Sodium channels play a diverse set of roles in maintaining normal and pathological states, including the long recognized role that voltage gated sodium channels play in the generation of abnormal neuronal activity and neuropathic or pathological pain. Damage to peripheral nerves following trauma or disease can result in changes to sodium channel activity and the development of abnormal afferent activity including ectopic discharges from axotomised afferents and spontaneous activity of sensitized intact nociceptors. These changes can produce long-lasting abnormal hypersensitivity to normally innocuous stimuli, or allodynia. Examples of neuropathic pain include, but are not limited to, post-herpetic neuralgia, trigeminal neuralgia, diabetic neuropathy, chronic lower back pain, phantom limb pain, and pain resulting from cancer and chemotherapy, chronic pelvic pain, complex regional pain syndrome and related neuralgias.
There have been some advances in treating neuropathic pain symptoms by using medications, such as gabapentin, and more recently pregabalin, as short-term, first-line treatments. However, pharmacotherapy for neuropathic pain has generally had limited success with little response to commonly used pain reducing drugs, such as NSAIDS and opiates. Consequently, there is still a considerable need to explore novel treatment modalities.
There remain a limited number of potent effective sodium channel blockers with a minimum of adverse events in the clinic. There is also an unmet medical need to treat neuropathic pain and other sodium channel associated pathological states effectively and without adverse side effects. PCT Published Patent Application No. WO 2006/110917, PCT Published Patent Application No. WO 2010/045251 , PCT Published Patent Application No. WO 2010/045197, PCT Published Patent Application No. WO 2011/047174 and PCT Published Patent Application No. WO 2011/002708 discloses certain spiro-oxindole compounds. These compounds are disclosed therein as being useful for the treatment of sodium channel-mediated diseases, preferably diseases related to pain, central nervous conditions such as epilepsy, anxiety, depression and bipolar disease;
cardiovascular conditions such as arrhythmias, atrial fibrillation and ventricular fibrillation; neuromuscular conditions such as restless leg syndrome; neuroprotection against stroke, neural trauma and multiple sclerosis; and channelopathies such as erythromelalgia and familial rectal pain syndrome.
Methods of preparing these compounds and pharmaceutical compositions containing them are also disclosed in PCT Published Patent Application No. WO 2006/110917, PCT Published Patent Application No. WO 2010/045251 , PCT
Published Patent Application No. WO 2010/045197, PCT Published Patent Application No. WO 2011/047174 and PCT Published Patent Application No. WO 2011/002708.
Postherpetic neuralgia (PHN) is a rare disorder that is defined as significant pain or abnormal sensation 120 days or more after the presence of the initial rash caused by shingles. This pain persists after the healing of the associated rash. Generally, this affliction occurs in older individuals and individuals suffering from immunosuppression. There are about one million cases of shingles in the US per year, of which 10–20% will result in PHN.
Topical analgesics such as lidocaine and capsaicin are traditionally used to treat this disorder. Both lidocaine and TV-45070 have a mechanism of action that involves the inhibition of voltage-gated sodium ion channels.
TV-45070 (formerly XEN-402) was in-licensed by Teva from Xenon Pharmaceuticals and is reported to be an antagonist of the Nav1.7 sodium ion channel protein.
It is currently in Phase II clinical trials for PHN. Interestingly, the loss of function of the Nav1.7 sodium ion channel was reported to result in the inability to experience pain as a hereditary trait in certain individuals.
CONTD…….
The (S)-enantiomer of the invention and the corresponding (R)-enantiomer are prepared by the resolution of the compound of formula (I), as set forth above in the Summary of the Invention, using either chiral high pressure liquid chromatography methods or by simulated moving bed chromatography methods, as described below in the following Reaction Scheme wherein “chiral HPLC” refers to chiral high pressure liquid chromatography and “SMB” refers to simulated moving bed chromatography:
The compound of formula (I) can be prepared by the methods disclosed in PCT Published Patent Application No. WO 2006/110917, by methods disclosed herein, or by methods known to one skilled in the art.
One of ordinary skill in the art would recognize variations in the above Reaction Scheme which are appropriate for the resolution of the individual enantiomers.
Alternatively, the (S)-enantiomer of formula (I-S) and the (R)-enantiomer of formula (I-R), can be synthesized from starting materials which are known or readily prepared using process analogous to those which are known.
Preferably, the (S)-enantiomer of the invention obtained by the resolution methods disclosed herein is substantially free of the (R)-enantiomer or contains only traces of the (R)-enantiomer.
The following Synthetic Examples serve to illustrate the resolution methods disclosed by the above Reaction Schemes and are not intended to limit the scope of the invention.
