
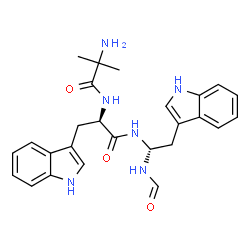
Macimorelin
- Molecular FormulaC26H30N6O3
- Average mass474.555 Da
CAS 381231-18-1
Chemical Formula: C26H30N6O3
Exact Mass: 474.23794
Molecular Weight: 474.55480
Elemental Analysis: C, 65.80; H, 6.37; N, 17.71; O, 10.11
2-Methylalanyl-N-[(1R)-1-formamido-2-(1H-indol-3-yl)ethyl]-D-tryptophanamide
8680B21W73
9073
D-Tryptophanamide, 2-methylalanyl-N-[(1R)-1-(formylamino)-2-(1H-indol-3-yl)ethyl]-
CAS 945212-59-9 (Macimorelin acetate)
(2R)-2-(2-amino-2-methylpropanamido)-3-(1H-indol-3-yl)-N-[(1R)-2-(1H-indol-3-yl)-1-formamidoethyl]propanamide; acetic acid
AEZS-130
ARD-07
D-87875
EP-01572
EP-1572
JMV-1843
USAN (ab-26)
MACIMORELIN ACETATE
AQZ1003RMG
ARD 07
D-87575
D-Tryptophanamide, 2-methylalanyl-N-[(1R)-1-(formylamino)-2-(1H-indol-3-yl)ethyl]-, acetate (1:1) [ACD/Index Name]
EP 1572
THERAPEUTIC CLAIM
Diagnostic agent for adult growth hormone deficiency (AGHD)
CHEMICAL NAMES
1. D-Tryptophanamide, 2-methylalanyl-N-[(1R)-1-(formylamino)-2-(1H-indol-3-yl)ethyl]-, acetate (1:1)
2. N2-(2-amino-2-methylpropanoyl-N1-[(1R)-1-formamido-2-(1H-indol-3-yl)ethyl]- D-tryptophanamide acetate
MOLECULAR FORMULA
C26H30N6O3.C2H4O2
MOLECULAR WEIGHT
534.6
SPONSOR
Aeterna Zentaris GmbH
CODE DESIGNATIONS
D-87575, EP 1572, ARD 07
CAS REGISTRY NUMBER
945212-59-9
Macimorelin (also known as AEZS-130, EP-1572) is a novel synthetic small molecule, acting as a ghrelin agonist, that is orally active and stimulates the secretion of growth hormone (GH). Based on results of Phase 1 studies, AEZS-130 has potential applications for the treatment of cachexia, a condition frequently associated with severe chronic diseases such as cancer, chronic obstructive pulmonary disease and AIDS. In addition to the therapeutic application, a Phase 3 trial with AEZS-130 as a diagnostic test for growth hormone deficiencies in adults has been completed.
http://www.ama-assn.org/resources/doc/usan/macimorelin-acetate.pdf
QUEBEC, Nov. 5, 2013 /PRNewswire/ – Aeterna Zentaris Inc. (the “Company”) today announced that it has submitted a New Drug Application (“NDA”) to the U.S. Food and Drug Administration (“FDA”) for its ghrelin agonist, macimorelin acetate (AEZS-130). Phase 3 data have demonstrated that the compound has the potential to become the first orally-approved product that induces growth hormone release to evaluate adult growth hormone deficiency (“AGHD”), with accuracy comparable to available intravenous and intramuscular testing procedures. read at
http://www.drugs.com/nda/macimorelin_acetate_131105.html
http://www.ama-assn.org/resources/doc/usan/macimorelin-acetate.pdf
macimorelin (JMV 1843), a ghrelin-mimetic growth hormone secretagogue in Phase III for adult growth hormone deficiency (AGHD)
Macimorelin, a growth hormone modulator, is currently awaiting registration in the U.S. by AEterna Zentaris as an oral diagnostic test of adult growth hormone deficit disorder. The company is also developing the compound in phase II clinical trials for the treatment of cancer related cachexia. The compound was being codeveloped by AEterna Zentaris and Ardana Bioscience; however, the trials underway at Ardana were suspended in 2008 based on a company strategic decision. AEterna Zentaris owns the worldwide rights of the compound. In 2007, orphan drug designation was assigned by the FDA for the treatment of growth hormone deficit in adults.
