
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
VILAZODONE SPECTRAL DATA


VILAZODONE
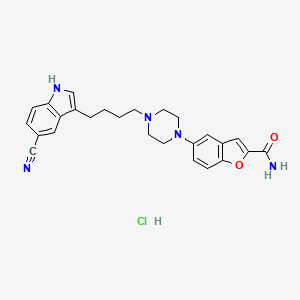
Vilazodone hydrochloride; 163521-08-2; Vilazodone HCl; Viibryd; UNII-U8HTX2GK8J; EMD-68843
NO SYNTHESIS IS THIS POST, ONLY SPECTRAL DATA DISCUSSED
SEE MORE SPECTROSCOPY DATA AT………..http://orgspectroscopyint.blogspot.in/2015/06/vilazodone.html
ENJOY THE INTERPRETATIONS
Vilazodone (United States trade name Viibryd veye-brid) is a serotonergic antidepressant developed by Clinical Data for the treatment of major depressive disorder. The chemical compound was originally developed by Merck KGaA (Germany).[2] Vilazodone was approved by the FDA for use in the United States to treat major depressive disorder in 2011.[3][4][5]
Medical uses
According to two eight-week, randomized, double-blind, placebo-controlled trials in adults, vilazodone elicits an antidepressant response after one week of treatment. After eight weeks, subjects assigned to vilazodone 40 mg daily dose (titrated over 2 weeks) experienced a significantly higher response rate than the group given placebo (44% vs 30%, P = .002). Remission rates for vilazodone were not significantly different versus placebo.[6]
According to an article on the United States approval of vilazodone written by FDA staff, “it is unknown whether [vilazodone] has any advantages compared to other drugs in the antidepressant class.”[7]
PAPER FROM OPRD
Scale-Up Synthesis of Antidepressant Drug Vilazodone

A scale-up synthesis of antidepressant drug vilazodone was accomplished in five steps. Friedel–Crafts acylation of 1-tosyl-1H-indole-5-carbonitrile with 4-chlorobutyryl chloride, selective deoxygenation in NaBH4/CF3COOH system coupled with ethyl 5-(piperazin-1-yl)-benzofuran-2-carboxylate hydrochloride, one-step deprotection and esterolysis, and the final ammonolysis led to the target molecule vilazodone in 52.4% overall yield and 99.7% purity. This convenient and economical procedure is remarkably applicable for scale-up production.
5-(4-(3-(5-Cyano-1H-indol-3-yl)butyl)piperazin-1-yl)benzofuran-2-carboxamide (1)

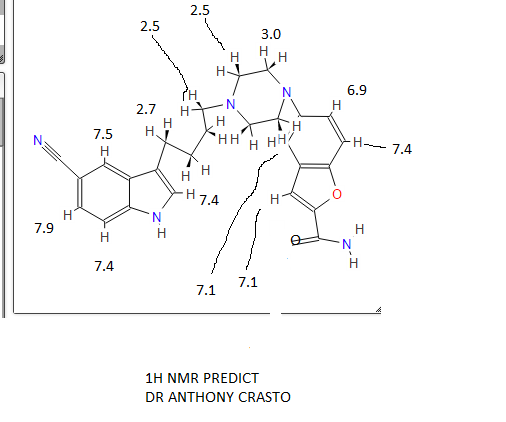
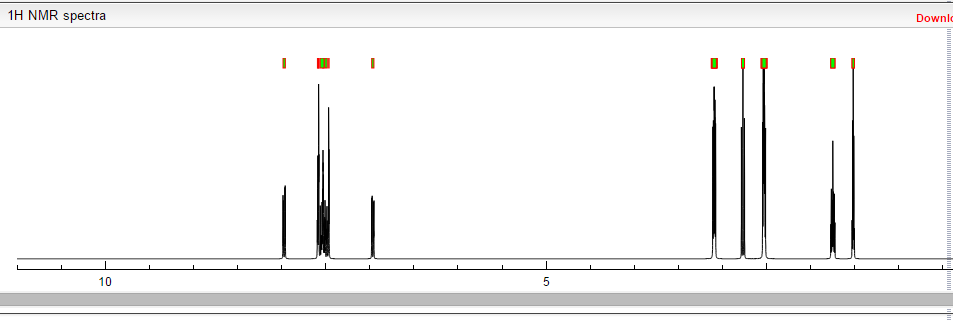
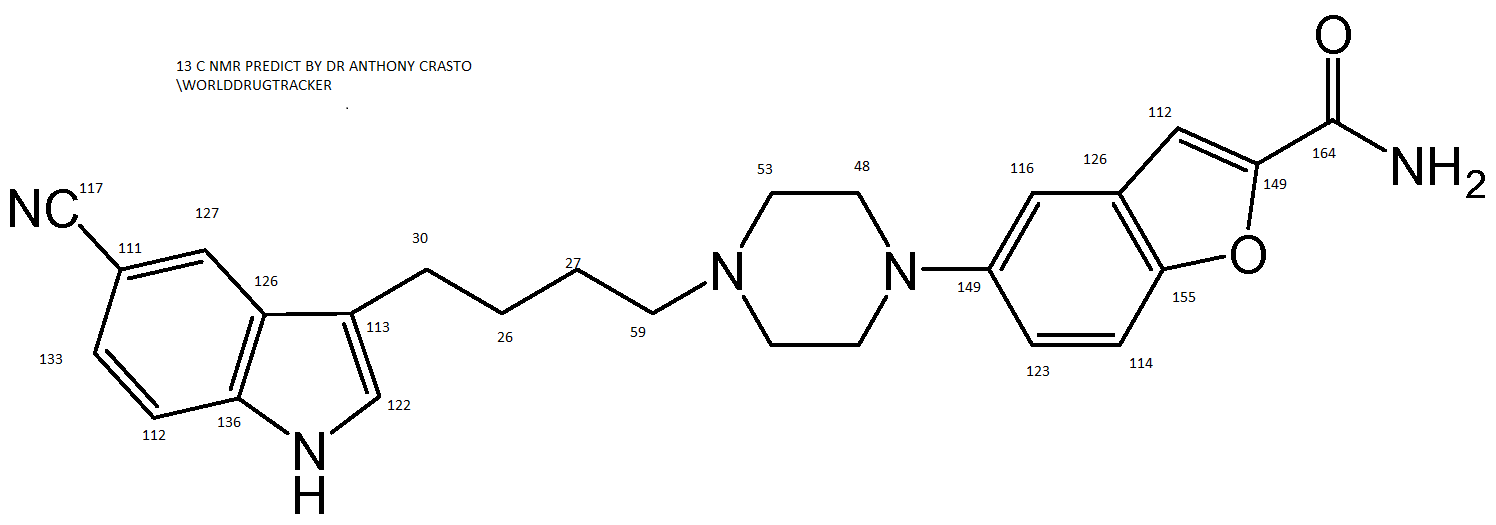

…………………………
SEE MORE SPECTROSCOPY DATA AT………..http://orgspectroscopyint.blogspot.in/2015/06/vilazodone.html
VIIBRYD Tablets for oral administration contain polymorph Form IV vilazodone hydrochloride (HCl), a selective serotonin reuptake inhibitor and a 5HT1A receptor partial agonist.
Vilazodone HCl is 2-benzofurancarboxamide, 5-[4-[4-(5cyano-1H-indol-3-yl)butyl]-1-piperazinyl]-, hydrochloride (1:1). Its molecular weight is 477.99. The structural formula is:
 |
In addition to the active ingredient, VIIBRYD Tablets contain lactose monohydrate, microcrystalline cellulose, magnesium stearate, colloidal silicon dioxide, polyvinyl alcohol, titanium dioxide, polyethylene glycol, talc, FD&C Blue #1 (40 mg only), FD&C Yellow #6 (20 mg only) and FD&C Red #40 (10 mg only).


- “VIIBRYD (vilazodone hydrochloride) tablet VIIBRYD (vilazodone hydrochloride) kit [Forest Laboratories, Inc.]”. DailyMed. Forest Laboratories, Inc. December 2012. Retrieved28 October 2013.
- “Clinical Data’s Vilazodone Patient Enrollment Over One Third Complete”. Business Wire. Berkshire Hathaway. 17 August 2006. Retrieved 12 April 2014.
- “FDA approves Clinical Data Inc’s antidepressant”. Reuters. January 22, 2011.
- “FDA approves Clinical Data Inc’s antidepressant”. Reuters. January 22, 2011. Retrieved 12 April 2014.
- “Clinical Data, Inc. – Clinical Data, Inc. Submits New Drug Application for Vilazodone for the Treatment of Major Depressive Disorder”. Retrieved 12 April 2014.
- Wang, SM; Han, C; Lee, SJ; Patkar, AA; Masand, PS; Pae, CU (August 2013). “A review of current evidence for vilazodone in major depressive disorder.”. International Journal of Psychiatry in Clinical Practice 17 (3): 160–9. doi:10.3109/13651501.2013.794245. PMID 23578403.
- Laughren TP, Gobburu J, Temple RJ, Unger EF, Bhattaram A, Dinh PV, Fossom L, Hung HM, Klimek V, Lee JE, Levin RL, Lindberg CY, Mathis M, Rosloff BN, Wang SJ, Wang Y, Yang P, Yu B, Zhang H, Zhang L, Zineh I (September 2011). “Vilazodone: clinical basis for the US Food and Drug Administration’s approval of a new antidepressant”. The Journal of Clinical Psychiatry 72 (9): 1166–73. doi:10.4088/JCP.11r06984. PMID 21951984.
Honokiol, from magnolia bark, shuts down cancer cells in lab
 |
|
| Magnolia virginiana |
Honokiol, from magnolia bark, shuts down cancer cells in lab
Compound in magnolia may combat head and neck cancers
Honokiol, from magnolia bark, shuts down cancer cells in lab


Magnolias are prized for their large, colorful, fragrant flowers. Does the attractive, showy tree also harbor a potent cancer fighter?
Yes, according to a growing number of studies, including one from VA and the University of Alabama at Birmingham that is now online in the journal Oncotarget.
The study focused on squamous cell head and neck cancers, a scourge among those who use tobacco and alcohol. According to the National Cancer Institute, at least 3 in 4 head and neck cancers are caused by the use of tobacco and alcohol. The cancers have only a 50 percent survival rate, killing some 20,000 Americans each year.
Enter honokiol–chemical formula C18H18O2. As one of the major active compounds in magnolia extract, the phytochemical has been used for centuries in traditional Chinese and Japanese medicine to treat anxiety and other conditions. More recently, scientists have been discovering that the compound, found in magnolia bark, is a wily and versatile adversary of cancer. It seems to exploit many biochemical pathways to shrink tumors of various types, or to keep them from growing in the first place.

The Alabama scientists have now shown how it works against head and neck cancers: It blocks a protein called epidermal growth factor receptor, or EGFR. Prior research has found that almost all head and neck cancer cells display an over-abundance of the protein, and it had been suggested in the literature as a potential target.
The VA-UAB team says, based on its lab studies, that honokiol binds more strongly with EGFR than does the drug gefitinib (sold as Iressa), which is commonly used to treat head and neck cancers.
The researchers tested honokiol on cell lines derived from human cancers of the oral cavity, larynx, tongue, and pharynx. In all cases, the botanical shut down the aberrant cells. The team also tested it against tumors implanted into mice, with similar results.

Senior author Dr. Santosh K. Katiyar and his colleagues wrote, “Conclusively, honokiol appears to be an attractive bioactive small molecule phytochemical for the management of head and neck cancer which can be used either alone or in combination with other available therapeutic drugs.”
Katiyar has published extensively in the past on other natural substances that work against tumors, especially skin cancer. Some of his recent work has focused on compounds in green tea, for example, and grape seed proanthocyanidins.
Purification
There are several methods for purifying and isolating honokiol. In nature, honokiol exists with its structural isomer magnolol, which differs from honokiol only by the position of onehydroxyl group. Because of the very similar properties of magnolol and honokiol, purification has often been limited to a HPLC or electromigration. However, methods developed in 2006 by workers in the lab of Jack L. Arbiser, took advantage of the proximity of the phenolic hydroxyl groups in magnolol, which form a protectable diol, to generate amagnolol acetonide (Figure 1), with a subsequent simple purification via flash chromatography over silica.[4]
Figure 1

Magnolol and Honokiol are normally inseparable. Honokiol is easily separable from the protected magnolol acetonide
Additionally a rapid separation approach was published in the Journal of Chromatography A in 2007. The process uses high-capacity high-speed countercurrent chromatography(high-capacity HSCCC).[5] Through this method honokiol can be separated and purified to above 98% purity with a high yield in under an hour.
Honokiol is a lignan isolated from the bark, seed cones, and leaves of trees belonging to the genus Magnolia. It has been identified as one the chemical compounds in some traditional eastern herbal medicines along with magnolol, 4-O-methylhonokiol, andobovatol.
Traditional medicine

Seed Cone
Extracts from the bark or seed cones of the Magnolia tree have been widely used in traditional medicine in China, Korea, and Japan.[2]
Houpu has traditionally been used in Eastern medicine as analgesic and to treat anxiety and mood disorders.[2][6] However, it has been shown to treat a number of other conditions. In China, magnolia bark is called Houpu and is most commonly taken from the Magnolia obovata and the Magnolia officinalis species.[7] Some Chinese traditional formulas containing Houpu include Banxia Houpu Tang (半夏厚朴丸), Xiao Zhengai Tang, Ping Wei San(平胃散) and Shenmi Tang.[2] Japanese Kampo formulas include, Hange-koboku-to (半夏厚朴湯) and Sai-boku-to (柴朴湯).[2][6]

Seeds
Modern medicine
In the late 1990s, honokiol saw a revival in interest as a potent and highly tolerable antitumorigenic and neurotrophiccompound.
Alternative medicine
Currently there are a large number of supplements containing honokiol on the market, and its use has been widely well received among practitioners of new age, homeopathic, and holistic medicine
|
Stereo image
|
||
|
||
|
||
|
||
|
||
| Mature Magnolia fruit just starting to open, with a few seeds visible |

 |
|
| Names | |
|---|---|
| IUPAC name
2-(4-hydroxy-3-prop-2-enyl-phenyl)- 4-prop-2-enyl-phenol
|
|
| Other names
houpa, hnk
|
|
| Identifiers | |
| 35354-74-6 |
|
| ChEMBL | ChEMBL16901 |
| ChemSpider | 65254 |
| Jmol-3D images | Image |
| KEGG | C10630 |
| PubChem | 72303 |
| Properties | |
| C18H18O2 | |
| Molar mass | 266.334 g/mol |
| Appearance | White solid |
| sparingly (25 °C) | |
| Related compounds | |
|
Related biphenols
|
diethylstilbestrol, dihydroxyeugenol |
|
Related compounds
|
magnolol. 4-O-Methylhonokiol |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
.JPG)
Magnolia seeds and fruit on a tree in northern Argentina

