
Home » fda 2021
Category Archives: fda 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tixagevimab
| (Heavy chain) QMQLVQSGPE VKKPGTSVKV SCKASGFTFM SSAVQWVRQA RGQRLEWIGW IVIGSGNTNY AQKFQERVTI TRDMSTSTAY MELSSLRSED TAVYYCAAPY CSSISCNDGF DIWGQGTMVT VSSASTKGPS VFPLAPSSKS TSGGTAALGC LVKDYFPEPV TVSWNSGALT SGVHTFPAVL QSSGLYSLSS VVTVPSSSLG TQTYICNVNH KPSNTKVDKR VEPKSCDKTH TCPPCPAPEF EGGPSVFLFP PKPKDTLYIT REPEVTCVVV DVSHEDPEVK FNWYVDGVEV HNAKTKPREE QYNSTYRVVS VLTVLHQDWL NGKEYKCKVS NKALPASIEK TISKAKGQPR EPQVYTLPPS REEMTKNQVS LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSKLTVDK SRWQQGNVFS CSVMHEALHN HYTQKSLSLS PGK (Light chain) EIVLTQSPGT LSLSPGERAT LSCRASQSVS SSYLAWYQQK PGQAPRLLIY GASSRATGIP DRFSGSGSGT DFTLTISRLE PEDFAVYYCQ HYGSSRGWTF GQGTKVEIKR TVAAPSVFIF PPSDEQLKSG TASVVCLLNN FYPREAKVQW KVDNALQSGN SQESVTEQDS KDSTYSLSST LTLSKADYEK HKVYACEVTH QGLSSPVTKS FNRGEC (Disulfide bridge: H22-H96, H101-H106, H150-H206, H216-L216, H232-H’232, H235-H’235, H267-H327, H373-H431, H’22-H’96, H’101-H’106, H’150-H’206, H’226-L’216, H’267-H’327, H’373-H’431, L23-L89, L136-L196, L’23-L’89, L’136-L’196) |
Tixagevimab
FDA 2021, 2021/12/8
ANTI VIRAL, CORONA VIRUS, PEPTIDE
Monoclonal antibody
Treatment and prevention of SARS-CoV-2 infection
| Formula | C6488H10034N1746O2038S50 |
|---|---|
| CAS | 2420564-02-7 |
| Mol weight | 146704.817 |
- 2196
- AZD-8895
- AZD8895
- COV2-2196
- Tixagevimab
- Tixagevimab [INN]
- UNII-F0LZ415Z3B
- WHO 11776
- OriginatorVanderbilt University
- DeveloperAstraZeneca; INSERM; National Institute of Allergy and Infectious Diseases
- ClassAntivirals; Monoclonal antibodies
- Mechanism of ActionVirus internalisation inhibitors
- RegisteredCOVID 2019 infections
- 24 Dec 2021Pharmacodynamics data from a preclinical trial in COVID-2019 infections released by AstraZeneca
- 16 Dec 2021Pharmacodynamics data from a preclinical trial in COVID-2019 infections released by AstraZeneca
- 10 Dec 2021Registered for COVID-2019 infections (In the elderly, Prevention, In adults) in USA (IM) – Emergency Use Authorization
Tixagevimab/cilgavimab is a combination of two human monoclonal antibodies, tixagevimab (AZD8895) and cilgavimab (AZD1061) targeted against the surface spike protein of SARS-CoV-2[4][5] used to prevent COVID-19. It is being developed by British-Swedish multinational pharmaceutical and biotechnology company AstraZeneca.[6][7] It is co-packaged and given as two separate consecutive intramuscular injections (one injection per monoclonal antibody, given in immediate succession).[2]
/////////////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Development
In 2020, researchers at Vanderbilt University Medical Center discovered particularly potent monoclonal antibodies, isolated from COVID-19 patients infected with a SARS-CoV-2 circulating at that time. Initially designated COV2-2196 and COV2-2130, antibody engineering was used to transfer their SARS-CoV-2 binding specificity to IgG scaffolds that would last longer in the body, and these engineered antibodies were named AZD8895 and AZD1061, respectively (and the combination was called AZD7442).[8]
To evaluate the antibodies’ potential as monoclonal antibody based prophylaxis (prevention), the ‘Provent’ clinical trial enrolled 5,000 high risk but not yet infected individuals and monitored them for 15 months.[9][10] The trial reported that those receiving the cocktail showed a 77% reduction in symptomatic COVID-19 and that there were no severe cases or deaths. AstraZeneca also found that the antibody cocktail “neutralizes recent emergent SARS-CoV-2 viral variants, including the Delta variant“.[7]
In contrast to pre-exposure prophylaxis, the Storm Chaser study of already-exposed people (post-exposure prophylaxis) did not meet its primary endpoint, which was prevention of symptomatic COVID-19 in people already exposed. AZD7442 was administered to 1,000 volunteers who had recently been exposed to COVID.[9]
Regulatory review
In October 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of tixagevimab/cilgavimab, which is being developed by AstraZeneca AB, for the prevention of COVID-19 in adults.[11]
Also in October 2021, AstraZeneca requested Emergency Use Authorization for tixagevimab/cilgavimab to prevent COVID-19 from the U.S. Food and Drug Administration (FDA).[12][13]
Emergency use authorization
On 14 November 2021, Bahrain granted emergency use authorization.[14]
On 8 December 2021, the U.S. Food and Drug Administration (FDA) granted emergency use authorization of this combination to prevent COVID-19 (before exposure) in people with weakened immunity or who cannot be fully vaccinated due to a history of severe reaction to coronavirus vaccines.[15] The FDA issued an emergency use authorization (EUA) for AstraZeneca’s Evusheld (tixagevimab co-packaged with cilgavimab and administered together) for the pre-exposure prophylaxis (prevention) of COVID-19 in certain people aged 12 years of age and older weighing at least 40 kilograms (88 lb).[2] The product is only authorized for those individuals who are not currently infected with the SARS-CoV-2 virus and who have not recently been exposed to an individual infected with SARS-CoV-2.[2]
References
- ^ “Evusheld- azd7442 kit”. DailyMed. Retrieved 4 January 2022.
- ^ Jump up to:a b c d “Coronavirus (COVID-19) Update: FDA Authorizes New Long-Acting Monoclonal Antibodies for Pre-exposure Prevention of COVID-19 in Certain Individuals”. U.S. Food and Drug Administration (FDA) (Press release). 8 December 2021. Retrieved 9 December 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ O’Shaughnessy, Jacqueline A. (20 December 2021). “Re: Emergency Use Authorization 104” (PDF). Food and Drug Administration. Letter to AstraZeneca Pharmaceuticals LP | Attention: Stacey Cromer Berman, PhD. Archived from the original on 29 December 2021. Retrieved 18 January 2022.
- ^ “IUPHAR/BPS Guide to PHARMACOLOGY”. IUPHAR. 27 December 2021. Retrieved 27 December 2021.
- ^ “IUPHAR/BPS Guide to PHARMACOLOGY”. IUPHAR. 27 December 2021. Retrieved 27 December 2021.
- ^ Ray, Siladitya (21 August 2021). “AstraZeneca’s Covid-19 Antibody Therapy Effective In Preventing Symptoms Among High-Risk Groups, Trial Finds”. Forbes. ISSN 0015-6914. Archived from the original on 21 August 2021. Retrieved 18 January 2022.
- ^ Jump up to:a b Goriainoff, Anthony O. (20 August 2021). “AstraZeneca Says AZD7442 Antibody Phase 3 Trial Met Primary Endpoint in Preventing Covid-19”. MarketWatch. Archived from the original on 21 August 2021. Retrieved 18 January 2022.
- ^ Dong J, Zost SJ, Greaney AJ, Starr TN, Dingens AS, Chen EC, et al. (October 2021). “Genetic and structural basis for SARS-CoV-2 variant neutralization by a two-antibody cocktail”. Nature Microbiology. 6 (10): 1233–1244. doi:10.1038/s41564-021-00972-2. ISSN 2058-5276. PMC 8543371. PMID 34548634.
- ^ Jump up to:a b Haridy, Rich (23 August 2021). “”Game-changing” antibody cocktail prevents COVID-19 in the chronically ill”. New Atlas. Retrieved 23 August 2021.
- ^ “AZD7442 PROVENT Phase III prophylaxis trial met primary endpoint in preventing COVID-19”. AstraZeneca (Press release). 20 August 2021. Retrieved 15 October 2021.
- ^ “EMA starts rolling review of Evusheld (tixagevimab and cilgavimab)”. European Medicines Agency. 14 October 2021. Retrieved 15 October 2021.
- ^ “AZD7442 request for Emergency Use Authorization for COVID-19 prophylaxis filed in US”. AstraZeneca US (Press release). 5 October 2021. Retrieved 15 October 2021.
- ^ “AZD7442 request for Emergency Use Authorization for COVID-19 prophylaxis filed in US”. AstraZeneca (Press release). 5 October 2021. Retrieved 15 October 2021.
- ^ Abd-Alaziz, Moaz; Elhamy, Ahmad (14 November 2021). Macfie, Nick (ed.). “Bahrain authorizes AstraZeneca’s anti-COVID drug for emergency use”. Reuters. Archived from the original on 23 November 2021. Retrieved 18 January 2022.
- ^ Mishra, Manas; Satija, Bhanvi (8 December 2021). Dasgupta, Shounak (ed.). “U.S. FDA authorizes use of AstraZeneca COVID-19 antibody cocktail”. Reuters. Archived from the original on 13 January 2022. Retrieved 18 January 2022.
External links
“Tixagevimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Cilgavimab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT04625972 for “Phase III Double-blind, Placebo-controlled Study of AZD7442 for Post-exposure Prophylaxis of COVID-19 in Adults (STORM CHASER)” at ClinicalTrials.gov
- Clinical trial number NCT04625725 for “Phase III Double-blind, Placebo-controlled Study of AZD7442 for Pre-exposure Prophylaxis of COVID-19 in Adult. (PROVENT)” at ClinicalTrials.gov
| Tixagevimab (teal, right) and cilgavimab (purple, left) binding the spike protein RBD. From PDB: 7L7E. | |
| Combination of | |
|---|---|
| Tixagevimab | Monoclonal antibody |
| Cilgavimab | Monoclonal antibody |
| Clinical data | |
| Trade names | Evusheld |
| Other names | AZD7442 |
| License data | US DailyMed: Tixagevimab |
| Routes of administration | Intramuscular |
| ATC code | J06BD03 (WHO) |
| Legal status | |
| Legal status | US: ℞-only via emergency use authorization[1][2][3] |
| Identifiers | |
| KEGG | D12262 |
| Clinical data | |
|---|---|
| Drug class | Antiviral |
| ATC code | None |
| Identifiers | |
| CAS Number | 2420564-02-7 |
| DrugBank | DB16394 |
| UNII | F0LZ415Z3B |
| KEGG | D11993 |
| Chemical and physical data | |
| Formula | C6488H10034N1746O2038S50 |
| Molar mass | 146706.82 g·mol−1 |
| Clinical data | |
|---|---|
| Drug class | Antiviral |
| ATC code | None |
| Identifiers | |
| CAS Number | 2420563-99-9 |
| DrugBank | DB16393 |
| UNII | 1KUR4BN70F |
| KEGG | D11994 |
| Chemical and physical data | |
| Formula | C6626H10218N1750O2078S44 |
| Molar mass | 149053.44 g·mol−1 |
/////////////////Tixagevimab, ANTI VIRAL, CORONA VIRUS, PEPTIDE, Monoclonal antibody, SARS-CoV-2 , WHO 11776, 2196, AZD-8895, AZD 8895, COV2-2196, COVID 19
NEW DRUG APPROVALS
ONE TIME
$10.00
Tezepelumab-ekko

(Heavy chain)
QMQLVESGGG VVQPGRSLRL SCAASGFTFR TYGMHWVRQA PGKGLEWVAV IWYDGSNKHY
ADSVKGRFTI TRDNSKNTLN LQMNSLRAED TAVYYCARAP QWELVHEAFD IWGQGTMVTV
SSASTKGPSV FPLAPCSRST SESTAALGCL VKDYFPEPVT VSWNSGALTS GVHTFPAVLQ
SSGLYSLSSV VTVPSSNFGT QTYTCNVDHK PSNTKVDKTV ERKCCVECPP CPAPPVAGPS
VFLFPPKPKD TLMISRTPEV TCVVVDVSHE DPEVQFNWYV DGVEVHNAKT KPREEQFNST
FRVVSVLTVV HQDWLNGKEY KCKVSNKGLP APIEKTISKT KGQPREPQVY TLPPSREEMT
KNQVSLTCLV KGFYPSDIAV EWESNGQPEN NYKTTPPMLD SDGSFFLYSK LTVDKSRWQQ
GNVFSCSVMH EALHNHYTQK SLSLSPGK
(Light chain)
SYVLTQPPSV SVAPGQTARI TCGGNNLGSK SVHWYQQKPG QAPVLVVYDD SDRPSWIPER
FSGSNSGNTA TLTISRGEAG DEADYYCQVW DSSSDHVVFG GGTKLTVLGQ PKAAPSVTLF
PPSSEELQAN KATLVCLISD FYPGAVTVAW KADSSPVKAG VETTTPSKQS NNKYAASSYL
SLTPEQWKSH RSYSCQVTHE GSTVEKTVAP TECS
(Disulfide bridge: H22-H96, H136-L213, H149-H205, H224-H’224, H225-H’225, H228-H’228, H231-H’231, H262-H322, H368-H426, H’22-H’96, H’136-L’213, H’149-H’205, H’262-H’322, H’368-H’426, L22-L87, L136-L195, L’22-L’87, L’136-L’195)
Tezepelumab-ekko
テゼペルマブ (遺伝子組換え)
| Formula | C6400H9844N1732O1992S52 |
|---|---|
| CAS | 1572943-04-4 |
| Mol weight | 144588.4306 |
PEPTIDE
UD FDA APPROVED, 12/17/2021, To treat severe asthma as an add-on maintenance therapy , Tezspire
Monoclonal antibody
Treatment of asthma and atopic dermatitis
Tezepelumab, sold under the brand name Tezspire, is a human monoclonal antibody used for the treatment of asthma.[4][5]
It blocks thymic stromal lymphopoietin (TSLP),[2] an epithelial cytokine that has been suggested to be critical in the initiation and persistence of airway inflammation.[6]
It was approved for medical use in the United States in December 2021.[2][3]
Medical uses
Tezepelumab is indicated for the add-on maintenance treatment of people aged twelve years and older with severe asthma.[2]
Research
In Phase III trials, tezepelumab demonstrated efficacy compared to placebo for patients with severe, uncontrolled asthma.[7][8]
Structural studies by X-ray crystallography showed that Tezepelumab competes against a critical part of the TSLPR binding site on TSLP.[1]
It is being studied for the treatment of chronic obstructive pulmonary disease, chronic rhinosinusitis with nasal polyps, chronic spontaneous urticaria and eosinophilic esophagitis (EoE).[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b Verstraete K, Peelman F, Braun H, Lopez J, Van Rompaey D, Dansercoer A, et al. (April 2017). “Structure and antagonism of the receptor complex mediated by human TSLP in allergy and asthma”. Nature Communications. 8 (1): 14937. Bibcode:2017NatCo…814937V. doi:10.1038/ncomms14937. PMC 5382266. PMID 28368013.
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761224s000lbl.pdf
- ^ Jump up to:a b c “Tezspire (tezepelumab) approved in the US for severe asthma”. AstraZeneca (Press release). 17 December 2021. Retrieved 17 December 2021.
- ^ Marone G, Spadaro G, Braile M, Poto R, Criscuolo G, Pahima H, et al. (November 2019). “Tezepelumab: a novel biological therapy for the treatment of severe uncontrolled asthma”. Expert Opinion on Investigational Drugs. 28 (11): 931–940. doi:10.1080/13543784.2019.1672657. PMID 31549891. S2CID 202746054.
- ^ Matera MG, Rogliani P, Calzetta L, Cazzola M (February 2020). “TSLP Inhibitors for Asthma: Current Status and Future Prospects”. Drugs. 80 (5): 449–458. doi:10.1007/s40265-020-01273-4. PMID 32078149. S2CID 211194472.
- ^ “Tezepelumab granted Breakthrough Therapy Designation by US FDA”. AstraZeneca (Press release). 7 September 2018.
- ^ “Studies found for: Tezepelumab”. ClinicalTrials.Gov. National Library of Medicine, National Institutes of Health, U.S. Department of Health and Human Services.
- ^ Menzies-Gow A, Corren J, Bourdin A, Chupp G, Israel E, Wechsler ME, et al. (May 2021). “Tezepelumab in Adults and Adolescents with Severe, Uncontrolled Asthma”. New England Journal of Medicine. 384 (19): 1800–09. doi:10.1056/NEJMoa2034975. PMID 33979488. S2CID 234484931.
External links
- “Tezepelumab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02054130 for “Study to Evaluate the Efficacy and Safety of MEDI9929 (AMG 157) in Adult Subjects With Inadequately Controlled, Severe Asthma” at ClinicalTrials.gov
- Clinical trial number NCT03347279 for “Study to Evaluate Tezepelumab in Adults & Adolescents With Severe Uncontrolled Asthma (NAVIGATOR)” at ClinicalTrials.gov
| Structural basis for inhibition of TSLP-signaling by Tezepelumab (PDB 5J13)[1] | |
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | thymic stromal lymphopoietin (TSLP) |
| Clinical data | |
| Trade names | Tezspire |
| Other names | MEDI9929, AMG 157, tezepelumab-ekko |
| License data | US DailyMed: Tezepelumab |
| Routes of administration | Subcutaneous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2][3] |
| Identifiers | |
| CAS Number | 1572943-04-4 |
| DrugBank | DB15090 |
| ChemSpider | None |
| UNII | RJ1IW3B4QX |
| KEGG | D11771 |
| Chemical and physical data | |
| Formula | C6400H9844N1732O1992S52 |
| Molar mass | 144590.40 g·mol−1 |
////////////Tezepelumab-ekko, Tezspire, PEPTIDE, APPROVALS 2021, FDA 2021, Monoclonal antibody
, asthma, atopic dermatitis, ANTI INFLAMATORY, テゼペルマブ (遺伝子組換え)

NEW DRUG APPROVALS
ONE TIME
$10.00
Efgartigimod alfa-fcab
DKTHTCPPCP APELLGGPSV FLFPPKPKDT LYITREPEVT CVVVDVSHED PEVKFNWYVD
GVEVHNAKTK PREEQYNSTY RVVSVLTVLH QDWLNGKEYK CKVSNKALPA PIEKTISKAK
GQPREPQVYT LPPSRDELTK NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS
DGSFFLYSKL TVDKSRWQQG NVFSCSVMHE ALKFHYTQKS LSLSPGK
(Disulfide bridge: 6-6′, 9-9′, 41-101, 147-205, 41′-101′, 147′-205′)
Efgartigimod alfa-fcab
| Formula | C2310H3554N602O692S14 |
|---|---|
| CAS | 1821402-21-4 |
| Mol weight | 51279.464 |
US FDA APPROVED 12/17/2021, To treat generalized myasthenia gravis
Press Release, Vyvgart, BLA 761195
| エフガルチギモドアルファ (遺伝子組換え) |
PEPTIDE
Treatment of IgG-driven autoimmune diseases

