analgesic effects of the drugs. There is no evidence that Reuben colluded with Merck in falsifying his data. Reuben was also a former paid spokesperson for the drug company Pfizer (which owns the intellectual property rights for marketing celecoxib in the United States). The retracted studies were not submitted to either the FDA or the European Union’s regulatory agencies prior to the drug’s approval. Drug manufacturer Merckhad no comment on the disclosure.[10]
Adverse drug reactions

Aside from the reduced incidence of gastric ulceration, rofecoxib exhibits a similar adverse effect profile to other NSAIDs.
Prostaglandin is a large family of lipids. Prostaglandin I2/PGI2/prostacyclin is just one member of it. Prostaglandins other than PGI2 (such as PGE2) also play important roles in vascular tone regulation. Prostacyclin/thromboxane are produced by both COX-1 and COX-2, and rofecoxib suppresses just COX-2 enzyme, so there is no reason to believe that prostacyclin levels are significantly reduced by the drug. And there is no reason to believe that only the balance between quantities of prostacyclin and thromboxane is the determinant factor for vascular tone.[11] Indeed Merck has stated that there was no effect on prostacyclin production in blood vessels in animal testing.[12] Other researchers have speculated that the cardiotoxicity may be associated with maleic anhydride metabolites formed when rofecoxib becomes ionized under physiological conditions. (Reddy & Corey, 2005)
Adverse cardiovascular events
VIGOR study and publishing controversy
The VIGOR (Vioxx GI Outcomes Research) study, conducted by Bombardier, et al., which compared the efficacy and adverse effect profiles of rofecoxib and naproxen, had indicated a significant 4-fold increased risk of acute myocardial infarction (heart attack) in rofecoxib patients when compared with naproxen patients (0.4% vs 0.1%, RR 0.25) over the 12 month span of the study. The elevated risk began during the second month on rofecoxib. There was no significant difference in the mortality from cardiovascular events between the two groups, nor was there any significant difference in the rate of myocardial infarction between the rofecoxib and naproxen treatment groups in patients without high cardiovascular risk. The difference in overall risk was by the patients at higher risk of heart attack, i.e. those meeting the criteria for low-dose aspirin prophylaxis of secondary cardiovascular events (previous myocardial infarction, angina, cerebrovascular accident, transient ischemic attack, or coronary artery bypass).
Merck’s scientists interpreted the finding as a protective effect of naproxen, telling the FDA that the difference in heart attacks “is primarily due to” this protective effect (Targum, 2001). Some commentators have noted that naproxen would have to be three times as effective as aspirin to account for all of the difference (Michaels 2005), and some outside scientists warned Merck that this claim was implausible before VIGOR was published.[13] No evidence has since emerged for such a large cardioprotective effect of naproxen, although a number of studies have found protective effects similar in size to those of aspirin.[14][15] Though Dr. Topol’s 2004 paper criticized Merck’s naproxen hypothesis, he himself co-authored a 2001 JAMA article stating “because of the evidence for an antiplatelet effect of naproxen, it is difficult to assess whether the difference in cardiovascular event rates in VIGOR was due to a benefit from naproxen or to a prothrombotic effect from rofecoxib.” (Mukherjee, Nissen and Topol, 2001.)
The results of the VIGOR study were submitted to the United States Food and Drug Administration (FDA) in February 2001. In September 2001, the FDA sent a warning letter to the CEO of Merck, stating, “Your promotional campaign discounts the fact that in the VIGOR study, patients on Vioxx were observed to have a four to five fold increase in myocardial infarctions (MIs) compared to patients on the comparator non-steroidal anti-inflammatory drug (NSAID), Naprosyn (naproxen).”[16] This led to the introduction, in April 2002, of warnings on Vioxx labeling concerning the increased risk of cardiovascular events (heart attack and stroke).
Months after the preliminary version of VIGOR was published in the New England Journal of Medicine, the journal editors learned that certain data reported to the FDA were not included in the NEJM article. Several years later, when they were shown a Merck memo during the depositions for the first federal Vioxx trial, they realized that these data had been available to the authors months before publication. The editors wrote an editorial accusing the authors of deliberately withholding the data.[17] They released the editorial to the media on December 8, 2005, before giving the authors a chance to respond. NEJM editor Gregory Curfman explained that the quick release was due to the imminent presentation of his deposition testimony, which he feared would be misinterpreted in the media. He had earlier denied any relationship between the timing of the editorial and the trial. Although his testimony was not actually used in the December trial, Curfman had testified well before the publication of the editorial.[18]
The editors charged that “more than four months before the article was published, at least two of its authors were aware of critical data on an array of adverse cardiovascular events that were not included in the VIGOR article.” These additional data included three additional heart attacks, and raised the relative risk of Vioxx from 4.25-fold to 5-fold. All the additional heart attacks occurred in the group at low risk of heart attack (the “aspirin not indicated” group) and the editors noted that the omission “resulted in the misleading conclusion that there was a difference in the risk of myocardial infarction between the aspirin indicated and aspirin not indicated groups.” The relative risk for myocardial infarctions among the aspirin not indicated patients increased from 2.25 to 3 (although it remained statitistically insignificant). The editors also noted a statistically significant (2-fold) increase in risk for serious thromboembolic events for this group, an outcome that Merck had not reported in the NEJM, though it had disclosed that information publicly in March 2000, eight months before publication.[19]
The authors of the study, including the non-Merck authors, responded by claiming that the three additional heart attacks had occurred after the prespecified cutoff date for data collection and thus were appropriately not included. (Utilizing the prespecified cutoff date also meant that an additional stroke in the naproxen population was not reported.) Furthermore, they said that the additional data did not qualitatively change any of the conclusions of the study, and the results of the full analyses were disclosed to the FDA and reflected on the Vioxx warning label. They further noted that all of the data in the “omitted” table were printed in the text of the article. The authors stood by the original article.[20]
NEJM stood by its editorial, noting that the cutoff date was never mentioned in the article, nor did the authors report that the cutoff for cardiovascular adverse events was before that for gastrointestinal adverse events. The different cutoffs increased the reported benefits of Vioxx (reduced stomach problems) relative to the risks (increased heart attacks).[19]
Some scientists have accused the NEJM editorial board of making unfounded accusations.[21][22] Others have applauded the editorial. Renowned research cardiologist Eric Topol,[23] a prominent Merck critic, accused Merck of “manipulation of data” and said “I think now the scientific misconduct trial is really fully backed up”.[24] Phil Fontanarosa, executive editor of the prestigious Journal of the American Medical Association, welcomed the editorial, saying “this is another in the long list of recent examples that have generated real concerns about trust and confidence in industry-sponsored studies”.[25]
On May 15, 2006, the Wall Street Journal reported that a late night email, written by an outside public relations specialist and sent to Journal staffers hours before the Expression of Concern was released, predicted that “the rebuke would divert attention to Merck and induce the media to ignore the New England Journal of Medicine‘s own role in aiding Vioxx sales.”[26]
“Internal emails show the New England Journal’s expression of concern was timed to divert attention from a deposition in which Executive Editor Gregory Curfman made potentially damaging admissions about the journal’s handling of the Vioxx study. In the deposition, part of the Vioxx litigation, Dr. Curfman acknowledged that lax editing might have helped the authors make misleading claims in the article.” The Journal stated that NEJM‘s “ambiguous” language misled reporters into incorrectly believing that Merck had deleted data regarding the three additional heart attacks, rather than a blank table that contained no statistical information; “the New England Journal says it didn’t attempt to have these mistakes corrected.”[26]
Alzheimer’s studies
In 2000 and 2001, Merck conducted several studies of rofecoxib aimed at determining if the drug slowed the onset of Alzheimer’s disease. Merck has placed great emphasis on these studies on the grounds that they are relatively large (almost 3000 patients) and compared rofecoxib to a placebo rather than to another pain reliever. These studies found an elevated death rate among rofecoxib patients, although the deaths were not generally heart-related. However, they did not find any elevated cardiovascular risk due to rofecoxib.[27] Before 2004, Merck cited these studies as providing evidence, contrary to VIGOR, of rofecoxib’s safety.
APPROVe study
In 2001, Merck commenced the APPROVe (Adenomatous Polyp PRevention On Vioxx) study, a three-year trial with the primary aim of evaluating the efficacy of rofecoxib for theprophylaxis of colorectal polyps. Celecoxib had already been approved for this indication, and it was hoped to add this to the indications for rofecoxib as well. An additional aim of the study was to further evaluate the cardiovascular safety of rofecoxib.
The APPROVe study was terminated early when the preliminary data from the study showed an increased relative risk of adverse thrombotic cardiovascular events (includingheart attack and stroke), beginning after 18 months of rofecoxib therapy. In patients taking rofecoxib, versus placebo, the relative risk of these events was 1.92 (rofecoxib 1.50 events vs placebo 0.78 events per 100 patient years). The results from the first 18 months of the APPROVe study did not show an increased relative risk of adverse cardiovascular events. Moreover, overall and cardiovascular mortality rates were similar between the rofecoxib and placebo populations.[28]
In summary, the APPROVe study suggested that long-term use of rofecoxib resulted in nearly twice the risk of suffering a heart attack or stroke compared to patients receiving a placebo.
Other studies
Pre-approval Phase III clinical trials, like the APPROVe study, showed no increased relative risk of adverse cardiovascular events for the first eighteen months of rofecoxib usage (Merck, 2004). Others have pointed out that “study 090,” a pre-approval trial, showed a 3-fold increase in cardiovascular events compared to placebo, a 7-fold increase compared to nabumetone (another [NSAID]), and an 8-fold increase in heart attacks and strokes combined compared to both control groups.[29][30] Although this was a relatively small study and only the last result was statistically significant, critics have charged that this early finding should have prompted Merck to quickly conduct larger studies of rofecoxib’s cardiovascular safety. Merck notes that it had already begun VIGOR at the time Study 090 was completed. Although VIGOR was primarily designed to demonstrate new uses for rofecoxib, it also collected data on adverse cardiovascular outcomes.