The root and stem bark of Magnolia has been used as a traditional Chinese medicine for the treatment of thrombotic stroke, gastrointestinal complaints, and anxiety. Honokiol (HNK), a substituted biphenyl and an active component isolated and purified from Magnolia, has anti-oxidant, antithrombosis, antibacterial, neurotrophic, xanthine oxidase inhibitory, and anxiolytic effects (Taira et al., Free Radic Res Commun. 1993;19 Suppl l:S71-77; Teng et al. Thromb Res. 1988;50:757-765; Clark et al., J. Pharm. Sci. 1981;70:951-952; Chang et al., Anticancer Res. 1994;14:501-506; Kuribara et al., J. Pharm Pharmacol. 1998;50:819-826; Esumi et al., Bioorg & Medicinal Chem Let 2004, 14: 2621-25).
In the early 1990s, reports of HNK’s anticancer effects were published. In 1994, Hirano et al (Life Sci. 1994;55(13): 1061-9) examined the anti leukemic-cell efficacy of 28 naturally occurring and synthetic flavonoids and 11 naturally occurring ligands on human promyelocytic leukemic cell line HL-60, and cytotoxicity of these compounds was compared with four clinical anti-cancer agents. HNK was identified as one of the most potent compounds in this screen, with an IC50 value less than 100 ng/ml. In 1998, Hibasami et al. demonstrated that HNK induced apoptosis in human lymphoid leukemia Molt 4B cells (Hibasami et al., Int. J. MoI. Med. 1998).
HNK has also been found to induce apoptosis in human squamous cell lung cancer CH27 cells (Yang SE, et al Biochem Pharmacol. 2002;63:1641-1651) and in human colorectal RKO cells (Wang et al World J Gastroenterol. 2004; 10:2205-2208). In 2004, Chen et al. (World J Gastroenterol. 2004; 10: 3459-3463) reported that HNK was effective in an in vivo animal model of human colon cancer by inhibiting tumor growth and prolonging the lifespan of tumor bearing mice.
Honokiol is an inhibitor of angiogenesis and antitumor activity in vivo. HNK can cause apoptosis in tumor cells and inhibit angiogenesis through blocking phosphorylation of vascular endothelial growth factor receptor 2 (VEGFR2), the major mitogenic and chemoattractant endothelial growth factor (Bai et al. (2003) J. Biol. Chem. 278, 35501- 35507). Honokiol also exhibits direct antitumor activity through induction of apoptosis through tumor necrosis factor apoptosis-inducing ligand (TRAIL/ Apo2L) signaling and has been found to be highly effective against angiosarcoma in nude mice in vivo (Bai et al. (2003) J. Biol. Chem. 278, 35501-35507).
Esumi et al. (Biorganic & Medicinal Chemistry Letters (2004) 14: 2621-2625) describe a synthesis method to produce HNK. This report also evaluates the structure activity relationship of O-methylated and/or its hydrogenated analogs of HNK in an in vitro neurotrophic assay. Esumi et al. conclude that the 5-allyl and 4′-hydroxyl groups are essential for the neurotrophic activity of HNK.
PCT Publication No. WO 02/076393 and U.S. Publication No. 2004/0105906 to Emory University describe pharmaceutical compositions and methods of treating conditions such angiogenic-, neoplastic-, and cancer-related conditions and skin conditions by administration of honokiol-type and/or magnolol-type compounds, as shown in Figures 1-4. For example, such compositions comprise at least one compound of formula Al :

AI wherein R1, R2, R3, R4, R5, R1, R2, R3, R4, and R’5 can be independently selected from groups that include, but are not limited to, hydrogen, hydroxyl groups, amides, amines, hydrocarbons, halogenated hydrocarbons, cyclic hydrocarbons, cyclic heterocarbons, halogenated cyclic heterocarbons, benzyl, halogenated benzyl, organo selenium compounds, sulfides, carbonyl, thiol, ether, dinitrogen ring compounds, thiophenes, pyridines, pyrroles, imidazoles, and pyrimidines. Honokiol-type and magnolol-type compounds are shown to inhibit SVR cell proliferation.
In November of 2004, Arbiser et al. reported that honokiol inhibited the growth of multuple myeloma cell lines via induction of Gl growth arrest, followed by apoptosis with IC50 values at 48h of 5 to 10 μg/mL. It was also reported that honokiol inhibited growth of doxorubin (Dox)-resistant (RPMI-Dox40), mephalan resistant (RPMI-LR5) and dexamethasone (Dex)-resistant (MM. IR) cell lines. It was suggested that the mechanism of honokiol triggered cytotoxicity is the honokiol induced increased expressin of Bax and Bad, down-regulated Mc-I protein expression, followed by caspase-8/9/3 cleavage, (Arbiser, J. et al. Poster at the American Society of Hematology Annual Meeting, 2004. Abstract published online November 4, 2004).
In July of 2005, Battle et al. reported that honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells (Blood. July 2005; 106:690- 697). Honokiol induced caspase-dependent cell death in all of the B-CLL cells examined, which were primary tumor cells derived from B-CLL patients, and was more toxic toward B- CLL cells than to normal mononuclear cells. The honokiol-induced apoptosis was characterized by the activation of caspase-3, -8, and -9 and cleavage of poly(adenosine diphosphate-ribose) polymerase (PARP). It was also reported that honokiol enhanced cytotoxicity induced by fludarabine, cladribine, or chlorambucil.
In September 2005, Ishitsuka et al reported that honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and – independent apoptosis (Blood, 1 September 2005, Vol. 106, No. 5, pp.1794-1800). HNK induced cytotoxicity in human multiple myeloma (MM) cell lines and tumor cells from patients with relapsed refractory MM through induction of apoptosis via both caspase- dependent and -independent pathways. HNK also enhanced MM cell cytotoxicity and apoptosis induced by bortezomib.
It is an object of the present invention to provide new compounds, compositions, methods and uses for the treatment of disorders associated with angiogenesis, cell proliferation, tumor growth, tumorogenesis, and myeloma.

the intermediates for the synthesis of honokiol are 3-allyl-4- hydroxybenzeneboronate 5 and 4-allyl-2-bromophenol 9. The boronate 5 can be prepared from 2-iodophenol 1 by bromination, followed by Suzuki coupling to introduce the allyl group, and boronation under Suzuki conditions. Compound 9 can be prepared from 4- iodophenol 6 by bromination and allylation (Suzuki coupling). The coupling of 5 and 9 under Suzuki conditions can yield honokiol from Suzuki coupling, not other allyl-oriented products from the Heck reaction, as it was shown that Suzuki coupling can succeed in the presence of C=C double bond (see Miyaura, N.; Suzuki, A. (1995), Chem. Rev. 95, 2457- 2483; and Suzuki, A. (1999), J. Organometal. Chem. 576, 147-168, and the references cited therein). Thus, honokiol and derivatives can be synthesized from commercially available starting materials in 6 steps (Scheme 1). Scheme 1

Treatment of honokiol with TMS-diazomethane in methanol results in mono- and di- methylated compounds I-III, and hydrogenation of honokiol with Wilkinson’s catalyst yields di- and tetrahydrohonokiols VI-VIII, as reported by Esumi, T. et al. (2004), Bioorg. Med. Chern. Lett. 14, 2621-2625. The amino and fluoro analogues (IV and V) can be constructed from iodoacetanilide under Suzuki coupling conditions. From 2-iodoacetanilide 10, after bromination, allylation, and boronation, the boronated intermediate 13 can be prepared. The other bromo intermediate 16 can be prepared from 4-iodoacetanilide 14 via bromination and allylation. The coupling of boronate 13 and bromide 16 under Suzuki conditions can afford, after deprotection, the compound IV. Diazotization followed by Schiemann reaction can convert the amino analogue TV to fluoro analogue V (Scheme 2).

The dimethoxy honokiol derivative, III, can also be prepared, for example, by the treatment of honokiol with potassium carbonate, iodomethane. (Scheme 2a). The hydrogenated honokiol analog can alternatively be prepared by the hydrogenation of honokiol with sodium borohydride and nickel(II) chloride to yields tetrahydrohonokiols VI- Vπi. (Scheme 2a). Scheme 2a

The preparation of the vinyl analogue IX is based on combining the Wittig reaction with Suzuki coupling. The intermediate aldehyde 18 can be prepared from 4-iodophenol 17 via the Reimer-Tiemann reaction, while 3-bromo-4-hydroxybenzenealdehyde 23 can be prepared from para-hydroxybenzoic ester 21 via bromination and reduction. The Wittig reaction of these two aldehydes can yield the corresponding vinyl substituted benzenes 19 and 24. Compound 19 can afford the boronate 20, which can be coupled with 24, to yield the compound IX (Scheme 3).

Reagents and conditions: (a) CHCl3, aq. NaOH, 70 °C; (b) Ph3PCH3Br1 n-BuLi, THF; (c) PdC!2(dppf), dppf, KOAc, dioxane, bis(pinacoato)diboron, 80 °C; (d) DIBALH, -70 °C; (e) PdCI2(dppf), dppf, K3PO4, dioxane, reflux.
For the synthesis of honokiol analogues with changed positions of the allyl or hydroxyl groups, the boronate 5, and the bromophenols 4 and 9 can be used as intermediates. Suzuki coupling of one of these intermediates with an appropriate halide or boronate can provide the compounds X-XVII. Compounds X-XII and XTV-XV can be prepared by Suzuki coupling of boronate 5 with an appropriate halide. Halide 25, needed for compound X, can be prepared from 2-bromo-6-iodophenol 2 via allylation, while the intermediate, 5-allyl-2- bromophenol 29 for compound XI, can be furnished from 3-iodophenol 26 via bromination and allylation. The preparation of halide 5-allyl-3-bromophenol 33, an intermediate for the synthesis of compound XIV, requires an organothallium reagent. The thallation of 3- bromophenol 30 followed by treatment with iodide can yield 3-bromo-5-iodophenol 32. After allylation, the allyl-substituted intermediate 33 can be prepared. The synthesis of compound XII can begin with 2-iodoacetanilide 10, via sulfonation, nitration, and reduction to obtain the intermediate 36. Aniline 36, after diazotization, followed by acid and base treatments, will afford 2-amino-3-iodophenol 37. Diazotization, Sandmeyer reaction, and allylation of compound 37 will yield halide 39. By a coupling reaction of these halides (25, 29, 33, and 39) with boronate 5, these compounds (X-XII, and XTV) can be prepared. Compound XV can be synthesized by Suzuki coupling of halide 4 with boronate 5 (Scheme 4).
Scheme 4

Alternatively, compounds X, XV, and XVII can be synthesized by an allylation- Claisen pathway. Biphenol compounds can be reacted first with potassium carbonate and allyl bromide, followed by reaction with BCl3 to yield honokiol-like compounds, for example, X, XV, and XVII. (Scheme 4a). To a cooled solution (O0C with an ice bath) of diallyl starting material (1 eq.) in dry diehloromethane (Concentration of the solution : 0.1 mol.L“1) was added dropwise a solution Of BCl3 (IM in diehloromethane; 1.5 eq. = 0.75 eq for each allyl group). The reaction is then stirred at O0C until disappearance of the starting material on TLC (If after 15 minutes, the reaction is not complete, 1 more equivalent of BCl3 can be added). After hydrolysis with water (about same volume than diehloromethane), the two layers are separated. The organic layer is washed again with water, dried under MgSO4 then evaporated under vacuum. The residue is finally purified by column chromatography to give the di- hydroxy derivative . Scheme 4a

Bromide 9 is also a useful intermediate for coupling with some boronates. For example, Suzuki coupling of bromide 9 with boronate 42, which is prepared from 4-bromo-3- iodophenol 40 via allylation and boronation, can yield the compound XIII. Similarly, the coupling between bromide 9 and boronate 43 can afford the compound XVT. The compound XVII can be prepared from 4-allyl-2-bromophenol 9 via boronation followed by Suzuki coupling with 2-allyl-6-bromophenol 25 (Scheme 5).

The compounds XVIII and XIX can be synthesized from commercially available bisphenol 45 and the dihydroxynaphthalene-disulfuric acid salt 47. Thus, the bisphenol 45, through the Williamson reaction and Claisen rearrangement, can be converted to compound XVπi. Similarly, desulfonation of dihydroxynaphthalene-disulfuric acid salt .47, followed by the Williamson reaction and Claisen rearrangement, can produce the compound XIX (Scheme 6).Scheme 6

Dioxolane compounds can be prepared from magnoliol by reaction of magnoliol with 2,2′-dimethoxypropane and p-toluenesulfonic acid. (Scheme 7). This synthesis also provides a method of separating mixtures of honokiol and magnoliol. Scheme 7

The following examples are offered by way of illustration and not by way of limitation.
……..
http://www.google.com/patents/US20080300298

BOCEPREVIR, Боцепревир ,بوسيبريفير , 波普瑞韦



Boceprevir (INN, trade name Victrelis) is a protease inhibitor used as a treatment hepatitis caused by hepatitis C virus (HCV) genotype 1.[2][3] It binds to HCV nonstructural 3 NS3 (HCV) active site.[4]
It was being developed by Schering-Plough,[5] but is now being developed by Merck since Schering was acquired in 2009. It was approved by the FDA on May 13, 2011.[6]


Efforts toward the synthesis and process optimization of boceprevir 1 are described. Boceprevir synthesis was optimized by telescoping the first three steps and last two steps of the five-step process. Optimization of oxidation, which is one of the critical steps in the total synthesis, is discussed. A control strategy for the three impurities is described. A novel process for the synthesis of fragment A (2) has been developed, which is the key starting material for the synthesis of boceprevir.
…………………


WO 2015004685
( 1 R,5S)-N-[3-Amino- 1 -(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[( 1 , 1 -dimethylethyl) amino]carbonyl]amino]-3,3-dimethyl-l-oxobutyl]-6,6-dimethyl-3-azabicyclo [3.1.0]hexan-2(S)-carboxamide (Boceprevir); having formula I. It is a hepatitis C virus (“HCV”) protease inhibitor, developed by Merck & Co and marketed under the brand name of VICTRELIS.

Formula I
U.S. patent number 6,992,220, U.S. patent application numbers 201 1034705, U.S. 20050249702 and U.S. 201001 13821 are disclosed process for the preparation of Boceprevir.
U.S. patent number 7,326,795 claims Boceprevir bisulfate adduct as a product. Advanced Organic Chemistry, 4th ed., Jerry March Ed., John Wiley and Sons, 1972 disclosed purification methods from bisulfate adduct to provide the compound in a pure form.
U.S. patent number 8,222,427 claims a process for the purification of Boceprevir through a corresponding bisulfite adduct, wherein the compound of Formula I is dissolved in organic solvent, which is treated with an aqueous phase comprising bisulfite, thereby forming an aqueous solution of the bisulfite adduct of the compound of Formula I, which is subsequently regenerated from the aqueous phase without isolating the bisulfite adduct.

Examples:
Example 1:
183.7 gm of l-Dimethylaminopropyl-3-ethylcarbodiimide hydrochloride and 500 ml of dimethylsulfoxide were taken at 23-25 °C and to this 500 ml of ethyl acetate was added then cooled to 2-8 °C. 3-[2-(3-Tert-butylureido)-3,3-dimethyl-butyryl]-6,6-dimethyl-3-azabicyclo[3.1.0] hexane-2 carboxylic acid(2-carbamoyl-l-cyclobutyl-(methyl-2-hydroxy-ethyl)amide (Hydroxy Boceprevir) 100 gm was added to the reaction mixture under stirring at same temperature followed by 86.5 gm of dichloroacetic acid and continued stirring for 1-2 hrs. After completion of the reaction, 2500 mL of water was added to the reaction mixture at 2-10 °C and the reaction mixture temperature was raised to 15-20 °C. Ethyl acetate 600 ml was added to the reaction mass and the organic layer was separated. The product was extracted from aqueous layer with ethyl acetate. The organic layer was washed with 5% w/w hydrochloric acid followed by water. To the organic layer, aqueous solution of sodium bisulfite (300 gm in 600 ml) was added and stirred for 2 hrs. The layers were separated and organic layer was extracted with water. Thereafter, extracted aqueous layer was washed with ethyl acetate. To the aqueous layer sodium bisulfite (5.1 gm in 17 ml of water) was added and stirred for 30 min. The obtained solution was degassed and the pH was adjusted to 1.0 to 2.5 with dilute hydrochloric acid (15 ml of 35% w/w hydrochloric acid and 15 ml of water) and cooled to 10-15 °C. The obtained solid was filtered and washed with water to yield pure Boceprevir.
Exam le 2:
202 gm of l-Dimethylaminopropyl-3-ethylcarbodiimide hydrochloride and 500 ml of dimethylsulfoxide were taken at 23-25 °C and stirred, to this reaction mixture 500 ml of ethyl acetate was added; stirred and cooled to 2-8 °C. Hydroxy Boceprevir 100 gm was added under stirring at same temperature followed by 92.7 gm of dichloroacetic acid and continued stirring for 2-4 hrs. After completion of the reaction, 2500 mL of water was added to the reaction mixture at 2-10 °C and temperature was raised to 20-25 °C. Ethyl acetate 600 ml was added to the reaction mass and the organic layer was separated. The product was extracted from aqueous layer with ethyl acetate. The both organic layers were combined and stirred with dilute hydrochloric acid solution (prepared by mixing 50 ml of ~35% w/w of hydrochloric acid and 950 mL of water). The organic layer containing the product was separated and washed with water. The organic layer was cooled to 1-5 °C. To the organic layer, aqueous solution of sodium bisulfite (300 gm in 600 ml) was added and stirred for 2 hrs at 5- 9 °C. The organic layer was cooled without agitation and added precooled water at 5-10 °C. The aqueous layer containing the product was collected. The aqueous layer filtered through hyflo and washed with precooled water. Further the aqueous layer was diluted with precooled water, and adjusted the pH to 2 – 2.8 with dilute hydrochloric acid. Vacuum was applied to the aqueous layer and the temperature was slowly raised to less than 23 °C under reduced pressure. The separated solid was filtered at 22-30 °C and washed with water. Further, the filtered solid was washed with water having pH 1.8-2.4 (The pH of the water was adjusted with HC1). The product was dried at 24-28 °C under reduced pressure to yield pure Boceprevir.
Example 7:
100 gm of Crude Boceprevir was added to 300 mL of ethanol-isopropyl alcohol (1 : 1) at 22-30 °C and contents were stirred for about 40 minutes. The resulting solution was added to water slowly at 5-10 °C and stirred for 2-4 hrs at the same temperature. The product was filtered, washed with water and dried at 25-30°C under reduced pressure.

