
Home » fda 2021 (Page 2)
Category Archives: fda 2021
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lonapegsomatropin
FPTIPLSRLF DNAMLRAHRL HQLAFDTYQE FEEAYIPKEQ KYSFLQNPQT SLCFSESIPT
PSNREETQQK SNLELLRISL LLIQSWLEPV QFLRSVFANS LVYGASDSNV YDLLKDLEEG
IQTLMGRLED GSPRTGQIFK QTYSKFDTNS HNDDALLKNY GLLYCFRKDM DKVETFLRIV
QCRSVEGSCG F
(Disulfide bridge: 53-165, 182-189)

Lonapegsomatropin, ロナペグソマトロピン
FDA APPROVED, 25/8/21, Skytrofa, Treatment of growth hormone deficiency
To treat short stature due to inadequate secretion of endogenous growth hormone
1934255-39-6 CAS, UNII: OP35X9610Y
Molecular Formula, C1051-H1627-N269-O317-S9[-C2-H4-O]4n
ACP 001; ACP 011; lonapegsomatropin-tcgd; SKYTROFA; TransCon; TransCon growth hormone; TransCon hGH; TransCon PEG growth hormone; TransCon PEG hGH; TransCon PEG somatropin,
WHO 10598
PEPTIDE
Biologic License Application (BLA): 761177
Company: ACENDIS PHARMA ENDOCRINOLOGY DIV A/S
SKYTROFA is a human growth hormone indicated for the treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH) (1).
- OriginatorAscendis Pharma
- DeveloperAscendis Pharma; VISEN Pharmaceuticals
- ClassGrowth hormones; Hormonal replacements; Polyethylene glycols
- Mechanism of ActionSomatotropin receptor agonists
- Orphan Drug StatusYes – Somatotropin deficiency
- RegisteredSomatotropin deficiency
- 25 Aug 2021Registered for Somatotropin deficiency (In children, In infants) in USA (SC)
- 27 May 2021Ascendis Pharma expects European Commission decision on the Marketing Authorisation Application (MAA) for Somatotropin deficiency (In children, In infants, In neonates) in fourth quarter of 2021
- 27 May 2021Phase-III clinical trials in Somatotropin deficiency (In children, Treatment-naive) in Japan (SC)
Ascendis Pharma A/S Announces U.S. Food and Drug Administration Approval of SKYTROFA® (lonapegsomatropin-tcgd), the First Once-weekly Treatment for Pediatric Growth Hormone Deficiency
SKYTROFA, the first FDA approved treatment utilizing TransCon™ technology, is a long-acting prodrug of somatropin that releases the same somatropin used in daily therapies –
– Once weekly SKYTROFA demonstrated higher annualized height velocity (AHV) at week 52 compared to a daily growth hormone with similar safety and tolerability –
– Availability in the U.S. expected shortly supported by a full suite of patient support programs –
– Ascendis Pharma to host investor conference call today, Wednesday, August 25 at 4:30 p.m. E.T. –
COPENHAGEN, Denmark, Aug. 25, 2021 (GLOBE NEWSWIRE) — Ascendis Pharma A/S (Nasdaq: ASND), a biopharmaceutical company that utilizes its innovative TransCon technologies to potentially create new treatments that make a meaningful difference in patients’ lives, today announced that the U.S. Food and Drug Administration (FDA) has approved SKYTROFA (lonapegsomatropin-tcgd) for the treatment of pediatric patients one year and older who weigh at least 11.5 kg (25.4 lb) and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
As a once-weekly injection, SKYTROFA is the first FDA approved product that delivers somatropin (growth hormone) by sustained release over one week.
“Today’s approval represents an important new choice for children with GHD and their families, who will now have a once-weekly treatment option. In the pivotal head-to-head clinical trial, once-weekly SKYTROFA demonstrated higher annualized height velocity at week 52 compared to somatropini,” said Paul Thornton, M.B. B.Ch., MRCPI, a clinical investigator and pediatric endocrinologist in Fort Worth, Texas. “This once-weekly treatment could reduce treatment burden and potentially replace the daily somatropin therapies, which have been the standard of care for over 30 years.”
Growth hormone deficiency is a serious orphan disease characterized by short stature and metabolic complications. In GHD, the pituitary gland does not produce sufficient growth hormone, which is important not only for height but also for a child’s overall endocrine health and development.
The approval includes the new SKYTROFA® Auto-Injector and cartridges which, after first removed from a refrigerator, allow families to store the medicine at room temperature for up to six months. With a weekly injection, patients switching from injections every day can experience up to 86 percent fewer injection days per year.
“SKYTROFA is the first product using our innovative TransCon technology platform that we have developed from design phase through non-clinical and clinical development, manufacturing and device optimization, and out to the patients. It reflects our commitment and dedication to addressing unmet medical needs by developing a pipeline of highly differentiated proprietary products across multiple therapeutic areas,” said Jan Mikkelsen, Ascendis Pharma’s President and Chief Executive Officer. “We are grateful to the patients, caregivers, clinicians, clinical investigators, and our employees, who have all contributed to bringing this new treatment option to children in the U.S. with GHD.”
In connection with the commercialization of SKYTROFA, the company is committed to offering a full suite of patient support programs, including educating families on proper injection procedures for SKYTROFA as the first once-weekly treatment for children with GHD.
“It is wonderful that patients and their families now have the option of a once-weekly growth hormone therapy,” said Mary Andrews, Chief Executive Officer and co-founder of the MAGIC Foundation, a global leader in endocrine health, advocacy, education, and support. “GHD is often overlooked and undertreated in our children and managing it can be challenging for families. We are excited about this news as treating GHD is important, and children have a short time to grow.”
The FDA approval of SKYTROFA was based on results from the phase 3 heiGHt Trial, a 52-week, global, randomized, open-label, active-controlled, parallel-group trial that compared once-weekly SKYTROFA to daily somatropin (Genotropin®) in 161 treatment-naïve children with GHDii. The primary endpoint was, AHV at 52 weeks for weekly SKYTROFA and daily hGH treatment groups. Other endpoints included adverse events, injection-site reactions, incidence of anti-hGH antibodies, annualized height velocity, change in height SDS, proportion of subjects with IGF-1 SDS (0.0 to +2.0), PK/PD in subjects < 3 years, and preference for and satisfaction with SKYTROFA.
At week 52, the treatment difference in AHV was 0.9 cm/year (11.2 cm/year for SKYTROFA compared with 10.3 cm/year for daily somatropin) with a 95 percent confidence interval [0.2, 1.5] cm/year. The primary objective of non-inferiority in AHV was met for SKYTROFA in this trial and further demonstrated a higher AHV at week 52 for lonapegsomatropin compared to daily somatropin, with similar safety, in treatment-naïve children with GHD.
No serious adverse events or discontinuations related to SKYTROFA were reported. Most common adverse reactions (≥ 5%) in pediatric patients include: infection, viral (15%), pyrexia (15%), cough (11%), nausea and vomiting (11%), hemorrhage (7%), diarrhea (6%), abdominal pain (6%), and arthralgia and arthritis (6%)ii. In addition, both arms of the study reported low incidences of transient, non-neutralizing anti-hGH binding antibodies and no cases of persistent antibodies.
Conference Call and Webcast Information
| Date | Wednesday, August 25, 2021 |
| Time | 4:30 p.m. ET/1:30 p.m. Pacific Time |
| Dial In (U.S.) | 844-290-3904 |
| Dial In (International) | 574-990-1036 |
| Access Code | 8553236 |
A live webcast of the conference call will be available on the Investors and News section of the Ascendis Pharma website at www.ascendispharma.com. A webcast replay will be available on this website shortly after conclusion of the event for 30 days.
The Following Information is Intended for the U.S. Audience Only
INDICATION
SKYTROFA® is a human growth hormone indicated for the treatment of pediatric patients 1 year and older who weigh at least 11.5 kg and have growth failure due to inadequate secretion of endogenous growth hormone (GH).
IMPORTANT SAFETY INFORMATION
- SKYTROFA is contraindicated in patients with:
- Acute critical illness after open heart surgery, abdominal surgery or multiple accidental trauma, or if you have acute respiratory failure due to the risk of increased mortality with use of pharmacologic doses of somatropin.
- Hypersensitivity to somatropin or any of the excipients in SKYTROFA. Systemic hypersensitivity reactions have been reported with post-marketing use of somatropin products.
- Closed epiphyses for growth promotion.
- Active malignancy.
- Active proliferative or severe non-proliferative diabetic retinopathy.
- Prader-Willi syndrome who are severely obese, have a history of upper airway obstruction or sleep apnea or have severe respiratory impairment due to the risk of sudden death.
- Increased mortality in patients with acute critical illness due to complications following open heart surgery, abdominal surgery or multiple accidental trauma, or those with acute respiratory failure has been reported after treatment with pharmacologic doses of somatropin. Safety of continuing SKYTROFA treatment in patients receiving replacement doses for the approved indication who concurrently develop these illnesses has not been established.
- Serious systemic hypersensitivity reactions including anaphylactic reactions and angioedema have been reported with post-marketing use of somatropin products. Do not use SKYTROFA in patients with known hypersensitivity to somatropin or any of the excipients in SKYTROFA.
- There is an increased risk of malignancy progression with somatropin treatment in patients with active malignancy. Preexisting malignancy should be inactive with treatment completed prior to starting SKYTROFA. Discontinue SKYTROFA if there is evidence of recurrent activity.
- In childhood cancer survivors who were treated with radiation to the brain/head for their first neoplasm and who developed subsequent growth hormone deficiency (GHD) and were treated with somatropin, an increased risk of a second neoplasm has been reported. Intracranial tumors, in particular meningiomas, were the most common of these second neoplasms. Monitor all patients with a history of GHD secondary to an intracranial neoplasm routinely while on somatropin therapy for progression or recurrence of the tumor.
- Because children with certain rare genetic causes of short stature have an increased risk of developing malignancies, practitioners should thoroughly consider the risks and benefits of starting somatropin in these patients. If treatment with somatropin is initiated, carefully monitor these patients for development of neoplasms. Monitor patients on somatropin therapy carefully for increased growth, or potential malignant changes of preexisting nevi. Advise patients/caregivers to report marked changes in behavior, onset of headaches, vision disturbances and/or changes in skin pigmentation or changes in the appearance of preexisting nevi.
- Treatment with somatropin may decrease insulin sensitivity, particularly at higher doses. New onset type 2 diabetes mellitus has been reported in patients taking somatropin. Undiagnosed impaired glucose tolerance and overt diabetes mellitus may be unmasked. Monitor glucose levels periodically in all patients receiving SKYTROFA. Adjust the doses of antihyperglycemic drugs as needed when SKYTROFA is initiated in patients.
- Intracranial hypertension (IH) with papilledema, visual changes, headache, nausea, and/or vomiting has been reported in a small number of patients treated with somatropin. Symptoms usually occurred within the first 8 weeks after the initiation of somatropin and resolved rapidly after cessation or reduction in dose in all reported cases. Fundoscopic exam should be performed before initiation of therapy and periodically thereafter. If somatropin-induced IH is diagnosed, restart treatment with SKYTROFA at a lower dose after IH-associated signs and symptoms have resolved.
- Fluid retention during somatropin therapy may occur and is usually transient and dose dependent.
- Patients receiving somatropin therapy who have or are at risk for pituitary hormone deficiency(s) may be at risk for reduced serum cortisol levels and/or unmasking of central (secondary) hypoadrenalism. Patients treated with glucocorticoid replacement for previously diagnosed hypoadrenalism may require an increase in their maintenance or stress doses following initiation of SKYTROFA therapy. Monitor patients for reduced serum cortisol levels and/or need for glucocorticoid dose increases in those with known hypoadrenalism.
- Undiagnosed or untreated hypothyroidism may prevent response to SKYTROFA. In patients with GHD, central (secondary) hypothyroidism may first become evident or worsen during SKYTROFA treatment. Perform thyroid function tests periodically and consider thyroid hormone replacement.
- Slipped capital femoral epiphysis may occur more frequently in patients undergoing rapid growth. Evaluate pediatric patients with the onset of a limp or complaints of persistent hip or knee pain.
- Somatropin increases the growth rate and progression of existing scoliosis can occur in patients who experience rapid growth. Somatropin has not been shown to increase the occurrence of scoliosis. Monitor patients with a history of scoliosis for disease progression.
- Cases of pancreatitis have been reported in pediatric patients receiving somatropin. The risk may be greater in pediatric patients compared with adults. Consider pancreatitis in patients who develop persistent severe abdominal pain.
- When SKYTROFA is administered subcutaneously at the same site over a long period of time, lipoatrophy may result. Rotate injection sites when administering SKYTROFA to reduce this risk.
- There have been reports of fatalities after initiating therapy with somatropin in pediatric patients with Prader-Willi syndrome who had one or more of the following risk factors: severe obesity, history of upper airway obstruction or sleep apnea, or unidentified respiratory infection. Male patients with one or more of these factors may be at greater risk than females. SKYTROFA is not indicated for the treatment of pediatric patients who have growth failure due to genetically confirmed Prader-Willi syndrome.
- Serum levels of inorganic phosphorus, alkaline phosphatase, and parathyroid hormone may increase after somatropin treatment.
- The most common adverse reactions (≥5%) in patients treated with SKYTROFA were: viral infection (15%), pyrexia (15%), cough (11%), nausea and vomiting (11%), hemorrhage (7%), diarrhea (6%), abdominal pain (6%), and arthralgia and arthritis (6%).
- SKYTROFA can interact with the following drugs:
- Glucocorticoids: SKYTROFA may reduce serum cortisol concentrations which may require an increase in the dose of glucocorticoids.
- Oral Estrogen: Oral estrogens may reduce the response to SKYTROFA. Higher doses of SKYTROFA may be required.
- Insulin and/or Other Hypoglycemic Agents: SKYTROFA may decrease insulin sensitivity. Patients with diabetes mellitus may require adjustment of insulin or hypoglycemic agents.
- Cytochrome P450-Metabolized Drugs: Somatropin may increase cytochrome P450 (CYP450)-mediated antipyrine clearance. Carefully monitor patients using drugs metabolized by CYP450 liver enzymes in combination with SKYTROFA.
You are encouraged to report side effects to FDA at (800) FDA-1088 or www.fda.gov/medwatch. You may also report side effects to Ascendis Pharma at 1-844-442-7236.
Please click here for full Prescribing Information for SKYTROFA.
About SKYTROFA® (lonapegsomatropin-tcgd)
SKYTROFA® is a once-weekly prodrug designed to deliver somatropin over a one-week period. The released somatropin has the same 191 amino acid sequence as daily somatropin.
SKYTROFA single-use, prefilled cartridges are available in nine dosage strengths, allowing for convenient dosing flexibility. They are designed for use only with the SKYTROFA® Auto-Injector and may be stored at room temperature for up to six months. The recommended dose of SKYTROFA for treatment-naïve patients and patients switching from daily somatropin is 0.24 mg/kg body weight, administered once weekly. The dose may be adjusted based on the child’s weight and insulin-like growth factor-1 (IGF-1) SDS.
SKYTROFA has been studied in over 300 children with GHD across the Phase 3 program which consists of the heiGHt Trial (for treatment-naïve patients), the fliGHt Trial (for treatment-experienced patients), and the enliGHten Trial (an ongoing long-term extension trial). Patients who completed the heiGHt Trial or the fliGHt Trial were able to continue into the enliGHten Trial and some have been on SKYTROFA for over four years.
SKYTROFA is being evaluated for pediatric GHD in Phase 3 trials in Japan and Greater China, including the People’s Republic of China, Hong Kong, Macau and Taiwan. Ascendis Pharma is also conducting the global Phase 3 foresiGHt Trial in adults with GHD. SKYTROFA has been granted orphan designation for GHD in both the U.S. and Europe.
About TransCon™ Technologies
TransCon refers to “transient conjugation.” The proprietary TransCon platform is an innovative technology to create new therapies that are designed to potentially optimize therapeutic effect, including efficacy, safety and dosing frequency. TransCon molecules have three components: an unmodified parent drug, an inert carrier that protects it, and a linker that temporarily binds the two. When bound, the carrier inactivates and shields the parent drug from clearance. When injected into the body, physiologic conditions (e.g., pH and temperature) initiate the release of the active, unmodified parent drug in a predictable manner. Because the parent drug is unmodified, its original mode of action is expected to be maintained. TransCon technology can be applied broadly to a protein, peptide or small molecule in multiple therapeutic areas, and can be used systemically or locally.
About Ascendis Pharma A/S
Ascendis Pharma is applying its innovative platform technology to build a leading, fully integrated biopharma company focused on making a meaningful difference in patients’ lives. Guided by its core values of patients, science and passion, the company utilizes its TransCon technologies to create new and potentially best-in-class therapies.
Ascendis Pharma currently has a pipeline of multiple independent endocrinology rare disease and oncology product candidates in development. The company continues to expand into additional therapeutic areas to address unmet patient needs.
Ascendis is headquartered in Copenhagen, Denmark, with additional facilities in Heidelberg and Berlin, Germany, in Palo Alto and Redwood City, California, and in Princeton, New Jersey.
Please visit www.ascendispharma.com (for global information) or www.ascendispharma.us (for U.S. information).

NEW DRUG APPROVALS
ONE TIME
$10.00
///////////Lonapegsomatropin, Skytrofa, APPROVALS 2021, FDA 2021, PEPTIDE, ロナペグソマトロピン , ACP 00, ACP 011, lonapegsomatropin-tcgd, TransCon, TransCon growth hormone, TransCon hGH, TransCon PEG growth hormone, TransCon PEG hGH, TransCon PEG somatropin, ORPHAN DRUG
Belzutifan
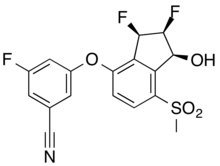
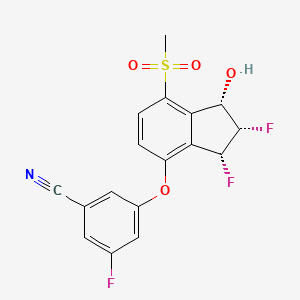
Belzutifan
CAS 1672668-24-4
383.34 g·mol−1 C17H12F3NO4S
3-[[(1S,2S,3R)-2,3-difluoro-1-hydroxy-7-methylsulfonyl-2,3-dihydro-1H-inden-4-yl]oxy]-5-fluorobenzonitrile
MK-6482, PT-2977, UNII-7K28NB895L, 7K28NB895L
3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile
GTPL11251, PT 2977 [WHO-DD], BDBM373040
FDA APPROVED 8/13/2021, Welireg
To treat von Hippel-Lindau disease under certain conditions
EMA Drug Information
| Disease/Condition | Treatment of von Hippel-Lindau disease |
|---|---|
| Active Substance | 3-(((1S,2S,3R)-2,3-difluoro-1-hydroxy-7-(methylsulfonyl)-2,3-dihydro-1H-inden-4-yl)oxy)-5-fluorobenzonitrile |
| Status of Orphan Designation | Positive |
| Decision Date | 2020-08-21 |
FDA approves belzutifan for cancers associated with von Hippel-Lindau disease
On August 13, 2021, the Food and Drug Administration approved belzutifan (Welireg, Merck), a hypoxia-inducible factor inhibitor for adult patients with von Hippel-Lindau (VHL) disease who require therapy for associated renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, or pancreatic neuroendocrine tumors (pNET), not requiring immediate surgery.
Belzutifan was investigated in the ongoing Study 004 (NCT03401788), an open-label clinical trial in 61 patients with VHL-associated RCC (VHL-RCC) diagnosed based on a VHL germline alteration and with at least one measurable solid tumor localized to the kidney. Enrolled patients had other VHL-associated tumors, including CNS hemangioblastomas and pNET. Patients received belzutifan 120 mg once daily until disease progression or unacceptable toxicity.
The primary efficacy endpoint was overall response rate (ORR) measured by radiology assessment, as assessed by an independent review committee using RECIST v1.1. Additional efficacy endpoints included duration of response (DoR), and time- to- response (TTR). An ORR of 49% (95% CI:36, 62) was reported in patients with VHL-associated RCC. All patients with VHL-RCC with a response were followed for a minimum of 18 months from the start of treatment. The median DoR was not reached; 56% of responders had DoR ≥ 12 months and a median TTR of 8 months. In patients with other VHL-associated non-RCC tumors, 24 patients with measurable CNS hemangioblastomas had an ORR of 63% and 12 patients with measurable pNET had an ORR of 83%. Median DoR was not reached, with 73% and 50% of patients having response durations ≥ 12 months for CNS hemangioblastomas and pNET, respectively.
The most common adverse reactions, including laboratory abnormalities, reported in ≥ 20% of patients who received belzutifan were decreased hemoglobin, anemia, fatigue, increased creatinine, headache, dizziness, increased glucose, and nausea. Anemia and hypoxia from belzutifan use can be severe. In Study 004, anemia occurred in 90% of patients and 7% had Grade 3 anemia. Patients should be transfused as clinically indicated. The use of erythropoiesis stimulating agents for treatment of anemia is not recommended in patients treated with belzutifan. In Study 004, hypoxia occurred in 1.6% of patients. Belzutifan can render some hormonal contraceptives ineffective, and belzutifan exposure during pregnancy can cause embryo-fetal harm.
The recommended belzutifan dosage is 120 mg administered orally once daily with or without food.
View full prescribing information for Welireg.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Australian Therapeutic Goods Administration (TGA), Health Canada, and the Medicines and Healthcare products Regulatory Agency (MHRA) of the United Kingdom. The application reviews are ongoing at the other regulatory agencies.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, as well as the Assessment Aid and the Product Quality Assessment Aid, voluntary submissions from the applicant to facilitate the FDA’s assessment. The FDA approved this application approximately 1 month ahead of the FDA goal date.
This application was granted priority review for this indication. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Belzutifan, sold under the brand name Welireg, is a medication used for the treatment of von Hippel–Lindau disease-associated renal cell carcinoma.[1][2][3][4][5][6] It is taken by mouth.[1]
The most common side effects include decreased hemoglobin, anemia, fatigue, increased creatinine, headache, dizziness, increased glucose, and nausea.[2]
Belzutifan is an hypoxia-inducible factor-2 alpha (HIF-2α) inhibitor.[1][2][7]
Belzutifan is the first drug to be awarded an “innovation passport” from the UK Medicines and Healthcare products Regulatory Agency (MHRA).[8][4] Belzutifan was approved for medical use in the United States in August 2021.[2][9] Belzutifan is the first hypoxia-inducible factor-2 alpha inhibitor therapy approved in the U.S.[9]
Medical uses
Belzutifan is indicated for treatment of adults with von Hippel-Lindau (VHL) disease who require therapy for associated renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, or pancreatic neuroendocrine tumors (pNET), not requiring immediate surgery.[2]
PATENT
WO 2019191227
https://patents.google.com/patent/WO2019191227A1/en

PATENT
WO 2015035223
https://patents.google.com/patent/WO2015035223A1/enScheme 9


[01237] 3-r(15,25.3 ?‘)-2.3-difluoro-l-hvdroxy-7-methylsulfonyl-indan-4- νΠοχν-5-fluoro-benzonitrile (Compound 289)[01238] Step A: r(15.2/?V4- -cvano-5-fluoro-phenoxy)-2-fluoro-7- methylsulfonyl-indan-l -vH acetate: To a stirred solution of 3-fluoro-5-[(15,27?)-2-fluoro-l – hydroxy-7-methylsulfonyl-indan-4-yl]oxy-benzonitrile (2.00 g, 5.47 mmol) in DCM (27 mL) was added 4-(dimethylamino)pyridine (0.2 g, 1.64 mmol) and triethylamine (1.53 mL, 10.9 mmol). Acetic anhydride (1.00 mL, 10.9 mmol) was added dropwise at 0 °C under nitrogen. The reaction mixture was stirred at ambient temperature overnight. The reaction mixture was diluted with DCM, washed with saturated aqueous NaHC03 and brine, dried andconcentrated. The residue was purified by flash chromatography on silica gel (20-40% EtOAc/hexane) to give [(lS,2/?)-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl- indan-l-yl] acetate (1.95 g, 87%). LCMS ESI (+) m/z 408 (M+H).[01239] Step B: Γ( 1 .25.35)-3-bromo-4-(3-cvano-5-fluoro-Dhenoxy)-2-fluoro- 7-methylsulfonyl-indan-l-yll acetate and f(15.25,3/?)-3-bromo-4-(3-cyano-5-fluoro- phenoxy)-2-fluoro-7-methylsulfonyl-indan-l -yl1 acetate: To a stirred solution of [(15,2/?)-4- (3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl-indan-] -yl] acetate (1.95 g, 4.79 mmol) in 1 ,2-dichloroethane (24 mL) was added N-bromosuccinimide (0.94 g, 5.27 mmol) and 2,2′-azobisisobutyronitrile (8 mg, 0.05 mmol). The reaction mixture was heated at 80 °C for 3 hours. After cooling, the reaction mixture was diluted with DCM, washed with saturated aqueous NaHC03 and brine, dried and concentrated. The residue was purified by column chromatography on silica gel (20-30% EtOAc hexane) to give [(lS,2S,3S)-3-bromo- 4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl-indan-l-yl] acetate (1 .52 g, 65%). LCMS ESI (+) m/z 486, 488 (M+H). Further elution with 30-50% EtOAc/hexane gave the more polar product [(lS,2S,3/?)-3-bromo-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7- methylsulfonyl-indan-l -yl] acetate (0.583 g, 25%). LCMS ESI (+) m/z 486, 488 (M+H). [01240] Step C: rd5.2^.3 V4-(3-cvano-5-fluoro-phenoxy)-2-fluoro-3- hvdroxy-7-methylsulfonyl-indan- 1 -yll acetate: To a combined mixture of [(1 ,25,35)-3- bromo-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl-indan-l -yl] acetate and [( 15,2S,3/?)-3-bromo-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-7-methylsulfonyl-indan- 1 -yl] acetate prepared in Step B (2.05 g, 4.22 mmol) were added 1 ,2-dimethoxyethane (28 mL) and water (0.050 mL) followed by silver perchlorate hydrate (1.42 g, 6.32 mmol). The reaction mixture was heated at 70 °C for 2 hours. After cooling, the reaction mixture was diluted with EtOAc and filtered through Celite. The filtrate was washed with water and brine, dried and concentrated. The residue was purified by flash chromatography on silica gel (20-50%) to give [(15,2/?,35)-4-(3-cyano-5-fluoro-phenoxy)-2-fluoro-3-hydroxy-7-methylsulfonyl-indan- 1 -yl] acetate (0.416 g, 23%) as the less polar product. LCMS ESI (+) m/z 441 (M+NH4+). Further elution with 60% EtOAc/hexane gave [(15,2/?,3R)-4-(3-cyano-5-fluoro-phenoxy)-2- fluoro-3-hydroxy-7-methylsulfonyl-indan-l-yl] acetate (0.58 g, 32 %). LCMS ESI (+) m/z 441 (M+NH4+).[01241] Step D: r(15.25.3/? -4-(3-cvano-5-fluoro-phenoxyV2.3-difluoro-7- methylsulfonyl-indan-l-vH acetate: To a stirred solution of [(15,2/?,35)-4-(3-cyano-5-fluoro- phenoxy)-2-fluoro-3-hydroxy-7-methylsulfonyl-indan-l-yl] acetate (416 mg, 0.98 mmol) in DCM (10 mL) was added (diethylamino)sulfur trifluoride (DAST) (0.26 mL, 2.0 mmol) at – 78 °C under nitrogen. The reaction mixture was allowed to warm to 0 °C and stirred for 15 minutes. The reaction was quenched by saturated aqueous NaHC03. The mixture was partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried and concentrated. The residue was purified by flash chromatography on silica gel (20-40% EtOAc/hexane) to give [(15,25,3/?)- 4-(3-cyano-5-fluoro-phenoxy)-2,3-difluoro-7-methylsulfonyl-indan-l -yl] acetate (310 mg, 74%). LCMS ESI (+) m/z 426 (M+H).[01242] Step E: 3-r(15.25.3^)-2.3-difluoro-l-hvdroxy-7-methylsulfonyl-indan-4-vnoxy-5-fluoro-benzonitrile (Compound 289): Prepared as described in Example 288 Step F substituting [(l ?)-4-(3-cyano-5-fluoro-phenoxy)-3,3-difluoro-7-methylsulfonyl-indan- 1-yl] acetate with [(15,25,3/?)-4-(3-cyano-5-fluoro-phenoxy)-2,3-difluoro-7-methylsulfonyl- indan-l-yl] acetate. LCMS ESI (+) m/z 384 (M+H); Ή NMR (400 MHz, CDC13): δ 8.13 (d, 1H), 7.31-7.25 (m, 1 H), 7.23-7.19 (m, 1 H), 7.14-7.09 (m, 1H), 7.04 (d, 1H), 6.09-5.91 (m, 1 H), 5.87-5.80 (m, 1 H), 5.25-5.05 (m, 1H), 3.32 (s, 3H), 2.95 (d, 1H).
PatentWO 2016145032https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016145032&tab=PCTDESCRIPTIONCOMPD 289
PATENTWO 2016145045WO 2016168510WO 2016057242WO 2019191227
| PMID | Publication Date | Title | Journal |
|---|---|---|---|
| 31282155 | 2019-08-08 | 3-[(1S,2S,3R)-2,3-Difluoro-1-hydroxy-7-methylsulfonylindan-4-yl]oxy-5-fluorobenzonitrile (PT2977), a Hypoxia-Inducible Factor 2α (HIF-2α) Inhibitor for the Treatment of Clear Cell Renal Cell Carcinoma | Journal of medicinal chemistry |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2020146758-A1 | Methods to treat mitochondrial-associated dysfunctions or diseases | 2019-01-10 | |
| WO-2020092100-A1 | Solid dispersions and pharmaceutical compositions comprising a substituted indane and methods for the preparation and use thereof | 2018-10-30 | |
| TW-202003430-A | Methods of reducing inflammation of the digestive system with inhibitors of HIF-2-alpha | 2018-03-28 | |
| WO-2019191227-A1 | Methods of reducing inflammation of the digestive system with inhibitors of hif-2-alpha | 2018-03-28 | |
| US-2019151347-A1 | Compositions and methods of modulating hif-2a; to improve muscle generation and repair | 2017-11-20 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2019048421-A1 | Biomarkers of response to hif-2-alpha inhibition in cancer and methods for the use thereof | 2015-09-21 | |
| WO-2017053192-A1 | Biomarkers of response to hif-2-alpha inhibition in cancer and methods for the use thereof | 2015-09-21 | |
| US-10335388-B2 | Combination therapy of a HIF-2-alpha inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 | 2019-07-02 |
| US-2018140569-A1 | Combination therapy of a hif-2-alpha inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 | |
| US-2019282535-A1 | Combination therapy of a hif-2-alpha inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| WO-2016168510-A1 | Combination therapy of a hif-2-alpha inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 | |
| US-10786480-B2 | Combination therapy of a HIF-2-α inhibitor and an immunotherapeutic agent and uses thereof | 2015-04-17 | 2020-09-29 |
| US-10278942-B2 | Compositions for use in treating pulmonary arterial hypertension | 2015-03-11 | 2019-05-07 |
| US-10512626-B2 | Compositions for use in treating glioblastoma | 2015-03-11 | 2019-12-24 |
| US-2018042884-A1 | Compositions for use in treating glioblastoma | 2015-03-11 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2018177754-A1 | Compositions for use in treating pulmonary arterial hypertension | 2015-03-11 | |
| US-2019015377-A1 | Compositions for Use in Treating Pulmonary Arterial Hypertension | 2015-03-11 | |
| WO-2016145032-A1 | Compositions for use in treating pulmonary arterial hypertension | 2015-03-11 | |
| WO-2016145045-A1 | Compositions for use in treating glioblastoma | 2015-03-11 | |
| US-10098878-B2 | HIF-2α inhibitors for treating iron overload disorders | 2014-10-10 | 2018-10-16 |
| Publication Number | Title | Priority Date | Grant Date |
|---|---|---|---|
| US-2020190031-A1 | Aryl ethers and uses thereof | 2013-09-09 | |
| US-9896418-B2 | Aryl ethers and uses thereof | 2013-09-09 | 2018-02-20 |
| US-9908845-B2 | Aryl ethers and uses thereof | 2013-09-09 | 2018-03-06 |
| US-9969689-B2 | Aryl ethers and uses thereof | 2013-09-09 | 2018-05-15 |
| WO-2015035223-A1 | Aryl ethers and uses thereof | 2013-09-09 |
Merck Team Wins 2021 Pete Dunn Award
05-17-2021 10:52 AM