Macimorelin (INN), or Macrilen (trade name) is a drug being developed by Æterna Zentaris for use in the diagnosis of adult growth hormone deficiency. Macimorelin acetate, the salt formulation, is a synthetic growth hormone secretagogue receptor agonist.[1]Macimorelin acetate is described chemically as D-Tryptophanamide, 2-methylalanyl-N-[(1R)-1-(formylamino)-2-(1H-indol-3-yl)ethyl]-acetate.
As of January 2014, it was in Phase III clinical trials.[2] The phase III trial for growth hormone deficiency is expected to be complete in December 2016.[3]
As of December 2017, it became FDA-approved as a method to diagnose growth hormone deficiency.[4] Traditionally, growth hormone deficiency was diagnosed via means of insulin tolerance test (IST) or glucagon stimulation test (GST). These two means are done parenterally, whereas Macrilen boasts an oral formulation for ease of administration for patients and providers.
Macimorelin is a growth hormone secretagogue receptor (ghrelin receptor) agonist causing release of growth hormone from the pituitary gland.[5][6][7]
Macimorelin, a novel and orally active ghrelin mimetic that stimulates GH secretion, is used in the diagnosis of adult GH deficiency (AGHD). More specifically, macimorelin is a peptidomimetic growth hormone secretagogue (GHS) that acts as an agonist of GH secretagogue receptor, or ghrelin receptor (GHS-R1a) to dose-dependently increase GH levels [3]. Growth hormone secretagogues (GHS) represent a new class of pharmacological agents which have the potential to be used in numerous clinical applications. They include treatment for growth retardation in children and cachexia associated with chronic disease such as AIDS and cancer.
Growth hormone (GH) is classically linked with linear growth during childhood. In deficiency of this hormone, AGHD is commonly associated with increased fat mass (particularly in the abdominal region), decreased lean body mass, osteopenia, dyslipidemia, insulin resistance, and/or glucose intolerance overtime. In addition, individuals with may be susceptible to cardiovascular complications from altered structures and function [5]. Risk factors of AGHD include a history of childhood-onset GH deficiency or with hypothalamic/pituitary disease, surgery, or irradiation to these areas, head trauma, or evidence of other pituitary hormone deficiencies [3]. While there are various therapies available such as GH replacement therapy, the absence of panhypopituitarism and low serum IGF-I levels with nonspecific clinical symptoms pose challenges to the detection and diagnosis of AGHD. The diagnosis of AGHD requires biochemical confirmation with at least 1 GH stimulation test [3]. Macimorelin is clinically useful since it displays good stability and oral bioavailability with comparable affinity to ghrelin receptor as its endogenous ligand. In clinical studies involving healthy subjects, macimorelin stimulated GH release in a dose-dependent manner with good tolerability [3].
Macimorelin, developed by Aeterna Zentaris, was approved by the FDA in December 2017 under the market name Macrilen for oral solution.
New active series of growth hormone secretagogues
J Med Chem 2003, 46(7): 1191
WO 2001096300
WO 2007093820
PAPER
J Med Chem 2003, 46(7): 1191
http://pubs.acs.org/doi/full/10.1021/jm020985q


Synthetic Pathway for JMV 1843 and Analoguesa
a Reagents and conditions: (a) IBCF, NMM, DME, 0 °C; (b) NH4OH; (c) H2, Pd/C, EtOH, HCl; (d) BOP, NMM, DMF, Boc-(d)-Trp-OH; (e) Boc2O, DMAP cat., anhydrous CH3CN; (f) BTIB, pyridine, DMF/H2O; (g) 2,4,5-trichlorophenylformate, DIEA, DMF; (h) TFA/anisole/thioanisole (8:1:1), 0 °C; (i) BOP, NMM, DMF, Boc-Aib-OH; (j) TFA/anisole/thioanisole (8:1:1), 0 °C; (k) RP preparative HPLC.