…………………
SCHERING CORPORATION Patent: WO2008/76316 A2, 2008 ; Location in patent: Page/Page column 27 ;
or eq https://www.google.co.in/patents/EP2121604A2?cl=en
Hepatitis C virus (HCV) is a (+)-sense single-stranded RNA virus that has been implicated as the major causative agent in non-A, non-B hepatitis; an HCV protease necessary for polypeptide processing and viral replication has been identified. U.S. Patent No. 7,012,066 discloses a genus of HCV protease inhibitor compounds that includes the compound of Formula I, (1 R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3- dioxopropyl]-3-[2(S)-[[[(1 , 1 -dimethylethyl)amino]-carbonyl]amino]-3,3-dimethyl-1 – oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide.

Formula I
US2005/0059800, published March 17, 2005, discloses a process for preparing the compound of Formula I and discloses a bisulfite adduct of Formula I which can be used to provide the compound in a pure form in accordance with the methods taught in Advanced Organic Chemistry, 4th ed., Jerry March Ed., John Wiley and Sons, 1972.
US2005/0020689, filed January 27, 2005, discloses processes for preparing an intermediate useful in preparing the compound of Formula I. Methods for preparing diastereomers of the compound of Formula I are disclosed in US2005/0249702, filed November 10, 2005. Published US Patent Application No. 2007/0149459, filed November 13, 2006, discloses oxidation processes for preparing the compound of Formula I.
Purification of the compound of Formula I is difficult for several reasons. The compound Formula I is an alpha-keto amide that is unstable and forms dimers, especially under basic conditions. Also, the compound of Formula I is amorphous, thus it does not crystallize and precipitation does not improve the purity of the solid —
Previously published procedures for preparing the compound of Formula I resulted in about 63 to about 98.5% purity.
Historically, aldehydes and ketones have been purified by preparing their bisulfite adduct. Bisulfite purification of these types of compounds was performed through isolation of a solid bisulfite adduct intermediate from aqueous alcoholic solution by filtration. Regeneration of an aldehyde or ketone from an isolated bisulfite adduct is accomplished using a base or a strong acid. Examples appearing in the literature of regeneration using bases includes: Na2Cθ3 in Org. Synthesis Coll. Vol. 4, 903 (1963); NaOH in WO 2006/074270 A2; and K2CO3 in Tetrahedron Lett., 45, 3219 (2004). Examples of regeneration using acids include: H2SO4 in J. Am. Chem. Soc, 70, 1748 (1948); and HCI in WO 99/57123.
For the preparation of a purified product, isolation of an intermediate solid bisulfite adduct is not preferred since filtration of the adduct is required. In addition, base regeneration of the adduct to yield the substrate is not appropriate in those cases wherein the regenerated product is unstable in basic conditions, for example, where the regenerated product is the compound of Formula I. When acid conditions are used to regenerate the substrate compound from a bisulfite adduct, generally strongly acidic conditions and heating are necessary (see references above).
Published international application no. WO 99/57123 reports using non- alcoholic solvent in a process for forming a bisulfite adduct, however the process required isolation of a solid bisulfite adduct and regeneration the substrate from the adduct using NaOH.
A non-aqueous method for regeneration of a substrate from the corresponding bisulfite adduct was reported in J. Org. Chem., 64, 5722 (1999) as a means to overcome side-reactions such as degradation and hydrolysis during regeneration of aldehyde/ketone with a base or an acid. In this method, trimethylsilyl chloride (TMSCI) or its equivalent was employed in acetonitrile. During the process TMS2O, NaCI1 SO2 and HCI were generated as co-products when TMSCI was used.
Removal of the co-products required the process steps of filtration (for NaCI), aqueous work-up (for NaCI and excess TMSCI) and distillation (for TMS2O), which requires use of a high boiling solvent. Regeneration of aldehydes from the corresponding bisulfite adducts with ammonium acetate in solvent-free conditions was reported in J. of Chem. Research, 237 (2004), however this process requires microwave irradiation.
Published international application no. WO 2006/076415 describes regeneration of an aldehyde from a corresponding bisulfite adduct isolated from an alcoholic solvent system using a carbonate base with a lower alkyl carbonyl compound, for example, acetone and glyoxylic acid.
SCHEME Il
solv


Bisulfite Adduct

Formula I in water Formula I
SCHEME III

Formula I

Published U.S. patent application no. 2007/0149459, published June 28, 2007, discloses several alternate procedures for oxidizing the intermediate compound of the Formula II:

Formula II, to obtain the compound of Formula I.
HPLC Determination of Purity
The purity of the compound of Formula I is determined by HPLC according to the methods described below:

alternatively, the following equipment and conditions are used:

Example 1
(Purification Process of Scheme III, Regeneration Option “a”)
Preparation of Compound: To a reactor was charged (16.5 kg) of the compound of Formula II,

Formula Il24.3 Kg of EDCI1 and 190 L of EtOAc. The batch temperature was adjusted between 15 and 250C. At the same temperature, Et3N (9.60 kg, 3 eq) followed by EtOAc rinse (8 L) was charged. To the resultant mixture was charged DMSO (83 L) while maintaining the temperature of the batch between 150C and 250C. CH3SO3H (10.89 kg) was charged while maintaining the reaction mixture between 150C and 30° C. After agitating at the reaction mixture for 1.5 hours while maintaining the reaction mixture between 200C and 300C, the reaction mixture was cooled to a temperature between -50C and 50C.
Purification of the Compound of Formula I
In a separate reactor was charged 165 L of water and 33 L of EtOAc, and the mixture was cooled below 50C. The reaction mixture containing the compound was transferred into the mixture of cold water/EtOAc at 0 to 100C. The organic layer was separated and washed with water (99 L) three times. Step 1 : To the resulting organic solution was added NaHSθ3 aqueous solution
(prepared from 49.5 kg of NaHSO3 and 109 L of water). The whole was agitated for 3 h at 20-300C. The aqueous NaHSO3 layer was separated and saved. The organic layer was concentrated to about 116 L of volume and diluted with MTBE (220 L). The separated aqueous NaHSO3 layer was added to the organic layer. The resultant mixture was agitated for 3 h at 20-30 0C. The organic layer was separated and cooled to 0-10 0C.
Step 2: To the cooled organic layer of Step 1 was added cold water (165 L, 0-100C) without agitation, and the whole was agitated for 5 min. The aqueous layer was separated, and a solution of water (2 L) containing NaHSO3 (0.71 kg) was added to the water layer. The water layer was distilled to the final volume of about 171 L under vacuum below 25 0C to remove volatiles.
Step 3: (Regeneration method a): The resultant water layer of Step 2 was added into a slurry of NaCI (49.5 kg) in acetone (83 L) at 20-300C. The separated acetone layer followed by acetone rinse (8 L) was added through a 0.2 micron filter to water (347 L) over 20 min at 15-25 0C. After agitation for about 1 h, the precipitate was filtered and washed with water (83 L). The wet cake was dried under vacuum at 30-400C to produce 13.0 kg (79%) of the purified compound as a white solid.
………………..
US2007/149459
http://www.google.co.in/patents/US20070149459
EXAMPLESPreparation of (1R,2S,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-{N-[(tert-butylamino)carbonyl]-3-methyl-L-valyl}-6,6-dimethyl-3-azabicyclo-[3.1.0]hexane-2-carboxamide (the Compound of Structure 2 in Scheme A, Below)

Example 1Preparation of Compound 2 Using Aqueous Acetic Acid in the Reaction Mixture
Into a 1 L, three necked flask is placed KBr (10 g, 84 mmol), NaOAc (10 g, 122 mmol), Compound 1 (50 g, 96 mmol), and TEMPO (15 g, 96 mmol), followed by 500 mL of MTBE. The reaction mixture is stirred at 350-400 rpm and the temperature is maintained at a temperature of from 10° C. to 20° C. Acetic acid (50 mL, 874 mmol), and water (5 mL) are added to the reaction mixture and the two phase mixture is agitated for 15 minutes. Continuously, over a two hour period, to the reaction mixture is added 158 mL of a 0.82 M solution of NaOCl (130 mmol). When all of the NaOCl solution is added, the reaction mixture is stirred for an additional 3 hours while maintaining the temperature. Water (50 mL) is added.
The layers are separated and the organic layer is washed twice with water (2×250 mL). A solution of ascorbic acid, which is prepared from 50 g of sodium ascorbate, 200 mL of water, and 50 mL of 4N HCl, is added to the organic layer and the mixture is stirred for about 1 hour. After the layers are separated, the organic layer is washed twice with water (2×250 mL). The organic layer is concentrated by distilling off solvent at low temperature (0-5° C.) until the total volume is about 350 mL. The concentrated organic layer is added dropwise over 30 minutes into a 3 L flask containing 2 L of n-heptane at about 0° C. providing a white precipitate. The white precipitate is collected by filtration, washed with n-heptane (400 mL) and dried in a vacuum oven (2 hr at 25° C., 8 hr at 350, and 8° C. at 45° C.). The product is obtained as a white powder (typically 94-96% yield).
1H NMR, δ 0.84 (d, J=2.3 Hz, 3H), 0.90-1.02 (m, 9H), 0.99 (d, J=4.0 Hz, 3H), 1.24 (s, 9H), 1.40-1.86 (m, 7H), 1.90-2.10 (m, 3H), 2.25-2.40 (m, 1H), 3.75 (dd, J=5.3 and 10.4 Hz, 1H), 4.10 (dd, J=6.8 and 10.4 Hz, 1H), 4.4 (dd, J=3.0 and 5.3 Hz, 2H), 5.17 (dddd, J=4.6, 8.1, 8.1, and 10.4 Hz, 1H), 5.3 (br s, 2H), 6.71 (d, J=14.7 Hz, 1H), 6.90 (dd, J=2.3 and 19.0 Hz, 1H), and 7.34 (dd, J=7.1 and 20.2 Hz, 1H).
Example 2Preparation of Compound 2 Using Glacial Acetic Acid in the Reaction Mixture
Into a 2 L, three necked flask was charged KBr (20 g, 168 mmol), NaOAc (20 g, 243 mmol), Compound 1 (100 g, 192 mmol), and TEMPO (30 g, 192 mmol), followed by 800 mL of MTBE. The reaction mixture was stirred at 350400 rpm while the temperature of the reaction mixture was maintained at a temperature of from 10° C. to 20° C. Acetic acid (70 mL, 1223 mmol, used as received), was added and the mixture was agitated for 15 minutes additional. Continuously, over a two hour period, 315 ml of a 0.73M solution of NaOCl (230 mmol) was added to the reaction mixture. When all of the NaOCl solution had been added, agitation was continued for an additional 3 hours. Water (100 mL) was added to the reaction mixture at the end of 3 hours. The layers were separated and the organic layer was washed once with water (500 mL).
A solution of ascorbic acid, which was prepared from 100 g of sodium ascorbate, 456 mL of water, and 44 mL of 36% HCl, was added to the organic layer and the mixture was stirred for about 2 hours. The layers were separated and then a solution of 3.5N HCL was added and stirred about 30 minutes. After the layers were separated, the organic layer was washed three times with water (3×500 mL). This organic layer was then added drop-wise over 30 minutes into a 5 L flask containing 3 L of n-heptane at about −10 to about 0° C. The white precipitate was filtered, washed with n-heptane (600 mL) and dried in a vacuum oven (2 hr at 25° C., 8 hr at 350, and 8° C. at 45° C.). The product was obtained as a white powder (93% yield).
1H NMR, δ 0.84 (d, J=2.3 Hz, 3H), 0.90-1.02 (m, 9H), 0.99 (d, J=4.0 Hz, 3H), 1.24 (s, 9H), 1.40-1.86 (m, 7H), 1.90-2.10 (m, 3H), 2.25-2.40 (m, 1H), 3.75 (dd, J=5.3 and 10.4 Hz, 1H), 4.10 (dd, J=6.8 and 10.4 Hz, 1H), 4.4 (dd, J=3.0 and 5.3 Hz, 2H), 5.17 (dddd, J=4.6, 8.1, 8.1, and 10.4 Hz, 1H), 5.3 (br s, 2H), 6.71 (d, J=14.7 Hz, 1H), 6.90 (dd, J=2.3 and 19.0 Hz, 1H), and 7.34 (dd, J=7.1 and 20.2 Hz, 1H).
Boceprevir
…………………


Chinese journal of medicinal chemistry 2011, 21, 5 , pg 409-10
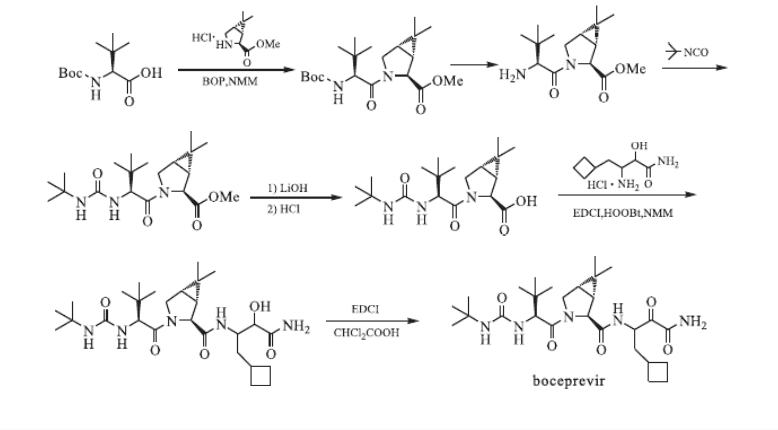

……………………
J Med Chem,2006,49(20):6074-6086.
……………………………………….
WO2004/113294 A1
…………..
MSN LABORATORIES LIMITED; THIRUMALAI RAJAN, Srinivasan; ESWARAIAH, Sajja; VENKAT REDDY, Ghojala; SAHADEVA REDDY, Maramreddy Patent: WO2014/61034 A1, 2014 ;
………………………..
MERCK SHARP and DOHME CORP.; WU, George, G.; ITOH, Tetsuji; MCLAUGHLIN, Mark; LIU, Zhijian; QIAN, Gang Patent:WO2013/66734 A1, 2013 ;
Example 1: Cyclobutylacetonitrile
Step 1 : Cyclobutylmethyl methanesulfonate
A 50-L jacket vessel was charged with DCM (20 L) (KF 34 ppm), and cyclobutylmethyl alcohol (5.0 kg, 58.0 mol) followed by TEA (8850 mL, 63.5 mol). The reaction mixture was cooled to approximately -10°C, and MsCl (4735 mL, 60.8 mol) was added via an addition funnel dropwise over approximately 3 hours, while the temperature was maintained below -5°C. The reaction resulted in a yellow slurry after 70 minutes of aging. H20 (8 L) was added to give a clear solution, which was agitated for 15 minutes. Then, the organic layer was separated. H20 (8 L) was charged to the organic layer. The mixture was agitated for 20 minutes, and then the organic layer was separated. Brine (10% solution, 4 L) was charged to the organic layer. The mixture was agitated for 20 minutes, and then the organic layer was separated. The organic phase was concentrated by vacuum distillation at approximately 30°C to 40°C and 28 inches Hg, resulting in a light brown residue (10.0 kg crude, approximately 9.5 kg product assumed, 58.0 mol, approximately 100% yield). A portion of the material was purified by distillation for characterization.
1H NMR (CDC13, 400 MHz): δ 4.18 (d, J = 6.8 Hz, 2H), 3.00 (s, 3H), 2.71 (m, 1H), 2.11 (m, 2H), 2.00-1.80 (m, 4H).
Step 2: Cyclobutylacetonitrile

