
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Imipenem, イミペネム水和物
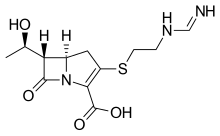
Imipenem
イミペネム水和物
Cas 74431-23-5
- Molecular FormulaC12H19N3O5S
- Average mass317.361 Da
(5R,6S)-3-((2-(Formimidoylamino)ethyl)thio)-6-((R)-1-hydroxyethyl)-7-oxo-1-azabicyclo(3.2.0)hept-2-ene-2-carboxylic acid monohydrate
Antibacterial, Cell wall biosynthesis inhibitor
Imipenem (Primaxin among others) is an intravenous β-lactam antibiotic discovered by Merck scientists Burton Christensen, William Leanza, and Kenneth Wildonger in the mid-1970s.[1] Carbapenems are highly resistant to the β-lactamase enzymes produced by many multiple drug-resistant Gram-negative bacteria,[2] thus play a key role in the treatment of infections not readily treated with other antibiotics.[3]
Imipenem was patented in 1975 and approved for medical use in 1985.[4] It was discovered via a lengthy trial-and-error search for a more stable version of the natural product thienamycin, which is produced by the bacterium Streptomyces cattleya. Thienamycin has antibacterial activity, but is unstable in aqueous solution, so impractical to administer to patients.[5] Imipenem has a broad spectrum of activity against aerobic and anaerobic, Gram-positive and Gram-negative bacteria.[6] It is particularly important for its activity against Pseudomonas aeruginosa and the Enterococcus species. It is not active against MRSA, however.
Medical uses
Spectrum of bacterial susceptibility and resistance
Acinetobacter anitratus, Acinetobacter calcoaceticus, Actinomyces odontolyticus, Aeromonas hydrophila, Bacteroides distasonis, Bacteroides uniformis, and Clostridium perfringens are generally susceptible to imipenem, while Acinetobacter baumannii, some Acinetobacter spp., Bacteroides fragilis, and Enterococcus faecalis have developed resistance to imipenem to varying degrees. Not many species are resistant to imipenem except Pseudomonas aeruginosa (Oman) and Stenotrophomonas maltophilia.[7]
Coadministration with cilastatin
Imipenem is rapidly degraded by the renal enzyme dehydropeptidase 1 when administered alone, and is almost always coadministered with cilastatin to prevent this inactivation[8]
Adverse effects
Common adverse drug reactions are nausea and vomiting. People who are allergic to penicillin and other β-lactam antibiotics should take caution if taking imipenem, as cross-reactivity rates are high. At high doses, imipenem is seizurogenic.[9]
Mechanism of action
Imipenem acts as an antimicrobial through inhibiting cell wall synthesis of various Gram-positive and Gram-negative bacteria. It remains very stable in the presence of β-lactamase (both penicillinase and cephalosporinase) produced by some bacteria, and is a strong inhibitor of β-lactamases from some Gram-negative bacteria that are resistant to most β-lactam antibiotics.
SYM
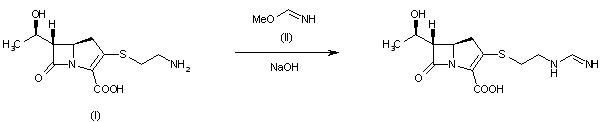
By reaction of thienamycin (I) with methyl formimidate (II) by means of NaOH in water.
| DE 2652679; FR 2332012; GB 1570990; NL 7612939 |
SYN 2
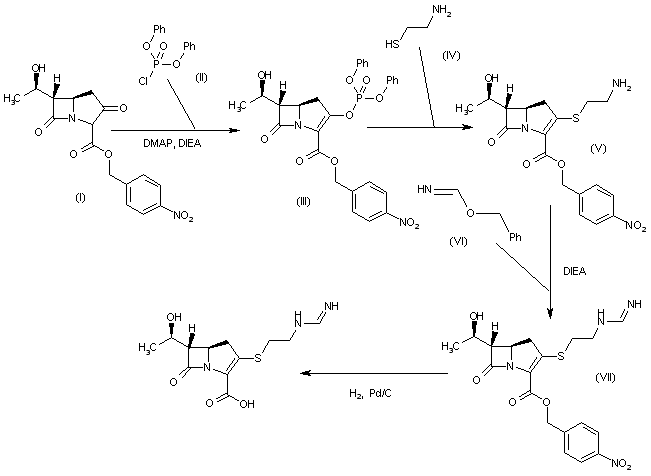
WO 0294828
The reaction of (3R,5R,6S)-6-(1(R)-hydroxyethyl)-2-oxo-1-carbapenem-3-carboxylic acid p-nitrobenzyl ester (I) with diphenyl chlorophosphate by (II) means of DMAP and DIEA in DMA/dichloromethane gives the enol phosphate (III), which is condensed with 2-aminoethanethiol (IV) in DMA to yield the 2-aminoethylsulfanyl derivative (V). The reaction of (V) with benzyl formimidate (VI) by means of DIEA in DMA affords the intermediate p-nitrobenzyl ester (VII), which is finally hydrogenated with H2 over Pd/C in water/isopropanol/N-methylmorpholine to provide the target Imipemide.
SYN 3
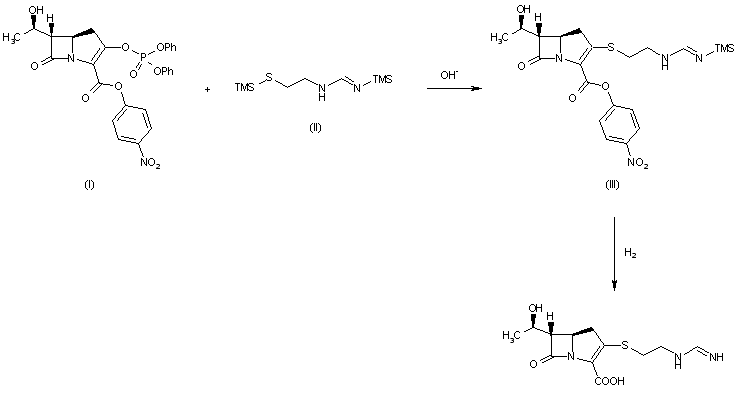
Tetrahedron Lett 1982,23(47),4903
The condensation of 7-oxo-6-(1-hydroxyethyl)-3-(diphenoxyphosphate)-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid p-nitrophenyl ester (I) with the bis(trimethylsilyl) derivative of 2-(iminomethylamino)ethanethiol (II) in the presence of base gives p-nitrophenyl ester of MK-0787, protected with a trimethylsilyl group (III), which is finally deprotected by hydrogenolysis.
CLIP

Synthesis Path
References
- ^ U.S. Patent 4,194,047
- ^ Clissold, SP; Todd, PA; Campoli-Richards, DM (Mar 1987). “Imipenem/cilastatin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy”. Drugs. 33 (3): 183–241. doi:10.2165/00003495-198733030-00001. PMID 3552595.
- ^ Vardakas, KZ; Tansarli, GS; Rafailidis, PI; Falagas, ME (Dec 2012). “Carbapenems versus alternative antibiotics for the treatment of bacteraemia due to Enterobacteriaceae producing extended-spectrum β-lactamases: a systematic review and meta-analysis”. The Journal of Antimicrobial Chemotherapy. 67 (12): 2793–803. doi:10.1093/jac/dks301. PMID 22915465.
- ^ Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 497. ISBN 9783527607495.
- ^ Kahan, FM; Kropp, H; Sundelof, JG; Birnbaum, J (Dec 1983). “Thienamycin: development of imipenen-cilastatin”. The Journal of Antimicrobial Chemotherapy. 12 Suppl D: 1–35. doi:10.1093/jac/12.suppl_d.1. PMID 6365872.
- ^ Kesado, Tadataka; Hashizume, Terutaka; Asahi, Yoshinari (1980). “Antibacterial activities of a new stabilized thienamycin, N-formimidoyl thienamycin, in comparison with other antibiotics”. Antimicrobial Agents and Chemotherapy. 17 (6): 912–7. doi:10.1128/aac.17.6.912. PMC 283902. PMID 6931548.
- ^ “Imipenem spectrum of bacterial susceptibility and Resistance” (PDF). Retrieved 4 May 2012.
- ^ “IMIPENEM/CILASTATIN”. livertox.nih.gov. Retrieved 2019-03-08.
- ^ Cannon, Joan P.; Lee, Todd A.; Clark, Nina M.; Setlak, Paul; Grim, Shellee A. (2014-08-01). “The risk of seizures among the carbapenems: a meta-analysis”. Journal of Antimicrobial Chemotherapy. 69 (8): 2043–2055. doi:10.1093/jac/dku111. ISSN 0305-7453.
Further reading
- Clissold, SP; Todd, PA; Campoli-Richards, DM (1987). “Imipenem/cilastatin. A review of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy”. Drugs. 33(3): 183–241. doi:10.2165/00003495-198733030-00001. PMID 3552595.
- Buckley, MM; Brogden, RN; Barradell, LB; Goa, KL (1992). “Imipenem/cilastatin. A reappraisal of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy”. Drugs. 44 (3): 408–44. doi:10.2165/00003495-199244030-00008. PMID 1382937.
External links
- Imipenem bound to proteins in the PDB
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Primaxin |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686013 |
| Pregnancy category |
|
| Routes of administration |
IM, IV |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Protein binding | 20% |
| Metabolism | Renal |
| Elimination half-life | 38 minutes (children), 60 minutes (adults) |
| Excretion | Urine (70%) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.058.831 |
| Chemical and physical data | |
| Formula | C12H17N3O4S |
| Molar mass | 299.347 g/mol g·mol−1 |
| 3D model (JSmol) | |
-
- Synonyms:Imipemide
- ATC:J01DH51
- Use:carbapenem antibiotic
- Chemical name:[5R-[5α,6α(R*)]]-6-(1-hydroxyethyl)-3-[[2-[(iminomethyl)amino]ethyl]thio]-7-oxo-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
- Formula:C12H17N3O4S
- MW:299.35 g/mol
- CAS-RN:64221-86-9
- InChI Key:ZSKVGTPCRGIANV-ZXFLCMHBSA-N
- InChI:InChI=1S/C12H17N3O4S/c1-6(16)9-7-4-8(20-3-2-14-5-13)10(12(18)19)15(7)11(9)17/h5-7,9,16H,2-4H2,1H3,(H2,13,14)(H,18,19)/t6-,7-,9-/m1/s1
- EINECS:264-734-5
- LD50:1660 mg/kg (M, i.v.); >5 g/kg (M, p.o.);
1972 mg/kg (R, i.v.); >5 g/kg (R, p.o.)
Derivatives, monohydrate
- Formula:C12H17N3O4S • H2O
- MW:317.37 g/mol
- CAS-RN:74431-23-5
References
-
-
Leanza, W.J. et al.: J. Med. Chem. (JMCMAR) 22, 1435 (1979).
-
a Salzmann, T.L. et al.: J. Am. Chem. Soc. (JACSAT) 102, 6161-6163 (1980).
-
Reider, P.J.; Grabowski, E.J.J.: Tetrahedron Lett. (TELEAY) 23, 2293-2296 (1982).
-
Grabowski, E.J.J.: Chirality (CHRLEP) 17, 249-259 (2005).
-
US 4 194 047 (Merck & Co.; 18.3.1980; prior. 21.11.1975).
-
DOS 2 652 679 (Merck & Co.; appl. 19.11.1976; USA-prior. 21.11.1975).
-
b US 5 998 612 (Merck & Co.; 7.12.1999; appl. 12.6.1992; prior. 23.10.1981).
-
c US 4 981 992 (Takasago; 27.1.1998; appl. 13.5.1996; J-prior. 11.5.1995).
-
US 5 204 460 (Takasago; 20.4.1993; appl. 8.11.1991; J-prior. 8.11.1990).
-
US 5 204 462 (Takasago; 20.4.1993; appl. 8.11.1991; J-prior. 8.11.1990).
-
US 5 712 388 (Takasago; 27.1.1998; appl. 13.5.1996; J-prior. 11.5.1995).
-
US 5 081 239 (Takasago; 14.1.1992; appl. 29.11.1989; J-prior. 29.11.1988).
-
-
Acetoxylation of 2-azetidinones in 4-position:
-
Noyori, R. et al.: J. Am. Chem. Soc. (JACSAT) 111, 9134-9135 (1989).
-
Noyori, R. et al.: Angew. Chem. (ANCEAD) 114, 2108-2123 (2002).
-
US 5 288 862 (Takasago; 22.2.1994; appl. 16.4.1992; J-prior. 18.4.1991).
-
US 5 606 052 (Takasago; 25.2.1997; appl. 16.4.1992; J-prior. 18.4.1991).
-
-
Noyori-catalyst:
-
US 4 739 084 (Takasago; 19.4.1988; appl. 15.4.1987; J-prior. 13.5.1986).
-
-
d process of Nippon Soda (Nisso):
-
US 5 026 844 (Suntory & Nippon Soda; 25.6.1991; appl. 13.10.1989; J-prior. 19.10.1988).
-
US 5 792 861 (Tanabe Seiyaku & Nippon Soda; 11.8.1998; appl. 29.6.1994, 4.11.1996; J-prior. 30.6.1993).
-
US 5 808 055 (Suntory & Nippon Soda; 15.9.1998; appl. 30.3.1993, 5.7.1995; J-prior. 30.3.1993).
-
e US 4 791 198 (Kanegafuchi; 13.12.1988; appl. 1.7.1985, 6.1.1987; J-prior. 5.7.1984, 14.1.1986).
-
US 4 861 877 (Kanegafuchi; 29.8.1989; appl. 1.7.1985, 6.1.1987; J-prior. 5.7.1984, 14.1.1985, 14.1.1986).
-
US 5 061 817 (Kanegafuchi; 29.10.1991; appl. 1.7.1985, 6.1.1987, 31.5.1988; J-prior. 5.7.1984, 14.1.1986).
-
US 4 914 200 (Kanegafuchi; 3.4.1990; appl. 28.4.1987, 14.2.1989; J-prior. 30.4.1986, 13.11.1986, 9.2.1987).
-
-
Enzymatic reduction of alkyl-2-(N-benzoylamino)methyl-3-oxobutyrates with bakers yeast:
-
US 5 463 047 (Ciba-Geigy; 31.10.1995; appl. 15.9.1994; CH-prior. 4.5.1987).
-
-
Further synthesis processes of Merck & Co. for thienamycin:
-
Johnston, D.B.R. et al.: J. Am. Chem. Soc. (JACSAT) 100, 313-315 (1978).
-
Mellilo, D.G. et al.: Tetrahedron Lett. (TELEAY) 21, 2783 (1980).
-
Melillo, D.G. et al.: J. Org. Chem. (JOCEAH) 51, 1498-1504 (1986).
-
Karady, S. et al.: J. Am. Chem. Soc. (JACSAT) 103, 6765-6767 (1981).
-
US 4 269 772 (Merck & Co.; 26.5.1981; appl. 14.1.1980).
-
US 4 282 148 (Merck & Co.; 4.8.1981; appl. 14.1.1980).
-
US 4 287 123 (Merck & Co.; 1.9.1981; appl. 14.1.1980).
-
US 4 290 947 (Merck & Co.; 22.9.1981; appl. 29.5.1980).
-
US 4 360 684 (Merck & Co.; 23.11.1982; appl. 8.4.1981).
-
US 4 206 219 (Merck & Co.; 3.6.1980; appl. 24.10.1978).
-
US 4 348 320 (Merck & Co.; 7.9.1982; appl. 20.8.1980; USA-prior. 19.11.1976).
-
US 4 460 507 (Merck & Co.; 17.7.1984; appl. 29.4.1982; USA-prior. 10.10.1980).
-
US 5 055 573 (Merck & Co.; 8.10.1991, appl. 24.8.1990; USA-prior. 19.11.1976).
-
US 5 037 974 (Merck & Co.; 6.8.1991; appl. 14.8.1990; prior. 23.5.1988, 10.4.1990).
-
-
Review of thienamycin syntheses:
-
Nicolaou, K.C.; Sorensen, E.J.: Classics in Total Synthesis, VCH 1996, Weinheim & New York, chapter 16, p. 249-263.
-
Berks, A.H.: Tetrahedron (TETRAB) 52, 331-375 (1996).
-
-
Alternative 2-azetidinone ring closure with chlorosulfonyl isocyanate:
-
US 4 350 631 (Merck & Co.; 21.9.1982; appl. 18.3.1981; prior. 18.12.1980).
-
-
Thienamycin (by fermentation of S. cattleya):
-
US 3 950 357 (Merck & Co.; 13.4.1976; appl. 25.11.1974).
-
DOS 2 552 638 (Merck & Co.; appl. 24.11.1975; USA-prior. 25.11.1974).
-
-
Combination with cilastatin:
-
EP 48 301 (Merck & Co.; appl. 24.9.1980).
-
/////////////Imipenem, イミペネム水和物 , MK-787,
SK1-I , BML 258