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-myasthenia-gravis
FDA Approves New Treatment for Myasthenia Gravis
Approval is the First of a New Class of Medication for this Rare, Chronic, Autoimmune, Neuromuscular DiseaseFor Immediate Release:December 17, 2021
The U.S. Food and Drug Administration today approved Vyvgart (efgartigimod) for the treatment of generalized myasthenia gravis (gMG) in adults who test positive for the anti-acetylcholine receptor (AChR) antibody.
Myasthenia gravis is a chronic autoimmune, neuromuscular disease that causes weakness in the skeletal muscles (also called voluntary muscles) that worsens after periods of activity and improves after periods of rest. Myasthenia gravis affects voluntary muscles, especially those that are responsible for controlling the eyes, face, mouth, throat, and limbs. In myasthenia gravis, the immune system produces AChR antibodies that interfere with communication between nerves and muscles, resulting in weakness. Severe attacks of weakness can cause breathing and swallowing problems that can be life-threatening.
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” said Billy Dunn, M.D., director of the Office of Neuroscience in the FDA’s Center for Drug Evaluation and Research. “Today’s approval is an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases.”
Vyvgart is the first approval of a new class of medication. It is an antibody fragment that binds to the neonatal Fc receptor (FcRn), preventing FcRn from recycling immunoglobulin G (IgG) back into the blood. The medication causes a reduction in overall levels of IgG, including the abnormal AChR antibodies that are present in myasthenia gravis.
The safety and efficacy of Vyvgart were evaluated in a 26-week clinical study of 167 patients with myasthenia gravis who were randomized to receive either Vyvgart or placebo. The study showed that more patients with myasthenia gravis with antibodies responded to treatment during the first cycle of Vyvgart (68%) compared to those who received placebo (30%) on a measure that assesses the impact of myasthenia gravis on daily function. More patients receiving Vyvgart also demonstrated response on a measure of muscle weakness compared to placebo.
The most common side effects associated with the use of Vyvgart include respiratory tract infections, headache, and urinary tract infections. As Vyvgart causes a reduction in IgG levels, the risk of infections may increase. Hypersensitivity reactions such as eyelid swelling, shortness of breath, and rash have occurred. If a hypersensitivity reaction occurs, discontinue the infusion and institute appropriate therapy. Patients using Vyvgart should monitor for signs and symptoms of infections during treatment. Health care professionals should administer appropriate treatment and consider delaying administration of Vyvgart to patients with an active infection until the infection is resolved.
The FDA granted this application Fast Track and Orphan Drug designations. The FDA granted the approval of Vyvgart to argenx BV.
///////////efgartigimod alfa-fcab, Vyvgart, FDA 2021,APPROVALS 2021, myasthenia gravis, argenx BV, Fast Track, Orphan Drug, PEPTIDE,
| エフガルチギモドアルファ (遺伝子組換え) |

NEW DRUG APPROVALS
one time
$10.00
Maribavir

Maribavir
- Molecular FormulaC15H19Cl2N3O4
- Average mass376.235 Da
FDA APROVED 11/23/2021, Livtencity1263 W94, 1263W94
176161-24-3[RN]
1H-Benzimidazol-2-amine, 5,6-dichloro-N-(1-methylethyl)-1-β-L-ribofuranosyl-
UNII-PTB4X93HE1, марибавир , ماريبافير ,马立巴韦 , BW-1263W94
Camvia, D04859, G1263, GW257406X
1263W94; BW-1263W94; GW-1263; GW-257406X; SHP-620; VP-41263
Company:GlaxoSmithKline (Originator) , Shire
MOA:UL97 kinase inhibitorIndication:CMV prophylaxis
To treat post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV
Press Release
Reference:1. WO9601833A1.
Syn
US 6204249
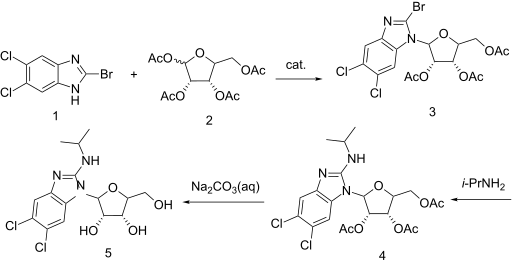

https://patents.google.com/patent/WO2001077083A1/enExample 7: 5,6-Dichloro-2-(isoproylamino)-1-(β-L-ribofuranosyl)-1 H-benzimidazolesoprylamino (10 mL) and 2-bromo-5,6-dichloro-1-(2,3,5-tri-0-acetyl-β-L- ribofuranosyl)-1 H-benzimidazole (1.0 g, 1.9 mmol) were combined with absolute ethanol (20 mL) and stirred at 75°C for 48 h. The reaction mixture was concentrated and purified on a silica gel column (2.5 vm x 16 cm, 230-400 mesh) with 1 :20 methanol: dichloromethane to give product contaminated with a small amount of higher Rf material. This was repurified on a chromatotron, fitted with a 2 mm silica gel rotor, with 1 :25 methanol.dichloromethane to give a white solid (0.43 g, 1.15 mmol, 60o/o); [a]20D=(-)22.4 (c=0.5 DMF); UVλ™* (E): pH 7.0:304 nm (95,00), 275 (1 ,800) 260 (8,300); 0.1 NaOH: 304 nm (9,900), 275 (19,00), 260 (8,100); MS (Cl): m/z (re/, intensity) 376 (100, M+1); ‘H NMR (DMSO-de) d 7.59 (s, 1 H, Ar-H), 7.35 (s, 1 H, Ar- H), 6.90 (d, 1 H, NH, J=7.8 Hz), 5.73 (d, 1 H, H-1′, J=6.5 Hz), 5.62 (t, 1 H, OH, J=4.2 Hz), 5.27-5.23 (m, 2H, OH), 4.27 (apparent dd, 1 H, J=13.4 Hz, J=7.6 Hz), 4.11 -3.99 (m, 2H), 3.97 (br. s, 1 H), 3.72-3.61 (m, 2H, H-5’), 1.18 (d, 6H, CH(CH3)2, J=6.6 Hz).Anal. Calcd. for

H2O: C, 45.70; H, 5.37; N, 10.66. Found: C, 45.75; H, 4.98; N, 10.50.
Maribavir was in phase II clinical trials for the treatment of cytomegalovirus (CMV) infection. It was granted orphan drug designation by the FDA for the indication.
The drug was originally developed by the University of Michigan and was licensed to GlaxoSmithKline. ViroPharma (now subsidiary of Shire) acquired worldwide rights to the drug from GlaxoSmithKline in 2003.
Maribavir, sold under the brand name Livtencity, is an antiviral medication that is used to treat post-transplant cytomegalovirus (CMV).[1][2]
The most common side effects include taste disturbance, nausea, diarrhea, vomiting and fatigue.[2]
Maribavir is a cytomegalovirus pUL97 kinase inhibitor that works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.[2]
Maribavir was approved for medical use in the United States in November 2021.[2][3]
Medical uses
Maribavir is indicated to treat people twelve years of age and older and weighing at least 35 kilograms (77 lb) with post-transplant cytomegalovirus infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for cytomegalovirus.[2]
Contraindications
Maribavir may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these medications is not recommended.[2]
History
Maribavir is licensed by ViroPharma from GlaxoSmithKline in 2003, for the prevention and treatment of human cytomegalovirus (HCMV) disease in hematopoietic stem cell/bone marrow transplant patients. The mechanism by which maribavir inhibits HCMV replication is by inhibition of an HCMV encoded protein kinase enzyme called UL97 or pUL97.[4] Maribavir showed promise in Phase II clinical trials and was granted fast track status, but failed to meet study goals in a Phase III trial.[5] However, the dosage used in the Phase III trial may have been too low to be efficacious.[6]
A Phase II study with maribavir demonstrated that prophylaxis with maribavir displayed strong antiviral activity, as measured by statistically significant reduction in the rate of reactivation of CMV in recipients of hematopoietic stem cell/bone marrow transplants.[7] In an intent-to-treat analysis of the first 100 days after the transplant, the number of subjects who required pre-emptive anti-CMV therapy was statistically significantly reduced with maribavir compared to placebo.
ViroPharma conducted a Phase III clinical study to evaluate the prophylactic use for the prevention of cytomegalovirus disease in recipients of allogeneic stem cell transplant patients. In February 2009, ViroPharma announced that the Phase III study failed to achieve its goal, showing no significant difference between maribavir and a placebo at reducing the rate at which CMV DNA levels were detected in patients.[8]
The safety and efficacy of maribavir were evaluated in a Phase III, multicenter, open-label, active-controlled trial that compared maribavir with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat cytomegalovirus: ganciclovir, valganciclovir, foscarnet, or cidofovir.[2] In the study, 352 transplant recipients with cytomegalovirus infections who did not respond (with or without resistance) to treatment randomly received maribavir or treatment assigned by a researcher for up to eight weeks.[2] The study compared the two groups’ plasma cytomegalovirus DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable.[2] Of the 235 participants who received maribavir, 56% had levels of cytomegalovirus DNA below what was measurable versus 24% of the 117 participants who received an investigator-assigned treatment.[2]
The U.S. Food and Drug Administration (FDA) granted the application for maribavir orphan drug, breakthrough therapy and priority review designations.[2][3][9][10] The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.[2][3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs
Approval is for Cytomegalovirus, a Type of Herpes Virus
https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-common-type-post-transplant-infection-resistant-other-drugsFor Immediate Release:November 23, 2021
Today, the U.S. Food and Drug Administration approved Livtencity (maribavir) as the first drug for treating adults and pediatric patients (12 years of age and older and weighing at least 35 kilograms) with post-transplant cytomegalovirus (CMV) infection/disease that does not respond (with or without genetic mutations that cause resistance) to available antiviral treatment for CMV. Livtencity works by preventing the activity of human cytomegalovirus enzyme pUL97, thus blocking virus replication.
“Transplant recipients are at a much greater risk for complications and death when faced with a cytomegalovirus infection,” said John Farley, M.D., M.P.H., director of the Office of Infectious Diseases in the FDA’s Center for Drug Evaluation and Research. “Cytomegalovirus infections that are resistant or do not respond to available drugs are of even greater concern. Today’s approval helps meet a significant unmet medical need by providing a treatment option for this patient population.”
CMV is a type of herpes virus that commonly causes infection in patients after a stem cell or organ transplant. CMV infection can lead to CMV disease and have a major negative impact on transplant recipients, including loss of the transplanted organ and death.
Livtencity’s safety and efficacy were evaluated in a Phase 3, multicenter, open-label, active-controlled trial that compared Livtencity with a treatment assigned by a researcher running the study, which could include one or two of the following antivirals used to treat CMV: ganciclovir, valganciclovir, foscarnet or cidofovir. In the study, 352 transplant recipients with CMV infections who did not respond (with or without resistance) to treatment randomly received Livtencity or treatment assigned by a researcher for up to eight weeks.
The study compared the two groups’ plasma CMV DNA concentration levels at the end of the study’s eighth week, with efficacy defined as having a level below what is measurable. Of the 235 patients who received Livtencity, 56% had levels of CMV DNA below what was measurable versus 24% of the 117 patients who received an investigator-assigned treatment.
The most common side effects of Livtencity include taste disturbance, nausea, diarrhea, vomiting and fatigue. Livtencity may reduce the antiviral activity of ganciclovir and valganciclovir, so coadministration with these drugs is not recommended. Virologic failure due to resistance can occur during and after treatment with Livtencity, therefore CMV DNA levels should be monitored and Livtencity resistance should be checked if the patient is not responding to treatment or relapses.
Livtencity received Breakthrough Therapy and Priority Review designations for this indication. Breakthrough Therapy designation is a process designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s). Priority Review designation directs overall attention and resources to the evaluation of applications for drugs that, if approved, would be significant improvements in the safety or effectiveness of the treatment, diagnosis or prevention of serious conditions when compared to standard applications.
The FDA granted the approval of Livtencity to Takeda Pharmaceuticals Company Limited.
Related Information
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215596lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l m “FDA Approves First Treatment for Common Type of Post-Transplant Infection that is Resistant to Other Drugs”. U.S. Food and Drug Administration (FDA) (Press release). 23 November 2021. Retrieved 23 November 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “Takeda’s Livtencity (maribavir) Approved by U.S. FDA as the First and Only Treatment for People Ages 12 and Older with Post-Transplant Cytomegalovirus (CMV), Refractory (With or Without Genotypic Resistance) to Conventional Antiviral Therapies”. Takeda (Press release). 23 November 2021. Retrieved 26 November 2021.
- ^ Biron KK, Harvey RJ, Chamberlain SC, Good SS, Smith AA, Davis MG, et al. (August 2002). “Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L-riboside with a unique mode of action”. Antimicrobial Agents and Chemotherapy. 46 (8): 2365–72. doi:10.1128/aac.46.8.2365-2372.2002. PMC 127361. PMID 12121906.
- ^ Marty FM, Ljungman P, Papanicolaou GA, Winston DJ, Chemaly RF, Strasfeld L, et al. (April 2011). “Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial”. The Lancet. Infectious Diseases. 11 (4): 284–92. doi:10.1016/S1473-3099(11)70024-X. PMID 21414843.
- ^ Snydman DR (April 2011). “Why did maribavir fail in stem-cell transplants?”. The Lancet. Infectious Diseases. 11 (4): 255–7. doi:10.1016/S1473-3099(11)70033-0. PMID 21414844.
- ^ Phase 2 Data Shows Maribavir Markedly Reduced Rate Of Cytomegalovirus Infection And Disease In Bone Marrow Transplant Patients, Medical News Today, Jun 2, 2008
- ^ ViroPharma:Maribavir Phase III Study Missed Goal;Shares Plunge, CNN Money, February 09, 2009
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 1 February 2007. Retrieved 26 November 2021.
- ^ “Maribavir Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 7 June 2011. Retrieved 26 November 2021.
External links
- “Maribavir”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02931539 for “Efficacy and Safety Study of Maribavir Treatment Compared to Investigator-assigned Treatment in Transplant Recipients With Cytomegalovirus (CMV) Infections That Are Refractory or Resistant to Treatment With Ganciclovir, Valganciclovir, Foscarnet, or Cidofovir” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Livtencity |
| Other names | 1263W94 |
| License data | USDailyMed: Maribavir |
| Routes of administration | By mouth |
| ATC code | J05AX10 (WHO) |
| Legal status | |
| Legal status | US:℞-only[1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 176161-24-3 |
| PubChemCID | 471161 |
| DrugBank | DB06234 |
| ChemSpider | 413807 |
| UNII | PTB4X93HE1 |
| ChEMBL | ChEMBL515408 |
| NIAID ChemDB | 070966 |
| CompTox Dashboard (EPA) | DTXSID60170091 |
| Chemical and physical data | |
| Formula | C15H19Cl2N3O4 |
| Molar mass | 376.23 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////Maribavir, APPROVALS 2021, FDA 2021, Livtencity, Takeda, Breakthrough Therapy, Priority Review , ORPHAN, UNII-PTB4X93HE1, марибавир , ماريبافير ,马立巴韦 , BW-1263W94, Camvia, D04859, G1263, GW257406X, 1263W94, BW-1263W94, GW-1263, GW-257406X, SHP-620, VP-41263,

NEW DRUG APPROVALS
ONE TIME
$10.00
Pafolacianine

Pafolacianine
OTL-38
- Molecular FormulaC61H67N9O17S4
- Average mass1326.495 Da
FDA APPROVED NOV 2021
2-{(E)-2-[(3E)-2-(4-{2-[(4-{[(2-Amino-4-oxo-3,4-dihydro-6-pteridinyl)methyl]amino}benzoyl)amino]-2-carboxyethyl}phenoxy)-3-{(2E)-2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene ]ethylidene}-1-cyclohexen-1-yl]vinyl}-3,3-dimethyl-1-(4-sulfobutyl)-3H-indolium-5-sulfonate OTL-38Tyrosine, N-[4-[[(2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl]amino]benzoyl]-O-[(6E)-6-[(2E)-2-[1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene]ethylidene]-2-[(E)-2-[3,3-dimethy l-5-sulfo-1-(4-sulfobutyl)-3H-indolium-2-yl]ethenyl]-1-cyclohexen-1-yl]-, inner salt
2-(2-(2-(4-((2S)-2-(4-(((2-amino-4-oxo-3,4-dihydropteridin-6-yl)methyl)amino)benzamido)-2-carboxyethyl)phenoxy)-3-(2-(3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2H-indol-2-ylidene)ethylidene)cyclohex-1-en-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-3H-indolium inner salt,sodium salt (1:4)
- 3H-Indolium, 2-(2-(2-(4-((2S)-2-((4-(((2-amino-3,4-dihydro-4-oxo-6-pteridinyl)methyl)amino)benzoyl)amino)-2-carboxyethyl)phenoxy)-3-(2-(1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene)ethylidene)-1-cyclohexen-1-yl)ethenyl)-3,3-dimethyl-5-sulfo-1 (4-sulfobutyl)-, inner salt,sodium salt (1:4)
1628423-76-6 [RN]
Pafolacianine sodium [USAN]
RN: 1628858-03-6
UNII: 4HUF3V875C
C61H68N9Na4O17S4+5
- Intraoperative Imaging and Detection of Folate Receptor Positive Malignant Lesions
Pafolacianine, sold under the brand name Cytalux, is an optical imaging agent.[1][2]
The most common side effects of pafolacianine include infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity.[2]
It was approved for medical use in the United States in November 2021.[2][3]
Pafolacianine is a fluorescent drug that targets folate receptor (FR).[1]
Medical uses
Pafolacianine is indicated as an adjunct for intraoperative identification of malignant lesions in people with ovarian cancer.[1][2]
History
The safety and effectiveness of pafolacianine was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.[2] Of the 134 women (ages 33 to 81 years) who received a dose of pafolacianine and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.[2]
The U.S. Food and Drug Administration (FDA) granted the application for pafolacianine orphan drug, priority review, and fast track designations.[2][4] The FDA granted the approval of Cytalux to On Target Laboratories, LLC.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