A 100-L RB flask was set up with a mechanical stirrer, a thermocouple, an addition funnel, a N2 inlet, and a condenser that is connected to a scrubber (11 L bleach and 5 L 2N NaOH). DMSO (30.3 L) (KF approximately 680 ppm) and NaCN (3030 g, 61.8 mol) were charged to the flask. The mixture was heated to approximately 75 °C by steam to dissolve most chunks of NaCN, resulting in a turbid solution. The product of Step 1 (9476 g, 57.7 mol) in DMSO (4 L) was added dropwise in 1 hour, 40 minutes while the temperature was maintained below approximately 87°C. The reaction was aged at approximately 85°C for 3 hours and cooled down to RT. H20 (24 L) and MTBE (24 L) were charged. The mixture was agitated, and the organic layer was separated. The aqueous layer was extracted with MTBE (18 L), and the combined organic layer was agitated with H20 (12 L) and separated. The organic layer was washed with 10% brine (4 L and 2 L), and concentrated by vacuum distillation at approximately 45°C and approximately 20 inches Hg, giving a light brown liquid (7.235 kg crude, 73.3% by GC assay, 5.30 kg product assay, 55.7 mol, 96.5% for two steps).
Ή NMR (CDCI3, 400 MHz): δ 2.65 (m, 1H), 2.41 (d, J – 5.2 Hz , 2H), 2.18 (t, J = 6.8 Hz, 2H), 2.00-1.80 (m, 4H).
Example 2: Ethyl 4-cyclobutyl-3-oxobutanoate

THF (20 L) and zinc dust (2.75 kg, 42.0 mol) were charged under N2 to a 50-L jacketed vessel with a thermocouple, an addition funnel and a condenser. The mixture was stirred, and chlorotrimethylsilane (0.571 kg, 5.26 mol) was added at RT. The mixture was heated at 67°C for 30 minutes. Cyclobutylacetonitrile (2.5 kg, 26.3 mol, product of Example 1) was added at 67°C. Ethyl bromoacetate (6.108 kg, 36.6 mol) was added to the mixture at approximately 67°C to 70°C for over 3 hours. After the addition, the mixture was heated at approximately 70°C for 1 hour and then cooled to approximately 0°C to 5°C. 10% H2S04 (aq.) (35 L, 33.9 mol, approximately 1.3 eq.) was added slowly. The mixture was aged at RT for 1 hour. The organic layer was separated and subsequently washed with 10% aqueous citric acid (15 L, 7.88 mol, 0.3 eq.), 10% aqueous Na2S205 (25 L), 10% Na2S205 (aq.) (10 L), and 10% brine (10 L). The organic layer was concentrated in vacuo to afford the crude product (4.08 kg assay, 22.15 mol) in 84% yield. A part of the material was purified by distillation for characterization (with NMR in CDC13, approximately 10-15% enol-form of the compound was observed, major keto-form as shown.)
1H NMR (CDC13, 400 MHz): δ 4.19 (q, J = 7.1 Hz, 2 H), 3.38 (s, 2 H), 2.75-2.65 (m, 1H), 2.65-2.63 (m, 2 H), 2.19-2.08 (m, 2 H), 1.95-1.79 (m, 2 H), 1.73-1.60 (m, 2 H), 1.27 (t, J = 7.1 Hz, 3 H).
13C NMR (CDC13, 400 MHz): δ 202.2, 167.2, 61.3, 50.0, 49.3, 31.1, 28.4, 18.7,
14.1.
Example 3: Ethyl 2-chloro-4-c clobut l-3-oxobutanoate

Methyl t-butyl ether (30.2 L), and the crude product of Example 2 (3.78 kg assay,
20.52 mol) were charged to a 100-L RB flask with an overhead stirrer, an addition funnel, a thermometer, and an acid scrubber (with 2N NaOH at RT under N2). Sulfuryl chloride (2.98 kg,
22.06 mol) was added at approximately 20°C to 23 °C over 1.5 hours. After addition, the mixture was cooled to approximately 5°C and then quenched with 1M K3P04 (aq.) (23.6 L). The organic layer was separated and concentrated under vacuum to afford the crude chloride (4.487 kg, assume 100% yield, 20.52 mol), which was used in the next reaction without purification. A part of the material was purified by distillation for characterization (with NMR in CDC13,
approximately 10% enol-form of the compound was observed, major keto-form was shown below).
1H NMR (CDCI3, 400 MHz): δ 4.73 (s, 1 H), 4.29 (q, J = 7.1 Hz, 2 H), 2.89-2.79 (m, 2 H), 2.79-2.69 (m, 1 H), 2.20-2.07 (m, 2 H), 1.98-1.78 (m, 2 H), 17.3-1.61 (m, 2 H), 1.32 (t, J = 7.1 Hz, 3 H).
13C NMR (CDC13, 400 MHz): δ 198.1, 165.0, 63.1, 60.9, 45.7, 31.0, 28.3, 18.7, 13.9. Example 4: -C clobut l-l-ethox -l,3-dioxobutan-2-yl 4-methoxybenzoate

The crude chloride product of Example 3 (4.487 kg assumed, 20.52 mol) and Ν,Ν-dimethylformamide (11.2 L) were charged to a 50-L jacketed vessel with a thermocouple and a condenser at RT under N2. -Methoxybenzoic acid (3.75 kg, 24.62 mol) and TEA (2.285 kg, 22.57 mol) were added to the mixture. The mixture was heated at 55°C for 14 hours. The mixture was cooled to approximately 10°C, diluted with methyl tert-butyl ether (24 L), quenched with ¾0 (24 L). The organic layer was separated and subsequently washed with IN NaHC03 (20 L), then H20 (18 L) with NaCl (0.90 kg) and NaHC03 (0.45 kg). The organic layer was separated and concentrated in vacuo to afford the product (6.07 kg, 18.15 mol) in 88% assay yield. A part of the material was purified by distillation for characterization.
1H NMR (CDCI3, 400 MHz): δ 8.09 (dt, J = 2.1, 9.0 Hz, 2 H), 6.96 (dt, J = 2.1, 9.0 Hz, 2 H), 5.66 (s, 1 H), 4.31 (q, J = 7.1 Hz, 2 H), 3.88 (s, 3 H), 2.86 (dd, J = 5.7, 7.6 Hz, 2 H, 2.83-2.74 (m, 1 H), 2.23-2.12 (m, 2H), 1.98-1.80 (m, 2 H), 1.74-1.65 (m, 2 H), 1.32 (t, J = 7.1 Hz, 3 H).
Example 5: (2 -3-Amino-4-cyclobutyl-l-ethoxy-l-oxobut-2-en-2-yl 4-methoxybenzoate

The crude product of Example 4 (5.97 kg, 17.85 mol), 1-propanol (12 L), and EtOH (12 L) were charged to a 100-L RB flask with an overhead stirrer and a thermometer at RT under N2. NH4OAc (4.82 kg, 62.5 mol) was added to the mixture. The mixture was heated at 50°C for 1 hour. The mixture was concentrated in vacuo to remove H20 azeotropically with continuous addition of 1-propanol (total approximately 24 L). The mixture was solvent-switched to i‘PrOAc (24 L) under vacuum. The mixture was quenched with 2M K3P04 (aq.) (17.85 L). The organic layer was separated and washed with 15% brine (18 L) twice. The organic layer was concentrated in vacuo to afford crude enamine product (5.95 kg, assume 100% yield, 17.85 mol).
1H NMR (CDC13, 400 MHz): δ 8.12 (d, J= 8.0 Hz, 2H), 6.98 (d, J= 8.0 Hz, 2H),
6.02 (s, 2H), 4.15 (q, J= 8 Hz, 2H), 3.89 (s, 3H), 2.60-2.53 (m, 1H), 2.33 (s, 2H), 2.13-2.06 (m,
2H), 1.91-169 (m, 4H), 1.20 (t, J = 8 Hz, 3H).
13C NMR (CDC13, 400 MHz): δ 165.7, 167.6, 163.6, 153.9, 132.1, 122.2, 113.9,
113.7, 112.5, 59.6, 44.5, 37.8, 33.9, 28.5, 28.4, 18.5, 14.4.
Example 6A: 3-[(tert-Butoxycarbonyl)amino]-4-cyclobutyl-l-ethoxy-l-oxobut-2-yl 4- methoxybenzoate

The crude product of Example 5 (5.92 kg, 17.75 mol) and MeOH (23.7 L) were charged to a 100-L RB flask with an overhead stirrer, a thermocouple, and an addition funnel at RT under N2. Di-tert-butyl dicarbonate (5.81 kg, 26.6 mol) and sodium cyanoborohydride
(1.171 kg, 18.64 mol) were charged to the mixture. A solution of glycolic acid (1.485 kg, 19.53 mol) in MeOH (3.55 L) was added to the mixture drop wise at a rate to maintain the temperature at approximately 15°C to 22°C. The mixture was aged at approximately 20°C for approximately 8-10 hours. EtOAc (3.49 L, 35.5 mol) and a solution of glycine (0.866 kg, 11.4 mol) in H20 (11 L) were added to the mixture at RT. Then, 2M K3P04 (aq ) solution (17.75 L) was added. The mixture was aged for 20 minutes. The mixture was extracted with methyl tert-butyl ether (28 L). The organic layer was separated and washed subsequently with 2M K3P04 (aq.) solution (17.75 L), 10% brine (17.75 L, twice). The organic layer was concentrated under vacuum to afford the desired two diastereoisomers in almost 1 : 1 ratio (7.30 kg, 16.76 mol) in 94% assay yield.
1H NMR (CDCI3, 400 MHz): δ 8.02 (d, J= 8.0 Hz, 2H), 6.94 (d, J= 8.0 Hz, 1H),
6.93 (d, J= 8.0 Hz, 1H), 5.30 (d, J= 4.0 Hz, 0.5H), 5.17 (d, J= 4.0 Hz, 0.5H), 4.80 (d, J= 8.0 Hz, 0.5H), 4.63 (d, J = 8.0 Hz, 0.5H), 4.27-4.18 (m, 3H), 3.86 (s, 3H), 2.50-2.30 (m, 1H), 2.15- 2.00 (m, 2H), 1.89-1.60 (m, 6H), 1.43 -1.42 (m, 9H), 1.27 (t, J= 8.0 Hz, 3H).
Example 6B: 3-[(tert-Butoxycarbonyl)amino]-4-cyclobutyl-l-ethoxy-l-oxobut-2-yl 4- methoxybenzoate (First alternate procedure)

The crude product of Example 5 (19.2 g, 58.0 mmol) and MeOH (100 mL) were charged to an autoclave with a thermocouple at RT. Di-tert-butyl dicarbonate (19.0 g, 87.0 mmol) and 5% Ir/CaC03 (10.0 g) were charged to the mixture. The mixture was heated to 40°C under sealed conditions, where H2 was transferred until the internal pressure became
approximately 200 psig. The mixture was heated at 40°C at approximately 200 psig for 20 hours. The reaction mixture was cooled to RT and filtered to remove the solid to afford a clear solution. EtOAc (5.7 mL, 58 mmol) and a solution of glycine (2.8 g, 38 mmol) in H20 (37 mL) were added to the mixture at RT. Then, 2M K3P04 (aq ) solution (58 mL) was added. The mixture was aged for 20 minutes. The mixture was extracted with methyl tert-butyl ether (130 mL). The organic layer was separated and washed subsequently with 2M 3P04 (aq.) solution (58 mL), 10% brine (58 mL, twice). The organic layer was concentrated under vacuum to afford the desired two diastereoisomers in almost 1 :1 ratio (23 g, 52 mmol) in a 90% assay yield.
1H NMR (CDC13, 400 MHz): δ 8.02 (d, J= 8.0 Hz, 2H), 6.94 (d, J= 8.0 Hz, 1H), 6.93 (d, J= 8.0 Hz, 1H), 5.30 (d, J= 4.0 Hz, 0.5H), 5.17 (d, J- 4.0 Hz, 0.5H), 4.80 (d, J= 8.0 Hz, 0.5H), 4.63 (d, J= 8.0 Hz, 0.5H), 4.27-4.18 (m, 3H), 3.86 (s, 3H), 2.50-2.30 (m, 1H), 2.15- 2.00 (m, 2H), 1.89-1.60 (m, 6H), 1.43 -1.42 (m, 9H), 1.27 (t, J= 8.0 Hz, 3H). Example 6C: 3-[(tert-Butoxycarbonyl)amino]-4-cyclobutyl-l-ethoxy-l-oxobut-2-yl 4- methoxybenzoate (Second alternate procedure)

NaBH4 (0.23 g, 6 mmol) and THF (5 mL) were charged to a 100-ml RB flask. The mixture was cooled to -10°C. Methanesulfonic acid (0.78 mL, 12 mmol) was charged slowly into the mixture at less than -8°C and the mixture was agitated for 15 minutes. A 0.3M solution of the crude product of Example 5 (1 g, 3 mmol) in THF was charged slowly into the mixture at below -8°C. The mixture was agitated for 16 hours. H20 (1 ml) was charged slowly into the mixture at 0°C, and the mixture was warmed to RT. Di-tert-butyl dicarbonate (1.31 g, 6 mmol) and 2M aqueous NaOH (3.75 ml) were charged into the mixture. The mixture was agitated for 2 hours at RT. An assay of the reaction mixture gave the product (1.23 g, 94%). Example 7A: Ethyl 3-f(tert-buyoxycarbonyl)aminoJ-4-cyclobutyl-2-hydroxybutanoate

The crude product of Example 6A (6.0 kg, 13.78 mol) and MeOH (24 L) were charged into a 10-gallon autoclave at RT. The mixture was heated to 70°C under sealed conditions, where NH4 was transferred until the internal pressure became approximately 80 psig. The mixture was heated at 70°C at approximately 80 psig for 22 hours. The mixture was cooled to RT. NH4 was vented at RT. DMSO (5.4 L) was added to the mixture, and the mixture was aged at RT for 1 hour. The mixture was transferred into a 100-L RB flask with an overhead stirrer and a thermometer. The autoclave was rinsed with MeOH, and the mixture and rinse liquid were combined. This combined mixture was concentrated to remove MeOH under vacuum. Then, the flask was rinsed with DMSO (2.6 L) to wash the walls. Total DMSO volume was 8.0 L. The mixture was heated to 70°C to dissolve the solid to afford a clear solution, which was cooled to RT slowly to afford a slurry. ¾0 (32.0 L) was charged for approximately 1.5 hours at 20°C to 27°C. After addition of H20, the mixture was aged at RT overnight and then cooled to 0°C to 5°C for 4 more hours. The mixture was filtered to collect the solid, which was washed with cold H20 (12 L). The solid was dried at 40°C in a vacuum oven with N2 sweep (approximately 150 torr) to afford the crude product 5.63 kg (3.75 kg).
1H NMR (DMSO-d6, 400 MHz): δ 7.20-7.15 (m, 2H), 7.25 (d, J= 12.0 Hz, 0.5H), 5.92 (d, J= 12.0 Hz, 0.44H), 5.52-5.44 (m, 1H), 3.83-3.81 (m, 0.5H), 3.74-3.62 (m, 1.5H), 2.29- 2.22 (m, 1H), 2.03-1.92 (m, 2H), 1.83-1.70 (m, 2H), 1.62-1.24 (m, 13H).
13C NMR (DMSO-d6, 400 MHz) δ 175.2, 174.6, 155.5, 155.4, 78.0, 77.9, 74.4, 72.7, 51.9, 51.8, 38.8, 35.8, 33.3, 33.2, 33.0, 28.8, 28.7, 28.6, 28.5, 28.4, 28.2, 18.6, 18.5.
Example 7B: Ethyl 3-[(tert-buyoxycarbonyl)amino]-4-cyclobutyl-2-hydroxybutanoate

