
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Casirivimab
(Heavy chain)
QVQLVESGGG LVKPGGSLRL SCAASGFTFS DYYMSWIRQA PGKGLEWVSY ITYSGSTIYY
ADSVKGRFTI SRDNAKSSLY LQMNSLRAED TAVYYCARDR GTTMVPFDYW GQGTLVTVSS
ASTKGPSVFP LAPSSKSTSG GTAALGCLVK DYFPEPVTVS WNSGALTSGV HTFPAVLQSS
GLYSLSSVVT VPSSSLGTQT YICNVNHKPS NTKVDKKVEP KSCDKTHTCP PCPAPELLGG
PSVFLFPPKP KDTLMISRTP EVTCVVVDVS HEDPEVKFNW YVDGVEVHNA KTKPREEQYN
STYRVVSVLT VLHQDWLNGK EYKCKVSNKA LPAPIEKTIS KAKGQPREPQ VYTLPPSRDE
LTKNQVSLTC LVKGFYPSDI AVEWESNGQP ENNYKTTPPV LDSDGSFFLY SKLTVDKSRW
QQGNVFSCSV MHEALHNHYT QKSLSLSPGK
(Light chain)
DIQMTQSPSS LSASVGDRVT ITCQASQDIT NYLNWYQQKP GKAPKLLIYA ASNLETGVPS
RFSGSGSGTD FTFTISGLQP EDIATYYCQQ YDNLPLTFGG GTKVEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H147-H203, H223-L214, H229-H’229, H232-H’232, H264-H324, H370-H428, H’22-H’96, H’147-H’203, H’223-L’214, H’264-H’324, H’370-H’428, L23-L88, L134-L194, L’23-L’88, L’134-L’194)
Casirivimab
カシリビマブ;
- Immunoglobulin G1, anti-(severe acute respiratory syndrome coronavirus 2 spike glycoprotein) (human monoclonal REGN10933 γ1-chain), disulfide with human monoclonal REGN10933 κ-chain, dimer
| Formula | C6454H9976N1704O2024S44 |
|---|---|
| CAS | 2415933-42-3 |
| Mol weight | 145233.3296 |
Monoclonal antibody
Treatment and prophylaxis of SARS-CoV-2 infection (COVID-19)
SARS-CoV-2 spike glycoprotein
- Protein Sequence
- Sequence Length: 1328, 450, 450, 214, 214
- REGN 10933
- RG 6413
https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-authorizes-monoclonal-antibodies-treatment-covid-19 November 21, 2020
Today, the U.S. Food and Drug Administration issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 years of age or older weighing at least 40 kilograms [about 88 pounds]) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19. This includes those who are 65 years of age or older or who have certain chronic medical conditions.
In a clinical trial of patients with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in patients at high risk for disease progression within 28 days after treatment when compared to placebo. The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.
Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.
Casirivimab and imdevimab are not authorized for patients who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19. A benefit of casirivimab and imdevimab treatment has not been shown in patients hospitalized due to COVID-19. Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized patients with COVID-19 requiring high flow oxygen or mechanical ventilation.
“The FDA remains committed to advancing the nation’s public health during this unprecedented pandemic. Authorizing these monoclonal antibody therapies may help outpatients avoid hospitalization and alleviate the burden on our health care system,” said FDA Commissioner Stephen M. Hahn, M.D. “As part of our Coronavirus Treatment Acceleration Program, the FDA uses every possible pathway to make new treatments available to patients as quickly as possible while continuing to study the safety and effectiveness of these treatments.”
Monoclonal antibodies are laboratory-made proteins that mimic the immune system’s ability to fight off harmful pathogens such as viruses. Casirivimab and imdevimab are monoclonal antibodies that are specifically directed against the spike protein of SARS-CoV-2, designed to block the virus’ attachment and entry into human cells.
“The emergency authorization of these monoclonal antibodies administered together offers health care providers another tool in combating the pandemic,” said Patrizia Cavazzoni, M.D., acting director of the FDA’s Center for Drug Evaluation and Research. “We will continue to facilitate the development, evaluation and availability of COVID-19 therapies.”
The issuance of an EUA is different than an FDA approval. In determining whether to issue an EUA, the FDA evaluates the totality of available scientific evidence and carefully balances any known or potential risks with any known or potential benefits of the product for use during an emergency. Based on the FDA’s review of the totality of the scientific evidence available, the agency has determined that it is reasonable to believe that casirivimab and imdevimab administered together may be effective in treating patients with mild or moderate COVID-19. When used to treat COVID-19 for the authorized population, the known and potential benefits of these antibodies outweigh the known and potential risks. There are no adequate, approved and available alternative treatments to casirivimab and imdevimab administered together for the authorized population.
The data supporting this EUA for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms. Of these patients, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline. Viral load reduction in patients treated with casirivimab and imdevimab was larger than in patients treated with placebo at day seven. However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment. For patients at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated patients on average compared to 9% in placebo-treated patients. The effects on viral load, reduction in hospitalizations and ER visits were similar in patients receiving either of the two casirivimab and imdevimab doses.
Under the EUA, fact sheets that provide important information about using casirivimab and imdevimab administered together in treating COVID-19 as authorized must be made available to health care providers and to patients and caregivers. These fact sheets include dosing instructions, potential side effects and drug interactions. Possible side effects of casirivimab and imdevimab include: anaphylaxis and infusion-related reactions, fever, chills, hives, itching and flushing.
The EUA was issued to Regeneron Pharmaceuticals Inc.
The FDA, an agency within the U.S. Department of Health and Human Services, protects the public health by assuring the safety, effectiveness, and security of human and veterinary drugs, vaccines and other biological products for human use, and medical devices. The agency also is responsible for the safety and security of our nation’s food supply, cosmetics, dietary supplements, products that give off electronic radiation, and for regulating tobacco products.
Related Information
- Casirivimab and Imdevimab EUA Letter of Authorization
- Frequently Asked Questions on the Emergency Use Authorization for Casirivimab and Imdevimab
- Emergency Use Authorization: Therapeutics
- Coronavirus Disease (COVID-19)
Casirivimab/imdevimab, sold under the brand name REGEN-COV,[1] is an experimental medicine developed by the American biotechnology company Regeneron Pharmaceuticals. It is an artificial “antibody cocktail” designed to produce resistance against the SARS-CoV-2 coronavirus responsible for the COVID-19 pandemic.[3][4] It consists of two monoclonal antibodies, casirivimab (REGN10933) and imdevimab (REGN10987) that must be mixed together.[1][5][6] The combination of two antibodies is intended to prevent mutational escape.[7]
Trials
In a clinical trial of people with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in people at high risk for disease progression within 28 days after treatment when compared to placebo.[2] The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continues to be evaluated.[2]
The data supporting the emergency use authorization (EUA) for casirivimab and imdevimab are based on a randomized, double-blind, placebo-controlled clinical trial in 799 non-hospitalized adults with mild to moderate COVID-19 symptoms.[2] Of these participants, 266 received a single intravenous infusion of 2,400 milligrams casirivimab and imdevimab (1,200 mg of each), 267 received 8,000 mg casirivimab and imdevimab (4,000 mg of each), and 266 received a placebo, within three days of obtaining a positive SARS-CoV-2 viral test.[2]
The prespecified primary endpoint for the trial was time-weighted average change in viral load from baseline.[2] Viral load reduction in participants treated with casirivimab and imdevimab was larger than in participants treated with placebo at day seven.[2] However, the most important evidence that casirivimab and imdevimab administered together may be effective came from the predefined secondary endpoint of medically attended visits related to COVID-19, particularly hospitalizations and emergency room visits within 28 days after treatment.[2] For participants at high risk for disease progression, hospitalizations and emergency room visits occurred in 3% of casirivimab and imdevimab-treated participants on average compared to 9% in placebo-treated participants.[2] The effects on viral load, reduction in hospitalizations and ER visits were similar in participants receiving either of the two casirivimab and imdevimab doses.[2]
As of September 2020, REGEN-COV is being evaluated as part of the RECOVERY Trial.[8]
On 12 April 2021, Roche and Regeneron announced that the Phase III clinical trial REGN-COV 2069 met both primary and secondary endpoints, reducing risk of infection by 81% for the non-infected patients, and reducing time-to-resolution of symptoms for symptomatic patients to one week vs. three weeks in the placebo group.[9]
Authorization
On 21 November 2020, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for casirivimab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in people twelve years of age or older weighing at least 40 kilograms (88 lb) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19.[2][10][11] This includes those who are 65 years of age or older or who have certain chronic medical conditions.[2] Casirivimab and imdevimab must be administered together by intravenous (IV) infusion.[2]
Casirivimab and imdevimab are not authorized for people who are hospitalized due to COVID-19 or require oxygen therapy due to COVID-19.[2] A benefit of casirivimab and imdevimab treatment has not been shown in people hospitalized due to COVID-19.[2] Monoclonal antibodies, such as casirivimab and imdevimab, may be associated with worse clinical outcomes when administered to hospitalized people with COVID-19 requiring high flow oxygen or mechanical ventilation.[2]
The EUA was issued to Regeneron Pharmaceuticals Inc.[2][10][12]
On 1 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) started a rolling review of data on the REGN‑COV2 antibody combination (casirivimab/imdevimab), which is being co-developed by Regeneron Pharmaceuticals, Inc. and F. Hoffman-La Roche, Ltd (Roche) for the treatment and prevention of COVID‑19.[13][14] In February 2021, the CHMP concluded that the combination, also known as REGN-COV2, can be used for the treatment of confirmed COVID-19 in people who do not require supplemental oxygen and who are at high risk of progressing to severe COVID-19.[15]
The Central Drugs Standards Control Organisation (CDSCO) in India, on 5 May 2021, granted an Emergency Use Authorisation to Roche (Genentech)[16] and Regeneron[17] for use of the casirivimab/imdevimab cocktail in the country. The announcement came in light of the second wave of the COVID-19 pandemic in India. Roche India maintains partnership with Cipla, thereby permitting the latter to market the drug in the country.[18]
Deployment
Although Regeneron is headquartered in Tarrytown, New York (near New York City), REGEN-COV is manufactured at the company’s primary U.S. manufacturing facility in Rensselaer, New York (near the state capital at Albany).[19] In September 2020, to free up manufacturing capacity for REGEN-COV, Regeneron began to shift production of its existing products from Rensselaer to the Irish city of Limerick.[20]
Regeneron has a deal in place with Roche (Genentech)[21]to manufacture and market REGEN-COV outside the United States.[10][22]
On 2 October 2020, Regeneron Pharmaceuticals announced that US President Donald Trump had received “a single 8 gram dose of REGN-COV2” after testing positive for SARS-CoV-2.[23][24] The drug was provided by the company in response to a “compassionate use” (temporary authorization for use) request from the president’s physicians.[23]
References
- ^ Jump up to:a b c “REGEN-COV- casirivimab and imdevimab kit”. DailyMed. Retrieved 18 March 2021.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Coronavirus (COVID-19) Update: FDA Authorizes Monoclonal Antibodies for Treatment of COVID-19”. U.S. Food and Drug Administration (FDA) (Press release). 21 November 2020. Retrieved 21 November 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Kelland K (14 September 2020). “Regeneron’s antibody drug added to UK Recovery trial of COVID treatments”. Reuters. Retrieved 14 September 2020.
- ^ “Regeneron’s COVID-19 Response Efforts”. Regeneron Pharmaceuticals. Retrieved 14 September 2020.
- ^ Morelle R (14 September 2020). “Antibody treatment to be given to Covid patients”. BBC News Online. Retrieved 14 September2020.
- ^ “Safety, Tolerability, and Efficacy of Anti-Spike (S) SARS-CoV-2 Monoclonal Antibodies for Hospitalized Adult Patients With COVID-19”. ClinicalTrials. 3 September 2020. Retrieved 14 September2020.
- ^ Baum A, Fulton BO, Wloga E, Copin R, Pascal KE, Russo V, et al. (August 2020). “Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies”. Science. 369 (6506): 1014–1018. Bibcode:2020Sci…369.1014B. doi:10.1126/science.abd0831. PMC 7299283. PMID 32540904.
- ^ “RECOVERY COVID-19 phase 3 trial to evaluate Regeneron’s REGN-COV2 investigational antibody cocktail in the UK”. Recovery Trial. Retrieved 14 September 2020.
- ^ “Phase III prevention trial showed subcutaneous administration of investigational antibody cocktail casirivimab and imdevimab reduced risk of symptomatic COVID-19 infections by 81%”. streetinsider.com. Archived from the original on 2021-04-12. Retrieved 2021-04-12.
- ^ Jump up to:a b c “Regeneron Reports Positive Interim Data with REGEN-COV Antibody Cocktail used as Passive Vaccine to Prevent COVID-19”(Press release). Regeneron Pharmaceuticals. 26 January 2021. Retrieved 19 March 2021 – via PR Newswire.
- ^ “Fact Sheet For Health Care Providers Emergency Use Authorization (EUA) Of Casirivimab And Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
- ^ “Casirivimab and Imdevimab”. Regeneron Pharmaceuticals. Retrieved 19 March 2021.
- ^ “EMA starts rolling review of REGN‑COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 1 February 2021. Retrieved 1 February 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “EMA reviewing data on monoclonal antibody use for COVID-19” (Press release). European Medicines Agency (EMA). 4 February 2021. Retrieved 4 March 2021.
- ^ “EMA issues advice on use of REGN-COV2 antibody combination (casirivimab / imdevimab)” (Press release). European Medicines Agency (EMA). 26 February 2021. Retrieved 5 March 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ https://timesofindia.indiatimes.com/india/india-approves-roche/regeneron-antibody-cocktail-to-treat-covid-19/articleshow/82407551.cms
- ^ “Roche receives Emergency Use Authorisation in India for its investigational Antibody Cocktail (Casirivimab and Imdevimab) used in the treatment of Covid-19 | Cipla”. http://www.cipla.com. Retrieved 2021-05-06.
- ^ Williams, Stephen (3 October 2020). “Experimental drug given to President made locally”. The Daily Gazette.
- ^ Stanton, Dan (11 September 2020). “Manufacturing shift to Ireland frees up US capacity for Regeneron’s COVID antibodies”. BioProcess International.
- ^https://www.businesswire.com/news/home/20200818005847/en/Genentech-and-Regeneron-Collaborate-to-Significantly-Increase-Global-Supply-of-REGN-COV2-Investigational-Antibody-Combination-for-COVID-19
- ^ “Roche and Regeneron link up on a coronavirus antibody cocktail”. CNBC. 19 August 2020. Retrieved 14 September 2020.
- ^ Jump up to:a b Thomas K (2 October 2020). “President Trump Received Experimental Antibody Treatment”. The New York Times. ISSN 0362-4331. Retrieved 2 October 2020.
- ^ Hackett DW (3 October 2020). “8-Gram Dose of COVID-19 Antibody Cocktail Provided to President Trump”. http://www.precisionvaccinations.com. Archived from the original on 3 October 2020.
External links
- “Casirivimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Imdevimab”. Drug Information Portal. U.S. National Library of Medicine.
- “Casirivimab and Imdevimab EUA Letter of Authorization” (PDF). U.S. Food and Drug Administration (FDA).
- “Frequently Asked Questions on the Emergency Use Authorization of Casirivimab + Imdevimab” (PDF). U.S. Food and Drug Administration (FDA).
| REGN10933 (blue) and REGN10987 (orange) bound to SARS-CoV-2 spike protein (pink). From PDB: 6VSB, 6XDG. | |
| Combination of | |
|---|---|
| Casirivimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Imdevimab | Monoclonal antibody against spike protein of SARS-CoV-2 |
| Clinical data | |
| Trade names | REGEN-COV |
| Other names | REGN-COV2 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Casirivimab |
| Routes of administration | Intravenous |
| ATC code | None |
| Legal status | |
| Legal status | US: Unapproved (Emergency Use Authorization)[1][2] |
| Identifiers | |
| DrugBank | DB15691 |
| KEGG | D11938 |
//////////// Casirivimab, ANTI VIRAL, PEPTIDE, SARS-CoV-2, MONOCLONAL ANTIBODY, FDA 2020, 2020APPROVALS, CORONA VIRUS, COVID 19, カシリビマブ, REGN-COV2, REGN10933+REGN10987 combination therapy, REGN 10933, RG 6413
NEW DRUG APPROVALS
ONE TIME
$10.00
OCID 5090, Enmetazobactam
OCID 5090
Enmetazobactam
Beta-lactamase inhibitor.
AAI-101
RN: 1001404-83-6
UNII: 80VUN7L00C
2/22/2024 FDA APPROVED, To treat complicated urinary tract infections, Exblifep
Molecular Formula, C11-H14-N4-O5-S, Molecular Weight, 314.3206
(2S,3S,5R)-3-Methyl-3-((3-methyltriazol-3-ium-1-yl)methyl)-4,4,7-trioxo-4^6-thia-1-azabicyclo(3.2.0)heptane-2-carboxylate
- 1H-1,2,3-Triazolium, 3-(((2S,3S,5R)-2-carboxy-3-methyl-4,4-dioxido-7-oxo-4-thia-1-azabicyclo(3.2.0)hept-3-yl)methyl)-1-methyl-, inner salt
- Enmetazobactam
The Board of directors of Orchid Pharma Ltd has announced that the company had developed a new molecule known as OCID-5090, which was licensed to a company named Allecra Therapeutics, this molecule was undergoing the clinical trials and the company is happy to announce that the molecule has cleared the Phase 3 clinical trials.
Allecra Therapeutics would now either directly or through out license file for NDA of this molecule. Allecra has already out licensed the product to Haini Pharmaceuticals, China for the Chinese Territory at a value of $78mn plus royalties.
As per the IP Agreement between Orchid Pharma Limited and Allecra Therapeutics, Orchid is entitled to receive a Royalty of 6-8% on the worldwide sales of the product. Therefore, once the molecule is commercialised, Orchid can expect a regular stream of Royalty from Allecra. Further, the rights to develop and commercialise the molecule in India (which is under patent protection) remain with Orchid Pharma Limited, and the company is evaluating the various options to commercialise the product.
Orchid had developed a new molecule known as OCID-5090, which was licensed to a company named Allecra Therapeutics, this molecule was undergoing the clinical trials and the molecule has cleared the Phase 3 clinical trials.
Allecra Therapeutics would now either directly or through out license file for NDA of this molecule. Allecra has already out licensed the product to Haini Pharmaceuticals, China for the Chinese Territory at a value of $78mn plus royalties.
As per the IP Agreement between Orchid Pharma Limited and Allecra Therapeutics, Orchid is entitled to receive a Royalty of 6-8% on the worldwide sales of the product. Therefore, once the molecule is commercialised, Orchid can expect a regular stream of Royalty from Allecra. Further, the rights to develop and commercialise the molecule in India (which is under patent protection) remain with Orchid Pharma Limited, and the company is evaluating the various options to commercialise the product.
INVENTOR