SK1-I , BML 258
Sphingosine kinase 1 (SphK1) inhibitor; antiproliferative
- (1E)-1,2,4-Trideoxy-4-(methylamino)-1-(4-pentylphenyl)-D-erythro-pent-1-enitol
- (E,2R,3S)-2-(Methylamino)-5-(4-pentylphenyl)pent-4-ene-1,3-diol
- D-erythro-Pent-1-enitol, 1,2,4-trideoxy-4-(methylamino)-1-(4-pentylphenyl)-, (1E)-
| Name: | (2R,3S,4E)-N-methyl-5-(4′-pentylphenyl)-2-aminopent-4-ene-1,3-diol . HCl |
| Formula: | C17H27NO2 . HCl |
| MW: | 313.9 |
| CAS: | 1072443-89-0
|
- Originator Enzo Biochem; Virginia Commonwealth University
- Developer Enzo Biochem
- Class Antineoplastics; Small molecules
- Mechanism of Action Sphingosine kinase inhibitors
- Preclinical Autoimmune hepatitis; Haematological malignancies; Liver cancer; Solid tumours
- 07 May 2019 Preclinical trials in Liver cancer in USA (unspecified route)
- 03 Dec 2018 SK1 I is available for licensing as of 03 Dec 2018. http://www.enzo.com/
- 03 Dec 2018 Enzo Biochem has patent pending for SK1 I worldwide
SK1 I, a small molecule that specifically inhibits sphingosine kinase 1, is being developed by Enzo Biochem for the treatment of cancer and autoimmune diseases. Preclinical development is underway for the treatment of solid tumours, liver cancer, haematological malignancies and autoimmune hepatitis in the US.
As at December 2018, Enzo Biochem seeks partners for the development of SK1
SK1-I is a sphingosine analog and a sphingosine competitive inhibitor specific for sphingosine kinase 1 (SK1), with ki~10µM and excellent water solubility. It is not to be confused with SKI-I, 5-naphthalen-2-yl-2H-pyrazole-3-carboxylic acid (2-hydroxy-naphthalen-1-ylmethylene)-hydrazide, CAS 306301-68-8, a noncompetitive inhibitor of both SK1 and SK2 with poor water solubility (K.J. French, et al., 2006; N.J. Pyne and S. Pyne, 2010). SK1-I does not inhibit SK2, PKCα, PKCδ, PKA, AKT1, ERK1, EGFR, CDK2, IKKβ or CamK2β. Not only does it decrease sphingosine-1-phosphate levels, it also causes an accumulation of its proapoptotic precursor ceremide. Inhibits tumor cell growth in vitro and in vivo.
PATENTS
US 20100035959
WO 2010127093
US 20100278741
WO 2011025545
Patent
US-10364211
This patent was granted in July 30, 2019 and set to expire on October 24, 2038. Claims methods for synthesizing the compound (2R,3S,4E)-N-methyl-5-(4′-pentylphenyl)-2-aminopent-4-ene-1,3-diol (also known as SK1-I and BML-258 (as HCl salt)) and its intermediates.
(2R,3S,4E)-N-methyl-5-(4′-pentylphenyl)-2-aminopent-4-ene-1,3-diol, also known as SK1-I and BML-258 (as HCl salt), is a pharmaceutical inhibitor of sphingosine kinase 1 initially described in Paugh et al., Blood. 2008 Aug. 15; 112(4): 1382-1391. An existing method for synthesizing SK1-I is disclosed in U.S. Pat. No. 8,314,151.
|
and |
|
|
N-Boc-(D)-Serine Methyl Ester
Protection of N-Boc-(D)-Serine Methyl Ester
(R)—Garner Aldehyde
Addition of 4-Pentylphenyl Acetylene to the Above Aldehyde
Deprotection of the Above Oxazolidine
Reduction of the Above Alcohol
Deprotection to SK1-I (BML-258)
PATENT
WO2018237379 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018237379
claiming sphingosine pathway modulating compounds for the treatment of cancers, assigned to Enzo Biochem Inc , naming different team
Sphingosine- 1 -phosphate (SIP) was discovered to be a bioactive signaling molecule over 20 years ago. Studies have since identified two related kinases, sphingosine kinase 1 and 2 (a/k/a sphingosine kinase “type I” and “type II” respectively, and SphKl and SphK2 respectively), which catalyze the phosphorylation of sphingosine to SIP. Extracellular SIP can bind to and activate each of five S IP-specific, G protein-coupled receptors (designated S IPR1-5) to regulate cellular and physiological processes in an autocrine or paracrine manner. Selective inhibitors of each of sphingosine kinase 1 and 2, as well as both nonselective and selective agonists of SlPRs, have been developed and are known in the art.
Product Literature References
General Literature References
/////////SK1-I , SK1I , SK1 I , BML 258, Enzo Biochem, Virginia Commonwealth, Preclinical, solid tumours, liver cancer, haematological malignancies, autoimmune hepatitis,
CCCCCC1=CC=C(/C=C/[C@H](O)[C@H](NC)CO)C=C1.Cl
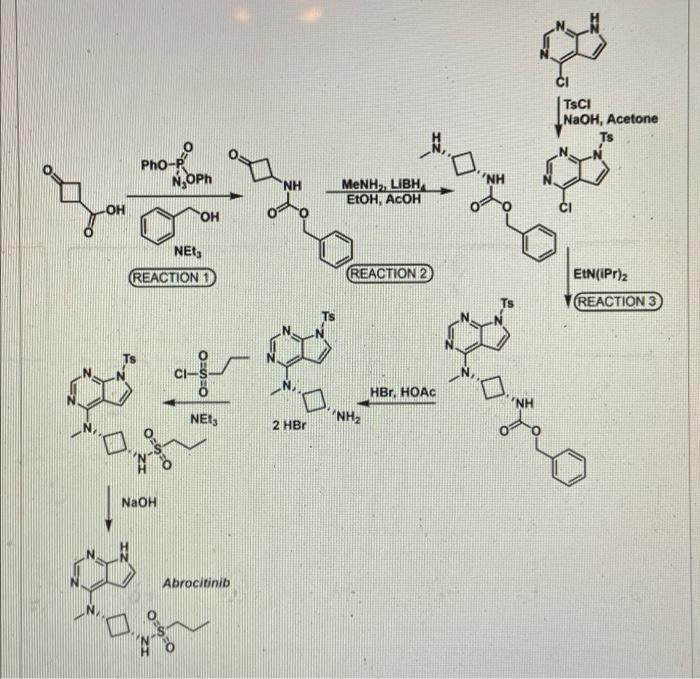
PF 04965842, Abrocitinib

