
Home » x ray contrast agent
Category Archives: x ray contrast agent
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Gadoquatrane
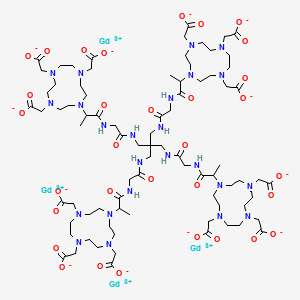
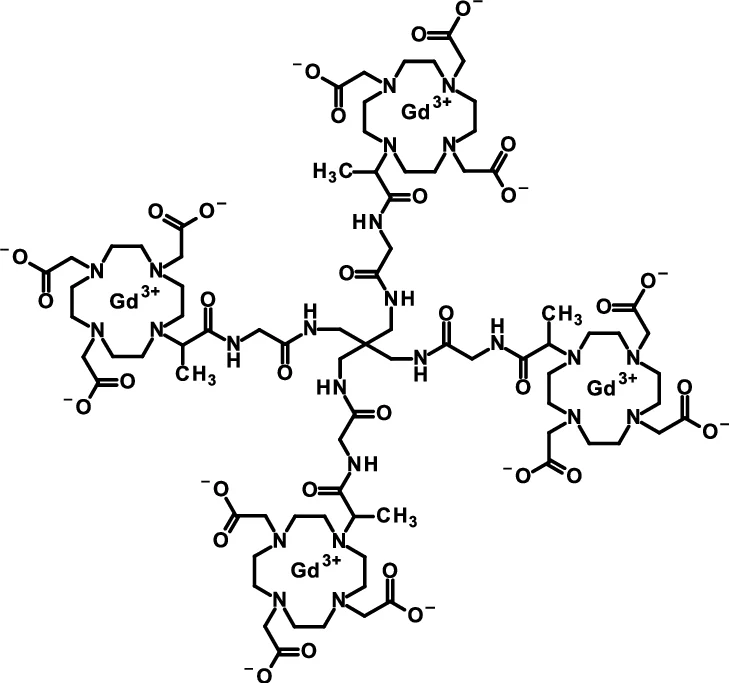
Gadoquatrane
CAS2048221-65-2MW2579.0 g/mol
FDA 2026, APPROVALS 2026, Ambelvist, OZG7J613HK, BAY-1747846, BAY 1747846
2-[4,10-bis(carboxylatomethyl)-7-[1-oxo-1-[[2-oxo-2-[[3-[[2-[2-[4,7,10-tris(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]propanoylamino]acetyl]amino]-2,2-bis[[[2-[2-[4,7,10-tris(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]propanoylamino]acetyl]amino]methyl]propyl]amino]ethyl]amino]propan-2-yl]-1,4,7,10-tetrazacyclododec-1-yl]acetate;tetrakis(gadolinium(3+))
To detect and visualize lesions with abnormal vascularity, in conjunction with MRI
Gadoquatrane (marketed as AMBELVIST®) is a low-dose, macrocyclic gadolinium-based contrast agent (GBCA) developed by Bayer for use in magnetic resonance imaging (MRI). It is designed to enhance the visualization of lesions in the central nervous system (CNS) and other body regions in adult and pediatric patients.
Core Highlights:
- Lower Gadolinium Exposure: It requires a dose of 0.04 mmol/kg, which results in 60% less gadolinium exposure compared to standard macrocyclic GBCAs.
- Regulatory Approval: The FDA approved it in June 2026 for use in adults and pediatric patients, including term neonates. It was also approved in Japan in March 2026.
- Efficacy & Safety: Phase III clinical trials (the QUANTI studies) showed it effectively detects lesions with abnormal vascularity while maintaining an efficacy and safety profile comparable to other standard macrocyclic agents.
- Structure: Gadoquatrane features a tetrameric, macrocyclic structure that gives it high relaxivity and stability in the body
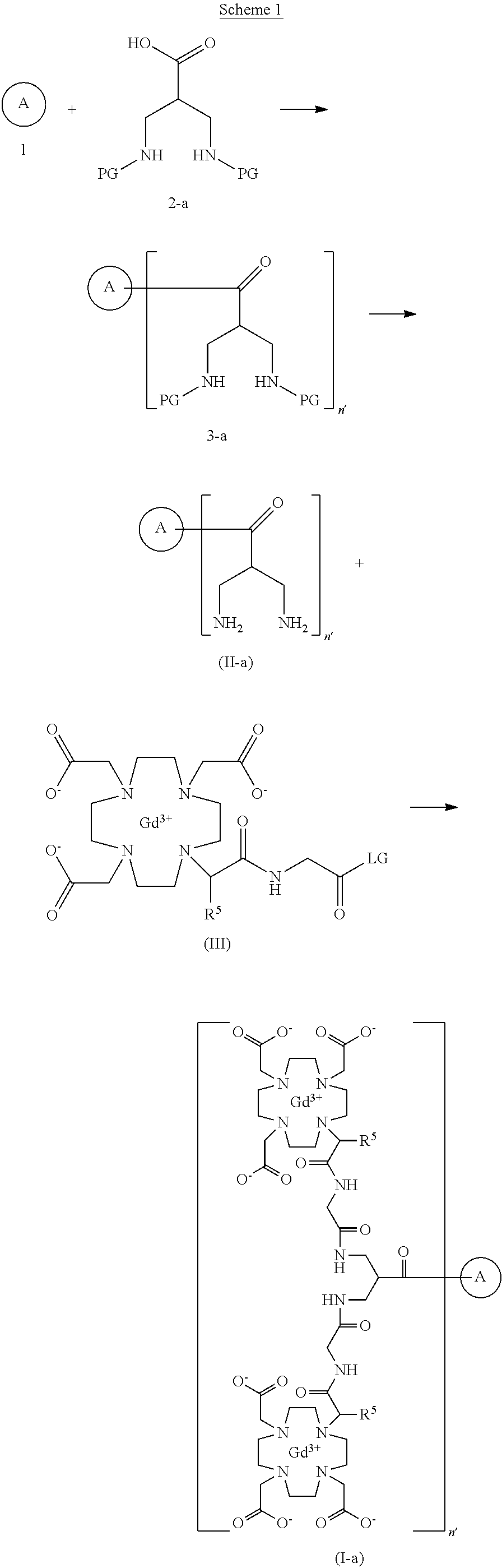
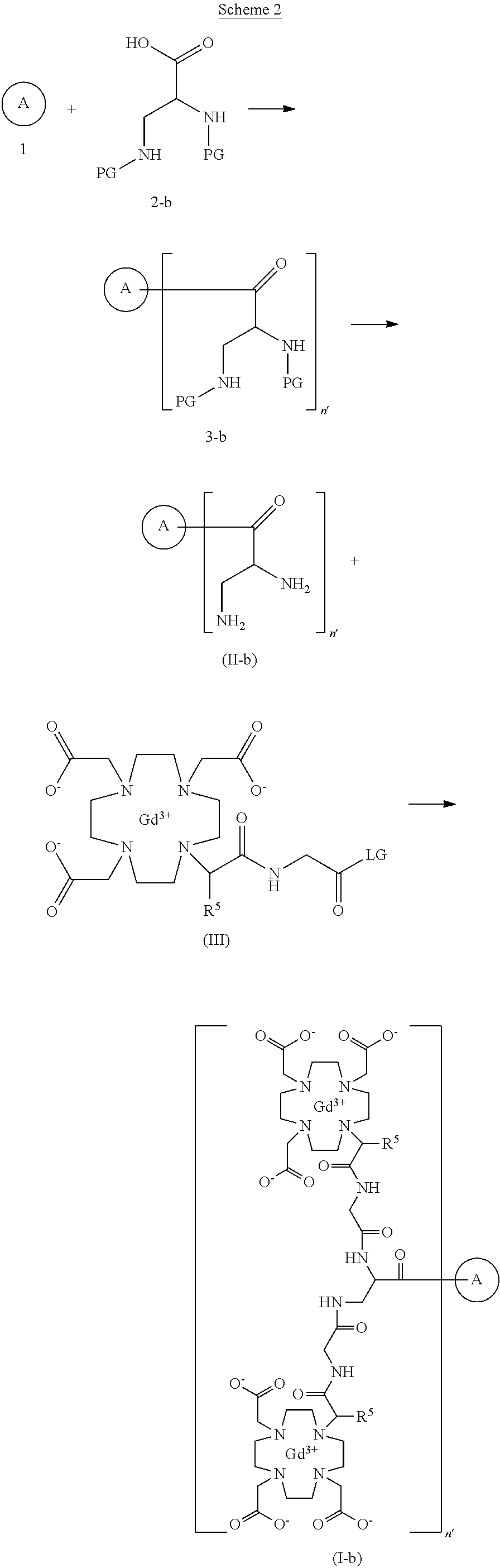
SYN
https://patents.google.com/patent/US20250114485A1
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma



AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

PAT
- Gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: US-11491245-B2Priority Date: 2015-06-04Grant Date: 2022-11-08
- Gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: US-2025114485-A1Priority Date: 2015-06-04