Synthetic Example 1Synthesis of 1-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-f][1,3]benzodioxole-7,3′-indol]-2′(1′H)-one (Compound of formula (I))
To a suspension of spiro[furo[2,3-f][1,3]benzodioxole-7,3′-indol]-2′(1′H)-one (1.0 g, 3.6 mmol), which can be prepared according to the methods disclosed in PCT Published Patent Application No. WO 2006/110917, and cesium carbonate (3.52 g, 11 mmol) in acetone (50 mL) was added 2-bromomethyl-5-trifluoromethylfuran (1.13 g, 3.9 mmol) in one portion and the reaction mixture was stirred at 55-60° C. for 16 hours. Upon cooling to ambient temperature, the reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The residue was subjected to column chromatography, eluting with ethyl acetate/hexane (1/9-1/1) to afford 1′-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-f][1,3]benzodioxole-7,3′-indol]-2′(1 ′H)-one, i.e., the compound of formula (I), (1.17 g, 76%) as a white solid: mp 139-141° C.;
The compound of formula (I) was resolved into the (S)-enantiomer of the invention and the corresponding (R)-enantiomer by chiral HPLC under the following conditions:
Column: Chiralcel® OJ-RH; 20 mm I.D.×250 mm, 5 mic; Lot: OJRH CJ-EH001 (Daicel Chemical Industries, Ltd)
Eluent: Acetonitrile/Water (60/40, v/v, isocratic)
Flow rate: 10 mL/min
Run time: 60 min
Loading: 100 mg of compound of formula (I) in 1 mL of acetonitrileTemperature: Ambient
Under the above chiral HPLC conditions, the (R)-enantiomer of the compound of formula (I), i.e., (R)-1′-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-f][1,3]-benzodioxole-7,3′-indol]-2′(1′H)-one, was isolated as the first fraction as a white solid; ee (enantiomeric excess)>99% (analytical OJ-RH, 55% acetonitrile in water); mp 103-105° C.; 1H NMR (300 MHz, DMSO-d6) δ 7.32-6.99 (m, 5H), 6.71 (d, J=3.4 Hz, 1H), 6.67 (s, 1H), 6.05 (s, 1H), 5.89 (d, J=6.2 Hz, 2H), 5.13, 5.02 (ABq, JAB=16.4 Hz, 2H), 4.82, 4.72 (ABq, JAB=9.4 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 177.2, 155.9, 152.0, 149.0, 142.4, 142.0, 141.3, 132.0, 129.1, 123.9, 120.6, 119.2, 117.0, 112.6, 109.3, 108.9, 103.0, 101.6, 93.5, 80.3, 58.2, 36.9; MS (ES+) m/z 430.2 (M+1), [α]D−17.46° (c 0.99, DMSO).
The (S)-enantiomer of the compound of formula (I), i.e., (S)-1′-{[5-(trifluoromethypfuran-2-yl]methyl}spiro-[furo[2,3-f][1,3]benzodioxole-7,3′-indol]-2′(1′H)-one was isolated as the second fraction as a white solid; ee >99% (analytical OJ-RH, 55% acetonitrile in water); mp 100-102° C.; 1H NMR (300 MHz, DMSO-d6) δ 7.32-6.99 (m, 5H), 6.71 (d, J=3.4 Hz, 1H), 6.67 (s, 1H), 6.05 (s, 1H), 5.89 (d, J=6.3 Hz, 2H), 5.12, 5.02 (ABq, JAB=16.4 Hz, 2H), 4.82, 4.72 (ABq, JAB=9.4 Hz, 2H); 13C NMR (75MHz, CDCl3) δ 177.2, 155.9, 152.0, 149.0, 142.4, 142.0, 141.3, 132.0, 129.1, 123.9, 120.6, 119.2, 117.0, 112.6, 109.3, 108.9, 103.0, 101.6, 93.5, 80.3, 58.2, 36.9; MS (ES+) m/z 430.2 (M+1), [α]D+14.04° (c 0.99, DMSO)
Synthetic Example 3Resolution of Compound of Formula (I) by SMB Chromatography
The compound of formula (I) was resolved into the (S)-enantiomer of the invention and the corresponding (R)-enantiomer by SMB chromatography under the following conditions:
Extract: 147.05 mL/min, Raffinate: 76.13 mL/min Eluent: 183.18 mL/min Feed: 40 mL/min Recycling: 407.88 mL/min Run Time: 0.57 min Temperature: 25° C. Pressure: 46 bar
The feed solution (25 g of compound of formula (I) in 1.0 L of mobile phase (25:75:0.1 (v:v:v) mixture of acetonitrile/methanol/trifluoroacetic acid)) was injected continuously into the SMB system (Novasep Licosep Lab Unit), which was equipped with eight identical columns in 2-2-2-2 configuration containing 110 g (per column, 9.6 cm, 4.8 cm I.D.) of ChiralPAK-AD as stationary phase. The first eluting enantiomer (the (R)-enantiomer of the compound of formula (I)) was contained in the raffinate stream and the second eluting enantiomer (the (S)-enantiomer of the compound of formula (I)) was contained in the extract stream. The characterization data of the (S)-enantiomer and the (R)-enantiomer obtained from the SMB resolution were identical to those obtained above utilizing chiral HPLC.
The compound of formula (I) was resolved into its constituent enantiomers on a Waters preparative LCMS autopurification system. The first-eluting enantiomer from the chiral column was brominated (at a site well-removed from the stereogenic centre) to give the corresponding 5′-bromo derivative, which was subsequently crystallized to generate a single crystal suitable for X-ray crystallography. The crystal structure of this brominated derivative of the first-eluting enantiomer was obtained and its absolute configuration was found to be the same as the (R)-enantiomer of the invention. Hence, the second-eluting enantiomer from the chiral column is the (S)-enantiomer of the invention. Moreover, the material obtained from the extract stream of the SMB resolution had a specific optical rotation of the same sign (positive, i.e. dextrorotatory) as that of the material obtained from the aforementioned LC resolution.
