PROUD Indian WORLD RECORD VIEWS holder on THIS BLOG, ………live, by DR ANTHONY MELVIN CRASTO, Worldpeaceambassador, Worlddrugtracker, Helping millions, 100 million hits on google, pushing boundaries,2.5 lakh plus connections worldwide, 45 lakh plus VIEWS on this blog in 227 countries, 7 CONTINENTS ……A 90 % paralysed man in action for you, I am suffering from transverse mylitis and bound to a wheel chair, [THIS BLOG HOLDS WORLD RECORD VIEWS ] A PROUD INDIAN
DR ANTHONY MELVIN CRASTO, Born in Mumbai in 1964 and graduated from Mumbai University, Completed his Ph.D from ICT, 1991,Matunga, Mumbai, India, in Organic Chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with AFRICURE PHARMA, ROW2TECH, NIPER-G, Department of Pharmaceuticals, Ministry of Chemicals and Fertilizers, Govt. of India as ADVISOR, earlier assignment was
with GLENMARK LIFE SCIENCES LTD, as CONSUlTANT, Retired from GLENMARK in Jan2022 Research Centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Total Industry exp 32 plus yrs, Prior to joining Glenmark, he has worked with major multinationals like Hoechst Marion Roussel, now Sanofi, Searle India Ltd, now RPG lifesciences, etc. He has worked with notable scientists like Dr K Nagarajan, Dr Ralph Stapel, Prof S Seshadri, etc, He did custom synthesis for major multinationals in his career like BASF, Novartis, Sanofi, etc., He has worked in Discovery, Natural products, Bulk drugs, Generics, Intermediates, Fine chemicals, Neutraceuticals, GMP, Scaleups, etc, he is now helping millions, has 9 million plus hits on Google on all Organic chemistry websites. His friends call him Open superstar worlddrugtracker. His New Drug Approvals, Green Chemistry International, All about drugs, Eurekamoments, Organic spectroscopy international,
etc in organic chemistry are some most read blogs He has hands on experience in initiation and developing novel routes for drug molecules
and implementation them on commercial scale over a 32 PLUS year tenure till date Feb 2023, Around 35 plus products in his career. He has good knowledge of IPM, GMP, Regulatory aspects, he has several International patents published worldwide . He has good proficiency in Technology transfer, Spectroscopy, Stereochemistry, Synthesis, Polymorphism etc., He suffered a paralytic stroke/ Acute Transverse mylitis in Dec 2007 and is 90 %Paralysed, He is bound to a wheelchair, this seems to have injected feul in him to help chemists all around the world, he is more active than before and is pushing boundaries, He has 100 million plus hits on Google, 2.5 lakh plus connections on all networking sites, 100 Lakh plus views on dozen plus blogs, 227 countries, 7 continents, He makes himself available to all, contact him on +91 9323115463, email amcrasto@gmail.com, Twitter, @amcrasto , He lives and will die for his family, 90% paralysis cannot kill his soul., Notably he has 38 lakh plus views on New Drug Approvals Blog in 227 countries......https://newdrugapprovals.wordpress.com/ , He appreciates the help he gets from one and all, Friends, Family, Glenmark, Readers, Wellwishers, Doctors, Drug authorities, His Contacts, Physiotherapist, etc
He has total of 32 International and Indian awards
launched 2012, as forxiga in EU, FDA 2014, JAPAN PMDA 2014
Dapagliflozin propanediol monohydrate was first approved by European Medicine Agency (EMA) on November 12, 2012, then approved by the U.S. Food and Drug Administration (FDA) on January 8, 2014, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on March 24, 2014. It was co-developed and co-marketed as Forxiga® by Bristol-Myers Squibb and AstraZeneca in EU.
Dapagliflozin propanediol monohydrate is a sodium-glucose co-transporter 2 (SGLT2) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Forxiga® is available as tablet for oral use, containing 5 mg or 10 mg of free Dapagliflozin. The recommended starting dose is 5 mg once daily in the morning.
Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.
Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
1. A process for the preparation of dapagliflozin in amorphous form, the process comprising:
(a) reducing a compound of formula II to a compound of formula ΠΙ in the presence of a Lewis acid;
(b) silylating a compound of formula IV with hexamethyldisilazane to form a compound of formula V;
(c) reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;
(d) converting the compound of formula VII to dapagliflozin;
(e) acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;
(f) optionally, purifying the compound of formula VIII with a solvent selected from halogenated hydrocarbons, alcohols, ethers, or mixtures thereof; (g) hydrolyzing the compound of formula VIII obtained in step (f) to give dapagliflozin;
EXAMPLE 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
To a suspension of 5-bromo-2-chiorobenzoic acid (lOg) in methylene di chloride (40niL), dimethylformamide (0.2g) and thionyl chloride were added and the reaction mixture was refluxed for about 2h. After completion of reaction, the solvent was distilled out. The mass obtained was degassed under vacuum followed by stripping with cyclohexane to give crude 5-bromo-2-chlorobenzoyl chloride (10.8g).
[0189] EXAMPLE 2: Preparation of 5-bromo-2-chloro-4′-ethoxybenzophenone (compound of Formula II)
5-bromo-2-chlorobenzoyl chloride (10.7g) was dissolved in methylene dichloride (40mL) and the reaction mixture was cooled to about -8°C to about -12°C under inert atmosphere. Aluminum chloride (5.65g) was added to the reaction mixture followed by addition of a solution of ethoxybenzene in methylene dichloride. The reaction mixture was stirred for about lh at about -8°C to -12°C and then quenched in dilute hydrochloric acid followed by extraction with methylene dichloride. The organic layer was washed with sodium bicarbonate solution and concentrated. The residue obtained was crystallized from methanol to give 5-bromo-2-chloro-4′-ethoxybenzophenone (8.5g). HPLC purity: 99.34%
[0190] EXAMPLE 3: Preparation of 5-bromo-2-chloro-4′-ethoxydiphenylmethane (compound of formula III)
To a mixture of 5-bromo-2-chloro-4′-ethoxybenzophenone (lOg) and methylene dichloride (50mL), cooled to about 0°C to about 5°C, triethylsilane (11.98g) and titanium chloride (22.3g) were added. The reaction mixture was stirred for about 3h at about 10°C to about 15°C. The reaction mixture was quenched into chilled water. The organic layer was separated, washed with water and sodium bicarbonate solution and concentrated under vacuum followed by stripping with toluene. The residue obtained was stirred with methanol, filtered and dried to give 5-bromo-2-chloro-4′-ethoxydiphenylmethane (9g). HPLC purity: 99.4%
[0191] EXAMPLE 4: Preparation of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5- lactone (compound of Formula V)
To a mixture of D-glucono- 1,5 -lactone (lOg) and iodine (0.28g) in methylene dichloride (80mL), hexamethyldisilazane (36.1g) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed to give 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5-lactone as liquid (25g). HPLC purity: 95%
[0192] EXAMPLE 5: Preparation of D-glucopyranoside, methyl l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (compound of Formula VII wherein R is methyl)
To a mixture of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l ,5-lactone (25g) and 5- bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) in tetrahydrofuran (174mL), cooled to about -75°C to about -88 °C under nitrogen atmosphere, n-butyl lithium in hexane (50mL) was slowly added. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. The residue was purified with a mixture of toluene and cyclohexane. Yield: 1 lg as thick mass with 80-85% HPLC purity.
reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;
[0193] EXAMPLE 6: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl) methyl] phenyl]
To a mixture of D-glucopyranoside, methyl l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] in methylene di chloride (40mL) and acetonitrile (40mL), cooled to about -40°C to about -45°C, triethylsilane (8.74g) was added followed by addition of boron trifluoride etherate (10.67g) maintaining the temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum to give title compound (1 lg) as thick residue with 80-85% HPLC purity.
[0194] EXAMPLE 7: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (lS)-
To a cooled solution of D-glucitol, l,5-anhydro-l -C-[4-chloro-3-[(4-ethoxyphenyl) methyl] phenyl]- (l lg) in methylene dichloride (55mL) at about 0°C to about 5°C, diisopropylethylamme, dmiethylaminopyridine and acetic anhydride were added and the reaction mixture was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol. The residue was purified with methanol and charcoal, followed by diisopropyl ether and methanol crystallization. Yield: lOg; HPLC purity: 99.6%
acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;
[0195] EXAMPLE 8: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (Dapagliflozin)
To a stirred solution of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, (lOg) in THF: methanol: water mixture (50mL: 50mL:30mL), sodium hydroxide was added and the reaction mixture was stirred. After completion of reaction, the solvents were distilled out under vacuum and the residue obtained was dissolved in methylene dichloride and washed with water and brine and dried over sodium sulfate. The reaction mixture was concentrated and degassed to give off- white to white solids of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]- (dapagliflozin) Yield: 7g (XRD matches with amorphous form) HPLC purity: 99.8%
[0197] EXAMPLE 10: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (IS)- from D-glucono-1,5- lactone (One-pot Synthesis)
To a mixture of D-glucono-l,5-lactone (lOg) in methylene dichloride (80mL), hexamethyldisilazane (36. lg) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed. The residue obtained was dissolved in tetrahydrofuran. 5-Bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) was added to the reaction mixture which was cooled to about -75°C to about-85°C under nitrogen atmosphere. n-Butyl lithium in hexane (50mL) was slowly added to the reaction mixture maintaining the temperature between -75°C to about -85°C. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. This residue was purified by a mixture of toluene and cyclohexane. To the product obtained, methylene dichloride and acetonitrile were added and the reaction mixture was cooled to about -40°C to about -45°C. Triethylsilane (8.74g) was added to the reaction mixture followed by addition of boron trifluoride etherate (10.67g) maintaining temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution. The aqueous layer was separated and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum. The thick residue obtained was dissolved in methylene dichloride and cooled to about 0°C to about 5°C. Diisopropylethylamine, dimethylaminopyridine and acetic anhydride were added to the reaction mixture which was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol The residue obtained was recrystallized with methanol and charcoal to give title compound (iOg) with 99.7% HPLC purity.
The cardiovascular complications were highly prevalent in type 2 diabetes mellitus (T2DM), even at the early stage of T2DM or the state of intensive glycemic control. Therefore, there is an urgent need for the intervention of cardiovascular complications in T2DM. Herein, the new hybrids of NO donor and SGLT2 inhibitor were design to achieve dual effects of anti-hyperglycemic and anti-thrombosis. As expected, the preferred hybrid 2 exhibited moderate SGLT2 inhibitory effects and anti-platelet aggregation activities, and its anti-platelet effect mediated by NO was also confirmed in the presence of NO scavenger. Moreover, compound 2 revealed significantly hypoglycemic effects and excretion of urinary glucose during an oral glucose tolerance test in mice. Potent and multifunctional hybrid, such as compound 2, is expected as a potential candidate for the intervention of cardiovascular complications in T2DM.
Graphical abstract
Scheme 1. Synthesis of target compounds 1-3. Reagents and conditions: (a) TMSCl, NMM, THF, 35 °C; (b) (COCl)2, CH2Cl2, DMF, then phenetole, AlCl3, 0 °C; (c) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, 25 °C; (d) n-BuLi, THF, toluene, -78 °C, then 2a followed by MeOH, CH3SO3H; (e) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, -10 °C; (f) Ac2O, pyridine, CH2Cl2, DMAP;
Dapagliflozin, also known as SGLT2 inhibitor, chemical name is (2S,3R,4R,5S,6R)-2-[3-(4-ethoxybenzyl)-4-chlorophenyl]-6- Hydroxymethyltetrahydro-2H-pyran-3,4,5-triol, a sodium-glucose cotransporter 2 inhibitor, announced by the U.S. Food and Drug Administration (FDA) on January 8, 2014 , approved the use of dapagliflozin for the treatment of type 2 diabetes, the specific structural formula is as follows:
Dapagliflozin works by inhibiting sodium-glucose transporter 2 (SGLT2), a protein in the kidney that allows glucose to be reabsorbed into the blood. This allows excess glucose to be excreted through the urine, thereby improving blood sugar control without increasing insulin secretion.
At present, there are two main methods for synthesizing dapagliflozin. One uses 5-bromo-2-chlorobenzoic acid as the starting material, which is chlorinated, Falk acylated, reduced, and then combined with 2,3,4 ,6-tetra-0-trimethylsilyl-D-glucopyranosic acid 1,5-lactone is condensed, methyl etherified, and demethoxylated to obtain dapagliflozin. The specific process route is as follows:
The method has expensive starting materials and too many process steps, so it is not suitable for industrial production, and dangerous n-butyllithium needs to be used in the reaction process, so the requirements for experimental conditions are too high;
Another method is to use o-toluidine as the starting material, undergo bromination, diazotization, chlorination, and alkylation reactions, and then react with 2,3,4,6-tetra-0-trimethylsilyl -D-glucopyranosic acid 1,5-lactone is condensed, then methyl etherified and demethoxylated to obtain dapagliflozin. The specific process route is as follows:
AIBN will be used in this reaction, which will produce highly toxic cyanide, which will seriously pollute the environment, and also requires the use of n-butyllithium, which requires high experimental conditions and is dangerous to operate, and is not suitable for large-scale production.
Example 1
Weigh 16g (0.8mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodobenzene to it 2000mL of THF solution (total 2000mL: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsided, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene.
433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 423.6 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 93%.
Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 2500mL of toluene, then add 5.6g of europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 259.49 g of solid with a yield of 85%.
The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:3 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 147.1 g of white rice-like crystals with a yield of 80.2% and a purity of 99.2% (determined by high performance liquid chromatography, external standard method).
Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene .
433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 272.75mL of 33% hydrogen bromide in acetic acid solution (1.1mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 381.5 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 83.8%.
Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , dried to obtain solid 281.2g, yield 92.1%.
The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 150.9 g of white rice-like crystals with a yield of 88.2% and a purity of 99.5% (determined by high performance liquid chromatography, external standard method).
Example 3
Weigh 37.4g (1.862mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene .
433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 818.25mL of 33% hydrogen bromide in acetic acid solution (3.3mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 375.3 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 82.4%.
Weigh 217.83g 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 7500mL toluene, then add 5.6g europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 273.25 g of solid with a yield of 89.5%.
The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:5 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 152.7 g of white rice-like crystals with a yield of 82.9% and a purity of 99.4% (determined by high performance liquid chromatography, external standard method).
Example 4
Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene .
433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 425.1 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose in 94% yield.
Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, control the temperature at -20~-15°C, after the dropwise addition, raise the temperature to 5°C for 2 hours, concentrate in vacuo to an oily substance, then add to it 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, then 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 278.6 g of solid with a yield of 91.2%.
The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was completed, and 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 155.62 g of white rice-like crystals with a yield of 84.5% and a purity of 99.7% (determined by high performance liquid chromatography, external standard method).
Sodium-glucose co-transporter-2 (SGLT2) inhibitors are a group of oral medicines used for treating diabetes that have been approved since 2013. SGLT2 inhibitors prevent the kidneys from re-absorbing glucose back into the blood by passing into the bladder. Glucose is re-absorbed back into the blood via the renal proximal tubules. SGLT2 is a protein predominantly expressed in the renal proximal tubules and is likely to be major transporter responsible for this uptake. Glucose-lowering effect of SGLT-2 inhibitors occurs via an insulin-independent mechanism mostly through glucosuria by increasing the urinary excretion of glucose.
It has been shown that the treatment with SGLT2 inhibitors in patients with type II diabetes lowers HbAlc, reduces body weight, lowers systemic blood pressure (BP) and induces a small increase in LDL-C and HDL-C levels.
SGLT2 inhibitors inhibit the reabsorption of sodium and glucose from the tubule and hence, more sodium is delivered in the macula densa causing arteriole dilation, reduced intraglomerular pressure and decreased hyperfiltration. SGLT2 inhibitors cause natriuresis and volume depletion, and an increase in circulating levels of renin, angiotensin and aldosterone. They also reduce albuminuria and slow GFR loss through mechanisms that appear independent of glycemia.
Dapagliflozin is a highly potent and reversible SGLT2 inhibitor, which increases the amount of glucose excreted in the urine and improves both fasting and post-prandial plasma glucose levels in patients with type 2 diabetes. Dapagliflozin has also been shown to tend to reduce liver fat content in some studies in a diabetic population.
Dapagliflozin is available on the market in the form of dapagliflozin propanediol monohydrate and is sold under trade name Forxiga or Farxiga in the form of fdm-coated tablets. Further it is available on the market as a combination product with metformin hydrochloride which is sold under trade name Xigduo IR or Xigduo XR in the form of film-coated tablets. In addition, it is available on the market as a combination product with saxagliptin hydrochloride which is sold under trade name Qtem in the form of film-coated tablets. Moreover, it is available on the market as a combination product with saxagliptin hydrochloride and metformin hydrochloride which is sold under trade name Qtemmet XR in the form of film-coated tablets.
Dapagliflozin as a monotherapy and in a combination with other active substances has demonstrated its efficacy in improving glycaemic control and reducing body weight and blood pressure in a broad spectrum of patients with type II diabetes, including those with high baseline HbAlc and the elderly. A sustained reduction in serum uric acid concentration was also observed. Dapagliflozin provides significant improvement in HbAlc, reduction in insulin dose and reduction in body weight in patients with type 1 diabetes as adjunct therapy to adjustable insulin.
Dapagliflozin can be in its free form or any stereoisomer or any pharmaceutically acceptable salt or co crystal complex or a hydrate or a solvate thereof and in any polymorphic forms and any mixtures thereof.
Dapagliflozin as a substance was first disclosed in US 6,515,117. The process for the preparation of dapagliflozin involves the reaction of 4-bromo- 1 -chloro-2-(4-ethoxybenyl)benzene with 2,3,4,6-tetra-O-trimethyl silyl -D-gluconolactone, the obtained compound 3 on demethoxylation yields diastereomeric mixture of Dapagliflozin. Hie diastereomeric mixture of dapagliflozin is further acetylated with acetic anhydride in the presence of pyridine and dimethylaminopyridine yields, then recrystallized from absolute ethanol to yield the desired tetra-acetyJated b-C-glucoside as a white solid. Compound tetra-acetylated b-C-glucoside is treated with lithium hydroxide hydrate which undergoes deprotection to yield the compound dapagliflozin.
Several other documents, patents and applications disclose the process for the preparation of dapagliflozin such as for example WOO 127128, WO03099836, W02004063209, W02006034489,
Prior art documents already provided some compositions of SGLT2 inhibitor dapagliflozin.
W02008116179 discloses immediate release formulation in the form of a stock granulation or in the form of a capsule or a tablet which comprises dapagliflozin propylene glycol hydrate, one or more bulking agent, one or more binder and one or more disintegrant.
WO2011060256 describes the bilayer tablet comprising dapagliflozin having sustained release profde in one layer and metformin in another layer while WO2011060290 describes immediate release formulation of dapagliflozin and metformin.
WO2012163546 discloses the pharmaceutical composition comprising cyclodextrin and dapagliflozin.
Co-crystals of dapagliflozin with lactose are described in WO2014178040.
Solid dispersion compositions comprising amorphous dapagliflozin and at least one polymer are disclosed in W02015011113 and in WO2015128853.
CN103721261 discloses the combination of SGLT2 inhibitor with vitamins such as vitamin B.
Pharmaceutical composition preparation comprising dapagliflozin L-proline and metformin and/or DPP-IV inhibitor is disclosed in WO2018124497.
EP2252289A1 provides a combination of SGLT inhibitor with DPP4 inhibitor showing synergistic effect in increasing plasma active GLP-1 level in a patient over that provided by administration of the SGLT inhibitor or the DPP4 inhibitor alone.
EP2395983A1 relates to a pharmaceutical composition comprising a SGLT2 inhibitor, a DPP4 inhibitor and a third antidiabetic agent which is suitable in the treatment or prevention of one or more conditions selected from type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance and hyperglycemia.
Example A: HPLC method
The purity of Dapagliflozine in general may be determined with the following HPLC method: column: XBridge C18, 150×4.6mm, 3.5; flow-rate: 0.9ml/min; column temperature: 50°C, wavelength: UV 225 nm; mobile phase: eluent A: 0.1% H3PO4, Eluent B: methanol; gradient:
Sample preparation: Accurately weigh about 40mg of sample and dissolve in 50 ml of solvent. Calculation: Use area per cent method. Do not integrate solvent peaks.
Example 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
5-bromo-2-chlorobenzoic acid (450 g) was suspended in dichloromethane (2.25 L) and dimethylformamide (0.74 ml). At 15 – 30°C oxalyl chloride (180.3 ml) was slowly added. During addition gas evolution of HC1 and CO2 occurred. The reaction was performed at 20-30°C. The reaction was considered to be complete if 2-chloro-5-bromobenzoic acid was below 1% (area percent purity). The mixture was concentrated at elevated temperature until oily residue was obtained.
Example 2: Preparation of (5-bromo-2-chlorophenyl)(4-ethoxyphenyl)methanone
Dichloromethane (900 ml) was charged into reactor and then aluminum chloride (267.6 g) was added. The reaction mixture was cooled below 5°C and ethoxybenzene (256.1 ml) was slowly added. After complete addition, the mixture was gradually cooled below -5°C. In a separate reactor, 5-bromo-2-chlorobenzoyl chloride (485g) was dissolved in dichloromethane (900 ml). This solution was slowly added to the mixture of aluminum chloride and ethoxybenzene with such rate that temperature was kept below -5°C. After complete addition the mixture was stirred below -5°C until reaction was finished. The reaction was considered to be complete if methyl ester was below 1 % (the reaction mixture is sampled in methanol). After reaction was completed the reaction mixture was slowly added into cooled 1M HC1 solution and flushed with of dichloromethane (450 ml). The organic phase was separated and water phase was washed again with dichloromethane. Organic phases were combined and washed with water and NaHC03 solution. So obtained organic phase was concentrated to oily residue and dissolved in methanol ethyl acetate mixture in 10 to 1 ratio at reflux temperature. The clear solution was gradually cooled down to 35-45 °C and seeded with pure 5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone. The reaction mass was gradually cooled down to 0-10°C and stirred at that temperature up to 4 hours. The precipitate was isolated and washed with precooled methanol. The product was dried to a final LOD (Loss on drying) content of less than 1.0% with a yield of 564g (87% mass yield).
Example 3: Preparation of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene
5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone (400g) was dissolved in 1.62L tetrahydrofuran. Into solution NaBTL (53.5g ) was added. After addition, the mixture was stirred at ambient temperature for 30-60 min followed by cooling of reaction mixture below -5°C. Aluminum chloride (314g) was added in portion and reaction mixture maintained below 5°C. After addition, the reaction mixture was gradually heated to reflux temperature and stirred until reaction was complete. Reaction mixture was cooled to ambient temperature and mixture of THF/water was slowly added into reaction mixture followed by addition of water and stirred at ambient temperature. Organic phase was collected and washed with saturated NaCl solution. Organic phase was concentrated to oily residue and dissolved in ethanol (800ml) at elevated temperature. Solution was cooled to 25-30°C and seeded with pure 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene. The reaction mass was gradually cooled to -2 to 10°C and stirred at that temperature. The product was isolated and washed with precooled ethanol and dried until final LOD (Loss on drying) content was less than 1.0%. Yield was 322 g (89%).
Example 4: Preparation of 3R,4S,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (97.5g ) and toluene (1.46L) was charge into reactor. Solution was heated to reflux temperature and approximately half of the solvent was distilled out. Tetrahydrofuran (195 mL) was charged into the solution and mixture was cooled below -70°C. Solution of 15% «-Buli in hexane (227.5 ml) was slowly added and temperature was kept below -70°C. After complete addition solution was stirred at temperature below -70°C to complete reaction. Solution of 2,3,4,6-tetra-O-trimethylsilyl-D-gluconolactone (182 g) in toluene (243 mL) was added into reaction mixture at temperature below -70°C. After complete addition, the mixture was stirred below -70°C, warmed to approximately -65°C and then mixture of 57.6 g methanesulfonic acid in 488 ml methanol was added. After addition, the mixture was gradually warmed to ambient temperature and stirred until reaction was complete. After reaction was finished reaction mixture was slowly added into saturated NaHCCL solution (630ml) and stirred. Into quenched mixture 975 ml of heptane and 585 ml methanol was added. The mixture was stirred for additional 15 min. Organic phase was washed with water/methanol mixture several times. Water phases were combined and distilled to remove organic solvents. Into the residual water phase, toluene was added to perform extraction. Organic phases were combined and washed with water. Organic phase was distillated at elevated temperature until oily residue was obtained.
Example 5: Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl) tetrahydro2H-pyran-3,4,5-triol – Dapagliflozin
Dichloromethane (656 mL) was charged into oily residue from step 4 and stirred at ambient temperature until clear solution was obtained. Triethysilane (122 mL) was added into the so obtained solution. Reaction mixture was cooled below -30°C and 94.2 mL of boron trifluoride etherate was slowly added at temperatures below -30°C. After complete addition, the mixture was stirred below -30°C for one hour and gradually warmed to -5 to 0°C until reaction was completed. After reaction was finished saturated NaHCCL solution (468 mL) was slowly added. Reaction mixture was distilled to remove organic solvents followed by addition ethyl acetate into the residue. Organic phase was collected and washed again with saturated NaHC03 and water. So obtained organic phase was distillated at elevated temperature until oily residue is obtained.
Example 6: Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate.
Oily residual from example 5 was dissolved in dichloromethane (602 mL) at ambient temperature followed by addition of DMAP (6.22g). Reaction mixture was cooled to 0 – 10°C and 144.3 mL of acetic anhydride was added at temperatures below 10°C. Reaction mixture was gradually warmed to ambient temperature and stirred until reaction was completed. Reaction mass was washed with water with saturated NaHC03. Organic phase was collected and concentrated to oily residue to which ethanol (1.68L) was charged and approximately 300 ml ethanol was removed by distillation. The clear solution was gradually cooled to 60-65 °C and seeded. The reaction mass was gradually cooled to 20-25 °C and product was isolated. The product was dried at 50°C in vacuum until LOD (Loss on drying) below 1.0% 123g of product was obtained with yield 71%. HPLC purity: 99.97 %.
Formula 9 Formula 2
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (740 g) as prepared according to process described in examples 1 to 6 was charged into solution of methanol (2.27L), water (0.74L) and NaOH (23 lg) at 35-45°C and stirred at 35-45°C until reaction was completed. After reaction was finished 1M HC1 (1.63L) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. Combined organic phases were washed with water and concentrated at elevated temperature until oily residue was obtained. Content of impurity IMP A was below 0.02%.
Example 7a: Preparation of amorphous dapagliflozin.
Oily residue as prepared according to example 7 comprising approximately 262 g of dapagliflozin was dissolved in toluene (2.5L) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (5.3L) at temperature between 10 to 15°C and stirring rate with P/V at 4 W/m3. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was 14 · 104 m/s. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of impurity IMP A was below 0.02% and residual heptane and toluene were 1673 ppm and below 89 ppm.
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (30 g) of 4 different qualities obtained by the process known in the prior art was charged into solution of methanol (90 mL), water (30 mL) and NaOH (9.36 g) and stirred at 35-45°C until reaction was completed and sampled for HPLC analysis (Sample 1).
After reaction was finished 1M HC1 (66 mL) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. To the combined organic phases 68ml of 1M NaOH was added and pH was set to 12.5 to 13.5. Phases were separated and organic phase is washed again with 68ml of water without pH correction. So obtained organic phase was sampled for HPLC analysis (Sample 2) and concentrated at elevated temperature until oily residue was obtained.