PF-04965842
PF 04965842, Abrocitinib
UNII: 73SM5SF3OR
CAS Number 1622902-68-4, Empirical Formula C14H21N5O2S, Molecular Weight 323.41
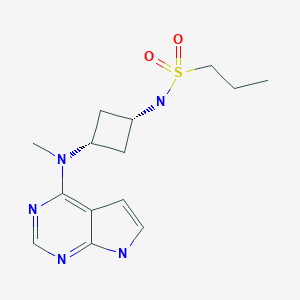
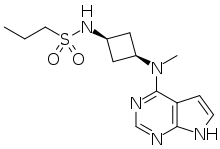
N-[cis-3-(Methyl-7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)cyclobutyl]-1-propanesulfonamide,
N-((1s,3s)-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)cyclobutyl)propane-1-sulfonamide
1-Propanesulfonamide, N-(cis-3-(methyl-7H-pyrrolo(2,3-d)pyrimidin-4-ylamino)cyclobutyl)-
N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane-1-sulfonamide
PHASE 3, for the potential oral treatment of moderate-to-severe atopic dermatitis (AD)
Jak1 tyrosine kinase inhibitor
UPDATE…… JAPAN APPROVED, 2021, 2021/9/27, CIBINQO
ALSO
fda 2022, APPROVALS 2022, 1/14/2022
THE US
In February 2018, the FDA granted Breakthrough Therapy designation for the treatment of patients with moderate-to-severe AD
PHASEIII
In December 2017, a randomized, double-blind, placebo-controlled, parallel-group, phase III trial (NCT03349060; JADE Mono-1; JADE; B7451012; 2017-003651-29) of PF-04965842 began in patients aged 12 years and older (expected n = 375) with moderate-to-severe AD
PRODUCT PATENT
| Pub. No.: | WO/2014/128591 | International Application No.: | PCT/IB2014/058889 | |||
| Publication Date: | 28.08.2014 | International Filing Date: | 11.02.2014 |
EXPIRY Roughly 2034
| form | powder |
| color | white to beige |
| solubility | DMSO: 10 mg/mL, clear |
| storage temp. | room temp |
- Biochem/physiol Actions
-
- PF-04965842 is a Janus Kinase (JAK) inhibitor selective for JAK1 with an IC50value of 29 nM for JAK1 compared to 803 nM for JAK2, >10000 nM for JAK3 and 1250 nM for Tyk2. JAKs mediate cytokine signaling, and are involved in cell proliferation and differentiation. PF-04965842 has been investigated as a possible treatment for psoriasis.
- Originator Pfizer
- Class Skin disorder therapies; Small molecules
- Mechanism of Action Janus kinase 1 inhibitors
Highest Development Phases
- Phase IIIAtopic dermatitis
- DiscontinuedLupus vulgaris; Plaque psoriasis
Most Recent Events
- 08 Mar 2018Phase-III clinical trials in Atopic dermatitis (In children, In adults, In adolescents) in USA (PO) (NCT03422822)
- 14 Feb 2018PF 4965842 receives Breakthrough Therapy status for Atopic dermatitis in USA
- 06 Feb 2018Pfizer plans the phase III JADE EXTEND trial for Atopic Dermatitis (In children, In adults, In adolescents) in March 2018 (PO) (NCT03422822)
This compound was developed by Pfizer for Kinase Phosphatase Biology research. To learn more about Sigma′s partnership with Pfizer and view other authentic, high-quality Pfizer compounds,
PF-04965842 is an oral Janus Kinase 1 inhibitor being investigated for treatment of plaque psoriasis.
Protein kinases are families of enzymes that catalyze the phosphorylation of specific residues in proteins, broadly classified into tyrosine and serine/threonine kinases. Inappropriate kinase activity, arising from mutation, over-expression, or inappropriate regulation, dys-regulation or de-regulation, as well as over- or under-production of growth factors or cytokines has been i mplicated in many diseases, including but not limited to cancer, cardiovascular diseases, allergies, asthma and other respiratory diseases, autoimmune d iseases, inflammatory diseases, bone diseases, metabolic disorders, and neurological and neurodegenerative disorders such as Alzheimer’s disease. Inappropriate kinase activity triggers a variety of biological cellular responses relating to cell growth, cell differentiation , survival, apoptosis, mitogenesis, cell cycle control, and cel l mobility implicated in the aforementioned and related diseases.
Thus, protein kinases have emerged as an important class of enzymes as targets for therapeutic intervention. In particular, the JAK family of cellular protein tyrosine kinases (JAK1, JAK2, JAK3, and Tyk2) play a central role in cytoki ne signaling (Kisseleva et al., Gene, 2002, 285 , 1; Yamaoka et al. Genome Biology 2004, 5, 253)). Upon binding to their receptors, cytokines activate JAK which then phosphorylate the cytokine receptor, thereby creating docking sites for signaling molecules, notably, members of the signal transducer and activator of transcription (STAT) family that ultimately lead to gene expression. Numerous cytokines are known to activate the JAK family. These cytokines include, the IFN family (IFN-alpha, IFN-beta, IFN-omega, Limitin, IFN-gamma, IL- 10, IL- 19, IL-20, IL-22), the gp 130 family (IL-6, IL- 11, OSM, LIF, CNTF, NNT- 1//SF-3, G-CSF, CT- 1, Leptin, IL- 12 , I L-23), gamma C family (IL-2 , I L-7, TSLP, IL-9, IL- 15 , IL-21, IL-4, I L- 13), IL-3 family (IL-3 , IL-5 , GM-CSF), single chain family (EPO, GH, PRL, TPO), receptor tyrosine kinases (EGF, PDGF, CSF- 1, HGF), and G-protein coupled receptors (ATI).
Abrocitinib, sold under the brand name Cibinqo, is a Janus kinase inhibitor medication used for the treatment of atopic dermatitis (eczema).[2] It was developed by Pfizer.[2]
Medical uses
Abrocitinib is indicated for the treatment of moderate-to-severe atopic dermatitis in adults who are candidates for systemic therapy.[2]
Side effects
The most common adverse effects in studies were upper respiratory tract infection, headache, nausea, and diarrhea.[3]
Pharmacology
Mechanism of action
It is a selective inhibitor of the enzyme janus kinase 1 (JAK1).[3]
Pharmacokinetics
Abrocitinib is quickly absorbed from the gut and generally reaches highest blood plasma concentrations within one hour. Only 1.0 to 4.4% of the dose are found unmetabolized in the urine.[4]
History
- April 2016: initiation of Phase 2b trial
- December 2017: initiation of JADE Mono-1 Phase 3 trial[5]
- May 2018: Results of Phase 2b trial posted
- October 2019: Results of Phase 3 trial presented[6]
- June 2020: Results of second Phase 3 trial published[7]
Society and culture
Legal status
In October 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Cibinqo, intended for the treatment of atopic dermatitis.[8] The applicant for this medicinal product is Pfizer Europe MA EEIG.[8] In December 2021, the European Commission approved abrocitinib for the treatment of atopic dermatitis.[2][9]
In January 2022, the United States Food and Drug Administration (FDA) approved abrocitinib for adults with moderate-to-severe atopic dermatitis.[10]
////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
EU
Click to access cibinqo-epar-public-assessment-report_en.pdf
Introduction
The finished product is presented as immediate release film-coated tablets containing 50 mg, 100 mg
or 200 mg of abrocitinib as active substance.
Other ingredients are:
Tablet core: microcrystalline cellulose (E460i), anhydrous dibasic calcium phosphate (E341ii), sodium
starch glycolate and magnesium stearate (E470b).
Film-coat: hypromellose (E464), titanium dioxide (E171), lactose monohydrate, macrogol (E1521),
triacetin (E1518) and red iron oxide (E172).
The product is available in high-density polyethylene (HDPE) bottles with polypropylene closure or
polyvinylidene chloride (PVDC) blisters with aluminium foil lidding film, as described in section 6.5 of
the SmPC.
The chemical name of abrocitinib is N-((1S,3S)-3-(methyl(7H-pyrrolo[2,3-d]pyrimidin-4-
yl)amino)cyclobutyl)propane-1-sulfonamide corresponding to the molecular formula C14H21N5O2S. It
has a relative molecular mass of 323.42 Daltons and the following structure depicted in Figure 1:

The chemical structure of abrocitinib was elucidated by a combination of UV/VIS and IR spectroscopy,
mass spectrometry, NMR spectroscopy and X-ray diffraction.
The active substance is a white to pale-purple or pale pink crystalline powder. It is non-hygroscopic
and its solubility is pH dependent. Abrocitinib is classified as BCS Class II. The impact of particle size
on finished product uniformity of dosage units and dissolution has been studied (see finished product
section). Based on the abrocitinib finished product biopharmaceutics performance, stability, and
manufacturing experience, the active substance particle size specification was established.
Abrocitinib is an achiral molecule, but with 2 stereocentres.
Only one crystalline anhydrous form (Form 1) of abrocitinib has been identified. This form has been the
only form used in all toxicology and clinical studies. Extensive polymorph and hydrate screening have
been conducted to investigate if additional solid forms of abrocitinib could be discovered. Abrocitinib,
Form 1 was the only anhydrous crystalline form identified from these studies. No new anhydrous
polymorphs, hydrates or amorphous solids of abrocitinib were isolated from these screens.
Experiments with 1,4 dioxane and dimethyl sulfoxide yielded solvated forms of abrocitinib. When these
solvated structures were subjected to high temperature, these materials desolvated and converted to
Form 1, free base anhydrous form of abrocitinib. However, these are not relevant since the commercial
crystallisation step does not utilise either of these solvent systems.
It has been confirmed that the manufacturing process consistently yields polymorphic form I. This form
is physically and chemically stable under normal manufacturing and storage conditions as well as
under accelerated conditions. Hence the absence of control of form I is justified.
FDA
U.S. FDA Approves Pfizer’s CIBINQO® (abrocitinib) for Adults with Moderate-to-Severe Atopic Dermatitis
CIBINQO is a once-daily oral treatment with proven efficacy to manage symptoms for adults who have not yet found relief with current options
NEW YORK–(BUSINESS WIRE)– Pfizer Inc. (NYSE: PFE) announced today that the United States (U.S.) Food and Drug Administration (FDA) approved CIBINQO® (abrocitinib), an oral, once-daily, Janus kinase 1 (JAK1) inhibitor, for the treatment of adults living with refractory, moderate-to-severe atopic dermatitis (AD) whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
CIBINQO is approved at the recommended doses of 100 mg and 200 mg, with the 200 mg dose being recommended for patients who are not responding to the 100 mg dose. Additionally, a 50 mg dose was approved to treat moderate-to-severe AD specifically in patients with moderate renal impairment (kidney failure), certain patients receiving treatment with inhibitors of cytochrome P450 (CYP) 2C19, or patients who are known or suspected to be poor metabolizers of CYP2C19. For patients with moderate renal impairment who are not responding to 50 mg once daily, 100 mg once daily may also be prescribed.
“The reality for patients living with chronic inflammatory skin disease such as moderate-to-severe atopic dermatitis is that many experience debilitating symptoms that are not managed by current treatment options. Today’s approval of CIBINQO will provide an important new oral option that could help those who have yet to find relief,” said Jonathan Silverberg, MD, PhD, MPH, Department of Dermatology, The George Washington University School of Medicine and Health Sciences. “In multiple large-scale clinical trials, CIBINQO demonstrated strong efficacy at clearing skin, improving itch, and managing the extent and severity of eczema, offering a benefit-risk profile that supports the use of this treatment in the FDA-approved patient population.”
The FDA approval was based on results of five clinical trials from a large-scale clinical trial program of more than 1,600 patients. The safety and efficacy of CIBINQO was evaluated in three randomized, placebo-controlled, Phase 3 trials. Additionally, safety was evaluated through a randomized, placebo-controlled, dose-ranging trial and an ongoing long-term open-label extension trial. Across the trials, CIBINQO demonstrated a consistent safety profile and profound improvements in skin clearance, extent of disease, and severity, as well as rapid improvement in itch after two weeks, for some people living with AD versus placebo. In addition, a higher proportion of subjects treated with CIBINQO in two monotherapy trials achieved improvement in itching at week 12 compared to placebo.
“The FDA’s approval offers hope to the millions of patients across the U.S. who are suffering daily with an immuno-inflammatory condition that can cause intense and persistent itching, pain, discomfort, and distress if left uncontrolled,” said Mike Gladstone, Global President of Pfizer Inflammation & Immunology. “CIBINQO, an efficacious once-daily pill, is a medical breakthrough made possible by Pfizer researchers and the people living with moderate-to-severe atopic dermatitis who participated in our clinical trials.”
“Atopic dermatitis is so much more than just a rash, and it goes beyond the surface of the skin. It’s a chronic condition that can both significantly disrupt patients’ daily lives and negatively impact their emotional well-being,” said Julie Block, President and CEO, National Eczema Association. “We appreciate Pfizer’s commitment to this resilient patient community and eagerly await the positive impact CIBINQO could have on the treatment landscape for moderate-to-severe atopic dermatitis.”
The most common adverse events reported in ≥5% of patients with CIBINQO included nasopharyngitis (12.4% with CIBINQO 100 mg, 8.7% with CIBINQO 200 mg, and 7.9%, with placebo), nausea (6%, 14.5%, and 2.1%, respectively), and headache (6%, 7.8%, and 3.5%, respectively).
The full prescribing information for CIBINQO can be found here. CIBINQO will be made available in the coming weeks.
Additional Details on the CIBINQO Clinical Trial Program
Five clinical trials in the CIBINQO JAK1 Atopic Dermatitis Efficacy and Safety (JADE) global development program were included in the New Drug Application (NDA) to support the FDA approval.
The safety and efficacy of CIBINQO was evaluated in three Phase 3, randomized, placebo-controlled clinical trials. The trials evaluated measures of improvements in skin clearance, itch, disease extent, and severity, including the Investigator Global Assessment (IGA), Eczema Area and Severity Index (EASI), and Peak Pruritus Numerical Ratings Scale (PP-NRS). In each of the trials, over 40% of patients had prior exposure to a systemic therapy:
- JADE MONO-1 and JADE MONO-2: A pair of randomized, double-blind, placebo-controlled trials designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO monotherapy in 778 patients 12 years of age and older with moderate-to-severe AD. The trials assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
- JADE COMPARE: A randomized, double-blind, placebo-controlled trial designed to evaluate the efficacy and safety of two doses (100 mg and 200 mg once daily) of CIBINQO in 837 adult patients with moderate-to-severe AD on background topical medicated therapy. The trial also included an active control arm with dupilumab, a biologic treatment administered by subcutaneous injection, compared with placebo. The trial assessed the co-primary endpoints of IGA and EASI-75 responses at Week 12.
Select findings for CIBINQO 100 mg, 200 mg, and placebo follow (*p<0.01 or **p<0.001):
- JADE MONO-1:
- IGA Response Rate (Week 12): 24%*, 44%**, and 8%, respectively
- EASI-75 Response Rate (Week 12): 40%**, 62%**, and 12%, respectively
- JADE MONO-2
- IGA Response Rate (Week 12): 28%**, 38%**, and 9%, respectively
- EASI-75 Response Rate (Week 12): 44%**, 61%**, and 10%, respectively
- JADE COMPARE
- IGA Response Rate (Week 12): 36%**, 47%**, and 14%, respectively
- EASI-75 Response Rate (Week 12): 58%**, 68%**, and 27%, respectively
Safety was additionally evaluated through a randomized dose-ranging trial and a long-term, open-label, extension trial (JADE EXTEND).
U.S. IMPORTANT SAFETY INFORMATION
WARNING: SERIOUS INFECTIONS, MORTALITY, MALIGNANCY, MAJOR ADVERSE CARDIOVASCULAR EVENTS, AND THROMBOSIS
Serious Infections
Patients treated with CIBINQO may be at increased risk for developing serious infections that may lead to hospitalization or death. The most frequent serious infections reported with CIBINQO were herpes simplex, herpes zoster, and pneumonia.
If a serious or opportunistic infection develops, discontinue CIBINQO and control the infection.
Reported infections from Janus kinase (JAK) inhibitors used to treat inflammatory conditions:
- Active tuberculosis, which may present with pulmonary or extrapulmonary disease. Test for latent TB before and during therapy; treat latent TB prior to use. Monitor all patients for active TB during treatment, even patients with initial negative, latent TB test.
- Invasive fungal infections, including cryptococcosis and pneumocystosis. Patients with invasive fungal infections may present with disseminated, rather than localized, disease.
- Bacterial, viral (including herpes zoster), and other infections due to opportunistic pathogens.
Avoid use of CIBINQO in patients with an active, serious infection, including localized infections. The risks and benefits of treatment with CIBINQO should be carefully considered prior to initiating therapy in patients with chronic or recurrent infections or those who have resided or traveled in areas of endemic tuberculosis or endemic mycoses.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with CIBINQO, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy.
Consider yearly screening for patients in highly endemic areas for TB. CIBINQO is not recommended for use in patients with active TB. For patients with a new diagnosis of latent TB or prior untreated latent TB, or for patients with a negative test for latent TB but who are at high risk for TB infection, start preventive therapy for latent TB prior to initiation of CIBINQO.
Viral reactivation, including herpes virus reactivation (eg, herpes zoster, herpes simplex), was reported in clinical studies with CIBINQO. If a patient develops herpes zoster, consider interrupting CIBINQO until the episode resolves. Hepatitis B virus reactivation has been reported in patients receiving JAK inhibitors. Perform viral hepatitis screening and monitoring for reactivation in accordance with clinical guidelines before starting therapy and during therapy with CIBINQO. CIBINQO is not recommended for use in patients with active hepatitis B or hepatitis C.
Mortality
In a large, randomized postmarketing safety study in rheumatoid arthritis (RA) patients 50 years of age and older with at least one cardiovascular risk factor comparing another JAK inhibitor to TNF blocker treatment, a higher rate of all-cause mortality (including sudden cardiovascular death) was observed with the JAK inhibitor. CIBINQO is not approved for use in RA patients.
Malignancies
Malignancies, including non-melanoma skin cancer (NMSC), were reported in patients treated with CIBINQO. Lymphoma and other malignancies have been observed in patients receiving JAK inhibitors used to treat inflammatory conditions. Perform periodic skin examination for patients who are at increased risk for skin cancer. Exposure to sunlight and UV light should be limited by wearing protective clothing and using broad-spectrum sunscreen.
In a large, randomized postmarketing safety study of another JAK inhibitor in RA patients, a higher rate of malignancies (excluding non-melanoma skin cancer [NMSC]) was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. CIBINQO is not approved for use in RA patients. A higher rate of lymphomas was observed in patients treated with the JAK inhibitor compared to those treated with TNF blockers. A higher rate of lung cancers was observed in current or past smokers treated with the JAK inhibitor compared to those treated with TNF blockers. Patients who are current or past smokers are at additional increased risk.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients with a known malignancy (other than a successfully treated NMSC), patients who develop a malignancy when on treatment, and patients who are current or past smokers.
Major Adverse Cardiovascular Events
Major adverse cardiovascular events were reported in patients treated with CIBINQO. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of major adverse cardiovascular events (MACE) (defined as cardiovascular death, myocardial infarction, and stroke), was observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients. Patients who are current or past smokers are at additional increased risk. Discontinue CIBINQO in patients that have experienced a myocardial infarction or stroke.
Consider the benefits and risks for the individual patient prior to initiating or continuing therapy with CIBINQO, particularly in patients who are current or past smokers and patients with other cardiovascular risk factors. Patients should be informed about the symptoms of serious cardiovascular events and the steps to take if they occur.
Thrombosis
Deep vein thrombosis (DVT) and pulmonary embolism (PE) have been reported in patients treated with CIBINQO. Thrombosis, including PE, DVT, and arterial thrombosis have been reported in patients receiving JAK inhibitors used to treat inflammatory conditions. Many of these adverse reactions were serious and some resulted in death. In RA patients 50 years of age and older with at least one cardiovascular risk factor treated with another JAK inhibitor, a higher rate of overall thrombosis, DVT, and PE were observed when compared with TNF blockers. CIBINQO is not approved for use in RA patients.
Avoid CIBINQO in patients that may be at increased risk of thrombosis. If symptoms of thrombosis occur, discontinue CIBINQO and treat patients appropriately.
Contraindication
CIBINQO is contraindicated in patients taking antiplatelet therapies, except for low-dose aspirin (≤81 mg daily), during the first 3 months of treatment.
Laboratory Abnormalities
Hematologic Abnormalities: Treatment with CIBINQO was associated with an increased incidence of thrombocytopenia and lymphopenia. Prior to CIBINQO initiation, perform a complete blood count (CBC). CBC evaluations are recommended at 4 weeks after initiation and 4 weeks after dose increase of CIBINQO. Discontinuation of CIBINQO therapy is required for certain laboratory abnormalities.
Lipid Elevations: Dose-dependent increase in blood lipid parameters were reported in patients treated with CIBINQO. Lipid parameters should be assessed approximately 4 weeks following initiation of CIBINQO therapy, and thereafter patients should be managed according to clinical guidelines for hyperlipidemia. The effect of these lipid parameter elevations on cardiovascular morbidity and mortality has not been determined.
Immunizations
Prior to initiating CIBINQO, complete all age-appropriate vaccinations as recommended by current immunization guidelines, including prophylactic herpes zoster vaccinations. Avoid vaccination with live vaccines immediately prior to, during, and immediately after CIBINQO therapy.
Renal Impairment
Avoid use in patients with severe renal impairment or end stage renal disease, including those on renal replacement therapy.
Hepatic Impairment
Avoid use in patients with severe hepatic impairment.
Adverse Reactions
Most common adverse reactions (≥1%) in subjects receiving 100 mg and 200 mg include: nasopharyngitis, nausea, headache, herpes simplex, increased blood creatinine phosphokinase, dizziness, urinary tract infection, fatigue, acne, vomiting, oropharyngeal pain, influenza, gastroenteritis.
Most common adverse reactions (≥1%) in subjects receiving either 100 mg or 200 mg also include: impetigo, hypertension, contact dermatitis, upper abdominal pain, abdominal discomfort, herpes zoster, and thrombocytopenia.
Use in Pregnancy
Available data from pregnancies reported in clinical trials with CIBINQO are not sufficient to establish a drug-associated risk for major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Advise females of reproductive potential that CIBINQO may impair fertility.
There will be a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to CIBINQO during pregnancy. Pregnant women exposed to CIBINQO and health care providers are encouraged to call 1-877-311-3770.
Lactation
Advise women not to breastfeed during treatment with CIBINQO and for one day after the last dose.
Indication
CIBINQO is indicated for the treatment of adults with refractory, moderate to severe atopic dermatitis whose disease is not adequately controlled with other systemic drug products, including biologics, or when use of those therapies is inadvisable.
Limitations of Use: CIBINQO is not recommended for use in combination with other JAK inhibitors, biologic immunomodulators, or with other immunosuppressants.
About CIBINQO® (abrocitinib)
CIBINQO is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD, including interleukin IL-4, IL-13, IL-31, IL-22, and thymic stromal lymphopoietin (TSLP).
In addition to receiving regulatory approval in the U.S., CIBINQO has received marketing authorization in the European Union, Great Britain, Japan, Korea, the United Arab Emirates, Norway, Iceland, and Singapore.
About Atopic Dermatitis
AD is a chronic skin disease characterized by inflammation of the skin and skin barrier defects.i,ii Most people know AD is a skin condition. But many don’t realize it can be caused in part by an abnormal immune response beneath the skin. This dysregulated immune response is thought to contribute to inflammation within the skin and the signs of AD on the surface. Lesions of AD are characterized by erythema (red/pink or discolored skin patches, depending on normal skin color), itching, lichenification (thick/leathery skin), induration (hardening)/papulation (formulation of papules), and oozing/crusting.i,ii
AD is one of the most common inflammatory skin diseases, affecting approximately 5-10% of adults in the U.S.iii,iv Approximately 1 in 3 adults with AD have moderate-to-severe disease.v,vi
About Pfizer Inflammation & Immunology
At Pfizer Inflammation & Immunology, we strive to deliver breakthroughs that enable freedom from day-to-day suffering for people living with autoimmune and chronic inflammatory diseases, which can be debilitating, disfiguring and distressing, dramatically affecting what they can do. With a focus on immuno-inflammatory conditions in Rheumatology, Gastroenterology and Medical Dermatology, our current portfolio of approved medicines and investigational molecules spans multiple action and delivery mechanisms, from topicals to small molecules, biologics and biosimilars. The root cause of many immunological diseases is immuno-inflammation, which requires specifically designed agents. Our differentiated R&D approach resulted in one of the broadest pipelines in the industry, where we purposefully match molecules to diseases where we believe they can make the biggest difference. Building on our decades-long commitment and pioneering science, we continue to advance the standard of care for patients living with immuno-inflammatory diseases and are working hand-in-hand with patients, caregivers and the broader healthcare community on healthcare solutions for the many challenges of managing chronic inflammatory diseases, allowing patients to live their best lives.
Pfizer Inc.: Breakthroughs that Change Patients’ Lives
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety, and value in the discovery, development, and manufacture of health care products, including innovative medicines and vaccines. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments, and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments, and local communities to support and expand access to reliable, affordable health care around the world. For more than 170 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
There remains a need for new compounds that effectively and selectively inhibit specific JAK enzymes, and JAK1 in particular, vs. JAK2. JAK1 is a member of the Janus family of protein kinases composed of JAK1, JAK2, JAK3 and TYK2. JAK1 is expressed to various levels in all tissues. Many cytokine receptors signal through pairs of JAK kinases in the following combinations: JAK1/JAK2, JAK1/JAK3, JAK1/TYK2 , JAK2/TYK2 or JAK2/JAK2. JAK1 is the most broadly
paired JAK kinase in this context and is required for signaling by γ-common (IL-2Rγ) cytokine receptors, IL—6 receptor family, Type I, II and III receptor families and IL- 10 receptor family. Animal studies have shown that JAK1 is required for the development, function and homeostasis of the immune system. Modulation of immune activity through inhibition of JAK1 kinase activity can prove useful in the treatment of various immune disorders (Murray, P.J.
J. Immunol., 178, 2623-2629 (2007); Kisseleva, T., et al., Gene, 285 , 1-24 (2002); O’Shea, J . J., et al., Ceil , 109, (suppl .) S121-S131 (2002)) while avoiding JAK2 dependent erythropoietin (EPO) and thrombopoietin (TPO) signaling (Neubauer H., et al., Cell, 93(3), 397-409 (1998);
Parganas E., et al., Cell, 93(3), 385-95 (1998)).

