
Home » Solid tumours
Category Archives: Solid tumours
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
SY 5609
[ Fig. 0001] [ Fig. 0002]
[ Fig. 0003]
[ Fig. 0004]
SY 5609
CAS 2519828-12-5
Cancer, solid tumor
PHASE 1
A highly selective and potent oral inhibitor of cyclin-dependent kinase 7 (CDK7) for potential treatment of advanced solid tumors that harbor the Rb pa thway alterations (Syros Pharmaceuticals, Inc., Cambridge, Massachusetts, USA)
SY-5609 is an oral non-covalent CDK7 inhibitor in early clinical development at Syros Pharmaceuticals for the treatment of patients with advanced breast, colorectal, lung or ovarian cancer, or with solid tumors of any histology that harbor Rb pathway alterations.
- OriginatorSyros Pharmaceuticals
- ClassAntineoplastics; Small molecules
- Mechanism of ActionCyclin-dependent kinase-activating kinase inhibitors
- Phase IBreast cancer; Solid tumours
- 05 Aug 2021Roche plans the phase I/Ib INTRINSIC trial in Colorectal cancer (Combination therapy, Metastatic disease) in USA, Canada, Italy, South Korea, Spain and United Kingdom (NCT04929223)
- 05 Aug 2021Roche and Syros Pharmaceuticals enters into a clinical trial collaboration to evaluate atezolizumab in combination with SY 5609 in a clinical trial
- 05 Aug 2021Syros Pharmaceuticals plans a phase I trial in Cancer in second half of 2021
- NCT04247126
- https://clinicaltrials.gov/ct2/show/NCT04247126
At #ESMO21, we will be presenting new preclinical and clinical data on SY-5609, our highly selective and potent oral CDK7 inhibitor. #oncology #biotech Learn more: https://lnkd.in/gqYmWYhb
A Promising Approach for Difficult-to-Treat Cancers
SY-5609 is a highly selective and potent oral inhibitor of the cyclin-dependent kinase 7 (CDK7) in a Phase 1 dose-escalation trial in patients with advanced breast, colorectal, lung, ovarian or pancreatic cancer, or with solid tumors of any histology that harbor Rb pathway alterations.
SY-5609 represents a new approach to treating cancer that we believe has potential in a range of difficult-to-treat cancers. It has shown robust anti-tumor activity, including complete regressions, in preclinical models of breast, colorectal, lung and ovarian cancers at doses below the maximum tolerated dose. In preclinical studies of breast, lung and ovarian cancers, deeper and more sustained responses were associated with the presence of Rb pathway alterations. SY-5609 has also shown substantial anti-tumor activity in combination with fulvestrant in treatment-resistant models of estrogen receptor-positive breast cancer, including those resistant to both fulvestrant and a CDK4/6 inhibitor. Early dose-escalation data demonstrated proof-of-mechanism at tolerable doses.
Syros to Present New Data from Phase 1 Clinical Trial of SY-5609 in Oral Presentation at ESMO Congress 2021SEPTEMBER 13, 2021
Management to Host Conference Call on Monday, September 20, 2021 at 4:00 p.m. ET
CAMBRIDGE, Mass.–(BUSINESS WIRE)– Syros Pharmaceuticals (NASDAQ:SYRS), a leader in the development of medicines that control the expression of genes, today announced that it will present new data from the dose-escalation portion of the Phase 1 clinical trial of SY-5609, its highly selective and potent oral cyclin-dependent kinase 7 (CDK7) inhibitor, at the ESMO Congress 2021, taking place virtually September 16-21, 2021. The oral presentation will include safety, tolerability, and initial clinical activity data for SY-5609 in patients with breast, colorectal, lung, ovarian and pancreatic cancers, as well as in patients with solid tumors of any histology harboring Rb pathway alterations.
In separate poster presentations, Syros will present new preclinical data evaluating the antitumor and pharmacodynamic activity of intermittent dosing regimens for SY-5609 in ovarian cancer models, as well as new preclinical data evaluating antitumor activity of SY-5609 as a single agent and in combination with chemotherapy in KRAS-mutant models.
The abstracts for the two poster presentations are now available online on the ESMO conference website at: https://www.esmo.org/meetings/esmo-congress-2021/abstracts, and the presentations will become available for on-demand viewing starting September 16 at 08:30 CEST (September 16 at 2:30 a.m. ET). The abstract for the oral presentation on the Phase 1 dose-escalation data will remain embargoed until September 17 at 00:05 CEST (September 16 at 6:05 p.m. ET).
Details of the oral presentation are as follows:
Presentation Title: Tolerability and Preliminary Clinical Activity of SY-5609, a Highly Potent and Selective Oral CDK7 Inhibitor, in Patients with Advanced Solid Tumors
Session Date & Time: Monday, September 20, 17:30-18:30 CEST (11:30-12:30 p.m. ET)
Presentation Time: 17:55-18:00 CEST (11:55-12:00 p.m. ET)
Session Title: Mini Oral Session: Developmental Therapeutics
Presenter: Manish Sharma, M.D., START Midwest
Abstract Number: 518MO
Details of the poster presentations are as follows:
Presentation Title: Preclinical Evaluation of Intermittent Dosing Regimens on Antitumor and PD Activity of SY-5609, a Potent and Selective Oral CDK7 Inhibitor, in Ovarian Cancer Xenografts
Abstract Number: 14P
Presentation Title: SY-5609, a Highly Potent and Selective Oral CDK7 inhibitor, Exhibits Robust Antitumor Activity in Preclinical Models of KRAS Mutant Cancers as a Single Agent and in Combination with Chemotherapy
Abstract Number: 13P
Conference Call Information
Syros will host a conference call on Monday, September 20, 2021 at 4:00 p.m. ET to discuss the new clinical and preclinical data for SY-5609, which will be presented at the ESMO Congress 2021.
To access the live conference call, please dial 866-595-4538 (domestic) or 636-812-6496 (international) and refer to conference ID 4648345. A webcast of the call will also be available on the Investors & Media section of the Syros website at www.syros.com. An archived replay of the webcast will be available for approximately 30 days following the conference call.
About Syros Pharmaceuticals
Syros is redefining the power of small molecules to control the expression of genes. Based on its unique ability to elucidate regulatory regions of the genome, Syros aims to develop medicines that provide a profound benefit for patients with diseases that have eluded other genomics-based approaches. Syros is advancing a robust clinical-stage pipeline, including: tamibarotene, a first-in-class oral selective RARα agonist in RARA-positive patients with higher-risk myelodysplastic syndrome and acute myeloid leukemia; SY-2101, a novel oral form of arsenic trioxide in patients with acute promyelocytic leukemia; and SY-5609, a highly selective and potent oral CDK7 inhibitor in patients with select solid tumors. Syros also has multiple preclinical and discovery programs in oncology and monogenic diseases.