Oily residue comprising approximately 21 g of dapagliflozin was dissolved in toluene (210 mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (420 mL) at temperature between 10 to 15°C and stirring rate with P/V as defined in Table 1. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was as defined in Table 1. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Amorphous dapagliflozin with content of impurity IMP A as shown in Table 1 and residual heptane and toluene as shown in Table 1 was obtained for each cases.
Table 1 : Process parameters used in the preparation of four different starting materials (cases).
As it is evident from Table 2 the final amorphous dapagliflozin prepared by the extraction process according to the present invention contains less than 0.02% of impurity IMP A irrespective of the level of impurity IMP A present in the starting material.
Table 2: Content of impurity IMP A in the final amorphous dapagliflozin obtained with and without extraction.
Example 9
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 25 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 10
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 40 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 11
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -10°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1480 ppm and 732 ppm.
Example 12
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 20°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 5°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled hcptanc Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2873 ppm and 639 ppm.
Comparative Example 1
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at -5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1940 ppm and 1557 ppm.
Comparative Example 2
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 25°C and stirring rate with P V at 16 W/m3. After complete addition, the suspension was cooled to 20°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 3663 ppm and 2047 ppm.
Comparative Example 3
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 30°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2425 ppm and 1812 ppm.
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Daggliflozin (English name: Dapagliflozin) is a new Sodium glucose co-transporters 2 (SGLT-2) inhibitor developed by Bristol-Myers Squibb and AstraZeneca. Approved by the European Commission on November 14, 2012, and marketed in the United States on January 8, 2014, to improve glycemic control in adult patients with type 2 diabetes by combining diet and exercise; the trade name is Farxiga, currently offering 5 mg and 10 mg tablets. At the same time, a combination of dapagliflozin and metformin hydrochloride has also been marketed.The chemical name of dapagliflozin is (2S,3R,4R,5S,6R)-2-(3-(4-ethoxybenzyl)-4-chlorophenyl)-6-hydroxymethyltetrahydro-2H – pyran-3,4,5-triol, the chemical formula is C 21 H 25 ClO 6 , CAS No. 461432-26-8, the structural formula is shown as 2, clinically used as a pharmaceutical for dapagliflozin (S) -1,2-propanediol monohydrate, the structural formula is as shown in 1.
The synthesis of β-type C-aryl glycosidic bonds is a key point in the synthetic route during the preparation of dapagliflozin. At present, there are four synthetic methods for the synthesis of dapagliflozin reported in the literature and patents.Route 1: The synthetic route of dapagliflozin reported in patent WO03099836A1 is as follows:
The route uses 2-chloro-5-bromobenzoic acid (12) as raw material to react with phenethyl ether to form intermediate 11 and then triethylsilane to obtain intermediate 10; intermediate 10 and n-butyl The lithium is reacted at -78 ° C, and then subjected to a nucleophilic addition reaction with the intermediate 9, and then methoxylated to obtain the intermediate 8; the intermediate 8 is subjected to acylation reduction and deprotection to obtain the intermediate 2. The disadvantage of this method is that the β-type C-aryl glycosidic bond synthesis of the compound is carried out at a low temperature of -78 ° C, which is obviously difficult to meet the needs of industrial production; and, through nucleophilic addition, methoxylation, The five-step reaction of acetylation, reduction and hydrolysis can synthesize the β-type C-aryl glycosidic bond. The procedure is relatively long, and the purity of the intermediate 2 is only 94%.Route 2: The synthetic route of dapagliflozin reported in the literature OrgLett.2012, 14, 1480 is as follows:
The intermediate 14 of the route is reacted with di-n-butyl-n-hexylmagnesium for 48 hours at 0 ° C, and then reacted with zinc bromide to prepare an organozinc reagent by Br/Mg/Zn exchange reaction, and then with intermediate 4 Intermediate 3 was prepared by nucleophilic substitution reaction; finally, intermediate 2 was obtained by deprotection with sodium methoxide. The synthesis method is relatively novel, and the synthesis step is short. However, the research experiment is conducted only as a synthesis method, and the post treatment of the intermediate 3 is performed by column chromatography. The purity of the intermediate 2 produced was not reported. Moreover, the di-n-butyl-n-hexylmagnesium reagent used in the route is not a commonly used reagent, and is not commercially available in China. It can only be prepared by reacting dibutylmagnesium with n-hexyllithium reagent before the test, and the operation is cumbersome and difficult to mass. use.Route 3: The synthetic route of dapagliflozin reported in patent WO2013068850A2 is as follows:
The route uses 1,6-anhydroglucose (20) as a raw material, protects the 2,4-hydroxyl group by tert-butyldiphenylchlorosilane, and then protects the 3-position hydroxyl group with phenylmagnesium bromide. Intermediate 18. The intermediate 14 is subjected to an Br/Mg/Al exchange reaction to prepare an organoaluminum reagent 16, which is reacted with an intermediate 18 to form an intermediate 15, and finally, deprotected to obtain an intermediate 2. The synthesis method is very novel and is also used as a synthetic methodological study. The purification of the intermediates is carried out by column chromatography. The 1,6-anhydroglucose (20) used in the route is very expensive; and the multi-step reaction in the route uses a format reagent, a preparation format reagent or an organoaluminum reagent, which is cumbersome and cumbersome to perform, and is difficult to scale synthesis. The purity of the intermediate 2 produced was not reported.Route 4: The synthetic route of dapagliflozin reported in patent WO2013152476A1 is as follows:
The route uses 2-chloro-5-iodobenzoic acid (24) as raw material to form intermediate 22 by Friedel acylation and reduction reaction, and exchange with I-Mg at -5 ° C with isopropyl magnesium chloride lithium chloride. The intermediate 8 is obtained by nucleophilic addition and methoxylation with the intermediate 9, and then the intermediate 2 is obtained by reduction with triethylsilane, and the intermediate 2 is further purified by co-crystallizing with L-valine. Finally, The pure intermediate 2 was obtained by removing L-valine. This route is a modified route of Route 1, which replaces n-butyllithium with isopropylmagnesium chloride chloride to raise the reaction temperature of the reaction from -78 °C to -5 °C. However, the problem of a long step of synthesizing a β-type C-aryl glycosidic bond still exists. The obtained intermediate 2 is not optically pure, and needs to be purified by co-crystallizing with L-valine, and the work amount of post-treatment is increased, and finally the purity of the intermediate 2 is 99.3%.Among the four synthetic routes described above for dapagliflozin, route one and route four are commonly used synthetic methods for β-type C-aryl glycosidic bonds, and the route is long, and the optical purity of the obtained product is not high, and further purification is required. Post processing is cumbersome. Moreover, the reaction required at -78 °C in Route 1 requires high equipment and high energy consumption, which undoubtedly increases the cost. Although both Route 2 and Route 3 are new methods, most of the purification of intermediates used is column chromatography. Such a process is not suitable for scale production in factories; and some of the synthetic routes are used. Reagents are not commercially available or expensive, and there is no advantage in such route costs. Therefore, there is an urgent need to find a new method for the synthesis of dapagliflozin, and to enable industrial production, and the route has a cost advantage.Repeating the procedure reported in the literature in Equation 2, the yield of Intermediate 3 was only 46%. The organic zinc reagent is prepared by Br/Mg/Zn exchange reaction, and the exchange reaction yield is 78%; and the raw material is prepared by X/Li/Zn exchange reaction to prepare an organic zinc reagent, and the exchange reaction yield is 98.5%, which is also the two Different reaction pathways lead to the essential reason for the different yields of intermediate 3. Moreover, the price of commercially available 1.0 mol/L di-n-butyl magnesium n-heptane solution 500 mL is 1380 yuan, and the price of 1.6 mol/L n-hexyl lithium n-hexane solution 500 mL is 950 yuan, and 2.5 mol/L n-butyl lithium. The price of 500 mL of n-hexane solution is only 145 yuan. Therefore, the method for preparing dapagliflozin by preparing an organozinc reagent by X/Li/Zn and then synthesizing the β-type C-aryl glycosidic bond designed by the invention has the advantages of cost, ease of operation and industrialization. Very obvious advantage.In order to solve this problem, the original compound company uses a eutectic method in the production of dapagliflozin to make dapagliflozin together with a solvent or an amino acid compound, since the compound 2 sugar ring structure contains four hydroxyl groups and is easy to absorb moisture and deteriorate. The crystal is made into a relatively stable solid, easy to store, stable and controllable in quality, and easy to prepare. Among them, the marketed dapagliflozin forms a stable eutectic with (S)-1,2-propanediol and water (1). The original crystal form patent (CN101479287B, CN103145773B) reported that all 11 crystal forms are dapagliflozin solvate or dapagliflozin. Crystal. Among them, there are two preparation methods for the da forme (S)-1,2-propanediol monohydrate (1) having a crystal structure of type Ia:Method 1: The preparation method is as follows:
Compound 7 is deprotected with sodium hydroxide to obtain compound 2, then compound 2 is extracted with isopropyl acetate, (S)-1,2-propanediol ((S)-PG) is added, and seed crystal of compound 1 is added. Then, cyclohexane was added to crystallize and separated to obtain a eutectic of the compound (1) of the type Ia.Method 2: The preparation method is as follows:
Compound 8 is subjected to reduction of methoxy group by triethylsilane and boron trifluoride diethyl ether complex, and then the reaction solution is extracted with methyl tert-butyl ether (MTBE), and (S)-1,2-propanediol ( (S)-PG), a seed crystal of the compound 1 is added, and then cyclohexane is added to crystallize, and the mixture is separated and dried to obtain a eutectic of the compound (1) of the type Ia.The above two methods for preparing the eutectic are all used in the cyclohexane solvent, which is listed in the appendix of the 2015 edition of the Pharmacopoeia (four parts) as the second type of solvent that should be restricted, with a residual limit of 0.388%. The solvent residue of the final product obtained must reach the specified limit, and the post-treatment process is complicated, time-consuming and labor-intensive, and the production cost is correspondingly increased. The invention finds a suitable solvent on the basis of the synthetic route to prepare a medicinal crystal form, and has obvious advantages in both the method and the process operation steps.The synthetic route is as follows:
Comparative Example 1, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4- Preparation of chlorophenyl]glucosamine (Compound 3)Under nitrogen protection, 1.0 mol/L di-n-butylmagnesium-n-heptane solution (16 mL) was cooled to 0 ° C, and 1.6 mol/L n-hexane lithium n-hexane solution (10 mL) was slowly added dropwise. After the addition was completed, 0 ° C After stirring for 15 h, dry n-butyl ether (2.5 mL) was added to prepare a solution of di-n-butyl-n-hexylmagnesium lithium solution, which was calibrated with iodine and stored for use.Zinc bromide (2.7 g) and lithium bromide (1.04 g) were added with n-butyl ether (20 mL), heated to 50 ° C for 4 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (8 mL) and n-butyl ether (5 mL) under nitrogen, cooled to 0 ° C, and 0.61 mol/L was added dropwise. n-Butyl-n-hexylmagnesium lithium solution (13.1 mL), after the addition is completed, the reaction was kept at 0 ° C for 48 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution were added, and the reaction was kept at 0 ° C for 1 h, and added 2 , 3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (25 mL), heated to 100 ° C to stir the reaction, after TLC detection reaction, add 1 mol / L diluted hydrochloric acid (60 mL), taken after stirring extraction, the organic phase was washed with water (40 mL), then washed with saturated brine (40 mL), dried over anhydrous Na 2 SO 4, concentrated under reduced pressure, column chromatography (petroleum ether / Ethyl acetate = 20:1) 10.38 g of Compound 3 as a pale yellow oil. Yield: 46%. Purity: 99.02%. The organozinc reagent prepared by the method has an iodine calibration yield of 78%.The calibration method of the concentration of the prepared organic zinc reagent: accurately weighed iodine (1 mmol), placed in a three-necked flask, replaced nitrogen, and added anhydrous 0.5 mol/L LiCl tetrahydrofuran solution (5 mL), stirred and dissolved, and cooled to 0 ° C. The prepared organozinc reagent was slowly added dropwise until the color of the brownish yellow solution disappeared.Example 2 (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (2.25 g) and lithium bromide (0.87 g) were added with n-butyl ether (30 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and n-butyl ether (10 mL) under nitrogen, cooled to -20 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (14mL), control the internal temperature does not exceed -10 ° C, after the completion of the addition, the temperature is incubated at -20 ° C for 0.5 h, adding the above-mentioned spare zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at 20 ° C for 3 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (11.59g) toluene (50mL) solution, heat to 120 ° C and stir the reaction for 4h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (40 mL), water (20 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.8 g of Compound 3 as a white solid was obtained in a yield: 72.42%. Purity: 99.47%. Melting point: 99.5 to 101.6 °C. (The organic zinc reagent prepared by this method was iodine-calibrated in a yield of 98.5%.) ESI-MS (m/z): 767.30 [M+Na] + . 1 H-NMR (400 MHz, CDCl 3 ): δ 7.33 (1H, d), 7.14-7.17 (2H, m), 7.05 (2H, d), 6.79-6.81 (2H, dd), 5.39 (1H, t ), 5.21-5.31 (2H, m), 4.33 (1H, d), 4.17-4.20 (1H, dd), 3.94-4.11 (5H, m), 3.79-3.83 (1H, m), 1.39 (3H, t ), 1.20 (9H, s), 1.16 (9H, s), 1.11 (9H, s), 0.86 (9H, s).Example 3, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3) PrepareZinc bromide (3.38 g) and lithium bromide (1.3 g) were added with n-butyl ether (40 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (20 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -50 ° C, and slowly added dropwise 2.5 mol / L-butyllithium hexane solution (8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -50 ° C for 10 h, adding the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at -20 ° C for 10 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (34.77g) toluene (80mL) solution, heat to 100 ° C and stir the reaction for 24h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (60 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.854 g of Compound 3 as a white solid. Yield: 72.81%. Purity: 99.53%.Example 4, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)N-butyl ether (50 mL) was added to zinc iodide (3.19 g) and lithium iodide (1.34 g), and the mixture was heated to 50 ° C for 1.5 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -60 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -60 ° C for 5 h, and the above-mentioned alternate zinc iodide and lithium iodide n-butyl ether solution is added. The reaction was stirred at 25 ° C for 1 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (23.2g) toluene (50mL) solution, heat to 140 ° C reflux reaction for 0.5h, after TLC detection reaction was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous SO 4 Na 2, concentrated by weight of n-heptane (15mL) and methanol (60 mL) Crystallization gave 10.51 g of Compound 3 as a white solid, yield 70.5%. Purity: 99.41%.Example 5, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)To the zinc bromide (2.25 g) and lithium bromide (0.87 g), cyclopentyl methyl ether (30 mL) was added, and the mixture was heated to 50 ° C for 3 hours, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and cyclopentyl methyl ether (10 mL) under nitrogen, cooled to -5 ° C, and slowly added dropwise. Mol / L n-hexyl lithium n-hexane solution (12.5mL), control the internal temperature does not exceed 0 ° C, after the addition is completed, the reaction is kept at -5 ° C for 3 h, adding the above-mentioned spare zinc bromide and lithium bromide cyclopentyl methyl ether The solution was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (40 mL) was added and heated to 80 ℃ reaction was stirred 6h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous 2 SO 4 Na, and concentrated under reduced pressure, Recrystallization of n-heptane (15 mL) and methanol (60 mL) gave 8.15 g of Compound 3 as a white solid. Purity: 99.39%.Example 6, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (4.5 g) and lithium bromide (1.74 g) were added with n-butyl ether (60 mL), heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -30 ° C, and slowly added dropwise 2.5 mol / L-butyllithium n-hexane solution (8.4mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -30 ° C for 3 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution is added. The reaction was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (50 mL) was added and heated to 120 ° C for stirring. the reaction 4h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure, n-heptyl Recrystallization of the alkane (15 mL) and methanol (60 mL) gave 10.38 g of Compound 3 as a white solid. Purity: 99.54%.Example 7, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Methyl bromide (40 mL) was added to zinc bromide (2.25 g) and lithium bromide (0.87 g), and the mixture was heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL), methyl tert-butyl ether (15 mL), cooled to -40 ° C, and slowly added dropwise. 1.6mol/L n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -40 ° C for 4 h, and the above-mentioned alternate zinc bromide and lithium bromide are added. The butyl ether solution was incubated at 5 ° C for 7 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (50 mL) was added and heated. to 90 deg.] C the reaction was stirred 8h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (40 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure Recrystallization from n-heptane (15 mL) and methanol (60 mL) gave 9.41 g of Compound 3 as a white solid. Purity: 99.42%. Example 8. Preparation of dapagliflozin (S)-1,2-propanediol monohydrate eutectic (Compound 1)To the compound 3 (37.27 g), methanol (190 mL) was added, and sodium methoxide (10.8 g) was added thereto, and the mixture was heated under reflux for 3 hours. After the TLC reaction was completed, methanol was concentrated, and isopropyl acetate (100 mL) was added to the residue, and water was added. (60 mL), extracted with stirring and the organic phase washed with water (50 mL). (S)-1,2-propanediol (3.8g) and water (0.9g) were added to the organic phase, stirred until it was dissolved, and n-heptane (200 mL) was added, and the mixture was stirred for 2 hours under ice-cooling, suction filtration, filter cake Washing with n-heptane and drying at 30 ° C gave 23.89 g of Compound 1 as a white solid. Yield: 95%. Purity: 99.79%. Melting point: 69.1 to 75.6 °C. The product obtained was subjected to KF = 3.74% (theoretical value: 3.58%). ESI-MS (m/z): 431.22 [M+Na] + . 1 H-NMR (400 MHz, CD 3 OD): δ 7.33 – 7.37 (2H, m), 7.28-7.30 (1H, dd), 7.11 (2H, d), 6.80-6.83 (2H, dd), 4.1 ( 1H, d), 3.98-4.05 (4H, m), 3.88-3.91 (1H, dd), 3.74-3.82 (1H, m), 3.68-3.73 (1H, m), 3.37-3.49 (5H, m), 3.28-3.34 (1H, m), 1.37 (1H, t), 1.15 (3H, d).The crystal form of the obtained product was subjected to thermogravimetric analysis (TGA) by a Universal V4.7A TA instrument, and the TGA curve (Fig. 1) showed a weight loss of about 18.52% from about room temperature to about 240 ° C. The original form Ia crystal form The TGA plot shows a value of 18.7%.The crystal form of the obtained product was subjected to differential scanning calorimetry (DSC) by a Universal V4.7A TA instrument, and the DSC curve (Fig. 2) showed endotherm in the range of about 60 ° C to 85 ° C. The DSC plot shows a range of approximately 50 ° C to 78 ° C.
The crystal form of the obtained product was examined by a Bruker D8advance instrument for powder X-ray diffraction (PXRD), and the 2X value of the PXRD pattern (Fig. 3) (CuKα).
There are characteristic peaks at 3.749°, 7.52°, 7.995°, 8.664°, 15.134°, 15.708°, 17.069°, 18.946°, 20.049°, which are completely consistent with the characteristic peaks of the PXRD pattern of the Ia crystal form in the original patent.In combination with the nuclear magnetic data and melting point of the prepared crystal form, the crystal form of the product (Compound 1) obtained by the present invention is consistent with the pharmaceutically acceptable crystalline form Ia reported in the original patent.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleCN101479287A *2006-06-282009-07-08布里斯托尔-迈尔斯斯奎布公司Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetesCN104496952A *2014-11-282015-04-08深圳翰宇药业股份有限公司Synthesis method of dapagliflozinCN105153137A *2015-09-172015-12-16上海应用技术学院Preparation method of empagliflozinFamily To Family CitationsCN104829572B *2014-02-102019-01-04江苏豪森药业集团有限公司Dapagliflozin novel crystal forms and preparation method thereofCN105399735A *2015-12-292016-03-16上海应用技术学院Empagliflozin intermediate, and preparation method and application thereof* Cited by examiner, † Cited by third party
Non-Patent Citations
TitleCHEN DEJIN ET AL., CHINA MASTER’S THESES FULL-TEXT DATABASE, ENGINEERING TECHNOLOGY I, vol. B016-731, no. 3, 15 March 2016 (2016-03-15) *LEMAIRE S. ET AL.: “Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents”, PURE APPL. CHEM., vol. 86, no. 3, 4 March 2014 (2014-03-04), pages 329 – 333 *LEMAIRE S. ET AL.: “Stereoselective C-Glycosylation Reactions with Arylzinc Reagents”, ORGANIC LETTERS, vol. 14, no. 6, 2 March 2012 (2012-03-02), pages 1480 – 1483, XP055069093 ** Cited by examiner, † Cited by third partyCLIP
Chemical Synthesis
Dapagliflozin propanediol hydrate, an orally active sodium glucose cotransporter type 2 (SGLT-2) inhibitor, was developed by Bristol-Myers Squibb (BMS) and AstraZeneca for the once-daily treatment of type 2 diabetes. As opposed to competitor SGLT-2 inhibitors, dapagliflozin was not associated with renal toxicity or long-term deterioration of renal function in phase III clinical trials. The drug exhibits excellent SGLT2 potency with more than 1200 fold selectivity over the SGLT1 enzyme.
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7
dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .
formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2
formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
PATENT
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
HPLC
HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method: Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm; Column temperature: 25 °C – Eluent A: 0.1 % formic acid in water – Eluent B: 0.1 % formic acid in acetonitrile – Injection volume: 3 mL – Flow: 0.7 mL/min – Gradient:Time [min][%] B0.02525.06526.07029.07029.52535.025……………………..
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))
[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)
III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
LINK>>> (WO2016147197) A NOVEL PROCESS FOR PREPARING (2S,3R,4R,5S,6R)-2-[4-CHLORO-3-(4-ETHOXYBENZYL)PHENY 1] -6-(HY DROXY METHYL)TETRAHYDRO-2H-PY RAN-3,4,5-TRIOL AND ITS AMORPHOUS FORM
PATENT
PATENT
WO2016018024, CRYSTALLINE COMPOSITE COMPRISING DAPAGLIFLOZIN AND METHOD FOR PREPARING SAME
HANMI FINE CHEMICAL CO., LTD. [KR/KR]; 59, Gyeongje-ro, Siheung-si, Gyeonggi-do 429-848 (KR)
Dapagliflozin, sold under the brand name Farxiga among others, is a medication used to treat type 2 diabetes and, with certain restrictions, type 1 diabetes.[2] It is also used to treat adults with certain kinds of heart failure.[3][4][5]
It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. In 2018, it was the 227th most commonly prescribed medication in the United States, with more than 2 million prescriptions.[10][11]
Medical uses
Dapagliflozin is used along with diet and exercise to improve glycemic control in adults with type 2 diabetes and to reduce the risk of hospitalization for heart failure among adults with type 2 diabetes and known cardiovascular disease or other risk factors.[12][3] It appears more useful than empagliflozin.[13][verification needed]
In addition, dapagliflozin is indicated for the treatment of adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3][4][5] It is also indicated to reduce the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14]
In the European Union it is indicated in adults:
for the treatment of insufficiently controlled type 2 diabetes mellitus as an adjunct to diet and exercise:
as monotherapy when metformin is considered inappropriate due to intolerance;
in addition to other medicinal products for the treatment of type 2 diabetes;
for the treatment of insufficiently controlled type 1 diabetes mellitus as an adjunct to insulin in patients with BMI ≥ 27 kg/m2, when insulin alone does not provide adequate glycaemic control despite optimal insulin therapy; and
for the treatment of heart failure with reduced ejection fraction.[5]
Adverse effects
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Rarely, use of an SGLT2 drug, including dapagliflozin, is associated with necrotizing fasciitis of the perineum, also called Fournier gangrene.[15]
Dapagliflozin can cause dehydration, serious urinary tract infections and genital yeast infections.[3] Elderly people, people with kidney problems, those with low blood pressure, and people on diuretics should be assessed for their volume status and kidney function.[3] People with signs and symptoms of metabolic acidosis or ketoacidosis (acid buildup in the blood) should also be assessed.[3] Dapagliflozin can cause serious cases of necrotizing fasciitis of the perineum (Fournier gangrene) in people with diabetes and low blood sugar when combined with insulin.[3]
To lessen the risk of developing ketoacidosis (a serious condition in which the body produces high levels of blood acids called ketones) after surgery, the FDA has approved changes to the prescribing information for SGLT2 inhibitor diabetes medicines to recommend they be stopped temporarily before scheduled surgery. Canagliflozin, dapagliflozin, and empagliflozin should each be stopped at least three days before, and ertugliflozin should be stopped at least four days before scheduled surgery.[17]
Symptoms of ketoacidosis include nausea, vomiting, abdominal pain, tiredness, and trouble breathing.[17]
Use is not recommended in patients with eGFR < 45ml/min/1.73m2, though data from 2021 shows the reduction in the kidney failure risks in people with chronic kidney disease using dapagliflozin.[18]
Mechanism of action
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2) which are responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter mechanism causes blood glucose to be eliminated through the urine.[19] In clinical trials, dapagliflozin lowered HbA1c by 0.6 versus placebo percentage points when added to metformin.[20]
Regarding its protective effects in heart failure, this is attributed primarily to haemodynamic effects, where SGLT2 inhibitors potently reduce intravascular volume through osmotic diuresis and natriuresis. This consequently may lead to a reduction in preload and afterload, thereby alleviating cardiac workload and improving left ventricular function.[21]
Selectivity
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/L), so that the drug does not interfere with intestinal glucose absorption.[22]
In July 2016, the fixed-dose combination of saxagliptin and dapagliflozin was approved for medical use in the European Union and is sold under the brand name Qtern.[28] The combination drug was approved for medical use in the United States in February 2017, where it is sold under the brand name Qtern.[29][30]
In May 2019, the fixed-dose combination of dapagliflozin, saxagliptin, and metformin hydrochloride as extended-release tablets was approved in the United States to improve glycemic control in adults with type 2 diabetes when used in combination with diet and exercise. The FDA granted the approval of Qternmet XR to AstraZeneca.[31] The combination drug was approved for use in the European Union in November 2019, and is sold under the brand name Qtrilmet.[32]
Dapagliflozin was found effective in several studies in participants with type 2 and type 1 diabetes.[5] The main measure of effectiveness was the level of glycosylated haemoglobin (HbA1c), which gives an indication of how well blood glucose is controlled.[5]
In two studies involving 840 participants with type 2 diabetes, dapagliflozin when used alone decreased HbA1c levels by 0.66 percentage points more than placebo (a dummy treatment) after 24 weeks.[5] In four other studies involving 2,370 participants, adding dapagliflozin to other diabetes medicines decreased HbA1c levels by 0.54-0.68 percentage points more than adding placebo after 24 weeks.[5]
In a study involving 814 participants with type 2 diabetes, dapagliflozin used in combination with metformin was at least as effective as a sulphonylurea (another type of diabetes medicines) used with metformin.[5] Both combinations reduced HbA1c levels by 0.52 percentage points after 52 weeks.[5]
A long-term study, involving over 17,000 participants with type 2 diabetes, looked at the effects of dapagliflozin on cardiovascular (heart and circulation) disease.[5] The study indicated that dapagliflozin’s effects were in line with those of other diabetes medicines that also work by blocking SGLT2.[5]
In two studies involving 1,648 participants with type 1 diabetes whose blood sugar was not controlled well enough on insulin alone, adding dapagliflozin 5 mg decreased HbA1c levels after 24 hours by 0.37% and by 0.42% more than adding placebo.[5]
Dapagliflozin was approved for medical use in the European Union in November 2012.[5] It is marketed in a number of European countries.[33]
Dapagliflozin was approved for medical use in the United States in January 2014.[34][14]
In 2020, the U.S. Food and Drug Administration (FDA) expanded the indications for dapagliflozin to include treatment for adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3] It is the first in this particular drug class, sodium-glucose co-transporter 2 (SGLT2) inhibitors, to be approved to treat adults with New York Heart Association’s functional class II-IV heart failure with reduced ejection fraction.[3]
Dapagliflozin was shown in a clinical trial to improve survival and reduce the need for hospitalization in adults with heart failure with reduced ejection fraction.[3] The safety and effectiveness of dapagliflozin were evaluated in a randomized, double-blind, placebo-controlled study of 4,744 participants.[3] The average age of participants was 66 years and more participants were male (77%) than female.[3] To determine the drug’s effectiveness, investigators examined the occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visits.[3] Participants were randomly assigned to receive a once-daily dose of either 10 milligrams of dapagliflozin or a placebo (inactive treatment).[3] After about 18 months, people who received dapagliflozin had fewer cardiovascular deaths, hospitalizations for heart failure, and urgent heart failure visits than those receiving the placebo.[3]