Tofacitinib (1), baricitinib (2), and ruxolitinib (3)
SYNTHESIS 5+1 =6 steps
Main synthesis
Journal of Medicinal Chemistry, 61(3), 1130-1152; 2018

INTERMEDIATE
CN 105732637
ONE STEP

CAS 479633-63-1, 7H-Pyrrolo[2,3-d]pyrimidine, 4-chloro-7-[(4- methylphenyl)sulfonyl]-
![]()
Pfizer Receives Breakthrough Therapy Designation from FDA for PF-04965842, an oral JAK1 Inhibitor, for the Treatment of Patients with Moderate-to-Severe Atopic Dermatitis
Dateline:
Public Company Information:
NEW YORK–(BUSINESS WIRE)–Pfizer Inc. (NYSE:PFE) today announced its once-daily oral Janus kinase 1 (JAK1) inhibitor PF-04965842 received Breakthrough Therapy designation from the U.S. Food and Drug Administration (FDA) for the treatment of patients with moderate-to-severe atopic dermatitis (AD). The Phase 3 program for PF-04965842 initiated in December and is the first trial in the J AK1 A topic D ermatitis E fficacy and Safety (JADE) global development program.
“Achieving Breakthrough Therapy Designation is an important milestone not only for Pfizer but also for patients living with the often devastating impact of moderate-to-severe atopic dermatitis, their providers and caregivers,” said Michael Corbo, Chief Development Officer, Inflammation & Immunology, Pfizer Global Product Development. “We look forward to working closely with the FDA throughout our ongoing Phase 3 development program with the hope of ultimately bringing this important new treatment option to these patients.”
Breakthrough Therapy Designation was initiated as part of the Food and Drug Administration Safety and Innovation Act (FDASIA) signed in 2012. As defined by the FDA, a breakthrough therapy is a drug intended to be used alone or in combination with one or more other drugs to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If a drug is designated as a breakthrough therapy, the FDA will expedite the development and review of such drug.1
About PF-04965842 and Pfizer’s Kinase Inhibitor Leadership
PF-04965842 is an oral small molecule that selectively inhibits Janus kinase (JAK) 1. Inhibition of JAK1 is thought to modulate multiple cytokines involved in pathophysiology of AD including interleukin (IL)-4, IL-13, IL-31 and interferon gamma.
Pfizer has established a leading kinase research capability with multiple unique kinase inhibitor therapies in development. As a pioneer in JAK science, the Company is advancing several investigational programs with novel selectivity profiles, which, if successful, could potentially deliver transformative therapies for patients. Pfizer has three additional kinase inhibitors in Phase 2 development across multiple indications:
- PF-06651600: A JAK3 inhibitor under investigation for the treatment of rheumatoid arthritis, ulcerative colitis and alopecia areata
- PF-06700841: A tyrosine kinase 2 (TYK2)/JAK1 inhibitor under investigation for the treatment of psoriasis, ulcerative colitis and alopecia areata
- PF-06650833: An interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitor under investigation for the treatment of rheumatoid arthritis
Working together for a healthier world®
At Pfizer, we apply science and our global resources to bring therapies to people that extend and significantly improve their lives. We strive to set the standard for quality, safety and value in the discovery, development and manufacture of health care products. Our global portfolio includes medicines and vaccines as well as many of the world’s best-known consumer health care products. Every day, Pfizer colleagues work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. Consistent with our responsibility as one of the world’s premier innovative biopharmaceutical companies, we collaborate with health care providers, governments and local communities to support and expand access to reliable, affordable health care around the world. For more than 150 years, we have worked to make a difference for all who rely on us. We routinely post information that may be important to investors on our website at www.pfizer.com. In addition, to learn more, please visit us on www.pfizer.com and follow us on Twitter at @Pfizer and @Pfizer_News, LinkedIn, YouTube and like us on Facebook at Facebook.com/Pfizer.
DISCLOSURE NOTICE: The information contained in this release is as of February 14, 2018. Pfizer assumes no obligation to update forward-looking statements contained in this release as the result of new information or future events or developments.
This release contains forward-looking information about PF-04965842 and Pfizer’s ongoing investigational programs in kinase inhibitor therapies, including their potential benefits, that involves substantial risks and uncertainties that could cause actual results to differ materially from those expressed or implied by such statements. Risks and uncertainties include, among other things, the uncertainties inherent in research and development, including the ability to meet anticipated clinical trial commencement and completion dates and regulatory submission dates, as well as the possibility of unfavorable clinical trial results, including unfavorable new clinical data and additional analyses of existing data; risks associated with preliminary data; the risk that clinical trial data are subject to differing interpretations, and, even when we view data as sufficient to support the safety and/or effectiveness of a product candidate, regulatory authorities may not share our views and may require additional data or may deny approval altogether; whether regulatory authorities will be satisfied with the design of and results from our clinical studies; whether and when drug applications may be filed in any jurisdictions for any potential indication for PF-04965842 or any other investigational kinase inhibitor therapies; whether and when any such applications may be approved by regulatory authorities, which will depend on the assessment by such regulatory authorities of the benefit-risk profile suggested by the totality of the efficacy and safety information submitted, and, if approved, whether PF-04965842 or any such other investigational kinase inhibitor therapies will be commercially successful; decisions by regulatory authorities regarding labeling, safety and other matters that could affect the availability or commercial potential of PF-04965842 or any other investigational kinase inhibitor therapies; and competitive developments.
A further description of risks and uncertainties can be found in Pfizer’s Annual Report on Form 10-K for the fiscal year ended December 31, 2016 and in its subsequent reports on Form 10-Q, including in the sections thereof captioned “Risk Factors” and “Forward-Looking Information and Factors That May Affect Future Results”, as well as in its subsequent reports on Form 8-K, all of which are filed with the U.S. Securities and Exchange Commission and available at www.sec.gov and www.pfizer.com .

# # # # #
1 Food and Drug Administration Fact Sheet Breakthrough Therapies at https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDASIA/ucm329491.htmaccessed on January 25, 2018
PATENT
CA 2899888
PATENT
WO 2014128591
PFIZER INC. [US/US]; 235 East 42nd Street New York, New York 10017 (US)
BROWN, Matthew Frank; (US).
FENWICK, Ashley Edward; (US).
FLANAGAN, Mark Edward; (US).
GONZALES, Andrea; (US).
JOHNSON, Timothy Allan; (US).
KAILA, Neelu; (US).
MITTON-FRY, Mark J.; (US).
STROHBACH, Joseph Walter; (US).
TENBRINK, Ruth E.; (US).
TRZUPEK, John David; (US).
UNWALLA, Rayomand Jal; (US).
VAZQUEZ, Michael L.; (US).
PARIKH, Mihir, D.; (US)
COMPD 2

Example 2 : N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane- l -sulƒonamide
This compound was prepared using 1-propanesulfonyl chloride. The crude compound was purified by chromatography on silica gel eluting with a mixture of dichloromethane and methanol (93 : 7) to afford the title compound as a tan sol id (78% yield). 1H NMR (400 MHz, DMSO-d6): δ 11.60 (br s, 1 H), 8.08 (s, 1 H), 7.46 (d, 1 H), 7.12 (d, 1 H), 6.61 (d, 1 H), 4.81-4.94 (m, 1 H), 3.47-3.62 (m, 1 H), 3.23 (s, 3 H), 2.87-2.96 (m, 2 H), 2.52-2.63 (m, 2 H), 2.14-2.27 (m, 2 H) 1.60- 1.73 (m, 2 H) 0.96 (t, 3 H). LC/MS (exact mass) calculated for C14H21N5O2S;
323.142, found (M + H+); 324.1.
PAPER
Journal of Medicinal Chemistry (2018), 61(3), 1130-1152.