WO 2014149073
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014149073
In another aspect of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0029] In a fourth embodiment of the invention, this disclosure provides a method of synthesizing a compound having the formula
[0030]
[0032] wherein C is any carbon isotope. In this embodiment, the amino acid linker is selected from a group consisting of methyl 2-di-tert-butyl dicarbonate-amino-3-(4-phenyl)propanoate, 3-(4-hydroxyphenyl)-2-(di-tert-butyl-dicarbonate methylamino)propanoic acid, 2-amino-4-(4-hydroxyphenyl)butanoic acid, and Tert-butyl (2-di-tert-butyl dicarbonate- amino)-3-(4-hydroxyphenyl)propanoate . In a particular embodiment, the aqueous base is potassium hydroxide (KOH). The method of this embodiment may also further include purifying the compound by preparatory HPLC.
EXAMPLE 1 : General synthesis of Pte – L Tyrosine – S0456 (OTL-0038)
[0088] Scheme:
C33H37CIF3N
Reactants for Step I:
[0089] A 500 mL round bottom flask was charged with a stirring bar, pteroic acid
(12.0 g, 29.40 mmol, 1 equiv), (L)-Tyr(-OfBu)-OfBu- HCI (1 1 .63 g, 35.28 mmol, 1 .2
equiv) and HATU (13.45 g, 35.28 mmol, 1 .2 equiv) then DMF (147 mL) was added to give a brown suspension [suspension A]. DIPEA (20.48 mL, 1 17.62 mmol, 4.0 equiv) was added slowly to suspension A at 23 °C, over 5 minutes. The suspension turned in to a clear brown solution within 10 minutes of addition of DIPEA. The reaction was stirred at 23 °C for 2.5 h. Reaction was essentially complete in 30 minutes as judged by LC/MS but was stirred further for 2.5 h. The formation of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI (Figure 12) was confirmed by LC/MS showing m/z 409→m/z 684. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column . The reaction mixture was cannulated as a steady stream to a stirred solution of aq. HCI (2.0 L, 0.28 M) over the period of 30 minutes to give light yellow precipitate of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The precipitated Pte_N 10(TFA)_L_Tyr(- OfBu)-OfBu HCI was filtered using sintered funnel under aspirator vacuum, washed with water (8 * 300 mL) until the pH of the filtrate is between 3 and 4. The wet solid was allowed to dry under high vacuum for 12 hours on the sintered funnel. In a separate batch, where this wet solid (3) was dried under vacuum for 48 hours and then this solid was stored at -20 0 C for 48 h. However, this brief storage led to partial decomposition of 3. The wet cake (58 g) was transferred to a 500 mL round bottom flask and was submitted to the next step without further drying or purification.
Reactants for Step II:
The wet solid (58 g) was assumed to contain 29.40 mmol of the desired compound (3) (i. e. quantitative yield for the step I ).
[0090] A 500 mL round bottom flask was charged with a stirring bar, Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI as a wet cake (58 g, 29.40 mmol, 1 equiv). A solution of TFA:TIPS:H20 (95:2.5:2.5, 200 mL) was added at once to give a light brown suspension. The reaction content was stirred at 23°C for 1 .5 hours and was monitored by LC/MS. The suspension became clear dull brown solution after stirring for 5 minutes. LC/MS method: 0-50% acetonitrile in 20 mM aqueous NH4OAc for 5 min using Aquity UPLC-BEH C18, 1 .7μιη 2.1 * 50 mm column. The formation of Pte_TFA_L_Tyr (Figure 12) was confirmed by showing m/z 684→m/z 572. Reaction time varies from 30 min to 1 .5 hours depending on the water content of Pte_N10(TFA)_L_Tyr(-OfBu)-OfBu HCI. The reaction mixture was cannulated as a steady stream to a stirred MTBE (1 .8 L) at 23 °C or 100 °C to give light yellow precipitate of Pte_TFA_L_Tyr. The precipitated Pte_TFA_L_Tyr was filtered using sintered funnel under aspirator vacuum, washed with MTBE (6 * 300 mL) and dried under high vacuum for 8 hours to obtain Pte_TFA_L_Tyr (14.98 g, 83.98% over two steps) as a pale yellow solid. The MTBE washing was tested for absence of residual TFA utilizing wet pH paper (pH between 3-4). The yield of the reaction was between 80-85% in different batches. The deacylated side product was detected in 3.6% as judged by LC/MS. For the different batches this impurity was never more than 5%.
Reactants for Step III:
[0091] A 200 mL round bottom flask was charged with a stirring bar and Pte_TFA_L_Tyr (13.85 g, 22.78 mmol, 1 equiv), then water (95 mL) was added to give a yellow suspension [suspension B]. A freshly prepared solution of aqueous 3.75 M NaOH (26.12 mL, 97.96 mmol, 4.30 equiv), or an equivalent base at a corresponding temperature using dimethylsulfoxide (DMSO) as a solvent (as shown in Table 1 ), was added dropwise to suspension B at 23 °C, giving a clear dull yellow solution over 15 minutes [solution B]. The equivalence of NaOH varied from 3.3 to 5.0 depending on the source of 4 (solid or liquid phase synthesis) and the residual TFA. Trianion 5 (Figure 12) formation was confirmed by LC/MS showing m/z 572→m/z 476 while the solution pH was 9-10 utilizing wet pH paper. The pH of the reaction mixture was in the range of 9-10. This pH is crucial for the overall reaction completion. Notably, pH more than 10 leads to hydrolysis of S0456. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. The presence of hydrolysis by product can be visibly detected by the persistent opaque purple/blue to red/brown color.
TABLE 1 : Separate TFA deprotection via trianion formation; S0456
[0092] The precipitated OTL-0038 product could also be crashed out by adding the reaction solution steady dropwise to acetone, acetonitrile, isopropanol or ethyl acetate/acetone mixture. Acetone yields optimal results. However, viscous reactions could be slower due to partial insolubility and/or crashing out of S0456. In this reaction, the equivalence of the aqueous base is significant. Excess base will efficiently drive reaction forward with potential hydrolysis of S0456. This solution phase synthesis provides Pte_N10(TFA)_Tyr-OH »HCI salt and desires approximately 4.1 to approximately 4.8 equiv base as a source to hydrolyze the product. Particularly, precipitation of Pte_Tyr_S0456 was best achieved when 1 mL of reaction mixture is added dropwise to the stirred acetone (20 mL). Filtration of the precipitate and washing with acetone (3 x10 mL) gave the highest purity as judged from LC/MS chromatogram.
[0093] During experimentation of this solution-phase synthesis of Pte – L Tyrosine -S0456 (OTL-0038) at different stages, some optimized conditions were observed:
Mode of addition: Separate TFA deprotection via trianion formation; S0456 @ 23 °C; reflux.
Stability data of Pte – L Tyrosine – S0456 (OTL-0038):
Liquid analysis: At 40 °C the liquid lost 8.6% at 270 nm and 1 % at 774 nm. At room temperature the liquid lost about 1 .4% at 270 nm and .5% at 774 nm. At 5 °C the
270 nm seems stable and the 774 nm reasonably stable with a small degradation purity.
Source Purity Linker S0456 Base Solvent Duration % Conversion
4.3-4.6
Solution 0.95
95% 1 equiv equiv H20 15 min 100% phase equiv
K2C03
PATENT
US 20140271482
FDA approves pafolacianine for identifying malignant ovarian cancer lesions
On November 29, 2021, the Food and Drug Administration approved pafolacianine (Cytalux, On Target Laboratories, LLC), an optical imaging agent, for adult patients with ovarian cancer as an adjunct for interoperative identification of malignant lesions. Pafolacianine is a fluorescent drug that targets folate receptor which may be overexpressed in ovarian cancer. It is used with a Near-Infrared (NIR) fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
Efficacy was evaluated in a single arm, multicenter, open-label study (NCT03180307) of 178 women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer scheduled to undergo primary surgical cytoreduction, interval debulking, or recurrent ovarian cancer surgery. All patients received pafolacianine. One hundred and thirty-four patients received fluorescence imaging evaluation in addition to standard of care evaluation which includes pre-surgical imaging, intraoperative palpation and normal light evaluation of lesions. Among these patients, 36 (26.9%) had at least one evaluable ovarian cancer lesion detected with pafolacianine that was not observed by standard visual or tactile inspection. The patient-level false positive rate of pafolacianine with NIR fluorescent light with respect to the detection of ovarian cancer lesions confirmed by central pathology was 20.2% (95% CI 13.7%, 28.0%).
The most common adverse reactions (≥1%) occurring in patients were nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, pruritus, and hypersensitivity.
The recommended pafolacianine dose is 0.025 mg/kg administered intravenously over 60 minutes, 1 to 9 hours before surgery. The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of pafolacianine.
View full prescribing information for Cytalux.
This application was granted priority review, fast track designation, and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
USFDA approves new drug to help identify cancer lesions
This drug is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery.By The Health Master -December 2, 2021
The U.S. Food and Drug Administration (USFDA) has approved Cytalux (pafolacianine), an imaging drug intended to assist surgeons in identifying ovarian cancer lesions. The drug is designed to improve the ability to locate additional ovarian cancerous tissue that is normally difficult to detect during surgery.
Cytalux is indicated for use in adult patients with ovarian cancer to help identify cancerous lesions during surgery. The drug is a diagnostic agent that is administered in the form of an intravenous injection prior to surgery.
Alex Gorovets, M.D., deputy director of the Office of Specialty Medicine in the FDA’s Center for Drug Evaluation and Research said, “The FDA’s approval of Cytalux can help enhance the ability of surgeons to identify deadly ovarian tumors that may otherwise go undetected.
By supplementing current methods of detecting ovarian cancer during surgery, Cytalux offers health care professionals an additional imaging approach for patients with ovarian cancer.”
The American Cancer Society estimates there will be more than 21,000 new cases of ovarian cancer and more than 13,000 deaths from this disease in 2021, making it the deadliest of all female reproductive system cancers.
Conventional treatment for ovarian cancer includes surgery to remove as many of the tumors as possible, chemotherapy to stop the growth of malignant cells or other targeted therapy to identify and attack specific cancer cells.
Ovarian cancer often causes the body to overproduce a specific protein in cell membranes called a folate receptor. Following administration via injection, Cytalux binds to these proteins and illuminates under fluorescent light, boosting surgeons’ ability to identify the cancerous tissue.
Currently, surgeons rely on preoperative imaging, visual inspection of tumors under normal light or examination by touch to identify cancer lesions. Cytalux is used with a Near-Infrared fluorescence imaging system cleared by the FDA for specific use with pafolacianine.
The safety and effectiveness of Cytalux was evaluated in a randomized, multi-center, open-label study of women diagnosed with ovarian cancer or with high clinical suspicion of ovarian cancer who were scheduled to undergo surgery.
Of the 134 women (ages 33 to 81 years) who received a dose of Cytalux and were evaluated under both normal and fluorescent light during surgery, 26.9% had at least one cancerous lesion detected that was not observed by standard visual or tactile inspection.
The most common side effects of Cytalux were infusion-related reactions, including nausea, vomiting, abdominal pain, flushing, dyspepsia, chest discomfort, itching and hypersensitivity. Cytalux may cause fetal harm when administered to a pregnant woman.
The use of folate, folic acid, or folate-containing supplements should be avoided within 48 hours before administration of Cytalux. There is a risk of image interpretation errors with the use of Cytalux to detect ovarian cancer during surgery, including false negatives and false positives.
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/214907s000lbl.pdf
- ^ Jump up to:a b c d e f g h i “FDA Approves New Imaging Drug to Help Identify Ovarian Cancer Lesions”. U.S. Food and Drug Administration (FDA) (Press release). 29 November 2021. Retrieved 30 November 2021. This article incorporates text from this source, which is in the public domain.
- ^ “On Target Laboratories Announces FDA Approval of Cytalux (pafolacianine) injection for Identification of Ovarian Cancer During Surgery”. On Target Laboratories. 29 November 2021. Retrieved 30 November 2021 – via PR Newswire.
- ^ “Pafolacianine Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 23 December 2014. Retrieved 30 November 2021.
External links
- “Pafolacianine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Cytalux |
| Other names | OTL-0038 |
| License data | US DailyMed: Pafolacianine |
| Pregnancy category | Not recommended |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1628423-76-6 |
| PubChem CID | 135565623 |
| DrugBank | DB15413 |
| ChemSpider | 64880249 |
| UNII | F7BD3Z4X8L |
| ChEMBL | ChEMBL4297412 |
| Chemical and physical data | |
| Formula | C61H67N9O17S4 |
| Molar mass | 1326.49 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Pafolacianine, FDA 2021, APPROVALS 2021, Cytalux, OVARIAN CANCER, OTL 38,
[Na+].[Na+].[Na+].[Na+].CC1(C)\C(=C/C=C/2\CCCC(=C2Oc3ccc(C[C@H](NC(=O)c4ccc(NCc5cnc6N=C(N)NC(=O)c6n5)cc4)C(=O)O)cc3)\C=C\C7=[N](CCCCS(=O)(=O)O)c8ccc(cc8C7(C)C)S(=O)(=O)O)\N(CCCCS(=O)(=O)O)c9ccc(cc19)S(=O)(=O)O

NEW DRUG APPROVALS
ONE TIME
$10.00
Ropeginterferon alfa-2b
PCDLPQTHSL GSRRTLMLLA QMRRISLFSC LKDRHDFGFP QEEFGNQFQK AETIPVLHEM
IQQIFNLFST KDSSAAWDET LLDKFYTELY QQLNDLEACV IQGVGVTETP LMKEDSILAV
RKYFQRITLY LKEKKYSPCA WEVVRAEIMR SFSLSTNLQE SLRSKE
(Disulfide bridge: 2-99, 30-139)
Ropeginterferon alfa-2b
- AOP2014
CAS 1335098-50-4
UNII981TME683S
FDA APPROVED, 2021/11/12, BESREMI
PEPTIDE, Antineoplastic, Antiviral
Polycythemia vera (PV) is the most common Philadelphia chromosome-negative myeloproliferative neoplasm (MPN), characterized by increased hematocrit and platelet/leukocyte counts, an increased risk for hemorrhage and thromboembolic events, and a long-term propensity for myelofibrosis and leukemia.1,2 Interferon alfa-2b has been used for decades to treat PV but requires frequent dosing and is not tolerated by all patients.2 Ropeginterferon alfa-2b is a next-generation mono-pegylated type I interferon produced from proline-IFN-α-2b in Escherichia coli that has high tolerability and a long half-life.4,6 Ropeginterferon alfa-2b has shown efficacy in PV in in vitro and in vivo models and clinical trials.3,4
Ropeginterferon alfa-2b was approved by the FDA on November 12, 2021, and is currently marketed under the trademark BESREMi by PharmaEssentia Corporation.6
Ropeginterferon alfa-2b, sold under the brand name Besremi, is a medication used to treat polycythemia vera.[1][2][3][4] It is an interferon.[1][3] It is given by injection.[1][3]
The most common side effects include low levels of white blood cells and platelets (blood components that help the blood to clot), muscle and joint pain, tiredness, flu-like symptoms and increased blood levels of gamma-glutamyl transferase (a sign of liver problems).[3] Ropeginterferon alfa-2b can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness.[2] Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).[2]
It was approved for medical use in the European Union in February 2019,[3] and in the United States in November 2021.[2][5] Ropeginterferon alfa-2b is the first medication approved by the U.S. Food and Drug Administration (FDA) to treat polycythemia vera that people can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.[2]
https://www.fda.gov/news-events/press-announcements/fda-approves-treatment-rare-blood-disease#:~:text=FDA%20NEWS%20RELEASE-,FDA%20Approves%20Treatment%20for%20Rare%20Blood%20Disease,FDA%2DApproved%20Option%20Patients%20Can%20Take%20Regardless%20of%20Previous%20Therapies,-ShareFor Immediate Release:November 12, 2021
Today, the U.S. Food and Drug Administration approved Besremi (ropeginterferon alfa-2b-njft) injection to treat adults with polycythemia vera, a blood disease that causes the overproduction of red blood cells. The excess cells thicken the blood, slowing blood flow and increasing the chance of blood clots.
“Over 7,000 rare diseases affect more than 30 million people in the United States. Polycythemia vera affects approximately 6,200 Americans each year,” said Ann Farrell, M.D., director of the Division of Non-Malignant Hematology in the FDA’s Center for Drug Evaluation and Research. “This action highlights the FDA’s commitment to helping make new treatments available to patients with rare diseases.”
Besremi is the first FDA-approved medication for polycythemia vera that patients can take regardless of their treatment history, and the first interferon therapy specifically approved for polycythemia vera.
Treatment for polycythemia vera includes phlebotomies (a procedure that removes excess blood cells though a needle in a vein) as well as medicines to reduce the number of blood cells; Besremi is one of these medicines. Besremi is believed to work by attaching to certain receptors in the body, setting off a chain reaction that makes the bone marrow reduce blood cell production. Besremi is a long-acting drug that patients take by injection under the skin once every two weeks. If Besremi can reduce excess blood cells and maintain normal levels for at least one year, then dosing frequency may be reduced to once every four weeks.
The effectiveness and safety of Besremi were evaluated in a multicenter, single-arm trial that lasted 7.5 years. In this trial, 51 adults with polycythemia vera received Besremi for an average of about five years. Besremi’s effectiveness was assessed by looking at how many patients achieved complete hematological response, which meant that patients had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots. Overall, 61% of patients had a complete hematological response.
Besremi can cause liver enzyme elevations, low levels of white blood cells, low levels of platelets, joint pain, fatigue, itching, upper airway infection, muscle pain and flu-like illness. Side effects may also include urinary tract infection, depression and transient ischemic attacks (stroke-like attacks).
Interferon alfa products like Besremi may cause or worsen neuropsychiatric, autoimmune, ischemic (not enough blood flow to a part of the body) and infectious diseases, which could lead to life-threatening or fatal complications. Patients who must not take Besremi include those who are allergic to the drug, those with a severe psychiatric disorder or a history of a severe psychiatric disorder, immunosuppressed transplant recipients, certain patients with autoimmune disease or a history of autoimmune disease, and patients with liver disease.
People who could be pregnant should be tested for pregnancy before using Besremi due to the risk of fetal harm.
Besremi received orphan drug designation for this indication. Orphan drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted the approval of Besremi to PharmaEssentia Corporation.
Medical uses
In the European Union, ropeginterferon alfa-2b is indicated as monotherapy in adults for the treatment of polycythemia vera without symptomatic splenomegaly.[3] In the United States it is indicated for the treatment of polycythemia vera.[1][2][5]
History
The effectiveness and safety of ropeginterferon alfa-2b were evaluated in a multicenter, single-arm trial that lasted 7.5 years.[2] In this trial, 51 adults with polycythemia vera received ropeginterferon alfa-2b for an average of about five years.[2] The effectiveness of ropeginterferon alfa-2b was assessed by looking at how many participants achieved complete hematological response, which meant that participants had a red blood cell volume of less than 45% without a recent phlebotomy, normal white cell counts and platelet counts, a normal spleen size, and no blood clots.[2] Overall, 61% of participants had a complete hematological response.[2] The U.S. Food and Drug Administration (FDA) granted the application for Ropeginterferon_alfa-2b orphan drug designation and granted the approval of Besremi to PharmaEssentia Corporation[2]
REF
- Bartalucci N, Guglielmelli P, Vannucchi AM: Polycythemia vera: the current status of preclinical models and therapeutic targets. Expert Opin Ther Targets. 2020 Jul;24(7):615-628. doi: 10.1080/14728222.2020.1762176. Epub 2020 May 18. [Article]
- How J, Hobbs G: Use of Interferon Alfa in the Treatment of Myeloproliferative Neoplasms: Perspectives and Review of the Literature. Cancers (Basel). 2020 Jul 18;12(7). pii: cancers12071954. doi: 10.3390/cancers12071954. [Article]
- Verger E, Soret-Dulphy J, Maslah N, Roy L, Rey J, Ghrieb Z, Kralovics R, Gisslinger H, Grohmann-Izay B, Klade C, Chomienne C, Giraudier S, Cassinat B, Kiladjian JJ: Ropeginterferon alpha-2b targets JAK2V617F-positive polycythemia vera cells in vitro and in vivo. Blood Cancer J. 2018 Oct 4;8(10):94. doi: 10.1038/s41408-018-0133-0. [Article]
- Gisslinger H, Zagrijtschuk O, Buxhofer-Ausch V, Thaler J, Schloegl E, Gastl GA, Wolf D, Kralovics R, Gisslinger B, Strecker K, Egle A, Melchardt T, Burgstaller S, Willenbacher E, Schalling M, Them NC, Kadlecova P, Klade C, Greil R: Ropeginterferon alfa-2b, a novel IFNalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood. 2015 Oct 8;126(15):1762-9. doi: 10.1182/blood-2015-04-637280. Epub 2015 Aug 10. [Article]
- EMA Approved Products: Besremi (ropeginterferon alfa-2b ) solution for injection [Link]
- FDA Approved Drug Products: BESREMi (ropeginterferon alfa-2b-njft) injection [Link]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
References
- ^ Jump up to:a b c d e https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761166s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j k l “FDA Approves Treatment for Rare Blood Disease”. U.S. Food and Drug Administration (FDA) (Press release). 12 November 2021. Retrieved 12 November 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g “Besremi EPAR”. European Medicines Agency (EMA). Retrieved 14 November 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Wagner SM, Melchardt T, Greil R (March 2020). “Ropeginterferon alfa-2b for the treatment of patients with polycythemia vera”. Drugs of Today. Barcelona, Spain. 56 (3): 195–202. doi:10.1358/dot.2020.56.3.3107706. PMID 32282866. S2CID 215758794.
- ^ Jump up to:a b “U.S. FDA Approves Besremi (ropeginterferon alfa-2b-njft) as the Only Interferon for Adults With Polycythemia Vera” (Press release). PharmaEssentia. 12 November 2021. Retrieved 14 November 2021 – via Business Wire.
External links
- “Ropeginterferon alfa-2b”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01193699 for “Safety Study of Pegylated Interferon Alpha 2b to Treat Polycythemia Vera (PEGINVERA)” at ClinicalTrials.gov
- Clinical trial number NCT02218047 for “AOP2014 vs. BAT in Patients With Polycythemia Vera Who Previously Participated in the PROUD-PV Study. (CONTI-PV)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Besremi |
| Other names | AOP2014, ropeginterferon alfa-2b-njft |
| License data | EU EMA: by INNUS DailyMed: Ropeginterferon_alfa |
| Pregnancy category | Contraindicated |
| Routes of administration | Subcutaneous |
| Drug class | Interferon |
| ATC code | L03AB15 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2]EU: Rx-only [3] |
| Identifiers | |
| CAS Number | 1335098-50-4 |
| DrugBank | DB15119 |
| UNII | 981TME683S |
| KEGG | D11027 |
/////////Ropeginterferon alfa-2b, FDA 2021, APPROVALS 2021, BESREMI, PEPTIDE, Antineoplastic, Antiviral, AOP 2014, PharmaEssentia