The ACS Green Chemistry Institute (GCI) Pharmaceutical Roundtable honors the work of Stephen Dalby, François Lévesque, Cecilia Bottecchia and Jonathan McMullen at Merck with the 2021 Peter J. Dunn Award for Green Chemistry & Engineering Impact in the Pharmaceutical Industry. The team’s innovation is titled, “Greener Manufacturing of Belzutifan (MK-6482) Featuring a Photo-Flow Bromination.”
Belzutifan is an important new drug used in the treatment of cancer and other non-oncology diseases. Acquired by Merck in 2019 through the purchase of Peloton Therapeutics, a new, greener manufacturing process for its synthesis was needed. Over the next 18 months, the team developed a more direct route from commodity chemical to API, employed new reaction conditions, particularly in the oxidation sequence, and incorporated new technology, photo-flow.
Despite this accelerated timeline, the team achieved a five-fold improvement in overall yield with a commensurate 73% reduction in process mass intensity (PMI) compared to the original route. Notably, the Merck team also developed a visible light-initiated radical bromination performed in flow. According to the L.-C. Campeau, Executive Director and Head of Process Chemistry and Discovery Process Chemistry at Merck, this is the “first example of a photo-flow reaction run on manufacturing scale at Merck and represents the linchpin of the synthesis.”
The improved process for Belzutifan, which is expected to launch this year, will reduce the waste associated with its manufacture and is aligned with Merck’s corporate sustainability goals.
“The Merck team delivered an excellent example of the application of innovative technologies to develop a more sustainable synthesis of the pharmaceutically-active compound, Belzutifan,” comments Paul Richardson, Director of Oncology and Chemical Synthesis at Pfizer and Co-Chair of the ACS GCI Pharmaceutical Roundtable. “Using the guiding principles of green chemistry, for example, in the use of catalysis and a relatively benign reaction media, further illustrate the Merck team’s work as worthy of recognition for the 2021 Peter Dunn Award.”
The award will be presented at the June 11 GC&E Friday, part of the 25th Annual Green Chemistry & Engineering Conference. During this session from 10 a.m. – 1 p.m., Stephen Dalby & Jon MacMullen will be discussing the details of this innovative process.
References
- ^ Jump up to:a b c d https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215383s000lbl.pdf
- ^ Jump up to:a b c d e f “FDA approves belzutifan for cancers associated with von Hippel-Lindau”. U.S. Food and Drug Administration (FDA). 13 August 2021. Retrieved 13 August 2021.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Belzutifan”. SPS – Specialist Pharmacy Service. 18 March 2021. Retrieved 25 April 2021.
- ^ Jump up to:a b “MHRA awards first ‘innovation passport’ under new pathway”. RAPS (Press release). Retrieved 25 April 2021.
- ^ “Merck Receives Priority Review From FDA for New Drug Application for HIF-2α Inhibitor Belzutifan (MK-6482)” (Press release). Merck. 16 March 2016. Retrieved 25 April 2021 – via Business Wire.
- ^ “FDA Grants Priority Review to Belzutifan for von Hippel-Lindau Disease–Associated RCC”. Cancer Network. Retrieved 26 April 2021.
- ^ {{cite journal |vauthors=Choueiri TK, Bauer TM, Papadopoulos KP, Plimack ER, Merchan JR, McDermott DF, Michaelson MD, Appleman LJ, Thamake S, Perini RF, Zojwalla NJ, Jonasch E | display-authors=6 |title=Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis |journal=Nat Med |volume= |issue= |pages= |date=April 2021 |pmid=33888901 |doi=10.1038/s41591-021-01324-7 }
- ^ “First Innovation Passport awarded to help support development and access to cutting-edge medicines”. Medicines and Healthcare products Regulatory Agency (MHRA) (Press release). 26 February 2021. Retrieved 14 August 2021.
- ^ Jump up to:a b “FDA Approves Merck’s Hypoxia-Inducible Factor-2 Alpha (HIF-2α) Inhibitor Welireg (belzutifan) for the Treatment of Patients With Certain Types of Von Hippel-Lindau (VHL) Disease-Associated Tumors” (Press release). Merck. 13 August 2021. Retrieved 13 August 2021 – via Business Wire.
External links
- “Belzutifan”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT04195750 for “A Study of Belzutifan (MK-6482) Versus Everolimus in Participants With Advanced Renal Cell Carcinoma (MK-6482-005)” at ClinicalTrials.gov
- Clinical trial number NCT03401788 for “A Phase 2 Study of Belzutifan (PT2977, MK-6482) for the Treatment of Von Hippel Lindau (VHL) Disease-Associated Renal Cell Carcinoma (RCC) (MK-6482-004)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Pronunciation | bell-ZOO-ti-fan |
| Trade names | Welireg |
| Other names | MK-6482, PT2977 |
| License data | US DailyMed: Belzutifan |
| Routes of administration | By mouth |
| Drug class | Antineoplastic |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1672668-24-4 [KEGG] |
| PubChem CID | 117947097 |
| ChemSpider | 59053536 |
| UNII | 7K28NB895L |
| KEGG | D11954 |
| ChEMBL | ChEMBL4585668 |
| PDB ligand | 72Q (PDBe, RCSB PDB) |
| Chemical and physical data | |
| Formula | C17H12F3NO4S |
| Molar mass | 383.34 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////Belzutifan, Welireg, FDA 2021, APPROVALS 2021, MK 6482, PT 977, Antineoplastic
CS(=O)(=O)C1=C2C(C(C(C2=C(C=C1)OC3=CC(=CC(=C3)C#N)F)F)F)O
NEW DRUG APPROVALS
ONE TIME
$10.00
Avalglucosidase alfa
QQGASRPGPR DAQAHPGRPR AVPTQCDVPP NSRFDCAPDK AITQEQCEAR GCCYIPAKQG
LQGAQMGQPW CFFPPSYPSY KLENLSSSEM GYTATLTRTT PTFFPKDILT LRLDVMMETE
NRLHFTIKDP ANRRYEVPLE TPRVHSRAPS PLYSVEFSEE PFGVIVHRQL DGRVLLNTTV
APLFFADQFL QLSTSLPSQY ITGLAEHLSP LMLSTSWTRI TLWNRDLAPT PGANLYGSHP
FYLALEDGGS AHGVFLLNSN AMDVVLQPSP ALSWRSTGGI LDVYIFLGPE PKSVVQQYLD
VVGYPFMPPY WGLGFHLCRW GYSSTAITRQ VVENMTRAHF PLDVQWNDLD YMDSRRDFTF
NKDGFRDFPA MVQELHQGGR RYMMIVDPAI SSSGPAGSYR PYDEGLRRGV FITNETGQPL
IGKVWPGSTA FPDFTNPTAL AWWEDMVAEF HDQVPFDGMW IDMNEPSNFI RGSEDGCPNN
ELENPPYVPG VVGGTLQAAT ICASSHQFLS THYNLHNLYG LTEAIASHRA LVKARGTRPF
VISRSTFAGH GRYAGHWTGD VWSSWEQLAS SVPEILQFNL LGVPLVGADV CGFLGNTSEE
LCVRWTQLGA FYPFMRNHNS LLSLPQEPYS FSEPAQQAMR KALTLRYALL PHLYTLFHQA
HVAGETVARP LFLEFPKDSS TWTVDHQLLW GEALLITPVL QAGKAEVTGY FPLGTWYDLQ
TVPIEALGSL PPPPAAPREP AIHSEGQWVT LPAPLDTINV HLRAGYIIPL QGPGLTTTES
RQQPMALAVA LTKGGEARGE LFWDDGESLE VLERGAYTQV IFLARNNTIV NELVRVTSEG
AGLQLQKVTV LGVATAPQQV LSNGVPVSNF TYSPDTKVLD ICVSLLMGEQ FLVSWC
(Disulfide bridge:26-53, 36-52, 47-71, 477-502, 591-602, 882-896)
Avalglucosidase alfa
アバルグルコシダーゼアルファ (遺伝子組換え)
Avalglucosidase alfa (USAN/INN);
Avalglucosidase alfa (genetical recombination) (JAN);
Avalglucosidase alfa-ngpt
To treat late-onset Pompe disease
| Formula | C4490H6818N1197O1299S32 |
|---|---|
| CAS | 1802558-87-7 |
| Mol weight | 99375.4984 |
FDA APPROVED Nexviazyme, 2021/8/6, Enzyme replacement therapy product
Treatment of Pompe disease
Biologic License Application (BLA): 761194
Company: GENZYME CORP
https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-pompe-diseaseFor Immediate Release:August 06, 2021
Today, the U.S. Food and Drug Administration approved Nexviazyme (avalglucosidase alfa-ngpt) for intravenous infusion to treat patients 1 year of age and older with late-onset Pompe disease.
Patients with Pompe disease have an enzyme deficiency that leads to the accumulation of a complex sugar, called glycogen, in skeletal and heart muscles, which cause muscle weakness and premature death from respiratory or heart failure. Normally, glycogen—the stored form of glucose—breaks down to release glucose into the bloodstream to be used as fuel for the cells.
“Pompe disease is a rare genetic disease that causes premature death and has a debilitating effect on people’s lives,” said Janet Maynard, M.D., deputy director of the Office of Rare Diseases, Pediatrics, Urologic and Reproductive Medicine in the FDA’s Center for Drug Evaluation and Research. “Today’s approval brings patients with Pompe disease another enzyme replacement therapy option for this rare disease. The FDA will continue to work with stakeholders to advance the development of additional new, effective and safe therapies for rare diseases, including Pompe disease.”
Nexviazyme, an enzyme replacement therapy, is an intravenous medication that helps reduce glycogen accumulation. The effectiveness of Nexviazyme for the treatment of Pompe disease was demonstrated in a study of 100 patients who were randomized to take Nexviazyme or another FDA-approved enzyme replacement therapy for Pompe disease. Treatment with Nexviazyme improved lung function similar to the improvement seen with the other therapy.
The most common side effects included headache, fatigue, diarrhea, nausea, joint pain (arthralgia), dizziness, muscle pain (myalgia), itching (pruritus), vomiting, difficulty breathing (dyspnea), skin redness (erythema), feeling of “pins and needles” (paresthesia) and skin welts (urticaria). Serious reactions included hypersensitivity reactions like anaphylaxis and infusion-associated reactions, including respiratory distress, chills and raised body temperature (pyrexia). Patients susceptible to fluid volume overload or with compromised cardiac or respiratory function may be at risk for serious acute cardiorespiratory failure.
The FDA granted this application Fast Track, Priority Review and Breakthrough Therapy designations. Nexviazyme also received an orphan drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Nexviazyme to Genzyme Corporation.
###
NEW DRUG APPROVALS
one time
$10.00
FDA grants priority review for avalglucosidase alfa, a potential new therapy for Pompe disease
- The FDA decision date for avalglucosidase alfa, an investigational enzyme replacement therapy, is set for May 18, 2021
- Regulatory submission based on positive data from two trials in patients with late-onset and infantile-onset Pompe disease, respectively
- Avalglucosidase alfa received FDA Breakthrough Therapy and Fast Track designations for the treatment of people with Pompe Disease
- Pompe disease, a rare degenerative muscle disorder, affects approximately 3,500 people in the U.S.
- Milestone reinforces 20+year commitment to Pompe disease community
PARIS – November 18, 2020 – The U.S. Food and Drug Administration (FDA) has accepted for priority review the Biologics License Application (BLA) for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease (acid α-glucosidase deficiency). The target action date for the FDA decision is May 18, 2021.
Avalglucosidase alfa is an investigational enzyme replacement therapy designed to improve the delivery of acid alpha-glucosidase (GAA) enzyme to muscle cells, and if approved, would offer a potential new standard of care for patients with Pompe disease.
In October, the European Medicines Agency accepted for review the Marketing Authorization Application for avalglucosidase alfa for long-term enzyme replacement therapy for the treatment of patients with Pompe disease. The Medicines and Healthcare Products Regulatory Agency in the UK has granted Promising Innovative Medicine designation for avalglucosidase alfa.
“The hallmarks of Pompe disease are the relentless and debilitating deterioration of the muscles, which causes decreased respiratory function and mobility,” said Karin Knobe, Head of Development for Rare Diseases and Rare Blood Disorders at Sanofi. “Avalglucosidase alfa is specifically designed to deliver more GAA enzyme into the lysosomes of the muscle cells. We have been greatly encouraged by positive clinical trial results in patients with late-onset and infantile-onset Pompe disease.”
Pompe disease is a rare, degenerative muscle disorder that can impact an individual’s ability to move and breathe. It affects an estimated 3,500 people in the U.S. and can manifest at any age from infancy to late adulthood.i
The BLA is based on positive data from two trials:
- Pivotal Phase 3, double-blind, global comparator-controlled trial (COMET), which evaluated the safety and efficacy of avalglucosidase alfa compared to alglucosidase alfa (standard of care) in patients with late-onset Pompe disease. Results from this trial were presented during a Sanofi-hosted virtual scientific session in June 2020 and in October 2020 at World Muscle Society and the American Association of Neuromuscular and Electrodiagnostic Medicine.
- The Phase 2 (mini-COMET) trial evaluated the safety and exploratory efficacy of avalglucosidase alfa in patients with infantile-onset Pompe disease previously treated with alglucosidase alfa. Results from this trial were presented at the WORLDSymposium, in February 2020.
Delivery of GAA to Clear Glycogen
Pompe disease is caused by a genetic deficiency or dysfunction of the lysosomal enzyme GAA, which results in build-up of complex sugars (glycogen) in muscle cells throughout the body. The accumulation of glycogen leads to irreversible damage to the muscles, including respiratory muscles and the diaphragm muscle supporting lung function, and other skeletal muscles that affect mobility.
To reduce the glycogen accumulation caused by Pompe disease, the GAA enzyme must be delivered into the lysosomes within muscle cells. Research led by Sanofi has focused on ways to enhance the delivery of GAA into the lysosomes of muscle cells by targeting the mannose-6-phosphate (M6P) receptor that plays a key role in the transport of GAA.
Avalglucosidase alfa is designed with approximately 15-fold increase in M6P content, compared to standard of care alglucosidase alfa, and aims to help improve cellular enzyme uptake and enhance glycogen clearance in target tissues.ii The clinical relevance of this difference has not been confirmed.
Avalglucosidase alfa is currently under clinical investigation and its safety and efficacy have not been evaluated by any regulatory authority worldwide.
| About Sanofi Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe. Sanofi, Empowering Life |
/////////Avalglucosidase alfa, FDA 2021, Nexviazyme, APPROVALS 2021, PEPTIDE, Enzyme replacement therapy , Pompe disease, アバルグルコシダーゼアルファ (遺伝子組換え), Fast Track, Priority Review, Breakthrough Therapy, orphan drug designation, genzyme, sanofi
Anifrolumab
(Heavy chain)
EVQLVQSGAE VKKPGESLKI SCKGSGYIFT NYWIAWVRQM PGKGLESMGI IYPGDSDIRY
SPSFQGQVTI SADKSITTAY LQWSSLKASD TAMYYCARHD IEGFDYWGRG TLVTVSSAST
KGPSVFPLAP SSKSTSGGTA ALGCLVKDYF PEPVTVSWNS GALTSGVHTF PAVLQSSGLY
SLSSVVTVPS SSLGTQTYIC NVNHKPSNTK VDKRVEPKSC DKTHTCPPCP APEFEGGPSV
FLFPPKPKDT LMISRTPEVT CVVVDVSHED PEVKFNWYVD GVEVHNAKTK PREEQYNSTY
RVVSVLTVLH QDWLNGKEYK CKVSNKALPA SIEKTISKAK GQPREPQVYT LPPSREEMTK
NQVSLTCLVK GFYPSDIAVE WESNGQPENN YKTTPPVLDS DGSFFLYSKL TVDKSRWQQG
NVFSCSVMHE ALHNHYTQKS LSLSPGK
(Lihgt chain)
EIVLTQSPGT LSLSPGERAT LSCRASQSVS SSFFAWYQQK PGQAPRLLIY GASSRATGIP
DRLSGSGSGT DFTLTITRLE PEDFAVYYCQ QYDSSAITFG QGTRLEIKRT VAAPSVFIFP
PSDEQLKSGT ASVVCLLNNF YPREAKVQWK VDNALQSGNS QESVTEQDSK DSTYSLSSTL
TLSKADYEKH KVYACEVTHQ GLSSPVTKSF NRGEC
(Disulfide bridge: H22-96, H144-H200, H220-L215, H226-H’226, H229-H’229, H261-H321, H367-H425, H’22-H’96, H’144-H’200, H’220-L’215, H’261-H’321, H’367-H’425, L23-L89, L135-L195, L’23-L’89, L’135-L’195)
Anifrolumab
| アニフロルマブ (遺伝子組換え) |
FDA APPROVED 2021/7/30, Saphnelo
- MEDI 546
| Formula | C6444H9964N1712O2018S44 |
|---|---|
| Cas | 1326232-46-5 |
| Mol weight | 145117.1846 |
| Immunomodulator, Anti-IFN-type 1 receptor antibody | |
| Disease | Systemic lupus erythematosus |
|---|
Monoclonal antibody
Treatment of systemic lupus erythematosus (SLE)
- OriginatorMedarex
- DeveloperAstraZeneca; Medarex; MedImmune
- ClassAntirheumatics; Monoclonal antibodies; Skin disorder therapies
- Mechanism of ActionInterferon alpha beta receptor antagonists
- RegisteredSystemic lupus erythematosus
- Phase IILupus nephritis
- DiscontinuedRheumatoid arthritis; Scleroderma
- 02 Jul 2021Phase-III clinical trials in Systemic lupus erythematosus in USA (SC) (NCT04877691)
- 25 Jun 2021AstraZeneca plans a phase III trial in Systemic lupus erythematosus (Adjunctive treatment) in the China, Hong Kong, South Korea, Philipines, Taiwan and Thailand (IV, Infusion), in July 2021 (NCT04931563)
- 02 Jun 2021Pharmacokinetic, efficacy and adverse events data from a phase II TULIP-LN1 trial in Lupus nephritis presented at the 22nd Annual Congress of the European League Against Rheumatism (EULAR-2021)
Anifrolumab, sold under the brand name Saphnelo, is a monoclonal antibody used for the treatment of systemic lupus erythematosus (SLE).[1][2] It binds to the type I interferon receptor, blocking the activity of type I interferons such as interferon-α and interferon-β.[medical citation needed]
Anifrolumab was approved for medical use in the United States in August 2021.[1][3][4][5]
Anifrolumab is a monoclonal antibody that inhibits type 1 interferon receptors, indicated in the treatment of moderate to severe systemic lupus erythematosus.
Anifrolumab, or MEDI-546, is a type 1 interferon receptor (IFNAR) inhibiting IgG1κ monoclonal antibody indicated in the treatment of adults with moderate to severe systemic lupus erythematosus.7,11 The standard therapy for systemic lupus erythematosus consists of antimalarials like hydroxychloroquine, glucocorticoids like dexamethasone, and disease modifying antirheumatic drugs like methotrexate.8,11
Three monoclonal antibodies (anifrolumab, rontalizumab, and sifalimumab) that target the type 1 interferon pathway entered clinical trials as potential treatments for systemic lupus erythematosus, but so far only anifrolumab has been approved.3
The design of early clinical trials of anti-interferon treatments such as anifrolumab, rontalizumab, and sifalimumab have come under criticism.3 The design of the clinical trials use different definitions of autoantibody positivity, making comparison between trials difficult; all trials involve large portions of patients also using corticosteroids, which may alter patient responses in the experimental and placebo groups; and patient populations were largely homogenous, which may have increased the odds of success of the trial.3
Anifrolumab has also been investigated for the treatment of Scleroderma.1
Anifrolumab was granted FDA approval on 30 July 2021.11
Adverse effects
The most common adverse effect was shingles, which occurred in 5% of patients in the low-dose group, to 10% in the high-dose group, and to 2% in the placebo group. Overall adverse effect rates were comparable in all groups.[6]
History
The drug was developed by MedImmune, a unit of AstraZeneca, which chose to move anifrolumab instead of sifalimumab into phase III trials for lupus in 2015.[7][8][9]
Clinical trial results
Anifrolumab failed to meet its endpoint of significant reduction in disease as assessed by the SLE Responder Index 4 instrument in the TULIP 1 phase III trial.[10] This multi-center, double-blind, placebo-controlled study followed adults with moderate to severe SLE over the course of one year. Preliminary results were announced on 31 August 2018.
Names
Anifrolumab is the international nonproprietary name (INN).[11]
References
- ^ Jump up to:a b chttps://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761123s000lbl.pdf
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Anifrolumab, American Medical Association.
- ^https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/761123Orig1s000ltr.pdf
- ^ https://www.astrazeneca.com/media-centre/press-releases/2021/saphnelo-approved-in-the-us-for-sle.html
- ^ “Saphnelo (anifrolumab) Approved in the US for Moderate to Severe Systemic Lupus Erythematosus” (Press release). AstraZeneca. 2 August 2021. Retrieved 2 August 2021 – via Business Wire.
- ^ Spreitzer H (29 August 2016). “Neue Wirkstoffe – Anifrolumab”. Österreichische Apothekerzeitung (in German) (18/2016).
- ^ “Press release: New Hope for Lupus Patients”. MedImmune. 11 August 2015. Archived from the original on 31 July 2017.
- ^ “Anifrolumab”. NHS Specialist Pharmacy Service. Retrieved 31 July 2017.
- ^ “Anifrolumab”. AdisInsight. Retrieved 31 July 2017.
- ^ “Update on TULIP 1 Phase III trial for anifrolumab in systemic lupus erythematosus”. http://www.astrazeneca.com. Retrieved 2019-02-05.
- ^ World Health Organization (2014). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 71”. WHO Drug Information. 28 (1). hdl:10665/331151.
Further reading
- Anderson E, Furie R (April 2020). “Anifrolumab in systemic lupus erythematosus: current knowledge and future considerations”. Immunotherapy. 12 (5): 275–86. doi:10.2217/imt-2020-0017. PMID 32237942.
External links
- “Anifrolumab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT01438489 for “A Study of the Efficacy and Safety of MEDI-546 in Systemic Lupus Erythematosus” at ClinicalTrials.gov
- Clinical trial number NCT02446912 for “Efficacy and Safety of Two Doses of Anifrolumab Compared to Placebo in Adult Subjects With Active Systemic Lupus Erythematosus” at ClinicalTrials.gov
- Clinical trial number NCT02446899 for “Efficacy and Safety of Anifrolumab Compared to Placebo in Adult Subjects With Active Systemic Lupus Erythematosus” at ClinicalTrials.gov
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Interferon α/β receptor |
| Clinical data | |
| Trade names | Saphnelo |
| Other names | MEDI-546, anifrolumab-fnia |
| License data | US DailyMed: Anifrolumab |
| Routes of administration | Intravenous |
| Drug class | type I interferon receptor antagonist (IFN) |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1326232-46-5 |
| DrugBank | DB11976 |
| ChemSpider | none |
| UNII | 38RL9AE51Q |
| KEGG | D11082 |
| Chemical and physical data | |
| Formula | C6444H9964N1712O2018S44 |
| Molar mass | 145119.20 g·mol−1 |
- Goldberg A, Geppert T, Schiopu E, Frech T, Hsu V, Simms RW, Peng SL, Yao Y, Elgeioushi N, Chang L, Wang B, Yoo S: Dose-escalation of human anti-interferon-alpha receptor monoclonal antibody MEDI-546 in subjects with systemic sclerosis: a phase 1, multicenter, open label study. Arthritis Res Ther. 2014 Feb 24;16(1):R57. doi: 10.1186/ar4492. [Article]
- Peng L, Oganesyan V, Wu H, Dall’Acqua WF, Damschroder MM: Molecular basis for antagonistic activity of anifrolumab, an anti-interferon-alpha receptor 1 antibody. MAbs. 2015;7(2):428-39. doi: 10.1080/19420862.2015.1007810. [Article]
- Massarotti EM, Allore HG, Costenbader K: Editorial: Interferon-Targeted Therapy for Systemic Lupus Erythematosus: Are the Trials on Target? Arthritis Rheumatol. 2017 Feb;69(2):245-248. doi: 10.1002/art.39985. [Article]
- Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, Illei GG, Drappa J, Wang L, Yoo S: Anifrolumab, an Anti-Interferon-alpha Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017 Feb;69(2):376-386. doi: 10.1002/art.39962. [Article]
- Tummala R, Rouse T, Berglind A, Santiago L: Safety, tolerability and pharmacokinetics of subcutaneous and intravenous anifrolumab in healthy volunteers. Lupus Sci Med. 2018 Mar 23;5(1):e000252. doi: 10.1136/lupus-2017-000252. eCollection 2018. [Article]
- Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, Vainshtein I, Farmer E, Rosenthal K, Morehouse C, de Los Reyes M, Schifferli K, Liang M, Sanjuan MA, Sims GP, Kolbeck R: Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med. 2018 Apr 5;5(1):e000261. doi: 10.1136/lupus-2018-000261. eCollection 2018. [Article]
- Bui A, Sanghavi D: Anifrolumab . [Article]
- Trindade VC, Carneiro-Sampaio M, Bonfa E, Silva CA: An Update on the Management of Childhood-Onset Systemic Lupus Erythematosus. Paediatr Drugs. 2021 Jul;23(4):331-347. doi: 10.1007/s40272-021-00457-z. Epub 2021 Jul 10. [Article]
- Ryman JT, Meibohm B: Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacometrics Syst Pharmacol. 2017 Sep;6(9):576-588. doi: 10.1002/psp4.12224. Epub 2017 Jul 29. [Article]
- Koh JWH, Ng CH, Tay SH: Biologics targeting type I interferons in SLE: A meta-analysis and systematic review of randomised controlled trials. Lupus. 2020 Dec;29(14):1845-1853. doi: 10.1177/0961203320959702. Epub 2020 Sep 22. [Article]
- FDA Approved Drug Products: Saphnelo (Anifrolumab-fnia) Intravenous Injection [Link]
 //////////Anifrolumab, Saphnelo, FDA 2021, APPROVALS 2021, peptide, Monoclonal antibody, アニフロルマブ (遺伝子組換え) , MEDI 546, AstraZeneca, Medarex, MedImmune
//////////Anifrolumab, Saphnelo, FDA 2021, APPROVALS 2021, peptide, Monoclonal antibody, アニフロルマブ (遺伝子組換え) , MEDI 546, AstraZeneca, Medarex, MedImmune
NEW DRUG APPROVALS
one time
$10.00
BELUMOSUDIL

BELUMOSUDIL
MW 452.5
911417-87-3, SLx-2119, KD-025, KD 025, WHO 11343
2-[3-[4-(1H-indazol-5-ylamino)quinazolin-2-yl]phenoxy]-N-propan-2-ylacetamide
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
Belumosudil mesylate
KD025 mesylate
2109704-99-4
UPDATE FDA APPROVED 7/16/2021 To treat chronic graft-versus-host disease after failure of at least two prior lines of systemic therapy, Rezurock
New Drug Application (NDA): 214783
Company: KADMON PHARMA LLC
200 MG TABLET
FDA approves belumosudil for chronic graft-versus-host disease
On July 16, 2021, the Food and Drug Administration approved belumosudil (Rezurock, Kadmon Pharmaceuticals, LLC), a kinase inhibitor, for adult and pediatric patients 12 years and older with chronic graft-versus-host disease (chronic GVHD) after failure of at least two prior lines of systemic therapy.
Efficacy was evaluated in KD025-213 (NCT03640481), a randomized, open-label, multicenter dose-ranging trial that included 65 patients with chronic GVHD who were treated with belumosudil 200 mg taken orally once daily.
The main efficacy outcome measure was overall response rate (ORR) through Cycle 7 Day 1 where overall response included complete response (CR) or partial response (PR) according to the 2014 criteria of the NIH Consensus Development Project on Clinical Trials in Chronic Graft-versus-Host Disease. The ORR was 75% (95% CI: 63, 85); 6% of patients achieved a CR, and 69% achieved a PR. The median time to first response was 1.8 months (95% CI: 1.0, 1.9). The median duration of response, calculated from first response to progression, death, or new systemic therapies for chronic GVHD, was 1.9 months (95% CI: 1.2, 2.9). In patients who achieved response, no death or new systemic therapy initiation occurred in 62% (95% CI: 46, 74) of patients for at least 12 months since response.
The most common adverse reactions (≥ 20%), including laboratory abnormalities, were infections, asthenia, nausea, diarrhea, dyspnea, cough, edema, hemorrhage, abdominal pain, musculoskeletal pain, headache, phosphate decreased, gamma glutamyl transferase increased, lymphocytes decreased, and hypertension.
The recommended dosage of belumosudil is 200 mg taken orally once daily with food.
View full prescribing information for Rezurock.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Australia’s Therapeutic Goods Administration, Health Canada, Switzerland’s Swissmedic, and the United Kingdom’s Medicines and Healthcare products Regulatory Agency.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application 6 weeks ahead of the FDA goal date.
This application was granted priority review and breakthrough therapy designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Belumosudil mesylate is an orally available rho kinase 2 (ROCK 2) inhibitor being developed at Kadmon. In 2020, the drug candidate was submitted for a new drug application (NDA) in the U.S., under a real-time oncology review pilot program, for the treatment of chronic graft-versus-host disease (cGVHD). The compound is also in phase II clinical development for the treatment of idiopathic pulmonary fibrosis and diffuse cutaneous systemic sclerosis. Formerly, the company had also been conducting clinical research for the treatment of psoriasis and non-alcoholic steatohepatitis (NASH); however, no further development has been reported for these indications. Originally developed by Nano Terra, the product was licensed to Kadmon on an exclusive global basis in 2011. In 2019, Kadmon entered into a strategic partnership with BioNova Pharmaceuticals and established a joint venture, BK Pharmaceuticals, to exclusively develop and commercialize KD-025 for the treatment of graft-versus-host disease in China. The compound has been granted breakthrough therapy designation in the U.S. for the treatment of cGVHD and orphan drug designations for cGVHD and systemic sclerosis. In the E.U. belumosudil was also granted orphan drug status in the E.U. for the treatment of cGVHD.
Kadmon , under license from NT Life Sciences , is developing belumosudil as mesylate salt, a ROCK-2 inhibitor, for treating IPF, chronic graft-versus-host disease, hepatic impairment and scleroderma. In July 2021, belumosudil was reported to be in pre-registration phase.
Belumosudil (formerly KD025 and SLx-2119) is an experimental drug being explored for the treatment of chronic graft versus host disease (cGvHD), idiopathic pulmonary fibrosis (IPF), and moderate to severe psoriasis. It is an inhibitor of Rho-associated coiled-coil kinase 2 (ROCK2; ROCK-II).[1] Belumosudil binds to and inhibits the serine/threonine kinase activity of ROCK2. This inhibits ROCK2-mediated signaling pathways which play major roles in pro- and anti-inflammatory immune cell responses. A genomic study in human primary cells demonstrated that the drug also has effects on oxidative phosphorylation, WNT signaling, angiogenesis, and KRAS signaling.[2] Originally developed by Surface Logix, Inc,[1] Belumosudil was later acquired by Kadmon Corporation. As of July 2020 the drug was in completed or ongoing Phase II clinical studies for cGvHD, IPF and psoriasis.[3]
cGvHD is a complication that can follow stem cell or hematopoietic stem cell transplantation where the transplanted cells (graft) attack healthy cells (host). This causes inflammation and fibrosis in multiple tissues. Two cytokines controlled by the ROCK2 signaling pathway, IL-17 and IL-21, have a major role in the cGvHD response. In a 2016 report using both mouse models and a limited human clinical trial ROCK2 inhibition with belumosudil targeted both the immunologic and fibrotic components of cGvHD and reversed the symptoms of the disease.[4] In October 2017 KD025 was granted orphan drug status in the United States for treatment of patients with cGvHD.[5]
IPF is a progressive fibrotic disease where the lining of the lungs become thickened and scarred.[6] Increased ROCK activity has been found in the lungs of humans and animals with IPF. Treatment with belumosudil reduced lung fibrosis in a bleomycin mouse model study.[7] Belumosudil may have a therapeutic benefit in IPF by targeting the fibrotic processes mediated by the ROCK signaling pathway.
Psoriasis is an inflammatory skin condition where patients experiences eruptions and remissions of thickened, erythematous, and scaly patches of skin. Down-regulation of pro-inflammatory responses was observed with KD025 treatment in Phase 2 clinical studies in patients with moderate to severe psoriasis.[8]
“Substance Name:Substance Name: Belumosudil [USAN]”.
PATENT
| WO2012040499 |
https://patents.google.com/patent/WO2012040499A2/en
PATENT
| CN106916145 |
https://patents.google.com/patent/CN106916145A/en