TFA, H-Aib-(d)-Trp-(d)-gTrp-CHO (7). 6 (1 g, 1.7 mmol) was dissolved in a mixture of trifluoroacetic acid (8 mL), anisole (1 mL), and thioanisole (1 mL) for 30 min at 0 °C. The solvents were removed in vacuo, the residue was stirred in ether, and the precipitated TFA, H-Aib-(d)-Trp-(d)-gTrp-CHO was filtered. 7 was purified by preparative HPLC and obtained in 52% yield. 1H NMR (400 MHz, DMSO-d6) + correlation 1H−1H: δ 1.21 (s, 3H, CH3 (Aib)), 1.43 (s, 3H, CH3(Aib)), 2.97 (m, 2H, (CH2)β), 3.1 (m, 2H, (CH2)β‘), 4.62 (m, 1H, (CH)αA and (CH)αB), 5.32 (q, 0.4H, (CH)α‘B), 5.71 (q, 0.6H, (CH)α‘A), 7.3 (m, 4H, H5 and H6(2 indoles)), 7.06−7.2 (4d, 2H, H2A and H2B (2 indoles)), 7.3 (m, 2H, H4 or H7 (2 indoles)), 7.6−7.8 (4d, 2H, H4A and H4B or H7A and H7B), 7.97 (s, 3H, NH2 (Aib) and CHO (formyl)), 8.2 (d, 0.4H, NH1B (diamino)), 8.3 (m,1H, NHA and NHB), 8.5 (d, 0.6H, NH1A (diamino)), 8.69 (d, 0.6H, NH2A (diamino)), 8.96 (d, 0.4H, NH2B(diamino)), 10.8 (s, 0.6H, N1H1A (indole)), 10.82 (s, 0.4H, N1H1B (indole)), 10.86 (s, 0.6H, N1H2A (indole)), 10.91 (s, 0,4H, N1H2B (indole)). MS (ES), m/z: 475 [M + H]+, 949 [2M + H]+. HPLC tR: 16.26 min (conditions A).
PATENTS
http://www.google.com/patents/US8192719
The inventors have now found that the oral administration of growth hormone secretagogues (GHSs) EP 1572 and EP 1573 can be used effectively and reliably to diagnose GHD.
EP 1572 (Formula I) or EP 1573 (Formula II) are GHSs (see WO 01/96300, Example 1 and Example 58 which are EP 1572 and EP 1573, respectively) that may be given orally.
EP 1572 and EP 1573 can also be defined as H-Aib-D-Trp-D-gTrp-CHO and H-Aib-D-Trp-D-gTrp-C(O)NHCH2CH3. Wherein, His hydrogen, Aib is aminoisobutyl, D is the dextro isomer, Trp is tryptophan and gTrp is a group of Formula III:
PATENT
http://www.google.com/patents/US6861409
H-Aib-D-Trp-D-gTrp-CHO: 
Example 1 H-Aib-D-Trp-D-gTrp-CHO
Total synthesis (percentages represent yields obtained in the synthesis as described below):
Z-D-Tr-NH2
Z-D-Trp-OH (8.9 g; 26 mmol; 1 eq.) was dissolved in DME (25 ml) and placed in an ice water bath to 0° C. NMM (3.5 ml; 1.2 eq.), IBCF (4.1 ml; 1.2 eq.) and ammonia solution 28% (8.9 ml; 5 eq.) were added successively. The mixture was diluted with water (100 ml), and the product Z-D-Trp-NH2 precipitated. It was filtered and dried in vacuo to afford 8.58 g of a white solid.
Yield=98%.
C19H19N3O3, 337 g.mol−1.
Rf=0.46 {Chloroform/Methanol/Acetic Acid (180/10/5)}.