Senthilkumar U P
ORCHID
Summary of Profile of Dr. U. P. Senthilkumar, R&D Centre, Orchid Pharma Ltd.
Dr. U. P. Senthilkumar Ph.D., the principal inventor of novel beta-lactamase inhibitor, OCID5090, is currently serving as the senior vice-president at Orchid’s Research and Development Centre, Chennai.
With illustrious credentials — top ranks in B.Sc. and M.Sc. degrees, first rank in the Graduate Aptitude Test in Engineering (GATE), UGC-CSIR Junior Research Fellowship (JRF) and the prestigious Dr. K.S. Krishnan Fellowship from the Department of Atomic Energy (DAE) and publication of M.Sc. project work in the Indian Journal of Chemistry in 1987 — Mr. Senthilkumar chose to pursue his doctoral research in synthetic organic chemistry with his mentor Prof.
Ramasubbu Jeyaraman at Bharathidasan University, Tiruchirapalli. His research focus on the conformational preferences of sterically challenged novel N-Nitroso heterocycles and their conformation dependent anti-cancer properties, led to the publication of 9 articles in reputed peer-reviewed international journals – a commendable accomplishment in the 90s.
After a brief post-doctoral stint on fluorescent dicyclopentapyridines, Dr. U. P. Senthilkumar joined Torrent Research Centre at Ahmedabad and started his new endeavor of drug discovery on ACE inhibitors. At the process research and development laboratory, he was actively involved in asymmetric and stereo-selective synthesis of Active Pharmaceutical Ingredients (APIs), and exploited the full potential of chiral prep-HPLC to realize the target molecules.
After joining Orchid Pharma Ltd., Chennai, Dr. Senthilkumar led the efforts in the development of differentiated and patentable manufacturing processes for APIs related to both non-antibiotics and beta-lactam antibiotics. He played a significant role in successfully implementing the manufacturing processes overcoming several challenging problems. In addition, his scientific insights and breath of understanding on the patent landscape were invaluable and impactful in
creating significant value to the organization and growth of the company in realizing the mission to become a leader in the pharmaceutical generic business.
One finds more than 100 articles/patents/publications to his credit, which include inventions on new drugs, drug-intermediates, products, processes, new synthetic routes, rearrangements and novel polymorphs. As a Leadership Persona of the IP management team, he had exhibited a thoroughness of the science/invention and meticulously executed the task of prosecution of few hundred patents in many countries from both New Drug Discovery and Process Chemistry space.
All the successful effort earned Orchid Pharma Ltd the National Intellectual Property Award from the Department of Industrial Policy and Promotion, Ministry of Commerce and Industry,Government of India.
Through his executive and decision-making skills combined with scientific rationale and clarity, Dr. Senthilkumar played significant role in the selection of products and creation of generic product portfolio for Orchid, with unique IP strategies, analysis of patents, patent mapping, designing & developing invalidation/non-infringing positions, and early launch opportunity, including first-to-file (FTF) positioning. His appearance in the US courts, for deposition in couple of patent litigations, and successful accomplishment of the same are testimony to his depth, thoroughness of science and the ability to defend the invention with grit and professionalism.
Additionally, his effectual role in the first-to-launch of one of the large volume sterile penicillins with regulatory exclusivity, achieved successfully by overcoming the citizen petition process in the regulatory pathway, is another shining example of his leadership and scientific strength.
To support in-house projects as well as multinational pharma majors, Dr. Senthilkumar has taken up CRAMS (Contract Research and Manufacturing Services) and CMC (Chemistry, Manufacturing and Control) for new chemical entities. Besides, he passionately focused on novel beta-lactamase inhibitors and their antibiotic combinations that were envisaged by him to exhibit potent activity against multi-drug resistant bacteria. His dedicated effort brought a novel extended spectrum beta-lactamase inhibitor, OCID5090, which was out-licensed to Allecra Inc. OCID5090/cefepime combination has completed successfully the Phase III clinical trials for treating complicated urinary-track-infections (cUTI), including acute pyelonephritis (AP), and rightfully, OCID5090 has gotten the US FDA fast track designation as a Qualified Infectious Disease product (QIDP) that provides a five-year additional market exclusivity and priority review.
His never-ending passion for research is infectious and roped him with academic institutions to explore novel technologies including electron-beam irradiated heterogeneous catalysis. His commendable knowledge on intellectual property is being utilized by the IP Cells of various institutions as well as the Tamil Nadu State Technology Development and Promotion Centre.
A sincere student he is, Dr. Senthilkumar is also a founder-member of Prof. Ramasubbu Jeyaraman Science Foundation (RJSF). Since 2011, he has been playing a significant role in rganizing several academic events (seminars, work-shops, invited lectures, state-level proficiency tests, and research-orientation programs) for post-graduate chemistry students to create passion for research. His concern and help for poor and rural students show his human face.
SYN

PATENT
US 20080015156
https://patents.google.com/patent/US20080015156A1/en
- [0050]
- [0051]To a suspension of (2S,3S,5R)-3-methyl-7-oxo-3-(1H-1,2,3-triazol-1-ylmethyl)-4-thia-1-azabicyclo-[3.2.0]heptane-2-carboxylic acid 4,4-dioxide (25 g) in acetone (100 mL) at 25-30° C. was added slowly N,O-bis(silylacetamide) (18.6 g) with stirring. The reaction mixture was stirred at this temperature (25-30° C.) for 15-20 min. To the clear solution obtained, methyl iodide (100 mL) was added over a period of 15 min. and stirred at 25-30 min. for 24 h. The precipitated solid was separated by filtration and washed with acetone (25 mL). Wet weight of the solid obtained was 30 g.
- [0052]The above wet solid was stirred with purified water (300 mL) at 10-15° C. for 2.5 h. To the resulted reaction mixture was added sodium thiosulfate (0.1 g) and stirred at 10-15° C. for 10-15 min. To the reaction mixture, dichloromethane (300 mL) was added, stirred and the organic layer separated. The aqueous layer was washed with a solution of Amberlite LA-2 resin (5% solution in dichloromethane twice, followed by dichloromethane twice. To the aqueous solution, activated carbon (1 g) was added, stirred for 15 min, filtered and washed with purified water (25 mL). The solution was filtered and lyophilized to get the title compound in pure form (10 g). 1H NMR (400 MHz, DMSO) δ ppm: 1.39 (s, 3H), 3.14 (dd, J=16.0, 1.3 Hz, 1H), 3.55 (dd, J=16.0, 4.2 Hz, 1H), 3.97 (s, 1H), 4.34 (s, 3H), 5.05 (dd, J=4.2, 1.3 Hz, 1H), 5.29 (d, J=14.7 Hz, 1H), 5.42 (d, J=14.7 Hz, 1H), 8.91 (d, J=1.3 Hz, 1H), 8.99 (d, J=1.3 Hz, 1H). Mass m/z: M+1 peak at 315. Alternatively the solution could be subjected to spray-drying to yield the title compound.
PATENT
WO 2012070071
IN 2010CH03555
US 20140057888
PATENT
WO 2015173378
Scheme 1
Examples
Synthesis of (2535.5R)-3-methyl-3-((3-methyl-lH-1.2 -triazol-3-ium-l-yl)methvn-7-oxo-4-thia-l-azabicyclor3.2.01heptane-2-carboxylate 4,4-dioxide (4),
Compound (4) was prepared according to Scheme 2.
Scheme 2
i) Ν,Ο-bis-trimethylsilylacetamide, CH2CI2; ii) CH3OTf; iii) Na 2-ethylhexanoate
In a round bottom flask under nitrogen flow 100 g of Tazobactam acid (1) and 500 mL of Dichloromethane are loaded. The temperature is adjusted to +30/35°C then 37 g of Ν,Ο-Bis(trimethylsilyl) acetamide are loaded in 15-20 minutes maintaining the temperature to +35/42°C. The mixture is heated to reflux (+40/42°C) for 60 minutes. If the solution is not clear, N,0-Bis(trimethylsilyl) acetamide is loaded in small portions (0,5-1.0 g each) waiting 15 minutes every time till a clear solution containing intermediate (2) is obtained. 0.55 moles of N,0-Bis(trimethylsilyl) acetamide is used, with further 0.1-0.2 equivalents being added if the reaction is not complete.
Then the temperature is cooled down to 0/+5°C and 70 g of Methyl trifluoromethanesulfonate are loaded in 60-90 minutes maintaining the temperature at 0/+5°C. After 30 minutes the reaction is monitored by HPLC to control the disappearance of intermediate (2) and formation of intermediate (3). The reaction is monitored every 30 minutes until completion.
In a round bottom flask, under nitrogen, are loaded 500 mL of Ethanol and 55 g of Sodium 2-Ethylhexanoate and the temperature is adjusted to +20/25°C, then the reaction solution containing intermediate (3) is added in 60-90 minutes maintaining the temperature of +20/25 °C under vigorous stirring. The suspension is stirred for 30 minutes then is filtered and washed with 300 mL of Ethanol followed by 500 mL of Dichloromethane under nitrogen. The crude product (4) is dried under nitrogen flow till constant weight (150 g) is obtained. The crude product compound (4) was isolated as a solid product (HPLC assay = 70%, yield = 80%).
Purification of (2tS’,3^5^)-3-methyl-3-((3-methyl-lH-l,2,3-triazol-3-ium-l-yl)methyl)-7-oxo-4-thia-l-azabicyclor3.2.01heptane-2-carboxylate 4,4-dioxide (4)
In a round bottom flask 800 mL of Dimethylformamide are loaded, the temperature is adjusted to +20/25°C then crude Compound 4 (150g) obtained above is loaded using 100 mL of Dimethylformamide to facilitate the transfer. The mixture is stirred for 5 minutes and a solution is obtained, then and after a few minutes crystallization takes place. The suspension is stirred for about 3 hours, then is cooled to 0/+5°C and stirred for another 3 hours.
The solid is filtered and washed with 300 mL of Dimethylformamide pre-cooled to 0/+5°C. Compound 4 is then suspended in 700 mL of Ethyl acetate and the temperature is adjusted to +40/45°C. The suspension is stirred for 30 minutes then the solid is filtered and washed with 150 mL of Ethyl acetate pre-heated to +40/45°C. The suspension with
Ethyl acetate is repeated twice. Finally Compound 4 is dried under vacuum at +40°C till constant weight is achieved (66 g, HPLC assay = 99%, yield = 76%).
Compound 4 Sterile filtration and recrystallization Procedure
In a round bottom flask 350 mL of Methanol are loaded, the temperature is adjusted to +30/35°C then 100 g of Compound 4 are loaded and finally the flask is washed with 60 mL of Methanol. After 5-10 minutes a solution is obtained. The solution is diluted with 330 mL of acetone adjusting the temperature to +20/+25°C. The obtained solution is treated with 2,2 g of charcoal for 20 minutes then filtered using a 0.22microM filter and the filter is washed with a mixture of 13 mL of Methanol and 110 mL of Acetone. The temperature of the solution is adjusted to +30/35°C and under vigorous stirring 830 mL of Acetone are loaded in about 15-20 minutes. After stirring for 60 minutes at temperature of +30/35°C 1170 mL of Acetone are loaded in 45-60 minutes. Then the temperature is adjusted to +20/25 °C in about 30-60 minutes and maintained for 30 minutes. The obtained crystalline solid is filtered and washed with 430 mL of Acetone. Finally the product is dried under vacuum at +40°C till constant weight is achieved (83 g of Compound 4) are obtained with an HPLC assay = 98-99%, yield =t 80%).

Mr. Ram Gopal Agarwal
Chairman and Non-Executive Director
- Mr. Ram Gopal Agarwal is Founder Chairman of Dhanuka Group.
- He is a decisive and action oriented visionary who took over a sick pesticide Company named Northern Mineral Pvt. Ltd. in 1980 and transformed it today into a Rs 1000 Crore organization called Dhanuka Agritech Ltd.
- His deep commitment and inspiring leadership in initial turbulent days is an example worth inculcating and his passion to contribute to Indian Agriculture is commendable.
- His ability to prioritize and deal effectively with a number of tasks simultaneously reinforced with the skills to make effective decisions, has metamorphosed the business venture into one of the fastest growing Agrochemical Company in India which has thrice been rated as ‘Best under a Billion Company’ by Forbes Magazine.
- In order to achieve his aspiration of “Transforming India through Agriculture” he has dedicated himself to bring changes in Agrochemicals Industry and the farming community. His contribution for adopting newer farming techniques at the grass root level, judicious use of agro chemicals in farming and imparting knowledge through his nationwide network of distributors and Dhanuka Doctors in field has resulted in the overall prosperity of farmers.
- Mr. Ram Gopal Agarwal has been the past Chairman of CCFI, (Crop Care Federation of India) the apex Chamber of all Indian Agrochemical majors. He is also Chairman of Advisory Committee of AGRO Chemicals Federation of India.
- Mr. Ram Gopal Agarwal, Group Chairman, has been bestowed with many Awards for his tremendous contribution in Agro Industry like “Life Time Achievement Award” by Agri Business Summit and Agri Awards 2019, “Distinguished Contribution to Indian Agrochemicals Industry” during India Chem 2016 International Conference organised by FICCI etc.

Mr. Manish Dhanuka
Managing Director
- Mr. Manish Dhanuka is the Director of Orchid Pharma Limited; he has the vision to rejuvenate Orchid Pharma Ltd. and take it on a fruitful path. His wide-ranging experience of handling operations, commercial, marketing and finance in the manufacturing industry provides for his analytical and decision-making skills facilitating the restoration of the company to its glorious past and to achieve even greater heights.
- He excels in creating economical Pharmaceutical technologies and accelerated evaluation process for improving healthcare. Experience of 25 years in research, evaluation, and teaching in the pharmaceutical industry equips him with the expertise in innovative pharmaceutical technologies…
- He holds a B.Tech in Chemical Engineering from IIT, New Delhi, and M.S in Chemical Engineering from the University of Akron, USA.
- Before establishing Dhanuka Laboratories Ltd. in 1993, he began his career at Ranbaxy Labs Ltd. in New Delhi and worked there for 5 years. His vision and strategy to grow the Pharmaceutical industry in the Indian sub-continent, have helped the Dhanuka Group of companies enhance its Bulk Drugs manufacturing arm exponentially. He spearheaded the acquisition of Synmedic Laboratories in the year 2013 which is involved in pharmaceutical formulations. This entrepreneurial vigor enabled him to take over the operations of Orchid Pharma Ltd. in March 2020.
- Outside of work, he likes to travel for wildlife adventures.

