
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Onasemnogene abeparvovec オナセムノジーンアベパルボベック
Onasemnogene abeparvovec
オナセムノジーンアベパルボベック
DNA (synthetic adeno-associated virus 9 vector scAAV9.CB.hSMN human survivor motor neuron protein-specifying)
Zolgensma
FDA 2019/5/24 APPROVED
CAS: 1922968-73-7
AVXS-101
Spinal muscular atrophy treatment
Treatment of Spinal Muscular Atrophy (SMA) Type 1
Gene therapy product
![]()
Onasemnogene abeparvovec, sold under the trade name Zolgensma, is a gene therapy medication used to treat spinal muscular atrophy (SMA).
SMA is a neuromuscular disorder caused by a mutation in the SMN1 gene, which in turn reduces the amount of SMN protein necessary for survival of motor neurons. Onasemnogene abeparvovec is a biologic drug consisting of AAV9 virus capsids that have been deprived of the original viral DNA and instead contain a SMN1 transgene along with promoters. The drug is administered intravenously or intrathecally. Upon administration, the AAV9 viral vector delivers the SMN1 transgene to cell nuclei where the transgene begins encoding SMN protein, thus addressing the root cause of the disease. Since motor neurons do not divide, it is thought that a single dose of the drug will have a lifelong effect.[1]
The medication was developed by a US biotechnology company AveXis, a subsidiary of Novartis,[2] based on an earlier discovery by French researchers.[3] The intravenous formulation was approved in May 2019 in the United States for use in children under 2 years.[4]It carries a list price of US$ 2.125 million per dose (one-time treatment), making it the most expensive medication in the world as of 2019.[5]
Terminology
Onasemnogene abeparvovec is the international nonproprietary name (INN) and US adopted name (USAN).[6] It was previously known under compound name AVXS-101.
FDA approves a gene therapy that is the most expensive drug in the world
FDA on Friday approved onasemnogene abeparvovec-xioi (Zolgensma—AveXis), a one-time gene therapy for the treatment of spinal muscular atrophy (SMA).
FDA on Friday approved onasemnogene abeparvovec-xioi (Zolgensma—AveXis), a one-time gene therapy for the treatment of spinal muscular atrophy (SMA). The ultrarare disease affects infants. In announcing the approval, Novartis—which acquired AveXis last year—also disclosed the price of the drug, $2.1 million. The company noted that it would provide rebates to insurance companies if the drug is not successful, though it did not offer details about what would be considered failure. Novartis also said it will set up 5-year payment plans for states, small insurance firms, and self-insured employers. Another drug, nusinersen (Spinraza—Biogen) is already available for the treatment of SMA; however, that drug must continue to be injected into patients’ spines throughout their lives, at a cost of $750,000 in the first year and $375,000 a year after that. “Patients with SMA now have another treatment option to minimize the progression of SMA and improve survival,” said Peter Marks, director of FDA’s Center for Biologics Evaluation and Research.
References
- ^ “Novartis announces FDA filing acceptance and Priority Review of AVXS-101, a one-time treatment designed to address the genetic root cause of SMA Type 1 | Novartis”. Novartis. Retrieved 2018-12-04.
- ^ “Novartis successfully completes acquisition of AveXis, Inc. | Novartis”. Novartis. Retrieved 2018-10-06.
- ^ “AveXis receives FDA approval for Zolgensma®, the first gene therapy for paediatric patients with SMA”. SMA Europe. 2015-05-25. Retrieved 2019-05-25.
- ^ “FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality”. FDA. 2019-05-24. Retrieved 2019-05-24.
- ^ Reuters (2019-05-25). “$2.1m Novartis gene therapy to become world’s most expensive drug”. The Guardian. ISSN 0261-3077. Retrieved 2019-05-25.
- ^ “Onasemnogene abeparvovec – AveXis – AdisInsight”. adisinsight.springer.com. Retrieved 2018-10-06.
| Clinical data | |
|---|---|
| Trade names | Zolgensma |
| Synonyms | AVXS-101 |
| License data | |
| Routes of administration |
Intravascular |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Duration of action | lifetime (?) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| KEGG | |
/////////Onasemnogene abeparvovec, Zolgensma, FDA 2019, オナセムノジーンアベパルボベック ,Spinal muscular atrophy, Gene therapy product, AVXS-101
FDA approves first PI3K inhibitor Piqray (alpelisib) for breast cancer

FDA approves first PI3K inhibitor for breast cancer
syn https://newdrugapprovals.org/2018/06/25/alpelisib-byl-719/
Today, the U.S. Food and Drug Administration approved Piqray (alpelisib) tablets, to be used in combination with the FDA-approved endocrine therapy fulvestrant, to treat postmenopausal women, and men, with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer (as detected by an FDA-approved test) following progression on or after an endocrine-based regimen.
The FDA also approved the companion diagnostic test, therascreen PIK3CA RGQ PCR Kit, to detect the PIK3CA mutation in a tissue and/or a liquid biopsy. Patients who are negative by
- May 24, 2019
Today, the U.S. Food and Drug Administration approved Piqray (alpelisib) tablets, to be used in combination with the FDA-approved endocrine therapy fulvestrant, to treat postmenopausal women, and men, with hormone receptor (HR)-positive, human epidermal growth factor receptor 2 (HER2)-negative, PIK3CA-mutated, advanced or metastatic breast cancer (as detected by an FDA-approved test) following progression on or after an endocrine-based regimen.
The FDA also approved the companion diagnostic test, therascreen PIK3CA RGQ PCR Kit, to detect the PIK3CA mutation in a tissue and/or a liquid biopsy. Patients who are negative by the therascreen test using the liquid biopsy should undergo tumor biopsy for PIK3CA mutation testing.
“Piqray is the first PI3K inhibitor to demonstrate a clinically meaningful benefit in treating patients with this type of breast cancer. The ability to target treatment to a patient’s specific genetic mutation or biomarker is becoming increasingly common in cancer treatment, and companion diagnostic tests assist oncologists in selecting patients who may benefit from these targeted treatments,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “For this approval, we employed some of our newer regulatory tools to streamline reviews without compromising the quality of our assessment. This drug is the first novel drug approved under the Real-Time Oncology Review pilot program. We also used the updated Assessment Aid, a multidisciplinary review template that helps focus our written review on critical thinking and consistency and reduces time spent on administrative tasks.”
Metastatic breast cancer is breast cancer that has spread beyond the breast to other organs in the body (most often the bones, lungs, liver or brain). When breast cancer is hormone-receptor positive, patients may be treated with anti-hormonal treatment (also called endocrine therapy), alone or in combination with other medicines, or chemotherapy.
The efficacy of Piqray was studied in the SOLAR-1 trial, a randomized trial of 572 postmenopausal women and men with HR-positive, HER2-negative, advanced or metastatic breast cancer whose cancer had progressed while on or after receiving an aromatase inhibitor. Results from the trial showed the addition of Piqray to fulvestrant significantly prolonged progression- free survival (median of 11 months vs. 5.7 months) in patients whose tumors had a PIK3CA mutation.
Common side effects of Piqray are high blood sugar levels, increase in creatinine, diarrhea, rash, decrease in lymphocyte count in the blood, elevated liver enzymes, nausea, fatigue, low red blood cell count, increase in lipase (enzymes released by the pancreas), decreased appetite, stomatitis, vomiting, weight loss, low calcium levels, aPTT prolonged (blood clotting taking longer to occur than it should), and hair loss.
Health care professionals are advised to monitor patients taking Piqray for severe hypersensitivity reactions (intolerance). Patients are warned of potentially severe skin reactions (rashes that may result in peeling and blistering of skin or mucous membranes like the lips and gums). Health care professionals are advised not to initiate treatment in patients with a history of severe skin reactions such as Stevens-Johnson Syndrome, erythema multiforme, or toxic epidermal necrolysis. Patients on Piqray have reported severe hyperglycemia (high blood sugar), and the safety of Piqray in patients with Type 1 or uncontrolled Type 2 diabetes has not been established. Before initiating treatment with Piqray, health care professionals are advised to check fasting glucose and HbA1c, and to optimize glycemic control. Patients should be monitored for pneumonitis/interstitial lung disease (inflammation of lung tissue) and diarrhea during treatment. Piqray must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
Piqray is the first new drug application (NDA) for a new molecular entity approved under the Real-Time Oncology Review (RTOR) pilot program, which permits the FDA to begin analyzing key efficacy and safety datasets prior to the official submission of an application, allowing the review team to begin their review and communicate with the applicant earlier. Piqray also used the updated Assessment Aid (AAid), a multidisciplinary review template intended to focus the FDA’s written review on critical thinking and consistency and reduce time spent on administrative tasks. With these two pilot programs, today’s approval of Piqray comes approximately three months ahead of the Prescription Drug User Fee Act (PDUFA) VI deadline of August 18, 2019.
The FDA granted this application Priority Review designation. The FDA granted approval of Piqray to Novartis. The FDA granted approval of the therascreen PIK3CA RGQ PCR Kit to QIAGEN Manchester, Ltd.
//////////////FDA, PI3K inhibitor, breast cancer, fda 2019, Piqray, alpelisib, therascreen PIK3CA RGQ PCR Kit, QIAGEN Manchester, Priority Review, BYL719, BYL 719
CT 1812
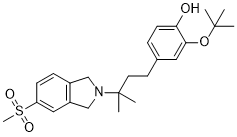
CT-1812
Elayta
Condition(s): Alzheimer’s Disease
U.S. FDA Status: Alzheimer’s Disease (Phase 2)
Company: Cognition Therapeutics Inc.
CAS: 1802632-22-9
Chemical Formula: C24H33NO4S
Molecular Weight: 431.591
2-(tert-butoxy)-4-(3-methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol
Phenol, 4-[3-[1,3-dihydro-5-(methylsulfonyl)-2H-isoindol-2-yl]-3-methylbutyl]-2-(1,1-dimethylethoxy)-
- Originator Cognition Therapeutics
- Class Antidementias; Neuroprotectants; Nootropics; Small molecules
- Mechanism of Action Sigma-2 receptor antagonists
- Phase II Alzheimer’s disease
- Phase I Cognition disorders
- 21 Feb 2019 Cognition Therapeutics receives patent for a composition of matter patent covering Elayta™ in Europe
- 19 Feb 2019 Pharmacokinetics and adverse events data from a phase I trial in Cognition disorders released by Cognition Therapeutics
- 22 Oct 2018 CTP push 289675: Updated KDM, forwarded USA line from PI/II to PII
CT-1812 is a first-in-class, orally available sigma-2/PGRMC1 antagonist (alpha beta oligomer receptor antagonist), is being developed by Cognition. sCT-1812 is a novel therapeutic candidate for Alzheimer’s disease
SYN

