
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
FDA approves first treatment Dupixent (Dupilumab) for chronic rhinosinusitis with nasal polyps
The U.S. Food and Drug Administration today approved Dupixent (dupilumab) to treat adults with nasal polyps (growths on the inner lining of the sinuses) accompanied by chronic rhinosinusitis (prolonged inflammation of the sinuses and nasal cavity). This is the first treatment approved for inadequately controlled chronic rhinosinusis with nasal polyps.
“Nasal polyps can lead to loss of smell and often patients require surgery to remove the polyps,” said Sally Seymour, M.D., Director of the Division of Pulmonary, Allergy and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research. “Dupixent provides an important treatment option for patients whose nasal polyps are not …
- June 26, 2019
The U.S. Food and Drug Administration today approved Dupixent (dupilumab) to treat adults with nasal polyps (growths on the inner lining of the sinuses) accompanied by chronic rhinosinusitis (prolonged inflammation of the sinuses and nasal cavity). This is the first treatment approved for inadequately controlled chronic rhinosinusis with nasal polyps.
“Nasal polyps can lead to loss of smell and often patients require surgery to remove the polyps,” said Sally Seymour, M.D., Director of the Division of Pulmonary, Allergy and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research. “Dupixent provides an important treatment option for patients whose nasal polyps are not adequately controlled with intranasal steroids. It also reduces the need for nasal polyp surgery and oral steroids.”
Dupixent is given by injection. The efficacy and safety of Dupixent were established in two studies with 724 patients, 18 years and older with chronic rhinosinusitis with nasal polyps who were symptomatic despite taking intranasal corticosteroids. Patients who received Dupixent had statistically significant reductions in their nasal polyp size and nasal congestion compared to the placebo group. Patients taking Dupixent also reported an increased ability to smell and required less nasal polyp surgery and oral steroids.
Dupixent may cause serious allergic reactions and eye problems, such as inflammation of the eye (conjunctivitis) and inflammation of the cornea (keratitis). If patients experience new or worsening eye symptoms, such as redness, itching, pain or visual changes, they should consult their health care professional. The most common side effects reported include injection site reactions as well as eye and eyelid inflammation, which included redness, swelling and itching. Patients receiving Dupixent should avoid receiving live vaccines.
Dupixent was originally approved in 2017 for patients 12 and older with eczema that is not controlled adequately by topical therapies or when those therapies are not advisable. In 2018, Dupixent was approved as an add-on maintenance treatment for patients 12 years and older with moderate-to-severe eosinophilic asthma or with oral corticosteroid-dependent asthma.
The FDA granted this application Priority Review. The approval of Dupixent was granted to Regeneron Pharmaceuticals.
///////////Dupixent, dupilumab, fda 2019, nasal polyps, chronic rhinosinusitis, Priority Review, Regeneron Pharmaceuticals,
Octamoxin, октамоксин , أوكتاموكسين , 奥他莫辛 ,
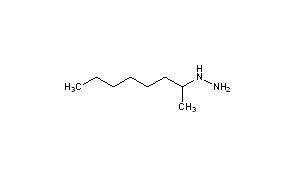
- Molecular FormulaC8H20N2
- Average mass144.258 Da

References
- ^ “Octamoxin – Compound Summary”. USA: National Center for Biotechnology Information. 26 March 2005. Identification and Related Records. Retrieved 31 May 2012.
- ^ “Dictionary of pharmacological agents – Google Books”.
- ^ “13-06781. Octamoxin [Archived]: The Merck Index”.
- ^ Levy J, Michel-Ber E (1966). “[Relations between the antidepressive effects of octamoxine revealed by 3 pharmacological tests and inhibition of cerebral monoamine oxidase in mice]”. Thérapie (in French). 21 (4): 929–45. PMID 5925088.
- ^ Gayral L, Stern H, Puyuelo R (1966). “[Indications and results of the treatment of mental depression by octamoxine (ximaol)]”. Thérapie (in French). 21 (5): 1183–90. PMID 5976767.
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
1-Methylheptylhydrazine[citation needed]
|
|
| Systematic IUPAC name
Octan-2-ylhydrazine[1]
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C8H20N2 | |
| Molar mass | 144.262 g·mol−1 |
| Density | 0.831 g/mL |
| Boiling point | 228 °C (442 °F; 501 K) |
| Pharmacology | |
| Oral | |
| Related compounds | |
|
Related compounds
|
Tuaminoheptane |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
CK-101
![N-[3-[2-[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]anilino]quinazolin-8-yl]phenyl]prop-2-enamide.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=117909640&t=l)

CK-101, RX-518
CAS 1660963-42-7
N-[3-[2-[[2,3-Difluoro-4-[4-(2-hydroxyethyl)piperazin-1-yl]phenyl]amino]quinazolin-8-yl]phenyl]acrylamide
N-(3-(2-((2,3-Difluoro-4-(4-(2-hydroxyethyl)piperazin-1-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide
EGFR-IN-3
UNII-708TLB8J3Y
Suzhou NeuPharma (Originator)
Checkpoint Therapeutics
Non-Small Cell Lung Cancer Therapy
Solid Tumors Therapy
PHASE 2 Checkpoint Therapeutics, Cancer, lung (non-small cell) (NSCLC), solid tumour
RX518(CK-101) is an orally available third-generation and selective inhibitor of certain epidermal growth factor receptor (EGFR) activating mutations, including the resistance mutation T790M, and the L858R and exon 19 deletion (del 19) mutations, with potential antineoplastic activity.
In August 2019, Suzhou Neupharma and its licensee Checkpoint Therapeutics are developing CK-101 (phase II clinical trial), a novel third-generation, covalent, EGFR inhibitor, as a capsule formulation, for the treatment of cancers including NSCLC and other advanced solid tumors. In September 2017, the FDA granted Orphan Drug designation to this compound, for the treatment of EGFR mutation-positive NSCLC; in January 2018, the capsule was being developed as a class 1 chemical drug in China.
CK-101 (RX-518), a small-molecule inhibitor of epidermal growth factor receptor (EGFR), is in early clinical development at Checkpoint Therapeutics and Suzhou NeuPharma for the potential treatment of EGFR-mutated non-small cell lung cancer (NSCLC) and other advanced solid malignancies.
In 2015, Suzhou NeuPharma granted a global development and commercialization license to its EGFR inhibitor program, excluding certain Asian countries, to Coronado Biosciences (now Fortress Biotech). Subsequently, Coronado assigned the newly acquired program to its subsidiary Checkpoint Therapeutics.
In 2017, the product was granted orphan drug designation in the U.S. for the treatment of EGFR mutation-positive NSCLC.
There are at least 400 enzymes identified as protein kinases. These enzymes catalyze the phosphorylation of target protein substrates. The phosphorylation is usually a transfer reaction of a phosphate group from ATP to the protein substrate. The specific structure in the target substrate to which the phosphate is transferred is a tyrosine, serine or threonine residue. Since these amino acid residues are the target structures for the phosphoryl transfer, these protein kinase enzymes are commonly referred to as tyrosine kinases or serine/threonine kinases.
[0003] The phosphorylation reactions, and counteracting phosphatase reactions, at the tyrosine, serine and threonine residues are involved in countless cellular processes that underlie responses to diverse intracellular signals (typically mediated through cellular receptors), regulation of cellular functions, and activation or deactivation of cellular processes. A cascade of protein kinases often participate in intracellular signal transduction and are necessary for the realization of these cellular processes. Because of their ubiquity in these processes, the protein kinases can be found as an integral part of the plasma membrane or as cytoplasmic enzymes or localized in the nucleus, often as components of enzyme complexes. In many instances, these protein kinases are an essential element of enzyme and structural protein complexes that determine where and when a cellular process occurs within a cell.
[0004] The identification of effective small compounds which specifically inhibit signal transduction and cellular proliferation by modulating the activity of tyrosine and serine/threonine kinases to regulate and modulate abnormal or inappropriate cell proliferation, differentiation, or metabolism is therefore desirable. In particular, the identification of compounds that specifically inhibit the function of a kinase which is essential for processes leading to cancer would be beneficial.
[0005] While such compounds are often initially evaluated for their activity when dissolved in solution, solid state characteristics such as polymorphism are also important. Polymorphic forms of a drug substance, such as a kinase inhibitor, can have different physical properties, including melting point, apparent solubility, dissolution rate, optical and mechanical properties, vapor pressure, and density. These properties can have a direct effect on the ability to process or manufacture a drug substance and the drug product. Moreover, differences in these properties
can and often lead to different pharmacokinetics profiles for different polymorphic forms of a drug. Therefore, polymorphism is often an important factor under regulatory review of the ‘sameness’ of drug products from various manufacturers. For example, polymorphism has been evaluated in many multi-million dollar and even multi-billion dollar drugs, such as warfarin sodium, famotidine, and ranitidine. Polymorphism can affect the quality, safety, and/or efficacy of a drug product, such as a kinase inhibitor. Thus, there still remains a need for polymorphs of kinase inhibitors. The present disclosure addresses this need and provides related advantages as well.
PATENT
WO2015027222
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015027222