Mr. Mridul Dhanuka
Whole-Time Director
He is associated with Dhanuka Group Ltd. since 2005. He was responsible in successfully realigning the entire supply chain vertical from procurement to sales. At Orchid, he hopes to replicate the Group’s success and put another feather in Dhanuka cap.
CLIP

Orchid Chemicals & Pharmaceuticals, or Orchid Pharma since its recent name change in 2015, was established in 1992 in Chennai to manufacture antibiotics, and entered drug discovery in 2001 with projects in the areas of anti-infectives and treatments for pain.32, 197 In 2002, the company engaged in a joint venture to develop US-based firm Bexel Biotechnology’s BLX-1002, an oral, non-PPAR AMPK activator for the treatment of diabetes,198 later repositioned for NASH (2012), but no further progress has been reported recently.197 In 2008, Orchid invested in Diakron Pharmaceuticals, a US-based company that had an exclusive license to MSD′s investigational oral anticoagulant drug, a direct thrombin inhibitor later known as DPOC-4088 (or DP-4088),199 which reached Phase 1 clinical studies in Europe in 2012 (Supporting Information Table 6b, entries 5–6).200 The company’s own internal discovery efforts had a broad therapeutic focus, covering infectious diseases, inflammation, pain, oncology, metabolic disorders, and CNS diseases. OCID-2987,197, 201 a PDE4 inhibitor for the treatment of inflammatory disorders such as COPD, completed successfully Phase 1 studies in Europe in 2012, and OCID-4681 29,202, 203 a histone deacetylase (HDAC) inhibitor for cancer had received approval in 2011 for Phase 1 studies for solid tumors in India, but we assume both have been abandoned, as cancer and inflammation are not mentioned in the company’s latest annual reports.197 Two additional compounds were abandoned at the preclinical stage: OCID-5005, a STAT-3/IL-6 inhibitor for oncology, and a unnamed Th1/Th2 cytokine synthesis inhibitor for inflammation (Supporting Information Table 2a, entries 134–138).197 Financial issues led Orchid, as of 2009, to sell parts of its business to Hospira (now part of Pfizer). As a consequence, no progress has been reported on its discovery programs since 2010, and no further NCE patent application has been published since 2012. However, in 2013 Orchid licensed its broad-spectrum β-lactamase inhibitor OCID-5090, a zwitterionic N-methylated tazobactam derivative, to the German Allecra Therapeutics for a 20 % stake in the company, for use in combination with antibiotics to treat multidrug-resistant gram negative bacteria.204–207 Allecra’s lead compound AAI202, a combination of cefepime and AAI101/OCID-5090 30, is currently in Phase 1 studies in France.208, 209
NEW DRUG APPROVALS
ONE TIME
$10.00

Dr. B. Gopalan
Scientific Advisor
Dr. Gopalan is a synthetic organic chemist with extensive experience in the field of drug discovery and development. After completing his PhD from University of Madras, he went to Harvard University where he worked with the Nobel Laureate, Prof. E.J. Corey, as a post-doctoral fellow. Subsequent to this he joined Syntex Research Inc. in California to work on the synthesis of unnatural amino acids. After a year, he moved to Bristol-Meyers Squibb, Princeton, New Jersey, to contribute to their program on novel antibiotics and ACE inhibitors. Dr. Gopalan then moved back to India in 1982 to join the Drug Discovery Research Division of Boots Pharmaceuticals (India) Ltd. in Mumbai. Over his decade long stint there he contributed extensively to their drug discovery program, and one of the product candidates that he developed went up to Phase-2 clinical trials in both USA and UK. He then moved to Sun Pharma Advanced Research Center as Vice-President and, after a year, took up the position as General Manager at Glaxo (India) Ltd. in 1993. Here, he worked in a broad range of areas that included process development, synthesis of impurities of APIs, and generation of small molecule libraries to support drug discovery efforts to Glaxo, France. In 1999 he took over as Senior Vice President of the Drug Discovery Chemistry Division of Glenmark Pharmaceuticals Ltd. where he was involved in the design and development of inhibitors for PDE IV and DPP IV, as well as agonists for CB2. After a 6-year stint at Glenmark, Dr. Gopalan joined Matrix Laboratories Ltd. as CSO and Executive Vice-President, where he successfully helped to develop novel and selective inhibitors for PDE4 and DPP4. Five years later he became CSO and Executive Director of Orchid Pharmaceuticals Ltd in Chennai. He served in this capacity for close to a decade, contributing extensively to drug design and development in the broad segments of oncology, anti-infectives, and anti-inflammatory & metabolic disorders. Since 2017, Dr. Gopalan has been associated with CSIR-Indian Institute of Chemical Technology as a Scientific Advisor.
Dr. Gopalan’s illustrious career is endowed with numerous successes. He has been inventor, or co-inventor, of several drugs or candidate drugs. These include the novel potassium channel blockers BTS-67582 (BTI-2927) for tpe-2 diabetes, the PDE IV inhibitors Oglemilast (COPD) and Revamilast (RA); DPP IV inhibitor Melogliptin; a selective Cannaboid-2 agonist Tedalinib (Neuropathic pain); a Beta lactamase inhibitor Enmetazobactum (OCID-5090); OCID-18034 (an inhibitor of KPC enzyme); and OCID-18174 (an inhibitor of P. arugenosa). Most of these compounds were out-licensed to major international pharmaceutical companies such as Forest Laboratories Inc. USA, Teijin of Japan, Merck KGaA of Germany, Allecra of Switzerland, and Merck & Co. USA. Dr.Gopalan has 34 publications in National and International Journals, has contributed a Chapter,Co-authored with Professor K.K.Balasubramanian (IITM) on Applications of Click Chemistry in Drug Discovery and Development in a Book on Click reaction in Organic Synthesis, published by Wiley-VCH VERLAG GmbH &Co,KGaA, Weinheim,Germany,Chapter 2, p 25-70,2016, edited by Prof. S. Chandrasekharan (IISc,Bangalore) & 51 Patents.
Commensurate with his achievements, Dr. Gopalan has also received many awards. The more prominent of these include Inventor’s award by Glenmark (2004), Ranbaxy Science Foundation Award in Pharmaceutical Sciences (2005), and the Lifetime Achievement Award in the Field of Chemistry from Vels University (2011).
//////////OCID 5090, AAI-101, AAI 101, Enmetazobactam, ORCHID, Allecra Therapeutics, PHASE 3
C[n+]1ccn(C[C@@]2(C)[C@@H](N3[C@@H](CC3=O)S2(=O)=O)C(=O)[O-])n1
Idecabtagene vicleucel
Idecabtagene vicleucel
CAS 2306267-75-2
STN: BLA 125736
An autologous T lymphocyte-enriched cell transduced ex vivo with an anti-BCMA CAR lentiviral vector encoding a chimeric antigen receptor CAR, comprising a CD8 hinge and TM domain, 4-1BB costimulatory domain and CD3ζ signaling domain, targeting human B cell maturation antigen for cancer immunotherapy (Celgene Corp., NJ)
- Bb2121
| Name | Idecabtagene vicleucel (USAN); Abecma (TN) |
|---|---|
| Product | ABECMA (Celgene Corporation) |
| CAS | 2306267-75-2 |
| Efficacy | Antineoplastic, Anti-BCMA CAR-T cell |
| Disease | Multiple myeloma [DS:H00010] |
| Comment | Cellular therapy product |
USFDA 2021/4/21 APPROVED
Dendritic cells (DCs) are antigen-presenting cells (APCs) that process antigens and display them to other cells of the immune system. Specifically, dendritic cells are capable of capturing and presenting antigens on their surfaces to activate T cells such as cytotoxic T cells (CTLs). Further, activated dendritic cells are capable of recruiting additional immune cells such as macrophages, eosinophils, natural killer cells, and T cells such as natural killer T cells.
Despite major advances in cancer treatment, cancer remains one of the leading causes of death globally. Hurdles in designing effective therapies include cancer immune evasion, in which cancer cells escape destructive immunity, as well as the toxicity of many conventional cancer treatments such as radiation therapy and chemotherapy, which significantly impacts a patient’s ability to tolerate the therapy and/or impacts the efficacy of the treatment.
Given the important role of dendritic cells in immunity, derailed dendritic cell functions have been implicated in diseases such as cancer and autoimmune diseases. For example, cancer cells may evade immune detection and destruction by crippling dendritic cell functionality through prevention of dendritic cell recruitment and activation. In addition, dendritic cells have been found in the brain during central nervous system inflammation and may be involved in the pathogenesis of autoimmune diseases in the brain.
One mechanism by which cancers evade immune detection and destruction is by crippling dendritic cell functionality through prevention of dendritic cell (DC) recruitment and activation. Accordingly, there remains a need for cancer therapies that can effectively derail tumor evasion and enhance anti-tumor immunity as mediated, for example, by dendritic cells.
NEW DRUG APPROVALS
ONE TIME
$10.00
DESCRIPTION
ABECMA is a BCMA-directed genetically modified autologous T cell immunotherapy product consisting of a patient’s own T cells that are harvested and genetically modified ex vivo through transduction with an anti-BCMA02 chimeric antigen receptor (CAR) lentiviral vector (LVV). Autologous T cells transduced with the anti-BCMA02 CAR LVV express the anti-BCMA CAR on the T cell surface. The CAR is comprised of a murine extracellular single-chain variable fragment (scFv) specific for recognizing B cell maturation antigen (BCMA) followed by a human CD8α hinge and transmembrane domain fused to the T cell cytoplasmic signaling domains of CD137 (4-1BB) and CD3ζ chain, in tandem. Binding of ABECMA to BCMA-expressing target cells leads to signaling initiated by CD3ζ and 4-1BB domains, and subsequent CAR-positive T cell activation. Antigen-specific activation of ABECMA results in CAR-positive T cell proliferation, cytokine secretion, and subsequent cytolytic killing of BCMA-expressing cells.
ABECMA is prepared from the patient’s peripheral blood mononuclear cells (PBMCs), which are obtained via a standard leukapheresis procedure. The mononuclear cells are enriched for T cells, through activation with anti-CD3 and anti-CD28 antibodies in the presence of IL-2, which are then transduced with the replication-incompetent lentiviral vector containing the anti-BCMA CAR transgene. The transduced T cells are expanded in cell culture, washed, formulated into a suspension, and cryopreserved. The product must pass a sterility test before release for shipping as a frozen suspension in one or more patient-specific infusion bag(s). The product is thawed prior to infusion back into the patient [see DOSAGE AND ADMINISTRATION and HOW SUPPLIED/Storage And Handling].
The ABECMA formulation contains 50% Plasma-Lyte A and 50% CryoStor® CS10, resulting in a final DMSO concentration of 5%.
FDA approves idecabtagene vicleucel for multiple myeloma
On March 26, 2021, the Food and Drug Administration approved idecabtagene vicleucel (Abecma, Bristol Myers Squibb) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen (BCMA)-directed genetically modified autologous chimeric antigen receptor (CAR) T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies; 88% had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel in the dose range of 300 to 460 x 106 CAR-positive T cells. Efficacy was established based on overall response rate (ORR), complete response (CR) rate, and duration of response (DOR), as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The ORR was 72% (95% CI: 62%, 81%) and CR rate was 28% (95% CI 19%, 38%). An estimated 65% of patients who achieved CR remained in CR for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome (CRS), neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. The most common side effects of idecabtagene vicleucel include CRS, infections, fatigue, musculoskeletal pain, and hypogammaglobulinemia.
Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities that dispense the therapy must be specially certified to recognize and manage CRS and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
The recommended dose range for idecabtagene vicleucel is 300 to 460 × 106 CAR-positive T cells. View full prescribing information for Abecma.
This application was granted breakthrough therapy designation and orphan drug designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
FDA D.I.S.C.O. Burst Edition: FDA approval of ABECMA (idecabtagene vicleucel) the first FDA approved cell-based gene therapy for the treatment of adult patients with relapsed or refractory multiple myeloma
Welcome back to the D.I.S.C.O., FDA’s Drug Information Soundcast in Clinical Oncology, Burst Edition, brought to you by FDA’s Division of Drug Information in partnership with FDA’s Oncology Center of Excellence. Today we have another quick update on a recent FDA cancer therapeutic approval.
On March 26, 2021, the FDA approved idecabtagene vicleucel (brand name Abecma) for the treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy, including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody. This is the first FDA-approved cell-based gene therapy for multiple myeloma.
Idecabtagene vicleucel is a B-cell maturation antigen-directed genetically modified autologous chimeric antigen receptor T-cell therapy. Each dose is customized using a patient’s own T-cells, which are collected and genetically modified, and infused back into the patient.
Safety and efficacy were evaluated in a multicenter study of 127 patients with relapsed and refractory multiple myeloma who received at least three prior lines of antimyeloma therapies, 88% of whom had received four or more prior lines of therapies. Efficacy was evaluated in 100 patients who received idecabtagene vicleucel and was established based on overall response rate, complete response rate, and duration of response, as evaluated by an Independent Response committee using the International Myeloma Working Group Uniform Response Criteria for Multiple Myeloma.
The overall response rate was 72% and complete response rate was 28%. An estimated 65% of patients who achieved complete response remained in complete response for at least 12 months.
The idecabtagene vicleucel label carries a boxed warning for cytokine release syndrome, neurologic toxicities, hemophagocytic lymphohistiocytosis/ macrophage activation syndrome, and prolonged cytopenias. Idecabtagene vicleucel is approved with a risk evaluation and mitigation strategy requiring that healthcare facilities dispensing the therapy must be specially certified to recognize and manage cytokine release syndrome and nervous system toxicities. To evaluate long-term safety, the FDA is requiring the manufacturer to conduct a post-marketing observational study involving patients treated with idecabtagene vicleucel.
Full prescribing information for this approval can be found on the web at www.fda.gov, with key word search “Approved Cellular and Gene Therapy Products”.
Health care professionals should report serious adverse events to FDA’s MedWatch Reporting System at www.fda.gov/medwatch.
Follow the Division of Drug Information on Twitter @FDA_Drug_InfoExternal Link Disclaimer and the Oncology Center of Excellence @FDAOncologyExternal Link Disclaimer. Send your feedback via email to FDAOncology@fda.hhs.gov. Thanks for tuning in today to the DISCO Burst Edition.
PAT
WO 2019148089
In various aspects, the present invention relates to XCR1 binding agents having at least one targeting moiety that specifically binds to XCR1. In various embodiments, these XCR1 binding agents bind to, but do not functionally modulate ( e.g . partially or fully neutralize) XCR1. Therefore, in various embodiments, the present XCR1 binding agents have use in, for instance, directly or indirectly recruiting a XCR1-expressing cell to a site of interest while still allowing the XCR1-expressing cell to signal via XCR1 (i.e. the binding of the XCR1 binding agent does not reduce or eliminate XCR1 signaling at the site of interest). In various embodiments, the XCR-1 binding agent functionally modulates XCR1. In an embodiment, the targeting moiety is a single domain antibody (e.g. VHH, HUMABODY, scFv, on antibody). In various embodiments, the XCR1 binding agent further comprises a signaling agent, e.g., without limitation, an interferon, an interleukin, and a tumor necrosis factor, that may be modified to attenuate activity. In various embodiments, the XCR1 binding agent comprises additional targeting moieties that bind to other targets (e.g. antigens, receptor) of interest. In an embodiment, the other targets (e.g. antigens, receptor) of interest are present on tumor cells. In another embodiment, the other targets (e.g. antigens, receptor) of interest are present on immune cells. In some embodiments, the present XCR1 binding agent may directly or indirectly recruit an immune cell (e.g. a dendritic cell) to a site of action (such as, by way of non-limiting example, the tumor microenvironment). In some embodiments, the present XCR1 binding agent facilitates the presentation of antigens (e.g., tumor antigens) by dendritic cells.
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises the heavy chain of SEQ ID NO: 223 and/or the light chain of SEQ ID NO: 224, or a variant thereof (e.g. an amino acid sequence having at least about 90%, or at least about 93%, at least about 95%, at least about 97%, at least about 98%, at least about 99%, identity with SEQ ID NO: 223 and/or SEQ ID NO: 224).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227) and/or a light chain CDR 1 of RSSLGLVHRNGNTYLH (SEQ ID NO: 228), light chain CDR 2 of KVSHRFS (SEQ ID NO: 229), and light chain CDR 3 of SQSTFIVPWT (SEQ ID NO: 230), or a variant thereof (e.g. with four or fewer amino acid substitutions, or with three or fewer amino acid substitutions, or with two or fewer amino acid substitutions, or with one amino acid substitution).
In various embodiments, the present XCR binding agent or targeting moiety of the present chimeric proteins comprises a heavy chain CDR 1 of SHNLH (SEQ ID NO: 225), heavy chain CDR 2 of AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), and heavy chain CDR 3 of WGSVVGDWYFDV (SEQ ID NO: 227).
Illustrative Disease Modifying Therapies
EXAMPLES
Example 1. Identification and Characterization of Human XCR1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 5G7 antibody and human IFNa2 with an R149A mutation.
AFNs were made based on the 5G7 anti-hXcr1 Ab using the intact (full) Ab or a scFv format.
The 5G7 heavy chain is:
QAYLQQSGAELVRPGASVKMSCKASGYTFTSHNLHWVKQTPRQGLQWIGAIYPGNGNTAYNQKFKGKATLTVD
KSSSTAYMQLSSLTSDDSAVYFCARWGSVVGDWYFDVWGTGTTVTVSSASTKGPSVFPLAPCSRSTSESTAAL
GCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSWTVPSSNFGTQTYTCNVDHKPSNTKVDKTVE
RKCCVECPPCPAPPAAAPSVFLFPPKPKDTLMISRTPEVTCVWDVSHEDPEVQFNWYVDGVEVHNAKTKPREE
QFNSTFRVVSVLTWHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLV
KGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLS
LSPGK (SEQ ID NO: 223)
The 5G7 light chain is:
DWMTQTPLSLPVTLGNQASIFCRSSLGLVHRNGNTYLHWYLQKPGQSPKLLIYKVSHRFSGVPDRFSGSGSGT DFTLKISRVEAEDLGVYFCSQSTHVPWTFGGGTKLEIKRTVAAPSVFIFPPSDEQLKSGTASWCLLNNFYPREAK VQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC (SEQ ID NO: 224)
5G7 Heavy chain CDR 1 is SHNLH (SEQ ID NO: 225), Heavy chain CDR 2 is AIYPGNGNTAYNQKFKG (SEQ ID NO: 226), Heavy chain CDR 3 is WGSVVGDWYFDV (SEQ ID NO: 227). 5G7 Light chain CDR 1 is RSSLGLVHRNGNTYLH (SEQ ID NO: 228), Light chain CDR 2 is KVSHRFS (SEQ ID NO: 229), and Light chain CDR 3 is SQSTHVPWT (SEQ ID NO: 230).
The sequence of hulFNa2(R149A) is:
CDLPQTHSLGSRRTLMLLAQMRKISLFSCLKDRHDFGFPQEEFGNQFQKAETIPVLHEMIQQIFNLFSTKDSSAA WDETLLDKFYTELYQQLNDLEACVIQGVGVTETPLMKEDSILAVRKYFQRITLYLKEKKYSPCAWEVVRAEIMASF SLSTNLQESLRSKE (SEQ ID NO: 231).
In case of the intact Ab AFN, the 5G7 Ab heavy chain was fused to h I FN a2_R149A (human IFNal with a R149A mutation) via a flexible (GGS)2oG-linker and co-expressed with the 5G7 Ab light chain (sequences shown below). 5G7 scFv-AFN was constructed by linking the Ab VL and VH domains via a (GGGS)4 linker and followed by a (GGS)2o-linker and the sequence encoding hlFNa2_R149A. Recombinant proteins, cloned in the pcDNA3.4 expression-vector, were produced in ExpiCHO cells (Thermo Fisher Scientific) and purified on HisPUR spin plates (Thermo Fisher Scientific) according to the manufacturer’s instructions.
To test binding of the AFNs, parental HL1 16 and HL1 16 cells stably expressing hXcrl (HL116-hXcr1) were incubated with a serial dilution AFN for two hours at 4°C. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figures 1A and 1 B clearly show that both 5G7 Ab-AFN and 5G7 scFv bind specifically to hXcrl expressing cells.
Biological activity was measured on parental HL1 16 cells (an IFN responsive cell-line stably transfected with a p6-16 luciferase reporter) and the derived HL116-hXcr1 cells. Cells were seeded overnight and stimulated for 6 hours with a serial dilution 5G7 AFNs. Luciferase activity was measured on an EnSight Multimode Plate Reader (Perkin Elmer). Data in Figures 2A and 2B clearly illustrate that 5G7 AFNs, in the intact Ab format or as scFv, are clearly more active on cells expressing hXcrl compared to parental cells, illustrating that it is possible to restore signaling of an IFNa2 mutant by specific targeting to hXcrl .
Example 2. Identification and Characterization of Mouse Xcr1 Ab AFNs
As used in this Example and associated figures,“AFN” is a chimera of the anti-Xcr1 MAARX10 antibody and human IFNa2 with Q124R mutation.
Similar to the anti-human Xcr1 Ab, AFNs based on the MARX10 anti-mouse Xcr1 Ab were made, as intact Ab or as scFv. In case of the intact Ab AFN, the MARX10 Ab heavy chain was fused to hlFNa2_Q124R (human IFNa2 with Q124R mutation) via a flexible (GGS)2oG-linker and co-expressed with the MARX10 Ab light chain. scFv-AFN was constructed by linking the Ab VL and VH domains, in VH-VL (scFv(1 )) or VL-VH (scFv(2)) orientation, via a (GGGS)4 linker and followed by a (GGS)2o-linker and h I FN a2_Q 124R.
Selectivity of AFNs (produced and purified as described above for the human Xcr1 Ab AFNs) was tested by comparing binding at 2.5 pg/ml to MOCK or mouse Xcr1 transfected Hek293T cells. Binding was detected using THE™ HIS antibody-FITC (GenScript) and measured on a MACSQuant X instrument (Miltenyi Biotec) and analysed using the FlowLogic software (Miltenyi Biotec). Data in Figure 3 clearly show that all three specifically bind to mXcrl expressing cells.
REF
New England Journal of Medicine (2021), 384(8), 705-716
https://www.rxlist.com/abecma-drug.htm#indications
///////////Idecabtagene vicleucel, breakthrough therapy designation, orphan drug designation, FDA 2021, APPROVALS 2021, Bb2121, Bb , ABECMA
Manufacturer: Celgene Corporation, a Bristol-Myers Squibb Company
Indications:
- Treatment of adult patients with relapsed or refractory multiple myeloma after four or more prior lines of therapy including an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 monoclonal antibody.
Product Information
- Package Insert – ABECMA
- Demographic Subgroup Information – idecabtagene vicleucel [ABECMA]
Refer to Section 1.1 of the Clinical Review Memo for information about participation in the clinical trials and any analysis of demographic subgroup outcomes that is notable.
Supporting Documents
Loncastuximab tesirine