View source version on businesswire.com: https://www.businesswire.com/news/home/20210913005090/en/
PATENT
CN(C)C\C=C\C(=O)Nc1ccc(cc1)C(=O)Nc1cccc(c1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21
THZ1; 1604810-83-4; THZ-1; HY-80013
CLIP
SY 1365 MEVOCICLIB, CAS 1816989-16-8
CN(C)C\C=C\C(=O)Nc1ccc(nc1)C(=O)N[C@]1(C)C[C@@H](CCC1)Nc1ncc(Cl)c(n1)c1c[NH]c2ccccc21
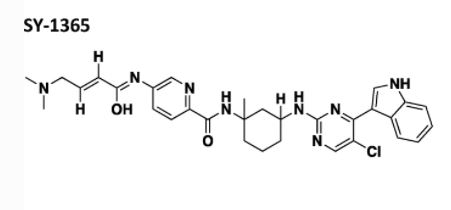
PATENT
PATENT
3-fluoro-4-(methylamino)-N-[(1S,3R)-1-methyl-3-[[4-(7-methyl-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]cyclohexyl]benzamide (Compound 130)
3-chloro-4-[[4-(dimethylamino)-3-hydroxy-butanoyl]amino]-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 129)
4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)-1-methylcyclohexyl)benzamide (Compound 128)
4-amino-3-fluoro-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 127)
4-amino-N-((1S,3R)-3-((5-chloro-4-(2-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 126)
4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 124)
Example 25 Synthesis of N1-(4-(((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)carbamoyl)phenyl)oxalamide (Compound 113)
Example 24 Synthesis of N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)-4-(4-(dimethylamino)butanamido)benzamide (Compound 105)
PATENT
4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)tricyclo[3.3.1.13,7]decanyl)benzamide (Compound 100).
+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-5 hydroxycyclohexyl)benzamide (Compound 101)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 102)
(1S,3R)-N-(4-aminophenyl)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexanecarboxamide (Compound 106)
4-amino-N-((1S,3R)-3-(5-cyclopropyl-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 103)
4-amino-N-((1S,3R)-3-(5-chloro-4-(pyridin-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 108)
4-amino-N-((1S,3R)-3-(5-cyano-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide (Compound 107)
(+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-5-fluorocyclohexyl)benzamide (Compound 110)
4-amino-N-(5-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)bicyclo[3.1.1]heptan-1-yl)benzamide (Compound 104)
4-amino-N4(1R,5S)-5-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-3,3-difluorocyclohexyl)benzamide (Compound 115)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzenesulfonamide (Compound 109).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-2-fluorobenzamide (Compound 112)
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-3-fluorobenzamide (Compound 111).
(+/−)-4-amino-N-(3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)-1-methylcyclohexyl)benzamide (Compound 116).
N-((1S,3R)-3-(4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-4-aminobenzamide (Compound 114).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)-2-morpholinobenzamide(Compound 117).
4-amino-N-((1S,3R)-3-(5-chloro-4-(1H-indol-3-yl)pyridin-2-ylamino)cyclohexyl)benzamide (Compound 118).
3-amino-N-(trans-4-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 119).
(1S,3R)-N1-(R)-1-(4-aminophenyl)-2,2,2-trifluoroethyl)-N3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)cyclohexane-1,3-diamine (Compound 120).
(1S,3R)-N1-(4-aminobenzyl)-N3-(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)-N1-methylcyclohexane-1,3-diamine.HCl (Compound 122).
4-amino-N-((1S,3R)-3-(5-chloro-4-(pyrazolo[1,5-a]pyridin-3-yl)pyrimidin-2-ylamino)cyclohexyl)benzamide.HCl (Compound 123).
Synthesis of 5-amino-N-((1S,3R)-3-(5-chloro-4-(1-methyl-1H-indol-3-yl)pyrimidin-2-ylamino)cyclohexyl)picolinamide (Compound 125)
Synthesis of N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)-4-(4-(dimethylamino)butanamido)benzamide (Compound 105)
Synthesis of N1-(4-(((1S,3R)-3-)(5-chloro-4-(1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)carbamoyl)phenyl)oxalamide (Compound 113)
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indazol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 124)
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(2-methyl-1H-indol-3-yl)pyrimidin-2-yl)amino)cyclohexyl)benzamide (Compound 126)
Synthesis of 4-amino-3-fluoro-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 127).
Synthesis of 4-amino-N-((1S,3R)-3-((5-chloro-4-(1H-indol-3-yl) pyrimidin-2-yl)amino)-1-methylcyclohexyl)benzamide (Compound 128)
Synthesis of 3-chloro-4-[[4-(dimethylamino)-3 hydroxy-butanoyl]amino]-N-[(1S,3R)-3-[[4-(1H-indazol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]-1-methyl-cyclohexyl]benzamide (Compound 129).
Synthesis of 3-fluoro-4-(methylamino)-N-[(1S,3R)-1-methyl-3-[[4-(7-methyl-1H-indol-3-yl)-5-(trifluoromethyl)pyrimidin-2-yl]amino]cyclohexyl]benzamide (Compound 130)
//////////////SY 5609, 2519828-12-5, Cancer, solid tumor, PHASE 1, SYROS

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
CC-90010
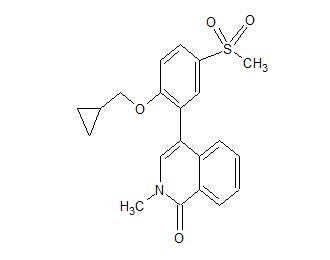
CC-90010
C21 H21 N O4 S, 383.46
CAS 1706738-98-8
1(2H)-Isoquinolinone, 4-[2-(cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]-2-methyl-
- 4-[2-(Cyclopropylmethoxy)-5-(methylsulfonyl)phenyl]-2-methyl-1(2H)-isoquinolinone
- 4-[2-(Cyclopropylmethoxy)-5-(methanesulfonyl)phenyl]-2-methylisoquinolin-1(2H)-one
- 4-[2-(Cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one
Quanticel Pharmaceuticals Inc, Michael John BennettJuan Manuel BetancortAmogh BoloorStephen W. KaldorJeffrey Alan StaffordJames Marvin Veal
![]()
Celgene (now a wholly owned subsidiary of Bristol-Myers Squibb ) , following its acquisition of Quanticel , is developing CC-90010, an oral inhibitor of BET (bromodomain and extraterminal) proteins, for the potential treatment of solid tumors and non-Hodgkin’s lymphoma. In August 2019, a phase I trial for diffuse astrocytoma, grade III anaplastic astrocytoma and recurrent glioblastoma was planned
PATENT
WO2018075796 claiming solid composition comprising a bromodomain inhibitor, preferably 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one in crystalline form A.
PATENT
WO2015058160 (compound 89, page 103).

Example 89: 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
Step 1 : 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one
[00344] A suspension of 4-bromo-2-methylisoquinolin-l-one (100 mg, 0.42 mmol), bis(pinacolato)diboron (214 mg, 0.84 mmol), Pd(dppf)Cl2 (31 mg, 0.04 mmol) and potassium acetate (104 mg, 1.05 mmol) in dioxane (2 mL) under nitrogen was warmed up to 90 °C for 135 minutes. It was then cooled down to room temperature and diluted with ethyl acetate (8 mL). The mixture was washed with aqueous saturated solution of NaHC03 (8 mL) and brine (8 mL). The organic phase was separated, dried over Na2S04, filtered and concentrated under reduced pressure. The residue was purifed by normal phase column chromatography (10-90% EtOAc/Hexanes) to give the title compound (44 mg, 37%). 1H NMR (CDC13, 400 MHz) δ 8.43 (d, J = 7.9 Hz, 1 H), 8.40 (dd, J = 8.2 Hz, 0.9 Hz, 1 H), 7.68 (s, 1 H), 7.65 (ddd, J = 8.2, 8.2, 1.1 Hz, 1 H), 7.46 (t, J = 7.5 Hz, 1 H), 3.63 (s, 3H), 1.38 (s, 12H). LCMS (M+H)+ 286. Step 2: 4-[2-(cyclopropylmethox -5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
[00345] The title compound was prepared in a manner similar to Example 18, step 3, substituting 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene for 4-bromo-2-methylisoquinolin-l(2H)-one and 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one for N-benzyl-2-methoxy-5-(tetramethyl-l,3,2-dioxaborolan-2-yl)benzamide. 1H NMR (DMSO-d6, 400 MHz) δ 0.09 (m, 2 H), 0.29 (m, 1H), 0.35 (m, 1H),
0.94 (m, 1H), 3.22 (s, 3H), 3.57 (s, 3H), 3.95 (m, 2H), 7.16 (d, J = 7.9 Hz, 1H), 7.37 (d, J =
8.8 Hz, 1H), 7.53 (m, 2H), 7.65 (t, J = 7.6 Hz, 1H), 7.81 (d, J = 2.4 Hz, 1H), 7.97 (dd, J = 8.8,
2.4 Hz, 1H), 8.30 (d, J = 8.1 Hz, 1H). LCMS (M+H)+ 384.
[00346] Alternatively, 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one can be prepared as described below.