PATENT
WO-2019157225
Crystalline form II-VIII of the compound presumed to be CK-101 (first disclosed in WO2015027222 ), for treating a disorder mediated by epidermal growth factor receptor (EGFR) eg cancer.
SCHEME A
Scheme B
General Procedures
Example 1: Preparation of the compound of Formula I (N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide)
[0253] To a solution of l,2,3-trifluoro-4-nitrobenzene (2.5 g, 14 mmol, 1.0 eq.) in DMF (20 mL) was added K2C03 (3.8 g, 28 mmol, 2.0 eq.) followed by 2-(piperazin-l-yl)ethanol (1.8 g, 14 mmol, 1.0 eq.) at 0 °C and the mixture was stirred at r.t. overnight. The mixture was poured into ice-water (200 mL), filtered and dried in vacuo to afford 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 67.5%).
[0254] To a solution of 2-(4-(2,3-difluoro-4-nitrophenyl)piperazin-l-yl)ethanol (2.7 g, 9.0 mmol) in MeOH (30 mL) was added Pd/C (270 mg) and the resulting mixture was stirred at r.t.
overnight. The Pd/C was removed by filtration and the filtrate was concentrated to afford 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (2.39 g, 99% yield) as off-white solid.
[0255] To a solution of 8-bromo-2-chloroquinazoline (15.4 g, 63.6 mmol, 1 eq. ) and (3-aminophenyl)boronic acid (8.7 g, 63.6 mmol, 1 eq.) in dioxane/H20 (200 mL/20 mL) was added Na2C03 (13.5 g, 127.2 mmol, 2 eq.), followed by Pd(dppf)Cl2 (2.6 g, 3.2 mmol, 0.05 eq.) under N2, then the mixture was stirred at 80 °C for 12 h. Then the solution was cooled to r.t.,
concentrated and the residue was purified via column chromatography (PE/EA=3 :2, v/v) to afford 3-(2-chloroquinazolin-8-yl)aniline as yellow solid (8.7 g, 53.7% yield).
[0256] To a solution of 3-(2-chloroquinazolin-8-yl)aniline (8.7 g, 34 mmol, 1 eq.) in DCM ( 200 mL ) cooled in ice-bath was added TEA (9.5 mL, 68 mmol, 2 eq. ), followed by acryloyl chloride (4.1 mL, 51 mmol, 1.5 eq.) dropwise. The resulting mixture was stirred at r.t. for 1 h, then washed with brine, dried over anhydrous N2S04 concentrated and the residue was purified via column chromatography (PE/EA=l : 1, v:v) to afford N-(3-(2-chloroquinazolin-8-yl)phenyl)acryl amide as yellow solid(6.6 g, 65% yield).
[0257] To a suspension of 2-(4-(4-amino-2,3-difluorophenyl)piperazin-l-yl)ethanol (83 mg,
0.32 mmol, 1 eq.) and N-(3-(2-chloroquinazolin-8-yl)phenyl)acrylamide (100 mg, 0.32 mmol, 1 eq.) in n-BuOH (5 mL) was added TFA (68 mg, 0.64 mmol, 2 eq.) and the resulting mixture was stirred at 90 °C overnight. The mixture was concentrated, diluted with DCM (20 mL) , washed with Na2C03 solution (20 mL), dried over anhydrous Na2S04, concentrated and the residue was purified via column chromatography (MeOH/DCM=l/30, v:v) to afford N-(3-(2-((2,3-difluoro-4-(4-(2-hydroxyethyl)piperazin-l-yl)phenyl)amino)quinazolin-8-yl)phenyl)acrylamide as a yellow solid(l6.3 mg, 9.5% yield). LRMS (M+H+) m/z calculated 531.2, found 531.2. 1H NMR
(CD3OD, 400 MHz) d 9.21 (s, 1 H), 7.19-8.01 (m, 10 H), 8.90 (s, 1 H), 6.41-6.49 (m, 3 H), 5.86 (m, 1 H), 3.98-4.01 (m, 3 H), 3.70-3.76 (m, 3 H), 3.40-3.49 (m, 2 H), 3.37-3.39 (m, 4 H), 3.18 (m, 2H).
Example 2. Preparation of Form I of the compound of Formula I
[0258] Crude compound of Formula I (~30 g, 75% of weight based assay) was dissolved in ethyl acetate (3 L) at 55-65 °C under nitrogen. The resulting solution was filtered via silica gel pad and washed with ethyl acetate (3 L><2) at 55-65 °C. The filtrate was concentrated via vacuum at 30-40 °C to ~2.4 L. The mixture was heated up to 75-85 °C and maintained about 1 hour.
Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and the mixture was then cooled down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with ethyl acetate (60 mL><2). The wet cake was dried via vacuum at 30-40 °C to get (about 16 g) of the purified Form I of the compound of Formula I.
Example 3. Preparation of Form III of the compound of Formula I
[0259] The compound of Formula I (2 g) was dissolved in EtOH (40 mL) at 75-85 °C under nitrogen. n-Heptane (40 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 75-85 °C for 1 hour. Then cooled down to 50-60 °C and maintained about 2 hours. The heat-cooling operation was repeated again and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. The resulting mixture was filtered and washed with EtOH/n-Heptane (1/1, 5 mL><2). The wet cake was dried via vacuum at 30-40 °C to get the purified Form III of the compound of Formula I (1.7 g).
Example 4. Preparation of Form IV of the compound of Formula I The crude compound of Formula I (15 g) was dissolved in ethyl acetate (600 mL) at 75-85 °C under nitrogen and treated with anhydrous Na2S04, activated carbon, silica metal scavenger for 1 hour. The resulting mixture was filtered via neutral Al203 and washed with ethyl acetate (300 mL><2) at 75-85 °C. The filtrate was concentrated under vacuum at 30-40 °C and swapped with DCM (150 mL). n-Heptane (75 mL) was added into this DCM solution at 35-45 °C, and then the mixture was cooled down to 20-30 °C slowly. The resulting mixture was filtered and washed with DCM/n-Heptane (2/1, 10 mL><3). The wet cake was dried via vacuum at 35-40 °C to get the purified Form IV of the compound of Formula I (9.6 g).
Example 5. Preparation of Form V of the compound of Formula I
[0260] Polymorph Form III of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form V.
Example 6. Preparation of Form VI of the compound of Formula I
[0261] The compound of Formula I (1 g) was dissolved in IPA (20 mL) at 75-85 °C under nitrogen. n-Heptane (20 mL) was added dropwise into reaction at 75-85 °C. The mixture was stirred at 45-55 °C for 16 hours. Then heated up to 75-85 °C and maintained about 0.5 hour.
Then cooled down to 45-55 °C for 0.5 hour and continued to cool the mixture down to 20-30 °C and stirred for 3 hours. Filtered and washed with IPA/n-Heptane (1/1, 3 mL><2). The wet cake was dried via vacuum at 75-80 °C for 2 hours to get the purified Form VI of the compound of Formula I.
Example 7. Preparation of Form VIII of the compound of Formula I
[0262] The polymorph Form VI of the compound of Formula I was dried in oven at 80 °C for 2 days to obtain the polymorph Form VIII.
Example 8. X-ray powder diffraction (XRD)
[0263] X-ray powder diffraction (XRD) patterns were obtained on a Bruker D8 Advance. A CuK source (=1.54056 angstrom) operating minimally at 40 kV and 40 mA scans each sample between 4 and 40 degrees 2-theta. The step size is 0.05°C and scan speed is 0.5 second per step.
Example 9. Thermogravimetric Analyses (TGA)
[0264] Thermogravimetric analyses were carried out on a TA Instrument TGA unit (Model TGA 500). Samples were heated in platinum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 60mL/min (sample purge) and 40mL/min (balance purge). The TGA temperature was calibrated with nickel standard, MP=354.4 °C. The weight calibration was performed with manufacturer-supplied standards and verified against sodium citrate dihydrate desolvation.
Example 10. Differential scanning calorimetry (DSC)
[0265] Differential scanning calorimetry analyses were carried out on a TA Instrument DSC unit (Model DSC 1000 or 2000). Samples were heated in non-hermetic aluminum pans from ambient to 300 °C at 10 °C/min with a nitrogen purge of 50mL/min. The DSC temperature was calibrated with indium standard, onset of l56-l58°C, enthalpy of 25-29J/g.
Example 11. Hygroscopicity (DVS)
[0266] The moisture sorption profile was generated at 25°C using a DVS Moisture Balance Flow System (Model Advantage) with the following conditions: sample size approximately 5 to 10 mg, drying 25°C for 60 minutes, adsorption range 0% to 95% RH, desorption range 95% to 0% RH, and step interval 5%. The equilibrium criterion was <0.01% weight change in 5 minutes for a maximum of 120 minutes.
Example 12: Microscopy
[0267] Microscopy was performed using a Leica DMLP polarized light microscope equipped with 2.5X, 10X and 20X objectives and a digital camera to capture images showing particle shape, size, and crystallinity. Crossed polars were used to show birefringence and crystal habit for the samples dispersed in immersion oil.
Example 13: HPLC
[0256] HPLCs were preformed using the following instrument and/or conditions.
///////////////CK-101 , CK 101 , CK101 , phase II , Suzhou Neupharma, Checkpoint Therapeutics , Orphan Drug designation, EGFR mutation-positive NSCLC, NSCLC, CANCER, SOLID TUMOUR, China, RX-518, AK543910
OCCN1CCN(CC1)c5ccc(Nc2nc3c(cccc3cn2)c4cccc(NC(=O)C=C)c4)c(F)c5F
Labetalol Hydrochloride, ラベタロール ,

Labetalol
ラベタロール;
- Molecular FormulaC19H24N2O3
- Average mass328.405 Da
Labetalol hydrochloride, AH-5158A, Sch-15719W, Amipress, Trandate, Normodyne
Labetalol was granted FDA approval on 1 August 1984
Presolol; (RS)-2-Hydroxy-5-{1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl}benzamide; 5-[1-Hydroxy-2-[(1-methyl-3-phenyl propyl)amino]ethyl]salicylamide
A salicylamide derivative that is a non-cardioselective blocker of BETA-ADRENERGIC RECEPTORS and ALPHA-1 ADRENERGIC RECEPTORS.
- AH 5158
- Albetol
- EC 253-258-3
- EINECS 253-258-3
- HSDB 6537
- Ibidomide
- Labetalol
- Labetalolum
- Labetalolum [INN-Latin]
- Labetolol
- SCH 15719W
- UNII-R5H8897N95
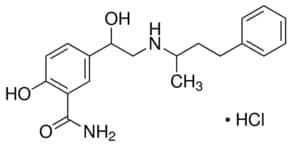
Labetalol hydrochloride
- CAS Number 32780-64-6,
- Empirical Formula (Hill Notation) C19H24N2O3 · HCl,
- Molecular Weight 364.87
REF https://www.accessdata.fda.gov/drugsatfda_docs/anda/98/74787_Labetalol%20Hydrochloride_Chemr.pdf

