
Home » ANTI ACNE
Category Archives: ANTI ACNE
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
ADAPALENE

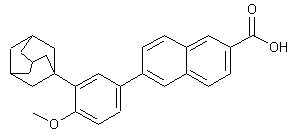
ADAPALENE
- Molecular FormulaC28H28O3
- Average mass412.520 Da
- CD 271
- CD-271
CD-271, Differin, Differine106685-40-9[RN]
2-Naphthalenecarboxylic acid, 6-(4-methoxy-3-tricyclo[3.3.1.13,7]dec-1-ylphenyl)-
6-[3-(Adamantan-1-yl)-4-methoxyphenyl]-2-naphthoic acid
6-[4-methoxy-3-(tricyclo[3.3.1.13,7]dec-1-yl)phenyl]naphthalene-2-carboxylic acid AdapaleneCAS Registry Number: 106685-40-9
CAS Name: 6-(4-Methoxy-3-tricyclo[3.3.1.13,7]dec-1-ylphenyl)-2-naphthalenecarboxylic acid
Additional Names: 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid
Manufacturers’ Codes: CD-271
Trademarks: Differin (Galderma)
Molecular Formula: C28H28O3
Molecular Weight: 412.52
Percent Composition: C 81.52%, H 6.84%, O 11.64%
Literature References: Retinoid selective for retinoic acid receptor (RAR) subtypes b and g. Prepn: B. Shroot et al.,EP199636; eidem,US4717720 (1986, 1988 both to Cent. Int. Recher. Dermatol.); and structure-activity study: B. Charpentier et al.,J. Med. Chem.38, 4993 (1995). Pilot-scale synthesis: Z. Liu, J. Xiang, Org. Process Res. Dev.10, 285 (2006). HPLC determn in plasma and tissue: R. Ruhl, H. Nau, Chromatographia45, 269 (1997). Clinical pharmacology: C. E. M. Griffiths et al.,J. Invest. Dermatol.101, 325 (1993). Clinical trial in acne: A. Shalita et al.,J. Am. Acad. Dermatol.34, 482 (1996). Reviews of pharmacology and clinical potential: B. A. Bernard, Skin Pharmacol.6, Suppl. 1, 61-69 (1993); R. N. Brogden, K. L. Goa, Drugs53, 511-519 (1997); of clinical use in acne vulgaris: J. Waugh et al.,Drugs64, 1465-1478 (2004).
Properties: White crystals from THF and ethyl acetate, mp 319-322°. pK 4.2. Stable to light.
Melting point: mp 319-322°
pKa: pK 4.2
Therap-Cat: Antiacne.
Keywords: Antiacne.
Adapalene is a third-generation topical retinoid primarily used in the treatment of mild-moderate acne, and is also used off-label to treat keratosis pilaris as well as other skin conditions.[1] Studies have found adapalene is as effective as other retinoids, while causing less irritation.[2] It also has several advantages over other retinoids. The adapalene molecule is more stable compared to tretinoin and tazarotene, which leads to less concern for photodegradation.[2] It is also chemically more stable compared to the other two retinoids, allowing it to be used in combination with benzoyl peroxide.[2] Due to its effects on keratinocyte proliferation and differentiation, adapalene is superior to tretinoin for the treatment of comedonal acne and is often used as a first-line agent. [3]
Adapalene is a third-generation topical retinoid with anti-comedogenic, comedolytic, and anti-inflammatory properties used to treat acne vulgaris in adolescents and adults.

SYN
AU 9047961; EP 0199636; US 4717720; US 5098895; US 5183889
J Med Chem 1995,38(26),4993
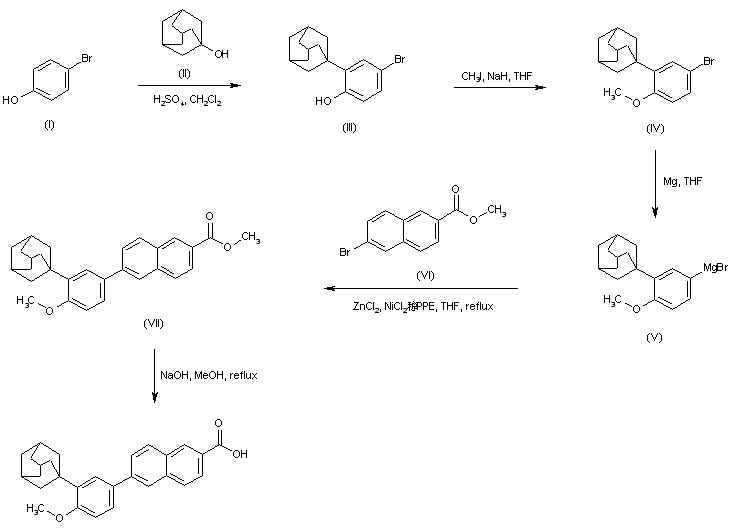
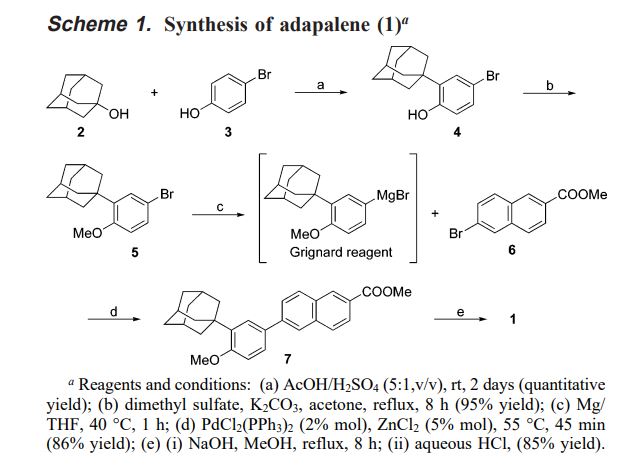
Friedel-Crafts condensation of 4-bromophenol (I) with 1-adamantanol (II) in the presence of H2SO4 yielded the adamantyl phenol (III). Subsequent alkylation of the sodium phenoxide of (III) with iodomethane produced the methyl ether (IV). The Grignard reagent (V), prepared from aryl bromide (IV), was converted to the organozincate derivative, and then subjected to a nickel-catalyzed cross-coupling with methyl 6-bromo-2-naphthoate (VI) to furnish adduct (VII). The target carboxylic acid was finally obtained by saponification of the methyl ester (VII).
SYN
CA 2021550; EP 0409740; FR 2649976; JP 1991063246; US 5073361; US 5149631
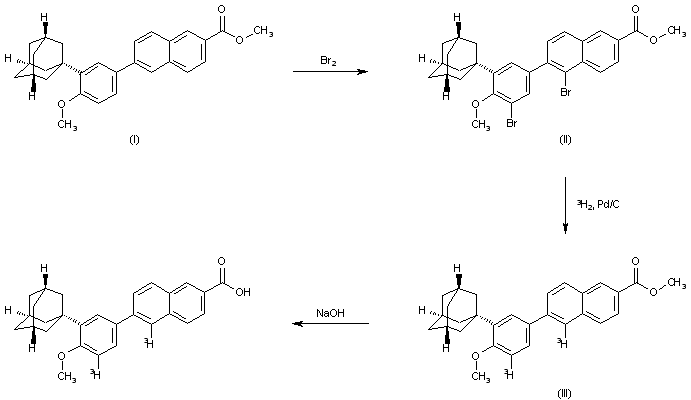
The bromination of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid methyl ester (I) with Br2 in dichloromethane gives the dibromo derivative (II), which is hydrogenated with tritium gas over Pd/C in THF containing TEA to yield the bis tritiated ester (III). Finally, ester (III) is hydrolyzed with NaOH in refluxing methanol to afford the target tritiated naphthoic acid.
SYN
doi:10.1071/CH9732303c US4717720
SYN
Adapalene (CAS NO.: 106685-40-9), with its systematic name of 2-Naphthalenecarboxylic acid, 6-(4-methoxy-3-tricyclo(3.3.1.1(sup 3,7))dec-1-ylphenyl)-, could be produced through many synthetic methods.
Following is one of the synthesis routes:
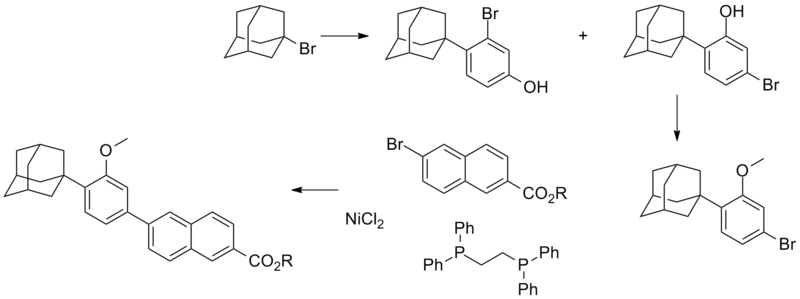
Firstly, Friedel-Crafts condensation of 4-bromophenol (I) with 1-adamantanol (II) in the presence of H2SO4 yields the adamantyl phenol (III). Next, subsequent alkylation of the sodium phenoxide of (III) with iodomethane produces the methyl ether (IV). The Grignard reagent (V), prepared from aryl bromide (IV), is converted to the organozincate derivative, and then subjects to a nickel-catalyzed cross-coupling with methyl 6-bromo-2-naphthoate (VI) to furnish adduct (VII). Finally, the target carboxylic acid is obtained by saponification of the methyl ester (VII).