- Formulation of contrast media and process of preparation thereofPublication Number: US-2024252690-A1Priority Date: 2018-11-23
- Formulation of contrast media and process of preparation thereofPublication Number: US-12303573-B2Priority Date: 2018-11-23Grant Date: 2025-05-20
- New gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: EP-3611169-A1Priority Date: 2015-06-04
- New gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: US-2023113481-A1Priority Date: 2015-06-04
- New gadolinium chelate compounds for use in magnetic resonance imagingPublication Number: EP-3303307-B1Priority Date: 2015-06-04Grant Date: 2019-09-04
- Generation of artificial contrast-enhanced computed tomography imagesPublication Number: US-2024346718-A1Priority Date: 2023-04-13
- Generation of artificial contrast-enhanced radiological imagesPublication Number: WO-2024100233-A1Priority Date: 2022-11-12
- Generation of artificial contrast-enhanced radiological imagesPublication Number: EP-4616420-A1Priority Date: 2022-11-12
- Automated analysis of radiological imagesPublication Number: WO-2024083466-A1Priority Date: 2022-10-17
- Formulation of contrast media and process of preparation thereofPublication Number: US-11944690-B2Priority Date: 2018-11-23Grant Date: 2024-04-02
/////////gadoquatrane, anax labs, FDA 2026, APPROVALS 2026, Ambelvist, OZG7J613HK, BAY-1747846, BAY 1747846
Gadosircoclamide
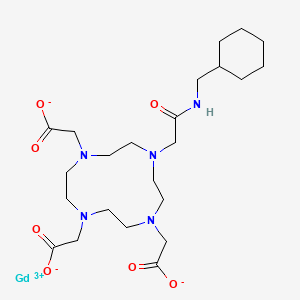
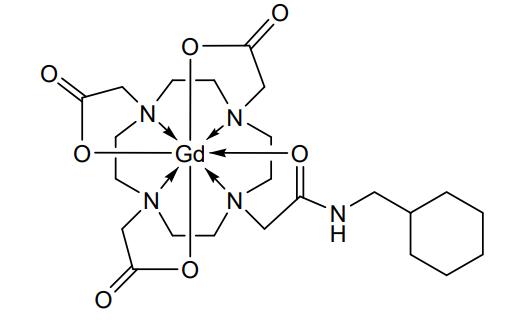
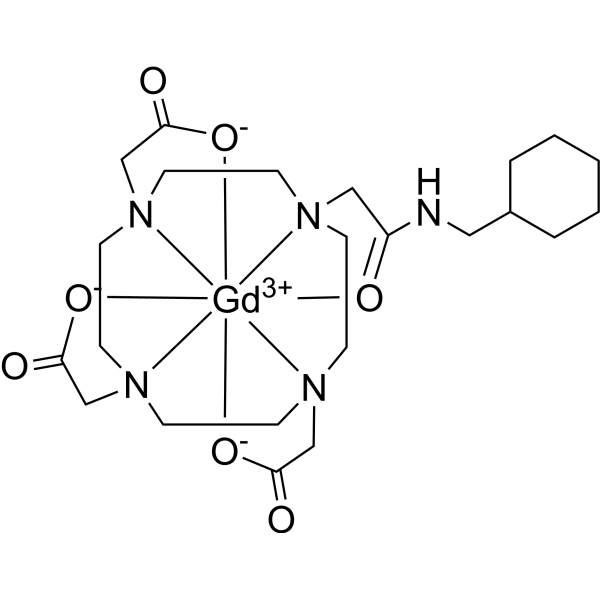
Gadosircoclamide
CAS 1801159-68-1
MF C23H38GdN5O7. MW653.8 g/mol
2-[4,7-bis(carboxylatomethyl)-10-[2-(cyclohexylmethylamino)-2-oxoethyl]-1,4,7,10-tetrazacyclododec-1-yl]acetate;gadolinium(3+)
- [10-[2-[(Cyclohexylmethyl)amino]-2-(oxo-kappaO)ethyl]-1,4,7,10-tetraazacyclododecane-1,4,7-triacetato(3-)-kappaN1,kappaN4,kappaN7,kappaN10,kappaO1,kappaO4,kappaO7]gadolinium
- [2,2′,2”-(10-{2-[(cyclohexylmethyl)amino]-2-oxo-kappaOethyl}-1,4,7,10-tetraazacyclododecane-1,4,7-triyl-kappa4N1,N4,N7,N10)tri(acetato-kappaO)]gadolinium
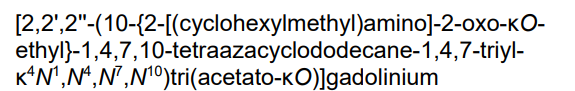
radiodiagnostic agent, 7V6P6PCM4U
Gadosircoclamide (CAS # 1801159-68-1) is a specialized gadolinium-based coordination complex used primarily as a magnetic resonance imaging (MRI) contrast agent. It is designed to enhance image contrast, help visualize lesions, and accurately track abnormalities during diagnostic scans.