Patent
WO 2013154712
EXAMPLE 8
Synthesis of (7S)-1 ‘-{[5-(trifluoromethyl)furan-2- yllmethylJspirotfurop.S-flll .Sl enzoclioxole-y.S’-indoll-Zil ‘Wi-one
Compound of formula (ia1 )

To a cooled (0 °C) solution of (3S)-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-3- (hydroxymethyl)-1-{[5-(trifluoromethyl)furan-2-yl]methyl}-1 ,3-dihydro-2H-indol-2-one prepared according to the procedure described in Example 7 (16.4 mmol) and 2- (diphenylphosphino)pyridine (5.2 g, 20 mmol) in anhydrous tetrahydrofuran (170 mL) was added di-ferf-butylazodicarboxylate (4.5 g, 20 mmol). The mixture was stirred for 2 h at 0 °C, then the reaction was diluted with ethyl acetate (170 mL), washed with 3 N hydrochloric acid (7 x 50 mL) and brine (2 x 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was dissolved in ethanol (80 mL), decolorizing charcoal (15 g) was added and the mixture was heated at reflux for 1 h. The mixture was filtered while hot through a pad of diatomaceous earth. The filtrate was concentrated in vacuo and the residue triturated in a mixture of diethyl ether/hexanes to afford (7S)-1 ‘-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro- [furo[2,3-/][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘H)-one (1.30 g) as a colorless solid in 18% yield. The mother liquor from the trituration was concentrated in vacuo, trifluoroacetic acid (20 mL) was added and the mixture stirred for 3 h at ambient temperature. The mixture was diluted with ethyl acetate (100 mL), washed with saturated aqueous ammonium chloride (100 mL), 3 N hydrochloric acid (4 x 60 mL) and brine (2 x 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was purified by column chromatography, eluting with a gradient of ethyl acetate in hexanes to afford further (7S)-1 ‘-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro- [furo[2,3- ][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘H)-one (2.6 g) as a colorless solid (37% yield, overall yield 55% over 2 steps): H NMR (300 MHz, CDCI3) £7.29-6.96 (m, 4H), 6.73 (s, 1 H), 6.50 (s, 1 H), 6.38 (s, 1 H), 6.09 (s, 1 H), 5.85 (br s, 2H), 5.06 (d, J = 16.0 Hz, 1 H), 4.93-4.84 (m, 2H), 4.68-4.65 (m, 1 H); MS (ES+) m/z 429.8 (M + 1 ); ee (enantiomeric excess) >99.5% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl tert- butyl ether).
EXAMPLE 9
Synthesis of 1-(diphenylmethyl)-1 H-indole-2,3-dione
Compound of formula (15a)
A. To a suspension of hexanes-washed sodium hydride (34.0 g, 849 mmol) in anhydrous Λ/,/V-dimethylformamide (400 mL) at 0 °C was added a solution of isatin (99.8 g, 678 mmol) in anhydrous Λ/,/V-dimethylformamide (400 mL) dropwise over 30 minutes. The reaction mixture was stirred for 1 h at 0 °C and a solution of benzhydryl bromide (185 g, 745 mmol) in anhydrous N-dimethylformamide (100 mL) was added dropwise over 5 minutes. The reaction mixture was allowed to warm to ambient temperature, stirred for 16 h and heated at 60 °C for 2 h. The mixture was cooled to 0 °C and water (500 mL) was added. The mixture was poured into water (2 L), causing a precipitate to be deposited. The solid was collected by suction filtration and washed with water (2000 mL) to afford 1-(diphenylmethyl)-1H-indole-2,3- dione (164 g) as an orange solid in 77% yield.
B. Alternatively, to a mixture of isatin (40.0 g, 272 mmol), cesium carbonate (177 g, 543 mmol) and A/./V-dimethylformamide (270 mL) at 80 °C was added dropwise a solution of benzhydryl bromide (149 g, 544 mmol) in N,N- dimethyiformamide (200 mL) over 30 minutes. The reaction mixture was heated at 80 °C for 3 h, allowed to cool to ambient temperature and filtered through a pad of diatomaceous earth. The pad was rinsed with ethyl acetate (1000 mL). The filtrate was washed with saturated aqueous ammonium chloride (4 x 200 mL), 1 N
hydrochloric acid (200 mL) and brine (4 x 200 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with diethyl ether to afford 1 -(diphenylmethyl)-1 H-indole-2,3-dione (59.1 g) as an orange solid in 69% yield. The mother liquor from the trituration was concentrated in vacuo and the residue triturated in diethyl ether to afford a further portion of 1-(diphenylmethyl)-1 H- indole-2,3-dione (8.2 g) in 10% yield: 1H NMR (300 MHz, CDCI3) £7.60 (d, J = 7.4 Hz, 1 H), 7.34-7.24 (m, 1 1 H), 7.05-6.97 (m, 2H), 6.48 (d, J = 8.0 Hz, 1 H); MS (ES+) m/z 313.9 (M + 1 ).
C. Alternatively, a mixture of isatin (500 g, 3.4 mol) and anhydrous N,N- dimethylformamide (3.5 L) was stirred at 15-35 °C for 0.5 h. Cesium carbonate (2.2 kg, 6.8 mol) was added and the mixture stirred at 55-60 °C for 1 h. A solution of benzhydryl bromide (1.26 kg, 5.1 mol) in anhydrous N, A/-dimethylformamide (1.5 L) was added and the resultant mixture stirred at 80-85 °C for 1 h, allowed to cool to ambient temperature and filtered. The filter cake was washed with ethyl acetate (12.5 L). To the combined filtrate and washes was added 1 N hydrochloric acid (5 L). The phases were separated and the aqueous phase was extracted with ethyl acetate (2.5 L). The combined organic extracts were washed with 1 N hydrochloric acid (2 * 2.5 L) and brine (3 χ 2.5 L) and concentrated in vacuo to a volume of approximately 750 mL. Methyl ferf-butyl ether (2 L) was added and the mixture was cooled to 5-15 °C, causing a solid to be deposited. The solid was collected by filtration, washed with methyl ferf- butyl ether (250 mL) and dried in vacuo at 50-55 °C for 16 h to afford 1- (diphenylmethyl)-1 H-indole-2,3-dione (715 g) as an orange solid in 67% yield: 1H NMR (300 MHz, CDCI3) 7.60 (d, J = 7.4 Hz, H), 7.34-7.24 (m, 1 H), 7.05-6.97 (m, 2H), 6.48 (d, J = 8.0 Hz, 1 H); MS (ES+) m/z 313.9 (M + 1 ).