1H NMR (250 MHZ, DMSO-d6): δ 2.9 (dd, 1H, Hβ, Jββ′=14.5 Hz; Jβα=9.8 Hz); 3.1 (dd, 1H, Hβ′, Jβ′β=14.5 Hz; Jβ′α=4.3 Hz); 4.2 (sextuplet, 1H, Hα); 4.95 (s, 2H, CH2(Z); 6.9-7.4 (m, 11H); 7.5 (s, 1H, H2); 7.65 (d, 1H, J=7.7 Hz); 10.8 (s, 1H, N1H).
Mass Spectrometry (Electrospray), m/z 338 [M+H]+, 360 [M+Na]+, 675 [2M+H]+, 697 [2M+Na]+.
Boc-D-Trp-D-Trp-NH2
Z-D-Trp-NH2 (3 g; 8.9 mmol; 1 eq.) was dissolved in DMF (100 ml). HCl 36% (845 μl; 1.1 eq.), water (2 ml) and palladium on activated charcoal (95 mg, 0.1 eq.) were added to the stirred mixture. The solution was bubbled under hydrogen for 24 hr. When the reaction went to completion, the palladium was filtered on celite. The solvent was removed in vacuo to afford HCl, H-D-Trp-NH2as a colorless oil.
In 10 ml of DMF, HCl, H-D-Trp-NH2 (8.9 mmol; 1 eq.), Boc-D-Trp-OH (2.98 g; 9.8 mmol; 1.1 eq.), NMM (2.26 ml; 2.1 eq.) and BOP (4.33 g; 1.1 eq.) were added successively. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (100 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo to afford 4.35 g of Boc-D-Trp-D-Trp-NH2 as a white solid.
Yield=85%.
C27H31N5O4, 489 g.mol−1.
Rf=0.48 {Chloroform/Methanol/Acetic Acid (85/10/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.28 (s, 9H, Boc); 2.75-3.36 (m, 4H, 2 (CH2)β; 4.14 (m, 1H, CHα); 4.52 (m, 1H, CHα′); 6.83-7.84 (m, 14H, 2 indoles (10H), NH2, NH (urethane) and NH (amide)); 10.82 (d, 1H, J=2 Hz, N1H); 10.85 (d, 1H, J=2 Hz, N1H).
Mass Spectrometry (Electrospray), m/z 490 [M+H]+, 512 [M+Na]+, 979 [2M+H]+.
Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2
Boc-D-Trp-D-Trp-NH2 (3 g; 6.13 mmol; 1 eq.) was dissolved in acetonitrile (25 ml).
To this solution, di-tert-butyl-dicarbonate (3.4 g; 2.5 eq.) and 4-dimethylaminopyridine (150 mg; 0.2 eq.) were successively added. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate/hexane {5/5} to afford 2.53 g of Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2 as a white solid.
Yield=60%.
C37H47N5O8, 689 g.mol−1.
Rf=0.23 {ethyl acetate/hexane (5/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.25 (s, 9H, Boc); 1.58 (s, 9H, Boc); 1.61 (s, 9H, Boc); 2.75-3.4 (m, 4H, 2 (CH2)β); 4.2 (m, 1H, CHα′); 4.6 (m, 1H, CHα); 7.06-8 (m, 14H, 2 indoles (10H), NH (urethane), NH and NH2 (amides)).
Mass Spectrometry (Electrospray), m/z 690 [M+H]+, 712 [M+Na]+, 1379 [2M+H]+, 1401 [2M+Na]+.
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-H
Boc-D-(NiBoc)Trp-D-(NiBoc)Trp-NH2 (3 g; 4.3 mmol; 1 eq.) was dissolved in the mixture DMF/water (18 ml/7 ml). Then, pyridine (772 μl; 2.2 eq.) and Bis(Trifluoroacetoxy)IodoBenzene (2.1 g; 1.1 eq.) were added. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and aqueous saturated sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. Boc-D-NiBoc)Trp-D-g(NiBoc)Trp-H was used immediately for the next reaction of formylation.