SYN
R=H
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015105352&_cid=P11-MPXG49-92326-1
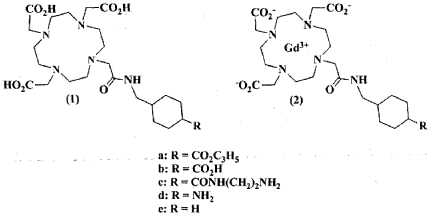
4) Synthesis of le.
DG3A-(f BuO)3 (3.0 g, 5.8 mmol) was added to a solution of 2-chlorocyclohexylmethylacetamid (1.2 g, 6.4 mmol) in acetanitrile (30 mL) prepared according to the conventional literature method (Cho, SD; Song, SY; Kim, . H.; Zhao, BX; Ahn, C; J oo, WH; Yoon, YJ; Falck, JR; Shin, DS B / (or. Chem. St. 2004, 25, 415) . The solution was stirred at room temperature for 24 hours. Solid impurities were removed by filtration, and the filtrate was evaporated under vacuum to obtain an oil phase residue. Subsequently, column chromatography on a silica phase (gradient elution: CH₂C1₂ to 10 % MeOH -CH₂Cl₂ , R f = 0.4 ( MeOH/ CH₂Cl₂ = A 1:9 mixture was performed and evaporated under reduced pressure to obtain a yellowish-white solid. As described in the preparation of the above Id, deprotection with TFA was performed to obtain a yellowish-white solid as a product. Yield: 2.4 g (82%). 1H R ( O): δ = 3.74/3.57 (m, 8H, -NCH₂CO₂- ) , 3.30 (m, 10H , overlapped -NCH₂CH₂N- ( 8H) & -CONHCH₂- ( 2H )), 3.10 (m, 8H, -NCH₂CH₂N- ) , 1.98/1.44/1.27 (in, 4H, -CH₂- , cyclohexyl ) , 1.88 (m, 1H, -NHCH₂CH- ) . Anal . Calculated for C₂₂H₃₅N₅₀ 0 7 · 3CF 3 C00H ■ 3H 2 0 : C, 38.14; H, 5.49; N, 7.94. Found: C, 37.83; H, 5.76; N, 8.44. MALDI-T0F MS (m/z): Calcd for C22H39N5O7,: 485.28, Found: 486.42 ([MH] + ), 508.44 ([MNa] + ).
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
[1].
Kim T, et al., Gadolinium complex comprising do3a-tranexamic acid conjugate. WO2015105352
////////////gadosircoclamide. anax labs, radiodiagnostic agent, 7V6P6PCM4U
Flormotridazum (18F)
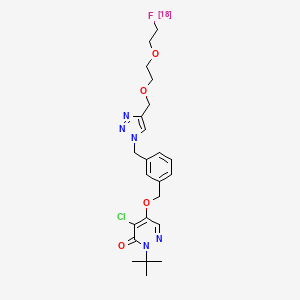
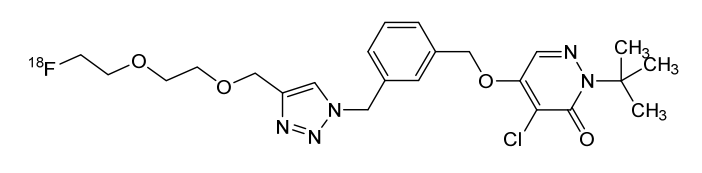
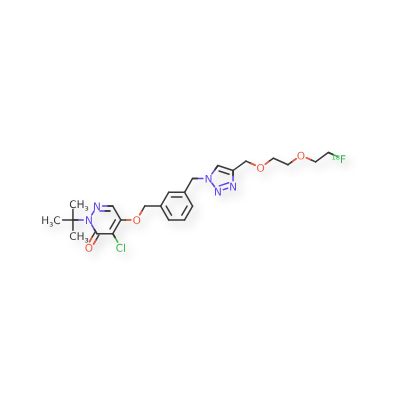
Flormotridazum (18F)
CAS 2798832-03-6
MF C23H29Cl18FN5O4 MW492.961
2-tert-butyl-4-chloro-5-[(3-{[4-({2-[2-(18F)fluoroethoxy]ethoxy}methyl)-1H-1,2,3-triazol-1-yl]methyl}phenyl)methoxy]pyridazin-3(2H)-one
| 3(2H)-Pyridazinone, 4-chloro-2-(1,1-dimethylethyl)-5-[[3-[[4-[[2-[2-(fluoro-18F)ethoxy]ethoxy]methyl]-1H-1,2,3-triazol-1-yl]methyl]phenyl]methoxy]- |
2-tert-butyl-4-chloro-5-[(3-{[4-({2-[2-(18F)fluoroethoxy]ethoxy}methyl)-1H-1,2,3-triazol-1-yl]methyl}phenyl)methoxy]pyridazin-3(2H)-one
imaging agent, 7AR6ZH8YUU
Flormotridaz (18F) (also referred to by its International Nonproprietary Name, flormotridazum) is an advanced radiopharmaceutical compound utilized in nuclear medicine. It is specifically engineered as a radioactive diagnostic tracer containing the fluorine-18 positron-emitting isotope.