NEW DRUG APPROVALS
ONE TIME
$10.00
Tisotumab vedotin

Tisotumab vedotin
チソツマブベドチン (遺伝子組換え)Immunoglobulin G1, anti-(human blood-coagulation factor III) (human monoclonal HuMax-TF heavy chain), disulfide with human monoclonal HuMax-TF κ-chain, dimer, tetrakis(thioether) with N-[[[4-[[N-[6-(3-mercapto-2,5-dioxo-1-pyrrolidinyl)-1-oxohexyl]-L-valyl-N5-(aminocarbonyl)-L-ornithyl]amino]phenyl]methoxy]carbonyl]-N-methyl-L-valyl-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-hydroxy-1-methyl-2-phenylethyl]amino]-1-methoxy-2-methyl-3-oxopropyl]-1-pyrrolidinyl]-2-methoxy-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-L-valinamide
- HuMax-TF-ADC
- Immunoglobulin G1, anti-(human tissue factor) (human monoclonal HuMax-TF heavy chain), disulfide with human monoclonal HuMax-TF κ-chain, dimer, tetrakis(thioether) with N-[[[4-[[N-[6-(3-mercapto-2,5-dioxo-1-pyrrolidinyl)-1-oxohexyl]-L-valyl-N5-(aminocarbonyl)-L-ornithyl]amino]phenyl]methoxy]carbonyl]-N-methyl-L-valyl-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-hydroxy-1-methyl-2-phenylethyl]amino]-1-methoxy-2-methyl-3-oxopropyl]-1-pyrrolidinyl]-2-methoxy-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-L-valinamide
Protein Sequence
Sequence Length: 1324, 448, 448, 214, 214multichain; modified (modifications unspecified)
| Formula | C6418H9906N1710O2022S44.(C68H106N11O15)n |
|---|---|
| Efficacy | Antineoplastic |
| Disease | Cervical cancer |
| Comment | Antibody-drug conjugateCAS:1418731-10-8 |
- HuMax-TF-ADC
- Tisotumab vedotin
- Tisotumab vedotin [WHO-DD]
- UNII-T41737F88A
- WHO 10148
US FDA APPROVED 2021/9/20 , TIVDAK

FDA grants accelerated approval to tisotumab vedotin-tftv for recurrent or metastatic cervical cancer……….. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-tisotumab-vedotin-tftv-recurrent-or-metastatic-cervical-cancer
On September 20, 2021, the Food and Drug Administration granted accelerated approval to tisotumab vedotin-tftv (Tivdak, Seagen Inc.), a tissue factor-directed antibody and microtubule inhibitor conjugate, for adult patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy.
Approval was based on innovaTV 204, an open-label, multicenter, single-arm clinical trial (NCT03438396). Efficacy was evaluated in 101 patients with recurrent or metastatic cervical cancer who had received no more than two prior systemic regimens in the recurrent or metastatic setting, including at least one prior platinum-based chemotherapy regimen. Sixty-nine percent of patients had received bevacizumab as part of prior systemic therapy. Patients received tisotumab vedotin-tftv 2 mg/kg every 3 weeks until disease progression or unacceptable toxicity.
The main efficacy outcome measures were confirmed objective response rate (ORR) as assessed by an independent review committee (IRC) using RECIST v1.1 and duration of response (DOR). The ORR was 24% (95% CI: 15.9%, 33.3%) with a median response duration of 8.3 months (95% CI: 4.2, not reached).
The most common adverse reactions (≥25%), including laboratory abnormalities, were hemoglobin decreased, fatigue, lymphocytes decreased, nausea, peripheral neuropathy, alopecia, epistaxis, conjunctival adverse reactions, hemorrhage, leukocytes decreased, creatinine increased, dry eye, prothrombin international normalized ratio increased, activated partial thromboplastin time prolonged, diarrhea, and rash. Product labeling includes a boxed warning for ocular toxicity.
The recommended dose is 2 mg/kg (up to a maximum of 200 mg for patients ≥100 kg) given as an intravenous infusion over 30 minutes every 3 weeks until disease progression or unacceptable toxicity.
View full prescribing information for Tivdak.
This review used the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
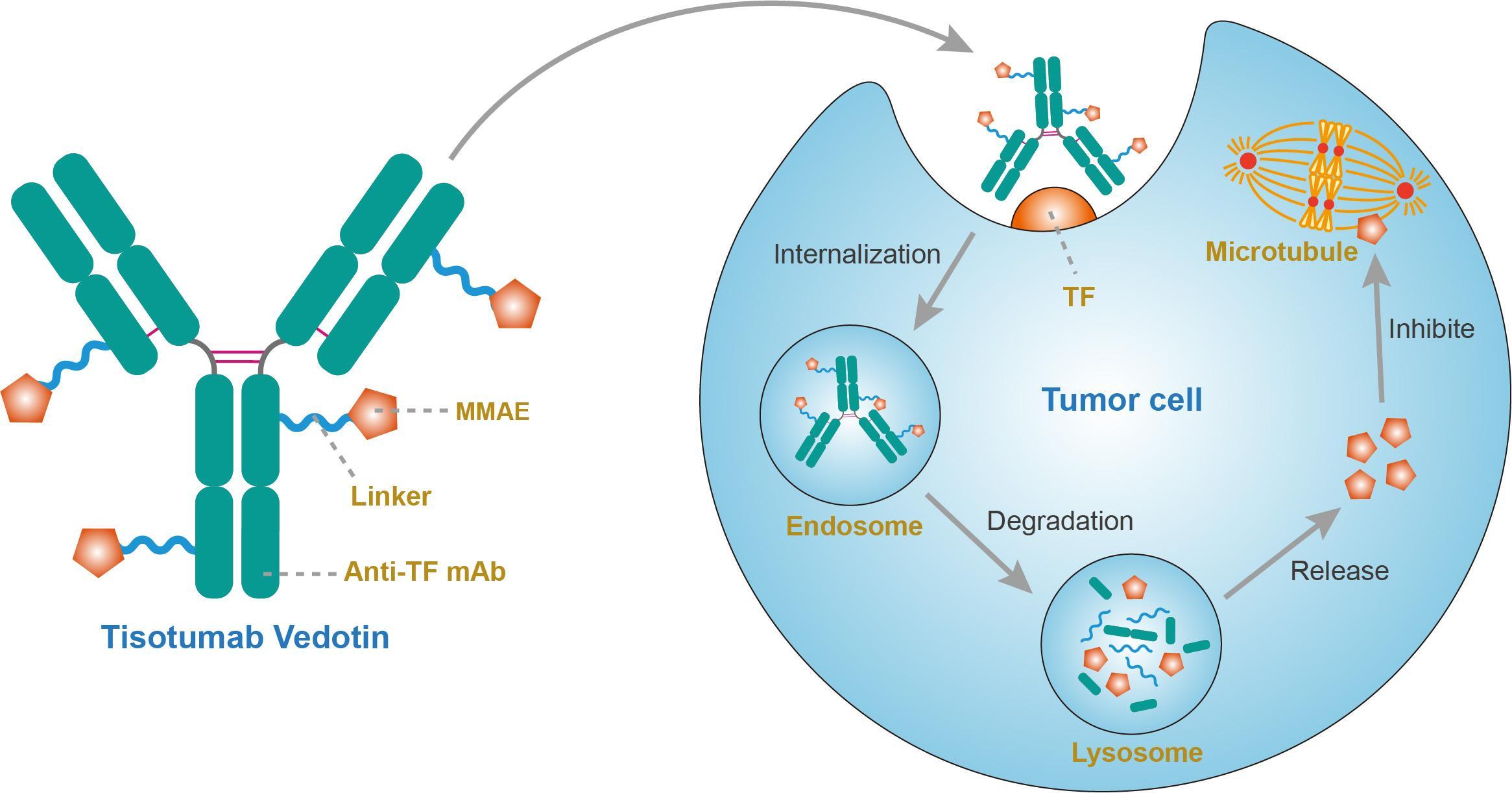
A fully human monoclonal antibody specific for tissue factor conjugated to the microtubule-disrupting agent monomethyl auristatin E (MMAE) via a protease-cleavable valine-citrulline linker.
Tisotumab vedotin, sold under the brand name Tivdak is a human monoclonal antibody used to treat cervical cancer.[1]
Tisotumab vedotin was approved for medical use in the United States in September 2021.[1][2]
Tisotumab vedotin is the international nonproprietary name (INN).[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto

NEW DRUG APPROVALS
one time
$10.00
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761208s000lbl.pdf
- ^ “Seagen and Genmab Announce FDA Accelerated Approval for Tivdak (tisotumab vedotin-tftv) in Previously Treated Recurrent or Metastatic Cervical Cancer”. Seagen. 20 September 2021. Retrieved 20 September 2021 – via Business Wire.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 75”. WHO Drug Information. 30 (1): 159–60. hdl:10665/331046.
External links
- “Tisotumab vedotin”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03438396 for “A Trial of Tisotumab Vedotin in Cervical Cancer” at ClinicalTrials.gov
- Clinical trial number NCT03245736 for “Tisotumab Vedotin Continued Treatment in Patients With Solid Tumors” at ClinicalTrials.gov
- Clinical trial number NCT02001623 for “Tisotumab Vedotin (HuMax-TF-ADC) Safety Study in Patients With Solid Tumors” at ClinicalTrials.gov
- Clinical trial number NCT02552121 for “Tisotumab Vedotin (HuMax-TF-ADC) Safety Study in Patients With Solid Tumors” at ClinicalTrials.gov
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Tissue factor (TF) |
| Clinical data | |
| Trade names | Tivdak |
| Other names | Tisotumab vedotin-tftv |
| License data | US DailyMed: Tisotumab_vedotin |
| Pregnancy category | Contraindicated[1] |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1418731-10-8 |
| UNII | T41737F88A |
| KEGG | D11814 |
//////////Tisotumab vedotin, チソツマブベドチン (遺伝子組換え) , FDA 2021, APPROVALS 2021, Antineoplastic, CERVICAL CANCER, CANCER, MONOCLONAL ANTIBODY, UNII-T41737F88A, WHO 10148
Mobocertinib

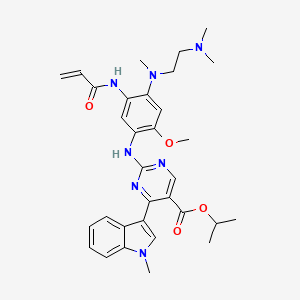
Mobocertinib
1847461-43-1
MF C32H39N7O4
MW 585.70
propan-2-yl 2-[4-[2-(dimethylamino)ethyl-methylamino]-2-methoxy-5-(prop-2-enoylamino)anilino]-4-(1-methylindol-3-yl)pyrimidine-5-carboxylate
TAK-788, AP32788, TAK788, UNII-39HBQ4A67L, AP-32788, 39HBQ4A67L
US10227342, Example 10, MFCD32669806, NSC825519, s6813, TAK-788;AP32788, WHO 11183
NSC-825519, example 94 [WO2015195228A1], GTPL10468, BDBM368374, BCP31045, EX-A3392
US FDA APPROVED 9/15/2021, Exkivity, To treat locally advanced or metastatic non-small cell lung cancer with epidermal growth factor receptor exon 20 insertion mutation

Mobocertinib succinate Chemical Structure
CAS No. : 2389149-74-8
| Molecular Weight | 703.78 |
|---|---|
| Formula | C₃₆H₄₅N₇O₈ |
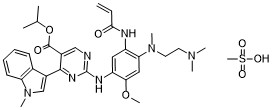
Mobocertinib mesylateCAS# 2389149-85-1 (mesylate)C33H43N7O7S
Molecular Weight: 681.809
CAS #: 2389149-85-1 (mesylate) 1847461-43-1 (free base) 2389149-74-8 (succinate) 2389149-76-0 (HBr) 2389149-79-3 (HCl) 2389149-81-7 (sulfate) 2389149-83-9 (tosylate) 2389149-87-3 (oxalate) 2389149-89-5 (fumarate)
JAPANESE ACCEPTED NAME
Mobocertinib Succinate
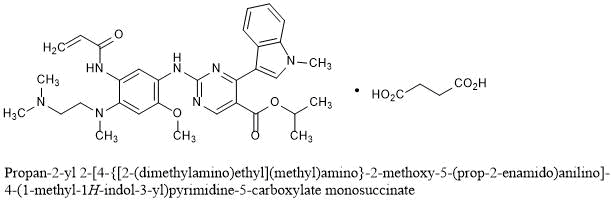
Propan-2-yl 2-[4-{[2-(dimethylamino)ethyl](methyl)amino}-2-methoxy-5-(prop-2-enamido)anilino]-4-(1-methyl-1H-indol-3-yl)pyrimidine-5-carboxylate monosuccinate
C32H39N7O4▪C4H6O4 : 703.78
[2389149-74-8]
FDA grants accelerated approval to mobocertinib for metastatic non-small cell lung cancer with EGFR exon 20 insertion mutations……. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-mobocertinib-metastatic-non-small-cell-lung-cancer-egfr-exon-20
On September 15, 2021, the Food and Drug Administration granted accelerated approval to mobocertinib (Exkivity, Takeda Pharmaceuticals, Inc.) for adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.
Today, the FDA also approved the Oncomine Dx Target Test (Life Technologies Corporation) as a companion diagnostic device to select patients with the above mutations for mobocertinib treatment.
Approval was based on Study 101, an international, non-randomized, open-label, multicohort clinical trial (NCT02716116) which included patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. Efficacy was evaluated in 114 patients whose disease had progressed on or after platinum-based chemotherapy. Patients received mobocertinib 160 mg orally daily until disease progression or intolerable toxicity.
The main efficacy outcome measures were overall response rate (ORR) according to RECIST 1.1 as evaluated by blinded independent central review (BICR) and response duration. The ORR was 28% (95% CI: 20%, 37%) with a median response duration of 17.5 months (95% CI: 7.4, 20.3).
The most common adverse reactions (>20%) were diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain. Product labeling includes a boxed warning for QTc prolongation and Torsades de Pointes, and warnings for interstitial lung disease/pneumonitis, cardiac toxicity, and diarrhea.
The recommended mobocertinib dose is 160 mg orally once daily until disease progression or unacceptable toxicity.
View full prescribing information for mobocertinib.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), the Brazilian Health Regulatory Agency (ANVISA), and United Kingdom’s Medicines & Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 6 weeks ahead of the FDA goal date.
This application was granted priority review, breakthrough therapy designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.Takeda’s EXKIVITY™ (mobocertinib) Approved by U.S. FDA as the First Oral Therapy Specifically Designed for Patients with EGFR Exon20 Insertion+ NSCLC…….. https://www.takeda.com/newsroom/newsreleases/2021/takeda-exkivity-mobocertinib-approved-by-us-fda/September 15, 2021
- Approval based on Phase 1/2 trial results, which demonstrated clinically meaningful responses with a median duration of response (DoR) of approximately 1.5 years
- Next-generation sequencing (NGS) companion diagnostic test approved simultaneously to support identification of patients with EGFR Exon20 insertion mutations
OSAKA, Japan, and CAMBRIDGE, Mass. September 15, 2021 – Takeda Pharmaceutical Company Limited (TSE:4502/NYSE:TAK) (“Takeda”) today announced that the U.S. Food and Drug Administration (FDA) has approved EXKIVITY (mobocertinib) for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy. EXKIVITY, which was granted priority review and received Breakthrough Therapy Designation, Fast Track Designation and Orphan Drug Designation from the FDA, is the first and only approved oral therapy specifically designed to target EGFR Exon20 insertion mutations. This indication is approved under Accelerated Approval based on overall response rate (ORR) and DoR. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
“The approval of EXKIVITY introduces a new and effective treatment option for patients with EGFR Exon20 insertion+ NSCLC, fulfilling an urgent need for this difficult-to-treat cancer,” said Teresa Bitetti, president, Global Oncology Business Unit, Takeda. “EXKIVITY is the first and only oral therapy specifically designed to target EGFR Exon20 insertions, and we are particularly encouraged by the duration of the responses observed with a median of approximately 1.5 years. This approval milestone reinforces our commitment to meeting the needs of underserved patient populations within the oncology community.”
The FDA simultaneously approved Thermo Fisher Scientific’s Oncomine Dx Target Test as an NGS companion diagnostic for EXKIVITY to identify NSCLC patients with EGFR Exon20 insertions. NGS testing is critical for these patients, as it can enable more accurate diagnoses compared to polymerase chain reaction (PCR) testing, which detects less than 50% of EGFR Exon20 insertions.
“EGFR Exon20 insertion+ NSCLC is an underserved cancer that we have been unable to target effectively with traditional EGFR TKIs,” said Pasi A. Jänne, MD, PhD, Dana Farber Cancer Institute. “The approval of EXKIVITY (mobocertinib) marks another important step forward that provides physicians and their patients with a new targeted oral therapy specifically designed for this patient population that has shown clinically meaningful and sustained responses.”
“Patients with EGFR Exon20 insertion+ NSCLC have historically faced a unique set of challenges living with a very rare lung cancer that is not only underdiagnosed, but also lacking targeted treatment options that can improve response rates,” said Marcia Horn, executive director, Exon 20 Group at ICAN, International Cancer Advocacy Network. “As a patient advocate working with EGFR Exon20 insertion+ NSCLC patients and their families every day for nearly five years, I am thrilled to witness continued progress in the fight against this devastating disease and am grateful for the patients, families, healthcare professionals and scientists across the globe who contributed to the approval of this promising targeted therapy.”
The FDA approval is based on results from the platinum-pretreated population in the Phase 1/2 trial of EXKIVITY, which consisted of 114 patients with EGFR Exon20 insertion+ NSCLC who received prior platinum-based therapy and were treated at the 160 mg dose. Results were presented at the 2021 American Society of Clinical Oncology (ASCO) Annual Meeting from the Phase 1/2 trial and demonstrated a confirmed ORR of 28% per independent review committee (IRC) (35% per investigator) as well as a median DoR of 17.5 months per IRC, a median overall survival (OS) of 24 months and a median progression-free survival (PFS) of 7.3 months per IRC.
The most common adverse reactions (>20%) were diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain. The EXKIVITY Prescribing Information includes a boxed warning for QTc prolongation and Torsades de Pointes, and warnings and precautions for interstitial lung disease/pneumonitis, cardiac toxicity, and diarrhea.
The FDA review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE), which provides a framework for concurrent submission and review of oncology products among international partners. We look forward to continuing our work with regulatory agencies across the globe to bring mobocertinib to patients.
About EXKIVITY (mobocertinib)
EXKIVITY is a first-in-class, oral tyrosine kinase inhibitor (TKI) specifically designed to selectively target epidermal growth factor receptor (EGFR) Exon20 insertion mutations.
EXKIVITY is approved in the U.S. for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with EGFR exon 20 insertion mutations as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.
Results from the Phase 1/2 trial of mobocertinib have also been accepted for review by the Center for Drug Evaluation (CDE) in China for locally advanced or metastatic NSCLC patients with EGFR Exon20 insertion mutations who have been previously treated with at least one prior systemic chemotherapy.
For more information about EXKIVITY, visit http://www.EXKIVITY.com. For the Prescribing Information, including the Boxed Warning, please visit https://takeda.info/Exkivity-Prescribing-Information.
About EGFR Exon20 Insertion+ NSCLC
Non-small cell lung cancer (NSCLC) is the most common form of lung cancer, accounting for approximately 85% of the estimated 2.2 million new cases of lung cancer diagnosed each year worldwide, according to the World Health Organization.1,2 Patients with epidermal growth factor receptor (EGFR) Exon20 insertion+ NSCLC make up approximately 1-2% of patients with NSCLC, and the disease is more common in Asian populations compared to Western populations.3-7 This disease carries a worse prognosis than other EGFR mutations, as EGFR TKIs – which do not specifically target EGFR Exon20 insertions – and chemotherapy provide limited benefit for these patients.
Takeda is committed to continuing research and development to meet the needs of the lung cancer community through the discovery and delivery of transformative medicines.
EXKIVITY IMPORTANT SAFETY INFORMATION
QTc Interval Prolongation and Torsades de Pointes: EXKIVITY can cause life-threatening heart rate-corrected QT (QTc) prolongation, including Torsades de Pointes, which can be fatal, and requires monitoring of QTc and electrolytes at baseline and periodically during treatment. Increase monitoring frequency in patients with risk factors for QTc prolongation. Avoid use of concomitant drugs which are known to prolong the QTc interval and use of strong or moderate CYP3A inhibitors with EXKIVITY, which may further prolong the QTc. Withhold, reduce the dose, or permanently discontinue EXKIVITY based on the severity of QTc prolongation.
Interstitial Lung Disease (ILD)/Pneumonitis: Monitor patients for new or worsening pulmonary symptoms indicative of ILD/pneumonitis. Immediately withhold EXKIVITY in patients with suspected ILD/pneumonitis and permanently discontinue EXKIVITY if ILD/pneumonitis is confirmed.
Cardiac Toxicity: Monitor cardiac function, including left ventricular ejection fraction, at baseline and during treatment. Withhold, resume at reduced dose or permanently discontinue based on severity.
Diarrhea: Diarrhea may lead to dehydration or electrolyte imbalance, with or without renal impairment. Monitor electrolytes and advise patients to start an antidiarrheal agent at first episode of diarrhea and to increase fluid and electrolyte intake. Withhold, reduce the dose, or permanently discontinue EXKIVITY based on the severity.
Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective non-hormonal contraception.
Mobocertinib, sold under the brand name Exkivity, is used for the treatment of non-small cell lung cancer.[2][3]
The most common side effects include diarrhea, rash, nausea, stomatitis, vomiting, decreased appetite, paronychia, fatigue, dry skin, and musculoskeletal pain.[2]
Mobocertinib is a small molecule tyrosine kinase inhibitor. Its molecular target is epidermal growth factor receptor (EGFR) bearing mutations in the exon 20 region.[4][5]
Mobocertinib was approved for medical use in the United States in September 2021.[2][3] It is a first-in-class oral treatment to target EGFR Exon20 insertion mutations.[3]
Medical uses
Mobocertinib is indicated for adults with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.[2]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter a
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
PATENT
WO 2019222093
https://patents.google.com/patent/WO2019222093A1