EXAMPLE 10
Synthesis of 1-(diphenylmethyl)-3-hydroxy-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-1 ,3- dihydro-2H-indol-2-one
Compound of formula (16a1 )
A. To a solution of sesamol (33.1 g, 239 mmol) in anhydrous
tetrahydrofuran (500 mL) at 0 °C was added dropwise a 2 M solution of
isopropylmagnesium chloride in tetrahydrofuran (104 mL, 208 mmol), followed by 1 – (diphenylmethyl)-1H-indole-2,3-dione (50.0 g, 160 mmol) and tetrahydrofuran (100 mL). The reaction mixture was stirred at ambient temperature for 5 h, diluted with ethyl acetate (1500 mL), washed with saturated aqueous ammonium chloride (400 mL) and brine (2 x 400 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with a mixture of diethyl ether and hexanes to afford 1- (diphenylmethyl)-3-hydroxy-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-1 ,3-dihydro-2H-in
2- one (70.7 g) as a colorless solid in 98% yield: 1H NMR (300 MHz, CDCI3) <59.12 (br s, 1 H), 7.45-7.43 (m, 1 H), 7.30-7.22 (m, 10H), 7.09-7.07 (m, 2H), 6.89 (s, 1 H), 6.56- 6.55 (m, 1 H), 6.47-6.46 (m, 1 H), 6.29-6.28 (m, 1 H), 5.86 (s, 2H), 4.52 (br s, 1 H); MS (ES+) m/z 433.7 (M – 17).
B. Alternatviely, a mixture of sesamol (0.99 kg, 7.2 mol) and anhydrous tetrahydrofuran (18 L) was stirred at 15-35 °C for 0.5 h and cooled to -5-0 °C.
Isopropyl magnesium chloride (2.0 M solution in tetrahydrofuran, 3.1 L, 6.2 mol) was added, followed by 1-(diphenylmethyl)-1 H-indole-2,3-dione (1.50 kg, 4.8 mol) and further anhydrous tetrahydrofuran (3 L). The mixture was stirred at 15-25 °C for 5 h. Ethyl acetate (45 L) and saturated aqueous ammonium chloride (15 L) were added. The mixture was stirred at 15-25 °C for 0.5 h and was allowed to settle for 0.5 h. The phases were separated and the organic phase was washed with brine (2.3 L) and concentrated in vacuo to a volume of approximately 4 L. Methyl ferf-butyl ether (9 L) was added and the mixture concentrated in vacuo to a volume of approximately 4 L. Heptane (6 L) was added and the mixture was stirred at 15-25 °C for 2 h, causing a solid to be deposited. The solid was collected by filtration, washed with methyl tert- butyl ether (0.3 L) and dried in vacuo at 50-55 °C for 7 h to afford 1-(diphenylmethyl)-3- hydroxy-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-1 ,3-dihydro-2H-indol-2-one (2.12 kg) as an off-white solid in 98% yield: 1H NMR (300 MHz, CDCI3) 9.12 (br s, 1 H), 7.45-7.43 (m, 1 H), 7.30-7.22 (m, 10H), 7.09-7.07 (m, 2H), 6.89 (s, 1 H), 6.56-6.55 (m, 1 H), 6.47-6.46 (m, 1 H), 6.29-6.28 (m, 1 H), 5.86 (s, 2H), 4.52 (br s, 1 H); MS (ES+) m/z 433.7 (M – 17).
EXAMPLE 1 1
Synthesis of 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1-(diphenylmethyl)-3-hydroxy-1 ,3- dihydro-2H-indol-2-one
Compound of formula (17a1)
A. A mixture of 1-(diphenylmethyl)-3-hydroxy-3-(6-hydroxy-1 ,3- benzodioxol-5-yl)-1 ,3-dihydro-2H-indol-2-one (30.0 g, 66.5 mmol), benzyl bromide (8.3 mL, 70 mmol), and potassium carbonate (18.4 g, 133 mmol) in anhydrous N,N- dimeihylformamide (100 mL) was stirred at ambient temperature for 16 h. The reaction mixture was filtered and the solid was washed with /V,A/-dimethylformamide (100 mL). The filtrate was poured into water (1000 mL) and the resulting precipitate was collected by suction filtration and washed with water to afford 3-[6-(benzyloxy)-1 ,3-benzodioxol- 5-yl]-1-(diphenylmethyl)-3-hydroxy-1 ,3-dihydro-2H-indol-2-one (32.0 g) as a beige solid in 83% yield: 1H NMR (300 MHz, CDCI3) 7.42-7.28 (m, 9H), 7.22-7.14 (m, 6H), 7.10- 6.93 (m, 3H), 6.89-6.87 (m, 2H), 6.53 (d, J = 7.6 Hz, 1 H), 6.29 (br s, 1 H), 5.88 (s, 1 H), 5.85 (s, 1 H), 4.66 (d, J = 14.2 Hz, 1 H), 4.51 (d, J = 14.1 Hz, 1 H), 3.95 (s, 1 H); MS (ES+) m/z 542.0 (M + 1), 523.9 (M – 17).
B. Alternatively, to a solution of 1-(diphenylmethyl)-3-hydroxy-3-(6- hydroxy-1 ,3-benzodioxol-5-yl)-1 ,3-dihydro-2H-indol-2-one (2.1 kg, 4.6 mol) in anhydrous A/,A/-dimethylformamide (8.4 L) at 20-30 °C was added potassium carbonate (1.3 kg, 9.2 mol), followed by benzyl bromide (0.58 L, 4.8 mol). The mixture was stirred at 20-30 °C for 80 h and filtered. The filter cake was washed with
A/,/V-dimethylformamide (0.4 L) and the filtrate was poured into water (75 L), causing a solid to be deposited. The mixture was stirred at 15-25 °C for 7 h. The solid was collected by filtration, washed with water (2 L) and dried in vacuo at 50-60 °C for 48 h to afford 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1-(diphenylmethyl)-3-hydroxy-1 ,3- dihydro-2H-indol-2-one (2.1 1 kg) as an off-white solid in 84% yield; 1H NMR (300
MHz, CDCI3) £7.42-7.28 (m, 9H), 7.22-7.14 (m, 6H), 7.10-6.93 (m, 3H), 6.89-6.87 (m, 2H), 6.53 (d, J = 7.6 Hz, 1 H), 6.29 (br s, 1 H), 5.88 (s, 1 H), 5.85 (s, 1 H), 4.66 (d, J = 14.2 Hz, 1 H), 4.51 (d, J = 14.1 Hz, 1 H), 3.95 (s, 1 H); MS (ES+) m/z 542.0 (M + 1 ).