Several very large observational studies have also found elevated risk of heart attack from rofecoxib. For example, a recent retrospective study of 113,000 elderly Canadians suggested a borderline statistically significant increased relative risk of heart attacks of 1.24 from Vioxx usage, with a relative risk of 1.73 for higher-dose Vioxx usage. (Levesque, 2005). Another study, using Kaiser Permanente data, found a 1.47 relative risk for low-dose Vioxx usage and 3.58 for high-dose Vioxx usage compared to current use of celecoxib, though the smaller number was not statistically significant, and relative risk compared to other populations was not statistically significant. (Graham, 2005).
Furthermore, a more recent meta-study of 114 randomized trials with a total of 116,000+ participants, published in JAMA, showed that Vioxx uniquely increased risk of renal (kidney) disease, and heart arrhythmia.[31]
Other COX-2 inhibitors
Any increased risk of renal and arrhythmia pathologies associated with the class of COX-2 inhibitors, e.g. celecoxib (Celebrex), valdecoxib (Bextra), parecoxib (Dynastat),lumiracoxib, and etoricoxib is not evident,[31] although smaller studies[32][33] had demonstrated such effects earlier with the use of celecoxib, valdecoxib and parecoxib.
Nevertheless, it is likely that trials of newer drugs in the category will be extended in order to supply additional evidence of cardiovascular safety. Examples are some more specific COX-2 inhibitors, including etoricoxib (Arcoxia) and lumiracoxib (Prexige), which are currently (circa 2005) undergoing Phase III/IV clinical trials.
Besides, regulatory authorities worldwide now require warnings about cardiovascular risk of COX-2 inhibitors still on the market. For example, in 2005, EU regulators required the following changes to the product information and/or packaging of all COX-2 inhibitors:[34]
- Contraindications stating that COX-2 inhibitors must not be used in patients with established ischaemic heart disease and/or cerebrovascular disease (stroke), and also in patients with peripheral arterial disease
- Reinforced warnings to healthcare professionals to exercise caution when prescribing COX-2 inhibitors to patients with risk factors for heart disease, such as hypertension, hyperlipidaemia (high cholesterol levels), diabetes and smoking
- Given the association between cardiovascular risk and exposure to COX-2 inhibitors, doctors are advised to use the lowest effective dose for the shortest possible duration of treatment
Other NSAIDs
Since the withdrawal of Vioxx it has come to light that there may be negative cardiovascular effects with not only other COX-2 inhibitiors, but even the majority of other NSAIDs. It is only with the recent development of drugs like Vioxx that drug companies have carried out the kind of well executed trials that could establish such effects and these sort of trials have never been carried out in older “trusted” NSAIDs such as ibuprofen, diclofenac and others. The possible exceptions may be aspirin and naproxen due to their anti-platelet aggregation properties.
Withdrawal
Due to the findings of its own APPROVe study, Merck publicly announced its voluntary withdrawal of the drug from the market worldwide on September 30, 2004.[35]
In addition to its own studies, on September 23, 2004 Merck apparently received information about new research by the FDA that supported previous findings of increased risk of heart attack among rofecoxib users (Grassley, 2004). FDA analysts estimated that Vioxx caused between 88,000 and 139,000 heart attacks, 30 to 40 percent of which were probably fatal, in the five years the drug was on the market.[36]
On November 5, the medical journal The Lancet published a meta-analysis of the available studies on the safety of rofecoxib (Jüni et al., 2004). The authors concluded that, owing to the known cardiovascular risk, rofecoxib should have been withdrawn several years earlier. The Lancet published an editorial which condemned both Merck and the FDA for the continued availability of rofecoxib from 2000 until the recall. Merck responded by issuing a rebuttal of the Jüni et al. meta-analysis that noted that Jüni omitted several studies that showed no increased cardiovascular risk. (Merck & Co., 2004).
In 2005, advisory panels in both the U.S. and Canada encouraged the return of rofecoxib to the market, stating that rofecoxib’s benefits outweighed the risks for some patients. The FDA advisory panel voted 17-15 to allow the drug to return to the market despite being found to increase heart risk. The vote in Canada was 12-1, and the Canadian panel noted that the cardiovascular risks from rofecoxib seemed to be no worse than those from ibuprofen—though the panel recommended that further study was needed for all NSAIDs to fully understand their risk profiles. Notwithstanding these recommendations, Merck has not returned rofecoxib to the market.[37]
In 2005, Merck retained Debevoise & Plimpton LLP to investigate Vioxx study results and communications conducted by Merck. Through the report, it was found that Merck’s senior management acted in good faith, and that the confusion over the clinical safety of Vioxx was due to the sales team’s overzealous behavior. The report that was filed gave a timeline of the events surrounding Vioxx and showed that Merck intended to operate honestly throughout the process. Any mistakes that were made regarding the mishandling of clinical trial results and withholding of information was the result of oversight, not malicious behavior. The Martin Report did conclude that the Merck’s marketing team exaggerated the safety of Vioxx and replaced truthful information with sales tactics.[citation needed] The report was published in February 2006, and Merck was satisfied with the findings of the report and promised to consider the recommendations contained in the Martin Report. Advisers to the US Food and Drug Administration (FDA) have voted, by a narrow margin, that it should not ban Vioxx — the painkiller withdrawn by drug-maker Merck.
They also said that Pfizer’s Celebrex and Bextra, two other members of the family of painkillers known as COX-2 inhibitors, should remain available, despite the fact that they too boost patients’ risk of heart attack and stroke. url = http://www.nature.com/drugdisc/news/articles/433790b.html The recommendations of the arthritis and drug safety advisory panel offer some measure of relief to the pharmaceutical industry, which has faced a barrage of criticism for its promotion of the painkillers. But the advice of the panel, which met near Washington DC over 16–18 February, comes with several strings attached.
For example, most panel members said that manufacturers should be required to add a prominent warning about the drugs’ risks to their labels; to stop direct-to-consumer advertising of the drugs; and to include detailed, written risk information with each prescription. The panel also unanimously stated that all three painkillers “significantly increase the risk of cardiovascular events”.
The panel voted 17 to 15 against banning Vioxx (rofecoxib) entirely; the vote on Bextra (valdecoxib) was 17 to 13 with 2 abstentions; Celebrex (celecoxib) was endorsed 31 to 1. Shares of Merck, based in Whitehouse Station, New Jersey, and New York-based Pfizer closed up 13% and 7% respectively on 18 February, 2013, the day of the votes.
The FDA is expected to act on the recommendations within weeks. Although the agency usually follows the recommendations of its outside advisers, it is not bound to do so. A top official said that, in light of the closeness of some of the votes, the agency will examine the panel members’ comments in detail before deciding what to do.
An official from Merck said during the meeting that it would consider reintroducing Vioxx, which it withdrew in September 2004. On April 7, 2005, Pfizer withdrew Bextra from the U.S. market on recommendation by the FDA. Pfizer’s other painkiller, Celebrex, is still on the market.
Litigation
As of March 2006, there had been over 10,000 cases and 190 class actions filed against Merck[citation needed] over adverse cardiovascular events associated with rofecoxib and the adequacy of Merck’s warnings. The first wrongful death trial, Rogers v. Merck, was scheduled in Alabama in the spring of 2005, but was postponed after Merck argued that the plaintiff had falsified evidence of rofecoxib use.[1]
On August 19, 2005, a jury in Texas voted 10-2 to hold Merck liable for the death of Robert Ernst, a 59-year-old man who allegedly died of a rofecoxib-induced heart attack. The plaintiffs’ lead attorney was Mark Lanier. Merck argued that the death was due to cardiac arrhythmia, which had not been shown to be associated with rofecoxib use. The jury awarded Carol Ernst, widow of Robert Ernst, $253.4 million in damages. This award will almost certainly be capped at no more than US$26.1 million because of punitive damages limits under Texas law.[2] As of March 2006, the plaintiff had yet to ask the court to enter a judgment on the verdict; Merck has stated that it will appeal.
On November 3, 2005, Merck won the second case Humeston v. Merck, a personal injury case, in Atlantic City, New Jersey. The plaintiff experienced a mild myocardial infarction and claimed that rofecoxib was responsible, after having taken it for two months. Merck argued that there was no evidence that rofecoxib was the cause of Humeston’s injury and that there is no scientific evidence linking rofecoxib to cardiac events with short durations of use. The jury ruled that Merck had adequately warned doctors and patients of the drug’s risk.[3]
The first federal trial on rofecoxib, Plunkett v. Merck, began on November 29, 2005 in Houston. The trial ended in a hung jury and a mistrial was declared on December 12, 2005. According to the Wall Street Journal, the jury hung by an eight to one majority, favoring the defense. Upon retrial in February 2006 in New Orleans, where the Vioxx multidistrict litigation (MDL) is based, a jury found Merck not liable, even though the plaintiffs had the NEJM editor testify as to his objections to the VIGOR study.
On January 30, 2006, a New Jersey state court dismissed a case brought by Edgar Lee Boyd, who blamed Vioxx for gastrointestinal bleeding that he experienced after taking the drug. The judge said that Boyd failed to prove the drug caused his stomach pain and internal bleeding.
In January 2006, Garza v. Merck began trial in Rio Grande City, Texas. The plaintiff, a 71-year-old smoker with heart disease, had a fatal heart attack three weeks after finishing a one-week sample of rofecoxib. On April 21, 2006 the jury awarded the plaintiff $7 million compensatory and $25 million punitive. The Texas state court of appeals in San Antonio later rules Garza’s fatal heart attack probably resulted from pre-existing health conditions unrelated to his taking of Vioxx, thus reversing the $32 million jury award.[4]
On April 5, 2006, the jury held Merck liable for the heart attack of 77-year-old John McDarby, and awarded Mr McDarby $4.5 million in compensatory damages based on Merck’s failure to properly warn of Vioxx safety risks. After a hearing on April 11, 2006, the jury also awarded Mr McDarby an additional $9 million in punitive damages. The same jury found Merck not liable for the heart attack of 60-year-old Thomas Cona, a second plaintiff in the trial, but was liable for fraud in the sale of the drug to Cona.
Merck has reserved $970 million to pay for its Vioxx-related legal expenses through 2007, and have set aside $4.85bn for legal claims from US citizens. Patients who claim to have suffered as a result of taking Vioxx in countries outside the US are campaigning for this to be extended.
In March 2010, an Australian class-action lawsuit against Merck ruled that Vioxx doubled the risk of heart attacks, and that Merck had breached the Trade Practices Act by selling a drug which was unfit for sale.[38]
In November 2011, Merck announced a civil settlement with the US Attorney’s Office for the District of Massachusetts, and individually with 43 US states and the District of Columbia, to resolve civil claims relating to Vioxx.[5] Under the terms of the settlement, Merck agreed to pay two-thirds of a previously recorded $950 million reserve charge in exchange for release from civil liability. Litigation with seven additional states remains outstanding. Under separate criminal proceedings, Merck plead guilty to a federal misdemeanor charge relating to the marketing of the drug across state lines, incurring a fine of $321.6 million.[6]
Other effects
Rofecoxib was shown to improve premenstrual acne vulgaris in a placebo controlled study.[39]
Synthesis