Rf=0.14 {ethyl acetate/hexane (7/3)}.
C36H47N5O7, 661 g.mol−1.
1H NMR (200 MHZ, DMSO-d6): δ 1.29 (s, 9H, Boc); 1.61 (s, 18H, 2 Boc); 2.13 (s, 2H, NH2 (amine)); 3.1-2.8 (m, 4H, 2 (CH2)β); 4.2 (m, 1H, CHα′); 4.85 (m, 1H, CHα); 6.9-8 (m, 12H, 2 indoles (10H), NH (urethane), NH (amide)).
Mass Spectrometry (Electrospray), m/z 662 [M+H]+, 684 [M+Na]+.
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-H (4.3 mmol; 1 eq.) was dissolved in DMF (20 ml). Then, N,N-diisopropylethylamine (815 μl; 1.1 eq.) and 2,4,5-trichlorophenylformate (1.08 g; 1.1 eq.) were added. After 30 minutes, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate/hexane {5/5} to afford 2.07 g of Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO as a white solid.
Yield=70%.
C37H47N5O8, 689 g.mol−1.
Rf=0.27 {ethyl acetate/hexane (5/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.28 (s, 9H, Boc); 1.6 (s, 9H, Boc); 1.61 (s, 9H, Boc); 2.75-3.1 (m, 4H, 2 (CH2)β); 4.25 (m, 1H, (CH)αA&B); 5.39 (m, 0.4H, (CH)α′B); 5.72 (m, 0.6H, (CH)α′A); 6.95-8.55 (m, 14H, 2 indoles (10H), NH (urethane), 2 NH (amides), CHO (formyl)).
Mass Spectrometry (Electrospray), m/z 690 [M+H]+, 712 [M+Na]+, 1379 [2M+H]+.
Boc-Aib-D-Trp-D-gTrp-CHO
Boc-D-(NiBoc)Trp-D-g(NiBoc)Trp-CHO (1.98 g; 2.9 mmol; 1 eq.) was dissolved in a -mixture of trifluoroacetic acid (16 ml), anisole (2 ml) and thioanisole (2 ml) for 30 minutes at 0° C. The solvents were removed in vacuo, the residue was stirred with ether and the precipitated TFA, H-D-Trp-D-gTrp-CHO was filtered.
TFA, H-D-Trp-D-gTrp-CHO (2.9 mmol; 1 eq.), Boc-Aib-OH (700 mg; 1 eq.), NMM (2.4 ml; 4.2 eq.) and BOP (1.53 g; 1.2 eq.) were successively added in 10 ml of DMF. After 1 hr, the mixture was diluted with ethyl acetate (100 ml) and washed with saturated aqueous sodium hydrogen carbonate (200 ml), aqueous potassium hydrogen sulfate (200 ml, 1M), and saturated aqueous sodium chloride (200 ml). The organic layer was dried over sodium sulfate, filtered and the solvent removed in vacuo. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate to afford 1.16 g of Boc-Aib-D-Trp-D-gTrp-CHO as a white solid.
Yield=70%.
C31H38N6O5, 574 g.mol−1.
Rf=0.26 {Chloroform/Methanol/Acetic Acid (180/10/5)}.
1H NMR (200 MHZ, DMSO-d6): δ 1.21 (s, 6H, 2 CH3(Aib)); 1.31 (s, 9H, Boc); 2.98-3.12 (m, 4H, 2 (CH2)β); 4.47 (m, 1H, (CH)αA&B); 5.2 (m, 0.4H, (CH)α′B); 5.7 (m, 0.6H, (CH)α′A); 6.95-8.37 (m, 15H, 2 indoles (10H), 3 NH (amides), 1 NH (urethane) CHO (formyl)); 10.89 (m, 2H, 2 N1H (indoles)).
Mass Spectrometry (Electrospray), ml/z 575 [M+H]+, 597 [M+Na]+, 1149 [2M+H]+, 1171 [2M+Na]+.