Scheme I



Example 1 Procedure for the preparation of isopropyl 2-((5-acrylamido-4-((2- (dimethylamino)ethyl) (methyl)amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indol-3- yl)pyrimidine-5-carboxylate (Compound (A)).

[00351] Step 1 : Preparation of isopropyl 2-chloro-4-(l -methyl- lH-indo 1-3 -yl)pyrimidine-5- carboxylate.

[00352] To a 2 L Radley reactor equipped with a mechanical stirrer, a thermometer, and a refluxing condenser was charged isopropyl 2,4-dichloropyrimidine-5-carboxylate (100 g, 42.5 mmol, 1.00 eq.) andl,2-dimethoxyethane (DME, 1.2 L, 12 vol) at RT. The mixture was cooled to 3 °C, and granular AlCb (65.5 g, 49.1 mmol, 1.15 eq.) was added in 2 portions (IT 3-12 °C, jacket set 0 °C). The white slurry was stirred 15-25 °C for 60 minutes. 1 -Methylindole (59 g, 44.9 mmol, 1.06 eq.) was added in one portion (IT 20-21°C). DME (100 mL) was used to aid 1- Methylindole transfer. The reaction mixture was aged for at 35 °C for 24 h. Samples (1 mL) were removed at 5 h and 24 h for HPLC analysis (TM1195).[00353] At 5 h the reaction had 70 % conversion, while after 24 h the desired conversion was attained (< 98%).[00354] The reaction mixture was cooled to 0 °C to 5 °C and stirred for 1 h. The solids were collected via filtration and washed with DME (100 mL). The solids (AlCb complex) were charged back to reactor followed by 2-MeTHF (1 L, 10 vol), and water (400 mL, 4 vol). The mixture was stirred for 10 minutes. The stirring was stopped to allow the layers to separate.The organic phase was washed with water (200 mL, 2 vol). The combined aqueous phase was re-extracted with 2-MeTHF (100 mL, 1 vol).[00355] During workup a small amount of product title compound started to crystallize.Temperature during workup should be at about 25-40 °C.[00356] The combined organic phase was concentrated under mild vacuum to 300-350 mL (IT 40-61 °C). Heptane (550 mL) was charged while maintaining the internal temperature between 50 °C and 60 °C. The resulting slurry was cooled at 25 °C over 15 minutes, aged for 1 h (19-25 °C) and the resulting solids isolated by filtration.[00357] The product was dried at 50 °C under vacuum for 3 days to yield 108.1 g (77 % yield) of the title compound, in 100% purity (AUC) as a yellow solid.‘H NMR (400 MHz, DMSO-i/e) d ppm 1.24 (d, J= 6.53 Hz, 6 H) 3.92 (s, 3 H) 5.19 (spt, J=6.27 Hz, 1 H) 7.25 – 7.35 (m, 2 H) 7.59 (d, J=8.03 Hz, 1 H) 8.07 (s, 1 H) 8.16 (d, J= 7.53 Hz, 1 H) 8.82 (s, 1 H).[00358] Step 2: Preparation of isopropyl 2-((4-fhioro-2-methoxy-5-nitrophenyl)amino)-4-(l- methyl-lH-indol-3-yl)pyrimidine-5-carboxylate.

[00359] A mixture of the product of step 1 (85.0 g, 258 mmol, 1.0 eq.), 4-fluoro-2-methoxy- 5nitroaniline (57.0 g, 306 mmol, 1.2 eq.) and PTSA monohydrate (13.3 g, 70.0 mmol, 0.27 eq.) in acetonitrile (1.4 L, 16.5 v) was heated to 76-81 °C under nitrogen in a 2 L Radley reactor. IPC at 19 h indicated that the reaction was complete.[00360] The reaction mixture was cooled to 25 °C and water (80 mL) was charged in one portion (IT during charge dropped from 25 °C to 19 °C). The reaction mixture was aged for 1 h at 21 °C and then the resulting solids were isolated by filtration. The product was washed with EtOAc (2 x 150 mL) and dried in high vacuum at 50 °C to 60 °C for 44 h to give 121.5 g of the title compound (98% yield), HPLC purity 100 % a/a; NMR indicated that PTSA was purged.¾ NMR (400 MHz, DMSO-7,) d ppm 1.21 (d, 7=6.02 Hz, 6 H) 3.91 (s, 3 H) 4.02 (s, 3 H) 5.09 (spt, 7=6.27 Hz, 1 H) 7.10 (t, 7=7.53 Hz, 1 H) 7.26 (t, 7=7.58 Hz, 1 H) 7.42 (d, 7=13.05 Hz, 1 H) 7.55 (d, 7=8.53 Hz, 1 H) 7.90 (br d, 7=7.53 Hz, 1 H) 7.98 (s, 1 H) 8.75 (s, 1 H) 8.88 (d, 7=8.03 Hz, 1 H) 9.03 (s, 1 H).[00361] Step 3: Preparation of isopropyl 2-((4-((2-(dimethylamino)ethyl(methyl)amino)-2- methoxy-5-nitrophenyl)amino)-4-(l-methyl-lH-indol-3-yl)pyrimidine-5-carboxylate.

[00362] A 50 L flask was charged 1.500 kg of the product of step 2 (3.1 moles, l.O equiv.), 639.0 g A,A,A-trimethylethylenediamine (6.3 mol, 2 equiv.), and 21 L MeCN. The resulting slurry was mixed for 7 hours at reflux. The reaction was cooled overnight. Water (16.5 L) was added before the solids were isolated. After isolation of the solids, a wash of 2.25 L MeCN in 2.25 L water was conducted to provide the title compound. The solids were dried, under vacuum, at 75 °C. HPLC purity a/a % of the dry solid was 99.3%.¾ NMR (400 MHz, DMSO-7,) d ppm 1.22 (d, 7=6.02 Hz, 6 H) 2.09 – 2.13 (m, 1 H) 2.19 (s, 6 H) 2.49 – 2.52 (m, 1 H) 2.89 (s, 3 H) 3.29 – 3.35 (m, 2 H) 3.89 (s, 3 H) 3.94 (s, 3 H) 5.10 (spt, 7=6.19 Hz, 1 H) 6.86 (s, 1 H) 7.07 (br t, 7=7.53 Hz, 1 H) 7.24 (t, 7=7.28 Hz, 1 H) 7.53 (d, 7=8.53Hz, 1 H) 7.86 – 8.02 (m, 2 H) 8.36 (s, 1 H) 8.69 (s, 1 H) 8.85 (s, 1 H).[00363] Step 4: Preparation of isopropyl 2-((5-amino-4-((2-(dimethylamino)ethyl)(methyl)- amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indo 1-3 -yl)pyrimidine-5-carboxy late.

[00364] To a mixture of the product of step 3 (1.501 kg, 2.67 mol, 1.00 eq.) and 10% Pd/C (64 % wet, 125.0 g, 0.01 1 eq.) was added 2-MeTHF (17.7 L) in a 20 L pressure reactor. The mixture was hydrogenated at 6- 10 psi ¾ and at 40 °C until IPC indicated complete conversion (1 1 h, the reaction product 99.0%). The reaction mixture was filtered (Celite), and the pad rinsed with MeTHF (2.5 L total). The filtrate was stored under N2 in a refrigerator until crystallization.[00365] Approximately 74% of 2-MeTHF was evaporated under reduced pressure while maintaining IT 23-34 °C (residual volume in the reactor was approximately 4.8 L). To the mixture was added n-heptane (6 L) over 15 min via dropping funnel. The resulting slurry was aged at room temperature overnight. The next day the solids on the walls were scraped to incorporate them into the slurry and the solids were isolated by filtration. The isolated solids were washed with n-heptane containing 5% MeTFlF (2 x 750 mL), and dried (75 °C, 30 inch Flg) to yield 1287 g (91 % yield) of the title compound as a yellow solid. F1PLC purity: 99.7% pure.[00366] ¾ NMR (400 MHz, DMSO- ) d ppm 1.20 (d, .7=6.02 Hz, 6 H) 2.21 (s, 6 H) 2.37 -2.44 (m, 2 H) 2.68 (s, 3 H) 2.93 (t, .7=6.78 Hz, 2 H) 3.74 (s, 3 H) 3.90 (s, 3 H) 4.60 (s, 2 H) 5.08 (spt, 7=6.19 Hz, 1 H) 6.80 (s, 1 H) 7.08 – 7.15 (m, 1 H) 7.19 – 7.26 (m, 2 H) 7.52 (d, .7=8.03 Hz, 1 H) 7.94 – 8.01 (m, 2 H) 8.56 (s, 1 H) 8.66 (s, 1 H).[00367] Step 5: Preparation of isopropyl 2-((4-((2-(dimethylamino)ethyl)(methyl)amino)-2- methoxy-5 -(3 -(phenylsulfonyl)propanamido)phenyl)amino)-4-(l -methyl- lH-indol-3- yl)pyrimidine-5-carboxylate.

lnt-5[00368] A mixture of the product of step 4 (1.284 kg, 2.415 mol, 1.0 eq.) and 3- (phenylsulfonyl)propionic acid (0.5528 kg, 2.580 mol, 1.07 eq.) in anhydrous DCM (8.5 L) was cooled to 2 °C, and treated with DIEA (0.310 kg, 2.399 mol, 1.0 eq.). To the reaction mixture was charged over 40 min, 50 % w/w T3P in MeTHF (1.756 kg, 2.759 mol, 1.14 eq.) while maintaining the internal temperature between 0 °C and 8 °C. The mixture was stirred at 0 °C to 5 °C for 15 minutes and then warmed over 30 min to 15 °C then held at 15 °C to 30 °C for 60 min.[00369] The reaction was quenched with water (179 mL). The reaction mixture was stirred at ambient temperature for 30 min then DIEA (439 g) was charged in one portion. The resulting mixture was aged for 15 min, and then treated with 5% aqueous K2CO3 (7.3 L) at 22-25 °C. The organic layer was separated and the aqueous layer back extracted with DCM (6.142 L). The combined organic extract was washed with brine (2 x 5.5 L).[00370] The organic extract was concentrated to 6.5 L, diluted with EtOFl, 200 Proof (14.3 kg), and the mixture concentrated under vacuum (23-25 inch Flg/IT40-60 °C) to a residual volume of 12.8 L.[00371] The residual slurry was treated with EtOFl, 200 Proof (28.8 Kg), and heated to 69 °C to obtain a thin slurry. The reaction mixture was cooled to 15 °C over 2 h, and stored overnight at 15 °C under nitrogen.[00372] The next day, the mixture was cooled to 5 °C, and aged for 30 minutes. The resulting solid was isolated by filtration, washed with EtOFl (2 x 2.16 kg) and dried to give 1.769 kg (100% yield) of the title compound. F1PLC purity 99.85%.‘H NMR (400 MHz, DMSO-i¾ d ppm 1.08 – 1.19 (m, 8 H) 2.15 (s, 6 H) 2.32 (t, J= 5.77 Hz, 2 H) 2.66 – 2.76 (m, 5 H) 2.88 (br t, J= 5.52 Hz, 2 H) 3.48 (qd, J= 7.03, 5.02 Hz, 1 H) 3.60 – 3.69 (m, 2 H) 3.83 (s, 3 H) 3.89 (s, 3 H) 4.40 (t, J=5.02 Hz, 1 H) 5.04 (quin, J=6.27 Hz, 1 H) 7.01 – 7.09 (m, 2 H) 7.22 (t, J= 7.53 Hz, 1 H) 7.52 (d, J= 8.53 Hz, 1 H) 7.67 – 7.82 (m, 4 H) 7.97 (s, 1 H) 7.98 – 8.00 (m, 1 H) 8.14 (s, 1 H) 8.61 – 8.70 (m, 3 H) 10.09 (s, 1 H).[00373] Step 6: Preparation of isopropyl 2-((5-acrylamido-4-((2-(dimethylamino)ethyl) (methyl)amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indol-3-yl)pyrimidine-5-carboxylate (Compound (A)).

compound (A)[00374] The product of step 5 (1.600 kg, 2.198 mol, 1.0 equiv.) was dissolved in anhydrous THF (19.5 kg) and was treated at -1 °C to 1 °C with 2M KOSi(CH3)3 in THF (2.72 L, 5.44 mol, 2.47 equiv.). KOSi(CFb)3 was added over 5 minutes, reactor jacket set at -5 °C to 10 °C. 2 M KOSi(CFh)3 solution was prepared by dissolving 871 g of KOSi(CFh)3 technical grade (90%) in 3.056 L of anhydrous TF1F.[00375] The reaction mixture was aged for 60 minutes. Potable water (22 L) was charged to the reaction mixture over 1 10 minutes, while maintaining temperature at 2-7 °C. The resulting suspension was aged at 3-7 °C for 60 minutes; the product was isolated by filtration (the filtration rate during crude product isolation was (1.25 L/min), washed with potable water (2 x 1.6 L) and air dried overnight and then in high vacuum for 12 h at 45 °C to give 1.186 kg of crude title compound (92% yield).‘H NMR (500 MHz, DMSO-i¾ d ppm 1.05 (t, J= 7.09 Hz, 2 H) 1.1 1 (d, J= 6.36 Hz, 6 H) 2.1 1 (s, 6 H) 2.28 (br t, .7=5.38 Hz, 3 H) 2.55 – 2.67 (m, 3 H) 2.69 (s, 3 H) 2.83 (br t, .7=5.38 Hz, 3 H) 3.31 (s, 3 H) 3.36 – 3.51 (m, 2 H) 3.54 – 3.70 (m, 3 H) 3.75 – 3.82 (m, 3 H) 4.33 (t, .7=5.14 Hz, 1 H) 4.99 (dt, 7=12.35, 6.30 Hz, 2 H) 5.75 (s, 1 H) 6.95 – 7.07 (m, 2 H) 7.17 (br t, .7=7.58 Hz, 2 H) 7.48 (d, 7=8.31 Hz, 2 H) 7.62 – 7.71 (m, 3 H) 7.71 – 7.83 (m, 2 H) 7.93 (d, .7=7.83 Hz, 3 H) 8.09 (s, 2 H) 8.53 – 8.67 (m, 3 H) 10.03 (s, 2 H).[00376] Step 7: Preparation of polymorphic Form-I of isopropyl 2-((5-acrylamido-4-((2- (dimethylamino)ethyl) (methyl)amino)-2-methoxyphenyl)amino)-4-(l -methyl- lH-indol-3- yl)pyrimidine-5-carboxylate (Free base Compound (A)).[00377] Method 1 : The crude product of step 6 (1.130 kg) was recrystallized by dissolving it in EtOAc (30.1 kg) at 75 °C, polish filtered (1.2 pm in-line filter), followed by concentration of the filtrate to 14 L of residue (IT during concentration is 58-70 °C). The residual slurry was cooled to 0 °C over 70 minutes and then aged at 0-2 °C for 30 minutes. Upon isolation the product was dried to a constant weight to give 1.007 kg (89% recovery) of the title compound as polymorphic Form-I. Purity (HPLC, a/a %, 99.80%).
PATENT
WO 2015195228
https://patents.google.com/patent/WO2015195228A1/en
PATENT
https://patents.google.com/patent/US10227342
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215310s000lbl.pdf
- ^ Jump up to:a b c d e “FDA grants accelerated approval to mobocertinib for metastatic non-sma”. U.S. Food and Drug Administration (FDA). 16 September 2021. Retrieved 16 September 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “Takeda’s Exkivity (mobocertinib) Approved by U.S. FDA as the First Oral Therapy Specifically Designed for Patients with EGFR Exon20 Insertion+ NSCLC” (Press release). Takeda Pharmaceutical Company. 15 September 2021. Retrieved 16 September 2021 – via Business Wire.
- ^ “TAK-788 as First-line Treatment Versus Platinum-Based Chemotherapy for Non-Small Cell Lung Cancer (NSCLC) With EGFR Exon 20 Insertion Mutations”. Clinicaltrials.gov. Retrieved 17 February 2021.
- ^ Zhang SS, Zhu VW (2021). “Spotlight on Mobocertinib (TAK-788) in NSCLC with EGFR Exon 20 Insertion Mutations”. Lung Cancer. Auckland, N.Z. 12: 61–65. doi:10.2147/LCTT.S307321. PMC 8286072. PMID 34285620.
External links
- “Mobocertinib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02716116 for “A Study of TAK-788 in Adults With Non-Small Cell Lung Cancer” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Exkivity |
| Other names | TAK-788 |
| License data | US DailyMed: Mobocertinib |
| Pregnancy category | Contraindicated[1] |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1847461-43-12389149-74-8 |
| PubChem CID | 118607832 |
| DrugBank | DB16390DBSALT003192 |
| ChemSpider | 84455481 |
| UNII | 39HBQ4A67L |
| KEGG | D12001D11969 |
| ChEMBL | ChEMBL4650319 |
| Chemical and physical data | |
| Formula | C32H39N7O4 |
| Molar mass | 585.709 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////mobocertinib, Exkivity, TAK 788, AP32788, fda 2021, approvals 2021, cancer
CC(C)OC(=O)C1=CN=C(N=C1C2=CN(C3=CC=CC=C32)C)NC4=C(C=C(C(=C4)NC(=O)C=C)N(C)CCN(C)C)OC

