
Home » EU 2016
Category Archives: EU 2016
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Selexipag, セレキシパグ ,селексипаг , سيليكسيباق ,

Selexipag
- Molecular FormulaC26H32N4O4S
- Average mass496.622 Da
Selexipag, Uptravi
475086-01-2 CAS
(C26H32N4O4S, Mr = 496.6 g/mol)
A prostacyclin receptor (PGI2) agonist used to treat pulmonary arterial hypertension (PAH).
NIPPON SHINYAKU….INNOVATOR
セレキシパグ
Selexipag (brand name Uptravi) is a drug developed by Actelion for the treatment of pulmonary arterial hypertension (PAH). Selexipag and its active metabolite, ACT-333679 (MRE-269) (the free carboxylic acid), are agonists of the prostacyclin receptor, which leads to vasodilation in the pulmonary circulation.[1]
FDA approves new orphan drug to treat pulmonary arterial hypertension
December 22, 2015
On December 21, the U.S. Food and Drug Administration approved Uptravi (selexipag) tablets to treat adults with pulmonary arterial hypertension (PAH), a chronic, progressive, and debilitating rare lung disease that can lead to death or the need for transplantation.
“Uptravi offers an additional treatment option for patients with pulmonary arterial hypertension,” said Ellis Unger, M.D., director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research. “The FDA supports continued efforts to provide new treatment options for rare diseases.”
PAH is high blood pressure that occurs in the arteries that connect the heart to the lungs. It causes the right side of the heart to work harder than normal, which can lead to limitations on exercise ability and shortness of breath, among other more serious complications.
Uptravi belongs to a class of drugs called oral IP prostacyclin receptor agonists. The drug acts by relaxing muscles in the walls of blood vessels to dilate (open) blood vessels and decrease the elevated pressure in the vessels supplying blood to the lungs.
Uptravi’s safety and efficacy were established in a long-term clinical trial of 1,156 participants with PAH. Uptravi was shown to be effective in reducing hospitalization for PAH and reducing the risks of disease progression compared to placebo. Participants were exposed to Uptravi in this trial for a median duration of 1.4 years.
Common side effects observed in those treated with Uptravi in the trial include headache, diarrhea, jaw pain, nausea, muscle pain (myalgia), vomiting, pain in an extremity, and flushing.
Uptravi was granted orphan drug designation. Orphan drug designation provides incentives such as tax credits, user fee waivers, and eligibility for exclusivity to assist and encourage the development of drugs for rare diseases.
Uptravi is marketed by San Francisco-based Actelion Pharmaceuticals US, Inc.
The US FDA granted it Orphan Drug status[2] (for PAH). It was approved by the U.S. FDA on 22 December 2015.[2]
In 2016, the EMA granted marketing authorization in the E.U. for this indication and launch took place shortly after in Germany and the United Kingdom. In Japan, Nippon Shinyaku received approval for the treatment of PAH in 2016.
Selexipag was approved by the U.S. Food and Drug Administration (FDA) on Dec 21, 2015, approved by European Medicine Agency (EMA) on May 12, 2016. It was originally developed by Nippon Shinyaku and then it was licensed to Actelion for co-development. It is marketed as Uptravi® by Actelion in US and EU.
Selexipag is a prostacyclin receptor (PGI2) agonist, which leads to vasodilation in the pulmonary circulation. It is indicated for the treatment of pulmonary arterial hypertension (PAH).
Uptravi® is available as tablets for oral use, containing 200, 400, 600, 800, 1000, 1200, 1400, or 1600 mcg of selexipag. The initial dose is 200 mcg twice daily, and increase the dose by 200 mcg twice daily at weekly intervals to the highest tolerated dose up to 1600 mcg twice daily.

ACT-333679 or MRE-269, the active metabolite of selexipag
SYNTHESIS DEPICT



PATENT
US2012/101276
http://www.google.st/patents/US20120101276?hl=pt-PT&cl=en
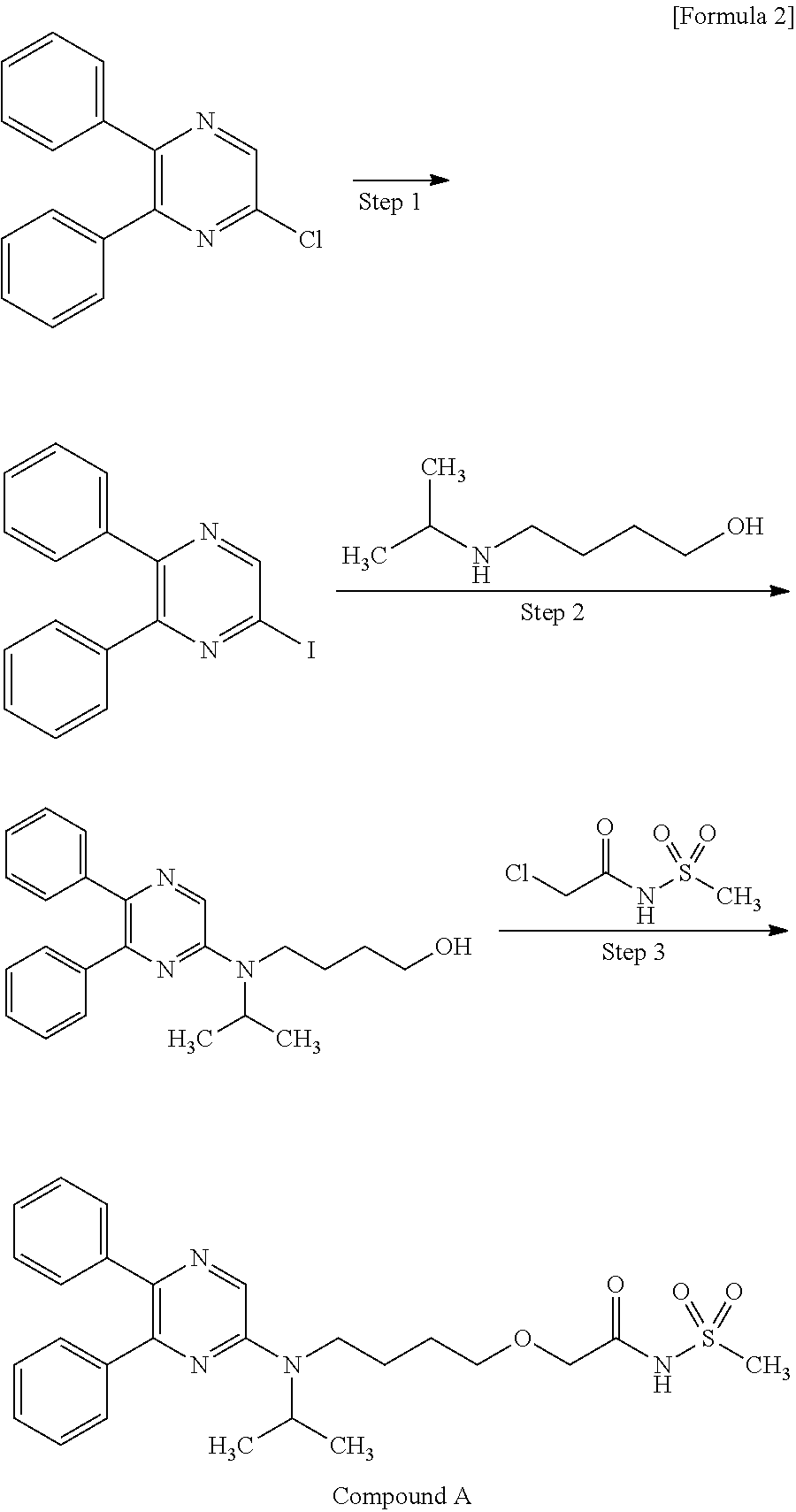
The present invention relates to a crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide (hereinafter referred to as “compound A”).

BACKGROUND OF THE INVENTION
Compound A has an excellent PGI2 agonistic effect and shows a platelet aggregation inhibitory effect, a vasodilative effect, a bronchodilative effect, a lipid deposition inhibitory effect, a leukocyte activation inhibitory effect, etc. (see, for example, in WO 2002/088084 (“WO ‘084”)).
Specifically, compound A is useful as preventive or therapeutic agents for transient ischemic attack (TIA), diabetic neuropathy, diabetic gangrene, peripheral circulatory disturbance (e.g., chronic arterial occlusion, intermittent claudication, peripheral embolism, vibration syndrome, Raynaud’s disease), connective tissue disease (e.g., systemic lupus erythematosus, scleroderma, mixed connective tissue disease, vasculitic syndrome), reocclusion/restenosis after percutaneous transluminal coronary angioplasty (PTCA), arteriosclerosis, thrombosis (e.g., acute-phase cerebral thrombosis, pulmonary embolism), hypertension, pulmonary hypertension, ischemic disorder (e.g., cerebral infarction, myocardial infarction), angina (e.g., stable angina, unstable angina), glomerulonephritis, diabetic nephropathy, chronic renal failure, allergy, bronchial asthma, ulcer, pressure ulcer (bedsore), restenosis after coronary intervention such as atherectomy and stent implantation, thrombocytopenia by dialysis, the diseases in which fibrosis of organs or tissues is involved [e.g., Renal diseases (e.g., tuburointerstitial nephritis), respiratory diseases (e.g., interstitial pneumonia (pulmonary fibrosis), chronic obstructive pulmonary disease), digestive diseases (e.g., hepatocirrhosis, viral hepatitis, chronic pancreatitis and scirrhous stomachic cancer), cardiovascular diseases (e.g, myocardial fibrosis), bone and articular diseases (e.g, bone marrow fibrosis and rheumatoid arthritis), skin diseases (e.g, cicatrix after operation, scalded cicatrix, keloid, and hypertrophic cicatrix), obstetric diseases (e.g., hysteromyoma), urinary diseases (e.g., prostatic hypertrophy), other diseases (e.g., Alzheimer’s disease, sclerosing peritonitis; type I diabetes and organ adhesion after operation)], erectile dysfunction (e.g., diabetic erectile dysfunction, psychogenic erectile dysfunction, psychotic erectile dysfunction, erectile dysfunction associated with chronic renal failure, erectile dysfunction after intrapelvic operation for removing prostata, and vascular erectile dysfunction associated with aging and arteriosclerosis), inflammatory bowel disease (e.g., ulcerative colitis, Crohn’s disease, intestinal tuberculosis, ischemic colitis and intestinal ulcer associated with Behcet disease), gastritis, gastric ulcer, ischemic ophthalmopathy (e.g., retinal artery occlusion, retinal vein occlusion, ischemic optic neuropathy), sudden hearing loss, avascular necrosis of bone, intestinal damage caused by administration of a non-steroidal anti-inflammatory agent (e.g., diclofenac, meloxicam, oxaprozin, nabumetone, indomethacin, ibuprofen, ketoprofen, naproxen, celecoxib) (there is no particular limitation for the intestinal damage so far as it is damage appearing in duodenum, small intestine and large intestine and examples thereof include mucosal damage such as erosion and ulcer generated in duodenum, small intestine and large intestine), and symptoms associated with lumbar spinal canal stenosis (e.g., paralysis, dullness in sensory perception, pain, numbness, lowering in walking ability, etc. associated with cervical spinal canal stenosis, thoracic spinal canal stenosis, lumbar spinal canal stenosis, diffuse spinal canal stenosis or sacral stenosis) etc. (see, for example, in WO ‘084, WO 2009/157396, WO 2009/107736, WO 2009/154246, WO 2009/157397, and WO 2009/157398).
In addition, compound A is useful as an accelerating agent for angiogenic therapy such as gene therapy or autologous bone marrow transplantation, an accelerating agent for angiogenesis in restoration of peripheral artery or angiogenic therapy, etc. (see, for example, in WO ‘084).
Production of Compound A
Compound A can be produced, for example, according to the method described in WO ‘084, and, it can also be produced according to the production method mentioned below.

Step 1:
6-Iodo-2,3-diphenylpyrazine can be produced from 6-chloro-2,3-diphenylpyrazine by reacting it with sodium iodide. The reaction is carried out in the presence of an acid in an organic solvent (e.g., ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or their mixed solvent). The acid to be used is, for example, acetic acid, sulfuric acid, or their mixed acid. The amount of sodium iodide to be used is generally within a range of from 1 to 10 molar ratio relative to 6-chloro-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the acid to be used, but may be generally within a range of from 60° C. to 90° C. The reaction time varies depending on the kinds of the solvent and the acid to be used and on the reaction temperature, but may be generally within a range of from 9 hours to 15 hours.
Step 2:
5,6-Diphenyl-2-[(4-hydroxybutyl(isopropyl)amino]pyrazine can be produced from 6-iodo-2,3-diphenylpyrazine by reacting it with 4-hydroxybutyl(isopropyl)amine. The reaction is carried out in the presence of a base in an organic solvent (e.g., sulfolane, N-methylpyrrolidone, N,N-dimethylimidazolidinone, dimethyl sulfoxide or their mixed solvent). The base to be used is, for example, sodium hydrogencarbonate, potassium hydrogencarbonate, potassium carbonate, sodium carbonate or their mixed base. The amount of 4-hydroxybutyl(isopropyl)amine to be used may be generally within a range of from 1.5 to 5.0 molar ratio relative to 6-iodo-2,3-diphenylpyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from 170° C. to 200° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 5 hours to 9 hours.
Step 3:
Compound A can be produced from 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine by reacting it with N-(2-chloroacetyl)methanesulfonamide. The reaction is carried out in the presence of a base in a solvent (N-methylpyrrolidone, 2-methyl-2-propanol or their mixed solvent). The base to be used is, for example, potassium t-butoxide, sodium t-butoxide or their mixed base. The amount of N-(2-chloroacetyl)methanesulfonamide to be used may be generally within a range of from 2 to 4 molar ratio relative to 5,6-diphenyl-2-[4-hydroxybutyl(isopropyl)amino]pyrazine, preferably within a range of from 2 to 3 molar ratio. The reaction temperature varies depending on the kinds of the solvent and the base to be used, but may be generally within a range of from −20° C. to 20° C. The reaction time varies depending on the kinds of the solvent and the base to be used and on the reaction temperature, but may be generally within a range of from 0.5 hours to 2 hours.
The compounds to be used as the starting materials in the above-mentioned production method for compound A are known compounds, or can be produced by known methods.
PATENT
WO 2002088084
and
http://www.google.fm/patents/WO2009157398A1?cl=en
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 21 p. 6692 – 6704
compd 31
PAPER
Bioorganic and Medicinal Chemistry, 2007 , vol. 15, 24 p. 7720 – 7725
 2a is the drug
2a is the drug
N-Acylsulfonamide and N-acylsulfonylurea derivatives of the carboxylic acid prostacyclin receptor agonist 1 were synthesized and their potential as prodrug forms of the carboxylic acid was evaluated in vitro and in vivo. These compounds were converted to the active compound 1 by hepatic microsomes from rats, dogs, monkeys, and humans, and some of the compounds were shown to yield sustained plasma concentrations of 1 when they were orally administered to monkeys. These types of analogues, including NS-304 (2a), are potentially useful prodrugs of 1.
http://www.sciencedirect.com/science/article/pii/S0968089607007614
PATENT
Compound A Compound A can be produced , for example, by the method described in Patent Document 1, but can also be produced by the production method described below.
[
6-iodo-2,3-diphenylpyrazine can be produced by reacting 6-chloro-2,3-diphenylpyrazine with sodium iodide. This reaction is carried out in an organic solvent (for example, ethyl acetate, acetonitrile, acetone, methyl ethyl ketone, or a mixed solvent thereof) in the presence of an acid. As the acid to be used, for example, acetic acid, sulfuric acid, or a mixed acid thereof can be mentioned. The amount of sodium iodide used is, for example, suitably in the range of 1 mole to 10 moles, preferably in the range of 2 time moles to 3 times the amount of 1 mole of 6-chloro-2,3-diphenylpyrazine . The reaction temperature varies depending on the raw materials used and the type of acid, but is usually carried out within the range of 60 ° C. to 90 ° C. The reaction time varies depending on the starting materials used, the type of acid and the reaction temperature, but it is usually within the range of 9 hours to 15 hours.Step 2
5,6-diphenyl-2- [4-hydroxybutyl (isopropyl) amino] pyrazine can be prepared by reacting 6-iodo-2,3-diphenylpyrazine with 4-hydroxybutyl (isopropyl) amine. This reaction is carried out in an organic solvent (for example, sulfolane, N-methylpyrrolidone, N, N-dimethylimidazolidinone, dimethylsulfoxide or a mixed solvent thereof) in the presence of a base. Examples of the base used include sodium hydrogencarbonate, potassium hydrogen carbonate, potassium carbonate, sodium carbonate, and mixed bases thereof. The amount of 4-hydroxybutyl (isopropyl) amine to be used is, for example, suitably in the range of 1.5 mol to 5.0 mol per 1 mol of 6-iodo-2,3-diphenylpyrazine, It is within the range of 2 mol to 3 mol. The reaction temperature varies depending on the type of raw material and base used, but is usually carried out within the range of 170 ° C. to 200 ° C. The reaction time varies depending on the type of raw materials and base used and the reaction temperature, but it is usually within the range of 5 hours to 9 hours.Step 3
Compound A can be prepared by reacting 5,6-diphenyl-2- [4-hydroxybutyl (isopropyl) amino] pyrazine with N- (2-chloroacetyl) -methanesulfonamide. This reaction is carried out in an organic solvent (N-methylpyrrolidone, 2-methyl-2-propanol or a mixed solvent thereof) in the presence of a base. Examples of the base to be used include potassium t-butoxide, sodium t-butoxide or mixed bases thereof. The amount of N- (2-chloroacetyl) -methanesulfonamide used is, for example, 2 to 4 mol per 1 mol of 5,6-diphenyl-2- [4-hydroxybutyl (isopropyl) amino] It is suitable within the range, and preferably within the range of 2 mol to 3 mol. The reaction temperature varies depending on the type of raw material and base used, but is usually carried out within the range of -20 ° C. to 20 ° C. The reaction time varies depending on the kinds of raw materials and bases used and the reaction temperature, but it is usually within the range of 0.5 hour to 2 hours.Each compound used as a raw material in the above-mentioned production method of compound A is a known compound or can be produced according to a known method.
present invention can be obtained, for example, by the following method.
The salt of the present invention can be prepared by dissolving the compound A in an appropriate solvent (for example, an ether solvent (for example, dimethoxyethane, tetrahydrofuran), an ester solvent (for example, isopropyl acetate), an aromatic hydrocarbon (for example, toluene), acetonitrile After dissolving and adding a desired base, if necessary, the mixed solution is left to stand at room temperature or under cooling in the state of concentrating or stirring or leaving it stationary. The precipitate formed is collected by filtration , Followed by washing with an appropriate solvent to obtain the desired salt of the present invention. When cooling, not only cooling but also gradual cooling or rapid cooling may be effective in obtaining good crystals. It is also effective to obtain good crystals by adding an ether solvent (for example, t-butyl methyl ether), an ester solvent (for example, ethyl acetate), and an aromatic hydrocarbon (for example, toluene) There are cases.The amount of the solvent used for dissolving the compound A is suitably in the range of 10 ml to 300 ml with respect to the compound A 1 g, for example.
The amount of the base to be used for preparing the salt of the present invention is suitably in the range of 0.5 mol to 1.2 mol with respect to the mol of the compound A 1.
Further, the salt of the present invention, which is a crystal, can be obtained by, for example, the method described in Examples described later.
Example 1 t- butylamine Form I crystal of the salt
Compound A (40 mg) with 0.5mL dimethoxyethane (hereinafter, referred to as. “DME”) was dissolved in, and t- butylamine (1.1 eq) were added, 25 1 ° C. at 8 it was stirred for hours. Thereafter, the reaction solution was added t- butyl methyl ether (1mL), at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, I-form crystals of t- butylamine salt ( 3 to afford 9.9mg). B Powder X-ray diffraction spectrum of type I crystal obtained t- butylamine salt using the apparatus shown in Figure 1.
Melting point: 152.5 ℃
elemental analysis (C 3 0 H 4 3 N 5 O 4 S + 0.0 3 H 2 as O)
calculated value (%) C: 6 3 .1 8 H: 7 . 6 1 N: 12 .2 8 measured value (%) C: 6 2. 8 5 H: 7 . 6 4 N: 12.52 1 H-NMR (DMSO-D 6 ): delta 8 .15 (s, 1H), 7 .55 – 7 . 8 0 (M, 2H), 7 .10- 7 . .45 (M, 10H), 4 7 . 0-4 8 5 (M, 1H), 3 . 6 6 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 3 (s, 3 H), 1.50-1. 7 5 (M, 4H), 1.2 3 (s, 9H), 1.22 (D, 6 H)
Example 2 I-form crystal of the potassium salt
Compound A tetrahydrofuran with (40mg) 12mL (hereinafter, referred to as. “THF”) was dissolved in, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added, 40 ℃ It was heated and stirred in for 15 minutes. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. After repeated two more times this step, at -20 ° C. 3 and held hours. The resulting precipitated crystals were collected by filtration under reduced pressure, and dried to obtain Form I crystal of the potassium salt. B Powder X-ray diffraction spectrum of type I crystal of the obtained potassium salt using the apparatus shown in Fig. 1 H-NMR (DMSO-D 6 ): delta 8 .14 (s, 1H), 7 .1 8 – 7 . 3 8 . (M, 10H), 4 7 . 2-4 8 4 (M, 1H) , 3 . 6 5 (s, 2H), 3 .4 7 (t, 2H), 3 .45 (t, 2H), 2. 7 2 (s, 3 H), 1.55-1. 7 0 ( M, 4H), 1.2 3 (D, 6 H)
Example 3 II-form crystals of the potassium salt
Compound A with (40mg) was dissolved in THF and 12mL, 0.1M aqueous potassium hydroxide solution (1.1 eq) was added and heated with stirring for 15 min at 40 ℃. After that, it was evaporated under reduced pressure, the solvent. The residue it was added ethyl acetate (200μL). While shaking the mixture heated to 50 ° C. 8 was allowed to cool to 25 ℃ over hours. This operation was repeated two more times, at -20 ° C. 3 and held hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, after drying, 40 ℃, relative humidity 7 while 5% of thermo-hygrostat 7 left for days to give crystalline Form II of the potassium salt. B Powder X-ray diffraction spectrum of crystalline Form II of the resulting potassium salt using the apparatus Fig 3 is shown in.
Example 4 III type crystal of the potassium salt
Compound A , in addition to (100mg) acetonitrile (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (1.1 eq) was added and stirred for 200 minutes at 20 ℃. While stirring the mixture 7 after a heated stirring for 1 hour to 0 ° C., and then cooled to 10 ℃ over 10 hours. Further heated while the mixture 6 is heated to 0 ℃, t- butyl methyl ether (0. 3 after adding mL), cooled to 20 ℃ over 10 hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried, III type crystal of the potassium salt ( 7 to afford 5mg). The powder X-ray diffraction spectrum of the type III crystal of the obtained potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, of about 7 endothermic peak was observed at around 4 ° C..
Elemental analysis (C 2 6 H 3 1 N 4 O 4 . SK + 0 7 8 H 2 as O)
calculated value (%) C: 5 6 .91 H: 5.9 8 N: 10.21
measured value (%) C: 5 6 . 6 1 H: 5.55 N:. 10 3 6
EXAMPLE 5 IV-type crystal of the potassium salt
Compound A , in addition to (50mg) and ethyl acetate (1mL), and stirred with heating, Compound A was dissolved, followed by cooling to 20 ℃. To a solution 3 .5M potassium hydroxide / ethanol solution (2.2 eq) was added and 2 at 20 ° C. 3 and stirred for hours. It was collected by filtration the precipitated crystals produced, under reduced pressure, and dried to obtain Form IV crystal of the potassium salt (41mg). The powder X-ray diffraction spectrum of crystalline Form IV of the resulting potassium salt using R unit is shown in FIG. Furthermore, in differential scanning calorimetry, an endothermic peak was observed at around approximately 91 ℃.
Paper
J Med Chem 2015, 58(18): 7128
PATENT
WO 2018008042
https://patents.google.com/patent/WO2018008042A1/en
The present invention relates to an improved and novel processes for the preparation of 2- {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy} -N-(methylsulfonyl) acetamide compound of formula- 1 , which is represented by the following structural formula- l .

Formula-
The present invention also relates to novel crystalline forms of the compound of formula- 1 and process for the preparation thereof.
Background of the Invention:
2- {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl) acetamide is known as Selexipag. It is developed by Nippon Shinyaku under the brand name of Uptravi®, for the treatment of pulmonary arterial hypertension.
2- {4-[(5,6-diphenylpyTazin-2-yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl) acetamide was firstly described in US7205302B2 herein after referred as US ‘302. The said patent also describes its process for the preparation. According to this process the final product was obtained with low yield and purity.
US8791 122 (herein after referred as US’ 122) patent describes crystalline form-I, II and III of 2- {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy} -N-(methylsulfonyl) acetamide. Because of drug compounds having, for example, improved stability, solubility, shelf life and in vivo pharmacology, are consistently sought, there is an ongoing need for new or pure salts, hydrates, solvates and polymorphic forms of existing drug molecules. The novel crystalline forms of 2- {4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy} -N- (methylsulfonyl) acetamide described herein help meet this requirement.
US ‘ 122 patent describes amorphous form of the compound of formula- 1 . This patent does not disclose any detailed process for amorphous form and PXRD pattern of amorphous compound of formula- 1 .