In July 2020, the FDA granted AstraZeneca a Fast Track Designation in the US for the development of dapagliflozin to reduce the risk of hospitalisation for heart failure or cardiovascular death in adults following a heart attack.[35]
In August 2020, it was reported that detailed results from the Phase III DAPA-CKD trial showed that AstraZeneca’s FARXIGA® (dapagliflozin) on top of standard of care reduced the composite measure of worsening of renal function or risk of cardiovascular (CV) or renal death by 39% compared to placebo (p<0.0001) in patients with chronic kidney disease (CKD) Stages 2-4 and elevated urinary albumin excretion. The results were consistent in patients both with and without type 2 diabetes (T2D)[36]
In April 2021, the FDA expanded the indications for dapagliflozin (Farxiga) to include reducing the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14] The efficacy of dapagliflozin to improve kidney outcomes and reduce cardiovascular death in people with chronic kidney disease was evaluated in a multicenter, double-blind study of 4,304 participants.[14]
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Dapagliflozin is an approved drug by U.S. Food and Drug Administration (FDA). Dapagliflozin is a representative of SGLT-2 inhibitors, actively considered to cure diabetes type 2. Thus, methodology of dapagliflozin synthesis has rarely published (Ellsworth et al. 2002; Meng 2008). Scheme 1 have shown the general synthetic route for the synthesis of dapagliflozin. Gluconolactone 3 which was protected by trimethylsilyl TMS was treated with aryl lithium. Aryl lithium was obtained by reacting aryl bromide 2 (exchange of Li/Br) with n-BuLi. Methyl C-aryl glucoside 4 was produced by treatment of resulting mixture with methane sulfonic acid in the presence of methanol. Compound 4 was subjected to acetylation in the presence of Ac2O, resulted in the formation of 5 followed by reduction of 5 to 6 with the help of Et3SiH and BF3.OEt2. Finally, dapagliflozin 1 was produced via hydrolysis of 6 by LiOH (Deshpande et al. 2008; Meng 2008).
Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN (2008) “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. ” Journal of Medicinal Chemistry 51(5): 1145–1149. https://doi.org/10.1021/jm701272q
Deshpande PP, Ellsworth BA, Singh J, Denzel TW, Lai C, Crispino G, Randazzo ME, Gougoutas JZ (2008) Methods of producing C-aryl glucoside SGLT2 inhibitors, Google Patents. https://patents.google.com/patent/US7375213B2/en
Jun et al. has reported a few improvements to the scheme 1. In the improved methodology scheme 2, trimethylsilyl chloride was added to gluconolactone 7 in the presence of N-methylmorpholine and tetrahydrofuran THF (Horton et al. 1981), followed by the formation of persilyated lactone 3. After completing the reaction of aryl bromide 2 with n-BuLi, added to persilyated lactone 3. Intermediate lactol 8 was produced by treating resulting reaction mixture with trifluoroacetic acid in aqueous form. Then ethyl C-aryl glycoside 9 yielded when subsequently compound 8 was subjected to methane-sulfonic acid in ethyl alcohol. Crude product 9 in the form of oil was secured after the screening of solvents. Jun et al. proposed that more than 98% pure 9 was collected as crystalline solvate after the crystallization of crude oil from n-propanol and n-heptane mixture (Yu et al. 2019). Moreover, Wang et al. proposed that a high extent of diastereoselectivity obtained after the reduction of tetra-O-unprotected methyl C-aryl glucoside by utilizing Et3SiH and BF3.Et2O (Wang et al. 2014). The nature of active pharmaceutical ingredient is amorphous foam which is isolated after the reduction of 9. Production of cocrystalline complex facilitate the isolation and purification of API (Deng et al. 2017). It is concluded that more than 99.7% pure dapagliflozin produced in overall 79% yield, after the crystallization of a mixture consists of n-heptane and ethyl acetate (Yu et al. 2019).
Zheng et al. designed the production methodology of dapagliflozin by introducing NO donor group at the last steps of general route of dapagliflozin synthesis (scheme 1). Novel hybrids achieved by the combination of dapagliflozin and NO donor, having excellent dual characteristics of anti-hyperglycemic and anti-thrombosis. The figure 2 represent the modifiable site (4-position) of dapagliflozin.
Horton D, Priebe W (1981) “Synthetic routes to higher-carbon sugars. Reaction of lactones with 2-lithio-, 3-dithiane. ” Carbohydrate Research 94(1): 27–41. https://doi.org/10.1016/S0008-6215(00)85593-7
Yu J, Cao Y, Yu H, Wang JJ (2019) “A Concise and Efficient Synthesis of Dapagliflozin. ” Organic Process Research & Development 23(7): 1458–1461. https://doi.org/10.1021/acs.oprd.9b00141
Wang X-j, Zhang L, Byrne D, Nummy L, Weber D, Krishnamurthy D, Yee N, Senanayake CH (2014) “Efficient synthesis of empagliflozin, an inhibitor of SGLT-2, utilizing an AlCl3-promoted silane reduction of a β-glycopyranoside. ” Organic Letters 16(16): 4090–4093. https://doi.org/10.1021/ol501755h
Deng J-H, Lu T-B, Sun CC, J-M Chen (2017) “Dapagliflozin-citric acid cocrystal showing better solid state properties than dapagliflozin. ” European Journal of Pharmaceutical Sciences 104: 255–261. https://doi.org/10.1016/j.ejps.2017.04.008
Scheme 3 has shown the formation strategy of new hybrids of nitric oxide with dapagliflozin. During the synthesis process, the compound 6 was treated with BBr3 that result in the formation of phenol 10, which was further subjected to condensation with bromoalkane and then undergo hydrolysis and produce 11 intermediates. Target compound was obtained by the reacting 11 with silver nitrate in acetonitrile (Li et al. 2018).
Li Z, Xu X, Deng L, Liao R, Liang R, Zhang B, Zhang LJB (2018) Design, synthesis and biological evaluation of nitric oxide releasing derivatives of dapagliflozin as potential anti-diabetic and anti-thrombotic agents. Bioorganic & Medicinal Chemistry 26(14): 3947–3952. https://doi.org/10.1016/j.bmc.2018.06.017
Lin et al. fabricated green route (scheme 4) for the production of dapagliflozin. 5-bromo-2-chlorobenzoic acid 12 and gluconolactone were utilized to initiate the synthesis. By taking BF3.Et2O in catalytic amount to produce 13, overall yield of 76% was obtained in one-pot way via considering the Friedel-Craft acylation and ketallization. There was no need to do work-up operations to separate the diaryl ketal 13 as it was easily crystallized from the mixture. Compound 14 was produce as a result of condensation between 13 and 3. Overall yield of 68% of compound 15 was produced by the deprotection of silyl group in ethyl alcohol media. In THF presence, single crystals of 15 was achieved and characterized by XRD-analysis. High yield of dapagliflozin was obtained after the reduction of 15 that was carried out by triethylsilane in the presence of boron trifluoride diethyl etherate in dichloromethane. Upon crystallization from the mixture having heptane and ethyl acetate, greater than 98% pure dapagliflozin was produced by green synthetic pathway (Hu et al. 2019).
Hu L, Zou P, Wei W, Yuan X-M, Qiu X-L, Gou S-H (2019) “Facile and green synthesis of dapagliflozin. ” Synthetic Communications 49(23): 3373–3379. https://doi.org/10.1080/00397911.2019.1666283
PAPER
A Concise and Efficient Synthesis of Dapagliflozin
Cite this: Org. Process Res. Dev.2019, 23, 7, 1458–1461
A concise and efficient synthesis of the SGLT-2 inhibitor dapagliflozin (1) has been developed. This route involves ethyl C-aryl glycoside 9 as the key intermediate, which is easily crystallized and purified as the crystalline n-propanol solvate with high purity (>98.5%). The tetra-O-unprotected compound 9 could be directly reduced to crude dapagliflozin with high diastereoselectivity. The final pure API product 1 was isolated and purified with high purity (>99.7%). The process has been implemented on a multikilogram scale.
A general, transition-metal-free, highly stereoselective cross-coupling reaction between glycosyl bromides and various arylzinc reagents leading to β-arylated glycosides is reported. The stereoselectivity of the reaction is explained by invoking anchimeric assistance via a bicyclic intermediate. Stereochemical probes confirm the participation of the 2-pivaloyloxy group. Finally, this new method was applied to a short and efficient stereoselective synthesis of Dapagliflozin and Canagliflozin.
One study found that it had no benefit on heart disease risk or overall risk of death in people with diabetes.[37] Another study found that in heart failure with a reduced ejection fraction, dapagliflozin reduced the risk of worsening of heart failure or progression to death from cardiovascular causes, irrespective of diabetic status.[38]
^ Ptaszynska, Agata; Johnsson, Kristina M.; Parikh, Shamik J.; De Bruin, Tjerk W. A.; Apanovitch, Anne Marie; List, James F. (2014). “Safety Profile of Dapagliflozin for Type 2 Diabetes: Pooled Analysis of Clinical Studies for Overall Safety and Rare Events”. Drug Safety. 37 (10): 815–829. doi:10.1007/s40264-014-0213-4. PMID25096959. S2CID24064402.
^ Zelniker TA, Wiviott SD, Raz I, et al. (January 2019). “SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials”. Lancet. 393(10166): 31–9. doi:10.1016/S0140-6736(18)32590-X. PMID30424892. S2CID53277899. However, in patients with atherosclerotic cardiovascular disease, the effect of empagliflozin on cardiovascular death was more pro-nounced than that of canagliflozin or dapagliflozin
Clinical trial number NCT00528372 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled With Diet and Exercise” at ClinicalTrials.gov
Clinical trial number NCT00643851 for “An Efficacy & Safety Study of BMS-512148 in Combination With Metformin Extended Release Tablets” at ClinicalTrials.gov
Clinical trial number NCT00859898 for “Study of Dapagliflozin in Combination With Metformin XR to Initiate the Treatment of Type 2 Diabetes” at ClinicalTrials.gov
Clinical trial number NCT00528879 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled on Metformin Alone” at ClinicalTrials.gov
Clinical trial number NCT00660907 for “Efficacy and Safety of Dapagliflozin in Combination With Metformin in Type 2 Diabetes Patients” at ClinicalTrials.gov
Clinical trial number NCT00680745 for “Efficacy and Safety of Dapagliflozin in Combination With Glimepiride (a Sulphonylurea) in Type 2 Diabetes Patients” at ClinicalTrials.gov
Clinical trial number NCT01392677 for “Evaluation of Safety and Efficacy of Dapagliflozin in Subjects With Type 2 Diabetes Who Have Inadequate Glycaemic Control on Background Combination of Metformin and Sulfonylurea” at ClinicalTrials.gov
Clinical trial number NCT00673231 for “Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin” at ClinicalTrials.gov
Clinical trial number NCT02229396 for “Phase 3 28-Week Study With 24-Week and 52-week Extension Phases to Evaluate Efficacy and Safety of Exenatide Once Weekly and Dapagliflozin Versus Exenatide and Dapagliflozin Matching Placebo” at ClinicalTrials.gov
Clinical trial number NCT02413398 for “A Study to Evaluate the Effect of Dapagliflozin on Blood Glucose Level and Renal Safety in Patients With Type 2 Diabetes (DERIVE)” at ClinicalTrials.gov
Clinical trial number NCT01730534 for “Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI58)” at ClinicalTrials.gov
Clinical trial number NCT03036124 for “Study to Evaluate the Effect of Dapagliflozin on the Incidence of Worsening Heart Failure or Cardiovascular Death in Patients With Chronic Heart Failure (DAPA-HF)” at ClinicalTrials.gov
Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;
Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;
Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ;
Hecheng Huaxue , 18 (3), 389-392; 20105) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,.
WO20100223136) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,.
WO20100092437) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,.
WO20090265378) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ;
Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,.
WO200800282410) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;..
Icosapent ethyl or ethyl eicosapentaenoic acid is a synthetic derivative of the omega-3 fatty acid eicosapentaenoic acid (EPA). It is used as adjunct therapy for severe hypertriglyceridemia (TG levels > 500 mg/dL). FDA approved on July 26, 2012.
In 2000, Amarin licensed exclusive U.S. rights to icosapent ethyl ester from the Scottish company Laxdale, and acquired the company in July 2004. In 2015, the product was licensed to Eddingpharm by Amarin for the development and commercialization in China, Hong Kong and Taiwan. Fast-track status has been granted in the U.S. for the treatment of HD. Orphan drug designation was assigned to the compound for this indication in both the U.S. and E.U.
fda
IND 107616 was submitted on 25 March 2010 for the indication of severe hypertriglyceridemia; Epanova had been previously investigated for the treatment of Crohn’s Disease under IND in the Division of Gastroenterology Products. An end-of-phase 2 (EOP2) meeting was held on 02 June 2010. Regarding the indication under consideration at this time, a special protocol assessment (SPA) for the single phase 3 trial OM-EPA-003 (also known as “EVOLVE”) was submitted 02 July 2010 and ultimately agreed upon, after amendments, on 22 October 2010. On 25 April 2012, the applicant proposed an alternative to conducting a thorough QTc study by assessing ECGs recorded during OM-EPA-003; this was found acceptable. A clinical pre-NDA meeting was held on 14 November 2012. The nonclinical development strategy was found reasonable. A clinical package containing OM-EPA-003 (pivotal) and OMEPA-004 (a 6-week phase 3 trial , with long-term safety supported by data from the former Crohn’s disease program (“EPIC” trials), was found adequate for submission. Agreement was reached regarding the clinical pharmacology portion of the submission. Details regarding data pooling for the Integrated Summary of Safety (ISS) were found acceptable
from the former Crohn’s disease program (“EPIC” trials), was found adequate for submission. Agreement was reached regarding the clinical pharmacology portion of the submission. Details regarding data pooling for the Integrated Summary of Safety (ISS) were found acceptable
CMC Drug Substance & Drug Product Chemistry, manufacturing, and controls data related to both the drug substance (omega-3- carboxylic acids) and drug product (Epanova Capsules 1 g) are detailed in the review by Martin Haber, PhD, and Xavier Ysern, PhD. They recommend the NDA for approval. There are no pending CMC issues. The drug substance at sites in Nova Scotia and Prince Edward Island, Canada, from crude fish oil obtained from fish It is a complex mixture of PUFAs, predominantly the omega-3 acids EPA (55%), DHA (20%), and docosapentaenoic acid %). It consistently contains omega-3 and omega-6 PUFA components: total omega-3 fatty acids are limited to not less than % and total omega-6 fatty acids are limited to not more than %. The drug substance also contains 0.3% (m/m) α-tocopherol as . During purification, . Environmental pollutants (heavy metals, pesticides, are controlled by specific tests on the drug substance . Drug substance specifications include tests for acid value, saponification value, ester value, peroxide value, p-anisidine value, total oxidation value, cholesterol, oligomers, , fatty acid composition (PUFAs, EPA, DHA, DPA, total omega-3 fatty acids, total omega-6 fatty acids, other polyunsaturated fatty acids, As described in the review by Drs. Haber and Ysern, the qualitative identify of the drug substance was developed by examining consistencies of peak patterns across 21 discrete lots: there are omega-3 and omega-6 PUFA peaks consistently present in the GC chromatograms (although not necessarily always above the limit of quantitation), which can be used to establish the fingerprint identity of omega-3-carboxylic acids . The quantitative fatty acid composition is given in the table below, excerpted from p. 25 of their review:
Ethyl eicosapentaenoic acid (E-EPA, icosapent ethyl) is a derivative of the omega-3 fatty acideicosapentaenoic acid (EPA) that is used in combination with changes in diet to lower triglyceride levels in adults with severe (≥ 500 mg/dL) hypertriglyceridemia. This was the second class of fish oil-based drug to be approved for use as a drug and was approved by the FDA in 2012. These fish oil drugs are similar to fish oil dietary supplements but the ingredients are better controlled and have been tested in clinical trials.
The company that developed this drug, Amarin Corporation, challenged the FDA’s ability to limit its ability to market the drug for off-label use and won its case on appeal in 2012, changing the way the FDA regulates pharmaceutical marketing.
Intake of large doses (2.0 to 4.0 g/day) of long-chain omega-3 fatty acids as prescription drugs or dietary supplements are generally required to achieve significant (> 15%) lowering of triglycerides, and at those doses the effects can be significant (from 20% to 35% and even up to 45% in individuals with levels greater that 500 mg/dL). It appears that both eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) lower triglycerides, however, DHA alone appears to raise low-density lipoprotein (the variant which drives atherosclerosis; sometimes very inaccurately called: “bad cholesterol”) and LDL-C values (always only a calculated estimate; not measured by labs from person’s blood sample for technical and cost reasons), whilst EPA alone, does not and instead lowers the parameters aforementioned.[2]
Other fish-oil based drugs
There are other omega-3 fish oil based drugs on the market that have similar uses and mechanisms of action.[3]
There are many fish oil dietary supplements on the market.[8] There appears to be little difference in effect between dietary supplements and prescription forms of omega-3 fatty acids, but EPA and DHA ethyl esters (prescription forms) work less well when taken on an empty stomach or with a low-fat meal.[2] The ingredients of dietary supplements are not as carefully controlled as prescription products and have not been fixed and tested in clinical trials, as prescription drugs have,[9] and the prescription forms are more concentrated, requiring fewer capsules to be taken and increasing the likelihood of compliance.[8]
Side effects
Special caution should be taken with people who have with fish and shellfish allergies.[1] In addition, as with other omega-3 fatty acids, taking E-EPA puts people who are on anticoagulants at risk for prolonged bleeding time.[1][2] The most commonly reported side effect in clinical trials has been joint pain; some people also reported pain in their mouth or throat.[1] E-EPA has not been tested in pregnant women is rated pregnancy category C; it is excreted in breast milk and the effects on infants are not known.[1]
Pharmacology
After ingestion, E-EPA is metabolized to EPA. EPA is absorbed in the small intestine and enters circulation. Peak plasma concentration occurs about 5 hours after ingestion and the half-life is about 89 hours. EPA is metabolized mostly in the liver like other dietary fatty acids.[1]
Mechanism of action
EPA, the active metabolite of E-EPA, like other omega-3 fatty acid based drugs, appears to reduce production of triglycerides in the liver, and to enhance clearance of triglycerides from circulating very low-density lipoprotein (VLDL) particles; the way it does that is not clear, but potential mechanisms include increased breakdown of fatty acids; inhibition of diglyceride acyltransferase which is involved in biosynthesis of triglycerides in the liver; and increased activity of lipoprotein lipase in blood.[1][3]
E-EPA was the second fish-oil drug to be approved, after omega-3 acid ethyl esters (GlaxoSmithKline‘s Lovaza which was approved in 2004[11]) and sales were not as robust as Amarin had hoped. The labels for the two drugs were similar, but doctors prescribed Lovaza for people who had triglycerides lower than 500 mg/dL based on some clinical evidence. Amarin wanted to actively market E-EPA for that population as well which would have greatly expanded its revenue, and applied to the FDA for permission to do so in 2013, which the FDA denied.[12] In response, in May 2015 Amarin sued the FDA for infringing its First Amendment rights,[13] and in August 2015 a judge ruled that the FDA could not “prohibit the truthful promotion of a drug for unapproved uses because doing so would violate the protection of free speech.”[14] The ruling left open the question of what the FDA would allow Amarin to say about E-EPA, and in March 2016 the FDA and Amarin agreed that Amarin would submit specific marketing material to the FDA for the FDA to review, and if the parties disagreed on whether the material was truthful, they would seek a judge to mediate.[15]
Synthesis of Fatty-Acid Ethanolamides from Linum catharticum Oils and Cololabis saira Fats
Chemistry of Natural Compounds (Translation of Khimiya Prirodnykh Soedinenii) (2004), 40, (3), 222-226
PAPER
Journal of Molecular Catalysis B: Enzymatic, 84, 173-176; 2012
[0041] A method for preparing a concentrated fish oil fatty acid glycerides, the process steps shown in Figure 1, comprising the steps of:
[0042] S11 using crude enzyme preparation of deep sea fish art: the ratio: (m m) of deep-sea fish through the machine crushed bone formation minced, weighed 600g yue meat, meat by:: water = 0 5.1 water was added seal, in the dark, under nitrogen flow, at 75 ° C cooking lh.Using NaOH to adjust pH to 8.0.Mass fraction of 2% trypsin (trypsin: food grade, Zhengzhou Hong Cheng Chemical Products Limited), in the dark, enzyme 17h at 20 ° C.After 20min by centrifugation 3000r / min, the upper layer was enzymolysis, namely crude fish oil;
[0043] S12 is prepared refined fish oil: Crude fish oil prepared in Step S11 is added a volume ratio of 0.5% phosphoric acid: degummed (crude phosphoric acid fish oil), a concentration of 70% phosphoric acid, followed by centrifugation speed of 3000 rpm / min, and then add a volume ratio of 1% deacidification NaOH, the NaOH concentration is 20%, after centrifugation, the rotational speed of 3000- rpm / min, to obtain refined fish oil;
. [0044] S13 of the refined fish oil fatty acid ethyl ester prepared by esterification process: step S12 is added to the fish oil refining prepared in mass ratio of 0.5% of sodium ethoxide, and a mass ratio of 0.5 in ethanol (ethanol: fish oil refining ), 40 ° C water bath for 1 hour, 1% (by mass) citric acid (citric acid: fish oil refining), standing layer, the upper layer and the liquid was washed with hot deionized water, standing layered repeated three times to give fatty acid ethyl ester.
. [0045] S14 of the fatty acid ethyl ester was extracted Separation: fatty acid ethyl ester obtained in step S13 is subjected to supercritical fluid extraction (extraction process of separation vessel as a rectification column I – separation kettle II), extraction conditions: a rectification column temperature 25-30-35-40 ° C, a pressure of 6 MPa rectification column, separation kettle I temperature 25 ° C, pressure in the separator tank I is 6 MPa, the temperature in the separation tank II 30-45 ° C, C0 2 flow rate of 151,711;
. [0046] S15 of the fatty acid ethyl ester after enzymatic extraction separation processing: The fatty acid ethyl ester obtained in step S14 using Penicillium expansum lipase enzyme, 4% of the amount of enzyme added,, reaction temperature 40 ° C , reaction pH of 10, speed 150 revolutions / min, hydrolysis time 4h, to obtain fatty acid glycerides.
[0047] Example 2
[0048] A process for preparing concentrated fish oil fatty acid glycerides, comprising the steps of:
. [0049] S21 using crude enzyme preparation of deep sea fish art: The procedure of Example 1 with reference to embodiment 11, wherein the cooking temperature is 85 ° C, hydrolysis temperature 25 ° C, centrifuge speed is 4000r / min;
. [0050] S22 refined fish oil preparation: The procedure of Example 1 with reference to embodiment 12; wherein, phosphate: the crude fish oil volume ratio is 1.5%, the phosphoric acid concentration of 75%; K0H: crude fish oil volume ratio of 3%, K0H the concentration of 30%, a centrifugal speed of 4000r / min;
. [0051] S23 of the refined fish oil fatty acid ethyl ester prepared by esterification process: The procedure of Example 1 with reference to embodiment 13; wherein, potassium ethoxide: refined fish oil mass ratio of 1 billion% ethanol: refined fish oil mass ratio of 2.0 , heat the water bath 60 ° C for 3 hours, and acetic acid is acetic acid: refined fish oil mass ratio of 3.0%;
. [0052] S24 was extracted to separate fatty acid ethyl ester: The procedure of Example 1 with reference to embodiment 14; wherein the extraction conditions: temperature rectification column 30-35-40-45 ° C, a pressure rectification column is 15 megabytes Pa, temperature of separation vessel I 35 ° C, pressure in the separator tank I is 8 MPa, the temperature in the separation tank II was 40 ° C, C0 2 flow rate of 171,711;
. [0053] S25 of the fatty acid ethyl ester after enzymatic extraction is carried out the separation treatment: The procedure of Example 1 with reference to embodiment 15; wherein 10% of the amount of enzyme added, reaction temperature 50 ° C, pH 8 hydrolysis, speed 300 rpm / min, hydrolysis time 12h, to obtain fatty acid glycerides.
[0054] Example 3
[0055] – Preparation Method Species of concentrated fish oil fatty acid glycerides, comprising the steps of:
. [0056] S31 using crude enzyme preparation of deep sea fish art: The procedure of Example 1 with reference to embodiment 11, wherein the cooking temperature is 90 ° C, hydrolysis temperature 35 ° C, centrifuge speed is 5000r / min;
. [0057] S32 prepared fine fish oil: The procedure of Example 1 with reference to embodiment 12; wherein, phosphate: the crude fish oil volume ratio of 3% phosphoric acid concentration of 85%; NaOH: crude fish oil volume ratio of 6% and the concentration of NaOH 50%, a centrifugal speed of 5000r / min;
. [0058] S33 of the refined fish oil fatty acid ethyl ester prepared by esterification process: The procedure of Example 1 with reference to embodiment 13; wherein, potassium ethoxide: refined fish oil mass ratio of 1.5%, ethanol: refined fish oil mass ratio of 4.0 heat treatment is 80 ° C water bath for 5 hours, citric acid and citric acid are added: refined fish oil mass ratio of 5.0%;
. [0059] S34 was extracted to separate fatty acid ethyl ester: The procedure of Example 1 with reference to embodiment 14; wherein the extraction conditions: temperature rectification column 30-35-40-45 ° C, pressure column 17 trillion Pa, I of separation vessel temperature 40 ° C, pressure in the separator tank I is 10 MPa, the temperature in the separation tank II is 45 ° C, C0 2 flow rate is?L / h;
. [0060] S35 of the fatty acid ethyl ester after enzymatic extraction separation processing: The procedure of Example 1 with reference to embodiment 15; wherein 20% of the amount of enzyme added, reaction temperature 60 ° C, a pH of 6.5 hydrolysis, speed 300 rpm / min, hydrolysis time 24h, to obtain fatty acid glycerides.
[0061] Comparative Example
[0062] S1 • obtaining crude fish: The procedure of Example 1 with reference to embodiment 11;
. [0063] S2 refined fish oil preparation: see Example 1, Step 12;
. [0064] S3 of refined fish oil fatty acid ethyl ester prepared by esterification process: Step 1, Example 13 process embodiment with reference, to obtain fatty acid ethyl ester.
In recent years, highly unsaturated fatty acids such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have been clarified for their pharmacological effects and are used as raw materials for pharmaceuticals and health foods. Since these polyunsaturated fatty acids have a plurality of double bonds, it is not easy to obtain them by chemical synthesis. Therefore, most of industrially used highly unsaturated fatty acids are produced by extraction or purification from marine organism-derived materials rich in polyunsaturated fatty acids, such as fish oil, etc. However, the content of highly unsaturated fatty acid is not necessarily high, because the biological material is a mixture of various kinds of fatty acids having different numbers of carbon atoms, number and position of double bonds, constitutional ratio of stereoisomers, and the like. For this reason, conventionally, it has been required to selectively purify a target highly unsaturated fatty acid from a biological raw material.
Patent Document 1 discloses a supercritical gas extraction method after a thin film distillation method when a raw material containing a highly unsaturated fatty acid or an alkyl ester thereof is treated by a thin film distillation method, a supercritical gas extraction method and a urea addition method A method for purifying a highly unsaturated fatty acid or an alkyl ester thereof is described.
In Patent Document 2, a raw material containing a highly unsaturated fatty acid such as EPA is subjected to vacuum precision distillation treatment, and the resulting EPA or a fraction containing a lower alcohol ester thereof is mixed with an aqueous silver nitrate solution, whereby a high purity eicosapentaene A method of purifying an acid or a lower alcohol ester thereof is described. It is described that the condition of the vacuum precision distillation is a pressure of 5 mmHg (665 Pa) or less, preferably 1 mmHg (133 Pa) or less, 215 ° C. or less, preferably 210 ° C. or less.
Further, Patent Document 3 discloses a process for producing eicosapentaenoic acid or an ester thereof having a concentration of 80% or more by gradually distilling a raw material containing a highly unsaturated fatty acid or an alkyl ester thereof using a distillation tower having three or more stages Is described. It is described that the condition of the distillation is 10 Torr (1330 Pa) or less, preferably 0.1 Torr (13.3 Pa) or less, 210 ° C. or less, preferably 195 ° C. or less.
However, highly unsaturated fatty acids having higher concentrations and purities than those obtained by the above-mentioned conventional methods are required as raw materials for pharmaceuticals and health foods.
There are cis and trans isomers in highly unsaturated fatty acids. Most of the highly unsaturated fatty acids in vivo are cis, however, they may be converted from cis form to trans form by heating or the like at the stage of purification from biological origin materials (Non-Patent Document 1). Therefore, polyunsaturated fatty acids conventionally purified industrially from biologically derived raw materials contain a certain amount of trans isomer. However, trans fatty acids have been reported to increase health risks, especially LDL cholesterol levels, and increase the risk of cardiovascular disease. In the United States and Canada, foods are obliged to indicate the content of trans fatty acids.
Therefore, there is a need for a highly unsaturated fatty acid-containing composition which not only contains the targeted highly unsaturated fatty acid at a high concentration as a raw material for pharmaceuticals and health foods but also contains a trans fatty acid content as low as possible . However, conventionally, purification of highly unsaturated fatty acids has not been conducted focusing on the stereoisomer ratio.
Patent Document 1: Japanese Patent Application Laid-Open No. 10-95744
Patent Document 2: Japanese Patent Application Laid-Open No. 7-242895
Patent Document 3: Japanese Patent No. 3005638
Non-patent literature
[0010]
Non-patent document 1: Journal of the American Oil Chemists’ Society, 1989, 66 (12): 1822-1830
Example
[0035]
Hereinafter, the present invention will be described in more detail by way of examples, but the present invention is not limited to only these examples.
[0036]