RR
CAS 75659-07-3
- (R,R)-Labetalol
- Dilevalol
- Dilevalolum
- Dilevalolum [Latin]
- UNII-P6629XE33T
Labetalol is a racemic mixture of 2 diastereoisomers where dilevalol, the R,R’ stereoisomer, makes up 25% of the mixture.8 Labetalol is formulated as an injection or tablets to treat hypertension
Labetalol is a medication used to treat high blood pressure and in long term management of angina.[1][2] This includes essential hypertension, hypertensive emergencies, and hypertension of pregnancy.[2] In essential hypertension it is generally less preferred than a number of other blood pressure medications.[1] It can be given by mouth or by injection into a vein.[1]
Common side effects include low blood pressure with standing, dizziness, feeling tired, and nausea.[1] Serious side effects may include low blood pressure, liver problems, heart failure, and bronchospasm.[1] Use appears safe in the latter part of pregnancy and it is not expected to cause problems during breastfeeding.[2][3] It works by blocking the activation of β-receptors and α-receptors.[1]
Labetalol was patented in 1966 and came into medical use in 1977.[4] It is available as a generic medication.[2] A month supply in the United Kingdom costs the NHS about 8 £ as of 2019.[2] In the United States the wholesale cost of this amount is about US$12.[5] In 2016 it was the 233rd most prescribed medication in the United States with more than 2
Medical uses
Labetalol is effective in the management of hypertensive emergencies, postoperative hypertension, pheochromocytoma-associated hypertension, and rebound hypertension from beta blocker withdrawal. [7]
It has a particular indication in the treatment of pregnancy-induced hypertension which is commonly associated with pre-eclampsia. [8]
It is also used as an alternative in the treatment of severe hypertension.[7]
Special populations
Pregnancy: studies in lab animals showed no harm to the baby. However, a comparable well-controlled study has not been performed in pregnant women.[9]
Nursing: breast milk has been shown to contain small amounts of labetalol (0.004% original dose). Prescribers should be cautious in the use of labetalol for nursing mothers.[9]
Pediatric: no studies have established safety or usefulness in this population.[9]
Geriatric: the elderly are more likely to experience dizziness when taking labetalol. Labetalol should be dosed with caution in the elderly and counseled on this side effect.[9]
Side effects
Common
- Neurologic: headache (2%), dizziness (11%) [9]
- Gastrointestinal: nausea (6%), dyspepsia (3%) [9]
- Cholinergic: nasal congestion (3%), ejaculation failure (2%) [9]
- Respiratory: dyspnea (2%) [9]
- Other: fatigue (5%), vertigo (2%), orthostatic hypotension [9]
Low blood pressure with standing is more severe and more common with IV formulation (58% vs 1%[9]) and is often the reason larger doses of the oral formulation cannot be used.[10]
Rare
- Fever [9]
- Muscle cramps [9]
- Dry eyes [9]
- Heart block [9]
- Hyperkalemia [9]
- Hepatotoxicity [9]
- Drug eruption similar to lichen planus[11]
- Hypersensitivity – which may result in a lethal respiratory distress[9]
Contraindications
Labetalol is contraindicated in people with overt cardiac failure, greater-than-first-degree heart block, severe bradycardia, cardiogenic shock, severe hypotension, anyone with a history of obstructive airway disease including asthma, and those with hypersensitivity to the drug.[12]
Chemistry
The minimum requirement for adrenergic agents is a primary or secondary amine separated from a substituted benzene ring by one or two carbons.[13] This configuration results in strong agonist activity. As the size of the substituent attached to the amine becomes greater, particularly with respect to a t-butyl group, then the molecule typically is found to have receptor affinity without intrinsic activity, and is, therefore, an antagonist.[13] Labetalol, with its 1-methyl-3-phenylpropyl substituted amine, is greater in size relative to a t-butyl group and therefore acts predominantly as an antagonist. The overall structure of labetalol is very polar. This was created by substituting the isopropyl group in the standard beta-blocker structure with an aralkyl group, including a carboxamide group on the meta position, and by adding a hydroxyl group on the para position.[14]
Labetalol has two chiral carbons and consequently exists as four stereoisomers.[15] Two of these isomers, the (S,S)- and (R,S)- forms are inactive. The third, the (S,R)-isomer, is a powerful α1 blocker. The fourth isomer, the (R,R)-isomer which is also known as dilevalol, is a mixed nonselective β blocker and selective α1 blocker.[14] Labetalol is typically given as a racemic mixture to achieve both alpha and beta receptor blocking activity.[16]
| Stereoisomers of labetalol | |
|---|---|
-Labetalol_Structural_Formula_V1.svg) (R,R)-Labetalol CAS number: 75659-07-3 |
-Labetalol_Structural_Formula_V1.svg) (S,S)-Labetalol CAS number: 83167-24-2 |
-Labetalol_Structural_Formula_V1.svg) (R,S)-Labetalol CAS number: 83167-32-2 |
-Labetalol_Structural_Formula_V1.svg) (S,R)-Labetalol CAS number: 83167-31-1 |
Labetalol acts by blocking alpha and beta adrenergic receptors, resulting in decreased peripheral vascular resistance without significant alteration of heart rate or cardiac output.
The β:α antagonism of labetalol is approximately 3:1.[17][18]
It is chemically designated in International Union of Pure and Applied Chemistry (IUPAC) nomenclature as 2-hydroxy-5-[1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl]benzamide monohydrochloride.[16][19]
Pharmacology
Mechanism of action
Labetalol’s dual alpha and beta adrenergic antagonism has different physiological effects in short- and long-term situations. In short-term, acute situations, labetalol decreases blood pressure by decreasing systemic vascular resistance with little effect on stroke volume, heart rate and cardiac output.[20] During long-term use, labetalol can reduce heart rate during exercise while maintaining cardiac output by an increase in stroke volume.[21]
Labetalol is a dual alpha (α1) and beta (β1/β2) adrenergic receptor blocker and competes with other Catecholamines for binding to these sites.[22] Its action on these receptors are potent and reversible.[12] Labetalol is highly selective for postsynaptic alpha1- adrenergic, and non-selective for beta-adrenergic receptors. It is about equipotent in blocking both beta1- and beta2- receptors.[14]
The amount of alpha to beta blockade depends on whether labetalol is administered orally or intravenously (IV). Orally, the ratio of alpha to β blockade is 1:3. Intravenously, alpha to β blockade ratio is 1:7.[14][12] Thus, the labetalol can be thought to be a beta-blocker with some alpha-blocking effects.[12][22][23] By comparison, labetalol is a weaker β-blocker than propranolol, and has a weaker affinity for alpha-receptors compared to Phentolamine.[14][22]
Labetalol possesses intrinsic sympathomimetic activity.[23] In particular, it is a partial agonist at beta2- receptors located in the vascular smooth muscle. Labetalol relaxes vascular smooth muscle by a combination of this partial beta2- agonism and through alpha1- blockade.[23][24] Overall, this vasodilatory effect can decrease blood pressure.[25]
Similar to local anesthetics and sodium channel blocking antiarrhythmics, labetalol also has membrane stabilizing activity.[23][26] By decreasing sodium entry, labetalol decreases action potential firing and thus has local anesthetic activity.[27]
Physiological action
The physiological effects of labetalol when administered acutely (intravenously) are not predictable solely by their receptor blocking effect, i.e. blocking beta1- receptors should decrease heart rate, but labetalol does not. When labetalol is given in acute situations, it decreases the peripheral vascular resistance and systemic blood pressure while having little effect on the heart rate, cardiac output and stroke volume, despite its alpha1-, beta1- and beta2- blocking mechanism.[20][21] These effects are mainly seen when the person is in the upright position.[25]
Long term labetalol use also has different effects from other beta-blocking drugs. Other beta-blockers, such as propranolol, persistently reduce cardiac output during exercise. The peripheral vascular resistance decreases when labetalol is first administered. Continuous labetalol use further decreases peripheral vascular resistance. However, during exercise, cardiac output remains the same due to a compensatory mechanism that increases stroke volume. Thus, labetalol is able to reduce heart rate during exercise while maintaining cardiac output by the increase in stroke volume.[21]
Pharmacokinetics
Labetalol, in animal models, was found to cross the blood-brain-barrier in only negligible amounts.[28]
History
Labetalol was the first drug created that combined both alpha- and beta- adrenergic receptor blocking properties. It was created to potentially fix the compensatory reflex issue that occurred when blocking a single receptor subtype, i.e. vasoconstriction after blocking beta-receptors or tachycardia after blocking alpha receptors. Because the reflex from blocking the single receptor subtypes acted to prevent the lowering of blood pressure, it was postulated that weak blocking of both alpha- and beta- receptors could work together to decrease blood pressure.[14][21]
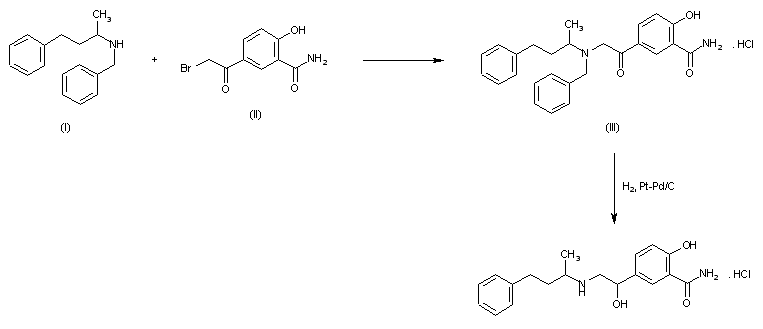
Syn 1
Drugs Fut 1976,1(3),125
DE 1643224; FR 1557677; FR 8010M; GB 1200886; US 3642896; US 3644353; US 3705233
Condensation of 5-bromoacetylsalicylamide (I) with N-benzyl-N-(1-methyl-3-phenylpropyl)amine (II) in refluxing butanone to 5-(N-benzyl-N-(1-methyl-3-phenylpropyl) glycyl)salicylamide hydrochloride (III), m.p. 139-141 C, which is reduced with H2 over Pt-Pd/C in ethanol.
SYN 2
Reductocondensation of 5-(N,N-dibenzylglycyl)salicylamide (IV) and benzylace-tone (V) with H2 over Pd-Pt/C in methanol – acetic acid.
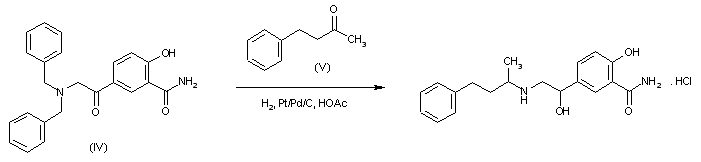
SYN 3
Reaction of methyl 5-(2-amino-1-hydroxyethyl)salicylate hydrochloride (VI) with NH3 to 5-(2-amino-1-hydroxyethyl)salicylamide hydrochloride (VII), m.p. >360 C, which is finally condensed with benzylacetone (V) and reduced with H2 over Pd-Pt/C in methanol.
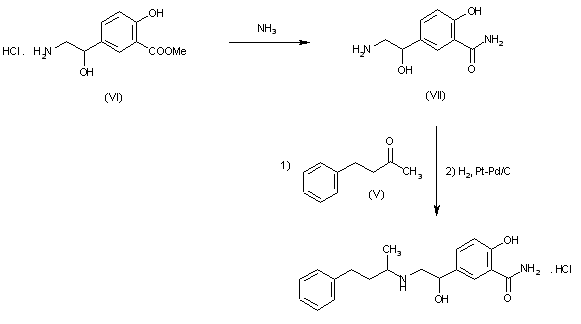
SYN 4

SYN 5
2-hydroxy-5-(1-hydroxy-2-((1-methyl-3-phenylpropyl)amino)ethyl)-, monohydrochloride, could be produced through many synthetic methods.
Following is one of the synthesis routes: 5-Bromoacetylsalicylamide (I) with N-benzyl-N-(1-methyl-3-phenylpropyl)amine (II) is condensed in the presence of refluxing butanone to produce 5-(N-benzyl-N-(1-methyl-3-phenylpropyl) glycyl)salicylamide hydrochloride (III), m.p. 139-141 C, and next the yielding compound is reduced with H2 over Pt-Pd/C in ethanol.

SYN 6
https://patents.google.com/patent/WO2017098520A1/en
aration of Labetaiol Hydrochloride of

Scheme -I illustrates the process for preparation of Labetaiol Hydrochloride of formula (I).

30% NaOH
Step – Sodium borohydride

Pure Labetaiol Hydrochloride (I)
aration of Labetaiol Hydrochloride of
Scheme -I illustrates the process for preparation of Labetaiol Hydrochloride of formula (I).
30% NaOH
Step – Sodium borohydride
Pure Labetaiol Hydrochloride (I)
SYN
https://patents.google.com/patent/EP0009702A1/en
-
The substance labetalol is known from British patent specification 1,266,058 and U.S.P. 4,012,444. Its pharmacological properties are discussed by Farmer et. al. in British Journal of Pharmacology, 45: 660-675 (1972), who designate it AH5158; it is shown to block a- and β-adrenergic receptors, suggesting that it would be useful in the treatment of arrhythmia, hypertension and angina pectoris.
- [0003]
The unique pharmacological properties of labetalol and its use as an antihypertensive agent are said to be largely a function of the exquisite balance of its a- and a-blocking activities. The file history of U.S.P. 4,012,444 indeed indicates that slight changes in the chemical structure of labetalol deleteriously affect this balance, and, even in the few analogous compounds where the balance is retained, the absolute potencies of these compounds are shown to be too low for them to be useful antihypertensive agents. Therefore, in the treatment of hypertension, labetalol is the compound of choice among those disclosed in British patent specification 1,266,058 and U.S.P. 4,012,444.
- [0004]
Labetalol has two asymmetrically substituted carbon atoms and therefore can exist as two diastereoisomers and four optical isomers. Indeed, British patent specification 1,266,058 and U.S.P. 4,012,444 disclose that compounds such as labetalol have optically active forms, but give no example of an optically active form. These patent specifications .teach that “the racemic mixtures may be resolved by conventional methods, for example by salt formation with an optically active acid, followed by fractional crystallization”, but give no method of resolution. Example 14 of each specifi– cation does indeed describe the separation of labetalol into two diastereoisomers “1” and “2”, using benzoic acid, but this is not an optical resolution. In British patent specifications 1,541,932 and 1,541,933, “isomer 1” is designated “diastereoisomer A” and is characterised as that diastereoisomer whose hydrochloride salt has the higher melting point. These two British patent specifications also disclose that diastereoisomer A is a valuable antiarrhythmic agent since it has strongly reduced β-adrenergic blocking activity and is therefore useful in the treatment of people who have suffered myocardial infarction.
- [0005]
We have now discovered that diastereoisomer A is composed of the (S,R) and (R,S) optical isomers of labetalol, whereas diastereoisomer B is composed of the (S,S) and (R,R) optical isomers. We have also-surprisingly found that the novel (R,R) optical isomer of labetalol exhibits, in comparison with labetalol itself, both an unexpectedly high increase in β-adrenergic blocking potency and a decrease in a-adrenergic blocking potency. Thus, when the (R,R) optical isomer is compared with labetalol, the ratio of the β-adrenergic blocking potency to the a-adrenergic blocking potency is found to be greatly and unexpectedly increased. In particular, animal tests have indicated that the (R,R) optical isomer has about twelve times the β-blocking potency of labetalol, but only about one third of the a-blocking potency of labetalol. These. properties could in no way have been predicted theoretically, especially as the β-blocking potency of diastereoisomer B is not significantly different from that of labetalol and the a-blocking potency of diastereoisomer B is half that of labetalol. Indeed, it is clear, when the activities of the four optical isomers of labetalol are compared, that the activities of the diastereoisomers A and B and indeed of labetalol itself cannot be calculated from the activities of their components. One can put this the other way around by saying that the α-and β-blocking activities of the four optical isomers of labetalol do not merely average to give the a- and β-blocking activites of labetalol and of its diastereoisomers A and B. Some of the activities are much greater than could ever have been expected on a simple basis of mathematical proportions, in particular the high β-blocking activity of the (R,R) optical isomer: this activity is much higher than the β-blocking activity of diastereoisomer B so that antagonism evidently exists between the (S,S) and (R,R) optical isomers with respect to the β-blocking activity. This degree of antagonism could in no way have been foreseen. In the absence of this antagonism, the (R,R) optical isomer shows a balance of properties that make it the optical isomer of choice in the treatment of hypertension. In particular, the (R,R) optical isomer possesses potent antihypertensive activity and rapid onset of activity while substantially lacking the undesirable side-effects usually associated with a-blockade, e.g. postural hypotension.
-
The following Table shows the relationships between labetalol, its diastereoisomersA and B and the four pure optical isomers; below each compound are given its potencies as an a-blocking and then as a β-blocking agent, all relative to the values for labetalol (assigned values 1.0 for each blocking activity):

This table clearly shows the unexpectedly high β-blocking activity and ratio of β-:α-blocking activities possessed by the (R,R)-optical isomer. Additionally, the (R,R)–optical isomer has been found to possess greater direct peripheral vasodilation activity than labetalol, and this also contributes to its anti-hypertensive activity. Moreover, the (R,R)-optical isomer is substantially non-toxic at therapeutic doses.
- [0007]
According to the invention therefore we provide the (R,R)-optical isomer of labetalol, namely 5- {(R)–1-hydroxy-2-[(R)-(1-methyl-3-phenylpropyl)amino]ethyl} salicylamide, which can be characterised by means of its hydrochloride salt which is dimorphic with m.pts. of about 133-134°C. and about 192-193.5°C. and an [α]D 26 of about -30.6° (conc. 1 mg./ml., ethanol), said (R,R) optical isomer being substantially free of the corresponding (R,S), (S,R) and (S,S) optical isomers
reaction scheme:

- E. (-)-5- { (R)-l-Hydroxy-2-[(R)-(l-methyl-3-phenylpropyl)-amino]ethyl} salicylamide hydrochloride salt (9)
- [0032]
Treat a solution of 3.0 g. (0.0059 mol.) of 2-0-benzyl-5-{(R) -1-hydroxy-2-[(R)-(1-methyl-3-phenylpropyl)benzylamino]ethyl} salicylamide in 30 ml. of ethyl ether with 2N ethereal hydrogen chloride until no further precipitation occurs. Wash the precipitated 2-0-benzyl-5-{(R)-1-hydroxy-2-[(R)-(1-methyl–3-phenylpropyl)benzylamino]ethyl} salicylamide hydrochloride with ether to remove excess hydrogen chloride and dissolve it in 100 ml. ethanol. To the ethanol solution add 300 mg. of a 20% palladium hydroxide on carbon catalyst and hydrogenate (3 atm.; 3.1 kg. cm.-2) in a Paar apparatus with shaking at room temperature for 3 hours. Filter off the catalyst, evaporate, and triturate the solid residue with isopropanol. Dissolve the solid in 11 ml. of 1N sodium hydroxide, adjust the pH to about 8 and precipitate the free base by bubbling in carbon dioxide. Collect the free base, wash it with water and dry it in vacuo at 40°C. Chromatograph the free base on 450 g. of silica gel and dissolve the pure product in 20 ml. of boiling acetonitrile. Cool the solution and carefully acidify with 2N ethereal HC1 to about pH2. Solidify the gum which precipitates by refluxing the mixture for 10 minutes, filter off the solid, wash it with ethyl ether and recrystallize it from ethanol to obtain analytically pure product (9), m.p. 192-193.5°C.(dec.), [α]D26 = -30.6° (c=1.0, ethanol).
Dilevalol
Synonyms:(R,R)-Labetalol
ATC:C02CB
- Use:α- and β-adrenoceptor antagonist, α- and β-blocker, isomer of labetalol, antihypertensive
- Chemical name:[R-(R*,R*)]-2-hydroxy-5-[1-hydroxy-2-[(1-methyl-3-phenylpropyl)amino]ethyl]benzamide
- Formula:C19H24N2O3
- MW:328.41 g/mol
- CAS-RN:75659-07-3
- LD50:1719 mg/kg (M, p.o.);
1228 mg/kg (R, p.o.)
Derivatives
Monohydrochloride
- Formula:C19H24N2O3 • HCl
- MW:364.87 g/mol
- CAS-RN:75659-08-4
- LD50:1079 mg/kg (M, p.o.);
82 mg/kg (R, i.v.); 1026 mg/kg (R, p.o.)
Synthesis Path
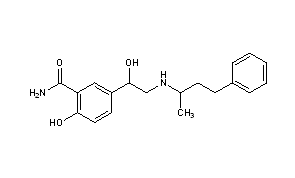
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ləˈbɛtəlɔːl/ |
| Trade names | Normodyne, Trandate, others |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a685034 |
| Pregnancy category |
|
| Routes of administration |
By mouth, intravenous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 25% |
| Protein binding | 50% |
| Metabolism | Liver pass metabolism, |
| Elimination half-life | Tablet: 6-8 hours; IV: 5.5 hours |
| Excretion | Excreted in urine, not removed by hemodialysis |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.048.401 |
| Chemical and physical data | |
| Formula | C19H24N2O3 |
| Molar mass | 328.412 g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
References
- ^ Jump up to:a b c d e f “Labetalol Hydrochloride Monograph for Professionals”. Drugs.com. American Society of Health-System Pharmacists. Retrieved 3 March 2019.
- ^ Jump up to:a b c d e British national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 147–148. ISBN 9780857113382.
- ^ “Labetalol Use During Pregnancy”. Drugs.com. Retrieved 11 March 2019.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 463. ISBN 9783527607495.
- ^ “NADAC as of 2019-02-27”. Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- ^ “The Top 300 of 2019”. clincalc.com. Retrieved 22 December 2018.
- ^ Jump up to:a b Koda-Kimble, Mary A.; Alldredge, Brian K. (2013). “21”. Koda-Kimble and Young’s Applied Therapeutic: The Clinical Use of Drugs. Philadelphia: Philadelphia: Lippincott Williams & Wilkins. ISBN 978-1-60913-713-7.
- ^ Arulkumaran, N; Lightstone, L (December 2013). “Severe pre-eclampsia and hypertensive crises”. Best Practice & Research. Clinical Obstetrics & Gynaecology. 27 (6): 877–84. doi:10.1016/j.bpobgyn.2013.07.003. PMID 23962474.
- ^ Jump up to:a b c d e f g h i j k l m n o p q “Trandate” (PDF). Prometheus Laboratories Inc. November 2010. Retrieved 3 November 2015.
- ^ “Labetalol hydrochloride” (PDF). Hospira. May 2015. Retrieved 3 November 2015.
- ^ Shiohara T, Kano Y (2007). “Lichen planus and lichenoid dermatoses”. In Bolognia JL (ed.). Dermatology. St. Louis: Mosby. p. 161. ISBN 978-1-4160-2999-1.
- ^ Jump up to:a b c d “Labetalol [package insert]. Spring Valley, NY: Par Pharmaceutical; 2011” (PDF). Retrieved 2015-11-03.
- ^ Jump up to:a b Medicinal Chemistry of Adrenergics and Cholinergics
- ^ Jump up to:a b c d e f Louis, W.J.; McNeill, JJ; Drummer, OH (1988). Doyle, AE (ed.). Labetalol and other vasodilator/Beta-blocking drugs. IN: Handbook of Hypertension. Amsterdam, Netherlands: Elsevier Sciences Publishing Co. pp. 246–273. ISBN 978-0-444-90469-0.
- ^ Riva E, Mennini T, Latini R (December 1991). “The alpha- and beta-adrenoceptor blocking activities of labetalol and its RR-SR (50:50) stereoisomers”. Br. J. Pharmacol. 104 (4): 823–8. doi:10.1111/j.1476-5381.1991.tb12513.x. PMC 1908821. PMID 1687367.
- ^ Jump up to:a b Robertson D, Biaggioni, I. Adrenoceptor Antagonist Drugs. In: Katzung BG, Masters SB, Trevor AJ, eds. Basic & Clinical Pharmacology. 12th ed. San Francisco, CA: McGraw Hill Lange Medical; 2012: 151-168. ISBN 978-0-07-176401-8.
- ^ Katzung, Bertram G. (2006). Basic and clinical pharmacology. New York: McGraw-Hill Medical. p. 170. ISBN 978-0-07-145153-6.
- ^ D A Richards; J Tuckman; B N Prichard (October 1976). “Assessment of alpha- and beta-adrenoceptor blocking actions of labetalol”. Br J Clin Pharmacol. 3 (5): 849–855. doi:10.1111/j.1365-2125.1976.tb00637.x. PMC 1428931. PMID 9968.
- ^ “labetalol | C19H24N2O3 – PubChem”. pubchem.ncbi.nlm.nih.gov. Retrieved 2015-11-04.
- ^ Jump up to:a b MacCarthy, E. P.; Bloomfield, S. S. (1983-08-01). “Labetalol: a review of its pharmacology, pharmacokinetics, clinical uses and adverse effects”. Pharmacotherapy. 3(4): 193–219. doi:10.1002/j.1875-9114.1983.tb03252.x. ISSN 0277-0008. PMID 6310529.
- ^ Jump up to:a b c d Louis, W. J.; McNeil, J. J.; Drummer, O. H. (1984-01-01). “Pharmacology of combined alpha-beta-blockade. I”. Drugs. 28 Suppl 2: 16–34. doi:10.2165/00003495-198400282-00003. ISSN 0012-6667. PMID 6151889.
- ^ Jump up to:a b c Robertson, D; Biaggioni, I (2012). Katzung, BG (ed.). Adrenoceptor Antagonist Drugs IN: Basic & Clinical Pharmacology (12 ed.). San Francisco: McGraw Hill Lange Medical. pp. 151–168. ISBN 978-0-07-176401-8.
- ^ Jump up to:a b c d Westfall, David P (2004). Craig, Charles R (ed.). Adrenoreceptor Antagonists IN: Modern Pharmacology with Clinical Applications (6th ed.). Baltimore, MD: Lippincott Williams & Wilkins. pp. 109–117. ISBN 978-0781737623.
- ^ Lund-Johansen, P. (1988-01-01). “Hemodynamic effects of beta-blocking compounds possessing vasodilating activity: a review of labetalol, prizidilol, and dilevalol”. Journal of Cardiovascular Pharmacology. 11 Suppl 2: S12–17. doi:10.1097/00005344-198800000-00004. ISSN 0160-2446. PMID 2464093.
- ^ Jump up to:a b Lund-Johansen, P. (1984-01-01). “Pharmacology of combined alpha-beta-blockade. II. Haemodynamic effects of labetalol”. Drugs. 28 Suppl 2: 35–50. doi:10.2165/00003495-198400282-00004. ISSN 0012-6667. PMID 6151890.
- ^ Mottram, Allan R.; Erickson, Timothy B. (2009). Field, John (ed.). Toxicology in Emergency Cardiovascular Care IN: The Textbook of Emergency Cardiovascular Care and CPR. Philadelphia, PA: Lippincott WIlliams & Wilkins. pp. 443–452. ISBN 978-0-7817-8899-1.
- ^ Exam Zone (1 January 2009). Elsevier Comprehensive Guide. Elsevier India. pp. 449–. ISBN 978-81-312-1620-0.
- ^ Detlev Ganten; Patrick J. Mulrow (6 December 2012). Pharmacology of Antihypertensive Therapeutics. Springer Science & Business Media. pp. 147–. ISBN 978-3-642-74209-5.
External links
References
-
- EP 9 702 (Schering Corp.; appl. 17.9.1979; USA-prior. 20.9.1978).
-
Improvement of diastereomer separation:
- DOS 2 616 403 (Scherico; appl. 14.4.1976; USA-prior. 17.4.1975).
- US 4 173 583 (Schering Corp.; 6.11.1979; appl. 21.9.1978; prior. 17.4.1975).
-
Synthesis without chromatographic purification:
- EP 92 787 (Schering Corp.; appl. 20.4.1983; USA-prior. 26.4.1982).
-
Chiral reduction of IV:
- Clifton, J.E. et al.: J. Med. Chem. (JMCMAR) 25, 670 (1982).
- Gold, E.H. et al.: J. Med. Chem. (JMCMAR) 25, 1363 (1982).
- EP 382 157 (Schering Corp.; appl. 6.2.1990; USA-prior. 10.2.1989, 26.9.1989).
- US 4 948 732 (Schering Corp.; 14.8.1990; prior. 26.9.1989, 10.2.1989).
FDA approves new add-on drug Nourianz (istradefylline) to treat off episodes in adults with Parkinson’s disease
READ AT https://newdrugapprovals.org/2016/04/25/istradefylline/
FDA approves new add-on drug Nourianz (istradefylline) to treat off episodes in adults with Parkinson’s disease
The U.S. Food and Drug Administration today approved Nourianz (istradefylline) tablets as an add-on treatment to levodopa/carbidopa in adult patients with Parkinson’s disease (PD) experiencing “off” episodes. An “off” episode is a time when a patient’s medications are not working well, causing an increase in PD symptoms, such as tremor and difficulty walking.
“Parkinson’s disease is a debilitating condition that profoundly impacts patients’ lives,” said Eric Bastings, M.D., acting director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to helping make additional treatments for Parkinson’s disease available to patients.”
According to the National Institutes of Health, PD is the second-most common neurodegenerative disorder in the U.S. after Alzheimer’s disease. An estimated 50,000 Americans are diagnosed with PD each year, and about one million Americans have the condition. The neurological disorder typically occurs in people over age 60, although it can occur earlier. It happens when cells in the brain, which produce a chemical called dopamine, become impaired or die. Dopamine helps transmit signals between the areas of the brain that produce smooth, purposeful movements – such as eating, writing, and shaving. Early symptoms of the disease are subtle and typically worsen gradually; however, the disease progresses more quickly in some people than in others.
The effectiveness of Nourianz in treating “off” episodes in patients with PD who are already being treated with levodopa/carbidopa was shown in four 12-week placebo-controlled clinical studies that included a total of 1,143 participants. In all four studies, patients treated with Nourianz experienced a statistically significant decrease from baseline in daily “off” time compared to patients receiving a placebo.
The most common adverse reactions observed in patients taking Nourianz were involuntary muscle movement (dyskinesia), dizziness, constipation, nausea, hallucination and sleeplessness (insomnia). Patients should be monitored for development of dyskinesia or exacerbation of existing dyskinesia. If hallucinations, psychotic behavior, or impulsive/compulsive behavior occurs, a dosage reduction or stoppage of Nourianz should be considered. Use of Nourianz during pregnancy is not recommended. Women of childbearing potential should be advised to use contraception during treatment.
The FDA granted approval of Nourianz to Kyowa Kirin, Inc.
////// Nourianz, istradefylline, Kyowa Kirin, FDA 2019, Parkinson’s disease
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
Today, the U.S. Food and Drug Administration approved Inrebic (fedratinib) capsules to treat adult patients with certain types of myelofibrosis.
“Prior to today, there was one FDA-approved drug to treat patients with myelofibrosis, a rare bone marrow disorder. Our approval today provides another option for patients,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The FDA is committed to encouraging the development of treatments for patients with rare diseases and providing alternative options, as not all patients respond in the same way.”
Myelofibrosis is a chronic disorder where scar tissue forms in the bone marrow and the production of the blood cells moves from the bone marrow to the spleen and liver, causing organ enlargement. It can cause extreme fatigue, shortness of breath, pain below the ribs, fever, night sweats, itching and bone pain. When myelofibrosis occurs on its own, it is called primary myelofibrosis. Secondary myelofibrosis occurs when there is excessive red blood cell production (polycythemia vera) or excessive platelet production (essential thrombocythemia) that evolves into myelofibrosis.
Jakafi (ruxolitinib) was approved by the FDA in 2011. The approval of Inrebic for intermediate-2 or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) myelofibrosis was based on the results of a clinical trial where 289 patients with myelofibrosis were randomized to receive two different doses (400 mg or 500 mg daily by mouth) of fedratinib or placebo. The clinical trial showed that 35 of 96 patients treated with the fedratinib 400 mg daily dose (the dose recommended in the approved label) experienced a significant therapeutic effect (measured by greater than or equal to a 35% reduction from baseline in spleen volume at the end of cycle 6 (week 24) as measured by an MRI or CT scan with a follow-up scan four weeks later). As a result of treatment with Inrebic, 36 patients experienced greater than or equal to a 50% reduction in myelofibrosis-related symptoms, such as night sweats, itching, abdominal discomfort, feeling full sooner than normal, pain under ribs on left side, and bone or muscle pain.
The prescribing information for Inrebic includes a Boxed Warning to advise health care professionals and patients about the risk of serious and fatal encephalopathy (brain damage or malfunction), including Wernicke’s, which is a neurologic emergency related to a deficiency in thiamine. Health care professionals are advised to assess thiamine levels in all patients prior to starting Inrebic, during treatment and as clinically indicated. If encephalopathy is suspected, Inrebic should be immediately discontinued.
Common side effects for patients taking Inrebic are diarrhea, nausea, vomiting, fatigue and muscle spasms. Health care professionals are cautioned that patients may experience severe anemia (low iron levels) and thrombocytopenia (low level of platelets in the blood). Patients should be monitored for gastrointestinal toxicity and for hepatic toxicity (liver damage). The dose should be reduced or stopped if a patient develops severe diarrhea, nausea or vomiting. Treatment with anti-diarrhea medications may be recommended. Patients may develop high levels of amylase and lipase in their blood and should be managed by dose reduction or stopping the mediation. Inrebic must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
The FDA granted this application Priority Review designation. Inrebic also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Inrebic to Impact Biomedicines, Inc., a wholly-owned subsidiary of Celgene Corporation.
LINK
///////Inrebic , fedratinib, FDA 2019, Priority Review , Orphan Drug, Biomedicines, Celgene , bone marrow disorder
FDA approves third oncology drug Rozlytrek (entrectinib) that targets a key genetic driver of cancer, rather than a specific type of tumor
FDA also approves drug for second indication in a type of lung cancer
The U.S. Food and Drug Administration today granted accelerated approval to Rozlytrek (entrectinib), a treatment for adult and adolescent patients whose cancers have the specific genetic defect, NTRK (neurotrophic tyrosine receptor kinase) gene fusion and for whom there are no effective treatments.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” said FDA Acting Commissioner Ned Sharpless, M.D. “Using the FDA’s expedited review pathways, including breakthrough therapy designation and accelerated approval process, we’re supporting this innovation in precision oncology drug development and the evolution of more targeted and effective treatments for cancer patients. We remain committed to encouraging the advancement of more targeted innovations in oncology treatment and across disease types based on our growing understanding of the underlying biology of diseases.”
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than the location in the body where the tumor originated. The approval marks a new paradigm in the development of cancer drugs that are “tissue agnostic.” It follows the policies that the FDA developed in a guidance document released in 2018. The previous tissue agnostic indications approved by the FDA were pembrolizumab for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib for NTRK gene fusion tumors in 2018.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK-fusion-positive tumors by relying on efficacy information obtained primarily in adults. The FDA continues to encourage the inclusion of adolescents in clinical trials. Traditionally, clinical development of new cancer drugs in pediatric populations is not started until development is well underway in adults, and often not until after approval of an adult indication,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
The ability of Rozlytrek to shrink tumors was evaluated in four clinical trials studying 54 adults with NTRK fusion-positive tumors. The proportion of patients with substantial tumor shrinkage (overall response rate) was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for nine months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid and colon/rectum.
Rozlytrek was also approved today for the treatment of adults with non-small cell lung cancer whose tumors are ROS1-positive (mutation of the ROS1 gene) and has spread to other parts of the body (metastatic). Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
Rozlytrek’s common side effects are fatigue, constipation, dysgeusia (distorted sense of taste), edema (swelling), dizziness, diarrhea, nausea, dysesthesia (distorted sense of touch), dyspnea (shortness of breath), myalgia (painful or aching muscles), cognitive impairment (confusion, problems with memory or attention, difficulty speaking, or hallucinations), weight gain, cough, vomiting, fever, arthralgia and vision disorders (blurred vision, sensitivity to light, double vision, worsening of vision, cataracts, or floaters). The most serious side effects of Rozlytrek are congestive heart failure (weakening or damage to the heart muscle), central nervous system effects (cognitive impairment, anxiety, depression including suicidal thinking, dizziness or loss of balance, and change in sleep pattern, including insomnia and excessive sleepiness), skeletal fractures, hepatotoxicity (damage to the liver), hyperuricemia (elevated uric acid), QT prolongation (abnormal heart rhythm) and vision disorders. Health care professionals should inform females of reproductive age and males with a female partner of reproductive potential to use effective contraception during treatment with Rozlytrek. Women who are pregnant or breastfeeding should not take Rozlytrek because it may cause harm to a developing fetus or newborn baby.
Rozlytrek was granted accelerated approval. This approval commits the sponsor to provide additional data to the FDA. Rozlytrek also received Priority Review, Breakthrough Therapy and Orphan Drug designation. The approval of Rozlytrek was granted to Genentech, Inc.
///////////////Rozlytrek, entrectinib, accelerated approval, priority Review, Breakthrough Therapy, Orphan Drug designation, fda 2019, Genentech, cancer
AK 3280
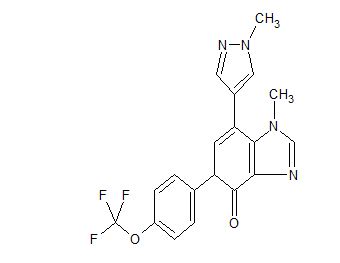
AK-3280
AK 3280; GDC3280; RG 6069
Ci8Hi4N502F3, mass 389.3 g/mol),
ROCHE,
Ark Biosciences , under license from Roche , is developing AK-3280, an antifibrotic agent, for the potential oral treatment of IPF. In July 2018, Ark intended to further clinical development of the drug, for IPF. In June 2019, a phase I trial was planned in Sweden.
- Originator Genentech
- Mechanism of Action Undefined mechanism
- Phase I Interstitial lung diseases
- 19 Jun 2019Ark Biosciences plans a phase I trial for Idiopathic pulmonary fibrosis (In volunteers) in Sweden (PO, Tablet), in August 2019 , (NCT03990688)
- 28 Sep 2018GDC 3280 is still in phase I trials for Interstitial lung diseases (Genentech pipeline, September 2018)
- 28 Jun 2018No recent reports of development identified for phase-I development in Fibrosis(In volunteers) in United Kingdom (PO)
Introduction
GDC 3280 (also known as RG 6069), an orally administered drug, is being developed by Genentech, for the treatment of interstitial lung diseases. Early stage clinical development is underway in the UK.
Company Agreements
In September 2018, Genentech licensed exclusive worldwide development and commercialisation rights of GDC 3280 to Ark Biosciences, for the treatment of idiopathic pulmonary fibrosis
Key Development Milestones
As at September 2018, GDC 3280 is still in phase I development for interstitial lung disease (Genentech pipeline, September 2018).