Step 1 : 2-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)isoquinolin-l-one
[00347] A mixture of 4-bromo-2-methylisoquinolin-l-one (8.0 g, 33.6 mmol),
bis(pinacolato)diboron (17.1 g, 67.2 mmol), KOAc (6.6 g, 67.2 mmol), Pd2(dba)3 (3.1 g, 3.36 mmol) and X-Phos (1.6 g, 3.36 mmol) in anhydrous dioxane (200 mL) was stirred at 60 °C for 12 h. The reaction mixture was concentrated and the residue was purified by column chromatography on silica gel (PE : EA = 15 : 1) to give the title compound (6.0 g, 62 %) as a solid.
Step 2: 4-[2-(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-l-one
[00348] The title compound from Step 1 (5.0 g, 17.5 mmol), 2-bromo-l-(cyclopropylmethoxy)-4-methylsulfonylbenzene (6.4 g, 21 mmol), K3PO4 (9.3 g, 43.9 mmol) and Pd(dppf)Cl2 (1.4 g, 1.75 mmol) in a dioxane/water (100 mL / 10 mL) mixture were stirred at 60 °C for 12 hrs. The reaction mixture was concentrated under reduced pressure and the residue was purified by column chromatography on silica gel (EA : DCM = 1 : 4).
Appropriate fractions were combined and concentrated under reduce pressure. The resultant solid was recrystallized from DCM / MTBE (1 : 1, 50 mL) to give the title compound (4.0 g, 60 %) as a white solid. 1H NMR: (CDC13, 400 MHz) δ 8.51 (dd, Ji = 8.0 Hz, J2 = 0.8 Hz, 1 H), 7.98 (dd, Ji = 8.4 Hz, J2 = 2.4 Hz, 1 H), 7.86 (d, J = 2.4 Hz, 1 H), 7.53 (m, 2 H), 7.16 (d, J = 7.6 Hz, 1 H), 7.10 (m, 2 H), 3.88 (m, 2 H), 3.66 (s, 3 H), 3.09 (s, 3 H), 1.02-0.98 (m, 1 H), 0.44-0.38 (m, 2 H), 0.11-0.09 (m, 2 H). LCMS: 384.1 (M+H)+
Patent
WO-2020023438
A process for preparing bromodomain inhibitor, particularly 4-[2(cyclopropylmethoxy)-5-methylsulfonylphenyl]-2-methylisoquinolin-1-one (having HPLC purity of 99%; compound 1; (hereafter referred to as C-90010)) and its hydrates, solvates, prodrugs and salts comprising the reaction of a substituted 4-(methylsulfonyl)phenol compound with a quinoline derivative, followed by purification is claimed. Also claimed are novel intermediates of CC-90010 and their processes for preparation. Further claimed are novel crystalline form of CC-90010. CC-90010 is known and disclosed to be a bromodomain containing protein inhibitor, useful for treating cancer.
Scheme 10: Synthesis of Compound 1
[0090] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compound 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223g, l.OSmol). Agitation
was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum purged three times with nitrogen. The batch was heated to about 65 to about 75 °C (or any temperature in between and including these two values) and contents stirred for at least about 16 hours until reaction was complete by HPLC analysis. The batch was cooled to about 60 to about 70 °C (or any temperature in between and including these two values), agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at about 60 to about 70°C (or any temperature in between and including these two values). Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at about 60 to about 70°C (or any temperature in between and including these two values). After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at about 60 to about 70°C (or any temperature in between and including these two values). The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to about 60 to about 65°C (or any temperature in between and including these two values) and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at about 60 to about 65°C (or any temperature in between and including these two values). The slurry was cooled to about 15 to about 25°C (or any temperature in between and including these two values) over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at about 40°C with nitrogen purge. Yield: 139g of 1.
[0091] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox et al., Chem. Soc. Rev., 43: 412 (2014). Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used.
Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used.
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCk(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cb (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 fCC-900101 bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1); cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCh(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5);
Pd(PPh3)2Cl2 (Cas# 13965-03-2). Examples of catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
VI. Purification of Compound 1 (CC-90010! bv crystallization from formic acid and water
[0092] Described herein are methods of purifying Compound 1 by crystallization from formic acid and water. Also described are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1 and two metastable forms, Form 4
33 -a
and Form 5. Supporting data (XRPD, DSC, photomicroscopy) for all three forms is provided in the examples below.
[0093] The stmcture of Compound 1 (CC-90010) is shown below:
Example 1: Synthesis of Compound 1
[0217] Synthesis of compound 1 was accomplished according to Scheme 1 below. Referring to Scheme 1, synthesis commenced with bromination of starting material 4-(methylsulfonyl)phenol 4, to produce compound 5. Compound 5 was O-alkylated with (bromomethyl)cyclopropane to produce compound 6. Boronate Compound 2 was then formed by borylation of Compound 6 with Pd catalyst and bis(pinacolato)diboron to produce transient Compound 7, which was subsequenctly treated with diethanolamine (DBA) to afford cross-coupling partner Compound 2. Cross-coupling partner Compound 3 was formed in one pot starting from commercially available Compound 8. Compound 8 was N-methylated and brominated to afford Compound 3. Compounds 2 and 3 were cross-coupled (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)) to afford the target compound 1.
Scheme 1: Synthesis of compound 1
1.1: Bromination of 4
[0218] The bromination of Compound 4 to produce Compound 5 itself is simple, however stopping at the mono-brominated Compound 5 was challenging. The bis-brominated Compound 5-a (see Scheme 2 below) is a particularly pernicious impurity as it couples downstream to form a di ffi cult-to-purge impurity.
Scheme 2: Bromination of Compound 4
[0219] The key to high purity with reasonable yield was to exploit the solubility differences of the starting material Compound 4 (46 mg/ml at 20 °C) and the product Compound 5 (8 mg/ml) in CH2CI2. These solubility differences are summarized in Table 3 below.
[0220] This solubility difference is exploited by performing the reaction at a high
concentration to drive Compound 5 out of solution once formed, thereby minimizing its ability to react further with the brominating reagent to form Compound 5-a diBr. The reaction is seeded with Compound 5 to initiate its crystallization.
[0221] In Fig. 22 (Conversion of Compound 4 to Compound 5: Effect of Sulfuric Acid) it can be seen that in the absence of acid the initial reaction to Compound 5 is rapid, however the conversion plateaus at about 30% Compound 5. The main side product was found to be the impurity Compound 5-a diBr (see Fig. 23: Conversion of Compound 5 and Compound 5-a diBr: No H2SO4). Addition of increasing amounts of sulfuric acid leads to a higher conversion to desired Compound 5.