The crude product of Example 6A (6.0 g, 84 wt%, 11.57 mmol) and CaCl2 (1.413 g, 12.73 mmol) and 7N NH3 in MeOH (60 mL, 420 mmol) were charged into a 40 mL vial. The mixture was aged at approximately 33°C for 3 hours. The mixture was concentrated under reduced pressure to afford the product (7.8 g crude, assume 100% yield) as a tan solid. Example 8: Ethyl 3-amino-4-cyclobutyl-2-hydroxybutanoate hydrochloride

IP A (13.8 L) was charged into a 100-L RB flask with a mechanical stirrer, dry and clean with a thermometer and an addition funnel, followed by addition of the product of Example 7 (3.46 kg assay, 12.70 mol). HCI in IPA (5-6 M 13.8 L, 69 mol) was slowly added into the reaction mixture. The reaction mixture was heated at 50°C for 4 hours. The mixture was cooled to RT. Then, MTBE (28 L) was added to the mixture over 30 minutes. The reaction mixture was cooled to 0°C to 5°C by MeOH/ice bath for 1.5 hour. The mixture was filtered to collect the solid, which was washed with MTBE (7 L) twice. The wet cake was dried under vacuum with N2 and sweep overnight to afford the product as an off-white solid (2.15 kg, 10.30 mol) in 76.6% overall yield for Examples 5-8.
1H NMR (DMSO-d6, 400 MHz): δ 8.20-7.95 (m, 3H), 7.54-7.44 (m, 2H), 6.46 (d, J= 4.0 Hz, 0.5H), 6.26 (d, J= 8.0 Hz, 0.5H), 4.22 (s, 0.5H), 3.98 (s, 0.5H), 3.26 (s, 0.5H), 3.10 (d, J= 4.0 Hz, 0.5H), 2.45-2.36 (m, 1H), 2.00-1.96 (m, 2H), 1.81-1.39 (m, 6H).
13C NMR (DMSO-d6, 400 MHz) δ 174.1, 173.6, 71.2, 69.8, 51.7, 51.5, 36.0, 34.6,
31.7, 31.5, 28.0, 27.8, 27.7, 18.3, 18.1.
Exam le 9: Ethyl 3-amino-4-cyclobutyl-2-hydroxybutanoate hydrochloride (Recrystallization)

H20 (3.0 L), CH3CN (6 L) and the product of Example 8 (2.00 kg, 9.58 mol) were charged to a 100-L RB flask with an overhead stirrer, a thermocouple and a condenser at RT under N2. The mixture was heated to 65°C to get a clear solution. The mixture was cooled to 50°C to get a thin slurry. CH3CN (6.0 L) was added at 50°C for over 1 hour. The mixture was cooled to 40°C. CH3CN (9.0 L) was added at 40°C for over 1 hour. The mixture was cooled to 30°C. CH3CN (18 L) was added at 30°C. The mixture was cooled to approximately 0°C to 5°C and stirred for 1 hour before filtration. The mixture was filtered, washed with CH3CN (4 L) twice, and dried with N2 stream to afford the recrystallized product as a white solid (1.887 kg, 9.04 mol, 94% isolated yield).
Ή NMR (DMSO-d6, 400 MHz): δ 8.20-7.95 (m, 3H), 7.54-7.44 (m, 2H), 6.46 (d, J= 4.0 Hz, 0.5H), 6.26 (d, J= 8.0 Hz, 0.5H), 4.22 (s, 0.5H), 3.98 (s, 0.5H), 3.26 (s, 0.5H), 3.10 (d, J= 4.0 Hz, 0.5H), 2.45-2.36 (m, 1H), 2.00-1.96 (m, 2H), 1.81-1.39 (m, 6H).
13C NMR (DMSO-d6, 400 MHz): δ 174.1, 173.6, 71.2, 69.8, 51.7, 51.5, 36.0, 34.6, 31.7, 31.5, 28.0, 27.8, 27.7, 18.3, 18.1.
Example 10: (lR,2S,5S)-N-(4-amino-l-cyclobutyl-3-hydroxy-4-oxobutan-2-yl)-3-[N-(t rt- butylcarbamoyl)-3-methyl-I^valyl]-6,6-dimethyl-3-azabicyclo[3A ]h

Hydroxybenzotiazole (HOBT, 4.83 g, 31.5 mmol), water (4.5 mL), (1R,2S,5S)-N- (4-amino- 1 -cyclobutyl-3 -hydroxy-4-oxobutan-2-yl)-3- [N-(tertbutylcarbamoyl)-3 -methylvalyl] – 6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2-carboxamide (30 g, 60.6 mmol), HCl salt product of Example 9 (13.79 g, 66.1 mmol), ethyl acetate (120 mL) and N-methyl-2-pyrrolidone (NMP, 30 mL) were added at 19°C to a three-necked 500mL RB flask equipped with an overhead stirrer and a thermocouple. N-methylmorpholine (13.3 mL, 121 mmol) was added to the mixture at 19°C. l-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI, 15.0 g, 78.0 mmol) was added to the mixture at 21°C. Ethyl acetate (30 mL) was then added to the mixture at 18°C.
The mixture was agitated at approximately 20°C to 24°C for about 16 hours. After the reaction was complete, ethyl acetate (120 mL) was added at 23°C. The mixture was washed with 10% aqueous potassium carbonate solution (180 mL) twice at approximately 20°C to 24°C. Then, the organic layer was washed with 3.3% aqueous HCl (180 mL) twice at approximately 12°C to 18°C. The organic layer then was washed with 10% aqueous potassium carbonate solution (180 mL) and water (180 mL). The organic layer was concentrated to approximately 100 mL volume and was added to heptane (900 mL) dropwise at approximately -10°C to -5°C to precipitate the product. The mixture was filtered and washed with heptane. The solid was dried in vacuo at approximately 50°C to 60°C overnight. 31.3 g of the product compound was obtained as a white solid in 99% yield. The above procedure is in accordance with the processes disclosed in U.S. Patent Application Publication No. US2010/519485 Al, the disclosures of which are herein
incorporated by reference. It will be appreciated that the processes disclosed therein can be modified without undue experimentation to prepare specifically desired materials. The results of H NMR and C NMR for the above procedure were consistent with those reported in U.S. Patent Application Publication No. US2010/519485 Al .
Example 11: (lR,5S)-N-[3-Amino-l-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(l,l- dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-l-oxobutyl]-6,6-dimazabicyclo[3.1.0]hexan-2(S)-carboxamide