H-Aib-D-Trp-D-gTrT-CHO
Boc-Aib-D-Trp-D-gTrp-CHO (1 g; 1.7 nmmol) was dissolved in a mixture of trifluoroacetic acid (8 ml), anisole (1 ml) and thioanisole (1 ml) for 30 minutes at 0° C. The solvents were removed in vacuo, the residue was stirred with ether and the precipitated TFA, H-Aib-D-Trp-D-gTrp-CHO was filtered.
The product TFA, H-Aib-D-Trp-D-gTrp-CHO was purified by preparative HPLC (Waters, delta pak, C18, 40×100 mm, 5 μm, 100 A).
Yield=52%.
C26H30N6O3, 474 g.mol−1.
1H NMR (400 MHZ, DMSO-d6)+1H/1H correlation: δ 1.21 (s, 3H, CH3 (Aib)); 1.43 (s, 3H, CH3 (Aib)); 2.97 (m, 2H, (CH2)β); 3.1 (m, 2H, (CH2)β′); 4.62 (m, 1H, (CH)αA&B); 5.32 (q, 0.4H, (CH)α′B); 5.71 (q, 0.6H, (CH)α′A); 7.3 (m, 4H5 and H6(2 indoles)); 7.06-7.2 (4d, 2H, H2A et H2B (2 indoles)); 7.3 (m, 2H, H4 or H7 (2 indoles)); 7.6-7.8 (4d, 2H, H4A and H4B or H7A et H7B); 7.97 (s, 3H, NH2 (Aib) and CHO (Formyl));8.2 (d, 0.4H, NH1B (diamino)); 8.3 (m,1H, NHA&B); 8.5 (d, 0.6H, NH1A (diamino)); 8.69 (d, 0.6H, NH2A (diamino)); 8.96 (d, 0.4H, NH2B (diamino)); 10.8 (s, 0.6H, N1H1A (indole)); 10.82 (s, 0.4H, N1H1B (indole)); 10.86 (s, 0.6H, N1H2A (indole)); 10.91 (s, 0.4, N1H2B (indole)).
Mass Spectrometry (Electrospray), m/z 475 [M+H]+, 949 [2M+H]+.
CLIP

CLIP

CLIP
UPDATED INFO AS ON JAN 6 2014
Aeterna Zentaris NDA for Macimorelin Acetate in AGHD Accepted for Filing by the FDA
Quebec City, Canada, January 6, 2014 – Aeterna Zentaris Inc. (NASDAQ: AEZS) (TSX: AEZS) (the “Company”) today announced that the U.S. Food and Drug Administration (“FDA”) has accepted for filing the Company’s New Drug Application (“NDA”) for its ghrelin agonist, macimorelin acetate, in Adult Growth Hormone Deficiency (“AGHD”). The acceptance for filing of the NDA indicates the FDA has determined that the application is sufficiently complete to permit a substantive review.
The Company’s NDA, submitted on November 5, 2013, seeks approval for the commercialization of macimorelin acetate as the first orally-administered product that induces growth hormone release to evaluate AGHD. Phase 3 data have demonstrated the compound to be well tolerated, with accuracy comparable to available intravenous and intramuscular testing procedures. The application will be subject to a standard review and will have a Prescription Drug User Fee Act (“PDUFA”) date of November 5, 2014. The PDUFA date is the goal date for the FDA to complete its review of the NDA.
David Dodd, President and CEO of Aeterna Zentaris, commented, “The FDA’s acceptance of this NDA submission is another significant milestone in our strategy to commercialize macimorelin acetate as the first approved oral product for AGHD evaluation. We are finalizing our commercial plan for this exciting new product. We are also looking to broaden the commercial application of macimorelin acetate in AGHD for use related to traumatic brain injury victims and other developmental areas, which would represent significant benefit to the evaluation of growth hormone deficiency, while presenting further potential revenue growth opportunities for the Company.”