In December 2015, Genentech completed a phase I trial that evaluated the safety, pharmacokinetics and tolerability of GDC 3280 in healthy volunteers, compared with placebo (GB29751; EudraCT2015-000560-33; NCT02471859). The randomised, double-blind, single and multiple oral dose trial was initiated in June 2015 and enrolled eight volunteers in the UK .
PATENT
WO-2019152863
Novel crystalline salt forms of 1-methyl-7-(1-methyl-lH-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-1,5-dihydro-4H-imidazo[4,5-c]pyridin-4-one (compound I; presumed to be AK-3280 ), processes for their preparation and compositions comprising them are claimed.
Compound I is an orally available small molecule having the structure:
[0004] Compound I has therapeutic value in several different indications that display fibrotic pathophysiology, including idiopathic pulmonary fibrosis (IPF).
[0005] Idiopathic pulmonary fibrosis is a disease of unknown etiology that occurs mainly in middle-aged and elderly patients, which is characterized by progressive fibrosis of the lung, leading to pulmonary insufficiency and death. Because fibrosis has long been considered to be a clinically irreversible process, treatments have traditionally been focused on managing the symptoms and complications, with little hope of significantly slowing progression of the condition. For many years, mainstay treatments have been typically anti inflammatory, immunosuppressive, and anti-oxidant agents. The effectiveness of these therapies in the treatment of IPF and other fibrotic conditions appears to be minimal and variable, and their side effects are often poorly tolerated by patients.
[0006] New treatment options have only recently become available. Both pirfenidone and nintedanib have been approved for use in the treatment of IPF. Current research efforts to develop new anti-fibrotic agents are targeting multiple mechanisms proposed to be linked to the underlying molecular pathogenic processes. This changing landscape has raised hopes and expectations for what might be achievable with new single agents or combination therapies targeting additional pathways.
Preparation of Compound I and its salts
[0045] A synthesis of Compound I and its tosylate salt is shown in the scheme below:
[0046] l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4H-imidazo[4,5-c]pyridin-4-one (5) was synthesized in 4 steps, including a copper-catalyzed coupling reaction e.g., a Goldberg-Ullmann coupling reaction. In another aspect of the invention, intermediate (5) is synthesized using any transition metal-catalyzed coupling reaction. The skilled chemist would know that intermediate (5) could be synthesized from intermediate (4) and compounds
LG
of the general formula: OCF3 , wherein the leaving group“LG” includes but is not limited to halogen, tosylate, mesylate, triflate, etc.
[0047] Compound I was synthesized in 6 steps, using a transition metal cross-coupling reaction, e.g., a Suzuki reaction. In another aspect of the invention, Compound I is synthesized using any cross -coupling reaction. Compound I is synthesized from intermediate 6 containing any leaving group. For example, the skilled chemist would use compounds of
the general formula:
, wherein the leaving group“LG” includes but is not limited to halogen, tosylate, mesylate, triflate, etc.
An alternative synthesis of Compound I and its salts is shown in the scheme below:
Example 13 – Synthesis of Compound I Tosylate Salt
[00183] A process for the formation of mono- and di-tosylate salts of Compound I was developed and a batch was performed to successfully produce the mono-tosylate salt.
Step 1 : Synthesis of2-chloro-N-methyl-3-nitropyridin-4-amine
[00184] A reactor was charged with 2,4-dichloro-3-nitropyridine and 3.0 volumes of DMF. The solution was stirred at 20-25 °C until a clear solution was obtained. The solution was then cooled to 0-5 °C, and 2.1 equivalents of 40% methylamine in water were slowly added over at least 2 hours at 0-5 °C. The reaction mixture was stirred for at least 2 hours at 0-5 °C until conversion to the product was 95% (as measured by HPLC). The reaction mixture was diluted by slowly adding 10 volumes of water over at least 30 minutes at 0-5 °C. The obtained suspension was stirred for at least 60 minutes at 0-5 °C. The precipitate was collected by filtration, and the filter cake was rinsed via the reactor with 10 volumes of water at 0-5 °C. The damp filter cake was then dried in a flow of dry nitrogen to yield 2-chloro-A-methyl-3-nitropyridin-4-amine in 78% yield.
Step 2: Synthesis of 2-chloro-N4 -methylpyridine-3, 4-diamine
[00185] A reactor was charged with catalyst [2% Pt on charcoal, 59 %wt. water] (0.0004 equivalents Pt), damp 2-chloro-/V-methyl-3-nitropyridin-4-amine from step 1 and 9.4 volumes of THF. The solution was stirred, and then the suspension was transferred from the glass-reactor to an autoclave. The line was rinsed with 1.2 volumes of THF into the autoclave, and the autoclave was purged with nitrogen for 15 minutes at 50 rpm, followed by hydrogen for 15 minutes at 150 rpm. The autoclave was closed, and the hydrogen pressure was adjusted to 2 bar at 20-30 °C. The reaction mixture was stirred for 4-8 hours at 2 bar and 20-30 °C.
[00186] Next, the autoclave was released to atmospheric pressure and purged with nitrogen for at least 15 minutes. Conversion to the product was verified by HPLC, and then the catalyst was removed by filtration. The filtered catalyst was rinsed with 1.3 volumes of THF and the filtrates were combined. The combined filtrates were charged to a second reactor via a particle filter, and the line was rinsed with 0.5 volumes of THF. The solution was concentrated to a final volume of 2.5 volumes by distillation under reduced pressure at 40-45 °C.
[00187] The solution was then diluted with 10 volumes of THF in portions while concentrating the solution to a final volume of 2.5 volumes by distillation under reduced pressure at 45-50 °C. The reactor was purged with nitrogen to atmospheric pressure, and 5.0 volumes of heptane were added to the residue at 40-50 °C. The reaction mixture was cooled over 2 hours to 20-25 °C, and stirring was continued for 1 hour. The reaction mixture was then further cooled to 0-5 °C over 1 hour, and stirring was continued for 1 hour. The precipitated product was collected by filtration, rinsed via the reactor with 5.0 volumes of heptane, and the damp filter cake was dried in a vacuum drying oven at max. 40 °C until loss on drying was < 2 % weight, giving 2-chloro-/V4-methylpyridine-3, 4-diamine in 85% yield.
Step 3 : Synthesis of -inelhyl- 1 ,5-dihvdro-4H-iinidazoi4,5-c h yridin-4-one
[00188] A reactor was charged with 2-chloro-/V4-methylpyridine-3, 4-diamine and 4 volumes of formic acid. The reaction mixture was heated to smooth reflux within one hour, and reflux was maintained for 6 hours. The reaction mixture was then cooled to
approximately 60 °C, and conversion to the product was verified by HPLC.
[00189] The reaction mixture was then concentrated by distillation under reduced pressure at 60-80 °C to a final volume of 2 volumes. The temperature of the solution was adjusted to 60 °C, maintaining the temperature above 50 °C to avoid precipitation.
[00190] Next, a second reactor was charged with 10 volumes of acetone, and heated to gentle reflux. The product solution from the first reactor was slowly transferred to the acetone in the second reactor over 20 minutes, and the line was rinsed with approximately 0.05 volumes of formic acid. Reflux of the obtained suspension was maintained for 15 minutes. The slurry was cooled to 0 °C within 1 hour, and stirring was continued for 1 hour at that temperature. The precipitate was collected by filtration, and the filter cake was rinsed via the reactor with 3.7 volumes of cold acetone at 0-10 °C. The filter cake was dried in a flow of dry nitrogen or in a vacuum drying oven at 50 °C until loss on drying was < 2% of weight, giving 1 -methyl- 1 ,5-dihydiO-4/7-imidazo[4,5-c]pyndin-4-onc in 95% yield.
Step 4: Synthesis of l-methyl-5-(4-(trifluoromethoxy)phenyl)-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one
[00191] A first reactor (Reactor A) was charged with 1 -methyl- 1 ,5-dihydro-4/7-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalent), Cu(0Ac)2 H20 (0.1 mol equivalents), and K2C03 (1.1 mol equivalents). The reactor was closed and the atmosphere replaced with nitrogen.
[00192] Next, l-bromo-4-(trifluoromethoxy)benzene (1.5 mol equivalents) and N-methylpyrrolidinone (5.4 volume equivalents) were added, whereupon a suspension was formed. The suspension was stirred until the temperature had fallen again to approximately 20-25 °C and gas evolution had slowed. The reaction mixture was heated to approximately 130-150 °C at which time a blue/green color was observed, changing to dark brown after some time. The reaction was stirred at 130-150 °C for at least 40 hours. Stirring times of 40 hours up to 72 hours were required to reach an acceptable level of conversion. In general, higher reaction temperatures supported faster conversion.
[00193] Next, the reaction mixture was cooled to approximately 20-30 °C, and 25% aqueous NH3 (0.7 volume equivalents) was added, followed by water (3.5 volume equivalents). The resulting suspension was transferred into a second reactor (Reactor B). Additional water was added (18.1 volume equivalents) to the reaction mixture via Reactor A, followed by n-heptane (3.2 volume equivalents). The resulting suspension was cooled to approximately 0-5 °C, and stirred for approximately 2 hours.
[00194] The suspension was filtered, and the filter cake was washed with water (9.7 volume equivalents). The filter cake was then dissolved in dichloromethane (14.1 volume equivalents) and transferred back into reactor B. To this solution was added water (5.7 volume equivalents) via the filter, followed by 25% aq. NH3(1.6 volume equivalents). The mixture was stirred for approximately 1 hour at approximately 15-25 °C.
[00195] Next, the layers were separated, and dichloromethane was added (3.6 volume equivalents) to the aqueous layer. The biphasic mixture was stirred at approximately 15-25 °C for approximately 20-30 minutes. The layers were separated over a period of at least 1 hour, and to the combined organic layers was added a solution of NH4Cl (2.5 mol equivalents) in water (7.0 volume equivalents). The biphasic mixture was stirred at approximately 15-25 °C for about 20-30 minutes, then the layers were separated over the course of 1 hour.
[00196] The lower organic layer was filtered through a particle filter and diluted with toluene (7.1 volume equivalents) via the filter. The organic layer was concentrated under ambient pressure at approximately 80 °C, until no further liquid was seen to evaporate and a precipitate began to form. Toluene was added (16.6 volume equivalents), then concentrated in vacuo, followed by addition of more toluene (7.1 volume equivalents) and again concentrated in vacuo. The suspension was cooled to approximately 0-5 °C, stirred for approximately 2 hours, and filtered. The filter cake was washed with toluene (2.9 volume equivalents), and dried in vacuo at approximately 50 °C until the loss on drying was 0.5% of the weight to give l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one as a beige-colored solid in 83.1% yield.
Step 5 : Synthesis of 7-bromo- 1 -methyl-5-(4-( trifluoromethoxy Iphenyl )- l,5- 4H-
imidaz.o[4,5-clpyridin-4-one
[00197] A first reactor (Reactor A) was charged with water (1.8 volume equivalents) and cooled to approximately 0-5 °C, to which was slowly added 96% sulfuric acid (14 mol. equivalents) at approximately 0-20 °C. The temperature of the solution was adjusted to approximately 0-5 °C, and l -mcthyl-5-(4-(tnfluoromcthoxy)phcnyl)-l ,5-dihydro-4/7-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalent) was added in 3-4 portions at approximately 0-5 °C. The temperature of the mixture was adjusted to approximately 0-5 °C, and N-bromosuccinimide (1.0 mol equivalents) was slowly added in 3-4 portions, while maintaining the temperature at approximately 0-5 °C.
[00198] The reaction mixture was stirred for about 1 hour at approximately 0-5 °C, and then for an additional 4-16 hours at approximately 0-22 °C. Conversion to the product was confirmed by HPLC, then the reaction mixture was cooled to approximately 0-5 °C.
[00199] A second reactor (Reactor B) was charged with water (42.7 volume equivalents) and cooled to approximately 0-5 °C. The reaction mixture from Reactor A was transferred into the pre-cooled water in Reactor B at a temperature below 30 °C over 2 hours. The reaction was rinsed with water (1.6 volume equivalents), and 50% aqueous sodium hydroxide (25 mol. equivalents) was carefully added at approximately 0-30 °C over about 2 hours until the pH reached 2-5.
[00200] Next, MTBE (6.5 volume equivalents) was added at approximately 0-20 °C, and the mixture was stirred for about 5 minutes. Additional 50% aqueous sodium hydroxide (2 mol. equivalents) was added at approximately 0-30 °C until the pH of the solution was in the range of 10-14. The reaction was stirred for at least 1.5 hours at approximately 15-25 °C, and then the layers were allowed to separate over a period of at least 1 hour. The suspension was filtered, taking care to capture the product, which accumulated at the interface of the aqueous and organic layers. The filter cake was washed with MTBE (1.7 volume equivalents), water (3.0 volume equivalents), and then MTBE again (3.0 volume equivalents). The product was dried in vacuo at below 50 °C until the loss on drying was < 1% of the weight, giving 7-bromo-l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one as a pale beige-colored solid in 97.6% yield.
Step 6: Synthesis of 1 -methyl-7 -( 1 -methyl-lH-pyraz.ol-4-yl )-5-(4-( trifluoromethoxy )pheml )-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one (Compound /)
[00201] A reactor was charged with 7-bromo-l-methyl-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one (1.0 mol equivalents), ( 1 -methyl- 1 //-pyrazol-4-yl)boronic acid pinacol ester (l-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-l//-pyrazole, 1.6 mol equivalents), Pd[Ph3]4 (0.025 mol equivalents, and K2C03 (2.0 mol equivalents), to which were added acetonitrile (10.0 volume equivalents) and water (3.0 volume equivalents). The reaction mixture was stirred for approximately 10-20 minutes at about 20-25 °C to form a suspension.
[00202] The mixture was heated to slight reflux, whereupon a biphasic, yellow solution formed. The mixture was stirred at slight reflux for at least 10 hours. The reaction mixture was cooled to between 30-50 °C, then passed through a particle filter. The filter was washed with acetonitrile (2.6 volume equivalents), the filtrates were combined, and the solution was concentrated to a final volume of approximately 120 mL (4.8 volume equivalents) under reduced pressure at below 60 °C.
[00203] To the resulting suspension was added water (1.9 volume equivalents), methanol (26 mL, 1.0 volume equivalents), and dichloromethane (14.8 volume equivalents). The mixture was warmed to about 30-35 °C and stirred until two clear layers were observed. The layers were allowed to separate without stirring at about 30-35 °C, and additional dichloromethane (3.7 volume equivalents) was added to the aqueous layer. The mixture was warmed to approximately 30-35 °C and stirred for about 5 minutes, and then the layers were allowed to separate at approximately 30-35 °C.
[00204] To the combined organic layers was added water (1.9 volume equivalents), and the mixture was warmed to approximately 30-35 °C and stirred for about 5 minutes. The layers were separated at approximately 30-35 °C. Charcoal was added to the combined organic layers and stirred for 30-60 minutes at approximately 30-35 °C. The charcoal was removed by filtration, and the filter was washed with dichloromethane (39 mL, 1.6 volume equivalents).
[00205] The solution was concentrated to approximately 4.0 volume equivalents at ambient pressure and at below 50 °C, then diluted with methanol (5.0 volume equivalents). The solution was again concentrated to approximately 4.0 volume equivalents at ambient pressure and below 60 °C, diluted with methanol (5.0 volume equivalents), and concentrated to a final volume of approximately 3.0 volume equivalents under reduced pressure below 60 °C.
[00206] To the resulting suspension was added methanol (2.9 volume equivalents), and the suspension was warmed to approximately 45-55 °C and stirred for about 1 hour. The suspension was cooled to approximately 0-5 °C within approximately 1 hour, stirred for 1 hour at approximately 0-5 °C, and then filtered. The filter cake was washed with cold methanol (pre-cooled to approximately 0-10 °C, 2.9 volume equivalents), and the product was dried under a stream of nitrogen and in vacuo at below 60 °C until the loss on drying was < 1% by weight, giving Compound I (l-methyl-7-(l-methyl-l -pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one) as a white solid in 88.5% yield.
Step 7: Recrystallization of 1 -methyl-7 -(1 -methyl- lH-pyraz.ol-4-yl)-5-( 4-(trifluoromethoxy)phenyl)-J5-dihvdro-4H-imidaz.o[4,5-c]pyridin-4-one (Compound /)
[00207] A reactor was charged with crude l-methyl-7-(l -methyl- l//-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one from step 6, and to this was added glacial acetic acid (1.5 volume equivalents). The suspension was warmed to approximately 50-60 °C and stirred until a clear solution was obtained, approximately 10-20 minutes. The warm solution was passed through a particle filter into a second reactor.
[00208] To this solution was added ethanol (10.0 volume equivalents) at approximately 45-55 °C over 2 hours. The suspension was stirred for approximately 30 minutes at approximately 45-55 °C, then cooled to approximately 0-5 °C over about 4 hours. The suspension was then stirred for approximately 4-16 hours at about 0-5 °C.
[00209] Next, the suspension was filtered and the filter cake was washed with cold isopropanol (4.2 volume equivalents) at approximately 0-20 °C. The product was dried under a nitrogen stream and in vacuo at below 60 °C until the loss on drying was < 1% by weight, giving Compound I ( 1 – mcthyl-7-( 1 -methyl- 1 /7-pyrazol-4-yl)-5-(4-(tnfluoromcthoxy)phcnyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one) as a white solid in 93.0% yield.
Step 8 : Synthesis of 1 -methyl-7 -( 1 -methyl- 1 H-pyrazol-4-yl )-5-(4-( trifluoromethoxy )phenyl )- 1 ,5-dihvdro-4H-imidaz.oi 4,5-clpyridin-4-one, mono – mono -tosylate
salt)
[00210] A reactor was charged with Compound I ( 1 -mcthyl-7-( 1 -methyl- 1 /7-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-4//-imidazo[4,5-c]pyridin-4-one, 1.00 mol equivalent), para-toluenesulfonic acid monohydrate (1.05 mol equivalents), acetone (6.75 volume equivalents), and water (0.75 volume equivalents). The mixture was stirred at 15-25 °C until a clear solution formed, and then this solution was filtered through a particle filter into a second reactor.
[00211] The filter was washed with acetone (2.5 volume equivalents), and to the combined filtrates was added MTBE (7.5 volume equivalents) at 15-25 °C and Compound I mono-tosylate seeding crystals (0.001 mol equivalents).
[00212] The resulting suspension was stirred at 15-25 °C for approximately 30-60 minutes, and MTBE was added (22.5 volume equivalents) at 15-25 °C during a period of
approximately 30 minutes. Stirring was continued at 15-25 °C for approximately 30-60 minutes, and then the suspension was filtered. The filter was washed with MTBE (2.5 volume equivalents), and the material was dried in vacuo at below 55 °C to give Compound I mono-tosylate salt (l-methyl-7-(l-methyl-l//-pyrazol-4-yl)-5-(4-(trifluoromethoxy)phenyl)-l,5-dihydro-47/-imidazo[4,5-c]pyridin-4-one, mono-tosylate salt) as a white, crystalline solid in 93% yield.
PATENT
WO2018102323 ,
claiming use of a specific compound, orally administered, in combination with food (eg low, medium or high fat meal) for treating fibrotic, inflammatory or autoimmune disorders eg idiopathic pulmonary fibrosis IPF, assigned to Genentech Inc ,
References
-
Roche licenses IPF candidate to Ark Biosciences. Internet-Doc 2019;.
Available from: URL: https://scrip.pharmaintelligence.informa.com/deals/201820364
-
Roche Q3 2018. Internet-Doc 2018;.
Available from: URL: https://www.roche.com/dam/jcr:f9cad8fc-8655-4692-9a85-efbe1cf7a59b/en/irp181017.pdf
-
A Phase 1, Randomized, Double-Blind, Placebo-Controlled, Ascending, Single- and Multiple-Oral-Dose, Safety, Tolerability, and Pharmacokinetic Study of GDC-3280 in Healthy Subjects
// AK-3280, AK 3280, AK3280, GDC 3280, RG 6069, PHASE 1, Idiopathic pulmonary fibrosis
Novobiocin, ノボビオシン;