[0222] Fig. 24 (Compound 4 to Compound 5 Reaction Profile: Portion-wise Addition of NBS, Seeding) depicts further reaction control. The portion-wise addition ofNBS after addition of catalytic sulfuric acid minimizes the temperature rise, and the addition of Compound 5 after an initial NBS charge promotes the reactive crystallization of Compound 5. After about 6 to 7 hours of reaction it can be seen that the major product is Compound 5, with only a small (<5%) of the di-brominated impurity formed. In contrast, in a reaction where Compound 4 and all of the NBS were charged followed by the addition of 4 volumes of methylene chloride, a rapid exotherm resulted and undesired Compound 5-a diBr was found to be the major product.
[0223] Thus, the reaction was run under a high concentration in CH2CI2 with a portion-wise solid addition of NBS (to control both availability of the electrophile and the exotherm). An end of reaction slurry sample typically showed not more than 5% of the starting material Compound 4 remaining. After filtration the crude cake was washed with cold CH2CI2 and the OkCk-washed filter cake contained not more than 0.5% by weight dibrominated Compound 5-a. It also contained a large amount of HPLC-silent succinimide.
[0224] The following procedure was carried out: Compound 4 (25g, 145mmol) followed by CH2CI2 (lOOmL) were added to a reaction vessel and agitated. The batch was adjusted to 17 °C to 23 °C. Sulfuric acid was charged (2.7mL, Slmmol) to the batch maintaining 17 °C to 23°C. The batch was stirred at 17 °C to 23 °C for 10 minutes to 20 minutes. The first portion of A-bromosuccimide (NBS) was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The second portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23°C and stirred for at least 30 min. The batch was seeded with
Compound 5 (0.02wt) and stirred for ca. 30 min at 17 °C to 23 °C to induce crystallization.
[0225] The third portion of NBS was charged (6.5g, 36.5 mmol) to the batch at 17 °C to 23 °C and stirred for at least 30 min. NBS (6.5g, 36.5 mmol) was charged to the batch at 17 °C to 23 °C and stirred for at least 30 min. Additional CH2CI2 was charged (50mL) to the batch while maintaining 17 °C to 23 °C to aid in agitation and transfer for filtration. The batch was stirred at 17 °C to 23 °C until complete by HPLC analysis (~20 – 40 h). The product was collected by suction filtration. The filter cake was slurry washed with CH2CI2 (3 x 50mL) at 17 °C to 23 °C (target 20 °C). The filter cake was slurry washed with purified water (3.0vol) at 65 °C to 75 °C for 2 to 3 hours. Then, the filter cake was slurry washed with purified water (3 x 1.0 vol, 3 x 1.0 wt) at 17 °C to 23°C. The wet cake was dried under vacuum with nitrogen bleed at 60 °C. Yield: 27g 5 (74% molar) >97% by weight. ¾ NMR (500 MHz, de-DMSO) 8.01 (1H, d, 4J = 2.1 Hz, RO-Ar meta- H ), 7.76 (1H, dd, J = 8.6 and 4J = 2.1 Hz, RO-Ar meta-H ), 7.14 (1H, d, J = 8.6 Hz, RO-Ar ortho- H), 3.38 (1H, br s, OH), 3.20 (3H, s,
CHJ); MS (ES-) calc. 249/251; found 249/251. Melting point (MP): (DSC) 188 °C.
[0226] The above procedure allowed for the following modifications. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as acetonitrile, tetrahydrofuran, or 2- methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from about 2X vol to about 20 X vol (with respect to Compound 4). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 1.9 equiv. Bromination reagent addition: The brominating reagent can be added all at once, portion wise in about 2 to about 20 portions, or continuously. The addition times can vary from about 0 to about 72 hours. Temperature: Reaction temperatures from about 0 °C to about 40 °C could be used. Acids: Different acids can be envisioned, including benzenesulfonic acid, para-toluenesulfonic acid, triflic acid, hydrobromic acid, and trifluoroacetic acid. Isolation: Instead of directly filtering the product and washing with methylene chloride and water, at the end of reaction an organic solvent capable of dissolving Compound 5 could be charged, followed by an aqueous workup to remove succinimide, and addition of an antisolvent or solvent exchange to an appropriate solvent to crystallize Compound 4. Drying: A temperature range of about 10 to about 60 °C could be used for drying.
[0227] An alternative process to Compound 5 has also been developed. This process is advantageous in that it does not use a chlorinated solvent, and provides additional controls over the formation of the Compound 5-a dibromo impurity. See Oberhauser, T. J Org. Chem 1997, 62, 4504-4506. The process is as follows. Compound 4 (10 g, 58 mmol) and acetonitrile (100 ml) were charged to the reactor and agitated. The batch was cooled to -20 °C. Triflic acid (CF3SO3H or TfOH, 5.5 mL, 62 mmol) was charged while maintaining a batch temperature of -10 to -25 °C. N-bromosuccinimide was charged (NBS, 11.4 g, 64 mmol), stirred at -10 to -25 °C for 30 minutes, then warmed to 20 °C over 3 to 4 hours. Agitation was continued at 15 °C to 25 °C until reaction completion. If the reaction conversion plateaued before completion, the reaction was cooled to -5 to -15 °C, and additional NBS was added, the amount based off of unreacted starting material, followed by warming to 15 °C to 25 °C and reacting until complete.
[0228] After reaction completion, the batch was warmed to 40 °C to 50 °C and concentrated under reduced pressure to 40 mL. The batch was cooled to -5 °C to -15 °C and the resulting product solids were filtered off. The solids were slurry washed three times, each with 20 mL water, for at least 15 minutes. The final cake was dried at 50 °C to 60 °C under reduced pressure to furnish 10 g of 5 containing less than 0.1% MeCN, 0.07% water, and 0.1% triflic acid (TfOH) by weight.
[0229] Alternatives to the above procedure employing MeCN and TfOH are as follows. Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination Reagent Stoichiometry: Different amounts of the brominating reagent can be used, from about 0.8 equiv to about 2 equiv. Drying: A temperature range of about 10 °C to about 60 °C could be used for drying.
[0230] The impurity 5-a is was prepared and characterized as follows. 10 g of Compound 4 and sulfuric acid (35 mol%) were dissolved in MeOH (10 vol). The mixture was set to stir at 20 °C to 25 °C for 5-10 min and 2.0 equivalents of NBS were charged in one portion. The resulting yellow mixture was stirred for three days at 20-25 °C. The batch was concentrated under reduced pressure and the resulting solid was slurried in water at 95-100 °C for 3 hours. After a second overnight slurry in CH2CI2 at room temperature, the batch was filtered and dried to give a white solid 5-a (15.0 g, 78%). ¾ NMR (500 MHz, de-DMSO), 8.05 (2H, s, ArH), 3.40 (1H, br s, HO-Ar), 3.28 (3H, s, CH3); MS (ES‘) calc. 327/329/331; found
327/329/331; MP (DSC): 226 °C (onset 221 °C, 102 J/g); lit. 224-226 °C.
1.2: O-alkylation of 5 to produce 6
[0231] Compound 6 was prepared according to Scheme 7 below.
Scheme 7: O-alkylation of 5 to produce 6
[0232] Compound 5 (100 g, 398 mmol) and methyl ethyl ketone (MEK, 700 mL) were charged to the reaction vessel and agitated. Potassium carbonate (K2CO3, 325 mesh 82.56 g, 597 mmol) was then charged to the stirred reaction vessel at 15 °C to 25 °C.