About Macimorelin Acetate
Macimorelin acetate, a ghrelin agonist, is a novel orally-active small molecule that stimulates the secretion of growth hormone. The Company has completed a Phase 3 trial for use in evaluating AGHD, and has filed an NDA to the FDA in this indication. Macimorelin acetate has been granted orphan drug designation by the FDA for use in AGHD. Furthermore, macimorelin acetate is in a Phase 2 trial as a treatment for cancer-induced cachexia. Aeterna Zentaris owns the worldwide rights to this novel patented compound.
About AGHD
AGHD affects about 75,000 adults across the U.S., Canada and Europe. Growth hormone not only plays an important role in growth from childhood to adulthood, but also helps promote a hormonally-balanced health status. AGHD mostly results from damage to the pituitary gland. It is usually characterized by a reduction in bone mineral density, lean mass, exercise capacity, and overall quality of life.
About Aeterna Zentaris
Aeterna Zentaris is a specialty biopharmaceutical company engaged in developing novel treatments in oncology and endocrinology. The Company’s pipeline encompasses compounds from drug discovery to regulatory approval.
References
- ^ “Macrilen Prescribing Information” (PDF). Retrieved 2018-07-25.
- ^ “Aeterna Zentaris NDA for Macimorelin Acetate in AGHD Accepted for Filing by the FDA”. Wall Street Journal. January 6, 2014.
- ^ https://clinicaltrials.gov/ct2/show/NCT02558829
- ^ Research, Center for Drug Evaluation and. “Drug Approvals and Databases – Drug Trials Snapshots: Marcrilen”. http://www.fda.gov. Retrieved 2018-07-25.
- ^ “Macimorelin”. NCI Drug Dictionary. National Cancer Institute.
- ^ Koch, Linda (2013). “Growth hormone in health and disease: Novel ghrelin mimetic is safe and effective as a GH stimulation test”. Nature Reviews Endocrinology. 9 (6): 315. doi:10.1038/nrendo.2013.89.
- ^ Garcia, J. M.; Swerdloff, R.; Wang, C.; Kyle, M.; Kipnes, M.; Biller, B. M. K.; Cook, D.; Yuen, K. C. J.; Bonert, V.; Dobs, A.; Molitch, M. E.; Merriam, G. R. (2013). “Macimorelin (AEZS-130)-Stimulated Growth Hormone (GH) Test: Validation of a Novel Oral Stimulation Test for the Diagnosis of Adult GH Deficiency”. Journal of Clinical Endocrinology & Metabolism. 98 (6): 2422. doi:10.1210/jc.2013-1157. PMC 4207947.
Macimorelin
|
| Names |
| IUPAC name
2-Amino-N-[(2R)-1-[[(1R)-1-formamido-2-(1H-indol-3-yl)ethyl]amino]-3-1H-indol-3-yl)-1-oxopropan-2-yl]-2-methylpropanamide
|
| Other names
Aib-Trp-gTrp-CHO; AEZS-130; JMV 1843; Macimorelin acetate
|
| Identifiers |
|
|
|
|
|
|
| ChemSpider |
|
| KEGG |
|
|
|
|
| UNII |
|
|
|
|
|
| Properties |
|
|
C26H30N6O3 |
| Molar mass |
474.565 g·mol−1 |
| Pharmacology |
|
|
V04CD06 (WHO) |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
FDA
https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/205598Orig1s000ChemR.pdf

///////////macimorelin, FDA 2017, Aeterna Zentaris, AEZS-130, ARD-07, D-87875, EP-01572, EP-1572, JMV-1843, USAN (ab-26), MACIMORELIN ACETATE, orphan drug designation
CC(O)=O.CC(C)(N)C(=O)N[C@H](CC1=CNC2=CC=CC=C12)C(=O)N[C@H](CC1=CNC2=CC=CC=C12)NC=O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....



