EXAMPLE 12
Synthesis of 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1 -(diphenylmethyl)-l ,3-dihydro-2H- indol-2-one
Compound of formula (18a1 )
A. To a solution of 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1- (diphenylmethyl)-3-hydroxy-1 ,3-dihydro-2H-indol-2-one (32.0 g, 57.7 mmol) in dichloromethane (100 mL) was added trifluoroacetic acid (50 mL) followed by triethylsilane (50 mL). The reaction mixture was stirred at ambient temperature for 2 h and concentrated in vacuo. The residue was dissolved in ethyi acetate (250 mL), washed with saturated aqueous ammonium chloride (3 x 100 mL) and brine (3 x 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with diethyl ether to afford 3-[6-(benzyloxy)-1 ,3-benzodioxol-5- yl]-1-(diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one (19.0 g) as a colorless solid in 61 % yield: 1H NMR (300 MHz, CDCI3) 7.31 -7.23 (m, 15H), 7.10-6.88 (m, 4H), 6.50-6.45 (m, 3H), 5.86 (s, 2H), 4.97-4.86 (m, 3H); MS (ES+) m/z 525.9 (M + 1).
B. Alternatively, to a solution of 3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-1- (diphenylmethyl)-3-hydroxy-1 ,3-dihydro-2H-indol-2-one (2.0 kg, 3.7 mol) in
dichloromethane (7 L) at 20-30 °C was added trifluoracetic acid (2.5 L), followed by triethylsilane (3.1 L). The mixture was stirred at 15-35 °C for 4 h and concentrated in vacuo to dryness. To the residue was added ethyl acetate (16 L) and the mixture was stirred at 15-35 °C for 0.5 h, washed with saturated aqueous ammonium chloride (3 x 7 L) and brine (3 χ 7 L) and concentrated in vacuo to a volume of approximately 7 L. Methyl ferf-butyl ether (9 L) was added and the mixture concentrated in vacuo to a volume of approximately 9 L and stirred at 10-20 °C for 2.5 h, during which time a solid was deposited. The solid was collected by filtration, washed with methyl te/t-butyl ether (0.4 L) and dried in vacuo at 50-55 °C for 7 h to afford 3-[6-(benzyloxy)-1 ,3- benzodioxol-5-yl]-1-(diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one (1 .26 kg) as an off-white solid in 65% yield: 1H NMR (300 MHz, CDCI3) £7.31 -7.23 (m, 15H), 7.10- 6.88 (m, 4H), 6.50-6.45 (m, 3H), 5.86 (s, 2H), 4.97-4.86 (m, 3H); MS (ES+) m/z 525.9 (M + 1).
EXAMPLE 13
Synthesis of (3S)-3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-3-[(benzyloxy)methyl]-1 –
(diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one
Compound of formula (19a1 )
A. To a nitrogen-degassed mixture of 50% w/w aqueous potassium hydroxide (69.6 mL, 619 mmol), toluene (100 mL), and (9S)-1 -(anthracen-9- ylmethyl)cinchonan-1 -ium-9-ol chloride (0.50 g, 0.95 mmol) cooled in an ice/salt bath to an internal temperature of -18 °C was added a nitrogen-degassed solution of 3-[6- (benzyloxy)-l ,3-benzodioxol-5-yl]-1 -(diphenylmethyl)-l ,3-dihydro-2H-indol-2-one (10.0 g, 19.0 mmol) and benzyl chloromethyl ether (2.9 mL, 21 mmol) in
toluene/tetrahydrofuran (1 :1 v/v, 80 mL) dropwise over 1 h. The reaction mixture was stirred for 3.5 h and diluted with ethyl acetate (80 mL). The organic phase was washed with 1 N hydrochloric acid (3 x 150 mL) and brine (2 x 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo to afford (3S)-3-[6-(benzyloxy)-1 ,3- benzodioxol-5-yl]-3-[(benzyloxy)methyl]-1-(diphenylmethyl)-1 ,3-dihydro-2/-/-indol-2-one (12.6 g) as a colorless solid in quantitative yield: 1H NMR (300 MHz, CDCI3) 7.42 (d, 2H), 7.24-6.91 (m, 21 H), 6.69-6.67 (m, 2H), 6.46 (d, J = 7.7 Hz, 1 H), 6.15 (s, 1 H), 5.83- 5.81 (m, 2H), 4.53-4.31 (m, 3H), 4.17-4.09 (m, 3H); MS (ES+) m/z 646.0 (M + 1); ee (enantiomeric excess) 90% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl tert-butyl ether).