Core Characteristics & Chemical Profile
- Substance Classification: Radioactive Diagnostic Agent / Small Molecule.
- Mechanism Basis: It shares core structural similarities and structural lineage with pyridazinone-based mitochondrial complex 1 (MC-1) inhibitors, heavily linking its functionality to target-specific tissues with high metabolic or mitochondrial activity.
Mechanism and Clinical Application
Like related fluorine-18 labeled pyridazinone analogues, this agent is designed for Positron Emission Tomography (PET) imaging workflows. [1]
- Administration: The agent is administered intravenously as a sterile unit dose before scanning.
- Cellular Targeting: It binds selectively to specific intracellular molecular targets (such as mitochondrial pathways) within highly active tissues.
- PET Imaging: As the Fluorine-18 radioisotope decays, it emits positrons. These positrons encounter electrons to produce gamma rays, which the PET scanner captures to map high-resolution, three-dimensional metabolic layouts of internal organ systems.
Contextual Comparison
In clinical nuclear medicine, molecular tracers tagged with Fluorine-18 offer significant clinical benefits over older Single-Photon Emission Computed Tomography (SPECT) agents. Their 110-minute half-life allows them to be manufactured at centralized cyclotron facilities and distributed directly to regional medical centres as ready-to-use unit doses, eliminating the need for an on-site cyclotron
Flormotridaz (\(^{18}\text{F}\)):
- CN112807276B: “Preparation method and application of a pyridazinone myocardial perfusion PET radiopharmaceutical” (Covers the definitive radiosynthesis scheme).
- CN115947775A: “Method for preparing compound (I), compound (I), and uses thereof”.
- WO2024008073A1 / CN114832118B: “Compound I liquid composition, preparation method and use thereof” (Covers final formulation stabilization utilizing vitamin C and gentisic acid)
PAT
https://patents.google.com/patent/WO2024008073A1/zh
Compound I, chemically named 2-tert-butyl-4-chloro-5-((3-((4-((2-(2-fluoro[ 18F ]ethoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)benzyl)oxy)pyridazine-3(2H)-one. Chemical structural formula:Molecular formula : C₂₃H₂₉Cl₁₈FN₅O₄
Molecular weight: 492.97The mechanism of action of compound I as a myocardial perfusion PET imaging agent: Once compound I enters cardiomyocytes, it can rapidly interact with respiratory chain complex I (MC-I) in mitochondria and remain in the myocardium for a long time. Preliminary animal studies showed that it has high cardiac uptake and low hepatic uptake 15 minutes after injection, and maintains a good heart-liver ratio 60 minutes after injection, showing good potential for myocardial perfusion imaging.In this application, Compound I liquid composition or Compound I is used as a myocardial perfusion PET imaging agent.Precursor of Compound I: Chemical name is methyl 2-(2-((1-(3-(((1-(tert-butyl)-5-chloro-6-oxo-1,6-dihydropyridazin-4-yl)oxy)methyl)benzyl)-1H-1,2,3-triazol-4-yl)methoxy)ethoxy)ethyl-4-methylbenzenesulfonate, chemical structural formula is:Molecular formula : C30H36ClN5O7S
Molecular weight: 646.16Amino polyethers (K222 ) are tribridged crown ether molecules with cavitary structures, and are typical nitrogen-containing cavitary ethers, belonging to the category of cavitary ethers. Due to their unique coordination properties, nitrogen-containing cavitary ethers can effectively and selectively complex transition metal and heavy metal cations, forming more stable complexes. Furthermore, they possess both lipophilic and hydrophilic properties, thus showing promising research potential.In existing technologies, the classic synthetic method for amino polyether (K
222 ) is the highly diluted method proposed by Lehn et al., which is a typical non-template ion synthesis method. The specific steps involve dissolving the starting materials 1,8-diamino-3,6-dioxane and 1,8-diacyl chloride-3,6-dioxane in a large amount of benzene solvent and heating the reaction for 8 hours. Then, a reduction reaction with lithium aluminum hydride is performed for 24 hours, followed by column chromatography separation and recrystallization to obtain amino polyether (K
222 ). This method requires a large amount of solvent, such as benzene, has a long synthetic route, is complex, has a low yield, and is not economically efficient. Besides the highly diluted method, another classic synthetic method for amino polyether (K222 ) is proposed by Kulstad and Malmsten, which uses Na 2CO 3
as a template to obtain a sodium iodide complex of amino polyether (K 222 ) in acetonitrile , and then decomplexes it using a resin to obtain amino polyether (K 222 ). The specific steps are as follows: 1,2-bis(2-iodoethoxy)ethane and benzylamine are refluxed in acetonitrile solution for 3 days. An intermediate is then obtained through post-processing. This intermediate is recrystallized from acetone and filtered to obtain a NaI complex. This complex is then decomplexed under acidic conditions using cation exchange resins and anion exchange resins to prepare amino polyether (K222 ) . This method uses simple equipment, requires little solvent, and has relatively mild reaction conditions. However, the applicant has found that the decomplexing method using ion exchange resins fails to proceed when the sodium ion content decreases to a certain level, resulting in a low yield.
