
Home » anaesthetics
Category Archives: anaesthetics
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Cipepofol
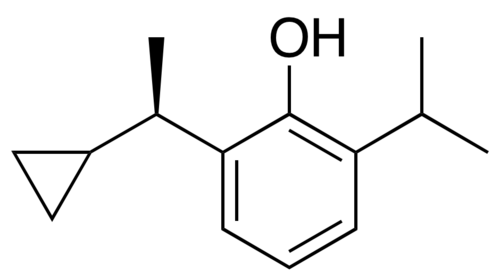
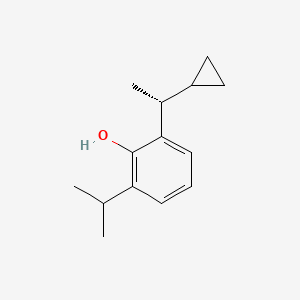
Cipepofol
CAS1637741-58-2
MW 204.31 g/mol MF C14H20O
2-[(1R)-1-cyclopropylethyl]-6-propan-2-ylphenol
FDA 2026, APPROVALS 2026, Cypsedo, HSK 3486, CS-0064163, GTPL 10812, HSK-3486, HY-116152, M3WGS532VY
- OriginatorSichuan Haisco Pharmaceutical
- ClassCyclopropanes; General anaesthetics; Phenols; Small molecules
- Mechanism of ActionGABA A receptor agonists
- RegisteredAnaesthesia; Sedation
- 10 Apr 2026Sichuan Haisco Pharmaceutical plans a phase III trial for Anesthesia (In Children, In adolescents) (IV) in May 2026 (NCT07510945)
- 28 Aug 2024No recent reports of development identified for preclinical development in Sedation in USA (IV, Infusion)
- 01 Aug 2024Zhongda Hospital plans a clinical trial for Sedation (IV) in August 2024 (NCT06538883)
To induce general anesthesia in adults undergoing surgery
Cipepofol (also known as ciprofol or HSK3486) is a novel, short-acting intravenous anesthetic and sedative. As a structural analog of propofol, it targets \(GABA_{A}\) receptors but is 4 to 6 times more potent. It offers faster recovery, improved cardiovascular stability, and significantly less injection pain than propofol.
Key Clinical Advantages
- Superior Efficacy: Requires a lower dose to achieve the same sedative depth as propofol.
- Better Safety Profile: Associated with a lower incidence of injection pain, reduced respiratory depression, and better hemodynamic (blood pressure) stability.
- Fast Acting: Characterized by rapid onset and quick recovery times, making it ideal for procedures like gastrointestinal endoscopy, bronchoscopy, and general anesthesia induction.
Recent Developments
- FDA Approval: Cipepofol (sold under the brand name CYPSEDO) officially received U.S. FDA marketing approval, becoming the first China-originated innovative intravenous anesthetic to enter the global market.
- Ongoing Trials: Clinical trials and post-marketing studies are actively evaluating its safety in specific populations, such as elderly patients and children.
Cipepofol (INNTooltip International Nonproprietary Name, USANTooltip United States Adopted Name), also known as ciprofol or by its developmental code name HSK3486, is a general anesthetic related to propofol which is used for anesthesia and sedation.[1][2][3][4] The drug is used by intravenous infusion.[1] A short-acting and highly selective γ-aminobutyric acid positive allosteric modulator,[5] ciprofol is 4 to 6 times more potent than other phenol derivatives such as propofol or fospropofol.[6]
In May 2026, cipepofol was approved by the US FDA.[7] Manufactured by Haisco Pharmaceutical Group of Chengdu, Sichuan, China, ciprofol underwentphase I and II trials in Australia and China.[8][9][10] In these early studies, ciprofol was comparable in efficacy to propofol and was associated with fewer adverse events.[4][6][11][12][13][14][15][16][17][18]
Physical properties
Ciprofol is an optically active 2,6-disubstituted alkylphenol with a cyclopropylethyl group incorporated at the second carbon atom. This cyclopropyl group increases the steric effects and introduces stereoselective effects over its anesthetic properties. These properties appear to increase the anesthetic potency of ciprofol, when compared with propofol.[9]
Medical use
Ciprofol is used for the intravenous induction of general anesthesia.[3][4] Studies published in 2022 and 2023 found it was efficacious as a general anesthetic in patients undergoing gynecological surgery[6][11] and kidney transplantation,[19] as well as for endoscopic procedures such as bronchoscopy,[15][20] esophagogastroduodenoscopy and colonoscopy.[21][22]
Ciprofol has also been used for sedation of critically ill patients undergoing mechanical ventilation in the intensive care unit,[23] as well as for the treatment of agitation and delirium in that patient population.[24] When combined with mild therapeutic hypothermia, ciprofol may also be useful as a cerebral protective agent in the setting of cerebral ischemia-reperfusion injury.[25]
Experimental use
In experimental models of isoproterenol-induced myocardial infarction (using mice as subjects), ciprofol appears to protect the heart against oxidative damage, inflammation and apoptosis of cardiac muscle cells.[26]
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US428011434&_cid=P12-MPW0XO-91017-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014180305&_cid=P12-MPW0R4-87054-1
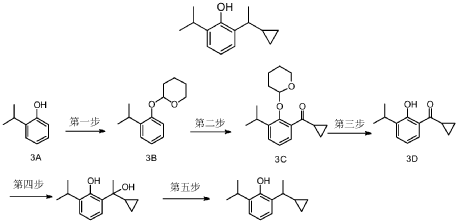
Example 16
[-cyclopropylethyl] -6 -isopropylphenol (compound 16)
2- [(lR)-l-cyclopropylethyl]-6-isopropyl -phenol
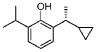
Preparation methods of Examples 16-17:
2-(1-Cyclopropylethyl-6-isopropylphenol (compound 3) 600 mg was used for resolution. Preparation conditions: (Instrument: Agilent 1260/CH-Y-J0404; Column: CHIRALPAK OJ-H (4.6 mm < 250 mm, 5 μm) No.: OJ-H-27; Mobile phase: A: isopropanol, B: n-hexane; Flow rate: 1.0 mL/min; Back pressure: 100 bar; Column temperature: 35°C; Wavelength: 210 nm; Period: 10 min)
Two optical isomers were obtained after separation: peak 1 (retention time: 10.72 min, 280 mg, pale yellow liquid, ee%=99%) and peak 2 (retention time: 13.58 min, 280 mg, pale yellow liquid, ee%=99%).
峰 1 : MS m/z(ESI): 203.1(Ml).
toMR (400 MHz,CDCl3 ) : δ 7.14(dd, 1H), δ 7.08(dd, 1H), 6.91 (t, 1H), 4.93 (s, 1H), 3.22-3.14(m, 1H), 2.55-2.48 (m, 1H), 1.33 (d, 6H), 1.28 (d, 3H), 1.10-1.05 (m, 1H), 0.60-0.58 (m, 1H), 0.49-0.46 (m, 1H), 0.25-0.18 (m, 2H).
峰 2: MS m/z(ESI): 203.1(Ml).
iHNMR (400 MHz,CDCl3) : 57.14(dd, 1H), δ 7.08(dd, 1H), 6.93 (t, 1H), 4.93 (s 1H), 3.22-3.15(m, 1H), 2.55-2.48 (m, 1H), 1.32 (d, 6H), 1.28 (d, 3H), 1.10-1.04 (m, 1H), 0.60-0.58 (m, 1H), 0.49-0.46 (m, 1H), 0.25-0.18 (m, 2H).
PAT
- Phenol derivative compound, methods of preparing said compound, pharmaceutical composition comprising said compound and use thereofPublication Number: BR-112015028212-B1Priority Date: 2013-05-09
- Phenol derivative and its preparation method and application in medicinePublication Number: CN-104507899-APriority Date: 2013-05-09
- Phenol derivative and preparation method and use in medicine thereofPublication Number: US-9517988-B2Priority Date: 2013-05-09Grant Date: 2016-12-13
- Phenol derivative and preparation method and use in medicine thereofPublication Number: EP-2995604-B1Priority Date: 2013-05-09Grant Date: 2019-07-10
- Phenol derivative, its production method and medicinal applicationPublication Number: JP-6431155-B2Priority Date: 2013-05-09Grant Date: 2018-11-28
- Phenol derivative, preparation method and medical application thereofPublication Number: CN-104507899-BPriority Date: 2013-05-09Grant Date: 2016-11-30
- Phenol derivative and method of preparation and medical use thereofPublication Number: ES-2746987-T3Priority Date: 2013-05-09Grant Date: 2020-03-09
- Phenol derivative and preparation method and use in medicine thereofPublication Number: US-2016060197-A1Priority Date: 2013-05-09
- Isopropyl phenol derivative and preparation method thereofPublication Number: WO-2016026459-A1Priority Date: 2014-08-22
- Phenol derivative and preparation method and use in medicine thereofPublication Number: AU-2014264103-B2Priority Date: 2013-05-09Grant Date: 2018-03-22
- Phenol derivative and preparation method and use in medicine thereofPublication Number: AU-2014264103-A1Priority Date: 2013-05-09
- Phenol derivative and preparation method and use in medicine thereofPublication Number: AU-2014264103-C1Priority Date: 2013-05-09Grant Date: 2018-08-02
- Phenol derivative and preparation method and use in medicine thereofPublication Number: EP-2995604-A1Priority Date: 2013-05-09
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

References
References
- “Sichuan Haisco Pharmaceutical”. AdisInsight. 28 August 2024. Retrieved 1 October 2025.
- “Ciprofol (Cipepofol): A γ-Aminobutyric Acid Receptor Agonist for Induction of Anesthesia”. Chemistry and Pharmacology of Drug Discovery. Wiley. 2024. pp. 251–274. doi:10.1002/9781394225156.ch12. ISBN 978-1-394-22512-5. Retrieved 1 October 2025.
- Wang X, Wang X, Liu J, Zuo YX, Zhu QM, Wei XC, et al. (March 2022). “Effects of ciprofol for the induction of general anesthesia in patients scheduled for elective surgery compared to propofol: a phase 3, multicenter, randomized, double-blind, comparative study”. European Review for Medical and Pharmacological Sciences. 26 (5): 1607–1617. PMID 35302207.
- Zeng Y, Wang DX, Lin ZM, Liu J, Wei XC, Deng J, et al. (February 2022). “Efficacy and safety of HSK3486 for the induction and maintenance of general anesthesia in elective surgical patients: a multicenter, randomized, open-label, propofol-controlled phase 2 clinical trial”. European Review for Medical and Pharmacological Sciences. 26 (4): 1114–1124. PMID 35253166.
- Liao J, Li M, Huang C, Yu Y, Chen Y, Gan J, et al. (2022). “Pharmacodynamics and Pharmacokinetics of HSK3486, a Novel 2,6-Disubstituted Phenol Derivative as a General Anesthetic”. Frontiers in Pharmacology. 13 830791. doi:10.3389/fphar.2022.830791. PMC 8851058. PMID 35185584.
- Chen BZ, Yin XY, Jiang LH, Liu JH, Shi YY, Yuan BY (August 2022). “The efficacy and safety of ciprofol use for the induction of general anesthesia in patients undergoing gynecological surgery: a prospective randomized controlled study”. BMC Anesthesiology. 22 (1) 245. doi:10.1186/s12871-022-01782-7. PMC 9347095. PMID 35922771.
- “Novel Drug Approvals for 2026”. U.S. Food and Drug Administration. 29 May 2026. Retrieved 31 May 2026.
- Lu M, Liu J, Wu X, Zhang Z (2023). “Ciprofol: A Novel Alternative to Propofol in Clinical Intravenous Anesthesia?”. BioMed Research International. 2023 7443226. doi:10.1155/2023/7443226. PMC 9879693. PMID 36714027.
- Qin L, Ren L, Wan S, Liu G, Luo X, Liu Z, et al. (May 2017). “Design, Synthesis, and Evaluation of Novel 2,6-Disubstituted Phenol Derivatives as General Anesthetics”. Journal of Medicinal Chemistry. 60 (9): 3606–3617. doi:10.1021/acs.jmedchem.7b00254. PMID 28430430.
- Nair A, Seelam S (2022). “Ciprofol- a game changing intravenous anesthetic or another experimental drug!”. Saudi Journal of Anaesthesia. 16 (2): 258–259. doi:10.4103/sja.sja_898_21. PMC 9009555. PMID 35431734.
- Man Y, Xiao H, Zhu T, Ji F (March 2023). “Study on the effectiveness and safety of ciprofol in anesthesia in gynecological day surgery: a randomized double-blind controlled study”. BMC Anesthesiology. 23 (1) 92. doi:10.1186/s12871-023-02051-x. PMC 10039513. PMID 36964501.
- Chen X, Guo P, Yang L, Liu Z, Yu D (2022). “Comparison and Clinical Value of Ciprofol and Propofol in Intraoperative Adverse Reactions, Operation, Resuscitation, and Satisfaction of Patients under Painless Gastroenteroscopy Anesthesia”. Contrast Media & Molecular Imaging. 2022 9541060. doi:10.1155/2022/9541060. PMC 9314164. PMID 35935320.
- Zhong J, Zhang J, Fan Y, Zhu M, Zhao X, Zuo Z, et al. (May 2023). “Efficacy and safety of Ciprofol for procedural sedation and anesthesia in non-operating room settings”. Journal of Clinical Anesthesia. 85 111047. doi:10.1016/j.jclinane.2022.111047. PMID 36599219. S2CID 255468218.
- Liang P, Dai M, Wang X, Wang D, Yang M, Lin X, et al. (June 2023). “Efficacy and safety of ciprofol vs. propofol for the induction and maintenance of general anaesthesia: A multicentre, single-blind, randomised, parallel-group, phase 3 clinical trial”. European Journal of Anaesthesiology. 40 (6): 399–406. doi:10.1097/EJA.0000000000001799. PMC 10155686. PMID 36647565.
- Luo Z, Tu H, Zhang X, Wang X, Ouyang W, Wei X, et al. (March 2022). “Efficacy and Safety of HSK3486 for Anesthesia/Sedation in Patients Undergoing Fiberoptic Bronchoscopy: A Multicenter, Double-Blind, Propofol-Controlled, Randomized, Phase 3 Study”. CNS Drugs. 36 (3): 301–313. doi:10.1007/s40263-021-00890-1. PMC 8927014. PMID 35157236.
- Hu C, Ou X, Teng Y, Shu S, Wang Y, Zhu X, et al. (November 2021). “Sedation Effects Produced by a Ciprofol Initial Infusion or Bolus Dose Followed by Continuous Maintenance Infusion in Healthy Subjects: A Phase 1 Trial”. Advances in Therapy. 38 (11): 5484–5500. doi:10.1007/s12325-021-01914-4. PMC 8523013. PMID 34559359.
- Teng Y, Ou M, Wang X, Zhang W, Liu X, Liang Y, et al. (September 2021). “Efficacy and safety of ciprofol for the sedation/anesthesia in patients undergoing colonoscopy: Phase IIa and IIb multi-center clinical trials”. European Journal of Pharmaceutical Sciences. 164 105904. doi:10.1016/j.ejps.2021.105904. PMID 34116176.
- Zhu Q, Luo Z, Wang X, Wang D, Li J, Wei X, et al. (April 2023). “Efficacy and safety of ciprofol versus propofol for the induction of anesthesia in adult patients: a multicenter phase 2a clinical trial”. International Journal of Clinical Pharmacy. 45 (2): 473–482. doi:10.1007/s11096-022-01529-x. PMC 10147789. PMID 36680620.
- Qin K, Qin WY, Ming SP, Ma XF, Du XK (July 2022). “Effect of ciprofol on induction and maintenance of general anesthesia in patients undergoing kidney transplantation”. European Review for Medical and Pharmacological Sciences. 26 (14): 5063–5071. PMID 35916802.
- Wu B, Zhu W, Wang Q, Ren C, Wang L, Xie G (2022). “Efficacy and safety of ciprofol-remifentanil versus propofol-remifentanil during fiberoptic bronchoscopy: A prospective, randomized, double-blind, non-inferiority trial”. Frontiers in Pharmacology. 13 1091579. doi:10.3389/fphar.2022.1091579. PMC 9812563. PMID 36618929.
- Li J, Wang X, Liu J, Wang X, Li X, Wang Y, et al. (August 2022). “Comparison of ciprofol (HSK3486) versus propofol for the induction of deep sedation during gastroscopy and colonoscopy procedures: A multi-centre, non-inferiority, randomized, controlled phase 3 clinical trial”. Basic & Clinical Pharmacology & Toxicology. 131 (2): 138–148. doi:10.1111/bcpt.13761. PMC 9543620. PMID 35653554.
- Long YQ, Feng CD, Ding YY, Feng XM, Liu H, Ji FH, et al. (2022). “Esketamine as an Adjuvant to Ciprofol or Propofol Sedation for Same-Day Bidirectional Endoscopy: Protocol for a Randomized, Double-Blind, Controlled Trial With Factorial Design”. Frontiers in Pharmacology. 13 821691. doi:10.3389/fphar.2022.821691. PMC 8975265. PMID 35370640.
- Liu Y, Yu X, Zhu D, Zeng J, Lin Q, Zang B, et al. (May 2022). “Safety and efficacy of ciprofol vs. propofol for sedation in intensive care unit patients with mechanical ventilation: a multi-center, open label, randomized, phase 2 trial”. Chinese Medical Journal. 135 (9): 1043–1051. doi:10.1097/CM9.0000000000001912. PMC 9276409. PMID 34924506.
- Liu GL, Wu GZ, Ge D, Zhou HJ, Cui S, Gao K, et al. (2023). “Efficacy and safety of ciprofol for agitation and delirium in the ICU: A multicenter, single-blind, 3-arm parallel randomized controlled trial study protocol”. Frontiers in Medicine. 9 1024762. doi:10.3389/fmed.2022.1024762. PMC 9868613. PMID 36698817.
- Wang YC, Wu MJ, Zhou SL, Li ZH (January 2023). “Protective effects of combined treatment with ciprofol and mild therapeutic hypothermia during cerebral ischemia-reperfusion injury”. World Journal of Clinical Cases. 11 (3): 487–492. doi:10.12998/wjcc.v11.i3.487. PMC 9923870. PMID 36793629.
- Yang Y, Xia Z, Xu C, Zhai C, Yu X, Li S (2022). “Ciprofol attenuates the isoproterenol-induced oxidative damage, inflammatory response and cardiomyocyte apoptosis”. Frontiers in Pharmacology. 13 1037151. doi:10.3389/fphar.2022.1037151. PMC 9723392. PMID 36483733.
- Vittori A, Di Fabio C, Cascella M, Marinangeli F, Francia E, Mascilini I, et al. (January 2026). “Advantages of Ciprofol with Special Consideration of Pediatric Anesthesia”. Children (Basel, Switzerland). 13 (2). doi:10.3390/children13020188. PMC 12939459. PMID 41749542.
- Liu SB, Yao X, Tao J, Yang JJ, Zhao YY, Liu DW, et al. (March 2023). “Population total and unbound pharmacokinetics and pharmacodynamics of ciprofol and M4 in subjects with various renal functions”. British Journal of Clinical Pharmacology. 89 (3): 1139–1151. doi:10.1111/bcp.15561. PMID 36217805. S2CID 252818288.
- Hu Y, Li X, Liu J, Chen H, Zheng W, Zhang H, et al. (December 2022). “Safety, pharmacokinetics and pharmacodynamics of a novel γ-aminobutyric acid (GABA) receptor potentiator, HSK3486, in Chinese patients with hepatic impairment”. Annals of Medicine. 54 (1): 2769–2780. doi:10.1080/07853890.2022.2129433. PMC 9559057. PMID 36217101.
- Li X, Yang D, Li Q, Wang H, Wang M, Yan P, et al. (2021). “Safety, Pharmacokinetics, and Pharmacodynamics of a Single Bolus of the γ-aminobutyric Acid (GABA) Receptor Potentiator HSK3486 in Healthy Chinese Elderly and Non-elderly”. Frontiers in Pharmacology. 12 735700. doi:10.3389/fphar.2021.735700. PMC 8430033. PMID 34512361.
- Ding YY, Long YQ, Yang HT, Zhuang K, Ji FH, Peng K (December 2022). “Efficacy and safety of ciprofol for general anaesthesia induction in elderly patients undergoing major noncardiac surgery: A randomised controlled pilot trial”. European Journal of Anaesthesiology. 39 (12): 960–963. doi:10.1097/EJA.0000000000001759. PMID 36214498. S2CID 252779399.
- Duan G, Lan H, Shan W, Wu Y, Xu Q, Dong X, et al. (April 2023). “Clinical effect of different doses of ciprofol for induction of general anesthesia in elderly patients: A randomized, controlled trial”. Pharmacology Research & Perspectives. 11 (2) e01066. doi:10.1002/prp2.1066. PMC 9944862. PMID 36811327. S2CID 257098376.
- Yang Y, Xia Z, Xu C, Zhai C, Yu X, Li S (2022). “Ciprofol attenuates the isoproterenol-induced oxidative damage, inflammatory response and cardiomyocyte apoptosis”. Frontiers in Pharmacology. 13 1037151: 1037151. doi:10.3389/fphar.2022.1037151. PMC 9723392. PMID 36483733.
- Bian Y, Zhang H, Ma S, Jiao Y, Yan P, Liu X, et al. (January 2021). “Mass balance, pharmacokinetics and pharmacodynamics of intravenous HSK3486, a novel anaesthetic, administered to healthy subjects”. British Journal of Clinical Pharmacology. 87 (1): 93–105. doi:10.1111/bcp.14363. PMID 32415708. S2CID 218658207.
Further reading
- Bajwa SJ, Vinayagam S, Shinde S, Dalal S, Vennel J, Nanda S (January 2023). “Recent advancements in total intravenous anaesthesia and anaesthetic pharmacology”. Indian Journal of Anaesthesia. 67 (1): 56–62. doi:10.4103/ija.ija_1022_22. PMC 10034929. PMID 36970470.
- Skiljic S, Budrovac D, Cicvaric A, Neskovic N, Kvolik S (February 2023). “Advances in Analgosedation and Periprocedural Care for Gastrointestinal Endoscopy”. Life. 13 (2): 473. Bibcode:2023Life…13..473S. doi:10.3390/life13020473. PMC 9962362. PMID 36836830.
- Wei A, Yang L, Ma S, Jin G, Yang M, Zhou J (November 2022). “A case report of ciprofol overdose during anesthesia/analgesia and literature review: clinical presentation, blood pressure, and management”. The Journal of International Medical Research. 50 (11) 3000605221132466. doi:10.1177/03000605221132466. PMC 9659933. PMID 36366740.
| Clinical data | |
|---|---|
| Other names | Ciprofol; CS-0064163; CS0064163; GTPL10812; GTPL-10812; HSK-3486; HSK3486; HY-116152; HY116152; (R)-2-(1-Cyclopropylethyl)-6-isopropylphenol |
| Routes of administration | Intravenous infusion[1] |
| Drug class | GABAA receptor positive allosteric modulator |
| Pharmacokinetic data | |
| Metabolism | Liver glucuronidation |
| Excretion | Kidney |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1637741-58-2 |
| PubChem CID | 86301664 |
| DrugBank | DB16295 |
| ChemSpider | 76794458 |
| UNII | M3WGS532VY |
| KEGG | D12449 |
| ChEMBL | ChEMBL4094894 |
| Chemical and physical data | |
| Formula | C14H20O |
| Molar mass | 204.313 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
////////cipepofol, FDA 2026, APPROVALS 2026, Cypsedo, HSK 3486, CS-0064163, GTPL 10812, HSK-3486, HY-116152, M3WGS532VY, ANAESTHETIC
PRILOCAINE