B. Alternatively, a mixture of 50% w/v aqueous potassium hydroxide (4.2 kg), toluene (12 L) and (9S)-1 -(anthracen-9-ylmethyl)cinchonan-1 -ium-9-ol chloride (0.06 kg, 0.1 mol) was degassed with dry nitrogen and cooled to -18 to -22 °C. To this mixture was added a cold (-18 to -22 °C), nitrogen-degassed solution of 3-[6-
(benzyloxy)-l ,3-benzodioxol-5-yl]-1 ~(diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one (1.2 kg, 2.3 mol) and benzyl chloromethyl ether (0.43 kg, 2.8 mol) in toluene (10 L) and tetrahydrofuran (10 L) at -18 to 22 °C over 3 h. The mixture was stirred at -18 to -22 °C for 5 h, allowed to warm to ambient temperature and diluted with ethyl acetate (10 L). The phases were separated and the organic layer was washed with 1 N
hydrochloric acid (3 χ 8 L) and brine (2 χ 12 L) and concentrated in vacuo to dryness to afford (3S)-3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-3-[(benzyloxy)methyl]-1- (diphenylmethyl)-1 ,3-dihydro-2H-indol-2-one (1.5 kg) as a colorless solid in quantitative yield: 1H NMR (300 MHz, CDCI3) £7.42 (d, 2H), 7.24-6.91 (m, 21 H), 6.69-6.67 (m, 2H), 6.46 (d, J = 7.7 Hz, 1 H), 6.15 (s, 1 H), 5.83-5.81 (m, 2H), 4.53-4.31 (m, 3H), 4.17- 4.09 (m, 3H); MS (ES+) m/z 646.0 (M + 1); ee (enantiomeric excess) 90% (HPLC, ChiralPak IA). EXAMPLE 14
Synthesis of (3S)-1-(diphenylmethyl)-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-3- (hydroxymethyl)-1 ,3-dihydro-2/-/-indol-2-one
Compound of formula (20a1)

A. A mixture of (3S)-3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-3- [(benzyloxy)methyl]-1 -(diphenylmethyl)-1 ,3-dihydro-2/-/-indol-2-one (8.8 g, 14 mmol), 10% w/w palladium on carbon (50% wetted powder, 3.5 g, 1.6 mmol), and acetic acid (3.9 ml_, 68 mmol) in a nitrogen-degassed mixture of ethanol/tetrahydrofuran (1 : 1 v/v, 140 mL) was stirred under hydrogen gas (1 atm) at ambient temperature for 4 h. The reaction mixture was filtered through a pad of diatomaceous earth and the pad was rinsed with ethyl acetate (100 mL). The filtrate was concentrated in vacuo to afford (3S)-1-(diphenylmethyl)-3-(6-hydroxy-1 ,3-benzodioxol-5-yl)-3-(hydroxymethyl)-1 ,3- dihydro-2H-indol-2-one as a colorless solid that was carried forward without further purification: H NMR (300 MHz, CDCI3) 9.81 (br s, 1 H), 7.35-7.24 (m, 1 1 H), 7.15- 7.01 (m, 3H), 6.62 (s, 1 H), 6.54-6.47 (m, 2H), 5.86-5.84 (m, 2H), 4.76 (d, J = 1 1.0 Hz, 1 H), 4.13-4.04 (m, 1 H), 2.02 (s, 1 H); MS (ES+) m/z 465.9 (M + 1); ee (enantiomeric excess) 93% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl ie t-butyl ether).
B. Alternatively, a glass-lined hydrogenation reactor was charged with (3S)-3-[6-(benzyloxy)-1 ,3-benzodioxol-5-yl]-3-[(benzyloxy)methyl]-1 -(diphenylmethyl)- 1 ,3-dihydro-2H-indol-2-one (0.1 kg, 0.15 mol), tetrahydrofuran (0.8 L), ethanol (0.4 L), acetic acid (0.02 L) and 20% w/w palladium (li) hydroxide on carbon (0.04 kg). The reactor was purged three times with nitrogen. The reactor was then purged three times with hydrogen and was then pressurized to 50-55 lb/in2 with hydrogen. The mixture was stirred at 20-30 °C for 5 h under a 50-55 lb/in2 atmosphere of hydrogen. The reactor was purged and the mixture was filtered. The filtrate was concentrated in vacuo to a volume of approximately 0.2 L and methyl te/t-butyl ether (0.4 L) was added. The mixture was concentrated in vacuo to a volume of approximately 0.2 L and methyl ie/t-butyl ether (0.2 L) was added, followed by heptane (0.25 L). The mixture was stirred at ambient temperature for 2 h, during which time a solid was deposited. The solid was collected by filtration, washed with heptane (0.05 L) and dried in vacuo at a temperature below 50 °C for 8 h to afford (3S)-1 -(diphenylmethyl)-3-(6-hydroxy- 1 ,3-benzodioxol-5-yl)-3-(hydroxymethyl)-1 ,3-dihydro-2H-indol-2-one (0.09 kg) as a colorless solid in 95% yield: 1H NMR (300 MHz, CDCI3) 9.81 (br s, 1 H), 7.35-7.24 (m, 1 1 H), 7.15-7.01 (m, 3H), 6.62 (s, 1 H), 6.54-6.47 (m, 2H), 5.86-5.84 (m, 2H), 4.76 (d, J = 1 1.0 Hz, 1 H), 4.13-4.04 (m, 1 H), 2.02 (s, 1 H); MS (ES+) m/z 465.9 (M + 1); ee (enantiomeric excess) 91% (HPLC, ChiralPak IA).
EXAMPLE 15
Synthesis of (7S)-1′-(diphenylmethyl)spiro[furo[2,3-/][1 ,3]benzodioxole-7,3′-indol]-
2′(1 ‘tf)-one
Compound of formula (21 a1 )
A. To a cooled (0 °C) solution of (3S)-1 -(diphenylmethyl)-3-(6-hydroxy-1 ,3- benzodioxol-5-yl)-3-(hydroxymethyl)-1 ,3-dihydro-2H-indol-2-one prepared according to the procedure described in Example 14 (13.6 mmol) and 2-
(diphenylphosphino)pyridine (4.3 g, 16 mmol) in anhydrous tetrahydrofuran (140 mL) was added di-tert-butylazodicarboxylate (3.8 g, 17 mmol). The reaction mixture was stirred at 0 °C for 3 h, diluted with ethyl acetate (140 mL), washed with 3 N
hydrochloric acid (6 * 50 mL) and brine (2 χ 100 mL), dried over anhydrous sodium sulfate, filtered and concentrated in vacuo. The residue was triturated with a mixture of diethyl ether and hexanes to afford (7S)-1 ‘-(diphenylmethyl)spiro[furo[2,3- ][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘H)-one (4.55 g) as a colorless solid in a 75% yield over 2 steps: 1H NMR (300 MHz, CDCI3) 7.34-7.24 (m, 10H), 7.15-7.13 (m, 1 H), 7.04 (s, 1 H), 6.99-6.95 (m, 2H), 6.50-6.48 (m, 2H), 6.06 (s, 1 H), 5.85-5.83 (m, 2H), 4.96 (d, J = 8.9 Hz, 1 H), 4.69 (d, J = 8.9 Hz, 1 H); MS (ES+) m/z 447.9 (M + 1); ee
(enantiomeric excess) 93% (HPLC, Chiraipak IA, 2.5% acetonitrile in methyl te/f-butyl ether).