Rofecoxib synthesis.[40]
,,,,,,,,,,,,,,,,,
The oxidation of 4- (methylsulfanyl) acetophenone (X) with monoperoxyphthalic acid (MMPP) in dichloro-methane / methanol gives the corresponding sulfone (XI), which is brominated with Br2 / AlCl3 in chloroform, yielding the expected phenacyl bromide ( XII). Finally, this compound is cyclocondensed with phenylacetic acid (I) by means of 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU) and triethylamine in acetonitrile. 5) Reaction of [4- (methylsulfonyl ) phenyl] phenylacetyl-ene (XIII) with CO catalyzed by Rh4 (CO) 12 in THF at 100 C in a stainless steel autoclave at 100 Atm pressure, followed by a chromatographic separation in a silicagel column to eliminate the undesired regioisomer.

……………….
The synthesis of rofecoxib can be performed by several different ways: 1) The condensation of phenylacetic acid (I) with ethyl bromoacetate (II) by means of triethylamine in THF yields 2- (phenylacetoxy) acetic acid ethyl ester (III), which is cyclized to the hydroxyfuranone (IV) by means of potassium tert-butoxide in tert-butanol. The reaction of (IV) with triflic anhydride and diisopropylethylamine in dichloro-methane affords the corresponding triflate (V), which by reaction with LiBr in hot acetone yields the bromofuranone (VI) The condensation of (VI) with 4- (methylsulfanyl) phenylboronic acid (VII) by means of Na2CO3 and Pd (Ph3P) 4 in hot toluene gives 4- [4- (methylsulfanyl) -phenyl]. – 3-phenylfuran-2 (5H) -one (VIII), which is finally oxidized with 2KHSO5.KHSO4.K2SO4 (oxone). 2) The intermediate (VIII) can also be obtained by condensation of triflate (V) with boronic acid ( VII) by means of Na2CO3 and Pd (Ph3P) 4 in hot toluene. 3) The intermediate (VIII) can also be synthesized by the reaction of triflate (V) with tetramethylammonium chloride, giving the chlorofuranone (IX), which is then condensed with boronic acid (VII) as before.

Footnotes
- Jump up^ http://www.npr.org/templates/story/story.php?storyId=4054991
- Jump up^ “Up to 140,000 heart attacks linked to Vioxx.”. New Scientist. 2005-01-25. p. 1.
- Jump up^ “Merck Sees Slightly Higher 2007 Earnings”. New York Times. Reuters. 2006-12-07. p. A1.
- Jump up^ Bombardier, C.; Laine, L.; Reicin, A.; Shapiro, D.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M. B.; Hawkey, C. J.; Hochberg, M. C.; Kvien, T. K.; Schnitzer, T. J.; Vigor Study, G. (2000). “Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis”. New England Journal of Medicine 343 (21): 1520–1528, 2 1528 following 1528. doi:10.1056/NEJM200011233432103. PMID 11087881. edit
- ^ Jump up to:a b c d Merck & Co. VIOXX (rofecoxib tablets and oral suspension). Accessed at: http://www.merck.com/product/usa/pi_circulars/v/vioxx/vioxx_pi.pdf 01 Feb 2010
- ^ Jump up to:a b Gold Standard Inc. Rofecoxib Vioxx Accessed at: http://www.mdconsult.com/das/pharm/body/181267313-3/946823742/full/2399 01 Feb 2010
- ^ Jump up to:a b Davies, N. M.; Teng, X. W.; Skjodt, N. M. (2003). “Pharmacokinetics of rofecoxib: a specific cyclo-oxygenase-2 inhibitor”. Clinical pharmacokinetics 42 (6): 545–556.PMID 12793839. edit
- Jump up^ Padi, S.; Kulkarni, S. (2004). “Differential effects of naproxen and rofecoxib on the development of hypersensitivity following nerve injury in rats”. Pharmacology, Biochemistry, and Behavior 79 (2): 349–358. doi:10.1016/j.pbb.2004.08.005. PMID 15501312. edit
- ^ Jump up to:a b Scott, L. J.; Lamb, H. M. (1999). “Rofecoxib”. Drugs 58 (3): 499–505; discussion 506–7. doi:10.2165/00003495-199958030-00016. PMID 10493277. edit
- Jump up^ Winstein, Keith J. (March 11, 2009). “Top Pain Scientist Fabricated Data in Studies, Hospital Says”. The Wall Street Journal.
- Jump up^ Vane, J.; Bakhle, Y.; Botting, R. (1998). “Cyclooxygenases 1 and 2”. Annual review of pharmacology and toxicology 38: 97–120. doi:10.1146/annurev.pharmtox.38.1.97.PMID 9597150. edit
- Jump up^ sfgate.com
- Jump up^ www.saferdrugsnow.org
- Jump up^ Karha, J.; Topol, E. J. (2004). “The sad story of Vioxx, and what we should learn from it”. Cleveland Clinic journal of medicine 71 (12): 933–934, 936, 934–9.doi:10.3949/ccjm.71.12.933. PMID 15641522. edit
- Jump up^ Solomon, D. H.; Glynn, R. J.; Levin, R.; Avorn, J. (2002). “Nonsteroidal anti-inflammatory drug use and acute myocardial infarction”. Archives of Internal Medicine 162 (10): 1099–1104.doi:10.1001/archinte.162.10.1099. PMID 12020178. edit
- Jump up^http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/EnforcementActivitiesbyFDA/WarningLettersandNoticeofViolationLetterstoPharmaceuticalCompanies/UCM166383.pdf
- Jump up^ Curfman, G.; Morrissey, S.; Drazen, J. (2005). “Expression of concern: Bombardier et al., “Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis,” N Engl J Med 2000;343:1520-8″. The New England Journal of Medicine 353 (26): 2813–2814. doi:10.1056/NEJMe058314. PMID 16339408. edit
- Jump up^ http://www.forbes.com/work/feeds/ap/2006/02/13/ap2523250.html. Missing or empty
|title=(help)[dead link] - ^ Jump up to:a b Curfman, G.; Morrissey, S.; Drazen, J. (2006). “Expression of concern reaffirmed”. The New England Journal of Medicine 354 (11): 1193. doi:10.1056/NEJMe068054.PMID 16495386. edit
- Jump up^ Bombardier, C.; Laine, L.; Burgos-Vargas, R.; Davis, B.; Day, R.; Ferraz, M.; Hawkey, C.; Hochberg, M.; Kvien, T.; Schnitzer, T. J.; Weaver, A. (2006). “Response to expression of concern regarding VIGOR study”. The New England Journal of Medicine 354 (11): 1196–1199. doi:10.1056/NEJMc066096. PMID 16495387. edit
- Jump up^ http://pipeline.corante.com/archives/2006/02/22/nejm_vs_its_contributors_round_two.php
- Jump up^ http://dimer.tamu.edu/simplog/archive.php?blogid=3&pid=3293
- Jump up^ http://genetics.case.edu/faculty2.php?fac=ejt9
- Jump up^ http://www.medicinenet.com/script/main/art.asp?articlekey=56384&page=2
- Jump up^ http://www.beasleyallen.com/news/vioxx-plaintiffs-seek-mistrial-after-allegation-on-merck-study/
- ^ Jump up to:a b David Armstrong (2006-05-15). “How the New England Journal Missed Warning Signs on Vioxx”. Wall Street Journal. p. A1.
- Jump up^ Konstam, M. A.; Weir, M. R.; Reicin, A.; Shapiro, D.; Sperling, R. S.; Barr, E.; Gertz, B. J. (2001). “Cardiovascular thrombotic events in controlled, clinical trials of rofecoxib”. Circulation104 (19): 2280–2288. doi:10.1161/hc4401.100078. PMID 11696466. edit
- Jump up^ Bresalier, R.; Sandler, R.; Quan, H.; Bolognese, J.; Oxenius, B.; Horgan, K.; Lines, C.; Riddell, R.; Morton, D.; Lanas, A.; Konstam, M. A.; Baron, J. A.; Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators (2005). “Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial”. The New England Journal of Medicine 352(11): 1092–1102. doi:10.1056/NEJMoa050493. PMID 15713943. edit
- Jump up^ http://www.fda.gov/ohrms/dockets/ac/01/briefing/3677b2_06_cardio.pdf
- Jump up^ Wolfe, M. M. (2004). “Rofecoxib, Merck, and the FDA”. The New England Journal of Medicine 351 (27): 2875–2878; author 2878 2875–2878. doi:10.1056/NEJM200412303512719.PMID 15625749. edit
- ^ Jump up to:a b Zhang, J.; Ding, E.; Song, Y. (2006). “Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: meta-analysis of randomized trials”. Journal of the American Medical Association 296 (13): 1619–1632. doi:10.1001/jama.296.13.jrv60015. PMID 16968832. edit
- Jump up^ Solomon, S.; McMurray, J.; Pfeffer, M.; Wittes, J.; Fowler, R.; Finn, P.; Anderson, W.; Zauber, A.; Hawk, E.; Bertagnolli, M.; Adenoma Prevention with Celecoxib (APC) Study Investigators (2005). “Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention”. The New England Journal of Medicine 352 (11): 1071–1080.doi:10.1056/NEJMoa050405. PMID 15713944. edit
- Jump up^ Nussmeier, N.; Whelton, A.; Brown, M.; Langford, R.; Hoeft, A.; Parlow, J.; Boyce, S.; Verburg, K. (2005). “Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery”. The New England Journal of Medicine 352 (11): 1081–1091. doi:10.1056/NEJMoa050330. PMID 15713945. edit
- Jump up^ “European Medicines Agency concludes action on COX-2 inhibitors” (pdf). European Medicines Agency. Retrieved 2008-04-16.
- Jump up^ “Merck Announces Voluntary Worldwide Withdrawal of VIOXX” (pdf). Retrieved 2008-04-16.
- Jump up^ “Congress Questions Vioxx, FDA”. PBS NewsHour. 2004-11-18. Retrieved 2013-06-03.
- Jump up^ “SUMMARY: Report of the Expert Advisory Panel on the Safety of Cox-2 Selective Non-steroidal Anti-Inflammatory Drugs (NSAIDs)”. Health Canada. 2005-07-06. Retrieved 2011-06-04.
- Jump up^ Drug unfit for sale, says judge in compo case The Age, March 6, 2010
- Jump up^ http://bioline.utsc.utoronto.ca/archive/00002693/01/dv04120.pdf#search=%22acne%20rofecoxib%22
- Jump up^ http://vioxxlawyer.org/rofecoxib-synthesis/
References
- FDA (2005). “Summary minutes for the February 16, 17 and 18, 2005, Joint meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee.” Published on the internet, March 2005. Link
- Fitzgerald GA, Coxibs and Cardiovascular Disease, N Engl J Med 2004;351(17): 1709–1711. PMID 15470192.
- Grassley CE (15 Oct 2004). Grassley questions Merck about communication with the FDA on Vioxx. Press Release.
- Jüni P, Nartey L, Reichenbach S, Sterchi R, Dieppe PA, Egger M (2004). Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet (published online; see also Merck response below)
- Karha J and Topol EJ. The sad story of Vioxx, and what we should learn from it Cleve Clin J Med 2004; 71(12):933-939. PMID 15641522
- Michaels, D. (June 2005) DOUBT Is Their Product, Scientific American, 292 (6).
- Merck & Co., (5 Nov 2004). Response to Article by Juni et al. Published in The Lancet on Nov. 5. Press Release.
- Merck & Co (30 Sep 2004) Merck Announces Voluntary Worldwide Withdrawal of VIOXX. Press release [7].
- D. M. Mukherjee, S. E. Nissen, and E. J. Topol, “Risk of Cardiovascular Events Associated with Selective COX-2 Inhibitors,” Journal of the American Medical Association 186 (2001): 954–959.
- Nussmeier NA, Whelton AA, Brown MT, Langford RM, Hoeft A, Parlow JL, et al. Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N Engl J Med 2005;352(11):1081-91. PMID 15713945
- Okie, S (2005) “Raising the safety bar–the FDA’s coxib meeting.” N Engl J Med. 2005 Mar 31;352(13):1283-5. PMID 15800221.
- Leleti Rajender Reddy, Corey EJ. Facile air oxidation of the conjugate base of rofecoxib (Vioxx), a possible contributor to chronic human toxicity Tetrahedron Lett 2005, 46: 927. doi:10.1016/j.tetlet.2004.12.055
- Swan SK et al., Effect of Cyclooxygenase-2 Inhibition on Renal Function in Elderly Persons Receiving a Low-Salt Diet. Annals of Int Med 2000; 133:1–9
- Targum, SL. (1 Feb. 2001) Review of cardiovascular safety database. FDA memorandum. [8]
- Wolfe, MM et al., Gastrointestinal Toxicity of Nonsteroidal Anti-anflamattory Drugs, New England Journal of Medicine. 1999; 340; 1888-98.
External links
- National Public Radio 2004 Q&A on the case, following withdrawal announcement
- Court TV’s full coverage of the Vioxx civil trials
- Merck website on Vioxx litigation
- FDA Public Health Advisory on Vioxx
- David Michaels. Doubt is Their Product Scientific American, June 2004, p. 96-101
- JURIST, Much Pain, Much Gain: Skeptical Ruminations on the Vioxx Litigation
- Ted Frank, American Enterprise Institute, The Vioxx Litigation, Part I and Part II, December 2005
- briandeer.com – Vioxx: the UK connection
- Campaign for compensation for Vioxx victims outside the US
IMPs: How do GDP Guidelines apply?
Is distribution of Investigational Medicinal Products covered by the new Guidelines on Good Distribution Practice (GDP)? What needs to be considered can be found here.
GMP News: IMPs: How do GDP Guidelines apply?
The 2013 Guidelines on Good Distribution Practice (2013/C 343/01) apply to medicinal products for human use. Investigational Medicinal Products (IMPs) are also medicinal products for human use. But is IMP distribution really covered by the new Guidelines? The Guidelines focus on wholesale distribution of medicinal products. And IMPs are normally not distributed via wholesalers. However IMPs are not particularly excluded. The Guideline may therefore give some guidance on how to supply clinical trial material. Better guidance might be given by the Questions and Answers documents of the European Medicines Agency (EMA). In the part on supplementary requirements, Annex 13, a few Q&As are dealing with storage and transportation of IMPs.
When it comes to transport of IMPs from the manufacturer to the distributor or investigator sites, the sponsor is responsible for controlling the distribution chain and assuring “that IMPs are stored, transported, and handled in a suitable manner”. The responsibility for storage and transportation lies with the manufacturer or an importer, when the IMP comes from outside the EU. To define the specific responsibilities of the parties involved, a contract should be in place.
During storage and transportation, conditions should at least be monitored. The sponsor should define the applicable storage (and transport) conditions for the IMPs. When the IMP arrives at the investigator site, IMPs should be stored in a restricted area where appropriate, with ongoing monitoring. Everything should be defined in SOPs.
-
Definition of Investigational Medicinal Products (IMPs …
ec.europa.eu/health/files/pharmacos/…/def_imp_2006_07_27_en.pdfDefinition of Non Investigational Medicinal Products. (NIMPs). To be included in. The rules governing medicinal products in the European Union. Volume 10.
-
[PDF]
Non Investigational Medicinal Products – European …
ec.europa.eu/health/files/eudralex/vol-10/imp_03-2011.pdfMar 18, 2011 – Guidance on investigational medicinal products (IMPs) and othermedicinal products used in clinical trials (2007). Description of changes:.
-
Clinical Trials Toolkit | Investigational Medicinal Product (IMP)
http://www.ct-toolkit.ac.uk/glossary/investigational–medicinal–product-impInvestigational Medicinal Product (IMP). A pharmaceutical form of an active substance or placebo being tested, or to be tested, or used, or to be used, as a …
-
[PDF]
Guidance on Investigational Medicinal Products (IMPs) and …
Guidance on Investigational Medicinal Products (IMPs) and other medicinal products used in Clinical Trials. To be included in. The rules governing medicinal …
-
Good Manufacturing Practice: Investigational medicinal …
May 23, 2013 – My investigational medicinal product (IMP) unit is engaged upon reconstituting sterile injections and then giving them to the clinical trial …
-
Clinical trials for medicines: Is a clinical trial authorisation …
Apr 2, 2013 – Is the product an investigational medicinal product (IMP) or a non-investigational medicinal product (NIMP)? Clinical trials in the UK are …
-
Is my study a Clinical Trial of an Investigational Medicinal …
Clinical Trials of Investigational Medicinal Products (CTIMPs) must also adhere to the European Clinical Trials Directive (2001/20/EC) , the Medicines for Human …
-
Investigational Medicinal Products IMP – Clinical Trials Finland
Based on Fimea Regulation 1/2007 ann 2/2012 (2012 only in FInnish) Clinical Trials onMedicinal Products in Human Subjects, an investigational medicinal …
-
Clinical trials of investigational medicinal products (CTIMPs)
Under the EU Clinical Trials Directive 2001/20/EC and the Medicines for Human Use (Clinical Trials) Regulations 2004, which transposed the Directive into UK …
-
Investigational Medicinal Product Dossier (IMPD)
The Investigational Medicinal Product Dossier is the basis for approval of clinical trials by the competent authorities in the EU. The Clinical Trials Directive …
Draft USP Chapter 1223 Validation of Alternative Microbiological Methods published