PRILOCAINE
- Molecular FormulaC13H20N2O
- Average mass220.311 Da
Propanamide, N-(2-methylphenyl)-2-(propylamino)-
прилокаин [Russian] [INN]
بريلوكائين [Arabic] [INN]
丙胺卡因 [Chinese] [INN]
1641
211-957-0[EINECS]
721-50-6[RN]
N-(2-Methylphenyl)-2-(propylamino)propanamid
N-(2-méthylphényl)-2-(propylamino)propanamide
PrilocaineCAS Registry Number: 721-50-6
CAS Name:N-(2-Methylphenyl)-2-(propylamino)propanamide
Additional Names: 2-(propylamino)-o-propionotoluidide; N-(a-propylaminopropionyl)-o-toluidine; a-propylamino-2-methylpropionanilide; propitocaine
Molecular Formula: C13H20N2O
Molecular Weight: 220.31
Percent Composition: C 70.87%, H 9.15%, N 12.72%, O 7.26%
Literature References: Prepn: N. Löfgren, C. Tegner, Acta Chem. Scand.14, 486 (1960); GB839943; N. Löfgren, C. Tegner, US3160662 (1960, 1964 both to Astra).
Properties: Needles, mp 37-38°. bp0.1 159-162°. nD20 1.5298.
Melting point: mp 37-38°
Boiling point: bp0.1 159-162°
Index of refraction:nD20 1.5298 Derivative Type: Hydrochloride
CAS Registry Number: 1786-81-8
Manufacturers’ Codes: L-67
Trademarks: Citanest (AstraZeneca); Xylonest (AstraZeneca)
Molecular Formula: C13H20N2O.HCl
Molecular Weight: 256.77
Percent Composition: C 60.81%, H 8.24%, N 10.91%, O 6.23%, Cl 13.81%
Properties: Crystals from ethanol + isopropyl ether, mp 167-168°. Readily sol in water.
Melting point: mp 167-168° Therap-Cat: Anesthetic (local).Keywords: Anesthetic (Local).
- ASTRA 1512
- ASTRA 1515
- ASTRA-1512
- ASTRA-1515
- L 67
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Prilocaine hydrochloride | MJW015BAPH | 1786-81-8 | BJPJNTKRKALCPP-UHFFFAOYSA-N |
Agoneaze, Anodyne Lpt, Citanest, Citanest Forte, Dermacinrx Prikaan, Dermacinrx Prizopak, Emla, Fortacin, Lido Bdk, Lido-prilo Caine Pack, Lidopril, Oraqix, Prilolid, Prizotral, Relador
Prilocaine is a local anesthetic used in dental procedures.
A local anesthetic that is similar pharmacologically to lidocaine. Currently, it is used most often for infiltration anesthesia in dentistry. (From AMA Drug Evaluations Annual, 1992, p165)
Prilocaine (/ˈpraɪləˌkeɪn/[1]) is a local anesthetic of the amino amide type first prepared by Claes Tegner and Nils Löfgren. In its injectable form (trade name Citanest), it is often used in dentistry. It is also often combined with lidocaine as a topical preparation for dermal anesthesia (lidocaine/prilocaine or EMLA), for treatment of conditions like paresthesia. As it has low cardiac toxicity, it is commonly used for intravenous regional anaesthesia (IVRA).
Contraindications
In some patients, ortho-toluidine, a metabolite of prilocaine, may cause methemoglobinemia, which may be treated with methylene blue. Prilocaine may also be contraindicated in people with sickle cell anemia, anemia, or symptomatic hypoxia.[2]
Combinations
It is given as a combination with the vasoconstrictor epinephrine under the trade name Citanest Forte. It is used as an eutectic mixture with lidocaine, 50% w/w, as lidocaine/prilocaine. The mixture is an oil with a melting point of 18 °C (64 °F). A 5% emulsion preparation, containing 2.5% each of lidocaine/prilocaine, is marketed by APP Pharmaceuticals under the trade name EMLA (an abbreviation for eutectic mixture of local anesthetics).[3]
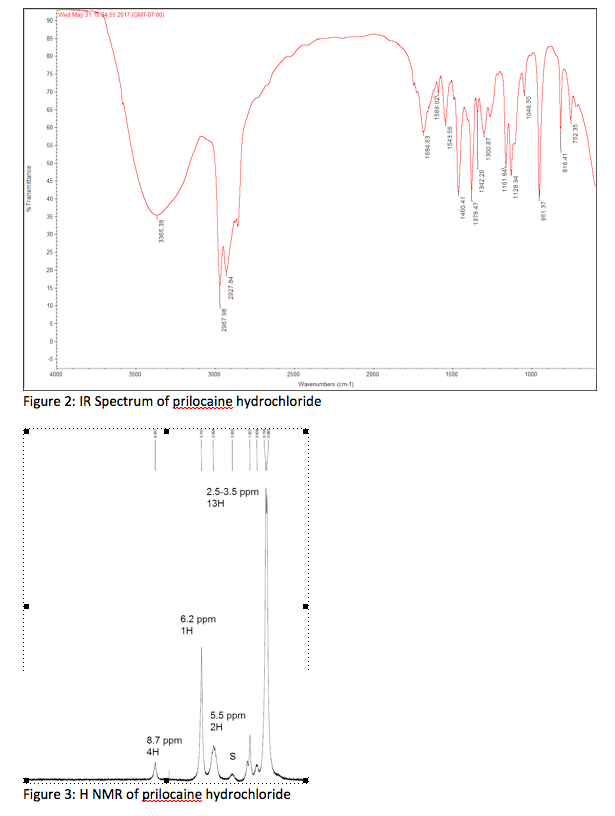
NMR
![1 H-nuclear magnetic resonance ( 1 H-NMR) spectra of prilocaine solution after sterilization with the assignment of the prilocaine hydrogens. [Prilocaine] = 5 mM, 20°C, 500 MHz.](https://www.researchgate.net/profile/Francisco-Groppo/publication/23458262/figure/fig3/AS:394557704425486@1471081295276/1-H-nuclear-magnetic-resonance-1-H-NMR-spectra-of-prilocaine-solution-after.png) 1 H-nuclear magnetic resonance ( 1 H-NMR) spectra of prilocaine solution after sterilization with the assignment of the prilocaine hydrogens. [Prilocaine] = 5 mM, 20°C, 500 MHz.
1 H-nuclear magnetic resonance ( 1 H-NMR) spectra of prilocaine solution after sterilization with the assignment of the prilocaine hydrogens. [Prilocaine] = 5 mM, 20°C, 500 MHz.
Compendial status

Table 1 The common types of local anesthetics
| COMPOUND | STRUCTURE | TIME TO MARKET | APPLICATION METHODS |
|---|---|---|---|
| Procaine | 1904 | Infiltration anesthesia, conduction anesthesia, subarachnoid anesthesia and epidural anesthesia | |
| Chloroprocaine | 1952 | Infiltration anesthesia, epidural anesthesia and conduction anesthesia | |
| Hydroxyprocaine | 1960 | Infiltration anesthesia | |
| Tetracaine | 1988 | Conduction anesthesia, subarachnoid anesthesia and epidural anesthesia | |
| Oxybuprocaine | 1975 | Topical anesthesia | |
| Tutocaine | 1976 | Topical anesthesia and infiltration anesthesia | |
| Butacaine | 1976 | Topical anesthesia and infiltration anesthesia | |
| Dimethocaine | 1938 | Topical anesthesia and infiltration anesthesia | |
| Thiocaine | Halt sales | Topical anesthesia and infiltration anesthesia | |
| Lidocaine | 1948 | Conduction anesthesia and epidural anesthesia | |
| Mepivacaine | 1986 | Infiltration anesthesia, conduction anesthesia, epidural anesthesia and topical anesthesia | |
| Bupivacaine | 2000 | Infiltration anesthesia, conduction anesthesia and epidural anesthesia | |
| Ropivacaine | 1996 | Infiltration anesthesia, conduction anesthesia and epidural anesthesia | |
| Trimecaine | 1965 | Infiltration anesthesia, surface anesthesia and epidural anesthesia | |
| Prilocaine | 1993 | Infiltration anesthesia, topical anesthesia and epidural anesthesia | |
| Etidocaine | 1976 | Epidural anesthesia | |
| Pyrrocaine | 1964 | Conduction anesthesia and epidural anesthesia | |
| Butanilicaine | 1982 | Infiltration anesthesia and conduction anesthesia | |
| Cinchocaine | 1985 | Topical anesthesia, subarachnoid anesthesia and epidural anesthesia | |
| Articaine | 2002 | Infiltration anesthesia and subarachnoid anesthesia | |
| Dyclonine | 1956 | Topical anesthesia | |
| Falicaine | 1957 | Topical anesthesia | |
| Quinisocaine | 1957 | Topical anesthesia | |
| Pramocaine | 1977 | Topical anesthesia | |
| Diperodon | 1980 | Topical anesthesia | |
| Heptacaine | 1984 | Infiltration anesthesia |
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Synthesis Reference
SYN
English: N. Lofgren and C. Tegner, Acta Chem. Scand., 14, 486 (1960). DOI number: 10.3891/acta.chem.scand.14-0486
SYN
SUN
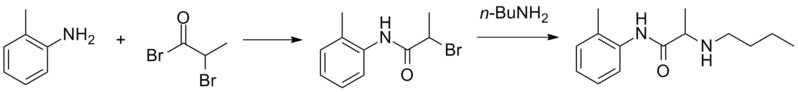
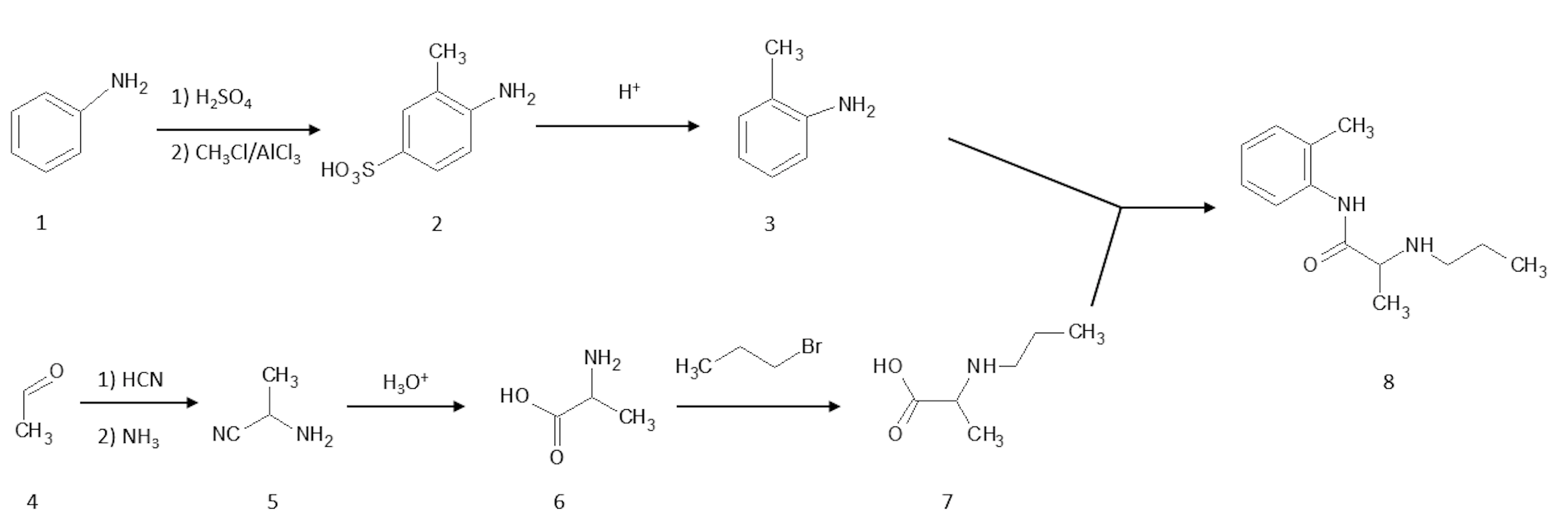
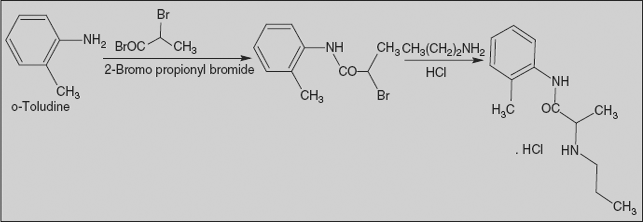
Prilocaine, 2-(propylamino)-o-propiontoluidine (2.2.14), is structurally related to the exact same group as ethidocaine, yet it differs structurally in that during synthesis, o-toluidine is used instead of 2,6-dimethylaniline, and instead of a butyric acid, a fragment of propionic acid, and a terminal propylethylamine group is replaced with a propylamine group. In order to synthesize prilocaine, o-toluidine is reacted with bromopropionyl bromide, and the resulting bromopropionyltoluidide (2.2.13) is then reacted with propylamine, which gives prilocaine [22,23].
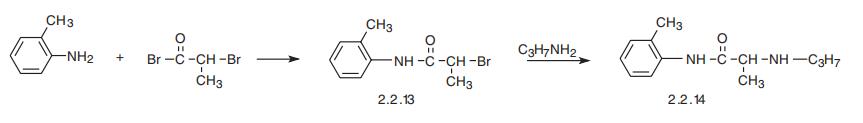
SYN

SYN
| Clinical data | |
|---|---|
| Trade names | Citanest |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a603026 |
| License data | EU EMA: by INNUS DailyMed: Prilocaine |
| Pregnancy category | AU: A |
| Routes of administration | Subcutaneous |
| ATC code | N01BB04 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Protein binding | 55% |
| Metabolism | Liver and kidney |
| Elimination half-life | 10-150 minutes, longer with impaired liver or kidney function |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 721-50-6 |
| PubChem CID | 4906 |
| IUPHAR/BPS | 7276 |
| DrugBank | DB00750 |
| ChemSpider | 4737 |
| UNII | 046O35D44R |
| KEGG | D00553 as HCl: D01243 |
| ChEBI | CHEBI:8404 |
| ChEMBL | ChEMBL1194 |
| CompTox Dashboard (EPA) | DTXSID7031955 |
| ECHA InfoCard | 100.010.871 |
| Chemical and physical data | |
| Formula | C13H20N2O |
| Molar mass | 220.316 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| Melting point | 37 to 38 °C (99 to 100 °F) |
| showSMILES | |
| showInChI | |
| (verify) |
References
- ^ “Prilocaine”. Merriam-Webster Dictionary. Retrieved 2016-01-21.
- ^ Patel V, Morrissey J (2011-09-15). Practical and Professional Clinical Skills. Oxford University Press. p. 267. ISBN 9780199585618.
- ^ “Topical Anesthesia Use in Children: Eutectic Mixture of Local Anesthetics”. Medscape.com. Retrieved 2014-01-07.
- ^ The United States Pharmacopeial Convention, Revision Bulletin: Lidocaine and Prilocaine Cream–Revision to Related Compounds Test, archived from the original on 5 July 2010, retrieved 10 July 2009
External links
- “Prilocaine”. Drug Information Portal. U.S. National Library of Medicine.
- “Prilocaine hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
//////////PRILOCAINE, Anesthetic, ASTRA 1512, ASTRA 1515, ASTRA-1512, ASTRA-1515, L 67,
CCCNC(C)C(=O)NC1=CC=CC=C1C

NEW DRUG APPROVALS
ONE TIME
$10.00
BUPIVACAINE
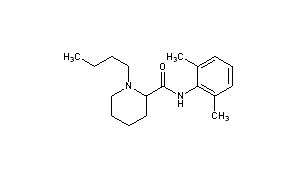
Bupivacaine
cas 38396-39-3, MF C18H28N2O, Average: 288.4277
1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide
- AH 250
- DUR-843
- LAC-43
- SKY 0402
- SKY-0402
- SKY0402
- Win 11318
2-Piperidinecarboxamide, 1-butyl-N-(2,6-dimethylphenyl)-, hydrochloride, hydrate (1:1:1), cas 73360-54-0
Molecular Formula, C18H28N2O.ClH.H2O
Bupivan (Sun) / Carbostesin (AstraZeneca) / Marcain (AstraZeneca) / Marcaina (AstraZeneca) / Posimir (Durect) / Sensorcaine-MPF (Astra Zeneca) / Xaracoll (Innocoll Holdings Limited)
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Bupivacaine hydrochloride | 7TQO7W3VT8 | 73360-54-0 | HUCIWBPMHXGLFM-UHFFFAOYSA-N |
| Bupivacaine hydrochloride anhydrous | AKA908P8J1 | 18010-40-7 | SIEYLFHKZGLBNX-UHFFFAOYSA-N |
BupivacaineCAS Registry Number: 2180-92-9
CAS Name: 1-Butyl-N-(2,6-dimethylphenyl)-2-piperidinecarboxamide
Additional Names:dl-1-butyl-2¢,6¢-pipecoloxylidide; 1-n-butyl-2¢,6¢-dimethyl-2-piperidinecarboxanilide; dl-N-n-butylpipecolic acid 2,6-xylidide; 1-butyl-2-(2,6-xylylcarbamoyl)piperidine; dl-1-n-butylpiperidine-2-carboxylic acid 2,6-dimethylanilide
Molecular Formula: C18H28N2O
Molecular Weight: 288.43
Percent Composition: C 74.95%, H 9.78%, N 9.71%, O 5.55%
Literature References: Prepn: B. Ekenstam et al.,Acta Chem. Scand.11, 1183 (1957); B. T. Ekenstam, B. G. Pettersson, US2955111 (1960 to AB Bofors). Resolution of isomers: B. F. Tullar, J. Med. Chem.14, 891 (1971). Stereospecific synthesis: B. Adger et al.,Tetrahedron Lett.37, 6399 (1996).Pharmacology of racemate: F. Henn, R. Brattsand, Acta Anaesthesiol. Scand. Suppl.21, 9 (1966), C.A.66, 17863u (1967); of isomers: F. P. Luduena et al.,Arch. Int. Pharmacodyn.200, 359 (1972). Clinical pharmacokinetics: D. W. Blake et al.,Anaesth. Intensive Care22, 522 (1994). Comprehensive description: T. D. Wilson, Anal. Profiles Drug Subs.19, 59-94 (1990). Review of use in spinal anesthesia: Acta Anaesthesiol. Scand.35, 1-10 (1991). Review of pharmacology and clinical efficacy of levobupivacaine: K. J. McClellan, C. M. Spencer, Drugs56, 355-362 (1998).Properties: mp 107.5-108°. pKa 8.09; also reported as 8.17. Partition coefficient: (oleyl alcohol/water) 1565; (n-heptane/pH 7.4 buffer) 27.5.
Melting point: mp 107.5-108°
pKa: pKa 8.09; also reported as 8.17
Log P: Partition coefficient: (oleyl alcohol/water) 1565; (n-heptane/pH 7.4 buffer) 27.5
Derivative Type: Hydrochloride monohydrate
CAS Registry Number: 14252-80-3
Manufacturers’ Codes: AH-2250; LAC-43
Trademarks: Carbostesin (AstraZeneca); Marcaine (AstraZeneca); Sensorcaine (AstraZeneca)
Molecular Formula: C18H28N2O.HCl.H2O
Molecular Weight: 342.90
Percent Composition: C 63.05%, H 9.11%, N 8.17%, O 9.33%, Cl 10.34%
Properties: White, odorless crystalline powder. mp 258.5°. Slightly sol in acetone, chloroform, ether. Soly (mg/ml): water 40; alcohol 125. LD50 in mice (mg/kg): 7.8 i.v., 82 s.c. (Henn, Brattsand).
Melting point: mp 258.5°
Toxicity data: LD50 in mice (mg/kg): 7.8 i.v., 82 s.c. (Henn, Brattsand)
Derivative Type: (-)-Form
CAS Registry Number: 27262-47-1
Additional Names: Levobupivacaine; (S)-bupivacaine
Properties: Crystals from isopropanol, mp 135-137°. [a]D25 -80.9° (c = 5 in methanol).
Melting point: mp 135-137°
Optical Rotation: [a]D25 -80.9° (c = 5 in methanol)
Derivative Type: (-)-Form hydrochloride
CAS Registry Number: 27262-48-2
Trademarks: Chirocaine (Abbott)
Molecular Formula: C18H28N2O.HCl
Molecular Weight: 324.89
Percent Composition: C 66.54%, H 9.00%, N 8.62%, O 4.92%, Cl 10.91%
Properties: mp 255-257°. [a]D25 -12.3° (c = 2 in water).
Melting point: mp 255-257°
Optical Rotation: [a]D25 -12.3° (c = 2 in water)
Therap-Cat: Anesthetic (local).
Keywords: Anesthetic (Local).
Other Names for this Substance
- 2-Piperidinecarboxamide, 1-butyl-N-(2,6-dimethylphenyl)-, hydrochloride, hydrate (1:1:1)
- 2-Piperidinecarboxamide, 1-butyl-N-(2,6-dimethylphenyl)-, monohydrochloride, monohydrate
- Bupivacaine hydrochloride monohydrate
- Marcain Heavy
- Marcain
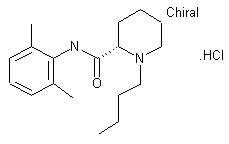
(-)-Bupivacaine hydrochloride, Levobupivacaine hydrochloride, Chirocaine
Synthesis Reference
Thuresson, B. and Egner, B.P.H.; U.S. Patent 2,792,399; May 14, 1957; assigned to AB Bofors, Sweden. Thuresson, B. and Pettersson, B.G.; US. Patent 2,955.1 11; October 4,1960; assigned to AB Bofors, Sweden., US2955111
SYN
British Patent 869,978 (1959).