NEW DRUG APPROVALS
one time to maintain this blog
$10.00
Plasminogen
Plasminogen
FDA APPROVED 2021, Ryplazim, 2021/6/4
Plasminogen;
Glu-plasminogen;
Plasminogen, human-tvmh;
Ryplazim (TN)
RYPLAZIM (plasminogen, human-tvmh)
Enzyme replacement (plasminogen), Plasminogen deficiency type 1
CAS: 9001-91-6
STN: 125659
Proper Name: plasminogen, human-tvmh
Tradename: RYPLAZIM
Manufacturer: Prometic Biotherapeutics Inc.
Indication:
For the treatment of patients with plasminogen deficiency type 1 (hypoplasminogenemia)
READ https://diapharma.com/plasminogen-plg/
On August 11, 2017 Prometic Biotherapeutics submitted a BLA (STN 125659) for a Drug Product (DP) RYPLAZIM, Plasminogen (Human). This drug product is indicated for replacement therapy in children and adults with plasminogen deficiency.
Plasmin is an important enzyme (EC 3.4.21.7) present in blood that degrades many blood plasma proteins, including fibrin clots. The degradation of fibrin is termed fibrinolysis. In humans, the plasmin protein is encoded by the PLG gene.[5]
Function
Fibrinolysis (simplified). Blue arrows denote stimulation, and red arrows inhibition.
Plasmin is a serine protease that acts to dissolve fibrin blood clots. Apart from fibrinolysis, plasmin proteolyses proteins in various other systems: It activates collagenases, some mediators of the complement system, and weakens the wall of the Graafian follicle, leading to ovulation. Plasmin is also integrally involved in inflammation.[6] It cleaves fibrin, fibronectin, thrombospondin, laminin, and von Willebrand factor. Plasmin, like trypsin, belongs to the family of serine proteases.
Plasmin is released as a zymogen called plasminogen (PLG) from the liver into the systemic circulation. Two major glycoforms of plasminogen are present in humans – type I plasminogen contains two glycosylation moieties (N-linked to N289 and O-linked to T346), whereas type II plasminogen contains only a single O-linked sugar (O-linked to T346). Type II plasminogen is preferentially recruited to the cell surface over the type I glycoform. Conversely, type I plasminogen appears more readily recruited to blood clots.
In circulation, plasminogen adopts a closed, activation-resistant conformation. Upon binding to clots, or to the cell surface, plasminogen adopts an open form that can be converted into active plasmin by a variety of enzymes, including tissue plasminogen activator (tPA), urokinase plasminogen activator (uPA), kallikrein, and factor XII (Hageman factor). Fibrin is a cofactor for plasminogen activation by tissue plasminogen activator. Urokinase plasminogen activator receptor (uPAR) is a cofactor for plasminogen activation by urokinase plasminogen activator. The conversion of plasminogen to plasmin involves the cleavage of the peptide bond between Arg-561 and Val-562.[5][7][8][9]
Plasmin cleavage produces angiostatin.
Mechanism of plasminogen activation
Full length plasminogen comprises seven domains. In addition to a C-terminal chymotrypsin-like serine protease domain, plasminogen contains an N-terminal Pan Apple domain (PAp) together with five Kringle domains (KR1-5). The Pan-Apple domain contains important determinants for maintaining plasminogen in the closed form, and the kringle domains are responsible for binding to lysine residues present in receptors and substrates.
The X-ray crystal structure of closed plasminogen reveals that the PAp and SP domains maintain the closed conformation through interactions made throughout the kringle array .[9] Chloride ions further bridge the PAp / KR4 and SP / KR2 interfaces, explaining the physiological role of serum chloride in stabilizing the closed conformer. The structural studies also reveal that differences in glycosylation alter the position of KR3. These data help explain the functional differences between the type I and type II plasminogen glycoforms.[citation needed]
In closed plasminogen, access to the activation bond (R561/V562) targeted for cleavage by tPA and uPA is blocked through the position of the KR3/KR4 linker sequence and the O-linked sugar on T346. The position of KR3 may also hinder access to the activation loop. The Inter-domain interactions also block all kringle ligand-binding sites apart from that of KR-1, suggesting that the latter domain governs pro-enzyme recruitment to targets. Analysis of an intermediate plasminogen structure suggests that plasminogen conformational change to the open form is initiated through KR-5 transiently peeling away from the PAp domain. These movements expose the KR5 lysine-binding site to potential binding partners, and suggest a requirement for spatially distinct lysine residues in eliciting plasminogen recruitment and conformational change respectively.[9]
Mechanism of plasmin inactivation
Plasmin is inactivated by proteins such as α2-macroglobulin and α2-antiplasmin.[10] The mechanism of plasmin inactivation involves the cleavage of an α2-macroglobulin at the bait region (a segment of the aM that is particularly susceptible to proteolytic cleavage) by plasmin. This initiates a conformational change such that the α2-macroglobulin collapses about the plasmin. In the resulting α2-macroglobulin-plasmin complex, the active site of plasmin is sterically shielded, thus substantially decreasing the plasmin’s access to protein substrates. Two additional events occur as a consequence of bait region cleavage, namely (i) a h-cysteinyl-g-glutamyl thiol ester of the α2-macroglobulin becomes highly reactive and (ii) a major conformational change exposes a conserved COOH-terminal receptor binding domain. The exposure of this receptor binding domain allows the α2-macroglobulin protease complex to bind to clearance receptors and be removed from circulation.
Pathology
Plasmin deficiency may lead to thrombosis, as the clots are not adequately degraded. Plasminogen deficiency in mice leads to defective liver repair,[11] defective wound healing, reproductive abnormalities.[citation needed]
In humans, a rare disorder called plasminogen deficiency type I (Online Mendelian Inheritance in Man (OMIM): 217090) is caused by mutations of the PLG gene and is often manifested by ligneous conjunctivitis.
Interactions
Plasmin has been shown to interact with Thrombospondin 1,[12][13] Alpha 2-antiplasmin[14][15] and IGFBP3.[16] Moreover, plasmin induces the generation of bradykinin in mice and humans through high-molecular-weight kininogen cleavage.[17]
References
- ^ Jump up to:a b c GRCh38: Ensembl release 89: ENSG00000122194 – Ensembl, May 2017
- ^ Jump up to:a b c GRCm38: Ensembl release 89: ENSMUSG00000059481 – Ensembl, May 2017
- ^ “Human PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ “Mouse PubMed Reference:”. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ Jump up to:a b “Entrez Gene: plasminogen”.
- ^ Atsev S, Tomov N (December 2020). “Using antifibrinolytics to tackle neuroinflammation”. Neural Regeneration Research. 15(12): 2203–2206. doi:10.4103/1673-5374.284979. PMC 7749481. PMID 32594031.
- ^ Miyata T, Iwanaga S, Sakata Y, Aoki N (October 1982). “Plasminogen Tochigi: inactive plasmin resulting from replacement of alanine-600 by threonine in the active site”. Proc. Natl. Acad. Sci. U.S.A. 79 (20): 6132–6. Bibcode:1982PNAS…79.6132M. doi:10.1073/pnas.79.20.6132. PMC 347073. PMID 6216475.
- ^ Forsgren M, Råden B, Israelsson M, Larsson K, Hedén LO (March 1987). “Molecular cloning and characterization of a full-length cDNA clone for human plasminogen”. FEBS Lett. 213 (2): 254–60. doi:10.1016/0014-5793(87)81501-6. PMID 3030813. S2CID 9075872.
- ^ Jump up to:a b c Law RH, Caradoc-Davies T, Cowieson N, Horvath AJ, Quek AJ, Encarnacao JA, Steer D, Cowan A, Zhang Q, Lu BG, Pike RN, Smith AI, Coughlin PB, Whisstock JC (2012). “The X-ray crystal structure of full-length human plasminogen”. Cell Rep. 1 (3): 185–90. doi:10.1016/j.celrep.2012.02.012. PMID 22832192.
- ^ Wu, Guojie; Quek, Adam J.; Caradoc-Davies, Tom T.; Ekkel, Sue M.; Mazzitelli, Blake; Whisstock, James C.; Law, Ruby H.P. (2019-03-05). “Structural studies of plasmin inhibition”. Biochemical Society Transactions. 47 (2): 541–557. doi:10.1042/bst20180211. ISSN 0300-5127. PMID 30837322.
- ^ Bezerra JA, Bugge TH, Melin-Aldana H, Sabla G, Kombrinck KW, Witte DP, Degen JL (December 21, 1999). “Plasminogen deficiency leads to impaired remodeling after a toxic injury to the liver”. Proc. Natl. Acad. Sci. U.S.A. Proceedings of the National Academy of Sciences of the United States of America. 96 (26): 15143–8. Bibcode:1999PNAS…9615143B. doi:10.1073/pnas.96.26.15143. PMC 24787. PMID 10611352.
- ^ Silverstein RL, Leung LL, Harpel PC, Nachman RL (November 1984). “Complex formation of platelet thrombospondin with plasminogen. Modulation of activation by tissue activator”. J. Clin. Invest. 74 (5): 1625–33. doi:10.1172/JCI111578. PMC 425339. PMID 6438154.
- ^ DePoli P, Bacon-Baguley T, Kendra-Franczak S, Cederholm MT, Walz DA (March 1989). “Thrombospondin interaction with plasminogen. Evidence for binding to a specific region of the kringle structure of plasminogen”. Blood. 73 (4): 976–82. doi:10.1182/blood.V73.4.976.976. PMID 2522013.
- ^ Wiman B, Collen D (September 1979). “On the mechanism of the reaction between human alpha 2-antiplasmin and plasmin”. J. Biol. Chem. 254 (18): 9291–7. doi:10.1016/S0021-9258(19)86843-6. PMID 158022.
- ^ Shieh BH, Travis J (May 1987). “The reactive site of human alpha 2-antiplasmin”. J. Biol. Chem. 262 (13): 6055–9. doi:10.1016/S0021-9258(18)45536-6. PMID 2437112.
- ^ Campbell PG, Durham SK, Suwanichkul A, Hayes JD, Powell DR (August 1998). “Plasminogen binds the heparin-binding domain of insulin-like growth factor-binding protein-3”. Am. J. Physiol. 275 (2 Pt 1): E321-31. doi:10.1152/ajpendo.1998.275.2.E321. PMID 9688635.
- ^ Marcos-Contreras OA, Martinez de Lizarrondo S, Bardou I, Orset C, Pruvost M, Anfray A, Frigout Y, Hommet Y, Lebouvier L, Montaner J, Vivien D, Gauberti M (August 2016). “Hyperfibrinolysis increases blood brain barrier permeability by a plasmin and bradykinin-dependent mechanism”. Blood. 128 (20): 2423–2434. doi:10.1182/blood-2016-03-705384. PMID 27531677.
Further reading
- Shanmukhappa K, Mourya R, Sabla GE, Degen JL, Bezerra JA (July 2005). “Hepatic to pancreatic switch defines a role for hemostatic factors in cellular plasticity in mice”. Proc. Natl. Acad. Sci. U.S.A. 102 (29): 10182–7. Bibcode:2005PNAS..10210182S. doi:10.1073/pnas.0501691102. PMC 1177369. PMID 16006527.
- Anglés-Cano E, Rojas G (2002). “Apolipoprotein(a): structure-function relationship at the lysine-binding site and plasminogen activator cleavage site”. Biol. Chem. 383 (1): 93–9. doi:10.1515/BC.2002.009. PMID 11928826. S2CID 29248198.
- Ranson M, Andronicos NM (2003). “Plasminogen binding and cancer: promises and pitfalls”. Front. Biosci. 8 (6): s294-304. doi:10.2741/1044. PMID 12700073.
External links
- The MEROPS online database for peptidases and their inhibitors: S01.233
- Plasmin at the US National Library of Medicine Medical Subject Headings (MeSH)
| PLG | ||
|---|---|---|
| Available structuresPDBOrtholog search: PDBe RCSBshowList of PDB id codes | ||
| Identifiers | ||
| Aliases | PLG, plasminogen, plasmin, HAE4 | |
| External IDs | OMIM: 173350 MGI: 97620 HomoloGene: 55452 GeneCards: PLG | |
| showGene location (Human) | ||
| showGene location (Mouse) | ||
| showRNA expression pattern | ||
| showGene ontology | ||
| Orthologs | ||
| Species | Human | Mouse |
| Entrez | 5340 | 18815 |
| Ensembl | ENSG00000122194 | ENSMUSG00000059481 |
| UniProt | P00747 | P20918 |
| RefSeq (mRNA) | NM_001168338 NM_000301 | NM_008877 |
| RefSeq (protein) | NP_000292 NP_001161810 | NP_032903 |
| Location (UCSC) | Chr 6: 160.7 – 160.75 Mb | Chr 17: 12.38 – 12.42 Mb |
| PubMed search | [3] | [4] |
| Wikidata | ||
| View/Edit HumanView/Edit Mouse |
///////////Plasminogen, FDA 2021, APPROVALS 2021, Ryplazim

NEW DRUG APPROVALS
ONE TIME
$10.00
Difelikefalin acetate
Difelikefalin acetate
ジフェリケファリン酢酸塩
CAS 1024829-44-4
| Formula | C36H53N7O6. (C2H4O2)x |
|---|
D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4- carboxylic acid)]-OH
FDA APPROVED, 2021/8/23, FORSUVA
Analgesic, Antipruritic, Opioid receptor agonist
Treatment of moderate-to-severe pruritus associated with chronic kidney disease in adults undergoing hemodialysis
Difelikefalin, CR-845; MR-13A-9; MR-13A9
4-amino-1- (D-phenylalanyl-D-phenylalanyl-D-leucyl-D-lysyl) piperidine-4-carboxylic acid
C36H53N7O6, 679.40573
| ORIGINATOR | Ferring Pharmaceuticals |
|---|---|
| DEVELOPER | Cara Therapeutics |
| CLASS | Analgesic drugs (peptides) |
| MECHANISM OF ACTION | Opioid kappa receptor agonists |
| WHO ATC CODES | D04A-X (Other antipruritics), N02A (Opioids) |
| EPHMRA CODES | D4A (Anti-Pruritics, Including Topical Antihistamines, Anaesthetics, etc), N2A (Narcotics) |
| INDICATION | Pain, Osteoarthritis, Pruritus |
Difelikefalin, sold under the brand name Korsuva , is an analgesic opioid peptide used for the treatment of moderate-to-severe pruritus. It acts as a peripherally specific, highly selective agonist of the κ-opioid receptor (KOR).[3][4][5][6]
Difelikefalin was approved for medical use in the United States in August 2021.[2][7][8]
Difelikefalin acts as an analgesic by activating KORs on peripheral nerve terminals and KORs expressed by certain immune system cells.[3] Activation of KORs on peripheral nerve terminals results in the inhibition of ion channels responsible for afferent nerve activity, causing reduced transmission of pain signals, while activation of KORs expressed by immune system cells results in reduced release of proinflammatory, nerve-sensitizing mediators (e.g., prostaglandins).[3]