WO 2014055996, WO 2015157556



Patent
WO-2021129589
Novel crystalline polymorphic forms (N1, N2 and N15) of KD-025 (also known as belumosudil ), useful as a Rho A kinase 2 (ROCK-2) inhibitor for treating multiple sclerosis, psoriasis, rheumatoid arthritis, idiopathic pulmonary fibrosis (IPF), atherosclerosis, non-alcoholic fatty liver and systemic sclerosis. Represents the first filing from Sunshine Lake Pharma or its parent HEC Pharm that focuses on belumosudil.KD-025 is a selective ROCK2 (Rho-associated protein kinase 2, Rho-related protein kinase 2) inhibitor. It has multiple clinical indications such as the treatment of multiple sclerosis, psoriasis, rheumatoid arthritis, and Primary pulmonary fibrosis, atherosclerosis, non-alcoholic fatty liver, etc., among which many indications are in clinical phase I, and psoriasis and systemic sclerosis are in clinical phase II.
The structure of KD-025 is shown in the following formula (1).
Example 1 Preparation method of crystal form N1 of KD-025[0222]300mg of KD-025 solid was suspended and stirred in 10mL methanol at room temperature. After 22h, it was filtered, suction filtered and placed in a drying oven at 50°C under vacuum overnight to obtain 262mg of powder. The obtained crystal was detected by XPRD and confirmed to be KD-025 crystal form N1; its X-ray powder diffraction pattern was basically the same as that of Fig. 1, its DSC pattern was basically the same as that of Fig. 2, and the TGA pattern was basically the same as that of Fig. 3.
PATENT
WO2006105081 ,
Belumosudil product pat,
protection in the EU states until March 2026, expires in the US in May 2029 with US154 extension.
Example 82
2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide
[0257] A suspension of 2-(3-(4-(lH-indazol-5-ylamino)qumazolin-2-yl)ρhenoxy)acetic acid (70 mg, 0.14 mmol), PyBOP® (40 mg, 0.077 mmol), DlEA (24 μL, 0.14 mmol) in dry CH2Cl2 : DMF (2 : 0.1 mL) was stirred at RT for 15 minutes. To this solution of activated acid was added propan-2-amine (5.4 mg, 0.091 mmol). After 30 minutes, 1.0 equivalent of DIEA and 0.55 equivalents of PyBOP® were added. After stirring the solution for 15 minutes, 0.65 equivalents of propan-2-aminewere added and the mixture was stirred for an additional 30 minutes. The solvent was removed in vacuo and the crude product was purified using prep HPLC (25-50 90 rnins) to afford 2-(3-(4-(lH-indazol-5-ylamino)quinazolin-2-yl)phenoxy)-N-isopropylacetamide. (40 mg, 0.086 mmol, 61 %).
References
- ^ Jump up to:a b Boerma M, Fu Q, Wang J, Loose DS, Bartolozzi A, Ellis JL, et al. (October 2008). “Comparative gene expression profiling in three primary human cell lines after treatment with a novel inhibitor of Rho kinase or atorvastatin”. Blood Coagulation & Fibrinolysis. 19 (7): 709–18. doi:10.1097/MBC.0b013e32830b2891. PMC 2713681. PMID 18832915.
- ^ Park J, Chun KH (5 May 2020). “Identification of novel functions of the ROCK2-specific inhibitor KD025 by bioinformatics analysis”. Gene. 737: 144474. doi:10.1016/j.gene.2020.144474. PMID 32057928.
- ^ “KD025 – Clinical Trials”. ClinicalTrials.gov. Retrieved 25 July 2020.
- ^ Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. (April 2016). “Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism”. Blood. 127 (17): 2144–54. doi:10.1182/blood-2015-10-678706. PMC 4850869. PMID 26983850.
- ^ Shanley M (October 6, 2017). “Therapy to Treat Transplant Complications Gets Orphan Drug Designation”. RareDiseaseReport. Retrieved 25 July 2018.
- ^ “Pulmonary Fibrosis”. The Mayo Clinic. Retrieved July 25, 2018.
- ^ Semedo D (June 5, 2016). “Phase 2 Study of Molecule Inhibitor for Idiopathic Pulmonary Fibrosis Begins”. Lung Disease News. BioNews Services, LLC. Retrieved 25 July 2018.
- ^ Zanin-Zhorov A, Weiss JM, Trzeciak A, Chen W, Zhang J, Nyuydzefe MS, et al. (May 2017). “Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10”. Journal of Immunology. 198 (10): 3809–3814. doi:10.4049/jimmunol.1602142. PMC 5421306. PMID 28389592.
| Clinical data | |
|---|---|
| Routes of administration |
Oral administration (tablets or capsules) |
| ATC code | None |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 911417-87-3 |
| PubChem CID | 11950170 |
| UNII | 834YJF89WO |
| CompTox Dashboard (EPA) | DTXSID80238425 |
| Chemical and physical data | |
| Formula | C26H24N6O2 |
| Molar mass | 452.518 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////BELUMOSUDIL, SLx-2119, KD-025, KD 025, WHO 11343, PHASE 2, cGvHD, IPF, psoriasis, Breakthrough Therapy, Orphan Drug Designation
CC(C)NC(=O)COC1=CC=CC(=C1)C2=NC3=CC=CC=C3C(=N2)NC4=CC5=C(C=C4)NN=C5
NEW DRUG APPROVALS
ONE TIME
$10.00
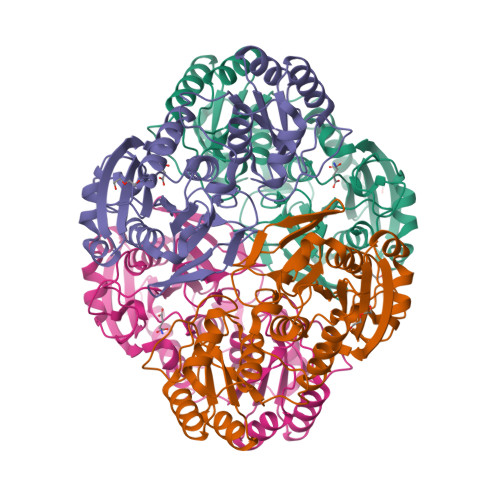
Asparaginase erwinia chrysanthemi (recombinant)-rywn
Sequence:
1ADKLPNIVIL ATGGTIAGSA ATGTQTTGYK AGALGVDTLI NAVPEVKKLA51NVKGEQFSNM ASENMTGDVV LKLSQRVNEL LARDDVDGVV ITHGTDTVEE101SAYFLHLTVK SDKPVVFVAA MRPATAISAD GPMNLLEAVR VAGDKQSRGR151GVMVVLNDRI GSARYITKTN ASTLDTFKAN EEGYLGVIIG NRIYYQNRID201KLHTTRSVFD VRGLTSLPKV DILYGYQDDP EYLYDAAIQH GVKGIVYAGM251GAGSVSVRGI AGMRKAMEKG VVVIRSTRTG NGIVPPDEEL PGLVSDSLNP301AHARILLMLA LTRTSDPKVI QEYFHTY
>Protein sequence for asparaginase (Erwinia chrysanthemi) monomer ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGEQFSNM ASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTVKSDKPVVFVAA MRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSARYITKTNASTLDTFKAN EEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKVDILYGYQDDPEYLYDAAIQH GVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEELPGLVSDSLNP AHARILLMLALTRTSDPKVIQEYFHTY
References:
- Therapeutic Targets Database: TTD Biologic drug sequences in fasta format [Link]
Asparaginase erwinia chrysanthemi (recombinant)-rywn
JZP458-201
JZP458
CAS Registry Number 1349719-22-7
Protein Chemical FormulaC1546H2510N432O476S9
Protein Average Weight 140000.0 Da
Rylaze, FDA APPROVED 6/30/2021, BLA 761179
L-Asparaginase (ec 3.5.1.1, L-asparagine amidohydrolase) erwinia chrysanthemi tetramer alpha4Asparaginase (Dickeya chrysanthemi subunit)
Other Names
- Asparaginase Erwinia chrysanthemi
- Crisantaspase
- Cristantaspase
- Erwinase
- Erwinaze
- L-Asparagine amidohydrolase (Erwinia chrysanthemi subunit)
Asparaginase erwinia chrysanthemi [USAN]
L-Asparaginase, erwinia chrysanthemi
Asparaginase (erwinia chrysanthemi)
Asparaginase erwinia chrysanthemi
L-Asparaginase (ec 3.5.1.1, L-asparagine amidohydrolase) erwinia chrysanthemi tetramer alpha4
Asparaginase erwinia chrysanthemi (recombinant) [USAN]
Asparaginase erwinia chrysanthemi (recombinant)
A hydrolase enzyme that converts L-asparagine and water to L-aspartate and NH3.
NCI: Asparaginase Erwinia chrysanthemi. An enzyme isolated from the bacterium Erwinia chrysanthemi (E. carotovora). Asparagine is critical to protein synthesis in leukemic cells, which cannot synthesize this amino acid due to the absence of the enzyme asparagine synthase. Asparaginase hydrolyzes L-asparagine to L-aspartic acid and ammonia, thereby depleting leukemic cells of asparagine and blocking protein synthesis and tumor cell proliferation, especially in the G1 phase of the cell cycle. This agent also induces apoptosis in tumor cells. The Erwinia-derived product is often used for those patients who have experienced a hypersensitivity reaction to the E. Coli formulation. (NCI Thesaurus)
- Treatment of Acute Lymphoblastic Leukemia (ALL)
- Antineoplastic Agents
| 10MG/0.5ML | INJECTABLE;INTRAMUSCULAR |
PATENT
WO 2011003633
https://patents.google.com/patent/WO2011003633A1/en
The present invention concerns a conjugate of a protein having substantial L-asparagine aminohydrolase activity and polyethylene glycol, particularly wherein the polyethylene glycol has a molecular weight less than or equal to about 5000 Da, particularly a conjugate wherein the protein is a L-asparaginase from Erwinia, and its use in therapy.Proteins with L-asparagine aminohydrolase activity, commonly known as L- asparaginases, have successfully been used for the treatment of Acute Lymphoblastic Leukemia(ALL) in children for many years. ALL is the most common childhood malignancy (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393).[0003] L-asparaginase has also been used to treat Hodgkin’s disease, acute myelocytic leukemia, acute myelomonocytic leukemia, chronic lymphocytic leukemia, lymphosarcoma, reticulosarcoma, and melanosarcoma (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).The anti-tumor activity of L-asparaginase is believed to be due to the inability or reduced ability of certain malignant cells to synthesize L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669). These malignant cells rely on an extracellular supply of L-asparagine. However, the L-asparaginase enzyme catalyzes the hydrolysis of L-asparagine to aspartic acid and ammonia, thereby depleting circulating pools of L-asparagine and killing tumor cells which cannot perform protein synthesis without L-asparagine (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0004] L-asparaginase from E. coli was the first enzyme drug used in ALL therapy and has been marketed as Elspar® in the USA or as Kidrolase® and L-asparaginase Medac® in Europe. L- asparaginases have also been isolated from other microorganisms, e.g., an L-asparaginase protein from Erwinia chrysanthemi, named crisantaspase, that has been marketed as Erwinase® (Wriston Jr., J.C. (1985) “L-asparaginase” Meth. Enzymol. 113, 608-618; Goward, CR. et al. (1992) “Rapid large scale preparation of recombinant Erwinia chrysanthemi L-asparaginase”, Bioseparation 2, 335-341). L-asparaginases from other species of Erwinia have also been identified, including, for example, Erwinia chrysanthemi 3937 (Genbank Accession#AAS67028), Erwinia chrysanthemi NCPPB 1125 (Genbank Accession #CAA31239), Erwinia carotovora (Genbank Accession #AAP92666), and Erwinia carotovora subsp. Astroseptica (Genbank Accession #AAS67027). These Erwinia chrysanthemi L-asparaginases have about 91-98% amino acid sequence identity with each other, while the Erwinia carotovora L- asparaginases have approximately 75-77% amino acid sequence identity with the Erwinia chrysanthemi L-asparaginases (Kotzia and Labrou, J. Biotechnol. 127 (2007) 657-669).[0005] L-asparaginases of bacterial origin have a high immunogenic and antigenic potential and frequently provoke adverse reactions ranging from mild allergic reaction to anaphylactic shock in sensitized patients (Wang, B. et al. (2003) “Evaluation of immunologic cross reaction of anti- asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),Leukemia 17, 1583-1588). E. coli L-asparaginase is particularly immunogenic, with reports of the presence of anti-asparaginase antibodies to E. coli L-asparaginase following i.v. or i.m. administration reaching as high as 78% in adults and 70% in children (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0006] L-asparaginases from Escherichia coli and Erwinia chrysanthemi differ in their pharmacokinetic properties and have distinct immunogenic profiles, respectively (Klug Albertsen, B. et al. (2001) “Comparison of intramuscular therapy with Erwinia asparaginase and asparaginase Medac: pharmacokinetics, pharmacodynamics, formation of antibodies and influence on the coagulation system” Brit. J. Haematol. 115, 983-990). Furthermore, it has been shown that antibodies that developed after a treatment with L-asparaginase from E. coli do not cross react with L-Asparaginase from Erwinia (Wang, B. et al., Leukemia 17 (2003) 1583-1588). Thus, L-asparaginase from Erwinia (crisantaspase) has been used as a second line treatment of ALL in patients that react to E. coli L-asparaginase (Duval, M. et al. (2002) “Comparison of Escherichia co/z-asparaginase with £Vwzm‘α-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment ofCancer, Children’s Leukemia Group phase 3 trial” Blood 15, 2734-2739; Avramis and Panosyan,Clin. Pharmacokinet. (2005) 44:367-393).[0007] In another attempt to reduce immunogenicity associated with administration of microbial L-asparaginases, an E. coli L-asparaginase has been developed that is modified with methoxy- polyethyleneglycol (mPEG). This method is commonly known as “PEGylation” and has been shown to alter the immunological properties of proteins (Abuchowski, A. et al. (1977) “Alteration of Immunological Properties of Bovine Serum Albumin by Covalent Attachment of Polyethylene Glycol,” J.Biol.Chem. 252 (11), 3578-3581). This so-called mPEG-L- asparaginase, or pegaspargase, marketed as Oncaspar® (Enzon Inc., USA), was first approved in the U.S. for second line treatment of ALL in 1994, and has been approved for first- line therapy of ALL in children and adults since 2006. Oncaspar® has a prolonged in vivo half-life and a reduced immunogenicity/antigenicity.[0008] Oncaspar® is E. coli L-asparaginase that has been modified at multiple lysine residues using 5 kDa mPEG-succinimidyl succinate (SS-PEG) (U.S. Patent No. 4,179,337). SS-PEG is aPEG reagent of the first generation that contains an instable ester linkage that is sensitive to hydro lysis by enzymes or at slightly alkaline pH values (U.S. Patent No. 4,670,417; Makromol. Chem. 1986, 187, 1131-1144). These properties decrease both in vitro and in vivo stability and can impair drug safety.[0009] Furthermore, it has been demonstrated that antibodies developed against L-asparaginase from E. coli will cross react with Oncaspar® (Wang, B. et al. (2003) “Evaluation of immunologic cross-reaction of anti-asparaginase antibodies in acute lymphoblastic leukemia (ALL and lymphoma patients),” Leukemia 17, 1583-1588). Even though these antibodies were not neutralizing, this finding clearly demonstrated the high potential for cross-hypersensitivity or cross-inactivation in vivo. Indeed, in one report 30-41% of children who received pegaspargase had an allergic reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588).[0010] In addition to outward allergic reactions, the problem of “silent hypersensitivity” was recently reported, whereby patients develop anti-asparaginase antibodies without showing any clinical evidence of a hypersensitivity reaction (Wang, B. et al. (2003) Leukemia 17, 1583-1588). This reaction can result in the formation of neutralizing antibodies to E. coli L-asparaginase and pegaspargase; however, these patients are not switched to Erwinia L-asparaginase because there are not outward signs of hypersensitivity, and therefore they receive a shorter duration of effective treatment (Holcenberg, J., J. Pediatr. Hematol. Oncol. 26 (2004) 273-274).[0011] Erwinia chrysanthemi L-asparaginase treatment is often used in the event of hypersensitivity to E. co/z-derived L-asparaginases. However, it has been observed that as many as 30-50% of patients receiving Erwinia L-asparaginase are antibody-positive (Avramis andPanosyan, Clin. Pharmacokinet. (2005) 44:367-393). Moreover, because Erwinia chrysanthemi L-asparaginase has a significantly shorter elimination half-life than the E. coli L-asparaginases, it must be administered more frequently (Avramis and Panosyan, Clin. Pharmacokinet. (2005) 44:367-393). In a study by Avramis et al., Erwinia asparaginase was associated with inferior pharmacokinetic profiles (Avramis et al., J. Pediatr. Hematol. Oncol. 29 (2007) 239-247). E. coli L-asparaginase and pegaspargase therefore have been the preferred first-line therapies for ALL over Erwinia L-asparaginase.[0012] Numerous biopharmaceuticals have successfully been PEGylated and marketed for many years. In order to couple PEG to a protein, the PEG has to be activated at its OH terminus. The activation group is chosen based on the available reactive group on the protein that will bePEGylated. In the case of proteins, the most important amino acids are lysine, cysteine, glutamic acid, aspartic acid, C-terminal carboxylic acid and the N-terminal amino group. In view of the wide range of reactive groups in a protein nearly the entire peptide chemistry has been applied to activate the PEG moiety. Examples for this activated PEG-reagents are activated carbonates, e.g., p-nitrophenyl carbonate, succinimidyl carbonate; active esters, e.g., succinimidyl ester; and for site specific coupling aldehydes and maleimides have been developed (Harris, M., Adv. Drug – A -DeI. Rev. 54 (2002), 459-476). The availability of various chemical methods for PEG modification shows that each new development of a PEGylated protein will be a case by case study. In addition to the chemistry the molecular weight of the PEG that is attached to the protein has a strong impact on the pharmaceutical properties of the PEGylated protein. In most cases it is expected that, the higher the molecular weight of the PEG, the better the improvement of the pharmaceutical properties (Sherman, M. R., Adv. Drug Del. Rev. 60 (2008), 59-68; Holtsberg, F. W., Journal of Controlled Release 80 (2002), 259-271). For example, Holtsberg et al. found that, when PEG was conjugated to arginine deaminase, another amino acid degrading enzyme isolated from a microbial source, pharmacokinetic and pharmacodynamic function of the enzyme increased as the size of the PEG attachment increased from a molecular weight of 5000Da to 20,000 Da (Holtsberg, F.W., Journal of Controlled Release 80 (2002), 259-271).[0013] However, in many cases, PEGylated biopharmaceuticals show significantly reduced activity compared to the unmodified biopharmaceutical (Fishburn, CS. (2008) Review “The Pharmacology of PEGylation: Balancing PD with PK to Generate Novel Therapeutics” J. Pharm. Sd., 1-17). In the case of L-asparaginase from Erwinia carotovora, it has been observed that PEGylation reduced its in vitro activity to approximately 57% (Kuchumova, A.V. et al. (2007) “Modification of Recombinant asparaginase from Erwinia carotovora with Polyethylene Glycol 5000” Biochemistry (Moscow) Supplement Series B: Biomedical Chemistry, 1, 230-232). The L-asparaginase from Erwinia carotovora has only about 75% homology to the Erwinia chrysanthemi L-asparaginase (crisantaspase). For Oncaspar® it is also known that its in vitro activity is approximately 50% compared to the unmodified E. coli L-asparaginase.[0014] The currently available L-asparaginase preparations do not provide alternative or complementary therapies— particularly therapies to treat ALL— that are characterized by high catalytic activity and significantly improved pharmacological and pharmacokinetic properties, as well as reduced immunogenicity. L-asparaginase protein has at least about 80% homology or identity with the protein comprising the sequence of SEQ ID NO:1, more specifically at least about 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99%, or 100% homology or identity with the protein comprising the sequence of SEQ ID NO:1. SEQ ID NO:1 is as follows:ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGE QFSNMASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTV KSDKPVVFVAAMRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSA RYITKTNASTLDTFKANEEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKV DILYGYQDDPEYLYDAAIQHGVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEELPGLVSDSLNPAHARILLMLALTRTSDPKVIQEYFHTY (SEQ ID NO:1) [0048] The term “comprising the sequence of SEQ ID NO:1” means that the amino-acid sequence of the protein may not be strictly limited to SEQ ID NO:1 but may contain additional amino-acids.ExamplesExample 1 : Preparation of Recombinant Crisantaspase [0100] The recombinant bacterial strain used to manufacture the naked recombinant Erwinia chrysanthemi L-asparaginase protein (also referred to herein as “r-crisantaspase”) was an E. coli BL21 strain with a deleted ansB gene (the gene encoding the endogenous E. coli type II L- asparaginase) to avoid potential contamination of the recombinant Erwinia chrysanthemi L- asparaginase with this enzyme. The deletion of the ansB gene relies on homologous recombination methods and phage transduction performed according to the three following steps:1) a bacterial strain (NMI lOO) expressing a defective lambda phage which supplies functions that protect and recombine electroporated linear DNA substrate in the bacterial cell was transformed with a linear plasmid (kanamycin cassette) containing the kanamycin gene flanked by an FLP recognition target sequence (FRT). Recombination occurs to replace the ansB gene by the kanamycin cassette in the bacterial genome, resulting in a ΛansB strain; 2) phage transduction was used to integrate the integrated kanamycin cassette region from the ΛansB NMI lOO strain to the ansB locus in BL21 strain. This results in an E. coli BL21 strain with a deleted ansB gene and resistant to kanamycin; 3) this strain was transformed with a FLP -helper plasmid to remove the kanamycin gene by homologous recombination at the FRT sequence. The genome of the final strain (BL21 ΛansB strain) was sequenced, confirming full deletion of the endogenous ansB gene.[0101] The E. co/z‘-optimized DNA sequence encoding for the mature Erwinia chrysanthemi L- asparaginase fused with the ENX signal peptide from Bacillus subtilis was inserted into an expression vector. This vector allows expression of recombinant Erwinia chrysanthemi L- asparaginase under the control of hybrid T5/lac promoter induced by the addition of Isopropyl β- D-1-thiogalactopyranoside (IPTG) and confers resistance to kanamycin.[0102] BL21 ΛansB strain was transformed with this expression vector. The transformed cells were used for production of the r-crisantaspase by feed batch glucose fermentation in Reisenberg medium. The induction of the cell was done 16h at 23°C with IPTG as inducer. After cell harvest and lysis by homogenization in 1OmM sodium phosphate buffer pH6 5mM EDTA (Buffer A), the protein solution was clarified by centrifugation twice at 1500Og, followed by 0.45μm and 0.22μm filtration steps. The recombinant Erwinia chrysanthemi L-asparaginase was next purified using a sequence of chromatography and concentration steps. Briefly, the theoretical isoelectric point of the Erwinia chrysanthemi L-asparaginase (7.23) permits the recombinant enzyme to adsorb to cation exchange resins at pH6. Thus, the recombinant enzyme was captured on a Capto S column (cation exchange chromatography) and eluted with salt gradient in Buffer A. Fractions containing the recombinant enzyme were pooled. The pooled solution was next purified on Capto MMC column (cation exchange chromatography) in Buffer A with salt gradient. . The eluted fractions containing Erwinia chrysanthemi L-asparaginase were pooled and concentrated before protein separation on Superdex 200pg size exclusion chromatography as polishing step. Fractions containing recombinant enzymes were pooled, concentrated, and diafiltered against 10OmM sodium phosphate buffer pH8. The purity of the final Erwinia chrysanthemi L-asparaginase preparation was evaluated by SDS-PAGE (Figure 1) and RP-HPLC and was at least 90%. The integrity of the recombinant enzyme was verified byN-terminal sequencing and LC-MS. Enzyme activity was measured at 37°C using Nessler’s reagent. The specific activity of the purified recombinant Erwinia chrysanthemi L-asparaginase was around 600 U/mg. One unit of enzyme activity is defined as the amount of enzyme that liberates lμmol of ammonia from L-asparagine per minute at 37°C. Example 2: Preparation of 10 kDa mPEG-L- Asparaginase Conjugates[0103] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration between 2.5 and 4 mg/mL, in the presence of 150 mg/mL or 36 mg/mL 10 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 10 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 10 kDa mPEG-L-asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) residues being conjugated corresponding to PEGylation of 78% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (39% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 50% of accessible amino groups (e.g., lysine residues and/or the N-terminus)) . SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 3: Preparation of 5 kDa mPEG-L-Asparaginase Conjugates[0104] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer at pH 8.0, at a protein concentration of 4 mg/mL, in the presence of 150 mg/mL or 22.5 mg/mL 5 kDa mPEG-NHS, for 2 hours at 22°C. The resulting crude 5 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein-containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 5 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as a reference, one corresponding to full PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N-terminus) being conjugated corresponding to PEGylation of 84% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (36% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 43% of accessible amino groups (e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 4: Preparation of 2 kDa mPEG-L-Asparaginase Conjugates[0105] A solution of L-asparaginase from Erwinia chrysanthemi was stirred in a 100 mM sodium phosphate buffer pH 8.0 at a protein concentration of 4 mg/mL in the presence of150 mg/mL or 22.5 mg/mL 2 kDa mPEG-NHS for 2 hours at 22°C. The resulting crude 2 kDa mPEG-L-asparaginase was purified by size exclusion chromatography using a Superdex 200 pg column on an Akta purifier UPC 100 system. Protein containing fractions were pooled and concentrated to result in a protein concentration between 2 and 8 mg/mL. Two 2 kDa mPEG-L- asparaginase conjugates were prepared in this way, differing in their degree of PEGylation as determined by TNBS assay with unmodified L-asparaginase as reference, one corresponding to maximum PEGylation (100% of accessible amino groups (e.g., lysine residues and/or the N- terminus) being conjugated corresponding to PEGylation of 86% of total amino groups (e.g., lysine residues and/or the N-terminus)); the second one corresponding to partial PEGylation (47% of total amino groups (e.g., lysine residues and/or the N-terminus) or about 55% of accessible amino groups {e.g., lysine residues and/or the N-terminus)). SDS-PAGE analysis of the conjugates is shown in Figure 2. The resulting conjugates appeared as an essentially homogeneous band and contained no detectable unmodified r-crisantaspase.Example 5: Activity of mPEG-r-Crisantaspase Conjugates[0106] L-asparaginase aminohydrolase activity of each conjugate described in the proceeding examples was determined by Nesslerization of ammonia that is liberated from L-asparagine by enzymatic activity. Briefly, 50μL of enzyme solution were mixed with 2OmM of L-asparagine in a 50 mM Sodium borate buffer pH 8.6 and incubated for 10 min at 37°C. The reaction was stopped by addition of 200μL of Nessler reagent. Absorbance of this solution was measured at 450 nm. The activity was calculated from a calibration curve that was obtained from Ammonia sulfate as reference. The results are summarized in Table 2, below:Table 2: Activity of mPEG-r-crisantaspase conjugates