Synthesis Reference
Graziano Castaldi, Pietro Allegrini, Gabriele Razzetti, Mauro Ercoli, “Process for the preparation of adapalene.” U.S. Patent US20060229465, issued October 12, 2006.
PATENT
https://patents.google.com/patent/US8119834B2/enThe chemical name for adapalene is 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid, which is represented by Compound I (below):

Adapalene has been approved by the FDA as a cream, a gel, a solution and pledgets for the topical treatment of acne vulgaris and is marketed under the tradename of DIFFERIN®.U.S. Pat. No. 4,717,720 (“the ‘720 patent”) discloses benzonaphthalene derivatives, including adapalene. The ‘720 patent describes a process for preparing adapalene (i.e., according to example 9c followed by example 10) that involves two reaction steps.The first step for preparing adapalene according to the ‘720 patent involves the preparation of the methyl ester of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid. According to example 9c of the ‘720 patent, 2-(1-adamantyl)-4-bromoanisole (also known as 1-(5-bromo-2-methoxyphenyl)adamantane) is converted to its organomagnesium derivative and then into its organozinc derivative. The organozinc derivative is next coupled to methyl 6-bromo-2-naphthoate by adding a catalytic amount of NiCl2/DPPE complex (also known as [bis(diphenylphosphino) ethane]dichloronickel(II)). Upon completion of the reaction, the mixture is poured into water, extracted with dichloromethane, and then dried. The product is next isolated by column chromatography by eluting with a mixture of heptane (70%) and dichloromethane (30%). The resulting product is then recrystallized in ethyl acetate (yield: 78%).The second step for preparing adapalene according to the ‘720 patent involves hydrolyzing the product of step 1 (above). According to example 10 of the ‘720 patent, the ester obtained in Example 9c can be treated with a solution of soda in methanol followed by heating at reflux for 48 hours. The solvents are then evaporated and the resulting residue is taken up in water and acidified with concentrated HCl to neutralize the resulting adapalene sodium salt. The resulting solid is next filtered and dried under vacuum over phosphoric anhydride and then recrystallized in a mixture of tetrahydrofuran and ethyl acetate to yield adapalene (yield: 81%).The process of preparing adapalene according to the ‘720 patent is both difficult and uneconomical to conduct on an industrial scale. Regarding step 1, the use of dichloromethane is both toxic and hazardous for the environment. Additionally, purification of the intermediate product by column chromatography, followed by recrystallization, in order to obtain a crystalline product of acceptable purity is both expensive and laborious. Moreover, the step 1 process produces as a biaryllic C—C bond, and the catalytic coupling is noticeably exothermic. Regarding step 2, the synthesis of adapalene and/or its sodium salt requires a long reaction time (i.e., 48 hours) at methanol reflux and further requires a high ratio of solvent (volume) to product (mass).Additionally, according to the prior art, the manufacture of adapalene is not satisfactory for industrial implementation because the presence of high amounts of undesired by-products makes it necessary to use uneconomical purification procedures to isolate the product according to quality specifications. One significant undesired by-product produced during the Grignard reaction of step 1 in the synthesis of adapalene is 3,3′-diadamantyl-4,4′-dimethoxybiphenyl, which has not been previously described in the literature and which is represented by Compound VI (below):

The level of the by-product in a sample of adapalene, adapalene methyl ester and/or an adapalene salt can be determined using standard analytical techniques known to those of ordinary skill in the art. For example, the level can be determined by HPLC. A specific method for determining the level of this impurity is provided herein.Since the solubility of the dimeric by-product is very low in most solvents, the design of an economical industrial process that yields pure adapalene without the use of expensive chromatographic methods requires the selection of the proper solvents and conditions to inhibit formation of the by-product during the manufacturing process.Additionally, adapalene has been described as being white (see, e.g., Merck Index, 13th ed., p. 29). It has been observed that adapalene has a tendency to yellow under certain synthetic conditions or due to the quality of the starting materials used in its preparation. In this regard, color must be attributed to the presence of some specific impurities that may or may not be detectable by conventional methods such as HPLC.

ExampleStep 2: Preparation of 6-[3-(1-adamantyl)-4-methoxy phenyl]-2-naphthoic acid-potassium Salt (i.e., Adapalene Potassium Salt)In a 2 L, five necked cylindrical reaction vessel equipped with reflux condenser, distillation kit, heat-transfer jacket, anchor impeller and purged with nitrogen, were added 48.38 g (dry equivalent amount) of methyl 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoate (1.134×10−1 mol), wet with methanol, 2.73 g of tetrabutylammonium bromide (8.47×10−3 mol), 18.39 g of potassium hydroxide (85% alkali content, freshly titrated. 2.79×10−1 mol) and 581 mL of toluene. The mixture was heated to reflux temperature, and the methanol/water was removed by distillation. The distilled mixture was replaced by pure toluene and the mixture was stirred at reflux for approximately three hours (including the time required for the distillation). The solution was then cooled to approximately 20-25° C., filtered and the resulting solid was washed with toluene.The solid was next suspended in 187 mL of tetrahydrofuran and stirred for approximately 30 minutes. Then, 375 mL of toluene was added, and the mixture was heated to reflux and maintained at that temperature for approximately 1 hour. The solution was then cooled to approximately 20-25° C., filtered, and the resulting solid washed with toluene. The toluene-wet product was then suspended in 256 mL of methanol, heated to reflux for approximately 30 minutes and cooled to 50-60° C. After cooling, 409 mL of water was added dropwise. The mixture was then again heated to reflux for approximately 15 additional minutes, cooled to room temperature and filtered. The resulting solid was washed with water to yield 50.69 g (wet) of adapalene potassium salt (1.12×10−1 mol, dry equivalent amount calculated from loss on drying; yield: 99.18%). Analytical data: HPLC Purity (HPLC at 272 nm): 99.86%; Impurity (i.e., 3,3′-diadamantyl-4,4′-dimethoxybiphenyl) area percent (HPLC at 272 nm): not detected; 1H-NMR (300 MHz, CD3OD): δ 1.83 (broad s, 6H), 2.08 (broad s, 3H), 2.21 (broad s, 6H), 3.88 (s, 3H), 7.04 (d, 1H, J=8.4 Hz), 7.56 (overlapped, 1H, J=2.4, 9.6 Hz), 7.57 (overlapped s, 1H), 7.74 (dd, 1H, J=8.7, 1.8 Hz), 7.87 (d, 1H, J=9.0 Hz), 7.97 (d, 1H, J=8.7 Hz), 8.00 (broad d, 1H, J=0.9 Hz), 8.06 (dd, 1H, 8.4, J=1.8 Hz), 8.47 (broad d, 1H, J=0.9 Hz); 13C-NMR (75.4 MHz, CD3OD): δ 30.6, 38.3, 41.8, 55.5, 113.3, 125.3, 126.4, 126.6, 127.8, 128.3, 130.0, 130.4, 133.0, 134.2, 136.1, 136.3, 139.7, 141.1, 159.9, 175.4.
ExampleStep 3: Preparation of 6-[3-(1-adamantyl)-4-methoxy phenyl]-2-naphthoic Acid (i.e., Adapalene)In 500 mL of methanol was added 49.59 g (1.10×10−1 mol, dry equivalent amount) of the wet solid obtained in Example/Step 2, and the mixture was heated to reflux for 30 minutes and cooled to approximately 40° C. Next, 33.17 g of concentrated HCl was slowly added over approximately 1 hour with gentle stirring in order to ensure homogeneity, followed by the slow addition of 248 mL of water. The resulting mixture was stirred for approximately 30 additional minutes at approximately 40° C. and then cooled to room temperature, filtered and washed with methanol. The wet solid was then suspended with 1020 mL of tetrahydrofuran and heated to reflux for approximately 10 minutes or until complete dissolution. The solution was then cooled to approximately 35° C., the solid particles were removed by filtration, and the filter was washed with tetrahydrofuran.The collected mother liquors were heated to reflux, and 654 g of tetrahydrofuran was removed by distillation. The mixture was then cooled to approximately 55-60° C. Thereafter, 650 mL of methanol was added over approximately 10 minutes, and the mixture heated to reflux for approximately 30 minutes, cooled, and filtered. The resulting solid was filtered with methanol and dried at 80° C. in a vacuum oven to yield 40.54 g of adapalene (9.83×10−2 mol; yield: 89.29% (from adapalene potassium salt); 88.56% (from adapalene methyl ester); and 78.67% (from methyl 6-bromo-2-naphthoate)). Analytical data: HPLC Purity (HPLC at 272 nm): 100.00%; Assay: 99.99%; Residue on Ignition: 0.02%; IR: matches reference.Table 1 (below) lists the peak assignments of the X-ray powder diffractogram of the adapalene obtained and are illustrated in FIG. 1.
| TABLE 1 | |||
| peak | peak_position | peak_intensity | background |
| 1 | 9.94547 | 175.32198 | 42.94638 |
| 2 | 13.18338 | 239.32156 | 48.88440 |
| 3 | 14.87487 | 234.32591 | 47.91444 |
| 4 | 15.28319 | 573.40082 | 53.73505 |
| 5 | 16.37472 | 1207.21631 | 69.64595 |
| 6 | 16.54000 | 882.00000 | 68.42529 |
| 7 | 17.39657 | 110.88804 | 58.39248 |
| 8 | 17.93203 | 114.02068 | 55.36037 |
| 9 | 19.44575 | 285.34473 | 113.52401 |
| 10 | 19.94692 | 569.60516 | 153.63921 |
| 11 | 22.43198 | 2846.14307 | 110.81189 |
| 12 | 24.02238 | 140.20882 | 85.37505 |
| 13 | 25.04586 | 925.64282 | 121.97974 |
| 14 | 25.41035 | 240.42351 | 102.81077 |
| 15 | 26.68556 | 362.45480 | 68.05973 |
| 16 | 27.71646 | 141.77916 | 72.53469 |
| 17 | 40.51307 | 133.00453 | 43.44914 |
| 18 | 46.52728 | 130.31587 | 50.16773 |
ExampleStep 4: Preparation of 3,3′-diadamantyl-4,4′-dimethoxybiphenylTo a 100 mL rounded bottom reaction vessel equipped with a magnetic stirrer, thermometer, reflux condenser, pressure compensated addition funnel, were added 0.15 g of 1-(5-bromo-2-methoxyphenyl)adamantane, 0.47 g of magnesium turnings and 7 mL of tetrahydrofuran. The mixture was heated to approximately 35° C., and 0.13 mL of 1,2-dibromoethane were added to the mixture. Reaction exothermy self-heated the mixture. Next, a solution of 4.85 g of 1-(5-bromo-2-methoxyphenyl)adamantane and 28 mL of tetrahydrofuran was added to the mixture dropwise. During this addition, the temperature of the mixture dropped from reflux temperature to approximately 45° C. The reaction was then stirred for approximately 45 additional minutes at approximately 45° C. and was permitted to cool to approximately 22° C. Next, 2.3 g of ZnCl2 was added to the mixture, resulting in an exothermic reaction that raised the temperature of the mixture to approximately 38° C. The mixture was then permitted to cool to approximately 22° C. and was stirred for approximately 1 hour at this temperature.Next, 0.03 g of Pd(OAc)2 and 3.5 g of 1-(5-bromo-2-methoxyphenyl) adamantane were added to the mixture, followed by 25 mL of tetrahydrofuran in order to improve agitation, and the mixture was heated at reflux for approximately 24 hours. The resulting mixture was then evaporated to dryness and poured into 103 mL of 0.015 N HCl. Next, 150 mL of dichloromethane and 100 mL of water were added to yield a mixture consisting of a solid, an aqueous layer and an organic layer. The mixture was then filtered to separate the solid, the aqueous layer was discarded, and the organic layer was washed with 200 mL of water and decanted again. This process was repeated twice on the filtered solid. The three collected organic layers were evaporated to dryness, washed in methanol, and dried to yield 2.1 g of 3,3′-diadamantyl-4,4′-dimethoxybiphenyl (yield: 39.9%). Analytical data: Melting point: 288.1-289.1° C.; Elemental analysis: C 83.63%, H 8.73%; 1H-NMR (300 MHz, CDCl3): δ 1.78 (broad s, 12H), 2.08 (broad s, 6H), 2.15 (broad s, 12H), 3.86 (s, 6H), 6.92 (dm, 2H, J=8.1 Hz), 7.34 (dd, 2H, J=2.4, 8.1 Hz), 7.39 (d, 2H, J=2.4 Hz); 13C-NMR (75.4 MHz, CDCl3): δ 29.2, 37.1, 37.2, 40.6, 55.1, 111.9, 125.0, 125.5, 134.0, 138.5, 157.8; MS (EI, 70 eV): m/z=484 (6), 483 (36), 412 (1,100), 410 (5), 347 (8), 135 (22), 107 (7), 93 (14), 79 (17), 67 (9), 55 (6); IR (Selected absorption bands): 2992, 2964, 2898, 2850, 1603 cm−1.
PATENT
https://patents.google.com/patent/WO2008126104A2/en
The compound 6-[3-(l – Adamantyl) – 4 – methoxy phenyl] – 2 – naphthoic acid of Formula – I known as Adapalene is used in dermatology, particularly in the treatment of acne vulgaris and psoriasis.