In the following examples, the method of composition analysis of highly unsaturated fatty acids and the method of quantitating stereoisomers are as follows.
9 μL of the measurement sample was diluted to 1.5 mL of n-hexane, and the content ratio of each fatty acid and the content ratio of isomers were analyzed using a gas chromatography analyzer (Type 6890 GC, manufactured by Agilent Technologies) under the following conditions did. The results are expressed as mass% converted from the area of the chromatogram.
<Column condition>
Column: DB-WAX 0.25 mm × 30 m manufactured by J & W Co., column temperature: 210 ° C.
He flow rate: 1.0 ml / min, He pressure: 134 kPa
<Detection condition>
H 2 flow rate: 30 ml / min, Air flow rate : 400 ml / min
He flow rate: 10 ml / min, DET temperature: 260 ° C.
The isomer ratio in the target highly unsaturated fatty acid was obtained by the following formula.
[0037]
[Expression 1]
[0038]
(Example 1)
Raw material: 1000 mL of anhydrous ethanol solution in which 50 g of sodium hydroxide was dissolved was added to 1 kg of sardine oil, mixed and stirred at 70 to 80 ° C. for 1 hour, then 500 mL of water was added and mixed well, 1 It was left standing for a while. The separated aqueous phase was removed and the oil phase was washed several times with water to neutralize the washings to give 820 g of ethyl esterified sardine oil.
As shown in Table 1, the composition of the sardine oil was 44.09% (mass%, hereinafter the same) of eicosapentaenoic acid (EPA), 1.52% of eicosatetraenoic acid (ETA), 1.52% of arachidonic acid (AA) 1.77%, docosahexaenoic acid (DHA) 6.92%. Also, the trans isomer ratio in EPA was 1.23%.
Step (1) 160 ml of n-hexane was added to 300 g of the ethyl esterified sardine oil prepared above, and the mixture was stirred well and dissolved. To this was added 500 mL of an aqueous solution containing 50% by weight of silver nitrate, and the mixture was stirred under conditions of 5 to 30 ° C. After standing, the separated n-hexane phase was removed, and the aqueous phase was recovered.
Step (2): 2000 mL of fresh n-hexane was added to the aqueous phase obtained in the step (1), and the mixture was sufficiently stirred at 50 to 69 ° C. to extract the fatty acid ethyl ester into n-hexane. After standing, the separated aqueous phase was removed and the n-hexane phase was concentrated. The crude fatty acid ethyl ester crude product contained in this n-hexane phase contained 74.54% EPA, 0.32% ETA, 0.17% AA and 14.87% DHA in total fatty acids as shown in Table 1 It was. Also, the trans isomer ratio in EPA was 0.19%.
Step (3): The n-hexane phase containing the fatty acid ethyl ester obtained in the step (2) was maintained under conditions of a top vacuum degree of 1 Pa or less and a distillation temperature of 170 to 190 ° C. using a packed tower precision distillation apparatus While performing vacuum distillation to obtain a highly purified EPA ethyl ester-containing composition in a yield of about 60%. As shown in Table 1, this EPA ethyl ester-containing composition contained 98.25% of EPA, 0.43% of ETA, 0.21% of AA, and 0.05% of DHA in total fatty acids. Also, the trans isomer ratio in EPA was 0.45%.
The yield of EPA in this example in which the steps were performed in the order of (1), (2), (3) was about 53%.
[0039]
Example 2 The
steps (1), (2) and (3) were carried out in the same manner as in Example 1 except that the step (3) was carried out while maintaining the distillation temperature of 180 to 185 ° C., EPA ethyl ester-containing composition was obtained in a yield of about 58%. As shown in Table 1, this EPA ethyl ester-containing composition contained 98.29% of EPA, 0.40% of ETA, 0.32% of AA, and 0.05% of DHA in total fatty acids. Also, the trans isomer ratio in EPA was 0.28%, and the trans isomer was extremely small.
Comparative Example 1 An
EPA ethyl ester-containing composition was obtained in the same manner as in Example 1, except that the top vacuum degree was set to 13.3 Pa (0.1 Torr) in the step (3). As shown in Table 1, the composition contained EPA content ratio as high as 97.44% in the total fatty acid, but the trans isomer ratio in EPA was high (1.37%).
[0040]
Comparative Example 2 The
EPA ethyl ester-containing composition was obtained by performing vacuum distillation (step (3)) of ethyl esterified sardine oil and then steps (1) and (2). The conditions of each step were the same as in Example 1. As shown in Table 1, this composition contained 95.05% EPA, 0.72% ETA, 0.50% AA, 0.21% DHA in total fatty acids, the trans isomer ratio in EPA was 1.55% Met. The yield of EPA in this comparative example in which the steps were carried out in the order of (3), (1) and (2) was about 31%, and the EPA yield greatly decreased as compared with Example 1.
By changing the condition of the vacuum distillation in this Comparative Example (0.5 Pa, 185 to 195 ° C.), it was possible to raise the content of EPA in the total fatty acids in the composition to 98.12%, however, The rate further declined and the trans isomer ratio in EPA was 2.01%, further increased.
[0041]
[table 1]
[0042]
Examples 3 to 4 and Comparative Example 3 In the
step (3), the distillation temperature was 180 ° C. (Example 3), 190 ° C. (Example 4), 200 ° C. (Comparative Example 3), and the vacuum distillation time was A highly purified EPA ethyl ester-containing composition was obtained in the same manner as in Example 1 except that various changes were made and the trans isomer ratio of EPA in the composition was determined. The results are shown in Fig. 1. 1, in Examples 3 to 4 having a distillation temperature of 190 ° C. or less, the trans isomer ratio was less than 1% by mass, but in Comparative Example 3 having a distillation temperature of 200 ° C., the trans isomer The ratio exceeds 1% by mass.
^ Jump up to:abcJacobson TA, et al, NLA Expert Panel. National Lipid Association Recommendations for Patient-Centered Management of Dyslipidemia: Part 2. J Clin Lipidol. 2015 Nov-Dec;9(6 Suppl):S1-S122.e1. PMID26699442Free full text
The applicant Janssen-Cilag International N.V. submitted on 28 August 2012 an application for Marketing Authorisation to the European Medicines Agency (EMA) for SIRTURO, through the centralised procedure falling within the Article 3(1) and point 4 of Annex of Regulation (EC) No 726/2004. The eligibility to the centralised procedure was agreed upon by the EMA/CHMP on 21 July 2011. SIRTURO was designated as an orphan medicinal product EU/3/05/314 on 26 August 2005. SIRTURO was designated as an orphan medicinal product in the following indication: treatment of tuberculosis. The applicant applied for the following indication: SIRTURO is indicated in adults (≥ 18 years) as part of combination therapy of pulmonary tuberculosis due to multi-drug resistant Mycobacterium tuberculosis.
Disease to be treated About a third of the global population, more than 2 billion people, is infected with M. tuberculosis, of which the majority is latent. The life time risk to fall ill in overt TB is around 10% in general, but many times higher (around 10% annual risk) in untreated HIV-positive individuals. Tuberculosis is the leading cause of death in the latter population. It was estimated that a total of 8.8 million new TB cases occurred in 2010, including 1.1 million people co infected with HIV, and that about 1.45 million people died due to TB. During more recent years the burden of TB resistant to first line therapy has increased rapidly. Such multidrug resistant tuberculosis (defined later in this assessment report) has been reported in all regions of the world. Presently around 500.000 of new MDR cases are estimated to emerge every year, which is close to 5% of all new TB cases. China and India carried nearly 50% of the total burden of incident MDR-TB cases in 2008, followed by the Russian Federation (9%). The incidence of MDR-TB in US and EU was reported to be 1.1% and 2.4%, respectively. Within the EU, the incidence is much higher in certain Eastern European countries, with the largest burden in Romania, Latvia and Lithuania. MDR TB is an orphan disease in the EU, US and in Japan.
Current TB therapy and definitions Treatment of pulmonary drug susceptible TB typically takes 6 months resulting in cure rates in well over 90% of cases with good treatment adherence. The two most important drugs in this treatment are isoniazid (INH) and rifampicin (RIF). TB with resistance to at least both INH and RIF is called multidrug resistant (MDR) TB. The two most important “classes” of second-line TB drugs to be used in such cases are injectable drugs (the aminoglycosides amikacin and kanamycin, and the related agent capreomycin) and fluoroquinolones. Apart from these agents a number of miscellaneous drugs are used in addition, as part of combination therapy. The effectiveness of these latter miscellaneous drugs is generally lower, the tolerability is problematic and established breakpoints for resistance determination are lacking.
The term pre-XDR (pre-extensively drug resistant) TB is used when resistance is present also to one of the two main second-line class agents (injectables or any of the fluoroquinolones), and XDR-TB when resistance is present to INH+RIF + injectables + fluoroquinolones. The WHO standard treatment for MDR-TB is commonly divided into 2 phases: • a 4 to 6-month intensive treatment phase in which an injectable drug plus 3-4 other drugs, including a fluoroquinolone, • a continuation phase without the injectable drug and often without pyrazinamide (PZA) for a total duration of 18-24 months. Using this approach, cure rates in MDR-TB are much lower than those seen in DS-TB (ranging from less than 50% to around 75%), despite the higher number of agents and longer treatment duration. Hence, MDR TB is associated with a high mortality and is considered an important major threat to public health. More recent approaches to evaluate various MDR TB regimens have yielded somewhat more optimistic outcomes, despite shorter treatment durations. In these non-randomised studies (with low number of patients) cure rates in the range of 90% were achieved by including a fourth generation fluoroquinolone and by increasing the number of agents even further, to include up to 7 agents in the intensive phase, and still 4-5 agents in a second phase.
About the product SIRTURO (bedaquiline, formerly known as TMC 207) is a new agent of a unique class, specific for mycobacteria, and seemingly without cross-resistance to available TB agents. A large number of pre-clinical studies showed promising results for bedaquiline. For example, in animal models bedaquiline + pyrazinamide cured TB at a higher rate than the traditional first line combination, even when therapy was shortened for the former combination. The clinical program for bedaquiline has been aimed at treating MDR-TB, and data is now available from phase 2b studies of moderate size, both placebo-controlled and non-controlled studies. The treatments given in these studies were similar to those recommended by the WHO, although the number of agents used was slightly higher (five agents in the preferred background regimens). Bedaquiline (versus placebo in the controlled study) was added during the first (intensive) treatment phase, while the background regimens were generally unchanged throughout the complete course of therapy (18-24 months). On the basis of these studies, the applicant submitted an application for a conditional approval for bedaquiline, with the proposed indication: treatment of adult patients infected with pulmonary tuberculosis due to MDR M. tuberculosis, as part of combination therapy. In line with the approach in the phase 2 studies, Sirturo is only to be used during the first 6 months of therapy. However the planned pivotal study (as a specific obligation) will test for 40 weeks of bedaquiline treatment.
In 2009, the drug candidate was licensed to Global Alliance TB Drug Development by Tibotec worldwide for the treatment of tuberculosis.
Bedaquiline (INN) is chemically designated as (1R,2S)-1-(6-bromo-2-methoxy-3-quinolinyl)-4- (dimethylamino)-2-(1-naphthalenyl)-1-phenyl-2-butanol with fumaric acid (1:1), and has the following structure:
Bedaquiline fumarate is a white to almost white powder. It contains two asymmetric carbon atoms, C-1 (R), C-2 (S) and exhibits ability to rotate the orientation of linearly polarized light (optical rotation). The substance is non-hygroscopic. It is practically insoluble in aqueous media over a wide pH range and very slightly soluble in 0.01 N HCl. The substance is soluble in a variety of organic solvents. Due to the low solubility Log KD (log P) could not be determined experimentally. In Biopharmaceutics Classification System (BCS) bedaquiline is classified as a Class 2 compound (expressing low solubility and high permeability). Bedaquiline exists in only one non-solvated crystalline form: Form A. In addition 2 pseudopoly-morphs were found: Form B and Form C. The substance can also be made amorphous. Sufficient evidence was provided to demonstrate that Form A is obtained by the employed manufacturing process of the active substance. Particle size was considered a critical quality attribute of the active substance as bedaquiline is not dissolved in the dosage form. Therefore an appropriate test on particle size determination was included in the active substance specification. The acceptance criteria are based upon the capabilities of the milling process, batch and stability data, and the known impact of the particle size on manufacturability, in-vitro release, and in-vivo performance
Bedaquiline is a bactericidal antimycobacterial drug. Chemically it is a diarylquinoline. FDA approved on December 28, 2012.
Bedaquiline is indicated as part of combination therapy in adults (≥ 18 years) with pulmonary multi-drug resistant tuberculosis (MDR-TB).
Bedaquiline was approved for medical use in the United States in 2012.[1] It is on the World Health Organization’s List of Essential Medicines, the most effective and safe medicines needed in a health system.[7] The cost for six months is approximately $900 USD in low income countries, $3,000 USD in middle income countries, and $30,000 USD in high income countries.[5]
SIRTURO (bedaquiline) for oral administration is available as 100 mg strength tablets. Each tablet contains 120.89 mg of bedaquiline fumarate drug substance, which is equivalent to 100 mg of bedaquiline. Bedaquiline is a diarylquinoline antimycobacterial drug.
Bedaquiline fumarate is a white to almost white powder and is practically insoluble in aqueous media. The chemical name of bedaquiline fumarate is (1R, 2S)-1-(6-bromo-2-methoxy-3-quinolinyl)-4- (dimethylamino)-2-(1-naphthalenyl)-1-phenyl-2-butanol compound with fumaric acid (1:1). It has a molecular formula of C32H31BrN2O2 · C4H4O4 and a molecular weight of 671.58 (555.50 + 116.07). The molecular structure of bedaquiline fumarate is the following:
SIRTURO (bedaquiline) contains the following inactive ingredients: colloidal silicon dioxide, corn starch, croscarmellose sodium, hypromellose 2910 15 mPa.s, lactose monohydrate, magnesium stearate, microcrystalline cellulose, polysorbate 20, purified water (removed during processing).
As of 2013 Both the World Health Organization (WHO) and US Centers for Disease Control (CDC) have recommended (provisionally) that bedaquiline be reserved for patients with multidrug-resistant tuberculosis when an otherwise recommended regimen cannot be designed.[9][10]
Clinical trials
Bedaquiline has been studied in phase IIb studies for the treatment of multidrug-resistant tuberculosis while phase III studies are currently underway.[11] It has been shown to improve cure rates of smear-positive multidrug-resistant tuberculosis, though with some concern for increased rates of death (further detailed in the Adverse effects section).[12]
The most common side effects of bedaquiline in studies were nausea, joint and chest pain, and headache. The drug also has a black-box warning for increased risk of death and arrhythmias, as it may prolong the QT interval by blocking the hERG channel.[15] All patients on bedaquiline should have monitoring with a baseline and repeated ECGs.[16] If a patient has a QTcF of > 500ms or a significant ventricular arrythmia, bedaquiline and other QT prolonging drugs should be stopped.
There is considerable controversy over the approval for the drug, as one of the largest studies to date had more deaths in the group receiving bedaquiline that those receiving placebo.[17] 10 deaths occurred in the bedaquiline group out of 79, while 2 occurred in the placebo group, out of 81.[12] Of the 10 deaths on bedaquiline, 1 was due to a motor vehicle accident, 5 were judged as due to progression of the underlying tuberculosis and 3 were well after the patient had stopped receiving bedaquiline.[17] However, there is still significant concern for the higher mortality in patients treated with bedaquiline, leading to the recommendation to limit its use to situations where a 4 drug regimen cannot otherwise be constructed, limit use with other medications that prolong the QT interval and the placement of a prominent black box warning.[17][11]
Drug interactions
Bedaquiline should not be co-administered with other drugs that are strong inducers or inhibitors of CYP3A4, the hepatic enzyme responsible for oxidative metabolism of the drug.[16] Co-administration with rifampin, a strong CYP3A4 inducer, results in a 52% decrease in the AUC of the drug. This reduces the exposure of the body to the drug and decreases the antibacterial effect. Co-administration with ketoconazole, a strong CYP3A4 inhibitor, results in a 22% increase in the AUC, and potentially an increase in the rate of adverse effects experienced[16]
Since bedaquiline can also prolong the QT interval, use of other QT prolonging drugs should be avoided.[9] Other medications for tuberculosis that can prolong the QT interval include fluoroquinolones and clofazimine.
Mode of action
Bedaquiline blocks the proton pump for ATP synthase of mycobacteria. ATP production is required for cellular energy production and its loss leads to cell death, even in dormant or nonreplicating mycobacteria.[18] It is the first member of a new class of drugs called the diarylquinolines.[18] Bedaquiline is bactericidal.[18]
Resistance
The specific part of ATP synthase affected by bedaquiline is subunit c which is encoded by the gene atpE. Mutations in atpE can lead to resistance. Mutations in drug efflux pumps have also been linked to resistance.[19]
History
Bedaquiline was described for the first time in 2004 at the Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC) meeting, after the drug had been in development for over seven years.[20] It was discovered by a team led by Koen Andries at Janssen Pharmaceutica.[21]
Bedaquiline was approved for medical use in the United States in 2012.[1]
It is manufactured by Johnson & Johnson (J&J), who sought accelerated approval of the drug, a type of temporary approval for diseases lacking other viable treatment options.[22] By gaining approval for a drug that treats a neglected disease, J&J is now able to request expedited FDA review of a future drug.[23]
When it was approved by the FDA on the 28th December 2012, it was the first new medicine for TB in more than forty years.[24][25]
Bedaquiline, formally called (1R, 2S)-1-(6-Bromo-2-methoxy-3-quinolinyl)-4-(dimethylamino)-2-(1-naphthyl)-1-phenyl-2-butanol in chemistry and known as Sirturo in commercial, is a new anti-mycobacterial medicine of diarylquinolines. It impinges on the
ATP synthesis of Mycobacterium tuberculosis by inhibiting the activity of proton pump on the cell’s ATP synthetase, and thereby eliminates M. tuberculosis (TB). It’s used for adult multi-drug resistant tuberculosis (MDR-PTB).
The chemical name of beidaquinoline is (1R,2S)-1-(6-bromo-2-methoxy-3-quinolinyl)-4-dimethylamino-2-(1-naphthyl)-1 -Phenyl-2-butanol, the first drug developed by Johnson & Johnson in the United States to inhibit mycobacterium adenosine triphosphate (ATP) synthetase, was first introduced in the United States in December 2012 for the treatment of adult multidrug-resistant tuberculosis. The trade name is Sirturo. Beidaquinoline shows strong selectivity for Mycobacterium tuberculosis ATP synthase. Its novel mechanism of action makes it not cross-resistance with other anti-tuberculosis drugs, which will greatly reduce the drug resistance of Mycobacterium tuberculosis. It shows good activity against MDR-TB in macrophages, suggesting that it has the effect of shortening treatment time.
The synthesis of beidaquinoline has been reported in the literature. The specific synthesis route is as follows:
The patent WO2004011436 mentions the use of column chromatography to separate and purify the crude product, but this method is not conducive to industrialization; in addition, a method for isolating and purifying beraquinoline diastereomer A is disclosed in Step C of the Example of WO2006125769. . However, although the purity of the diastereomer A obtained by the separation and purification method disclosed in this patent is 82%, it is actually only possible to achieve the reaction conversion rate of more than 80%. The actual study found that due to the difficult control of the reaction conditions for the preparation of bedaquino, the control conditions for water, temperature, and drip rate are harsh and the reaction is unstable, and it cannot be ensured that the conversion rate reaches more than 80% per batch, and the conversion is usually When the rate is between 60-80%, the ratio of diastereomer B to diastereomer A obtained by this method is between 1:1 and 1:3, and the next step is chiral separation. It has an impact; even the conversion rate is sometimes as low as about 50%. When the conversion rate is as low as 50%, since the amount of the product in the reaction liquid is small, as in the method using patent WO 2006125769, the isolated product can hardly be purified even if the product is separated and purified by the purification method disclosed in this patent. The resulting diastereomer A is also of low purity.
Example 1
Reaction material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA (20g) reaction, one-step reaction to obtain a racemic bedaquiline reaction solution. The conversion of this reaction by HPLC analysis was 56%. After quenching the reaction, n-heptane (40 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 0° C. and filtered to remove diastereoisomer B. The resulting filtrate was washed with 50% acetic acid aqueous solution to remove 3-dimethylamino-1-(naphthalene-5-yl)acetone as a raw material, and 15% hydrochloric acid aqueous solution was added to the organic layer for stirring to make the product salified in the aqueous layer. In the middle. After filtration, the filtrate was separated and the product was transferred to the aqueous layer. The raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by layering the filtrate, adjusted to alkaline with aqueous ammonia, extracted with toluene and free, and then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure to obtain a product that is not correct. Enantiomer A (4.9 g), purity 89%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetylene (40 ml), DMSO (4.9 ml), and R-binaphthol phosphate (2.62 g) were added to diastereomer A (4.9 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. After cooling, precipitates are separated out; at room temperature, the filter cake is washed with acetone and dried under vacuum at 50-60° C. to give a resolution salt (2.07 g);
Split salt (2.07g), toluene (37ml), potassium carbonate (1.51g) and water (13ml) were mixed, heated to 90°C and stirred until completely dissolved. While hot stratified, organic layer was treated with 10% potassium carbonate aqueous solution ( (5ml) was washed once, at this time organic layer TLC monitoring; washed with purified water to neutral pH (20ml × 3 times); organic layer was concentrated under reduced pressure to give a colorless oil (1.5g); add toluene (1ml) to heat the whole Dissolve, add ethanol (12ml) and stir at room temperature for 0.5h. Precipitate the solid, and stir in ice water bath for 1h. Filter and wash the filter cake with ethanol. Dry it in vacuo at 50-60°C to give bedaquinoline (1.07g). The HPLC purity is >99%. .
Example 2
Starting material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA ( 20g) Reaction, one-step reaction to obtain a racemic bedaquiline reaction solution. The conversion of this reaction by HPLC analysis was 65%. After quenching the reaction, diisopropyl ether (160 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 5° C. and filtered to remove diastereoisomer B. The resulting filtrate was washed with 10% aqueous formic acid to remove 3-dimethylamino-1-(naphthalene-5-yl)acetone as a raw material, and 5% aqueous sulfuric acid solution was added to the organic layer for stirring to make the product salified in the aqueous layer. In the middle. Filtration, filtration of the filtrate, the product was transferred to the aqueous layer, the raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer, and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by the layering of the filtrate, adjusted to be weakly alkaline with sodium hydroxide, extracted with dichloromethane, and washed, then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure. The product was diastereoisomer A (5.7 g), purity 92%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetate (45 ml), DMSO (5.7 ml), and R-binaphthol phosphate (3.04 g) were added to diastereomer A (5.7 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. Cooling, precipitated salt precipitation; filtered at room temperature, washed with acetone cake, 50-60 ° C vacuum drying salt (2.6g);
The resolved salt (2.41 g), toluene (39 ml), potassium carbonate (1.58 g) and water (14 ml) were mixed, heated to 90°C and stirred until completely dissolved. While hot stratified, the organic layer was treated with 10% aqueous potassium carbonate solution ( (5ml) was washed once, washed with purified water until the pH was neutral (20ml × 3 times); the organic layer was concentrated under reduced pressure to give a colorless oil (1.6g); toluene was added (1ml) to heat the solution and ethanol was added (12ml) The precipitated solid was stirred at room temperature for 0.5 h, stirred in an ice-water bath for 1 h, filtered, washed with ethanol, and dried in vacuo at 50-60° C. to give bedalquinoline (1.19 g) with an HPLC purity of >99%.
Example 3
Starting material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA ( 20g) Reaction, one-step reaction to obtain a racemic bedaquiline reaction solution. The conversion of this reaction by HPLC analysis was 75%. After quenching the reaction, diisopropyl ether (400 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 2° C. and filtered to remove diastereoisomer B. The resulting filtrate was washed with 60% aqueous solution of propionic acid to remove 3-dimethylamino-1-(naphthalen-5-yl)acetone as a raw material, and 40% methanesulfonic acid aqueous solution was added to the organic layer for stirring to make the product salified. Precipitated in the water layer. After filtration, the filtrate was separated and the product was transferred to the aqueous layer. The raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by the layering of the filtrate, adjusted to be weakly alkaline with sodium hydroxide, extracted with dichloromethane, and washed, then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure. Obtained product diastereomer A (6.0 g), purity 94%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetate (48 ml), DMSO (6.0 ml), and R-binaphthol phosphate (3.09 g) were added to diastereomeric A (6.0 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. After cooling, precipitated salt precipitated; it was filtered at room temperature, and the filter cake was washed with acetone and dried under vacuum at 50-60° C. to give the resolved salt (2.59 g).
The resolved salt (2.59g), toluene (40ml), potassium carbonate (1.60g) and water (14ml) were mixed, heated to 90°C and stirred until completely dissolved; while hot stratified, the organic layer was treated with 10% potassium carbonate aqueous solution ( (5 ml) was washed once, washed with purified water until the pH was neutral (20 ml × 3 times); the organic layer was concentrated under reduced pressure to give a colorless oil (1.7 g); toluene (1 ml) was added and heated to complete dissolution, and ethanol (12 ml) was added. The precipitated solid was stirred at room temperature for 0.5 h, stirred in an ice-water bath for 1 h, filtered, washed with ethanol, and dried in vacuo at 50-60° C. to give bedaquinoline (1.20 g) with an HPLC purity of >99%.
Example 4
Starting material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA ( 20g) Reaction, one step reaction to obtain the racemic bedaquiline reaction solution. The conversion of this reaction by HPLC analysis was 70%. After quenching the reaction, petroleum ether (16 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 3° C. and filtered to remove diastereoisomer B. The obtained filtrate was washed with 30% acetic acid aqueous solution to remove 3-methylamino-1-(naphthalen-5-yl)acetone as a raw material, and 25% phosphoric acid aqueous solution was added to the organic layer for stirring to make the product salified in the aqueous layer. In the middle. After filtration, the filtrate was separated and the product was transferred to the aqueous layer. The raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by the layering of the filtrate, adjusted to be slightly alkaline with sodium hydroxide, extracted with dichloromethane, and washed, and then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure. Obtained product diastereomer A (5.72 g), purity 88%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetylene (45 ml), DMSO (5.7 ml), and R-binaphthol phosphate (3.04 g) were added to diastereomer A (5.72 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. After cooling, precipitated salt precipitated out; it was filtered at room temperature, and the filter cake was washed with acetone and dried under vacuum at 50-60° C. to give a resolution salt (2.43 g);
Split salt (2.43g), toluene (40ml), potassium carbonate (1.60g) and water (14ml) were mixed, heated to 90°C and stirred until completely dissolved. While hot stratified, the organic layer was treated with 10% potassium carbonate aqueous solution ( (5 ml) was washed once, washed with purified water until the pH was neutral (20 ml x 3 times); the organic layer was concentrated under reduced pressure to give a colorless oil (1.5 g); toluene (1 ml) was added for heating and ethanol was added (12 ml) The precipitated solid was stirred at room temperature for 0.5 h, stirred in an ice-water bath for 1 h, filtered, and the filter cake was washed with ethanol. Drying in vacuo at 50-60° C. gave bedaquinoline (1.16 g) with an HPLC purity of >99%.
Example 5
Starting material 3-benzyl-6-bromo-2-methoxyquinoline (10 g) and 3-dimethylamino-1-(naphthalene-5-yl)propanone (10 g) in tetrahydrofuran (80 ml) with LDA ( 20g) Reaction, one step reaction to obtain the racemic bedaquiline reaction solution. The conversion of this reaction was 80% by HPLC analysis. After quenching the reaction, n-hexane (80 ml) was added to the reaction solution. Undesired diastereomer B was precipitated in an ice-water bath at 1° C. and filtered to remove diastereoisomer B. The resulting filtrate was washed with 40% aqueous acetic acid to remove 3-dimethylamino-1-(naphthalen-5-yl)acetone as starting material, and 20% aqueous hydrochloric acid solution was added to the organic layer for stirring to make the product salified in the aqueous layer. In the middle. After filtration, the filtrate was separated and the product was transferred to the aqueous layer. The raw material 3-benzyl-6-bromo-2-methoxyquinoline was left in the organic layer and the organic layer was discarded. The filtered product salt solid is combined with the aqueous layer obtained by the layering of the filtrate, adjusted to be weakly alkaline with sodium hydroxide, extracted with dichloromethane, and washed, then the organic layer is washed with water to neutrality, and the organic layer is concentrated under reduced pressure. Obtained product diastereomer A (6.1 g), purity 96%.