Bromomethylcyclopropane (64.4 mL, 664 mmol) was charged to the reaction vessel over at least 1 hour, maintaining the temperature between 15 °C to 25 °C. MEK (200 mL) was added into the reactor and the reactor heated to 65 to 75 °C. The contents of the reaction vessel were stirred at 65 to 75°C for approximately 10 hours until reaction was complete by HPLC analysis. Water (3.0 vol, 3.0wt) was charged to the vessel maintaining the temperature at 65 to 75 °C. The batch was stirred at 65 to 75 °C. The phases were allowed to separate at 65°C to 75 °C and the lower aqueous phase was removed. Water (300 mL) was charged to the vessel maintaining the temperature at 65 °C to 75 °C. The batch was agitated for at least 10 minutes at 65 to 75 °C. The phases were allowed to separate at 65 °C to 75 °C and the lower aqueous phase was removed. The water wash was repeated once. The temperature was adjusted to 40 to 50°C. The mixture was concentrated to car. 500 mL under reduced pressure. The mixture was distilled under reduced pressure at up to 50 °C with MEK until the water content was <1.0% w/w. n-heptane (500mL) was charged to the vessel maintaining the temperature at 40 to 50 °C. The mixture was continuously distilled under vacuum with n-heptane (300mL), maintaining a 1L volume in the reaction vessel. Compound 6 seeds (0.0 lwt) were added at 40 to 50 °C. The mixture was continuously distilled under reduced pressure at up to 50 °C with n-heptane (300mL) while maintaining 1L volume in the reactor. The batch was cooled to 15 to 25 °C and aged for 2 hours. The product was collected by suction filtration. The filter cake was washed with a solution of 10% MEK in n-heptane (5vol) at 15 to 25°C. The filter cake was dried under reduced pressure at up to 40 °C under vacuum with nitrogen flow to afford 95g of 6. 1H NMR (500 MHz, de-DMSO) 8.07 (1H, d, 4J = 2.2 Hz, ArH), 7.86 (1H, d, J = 8.7 Hz, meta-ArH), 7.29 (1H, d, J = 8.8 Hz, ortho-AiK),
4.04 (2H, d, J = 6.9 Hz, OCH2CH), 3.21 (3H, s, CH3), 1.31-1.24 (1H, m, OCH), 0.62- 0.58 (2H, m, 2 x CHCHaHb), 0.40-0.37 (2H, m, 2 x CHC¾Hb); MS (ES+) calc. 305/307; found 305/307; MP: (DSC) 93 °C.
[0233] The following modifications of the above reaction, synthesis of 6 from 5, may be employed as well. Solvent: Different solvents could be used, for example acetone, methyl isobutyl ketone, ethyl acetate, isopropyl acetate, acetonitrile, or 2-methyl tetrahydrofuran. Reaction volume: Reaction volumes of 3 to 30 volumes with respect to 3 could be used. Base: Different inorganic bases, such as cesium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Also, organic bases, such as trimethylamine or diisopropyldiimide could be used. Base particle size: Different particle sizes of potassium carbonate from 325 mesh could be used. Reaction temperature: A lower temperature, such
as 50 °C could be used. A higher temperature, such as about 100 °C could be used. Any temperature above the boiling point of the solvent could be run in a pressure vessel.
Isolation: Different solvent ratios of MEK to n-heptane could be used. Different amounts of residual water can be left. Different amounts of seeds, from 0 to 50% could be used.
Seeding could take place later in the process and/or at a lower temperature. An un-seeded crystallization can be employed. A different isolation temperature, from 0 °C to 50 °C could be used. A different wash could be used, for example a different ratio of MEK to n-heptane. A different antisolvent from n-heptane could be used, such as hexane, pentane, or methyl tert-butyl ether. Alternatively, the batch could be solvent exchanged into a solvent where Compound 3 has a solubility of less than 100 mg/ml and isolated from this system. Drying: A temperature range of 10 to 60 °C could be used for drying.
[0234] Compound 10, shown below may also be formed as a result of O-alkylation of unreacted 4 present in product 5, or alternatively from or via a palladium mediated proteodesbromination or proteodesborylation in subsequent chemistry discussed in Example 1.3 below.
[0235] Preparation of methylsulfonylphenyl(cyclopropylmethyl) ether 10: Compound 4 (0.86 g, 5.0 mmol) and K2CO3 (1.04 g, 7.5 mmol) were slurried in acetone (17 mL, 20 vols). Cyclopropylmethyl bromide (0.73 mL, 7.5 mmol) was added in several small portions over ~1 minute and the reaction mixture heated to 50 °C for 48 hours, then cooled to 25 °C. Water (5.0 mL) was added with stirring and the acetone was evaporated on a rotary evaporator from which a fine white solid formed which was filtered off and returned to a vessel as a damp paste. A 1 : 1 mixture of MeOH/ water (8 mL) was added and heated to 40 °C with stirring. After 1 hour, the white solid was filtered off. Some residual solid was washed out with fresh water that was also rinsed through the cake, which was then isolated and left to air dry over the two days to give a dense white solid 10 (1.00 g, 88%). ¾ NMR (500 MHz, CDCb) 7.85
(2H, d, J = 8.8 Hz, RO-Ar ortho-H), 7.00 (2H, d, J = 8.8 Hz, RO-Ar meta- H), 3.87 (2H, d, J = 7.0 Hz, OCH2CH), 3.02 (3H, s, CHs), 1.34-1.23 (1H, m, OCH2CH), 0.72-0.60 (2H, m, 2 x CHCHflHb), 0.42-0.31 (2H, m, 2 x CHCH^.
1.3: Synthesis and Isolation Coupling Partner Boronic Ester 2
[0236] The final bond forming step to Compound 1 is a Suzuki-Miyaura coupling between Compounds 2 and 3, as shown in Scheme 3 below (Norio, M. and Suzuki, A., Chem. Rev., 95(7), 2457-2483 (1995)). Early studies demonstrated that the boronic ester of the isoquinolinone Compound 3-a had poor physical attributes and solid phase stability (Kaila, N. et al., J. Med Chem., 57: 1299-1322 (2014)). The pinacolatoboronate of the O-alkyl phenol, Compound 7, had acceptable solid phase stability and could be isolated via crystallization.
Scheme 3: Suzuki-Miyaura coupling between 2 and 3
[0237] Process robustness studies for the isolation of Compound 7, however, indicated that Compound 7 has poor solution stability, decomposing primarily to the proteodeborylated compound 10, as shown in Scheme 4 below. This was particularly problematic as the isolation process involved a solvent exchange from 2-MeTHF (2-methyl tetrahydrofuran) to iPrOAc (isopropyl acetate), which is not a fast unit operation on scale.
Scheme 4: Modification of 7
[0238] A search for a more stable boronic ester was undertaken. Early attempts targeted making N-methyliminodiacetic acid (MID A) boronate Compound 2-a (E. Gilis and M. Burke,“Multi step Synthesis of Complex Boronic Acids from Simple MIDA Boronates,” J Am. Chem. Soc., 750(43): 14084-14085 (2008)), however, all attempts resulted in product decomposition. Applicant then turned to a relatively obscure boronate formed by the addition of diethanolamine to Compound 7 (Bonin et al., Tetrahedron Lett., 52: 1132-1135 (2011)). Addition of diethanolamine to a solution of Compound 7 led to rapid ester formation and concomitant crystallization of Compound 2.