SYN
Bupivacaine, N-2,6-(dimethyl)1-butyl-2-piperidincarboxamide (2.2.7), is chemically similar to mepivacaine and only differs in the replacement of the N-methyl substituent on the piperidine ring with an N-butyl substituent. There are also two suggested methods of synthesis. The first comes from α-picolin-2,6-xylidide (2.2.4). The alkylation of the last with butyl bromide gives the corresponding pyridine salt (2.2.6). Finally, it is reduced by hydrogen using platinum oxide as a catalyst into a piperidine derivative—bupivacaine [13,16].

The other method results directly from the piperidine-2-carboxylic acid chloride, which is reacted with 2,6-dimethylaniline. The resulting amide (2.2.8) is further alkylated with butyl bromide to bupivacaine [17–19].

Like lidocaine and mepivacaine, bupivacaine is used in infiltration, spinal, and epidural anesthesia in blocking nerve transmission. Its most distinctive property is its long-lasting action. It is used for surgical intervention in urology and in lower thoracic surgery from 3 to 5 h in length, and in abdominal surgery lasting from 45 to 60 min. It is used to block the trifacial nerve, the sacral and brachial plexuses, in resetting dislocations, in epidural anesthesia, and during Cesarian sections. The most common synonym for bupivacaine is marcaine.
SYN
3.7 Bupivacaine (21293) and Levobupivacaine (1976)
Bupivacaine (3.1.41) (Marcaine) is a local anesthetic of great potency and long duration that has been widely used for years, but it has cardio and CNS toxic sideeffects. For many years it was nearly the only local anesthetic applicable to almost all kinds of loco-regional anesthetic techniques, and nowadays, in many occasions, it is still the only alternative available [61–64].
Bupivacaine is currently used in racemic form. At high doses, however, the racemate is potentially hazardous due to toxicity problems.
Currently, racemic bupivacaine (3.1.41) is produced from picolinic acid (3.1.38) either by reduction to pipecolic acid (3.1.39) and then, after conversion to corresponding acid chloride (3.1.40) coupling with 2,6-xylidine to give pipecolic acid-2,6-xylidide (3.1.33), or by reducing the pyridyl amide (3.1.43) prepared from picolinic acid chloride (3.1.42) over platinum oxide. The amide intermediate (3.1.33), which can also be used to prepare the anesthetics ropivacaine (3.1.37) and mepivacaine (3.1.31), was transformed to desired bupivacaine (3.1.41) either by direct alkylation using butyl bromide and potassium carbonate or by reductive amination using butyraldehyde [45,59,65–69] (Scheme 3.7).

Enantiomers of bupivacaine can be prepared via diastereomeric salt resolution with tartaric acid or by resolution of the amide (3.1.33) with O,O-dibenzoyl tartaric acid followed by alkylation [47,70].
One of enantiomers, S(–) isomer of the racemic bupivacaine (levobupivacaine), has equal potency but less cardiotoxic and CNS effects in comparison with both R(+) bupivacaine and bupivacaine racemate. The reduced toxicity of levobupivacaine (3.1.48) gives a wider safety margin in clinical practice [71,72].
Stereospecific synthesis of levobupivacaine from (S)-lysine have been proposed (Scheme 3.8).

Treatment of N-CBZ (S)-lysine (3.1.44) with sodium nitrite in acetic acid yields the acetate (3.1.45). The prepared acetate (3.1.45) was then coupled with dimethyl aniline using N,N′-dicyclohexylcarbodiimide to give the amide (3.1.46) in good yield. The acetate group was then converted into the tosylate (3.1.47), which was deprotected and cyclized stereospecifically in one-pot reaction to give the amide (3.1.33) in high yield. Alkylation is easily achieved using an alkyl bromide and K2CO3 without any racemization. Alkylation can also be carried out using butyraldehyde/formic acid although the former is a much simpler process [73] (Scheme 3.8).
SYN
WO 9611181
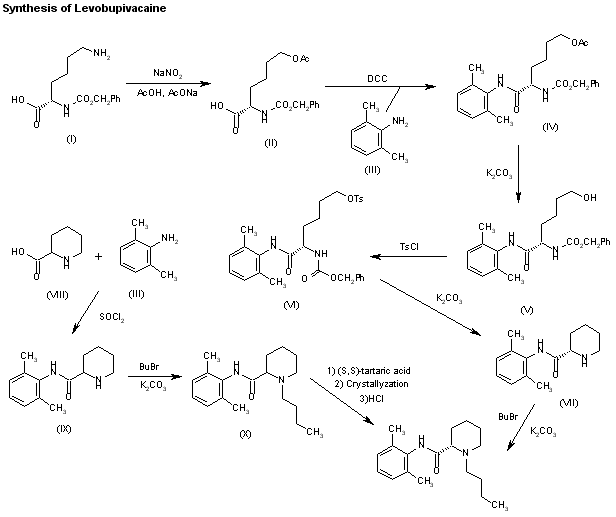
Levobupivacaine has been obtained by two different ways: 1) The deamination of N-benzoyloxycarbonyl-L-lysine (I) with NaNO2/acetic acid gives 6-acetoxy-2(S)-(benzyl-oxycarbonylamino)hexanoic acid (II), which is amidated with 2,6-dimethylaniline (III) and dicyclohexylcarbodiimide (DCC) to the expected amide (IV). The deacetylation of (IV) with K2CO3 in methanol affords compound (V), which is tosylated as usual with tosyl chloride giving intermediate (VI), which is stereospecifically cyclized by means of K2CO3 in ethanol yielding N-(2,6-dimethyl-phenyl)piperidine-2 (S)-carboxamide (VII). Finally, this compound is alkylated with butyl bromide and K2CO3 or by reductoalkylation with butyraldehyde. 2) The amidation of piperidine-2-carboxylic acid (VIII) with 2,6-dimethylaniline (III) by means of SOCl2 in toluene gives the corresponding amide (IX), which is alkylated with butyl bromide as before yielding racemic bupivacaine (X) (3). This compound is then submitted to optical resolution by treatment with (S,S)-(?-tartaric acid followed by crystallization of the resulting tartrate and acidification with HCl in isopropanol.
SYN
| Org Process Res Dev 2000,4(6),530 |
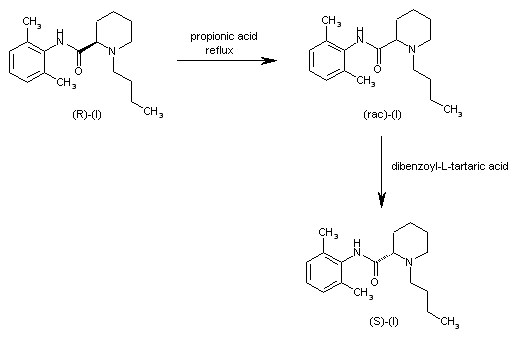
Improved yield in the synthesis of levobupivacaine. An improved yield in the synthesis of levobupivacaine can be obtained by recovering the unwanted (R)-enantiomer side product in the optical resolution of the racemic bupivacaine. The treatment of (R)-(I) with refluxing propionic acid causes its racemization, yielding racemic-(I) (bupivacaine), which is then submitted to a new optical resolution process using dibenzoyl-L-tartaric acid.
Literatures:
Acta Chemica Scandinavica (1947-1973), , vol. 11, p. 1183,1184
Literatures:
BRIDGE PHARMA, INC. Patent: WO2008/88756 A1, 2008 ; Location in patent: Page/Page column 30-31 ;
Yield: ~94%
nmr
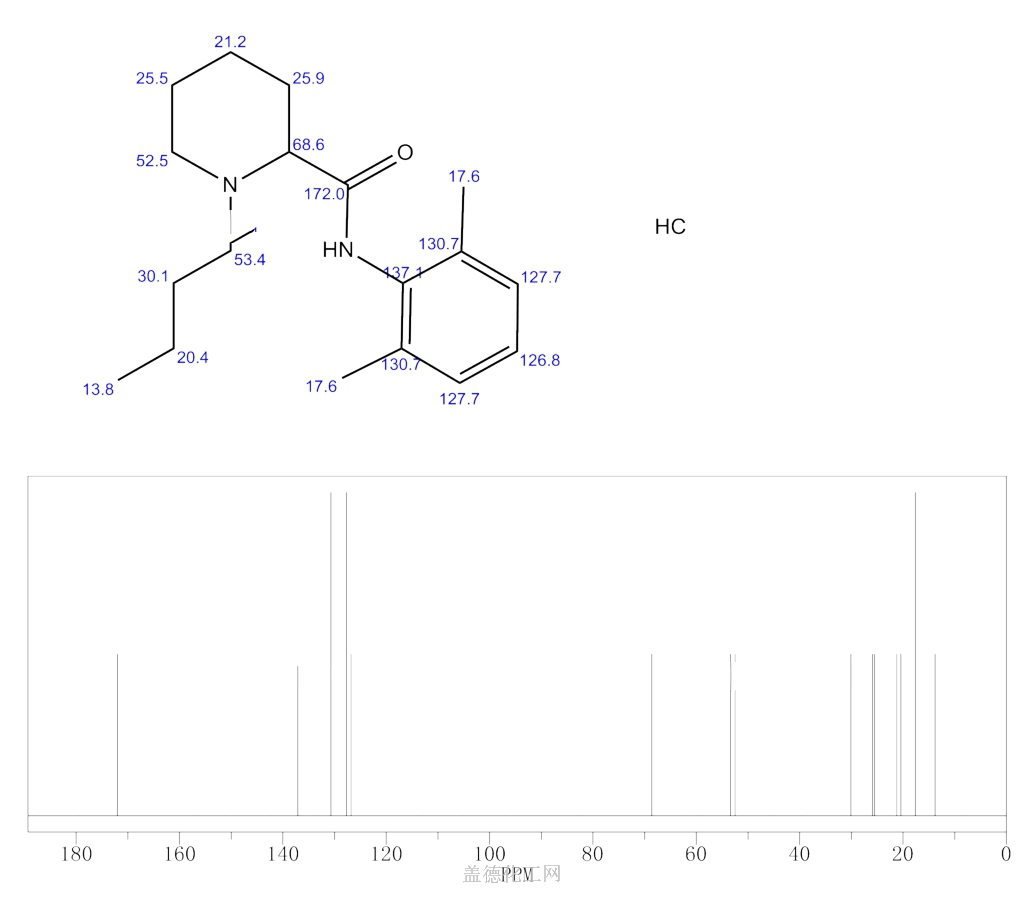
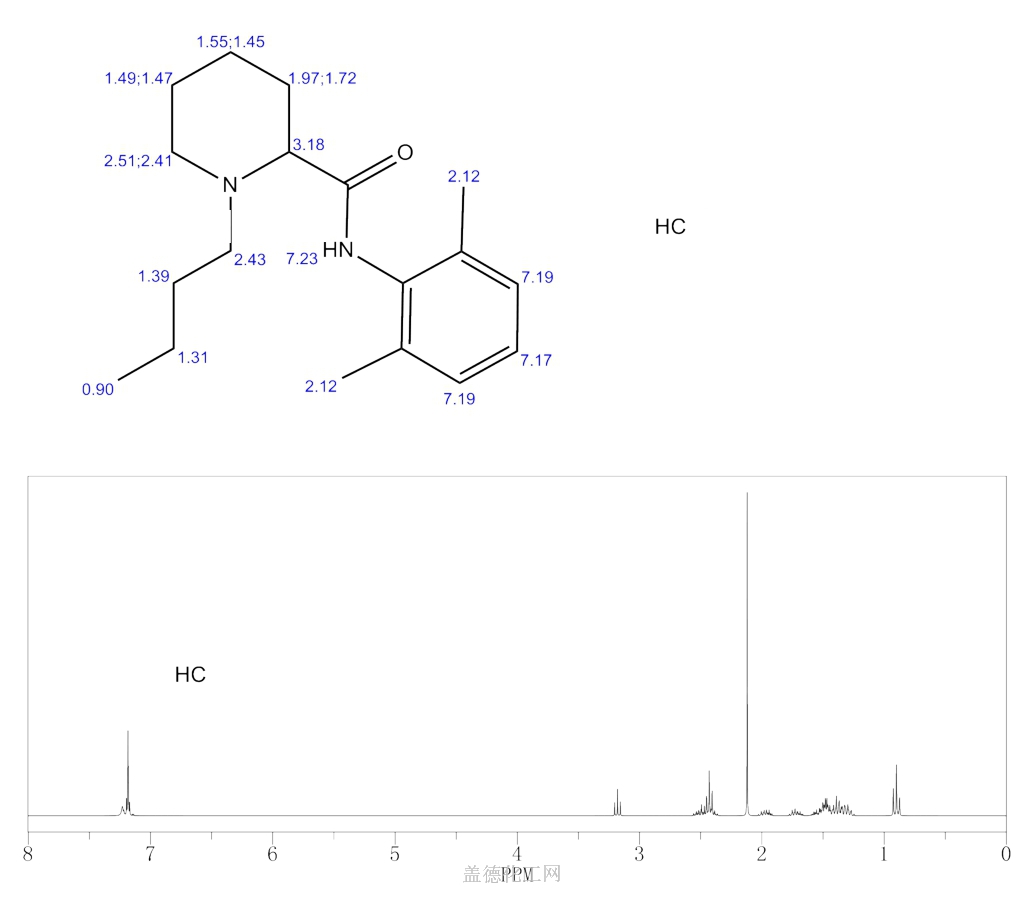
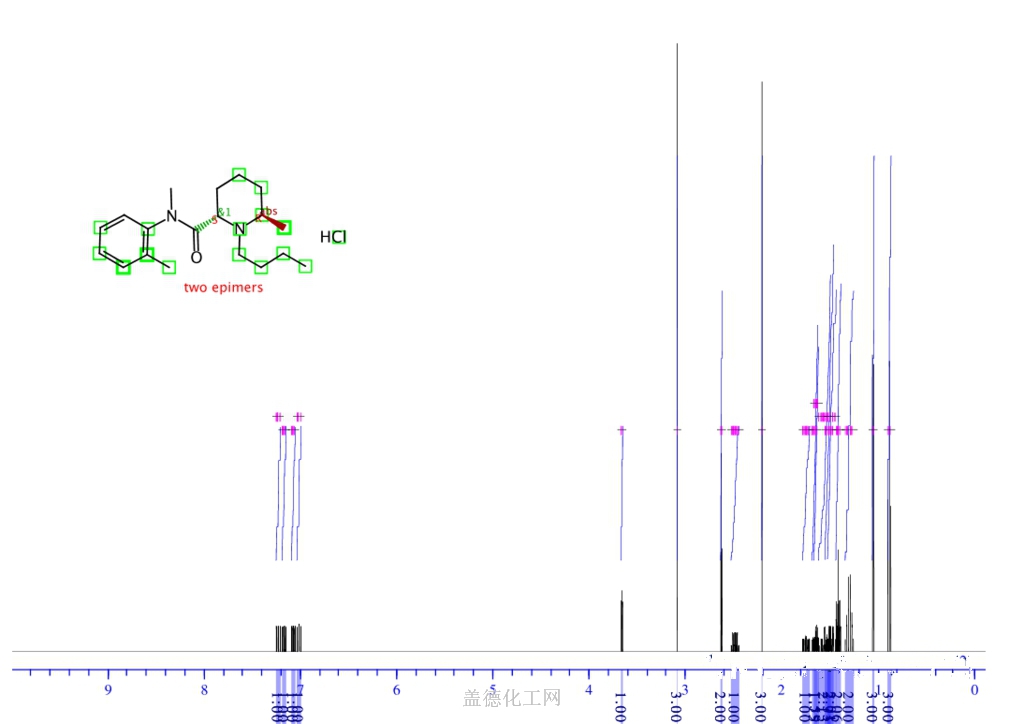
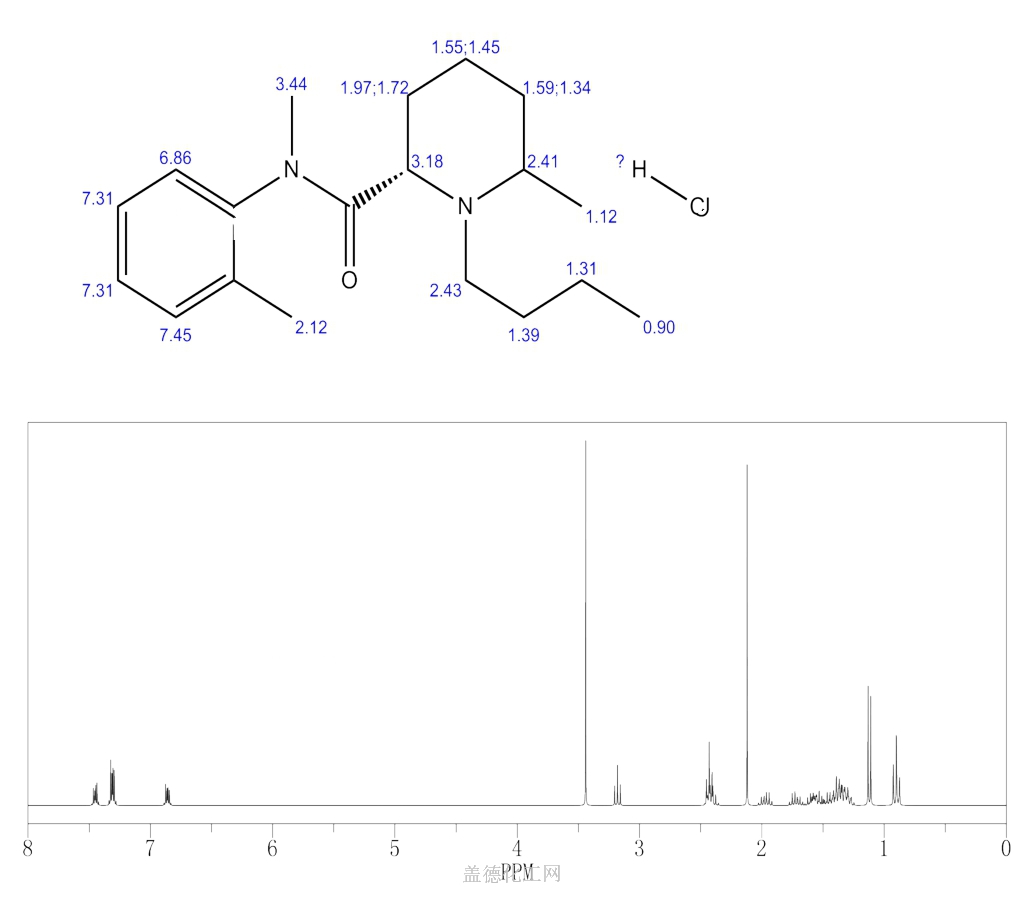
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Bupivacaine is a local anesthetic used in a wide variety of superficial and invasive procedures.
Bupivacaine, marketed under the brand name Marcaine among others, is a medication used to decrease feeling in a specific area.[4] In nerve blocks, it is injected around a nerve that supplies the area, or into the spinal canal’s epidural space.[4] It is available mixed with a small amount of epinephrine to increase the duration of its action.[4] It typically begins working within 15 minutes and lasts for 2 to 8 hours.[4][5]
Possible side effects include sleepiness, muscle twitching, ringing in the ears, changes in vision, low blood pressure, and an irregular heart rate.[4] Concerns exist that injecting it into a joint can cause problems with the cartilage.[4] Concentrated bupivacaine is not recommended for epidural freezing.[4] Epidural freezing may also increase the length of labor.[4] It is a local anaesthetic of the amide group.[4]
Bupivacaine was discovered in 1957.[6] It is on the World Health Organization’s List of Essential Medicines.[7] Bupivacaine is available as a generic medication.[4][8] An implantable formulation of bupivacaine (Xaracoll) was approved for medical use in the United States in August 2020.[9][10][11]
Medical uses
Bupivacaine is indicated for local infiltration, peripheral nerve block, sympathetic nerve block, and epidural and caudal blocks. It is sometimes used in combination with epinephrine to prevent systemic absorption and extend the duration of action. The 0.75% (most concentrated) formulation is used in retrobulbar block.[12] It is the most commonly used local anesthetic in epidural anesthesia during labor, as well as in postoperative pain management.[13] Liposomal formulations of bupivacaine (brand name EXPAREL) have shown to be more effective in providing pain relief than plain solutions of bupivacaine.[14][15]
The fixed-dose combination of bupivacaine with Type I collagen (brand name Xaracoll) is indicated for acute postsurgical analgesia (pain relief) for up to 24 hours in adults following open inguinal hernia repair.[10][11]
Bupivacaine (Posimir) is indicated in adults for administration into the subacromial space under direct arthroscopic visualization to produce post-surgical analgesia for up to 72 hours following arthroscopic subacromial decompression.[16][17]
Contraindications
Bupivacaine is contraindicated in patients with known hypersensitivity reactions to bupivacaine or amino-amide anesthetics. It is also contraindicated in obstetrical paracervical blocks and intravenous regional anaesthesia (Bier block) because of potential risk of tourniquet failure and systemic absorption of the drug and subsequent cardiac arrest. The 0.75% formulation is contraindicated in epidural anesthesia during labor because of the association with refractory cardiac arrest.[18]
Adverse effects
Compared to other local anaesthetics, bupivacaine is markedly cardiotoxic.[19] However, adverse drug reactions (ADRs) are rare when it is administered correctly. Most ADRs are caused by accelerated absorption from the injection site, unintentional intravascular injection, or slow metabolic degradation. However, allergic reactions can rarely occur.[18]
Clinically significant adverse events result from systemic absorption of bupivacaine and primarily involve the central nervous system (CNS) and cardiovascular system. CNS effects typically occur at lower blood plasma concentrations. Initially, cortical inhibitory pathways are selectively inhibited, causing symptoms of neuronal excitation. At higher plasma concentrations, both inhibitory and excitatory pathways are inhibited, causing CNS depression and potentially coma. Higher plasma concentrations also lead to cardiovascular effects, though cardiovascular collapse may also occur with low concentrations.[20] Adverse CNS effects may indicate impending cardiotoxicity and should be carefully monitored.[18]
- CNS: circumoral numbness, facial tingling, vertigo, tinnitus, restlessness, anxiety, dizziness, seizure, coma
- Cardiovascular: hypotension, arrhythmia, bradycardia, heart block, cardiac arrest[13][18]
Toxicity can also occur in the setting of subarachnoid injection during high spinal anesthesia. These effects include: paresthesia, paralysis, apnea, hypoventilation, fecal incontinence, and urinary incontinence. Additionally, bupivacaine can cause chondrolysis after continuous infusion into a joint space.[18]
Bupivacaine has caused several deaths when the epidural anaesthetic has been administered intravenously accidentally.[21]
Treatment of overdose
Further information: Lipid rescue
Animal evidence[22][23] indicates intralipid, a commonly available intravenous lipid emulsion, can be effective in treating severe cardiotoxicity secondary to local anaesthetic overdose, and human case reports of successful use in this way.[24][25] Plans to publicize this treatment more widely have been published.[26]
Pregnancy and lactation
Bupivacaine crosses the placenta and is a pregnancy category C drug. However, it is approved for use at term in obstetrical anesthesia. Bupivacaine is excreted in breast milk. Risks of discontinuing breast feeding versus discontinuing bupivacaine should be discussed with the patient.[18]
Postarthroscopic glenohumeral chondrolysis
Bupivacaine is toxic to cartilage and its intra-articular infusions may lead to postarthroscopic glenohumeral chondrolysis.[27]
Pharmacology
Pharmacodynamics
Bupivacaine binds to the intracellular portion of voltage-gated sodium channels and blocks sodium influx into nerve cells, which prevents depolarization. Without depolarization, no initiation or conduction of a pain signal can occur.
Pharmacokinetics
The rate of systemic absorption of bupivacaine and other local anesthetics is dependent upon the dose and concentration of drug administered, the route of administration, the vascularity of the administration site, and the presence or absence of epinephrine in the preparation.[28]
- Onset of action (route and dose-dependent): 1-17 min
- Duration of action (route and dose-dependent): 2-9 hr
- Half life: neonates, 8.1 hr, adults: 2.7 hr
- Time to peak plasma concentration (for peripheral, epidural, or caudal block): 30-45 min
- Protein binding: about 95%
- Metabolism: hepatic
- Excretion: renal (6% unchanged)[18]
Chemical structure
Like lidocaine, bupivacaine is an amino-amide anesthetic; the aromatic head and the hydrocarbon chain are linked by an amide bond rather than an ester as in earlier local anesthetics. As a result, the amino-amide anesthetics are more stable and less likely to cause allergic reactions. Unlike lidocaine, the terminal amino portion of bupivacaine (as well as mepivacaine, ropivacaine, and levobupivacaine) is contained within a piperidine ring; these agents are known as pipecholyl xylidines.[13]
Society and culture
Legal status
On 17 September 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Exparel, intended for the treatment of post-operative pain.[29] The applicant for this medicinal product is Pacira Ireland Limited.[29] Exparel liposomal was approved for medical use in the European Union in November 2020.[30]
Economics
Bupivacaine is available as a generic medication.[4][8]
Research
Levobupivacaine is the (S)-(–)-enantiomer of bupivacaine, with a longer duration of action, producing less vasodilation. Durect Corporation is developing a biodegradable, controlled-release drug delivery system for after surgery. It has currently[when?] completed a phase-III clinical trial.[31]
References
- ^ “Bupivacaine Use During Pregnancy”. Drugs.com. 13 April 2020. Retrieved 21 September 2020.
- ^ “Marcaine- bupivacaine hydrochloride injection, solution Marcaine with epinephrine- bupivacaine hydrochloride and epinephrine bitartrate injection, solution”. DailyMed. Retrieved 13 February2021.
- ^ “Sensorcaine MPF- bupivacaine hydrochloride injection, solution”. DailyMed. Retrieved 13 February 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “Bupivacaine Hydrochloride”. The American Society of Health-System Pharmacists. Archived from the original on 2015-06-30. Retrieved August 1, 2015.
- ^ Jump up to:a b Whimster, David Skinner (1997). Cambridge textbook of accident and emergency medicine. Cambridge: Cambridge University Press. p. 194. ISBN 9780521433792. Archived from the original on 2015-10-05.
- ^ Egan, Talmage D. (2013). Pharmacology and physiology for anesthesia : foundations and clinical application. Philadelphia, PA: Elsevier/Saunders. p. 291. ISBN 9781437716795. Archivedfrom the original on 2016-05-12.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
- ^ Jump up to:a b Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 22. ISBN 9781284057560.
- ^ “Xaracoll: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 2 September 2020.
- ^ Jump up to:a b “FDA approval letter” (PDF). U.S. Food and Drug Administration (FDA). 28 August 2020. Retrieved 2 September2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “FDA Approves Xaracoll (bupivacaine HCl) Implant, a Non-opioid, Drug-device Treatment Option for Acute Postsurgical Pain Relief for up to 24 Hours Following Open Inguinal Hernia Repair in Adults” (Press release). Innocoll Pharmaceuticals. 31 August 2020. Retrieved 2 September 2020 – via PR Newswire.
- ^ Lexicomp. “Bupivacaine (Lexi-Drugs)”. Archived from the original on 2014-04-10. Retrieved 20 April 2014.
- ^ Jump up to:a b c Miller, Ronald D. (November 2, 2006). Basics of Anesthesia. Churchill Livingstone.
- ^ Ma, Ting-Ting, et al. (2017). “Liposomal bupivacaine versus traditional bupivacaine for pain control after total hip arthroplasty: A meta-analysis”. Medicinevol. 96 (96, 25 (2017): e7190): e7190. doi:10.1097/MD.0000000000007190. PMC 5484209. PMID 28640101.
- ^ Mont, M. A., Beaver, W. B., Dysart, S. H., Barrington, J. W., & Gaizo, D. J. (2018). “Local Infiltration Analgesia With Liposomal Bupivacaine Improves Pain Scores and Reduces Opioid Use After Total Knee Arthroplasty: Results of a Randomized Controlled Trial”. The Journal of Arthroplasty. 33 (1): 33(1), 90–96. doi:10.1016/j.arth.2017.07.024. PMID 28802777.
- ^https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/204803Orig1s000ltr.pdf
- ^ “Durect Corporation Announces U.S. FDA Approval of Posimir For Post-Surgical Pain Reduction for up to 72 Hours Following Arthroscopic Subacromial Decompression” (Press release). Durect Corporation. 2 February 2021. Retrieved 13 February 2021 – via PR Newswire.
- ^ Jump up to:a b c d e f g “Bupivacaine (Lexi-Drugs)”. Archived from the original on 2014-04-10. Retrieved 20 April 2014.
- ^ de La Coussaye, J. E.; Eledjam, J. J.; Brugada, J.; Sassine, A. (1993). “[Cardiotoxicity of local anesthetics]”. Cahiers d’Anesthésiologie. 41 (6): 589–598. ISSN 0007-9685. PMID 8287299.
- ^ Australian Medicines Handbook. Adelaide. 2006. ISBN 978-0-9757919-2-9.
- ^ ABS-CBN Interactive: Filipino nurse dies in UK due to wrong use of anaesthetic
- ^ Weinberg, GL; VadeBoncouer, T; Ramaraju, GA; Garcia-Amaro, MF; Cwik, MJ. (1998). “Pretreatment or resuscitation with a lipid infusion shifts the dose-response to bupivacaine-induced asystole in rats”. Anesthesiology. 88 (4): 1071–5. doi:10.1097/00000542-199804000-00028. PMID 9579517. S2CID 1661916.
- ^ Weinberg, G; Ripper, R; Feinstein, DL; Hoffman, W. (2003). “Lipid emulsion infusion rescues dogs from bupivacaine-induced cardiac toxicity”. Regional Anesthesia and Pain Medicine. 28 (3): 198–202. doi:10.1053/rapm.2003.50041. PMID 12772136. S2CID 6247454.
- ^ Rosenblatt, MA; Abel, M; Fischer, GW; Itzkovich, CJ; Eisenkraft, JB (July 2006). “Successful use of a 20% lipid emulsion to resuscitate a patient after a presumed bupivacaine-related cardiac arrest”. Anesthesiology. 105 (1): 217–8. doi:10.1097/00000542-200607000-00033. PMID 16810015.
- ^ Litz, RJ; Popp, M; Stehr, S N; Koch, T. (2006). “Successful resuscitation of a patient with ropivacaine-induced asystole after axillary plexus block using lipid infusion”. Anaesthesia. 61 (8): 800–1. doi:10.1111/j.1365-2044.2006.04740.x. PMID 16867094. S2CID 43125067.
- ^ Picard, J; Meek, T (February 2006). “Lipid emulsion to treat overdose of local anaesthetic: the gift of the glob”. Anaesthesia. 61(2): 107–9. doi:10.1111/j.1365-2044.2005.04494.x. PMID 16430560. S2CID 29843241.
- ^ Gulihar, Abhinav; Robati, Shibby; Twaij, Haider; Salih, Alan; Taylor, Grahame J.S. (December 2015). “Articular cartilage and local anaesthetic: A systematic review of the current literature”. Journal of Orthopaedics. 12 (Suppl 2): S200–S210. doi:10.1016/j.jor.2015.10.005. PMC 4796530. PMID 27047224.
- ^ “bupivacaine hydrochloride (Bupivacaine Hydrochloride) injection, solution”. FDA. Archived from the original on 21 April 2014. Retrieved 20 April 2014.
- ^ Jump up to:a b “Exparel: Pending EC decision”. European Medicines Agency (EMA). 17 September 2020. Retrieved 21 September 2020.Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Exparel liposomal EPAR”. European Medicines Agency (EMA). 15 September 2020. Retrieved 11 December 2020.
- ^ “Bupivacaine Effectiveness and Safety in SABER Trial (BESST)”. ClinicalTrials.gov. 20 January 2010. Archived from the original on 2011-12-27. Retrieved 2012-03-01.
External links
- “Bupivacaine”. Drug Information Portal. U.S. National Library of Medicine.
///////////Bupivacaine, AH 250, DUR-843, LAC-43, SKY 0402, SKY-0402, SKY0402, Win 11318, ANAESTHETIC
- CCCCN1CCCCC1C(=O)NC1=C(C)C=CC=C1C