Novobiocin
ノボビオシン;
- Molecular FormulaC31H36N2O11
- Average mass612.624 Da
Monoisotopic: 612.231910004
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Novobiocin sodium | Q9S9NQ5YIY | 1476-53-5 | WWPRGAYLRGSOSU-RNROJPEYSA-M |
Reata Pharmaceuticals Inc
Abgentis is investigating a novobiocin analog, GYR-12 (discovery), as a re-engineered, previously-marketed-but-uncompetitive (undisclosed) antibacterial compound inhibiting ATPase activity of DNA supercoiling GyrB/ParE, for the potential broad-spectrum treatment of bacterial infections, including multi-drug resistant Gram-negative infections. In April 2017, development was underway [1924695].
Novobiocin, also known as albamycin or cathomycin, is an aminocoumarin antibiotic that is produced by the actinomycete Streptomyces niveus, which has recently been identified as a subjective synonym for S. spheroides[1] a member of the order Actinobacteria. Other aminocoumarin antibiotics include clorobiocin and coumermycin A1.[2] Novobiocin was first reported in the mid-1950s (then called streptonivicin).[3][4]
It is active against Staphylococcus epidermidis and may be used to differentiate it from the other coagulase-negative Staphylococcus saprophyticus, which is resistant to novobiocin, in culture.
Novobiocin was licensed for clinical use under the tradename Albamycin (Pharmacia And Upjohn) in the 1960s. Its efficacy has been demonstrated in preclinical and clinical trials.[5][6] The oral form of the drug has since been withdrawn from the market due to lack of efficacy.[7] Novobiocin is an effective antistaphylococcal agent used in the treatment of MRSA.[8]
Mechanism of action
The molecular basis of action of novobiocin, and other related drugs clorobiocin and coumermycin A1 has been examined.[2][9][10][11][12] Aminocoumarins are very potent inhibitors of bacterial DNA gyrase and work by targeting the GyrB subunit of the enzyme involved in energy transduction. Novobiocin as well as the other aminocoumarin antibiotics act as competitive inhibitors of the ATPase reaction catalysed by GyrB. The potency of novobiocin is considerably higher than that of the fluoroquinolones that also target DNA gyrase, but at a different site on the enzyme. The GyrA subunit is involved in the DNA nicking and ligation activity.
Novobiocin has been shown to weakly inhibit the C-terminus of the eukaryotic Hsp90 protein (high micromolar IC50). Modification of the novobiocin scaffold has led to more selective Hsp90 inhibitors.[13] Novobiocin has also been shown to bind and activate the Gram-negative lipopolysaccharide transporter LptBFGC.[14][15]
Structure
Novobiocin is an aminocoumarin. Novobiocin may be divided up into three entities; a benzoic acid derivative, a coumarin residue, and the sugar novobiose.[9] X-ray crystallographic studies have found that the drug-receptor complex of Novobiocin and DNA Gyrase shows that ATP and Novobiocin have overlapping binding sites on the gyrase molecule.[16] The overlap of the coumarin and ATP-binding sites is consistent with aminocoumarins being competitive inhibitors of the ATPase activity.[17]
Structure–activity relationship
In structure activity relationship experiments it was found that removal of the carbamoyl group located on the novobiose sugar lead to a dramatic decrease in inhibitory activity of novobiocin.[17]
Biosynthesis
This aminocoumarin antibiotic consists of three major substituents. The 3-dimethylallyl-4-hydroxybenzoic acid moiety, known as ring A, is derived from prephenate and dimethylallyl pyrophosphate. The aminocoumarin moiety, known as ring B, is derived from L-tyrosine. The final component of novobiocin is the sugar derivative L-noviose, known as ring C, which is derived from glucose-1-phosphate. The biosynthetic gene cluster for novobiocin was identified by Heide and coworkers in 1999 (published 2000) from Streptomyces spheroidesNCIB 11891.[18] They identified 23 putative open reading frames (ORFs) and more than 11 other ORFs that may play a role in novobiocin biosynthesis.
The biosynthesis of ring A (see Fig. 1) begins with prephenate which is a derived from the shikimic acid biosynthetic pathway. The enzyme NovF catalyzes the decarboxylation of prephenate while simultaneously reducing nicotinamide adenine dinucleotide phosphate (NADP+) to produce NADPH. Following this NovQ catalyzes the electrophilic substitution of the phenyl ring with dimethylallyl pyrophosphate (DMAPP) otherwise known as prenylation.[19] DMAPP can come from either the mevalonic acid pathway or the deoxyxylulose biosynthetic pathway. Next the 3-dimethylallyl-4-hydroxybenzoate molecule is subjected to two oxidative decarboxylations by NovR and molecular oxygen.[20] NovR is a non-heme iron oxygenase with a unique bifunctional catalysis. In the first stage both oxygens are incorporated from the molecular oxygen while in the second step only one is incorporated as determined by isotope labeling studies. This completes the formation of ring A.