The USP published the draft of the revised chapter 1223 “Validation of Alternative Microbiologiocal Methods.” Read more.
GMP News: Draft USP Chapter 1223 Validation of Alternative Microbiological Methods published
With the GMP News from 21. November 2012 we informed you about the status of the revision of USP chapter 1223, Validation of Alternative Microbiological Methods. Now the USP published a draft of the document.
The draft provides guidance with regard to the selection and implementation of appropriate methods, i.e. important steps for the evaluation of possible methods, for the selection of the analytical technology and finally for the qualification with regard to a current product. In this context the chapter includes information about demonstration that the new method is comparable to the compendial method, and about applicability as a replacement for an existing method. Furthermore the document provides information about the qualification of a method in the laboratory.
The public can provide comments on the draft chapter 1223 until September 30, 2014. The draft chapter can be found at USP website. You are required to register to gain access to the USP PF online, but this is a free service to the pharmaceutical community.
-
Validation of Alternative Microbiology Methods for Product …
www.microbiologynetwork.com/…/PharmTech_2005_Validation-of-Alt…by S Sutton – 2005 – Cited by 12 – Related articleshe validation of alternative microbiological methods for pharmaceutical testing is a very confusing topic, the more so as people use phrases such as “alternate …
-
<1223> validation of alternative microbiological methods
The purpose of this chapter is to provide guidance for validating methods for use asalternatives to the official compendial microbiological methods. For microbial …
-
[PDF]
Alternative methods for control of microbiological quality …
Specific validation recommendations …. The role of rapid microbiological methodswithin the process analytical … “Validation studies of alternate microbiological.
-
[PDF]
Method Validation of U.S. EPA Microbiological Methods of …
http://www.epa.gov/fem/…/final_microbiology_method_guidance_110409.pd…by S Parshionikar – 2009 – Cited by 1 – Related articles1.0 Alternate Method Approval Processes. 1.1 Alternate Test … guidance for thevalidation of microbiological methods likely to be used in future EPA methods.
-
Validation of Alternative Methods for the Analysis of Drinking …
by A Boubetra – 2011 – Cited by 10 – Related articlesIn this respect, the ISO 16140 standard (ISO, ISO 16140. Microbiology of Food and Animal Feeding Stuffs—Protocol for the Validation of Alternative Methods, …
-
[PDF]
Protocol for the validation of alternative microbiological …
NordVal c/o National Veterinary Institute. P.O.Box 750 Sentrum. N–0106 Oslo. Norway. Protocol for the validation of alternative microbiological methods.
-
Alternative Microbiological Methods | U.S. Pharmacopeial …
This workshop will focus on new and revised information on the validation of alternative microbiological methods, validation of alternative methods to antibiotic …
-
[PDF]
Guidelines for the Validation of Analytical Methods for the …
Sep 8, 2011 – be fulfilled in the evaluation of microbiological methods to be used in our …… Validation of an Alternative method: Demonstration that adequate …
-
[PPT]
A Regulatory View of Rapid Microbiology Methods – Food …
Rapid Microbiology Methods … Traditional Methods (plate counts, mpn) … USP Draft Chapter <1223> – Validation of Alternative Microbiological Methods.
-
usp 1223 validation of alternative microbiological methods
wenku.baidu.com/view/6db6520502020740be1e9b9d1223 VALIDATION OF ALTERNATIVE MICROBIOLOGICAL METHODSINTRODUCTION The purpose of this chapter is to provide guidance for validating …
-
PDA Microbiology Conference Update: Revision of USP 1223
blog.rapidmicromethods.com/2012/…/usp-chapter–1223-validation-of.ht…Oct 23, 2012 – USP Chapter 1223, Validation of Alternative Microbiological Methods, has been under revision for at least the past year. During today’s …
-
GMP News: Revision of USP 1223 – current Status
GMP News 21/11/2012. Revision of USP 1223 – current Status. The chapter <1223> Validation of Alternative Microbiological Methods of the USP is still under …
-
GMP News: Draft USP Chapter 1223 Validation of …
http://www.gmp-compliance.org/enews_4397_Draft-USP-Chapter–1223-Valid…17 hours ago – November 2012 we informed you about the status of the revision of USPchapter 1223, Validation of Alternative Microbiological Methods.
 1223
1223 VALIDATION OF ALTERNATIVE MICROBIOLOGICAL METHODS
VALIDATION OF ALTERNATIVE MICROBIOLOGICAL METHODS
INTRODUCTIONThe purpose of this chapter is to provide guidance for validating methods for use as alternatives to the official compendial microbiological methods. For microbial recovery and identification, microbiological testing laboratories sometimes use alternative test methods to those described in the general chapters for a variety of reasons, such as economics, throughput, and convenience. Validation of these methods is required. Some guidance on validation of the use of alternate methods is provided in the Tests and Assays section in the General Notices and Requirements. This section also notes that in the event of a dispute, only the result obtained by the compendial test is conclusive.Validation studies of alternate microbiological methods should take a large degree of variability into account. When conducting microbiological testing by conventional plate count, for example, one frequently encounters a range of results that is broader (%RSD 15 to 35) than ranges in commonly used chemical assays (%RSD 1 to 3). Many conventional microbiological methods are subject to sampling error, dilution error, plating error, incubation error, and operator error.Validation of Compendial Procedures1225 defines characteristics such as accuracy, precision, specificity, detection limit, quantification limit, linearity, range, ruggedness, and robustness in their application to analytical methods. These definitions are less appropriate for alternate microbiological method validation as “at least equivalent to the compendial method” given the comparative nature of the question (see the Tests and Assays—Procedures section in General Notices and Requirements). The critical question is whether or not the alternate method will yield results equivalent to, or better than, the results generated by the conventional method.Other industry organizations have provided guidance for the validation of alternate microbiological methods.* The suitability of a new or modified method should be demonstrated in a comparison study between the USP compendial method and the alternate method. The characteristics defined in this chapter may be used to establish this comparison.
TYPES OF MICROBIOLOGICAL TESTSIt is critical to the validation effort to identify the portion of the test addressed by an alternate technology. For example, there is a variety of technologies available to detect the presence of viable cells. These techniques may have application in a variety of tests (e.g., bioburden, sterility test) but may not, in fact, replace the critical aspects of the test entirely. For example, a sterility test by membrane filtration may be performed according to the compendial procedure up to the point of combining the processed filter with the recovery media, and after that the presence of viable cells might then be demonstrated by use of some of the available technologies. Validation of this application would, therefore, require validation of the recovery system employed rather than the entire test.There are three major types of determinations specific to microbiological tests. These include tests to determine whether microorganisms are present in a sample, tests to quantify the number of microorganisms (or to enumerate a specific subpopulation of the sample), and tests designed to identify microorganisms. This chapter does not address microbial identification.Qualitative Tests for the Presence or Absence of MicroorganismsThis type of test is characterized by the use of turbidity in a liquid growth medium as evidence of the presence of viable microorganisms in the test sample. The most common example of this test is the sterility test. Other examples of this type of testing are those tests designed to evaluate the presence or absence of a particular type of microorganism in a sample (e.g., coliforms in potable water and E. coli in oral dosage forms).Quantitative Tests for MicroorganismsThe plate count method is the most common example of this class of tests used to estimate the number of viable microorganisms present in a sample. The membrane filtration and Most Probable Number (MPN) multiple-tube methods are other examples of these tests. The latter was developed as a means to estimate the number of viable microorganisms present in a sample not amenable to direct plating or membrane filtration.General ConcernsValidation of a microbiological method is the process by which it is experimentally established that the performance characteristics of the method meet the requirements for the intended application, in comparison to the traditional method. For example, it may not be necessary to fully validate the equivalence of a new quantitative method for use in the antimicrobial efficacy test by comparative studies, as the critical comparison is between the new method of enumeration and the plate count method (the current method for enumeration). As quantitative tests, by their nature, yield numerical data, they allow for the use of parametric statistical techniques. In contrast, qualitative microbial assays, e.g., the sterility test in the example above, may require analysis by nonparametric statistical methods. The validation of analytical methods for chemical assays follows well-established parameters as described in Validation of Compendial Procedures1225. Validation of microbiological methods shares some of the same concerns, although consideration must be given to the unique nature of microbiological assays (see Table 1).Table 1. Validation Parameters by Type of Microbiological Test Parameter
ParameterQualitative
TestsQuantitative
TestsAccuracy No Yes Precision No Yes Specificity Yes Yes Detection limit Yes Yes Quantification limit No Yes Linearity No Yes Operational range No Yes Robustness Yes Yes Repeatability Yes Yes Ruggedness Yes Yes
VALIDATION OF QUALITATIVE TESTS FOR DEMONSTRATION OF VIABLE MICROORGANISMS IN A SAMPLESpecificityThe specificity of an alternate qualitative microbiological method is its ability to detect a range of microorganisms that may be present in the test article. This concern is adequately addressed by growth promotion of the media for qualitative methods that rely upon growth to demonstrate presence or absence of microorganisms. However, for those methods that do not require growth as an indicator of microbial presence, the specificity of the assay for microbes assures that extraneous matter in the test system does not interfere with the test.Limit of DetectionThe limit of detection is the lowest number of microorganisms in a sample that can be detected under the stated experimental conditions. A microbiological limit test determines the presence or absence of microorganisms, e.g., absence of Salmonella spp. in 10 g. Due to the nature of microbiology, the limit of detection refers to the number of organisms present in the original sample before any dilution or incubation steps; it does not refer to the number of organisms present at the point of assay.One method to demonstrate the limit of detection for a quantitative assay would be to evaluate the two methods (alternative and compendial) by inoculation with a low number of challenge microorganisms (not more than 5 cfu per unit) followed by a measurement of recovery. The level of inoculation should be adjusted until at least 50% of the samples show growth in the compendial test. It is necessary to repeat this determination several times, as the limit of detection of an assay is determined from a number of replicates (not less than 5). The ability of the two methods to detect the presence of low numbers of microorganisms can be demonstrated using the Chi square test. A second method to demonstrate equivalence between the two quantitative methods could be through the use of the Most Probable Number technique. In this method, a 5-tube design in a ten-fold dilution series could be used for both methods. These would then be challenged with equivalent inoculums (for example, a 10–1, 10–2, and 10–3 dilution from a stock suspension of approximately 50 cfu per mL to yield target inocula of 5, 0.5, and 0.05 cfu per tube) and the MPN of the original stock determined by each method. If the 95% confidence intervals overlapped, then the methods would be considered equivalent.RuggednessThe ruggedness of a qualitative microbiological method is the degree of precision of test results obtained by analysis of the same samples under a variety of normal test conditions, such as different analysts, instruments, reagent lots, and laboratories. Ruggedness can be defined as the intrinsic resistance to the influences exerted by operational and environmental variables on the results of the microbiological method. Ruggedness is a validation parameter best suited to determination by the supplier of the test method who has easy access to multiple instruments and batches of components.RobustnessThe robustness of a qualitative microbiological method is a measure of its capacity to remain unaffected by small but deliberate variations in method parameters, and provides an indication of its reliability during normal usage. Robustness is a validation parameter best suited to determination by the supplier of the test method. As there are no agreed upon standards for current methods, acceptance criteria are problematic and must be tailored to the specific technique. It is essential, however, that an estimate of the ruggedness of the alternate procedure be developed. The measure of robustness is not necessarily a comparison between the alternate method and the traditional, but rather a necessary component of validation of the alternate method so that the user knows the operating parameters of the method.
VALIDATION OF QUANTITATIVE ESTIMATION OF VIABLE MICROORGANISMS IN A SAMPLEAs colony-forming units follow a Poisson distribution, the use of statistical tools appropriate to the Poisson rather than those used to analyze normal distributions is encouraged. If the user is more comfortable using tools geared towards normally distributed data, the use of a data transformation is frequently useful. Two techniques are available and convenient for microbiological data. Raw counts can be transformed to normally distributed data either by taking the log10 unit value for that count, or by taking the square root of count +1. The latter transformation is especially helpful if the data contain zero counts.AccuracyThe accuracy of this type of microbiological method is the closeness of the test results obtained by the alternate test method to the value obtained by the traditional method. It should be demonstrated across the operational range of the test. Accuracy is usually expressed as the percentage of recovery of microorganisms by the assay method.Accuracy in a quantitative microbiological test may be shown by preparing a suspension of microorganisms at the upper end of the range of the test, that has been serially diluted down to the lower end of the range of the test. The operational range of the alternate method should overlap that of the traditional method. For example, if the alternate method is meant to replace the traditional plate count method for viable counts, then a reasonable range might be from 100 to 106 cfu per mL. At least 5 suspensions across the range of the test should be analyzed for each challenge organism. The alternate method should provide an estimate of viable microorganisms not less than 70% of the estimate provided by the traditional method, or the new method should be shown to recover at least as many organisms as the traditional method by appropriate statistical analysis, an example being an ANOVA analysis of the log10 unit transforms of the data points. Note that the possibility exists that an alternate method may recover an apparent higher number of microorganisms if it is not dependent on the growth of the microorganisms to form colonies or develop turbidity. This is determined in the Specificity evaluation.PrecisionThe precision of a quantitative microbiological method is the degree of agreement among individual test results when the procedure is applied repeatedly to multiple samplings of suspensions of laboratory microorganisms across the range of the test. The precision of a microbiological method is usually expressed as the standard deviation or relative standard deviation (coefficient of variation). However, other appropriate measures may be applied.One method to demonstrate precision uses a suspension of microorganisms at the upper end of the range of the test that has been serially diluted down to the lower end of the range of the test. At least 5 suspensions across the range of the test should be analyzed. For each suspension at least 10 replicates should be assayed in order to be able to calculate statistically significant estimates of the standard deviation or relative standard deviation (coefficient of variation). Generally, a RSD in the 15% to 35% range would be acceptable. Irrespective of the specific results, the alternate method should have a coefficient of variation that is not larger than that of the traditional method. For example, a plate count method might have the RSD ranges as shown in the following table.Table 2. Expected RSD as a Function of cfu per Platecfu per Plate Expected RSD 30–300 <15% 10–30 <25% <10 <35% SpecificityThe specificity of a quantitative microbiological method is its ability to detect a panel of microorganisms suitable to demonstrate that the method is fit for its intended purpose. This is demonstrated using the organisms appropriate for the purpose of the alternate method. It is important to challenge the alternate technology in a manner that would encourage false positive results (specific to that alternate technology) to demonstrate the suitability of the alternate method in comparison to the traditional method. This is especially important with those alternate methods that do not require growth for microbial enumeration (for example, any that do not require enrichment or can enumerate microorganisms into the range of 1–50 cells).Limit of QuantificationThe limit of quantification is the lowest number of microorganisms that can be accurately counted. As it is not possible to obtain a reliable sample containing a known number of microorganisms, it is essential that the limit of quantification of an assay is determined from a number of replicates (n > 5) at each of at least 5 different points across the operational range of the assay. The limit of quantification should not be a number greater than that of the traditional method. Note that this may have an inherent limit due to the nature of bacterial enumeration and the Poisson distribution of bacterial counts (see Validation of Microbial Recovery from Pharmacopeial Articles1227). Therefore, the alternate method need only demonstrate that it is at least as sensitive as the traditional method to similar lower limits.LinearityThe linearity of a quantitative microbiological test is its ability to produce results that are proportional to the concentration of microorganisms present in the sample within a given range. The linearity should be determined over the range of the test. A method to determine this would be to select at least 5 concentrations of each standard challenge microorganism and conduct at least 5 replicate readings of each concentration. An appropriate measure would be to calculate the square of the correlation coefficient, r2, from a linear regression analysis of the data generated above. While the correlation coefficient does not provide an estimate of linearity, it is a convenient and commonly applied measure to approximate the relationship. The alternate method should not have an r2 value less than 0.95.Limit of DetectionSee Limit of Detection under Validation of Qualitative Tests for Demonstration of Viable Microorganisms in a Sample.RangeThe operational range of a quantitative microbiological method is the interval between the upper and lower levels of microorganisms that have been demonstrated to be determined with precision, accuracy, and linearity.RuggednessSee Ruggedness under Validation of Qualitative Tests for Demonstration of Viable Microorganisms in a Sample.RobustnessSee Robustness under Validation of Qualitative Tests for Demonstration of Viable Microorganisms in a Sample.
* PDA Technical Report No. 33. The Evaluation, Validation and Implementation of New Microbiological Testing Methods. PDA Journal of Pharmaceutical Science & Technology. 54 Supplement TR#33 (3) 2000 and Official Methods Programs of AOAC International.
European Pharmacopoeia Commission announces Strategy for Implementation of ICH Q3D
![]()
The publication of the final ICH Q3D guideline, which has been announced for September of this year, will lead to extensive revisions to chapters and monographs in the European Pharmacopoeia. Find out in what steps the Pharmacopoeia Commission will proceed.
GMP News: European Pharmacopoeia Commission announces Strategy for Implementation of ICH Q3D
In a press release from 7 July 2014, the ICH Steering Committee announced that the finalisation of the ICH Q3D Guideline on Elemental Impurities is planned for September 2014. A press release of the European Pharmacopoeia Commission entitled “The European Pharmacopoeia Commission validates its strategy regarding elemental impurities and the implementation plan of the upcoming ICH Q3D guideline” was released 11 days later. In this release, the Commission explains their approach with regard to the integration of the content of ICH Q3D in the European Pharmacopoeia. This is supposed to be done in the following steps:
- Chapter 5.20 of the Pharmacopoeia (“Metal catalysts or metal reagent residues”), which so far includes a literal rendition of the EMA Guideline “Specification limits for residues of metal catalysts or metal reagents“, will be replaced by the wording of the ICH Q3D Guideline, as soon as it is published as Step 4 document.
- Chapter 5.20 will only become legally binding when it is referenced in a pharmacopoeia monograph. For this purpose references to Chapter 5.20 are supposed to be inserted in the general monographs 2034 (“Substances for pharmaceutical use’) and 2619 (“Pharmaceutical preparations”). The time at which this will take place, has not yet been fixed and depends on the CHMP, which must formally decide to replace the EMA guideline by ICH Q3D in Chapter 5.20.
- In all individual monographs (except in those that relate to substances for veterinary medicinal products) references to Chapter 2.4.8 will be removed. This Chapter still describes wet chemical tests for heavy metals. A list of the affected monographs will appear in the January 2015 issue of the journal “Pharmeuropa”. The publication of the revised monographs is intended for the 9th edition of the European Pharmacopoeia with an implementation date of 1 January 2017.
-
- Chapter 2.4.20 (“Determination of metal catalyst and metal reagent residues”) covering the topics of “sample preparation” and “method suitability”, will be reviewed and adapted to the requirements according to ICH Q3D.
After the revision of the individual chapters and monographs it is at the discretion of the responsible quality control laboratories to choose an appropriate analytical strategy in accordance with the requirements of ICH Q3D.
………………..
clippings
-
Description:
The Q3D draft Guideline has been relased for consultation under Step 2B of the ICH process in July 2013.
This new guidance is proposed to provide a global policy for limiting metal impurities qualitatively and quantitatively in drug products and ingredients. The existing ICH Q3A Guideline classifies impurities as organic, inorganic, and residual solvents. The Q3A and Q3B Guidelines effectively address the requirements for organic impurities. An additional Guideline Q3C was developed to provide clarification of the requirements for residual solvents. The proposed new Guideline Q3D would provide similar clarification of the requirements for metals, which are included in the ICH inorganic impurities classification.
Status:Step 2b
EU:
Transmission to CHMP in June 2013, issued as EMA/CHMP/ICH/353369/2013. Deadline for comments: 31 December 2013
MHLW:
Released for consultation, 4 October 2013, PFSB/ELD. Deadline for comments: 29 November 2013
FDA:
Published in the Federal Register 23 October 2013, Vol. 78, No. 205, p. 63219-20. Deadline for comments: 23 December 2013