BACKGROUND
CT1812 is a small-molecule antagonist of the sigma2 receptor, also known as the progesterone receptor membrane component 1. The rationale behind this therapeutic approach is that ligands for the sigma2/PGRMC1 receptor will compete with oligomeric Aβ binding to this receptor and thus interfere with Aβ-induced synaptic toxicity. CT1812 grew out of screening programs at Cognition Therapeutics. Company scientists have reported that compounds in this series not only block binding of a range of different Aβ species to this receptor but also displace it when applied after Aβ has bound (Dec 2014 conference news).
The structure of CT1812 has not been disclosed, but similar compounds in the series have been reported to enter the brain, occupy up to 80 percent of sigma2/PGRMC1 receptors, and restore behavioral deficits in APP transgenic mice (Izzo et al., 2014; Izzo et al., 2014).
FINDINGS
From September 2015 to May 2016, Cognition Therapeutics ran a Phase 1 trial in 80 healthy volunteers aged 18 to 75 in Melbourne, Australia; target enrollment was originally listed as 114. Single-ascending-dose administration was followed by multiple ascending doses given once daily for two weeks. The dose range in this trial spanned 10 to 650 mg; if this would not generate data to set a maximum tolerated dose, doses up to 1,350 mg were to be tried. Outcome measures included safety, tolerability, plasma pharmacokinetics, and CSF CT1812 concentration. At the 2016 and 2017 AAIC conferences, company scientists reported that single doses up to 1,120 mg were given, as were multiple doses of up to 840 mg in young and up to 560 mg in elderly volunteers. The drug was reported to be well-tolerated, with suitable pharmacokinetics, sufficient brain penetrance and target exposure, and minimal drug-drug interactions affecting cytochrome P450 activity (Catalano et al., 2016; Catalano et al., 2017).
From September 2016 to August 2017, Cognition Therapeutics ran a Phase 1/2 trial at four sites in Australia, enrolling 19 participants with mild to moderate Alzheimer’s disease supported by a recent MRI. It compared a four-week course of 90, 280, or 560 mg of CT 1812 to placebo, taken once daily, on safety and tolerability parameters. At the subsequent CTAD conference, Elayta was reported to have been generally safe and well tolerated, though there were four cases of lymphocytopenia. Exploratory measures such as ADAS-Cog14, verbal or category fluency tests recorded no difference between groups, but exploratory biomarker analyses yielded possible signals of synapse protection (Dec 2017 conference news).
In April 2018, a Phase 1/2 study started enrolling 21 people whose mild to moderate AD was confirmed by amyloid PET or CSF testing. Conducted at Yale University School of Medicine and dubbed COG0105 or SPARC, this trial will compare a six-month course of 100 or 300 mg of Elayta, or placebo. The primary outcome is cognition as assessed by the Alzheimer’s Disease Clinical Study Activities of Daily Living (ADCS-ADL), but the trial will also use the investigational PET tracer UCB-J, which binds to the synaptic vesicle glycoprotein 2A, in an attempt to monitor synapse density before and after treatment (see company press release; Jul 2016 news).
In summer 2018, a Phase 1b target engagement study at the University of Pennsylvania will start enrolling 18 people whose mild to moderate AD is confirmed by amyloid PET. Called COG0104 or SNAP, it will compare single injections of 90, 280, or 560 mg of Elayta or placebo for their ability to displace Aβ oligomers and clear them into the CSF, as measured by a CSF Aβ oligomer assay.
Also in summer 2018, a Phase 2 multi-center study is expected to begin enrolling 24 people with mild to moderate AD as confirmed by amyloid PET for a six-month course of 100 or 300 mg of Elayta, or placebo. As of May 22, 2018, this trial lists CT1812 pharmacodynamic effects on CSF biomarkers, specifically as assessed by CSF neurogranin levels, as primary outcome.
For all trials of this compound, see clinicaltrials.gov.
PATENT
WO 2015116923
https://patents.google.com/patent/WO2015116923A1
There are only five medications currently FDA-approved for the treatment of Alzheimer’s Disease (AD). Four are cholinesterase inhibitors: tacrine (COGNEX®; Sciele), donepezil (ARICEPT®; Pfizer), rivastigmine (EXELON®; Novartis), and galantamine (RAZADYNE®; Ortho-McNeil-Janssen). Donepezil, rivastigmine, and galantamine are successors to tacrine, a first generation compound rarely prescribed because of the potential for hepatotoxicity; they are roughly equally efficacious at providing symptomatic improvement of cognition and function at all stages of AD. The fifth approved medication is memantine (NAMENDA®; Forest), a low-affinity, use dependent N-methyl-D-aspartate glutamate receptor antagonist that offers similar benefits, but only in moderate to severe AD. The clinical effects of these compounds are small and impermanent, and currently available data are inconclusive to support their use as disease modifying agents. See, e.g., Kerchner et al, 2010, Bapineuzumab, Expert Opin Biol Ther., 10(7): 1121-1130. Clearly, alternative approaches to treatment of AD are required.
[004] Certain isoindoline compounds are provided that act as sigma-2 receptor functional antagonists and inhibit the deleterious effects of soluble Αβ oligomers. In some embodiments, isoindoline sigma-2 receptor antagonist compounds and compositions are used to treat or prevent synaptic dysfunction in a subject.
Example 21 illustrates representative preparation of 2-(Tert-butoxy)-
4-(3-methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol, Example Compound 62, as shown in Scheme 17.


10 Compound 62
[0534] Scheme 17: Procedure for preparation of 2-(Tert-butoxy)-4-(3- methyl-3-(5-(methylsulfonyl)isoindolin-2-yl)butyl)phenol, Example Compound 62.
[0535] Preparation of compound l(Scheme 17): To a glass pressure -bottle at -30 °C containing a mixture of catechol (50.0 g, 454 mmol, 1.0 eq), concentrated sulfuric acid (0.3 mL) in dichloromethane (200 mL), isobutene (152.6 g, 2.72 mol, 6.0 eq) was condensed. After sealing the pressure-bottle with a threaded Teflon cap tipped with a Teflon-protected rubber O-ring, the mixture was heated at 35 °C for 3 h until a clear solution was obtained. After cooling (-30 °C), triethylamine (1.5 mL, 10.8 mmol) was added and the mixture was concentrated. The residue was suspended in 0.5 M NaOH (1 L) and stirred for 10 min. The dark-green colored solution was washed with petroleum ether (2x 100 mL) and the washing layers were reextracted with 0.5 M NaOH (3x 100 mL). The combined aqueous layers were brought to pH 7-8 with 2 N HCl (400 mL), and extracted with ethyl acetate (2* 1 L), dried over sodium sulfate and concentrated to afford product 1 (67.7 g, 90%) as a colorless oil, which was used directly for the next step reaction without further purification. TLC: PE/EA = 50/1 ; Rf (Catechol) = 0.1 ; Rf (Compound 1) = 0.6.
[0536] Preparation of compound 2 (Scheme 17): To a stirred solution of compound 1 (1 12.2 g, 676 mmol, 1.2 eq) and potassium iodide (1 12.2 g, 676 mmol, 1.0 eq) in methanol (2 L) at 0 °C was slowly added sodium hydroxide (27.0 g, 676 mmol, 1.0 eq), followed with aqueous sodium chlorite (7% aq., 718.8 mL, 710 mmol, 1.05 eq) dropwise over 3 h while keeping the reaction below 0 °C. The mixture was stirred at 0 °C for another 30 min and neutralized by adding 2 N HCl at 0 °C till pH 7, extracted with DCM (2 x 1 L). The organic layers were dried over sodium sulfate and concentrated to afford product 2 (179.8 g, 91%). TLC: PE/EA = 50/1; Rf(Compound 1) = 0.6 ; Rf (Compound 2) = 0.6.
[0537] Preparation of compound 3(Scheme 17): To a stirred solution of compound 2 (179.8 g, 616 mmol, 1.0 eq) and triethylamine (186.6 g, 1.85 mol, 3.0 eq) in dichloromethane (2 L) at 0 °C was slowly added acetyl chloride (53.2 g, 677 mmol, 1.1 eq). The mixture was stirred at 0 °C for another 30 min, and warmed up to rt, and stirred at rt for 3 h, water (1 L) was added into the reaction mixture and the organic layer was washed with brine, dried over sodium sulfate and concentrated to afford product 3 (206 g, 100%), which was used directly to the next step without further purification. TLC: PE/EA = 50/1; Rf (Compound 2) = 0.6; Rf (Compound 3) = 0.5.
[0538] Preparation of compound 4 (Scheme 17): To a stirred solution of compound 3 (206 g, 616 mmol, 1.0 eq) in triethylamine (4.0 L) was added 2- methylbut-3-yn-2-amine (102.5 g, 1.23 mol, 2.0 eq), Pd(PPh3)2Cl2 (15.1 g, 18.5 mmol, 0.03 eq) and copper(I) iodide (5.9 g, 31 mmol, 0.05 eq) and resulting mixture was stirred at rt for 17 h. The solvent was removed under reduced pressure and the crude product was purified by silica gel chromatography to afford the title compound 4 (132.7 g, 74%). TLC: PE/EA = 1/1; Rf (Compound 3) = 0.9; Rf (Compound 4) = 0.3. [0539] Preparation of compound 5(Scheme 17): To a stirred solution of compound 4 (104.5 g, 0.36 mol) in ethanol (1.5 L) was added Pd/C (10% wt, 10.5 g). The mixture was stirred under hydrogen (balloon) overnight, and filtered. The filtrate was evaporated to dryness to afford compound 5 (106.3 g, 100%), which was used directly to the next step without further purification. TLC: PE/EA = 1/1; Rf(Compound 4) = 0.3 ; Rf (Compound 5) = 0.3.
[0540] Preparation of compound 6 (Scheme 17): To a solution of o-xylene
(115.7 g, 1.09 mol, 1.0 eq) in chloroform (1.0 L) at 0 °C was added C1S03H (254 g, 2.18 mol, 2.0 eq) dropwise. After the addition, the reaction mixture was stirred at room temperature for 2 days, and poured into ice. The crude mixture was extracted with dichloromethane (3 x 1.0 L). The organic layers were combined, dried over anhydrous sodium sulfate, concentrated to afford the crude compound 6 (161.5 g, 80%) as a white solid, which was used directly to the next step without further purification. TLC: PE/EA = 5/1; Rf (Compound 6) = 0.7.
[0541] General procedure for the preparation of compound 7 (Scheme
17): To a stirred solution of compound 6 (161.5 g, 0.87 mol, 1.0 eq) in saturated sodium sulfite solution (273 g, 2.17 mol, 2.5 eq, in 2.0 L of water) was added dropwise 32% NaOH (69.4 g, 1.73 mol, 2.0 eq) till the solution reached pH 9. After stirring at rt overnight, the reaction mixture was acidified with cone. HC1 in ice- cooling bath till pH 1. The precipitate was filtered, and washed with ice-water (2x), dried in vacuo to afford the crude product 7 (131 g, 88%), which was used directly for next step without further purification. TLC: PE/EA = 5/1; Rf (Compound 6) = 0.7; Rf (Compound 7) = 0.6.
[0542] Preparation of compound 8 (Scheme 17): To a stirred solution of compound 7 (130 g, 0.76 mol, 1.0 eq) and potassium carbonate (211 g, 1.53 mol, 2.0 eq) in DMF (300 mL) was added iodomethane (96 mL, 1.53 mol, 2.0 eq). The reaction was stirred at 40 °C overnight. The reaction mixture was evaporated to dryness, extracted with ethyl acetate. The organic layers were washed with water and brine, dried over sodium sulfate and concentrated, purified by flash column chromatography (PE: EA,10: 1 ~ 5: 1) to afford compound 8 (85.2 g, 61%). TLC: PE/EA = 5/1; Rf (Compound 7) = 0.6; Rf (Compound 8) = 0.3. [0543] Preparation of compound 9 (Scheme 17):To a stirred solution of compound 8 (78.2 g, 424 mmol, 1.0 eq) in 1 ,2-dichloroethane (1.2 L), were added N-bromosuccinimide (166 g, 934 mmol, 2.2 eq) and AIBN (6.9 g, 42.4 mmol, 0.1 eq). The reaction was stirred at reflux overnight. The reaction was diluted with water and dichloromethane. The organic layer was collected, and dried over sodium sulfate and concentrated, purified by flash column chromatography to afford compound 9, which was further recrystallized from hot methanol to afford the pure product 8 (75 g, 52%). TLC: PE/EA = 5/1; Rf (Compound 8) = 0.3; Rf (Compound 9) = 0.2.
[0544] Preparation of compound 10 (Scheme 17):To a stirred solution of compound 5 (46 g, 157 mmol, 1.0 eq) and compound 9 (53.5 g, 157 mmol, 1.0 eq) in THF (460 mL) was added triethylamine (47.7 g, 472 mmol, 3.0 eq). The reaction was stirred at 40 °C overnight, filtered and the filtrate was evaporated to dryness and purified by flash column chromatography to afford compound 10 (45 g, 63%). TLC: PE/EA = 1/1; Rf (Compound 5) = 0.3; Rf (Compound 9) = 1.0; Rf (Compound 10) = 0.4.
[0545] Preparation of Compound 62 (Scheme 17):To a stirred solution of compound 10 (45 g, 98.4 mmol) in methanol (300 mL) was added sodium methoxide (844 mg, 15.6 mmol, 0.16 eq) in one portion. The solution was stirred at rt overnight. Water (250 mL) was added dropwise into the reaction mixture over 1 h, the mixture was stirred at rt for 2 h, and filtered. The white solid was collected and dried on vacuum overnight to afford pure example Compound 62 base (38 g, 89%>). TLC: PE/EA = 1/1; Rf (Compound 10) = 0.4; Rf (Compound 62) = 0.4; ESI-MS: 432 (M+l)+; 1H NMR (400 MHz, CDC13) δ 7.80-7.78 (m, 2H). 7.40-7.38 (m, 1H), 6.87-6.79 (m, 3H), 5.58 (s, 1H), 4.11 (s, 4H), 3.05 (s, 3H), 2.61-2.57 (m, 2H), 1.76- 1.72 (m, 2H), 1.48 (s, 9H), 1.18 (s, 6H). Example 22: Preparation of (2-(4-(4-Hydroxy-3-methoxyphenyl)-2- methylbutan-2-yl)isoindolin-4-yl)(piperazin-l-yl)methanone,
REFERENCES
1: Grundman M, Morgan R, Lickliter JD, Schneider LS, DeKosky S, Izzo NJ,
Guttendorf R, Higgin M, Pribyl J, Mozzoni K, Safferstein H, Catalano SM. A phase
1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a
novel therapeutic candidate for Alzheimer’s disease. Alzheimers Dement (N Y).
2019 Jan 23;5:20-26. doi: 10.1016/j.trci.2018.11.001. eCollection 2019. PubMed
PMID: 30723776; PubMed Central PMCID: PMC6352291.
Paper Citations
- Catalano S, Grundman M, Schneider LS, DeKosky S, Morgan R, Higgin M, Pribyl J, Mozzoni K, Izzo NJ, Safferstein H, Lickliter J. A Two-Part, Double-Blind, Placebo-Controlled, Phase 1 Study of the Safety and Pharmacokinetics of Single and Multiple Ascending Doses of Ct1812 in Healthy Volunteers. Alzheimer’s & Dementia, July 2016, Volume 12, Issue 7, Supplement
- Catalano S, Grundman M, Schneider LS, DeKosky S, Morgan R, Guttendorf R, Higgin M, Pribyl J, Mozzoni K, Izzo NJ, Safferstein H. A Phase 1 Safety Trial of the aβ Oligomer Receptor Antagonist CT1812. Alzheimer’s & Dementia, July 2017, Volume 13, Issue 7
- Izzo NJ, Staniszewski A, To L, Fa M, Teich AF, Saeed F, Wostein H, Walko T 3rd, Vaswani A, Wardius M, Syed Z, Ravenscroft J, Mozzoni K, Silky C, Rehak C, Yurko R, Finn P, Look G, Rishton G, Safferstein H, Miller M, Johanson C, Stopa E, Windisch M, Hutter-Paier B, Shamloo M, Arancio O, LeVine H 3rd, Catalano SM. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits. PLoS One. 2014;9(11):e111898. Epub 2014 Nov 12 PubMed.
- Izzo NJ, Xu J, Zeng C, Kirk MJ, Mozzoni K, Silky C, Rehak C, Yurko R, Look G, Rishton G, Safferstein H, Cruchaga C, Goate A, Cahill MA, Arancio O, Mach RH, Craven R, Head E, LeVine H 3rd, Spires-Jones TL, Catalano SM. Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS One. 2014;9(11):e111899. Epub 2014 Nov 12PubMed.
/////CT-1812, CT 1812, CT1812, Alzheimers , Cognition Therapeutics, Elayta, phase 2, Cognition disorders
OC1=CC=C(CCC(C)(N2CC3=C(C=C(S(=O)(C)=O)C=C3)C2)C)C=C1OC(C)(C)C
Dipivefrine, дипивефрин , ديبيفيفرين , 地匹福林 , ジピベフリン
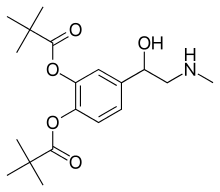
Dipivefrine
- Molecular FormulaC19H29NO5
- Average mass351.437 Da