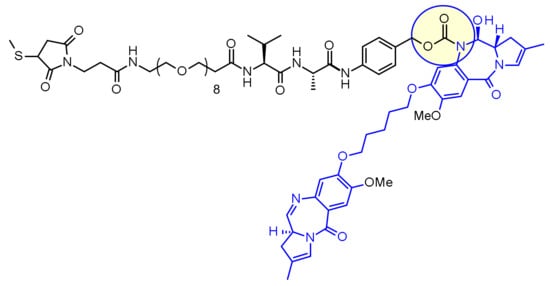
Loncastuximab tesirine
ZYNLONTA FDA APPROVED 2021/4/23
| Formula | C6544H10048N1718O2064S52 |
|---|---|
| Exact mass | 147387.9585 |
| CAS | 1879918-31-6 |
| Efficacy | Antineoplasitc, Anti-CD19 antibody |
| Disease | Diffuse large B-cell lymphoma not otherwise specified [DS:H02434] |
| Comment | Antibody-drug conjugate Treatment of hematological cancers |
ロンカスツキシマブテシリン; ADCT-402, ADCX 19
Immunoglobulin G1, anti-(human CD19 antigen) (human-Mus musculus monoclonal RB4v1.2 γ1-chain), disulfide with human-Mus musculus monoclonal RB4v1.2 κ-chain, dimer, bis(thioether) with N-[31-(3-mercapt-2,5-dioxo-1-pyrrolidinyl)-1,29-dioxo-4,7,10,13,16,19,22,25-octaoxa-28-azahentriacont-1-yl]-L-valyl-N-[4-[[[[(11S,11aS)-8-[[5-[[(11aS)-5,11a-dihydro-7-methoxy-2-methyl-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-8-yl]oxy]pentyl]oxy]-11,11a-dihydro-11-hydroxy-7-methoxy-2-methyl-5-oxo-1H-pyrrolo[2,1-c][1,4]benzodiazepin-10(5H)-yl]carbonyl]oxy]methyl]phenyl]-L-alaninamide
NEW DRUG APPROVALS
ONETIME
$10.00
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | CD19 |
| Clinical data | |
| Trade names | Zynlonta |
| Other names | ADCT-402, loncastuximab tesirine-lpyl |
| License data | US DailyMed: Loncastuximab_tesirine |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| CAS Number | 1879918-31-6 |
| DrugBank | DB16222 |
| ChemSpider | none |
| UNII | 7K5O7P6QIU |
| KEGG | D11338 |
| Chemical and physical data | |
| Formula | C6544H10048N1718O2064S52 |
| Molar mass | 147481.45 g·mol−1 |
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Zynlonta | Injection, powder, lyophilized, for solution | 5 mg/1mL | Intravenous | ADC Therapeutics America, Inc. | 2021-04-30 | Not applicable |  |
Loncastuximab tesirine-lpyl is a CD19-directed antibody and alkylating agent conjugate, consisting of a humanized IgG1 kappa monoclonal antibody conjugated to SG3199, a pyrrolobenzodiazepine (PBD) dimer cytotoxic alkylating agent, through a protease-cleavable valine–alanine linker. SG3199 attached to the linker is designated as SG3249, also known as tesirine.
|
Loncastuximab tesirine-lpyl has an approximate molecular weight of 151 kDa. An average of 2.3 molecules of SG3249 are attached to each antibody molecule. Loncastuximab tesirine-lpyl is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells, and the small molecule components are produced by chemical synthesis.
ZYNLONTA (loncastuximab tesirine-lpyl) for injection is supplied as a sterile, white to off-white, preservative-free, lyophilized powder, which has a cake-like appearance, for intravenous infusion after reconstitution and dilution. Each single-dose vial delivers 10 mg of loncastuximab tesirine-lpyl, L-histidine (2.8 mg), L-histidine monohydrochloride (4.6 mg), polysorbate 20 (0.4 mg), and sucrose (119.8 mg). After reconstitution with 2.2 mL Sterile Water for Injection, USP, the final concentration is 5 mg/mL with a pH of approximately 6.0.
Loncastuximab tesirine , sold under the brand name Zynlonta, is used for the treatment of large B-cell lymphoma. It is an antibody-drug conjugate (ADC) composed of a humanized antibody targeting the protein CD19, which is expressed in a wide range of B cell hematological tumors.[2] The experimental drug, developed by ADC Therapeutics is being tested in clinical trials for the treatment of B-cell non-Hodgkin lymphoma (NHL) and B-cell acute lymphoblastic leukemia (ALL).
On April 23, 2021, the Food and Drug Administration granted accelerated approval to loncastuximab tesirine-lpyl (Zynlonta, ADC Therapeutics SA), a CD19-directed antibody and alkylating agent conjugate, for adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, DLBCL arising from low grade lymphoma, and high-grade B-cell lymphoma.
Approval was based on LOTIS-2 (NCT03589469), an open-label, single-arm trial in 145 adult patients with relapsed or refractory DLBCL or high-grade B-cell lymphoma after at least two prior systemic regimens. Patients received loncastuximab tesirine-lpyl 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles. Patients received treatment until progressive disease or unacceptable toxicity.
The main efficacy outcome measure was overall response rate (ORR), as assessed by an independent review committee using Lugano 2014 criteria. The ORR was 48.3% (95% CI: 39.9, 56.7) with a complete response rate of 24.1% (95% CI: 17.4, 31.9). After a median follow-up of 7.3 months, median response duration was 10.3 months (95% CI: 6.9, NE). Of the 70 patients who achieved objective responses, 36% were censored for response duration prior to 3 months.
Most common (≥20%) adverse reactions in patients receiving loncastuximab tesirine-lpyl, including laboratory abnormalities, are thrombocytopenia, increased gamma-glutamyltransferase, neutropenia, anemia, hyperglycemia, transaminase elevation, fatigue, hypoalbuminemia, rash, edema, nausea, and musculoskeletal pain.
The prescribing information provides warnings and precautions for adverse reactions including edema and effusions, myelosuppression, infections, and cutaneous reactions.
The recommended loncastuximab tesirine-lpyl dosage is 0.15 mg/kg every 3 weeks for 2 cycles, then 0.075 mg/kg every 3 weeks for subsequent cycles, by intravenous infusion over 30 minutes on day 1 of each cycle (every 3 weeks). Patients should be premedicated with dexamethasone 4 mg orally or intravenously twice daily for 3 days beginning the day before loncastuximab tesirine-lpyl.
Technology
The humanized monoclonal antibody is stochastically conjugated via a valine-alanine cleavable, maleimide linker to a cytotoxic (anticancer) pyrrolobenzodiazepine (PBD) dimer. The antibody binds to CD19, a protein which is highly expressed on the surface of B-cell hematological tumors[3] including certain forms of lymphomas and leukemias. After binding to the tumor cells the antibody is internalized, the cytotoxic drug PBD is released and the cancer cells are killed. PBD dimers are generated out of PBD monomers, a class of natural products produced by various actinomycetes. PBD dimers work by crosslinking specific sites of the DNA, blocking the cancer cells’ division that cause the cells to die. As a class of DNA-crosslinking agents they are significantly more potent than systemic chemotherapeutic drugs.[4]
Clinical trials
Two phase I trials are evaluating the drug in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma and relapsed or refractory B-cell acute lymphoblastic leukemia.[5] At the 14th International Conference on Malignant Lymphoma interim results from a Phase I, open-label, dose-escalating study designed to evaluate the treatment of loncastuximab tesirine in relapsed or refractory non-Hodgkin’s lymphoma were presented.[6] Among the patients enrolled at the time of the data cutoff the overall response rate was 61% in the total patient population (42% complete response and 19% partial response) and in patients with relapsing or refractory diffuse large B-cell lymphoma (DLBCL) the overall response rate was 57% (43% complete response and 14% partial response).[7][8]
Orphan drug designation
Loncastuximab tesirine was granted Orphan Drug Designation by the U.S. Food and Drug Administration (FDA) for the treatment of diffuse large B-cell lymphoma and mantle cell lymphoma.[9]
References
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf
- ^ WHO Drug Information: International Nonproprietary Names for Pharmaceutical Substances
- ^ Wang K, Wei G, Liu D (November 2012). “CD19: a biomarker for B cell development, lymphoma diagnosis and therapy”. Experimental Hematology & Oncology. 1 (1): 36. doi:10.1186/2162-3619-1-36. PMC 3520838. PMID 23210908.
- ^ “Pyrrolobenzodiazepine”. ADC Review.
- ^ Clinical trial number NCT02669017 for “ADCT-402 in B-NHL” at ClinicalTrials.gov
- ^ Kahl B, Hamadani M, Caimi PF, Reid EG, Havenith K, He S, Feingold JM, O’Connor O (June 2017). “First clinical results of ADCT‐402, a novel pyrrolobenzodiazepine-based antibody drug conjugate (ADC), in relapsed/refractory B‐cell linage NHL” (PDF). Hematol Oncol. 35 (S2): 49–51. doi:10.1002/hon.2437_33.
- ^ “First clinical results of ADCT-402”. ADC Review.
- ^ Bainbridge K. “Grandfather fighting deadly cancer reveals scans of tumors after testing new drug”. Mirror.
- ^ “ADCT-402 Orphan Drug Designation” (PDF). ADC Therapeutics press release.
External links
- “Loncastuximab tesirine”. Drug Information Portal. U.S. National Library of Medicine.
/////////Loncastuximab tesirine, FDA 2021, APPROVALS 2021, ZYNLONTA, ロンカスツキシマブテシリン, ORPHAN DRUG, ADCT-402, priority review, ADCX 19
ZyCoV-D