[0239] The discovery of boronic ester Compound 2 allowed for a simple, fast, high-yielding, high-purity process comprising the following procedure. Tetrahydrofuran (THF, 1500mL) was charged to a flask containing Compound 6 (100g, 328 mmol), bis(pinacolato)diboron (90.7g, 357 mmol) and cesium acetate (CsOAc, 158g, 822 mmol). The system was vacuum purged three times with nitrogen. Pd(PPh3)2Cl2 (13.8g, 20 mmol) was charged to the reaction and the system was vacuum purged three times with nitrogen. The reaction was then heated to 55 to 65°C.
[0240] The batch was stirred for approximately 8 hours until reaction was complete by HPLC analysis. The batch was cooled to 15 to 25 °C (target 20 °C ) and charged with silica gel (20g) and Ecosorb C-941 (20g). After lh, the mixture was filtered to remove solid. The residual solids were washed twice, each with THF (300mL). The filtrate and washes were combined. In a separate vessel, diethanolamine (34.5mL, 360 mmol) was dissolved in THF (250 mL). The diethanolamine solution in THF (25mL) was then charged to the batch. After 10 minutes, the batch was seeded with 2 (1 g) and aged for 1 to 2 hours. The remaining of the diethanolamine solution in THF was charged to the batch over at least 2 hours and the slurry was stirred for at least 2 hours. The product 2 was collected by suction filtration. The wet cake was washed thrice with THF (200mL). The material was dried under vacuum at 40 °C with nitrogen purge yielding 94.6g of 2.
[0241] The reaction to synthesize Compound 2 from Compound 6 described above may be modified as follows. Solvent: Different solvents from THF could be used, such as 1,4 dioxane or 2-methyltetrahydrofuran. Reaction volume: The reaction volume can be varied from 4 to 50 volumes with respect to compound 2. Catalyst and base: Different palladium catalyst and bases can be used for the borylation. Examples can be found in Chow et al., RSC Adv., 3 : 12518-12539 (2013). Borylation reaction temperature: Reaction temperatures from room temperature (20 °C) to solvent reflux can be used. Carbon/ Silica treatment:
The treatment can be performed without silica gel. The process can be performed without a carbon treatment. Different carbon sources from Ecosorb C-941 can be used. Different amounts of silica, from 0.01X to IX weight equivalents, can be used. Different amounts of Ecosorb C-941, from 0.01X to IX weight equivalents, can be used. Crystallization: A different addition rate of diethanolamine can be used. Different amounts of diethanolamine, from 1.0 to 3.0 molar equivalents can be used. A different cake wash with more or less THF can be used. Different amount of seeds from 0.0001X wt to 50X wt can be used.
Alternatively, the process can be unseeded. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
[0242] The subsequent Suzuki-Miyaura coupling between Compounds 2 and 3 also proceeded well, providing over 20 kg of crude compound 1 with an average molar yield of 80% and LCAP of 99.7%.
1.4: Synthesis of Coupling Partner 3
[0243] Cross-coupling partner 3 was prepared by two different processes corresponding to Schemes 8 and 9 shown below.
Scheme 8: Process A for preparation of 3
[0244] According to Process A, Compound 9 (100g, 628 mmol) was dissolved in acetonitrile (450 mL) at room temperature. In a separate vessel, N-bromosuccinimide (NBS, 112g, 628 mmol) was suspended in acetonitrile (1 L). Compound 9 in acetonitrile was charged to the NBS slurry over at least 45 minutes. The contents of the reaction vessel were warmed to 45 °C to 55 °C and the batch stirred until the reaction was complete by HPLC analysis. The batch was cooled to 35 °C to 45 °C and ensured dissolution. Norit SX plus carbon (lOg) was charged to the mixture and the reaction mixture adjusted to 55 °C to 60 °C. The mixture was stirred at 55 °C to 60 °C for about lh and the mixture filtered at 55 °C to 60 °C to remove solids. The solids were washed with acetonitrile (500mL) at 55 °C to 60 °C. The volume of the combined filtrate was reduced to 900 mL by distilling off acetonitrile under reduced pressure. The batch with Compound 3 (lg) and stirred at 35 °C to 45 °C for at least 60 minutes. The contents of the reaction vessel were cooled to 15 °C to 25 °C over at least 1 hour. Water (2000 mL) was charged to the reaction vessel over at least 90 minutes and the slurry aged for at least 60 minutes. The product was collected by suction filtration. The cake was washed with a premixed 5% solution of acetonitrile in water (300mL). The wet cake was dried under vacuum at 40 °C with nitrogen purge. Yield: 120g of 3.
[0245] The above procedure, Process A for this synthesis of 3, may be practiced with alternative reagents and conditions as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 9). Brominating agents: Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent Stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 °C to 60 °C could be used for drying.
Scheme 9: Process B for preparation of 3
[0246] According to Process B, Compound 3 can be formed starting from 8 via non-isolated compound 9 as follows. Compound 8 (80 g, 55 mmol), cesium carbonate (CS2CO3, 215 g, 66 mmol), and acetonitrile (800 mL) were charged to the reactor. The temperature was adjusted from 15 to 25 °C and iodomethane charged to the reactor (Mel, 86 g, 0.61 mol) while maintaining a batch temperature below 25 °C. The batch was heated to 40 °C and agitated for 10 hours to form Compound 9. The batch was cooled to 25 °C, filtered into a fresh reactor to remove solids, and the solids washed twice with acetonitrile. The combined organic layers were concentrated via atmospheric distillation to about 320 mL.
[0247] In a separate reactor N-bromosuccinimide (NBS, 98.1 g, 0.55 mol) was charged to acetonitrile (800 mL) and agitated. The batch containing Compound 9 was transferred to the NBS solution while maintaining a batch temperature of 15 to 25 °C. The batch was heated to 45 to 55 °C and agitated for at least 4 hours to allow for reaction completion to Compound 3. Upon reaction completion, Norit SX Plus activated carbon (8 g) was charged, and agitated at 45 to 55 °C for one hour. The batch was filtered into a fresh vessel, the Norit SX plus cake was washed with 400 ml of 45 to 55 °C acetonitrile. The acetonitrile layers were combined, cooled to 35 to 45 °C, and distilled under reduced pressure to 720 mL. The batch was adjusted to a temperature of 40 °C, charged with Compound 3 seeds (0.8 g), agitated for one hour, cooled to 15 to 25 °C over at least on hour, then charged with water (1600 mL) over at least two hours. The mixture was agitated for an additional one to two hours, filtered, the cake washed with a premixed 5% solution of acetonitrile in water (240 mL). The wet cake was dried under vacuum at 40°C with nitrogen purge. Yield: 52 g of 3.
[0248] Process B to synthesize Compound 3, described above, may be modified as follows. Solvents: Alternative solvents could be used. Examples include chlorinated solvents, such as methylene chloride, chloroform or 1,2 dichloroethane, and non-chlorinated solvents such as tetrahydrofuran, or 2-methyltetrahydrofuran. Reaction concentration: The reaction concentration can be varied from 2X vol to 40 X vol (with respect to Compound 8).
Alkylating reagent: Alternative methylating reagents to methyl iodide can be used such as dimethylsulfate. Alkylating reagent stoichiometry: 1 to 10 molar equivalents of methyl iodide may be used. Base: Different inorganic bases, such as potassium carbonate or phosphate bases (sodium, potassium, or cesium) could be used. Brominating agents:
Additional brominating reagents include bromine and l,3-dibromo-5,5-dimethylhydantoin. Bromination reagent stoichiometry: Different amounts of the brominating reagent can be used, from 0.8 equiv to 2 equiv. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. Seeding levels from 0.0001% to 50% can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying.