Acetic acid (27.0 mL, 472 mmol) and MTBE (240 mL) at RT were added to a three-necked 1L RB flask equipped with an overhead stirrer, a thermocouple and a chiller. The mixture was cooled to approximately 14°C, then the product from Example 10 (30.0 g, 57.5 mmol) was charged at approximately 14°C. The mixture was cooled to approximately 11°C. 2,2,6,6-Tetramethylpiperidin-l-yl)oxyl (TEMPO, 9.97 g, 63.8 mmol) was added to the mixture. A pre-mixed solution containing 40% aqueous sodium permanganate (17.02 g, 48.0 mmol) and water (99 mL) at approximately 12°C to 14°C was added to the reaction mixture over about 2 hours. The mixture was agitated at approximately 12°C until completion.
After the reaction was complete, the mixture was cooled to approximately 1°C. Water (30 mL) was added, then aqueous layer was separated. The organic layer was then washed with water (150 mL) at approximately 0°C to 10°C, and then washed with a pre-mixed solution of sodium ascorbate (30.0 g, 151 mmol) in water (150 mL) and concentrated HCl (12.42 mL, 151 mmol) at approximately 5°C to 15°C. The mixture was agitated at approximately 5°C to 10°C for 2 hours; then aqueous layer was separated. The organic layer was further washed with 2.5 N HCl (120 mL) at approximately 0°C to 10°C and with water (150 mL) at
approximately 0°C to 10°C four times. The organic layer (approximately 170 mL) was then added dropwise to heptane (720 mL) at approximately -20°C to -15°C to precipitate the product. The mixture was then warmed to -5°C and filtered to collect the solid. The solid was washed with heptane, dried in a vacuum oven with nitrogen sweep at room temperature to afford 27.1 g of desired product of Formula II as a white solid in 91% yield.
The above procedure is in accordance with the processes disclosed in U.S.
Provisional Patent Application No.61/482,592 (unpublished), the disclosures of which are herein incorporated by reference. It will be appreciated that the processes disclosed therein can be modified without undue experimentation to prepare specifically desired materials. The results of 1H NMR and 13C NMR for the above procedure were consistent with those reported in U.S. Provisional Patent Application No.61/482,592 (unpublished).
……………………………………………………………….
EXTRAS some images are not visible…….see………..http://www.allfordrugs.com/2015/08/02/boceprevir-%D0%B1%D0%BE%D1%86%D0%B5%D0%BF%D1%80%D0%B5%D0%B2%D0%B8%D1%80-%D8%A8%D9%88%D8%B3%D9%8A%D8%A8%D8%B1%D9%8A%D9%81%D9%8A%D8%B1-%E6%B3%A2%E6%99%AE%E7%91%9E%E9%9F%A6/
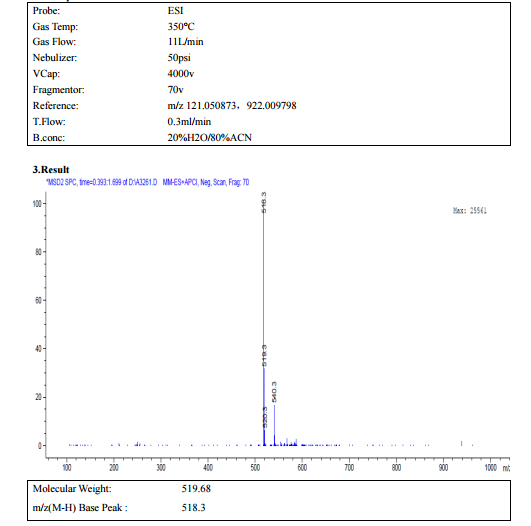
HPLC
MASS SPECTROSCOPY
IR GRAPH
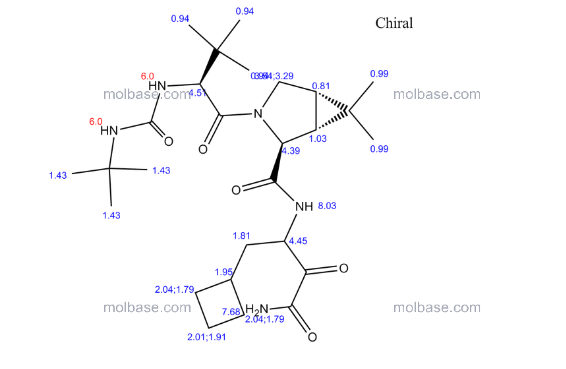
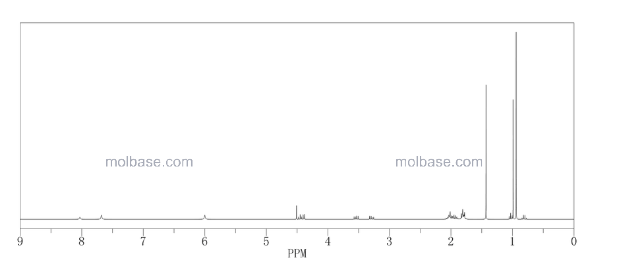
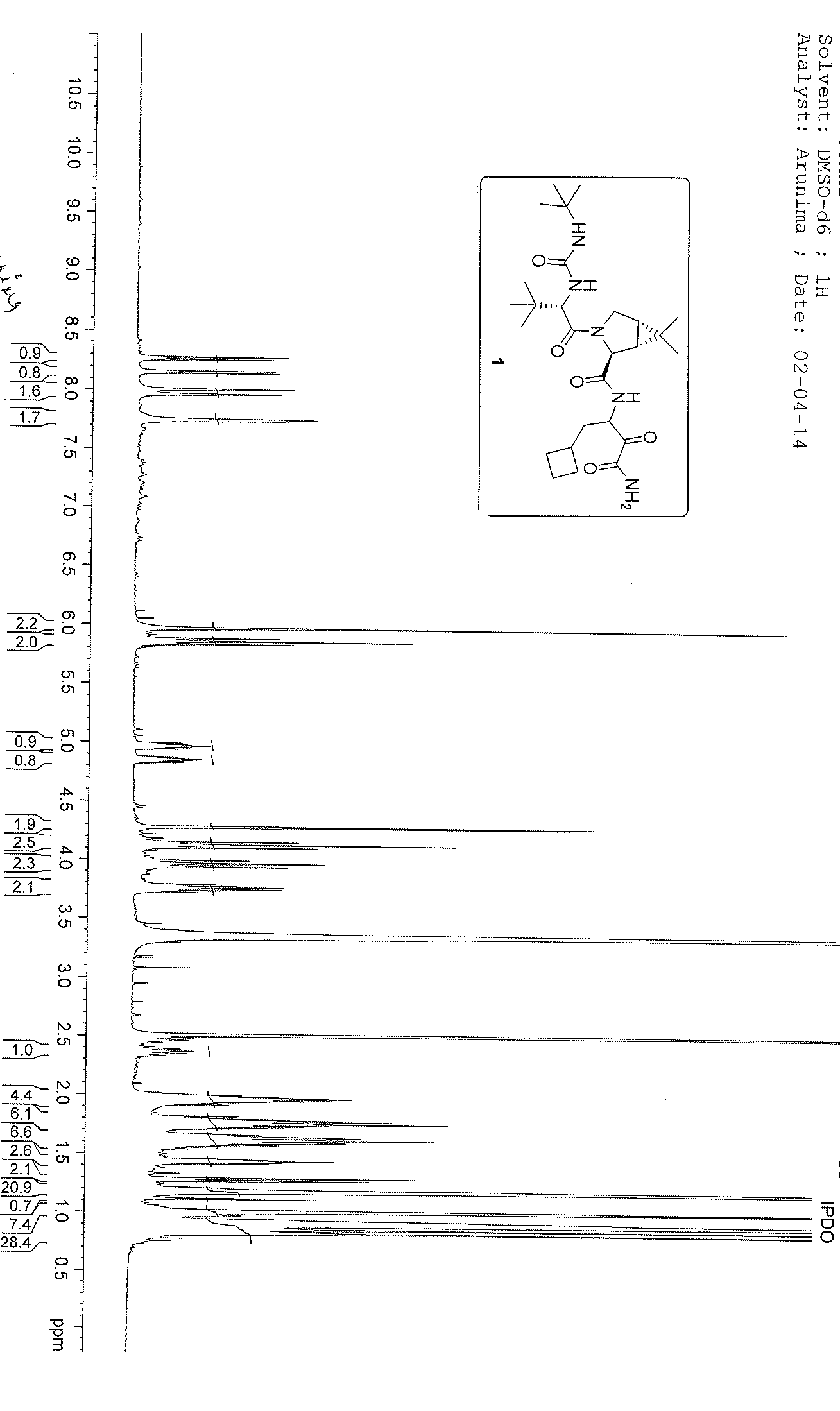
1H NMR GRAPH
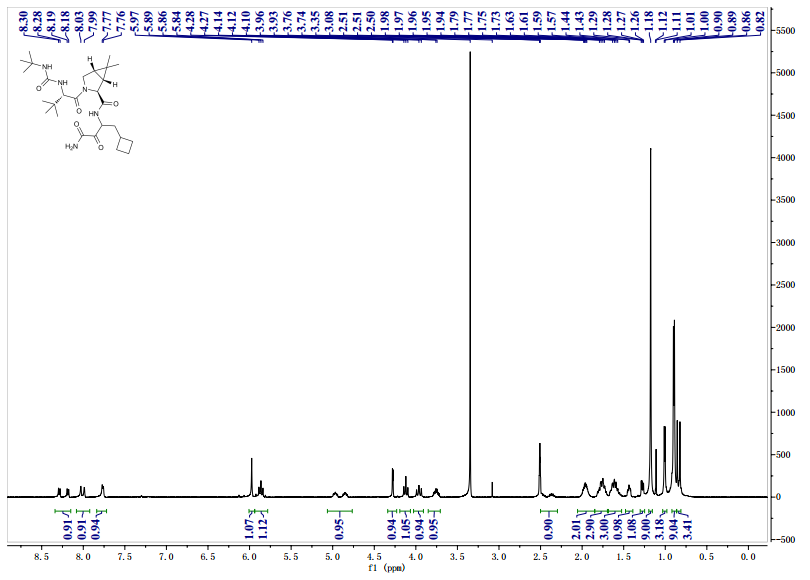
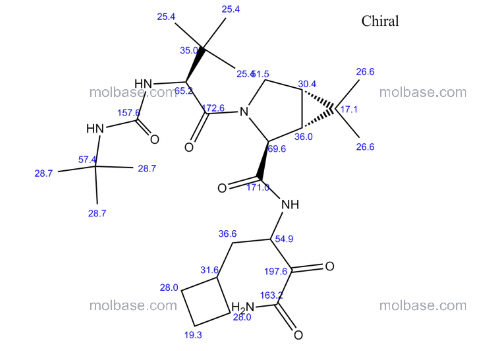
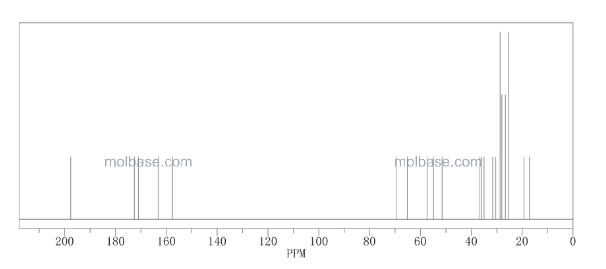
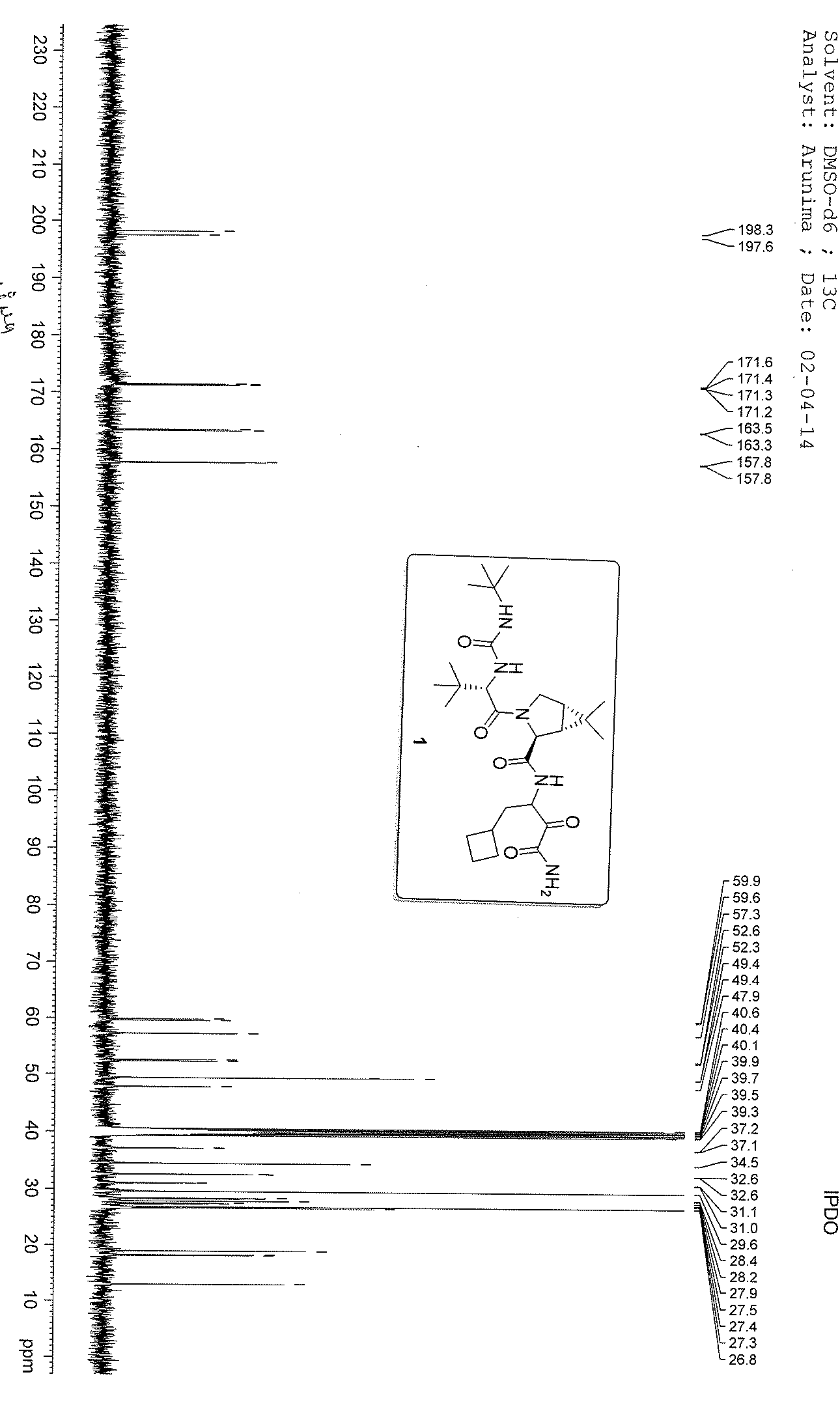
13 C NMR GRAPH
WILL BE UPDATED![[14C]-Boceprevir NMR spectra analysis, Chemical CAS NO. 394730-60-0 NMR spectral analysis, [14C]-Boceprevir H-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-29/002/493/2493725_1h.png)
13C NMR PREDICT
![[14C]-Boceprevir NMR spectra analysis, Chemical CAS NO. 394730-60-0 NMR spectral analysis, [14C]-Boceprevir C-NMR spectrum](https://i0.wp.com/pic11.molbase.net/nmr/nmr_image/2014-11-29/002/493/2493725_13c.png)
| WO2010138889A1* | 28 May 2010 | 2 Dec 2010 | Concert Pharmaceuticals, Inc. | Peptides for the treatment of hcv infections |
| WO2011125006A2* | 31 Mar 2011 | 13 Oct 2011 | Pfizer Inc. | Novel sultam compounds |
| US20110034705 * | 17 Dec 2008 | 10 Feb 2011 | Schering-Plough Corporation | Process For the Synthesis of 3- Amino-3-Cyclobuthylmethyl-2-Hydroxypropionamide or Salts Thereof |
| US8188137 | 14 Aug 2009 | 29 May 2012 | Avila Therapeutics, Inc. | HCV protease inhibitors and uses thereof |
| US8524760 | 10 Apr 2012 | 3 Sep 2013 | Celgene Avilomics Research, Inc. | HCV protease inhibitors and uses thereof |
| EP2704570A1 * | 2 May 2012 | 12 Mar 2014 | Merck Sharp & Dohme Corp. | Drug substances, pharmeceutical compositions and methods for preparing the same |
| WO2014061034A1* | 17 Oct 2013 | 24 Apr 2014 | Msn Laboratories Limited | Process for preparation of boceprevir and intermediates thereof |
References
- 1 Kiser JJ, Burton JR, Anderson PL, Everson GT (May 2012). “Review and Management of Drug Interactions with Boceprevir and Telaprevir”. Hepatology 55 (5): 1620–8. doi:10.1002/hep.25653. PMC 3345276. PMID 22331658.
- 2
- Degertekin B, Lok AS (May 2008). “Update on viral hepatitis: 2007″. Curr. Opin. Gastroenterol. 24 (3): 306–11.doi:10.1097/MOG.0b013e3282f70285. PMID 18408458.
- 3
- Njoroge FG, Chen KX, Shih NY, Piwinski JJ (January 2008). “Challenges in modern drug discovery: a case study of boceprevir, an HCV protease inhibitor for the treatment of hepatitis C virus infection”. Acc. Chem. Res. 41 (1): 50–9. doi:10.1021/ar700109k.PMID 18193821.
- 4
- “Boceprevir – FDA Antiviral Drugs”. FDA. FDA. April 2011. Retrieved April 2014.
- 5
- “Interim Results from Boceprevir Phase II Study in Genotype 1 Treatment-Naive Hepatitis C Patients Presented At EASL – Forbes.com” (Press release). Forbes.com. Retrieved 2008-05-19.
- 6
- “FDA Approves Merck’s VICTRELIS™ (boceprevir), First-in-Class Oral Hepatitis C Virus (HCV) Protease Inhibitor” (Press release). Merck & Co. Retrieved 2011-05-14.
- 7
- http://www.hcvadvocate.org/news/reports/EASL_2009/Advocate_EASL_2009_Coverage.htm
- 8
- Poordad, F et al. (March 2011). “Boceprevir for Untreated Chronic HCV Genotype 1 Infection”. N Engl J Med. 364 (13): 1195–206. doi:10.1056/NEJMoa1010494. PMID 21449783.
- 9
- Jensen, D (March 2011). “A New Era of Hepatitis C Therapy Begins”. N Engl J Med. 364 (13): 1272–1273.doi:10.1056/NEJMe1100829. PMID 21449791.
- 10
Bacon, B et al. (March 2011). “Boceprevir for Previously Treated Chronic HCV Genotype 1 Infection”. N Engl J Med. 364 (13): 1207–17.doi:10.1056/NEJMoa1009482. PMC 3153125. PMID 21449784.
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| (1R,5S)-N-[3-Amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3-[2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2(S)-carboxamide | |
| CLINICAL DATA | |
| TRADE NAMES | Victrelis |
| AHFS/DRUGS.COM | Consumer Drug Information |
| MEDLINEPLUS | a611039 |
| LICENCE DATA | US FDA:link |
|
|
|
|
| Oral | |
| PHARMACOKINETIC DATA | |
| PROTEIN BINDING | 75% [1] |
| HALF-LIFE | 3.4 hours [1] |
| IDENTIFIERS | |
| 394730-60-0 |
|
| J05AE12 | |
| PUBCHEM | CID 10324367 |
| CHEMSPIDER | 8499830 |
| UNII | 89BT58KELH |
| CHEMBL | CHEMBL218394 |
| NIAID CHEMDB | 398493 |
| CHEMICAL DATA | |
| FORMULA | C27H45N5O5 |



////
Practical Process Research and development; Development..Optimizing the Reaction by Minimizing Impurities


2000, Pages 165–184

Chapter 8 – Optimizing the Reaction by Minimizing Impurities
- Process Solutions L.L.C., Nicasio, California
The goals of process optimization change with the successful development of a project from early process research through scale-up into dedicated manufacturing. This general order of optimization may differ according to the nature of the process being considered; for instance, a process generating an inordinate amount of waste may be optimized to decrease waste before scaling up to the pilot plant. The initial goal of all process research and development is to maximize the amount of product generated under the reaction conditions. This is done by driving the reaction to completion, that is, by consuming any starting material that is charged in limiting amounts and by generating product with a minimal amount of by-products. Once the in-process yield has been optimized, the maximum yield of isolated product is expected. Rapid optimization is possible by judiciously changing solvents, reagents, catalysts, and ligands; investigations in this area allow the chemist considerable room for creativity and simplifying a process. Such changes may generate different impurities in the isolated intermediates, and it may be necessary to examine the tolerance of subsequent processes for the new impurities.
-
Chapter 8 – Optimizing the Reaction by Minimizing Impurities
-

Neal G. Anderson, PhD
For the past 17 years Neal has consulted to the pharmaceutical and fine chemical industries on chemical process research and development. He has presented short courses internationally on process R&D for “small molecules” to over 1400 participants from more than 160 companies. Keys to efficient scale-up are anticipating and then avoiding diffiulcties. Prior to consulting he worked at Bristol-Myers Squibb for 17 years. During that time, he had extensive hands-on experience with chemical process R&D in the lab, pilot plant, and manufacturing sites, including 12 manufacturing start-ups and process development for four major drugs. He is the author of Practical Process Research & Development(Academic Press; 2nd edition 2012).
Education & Training
- 1977-1979, post-doctoral studies, McNeil Laboratories
- 1977, Ph.D., Medicinal Chemistry, University of Michigan
- 1972, B.Sc., Honors Biology, University of Illinois
I consult to the pharmaceutical and fine chemical industries on developing and trouble-shooting processes to efficiently prepare drug substances and intermediates on large scale. Anticipating and avoiding problems are key for effective and efficient scale-up. For 17 years I have been consulting and presenting short courses internationally on process chemistry R & D for “small molecules” (over 1400 participants from more than 160 companies). Prior to consulting I worked at Bristol-Myers Squibb for 17 years. During that time I had extensive hands-on experience with chemical process development in the lab, pilot plant, and manufacturing sites, including 12 manufacturing start-ups and process development for four major drugs and many new drug candidates. I wrote Practical Process Research & Development (Academic Press, 2000; 2nd edition 2012).
Practical Process Research & Development describes the development of chemical processes for the pharmaceutical and fine chemicals industries. It provides a comprehensive, step-by-step approach to process R & D, and it is designed for those who want insights into generating rugged, practical, cost-effective processes. Guidelines for industrial process R & D are rarely taught in academia, although this book has been used as a textbook. It is primarily used by those in industry.
The second edition updates the first edition and includes topics not covered in the first edition , such as genotoxins, biocatalysis, green solvents, predicting effective solvent combinations, and process validation. Almost 85% of the references cited were published after the first edition was published, and virtually all examples in the Figures are new. Trevor Laird kindly wrote a forward for this edition.
, such as genotoxins, biocatalysis, green solvents, predicting effective solvent combinations, and process validation. Almost 85% of the references cited were published after the first edition was published, and virtually all examples in the Figures are new. Trevor Laird kindly wrote a forward for this edition.
The second edition has been translated into Japanese and graced with a handsome cover. Noriaki Murase was the translation supervisor, and the translators were Shohei Imachi, Koreaki Imura, Dai Tatsuta, Taro Tsukude, Toyoharu Numata, Yujiro Furuya, Akira Manaka, and Noriaki Murase. Sayaka Nukatsuka was the editor. I am very grateful to these people for their hard work to translate my book.
I am grateful to Barry Sharpless and Jerry Moniot for writing forwards to the first edition I am also grateful to the following people for their translations of the first edition of my book. Noriaki Murase, Yoshinori Murata, Toyoharu Numata, Mio Sakai, and Tatsuo Ueki translated Practical Process Research & Development into Japanese. Kwang-Hyun Ahn, Yeung-Ho Park, and Sung-Kwan Hwang translated Practical Process Research & Development into Korean. Zhinong Gao and Wenhao Hu translated Practical Process Research & Development into Chinese.
")

")
- Evaluate the existing synthesis and identify steps, or sequences in the route that may pose a problem for large scale synthesis
- Propose alternatives to any problematic steps or sequences and then implement these alternatives bases upon laboratory experimentation using Ph.D. level chemists with process research expertise
- Ensure the synthesis is suitable for the immediate needs of the project, which maybe for only a few kilograms of API
- Ensure the synthesis is suitable for long term, large scale manufacturing
- Optimize reagent charges, operating temperatures, concentrations, work-up conditions and volumes, and solvent use in general
- Identify which steps can be combined to result in a “through process” and implement the through process
- Optimize purification schemes by identifying key crystalline intermediates and remove chromatographies from the synthesis
- Optimize recrystallization parameters to ensure consistently high purity with similar impurity profiles from batch to batch, with low mother liquor losses
- Institute appropriate analytical controls for in-process assays, end of reaction specifications, and acceptable intermediate or API purity
- The process research team works closely with the analytical team to integrate the chemistry and analytical controls into the process at an early stage of the development cycle. The process research is then documented into a JACS style development report that outlines the chemistry and synthetic approaches that were tried as part of the synthetic development effort. This development report also includes a detailed experimental with supporting analytical data for the successful chemistry that results from our effort.The experimental that is part of these development reports is much more detailed than any journal publication. When coupled with our analytical and cGMP capabilities, the process research we provide is an essential groundwork for any compound that is just advancing from nomination at the discovery phase into clinical trial development. The process we develop provides the foundation of the ultimate manufacturing process, and should not need any changes (at a later date), to the synthetic strategy or bond forming steps used to prepare the API.