ZyCoV-D
CAS 2541524-47-2
DNA vaccine construct encoding a spike protein antigen of SARS-CoV-2 virus (Zydus-Cadila)
UPDATE. APPROVED IN INDIA AUG 2021
http://ctri.nic.in/Clinicaltrials/showallp.php?mid1=51254&EncHid=&userName=ZyCoV-D
bioRxiv (2021), 1-26.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7423510/
| ZyCoV-D | (CTRI/2020/07/026352, 2020, CTRI/2020/07/026352, 2020; Myupchar, 2020) | ZYDUS CADILA |
ZyCoV-D is a genetically engineered DNA plasmid based vaccine encoding for the membrane proteins of the virus. The clinical trials to study the immunogenicity, and safety of the vaccine, will administer three doses at an interval of 28 days in 1048 individuals.
Phase 1/2: CTRI/2020/07/026352
| Vaccine description | |
|---|---|
| Target | SARS-CoV-2 |
| Vaccine type | DNA |
| Clinical data | |
| Routes of administration |
Intradermal |
| ATC code | None |
| Identifiers | |
| DrugBank | DB15892 |
| Part of a series on the |
| COVID-19 pandemic |
|---|
| SARS-CoV-2 (virus)COVID-19 (disease) |
| showTimeline |
| showLocations |
| showInternational response |
| showMedical response |
| showImpact |
| COVID-19 portal |
ZyCoV-D is a DNA plasmid based COVID-19 vaccine being developed by Cadila Healthcare with support from the Biotechnology Industry Research Assistance Council.
The ZYCOV-D vaccine candidate was developed by Cadila Healthcare Ltd. based in India1. The vaccine was developed using a DNA vaccine platform with a non-replicating and non-integrating plasmid carrying the gene of interest3. Once the plasmid DNA is introduced into host cells and the viral protein is translated, it elicits a strong immune response, stimulating the humoral and cellular components of the immune system3. The DNA vaccine platform offers minimal biosafety requirements, more improved vaccine stability, and lower cold chain requirements3. Phase I clinical trials of this vaccine candidate were completed in July 2020, with the company reporting successful dosing and tolerance1,2. As of August, 2020 the candidate is in Phase II clinical trials1.
NEW DRUG APPROVALS
ONE TIME
$10.00
Clinical research
Phase I and II trials
In February 2020, Cadila Healthcare decided to develop a DNA plasmid based COVID-19 vaccine at their Vaccine Technology Centre (VTC) in Ahmedabad.[1] The vaccine candidate was able to pass the pre-clinical trials on animal models successfully. A report of the study was made available via bioRxiv.[2] Thereafter, human trials for Phase I and II were approved by the regulator.[3]
The Phase II trials of the vaccine candidate were conducted in over 1,000 volunteers as part of the adaptive Phase I/II multi-centric, dose escalation, randomised, double-blind placebo controlled method.[4][5]
Phase III trials
In November 2020, the company announced it would test the vaccine candidate on 30,000 patients in Phase III trials.[6] The vaccine would be given out in three doses at five sites across four cities of India.[7] In January 2021, the Drugs Controller General of India (DCGI) granted permission to conduct the Phase III clinical trials for 28,216 Indian participants.[8][9]
In April 2021, the company reported that they expected to have initial data for the Phase III trials by May 2021.[10]
Production
On 23 April 2021, production of the ZyCoV-D vaccine was started, with a yearly capacity of 240 million doses. It is expected to get emergency use authorization in May or June.[11]
References
- ^ “Zydus Cadila launches a fast tracked programme to develop vaccine for the novel coronavirus, 2019-nCoV (COVID-19)”(PDF). http://www.zyduscadila.com. Cadila Healthcare.
- ^ Dey A, Rajanathan C, Chandra H, Pericherla HP, Kumar S, Choonia HS, et al. (26 January 2021). “Immunogenic Potential of DNA Vaccine candidate, ZyCoV-D against SARS-CoV-2 in Animal Models”. bioRxiv: 2021.01.26.428240. doi:10.1101/2021.01.26.428240. S2CID 231777527.
- ^ “A prospective, randomized, adaptive, phase I/II clinical study to evaluate the safety and immunogenicity of Novel Corona Virus −2019-nCov vaccine candidate of M/s Cadila Healthcare Limited by intradermal route in healthy subjects”. ctri.nic.in. Clinical Trials Registry India. 15 December 2020. CTRI/2020/07/026352. Archived from the original on 22 November 2020.
- ^ “Zydus Cadila’s ZyCov-D vaccine found to be ‘safe and immunogenic'”. @businessline. The Hindu. 24 December 2020.
- ^ Rawat K, Kumari P, Saha L (February 2021). “COVID-19 vaccine: A recent update in pipeline vaccines, their design and development strategies”. European Journal of Pharmacology. 892: 173751. doi:10.1016/j.ejphar.2020.173751. PMC 7685956. PMID 33245898.
- ^ Thacker T (7 November 2020). “Zydus Cadila to test ZyCoV-D on 30,000 patients in Phase-3 trials”. The Economic Times.
- ^ “Covid 19 vaccine in India: Zydus Cadila begins enrolment for Phase 3 trial of ZyCoV-D in 4 cities”. The Financial Express. 22 January 2021.
- ^ “DBT-BIRAC supported indigenously developed DNA Vaccine Candidate by Zydus Cadila, approved for Phase III clinical trials”. pib.gov.in. Press Information Bureau. 3 January 2021.
- ^ “Novel Corona Virus-2019-nCov vaccine by intradermal route in healthy subjects”. ctri.nic.in. Clinical Trials Registry – India. Retrieved 10 April 2021.
- ^ Das, Sohini (22 April 2021). “Cadila Healthcare testing two-shot regimen for ZyCoV-D, data likely by May”. Business Standard India.
- ^ Writer, Staff (24 April 2021). “Cadila Healthcare starts production of Covid vaccine candidate”. mint. Retrieved 27 April 2021.
Zydus Cadila Covid vaccine close to getting approved in India, says MD Sharvil Patel
In an exclusive interview with India Today TV, Managing Director of Zydus Cadila Dr Sharvil Patel said the company’s Covid vaccine candidate ZyCoV-D against the Covid-19 infection is very close to getting approved in India. They are likely to apply for emergency use authorisation this month.
Ahmedabad-based pharmaceutical company Zydus Cadila is likely to submit the application for emergency use authorisation of its Covid-19 vaccine candidate ‘ZyCoV-D’ in India this month. The company is confident that the vaccine will be approved in May itself. The company plants to produce one crore doses of its ‘painless’ Covid-19 vaccine per month.
If approved, ZyCoV-D will be the fourth vaccine to be used in India’s Covid-19 vaccination drive. Made in India, the company plans to ramp up the vaccine’s production to 3-4 crore doses per month and is already in talks with two other manufacturing companies for the same
Although the vaccine should ideally be stored between 2 and 8 degrees Celsius, it remains stable even at room temperature conditions at 25 degrees Celsius. It is easy to administer, the developers said, and will be administered via intradermal injection.
If approved for emergency use, ZyCoV-D could help India fill the vacuum of vaccine doses currently being experienced in the country’s immunisation drive.
Earlier in April, Zydus Cadila announced that its drug Virafin had received restricted emergency use approval from the Drug Controller General of India for the treatment of mild cases of Covid-19.
In an exclusive interview with India Today TV, Sharvil Patel sheds details on all aspects of the Covid-19 vaccine ZyCoV-D.
When asked the status of Covid vaccine candidate ZyCoV-D and when exactly Zydus Cadila would apply for emergency use authorisation in India, Dr Sharvil Patel said the vaccine was getting very close to getting approved in the country.
“I am very happy to say that India’s first indigenously developed DNA vaccine candidate against Covid, which is our ZyCoV-D, is getting very close to approval,” he said.
“We have almost completed all our recruitment for the clinical trials. We have, by far, recruited the largest number of patients for a Covid vaccine trial in India. The number of volunteers who have been vaccinated as a part of the trial is 28,000,” Sharvil Patel said.
Sharvil Patel also said that his company has also included children in the 12-17 age group for the vaccine trials.
He said, “The recruitment holds very important milestones in terms of cohorts because not only have we included the elderly and those with co-morbidities, but also children in the age group of 12 to 17 years.”
Sharvil Patel said as soon as the efficacy data is obtained, Sydus Cadila will file for emergency use authorisation. As soon as the approval is granted, Zydus Cadila will start production of Covid-19 vaccines from July, he said.
“We hope to see our efficacy data in the middle of May. As soon as we see strong efficacy which correlates to the vaccine’s strong immunogenicity in Phase 2, we will file for emergency use authorization. We hope to produce a good quantity of the vaccine from July onwards to make sure it is available to the people. That is the need of the hour right now,” Sharvil Patel said.
He said by May the company will be in a position to talk to the regulators about the restricted use of the Covid-19 vaccine. “The regulatory process is a rolling one. I believe the regulators look at the data in a short period of time,” Sharvil Patel said.
“We have submitted a lot of data already so that it will aid the regulators once we provide them with the efficacy results. We are, hence, expecting to get the approval in May itself,” Sharvil Patel said.
///////////ZyCoV-D, COVID 19, CORONA VIRUS, VACCINE, INDIA 2021, APPROVALS 2021, SARS-CoV-2
Dostarlimab
(Heavy chain)
EVQLLESGGG LVQPGGSLRL SCAASGFTFS SYDMSWVRQA PGKGLEWVST ISGGGSYTYY
QDSVKGRFTI SRDNSKNTLY LQMNSLRAED TAVYYCASPY YAMDYWGQGT TVTVSSASTK
GPSVFPLAPC SRSTSESTAA LGCLVKDYFP EPVTVSWNSG ALTSGVHTFP AVLQSSGLYS
LSSVVTVPSS SLGTKTYTCN VDHKPSNTKV DKRVESKYGP PCPPCPAPEF LGGPSVFLFP
PKPKDTLMIS RTPEVTCVVV DVSQEDPEVQ FNWYVDGVEV HNAKTKPREE QFNSTYRVVS
VLTVLHQDWL NGKEYKCKVS NKGLPSSIEK TISKAKGQPR EPQVYTLPPS QEEMTKNQVS
LTCLVKGFYP SDIAVEWESN GQPENNYKTT PPVLDSDGSF FLYSRLTVDK SRWQEGNVFS
CSVMHEALHN HYTQKSLSLS LGK
(Light chain)
DIQLTQSPSF LSAYVGDRVT ITCKASQDVG TAVAWYQQKP GKAPKLLIYW ASTLHTGVPS
RFSGSGSGTE FTLTISSLQP EDFATYYCQH YSSYPWTFGQ GTKLEIKRTV AAPSVFIFPP
SDEQLKSGTA SVVCLLNNFY PREAKVQWKV DNALQSGNSQ ESVTEQDSKD STYSLSSTLT
LSKADYEKHK VYACEVTHQG LSSPVTKSFN RGEC
(Disulfide bridge: H22-H96, H130-L214, H143-H199, H222-H’222, H225-H’225, H257-H317, H363-H421, H’22-H’96, H’130-L’214, H’143-H’199, H’257-H’317, H’363-H’421, L23-L88, L134-L194, L’23-L’88, L’194-L’134)
>Heavy Chain EVQLLESGGGLVQPGGSLRLSCAASGFTFSSYDMSWVRQAPGKGLEWVSTISGGGSYTYY QDSVKGRFTISRDNSKNTLYLQMNSLRAEDTAVYYCASPYYAMDYWGQGTTVTVSSASTK GPSVFPLAPCSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYS LSSVVTVPSSSLGTKTYTCNVDHKPSNTKVDKRVESKYGPPCPPCPAPEFLGGPSVFLFP PKPKDTLMISRTPEVTCVVVDVSQEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTYRVVS VLTVLHQDWLNGKEYKCKVSNKGLPSSIEKTISKAKGQPREPQVYTLPPSQEEMTKNQVS LTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSRLTVDKSRWQEGNVFS CSVMHEALHNHYTQKSLSLSLGK
>Light Chain DIQLTQSPSFLSAYVGDRVTITCKASQDVGTAVAWYQQKPGKAPKLLIYWASTLHTGVPS RFSGSGSGTEFTLTISSLQPEDFATYYCQHYSSYPWTFGQGTKLEIKRTVAAPSVFIFPP SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLT LSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
References:
- Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
Dostarlimab
Immunoglobulin G4, anti-(programmed cell death protein 1 (PDCD1)) (humanized clone ABT1 γ4-chain), disulfide with humanized clone ABT1 κ-chain, dimer
Protein Sequence
Sequence Length: 1314, 443, 443, 214, 214multichain; modified (modifications unspecified)
- GSK-4057190
- GSK4057190
- TSR 042
- TSR-042
- WBP-285
- ANB 011
| Formula | C6420H9832N1680O2014S44 |
|---|---|
| CAS | 2022215-59-2 |
| Mol weight | 144183.6677 |
Jemperli FDA 2021/4/22 AND EMA 2021/4/21
NEW DRUG APPROVALS
ONE TIME
$10.00
Dostarlimab, sold under the brand name Jemperli, is a monoclonal antibody medication used for the treatment of endometrial cancer.[1][2][3][4]
The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation.[1][2] The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased.[1][2]
Dostarlimab is a programmed death receptor-1 (PD-1)–blocking antibody.[1][2]
Dostarlimab was approved for medical use in the United States in April 2021.[1][2][5]
| NAME | DOSAGE | STRENGTH | ROUTE | LABELLER | MARKETING START | MARKETING END | ||
|---|---|---|---|---|---|---|---|---|
| Jemperli | Injection | 50 mg/1mL | Intravenous | GlaxoSmithKline LLC | 2021-04-22 | Not applicable | |
Medical uses
Dostarlimab is indicated for the treatment of adults with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following prior treatment with a platinum-containing regimen.[1][2]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC) for adult patients with mismatch repair deficient (dMMR) recurrent or advanced endometrial cancer, as determined by an FDA-approved test, that has progressed on or following a prior platinum-containing regimen.
Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors. The efficacy population consisted of 71 patients with dMMR recurrent or advanced endometrial cancer who progressed on or after a platinum-containing regimen. Patients received dostarlimab-gxly, 500 mg intravenously, every 3 weeks for 4 doses followed by 1,000 mg intravenously every 6 weeks.
The main efficacy endpoints were overall response rate (ORR) and duration of response (DOR), as assessed by blinded independent central review (BICR) according to RECIST 1.1. Confirmed ORR was 42.3% (95% CI: 30.6%, 54.6%). The complete response rate was 12.7% and partial response rate was 29.6%. Median DOR was not reached, with 93.3% of patients having durations ≥6 months (range: 2.6 to 22.4 months, ongoing at last assessment).
Serious adverse reactions occurred in 34% of patients receiving dostarlimab-gxly. Serious adverse reactions in >2% of patients included sepsis , acute kidney injury , urinary tract infection , abdominal pain , and pyrexia . The most common adverse reactions (≥20%) were fatigue/asthenia, nausea, diarrhea, anemia, and constipation. The most common grade 3 or 4 adverse reactions (≥2%) were anemia and transaminases increased. Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.
The recommended dostarlimab-gxly dose and schedule (doses 1 through 4) is 500 mg every 3 weeks. Subsequent dosing, beginning 3 weeks after dose 4, is 1,000 mg every 6 weeks until disease progression or unacceptable toxicity. Dostarlimab-gxly should be administered as an intravenous infusion over 30 minutes.
View full prescribing information for Jemperli.
This indication is approved under accelerated approval based on tumor response rate and durability of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
FDA also approved the VENTANA MMR RxDx Panel as a companion diagnostic device for selecting endometrial cancer patients for treatment with dostarlimab-gxly.
This review used the Real-Time Oncology Review (RTOR) pilot program, which streamlined data submission prior to the filing of the entire clinical application, and the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment.
This application was granted priority review, and breakthrough therapy designation. A description of FDA expedited programs is in the Guidance for Industry: Expedited Programs for Serious Conditions-Drugs and Biologics.
Side effects
Serious adverse reactions in >2% of patients included sepsis, acute kidney injury, urinary tract infection, abdominal pain, and pyrexia.[1][2]
Immune-mediated adverse reactions can occur including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis.[1][2]
History
Like several other available and experimental monoclonal antibodies, it is a PD-1 inhibitor. As of 2020, it is undergoing Phase I/II and Phase III clinical trials.[6][7][8] The manufacturer, Tesaro, announced prelimary successful results from the Phase I/II GARNET study.[6][9][10]
In 2020, the GARNET study announced that Dostarlimab was demonstrating potential to treat a subset of women with recurrent or advanced endometrial cancer.[11]
April 2021, Dostarlimab is approved for the treatment of recurrent or advanced endometrial cancer with deficient mismatch repair (dMMR), which are genetic anomalies abnormalities that disrupt DNA repair.[12]
On April 22, 2021, the Food and Drug Administration granted accelerated approval to dostarlimab-gxly (Jemperli, GlaxoSmithKline LLC).[1] Efficacy was evaluated based on cohort (A1) in GARNET Trial (NCT02715284), a multicenter, multicohort, open-label trial in patients with advanced solid tumors.[1]
Society and culture
Legal status
On 25 February 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Jemperli, intended for the treatment of certain types of recurrent or advanced endometrial cancer.[13] The applicant for this medicinal product is GlaxoSmithKline (Ireland) Limited.[13]
References[
- ^ Jump up to:a b c d e f g h i j k “FDA grants accelerated approval to dostarlimab-gxly for dMMR endometri”. U.S. Food and Drug Administration(FDA) (Press release). 22 April 2021. Retrieved 22 April 2021. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c d e f g h i “Jemperli- dostarlimab injection”. DailyMed. Retrieved 28 April 2021.
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Dostarlimab, American Medical Association.
- ^ World Health Organization (2018). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 119” (PDF). WHO Drug Information. 32 (2).
- ^ “FDA grants accelerated approval for GSK’s Jemperli (dostarlimab-gxly) for women with recurrent or advanced dMMR endometrial cancer” (Press release). GlaxoSmithKline. 22 April 2021. Retrieved 22 April 2021 – via PR Newswire.
- ^ Jump up to:a b Clinical trial number NCT02715284 for “A Phase 1 Dose Escalation and Cohort Expansion Study of TSR-042, an Anti-PD-1 Monoclonal Antibody, in Patients With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
- ^ Clinical trial number NCT03981796 for “A Study of Dostarlimab (TSR-042) Plus Carboplatin-paclitaxel Versus Placebo Plus Carboplatin-paclitaxel in Patients With Recurrent or Primary Advanced Endometrial Cancer (RUBY)” at ClinicalTrials.gov
- ^ Clinical trial number NCT03602859 for “A Phase 3 Comparison of Platinum-Based Therapy With TSR-042 and Niraparib Versus Standard of Care Platinum-Based Therapy as First-Line Treatment of Stage III or IV Nonmucinous Epithelial Ovarian Cancer (FIRST)” at ClinicalTrials.gov
- ^ “Data from GARNET study indicates robust activity of dostarlimab in patients with advanced or recurrent endometrial cancer”. Tesaro (Press release). Retrieved 1 January 2020.
- ^ Scalea B (28 May 2019). “Dostarlimab Effective in Endometrial Cancer Regardless of MSI Status”. Targeted Oncology. Retrieved 1 January 2020.
- ^ “GSK Presents New Data from the GARNET Study Demonstrating Potential of Dostarlimab to Treat a Subset of Women with Recurrent or Advanced Endometrial Cancer – Drugs.com MedNews”. Drugs.com. Retrieved 29 April 2020.
- ^ “FDA Approves New Immunotherapy for Endometrial Cancer”. Medscape. Retrieved 23 April 2021.
- ^ Jump up to:a b “Jemperli: Pending EC decision”. European Medicines Agency (EMA) (Press release). 25 February 2021. Retrieved 22 April 2021.
External links
- “Dostarlimab”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02715284 for “Study of TSR-042, an Anti-programmed Cell Death-1 Receptor (PD-1) Monoclonal Antibody, in Participants With Advanced Solid Tumors (GARNET)” at ClinicalTrials.gov
- Kaplon H, Muralidharan M, Schneider Z, Reichert JM: Antibodies to watch in 2020. MAbs. 2020 Jan-Dec;12(1):1703531. doi: 10.1080/19420862.2019.1703531. [Article]
- Temrikar ZH, Suryawanshi S, Meibohm B: Pharmacokinetics and Clinical Pharmacology of Monoclonal Antibodies in Pediatric Patients. Paediatr Drugs. 2020 Apr;22(2):199-216. doi: 10.1007/s40272-020-00382-7. [Article]
- Green AK, Feinberg J, Makker V: A Review of Immune Checkpoint Blockade Therapy in Endometrial Cancer. Am Soc Clin Oncol Educ Book. 2020 Mar;40:1-7. doi: 10.1200/EDBK_280503. [Article]
- Deshpande M, Romanski PA, Rosenwaks Z, Gerhardt J: Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers (Basel). 2020 Nov 10;12(11). pii: cancers12113319. doi: 10.3390/cancers12113319. [Article]
- FDA Approved Drug Products: Jemperli (dostarlimab-gxly) for intravenous injection [Link]
- FDA News Release: FDA grants accelerated approval to dostarlimab-gxly for dMMR endometrial cancer [Link]
- Statement on a Nonproprietary Name Adopted by the USAN Council: Dostarlimab [Link]
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | PCDP1 |
| Clinical data | |
| Trade names | Jemperli |
| Other names | TSR-042, WBP-285, dostarlimab-gxly |
| License data | US DailyMed: Dostarlimab |
| Routes of administration | Intravenous |
| Drug class | Antineoplastic |
| ATC code | L01XC40 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| CAS Number | 2022215-59-2 |
| PubChem SID | 384585344 |
| DrugBank | DB15627 |
| UNII | P0GVQ9A4S5 |
| KEGG | D11366 |
| Chemical and physical data | |
| Formula | C6420H9832N1690O2014S44 |
| Molar mass | 144325.73 g·mol−1 |
/////////Dostarlimab, PEPTIDE, ANTINEOPLASTIC, CANCER, ドスタルリマブ , GSK 4057190, GSK4057190, TSR 042, TSR-042, WBP-285, FDA 2021, EU 2021
2-Deoxy-D-glucose