NEW DRUG APPROVALS
ONE TIME
$10.00
ROPIVACAINE

Ropivacaine
CAS No.84057-95-4 (Ropivacaine);
- Molecular FormulaC17H26N2O
- Average mass274.401 Da
HCL SALTCAS Registry Number: 98717-15-8
HCL MONOHYDRATE
Molecular Weight328.88, FormulaC17H26N2O • HCl • H2O
132112-35-7 (Ropivacaine HCl Monohydrate);
Chemical Name S-(-)-1-propyl-2′,6′-pipecoloxylidide hydrochloride monohydrate(S)-(-)-1-Propyl-2′,6′-pipecoloxylidide
(S)-N-(2,6-dimethylphenyl)-1-propyl-2-piperidinecarboxamide
2-Piperidinecarboxamide, N-(2,6-dimethylphenyl)-1-propyl-, (2S)-
5376
5421606[Beilstein]
7IO5LYA57N
84057-95-4[RN]
854056-07-8[RN]
(S)-ropivacaine
(2S)-N-(2,6-Dimethylphenyl)-1-propyl-2-piperidinecarboxamide
ропивакаин [Russian] [INN]
روبيفاكائين [Arabic] [INN]
罗哌卡因 [Chinese] [INN]
Drug Name:Ropivacaine Hydrochloride Hydrate
Research Code:LEA-103; NA-001; (-)-LEA-103;
Trade Name:Naropin® / Anapeine®
MOA:Sodium channels blockers
Indication:Anaesthetic
Company:AstraZeneca (Originator) , Fresenius Kabi
ATC Code:N01BB09APPROVED
- US
- JP
- CN
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1996-09-26 | First approval | Naropin | Anaesthetic | Injection | 2 mg/ml; 5 mg/ml; 7.5 mg/ml; 10 mg/ml | APP Pharmaceuticals |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2001-04-04 | First approval | Anapeine | Anaesthetic | Injection | 2 mg/ml; 7.5 mg/ml; 10 mg/ml | AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-02-11 | Marketing approval | 耐乐品/Naropin | Anaesthetic | Injection | 20 mg/10 ml;100 mg/10 ml; 75 mg/10 ml; 50 mg/10 ml | AstraZeneca | |
| 2010-02-03 | Marketing approval | 耐乐品/Naropin | Anaesthetic | Injection | 2 mg/mL | AstraZeneca |
| No. | NDA No. | Major Technical Classification | Patent No. | Estimated Expiry Date | Drug Substance Claim | Drug Product Claim | Patent Use Code (All list) |
| 1 | N020533 | Uses(Indication) | 5670524 | 2014-09-23 | Y | Y | U – 833 |
| 2 | N020533 | Device | 7828787 | 2025-10-18 | Y | ||
| 3 | N020533 | Device | 7857802 | 2026-11-28 | Y | ||
| 4 | N020533 | Device | 8118802 | 2023-05-18 | Y | ||
| 5 | N020533 | Device | 8162915 | 2024-05-23 | Y |
Ropivacaine
CAS Registry Number: 84057-95-4
CAS Name: (2S)-N-(2,6-dimethylphenyl)-1-propyl-2-piperidinecarboxamide
Additional Names: (S)-(-)-1-propyl-2¢,6¢-pipecoloxylidide; l-N-n-propylpipecolic acid-2,6-xylidide
Manufacturers’ Codes: LEA-103
Molecular Formula: C17H26N2O, Molecular Weight: 274.40
Percent Composition: C 74.41%, H 9.55%, N 10.21%, O 5.83%
Literature References: Prepn: A. F. Thuresson, C. Bovin, WO8500599 (1985 to Apothekernes); H.-J. Federsel et al.,Acta Chem. Scand.B41, 757 (1987).Physicochemical properties: G. R. Strichartz et al.,Anesth. Analg.71, 158 (1990).HPLC determn in human plasma: Z. Yu et al.,J. Chromatogr. B654, 221 (1994). In vitro metabolism: Y. Oda et al.,Anesthesiology82, 214 (1995). Clinical pharmacokinetics: D. J. Kopacz et al.,ibid.81, 1139 (1994). Toxicity study in sheep: A. C. Santos et al.,ibid.82, 734 (1995). Clinical evaluation in relief of surgical pain: I. Cederholm et al.,Reg. Anesth.19, 18 (1994); B. Johansson et al.,Anesth. Analg.78, 210 (1994); labor pain: R. Stienstra et al.,ibid.80, 285 (1995).
Properties: Crystals from toluene, mp 144-146°. [a]D25 -82.0° (c = 2 in methanol). pKa 8.16. Distribution coefficient (1-octanol/aq buffer, pH 7.4): 115.0.
Melting point: mp 144-146°
pKa: pKa 8.16
Optical Rotation: [a]D25 -82.0° (c = 2 in methanol)
Derivative Type: Hydrochloride
CAS Registry Number: 98717-15-8
Trademarks: Naropin (AstraZeneca)
Molecular Formula: C17H26N2O.HCl, Molecular Weight: 310.86
Percent Composition: C 65.68%, H 8.75%, N 9.01%, O 5.15%, Cl 11.40%
Properties: Crystals from isopropyl alcohol, mp 260-262°. [a]D25 -6.6° (c = 2 in water).
Melting point: mp 260-262°
Optical Rotation: [a]D25 -6.6° (c = 2 in water)
Derivative Type: Hydrochloride monohydrate
CAS Registry Number: 132112-35-7
Properties: Crystals from acetone + water, mp 269.5-270.6°. [a]D20 -7.28° (c = 2 in water).
Melting point: mp 269.5-270.6°
Optical Rotation: [a]D20 -7.28° (c = 2 in water)
Therap-Cat: Anesthetic (local).
Keywords: Anesthetic (Local).Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Ropivacaine hydrochloride | V910P86109 | 132112-35-7 | VSHFRHVKMYGBJL-CKUXDGONSA-N |
| Ropivacaine hydrochloride anhydrous | 35504LBE2T | 98717-15-8 | NDNSIBYYUOEUSV-RSAXXLAASA-N |
Ropivacaine is an analgesic drug used for local or regional anesthesia for surgery and short-term management of pain.Ropivacaine is an aminoamide local anaesthetic drug commonly marketed by AstraZeneca under the trade name Naropin. It is present as a racemic mixture of the enantiomers containing equal proportions of the “S” and “R” forms. The marketed form contains the single S-enantiomer as the active ingredient.
Ropivacaine hydrochloride hydrate was first approved by the U.S. Food and Drug Administration (FDA) on September 26, 1996, then approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) in April 4, 2001. It was developed by AstraZeneca, then marketed as Naropin® by APP Pharmaceuticals, LLC. in the US and as Anapeine® by AstraZeneca in JP.
Ropivacaine is a local anaesthetic drug belonging to the amino amide group. It is indicated for the production of local or regional anesthesia for surgery and for acute pain management.
Naropin® is available as injection solution for intravenous use, containing 2, 5, 7.5 or 10 mg of Ropivacaine hydrochloride one mL. Common concentration is 7.5 mg/mL, and the maximum single dose is 200 mg.
Ropivacaine (rINN) /roʊˈpɪvəkeɪn/ is a local anaesthetic drug belonging to the amino amide group. The name ropivacaine refers to both the racemate and the marketed S–enantiomer. Ropivacaine hydrochloride is commonly marketed by AstraZeneca under the brand name Naropin.
Table 1 The common types of local anesthetics
Syn
Synthesis Reference
Peter Jaksch, “Process for the preparation of ropivacaine hydrochloride monohydrate.” U.S. Patent US5959112, issued February, 1970.
US5959112Route 1
Reference:1. US2799679A.
Reference:1. WO8500599A1.
https://patents.google.com/patent/WO1985000599A1/enA large variety of N-alkyl-pipecolic acid amides have been synthesized. A number of these compounds have found use as local anesthetics, such as Mepivacaine, namely the racemate of N-methylpipecolic–acid-2,6-xylidide:

and Bupivacaine, namely the racemate of N-butylpipecolic- acid-2,6-xylidide:

References disclosing homologs of this series of compounds include U.S. Patent 2,799,679; British Patent 775,749; British Patent 775,750; British Patent 800,565; British Patent 824,542; British Patent 869,978; British Patent 949,729; U.S. Patent 4,110,331; and U.S. Patent 4,302,465.There is a summary paper dealing with these types of anesthetics, and related compounds in a paper in Acta Che ica Scandinavica 11, (1957) No. 7 pp. 1183-1190 by Bo Thuresson af Ekenstam et al.There is a discussion of the“ effect of optical isomers in related compounds in J. Med. Chem., 14 (1971) pp. 891-892 entitled “Optical Isomers Of Mepivacaine And Bupivacaine” by Benjamin F. Tullar; Acta Pha m. Suecica, 8 (1971) pp. 361- 364 entitled “Some Physicochemical Properties Of The Racemates And The Optically Active Isomers Of Two Local Anaesthetic Compounds”, by . Friberger et al -.; Acta Pharmacol et Toxicol, 31 (1972). pp. 273-286 entitled “Toxicological And Local Anaesthetic Effects Of Optically Active Isomers Of Two Local Anaesthetic Compounds”, by G. Aberg; Annual Review Of Pharmacology, 9 (1969) pp. 5Q3-520 entitled “Duration Of Local Anaesthesia”, by F.P. Luduena and Acta Pharmacol, et Toxicol, 41 (1977). pp. 432-443 entitled “Studies On The Duration Of Local Anaesthesia: Structure/Activity Relationships In A Series Of Homologous Local Anaesthetics”, by G. Aberg et al.