Formula – 1Adapalene was first time disclosed in the US patent No. 4,717,720 (herein after referred as ‘720) describe the preparation of compound of Formula – I using Negishi cross Coupling. In this reaction, 2-(l-adamantyl)-4-bromoanisole is converted to its organomagnesium compound followed by conversion to organozinc compound using zinc chloride and reacted with 6-bromo-2-methylnaphthoate employing transition metal as reaction catalyst such as palladium or nickel or one of its complexes with various phosphines. The reaction sequence is as shown in scheme – 1 below:

Scheme – 1 Another US patent No. 5,015,758 describe the process for preparation of 6[3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoate a penultimate step for preparation of Adapalene using Friedel – Crafts alkylation by reacting 1 – acetoxy adamantane with methyl – 6 – (4 – hydroxyphenyl) – 2 – naphthoate in presence of cone. Sulfuric acid in solvent n – heptane.Another improved process was published in the journal, Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. The process involves the preparation of intermediates followed by Negishi cross Coupling, where in 2-(l-adamantyl)-4-bromophenol was prepared using 1 – adamentol and 4- bromo phenol in presence of 98% sulphuric acid and acetic acid, which on methylation with dimethyl sulfate and potassium carbonate in dry acetone yields 2-(l -adamantyl)-4-bromoanisole. The compound is reacted with magnesium to form Grignard reagent and then coupled with 6-bromo-2-methylnaphthoate in presence of novel Pd – Zn double metal catalyst to yield ester, which on saponification followed by treatment with acid yields Adapalene.The recent published application WO 2006/108717 describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – 1. The application describes the preparation of 3-adamantyl-4-methoxyphenyl boronic acid from 2-(l-adamantyl)-4- bromoanisole using n-Butyl Lithium and triisopropyl borate in solvent tetrahydrofuran. Finally 3-adamantyl-4-methoxyphenyl boronic acid is reacted with 6-bromo-2-naphthoic acid involving Suzuki coupling in presence of Palladium acetate catalyst, a ligand 2 – (dicyclohexyl – phosphino) biphenyl, an inorganic base in solvent to get the compound adapalene.Some of the drawbacks of the prior art processes include:- The reported process in US patent 4717720, using Negishi cross coupling involves Grignard reaction. This requires anhydrous condition and a possibility of runaway reaction during Grignard reagent formation. Also the reaction involves the addition of fused ZnC12 and the preparation of the catalyst NiC12 (DPPE) complex, which needs to be freshly prepared increases the reaction step and has to be thoroughly dried before its use for coupling. Further the coupling reaction, results in the formation of dimer impurities during the organozinc compound reaction, with 2-(I -adamantyl)-4-bromoanisole and 6-bromo-2-methylnaphthoate respectively, which are difficult to remove. All these operations make the entire synthesis extremely sensitive and difficult to handle.Some of the above drawbacks were addressed by the authors in the article published in Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. But the use of Pd catalyst with the ligand like PdCl2 (PPh3)2 for the direct conversion of Grignard reagent employing ZnCYl in catalytic amount has its own limitations. The use of Grignard reagent, palladium catalyst with ligand and hygroscopic ZnCl2 demerits this process for industrial application.The recent published application WO 2006/108717; describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – I. The use of organo boronic acids for the Suzuki reaction has some limitations because of the indeterminate stoichiometry associated with the use of boronic acid, and its difficulty in purification and the byproducts formed during the reaction.Therefore there remains a need for an improved process for preparing adapalene that eliminates or substantially reduces the impurities, decreases the number of steps, and employs a more robust process which is convenient and cost efficient.

Examples:Example 1: Preparation of 3 – Adamantyl – 4 – methoxy phenyl potassium trifluoroborate:In a 2.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 100.0 gm of 2-(l- adamantyl) 4-bromo anisole was charged in 600 ml tetrahydrofuran. The reaction mixture was cooled to -55 ± 50C and 302 ml of 1.6 M n – butyl Lithium was slowly added and stirred. 87 ml of tri isopropyl borate was then charged and stirring was continued for 30 minutes at -55 ± 5°C. Cooling was removed and the temperature raised slowly to 25 – 300C. 1.0 L of 1.2N hydrochloric acid was then charged and reaction mass was stirred for 30 minutes and separated the organic layer. The organic layer was charged in 1.0 L round bottom flask and freshly prepared aqueous solution of potassium hydrogen difluoride (230 gm, in 700 ml water) was added at 25 – 300C and stirring was maintained till white precipitate is obtained. The mixture was continued under stirring and cooled to 0 – 50C. The product, 3 – adamantyl – 4 – methoxyphenyl potassium trifluoroborate obtained was filtered, washed with 100 ml of ethyl acetate. The product was dried at 60 – 65°C till constant weight. Yield: 90.5 gm (83%), Purity: 99.0 % by HPLC.Example 2: Preparation of 6 – [3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid:In a 1.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 50.0 gm of 3 – Adamantyl – 4 – methoxyphenyl potassium trifluoroborate, 23 gm of 6- bromo -2-methyl napthoate in 300 ml tetrahydrofuran (THF) was charged. Stirred for 15 min and charged 3.0 gm of 5% Pd / C was and aqueous potassium hydroxide solution (50.0 gm in 300 ml water). Stirring was continued and the temperature was raised to reflux. The reaction mass was maintained for 10 hours at reflux and after the completion of the reaction, 200 ml of tetrahydrofuran: water (1 : 1) mixture was added and then filtered through hyflow bed at 45-500C. The hyflow bed was washed with tetrahydrofuran: water (1 : 1) mixture at 45-500C. 500 ml water was charged and the reaction mass was stirred. The aqueous layer was acidified with 1.2N hydrochloric acid. The precipitated mass was filtered, washed with water till neutral pH. The solid product obtained was dried at 70 – 75°C till constant weight to get 6 – [3-(l- adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid.The dried product was taken in 300 ml of tetrahydrofuran and stirred. The temperature was raised to reflux and was maintained for 30 minutes. The heating was stopped and cooled the reaction mass to 25 – 300C. 500 ml of n – heptane was charged to the reaction mass and stirred for 30 minutes. The reaction mass was then chilled to 0 – 5°C and maintained stirring at 0 – 5°C temperature for 2.0 hours. The precipitated solid was filtered and washed with n – heptane. The pure crystalline 6 – [3-(l- adamantyl) – 4 methoxyphenyl] – 2 – naphthoic acid thus obtained was then dried till constant weight. Yield = 40 – 42 gms (68 – 72 %)
PATENT
https://pubs.acs.org/doi/10.1021/op050223f
Strategies that were adopted during the process development of adapalene to achieve a cost-effective commercial-scale synthesis are described herein. These included (1) the use of AcOH/H2SO4 to afford 2-(1-adamantyl)-4-bromophenol in quantitative yield; (2) the dimethyl sulfate methylation to enhance the yield of methylation to 95%; (3) direct conversion of the Grignard reagent into methyl 6-(3-(1-adamantyl)-4-methoxyphenyl)-2-naphthoate by the catalysis of both PdCl2(PPh3)2 and ZnCl2 in high yield; (4) the use of EDTA-disodium salt dihydrate to ensure the heavy metal’s content within acceptable limits; (5) the use of toluene to simplify the original chromatographic purification to recrystallization. The pilot-scale synthesis of adapalene is described in detail in the Experimental Section.