2-Deoxy-D-glucose
- Molecular FormulaC6H12O5
- Average mass164.156 Da
2-Deoxy-D-glucose
(4R,5S,6R)-6-(Hydroxymethyl)tetrahydro-2H-pyran-2,4,5-triol(4R,5S,6R)-6-(Hydroxyméthyl)tétrahydro-2H-pyran-2,4,5-triol
154-17-6[RN]
- 2-Deoxy-D-arabino-hexose
- 2 DG
- 2-Deoxy-D-glucose
- 2-Deoxy-D-mannose
- 2-Deoxyglucose
- 2-Desoxy-D-glucose
- Ba 2758
- D-Glucose, 2-deoxy-
- NSC 15193
2-Deoxy-D-arabino-hexopyranose2-deoxy-D-glucopyranose2-deoxyglucose
2-DGD-arabino-2-DesoxyhexoseD-arabino-Hexopyranose, 2-deoxy- [(4R,5S,6R)-6-(Hydroxymethyl)oxane-2,4,5-triol2-deoxyglucopyranose2-deoxymannopyranose2-dGlc
D-arabino-2-Deoxyhexoseglucitol, 2,5-anhydro-
2-Deoxy-D-glucose
CAS Registry Number: 154-17-6
CAS Name: 2-Deoxy-D-arabino-hexose
Additional Names: D-arabino-2-desoxyhexose; 2-deoxyglucose; 2-DGManufacturers’ Codes: Ba-2758Molecular Formula: C6H12O5Molecular Weight: 164.16Percent Composition: C 43.90%, H 7.37%, O 48.73%Literature References: Antimetabolite of glucose, q.v., with antiviral activity.
Synthesis: M. Bergmann et al.,Ber.55, 158 (1922); 56, 1052 (1923); J. C. Sowden, H. O. L. Fischer, J. Am. Chem. Soc.69, 1048 (1947); H. R. Bolliger, Helv. Chim. Acta34, 989 (1954); H. R. Bolliger, M. D. Schmid, ibid. 1597, 1671; H. R. Bolliger, “2-Deoxy-D-arabino-hexose (2-Deoxy-D-glucose)” in Methods in Carbohydrate Chemistryvol. I, R. L. Whistler, M. L. Wolfrom, Eds. (Academic Press, New York, 1962) pp 186-189.
Inhibition of influenza virus multiplication: E. D. Kilbourne, Nature183, 271 (1959).
Effects on herpes simplex virus: R. J. Courtney et al.,Virology52, 447 (1973). Mechanism of action studies: M. R. Steiner et al.,Biochem. Biophys. Res. Commun.61, 745 (1974); E. K. Ray et al.,Virology58, 118 (1978). Use in human genital herpes infections: H. A. Blough, R. L. Giuntoli, J. Am. Med. Assoc.241, 2798 (1979); L. Corey, K. K. Holmes, ibid.243, 29 (1980). Effect vs respiratory syncytial viral infections in calves: S. B. Mohanty et al.,Am. J. Vet. Res.42, 336 (1981).
Properties: Cryst from acetone or butanone, mp 142-144°. [a]D17.5 +38.3° (35 min) ®+45.9° (c = 0.52 in water); +22.8° (24 hrs) ® +80.8° (c = 0.57 in pyridine).
Melting point: mp 142-144°
Optical Rotation: [a]D17.5 +38.3° (35 min) ®+45.9° (c = 0.52 in water); +22.8° (24 hrs) ® +80.8° (c = 0.57 in pyridine) Derivative Type: a-Form
Properties: Cryst from isopropanol, mp 134-136°. [a]D26 +156° ® +103° (c = 0.9 in pyridine).Melting point: mp 134-136°Optical Rotation: [a]D26 +156° ® +103° (c = 0.9 in pyridine) Use: Exptlly as an antiviral agent.


Source Temperature: 210 °C Sample Temperature: 150 °C Direct, 75 eV
14.0 2.2
15.0 11.5
17.0 3.9
18.0 19.4
19.0 13.7
26.0 2.5
27.0 12.1
28.0 21.9
29.0 31.2
30.0 4.6
31.0 41.3
32.0 12.4
39.0 5.9
40.0 2.1
41.0 10.9
42.0 12.4
43.0 46.3
44.0 31.5
45.0 34.3
46.0 2.8
47.0 4.1
53.0 1.5
54.0 2.0
55.0 14.4
56.0 35.3
57.0 55.7
58.0 11.4
59.0 2.0
60.0 100.0
61.0 31.1
62.0 2.3
68.0 4.6
69.0 12.2
70.0 3.0
71.0 34.9
72.0 7.0
73.0 25.3
74.0 46.6
75.0 5.1
81.0 1.5
82.0 2.4
83.0 1.3
84.0 1.3
85.0 18.1
86.0 55.3
87.0 4.6
89.0 1.2
91.0 1.5
97.0 3.6
98.0 2.9
99.0 1.7
100.0 3.5
102.0 1.1
103.0 19.8
104.0 1.4
111.0 1.6
115.0 25.2
116.0 3.0
117.0 2.1
120.0 3.3
128.0 1.0
129.0 2.5
133.0 1.8
147.0 2.2
1H NMR DMSO D6


1H NMR D20

IR NUJOL MULL

IR KBR

PAPERCollection of Czechoslovak Chemical Communications (1955), 20, 42-5. http://cccc.uochb.cas.cz/20/1/0042/
Preparation of 2-deoxy-D-glucose
By: Stanek, Jaroslav; Schwarz, Vladimir
Triacetyl-D-glucal (I) adds (BzO)2IAg and (BzO)2BrAg, to give 1-benzoyl-3,4,6-triacetyl-2-deoxy-2-iodo-α-D-glucopyranose (II) and 1-benzoyl-3,4,6-triacetyl-2-deoxy-2-bromo-α-D-glucopyranose (III), resp. Both halogen derivs. give 2-deoxy-D-glucose (IV) by reduction. Adding a C6H6 soln. of 16.7 g. iodine into a suspension of 33.6 g. dry BzOAg in 200 ml. C6H6, treating the mixt. with a soln. of 20 g. I in 200 ml. C6H6, heating the mixt. 7 hrs. on the steam bath, removing the AgI, evapg. the solvent, and crystg. the residue from EtOH gave 20.8 g. (54.7%) II, m. 129-30°, [α]21D 21.7°. Analogous procedure with 13.4 g. BzOAg, 4.6 g. Br, and 8 g. I gave 3.9 g. (33%) III, m. 139-40°, [α]17D 33.5°. The same compd. (3 g.), m. 140°, [α]18D 33.6°, was obtained by adding 3.2 g. Br to a soln. of 5.44 g. I in 50 ml. CCl4, by refluxing the mixt. 2 hrs. with 6 g. BzOAg, filtering off the AgBr, and evapg. the solvent. Reducing 8 g. II or an equiv. III in 150 ml. MeOH with 60 g. Zn activated by 1 hr. immersion in a soln. of 60 g. CuSO4 in 1500 ml. H2O, removing Zn after 8 hrs., evapg. the MeOH, and sapong. the residue with Ba(OH)2 yielded 0.42 g. (20%) IV, m. 145°, [α]18D 46.1°.
Wavlen: 589.3 nm; Temp: 18 °C, +46.1 ° ORD
PATENT
https://patents.google.com/patent/WO2004058786A1/enThe present invention relates to a process for the synthesis of 2-deoxy-D-glucose. Background of the invention 2-deoxy-D-glucose is useful in control of respiratory infections and for application as an antiviral agent for treatment of human genital herpes.Prior art for preparation of 2-deoxy-D-glucose while operable, tend to be expensive and time consuming. Reference may be made to Bergmann, M., Schotte, H., Lechinsky, W., Ber, 55, 158 (1922) and Bergmann, M., Schotte, H., Lechinsky, W., Ber 56, 1052 (1923) which disclose the preparation of 2-deoxy-D-glucose in low yield by mineral acid catalyzed addition of water to D-glucal. Another method of producing 2-deoxy-D-glucose is from diethyldithioacetal derivative of D-glucose (Bolliger, H.R. Schmid, M.D., Helv. Chim. Ada 34, 989 (1951); Bolliger, H.R., Schmid, M.D., Helv. Chim. A a 34, 1597 (1951); Bolliger, H.R. Schmid, M.D., Helv. Chim. Ada 34, 1671 (1951) and from D-arabhiose by reaction with nitromethane followed by acetylation, reduction and hydrolysis (Sowden, J.C, Fisher, H.O.L., J. Am. Chem., 69, 1048 (1947). However these methods result in the formation of 2- deoxy-D-glucose in low yield and of inferior purity due to the formation of several byproducts and involve use of toxic reagents such as ethanethiol and nitromethane. As a result purification of 2-deoxy-D-glucose has to be done by recrystallisation which is tedious, time consuming and difficult.Accordingly it is important to develop a process for synthesis of 2-deoxy-D-glucose which obviates the drawbacks as detailed above and results in good yield and good purity. Objects of the inventionThe main object of the present invention is to provide a process for the synthesis of 2- deoxy-D-glucose resulting in good yield and with good purity.Another object of the invention is to provide an economical process for the synthesis of 2-deoxy-D-glucose. Summary of the inventionA process that would produce 2-deoxy-D-glucose economically and with desired purity, is a welcome contribution to the art. This invention fulfills this need efficiently.Accordingly the present invention relates to a process for the synthesis of 2-deoxy- D-glucose comprising haloalkoxylation of R-D-Glucal wherein R is selected from H and 3, 4, 6-tri-O-benzyl, to obtain alkyl 2-deoxy-2-halo-R-α/ -D-gluco/mannopyranoside, converting alkyl 2-deoxy-2-halo-R-α/β-D-gluco/mannopyranoside by reduction to alkyl 2- deoxy-α/β-D-glucopyranoside, hydrolysing alkyl 2-deoxy-α/β-D-glucopyranoside to 2- deoxy-D-glucose.In one embodiment of the invention, the alkyl 2-deoxy-α/β-D-glucopyranoside is obtained by (a) haloalkoxylating 3,4,6,-tri-O-benzyl-D-glucal to alkyl 2-deoxy-2-halo-3,4,6-tri-O- benzyl-α/β-D-gluco-/mannopyranoside; (b) subjecting alkyl 2-deoxy-2-halo-3,4,6-tri-O-benzyl-α/β-D-gluco/mannopyranoside to reductive dehalogenation and debenzylation to obtain alkyl 2-deoxy -α/β-D- glucopyranoside. In another embodiment of the invention, in step (a) haloalkoxylation of 3,4,6-tri-O- benzyl-D-glucal is carried out by reaction with a haloalkoxylating agent selected from a N- halosuccinimide and a N-haloacetamide, and alcohol.The reaction scheme for the reactions involved in the process of the invention are also given below:

in R’=CH3I R=C6H5CH2 H R=C6H5CH2, X=Br, R’=CH3 IV R=H V R=CH3, C2HSJ C6H5CH3, iPr, X=Br

Such overall synthesis may be depicted as follows where R=H, CH3, C2H5, (CH3)2CH, C6H5CH ; RX-CH3; X-CL, Br.Example 1 To a solution of 3,4,6-tri-O-benzyl-D-glucal (39 g, 0.09 mol) in dichloromethane (20ml) and methanol (100 ml) was added N-bromosuccinimide (18.7 g, 0.09 mil) during 10 min. at room temperature and stirred for 4 h. After completion of the reaction solvent was distilled off. The resultant residue extracted into carbon tetrachloride (2×100 ml) and organic phase concentrated to obtain methyl 2-bromo 2-deoxy-3,4,6-tri-O-benzyl-α/β-D-gluco- /mannopyranoside as a syrup. Quantity obtained 50 g. 1H NMR (200 MHz, CDC13) 3.40-4.00 (m, 7H, H-2,5,6,6′ and OCH3) 4.30-5.10 (m, 9H, H-1,3,4 and 3xPhCH2O), 7.10-7.60 (m, 15H, Ar-H). A solution of methyl 2-bromo-2-deoxy-3,4,6-tri-O-benzyl-α/β-D-gluco- /mannopyranoside (50 g) in methanol (300) was charged into one litre autoclave along with Raney nickel (10 ml) Et3N (135 ml) and subjected to hydrogenation at 120 psi pressure at 50°C for 8 h. After completion of the reaction the catalyst was filtered off and the residue washed with methanol (25 ml). The filtrate was concentrate to obtain methyl 2-deoxy-3,4,6- tri-O-benzyl-α/β-D-glucopyranoside as a syrup (37.9 g, 89%). 1H NMR (200 MHz, CDC13): δ 1.50-2.40 (m,2H,H-2,2′)5 3.32, 3.51 (2s, 3H, OCH3) 3.55-4.00 (m, 5H, H-3,4,5,6,6′), 4.30-5.00 (m, 7H, 3xPhCH2, H-l), 7.10-7.45 (m, 15H, Ar-H). The syrup of methyl 2-deoxy-3,4,6- tri-O-benzyl-α/β-D-glucopyranoside (37.9g) was dissolved in methanol (200 ml). 1 g of 5%Pd/C was added and hydrogenated at 150 psi pressure at room temperature. After 5 hours catalyst was filtered off and solvent evaporated. Quantity of the methyl 2-deoxy-α/β-D- glucopyranoside obtained 10.5 g (70%). [ ]D + 25.7° (c 1.0, MeOH), 1H NMR (200 MHz, D2O); δ 1.45-2.40 (m, 2H, H-2,2′) 3.20-4.80, (m 9H, H- 1,3,4,5,6,6′ – OCH3).Example 2 To a solution of D-glucal (64.6g, 0.44 mol) in methanol (325 ml) at 10°C was addedN-bromosuccinimide (78.7 g, 0.44 mol) during 40 min. maintaining the temperature between 10-15°C during the addition. The reaction mixture was stirred at room temperature. After 5 hours solvent was evaporated to obtain a residue which was refluxed in ethyl acetate (100 ml). Ethyl acetate layer was discarded to leave a residue of methyl 2-bromo-2-deoxy-α/β-D- gluco/mannopyranoside (105 g) as a syrup. [α]D + 36° (c 1.0, MeOH). 1H NMR (200 MHz, D2O): δ 3.47, 3.67 (2s, 3H, OCH3), 3.70-4.05 (m, 6h, H-23,4,5,6,6′), 4.48-5.13 (2s, 1H, H-l). The syrupy methyl 2-bromo-2-deoxy-α/β-D-gluco-/mannopyranoside was dissolved in methanol (400 ml), a slurry of 80 g Raney nickel (a 50% slurry in methanol), Et3N (30 ml) and hydrogenated in a Parr apparatus at 120 psi. After 8-9 hours, the reaction mixture was filtered through a Celite filter pad and washed with MeOH. The washings and filtrate were combined and triturated with hexane to separate and remove by filtration insoluble triethylamine hydrobromide and traces of succinimide. The filtrate was concentrated to a residue. The isolated yield of methyl 2-deoxy-α/β-D-glucopyranoside was 89%. Ethyl 2-bromo-2deoxy-α/β-D-gluco-/mannopyranoside: When solvent was ethanol instead of methanol the compound obtained was ethyl 2- bromo-2-deoxy-α/β-D-gluco-/mannopyranoside. 1HNMR (200 MHz, D2O): δ 1.10-1.32 (m, 3H, CH3), 2.80 (s, 4H, -CO(CH2)2CO-NH-), 3.40-4.10 (m, 8H, H-2,3,4,5,6,6′, CH2), 4.40, 5.20 (2s 1H, H-l α/β).Isopropyl 2-bromo-2-deoxy- /β-D-gluco-/mannopyranoside: When isopropanol instead of methanol was used as a solvent the compound obtained was isopropyl 2-bromo-2-deoxy-α/β-D-gluco/mannopyranoside. 1H NMR (200 MHz, D2O): δ 1.10-1.30 (m, 6H, 2xCH3) 2.80 (s, 4H, -CO(CH2)2CO-NH-), 3.60-4.60 (m 8H,H- 2,3,4,5,6,6′, CH2) 4.40, 5.30 (2s, 1H, H-l, α/β).Example 3 A mixture of D-glucal (64.6 g), methanol (400 ml), N-bromosuccinimide (79 g) were stirred at 15 C for 6 h. The reaction mixture was hydrogenated in a Parr apparatus in presence of 60 g of Raney nickel catalyst (a 50% slurry in methanol) and triethylamine (62 ml). After 8-9 h, the reaction mixture was filtered on a Celite filter pad. The Celite pad was washed with methanol. The washings and filtrate were combined, concentrated to a thick heavy syrup, dissolve in chloroform (500 ml), pyridine (400 ml) and acetic anhydride (251 ml) was added while stirring, maintaining the temperature between 5-10°C. After 12 hours, the reaction mixture was diluted with CHC13 (500 ml) transferred to a separating funnel and organic phase was washed with water. The organic phase was separated, dried (Na2SO4) and concentrated to obtain methyl 2-deoxy-3,4,6-tri-O-acetyl-2 deoxy-α/β-D-glucopyranoside as a syrup (163.43 g, 87%). [α]D + 65.0° (c 1.0, CHC13) 1H NMR (200 MHz, CDC13): δ 1.55-1.90 (m, 2H, H-2,2′), 2.01, 2.04,2.11, 2.15, (4s, 9H, 3xOCOCH3), 2.18,3.40 (2s, 3H, OCH3), 3.45-50 (m, 3H, H-5, 6,6′) 4.80-5.40 (m, 3H,H-1,3,4). The syrup was dissolved in methanol (600 ml) IN NaOMe in methanol (25ml) was added and left at room temperature. After 6-10 h, dry CO2 gas was passed into the reaction mixture, solvent was evaporated to obtain a syrupy residue. The residue was once again extracted into dry methanol and concentrated to obtain methyl 2-deoxy-α/β-D-glucopyranoside as syrup. Quantity obtained 81 g (92%).Example 4 A 500 ml round bottom flask equipped with magnetic stir bar was charged with a solution of D-glucal (32.3 g) in methanol (175 ml), cooled to 15°C, N-bromosucci-t imide (NBS) (39.4 g) was added and stirred for 6 hours at 15°C. The reaction mixture was concentrated to half the volume, cooled to 0°C and separated succinimide was removed by filtration. To the filtrate was added a slurry of 30 g Raney nickel (a 50% slurry in methanol) Et3N (32 ml) and hydrogenated in a Parr apparatus at 120 psi. After 7-8 hours, the reaction mixture was filtered through a Celite filter pad, and washed with MeOH. The washings and filtrate were combined and triturate with hexane to separate and remove by filtration insoluble triethylamine hydrobromide and succinimide. The filtrate was concentrated to a residue, dissolved in methanol and triturated with hexane to remove most of the triethylamine hydrobromide and succinimide. The filtrate was concentrated to obtain methyl 2-deoxy-α/β- D-glucopyranoside (85%).Example 5 To a stirred solution of methyl 3,4,6-tri-O-acetyl-2-deoxy-α/β-D-glucopyranoside (47 g) (from example 3) in acetic acid (40 ml) and acetic anhydride (110 ml) was added concentrated sulphuric acid (0.94 ml) at 0°. The reaction mixture was brought to room temperature and stirred. After 2 hours the reaction mixture was diluted with water (50 ml) and extracted into CH2C12 (3×150 ml). The organic phase was separated, washed with saturated NaHCO3 solution, H2O dried over Na2SO and concentrated to obtain 2-deoxy- 1,3,4,6-tetra-O-acetyl-α/β-D-glucopyranoside as a crystalline compound, mp. 115-118°C. Quantity obtained 44.5 g (86%). [α]D + 21.5° (c 1.0, CHC13). 1H NMR (200 MHz, CDC13): δ 1.50-2.45 (m, 14H, H-2,2′, 4xOCOCH3), 3.85-5.40, (m, 5H, H-3,4,5,6,6′), 5.75-6.20 (m, 1H, H-l,α/ β). To a heterogeneous mixture of l,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D- glucopyranoside (10 g) in water (100 ml) was added acetyl chloride (10 ml) and heated to 80°C. After 6 hours the reaction mixture was cooled to room temperature, neutralised with saturated aq. Ba(OH)2, concentrated to half the volume and filtered on a Celite pad. Filtrate was concentrated on a rotary evaporator and dried over anhydrous P2O5 to obtain a residue which was dissolved in hot isopropyl alcohol and filtered on a pad of Celite to obtain a clear filtrate. The filtrate was concentrated to a residue, dissolved in hot isopropyl alcohol (50 ml), acetone (75 ml) and seeded with a few crystals of 2-deoxy-D-glucose. After 15-18 hours at 5°C crystalline title product was filtered. Quantity obtained 3.21 g (64.9%) m.p. 148-149°C.Example 6 A heterogeneous mixture of l,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (9 g) (from example 5), water (30 ml) and 11% aq. H2SO (0.3 ml) was stirred at 85°C for 7 h to obtain a homogenous solution. The reaction mixture was cooled, neutralised with aq. Ba(OH)2 solution and filtered. The filtrate obtained was concentrated to half the volume and solids separated were filtered. To the filtrate was added activated carbon (1 g) and filtered. The filtrate was concentrated on a rotary evaporator and dried over P2O5 to obtain 2-deoxy- D-glucose that was crystallized from methyl alcohol (27 ml) and acetone (54 ml). Quantity obtained 2.4 g. mp. 146-149°C.Example 7A heterogeneous mixture of l,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside(25g) (from example 5), H2O (250 ml), toluene (250 ml) and glacial acetic acid (1.25 ml) was heated to reflux for 10-12 hours, while it was connected to a Dean- Stark azeotropic distillation apparatus. An azeotropic mixture of acetic acid, toluene was collected to remove acetic acid and every one hour fresh toluene (50 ml) was introduced. After completion of the reaction, toluene was removed by distillation from the reaction mixture to obtain a residue that was dissolved in methanol, treated with charcoal and filtered. The filtrate was separated, concentrated to a residue and crystallized from isopropyl alcohol and acetone to obtain 2- deoxy-D-glucose (7.33 g, 59%). mp. 148-151°C.Example 8 A heterogeneous mixture of l,3,4,5-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (lOg) (from example 5), H2O (200 ml) cone. HC1 (0.3 ml) and glacial acetic acid (0.5 ml) was heated to 85°C. After 6 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered on a pad of Celite. Filtrate was separated, treated with charcoal and filtered. The filtrate was concentrated to a residue and crystallized from MeOH, acetone to obtain the product. Quantity obtained 2.75 g. mp. 147-148°C.Example 9 A heterogeneous mixture of l,3,4,5-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside(lOg) (from example 3) water (100 ml) and cone. HCI (0.5ml) was heated to 80°C. After 2-5 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered on a pad of Celite. The filtrate was concentrated to a residue, dissolved in ethanol, treated with charcoal and filtered. The filtrate was concentrated to a solid residue andcrystallized from methanol-acetone to obtain the title product. Quantity obtained 3.15g mp. 148-151°C.Example 10A solution of methyl 2-deoxy-α/β-D-glucopyranoside (30g) (from example 2) water(15 ml) and cone. HCI (1.5 ml) was heated to 80-85°C. After 3-5 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered to remove insoluble salts. The filtrate was concentrated to a residue, crystallized from MeOH, acetone and hexane to obtain 2-deoxy-D-glucose (11.77 g) mp. 149-151°C.Example 11A solution of methyl 2-deoxy-α/β-D-glucopyranoside (30g) (from example 2) water (195 ml) and cone. H2SO (5.9 ml) was heated to 80°C. After 2-3 hours the reaction mixture was cooled, neutralized with aq. Ba(OH)2 and filtered. The filtrate was separated, treated with charcoal and filtrate. The Filtrate was concentrated to a residue and crystallized from isopropyl alcohol to obtain the title product. Quantity obtained 5.2 g. mp. 152-154°C.Example 12 A mixture of methyl 2-deoxy-α/β-D-glucopyranoside (24g) (from example 2) water(125 ml) and IR 120 H+ resin (7.5 ml) was heated to 90-95°C for 2h. The reaction mixture was cooled to room temperature, filtered and the resin was washed with water (20 ml). The filtrate was concentrated to residue and crystallized from ethanol to obtain 2-deoxy-D- glucose (8.8 g), mp. 150-152°C. The main advantages of the present invention are:-1). It does not involve the use of toxic mercaptans like ethane thiol. 2). This process does not involve reaction of D-glucal with mineral acid, thereby avoiding the formation of Ferrier by-products.
2-Deoxy-d-glucose is a glucose molecule which has the 2-hydroxyl group replaced by hydrogen, so that it cannot undergo further glycolysis. As such; it acts to competitively inhibit the production of glucose-6-phosphate from glucose at the phosphoglucoisomerase level (step 2 of glycolysis).[2] In most cells, glucose hexokinase phosphorylates 2-deoxyglucose, trapping the product 2-deoxyglucose-6-phosphate intracellularly (with exception of liver and kidney)[; thus, labelled forms of 2-deoxyglucose serve as a good marker for tissue glucose uptake and hexokinase activity. Many cancers have elevated glucose uptake and hexokinase levels. 2-Deoxyglucose labeled with tritium or carbon-14 has been a popular ligand for laboratory research in animal models, where distribution is assessed by tissue-slicing followed by autoradiography, sometimes in tandem with either conventional or electron microscopy.
2-DG is uptaken by the glucose transporters of the cell. Therefore, cells with higher glucose uptake, for example tumor cells, have also a higher uptake of 2-DG. Since 2-DG hampers cell growth, its use as a tumor therapeutic has been suggested, and in fact, 2-DG is in clinical trials. [3] A recent clinical trial showed 2-DG can be tolerated at a dose of 63 mg/kg/day, however the observed cardiac side-effects (prolongation of the Q-T interval) at this dose and the fact that a majority of patients’ (66%) cancer progressed casts doubt on the feasibility of this reagent for further clinical use.[4] However, it is not completely clear how 2-DG inhibits cell growth. The fact that glycolysis is inhibited by 2-DG, seems not to be sufficient to explain why 2-DG treated cells stop growing.[5] Because of its structural similarity to mannose, 2DG has the potential to inhibit N-glycosylation in mammalian cells and other systems, and as such induces ER stress and the Unfolded Protein Response (UPR) pathway.[6][7][8]
Clinicians have noted that 2-DG is metabolised in the pentose phosphate pathway in red blood cells at least, although the significance of this for other cell types and for cancer treatment in general is unclear.
Work on the ketogenic diet as a treatment for epilepsy have investigated the role of glycolysis in the disease. 2-Deoxyglucose has been proposed by Garriga-Canut et al. as a mimic for the ketogenic diet, and shows great promise as a new anti-epileptic drug.[9][10] The authors suggest that 2-DG works, in part, by increasing the expression of Brain-derived neurotrophic factor (BDNF), Nerve growth factor (NGF), Arc (protein) (ARC), and Basic fibroblast growth factor (FGF2).[11] Such uses are complicated by the fact that 2-deoxyglucose does have some toxicity.
A study found that by combining the sugar 2-deoxy-D-glucose (2-DG) with fenofibrate, a compound that has been safely used in humans for more than 40 years to lower cholesterol and triglycerides, an entire tumor could effectively be targeted without the use of toxic chemotherapy.[12][13]
2-DG has been used as a targeted optical imaging agent for fluorescent in vivo imaging.[14][15] In clinical medical imaging (PET scanning), fluorodeoxyglucose is used, where one of the 2-hydrogens of 2-deoxy-D-glucose is replaced with the positron-emitting isotope fluorine-18, which emits paired gamma rays, allowing distribution of the tracer to be imaged by external gamma camera(s). This is increasingly done in tandem with a CT function which is part of the same PET/CT machine, to allow better localization of small-volume tissue glucose-uptake differences.
Resistance to 2-DG has been reported in HeLa cells [16] and in yeast;[17][8] in the latter, it involves the detoxification of a metabolite derived from 2-DG (2DG-6-phosphate) by a phosphatase. Despite the existence of such a phosphatase in human (named HDHD1A) However it is unclear whether it contributes to the resistance of human cells to 2DG or affects FDG-based imaging.
SYN
Indian Pat. Appl., 2004DE02075,

SYN
CN 106496288,
STARTING MATERIAL CAS 69515-91-9
C14 H20 O9, 332.30
D-arabino-Hexopyranose, 2-deoxy-, 1,3,4,6-tetraacetate
SYN
Bioorganic & Medicinal Chemistry Letters, 22(10), 3540-3543; 2012
https://www.sciencedirect.com/science/article/abs/pii/S0960894X12004258
PATENT
https://patents.google.com/patent/US6933382B2/en2-deoxy-D-glucose is useful in control of respiratory infections and for application as an antiviral agent for treatment of human genital herpes.Prior art for preparation of 2-deoxy-D-glucose while operable, tend to be expensive and time consuming. Reference may be made to Bergmann M., Schotte, H., Lechinsky, W., Ber, 55, 158 (1922) and Bergmann, M., Schotte, H., Lechinsky, W., Ber 56, 1052 (1923) which disclose the preparation of 2-deoxy-D-glucose in low yield by mineral acid catalyzed addition of water to D-glucal. Another method of producing 2-deoxy-D-glucose is from diethyldithioacetal derivative of D-glucose (Bolliger, H. R. Schmid, M. D., Helv. Chim. Acta 34, 989 (1951); Bolliger, H. R., Schmid, M. D., Helv, Chim. Acta 34, 1597 (1951); Bolliger, H. R Schmid, M. D., Helv. Chim. Acta 34, 1671 (1951) and from D-arabinose by reaction with nitromethane followed by acetylation, reduction and hydrolysis (Sowden, J. C., Fisher, H. O. L., J. Am. Chem., 69, 1048 (1947). However these methods result in the formation of 2-deoxy-D-glucose in low yield and of inferior purity due to the formation of several by-products and involve use of toxic reagents such as ethanethiol and nitromethane. As a result purification of 2-deoxy-D-glucose has to be done by recrystallisation which is tedious, time consuming and difficult.