PAT
https://patents.google.com/patent/CN114773179B/en
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
//////////flormotridazum (18F), anax labs, imaging agent, 7AR6ZH8YUU
Florcicaper (18F)
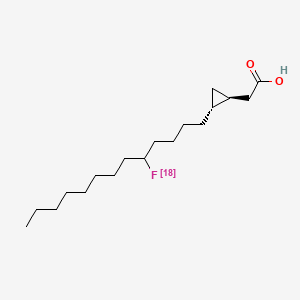
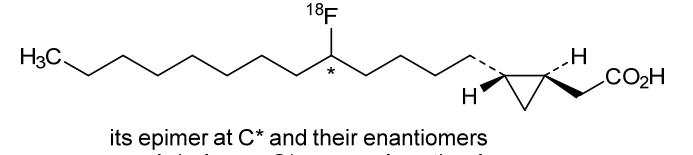
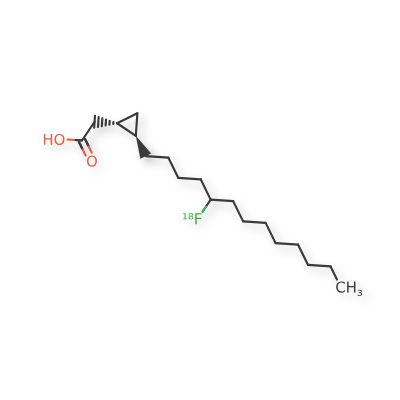
Florcicaper (18F)
CAS 855927-17-2
MF C18H3318FO2, MW 299.4544
2-[(1S,2R)-2-(5-(18F)fluorotridecyl)cyclopropyl]acetic acid
| 2-((1S,2R)-2-(5-(FLUORO-18F)TRIDECYL)CYCLOPROPYL)ACETIC ACID |
| TRANS-9(RS)-18F-FLUORO-3,4(RS,RS)-METHYLENEHEPTADECANOIC ACID |
rac-{(1R,2S)-2-[(5RS)-5-(18F)fluorotridecyl]cyclopropyl}aceticacid
imaging agent, CARDIOPET, (18F FCPHA), FDG79C95XB
CardioPET is: An F-18 labeled, modified fatty acid that provides insight into regions with decreased blood flow or metabolic insufficiency in the myocardium.; CardioPET may be used to: Identify patients that will benefit from PCI or revascularization and guide intervention, Assess myocardial viability, Evaluate CAD in patients that cannot exercise.; Agent: Muscle State Imaging Agent, Type: Fatty Acid (Labeled with Fluorine 18), Condition: Coronary Artery Disease, Status: completed enrollment.;This imaging agent exploits the dietary needs of the heart as it relates to glucose and fatty acids. By introducing a radio-labeled analog to the natural fatty acids utilized as an energy source by the heart we can visualize the anatomic location and state of the muscle within the areas defined by the specific coronary artery blood flow distribution and detect problems in advance of symptoms that would lead to a stress test.
Cardiopet is under investigation in clinical trial NCT01826773 (Cardiopet as PET Imaging Agent to Assess Myocardial Perfusion and Fatty Acid Uptake in Known or Suspected CAD Subjects).
A Phase I Study in Healthy Volunteers to Evaluate the Safety of CardioPET™ in Detection of Coronary Artery Disease
CTID: NCT00413647
Phase: Phase 1
Status: Completed
Date: 2013-06-12
PATENTS
CA-2876139-A1 CN-104684546-A CN-114736112-A CN-115141087-A CN-115141087-B CN-115141125-A CN-115141125-B CN-115181013-A CN-115181013-B CN-115850224-A CN-115850224-B CN-115959978-A CN-115959978-B CN-116041169-A CN-116199658-A CN-116217356-A EP-2858630-A1 EP-4133284-A1 US-10533059-B2 US-11701429-B2 US-2015361110-A1 US-2017014528-A1 US-2020199249-A1 US-2020297854-A1 US-20230314449-A1 US-20230381092-A1 US-9409927-B2 WO-2013185032-A1 WO-2022082327-A1 WO-2023085674-A1 WO-2023236978-A1 WO-2023237092-A1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2013185032&_cid=P11-MPJ5UV-89230-1
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2022082327&_cid=P11-MPJ5WJ-90048-1
ADVT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect
Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
References
- Nanotherapeutics for drug targetingPublication Number: WO-2013185032-A1Priority Date: 2012-06-07
- Nanotherapeutics for drug targetingPublication Number: EP-2858630-A1Priority Date: 2012-06-07
- Super lewis acidic borate esters as 18F-labeled PET probesPublication Number: US-9409927-B2Priority Date: 2013-01-24Grant Date: 2016-08-09
- Super lewis acidic borate esters as 18f-labeled pet probesPublication Number: US-2015361110-A1Priority Date: 2013-01-24
- Nanotherapeutics for drug targetingPublication Number: CA-2876139-A1Priority Date: 2012-06-07
- Nanotherapeutics for drug targetingPublication Number: US-2020297854-A1Priority Date: 2012-06-07
- Nanotherapeutics for drug targetingPublication Number: CN-104684546-APriority Date: 2012-06-07
- Diagnosis of stage b2 dmvdPublication Number: US-2023314449-A1Priority Date: 2020-04-07
- Targeted drug delivery through affinity based linkersPublication Number: US-2017014528-A1Priority Date: 2014-03-12
- Targeted drug delivery through affinity based linkersPublication Number: US-2020199249-A1Priority Date: 2014-03-12
- Targeted drug delivery through affinity based linkersPublication Number: US-10533059-B2Priority Date: 2014-03-12Grant Date: 2020-01-14
- Targeted drug delivery through affinity based linkersPublication Number: US-11701429-B2Priority Date: 2014-03-12Grant Date: 2023-07-18
//////////florcicaper (18F), anax labs, imaging agent, CARDIOPET, (18F FCPHA), FDG79C95XB
Teprosulvose
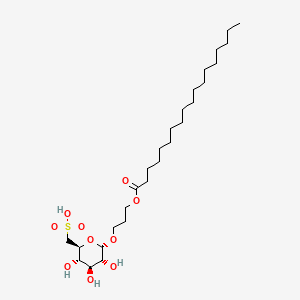
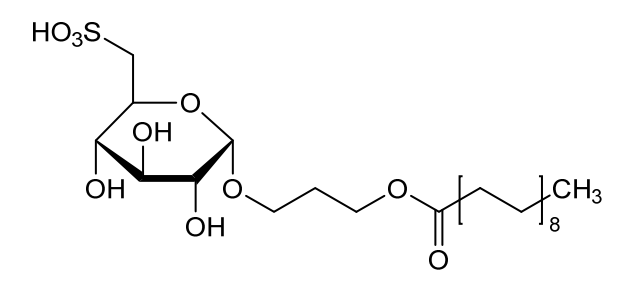
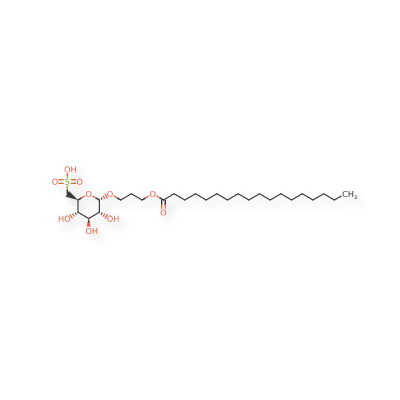
Teprosulvose
CAS 1983131-47-0
MF C27H52O10S MW568.761
| Sulfoquynovosylacylpropanediol [(2S,3S,4S,5R,6S)-3,4,5-trihydroxy-6-(3-octadecanoyloxypropoxy)oxan-2-yl]methanesulfonic acid |
3-(octadecanoyloxy)propyl 6-deoxy-6-sulfo-α-D-glucopyranoside
radiosensitizer (veterinary use), WV7377RGM8, SQAP
Teprosulvose (CAS 1983131-47-0) is a novel synthetic glycolipid, specifically a sulfoquinovosylacylpropanediol (SQAP). It is primarily developed for use in veterinary medicine as a radiosensitizer, intended to enhance the effectiveness of radiation therapy in treating malignant tumors.