Dipivefrine (INN) or dipivefrin (USAN), trade name Propine among others, is a prodrug of epinephrine, and is used to treat open-angle glaucoma.[1][2] It is available as a 0.1% ophthalmic solution. It is no longer available in the United States.[3]
Dipivefrin is a prodrug with little or no pharmacologically activity until it is hydrolyzed into epinephrine inside the human eye. The liberated epinephrine, an adrenergic agonist, appears to exert its action by stimulating α -and/or β2-adrenergic receptors, leading to a decrease in aqueous production and an enhancement of outflow facility. The dipivefrin prodrug delivery system is a more efficient way of delivering the therapeutic effects of epinephrine, with fewer side effects than are associated with conventional epinephrine therapy. Dipivefrin is used as initial therapy for the control of intraocular pressure in chronic open-angle glaucoma.

Contraindications
Use in narrow-angle glaucoma may be dangerous because it could make the eye susceptible to an attack of angle closure,[2] causing an increase in pressure and pain, and possibly loss of vision.
Side effects
The most common side effects of dipivefrine are burning, stinging and other irritations of the eye. Possible, but uncommon, side effects are those of epinephrine: tachycardia (fast heartbeat), hypertension (high blood pressure) and arrhythmias (irregular heartbeat).[2]
Pharmacology
Dipivefrine penetrates the cornea and is then hydrolysed to epinephrine by esterase enzymes. It increases outflow of the aqueous humour and also reduces its formation (mediated by its action on α1 and α2 receptors), thus reducing pressure inside the eye. It also increases the conductivity of trabecular filtering cells (a β2 receptor mediated action). It is preferred to epinephrine because it is longer acting, more consistent in its action and better tolerated.[1]
Patent
https://patents.google.com/patent/CN102153485A/en

Example 1 [0023] Embodiment
[0024] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 5 ° C, was added 666g (6. 58mol) of triethylamine, and then was added dropwise 784g (6. 5mol) pivaloyl chloride was added dropwise and stirring was continued after the pool. Filtered off with suction, the filtrate by rotary evaporation; to give 990g yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 96.2%. [0025] The 35mol) N- methyl amine section, 370g (3. 66mol) of triethylamine, 25g (0. 15mol) KI, 3L DMF was added 4-neck flask of the IOL. Cooled to 0 ° C, was added dropwise 990g (2. 8mol) 4- (2- chloroacetyl) -I, DMF solution tank 2-phenyl pivalate ester. At room temperature was stirred for 4h.
[0026] suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 923. Og. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 96.5%.
[0027] Take 625g (1. 422mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 65g (1.71mol) of sodium borohydride. At room temperature was stirred for 4h. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 552. 5g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.2%.
[0028] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 13g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0029] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0030] After the solution was washed with methanol hydrochloride salt to give an off-white solid 119. 9g, dipivefrin i.e., the content of 98.9%.
[0031] m.p. 161 ~162 ° C;
[0032] 1H NMR (CDCl3) δ: 1. 35 (s, 18Η), 2 68 (s, 3Η), 3 07-3 13 (m, 2Η), 5 36-5 39 (m….. , 1H),
[0033] 7. 06-7. 30 (m, 3H), 8. 61 (s, 1H), 9. 48 (s, 1H)
Dipivefrin prepared: Example 2 [0034] Embodiment
[0035] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 10 ° C, was added 666g (6. 58mol) of triethylamine, and then dropwise 78½ (6. 5mol) pivaloyl chloride was added dropwise and stirring was continued after the pool. Filtered off with suction, the filtrate by rotary evaporation; 978. 2g to give yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 96. 2% o
[0036] The 35mol) N- methyl amine section, 370g (3. 66mol) of triethylamine, 25g (0. 15mol) KI, 3L DMF was added 4-neck flask of the IOL. Cooled to O0C, dropwise 978. 2g (2. 77mol) 4- (2- chloroacetyl) of DMF solution tank Laid-1,2-phenyl valerate. At room temperature was stirred for 4h.
[0037] suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 910. 2g. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 96.3%.
[0038] Take 625g (1. 422mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 97g (l. SOmol) potassium borohydride. Stirred cell at room temperature. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 532. 7g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.0%.
[0039] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 15g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0040] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0041] After the solution was washed with methanol hydrochloride salt to give an off-white solid was 112. 8g, i.e., dipivefrin, content 98.6%.
3 [0042] Example 2: Preparation of dipivefrin
[0043] A 600g (3. 21mol) 4_ chloroacetyl catechol, the IOL 6L methylene chloride was added 4-neck flask, the system was cooled to 5 ° C, was added 897g (6. 5mol) of potassium carbonate, and then drops was added 784g (6. 5mol) pivaloyl chloride addition was completed stirring was continued Syndrome. Filtered off with suction, the filtrate by rotary evaporation; to give 900g yellow-brown solid, 4- (2-chloroacetyl) -1,2-pivalate phenyl ester, the content of 95.6%.
[0044] A 526g (4. 35mol) N_ methylbenzylamine, 414g (3. Omol) of potassium carbonate, 25g (0. 15mol) KI, 3L DMF force Λ IOL of four port flask. Cooled to O0C, was added dropwise 900g (2. 55mol) 4- (2- chloroacetyl) of DMF solution of 1,2-Shan Laid phenyl valerate. It was stirred at room temperature Mi.
[0045] The suction filtration, washed with water IOL filtrate was added 3 times, the organic phase was separated, the organic phase by rotary evaporation to give a yellow-brown oil; frozen stirring, the precipitated solid was suction filtered to give a solid 820g. I.e., 1- (3,4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) -1-one content of 95.6%.
[0046] Take 625g (1. 42mol) 1_ (3,4- two pivaloyloxymethyl phenyl) _2_ (N- benzyl-methylamino) ketone, 6L IOL of absolute ethanol was added 4-neck flask. Under cooling, was added 65g (1.71mol) of sodium borohydride. Stirred cell at room temperature. 500mL of water was slowly added to the system, then add ethyl acetate extract products. After solvent removal to give 512. 5g of solid particles, i.e. 1_ (3, 4-pivaloyloxymethyl-phenyl) -2- (N- benzyl-methylamino) ethanol, the content of 98.0%.
[0047] 1828 was added to the beaker (0.41211101) of 1- (3,4-pivaloyloxymethyl-phenyl) -2 – (^ -benzyl methylamino) ethanol, with ethanol and dissolved IL; to 2L autoclave was charged with 16g 5% palladium on carbon, infiltration system with IOOml ethanol, then added to the solution in a closed system. Through hydrogenation under hydrogen 2MPa pool.
[0048] suction filtered to remove palladium on carbon. The filtrate was twice filtered off with suction, the filtrate by rotary evaporation to give a yellow-brown oil; standing crystallization, the precipitated pale yellow solid was suction filtered to give a solid crude product.
[0049] After the solution was washed with methanol hydrochloride salt to give an off-white solid was 109. 8g, i.e., dipivefrin, content 98.5%.
SYN
SYN
2-chloro-3′,4′-dihydroxyacetophenone, 99-40-1
3′,4′-dihydroxy-2-methylaminoacetophenone, 99-45-6
2,2-dimethylpropanoic acid 4-[(methylamino)acetyl]-1,2-phenylene ester, 52245-00-8
Pivaloyl chloride, 3282-30-2
Trimethylacetyl chloride, 3282-30-2