6-(3-(1-Adamantyl)-4-methoxyphenyl)-2-naphthoic Acid (Adapalene, 1). Compound 7 (213 g, 0.5 mol) was treated with 2 N NaOH solution (8 L) in methanol under reflux for 8 h. After evaporation of methanol (7 L) and addition of water (1.5 L), the mixture was acidified until pH 1 with 6 N HCl and filtrated through Celite. The residue was washed with water (3 × 5 L), and recrystallized twice in THF (194 g/2 L/time) to give pure (99% HPLC) 1 (177 g, 85%), mp 320-322 °C.1 H NMR (400 MHz, DMSO-d6) δ 1.77 (6 H,s, H on 1-adamantyl), 2.07 (3 H, s, H on 1-adamantyl), 2.14 (6 H, s, H on 1-adamantyl), 3.87 (3 H, s, H on ArOCH3), 7.12 (1 H, d, J ) 8.4 Hz, 5-phenyl H), 7.58 (1 H, d, J ) 2.0 Hz, 2-phenyl H), 7.65 (1 H, dd, J ) 8.4 Hz, J ) 2.0 Hz, 6-phenyl H), 7.89 (1 H, d, J ) 8.8 Hz, 7-naphthyl H), 7.98 (1 H, d, J ) 8.8 Hz, 4-naphthyl H), 8.08 (1 H, d, J ) 8.8 Hz, 8-naphthyl H), 8.15 (1 H, d, J ) 8.8 Hz, 3-naphthyl H), 8.22 (1 H, s, 5-naphthyl H), 8.60 (1 H, s, 1-naphthyl H), 13.05 (1 H, s, -COOH); 13C NMR (100 MHz, DMSO-d6) δ 28.32, 36.47, 40.09, 55.28, 112.68, 123.99, 124.99, 125.38, 125.68, 125.85, 127.55, 128.25, 129.72, 130.13, 130.83, 131.46, 135.38, 138.00, 140.13, 158.53, 167.34.
PATENT
https://patents.google.com/patent/US7345189B2/en
Adapalene, namely 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid, having the following chemical formula:

is disclosed in U.S. Pat. No. 4,717,720 and used in dermatology, in particular for the treatment of acne vulgaris and psoriasis.According to U.S. Pat. No. 4,717,720 the synthesis is carried out by a coupling reaction between a magnesium, lithium or zinc derivative of a compound of formula (A) and a compound of formula (B), wherein X and Y are Cl, Br, F or I; R is hydrogen or alkyl; and Ad is 1-adamantyl

in an anhydrous solvent, in the presence of a metal transition or a complex thereof as a catalyst.A number of alternative synthetic approaches have been suggested in order to reduce the preparation costs. Surprisingly, particularly advantageous proved the alternative synthesis of the invention, which makes use of easily-available, low-cost 6-hydroxy-2-naphthoic acid alkyl esters as intermediates, and provides good yields.EXAMPLE 1Synthesis of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid methyl ester [adapalene methyl ester]A round-bottom flask is loaded with nickel (II) chloride (0.158 g; 1.2 mmol) and THF (20 ml), and tris(hydroxypropyl)phosphine (1.53 g; 7.3 mmol) is added to the mixture, which is refluxed for an hour, then cooled to a temperature of 50° C. and added in succession with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), potassium phosphate (10.38 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (7-g; 24.4 mmol), water (0.88 g; 48.8 mmol) and THF (50 ml). The mixture is heated under reflux for 24 hours, then cooled to a temperature ranging from 50 to 55° C. and added with water, adjusting pH to a value below 7 with acetic acid. After cooling to a temperature of 15° C., the resulting product is filtered, thereby obtaining crystalline adapalene methyl ester (8.5 g; 20.08 mmol) in 82% yield.1H NMR: (300 MHz, DMSO), δ 8.6 (s, 1H), δ 8.3-7.8 (m, 6H), δ 7.7-7.5 (m, 2H), δ 7.1 (d, 1H), δ 3.9 (s, 3H), δ 3.85 (s, 3H), δ 2 (m, 9H), δ 1.7 (m, 6H).EXAMPLE 2Synthesis of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid sodium salt [adapalene sodium salt]A round-bottom flask is loaded with adapalene methyl ester (7 g; 16.41 mmol), THF (42 ml), water (7 ml) and a 50% w/w sodium hydroxide aqueous solution (1.44 g; 18.05 mmol). The mixture is refluxed for 6 hours, then added with water (133 ml) and THF is distilled off to a residual content of approx. 5% w/w, heated to a temperature of about 80° C. until complete dissolution of the solid, then cooled to 15° C. The crystallized product is filtered and dried under vacuum in a static dryer at a temperature of 50° C., thereby obtaining adapalene sodium salt (6.7 g; 15.42 mmol) in 94% yield.EXAMPLE 3Synthesis of 6-[3-(1-adamantyl)-4-methoxyphenyl]-2-naphthoic acid [adapalene]A round-bottom flask is loaded with adapalene sodium salt (6.7 g; 15.42 mmol), THF (40 ml) and water (7 ml) and the mixture is refluxed until complete dissolution of the solid. The resulting solution is dropped into a 3% w/w acetic acid aqueous solution, keeping the temperature above 60-70° C., to precipitate adapalene acid (6.3 g; 15.27 mmol), which is filtered and dried under vacuum at a temperature of 50-60° C. The yield is 95%.EXAMPLE 4Synthesis of adapalene methyl esterA round-bottom flask is loaded with nickel (II) chloride (0.158 g; 1.2 mmol) and THF (20 ml), and tris(hydroxypropyl)phosphine (1.53 g; 7.3 mmol) is added. The mixture is refluxed for an hour, then cooled to a temperature of 50° C. and added in succession with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), potassium phosphate (10.38 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (9.1 g; 31.8 mmol), water (10.53 g; 585.3 mmol) and THF (50 ml). The mixture is refluxed for 24 hours, then cooled to a temperature ranging from 50 to 55° C., added with water, and adjusted to pH lower than 7 with acetic acid. After cooling to 15° C., the resulting product is filtered, thereby obtaining adapalene methyl ester (9 g; 21.2 mmol) in 86% yield.EXAMPLE 5Synthesis of adapalene methyl esterA round-bottom flask is loaded with nickel (II) chloride (0.158 g; 1.2 mmol) and THF (15 ml), and tris(hydroxypropyl)phosphine (1.53 g; 7.3 mmol) is added. The mixture is refluxed for an hour, then cooled to a temperature of 50° C. and added in succession with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), potassium carbonate (6.75 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (9.1 g; 31.8 mmol), water (8.11 g; 450.5 mmol) and THF (30 ml). The mixture is refluxed for 24 hours, then cooled to a temperature ranging from 50 to 55° C., added with water, and adjusted to pH lower than 7 with acetic acid. After cooling to 15° C., the resulting product is filtered, thereby obtaining adapalene methyl ester (9.37 g; 21.96 mmol) in 90% yield.EXAMPLE 6Synthesis of adapalene methyl esterA round-bottom flask is loaded with methyl 6-tosyl-naphthalene-2-carboxylate (8.7 g; 24.4 mmol), THF (70 ml), potassium phosphate (10.38 g; 48.8 mmol), 4-methoxy-3-adamantyl-phenylboronic acid (7 g; 24.4 mmol), nickel chloride complexed with tri(cyclohexyl)phosphine (0.83 g; 1.2 mmol) and tri(cyclohexyl)phosphine (1.37 g; 4.88 mmol). The mixture is refluxed for 24 hours, then cooled to a temperature ranging from 50 to 55° C. and added with water, then cooled to 15° C. The resulting product is filtered, thereby obtaining adapalene methyl ester (8.1 g; 19.0 mmol) in 78% yield.
PATENThttps://patents.google.com/patent/WO2008126104A2/en
The compound 6-[3-(l – Adamantyl) – 4 – methoxy phenyl] – 2 – naphthoic acid of Formula – I known as Adapalene is used in dermatology, particularly in the treatment of acne vulgaris and psoriasis.
Formula – 1Adapalene was first time disclosed in the US patent No. 4,717,720 (herein after referred as ‘720) describe the preparation of compound of Formula – I using Negishi cross Coupling. In this reaction, 2-(l-adamantyl)-4-bromoanisole is converted to its organomagnesium compound followed by conversion to organozinc compound using zinc chloride and reacted with 6-bromo-2-methylnaphthoate employing transition metal as reaction catalyst such as palladium or nickel or one of its complexes with various phosphines. The reaction sequence is as shown in scheme – 1 below:
Scheme – 1 Another US patent No. 5,015,758 describe the process for preparation of 6[3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoate a penultimate step for preparation of Adapalene using Friedel – Crafts alkylation by reacting 1 – acetoxy adamantane with methyl – 6 – (4 – hydroxyphenyl) – 2 – naphthoate in presence of cone. Sulfuric acid in solvent n – heptane.Another improved process was published in the journal, Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. The process involves the preparation of intermediates followed by Negishi cross Coupling, where in 2-(l-adamantyl)-4-bromophenol was prepared using 1 – adamentol and 4- bromo phenol in presence of 98% sulphuric acid and acetic acid, which on methylation with dimethyl sulfate and potassium carbonate in dry acetone yields 2-(l -adamantyl)-4-bromoanisole. The compound is reacted with magnesium to form Grignard reagent and then coupled with 6-bromo-2-methylnaphthoate in presence of novel Pd – Zn double metal catalyst to yield ester, which on saponification followed by treatment with acid yields Adapalene.The recent published application WO 2006/108717 describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – 1. The application describes the preparation of 3-adamantyl-4-methoxyphenyl boronic acid from 2-(l-adamantyl)-4- bromoanisole using n-Butyl Lithium and triisopropyl borate in solvent tetrahydrofuran. Finally 3-adamantyl-4-methoxyphenyl boronic acid is reacted with 6-bromo-2-naphthoic acid involving Suzuki coupling in presence of Palladium acetate catalyst, a ligand 2 – (dicyclohexyl – phosphino) biphenyl, an inorganic base in solvent to get the compound adapalene.Some of the drawbacks of the prior art processes include:- The reported process in US patent 4717720, using Negishi cross coupling involves Grignard reaction. This requires anhydrous condition and a possibility of runaway reaction during Grignard reagent formation. Also the reaction involves the addition of fused ZnC12 and the preparation of the catalyst NiC12 (DPPE) complex, which needs to be freshly prepared increases the reaction step and has to be thoroughly dried before its use for coupling. Further the coupling reaction, results in the formation of dimer impurities during the organozinc compound reaction, with 2-(I -adamantyl)-4-bromoanisole and 6-bromo-2-methylnaphthoate respectively, which are difficult to remove. All these operations make the entire synthesis extremely sensitive and difficult to handle.Some of the above drawbacks were addressed by the authors in the article published in Organic Process Research & Development, 2006, 10, 285 – 288 for the preparation of Adapalene. But the use of Pd catalyst with the ligand like PdCl2 (PPh3)2 for the direct conversion of Grignard reagent employing ZnCYl in catalytic amount has its own limitations. The use of Grignard reagent, palladium catalyst with ligand and hygroscopic ZnCl2 demerits this process for industrial application.The recent published application WO 2006/108717; describes the use of Suzuki coupling for the synthesis of adapalene the compound of formula – I. The use of organo boronic acids for the Suzuki reaction has some limitations because of the indeterminate stoichiometry associated with the use of boronic acid, and its difficulty in purification and the byproducts formed during the reaction.Therefore there remains a need for an improved process for preparing adapalene that eliminates or substantially reduces the impurities, decreases the number of steps, and employs a more robust process which is convenient and cost efficient.The present inventors have come out with a novel process which ameliorates the problems in the prior art with a one – pot process for the preparation of adapalene by employing Suzuki – Miyaura coupling involving the use of novel reactant 3-adamantyl-4- methoxyphenyl potassium trifiuoroborate.The novel compound 3 – Adamantyl – 4 – methoxy phenyl potassium trifiuoroborate, exhibit superb behavior in the Suzuki-Miyaura reaction and provides a powerful method for the preparation of 6 – [3-(I – Adamantyl) – 4 – methoxy phenyl] – 2 – naphthoic acid, the compound of Formula – I.