With reference to the method of patent WO2006125769, the obtained diastereoisomer A is resolved to obtain the desired bedaquiline, the specific method is as follows:
Acetate (48 ml), DMSO (6.1 ml), and R-binaphthol phosphate (3.09 g) were added to diastereomer A (6.1 g) of the obtained bedaquinoline, and the mixture was heated under reflux for 2 hours. After cooling, precipitated salt precipitated out; it was filtered at room temperature, and the filter cake was washed with acetone and dried under vacuum at 50-60° C. to give the resolution salt (2.69 g).
The resolved salt (2.69g), toluene (40ml), potassium carbonate (1.60g) and water (14ml) were mixed, heated to 90°C and stirred until completely dissolved; while hot stratified, the organic layer was treated with 10% potassium carbonate aqueous solution ( (5ml) was washed once, washed with purified water until the pH was neutral (20ml×3 times); the organic layer was concentrated under reduced pressure to give a colorless oil (1.8g); toluene (1ml) was added to heat to dissolve and ethanol (12ml) was added. The precipitated solid was stirred for 0.5 h at room temperature, stirred in an ice-water bath for 1 h, filtered, washed with ethanol, and dried in vacuo at 50-60° C. to give bedalquinoline (1.28 g) with an HPLC purity of >99%.
⊥ Institute for Tuberculosis Research, College of Pharmacy, University of Illinois at Chicago, 833 South Wood Street, Chicago, Illinois 60612, United States
Bedaquiline (1) is a new drug for tuberculosis and the first of the diarylquinoline class. It demonstrates excellent efficacy against TB but induces phospholipidosis at high doses, has a long terminal elimination half-life (due to its high lipophilicity), and exhibits potent hERG channel inhibition, resulting in clinical QTc interval prolongation. A number of structural ring A analogues of bedaquiline have been prepared and evaluated for their anti-M.tb activity (MIC90), with a view to their possible application as less lipophilic second generation compounds. It was previously observed that a range of 6-substituted analogues of 1 demonstrated a positive correlation between potency (MIC90) toward M.tb and drug lipophilicity. Contrary to this trend, we discovered, by virtue of a clogP/M.tb score, that a 6-cyano (CN) substituent provides a substantial reduction in lipophilicity with only modest effects on MIC values, suggesting this substituent as a useful tool in the search for effective and safer analogues of 1.
Multi-drug resistant tuberculosis (MDR-TB) is of growing global concern and threatens to undermine increasing efforts to control the worldwide spread of tuberculosis (TB). Bedaquiline has recently emerged as a new drug developed to specifically treat MDR-TB. Despite being highly effective as a result of its unique mode of action, bedaquiline has been associated with significant toxicities and as such, safety concerns are limiting its clinical use. In order to access pharmaceutical agents that exhibit an improved safety profile for the treatment of MDR-TB, new synthetic pathways to facilitate the preparation of bedaquiline and analogues thereof have been discovered.
The first asymmetric synthesis of a very promising antituberculosis drug candidate, R207910, was achieved by developing two novel catalytic transformations; a catalytic enantioselective proton migration and a catalytic diastereoselective allylation of an intermediate α-chiral ketone. Using 2.5 mol % of a Y-catalyst derived from Y(HMDS)3 and the new chiral ligand 9, 1.25 mol % of p-methoxypyridine N-oxide (MEPO), and 0.5 mol % of Bu4NCl, α-chiral ketone 3 was produced from enone 4 with 88% ee. This reaction proceeded through a catalytic chiral Y-dienolate generation via deprotonation at the γ-position of 4, followed by regio- and enantioselective protonation at the α-position of the resulting dienolate. Preliminary mechanistic studies suggested that a Y: 9: MEPO = 2: 3: 1 ternary complex was the active catalyst. Bu4NCl markedly accelerated the reaction without affecting enantioselectivity. Enantiomerically pure 3 was obtained through a single recrystallization. The second key catalytic allylation of ketone 3 was promoted by CuF•3PPh3•2EtOH (10 mol %) in the presence of KOtBu (15 mol %), ZnCl2 (1 equiv), and Bu4PBF4 (1 equiv), giving the desired diastereomer 2 in quantitative yield with a 14: 1 ratio without any epimerization at the α-stereocenter. It is noteworthy that conventional organometallic addition reactions did not produce the desired products due to the high steric demand and a fairly acidic α-proton in substrate ketone 3. This first catalytic asymmetric synthesis of R207910 includes 12 longest linear steps from commercially available compounds with an overall yield of 5%.
Dr.Chandrasekhar S Director CSIR-Indian Institute of Chemical Technology (Council of Scientific and Industrial Research)
Ministry of Science & Technology, Government of India
Tarnaka, Hyderabad-500007, Telangana, INDIA
(1R,2S)-1-(6-Bromo-2-methoxyquinolin-3-yl)-4-(dimethylamino)- 2-(naphthalen-1-yl)-1-phenylbut-an-2-ol (3a): A solution of 16a and 16b (6.0 g, 10.2 mmol) in Me2NH (200 mL, 8.0 m in THF) was stirred at 45 °C for 24 h. The solution was filtered and the filtrate concentrated under reduced pressure to afford the crude product which on purification by silica gel column chromatography (eluent: ethyl acetate/hexane = 1:6) furnished 3a and 3b as white solids (4.8 g, 90%) (1:1 w/w).
Jump up^Diacon AH, Pym A, Grobusch M, et al. (2009). “The diarylquinoline TMC207 for multidrug-resistant tuberculosis”. N Engl J Med. 360 (23): 2397–405. doi:10.1056/NEJMoa0808427. PMID19494215.
^ Jump up to:abCenters for Disease Control and Prevention (2013-10-25). “Provisional CDC guidelines for the use and safety monitoring of bedaquiline fumarate (Sirturo) for the treatment of multidrug-resistant tuberculosis”. MMWR. 62 (RR-09): 1–12. ISSN1545-8601. PMID24157696.
Jump up^de Jonge MR, Koymans LH, Guillemont JE, Koul A, Andries K (June 2007). “A computational model of the inhibition of Mycobacterium tuberculosis ATPase by a new drug candidate R207910”. Proteins. 67 (4): 971–80. doi:10.1002/prot.21376. PMID17387738.
Vismodegib is a Hedgehog Pathway Inhibitor. The mechanism of action of vismodegib is as a Smoothened Receptor Antagonist.
Hedgehog Antagonist GDC-0449 is an orally bioavailable small molecule with potential antineoplastic activity. Hedgehog antagonist GDC-0449 targets the Hedgehog signaling pathway, blocking the activities of the Hedgehog-ligand cell surface receptors PTCH and/or SMO and suppressing Hedgehog signaling. The Hedgehog signaling pathway plays an important role in tissue growth and repair; aberrant constitutive activation of Hedgehog pathway signaling and uncontrolled cellular proliferation may be associated with mutations in the Hedgehog-ligand cell surface receptors PTCH and SMO.
NMR from net
Vismodegib is an active pharmaceutical ingredient produced by Genentech (Roche) and sold under the trade name Erivedge® (which contains crystalline Vismodegib as the active ingre-dient). Erivedge® is an oral Hedgehog signaling pathway inhibitor approved for the treatment of basal-cell carcinoma (BCC).
Developed and launched by Roche and its subsidiary Genentech, under license from Curis. Family members of the product Patent of vismodegib (WO2006028958),
Vismodegib was first disclosed in WO Patent Publication No. 06/028959. Vismodegib, chem-ically 2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide, is represented by the following structure:
Vismodegib is indicated for patients with basal cell carcinoma (BCC) which has metastasized to other parts of the body, relapsed after surgery, or cannot be treated with surgery or radiation.[3][4]
Mechanism of action
The substance acts as a cyclopamine-competitive antagonist of the smoothened receptor (SMO) which is part of the hedgehog signaling pathway.[2] SMO inhibition causes the transcription factors GLI1 and GLI2 to remain inactive, which prevents the expression of tumor mediating genes within the hedgehog pathway.[5] This pathway is pathogenetically relevant in more than 90% of basal-cell carcinomas.[6]
In embryonic development, Hedgehog signaling in cell differentiation, tissue development and organogenesis play an important role.In the adult body, Hedgehog signaling pathway is mainly in slumber, but when abnormal tissue growth and self-healing, Hedgehog pathway may be activated.With the in-depth study of the tumor, the presence of numerous evidence of abnormal tumor occurrence and the close relationship between Hedgehog signaling pathway, such as sporadic basal cell carcinoma, medulloblastoma, small cell lung cancer and gastrointestinal cancer and other diseases, therefore Hedgehog signaling pathway targeted anti-cancer therapy inhibitors become hot.
Vismodegib chemical name 2_ chlorine -N_ (4_ chlorine _3_ (_2_ pyridyl) phenyl) _4_ (methylsulfonyl) benzamide, is by Roche’s Genentech (Genentech) Hedgehog pathway inhibitors developed, and can be inhibited by binding seven transmembrane protein Smoothened (Smo), thereby preventing signal transduction.Vismodegib capsule in January 2012 I was approved and listed by the US Food and Drug Administration, under the trade name Erivedge, for the treatment of adults with the most common type of skin cancer – basal cell carcinoma.This medicine is not intended for surgery or radiotherapy of cancer and basal cell skin cancer locally advanced patients have been transferred.This was the first drug approved for the treatment of basal cell carcinoma.
W02006028958 Vismodegib disclose the following synthesis route:
Route One Negishi coupling reactions
wherein, X1 is chloro, bromo or iodo; X2 is bromo, iodo or tosylate.The route to the 2-halo-pyridine as starting material an organic zinc compound, and then prepared by Negishi coupling reaction to give 2- (2-chloro-5-nitrophenyl) pyridine.2- (2-chloro-5-nitrophenyl) pyridine in turn through a reduction reaction with acylation reaction, to give the final product Vismodegib.The key coupling step of the route using an organic zinc reagent required to react under strict anhydrous, anaerobic conditions.
The second route Suzuki coupling reaction [0010]
wherein, X2 is bromo, iodo or tosylate.The route from 3-halo-4-chloro-nitrobenzene as raw material, and 2-chloro-5-nitrophenyl boronic acid pinacol ester, and then reacted with a 2-halo-pyridine was prepared to give 2- (2-chloro 5-nitrophenyl) pyridine.2- (2-chloro-5-nitrophenyl) pyridine then after reduction and acylation reaction, to give the final product Vismodegib.The key coupling step of the route using the Suzuki coupling reaction, organic boron reagent price to use expensive, high production costs.
The route three Suzuki coupling reaction
wherein, X2 is bromo, iodo or tosylate.Similar to the second route, the route is still critical coupling step using a Suzuki coupling reaction, the same need to use expensive organic boron reagents, higher production costs.
route four Stille coupling reaction
The route to 2-p-toluenesulfonyl pyridine as starting material, is reacted with an organotin reagent, prepared to give pyridin-2-yl trimethyltin, then by Stille coupling reaction, was prepared to give 2- (2-chloro – 5- nitrophenyl) pyridine, followed by reduction reaction, acylation prepared to give Vismodegib.The key step of the route using the Stille coupling reaction, this step need to use expensive and toxic organotin reagents, and the need to carry out the reaction under strict anhydrous, anaerobic conditions.
A process for preparing 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide, comprising: a compound of formula III was prepared as a compound of Formula II;
Then, the compound of formula II with a compound of formula I, to give 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide;
Wherein, R1 is halogen or hydroxy, preferably chlorine, or a hydroxyl group.
2. A process for preparing 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide, comprising:
Wherein, X is halogen, preferably bromo or iodo condition is halo or hydroxy, preferably chlorine, or a hydroxyl group.
3. A process for preparing 2-chloro -N- (4- chloro-3- (pyridin-2-yl) phenyl) -4- (methylsulfonyl) benzamide, comprising:
Wherein, X is halogen, preferably bromo or iodo condition is halo or hydroxy, preferably chlorine, or a hydroxyl group.
Method 2 or claim 3,
Example 1: N–oxo-2- (2-chloro-5-nitrophenyl) pyridine
[0108] To a 100mL three-necked flask were added 30mmoll- oxopyrido, 10mmol2- bromo-1-chloro-4-nitrobenzene, 12mmol potassium carbonate, 0.05mmol tri-butyl acetate button and 0.15mmol phosphorus tetrafluoroborate salt, 40ml of toluene, IS gas exchange three times, under argon at reflux for 2 days, then the reaction mixture was poured into 100mL of ethyl acetate, filtered, and the filtrate was washed with saturated brine, dried and the solvent was distilled off under reduced pressure, column chromatography (mobile phase V / V: methanol / dichloromethane = 1/50), fractions were collected and the solvent was distilled off under reduced pressure to give a pale yellow solid, yield 60%.
To a 100mL three-necked flask 30mmoll- oxopyrido, 10mmol2- bromo-1-chloro-4-nitrobenzene, 12mmol of potassium carbonate, 0.05mmol iodide and 0.1Ommoll, 10- Fei Luo Jie morpholine, 40ml of xylene, an argon gas exchange three times, under argon at reflux for 2 days, cooled to room temperature and then the reaction system was poured into 100mL methylene chloride, filtered and the filtrate washed with saturated brine, dried, filtered, The filtrate solvent was distilled off under reduced pressure, column chromatography (mobile phase V / V: methanol / dichloromethane = 1/50) to give a pale yellow solid, yield 42%..
3 Example: 2- (2-chloro-5-nitrophenyl) pyridine
After 3.0mmol N- oxo added to 100mL of Lord vial _2_ (2_ chloro _5_ nitrophenyl) pyrazole 唳, 15mmol phosphorus trichloride and 30ml of chloroform was heated at reflux for 12h, the reaction It was poured into 100mL of water and extracted with ethyl acetate (50ml X 2), and the combined organic phase was dried and the solvent was distilled off under reduced pressure, column chromatography (mobile phase V / V: petroleum ether / ethyl acetate = 20/1) , fractions were collected, the solvent was distilled off under reduced pressure to give a white solid, yield 95%.
To a vial was added 100mL of Lord 20mmol2- (2- chloro-5-nitrophenyl) pyridine 唳, 50ml of acetic acid, heated to 80 ° C and stirred, and then slowly added IOOmmol iron, reaction 0.5h The reaction solution was poured into 200ml water and extracted with dichloromethane (150ml X 3), the combined organic phases, the organic phase was washed with saturated sodium carbonate solution (50ml X 3), the organic phase was dried, evaporated under reduced pressure to give the crude product, n-propyl alcohol weight crystallized to give a pale yellow solid, yield 75%.
to 100mL of God-shaped flask 20mmol2_ (2_ chlorine _5_ nitrophenyl) pyridine Jie set, 50ml of methanol, Ig activated carbon, 2mmol FeOOH and 60mmol85% of hydrazine hydrate, heated to reflux and stirred for 6 ~ 8h, after the completion of the reaction, was filtered, spin-dry the solvent, dissolved in 150ml of dichloromethane, the organic phase was washed with saturated sodium bicarbonate solution (20ml X3), the organic phase was dried, evaporated under reduced pressure to give the crude product was recrystallized from n-propanol to give a pale yellow solid, yield 96%.
6 Example 2: Preparation 4_-chloro-3- (2-yl) aniline
20mmol N- oxo added to 100mL eggplant-shaped flask _2_ (2_ chloro _5_ nitrophenyl) pyridine, 50ml of acetic acid, heated to 80 ° C and stirred, and then iron powder was slowly added IOOmmol After 0.5h the reaction the reaction solution was poured into 200ml water and extracted with dichloromethane (150ml X3), the combined organic phases were washed with saturated sodium carbonate solution (50ml X3), the organic phase was dried, evaporated under reduced pressure to give the crude product, n-propanol recrystallized to give a white solid, yield 70%.
20mmol N- oxo added to 100mL eggplant type flask _2_ (2_ chloro _5_ nitrophenyl) pyridine, 50ml of methanol, Ig active carbon, 2mmol FeOOH 60mmol85% hydrazine hydrate and heated to reflux and stirred for 6 ~ 8h, after the completion of the reaction, was filtered, spin-dry the solvent, dissolved in 150ml of dichloromethane, washed with saturated aqueous sodium bicarbonate solution, the organic phase (20mlX3), the organic phase was dried, evaporated under reduced pressure to give the crude product, n-propyl alcohol weight crystallized to give a white solid, yield 82%.
Vismodegib Preparation: 8 Example
In the Lord 50ml vial, the 1.50mmol2- chloro-4-methanesulfonyl-chloride in 15ml of dry tetrahydrofuran, cooled to ice bath O ~ 10 ° C, a solution of 4-chloro-3 – (pyridin-2-yl) aniline in anhydrous tetrahydrofuran (1.47mmol / 10ml), triethylamine was added dropwise and then finished 2.5mmol of dropwise addition, the reaction at room temperature 4h, the reaction was completed, the reaction system was poured into 50ml water and stirred, precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 88%.
In 50ml vial of God, will 1.50mmol2_ chlorine _4_ methylsulfonyl benzoic acid, 1.47mmol4_ chlorine _3_ (batch 唳 2-yl) aniline and triethylamine were dissolved in 25ml 2.5mmol anhydrous tetrahydrofuran in an ice bath to cool to O ~ 10 ° C, was added in portions N, N ‘- dicyclohexyl carbodiimide (DCC) 1.50mmol, After the addition, the reaction at room temperature 6h, after the reaction, white solid was removed by filtration, the filtrate was poured into 50ml water and stirred, precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 84%.
Vismodegib Preparation: 10 [0141] Example
In 50ml eggplant-shaped flask, 1.50mmol2- chloro-4-methanesulfonyl-benzoic acid was dissolved in 15ml of dichloromethane, cooled to ice bath O ~ 5 ° C, thionyl chloride was added dropwise 3.0mmol After stirring at room temperature 30min, removed by rotary evaporation dichloromethane and excess thionyl chloride, 15ml of anhydrous tetrahydrofuran was added, the ice bath was cooled to O ~ 10 ° C, solution of 4-chloro-3- (pyridin-2- yl) aniline in anhydrous THF (1.47mmol / 10ml), triethylamine was added dropwise and then finished 2.5mmol of dropwise addition, the reaction at room temperature 4h, the reaction was completed, the reaction was poured into 50ml water system and stirring, the precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 88%.
In 50ml eggplant-shaped flask, 1.50mmol2- chloro-4-methanesulfonyl-benzoic acid was dissolved in 15ml of dichloromethane, cooled to ice bath O ~ 5 ° C, thionyl chloride was added dropwise 3.0mmol After stirring at room temperature 30min, removed by rotary evaporation dichloromethane and excess thionyl chloride, 15ml of anhydrous tetrahydrofuran was added, the ice bath was cooled to O ~ 10 ° C, solution of 4-chloro-3- (pyridin-2- yl) aniline in anhydrous THF (1.47mmol / 10ml), triethylamine was added dropwise and then finished 2.5mmol of dropwise addition, the reaction at room temperature 4h, the reaction was completed, the reaction was poured into 50ml water system and stirring, the precipitated solid was filtered, washed with water, and dried to give a white solid product, yield 88%.
Compounds of examples 2-51 were prepared according to the following general procedures.
A: Suzuki Coupling Procedure
2 M aq. Potassium carbonate (5.0 eq) and 4:1 toluene :ethanol mixture (2.5 mL) were added to a microwave vial charged with the appropriate boronate ester (2.6 eq), aryl halide (0.35 mmol, 1.0 eq), and Pd(PPh3)4 (0.04 eq). The vial was sealed and heated with stirring in the microwave to 160 0C for ten minutes. The solution was poured onto 2 M aq. Sodium hydroxide (20 mL), extracted with ethyl acetate (2 x 20 mL), dried (MgSO4), and concentrated. Purification of the crude product by chromatography on silica gel (conditions given below) afforded the desired product.
B: Negishi Coupling Procedure
X = I or Br R = H, 3-Me, 4-Me5 5-Me, 6-Me
Aryl zinc bromide (0.5 M in THF, 2.5 eq) was added to an oven-dried microwave vial charged with the appropriate aryl halide (1.0 eq) and Pd(PPh3)4 (0.04 eq). The vial was sealed and heated with stirring in the microwave to 140 0C for 10 minutes. The crude reaction mixture was concentrated and purified by chromatography on silica gel (conditions given below) to afford the desired product.
C: Iron Reduction of Aryl Nitro Group
R = I or pyridin-2-yl
The appropriate nitro aryl (1 mmol, 1 eq) in AcOH/EtOH (1:1, 0.42 M) was added slowly to a solution of Iron powder (6.0 eq) in AcOH/EtOH (1:2, 2 M) at 60 °C. The solution was stirred at 70 0C for 30-60 minutes. The reaction mixture was cooled to 23 0C, filtered through celite, washed with ethyl acetate, and concentrated. The oily residue was dissolved in ethyl acetate (30 mL), washed with saturated aq. NaHCO3 (2 x 15 rnL) and water (2 x 10 niL), dried (MgSO4), and concentrated. The oily residue was used with out further purification.
D: Amide Bond Formation
R = I or pyridin-2-yI
Acid chloride (1.05-1.1 eq) was added to a solution of aniline (1.0 eq) and TEA (1.1-1.5 eq) in methylene chloride at the indicated temperature. The solution was stirred for 0.5-3 hours, poured onto saturated aq. NaHCO3, extracted twice with methylene chloride, dried (MgSO4), and concentrated. Purification of the crude product by chromatography on silica gel (conditions given below) afforded the desired product.
E: EDC Amide Bond Formation
R = I or pyridin-2-yl
Carboxylic acid (1.1 eq) was added to a solution of aniline (1.0 eq) and EDC (1.4 eq) in methylene chloride (0.7 M in aniline). The solution was stirred at 23 0C for 2 hours, poured onto a 1 :1 mixture of saturated aq. NH4Cl and water, extracted twice with methylene chloride, dried (MgSO4), and concentrated. Purification of the crude product by chromatography on silica gel (conditions given below) afforded the desired product. F: addition of amines to 2-chloropyridine
Primary or secondary amine (5 eq) in either BuOH or a mixture of BuOH/ethylene gylcol was heated to 170 to 220 0C for 20 min in a sealed tube. The BuOH was removed under reduced pressure. In cases where ethylene glycol was used, the reaction was diluted with water, and the product was extracted into ethyl acetate, dried (MgSO^, and concentrated. The crude residue was purified by reverse phase HPLC to afford the desired product.
G: Amide bond coupling with HATU
HATU, DIPEA, DMF NaOH or NaHCO3
ethyl acetate extraction
Aniline (1.0 eq) was added to a mixture of carboxylic acid (1.1 eq), HATU (1.1 eq) and DIPEA (2 eq) in DMF (0.1 – 0.2 M). After stirring overnight, the reaction mixture was diluted with 0.1 N sodium hydroxide or saturated NaHCθ3, extracted into ethyl acetate and the combined organic layers were washed with brine. The organic layer was dried (MgSO4), concentrated and the crude mixture was purified by reverse phase HPLC. H: Preparation of sulfonamide benzoic acids
Chlororsulfonylbenzoic acid (1.0 eq) was added to a solution of amine (1.1 eq) in 10-20% DEPEA/methanol (1 M) at 4 0C. After 1 h, the reaction mixture was concentrated, and the crude residue was purified by reverse phase HPLC.
I : Stannylation of 2-pyridyl triflates
A solution of tetrakis-triphenylphosphinepalladium (0.04 eq.) in toluene (1 mL) was added to degassed solution of aryltriflate (1 eq), bis-trialkyltin (1.05 eq), and lithium chloride (3 eq) in dioxane. Heated to reflux for 2 hours, cooled to 23 0C, diluted with ethyl acetate, washed with 10% NH4θH(aq) and brine, dried (MgSO4) and concentrated. The crude material was used without further purification.
J: Stannylation of substituted pyridines
ιMmβco3 n-Butyl lithium (6 eq, 2.5 M in hexanes) was added dropwise to a solution of dimethylaminoethanol (3 eq) in hexane at 0 0C. The solution was stirred at 0 0C for thirty minutes before dropwise addition of the substituted pyridine (1 eq). The solution was stirred at 0 0C for an additional hour, then cooled to -78 0C. A solution of trialkyltin in hexane was added dropwise. The solution was stirred at -78 0C for thirty minutes, warmed to 0 0C, quenched with water, extracted twice with ether, dried (MgSO4), and concentrated. K: Stille Coupling
Palladium catalyst (0.02 eq) was added to a degassed solution of aryliodide (1 eq), arylstannane (2 eq), and triphenylphosphine (0.16 eq) in NMP. Heated in the microwave to 130 0C for 15 minutes. The reaction mixture was diluted with ethylacetate, washed with 10% NH4θH(aq) and brine, dried (MgSC>4), concentrated and purified by silica gel chromatography.
L: Synthesis of alky lethers
A solution of hydroxypyridine (1 eq), alkyliodide (excess), and cesium carbonate in NMP was heated in the microwave to 1000C for ten minutes. The reaction mixture was diluted with ethylacetate, washed with 10% NH4θH(aq) and brine, dried (MgSC^), concentrated and purified by silica gel chromatography.
M: Methyl Ester Saponification
The methyl ester (leq) was hydrolyzed with LiOH (2eq) in 50/50 THF/water mix. Upon completion of the reaction the THF was evaporated under reduced pressure and the solution is acidified with HCl to pH 2. The resultant solid was filtered and dried to give the pure acid.
N: Bromination in the presence of a free acid functionality
The paramethylbenzoic acid (leq) was combined with Benzoyl Peroxide (O.leq) and N- Bromosuccinimde (0.9eq) in a solution of 5%AcOH in Benzene and heated in the microwave at 120°C for 5-15minutes. The product was separated from the starting material and di-bromo product via ISCO flash chromatography with an ethyl acetate (with 1% AcOH) and hexanes solvent system.
O: Sodium Methanesulfinate displacement of Bromine
To the bromine starting material (leq) was added sodium methanesulfinate (2eq) in DMF and heated to 120°C in the microwave for 5 minutes. Alternatively, the reaction was heated to 60°C in an oil bath for several hours until completed. Reaction mixture was concentrated under reduced pressure and extracted in ethyl acetate and water. The organic layer was dried over Magnesium Sulfate, filtered and concentrated in vacuo to yield generic methylsulfone.
P: Amine displacement of Bromine
To the bromo starting material (leq) was added appropriate amine (3eq) in either DMSO or BuOH and stirred at room temperature until complete. For less nucleophilic amines or anilines, the reactions were forced to completion using microwave conditions ranging from 150°-170°C for 15 minutes. Crude reactions were concentrated to dryness and either extracted with ethyl acetate and saturated bicarbonate if the reaction resulted in an intermediate or purified via HPLC if the reaction resulted in a final product.
Q: Thiol displacement of halogen
The paramethylbromo benzoate (leq) was treated with Potassium (or Cesium) Carbonate (1.5eq) and appropriate thiol derivative (l,leq) in DMF (or CH3CN) and stirred overnight at room temperature. The DMF was evaporated in vacuo and the reaction was extracted with ethyl acetate and water. The organic layer was dried over Magnesium Sulfate , filtered and concentrated to yield the thiol or derivatized thiol compound.
R: Oxone Oxidation
oxone 2:1 MeOHTH2O
Derivatized thiol (leq) was dissolved in MeOH while Oxone (2eq) was seperately dissolved in half the amount of water. Once all the oxone was dissolved, the solution was added to the thiol in MeOH solution at once and stirred until complete. The MeOH was evaporated in vacuo and the remaining water was extracted twice with Ethyl Acetate. The organic layer was dried over Magnesium Sulfate and concentrated to yield the sulfone.
S: Thio lysis of epoxides at alumina surfaces
A mixture of epoxides (1.0 eq), thiophenol (1.5 eq) and neutral aluminum oxide (~70 eq) in diethyl ether was stirred for 3 h at room temperature while being monitored by TLC. The reaction mixture was filtered through Celite, washed with ethyl acetate and concentrated. Purified by silica gel chromatography (0-40% ethyl acetate/hexane) to yield β -hydroxysulfide product.
T: Conversion of nitrile group to carboxylic acid
R
A solution of benzonitrile (1.0 eq) and sodium hydroxide (2.0 eq) in H2O was heated to 120 ° C for 2h. The reaction mixture was cooled to room temperature and acidified with HCl to pH 2. The resulting solid was filtered to afford the pure acid product.
U. Alkylation of phenols
The phenol was dissolved in DMF (1.0 ml). Cesium carbonate (1.0 eq.) and an alkyl bromide or alkyl iodide (1.0 to 2.0 eq.) were added, and the reaction was stirred at room temperature for 18 hrs or 5O0C for 1 to 24 hours. The reaction was quenched in water, and extracted with ethyl acetate twice. The organic extracts were washed with water once, brine once, dried with MgSC>4, and evaporated to a crude oil which was purified on reverse phase HPLC.
V. Amide bond formation with an acid chloride and an aniline
The aniline was dissolved in THF (1.5 ml) and dichloromethane (1.5 ml). MP-Carbonate (1.5 eq.) and an acid chloride (1.1 eq.) were added, and the solution was stirred at room temperature for 18 hours. The reaction was diluted with methanol and dichloromethane, and filtered to remove the MP-Carbonate. The mother liquors were evaporated to a solid and purified by reverse phase HPLC.
W. Amidine formation from an imidate
A solution of freshly formed imidate in methanol was treated with a primary or secondary amine (1.5 eq.) at room temperature for 18 hours. The methanol was removed on a rotary evaporator and the residue purified by reverse phase HPLC.
Example 37 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide
Procedure G was used to couple 4-chloro-3-(pyridin-2-yl)aniline (50 mg) and 2-chloro-4- methylsulfonylbenzoic acid to produce 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4- (methylsulfonyl)benzamide. MS (Ql) 421.0 (M)+. The product was then dissolved in 1 Ν HCI solution followed by freebasing with 0.5 Ν NaOH solution (pH to 11). The resulting precipitate was filtered and vacuum-dry.
Procedure D may also be used to couple 4-chloro-3-(pyridin-2-yl)aniline and 2-chloro-4- (methylsulfonyl)benzoyl chloride to produce 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-
(methylsulfonyl)benzamide which is collected by suction filtration and the HCl salt is washed with
Et2O (or alternatively with MTBE). This material is freebased using EtOAc/aq NaHCO3 and the organics are dried and concentrated to the solid freebase. This material is then crystallized from acetone :EtOAc (80:20, approx lOmL/g) which is then finally recrystallized from hot slurry of iPrOAc. 2-chloro-N-(4-chloro-3-(pyridin-2-yl)phenyl)-4-(methylsulfonyl)benzamide HCl salt may also be dissolved in distilled water followed by freebasing with 0.5 N NaOH solution (pH to 11) and filtering and vacuum drying the precipitate.
The present invention primarily relates to multi-component crystals comprising a compound of formula 1 and a second compound selected from the group consisting of co-crystal formers and sol-vents. The invention is further related to pharmaceutical compositions comprising such multi-component crystals. Furthermore, the invention relates to processes for preparing said multi-component crystals. The invention also relates to several aspects of using said multi-component crystals or pharmaceutical compositions to treat a disease.
Developed and launched by Roche and its subsidiary Genentech, under license from Curis. Family members of the product Patent of vismodegib (WO2006028958),
Vismodegib was first disclosed in WO Patent Publication No. 06/028959. Vismodegib, chem-ically 2-Chloro-N-(4-chloro-3-pyridin-2-ylphenyl)-4-methylsulfonylbenzamide, is represented by the following structure:
formula 1
Vismodegib is an active pharmaceutical ingredient produced by Genentech (Roche) and sold under the trade name Erivedge® (which contains crystalline Vismodegib as the active ingre-dient). Erivedge® is an oral Hedgehog signaling pathway inhibitor approved for the treatment of basal-cell carcinoma (BCC).
The present invention primarily relates to multi-component crystals comprising a compound of formula 1 (cf. above) and a second compound selected from the group consisting of co-crystal formers and solvents.