https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.7b01598
N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}propane-1-sulfonamide (25)
Schmieder, G.; Draelos, Z.; Pariser, D.; Banfield, C.; Cox, L.; Hodge, M.; Kieras, E.; Parsons-Rich, D.; Menon, S.; Salganik, M.; Page, K.; Peeva, E. Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled study Br. J. Dermatol. 2017, DOI: 10.1111/bjd.16004
Compound 25, N-{cis-3-[Methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutyl}-propane-1-sulfonamide is available through MilliporeSigma (cat. no. PZ0304).
CLIP
REFERENCES
1: Schmieder GJ, Draelos ZD, Pariser DM, Banfield C, Cox L, Hodge M, Kieras E, Parsons-Rich D, Menon S, Salganik M, Page K, Peeva E. Efficacy and safety of the Janus Kinase 1 inhibitor PF-04965842 in patients with moderate to severe psoriasis: phase 2, randomized, double-blind, placebo-controlled study. Br J Dermatol. 2017 Sep 26. doi: 10.1111/bjd.16004. [Epub ahead of print] PubMed PMID: 28949012
2 Journal of Medicinal Chemistry (2018), 61(3), 1130-1152.
- Originator Pfizer
- Class Anti-inflammatories; Antipsoriatics; Pyrimidines; Pyrroles; Skin disorder therapies; Small molecules; Sulfonamides
- Mechanism of Action Janus kinase 1 inhibitors
- Phase III Atopic dermatitis
- Discontinued Lupus vulgaris; Plaque psoriasis
- 21 May 2019Pfizer initiates enrolment in a phase I trial in Healthy volunteers in USA (PO) (NCT03937258)
- 09 May 2019 Pfizer plans a phase I pharmacokinetic and drug-drug interaction trial in healthy volunteers in May 2019 (NCT03937258)
- 30 Apr 2019 Pfizer completes a phase I trial (In volunteers) in USA (PO) (NCT03626415)
References[
- ^ https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/213871s000lbl.pdf
- ^ Jump up to:a b c d e “Cibinqo EPAR”. European Medicines Agency (EMA). 11 October 2021. Retrieved 17 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. (October 2019). “Efficacy and Safety of Oral Janus Kinase 1 Inhibitor Abrocitinib for Patients With Atopic Dermatitis: A Phase 2 Randomized Clinical Trial”. JAMA Dermatology. 155 (12): 1371–1379. doi:10.1001/jamadermatol.2019.2855. PMC 6777226. PMID 31577341.
- ^ Peeva E, Hodge MR, Kieras E, Vazquez ML, Goteti K, Tarabar SG, et al. (August 2018). “Evaluation of a Janus kinase 1 inhibitor, PF-04965842, in healthy subjects: A phase 1, randomized, placebo-controlled, dose-escalation study”. British Journal of Clinical Pharmacology. 84 (8): 1776–1788. doi:10.1111/bcp.13612. PMC 6046510. PMID 29672897.
- ^ Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
- ^ “Pfizer Presents Positive Phase 3 Data at the 28th Congress of the European Academy of Dermatology and Venereology for Abrocitinib in Moderate to Severe Atopic Dermatitis”. Drugs.com. 12 October 2019.
- ^ Silverberg, J. I.; Simpson, E. L.; Thyssen, J. P.; Gooderham, M.; Chan, G.; Feeney, C.; Biswas, P.; Valdez, H.; Dibonaventura, M.; Nduaka, C.; Rojo, R. (3 June 2020). “Efficacy and Safety of Abrocitinib in Patients With Moderate-to-Severe Atopic Dermatitis: A Randomized Clinical Trial”. JAMA Dermatology. 156 (8): 863–873. doi:10.1001/jamadermatol.2020.1406. PMC 7271424. PMID 32492087.
- ^ Jump up to:a b “Cibinqo: Pending EC decision”. European Medicines Agency. 15 October 2021. Retrieved 15 October 2021. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “European Commission Approves Pfizer’s Cibinqo (abrocitinib) for the Treatment of Adults with Moderate-to-Severe Atopic Dermatitis”. Pfizer Inc. (Press release). 10 December 2021. Retrieved 17 December 2021.
- ^ “U.S. FDA Approves Pfizer’s Cibinqo (abrocitinib) for Adults with Moderate-to-Severe Atopic Dermatitis”. Pfizer Inc. (Press release). 14 January 2022. Retrieved 16 January 2022.
External links
- “Abrocitinib”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03349060 for “Study to Evaluate Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-1)” at ClinicalTrials.gov
- Clinical trial number NCT03575871 for “Study Evaluating Efficacy and Safety of PF-04965842 in Subjects Aged 12 Years And Older With Moderate to Severe Atopic Dermatitis (JADE Mono-2)” at ClinicalTrials.gov
- {{ClinicalTrialsGov|NCT03720470|Study Evaluating Efficacy and Safety of PF-04965842 and Dupilumab in Adult Subjects With Moderate to Severe Atopic Dermatitis on Background Topical Therapy (JADE Compare)}
 |
|
| Clinical data | |
|---|---|
| Trade names | Cibinqo |
| Other names | PF-04965842 |
| License data |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Elimination half-life | 2.8–5.2 h |
| Excretion | 1.0–4.4% unchanged in urine |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| ECHA InfoCard | 100.251.498 |
| Chemical and physical data | |
| Formula | C14H21N5O2S |
| Molar mass | 323.42 g·mol−1 |
| 3D model (JSmol) | |
/////////PF 04965842, Abrocitinib, Phase III, Atopic dermatitis, pfizer, fda 2022, APPROVALS 2022
CCCS(=O)(N[C@H]1C[C@@H](N(C)C2=C3C(NC=C3)=NC=N2)C1)=O
CCCS(=O)(=O)N[C@@H]1C[C@@H](C1)N(C)c2ncnc3[nH]ccc23
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

NEW DRUG APPROVALS
ONE TIME
$10.00
Ritlecitinib, PF 06651600
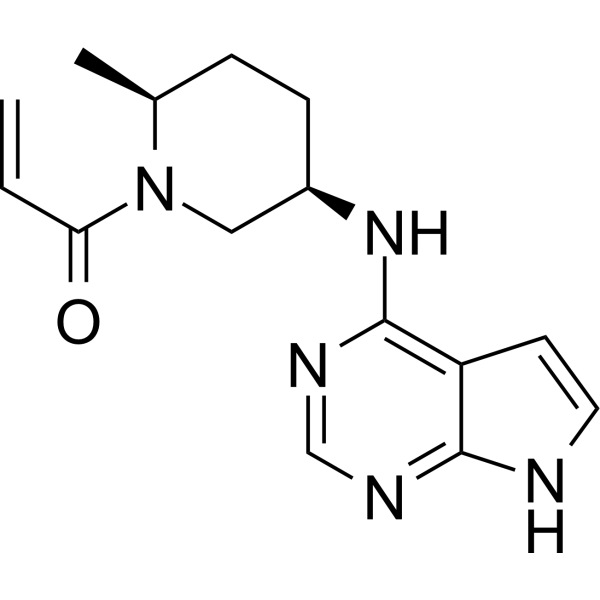


Ritlecitinib
PF-06651600
CAS 1792180-81-4
C₁₅H₁₉N₅O, 285.34, UNII-2OYE00PC25
Fda approved Litfulo, 6/23/2023, To treat severely patchy hair loss
Drug Trials Snapshot
1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one

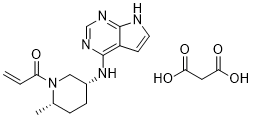
1-[(2S,5R)-2-Methyl-5-(7H-pyrrolo[2,3-d]pyrimidin-4-ylamino)-1-piperidinyl]-2-propen-1-one malonate
PF-06651600 malonate
CAS: 2140301-97-7 (malonate)
Chemical Formula: C18H23N5O5
Molecular Weight: 389.412
PHASE 2 alopecia areata, rheumatoid arthritis, Crohn’s disease, and ulcerative colitis.
Ritlecitinib, sold under the brand name Litfulo, is a medication used for the treatment of severe alopecia areata (hair loss).[6] Ritlecitinib is a kinase inhibitor which inhibits Janus kinase 3 and tyrosine kinase.[6][9][10]
The most common side effects include headache, diarrhea, acne, rashes, eczema, fever, mouth ulcers, dizziness, shingles rash, and abnormal findings in some laboratory test results.[11]
Ritlecitinib was approved for medical use in the United States in June 2023,[6][11][12] in the European Union in September 2023,[7] and in Canada in November 2023.[4]
Pfizer is developing ritlecitinib, an irreversible, covalent and selective dual JAK3/TEC inhibitor, for treating AA, RA, vitiligo and inflammatory bowel diseases, including UC and CD. In July 2021, this drug was reported to be in phase 3 clinical development.
PF-06651600 is a potent and selective JAK3 inhibitor. PF-06651600 is a potent and low clearance compound with demonstrated in vivo efficacy. The favorable efficacy and safety profile of this JAK3-specific inhibitor PF-06651600 led to its evaluation in several human clinical studies. JAK3 was among the first of the JAKs targeted for therapeutic intervention due to the strong validation provided by human SCID patients displaying JAK3 deficiencies
Pfizer has established a leading kinase research capability with multiple unique kinase inhibitors in development as potential medicines. PF-06651600 is a highly selective and orally bioavailable Janus Kinase 3 (JAK3) inhibitor that represents a potential immunomodulatory therapy. With the favorable efficacy, safety profile, and ADME properties, this JAK3-specific covalent inhibitor has been under clinical investigation for the treatment of alopecia areata, rheumatoid arthritis, Crohn’s disease, and ulcerative colitis. Supported by positive results from a Phase 2 study, 1 was granted Breakthrough Therapy designation by the FDA on Sept. 5, 2018 for treatment of alopecia areata.
SYN

PAPER
J. Med. Chem. 2017, 60 (5), 1971–1993, DOI: 10.1021/acs.jmedchem.6b01694
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b01694
Paper
Process Development and Scale Up of a Selective JAK3 Covalent Inhibitor PF-06651600,
Yong Tao*