………………………….
Q3D Elemental Impurities – Food and Drug Administration
Sep 30, 2013 – This document reached step 2B of the ICH Process on June 6, 2013. For questions … Q3D. Approval by the Steering Committee under Step 2b
………………
Guideline for Elemental Impurities – ICH
DRAFT CONSENSUS GUIDELINE. GUIDELINE FOR ELEMENTAL IMPURITIES. Q3D. Current Step 2b version dated 26 July 2013. At Step 2 of the ICH Process, …
5 August 2013
The ICH Q3D Guideline for Elemental Impurities reached Step 2b of the ICH Process in July 2013 and now enters the consultation period (Step 3).
This new Guideline is proposed to provide a global policy for limiting metal impurities qualitatively and quantitatively in drug products and ingredients. The existing ICH Q3A Guideline classifies impurities as organic, inorganic, and residual solvents. The Q3A and Q3B Guidelines effectively address the requirements for organic impurities.
An additional Guideline Q3C was developed to provide clarification of the requirements for residual solvents.
The proposed new Guideline Q3D would provide similar clarification of the requirements for metals, which are included in the ICH inorganic impurities classification.
The draft Guideline is now available for download under the Quality Guideline page. ローテンシルト通販ニクソン腕時計You are invited to provide comments on the draft Guideline by e-mailing the ICH Secretariat. More details under the Open Consultation page.
Note that stakeholders from EU, US and Japan are encouraged to submit their comments to their respective Regulatory Authorities.
Water Systems in FDA Warning Letters