Examples:
Example-1 Preparation of 4-((5)6-diphenylpyrazin-2-yl)(isopropyl)amino)butan-l-ol compound of formula-8
A mixture of 5-chloro-2,3-diphenylpyrazine (25 gm) compound of formula-7a and 4- (isopropyl amino)butan- 1 -ol (108 gm) was heated to 190-195°C and stirred the reaction i mixture for 10- 12 hours at same temperature. Cooled the reaction mixture to 25-35°C. To this reaction mixture n-heptane followed by water were added slowly at 25-30°C and stirred the reaction mixture for 2 hours at the same temperature. Filter the precipitated solid, washed with water and dried to get the title compound.
Yield: 30 gm.
Example-2: Preparation of tert-butyl 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino) butoxy)acetate
Potassium hydroxide solution (96.6 gm of potassium hydroxide dissolved in 175 ml of water) was added to the mixture of 4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butan- l -ol (25 gm) and toluene ( 175 ml) at 25-30°C and stirred the reaction mixture for 30 minutes at the same temperature. Cooled the reaction mixture to 0-5°C. Tert-butyl bromoacetate (94 gm) was slowly added to the reaction mixture at 0-5°C and stirred the reaction for 60 minutes at same temperature. Raised the temperature of the reaction mixture to 25-30°C and maintained for 60 minutes. Both the aqueous and organic layers were separated. The aqueous layer was extracted with toluene and combined the organic layers. Organic layer was washed with hydrochloric acid solution followed by with aqueous sodium bicarbonate solution. Organic layer was dried with sodium sulphate and distilled off the solvent completely from the organic layer under reduced pressure to get the title compound.
Yield: 29 gm.
Example-3: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butoxy) acetic acid compound of formula-6
Aqueous sodium hydroxide solution (7.5 gm of sodium hydroxide was dissolved in 80 ml of water) was added to the solution of tert-butyl 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl) amino)butoxy)acetate (30 gm) in methanol (290 ml) at 30-35°C. Heated the reaction mixture to reflux temperature and stirred for 3 hours at the same temperature. Distilled off solvent completely from the reaction mixture under reduced pressure and cooled the reaction mixture to 25-30°C. Water was added to the obtained compound and acidified the reaction mixture using diluted hydrochloric acid at the same temperature. Extracted the reaction mixture with ethyl acetate. The organic layer was washed with aqueous sodium chloride solution and dried with sodium sulphate. Distilled off the solvent from the organic layer under reduced pressure. Diisopropyl ether (60 ml) was added to the obtained compound at 25-30°C and stirred for 60 minutes at the same temperature. Filtered the precipitated solid, washed with diisopropyl ether and dried to get the title compound.
Yield: 19 gm.
Example-4: Preparation of 2-{4-[(5,6-diphenylpyrazin-2-yl)(isopropy.)amino]butoxy}- N-(methylsulfonyl) acetamide compound of formula-1
Triethylamine (9.6 gm) was added to the mixture of 2-(4-((5,6-diphenylpyrazin-2- yl)(isopropyl)amino)butoxy)acetic acid (10 gm), dichloro methane (100 ml), N,N- dicyclohexylcarbodiimide (4.9 gm), hydroxybenzotriazole (3.5 gm) and methane sulfonamide (3.39 gm) at 25-30°C and stirred the reaction mixture for 12 hours at the same temperature. Filtered the unwanted compounds from the reaction mixture and washed with dichloromethane. The organic layer was washed with water, followed by with aqueous citric acid solution and then washed with aqueous sodium chloride solution. Distilled off the solvent from the organic layer under reduced pressure. To this residue ethyl acetate (20 ml) and carbon (1 gm) were added at 25-30°C and stirred the reaction mixture for 30 minutes at the same temperature. Filtered the reaction mixture through hyflow bed and washed with ethyl acetate. The obtained filtrate was slowly added to the mixture of n-heptane and water at 25-30°C and stirred for 10 hours. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 4.5 gm.
Example-5: Preparation of 2-{4-f(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy}- N-(methylsulfonyl) acetamide compound of formula-1
Sodium t-butoxide (96.6 gm) was added to the mixture of n-methy pyrrolidinone (125 ml) and 4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butan-l-ol (25 gm) compound of formula-8 at 0-5°C and stirred the reaction for 20 minutes at the same temperature. 2-chloro- N-(methylsulfonyl)acetamide (23.7 gm) was slowly added to the reaction mixture at 0-5°C and raise the temperature of the reaction mixture to 25-30°C. Stirred the reaction mixture for 10-12 hours at 25-30°C and water was added to it at the same temperature. The reaction mixture was extracted with ethyl acetate. The organic layer was washed with aqueous sodium chloride solution and distilled off the solvent from the organic layer under reduced pressure. To this residue ethyl acetate (50 ml) and carbon (2.5 gm) were added at 25-30°C and stirred the reaction mixture for 30 minutes at the same temperature. Filtered the reaction mixture through hyflow bed and washed with ethyl acetate. The obtained filtrate was slowly added to the mixture of n-heptane and water at 25-30°C and stirred for 10 hours. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 14 gm.
Example-6: Preparation of 2-chIot*o- -(methylsulfonyl)acetamide
A mixture of methane sulfonamide (100 gm) and chloroacetyl chloride (356.4 gm) was heated to reflux temperature and stirred it for 10 hours at the same temperature. Cooled the reaction mixture to – 10 to -5°C and stirred it for 2 hours at the same temperature. Filtered the precipitated, solid, washed with toluene followed by n-heptane and dried to get the title compound.
Yield: 175 gm.
ExampIe-7: Purification of the compound of formula-1
Methanol (20 ml) was added to the compound of formula-1 (2 gm) at 25-30°C and heated to reflux temperature. Dichloromethane (3 ml) was added to the reaction mixture at reflux temperature and stirred for 15 minutes at the same temperature. Filtered the reaction mixture, distilled off the solvent from the filtrate under reduced pressure to get the title compound. Yield: 2 gm
Example-8: Preparation of N-isopropyI-5,6-diphenylpyrazin-2-amine (Formula-4) Isopropyl bromide (5.5 gm) was added to the mixture of 2-amino -5,6-diphenylpyrazine ( 10 gm), potassium tert-butoxide (9 gm) and dimethylformamide (50 ml) at 25-30°C, slowly heated to 80-85°C and stirred the reaction mixture for 6 hours at same temperature. The reaction mixture was cooled to 10- 15°C, diluted the reaction mixture with water and stirred it for 2 hours at the same temperature. Filtered the obtained solid and dried to get the title compound.
Yield: 9.5 gm
ExampIe-9: Preparation of N-isopropyl-5,6-diphenylpyrazin-2-amine (Formula-4)
A mixture of 5-chloro-2,3-diphenylpyrazine ( 10 gm), isopropyl amine (7.5 gm) and potassium carbonate (10.5 gm) and dioxane (50 ml) were heated to 40-45°C and stirred the reaction mixture for 12 hrs at the same temperature. The reaction mixture was cooled to 10- 15°C, diluted with water and extracted with dichloromethane. Combined the organic layers was washed with aqueous sodium hydrochloride solution and dried over anhydrous sodium sulphate. Distilled off the solvent completely from the organic layer under reduced pressure to provide the title compound.
Yield: 9 gm
Example-10: Preparation of 2-(4-chlorobutoxy)aceticacid (Formula-5a)
2-bromoaceticacid (10 gm) was slowly added to a mixture of l-chlorobutan-4-ol (7.2 gm), potassium carbonate (26.5 gm) and acetonitrile (50 ml) at 25-30°C. The reaction mixture was heated to 75-80°C and stirred the reaction mixture for 6 hours at same temperature. The reaction mixture was cooled to 25-30°C and diluted with , water. Acidified the reaction mixture using diluted hydrochloric acid at 25-30°C. The reaction mixture extracted with dichloromethane. Combined the organic layers was dried over anhydrous sodium sulphate and distilled off the solvent under reduced pressure to provide the title compound.
Yield: 10.5 gm.
Example-11: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino) butoxy)acetic acid (formula-6)
A mixture of N-isopropyl-5,6-diphenylpyrazin-2-amine (8 gm), potassium carbonate (7.5 gm) and acetonitrile (40 ml) was stirred for 1 hr at 25-30°C. A solution of 2-(4-chlorobutoxy) aceticacid (5.4 gm) in acetonitrile (15 ml) was slowly added to the reaction mixture at 25- 30°C. Heated the reaction mixture to reflux and stirred for 12 hours at the same temperature. The reaction mixture was cooled to 10-15°C and diluted with wateT. Acidified the reaction mixture using diluted hydrochloric acid and extracted the reaction mixture using ethyl acetate. Combined the organic layers and dried over sodium sulphate. Distilled off the solvent completely from the organic layer to get the title compound.
Yield: 8.5 gm
Example-12: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyt)amino)butoxy)- N-(methylsulfonyl)acetamide (formula-1)
A mixture of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butoxy)acetic acid (5 gm), HATU (5.4 gm), triethylamine (1.5 gm) and dimethylformamide (20 ml) was stirred for 1 hr at 5-10°C under nitrogen atmosphere. Methane sulfonamide (5.2 gm) was slowly added to the reaction mixture at 5-10°C and stirred for 12 hrs at the same temperature. The reaction mixture was diluted with water and stirred for 2 hrs. The precipitated solid was filtered and dried to get the title compound.
Yield: 4.5 gm
Example-13: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl) (isopropyl) amino) butoxy) acetonitrile (Formula-12)
To the mixture of 4-((5,6-diphenylpyrazin-2-yI)(isopropyl)amino)butan-l-ol ( 10 gm), tetrabutyl ammoniumbromide (0.2 gm), potassium carbonate (7.6 gm) and acetone (50 mL), chloroacetonitrile (3.2 gm) was added at 25-30°C. Heated the reaction mixture to reflux temperature and stirred the reaction mixture for 6 hrs at the same temperature. The reaction mixture was cooled to 10- 15°C and filtered the reaction mixture. Distilled off the solvent completely from the filtrate to get the tile compound.
Yield: 9 gm
Example-14: Preparation of 2-(4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butoxy) acetic acid (formula-6)
Sodium hydroxide (3.5 gm) was added to a solution of 2-(4-((5,6-diphenylpyrazin-2-yl) (isopropyl) amino) butoxy) acetonitrile (8 gm) in methanol (60 ml) and water (30 ml). The reaction mixture was heated to 65-70°C and maintained for 6 hrs. The reaction mixture was cooled to 10°C, acidified with diluted hydrochloric acid and stirred at same temperature for 2 hr. The obtained solid was filtered and dried to provide the title compound.
Yield: 7.5 gm
Example-15: Preparation of 2-chloro-N-(methylsulfonyl)acetamide (Formula-16)
The mixture of methane sulfonamide (50 gm) and chloroacetyl chloride (92 gm) was heated to 1 10-1 15°C and stirred the reaction mixture for 7 hours at the same temperature. The reaction mixture was cooled to 25-30°C and dichloromethane was added to the reaction mixture at the same temperature. Cooled the reaction mixture to 15-20°C and stirred for 1 hour at the same temperature. Filtered the precipitated solid and washed with dichloromethane. The obtained solid was recrystallized using dichloromethane to get pure title compound. Yield: 80 gm. M.R.: U0- 1 15°C. Purity by HPLC: 98.85%.
Example-16: Preparation of 4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butan-l-ol (Formula-8)
The mixture of 5-chloro-2,3-diphenylpyrazine ( 100 gm) and 4-(isopropylamino)butan-l -ol (245.5 gm) was heated to 190-195°G and stirred the reaction mixture for 12 hours at the same temperature. The reaction was cooled to 25-30°C and n-heptane was added to the reaction mixture. The reaction mixture was further cooled to 10-15°C, water was slowly added to the reaction mixture and stirred for 2 hours at the same temperature. Filtered the precipitated solid and washed with water. Dichloromethane (300 ml) was added to the obtained solid and stirred for 5 minutes. Both the organic and aqueous layers were separated. The organic layer was dried with sodium sulphate, distilled off the solvent from the organic layer completely under reduced pressure and co-distilled with n-heptane. 400 ml of n-heptane was added to the obtained compound at 25-30°C, heated the reaction mixture to 45-50°C and stirred for 30 minutes at the same temperature. The reaction mixture was cooled to 15-20°C and stirred for 2 hours at the same temperature. Filtered the solid, washed with n-heptane and dried to get the title compound.
Yield: 82 gm. M.R.: 100-105°C. Purity by HPLC: 95.4%.
Example-17: Purification of 4-((5,6-diphenylpyrazin-2-yl)(isopropyl)amino)butan-l-ol (Formula-8)
n-Heptane (750 ml) was slowly added to pre-cooled solution of 4-((5,6-diphenylpyrazin-2- yl)(isopropyl)amino)butan- l -ol (100 gm) in acetone (250 ml) was cooled to 0-5°C. Stirred the reaction mixture for 4 hours at the same tempereature. Filtered the precipitated solid, washed with n-heptane and dried to get the pure title compound.
Yield: 54 gm. Purity by HPLC: 99.92%.
Example-18: Preparation of crystalline form-L of compound of formula-1
Melting the compound of formula-1 (10 gm) at 140-145°C under reduced pressure for 15 minutes. The above obtained oily residue was added to 100 ml of pre-cooled n-heptane at 0- 5°C. Stirred the reaction mixture for 6 hr at 0-5°C. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound. Yield: 9 gm; PXRD of the obtained compound is depicted in figure- 10 and DSC thermogram is depicted in figure- 1 1. Example-19: Preparation of crystalline form-P of compound of formula-1
Melting the compound of formula-1 (10 gm) at 140-145°C under reduced pressure for 15 minutes. The above obtained oily residue was added to 100 ml of pre-cooled n-heptane at 0- 5°C. Stirred the reaction mixture for 36 hours at 0-5°C. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 9 gm; PXRD of the obtained compound is depicted in figure-7, its IR is depicted in figure-8 and its DSC is depicted in figure-9.
Example-20: Preparation of crystalline form-P of compound of formula-1
Melting the compound of formula-1 (10 gm) at 140-145°C under reduced pressure for 15 minutes. The above obtained oily residue was added to 100 ml of n-heptane at 30-40°C.
Stirred the reaction mixture for 36 hours at 30-40°C. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 9 gm; PXRD of the obtained compound is similar to the figure-7.
Example-21 : Preparation of amorphous form of compound of formula-1
Melting the compound of formula-1 ( 10 gm) at 140- 145°C under reduced pressure for 15 minutes and the above obtained oily residue was cooled to 0-5°C. Unload the obtained compound and dried to get the title compound. Yield: 9 gm; Purity by HPLC: 99.74%. PXRD of the obtained compound is depicted in figure-5 and IR is depicted in figure-6.
Exaniple-22: Preparation of crystalline form-I of compound of formula-1
Melting the compound of formula-1 (5 gm) at 140-145°C under reduced pressure for 15 minutes. 50 ml of n-heptane was added to the above obtained oily residue at 115-120°C.
Stirred the reaction mixture for 20 minutes at 1 15- 120°C. Cooled the reaction mixture to 25-
30°C and stirred for 60 minutes at the same temperature. Further cooled the reaction mixture to 0-5°C and stirred the reaction mixture for 60 minutes at the same temperature. Filtered the precipitated solid, washed with n-heptane and dried to get the title compound.
Yield: 4 gm; Purity by HPLC: 99.68%.
PATENT
CN 108675964
PATENT
CN 106316967
PATENT
WO 2017029594
PATENT
Form-I II III
https://patents.google.com/patent/US8791122B2/en
PATENT
https://patents.google.com/patent/WO2018022704A1/en
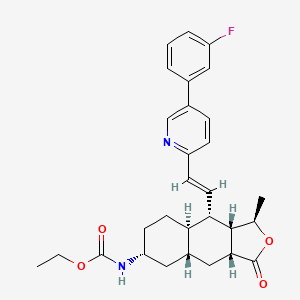
Selexipag has the chemical name 2-{4-[(5,6-diphenylpyrazin-2- yl)(isopropyl)amino]butoxy}-N-(methylsulfonyl)acetamide. Selexipag has the following chemical structure:

[0004] Selexipag is being developed by Actelion and Nippon Shinyaku for the treatment of arteriosclerosis obliterans, pulmonary hypertension and Raynaud’s disease secondary to systemic sclerosis.
[0005] Selexipag is disclosed in US 7,205,302. US 8,791,122, US 9,284,280 and US 2014- 0155414 disclose polymorphs of Selexipag, denominated forms I, II and III. WO
2017/040872 discloses form IV and V of Selexipag.
xample 1: Preparation of Selexipag
[00126] A. Route 1
[00127] Crude Selexipag can be obtained by any method known in the art, for example by the method described in US 7,205,302 or according to the following.
[00128] B. Route 2
[00129] Step a: Preparation of 4-((5,6-diphenyl-pyrazin-2-yl)(isopropyl)amino)butan-l-ol
[00130] To 50 g (0.161 mol) of 5-bromo-2,3-diphenylpyrazine, 116 g (0.884 mol, 5.5 eq/mol) of 4-(isopropylamino)-butan-l-ol and 13.33 g of KI (0.080 mol, 0.5 Eq/mol) were added. The reaction mixture was stirred, warmed and then heated up to 140°C for about 18- 20 hrs. The reaction was monitored by TLC up to completion (starting material about 1% by TLC). The reaction mixture was cooled down to room temperature. After the reaction was completed, the following work up step was performed:
[00131] Option 1 : Ethyl acetate was added (500 mL, 10 vol) and the organic phase was washed with water (150 mL, 3 vol). The organic phase was separated and aqueous phase was extracted with ethyl acetate (150 mL, 3 vol). The organic phases were joined and washed with water (200 mL, 2 vol) three times.
[00132] The solvent was distilled off under vacuum at not more than (“NMT”) 40°C until 1 vol (oil appearance).
[00133] Option 2: The material (mixture) was dissolved in acetone (250 mL, 5 vol), the solution obtained was cooled down to 0°C to 5°C and anti-solvent / water was added (1000 mL, 20 vol) for 40 minutes, then the suspension was stirred for about 30 minutes at about 0°C-5°C. The solid material was filtered and washed with water (200 mL, 4 vol). Crude wet product was obtained as yellow solid yielding 101.8 % WY (87 % MY), HPLC purity 90.8% on area at this stage.
[00134] The crude material, obtained in either of the above described options, was purified through crystallization from acetone :«-heptane as follows: to a solution of 4-((5,6-diphenyl- pyrazin-2-yl)(isopropyl)amino)butan-l-ol crude in acetone (175 mL, 3.5 vol) at 0°C – 5°C, hexane (600 mL, 12 vol) dropwise in about 120 min was added, then the precipitated mixture was cooled down to about -10°C and stirred for about 60 min. The product was filtered off and washed with hexane (250 mL, 5 vol) and dried under vacuum at 25°C. Pure product was obtained as yellowish solid yielding overall 77.2%, (66.5% MY), HPLC purity 98.2% on area.
[00135] Step b: Preparation (2-bromo-N-(methylsulfonyl)-acetamide)
[00136] To a suspension of 50 g (0.526 mol) of methanesulfonamide in toluene (625 mL, 12.5 vol) and isopropyl acetate (625 mL, 12.5 vol), 159.1 g (0.789 mol) of bromo-acetyl- bromide (“BAB”) was added under nitrogen atmosphere. The reaction mixture was heated up to about 90°C for about 8 hours under a nitrogen stream. The reaction was monitored by TLC up to completion (starting material about 1% by TLC). The reaction mixture was cooled down to about 40°C and concentrated under vacuum until 10 volumes. Subsequently, toluene was added (250 mL, 5 vol) and distilling off solvents is carried out at NMT 30°C until 10 volumes. Then was added dichloromethane (100 mL, 2 vol) and the mixture was cooled down at 0°C and is stirred for 90 min. The solid was filtered and washed with
dichloromethane (100 mL, 2 vol). Crude product was obtained as beige solid material yielding 187% WY (83% MY), HPLC purity 99.2% at this stage. [00137] The crude material (83 g) was purified through re-slurring with dichloromethane (166 mL, 2 vol; preferably 332 mL, 4 vol) by stirring at about 32°C for about 60 min. The crystallization mixture was cooled down to about 0°C-5°C and stirring for 30 min, filtered off and washed with dichloromethane (100 mL, 2 vol). Subsequently, the material was dried at 35°C for 24 hours. Pure and dried material was obtained as white off solid yielding overall 173%, (77% MY), HPLC purity 99.6 % on area.
[00138] Step c: Preparation of (2-[4-[(5,6-diphenyl-2-pyrazinyl)(l- methylethyl)amino]butoxy]-N-(methylsulfonyl)-acetamide) – Selexipag
[00139] To 10 g (0.028 mol) of 4-((5,6-diphenyl-pyrazin-2-yl)(isopropyl)amino) butan-l-ol was added a strong base (6.0 eq/mol), previously suspended in an appropriate solvent, within a range of from -10°C to 40°C under a nitrogen atmosphere and stirred for 60 min. Then, a solution of 17.9 g (3.0 eq/mol) of 2-bromo-N-(methylsulfonyl)-acetamide, previously dissolved in the same solvent, is added dropwise within a range of from 120 tol 80 min, controlling the exothermic temperature. The reaction was monitored by TLC up to completion. Subsequently, the mixture reaction was cooled down around 5°C and water is added by controlling the exotherm (NMT 15°C). Finally, an acetic acid solution was added and the suspension was stirred for about 60 min at 0°C -5°C. The product (crude) was filtered off and washed with water. An amorphous solid was obtained. The crude product was purified by crystallization from ethanol:THF.
[00140] Step d: Purification of Selexipag
[00141] Crude Selexipag can be purified by crystallization in an organic solvent for example alcohols such as ethanol, iso-amyl alcohol, iso-propyl alcohol, butanol; ethers such as tetrahydrofuran, hydrocarbons such as heptane and mixed solvents thereof.
[00142] C. Route 3
[00143] 33.3 g (0.297 mol, 6.0 eq/mol) of potassium tert-butoxide were dissolved in DMF (2.8 vol) in a flask (500 mL) under nitrogen atmosphere and stirred for 15 min. Then, a solution of 17.9 g (0.049 mol, 1.0 eq/mol) of 4-((5,6-diphenyl-pyrazin-2-yl)(isopropyl) amino) butan-l-ol (SLX-4) dissolved in DMF (1.2 vol) was added in one portion. The reaction mixture was stirred for 60 min within a temperature range from 20°C to 25°C at 150 rpm Then, a solution of 32.1 g (0.15 mol, 3.0 Eq/mol) of 2-bromo-N-(methylsulfonyl)- acetamide (SLX-9), previously dissolved in DMF (1.3 vol), was added dropwise for 120 minutes by controlling the temperature (exothermic process).
[00144] The reaction mixture was quenched with cool water (0.33 vol), transferred into a flask of more capacity (1000 mL) and placed in an ice bath. Cool water (38.32 vol) was added to the reaction mixture and the pH was adjusted to 5.0 with AcOH (0.33 vol). The mixture was stirred at 300 rpm for 40 min. Then, the flask with the reaction mixture was stored in the refrigerator at 8°C. After 8h, the solid was filtered and washed with cool water (5 vol, 2 times). The crude product (yellow solid) was drained (i.e. dried) for 30 min and was stored at 8°C.
Example 2: Preparation of crystalline Selexipag Form IV
[00145] A. Route 1
[00146] 3.0 g of Selexipag was dissolved in dimethylformamide (“DMF”) (12 mL, 4 vol). The obtained solution was added dropwise to a pre-cooled acetic acid solution (0.06 M, 120 mL, from 2°C to 8°C) to obtain a suspension. The suspension was stirred within a range of from 2°C to 8°C for 30 min; then the material was filtered, washed with water (10 mL, 3.3 vol) and drained (i.e. dried) for 10 minutes. The solid material (amorphous) was suspended in heptane (25 mL, 7.5 vol) and the obtained suspension was stirred for 30 minutes at room temperature. The material was filtered, washed with heptane (20 mL, 6.6 vol) and drained (i.e. dried) under vacuum for at least 30 minutes at room temperature to obtain the Form IV Crystal.
[00147] B. Route 2
[00148] Crude Selexipag (1.0 g, amorphous solid, obtained from the synthesis) was dissolved in ethyl acetate (5 vol, 5 mL), then water was added (10 vol, 10 mL) into the solution, the mixture was stirred for about 10 minutes and the pH was adjusted to a range of from 8.0 to 9.0 by titration with K2CO3 solution. The phases were separated; the pH of the aqueous phase was adjusted to a range of from 3.5 to 5.0 by titration with acetic acid. Then, ethyl acetate (10 vol, 10 mL) was added into the aqueous phase, the obtained mixture was stirred and the phases were separated. The organic phase was distilled off under reduced pressure (from 2 to 3 volumes), and a solution was obtained. The obtained concentrated solution was quickly added to a mixture (suspension) of Form IV in ^-heptane (17 mL, 17 vol), over a period of less than 5 minutes, (the suspension temperature was of from 15°C to 25°C), and a suspension was obtained. The obtained suspension was stirred (155rpm) for 90 minutes at a temperature of from 0°C to 5°C. The suspension was filtered, washed with heptane, squeezed for 15 minutes and dried at 25°C, under vacuum, for about 14 hours. The product was analyzed by PXRD – Form IV was obtained.
[00149] The above procedure can be performed by dissolving the crude amorphous starting material in any suitable organic solvent, for example ester solvent. Example 3: Preparation of (2-[4-[(5,6-diphenyl-2-pyrazinyl)(l- methylethyl)amino] butoxy] -N-(methylsulfonyl)-acetamide) – Selexipag

SLX-4 SLX-9 SLX-6
[00150] 9.2 grams (0.082 mol, 5.9 eq/mol) of potassium tert-butoxide were combined with DMF (2.7 vol, 13.5 mL) in a flask (50 mL) under nitrogen atmosphere and a suspension was formed and was stirred for 20 min. Then, 5.0 g (0.014 mol, 1.0 eq/mol) of 4-((5,6-diphenyl- pyrazin-2-yl)(isopropyl)amino)butan-l-ol (SLX-4) as solid powder was added under nitrogen atmosphere. The reaction mixture was stirred for 60 min within a temperature range from 20°C to 25°C and at 170 rpm. Then, a solution of 8.9 g (0.041 mol, 3.0 eq/mol) of 2-bromo- N-(methylsulfonyl)-acetamide (SLX-9), previously dissolved in DMF (1.3 vol, 6.5 mL), was added dropwise for 120 minutes by controlling the temperature (exothermic process). After the end of addition, the reaction was completed, and the reaction mixture was quenched with cold water (0.5 vol, 2.5 mL), subsequently transferred into a flask of more capacity (500 mL) and placed into an ice bath. Cold water (40 vol, 200 mL) was added into the suspension and the pH was adjusted within the range from 4.0 to 5.0 with acetic acid. The obtained mixture was stirred for 120 min. The crude amorphous product was collected by filtration and washed twice with cold water (5 vol, 25 mL). The product was drained (i.e. dried) for 30 min and isolated as a yellow-brown solid which was stored within the range from 2°C to 8°C for approximately 17 hours. Then, the crude amorphous material was dissolved in ethyl acetate (15 vol, 75 mL) and water was added into the solution (30 vol, 150 mL). The pH was adjusted from 8.0 to 9.0 by addition of potassium carbonate solution, the phases were separated and the aqueous phase was washed twice with ethyl acetate (7.5 vol, 37.5 mL). The pH of the final aqueous phase was adjusted to a range from 4.0 to 5.0 with acetic acid. Then, ethyl acetate was added (30 vol, 150 mL) and the phases were separated. The organic phase was washed twice with water (7.5 vol, 37.5 mL). The organic phase was distilled off under reduced pressure (from 6 to 7 volumes, or from 6 to 15 volumes) and a solution was obtained.
[00151] In a different flask (capacity of 250 mL with a PTFE stirrer blade), a suspension of 0.05 g of Selexipag Form IV in ^-heptane (30 volumes, 150 mL) was stirred for 60 minutes within the range 0°C to 5°C and this suspension was added into the above ethyl acetate concentrated solution at room temperature over a period of less than 5 minutes. The final suspension was cooled down to 0°C to 5°C and stirred (220 rpm) for 120 minutes. The solid product was filtered off and washed twice with cold heptane (5 vol, 25 mL). The product was drained (i.e. dried) overnight. The product was analyzed by PXRD – Form VI was obtained, PXRD pattern is depicted in Figure 1.
PATENT
CN105949135
https://patents.google.com/patent/CN105949135A/en