Critical Assessment of Pharmaceutical Processes, A Rationale for Changing the Synthetic Route

Critical Assessment of Pharmaceutical Processes A Rationale for Changing the Synthetic Route
A Rationale for Changing the Synthetic Route
Table of Contents
- 1. Introduction
- 2. Criteria for Process Assessment
-
- 2.1. Safety Issues2.1.1. Potential Safety Issues and Their Significance
- 2.1.2. Prediction and Assessment of Safety Issues
- 2.1.3. Options To Manage Safety Issues
- 2.1.4. Designing a Safer New Route
-
- 2.2. Environmental Issues
- 2.2.1. Potential Environmental Issues and Their Significance
- 2.2.2. Prediction and Assessment of Environmental Issues
- 2.2.3. Options To Manage Environmental Issues
- 2.2.4. Designing a New “Greener” Route
-
- 2.3. Legal Issues
- 2.3.1. Potential Legal Issues and Their Significance
- 2.3.2. Prediction and Assessment of Legal Issues Associated with Regulated Substances
- 2.3.3. Prediction and Assessment of Legal Issues Associated with Patent Infringement
- 2.3.4. Options To Manage Patent Issues
- 2.3.5. Designing a New Route with Freedom To Operate
-
- 2.4. Economic Issues
- 2.4.1. Potential Economic Issues and Their Significance
- 2.4.2. Prediction and Assessment of Economic Issues
- 2.4.3. Options To Manage Economic Issues
- 2.4.4. Designing a Cost-Effective New Route
-
- 2.5. Control Issues
- 2.5.1. Potential Control Issues and Their Significance
- 2.5.2. Prediction and Assessment of Control Issues
- 2.5.3. Options To Manage Control Issues
- 2.5.4. Designing a New Route with Adequate Control Measures
-
- 2.6. Throughput Issues
- 2.6.1. Potential Throughput Issues and Their Significance
- 2.6.2. Prediction and Assessment of Throughput Issues
- 2.6.3. Options To Manage Throughput Issues
- 2.6.4. Designing a New Route with High Throughput
- 3. Interrelationships between Process Issues
- 4. Conclusions
- 5. Acknowledgments
- 6. References
Pemirolast

Pemirolast (INN) is a mast cell stabilizer used as an anti-allergic drug therapy. It is marketed under the tradenames Alegysal and Alamast.
9-methyl-3-(1H-tetrazol-5-yl)-4H-pyrido-[1, 2-a]-pyrimidin-4-one
It has also been studied for the treatment of asthma.

https://www.google.com/patents/US9006431
Pemirolast is an orally-active anti-allergic drug which is used in the treatment of conditions such as asthma, allergic rhinitis and conjunctivitis. See, for example, U.S. Pat. No. 4,122,274, European Patent Applications EP 316 174 and EP 1 285 921, Yanagihara et al, Japanese Journal of Pharmacology, 51, 93 (1989) and Drugs of Today, 28, 29 (1992). The drug is presently marketed in e.g. Japan as the potassium salt under the trademark ALEGYSAL™.
Commercial pemirolast potassium has the disadvantage that it is known to give rise to sharp plasma concentration peaks in humans (see, for example, Kinbara et al, “Plasma Level and Urinary Excretion of TBX in Humans”, Japanese Pharmacology & Therapeutics, 18(3) (1990), and “Antiallergic agent—ALEGYSAL tablet 5 mg—ALEGYSAL tablet 10 mg—ALEGYSAL dry syrup”, Pharmaceutical Interview Form (IF), Revised in October 2007 (7th version), Standard Commodity Classification No.: 87449). The latter document also reports that the potassium salt of pemirolast is hygroscopic, which is believed to give rise to chemical instability, and possesses a bitter taste.
U.S. Pat. No. 4,122,274 describes a process for the production of salts of pemirolast, including potassium salts and (at Example 14) a sodium salt. As described herein, this technique produces a sodium salt that is physically unstable. Sodium salts of pemirolast are also mentioned (but a synthesis thereof not described) in international patent applications WO 2008/074975 and WO 2008/075028.
COMPARATIVE EXAMPLE 5Recrystallisation of Pemirolast Sodium According to the Method of U.S. Pat. No. 4,122,274
In U.S. Pat. No. 4,122,274, it is stated that the crude title product (pemirolast sodium) was recrystallised from water:ethanol to give pure title product. It is not clear from this level of detail what the ratio of water:ethanol employed was, so several experiments were performed with a view to reproducing the prior art technique.
- (i) Crude sodium salt of pemirolast (480 mg; from Example 4, method (I) above) was recrystallised from water and ethanol (95%) in a 1:1 ratio. The Na salt of pemirolast (480 mg, 1.92 mmol) was dissolved in H2O (8 mL) at 70° C. and EtOH 95% (8 mL) was added. The clear solution was allowed to reach room temperature and the solid material formed was filtered off, washed with a small amount of ethanol and dried in vacuum to give 316 mg of pure sodium salt.
- (ii) Crude sodium salt of pemirolast (500 mg; from Example 4, method (II) above) was dissolved in water (4.9 mL) at 70° C. Thereafter EtOH 95% (ca. 4.0 mL) was added at 70° C. until a solid started to form. Another 0.1 mL of water was added to get everything into solution. The solid material formed upon cooling was collected by filtration and dried under vacuum to give 348 mg of pure sodium salt.
- (iii) Crude sodium salt of pemirolast (300 mg; from Example 4, method (II) above) was recrystallised from water:ethanol (1:1 ratio; 10 mL) at 70° C. The solid material formed upon cooling was collected by filtration and dried under vacuum to give 174 mg of pure sodium salt.
- (iv) Crude sodium salt of pemirolast (300 mg; from Example 4, method (II) above) was recrystallised from water:ethanol (9:1 ratio, 4 mL) at 70° C. The solid material formed upon cooling was collected by filtration and dried under vacuum to give 219 mg of pure sodium salt.
All four samples of pure pemirolast sodium salt had the same physico-chemical properties (Raman spectra and NMR):
1H NMR (D2O) δ: 8.86-8.80 (m, 1H, CH), 8.57 (s, 1H, CH), 7.68-7.59 (m, 1H, CH), 7.22-7.13 (m, 1H, CH), 2.39 (s, 3H, CH3).
The PXRD pattern (measured in respect of Example 5(i) above) is shown in FIG. 3. It was concluded from this that this form of the sodium salt is an amorphous material mixed with a crystalline fraction.
The Raman spectrum was recorded directly after recrystallisation. All samples were then stored under ambient conditions on a shelf in a fume hood. About a month later, a Raman spectrum was recorded, which was significantly different to that recorded earlier. This is shown in FIG. 4, where the lower spectrum accords to the earlier measurement and the upper spectrum accords to the later measurement. In the light of these results, it was concluded that the prior art amorphous form of pemirolast sodium is physically unstable.
The amorphous material was also prepared by drying of the form obtained in accordance with Example 11 below at 40° C. and reduced pressure for 40 hours to yield 12 g of a pale yellow cotton-like amorphous solid.
………………………..
http://www.lookchem.com/Chempedia/Chemical-Technology/Organic-Chemical-Technology/18815.html

1) Firstly, 2-Amino-3-methylpyridine (I) is condensed with ethoxymethylenemalonodinitrile (II) to afford the monocyclic intermediate (III), which is in tautomeric equilibrium with the pyridopyrimidine derivative (IV). Next, the reaction of (IV) with aluminum azide (AlCl3.NaN3) in refluxing THF yields 4-imino-9-methyl-3-(1H-tetrazol-5-yl)-4H-pyrido[1,2-a]pyrimidine (V). Finally, this compound is first hydrolyzed with 1N HCl and then treated with KOH.
2) Compound (IV) can be converted to the final product by a one-pot reaction: (VI) is treated first with NaN3 in refluxing acetic acid, then hydrolyzed with HCl and finally treated with KOH.
………….
EXAMPLE 1
A suspension of 9-methyl-3-(1 H-tetrazol-5-yl)-4H-pyrido-[1,2-a]-pyrimidin-4-one (68.5 g; 0.3 mols) in methanol (420 ml) and water (210 ml) heated at 50° C. is added with a 40% N-methylamine aqueous solution (30 ml, 0.35 mols) to pH=10. The solution is heated at 68-70° C., and acidified with formic acid (21 ml) to pH=3. After completion of the addition the mixture is kept at 68-70° C. for about 15 minutes and then cooled to 20-25° C. The precipitate is filtered, washed with methanol and dried under vacuum at 40° C. to give 9-methyl-3-(1 H-tetrazol-5-yl)-4H-pyrido-[1,2-a]-pyrimidin-4-one with >99.8% HPLC purity (63 g, 92% yield).
EXAMPLE 2
9-Methyl-3-(1 H-tetrazol-5-yl)-4H-pyrido-[1,2-a]-pyrimidin-4-one (63 g, 0.28 mols) is suspended in methanol (1000 ml). The resulting suspension is kept at 45° C. and slowly added with a 45% potassium hydroxide aqueous solution to pH 9-9.5. The suspension is stirred at 45° C. for about 15 minutes and then cooled to 20° C. The precipitate is filtered, washed with methanol and dried under vacuum at 80° C., to obtain Pemirolast Potassium (71.9 g; 0.27 mols, 96% yield) with HPLC purity >99.8%. 1H NMR(D2O, TMS) d (ppm): 2.02 (s, 3H); 6.83 (t, 1H); 7.22 (d, 1H); 8.18 (s, 1H); 8.47 (d, 1H).
References
- Tinkelman DG, Berkowitz RB (February 1991). “A pilot study of pemirolast in patients with seasonal allergic rhinitis”. Ann Allergy 66 (2): 162–5. PMID 1994787.
- Kawashima T, Iwamoto I, Nakagawa N, Tomioka H, Yoshida S (1994). “Inhibitory effect of pemirolast, a novel antiallergic drug, on leukotriene C4 and granule protein release from human eosinophils”. Int. Arch. Allergy Immunol. 103 (4): 405–9. doi:10.1159/000236662. PMID 8130655.
- Abelson MB, Berdy GJ, Mundorf T, Amdahl LD, Graves AL (October 2002). “Pemirolast potassium 0.1% ophthalmic solution is an effective treatment for allergic conjunctivitis: a pooled analysis of two prospective, randomized, double-masked, placebo-controlled, phase III studies”. J Ocul Pharmacol Ther 18 (5): 475–88. doi:10.1089/10807680260362759. PMID 12419098.
- Kemp JP, Bernstein IL, Bierman CW et al. (June 1992). “Pemirolast, a new oral nonbronchodilator drug for chronic asthma”. Ann Allergy 68 (6): 488–91. PMID 1610024.
 |
|
| Systematic (IUPAC) name | |
|---|---|
|
9-methyl-3-(1H-tetrazol-5-yl)-4H-pyrido[1,2-a]pyrimidin-4-one
|
|
| Clinical data | |
| Trade names | Alamast |
| AHFS/Drugs.com | monograph |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral, ophthalmic |
| Identifiers | |
| CAS Registry Number | 69372-19-6 |
| ATC code | None |
| PubChem | CID: 57697 |
| IUPHAR/BPS | 7329 |
| DrugBank | DB00885 |
| ChemSpider | 51990 |
| UNII | 2C09NV773M |
| KEGG | D07476 |
| ChEMBL | CHEMBL1201198 |
| Chemical data | |
| Formula | C10H8N6O |
| Molecular mass | 228.21 g/mol |
| US4122274 * | May 25, 1977 | Oct 24, 1978 | Bristol-Myers Company | 3-Tetrazolo-5,6,7,8-substituted-pyrido[1,2-a]pyrimidin-4-ones |
| EP0316174A1 | Nov 10, 1988 | May 17, 1989 | Tokyo Tanabe Company Limited | Aqueous preparation of 9-methyl-3-(1H-tetrazol-5-yl)-4H-Pyrido[1,2-a]pyrimidin-4-one potassium salt |
| EP1285921A1 | Jun 25, 2002 | Feb 26, 2003 | Dinamite Dipharma S.p.A. | A process for the preparation of high purity pemirolast |
| JPH0374385A | Title not available | |||
| WO2008074975A1 | Nov 16, 2007 | Jun 26, 2008 | Cardoz Ab | New combination for use in the treatment of inflammatory disorders |
| WO2008075028A1 | Dec 18, 2007 | Jun 26, 2008 | Cardoz Ab | New combination for use in the treatment of inflammatory disorders |
| US4122274 | May 25, 1977 | Oct 24, 1978 | Bristol-Myers Company | 3-Tetrazolo-5,6,7,8-substituted-pyrido[1,2-a]pyrimidin-4-ones | |
| US5254688 * | Jun 19, 1991 | Oct 19, 1993 | Wako Pure Chemical Industries, Ltd. | Process for producing pyrido[1,2-a]pyrimidine derivative | |
| DE243821C | Title not available | ||||
| EP0462834A1 | Jun 20, 1991 | Dec 27, 1991 | Wako Pure Chemical Industries, Ltd | Process for producing pyrido [1,2-a]pyrimidine derivative | |
| WO1993025557A1 | Jun 7, 1993 | Dec 23, 1993 | Smithkline Beecham Plc | Process for the preparation of clavulanic acid |

Pemirolast Potassium (BMY 26517) cas100299-08-9is a histamine H1 antagonist and mast cell stabilizer that acts as an antiallergic agent.
Target: Histamine H1 Receptor
Pemirolast potassium (BMY 26517) is a new oral, nonbronchodilator antiallergy medication that is being evaluated for the therapy of asthma [1]. Pemirolast potassium (BMY 26517) inhibits chemical mediator release from tissue mast cells and is also shown to inhibit the release of peptides including substance P, Pemirolast potassium (BMY 26517) reduces kaolin intake by inhibition of substance P release in rats [2]. Pemirolast potently attenuates paclitaxel hypersensitivity reactions through inhibition of the release of sensory neuropeptides in rats [3]. Pemirolast potassium is used for the treatment of allergic conjunctivitis and prophylaxis for pulmonary hypersensitivity reactions to drugs such as paclitaxel [4].
Molecular formula: C10H7KN6O
Molecular Weight: 266.30
External links
- Mitsubishi Tanabe Pharma Corporation (2007). “ALEGYSAL (English)” (PDF). Retrieved 2008-09-02.
- “DailyMed Announcements”. U.S. National Library of Medicine. 2005. Retrieved 2008-09-02.
Necessity of Establishing Chemical Integrity of Polymorphs of Drug Substance Using a Combination of NMR, HPLC, Elemental Analysis, and Solid-State Characterization Techniques: Case Studies

Necessity of Establishing Chemical Integrity of Polymorphs of Drug Substance Using a Combination of NMR, HPLC, Elemental Analysis, and Solid-State Characterization Techniques: Case Studies
Moexipril


RS-10085-197
SPM-925
RS-10085 (free base)