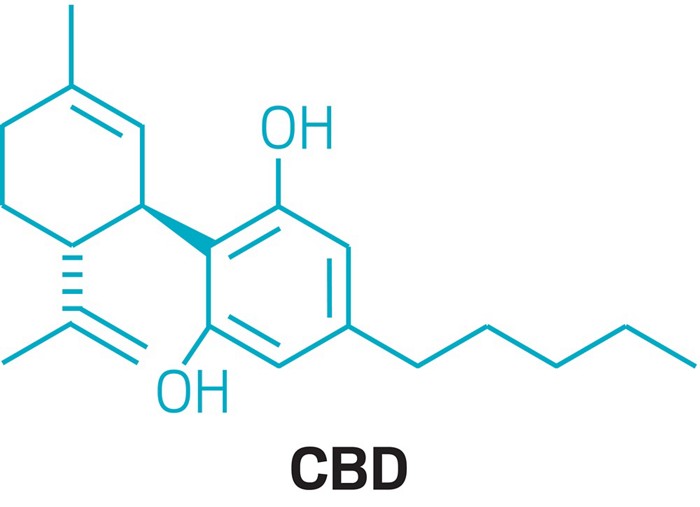
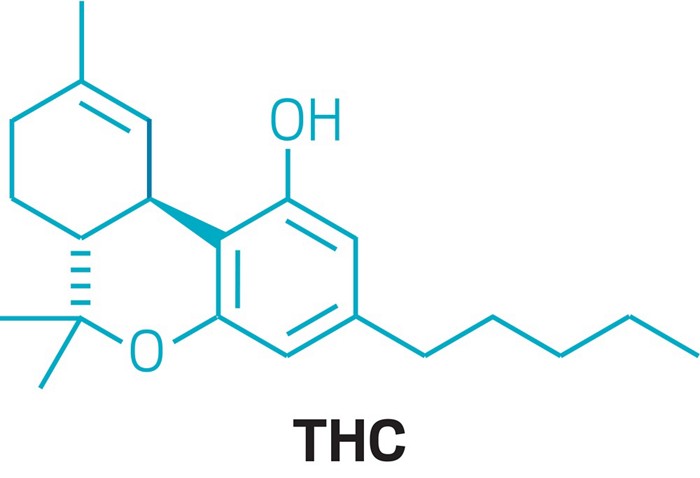





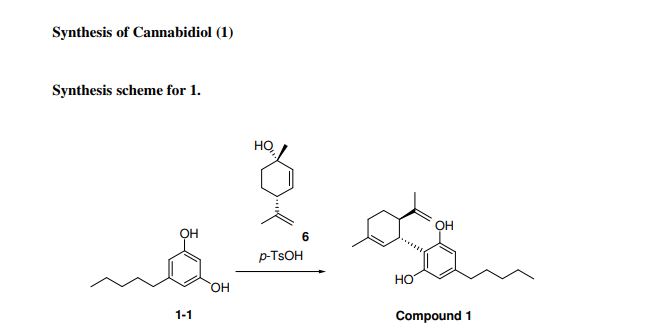
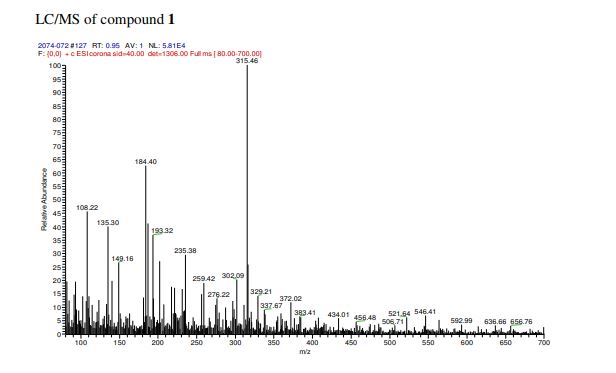























 One mechanistic explanation proposed by Lee and coworkers (1).
One mechanistic explanation proposed by Lee and coworkers (1).


{kind=link}
{kind=link}
{kind=link}