Example 1
[0027] A) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate:
[0028] 4 – [(tert-butoxycarbonyl) (isopropyl) amino] butanol -1_ (20 (^, 0.09111 〇1) and tert-butyl bromoacetate (21 · lg, 0 llmol) solution. The reaction was stirred for 2 hours to burn dichloromethane (90mL), was added tetrabutylammonium chloride (0.72g, 2.6mmol), potassium hydroxide (7.3g, 0.13mol) and water (12.0g), the reaction mixture was 25 ° C The reaction solution was concentrated under reduced pressure and rotary evaporated to dryness and extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethanol and recrystallized from isopropanol to give [4- (tert-butoxycarbonyl) (isopropyl yl) aminobutoxy] acetate, as a pale yellow oil (26.6 g of), a yield of 89.0%, the reaction formula of this step is as follows:
[0029]

[0030] B) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid:
[0031] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate (26. (^, 0.075! 11〇1) was dissolved in methanol (50 mL), was added sodium hydroxide solution (NaOH = 3 · 3g, 0 · 08mol; water 9 · 0g), was heated to 80 ° C for 6 hours, cooled to room temperature, after treatment and purification, to give [4- (tert-butoxycarbonyl) (isopropyl ) aminobutoxy] acetic acid (20.7 g of), a yield of 95.0%, the reaction formula of this step is as follows:

[0033] C) Preparation of 2- [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide:
[0034] [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid (20 (^, 0.07 11〇1) and a hoot “-.! P sitting carbonyldiimidazole (14.0g, 0.09mo 1 ) was dissolved in tetrahydro-thiopyran Misaki (70 mL), with stirring, was added methyl sulfonamide (7.9g, 0.08mol), the reaction mixture was 90 ° C the reaction stirred for 18 hours, the reaction solution was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide as an off-white solid (21.2 g), yield 83.7%, the reaction formula of this step is as follows:

[0036] D) Preparation of SIPA Seiler: 2- [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide (20 (^, 0.055111〇1. ) and dissolved in methanol (1101 ^), trifluoroacetic acid (6.88,0.06111〇1), 65 ° (: the reaction stirred for 6 hours to complete the reaction, the reaction was added dropwise to a stirred solution of water (200 mL), cooled to 0 ° C crystallization for 3 hours and filtered to give the intermediate compound (2- [4- (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide), and then dissolved in methanol (40 mL), was added 5 – chloro-2,3-diphenyl-pyrazine (16 · 0g, 0 · 06mol), N, N- diisopropylethylamine (15 · 5g, 0 · 12mol), the reaction mixture was stirred reactor 8 100 ° C hours, the reaction was cooled to room temperature, water (40 mL), cooled to -10 ° C crystallization for 3 hours and filtered to give SIPA game music, as a white solid (25.0 g of), a yield of 92.3%, the reaction step formula as follows:
[0037]

[0038] Example 2
[0039] A) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate:
[0040] 4 – [(tert-butoxycarbonyl) (isopropyl) amino] -1-butanol (23 (^, 0.10111 〇1) and tert-butyl bromoacetate (25 · 2g, 0 · 13mol) was dissolved. burning in 1,2-dichloroethane (110mL), was added tetrabutylammonium bromide (1 · lg, 3 · 5mmol), sodium hydroxide (6.4g, 0.16mol) and water (14.0g), the reaction mixture was 30 ° C The reaction was stirred for 3 hours, the reaction solution was concentrated by rotary evaporation to dryness under reduced pressure and extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethanol and recrystallized from isopropanol to give [4- (tert-butoxy butoxycarbonyl) (isopropyl) aminobutoxy] acetate, as a pale yellow oil (30.3 g of), a yield of 88.2%, the reaction of the present step is the same formula as in Example 1;
[0041] B) Preparation of [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid:
[0042] [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate (30. (^, 0.09! 11〇1) was dissolved in ethanol (85 mL), was added potassium hydroxide solution ( 1 (! = 5.78,0.10111〇1 01; 128 water), heated to 75 ° (: 7 hours, cooled to room temperature, after treatment and purification, to give [4- (tert-butoxycarbonyl) (isopropyl ) aminobutoxy] acetic acid, an off-white solid (23.5 g of), a yield of 93.7%, the reaction of the present step is the same formula as in Example 1;
[0043] C) Preparation of 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide:
[0044] [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid (23 (^, 0.08 11〇1) and Chi ^ -! Dicyclohexyl carbodiimide (22. lg, 0. llmol) was dissolved in chloroform (120 mL), with stirring, was added methyl sulfonamide (9.8g, 0. lOmol), the reaction mixture was 80 ° C the reaction stirred for 19 hours, the reaction solution was concentrated by rotary evaporation to dryness, ethyl acetate was added and extracted dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide, off-white the solid (24.8 g of), a yield of 85.0%, the reaction of the present step is the same formula as in Example 1;
[0045] D) Preparation of SIPA Seiler: 2- [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide (24 (^, 0.065111〇1. ) and dissolved in ethanol (1601 ^), trifluoroacetic acid (9 (^, 0.08111〇1.), 70 ° (: the reaction was stirred for 7 hours to complete the reaction, the reaction was added dropwise to a stirred solution of water (260 mL of), cooled crystallization to 0 ° C for 3 hours and filtered to give the intermediate compound (2- [4- (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide), and then dissolved in ethanol (90 mL) , 5-chloro-2,3-diphenyl-pyrazine (! 11〇1 21.8 8,0.08), triethylamine (14.98,0.15111〇1), the reaction mixture was 100 ° (: The reaction was stirred for 18 hours, the reaction solution cooled to room temperature, water (40 mL), cooled to -10 ° C crystallization for 3 hours and filtered to give SIPA game music, as a white solid (29.6 g of), a yield of 91.0%, the reaction of the present step is the same formula as in Example 1 .
[0046] Example 3
[0047] A) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate:
[0048] 4 – [(tert-butoxycarbonyl) (isopropyl) amino] -1-butanol (12g, 0.05mol) and t-butyl bromoacetate (12.1g, 0.06mol) was dissolved in chloroform (70mL), was added tetrabutylammonium iodide (0 · 5g, 1 · 3mmol), lithium hydroxide (1 · 7g, 0 · 07mol) and water (6.5 g of), the reaction mixture was stirred 20 ° C for 4 hours, the reaction solution under reduced pressure concentrated by rotary evaporation to dryness, extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethanol and recrystallized from isopropanol to give [4- (tert-butoxycarbonyl) (isopropyl) aminobutyrate oxygen yl] acetate, as a pale yellow oil (15.6 g of), a yield of 86.8%, the reaction of the present step is the same formula as in Example 1;
[0049] B) Preparation of [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid:
[0050] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate (15.0g, 0.04mol) was dissolved in isopropanol (40mL), was added a solution of lithium hydroxide (LiOH = 1 · 3g, 0 · 05mol; water 6 · 0g), was heated to 70 ° C for 8 hours, cooled to room temperature, after treatment and purification, to give [4- (tert-butoxycarbonyl) (isopropyl) aminobutyrate oxy] acetic acid as an off-white solid (11.7 g), 93.0% yield, this step is the same reaction scheme of Example 1;
[0051] C) Preparation of 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide:
[0052] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid (11 (^, 0.04! 11〇1) and 1- (3-dimethylaminopropyl) -3- ethylcarbodiimide (8.38,0.05111〇1) was dissolved in acetonitrile (4〇1111 ^), with stirring, was added methyl sulfonamide (5.] ^, 0.05mol), the reaction mixture was 95 ° C the reaction stirred for 22 hours, The reaction solution was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide as an off-white solid (11.7 g), yield 84.2%, the reaction of the present step is the same formula as in Example 1;
[0053] D) Preparation of SIPA Seiler: 2- [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide (11 (^, 0.03111〇1. ) and dissolved in dichloromethane (601 ^), trifluoroacetic acid (4.48,0.04111〇1), 50 ° (: the reaction was stirred for 10 hours to water (120 mL completion of the reaction, the reaction liquid was added to a stirred), cooled to crystallization 0 ° C for 3 hours and filtered to give the intermediate compound (2- [4- (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide), and then dissolved in tert-butanol (40 mL ), 5-chloro-2,3-diphenyl-pyrazine (9.68,0.036 11〇1), 4-dimethylaminopyridine (8.18,0.07111〇1), the reaction mixture was 110 ° (:! reaction was stirred for 14 hours the reaction was cooled to room temperature, water (15 mL), cooled to -10 ° C crystallization for 3 hours and filtered to give SIPA game music, as a white solid (13.5 g of), a yield of 90.5%, the reaction in this step is the same formula Example 1.
[0054] Example 4
[0055] A) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate:
[0056] 4 – [(tert-butoxycarbonyl) (isopropyl) amino] -1-butanol (15 (^, 0.065111〇1) and t-butyl bromoacetate (17.78,0.09111〇1) was dissolved in toluene (701] 11 ^), was added tetrabutylammonium hydrogen sulfate (0.888,2.61] 11] 1〇1), potassium carbonate (15.2 area, 0.1 lmol) and water (9.5 g of), the reaction mixture was stirred 40 ° C for 1.5 hours, the reaction solution was concentrated under reduced pressure and rotary evaporated to dryness and extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, a mixed solvent of ethanol and recrystallized from isopropanol to give [4- (tert-butoxycarbonyl) (isopropyl propyl) aminobutoxy] acetate, as a pale yellow oil (19.6 g of), in the same reaction formula in this step a yield of 87.5% in Example 1;
[0057] B) Preparation of [4_ (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid:
[0058] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetate (19 (^, 0.055! 11〇1) was dissolved in tert-butanol (60 mL), hydroxide solution of cesium (CsOH = 11. lg, 0.07mol; water, 8.0 g), the reaction was heated to 75 ° C for 6.5 hours cooled to room temperature, after treatment and purification, to give [4- (tert-butoxycarbonyl) (isopropyl ) aminobutoxy] acetic acid, an off-white solid (15.0 g of), a yield of 94.2%, the reaction of the present step is the same formula as in Example 1;
[0059] C) Preparation of 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide:
[0060] [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] acetic acid (15.0g, 0.05mol) and diazabicyclo 1,8_
[5.4.0] – | -7- dilute (9.5 region, 0.06111〇1) was dissolved in toluene (8〇1111 ^), with stirring, was added methyl sulfonamide (5.7 region, 0.06mo 1), the reaction mixture was 105 ° C for 16 hours, the reaction solution was concentrated by rotary evaporation to dryness, extracted with ethyl acetate, dried over magnesium sulfate, and concentrated by rotary evaporation to dryness, recrystallized from methanol to give 2- [4- (tert-butoxycarbonyl) (isopropyl ) aminobutoxy] -N- (methylsulfonyl) acetamide as an off-white solid (16.6 g of), 87.3% yield, this step is the same reaction scheme of Example 1;
[0061] D) Preparation of SIPA Seiler: 2- [4- (tert-butoxycarbonyl) (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide (16 (^, 0.04111〇. 1) and dissolved in ethyl acetate (^ 1,301,111), trifluoroacetic acid (5.78,0.051] 1〇1), 80 <€ the reaction was stirred for 5 hours to complete the reaction, the reaction was added dropwise to a stirred solution of water (150 mL), was cooled to 0 ° C crystallization for 3 hours and filtered to give the intermediate compound (2- [4- (isopropyl) aminobutoxy] -N- (methylsulfonyl) acetamide), then dissolved in isopropanol (50 mL), was added 5-chloro-2,3-diphenyl-pyrazine (13.58,0.05 11〇1!), a hoot dimethylaniline (12.28,0.10111〇1), the reaction mixture was 95 ° (: The reaction was stirred 12 hours, the reaction was cooled to room temperature, water (40 mL), cooled to -10 ° C crystallization for 3 hours and filtered to give SIPA game music, as a white solid (19.7 g of), a yield of 91.0%, the reaction step formula in Example 1.
//////////////
Selexipag (C26H32N4O4S, Mr = 496.6 g/mol) ist ein Diphenylpyrazin-Derivat. Es wird in der Leber zum aktiven Metaboliten ACT-333679 (MRE-269) biotransformiert. Selexipag unterscheidet sich strukturell von Prostazyklin und anderen Prostazylin-Rezeptor-Agonisten.

References
- 1 Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
- 2 New Drug Approved for Rare Lung Disorder. PPN. 23 Dec 2015 Has link to GRIPHON study results
- Kuwano et al. NS-304, an orally available and long-acting prostacyclin receptor agonist prodrug. J Pharmacol Exp Ther 2007;322:1181-1188.
- Kuwano et al. A long-acting and highly selective prostacyclin receptor agonist prodrug, NS-304, ameliorates rat pulmonary hypertension with unique relaxant responses of its active form MRE-269 on rat pulmonary artery. J Pharmacol Exp Ther 2008;326:691-699.
- Simonneau G, Lang I, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N. Selexipag, an oral, selective IP receptor agonist for the treatment of pulmonary arterial hypertension Eur Respir J 2012; 40: 874-880
- Mubarak KK. A review of prostaglandin analogs in the management of patients with pulmonary arterial hypertension. Respir Med 2010;104:9-21.
- Sitbon, O.; Morrell, N. (2012). “Pathways in pulmonary arterial hypertension: The future is here”. European Respiratory Review 21 (126): 321–327. doi:10.1183/09059180.00004812. PMID 23204120.
-
Publication numberPriority datePublication dateAssigneeTitleWO2016193994A12015-05-292016-12-08Megafine Pharma (P) Ltd.Amorphous selexipag and process for preparation thereofWO2017040872A12015-09-032017-03-09Teva Pharmaceuticals International GmbhSolid state forms of selexipagWO2017042731A12015-09-102017-03-16Lupin LimitedAmorphous form of selexipag and solid dispersion thereofWO2018008042A1 *2016-07-052018-01-11Maithri Drugs Private LimitedNovel process for the preparation of 2-{4-[(5,6-diphenyl pyrazin-2-yl)(isopropyl)amino]butoxy}-n-(methylsulfonyl)acetamide and novel polymorphs thereofWO2018015974A12016-07-202018-01-25Mylan Laboratories LimitedPolymorphic forms and amorphous solid dispersion of selexipagWO2018022704A12016-07-262018-02-01Teva Pharmaceuticals International GmbhCrystalline form vi of selexipagWO2018078383A12016-10-272018-05-03Cipla LimitedPharmaceutical composition comprising amorphous selexipagFamily To Family CitationsWO2017029594A1 *2015-08-172017-02-23Dr. Reddy’s Laboratories LimitedProcesses for preparation of selexipag and its amorphous formWO2017042828A3 *2015-09-102017-04-27Megafine Pharma (P) Ltd.Process for the preparation of selexipagWO2017168401A1 *2016-04-012017-10-05Honour (R&D)Process for the preparation of diphenylpyrazine derivativesEP3335699A12016-12-152018-06-20H e x a l AktiengesellschaftSelexipag formulation in liquisolid system
| Patent | Submitted | Granted |
|---|---|---|
| Methods of identifying critically ill patients at increased risk of development of organ failure and compounds for the treatment hereof [US8877710] | 2009-12-30 | 2014-11-04 |
| Form-I crystal of 2-{4-[N-(5,6-diphenylpyrazin-2-yl)-N-isopropylamino]butyloxy}-N-(methylsulfonyl)acetamide and method for producing the same [US8791122] | 2010-06-25 | 2014-07-29 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2015057325] | 2013-03-26 | 2015-02-26 |
| INHIBITION OF NEOVASCULARIZATION BY SIMULTANEOUS INHIBITION OF PROSTANOID IP AND EP4 RECEPTORS [US2014275200] | 2014-03-05 | 2014-09-18 |
| INHIBITION OF NEOVASCULARIZATION BY INHIBITION OF PROSTANOID IP RECEPTORS [US2014275238] | 2014-03-05 | 2014-09-18 |
| Fibrosis inhibitor [US8889693] | 2014-04-10 | 20 |
| Patent | Submitted | Granted |
|---|---|---|
| Heterocyclic compound derivatives and medicines [US7205302] | 2004-05-27 | 2007-04-17 |
| METHODS OF IDENTIFYING CRITICALLY ILL PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ORGAN FAILURE AND COMPOUNDS FOR THE TREATMENT HEREOF [US2014322207] | 2014-07-11 | 2014-10-30 |
| THERAPEUTIC COMPOSITIONS CONTAINING MACITENTAN [US2014329824] | 2014-07-18 | 2014-11-06 |
| Sustained Release Composition of Prostacyclin [US2014303245] | 2012-08-10 | 2014-10-09 |
| COMPOUNDS CAPABLE OF MODULATING/PRESERVING ENDOTHELIAL INTEGRITY FOR USE IN PREVENTION OR TREATMENT OF ACUTE TRAUMATIC COAGULOPATHY AND RESUSCITATED CARDIAC ARREST [US2013261177] | 2011-09-30 | 2013-10-03 |
| METHODS OF TREATMENT OF PATIENTS AT INCREASED RISK OF DEVELOPMENT OF ISCHEMIC EVENTS AND COMPOUNDS HEREOF [US2013040898] | 2011-04-29 | 2013-02-14 |
| Substituted Diphenylpyrazine Derivatives [US2013005742] | 2010-08-06 | 2013-01-03 |
| USE OF FORM-I CRYSTAL OF 2–N-(METHYLSULFONYL)ACETAMIDE [US2014148469] | 2014-01-22 | 2014-05-29 |
| CRYSTALS OF 2- {4- [N- (5,6-DIPHENYLPYRAZIN-2-YL) -N-ISOPROPYLAMINO]BUTYLOXY}-N- (METHYLSULFONYL) ACETAMIDE [US2014155414] | 2014-01-22 | 2014-06-05 |
| PROSTACYCLIN AND ANALOGS THEREOF ADMINISTERED DURING SURGERY FOR PREVENTION AND TREATMENT OF CAPILLARY LEAKAGE [US2014044797] | 2012-03-30 | 2014-02-13 |
 |
|
| Names | |
|---|---|
| IUPAC name
2-{4-[(5,6-diphenylpyrazin-2-yl)(propan-2-yl)amino]butoxy}-N-(methanesulfonyl)acetamide
|
|
| Other names
ACT-293987, NS-304
|
|
| Identifiers | |
| 475086-01-2 |
|
| ChEMBL | ChEMBL238804 |
| ChemSpider | 8089417 |
| 7552 | |
| Jmol interactive 3D | Image |
| KEGG | D09994 |
| PubChem | 9913767 |
| UNII | P7T269PR6S |
| Properties | |
| C26H32N4O4S | |
| Molar mass | 496.6 g·mol−1 |
//////////ACT-333679, MRE-269, Selexipag, セレキシパグ , UNII-5EXC0E384L, селексипаг , سيليكسيباق , Orphan Drug, fda 2015, NS 304, ACT 293987, Uptravi, EU 2016,
CC(C)N(CCCCOCC(=O)NS(=O)(=O)C)C1=CN=C(C(=N1)C2=CC=CC=C2)C3=CC=CC=C3
Selexipag (Uptravi)

-
24.Asaki, T.; Kuwano, K.; Morrison, K.; Gatfield, J.; Hamamoto, T.; Clozel, M. Selexipag: An Oral and Selective IP Prostacyclin Receptor Agonist for the Treatment of Pulmonary Arterial Hypertension J. Med. Chem. 2015,58, 7128– 7137 DOI: 10.1021/acs.jmedchem.5b00698
-
25.Skoro-Sajer, N.; Lang, I. M. Selexipag for the Treatment of Pulmonary Arterial Hypertension Expert Opin. Pharmacother. 2014, 15, 429– 436 DOI: 10.1517/14656566.2014.876007
-
26.Karmas, G.; Spoerri, P. E. The Preparation of Hydroxypyrazines and Derived Chloropyrazines J. Am. Chem. Soc. 1952, 74, 1580– 1584 DOI: 10.1021/ja01126a070
-
27.Asaki, T.; Hamamoto, T.; Sugiyama, Y.; Kuwano, K.; Kuwabara, K. Structure-activity Studies on Diphenylpyrazine Derivatives: a Novel Class of Prostacyclin Receptor Agonists Bioorg. Med. Chem. 2007,15, 6692– 6704 DOI: 10.1016/j.bmc.2007.08.010
VORAPAXAR SULPHATE

VORAPAXAR
Thrombosis, Antiplatelet Therapy, PAR1 Antagonists , MERCK ..ORIGINATOR
Ethyl N-[(3R,3aS,4S,4aR,7R,8aR,9aR)-4-[(E)-2-[5-(3-fluorophenyl)-2-pyridyl]vinyl]-3-methyl-1-oxo-3a,4,4a,5,6,7,8,8a,9,9a-decahydro-3H-benzo[f]isobenzofuran-7-yl]carbamate
Ethyl ((1R,3aR,4aR,6R,8aR,9S,9aS)-9-((1E)-2-(5-(3-fluorophenyl)pyridin-2-yl)ethenyl)- 1-methyl-3-oxododecahydronaphtho(2,3-c)furan-6-yl)carbamate
Carbamic acid, ((1R,3aR,4aR,6R,8aR,9S,9aS)-9-((1E)-2-(5-(3-fluorophenyl)-2- pyridinyl)ethenyl)dodecahydro-1-methyl-3-oxonaphtho(2,3-c)furan-6-yl)-, ethyl ester
618385-01-6 CAS NO FREE FORM
CAS Number: 705260-08-8 SULPHATE
Has antiplatelet activity.
- UNII-ZCE93644N2
- Zontivity
Registered – 2015 MERCK Thrombosis
Vorapaxar (formerly SCH 530348) is a thrombin receptor (protease-activated receptor, PAR-1) antagonist based on the natural product himbacine. Discovered by Schering-Plough and currently being developed by Merck & Co., it is an experimental pharmaceutical treatment for acute coronary syndrome chest pain caused by coronary artery disease.[1]
In January 2011, clinical trials being conducted by Merck were halted for patients with stroke and mild heart conditions.[2] In a randomized double-blinded trial comparing vorapaxar with placebo in addition to standard therapy in 12,944 patients who had acute coronary syndromes, there was no significant reduction in a composite end point of death from cardiovascular causes, myocardial infarction, stroke, recurrent ischemia with rehospitalization, or urgent coronary revascularization. However, there was increased risk of major bleeding.[3]
A trial published in February 2012, found no change in all cause mortality while decreasing the risk of cardiac death and increasing the risk of major bleeding.[4]
SCH-530348 is a protease-activated thrombin receptor (PAR-1) antagonist developed by Schering-Plough and waiting for approval in U.S. for the oral secondary prevention of cardiovascular events in patients with a history of heart attack and no history of stroke or transient ischemic attack. The drug candidate is being investigated to determine its potential to provide clinical benefit without the liability of increased bleeding; a tendency associated with drugs that block thromboxane or ADP pathways. In April 2006, SCH-530348 was granted fast track designation in the U.S. for the secondary prevention of cardiovascular morbidity and mortality outcomes in at-risk patients.
Vorapaxar was recommended for FDA approval on January 15, 2014.[5]
Vorapaxar is a protease-activated thrombin receptor (PAR-1) antagonist developed by Schering-Plough (now, Merck & Co.) and approved in the U.S. in 2014 for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction or with peripheral arterial disease. However, in 2018 Aralez discontinued U.S. commercial operations. In 2015, the product was approved in the E.U. for the reduction of atherothrombotic events in adult patients with a history of myocardial infarction. In April 2006, vorapaxar was granted fast track designation in the U.S. for the secondary prevention of cardiovascular morbidity and mortality outcomes in at-risk patients. In 2016, Aralez Pharmaceuticals acquired the U.S. and Canadian rights to the product pursuant to an asset purchase agreement entered into between this company and Merck & Co.
Merck & Co (following its acquisition of Schering-Plough) has developed and launched vorapaxar (Zontivity; SCH-530348; MK-5348), an oral antagonist of the thrombin receptor (protease-activated receptor-1; PAR1); the product is marketed in the US by Aralez Pharmaceuticals
WO-03089428, published in October 2003, claims naphtho[2,3-c]furan-3-one derivatives as thrombin receptor antagonists. WO-03033501 and WO-0196330, published in April 2003 and December 2001, respectively, claim himbacine analogs as thrombin receptor antagonists. WO-9926943 published in June 1999 claims tricyclic compounds as thrombin receptor antagonists
 VORAPAXAR
VORAPAXAR17 JAN 2014
FDA advisory panel votes to approve Merck & Co’s vorapaxar REF 6
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204886Orig1s000ChemR.pdf
Zontivity (vorapaxar) tablets NDA 204886