Formula – 1Potassium organotrifluoroborates are air and moisture-stable crystalline solids which can be stored for extended periods of time making it more industrial friendly to use on large scale production.The other advantage of the present invention is in the use of methyl ester of 6 – Bromo – 2 -naphthoic acid and isolating adapalane directly from the reaction instead of its methyl ester, the above process becomes more robust and eliminates the saponification step as reported in prior art. Also the use of readily and cheaply available Pd catalyst on carbon over the conventional and costlier Pd-catalyst with ligands offers further advantage to the current process.Examples:Example 1: Preparation of 3 – Adamantyl – 4 – methoxy phenyl potassium trifluoroborate:In a 2.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 100.0 gm of 2-(l- adamantyl) 4-bromo anisole was charged in 600 ml tetrahydrofuran. The reaction mixture was cooled to -55 ± 50C and 302 ml of 1.6 M n – butyl Lithium was slowly added and stirred. 87 ml of tri isopropyl borate was then charged and stirring was continued for 30 minutes at -55 ± 5°C. Cooling was removed and the temperature raised slowly to 25 – 300C. 1.0 L of 1.2N hydrochloric acid was then charged and reaction mass was stirred for 30 minutes and separated the organic layer. The organic layer was charged in 1.0 L round bottom flask and freshly prepared aqueous solution of potassium hydrogen difluoride (230 gm, in 700 ml water) was added at 25 – 300C and stirring was maintained till white precipitate is obtained. The mixture was continued under stirring and cooled to 0 – 50C. The product, 3 – adamantyl – 4 – methoxyphenyl potassium trifluoroborate obtained was filtered, washed with 100 ml of ethyl acetate. The product was dried at 60 – 65°C till constant weight. Yield: 90.5 gm (83%), Purity: 99.0 % by HPLC.Example 2: Preparation of 6 – [3-(l- Adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid:In a 1.0 L round bottom flask equipped with stirring and under nitrogen atmosphere 50.0 gm of 3 – Adamantyl – 4 – methoxyphenyl potassium trifluoroborate, 23 gm of 6- bromo -2-methyl napthoate in 300 ml tetrahydrofuran (THF) was charged. Stirred for 15 min and charged 3.0 gm of 5% Pd / C was and aqueous potassium hydroxide solution (50.0 gm in 300 ml water). Stirring was continued and the temperature was raised to reflux. The reaction mass was maintained for 10 hours at reflux and after the completion of the reaction, 200 ml of tetrahydrofuran: water (1 : 1) mixture was added and then filtered through hyflow bed at 45-500C. The hyflow bed was washed with tetrahydrofuran: water (1 : 1) mixture at 45-500C. 500 ml water was charged and the reaction mass was stirred. The aqueous layer was acidified with 1.2N hydrochloric acid. The precipitated mass was filtered, washed with water till neutral pH. The solid product obtained was dried at 70 – 75°C till constant weight to get 6 – [3-(l- adamantyl) – 4 – methoxyphenyl] – 2 – naphthoic acid.The dried product was taken in 300 ml of tetrahydrofuran and stirred. The temperature was raised to reflux and was maintained for 30 minutes. The heating was stopped and cooled the reaction mass to 25 – 300C. 500 ml of n – heptane was charged to the reaction mass and stirred for 30 minutes. The reaction mass was then chilled to 0 – 5°C and maintained stirring at 0 – 5°C temperature for 2.0 hours. The precipitated solid was filtered and washed with n – heptane. The pure crystalline 6 – [3-(l- adamantyl) – 4 methoxyphenyl] – 2 – naphthoic acid thus obtained was then dried till constant weight. Yield = 40 – 42 gms (68 – 72 %)
/////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Per the recommendations of the Global Alliance on Improving Outcomes of Acne, retinoids such as adapalene are considered first-line therapy in acne treatment and are to be used either independently or in conjunction with benzoyl peroxide and/or an antimicrobial agent, like clindamycin, for maximum efficacy.[4][5] Furthermore, adapalene, like other retinoids, increases the efficacy and penetration of other topical acne medications that are used in conjunction with topical retinoids as well as hastens the improvement of the post-inflammatory hyperpigmentation caused by acne.[4] In the long term, it can be used as maintenance therapy.[4]
Off-label uses
Adapalene has the unique ability to inhibit keratinocyte differentiation and decrease keratin deposition. This property makes adapalene an effective treatment for keratosis pilaris and callus. It may be used by men undergoing foreskin restoration to reduce excess keratin that forms a layer on the exterior of the human penis after circumcision. Other non-FDA approved indications that have been reported in the literature include treatment of warts, molluscum contagiosum, Darier disease, photoaging, pigmentary disorders, actinic keratoses and alopecia areata.[6]
Side effects
Adapalene is known to cause mild adverse effects such as photosensitivity, irritation, redness, dryness, itching, and burning.[2] It is common (between 1% and 10% of users)[7] to experience a brief sensation of warmth or stinging, as well as dry skin, peeling and redness during the first 2–4 weeks of using the medication.[4][8] These effects are considered mild and generally decrease over time.[4][8] Any serious allergic reaction is rare.[8] Furthermore, of the three topical retinoids, adapalene is often regarded as the most tolerable.[6]
In pregnancy
Use of topical adapalene in pregnancy has not been well studied, but has a theoretical risk of retinoid embryopathy.[9] Thus far, there is no evidence that the cream causes problems in the baby if used during pregnancy. Use is at the consumer’s own risk.[10]
According to the Drugs and Lactation Database, topical adapalene has poor systemic absorption and results in low blood levels (less than 0.025 mcg/L) despite long term use, suggesting that there is low risk of harm for a nursing infant.[11] However, it is recommended that the topical medication should not be applied to the nipple or any other area that may come into direct contact with the infant’s skin.[11]
Interactions
Adapalene has been shown to enhance the efficacy of topical clindamycin, although adverse effects are also increased.[12][13] Application of adapalene gel to the skin 3–5 minutes before application of clindamycin enhances penetration of clindamycin into the skin, which may enhance the overall efficacy of the treatment as compared to clindamycin alone.[14]
Pharmacology
Unlike the retinoid tretinoin (Retin-A), adapalene has also been shown to retain its efficacy when applied at the same time as benzoyl peroxide due to its more stable chemical structure.[15] Furthermore, photodegradation of the molecule is less of a concern in comparison to tretinoin and tazarotene.[6]
Pharmacokinetics
Absorption of adapalene through the skin is low. A study with six acne patients treated once daily for five days with two grams of adapalene cream applied to 1,000 cm2 (160 sq in) of skin found no quantifiable amounts, or less than 0.35 ng/mL of the drug, in the patients’ blood plasma.[16] Controlled trials of chronic users of adapalene have found drug levels in the patients’ plasma to be 0.25 ng/mL.[9]
Pharmacodynamics
Adapalene is highly lipophilic. When applied topically, it readily penetrates hair follicles and absorption occurs 5 minutes after topical application.[2] After penetration into the follicle, adapalene binds to nuclear retinoic acid receptors (namely retinoic acid receptor beta and gamma).[5][9] These complexes then bind to the retinoid X receptor which induces gene transcription by binding to specific DNA sites, thus modulating downstream keratinocyte proliferation and differentiation.[2][9] This results in normalization of keratinocyte differentiation, allowing for decreased microcomedone formation, decreased clogging of pores, and increased exfoliation by increasing cell turnover.[6][9][17] Adapalene is also regarded as an anti-inflammatory agent, as it suppresses the inflammatory response stimulated by the presence of Cutibacterium acnes,[6] and inhibits both lipoxygenase activity and the oxidative metabolism of arachidonic acid into prostaglandins.[9]
Adapalene selectively targets retinoic acid receptor beta and retinoic acid receptor gamma when applied to epithelial cells such as those found in the skin.[18] Its agonism of the gamma subtype is largely responsible for adapalene’s observed effects. In fact, when adapalene is applied in conjunction with a retinoic acid receptor gamma antagonist, adapalene loses clinical efficacy.[19]
Retinization is a common temporary phenomenon reported by patients when initiating use of retinols.[20] Within the initial period of treatment, skin can become red, irritated, dry and may burn or itch from retinol application; however, this tends to resolve within four weeks with once a day use.[20]
History
Adapalene is a research product of Galderma Laboratories, France.[21] Adapalene was approved in 1996 by the U.S. Food and Drug Administration (FDA) for use in the treatment of acne.[22]
Research
A study has concluded that adapalene can be used to treat plantar warts and may help clear lesions faster than cryotherapy.[23]
References
- ^ Rolewski SL (October 2003). “Clinical review: topical retinoids”. Dermatology Nursing. 15 (5): 447–50, 459–65. PMID 14619325.
- ^ Jump up to:a b c d e f Tolaymat, L; Zito, PM (January 2021). “Adapalene”. PMID 29494115.
- ^ Asai, Yuka; Baibergenova, Akerke; Dutil, Maha; Humphrey, Shannon; Hull, Peter; Lynde, Charles; Poulin, Yves; Shear, Neil H.; Tan, Jerry; Toole, John; Zip, Catherine (2 February 2016). “Management of acne: Canadian clinical practice guideline”. Canadian Medical Association Journal. 188 (2): 118–126. doi:10.1503/cmaj.140665. PMC 4732962. PMID 26573753.
- ^ Jump up to:a b c d e Kolli, Sree S.; Pecone, Danielle; Pona, Adrian; Cline, Abigail; Feldman, Steven R. (2019-01-23). “Topical Retinoids in Acne Vulgaris: A Systematic Review”. American Journal of Clinical Dermatology. 20 (3): 345–365. doi:10.1007/s40257-019-00423-z. ISSN 1179-1888. PMID 30674002. S2CID 59225325.
- ^ Jump up to:a b Xiang, Leihong Flora; Troielli, Patricia; Lozada, Vicente Torres; Tan, Jerry; Suh, Dae Hun; See, Jo-Ann; Piquero-Martin, Jaime; Perez, Montserrat; Orozco, Beatriz (2018-02-01). “Practical management of acne for clinicians: An international consensus from the Global Alliance to Improve Outcomes in Acne”. Journal of the American Academy of Dermatology. 78 (2): S1–S23.e1. doi:10.1016/j.jaad.2017.09.078. hdl:10067/1492720151162165141. ISSN 0190-9622. PMID 29127053. S2CID 31654121.
- ^ Jump up to:a b c d e Tolaymat, Leila; Zito, Patrick M. (2018), “Adapalene”, StatPearls, StatPearls Publishing, PMID 29494115, retrieved 2019-03-13
- ^ “Differin”. Swedish Drug Formulary. Retrieved 2017-12-11.
- ^ Jump up to:a b c “Adapalene Gel”. WebMD. Retrieved 2017-12-11.
- ^ Jump up to:a b c d e f Piskin, Suleyman; Uzunali, Erol (August 2007). “A review of the use of adapalene for the treatment of acne vulgaris”. Therapeutics and Clinical Risk Management. 3 (4): 621–624. ISSN 1176-6336. PMC 2374937. PMID 18472984.
- ^ “FDA approves Differin Gel 0.1% for over-the-counter use to treat acne”. July 8, 2016. Retrieved 14 July 2016.
- ^ Jump up to:a b “Adapalene”, Drugs and Lactation Database (LactMed), National Library of Medicine (US), 2006, PMID 30000483, retrieved 2019-03-13
- ^ Wolf JE, Kaplan D, Kraus SJ, Loven KH, Rist T, Swinyer LJ, Baker MD, Liu YS, Czernielewski J (September 2003). “Efficacy and tolerability of combined topical treatment of acne vulgaris with adapalene and clindamycin: a multicenter, randomized, investigator-blinded study”. Journal of the American Academy of Dermatology. 49 (3 Suppl): S211-7. doi:10.1067/S0190-9622(03)01152-6. PMID 12963897.
- ^ Jain, GauravK; Ahmed, FarhanJ (2007). “Adapalene pretreatment increases follicular penetration of clindamycin: In vitro and in vivo studies”. Indian Journal of Dermatology, Venereology and Leprology. 73 (5): 326–9. doi:10.4103/0378-6323.34010. ISSN 0378-6323. PMID 17921613.
- ^ Jain GK, Ahmed FJ (2007). “Adapalene pretreatment increases follicular penetration of clindamycin: in vitro and in vivo studies” (PDF). Indian Journal of Dermatology, Venereology and Leprology. 73 (5): 326–9. doi:10.4103/0378-6323.34010. PMID 17921613.
- ^ Martin B, Meunier C, Montels D, Watts O (October 1998). “Chemical stability of adapalene and tretinoin when combined with benzoyl peroxide in presence and in absence of visible light and ultraviolet radiation”. The British Journal of Dermatology. 139 Suppl 52: 8–11. doi:10.1046/j.1365-2133.1998.1390s2008.x. PMID 9990414. S2CID 43287596.
- ^ “DIFFERIN® (adapalene) Cream, 0.1% Label” (PDF). FDA. May 25, 2000. Retrieved 4 Oct 2011.
- ^ “DIFFERIN® (adapalene) Gel, 0.3%” (PDF). Retrieved March 12, 2019.
- ^ Mukherjee S, Date A, Patravale V, Korting HC, Roeder A, Weindl G (2006). “Retinoids in the treatment of skin aging: an overview of clinical efficacy and safety”. Clinical Interventions in Aging. 1 (4): 327–48. doi:10.2147/ciia.2006.1.4.327. PMC 2699641. PMID 18046911.
- ^ Michel S, Jomard A, Démarchez M (October 1998). “Pharmacology of adapalene”. The British Journal of Dermatology. 139 Suppl 52: 3–7. doi:10.1046/j.1365-2133.1998.1390s2003.x. PMID 9990413. S2CID 23084886.
- ^ Jump up to:a b “Differin Gel: An Over-the-Counter Retinoid for Acne”. http://www.differin.com. Retrieved 2019-03-25.
- ^ US Patent 4717720A, Shroot B, Eustache J, Bernardon J-M, “Benzonaphthalene derivatives and compositions”, published 1988-01-05, issued 1988-01-05, assigned to Galderma Research and Development SNC
- ^ “FDA approval of DIFFERIN® (adapalene) Solution, 0.1%”. FDA. May 31, 1996. Retrieved 29 May 2017.
- ^ Gupta, Ramji; Gupta, Sarthak (2015). “Topical Adapalene in the Treatment of Plantar Warts; Randomized Comparative Open Trial in Comparison with Cryo-Therapy”. Indian Journal of Dermatology. 60 (1): 102. doi:10.4103/0019-5154.147835. ISSN 0019-5154. PMC 4318023. PMID 25657417.
External links
- “Adapalene”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Differin, Pimpal, Gallet, Adelene, Adeferin |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a604001 |
| License data | US DailyMed: Adapalene |
| Pregnancy category | AU: D |
| Routes of administration | Topical |
| Drug class | Retinoids |
| ATC code | D10AD03 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) / S3CA: ℞-onlyUK: POM (Prescription only)US: OTC / Rx-only |
| Pharmacokinetic data | |
| Bioavailability | Very low[medical citation needed] |
| Excretion | Bile |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 106685-40-9 |
| PubChem CID | 60164 |
| IUPHAR/BPS | 5429 |
| DrugBank | DB00210 |
| ChemSpider | 54244 |
| UNII | 1L4806J2QF |
| KEGG | D01112 |
| ChEBI | CHEBI:31174 |
| ChEMBL | ChEMBL1265 |
| CompTox Dashboard (EPA) | DTXSID5046481 |
| ECHA InfoCard | 100.149.379 |
| Chemical and physical data | |
| Formula | C28H28O3 |
| Molar mass | 412.529 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
/////////////////ADAPALENE, CD 271, CD-271, ANTIACNE, Differin, Differine
COC1=C(C=C(C=C1)C1=CC2=C(C=C1)C=C(C=C2)C(O)=O)C12CC3CC(CC(C3)C1)C2