Among the FDA Warning Letters of the past two years there are every now and then letters citing deficiencies in water systems. On the bottom line the reason for objections is always the same. Read more.
GMP News: Water Systems in FDA Warning Letters
Taking a look at FDA Warning Letters from the past two years, objections with regard to pharmaceutical water systems are rather seldom. However, when there are complaints, it is the more interesting that the reason for these complaints is mostly the same: missing reliability. What the authority means in this case is the proof that the water system is capable of securely and reliably producing water in the required quality – taking into account for example the fluctuations in the feed water. The necessary means for this purpose are the validation of the water system and the establishment of a monitoring system which continuously verifies the function.
Excerpts from Warning Letters:
1. Failure to validate and monitor the water purification system to ensure that water is of appropriate quality. […] In your response to the observations noted during the 2012 inspection, you indicated your firm’s intention to conduct a comprehensive gap analysis of the purified water system. However, you have failed to indicate when you will initiate this gap analysis and when it will be completed. Your firm also failed to detail how you will determine the source(s) of high endotoxin and TOC in your purified water and how your firm will remedy identified problem(s). We note that, for example, your firm installed an endotoxin removal unit on your purified water system in January 2011 in response to the OOS results for endotoxin in the water used for API. However, your firm has not demonstrated that the water produced by the purified water system is now suitable for use in production. The operational parameters and effectiveness of the new endotoxin removal unit have not been qualified. Your firm does not monitor the microbial and chemical attributes of the feed water, and have no assurances that the purified water system is capable of consistently producing water that meets specifications for a given quality of feed water.
2. Your firm failed to assure that your water system is suitably designed and operated to produce appropriate water quality. Regarding the latter, your firm has not established and validated appropriate cleaning and sanitizing schedules for your purified water system. You have hired a water process subject matter expert and taken other steps to strengthen monitoring of the purified water system. Your response is not acceptable because you have not demonstrated that your purified water system is capable of operating in a continuing state of control.
3. […] your firm failed to subject the water to routine microbiological testing. Furthermore, your firm failed to validate the water system to ensure consistent water quality for drug production and implement procedures for maintaining or monitoring the quality of the water produced.
Cimicoxib

Cimicoxib
Uriach (Originator)
4-[4-Chloro-5-(3-fluoro-4-methoxyphenyl)-1H-imidazol-1-yl]benzenesulfonamide
IN PHASE 2
Cimicoxib (trade name Cimalgex) is a non-steroidal anti-inflammatory drug (NSAID) used in veterinary medicine to treat dogs for pain and inflammation associated with osteoarthritis and for the management of pain and inflammation associated with surgery.[1] It acts as a COX-2 inhibitor.
Cimicoxib is a selective COX-2 inhibitor being developed by Affectis as a treatment for depression and schizophrenia. If approved, Cimicoxib would be the first drug in decades to treat depression by a new mechanism of action
Cimicoxib, an imidazole derivative, is a selective cyclooxygenase-2 (COX-2) inhibitor. The product was in phase II development at Affectis Pharmaceuticals for the oral treatment of major depression, however, no recent development have been reported. Originally developed by Uriach, the compound was acquired by Palau Pharma, a spin-off created by Uriach in November 2006.
In 2007, Palau Pharma licensed global rights to cimicoxib to Affectis Pharmaceuticals for all CNS indications. Palau had been clinically evaluating the compound for the treatment of osteoarthritis, pain and rheumatoid arthritis, however, no recent development has been reported for these indications. The compound holds potential for the treatment of schizophrenia.


Treatment of 4-(acetylamino)phenylsulfonyl chloride (I) with tert-butylamine yields sulfonamide (II), which on deprotection with potassium hydroxide gives amine (III). Reaction of compound (III) with 4-methoxy-3-fluorobenz-aldehyde gives imine (IV), which is cyclized with tosylmethyl isocyanide to afford imidazole (V). Regioselective chlorination of compound (V) with N-chlorosuccinimide (NCS) to afford the chloroimidazole (VI) and then deprotection of the sulfonamide group of (VI) yields cimicoxib in 40% overall yield.