Figure 1. Biosynthetic scheme of benzamide portion of novobiocin (4-hydroxy-3-(3-methylbut-2-en-1-yl)benzoic acid)
The biosynthesis of ring B (see Fig. 2) begins with the natural amino acid L-tyrosine. This is then adenylated and thioesterified onto the peptidyl carrier protein (PCP) of NovH by ATPand NovH itself.[21] NovI then further modifies this PCP bound molecule by oxidizing the β-position using NADPH and molecular oxygen. NovJ and NovK form a heterodimer of J2K2 which is the active form of this benzylic oxygenase.[22] This process uses NADP+ as a hydride acceptor in the oxidation of the β-alcohol. This ketone will prefer to exist in its enol tautomer in solution. Next a still unidentified protein catalyzes the selective oxidation of the benzene (as shown in Fig. 2). Upon oxidation this intermediate will spontaneously lactonize to form the aromatic ring B and lose NovH in the process.

Figure 2. Biosynthesis of 3-amino-4,7-dihydroxy-2H-chromen-2-one component of novobiocin (ring B)
The biosynthesis of L-noviose (ring C) is shown in Fig. 3. This process starts from glucose-1-phosphate where NovV takes dTTP and replaces the phosphate group with a dTDP group. NovT then oxidizes the 4-hydroxy group using NAD+. NovT also accomplishes a dehydroxylation of the 6 position of the sugar. NovW then epimerizes the 3 position of the sugar.[23] The methylation of the 5 position is accomplished by NovU and S-adenosyl methionine (SAM). Finally NovS reduces the 4 position again to achieve epimerization of that position from the starting glucose-1-phosphate using NADH.

Figure 3. Biosynthesis of L-noviose component of novobiocin (ring C)
Rings A, B, and C are coupled together and modified to give the finished novobiocin molecule. Rings A and B are coupled together by the enzyme NovL using ATP to diphosphorylate the carboxylate group of ring A so that the carbonyl can be attacked by the amine group on ring B. The resulting compound is methylated by NovO and SAM prior to glycosylation.[24] NovM adds ring C (L-noviose) to the hydroxyl group derived from tyrosine with the loss of dTDP. Another methylation is accomplished by NovP and SAM at the 4 position of the L-noviose sugar.[25] This methylation allows NovN to carbamylate the 3 position of the sugar as shown in Fig. 4 completing the biosynthesis of novobiocin.