A scalable process for PF-06651600 (1) has been developed through successful enabling of the first generation syntheis. The synthesis highlights include the following: (1) replacement of costly PtO2 with a less expensive 5% Rh/C catalyst for a pyridine hydrogenation, (2) identification of a diasteroemeric salt crystallization to isolate the enantiomerically pure cis-isomer directly from a racemic mixture of cis/trans isomers, (3) a high yielding amidation via Schotten–Baumann conditions, and (4) critical development of a reproducible crystallization procedure for a stable crystalline salt (1·TsOH), which is suitable for long-term storage and tablet formulation. All chromatographic purifications, including two chiral SFC chromatographic separations, were eliminated. Combined with other improvements in each step of the synthesis, the overall yield was increased from 5% to 14%. Several multikilogram batches of the API have been delivered to support clinical studies.
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00198
1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop-2-en-1-one p-Toluenesulfonate (1·TsOH)
1·TsOH (4.41 kg, 9.64 mol) as a white powder in 89.6% yield (accounting for the amount of seed charged). Achiral HPLC purity: 99.6% with 0.22% of dimer 15. Chiral SFC purity: >99.7%. Mp 199 °C. Rotomers observed for NMR spectroscopies. 1H NMR (400 MHz, DMSO-d6): δ ppm 12.68 (brs, 1H), 9.22 (brs, 1H), 8.40 (s, 1H), 7.50 (d, J = 8.2 Hz, 2H), 7.45 (m, 1H), 7.12 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 1.2 Hz, 1H), 6.84 (m, 1H), 6.13 (m, 1H), 5.70 (m, 1H), 4.81 (m, 0.5H), 4.54 (m, 0.5H), 4.41 (m, 0.5H), 4.12 (m, 0.5H), 3.99 (m, 1H), 3.15 (m, 0.5H), 2.82 (m, 0.5H), 2.29 (s, 3H), 1.91–1.72 (m, 4H), 1.24–1.17 (m, 3H). 13C NMR (100 MHz, DMSO-d6): δ ppm 165.52, 165.13, 150.50, 145.64, 143.06, 138.48, 129.51, 129.24, 128.67, 127.99, 127.73, 125.97, 125.02, 102.30, 49.53, 48.92, 47.27, 43.83, 42.96, 29.37, 28.41, 25.22, 21.28, 16.97, 15.51. HRMS (ESI) m/z: calculated for C15H20N5O [M + H]+286.1668; observed 286.1692.
PAPER
PATENT
WO2015083028
PATENT
WO 2015083028
https://patents.google.com/patent/WO2015083028A1
PATENT
WO 2020084435
1 -((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one has the structural formula:
The synthesis of 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one is described in WO2015/083028, commonly assigned to the assignee of the present invention and which is incorporated herein by reference in its entirety. 1 -((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one is useful as an inhibitor of protein kinases, such as the enzyme Janus Kinase (JAK) and as such is useful therapy as an immunosuppressive agent for organ transplants, xeno transplantation, lupus, multiple sclerosis, rheumatoid arthritis, psoriasis, Type I diabetes and complications from diabetes, cancer, asthma, atopic dermatitis, autoimmune thyroid disorders, ulcerative colitis, Crohn’s disease, alopecia, vitiligo, Alzheimer’s disease, leukemia and other indications where immunosuppression would be desirable. See ACS Chem. Biol. , 2016, 11 (12), pp 3442-3451 . The present invention relates to a novel p-toluenesulfonic acid salt and crystalline solid form of the said salt of 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one that demonstrate improved properties for use in a pharmaceutical dosage form, particularly for oral dosage forms.
Preparations
Scheme 1. Synthesis of 1
Scheme 2. Alternate Synthesis of Intermediates 7 and 10
1. K2C03, MIBK/water 1. H2, H2O
2. EtOAc, aq. NaCI Pd(OH)2/C (wet)
3. MeOH, H20 2. NaOH, MeOH
7 + 8 – ► 9 – – 10 . H2O
89% 89%
Scheme 3. First Alternate Preparation of 1
Scheme 4. Second Alternate Preparation of 1
Preparation 1
ferf-Butyl (6-methylpyridin-3-yl)carbamate (3). To a 3000L reactor was charged 2 (72.00 kg, 665.8 mol) and THF (660 kg). A solution of NH4CI (1 .07 kg , 20 mol) in water (72 kg, 4000 mol) was added. The mixture was heated to 57 °C and Di-f-butyl dicarbonate (220.0 kg, 1003 mol) was added slowly with rinse of THF (45 kg) while maintaining the temperature between 55 – 60 °C. The mixture was stirred at 55 – 60 °C for 10 h. Upon reaction completion, the slurry was cooled to 20 °C and ethyl acetate (654 kg) and water (367 kg) were added. The organic phase was separated, washed by water (2 x 360 kg) and stirred with active carbon (22 kg) for 5 h. The mixture was filtered through a layer of diatomaceous earth (22 kg) with THF rinse and the filtrates were concentrated under vacuum at <40 °C to a residual volume of ~370 L. n-Heptane (500 kg) was added slowly over 1 h and the resulting slurry was cooled to 20 °C and stirred for 2 h. The solid was collected by centrifuge with an n-heptane wash (420 kg), then dried at 45 °C under vacuum for 20 h to give 3 (131 .15 kg, 629.7 mol) as a white powder in 94.5% yield. HPLC purity: 99.9%. 1H NMR (400 MHz, DMSO-c/6): d ppm 9.42 (brs, 1 H), 8.48 (d, J = 1 .9 Hz, 1 H), 7.75 (d, J = 8.6 Hz, 1 H), 7.13 (d, J = 8.6 Hz, 1 H), 2.38 (s, 3H), 1 .49 (s, 9H). 13C NMR (100 MHz, DMSO-d6y d ppm 153.34, 151 .56, 139.75, 134.13, 126.10, 123.09, 79.87, 28.56, 23.70. HRMS (ESI) m/z: calculated for C11H17N2O2 [M + H]+ 209.1290; observed 209.1285.
Preparation 2
ferf-Butyl (6-methylpiperidin-3-yl)carbamate (rac-4). To a 3000L reactor was charged 3 (137.0 kg, 667.8 mol), ethanol (988 kg) and acetic acid (139 kg). The reactor was purged with nitrogen three times and 5 wt% Rhodium on carbon (wet, 27.4 kg, 20 wt% loading relative to 3) was added. The reactor was purged with nitrogen three times and then with hydrogen three times. The hydrogen pressure was adjusted to 0.34 – 0.38 MPa and the reactor temperature was adjusted to 47 °C. The mixture was stirred at 45 – 60 °C under hydrogen pressure at 0.34 – 0.38 MPa for 10 h. Upon reaction completion, the reactor was cooled to 20 °C and flushed with nitrogen. The mixture was filtered through a layer of diatomaceous earth (20 kg) with an ethanol rinse (1320 kg) and the filtrates were concentrated under vacuum at <50 °C to a residual volume of ~350 L. n-Heptane (571 kg) was added and the mixture was concentrated under vacuum at <50 °C to a residual volume of~350 L. This operation was repeated twice until the residual acetic acid <8.0%. Ethanol (672 kg) was added and the mixture was concentrated under vacuum at <50 °C to a residual volume of ~350 L. This operation was repeated twice until the residual n-heptane was <0.2% and water was <0.2%. Ethanol (889 kg) was added and the solution (1254 kg) was transferred to drums for use in the subsequent classical resolution step. Achiral HPLC assay indicated that the solution contained 10.8 wt% of the total reduced product (rac-4) in 96% mass recovery and chiral SFC showed that the solution contained 36.3% of the desired stereoisomer cis-4.
Preparation 3
ferf-Butyl ((3R,6S)-6-methylpiperidin-3-yl)carbamate (R)-2-(3,5-dinitrobenzamido)-2-phenylacetic acid salt (15). To a 2000L reactor (R1 ) was charged rac-4 as a 10.8 wt% solution in ethanol (620.5 kg, ~312.7 mol. of all 4 isomers). The solution was concentrated under vacuum at <45 °C to a residual volume of ~210 L and then cooled to 20 °C. To a 3000 L reactor (R2) was charged (R)-2-(3,5-dinitrobenzamido)-2-phenylacetic acid 14 (47.0 kg, 136.1 mol) and ethanol (1 125 kg). With high speed agitation, reactor R2 was heated to 70 °C, stirred at 68 – 70 °C for ~2 h to dissolve all solid 14, and then seeded with crystalline 15 (1 1 g). The solution containing 4 in reactor R1 was slowly transferred to reactor R2 over 30 min with ethanol rinse (160 kg). Reactor R2 was stirred at ~74 °C for 3 h and then cooled to 22 °C with a linear cooling rate over a period of 5 h and stirred for 16 h. The solid was collected by centrifuge with ethanol wash (2 x 200 kg). The wet cake (with 97.1 % e.e.) was charged back to reactor R2. The slurry was heated to 74 °C and the mixture was stirred for 17 h. The mixture was then cooled to 22 °C with a linear cooling rate over a period of 5 h and stirred for 4 h. The solid was collected by centrifuge with ethanol wash (2 x 200 kg) and dried at 35 °C under vacuum for 25 h to give 15 (56.05 kg, 100.2 mol) as a white powder in 30.7% yield over 2 steps. Chiral HPLC purity: 99.1 %. 1H NMR (400 MHz, DMSO-d6): d ppm 9.46 (d, J = 7.0 Hz, 1 H), 9.07 (d, J = 2.2 Hz, 2H), 8.96 (t, J = 2.2 Hz, 1 H), 7.49 (d, J = 7.3 Hz, 2H), 7.30 (t, J = 7.3 Hz, 2H), 7.23 (t, J = 7.3, 1 H), 7.1 1 (m, 1 H), 5.31 (d, J = 7.0 Hz, 1 H), 3.66 (m, 1 H), 2.98 (m, 3H), 1 .63 (m, 2H), 1 .45 (m, 2H), 1 .40 (s, 9H), 1 .1 1 (d, J = 6.7 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 172.71 , 161 .71 , 155.42, 148.51 , 141 .27, 137.70, 128.29, 128.25, 128.02, 127.05, 121 .12, 78.49, 59.74, 50.66, 46.29, 43.34, 28.66, 26.88, 26.1 1 , 18.60.
Preparation 4
Benzyl (2S,5R)-5-amino-2-methylpiperidine-1 -carboxylate hydrochloride (7»HCI) -telescoped process. To a 2000L reactor was charged 15 (70.0 kg, 125 mol) and MTBE (500 kg). The mixture was cooled to 12 °C and 6.9 wt% aqueous NaOH solution (378 kg, 652 mol) was added slowly while maintaining the temperature between 10 – 25 °C. The mixture was stirred at 18 °C for 1 h . The organic phase was separated and washed with 3.8 wt% aqueous NaOH solution (2 x 221 kg) and then 25 wt% aqueous NaCI solution (2 x 220 kg). The organic layer (containing the free base cis-4) was concentrated under vacuum at <40 °C to a residual volume of ~300 L and then cooled to 20 °C. NaHCOs (53 kg, 632 mol) and water (200 kg) were added and the mixture was cooled to 7 °C. Benzyl chloroformate (32.30 kg, 189.3 mol) was added slowly while maintaining the temperature between 5 – 20 °C. The mixture was stirred at 17 °C for 20 h. Upon reaction completion, the mixture was cooled to 12 °C, 25 wt% aqueous ammonium hydroxide solution (79 kg, 1 160 mol) was added slowly while maintaining the temperature between 10 – 20 °C, and the mixture was stirred at 15 °C for 1 h. The organic phase was separated and washed with 25 wt% aqueous NaCI solution (3 x 90 kg). The organic layer (containing 5) was concentrated under vacuum at <45 °C to a residual volume of ~150 L. Isopropyl acetate (310 kg) was added and the mixture was concentrated under vacuum at <45 °C to a residual volume of ~150 L. This operation was repeated twice to meetthe criteria of water <0.1 % (by KF). Isopropyl acetate (130 kg) was then added and the mixture was cooled to -3 °C. 4-5N HCI in methanol (181 kg, ~730 mol) was added slowly while maintaining the temperature between -5 to 5 °C, and the mixture was stirred at 3 °C for 12 h. Upon reaction completion, the mixture was cooled to -3 °C and MTBE (940 kg) was added slowly while maintaining the temperature between -5 to 5 °C. The resulting slurry was stirred at 3 °C for 3 h. The solid was collected by centrifuge with MTBE washes (4 x 70 kg), and then dried at 45 °C under vacuum for 20 h to give 7»HCI (28.60 kg, 100.4 mol) as a white powder in 80.3% yield. Achiral HPLC purity: 100%. Chiral SFC purity: 99.8% e.e. 1H NMR (400 MHz, DMSO-d6): d ppm 8.36 (brs, 3H), 7.37 (m, 5H), 5.09 (s, 2H), 4.31 (m, 1 H), 4.16 (d, J = 8.2 Hz, 1 H), 3.00 (m, 2H), 1 .82 (m, 2H), 1 .59 (m, 2H), 1 .1 1 (d, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 154.71 , 137.24, 128.92, 128.34, 128.00, 66.89, 47.20, 45.66, 40.68, 28.16, 23.02, 15.67. HRMS (ESI) m/z. calculated for C H N O [M + H]+ 249.1603; observed 249.1598.