B. Alternativel, to a cooled (0-5 °C) solution of (3S)-1-(diphenylmethyl)-3- (6-hydroxy-1 ,3-benzodioxol-5-yl)-3-(hydroxymethyl)-1 ,3-dihydro-2 -/-indol-2-one (1 .0 kg, 2.1 mol) and 2-(diphenylphosphino)pyridine (0.66 kg, 2.5 mol) in anhydrous tetrahydrofuran (20 L) was added over 2 h a solution of di-terf-butylazodicarboxylate (0.62 kg, 2.7 mmol) in anhydrous tetrahydrofuran (5 L). The mixture was stirred for 4 h at 0-5 °C and was allowed to warm to ambient temperature. The mixture was diluted with ethyl acetate (20 L), washed with 3 N hydrochloric acid (6 * 8 L) and brine (2 x 12 L) and concentrated in vacuo to a volume of approximately 1.5 L. Methyl rert-butyl ether (4 L) was added and the mixture concentrated in vacuo to a volume of
approximately 1.5 L. Methyl terf-butyl ether (2 L) and heptane (2 L) were added and the mixture was stirred at ambient temperature for 2 h, during which time a solid was deposited. The solid was collected by filtration, washed with heptane (0.5 L) and dried in vacuo below 50 °C for 8 h to afford (7S)-1′-(diphenylmethyl)spiro[furo[2,3- f][1 ,3]benzodioxole-7,3′-indol]-2′(1’H)-one (0.76 kg) as a colorless solid in 79% yield: 1H NMR (300 MHz, CDCI3) 7.34-7.24 (m, 10H), 7.15-7.13 (m, 1 H), 7.04 (s, 1 H), 6.99- 6.95 (m, 2H), 6.50-6.48 (m, 2H), 6.06 (s, 1 H), 5.85-5.83 (m, 2H), 4.96 (d, J = 8.9 Hz, 1 H), 4.69 (d, J = 8.9 Hz, 1 H); MS (ES+) m/z 447.9 (M + 1 ); ee (enantiomeric excess) 92% (HPLC, ChiralPak IA).
EXAMPLE 16
Synthesis of (7S)-spiro[furo[2,3-f][1 ,3]benzodioxole-7,3′-indol]-2′(1 ‘H)-one
Compound of formula (22a1)
A. To a solution of (7S)-1′-(diphenylmethyl)spiro[furo[2,3- f][1 ,3]benzodioxole-7,3′-indol]-2′(1’H)-one (4.55 g, 10.2 mmol) in trifluoroacetic acid (80 ml_) was added triethylsilane (7 ml_). The reaction mixture was heated at reflux for 2.5 h, allowed to cool to ambient temperature and concentrated in vacuo. The residue was triturated with a mixture of diethyl ether and hexanes to afford
(7S)-spiro[furo[2,3-/][1 ,3]benzodioxole-7,3,-indol]-2′(1’W)-one (2.30 g) as a colorless solid in 80% yield: 1H NMR (300 MHz, CDCI3) £8.27 (br s, 1 H), 7.31-7.26 (m, 1 H), 7.17-7.15 (m, 1 H), 7.07-7.02 (m, 1 H), 6.96-6.94 (m, 1 H), 6.53-6.52 (m, 1 H), 6.24-6.23 (m, 1 H), 5.88-5.87 (m, 2H), 4.95 (d, J = 8.6 Hz, 1 H), 4.68 (d, J = 8.9 Hz, 1 H); MS (ES+) m/z 281.9 (M + 1 ); ee (enantiomeric excess) 99% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl fert-butyl ether). B. Alternatively, a mixture of (7S)-1 ‘-(diphenylmethyl)spiro[furo[2,3- /Kl^benzodioxole^-indol^ r^-one (0.70 kg, 1.6 mol), trifluoroacetic acid (12 L) and triethylsilane (1.1 L) was heated at reflux under nitrogen atmosphere for 3 h, allowed to cool to ambient temperature and concentrated in vacuo to dryness. To the residue was added ethyl acetate (0.3 L), methyl fert-butyl ether (1 L) and heptane (3.5 L), causing a solid to be deposited. The solid was collected by filtration, taken up in dichloromethane (3 L), stirred at ambient temperature for 1 h and filtered. The filtrate was concentrated in vacuo to dryness. The residue was taken up in ethyl acetate (0.3 L), methyl ferf-butyl ether (1 L) and heptane (3.5 L), causing a solid to be deposited. The solid was collected by filtration and dried in vacuo below 50 °C for 8 h to afford (7S)-spiro[furo[2,3- ][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘ -/)-one (0.40 kg) as a colorless solid in 91 % yield: 1H NMR (300 MHz, CDCI3) 8.27 (br s, 1 H), 7.31-7.26 (m, 1 H), 7.17-7.15 (m, 1 H), 7.07-7.02 (m, 1 H), 6.96-6.94 (m, 1 H), 6.53-6.52 (m, 1 H), 6.24-6.23 (m, 1 H), 5.88-5.87 (m, 2H), 4.95 (d, J = 8.6 Hz, 1 H), 4.68 (d, J = 8.9 Hz, 1 H); MS (ES+) m/z 281.9 (M + 1); ee (enantiomeric excess) 98.6% (HPLC, ChiralPak IA).