Reference:1. J. Labelled Compd. Rad. 1987, 24, 521-528.Route 4
Reference:1. CN104003930A.
https://patents.google.com/patent/CN104003930A/enRopivacaine (Ropivacaine) is the long-acting local anesthetics of amide derivatives of Novel pure levo form of Astra drugmaker of Sweden listing in 1996, there is analgesia and anesthesia dual function, be widely used in nerve block anesthesia, local infiltration anesthesia and epidural anesthesia , be particularly useful for Postoperative Analgesia After and obstetrical analgesia.On piperidine ring in ropivacaine structure, having a chiral carbon atom, is chipal compounds, and levoisomer is low compared with dextrorotatory isomer toxicity, and action effect is good.Ropivacaine HCL is the hydrochloride of ropivacaine, and chemistry is by name: (-)-(S)-N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-carboxamide hydrochloride, molecular formula is C 17 h 26 n 2 oHCl, structural formula:At present, in prior art, the synthetic method of ropivacaine mainly contains:Taking L-2-piperidine formyl chlorine as starting raw material, through phosphorus pentachloride or sulfur oxychloride acidylate, then with the condensation of 2,6-xylidine, and then react and obtain ropivacaine with n-propyl bromide.Although this method production technique is simple, reactions steps is also shorter, but commercially available L-2-piperidine carboxylic acid average price is 4~5 times of racemization Pipecolic Acid, raw materials cost is too high, and may there is racemization phenomenon in subsequent reactions process, affect optical purity of products, for example US Patent No. 4695576 and “Chinese Medicine magazine” o. 11th in 2012 “Ropivacaine HCL a synthetic” literary composition and Chinese patent CN201310041390.2 all adopt this kind of method.”synthetic chemistry” the 14th the 4th phase of volume “Synthesis of Ropivacaine Hydrochloride by Triphosgene” in 2006 and Hunan University’s Master’s thesis “synthesising process research of Ropivacaine HCL and” disclose the synthetic method of another ropivacaine, the Pipecolic Acid that adopts inexpensive racemization is raw material, prepare Ropivacaine HCL through reactions such as amidation, alkylations, use triphosgene or thionyl chloride to prepare acyl chlorides, but triphosgene danger in the time of storage and aftertreatment is larger, is not suitable for suitability for industrialized production; And partial condition in the latter’s method (reagent that the pH separating as intermediate (I) and reagent, catalyzer and recrystallization are used etc.) haves much room for improvement,under its test conditions, be difficult to take into account high purity and high yield simultaneously, according to prior art, the separation of ropivacaine raceme is also not ideal.The preparation method of embodiment 1, a kind of Ropivacaine HCL, comprises the steps:(1) preparation of intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea10.0g2-piperidine carboxylic acid, 160ml toluene are added in 500ml reaction flask.Pass into HCl gas, to pH2, be warming up to 48 ± 2 DEG C, add 1.5mlDMF, drip 11.2g (1.2 equivalent) sulfur oxychloride and 20ml toluene mixture liquid, drip and finish, be incubated 48 ± 2 DEG C of reaction 3h.Drip 2 of 4.0 equivalents, 6-xylidine and 20ml toluene mixture liquid, be incubated 58 ± 2 DEG C of reaction 3h.Filter, obtain yellow-green colour wet product 65g, dry to obtain gray solid 56g, solid is added in 280ml purified water, stir the molten reaction solution that obtains; 10%NaOH solution is slowly dropped in reaction solution, adjust pH to 4.5-5.0, use 100ml toluene wash , layering, retains water layer, continues to adjust pH to 9-10 with 10%NaOH solution, adds 100ml methylene dichloride.Layering, gets organic layer,and water layer continues to use 50ml dichloromethane extraction, merges organic layer, adds anhydrous sodium sulfate dehydration, 40 DEG C of concentrating under reduced pressure.Obtain pale yellow oily liquid body 15.5g, yield 86.2%, is intermediate (I) N-( 2,6-dimethyl benzene)-2-piperidyl urea.(2) preparation of intermediate (II) N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amideIntermediate 15.5 g of (the I) Dissolved in 60mlDMF IS, ADDS 8.9gK 2 cO . 3 , 8.2 g of drip (1.0 equivalent)-n-propyl bromide, and drip BE Finishing After Warming up to 78 ± 2 of DEG C, Insulation Reaction 2H; Ice Bath is down to room temperature, filters, and filtrate is added in 150ml frozen water, separates out a large amount of white solids, filter, dry, obtain white solid 17.4g, yield 95.0%, is intermediate (II) N-(2, 6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amide.(3) preparation of left-handed ropivacaine tartrate17.4g intermediate (II) is dissolved in 100ml Virahol, heats up 40 DEG C and stir molten; Treat entirely moltenly, add successively 1.80g (0.1 equivalent) titanium isopropylate, 1.91g (0.2 equivalent) D-tartrate, be warming up to backflow, after solution clarification, continue reaction 2h; Be cooled to 30 DEG C of crystallizatioies, filter, 75 DEG C of oven dry, obtain white solid 8.7g, and yield 39.2% is left-handed ropivacaine tartrate; After testing, ropivacaine purity 99.02%, dextrorotatory isomer per-cent 0.98%.(4) preparation of Ropivacaine HCL crude productLeft-handed 8.7g ropivacaine tartrate is joined in 50ml Virahol, be warming up to 50 DEG C, drip concentrated hydrochloric acid, surveying pH is 1~2, insulation reaction 2h.Be cooled to 0 DEG C of crystallization, separate out a large amount of white solids, filter, dry, obtain white solid 6.6g, yield 85.5%, is Ropivacaine HCL crude product.After testing, ropivacaine purity 99.11%, dextrorotatory isomer per-cent 0.89%.(5) refining6.6g crude product and 40ml dehydrated alcohol-concentrated hydrochloric acid mixed solution (20:1) are added in reaction flask, be heated to 50 DEG C and make to dissolve; Complete molten after, naturally cool to room temperature, ice-water bath is cooled to 0 DEG C, crystallization 2h; Filter, 5ml mixed solution washing for filter cake, obtains wet product, dries, and obtains white solid 6.0g, and yield 91.7%, is Ropivacaine HCL fine work.After testing, ropivacaine purity 99.91 %, dextrorotatory isomer per-cent 0.09%.The preparation method of embodiment 2, a kind of Ropivacaine HCLStep is as follows:(1) preparation of intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea100.0g2-piperidine carboxylic acid, 1600ml toluene are added in 3000ml reaction flask.Pass into HCl gas, to pH2 left and right, be warming up to 45~50 DEG C, add 15mlDMF, drip 111.5g (1.2 equivalent) sulfur oxychloride and 200ml toluene mixture liquid, drip and finish, be incubated 50-55 DEG C of reaction 3h.Drip 2 of 4.0 equivalents, 6-xylidine and 200ml toluene mixture liquid, be incubated 55~60 DEG C of reaction 2h.Filter, obtain the about 660g of yellow-green colour wet product, dry to obtain gray solid 545g, solid is added in 3000ml purified water, stir the molten reaction solution that obtains; 10%NaOH solution is slowly dropped in reaction solution, adjust pH to 4.5~5.0 , use 1000ml toluene wash, layering, retains water layer, continues to adjust pH to 9~10 with 10%NaOH solution, adds 1000ml methylene dichloride.Layering, gets organic layer,and water layer continues to use 750ml dichloromethane extraction, merges organic layer, adds anhydrous sodium sulfate dehydration, 40 DEG C of concentrating under reduced pressure.Obtain the about 151.8g of pale yellow oily liquid body, yield 84.5%, is intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea.(2) preparation of intermediate (II) N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amideIntermediate 150.0 g (the I) Dissolved in 600mlDMF IS, ADDS 86.5gK 2 cO . 3 , drip 95.4 g (1.2 equivalent)-n-propyl bromide, and drip BE Finishing After Warming up to 85 ~ 90 of DEG C, Insulation Reaction 2H; of Be Down to room temperature, filter, filtrate is added in 1500ml frozen water, separate out a large amount of white solids, filter, dry, obtain the about 167.6g of white solid, yield 94.6%, is intermediate (II) N-(2, 6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amide.(3) preparation of left-handed ropivacaine tartrate160.0g intermediate (II) is dissolved in 1000ml Virahol, heats up 50 DEG C and stir molten; Treat entirely moltenly, add successively 16.58g (0.1 equivalent) titanium isopropylate, 43.8g (0.5 equivalent) D-tartrate, be warming up to backflow, after solution clarification, continue reaction 3h; Cooling, is down to 30-35 DEG C of crystallization, filters, and 75 DEG C of oven dry, obtain white solid 84.2g, and yield 41.3% is left-handed ropivacaine tartrate; After testing, ropivacaine purity 98.97%, dextrorotatory isomer per-cent 1.03%.(4) preparation of Ropivacaine HCL crude productLeft-handed 80.0g ropivacaine tartrate is joined in 500ml Virahol, be warming up to 50 DEG C, drip concentrated hydrochloric acid, surveying pH is 1~2, insulation reaction 4h.Be cooled to 0~5 DEG C of crystallization, separate out a large amount of white solids, filter, dry, obtain the about 61.6g of white solid, yield 86.5%, is Ropivacaine HCL crude product.After testing, ropivacaine purity 99.07%, dextrorotatory isomer per-cent 0.93%.(5) refining60.0g crude product and 500ml dehydrated alcohol-concentrated hydrochloric acid mixed solution (20:1) are added in reaction flask, be heated to 50 DEG C and make to dissolve; Complete molten after, cooling crystallization, ice-water bath is cooled to 0-5 DEG C, crystallization 4h; Filter, a small amount of cold mixed solution washing for filter cake, obtains wet product, dries, and obtains white solid 55.6g, and yield 92.7%, is Ropivacaine HCL fine work.After testing, ropivacaine purity 99.87%, dextrorotatory isomer per-cent 0.13%.The preparation method of embodiment 3, a kind of Ropivacaine HCLStep is as follows:(1) preparation of intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea10.0g2-piperidine carboxylic acid, 160ml toluene are added in 500ml reaction flask.Pass into HCl gas, to pH3 left and right, be warming up to 48 ± 2 DEG C, add 1.5mlDMF, drip 9.3g (1.0 equivalent) sulfur oxychloride and 20ml toluene mixture liquid, drip and finish, be incubated 48 ± 2 DEG C of reaction 2h.Drip 2 of 4.0 equivalents, 6-xylidine and 20ml toluene mixture liquid, be incubated 58 ± 2 DEG C of reaction 3h.Filter, obtain yellow-green colour wet product 63.6g, dry to obtain gray solid 55g, solid is added in 280ml purified water, stir the molten reaction solution that obtains; 10%NaOH solution is slowly dropped in reaction solution, adjust pH to 4.5-5.0, use 100ml toluene wash, layering, retains water layer, continues to adjust pH to 9-10 with 10%NaOH solution, adds 100ml methylene dichloride.Layering, gets organic layer,and water layer continues to use 50ml dichloromethane extraction, merges organic layer, adds anhydrous sodium sulfate dehydration, 40 DEG C of concentrating under reduced pressure.Obtain the about 14.8g of pale yellow oily liquid body, yield 82.4%, is intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea.(2) preparation of intermediate (II) N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amideIntermediate 14.8 g of (the I) Dissolved in 60mlDMF IS, ADDS 8.5gK 2 cO . 3 , 7.8 g of drip (1.0 equivalent)-n-propyl bromide, and drip After Finishing of DEG BE Warming up to 75 C, Reaction Insulation 2H; IS Down Ice Bath to room temperature, filters, and filtrate is added in 150ml frozen water, separates out a large amount of white solids, filter, dry, obtain the about 16.0g of white solid, yield 91.5%, is intermediate (II) N-(2 ,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amide.(3) preparation of left-handed ropivacaine tartrate15g intermediate (II) is dissolved in 100ml Virahol, heats up 40 DEG C and stir molten; Treat entirely moltenly, add successively 1.72g (0.1 equivalent) titanium isopropylate, 1.82g (0.2 equivalent) D-tartrate, be warming up to backflow , after solution clarification, continue reaction 1h; Be cooled to 32 DEG C of crystallizatioies, filter, 75 DEG C of oven dry, obtain white solid 7.5g, and yield 39.2% is left-handed ropivacaine tartrate; After testing, ropivacaine purity 98.92 %, dextrorotatory isomer per-cent 0.99%.(4) preparation of Ropivacaine HCL crude productLeft-handed 7.5g ropivacaine tartrate is joined in 50ml Virahol, be warming up to 40 DEG C, drip concentrated hydrochloric acid, surveying pH is 1~2, insulation reaction 1h.Be cooled to 0 DEG C of crystallization, separate out a large amount of white solids, filter, dry, obtain the about 5.7g of white solid, yield 85.3%, is Ropivacaine HCL crude product.After testing, ropivacaine purity 99.12%, dextrorotatory isomer per-cent 0.96%.(5) refining5.7g crude product and 40ml dehydrated alcohol-concentrated hydrochloric acid mixed solution (20:1) are added in reaction flask, be heated to 50 DEG C and make to dissolve; Complete molten after, naturally cool to room temperature, ice-water bath is cooled to 0 DEG C, crystallization 2h; Filter, 10ml mixed solution washing for filter cake, obtains wet product, dries, and obtains white solid 5.5g, and yield 92.1%, is Ropivacaine HCL fine work.After testing, ropivacaine purity 99.92 %, dextrorotatory isomer per-cent 0.15%.The preparation method of embodiment 4, a kind of Ropivacaine HCLStep is as follows:(1) preparation of intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea10.0g2-piperidine carboxylic acid, 160ml toluene are added in 500ml reaction flask.Pass into HCl gas, to pH3 left and right, be warming up to 48 ± 2 DEG C, add 1.5mlDMF, drip 10.2g (1.1 equivalent) sulfur oxychloride and 20ml toluene mixture liquid, drip and finish, be incubated 48 ± 2 DEG C of reaction 6h.Drip 2 of 4.0 equivalents, 6-xylidine and 20ml toluene mixture liquid, be incubated 58 ± 2 DEG C of reaction 8h.Filter, obtain yellow-green colour wet product 64.2g, dry to obtain gray solid 55.6g, solid is added in 280ml purified water, stir the molten reaction solution that obtains; 10%NaOH solution is slowly dropped in reaction solution, adjust pH to 4.5-5.0 , use 100ml toluene wash, layering, retains water layer, continues to adjust pH to 9-10 with 10%NaOH solution, adds 100ml methylene dichloride.Layering, gets organic layer,and water layer continues to use 50ml dichloromethane extraction, merges organic layer, adds anhydrous sodium sulfate dehydration, 40 DEG C of concentrating under reduced pressure.Obtain the about 14.9g of pale yellow oily liquid body, yield 82.9%, is intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea.(2) preparation of intermediate (II) N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amideIntermediate 14.9 g of (the I) Dissolved in 60mlDMF IS, ADDS 8.5gK 2 cO . 3 , 7.8 g of drip (1.0 equivalent)-n-propyl bromide, and drip After Finishing of DEG BE Warming up to 75 C, Reaction Insulation 2H; IS Down Ice Bath to room temperature, filters, and filtrate is added in 150ml frozen water, separates out a large amount of white solids, filter, dry, obtain the about 16.1g of white solid, yield 92.0%, is intermediate (II) N-(2 ,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amide.(3) preparation of left-handed ropivacaine tartrate15g intermediate (II) is dissolved in 100ml Virahol, heats up 60 DEG C and stir molten; Treat entirely moltenly, add successively 1.72g (0.1 equivalent) titanium isopropylate, 1.82g (0.2 equivalent) D-tartrate, be warming up to backflow , after solution clarification, continue reaction 4h; Be cooled to 30 DEG C of crystallizatioies, filter, 75 DEG C of oven dry, obtain white solid 7.6g, and yield 39.7% is left-handed ropivacaine tartrate; After testing, ropivacaine purity 99.01 %, dextrorotatory isomer per-cent 1.05%.(4) preparation of Ropivacaine HCL crude productLeft-handed 7.6g ropivacaine tartrate is joined in 50ml Virahol, be warming up to 40 DEG C, drip concentrated hydrochloric acid, surveying pH is 1~2, insulation reaction 4h.Be cooled to 5 DEG C of crystallizatioies, separate out a large amount of white solids, filter, dry, obtain the about 5.7g of white solid, yield 85.3%, is Ropivacaine HCL crude product.After testing, ropivacaine purity 99.06%, dextrorotatory isomer per-cent 0.95%.(5) refining5.7g crude product and 40ml dehydrated alcohol-concentrated hydrochloric acid mixed solution (volume ratio 20:1) are added in reaction flask, be heated to 80 DEG C and make to dissolve; Complete molten after, naturally cool to room temperature, ice- water bath is cooled to 5 DEG C, crystallization 2h; Filter, 10ml mixed solution washing for filter cake, obtains wet product, dries, and obtains white solid 5.2g, and yield 91.2%, is Ropivacaine HCL fine work.After testing, ropivacaine purity 99.81%, dextrorotatory isomer per-cent 0.11%.The optical isomer method for detecting purity of left-handed ropivacaine tartrate, Ropivacaine HCL crude product and the Ropivacaine HCL purified product obtaining in above-described 1-4 is: measure according to high performance liquid chromatography (annex VD), with alpha- acid glycoprotein post (AGP, 100mm × 4.0mm, 5 μ m are suitable for); Agilent-1260 type high performance liquid chromatograph; (get potassium primary phosphate 2.72g with Virahol-phosphate buffered saline buffer, the 800ml that adds water dissolves, regulating pH value with 0.1mol/L sodium hydroxide solution is 7.1, be diluted with water to 1000ml) be (10:90) moving phase, detection wavelength is: 210nm, column temperature: 30 DEG C, flow velocity 1.0ml/min, limit is: dextrorotatory isomer must not be greater than 0.5%.
PATENThttps://patents.google.com/patent/CN109503465A/enThe embodiment of 1 intermediate (-) of-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamideL- piperidinecarboxylic acid hydrochloride (30.00g, 0.18mol), toluene are sequentially added in three mouthfuls of reaction flasks of 500ml cleaning N,N-Dimethylformamide (1ml), thionyl chloride (25.85g, 0.2 2mol) is added in (300ml) , stirring.It finishes, is warming up to 50~55 DEG C insulation reaction 3 hours.Snubber device is added to vacuumize 1 hour.The toluene solution of 2,6- dimethylaniline is added dropwise (2,6- dimethylanilines (109.75g , 0.91mol) are mixed with toluene (60ml)).It finishes, 60 DEG C of insulation reaction 2.0h.Cooling It to 20~30 DEG C, is added purified water (300ml), water phase is collected in layering; Fresh toluene (300ml), 10% hydrogen-oxygen is added in water phase Change sodium regulation system pH=6- 7, water phase is collected in layering; Water phase 10% sodium hydroxide regulation system pH=11~12,room temperature Stirring 4 hours, filter, purified water (150ml) elute filter cake, filter cake in 60 DEG C of air dry ovens it is dry 35.88g (yield 85%, HPLC purity 94.023% is calculated by areas of peak normalization method) .The purification of 2 intermediate (-) of-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide1 gained intermediate (-) of embodiment-(2S)-N- (2,6- diformazan is sequentially added in three mouthfuls of reaction flasks of 100ml cleaning Base phenyl) piperidines -2- formamide (5.00g, 21.52mmol), ether (50ml), stir and are warming up to reflux, flow back insulated and stirred 1 Hour, it is cooled to room temperature, insulated and stirred 1 hour, is filtered, ether (10ml) elutes filter cake, and filter cake is dry in 50 DEG C of air dry ovens Dry 2 hours 2.66g (yield 53.2% calculates HPLC purity 99.837% by areas of peak normalization method), map is shown in attached drawing 1.The purification of 3 intermediate (-) of-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide1 gained intermediate (-) of embodiment-(2S)-N- (2,6- diformazan is sequentially added in three mouthfuls of reaction flasks of 100ml cleaning Base phenyl) piperidines -2- formamide (5.00g, 21.52mmol), isopropyl ether (50ml), stir and are warming up to reflux, reflux heat preservation is stirred It mixes 1 hour, is cooled to room temperature, insulated and stirred 1 hour, filters, isopropyl ether (10m l) elutes filter cake, and filter cake is dry in 50 DEG C of air blast Dry 2 hours 3.45g of dry case (yield 69.0% calculates HPLC purity 99.332% by areas of peak normalization method).Map is shown in attached Fig. 2.The purification of 4 intermediate (-) of-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide1 gained intermediate (-) of embodiment-(2S)-N- (2,6- diformazan is sequentially added in three mouthfuls of reaction flasks of 100ml cleaning Base phenyl) piperidines -2- formamide (5.00g, 21.52mmol), methyl tertiary butyl ether(MTBE) (50ml), stir and are warming up to reflux, flow back Insulated and stirred 1 hour, be cooled to room temperature, insulated and stirred 1 hour, filter, methyl tertiary butyl ether(MTBE) (10ml) elutes filter cake, filter cake in Dry 2 hours 4.75g (yield 95% calculates HPLC purity 99.709% by areas of peak normalization method) of 50 DEG C of air dry ovens, Map is shown in attached drawing 3.5 intermediate (-) of embodiment-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide preparation and purificationA) the preparation of intermediate (-)-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamideL- piperidinecarboxylic acid hydrochloride (3kg, 18.1mol), toluene (30L) are added in 50L reaction kettle, N, N- is added in stirring Dimethylformamide (1L), thionyl chloride (2.59kg, 21.8mol). It finishes, is warming up to 50~55 DEG C of insulation reactions 3 hours. Snubber device is added to vacuumize 6 hours.Be added dropwise 2,6- dimethylaniline toluene solution (2,6- dimethylanilines (11kg, It 90.8mol) is mixed with toluene (6L)).It finishes, 60 DEG C of insulation reaction 2.0h. 20~30 DEG C are cooled to, is added purified water (30L), Water phase is collected in layering; Water phase is added fresh toluene (30L) , 10% sodium hydroxide regulation system pH=6-7, and water is collected in layering Phase; 10% sodium hydroxide regulation system pH=11~12 of water phase, are stirred at room temperature 4 hours, filter,purified water (15L) elution filter Cake, filter cake in 60 DEG C of air dry ovens it is dry 3.52kg (yield 84%, by areas of peak normalization method calculate HPLC purity 98.092%), map is shown in attached drawing 4.B) the purification of intermediate (-)-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamideIntermediate (-)-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide (3. is sequentially added in 50L reaction kettle 5kg, 15.1mol), methyl tertiary butyl ether(MTBE) (35L), stirring is warming up to reflux, flows back insulated and stirred 1 hour, be cooled to room temperature, guarantor Temperature stirring 1 hour, filters, and methyl tertiary butyl ether(MTBE) (7L) elutes filter cake, and filter cake is in the dry 8 hours 3.3kg of 50 DEG C of air dry ovens (yield 94% calculates HPLC purity 99.889% by areas of peak normalization method), map is shown in attached drawing 5.
Literatures:
Navinta LLC Patent: US2006/276654 A1, 2006 ; Location in patent: Page/Page column 5 ;
Yield: ~82%
Literatures:
US2006/276654 A1, ; Page/Page column 5 ;
Yield: null
SYN
https://pubs.rsc.org/en/content/articlehtml/2019/ra/c9ra09287k
Ropivacaine is the S-enantiomer of an N-alkyl pipecoloxylidine derivative, which is the first local anesthetic with chiral activity, and is widely used in clinical infiltration anesthesia, conduction anesthesia and epidural anesthesia. It has a long of local anesthesia and analgesic effect. However, ropivacaine also has serious safety risks in clinical practice. When the concentration of ropivacaine in human blood is too high, it may cause toxicity to the cardiovascular and central nervous system, and even cause allergic reactions in some patients. Thus far, the mechanism of the effect of ropivacaine on local anesthesia is not clear. Ropivacaine is a multitarget drug that acts on the gamma-aminobutyric acid a receptor (GABAA-R) and N-methyl-D-aspartate acid receptor (NDMA-R). Sodium (Na+) channels are a key target of local anesthetics and these two receptors regulate sodium channels. Previous studies on the structural modification of ropivacaine mainly focused on the substitution of –CH3 on the phenyl group or the substitution of –CH2CH2CH3 on piperidine with different alkyl groups. In 2017, Wen L. et al.69 reported the design and synthesis of ropivacaine analogues for local anesthesia. In the process of structural design, they used ropivacaine as the lead compound to design two series of compounds, 4a–4q (17 new substituted imines). In the first series of compounds, 4a–4i, different substituents were selected to replace –CH2CH2CH3 on piperidine. In the second series of compounds, 4j–4q, the methyl groups were replaced by –CF3 at the o-positions, m-positions and p-positions. Meanwhile, the –CH2CH2CH3 on piperidine ring was also substituted and modified. The process for the synthesis of the target compounds is shown in Scheme 8. The synthetic route takes piperic acid (compound 1) as the starting material, hydrochloric acid and sulfoxide chloride as additives, and toluene as the reaction solvent to convert compound 1 into acyl chloride salt (compound 2). Compound 2 was then treated with substituted aniline and reacted at 58 °C for 5 h to form compounds 3a–3i and 3j–3q. Finally, bromoalkyl and hydrochloric acid were used to treat compounds 3a–3i and 3j–3q. Potassium carbonate (K2CO3) was used as an acid dressing agent and dimethylformamide (DMF) as the reaction solvent in N-alkylation reaction. The N-alkylation reaction lasted 10 h at 80 °C, and the salt reaction lasted 5 min at room temperature to obtain the final target compounds 4a–4q. The total yield of the target compounds ranged from 17.5% to 87.7%. The synthetic route has the advantages of mild reaction conditions, cheap reagents and simple operation. However, using this synthetic route, the total yield of some products is too low, and too low yield will bring great problems to the synthesis cost, which needs to be further optimized in follow-up work. In the evaluation of the local anesthesia effect, sciatic nerve block activity, infiltration anesthesia activity, corneal anesthesia activity and spinal cord anesthesia activity were used as evaluation indexes. Ropivacaine was used as a positive control substance to test the local anesthesia activity in vitro. Firstly, the local anesthesia effect of all the target compounds 4a–4q was screened by a sciatic nerve block test in toads in vitro (Table 6). The preliminary screening results in vitro showed that these compounds increased the blocking effect of the sciatic nerve on electrical stimulation, with ED50 values ranging from 0.012 to 0.64 (positive control for ropivacaine was 0.013), with the highest activity shown by compound 4b. In terms of latent period, that of target compounds 4a–4q ranged from 27.7 to 59.4 min. Based on the results of the preliminary in vitro screening, compounds 4a, 4b, 4c, 4j and 4l were selected to test the efficacy of invasive anesthesia in guinea pigs. The results of the infiltration anesthesia test showed that the local anesthetic effect of compounds 4c and 4l was similar to that of the positive control ropivacaine, and the local anesthetic activity of other compounds was lower than that of the positive control. Furthermore, compounds 4a, 4b, 4c, 4j and 4l were used to test the local surface anesthesia effect of these compounds (Table 7). The results of the surface anesthesia test showed that compound 4l had a similar local anesthetic effect as the positive control ropivacaine, while the effect of the other compounds was poor in comparison with the positive control. Finally, compounds 4a, 4b, 4c, 4j and 4l were tested for spinal anesthesia in order to further study their local surface anesthesia effect. The experimental results showed that the ED50 produced by compounds 4l and 4b was 5.02 and 7.87, respectively, while the effects of compounds 4a, 4c and 4j were poor. The evaluation of local anesthesia in vitro found that compound 4l had the best activity, and thus molecular docking of compound 4l and ropivacaine was conducted to further study its local anesthesia mechanism. The molecular docking results showed that compound 4l interacts with receptor proteins of VGSC, GABAA-R and NDMA-R, which may help optimize and predict the activity of these ropivacaine analogues as potential local anesthetics.
| Scheme 8 Reagents and conditions: (a) (i) HCl,PhCH3, r.t., 1 h; (ii) PhCH3, SOCl2, 55 °C, 1 h; (b) substituted anilines, 58 °C, 5 h; and (c) (i) RBr, K2CO3, DMF, 80 °C, 10 h; (ii) HCl, r.t., 5 min. |
SYN
Prepn: A. F. Thuresson, C. Bovin, WO 8500599 (1985 to Apothekernes); H.-J. Federsel et al., Acta Chem. Scand. B41, 757 (1987).