* the numbers “40%” and “100%” indicate an approximate degree of PEGylation of respectively 40-55% and 100% of accessible amino groups (see Examples 2-4, supra).** the ratio mol PEG / mol monomer was extrapolated from data using TNBS assay, that makes the assumption that all amino groups from the protein (e.g., lysine residues and the N-terminus) are accessible.[0107] Residual activity of mPEG-r-crisantaspase conjugates ranged between 483 and 543 Units/mg. This corresponds to 78-87% of L-asparagine aminohydrolase activity of the unmodified enzyme. Example 6: L-Asparagine-Depleting Effect of Unmodified Crisantaspase
PAPER
Biotechnology and Applied Biochemistry (2019), 66(3), 281-289. |
https://iubmb.onlinelibrary.wiley.com/doi/10.1002/bab.1723
Crisantaspase is an asparaginase enzyme produced by Erwinia chrysanthemi and used to treat acute lymphoblastic leukemia (ALL) in case of hypersensitivity to Escherichia coli l-asparaginase (ASNase). The main disadvantages of crisantaspase are the short half-life (10 H) and immunogenicity. In this sense, its PEGylated form (PEG-crisantaspase) could not only reduce immunogenicity but also improve plasma half-life. In this work, we developed a process to obtain a site-specific N-terminal PEGylated crisantaspase (PEG-crisantaspase). Crisantaspase was recombinantly expressed in E. coli BL21(DE3) strain cultivated in a shaker and in a 2-L bioreactor. Volumetric productivity in bioreactor increased 37% compared to shaker conditions (460 and 335 U L−1 H−1, respectively). Crisantaspase was extracted by osmotic shock and purified by cation exchange chromatography, presenting specific activity of 694 U mg−1, 21.7 purification fold, and yield of 69%. Purified crisantaspase was PEGylated with 10 kDa methoxy polyethylene glycol-N-hydroxysuccinimidyl (mPEG-NHS) at different pH values (6.5–9.0). The highest N-terminal pegylation yield (50%) was at pH 7.5 with the lowest poly-PEGylation ratio (7%). PEG-crisantaspase was purified by size exclusion chromatography and presented a KM value three times higher than crisantaspase (150 and 48.5 µM, respectively). Nonetheless, PEG-crisantaspase was found to be more stable at high temperatures and over longer periods of time. In 2 weeks, crisantaspase lost 93% of its specific activity, whereas PEG-crisantaspase was stable for 20 days. Therefore, the novel PEG-crisantaspase enzyme represents a promising biobetter alternative for the treatment of ALL.
ADKLPNIVILATGGTIAGSAATGTQTTGYKAGALGVDTLINAVPEVKKLANVKGEQFSN
MASENMTGDVVLKLSQRVNELLARDDVDGVVITHGTDTVEESAYFLHLTVKSDKPVV
FVAAMRPATAISADGPMNLLEAVRVAGDKQSRGRGVMVVLNDRIGSARYITKTNAST
LDTFKANEEGYLGVIIGNRIYYQNRIDKLHTTRSVFDVRGLTSLPKVDILYGYQDDPEY
LYDAAIQHGVKGIVYAGMGAGSVSVRGIAGMRKAMEKGVVVIRSTRTGNGIVPPDEE
LPGLVSDSLNPAHARILLMLALTRTSDPKVIQEYFHTY
Figure S1 – Amino acid sequence of the enzyme crisantaspase without the signal peptide and with the lysines highlighted in red (Swiss-Prot/TrEMBL accession number: P06608|22-348 AA).
……………………………………………………………………………………………………………………………..
As a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in patients who are allergic to E. coli-derived asparaginase products
Press ReleaseFor Immediate Release:June 30, 2021
FDA Approves Component of Treatment Regimen for Most Common Childhood Cancer
Alternative Has Been in Global Shortage Since 2016
Today, the U.S. Food and Drug Administration approved Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) as a component of a chemotherapy regimen to treat acute lymphoblastic leukemia and lymphoblastic lymphoma in adult and pediatric patients who are allergic to the E. coli-derived asparaginase products used most commonly for treatment. The only other FDA-approved drug for such patients with allergic reactions has been in global shortage for years.
“It is extremely disconcerting to patients, families and providers when there is a lack of access to critical drugs for treatment of a life-threatening, but often curable cancer, due to supply issues,” said Gregory Reaman, M.D., associate director for pediatric oncology in the FDA’s Oncology Center of Excellence. “Today’s approval may provide a consistently sourced alternative to a pivotal component of potentially curative therapy for children and adults with this type of leukemia.”
Acute lymphoblastic leukemia occurs in approximately 5,700 patients annually, about half of whom are children. It is the most common type of childhood cancer. One component of the chemotherapy regimen is an enzyme called asparaginase that kills cancer cells by depriving them of substances needed to survive. An estimated 20% of patients are allergic to the standard E. coli-derived asparaginase and need an alternative their bodies can tolerate.
Rylaze’s efficacy was evaluated in a study of 102 patients who either had a hypersensitivity to E. coli-derived asparaginases or experienced silent inactivation. The main measurement was whether patients achieved and maintained a certain level of asparaginase activity. The study found that the recommended dosage would provide the target level of asparaginase activity in 94% of patients.
The most common side effects of Rylaze include hypersensitivity reactions, pancreatic toxicity, blood clots, hemorrhage and liver toxicity.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with Health Canada, where the application review is pending.
Rylaze received Fast Track and Orphan Drug designations for this indication. Fast Track is a process designed to facilitate the development and expedite the review of drugs to treat serious conditions and fulfill an unmet medical need. Orphan Drug designation provides incentives to assist and encourage drug development for rare diseases.
The FDA granted approval of Rylaze to Jazz Pharmaceuticals.
REF
DUBLIN, June 30, 2021 /PRNewswire/ — Jazz Pharmaceuticals plc (Nasdaq: JAZZ) today announced the U.S. Food and Drug Administration (FDA) approval of Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn) for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase.1 Rylaze is the only recombinant erwinia asparaginase manufactured product that maintains a clinically meaningful level of asparaginase activity throughout the entire duration of treatment, and it was developed by Jazz to address the needs of patients and healthcare providers with an innovative, high-quality erwinia-derived asparaginase with reliable supply.
“We are excited to bring this important new treatment to patients who are in critical need, and we are grateful to FDA for the approval of Rylaze based on its established safety and efficacy profile. We are pleased Rylaze was approved before the trial is complete and are diligently working to advance additional clinical trial data. We are committed to quickly engaging with FDA to evolve the Rylaze product profile with additional dosing options and an IV route of administration,” said Bruce Cozadd, chairman and CEO of Jazz Pharmaceuticals. “Thank you to our collaborators within the Children’s Oncology Group, the clinical trial investigators, patients and their families, and all of the other stakeholders who helped us achieve this significant milestone.”
Rylaze was granted orphan drug designation for the treatment of ALL/LBL by FDA in June 2021. The Biologics Licensing Application (BLA) approval followed review under the Real-Time Oncology Review (RTOR) program, an initiative of FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The company expects Rylaze will be commercially available in mid-July.
“The accelerated development and approval of Rylaze marks an important step in bringing a meaningful new treatment option for many ALL patients – most of whom are children – who cannot tolerate E. coli-derived asparaginase medicine,” said Dr. Luke Maese, assistant professor at the University of Utah, Primary Children’s Hospital and Huntsman Cancer Institute. “Before the approval of Rylaze, there was a significant need for an effective asparaginase medicine that would allow patients to start and complete their prescribed treatment program with confidence in supply.”
Recent data from a Children’s Oncology Group retrospective analysis of over 8,000 patients found that patients who did not receive a full course of asparaginase treatment due to associated toxicity had significantly lower survival outcomes – regardless of whether those patients were high risk or standard risk, slow early responders.2
About Study JZP458-201
The FDA approval of Rylaze, also known as JZP458, is based on clinical data from an ongoing pivotal Phase 2/3 single-arm, open-label, multicenter, dose confirmation study evaluating pediatric and adult patients with ALL or LBL who have had an allergic reaction to E. coli-derived asparaginases and have not previously received asparaginase erwinia chrysanthemi. The study was designed to assess the safety, tolerability and efficacy of JZP458. The determination of efficacy was measured by serum asparaginase activity (SAA) levels. The Phase 2/3 study is being conducted in two parts. The first part is investigating the intramuscular (IM) route of administration, including a Monday-Wednesday-Friday dosing schedule. The second part remains active to further confirm the dose and schedule for the intravenous (IV) route of administration.
The FDA approval of Rylaze was based on data from the first of three IM cohorts, which demonstrated the achievement and maintenance of nadir serum asparaginase activity (NSAA) greater than or equal to the level of 0.1 U/mL at 48 hours using IM doses of Rylaze 25 mg/m2. The results of modeling and simulations showed that for a dosage of 25 mg/m2 administered intramuscularly every 48 hours, the proportion of patients maintaining NSAA ≥ 0.1 U/mL at 48 hours after a dose of Rylaze was 93.6% (95% CI: 92.6%, 94.6%).1
The most common adverse reactions (incidence >15%) were abnormal liver test, nausea, musculoskeletal pain, fatigue, infection, headache, pyrexia, drug hypersensitivity, febrile neutropenia, decreased appetite, stomatitis, bleeding and hyperglycemia. In patients treated with the Rylaze, a fatal adverse reaction (infection) occurred in one patient and serious adverse reactions occurred in 55% of patients. The most frequent serious adverse reactions (in ≥5% of patients) were febrile neutropenia, dehydration, pyrexia, stomatitis, diarrhea, drug hypersensitivity, infection, nausea and viral infection. Permanent discontinuation due to an adverse reaction occurred in 9% of patients who received Rylaze. Adverse reactions resulting in permanent discontinuation included hypersensitivity (6%) and infection (3%).1
The company will continue to work with FDA and plans to submit additional data from a completed cohort of patients evaluating 25mg/m2 IM given on Monday and Wednesday, and 50 mg/m2 given on Friday in support of a M/W/F dosing schedule. Part 2 of the study is evaluating IV administration and is ongoing. The company also plans to submit these data for presentation at a future medical meeting.
Investor Webcast
The company will host an investor webcast on the Rylaze approval in July. Details will be announced separately.
About Rylaze™ (asparaginase erwinia chrysanthemi (recombinant)-rywn)
Rylaze, also known as JZP458, is approved in the U.S. for use as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia (ALL) or lymphoblastic lymphoma (LBL) in pediatric and adult patients one month and older who have developed hypersensitivity to E. coli-derived asparaginase. Rylaze has orphan drug designation for the treatment of ALL/LBL in the United States. Rylaze is a recombinant erwinia asparaginase that uses a novel Pseudomonas fluorescens expression platform. JZP458 was granted Fast Track designation by the U.S. Food and Drug Administration (FDA) in October 2019 for the treatment of this patient population. Rylaze was approved as part of the Real-Time Oncology Review program, an initiative of the FDA’s Oncology Center of Excellence designed for efficient delivery of safe and effective cancer treatments to patients.
The full U.S. Prescribing Information for Rylaze is available at: <http://pp.jazzpharma.com/pi/rylaze.en.USPI.pdf>
Important Safety Information
RYLAZE should not be given to people who have had:
- Serious allergic reactions to RYLAZE
- Serious swelling of the pancreas (stomach pain), serious blood clots, or serious bleeding during previous asparaginase treatment
RYLAZE may cause serious side effects, including:
- Allergic reactions (a feeling of tightness in your throat, unusual swelling/redness in your throat and/or tongue, or trouble breathing), some of which may be life-threatening
- Swelling of the pancreas (stomach pain)
- Blood clots (may have a headache or pain in leg, arm, or chest)
- Bleeding
- Liver problems
Contact your doctor immediately if any of these side effects occur.
Some of the most common side effects with RYLAZE include: liver problems, nausea, bone and muscle pain, tiredness, infection, headache, fever, allergic reactions, fever with low white blood cell count, decreased appetite, mouth swelling (sometimes with sores), bleeding, and too much sugar in the blood.
RYLAZE can harm your unborn baby. Inform your doctor if you are pregnant, planning to become pregnant, or nursing. Females of reproductive potential should use effective contraception (other than oral contraceptives) during treatment and for 3 months following the final dose. Do not breastfeed while receiving RYLAZE and for 1 week after the final dose.
Tell your healthcare provider if there are any side effects that are bothersome or that do not go away.
These are not all the possible side effects of RYLAZE. For more information, ask your healthcare provider.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch, or call 1-800-FDA-1088 (1-800-332-1088).
About ALL
ALL is a cancer of the blood and bone marrow that can progress quickly if not treated.3 Leukemia is the most common cancer in children, and about three out of four of these cases are ALL.4 Although it is one of the most common cancers in children, ALL is among the most curable of the pediatric malignancies due to recent advancements in treatment.5,6 Adults can also develop ALL, and about four of every 10 cases of ALL diagnosed are in adults.7 The American Cancer Society estimates that almost 6,000 new cases of ALL will be diagnosed in the United States in 2021.7 Asparaginase is a core component of multi-agent chemotherapeutic regimens in ALL.8 However, asparaginase treatments derived from E. coli are associated with the potential for development of hypersensitivity reactions.9
About Lymphoblastic Lymphoma
LBL is a rare, fast-growing, aggressive subtype of Non-Hodgkin’s lymphoma, most often seen in teenagers and young adults.8 LBL is a very aggressive lymphoma – also called high-grade lymphoma – which means the lymphoma grows quickly with early spread to different parts of the body.10,11
About Jazz Pharmaceuticals plc
Jazz Pharmaceuticals plc (NASDAQ: JAZZ) is a global biopharmaceutical company whose purpose is to innovate to transform the lives of patients and their families. We are dedicated to developing life-changing medicines for people with serious diseases – often with limited or no therapeutic options. We have a diverse portfolio of marketed medicines and novel product candidates, from early- to late-stage development, in neuroscience and oncology. We actively explore new options for patients including novel compounds, small molecules and biologics, and through cannabinoid science and innovative delivery technologies. Jazz is headquartered in Dublin, Ireland and has employees around the globe, serving patients in nearly 75 countries. For more information, please visit www.jazzpharmaceuticals.com and follow @JazzPharma on Twitter.
About The Children’s Oncology Group (COG)
COG (childrensoncologygroup.org), a member of the NCI National Clinical Trials Network (NCTN), is the world’s largest organization devoted exclusively to childhood and adolescent cancer research. COG unites over 10,000 experts in childhood cancer at more than 200 leading children’s hospitals, universities, and cancer centers across North America, Australia, and New Zealand in the fight against childhood cancer. Today, more than 90% of the 14,000 children and adolescents diagnosed with cancer each year in the United States are cared for at COG member institutions. Research performed by COG institutions over the past 50 years has transformed childhood cancer from a virtually incurable disease to one with a combined 5-year survival rate of 80%. COG’s mission is to improve the cure rate and outcomes for all children with cancer.
Caution Concerning Forward-Looking Statements
This press release contains forward-looking statements, including, but not limited to, statements related to Jazz Pharmaceuticals’ belief in the potential of Rylaze to provide a reliable therapeutic option for adult and pediatric patients to maximize their chance for a cure, plans for a mid-July 2021 launch of Rylaze, the availability of a reliable supply of Rylaze and other statements that are not historical facts. These forward-looking statements are based on Jazz Pharmaceuticals’ current plans, objectives, estimates, expectations and intentions and inherently involve significant risks and uncertainties. Actual results and the timing of events could differ materially from those anticipated in such forward-looking statements as a result of these risks and uncertainties, which include, without limitation, effectively launching and commercializing new products; obtaining and maintaining adequate coverage and reimbursement for the company’s products; delays or problems in the supply or manufacture of the company’s products and other risks and uncertainties affecting the company, including those described from time to time under the caption “Risk Factors” and elsewhere in Jazz Pharmaceuticals’ Securities and Exchange Commission filings and reports (Commission File No. 001-33500), including Jazz Pharmaceuticals’ Annual Report on Form 10-K for the year ended December 31, 2020 and future filings and reports by Jazz Pharmaceuticals. Other risks and uncertainties of which Jazz Pharmaceuticals is not currently aware may also affect Jazz Pharmaceuticals’ forward-looking statements and may cause actual results and the timing of events to differ materially from those anticipated. The forward-looking statements herein are made only as of the date hereof or as of the dates indicated in the forward-looking statements, even if they are subsequently made available by Jazz Pharmaceuticals on its website or otherwise. Jazz Pharmaceuticals undertakes no obligation to update or supplement any forward-looking statements to reflect actual results, new information, future events, changes in its expectations or other circumstances that exist after the date as of which the forward-looking statements were made.
Jazz Media Contact:
Jacqueline Kirby
Vice President, Corporate Affairs
Jazz Pharmaceuticals plc
CorporateAffairsMediaInfo@jazzpharma.com
Ireland, +353 1 697 2141
U.S. +1 215 867 4910
Jazz Investor Contact:
Andrea N. Flynn, Ph.D.
Vice President, Head, Investor Relations
Jazz Pharmaceuticals plc
investorinfo@jazzpharma.com
Ireland, +353 1 634 3211
References
- Rylaze (asparaginase erwinia chrysanthemi (recombinant)-rywn) injection, for intramuscular use Prescribing Information. Palo Alto, CA: Jazz Pharmaceuticals, Inc.
- Gupta S, Wang C, Raetz EA et al. Impact of Asparaginase Discontinuation on Outcome in Childhood Acute Lymphoblastic Leukemia: A Report From the Children’s Oncology Group. J Clin Oncol. 2020 Jun 10;38(17):1897-1905. doi: 10.1200/JCO.19.03024
- National Cancer Institute. Adult Acute Lymphoblastic Leukemia Treatment (PDQ®)–Patient Version. Available at www.cancer.gov/types/leukemia/patient/adult-all-treatment-pdq. Accessed June 29, 2021
- American Cancer Society. Key Statistics for Childhood Leukemia. Available at https://www.cancer.org/cancer/leukemia-in-children/about/key-statistics.html. Accessed June 29, 2021.
- American Cancer Society. Cancer Facts & Figures 2019. www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2019.html. Accessed June 29, 2021.
- Pui C, Evans W. A 50-Year Journey to Cure Childhood Acute Lymphoblastic Leukemia. Seminars in Hematology. 2013;50(3), 185-196.
- American Cancer Society. Key Statistics for Acute Lymphocytic Leukemia (ALL). Available at https://cancerstatisticscenter.cancer.org/?_ga=2.8163506.1018157754.1621008457-1989786785.1621008457#!/data-analysis/NewCaseEstimates. Accessed June 29, 2021.
- Salzer W, Bostrom B, Messinger Y et al. 2018. Asparaginase activity levels and monitoring in patients with acute lymphoblastic leukemia. Leukemia & Lymphoma. 59:8, 1797-1806, DOI: 10.1080/10428194.2017.1386305.
- Hijiya N, van der Sluis IM. Asparaginase-associated toxicity in children with acute lymphoblastic leukemia. Leuk Lymphoma. 2016;57(4):748–757. DOI: 10.3109/10428194.2015.1101098.
- Leukemia Foundation. Lymphoblastic Lymphoma. Available at https://www.leukaemia.org.au/disease-information/lymphomas/non-hodgkin-lymphoma/other-non-hodgkin-lymphomas/lymphoblastic-lymphoma/. Accessed June 29, 2021.
- Mayo Clinic. Acute Lymphocytic Leukemia Diagnosis. Available at https://www.mayoclinic.org/diseases-conditions/acute-lymphocytic-leukemia/diagnosis-treatment/drc-20369083. Accessed June 29, 2021.
SOURCE Jazz Pharmaceuticals plc
Related Links
CLIP
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4776285/

/////////////asparaginase erwinia chrysanthemi (recombinant)-rywn, Rylaze, Jazz Pharmaceuticals, JZP458-201, JZP458, FDA 2021, APPROVALS 2021, ORPHAN, Fast Track, Acute Lymphoblastic Leukemia, ALL, Antineoplastic Agents
https://chem.nlm.nih.gov/chemidplus/id/1349719227
https://go.drugbank.com/drugs/DB08886

NEW DRUG APPROVALS
ONE TIME
$10.00
Estetrol

Estetrol
エステトロール;
| Formula | C18H24O4 |
|---|---|
| CAS | 15183-37-6 |
| Mol weight | 304.3808 |
FDA 4/15/2021, To prevent pregnancy, Nextstellis
New Drug Application (NDA): 214154
Company: MAYNE PHARMA
PATENT
https://patents.google.com/patent/EP1562976B1/en
Estrogenic substances are commonly used in methods of Hormone Replacement Therapy (HRT) and methods of female contraception. These estrogenic substances can be divided in natural estrogens and synthetic estrogens. Examples of natural estrogens that have found pharmaceutical application include estradiol, estrone, estriol and conjugated equine estrogens. Examples of synthetic estrogens, which offer the advantage of high oral bioavailability include ethinyl estradiol and mestranol.Recently, estetrol has been found effective as an estrogenic substance for use in HRT, disclosure of which is given in the Applicant’s co-pending application WO 02/094276 . Estetrol is a biogenic estrogen that is endogeneously produced by the fetal liver during human pregnancy. Other important applications of estetrol are in the fields of contraception, therapy of auto-immune diseases, prevention and therapy of breast and colon tumors, enhancement of libido, skin care, and wound healing as described in the Applicant’s co-pending applications WO 02/094276 , WO 02/094279 , WO 02/094278 , WO 02/094275 , EP 1511496 A1 , EP 1511498 A1 , WO 03/041718 , WO 03/018026 , EP 1526856 A1 and WO 04/0278032 .[0004]The synthesis of estetrol and derivatives thereof on a laboratory scale basis is known in the art: Fishman J., Guzik H., J. Org. Chem. 33, 3133 – 3135 (1968); Nambara T. et al., Steroids 27, 111 – 121 (1976); or Suzuki E. et al., Steroids 60, 277 – 284(1995).[0005]
Fishman J., Guzik H., J. Org. Chem. 33, 3133 – 3135 (1968) discloses a successful synthesis of estetrol from an estrone derivative (compound (III); cf. for a synthesis of compound (III) Cantrall, E.W., Littell, R., Bernstein, S. J. Org. Chem 29, 214 – 217 (1964)). In a first step, the carbonyl group at C17 of compound (III) was reduced with LiAlH4 to estra-1,3,5(10),15-tetraene-3,17-diol (compound VIa) that was isolated as the diacetate (compound VIb). Compound VIb was subjected to cis-hydroxylation of the double bond of ring D by using OsO4 which resulted into the formation of estra-1,3,5(10)-triene-3,15α,16α,17β-tetraol-3,17-diacetate (compound Ib) that under heating with K2CO3 in methanol produces estetrol (Scheme 1).

[0006]
The overall yield of this three step process is, starting from estrone derivative III, only about 7%. It is worth noting that the protected derivative 17,17-ethylenedioxyestra-1,3,5(10),15-tetraene-3-ol-3-acetate (compound IV) could be cis-hydroxylated to its 15α,16α-diol derivative (compound Va), but that thereafter the dioxolane group could not be removed (p-toluene sulfonic acid in acetone at room temperature) or that the hydrolysis (aqueous sulfuric acid in warm dioxane) of the dioxolane group resulted in a mixture containing a multitude of products (Scheme 2).

[0007]Nambara T. et al., Steroids 27, 111 – 121 (1976) discloses another synthesis of estetrol wherein estrone is the starting material. The carbonyl group of estrone is first protected by treatment with ethylene glycol and pyridine hydrochloride followed by acetylation of the hydroxy group at C3. The next sequence of steps involved a bromination/base catalyzed dehydrobromination resulting into the formation of 17,17-ethylenedioxyestra-1,3,5(10),15-tetraene-3-ol (compound IVa). This compound IVa was subsequently acetylated which produced 17,17-ethylenedioxyestra-1,3,5(10),15-tetraene-3-ol-3-acetate (compound IVb). In a next step, the dioxolane group of compound IVb was hydrolysed by using p-toluene sulfonic acid to compound Vb, followed subsequently by reduction of the carbonyl group at C17 (compound Vc) and oxidation of the double bond of ring D thereby forming estra-1,3,5(10)-triene-3,15α,16α,17β-tetraol-3,17-diacetate (compound VIb). See Scheme 3.[0008]
Suzuki E. et al., Steroids 60, 277 – 284 (1995) also discloses the synthesis of estetrol by using compound Vb of Nambara T. et al. as starting material. The carbonyl group at C17 of this compound was first reduced followed by acetylation yielding estra-1,3,5(10),15-tetraene-3,17-diol-3,17-diacetate (compound 2b). The latter was subjected to oxidation with OsO4 which provided estra-1,3,5(10)-triene-3,15α,16α,17β-tetraol-3,17-diacetate (compound 3b) in 46% yield.

[0009]According to the Nambara T. et al. and Suzuki E. et al., the synthesis of estetrol can be performed with a yield of approximately 8%, starting from estrone.0010]
Poirier D., et al., Tetrahedron 47, 7751 – 7766 (1991) discloses the following compounds which were prepared according to methods that have been used to prepare similar compounds:

[0011]Dionne, P. et al., Steriods 62, 674 – 681 (1997) discloses the compound shown above wherein R is either methyl or t-butyldimethylsilyl.[0012]Magnus, P. et al., J. Am. Chem. Soc. 120, 12486 -12499 (1998) discloses that the main methods for the synthesis of α,β-unsaturated ketones from saturated ketones are (a) halogenation followed by dehydrohalogenation, (b) utilising sulphur or selenium derivatives, (c) DDQ and (d) utilizing palladium(II) complexes.[0013]Furthermore, it has also been found that by following the prior art methods mentioned above, estetrol of high purity was obtained only in low yield when using an acetyl group as a protecting group for the 3-hydroxy group of estra-1,3,5(10),15-tetraen-3-ol-17-one, in particular because its sensitivity to hydrolysis and solvolysis. In particular, the lability of the acetyl group lead not only to an increased formation of byproducts during the reactions, but also during chromatography and crystallisation for purification of intermediate products when protic solvents such as methanol were used. Therefore, it is difficult to isolate purified estetrol and intermediates thereof in good yield.
Example 7 3-Benzyloxy-estra-1,3,5 (10),15-tetraen-17-ol (compound 5; A = benzyl)
[0088]To a solution of 3-benzyl-dehydroestrone (compound 6; A = benzyl; 58 g, 162 mmol) in a mixture of MeOH (900 mL) and THF (200 mL) at room temperature was added CeCl3 heptahydrate (66.4 g, 178 mmol). After stirring for 1 h the mixture was cooled to 0-5°C using an ice/water bath. Then NaBH4 (12.2 g, 324 mmol) was added in small portions maintaining a temperature below 8°C. After stirring for 2 h at 0-5°C (TLC showed the reaction to be complete) 1 N NaOH (300 mL) and DCM (1 L) were added and the mixture was stirred for ½ h at room temperature. The layers were separated and the aqueous layer was extracted with DCM (200 mL). The organic layers were combined, dried (Na2SO4) and concentrated in vacuo to give an off-white solid (55.0 g, 152.8 mmol, 94%) TLC: Rf = 0.25 (heptanes/ethyl acetate = 4:1); HPLC-MS: 93% β-isomer, 2% α-isomer; DSC: Mp. 149.7°C, purity 96.6%; 1H-NMR (200 MHz, CDCl3) δ 7.48 (m, 5H), 7.27 (d, 1H, J = 8.4 Hz), 6.85 (dd, 1H, J1 = 2.8 Hz, J2 = 8.6 Hz), 6.81 (d, 1H, J = 2.4 Hz), 6.10 (d, 1H, J = 5.8 Hz), 5.79 (dd, 1H, J1 = 1.8 Hz, J2 = 3.4 Hz), 5.11 (s, 2H), 4.48 (d, 1H, J = 7.6), 2.96 (m, 2H), 2.46 – 1.64 (m, 9H), 0.93 (s, 3H) ppm.
Example 8 17-Acetyloxy-3-benzyloxy-estra-1,3,5 (10),15-tetraene (compound 4; A = benzyl, C = acetyl)
[0089]A solution of 3-Benzyloxy-estra-1,3,5 (10),15-tetraen-17-ol (compound 5; A = benzyl; 55.0 g, max. 153 mmol) in pyridine (400 mL) was treated with Ac2O (50 mL, 0.53 mol) and 4-dimethylaminopyridine (1.5 g, 12.3 mmol). The mixture was stirred for 2 h at room temperature (TLC showed the reaction to be complete). It was concentrated in vacuo. The residue was dissolved in EtOAc (400 mL), washed with water (200 mL) and brine (150 mL), dried (Na2SO4) and concentrated in vacuo to yield a yellow solid (54.0 g, 49.8 mmol, 88%). The product was purified by recrystallization from heptanes/ EtOAc/ EtOH (1:0.5:1) to afford a white solid (45.0 g, 112 mmol, 73%) TLC: Rf = 0.6 (heptanes/ethyl acetate = 4/1); HPLC-MS: 98% β-isomer, 1% α-isomer, 1.3% ß-estradiol; DSC: Mp. 122.8°C, purity 99.8%; 1H-NMR (200 MHz, CDCl3) δ 7.44 (m, 5H), 7.27 (d, 1H, J = 8.4 Hz), 6.86 (dd, 1H, J1 = 2.6 Hz, J2 = 8.4 Hz), 6.80 (d, 1H, J = 2.6 Hz), 6.17 (d, 1H, J = 5.8 Hz), 5.78 (dd, 1H, J1 = 1.4 Hz, J2 = 3.2 Hz), 5.45 (m, 1H), 5.11 (s, 2H), 2.96 (m, 2H), 2.40 – 1.54 (m, 10H), 2.18 (s, 3H), 0.93 (s, 3H) ppm.
Example 9 17-Acetyl-3-Benzyl estetrol (compound 3; A = benzyl, C = acetyl)
[0090]OsO4 on PVP (9 g, ~5% w/w OsO4 on PVP, prepared according to Cainelli et al. Synthesis, 45 – 47 (1989) was added to a solution of 17-Acetyloxy-3-benzyloxy-estra-1,3,5 (10),15-tetraene (compound 4; A = benzyl, C = acetyl; 45 g, 112 mmol) in THF (450 mL) and the mixture was heated to 50°C. Trimethylamine-N-oxide dihydrate (24.9 g, 224 mmol) was added portion-wise over 2 h. After stirring for 36 h at 50°C (TLC showed the reaction to be complete) the reaction mixture was cooled to room temperature. The solids were filtered off, washed with THF (100 mL) and the filtrate was concentrated. The residue was taken up in EtOAc (250 mL) and water (250 mL) was added. The aqueous layer was acidified with 1 N HCl (ca. 10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (150 mL). The organic layers were combined, dried (Na2SO4) and concentrated in vacuo. The residue was triturated with heptanes/EtOAc (1:1, 100 mL), stirred for 2 h and the resulting white precipitate was filtered off to give the product as a white solid (41 g, 94 mmol, 84%). The product was purified by recrystallization from heptanes/ ethyl acetate/ EtOH (2:1:1) three times to afford a white solid (21 g, 48.2 mmol, 43%). HPLC-MS: 99.5% βαα-isomer; DSC: Mp. 159.3°C, purity 98.7%; 1H-NMR (200 MHz, CDCl3) δ 7.49 (m, 5H), 7.27 (d, 1H, J = 8.4 Hz), 6.84 (dd, 1H, J1 = 2.6 Hz, J2 = 8.4 Hz), 6.81 (d, 1H, J = 2.4 Hz), 5.11 (s, 2H), 4.45 (d, 1H, J = 4.4), 4.11 (m, 3H), 3.12 (m, 1H) 2.95 (m, 2H), 2.46 -1.64 (m, 10H), 2.24 (s, 3H), 0.93 (s, 3H) ppm.
Example 10 17-Acetyl estetrol (compound 2; C = acetyl)
[0091]To a solution of 17-acetyl-3-benzyl estetrol (compound 3; A = benzyl, C = acetyl; 21 g, 48.2 mmol) in MeOH (600 mL, HPLC-grade) was added a preformed suspension of 10% Palladium on activated carbon (2 g) in methanol (50 mL). The mixture was placed under an atmosphere of H2 at 1 atm and stirred for 24 h (TLC showed the reaction to be completed) at room temperature. It was filtered over Celite® and the filter cake was washed with MeOH (200 mL). The filtrate was concentrated in vacuo to give 17-acetyl estetrol as a white solid (15 g, 43.4 mmol, 90%). TLC: Rf = 0.2 (heptanes/ethyl acetate = 1/1); HPLC-MS: 99.2%, DSC: Mp. 212.2°C, purity 98.9%; 1H-NMR (200 MHz, CD3OD) δ 7.14 (d, 1H, J = 8.0 Hz), 6.60 (dd, 1H, J1 = 2.6 Hz, J2 = 8.8 Hz), 6.56 (d, 1H, J = 2.4 Hz), 4.81 (dd, 1H, J1 = 3.4 Hz, J2 = 6.4 Hz), 4.07 (m, 3H), 3.12 (m, 1H), 2.85 (m, 2H), 2.37 – 1.37 (m, 10H), 2.18 (s, 3H), 0.91 (s, 3H) ppm.
Example 11 Estetrol
[0092]17-Acetyl-estetrol (compound 2; C = acetyl; 15 g, 43.4 mmol) and K2CO3 (6 g, 43.4 mmol) were suspended in MeOH (500 mL, HPLC-grade) and stirred for 4 h at room temperature (TLC showed the reaction to be complete). The solvents were evaporated in vacuo. Water (200 mL) and CHCl3 (70 mL) were added and the mixture was stirred and neutralized with 0.1 N HCl (50 mL). The product was collected by filtration, washed with water (100 mL) and CHCl3 (100 mL) to give estetrol as a white solid (12.2 g, 40.1 mmol, 92.5%, overall yield from estrone 10.8%) after drying at 40°C in an air-ventilated oven. TLC: Rf = 0.05 (heptanes/ethyl acetate = 1/1); HPLC-MS: 99.1%, DSC: Mp. 243.7°C, purity 99.5%; 1H-NMR (200 MHz, CD3OD) δ 7.14 (d, 1H, J = 8.6 Hz), 6.61 (dd, 1H, J1 = 2.6 Hz, J2 = 8.4 Hz), 6.56 (d, 1H, J = 2.4 Hz), 4.83 (m, 1H), 3.93 (m, 3H), 3.50 (d, 1H, J = 5.2), 3.38 (m, 2H), 2.84 (m, 2H), 2.32 (m, 3H), 1.97 (m, 1H), 1.68 – 1.24 (m, 5H), 0.86 (s, 3H) ppm.
SYN
https://www.tandfonline.com/doi/abs/10.1080/13697130802054078?journalCode=icmt20
Estetrol (E4), or oestetrol, is a weak estrogen steroid hormone, which is found in detectable levels only during pregnancy in humans.[1][2] It is produced exclusively by the fetal liver.[1] Estetrol is closely related to estriol (E3), which is also a weak estrogen that is found in high quantities only during pregnancy.[1][2] Along with estradiol (E2), estrone (E1), and E3, estetrol (E4) is a major estrogen in the body, although only during pregnancy.[1]
In addition to its role as a natural hormone, estetrol is under clinical development for use as a medication, for instance in hormonal contraception (in combination with drospirenone) and as menopausal hormone therapy; for information on estetrol as a medication, see the estetrol (medication) article.
Biological function
Estetrol is an estrogen and has estrogenic effects in various tissues.[1] Estetrol interacts with nuclear Estrogen Receptor (ERα) in a manner identical to that of the other estrogens and distinct from that observed with Selective Estrogen Receptor Modulators (SERMs).[3][4] So far the physiological function of estetrol is unknown. The possible use of estetrol as a marker for fetal well-being has been studied quite extensively. However, due to the large intra- and inter-individual variation of maternal estetrol plasma levels during pregnancy this appeared not to be feasible.[5][6][7][8][9]
Biological activity
Estetrol is an agonist of the estrogen receptors (ERs), and hence is an estrogen.[10][11] It has moderate affinity for ERα and ERβ, with Ki values of 4.9 nM and 19 nM, respectively.[10][12] As such, estetrol has 4- to 5-fold preference for the ERα over the ERβ.[10][12] The estrogen has low affinity for the ERs relative to estradiol, and both estetrol and the related estrogen estriol require substantially higher concentrations than estradiol to produce similar effects to estradiol.[10] The affinity of estetrol for the ERs is about 0.3% (rat) to 6.25% (human) of that of estradiol, and its in vivo potency in animals is about 2 to 3% of that of estradiol.[10] Estetrol shows high selectivity for the ERs.[10][12]
Biochemistry
Biosynthesis
Estetrol is synthesized during pregnancy only in the fetal liver from estradiol (E2) and estriol (E3) by the two enzymes 15α- and 16α-hydroxylase.[13][14][15] Alternatively, estetrol is synthesized with 15α-hydroxylation of 16α-hydroxy-DHEA sulfate as an intermediate step.[16] It appears in maternal urine at around week 9 of pregnancy.[2] After birth the neonatal liver rapidly loses its capacity to synthesize estetrol because these two enzymes are no longer expressed.
Estetrol reaches the maternal circulation through the placenta and was already detected at nine weeks of pregnancy in maternal urine.[17][18] During the second trimester of pregnancy high levels were found in maternal plasma, with steadily rising concentrations of unconjugated estetrol to about 1 ng/mL (>3 nM) towards the end of pregnancy.[1]
Distribution
In terms of plasma protein binding, estetrol is moderately bound to albumin, and is not bound to sex hormone-binding globulin (SHBG).[19][20]
Metabolism
Estetrol undergoes no phase I metabolism by CYP P450 enzymes.[10] It is conjugated via glucuronidation and to a lesser extent sulfation and then excreted.[10][21]
Excretion
Estetrol is excreted mostly or completely in urine.[21][10]
Chemistry
See also: List of estrogens
vteStructures of major endogenous estrogens Estrone (E1)Estradiol (E2)Estriol (E3)Estetrol (E4) Estrone (E1)Estradiol (E2)Estriol (E3)Estetrol (E4) |
Estetrol, also known as 15α-hydroxyestriol or as estra-1,3,5(10)-triene-3,15α,16α,17β-tetrol, is a naturally occurring estrane steroid and derivative of estrin (estratriene).[10][11] It has four hydroxyl groups, which explains the abbreviation E4.[10][11]
Synthesis
Chemical syntheses of estetrol have been published.[22]
History
Estetrol was discovered in 1965 by Egon Diczfalusy and coworkers at the Karolinska Institute in Stockholm, Sweden, via isolation from the urine of pregnant women.[10][23]
References
- ^ Jump up to:a b c d e f Holinka CF, Diczfalusy E, Coelingh Bennink HJ (May 2008). “Estetrol: a unique steroid in human pregnancy”. J. Steroid Biochem. Mol. Biol. 110 (1–2): 138–43. doi:10.1016/j.jsbmb.2008.03.027. PMID 18462934.
- ^ Jump up to:a b c Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management, 3rd ed., SSC Yen and RB Jaffe (eds.), pp. 936–981, Copyright Elsevier/Saunders 1991
- ^ Abot, Anne; Fontaine, Coralie; Buscato, Mélissa; Solinhac, Romain; Flouriot, Gilles; Fabre, Aurélie; Drougard, Anne; Rajan, Shyamala; Laine, Muriel; Milon, Alain; Muller, Isabelle (2014). “The uterine and vascular actions of estetrol delineate a distinctive profile of estrogen receptor α modulation, uncoupling nuclear and membrane activation”. EMBO Molecular Medicine. 6 (10): 1328–1346. doi:10.15252/emmm.201404112. ISSN 1757-4676. PMC 4287935. PMID 25214462.
- ^ Foidart, JM; et al. (2019). “30th Annual Meeting of The North America Menopause Society September 25 – 28, 2019, Chicago, IL”. Menopause. 26 (12): 1445–1481. doi:10.1097/GME.0000000000001456. ISSN 1530-0374.
- ^ J. Heikkilä, T. Luukkainen, Urinary excretion of estriol and 15a-hydroxyestriol in complicated pregnancies, Am. J. Obstet. Gynecol. 110 (1971) 509-521.
- ^ D. Tulchinsky, F.D. Frigoletto, K.J. Ryan, J. Fishman, Plasma estetrol as an index of fetal well-being, J. Clin. Endocrinol. Metab. 40 (1975) 560-567
- ^ A.D. Notation, G.E. Tagatz, Unconjugated estriol and 15a-hydroxyestriol in complicated pregnancies, Am. J. Obstet. Gynecol. 128 (1977) 747-756.
- ^ N. Kundu, M. Grant, Radioimmunoassay of 15a-hydroxyestriol (estetrol) in pregnancy serum, Steroids 27 (1976) 785-796.
- ^ N. Kundu, M. Wachs, G.B. Iverson, L.P. Petersen, Comparison of serum unconjugated estriol and estetrol in normal and complicated pregnancies, Obstet. Gynecol. 58 (1981) 276-281.
- ^ Jump up to:a b c d e f g h i j k l Coelingh Bennink HJ, Holinka CF, Diczfalusy E (2008). “Estetrol review: profile and potential clinical applications”. Climacteric. 11 Suppl 1: 47–58. doi:10.1080/13697130802073425. PMID 18464023.
- ^ Jump up to:a b c Visser M, Coelingh Bennink HJ (March 2009). “Clinical applications for estetrol” (PDF). J. Steroid Biochem. Mol. Biol. 114(1–2): 85–9. doi:10.1016/j.jsbmb.2008.12.013. PMID 19167495.
- ^ Jump up to:a b c Visser M, Foidart JM, Coelingh Bennink HJ (2008). “In vitro effects of estetrol on receptor binding, drug targets and human liver cell metabolism”. Climacteric. 11 Suppl 1: 64–8. doi:10.1080/13697130802050340. PMID 18464025.
- ^ J. Schwers, G. Eriksson, N. Wiqvist, E. Diczfalusy, 15a-hydroxylation: A new pathway of estrogen metabolism in the human fetus and newborn, Biochim. Biophys. Acta. 100 (1965) 313-316
- ^ J. Schwers, M. Govaerts-Videtsky, N. Wiqvist, E. Diczfalusy, Metabolism of oestrone sulphate by the previable human foetus, Acta Endocrinol. 50 (1965) 597-610.
- ^ S. Mancuso, G. Benagiano, S. Dell’Acqua, M. Shapiro, N. Wiqvist, E. Diczfalusy, Studies on the metabolism of C-19 steroids in the human foeto-placental unit, Acta Endocrinol. 57 (1968) 208-227.
- ^ Jerome Frank Strauss; Robert L. Barbieri (2009). Yen and Jaffe’s Reproductive Endocrinology: Physiology, Pathophysiology, and Clinical Management. Elsevier Health Sciences. pp. 262–. ISBN 1-4160-4907-X.
- ^ J. Heikkilä, H. Adlercreutz, A method for the determination of urinary 15α-hydroxyestriol and estriol, J. Steroid Biochem. 1 (1970) 243-253
- ^ J. Heikkilä, Excretion of 15α-hydroxyestriol and estriol in maternal urine during normal pregnancy, J. Steroid Biochem. 2 (1971) 83-93.
- ^ Visser M, Holinka CF, Coelingh Bennink HJ (2008). “First human exposure to exogenous single-dose oral estetrol in early postmenopausal women”. Climacteric. 11 Suppl 1: 31–40. doi:10.1080/13697130802056511. PMID 18464021.
- ^ Hammond GL, Hogeveen KN, Visser M, Coelingh Bennink HJ (2008). “Estetrol does not bind sex hormone binding globulin or increase its production by human HepG2 cells”. Climacteric. 11 Suppl 1: 41–6. doi:10.1080/13697130701851814. PMID 18464022.
- ^ Jump up to:a b Mawet M, Maillard C, Klipping C, Zimmerman Y, Foidart JM, Coelingh Bennink HJ (2015). “Unique effects on hepatic function, lipid metabolism, bone and growth endocrine parameters of estetrol in combined oral contraceptives”. Eur J Contracept Reprod Health Care. 20 (6): 463–75. doi:10.3109/13625187.2015.1068934. PMC 4699469. PMID 26212489.
- ^ Warmerdam EG, Visser M, Coelingh Bennink HJ, Groen M (2008). “A new route of synthesis of estetrol”. Climacteric. 11 Suppl 1: 59–63. doi:10.1080/13697130802054078. PMID 18464024.
- ^ Hagen AA, Barr M, Diczfalusy E (June 1965). “Metabolism of 17-beta-oestradiol-4-14-C in early infancy”. Acta Endocrinol. 49: 207–20. doi:10.1530/acta.0.0490207. PMID 14303250.
| Names | |
|---|---|
| Preferred IUPAC name(1R,2R,3R,3aS,3bR,9bS,11aS)-11a-Methyl-2,3,3a,3b,4,5,9b,10,11,11a-decahydro-1H-cyclopenta[a]phenanthrene-1,2,3,7-tetrol | |
| Other namesOestetrol; E4; 15α-Hydroxyestriol; Estra-1,3,5(10)-triene-3,15α,16α,17β-tetrol | |
| Identifiers | |
| CAS Number | 15183-37-6 |
| 3D model (JSmol) | Interactive image |
| ChEBI | CHEBI:142773 |
| ECHA InfoCard | 100.276.707 |
| KEGG | D11513 |
| PubChem CID | 27125 |
| UNII | ENB39R14VF |
| CompTox Dashboard (EPA) | DTXSID50164888 |
| showSMILES | |
| Properties | |
| Chemical formula | C18H24O4 |
| Molar mass | 304.386 g/mol |
| Solubility in water | 1.38 mg/mL |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references |
//////////estetrol, Nextstellis, fda 2021, approvals 2021