EXAMPLE 1To a solution of 3,4,6-tri-O-benzyl-D-glucal (39 g, 0.09 mmol) in dichloromethane (20 ml) and methanol (100 ml) was added N-bromosuccinimide (18.7 g, 0.09 mil) during 10 min. at room temperature and stirred for 4 h. After completion of the reaction solvent was distilled off. The resultant residue extracted into carbon tetrachloride (2×100 ml) and organic phase concentrated to obtain methyl 2-bromo 2-deoxy-3,4,6-tri-O-benzyl-α/β-D-gluco-/mannopyranoside as a syrup. Quantity obtained 50 g. 1H NMR (200 MHz, CDCl3) 3.40-4.00 (m, 7H, H-2,5,6,6′ and OCH3) 4.30-5.10 (m, 9H, H-1,3,4 and 3×PhCH2O), 7.10-7.60 (m 15H, Ar—H). A solution of methyl 2-bromo-2-deoxy-3,4,6-tri-O-benzyl/α/β-D-gluco-/mannopyranoside (50 g) in methanol (300) was charged into one liter autoclave along with Raney nickel (10 ml) Et3N (135 ml) and subjected to hydrogenation at 120 psi pressure at 50° C. for 8 h. After completion of the reaction the catalyst was filtered off and the residue washed with methanol (25 ml). The filtrate was concentrate to obtain methyl 2-deoxy-3,4,6-tri-O-benzyl-α/β-D-glucopyranoside as a syrup (37.9 g, 89%). 1H NMR (200 MHz CDCl3): δ 1.50-2.40 (m,2H,H-2,2′), 3.32, 3.51 (2s, 3H, OCH3) 3.55-4.00 (m, 5, H-3,4,5,6,6′) 4.30-5.00 (M 7H, 3×PhCH2, H-1), 7.10-7.45 (m, 15H, Ar—H). The syrup of methyl 2-deoxy-3,4, 6-tri-O-benzyl-α/β-D-glucopyranoside (37.9 g) was dissolved in methanol (200 ml). 1 g of 5% Pd/C was added and hydrogenated at 150 psi pressure at room temperature. After 5 hours catalyst was filtered off and solvent evaporated. Quantity of the methyl 2-deoxy-α/β-D-glucopyranoside obtained 10.5 g (70%). [α]D+25.7° (c 1.0, MeOH), 1H NMR (200 MHz, D2O); δ 1.45-2.40 (m, 2H, H-2,2′) 3.20-4.80, (m 9H, H-1,3,4,5,6,6′—OCH3).EXAMPLE 2To a solution of D-glucal (64.6 g, 0.44 mmol) in methanol (325 ml) at 10° C. was added N-bromosuccinimide (78.7 g, 0.44 mol) during 40 min. maintaining the temperature between 10-15° C. during the addition. The reaction mixture was stirred at room temperature. After 5 hours solvent was evaporated to obtain a residue which was refluxed in ethyl acetate (100 ml). Ethyl acetate layer was discarded to leave a residue of methyl 2-bromo-2-deoxy-α/β-D-gluco/mannopyranoside (105 g) as a syrup. [α]D+36° (c 1.0, MeOH). 1H NMR (200 MHz, D2O): δ 3.47, 3.67 (2s, 3H, OCH3), 3.70-4.05 (m, 6h, H-2,3,4,5,6,6′), 4.48-5.13 (28, 1H, 1H, H-1). The syrupy methyl 2-bromo-2-deoxy-α/β-D-gluco-/mannopyranoside was dissolved in methanol (400 ml), a slurry of 80 g Raney nickel (a 50% slurry in methanol), Et3N (30 ml) and hydrogenated in a Parr apparatus at 120 psi. After 8-9 hours, the reaction mixture was filtered through a Celite filter pad and washed with MeOH. The washings and filtrate were combined and triturated with hexane to separate and remove by filtration insoluble triethylamine hydrobromide and traces of succinimide. The filtrate was concentrated to a residue. The isolated yield of methyl 2-deoxy-α/β-D-glucopyranoside was 89%.Ethyl 2-bromo-2deoxy-α/β-D-gluco-/mannopyranoside:When solvent was ethanol instead of methanol the compound obtained was ethyl 2-bromo-2deoxy-α/β-D-gluco-/mannopyranoside. 1H NMR (200 MHz, D2O): δ 1.10-1.32 (m, 3H, CH3), 2.80 (s, 4H, —CO(CH2)2CO—NH—), 3.40-4.10 (m, 8H, H-2,3,4,5,6,6′, CH2), 4.40, 5.20 (2s 1H, H-1, α/β).Isopropyl 2-bromo-2-deoxy-α/β-D-gluco-/mannopyranoside:When isopropanol instead of methanol was used as a solvent the compound obtained was isopropyl 2-bromo-2-deoxy-α/β-D-gluco/mannopyranoside, 1H NMR (200 MHz, D2O): δ 1.10-1.30 (m, 6H, 2×CH3) 2.80 (s, 4H, —CO(CH2)2CO—NH—), 3.60-4.60 (m 8H,H-2,3,4,5,6,6′, CH2) 4.40, 5,30 (2s, 1H, H-1, α/β.EXAMPLE 3A mixture of D-glucal (64.6 g), methanol (400 ml), N-bromosuccinimide (79 g) were stirred at 15° C. for 6 h. The reaction mixture was hydrogenated in a Parr apparatus in presence of 60 g of Raney nickel catalyst (a 50% slurry in methanol) and triethylamine (62 ml). After 8-9 h, the reaction mixture was filtered on a Celite filter pad. The Celite pad was washed with methanol. The washings and filtrate were combined, concentrated to a thick heavy syrup, dissolve in chloroform (500 ml), pyridine (400 ml) and acetic anhydride (251 ml) was added while stirring, maintaining the temperature between 5-10° C. After 12 hours, the reaction mixture was diluted with CHCl3 (500 ml) transferred to a separating funnel and organic phase was washed with water. The organic phase was separated, dried (Na2SO4) and concentrated to obtain methyl 2-deoxy-3,4,6-tri-O-acetyl-2 deoxy-α/β-D-glucopyranoside as a syrup (163.43 g, 87%). [α]D+65.0° (c 1.0, CHCl3) 1H NMR (200 MHz, CDCl3): δ 1.55-1.90 (m, 2H, H-22′), 2.01, 2.04, 2.11, 2.15, (4s, 9H, 3×OCOCH3), 2.18, 3.40 (2s, 3H, OCH3), 3.45-50 (m, 3H, H-5, 6,6′) 4.80-5.40 (m, 3H,H-1,3,4). The syrup was dissolved in methanol (600 ml) 1N NaOMe in methanol (25 ml) was added and left at room temperature. After 6-10 h, dry CO2 gas was passed into the reaction mixture, solvent was evaporated to obtain a syrupy residue. The residue was once again extracted into dry methanol and concentrated to obtain methyl 2-deoxy-α/β-D-glucopyranoside as syrup. Quantity obtained 81 g (92%).EXAMPLE 4A 500 ml round bottom flask equipped with magnetic stir bar was charged with a solution of D-glucal (323 g) in methanol (175 ml), cooled to 15° C., N-bromosuccinimide (NIBS) (39.4 g) was added and stirred or 6 hours at 15° C., The reaction mixture was concentrated to half the volume, cooled to 0° C. and separated succinimide, was removed by filtration. To the filtrate was added a slurry of 30 g Raney nickel (a 50% slurry in Methanol) Et3N (32 ml) and hydrogenated in a Parr apparatus at 120 psi. After 7-8 hours, the reaction mixture was filtered through a Celite filter pad, and washed with MeOH. The washings and filtrate were combined and triturate with hexane to separate and remove by filtration insoluble triethylamine hydrobromide and succinimide. The filtrate was concentrated to a residue, dissolved in methanol and triturated with hexane to remove most of the triethylamine hydrobromide and succinimide. The filtrate was concentrated to obtain methyl 2-deoxy-α/β-D-glucopyranoside (85%).EXAMPLE 5To a stirred solution of methyl 3,4,6-tri-O-acetyl-2-deoxy-α/β-D-glucopyranoside (47 g) (from example 3) in acetic acid (40 ml) and acetic anhydride (110 ml) was added concentrated sulphuric acid (0.94 ml) at 0°. The reaction mixture was brought to room temperature and stirred. After 2 hours the reaction mixture was diluted with water (50 ml) and extracted into CH2Cl2 (3×150 ml). The organic phase was separated, washed with saturated NaHCO3 solution H2O dried over Na2SO4 and concentrated to obtain 2-deoxy-1,3,4,6-tetra-O-acetyl-α/β-D-glucopyranoside as a crystalline compound. mp. 115-118° C. Quantity obtained 44.5 g (86%). [α]D+21.5° (c 1.0, CHCl3). 1H NMR (200 MHz, CDCl3): δ 1.50-2.45 (m, 14H, H-2,2′, 4×OCOCH3), 3.85-5.40, (m, 5H, H-3,4,5,6,6′), 5.75-6.20 (m, 1H, H-1, α/β). To a heterogeneous mixture of 1,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (10 g) in water (100 ml) was added acetyl chloride (10 ml) and heated to 80° C. After 6 hours the reaction mixture was cooled to room temperature, neutralised with saturated aq. Ba(OH)2, concentrated to half the volume and filtered on a Celite pad, Filtrate was concentrated on a rotary evaporator and dried over anhydrous P2O5 to obtain a residue which was dissolved in hot isopropyl alcohol and filtered on a pad of Celite to obtain a clear filtrate. The filtrate was concentrated to a residue, dissolved in hot isopropyl alcohol (50 ml), acetone (75 ml) and seeded with a few crystals of 2-deoxy-D-glucose. After 15-18 hours at 5° C. crystalline title product was filtered. Quantity obtained 3.21 g (64.9%) m.p. 148-149° C.EXAMPLE 6A heterogeneous mixture of 1,3,4,6-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (9 g) (from example 5), water (30 ml) and 11% aq. H2SO4 (0.3 ml) was stirred at 85° C. for 7 h to obtain a homogenous solution. The reaction mixture was cooled, neutralised with aq. Ba(OH)2 solution and filtered. The filtrate obtained was concentrated to half the volume and solids separated were filtered. To the filtrate was added activated carbon (1 g) and filtered. The filtrate was concentrated on a rotary evaporator and dried over P2O5 to obtain 2-deoxy-D-glucose that was crystallized from methyl alcohol (27 ml) and acetone (54 ml). Quantity obtained 2.4 g. mp. 146-149° C.,EXAMPLE 7A heterogeneous mixture of 1,3,4,tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (25 g) (from example 5), H2O (250 ml), toluene (250 ml) and glacial acetic acid (1.25 ml) was heated to reflux for 10-12 hours, while it was connected to a Dean-Stark azeotropic distillation apparatus. An azeotropic mixture of acetic acid, toluene was collected to remove acetic acid and every one hour fresh toluene (50 ml) was introduced. After completion of the reaction, toluene was removed by distillation from the reaction mixture to obtain a residue that was dissolved in methanol, treated with charcoal and filtered. Be filtrate was separated, concentrated to a residue and crystallized from isopropyl alcohol and acetone to obtain 2-deoxy-D-glucose (7.33 g, 59%). mp. 148-151° C.EXAMPLE 8A heterogeneous mixture of 1,3,4,5-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (10 g) (tom example 5), H2O (200 ml) conc. HCl (0.3 ml) and glacial acetic acid (0.5 ml) was heated to 85° C. After 6 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered on a pad of Celite. Filtrate was separated, treated with charcoal and filtered. The filtrate was concentrated to a residue and crystallized from MeOH, acetone to obtain the product. Quantity obtained 275 g. mp. 147-148° C.EXAMPLE 9A heterogeneous mixture of 1,3,4,5-tetra-O-acetyl-2-deoxy-α/β-D-glucopyranoside (10 g) (from example 3) water (100 ml) and conc. HCl (0.5 ml) was heated to 80° C. After 2-5 hours the reaction mixture was cooled to room temperature, neutralized with aq. Ba(OH)2 and filtered on a pad of Celite. The filtrate was concentrated to a residue, dissolved in ethanol, treated with charcoal and filtered. The filtrate was concentrated to a solid residue and crystallized from methanol-acetone to obtain the title product. Quantity obtained 3.15 g mp. 148-151° C.,EXAMPLE 10A solution of methyl 2-deoxy-α/β-D-glucopyranoside (30 g) (from example 2) water (15 ml) and conc. HCl (1.5 ml) was heated to 80-85° C. After 3-5 hours the reaction mixture was cooled to room temperature, neutralize with aq. Ba(OH)2 and filtered to remove insoluble salts. The filtrate was concentrated to a residue, crystallized from MeOH, acetone and hexane to obtain 2-deoxy-D-glucose (11.77 g) mp. 149-151° C.EXAMPLE 11A solution of methyl 2-deoxy-α/β-D-glucopyranoside (30 g) (form example 2) water (195 ml) and conc. H2SO4 (5.9 ml) was heated to 80° C. After 2-3 hours the reaction mixture was cooled, neutralized with aq. Ba(OH)2 and filtered. The filtrate was separated, treated with charcoal and filtrate. The Filtrate was concentrated to a residue and crystallized from isopropyl alcohol to obtain the title product. Quantity obtained 5.2 g. mp. 152-154° C.EXAMPLE 12A mixture of methyl 2-deoxy-α/β-D-glucopyranoside (24 g) (from example 2) water (125 ml) and IR 120H+resin (7.5 ml) was heated to 90-95° C. for 2 h. The reaction mixture was cooled to room temperature, filtered and the resin was washed with water (20 ml). The filtrate was concentrated to residue and crystallized from ethanol to obtain 2-deoxy-D-glucose (8.8 g), mp. 150-152° C.CLIP
References
- ^ Merck Index, 11th Edition, 2886.
- ^ Wick, AN; Drury, DR; Nakada, HI; Wolfe, JB (1957). “Localization of the primary metabolic block produced by 2-deoxyglucose”(PDF). J Biol Chem. 224 (2): 963–969. doi:10.1016/S0021-9258(18)64988-9. PMID 13405925.
- ^ Pelicano, H; Martin, DS; Xu, RH; Huang, P (2006). “Glycolysis inhibition for anticancer treatment”. Oncogene. 25 (34): 4633–4646. doi:10.1038/sj.onc.1209597. PMID 16892078.
- ^ Raez, LE; Papadopoulos, K; Ricart, AD; Chiorean, EG; Dipaola, RS; Stein, MN; Rocha Lima, CM; Schlesselman, JJ; Tolba, K; Langmuir, VK; Kroll, S; Jung, DT; Kurtoglu, M; Rosenblatt, J; Lampidis, TJ (2013). “A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors”. Cancer Chemother. Pharmacol. 71 (2): 523–30. doi:10.1007/s00280-012-2045-1. PMID 23228990. S2CID 2990078.
- ^ Ralser, M.; Wamelink, M. M.; Struys, E. A.; Joppich, C.; Krobitsch, S.; Jakobs, C.; Lehrach, H. (2008). “A catabolic block does not sufficiently explain how 2-deoxy-D-glucose inhibits cell growth”. Proceedings of the National Academy of Sciences. 105 (46): 17807–17811. Bibcode:2008PNAS..10517807R. doi:10.1073/pnas.0803090105. PMC 2584745. PMID 19004802.
- ^ Kurtoglu, M.; Gao, N.; Shang, J.; Maher, J. C.; Lehrman, M. A.; Wangpaichitr, M.; Savaraj, N.; Lane, A. N.; Lampidis, T. J. (2007-11-07). “Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation”. Molecular Cancer Therapeutics. 6 (11): 3049–3058. doi:10.1158/1535-7163.mct-07-0310. ISSN 1535-7163. PMID 18025288.
- ^ Xi, Haibin; Kurtoglu, Metin; Liu, Huaping; Wangpaichitr, Medhi; You, Min; Liu, Xiongfei; Savaraj, Niramol; Lampidis, Theodore J. (2010-07-01). “2-Deoxy-d-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion”. Cancer Chemotherapy and Pharmacology. 67 (4): 899–910. doi:10.1007/s00280-010-1391-0. ISSN 0344-5704. PMC 3093301. PMID 20593179.
- ^ Jump up to:a b Defenouillère, Quentin; Verraes, Agathe; Laussel, Clotilde; Friedrich, Anne; Schacherer, Joseph; Léon, Sébastien (2019-09-03). “The induction of HAD-like phosphatases by multiple signaling pathways confers resistance to the metabolic inhibitor 2-deoxyglucose”. Science Signaling. 12 (597): eaaw8000. doi:10.1126/scisignal.aaw8000. ISSN 1945-0877. PMID 31481524. S2CID 201829818.
- ^ Garriga-Canut, Mireia; Schoenike, Barry; Qazi, Romena; Bergendahl, Karen; Daley, Timothy J.; Pfender, Rebecca M.; Morrison, John F.; Ockuly, Jeffrey; Stafstrom, Carl; Sutula, Thomas; Roopra, Avtar (2006). “2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CTBP–dependent metabolic regulation of chromatin structure”. Nature Neuroscience. 9 (11): 1382–1387. doi:10.1038/nn1791. PMID 17041593. S2CID 10175791.
- ^ Garriga-Canut, M.; Schoenike, B.; Qazi, R.; Bergendahl, K.; Daley, T. J.; Pfender, R. M.; Morrison, J. F.; Ockuly, J.; Stafstrom, C.; Sutula, T.; Roopra, A. (2006). “2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP–dependent metabolic regulation of chromatin structure”. Nature Neuroscience. 9 (11): 1382–1387. doi:10.1038/nn1791. PMID 17041593. S2CID 10175791.
- ^ Jia Yao, Shuhua Chen, Zisu Mao, Enrique Cadenas, Roberta Diaz Brinton “2-Deoxy-D-Glucose Treatment Induces Ketogenesis, Sustains Mitochondrial Function, and Reduces Pathology in Female Mouse Model of Alzheimer’s Disease”, PLOS ONE
- ^ Researchers develop novel, non-toxic approach to treating variety of cancers. ScienceDaily
- ^ Liu, Huaping; Kurtoglu, Metin; León-Annicchiarico, Clara Lucia; Munoz-Pinedo, Cristina; Barredo, Julio; Leclerc, Guy; Merchan, Jaime; Liu, Xiongfei; Lampidis, Theodore J. (2016). “Combining 2-deoxy-D-glucose with fenofibrate leads to tumor cell death mediated by simultaneous induction of energy and ER stress”. Oncotarget. 7 (24): 36461–36473. doi:10.18632/oncotarget.9263. PMC 5095013. PMID 27183907.
- ^ Kovar, Joy L.; Volcheck, William; Sevick-Muraca, Eva; Simpson, Melanie A.; Olive, D. Michael (2009). “Characterization and performance of a near-infrared 2-deoxyglucose optical imaging agent for mouse cancer models”. Analytical Biochemistry. 384(2): 254–262. doi:10.1016/j.ab.2008.09.050. PMC 2720560. PMID 18938129.
- ^ Cheng, Z., Levi, J., Xiong, Z., Gheysens, O., Keren, S., Chen, X., and Gambhir, S., Bioconjugate Chemistry, 17(3), (2006), 662-669
- ^ Barban, Stanley (December 1962). “Induced resistance to 2-deoxy-d-glucose in cell cultures”. Biochimica et Biophysica Acta. 65(2): 376–377. doi:10.1016/0006-3002(62)91065-x. ISSN 0006-3002. PMID 13966473.
- ^ Sanz, Pascual; Randez-Gil, Francisca; Prieto, José Antonio (September 1994). “Molecular characterization of a gene that confers 2-deoxyglucose resistance in yeast”. Yeast. 10 (9): 1195–1202. doi:10.1002/yea.320100907. ISSN 0749-503X. PMID 7754708. S2CID 9497505.
The Drugs Controller General of India (DCGI) has given permission for the emergency use of drug 2-deoxy-D-glucose (2-DG) as an adjunct therapy in moderate to severe Covid-19 cases, said Defence Research and Development Organisation on Saturday.
“Being a generic molecule and analogue of glucose, it can be easily produced and made available in plenty,” said the DRDO in a statement.
An adjunct therapy refers to an alternative treatment that is used together with the primary treatment. Its purpose is to assist the primary treatment.
“The drug has been developed by DRDO lab Institute of Nuclear Medicine and Allied Sciences in collaboration with Dr Reddy’s Laboratories. Clinical trial have shown that this molecule helps in faster recovery of hospitalized patients and reduces supplemental oxygen dependence,” the statement read.
According to DRDO, the patients treated with 2-DG showed faster symptomatic cure than Standard of Care (SoC) on various endpoints in the efficacy trends.
“A significantly favourable trend (2.5 days difference) was seen in terms of the median time to achieving normalization of specific vital signs parameters when compared to SOC,” it said.
The drug comes in powder form in sachets, which is taken orally by dissolving it in water.
“It accumulates in the virus-infected cells and prevents virus growth by stopping viral synthesis and energy production,” said the DRDO.
In April 2020, during the first wave of the Covid-19 pandemic, INMAS-DRDO scientists conducted laboratory experiments of 2-DG with the help of the Centre for Cellular and Molecular Biology (CCMB), Hyderabad.
They found that this molecule works effectively against the SARS-CoV-2 virus and inhibits viral growth.
Based on the results, the DCGI had in May 2020 permitted Phase-II clinical trial of 2-DG in Covid-19 patients.
In Phase-II trials (including dose-ranging) conducted from May to October 2020, the drug was found to be safe and showed significant improvement in the patients’ recovery.
“Phase IIa was conducted in 6 hospitals and Phase IIb (dose-ranging) clinical trial was conducted at 11 hospitals all over the country. Phase-II trial was conducted on 110 patients,” said the DRDO.
NEW DRUG APPROVALS
ONE TIME
$10.00
| Names | |
|---|---|
| IUPAC name(4R,5S,6R)-6-(hydroxymethyl)oxane-2,4,5-triol | |
| Other names2-Deoxyglucose 2-Deoxy-d-mannose 2-Deoxy-d-arabino-hexose 2-DG | |
| Identifiers | |
| CAS Number | 154-17-6 |
| 3D model (JSmol) | Interactive image |
| ChEMBL | ChEMBL2074932 |
| ChemSpider | 388402 |
| EC Number | 205-823-0 |
| IUPHAR/BPS | 4643 |
| PubChem CID | 108223 |
| UNII | 9G2MP84A8W |
| showInChI | |
| showSMILES | |
| Properties | |
| Chemical formula | C6H12O5 |
| Molar mass | 164.16 g/mol |
| Melting point | 142 to 144 °C (288 to 291 °F; 415 to 417 K) |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
////////////2-Deoxy-D-glucose, 2 dg, 2-dg, 2 DEOXY D GLUCOSE, COVID 19, CORONA VIRUS, INDIA 2021, DCGI, DRDO, DR REDDYS
C(C=O)C(C(C(CO)O)O)O

{kind=link}
{kind=link}
{kind=link}
{kind=link}