EP 1122243; JP 2002527508; WO 0023426, ES 2184633; WO 0316285
……………………………….
http://www.google.com/patents/EP1424329A1?cl=en
EXAMPLE 1
4-Amino-N- tert -butylbenzenesulfonamide Method A:
-
[0031]


a) N-tert-Butyl-4-nitrobenzenesulfonamide
-
[0032]To a solution of tert-butylamine (0.47 L, 6.4 mol) in THF (0.55 L) is slowly added, at 0 °C, a solution of 4-nitrobenzenesulfonyl chloride (50 g, 0.23 mol) in THF (0.55 L) and the resulting mixture is stirred for 24 h at room temperature. The solvent is removed and the residue is taken up in a CHCl3/0.5 N HCl mixture, the layers are separated and the aqueous phase is extracted with CHCl3. The combined organic extracts are washed with H2O and brine and dried over MgSO4. The solvent is removed, yielding 56.3 g of a yellowish solid which is directly used in the next reaction (yield: 97%).
Mp: 105-109°C; 1H-NMR (300 MHz, CDCl3) δ (TMS): 1.29 (s, 9 H), 5.07 (s, 1 H), 8.13 (d, J = 9 Hz, 2 H), 8.39 (d, J = 9 Hz, 2 H).
b) Title compound
-
[0033]A solution of N-tert-butyl-4-nitrobenzenesulfonamide (10.0 g, 39 mmol) in EtOH (100 mL) is stirred for 48 h under a H2 atmosphere in the presence of 10% Pd/C (1.50 g). The resulting mixture is filtered and concentrated to give the desired product as a slightly-coloured solid (8.7 g, yield: 98%).
Mp: 127 °C; 1H-NMR (300 MHz, CDCl3 + CD3OD) δ (TMS): 1.19 (s, 9 H), 3.74 (s, CD3OD + 1 H), 6.93 (d, J = 9 Hz, 2 H), 7.66 (d, J = 9 Hz, 2 H).
Method B:
-
[0034]


a) 4-Acetylamino-N-tert-butylbenzenesulfonamide
-
[0035]To a suspension of 4-acetylaminobenzenesulfonyl chloride (10 g, 43 mmol) in DME (103 mL) is added, at 0 °C, tert-butylamine (9 mL, 86 mmol) in DME (103 mL). Next, the reaction mixture is stirred for 4 h at reflux. The solvent is removed and CHCl3 is added. The resulting suspension is filtered and the solid is washed with CHCl3, H2O and Et2O. The solid obtained is dried in vacuo to give 8.0 g of the product as a white solid (yield: 68%).
Mp: 200-201 °C; 1H-NMR (300 MHz, CDCl3 + CD3OD) δ (TMS): 1.15 (s, 9 H), 2.12 (s, 3 H), 4.21 (s, 2H + CD3OD), 7.66 (d, J = 9 Hz, 2 H), 7.75 (d, J = 9 Hz, 2 H).
b) Title compound
-
[0036]A solution of 4-acetylamino-N-tert-butylbenzenesulfonamide (8.0 g, 29.6 mmol), KOH (8.30 g, 148 mmol), H2O (6 mL) and MeOH (24 mL) is heated at 100°C for 2 h. H2O (24 mL) is added and the mixture is heated for two more hours. It is allowed to cool, H2O is added and it is brought to pH 8 with 1N HCl. It is then extracted with EtOAc, dried over Na2SO4 and the solvent is removed, to give 6.0 g of the product as a white solid (yield: 89%).
EXAMPLE 2 N- tert -Butyl-4-[(3-fluoro-4-methoxybenzylidene)amino]benzenesulfonamide
-
[0037]

-
[0038]A mixture of 4-amino-N-tert-butylbenzenesulfonamide (52.3 g, 0.23 mol, obtained in example 1), 3-fluoro-4-methoxybenzaldehyde (35.3 g, 0.23 mol) and toluene (2.5 L) is heated at reflux in a Dean-Stark for 24 h. The solvent is removed, yielding 83.5 g of the title compound (yield: quantitative).
Mp: 129-131 °C; 1H-NMR (300 MHz, CDCl3) δ (TMS): 1.23 (s, 9 H), 3.98 (s, 3 H), 4.65 (s, 1 H), 7.04 (t, J = 8.1 Hz, 1 H), 7.21 (d, J = 6.7 Hz, 2 H), 7.58 (m, 1 H), 7.73 (dd, JH-F = 11.8 Hz, J = 2 Hz, 1 H), 7.90 (d, J = 6.7 Hz, 2 H), 8.33 (s, 1 H).

EXAMPLE 3 N-tert-Butyl-4-[5-(3-fluoro-4-methoxyphenyl)imidazol-1-yl]benzenesulfonamide
-
[0039]

-
[0040]A mixture of N-tert-butyl-4-[(3-fluoro-4-methoxybenzylidene)amino]benzenesulfonamide (41.5 g, 114 mmol, obtained in example 2), tosylmethylisocyanide (33.22 g, 171 mmol), K2CO3 (31.1 g, 228 mmol), DME (340 mL) and MeOH (778 mL) is heated at reflux for 3 h. The solvent is removed and the residue is taken up in a CHCl3/H2O mixture and the layers are separated. The aqueous phase is extracted with CHCl3 and the combined organic extracts are dried over MgSO4 and concentrated. A crude product is obtained, which is washed with Et2O several times to give 41.40 g of a creamy solid that is directly used in the next reaction (yield: 90%).
Mp: 229-232°C; 1H-NMR (300 MHz, CDCl3) δ (TMS): 1.24 (s, 9 H), 3.89 (s, 3 H), 4.51 (s, 1 H), 6.90 (m, 3 H), 7.23 (s, 1 H), 7.29 (d, J = 8.7 Hz, 2 H), 7.73 (s, 1 H), 7.94 (d, J = 8.7 Hz, 2 H).

EXAMPLE 4 N-tert-Butyl-4-[4-chloro-5-(3-fluoro-4-methoxyphenyl)imidazol-1-yl]benzenesulfonamide
-
[0041]

-
[0042]A mixture of N-tert-butyl-4-[5-(3-fluoro-4-methoxyphenyl)imidazol-1-yl]benzenesulfonamide (41.40 g, 103 mmol, obtained in example 3) and acetonitrile (840 mL) is heated at reflux and acetonitrile is added until complete dissolution (200 mL more). Next, N-chlorosuccinimide (15.0 g, 113 mmol) is added and the mixture is refluxed for 24 h. The solvent is removed and the residue is suspended in EtOAc and 1N HCl and is stirred for 10 min. The solid obtained is filtered and washed directly in the filter with 1N HCl, 1N NaOH, saturated NH4Cl solution, H2O and Et2O. A solid is obtained, which is dried in vacuo to give 37.0 g of the product as a creamy solid (yield: 82%).
Mp: 208-210 °C; 1H-NMR (300 MHz, CDCl3) δ (TMS): 1.24 (s, 9 H), 3.89 (s, 3 H), 4.51 (s, 1 H), 6.90 (m, 3 H), 7.23 (d, J = 8.7 Hz, 2 H), 7.63 (s, 1 H), 7.92 (d, J = 8.7 Hz, 2 H).

EXAMPLE 5 4-[4-Chloro-5-(3-fluoro-4-methoxyphenyl)imidazol-1-yl]benzenesulfonamide
-
[0043]

-
[0044]A mixture of N-tert-butyl-4-[4-chloro-5-(3-fluoro-4-methoxyphenyl)imidazol-1-yl]benzenesulfonamide (37.0 g, 85 mmol, obtained in example 4), concentrated HCl (200 mL) and H2O (200 mL) is heated at reflux for 3 h. The mixture is allowed to cool and is brought to pH 6 with 6N NaOH. A white precipitate appears, which is collected by filtration and washed with plenty of H2O and then with CHCl3. 31 g of the title compound of the example is obtained (yield: 97%), which are recrystallized from acetonitrile.
Mp: 211-212 °C; -
1H-NMR (300 MHz, CDCl3 + CD3OD) δ (TMS): 3.90 (s, 3 H), 4.16 (s, CD3OD + 2 H), 6.93 (m, 3 H), 7.30 (d, J = 8.6 Hz, 2 H), 7.73 (s, 1 H), 7.95 (d, J = 8.7 Hz, 2 H).

References
- “European Public Assessment Report: Cimalgex (cimicoxib)”. European Medicines Agency.
|
9-1-2013
|
Detection and quantification of cimicoxib, a novel COX-2 inhibitor, in canine plasma by HPLC with spectrofluorimetric detection: development and validation of a new methodology.
|
Journal of pharmaceutical and biomedical analysis
|
|
|
6-1-2013
|
Efficacy and safety of cimicoxib in the control of perioperative pain in dogs.
|
The Journal of small animal practice
|
|
|
4-5-2007
|
NO-donor COX-2 inhibitors. New nitrooxy-substituted 1,5-diarylimidazoles endowed with COX-2 inhibitory and vasodilator properties.
|
Journal of medicinal chemistry
|
|
|
10-21-2004
|
New water-soluble sulfonylphosphoramidic acid derivatives of the COX-2 selective inhibitor cimicoxib. A novel approach to sulfonamide prodrugs.
|
Journal of medicinal chemistry
|
|
|
7-31-2003
|
Synthesis and structure-activity relationship of a new series of COX-2 selective inhibitors: 1,5-diarylimidazoles.
|
Journal of medicinal chemistry
|
|
4-15-2005
|
Compositions of a cyclooxygenase-2 selective inhibitor and a serotonin-modulating agent for the treatment of central nervous system damage
|
|
|
4-8-2005
|
Compositions of a cyclooxygenase-2 selective inhibitor and an IKK inhibitor for the treatment of ischemic mediated central nervous system disorders or injury
|
|
1-9-2009
|
Process for the Preparation of 4-(imidazol-1-yl)benzenesulfonamide Derivatives
|
|
|
9-5-2008
|
Medicament that is Intended for Oral Administration, Comprising a Cyclooxygenase-2 Inhibitor, and Preparation Method Thereof
|
|
|
4-2-2008
|
Method of preparing 4-(imidazol-1-yl)benzenesulphonamide derivatives
|
|
|
6-29-2007
|
Compositions of a cyclooxygenase-2 selective inhibitor administered under hypothermic conditions for the treatment of ischemic mediated central nervous system disorders or injury
|
|
|
7-8-2005
|
Compositions of a cyclooxygenase-2 selective inhibitor and a neurotrophic factor-modulating agent for the treatment of central nervous system mediated disorders
|
|
|
5-27-2005
|
Compositions of a cyclooxygenase-2 selective inhibitor administered under hypothermic conditions for the treatment of ischemic mediated central nervous system disorders or injury
|
|
|
5-13-2005
|
Compositions of a cyclooxygenase-2 selective inhibitior and a non-NMDA glutamate modulator for the treatment of central nervous system damage
|
|
|
4-22-2005
|
Compositions of a cyclooxygenase-2 selective inhibitor and a low-molecular-weight heparin for the treatment of central nervous system damage
|
|
|
4-22-2005
|
Mediated central nervous system compositions of a cyclooxygenase-2 selective inhibitor and a corticotropin releasing factor antagonist for the treatment of ischemic disorders or injury
|
Tilmacoxib