NEW DRUG APPROVALS
one time
$10.00
Drospirenone
Drospirenone
FDA APPROVED 4/15/2021, To prevent pregnancy Nextstellis
New Drug Application (NDA): 214154
Company: MAYNE PHARMALabel (PDF)
Letter (PDF)
ReviewLabel (PDF)DrospirenoneCAS Registry Number: 67392-87-4
CAS Name: (2¢S,6R,7R,8R,9S,10R,13S,14S,15S,16S)-1,3¢,4¢,6,7,8,9,10,11,12,13,14,15,16,20,21-Hexadecahydro-10,13-dimethylspiro[17H-dicyclopropa[6,7:15,16]cyclopenta[a]phenanthrene-17,2¢(5¢H)-furan]-3,5¢(2H)-dione
Additional Names: 6b,7b,15b,16b-dimethylene-3-oxo-4-androstene-[17(b-1¢)-spiro-5¢]perhydrofuran-2¢-one; 6b,7b,15b,16b-dimethylen-3-oxo-17a-pregn-4-ene-21,17-carbolactone; dihydrospirorenone
Manufacturers’ Codes: ZK-30595
Molecular Formula: C24H30O3Molecular Weight: 366.49Percent Composition: C 78.65%, H 8.25%, O 13.10%
Literature References: Synthetic progestogen exhibiting antimineralocorticoid and antiandrogenic activity. Prepn: R. Wiechert et al.,DE2652761; eidem,US4129564 (both 1978 to Schering AG); D. Bittler et al.,Angew. Chem.94, 718 (1982). HPLC determn in human plasma: W. Krause, U. Jakobs, J. Chromatogr.230, 37 (1982). Pharmacological profile: P. Muhn et al.,Contraception51, 99 (1995). Review of synthesis: H. Laurent et al.,J. Steroid Biochem.19, 771-776 (1983); of pharmacology and clinical experience: W. Oelkers, Mol. Cell. Endocrinol.217, 255-261 (2004).
Properties: mp 201.3°. [a]D22 -182° (c = 0.5 in chloroform). uv (methanol): 265 nm (e 19000).
Melting point: mp 201.3°
Optical Rotation: [a]D22 -182° (c = 0.5 in chloroform)
Derivative Type: Mixture with ethinyl estradiolTrademarks: Angeliq (Schering AG); Yasmin (Schering AG)Literature References: Clinical trial as oral contraceptive: K. S. Parsey, A. Pong, Contraception61, 105 (2000); in treatment of menopausal symptoms: R. Schürmann et al., Climacteric7, 189 (2004).
Therap-Cat: Progestogen. In combination with estrogen as oral contaceptive and in treatment of menopausal symptoms.Keywords: Progestogen; Contraceptive (Oral).SYNhttps://www.sciencedirect.com/science/article/abs/pii/S0039128X15002135
Abstract
A general methodology for the synthesis of different steroidal 17-spirolactones is described. This method uses lithium acetylide of ethyl propiolate as the three carbon synthon and the method was successfully applied for the process development of drospirenone.
Graphical abstract

SYN
Steroid Hormones
Ruben Vardanyan, Victor Hruby, in Synthesis of Best-Seller Drugs, 2016
Drospirenone–Yaz
The synthesis of drospirenone (27.4.12) is believed to have been described for the first time in Wiechert et al [79], with a total yield of approximately 2 to 3% via the pathway presented in Scheme 27.4.

Each compound produced after each reaction step was purified by column chromatography.
Androsta-5,15-diene-3-ol-17-one was methylenated at the 15,16-position (27.4.13) and reacted with organometallic reagent (3,3-dimethoxypropyl)lithium prepared from 3-bromo-1,1-dimethoxypropane (27.4.14) and lithium in THF to produce the tertiary alcohol (27.4.15), which on short-term reflux with toluenesulfonic acid in acetone transformed to cyclic 21,17-hemiacetal (27.4.16). Oppenanuer oxidation with aluminium isopropoxide in excess of cyclohexanone in toluene was brought to mild oxidation of both secondary alcohol groups, and the simultaneous isomerization of the 5,6 double bond to the 4,5 position produced the compound (27.4.17). The last was oxidized with Jones reagent—chromic trioxide in diluted sulfuric acid—producing conjugated diene-one (27.4.18). Corey methylenation of the obtained product with dimethyloxosulfonium methylide in DMSO containing sodium hydride produced the final compound, the desired drospirenone (27.4.12).
The following patents and publications [80-83], which differ slightly from one another, disclose similar processes for preparing drospirenone and are presented in Scheme 27.5.

In Scheme 27.5, drospirenone (27.4.12) is prepared by converting the key starting compound (27.4.19) into the corresponding chloride (27.4.20) via reaction with triphenylphosphine and tetrachloromethane under mild conditions. Reductive dechlorination with Zn in acetic acid in THF tetrahydrofuran produced 5-hydroxy-15β,16β-methylene-3β-pivaloyloxy-5β-androst-6-en-17-one (27.4.21). The pivaloyl protecting group of the last was removed with the mixture of potassium hydroxide and sodium perchlorate in THF/methanol mixture to produce the diol (27.4.22). Simmons–Smith cyclopropanation reaction was applied to this compound. For that purpose, solution of (27.4.22) in dimethyl Cellosolve was stirred at 80°C with zinc-copper couple and methylene iodide, which produced the desired compound (27.4.23). The compound (27.4.23) underwent ethinylation with propargyl alcohol using potassium methylate in THF as a base to produce the 1,4-butindiol derivative (27.4.24). The triple bond of the 1,4-butindiol derivative (27.4.24) was hydrogenated in aTHF/methanol/pyridine mixture in the presence of palladium on carbon to produce the 1,4-butanediol derivative (27.4.25). The obtained compound underwent oxidation–lactonization at 50°C using a solution of CrO3 in water and pyridine to produce the desired drospirenone (27.4.12).
Several other synthetic routes for the production of drospirenone have been proposed [84-96], one of which [96] is presented in Scheme 27.6.

According to Scheme 27.6, a mixture of the key starting ketodiol (27.4.26), synthesis of which was described previously [84], with ethyl propiolate in THF was added to a solution of lithium hexamethyldisilylamide to produce, after quenching with acetic acid and saturated ammonium chloride solution, ethinyl alcohol (27.4.27). This product was hydrogenated on H2-Pd/C catalyst to produce ethyl 4-hydroxybutanoate (27.4.28). The 3-hydroxy group in the obtained product was oxidized to the keto group with (2,2,6,6-tetramethyl-piperidin-1-yl)oxyl, resulting in the compound (27.4.29). Treatment of the last with potassium hydroxide in a methanol–water mixture affects both hydrolysis of the ester group and dehydration of 5-hydroxy substituent. Acidification of the resulting intermediate results in drospirenone (27.4.12).
Drospirenone is a unique synthetic progestogen derived from 17α-spirolactone; it has a pharmacological profile very similar to that of endogenous progesterone. Drospirenone prevents ovulation and is used in contraceptive pills; it is also used as a postmenopausal hormone replacement. Drospirenone provides reliable and well-tolerated contraception and effective treatment of menopause. It has progestational, antialdosterone, and antiandrogenic properties, but is devoid of any estrogenic, androgenic, glucocorticoid, antiglucocorticoid, and mineralocorticoid activities. The affinity of drospirenone for the mineralocorticoid receptor makes it an antagonist of aldosterone, which is not only important in the renin–angiotensin–aldosterone system, but also means it acts directly on the cardiovascular system. It is progestin with antimineralocorticoid property that acts to suppress gonadotropins. It is thus able to prevent excessive sodium loss and regulate blood pressure. Drospirenone slightly decreases body weight and blood pressure and shares many pharmacodynamic properties with progesterone [97-110].
PATENT
https://patents.google.com/patent/US8334375B2/enDrospirenone is a synthetic steroid with progestin, anti-mineral corticoid and anti androgen activity. Drospirenone is currently being used as a synthetic progestin in oral contraceptive formulations. A regioselective synthesis for drospirenone has been described (see e.g., Angew. Chem. 94, 1982, 718) that uses the 17 keto derivative (1) as a key intermediate.

The synthesis of intermediate (1) and the transformation of intermediate (1) into drospirenone has been described in, for example, U.S. Published Patent Application Nos. 2009/0023914; 20080207575; 2008/0200668; 2008/0076915, 20070049747, and 20050192450; U.S. Pat. Nos. 6,933,395; 6,121,465, and 4,129,564, European Patent No. 0 075 189 and PCT Publication No. WO 2006/061309, all of which are incorporated herein by reference. Many of these routes introduce the required C3 side chain in the 17 position of intermediate (1). These conversions are usually carried out with carbanions, such as propargylalcohol, trimethylsulfoxonium iodide, or the use of the anion generated from a suitably protected derivative of 1-bromopropionaldehyde. After oxidation of the 3-hydroxy substituent to a 3-keto group, and the oxidative formation of the 17-spirolactone, the 3-keto-5-hydroxy-17-spirolactone is transformed via acid catalysis into drospireneone. If the oxidation is performed under acidic conditions at elevated temperatures, the oxidation and elimination can be run without isolation of the intermediate products.Most of these procedures rely on the acid-catalyzed elimination of the 5-hydroxy group in the last step of the synthesis. It has been documented that 15,16-methylene-17-spirolactones are prone to undergo rearrangement to generate the inverted 17-spirolactone under mild acidic conditions (see, for example, Tetrahedron Letters, Vol. 27, No 45, 5463-5466) in considerable amounts. This isomer has very similar physical chemical properties, and typically requires chromatographic separation or repeated fractional recrystallizations to purify the product. This isomerization can make these approaches less desirable from an economical point of view.FIG. 1Experimental Example

A solution of compound (1) (5 g; 15.2 mmol) and tert-butyldimethyl (2-propynyloxy)silane (2.83 g, 16.7 mmol) in 75 ml of dry THF was added dropwise through an addition funnel to a precooled slurry of potassium tert-butoxide (8.49 g, 75.7 mmol) at −10 C. A thick white precipitate is formed during the addition and the resulting mixture was stirred for an hour at 0 C. TLC analysis (70% EtOAc/Hexanes) showed completion of the reaction and showed a less polar product. The reaction was quenched by the addition of ice water (100 ml) and neutralized by adding acetic acid (4.3 ml). The THF layer was separated and the aqueous layer was extracted with EtOAc (2×50 ml). The combined organic layers were washed with water (2×100 ml), brine (100 ml) and dried over anhydrous sodium sulfate. The solvent was removed under vacuum to afford compound (2a) (7.5 g, 99.2%) as a solid which was used in the next step without any purification.NMR (CDCl3) δ 0.139 (s, 6H, S1—CH3), 0.385 (m, 1H), 0.628 (m, 1H), 0.857 (s, 18-Me), 0.896 (s, 19-Me), 0.918 (s, 3H, Si—CH3), 0.927 (s, 6H, Si—CH3), 4.05 (s, 1H), 4.428 (s, 2H, —O—CH2) FTIR (ATR): 3311, 3017, 2929, 2858, 2270, 1058 cm−1

Compound (2a) (5 g, 9.98 mmol) was dissolved in 100 ml of ethyl acetate in a Parr hydrogenation bottle and was mixed with 10% palladium on charcoal (1 g, 0.09 mmol). This mixture was hydrogenated on a Parr apparatus at a pressure of 20 psi for 90 minutes. The catalyst was filtered and washed with ethyl acetate. The solvent was removed in vacuo to afford compound (3a) as a colorless foam (5.01 g, 99%).NMR (CDCl3) δ 0.0758 (s, 6H, Si—CH3), 0.283 (m, 1H), 0.628 (m, 1H), 0.856 (s, 18-Me), 0.893 (s, 19-Me), 0.918 (s, 9H, Si—CH3), 3.69 (m, 2H), 4.05 (s, 1H). FTIR (ATR): 3374, 3017, 2929, 2858, 1259, 1091, 1049, 835 cm−1

Chromium trioxide (4.95 g, 49.5 mmol) was added to a solution of pyridine (7.83 g, 99.05 mmol) in anhydrous dichloromethane (100 ml). The resulting mixture was stirred for 15 minutes during which time the color changed to burgundy. A solution of compound (1a) (5 g, 9.90 mmol) in 50 ml of dichloromethane was added and the mixture was stirred at room temperature for 6 h. The excess oxidizing agent was quenched by adding isopropanol. The reaction mixture was diluted with MTBE (50 ml) and was passed through a short pad of Celite. The solid was washed again with 2:1 MTBE-CH2Cl2 (50 ml x2). The solvent was removed in vacuo to give a residue which was dissolved in 100 ml of EtOAc, was washed with water, and dried over anhydrous sodium sulfate. The solvent was removed in vacuo to afford compound (4a) as a pale yellow foam (4.5 g, 90.3%).NMR (CDCl3) δ 0.08 (s, 6H, Si—CH3), 0.31 (m, 1H), 0.914 (s, 9H, Si—CH3), 0.931 (s, 6H, 18-Me, 19-Me) 3.70 (m, 2H). FTIR (ATR): 3399, 3022, 2950, 2929, 2862, 1708, 1649, 1259, 1041 cm−1

A solution of compound (4a) (5 g, 9.94 mmol) in 50 ml of MeOH was refluxed with NaOH (397 mg, 9.94 mmol) for 3 h. When the reaction was over, as shown by TLC, the reaction mixture was cooled to room temperature and added to ice cold water (150 ml). The mixture was extracted with ethyl acetate (3×50 mL). The combined EtOAc layers were washed with water (100 ml) brine (50 ml) and dried over sodium sulfate. The solvent was removed in vacuo to afford compound (5) as a colorless amorphous solid (4.5 g, 92%).NMR (CDCl3) δ 0.05 (s, 6H, Si—CH3), 0.296 (m, 1H), 0.886 (s, 9H, Si—CH3), 0.908 (s, 3H, 18-Me), 1.07 (s, 3H, 19-Me), 3.68 (m, 2-H), 5.95 (s, 1H). FTIR (ATR): 3450, 3009, 2950, 2858, 1653, 1603, 1095 cm−1

A solution of compound (5a) (5 g, 9.94 mmol) in 30 ml of acetone was cooled to −15 C as a 2.7M solution of Jones reagent (3.68 ml, 9.94 mmol) was added drop wise. The reaction mixture was stirred at 0 C for 2 h, during this time TLC showed completion of the reaction. The reaction was quenched by adding isopropanol and diluted with water. The reaction mixture was extracted with EtOAc. The combined EtOAc layers were washed with water, sat. NaHCO3 and brine. The EtOAc layers were dried over sodium sulfate and solvent was removed by vacuum to afford crude drospirenone as a pale yellow foam (3 g, 82%) Recrystallization from acetone-hexane gave 1.5 g of pure drospirenone as white solid.NMR (CDCl3) δ 0.0548 (m, 1H), 0.88 (m, 1H), 1.008 (s, 3H, 18-Me), 1.11 (s, 3H, 18-Me), 6.03 (s, 1H). FTIR (ATR): 3025, 2971, 2942, 1763, 1654, 1590, 1186 cm−1FIG. 2Experimental Example

Lithium hexamethyldisilylamide (LiHMDS) 1.0 M/THF (75.7 mL, 75.7 mmol) was introduced into a 500 mL, 3-neck flask equipped with an addition funnel and a pierced septa for the introduction of a thermocouple probe. The mixture was diluted with THF (25 mL). The solution was stirred (Teflon paddle) and chilled to an internal temperature of −72° C. A THF (75 mL) solution of ketodiol (5 g, 15.13 mmol) containing ethyl propiolate (3.07 mL, 30.26 mmol) was added dropwise over 1 hour while not allowing the temperature to rise above −65° C. Upon completion of the addition the mixture was stirred for 3 hrs while allowing the temperature to warm slowly to −60° C. Finally, the mixture was warned to −40° C. over 1 hour.The mixture was quenched through the addition of acetic acid (4.25 mL)/water (5.0 mL) followed by the addition of saturated ammonium chloride solution (100 mL). The mixture was stirred for 3 min and then transferred to a separatory funnel. The layers were separated and the upper, THF layer was diluted with ethyl acetate (75 mL). The organic phase was washed with water (3×100 mL) and brine (1×100 mL). All the aqueous washes were extracted with ethyl acetate (2×30 mL). The combined organic extract was dried over sodium sulfate, filtered, and evaporated in vacuo (45° C.) to afford a thick oil. Dichloromethane (ca. 35 mL) was added and evaporated in vacuo. The flask was cooled slightly and dichloromethane (33 ml) was added to give a solid mass. The solid was broken up and stirred until a homogeneous slurry was obtained. Hexanes (35 mL) was added slowly to the stirred mixture and the mixture was stored at 2-4 C overnight. The solid was filtered, washed with 30% dichloromethane/hexanes, and dried in vacuo at ambient temperature for 4 hours to give (2b) 5.86 g (90.4%) of a white powder.
Compound (2b) (5.0 g, 11.67 mmol) was dissolved in THF (50 mL) and 5% Pd/C (622 mg, 0.29 mmol Pd) was added and the mixture was shaken at 15 psi H2 for 2 hours. The mixture was diluted with ethyl acetate (25 mL) and filtered through a pad of Celite. The filter pad was washed with ethyl acetate (3×25 mL) and the filtrate was evaporated to dryness to afford 5.0 g (99.1%) of triol (7b) as a stable foam.

Compound (7a) (5.0 g, 11.56 mmol) was dissolved in dichloromethane (50 mL) and the solution was stirred vigorously and chilled to −15° C. (NaCl/ice) and TEMPO (45.16 mg, 0.29 mmol, 2.5 mol %) was added. The mixture was treated dropwise over about 15-20 min. with a mixture of sodium hypochlorite (12.5%) (11.17 mL, 23.12 mmol) in water (8.0 mL) containing potassium bicarbonate (833 mg, 8.32 mmol). The mixture was allowed to warm to 0 C for 1.25 hrs. Analysis of the reaction by TLC (60% EtOAc/hex) shows the appearance of a slightly less polar product (ΔRf=0.8 cm). The mixture was chilled to −5 C and was quenched through the dropwise addition (ca 10-15 min) of a water (15.0 mL) solution of sodium phosphate (1.27 g, 7.75 mmol) and sodium metabisulfite (1.10 g, 5.78 mmol). The layers were separated and the dichloromethane solution was washed with water (2×) and brine. All aqueous washes were extracted with additional dichloromethane (2×15 mL). The combined dichloromethane extract was dried over sodium sulfate, filtered, and evaporated to give 4.88 g (98.08%) of ketone (8b) as a stable foam.

Compound (8b) was added to a methanol (10 mL) solution containing 8.0 M KOH solution (6.3 mL, 50.36 mmol) preheated to 60 C. The solution was heated at reflux for 2.5 hours. The mixture was chilled in an ice bath and treated with acetic acid (36 mL) and water (5.0 mL). The solution was stirred at 50-60 C for 15 hours. The volatiles were evaporated in vacuo and the acetic acid solution was poured into cold water (150 mL) to give a white precipitate. The aqueous mixture was extracted with ethyl acetate (2×100 mL). The ethyl acetate extracts were washed with water (2×), saturated sodium bicarbonate solution, and brine. The combined ethyl acetate extract was dried over sodium sulfate. Evaporation of the solvent gave a yellow foam. Trituration of the foam with acetone/hexane followed by evaporation gave 4.27 g (92.62%) of a light yellow solid. Recrystallization of the solid from acetone/hexanes gave 3.07 g of drospirenone with an HPLC purity of 99.66%. Evaporation of the mother liquor and recrystallization of the residue affords an additional 0.54 g of slightly impure drospirenone.FIG. 3Experimental Example

Lithium hexamethyldisilylamide (LiHMDS) 1.0 M/THF (75.7 mL, 75.7 mmol) was introduced into a 500 mL, 3-neck flask equipped with an addition funnel and a pierced septa for the introduction of a thermocouple probe. The mixture was diluted with THF (25 mL). The solution was stirred (Teflon paddle) and chilled to an internal temperature of −72° C. A THF (75 mL) solution of ketodiol (5 g, 15.13 mmol) containing ethyl propiolate (3.07 mL, 30.26 mmol) was added dropwise over 1 hour while not allowing the temperature to rise above −65° C. Upon completion of the addition the mixture was stirred for 3 hrs while allowing the temperature to warm slowly to −60° C. Finally, the mixture was warmed to −40° C. over 1 hour.The mixture was quenched through the addition of acetic acid (4.25 mL)/water (5.0 mL) followed by the addition of saturated ammonium chloride solution (100 mL). The mixture was stirred for 3 min and then transferred to a separatory funnel. The layers were separated and the upper, THF layer was diluted with ethyl acetate (75 mL). The organic phase was washed with water (3×100 mL) and brine (1×100 mL). All the aqueous washes were extracted with ethyl acetate (2×30 mL). The combined organic extract was dried over sodium sulfate, filtered, and evaporated in vacuo (45° C.) to afford a thick oil. Dichloromethane (ca. 35 mL) was added and evaporated in vacuo. The flask was cooled slightly and dichloromethane (33 ml) was added to give a solid mass. The solid was broken up and stirred until a homogeneous slurry was obtained. Hexanes (35 mL) was added slowly to the stirred mixture and the mixture was stored at 2-4 C overnight. The solid was filtered, washed with 30% dichloromethane/hexanes, and dried in vacuo at ambient temperature for 4 hours to give (2c) 5.86 g (90.4%) of a white powder.

Propiolate adduct (2c) (5.86 g, 13.67 mmol) was suspended in dichloromethane (60 mL). The mixture was stirred vigorously and chilled to −15° C. (NaCl/ice) and TEMPO (54 mg, 0.35 mmol, 2.5 mol %) was added. The mixture was treated dropwise over about 15-20 min. with a mixture of sodium hypochlorite (12.5%) (13.2 mL, 27.34 mmol) in water (8.0 mL) containing potassium bicarbonate (985 mg, 9.84 mmol). During the addition of the hypochlorite solution, a 5-8 C temperature rise was observed and the mixture became yellow. The mixture was allowed to warm to at 0 C for 2 hrs. Analysis of the reaction by TLC (60% EtOAc/hex) shows the appearance of a slightly less polar product (ΔRf=0.8 cm). The mixture was chilled to −5 C and was quenched through the dropwise addition (ca 10-15 min) of a water (150 mL) solution of sodium phosphate (1.50 g, 9.16 mmol) and sodium metabisulfite (1.30 g, 6.84 mmol). Once again, a temperature rise of 5-8 C was observed and the yellow color was quenched. The layers were separated and the dichloromethane solution was washed with water (2×) and brine. All aqueous washes were extracted with additional dichloromethane (2×15 mL). The combined dichloromethane extract was dried over sodium sulfate and the bulk of the solvent was evaporated in vacuo. Upon the observation of solids in the mixture during the evaporation, the evaporation was discontinued and the residue in the flask diluted with MTBE (35 mL). While stirring, the mixture was slowly diluted with hexanes (35 mL). The mixture was then chilled in an ice bath for 30 min. The solid was filtered, washed with 25% MTBE/hexane, and dried to give intermediate (9c) (4.98 g, 85.31%) as a white solid.NMR (CDCl3) δ 0.462 (q, 1H), 0.699 (m, 1H), 0.924 (s, 18-Me), 0.952 (s, 19-Me), 1.338 (t, J=7 Hz, OCH2CH 3), 2.517 (d, 1H), 3.021 (d, 1H), 4.269 (t, OCH 2CH3) ppm. FTIR (ATR): 3493, 3252, 2948, 2226, 1697, 1241 cm−1.
Alkynyl ketone (9c) (5.37 g, 12.59 mmol) was dissolved in THF (27 mL) in a 250 mL shaker bottle. 5% Pd/C (670 mg, 2.5 mol %) was added to the solution and the mixture was shaken under a hydrogen pressure of 15 psi. Over approximately 30 min, there was observed a rapid up take of hydrogen. The pressure was continually adjusted to 15 psi until the uptake of hydrogen ceased and was shaken for a total of 1.5 hrs. The mixture was diluted with a small amount of methanol and filtered through Celite. The filter pad was washed with methanol (ca. 3×25 mL).NMR (CDCl3) δ 0.353 (q, 1H), 0.704 (m, 2H), 0.930 (s, 18-Me), 0.933 (s, 19-Me), 1.279 (t, J=7 Hz, OCH2CH 3), 2.480 (d, 1H), 2.672 (m, 2H), 3.981 (d, 1H), 4.162 (t, OCH 2CH3) ppm. FTIR (ATR): 3465, 2946, 1712, cm−1.

The filtrate containing compound (8c) described above, was added in one portion to a methanol (10 mL) solution containing 8.0 M KOH solution (6.3 mL, 50.36 mmol) preheated to 60 C. The solution was heated at reflux for 2.5 hours. The mixture was chilled in an ice bath and treated with acetic acid (36 mL) and water (5.0 mL). The solution was stirred at 50-60 C for 15 hours. The volatiles were evaporated in vacuo and the acetic acid solution was poured into cold water (150 mL) to give a white precipitate. The aqueous mixture was extracted with ethyl acetate (2×100 mL). The ethyl acetate extracts were washed with water (2×), saturated sodium bicarbonate solution, and brine. The combined ethyl acetate extract was dried over sodium sulfate. Evaporation of the solvent gave a yellow foam. Trituration of the foam with acetone/hexane followed by evaporation gave 4.27 g (92.62%) of a light yellow solid. Recrystallization of the solid from acetone/hexanes gave 3.07 g of drospirenone with an HPLC purity of 99.66%. Evaporation of the mother liquor and recrystallization of the residue affords an additional 0.54 g of slightly impure drospirenone.FIG. 4Experimental Example

Lithium hexamethyldisilylamide (LiHMDS) 1.0 M/THF (75.7 mL, 75.7 mmol) was introduced into a 500 mL, 3-neck flask equipped with an addition funnel and a pierced septa for the introduction of a theiniocouple probe. The mixture was diluted with THF (25 mL). The solution was stirred (Teflon paddle) and chilled to an internal temperature of −72° C. A THF (75 mL) solution of ketodiol (5 g, 15.13 mmol) containing ethyl propiolate (3.07 mL, 30.26 mmol) was added dropwise over 1 hour while not allowing the temperature to rise above −65° C. Upon completion of the addition the mixture was stirred for 3 hrs while allowing the temperature to warm slowly to −60° C. Finally, the mixture was warmed to −40° C. over 1 hour.The mixture was quenched through the addition of acetic acid (4.25 mL)/water (5.0 mL) followed by the addition of saturated ammonium chloride solution (100 mL). The mixture was stirred for 3 min and then transferred to a separatory funnel. The layers were separated and the upper, THF layer was diluted with ethyl acetate (75 mL). The organic phase was washed with water (3×100 mL) and brine (1×100 mL). All the aqueous washes were extracted with ethyl acetate (2×30 mL). The combined organic extract was dried over sodium sulfate, filtered, and evaporated in vacuo (45° C.) to afford a thick oil. Dichloromethane (ca. 35 mL) was added and evaporated in vacuo. The flask was cooled slightly and dichloromethane (33 ml) was added to give a solid mass. The solid was broken up and stirred until a homogeneous slurry was obtained. Hexanes (35 mL) was added slowly to the stirred mixture and the mixture was stored at 2-4 C overnight. The solid was filtered, washed with 30% dichloromethane/hexanes, and dried in vacuo at ambient temperature for 4 hours to give (2d) 5.86 g (90.4%) of a white powder.