CLIP

CLIP

CLIP

PATENT
US-20190241599
Novel co-crystal forms of novobiocin and its analogs and proline, processes for their preparation and compositions comprising them are claimed. Also claims are methods for inhibiting heat shock protein 90 and treating or preventing neurodegenerative disorders, such as diabetic peripheral neuropathy.
References
- ^ Lanoot B, Vancanneyt M, Cleenwerck I, Wang L, Li W, Liu Z, Swings J (May 2002). “The search for synonyms among streptomycetes by using SDS-PAGE of whole-cell proteins. Emendation of the species Streptomyces aurantiacus, Streptomyces cacaoi subsp. cacaoi, Streptomyces caeruleus and Streptomyces violaceus”. International Journal of Systematic and Evolutionary Microbiology. 52 (Pt 3): 823–9. doi:10.1099/ijs.0.02008-0. PMID 12054245.
- ^ Jump up to:a b Alessandra da Silva Eustáquio (2004) Biosynthesis of aminocoumarin antibiotics in Streptomyces: Generation of structural analogues by genetic engineering and insights into the regulation of antibiotic production. DISSERTATION
- ^ Hoeksema H.; Johnson J. L.; Hinman J. W. (1955). “Structural studies on streptonivicin, a new antibiotic”. J Am Chem Soc. 77 (24): 6710–6711. doi:10.1021/ja01629a129.
- ^ Smith C. G.; Dietz A.; Sokolski W. T.; Savage G. M. (1956). “Streptonivicin, a new antibiotic. I. Discovery and biologic studies”. Antibiotics & Chemotherapy. 6: 135–142.
- ^ Raad I, Darouiche R, Hachem R, Sacilowski M, Bodey GP (November 1995). “Antibiotics and prevention of microbial colonization of catheters”. Antimicrobial Agents and Chemotherapy. 39 (11): 2397–400. doi:10.1128/aac.39.11.2397. PMC 162954. PMID 8585715.
- ^ Raad II, Hachem RY, Abi-Said D, Rolston KV, Whimbey E, Buzaid AC, Legha S (January 1998). “A prospective crossover randomized trial of novobiocin and rifampin prophylaxis for the prevention of intravascular catheter infections in cancer patients treated with interleukin-2”. Cancer. 82 (2): 403–11. doi:10.1002/(SICI)1097-0142(19980115)82:2<412::AID-CNCR22>3.0.CO;2-0. PMID 9445199.
- ^ “Determination That ALBAMYCIN (Novobiocin Sodium) Capsule, 250 Milligrams, Was Withdrawn From Sale for Reasons of Safety or Effectiveness”. The Federal Register. 19 January 2011.
- ^ Walsh TJ, Standiford HC, Reboli AC, John JF, Mulligan ME, Ribner BS, Montgomerie JZ, Goetz MB, Mayhall CG, Rimland D (June 1993). “Randomized double-blinded trial of rifampin with either novobiocin or trimethoprim-sulfamethoxazole against methicillin-resistant Staphylococcus aureus colonization: prevention of antimicrobial resistance and effect of host factors on outcome”. Antimicrobial Agents and Chemotherapy. 37 (6): 1334–42. doi:10.1128/aac.37.6.1334. PMC 187962. PMID 8328783.
- ^ Jump up to:a b Maxwell A (August 1993). “The interaction between coumarin drugs and DNA gyrase”. Molecular Microbiology. 9 (4): 681–6. doi:10.1111/j.1365-2958.1993.tb01728.x. PMID 8231802.
- ^ Maxwell A (February 1999). “DNA gyrase as a drug target”. Biochemical Society Transactions. 27 (2): 48–53. doi:10.1042/bst0270048. PMID 10093705.
- ^ Lewis RJ, Tsai FT, Wigley DB (August 1996). “Molecular mechanisms of drug inhibition of DNA gyrase”. BioEssays. 18 (8): 661–71. doi:10.1002/bies.950180810. PMID 8760340.
- ^ Maxwell A, Lawson DM (2003). “The ATP-binding site of type II topoisomerases as a target for antibacterial drugs”. Current Topics in Medicinal Chemistry. 3 (3): 283–303. doi:10.2174/1568026033452500. PMID 12570764.
- ^ Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BS (September 2005). “Hsp90 inhibitors identified from a library of novobiocin analogues”. Journal of the American Chemical Society. 127 (37): 12778–9. doi:10.1021/ja0535864. PMID 16159253.
- ^ Mandler MD, Baidin V, Lee J, Pahil KS, Owens TW, Kahne D (June 2018). “Novobiocin Enhances Polymyxin Activity by Stimulating Lipopolysaccharide Transport”. Journal of the American Chemical Society. 140 (22): 6749–6753. doi:10.1021/jacs.8b02283. PMC 5990483. PMID 29746111.
- ^ May JM, Owens TW, Mandler MD, Simpson BW, Lazarus MB, Sherman DJ, Davis RM, Okuda S, Massefski W, Ruiz N, Kahne D (December 2017). “The Antibiotic Novobiocin Binds and Activates the ATPase That Powers Lipopolysaccharide Transport”. Journal of the American Chemical Society. 139 (48): 17221–17224. doi:10.1021/jacs.7b07736. PMC 5735422. PMID 29135241.
- ^ Tsai FT, Singh OM, Skarzynski T, Wonacott AJ, Weston S, Tucker A, Pauptit RA, Breeze AL, Poyser JP, O’Brien R, Ladbury JE, Wigley DB (May 1997). “The high-resolution crystal structure of a 24-kDa gyrase B fragment from E. coli complexed with one of the most potent coumarin inhibitors, clorobiocin”. Proteins. 28 (1): 41–52. doi:10.1002/(sici)1097-0134(199705)28:1<41::aid-prot4>3.3.co;2-b. PMID 9144789.
- ^ Jump up to:a b Flatman RH, Eustaquio A, Li SM, Heide L, Maxwell A (April 2006). “Structure-activity relationships of aminocoumarin-type gyrase and topoisomerase IV inhibitors obtained by combinatorial biosynthesis”. Antimicrobial Agents and Chemotherapy. 50 (4): 1136–42. doi:10.1128/AAC.50.4.1136-1142.2006. PMC 1426943. PMID 16569821.
- ^ Steffensky M, Mühlenweg A, Wang ZX, Li SM, Heide L (May 2000). “Identification of the novobiocin biosynthetic gene cluster of Streptomyces spheroides NCIB 11891”. Antimicrobial Agents and Chemotherapy. 44 (5): 1214–22. doi:10.1128/AAC.44.5.1214-1222.2000. PMC 89847. PMID 10770754.
- ^ Pojer F, Wemakor E, Kammerer B, Chen H, Walsh CT, Li SM, Heide L (March 2003). “CloQ, a prenyltransferase involved in clorobiocin biosynthesis”. Proceedings of the National Academy of Sciences of the United States of America. 100 (5): 2316–21. Bibcode:2003PNAS..100.2316P. doi:10.1073/pnas.0337708100. PMC 151338. PMID 12618544.
- ^ Pojer F, Kahlich R, Kammerer B, Li SM, Heide L (August 2003). “CloR, a bifunctional non-heme iron oxygenase involved in clorobiocin biosynthesis”. The Journal of Biological Chemistry. 278 (33): 30661–8. doi:10.1074/jbc.M303190200. PMID 12777382.
- ^ Chen H, Walsh CT (April 2001). “Coumarin formation in novobiocin biosynthesis: beta-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI”. Chemistry & Biology. 8 (4): 301–12. doi:10.1016/S1074-5521(01)00009-6. PMID 11325587.
- ^ Pacholec M, Hillson NJ, Walsh CT (September 2005). “NovJ/NovK catalyze benzylic oxidation of a beta-hydroxyl tyrosyl-S-pantetheinyl enzyme during aminocoumarin ring formation in novobiocin biosynthesis”. Biochemistry. 44 (38): 12819–26. CiteSeerX 10.1.1.569.1481. doi:10.1021/bi051297m. PMID 16171397.
- ^ Thuy TT, Lee HC, Kim CG, Heide L, Sohng JK (April 2005). “Functional characterizations of novWUS involved in novobiocin biosynthesis from Streptomyces spheroides”. Archives of Biochemistry and Biophysics. 436 (1): 161–7. doi:10.1016/j.abb.2005.01.012. PMID 15752721.
- ^ Pacholec M, Tao J, Walsh CT (November 2005). “CouO and NovO: C-methyltransferases for tailoring the aminocoumarin scaffold in coumermycin and novobiocin antibiotic biosynthesis”. Biochemistry. 44 (45): 14969–76. doi:10.1021/bi051599o. PMID 16274243.
- ^ Freel Meyers CL, Oberthür M, Xu H, Heide L, Kahne D, Walsh CT (January 2004). “Characterization of NovP and NovN: completion of novobiocin biosynthesis by sequential tailoring of the noviosyl ring”. Angewandte Chemie. 43 (1): 67–70. doi:10.1002/anie.200352626. PMID 14694473.
External links
- Novobiocin bound to proteins in the PDB
 |
|
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
intravenous |
| ATCvet code | |
| Pharmacokinetic data | |
| Bioavailability | negligible oral bioavailability |
| Metabolism | excreted unchanged |
| Elimination half-life | 6 hours |
| Excretion | renal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.005.589 |
| Chemical and physical data | |
| Formula | C31H36N2O11 |
| Molar mass | 612.624 g·mol−1 |
| 3D model (JSmol) | |
4309-70-0 CAS
calcium;7-[(2R,3R,4S,5R)-4-carbamoyloxy-3-hydroxy-5-methoxy-6,6-dimethyloxan-2-yl]oxy-3-[[4-hydroxy-3-(3-methylbut-2-enyl)benzoyl]amino]-8-methyl-2-oxochromen-4-olate
///////// Novobiocin, ノボビオシン , Antibacterial, Antimicrobial, crystallinic acid, streptonivicin,
History
Novobiocin is a coumarin antibiotic obtained from Streptomyces niveus and other Streptomyces species. Novobiocin is useful primarily in infections involving staphylococci, and other gram-positive organisms. It acts by inhibiting the initiation of DNA replication in bacterial and mammanlian cells. Evidences indicated that Novobiocin blocks prokaryotic DNA gyrase and eukaryotic II topoisomerase, enzymes that relax super-coiled DNA and are crucial for DNA replication.1
Novobiocin

| UIPAC Name | 4-Hydroxy-3-4-hydroxy-3-(3-methylbut-2-enyl)benzamido-8-methylcoumarin-7-yl 3-O-carbamoyl-5,5-di-C-methyl-α-l-lyxofuranoside |
| CAS Number | 303-81-1 |
| Molecular Mass | 612.624 g / mol |
| Chemical Formular | C31H36N2O11 |
Biosynthesis
The substituted coumarin (ring B, red) and the 4-OH benzoyl moiety (ring A, aqua) in novobiocin were derived from -Tyr based on earlier labeling studies. β-OH-Tyr is proposed to be a common intermediate in these two biosynthetic pathways.2

NovH is a -Tyr specific didomain NRPS that generates the
-tyrosyl-S-NovH intermediate. NovH, isolated from E. coli is primed by a PPTase with CoA. The A domain activates
-Tyr as
-tyrosyl-AMP and then transfers the
-tyrosyl group to the HS-pant-PCP domain of NovH through thioester formation.3

-tyrosyl-S-NovH is then function as a cytochrome P450 monooxygenase that hydroxylates the β-carbon of the tethered
-tyrosyl group on NovH. While the substrate
-tyrosyl-S-NovH provides two electrons for a single round of the hydroxylation reaction, the other two electrons needed to reduce the oxygen atom are provided by NADPH via two-electron transfer effected by electron transfer proteins ferrodoxin (Fd) and ferrodoxin reductase (Fd Red).3 The electron transfer route is from NADPH→FAD in Fd Red→Fe–S center in Fd→Heme in NovI→oxygen.

Both NovJ and NovK are similar to 3-keto-ACP reductase and they may form a heterodimer and operate in the reverse direction to oxidize 3-OH to 3-keto. NovO is similar to some quinone C-methyltransferases 3 but the timing of methylation is not clear. NovC resembles flavin-dependent monooxygenases (35 and 32% similarity to dimethylaniline and cyclohexanone monooxygenases, respectively) 3 and is proposed to hydroxylate the ortho position of the phenyl ring. The nucleophilic attack of the ortho hydroxyl group on the thioester carbonyl center would release the coumarin ring and regenerate NovH. Ring B is then synthesized.

Synthesis





Mechanism of action
E.Coli DNA gyrase utilizes ATP to catalyze the negative supercoiling, or under-twisting, of duplex DNA. The energy coupling components of the supercoiling reaction includes 1) the DNA-dependent hydrolysis that converts ATP to ADP and Pi, and 2) the gyrase cleavage reaction that targets the specified DNA site. The two activities are induced by treating the stable gyrase-DNA complex trapped by the inihibitor oxolinic acid with sodium dodecyl sulfate (SDS or Sulphate). 4 Novobiocin competes with ATP in the ATPase and supercoiling assays, hence Novobiocin prevents the ATP from shifting the primary cleavage site on ColE1 DNA by places the site of action of the antibiotics at a reaction step prior to ATP hydrolysis and blocks the binding of ATP. 4 Such a simple mechanism of action represents for all effects of the drugs on DNA gyrase.
Clinical Use
Due to factors as low solubility, poor pharmacokinetics, and limited activity agasinst Gram-negative bacteria, the clinical usage of Novobiocin is not achieved. 5 Therefore, it is of interest to study the novobiocin biosynthetic pathway in order to generate analogs with enhanced solubility and pharmacokinetic properties while maintaining the gyrase inhibitory properties.
References
1 J.C. D’Halluin, M. Milleville, and P. Boulanger. “Effect of Novobiocin on adenovirus DNA synthesis and encapsidation”. Nucleic Acids Research 1980; 8: 1625-1641
2 M. Steffensky, S.M. Li and L. Heide, “Cloning, overexpression, and purification of novobiocic acid synthetase from Streptomyces spheroides ” NCIB 11891. J. Biol. Chem. 275 (2000), pp. 21754–21760.
3 Huawei Chen and Christopher T. Walsh, “Coumarin formation in novobiocin biosynthesis: β-hydroxylation of the aminoacyl enzyme tyrosyl-S-NovH by a cytochrome P450 NovI” Chemistry and Biology; 2001; 8: 301-312
4 K. Scheirer and N. P. Higgins. “The DAN Cleavage Reaction of DNA Gyrase ” The Journal of Biological Chemistry; 1997; 272 (43): 27202-27209
5 N Pi, C. L. F. Meyers, M. Pacholec, C. T. Walsh, and J. A. Leary. “Mass spectrometric characterization of a three-enzyme tandem reacton for assembly and modification of the novobiocin skeleton” PNAS 2004;101;10036-10041