NEW DRUG APPROVALS
ONE TIME
$10.00
Clascoterone
Clascoterone
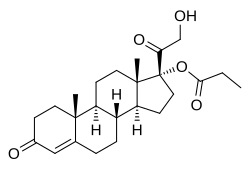
(1R,3aS,3bR,9aR,9bS,11aS)-1-(2-hydroxyacetyl)-9a,11a-dimethyl-7-oxo-1H,2H,3H,3aH,3bH,4H,5H,7H,8H,9H,9aH,9bH,10H,11H,11aH-cyclopenta[a]phenanthren-1-yl propanoate
| Formula |
C24H34O5
|
|---|---|
| CAS |
19608-29-8
|
| Mol weight |
402.5238
|
FDA APPROVED, 2020/8/26, Winlevi
|
クラスコステロン;
|
Anti-acne, Androgen receptor antagonist
Clascoterone, sold under the brand name Winlevi, is an antiandrogen medication which is used topically in the treatment of acne.[1][2][3] It is also under development for the treatment of androgen-dependent scalp hair loss.[2] The medication is used as a cream by application to the skin, for instance the face and scalp.[3]
Clascoterone is an antiandrogen, or antagonist of the androgen receptor (AR), the biological target of androgens such as testosterone and dihydrotestosterone.[4][5] It shows no systemic absorption when applied to skin.[3]
The medication, developed by Cassiopea and Intrepid Therapeutics,[2] was approved by the US Food and Drug Administration (FDA) for acne in August 2020.[6][7]
Medical uses
Clascoterone is indicated for the topical treatment of acne vulgaris in females and males age 12 years and older.[1][8] It is applied to the affected skin area in a dose of 1 mg cream (or 10 mg clascoterone) twice per day, once in the morning and once in the evening.[1] The medication should not be used ophthalmically, orally, or vaginally.[1]
Available forms
Clascoterone is available in the form of a 1% (10 mg/g) cream for topical use.[1]
Contraindications
Clascoterone has no contraindications.[1]
Side effects
The incidences of local skin reactions with clascoterone were similar to placebo in two large phase 3 randomized controlled trials.[1][9] Suppression of the hypothalamic–pituitary–adrenal axis (HPA axis) may occur during clascoterone therapy in some individuals due to its cortexolone metabolite.[1][8] HPA axis suppression as measured by the cosyntropin stimulation test was observed to occur in 3 of 42 (7%) of adolescents and adults using clascoterone for acne.[1][8] HPA axis function returned to normal within 4 weeks following discontinuation of clascoterone.[1][8] Hyperkalemia (elevated potassium levels) occurred in 5% of clascoterone-treated individuals and 4% of placebo-treated individuals.[1]
Pharmacology
Pharmacodynamics
Clascoterone is an steroidal antiandrogen, or antagonist of the androgen receptor (AR), the biological target of androgens such as testosterone and dihydrotestosterone (DHT).[1][4][5] In a bioassay, the topical potency of the medication was greater than that of progesterone, flutamide, and finasteride and was equivalent to that of cyproterone acetate.[10] Likewise, it is significantly more efficacious as an antiandrogen than other AR antagonists such as enzalutamide and spironolactone in scalp dermal papilla cells and sebocytes in vitro.[5]\
Pharmacokinetics
Steady-state levels of clascoterone occur within 5 days of twice daily administration.[1] At a dosage of 6 g clascoterone cream applied twice daily, maximal circulating levels of clascoterone were 4.5 ± 2.9 ng/mL, area-under-the-curve levels over the dosing interval were 37.1 ± 22.3 h*ng/mL, and average circulating levels of clascoterone were 3.1 ± 1.9 ng/mL.[1] In rodents, clascoterone has been found to possess strong local antiandrogenic activity, but negligible systemic antiandrogenic activity when administered via subcutaneous injection.[10] Along these lines, the medication is not progonadotropic in animals.[10]
The plasma protein binding of clascoterone is 84 to 89% regardless of concentration.[1]
Clascoterone is rapidly hydrolyzed into cortexolone (11-deoxycortisol) and this compound is a possible primary metabolite of clascoterone based on in-vitro studies in human liver cells.[1][8] During treatment with clascoterone, cortexolone levels were detectable and generally below or near the low limit of quantification (0.5 ng/mL).[1] Clascoterone may also produce other metabolites, including conjugates.[1]
The elimination of clascoterone has not been fully characterized in humans.[1]
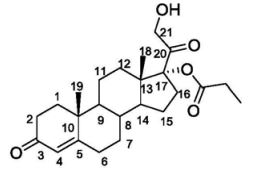
Chemistry
Clascoterone, also known as cortexolone 17α-propionate or 11-deoxycortisol 17α-propionate, as well as 17α,21-dihydroxyprogesterone 17α-propionate or 17α,21-dihydroxypregn-4-en-3,20-dione 17α-propionate, is a synthetic pregnane steroid and a derivative of progesterone and 11-deoxycortisol (cortexolone).[11] It is specifically the C17α propionate ester of 11-deoxycortisol.[10]
An analogue of clascoterone is 9,11-dehydrocortexolone 17α-butyrate (CB-03-04).[12]
History
C17α esters of 11-deoxycortisol were unexpectedly found to possess antiandrogenic activity.[10] Clascoterone, also known as cortexolone 17α-propionate, was selected for development based on its optimal drug profile.[10] The medication was approved by the US Food and Drug Administration (FDA) for the treatment of acne in August 2020.[6]
Two large phase 3 randomized controlled trials evaluated the effectiveness of clascoterone for the treatment of acne over a period of 12 weeks.[1][8][9] Clascoterone decreased acne symptoms by about 8 to 18% more than placebo.[1][9] The defined treatment success endpoint was achieved in about 18 to 20% of individuals with clascoterone relative to about 7 to 9% of individuals with placebo.[1][8][9] The comparative effectiveness of clascoterone between males and females was not described.[1][9]
A small pilot randomized controlled trial in 2011, found that clascoterone cream decreased acne symptoms to a similar or significantly greater extent than tretinoin 0.05% cream.[8][13] No active comparator was used in the phase III clinical trials of clascoterone for acne.[8] Hence, it’s unclear how clascoterone compares to other therapies used in the treatment of acne.[8]
The FDA approved clascoterone based on evidence from two clinical trials (Trial 1/NCT02608450 and Trial 2/NCT02608476) of 1440 participants 9 to 58 years of age with acne vulgaris.[14] The trials were conducted at 99 sites in the United States, Poland, Romania, Bulgaria, Ukraine, Georgia, and Serbia.[14]
Participants applied clascoterone or vehicle (placebo) cream twice daily for 12 weeks.[14] Neither the participants nor the health care providers knew which treatment was being given until after the trial was completed.[14] The benefit of clascoterone in comparison to placebo was assessed after 12 weeks of treatment using the Investigator’s Global Assessment (IGA) score that measures the severity of disease (on a scale from 0 to 4) and a decrease in the number of acne lesions.[14]
Society and culture
Names
Clascoterone is the generic name of the drug and its INN and USAN.[11][15]
Research
Clascoterone has been suggested as a possible treatment for hidradenitis suppurativa (acne inversa), an androgen-dependent skin condition.[16]
………………………………………………………………………….
PATENT
CN 112028956
https://patents.google.com/patent/CN112028956A/en