Propiolate adduct (2d) (5.86 g, 13.67 mmol) was suspended in dichloromethane (60 mL). The mixture was stirred vigorously and chilled to −15° C. (NaCl/ice) and TEMPO (54 mg, 0.35 mmol, 2.5 mol %) was added. The mixture was treated dropwise over about 15-20 min. with a mixture of sodium hypochlorite (12.5%) (13.2 mL, 27.34 mmol) in water (8.0 mL) containing potassium bicarbonate (985 mg, 9.84 mmol). During the addition of the hypochlorite solution, a 5-8 C temperature rise was observed and the mixture became yellow. The mixture was allowed to warm to at 0 C for 2 hrs. Analysis of the reaction by TLC (60% EtOAc/hex) shows the appearance of a slightly less polar product (ΔRf=0.8 cm). The mixture was chilled to −5 C and was quenched through the dropwise addition (ca 10-15 min) of a water (15.0 mL) solution of sodium phosphate (1.50 g, 9.16 mmol) and sodium metabisulfite (1.30 g, 6.84 mmol). Once again, a temperature rise of 5-8 C was observed and the yellow color was quenched. The layers were separated and the dichloromethane solution was washed with water (2×) and brine. All aqueous washes were extracted with additional dichloromethane (2×15 mL). The combined dichloromethane extract was dried over sodium sulfate and the bulk of the solvent was evaporated in vacuo. Upon the observation of solids in the mixture during the evaporation, the evaporation was discontinued and the residue in the flask diluted with MTBE (35 mL). While stirring, the mixture was slowly diluted with hexanes (35 mL). The mixture was then chilled in an ice bath for 30 min. The solid was filtered, washed with 25% MTBE/hexane, and dried to give intermediate (9d) (4.98 g, 85.31%) as a white solid.NMR (CDCl3) δ 0.462 (q, 1H), 0.699 (m, 1H), 0.924 (s, 18-Me), 0.952 (s, 19-Me), 1.338 (t, J=7 Hz, OCH2CH 3), 2.517 (d, 1H), 3.021 (d, 1H), 4.269 (t, OCH 2CH3) ppm. FTIR (ATR): 3493, 3252, 2948, 2226, 1697, 1241 cm−1.

Compound (9d) (5.0 g) was dissolved in methanol (50 mL) and treated with 1.0 N sulfuric acid (10 mL). The mixture was heated to reflux for 3 hours, cooled, and neutralized through the addition of saturated sodium bicarbonate solution. Most of the methanol was evaporated in vacuo at ambient temperature and diluted with water. The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water and brine, dried over sodium sulfate, filtered, and evaporated to give 4.95 g of unsaturated ketone (10d) as a stable foam.

Compound (10d) (5.0 g, 12.24 mmol) was dissolved in degassed benzene (50 mL) and treated with chlorotris(triphenylphosphine)rhodium (I) (283.1 mg, 0.31 mmol and the resulting mixture was stirred in a hydrogen atmosphere for 10 hours. The solution was evaporated, reconstituted in 50% ethyl acetate/hexanes, and passed through a short column of neutral alumina. Evaporation of the solvent gave 4.95 g of (11d) as a stable foam.

Compound (11d) (4.95 g, 12.01 mmol) was dissolved in 10% aqueous methanol (50 mL) and solid potassium carbonate (4.98 g, 36.04 mmol) was added. The mixture was stirred at room temperature for 30 min and the bicarbonate was neutralized through the addition of acetic acid (2.06 mL, 36.04 mmol). The methanol was evaporated in vacuo at ambient temperature and diluted with water. The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water and brine, dried over sodium sulfate, filtered, and evaporated to give 4.10 g (93%) of a semi solid. The material was dissolved in dichloromethane and evaporated in vacuo to give a stable foam. The foam was dissolved in ethyl acetate (5 mL) and allowed to stand overnight. The resulting solid was filtered, washed with cold ethyl acetate, and dried in vacuo to afford 2.86 g (66%) of pure drospirenone.FIG. 5Experimental Example 1

A dichloromethane (50 mL) solution of ketodiol (1) (5.0 g, 15.13 mmol) was treated with ethyl vinyl ether (7.24 mL, 75.65 mmol), followed by the addition of pyridinium tosylate (380 mg, 1.15 mmol). The solution was stirred at room temperature for 30 min. The dichloromethane solution was washed with water (2×), brine, and dried over sodium sulfate. Following filtration, evaporation of the solvent gave 6.14 g of the 3-protected compound (1e) as a stable foam.
Compound (1e) (6.14 g, 15.13 mmol) was dissolved in DMSO/THF (15 mL/15 mL), treated with trimethylsulfonium iodide (4.63 g, 22.70 mmol) and the mixture was chilled to −15 C. The mixture was treated portion wise with potassium t-butoxide (3.23 g, 28.82 mmol). The mixture was stirred at −15 C for 45 min and then poured into ice/water (200 mL). The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 6.21 g (98.42%) of oxirane (12e) as a stable foam.

A THF (30 mL) solution of di-isopropyl amine (12.02 mL, 85.03 mmol) was chilled to −40 C and treated with butyl lithium (2.5 M/hexanes, 34.01 mL, 85.03 mmol) and the mixture was stirred for 15 min. A THF (5 mL) solution of acetonitrile (4.7 mL, 90.79 mmol) was added dropwise to the in situ generated lithium di-isopropylamide (LDA) solution to give a slurry of the acetonitrile anion. After stirring for 15 min at −40° C., compound (12e) (6.21 g, 14.91 mmol) as a THF (25 mL) solution was added dropwise over 10 min. The mixture was stirred for 30 min and then quenched through the addition of saturated ammonium chloride solution (210 mL). The mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (3×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 7.07 g of the addition product (13e) as a tacky foam.

Compound 13e (5.0 g, 10.93 mmol) was dissolved in acetone (25 mL) and chilled to 0 C. The stirred solution was treated dropwise with 2.7M chromic acid (Jones Reagent) (7.0 mL, 18.91 mmol). After 1.5 hrs, the excess Cr (VI) was quenched through the addition of 2-propanol until the green color of Cr (IV) was evident. Water (300 mL) was added and the mixture was stirred until all the chromium salts were dissolved. The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 3.83 g (96%) of ketone (14e) as a stable foam.

Compound (14e) (3.83 g, 10.48 mmol) was dissolved in methanol (38 mL) and treated with 8.0 M KOH solution (7.0 mL, 56 mmol) and the mixture was heated at reflux for 5 hours. The mixture was cooled to 0 C and treated with acetic acid (15 mL) and water (6 mL) and the mixture was stirred at 50 C for 6 hours. The solvents were evaporated in vacuo and the residue was diluted with water (200 mL). The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 3.76 g (94%) of crude drospirenone as a stable foam. The crude drospirenone was dissolved in 60% ethyl acetate/hexanes and passed through a short column of neutral alumina (10× w/w) and the column was eluted with the same solvent. Following evaporation of the solvent, 2.58 g (65%) of crystalline drospirenone was obtained. Recrystallization from acetone/hexanes afforded pure drospirenone.FIG. 5Experimental Example 2

Intermediate (1) (5 g, 15.13 mmol) was dissolved in DMSO/THF (50 mL/50 mL), treated with trimethylsulfonium iodide (4.63 g, 22.70 mmol), and the mixture was chilled to −15 C. The mixture was treated portion wise with potassium t-butoxide (5.03 g, 43.88 mmol). The mixture was stirred at −15 C for 45 min and then poured into ice/water (200 mL). The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 5.11 g (98%) of oxirane (12f) as a stable foam.

A THF (30 mL) solution of di-isopropyl amine (12.02 mL, 85.03 mmol) was chilled to −40 C and treated with butyl lithium (2.5 M/hexanes, 34.01 mL, 85.03 mmol) and the mixture was stirred for 15 min. A THF (5 mL) solution of acetonitrile (4.7 mL, 90.79 mmol) was added dropwise to the above lithium di-isopropylamide (LDA) solution to give a slurry of the acetonitrile anion. After stirring for 15 min at −40 C, compound (12f) (5.11 g, 14.83 mmol) as a THF (75 mL) solution was added dropwise over 10 min. The mixture was stirred for 30 min and then quenched through the addition of saturated ammonium chloride solution (300 mL). The mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (3×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 5.75 g of addition product (13f) as a tacky foam.