CLIP
Ropivacaine hydrochloride was synthesized from L-2-pipecolic acid by successive reaction with SOCl2 and 2,6-dimethylaniline at 40 °C under ultrasonic irradiation to yield L-N-(2,6-dimethylphenyl)piperidin-2-carboxamide (4), and 4 was reacted with 1-bromopropane at 50 °C for 1 h under ultrasonic irradiation. The effects of reaction solvent, temperature and time under ultrasonic irradiation were investigated. Compared with conventional methods, present procedures have the advantages in milder conditions, shorter reaction time and higher yields. The total yield was 67.5%, [α]25 D= – 6.6°(c = 2, H2O).

SYN
Ropivacaine (3.1.37) (Naropin) is the pure S(–)-enantiomer of propivacaine released for clinical use in 1996. It is a long-acting, well tolerated local anesthetic agent and first produced as a pure enantiomer. Its effects and mechanism of action are similar to other local anesthetics working via reversible inhibition of sodium ion influx in nerve fibers. It may be a preferred option among other drugs among this class of compounds because of its reduced CNS and cardiotoxic potential and its lower propensity for motor block in the management of postoperative pain and labor pain [48–58].
The synthesis of ropivacaine (3.1.37) was carried out starting with l-pipecolic acid (3.1.34), prepared by a resolution of (±)-pipecolic acid with (+)-tartaric acid, which was dissolved in acetyl chloride and converted to acid chloride (3.1.35) with phosphorus pentachloride. The obtained compound (3.1.35) dissolved in toluene a solution of 2,6-xylidine (3.1.28) dissolved in the mixture of equal volumes of acetone, and N-methyl-2-pyrrolidone was added at 70°C to give (+)-l-pipecolic acid-2,6-xylidide (3.1.36). Reaction of this compound with propyl bromide in presence of potassium carbonate in i-PrOH/H2O gave the desired ropivacaine (3.1.37) [59] (Scheme 3.6).

Another approach for the synthesis of ropivacaine (3.1.37) was proposed via a resolution of enantiomers of chiral pipecolic acid-2,6-xylidide [60].
SYN

Scheme 21. Generation of ‘cation pool’ and its applications.
Reproduced from Yoshida, J.; Suga, S.; Suzuki, S.; et al. J. Am. Chem. Soc. 1999, 121, 9546–9549, and Shankaraiah, N.; Pilli, R. A.; Santos, L. S. Tetrahedron Lett. 2008, 49, 5098–5100.
CLIP
Process R&D under the magnifying glass: Organization, business model, challenges, and scientific context
Hans-Jürgen Federsel, in Bioorganic & Medicinal Chemistry, 2010
The synthesis of ropivacaine is achieved in only three steps, as in the previous example, comprised of a resolution of a racemic, commercially available starting material (pipecoloxylidide) followed by an N-alkylation and the final precipitation of the product as its HCl salt.14,24 Focusing on the middle step—the attachment of a propyl moiety onto the piperidine nitrogen—this reaction when developed in the laboratory and scaled up to maximum pilot plant volume (1000 L) behaved very well (Scheme 3). Thus, boiling the reaction mixture (reactants in a H2O/organic solvent mixture in the presence of a solid inorganic base) for an extended period of time (6 h) at high temperature (100 °C), the transformation was considered complete once a sample of the process solution showed <1% of remaining starting material. In preparation for launch, the method that had been thoroughly investigated and tested over a number of years and proven reliable on scale up had to be validated in the authentic 4000 L production equipment. Much to our surprise (and shock) we, however, found that the reaction came to a complete stand still long before reaching the expected end point. With a large amount of un-reacted starting material (30–40%) we were facing a situation that had never occurred during the lengthy development phase and this put the whole project in a very critical state as we were not able to reproduce the manufacturing method.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
History
Ropivacaine was developed after bupivacaine was noted to be associated with cardiac arrest, particularly in pregnant women. Ropivacaine was found to have less cardiotoxicity than bupivacaine in animal models.
Clinical use
Contraindications
Ropivacaine is contraindicated for intravenous regional anaesthesia (IVRA). However, new data suggested both ropivacaine (1.2-1.8 mg/kg in 40ml) and levobupivacaine (40 ml of 0.125% solution) be used, because they have less cardiovascular and central nervous system toxicity than racemic bupivacaine.[1]
Adverse effects
Adverse drug reactions (ADRs) are rare when it is administered correctly. Most ADRs relate to administration technique (resulting in systemic exposure) or pharmacological effects of anesthesia, however allergic reactions can rarely occur.
Systemic exposure to excessive quantities of ropivacaine mainly result in central nervous system (CNS) and cardiovascular effects – CNS effects usually occur at lower blood plasma concentrations and additional cardiovascular effects present at higher concentrations, though cardiovascular collapse may also occur with low concentrations. CNS effects may include CNS excitation (nervousness, tingling around the mouth, tinnitus, tremor, dizziness, blurred vision, seizures followed by depression (drowsiness, loss of consciousness), respiratory depression and apnea). Cardiovascular effects include hypotension, bradycardia, arrhythmias, and/or cardiac arrest – some of which may be due to hypoxemia secondary to respiratory depression.[2]
Postarthroscopic glenohumeral chondrolysis
Ropivacaine is toxic to cartilage and their intra-articular infusions can lead to Postarthroscopic glenohumeral chondrolysis.[3]
Treatment of overdose
As for bupivacaine, Celepid, a commonly available intravenous lipid emulsion, can be effective in treating severe cardiotoxicity secondary to local anaesthetic overdose in animal experiments[4] and in humans in a process called lipid rescue.[5][6][7]
References
- ^ (Basic of Anesthesia, Robert Stoelting, page 289)
- ^ Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006. ISBN 0-9757919-2-3
- ^ Gulihar A, Robati S, Twaij H, Salih A, Taylor GJ (December 2015). “Articular cartilage and local anaesthetic: A systematic review of the current literature”. Journal of Orthopaedics. 12 (Suppl 2): S200-10. doi:10.1016/j.jor.2015.10.005. PMC 4796530. PMID 27047224.
- ^ Weinberg G, Ripper R, Feinstein DL, Hoffman W (2003). “Lipid emulsion infusion rescues dogs from bupivacaine-induced cardiac toxicity”. Regional Anesthesia and Pain Medicine. 28 (3): 198–202. doi:10.1053/rapm.2003.50041. PMID 12772136. S2CID 6247454.
- ^ Picard J, Meek T (February 2006). “Lipid emulsion to treat overdose of local anaesthetic: the gift of the glob”. Anaesthesia. 61 (2): 107–9. doi:10.1111/j.1365-2044.2005.04494.x. PMID 16430560. S2CID 29843241.
- ^ Rosenblatt MA, Abel M, Fischer GW, Itzkovich CJ, Eisenkraft JB (July 2006). “Successful use of a 20% lipid emulsion to resuscitate a patient after a presumed bupivacaine-related cardiac arrest”. Anesthesiology. 105 (1): 217–8. doi:10.1097/00000542-200607000-00033. PMID 16810015.
- ^ Litz RJ, Popp M, Stehr SN, Koch T (August 2006). “Successful resuscitation of a patient with ropivacaine-induced asystole after axillary plexus block using lipid infusion”. Anaesthesia. 61 (8): 800–1. doi:10.1111/j.1365-2044.2006.04740.x. PMID 16867094. S2CID 43125067.
External links
- “Ropivacaine”. Drug Information Portal. U.S. National Library of Medicine.
- “Ropivacaine hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Naropin |
| AHFS/Drugs.com | Monograph |
| Pregnancy category | AU: B1 |
| Routes of administration | Parenteral |
| ATC code | N01BB09 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 87%–98% (epidural) |
| Metabolism | Liver (CYP1A2-mediated) |
| Elimination half-life | 1.6–6 hours (varies with administration route) |
| Excretion | Kidney 86% |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 84057-95-4 |
| PubChem CID | 175805 |
| IUPHAR/BPS | 7602 |
| DrugBank | DB00296 |
| ChemSpider | 153165 |
| UNII | 7IO5LYA57N |
| KEGG | D08490 as HCl: D04048 |
| ChEBI | CHEBI:8890 |
| ChEMBL | ChEMBL1077896 |
| CompTox Dashboard (EPA) | DTXSID4040187 |
| ECHA InfoCard | 100.128.244 |
| Chemical and physical data | |
| Formula | C17H26N2O |
| Molar mass | 274.408 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 144 to 146 °C (291 to 295 °F) |
| showSMILES | |
| showInChI | |
| (verify) |
Patent
Publication numberPriority datePublication dateAssigneeTitleUS4695576A *1984-07-091987-09-22Astra Lake Medel AktiebolagLNn-propylpipecolic acid-2,6-xylidideUS20050065345A1 *2001-09-102005-03-24Toshio TsuchidaMethod for producing pipecolamide derivativeCN103086954A *2013-02-042013-05-08Shandong Pharmaceutical Industry Research InstituteMethod for preparing ropivacaineCN104003930A *2014-06-132014-08-27Shandong Alura Pharmaceutical Research and Development Co., Ltd.Method for preparing hydrochloric acid ropivacaineCN107325041A *2017-06-202017-11-07Guangzhou Tonghui Pharmaceutical Co., Ltd.A kind of preparation method of Ropivacaine HCL
Non-Patent
TitleNAGULA SHANKARAIAH, etc.: “Enantioselective total syntheses of ropivacaine and its analogues”, “TETRAHEDRON LETTERS” *Liu Yi, et al.: “Synthesis of Ropivacaine Hydrochloride”, “Chinese Journal of Pharmaceutical Industry” *Ye Jiao, et al.: “Synthesis of Ropivacaine Hydrochloride by Triphosgene Method”, “Synthetic Chemistry” *Jiang Yao: “Study on the Synthetic Process of Ropivacaine Hydrochloride and Bupivacaine Hydrochloride”, “Engineering Science and Technology Series Ⅰ” *
/////////////Ropivacaine, Anesthetic, ропивакаин , روبيفاكائين , 罗哌卡因 , DRopivacaine Hydrochloride Hydrate, LEA-103, NA-001, (-)-LEA-103
CCCN1CCCC[C@H]1C(=O)NC1=C(C)C=CC=C1C

NEW DRUG APPROVALS
ONE TIME
$10.00
MIDAZOLAM


MIDAZOLAM
8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
59467-70-8 CAS NO OF FREE BASE
59467-94-6 MALEATE, Launched – 1982, Roche (Originator)
59467-96-8 (HCl)
A short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is used in dentistry, cardiac surgery, endoscopic procedures, as preanesthetic medication, and as an adjunct to local anesthesia. The short duration and cardiorespiratory stability makes it useful in poor-risk, elderly, and cardiac patients. It is water-soluble at pH less than 4 and lipid-soluble at physiological pH.
Midazolam (/mɪˈdæzəlæm/, marketed in English-speaking countries and Mexico under the trade names Dormicum, Hypnovel, andVersed,) is a short-acting drug in the benzodiazepine class developed by Hoffmann-La Roche in the 1970s. The drug is used for treatment of acute seizures, moderate to severe insomnia, and for inducing sedation and amnesia before medical procedures. It possesses profoundly potentanxiolytic, amnestic, hypnotic, anticonvulsant, skeletal muscle relaxant, and sedative properties.[6][7][8] Midazolam has a fast recovery time and is the most commonly used benzodiazepine as a premedication for sedation; less commonly it is used for induction and maintenance of anesthesia.Flumazenil, a benzodiazepine antagonist drug, can be used to treat an overdose of midazolam, as well as to reverse sedation.[7] However, flumazenil can trigger seizures in mixed overdoses and in benzodiazepine-dependent individuals, so is not used in most cases.[9][10]
midazolam
Administration of midazolam by the intranasal or the buccal route (absorption via the gums and cheek) as an alternative to rectally administereddiazepam is becoming increasingly popular for the emergency treatment of seizures in children. Midazolam is also used for endoscopyprocedural sedation and sedation in intensive care. The anterograde amnesia property of midazolam is useful for premedication before surgery to inhibit unpleasant memories. Midazolam, like many other benzodiazepines, has a rapid onset of action, high effectiveness and low toxicity level. Drawbacks of midazolam include drug interactions, tolerance, and withdrawal syndrome, as well as adverse events including cognitive impairment and sedation. Paradoxical effects occasionally occur, most commonly in children and the elderly, particularly after intravenous administration. The drug has also recently been hastily introduced for use in executions in the USA in combination with other drugs.
Midazolam is a short-acting benzodiazepine in adults with an elimination half-life of one to four hours; however, in the elderly, as well as young children and adolescents, the elimination half-life is longer. Midazolam is metabolised into an active metabolite alpha1-hydroxymidazolam. Age related deficits, renal and liver status affect the pharmacokinetic factors of midazolam as well as its active metabolite. However, the active metabolite of midazolam is minor and contributes to only 10 percent of biological activity of midazolam. Midazolam is poorly absorbed orally with only 50 percent of the drug reaching the bloodstream. Midazolam is metabolised by cytochrome P450 (CYP) enzymes and by glucuronide conjugation. The therapeutic as well as adverse effects of midazolam are due to its effects on the GABAA receptors; midazolam does not activate GABAA receptors directly but, as with other benzodiazepines, it enhances the effect of the neurotransmitter GABA on the GABAA receptors (↑ frequency of Cl− channel opening) resulting in neural inhibition. Almost all of the properties can be explained by the actions of benzodiazepines on GABAA receptors. This results in the following pharmacological properties being produced: sedation, hypnotic, anxiolytic, anterograde amnesia, muscle relaxation and anti-convulsant.Midazolam maleate is a benzodiazepine that is commercialized by Astellas Pharma and Roche as an intravenous or intramuscular injection for the long-term sedation of mechanically ventilated patients under intensive care. The drug is also available in a tablet formulation, and is currently distributed in various markets, including Germany, Japan, Switzerland and the U.K. In March 2002, two lots of a syrup formulation were recalled in the U.S. due to the potential presence of a crystalline precipitate of an insoluble complex of midazolam and saccharin. Subsequently, the injection and syrup formulations of the product were both withdrawn from the U.S. market. In 2010, a Pediatric Use Marketing Authorization (PUMA) was filed for approval in the E.U. by ViroPharma for the treatment of prolonged, acute, convulsive seizures in infants, toddlers, children and adolescents, from 3 months to less than 18 years. In 2011, a positive opinion was assigned to the PUMA and final approval was assigned in June 2011. The product was launched in the U.S. in November 2011. This product was filed for approval in Japan in 2013 by Astellas Pharma for the conscious sedation in dentistry and dental surgery. In the same year the product was approved for this indication.
In terms of clinical development, a nasal formulation of the drug is in phase III clinical trials at Upsher-Smith for rescue treatment of seizures in patients on stable anti-epileptic drug regimens who require control of intermittent bouts of increased seizure activity (seizure clusters). The Hopitaux de Paris had been developing a sublingual tablet formulation of midazolam to be used in combination with morphine for the treatment of pain in children following bone fractures; however, no recent development has been reported for this indication. NovaDel Pharma had been developing the compound preclinically for the treatment of generalized anxiety, however no recent developments have been reported.
Midazolam achieves its therapeutic effect through interaction with the gamma-aminobutyric acid benzodiazepine (GABA-BZ) receptor complex. Subunit modulation of the GABA-BZ receptor chloride channel macromolecular complex is hypothesized to be responsible for some of the pharmacological properties of benzodiazepines, which include sedative, anxiolytic, muscle relaxant, and anticonvulsive effects in animal models. GABA acts at inhibitory synapses in the brain by binding to specific transmembrane receptors in the plasma membrane of both pre- and post-synaptic neurons, opening ion channels and bringing about a hyperpolarization via either chloride or potassium ion flow.
In 2008, fast track designation was assigned to midazolam maleate in the U.S. for the treatment of seizure disorders.
In 2009, Orphan Drug Designation was received in the U.S. by for the treatment of seizure disorders in patients who require control of intermittent bouts of increased seizure activity (e.g. acute repetitive seizures, seizure clusters). This designation was assigned in the U.S. for the treatment of nerve agent-induced seizures.
In 2010, midazolam maleate was licensed to Upsher-Smith by Ikano Therapeutics for the treatment of acute repetitive seizure in patients with epilepsy. However, in 2010, Ikano closed and dissolved its business. Previously, Ikano had transferred to Upsher-Smith ownership of it nasal midazolam maleate program.
Midazolam is among about 35 benzodiazepines which are currently used medically, and was synthesised in 1975 by Walser and Fryer at Hoffmann-LaRoche, Inc in the United States.Owing to its water solubility, it was found to be less likely to cause thrombophlebitis than similar drugs.The anticonvulsant properties of midazolam were studied in the late 1970s, but not until the 1990s did it emerge as an effective treatment for convulsive status epilepticus. As of 2010, it is the most commonly used benzodiazepine in anesthetic medicine. In acute medicine, midazolam has become more popular than other benzodiazepines, such as lorazepam and diazepam, because it is shorter lasting, is more potent, and causes less pain at the injection site.Midazolam is also becoming increasingly popular in veterinary medicine due to its water solubility.
Midazolam is a water-soluble benzodiazepine available as a sterile, nonpyrogenic parenteral dosage form for intravenous or intramuscular injection. Each mL contains midazolam hydrochloride equivalent to 1 mg or 5 mg midazolam compounded with 0.8% sodium chloride and 0.01% edetate disodium with 1% benzyl alcohol as preservative, and sodium hydroxide and/or hydrochloric acid for pH adjustment. pH 2.9-3.7.
Midazolam is a white to light yellow crystalline compound, insoluble in water. The hydrochloride salt of midazolam, which is formed in situ, is soluble in aqueous solutions. Chemically, midazolam HCl is 8-chloro-6-(2-fluorophenyl)-1-methyl-4H– imidazo[1,5-a] [1,4] benzodiazepine hydrochloride. Midazolam hydrochloride has the molecular formula C18H13ClFN3•HCl, a calculated molecular weight of 362.25 and the following structural formula:
 |
In the Netherlands, midazolam is a List II drug of the Opium Law. Midazolam is a Schedule IV drug under the Convention on Psychotropic Substances. In the United Kingdom, midazolam is a Class C controlled drug. In the United States, midazolam (DEA number 2884) is on the Schedule IV list of the Controlled Substances Act as a non-narcotic agent with low potential for abuse.
midaolam hydrochloride NDA 018654, 075154
REF
U.S. Pat. No. 4,280,957
U.S. Pat. No. 5,693,795
U.S. Pat. No. 6,512,114
Midazolam Maleate
Drugs Fut 1978, 3(11): 822
Bioorganic and Medicinal Chemistry, 2012 , vol. 20, 18 pg. 5658 – 5667
Journal of Heterocyclic Chemistry, 1983 , vol. 20, 3 pg. 551 – 558.. 32 maleate
WO 2001070744
WO 2001002402
WO 2012075286
US2011/275799 A1… no 5
Journal of Organic Chemistry, 1978 , vol. 43, p. 936,942, mp free base, nmr
| US4280957 | May 15, 1978 | Jul 28, 1981 | Hoffmann-La Roche Inc. | Imidazodiazepines and processes therefor |
| US6262260 * | Mar 23, 2000 | Jul 17, 2001 | Abbott Laboratories | Process for the preparation of midazolam |
| US6512114 | Jun 30, 1999 | Jan 28, 2003 | Abbott Laboratories | Process for the preparation of Midazolam |
……………………….
introduction
4H-imidazo[1,5-a][1,4]benzodiazepines or, more simply, imidazobenzodiazepines, are a class of benzodiazepines having the general formula (I),

wherein the 1,4-diazepine ring is fused with a 1,3-imidazole ring. The main compounds part of the 4H-imidazo[1,5-a][1,4]benzodiazepines are Midazolam of formula (IV):

an active ingredient currently commercially available as a hydrochloride salt under the name of Versed or Hypnovel for anaesthetic and sedative use and the maleate salt currently commercially available under the name Dormicum or Flormidal.
Other important compounds are Climazolam of formula (VII):

Imidazenil of formula (VIII):

1-Hydroxymidazolam of formula (IX):

and Desmethyl midazolam of formula (X):

all these being biologically active substances and having psychotropic and sedative action.
The synthesis of the Midazolam as described in U.S. Pat. No. 4,280,957 of Hoffmann-La Roche provides for the decarboxylation reaction of the 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid of formula (VI) according to the following scheme:

The process for preparing the intermediate (VI) via basic hydrolysis of the corresponding ester is described in such patent publication and it is well known in the art.
The thermal decarboxylation reaction in high boiling solvent such as mineral oil at 230° C. for 5 min results in a mixture of products of Midazolam of formula (IV) and of Isomidazolam of formula (IV-bis), a non-pharmacologically active isomer, at a 80:20 ratio. The two products are separated by chromatography.
At industrial level, the formation of the Isomidazolam isomer impurity requires a further isomerisation reaction performed on the mixture of the two compounds to convert the isomer into the active product. The reaction mixture obtained from the thermal decarboxylation is thus subjected to basic treatment under the action of KOH in EtOH followed by an acid treatment which thus provides a mixture of Midazolam-Isomidazolam at a 95:5 ratio. The final removal of the Isomidazolam impurity from the product occurs through crystallisation of the product from AcOEt and EtOH. In order to limit this isomerisation treatment, in the subsequent U.S. Pat. No. 5,693,795 of Hoffmann-La Roche dated 1999, there is described a process for performing the decarboxylation of the compound of formula (VI) in n-butanol in a continuous tubular reactor with a 4 minutes permanence period with a yield between 47-77%. However, the reaction, performed at high temperature and pressure (280° C., 100 bars) results in the formation of a considerable percentage of Isomidazolam (85:15 Midazolam/Isomidazolam ratio) which still requires the basic isomerisation step.
Lastly, in U.S. Pat. No. 6,512,114 of Abbott Laboratories there is described the decarboxylation of the compound of formula (VI) in mineral oil or in N,N-Dimethylacetamide (DMA) at 160-230° C. for at least 3 hours obtaining a 3/1 to 6/1 Midazolam/Isomidazolam ratio with a yield of isolated product equal to just 54%.
Though performed using dedicated apparatus and in extreme conditions, the prior art processes do not allow selectively performing the decarboxylation reaction of the intermediate (VI) to Midazolam thus requiring a further synthetic passage followed by crystallisation with ensuing reduction of the overall yield.
Midazolam (8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine) is represented by the following structural formula (I):

Midazolam is a central nervous system (CNS) depressant, used for short term treatment of insomnia. Like other benzodiazepines, midazolam binds to benzodiazepine receptors in the brain and spinal cord and is thus used as a short-acting hypnotic-sedative drug with anxiolytic and amnestic properties. It is currently used in dentistry, cardiac surgery, endoscopic procedures, as a preanesthetic medication, as an adjunct to local anesthesia and as a skeletal muscle relaxant. Depending on the pH value, midazolam can exist in solution as a closed ring form (I) as well as an open ring form (IA), which are in equilibrium, as shown in Scheme 1:

The amount of the open ring form (IA) is dependent upon the pH value of the solution. At a pH value of about 3, the content of the open ring form (IA) can be 40%, while at pH value of 7.5, the closed ring form (I) can be more than 90%.
Clinical studies have demonstrated that there are no significant differences in the clinical activity between midazolam hydrochloride and midazolam maleate, however the use of intravenous midazolam hydrochloride has been associated, in some cases, with respiratory depression and arrest.
U.S Pat. No. 4,280,957 (hereinafter the ‘957 patent) describes a synthetic process for preparing midazolam, which is depicted in Scheme 2 below. This process includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) with acetic anhydride in dichloromethane to produce 2-acetamido-methyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (III). The latter is heated with polyphosphoric acid at 150° C. to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine of formula (IV), which is purified by column chromatography. Compound IV is then mixed with toluene and manganese dioxide and heated to reflux to afford midazolam base, which is crystallized from ether to yield a product with mp of 152-154° C.