VORAPAXAR SULPHATE

CAS Number: 705260-08-8 SULPHATE
Molecular Formula: C29H33FN2O4.H2O4S
Molecular Weight: 590.7
Chemical Name: Ethyl [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)pyridin-2- yl]ethenyl]-1-methyl-3-oxododecahydronaphtho[2,3-c]furan-6-yl]carbamate sulfate
Synonyms: Carbamic acid, [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-[(1E)-2-[5-(3-fluorophenyl)-2- pyridinyl]ethenyl]dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-,ethyl ester,sulfate; SCH-530348
Vorapaxar Sulfate (SCH 530348) a thrombin receptor (PAR-1) antagonist for the prevention and treatment of atherothrombosis.
POLYMORPH
U.S.Pat. No. 7,304,078 discloses Vorapaxar base. U.S.Pat. No. 7,235,567 discloses Polymorph I and II of vorapaxar sulphate
CN 106478608 provides a crystalline polymorph A
EMA
Atherosclerosis and ischemic cardiovascular (CV) diseases like coronary artery disease (CAD) are progressive systemic disorders in which clinical events are precipitated by episodes of vascular thrombosis. Patients with an established history of atherothrombotic or athero-ischemic disease are at particular risk of future cardiac or cerebral events, and vascular death. Anti-thrombotic therapy options in patients with stable atherosclerosis are not well-established. Long-term therapies to effectively modulate the key components responsible for atherothrombosis in secondary prevention of ischemic CV disease are therefore required. Vorapaxar is a first – in – class selective antagonist of the protease-activated receptor 1 (PAR-1), the primary thrombin receptor on human platelets, which mediates the downstream effects of this critical coagulation factor in hemostasis and thrombosis. Thrombin-induced platelet activation has been implicated in a variety of cardiovascular disorders including thrombosis, atherosclerosis, and restenosis following percutaneous coronary intervention (PCI). As an antagonist of PAR-1, vorapaxar blocks thrombin-mediated platelet aggregation and thereby has the potential to reduce the risk of atherothrombotic complications of coronary disease. The applicant has investigated whether a new class of antiplatelet agents, PAR-1 antagonists, can further decrease the risk of cardiovascular events in a population of established atherothrombosis when added to standard of care, in secondary prevention of ischemic diseases. The following therapeutic indication has been submitted for vorapaxar: Vorapaxar is indicated for the reduction of atherothrombotic events in patients with a history of MI. Vorapaxar has been shown to reduce the rate of a combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization. Vorapaxar will be contraindicated in patients with a history of stroke or TIA. The indication sought in the current application is supported by the efficacy results of the TRA 2P-TIMI, which is considered the pivotal trial for this indication. During the procedure, the applicant requested the possibility of extending the indication initially sought for, to extend it to the population of PAD patients. This request was discussed at the CHMP and not accepted by the Committee.
Introduction The finished product is presented as immediate release film-coated tablets containing 2.5 mg of vorapaxar sulfate as active substance per tablet, corresponding to 2.08 mg vorapaxar. Other ingredients are: lactose monohydrate, microcrystalline cellulose (E460), croscarmellose sodium (E468), povidone (E1201) , magnesium stearate (E572), hypromellose (E464), titanium dioxide (E171), triacetin (glycerol triacetate) (E1518), iron oxide yellow (E172), as described in section 6.1 of the SmPC. The product is available in Aluminium–Aluminium blisters (Alu-Alu) as described in section 6.5 of the SmPC.
General information The chemical name of the active substance vorapaxar sulfate is ethyl[(1R,3aR,4aR,6R,8aR,9S,9aS)- -9-{(1E)-2-[5-(3-fluorophenyl)pyridin-2-yl]ethen-1-yl}-1-methyl-3-oxododecahydronaphtho[2,3-c] furan-6-yl]carbamate sulfate, corresponding to the molecular formula C29H33FN2O4 • H2SO4 and has a relative molecular mass 590.7. It has the following structure:
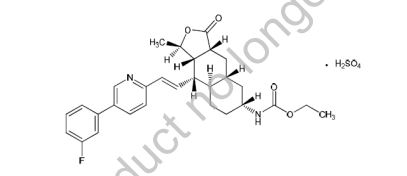
The structure of the active substance has been confirmed by mass spectrometry, infrared spectroscopy, 1H- and 13C-NMR spectroscopy and X-ray crystallography, all of which support the chemical structure elemental analysis. It appears as a white to off-white, slightly hygroscopic, crystalline powder. It is freely soluble in methanol and slightly soluble in ethanol and acetone but insoluble to practically insoluble in aqueous solutions at pH above 3.0. The highest solubility in aqueous solution can be achieved at pH 1.0 or in simulated gastric fluids at pH 1.4. The dissociation constant of vorapaxar sulfate was determined to be pKa = 4.7 and its partition coefficient LogP was determined to be 5.1. Vorapaxar sulfate contains seven chiral centers and a trans double bond. The seven chiral centres are defined by the manufacturing process of one of the intermediates in the vorapaxar synthesis and potential enantiomers are controlled by appropriate specifications. The cis-isomer of the double bond is controlled by a highly stereo-specific process reaction resulting in non-detectable levels of cis-isomer impurity. The cis-isomer impurity is controlled in one of the intermediates as an unspecified impurity. A single crystalline stable anhydrous form has been observed.
GENERAL INTRODUCTION
SIMILAR NATURAL PRODUCT
+ HIMBACINE


Himbacine is an alkaloid muscarinic receptor antagonist displaying more potent activity associated with M2 and M2 subtypes over M1 or M3. Observations show himbacine bound tightly to various chimeric receptors in COS-7 cells as well as possessed the ability to bind to cardiac muscarinic receptors allosterically. Recent studies have produced series of thrombin receptor (PAR1) antagonists derived from himbacine Himbacine is an inhibitor of mAChR M2 and mAChR M4.
| Physical State: | Solid |
| Derived from: | Australian pine Galbulimima baccata |
| Solubility: | Soluble in ethanol (50 mg/ml), methanol, and dichloromethane. Insoluble in water. |
| Storage: | Store at -20° C |
| Melting Point: | 132-134 °C |
| Boiling Point: | 469.65 °C at 760 mmHg |
| Density: | 1.08 g/cm3 |
| Refractive Index: | n20D 1.57 |
| Optical Activity: | α20/D +51.4º, c = 1.01 in chloroform |
| Application: | An alkaloid muscarinic receptor antagonist |
| CAS Number: | 6879-74-9 |
| Molecular Weight: | 345.5 |
| Molecular Formula: | C22H35NO2 |
General scheme:

PATENT
WO 2006076415
WO 2006076452
WO 2003089428
US 6063847
CN 107540564
WO 2008005344
CN 106749138
PATENT
Example 1:
[0027] The steel shed amide (300mg, 7. 93mmol) and 15 blood THF was added to 100 blood Ξ jar. The starting material II (2.OOg, 5. 89mmol) was dissolved in 15mL of THF dropwise via pressure-equalizing dropping funnel to the reaction system, the process temperature will produce a large number of bubbles -2 ~ 0 ° C, in the process, Lan mix of about 0.1 until no bubbles generate. THF solution containing 13 Blood Ship (0.75 Yap, 2. 95mmol) is transferred to a pressure-equalizing dropping funnel. It was slowly added dropwise to the reaction system. After the completion of dropwise continue to embrace mix ratio. After the treatment, at 0 ° C under 0.8 blood, Imol / L 1 fat slowly dropped into the embrace mixed reaction system, after adding the right amount of water, acetic acid extraction. The combined organic phase with Imol / L of 0H (17mLX3) washing the organic phase coating. Tu brine, dried over anhydrous sulfate steel, 25 ° C under reduced pressure to spin dry to give 1. 75g light yellow oil, yield 91%.
[0028] After the content was determined using the external standard method, first prepared by a qualified reference determine its content, W this as a standard substance, measuring the external standard method to get the content of 99%.
[0029] Zan NMR: (400MHz, CD3CN):… 5 46 of r, 1H), 4 70 (td, 1H), 4 03 based 2H), 3 69-3 57 (m, 2 Η).. , 3. 45-3. 32 (based, IH), 2. 77 (br, IH), 2. 61-2. 51 (m, IH), 2. 49-2. 39 (m, 1 field, 2 30 of r IH), 2 .12-1. 92 (m, IH), 1. 87 (dt, IH), 1. 81-1. 72 (m, IH), 1. 61-1. 50 ( …. m, IH), 1 48 (d, 3H), 1 23-1 09 (m, 7H), 1. 05-0 90 (m, 2H);
[0030] MS (ES +) m / z: 326. 24 [M + + field.
[Cited 00] Example 2:
[003 cited the steel shed amide (312mg, 8. 25mmol) and 16 blood THF was added to the lOOmL Ξ jar. The starting material II (2.OOg, 5. 89mmol) was dissolved in 15mL of THF dropwise via pressure-equalizing dropping funnel to the reaction system, the process temperature will produce a large number of bubbles -2 ~ -5 ° C, in the process and takes about 45min mix until no bubbles generate. The 13 ships of blood containing 60g, 2. 36mmol) in THF solution was transferred to a pressure-equalizing dropping funnel. It was slowly added dropwise to the reaction system. After the completion of dropwise continue to embrace mix ratio. After the treatment, at 0 ° C under 0.8 blood, Imol / L 1 fat slowly dropped into the embrace mixed reaction system, after adding the right amount of water, acetic acid extraction. The combined organic phase with llmol / L of 0H (17mLX3) washing the organic phase coating. Tu brine, dried over anhydrous sulfate steel, 25 ° C under reduced pressure to spin dry to give 1. 65g light yellow oil.
[0033] Determination of Reference Example 1 in an amount of 98.7%.
[0034] MS (ES +) m / z: 326. 24 [M + + field.
[003 cited Example 3:
[0036] 50 single jar of blood, condenser. Intermediate inb (l.〇〇g, 3. 07mmol) was dissolved in 10ml of dichloromethane burn during and after the blood was added to a 50-port flask, make dioxide of 32g, 3.68mmol), the reaction of reflux. After completion of the reaction by TLC, cooled to 20 ~ 25 ° C after suction filtration, the filter cake rinsed with methylene burning (the X3 3 blood), at 30 ° CW and the filtrate was concentrated to dryness. To the residue was added 5 blood acetic acid, at 20 ~ 25 ° C after mixing 0. embrace of suction, the resulting cake was vacuum dried at 30 ° C 10 ~ 12h. Give 0. 87g of white solid.
[0037] Electric NMR: (400MHz, CD3CN):. 9 74 oriented 1H), 5 40 of r, 1H), 4 77-4.66 (m, 1H), 4 09-3 98 (m, 2H…. ), 3. 49-3. 37 (m, IH), 2. 75-2. 64 (m, 2H), 2. 55-2. 48 (m, IH), 1. 95-1. 87 (m , 2H), 1. 89-1 .77 (m, 2H), 1. 61-1. 49 (m, IH), 1. 32-1. 13 (m, 9H), 1. 08-0. 82 (m, 2H);
[0038] MS (ES +) m / z: 324. 33 [M + + field.
PATENT
CN 106478608 crystal
https://patents.google.com/patent/CN106478608A/en
The present invention provides a crystalline polymorph A one kind of the compound of formula I:

In another embodiment, the present invention provides a method of preparing a crystalline polymorph of compound A I,

Which comprising, a) the compound II is dissolved in acetonitrile and stirred to form a mixture; b) heating the mixture to 50 ° C ~ 70 ° C; c) adding sulfuric acid to the heated mixture; d) evaluating the temperature was lowered to 0 ° C ~ 20 ° C, seeded and stirred to precipitate crystals.
Preparation [0042] A crystalline polymorph of the compound of Example 1 I

Compound II (1. 0g) was dissolved in 5. 0ml of acetonitrile, stirred and heated to 50 ° C ~ 70 ° C was added and this temperature was added 1.2ml 2N H2S04 / acetonitrile solution and then lowering the temperature of the system to 15 ° C ~ 20 ° C, the system was added to the appropriate amount of seed crystals and stirred for 2h, the precipitated solid was filtered and the cake washed twice with 2. 5ml of acetonitrile to give a white solid, the white solid was placed under 40 ° C desolventizing 2 hours and then dried at 80 ° C for vacuo to give a white solid 0. 83 g, 69. 3% yield, HPLC:. 99 94%. A powder X-ray diffraction spectrum shown in Figure 1, a DSC endothermic curve shown in Figure 2, which HPLC profile shown in Fig.
PATENT
https://patents.google.com/patent/CN106478608A/en
PATENT
WO 2009093972 synthesis
https://encrypted.google.com/patents/WO2009093972A1?cl=ko&hl=en&output=html_text
Clip
Vorapaxar sulfate (Zontivity)
Merck Sharp & Dohme successfully obtained approval in the EU in 2014 for vorapaxar sulfate, marketed as Zontivity. The drug is a first-in-class thrombin receptor (also referred to as a protease-activated or PAR-1) antagonist which, when used in conjunction with antiplatelet therapy, has been shown to reduce the chance of
myocardial infarction and stroke, particularly in patients with a history of cardiac events.277
Antagonism of PAR-1 allows for thrombin-mediated fibrin deposition while blocking thrombinmediated platelet activation.277 Although a variety of papers and patents describe the synthesis of vorapaxar sulfate (XXXVII),278–282 a combination of two patents describe the largest-scale synthesis reported in the literature, and this is depicted in Scheme 52.


Retrosynthetically, the drug can be divided into olefination partners 306 and 305.283,284 Lactone 305
is further derived from synthons 300 and 299, which are readily prepared from commercially available starting materials. Dienyl acid 300 was constructed in two steps starting from commercial vinyl bromide 307, which first undergoes a Heck reaction with methacrylate (308) followed by saponification of the ester to afford the desired acid 300 in 71% over two steps (Scheme 53).
The synthesis of alcohol 299 begins with tetrahydropyranyl (THP) protection of enantioenriched alcohol 295 to afford butyne 297 (Scheme 52). Lithiation of this system followed by trapping with (benzyloxy)chloroformate and Dowex work-up to remove the protective functionality provided acetyl ester 298. Hydrogenation of the alkyne with Lindlar’s catalyst delivered cis-allylic alcohol 299 in 93% yield. Acid 300 was then esterified with alcohol 299 by way of a 1,3-dicyclohexylcarbodiimide (DCC) coupling and, upon heating in refluxing xylenes, an intramolecular Diels–
Alder reaction occurred. Subsequent subjection to DBU secured the tricyclic system 301 in 38% over three steps as a single enantiomer.
Diastereoselective hydrogenation reduced the olefin with concomitant benzyl removal to give key fragment 302. Next, acidic revelation of the ketone followed by reductive amination with ammonium formate delivered primary amines 303a/303b as a mixture of diastereomers. These amines were then converted to the corresponding carbamates, and resolution by means of recrystallization yielded 50% of 304 as the desired diastereomer. Acid 304
was treated with oxalyl chloride and the resulting acid chloride was reduced to aldehyde 305 in 66% overall yield. Finally, deprotonation of phosphonate ester 306 (whose synthesis is described in Scheme 54) followed by careful addition of 305 and acidic quench delivered vorapaxar sulfate (XXXVII) in excellent yield over the
two-step protocol.
The preparation of vorapaxar phosponate ester 306 (Scheme 54)commenced from commercial sources of 5-(3-fluorophenyl)-2-methylpyridine (310). Removal of the methyl proton with LDA followed by quench with diethyl chlorophosphonate resulted in phosponate ester 306.

277. Frampton, J. E. Drugs 2015, 75, 797.
278. Chackalamannil, S.; Wang, Y.; Greenlee, W. J.; Hu, Z.; Xia, Y.; Ahn, H.; Boykow,G.; Hsieh, Y.; Palamanda, J.; Agans-Fantuzzi, J.; Kurowski, S.; Graziano, M.;Chintala, M. J. Med. Chem. 2008, 51, 3061.
279. Sudhakar, A.; Kwok, D.; Wu, G. G.; Green, M. D. WO Patent 2006076452A2,2006.
280. Wu, G. G.; Sudhakar, A.; Wang, T.; Ji, X.; Chen, F. X.; Poirier, M.; Huang, M.;Sabesan, V.; Kwok, D.; Cui, J.; Yang, X.; Thiruvengadam, T.; Liao, J.; Zavialov, I.;Nguyen, H. N.; Lim, N. K. WO Patent 2006076415A2, 2006.
281. Yong, K. H.; Zavialov, I. A.; Yin, J.; Fu, X.; Thiruvengadam, T. K. US Patent20080004449A1, 2008.
282. Chackalamannil, S.; Clasby, M.; Greenlee, W. J.; Wang, Y.; Xia, Y.; Veltri, E.;Chelliah, M. WO Patent 03089428A1, 2003.
283. Thiruven-Gadam, T. K.; Wang, T.; Liao, J.; Chiu, J. S.; Tsai, D. J. S.; Lee, H.; Wu,W.; Xiaoyong, F. WO Patent 2006076564A1, 2006.
284. Chackalamannil, S.; Asberon, T.;Xia, Y.; Doller, D.; Clasby, M. C.; Czarniecki,M. F. US Patent 6,063,847, 2000.
PRODUCT PATENT
SYNTHESIS
Inventor Samuel ChackalamannilMartin C. ClasbyWilliam J. GreenleeYuguang WangYan XiaEnrico P. VeltriMariappan ChelliahWenxue Wu
Original Assignee Schering Corporation
Priority date 2002-04-16
THE EXACT BELOW COMPD IS 14
Example 2
Step 1 :

Phosphonate 7, described in US 6,063,847, (3.27 g, 8.1 mmol) was dissolved in THF (12 ml) and C(O)Oled to 0 °C, followed by addition of 2.5 M n- BuLi (3.2 ml, 8.1 mmol). The reaction mixture was stirred at 0 °C for 10 min and warmed up to rt. A solution of aldehyde 6, described in US 6,063,847, in THF (12 ml) was added to the reaction mixture. The reaction mixture was stirred for 30 min. Standard aqueous work-up, followed by column chromatography (30-50% EtOAc in hexane) afforded product 8. 1HNMR (CDCI3): δ 0.92-1.38 (m, 31 H), 1.41 (d, J= 6 Hz, 3H), 1.40-1.55 (m, 2H), 1.70-1.80 (m, 2H), 1.81-1.90 (m, 2H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.89 (m, 4H), 4.75 (m, 1 H), 6.28-6.41 (m, 2H), 7.05-7.15 (m, 2H), 8.19 (br s, 1 H). Step 2:

Compound 8 (2.64 g, 4.8 mmol) was dissolved in THF (48 ml). The reaction mixture was C(O)Oled to 0 °C followed by addition of 1 M TBAF (4.8 ml). The reaction mixture was stirred for 5 min followed by standard aqueous work-up. Column chromatography (50% EtOAc/hexane) afforded product 9 (1.9 g, 100%). 1HNMR (CDCI3): δ 1.15-1.55 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.70-1.82 (m, 3H), 1.85-1.90 (m, 1 H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.18- 6.45 (m, 2H), 7.19 (br s, 2H), 8.19 (br s, 1 H). Step 3:

To a solution of compound 9 (250 mg, 0.65 mmol) in pyridine (5 ml) C(O)Oled to 0 °C was added Tf2O (295 μL, 2.1 mmol). The reaction mixture was stirred overnight at rt. Standard aqueous work-up followed by column chromatography afforded product 10 (270 mg, 80%). 1HNMR (CDCI3): δ 1.15-1.55 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.70-1.82 (m, 3H), 1.85-1.90 (m, 1 H), 2.36 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.42-6.68 (m, 2H), 7.25 (m, 1 H), 7.55 (m, 1 H), 8.49 (d, J= 2.8 Hz, 1 H).

Compound 10 (560 mg, 1.1 mmol), 3-fluorophenyl boronic acid (180 mg, 1.3 mmol) and K2CO3 (500 mg, 3.6 mmol) were mixed with toluene (4.4 ml), H2O (1.5 ml) and EtOH (0.7 ml) in a sealed tube. Under an atmosphere of N2, Pd(Ph3P)4 (110 mg, 0.13 mmol) was added. The reaction mixture was heated at 100 °C for 2 h under N2. The reaction mixture was C(O)Oled down to rt, poured to EtOAc (30 ml) and washed with water (2X20 ml). The EtOAc solution was dried with NaHCO3 and concentrated at reduced pressure to give a residue. Preparative TLC separation of the residue (50% EtOAc in hexane) afforded product 11 (445 mg, 89%). 1HNMR (CDCI3): δ 1.15-1.59 (m, 6H), 1.43 (d, J= 6 Hz, 3H), 1.70-1.79 (m, 2H), 1.82 (m, 1H), 1.91 (m, 2H), 2.41 (m, 2H), 2.69 (m, 1 H), 3.91 (m, 4H), 4.75 (m, 1 H), 6.52-6.68 (m, 2H), 7.15 (m, 1 H), 7.22 (m, 2H), 7.35 (m, 1 H), 7.44 (m, 1 H), 7.81 (m, 1 H), 8.77 (d, J= 1.2 Hz, 1 H). Step 5:

Compound 11 (445 mg, 0.96 mmol) was dissolved in a mixture of acetone (10 ml) and 1 N HCI (10 ml). The reaction mixture was heated at 50 °C for 1 h.
Standard aqueous work-up followed by preparative TLC separation (50% EtOAc in hexane) afforded product 12 (356 mg, 89%). 1HNMR (CDCI3): δ 1.21-1.45 (m, 2H), 1.47 (d, J= 5.6 Hz, 3H), 1.58-1.65 (m, 2H), 2.15 (m, 1 H), 2.18-2.28 (m, 2H), 2.35- 2.51 (m, 5H), 2.71 (m, 1 H), 4.79 (m, 1 H), 6.52-6.68 (m, 2H), 7.15 (m, 1 H), 7.22 (m, 2H), 7.35 (m, 1 H), 7.44 (m, 1 H), 7.81 (m, 1 H), 8.77 (d, J= 1.2 Hz, 1 H). Step 6:

Compound 12 (500 mg, 4.2 mmol) was dissolved in EtOH (40 ml) and CH2CI2 (15 ml) NH3 (g) was bubbled into the solution for 5 min. The reaction mixture was C(O)Oled to 0 °C followed by addition of Ti(O/‘Pr)4 (1.89 ml, 6.3 mmol). After stirring at 0 °C for 1 h, 1 M TiCI (6.3 ml, 6.3 mmol) was added. The reaction mixture was stirred at rt for 45 min and concentrated to dryness under reduced pressure. The residue was dissolved in CH3OH (10 ml) and NaBH3CN (510 mg, 8 mmol) was added. The reaction mixture was stirred overnight at rt. The reaction mixture was poured to 1 N NaOH (100 ml) and extracted with EtOAc (3x 100 ml). The organic layer was combined and dried with NaHC03. Removal of solvent and separation by PTLC (5% 2 M NH3 in CH3OH/ CH2CI2) afforded β-13 (spot 1 , 30 mg, 6%) and α-13 (spot 2, 98 mg, 20%). β-13: 1HNMR (CDCI3): δ 1.50-1.38 (m, 5H), 1.42 (d, J= 6 Hz, 3H), 1.51-1.75 (m, 5H), 1.84 (m, 2H), 2.38 (m, 1 H), 2.45 (m, 1 H), 3.38 (br s, 1 H), 4.78 (m, 1 H), 6.59 (m, 2H), 7.15 (m, 1 H), 7.26 (m, 2H), 7.36 (m, 1 H), 7.42 (m, 1 H), 7.82 (m, 1 H), 8.77 (d, J= 2 Hz, 1 H). α-13:1HNMR (CDCI3): δ 0.95 (m, 2H), 1.02-1.35 (m, 6H), 1.41 (d, J= 6 Hz, 3H), 1.82-1.95 (m, 4H), 2.37 (m; 2H), 2.69 (m, 2H), 4.71 (m, 1 H), 6.71 (m, 2H), 7.11 (m, 1 H), 7.25 (m, 2H), 7.38 (m, 1 H), 7.42 (m, 1 H), 7.80 (m, 1 H), 8.76 (d, J= 1.6 Hz, 1 H). Step 7:
Compound α-13 (300 mg, 0.71 mmol) was dissolved in CH2CI2 (10 ml) followed by addition of Et3N (0.9 ml). The reaction mixture was C(O)Oled to 0 °C and ethyl chloroformate (0.5 ml) was added. The reaction mixture was stirred at rt for 1 h. The reaction mixture was directly separated by preparative TLC (EtOAc/ hexane, 1 :1) to give the title compound (14) VORAPAXAR (300 mg, 86%). MS m/z 493 (M+1).
HRMS Calcd for C29H34N2O4F (M+1 ): 493.2503, found 493.2509.
PATENT
SYNTHESIS 1
http://www.google.com/patents/WO2006076564A1
VORAPAXAR= COMPD A
Example 6 – Preparation of Compound A

To a three-neck flask equipped with an agitator, thermometer and nitrogen inertion was added 7A (13.0 g), THF (30 mL). The mixture was cooled to below -200C after which lithium diisopropylamide (2M, 20 mL) was slowly added. The reaction mixture was agitated for an additional hour (Solution A). To another flask was added 6 (10.0 g) and THF (75 mL) . The mixture was stirred for about 30 minutes and then slowly transferred into the solution A while maintaining the temperature below 200C. The mixture was stirred at below -200C for an additional hour before quenching the reaction by adding 20 mL of water. The reaction mixture was warmed to 00C and the pH was adjusted to about 7 by addition of 25% HaSO4 (11 mL). The mixture was further warmed to 200C and then diluted with 100 mL of ethyl acetate and 70 mL of water. The two phases that had formed were separated and the aqueous layer was extracted with 50 mL of ethyl acetate. The solvents THF and ethyl acetate were then replaced with ethanol, and the Compound A was precipitated out as a crystalline solid from ethanol with seeding at 35 to 4O0C. After cooling to O0C, the suspension was stirred for an additional hour and then the product was filtered and washed with cold ethanol. The product was dried at 50 – 600C under vacuum to provide an off-white solid. VORAPAXAR
Yield: 12.7 g, (90%). m.p. 104.90C (DSC onset point).
1H NMR (CDCl3) δ 8.88 (d, J = 2.4 Hz, IH), 8.10 (dd, J = 8.2, 2.4 Hz, IH), 7.64 (IH), 7.61 (d, J = 8.8 Hz, IH), 7.55 (m, J = 8.2, 6.2 Hz, IH), 7.51 (d, J = 8.0 Hz, IH), 7.25 (dt, J = 9.0, 2.3 Hz, IH), 7.08 (d, J = 8.0 Hz, IH), 6.68 (dd, J = 15.4, 9.4 Hz, IH), 6.58 (d, J = 9.6 Hz, IH), 4.85 (dd, J = 14.2, 7.2 Hz, IH), 3.95 (dd, J = 14.2, 7.1 Hz, 2H), 3.29 (m, IH), 2.66 (m, J = 12.0, 6.4 Hz, IH), 2.33 (m, 2H), 1.76 (m, 4H), 1.30 (d, J = 5.6 Hz, 3H), 1.19 (m, 4H), 1.14 (t, J = 7.2 Hz, 3H), 0.98 (m, IH), 0.84 (m, IH). MS (EI) m/z: calcd. 492, found 492.
BISULPHATE SALT
Example 7 – Preparation of an Acid Salt (bisulfate) of Compound A:
Compound IA (5 g) was dissolved in about 25 mL of acetonitrile.
The solution was agitated for about 10 minutes and then heated to about 50 0C. About 6 mL of 2M sulfuric acid in acetonitrile was added into the heated reaction mixture. The solid salt of Compound A precipitated out during the addition of sulfuric acid in acetonitrile. After addition of sulfuric acid solution, the reaction mixture was agitated for 1 hour before cooling to room temperature. The precipitated solid was filtered and washed with about 30 mL of acetonitrile. The wet solid was dried under vacuum at room temperature for 1 hour and at 80 0C for about 12 hours to provide about 5 g white solid (yield 85%). m.p. 217.0 0C. 1H NMR (DMSO) 9.04 (s, IH), 8.60 (d, J = 8.1 Hz, IH), 8.10 (d, J = 8.2 Hz, IH), 7.76 (d, J = 10.4, IH), 7.71 (d, J = 7.8 Hz, IH), 7.60 (dd, J = 8.4, 1.8 Hz, IH), 7.34 (dd, 8.4, 1.8 Hz, IH), 7.08 (d, J = 8.0 Hz, IH), 7.02 (m, IH), 6.69 (d, J = 15.8 Hz, IH), 4.82 (m, IH), 3.94 (dd, J = 14.0, 7.0 Hz, 2H), 3.35 (brs, IH), 2.68 (m, IH), 2.38 (m, 2H), 1.80-1.70 (m, 4H), 1.27 (d, J = 5.8 Hz, 3H), 1.21 (m, 2H), 1.13 (t, J = 7.0 Hz, 3H), 0.95 (m, IH, 0.85 (m, IH). MS (EI) m/z calcd. 590, found 492.
INTERMEDIATE 6
Example 5- Preparation of Compound 6