1-(3,4-dipivaloyloxyphenyl)-2-(benzylmethylamino)ethan-1-one, 42146-03-2
SPECTROSCOPY

infrared spectral assignments for dipiveh hydrochloride
Wavelength (cm-1) Assignment
3255,2804,2475, 2397 RflHz+-NH stretch
2974-2875 sp3 C-H stretch
1273, 1258-1163 C-0-C stretch
3600-3400 0-H stretch
phenyl ester C=O stretch 1761
aromatic C-C stretch 1614, 1595, 1562, 1504
sp3 C-H bending and scissoring 1481, 1461, 1441, 1397
tert-butyl C-H bending1368, 1332
secondary alcohol C-0 stretch 1 124- 1028
out-of-plane bending for 1,substituted benzene ring 3,4 891,842

Ultraviolet absorption of dipivefrin hydrochloride
E (176, 1 cm)
Solvent 210 nm 264 Nn 270 nm
Acetonitrile 267.3 14.8 13.4
Ethanol 246.8 14.5 13.1
pH 3 Buffer 266.7 12.4 10.4
pH 7 Buffer 257.6 10.8 8.9
Water 278.0 18.0 16.2




References
- ^ Jump up to:a b KD Tripari. Essentials of Medical Pharmacology (5 ed.). Jaypee Brothers Medical Publishers(P) Ltd. p. 88. ISBN 81-8061-187-6.
- ^ Jump up to:a b c Dipivefrin FDA Professional Drug Information.
- ^ Zhang L, Weizer JS, Musch DC (2017). “Perioperative medications for preventing temporarily increased intraocular pressure after laser trabeculoplasty”. Cochrane Database Syst Rev. 2: CD010746. doi:10.1002/14651858.CD010746.pub2. PMC 5477062. PMID 28231380.
-
- Hussain, A.; Truelove, J.E.: J. Pharm. Sci. (JPMSAE) 65, 1510 (1976).
- US 3 839 584.
- a DOS 2 343 657 (Interx Res. Corp.; appl. 30.8.1973; USA-prior. 31.8.1972).
- US 3 809 714 (Interx; 7.5.1974; prior. 31.8.1972) also racemate resolution.
- b DOS 2 152 058 (Klinge; appl. 19.10.1971).
 |
|
| Clinical data | |
|---|---|
| Trade names | Propine, Pivalephrine |
| Synonyms | Dipivefrin |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686005 |
| Pregnancy category |
|
| Routes of administration |
Eye drops |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C19H29NO5 |
| Molar mass | 351.437 g/mol g·mol−1 |
| 3D model (JSmol) | |
//////////дипивефрин , ديبيفيفرين , 地匹福林 , Dipivefrine, antiglaucoma, GENERIC, ジピベフリン
Etosalamide, этосаламид , إيتوسالاميد , 依托柳胺 ,
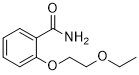
Cas 15302-15-5
Chemical Formula: C11H15NO3
Molecular Weight: 209.245
o-(2-Ethoxyethoxy)benzamide
Etosalamide, also known as Ethosalamide, is an antipyretic and analgesics agent
SYN

OR

CAS:592-55-2, 2-Bromoethyl ethyl ether
Cas, 611-20-1, 2-Hydroxybenzonitrile

PATENT
DE 1013643
PATENT
GB 774635
PATENT
US2822391
78 – 79 MP
PAPER
Journal of Chemical and Engineering Data (1962), 7, 265-6
70 – 71.5 MP
PATENT
WO 2004003198
US 20100226943
/////////Etosalamide, этосаламид , إيتوسالاميد , 依托柳胺 , ethosalamide
O=C(N)C1=CC=CC=C1OCCOCC
FDA approves first treatment Ruzurgi (amifampridine) for children with Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder

FDA approves first treatment Ruzurgi (amifampridine) for children with Lambert-Eaton myasthenic syndrome, a rare autoimmune disorder
The U.S. Food and Drug Administration today approved Ruzurgi (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in patients 6 to less than 17 years of age. This is the first FDA approval of a treatment specifically for pediatric patients with LEMS. The only other treatment approved for LEMS is only approved for use in adults.
“We continue to be committed to facilitating the development and approval of treatments for rare diseases, particularly those in children,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This approval will provide a much-needed treatment option for pediatric patients with LEMS who have significant weakness and fatigue that can often cause great difficulties with daily activities.”
LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. In people with LEMS, the body’s own immune system attacks the neuromuscular junction (the connection between nerves and muscles) and disrupts the ability of nerve cells to send signals to muscle cells. LEMS may be associated with …
- May 06, 2019
The U.S. Food and Drug Administration today approved Ruzurgi (amifampridine) tablets for the treatment of Lambert-Eaton myasthenic syndrome (LEMS) in patients 6 to less than 17 years of age. This is the first FDA approval of a treatment specifically for pediatric patients with LEMS. The only other treatment approved for LEMS is only approved for use in adults.
“We continue to be committed to facilitating the development and approval of treatments for rare diseases, particularly those in children,” said Billy Dunn, M.D., director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “This approval will provide a much-needed treatment option for pediatric patients with LEMS who have significant weakness and fatigue that can often cause great difficulties with daily activities.”
LEMS is a rare autoimmune disorder that affects the connection between nerves and muscles and causes weakness and other symptoms in affected patients. In people with LEMS, the body’s own immune system attacks the neuromuscular junction (the connection between nerves and muscles) and disrupts the ability of nerve cells to send signals to muscle cells. LEMS may be associated with other autoimmune diseases, but more commonly occurs in patients with cancer such as small cell lung cancer, where its onset precedes or coincides with the diagnosis of cancer. LEMS can occur at any age. The prevalence of LEMS specifically in pediatric patients is not known, but the overall prevalence of LEMS is estimated to be three per million individuals worldwide.
Use of Ruzurgi in patients 6 to less than 17 years of age is supported by evidence from adequate and well-controlled studies of the drug in adults with LEMS, pharmacokinetic data in adult patients, pharmacokinetic modeling and simulation to identify the dosing regimen in pediatric patients and safety data from pediatric patients 6 to less than 17 years of age.
The effectiveness of Ruzurgi for the treatment of LEMS was established by a randomized, double-blind, placebo-controlled withdrawal study of 32 adult patients in which patients were taking Ruzurgi for at least three months prior to entering the study. The study compared patients continuing on Ruzurgi to patients switched to placebo. Effectiveness was measured by the degree of change in a test that assessed the time it took the patient to rise from a chair, walk three meters, and return to the chair for three consecutive laps without pause. The patients that continued on Ruzurgi experienced less impairment than those on placebo. Effectiveness was also measured with a self-assessment scale for LEMS-related weakness that evaluated the feeling of weakening or strengthening. The scores indicated greater perceived weakening in the patients switched to placebo.
The most common side effects experienced by pediatric and adult patients taking Ruzurgi were burning or prickling sensation (paresthesia), abdominal pain, indigestion, dizziness and nausea. Side effects reported in pediatric patients were similar to those seen in adult patients. Seizures have been observed in patients without a history of seizures. Patients should inform their health care professional immediately if they have signs of hypersensitivity reactions such as rash, hives, itching, fever, swelling or trouble breathing.
The FDA granted this application Priority Review and Fast Track designations. Ruzurgi also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.
The FDA granted the approval of Ruzurgi to Jacobus Pharmaceutical Company, Inc.
/////////////////FDA 2019, Ruzurgi, amifampridine, Lambert-Eaton myasthenic syndrome, LEMS, RARE DISEASES, CHILDREN, Jacobus Pharmaceutical Company, Priority Review, Fast Track designations, Orphan Drug designation
Beperminogene perplasmid, ベペルミノゲンペルプラスミド
Beperminogene perplasmid
ベペルミノゲンペルプラスミド
HGF plasmid
- DNA (human hepatocyte growth factor plasmid pVAX1 cDNA)
- DNA (plasmid pVAX1HGF/MGBI)
- AMG-0001
DS-992
Nucleic Acid Sequence
Sequence Length: 51811342 a 1223 c 1314 g 1302 t
APPROVED, japan 2019, Collategene, 2019/3/29
Antiparkinsonian, Angiogenesis inducing agent
CAS: 627861-07-8
- Originator AnGes MG
- Developer AnGes MG; Osaka University Hospital
- Class Antiparkinsonians; Gene therapies; Ischaemic heart disorder therapies; Vascular disorder therapies
- Mechanism of Action Angiogenesis inducing agents; Gene transference; Hepatocyte growth factor expression stimulants
- Available For Licensing Yes – Ischaemic heart disorders; Lymphoedema; Parkinson’s disease
- Registered Peripheral arterial disorders
- Phase I/II Lymphoedema
- No development reported Arteriosclerosis obliterans; Ischaemic heart disorders; Parkinson’s disease; Thromboangiitis obliterans
- 26 Mar 2019 Registered for Peripheral arterial disorders in Japan (IM)
- 21 Feb 2019 The Pharmaceutical Affairs and Food Sanitation Council recommends conditional and time-limited approval of beperminogene perplasmid for the improvement of ulcers associated with chronic peripheral arterial disease
- 21 Feb 2019 AnGes plans a clinical study to assess the efficacy of beperminogene perplasmid in improvement of pain at rest in chronic peripheral arterial disorders
- In 2010, the product received fast track designation in the U.S. for the treatment of critical limb ischemia
HGF Plasmid (Beperminogene Perplasmid)Critical Limb Ischemia (Arteriosclerosis Obliterans & Buerger’s Disease) AMG0001 Injection, JAPAN AND US ALLIANCE Mitsubishi Tanabe Pharma
PATENT
WO 2017126488
US 20170283446
Expert Review of Cardiovascular Therapy (2014), 12(10), 1145-1156.
////////////Beperminogene perplasmid, japan 2019, ベペルミノゲンペルプラスミド , AnGes MG, Osaka University Hospital, Critical Limb Ischemia, Arteriosclerosis Obliterans, Buerger’s Disease, AMG0001, AMG-0001, DS-992 , HGF plasmid , fast track designation
BI-882370