Tilmacoxib
 ethoxymethyl-7-fluoro-3-oxo-1,2,3,5-tetrahydrobenzo(4,5)imidazo(1,2a)pyridine-4-N–(2-fluorophenyl)carboxamide
ethoxymethyl-7-fluoro-3-oxo-1,2,3,5-tetrahydrobenzo(4,5)imidazo(1,2a)pyridine-4-N–(2-fluorophenyl)carboxamideJapan Tobacco (JT) (Originator)
Tilmacoxib or JTE-522 is a COX-2 inhibitor and is an effective chemopreventive agent against rat experimental liver fibrosis.[1]
A member of the class of 1,3-oxazoles that is that is 1,3-oxazole which is substituted at positions 2, 4 and 5 by methyl, cyclohexyl, and 3-fluoro-4-sulfamoylphenyl groups, respectively.
………..
4-(4-Cycloalkyl/aryl-oxazol-5-yl)benzenesulfonamides as selective cyclooxygenase-2 inhibitors: Enhancement of the selectivity by introduction of a fluorine atom and identification of a potent, highly selective, and orally active COX-2 inhibitor JTE-522
J Med Chem 2002, 45(7): 1511
http://pubs.acs.org/doi/abs/10.1021/jm010484p
A series of 4-(4-cycloalkyl/aryl-oxazol-5-yl)benzenesulfonamide derivatives were synthesized and evaluated for their abilities to inhibit cyclooxygenase-2 (COX-2) and cyclooxygenase-1 (COX-1) enzymes. In this series, substituent effects at the ortho position to the sulfonamide group on the phenyl ring were examined. Most substituents reduced or lost both COX-2 and COX-1 activities. In contrast, introduction of a fluorine atom preserved COX-2 potency and notably increased COX1/COX-2 selectivity. This work led to the identification of a potent, highly selective, and orally active COX-2 inhibitor JTE-522 [9d, 4-(4-cyclohexyl-2-methyloxazol-5-yl)-2-fluorobenzenesulfonamide], which is currently in phase II clinical trials for the treatment of rheumatoid arthritis, osteoarthritis, and acute pain.
9d as a white solid: mp 166−167 °C; 1H NMR (CDCl3) δ 1.3−1.5 (m, 3H), 1.6−1.9 (m, 7H), 2.51 (s, 3H), 2.79 (tt, J = 3.7, 11.3 Hz, 1H), 5.11 (s, 2H), 7.36−44 (m, 2H), 7.94 (t, J = 7.9 Hz, 1H). Anal. (C16H19FN2O3S) C, H, N.
………………
WO 1996019463 OR http://www.google.com/patents/EP0745596A1?cl=en
Example 2
-
[0080]Synthesis of 5-(4-aminosulfonyl-3-fluorophenyl)-4-cyclohexyl-2-methyloxazole (formula (I); R=cyclohexyl, R1=4-aminosulfonyl-3-fluorophenyl, R2=methyl, Z=oxygen atom)
Step 10) Cyclohexyl 3-fluorobenzyl ketone (formula (IV’); R’=cyclohexyl, R1‘=3-fluorophenyl)
-
[0081]To a solution of tetrakis(triphenylphosphine)palladium (2.00 g) and zinc powder (17.98 g) in 1,2-dimethoxyethane (50 ml) was added a solution of cyclohexanecarbonyl chloride (20.00 g) in 1,2-dimethoxyethane (50 ml) at room temperature under a nitrogen atmosphere. A solution of 3-fluorobenzyl bromide (26.00 g) in 1,2-dimethoxyethane (100 ml) was gradually added dropwise to the mixture with stirring under ice-cooling. The mixture was stirred under ice-cooling for 30 minutes, and at room temperature for 2 hours. The insoluble matter was removed by filtration and the filtrate was concentrated under reduced pressure. Then, ethyl acetate (200 ml) was added to the residue, and the mixture was washed with 1N hydrochloric acid, and then with saturated aqueous sodium hydrogencarbonate solution and saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated to give 29.20 g of an oily crude product.
Step 16) 2-Cyclohexyl-1-(3-fluorophenyl)-2-oxoethyl acetate (formula (V”); R’=cyclohexyl, R1‘=3-fluorophenyl, R2‘=methyl, Z=oxygen atom)
-
[0082]Lead tetraacetate (75.00 g) was added to a solution of the compound (29.20 g) obtained in the above Step 10) in acetic acid (300 ml). The mixture was refluxed under heating for 1.5 hours, and the solvent was evaporated under reduced pressure. Ethyl acetate was added to the residue. The mixture was washed with water, a saturated aqueous sodium hydrogencarbonate solution and saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure, and the residue was purified by silica gel column chromatography (developing solvent; hexane:ethyl acetate=9:1) to give 18.30 g of the title compound as an oil (yield 50%).
Step 17) 4-Cyclohexyl-5-(3-fluorophenyl)-2-methyloxazole (formula (XIII); R’=cyclohexyl, R1‘=3-fluorophenyl, R2=methyl, Z=oxygen atom)
-
[0083]A solution of the compound (18.00 g) obtained in the above Step 16) and ammonium acetate (15.00 g) in acetic acid (100 ml) was refluxed under heating for 5 hours, and the solvent was evaporated under reduced pressure. Ethyl acetate was added to the residue. The mixture was washed with water, saturated aqueous sodium hydrogencarbonate solution and saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to give 17.20 g of an oily crude product. Step 15) 5-(4-Aminosulfonyl-3-fluorophenyl)-4-cyclohexyl-2-methyloxazole (formula (I); R=cyclohexyl, R1=4-aminosulfonyl-3-fluorophenyl, R2=methyl, Z=oxygen atom)

-
[0084]To a solution of the compound (17.00 g) obtained in the above Step 17) in chloroform (80 ml) was added dropwise chlorosulfonic acid (27 ml) with stirring under ice-cooling, and the mixture was heated at 100°C for 3 hours. The reaction mixture was cooled to room temperature, and dropwise added to ice-water (300 ml) with stirring. The organic layer was separated, washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to give 20.31 g of a crude product.
-
[0085]Aqueous ammonia (28%) was added to a solution of the obtained compound (10.00 g) in tetrahydrofuran (40 ml) with stirring at room temperature, and the mixture was stirred at room temperature for one hour. The solvent was evaporated under reduced pressure and ethyl acetate was added to the residue. The mixture was washed with water and saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated, and the residue was separated and purified by silica gel column chromatography (developing solvent; dichloromethane:ethyl acetate=6:1) to give 5.74 g of the title compound (yield 61%).




Example 2′
-
[0086]The compound of Example 2 (formula (I); R=cyclohexyl, R1=4-aminosulfonyl-3-fluorophenyl, R2=methyl, Z=oxygen atom) was synthesized according to another synthetic method.
Step 11) Cyclohexyl 3-fluorobenzyl ketone oxime (formula (XI); R’= cyclohexyl, R1‘=3-fluorophenyl)
-
[0087]To a solution of the compound (353 g) obtained according to a method similar to that of the above Example 2, Step 10) in ethanol (1300 ml) were added hydroxylamine hydrochloride (123 g) and sodium acetate (158 g). The mixture was refluxed under heating for 2 hours, and the solvent was evaporated under reduced pressure. Ethyl acetate was added to the residue. The mixture was washed with water, saturated aqueous sodium hydrogencarbonate solution and saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure, and the crude product was recrystallized from n-heptane to give 160 g of the title compound (yield 42%).
Step 14) 4-Cyclohexyl-5-(3-fluorophenyl)-2-methyloxazole (formula (XIII); R’=cyclohexyl, R1‘=3-fluorophenyl, R2=methyl, Z=oxygen atom)
-
[0088]Acetic anhydride (95 ml) was dropwise added to a solution of the compound (158 g) obtained in the above Step 11) in acetic acid (900 ml) with stirring at room temperature, and the mixture was refluxed under heating for 7 hours. The solvent was evaporated under reduced pressure and n-heptane was added to the residue. The mixture was washed with water, saturated aqueous sodium hydrogencarbonate solution, saturated brine and acetonitrile. The solvent was evaporated under reduced pressure to give 119 g of the title compound as an oil.
-
[0089]Then, the obtained compound (119 g) was reacted in the same manner as in the above Example 2, Step 15) to give a compound of Example 2 (formula (I); R=cyclohexyl, R1=4-aminosulfonyl-3-fluorophenyl, R2=methyl, Z=oxygen atom).


Example 3
-
[0090]Synthesis of 4-cyclohexyl-5-(3-fluoro-4-methylsulfonylphenyl)-2-methyloxazole (formula (I); R=cyclohexyl, R1=3-fluoro-4-methylsulfonylphenyl, R2=methyl, Z=oxygen atom)
Step 15) 4-Cyclohexyl-5-(3-fluoro-4-methylsulfonylphenyl)-2-methyloxazole (formula (I); R=cyclohexyl, R1=3-fluoro-4-methylsulfonylphenyl, R2=methyl, Z=oxygen atom)
-
[0091]To a solution of the compound (17.00 g) obtained in the above Example 2, Step 17) in chloroform (80 ml) was dropwise added chlorosulfonic acid (27 ml) with stirring under ice-cooling. The mixture was heated at 100°C for 3 hours. The reaction mixture was cooled to room temperature and dropwise added to ice-water (300 ml) with stirring. The organic layer was separated, washed with saturated brine, and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to give 20.31 g of a crude product.
-
[0092]Water (25 ml) was added to the obtained compound (3.66 g). To the mixture were added sodium sulfite (1.42 g) and sodium hydrogencarbonate (1.89 g) successively with stirring at room temperature. The mixture was heated at 70°C for 2 hours. Ethanol (25 ml) and methyl iodide (2.20 g) were added to the mixture, and the mixture was heated at 100°C for 2 hours. The mixture was cooled to room temperature and extracted with ethyl acetate. The extract was washed with saturated brine and dried over anhydrous sodium sulfate.
-
[0093]The solvent was evaporated under reduced pressure, and the residue was saparated and purified by silica gel column chromatography (developing solvent; hexane:ethyl acetate=2:1) to give 0.82 g of the title compound (yield 24%).

References
- Yamamoto, H., Kondo, M., Nakamori, S., Nagano, H., Wakasa, K., Sugita, Y., Chang-De, J., Kobayashi, S., Damdinsuren, B., Dono, K., Umeshita, K., Sekimoto, M., Sakon, M., Matsuura, N., Monden, M. (2003). “JTE-522, a cyclooxygenase-2 inhibitor, is an effective chemopreventive agent against rat experimental liver fibrosis1”. Gastroenterology 125 (2): 556–571. doi:10.1016/s0016-5085(03)00904-1. PMID 12891558.
-
3-28-20024-(4-cycloalkyl/aryl-oxazol-5-yl)benzenesulfonamides as selective cyclooxygenase-2 inhibitors: enhancement of the selectivity by introduction of a fluorine atom and identification of a potent, highly selective, and orally active COX-2 inhibitor JTE-522(1).Journal of medicinal chemistry
|
7-5-1999
|
The discovery of rofecoxib, [MK 966, Vioxx, 4-(4′-methylsulfonylphenyl)-3-phenyl-2(5H)-furanone], an orally active cyclooxygenase-2-inhibitor.
|
Bioorganic & medicinal chemistry letters
|