1.5: Cross-coupling of 2 and 3 to Produce Target Compound 1
[0249] 1 is synthesized by Suzuki cross-coupling of 3 and 2 according to Scheme 10 and as described below.
Scheme 10: Synthesis of 1
[0250] Acetonitrile (1.6L) was charged to a mixture of Compound 2 (156.7g, 460 mmol), Compovmd 3 (lOOg, 420 mmol) and potassium phosphate tribasic (223 g, l.OSmol). Agitation was begun and water (400mL) charged to the batch. The system was vacuum purged three times with nitrogen and charged with Pd(PPh3)2Cl2 (2.9g, 4 mmol) and the system vacuum
purged three times with nitrogen. The batch was heated to 65 to 75°C and contents stirred for at least 16 hours until reaction was complete by HPLC analysis. The batch was cooled to 60 to 70°C, agitation halted and the mixture allowed to settle. The bottom aqueous layer was removed. Water (150mL) and acetonitrile (700mL) were charged at 60 to 70°C. Ecosorb C-941 (15g) and Celite (lOg) were charged to the reaction vessel at 60 to 70°C. After lh, the mixture was filtered to remove solids. The solids were washed twice each with 18% water in acetonitrile (500 mL) at 60 to 70°C. The filtrates were combined and concentrated under atmospheric pressure to a final volume of 1.5L. The batch was cooled to 60 to 65°C and seeded with Compound 1 (1 g). After lh, water (500 mL) was charged over at least 1 hour at 60 to 65°C. The slurry was cooled to 15 to 25°C over 4 hours. The product was collected by suction filtration. The wet cake was washed with 45% water in acetonitrile (500mL) twice. The product was dried under vacuum at 40°C with nitrogen purge. Yield: 139g of 1.
[0251] The above procedure for coupling Compound 3 and Compound 2 to produce
Compound 1 may be modified in any of the ways that follow. Reaction solvents: Different reaction solvents from acetonitrile can be used, including tetrahydrofuran, 2-methyl tetrahydrofuran, toluene, and isopropanol. Boronic ester: Different boronic esters from Compound 2 can be used, including pinacolato ester compound 7, and the free boronic acid of Compound 2. Examples of boronic esters can be found in Lennox, Alister, J.J., Lloyd-Jones, Guy C. Chem. Soc. Rev., 2014, 43, 412. Carbon treatment: Different carbon treatments from Ecosorb C-941 could be used. Different amounts of carbon, from 0.01 to 0.5X weight can be used. The carbon can be eliminated. Different amounts of Celite, from 0.01 to 0.5X weight can be used. Crystallization: Different amounts of water, including 5 volumes to 50 volumes can be used. The crystallization can also proceed without the addition of seeds. Different water addition times and final hold times can be used. Different wash procedures can be used. Drying: A temperature range of 10 to 60 °C could be used for drying. Catalysts: Different metal and ligand combination could be used. Examples of metal/ligand combinations can be found in Maluenda, Irene; Navarro, Oscar, Molecules, 2015, 20, 7528. Various catalysts can be including: XPhos-3G (cas# 1445085-55-1);
cataCXium® A Pd 3G (CAS# 1651823-59-4); PdCk(DtBPF) (CAS# 95408-45-0); SPhos 3G (Cas# 1445085-82-4); AmPhos 3G (Cas# 1820817-64-8); PCy3 3G (Cas# 1445086-12-3); Pd PEPPSI IPent Cas#l 158652-41-5); Pd(PPh3)2Cl2 (Cas# 13965-03-2). Examples of
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compound 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
catalyst systems that have been demonstrated to afford Compound 1 are listed below in Table 4 using boronic esters 2 or 7 in coupling to 3.
Table 4: Catalyst screen summary
1.6: Crystallization of 1
[0252] The final isolation of Compoxmd 1 requires a polish filtration. For this, the batch must be completely soluble. Unfortunately, Compound 1 has low solubility in almost all
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Class 3 and common Class 2 (e.g. THF, MeCN) solvents (ICH
Harmonized Guideline“Impurities: Guideline for Residual Solvents Q3C(R6)” October 20, 2016). A reasonable solubility was obtained in a warm MeCN-water mix, but this is not an optimal system (requires a heated filtration, MeCN has a residual solvent limit of only 410 ppm). Additional solvents with reasonable solubility (>50 mg/ml) include N-methyl-2- pyrrolidone (NMP) and dimethylacetamide (DMAc); but the development of isolations from these solvents required large volumes and raised residual solvent limit concerns (530 ppm or less for NMT and 1090 ppm or less for DMAc).
[0253] Formic acid is one ICH Class 3 solvent in which Compound 1 is highly soluble, having a solubility greater than 250 mg/ml at 20 °C. The solubility curve of Compound 1 in formic acid-Water is quite steep (see Figure 7), which enables a volumetrically efficient process.
[0254] Initial attempts to recrystallize crude Compound 1 involved dissolving in formic acid, polish filtering, and charging polish filtered water to about 20% supersaturation, followed by seeding with the thermodynamically most stable form (Form 1), followed by slow addition of water to the final solvent ratio, filtration, washing, and drying. Applicant observed that during the initial water charge, if the batch self-seeded it formed a thick slurry. X-ray diffraction (XRD), differential scanning calorimetry (DSC), and photomicroscopy demonstrated that a metastable form was produced. Once seeded with Form 1, the batch converted to the desired form (Form 1) prior to the addition of the remaining water. This process worked well during multiple lab runs, consistently delivering the desired form and purity with about 85% yield.
[0255] Unfortunately, upon scale-up, the batch did not convert to Form 1 after seeding. Additional water was charged and the batch began to convert to the desired form (mix of Form 1 and the metastable form by X-ray powder diffraction (XRPD)). When additional water was charged, the XRPD indicated only the metastable form. After a few hours with no change, Applicant continued the water charge to the final solvent ratio, during which time the batch eventually converted to Form 1. This process is summarized in Figure 8.
[0256] It was subsequently found by closer analysis of the plant and laboratory retains that a new metastable form was formed during scale up, with a similar, but different XRPD pattern. This form (metastable B) could be reproduced in the laboratory, but only when the batch has a high formic acid:water ratio and is seeded with Form 1. Without Form 1 seeds, metastable A is the kinetic form. Both metastable forms converted to Form 1 with additional water and/or upon drying, leading Applicant to believe that the metastable forms are formic acid solvates. These findings are summarized in Figure 9.
[0257] While there is little risk in not being able to control the final form, there is a risk of forming a difficult-to-stir slurry which can lead to processing issues. The crystallization procedure was therefore modified to keep a constant formic acid-water ratio. This was performed by charging 2.4X wt. formic acid and 1.75X wt. water (final solvent composition)
to the crystallizer with 0.03X wt. Form 1 seeds, and performing a simultaneous addition of Compound 1 in 6. IX wt. formic acid and 4.4X wt. water. The batch filtered easily and was washed with formic acid/water, then water, and dried under reduced pressure to yield 8.9 kg of Compound 1 (92% yield) with 99.85% LCAP and N.D. formic acid.
Example 2: Exemplary high throughput experimentation reaction
[0258] The following procedure is an exemplary high throughput experimentation reaction.