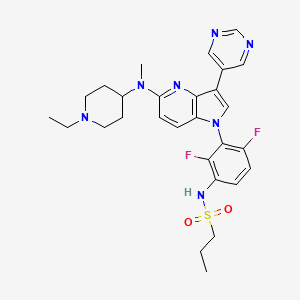
BI-882370
XP-102
N-(3-(5-((1-ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-1H-pyrrolo[3,2-b]pyridin-1-yl)-2,4-difluorophenyl)propane-1-sulfonamide
CAS 1392429-79-6
Chemical Formula: C28H33F2N7O2S
Molecular Weight: 569.68
Elemental Analysis: C, 59.03; H, 5.84; F, 6.67; N, 17.21; O, 5.62; S, 5.63
BI 882370 is a highly potent and selective RAF inhibitor that binds to the DFG-out (inactive) conformation of the BRAF kinase. BI 882370 inhibits proliferation of human BRAF-mutant melanoma cells with 100× higher potency (1-10 nmol/L) than vemurafenib.
Xynomic, under license from Boehringer Ingelheim , is investigating for treating BRAF mutant cancers, including colorectal cancer and melanoma; in October 2017, preclinical data were reported in the melanoma and colorectal cancer settings.
- Originator Boehringer Ingelheim
- Developer Boehringer Ingelheim; Xynomic Pharmaceuticals
- Class Antineoplastics; Piperidines; Pyridines; Pyrimidines; Pyrroles; Small molecules
- Mechanism of Action Proto oncogene protein b raf inhibitors
- Preclinical Colorectal cancer; Malignant melanoma
- 20 Dec 2018 Xynomic Pharma plans a phase Ib trial for Colorectal cancer (in combination with BI 860585) in third quarter of 2019
- 01 Jun 2018 Xynomic Pharmaceuticals plans a phase I trial for Colorectal cancer and Malignant melanoma in 2018 or 2019
- 06 Nov 2017 Chemical structure information added
-
US8889684
PATENT
WO2012104388