EXAMPLE 17
Synthesis of of (7S)-1 ‘-{[5-(trifluoromethyl)furan-2- yl]methyl}spiro[furo[2,3- ][1 ,3]benzodioxole-7,3’-indol]-2′(rH)-one
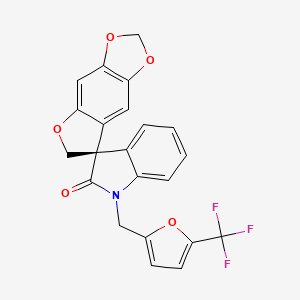
Compound of formula (Ia1)

A. To a mixture of (7S)-6H-spiro[[1 ,3]dioxolo[4,5-f]benzofuran-7,3′-indolin]- 2′-one (1.80 g, 6.41 mmol) and 2-(bromomethyl)-5-(trifluoromethyl)furan (1.47 g, 6.41 mmol) in acetone (200 mL) was added cesium carbonate (3.13 g, 9.61 mmol). The reaction mixture was heated at reflux for 2 h and filtered while hot through a pad of diatomaceous earth. The filtrate was concentrated in vacuo to afford (7S)-1′-{[5- (trifluoromethyOfuran^-yllmethy^spiroIfurop.S- ltl .Slbenzodioxole^.S’-indol^ rH)- one (2.71 g) as a colorless solid in quantitative yield (97% purity by HPLC). The product was crystallized from a mixture of methanol and hexanes to afford (7S)-1 ‘-{[5- (trifluoromethy furan^-yllmethylJspirotfuro^.S- lfl .Slbenzodioxole^.S’-indoll^ rH)- one (1.46 g) as colorless needles in 53% yield. The mother liquor was concentrated in vacuo and subjected to a second crystallization in methanol and hexanes to afford further (7S)-1 ‘-{[5-(trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-/][1 ,3]benzodioxole- 7,3’-indol]-2′(1 ‘H)-one (0.469 g) as a colorless solid in 17% yield (total yield 70%): 1H NMR (300 MHz, CDCI3) δ 7.29-6.96 (m, 4H), 6.73 (s, 1 H), 6.50 (s, 1 H), 6.38 (s, 1 H), 6.09 (s, 1 H), 5.85 (br s, 2H), 5.06 (d, J = 16.0 Hz, 1 H), 4.93-4.84 (m, 2H), 4.68-4.65 (m, 1 H); MS (ES+) m/z 429.8 (M + 1); ee (enantiomeric excess) >99.5% (HPLC, Chiralpak IA, 2.5% acetonitrile in methyl tert-butyl ether).
B. Alternatively, to a solution of (7S)-spiro[furoI2,3-f][1 ,3]benzodioxole-7,3′- indol]-2′(1’H)-one (0.40 kg, 1.4 mol) in anhydrous N, W-dimethylformamide (5 L) was added cesium carbonate (1.2 kg, 3.4 mol), followed by 2-(bromomethyl)-5- (trifluromethyl)furan (0.24 L, 1.7 mol). The mixture was heated at 80-85 °C for 3 h, allowed to cool to ambient temperature and filtered through a pad of diatomaceous earth. The pad was washed with ethyl acetate (8 L). The combined filtrate and washes were washed with water (4 L), saturated aqueous ammonium chloride (2 * 4 L) and brine (2 * 4 L) and concentrated in vacuo to dryness. The residue was purified by recrystallization from te/t-butyl methyl ether (0.4 L) and heptane (0.8 L), followed by drying of the resultant solid in vacuo at 40-50 °C for 8 h to afford (7S)-1 ‘-{[5- (trifluoromethyl)furan-2-yl]methyl}spiro[furo[2,3-f][1 ,3]benzodioxole-7,3’-indol]-2′(1 ‘H)- one (0.37 kg) as a colorless solid in 61% yield: 1H NMR (300 MHz, CDCI3) δ 7.29-6.96 (m, 4H), 6.73 (s, 1 H), 6.50 (s, 1 H), 6.38 (s, 1 H), 6.09 (s, 1 H), 5.85 (br s, 2H), 5.06 (d, J = 16.0 Hz,1 H), 4.93-4.84 (m, 2H), 4.68-4.65 (m, 1 H); MS (ES+) m/z 429.8 (M + 1 ); ee (enantiomeric excess) > 99% (HPLC, Chiralpak IA).
TV-45070 is a small-molecule lactam containing a chiral spiro-ether that has been reported as a potential topical therapy for pain associated with the Nav1.7 sodium ion channel encoded by the gene SCN9A. A pilot-scale synthesis is presented that is highlighted by an asymmetric aldol coupling at ambient temperature, used to create a quaternary chiral center. Although only a moderate ee is obtained, the removal of the undesired isomer is achieved through preferential precipitation of a near racemic mixture from the reaction, leaving the enantiopure isomer in solution. Cyclization to form the final API uses an uncommon diphenylphosphine-based leaving group which proved successful on the neopentyl system when other traditional leaving groups failed.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....






















































![[1,2,4]Triazolo[5,1-c][1,2,4]triazin-4(1H)-one, 7-(methylthio)-3-nitro-.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=3113817&t=l)






















 Next generation β-lactamase inhibitors recently approved or in clinical trials. A. Avibactam. B. Relebactam. C. Vaborbactam.
Next generation β-lactamase inhibitors recently approved or in clinical trials. A. Avibactam. B. Relebactam. C. Vaborbactam.












 Moxalactam synthesis
Moxalactam synthesis




{kind=link}