The invention is further related to pharmaceutical compositions comprising said multi-component crystals. Furthermore, the invention also relates to processes for preparing said multi-component crystals. The invention also relates to several aspects of using said multi-component crystals or pharmaceutical compositions to treat a disease. Further details as well as further aspects of the present invention will be described herein below.
Vismodegib is a BCS class II compound with a high permeability but a low solubility where enhanced solubility or dissolution rates can lead to a significant advantage in respect to bio-availability.
Vismodegib is known to exist as crystalline free base. Salts of Vismodegib are men-tioned in US 7,888,364 B2 but not specified. In particular, the HCI salt is mentioned as intermediate but not characterized. Co-crystals or solvates are not reported at all.
The solubility of Vismodegib is reported to be 0.1 μg/mL at pH 7 and 0.99 mg/mL at pH 1 for Erivedge®. The absolute bio-availability after single dose is reported to be 31.8 % and the ex-posure is not linear at single doses higher than 270 mg. Erivedge® capsules do not have a food label. The estimated elimination half-life (t1/2) after continuous once-daily dosing is 4 days and 12 days after a single dose treatment (Highlights of Prescribing Information: ERIVEDGE® (vismodegib) capsule for oral use; Revised: 01/2012).
The discovery and preparation of new co-crystals or solvates offer an opportunity to improve the performance profile of a pharmaceutical product. It widens the reservoir of techniques/materials that a formulation scientist can use for designing a new dosage form of an active pharmaceutical ingredient (API) with improved characteristics. One of the most important characteristics of an API such as Vismodegib is the bio-availability which is often determined by the aqueous solubility.
A compound like Vismodegib may give rise to a variety of crystalline forms having dis-tinct crystal structures and physical characteristics like melting point, X-ray diffraction pattern, infrared spectrum, Raman spectrum and solid state NMR spectrum. One crystalline form may give rise to thermal behavior different from that of another crystalline form. Thermal behavior can be measured in the laboratory by such techniques as capillary melting point, thermogravimetry (TG), and differential scanning calorimetry (DSC) as well as content of sol-vent in the crystalline form, which have been used to distinguish polymorphic forms.
Multi-component crystals comprising Vismodegib and selected co-crystal formers or solvents may improve the dissolution kinetic profile and allow to control the hygrosco-picity of Vismodegib.
Therefore, there is a need for multi-component crystals comprising Vismodegib that avoid the above disadvantages. In particular, it is an object of the present invention to provide multi-component crystals of Vismodegib with optimized manufacture, formula-tion, stability and/or biological efficacy
.
Example 1 :
314 mg Vismodegib and 86 mg maleic acid are suspended in toluene saturated with maleic acid for 2 d, filtered and dried.
TG data shows a mass loss of about 2.3 wt % between 100 and 1 18 °C which is attributed to rest solvent. DSC data shows a single endothermal peak with an onset of about 1 15 °C (99 J/g).
H-NMR spectroscopy indicates a molar ratio of Vismodegib to maleic acid of about 1 :1 .3. However single crystal X-ray data confirms a ratio of 1 :2 (Table 1 ).
update……………
Vismodegib Synthesis
WO2009126863A2: also see Ref. 1. It all started from here.
†Small Molecule Process Chemistry, ‡Small Molecule Analytical Chemistry, Genentech, A Member of the Roche Group, 1 DNA Way, South San Francisco, California 94080, United States
The development work toward the robust and efficient manufacturing process to vismodegib, the active pharmaceutical ingredient (API) in Erivedge, is described. The optimization of the four-stage manufacturing process was designed to produce the API with the required critical quality attributes: (1) the selective catalytic hydrogenation reduction of the nitro compound 3 to the corresponding aniline 4 while minimizing the formation of potential genotoxic (mutagenic) impurities; (2) the control of the polymorphic phase and multipoint specification for particle size distribution.
METHOD OF INHIBITING DYRK1B [US2014371251]2014-06-182014-12-18
USE OF SUBSTITUTED HEXITOLS INCLUDING DIANHYDROGALACTITOL AND ANALOGS TO TREAT NEOPLASTIC DISEASE AND CANCER STEM AND CANCER STEM CELLS INCLUDING GLIOBLASTOMA MULTIFORME AND MEDULLOBLASTOMA [US2014377336]2013-01-222014-12-25
SHH Regulation and Methods Thereof [US2012082623]2011-09-302012-04-05
NOVEL 2-PIPERIDIN-1-YL-ACETAMIDE COMPOUNDS FOR USE AS TANKYRASE INHIBITORS [US2015025070]2012-07-132015-01-22
Compositions and Methods for Modulating Neuron Degeneration and Neuron Guidance [US2011065645]2010-09-102011-03-17
SMOOTHENED ANTAGONISM FOR THE TREATMENT OF HEDGEHOG PATHWAY-RELATED DISORDERS [US2014200217]2014-01-242014-07-17
Mirabegron (YM-178, Astellas Pharma), is an orally active, first-in-class selective β₃-adrenoceptor agonist for the symptomatic treatment of overactive bladder (OAB), and has been approved for urinary frequency and urinary incontinence associated with OAB
Mirabegron (YM-178) is the first β3-adrenoceptor agonist that is clinically effective for overactive bladder. Mirabegron (0.3 and 1 mg/kg) inhibits mechanosensitive single-unit afferent activities (SAAs) of Aδ fibers in response to bladder filling. Mirabegron activates the β3 adrenergic receptor in the detrusor muscle in the bladder, which leads to muscle relaxation and an increase in bladder capacity. Mirabegron (YM-178) acts partly as an irreversible or quasi-irreversible metabolism-dependent inhibitor of CYP2D6. Mirabegron at a dose of 3 mg/kg i.v. decreased the frequency of rhythmic bladder contraction induced by intravesical filling with saline without suppressing its amplitude in anesthetized rats. Mirabegron decreases primary bladder afferent activity and bladder microcontractions in rats. Mirabegron (YM-178) also reduced non-micturition bladder contractions in an awake rat model of bladder outlet obstruction.
Mirabegron is a white crystalline powder, not hygroscopic and freely soluble in dimethyl sulfoxide, soluble in methanol and soluble in water between neutral to acidic pH. The chemical name is 2-(2- Amino-1,3-thiazol-4-yl)-N-[4-(2-{[(2R)-2-hydroxy-2- phenylethyl]amino}ethyl)phenyl]acetamide., Mirabegron exhibits stereoisomerism due to the presence of one chiral centre. The R enantiomer has been used in the manufacture of the finished product. The enantiomeric purity is controlled routinely by chiral HPLC-UV. Polymorphism has been observed for the active substance. The polymorphic form α is routinely and consistently produced by the synthetic process and it is used in the manufacture of the finished product…….http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002388/WC500137308.pdf
Mirabegron (formerly YM-178, trade name Myrbetriq, Betmiga in Spain) is a drug for the treatment of overactive bladder.[2] It was developed by Astellas Pharma and was approved in the United States in July 2012.[3]
Mirabegron activates the β3 adrenergic receptor in the detrusor muscle in the bladder, which leads to muscle relaxation and an increase in bladder capacity.[4]\
Journal of Chemical and Pharmaceutical Research, 2015, 7(4):1473-1478
In the first approach, the introduction of the chiral hydroxyl group was planned at the later stage (Scheme 1). Accordingly, 2-(4-nitrophenyl)ethyl amine 4 was protected as the Boc-derivative 5, followed by the reduction of the nitro group using stannous chloride to furnish corresponding aniline 6. Alternate reducing conditions such as hydrogenation in the presence of 10% Pd-C were also provided the desired 6 in good yield. Amide coupling of the aniline 6 with 2-(2-aminothiazol-4-yl) acetic acid 7 in the presence of EDC, HOBt/DIPEA furnished the desired amide 8. Interestingly, lower reactivity of 2-aminothiazole precluded any self-coupling of 7.
Removal of Boc-group in 8, set the stage for the critical step of introducing the chiral hydroxyl by means of stereocontrolled ring opening of the chiral (R)-styrene epoxide 10. Epoxide opening reaction of 10 was initially attempted with amine 9 in the presence of Et3N in MeOH as the solvent. Alternatively, epoxy opening was also performed under simple isopropanol reflux condition to get the desired 1. The desired product 1 was isolated in 27% yield after purification by column chromatography. This is due to the formation of N-alkylated derivatives of 1 by undesired reaction of 10 with amino functionalities of 1. However, the inefficiency of the epoxide opening reaction precluded a high purity of final product, Mirabegron 1. Since it is not practical to embark on repeated purifications at the last stage (which leads to poor yields), this route was not pursued for further optimization.
in 500mL three-necked flask, 2- (2-aminothiazol-4-yl) acetic acid 17.42g (0.086mol), N, N- dimethylformamide 180mL, then added H0BT15.12g (0.104 mol), was added (R) _2 _ ((4- aminophenyl) amino) phenyl-ethan-l-ol -1_ 20g (0.078mol), was added triethylamine 13.04g (0.13mol), was added portionwise EDCI21. 46g (0.104mol), under magnetic stirring, room temperature for 5h, TLC until the reaction was complete tracking.
After treatment: After the completion of the reaction, the reaction solution was poured into 900mL saturated saline water, and then extracted with 400mL of dichloromethane each time, and extracted three times, each time the organic phase is then washed with 200mL of saturated aqueous sodium carbonate solution, washed three times, each time with distilled water and then 200mL of water, washed three times, the organic phase was dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a white solid in methylene chloride was distilled off Mira Veron crude, the crude product was recrystallized from methanol solution, wherein the methanol solution of methanol and water, the volume ratio of 10: 4, and recrystallized to give 25.08g, yield 81.0%.
The present embodiment Mira Veron synthesized for testing and structural identification:
mp138 ~ 140 ° C (137 ~ 139 ° C)
[α] 20-18. ~ -22. (CH3OH)
chemical purity HPLC: 99.96%
Optical purity: 97.55ee%
HRMS (ES1-MS, m / z) calcd: for C21H25N4O2S [M + H] + 397.16.Found:. 397.16
1H Mffi (400MHz, DMS0) Sl0.00 (s, lH), 7.50 ( d, J = 8.5Hz, 2H), 7.30 (dd, J = 9.5,5.1Hz, 4H), 7.23 (dd, J = 6.0, 2.7Hz, 1H), 7.12 (d, J = 8.5Hz, 2H), 6.90 (s, 2H), 6.30 (s, 1H), 5.24 (s, 1H), 4.60 (s, 1H), 3.45 (s, 2H), 2.74 (dd, J = 9.8, 3.5Hz, 2H), 2.64 (m, 4H).
13C NMR (101MHz, DMSO) δ 168.69 (s), 168.26 (s), 146.35 (s), 145.03 (s), 137.66 (s), 135.51 (s), 129.24 (s ), 128.38 (s), 127.22 (s), 126.33 (s), 119.46 (s), 103.03 (s), 71.88 (s), 57.94 (s), 51.20 (s), 40.40 (s), 40.20 (s ), 39.99 (s), 39.78 (s), 39.57 (s), 35.77 (s)
By and O ° C under nitrogen protection temperature conditions, 7.3g (R) -2- amino _1_ benzeneethanol added 250mL three-necked flask, the stirring was dissolved in 50mL of dichloromethane Mira Veron Intermediate C was added dropwise to the reaction solution to form three-necked flask. Stirred for I hour under nitrogen, with stirring 4.12g of sodium borohydride was added to the reaction mixture. The reaction mixture was stirred (under TC 3 hours to TLC the reaction was complete. The reaction is complete the reaction mixture was added dropwise a saturated aqueous ammonium chloride solution IOmL quenched reaction was washed twice with 40mL of water, the organic phase was separated. The The organic phase at the conditions at 0 ° C was added concentrated sulfuric acid was stirred IOmL until TLC after 0.5 hours the reaction was complete, then was added 20mL of 20% aqueous sodium hydroxide solution to complete the reaction of the organic phase was adjusted to pH 10 and stirred for 15 minutes minutes solution. The organic phase first with 50mL saturated brine I times with IOg anhydrous sodium sulfate and concentrated to give crude product was recrystallized from methanol and water to give 18.7g of the final product Mira Veron purity of 99.33%, chiral purity of 99.01%, a yield of 88.12%.
Mira Veron use randomly selected samples prepared by the synthesis method of the present invention is detected by liquid chromatography.
Test conditions: Instrument: Agilent 1100 HPLC;
Column: Luna C18, 4.6mmX 250mm, 5 μ m;
Column temperature: 25 ° C;
flow rate: 1.0mL / min;
The detection wavelength: 2IOnm;
Injection volume: 5ul;
Mobile phase A: acetonitrile;
Mobile phase B: 0.1% phosphoric acid aqueous solution;
Running time: 40min.
FIG liquid chromatography after detection of the sample shown in Figure 1; results are shown in Table I.
Table 1: The Mira Veron chromatographic analysis sample preparation method of the present invention
Example 4 (Production of the α-form crystal from wet cake of the β-form crystal) :
The same procedures as in Example 2 were followed to obtain 23.42 kg of a wet cake of the β-form crystal of (R)-2-(2-aminothiazol-4-yl)-4′-[2-[(2-hydroxy-2-phenylethyl)amino]ethyl]acetanilide from 6.66 kg of (R)-2-[[2-(4-aminophenyl)ethyl]amino]-1-phenylethanol monohydrochloride. This cake was added with and dissolved in 92 L of water and 76 L of ethanol by heating at about 80°C, and the solution was cooled at a rate of about 10°C per hour, to which was then added 8.4 g of the α-form crystal at 55°C. Thereafter, the mixture was cooled to 20°C. A crystal was filtered and dried to obtain 6.56 kg of the α-form crystal of (R)-2-(2-aminothiazol-4-yl)-4′-[2-[(2-hydroxy-2-phenylethyl)amino]ethyl]acetanilide.
Powder X-ray diffraction diagram and thermal analysis diagram of the α-form crystal are shown in Fig. 4 and Fig. 5, respectively. 1H-NMR (DMSO-d6, 500 MHz) δ (ppm) = 1.60 (1H, s), 2.59 to 2.66 (4H, m), 2.68 to 2.80 (2H, m), 3.45 (2H, s), 4.59 (1H, br), 5.21 (1H, br), 6.30 (1H, s), 6.89 (2H, s), 7.11 (2H, d, J = 8.5 Hz), 7.19 to 7.23 (1H, m), 7.27 to 7.33 (4H, m), 7.49 (2H, d, J = 8.5 Hz), 9.99 (1H,s). FAB-MS m/z: 397 (M+H)+.
Sacco, E; Bientinesi, R et al. (Apr 2014). “Discovery history and clinical development of mirabegron for the treatment of overactive bladder and urinary incontinence”. Expert Opin Drug Discov9 (4): 433–48. doi:10.1517/17460441.2014.892923. PMID2455903.
Hepatic via (direct) glucuronidation, amide hydrolysis, and minimal oxidative metabolism in vivo byCYP2D6 and CYP3A4. Some involvement of butylcholinesterase[1]
Alpha-form or beta-form crystal of acetanilide derivative [US7342117]
2005-01-06
2008-03-11
Pharmaceutical composition for treating stress incontinence and/or mixed incontinence [US2006004105]
2006-01-05
Pharmaceutical composition comprising a beta-3-adrenoceptor agonist and a serotonin and/or norepinephrine reuptake inhibitor Pharmaceutical composition comprising a beta-3-adrenoceptor agonist and a serotonin and/or norepinephrine reuptake inhibitor [US2009012161]
2005-11-24
Pharmaceutical composition consisting of a beta-3-adrenoceptor agonist and alpha-agonist [US2005154041]
2005-07-14
Pharmaceutical composition consisting of a beta-3-adrenoceptor agonist and an active substance which influences prostaglandin metabolism [US2005119239]
2005-06-02
Pharmaceutical Composition For Treating Stress Incontinence And/Or Mixed Incontinence [US2007129435]
2007-06-07
Remedy for overactive bladder comprising acetic acid anilide derivative as the active ingredient [US7750029]
2006-06-01
2010-07-06
[alpha]-form or [beta]-form crystal of acetanilide derivative [US7982049]
2008-09-04
2011-07-19
BETA ADRENERGIC RECEPTOR AGONISTS FOR THE TREATMENT OF B-CELL PROLIFERATIVE DISORDERS [US2010009934]
2010-01-14
PHARMACEUTICAL COMPOSITION FOR IMPROVING LOWER URINARY TRACT SYMPTOMS [US2010261770]
2010-10-14
11 to 16 of 16
Patent
Submitted
Granted
PHARMACEUTICAL COMPOSITION FOR MODIFIED RELEASE [US2010144807]
2010-06-10
BENZYLAMINE DERIVATIVE OR PHARMACEUTICALLY ACCEPTABLE ACID ADDITION SALT THEREOF, AND USE THEREOF FOR MEDICAL PURPOSES [US8148427]
2010-04-22
2012-04-03
Pharmaceutical composition containing a beta-3-adrenoceptor agonist and an alpha antagonist and/or a 5-alpha reductase inhibitor [US2005101607]
2005-05-12
REMEDY FOR OVERACTIVE BLADDER COMPRISING ACETIC ACID ANILIDE DERIVATIVE AS THE ACTIVE INGREDIENT [US2009093529]
2009-04-09
PHARMACEUTICAL COMPOSITION FOR TREATING OVERACTIVE BLADDER [US2010240697]
2010-09-23
Pharmaceutical composition comprising beta-3-adrenoceptor-agonists and antimuscarinic agents [US2005261328]
2005-11-24
US Patent No
Patent Expiry
patent use
6346532
Oct 15, 2018
6562375
Aug 1, 2020
6699503
Sep 10, 2013
7342117
Nov 4, 2023
7750029
Dec 18, 2023
U-913
7982049
Nov 4, 2023
Exclusivity Code
Exclusivity Date
NCE
Jun 28, 2017
U-913……….TREATMENT OF OVERACTIVE BLADDER WITH SYMPTOMS OF URGE URINARY INCONTINENCE, URGENCY, AND FREQUENCY
//////Mirabegron, Overactive bladder, FDA 2012, ASTELLAS PHARMA, YM-178, Myrbetriq, Betmiga
Updates…….
Overactive bladder (OAB) is characterized by symptoms of urinary urgency, with or without urgency incontinence, usually with increased daytime frequency and nocturia.(1-3) Current guidelines recommend oral antimuscarinics drugs as the first-line pharmacologic therapy in the management of OAB despite the companion adverse effects.(4, 5) Mirabegron is an orally active β3 adrenoceptor agonist approved by the FDA for treatment of OAB in 2012, which is an important step toward the better treatment options for the management of OAB.(6)
(R)-Styrene oxide 1 and 4-nitrophenethylamine 2 were exploited as starting materials in the first synthesis of mirabegron (Scheme 1). Heating 1 and 2 in i-propanol afforded amino alcohol 3, and then the amino group was protected by di-tert-butyl dicarbonate (Boc2O), followed by a condensation with 2-aminothiazol-4-acetic acid. Deprotection of the condensation product 7 finally afforded mirabegron.(7-10) Although reactions in the whole process were all conventional reactions, optically pure 1 was not industrially available, which restricted its application in industry.
(R)-Mandelic 8 and 4-nitrophenethylamine hydrochloride 9 were exploited as starting materials in an alternate route (Scheme 2). Condensation of 8 and 9 in the presence of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDCI), 1-hydroxybenzotriazole (HOBt), and triethylamine in N,N-dimethylformamide (DMF) furnished the corresponding amide 10, which was further reduced in the presence of borane-tetrahydrofuran complex in a mixed solution of 1,3-dimethyl-2-imidazolidinone (DMI) and tetrahydrofuran (THF), affording amine 11. The nitro group of 11 was then reduced by hydrogenation affording aniline 12 which was further amidated by an aqueous EDCI coupling affording mirabegron. This route was rather concise with only four steps, in which the sole stereogenic center was introduced via a bulk starting material 8.(11-13) However, usage of the costly EDCI twice, especially in the first step, led to a high cost and more impurities.
2.Abrams, P.; Chapple, C.; Khoury, S.; Roehrborn, C.; de la Rosette, J.J. Urol.2009, 181, 1779, DOI: 10.1016/j.juro.2008.11.127
3.Jaiprakash, H.; Benglorkar, G. M.RJPBCS2014, 5 ( 3) 213
4.Lucas, M. G.; Ruud, J. L.; Bosch, R. J. L.; Burkhard, F. C.; Cruz, F.; Madden, T. B.; Nambiar, A. K.; Neisius,A.; de Ridder, D. J. M. K.; Tubaro, A.; Turner, W.; Pickard, R.Eur. Urol.2012, 62, 1130, DOI: 10.1016/j.eururo.2012.08.047
5.Gormley, E. A.; Lightner, D. J.; Burgio, K. L.; Chai, T. C.; Clemens, J. Q.; Culkin, D. J.; Das, A. K.; Foster, H. E.; Scarpero, H. M.; Tessier, C. D.; Vasavada, S. P.J. Urol.2012, 188, 2455, DOI: 10.1016/j.juro.2012.09.079
Pfizer Inc’s oral JAK inhibitor tofacitinib was approved on November 6, 2012 by US FDA for the treatment of rheumatoid arthritis.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Part of the Pfizer group responsible for Xeljanz: Front row, from left: Sally Gut Ruggeri, Chakrapani Subramanyam, Eileen Elliott Mueller, and Frank Busch. Second row, from left: Matthew Brown, Mark Flanagan, and Robert Dugger. Back row, from left: Elizabeth Kudlacz and Douglas Ball.
Credit: Pfizer
Mark Flanagan, who was on the team at Pfizer that discovered Xeljanz, (tofacitinib citrate), an oral treatment for rheumatoid arthritis, remembers testing the drug in a rat model and seeing the drug decrease the level of inflammation in the rats’ footpads. “What we look for is physical measurements of the size of the joint. In the control animals, there was quite a bit of inflammation in the joints, whereas animals treated with different doses of the drug showed a dose-dependent decrease in the size of the joint. “Tofacitinib showed robust efficacy in the first such study run. I can remember the excitement that this data generated on the team,” he says.
Tofacitinib, chemically known as (3R,4R)-4-methyl-3-(methyl-7H-pyrrolo [2,3- d]pyrimidin-4-ylamino)-B-oxo-l -piperidinepi panenitrile, is represented Formula I. Tofacitinib citrate, a janus kinase inhibitor, is approved as XELJANZ® tablets for treatment .of rheumatoid arthritis.
Various intermediates and processes for preparation of tofacitinib are disclosed in patents like US7301 023 and US8232394.
Formula I or isomers or a mixture of isomers thereof by following any method provided in the prior art, for example, by following Example 14 of U.S. Patent No. RE41,783 or by following Example 6 of U.S. Patent No. 7,301,023. Tofacitinib of Formula I or isomers of tofacitinib or a mixture of isomers thereof may be converted into a salt by following any method provided in the prior art, for example, by following Example 1 of U.S. Patent No. 6,965,027 or by following Example 1 or Example 8 of PCT Publication No. WO 2012/135338. The potential significance of JAK3 inhibition was first discovered in the laboratory of John O’Shea, an immunologist at the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH).[5] In 1994, Pfizer was approached by the NIH to form a public-private partnership in order to evaluate and bring to market experimental compounds based on this research.[5] Pfizer initially declined the partnership but agreed in 1996, after the elimination of an NIH policy dictating that the market price of a product resulting from such a partnership would need to be commensurate with the investment of public taxpayer revenue and the “health and safety needs of the public.”[5] The drug discovery, preclinical development, and clinical development of tofacitinib took place exclusively at Pfizer.[6] In November 2012, the U.S. Food and Drug Administration (FDA) approved tofacitinib for treatment of rheumatoid arthritis. Once on the market, rheumatologists complained that the $2,055 a month wholesale price was too expensive, though the price is 7% less than related treatments.[6] A 2014 study showed that tofacitinib treatment was able to convert white fat tissues into more metabolically active brown fat, suggesting it may have potential applications in the treatment of obesity.[7] It is an inhibitor of the enzyme janus kinase 1 (JAK1) and janus kinase 3 (JAK 3) , which means that it interferes with the JAK-STAT signaling pathway, which transmits extracellular information into the cell nucleus, influencing DNA transcription.[3] Recently it has been shown in a murine model of established arthritis that tofacitinib rapidly improved disease by inhibiting the production of inflammatory mediators and suppressing STAT1-dependent genes in joint tissue. This efficacy in this disease model correlated with the inhibition of both JAK1 and 3 signaling pathways, suggesting that tofacitinib may exert therapeutic benefit via pathways that are not exclusive to inhibition of JAK3.[4]
Preparation of 3-{(3R,4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile citrate salt (Tofacitinib citrate, Xeljanz, CP-690550-10)
To a round-bottomed flask fitted with a temperature probe, condenser, nitrogen source, and heating mantle, methyl-[(3R,4R)-4-methyl-piperidin-3-yl]-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (5.0 g, 20.4 mmol) was added followed by 1-butanol (15 mL), ethyl cyanoacetate (4.6 g, 40.8 mmol), and DBU (1.6 g, 10.2 mmol). The resulting amber solution was stirred at 40 °C for 20 h. Upon reaction completion, citric acid monohydrate (8.57 g, 40.8 mmol) was added followed by water (7.5 mL) and 1-butanol (39.5 mL). The mixture was heated to 81 °C and held at that temperature for 30 min. The mixture was then cooled slowly to 22 ºC and stirred for 2 h. The slurry was filtered and washed with 1-butanol (20 mL). The filter cake was dried in a vacuum oven at 80 °C to afford 9.6 g (93%) of tofacitinib citrate as an off-white solid.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
Anal. Calcd for C22H28N6O8: C, 52.38; H, 5.59; N, 16.66. Found: C, 52.32; H, 5.83; N, 16.30. For additional characterization of the monocitrate salt of 1 see WO 03/048162.
NMR PREDICT
References:
Weiling Cai, James L. Colony,Heather Frost, James P. Hudspeth, Peter M. Kendall, Ashwin M. Krishnan,Teresa Makowski, Duane J. Mazur, James Phillips, David H. Brown Ripin, Sally Gut Ruggeri, Jay F. Stearns, and Timothy D. White; Investigation of Practical Routes for the Kilogram-Scale Production of cis-3-Methylamino-4-methylpiperidines; Organic Process Research & Development 2005, 9, 51−56
Ripin, D. H.B.; 3-amino-piperidine derivatives and methods of manufacture, US patent application publication, US 2004/0102627 A1
Ruggeri, Sally, Gut;Hawkins, Joel, Michael; Makowski, Teresa, Margaret; Rutherford, Jennifer, Lea; Urban,Frank,John;Pyrrolo[2,3-d]pyrimidine derivatives: their intermediates and synthesis, PCT pub. No. WO 2007/012953 A 2, US20120259115 A1, United States Patent US8232393. Patent Issue Date: July 31, 2012
Kristin E. Price, Claude Larrive´e-Aboussafy, Brett M. Lillie, Robert W. McLaughlin, Jason Mustakis, Kevin W. Hettenbach, Joel M. Hawkins, and Rajappa Vaidyanathan; Mild and Efficient DBU-Catalyzed Amidation of Cyanoacetates, Organic Letters, 2009, vol.11, No.9, 2003-2006
COSY PREDICTसुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
A novel stereoselective synthesis of Tofacitinib (CP-690,550), a Janus tyrosine kinase (JAK3) specific inhibitor, has been achieved starting from (5S)-5-hydroxypiperidin-2-one in 10 steps from 2 with a 9.5% overall yield. The potentiality of this synthetic route is the obtention of tert-butyl-(3S,4R)-3-hydroxy-4-methylpiperidine-1-carboxylate (6b) as a new chiral precursor involved in the synthesis of CP690,550, in a three-step reaction, without epimerizations, rather than the 5 or more steps used in described reactions to achieve this compound from analogues of 6b.
Graphical abstract
…………………. Tofacitinib synthesis: US2001053782A1
Tofacitinib synthesis: WO2002096909A1
Tofacitinib synthesis: Org Process Res Dev 2014, 18(12), 1714-1720 (also from a chinese publication, same procedure just slight changes in reagents/conditions)
References:
1. Blumenkopf, T. A.; et. al. Pyrrolo[2,3-d]pyrimidine compounds. US2001053782A1
2. Flanagan, M. E.; et. al. Optical resolution of (1-benzyl-4-methylpiperidin-3-yl) -methylamine and the use thereof for the preparation of pyrrolo 2,3-pyrimidine derivatives as protein kinases inhibitors. WO2002096909A1
3. Das, A.; et. al. An Improved and Efficient Process for the Preparation of Tofacitinib Citrate. Org Process Res Dev2014, 18(12), 1714-1720.
PATENThttps://www.google.co.in/patents/WO2003048162A1?cl=en The crystalline form of the compound of this invention 3-{4-methyl-3-[methyl- (7H-pyrrolot2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo-propionitrile mono citrate salt is prepared as described below. Scheme 1
Scheme 2
Example 1 3-{(3R,4R)-4-methyl-3-rmethyl-(7H-pyrrolor2,3-dlpyrimidin-4-yl)-amino1- piperidin-1-yl}-3-oxo-propionitrile mono citrate salt Ethanol (13 liters), (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H-pyrrolo[2,3- d]pyrimidin-4-yl)-amine (1.3 kg), cyano-acetic acid 2,5-dioxo-pyrrolidin-1-yl ester (1.5 kg), and triethylamine (1.5 liters) were combined and stirred at ambient temperature. Upon reaction completion (determined by High Pressure Liquid Chromotography (HPLC) analysis, approximately 30 minutes), the solution was filtered, concentrated and azeotroped with 15 liters of methylene chloride. The reaction mixture was washed sequentially with 12 liters of 0.5 N sodium hydroxide solution, 12 liters of brine and 12 liters of water. The organic layer was concentrated and azeotroped with 3 liters of acetone (final pot temperature was 42°C). The resulting solution was cooled to 20°C to 25°C followed by addition of 10 liters of acetone. This solution was filtered and then aqueous citric acid (0.8 kg in 4 liters of water) added via in-line filter. The reaction mixture was allowed to granulate. The slurry was cooled before collecting the solids by filtration. The solids were dried to yield 1.9 kg (71 %) (3R, 4R)- 3-{4-Methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile mono citrate. This material was then combined with 15 liters of a 1:1 ratio of ethanol/water and the slurry was agitated overnight. The solids were filtered and dried to afford 1.7 kg (63% from (3R, 4R)-methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine) of the title compound as a white crystalline solid. 1H NMR (400 MH2)(D20) δ HOD: 0.92 (2H, d, J = 7.2 Hz), 0.96 (1H, d, J = 7.6 Hz),1.66 (1H, m), 1.80 (1H, m), 2.37 (1H, m), 2.58 (2H, 1/2 ABq, J = 15.4 Hz), 2.70 (2H, 3ABq, J = 154 Hz), 3.23 (2H, s), 3.25 (1H, s), 3.33 (1H, m), 3.46 (1H, m), 3.81 (4H, m),4.55 (1 H, m), 6.65 (1 H, d, J = 3.2 Hz), 7.20 (1 H, t, J = 3.2 Hz), 8.09 (1 H, m).
To a clean, dry, nitrogen-purged 2 L hydrogenation reactor were charged 20 wt% Pd(OH)2/C (24.0 g, 50% water wet), water (160 ml), isopropanol (640 ml), (1-benzyl-4-methyl-piperidin-3-yI)-methyi- (7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amine (160.0 g, 0.48 mol), and acetic acid (28.65 g, 0.48 mol). The reactor was purged with three times at 50 psi with nitrogen and three times at 50 psi with hydrogen. Once purging was complete, the reactor was heated to 45-55°C and pressurized to 50 psi with hydrogen through a continuous feed. The hydrogen uptake was monitored until no hydrogen was consumed for 1 hour. The reactor was cooled to 20-300C and purged three times at 50 psi with nitrogen. The reaction mixture was filtered through wet Celite and the filtrate was sent to a clean, dry, nitrogen-purged vessel. A solution of sodium hydroxide (39.33 g) in water (290 ml) was charged and the mixture was stirred for a minimum of 1 hour then heated to 75-900C. The isopropanol was removed by distillation. The reaction mixture was cooled to 20-30°C and 2-methyltetrahydrofuran (1.6 L) was added. The aqueous layer was drained off and the 2-methyltetrahydrofuran was displaced with toluene (1.6 L). The distillation was continued until the final volume was 800 ml. The slurry was cooled to 20-30°C and held for a minimum of 7 hours. The resulting solids were isolated by filtration and washed with toluene (480 ml). After drying under vacuum between 40-50DC for a minimum of 24 hours with a slight nitrogen bleed 102.3 g (87.3%) of the title compound were isolated. Mp 158.6-159.8°C. 1H NMR (400 MHz, CDCI3): δ 11.38 (bs, 1H), 8.30 (s, 1H), 7.05 (d, J=3.5 Hz, 1H), 6.54 (d, J=3.5 Hz, 1H), 4.89-4.87 (m, 1H), 3.39 (s, 3H), 3.27 (dd, J=12.0, 9.3 Hz, 1 H), 3.04 (dd, J=12.0, 3.9 Hz, 1H), 2.94 (td, J=12.6, 3.1 Hz, 1H0, 2.84 (dt, J=12.6, 4.3 Hz, 1H), 2.51-2.48 (m, 1H), 2.12 (bs, 2H), 1.89 (ddt, J=13.7, 10.6, 4 Hz, 1 H), 1.62 (dq, J=13.7, 4Hz, 1 H), 1.07 (d, J=7.3 Hz, 3H). 13C NMR (400 MHz, CDCI3): δ 157.9, 152.0, 151.0, 120.0, 103.0, 102.5, 56.3, 46.2, 42.4, 34.7, 33.4, 32.4, 14.3. KEY INT
Example 11 Preparation of 3-{(3R, 4R)-4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3- oxo-propionitrile….TOFACITINIB BASE