PATENT
Novel crystalline salts (monosuccinate salt), designated as Form A, of BI-882370 and their substantially anhydrous and non-solvated, processes for their preparation and compositions comprising them. Also claimed are their use as a RAF kinase Inhibitor, for the treatment of cancers and other diseases, such as infections, inflammations and autoimmune diseases.
The compound N-(3-(5-((l -ethylpiperidin-4-yl)(methyl)andno)-3-(pyrimidin-5-yl)-lH-pyrrolo [3, 2-Z>]pyri din- l-yl)-2,4-difluorophenyl)propane-l -sulfonamide (BI 882370), having Formula I:
I
is a RAF kinase inhibitor useful in the treatment of various diseases including cancer. The compound of Formula I, as well as its preparation and use, have been described in
WO/2012/104388, which is incorporated herein by reference in its entirety.
The RAS-RAF-MAPK (mitogen-activated protein kinase) signaling pathway plays a critical role in transmitting proliferation signals generated by the cell surface receptors and cytoplasmic signaling elements to the nucleus. Constitutive activation of this pathway is involved in malignant transformation by several oncogenes. Activating mutations in RAS
occur in approximately 15 % of cancers, and recent data has shown that B-RAF is mutated in about 7% of cancers (Wellbrock et al, “The RAF proteins take centre stage”, Nature Rev. Mol. Cell Biol., 2004, 5, 875-885), identifying it as another important oncogene in this pathway. In mammals, the RAF family of serine/threonine kinases comprises three members: A-RAF, B-RAF and C-RAF. However, activating mutations have so far been only identified in B-RAF underlining the importance of this isoform. It is believed that B-RAF is the main isoform that couples RAS to MEK, and that C-RAF and A-RAF signal to ERK only to fine-tune cellular responses (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885). The most common cancer mutation in B-RAF results in a valine to glutamic acid exchange at position 600 of the protein (V600E), which dramatically enhances B-RAF activity, presumably because its negative charge mimics activation loop phosphorylation (Wan et al , “Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF”, Cell, 2004, 116, 855-867). The highest incidence of B-RAF V600 mutations occurs in malignant melanoma (39%), thyroid cancer (46%), colorectal cancer (10%), biliary tract cancer (10%), prostate cancer (4%), ovary cancer (3%) and non-small cell lung cancer (2%), but they also occur at a low frequency in a wide variety of other cancers (frequencies of mutations according to COSMIC (Catalogue Of Somatic Mutations In Cancer; Wellcome Trust Sanger Institute) release v.53, 15th May 2011 ;
http://www.sanger.ac.uk/genetics/CGP/cosmic/). Literature supported the hypothesis that B-RA 600E mutated tumor cells seem to rely heavily on the continued activation of this pathway – a phenomenon termed “oncogene addiction” – whereas normal B-RAFwt cells use a broader range of signals. This provides an Achilles’ heel that can be exploited
therapeutically by treating patients with somatically mutated B-RAFV600E using orally available B-RAF inhibitors.
The key role of B-RAF V600E in aberrant ERK signaling and consequently oncogenesis has been demonstrated in several independent experimental approaches such as
overexpression of oncogenic/mutated B-RAF in vitro and in vivo (Wan et al., Cell, 2004, 116, 855-867; Wellbrock et al, Cancer Res. 2004, 64: 2338-2342), siRNA knock-down in vitro (Karasarides et al., Oncogene, “V599EB-RAF is an oncogene in melanocytes”, 2004, 23, 6292-6298) or in inducible short-hairpin RNA xenograft models where gain-of-function B-RAF signaling was found to be strongly associated with in vivo tumorigenicity (Hoeflich et al, “Oncogenic BRAF is required for tumor growth and maintenance in melanoma models”, Cancer Res., 2006, 66, 999-1006).
Treatment of B-RAFV600E mutated melanoma or colon carcinoma cells induces a B-RAF inhibition phenotype (e.g. reduction of phospho-MEK and phospho-ERK levels, reduction of cyclin D expression and induction of p27 expression). Consequently, these cells are locked in the Gl -phase of the cell cycle and do not proliferate.
Clinical proof of mechanism and proof of concept has been established for treating in cancer in B-RAFV600E mutated melanoma patients treated with Zelboraf®, B-RAF inhibitor (PLX-4032, vemurafenib, from Plexxikon/Daiichi Sankyo/Roche. Bollag et al., “Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma”, Nature, 2010, 467(7315), 596-9.; Flaherty et al, New Engl. J. Med., “Inhibition of Mutated, Activated BRAF in Metastatic Melanoma”, 2010, 363, 809-819; Chapman et al. “Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation”, New Engl. J. Med, 2011, 364:2507-2516. Favorable response rates were observed in both Phase I and Phase III clinical trials. It was reported, that melanoma patients carrying a B-RAFV600K mutation also do respond to therapy (Rubinstein et al, “Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032”, J. Transl. Med , 2010, 8, 67).
The most frequent B-RAF mutation is the exchange at amino acid position 600 from valine to glutamate with more than 90% frequency of all B-RAF mutations (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885), the second most frequent mutation is an alteration from valine to lysine, other mutations were found with lower frequency at that position (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885 and frequencies of mutations according to COSMIC (Catalogue Of Somatic Mutations In Cancer; Wellcome Trust Sanger Institute) release v53, 15th May 2011 ;
http://www.sanger.ac.uk/genetics/CGP/cosmic/). Additional mutations were found at e.g. the glycine rich loop (Wellbrock et al. Nature Rev. Mol. Cell Biol, 2004, 5, 875-885). Not all of these rather rare mutations seem to lead to direct activation of B-RAF (Wan et al. ,
“Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF”, Cell, 2004, 116, 855-867).
The compound of Formula I is a highly potent and selective RAF inhibitor that binds to the DFG-out (inactive) conformation of the B-RAF kinase. The compound inhibited proliferation of human B-RAF-mutant melanoma cells with 100 times higher potency (1-10 nmol/L) than vemurafenib, whereas wild-type cells were not affected at 1,000 nmol/L. A solution of the compound administered orally was efficacious in mouse models of B-RAF-mutant melanomas and colorectal carcinomas, and at 25 mg/kg twice daily showed superior efficacy compared with vemurafenib, dabrafenib, or trametinib. The compound was also active in A375 melanoma-bearing mice that were resistant to vemurafenib, particularly when dosed in combination with trametinib. Mice treated with the compound did not show any body weight loss or clinical signs of intolerability, and no pathologic changes were observed in several major organs investigated, including skin. Furthermore, in a pilot study in rats (up to 60 mg/kg daily for 2 weeks), the compound lacked toxicity in terms of clinical chemistry, hematology, pathology, and toxicogenomics. These results are described in Waizenegger et al., Mol. Cancer Ther., 2016, 75(3); 354-65, which is incorporated herein by reference in its entirety.
For the manufacture, purification, and formulation of a drug, it may be advantageous to employ a form of the drug having superior stability or other desirable formulation property exhibited by, for example, one or more salt or crystalline forms of the drug. Formation of salts of basic or acidic drugs can sometimes provide forms of the drug that have
advantageous properties such as solubility, non-hygroscopicity, crystallinity, and other physical properties that advantageous for formulating the drug. On the other hand, discovering a suitable salt or other crystalline form that is suitable for formulation is difficult, since there are numerous variables in the formation of a salt or crystalline form. These include the existence of numerous possible acids and bases that might be used as a counter-ion, various stoichiometric ratios that may be possible for combining a given basic or acid drug with an acid or base counter-ion, a wide variety of solvents and solvent systems
(including combinations of solvents) that potentially can be used to attempt to form salts or crystalline forms, and a variety of conditions (such as temperature or heating or cooling conditions) under which salts or crystalline forms may be generated. All of these variables of which may affect the properties of the salts or crystalline forms that might be obtained. Salts or solid forms may also have a variety of properties that render them unsuitable for drug development and formulation such as lack of crystallinity (amorphous forms), the presence or formation of multiple crystalline forms, which may interconvert and/or have different properties (polymorphism), lack of aqueous solubility, hygroscopicity, or stickiness of the solid. Furthermore, the formation of salts and crystalline forms and their properties are generally very unpredictable.
Accordingly, the crystalline salt forms of the compound of Formula I provided herein help satisfy the ongoing need for the development of a RAF kinase inhibitor for the treatment of serious diseases.
Preparation of A^-(3-(5-((l-ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo[3,2-Z>]pyridin-l- amide (BI 882370)
Step 1. 4-(6-Methyl-5-nitro-pyridin-2-yl)-piperazine-l-carboxylic acid tert-butyi ester
(3)
1 2 3
DIPEA (62.82 mL, 0.435 mol) is added to the solution of 6-chloro-3-nitro-2-methylpyridine (1) (50 g, 290 mmol) and N-Boc-piperazine (2) (53.95 g, 290 mmol) in dry MeCN (200 mL) and stirred for 4 h at 50 °C. After the reaction is finished the reaction mixture is diluted with MeCN and water and stirred for 30 min. The precipitated product is collected by filtration, washed with water and the solid is dried in vacuo.
Step 2. 4- [6-((£’)-2-Dimethylamino-vinyl)-5-nitro-pyridin-2-yl] -piperazine- 1-carboxylic acid
To a stirred solution of 4-(6-methyl-5-nitro-pyridin-2-yl)-piperazine- 1-carboxylic acid tert-butyl ester (3) (13 g, 40.3 mmol) in DMF (35 mL) is added N,N-dimethylformamide dimethylacetal (14.47 g, 121 mmol) and stirred in argon atmosphere for 36 h at 90 °C.
Additional 1.5 eq. of N^V-dimethylformamide dimethylacetal is added and stirred for 12 h at 90 °C. The reaction mixture is poured into water and extracted with DCM. The combined organic layers are washed with water, dried over anhydrous Na2S04 and concentrated in vacuo. The residue is used without further purification for the next step.
Step -(lH-pyrrolo[3,2-Z>]pyridin-5-yl)piperazine-l-carboxylic acid tert-butyl ester (5)
4 5
4-[6-((i?)-2-Dimethylairdno-vinyl)-5-nitro-pyridin-2-yl]-piperazine-l-carboxylic acid tert-butyl ester (36.4 g, 96 mmol) is taken up in MeOH, Pd/C (0.56 g, 10 %) is added and the mixture is hydrogenated in an autoclave at 60 psi for 16 h. The reaction mixture is filtered and concentrated under reduced pressure. The residue is purified by column chromatography viaNP MPLC. The product containing fractions of compound (5) (HPLC-MS method B: tRet. = 1.55 min.; MS (M+H)+ = 303) are combined and evaporated in vacuo.
Step 4. N- -Amino-2,6-difluorophenyl)acetamide (7)
6 7
Compound (6) (55.0 g, 254 mmol) is taken-up in MeOH (1.0 L). Pd/C (10.0 g, 10 %) is added and the mixture is hydrogenated in an autoclave at 200 psi for 3 h. The reaction mixture is filtered and concentrated under reduced pressure. The residue is purified by NP-MPLC on silica gel using DCM/MeOH (96:4) as eluent. The product containing fractions of the aniline intermediate (HPLC-MS method B: tRet. = 0.25 min.; MS (M-H)“ = 185) are combined and evaporated.
Step 5. N- -Difluoro-3-(propylsulfonamido)phenyl)acetamide (9)
To the aniline intermediate (35.0 g, 188 mmol) in DCM (100 mL) pyridine (6.6 mL, 75 mmol) and ^-propane sulfonyl chloride (8) (29.5 mL, 263 mmol) are added and the mixture is stirred at rt for 16 h. The reaction mixture is diluted with EtOAc (200 mL), washed with H2O and HC1 (aq., 1 N) and the layers are separated, dried over MgS04 and evaporated to yield the sulfonamide (9) which was used without further purification.
Step 6. N-
9 10
The sulfonylated aniline (9) (38.0 g, 130 mmol) is taken-up in EtOH (250 mL), H2O (200 mL) and concentrated hydrochloric acid (200 mL) and heated to 80 °C for 2 h. The reaction mixture is concentrated under reduced pressure, aqueous NaOH (4 N) is added until pH = 6 is reached and the mixture is extracted 2 x with DCM. The combined organic layer is washed with brine, dried over MgS04, filtered and evaporated to yield the deacylated aniline (10) (HPLC-MS method B: tRet. = 0.22 min.; MS (M-H)“ = 249) as a hydrochloride which was used without further purification.
Step 7. N-(2 -Difluoro-3-iodophenyl)propane-l-sulfonamide (11)
10 11
The hydrochloride of compound (10) is taken-up in DCM and extracted with NaHCCb solution. The organic layer is dried over MgSCn, filtered and evaporated. To the free base (10) (3.55 g, 14.21 mmol) in TFA (80 mL) at 0 °C is added NaNC (1.96 g, 28.4 mmol) in small portions and the mixture is stirred for 30 min. KI (23.83 g, 142 mmol) is added and stirring is continued for additional 15 min. The reaction mixture is diluted with Et^O and stirred for 1 h. Na2S203 solution (semiconc.) is added and the mixture is extracted 3 x with Et20. The combined organic layer is dried over MgSCn, filtered and concentrated in vacuo. The residue is purified by column chromatography via NP-MPLC. The product containing fractions of compound (11) (HPLC-MS method A: tRet. = 1.58 min.; MS (M-H)“ = 360) are combined and evaporated in vacuo.
Step 8. 4-((l-(2,6-Difluoro-3-(propylsulfonamido)phenyl)-lH-pyrrolo [3,2-b] pyridin-5-yl)
12
The lH-pyrrolo [3,2-*] pyridine (5) (10.0 g, 30.27 mmol), sulfonamide (11) (16.4 g,
45.4 mmol), Cul (576 mg, 3.03 mmol), ^^-(l ^^^-^N’-bismethyl-l^-cyclohexandiamine
(1.91 mL, 12.1 mmol) and CS2CO3 (29.6 g, 90.85 mmol) are taken-up in dry toluene (3 mL) and the resulting mixture is flushed with argon and stirred for 16 h at 120 °C. After the addition of further Cul (576 mg, 3.03 mmol), trans-(\R,2R)-N,N’-bismet y 1-1,2-cyclohexandiamine (1.91 mL, 12.1 mmol) and CS2CO3 (20.0 g, 60.0 mmol) the reaction mixture is stirred for further 24 h. The solvent is removed in vacuo, the residue is taken up in DCM and extracted with NaHCC solution (semiconc). The organic layer is dried over MgS04, filtered, the solvent is removed in vacuo and the residue is purified viaNP-MPLC. The product containing fractions of (12) (HPLC-MS method C: teet. = 1.62 mia; MS (M+H)+ = 564) are combined and the solvent is removed in vacuo.
Step 9. 4-((l-(2,6-Difluoro-3-(propylsulfonamido)phenyl)-3-iodo-lH-pyrrolo[3,2-b]pyridin-5 3)
To a solution of sulfonamide (12) (1.078 g, 1.9 mmol) in DMF (4 mL)/THF (100 μί) is added NIS (474 mg, 2.1 mmol) and the mixture is stirred for 1 h at rt. The reaction mixture is diluted with 30 mL DCM and extracted with NaHCCb solution (semiconc). The combined organic layer is dried over MgSCn, filtered and concentrated under reduced pressure. The residue is purified by column chromatography via RP HPLC. The product containing fractions of (13) (HPLC-MS method B: tRet. = 2.035 mia; MS (M+H)+ = 688) are freeze dried.
Step 10. 4-((l-(2,6-Difluoro-3-(propylsulfonamido)phenyl)-3-(pyrimidin-5-yl)-lH-pyrrolo[3,2-b]pyridin-5-yl)(methyl)amino)piperidine-l-carboxylic acid tert-butyi ester (15)
13 15
Sulfonamide (13) (770 mg, 1.12 mmol), pyrimidin-5-yl-boronic acid (14) (194 mg, 1.57 mmol), Pd(dppf)Cl2 (82 mg, 0.11 mmol), LiCl (142 mg, 3.35 mmol) and Na2C03 (294 mg, 2.8 mmol) are taken-up in dioxane/LhO (2: 1 mixture, 12 mL), and the resulting mixture is flushed with argon and stirred for 1 h at 100 °C. The reaction mixture is diluted with DCM and extracted with NaHCCb solution (semi-concentrated). The organic layer is dried over MgS04, filtered, Isolute® is added, the solvent is removed in vacuo and the residue is purified via RP HPLC. The product containing fractions of (15) (HPLC-MS method C: tRet. = 2.149 min.; MS (M+H)+ = 642) are freeze dried.
Step 11. N-(2,4-Difluoro-3-(5-(methyl(piperidin-4-yl)amino)-3-(pyrimidin-5-yl)- 1H-pyrrolo[3,2-b]pyridin-l-yl)phenyl)propane-l-sulfonamide
15 16
To a solution of example compound (15) (154 mg, 0.24 mmol) in DCM/MeOH (1 : 1, 4 mL) is added HC1 (in dioxane, 4 N, 2 mL) and the mixture is stirred for 3 h at rt. The solvent is removed in vacuo. Obtained compound (16) (HPLC-MS method B: tRet. = 1.02 min.; MS (M+H)+ = 542) is used without further purification.
Step 12. ^-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo [3,2-Z>] pyridin- l-yl)-2,4-diflu
Compound I was obtained from compound (16) by reductive alkylation with acetaldehyde (40% in iPrOH) in the presence of 1.5 eq. sodium acetoxyborohydride in iPrOH. The crude product was recrystallized from ethanol to obtain the title compound in 84% yield.
Scale-Up Synthesis of A/-(3-(5-((l-ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo[3,2-Z>]pyridin-l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide (BI 882370)
Step 1. N-(2,4-Difluoro-3-(5-(methyl(piperidin-4-yl)amino)-3-(pyrimidin-5-yl)-lH-pyrrolo[
15 16
Isopropanol (8.83 kg) and compound (15) (1.80 kg, 2.8 mol) were added into a reactor, and the mixture was stirred and heated to 55-60 °C. Concentrated hydrochloric acid (2.76 kg, 28 mol) was dropped into the reactor over than 20 min. at 60-65 °C. Then, the reaction mass was heated to 60-70 °C and held for 1 h. The conversion was monitored by HPLC, and reached about 99.5% after about 1 h.
The reaction mass was cooled and the isopropanol was removed by distillation under reduced pressure at not more than 50 °C. A brown oil was obtained, dissolved into water (6.75 kg) and washed by extraction with ethyl acetate (2.02 kg) at 20-30 °C. The water-phase was cooled to 15-20 °C. The pH was adjusted to 8.0-8.5 with 10% aqueous NaOH solution (-8.0 kg) at 20-30°C. The mixture was stirred for 3-4h at 20-30°C with the pH adjusted to 8.0-8.5 by addition of 10% NaOH solution every half-hour. The product was isolated by filtration and the cake washed with water (3.6 kg). The solid was dried under vacuum at 45-50 until the water content was not more than 5.5%. This provided about 1.64 kg of crude compound (16) (yield 108% of theoretical; the crude product containing water and NaCl detected). The crude product was used directly).
Step 12. ^-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)-lH-pyrr -Z>] pyridin- l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide (I)
Bl 878426 Bl 882370
Process:
Dichloromethane (19.88 kg) and compound (16) (1.5kg, 2.77mol) were added into a reactor, and the mixture was stirred and cooled to 0-10°C under a nitrogen atmosphere. Sodium triacetoxyborohydride (95%, 0.93 kg, 4.16 mol) was added into the mixture at 0-10°C. The mixture was stirred for 20-30 min. at 0- 10°C. Acetaldehyde in DCM (40%,
1.07 kg, 9.71 mol) added into the mixture slowly over 2 h at 0-10 °C. The reaction mixture was stirred at 0-10 °C under a nitrogen atmosphere for 0.5-lh. The conversion was monitored by HPLC, and reached about 99.5% after about 0.5-1 h.
Water (15 kg) was added into the reaction mass at a temperature below 15 °C. The mixture was stirred at 15-30 °C for 20-30 min. Aqueous ammonia (25%, 1.13 kg, 16.61 mol) was added into the mixture and the mixture was then stirred for 0.5 h. The organic phase was separated and then washed by extraction with water (15 kg) at 20-25 °C. Activated charcoal (0.15 kg) was added into the organic phase. The mixture was stirred for 1 h and then filtered. The filtrate was concentrated under reduced pressure at not more than 40°C, and compound (I) (1.58 kg, 100% yield) was obtained as a foamy solid.
Investigation of the Crystallinity of iV-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)- lH-pyrrolo [3,2-Z>] pyridin- l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide Free Base
Investigation of the crystallinity of N-(3-(5-((l-ethylpiperidin-4-yl)(methyl)amino)-3-(py rimidin-5-y 1)- lH-pyrrolo[3 ,2-b] pyridin- 1 -y l)-2,4-difluoropheny l)propane- 1 -sulfonamide free base, obtained by recrystallization from aqueous ethanol, which was used as a starting material to investigate salt formation showed that the compound had low crystallinity, as seen in FIG. 1.
Investigation of Salt forms of iV-(3-(5-((l-Ethylpiperidin-4-yl)(methyl)amino)-3-(pyrimidin-5-yl)- lH-pyrrolo [3,2-Z>] pyridin- l-yl)-2,4-difluorophenyl)propane- 1-sulfonamide
The compound N-(3-(5-((l-ethylpiperidin-4-yl)(methyl)andno)-3-(pyrimidin-5-yl)-lH-pyrrolo [3 ,2-Z>]pyri din- l-yl)-2,4-difluorophenyl)propane-l -sulfonamide was combined with various acids in various solvent systems.
A 96-well master plate was charged by dosing compound in MeOH (stock solution) with a concentration of approx. 40 mg/mL. This plate was placed in a vacuum oven for liquid removal to obtain the same amount of solid material in each well. Subsequently different solvents/solvent mixtures and the acids were added to the solid material in each well (approx. 500μί) and the whole plate was heated up to 50 °C for 2 hours while stirring (using a small stirring bar added to each well).
The acids used were as shown in Table 1. The solvents used were as shown in Table 2. Crystallinity of salts obtained either by the slurry experiment or crystallization by evaporation.
To investigate crystal formation by a slurry experiment, the plate was allowed to cool and the crystallinity of the resulting salts was investigated by XRPD. An image of the master plate showing the salts obtained is shown in FIG. 2A and images of XRPD performed on the salt from each of the master plate wells, showing the crystallinity of the salts formed, is shown in FIG. 2B.
To investigate crystal formation by an evaporation experiment, after the heating period, the solutions were filtered at the same temperature (50 °C) using a preheated filter plate to ensure that no non-dissolved material can be transferred into the other crystallization plates. The filtrate was dispensed into an evaporation plate (approx.. 200μί). The solvents were allowed to evaporate, and the crystallinity of the resulting salts was investigated by XRPD. An image of the master plate showing the salts obtained is shown in FIG. 3A and images of XRPD performed on the salt from each of the evaporation plate wells, showing the crystallinity of the salts formed, is shown in FIG. 3B.
Table 1. Salts Used for Salt Form Investigation
Table 2. Solvents Used for Salt Form Investigation
REFERENCES
1: Waizenegger IC, Baum A, Steurer S, Stadtmüller H, Bader G, Schaaf O, Garin-Chesa P, Schlattl A, Schweifer N, Haslinger C, Colbatzky F, Mousa S, Kalkuhl A, Kraut N, Adolf GR. A Novel RAF Kinase Inhibitor with DFG-Out-Binding Mode: High Efficacy in BRAF-Mutant Tumor Xenograft Models in the Absence of Normal Tissue Hyperproliferation. Mol Cancer Ther. 2016 Mar;15(3):354-65. doi: 10.1158/1535-7163.MCT-15-0617. Epub 2016 Feb 25. PubMed PMID: 26916115.
/////////////// BI-882370, BI 882370, BI882370, XP-102, Boehringer Ingelheim, Xynomic Pharmaceuticals, Preclinical, Colorectal cancer, Malignant melanoma
CCN1CCC(CC1)N(C)c3ccc4n(cc(c2cncnc2)c4n3)c5c(F)ccc(NS(=O)(=O)CCC)c5F
ACLIMOSTAT
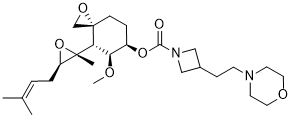
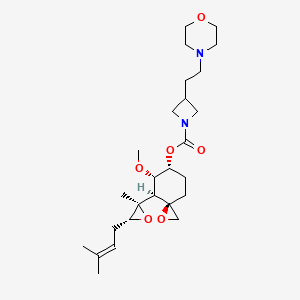
Aclimostat
CAS: 2082752-83-6
Chemical Formula: C26H42N2O6
Molecular Weight: 478.63
Elemental Analysis: C, 65.25; H, 8.85; N, 5.85; O, 20.06
ZGN-1061; ZGN1061; ZGN 1061; Aclimostat,
UNII-X150A3JK8R
X150A3JK8R
(3R,4S,5S,6R)-5-Methoxy-4-[(2R,3R)-2-methyl-3-(3- methylbut-2-en-1-yl)oxiran-2-yl]-1-oxaspiro[2.5]octan-6-yl 3-[2-(morpholin-4-yl)ethyl]azetidine-1-carboxylate
1-Azetidinecarboxylic acid, 3-[2-(4-morpholinyl)ethyl]-, (3R,4S,5S,6R)-5-methoxy-4-[(2R,3R)-2-methyl-3-(3-methyl-2-buten-1-yl)-2-oxiranyl]-1-oxaspiro[2.5]oct-6-yl ester
3R,4S,5S,6R)-5-methoxy-4-((2R,3R)-2-methyl-3-(3-methylbut-2-en-1-yl)oxiran-2-yl)-1- oxaspiro[2.5]octan-6-yl 3-(2-morpholinoethyl)azetidine-1-carboxylate
ZAFGEN, PHASE 2, DIABETES
Aclimostat, also known as ZGN-1061, is an anti-diabetic, anti-obesity MetAP2 inhibitor.
Over 1.1 billion people worldwide are reported to be overweight. Obesity is estimated to affect over 90 million people in the United States alone. Twenty-five percent of the population in the United States over the age of twenty is considered clinically obese. While being overweight or obese presents problems (for example restriction of mobility, discomfort in tight spaces such as theater or airplane seats, social difficulties, etc.), these conditions, in particular clinical obesity, affect other aspects of health, i.e., diseases and other adverse health conditions associated with, exacerbated by, or precipitated by being overweight or obese. The estimated mortality from obesity-related conditions in the United States is over 300,000 annually (O’Brien et al. Amer J Surgery (2002) 184:4S-8S; and Hill et al. (1998) Science, 280:1371). [0003] There is no curative treatment for being overweight or obese. Traditional pharmacotherapies for treating an overweight or obese subject, such as serotonin and noradrenergic re-uptake inhibitors, noradrenergic re-uptake inhibitors, selective serotonin re- uptake inhibitors, intestinal lipase inhibitors, or surgeries such as stomach stapling or gastric banding, have been shown to provide minimal short-term benefits or significant rates of relapse, and have further shown harmful side-effects to patients. [0004] MetAP2 encodes a protein that functions at least in part by enzymatically removing the amino terminal methionine residue from certain newly translated proteins such as glyceraldehyde-3-phosphate dehydrogenase (Warder et al. (2008) J. Proteome Res.7:4807). Increased expression of the MetAP2 gene has been historically associated with various forms of cancer. Molecules inhibiting the enzymatic activity of MetAP2 have been identified and have been explored for their utility in the treatment of various tumor types (Wang et al. (2003) Cancer Res.63:7861) and infectious diseases such as microsporidiosis, leishmaniasis, and malaria (Zhang et al. (2002) J. Biomed. Sci.9:34). Notably, inhibition of MetAP2 activity in obese and obese-diabetic animals leads to a reduction in body weight in part by increasing the oxidation of fat and in part by reducing the consumption of food (Rupnick et al. (2002) Proc. Natl. Acad. Sci. USA 99:10730).
[0005] Such MetAP2 inhibitors may be useful as well for patients with excess adiposity and conditions related to adiposity including type 2 diabetes, hepatic steatosis, and
cardiovascular disease (via e.g. ameliorating insulin resistance, reducing hepatic lipid content, and reducing cardiac workload). Accordingly, compounds capable of modulating MetAP2 are needed to address the treatment of obesity and related diseases as well as other ailments favorably responsive to MetAP2 modulator treatment.
Synthesis