Compound 13f (5.75 g, 14.95 mmol) was dissolved in acetone (25 mL) and chilled to 0 C. The stirred solution was treated dropwise with 2.7M chromic acid (Jones Reagent) until the orange color of Cr (VI) persisted. After 1.5 hrs, the excess Cr (VI) was quenched through the addition of 2-propanol until the green color of Cr (IV) was evident. Water (300 mL) was added and the mixture was stirred until all the chromium salts were dissolved. The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 5.5 g (96%) of ketone (14f) as a stable foam.
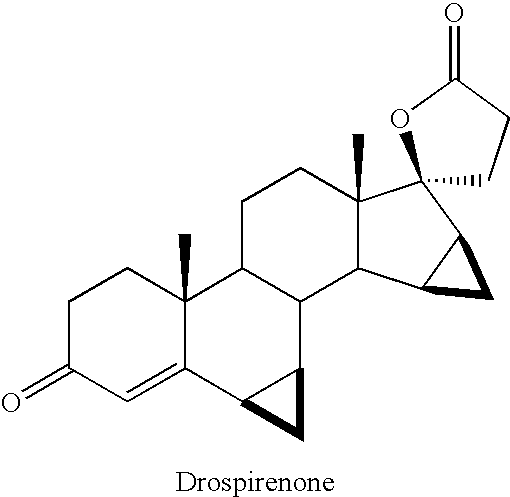
Compound (14f) (5.5 g, 14.38 mmol) was dissolved in methanol (50 mL) and treated with 8.0 M KOH solution (9.35 mL, 74.77 mmol) and the mixture was heated at reflux for 5 hours. The mixture was cooled to 0 C and treated with acetic acid (25 mL) and water (10 mL) and the mixture was stirred at 50 C for 6 hours. The solvents were evaporated in vacuo and the residue was diluted with water (300 mL). The aqueous mixture was extracted with ethyl acetate. The ethyl acetate extract was washed with water (2×) and brine, dried over sodium sulfate, and filtered. Evaporation of the solvent gave 4.95 g (94%) of crude drospirenone as a stable foam. The crude drospirenone was dissolved in 60% ethyl acetate/hexanes and passed through a short column of neutral alumina (10× w/w) and the column was eluted with the same solvent. Following evaporation of the solvent, 3.43 g (65%) of crystalline drospirenone was obtained. Recrystallization from acetone/hexanes afforded pure drospirenone.
Drospirenone is a progestin medication which is used in birth control pills to prevent pregnancy and in menopausal hormone therapy, among other uses.[1][7] It is available both alone under the brand name Slynd and in combination with an estrogen under the brand name Yasmin among others.[7][2] The medication is taken by mouth.[2][1]
Common side effects include acne, headache, breast tenderness, weight increase, and menstrual changes.[2] Rare side effects may include high potassium levels and blood clots, among others.[2][8] Drospirenone is a progestin, or a synthetic progestogen, and hence is an agonist of the progesterone receptor, the biological target of progestogens like progesterone.[1] It has additional antimineralocorticoid and antiandrogenic activity and no other important hormonal activity.[1] Because of its antimineralocorticoid activity and lack of undesirable off-target activity, drospirenone is said to more closely resemble bioidentical progesterone than other progestins.[9][10]
Drospirenone was patented in 1976 and introduced for medical use in 2000.[11][12] It is available widely throughout the world.[7] The medication is sometimes referred to as a “fourth-generation” progestin.[13][14] It is available as a generic medication.[15] In 2018, a formulation of drospirenone with ethinylestradiol was the 167th most commonly prescribed medication in the United States, with more than 3 million prescriptions.[16][17]
Medical uses
Drospirenone (DRSP) is used by itself as a progestogen-only birth control pill, in combination with the estrogens ethinylestradiol (EE) or estetrol (E4), with or without supplemental folic acid (vitamin B9), as a combined birth control pill, and in combination with the estrogen estradiol (E2) for use in menopausal hormone therapy.[2] A birth control pill with low-dose ethinylestradiol is also indicated for the treatment of moderate acne, premenstrual syndrome (PMS), premenstrual dysphoric disorder (PMDD), and dysmenorrhea (painful menstruation).[18][19] For use in menopausal hormone therapy, E2/DRSP is specifically approved to treat moderate to severe vasomotor symptoms (hot flashes), vaginal atrophy, and postmenopausal osteoporosis.[20][21][22] The drospirenone component in this formulation is included specifically to prevent estrogen-induced endometrial hyperplasia.[23] Drospirenone has also been used in combination with an estrogen as a component of hormone therapy for transgender women.[24][25]
Studies have found that EE/DRSP is superior to placebo in reducing premenstrual emotional and physical symptoms while also improving quality of life.[26][27] E2/DRSP has been found to increase bone mineral density and to reduce the occurrence of bone fractures in postmenopausal women.[28][23][29][30] In addition, E2/DRSP has a favorable influence on cholesterol and triglyceride levels and decreases blood pressure in women with high blood pressure.[29][30] Due to its antimineralocorticoid activity, drospirenone opposes estrogen-induced salt and water retention and maintains or slightly reduces body weight.[31]
Available forms
Drospirenone is available in the following formulations, brand names, and indications:[32][33]
- Drospirenone 4 mg (Slynd) – progestogen-only birth control pill[2]
- Drospirenone 3 mg and estetrol 14.2 mg (Nextstellis (US)) – combined birth control pill[34][35][36]
- Ethinylestradiol 30 μg and drospirenone 3 mg (Ocella, Syeda, Yasmin, Zarah, Zumandimine) – combined birth control pill[37][38][39][40]
- Ethinylestradiol 20 μg and drospirenone 3 mg (Gianvi, Jasmiel, Loryna, Lo-Zumandimine, Nikki, Vestura, Yaz) – combined birth control pill, acne, PMS, PMDD, dysmenorrhea[18]
- Ethinylestradiol 30 μg, drospirenone 3 mg, and levomefolate calcium 0.451 mg (Beyaz, Tydemy) – combined birth control pill with vitamin B9 supplementation, acne, PMS[41][42]
- Estetrol 15 mg and drospirenone 3 mg (Nextstellis (CA)) – combined birth control pill[43][44]
- Estradiol 0.5 or 1 mg and drospirenone 0.25 or 0.5 mg (Angeliq) – menopausal hormone therapy (menopausal syndrome, postmenopausal osteoporosis)[20]
Contraindications
Contraindications of drospirenone include renal impairment or chronic kidney disease, adrenal insufficiency, presence or history of cervical cancer or other progestogen-sensitive cancers, benign or malignant liver tumors or hepatic impairment, undiagnosed abnormal uterine bleeding, and hyperkalemia (high potassium levels).[2][45][46] Renal impairment, hepatic impairment, and adrenal insufficiency are contraindicated because they increase exposure to drospirenone and/or increase the risk of hyperkalemia with drospirenone.[2]
Side effects
Adverse effects of drospirenone alone occurring in more than 1% of women may include unscheduled menstrual bleeding (breakthrough or intracyclic) (40.3–64.4%), acne (3.8%), metrorrhagia (2.8%), headache (2.7%), breast pain (2.2%), weight gain (1.9%), dysmenorrhea (1.9%), nausea (1.8%), vaginal hemorrhage (1.7%), decreased libido (1.3%), breast tenderness (1.2%), and irregular menstruation (1.2%).[2]
High potassium levels
Drospirenone is an antimineralocorticoid with potassium-sparing properties, though in most cases no increase of potassium levels is to be expected.[45] In women with mild or moderate chronic kidney disease, or in combination with chronic daily use of other potassium-sparing medications (ACE inhibitors, angiotensin II receptor antagonists, potassium-sparing diuretics, heparin, antimineralocorticoids, or nonsteroidal anti-inflammatory drugs), a potassium level should be checked after two weeks of use to test for hyperkalemia.[45][47] Persistent hyperkalemia that required discontinuation occurred in 2 out of around 1,000 women (0.2%) with 4 mg/day drospirenone alone in clinical trials.[2]
Blood clots
Birth control pills containing ethinylestradiol and a progestin are associated with an increased risk of venous thromboembolism (VTE), including deep vein thrombosis (DVT) and pulmonary embolism (PE).[48] The incidence is about 4-fold higher on average than in women not taking a birth control pill.[48] The absolute risk of VTE with ethinylestradiol-containing birth control pills is small, in the area of 3 to 10 out of 10,000 women per year, relative to 1 to 5 out of 10,000 women per year not taking a birth control pill.[49][50] The risk of VTE during pregnancy is 5 to 20 in 10,000 women per year and during the postpartum period is 40 to 65 per 10,000 women per year.[50] The higher risk of VTE with combined birth control pills is thought to be due to the ethinylestradiol component, as ethinylestradiol has estrogenic effects on liver synthesis of coagulation factors which result in a procoagulatory state.[8] In contrast to ethinylestradiol-containing birth control pills, neither progestogen-only birth control nor the combination of transdermal estradiol and an oral progestin in menopausal hormone therapy is associated with an increased risk of VTE.[8][51]
Different progestins in ethinylestradiol-containing birth control pills have been associated with different risks of VTE.[8] Birth control pills containing progestins such as desogestrel, gestodene, drospirenone, and cyproterone acetate have been found to have 2- to 3-fold the risk of VTE of birth control pills containing levonorgestrel in retrospective cohort and nested case–control observational studies.[8][49] However, this area of research is controversial, and confounding factors may have been present in these studies.[8][49][52] Other observational studies, specifically prospective cohort and case control studies, have found no differences in risk between different progestins, including between birth control pills containing drospirenone and birth control pills containing levonorgestrel.[8][49][52][53] These kinds of observational studies have certain advantages over the aforementioned types of studies, like better ability to control for confounding factors.[53] Systematic reviews and meta-analyses of all of the data in the mid-to-late 2010s found that birth control pills containing cyproterone acetate, desogestrel, drospirenone, or gestodene overall were associated with a risk of VTE of about 1.3- to 2.0-fold compared to that of levonorgestrel-containing birth control pills.[54][55][49]
Androgenic progestins have been found to antagonize to some degree the effects of ethinylestradiol on coagulation.[56][57][58][59] As a result, more androgenic progestins, like levonorgestrel and norethisterone, may oppose the procoagulatory effects of ethinylestradiol and result in a lower increase in risk of VTE.[8][60] Conversely, this would be the case less or not at all with progestins that are less androgenic, like desogestrel and gestodene, as well as progestins that are antiandrogenic, like drospirenone and cyproterone acetate.[8][60]
In the early 2010s, the FDA updated the label for birth control pills containing drospirenone and other progestins to include warnings for stopping use prior to and after surgery, and to warn that such birth control pills may have a higher risk of blood clots.[46]
Breast cancer
Drospirenone has been found to stimulate the proliferation and migration of breast cancer cells in preclinical research, similarly to certain other progestins.[61][62] However, some evidence suggests that drospirenone may do this more weakly than certain other progestins, like medroxyprogesterone acetate.[61][62] The combination of estradiol and drospirenone has been found to increase breast density, an established risk factor for breast cancer, in postmenopausal women.[63][64][65]
Data on risk of breast cancer in women with newer progestins like drospirenone are lacking at present.[66] Progestogen-only birth control is not generally associated with a higher risk of breast cancer.[66] Conversely, combined birth control and menopausal hormone therapy with an estrogen and a progestogen are associated with higher risks of breast cancer.[67][66][68]
Overdose
These have been no reports of serious adverse effects with overdose of drospirenone.[2] Symptoms that may occur in the event of an overdose may include nausea, vomiting, and vaginal bleeding.[2] There is no antidote for overdose of drospirenone and treatment of overdose should be based on symptoms.[2] Since drospirenone has antimineralocorticoid activity, levels of potassium and sodium should be measured and signs of metabolic acidosis should be monitored.[2]
Interactions
Inhibitors and inducers of the cytochrome P450 enzyme CYP3A4 may influence the levels and efficacy of drospirenone.[2] Treatment for 10 days with 200 mg twice daily ketoconazole, a strong CYP3A4 inhibitor among other actions, has been found to result in a moderate 2.0- to 2.7-fold increase in exposure to drospirenone.[2] Drospirenone does not appear to influence the metabolism of omeprazole (metabolized via CYP2C19), simvastatin (metabolized via CYP3A4), or midazolam (metabolized via CYP3A4), and likely does not influence the metabolism of other medications that are metabolized via these pathways.[2] Drospirenone may interact with potassium-sparing medications such as ACE inhibitors, angiotensin II receptor antagonists, potassium-sparing diuretics, potassium supplements, heparin, antimineralocorticoids, and nonsteroidal anti-inflammatory drugs to further increase potassium levels.[2] This may increase the risk of hyperkalemia (high potassium levels).[2]
Pharmacology
Pharmacodynamics
Drospirenone binds with high affinity to the progesterone receptor (PR) and mineralocorticoid receptor (MR), with lower affinity to the androgen receptor (AR), and with very low affinity to the glucocorticoid receptor (GR).[1][69][70][4] It is an agonist of the PR and an antagonist of the MR and AR, and hence is a progestogen, antimineralocorticoid, and antiandrogen.[1][69][4][62] Drospirenone has no estrogenic activity and no appreciable glucocorticoid or antiglucocorticoid activity.[1][69][4][62]
Progestogenic activity
Drospirenone is an agonist of the PR, the biological target of progestogens like progesterone.[1][69] It has about 35% of the affinity of promegestone for the PR and about 19 to 70% of the affinity of progesterone for the PR.[1][3][62] Drospirenone has antigonadotropic and functional antiestrogenic effects as a result of PR activation.[1][69] The ovulation-inhibiting dosage of drospirenone is 2 to 3 mg/day.[72][73][1][74] Inhibition of ovulation occurred in about 90% of women at a dose of 0.5 to 2 mg/day and in 100% of women at a dose of 3 mg/day.[75] The total endometrial transformation dose of drospirenone is about 50 mg per cycle, whereas its daily dose is 2 mg for partial transformation and 4 to 6 mg for full transformation.[1][76][75] The medication acts as a contraceptive by activating the PR, which suppresses the secretion of luteinizing hormone, inhibits ovulation, and alters the cervical membrane and endometrium.[77][2]
Due to its antigonadotropic effects, drospirenone inhibits the secretion of the gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), and suppresses gonadal sex hormone production, including of estradiol, progesterone, and testosterone.[1][78][3] Drospirenone alone at 4 mg/day has been found to suppress estradiol levels in premenopausal women to about 40 to 80 pg/mL depending on the time of the cycle.[78] No studies of the antigonadotropic effects of drospirenone or its influence on hormone levels appear to have been conducted in men.[79][80][81] In male cynomolgus monkeys however, 4 mg/kg/day oral drospirenone strongly suppressed testosterone levels.[69]
Antimineralocorticoid activity
Drospirenone is an antagonist of the MR, the biological target of mineralocorticoids like aldosterone, and hence is an antimineralocorticoid.[69] It has about 100 to 500% of the affinity of aldosterone for the MR and about 50 to 230% of the affinity of progesterone for the MR.[1][3][71][62] Drospirenone is about 5.5 to 11 times more potent as an antimineralocorticoid than spironolactone in animals.[69][75][82] Accordingly, 3 to 4 mg drospirenone is said to be equivalent to about 20 to 25 mg spironolactone in terms of antimineralocorticoid activity.[83][2] It has been said that the pharmacological profile of drospirenone more closely resembles that of progesterone than other progestins due to its antimineralocorticoid activity.[69] Drospirenone is the only clinically used progestogen with prominent antimineralocorticoid activity besides progesterone.[1] For comparison to progesterone, a 200 mg dose of oral progesterone is considered to be approximately equivalent in antimineralocorticoid effect to a 25 to 50 mg dose of spironolactone.[84] Both drospirenone and progesterone are actually weak partial agonists of the MR in the absence of mineralocorticoids.[4][3][62]
Due to its antimineralocorticoid activity, drospirenone increases natriuresis, decreases water retention and blood pressure, and produces compensatory increases in plasma renin activity as well as circulating levels and urinary excretion of aldosterone.[3][85][1] This has been shown to occur at doses of 2 to 4 mg/day.[3] Similar effects occur during the luteal phase of the menstrual cycle due to increased progesterone levels and the resulting antagonism of the MR.[3] Estrogens, particularly ethinylestradiol, activate liver production of angiotensinogen and increase levels of angiotensinogen and angiotensin II, thereby activating the renin–angiotensin–aldosterone system.[3][1] As a result, they can produce undesirable side effects including increased sodium excretion, water retention, weight gain, and increased blood pressure.[3] Progesterone and drospirenone counteract these undesirable effects via their antimineralocorticoid activity.[3] Accumulating research indicates that antimineralocorticoids like drospirenone and spironolactone may also have positive effects on adipose tissue and metabolic health.[86][87]
Antiandrogenic activity
Drospirenone is an antagonist of the AR, the biological target of androgens like testosterone and dihydrotestosterone (DHT).[1][3] It has about 1 to 65% of the affinity of the synthetic anabolic steroid metribolone for the AR.[1][3][4][62] The medication is more potent as an antiandrogen than spironolactone, but is less potent than cyproterone acetate, with about 30% of its antiandrogenic activity in animals.[1][88][69][75] Progesterone displays antiandrogenic activity in some assays similarly to drospirenone,[3] although this issue is controversial and many researchers regard progesterone as having no significant antiandrogenic activity.[89][1][4]
Drospirenone shows antiandrogenic effects on the serum lipid profile, including higher HDL cholesterol and triglyceride levels and lower LDL cholesterol levels, at a dose of 3 mg/day in women.[3] The medication does not inhibit the effects of ethinylestradiol on sex hormone-binding globulin (SHBG) and serum lipids, in contrast to androgenic progestins like levonorgestrel but similarly to other antiandrogenic progestins like cyproterone acetate.[3][1][74] SHBG levels are significantly higher with ethinylestradiol and cyproterone acetate than with ethinylestradiol and drospirenone, owing to the more potent antiandrogenic activity of cyproterone acetate relative to drospirenone.[90] Androgenic progestins like levonorgestrel have been found to inhibit the procoagulatory effects of estrogens like ethinylestradiol on hepatic synthesis of coagulation factors, whereas this may occur less or not at all with weakly androgenic progestins like desogestrel and antiandrogenic progestins like drospirenone.[8][60][56][57][58][59]
Other activity
Drospirenone stimulates the proliferation of MCF-7 breast cancer cells in vitro, an action that is independent of the classical PRs and is instead mediated via the progesterone receptor membrane component-1 (PGRMC1).[91] Certain other progestins act similarly in this assay, whereas progesterone acts neutrally.[91] It is unclear if these findings may explain the different risks of breast cancer observed with progesterone and progestins in clinical studies.[66]
Pharmacokinetics
Absorption
The oral bioavailability of drospirenone is between 66 and 85%.[1][3][4] Peak levels occur 1 to 6 hours after an oral dose.[1][3][2][82] Levels are about 27 ng/mL after a single 4 mg dose.[2] There is 1.5- to 2-fold accumulation in drospirenone levels with continuous administration, with steady-state levels of drospirenone achieved after 7 to 10 days of administration.[1][2][3] Peak levels of drospirenone at steady state with 4 mg/day drospirenone are about 41 ng/mL.[2] With the combination of 30 μg/day ethinylestradiol and 3 mg/day drospirenone, peak levels of drospirenone after a single dose are 35 ng/mL, and levels at steady state are 60 to 87 ng/mL at peak and 20 to 25 ng/mL at trough.[3][1] The pharmacokinetics of oral drospirenone are linear with a single dose across a dose range of 1 to 10 mg.[2][3] Intake of drospirenone with food does not influence the absorption of drospirenone.[2]
Distribution
The distribution half-life of drospirenone is about 1.6 to 2 hours.[3][1] The apparent volume of distribution of drospirenone is approximately 4 L/kg.[2] The plasma protein binding of drospirenone is 95 to 97%.[2][1] It is bound to albumin and 3 to 5% circulates freely or unbound.[2][1] Drospirenone has no affinity for sex hormone-binding globulin (SHBG) or corticosteroid-binding globulin (CBG), and hence is not bound by these plasma proteins in the circulation.[1]
Metabolism
The metabolism of drospirenone is extensive.[3] It is metabolized into the acid form of drospirenone by opening of its lactone ring.[1][2] The medication is also metabolized by reduction of its double bond between the C4 and C5 positions and subsequent sulfation.[1][2] The two major metabolites of drospirenone are drospirenone acid and 4,5-dihydrodrospirenone 3-sulfate, and are both formed independently of the cytochrome P450 system.[2][3] Neither of these metabolites are known to be pharmacologically active.[2] Drospirenone also undergoes oxidative metabolism by CYP3A4.[2][3][5][6]
Elimination
Drospirenone is excreted in urine and feces, with slightly more excreted in feces than in urine.[2] Only trace amounts of unchanged drospirenone can be found in urine and feces.[2] At least 20 different metabolites can be identified in urine and feces.[3] Drospirenone and its metabolites are excreted in urine about 38% as glucuronide conjugates, 47% as sulfate conjugates, and less than 10% in unconjugated form.[3] In feces, excretion is about 17% glucuronide conjugates, 20% sulfate conjugates, and 33% unconjugated.[3]
The elimination half-life of drospirenone is between 25 and 33 hours.[2][3][1] The half-life of drospirenone is unchanged with repeated administration.[2] Elimination of drospirenone is virtually complete 10 days after the last dose.[3][2]
Chemistry
See also: Spirolactone, List of progestogens § Spirolactone derivatives, and List of steroidal antiandrogens § Spirolactone derivatives
vteChemical structures of spirolactones ProgesteroneSpirolactoneCanrenoneSpironolactoneDrospirenoneSpirorenone ProgesteroneSpirolactoneCanrenoneSpironolactoneDrospirenoneSpirorenone |
Drospirenone, also known as 1,2-dihydrospirorenone or as 17β-hydroxy-6β,7β:15β,16β-dimethylene-3-oxo-17α-pregn-4-ene-21-carboxylic acid, γ-lactone, is a synthetic steroidal 17α-spirolactone, or more simply a spirolactone.[7][92] It is an analogue of other spirolactones like spironolactone, canrenone, and spirorenone.[7][92] Drospirenone differs structurally from spironolactone only in that the C7α acetylthio substitution of spironolactone has been removed and two methylene groups have been substituted in at the C6β–7β and C15β–16β positions.[93]
Spirolactones like drospirenone and spironolactone are derivatives of progesterone, which likewise has progestogenic and antimineralocorticoid activity.[94][95][96] The loss of the C7α acetylthio group of spironolactone, a compound with negligible progestogenic activity,[97][98] appears to be involved in the restoration of progestogenic activity in drospirenone, as SC-5233, the analogue of spironolactone without a C7α substitution, has potent progestogenic activity similarly to drospirenone.[99]
History
Drospirenone was patented in 1976 and introduced for medical use in 2000.[11][12] Schering AG of Germany has been granted several patents on the production of drospirenone, including WIPO and US patents, granted in 1998 and 2000, respectively.[100][101] It was introduced for medical use in combination with ethinylestradiol as a combined birth control pill in 2000.[11] Drospirenone is sometimes described as a “fourth-generation” progestin based on its time of introduction.[13][14] The medication was approved for use in menopausal hormone therapy in combination with estradiol in 2005.[20] Drospirenone was introduced for use as a progestogen-only birth control pill in 2019.[2] A combined birth control pill containing estetrol and drospirenone was approved in 2021.[102]
Society and culture
Generic names
Drospirenone is the generic name of the drug and its INN, USAN, BAN, and JAN, while drospirénone is its DCF.[7] Its name is a shortened form of the name 1,2-dihydrospirorenone or dihydrospirenone.[7][92] Drospirenone is also known by its developmental code names SH-470 and ZK-30595 (alone), BAY 86-5300, BAY 98-7071, and SH-T-00186D (in combination with ethinylestradiol), BAY 86-4891 (in combination with estradiol), and FSN-013 (in combination with estetrol).[7][92][103][104][105][106][102]
Brand names
Drospirenone is marketed in combination with an estrogen under a variety of brand names throughout the world.[7] Among others, it is marketed in combination with ethinylestradiol under the brand names Yasmin and Yaz, in combination with estetrol under the brand name Nextstellis, and in combination with estradiol under the brand name Angeliq.[7][102]
Availability
See also: List of progestogens available in the United States
Drospirenone is marketed widely throughout the world.[7]
Generation
Drospirenone has been categorized as a “fourth-generation” progestin.[62]
Litigation
Many lawsuits have been filed against Bayer, the manufacturer of drospirenone, due to the higher risk of venous thromboembolism (VTE) that has been observed with combined birth control pills containing drospirenone and certain other progestins relative to the risk with levonorgestrel-containing combined birth control pills.[52]
In July 2012, Bayer notified its stockholders that there were more than 12,000 such lawsuits against the company involving Yaz, Yasmin, and other birth control pills with drospirenone.[107] They also noted that the company by then had settled 1,977 cases for US$402.6 million, for an average of US$212,000 per case, while setting aside US$610.5 million to settle the others.[107]
As of July 17, 2015, there have been at least 4,000 lawsuits and claims still pending regarding VTE related to drospirenone.[108] This is in addition to around 10,000 claims that Bayer has already settled without admitting liability.[108] These claims of VTE have amounted to US$1.97 billion.[108] Bayer also reached a settlement for arterial thromboembolic events, including stroke and heart attacks, for US$56.9 million.[108]
Research
See also: Estetrol/drospirenone and Ethinylestradiol/drospirenone/prasterone
A combination of ethinylestradiol, drospirenone, and prasterone is under development by Pantarhei Bioscience as a combined birth control pill for prevention of pregnancy in women.[109] It includes prasterone (dehydroepiandrosterone; DHEA), an oral androgen prohormone, to replace testosterone and avoid testosterone deficiency caused by suppression of testosterone by ethinylestradiol and drospirenone.[109] As of August 2018, the formulation is in phase II/III clinical trials.[109]
Drospirenone has been suggested for potential use as a progestin in male hormonal contraception.[79]
Drospirenone has been studied in forms for parenteral administration.[110][111][112][113]
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai ajak al Kuhl, H (2005). “Pharmacology of estrogens and progestogens: influence of different routes of administration”. Climacteric. 8 (sup1): 3–63. doi:10.1080/13697130500148875. ISSN 1369-7137. PMID 16112947. S2CID 24616324.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai ajak al am an ao ap aq ar as at au “Slynd- drospirenone tablet, film coated”. DailyMed. Retrieved 17 April 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ahKrattenmacher, Rolf (2000). “Drospirenone: pharmacology and pharmacokinetics of a unique progestogen”. Contraception. 62 (1): 29–38. doi:10.1016/S0010-7824(00)00133-5. ISSN 0010-7824. PMID 11024226.
- ^ Jump up to:a b c d e f g h i Stanczyk FZ, Hapgood JP, Winer S, Mishell DR (April 2013). “Progestogens used in postmenopausal hormone therapy: differences in their pharmacological properties, intracellular actions, and clinical effects”. Endocr. Rev. 34 (2): 171–208. doi:10.1210/er.2012-1008. PMC 3610676. PMID 23238854.
- ^ Jump up to:a b Bachmann, Gloria (2009). “Drospirenone/ethinyl estradiol 3 mg/20 μg (24/4 day regimen): hormonal contraceptive choices – use of a fourth-generation progestin”. Patient Preference and Adherence. 3: 259–64. doi:10.2147/PPA.S3901. ISSN 1177-889X. PMC 2778416. PMID 19936169.
- ^ Jump up to:a b Wiesinger, Herbert; Berse, Matthias; Klein, Stefan; Gschwend, Simone; Höchel, Joachim; Zollmann, Frank S.; Schütt, Barbara (2015). “Pharmacokinetic interaction between the CYP3A4 inhibitor ketoconazole and the hormone drospirenone in combination with ethinylestradiol or estradiol”. British Journal of Clinical Pharmacology. 80 (6): 1399–1410. doi:10.1111/bcp.12745. ISSN 0306-5251. PMC 4693482. PMID 26271371.
- ^ Jump up to:a b c d e f g h i j k “Drospirenone”.
- ^ Jump up to:a b c d e f g h i j Han L, Jensen JT (December 2015). “Does the Progestogen Used in Combined Hormonal Contraception Affect Venous Thrombosis Risk?”. Obstet. Gynecol. Clin. North Am. 42(4): 683–98. doi:10.1016/j.ogc.2015.07.007. PMID 26598309.
- ^ Oelkers W (December 2000). “Drospirenone–a new progestogen with antimineralocorticoid activity, resembling natural progesterone”. Eur J Contracept Reprod Health Care. 5 Suppl 3: 17–24. PMID 11246598.
- ^ Oelkers W (December 2002). “Antimineralocorticoid activity of a novel oral contraceptive containing drospirenone, a unique progestogen resembling natural progesterone”. Eur J Contracept Reprod Health Care. 7 Suppl 3: 19–26, discussion 42–3. PMID 12659403.
- ^ Jump up to:a b c Enrique Ravina (11 January 2011). The Evolution of Drug Discovery: From Traditional Medicines to Modern Drugs. John Wiley & Sons. pp. 193–. ISBN 978-3-527-32669-3.
- ^ Jump up to:a b Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 459. ISBN 9783527607495.
- ^ Jump up to:a b Robert Anthony Hatcher; Anita L. Nelson, M.D. (2007). Contraceptive Technology. Ardent Media. pp. 196–. ISBN 978-1-59708-001-9.
- ^ Jump up to:a b James Q. Del Rosso; Joshua A. Zeichner (20 April 2016). Advances in Acne Management, An Issue of Dermatologic Clinics, E-Book. Elsevier Health Sciences. pp. 160–. ISBN 978-0-323-41753-2.
- ^ “Generic Yasmin Availability”.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Drospirenone; Ethinyl Estradiol – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Jump up to:a b “Yaz- drospirenone and ethinyl estradiol kit”. DailyMed. Retrieved 17 April 2021.
- ^ Cerner Multum, Inc. (June 11, 2012). “drospirenone and ethinyl estradiol”. Auckland, New Zealand: Drugs.com. Retrieved October 24, 2011.
- ^ Jump up to:a b c “Angeliq- drospirenone and estradiol tablet, film coated”. DailyMed. Retrieved 17 April 2021.
- ^ Maclennan, A. H.; Broadbent, J. L.; Lester, S.; Moore, V. (18 October 2004). “Oral oestrogen and combined oestrogen/progestogen therapy versus placebo for hot flushes”. The Cochrane Database of Systematic Reviews (4): CD002978. doi:10.1002/14651858.CD002978.pub2. ISSN 1469-493X. PMC 7004247. PMID 15495039.
- ^ Torgerson, D. J.; Bell-Syer, S. E. (13 June 2001). “Hormone replacement therapy and prevention of nonvertebral fractures: a meta-analysis of randomized trials”. JAMA. 285 (22): 2891–2897. doi:10.1001/jama.285.22.2891. ISSN 0098-7484. PMID 11401611. S2CID 25078579.
- ^ Jump up to:a b Whitehead M (March 2006). “Hormone replacement therapy with estradiol and drospirenone: an overview of the clinical data”. J Br Menopause Soc. 12 Suppl 1: 4–7. doi:10.1258/136218006775992185. PMID 16513012. S2CID 38095916.
- ^ Majumder A, Sanyal D (2017). “Outcome and preferences in male-to-female subjects with gender dysphoria: Experience from Eastern India”. Indian J Endocrinol Metab. 21 (1): 21–25. doi:10.4103/2230-8210.196000. PMC 5240066. PMID 28217493.
- ^ Majumder, Anirban; Chatterjee, Sudip; Maji, Debasis; Roychaudhuri, Soumyabrata; Ghosh, Sujoy; Selvan, Chitra; George, Belinda; Kalra, Pramila; Maisnam, Indira; Sanyal, Debmalya (2020). “IDEA group consensus statement on medical management of adult gender incongruent individuals seeking gender reaffirmation as female”. Indian Journal of Endocrinology and Metabolism. 24 (2): 128–135. doi:10.4103/ijem.IJEM_593_19. ISSN 2230-8210. PMC 7333765. PMID 32699777.
- ^ Lanza di Scalea, Teresa (June 2017). “Premenstrual Dysphoric Disorder”. Psychiatric Clinics of North America. 40 (2): 201–206. doi:10.1016/j.psc.2017.01.002. PMID 28477648.
- ^ Lopez LM, Kaptein AA, Helmerhorst FM (February 2012). “Oral contraceptives containing drospirenone for premenstrual syndrome”. Cochrane Database Syst Rev (2): CD006586. doi:10.1002/14651858.CD006586.pub4. PMID 22336820.
- ^ Christiansen C (October 2005). “Effects of drospirenone/estrogen combinations on bone metabolism”. Climacteric. 8 Suppl 3: 35–41. doi:10.1080/13697130500330283. PMID 16203654. S2CID 42803561.
- ^ Jump up to:a b Archer DF (February 2007). “Drospirenone and estradiol: a new option for the postmenopausal woman”. Climacteric. 10 Suppl 1: 3–10. doi:10.1080/13697130601114859. PMID 17364592. S2CID 9221524.
- ^ Jump up to:a b “Drospirenone in HRT?”. Drug Ther Bull. 47 (4): 41–4. April 2009. doi:10.1136/dtb.2009.03.0011. PMID 19357298. S2CID 1909717.
- ^ Foidart JM, Faustmann T (December 2007). “Advances in hormone replacement therapy: weight benefits of drospirenone, a 17alpha-spirolactone-derived progestogen”. Gynecol. Endocrinol. 23 (12): 692–9. doi:10.1080/09513590701582323. PMID 18075844. S2CID 12572825.
- ^ “Drugs@FDA: FDA Approved Drug Products”. United States Food and Drug Administration. Retrieved 23 December 2019.
- ^ Research, Center for Drug Evaluation and. “Drug Safety and Availability – FDA Drug Safety Communication: Updated information about the risk of blood clots in women taking birth control pills containing drospirenone”. http://www.fda.gov. Retrieved 2017-11-07.
- ^https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214154s000lbl.pdf
- ^https://www.maynepharma.com/media/2507/mayne_pharma_drsp-e4_news_release_041421.pdf
- ^ https://www.maynepharma.com/media/2506/fda-approval-of-novel-oral-contraceptive-nextstellis.pdf
- ^ “Ocella- drospirenone and ethinyl estradiol kit”. DailyMed. Retrieved 17 April 2021.
- ^ “Syeda- drospirenone and ethinyl estradiol kit”. DailyMed. Retrieved 17 April 2021.
- ^ “Yasmin- drospirenone and ethinyl estradiol kit”. DailyMed. Retrieved 17 April 2021.
- ^ “Zarah- drospirenone and ethinyl estradiol kit”. DailyMed. Retrieved 17 April 2021.
- ^ “Beyaz- drospirenone/ethinyl estradiol/levomefolate calcium and levomefolate calcium kit”. DailyMed. Retrieved 17 April 2021.
- ^ “Tydemy- drospirenone, ethinyl estradiol and levomefolate calcium and levomefolate calcium kit”. DailyMed. Retrieved 17 April 2021.
- ^https://web.archive.org/web/20210413054728/https://pdf.hres.ca/dpd_pm/00060352.PDF
- ^ “Mithra and Searchlight Pharma Announce Nextstellis Approval in Canada”. Searchlight Pharma (Press release). 8 March 2021. Retrieved 17 April 2021.
- ^ Jump up to:a b c Bayer (March 25, 2013). “Summary of Product Characteristics (SPC): Yasmin”. London: electronic Medicines Compendium (eMC), Datapharm. Retrieved April 24, 2014.
4.3. Contraindications: • Severe chronic kidney disease or acute kidney failure. • Presence or history of severe hepatic disease as long as liver function values have not returned to normal.
- ^ Jump up to:a b Bayer (April 10, 2012). “Yasmin full prescribing information”(PDF). Silver Spring, Md.: Food and Drug Administration (FDA). Retrieved April 14, 2012.
4. Contraindications: • Renal impairment. • Adrenal insufficiency. • Liver disease.
- ^ Nelson, Anita L.; Cwiak, Carrie (2011). “Combined oral contraceptives (COCs)”. In Hatcher, Robert A.; Trussell, James; Nelson, Anita L.; Cates, Willard Jr.; Kowal, Deborah; Policar, Michael S. (eds.). Contraceptive Technology (20th revised ed.). New York: Ardent Media. pp. 249–341. ISBN 978-1-59708-004-0. ISSN 0091-9721. OCLC 781956734.
- ^ Jump up to:a b Heit JA, Spencer FA, White RH (2016). “The epidemiology of venous thromboembolism”. J. Thromb. Thrombolysis. 41 (1): 3–14. doi:10.1007/s11239-015-1311-6. PMC 4715842. PMID 26780736.
- ^ Jump up to:a b c d e Bateson D, Butcher BE, Donovan C, Farrell L, Kovacs G, Mezzini T, Raynes-Greenow C, Pecoraro G, Read C, Baber R (2016). “Risk of venous thromboembolism in women taking the combined oral contraceptive: A systematic review and meta-analysis”. Aust Fam Physician. 45 (1): 59–64. PMID 27051991.
- ^ Jump up to:a b “FDA Drug Safety Communication: Updated information about the risk of blood clots in women taking birth control pills containing drospirenone”. April 27, 2019. Archived from the original on 2019-04-27.
- ^ Vinogradova Y, Coupland C, Hippisley-Cox J (January 2019). “Use of hormone replacement therapy and risk of venous thromboembolism: nested case-control studies using the QResearch and CPRD databases”. BMJ. 364: k4810. doi:10.1136/bmj.k4810. PMC 6326068. PMID 30626577.
- ^ Jump up to:a b c Batur P, Casey PM (February 2017). “Drospirenone Litigation: Does the Punishment Fit the Crime?”. J Womens Health (Larchmt). 26 (2): 99–102. doi:10.1089/jwh.2016.6092. PMID 27854556.
- ^ Jump up to:a b Sitruk-Ware R (November 2016). “Hormonal contraception and thrombosis”. Fertil. Steril. 106 (6): 1289–1294. doi:10.1016/j.fertnstert.2016.08.039. PMID 27678035.
- ^ Oedingen C, Scholz S, Razum O (May 2018). “Systematic review and meta-analysis of the association of combined oral contraceptives on the risk of venous thromboembolism: The role of the progestogen type and estrogen dose”. Thromb. Res. 165: 68–78. doi:10.1016/j.thromres.2018.03.005. PMID 29573722.
- ^ Dragoman MV, Tepper NK, Fu R, Curtis KM, Chou R, Gaffield ME (June 2018). “A systematic review and meta-analysis of venous thrombosis risk among users of combined oral contraception”. Int J Gynaecol Obstet. 141 (3): 287–294. doi:10.1002/ijgo.12455. PMC 5969307. PMID 29388678.
- ^ Jump up to:a b Wiegratz I, Kuhl H (September 2006). “Metabolic and clinical effects of progestogens”. Eur J Contracept Reprod Health Care. 11(3): 153–61. doi:10.1080/13625180600772741. PMID 17056444. S2CID 27088428.
- ^ Jump up to:a b Kuhl H (May 1996). “Effects of progestogens on haemostasis”. Maturitas. 24 (1–2): 1–19. doi:10.1016/0378-5122(96)00994-2. PMID 8794429.
- ^ Jump up to:a b Sitruk-Ware R, Nath A (February 2013). “Characteristics and metabolic effects of estrogen and progestins contained in oral contraceptive pills”. Best Pract. Res. Clin. Endocrinol. Metab. 27 (1): 13–24. doi:10.1016/j.beem.2012.09.004. PMID 23384742.
- ^ Jump up to:a b Nelson AL (2015). “An update on new orally administered contraceptives for women”. Expert Opin Pharmacother. 16 (18): 2759–72. doi:10.1517/14656566.2015.1100173. PMID 26512437. S2CID 207481206.
- ^ Jump up to:a b c Farris M, Bastianelli C, Rosato E, Brosens I, Benagiano G (October 2017). “Pharmacodynamics of combined estrogen-progestin oral contraceptives: 2. effects on hemostasis”. Expert Rev Clin Pharmacol. 10 (10): 1129–1144. doi:10.1080/17512433.2017.1356718. PMID 28712325. S2CID 205931204.
- ^ Jump up to:a b Simoncini T, Genazzani AR (February 2010). “A review of the cardiovascular and breast actions of drospirenone in preclinical studies”. Climacteric. 13 (1): 22–33. doi:10.3109/13697130903437375. PMID 19938948. S2CID 4306359.
- ^ Jump up to:a b c d e f g h i j Africander D, Verhoog N, Hapgood JP (June 2011). “Molecular mechanisms of steroid receptor-mediated actions by synthetic progestins used in HRT and contraception”. Steroids. 76 (7): 636–52. doi:10.1016/j.steroids.2011.03.001. PMID 21414337. S2CID 23630452.
- ^ Palacios S, Mejía A (November 2016). “Progestogen safety and tolerance in hormonal replacement therapy”. Expert Opin Drug Saf. 15 (11): 1515–1525. doi:10.1080/14740338.2016.1223041. PMID 27548404. S2CID 31497860.
- ^ Caglayan EK, Caglayan K, Alkis I, Arslan E, Okur A, Banli O, Engin-Ustün Y (August 2015). “Factors Associated with Mammographic Density in Postmenopausal Women”. J Menopausal Med. 21 (2): 82–8. doi:10.6118/jmm.2015.21.2.82. PMC 4561745. PMID 26357645.
- ^ Hirschberg AL, Tani E, Brismar K, Lundström E (August 2019). “Effects of drospirenone and norethisterone acetate combined with estradiol on mammographic density and proliferation of breast epithelial cells-A prospective randomized trial”. Maturitas. 126: 18–24. doi:10.1016/j.maturitas.2019.04.205. PMID 31239112.
- ^ Jump up to:a b c d Trabert B, Sherman ME, Kannan N, Stanczyk FZ (September 2019). “Progesterone and breast cancer”. Endocr. Rev. 41 (2): 320–344. doi:10.1210/endrev/bnz001. PMC 7156851. PMID 31512725.
- ^ Collaborative Group on Hormonal Factors in Breast Cancer (September 2019). “Type and timing of menopausal hormone therapy and breast cancer risk: individual participant meta-analysis of the worldwide epidemiological evidence”. Lancet. 394 (10204): 1159–1168. doi:10.1016/S0140-6736(19)31709-X. PMC 6891893. PMID 31474332.
- ^ Sturdee DW (2013). “Are progestins really necessary as part of a combined HRT regimen?”. Climacteric. 16 Suppl 1: 79–84. doi:10.3109/13697137.2013.803311. PMID 23651281. S2CID 21894200.
- ^ Jump up to:a b c d e f g h i j Muhn P, Fuhrmann U, Fritzemeier KH, Krattenmacher R, Schillinger E (1995). “Drospirenone: a novel progestogen with antimineralocorticoid and antiandrogenic activity”. Ann. N. Y. Acad. Sci. 761 (3): 311–35. Bibcode:1995NYASA.761..311M. doi:10.1111/j.1749-6632.1995.tb31386.x. PMID 7625729. S2CID 36861309.
- ^ Fuhrmann, Ulrike; Krattenmacher, Rolf; Slater, Emily P.; Fritzemeier, Karl-Heinrich (1996). “The novel progestin drospirenone and its natural counterpart progesterone: Biochemical profile and antiandrogenic potential”. Contraception. 54 (4): 243–251. doi:10.1016/S0010-7824(96)00195-3. ISSN 0010-7824. PMID 8922878.
- ^ Jump up to:a b Hapgood JP, Africander D, Louw R, Ray RM, Rohwer JM (July 2014). “Potency of progestogens used in hormonal therapy: toward understanding differential actions”. J. Steroid Biochem. Mol. Biol. 142: 39–47. doi:10.1016/j.jsbmb.2013.08.001. PMID 23954501. S2CID 12142015.
- ^ Bastianelli C, Farris M, Rosato E, Brosens I, Benagiano G (November 2018). “Pharmacodynamics of combined estrogen-progestin oral contraceptives 3. Inhibition of ovulation”. Expert Rev Clin Pharmacol. 11 (11): 1085–1098. doi:10.1080/17512433.2018.1536544. PMID 30325245. S2CID 53246678.
- ^ Endrikat J, Gerlinger C, Richard S, Rosenbaum P, Düsterberg B (December 2011). “Ovulation inhibition doses of progestins: a systematic review of the available literature and of marketed preparations worldwide”. Contraception. 84 (6): 549–57. doi:10.1016/j.contraception.2011.04.009. PMID 22078182.
- ^ Jump up to:a b Kuhl H (2011). “Pharmacology of Progestogens” (PDF). J Reproduktionsmed Endokrinol. 8 (1): 157–177.
- ^ Jump up to:a b c d Elger W, Beier S, Pollow K, Garfield R, Shi SQ, Hillisch A (November 2003). “Conception and pharmacodynamic profile of drospirenone”. Steroids. 68 (10–13): 891–905. doi:10.1016/j.steroids.2003.08.008. PMID 14667981. S2CID 41756726.
- ^ Schindler AE, Campagnoli C, Druckmann R, Huber J, Pasqualini JR, Schweppe KW, Thijssen JH (December 2003). “Classification and pharmacology of progestins”. Maturitas. 46 Suppl 1: S7–S16. doi:10.1016/j.maturitas.2003.09.014. PMID 14670641.
- ^ “Drospirenone”. pubchem.ncbi.nlm.nih.gov.
- ^ Jump up to:a b Hadji P, Colli E, Regidor PA (December 2019). “Bone health in estrogen-free contraception”. Osteoporos Int. 30 (12): 2391–2400. doi:10.1007/s00198-019-05103-6. PMC 7203087. PMID 31446440.
- ^ Jump up to:a b Cornia, Paul B; Anawalt, Bradley D (2005). “Male hormonal contraceptives: a potentially patentable and profitable product”. Expert Opinion on Therapeutic Patents. 15 (12): 1727–1737. doi:10.1517/13543776.15.12.1727. ISSN 1354-3776. S2CID 83941717.
- ^ Nieschlag E (2010). “Clinical trials in male hormonal contraception” (PDF). Contraception. 82 (5): 457–70. doi:10.1016/j.contraception.2010.03.020. PMID 20933120.
- ^ Nieschlag, Eberhard; Behre, Hermann M.; Nieschlag, Eberhard; Behre, Hermann M.; Nieschlag, Susan (2012). “The essential role of testosterone in hormonal male contraception”. In Nieschlag, Eberhard; Behre, Hermann M; Nieschlag, Susan (eds.). Testosterone. pp. 470–493. doi:10.1017/CBO9781139003353.023. ISBN 9781139003353.
- ^ Jump up to:a b Stanczyk, Frank Z. (2007). “Structure –Function Relationships, Pharmacokinetics, and Potency of Orally and Parenterally Administered Progestogens”. Treatment of the Postmenopausal Woman. pp. 779–798. doi:10.1016/B978-012369443-0/50067-3. ISBN 9780123694430.
- ^ Hermann P.G. Schneider; Frederick Naftolin (22 September 2004). Climacteric Medicine – Where Do We Go?: Proceedings of the 4th Workshop of the International Menopause Society. CRC Press. pp. 133–. ISBN 978-0-203-02496-6.
- ^ Simon JA (December 1995). “Micronized progesterone: vaginal and oral uses”. Clinical Obstetrics and Gynecology. 38 (4): 902–14. doi:10.1097/00003081-199538040-00024. PMID 8616985.
- ^ Genazzani, Andrea R.; Mannella, Paolo; Simoncini, Tommaso (February 2007). “Drospirenone and its antialdosterone properties”. Climacteric. 10 (Supplement 1): 11–18. doi:10.1080/13697130601114891. PMID 17364593. S2CID 24872884. Retrieved November 26, 2011.
- ^ Infante, Marco; Armani, Andrea; Marzolla, Vincenzo; Fabbri, Andrea; Caprio, Massimiliano (2019). “Adipocyte Mineralocorticoid Receptor”. Vitamins and Hormones. Elsevier. 109: 189–209. doi:10.1016/bs.vh.2018.10.005. ISBN 9780128177822. ISSN 0083-6729. PMID 30678856.
- ^ Giordano A, Frontini A, Cinti S (June 2016). “Convertible visceral fat as a therapeutic target to curb obesity”. Nat Rev Drug Discov. 15(6): 405–24. doi:10.1038/nrd.2016.31. PMID 26965204. S2CID 2632187.
- ^ Sitruk-Ware R, Husmann F, Thijssen JH, Skouby SO, Fruzzetti F, Hanker J, Huber J, Druckmann R (September 2004). “Role of progestins with partial antiandrogenic effects”. Climacteric. 7 (3): 238–54. doi:10.1080/13697130400001307. PMID 15669548. S2CID 23112620.
- ^ Yeh YT, Chang CW, Wei RJ, Wang SN (2013). “Progesterone and related compounds in hepatocellular carcinoma: basic and clinical aspects”. Biomed Res Int. 2013: 290575. doi:10.1155/2013/290575. PMC 3581253. PMID 23484104.
- ^ Schindler, Adolf E. (2015). “Hormonal Contraceptives: Progestogen and Thrombotic Risk”. Frontiers in Gynecological Endocrinology. ISGE Series. pp. 69–75. doi:10.1007/978-3-319-09662-9_8. ISBN 978-3-319-09661-2. ISSN 2197-8735.
- ^ Jump up to:a b Neubauer H, Ma Q, Zhou J, Yu Q, Ruan X, Seeger H, Fehm T, Mueck AO (October 2013). “Possible role of PGRMC1 in breast cancer development”. Climacteric. 16 (5): 509–13. doi:10.3109/13697137.2013.800038. PMID 23758160. S2CID 29808177.
- ^ Jump up to:a b c d Martin Negwer; Hans-Georg Scharnow (4 October 2001). Organic-chemical drugs and their synonyms: (an international survey). Wiley-VCH. p. 2539. ISBN 978-3-527-30247-5.
- ^ Howard J.A. Carp (9 April 2015). Progestogens in Obstetrics and Gynecology. Springer. pp. 115–. ISBN 978-3-319-14385-9.
- ^ Ménard J (2004). “The 45-year story of the development of an anti-aldosterone more specific than spironolactone”. Mol. Cell. Endocrinol. 217 (1–2): 45–52. doi:10.1016/j.mce.2003.10.008. PMID 15134800. S2CID 19701784.
[Spironolactone] was synthesized after the demonstration of the natriuretic effect of progesterone (Landau et al., 1955).
- ^ J. Larry Jameson; Leslie J. De Groot (18 May 2010). Endocrinology – E-Book: Adult and Pediatric. Elsevier Health Sciences. pp. 2401–. ISBN 978-1-4557-1126-0.
[Spironolactone] is a potent antimineralocorticoid which was developed as a progestational analog […]
- ^ Aldosterone. Elsevier Science. 23 January 2019. p. 46. ISBN 978-0-12-817783-9.
In addition to spironolactone, which is a derivative of progesterone […]
- ^ Hu X, Li S, McMahon EG, Lala DS, Rudolph AE (2005). “Molecular mechanisms of mineralocorticoid receptor antagonism by eplerenone”. Mini Rev Med Chem. 5 (8): 709–18. doi:10.2174/1389557054553811. PMID 16101407.
- ^ Nakajima ST, Brumsted JR, Riddick DH, Gibson M (1989). “Absence of progestational activity of oral spironolactone”. Fertil. Steril. 52 (1): 155–8. doi:10.1016/s0015-0282(16)60807-5. PMID 2744183.
- ^ Hertz R, Tullner WW (1958). “Progestational activity of certain steroid-17-spirolactones”. Proc. Soc. Exp. Biol. Med. 99 (2): 451–2. doi:10.3181/00379727-99-24380. PMID 13601900. S2CID 20150966.
- ^ WO patent 9806738, Mohr, Jörg-Thorsten & Klaus Nickisch, “PROCESS FOR PRODUCING DROSPIRENONE (6ss,7ss;15ss,16ss-DIMETHYLENE-3-OXO-17 alpha -PREGN-4-EN-21,17-CARBOLACTONE, DRSP), AS WELL AS 7 alpha -(3-HYDROXY-1-PROPYL)-6ss,7ss;15ss,16ss-DIMETHYLENE-5ss-ANDROSTANE-3ss,5,17ss-TRIOL (ZK 92836) AND 6ss,7ss;15ss,16ss-DIMETHYLENE-5ss HYDROXY-5-OXO-17 alpha -ANDROSTANE-21, 17-CARBOLACTONE”, issued 1998-02-19, assigned to Shering AG
- ^ US patent 6121465, Mohr, Joerg-Thorston & Klaus Nickisch, “Process for production drospirenone and intermediate products of the process”, issued 2000-09-19, assigned to Scheiring AG and Bayer Schering Pharma
- ^ Jump up to:a b c “Drospirenone/Estetrol – Mithra Pharmaceuticals – AdisInsight”.
- ^ “Ethinylestradiol/drospirenone – AdisInsight”.
- ^ “Ethinylestradiol/drospirenone/folic acid – AdisInsight”.
- ^ “Drospirenone/ethinylestradiol low-dose – Bayer HealthCare Pharmaceuticals – AdisInsight”.
- ^ “Estradiol/drospirenone – Bayer HealthCare Pharmaceuticals – AdisInsight”.
- ^ Jump up to:a b Feeley, Jef; Kresge, Naomi (July 31, 2012). “Bayer’s Yasmin lawsuit settlements rise to $402.6 million”. Bloomberg News. New York. Retrieved November 11, 2012.
- ^ Jump up to:a b c d AG, Bayer. “Quarterly Reports of Bayer”. http://www.bayer.com.
- ^ Jump up to:a b c “Drospirenone/estradiol/prasterone – ANI Pharmaceuticals/Pantarhei Bioscience – AdisInsight”. adisinsight.springer.com.
- ^ Nippe S, General S (September 2011). “Parenteral oil-based drospirenone microcrystal suspensions-evaluation of physicochemical stability and influence of stabilising agents”. Int J Pharm. 416 (1): 181–8. doi:10.1016/j.ijpharm.2011.06.036. PMID 21729745.
- ^ Nippe S, General S (November 2012). “Combination of injectable ethinyl estradiol and drospirenone drug-delivery systems and characterization of their in vitro release”. Eur J Pharm Sci. 47 (4): 790–800. doi:10.1016/j.ejps.2012.08.009. PMID 22940138.
- ^ Nippe S, Preuße C, General S (February 2013). “Evaluation of the in vitro release and pharmacokinetics of parenteral injectable formulations for steroids”. Eur J Pharm Biopharm. 83 (2): 253–65. doi:10.1016/j.ejpb.2012.09.006. PMID 23116659.
- ^ Nippe S, General S (April 2015). “Investigation of injectable drospirenone organogels with regard to their rheology and comparison to non-stabilized oil-based drospirenone suspensions”. Drug Dev Ind Pharm. 41 (4): 681–91. doi:10.3109/03639045.2014.895375. PMID 24621345. S2CID 42932558.
Further reading
- Archer DF (February 2007). “Drospirenone and estradiol: a new option for the postmenopausal woman”. Climacteric. 10 Suppl 1: 3–10. doi:10.1080/13697130601114859. PMID 17364592. S2CID 9221524.
- Archer DF (February 2007). “Drospirenone-containing hormone therapy for postmenopausal women. Perspective on current data”. J Reprod Med. 52 (2 Suppl): 159–64. PMID 17477110.
- Archer DF (2007). “Drospirenone, a progestin with added value for hypertensive postmenopausal women”. Menopause. 14 (3 Pt 1): 352–4. doi:10.1097/gme.0b013e31804d440b. PMID 17414576.
- Batur P, Casey PM (February 2017). “Drospirenone Litigation: Does the Punishment Fit the Crime?”. J Womens Health (Larchmt). 26 (2): 99–102. doi:10.1089/jwh.2016.6092. PMID 27854556.
- Bitzer J, Paoletti AM (2009). “Added benefits and user satisfaction with a low-dose oral contraceptive containing drospirenone: results of three multicentre trials”. Clin Drug Investig. 29 (2): 73–8. doi:10.2165/0044011-200929020-00001. PMID 19133702. S2CID 10356578.
- Carranza-Lira S (2009). “Safety, efficacy and patient acceptability of drospirenone and estradiol in the treatment of menopausal vasomotor symptoms: a review”. Clin Interv Aging. 4: 59–62. doi:10.2147/CIA.S4117. PMC 2685225. PMID 19503766.
- Christiansen C (October 2005). “Effects of drospirenone/estrogen combinations on bone metabolism”. Climacteric. 8 Suppl 3: 35–41. doi:10.1080/13697130500330283. PMID 16203654. S2CID 42803561.
- Dickerson V (November 2002). “Quality of life issues. Potential role for an oral contraceptive containing ethinyl estradiol and drospirenone”. J Reprod Med. 47 (11 Suppl): 985–93. PMID 12497673.
- Fenton C, Wellington K, Moen MD, Robinson DM (2007). “Drospirenone/ethinylestradiol 3mg/20microg (24/4 day regimen): a review of its use in contraception, premenstrual dysphoric disorder and moderate acne vulgaris”. Drugs. 67 (12): 1749–65. doi:10.2165/00003495-200767120-00007. PMID 17683173. S2CID 46976925.
- Foidart JM (October 2005). “Added benefits of drospirenone for compliance”. Climacteric. 8 Suppl 3: 28–34. doi:10.1080/13697130500330309. PMID 16203653. S2CID 31883491.
- Foidart JM, Faustmann T (December 2007). “Advances in hormone replacement therapy: weight benefits of drospirenone, a 17alpha-spirolactone-derived progestogen”. Gynecol. Endocrinol. 23 (12): 692–9. doi:10.1080/09513590701582323. PMID 18075844. S2CID 12572825.
- Genazzani AR, Mannella P, Simoncini T (February 2007). “Drospirenone and its antialdosterone properties”. Climacteric. 10 Suppl 1: 11–8. doi:10.1080/13697130601114891. PMID 17364593. S2CID 24872884.
- Han L, Jensen JT (October 2014). “Expert opinion on a flexible extended regimen of drospirenone/ethinyl estradiol contraceptive”. Expert Opin Pharmacother. 15 (14): 2071–9. doi:10.1517/14656566.2014.949237. PMID 25186109. S2CID 25338932.
- Heinemann LA, Dinger J (2004). “Safety of a new oral contraceptive containing drospirenone”. Drug Saf. 27 (13): 1001–18. doi:10.2165/00002018-200427130-00003. PMID 15471507. S2CID 1773936.
- Idota N, Kobayashi M, Miyamori D, Kakiuchi Y, Ikegaya H (March 2015). “Drospirenone detected in postmortem blood of a young woman with pulmonary thromboembolism: A case report and review of the literature”. Leg Med (Tokyo). 17 (2): 109–15. doi:10.1016/j.legalmed.2014.10.001. PMID 25454533.
- Keam SJ, Wagstaff AJ (2003). “Ethinylestradiol/drospirenone: a review of its use as an oral contraceptive”. Treat Endocrinol. 2 (1): 49–70. doi:10.2165/00024677-200302010-00005. PMID 15871554. S2CID 209144694.
- Krattenmacher R (July 2000). “Drospirenone: pharmacology and pharmacokinetics of a unique progestogen”. Contraception. 62 (1): 29–38. doi:10.1016/S0010-7824(00)00133-5. PMID 11024226.
- Larivée N, Suissa S, Khosrow-Khavar F, Tagalakis V, Filion KB (September 2017). “Drospirenone-containing oral contraceptive pills and the risk of venous thromboembolism: a systematic review of observational studies”. BJOG. 124 (10): 1490–1499. doi:10.1111/1471-0528.14623. PMID 28276140.
- Lete I, Chabbert-Buffet N, Jamin C, Lello S, Lobo P, Nappi RE, Pintiaux A (2015). “Haemostatic and metabolic impact of estradiol pills and drospirenone-containing ethinylestradiol pills vs. levonorgestrel-containing ethinylestradiol pills: A literature review”. Eur J Contracept Reprod Health Care. 20 (5): 329–43. doi:10.3109/13625187.2015.1050091. PMID 26007631. S2CID 41601833.
- Li J, Ren J, Sun W (March 2017). “A comparative systematic review of Yasmin (drospirenone pill) versus standard treatment options for symptoms of polycystic ovary syndrome”. Eur. J. Obstet. Gynecol. Reprod. Biol. 210: 13–21. doi:10.1016/j.ejogrb.2016.11.013. PMID 27923166.
- Lopez LM, Kaptein AA, Helmerhorst FM (February 2012). “Oral contraceptives containing drospirenone for premenstrual syndrome”. Cochrane Database Syst Rev (2): CD006586. doi:10.1002/14651858.CD006586.pub4. PMID 22336820.
- Machado RB, Pompei Lde M, Giribela AG, Giribela CG (January 2011). “Drospirenone/ethinylestradiol: a review on efficacy and noncontraceptive benefits”. Womens Health (Lond). 7 (1): 19–30. doi:10.2217/whe.10.84. PMID 21175386.
- Mallareddy M, Hanes V, White WB (2007). “Drospirenone, a new progestogen, for postmenopausal women with hypertension”. Drugs Aging. 24 (6): 453–66. doi:10.2165/00002512-200724060-00002. PMID 17571911. S2CID 39236155.
- Motivala A, Pitt B (2007). “Drospirenone for oral contraception and hormone replacement therapy: are its cardiovascular risks and benefits the same as other progestogens?”. Drugs. 67 (5): 647–55. doi:10.2165/00003495-200767050-00001. PMID 17385938. S2CID 22985078.
- Oelkers W (December 2002). “Antimineralocorticoid activity of a novel oral contraceptive containing drospirenone, a unique progestogen resembling natural progesterone”. Eur J Contracept Reprod Health Care. 7 Suppl 3: 19–26, discussion 42–3. PMID 12659403.
- Oelkers W (December 2000). “Drospirenone–a new progestogen with antimineralocorticoid activity, resembling natural progesterone”. Eur J Contracept Reprod Health Care. 5 Suppl 3: 17–24. PMID 11246598.
- Oelkers W (March 2004). “Drospirenone, a progestogen with antimineralocorticoid properties: a short review”. Mol. Cell. Endocrinol. 217 (1–2): 255–61. doi:10.1016/j.mce.2003.10.030. PMID 15134826. S2CID 19936032.
- Oelkers W (February 2002). “The renin-aldosterone system and drospirenone”. Gynecol. Endocrinol. 16 (1): 83–7. doi:10.1080/gye.16.1.83.87. PMID 11915587. S2CID 32410408.
- Oelkers WH (October 2005). “Drospirenone in combination with estrogens: for contraception and hormone replacement therapy”. Climacteric. 8 Suppl 3: 19–27. doi:10.1080/13697130500330341. PMID 16203652. S2CID 42837148.
- Palacios S, Foidart JM, Genazzani AR (November 2006). “Advances in hormone replacement therapy with drospirenone, a unique progestogen with aldosterone receptor antagonism” (PDF). Maturitas. 55 (4): 297–307. doi:10.1016/j.maturitas.2006.07.009. hdl:2268/9932. PMID 16949774.
- Pérez-López FR (June 2008). “Clinical experiences with drospirenone: from reproductive to postmenopausal years”. Maturitas. 60 (2): 78–91. doi:10.1016/j.maturitas.2008.03.009. PMID 18468818.
- Rapkin AJ, Sorger SN, Winer SA (February 2008). “Drospirenone/ethinyl estradiol”. Drugs Today. 44 (2): 133–45. doi:10.1358/dot.2008.44.2.1191057. PMID 18389090. S2CID 32413831.
- Rapkin AJ, Winer SA (May 2007). “Drospirenone: a novel progestin”. Expert Opin Pharmacother. 8 (7): 989–99. doi:10.1517/14656566.8.7.989. PMID 17472544. S2CID 6954183.
- Rapkin RB, Creinin MD (October 2011). “The combined oral contraceptive pill containing drospirenone and ethinyl estradiol plus levomefolate calcium”. Expert Opin Pharmacother. 12 (15): 2403–10. doi:10.1517/14656566.2011.610791. PMID 21877996. S2CID 40231903.
- Rübig A (October 2003). “Drospirenone: a new cardiovascular-active progestin with antialdosterone and antiandrogenic properties”. Climacteric. 6 Suppl 3: 49–54. PMID 15018248.
- Scheinfeld NS (2007). “Yaz (3 mg drospirenone/20 microg ethinyl estradiol)”. Skinmed. 6 (6): 289. doi:10.1111/j.1540-9740.2007.07338.x. PMID 17975349.
- Sehovic N, Smith KP (May 2010). “Risk of venous thromboembolism with drospirenone in combined oral contraceptive products”. Ann Pharmacother. 44 (5): 898–903. doi:10.1345/aph.1M649. PMID 20371756. S2CID 8248469.
- Shulman LP (June 2006). “A review of drospirenone for safety and tolerability and effects on endometrial safety and lipid parameters contrasted with medroxyprogesterone acetate, levonorgestrel, and micronized progesterone”. J Womens Health (Larchmt). 15 (5): 584–90. doi:10.1089/jwh.2006.15.584. PMID 16796485.
- Simoncini T, Genazzani AR (February 2010). “A review of the cardiovascular and breast actions of drospirenone in preclinical studies”. Climacteric. 13 (1): 22–33. doi:10.3109/13697130903437375. PMID 19938948. S2CID 4306359.
- Sitruk-Ware R (October 2005). “Pharmacology of different progestogens: the special case of drospirenone”. Climacteric. 8 Suppl 3: 4–12. doi:10.1080/13697130500330382. PMID 16203650. S2CID 24205704.
- Thorneycroft IH (November 2002). “Evolution of progestins. Focus on the novel progestin drospirenone”. J Reprod Med. 47 (11 Suppl): 975–80. PMID 12497671.
- Toni I, Neubert A, Botzenhardt S, Gratzki N, Rascher W (September 2013). “Venous thromboembolism in adolescents associated with drospirenone-containing oral contraceptives – two case reports”. Klin Padiatr. 225 (5): 266–7. doi:10.1055/s-0033-1353169. PMID 23975850.
- White WB (February 2007). “Drospirenone with 17beta-estradiol in the postmenopausal woman with hypertension”. Climacteric. 10 Suppl 1: 25–31. doi:10.1080/13697130601114933. PMID 17364595. S2CID 9451771.
- Whitehead M (March 2006). “Hormone replacement therapy with estradiol and drospirenone: an overview of the clinical data”. J Br Menopause Soc. 12 Suppl 1: 4–7. doi:10.1258/136218006775992185. PMID 16513012. S2CID 38095916.
- Wu CQ, Grandi SM, Filion KB, Abenhaim HA, Joseph L, Eisenberg MJ (June 2013). “Drospirenone-containing oral contraceptive pills and the risk of venous and arterial thrombosis: a systematic review”. BJOG. 120 (7): 801–10. doi:10.1111/1471-0528.12210. PMID 23530659. S2CID 206904730.
- Zhao X, Zhang XF, Zhao Y, Lin X, Li NY, Paudel G, Wang QY, Zhang XW, Li XL, Yu J (September 2016). “Effect of combined drospirenone with estradiol for hypertensive postmenopausal women: a systemic review and meta-analysis”. Gynecol. Endocrinol. 32 (9): 685–689. doi:10.1080/09513590.2016.1183629. PMID 27176003. S2CID 9116138.
- “Drospirenone in HRT?”. Drug Ther Bull. 47 (4): 41–4. April 2009. doi:10.1136/dtb.2009.03.0011 (inactive 2021-05-06). PMID 19357298. S2CID 1909717.
External links
- “Drospirenone”. Drug Information Portal. U.S. National Library of Medicine.
//////////drospirenone, Nextstellis, ZK 30595, FDA 2021, APPROVALS 2021
NEW DRUG APPROVALS
ONE TIME
$10.00
Samidorphan
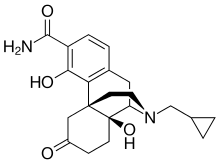
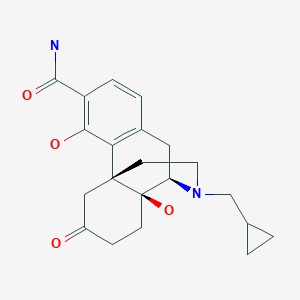
Samidorphan
サミドルファン;
| Formula | C21H26N2O4 |
|---|---|
| CAS | 852626-89-2 |
| Mol weight | 370.4421 |
FDA APPROVED 5/28/2021 Lybalvi
- ALKS 33
- ALKS-33
- RDC-0313
- RDC-0313-00
Product Ingredients