Abstract
Several 17α-monoesters of cortexolone and its Δ9-derivative are endowed with antiandrogenic activity. Their synthesis can be accomplished by means of a lipase-catalyzed chemoselective alcoholysis of the corresponding 17α,21-diesters.
Graphical abstract

1H NMR (500 MHz, CDCl3): selected data δ 5.78 (br s, 1H, H-4), 4.32 (dd, 1H, H-21, J18.3 and 4.9 Hz), 4.25 (dd, 1H, H-21, J18.3 and 4.9 Hz), 1.22 (s, 3H, CH3-19), 1.17 (t, 3H, CH3, J7.6 Hz), 0.72 (s, 3H, CH3-18) MP 133 °C (t-butylmethylether)
…………………………………………………………………..
PATENT
https://patents.google.com/patent/EP2503005B1/en
-
Cortexolone derivatives in which the hydroxyl group at position C-17α is esterified with short chain aliphatic or aromatic acids and the derivatives of the corresponding 9,11-dehydro derivative, are known to have an antiandrogenic effect.
- [0002]
EP 1421099 describes cortexolone 17α-propionate and 9,11-dehydro-cortexolone-17-α-butanoate regarding a high antiandrogenic biological activity demonstrated both “in vitro” and “in vivo” on the animal.
- [0003]
US3530038 discloses the preparation of a crystalline form of cortexolone-17α-propionate having a melting point of 126-129 °C and an IR spectrum with bands at (cm-1): 3500, 1732, 1713, 1655 and 1617.
- [0004]
A method for obtaining the above mentioned derivatives is described by Gardi et al. (Gazz. Chim. It. 63, 43 1,1963) and in the United States patent US3152154 providing for the transformation of cortexolone, or transformation of 9,11-dehydrocortexolone, in the intermediate orthoester using orthoesters available in the market as a mixture of aprotic solvents such as cyclohexane and DMF, in presence of acid catalysis (ex. PTSA.H20). The intermediate orthoester thus obtained can be used as is or upon purification by suspension in a solvent capable of solubilising impurities, preferably in alcohols. The subsequent hydrolysis in a hydroalcoholic solution, buffered to pH 4-5 preferably in acetate buffer, provides the desired monoester.
- [0005]
Such synthesis is indicated in the diagram 1 below

- [0006]
However, the monoesters thus obtained were, in the reaction conditions, unstable and, consequently hard to manipulate and isolate (R. Gardi et al Tetrahedron Letters, 448, 1961). The instability is above all due to the secondary reaction of migration of the esterifying acyl group from position 17 to position 21.
- [0007]
It is thus known that in order to obtain the above mentioned monoesters with a chemical purity in such a manner to be able to proceed to the biological tests, it is necessary to use, at the end of the synthesis, a purification process which is generally performed by means of column chromatography.
- [0008]
Furthermore, US3152154 describes how the hydrolysis of the diester in a basic environment is not convenient due to the formation of a mixture of 17α,21-diol, of 17- and 21 -monoesters, alongside the initial non-reacted product.
- [0009]
Now, it has been surprisingly discovered that an alcoholysis reaction using a lipase from Candida as a biocatalyst can be usefully applied during the preparation of 17α monoesters of cortexolone, or its 9,11-dehydroderivatives.
- [0010]
As a matter of fact, it has been discovered that such enzymatic alcoholysis of the 17,21-diester of the cortexolone, or of its derivative 9,11-dehydro, selectively occurs in position 21 moving to the corresponding monoester in position 17, as shown in diagram 2 below:

- [0011]
The chemoselectivity of the special enzymatic reaction in alcoholysis conditions, according to the present invention, opens new perspectives for preparation, at industrial level with higher yields, of 17α-monoesters with respect to the methods already indicated in literature.
- [0012]
The diesters serving as a substrate for the reaction of the invention can be prepared according to the prior art, for example following the one described in B.Turner, (Journal of American Chemical Society, 75, 3489, 1953) which provides for the esterification of corticosteroids with a linear carboxylic acid in presence of its anhydride and PTSA monohydrate.
EXAMPLES
-
- Example 1
Alcoholysis with CCL of cortexolone 17α, 21-dipropionate
-
-
- [0055]
Add butanol (0.4g, 5.45 mmoles) and CCL (17.4g, 3.86 U/mg, FLUKA) to a solution of cortexolone-17α,21-dipropionate (0.5g, 1.09 mmoles) in toluene (50ml). Maintain the mixture under stirring, at 30 °C, following the progress of the reaction in TLC (Toluene/ethyl acetate 6/4) until the initial material is dissolved (24h). Remove the enzyme by means of filtration using a Celite layer. Recover the cortexolone 17α-propionate (0.437, 99%) after evaporation under low pressure. Through crystallisation, from diisopropyl ether you obtain a product with a purity >99% in HPLC.
- [0056]
1H-NMR (500MHz, CDCl3) relevant signals δ (ppm) 5.78 (br s, 1 H, H-4), 4.32 (dd, 1 H, H-21), 4.25 (dd, 1H, H-21), 1.22 (s, 3H, CH3-19), 1.17 (t, 3H, CH3), 0.72 (s, 3H, CH3-18). P.f. 114 °C
- [0055]
-
Example 2 (comparative)
-
-
- [0057]
According to the method described in example 1 prepare cortexolone-17α-butanoate.
- [0058]
1H-NMR relevant signals δ (ppm) 5.78 (br s, 1H, H-4), 4.32 (dd, 1H, H-21), 4.26 (dd, 1H, H-21), 1.23 (s, 3H, CH3-19), 0.97 (t, 3H, CH3), 0.73 (s, 3H. CH3-18). P.F. 134-136 °C
- [0057]
-
Example 3 (comparative)
According to the method described in the example prepare cortexolone-17α-valerate.
-
-
- [0059]
1H-NMR relevant signals δ (ppm) 5.77 (br s, 1H, H-4), 4.32 (dd, 1H, H-21), 4.26 (dd, 1H, H-21), 1.22 (s, 3H, CH3-19), 0.95 (t, 3H, CH3), 0.72 (s, 3H, CH3-18). P.f. 114 °C (diisopropyl ether).
- [0059]
-
Example 4 (comparative)
According to the method described in the example prepare 9, 11-dehydro-cortexolone-17α-butanoate.
-
-
- [0060]
1H-NMR relevant signals δ (ppm) 5.77 (br s, 1H, H-4), 5.54 (m, 1H, H-9), 4.29 (dd, 1H, H-21), 4.24 (dd, 1H, H-21), 1.32 (s, 3H, CH3-19), 0.94(t, 3H, CH3), 0.68 (s, 3H, CH3-18). P.f. 135-136 °C (acetone/hexane).
- [0060]
-
Example 5
Alcoholysis with CALB of cartexolone-17α, 21-dipropionate
-
-
- [0061]
Dissolve cortexolone, 17α, 2-dipropionate (0.5g, 1.09 mmoles) in acetonitrile (40ml), add CALB (2.3g, 2.5 U/mg Fluka) and octanol (0.875ml). Leave the mixture under stirring, at 30 °C, for 76 hrs. Remove the enzyme by means of filtration using a paper filter. Once the solvents evaporate, recover a solid (0.4758) which upon analysis 1H-NMR shall appear made up of cortexolone-17α-propionate at 91%.
- [0061]
-
Example 6
Crystallisation
-
-
- [0062]
Add the solvent (t-butylmethylether or diisopropylether) to the sample according to the ratios indicated in Table 3. Heat the mixture to the boiling temperature of the solvent, under stirring, until the sample dissolves completely. Cool to room temperature and leave it at this temperature, under stirring, for 6 hours. Filter using a buchner funnel and maintain the solid obtained, under low pressure, at a room temperature for 15 hours and then, at 40°C, for 5 hours.
- [0062]
-
Example 7 (comparative)
Precipitation
-
-
- [0063]
Disslove the sample in the suitable solvent (dichloromethane, acetone, ethyl acetate or ethanol) according to the ratios indicated in table 3 and then add the solvent, hexane or water, according to the ratios indicated in table 3, maintaining the mixture, under stirring, at room temperature. Recover the precipitate by filtration using a buchner funnel and desiccate as in example 6.
- [0063]
-
Example 8.
Obtaining a pharmaceutical form containing the medication in a defined crystalline form.
- [0064]
Prepare a fluid cream containing 2 % cetylic alcohol, 16% glyceryl monostearate, 10% vaseline oil, 13 % propylene glycol, 10% polyethylenglycol with low polymerization 1.5% polysorbate 80 and 47.5 % purified water. Add 1 g of cortexolone 17α-propionate of crystalline form III to 100 g of this cream and subject the mixture to homogenisation by means of a turbine agitator until you obtain homogeneity. You obtain a cream containing a fraction of an active ingredient dissolved in the formulation vehicle and a non-dissolved fraction of an active ingredient, present as a crystal of crystalline form III. This preparation is suitable for use as a formulation vehicle for skin penetration tests on Franz cells, where a coefficient of penetration in the range of 0.04 to 0.03 cm/h is observed on the preparation.
References
- ^ Jump up to:a b c d e f g h i j k l m n o p q r s t u v w “Winlevi (clascoterone) cream, for topical use”(PDF). Cassiopea. Retrieved 9 September 2020.
- ^ Jump up to:a b c http://adisinsight.springer.com/drugs/800026561
- ^ Jump up to:a b c Kircik LH (July 2019). “What’s new in the management of acne vulgaris”. Cutis. 104(1): 48–52. PMID 31487336.
- ^ Jump up to:a b Rosette C, Rosette N, Mazzetti A, Moro L, Gerloni M (February 2019). “Cortexolone 17α-Propionate (Clascoterone) is an Androgen Receptor Antagonist in Dermal Papilla Cells In Vitro”. J Drugs Dermatol. 18 (2): 197–201. PMID 30811143.
- ^ Jump up to:a b c Rosette C, Agan FJ, Mazzetti A, Moro L, Gerloni M (May 2019). “Cortexolone 17α-propionate (Clascoterone) Is a Novel Androgen Receptor Antagonist that Inhibits Production of Lipids and Inflammatory Cytokines from Sebocytes In Vitro”. J Drugs Dermatol. 18 (5): 412–418. PMID 31141847.
- ^ Jump up to:a b “Cassiopea Receives FDA Approval for Winlevi (clascoterone cream 1%), First-in-Class Topical Acne Treatment Targeting the Androgen Receptor”. Cassiopea (Press release). Retrieved 2020-08-30.
- ^ “Winlevi: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 9 September 2020.
- ^ Jump up to:a b c d e f g h i j Barbieri, John S. (2020). “A New Class of Topical Acne Treatment Addressing the Hormonal Pathogenesis of Acne”. JAMA Dermatology. 156 (6): 619–620. doi:10.1001/jamadermatol.2020.0464. ISSN 2168-6068. PMID 32320045.
- ^ Jump up to:a b c d e Hebert A, Thiboutot D, Stein Gold L, Cartwright M, Gerloni M, Fragasso E, Mazzetti A (April 2020). “Efficacy and Safety of Topical Clascoterone Cream, 1%, for Treatment in Patients With Facial Acne: Two Phase 3 Randomized Clinical Trials”. JAMA Dermatol. 156 (6): 621–630. doi:10.1001/jamadermatol.2020.0465. PMC 7177662. PMID 32320027.
- ^ Jump up to:a b c d e f Celasco G, Moro L, Bozzella R, Ferraboschi P, Bartorelli L, Quattrocchi C, Nicoletti F (2004). “Biological profile of cortexolone 17alpha-propionate (CB-03-01), a new topical and peripherally selective androgen antagonist”. Arzneimittelforschung. 54 (12): 881–6. doi:10.1055/s-0031-1297043. PMID 15646372.
- ^ Jump up to:a b https://chem.nlm.nih.gov/chemidplus/rn/19608-29-8
- ^ Celasco G, Moroa L, Bozzella R, Ferraboschi P, Bartorelli L, Di Marco R, Quattrocchi C, Nicoletti F (2005). “Pharmacological profile of 9,11-dehydrocortexolone 17alpha-butyrate (CB-03-04), a new androgen antagonist with antigonadotropic activity”. Arzneimittelforschung. 55 (10): 581–7. doi:10.1055/s-0031-1296908. PMID 16294504.
- ^ Trifu V, Tiplica GS, Naumescu E, Zalupca L, Moro L, Celasco G (2011). “Cortexolone 17α-propionate 1% cream, a new potent antiandrogen for topical treatment of acne vulgaris. A pilot randomized, double-blind comparative study vs. placebo and tretinoin 0·05% cream”. Br. J. Dermatol. 165 (1): 177–83. doi:10.1111/j.1365-2133.2011.10332.x. PMID 21428978. S2CID 38404925.
- ^ Jump up to:a b c d e “Drug Trial Snapshot: Winlevi”. U.S. Food and Drug Administration (FDA). 26 August 2020. Retrieved 10 September 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3): 106. hdl:10665/330879.
- ^ Der Sarkissian SA, Sun HY, Sebaratnam DF (August 2020). “Cortexolone 17 α-proprionate for hidradenitis suppurativa”. Dermatol Ther: e14142. doi:10.1111/dth.14142. PMID 32761708.
External links
- “Clascoterone”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT02608450 for “A Study to Evaluate the Safety and Efficacy of CB-03-01 Cream, 1% in Subjects With Facial Acne Vulgaris (25)” at ClinicalTrials.gov
- Clinical trial number NCT02608476 for “A Study to Evaluate the Safety and Efficacy of CB-03-01 Cream, 1% in Subjects With Facial Acne Vulgaris (26)” at ClinicalTrials.gov
 |
|
| Clinical data | |
|---|---|
| Trade names | Winlevi |
| Other names | CB-03-01; Breezula; 11-Deoxycortisol 17α-propionate; 17α-(Propionyloxy)- deoxycorticosterone; 21-Hydroxy-3,20-dioxopregn-4-en-17-yl propionate |
| License data |
|
| Routes of administration |
Topical (cream) |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.210.810 |
| Chemical and physical data | |
| Formula | C24H34O5 |
| Molar mass | 402.531 g·mol−1 |
| 3D model (JSmol) | |
/////////Clascoterone, クラスコステロン , FDA 2020, 2020 APPROVALS, ANTI ACNE
[H][C@@]12CC[C@](OC(=O)CC)(C(=O)CO)[C@@]1(C)CC[C@@]1([H])[C@@]2([H])CCC2=CC(=O)CC[C@]12C

{kind=link}
{kind=link}