To a clean, dry, nitrogen-purged 1.0 L reactor were charged methyl-(4-methyl-piperidin-3-yI)-(7H- pyrroIo[2,3-d]pyrimidin-4-yl)-amine (32.0 g, 0.130 mol), toluene (160 ml), ethyl cyanoacetate (88.53 g, 0.783 mol) and triethyl amine (26.4 g, 0.261 mol). The reaction was heated to 1000C and held for 24 hours. The reaction was washed with water (160 ml). The organic layer concentrated to a volume of 10 ml and water (20 ml) was added. The residual toluene was removed by distillation and the mixture was cooled to room temperature. Acetone (224 ml) was added followed by citric acid (27.57 g, 0.144 mol) in water (76 ml). The resulting slurry was stirred for 7 hours. The solids were isolate by filtration, washed with acetone (96 ml), and dried under vacuum to afford 42.85 g (65.3%) of the title compound. Example 13 Preparation of 3-{(3R, 4R)~4-methyl-3-[methyl-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-amino]-piperidin-1-yl}-3-oxo- propionitrile citrate salt:…………..TOFACITINIB CITRATE To a clean, dry, nitrogen-purged 500 ml reactor were charged methyl-(4-methyl-piperidin-3-yl)-(7H- pyrrolo[2,3-d]pyrimidin-4-yl)-amine (25.0 g, 0.102 mol) and methylene chloride (250 ml). The mixture was stirred at room temperature for a minimum of 2.5 hours. To a clean, dry, nitrogen-purged 1 L reactor were charged cyanoacetic acid (18.2 g, 0.214 mol), methylene chloride (375 ml), and triethyl amine (30.1 ml, 0.214 mol). The mixture was cooled to -15.0— 5.00C over one hour and trimethylacetyl chloride (25.6 ml, 0.204 mol) was added at a rate to maintain the temperature below O0C. The reaction was held for a minimum of 2.5 hours, then the solution of the amine was added at a rate that maintained the temperature below O0C. After stirring for 1 hour, the mixture was warmed to room temperature and 1 M sodium hydroxide (125 ml) was added. The organic layer was washed with water (125 ml) The methylene chloride solution.was displaced with acetone until a volume of 500 ml and a temperature of 55-650C had been achieved. Water (75 ml) was charged to the mixture while maintaining the temperature at 55-65°C. A solution of citric acid (20.76 g, 0.107 mol) in water (25.0) was charged and the mixture was cooled to room temperature. The reactor was stirred for a minimum of 5 hours and then the resulting solids were isolated by filtration and washed with acetone (2×75 ml), which was sent to the filter. The salt was charged into a clean, dry, nitrogen-purged 1L reactor with 2B ethanol (190 ml) and water (190 ml). The slurry was heated to 75-850C for a minimum of 4 hours. The mixture was cooled to 20-300C and stirred for an additional 4 hours. The solids were isolated by filtration and washed with 2B ethanol (190 ml). After drying in a vacuum oven at 500C with a slight nitrogen bleed, 34.6 g (67.3%) of the title compound were isolated. 1H NMR (500 MHz, CZ6-DMSO): δ 8.14 (s, 1 H), 7.11 (d, J=3.6 Hz, 1 H), 6.57 (d, J=3.6 Hz, 1 H), 4.96 (q, J=6.0 Hz, 1 H), 4.00-3.90 (m, 2H), 3.80 (m, 2H), 3.51 (m, 1 H), 3.32 (s, 3H), 2.80 (Abq, J=15.6 Hz, 2H), 2.71 (Abq, J=15.6 Hz, 2H), 2.52-2.50 (m, 1 H), 2.45-2.41 (m, 1 H), 1.81 (m, 1 H), 1.69-1.65 (m, 1 H), 1.04 (d, J=6.9 Hz, 3H)
Phase II clinical trials tested the drug in rheumatoid arthritis patients that had not responded to DMARD therapy. In a tofacitinib monotherapy study, the ACR score improved by at least 20% (ACR-20) in 67% of patients versus 25% who received placebo; and a study that combined the drug with methotrexate achieved ACR-20 in 59% of patients versus 35% who received methotrexate alone. In a psoriasis study, the PASI score improved by at least 75% in between 25 and 67% of patients, depending on the dose, versus 2% in the placebo group.[8] The most important side effects in Phase II studies were increased blood cholesterol levels (12 to 25 mg/dl LDL and 8 to 10 mg/dl HDL at medium dosage levels) andneutropenia.[8] Phase III trials testing the drug in rheumatoid arthritis started in 2007 and are scheduled to run until January 2015.[9] In April 2011, four patients died after beginning clinical trials with tofacitinib. According to Pfizer, only one of the four deaths was related to tofacitinib.[10] By April 2011, three phase III trials for RA had reported positive results.[11] In November 2012, the U.S. FDA approved tofacitinib “to treat adults with moderately to severely active rheumatoid arthritis who have had an inadequate response to, or who are intolerant of, methotrexate.”[12]
Psoriasis
As of April 2011 a phase III trial for psoriasis is under way.[11]
Alopecia
In June 2014, scientists at Yale successfully treated a male patient afflicted with alopecia universalis. The patient was able to grow a full head of hair, eyebrows, eyelashes, facial, armpit, genitalia and other hair. No side effects were reported in the study.[13]
Ulcerative colitis
The OCTAVE study of Tofacitinib in Ulcerative Colitis started in 2012. It is currently enrolling patients, though the NIH trials page states that they expect the trial to close in June 2015.[14]
Vitiligo
In a June 2015 study, a 53-year-old woman with vitiligo showed noticeable improvement after taking tofacitinib for five months.[15]
Development of Safe, Robust, Environmentally Responsible Processes for New Chemical Entities
– Dr. V. Rajappa, Director & Head-Process R&D, Bristol-Myers Squibb, India
Kremer, J. M.; Bloom, B. J.; Breedveld, F. C.; Coombs, J. H.; Fletcher, M. P.; Gruben, D.; Krishnaswami, S.; Burgos-Vargas, R. N.; Wilkinson, B.; Zerbini, C. A. F.; Zwillich, S. H. (2009). “The safety and efficacy of a JAK inhibitor in patients with active rheumatoid arthritis: Results of a double-blind, placebo-controlled phase IIa trial of three dosage levels of CP-690,550 versus placebo”. Arthritis & Rheumatism60 (7): 1895–1905. doi:10.1002/art.24567. PMID19565475.edit
Ken Garber (9 January 2013). “Pfizer’s first-in-class JAK inhibitor pricey for rheumatoid arthritis market”. Nature Biotechnology31 (1): 3–4. doi:10.1038/nbt0113-3. PMID23302910.
Jump up^Moisan A, et al. White-to-brown metabolic conversion of human adipocytes by JAK inhibition. Nature Cell Biology, 8 December 2014. DOI 10.1038/ncb3075
Clinical trial number NCT00413699 for “Long-Term Effectiveness And Safety Of CP-690,550 For The Treatment Of Rheumatoid Arthritis” at ClinicalTrials.gov
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये।औकात बस इतनी देना,कि औरों का भला हो जाये।………..P.S. : The views expressed are my personal and in no-way suggest the views of the professional body or the company that I represent.
सुकून उतना ही देना प्रभू, जितने से जिंदगी चल जाये। औकात बस इतनी देना, कि औरों का भला हो जाये। DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed and bound to a wheel chair, Now I keep him as my source of inspiration and helping millions, thanks to millions of my readers who keep me going and help me to keep my son happy
सुकून उतना ही देना प्रभू, जितने से
जिंदगी चल जाये।
औकात बस इतनी देना,
कि औरों का भला हो जाये।
CAMBRIDGE, Mass.–Aug. 30, 2013–(BUSINESS WIRE)–Genzyme, a Sanofi company (EURONEXT: SAN and NYSE: SNY), announced today that the European Commission has granted marketing authorization for Aubagio® (teriflunomide) 14 mg, a once-daily, oral therapy indicated for the treatment of adult patients with relapsing remitting multiple sclerosis (RRMS).
Teriflunomide (trade name Aubagio, marketed by Sanofi, also known as A77 1726) is the active metabolite of leflunomide.[1]Teriflunomide was investigated in the Phase III clinical trial TEMSO as a medication for multiple sclerosis (MS). The study was completed in July 2010.[2] 2-year results were positive.[3] However, the subsequent TENERE head-to-head superiority trial reported that “although permanent discontinuations [of therapy] were substantially less common among MS patients who received teriflunomide compared with interferon beta-1a, relapses were more common with teriflunomide.”[4] The drug was approved by the FDA on September 13, 2012.[5]
Teriflunomide inhibits rapidly dividing cells, including activated T cells, which are thought to drive the disease process in MS. Teriflunomide may decrease the risk of infections compared to chemotherapy-like drugs because of its more-limited effects on the immune system.[7]
It has been found that teriflunomide blocks the transcription factor NF-κB. It also inhibits tyrosine kinase enzymes, but only in high doses not clinically used.[8]
Activation of leflunomide to teriflunomide
→↔
The structure which results from ring opening can interconvert between the E and Zenolic forms (and the corresponding keto-amide), with the Z enol being the most stable and therefore most predominant form.
Space filling model of the E isomer of teriflunomide
^ Magne D, Mézin F, Palmer G, Guerne PA (2006). “The active metabolite of leflunomide, A77 1726, increases proliferation of human synovial fibroblasts in presence of IL-1beta and TNF-alpha”. Inflamm. Res.55 (11): 469–75. doi:10.1007/s00011-006-5196-x. PMID17122964.
Teriflunomide is an immunosuppressant, acting as a tyrosine kinase inhibitor. It is also evaluated in the treatment of rheumatoid arthritis, autoimmune disease and multiple sclerosis. An oral film coated tablet containing teriflunomide as the active ingredient is marked in the United States by Sanofi Aventis US using brand AUBAGIO™. AUBAGIO is indicated for the treatment of patients with relapsing forms of multiple sclerosis.
U.S. Patent No. 5,679,709 appears to claim teriflunomide and its pharmaceutically acceptable salts, the same patent also further covers pharmaceutical composition and method of administering top a patients suffering from autoimmune disease.
U.S. Patent No. 5,494,91 I disclosesthe process for the preparation of teriflunomide by reacting 5-methylisoxazole-4-carbonyl chloride with trifluoromethyl aniline in the presence of acetonitrile to yield Leflunomide with on further hydrolysis with aqueous sodium hydroxide solution in methanol gives teriflunomide of formula I.
U.S. Patent No. 5,990,141 discloses the process for the preparation of teriflunomide by reacting 4-trifluoromethyl aniline with cyano acetic acid ethyl ester to yield cyanoaceto-(4-trifluromethyl)-aniline, with on further reacted with acetyl chloride in the presence of sodium hydride base and THF and acetonitrile solvent to give teriflunomide of formula I.
U.S. patent No. 6,365,626 discloses the process for the preparation of teriflunomide by reacting 4-trifluromethylaniline with cyanoacetic acid to give cyanoacet-(4- trifluoromethyl)anilide which on further reacted with acetyl chloride in the presence of sodium hydride to give teriflunomide of formula I.
U.S. Patent No. 6,894,184 discloses the process for the preparation of teriflunomide involves reacting 4-trifluromethylaniline with cyanoacetic acid to give cyanoacet-(4- trifluoromethyl)anilide which on further reacted with acetic anhydride in the presence of base to give teriflunomide of formula I.
International PCT application No. WO 2009/147624 discloses the process for the preparation of teriflunomide involves condensation of ethyl-2-cyano-3-hydroxybut-2-enoate and 4-(trifluoromethyl) aniline in presence of xylene solvent at reflux temperatures for 16 hours to give teriflunomide of formula I.
preparation of teriflunomide (I) comprises steps of;
1 ) condensation of cyanoacetic acid of formula (II) with 4-trifluoromethyl aniline of formula (III) in the presence of chlorinating agent to give 2-cyano-N-[4-(trifluromethyl)phenyl]acetamide of formula (IV);
(II I) (IV)
2) acetylation of 2-cyano-N-[4-(trifluromethyl)phenyl] acetamide of
formula (IV) with an acetylating agent in the presence of base and suitable solvents to yield teriflunomide of formula (I).
EXAMPLE 1 : Preparation of 2-cvano-N-f4-(trifluoromethyl> phenyl! acetamide (IV)
A round bottom flask is charged with cyanoacetic acid (100 g) and phosphorous pentachloride and tetrahydrofuran (300 ml) and the reaction mixture is stirred at room temperature for 4 hours. 4-trifluoromethyl aniline (161 g) dissolved in tetrahydrofuran (100 ml) is slowly added to the reaction mixture and stirred for completion of reaction. The resultant reaction mass is cooled and separated solid is filtered and washed with slurry of Isoproapnol and cyclohexane and dried under reduced pressure to afford the title compound. Weight: 196 gm.
Purity by HPLC: 98%
EXAMPLE 2: preparation of 2-cyano-3-hvdroxy-N-f4-( trifluoromethyl) phenyl] but-2-enamide (Teriflunomide crude)
A round bottom flask is charged with 2-cyano-N-[4-{trifluromethyl} phenyl] acetamide (100g), sodium hydroxide (70 gm) and dimethyl formamide is added and the reaction mixture is stirred for 30 minutes. Isopropenyl acetate (60 ml) is added slowly and the resultant mixture is stirred for about 4-5 hours at room temperature. After completion of the reaction, the resulting reaction mixture is diluted with water and acidified with Cone. HCI solution and stirred for solid separation. The separated solid is filtered and washed with water and dried under reduced pressure to afford Teriflunomide.
The obtained teriflunomide is charged in round bottom flask and aqueous solution of sodium hydroxide solution (29.6 g in 300 ml water) is added slowly at 25-35°C and stirred for 1 to 2 hours. The mixture is brought to 5 to 10°C and dichloromethane is added, the mixture is stirred for 15 minutes. The organic and the aqueous layer are separated, and the resultant aqueous layer is acidified with aq. Hcl and stirred. The separated solid is filtered and washed with water and dried under vacuum at 65-70°C for 10-12 hours to afford teriflunomide.
Weight: 101 gm
Purity by HPLC: 95%
EXAMPLE 3; Purification of Teriflunomide:
Teriflunomide (5 g) is charged into a flask followed by addition of acetonitrile (125 ml) and heated to reflux and stirred for 2 hours. The resultant reaction solution is filtered through highflow bed to obtain a clear solution and cooled to room temperature and stirred for solid separation. The separated solid is filtered, washed with Isopropanol (50 ml) and dried under vacuum to afford pure teriflunomide.
Weight: 3.8 gm
Purity by HPLC: 99.7%
…………………………………………………………………………………………………………
EP 0527736; JP 1993506425; JP 1999322700; JP 1999343285; US 5494911; US 5532259; WO 9117748
5-Methylisoxazole-4-carboxylic acid (I) was converted to the corresponding acid chloride (II) upon refluxing with SOCl2. Coupling of acid chloride (II) with 4-(trifluoromethyl)aniline (III) produced anilide (IV). Finally, isoxazole ring opening in the presence of NaOH gave rise to the title cyano amide.
Magne D, Mézin F, Palmer G, Guerne PA (2006). “The active metabolite of leflunomide, A77 1726, increases proliferation of human synovial fibroblasts in presence of IL-1beta and TNF-alpha”. Inflamm. Res.55 (11): 469–75. doi:10.1007/s00011-006-5196-x. PMID17122964.
Teriflunomide, a dihydroorotate dehydrogenase (DHODH) inhibitor, is the active metabolite of leflunomide a synthetic, low-molecular-weight drug currently used in the treatment of rheumatoid arthritis. The mechanisms by which teriflunomide exerts its antiinflammatory, antiproliferative and immunosuppressive effects are not yet completely understood, although inhibition of pyrimidine biosynthesis (via suppression of DHODH) and interference with tyrosine kinase activity both appear to be involved. Based on its efficacy shown in animal models of experimental allergic encephalomyelitis, teriflunomide was tested in a phase II study in patients with multiple sclerosis with relapses. Recruitment is ongoing for a phase III study to determine the efficacy of teriflunomide in reducing the frequency of relapses and accumulation of disability in multiple sclerosis patients.
The chemical name of Teriflunomide is 2-cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide and formula is C12H9F3N2O2 and molecular weight is 270.207.
Teriflunomide is used as Immunosupressant. It acts as tyrosine kinase inhibitor. It is used in treatment of rheumatoid arthritis, autoimmune disease and multiple sclerosis.
Teriflunomide was first disclosed and claimed in U.S. Pat. No. 5,679,709 but this patent does not mention any process of preparation for salt formation.
U.S. Pat. No. 5,494,911, U.S. Pat. No. 5,990,141 disclose various processes for preparing Teriflunomide. These patents do not disclose process for preparation Teriflunomide salts or mention any its polymorphic form.
EP 2280938 A2
HISTORY OF SYNTHESIS
The chemical name of Teriflunomide is
2-cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide and formula is Ci2H9 F3N2O2 and molecular weight is 270.207.
Teriflunomide is used as Immunosupressant. It acts as tyrosine kinase inhibitor. It is used in treatment of rheumatoid arthritis, autoimmune disease and multiple sclerosis.
Teriflunomide was first disclosed and claimed in US patent no. 5,679,709 but this application does not mention the process of preparation.
US patent no. 5,494,911 discloses a process for preparation of Teriflunomide as shown in given below
4-trifluoromethylaniline (IV) in acetonitrile to give leflunomide (VI). The subsequent hydrolysis with aqueous sodium hydroxide solution in methanol gives Teriflunomide (I). US patent 5,990,141 discloses a process for preparation of Teriflunomide as shown in given below
Teriflunomide (I)
The process involves reacting 4-trifluorometyl aniline (IV) with cyanoacetic acid ethyl ester (II) to give cyanoacet-(4-trifluoromethyl)-anilide (VII). This compound is further reacted first with sodium hydride in acetonitrile and then with acetylchloride in THF to give Teriflunomide (I).
US patent no. 6,365,626 discloses a process for preparation of Teriflunomide which is as given in below
Teriflunomide is used as Immunosupressant. It acts as tyrosine kinase inhibitor. It is used in treatment of rheumatoid arthritis, autoimmune disease and multiple sclerosis.
Teriflunomide was first disclosed and claimed in US patent no. 5,679,709 but this application does not mention the process of preparation.
[H] US patent no. 5,494,911 discloses a process for preparation of Teriflunomide in Example-4 as shown in given below scheme-I
(V) (IV) (VI) (D
Scheme-I
The proces; 5 involves re acting 5-metlr
4-trifluoromethylaniline (IV) in acetonitrile to give leflunomide (VI). The subsequent hydrolysis with aqueous sodium hydroxide solution in methanol gives Teriflunomide (I). US patent 5,990,141 discloses a process for preparation of Teriflunomide as shown in given below scheme-II.
Teriflunomide (I)
Scheme-II The process involves reacting 4-trifluorometyl aniline (IV) with cyanoacetic acid ethyl ester (II) to give cyanoacet-(4-trifluoromethyl)-anilide (VII). This compound is further reacted first with sodium hydride in acetonitrile and then with acetylchloride in THF to give Teriflunomide (I).
US patent no. 6,365,626 discloses a process for preparation of Teriflunomide in Fig. 19 which is as given in below scheme-Ill.
Teriflunomide
(I)
Scheme-Ill The process involves reacting 4-trifluoromethyl aniline (IV) with cyanoacetic acid (Ha) to give compound of formula (VII). This compound is further reacted first with sodium hydride and then with acetylchloride to give Teriflunomide (I)
………………………….
Example-1 Preparation of Ethyl-2-cyano-3-hydroxy-but-2-enoate (III) [77] Potassium carbonate (73.3 g) was added to the well stirred solution of Ethylcy- anoacetate (50 g) in Dimethylformamide (250 ml) and stirred for 15 minute at ambient temperature. Acetic anhydride (90.25 g) was added drop wise to the above well stirred solution during 2 to 3 hours at ambient temperature. Reaction mixture was stirred at ambient temperature for 15 to 20 hours. Reaction mixture was diluted with water (500 ml) and extracted with dichloromethane (3 xlOO ml). Combined organic layer was washed with saturated sodium carbonate solution (3x100ml). Aqueous carbonate layer was separated and acidified with 50% HCl solution and extracted with dichloromethane (3x100ml). Combined organic layer was washed with brine solution (100 ml), dried over sodium sulfate and evaporated to yield Ethyl 2-cyano-3-hydroxy-but-2-enoate (58 g).
Yield: 84.6%Example-2 ] Preparation of Teriflunomide (I) [82] Ethyl 2-cyano-3-hydroxybut-2-enoate (III) (50 g) and 4-(trifluoromethyl) aniline (51.9 g) in xylene (1000 ml) was refluxed for 48 hours. The reaction mixture was allowed to cool at room temperature. Separated solid was filtered and washed with xylene (2×100 ml). Solid was dried under vacuum at 700C to yield (62 g) of Teri- flunomide.
Currently, for the preparation of teriflunomide mainly in the following three categories:
The first synthetic methods: mainly 5-methyl-isoxazole-4-carboxylic acid starting materials or by Synthesis of 5-methyl-isoxazole-4-carboxylic acid intermediate, then reacted with 4- trifluoromethyl base – aniline was synthesized teriflunomide, specific synthetic route is as follows:
[0007]
The general reaction step above normal class methods, not easy to intermediate purification, total yield is low, and the synthesis process using a large number of chloride corrosion of equipment can easily produce large amounts of acid mist and acidic water, thus polluting the environment .
The second class of methods: 2-cyano-acetic acid derivatives and 4-trifluoromethyl aniline. Such methods will be first prepared as a 2-cyano acetic acid chloride, and then 4-trifluoromethyl-aniline to give the corresponding amide, and then acetyl chloride for
With, the condensation reaction between the molecules to give the desired product, the synthesis route is as follows:
This class methods used in the reaction process large amounts of chloride reagent for large equipment and environmental damage.
The third method: This method is quite similar to the second type of method, mainly in the 2-cyano-acetic acid derivatives and 4-trifluoromethyl-aniline; The method of the second type is different, In the last step with 1-methyl-2-chloro-propylene oxide as raw materials to build α, β-unsaturated nitrile of the enol structure, i.e., to give the desired product, the synthesis route is as follows:
Teriflunomide Preparation Example 18 [0185] Implementation
Example 17 was obtained as a pale yellow solid of 61.2g crude compound was used directly in the synthesis of teriflunomide. In a 2L round bottom flask was added compound 27.2g (0.32mol) having the structure shown in formula IV, dry dioxane (620mL), sodium hydride 4g (0.16mol, in g / mL count, mass volume ratio 60% saving in kerosene), calcium hydride
6.7g (0.16mol), 15 ° C was stirred for I h, then slowly added dropwise in Example 17 was obtained as a pale yellow solid compound 61.2g (0.32mol) embodiment of dioxane 200mL, approximately I hour addition was complete, After the addition was complete the reaction was heated to reflux, the reaction at 80 ° C for 24 hours, the reaction process using a nitrogen blanket. After completion of the reaction was added 500mL of ice water to quench the reaction, with 2mol / L of HCl (aq.) And the reaction solution was adjusted to neutral pH, and extracted with EtOAc three times each in an amount of 500mL, and the combined organic phase was washed with saturated aqueous NaCl solution 800mL, dried over anhydrous Na2SO4, concentrated under reduced pressure, the mixed solution was twice recrystallized from methanol i_PrOH, the volume ratio of 1-PrOH and methanol is 2: 1, by volume of each recrystallized with a mixed solution of methanol with i_PrOH for 600mL, the crystallization temperature of 10 ° C, to give 58.8g of white solid compound in a yield of 66%, the total yield of 54% ο
using mass spectrometry, nuclear magnetic resonance spectroscopy and NMR spectra of the resulting white solid carbon compound structures were identified. MS data [M-H +] = 269.1, H NMR data = 1H-NMR (DMSO-Cie) δ the white solid compound: 10.88 (s, 1Η), 10.07 (br, s, 1H), 7.79 ( d, 2H), 7.66 (d, 2H), 2.26 (s, 3H), carbon NMR spectral data for: 13C-NMR (DMS0-d6) δ: 23.5,80.2,119.1,119.9,120.3,122.4,122.0, 123.5,125.3,126.2,141.8,166.2,186.0. Structural analysis by a white solid compound obtained in the present embodiment example for teriflunomide. Cases detected by HPLC obtained teriflunomide the embodiment of purity, calculated based on the peak area normalization method available, the present embodiment obtained teriflunomide a purity of 99.9%.
Pure Teriflunomide ………………………………………….Crude Teriflunomide
xamples
Example- 1: Preparation of N-(4′-trifluoromethylphenyl)-5-methylisoxazole-4-carboxamide (Formula-2)
Methylene chloride (125 ml) and dimethyl formamide (2.87 gms) were added to 5-methylisoxazole-4-carboxylic acid (25 gms) at 25-30°C. Heated the reaction mixture to 35-40°C and thionyl chloride (47.59 gms) was slowly added and stirred for 4 hours at the same temperature. After completion of the reaction, distilled off the solvent completely from the reaction mixture. To the obtained compound, dichloromethane was added at 25-30°C. Distilled off the solvent completely from the reaction mixture. Acetonitrile (50 ml) was added to the obtained compound at 25-30°C and slowly added to a mixture of acetonitrile (300 ml) and 4-(trifluoromethyl)aniline (64.45 gms) at 25-30°C and stirred the reaction mixture for 5 hours at the same temperature. Filtered the reaction mixture and distilled off the solvent completely from the filtrate. Methanol (225 ml), followed by activated carbon (2.5 gms) were added to the obtained compound at 25-30°C and stirred for 30 minutes at the same temperature. Filtered the reaction mixture through hyflow bed and washed with methanol. Water (250 ml) was slowly added to the obtained filtrate at 25-30°C and stirred the reaction mixture for 2 hours. Filtered the precipitated solid, washed with water and dried to get the title compound. Yield: 39.8 gms; Melting point: 165-168°C. Purity by HPLC: 99.63%.
Example-2: Preparation of N-(4′-trifluoromethylphenyl)-5-methylisoxazoIe-4-carboxamide (FormuIa-2)
Methylene chloride (15 Its) and dimethyl formamide (40 ml) were added to 5-methylisoxazole-4-carboxylic acid (3 kgs) at 25-30°C. Thionyl chloride (5.70 kgs) was slowly added to the reaction mixture at 25-30°C. Heated the reaction mixture to 40-45°C and stirred for 4 hours at the same temperature. After completion of the reaction, distilled off the solvent completely from the reaction mixture. Cooled the reaction mixture to 25-30°C and dichloromethane was added at the same temperature. Distilled off the solvent completely from the reaction mixture. Cooled the reaction mixture to 25-30°C and dissolved the obtained compound in acetonitrile (6.0 Its) at the same temperature. Slowly added to a mixture of acetonitrile (36 Its) and 4-(trifluoromethyl)aniline (7.70 kgs) at 25-30°C and stirred the reaction mixture for 5 hours at the same temperature. After completion of the reaction, filtered the reaction mixture and distilled off the solvent completely from the filtrate. Methanol (27 Its), followed by activated carbon (30 gms) was added to obtained compound at 25-30°C and stirred for 30 minutes at the same temperature. Filtered the reaction mixture through hyflow bed and washed with methanol. Water (30 Its) was slowly added to the obtained filtrate at 25-30°C and stirred the reaction mixture for 2 hours. Filtered the precipitated solid, washed with water. To the obtained wet compound, toluene (9 Its) was added at 25-30°C. Heated the reaction mixture to 55-60°C and stirred for 30 minutes at the same temperature. Cooled the reaction mixture to 25-30°C and stirred for 3 hours at the same temperature. Filtered the solid, washed with toluene and dried to get the title compound. Yield: 4.7 kg.
Example-3: Preparation of (Z)-2-cyano-3-hydroxy-but-2-enoic acid-(4-trifluoromethyl phenyl)-amide (Formula-l)
Methanol (150 ml) was added to N-(4′-trifluoromethylphenyl)-5-methylisoxazole-4-carboxamide (50 gms) at 25-30°C. Cooled the reaction mixture to 0-5°C and aqueous sodium hydroxide solution was slowly added to the reaction mixture at the same temperature. Stirred the reaction mixture for 2 hours at 0-5°C. Water was added to the reaction mixture. Adjust the pH of the reaction mixture to 7.5 by using dilute hydrochloric acid at 25-30°C. Filtered the precipitated solid, washed with water and dried to get the title compound. Yield: 46.0 gms;
Example-4: Preparation of crystalline form-M of (Z)-2-cyano-3-hydroxy-but-2-enoic acid-(4-trifluoromethyl phenyl)-amide (Formula-1)
Dimethylformamide (300 ml) was added to (Z)-2-cyano-3-hydroxy-but-2-enoic acid-(4-trifluoromethylphenyl)-amide (50 gms) at 25-30°C. Heated the reaction mixture to 55-60°C and stirred for 30 minutes at the same temperature. Filtered the reaction mixture and washed with dimethyl formamide. To the obtained filtrate, methanol (350 ml) was added at 25-30°C. Cooled the reaction mixture to 10-15°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with chilled methanol and dried to get the title compound. Yield: 41 gms;
Melting point: 228-231°C; Water content: 0.06% w/w; Phenyl isoxazole impurity: 0.004%; Purity by HPLC: 99.97%.
Particle size distribution before micronisation: D10: 6.71 μιτι; D50: 34.4 μπι; D90: 109.8 μηι; Particle size distribution after micronisation: DIO: 1.35 μητ, D50: 4.52 μητ, D90: 10.26 μιη.
The P-XRD of the obtained compound is shown in figure- 1.
The DSC thermogram of the obtained compound is shown in figure-2.
Reference Example- 1: Preparation of (Z)-2-cyano-3-hydroxy-but-2-enoicacid-(4-trifluoromethylphenyl)-amide according to US5494911 (Formula-1)
Methanol (74 ml) was added to N-(4′-trifluoromethylphenyl)-5-methylisoxazole-4-carboxamide (20 gms) at 25-30°C. Cooled the reaction mixture to 0-5°C and aqueous sodium hydroxide solution {prepared by dissolving sodium hydroxide (3.26 gms) in water (74 ml)} was slowly added to the reaction mixture at the same temperature. Stirred the reaction mixture for 1 hour at 0-5°C. After completion of the reaction, 20% aqueous hydrochloric acid solution was added to the reaction mixture at 25-30°C and stirred for 2 hours at the same temperature. Filtered the precipitated solid, washed with water and dried to get the title compound. Yield: 8.7 gms.
The P-XRD pattern of the obtained compound is shown in figure-3.
The DSC thermogram of the obtained compound is shown in figure-4.
Example-1 Preparation of Ethyl-2-cyano-3-hydroxy-but-2-enoate (III) [77] Potassium carbonate (73.3 g) was added to the well stirred solution of Ethylcy- anoacetate (50 g) in Dimethylformamide (250 ml) and stirred for 15 minute at ambient temperature. Acetic anhydride (90.25 g) was added drop wise to the above well stirred solution during 2 to 3 hours at ambient temperature. Reaction mixture was stirred at ambient temperature for 15 to 20 hours. Reaction mixture was diluted with water (500 ml) and extracted with dichloromethane (3 xlOO ml). Combined organic layer was washed with saturated sodium carbonate solution (3x100ml). Aqueous carbonate layer was separated and acidified with 50% HCl solution and extracted with dichloromethane (3x100ml). Combined organic layer was washed with brine solution (100 ml), dried over sodium sulfate and evaporated to yield Ethyl 2-cyano-3-hydroxy-but-2-enoate (58 g).
Yield: 84.6% Example-2 Preparation of Teriflunomide (I) [82] Ethyl 2-cyano-3-hydroxybut-2-enoate (III) (50 g) and 4-(trifluoromethyl) aniline (51.9 g) in xylene (1000 ml) was refluxed for 48 hours. The reaction mixture was allowed to cool at room temperature. Separated solid was filtered and washed with xylene (2×100 ml). Solid was dried under vacuum at 700C to yield (62 g) of Teri- flunomide.
N-(4′-trifluoromethylphenyI)-5-methylisoxazole-4-carboxamide of formula-2:
Apparatus: A liquid chromatographic system equipped with variable wavelength UV- detector; Column: Cosmicsil APT CI 8, 100 x 4.6 mm, 3 μιη (or) equivalent; Flow rate: 1.5 ml/min; Wavelength: 210 nm; Column Temperature: 25°C; Injection volume: 20 μί; Run time: 40 min; Diluent: Mobile phase; Needle wash: Tetrahydrofuran; Elution: Isocratic; Mobile phase: 5 ml of triethyl amine into a 650 ml of water. Adjusted the pH to 3.4 with dil. Orthophosphoric acid and filter this solution through 0.22 μπι nylon membrane filter paper and sonicate to degas it. (Z)-2-cyano-3-hydroxy-but-2-enoicacid-(4-trifluoromethyl phenyl)-amide compound of formula- 1:
Apparatus: A liquid chromatographic system equipped with variable wavelength UV- detector; Column: Kromasil 100 C18, 250 x 4.6 mm, 5 μηι (or) equivalent; Flow rate: 1.0 ml/min; Wavelength: 250 nm; Column Temperature: 35°C; Injection volume: 5 μί; Run time: 37 min; Diluent: 0.01 M dipotassium hydrogen orthophosphate in 1000 ml of water; Elution: Gradient; Mobile phase-A: Buffer (100%); Mobile phase-B: Acetonitrile : Buffer (70:30 v/v); Buffer: 1 ml of ortho phosphoric acid into a 1000 ml of water and 3.0 grams of 1 -octane sulfonic acid sodium salt anhydrous. Adjust pH to 6.0 with potassium hydroxide solution and filtered through 0.22μηι Nylon membrane filter paper and sonicate to degas it……..http://www.google.com/patents/WO2015029063A2?cl=en