NEW DRUG APPROVALS
ONE TIME
$10.00
Research
It is under development by Cara Therapeutics as an intravenous agent for the treatment of postoperative pain.[3][4][6] An oral formulation has also been developed.[6] Due to its peripheral selectivity, difelikefalin lacks the central side effects like sedation, dysphoria, and hallucinations of previous KOR-acting analgesics such as pentazocine and phenazocine.[3][4] In addition to use as an analgesic, difelikefalin is also being investigated for the treatment of pruritus (itching).[3][4][5] Difelikefalin has completed phase II clinical trials for postoperative pain and has demonstrated significant and “robust” clinical efficacy, along with being safe and well tolerated.[4][6] It has also completed a phase III clinical trial for uremic pruritus in hemodialysis patients.[9]Kappa opioid receptors have been suggested as targets for intervention for treatment or prevention of a wide array of diseases and conditions by administration of kappa opioid receptor agonists. See for example, Jolivalt et al., Diabetologia, 49(11):2775-85; Epub Aug. 19, 2006), describing efficacy of asimadoline, a kappa receptor agonist in rodent diabetic neuropathy; and Bileviciute-Ljungar et al., Eur. J. Pharm. 494:139-46 (2004) describing the efficacy of kappa agonist U-50,488 in the rat chronic constriction injury (CCI) model of neuropathic pain and the blocking of its effects by the opioid antagonist, naloxone. These observations support the use of kappa opioid receptor agonists for treatment of diabetic, viral and chemotherapy- induced neuropathic pain. The use of kappa receptor agonists for treatment or prevention of visceral pain including gynecological conditions such as dysmenorrheal cramps and endometriosis has also been reviewed. See for instance, Riviere, Br. J. Pharmacol. 141:1331-4 (2004).[0004] Kappa opioid receptor agonists have also been proposed for the treatment of pain, including hyperalgesia. Hyperalgesia is believed to be caused by changes in the milieu of the peripheral sensory terminal occur secondary to local tissue damage. Tissue damage (e.g., abrasions, burns) and inflammation can produce significant increases in the excitability of polymodal nociceptors (C fibers) and high threshold mechanoreceptors (Handwerker et al. (1991) Proceeding of the VIth World Congress on Pain, Bond et al., eds., Elsevier Science Publishers BV, pp. 59-70; Schaible et al. (1993) Pain 55:5-54). This increased excitability and exaggerated responses of sensory afferents is believed to underlie hyperalgesia, where the pain response is the result of an exaggerated response to a stimulus. The importance of the hyperalgesic state in the post-injury pain state has been repeatedly demonstrated and appears to account for a major proportion of the post-injury/inflammatory pain state. See for example, Woold et al. (1993) Anesthesia and Analgesia 77:362-79; Dubner et al.(1994) In, Textbook of Pain, Melzack et al., eds., Churchill-Livingstone, London, pp. 225-242.[0005] Kappa opioid receptors have been suggested as targets for the prevention and treatment of cardiovascular disease. See for example, Wu et al. “Cardioprotection of Preconditioning by Metabolic Inhibition in the Rat Ventricular Myocyte – Involvement of kappa Opioid Receptor” (1999) Circulation Res vol. 84: pp. 1388-1395. See also Yu et al. “Anti-Arrhythmic Effect of kappa Opioid Receptor Stimulation in the Perfused Rat Heart: Involvement of a cAMP-Dependent Pathway”(1999) JMoI Cell Cardiol, vol. 31(10): pp. 1809-1819.[0006] It has also been found that development or progression of these diseases and conditions involving neurodegeneration or neuronal cell death can be prevented, or at least slowed, by treatment with kappa opioid receptor agonists. This improved outcome is believed to be due to neuroprotection by the kappa opioid receptor agonists. See for instance, Kaushik et al. “Neuroprotection in Glaucoma” (2003) J. Postgraduate Medicine vol. 49 (1): pp. 90-95. [0007] The presence of kappa opioid receptors on immune cells (Bidlak et al.,(2000) Clin. Diag. Lab. Immunol. 7(5):719-723) has been implicated in the inhibitory • action of a kappa opioid receptor agonist, which has been shown to suppress HIV-I expression. See Peterson PK et al, Biochem Pharmacol 2001, 61(19):1145-51. [0008] Walker, Adv. Exp. Med. Biol. 521: 148-60 (2003) appraised the antiinflammatory properties of kappa agonists for treatment of osteoarthritis, rheumatoid arthritis, inflammatory bowel disease and eczema. Bileviciute-Ljungar et al., Rheumatology 45:295-302 (2006) describe the reduction of pain and degeneration in Freund’s adjuvant-induced arthritis by the kappa agonist U-50,488.[0009] Wikstrom et al, J. Am. Soc. Nephrol. 16:3742-7 (2005) describes the use of the kappa agonist, TRK-820 for treatment of uremic and opiate-induced pruritis, and Ko et al., J. Pharmacol. Exp. Ther. 305: 173-9 (2003) describe the efficacy of U- 50,488 in morphine-induced pruritis in the monkey. [0010] Application of peripheral opioids including kappa agonists for treatment of gastrointestinal diseases has also been extensively reviewed. See for example, Lembo, Diges. Dis. 24:91-8 (2006) for a discussion of use of opioids in treatment of digestive disorders, including irritable bowel syndrome (IBS), ileus, and functional dyspepsia.[0011] Ophthalmic disorders, including ocular inflammation and glaucoma have also been shown to be addressable by kappa opioids. See Potter et ah, J. Pharmacol. Exp. Ther. 309:548-53 (2004), describing the role of the potent kappa opioid receptor agonist, bremazocine, in reduction of intraocular pressure and blocking of this effect by norbinaltorphimine (norBNI), the prototypical kappa opioid receptor antagonist; and Dortch-Carnes et al, CNS Drug Rev. 11(2): 195-212 (2005). U.S. Patent 6,191,126 to Gamache discloses the use of kappa opioid agonists to treat ocular pain. Otic pain has also been shown to be treatable by administration of kappa opioid agonists. See U.S. Patent 6,174,878 also to Gamache.[0012] Kappa opioid agonists increase the renal excretion of water and decrease urinary sodium excretion (i.e., produces a selective water diuresis, also referred to as aquaresis). Many, but not all, investigators attribute this effect to a suppression of vasopressin secretion from the pituitary. Studies comparing centrally acting and purportedly peripherally selective kappa opioids have led to the conclusion that kappa opioid receptors within the blood-brain barrier are responsible for mediating this effect. Other investigators have proposed to treat hyponatremia with nociceptin peptides or charged peptide conjugates that act peripherally at the nociceptin receptor, which is related to but distinct from the kappa opioid receptor (D. R. Kapusta, Life ScL, 60: 15-21, 1997) (U.S. Pat. No. 5,840,696). U.S. Pat Appl. 20060052284.
PATENTJpn. Tokkyo Koho, 5807140US 20090156508WO 2008057608
PATENTUS 20100075910https://patents.google.com/patent/US8236766B2/en


Example 2Synthesis of Compound (2): D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OHSee the scheme of FIG. 3 and Biron et al., Optimized selective N-methylation of peptides on solid support. J. Peptide Science 12: 213-219 (2006). The amino acid derivatives used were Boc-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Dde)-OH, and N-Boc-amino-(4-N-Fmoc-piperidinyl)carboxylic acid. HPLC and MS analyses were performed as described in the synthesis of compound (1) described above.The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (1.8 g, 0.9 mmol; Peptide International). Attachment of N-Boc-amino-(4-N-Fmoc-piperidinyl)carboxylic acid followed by peptide chain elongation and deprotection of Dde in D-Lys(Dde) at Xaa4 was carried out according to the procedure described in the synthesis of compound (1). See above. The resulting peptide resin (0.9 mmol; Boc-D-Phe-D-Phe-D-Leu-D-Lys-(N-Boc-amino-4-piperidinylcarboxylic acid)-[2-Cl-Trt resin]) was split and a portion of 0.3 mmol was used for subsequent cleavage. The peptide resin (0.3 mmol) was then treated with a mixture of TFA/TIS/H2O (15 ml, v/v/v=95:2.5:2.5) at room temperature for 90 minutes. The resin was then filtered and washed with TFA. The filtrate was evaporated in vacuo and the crude synthetic peptide amide (0.3 mmol; D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OH) was precipitated from diethyl ether.For purification, the crude synthetic peptide amide (0.3 mmol) was dissolved in 2% acetic acid in H2O (50 ml) and the solution was loaded onto an HPLC column and purified using TEAP buffer system with a pH 5.2 (buffers A=TEAP 5.2 and B=20% TEAP 5.2 in 80% ACN). The compound was eluted with a linear gradient of buffer B, 7% B to 37% B over 60 minutes. Fractions with purity exceeding 95% were pooled and the resulting solution was diluted with two volumes of water. The diluted solution was then loaded onto an HPLC column for salt exchange and further purification with a TFA buffer system (buffers A=0.1% TFA in H2O and B=0.1% TFA in 80% ACN/20% H2O) and a linear gradient of buffer B, 2% B to 75% B over 25 minutes. Fractions with purity exceeding 97% were pooled, frozen, and dried on a lyophilizer to yield the purified synthetic peptide amide as white amorphous powder (93 mg). HPLC analysis: tR=16.43 min, purity 99.2%, gradient 5% B to 25% B over 20 min; MS (MH+): expected molecular ion mass 680.4, observed 680.3.Compound (2) was also prepared using a reaction scheme analogous to that shown in FIG. 3 with the following amino acid derivatives: Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Boc)-OH, and Boc-4-amino-1-Fmoc-(piperidine)-4-carboxylic acid.The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (PS 1% DVB, 500 g, 1 meq/g). The resin was treated with Boc-4-amino-1-Fmoc-4-(piperidine)-4-carboxylic acid (280 g, 600 mmol) in a mixture of DMF, DCM and DIEA (260 mL of each) was added. The mixture was stirred for 4 hours and then the resin was capped for 1 h by the addition of MeOH (258 mL) and DIEA (258 mL).The resin was isolated and washed with DMF (3×3 L). The resin containing the first amino acid was treated with piperidine in DMF (3×3 L of 35%), washed with DMF (9×3 L) and Fmoc-D-Lys(Boc)-OH (472 g) was coupled using PyBOP (519 g) in the presence of HOBt (153 g) and DIEA (516 mL) and in DCM/DMF (500 mL/500 mL) with stiffing for 2.25 hours. The dipeptide containing resin was isolated and washed with DMF (3×3.6 L). The Fmoc group was removed by treatment with piperidine in DMF(3×3.6 L of 35%) and the resin was washed with DMF (9×3.6 L) and treated with Fmoc-D-Leu-OH (354 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL/500 mL) and stirred for 1 hour. Subsequent washing with DMF (3×4.1 L) followed by cleavage of the Fmoc group with piperidine in DMF (3×4.2 L of 35%) and then washing of the resin with DMF (9×4.2 L) provided the resin bound tripeptide. This material was treated with Fmoc-D-Phe-OH (387 g), DIC (157 mL) and HOBt (153 g) in DCM/DMF (500 mL/500 mL) and stirred overnight. The resin was isolated, washed with DMF (3×4.7 L) and then treated with piperidine in DMF (3×4.7 L of 35%) to cleave the Fmoc group and then washed again with DMF (9×4.7 L). The tetrapeptide loaded resin was treated with Fmoc-D-Phe-OH (389 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL/500 mL) and stirred for 2.25 hours. The resin was isolated, washed with DMF (3×5.2 L) and then treated piperidine (3×5.2 L of 35%) in DMF. The resin was isolated, and washed sequentially with DMF (9×5.2 L) then DCM (5×5.2 L). It was dried to provide a 90.4% yield of protected peptide bound to the resin. The peptide was cleaved from the resin using TFA/water (4.5 L, 95/5), which also served to remove the Boc protecting groups. The mixture was filtered, concentrated (⅓) and then precipitated by addition to MTBE (42 L). The solid was collected by filtration and dried under reduced pressure to give crude synthetic peptide amide.For purification, the crude synthetic peptide amide was dissolved in 0.1% TFA in H2O and purified by preparative reverse phase HPLC (C18) using 0.1% TFA/water—ACN gradient as the mobile phase. Fractions with purity exceeding 95% were pooled, concentrated and lyophilized to provide pure synthetic peptide amide (>95.5% pure). Ion exchange was conducted using a Dowex ion exchange resin, eluting with water. The aqueous phase was filtered (0.22 μm filter capsule) and freeze-dried to give the acetate salt of the synthetic peptide amide (2) with overall yield, 71.3%, >99% purity.Hydrochloride, hydrobromide and fumarate counterions were evaluated for their ability to form crystalline salts of synthetic peptide amide (2). Approximately 1 or 2 equivalents (depending on desired stoichiometry) of hydrochloric acid, hydrobromic acid or fumaric acid, as a dilute solution in methanol (0.2-0.3 g) was added to synthetic peptide amide (2) (50-70 mg) dissolved in methanol (0.2-0.3 g). Each individual salt solution was added to isopropyl acetate (3-5 mL) and the resulting amorphous precipitate was collected by filtration and dried at ambient temperature and pressure. Crystallization experiments were carried out by dissolving the 10-20 mg of the specific amorphous salt obtained above in 70:30 ethanol-water mixture (0.1-0.2 g) followed by the addition of ethanol to adjust the ratio to 90:10 (˜0.6-0.8 mL). Each solution was then seeded with solid particles of the respective precipitated salt. Each sample tube was equipped with a magnetic stir bar and the sample was gently stirred at ambient temperature. The samples were periodically examined by plane-polarized light microscopy. Under these conditions, the mono- and di-hydrochloride salts, the di-hydrobromide salt and the mono-fumarate salt crystallized as needles of 20 to 50 μm in length with a thickness of about 1 μm.PATENT
WO 2008057608
https://patents.google.com/patent/WO2008057608A2/en Compound (2): D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4- carboxylic acid)]-OH (SEQ ID NO: 2):