1. Chemical Identity and Structure
- USAN/INN Name: Teprosulvose
- Systemic Name: 3-(octadecanoyloxy)propyl 6-deoxy-6-sulfo-$\alpha$-D-glucopyranoside
- Molecular Weight: 568.76 g/mol
- Structure: It consists of a glucose derivative (6-deoxy-6-sulfo-$\alpha$-D-glucopyranoside) linked via a propyl bridge to a long-chain fatty acid (stearic acid/octadecanoic acid).
Regulatory Data
Teprosulvose is currently in the investigational stage, primarily focused on veterinary oncology.
- USAN/INN Status: The name “Teprosulvose” was officially adopted by the USAN Council in 2024 (File LM-156).
- Classification: Radiosensitizer.
- Target Application: Adjuvant therapy for malignant tumors in animals (e.g., canine or feline cancers).
- Current Status: It has not yet received full FDA or EMA approval for human use. In the U.S., it is typically handled under Investigational New Animal Drug (INAD) protocols for clinical trials in veterinary patients.
Note: Because it is a specialized veterinary investigative agent, detailed safety data (LD50, pharmacokinetics) is generally found in specific FDA Freedom of Information (FOI) summaries or peer-reviewed veterinary oncology journals rather than standard human drug databases.
INN List 131 (WHO): Teprosulvose was officially included in the World Health Organization’s International Nonproprietary Names (INN) list in 2024. This confirms its unique status as a distinct drug substance.
USAN Council: The United States Adopted Names Council assigned the name in 2024, classifying it as a radiosensitizer.
FDA Status: It is currently under investigation (INAD) for canine oral melanoma and other solid tumors in veterinary medicine. Human clinical trial data is not yet widely available as the primary focus remains on the “Veterinary First” pathway.
Mechanism of Action: It is a potent inhibitor of DNA polymerase $\alpha$ and $\beta$. By inhibiting the repair of radiation-induced DNA damage, it effectively “locks in” the damage to tumor cells while sparing normal tissue due to differential uptake.
PAT
US Patent 10,206,942 (and related continuations): Covers the use of SQAP compounds in combination with radiation.
WO 2017/023812: International filing regarding the composition and therapeutic application of these glycolipids.
PAT
PAT
- Sulfonated sugar compounds, pharmaceutical compositions which contain the same, and methods of treating tumors with the samePublication Number: US-7973145-B2Priority Date: 2007-07-20Grant Date: 2011-07-05
- Sulfonated sugar compounds, pharmaceutical compositions which contain the same, and methods of treating tumors with the samePublication Number: US-2009209475-A1Priority Date: 2007-07-20
- Novel sulfonated sugar derivative, and use thereof for medicinal agentPublication Number: EP-2130834-A1Priority Date: 2007-07-20
- Sulfonated sugar compounds, pharmaceutical compositions which contain the same, and methods of treating tumors with the samePublication Number: US-2010298246-A1Priority Date: 2007-07-20
- Novel sulfonated sugar derivatives and their use as pharmaceuticalsPublication Number: JP-4435861-B2Priority Date: 2007-07-20Grant Date: 2010-03-24
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma
AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
//////////teprosulvose, radiosensitizer (veterinary use), WV7377RGM8, SQAP
Filricianine




Filricianine
CAS 2140857-94-7
MF C45H52N2O12S3, 909.1
3H-Indolium, 3-(3-carboxypropyl)-2-[2-[3-[2-[1,3-dihydro-3,3-dimethyl-1-(3-sulfopropyl)-2H-indol-2-ylidene]ethylidene]-2-(4-sulfophenoxy)-1-cyclohexen-1-yl]ethenyl]-3-methyl-1-(3-sulfopropyl)-, inner salt
3-[(2Z)-3-(3-carboxypropyl)-2-[(2E)-2-[3-[(E)-2-[3,3-dimethyl-1-(3-sulfopropyl)indol-1-ium-2-yl]ethenyl]-2-(4-sulfophenoxy)cyclohex-2-en-1-ylidene]ethylidene]-3-methylindol-1-yl]propane-1-sulfonate
3-[(3RS)-3-(3-carboxypropyl)-2-{(1Ξ)-2-[(3Ξ)-3-{(2Ξ)-2-[3,3-dimethyl-1-(3-sulfopropyl)- 1,3-dihydro-2H-indol-2-ylidene]ethylidene}-2-(4-sulfophenoxy)cyclohex-1-en-1-yl]ethen-1-yl}-3-methyl-3H-indol-1-ium-1-yl]propane-1-sulfonate
diagnostic imaging agent, CI4MD9KLX8

AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
////////filricianine, diagnostic imaging agent, CI4MD9KLX8
Alizulatide vixocianine




Alizulatide vixocianine
CAS 2924859-51-6
MF C115H145N17O25S, 2,197.55
L-Serine, N-[6-[2-[7-[1,3-dihydro-1,1-dimethyl-3-(4-sulfobutyl)-2H-benz[e]indol-2-ylidene]-1,3,5-heptatrien-1-yl]-1,1-dimethyl-1H-benz[e]indolio]-1-oxohexyl]-L-α-glutamyl-L-α-glutamyl-L-α-aspartyl-3-cyclohexyl-L-alanyl-L-phenylalanyl-D-seryl-D-arginyl-L-tyrosyl-L-leucyl-L-tryptophyl-, inner salt
4-[2-[7-[3-[6-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2R)-1-[[(2R)-5-carbamimidamido-1-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(1S)-1-carboxy-2-hydroxyethyl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-1-oxopentan-2-yl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-cyclohexyl-1-oxopropan-2-yl]amino]-3-carboxy-1-oxopropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-6-oxohexyl]-1,1-dimethylbenzo[e]indol-3-ium-2-yl]hepta-2,4,6-trienylidene]-1,1-dimethylbenzo[e]indol-3-yl]butane-1-sulfonate

diagnostic imaging agent, 8M3Q8XZ6MJ
Alizulatide vixocianine is a polypeptide that can be discovered through polypeptide screening. Polypeptide screening is a research tool mainly based on immunoassay methods to identify active polypeptides. It can be applied to protein interaction, functional analysis, antigenic epitope screening, especially in the fields of active molecule research and development.