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....











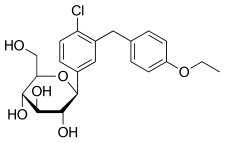

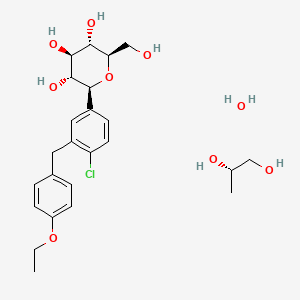











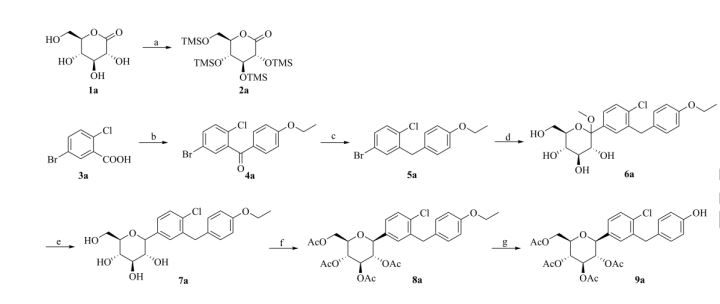












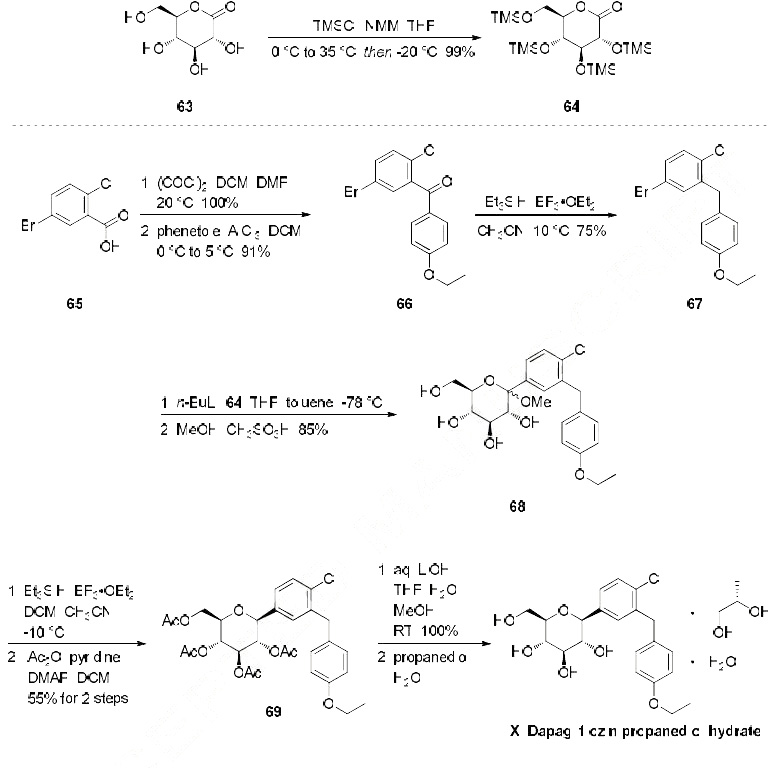
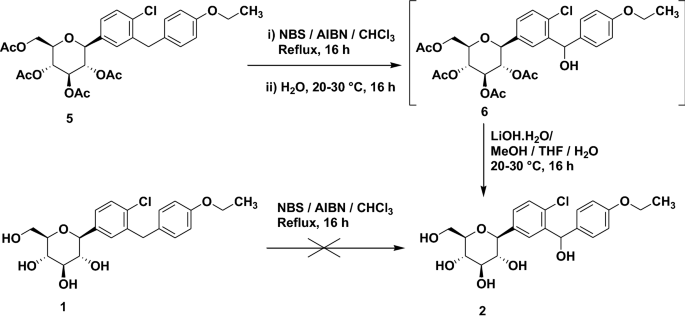
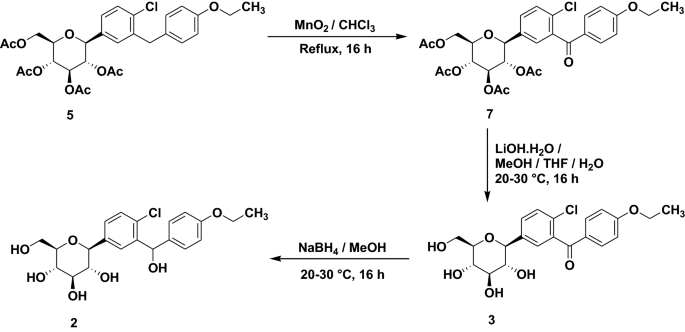











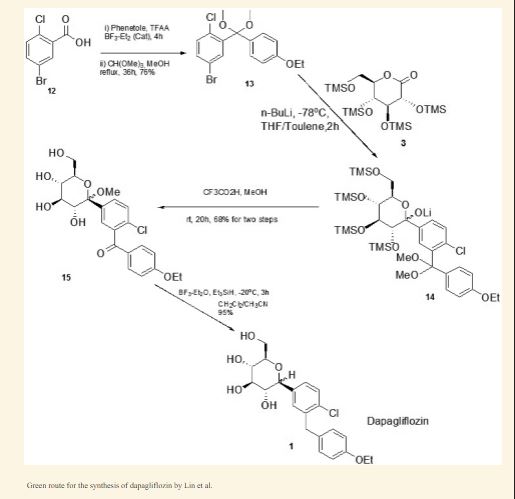

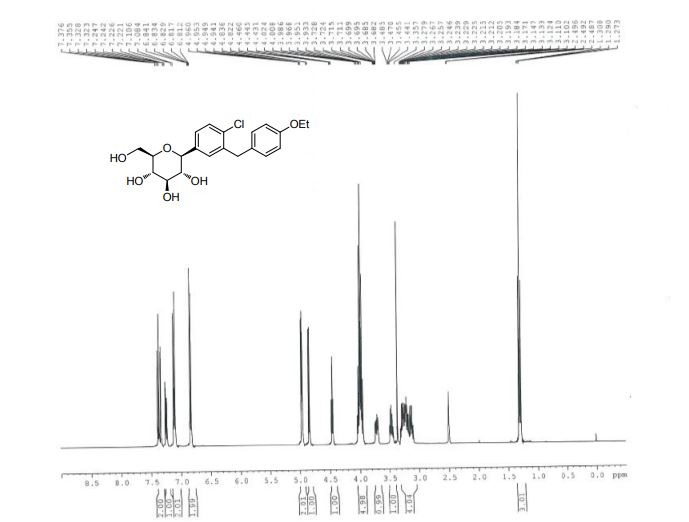
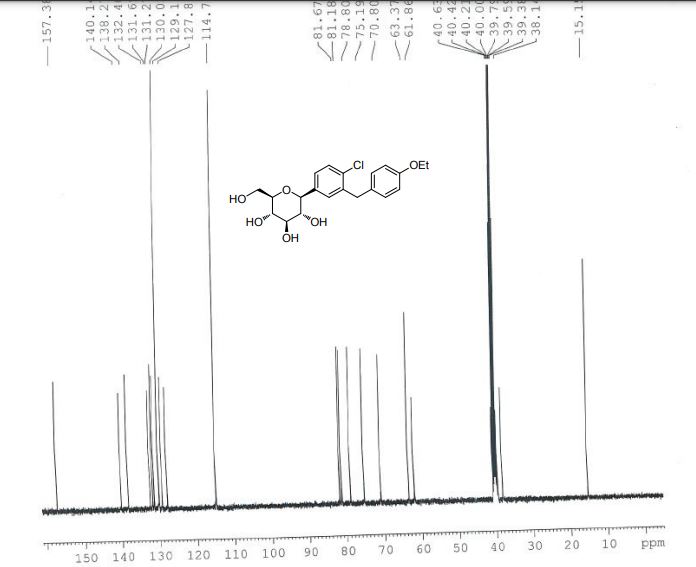
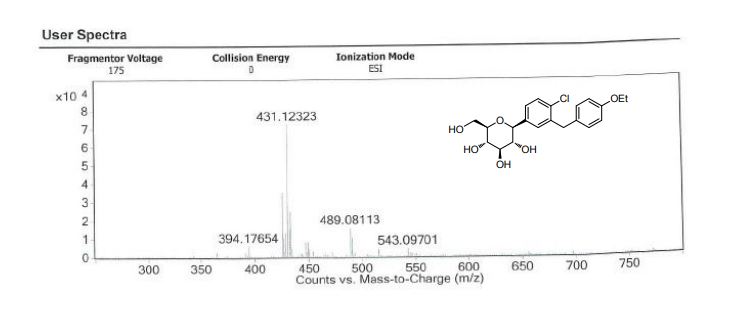
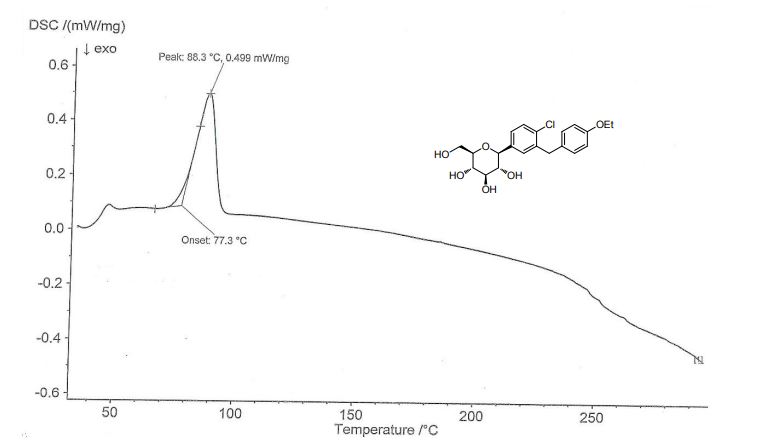




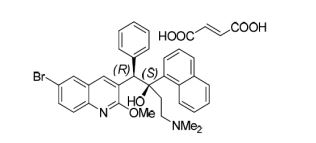









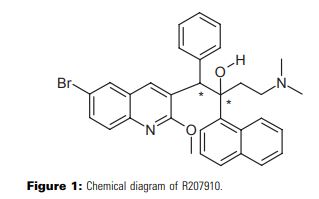
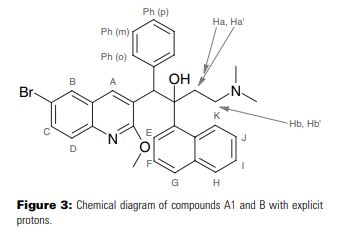















































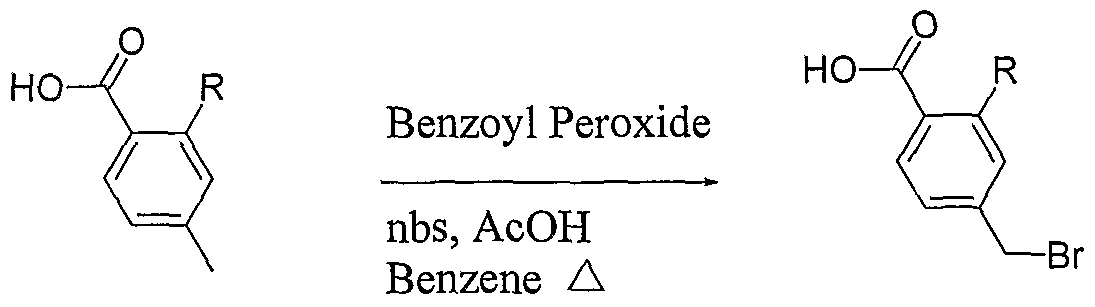























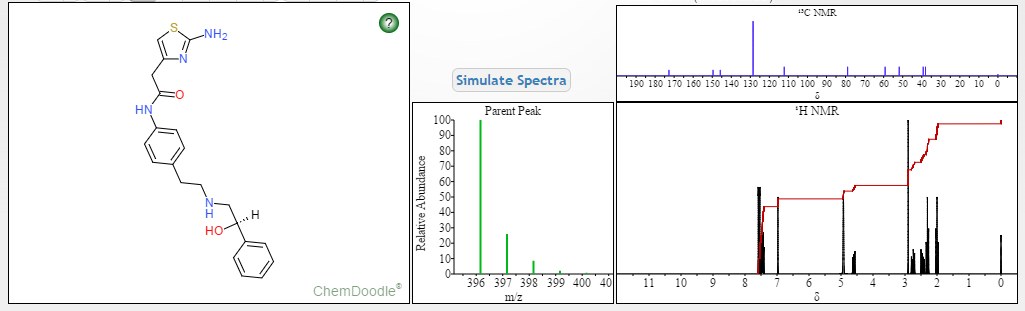
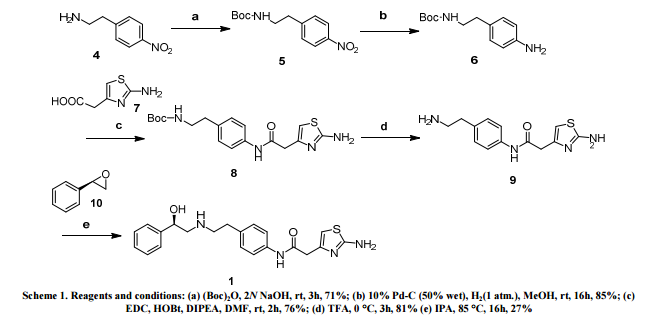
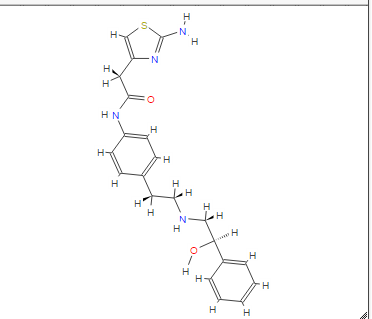
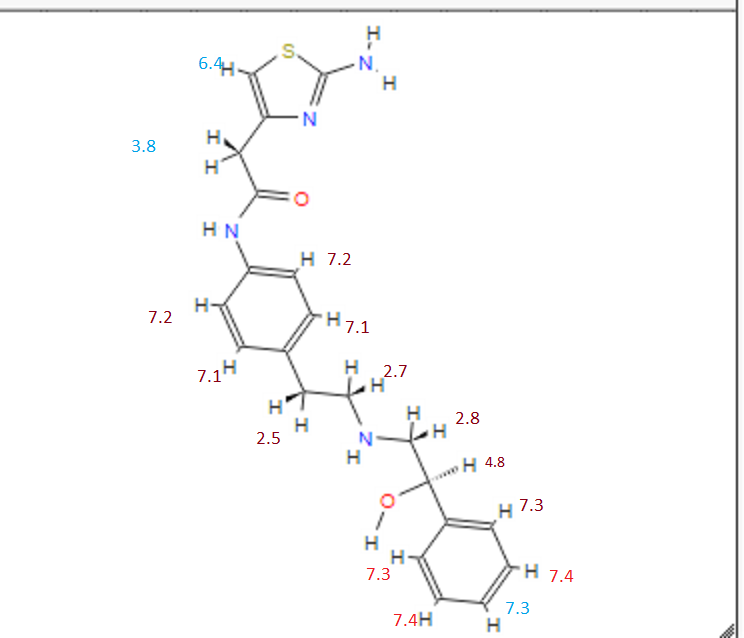
 CN 103896872
CN 103896872






























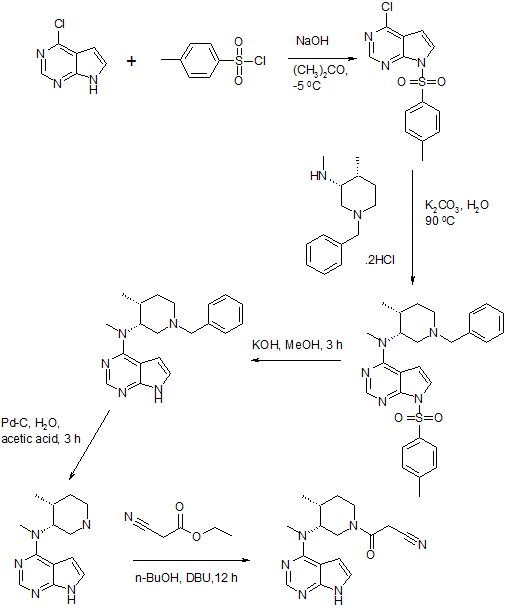









 ……………..
…………….. 

 The presentation will load below
The presentation will load below
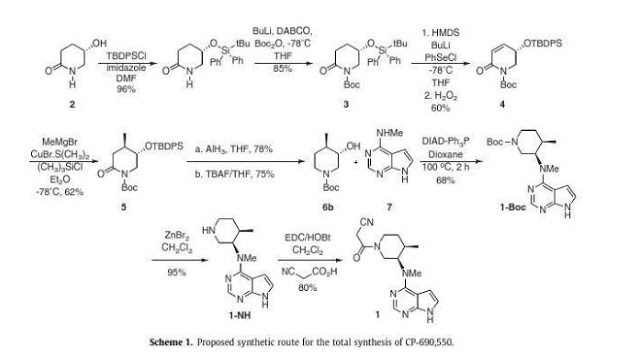







 LIONEL MY SON
LIONEL MY SON































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}