UNII0AJQ5N56E0
CAS Number1204592-75-5
WeightAverage: 504.536
Monoisotopic: 504.210780618
Chemical FormulaC25H32N2O9
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Samidorphan L-malate | 0AJQ5N56E0 | 1204592-75-5 | RARHXUAUPNYAJF-QSYGGRRVSA-N |
IUPAC Name(1R,9R,10S)-17-(cyclopropylmethyl)-3,10-dihydroxy-13-oxo-17-azatetracyclo[7.5.3.0^{1,10}.0^{2,7}]heptadeca-2,4,6-triene-4-carboxamide; (2S)-2-hydroxybutanedioic acid
MOA:mu-Opioid antagonist; delta-Opioid partial agonist; kappa-Opioid partial agonistsIndication:Alcohol dependence
New Drug Application (NDA): 213378
Company: ALKERMES INChttps://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213378s000lbl.pdfhttps://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/213378Orig1s000,%20Orig2s000ltr.pdf
To treat schizophrenia in adults and certain aspects of bipolar I disorder in adults
LYBALVI is a combination of olanzapine, an atypical antipsychotic, and samidorphan (as samidorphan L-malate), an opioid antagonist.
Olanzapine is 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine. The molecular formula of olanzapine is: C17H20N4S and the molecular weight is 312.44 g/mol. It is a yellow crystalline powder and has pKa values of 7.80 and 5.44. The chemical structure is:
 |
Samidorphan L-malate is morphinan-3-carboxamide, 17-(cyclopropylmethyl)-4, 14-dihydroxy-6-oxo-, (2S)-2-hydroxybutanedioate. The molecular formula of samidorphan L-malate is C21H26N2O4 • C4H6O5 and the molecular weight is 504.54 g/mol. It is a white to off-white crystalline powder and has pKa values of 8.3 (amine) and 10.1 (phenol). The chemical structure is:
 |
LYBALVI is intended for oral administration and is available as film-coated, bilayer tablets in the following strengths: 5 mg/10 mg, 10 mg/10 mg, 15 mg/10 mg, and 20 mg/10 mg of olanzapine and samidorphan (equivalent to 13.6 mg of samidorphan L-malate).
Inactive ingredients include colloidal silicon dioxide, crospovidone, lactose monohydrate, magnesium stearate, and microcrystalline cellulose. The film coating ingredients include hypromellose, titanium dioxide, triacetin, and color additives [iron oxide yellow (5 mg/10 mg); iron oxide yellow and iron oxide red (10 mg/10 mg); FD&C Blue No. 2/ indigo carmine aluminum lake (15 mg/10 mg); iron oxide red (20 mg/10 mg)].
- to treat schizophrenia
- alone for short-term (acute) or maintenance treatment of manic or mixed episodes that happen with bipolar I disorder
- in combination with valproate or lithium to treat manic or mixed episodes that happen with bipolar I disorder
Olanzapine is an effective atypical antipsychotic that, like other antipsychotics, is associated with weight gain, metabolic dysfunction, and increased risk of type II diabetes.5,6 Samidorphan is a novel opioid antagonist structurally related to naltrexone, with a higher affinity for opioid receptors, more potent μ-opioid receptor antagonism, higher oral bioavailability, and a longer half-life, making it an attractive candidate for oral dosing.1,5,11 Although antipsychotic-induced weight gain is incompletely understood, it is thought that the opioid system plays a key role in feeding and metabolism, such that opioid antagonism may be expected to ameliorate these negative effects. Samidorphan has been shown in animal models and clinical trials to ameliorate olanzapine-induced weight gain and metabolic dysfunction.5,6
Samidorphan was first approved as a variety of fixed-dose combination tablets with olanzapine by the FDA on May 28, 2021, and is currently marketed under the trademark LYBALVI™ by Alkermes Inc.11
Samidorphan (INN, USAN) (developmental code names ALKS-33, RDC-0313), also known as 3-carboxamido-4-hydroxynaltrexone,[2] is an opioid antagonist that preferentially acts as an antagonist of the μ-opioid receptor (MOR). It is under development by Alkermes for the treatment of major depressive disorder and possibly other psychiatric conditions.[3]
Development
Samidorphan has been investigated for the treatment of alcoholism and cocaine addiction by its developer, Alkermes,[4][5] showing similar efficacy to naltrexone but possibly with reduced side effects.
However, it has attracted much more attention as part of the combination product ALKS-5461 (buprenorphine/samidorphan), where samidorphan is combined with the mixed MOR weak partial agonist and κ-opioid receptor (KOR) antagonist buprenorphine, as an antidepressant. Buprenorphine has shown antidepressant effects in some human studies, thought to be because of its antagonist effects at the KOR, but has not been further developed for this application because of its MOR agonist effects and consequent abuse potential. By combining buprenorphine with samidorphan to block the MOR agonist effects, the combination acts more like a selective KOR antagonist, and produces only antidepressant effects, without typical MOR effects such as euphoria or substance dependence being evident.[6][7]
Samidorphan is also being studied in combination with olanzapine, as ALKS-3831 (olanzapine/samidorphan), for use in schizophrenia.[8] A Phase 3 study found that the addition of samidorphan to olanzapine significantly reduced weight gain compared to olanzapine alone.[9] The combination is now under review for approval by the US Food and Drug Administration.[10]
Pharmacology
Pharmacodynamics
The known activity profile of samidorphan at the opioid receptors is as follows:[11][12]
- μ-Opioid receptor (Ki = 0.052 nM; EC50 = N/A; Emax = 3.8%; IC50 = 0.88 nM; Imax = 92%)
- κ-Opioid receptor (Ki = 0.23 nM; EC50 = 3.3 nM; Emax = 36%; IC50 = 38 nM; Imax = 57%)
- δ-Opioid receptor (Ki = 2.6 nM; EC50 = 1.5 nM; Emax = 35%; IC50 = 6.9 nM; Imax = 56%)
As such, samidorphan is primarily an antagonist, or extremely weak partial agonist of the MOR.[11][12] In accordance with its in vitro profile, samidorphan has been observed to produce some side effects that are potentially consistent with activation of the KOR such as somnolence, sedation, dizziness, and hallucinations in some patients in clinical trials at the doses tested.[13]
SYNPATENT
WO2006052710A1.
https://patents.google.com/patent/WO2006052710A1/enExample 1 -Synthesis of 3-Carboxyamido-4-hvdroxy-naltrexone derivative 3

(A) Synthesis of 3-Carboxyamido-naltrexone 2[029] The triflate 11 of naltrexone was prepared according to the method of Wentland et al. (Bioorg. Med. Chem. Lett. 9, 183-187 (2000)), and the carboxamide 2 was prepared by the method described by Wentland et al. [(Bioorg. Med. Chem. Lett. ϋ, 623-626 (2001); and Bioorg. Med. Chem. Lett. 11, 1717-1721 (2001)] involving Pd-catalyzed carbonylation of the triflate 11 in the presence of ammonia and the Pd(O) ligand, DPPF ([l,l’-bis(diphenylρhosphino)ferrocene]) and DMSO.(B) Synthesis of 3-Carboxyamido-4-hydroxy-naltrexone derivative 3[030] Zinc dust (26 mg, 0.40 mmol) was added in portions to a solution of 2 (50 mg, 0.14 mmol) in HCl (37%, 0.2 mL) and AcOH (2 mL) at reflux. After heating at reflux for a further 15 min, the reaction was cooled by the addition of ice/water (10 mL) and basified (pH=9) with NH3/H2O, and the solution was extracted with EtOAc (3×10 mL). The organic extracts were washed with brine, dried, and concentrated. The residue was purified by column chromatography (SiO2, CH2Cl2, CH3OH : NH3/H2O = 15:1:0.01) to give compound 3 as a foam (25 mg, 50%). 1H NMR (CDC13) δl3.28(s, IH, 4-OH), 7.15(d, IH, J=8.1, H-2), 6.47(d, IH, J=8.4, H- 1), 6.10(br, IH, N-H), 4.35(br, IH, N-H), 4.04(dd,lH, J=I.8, 13.5, H-5), 3.11( d, IH, J=6), 2.99( d, IH, J=5.7), 2.94( s, IH), 2.86( d, IH, J= 6), 2.84-2.75(m, 2H), 2.65-2.61(m, 2H), 2.17-2.05(m, IH), 1.89-1.84(m, 2H), 0.85(m, IH), 0.56-0.50(m, 2H), 0.13-0.09(m, 2H). [α]D25= -98.4° (c=0.6, CH2Cl2). MS m/z (ESI) 371(MH+).
Paper
Bioorg. Med. Chem. Lett. 2000, 10, 183-187.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X99006708
Abstract
Opioid binding affinities were assessed for a series of cyclazocine analogues where the prototypic 8-OH substituent of cyclazocine was replaced by amino and substituted-amino groups. For μ and κ opioid receptors, secondary amine derivatives having the (2R,6R,11R)-configuration had the highest affinity. Most targets were efficiently synthesized from the triflate of cyclazocine or its enantiomers using Pd-catalyzed amination procedures.
PAPER
Bioorg. Med. Chem. Lett. 2001, 11, 1717-1721.
https://www.sciencedirect.com/science/article/abs/pii/S0960894X01002785
Abstract
In response to the unexpectedly high affinity for opioid receptors observed in a novel series of cyclazocine analogues where the prototypic 8-OH was replaced by a carboxamido group, we have prepared the corresponding 3-CONH2 analogues of morphine and naltrexone. High affinity (Ki=34 and 1.7 nM) for μ opioid receptors was seen, however, the new targets were 39- and 11-fold less potent than morphine and naltrexone, respectively.
Abstract
High-affinity binding to μ opioid receptors has been identified in a series of novel 3-carboxamido analogues of morphine and naltrexone.

References
- ^ Turncliff R, DiPetrillo L, Silverman B, Ehrich E (February 2015). “Single- and multiple-dose pharmacokinetics of samidorphan, a novel opioid antagonist, in healthy volunteers”. Clinical Therapeutics. 37 (2): 338–48. doi:10.1016/j.clinthera.2014.10.001. PMID 25456560.
- ^ Wentland MP, Lu Q, Lou R, Bu Y, Knapp BI, Bidlack, JM (April 2005). “Synthesis and opioid receptor binding properties of a highly potent 4-hydroxy analogue of naltrexone”. Bioorganic & Medicinal Chemistry Letters. 15 (8): 2107–10. doi:10.1016/j.bmcl.2005.02.032. PMID 15808478.
- ^ “Samidorphan”. Adis Insight. Springer Nature Switzerland AG.
- ^ Hillemacher T, Heberlein A, Muschler MA, Bleich S, Frieling H (August 2011). “Opioid modulators for alcohol dependence”. Expert Opinion on Investigational Drugs. 20 (8): 1073–86. doi:10.1517/13543784.2011.592139. PMID 21651459.
- ^ Clinical trial number NCT01366001 for “ALK33BUP-101: Safety and Pharmacodynamic Effects of ALKS 33-BUP Administered Alone and When Co-administered With Cocaine” at ClinicalTrials.gov
- ^ “ALKS 5461 drug found to reduce depressive symptoms in Phase 1/2 study”.
- ^ “Investigational ALKS 5461 Channels ‘Opium Cure’ for Depression”.
- ^ LaMattina J (15 January 2013). “Will Alkermes’ Antipsychotic ALKS-3831 Become Another Tredaptive?”. Forbes.
- ^ Correll, Christoph U.; Newcomer, John W.; Silverman, Bernard; DiPetrillo, Lauren; Graham, Christine; Jiang, Ying; Du, Yangchun; Simmons, Adam; Hopkinson, Craig; McDonnell, David; Kahn, René S. (2020-08-14). “Effects of Olanzapine Combined With Samidorphan on Weight Gain in Schizophrenia: A 24-Week Phase 3 Study”. American Journal of Psychiatry. 177 (12): 1168–1178. doi:10.1176/appi.ajp.2020.19121279. ISSN 0002-953X.
- ^ “FDA Panel: Some Risk OK for Olanzapine Combo With Less Weight Gain”. http://www.medpagetoday.com. 2020-10-09. Retrieved 2021-01-23.
- ^ Jump up to:a b Linda P. Dwoskin (29 January 2014). Emerging Targets & Therapeutics in the Treatment of Psychostimulant Abuse. Elsevier Science. pp. 398–399, 402–403. ISBN 978-0-12-420177-4.
- ^ Jump up to:a b Wentland MP, Lou R, Lu Q, Bu Y, Denhardt C, Jin J, et al. (April 2009). “Syntheses of novel high affinity ligands for opioid receptors”. Bioorganic & Medicinal Chemistry Letters. 19 (8): 2289–94. doi:10.1016/j.bmcl.2009.02.078. PMC 2791460. PMID 19282177.
- ^ McElroy SL, Guerdjikova AI, Blom TJ, Crow SJ, Memisoglu A, Silverman BL, Ehrich EW (April 2013). “A placebo-controlled pilot study of the novel opioid receptor antagonist ALKS-33 in binge eating disorder”. The International Journal of Eating Disorders. 46(3): 239–45. doi:10.1002/eat.22114. PMID 23381803.
External links
| Clinical data | |
|---|---|
| Other names | ALKS-33, RDC-0313; 3-Carboxamido-4-hydroxynaltrexone |
| Routes of administration | Oral |
| Pharmacokinetic data | |
| Elimination half-life | 7–9 hours[1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 852626-89-2 |
| PubChem CID | 11667832 |
| ChemSpider | 23259667 |
| UNII | 7W2581Z5L8 |
| KEGG | D10162 |
| Chemical and physical data | |
| Formula | C21H26N2O4 |
| Molar mass | 370.449 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////samidorphan, サミドルファン, ALKS 33, ALKS-33, RDC-0313, RDC-0313-00, APPROVALS 2021, FDA 2021, Lybalvi
SMILESO[C@@H](CC(O)=O)C(O)=O.NC(=O)C1=CC=C2C[C@H]3N(CC4CC4)CC[C@@]4(CC(=O)CC[C@@]34O)C2=C1O
NEW DRUG APPROVALS
one time
$10.00
Piflufolastat F 18 injection, Dcfpyl F-18

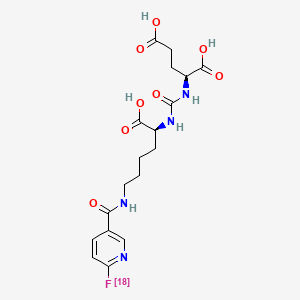
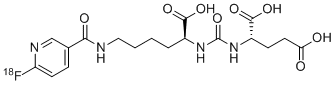
Piflufolastat F 18 injection
Dcfpyl F-18
CAS 207181-29-0
PLAIN F 1423758-00-2 WITHOUT RADIO LABELC18 H23 F N4 O8, 441.4L-Glutamic acid, N-[[[(1S)-1-carboxy-5-[[[6-(fluoro-18F)-3-pyridinyl]carbonyl]amino]pentyl]amino]carbonyl]-2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl) amino]-pentyl}ureido)-pentanedioic acid
Other Names
- N-[[[(1S)-1-Carboxy-5-[[[6-(fluoro-18F)-3-pyridinyl]carbonyl]amino]pentyl]amino]carbonyl]-L-glutamic acid
- [18F]DCFPyl
Dcfpyl F-18
(18F)Dcfpyl
UNII-3934EF02T7
18F-DCFPyL
3934EF02T7
Progenics Pharmaceuticals, Inc.
APPROVED 5/26/2021 fda, Pylarify
For positron emission tomography imaging of prostate-specific membrane antigen-positive lesions in men with prostate cancer
For positron emission tomography (PET) of prostatespecific membrane antigen (PSMA) positive lesions in men with prostate cancer: • with suspected metastasis who are candidates for initial definitive therapy. • with suspected recurrence based on elevated serum prostate-specific antigen (PSA) level.
- Originator Johns Hopkins University School of Medicine
- Developer Curium Pharma; Progenics Pharmaceuticals
- Class Amides; Carboxylic acids; Fluorinated hydrocarbons; Imaging agents; Pyridines; Radiopharmaceutical diagnostics; Radiopharmaceuticals; Small molecules; Urea compounds
- Mechanism of ActionPositron-emission tomography enhancers
- Orphan Drug StatusNo
- MarketedProstate cancer
- 28 May 2021Registered for Prostate cancer (Diagnosis) in USA (IV) – First global approval
- 28 May 2021Adverse events data from phase III CONDOR and phase II/III OSPREY trials in prostate cancer released by Lantheus Holdings
- 27 May 2021Lantheus Holdings intends to launch Fluorine-18 DCFPyL in USA at end of 2021
PYLARIFY contains fluorine 18 (F 18), radiolabeled prostate-specific membrane antigen inhibitor imaging agent. Chemically piflufolastat F 18 is 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl) amino]-pentyl}ureido)-pentanedioic acid. The molecular weight is 441.4 and the structural formula is:

The chiral purity of the unlabeled piflufolastat F 18 precursor is greater than 99% (S,S). PYLARIFY is a sterile, non-pyrogenic, clear, colorless solution for intravenous injection. Each milliliter contains 37 to 2,960 MBq (1 to 80 mCi) piflufolastat F 18 with ≤0.01 µg/mCi of piflufolastat at calibration time and date, and ≤ 78.9 mg ethanol in 0.9% sodium chloride injection USP. The pH of the solution is 4.5 to 7.0. PYLARIFY has a radiochemical purity of at least 95% up to 10 hours following end of synthesis, and specific activity of at least 1000 mCi/µmol at the time of administration.
PYLARIFY contains fluorine 18 (F 18), radiolabeled prostate-specific membrane antigen inhibitor imaging agent. Chemically piflufolastat F 18 is 2-(3-{1-carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)amino]-pentyl}ureido)-pentanedioic acid. The molecular weight is 441.4 and the structural formula is:
 |
The chiral purity of the unlabeled piflufolastat F 18 precursor is greater than 99% (S,S).
PYLARIFY is a sterile, non-pyrogenic, clear, colorless solution for intravenous injection. Each milliliter contains 37 to 2,960 MBq (1 to 80 mCi) piflufolastat F 18 with ≤0.01 μg/mCi of piflufolastat at calibration time and date, and ≤ 78.9 mg ethanol in 0.9% sodium chloride injection USP. The pH of the solution is 4.5 to 7.0.
PYLARIFY has a radiochemical purity of at least 95% up to 10 hours following end of synthesis, and specific activity of at least 1000 mCi/μmol at the time of administration.
Physical Characteristics
PYLARIFY is radiolabeled with fluorine 18 (F 18), a cyclotron produced radionuclide that decays by positron emission to stable oxygen 18 with a half-life of 109.8 minutes. The principal photons useful for diagnostic imaging are the coincident pair of 511 keV gamma photons, resulting from the interaction of the emitted positron with an electron (Table 3).
Table 3: Principal Radiation Produced from Decay of Fluorine 18
| Radiation Energy (keV) | Abundance (%) | |
| Positron | 249.8 | 96.9 |
| Gamma | 511 | 193.5 |
FDA
- Approval Letter(s) (PDF)
- Printed Labeling (PDF)
- Product Quality Review(s) (PDF)
- Multi-Discipline Review (PDF)
- Proprietary Name Review(s) (PDF)
- Officer/Employee List (PDF)
- Other Review(s) (PDF)
- Risk Assessment and Risk Mitigation Review(s) (PDF)
- Administrative and Correspondence Documents (PDF)
PATENT
WO 2016030329
WO 2017072200
PAPER
Journal of Labelled Compounds and Radiopharmaceuticals (2016), 59(11), 439-450
CLIP
https://ejnmmires.springeropen.com/articles/10.1186/s13550-016-0195-6
![Radiosynthesis of [ 18 F]DCFPyL](https://www.researchgate.net/publication/301830909/figure/fig4/AS:362696278069251@1463484939571/Radiosynthesis-of-18-FDCFPyL.png)
Radiosynthesis of [ 18 F]DCFPyL

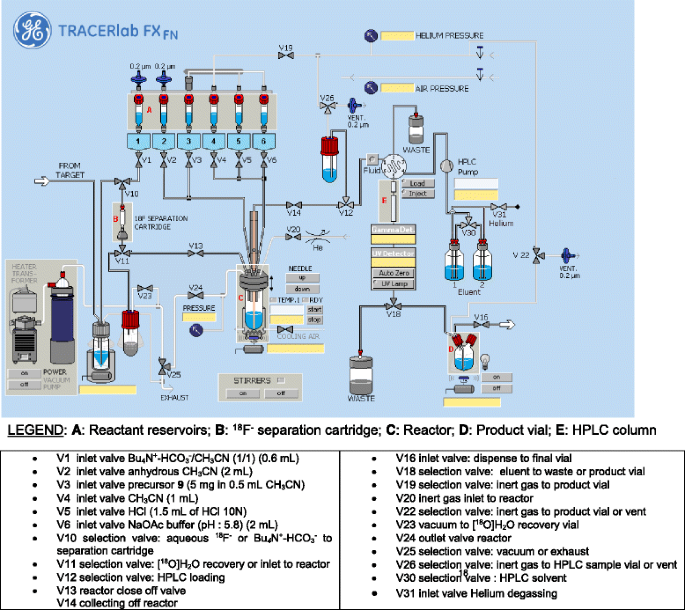
Structure of 18F-labeled small-molecule PSMA inhibitors
/////////piflufolastat F 18, injection, Orphan Drug , Prostate cancer, [18F]DCFPyL, 18F-DCFPYL, DCFPYL F-18, fda 2021, approvals 2021
- Product Quality Review(s) (PDF)
- Multi-Discipline Review (PDF)
- Proprietary Name Review(s) (PDF)
- Officer/Employee List (PDF)
- Other Review(s) (PDF)
- Risk Assessment and Risk Mitigation Review(s) (PDF)
- Administrative and Correspondence Documents (PDF)
- Label (PDF)
C1=CC(=NC=C1C(=O)NCCCCC(C(=O)O)NC(=O)NC(CCC(=O)O)C(=O)O)F

NEW DRUG APPROVALS
one time
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}