CONTD……………….

contd………………….

Tetrahedron, 73(30), 4371-4379; 2017

WO 2017027684
PATENT
WO 2017027684
https://patents.google.com/patent/WO2017027684A1/en
Example 1
(3R,4S,5S,6R)-5-methoxy-4-((2R,3R)-2-methyl-3-(3-methylbut-2-en-1-yl)oxiran-2-yl)-1- oxaspiro[2.5]octan-6-yl 3-(2-morpholinoethyl)azetidine-1-carboxylate
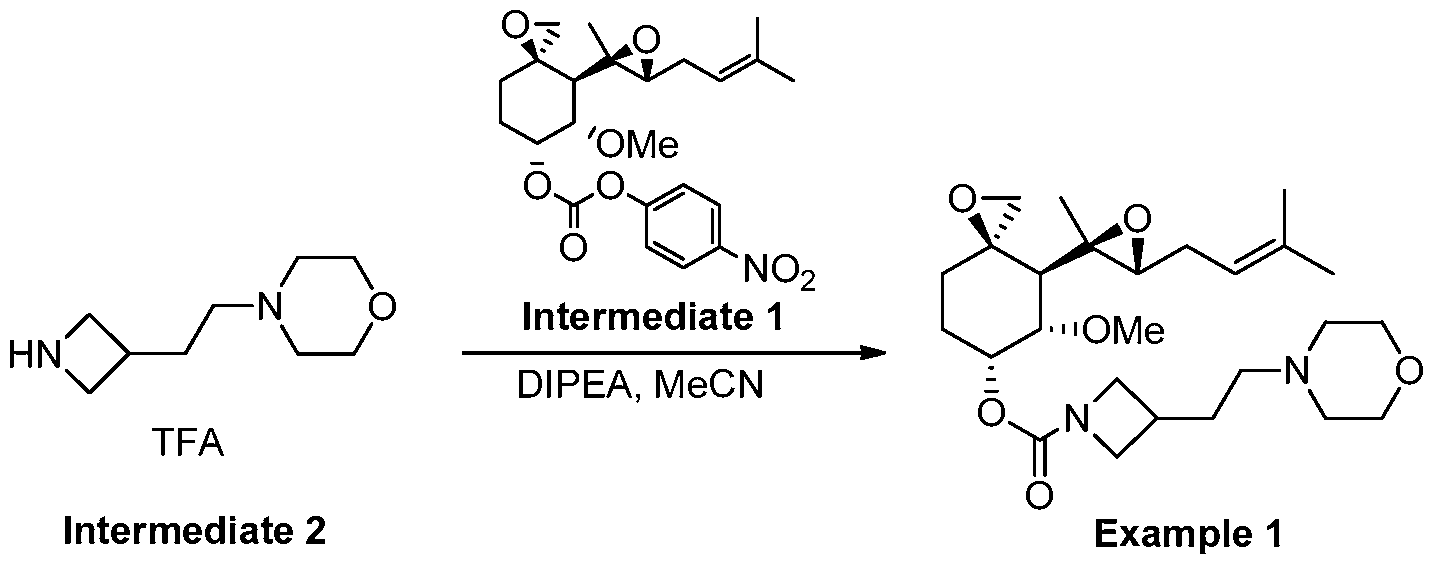
[00312] To a mixture of 4-(2-(azetidin-3-yl)ethyl)morpholine, trifluoroacetate (2.33 g, 3.7 mmol) in CH3CN (150 mL) was added DIPEA (2.9 mL, 17 mmol) drop-wise at 0-5oC. The mixture was then stirred at 0-5oC for 10 min, and carbonate Intermediate 1 (1.3 g, 2.9 mmol) was added to the mixture in portions at 0oC under a N2atmosphere. The reaction mixture was stirred at 25oC for 16 hrs. TLC (PE : EtOAc = 3 : 1) showed that the reaction was complete. The solvent was removed under vacuum below 40oC. The residue was diluted with DCM (60 mL), and the DCM solution was washed with ammonium acetate buffer (pH~4, 15 mL x 2). The combined aqueous layers were back-extracted with DCM (20 mL x 2). The combined organic layers were washed with aq. NaHCO3 solution (15 mL x 2, 5% wt), dried over Na2SO4 and concentrated. Purification by silica gel column chromatography (DCM: MeOH=100: 0~60: 1), followed by preparative HPLC (Method A, H2O (0.1% FA) / CH3CN) gave the title compound (1.15 g) as a light yellow syrup. LC-MS: m/z = 479 [M+H]+; 1H-NMR (400 MHz, CDCl3) δ 5.43 (br, 1H), 5.13 (t, J = 7.6 Hz, 1H), 3.87-4.15 (m, 2H), 3.63-3.65 (m, 4H), 3.52- 3.56 (m, 3H), 3.49 (s, 3H), 2.90 (d, J = 4.4 Hz, 1H), 2.46-2.54 (m, 3H), 2.19-2.36 (m, 7H), 1.97-2.13 (m, 2H), 1.78-1.89 (m, 5H), 1.73 (s, 3H), 1.62 (s, 3H), 1.13 (s, 3H), 0.99 (d, J = 13.6 Hz, 1H).
REFERENCES
1: Malloy J, Zhuang D, Kim T, Inskeep P, Kim D, Taylor K. Single and multiple dose evaluation of a novel MetAP2 inhibitor: Results of a randomized, double-blind, placebo-controlled clinical trial. Diabetes Obes Metab. 2018 Aug;20(8):1878-1884. doi: 10.1111/dom.13305. Epub 2018 Apr 23. PubMed PMID: 29577550; PubMed Central PMCID: PMC6055687.
2: Burkey BF, Hoglen NC, Inskeep P, Wyman M, Hughes TE, Vath JE. Preclinical Efficacy and Safety of the Novel Antidiabetic, Antiobesity MetAP2 Inhibitor ZGN-1061. J Pharmacol Exp Ther. 2018 May;365(2):301-313. doi: 10.1124/jpet.117.246272. Epub 2018 Feb 28. PubMed PMID: 29491038.
//////////////Aclimostat, ZGN-1061, ZAFGEN, PHASE 2, DIABETES
O=C(N1CC(CCN2CCOCC2)C1)O[C@H](CC3)[C@@H](OC)[C@H]([C@@]4(C)O[C@@H]4C/C=C(C)\C)[C@]53CO5
SRT 1720