The ‘957 patent further describes an alternative process which includes reacting 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-bezodiazepine (II) (optionally as a dimaleate salt) with triethylorthoacetate in ethanol and in the presence of p-toluenesulfonic acid to afford 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine (IV). This product is dissolved in xylene and treated with activated manganese dioxide to afford the crude base, which is reacted in situ with maleic acid in ethanol and crystallized by addition of ether to produce the midazolam maleate having melting point of 148-151° C. The process is depicted in Scheme 3 below.

The preparation of midazolam maleate from the isolated midazolam base is also described in a further example of the ‘957 Patent, wherein a warm solution of midazolam base in ethanol is combined with a warm solution of maleic acid in ethanol. The mixture is diluted with ether and at least part of the solvents is evaporated using a steam bath to obtain crystalline midazolam maleate having melting point of 148-151° C. The yield and the purity of the obtained midazolam maleate are not disclosed.
U.S. Pat. No. 6,512,114 (hereinafter the ‘114 patent) describes another synthetic process for preparing midazolam, which is depicted in Scheme 4 below. According to this Process, the starting material 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine-3-carboxylic acid (V) is heated in mineral oil for 3 hours at 230° C. until it is decarboxylated, followed by treatment with potassium tert-butoxide, to afford midazolm (I), isomidazolam (VI) and a midazolam dimmer (VII). Midazolam base is obtained in 54.5% yield after two re-crystallizations from ethyl acetate and heptane; however, the purity of the product is not disclosed.

The preparation of midazolam by conventional routes is liable to produce impurities such as isomidazolam (VI) and a midazolam dimmer (VII), and possibly other impurities. There is, therefore, a need in the art for a midazolam purification process that will provide highly pure midazolam containing minimal amounts of impurities produced. The present invention provides such a process.
This example describes the preparation of midazolam base as taught in the ‘957 patent.
16 g (0.03 mol) of 2-aminomethyl-7-chloro-5-(2-fluorophenyl)-2,3-dihydro-1H-1,4-bezodiazepine dimaleate was dissolved in 200 ml of toluene and 10 ml of 25% ammonium hydroxide solution was added and mixing was maintained for an hour. Then, the phases were separated and the toluene phase was dried by azeotropic distillation using a Dean Stark apparatus. 7 ml (0.038 mol) of triethylorthoacetate was added and the solution was heated to reflux for 4 hours, after which time the solution was left to cool to ambient temperature. 25 ml of methyl tert-butyl ether was added and the mixture was cooled overnight to produce 8-chloro-6-(2-fluorophenyl)-3a,4-dihydro-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine, which was isolated by filtration. The product was mixed with 200 ml of toluene and dried by azeotropic distillation using a Dean Stark apparatus. Then, 30 g of manganese dioxide was added and the mixture was heated to reflux for two hours. The excess manganese dioxide was filtered off to afford a solution of midazolam base in toluene, which was evaporated to obtain a product having 97.9% purity and containing 0.44% of impurity VIII and 1.14% of impurity IX (according to HPLC).
…………………………
EXAMPLE 28
2-Aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine dimaleate
A suspension of 17 g (0.05 m) of 7-chloro-1,3-dihydro-5-(2-fluorophenyl)-2-nitromethylene-2H-1,4-benzodiazepine-4-oxide in 200 ml of tetrahydrofuran and 100 ml of methanol was hydrogenated in presence of 17 g of Raney nickel at an initial pressure of 155 psi for 24 hrs. The catalyst was removed by filtration and the filtrate was evaporated. The residue was dissolved in 50 ml of 2-propanol and warmed on the steambath. A warm solution of 17 g of maleic acid in 60 ml of ethanol was added and the salt was allowed to crystallize by cooling in the ice bath. The final product consisted of yellow crystals with mp 196
EXAMPLE 14
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Acetic anhydride, 7 ml., was added to a solution of 6.16 g. of crude 2-aminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine in 200 ml. of methylene chloride. The solution was layered with 200 ml. of saturated aqueous sodium bicarbonate and the mixture was stirred for 20 minutes. The organic layer was separated, washed with sodium bicarbonate, dried over sodium sulfate and evaporated to leave 6.2 g. resinous 2-acetaminomethyl-7-chloro-2,3-dihydro-5-(2-fluorophenyl)-1H-1,4-benzodiazepine. This material was heated with 40 g. of polyphosphoric acid at 150 water, made alkaline with ammonia and ice and extracted with methylene chloride. The extracts were dried and evaporated and the residue (5.7 g.) was chromatographed over 120 g. of silica gel using 20% methanol in methylene chloride. The clean fractions were combined and evaporated to yield resinous 8-chloro-3a,4-dihydro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[ 1,5-a][1,4]benzodiazepine. A mixture of this material with 500 ml. of toluene and 30 g. of manganese dioxide was heated to reflux for 11/2 hours. The manganese dioxide was separated by filtration over celite. The filtrate was evaporated and the residue was crystallized from ether to yield a product with m.p. 152 was recrystallized from methylene chloride/hexane
EXAMPLE 49
8-Chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.625 g. (5.5 mmol), was added to a solution of 1.625 g. (5 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 nitrogen for 10 min. at -30 ml. of glacial acetic acid and was then partitioned between aqueous bicarbonate and toluene/methylene chloride (3:1 v/v). The organic layer was separated, dried and evaporated. The residue was chromatographed over 60 g. of silica gel using 25% (v/v) methylene chloride in ethyl acetate. The less polar product was eluted first and was crystallized from ethylacetate/hexane to yield product with m.p. 180
EXAMPLE 50
8-Chloro-6-(2-fluorphenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine
Potassium t-butoxide, 0.125 g. (1.1 mmol) was added to a solution of 0.325 g. (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-6H-imidazo[1,5-a][1,4]benzodiazepine in 20 ml. of dimethylformamide cooled to -30 -30 by addition of 0.2 ml. of glacial acetic acid and was partitioned between aqueous sodium bicarbonate and methylene chloridetoluene (1:3). The organic phase was washed with water, dried and evaporated. The residue was chromatographed over 20 g. of silica gel using ethyl acetate for elution. After elution of starting material, product was collected and crystallized from ether/hexane, m.p. 156
hyd and dihydrochloride
EXAMPLE 24
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine dihydrochloride
A solution of 0.32 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 5 ml of ethanol was treated with excess ethanolic hydrogen chloride. The salt was crystallized by addition of 2-propanol and ether. The colorless crystals were collected, washed with ether and dried to leave a final product with mp 290
EXAMPLE 258-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride
A solution of 0.325 g (1 mmol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 3 ml of ethanol was combined with a suspension of 0.4 g (1 mmol) of the dihydrochloride of this compound in 5 ml of ethanol. After filtration, the solution was treated with ether and heated on the steambath for 5 min to crystallize. The crystals were collected, washed with ether and dried to leave the monohydrochloride with mp 295
maleate
EXAMPLE 22
8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine maleate
A warm solution of 6.5 g (0.02 m) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine in 30 ml of ethanol was combined with a warm solution of 2.6 g (0.022 m) of maleic acid in 20 ml of ethanol. The mixture was diluted with 150 ml of ether and heated on the steam bath for 3 min. After cooling, the crystals were collected, washed with ether and dried in vacuo to yield a final product with mp 148
…
Synthesis
Midazolam, can be described according to scheme 4 indicated below:

 was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.
was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.For the reactions performed in the microreactor, the solutions containing the substrates to be decarboxylated were loaded into 5 and 10 mL gastight glass syringes (Hamilton, item n. 81527, 81627) mounted on syringe pumps (KD Scientifics, model KDS100). A pipe made of PTFE® (OD=1.58 mm, ID=0.8 mm, Supelco, item n. 58696-U) was used for making the reaction channel.A counterpressure valve sold by Swagelok (item n. SS-SS1-VH) was used for regulating the flow within the channel.Example 1Synthesis of the Compound of Formula (V)—Example of the Invention

50 g (0.135 mol) of 8-chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepin-3-carboxylic acid of formula (VI) and 250 mL of ethanol were loaded into a two-neck 500 mL flask, equipped with a magnetic stirrer. 40 mL of an aqueous solution of 1 M HCl are dripped in about 10 minutes. The open di-hydrochloride intermediate of formula (V) starts precipitating into the reaction environment already after 3 minutes from the beginning of the addition of the acid solution. The mixture is maintained stirred at RT for 3 hrs and then it is filtered on buckner washing the solid with ethanol. The moist product is dried in an oven under vacuum at 60° C. up to reaching a constant weight. A light yellow crystalline product is obtained (51.5 g, 83% yield). The crude product was used for the decarboxylation without further purifications.
ESI-MS [MeCN+0.1% HCOOH]: m/z 388 (V); 370 (VI).
1H-NMR (250 MHz, CD3OD): 2.52 (s, 3H); 4.27-4.41 (m, 2H); 7.22-8.1 (m, 7H). M.p.: 217° C.
Example 2
Synthesis of Midazolam of Formula (IV)—Performed in Batch—Example of the Invention

30 g (0.065 mol) of 5-(aminomethyl)-1-{(4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 90 mL of NMP are loaded into a three-neck 250 mL flask, equipped with a magnetic stirrer and coolant. The mass is heated using an oil bath at T=195-203° C. for one hour. Thus, 1 mL of solution is collected for performing HPLC analysis. The reaction product is Midazolam having 82% titre (w/w) (determined via HPLC titre correcting it using the solvent) and it contains 1% of Isomidazolam. The product is extracted using Isopropyl acetate after raising the pH to 10 by adding aqueous Na2CO3.
Example 3
Synthesis of Midazolam of Formula (IV)—Performed in a Micro-Reactor—Example of the Invention

3.22 g (7 mmol) of 5-(aminomethyl)-1-{4-chloro-2-[(2-fluorophenyl)carbonyl]phenyl}-2-methyl-1H-imidazole-4-carboxylic acid dihydrochloride of formula (V) and 10 mL of NMP are loaded into a 10 mL flask equipped with a magnetic stirrer. In order to facilitate the complete solubilisation of the substrate, it is necessary to slightly heat the reaction mixture (about 40° C.) for a few minutes. The solution thus obtained is transferred into a 10 mL gastight glass syringe mounted on a KDS100 syringe pump (FIG. 1) and the flow is regulated at 1.0 mL/h so as to set a residence period of 30 minutes at 200° C. The reaction product is Midazolam having an 89% titre (w/w) (determined via HPLC titre correcting it using the solvent) and containing 3% (w/w) of Isomidazolam.
Example 4Synthesis of Midazolam of formula (IV)—Comparison of the InventionA table is reported which summarises the results of the decarboxylation of the compound of formula (V) and (V-bis) (for the latter see Examples 6 and 7) obtained according to some embodiments of the invention and those obtained by way of experiment through the decarboxylation of the intermediate of formula (VI) (process of the prior art) both performed in 3 volumes of NMP at 200° C., both in batch method (Example 4) and in continuous method with the microreactor (MR) made of PTFE of FIG. 1. (Examples 4-1, 4-2, 4-3).
| Example | substrate | Mode | Solv. | T° C. | t min. | Midazolam (p/p) | Isomidaz. (P/P) |
| 2 | (V) | Batch | NMP | 200 | 60 | 82 | 1 |
| 3 | (V) | MR | NMP | 200 | 30 | 89 | 3 |
| 7 | (V-bis) | Batch | NMP | 200 | 60 | 68 | 3 |
| 4 | (VI) | Batch | NMP | 200 | 60 | 78 | 18 |
| 4-1 | (VI) | MR | NMP | 200 | 38 | 81 | 17 |
| 4-2 | (VI) | MR | NMP | 200 | 20 | 77 | 18 |
| 4-3 | (VI) | MR | NMP | 200 | 15 | 58 | 22 |
| U.S. Pat. No. | (VI) | Tubular | n-BuOH | 290 | 4 | 85 * | 15 * |
| 5,693,795 | reactor | ||||||
| U.S. Pat. No. | (VI) | Batch | Olio | 230 | 180 | 75 * | 25 * |
| 6,512,114 | min. | 87.5 * | 12.5 * | ||||
| or DMA | |||||||
| * = Midazolam/Isomidazolam ratio only (other impurities not considered). | |||||||
The product of the comparative experiments 4, 4-1, 4-2, 4-3 and of the two USA patents should be subjected to a further isomerisation process to reduce the high amount of Isomidazolam so as to be able to obtain Midazolam free of Isomidazolam after further crystallization, which would not be required for the product obtained according to the invention (examples 2 and 3).

A 4-neck RBF was charged under nitrogen flow with: 10 g of Midazolam (IV) (prepared according to example 2) and 40 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following solution: 3.72 g of maleic acid are dissolved in 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. The maleic acid solution is dropped in 30/40 minutes and keeping T=25/30° C. into the solution containing Midazolam. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 40 mL of cool Ethanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. 12.8 g of Midazolam Maleate as white solid were collected (Molar yield=94.5%). m.p.=149-152° C. (by DSC).

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 15 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. 5 mL of a ethanolic solution of Hydrochloric acid 2N were slowly added. 20 mL of Isopropanol were added over 30 minutes at RT. The slurry was cooled down at −15° C. in one hour and kept at that temperature for at least 2 hours. The slurry was then filtered and the cake was washed with 10 mL of cool isopropanol. The filter was discharged and the product was dried at 40° C. under vacuum for 2 hours and then at 60° C. for 8 hours. Midazolam dihydrochloride as white solid was collected.
MIDAZOLAM HYDROCHLORIDE
Example 10
Preparation of 8-Chloro-6-(2-fluorophenyl)-1-methyl-4H-imidazo[1,5-a][1,4]benzodiazepine hydrochloride (Midazolam hydrochloride)

A 4-neck RBF was charged under nitrogen flow with: 1 g of Midazolam (IV) (prepared according to example 2) and 10 mL of Ethanol. The slurry was stirred until complete dissolution at 25/30° C. In an other flask was prepared the following suspension: 1.22 g of Midazolam dihydrochloride (prepared according to example 9) and 15 mL of Ethanol. The Midazolam ethanolic solution was added to the Midazolam dihydrochloride suspension. After filtration, the solution was treated with MTBE and heated at 60° C. until crystallization. After cooling to RT, the slurry was filtered, the cake washed with MTBE and the product was dried to provide Midazolam (mono)hydrochloride as a white solid.
…..


NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
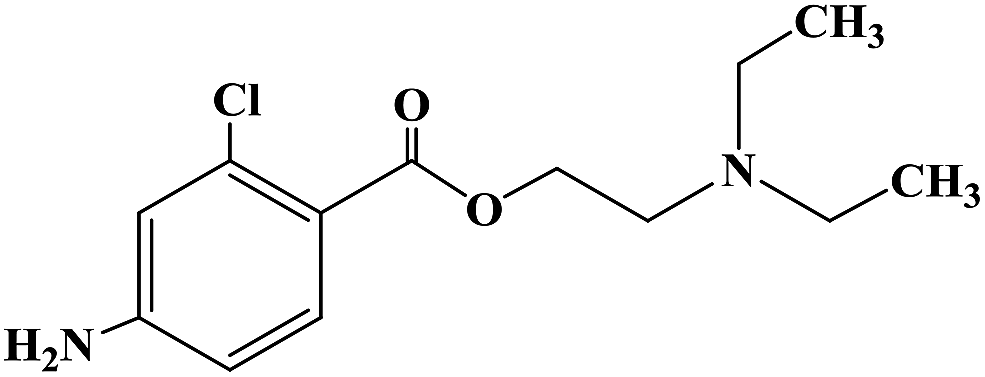{kind=link}
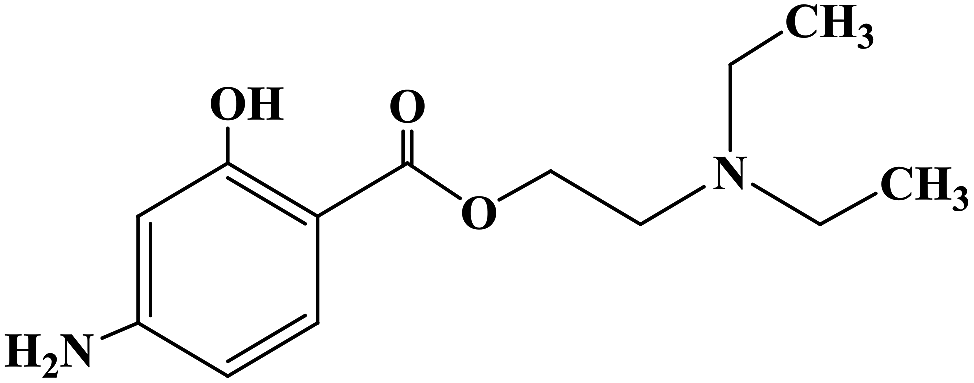{kind=link}
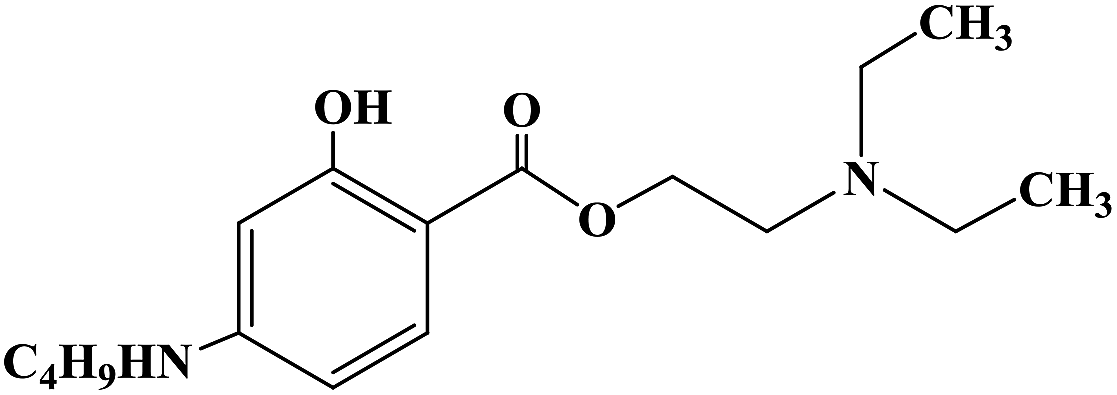{kind=link}
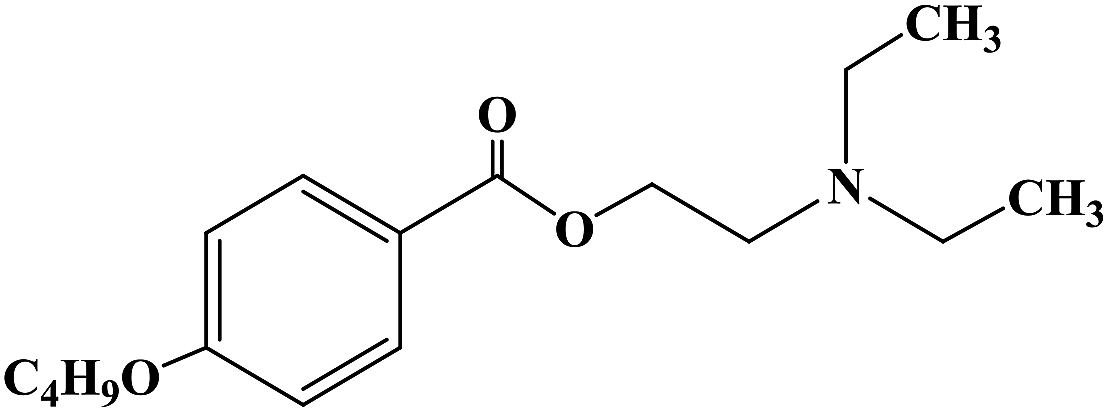{kind=link}
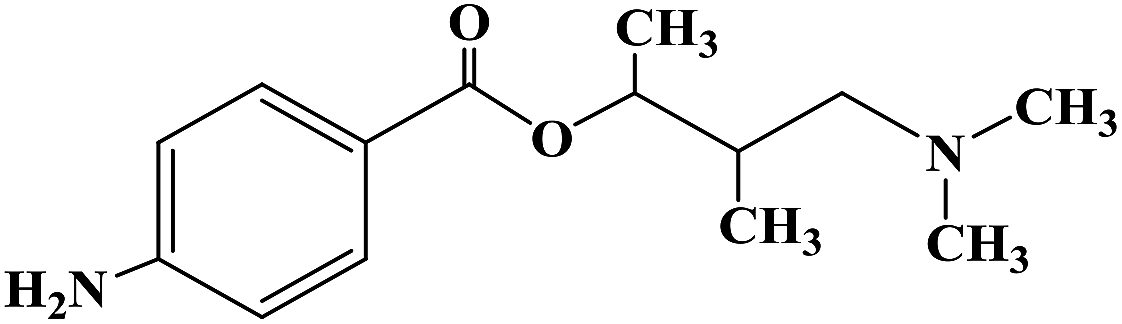{kind=link}
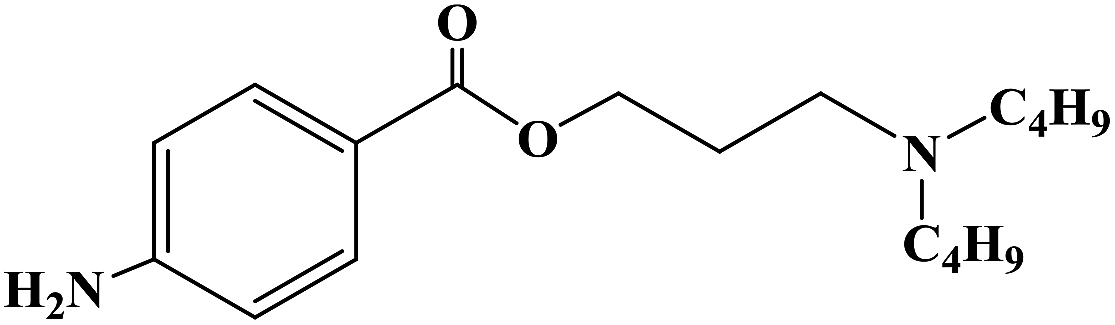{kind=link}
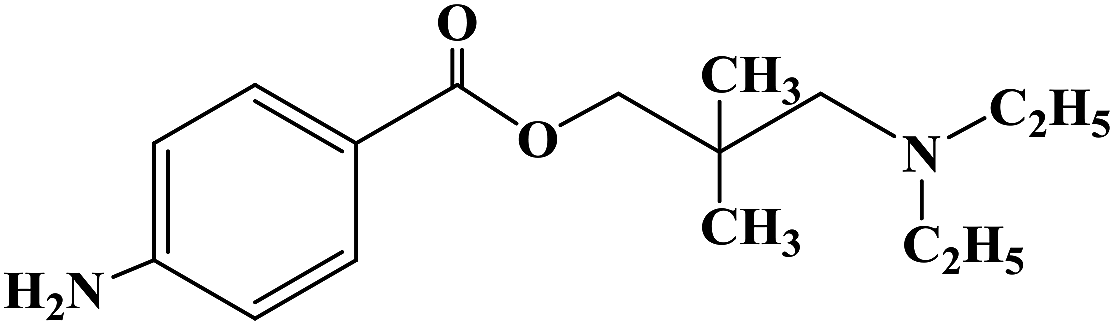{kind=link}
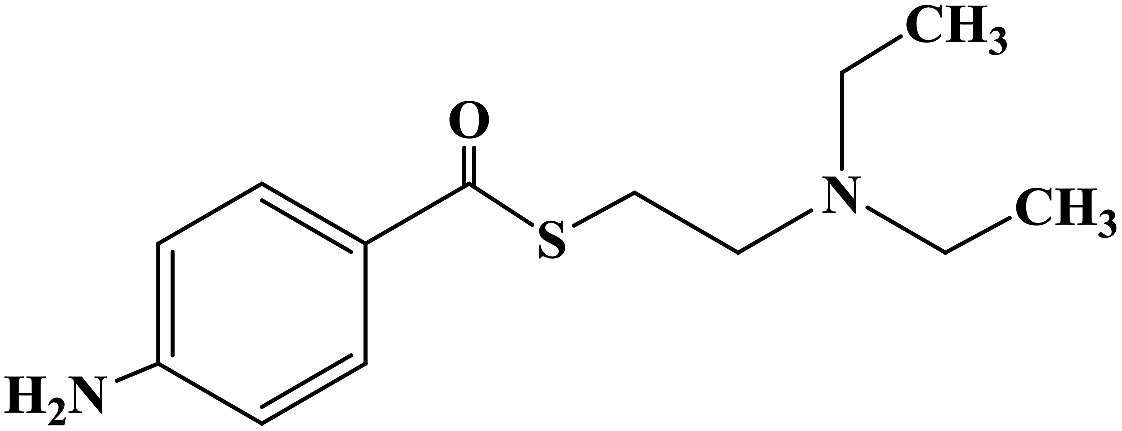{kind=link}
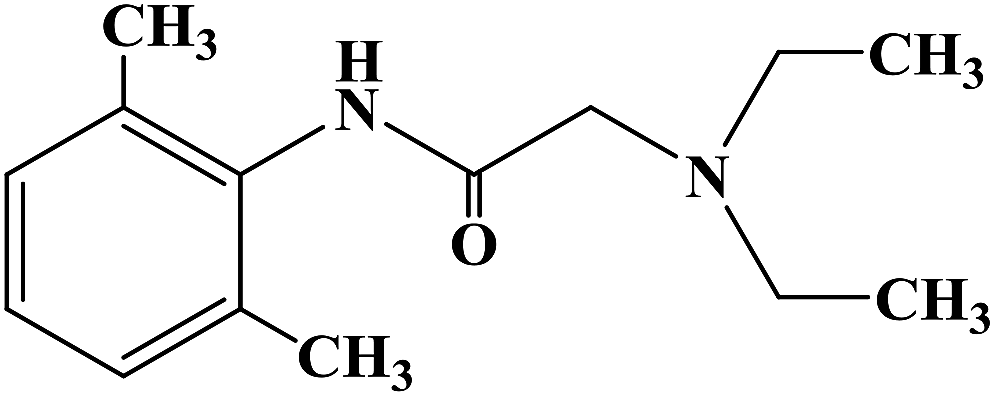{kind=link}
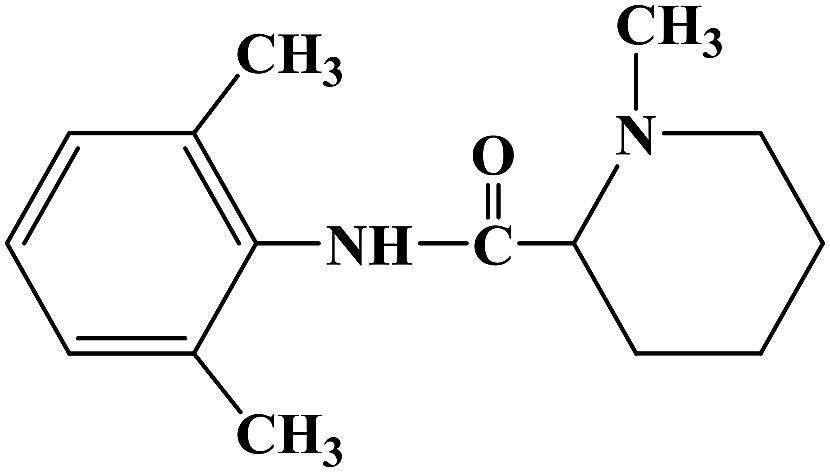{kind=link}
{kind=link}
{kind=link}
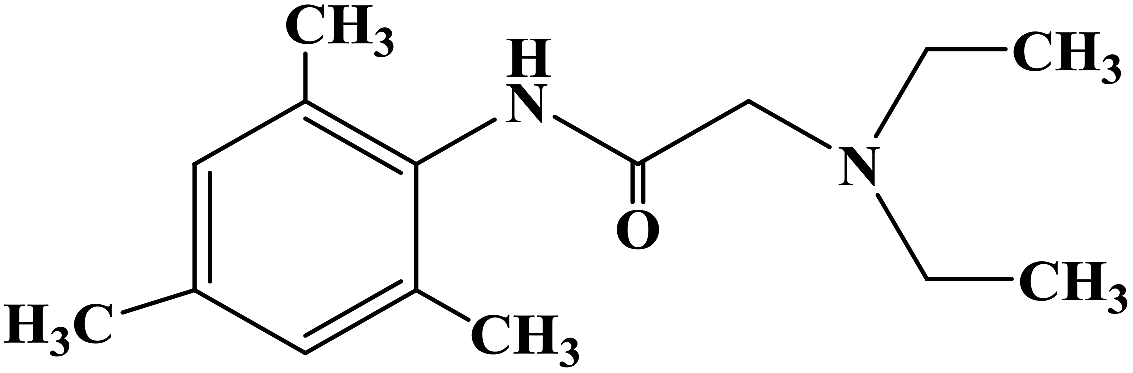{kind=link}
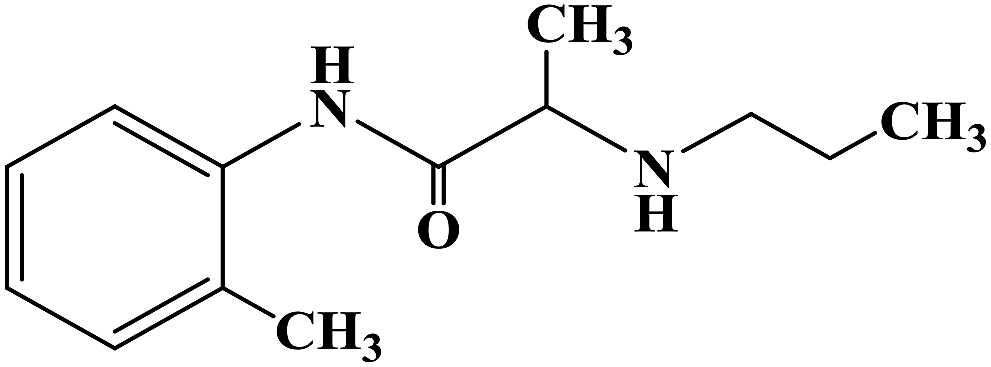{kind=link}
{kind=link}
{kind=link}
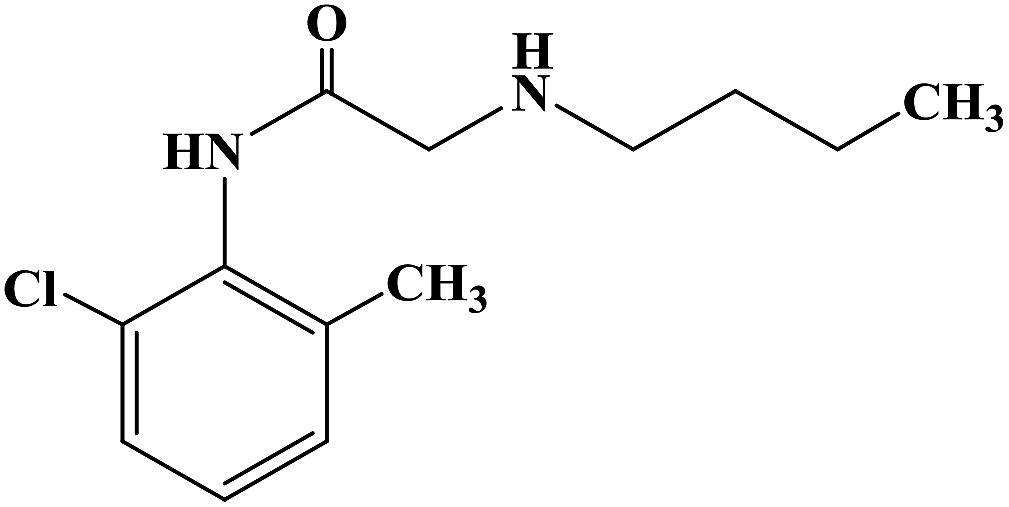{kind=link}
{kind=link}
{kind=link}
{kind=link}
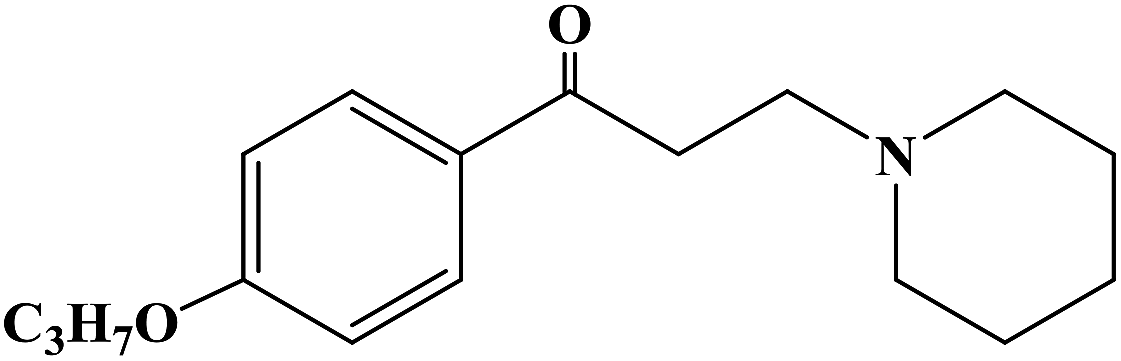{kind=link}
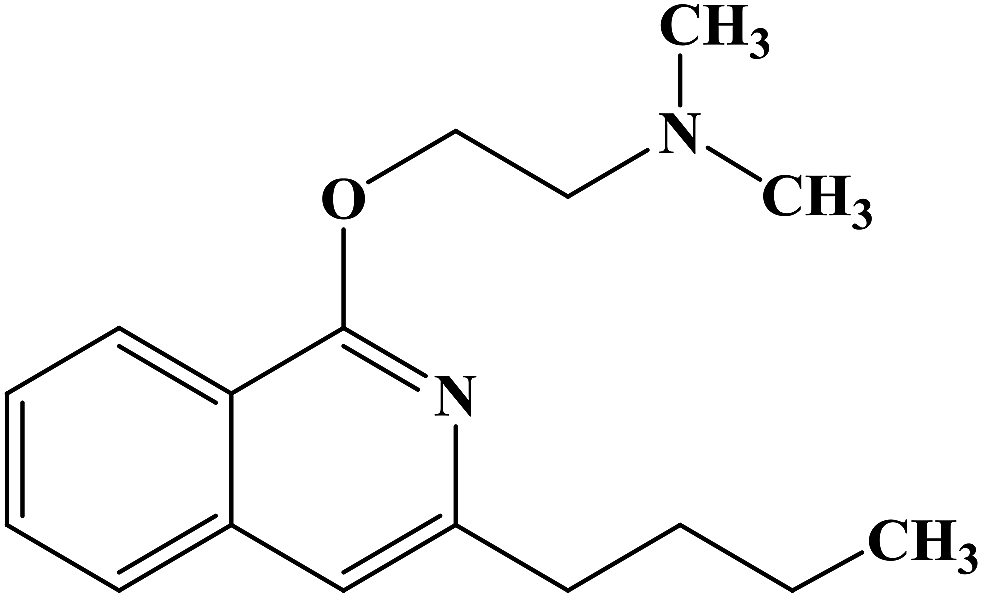{kind=link}
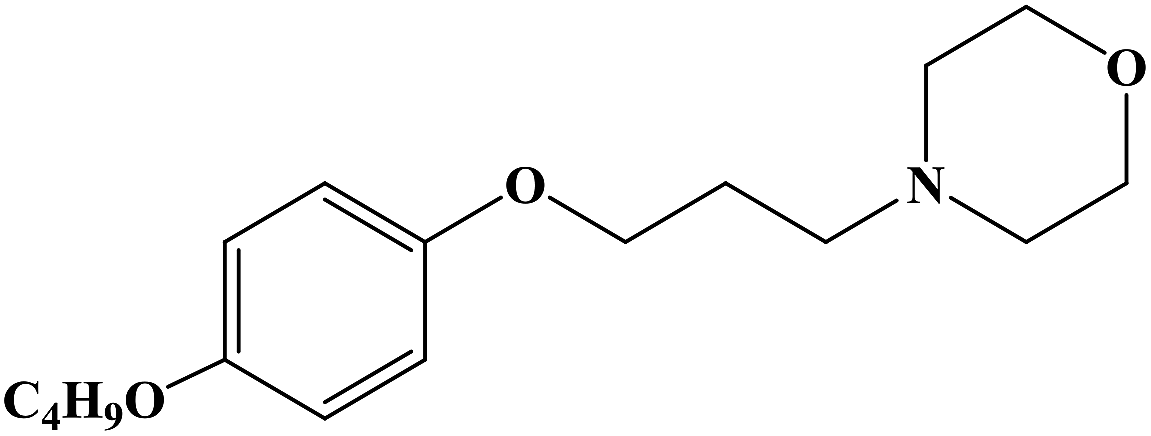{kind=link}
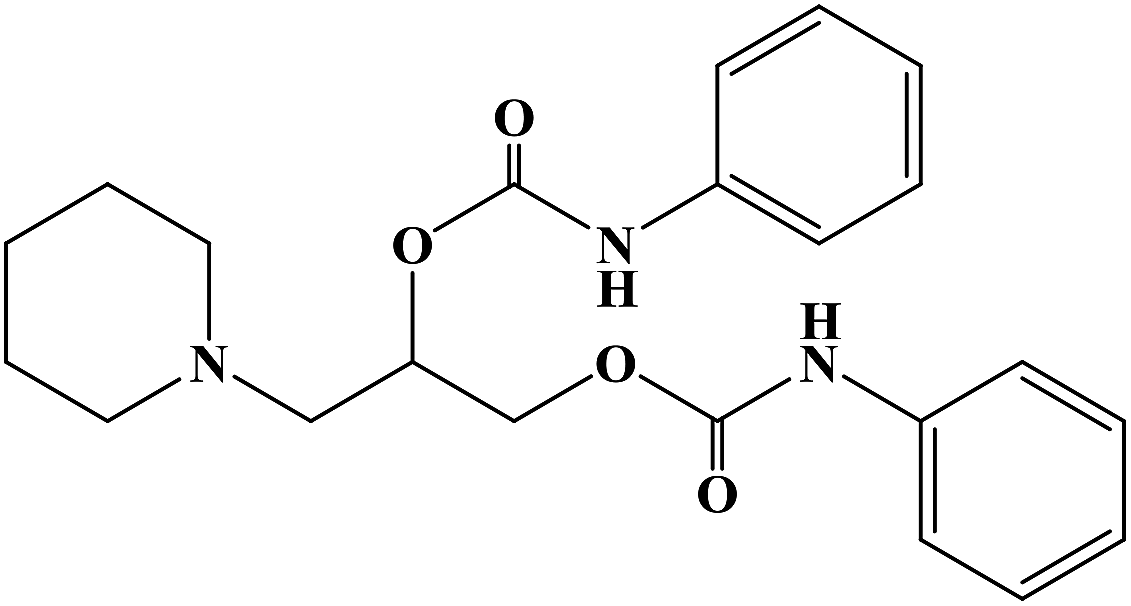{kind=link}
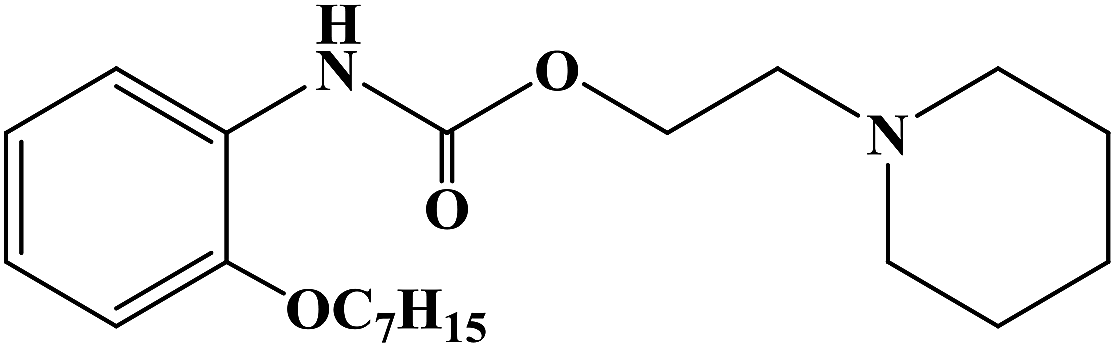{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
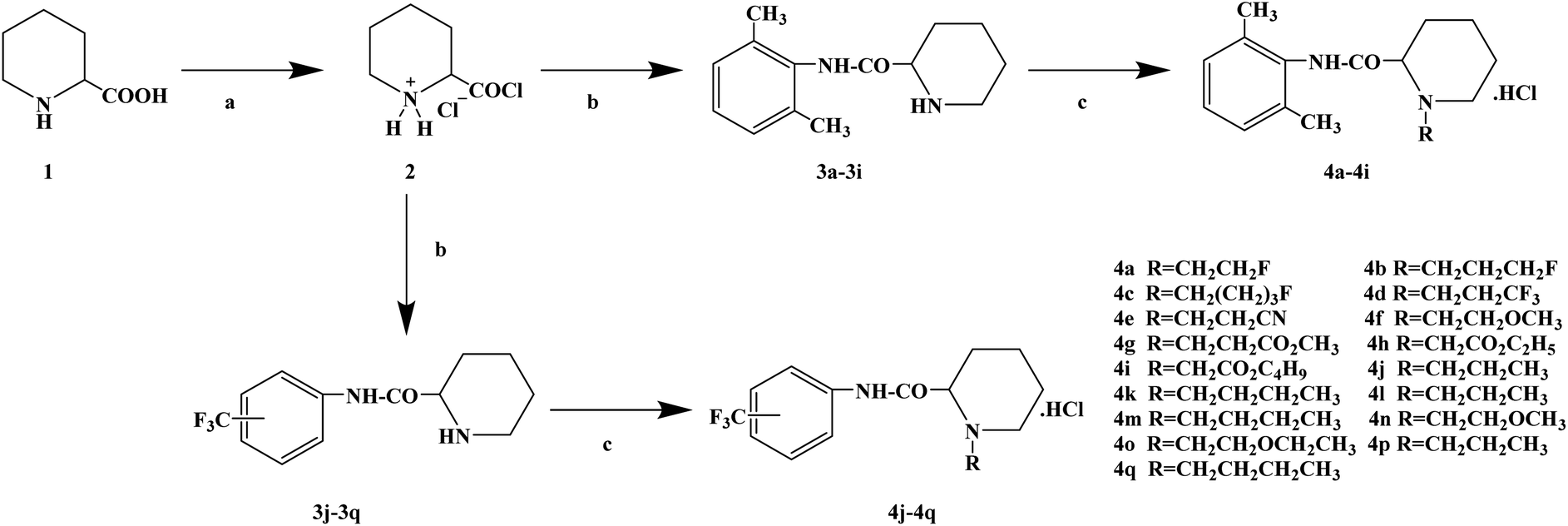{kind=link}
{kind=link}
{kind=link}