EXAMPLE 2: Synthesis of compound (2)[00288] D-Phe-D-Phe-D-Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OH (SEQ ID NO: 2):[00289] See the scheme of Figure 2 and B iron et al., Optimized selective N- methylation of peptides on solid support. J. Peptide Science 12: 213-219 (2006). The amino acid derivatives used were Boc-D-Phe-OH, Fmoc-D-Phe-OH, Fmoc-D-Leu- OH, Fmoc-D-Lys(Dde)-OH, and N-Boc-amino-(4-N-Fmoc-piperidinyl) carboxylic acid. HPLC and MS analyses were performed as described in the synthesis of compound (1) described above.[00290] The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (1.8 g, 0.9 mmol; Peptide International). Attachment of N-Boc-amino-(4-N-Fmoc-piperidinyl) carboxylic acid followed by peptide chain elongation and deprotection of Dde in D-Lys(Dde) at Xa^ was carried out according to the procedure described in the synthesis of compound (1). See above. The resulting peptide resin (0.9 mmol; Boc-D-Phe-D-Phe-D-Leu-D-Lys-(N- Boc-amino-4-piperidinylcarboxylic acid)-[2-Cl-Trt resin]) was split and a portion of 0.3 mmol was used for subsequent cleavage. The peptide resin (0.3 mmol) was then treated with a mixture of TFA/TIS/H2O (15 ml, v/v/v = 95:2.5:2.5) at room temperature for 90 min. The resin was then filtered and washed with TFA. The filtrate was evaporated in vacuo and the crude peptide (0.3 mmol; D-Phe-D-Phe-D- Leu-D-Lys-[ω(4-aminopiperidine-4-carboxylic acid)]-OH) was precipitated from diethyl ether.[00291] For purification, the crude peptide (0.3 mmol) was dissolved in 2% acetic acid in H2O (50 ml) and the solution was loaded onto an HPLC column and purified using TEAP buffer system with a pH 5.2 (buffers A = TEAP 5.2 and B = 20% TEAP 5.2 in 80% ACN). The compound was eluted with a linear gradient of buffer B, 7%B to 37%B over 60 min. Fractions with purity exceeding 95% were pooled and the resulting solution was diluted with two volumes of water. The diluted solution was then loaded onto an HPLC column for salt exchange and further purification with a TFA buffer system (buffers A = 0.1% TFA in H2O and B = 0.1% TFA in 80% ACN/20% H2O) and a linear gradient of buffer B, 2%B to 75%B over 25 min. Fractions with purity exceeding 97% were pooled, frozen, and dried on a lyophilizer to yield the purified peptide as white amorphous powder (93 mg). HPLC analysis: tR = 16.43 min, purity 99.2%, gradient 5%B to 25%B over 20 min; MS (M+H+): expected molecular ion mass 680.4, observed 680.3.[00292] Compound (2) was also prepared using a reaction scheme analogous to that shown in figure 2 with the following amino acid derivatives: Fmoc-D-Phe-OH, Fmoc-D-Leu-OH, Fmoc-D-Lys(Boc)-OH, and Boc-4-amino-l-Fmoc-(piperidine)-4- carboxylic acid.[00293] The fully protected resin-bound peptide was synthesized manually starting from 2-Chlorotrityl chloride resin (PS 1%DVB, 500 g, 1 meq/g). The resin was treated with Boc-4-amino-l-Fmoc-4-(piperidine)-4-carboxylic acid (280 g, 600 mmol) in a mixture of DMF, DCM and DIEA (260 mL of each) was added. The mixture was stirred for 4 hours and then the resin was capped for Ih by the addition of MeOH (258 mL) and DIEA[00294] (258 mL). The resin was isolated and washed with DMF (3 x 3 L). The resin containing the first amino acid was treated with piperidine in DMF (3 x 3 L of 35%), washed with DMF (9 x 3 L) and Fmoc-D-Lys(Boc)-OH (472 g) was coupled using PyBOP (519 g) in the presence of HOBt (153 g) and DIEA (516 mL) and in DCM/DMF (500 mL/ 500 mL) with stirring for 2.25 hours. The dipeptide containing resin was isolated and washed with DMF (3 x 3.6 L). The Fmoc group was removed by treatment with piperidine in DMF [00295] , (3 x 3.6 L of 35%) and the resin was washed with DMF (9 x 3.6 L) and treated with Fmoc-D-Leu-OH (354 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL / 500 mL) and stirred for 1 hour. Subsequent washing with DMF (3 x 4.1 L) followed by cleavage of the Fmoc group with piperidine in DMF (3 x 4.2 L of 35%) and then washing of the resin with DMF (9 x 4.2 L) provided the resin bound tripeptide. This material was treated with Fmoc-D-Phe-OH (387 g), DIC (157 mL) and HOBt (153 g) in DCM/DMF (500 mL / 500 mL) and stirred overnight. The resin was isolated, washed with DMF (3 x 4.7 L) and then treated with piperidine in DMF (3 x 4.7 L of 35%) to cleave the Fmoc group and then washed again with DMF (9 x 4.7 L). The tetrapeptide loaded resin was treated with Fmoc-D-Phe-OH (389 g), DIC (157 mL) and HOBt (154 g) in DCM/DMF (500 mL / 500 mL) and stirred for 2.25 hours. The resin was isolated, washed with DMF (3 x 5.2 L) and then treated piperidine (3 x 5.2 L of 35%) in DMF. The resin was isolated, and washed sequentially with DMF (9 x 5.2 L) then DCM (5 x 5.2 L). It was dried to provide a 90.4% yield of protected peptide bound to the resin. The peptide was cleaved from the resin using TFA/ water (4.5 L, 95/5), which also served to remove the Boc protecting groups. The mixture was filtered, concentrated (1/3) and then precipitated by addition to MTBE (42 L). The solid was collected by filtration and dried under reduced pressure to give crude peptide.[00296] For purification, the crude peptide was dissolved in 0.1% TFA in H2O and purified by preparative reverse phase HPLC (C 18) using 0.1% TF A/water – ACN gradient as the mobile phase. Fractions with purity exceeding 95% were pooled, concentrated and lyophilized to provide pure peptide (> 95.5% pure). Ion exchange was conducted using a Dowex ion exchange resin, eluting with water. The aqueous phase was filtered (0.22 μm filter capsule) and freeze-dried to give the acetate salt of the peptide (overall yield, 71.3%, >99% pure).
PATENT
κ opioid receptor agonists are known to be useful as therapeutic agents for various pain. Among, kappa opioid receptor agonist with high selectivity for peripheral kappa opioid receptors, are expected as a medicament which does not cause the central side effects. Such as peripherally selective κ opioid receptor agonist, a synthetic pentapeptide has been reported (Patent Documents 1 and 2). The following formula among the synthetic pentapeptide (A)
[Formula 1] Being Represented By Compounds Are Useful As Pain Therapeutics. The Preparation Of This Compound, Solid Phase Peptide Synthesis Methods In Patent Documents 1 And 2 Have Been Described.Document 1 Patent: Kohyo 2010-510966 JP
Patent Document 2: Japanese Unexamined Patent Publication No. 2013-241447 Compound (1) or a salt thereof and compound (A), for example as shown in the following reaction formula, 4-aminopiperidine-4-carboxylic acid, D- lysine (D-Lys), D- leucine (D-Leu) , it can be prepared by D- phenylalanine (D-Phe) and D- phenylalanine (D-Phe) sequentially solution phase peptide synthesis methods condensation.[Of 4]The present invention will next to examples will be described in further detail.Example
1 (1) Synthesis of Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3)
to the four-necked flask of 2L, α-Boc-Pic- OMe · HCl [α-Boc-4 – aminopiperidine-4-carboxylic acid methyl hydrochloride] were charged (2) 43.7g (148mmol), was suspended in EtOAc 656mL (15v / w). To the suspension of 1-hydroxybenzotriazole (HOBt) 27.2g (178mmol), while cooling with Cbz-D-Lys (Boc) -OH 59.2g (156mmol) was added an ice-bath 1-ethyl -3 – (3-dimethylcarbamoyl amino propyl) was added to the carbodiimide · HCl (EDC · HCl) 34.1g (178mmol). After 20 minutes, stirring was heated 12 hours at room temperature. After completion of the reaction, it was added and the organic layer was 1 N HCl 218 mL of (5.0v / w). NaHCO to the resulting organic layer 3 Aq. 218ML (5.0V / W), Et 3 N 33.0 g of (326Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 218ML 1N (5.0V / W), NaHCO 3 Aq. 218mL (5.0v / w), NaClaq . Was washed successively with 218ML (5.0V / W), Na 2 SO 4 dried addition of 8.74g (0.2w / w). Subjected to vacuum filtration, was concentrated under reduced pressure resulting filtrate by an evaporator, and pump up in the vacuum pump, the Cbz-D-Lys (Boc) -α-Boc-Pic-OMe (3) 88.9g as a white solid obtained (96.5% yield, HPLC purity 96.5%).[0033](2) D-Lys (Boc) Synthesis Of -Arufa-Boc-Pic-OMe (4)
In An Eggplant-Shaped Flask Of 2L, Cbz-D-Lys (Boc) -Arufa-Boc-Pic-OMe (3) 88.3g (142mmol) were charged, it was added and dissolved 441mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 17.7g (0.2w / w) was added, After three nitrogen substitution reduced pressure Atmosphere, Was Performed Three Times A Hydrogen Substituent. The Reaction Solution Was 18 Hours With Vigorous Stirring At Room Temperature To Remove The Pd / C And After The Completion Of The Reaction Vacuum Filtration. NaHCO The Resulting Filtrate 3 Aq. 441ML And (5.0V / W) Were Added For Liquid Separation, And The Organic Layer Was Extracted By The Addition Of EtOAc 200ML (2.3V / W) In The Aqueous Layer. NaHCO The Combined Organic Layer 3 Aq. 441ML And (5.0V / W) Were Added for liquid separation, and the organic layer was extracted addition of EtOAc 200mL (2.3v / w) in the aqueous layer. NaClaq the combined organic layers. 441mL and (5.0v / w) is added to liquid separation, was extracted by the addition EtOAc 200ML Of (2.3V / W) In The Aqueous Layer. The Combined Organic Layer On The Na 2 SO 4 Dried Addition Of 17.7 g of (0.2W / W), Then The Filtrate Was Concentrated Under Reduced Pressure Obtained Subjected To Vacuum Filtration By an evaporator, and pump up in the vacuum pump, D-Lys (Boc) -α-Boc-Pic- OMe (4) to give 62.7g (90.5% yield, HPLC purity 93.6%).(3) Cbz-D-Leu -D-Lys (Boc) -α-Boc-Pic-OMe synthesis of (5)
in the four-necked flask of 2L, D-Lys (Boc) -α-Boc-Pic-OMe (4) was charged 57.7 g (120 mmol), was suspended in EtOAc 576mL (10v / w). HOBt 19.3g (126mmol) to this suspension, was added EDC · HCl 24.2g (126mmol) while cooling in an ice bath added Cbz-D-Leu-OH 33.4g (126mmol). After 20 minutes, after stirring the temperature was raised 5 hours at room temperature, further the EDC · HCl and stirred 1.15 g (6.00 mmol) was added 16 h. After completion of the reaction, it was added liquid separation 1N HCl 576mL (10v / w) . NaHCO to the resulting organic layer 3 Aq. 576ML (10V / W), Et 3 N 24.3 g of (240Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 576ML 1N (10V / W), NaHCO 3 Aq. 576mL (10v / w), NaClaq . Was washed successively with 576ML (10V / W), Na 2 SO 4 dried addition of 11.5g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, the Cbz-D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe (5) 85.8g It was obtained as a white solid (98.7% yield, HPLC purity 96.9%).(4) D-Leu-D -Lys (Boc) -α-Boc-Pic-OMe synthesis of (6)
in an eggplant-shaped flask of 1L, Cbz-D-Leu- D-Lys (Boc) -α-Boc-Pic -OMe the (5) 91.9g (125mmol) were charged, was added and dissolved 459mL (5.0v / w) the EtOAc. The 5% Pd / C to the reaction solution 18.4g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 8 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 200mL (2.2v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 200mL (2.2v / w), NaClaq . It was sequentially added washed 200mL (2.2v / w). To the resulting organic layer Na 2 SO 4 dried added 18.4g (0.2w / w), to the filtrate concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and a pump-up with a vacuum pump. The resulting amorphous solid was dissolved adding EtOAc 200mL (2.2v / w), was crystallized by the addition of heptane 50mL (1.8v / w). Was filtered off precipitated crystals by vacuum filtration, the crystals were washed with a mixed solvent of EtOAc 120mL (1.3v / w), heptane 50mL (0.3v / w). The resulting crystal 46.1g to added to and dissolved EtOAc 480mL (5.2v / w), was crystallized added to the cyclohexane 660mL (7.2v / w). Was filtered off under reduced pressure filtered to precipitate crystals, cyclohexane 120mL (1.3v / w), and washed with a mixed solvent of EtOAc 20mL (0.2v / w), and 30 ° C. vacuum dried, D-Leu- as a white solid D-Lys (Boc) -α- Boc-Pic-OMe (6) to give 36.6 g (48.7% yield, HPLC purity 99.9%).(5) Synthesis of Cbz-D-Phe-D- Leu-D-Lys (Boc) -α-Boc-Pic-OMe (7)
to the four-necked flask of 1L, D-Leu-D- Lys (Boc) -α-Boc-Pic-OMe with (6) 35.8g (59.6mmol) was charged, it was suspended in EtOAc 358mL (10v / w). To this suspension HOBt 9.59g (62.6mmol), Cbz- D-Phe-OH 18.7g was cooled in an ice bath is added (62.6mmol) while EDC · HCl 12.0g (62.6mmol) It was added. After 20 minutes, a further EDC · HCl After stirring the temperature was raised 16 hours was added 3.09 g (16.1 mmol) to room temperature. After completion of the reaction, it was added and the organic layer was 1N HCl 358mL of (10v / w). NaHCO to the resulting organic layer 3 Aq. 358ML (10V / W), Et 3 N 12.1 g of (119Mmol) was stirred for 30 minutes, and the mixture was separated. The organic layer HCl 358ML 1N (10V / W), NaHCO 3 Aq. 358mL (10v / w), NaClaq . Was washed successively with 358ML (10V / W), Na 2 SO 4 dried addition of 7.16g (0.2w / w). After the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, and pump up in the vacuum pump, Cbz-D-Phe-D -Leu-D-Lys (Boc) -α-Boc-Pic-OMe (7) was obtained 52.5g as a white solid (yield quant, HPLC purity 97.6%).(6) D-Phe-D -Leu-D-Lys (Boc) synthesis of -α-Boc-Pic-OMe ( 8)
in an eggplant-shaped flask of 2L, Cbz-D-Phe- D-Leu-D-Lys ( Boc) -α-Boc-Pic- OMe (7) the 46.9g (53.3mmol) were charged, the 840ML EtOAc (18V / W), H 2 added to and dissolved O 93.8mL (2.0v / w) It was. The 5% Pd / C to the reaction mixture 9.38g (0.2w / w) was added, After three nitrogen substitution reduced pressure atmosphere, was performed three times a hydrogen substituent. The reaction solution was subjected to 10 hours with vigorous stirring at room temperature to remove the Pd / C and after the completion of the reaction vacuum filtration. NaHCO the resulting filtrate 3 Aq. 235mL (5.0v / w) were added to separate liquid, NaHCO to the organic layer 3 Aq. 235mL (5.0v / w), NaClaq . It was added sequentially cleaning 235mL (5.0v / w). To the resulting organic layer Na 2 SO 4 dried addition of 9.38g (0.2w / w), then the filtrate was concentrated under reduced pressure obtained subjected to vacuum filtration by an evaporator, pump up with a vacuum pump to D-Phe -D-Leu-D-Lys ( Boc) -α-Boc-Pic-OMe (7) was obtained 39.7g (yield quant, HPLC purity 97.3%).351mL was suspended in (10v / w). To this suspension HOBt 7.92g (51.7mmol), Boc-D-Phe-OH HCl HCl(8) D-Phe-D -Phe-D-Leu-D-Lys-Pic-OMe Synthesis Of Hydrochloric Acid Salt (1)
In An Eggplant-Shaped Flask Of 20ML Boc-D-Phe-D -Phe-D- Leu-D- lys (Boc) -α -Boc- Pic-OMe (9) and 2.00gg, IPA 3.3mL (1.65v / w), was suspended by addition of PhMe 10mL (5v / w). It was stirred at room temperature for 19 hours by addition of 6N HCl / IPA 6.7mL (3.35v / w). The precipitated solid was filtered off by vacuum filtration and dried under reduced pressure to a white solid of D-Phe-D-Phe- D- Leu-D-Lys-Pic- OMe 1.59ghydrochloride (1) (yield: 99 .0%, HPLC purity 98.2%) was obtained.(9) D-Phe-D -Phe-D-Leu-D-Lys-Pic-OMe Purification Of The Hydrochloric Acid Salt (1)
In An Eggplant-Shaped Flask Of 20ML-D-Phe-D- Phe D-Leu -D-Lys- pic-OMe hydrochloride crude crystals (1) were charged 200mg, EtOH: MeCN = 1: after stirring for 1 hour then heated in a mixed solvent 4.0 mL (20v / w) was added 40 ° C. of 5 , further at room temperature for 2 was time stirring slurry. Was filtered off by vacuum filtration, the resulting solid was dried under reduced pressure a white solid ((1) Purification crystals) was obtained 161 mg (80% yield, HPLC purity 99.2% ).(10) D-Phe-D -Phe-D-Leu-D-Lys-Pic Synthesis (Using Purified
(1)) Of (A) To A Round-Bottomed Flask Of 10ML D-Phe-D-Phe-D- -D-Lys Leu-Pic-OMe Hydrochloride Salt (1) Was Charged With Purified Crystal 38.5Mg (0.0488Mmol), H 2 Was Added And Dissolved O 0.2ML (5.2V / W). 1.5H Was Stirred Dropwise 1N NaOH 197MyuL (0.197mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 48.8μL (0.0488mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys- Pic (A) (yield: quant , HPLC purity 99.7%).
D-Phe-D-Phe- D-Leu-D-Lys-Pic-OMe (1) physical properties 1 H NMR (400 MHz, 1M DCl) [delta] ppm by: 0.85-1.02 (yd,. 6 H), 1.34-1.63 ( m, 5 H), 1.65-2.12 ( m, 5 H), 2.23-2.45 (m, 2 H), 2.96-3.12 (m, 4 H), 3.19 (ddt, J = 5.0 & 5.0 & 10.0 Hz), 3.33-3.62 (m, 1 H), 3.68-3.82 (m, 1 H), 3.82-3.95 (m, 4 H), 3.95-4.18 (m, 1 H), 4.25-4.37 (m, 2 H), 4.61-4.77 (M, 2 H), 7.21-7.44 (M, 10 H) 13 C NMR (400MHz, 1M DCl) Deruta Ppm: 21.8, 22.5, 24.8, 27.0, 30.5, 30.8, 31.0, 31.2, 31.7, 37.2 , 37.8, 38.4, 39.0, 39.8, 40.4, 40.6, 41.8, 42.3, 49.8, 50.2, 52.2, 52.6, 54.6, 55.2, 57.7, 57.9, 127.6, 128.4, 129.2, 129.6, 129.7, 129.8 dp 209.5 ℃Example 2
(Trifluoroacetic Acid (TFA)
Use) (1) D-Phe-D-Phe-D-Leu-D-Lys-Pic-OMe TFA Synthesis Of Salt (1)
TFA 18ML Eggplant Flask Of 50ML (18V / W) , 1- Dodecanethiol 1.6ML (1.6V / W), Triisopropylsilane 0.2ML (0.2V / W), H 2 Sequentially Added Stirring The O 0.2ML (0.2V / W) Did. The Solution To The Boc-D-Phe- D- Phe-D-Leu-D -Lys (Boc) -α-Boc-Pic-OMe the (9) 1.00g (1.01mmol) was added in small portions with a spatula. After completion of the reaction, concentrated under reduced pressure by an evaporator, it was added dropwise the resulting residue in IPE 20mL (20v / w). The precipitated solid was filtered off, the resulting solid was obtained and dried under reduced pressure to D-Phe-D-Phe- D-Leu -D-Lys-Pic-OMe · TFA salt as a white solid (1) (Osamu rate 93.0%, HPLC purity 95.2%).(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic synthesis of (A)
to a round-bottomed flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe TFA were charged salt (1) 83mg (0.0843mmol), was added and dissolved H2O 431μL (5.2v / w). Was 12h stirring dropwise 1N NaOH 345μL (0.345mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 84.3μL (0.0843mmol), to obtain a D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 95.4%).Example
3 (HCl / EtOAc
Use) (1) In An Eggplant-Shaped Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OMe (9) 1. It was charged with 00g (1.01mmol ), was added and dissolved EtOAc7.0mL (7.0v / w). 4N HCl / EtOAc 5.0mL (5.0v / w) was added after 24h stirring at room temperature, the precipitated solid was filtered off by vacuum filtration, washed with EtOAc 2mL (2.0v / w). The resulting solid D-Phe-D-Phe- D-Leu-D-Lys-Pic-OMe hydrochloride (1) was obtained 781mg of a white solid was dried under reduced pressure (the 96.7% yield, HPLC purity 95.4%).(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic (A) Synthesis of
eggplant flask of 10mL D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe hydrochloride were charged salt (1) 90 mg (0.112 mmol), H 2 was added and dissolved O 0.47mL (5.2v / w). Was 12h stirring dropwise 1N NaOH 459μL (0.459mmol) at room temperature. After completion of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.112μL (0.112mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) ( yield: quant, HPLC purity 93.1%).4 Example
Compound (1) Of The Compound By Hydrolysis Synthesis Of (The A) (Compound (1) Without
Purification) Eggplant Flask 10ML D-Phe-D-Phe -D-Leu-D-Lys-Pic-OMe (1) Charged Hydrochloride Were (Without Pre-Step Purification) 114.5Mg (0.142Mmol), H 2 Was Added And Dissolved O 595MyuL (5.2V / W). Was 14H Stirring Dropwise 1N NaOH 586MyuL (0.586Mmol) At Room Temperature. After Completion Of the reaction, concentrated under reduced pressure by an evaporator added 1N HCl 0.15μL (0.150mmol), was obtained D-Phe-D-Phe- D-Leu-D-Lys-Pic (A) (yield: quant, HPLC purity 95.2 %).Example 1 Comparative
Path Not Via The Compound (1) (Using Whole Guard Boc-D-Phe-D-Phe-D-Leu-D-Lys (Boc) -Alpha-Boc-Pic-OMe
(A)) (1) D–Boc Phe- D-Phe-D-Leu-D-Lys (Boc) -Arufa-Boc-Pic-OH Synthesis Of
Eggplant Flask Of 30ML Boc-D-Phe-D -Phe-D-Leu-D- Lys (Boc) -α- Boc-Pic -OMe (9) were charged 1.00g (1.00mmol), was added and dissolved MeOH 5.0mL (5.0v / w). After stirring for four days by the addition of 1N NaOH 1.1 mL (1.10mmol) at room temperature, further MeOH 5.0mL (5.0v / w), 1N NaOH 2.0mL the (2.0mmol) at 35 ℃ in addition 3h and the mixture was stirred. After completion of the reaction, 1 N HCl 6.1 mL was added, After distilling off the solvent was concentrated under reduced pressure was separated and the organic layer was added EtOAc 5.0mL (5.0mL) .NaClaq. 5.0mL (5.0v / w) Wash the organic layer was added, the organic layer as a white solid was concentrated under reduced pressure to Boc-D-Phe-D- Phe-D-Leu-D-Lys (Boc) – α-Boc-Pic-OH 975.1mg (99.3% yield, HPLC purity 80.8% )(2) D-Phe-D -Phe-D-Leu-D-Lys-Pic synthesis of (A)
to a round-bottomed flask of 20mL Boc-D-Phe-D -Phe-D-Leu-D-Lys (Boc) It was charged -α-Boc-Pic-OH ( 10) 959mg (0.978mmol), was added and dissolved EtOAc 4.9mL (5.0v / w). And 4h stirring at room temperature was added dropwise 4N HCl / EtOAc 4.9mL (5.0mL) at room temperature. After completion of the reaction, it was filtered under reduced pressure, a white solid as to give D-Phe-D-Phe- D-Leu-D-Lys-Pic the (A) (96.4% yield, HPLC purity 79.2%) . If not via the compound of the present invention (1), the purity of the compound obtained (A) was less than 80%.
PATENThttp://www.google.com/patents/US20110212882
References
- ^ Janecka A, Perlikowska R, Gach K, Wyrebska A, Fichna J (2010). “Development of opioid peptide analogs for pain relief”. Curr. Pharm. Des. 16 (9): 1126–35. doi:10.2174/138161210790963869. PMID 20030621.
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214916s000lbl.pdf
- ^ Jump up to:a b c d e f g h i j Raymond S. Sinatra; Jonathan S. Jahr; J. Michael Watkins-Pitchford (14 October 2010). The Essence of Analgesia and Analgesics. Cambridge University Press. pp. 490–491. ISBN 978-1-139-49198-3.
- ^ Jump up to:a b c d e Jeffrey Apfelbaum (8 September 2014). Ambulatory Anesthesia, An Issue of Anesthesiology Clinics. Elsevier Health Sciences. pp. 190–. ISBN 978-0-323-29934-3.
- ^ Jump up to:a b Alan Cowan; Gil Yosipovitch (10 April 2015). Pharmacology of Itch. Springer. pp. 307–. ISBN 978-3-662-44605-8.
- ^ Jump up to:a b c d Charlotte Allerton (2013). Pain Therapeutics: Current and Future Treatment Paradigms. Royal Society of Chemistry. pp. 56–. ISBN 978-1-84973-645-9.
- ^ “Korsuva: FDA-Approved Drugs”. U.S. Food and Drug Administration. Retrieved 24 August 2021.
- ^ “Vifor Pharma and Cara Therapeutics announce U.S. FDA approval of Korsuva injection for the treatment of moderate-to-severe pruritus in hemodialysis patients” (Press release). Vifor Pharma. 24 August 2021. Retrieved 24 August 2021 – via Business Wire.
- ^ Fishbane S, Jamal A, Munera C, Wen W, Menzaghi F (2020). “A phase 3 trial of difelikefalin in hemodialysis patients with pruritus”. N Engl J Med. 382 (3): 222–232. doi:10.1056/NEJMoa1912770. PMID 31702883.
External links
- “Difelikefalin”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03422653 for “A Study to Evaluate the Safety and Efficacy of CR845 in Hemodialysis Patients With Moderate-to-Severe Pruritus (KALM-1)” at ClinicalTrials.gov
- Clinical trial number NCT03636269 for “CR845-CLIN3103: A Global Study to Evaluate the Safety and Efficacy of CR845 in Hemodialysis Patients With Moderate-to-Severe Pruritus (KALM-2)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Korsuva |
| Other names | CR845, FE-202845, D-Phe-D-Phe-D-Leu-D-Lys-[γ-(4-N-piperidinyl)amino carboxylic acid][1] |
| License data | US DailyMed: Difelikefalin |
| Routes of administration | Intravenous |
| Drug class | Kappa opioid receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [2] |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV)[3] |
| Metabolism | Not metabolized[3] |
| Elimination half-life | 2 hours[3] |
| Excretion | Excreted as unchanged drug via bile and urine[3] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1024828-77-0 |
| PubChem CID | 24794466 |
| ChemSpider | 44208824 |
| UNII | NA1U919MRO |
| KEGG | D11111 |
| Chemical and physical data | |
| Formula | C36H53N7O6 |
| Molar mass | 679.863 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
//////////Difelikefalin acetate, FDA 2021, APPROVALS 2021, FORSUVA, ジフェリケファリン酢酸塩 , Difelikefalin, CR 845, MR 13A-9, MR-13A9, PEPTIDE

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}