SRT-1720 diHCl
CAY10559
CAS: 1001645-58-4 (di HCl) , 925434-55-5 (free base) 1001645-58-4 (HCl)
Chemical Formula: C25H25Cl2N7OS
Molecular Weight: 542.483
Elemental Analysis: C, 55.35; H, 4.65; Cl, 13.07; N, 18.07; O, 2.95; S, 5.91
SRT-1720 HCl, SRT-1720 hudrochloride; SRT1720; SRT-1720; SRT 1720; CAY10559; CAY-10559; CAY 10559; SIRT-1933; SIRT 1933; SIRT1933.
N-(2-(3-(piperazin-1-ylmethyl)imidazo[2,1-b]thiazol-6-yl)phenyl)quinoxaline-2-carboxamide dihydrochloride

- Molecular FormulaC25H23N7OS
- Average mass469.561 Da
SRT-1720, also known as CAY10559 and is a drug developed by Sirtris Pharmaceuticals intended as a small-molecule activator of the sirtuin subtype SIRT1. It has similar activity in the body to the known SIRT1 activator resveratrol, but is 1000x more potent. In animal studies it was found to improve insulin sensitivity and lower plasma glucose levels in fat, muscle and liver tissue, and increased mitochondrial and metabolic function. A study of SRT1720 conducted by the National Institute on Aging found that the drug may extend the lifespan of obese mice by 44% .
SRT1720 is an experimental drug that was studied by Sirtris Pharmaceuticals intended as a small-molecule activator of the sirtuinsubtype SIRT1. The compound has been studied in animals, but safety and efficacy in humans have not been established.
Animal research
In animal models of obesity and diabetes SRT1720 was found to improve insulin sensitivity and lower plasma glucose levels in fat, muscle and liver tissue, and increase mitochondrial and metabolic function.[1] In mice rendered obese and diabetic by feeding a high-fat, high-sugar diet, a study performed at the National Institute of Aging found that feeding chow infused with the highest dose of SRT1720 beginning at one year of age increased mean lifespan by 18%, and maximum lifespan by 5%, as compared to other short-lived obese, diabetic mice; however, treated animals still lived substantially shorter lives than normal-weight mice fed normal chow with no drug.[2] In a later study, SRT1720 increased mean lifespan of obese, diabetic mice by 21.7%, similar to the earlier study, but there was no effect on maximum lifespan in this study.[3] In normal-weight mice fed a standard rodent diet, SRT1720 increased mean lifespan by just 8.8%, and again had no effect on maximum lifespan.[3]
Since the discovery of SRT1720, the claim that this compound is a SIRT1 activator has been questioned[4][5][6] and further defended.[7][8]
Although SRT1720 is not currently undergoing clinical development, a related compound, SRT2104, is currently in clinical development for metabolic diseases.[9]
PAPER
Letters in Drug Design & Discovery, 10(9), 793-797; 2013
The Identification of the SIRT1 Activator SRT2104 as a Clinical Candidate
Author(s): Pui Yee Ng, Jean E. Bemis, Jeremy S. Disch, Chi B. Vu, Christopher J. Oalmann, Amy V. Lynch,David P. Carney, Thomas V. Riera, Jeffrey Song, Jesse J. Smith, Siva Lavu, Angela Tornblom, Meghan Duncan, Marie Yeager, Kristina Kriksciukaite, Akanksha Gupta, Vipin Suri, Peter J. Elliot, Jill C. Milne, Joseph J. Nunes, Michael R. Jirousek, George P. Vlasuk, James L. Ellis, Robert B. Perni.
Journal Name: Letters in Drug Design & Discovery
Volume 10 , Issue 9 , 2013

Paper
Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; Bemis, J.E.; Xie, R.; Disch, J.S.; Ng, P.Y.; Nunes, J.J.; Lynch, A.V.; Yang, H.; Galonek, H.; Israelian, K.; Choy, W.; Iffland, A.; Lavu, S.; Medvedik, O.; Sinclair, D.A.; Olefsky, J.M.; Jirousek, M.R.; Elliott, P.J.; Westphal, C.H.
Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes
Nature 2007, 450(7170): 712
PATENT
WO 2007019417
WO 2007019416
WO 2007019345
WO 2007019344
WO 2007019346
WO 2008115518
PAPER
Vu, Chi B.; Journal of Medicinal Chemistry 2009, VOL 52(5), PG 1275-1283
https://pubs.acs.org/doi/abs/10.1021/jm8012954

A series of imidazo[1,2-b]thiazole derivatives is shown to activate the NAD+-dependent deacetylase SIRT1, a potential new therapeutic target to treat various metabolic disorders. This series of compounds was derived from a high throughput screening hit bearing an oxazolopyridine core. Water-solubilizing groups could be installed conveniently at either the C-2 or C-3 position of the imidazo[1,2-b]thiazole ring. The SIRT1 enzyme activity could be adjusted by modifying the amide portion of these imidazo[1,2-b]thiazole derivatives. The most potent analogue within this series, namely, compound 29, has demonstrated oral antidiabetic activity in the ob/ob mouse model, the diet-induced obesity (DIO) mouse model, and the Zucker fa/fa rat model.
Discovery of Imidazo[1,2-b]thiazole Derivatives as Novel SIRT1 Activators
* To whom correspondence should be addressed. Phone: (617)-252-6920, extension 2129. Fax: (617)-252-6924. E-mail: cvu@sirtrispharma.com., †
Present address: Department of Medicine, Division of Endocrinology and Metabolism, University of California—San Diego, 9500 Gilman Drive, La Jolla, CA 92093.
Preparation of N-(2-(3-(Piperazin-1-ylmethyl)imidazo[2,1-b]thiazol-6-yl)phenyl)quinoxaline-2-carboxamide (29)
References
- ^ Milne JC; Lambert PD; Schenk S; Carney DP; Smith JJ; Gagne DJ; Jin L; Boss O; Perni RB; Vu CB; Bemis JE; Xie R; Disch JS; Ng PY; Nunes JJ; Lynch AV; Yang H; Galonek H; Israelian K; Choy W; Iffland A; Lavu S; Medvedik O; Sinclair DA; Olefsky JM; Jirousek MR; Elliott PJ; Westphal CH (November 2007). “Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes”. Nature. 450(7170): 712–6. doi:10.1038/nature06261. PMC 2753457. PMID 18046409.
- ^ Minor RK; Baur JA; Gomes AP; Ward TM; Csiszar A; Mercken EM; Abdelmohsen K; Shin YK; Canto C; Scheibye-Knudsen M; Krawczyk M; Irusta PM; Martín-Montalvo A; Hubbard BP; Zhang Y; Lehrmann E; White AA; Price NL; Swindell WR; Pearson KJ; Becker KG; Bohr VA; Gorospe M; Egan JM; Talan MI; Auwerx J; Westphal CH; Ellis JL; Ungvari Z; Vlasuk GP; Elliott PJ; Sinclair DA; de Cabo R (Aug 2011). “SRT1720 improves survival and healthspan of obese mice”. Scientific Reports. 1 (70): 70. doi:10.1038/srep00070. PMC 3216557. PMID 22355589. Retrieved 1 March 2014.
- ^ Jump up to:a b Mitchell SJ; Martin-Montalvo A; Mercken EM; et al. (Feb 2014). “The SIRT1 Activator SRT1720 Extends Lifespan and Improves Health of Mice Fed a Standard Diet”. Cell Reports. 6 (4): 836–43. doi:10.1016/j.celrep.2014.01.031. PMC 4010117. PMID 24582957. Retrieved 1 March 2014.
- ^ Pacholec M; Chrunyk BA; Cunningham D; Flynn D; Griffith DA; Griffor M; Loulakis P; Pabst B; Qiu X; Stockman B; Thanabal V; Varghese A; Ward J; Withka J; Ahn K (January 2010). “SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1”. J Biol Chem. 285 (11): 8340–8351. doi:10.1074/jbc.M109.088682. PMC 2832984. PMID 20061378.
- ^ Beher D; Wu J; Cumine S; Kim KW; Lu SC; Atangan L; Wang M (December 2009). “Resveratrol is not a direct activator of SIRT1 enzyme activity”. Chem Biol Drug Des. 74 (6): 619–24. doi:10.1111/j.1747-0285.2009.00901.x. PMID 19843076.
- ^ Zarse, K.; Schmeisser, S.; Birringer, M.; Falk, E.; Schmoll, D.; Ristow, M. (2010). “Differential Effects of Resveratrol and SRT1720 on Lifespan of AdultCaenorhabditis elegans”. Hormone and Metabolic Research. 42 (12): 837–839. doi:10.1055/s-0030-1265225. PMID 20925017.
- ^ Callaway E (2010-08-16). “GlaxoSmithKline strikes back over anti-ageing pills: Drugs do work as thought, says pharmaceutical giant”. Nature. doi:10.1038/news.2010.412.
- ^ Dai H; Kustigian L; Carney D; Case A; Considine T; Hubbard BP; Perni RB; Riera TV; Szczepankiewicz B; Vlasuk GP; Stein RL (August 2010). “SIRT1 activation by small molecules – kinetic and biophysical evidence for direct interaction of enzyme and activator”. J Biol Chem. 285 (43): 32695–32703. doi:10.1074/jbc.M110.133892. PMC 2963390. PMID 20702418.
- ^ “Sirtuin Pipeline”. Sirtris Pharmaceuticals.
 |
|
| Identifiers | |
|---|---|
| PubChem CID | |
| IUPHAR/BPS | |
| ChemSpider | |
| CompTox Dashboard(EPA) | |
| Chemical and physical data | |
| Formula | C25H23N7OS |
| Molar mass | 469.560 g/mol g·mol−1 |
| 3D model (JSmol) | |
////////////SRT-1720 DI HCl, obesity, diabetes, SRT 1720, Sirtris Pharmaceuticals, CAY10559, CAY 10559, Preclinical
O=C(NC1=CC=CC=C1C2=CN3C(SC=C3CN4CCNCC4)=N2)C5=NC6=CC=CC=C6N=C5.[H]Cl.[H]Cl