Pharmacology
Moexipril is available as a prodrug moexipril hydrochloride, and is metabolized in the liver to form the pharmacologically active compound moexiprilat. Formation of moexiprilat is caused by hydrolysis of an ethyl ester group.[5] Moexipril is incompletely absorbed after oral administration, and its bioavailability is low.[6] The long pharmacokinetic half-life and persistent ACE inhibition of moexipril allows once-daily administration.[7]
Moexipril is highly lipophilic,[2] and is in the same hydrophobic range as quinapril, benazepril, and ramipril.[7] Lipophilic ACE inhibitors are able to penetrate membranes more readily, thus tissue ACE may be a target in addition to plasma ACE. A significant reduction in tissue ACE (lung, myocardium, aorta, and kidney) activity has been shown after moexipril use.[8]
It has additional PDE4-inhibiting effects.[9]
Side effects
Moexipril is generally well tolerated in elderly patients with hypertension.[10] Hypotension, dizziness, increased cough, diarrhea, flu syndrome, fatigue, and flushing have been found to affect less than 6% of patients who were prescribed moexipril.[3][10]
Mechanism of action
As an ACE inhibitor, moexipril causes a decrease in ACE. This blocks the conversion of angiotensin I to angiotensin II. Blockage of angiotensin II limits hypertension within the vasculature. Additionally, moexipril has been found to possess cardioprotective properties. Rats given moexipril one week prior to induction of myocardial infarction, displayed decreased infarct size.[11] The cardioprotective effects of ACE inhibitors are mediated through a combination of angiotensin II inhibition and bradykininproliferation.[8][12] Increased levels of bradykinin stimulate in the production of prostaglandin E2[13] and nitric oxide,[12] which cause vasodilation and continue to exert antiproliferative effects.[8] Inhibition of angiotensin II by moexipril decreases remodeling effects on the cardiovascular system. Indirectly, angiotensin II stimulates of the production of endothelin 1 and 3 (ET1, ET3)[14] and the transforming growth factor beta-1 (TGF-β1),[15] all of which have tissue proliferative effects that are blocked by the actions of moexipril. The antiproliferative effects of moexipril have also been demonstrated by in vitro studies where moexipril inhibits the estrogen-stimulated growth of neonatal cardiac fibroblasts in rats.[12] Other ACE inhibitors have also been found to produce these actions, as well.
WO 2014202659
http://www.google.com/patents/WO2014202659A1?cl=en
US4344949
http://www.google.co.in/patents/US4344949
References
- Hochadel, Maryanne, ed. (2006). The AARP Guide to Pills. Sterling Publishing Company. p. 640. ISBN 978-1-4027-1740-6. Retrieved2009-10-09.
- Belal, F.F, K.M. Metwaly, and S.M. Amer. “Development of Membrane Electrodes for the Specific Determination of Moexipril Hydrochloride in Dosage Forms and Biological Fluids.” Portugaliae Electrochimica Acta. 27.4 (2009): 463-475.
- Rodgers, Katie, Michael C Vinson, and Marvin W Davis. “Breakthroughs: New drug approvals of 1995 — part 1.” Advanstar Communications, Inc. 140.3 (1996): 84.
- Dart, Richard C. (2004). Medical toxicology. Lippincott Williams & Wilkins. p. 647. ISBN 978-0-7817-2845-4. Retrieved 2009-10-09.
- Kalasz, H, G. Petroianu, K. Tekes, I. Klebovich, K. Ludanyi, et al. “Metabolism of moexipril to moexiprilat: determination of in vitro metabolism using HPLC-ES-MS.” Medicinal Chemistry. 3 (2007): 101-106.
- Jump up^ Chrysant, George S, PK Nguyen. “Moexipril and left ventricular hypertrophy.” Vascular Health Risk Management. 3.1 (2007): 23-30.
- Cawello W, H. Boekens, J. Waitzinger, et al. “Moexipril shows a long duration of action related to an extended pharmacokinetic half-life and prolonged ACE-inhibition.” Int J Clin Pharmacol Ther. 40 (2002): 9-17.
- ^ Jump up to:a b c Chrysant, SG. “Vascular remodeling: the role of angiotensin-converting enzyme inhibitors.” American Heart Journal. 135.2 (1998): 21-30.
- Jump up^ Cameron, RT; Coleman, RG; Day, JP; Yalla, KC; Houslay, MD; Adams, DR; Shoichet, BK; Baillie, GS (May 2013). “Chemical informatics uncovers a new role for moexipril as a novel inhibitor of cAMP phosphodiesterase-4 (PDE4)”. Biochemical Pharmacology 85 (9): 1297–1305. doi:10.1016/j.bcp.2013.02.026. PMC 3625111. PMID 23473803.
- White, WB, and M Stimpel. “Long-term safety and efficacy of moexipril alone and in combination with hydrochlorothiazide in elderly patients with hypertension.” Journal of human hypertension. 9.11 (1995): 879-884.
- Rosendorff, C. “The Renin-Angiotensin System and Vascular Hypertrophy.” Journal of the American College of Cardiology. 28 (1996): 803-812.
- Hartman, J.C. “The role of bradykinin and nitric oxide in the cardioprotective action of ACE inhibitors.” The Annals of Thoracic Surgery. 60.3 (1995): 789-792.
- Jaiswal, N, DI Diz, MC Chappell, MC Khosia, CM Ferrario. “Stimulation of endothelial cell prostaglandin production by angiotensin peptides. Characterization of receptors.” Hypertension. 19.2 (1992): 49-55.
- Phillips, PA. “Interaction between endothelin and angiotensin II.” Clinical and Experimental Pharmacology and Physiology. 26.7. (1999): 517-518.
- Youn, TJ, HS Kim, BH Oh. “Ventricular remodeling and transforming growth factor-beta 1 mRNA expression after nontransmural myocardial infarction in rats: effects of angiotensin converting enzyme inhibition and angiotensin II type 1 receptor blockade.” Basic research in cardiology. 94.4 (1999): 246-253.
////////////
| Systematic (IUPAC) name | |
|---|---|
|
(3S)-2-[(2S)-2-{[(2S)-1-ethoxy-1-oxo-4-phenylbutan-2-yl]amino}propanoyl]-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
|
|
| Clinical data | |
| Trade names | Univasc |
| AHFS/Drugs.com | monograph |
| MedlinePlus | a695018 |
| Pregnancy category |
|
| Legal status |
|
| Routes of administration |
Oral |
| Pharmacokinetic data | |
| Bioavailability | 13-22% |
| Protein binding | 90% |
| Metabolism | Hepatic (active metabolite, moexiprilat) |
| Biological half-life | 1 hour; 2-9 hours (active metabolite) |
| Excretion | 50% (faeces), 13% (urine) |
| Identifiers | |
| CAS Registry Number | 103775-10-6 |
| ATC code | C09AA13 |
| PubChem | CID: 91270 |
| IUPHAR/BPS | 6571 |
| DrugBank | DB00691 |
| ChemSpider | 82418 |
| UNII | WT87C52TJZ |
| KEGG | D08225 |
| ChEMBL | CHEMBL1165 |
| Chemical data | |
| Formula | C27H34N2O7 |
| Molecular mass | 498.568 g/mol |
9-(5-oxotetrahydrofuran-2-yl)nonanoic acid methyl ester
9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester |
 |
| Name | 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester | ||
| Synonyms | |||
| Name in Chemical Abstracts | 2-Furannonanoic acid, tetrahydro-5-oxo-, methyl ester | ||
| CAS No | 22623-86-5 | ||
| Molecular formula | C14H24O4 | ||
| Molecular mass | 256.35 | ||
| SMILES code | O=C1OC(CC1)CCCCCCCCC(=O)OC | ||
1H NMR

| 1H-NMR: 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester | |||
| 500 MHz, CDCl3 | |||
| delta [ppm] | mult. | atoms | assignment |
| 1.24-1.45 | m | 10 H | 4-H, 5-H, 6-H, 7-H, 8-H |
| 1.57 | m | 2 H | 3-H |
| 1.70 | m | 1 H | 9-H |
| 1.82 | m | 1 H | 9-H |
| 2.27 | t | 2 H | 2-H |
| 2.30 | m | 2 H | 3-H (ring) |
| 2.50 | m | 2 H | 4-H (ring) |
| 3.67 | s | 3 H | O-CH3 |
| 4.48 | m | 1 H | 2-H (ring) |

13C NMR

| 13C-NMR: 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester | |||
| 125.7 MHz, CDCl3 | |||
| delta [ppm] | assignment | ||
| 24.9 | C3 | ||
| 25.2 | C9 | ||
| 28.0-29.2 | C4, C5, C6, C7, C8, C3 (ring) | ||
| 34.0 | C2 | ||
| 35.5 | C4 (ring) | ||
| 51.4 | O-CH3 | ||
| 81.0 | C2 (ring) | ||
| 174.2 | C1 (O-C(=O)-) | ||
| 177.2 | C5 (O-C(=O)-, ring) | ||
| 76.5-77.5 | CDCl3 | ||

IR |

| IR: 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester | |||
| [Film, T%, cm-1] | |||
| [cm-1] | assignment | ||
| 2931, 2856 | aliph. C-H valence | ||
| 1776 | C=O valence, lactone | ||
| 1737 | C=O valence, ester | ||
| Cu |
  nonanoic acid methyl ester")  |
|
Synthesis of 9-(5-oxotetrahydrofuran-2-yl)nonanoic acid methyl ester |
| Reaction type: | addition to alkenes, radical reaction, ring closure reaction |
| Substance classes: | alkene, halogencarboxylic acid ester, lactone |
| Techniques: | working with cover gas, stirring with magnetic stir bar, heating under reflux, evaporating with rotary evaporator, filtering, recrystallizing, heating with oil bath |
| Degree of difficulty: | Easy |
Operating scheme
 Instructions
Instructions
http://www.oc-praktikum.de/nop/en/instructions/pdf/4005_en.pdf
Instruction (batch scale 100 mmol)
Equipment 250 mL two-neck flask, protective gas supply, reflux condenser, heatable magnetic stirrer, magnetic stir bar, rotary evaporator, Buechner funnel, suction flask, desiccator, oil bath Substances undecenoic acid methyl ester (bp 248 °C) 19.8 g (22.3 mL, 100 mmol) iodoacetic acid ethyl ester (bp 73-74 °C/ 21 hPa) 27.8 g (15.4 mL, 130 mmol) copper powder (finely powdered, >230 mesh ASTM) 30.5 g (480 mmol) tert-butyl methyl ether (bp 55 °C) 130 mL petroleum ether (bp 60-80 °C) 300 mL Reaction In a 250 mL two-neck flask with magnetic stir bar and a reflux condenser connected with a protective gas piping 19.8 g (22.3 mL, 100 mmol) undecenoic acid methyl ester and 27.8 g (15.4 mL, 130 mmol) iodoacetic acid ethyl ester are mixed with 30.5 g (480 mmol) copper powder under a protective gas atmosphere. Afterwards the reaction mixture is stirred at 130 °C oil bath temperature under protective gas for 4 hours. (Reaction monitoring see Analytics.)
Work up The reaction mixture is cooled down to room temperature, 30 mL tert-butyl methyl ether are added, the mixture is stirred for 5 minutes and filtered off. The copper powder on the filter is washed four times with 25 mL tert-butyl methyl ether each. Filtrates and wash solutions are combined, the solvent is evaporated at the rotary evaporator. A yellow oil remains as crude product. Crude yield: 25.4 g.
The crude product is dissolved in 300 mL petroleum ether under reflux. The solution is allowed to cool down to room temperature, then it is stored in the refrigerator over night for complete crystallization. The crystalline product is sucked off over a Buechner funnel and dried in the vacuum desiccator. The mother liquor is stored again in the refrigerator for a check of complete crystallization. Yield: 19.5 g (76.1 mmol, 76%); white solid, mp 34 °C Comments In order to achieve a quantitative reaction within 4 hours, a fivefold excess of copper is used.
Waste management Recycling The copper powder can be used three times.
Waste disposal Waste Disposal evaporated tert-butyl methyl ether (might contain iodoethane) organic solvents, containing halogen mother liquor from recrystallization organic solvents, containing halogen copper powder solid waste, free from mercury, containing heavy metals
Time 6-7 hours
Break After heating and before recrystallizing
Degree of difficulty Easy
Analytics Reaction monitoring with TLC Sample preparation: Using a Pasteur pipette, two drops of the reaction mixture are taken and diluted with 0.5 mL diethyl ether. TLC-conditions: adsorbant: TLC-aluminium foil (silica gel 60) eluent: petroleum ether (60/80) : acetic acid ethyl ester = 7 : 3 visualisation: The TLC-aluminium foil is dipped in 2 N H2SO4 and then dried with a hot air dryer. Reaction monitoring with GC Sample preparation: Using a Pasteur pipette, one drop of the reaction mixture is taken and diluted with 10 mL dichloromethane. From this solution, 0.2 µL are injected. 10 mg from the solid product are dissolved in 10 mL dichloromethane. From this solution, 0.2 µL are injected. GC-conditions: column: DB-1, 28 m, internal diameter 0.32 mm, film 0.25 µm inlet: on-column-injection carrier gas: hydrogen (40 cm/s) oven: 90 °C (5 min), 10 °C/min to 240 °C (40 min) detector: FID, 270 °C Percent concentration was calculated from peak areas.
Chromatogram |

| GC: crude product | |
| column | DB-1, L=28 m, d=0.32 mm, film=0.25 µm |
| inlet | on column injection, 0.2 µL |
| carrier gas | H2, 40 cm/s |
| oven | 90°C (5 min), 10°C/min –> 240°C (40 min) |
| detector | FID, 270°C |
| integration | percent concentration calculated from relative peak area |

| GC: pure product | |
| column | DB-1, L=28 m, d=0.32 mm, film=0.25 µm |
| inlet | on column injection, 0.2 µL |
| carrier gas | H2, 40 cm/s |
| oven | 90°C (5 min), 10°C/min –> 240°C (40 min) |
| detector | FID, 270°C |
| integration | percent concentration calculated from relative peak area |
Substances required |
| Batch scale: | 0.01 mol | 0.1 mol | 10-Undecenoic acid methyl ester |
| Educts | Amount | Risk | Safety | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 10-Undecenoic acid methyl ester |
|
19.8 g | H- EUH- | P- | |||||
| Iodoacetic acid ethyl ester |
|
27.8 g | H300 H314 EUH- | P264 P280 P305 + 351 + 338 P310 | |||||
| Reagents | Amount | Risk | Safety | ||||||
| Copper powder |
|
30.5 g | H400 EUH- | P273 | |||||
| Solvents | Amount | Risk | Safety | ||||||
| tert-Butyl methyl ether |
|
130 mL | H225 H315 | P210 | |||||
| Petroleum ether (60-80) |
|
300 mL | H225 H304 H315 H336 H411 EUH- | P210 P261 P273 P301 + 310 P331 | |||||
| Others | Amount | Risk | Safety | ||||||
| Sulfuric acid 2N |
|
H314 H290 EUH- | P280 P301 + 330 + 331 P305 + 351 + 338 P309 + 310 | ||||||
| Solvents for analysis | Amount | Risk | Safety | ||||||
| Petroleum ether (60-80) |
|
H225 H304 H315 H336 H411 EUH- | P210 P261 P273 P301 + 310 P331 | ||||||
| Acetic acid ethyl ester |
|
H225 H319 H336 EUH066 | P210 P261 P305 + 351 + 338 | ||||||
| Dichloromethane |
|
H351 H315 H319 H335 H336 H373 | P261 P281 P305 + 351 + 338 |
Substances produced |
| Batch scale: | 0.01 mol | 0.1 mol | 10-Undecenoic acid methyl ester |
| Products | Amount | Risk | Safety | |
|---|---|---|---|---|
| 9-(5-Oxotetrahydrofuran-2-yl)nonanoic acid methyl ester |
Equipment |
| Batch scale: | 0.01 mol | 0.1 mol | 10-Undecenoic acid methyl ester |
 |
two-necked flask 250 mL |  |
protective gas piping | |
 |
reflux condenser |  |
heatable magnetic stirrer with magnetic stir bar | |
 |
rotary evaporator |  |
suction filter | |
 |
suction flask |  |
exsiccator with drying agent | |
 |
oil bath |
Simple evaluation indices |
| Batch scale: | 0.01 mol | 0.1 mol | 10-Undecenoic acid methyl ester |
| Atom economy | 53.9 | % | |
| Yield | 76 | % | |
| Target product mass | 19.5 | g | |
| Sum of input masses | 370 | g | |
| Mass efficiency | 53 | mg/g | |
| Mass index | 19 | g input / g product | |
| E factor | 18 | g waste / g product |
………………
………
Aseptic Manufacturing Operation: Chinese Company Zhuhai United Laboratories does not comply with EU GMP
DRUG REGULATORY AFFAIRS INTERNATIONAL


While the focus of attention has been on Indian manufacturers during the last 2 years now also Chinese manufacturers are in the spot light. On 15 June 2015 the National Agency for Medicines and Medical Devices of Romania entered a GMP Non-Compliance Report for Zhuhai United Laboratories into EudraGMDP. Read more about the GMP deviations observed at Zhuhai United.

While the focus of attention has been on Indian manufacturers during the last 2 years now also Chinese manufacturers are again in the spot light. Just recently the EU found serious GMP deviations at an API manufacturer (Huzhou Sunflower Pharmaceuticals) and on 15 June 2015 the National Agency for Medicines and Medical Devices of Romania entered a GMP Non-Compliance Report for Zhuhai United Laboratories Co., LTD located at Sanzao Science &Technology Park, National Hi-Tech Zone, Zhuhai, Guangdong, 519040, China into EudraGMDP.
According to the report issued by the…
View original post 253 more words