To a three-neck flask equipped with an agitator, thermometer and nitrogen inert were added the crude product solution of Compound 5 (containing about 31 g. of Compound 5 in 300 mL solution) and anhydrous DMF (0.05 mL). After the mixture was agitated for 5 minutes, oxalyl chloride (12.2 mL) was added slowly while maintaining the batch temperature between 15 and 25°C. The reaction mixture was agitated for about an hour after the addition and checked by NMR for completion of reaction. After the reaction was judged complete, the mixture was concentrated under vacuum to 135 mL while maintaining the temperature of the reaction mixture below 300C. The excess oxalyl chloride was removed completely by two cycles of vacuum concentration at below 500C with replenishment of toluene (315 mL) each time, resulting in a final volume of 68 mL. The reaction mixture was then cooled to 15 to 25°C, after which THF (160 mL) and 2,6-lutidine (22 mL) were added. The mixture was agitated for 16 hours at 20 to 25°C under 100 psi hydrogen in the presence of dry 5% Pd/C (9.0 g). After the reaction was judged complete, the reaction mixture was filtered through celite to remove catalyst. More THF was added to rinse the hydrogenator and catalyst, and the reaction mixture was again filtered through celite. Combined filtrates were concentrated under vacuum at below 25°C to 315 mL. MTBE (158 mL) and 10% aqueous solution of phosphoric acid (158 mL) were added for a thorough extraction at 100C to remove 2,6- lutidine. Then phosphoric acid was removed by extracting the organic layer with very dilute aqueous sodium bicarbonate solution (about 2%), which was followed by a washing with dilute brine. The organic solution was concentrated atmospherically to a volume of 90 mL for solvent replacement. IPA (315 mL) was added to the concentrated crude product solution. The remaining residual solvent was purged to <_ 0.5% of THF (by GC) by repeated concentration under vacuum to 68 mL, with replenishment of IPA (315 mL) before each concentration. The concentrated (68 mL) IPA solution was heated to 50°C, to initiate crystallization. To this mixture n-heptane (68 mL) was added very slowly while maintaining the batch temperature at 50°C. The crystallizing mixture was cooled very slowly over 2.5 hours to 25°C. Additional n- heptane (34 mL) was added very slowly into the suspension mixture at 250C. The mixture was further cooled to 200C, and aged at that temperature for about 20 hours. The solid was filtered and washed with a solvent mixture of 25% IPA in n-heptane, and then dried to provide
19.5 g of a beige colored solid of Compound 6. (Yield: 66%) m.p. 169.30C. IH NMR (CD3CN) δ 9.74 (d, J = 3.03 Hz, IH), 5.42 (br, IH), 4.69 (m, IH), 4.03 (q, J = 7.02 Hz, 2H), 3.43 (qt, J = 3.80, 7.84 Hz, IH), 2.67 (m, 2H), 2.50 (dt, J = 3.00, 8.52 Hz, IH), 1.93 (d, J = 12.0 Hz, 2H), 1.82 (dt, J = 3.28, 9.75 Hz, 2H), 1.54 (qd, J = 3.00, 10.5 Hz, IH), 1.27 (d, J = 5.97 Hz, 3H), 1.20 (m, 6H), 1.03 – 0.92 (m, 2H). MS (ESI) m/z (M++1): calcd. 324, found 324.
INTERMEDIATE 7A
Example 4 – Preparation of Compound 7A
+ 1-Pr2NLi + (EtO)2POCI – + LiCI
![]()

7A
To a 10 L three-necked round bottomed flask equipped with an agitator, thermometer and a nitrogen inlet tube, was added 20Og of
Compound 8 (1.07 mol, from Synergetica, Philadelphia, Pennsylvania). THF (1000 mL) was added to dissolve Compound 8. After the solution was cooled to -80 0C to -50 0C, 2.0 M LDA in hexane/THF(1175 mL, 2.2 eq) was added while maintaining the batch temperature below -50 0C. After about 15 minutes of agitation at -800C to -50 0C, diethyl chlorophosphate (185 mL, 1.2 eq) was added while maintaining the batch temperature below -50 0C. The mixture was agitated at a temperature from -800C to – 50 0C for about 15 minutes and diluted with n-heptane (1000 mL). This mixture was warmed up to about -35 0C and quenched with aqueous ammonium chloride (400 g in 1400 mL water) at a temperature below -10 0C. This mixture was agitated at -150C to -10 0C for about 15 minutes followed by agitation at 150C to 25 0C for about 15 minutes. The aqueous layer was split and extracted with toluene (400 mL). The combined organic layers were extracted with 2N hydrochloric acid (700 mL) twice. The product-containing hydrochloric acid layers were combined and added slowly to a mixture of toluene (1200 mL) and aqueous potassium carbonate (300 g in 800 mL water) at a temperature below 30 0C. The aqueous layer was extracted with toluene (1200 mL). The organic layers were combined and concentrated under vacuum to about 600 ml and filtered to remove inorganic salts. To the filtrate was added n-heptane (1000 ml) at about 55 0C. The mixture was cooled slowly to 40 0C, seeded, and cooled further slowly to -10 0C. The resulting slurry was aged at about -10 0C for 1 h, filtered, washed with n- heptane, and dried under vacuum to give a light brown solid (294 g, 85% yield), m.p. 52 0C (DSC onset point).1H NMR (CDCl3) δ 8.73 (d, J = 1.5 Hz, IH), 7.85 (dd, Ji = 8.0 Hz, J2 = 1.5 Hz, IH), 7.49 (dd, Ji = 8.0 Hz, J2 = 1.3 Hz, IH), 7.42 (m, IH), 7.32 (d, J = 7.8 Hz, IH), 7.24 (m, IH), 7.08 (dt, Ji = 8.3 Hz, J2 = 2.3 Hz, IH), 4.09 (m, 4H), 3.48 (d, J = 22.0 Hz, 2H), 1.27 (t, J = 7.0 Hz, 6H). MS (ESI) for M+H calcd. 324, found 324.
Example 3 – Preparation of Compound 5:

4 5
To a three-necked round bottomed flask equipped with an agitator, thermometer and a nitrogen inlet tube was added a solution of Compound 4 in aqueous ethanol (100 g active in 2870 ml). The solution was concentrated to about 700 ml under reduced pressure at 350C to 40°C to remove ethyl alcohol. The resultant homogeneous mixture was cooled to 200C to 300C and its pH was adjusted to range from 12 to 13 with 250 ml of 25% sodium hydroxide solution while maintaining the temperature at 20-300C. Then 82 ml of ethyl chloroformate was slowly added to the batch over a period of 1 hour while maintaining the batch temperature from 200C to 300C and aged for an additional 30 minutes. After the reaction was judged complete, the batch was acidified to pH 7 to 8 with 10 ml of concentrated hydrochloric acid (37%) and 750 ml of ethyl acetate. The pH of the reaction mixture was further adjusted to pH 2 to 3 with 35% aqueous hydrochloric acid solution. The organic layer was separated and the aqueous layer was extracted again with 750 ml of ethyl acetate. The combined organic layers were washed twice with water (200 ml) . Compound 5 was isolated from the organic layer by crystallization from ethyl acetate and heptane mixture (1: 1 mixture, 1500 ml) at about 700C to 80 0C. The solid was filtered at 500C to 60 °C, washed with heptane and then dried to provide an off-white solid (yield 50%). m.p. 197.7°C. 1HNMR (CD3CN) δ 5.31 (brs, IH), 4.67 (dt, J = 16.1, 5.9 Hz, IH), 4.03 (q, J = 7.1 Hz, 2H), 3.41 (m, IH), 2.55 – 2.70 (m, 2H), 1.87 – 1.92 (m, IH), 1.32 – 1.42 (m, IH), 1.30 (d, J = 5.92 Hz, 3H), 1.30 – 1.25 (m, 6H), 0.98 (qt, J = 15.7, 3.18 Hz, 2H). MS (ESI) M+l m/z calculated 340, found 340.
Example 2 – Preparation of Compound 4;

3 4
7.4 kg of ammonium formate was dissolved in 9L of water at 15- 250C, and then cooled to 0-100C. 8.9 kg of Compound 3 was charged at 0-150C followed by an addition of 89L of 2B ethyl alcohol. The batch was cooled to 0-50C 0.9 kg of 10% Palladium on carbon (50% wet) and 9 L of water were charged. The batch was then warmed to 18-280C and agitated for 5 hours, while maintaining the temperature between 18-28 0C. After the reaction was judged complete, 7 IL of water was charged. The batch was filtered and the wet catalyst cake was then washed with 8OL of water. The pH of the filtrate was adjusted to 1-2 with 4N aqueous hydrochloric acid solution. The solution was used in the next process step without further isolation. The yield is typically quantiative. m.p. 216.40C. IH NMR (D2O+1 drop HCl) δ 3.15 (m, IH), 2.76 (m, IH), 2.62 (m, IH), 2.48 (dd,J-5.75Hz, IH), 1.94 (m, 2H), 1.78 (m, 2H), 1.38 (m, 2H), 1.20 (m, 6H), 1.18 (m, IH), 0.98 (q,J=2.99Hz, IH).
Example 1 – Preparation of Compound 3

2B 3
To a reactor equipped with an agitator, thermometer and nitrogen, were added about 10.5 kg of 2B, 68 L of acetone and 68 L of IN aqueous hydrochloric acid solution. The mixture was heated to a temperature between 50 and 600C and agitated for about 1 hour before cooling to room temperature. After the reaction was judged complete, the solution was concentrated under reduced pressure to about 42 L and then cooled to a temperature between 0 and 50C. The cooled mixture was agitated for an additional hour. The product 3 was filtered, washed with cooled water and dried to provide an off-white solid (6.9 kg, yield 76%). m.p. 2510C. Η NMR (DMSO) δ 12.8 (s, IH), 4.72 (m, J = 5.90 Hz, IH), 2.58 (m, 2H), 2.40 (m, J = 6.03 Hz, 2H), 2.21 (dd, J = 19.0, 12.8 Hz, 3H), 2.05 (m, IH), 1.87 (q, J = 8.92 Hz, IH), 1.75 (m, IH), 1.55 (m, IH), 1.35 (q, J = 12.6 Hz, IH), 1.27 (d, J = 5.88 Hz, 3H). MS (ESI) M+l m/z calcd. 267, found 267.
NOTE
Compound 7A may be prepared from Compound 8 by treating Compound 8 with diethylchlorophosphate:

Compound 8 may be obtained by the process described by Kyoku, Kagehira et al in “Preparation of (haloaryl)pyridines,” (API Corporation, Japan). Jpn. Kokai Tokkyo Koho (2004). 13pp. CODEN: JKXXAF JP
2004182713 A2 20040702. Compound 8 is subsequently reacted with a phosphate ester, such as a dialkyl halophosphate, to yield Compound 7A. Diethylchlorophosphate is preferred. The reaction is preferably conducted in the presence of a base, such as a dialkylithium amide, for example diisopropyl lithium amide.
Paper
J Med Chem 2008, 51(11): 3061
http://pubs.acs.org/doi/abs/10.1021/jm800180e
The discovery of an exceptionally potent series of thrombin receptor (PAR-1) antagonists based on the natural product himbacine is described. Optimization of this series has led to the discovery of 4 (SCH 530348), a potent, oral antiplatelet agent that is currently undergoing Phase-III clinical trials for acute coronary syndrome (unstable angina/non-ST segment elevation myocardial infarction) and secondary prevention of cardiovascular events in high-risk patients.
Ethyl [(3aR,4aR,8aR,9aS)-9(S)-[(E)-2-[5-(3-fluorophenyl)-2-
pyridinyl]ethenyl]dodecahydro-1(R)-methyl-3-oxonaphtho[2,3-c]furan-6(R)-yl]carbamate (4).
4 (300 mg, 86%). MS m/z 493 (M+1).
HRMS Calcd for C29H34N2O4F
(M+1): 493.2503, found 493.2509; mp125 °C;
[]D20 6.6 (c 0.5, MeOH).
1HNMR (CDCl3):
http://pubs.acs.org/doi/suppl/10.1021/jm800180e/suppl_file/jm800180e-file002.pdf
0.88-1.18 (m, 5 H), 1.22-1.30 (m, 3 H), 1.43 (d, J = 5.85 Hz, 3 H), 1.88-2.10 (m, 4 H), 2.33-2.42 (m, 2 H),
2.75-2.67 (m, 1 H), 3.52-3.60 (m, 1 H), 4.06-4.14 (m, 2 H), 4.54-4.80 (m, 1 H), 4.71-4.77 (m, 1 H),
6.55-6.63 (m, 2 H), 7.07-7.12 (m, 1 H), 7.26-7.29 (m, 2 H), 7.34 (d, J = 8.05 Hz, 1 H), 7.41-7.46 (m, 1 H), 7.80-7.82 (m, 1 H), 8.76-8.71 (m, 1 H).
PATENT
IN 201621010411
An improved process for preparation of Vorapaxar intermediates and a novel polymorphic form of Vorapaxar
ALEMBIC PHARMACEUTICALS LIMITED
Vorapaxar Sulfate is indicated for the reduction of thrombotic cardiovascular events in patients with a history of myocardial infarction (MI) or with peripheral arterial disease (PAD). ZONTIVITY has been shown to reduce the rate of a combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization (UCR).
According to present invention Vorapaxar sulfate is synthesized from compound of formula 1.
wherein R1 and R2 are each independently selected from the group consisting of H, alkyl, alkenyl, alkynyl, alkoxy, cycloalkyl, aryl, alkylaryl, arylalkyl, and heteroaryl groups. Process for the preparation of compound of formula 1 is disclosed in U.S Pat. No. 7,605,275. It disclosed preparation of compound of formula 1 via cyclization of compound 2 in presence of solvent selected from xylene, N-methylpyrrolidinone, Dimethylsulfoxide, diphenyl ether, dimethylacetamide. This cyclization step takes approximately 6-8 hrs.
There is need to develop a process which takes less time for cyclization step to prepare compound of formula 1. Therefore, our scientist works tenaciously to develop process which takes approximately 1-2 hrs for cyclization of compound 1.
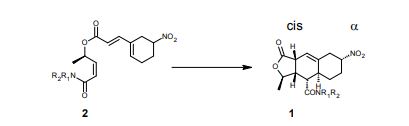
5 According to present invention Vorapaxar sulfate is synthesized from intermediate compound of formula-II.
Formula-II Compound of formula-II is critical intermediate in the preparation of Vorapaxar Sulfate.
10 Patent WO2006076415 discloses the process of preparation of above Formula-II in example 7, in which purification/crystallisation step involves treating the reaction mixture having compound of Formula-II with an ethanol/water mixture followed by azeotropic distillation of the mixture. This process yielded formula-II with low yields and with low purities. WO2009055416 (page 9, second paragraph) discloses that use of various solvent systems for
15 formula-II purification such as Methyl-tert-Butyl Ether (MTBE) and various solvent/antisolvent systems, for example, ethylacetate/heptane and toluene/heptane and by using these solvent systems, compound of formula-II are obtained as oil. These oils did not yield a reduced impurity profile in synthesis of the compound of Formula II, nor provide an improvement in the quality of the product compound of Formula II.
20 The inventors surprisingly found that using the process according to the invention provides formula-II with improved yield and high purity. Further, present invention provides a process for the preparation of novel crystalline form of Vorapaxar base. The present invention also relates to novel impurity and process for its preparation.
U.S.Pat. No. 7,304,078 discloses Vorapaxar base. U.S.Pat. No. 7,235,567 discloses Polymorph I and II of vorapaxar sulphate
Example 1- Preparation of compound 1a:
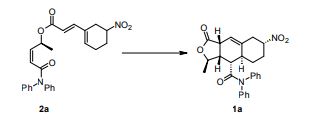
Process A: 5.0 g of compound 2a was suspended in 10.0 ml silicone oil at room temperature. The reaction mixture was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and 25 ml of cyclohexane was added to the reaction mass. The reaction mass was cooled slowly up to room temperature and stirred for 30 min.
15 The precipitated product was filtered off and washed with 5.0 ml Cyclohexane. Wet solid was suspended in mixture of 45.0 ml isopropyl alcohol and 20.0 ml denatured ethanol at 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried.
20 Process B: 5.0 g of compound 2a was suspended in 10.0 ml paraffin oil at room temperature. The reaction mixture was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and 25 ml of cyclohexane was added to the reaction mass. The reaction mass was cooled slowly up to room temperature and stirred for 30 min.
25 The precipitated product was filtered off and washed with 5.0 ml Cyclohexane. Wet solid was suspended in mixture of 45.0 ml isopropyl alcohol and 20.0 ml denatured ethanol at 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried. Yield: 4.3 g
Process C: 5.0 g of compound 2a was charged in reaction vessel at room temperature. The solid was then heated to 125°C and stirred for 30 min. Then reaction mass was further heated up to 150°C and stirred for 30 min. After completion of reaction, the reaction mass was cooled to 50-60°C and was added mixture of 45.0 ml isopropyl alcohol and 20.0 ml
5 denatured ethanol at 50-60°C. This was cooled to 40-45°C and further epimerized with 0.17 ml DBU. The crystallized solid was filtered off with suction, washed with mixture of 1.5 ml Isopropyl alcohol and 0.67 ml denatured ethanol and dried. Yield: 4.5 g Example 2: Preparation of Intermediate (Formula-II) of vorapaxar
10 Example 2(a): 50.0g of 1,3,3a,4,4a,5,6,7,8,9a-Decahydro-3-methyl-7-nitro-1-oxo-N,Ndiphenylnaphtho[2,3-c]furan-4-carboxamide compound was suspended in 300.0 ml THF, 15 g 10% Pd/C (50% wet) and 200 ml Process water at room temperature. The reaction mixture was heated to 45°C and drop wise formic acid (35 ml) was added and then stirred for 15 hrs. After completion of reaction, the reaction mass was cooled to 25-30°C and 100 ml THF was
15
added and pH was made acidic with 2M sulfuric acid solution. The reaction mass was filtered and washed with 150 ml THF, 150 ml water. Organic and aqueous layer were separated and aqueous layer was extracted with THF. Organic layers were combined and washed with water. The organic layer was cooled up to 5-10°C, 20 ml of TEA and 13 ml of Ethyl chloro formate were added. The reaction mass was stirred for 30 min. After completion of reaction,
20
reaction mass was washed with 2M sulfuric acid solution and distilled out reaction mass completely under vacuum. Acetonitrile (50 ml) was added to residue and heated up to 40- 45°C. Cooled the reaction mass up to 25-30°C and filtered the solid. Purity: 94-96% Example 2(b): Crystallization with Acetonitrile Acetonitrile (50 ml) was added to above obtained solid and heated to 40-45°C. Cooled the
25 reaction mass slowly up to 25-30°C and then up to 5-10°C. The reaction mass was stirred and the solid was filtered. XRD: Fig-1 Purity: 98-99% Example 2(c): Crystallization with Ethyl acetate To the solid obtained in example-1(a) Ethyl acetate (30 ml) was added. The reaction mass was heated up to 70-75°C and stirred for 10-15 min. The reaction mass was cooled slowly up 30 to 25-30°C and then up to 5-10°C. The reaction mass was stirred for 30 min. The solid was filtered and washed with Ethyl acetate. XRD: Fig-2 Purity: 98-99%
Example 3: Preparation of Amorphous Form of Vorapaxar base Vorapaxar base (10.0 g) was dissolved in 500 ml of 40% Ethyl acetate in Cyclohexane. The solvent was then completely removed under vacuum at 45-50o C to give a solid. Yield: 9.8 g
Example 3 (a): Preparation of crystalline vorapaxar base 5 (2-{[Ethyl (ethylperoxy)phosphory]methyl}-5-(3-fluorophenyl)pyridine) (10 g) was dissolved in THF (30ml) at 25±5°C under Nitrogen. Cool the reaction mass up to -30 to – 50°C. Add drop wise LDA (2.0 M solution in THF). After 1 hr add drop wise (N- [(1R,3aR,4aR,6R,8aR,9S,9aS)-9-formyl dodecahydro-1-methyl-3-oxonaphtho[2,3-c]furan-6- yl]-ethyl ester Carbamic acid) solution (10 g dissolved in 70 ml THF). After completion of 10 reaction mass quench the reaction mass to sulphuric acid solution. Separate the layers and distilled out organic layer under vacuum get foamy residue. (purity 82%) Add MIBK (10 ml) in above residue and stir it at 40-50°C till clear solution. Add drop wise n-Heptane (10 ml) and stir the reaction mass for 30 min. Gradually cool the reaction mass up to 25-30°C. Stir the reaction mass for 24 hrs. Filter the solid and washed it with n-Heptane (5.0 ml). Dry the 15 solid. Yield: 7.0 g. XRD: Fig-3 purity 96%
Example 3(b): Preparation of crystalline vorapaxar base Vorapaxar advance intermediate (2-{[Ethyl (ethylperoxy)phosphory]methyl}-5-(3- fluorophenyl)pyridine) (10 g) was dissolved in THF (30ml) at 25±5°C under Nitrogen. Cool the reaction mass up to -30 to -50°C. Add drop wise LDA (2.0 M solution in THF). After a 1
20 hr add drop wise VORA-Aldehyde (N-[(1R,3aR,4aR,6R,8aR,9S,9aS)-9-formyl dodecahydro1-methyl-3-oxonaphtho[2,3-c]furan-6-yl]-ethyl ester Carbamic acid) solution (10 g dissolved in 70 ml THF). After completion of reaction mass quench the reaction mass to sulphuric acid solution. Separate the layers and distilled out organic layer under vacuum get foamy residue (purity 82%). Add MTBE (10 ml) in above residue and stir it at 40-50°C till clear solution.
25 Add drop wise n-Heptane (30 ml) and stir the reaction mass for 30 min. Gradually cool the reaction mass up to 25-30°C. Stir the reaction mass for 24 hrs. Filter the solid and washed it with n-Heptane (5.0 ml). Dry the solid. Yield: 8.5.0 g. XRD: Fig-4 purity 97%
References
- Samuel Chackalamannil; Wang, Yuguang; Greenlee, William J.; Hu, Zhiyong; Xia, Yan; Ahn, Ho-Sam; Boykow, George; Hsieh, Yunsheng et al. (2008). “Discovery of a Novel, Orally Active Himbacine-Based Thrombin Receptor Antagonist (SCH 530348) with Potent Antiplatelet Activity”. Journal of Medicinal Chemistry 51 (11): 3061–4.doi:10.1021/jm800180e. PMID 18447380.
- Merck Blood Thinner Studies Halted in Select Patients, Bloomberg News, January 13, 2011
- Tricoci et al. (2012). “Thrombin-Receptor Antagonist Vorapaxar in Acute Coronary Syndromes”. New England Journal of Medicine 366 (1): 20–33.doi:10.1056/NEJMoa1109719. PMID 22077816.
- Morrow, DA; Braunwald, E; Bonaca, MP; Ameriso, SF; Dalby, AJ; Fish, MP; Fox, KA; Lipka, LJ; Liu, X; Nicolau, JC; Ophuis, AJ; Paolasso, E; Scirica, BM; Spinar, J; Theroux, P; Wiviott, SD; Strony, J; Murphy, SA; TRA 2P–TIMI 50 Steering Committee and, Investigators (Apr 12, 2012). “Vorapaxar in the secondary prevention of atherothrombotic events.”. The New England Journal of Medicine 366 (15): 1404–13. doi:10.1056/NEJMoa1200933.PMID 22443427.
- “Merck Statement on FDA Advisory Committee for Vorapaxar, Merck’s Investigational Antiplatelet Medicine”. Merck. Retrieved 16 January 2014.
- http://www.forbes.com/sites/larryhusten/2014/01/15/fda-advisory-panel-votes-in-favor-of-approval-for-mercks-vorapaxar/
- SCH-530348 (Vorapaxar) is an investigational candidate for the prevention of arterial thrombosis in patients with acute coronary syndrome and peripheral arterial disease. “Convergent Synthesis of Both Enantiomers of 4-Hydroxypent-2-ynoic Acid Diphenylamide for a Thrombin Receptor Antagonist Sch530348 and Himbacine Analogues.” Alex Zaks et al.: Adv. Synth. Catal. 2009, 351: 2351-2357 Full text;
- Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity
J Med Chem 2008, 51(11): 3061
- Stu Borman (2005). “Hopes Ride on Drug Candidates: Researchers reveal potential new medicines for thrombosis, anxiety, diabetes, and cancer”. Chemical & Engineering News 83 (16): 40–44.
PATENTS
- WO 2003089428
- WO 2006076452
- US 6063847
- WO 2006076565
- WO 2008005344
- WO2010/141525
- WO2008/5353
- US2008/26050
- WO2006/76564 mp, nmr
|
3-21-2012
|
EXO-SELECTIVE SYNTHESIS OF HIMBACINE ANALOGS
|
|
|
10-14-2011
|
EXO- AND DIASTEREO- SELECTIVE SYNTHESIS OF HIMBACINE ANALOGS
|
|
|
8-3-2011
|
Exo- and diastereo-selective syntheses of himbacine analogs
|
|
|
3-18-2011
|
COMBINATION THERAPIES COMPRISING PAR1 ANTAGONISTS WITH NAR AGONISTS
|
|
|
8-11-2010
|
Exo-selective synthesis of himbacine analogs
|
|
|
6-4-2010
|
SYNTHESIS Of DIETHYLPHOSPHONATE
|
|
|
5-12-2010
|
THROMBIN RECEPTOR ANTAGONISTS
|
|
|
3-31-2010
|
Synthesis of diethyl{[5-(3-fluorophenyl)-pyridine-2yl]methyl}phosphonate
|
|
|
12-4-2009
|
Local Delivery of PAR-1 Antagonists to Treat Vascular Complications
|
|
|
12-2-2009
|
SYNTHESIS OF HIMBACINE ANALOGS
|
|
10-21-2009
|
Exo- and diastereo- selective syntheses of himbacine analogs
|
|
|
6-31-2009
|
Synthesis of 3-(5-nitrocyclohex-1-enyl) acrylic acid and esters thereof
|
|
|
6-3-2009
|
Synthesis of himbacine analogs
|
|
|
1-23-2009
|
METHODS AND COMPOSITIONS FOR TREATING CARDIAC DYSFUNCTIONS
|
|
|
9-26-2008
|
REDUCTION OF ADVERSE EVENTS AFTER PERCUTANEOUS INTERVENTION BY USE OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
2-8-2008
|
IMMEDIATE-RELEASE TABLET FORMULATIONS OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
1-32-2008
|
SOLID DOSE FORMULATIONS OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
12-5-2007
|
Thrombin receptor antagonists
|
|
|
11-23-2007
|
THROMBIN RECEPTOR ANTAGONISTS
|
|
|
8-31-2007
|
THROMBIN RECEPTOR ANTAGONISTS AS PROPHYLAXIS TO COMPLICATIONS FROM CARDIOPULMONARY SURGERY
|
|
8-31-2007
|
CRYSTALLINE POLYMORPH OF A BISULFATE SALT OF A THROMBIN RECEPTOR ANTAGONIST
|
|
|
6-27-2007
|
Crystalline polymorph of a bisulfate salt of a thrombin receptor antagonist
|
|
|
8-4-2006
|
Preparation of chiral propargylic alcohol and ester intermediates of himbacine analogs
|
|
|
9-31-2004
|
Methods of use of thrombin receptor antagonists
|
| US6063847 * | Nov 23, 1998 | May 16, 2000 | Schering Corporation | Thrombin receptor antagonists |
| US6326380 * | Apr 7, 2000 | Dec 4, 2001 | Schering Corporation | Thrombin receptor antagonists |
| US20030216437 * | Apr 14, 2003 | Nov 20, 2003 | Schering Corporation | Thrombin receptor antagonists |
| US20040176418 * | Jan 9, 2004 | Sep 9, 2004 | Schering Corporation | Crystalline polymorph of a bisulfate salt of a thrombin receptor antagonist |
| WO2011128420A1 | Apr 14, 2011 | Oct 20, 2011 | Sanofi | Pyridyl-vinyl pyrazoloquinolines as par1 inhibitors |
//////////////fast track designation , VORAPAXAR, FDA 2014, EU 2016, Zontivity, NDA 204886, MERCK, VORAPAXAR SULPHATE
CCOC(=O)NC1CCC2C(C1)CC3C(C2C=CC4=NC=C(C=C4)C5=CC(=CC=C5)F)C(OC3=O)C
Pitolisant
Pitolisant
CAS 362665-56-3
FDA APPROVED 2019 AUG
1-(3-(3-(4-Chlorophenyl)propoxy)propyl)piperidine
MF C17H26ClNO
MW 295.1703
- HBS-101
- Pitolisant
- Tiprolisant
- UNII-4BC83L4PIY
(Wakix®)Approved EU 31/3/2016, Narcolepsy
A histamine H3 receptor antagonist/inverse agonist used to treat narcolepsy.
![]()
BF-2649; BF-2.649; FUB-649, Ciproxidine, Tiprolisant
CAS 362665-56-3, 362665-57-4 (oxalate)
CAS 903576-44-3(Pitolisant Hydrochloride)
APPROVED IN EU, European Medicine Agency (EMA) on Mar 31, 2016.
- BF 2.649
- BF 2649
- BF2.649
- Ciproxidine
- Pitolisant hydrochloride
- Tiprolisant
- UNII-YV33CH63HI
1-{3-[3-(4-Chlorophenyl)propoxy]propyl}piperidine hydrochloride (1:1)
| Molecular Weight | 332.31 |
| Formula | C17H26ClNO ● HCl |
![]()
Bioprojet INNOVATOR
Jean-Charles Schwartz, Jeanne-Marie Lecomte
- OriginatorBioprojet; Ferrer
- DeveloperAlium Medical; AOP Orphan Pharmaceuticals AG; Bioprojet; Ferox Therapeutics; Harmony Biosciences
- ClassNootropics; Piperidines; Sleep disorder therapies
- Mechanism of ActionHistamine H3 receptor antagonists; Histamine H3 receptor inverse agonists
- Orphan Drug StatusYes – Narcolepsy
- New Molecular EntityYes
Highest Development Phases
- MarketedNarcolepsy
- Phase IIIHypersomnia
- Phase IDrug abuse; Type 1 diabetes mellitus
- 15 Aug 2019Registered for Narcolepsy in USA (PO)
- 15 Aug 2019Harmony Biosciences intends to market pitolisant for excessive daytime sleepiness in patients with Narcolepsy in USA, in 4Q of 2019
- 19 Jun 2019Phase-I clinical trials in Type 1 diabetes mellitus in USA (PO) (NCT04026750)
- Pitolisant (INN), also known as tiprolisant (USAN),[1] is a medication in the United States that was approved by the FDA in August 2019. It was granted orphan designation for the treatment of narcolepsy, Fast Track designation for the treatment of excessive daytime sleepiness (EDS) and cataplexy in patients with narcolepsy, and Breakthrough Therapy designation for the treatment of cataplexy in patients with narcolepsy. Pitolisant, a first-in-class medication, is a potent and highly selective Histamine 3 (H₃) receptorantagonist/inverse agonist; it enhances the activity of histaminergic neurons in the brain that function to improve a patient’s wakefulness and inhibit attacks of cataplexy. It was designed and developed by Bioprojet, who has marketed the product in Europe since its approval by the European Medicines Agency in 2016. Pitolisant represents the first new therapy in the U.S. in over 15 years for the treatment of both EDS and cataplexy in adult patients with narcolepsy.The NDA (New Drug Submission) submission is based on results from the clinical development program in narcolepsy, which included over 300 patients, some of whom were treated for up to five years. It also included safety data in over 1500 patients across multiple patient populations. [1]It was developed by Jean-Charles Schwartz, Walter Schunack, and colleagues after the former discovered the H₃ receptor.[2] It was the first H₃ receptor inverse agonist to be tested in humans or introduced for clinical use.[2]
Pitolisant (INN) or tiprolisant (USAN) is a histamine receptor inverse agonist/antagonist selective for the H3 subtype.[1] It hasstimulant and nootropic effects in animal studies,[2] and may have several medical applications, having been researched for the treatment of narcolepsy, for which it has been granted orphan drug status in the EU and US.[3][4] It is currently in clinical trials forschizophrenia and Parkinson’s disease.[4][5][6]
Pitolisant hydrochloride was approved by European Medicine Agency (EMA) on Mar 31, 2016. It was developed and marketed as Wakix® by Bioprojet in EU.
Pitolisant is being developed by Bioprojet for the oral treatment of central nervous system disorders. Pitolisant is a selective histamine H3-receptor antagonist/inverse agonist which enhances the activity of histaminergic neurons. Pitolisant has been launched in several countries for the treatment of narcolepsy, and is approved in the US, EU, Iceland and Liechtenstein. Clinical development is underway for type-1 diabetes, hypersomnia and drug abuse in countries worldwide.
Phase III development was also conducted for the treatment of hypersomnia in Switzerland. Phase II development for attention-deficit hyperactivity disorder was conducted in France. However, there were no recent reports on development identified. Development in epilepsy and obesity has been discontinued.
Ferrer and Bioprojet appeared to have a co-development agreement for pitolisant that allowed the mutual use of both companies’ technical and scientific resources; however, as per Ferrer’s communication dated June 2016, the drug is no longer in its portfolio.