[0259] An overview of the reaction is shown below in Scheme 5:
Scheme 5: Reaction conditions tested for cross-coupling reaction of 2 and 3
[0260] Pd catalysts were dosed into the 24-well reactor vial as solutions (100 pL of 0.01 M solution in tetrahydrofuran (THF) or dichloroethane (DCE) depending upon the solubility of the ligand). Plates of these ligands are typically dosed in advance of the reaction, the solvent is removed by evacuation in an evaporative centrifuge and plates are stored in the glovebox. The catalysts screened in the coupling are the following: XPhos, SPhos, CataCXium A, APhos, P(Cy)3, PEPPSI-IPent. For the first five ligands, these were initially screened as the Buchwald Pd G2/G3 precatalysts.
[0261] To the plates was then added a stock solution of Compound 3 (10 pmol) and Compound 2 (12 pmol) dissolved in the following solvents: dimethylformamide (DMF),
tetrahydrofuran (THF), butanol (/r-BuOH), and toluene. The base was then added as a stock solution (30 mmol) in 20 mL of water.
[0262] A heatmap summarizing catalyst performance is shown in Figs. 10A and 10B. High performance liquid chromatography (HPLC) yields for this screening span from <5% up to -85%. Larger circles indicate higher yield. Lighter circles indicate higher cleanliness.
[0263] A similarly designed screening of base and solvent also indicate that a range of alcoholic solvents (methanol, ethanol, propanol, 2-butanol, 2-propanol, and /-amyl alcohol) are also all viable in this coupling chemistry. Bases such as potassium phosphate, potassium carbonate, potassium acetate, and potassium hydroxide were all successful in achieving the coupling. Fig. 10B shows a heatmap with HPLC yields ranging from -50 – 95%. Larger, darker circles indicate higher yield.
[0264] This chemistry from microvial screening has been scaled to a laboratory process. To a 3 -necked jacketed 250 mL flask equipped with overhead stirring, nitrogen inlet, and thermocouple was added Compound 3 (1.0 eq, 4.00 grams), Compound 2 (1.2 eq, 1.71 x wt), potassium carbonate (3.0 eq, 1.74 x wt). The reactor was inerted three times and then degassed 2-propanol (24 x vol.) followed by degassed water (6 x vol) was then added.
Stirring was then initiated at 300 rpms. The reactor was then stirred and blanketed with nitrogen for 1 hour. The catalyst was then added (0.01 eq, 0.028 x wt) and stirring continued (300 rpms) and the reactor was heated into the Tj = 65 °C.
[0265] After 2 hours, with full conversion confirmed analytically, trioctylphosphine (0.1 eq, 0.16 x wt) dosed, and reaction mixture allowed to cool slowly to room temperature hours.
The reaction mixture was then filtered, washed with 2-propanol (4 x vol), 2-propanol: water (4: 1, 4 x vol), and then with water (4 x vol). Note: If 2 is dimer present in cake, an additional ethyl acetate (EtOAc) wash (4 x vol) can be added for purging. The cake was then transferred to a vacuum oven to dry overnight at 40 °C, -40 cm Hg, under nitrogen flow. After transfer to a bottle, 6.03 grams of 1 were isolated, 98.6% assay, 91% overall yield.
Scheme 6: Alternative reagents and solvents for cross-coupling
[0266] Based on the previously delineated results, it was expected that a variety of monodentate (PPI13 [triphenylphosphine], PBu3 [tributylphosphine], etc) and bidentate phosphines (dppf [1,1 ‘-bis(diphenylphosphino)ferrocene], BINAP [2,2 -bis(diphenylphosphino)- 1 , 1 -binaphthyl], Xantphos [4,5-bis(diphenylphosphino)-9,9-dimethylxanthene], dppe [l,2-bis(diphenylphosphino)ethane], etc) ligated to any number of Pd sources (Pd halides, Pd(H) precatalyts, Pd(0) sources) could reasonably be employed to arrive at the Compound 1 crude material. A range of organic solvents ranging from non-polar (heptane, benzene), protic (alcohols), polar aprotic (dimethylsulfoxide, dimethylformamide, dimethylacetamide, acetonitrile) as well as a variety of esters and ketones (acetone, 2-butanone, ethylacetate) should also serve as effective solvents for this reactivity. Finally, inorganic bases of varying strength (phosphates, carbonates, acetates, etc) along with organic variants such as triethylamine, l,8-diazabicyclo(5.4.0)undec-7-ene, and others in a wide pKa range are viable as stoichiometric basic additives.
Example 3: Exemplary Compound 5 process
[0267] The purpose of this example was to describe an exemplary process for making Compound 5.
[0268] Charge 4 (lOg, 58mmol) and acetonitrile (lOOmL) to a reaction vessel and start the stirrer. Adjust the batch to -18 °C to -22 °C (target -20 °C). Charge triflic acid (5.5mL, 62mmol) to the batch maintaining -10 °C to -25 °C (target -20 °C). Stir the batch at -10 °C to -25 °C (target -20 °C) for 10 to 20 minutes. Charge NBS (11.38g, 64mmol) to the batch at -10 °C to -25 °C (target -20 °C) and stir for ca. 30 min at -10 °C to -25 °C (target -20 °C). Warm the batch to 20 °C over 3-4 hours (reaction will occur when internal temp is between 5 °C and 15 °C). Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and sample for reaction completion.
[0269] If Compound 4 relative to Compound 5 is more than 5%:
[0270] Cool the bath to -5 °C to -15 °C (target -10 °C) (cooling below 0 °C to ensure selectivity). Charge NBS to the batch according to the follow formula: Mass of NBS = (% Compound 4 x lOg). Warm the batch to 20 °C over 1-2 hours. Stir the batch at 15 °C to 25 °C (target 20 °C) for approximately 1 hour and check reaction for completion. Proceed to next line.
[0271] If Compound 4 relative to Compound 5 is less than 5%:
[0272] Warm the batch to 40 °C to 50 °C (target 48 °C). Concentrate the batch under reduced pressure to a final volume of ~40mL. Cool the batch to -15 °C to -5 °C (target -10 °C) and stir for ca. lh. Filter the batch by suction filtration. Slurry wash the filter cake with purified water (3 x 20mL) at 15 °C to 25 °C (target 20 °C) for 10 to 15 minutes each wash. Remove a sample of the filter cake for analysis by ¾ NMR. Continue washing cake until the residual succimide is below 1.0%mol% relative to 5. Dry the filter cake at up to 60°C under vacuum and nitrogen purge. Analyse the 5 by HPLC analysis (97%w/w to 99%w/w). Expected yield: 60-85% theory (90-110% w/w).
Example 4: Purification of Compound 1 (CC-90010) by crystallization from formic acid and water.
[0273] This example describes a method for the purification of Compound 1 by
crystallization from formic acid and water. Also detailed are methods for obtaining three different polymorphs of Compound 1, including the most stable form, Form 1.
[0274] Figure 11 shows XH NMR of Compound 1 (CC-90010). Solvent: d6DMSO; and Figure 12 shows microscopy of Compound 1 (CC-90010) Form I. Figure 13 shows XRPD of Compound 1 (CC-90010) Form I, with peak information detailed in Table 6:
PATENT
US 20190008852
WO 2018081475
US 20180042914
WO 2016172618
WO 2015058160
/////////CC-90010, solid tumors , non-Hodgkin’s lymphoma, PHASE 1, CANCER, QUANTICEL
CS(=O)(=O)c4cc(C1=CN(C)C(=O)c2ccccc12)c(OCC3CC3)cc4