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////alizulatide vixocianine, diagnostic imaging agent, 8M3Q8XZ6MJ
Zopocianine



Zopocianine
CAS 2206660-94-6, NA SALT 2206660-95-7
MF C74H93N7O27S4, 1,640.83
L-Tyrosine, N-[[[(1S)-1,3-dicarboxypropyl]amino]carbonyl]-L-g-glutamyl-3-[2-(2-aminoethoxy)ethoxy]propanoyl-L-phenylalanyl-O-[6-[2-[1,3-dihydro-3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-2H-indol-2-ylidene]ethylidene]-2-[2-[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-3H-indolium-2-yl]ethenyl]-1-cyclohexen-1-yl]-, inner salt
N-{[(1S)-1,3-dicarboxypropyl]carbamoyl}-L-γ-glutamyl3-[2-(2-aminoethoxy)ethoxy]propanoyl-L-phenylalanylO-[(6Ξ)-2-{(1Ξ)-2-[3,3-dimethyl-1-(4-sulfobutyl)-5-
sulfonato-3H-indol-1-ium-2-yl]ethen-1-yl}-6-{(2Ξ)-2-
[3,3-dimethyl-5-sulfo-1-(4-sulfobutyl)-1,3-dihydro-2Hindol-2-ylidene]ethylidene}cyclohex-1-en-1-yl]-Ltyrosine
diagnostic imaging agent, UD9V5S9M7A, OTL 0078, OTL 78
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////zopocianine, diagnostic imaging agent, UD9V5S9M7A, OTL 0078, OTL 78
Talogreptide mesaroxetan


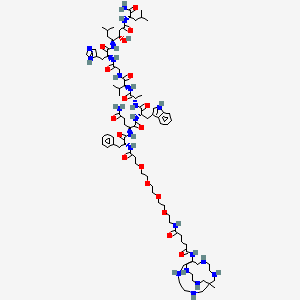
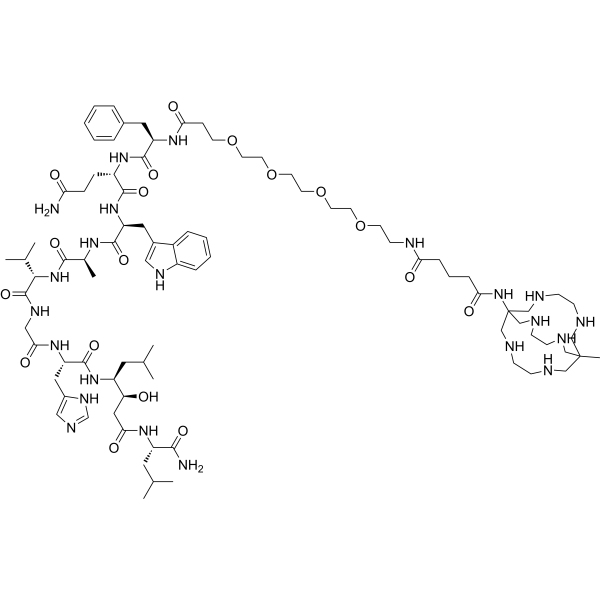
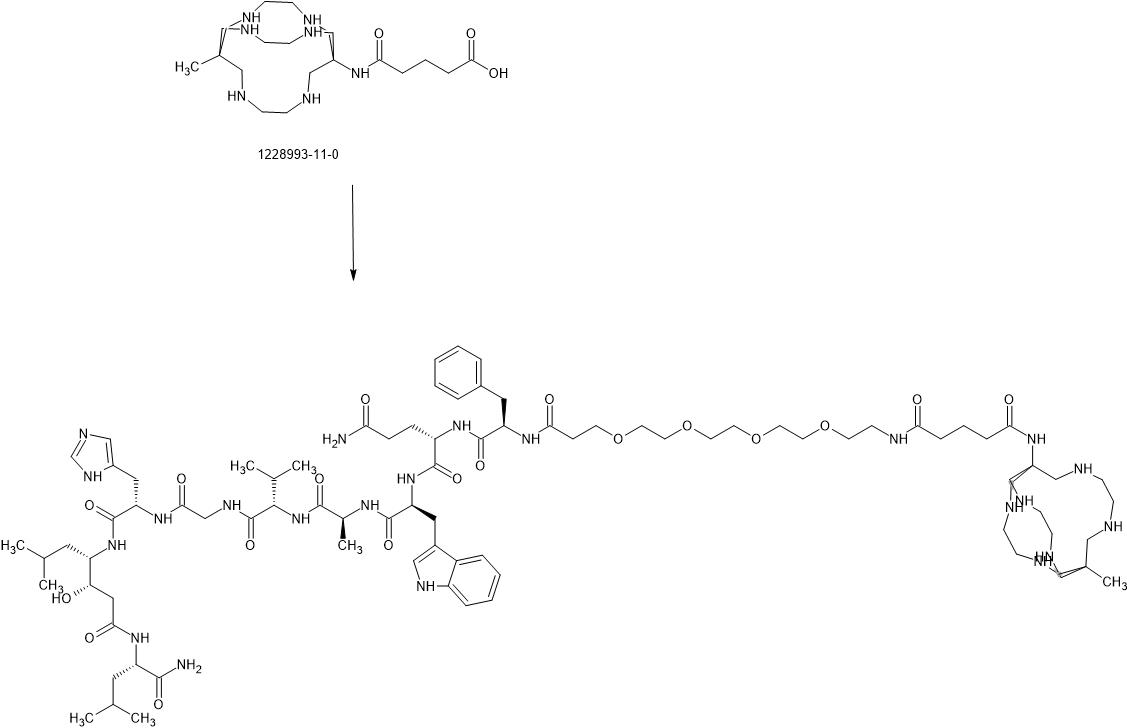
Talogreptide mesaroxetan
CAS 1801418-23-4
MF C86H140N22O18 MW1770.17
{MeCOSar}-PEG4-{d-Phe}-Gln-Trp-Ala-Val-Gly-His-{Sta}-Leu-NH2
(2S)-N-[(2S)-1-[[(2S)-1-[[(2S)-1-[[2-[[(2S)-1-[[(3S,4S)-1-[[(2S)-1-amino-4-methyl-1-oxopentan-2-yl]amino]-3-hydroxy-6-methyl-1-oxoheptan-4-yl]amino]-3-(1H-imidazol-5-yl)-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxopropan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]-2-[[(2R)-2-[3-[2-[2-[2-[2-[[5-[(8-methyl-3,6,10,13,16,19-hexazabicyclo[6.6.6]icosan-1-yl)amino]-5-oxopentanoyl]amino]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]-3-phenylpropanoyl]amino]pentanediamide
N-{21-[(8-methyl-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosan-1-yl)amino] -17,21-dioxo-4,7,10,13-tetraoxa-16-azahenicosan-1-oyl}-D-phenylalanyl-L-glutaminyl-L-tryptophyl-L-alanyl-Lvalylglycyl-L-histidyl-(3S,4S)-4-amino-3-hydroxy-6-methylheptanoyl-L-leucinamide
diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Talogreptide mesaroxetan (CAS 1801418-23-4) is a synthetic peptide, a complex molecule used as a diagnostic imaging agent with potential antitumor effects, targeting G-protein coupled receptors (GRPr) often overexpressed in cancers, allowing for specific tumor visualization in PET scans, particularly for metastatic disease detection, known for its high specificity and contrast for imaging tumors like those expressing GRPr.
Key Characteristics:
- Type: A peptide-based diagnostic agent, often labeled with radioisotopes like Copper-64 ($^{64}$Cu) for Positron Emission Tomography (PET) imaging, notes Patsnap Synapse.
- Structure: It’s a modified peptide sequence incorporating elements like PEG4 and specific amino acids, MedchemExpress.com.
- Function: Binds strongly to GRPr, helping to highlight tumors and metastatic sites.
- Application: Used in research to create high-contrast PET scans for better tumor detection and monitoring, showing promise in visualizing lymph node metastasis.
In Simple Terms:
Imagine it as a “smart tracer” that seeks out specific cancer cells. When attached to a radioactive tag, it lights up tumors on a PET scan, helping doctors see cancer more clearly, notes Patsnap Synapse.
Syn
WO2024086891
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2024086891&_cid=P10-MIZEJM-53111-1
67Cu radioisotope
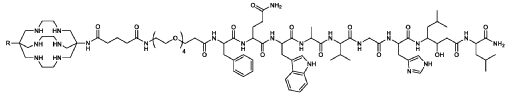
where R is CH3C(0)-;
(67CU-SAR-BBN)
Paper
Molecular Pharmaceutics (2015), 12(8), 2781-2790
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////Talogreptide mesaroxetan, diagnostic imaging agent, antineoplastic, ZUN64K4H2X, SAR-BBN
Mangaciclanol
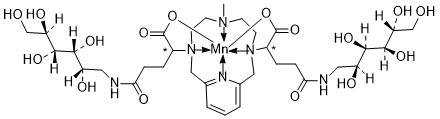
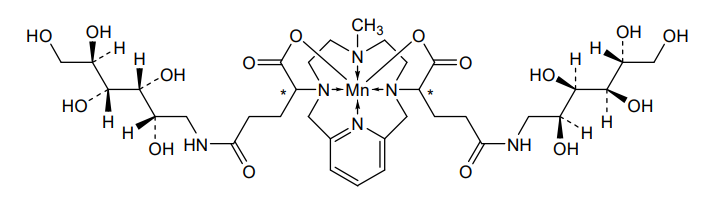
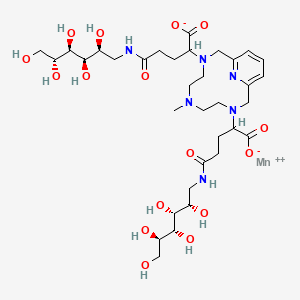
Mangaciclanol
Cas 2169771-05-3
MF C34H56MnN6O16 MW859.8 g/mol
- ANU6AE7NAP
- [[1,1′-[(6-Methyl-3,6,9,15-tetraazabicyclo[9.3.1]pentadeca-1(15),11,13-triene-3,9-diyl-kappaN3,kappaN6,kappaN9,kappaN15)bis[[4-(carboxy-kappaO)-1-oxo-4,1-butanediyl]imino]]bis[1-deoxy-D-glucitolato]](2-)]manganese
2-[9-[1-carboxylato-4-oxo-4-[[(2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl]amino]butyl]-6-methyl-3,6,9,15-tetrazabicyclo[9.3.1]pentadeca-1(15),11,13-trien-3-yl]-5-oxo-5-[[(2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl]amino]pentanoate;manganese(2+)

diagnostic imaging agent, ANU6AE7NAP
AS ON OCT2025 4.511 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
/////////mangaciclanol, diagnostic imaging agent, ANU6AE7NAP

{kind=link}