Pitolisant hydrochloride is an antagonist/inverse agonist of the histamine H3 receptor, which is indicated in adults for the treatment of narcolepsy with or without cataplexy.
Wakix® is available as tablet for oral use, containing 4.5 mg and 18 mg of Pitolisant hydrochloride. The initial dose of 9 mg (two 4.5 mg, tablets) per day, and it should be used at the lowest effective dose, depending on individual patient response and tolerance, according to an up-titration scheme, without exceeding the dose of 36 mg/day.
Pitolisant was developed by Jean-Charles Schwartz, Walter Schunack and colleagues after the former discovered H3 receptors.[7]Pitolisant was the first clinically used H3 receptor inverse agonist.
Pitolisant, also known as Tiprolisant, is a histamine receptor inverse agonist/antagonist selective for the H3 subtype. It has stimulant and nootropic effects in animal studies, and may have several medical applications, having been researched for the treatment of narcolepsy, for which it has been granted orphan drug status in the EU and US. It is currently in clinical trials for schizophrenia and Parkinson’s disease. Pitolisant was the first clinically used H3 receptor inverse agonist.

The European Medicines Agency (EMA) has recommended granting marketing authorization for pitolisant (Wakix, Bioprojet Pharma) for narcolepsy with or without cataplexy, the agency announced today.
Narcolepsy is a rare sleep disorder that affects the brain’s ability to regulate the normal sleep-wake cycle, leading to excessive daytime sleepiness, including the sudden urge to sleep, and disturbed night-time sleep. Some patients also experience sudden episodes of cataplexy, potentially causing dangerous falls and increasing the risks for accidents, including car accidents. Symptoms of narcolepsy can be severe and significantly reduce quality of life.
Pitolisant “will add to the available treatment options for narcolepsy. It is a first-in-class medicine that acts on histamine H3 receptors in the brain. This leads to increased histamine release in the brain, thereby enhancing wakefulness and alertness,” the EMA notes in a news release.
The EMA recommendation for approval of pitolisant is based on an evaluation of all available safety and efficacy data conducted by the Committee for Medicinal Products for Human Use (CHMP). The data include two pivotal placebo-controlled trials involving 259 patients, as well as one uncontrolled, open-label study involving 102 patients with narcolepsy and one supportive study in 105 patients.
The studies showed that pitolisant was effective in reducing excessive daytime sleepiness in patients with narcolepsy. The beneficial effect of the drug on cataplexy was demonstrated in one of the pivotal studies as well as in the supportive study.
No major safety concerns with pitolisant emerged in testing. Insomnia, headache, and nausea were among the most common adverse effects observed in the clinical trials, and the CHMP decided on measures to mitigate these risks, the EMA said. The CHMP also requested the company conduct a long-term safety study to further investigate the safety of the drug when used over long periods.
Pitolisant for narcolepsy received orphan designation from the Committee for Orphan Medicinal Products in 2007. Orphan designation provides medicine developers access to incentives, such as fee reductions for scientific advice, with the aim of encouraging the development of treatments for rare disorders.
The CHMP opinion will now be sent to the European Commission for the adoption of a decision on a European Union–wide marketing authorization. Once that has been granted, each member state will decide on price and reimbursement based on the potential role/use of this medicine in the context of its national health system.

Narcolepsy-cataplexy.
Narcolepsy-cataplexy, or Gelineau syndrome, is a rare but serious disorder characterized by excessive daytime sleepiness which can be an extreme hindrance to normal professional and social activities, and which is accompanied by more or less frequent attacks of cataplexy (a sudden loss of muscle tone triggered by emotions as varied as laughter or fear) and erratic episodes of REM sleep (during wakefulness and during sleep), sometimes associated with hypnagogic hallucinations. Moreover, individuals with narcolepsy have various degrees of cognitive impairment and tend to be obese (reviewed by Dauvilliers et al., Clin. Neurophysiol., 2003, 114, 2000; Baumann and Bassetti, Sleep Med. Rev., 2005, 9, 253).
The disorder is caused by the loss of a group of neurons in the brain which produce two peptides, orexins, also known as hypocretins, located in the anterior hypothalamus and projecting to the main groups of aminergic neurons which regulate wakefulness and sleep. Patients with the disorder generally have very low levels of orexins in cerebrospinal fluid. Orexin knock-out mice display many of the symptoms seen in narcoleptic subjects, confirming the role of these peptides and thereby providing an excellent animal model of the disease (Chemelli et al., Cell, 1999, 98, 437).
Several types of treatments which can improve the symptoms of narcolepsy already exist, although they do not completely relieve symptoms and, furthermore, can cause significant side effects limiting their usefulness.
For instance, amphetamines or analogues such as methylphenidate which release catecholamines are used to treated daytime sleepiness, but these agents induce a state of excessive excitation as well as cardiovascular disturbances and also carry a potential for drug addiction.
Modafinil, a drug whose mechanism of action is unclear, also improves daytime sleepiness without causing as many side effects as amphetamines. Nonetheless, its efficacy is limited and it can cause headaches and nausea, particularly at high doses. Moreover amphetamines and/or modafinil do not appear to improve some of the most disabling symptoms of the disease, particularly cataplexy attacks, cognitive deficits and weight gain. With regard to cataplexy, treatments include antidepressants and oxybate. Effectiveness of the former has not been demonstrated (Cochrane Database Syst. Rev., 2005, 20, 3), and the latter is a drug of illegal abuse and its use is restricted.
It has also been shown that histamine H3 receptor antagonists induce the activation of histaminergic neurons in the brain which release histamine, a neurotransmitter with a crucial role in maintaining wakefulness (Schwartz et al., Physiol. Rev. 1991, 71, 1).
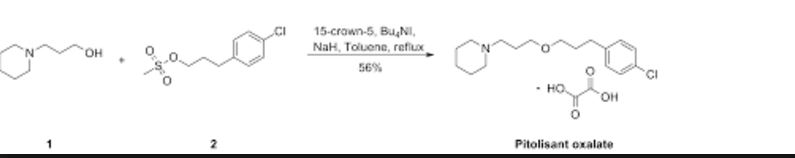
PATENT
Pharmaceutical products with histamine H3 receptor ligand properties and 0 subsequent pharmacological activities thereof are described in EP-980300. An especially important product among those disclosed is 1-[3-[3-(4- chlorophenyl)propoxy] propyl]-piperidine. This compound is disclosed as the free base and as the oxalate salt.
5 The use of 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine as the free base is limited because of its oily nature. On the contrary, 1-[3-[3-(4- chlorophenyl)propoxy]propyl]-piperidine oxalate is a crystalline substance but its low aqueous solubility (0.025 g/ml at 230C) also limits its use as a
pharmaceutical ingredient.
0
Subsequent patents EP-1100503 and EP-1428820 mention certain salts of 1- [3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine. However, the only one specifically described is the oxalate salt. The crystalline monohydrochloride salt is not described.
Example 1 : 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine
According to the method disclosed in EP-982300, Example 78, sodium 3-piperidinopropanolate (2.127 kg; 12.88 mol), 3-(4-chlorophenyl)propyl mesylate (1.121 kg; 4.51 mol) and 0.322 mol of 15-crown-5 in 4.5 kg of dry toluene were refluxed for 4 hours. The solvent was evaporated and the residue purified by column chromatography on silica gel (eluent: methylene chloride/methanol (90/10)). The obtained oil was distilled in a fractionating equipment at reduced pressure (0.3-0.7 mmHg) and with a heating jacket at 207-2100C. The head fractions and the distilled fraction at 0.001-0.010 mmHg with a jacket temperature of 180-2000C were collected. The obtained oil (1.0 kg; 3.38 mol) corresponds to 1-[3-[3-(4-chlorophenyl)propoxy] propyl]-piperidine. Yield 75%.
Example 2: 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine
monohydrochloride
Preparation
Distilled 1-[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine (1.0 kg) and anhydrous ethyl acetate (4.5 kg) are transferred to a 10-L glass vessel fitted with a cooling bath and a gas inlet. A stream of gaseous hydrogen chloride is bubbled in the reaction mixture at 20-250C.
The pH of the solution is checked by taking a 0.5 mL sample of the reaction mixture and diluting it with 5 mL of deionized water. The final pH must be about 3-4.
The mixture is cooled to -10°C-(-12°C) and stirred at this temperature for 1 h. The precipitate is filtered by using a sintered glass filter and washed with 0.5 L of anhydrous ethyl acetate previously cooled to 0-50C. The product is dried in a vacuum oven at 5O0C for a minimum period of 12 hours. The resulting crude 1 -[3-[3-(4-chlorophenyl)propoxy]propyl]-piperidine monohydrochloride weighs 1.10 kg.
Purification
A mixture of the above-described crude, 3.98 kg of anhydrous ethyl acetate and 0.35 kg of /-propanol is heated slowly at 55-6O0C in a 10-L glass vessel fitted with a heating and cooling system. When the solution has been completed, it is filtered through a heat-isolated sintered glass filter, keeping the temperature at 55-6O0C. The solution is transferred to a 10 L glass vessel and the mass is slowly cooled to 0-50C for about 1 hour. The mixture is stirred at this temperature for 1 hour and the precipitate is filtered through a sintered glass filter. The solid is washed with a mixture of 1.6 kg of anhydrous ethyl acetate and 0.14 kg of /-propanol cooled at 0-50C. The solid is dried in a vacuum oven at 5O0C for a minimum period of 12 hours. M. p. 117-1190C. Yield 80%.
IR spectrum (KBr): bands at 1112 and 1101 (C-O Ether/ St. asym), 2936 and 2868 (Alkane CH(CH2)) / St.), 1455 (Alkane CH(CH2)) / Deform.), 2647 and 2551 (Amine Salt / St.), 1492 (Amine / St.), 802 (Aromatic / Deform.) cm“1.
SEE
Eur. J. Pharm. Sci. 2001, 13, 249–259.
References
- Celanire S, Wijtmans M, Talaga P, Leurs R, de Esch IJ (December 2005). “Keynote review: histamine H3 receptor antagonists reach out for the clinic”. Drug Discov. Today. 10 (23-24): 1613–27. doi:10.1016/S1359-6446(05)03625-1. PMID 16376822.
- Ligneau X, Perrin D, Landais L, Camelin JC, Calmels TP, Berrebi-Bertrand I, Lecomte JM, Parmentier R, Anaclet C, Lin JS, Bertaina-Anglade V, la Rochelle CD, d’Aniello F, Rouleau A, Gbahou F, Arrang JM, Ganellin CR, Stark H, Schunack W, Schwartz JC. BF2.649 [1-{3-[3-(4-Chlorophenyl)propoxy]propyl}piperidine, hydrochloride], a nonimidazole inverse agonist/antagonist at the human histamine H3 receptor: Preclinical pharmacology. Journal of Pharmacology and Experimental Therapeutics. 2007 Jan;320(1):365-75. PMID 17005916
- Lin JS, Dauvilliers Y, Arnulf I, Bastuji H, Anaclet C, Parmentier R, Kocher L, Yanagisawa M, Lehert P, Ligneau X, Perrin D, Robert P, Roux M, Lecomte JM, Schwartz JC. An inverse agonist of the histamine H(3) receptor improves wakefulness in narcolepsy: studies in orexin-/- mice and patients. Neurobiology of Disease. 2008 Apr;30(1):74-83. PMID 18295497
- ^ Jump up to:a b Prous Science: Molecule of the Month September 2011
- Ligneau X, Landais L, Perrin D, Piriou J, Uguen M, Denis E, Robert P, Parmentier R, Anaclet C, Lin JS, Burban A, Arrang JM, Schwartz JC. Brain histamine and schizophrenia: potential therapeutic applications of H3-receptor inverse agonists studied with BF2.649. Biochemical Pharmacology. 2007 Apr 15;73(8):1215-24. PMID 17343831
- Stocking EM, Letavic MA (2008). “Histamine H3 antagonists as wake-promoting and pro-cognitive agents”. Current Topics in Medicinal Chemistry. 8 (11): 988–1002. doi:10.2174/156802608784936728. PMID 18673168.
- Schwartz, Jean-Charles (May 2011). “The histamine H3 receptor: from discovery to clinical trials with pitolisant”. BPJ. doi:10.1111/j.1476-5381.2011.01286.x.
References
- ^ http://adisinsight.springer.com/drugs/800029451
- ^ Jump up to:a b Schwartz JC (2011). “The histamine H3 receptor: from discovery to clinical trials with pitolisant”. Br. J. Pharmacol. 163(4): 713–21. doi:10.1111/j.1476-5381.2011.01286.x. PMC 3111674. PMID 21615387.
External links
REFERENCES
1: Leu-Semenescu S, Nittur N, Golmard JL, Arnulf I. Effects of pitolisant, a histamine H3 inverse agonist, in drug-resistant idiopathic and symptomatic hypersomnia: a chart review. Sleep Med. 2014 Jun;15(6):681-7. doi: 10.1016/j.sleep.2014.01.021. Epub 2014 Mar 18. PubMed PMID: 24854887.
2: Dauvilliers Y, Bassetti C, Lammers GJ, Arnulf I, Mayer G, Rodenbeck A, Lehert P, Ding CL, Lecomte JM, Schwartz JC; HARMONY I study group. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomised trial. Lancet Neurol. 2013 Nov;12(11):1068-75. doi: 10.1016/S1474-4422(13)70225-4. Epub 2013 Oct 7. PubMed PMID: 24107292.
3: Nirogi R, Ajjala DR, Kandikere V, Pantangi HR, Jonnala MR, Bhyrapuneni G, Muddana NR, Vurimindi H. LC-MS/MS method for the determination of pitolisant: application to rat pharmacokinetic and brain penetration studies. Biomed Chromatogr. 2013 Nov;27(11):1431-7. doi: 10.1002/bmc.2939. Epub 2013 Jun 13. PubMed PMID: 23760876.
4: Kasteleijn-Nolst Trenité D, Parain D, Genton P, Masnou P, Schwartz JC, Hirsch E. Efficacy of the histamine 3 receptor (H3R) antagonist pitolisant (formerly known as tiprolisant; BF2.649) in epilepsy: dose-dependent effects in the human photosensitivity model. Epilepsy Behav. 2013 Jul;28(1):66-70. doi: 10.1016/j.yebeh.2013.03.018. Epub 2013 May 8. PubMed PMID: 23665640.
5: Uguen M, Perrin D, Belliard S, Ligneau X, Beardsley PM, Lecomte JM, Schwartz JC. Preclinical evaluation of the abuse potential of Pitolisant, a histamine H₃ receptor inverse agonist/antagonist compared with Modafinil. Br J Pharmacol. 2013 Jun;169(3):632-44. doi: 10.1111/bph.12149. PubMed PMID: 23472741; PubMed Central PMCID: PMC3682710.
6: Brabant C, Charlier Y, Tirelli E. The histamine H₃-receptor inverse agonist pitolisant improves fear memory in mice. Behav Brain Res. 2013 Apr 15;243:199-204. doi: 10.1016/j.bbr.2012.12.063. Epub 2013 Jan 14. PubMed PMID: 23327739.
7: Zhang DD, Sisignano M, Schuh CD, Sander K, Stark H, Scholich K. Overdose of the histamine H₃ inverse agonist pitolisant increases thermal pain thresholds. Inflamm Res. 2012 Nov;61(11):1283-91. doi: 10.1007/s00011-012-0528-5. Epub 2012 Jul 21. PubMed PMID: 22820944.
8: Inocente C, Arnulf I, Bastuji H, Thibault-Stoll A, Raoux A, Reimão R, Lin JS, Franco P. Pitolisant, an inverse agonist of the histamine H3 receptor: an alternative stimulant for narcolepsy-cataplexy in teenagers with refractory sleepiness. Clin Neuropharmacol. 2012 Mar-Apr;35(2):55-60. doi: 10.1097/WNF.0b013e318246879d. PubMed PMID: 22356925.
9: Schwartz JC. The histamine H3 receptor: from discovery to clinical trials with pitolisant. Br J Pharmacol. 2011 Jun;163(4):713-21. doi: 10.1111/j.1476-5381.2011.01286.x. Review. PubMed PMID: 21615387; PubMed Central PMCID: PMC3111674.
 |
|
| Clinical data | |
|---|---|
| Trade names | Wakix |
| Synonyms | Tiprolisant; Ciproxidine; BF2.649 |
| License data | |
| Routes of administration |
Oral |
| Drug class | Histamine H3 receptor inverse agonists |
| ATC code | |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C17H26ClNO |
| Molar mass | 295.851 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////Pitolisant Hydrochloride, Wakix, histamine H3 receptor antagonist/inverse agonist, narcolepsy, orphan drug, tiprolisant, EU 2016, FDA 2019

Opicapone

Opicapone
2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-1,2,4-oxadiazol-3-yl)-4,6-dimethylpyridine-1-oxide
BIA-9-1067; ONO-2370; BIA-91067
CAS No.923287-50-7
MF C15H10Cl2N4O6
MW: 411.9977
TRADE NAME (Ongentys®)
Approved EU 2016-06-24 BIAL PORTELA
| PORTELA & CA. S.A. [PT/PT]; Av. Da Siderurgia Nacional, P-4745-457 S. Mamede do Coronado (PT) |
LEARMONTH, David Alexander; (PT).
KISS, Laszlo Erno; (PT).
LEAL PALMA, Pedro Nuno; (PT).
DOS SANTOS FERREIRA, Humberto; (PT).
ARAÚJO SOARES DA SILVA, Patrício Manuel Vieira; (PT)
MOA:Catechol-O-methyl transferase (COMT) inhibitor
Indication:Parkinson’s disease (PD)
A COMT inhibitor used as adjunctive therapy for parkinson’s disease.

Opicapone, also known as BIA 9-1067, is a novel potent and selective catechol-O-methyltransferase inhibitor (COMT inhibitor ) under clinical evaluation as an adjunct to L-Dopa therapy of Parkinson’s disease. Opicapone, a novel third generation COMT inhibitor, when compared to entacapone, provides a superior response upon the bioavailability of levodopa associated to more pronounced, long-lasting, and sustained COMT inhibition
Opicapone was approved by European Medicine Agency (EMA) on Jun 24, 2016. It was developed and marketed as Ongentys® by Bial – Portela in EU.
Opicapone is a selective and reversible COMT inhibitor, used as adjunctive therapy for Parkinson’s disease.
Ongentys® is available as hard capsules, containing 25 mg and 50 mg of opicapone. The recommended dose is 50 mg, taken once a day at bedtime, at least one hour before or after levodopa combination medicines.
Catechol-O-methyltransferase (COMTa) catalyzes the transfer of a methyl group from S-adenosyl-l-methionine to catecholic substrates such as endogenous catechol neurotransmitters(2)and xenobiotics including (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid (l-Dopa), the gold standard drug for treatment of Parkinson’s disease (PD). Coadministration of a peripheral amino acid decarboxylase (AADC) inhibitor prevents breakdown of l-Dopa in the periphery by blocking enzymatic decarboxylation, and inhibition of COMT further improves its bioavailability by reducing the formation of 3-O-methyl-l-Dopa (3-OMD).
Abbreviations: COMT, catechol-O-methyltransferase; PD, Parkinson’s disease; AADC, amino acid decarboxylase; SAR, structure−activity relationship; ADMET, absorption, distribution, metabolism, excretion, toxicity; l-Dopa, (S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid; 3-OMD, 3-O-methyl-l-Dopa.
![]()

Chemical structures of tolcapone 1, entacapone 2, and nebicapone 3.
Opicapone
A preferred method of treatment of Parkinson’s disease is the administration of a combination of levodopa and a peripherally selective aromatic amino acid decarboxylase inhibitor (AADCI) together with a catechol-O-methyltransferase (COMT) inhibitor. The currently employed COMT inhibitors are tolcapone and entacapone. However, some authorities believe that each of these COMT inhibitors have residual problems relating to pharmacokinetic or pharmacodynamic properties, or to clinical efficiency or safety. Hence, not all patients get most benefit from their levodopa/AADCI/COMT inhibitor therapy.
Favoured new COMT inhibitors were disclosed in L. E. Kiss et al, J. Med. Chem., 2010, 53, 3396-3411 (D1), WO 2007/013830 (D2) and WO 2007/117165 (D3) which are believed to have particularly desirable properties so that patients can benefit from enhanced therapy.
D1, D2 and D3 also disclosed methods of preparing the new COMT inhibitors. Those processes, although effective, would benefit from an increase in yields. Other benefits which would be appropriate include those selected from reduction in number of process steps, reduction in number of unit operations, reduction of cycle-times, increased space yield, increased safety, easier to handle reagents/reactants and/or increase in purity of the COMT inhibitor, especially when manufacture of larger quantities are envisaged. A process has now been discovered that proceeds via a new intermediate which is suitable for manufacture of commercially useful quantities of a particularly apt COMT inhibitor in good yield. Additional benefits occur such as those selected from a reduced number of process steps and number of unit operations, reduced cycle-times, increased space yield, increased safety, with easier to handle reagents/reactants, improved impurity profile and/or good purity.
CLINICAL
https://clinicaltrials.gov/show/NCT01851850


SYN1
Discovery of a Long-Acting, Peripherally Selective Inhibitor of Catechol-O-methyltransferase

1H : 11.07 (2H, br, OH), 8.11 (1H, d, J = 2 Hz, H6), 7.73 (1H, d, J = 2 Hz, H2), 2.66 (3H, s, H15),2.24 (3H, s, H14).
(C15H10Cl2N4O6) C, H, N, S: Calc: C, 43.60; H, 2.44; N, 13.56; Found: C, 44; H, 2.3; N, 13.6.

PATENT
LEARMONTH, David Alexander; (PT).
KISS, Laszlo Erno; (PT).
LEAL PALMA, Pedro Nuno; (PT).
DOS SANTOS FERREIRA, Humberto; (PT).
ARAÚJO SOARES DA SILVA, Patrício Manuel Vieira; (PT)
PATENT
The present invention in one aspect provides 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4,oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene and salts thereof, that is the compound of the formula (I):

and salts thereof.
Most aptly the compound of formula (I) is unsalted. However, salts of the hydroxy group with metal ions such as the alkali or alkaline earth metals, particularly the sodium and potassium salts are provided as well as those of highly basic organic compounds such as guanidine or the like.
Particularly suitably the compound of formula (I) or its salt is provided in a form suitable for use as a chemical intermediate. This may be, for example, in a form at least 50% pure, in crystalline form, in solid form or in an organic solvent or the like.
The compound of formula (I) is useful as an intermediate in the preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol i.e. the compound of formula II):

The compound of formula (II) may also be referred to as opicapone or 2,5-dichloro-3-(5-(3,4-dihydroxy-5-nitrophenyl)-[1,2,4]-oxadiazole-3-yl)-4,6-dimethylpyridine-1-oxide. Opicapone has been found to be more potent than tolcapone in inhibiting liver COMT both at 3 hours and 6 hours post oral administration to rats [ED50 in mg/kg, opicapone 0.87 at 3 hours and 1.12 at 6 hours as compared to tolcapone 1.28 at 3 hours and 2.08 at 6 hours]. Opicapone at a dose of 3 mg/kg was found to be more effective at inhibiting rat liver COMT with nearly complete inhibition occurring 2 to 6 hours post oral administration with only about 90% of enzyme activity recovered after 72 hours while tolcapone provided shorter duration of activity with about 84% recovery after only 9 hours. Both opicapone and tolcapone inhibit human recombinant S-COMT but opicapone has an inhibitory constant of 16pM being 10 fold lower than that for tolcapone. With respect to the desirable property of avoiding inhibition of COMT in the brain, opicapone following oral administration to the rat was found to be devoid of effect whereas tolcapone inhibited about 50% of enzyme activity over a period of 8 hours post administration.
Preparation 1

Cyanoacetamide (280g) was reacted with acetyl acetone (352.9g) in methanol (1015g) and morpholine (14.9g). The reaction was stirred under reflux at 65 °C until the reaction appeared complete. The resulting product suspension was filtered, washed with methanol and dried to provide the desired product about 97% yield.
Preparation 2

The product of Preparation 1 (159g) was suspended in acetonitrile (749.5g) and cooled to 0-5°C. Sulfuryl chloride (178.9g) was added and the reaction mixture warmed to room temperature and stirred until the reaction appeared complete.
The resulting suspension is cooled to 0-5°C and filtered. The solid was washed with acetonitrile, ethyl acetate and heptane. The product was then dried under vacuum at 50°C to yield the desired product (82%).
Preparation 3

Phosphoryl chloride (973.2g), tetramethylammonium chloride (67.3g) and compound of Preparation 2 (227.1g) were added to dichloromethane (500g). The suspension was heated to 85°C and stirred for 5 hours. Excess of phosphoryl chloride was removed by distillation in vacuo. The reaction mixture was cooled below 30°C and diluted with dichloromethane. The resulting solution was added to water (1350g) at room temperature and stirred for 30 minutes. The lower organic phase was separate and the aqueous phase extracted with dichloromethane. The organic phases were combined, washed with water and then treated with charcoal. The charcoal was filtered and a solvent swap to heptane was performed by distillation at atmospheric pressure. The solution was filtered at 50°C and then cooled to 30°C. On further cooling to 0°C
crystals were obtained. These were isolated by filtration, washed twice with heptane. After drying at 50°C the desired product was obtained typically at 88-91 % .
The above process was repeated with a reduction in dichloromethane during crystallisation and adding some methanol. The resulting plate-like crystals were more easily transferred for subsequent use.
Preparation 4a

Product of Preparation 3 (68.6g) and 1,10-phenanthroline monohydrate (0.9g) were suspended in methanol (240g) at room temperature. Water (518g) and a hydroxylamine solution (50% in water, 80.9g), were added and the mixture heated to 70-80°C and stirred for 5-6 hours. Water was added at 70-80 °C and the solution held for 1 hour to induce crystallization. Crystallization was completed by cooling to 15°C over 8 hours. The product was filtered off and washed twice with water and dried at 50°C under vacuum. The product was an off white to light yellow and the yield was 87.9% .
Preparation 4b

A suspension of 2,5-Dichloro-4,6-dimethyl-nicotinonitrile (45.0 kg) and 50% hydroxylamine (59.2 kg) in the presence of catalytic amount of 1,10-phenanthroline monohydrate (0.680 kg) in methanol / water (214 kg/362 kg) is heated to 70-80°C. The mixture is agitated at 70-80°C. Water (353 kg) is added slowly into the resulting solution while the temperature is maintained at > 79°C. The solution is cooled to 75 °C with stirring resulting in crystallization of (Z)-2,5-dichloro-N’-hydroxy-4,6-
dimethylnicotinimidamide. The suspension is further cooled to 20 °C, the solid is filtered off and the wet cake is washed with water (160 kg). (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide is dried under vacuum at max. 60°C until residual water level is max 0.15% (KF).
Example 1a
Preparation of 4-hydroxy-5-methoxy-3 nitrobenzoic acid

Vanillic acid (75g) was suspended in acetic acid (788g). The suspension was cooled to 10°C to 15°C and nitric acid (49g or 65% solution) was added over three hours at a rate which kept temperature between 10°C and 20°C. The resulting yellow orange was stirred for a further one hour at 18°C to 23°C. The suspension was filtered off, washed with acetic acid, then a mixture of acetic acid and water (1/2) and then water. Yield of 53% of a 87.9% pure product was obtained.
The above crude product was suspended in acetic acid and warmed to 105°C to 110°C until an orange brown solution is obtained. The solution was transferred to the crystallization vessel via a charcoal filter (or polish filtration) at a temperature above 85°C (optional step). The solution was then cooled to 80°C to 85°C. The mixture was stirred for one hour at 70°C to 80°C (optionally at 75°C) during which crystallization occurred. The product suspension was cooled to 20°C to 25°C for 17 hours or stirred for at least 12h at 20°C to 25 °C. The product suspension was filtered and washed with acetic acid, then acetic acid/ water (1/2) and finally water. The product was dried under vacuum at 50°C to 55°C. The yield of 70% corresponds to an overall yield of 44% for both parts of this preparation. The purity of the product assayed at 99.7% .
The preceding crystallization step is optional and the solution may be transferred to the crystallization vessel via polish filtration instead of via a charcoal filter.
The post crystallization suspension may be stirred for at least 12 hours at 20° C to 25 °C as an alternative to 17 hours.
Example 1b
Preparation of 4-hydroxy-5-methoxy-3 nitrobenzoic acid
A reactor was charged with 525 kg of glacial acetic acid and 50 kg vanillic acid. The mixture was heated with warm water gradually to 50°C in around 75 minutes. Temperature was set to 16°C. Nitric acid, 31.4 kg was then added gradually over a period of 3 hrs. When the administration was complete the mixture was allowed to stir for additional 3.5-4.5 hours.
The suspension was centrifuged whilst washed with 25 kg of acetic acid, 50 liter deionised water and 25 kg of acetic acid again. The wet crystalline material was suspended in 165 kg of acetic acid and heated at 91°C until complete dissolution. The solution was then cooled to 19.8°C and the mixture was allowed to stir for 1 hr. Centrifugation and washing with 15.2 kg acetic and 40 liter of deionised water was performed. The wet material was then dried in tray vacuum drier between 40-50°C until constant weight, for 72 hours. The dry material weight was 28.7 kg. The calculated yield was 45.4%.
Example 1c
Preparation of 4-h droxy-5-methoxy-3 nitrobenzoic acid

A suspension of vanillic acid (68.8 kg) in acetic acid (720 kg) is cooled to 17°C before an excess of a 65% nitric acid (44.0 kg) is added. After complete dosage of nitric acid the suspension is stirred for 2 hours. The suspension is filtered off and the wet cake is successively washed with acetic acid (80.0 kg), acetic acid/water (1:2 w/w – 105 kg) and finally water (80 kg – if necessary repeat). The solid is dried at 52°C for NMT 12 hours prior going to next step.
A suspension of the crude solid (650 kg) in acetic acid is warmed to 105 °C and stirred until complete dissolution of the crude solid. After polish filtration, the solution is cooled to 20°C over 3h resulting in crystallization and the suspension is stirred for 2h at 20°C. The solid is filtered off and the wet cake is successively washed with acetic acid (80 kg), acetic acid/water (1:2 w/w – 105 kg) and finally water (193 kg – if necessary repeat). 4-hydroxy-5-methoxy-3 nitrobenzoic acid pure is dried under vacuum at max. 55 °C until max 0.5% w/w residual acetic acid and max 0.2% w/w water is reached.
Example 2a
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoic acid
The process of Example la was scaled up to employ vanillic acid (375g) in acetic acid (3940g) to which was added nitric acid (65%, 245g) at 12°C over 3 hours followed by stirring for one hour. The overall yield was 40% of a 99.9% pure product.
Example 2b
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoic acid

Vanillic acid methyl ester (33g) and sodium nitrite (0.625g) are charged. Water (158mL) and 1,4-dioxane (158mL) are added at room temperature. The reaction mixture is heated to 40 °C. Nitric acid (65%) (15.75g) is added in the course of three hours and the resulting mixture is stirred for 4h after addition. The reaction mixture is sampled for completion.
The water/nitric-acid/dioxane azeotrope is distilled off in vacuum at 40 °C. The resulting product suspension is quenched by addition of sodium hydroxide solution (50% , 33.2 mL) and then stirred for 16h. The quench mixture is sampled for completion.
Then, HCl (18,5%, 70.2mL.) is added until the pH is below 1. The product is filtered off and washed with water (27.9mL). The product is then dried in vacuum at 50 °C. The overall yield was 81 % of a 97.3 % pure product.
Example 3a
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride

A suspension of compound of Example la (1.0 eq) in dioxane (approx 4.5 vol) was treated with thionyl chloride (1.5 eq) and heated to 80°C. A clear solution formed at approximately 75 °C. The mixture was stirred for 3 hours at 80°C. Unreacted thionyl chloride was distilled off and after distillation the residue was cooled to 10°C.
Example 3 b
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
A suspension of compound of Example la (1.0 eq) in DCM (approx 3.4 vol) is treated with thionyl chloride (1.0 – 1.2 eq, for example 1.1 eq) and catalytic amount (0.011 eq) of DMF and the mixture is stirred for 16 h at 40°C. DCM is distilled off (approx 2.7 vol) and the residue is diluted with THF (approx 1.8 vol). The excess of thionylchloride is distilled off with THF/DCM and the residue after distillation is cooled to 10°C.
Example 3c
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
A suspension of compound of Example la (1.0 eq) in DCM (approx 4.5 vol) is treated with thionyl chloride (1.0 – 1.2 eq, for example 1.1 eq) and catalytic amount (0.0055 eq) of DMF and the mixture is stirred for 16 h at reflux. Unreacted thionylchloride is distilled off with DCM and the residue after distillation is diluted with THF (approx 1.8 vol) and cooled to 10°C.
The amount of DCM may be approx 3.4 as an alternative to approx 4.5 vol.
The catalytic amount of DMF may be about 0.011 eq as an alternative to 0.0055 eq.
Example 3d
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
In a reactor 68 kg dichloromethane, 20 kg 5-nitro- vanillic acid of example 1b, 76 gram of N,N-dimethylformamide and 13.4 kg (8 L) thionyl chloride, was charged at 20.2°C.
The mixture was heated to 40°C until all the starting material dissolved and the evolution of HCl and SO2 stopped. When all the starting material was consumed 5-10 L dichloromethane was distilled off at normal pressure at 40°C then the mixture was cooled to 20-25 °C and the distillation was continued until dry under vacuum at 40°C.
The evaporation residue was dissolved in 36 kg dry THF. The THF solution was used in
Example 4d.
Example 3e
Preparation of 4-hydroxy-5-methoxy-3-nitrobenzoyl chloride
A suspension of product of example 1C (4-hydroxy-5-methoxy-3 nitrobenzoic acid -160g, 1eq) in 1,4-dioxane (720mL, 4.5vol) is treated with thionyl chloride (169.8g, 103.7mL,1.5eq) and heated to 80°C. A clear solution is formed at approx. 75 °C. The mixture is stirred at 80 °C (3 hours). Unreacted thionyl chloride is distilled off and the residue after distillation is cooled to 10°C.
Example 4a
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In this example the compound of formula (IV) is reacted with the compound of formula (V) to produce the compound of the formula (III).



Compound of formula (V) (1.24 eq) was suspended in 1,4-dioxane (approximately 4.5 vol) and the suspension cooled to 10°C. The acyl chloride (compound of formula (IV)) solution of Example 3a in 1,4-dioxane was added slowly maintaining the temperature below 20°C. A clear orange solution was formed. After complete addition, the reaction mixture was stirred at 20°C for one hour. Pyridine (approximately 8eq) was added and the reaction mixture heated slowly to 115°C. The mixture was stirred for 6 hours at 115°C and then cooled to 20°C.
The dioxane/pyridine was distilled off under vacuum at 70°C. The residue was kept at 80°C and ethanol (approx 8 vol) added to induce crystallization. The resulting yellow suspension was cooled to 0°C and stirred for two hours. The product was filtered off and washed with ethanol (2.5 vol) water (3.8 vol) and ethanol 2.5 vol). The product was dried under vacuum at 50 °C. Typical yields for this process are 82 to 85%.
In an optional variant, methanol was employed in place of ethanol to induce crystallization.
Example 4b
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In a different reactor, compound of formula (V) (1.1 eq) is dissolved in DM Ac (approx 5.8 vol) and the solution is cooled to 5°C. The benzoyl chloride solution of Example 3b in THF/DCM is then added slowly maintaining the temperature below 10°C. After complete addition, the reaction mixture is stirred at 20 ±5°C. Pyridine (1.3 to 1.6 eq, for example 1.5 eq) is charged and the reaction mixture is heated slowly to 110±5°C removing low boiling components by distillation. The mixture is stirred for additional 3 h at 110±5°C.
In a further reactor, concentrated HCl (23.8 eq) is diluted with water (approx. 8.5 vol) and cooled to 10 °C. The reaction mixture in pyridine is dosed slowly to diluted hydrochloric acid. After complete addition, the resulting suspension is stirred for additional 2 h and the solid is filtered off. The crude solid is washed once with water and pre-dried on funnel.
The crude solid is suspended in DCM (approx. 28.6 vol) and the suspension is heated to 40°C to reach a clear solution. Resulting solution is cooled to 20°C and extracted with water. After phase separation, the aqueous phase is re-extracted with DCM and combined organic phase are washed once with water. DCM is distilled off under vacuum followed by addition of ethanol. Resulting suspension is further distilled to reduce the amount of DCM, then cooled to 5°C and stirred for additional 2 h. Finally, the product is filtered off, washed once with cold ethanol and dried under vacuum at 45°C.
Example 4c
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In a second reactor, compound of formula (V) (1.1 eq) is dissolved in DMAc (approx. 7 vol) and the solution is cooled to 5°C. The benzoyl chloride solution of Example 3c in THF/DCM is added slowly maintaining the temperature below 10 °C. After complete addition, the reaction mixture is stirred at 20 ± 5°C for 30 min. Pyridine (6.9 to 7.3 eq, for example 7.14 eq) is charged and the reaction mixture is heated slowly to 110°C removing low boiling components by distillation. The mixture is stirred for additional 4 h at 110°C and cooled to 20°C.
In a third reactor an emulsion of diluted hydrochloric acid (prepared from cone. HCl (19.6 eq) and approx. 7.6 vol distilled water) and DCM (approx. 25.5 vol) is cooled to about 15 °C before the reaction mixture in pyridine is dosed slowly to the emulsion. After complete addition, the organic phase is separated and washed with water before DCM is distilled off under vacuum followed by addition of ethanol. The resulting suspension is further distilled to reduce the amount of DCM, then cooled to 5°C and stirred for additional 2 h.
Finally, the product is filtered off, washed once with cold ethanol and dried under vacuum at 45 °C.
Example 4d
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
140 kg Ν,Ν-dimethyl acetamide was charged into the reactor. 24.2 kg of amidoxime of Preparation 4 was dissolved in N,N-dimethyl acetamide while stirring at 21°C. The solution was cooled to 5-10°C. The THF solution of Example 3d was introduced slowly into the reaction mixture, 1.5-2 hrs, while the internal temperature was maintained at max. 9.5°C by external cooling. When the addition was complete the external cooling
was stopped. The internal temperature was allowed to raise to 21 °C in an hour. After stirring for 30 minutes, pyridine 53.0 kg was added to the mixture, while the temperature was in the range of 22.4°C – 20.6°C. Heating was started and the internal temperature raised to 105-115°C. The mixture started to reflux for 3h while the internal temperature managed to 113°C by partial distillation of some THF. The reaction mixture was then cooled and introduced to a mixture of 220 kg concentrated HCl and 170 kg of deionised water while the internal temperature was maintained between 14-16°C. The reactor was rinsed with 10 kg of Ν,Ν-dimethylacetamide and 20 kg deionised water. The rinse liquid was run to the mixture. The suspension was then further cooled to 5-10°C and stirred for 1.5-2.0 hours. The product was centrifuged and was washed 80 kg deionised water. Crude wet weight of the product was 88.6 kg.
The crude wet product, was dissolved in 460 kg (340 L) dichloromethane at max 40°C. When dissolved the temperature was set to 20-30°C and 120 kg deionised water was added. The organic phase was separated, the inorganic phase was extracted with 80 kg dichloromethane. The organic phase of 460 kg, was then washed with 200 kg deionised water and the phases were separated. The inorganic phase was extracted with the 80 kg dichloromethane and the organic phases were unified. The organic phase obtained so was concentrated in vacuum at 35°C to 200-240 Liter, then 260 kg ethanol 96% was continuously added and the evaporation was continued to a final 200-240 liter volume. Then the mixture was cooled to 5-10°C and was allowed to stir for 3 hrs. Centrifuging, washing with 20 kg ethanol resulted in 35.4 kg wet product. Vacuum drying for 16 hours at 45°C gave 34.09 kg dry product. The yield was 79.9%.
Example 4e
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
In a second vessel, (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotiriimidamide (201.2g, 1.24eq) is suspended in 1,4-dioxane (720mL, 4.5vol) and the suspension is cooled to 10°C. The residue of example 3e in 1,4-dioxane is added slowly maintaining the temperature below 20°C. A clear orange solution is formed. After complete addition, the reaction mixture is stirred at 20°C for 1 hour. Pyridine (483.7mL, 8eq) is then charged and the reaction mixture is heated slowly to 115°C. The mixture is stirred at 115°C for 6 hours. The solution is then cooled to 20°C. Dioxane/pyridine is distilled off.
After distillation, the pit is kept at 80 °C and ethanol (1.28L, 8vol) is added at this temperature to induce crystallization. The resulting yellow suspension is cooled to 75 °C and stirred for 1h at this temperature to allow crystal growth. The product suspension is then cooled to 0 °C and stirred for 2h at this temperature. The product is filtered off and washed subsequently with ethanol (400mL, 2.5vol), water (608mL, 3.8vol) and ethanol (400mL, 2.5vol). The product is dried under vacuum at 50°C until LOD is max 1% w/w.
Example 4f
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
A mixture of compound of formula (V) (11.7g, 50 mmol, 1.25eq), methyl 4-hydroxy-3-methoxy-5-nitrobenzoate (10g, 40 mmol, leq) and a catalytic amount of p-toluenesulfonic acid (0.76g, 4mmol, 0.1eq) in dimethyl acetamide was heated to 80°C. The reaction was followed by HPLC. After 23h, 6% of conversion was obtained.
Example 4g
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-y1]-2-hydroxy-3-methoxy-1-nitrobenzene
A mixture of compound of formula (V) (11.7g, 50 mmol, 1.25eq), methyl 4-hydroxy-3-methoxy-5-nitrobenzoate (10g, 40 mmol, 1eq) and a catalytic amount of aluminum chloride (0.53g, 4mmol, 0.1eq) in dimethyl acetamide was heated to 80°C. The reaction was followed by HPLC. After 20h, 10% of conversion was obtained.
Example 5a
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene

A solution of the product of Example 4a (24g) was dissolved in dichloromethane (388g) at 20-40°C. The yellow solution was cooled to 5°C and urea hydrogen-peroxide (UHP) (17.6g) and trifluoroacetic acid anhydride (37g) added and stirring continued for 12hr at 5°C. The reaction mixture was warmed to room temperature over one hour and stirring continued for a further five hours. The precipitate that formed was filtered off and washed with dichloromethane. The combined filtrates were diluted further with dichloromethane, all washed and concentrated at atmospheric pressure. Toluene was added and the resulting suspension concentrated under vacuum, to remove residual dichloromethane. Further toluene was added and the mixture checked to ensure less than 0.5% dichloromethane and less than 0.1% water was present. Formic acid was added to provide a 10-12% formic acid in toluene mixture. The resulting suspension was warmed to 90°C and stirred until complete dissolution of solid. Crude product was obtained by cooling the solution to 5-10°C until crystallization commenced. The suspension was agitated at 5-10°C until crystallization appeared complete. The solid was filtered off, washed with toluene and dried under a stream of nitrogen.
The crude product was suspended in 10-12% wt/wt solution of formic acid in toluene and warmed to 90°C until dissolution of the solid. The solution was cooled to 5°C and stirred at 5°C until crystallisation occurred. The solid was obtained by filtration and washed with toluene. This recrystallization was repeated until the product tested as containing less than 0.1 % of starting material. The pure product was dried under vacuum at 50°C.
Example 5b
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
After dissolution of the product of Example 4b (24g) in DCM (388g) at 20-40°C the yellow solution is cooled to 5°C before the temperature controlled addition of urea hydrogen peroxide complex (UHP)(17.6) and trifluoroacetic anhydride (TFAA) (37g). After addition of TFAA is complete stirring is continued for 12h at 5°C before the reaction mixture is warmed to room temperature (RT) within 1 h and stirring is continued for additional 5 h. The precipitate formed during the reaction is filtered and washed with DCM on the funnel filter. The combined filtrates are diluted with DCM (325g) and then repeatedly washed with water before concentrated at atmospheric pressure. DCM is replaced by toluene (170g) and the resulting suspension is concentrated again under vacuum to remove surplus DCM. Distillates are replaced by fresh toluene as before and the mixture is analyzed for residual water and DCM (Residual DCM after solvent switch max. 0.5%; residual water after solvent switch max. 0.1 %). Formic acid (24g) is charged resulting in an approx. 10-12 % w/w formic acid in toluene solvent mixture The resulting suspension is warmed to 90°C and stirred until compete dissolution of the solid is achieved. The crude product is crystallized by cooling of this solution to 5-10°C and subsequent agitation of the resulting suspension at 5-10°C. The solid is filtered of washed with toluene and then dried in a stream of nitrogen gas.
The crude product so obtained is suspended in an approx. 10-12 %w/w solution (176g) of formic acid in toluene. The suspension is warmed to 90°C and stirred until all product is dissolved. After cooling of this solution to 5°C and subsequent stirring at 5°C, crude product is isolated by filtration and subsequent washing of the wet product with toluene.
The re-crystallization of crude product is repeated (2 or more times). The pure product (11.8g) is dried at 50°C under vacuum.
Example 5c
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
After dissolution of the product of Example 4c (24g) in DCM (388g) at 20-40°C the yellow solution is cooled to 5°C prior to the temperature controlled addition of urea hydrogen peroxide complex (UHP) (17.6g) and trifluoroacetic acid anhydride (TFAA) (37g). After addition of TFAA is complete stirring is continued for 12h at 5°C before the reaction mixture is warmed to RT within 1 h and stirring is continued for additional 5 h. The precipitate formed during the reaction is filtered and the filter cake is washed with DCM. The combined filtrates are diluted with DCM (325g) and then repeatedly washed with water before concentrated at atmospheric pressure. DCM is replaced by toluene (170g) and the resulting suspension is concentrated again in vacuum in order to remove surplus DCM and water. Distillates are replaced by fresh toluene followed by addition of formic acid (24g). The resulting suspension is warmed to 80°C and stirring is continued in order to dissolve the solid. The product is crystallized by cooling of this solution to 5°C and subsequent agitation of the resulting suspension at 5°C. The solid is filtered, washed with toluene and then dried in a stream of nitrogen gas.
The product is suspended in a formic acid / toluene (18g/158g) mixture followed by warming of the reaction mixture to 80°C. After dissolution of the product the solution is cooled to 5°C whereby the product precipitates. After additional stirring at 5°C the suspension is filtered and the filter cake is washed with toluene.
The re-crystallization of the product is repeated. The product is used as a wet material in the next process step (12.1g product obtained if dried at max. 60°C).
Example 5d
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yI]-2-hydroxy-3-mefhoxy-1-nitrobenzene
550 kg (420 L) Dichloromethane was charged into a reactor. 34 kg of product of example 4d was added to in a short period at 20°C internal temperature. The solution was cooled to 6.5°C then 24.9 kg urea hydrogen peroxide complex (UHP) was added over a period of 20-40 minutes between 5-10°C. Stirring was continued for additional 20 minutes between 6.5-7.5°C. Trifluoroacetic anhydride, 53 kg, was administered into the reaction mixture, starting and maintaining the temperature at 6-7°C over a period of 2-3 hours. When the administration was complete the mixture was stirred for additional 30 minutes. Then the internal temperature was allowed to rise to a maximum of 25°C over a period of 1.5 hours. The internal temperature was maintained between 20-25°C and the mixture was allowed to react for additional 18-20 hrs. The reaction mixture was centrifuged and the fuge was washed with 45 kg dichloromethane. To the separated dichloromethane solution 460 kg (350 L) dichloromethane and 190 kg deionised water was added. The mixture was stirred for 10 minutes and the phases were separated for 30 minutes. The organic phase was washed again with 2×190 kg deionised water and separated as previously. Evaporation of the unified organic solution at max 35 °C under vacuum was done to a final volume of 100-120 L. Then a total of 105 kg acetonitrile was administered into the system while the distillation was continued to keep the volume at 100-120 L. When complete an additional 170 kg (220 L) acetonitrile was added to the mixture at normal pressure. This suspension was heated to 70-80°C at normal pressure while dichloromethane was distilled off continuously. The mixture was then kept stirred for an hour. The suspension was cooled to 20-25°C and was stirred for an additional 30 minutes. The suspension was then centrifuged and was washed with 30 kg acetonitrile. The wet material, 29.7 kg, was vacuum dried for 16 hrs at 30°C. Dried product yield was 81.5%.
27.7 kg product, 240 kg toluene and 29.2 kg formic acid was charged into reactor then heated to 90°C for complete dissolution for 1 hour. Then the solution was cooled to 7°C and then the suspension was kept at 7°C for additional 2 hrs. If necessary seeding was applied with 3-5 grams of pure product. The suspension was then centrifuged for 1 hour whilst washing with 28 kg cold toluene. The product was suspended in 225 kg toluene and 27.2 kg formic acid was charged. The mixture then was heated to 90°C for complete dissolution for 1 hour. Then the solution was cooled to 20-25 °C, then the suspension was kept between 15-25°C for additional 2 hrs, seeded if necessary. The suspension then was centrifuged for 60 minutes whilst washed with 28 kg cold toluene. The recrystallization process may be repeated 2-3 more times.
Drying for 24 hrs at 38-41°C under vacuum was conducted until constant weight. This resulted in 16.34 kg (58.8%) dry material.
Example 5e
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene
After dissolution of the product of Example 4e (150g) in DCM (2.43kg) at reflux, the yellow solution is cooled to 5°C prior to the temperature controlled addition of carbamide peroxide (UHP – urea hydrogen peroxide) ( 110g) and trifluoroacetic acid anhydride (TFAA) (155.1 ml in 4 portions within 2 hours). The mixture is stirred for 12h at 5°C then the reaction mixture is warmed to 25 °C over 1.5 hours and stirred for 5 hours. The precipitate formed during the reaction is filtered and the filter cake is washed with DCM (0.36 kg). The combined filtrates are warmed to 30°C and diluted with water (300g). 10% sodium hydroxide is added until pH= 4 is reached. The biphasic system is stirred for 10 minutes at 30°C and the mixture is then allowed to separate. The organic layer is then successively washed with a mixture water (750g) and 10% sodium hydroxide (7.5g) (until pH=4), 3.2% HCl solution (300g). DCM is distilled at atmospheric pressure and then replaced by toluene (1035g) applying vacuum (150mbar) and keeping internal temperature at 45°C. Formic acid (300g) and toluene (900g) are added keeping the internal temperature above 40°C. The resulting solution is distilled under vacuum (150 mbar, 45°C internal temperature) until distillation ceases. After seeding at 45°C, the slurry is stirred for 1 hour at 45°C then is cooled to 5°C over 2 hours. The suspension is stirred for at least 2 hours at 5°C and then filtered. The wet cake is washed with toluene (195g) and dried in a stream of nitrogen gas (Chemical purity of crude product min. 92 % area).
A suspension of crude product in formic acid (388g, 2wt) is warmed to 55°C and stirred until complete dissolution of the crude product. Toluene (1242g, 6.4wt) is added maintaining the internal temperature above 50 °C. The reaction is stirred at 150mBar and internal temperature 45 °C until distillation ceases. The vacuum and distillation is stopped and then seed is added at 45°C. The slurry is stirred for 1 hour at 45°C and cooled to 5°C in 2 hours. The resulting suspension is stirred for at least 2 hours at 5°C then filtered. The wet cake is washed with toluene (260g, 1.34wt). The wet cake is collected and charged into the reactor. This procedure is repeated at least twice until 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-y1]-2-hydroxy-3-methoxy-1-nitrobenzene level max is 0.1 % (a/a) prior to dry at 25°C max under vacuum.
Example 6
Example 5a was repeated on a larger scale employing product of Example 3 (82g), dichloromethane (1325g), urea peroxide (60.1g) and trifuoroacetic acid anhydride
(128g).
Example 7a
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.

(Π)
Product of Example 6 (15g) was suspended in N-methyl pyrrolidone (NMP) (131.5g) and cooled to 5°C. Aluminium chloride (6.2g) and pyridine (12g) were added while maintaining the temperature at 5°C. After the addition of pyridine was complete the reaction mixture was warmed to 60 °C and maintained for 2 hours. After confirmation that less than 0.5 starting material remained, the reaction mixture was cooled, and aqueous HCl (water 233g, HCl 123g, 37%) added. The resulting yellow solid was isolated by suction filtration. The resulting wet product was washed with water and propan-2-ol (67g) and dried under vacuum.
Optionally, the crude product was suspended in ethanol (492g) and warmed to reflux. The suspension was stirred for 1 hour under reflux and then cooled to room temperature. The solid was obtained by filtration, washed with ethanol and dried under vacuum at 50°C. A typical yield of 85% was achieved.
If desired either the final ethanol crystallised material or the initially produced product after washing with propan-2-ol may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7b
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-y1)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
An approx. 11 % w/w suspension of the product of example 5b (20g) in NMP (150g) is cooled to 5°C followed by a consecutive temperature controlled addition of aluminium chloride (8g) and pyridine (15.3g). After addition of pyridine is complete the reaction mixture is warmed to 60°C followed by additional 2 h reaction time. After complete conversion of the product of example 5b the reaction mixture is cooled before an aqueous diluted hydrochloric acid (water 293g, HCl 177g, 34%) is dosed. By addition of the hydrochloric acid, crude product precipitates from the NMP/water matrix as a yellow solid which is isolated by suction filtration. The resulting wet product is washed with water and 2-propanol in a replacement wash followed by drying of the wet crude product under vacuum.
The crude product is suspended in ethanol (282g) followed by warming of the mixture to reflux. The suspension is stirred for 1 h at reflux conditions followed by cooling to room temperature. The product is isolated by filtration of the suspension. The wet product is washed with ethanol and subsequently dried in vacuo at approx 50°C (typically weight corrected yield was 85%).
The product (20g) is suspended in formic acid (725g) before the resulting suspension is warmed to max. 67°C. Stirring is continued until complete dissolution of the product is achieved. The hot solution is filtered and the filtrate is cooled to 40 – 45°C before the product is precipitated first by concentration of the solution to approx. 40% (v/v) of its original volume followed by addition of the anti solvent 2-propanol (390g). After addition of 2-propanol is finished the resulting suspension is kept at 55-60°C for crystal ripening followed by cooling to RT and filtration. The filter cake is washed with 2-propanol followed by drying of the material at max. 58°C until loss on drying (LOD) max. 0.5% . Typically, a yield of 97-98% was obtained.
If desired the product may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7c
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
A suspension of the product of example 5c (20g) or of example 6 (20g) in NMP (153g) is cooled to 5°C followed by a consecutive temperature controlled addition of aluminium chloride (8.2g) and pyridine (15.4g). After addition of pyridine is complete the reaction mixture is warmed to 60°C followed by additional 3 h reaction time. After complete conversion of the product of example 5c or of example 6 the crude product is
precipitated by a temperature controlled addition of an aqueous hydrochloric acid solution (water 296g, HCl 179g, 34%). Filtration of the solid followed by washing of the wet filter cake with water and 2-propanol yields a crude product wet material which is immediately dissolved in formic acid (536g). After polish filtration the filtrate is concentrated under vacuum followed by addition of the anti-solvent 2-propanol (318g). After aging of the resulting suspension at 55-60°C the suspension is cooled to RT and filtered. The wet filter cake is washed with 2-propanol. The wet product is dried under vacuum at max. 58°C until LOD max. 0.5%. The yield was in the range of 70-95%
If desired the product may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7d
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
132 kg (147 L) N-methylpyrrolidone was charged into a 1000 L reactor. 16.3 kg of product of example 5d was then added. The suspension was cooled to 5-7°C and 6.5 kg of sublimed aluminium chloride was added in portions keeping the internal temperature between 5-10°C. The mixture was stirred for 10 minutes then 12.6 kg pyridine was added maintaining the internal temperature between 5-10°C. The mixture was warmed with water in the jacket to 20-25°C and the mixture was stirred for 30 minutes. Then the mixture is heated to 58-62 °C and reacted for around 2 hours. In a separate reactor a mixture of 240.5 kg deionised water and 146.4 kg concentrated HCl was mixed. This was cooled to 15-20°C. The reaction mixture from the demethylation was introduced into the diluted hydrochloric acid between 20-25°C. Optionally, 51.2 kg dichloromethane was added to the suspension, stirred for 30 minutes and was centrifuged, washed with 60 kg deionised water and 20 kg isopropanol. Drying gave 15.9 kg of product.
The product was suspended in 185.3 kg of ethanol. The mixture was then stirred at 78°C for an hour, then cooled to 20-25°C and stirred for 1 hour. The suspension was then centrifuged and the filtercake was washed with 44.5 kg ethanol, 96% . The solid material was dried at 50°C in vacuum in a stainless steel tray drier. 14.35 kg (90.3% yield) dry product was obtained.
A reactor was charged with 317.2 kg formic acid and dry product. The mixture was heated to 65 °C until all the solid dissolves. The hot solution was then filtered to an empty 1000 L reactor, was rinsed with 20 kg formic acid, then the formic acid solution was distilled partially off under vacuum to around 80-100L. 260 kg isopropanol was then introduced at 50-60°C and stirred for 30-35 minutes. The mixture was then cooled to 20-25°C with water in the jacket and was allowed to stir min 2 hours. The suspension was then centrifuged and was washed with 25 kg isopropanol. The wet material was removed from the fuge and was transferred into vacuum tray drier and was dried until constant weight under vacuum at 45-50°C resulting in 13.6 kg product, with a yield of 95.3% .
If desired the product may be employed in preparation of micronized material for use in pharmaceutical compositions.
Example 7e
Preparation of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
A suspension of product of Example 5e (34.1kg) in N-Methyl pyrrolidone (NMP) (182kg) is warmed to 50 °C until dissolution and then cooled to 5°C followed by a consecutive temperature controlled addition of aluminium chloride (9.8 kg) and pyridine (18.2kg). After addition of pyridine is complete the reaction mixture is warmed to 60°C and stirred for at least 2 hours. The reaction mixture is cooled to 10-16°C (e.g. 11, 13, 15°C) before an aqueous diluted hydrochloric acid (4M solution, 283L) is dosed maintaining the temperature below 25 °C. During the addition of the hydrochloric acid the crude product is precipitated from the NMP/water matrix as a yellow solid. The yellow solid is filtered and subsequently washed with water (179kg), 2-propanol (105kg). The wet solid is dried under vacuum at 55°C.
A suspension of wet product (25.1kg) in formic acid (813kg) is warmed to max. 67°C. The mixture is stirred at 67°C until complete dissolution of the product is achieved. The hot solution is filtered and the filtrate is cooled to 40 – 45°C before the product is precipitated first by concentration of the solution to approx. 40% (v/v) of its original volume followed by addition of the anti solvent 2-propanol (380kg). After addition of 2-propanol the resulting suspension is stirred at 55-60°C for crystal ripening followed by cooling to RT and filtration. The filter cake is washed with 2-propanol (38kg) and then dried at max. 58°C until LOD max. 0.5%). The product may be milled (for example using the method of Example 8).
Example 8
Micronization of 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol with MC JETMILL® type 200 milling equipment (micronization through spiral jet mills)
Equipment:
Mill: MC JETMILL® 200
Dosing unit: K-Tron T 35
Cyclone: type 600
Each micronization trial was performed on at least 2 kg of 5-(3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol.
The following working parameters have been defined for the micronization:
Feed rate range: 24.0-48.0 kg/h (200-400 g/30sec.)
Mill pressure range: 3.0-4.0 bar
Venturi pressure range: 3.0-4.0 bar; preferably the Venturi pressure is the same as the mill pressure
Using the above equipment and working parameters the microparticles of 5-(3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol comply with the following particle size specification (particle size determined by optical microscopy): D10 (EDC) is not less than 4 or 5 μm (for example not less than 5 μm), the D50 (EDC) is 10-45 or 15-30 μm (for example 15-30 μm) and the D95 (EDC) is not more than 60 or 70 μm (for example not more than 60 μm).
Example 9 (Figure 5)
2,5-Dichloro-4,6-dimethyl-nicotinonitrile is reacted with hydroxylamine in the presence of catalytic amounts of 1,10-phenanthroline monohydrate to yield the aldoxime (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide which represents the first coupling partner towards the synthesis of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene. The second coupling partner 5-nitro-vanillic acid pure is synthesized from vanillic acid by nitration with 65 % nitric acid followed by re-crystallization of the crude 5-nitro-vanillic acid intermediate from acetic acid. The convergent assembly of the oxadiazole moiety in 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene is achieved by first activation of 5-nitro-vanillic acid as its acid chloride and subsequent coupling with the aldoxime (Z)-2,5-dichloro-N’-hydroxy-4,6-dimethylnicotinimidamide. Cyclisation of the initially formed coupling product is achieved thermally to give the oxadiazole moiety by elimination of water. The reaction
mixture of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene, after ring closure reaction, is concentrated and product isolated from 1,4-dioxane/ethanol mixture in one step. Oxidation of the pyridine ring to the corresponding aryl-N-oxide (5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene) is achieved with trifluoroperoxoacetic acid which is formed in situ from UHP (Urea hydrogen peroxide complex) and trifluoroacetic acid anhydride. Unreacted 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene is subsequently removed from 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene by repeated re-crystallisation from formic acid/toluene. The analogue intermediate 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene pure with a level of 5-[3-(2,5-dichloro-4,6-dimethyl-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-2-hydroxy-3-methoxy-1-nitrobenzene below 0.10 %area is converted to 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol crude analogue by ether cleavage in the presence of a stoichiometric amount of aluminium chloride and pyridine. After completion of the reaction, the crude product is isolated by precipitation with an aqueous hydrochloric acid followed by dissolution of the precipitate in formic acid. After polish filtration of the resulting solution and partial solvent switch from formic acid to isopropanol, 5-[3-(2,5-dichloro-4,6-dimethyl-1-oxy-pyridin-3-yl)-[1,2,4]oxadiazol-5-yl]-3-nitrobenzene-1,2-diol is crystallized from the resulting formic acid/IPA crystallization matrix and finally optionally milled to the desired particle size.

PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2012107708
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007117165
scheme 1 depicts example how to produce a compound of the general formula IIB from a compound of the general formula IVB:

iii.

HB IVB
Scheme 1. Reagents: i. Piperidine, ethanol, reflux; ii. SO2Cl2, CCl4, reflux; iii. POCI3, 120 0C, 18 h; iv. 50% H2NOH, MeOH-H20, 1.25 mol % 1,10-phenanthroline hydrate.
The following reaction scheme 2 depicts an example how to produce certain compounds of general formula III:

I, R8 = methyl III, R8 = R9 = H
iv.
R9 = H

III, R8 = R9 = benzyl
Scheme 2. Reagents: i. 65 % HNO3, AcOH; ii. 48 % HBr (aq), 140 0C; iii. MeOH, HCl(g); iv. BnBr, K2CO3, CH3CN, reflux; v. 3N NaOH, MeOH/H2O.
The following reaction scheme 3 depicts an example how to produce the compound A, by activation of a compound according to general formula III followed by cyclisation involving condensation with a compound according to formula HB, dehydration and deprotection of the methyl residue protecting the hydroxyl group;
0C

compound A
| Cited Patent | Filing date | Publication date | Applicant | Title |
|---|---|---|---|---|
| WO2007013830A1 | Jul 26, 2006 | Feb 1, 2007 | Portela & Ca. S.A. | Nitrocatechol derivatives as comt inhibitors |
| WO2007117165A1 | Apr 10, 2007 | Oct 18, 2007 | Bial – Portela & Ca, S.A. | New pharmaceutical compounds |
| WO2008094053A1 * | Oct 10, 2007 | Aug 7, 2008 | Bial-Portela & Ca, S.A. | Dosage regimen for comt inhibitors |
| WO2012107708A1 * | Oct 21, 2011 | Aug 16, 2012 | Bial – Portela & Ca, S.A. | Administration regime for nitrocatechols |
| US20100168113 * | Apr 10, 2007 | Jul 1, 2010 | David Alexander Learmonth | Pharmaceutical Compounds |
| Reference | ||
|---|---|---|
| 1 | * | “[1,2,4]-oxadiazolyl nitrocatechol derivatives“, IP.COM JOURNAL, IP.COM INC., WEST HENRIETTA, NY, US, 3 May 2012 (2012-05-03), XP013150541, ISSN: 1533-0001 |
| 2 | * | KISS L E ET AL: “Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase“, JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY, US, vol. 53, no. 8, 22 April 2010 (2010-04-22), pages 3396 – 3411, XP002594266, ISSN: 0022-2623, [retrieved on 20100324], DOI: 10.1021/JM1001524 |
| 3 | L. E. KISS ET AL., J. MED. CHEM., vol. 53, 2010, pages 3396 – 3411 | |
| 4 | * | RASENACK N ET AL: “MICRON-SIZE DRUG PARTICLES: COMMON AND NOVEL MICRONIZATION TECHNIQUES“, PHARMACEUTICAL DEVELOPMENT AND TECHNOLOGY, NEW YORK, NY, US, vol. 9, no. 1, 1 January 2004 (2004-01-01), pages 1 – 13, XP009055393, ISSN: 1083-7450, DOI: 10.1081/PDT-120027417 |
REFERENCES
1: Bicker J, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. A new PAMPA model using an in-house brain lipid extract for screening the blood-brain barrier permeability of drug candidates. Int J Pharm. 2016 Jan 30. pii: S0378-5173(16)30072-2. doi: 10.1016/j.ijpharm.2016.01.074. [Epub ahead of print] PubMed PMID: 26836708.
2: Devos D, Moreau C. Opicapone for motor fluctuations in Parkinson’s disease. Lancet Neurol. 2015 Dec 22. pii: S1474-4422(15)00346-4. doi: 10.1016/S1474-4422(15)00346-4. [Epub ahead of print] PubMed PMID: 26725545.
3: Ferreira JJ, Lees A, Rocha JF, Poewe W, Rascol O, Soares-da-Silva P; Bi-Park 1 investigators. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: a randomised, double-blind, controlled trial. Lancet Neurol. 2015 Dec 22. pii: S1474-4422(15)00336-1. doi: 10.1016/S1474-4422(15)00336-1. [Epub ahead of print] PubMed PMID: 26725544.
4: Rascol O, Perez-Lloret S, Ferreira JJ. New treatments for levodopa-induced motor complications. Mov Disord. 2015 Sep 15;30(11):1451-60. doi: 10.1002/mds.26362. Epub 2015 Aug 21. Review. PubMed PMID: 26293004.
5: Gonçalves D, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. Development of a liquid chromatography assay for the determination of opicapone and BIA 9-1079 in rat matrices. Biomed Chromatogr. 2016 Mar;30(3):312-22. doi: 10.1002/bmc.3550. Epub 2015 Aug 17. PubMed PMID: 26147707.
6: Ferreira JJ, Rocha JF, Falcão A, Santos A, Pinto R, Nunes T, Soares-da-Silva P. Effect of opicapone on levodopa pharmacokinetics, catechol-O-methyltransferase activity and motor fluctuations in patients with Parkinson’s disease. Eur J Neurol. 2015 May;22(5):815-25, e56. doi: 10.1111/ene.12666. Epub 2015 Feb 4. PubMed PMID: 25649051.
7: Bonifácio MJ, Torrão L, Loureiro AI, Palma PN, Wright LC, Soares-da-Silva P. Pharmacological profile of opicapone, a third-generation nitrocatechol catechol-O-methyl transferase inhibitor, in the rat. Br J Pharmacol. 2015 Apr;172(7):1739-52. doi: 10.1111/bph.13020. Epub 2015 Jan 20. PubMed PMID: 25409768; PubMed Central PMCID: PMC4376453.
8: Kiss LE, Soares-da-Silva P. Medicinal chemistry of catechol O-methyltransferase (COMT) inhibitors and their therapeutic utility. J Med Chem. 2014 Nov 13;57(21):8692-717. doi: 10.1021/jm500572b. Epub 2014 Sep 2. PubMed PMID: 25080080.
9: Rocha JF, Falcão A, Santos A, Pinto R, Lopes N, Nunes T, Wright LC, Vaz-da-Silva M, Soares-da-Silva P. Effect of opicapone and entacapone upon levodopa pharmacokinetics during three daily levodopa administrations. Eur J Clin Pharmacol. 2014 Sep;70(9):1059-71. doi: 10.1007/s00228-014-1701-2. Epub 2014 Jun 14. PubMed PMID: 24925090.
10: Rocha JF, Santos A, Falcão A, Lopes N, Nunes T, Pinto R, Soares-da-Silva P. Effect of moderate liver impairment on the pharmacokinetics of opicapone. Eur J Clin Pharmacol. 2014 Mar;70(3):279-86. doi: 10.1007/s00228-013-1602-9. Epub 2013 Nov 24. PubMed PMID: 24271646.
11: Bonifácio MJ, Sutcliffe JS, Torrão L, Wright LC, Soares-da-Silva P. Brain and peripheral pharmacokinetics of levodopa in the cynomolgus monkey following administration of opicapone, a third generation nitrocatechol COMT inhibitor. Neuropharmacology. 2014 Feb;77:334-41. doi: 10.1016/j.neuropharm.2013.10.014. Epub 2013 Oct 19. PubMed PMID: 24148813.
12: Gonçalves D, Alves G, Fortuna A, Soares-da-Silva P, Falcão A. An HPLC-DAD method for the simultaneous quantification of opicapone (BIA 9-1067) and its active metabolite in human plasma. Analyst. 2013 Apr 21;138(8):2463-9. doi: 10.1039/c3an36671e. PubMed PMID: 23476919.
13: Rocha JF, Almeida L, Falcão A, Palma PN, Loureiro AI, Pinto R, Bonifácio MJ, Wright LC, Nunes T, Soares-da-Silva P. Opicapone: a short lived and very long acting novel catechol-O-methyltransferase inhibitor following multiple dose administration in healthy subjects. Br J Clin Pharmacol. 2013 Nov;76(5):763-75. doi: 10.1111/bcp.12081. PubMed PMID: 23336248; PubMed Central PMCID: PMC3853535.
14: Almeida L, Rocha JF, Falcão A, Palma PN, Loureiro AI, Pinto R, Bonifácio MJ, Wright LC, Nunes T, Soares-da-Silva P. Pharmacokinetics, pharmacodynamics and tolerability of opicapone, a novel catechol-O-methyltransferase inhibitor, in healthy subjects: prediction of slow enzyme-inhibitor complex dissociation of a short-living and very long-acting inhibitor. Clin Pharmacokinet. 2013 Feb;52(2):139-51. doi: 10.1007/s40262-012-0024-7. PubMed PMID: 23248072.
15: Palma PN, Bonifácio MJ, Loureiro AI, Soares-da-Silva P. Computation of the binding affinities of catechol-O-methyltransferase inhibitors: multisubstate relative free energy calculations. J Comput Chem. 2012 Apr 5;33(9):970-86. doi: 10.1002/jcc.22926. Epub 2012 Jan 25. PubMed PMID: 22278964.
16: Gonçalves D, Alves G, Soares-da-Silva P, Falcão A. Bioanalytical chromatographic methods for the determination of catechol-O-methyltransferase inhibitors in rodents and human samples: a review. Anal Chim Acta. 2012 Jan 13;710:17-32. doi: 10.1016/j.aca.2011.10.026. Epub 2011 Oct 20. Review. PubMed PMID: 22123108.
17: Kiss LE, Ferreira HS, Torrão L, Bonifácio MJ, Palma PN, Soares-da-Silva P, Learmonth DA. Discovery of a long-acting, peripherally selective inhibitor of catechol-O-methyltransferase. J Med Chem. 2010 Apr 22;53(8):3396-411. doi: 10.1021/jm1001524. PubMed PMID: 20334432.
////////BIA-9-1067, ONO-2370, BIA-91067, 923287-50-7, Opicapone, Catechol-O-methyl transferase, COMT inhibitor, Parkinson’s disease, PD, BIA 9-1067, BIA 91067, BIA-91067, BIA91067, EU 2016
OC1=CC(C2=NC(C3=C(Cl)[N+]([O-])=C(C)C(Cl)=C3C)=NO2)=CC([N+]([O-])=O)=C1O