Preparation 5
Benzyl (2S, 5R)-5-((2-chloro-7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2 -methyl-piperidine-1 -carboxylate (9). To a 2000L reactor was charged 7»HCI (88.6 kg, 31 1 .12 mol), 8 (56.0 kg, 298 mol), K2C03 (133.0 kg, 962.3 mol), water (570 kg) and MIBK (101 kg). The mixture was heated to 90 °C and stirred at this temperature for 22 h. Upon reaction completion, the mixture was cooled to 56 °C and ethyl acetate (531 kg) was added. After cooling the mixture to 22 °C, the organic phase was separated, washed with water (570 kg) and concentrated under vacuum at <40 °C to a residual volume of ~220 L. Methanol (360 kg) was added slowly over a period of 1 h and the mixture concentrated under vacuum at <50 °C to a residual volume of ~220 L. This operation was repeated three times until residual MIBK reached <5 wt%. Methanol (270 kg) was added, followed by seeding with 9 (120 g). The mixture was stirred at 22 °C for >4 h and water (286 kg) was added slowly over 4 h. The slurry was stirred for 10 h and the solid was then collected by centrifuge. The wet cake (165.6 kg) was charged back to a clean reactor and water (896 kg) was added. The slurry was heated to 55 °C and stirred at this temperature for 7 h; and then cooled to 22 °C and stirred at this temperature for 2 h. The solid was collected by centrifuge with water wash (3 x 170 kg) and dried at 55 °C under vacuum for 20 h to give 9 (106.62 kg, 266.6 mol) as a white powder in 89.5% yield. Achiral HPLC purity: 99.7%. 1H NMR (400 MHz, DMSO-d6): d ppm 1 1 .71 (brs, 1 H), 7.72 (d, J = 7.9 Hz, 1 H), 7.38 (m, 5H), 7.10 (s, 1 H), 6.57 (d, J = 2.7 Hz, 1 H), 5.1 1 (m, 2H), 4.39 (m, 1 H), 4.17 (m, 1 H), 4.01 (m, 1 H), 3.36 (s, 2H), 2.77 (m, 1 H), 1 .73-1 .81 (m, 4H), 1 .16 (d, J = 6.6 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 156.65, 154.74, 153.04, 151 .31 , 137.43, 128.89, 128.27, 127.96, 122.13, 101 .65, 99.51 , 66.75, 49.10, 47.32, 45.64, 42.98, 29.05, 25.08. HRMS (ESI) m/z. calculated for C20H22CIN5O2 [M + H]+ 400.1540; observed 400.1535.
Preparation 6
N-((3R,6S)-6-methylpiperidin-3-yl)-7H-pyrrolo[2,3-dlpyrimidin-4-amine monohydrate (10·H2O) To a 1600L reactor was charged water (570 kg). The reactor was purged with nitrogen three times. 10% Pd(OH)2/C (wet, 3.2 kg) and 9 (53.34 kg, 133.2 mol) were added with water rinses (2 x 55 kg). The reactor was purged with nitrogen three times and then with hydrogen three times. The hydrogen pressure was adjusted to 0.34 – 0.38 MPa and the reactor temperature was adjusted to 77 °C. The mixture was stirred at 75 – 80 °C under a hydrogen pressure of 0.34 -0.38 MPa for 10 h. Upon reaction completion, the reactor was cooled to 20 °C and purged with nitrogen. The mixture was filtered through a layer of diatomaceous earth (8 kg) with a water rinse (460 kg), and the filtrates were transferred to a 3000L reactor. Methanol (260 kg) was added, followed by slow addition of 50 wt% aqueous sodium hydroxide (12.0 kg , 150 mol) while maintaining the temperature between 15 – 25 °C. The slurry was heated to 55 °C and stirred for 2 h; then cooled to 22 °C and stirred for 10 h. The solid was collected by centrifuge with a 10:1 water/methanol wash (3 x 1 10 kg) and then dried at 55 °C under vacuum for 20 h to give 10·H2O (30.90 kg, 266.6 mol) as a white powder in 89.1 % yield. Achiral HPLC purity: 99.7%. Chiral SFC
purity: 99.8% e.e. 1H NMR (400 MHz, DMSO-d6): d ppm 1 1 .48 (brs, 1 H), 8.08 (s, 1 H), 7.07 (s, 1 H), 6.85 (d, J = 7.3 Hz, 1 H), 6.64 (s, 1 H), 4.16 (m, 1 H), 3.35 (brs, 2H), 2.96 (d, J = 12.7 Hz, 1 H), 2.82 (d, J = 12.7 Hz, 1 H), 2.67 (m, 1 H), 2.04 (brs, 1 H), 1 .92 (m, 1 H), 1 .63 (m, 1 H), 1 .44 (m, 1 H), 1 .33 (m, 1 H), 1 .03 (d, J = 6.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): d ppm 155.95, 151 .87, 150.74, 121 .20, 102.97, 99.20, 51 .27, 49.94, 44.78, 29.97, 28.69, 22.35. HRMS (ESI) m/z\ calculated for C12H17N5 [M + H]+ 232.1562; observed 232.1558.
Preparation 7
1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one (1 ). To a 100L reactor was charged water (18.0 L), 10·H2q (3.60 kg, 14.4 mol) and THF (36.0 L). The mixture was heated to 53 °C and stirred for 15 min to dissolve all the solids. The solution was then cooled to 18 °C and K3PO4 (6.38 kg, 30.1 mol) was added. The mixture was stirred at 18 °C for 10 min to dissolve all the solids, and then cooled to 10 °C. 3-Chloropropionyl chloride (2.20 kg, 17.3 mol) was added while maintaining the temperature <20 °C. The mixture was then stirred at 20 °C for 2 h. Upon reaction completion, 2 N aqueous NaOH solution (23.50 kg, 43.76 mol) was added while maintaining the temperature <25 °C. The mixture was stirred at 22 °C for >12 h until the elimination reaction was complete (11 <0.2%). KH2PO4 (10.32 kg, 75.8 mol) was added and the mixture was stirred at 20 °C for 10 min. The organic phase was separated and then washed with 23.5 wt% aqueous NaCI solution (2 x 8.5 kg). The isolated organic phase was concentrated under vacuum at <30 °C to a residual volume of ~10 L, whereupon MEK (39.6 L) was added. This operation was repeated once or twice until residual THF was <1 % and water was <2%. MgS04 (0.96 kg), Silica gel (4.90 kg) and Darco™ G-60 (0.48 kg) were added to the MEK solution, and the mixture was stirred at 20 °C for 1 h, then filtered through a layer of Diatomaceous Earth with a MEK rinse (76 L). The combined filtrates were concentrated under vacuum at <30 °C to a residual volume of ~8 L. The concentration of the residual solution was measured by qNMR, and the solution was transferred to a container with a rinse using the calculated amount of MEK to adjust the final concentration to 30 wt%. Thus, a 30 wt% solution of 1 in MEK (1 1 .09 kg, 1 1 .66 mol of 1) with 98.7% purity was obtained in 81 % yield, which was stored in a cold room (2 – 8 °C) for the next step.
Preparation 8
1 -((2S,5R)-5-((7H-pyrrolo[2,3-cdpyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one p-toluenesulfonate (1»TsOH). To a 20L reactor was charged a 30 wt% solution of 1 in MEK (9.80 kg, 10.30 mol of 1) and silica gel (0.74 kg). The mixture was stirred at 22 °C for 15 min and filtered through a 0.45 micron Teflon cartridge filter with a MEK rinse (7.89 kg, 9.8 L), collecting in a 100L reactor. Water (1 .27 L) was added, followed by a solution of p-toluenesulfonic acid monohydrate (2.18 kg, 1 1 .3 mol) in MEK (4.75 kg, 5.9 L) with a MEK rinse (3.14 kg, 3.9 L), followed by the addition of 1 »TsOH seed (188 g, 0.41 mol). The mixture was stirred at 22 °C for
4 h to form a slurry and MEK (31 .56 kg, 39.2 L) was added slowly over a period of 3 h. The slurry was stirred at 22 °C for an additional 2 h and then filtered. The cake was washed with MEK (4.02 kg, 5 L) and then dried at 50 °C under vacuum for 10 h to give 1 »TsOH (4.41 kg, 9.64 mol) as a white powder in 89.6% yield (accounting for the amount of seed charged). Achiral HPLC purity: 99.6% with 0.22% of dimer 15. Chiral SFC purity: >99.7%. m.p. 199 °C. Rotomers observed for NMR spectroscopies. Ή NMR (400 MHz, DMSO-d6): d ppm 12.68 (brs, 1 H), 9.22 (brs, 1 H), 8.40 (s, 1 H), 7.50 (d, J = 8.2 Hz, 2H), 7.45 (m, 1 H), 7.12 (d, J = 8.2 Hz, 2H), 6.94 (d, J = 1 .2 Hz, 1 H), 6.84 (m, 1 H), 6.13 (m, 1 H), 5.70 (m, 1 H), 4.81 (m, 0.5H), 4.54 (m, 0.5H), 4.41 (m, 0.5H), 4.12 (m, 0.5H), 3.99 (m, 1 H), 3.15 (m, 0.5H), 2.82 (m, 0.5H), 2.29 (s, 3H), 1 .91 -1 .72 (m, 4H), 1 .24-1 .17 (m, 3H). 13C NMR (100 MHz, DMSO-c/6): d ppm 165.52, 165.13, 150.50, 145.64, 143.06, 138.48, 129.51 , 129.24, 128.67, 127.99, 127.73, 125.97, 125.02, 102.30, 49.53, 48.92, 47.27, 43.83, 42.96, 29.37, 28.41 , 25.22, 21 .28, 16.97, 15.51 . HRMS (ESI) m/z: calculated for Ci5H2oN50 [M + H]+ 286.1668; observed 286.1692.
Comparative Example
Preparation of 1 -((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methyl-piperidin-1 -yl)prop-2-en-1 -one Malonic Acid Salt (Form 1 )
A 250 ml_ round bottom flask was charged with 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one (4.10 g, 14.4 mmol), MEK (Methyl Ethyl Ketone (15.0 ml_/g, 687 mmol, 49.5 g, 61 .5 ml_)). To the solution, malonic acid (0.950 equiv. 13.7 mmol, 1 .42 g) was added in one portion. The mixture was heated to 50 °C and stirred at 50 °C for 15min. The heating was turned off and the slurry was stirred for 16 hours. The resulting white slurry was filtered. The filter cake was washed with MEK (2 X 5 ml_) and dried in a vacuum oven (40 °C) for 2 hours give 1 -((2S,5R)-5-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1 -yl)prop-2-en-1 -one malonic acid salt (Form 1) (4.48 g, 1 1 .5 mmol, 4.48 g, 80.1 % Yield) as white powder.
PATENT
WO-2021136482
Improved process for preparing ritlecitinib (PF06651600), useful for treating alopecia areata (AA), rheumatoid arthritis (RA), Crohn’s disease (CD) and ulcerative colitis (UC). Also claims novel intermediates of ritlecitinib and their preparation method.
PATENT
CN111732591
crystalline polymorphic form of ritlecitinib L-tartrate. Jiangsu Alicorn focuses on developing generic, innovative and first-line drugs, whose website lists an undisclosed JAK inhibitor under its product’s list.
REFERENCES
1: D’Amico F, Fiorino G, Furfaro F, Allocca M, Danese S. Janus kinase inhibitors for the treatment of inflammatory bowel diseases: developments from phase I and phase II clinical trials. Expert Opin Investig Drugs. 2018 Jul;27(7):595-599. doi: 10.1080/13543784.2018.1492547. Epub 2018 Jul 6. Review. PubMed PMID: 29938545.
2: Robinette ML, Cella M, Telliez JB, Ulland TK, Barrow AD, Capuder K, Gilfillan S, Lin LL, Notarangelo LD, Colonna M. Jak3 deficiency blocks innate lymphoid cell development. Mucosal Immunol. 2018 Jan;11(1):50-60. doi: 10.1038/mi.2017.38. Epub 2017 May 17. PubMed PMID: 28513593; PubMed Central PMCID: PMC5693788.
3: Thorarensen A, Dowty ME, Banker ME, Juba B, Jussif J, Lin T, Vincent F, Czerwinski RM, Casimiro-Garcia A, Unwalla R, Trujillo JI, Liang S, Balbo P, Che Y, Gilbert AM, Brown MF, Hayward M, Montgomery J, Leung L, Yang X, Soucy S, Hegen M, Coe J, Langille J, Vajdos F, Chrencik J, Telliez JB. Design of a Janus Kinase 3 (JAK3) Specific Inhibitor 1-((2S,5R)-5-((7H-Pyrrolo[2,3-d]pyrimidin-4-yl)amino)-2-methylpiperidin-1-yl)prop -2-en-1-one (PF-06651600) Allowing for the Interrogation of JAK3 Signaling in Humans. J Med Chem. 2017 Mar 9;60(5):1971-1993. doi: 10.1021/acs.jmedchem.6b01694. Epub 2017 Feb 16. PubMed PMID: 28139931.
4: Telliez JB, Dowty ME, Wang L, Jussif J, Lin T, Li L, Moy E, Balbo P, Li W, Zhao Y, Crouse K, Dickinson C, Symanowicz P, Hegen M, Banker ME, Vincent F, Unwalla R, Liang S, Gilbert AM, Brown MF, Hayward M, Montgomery J, Yang X, Bauman J, Trujillo JI, Casimiro-Garcia A, Vajdos FF, Leung L, Geoghegan KF, Quazi A, Xuan D, Jones L, Hett E, Wright K, Clark JD, Thorarensen A. Discovery of a JAK3-Selective Inhibitor: Functional Differentiation of JAK3-Selective Inhibition over pan-JAK or JAK1-Selective Inhibition. ACS Chem Biol. 2016 Dec 16;11(12):3442-3451. Epub 2016 Nov 10. PubMed PMID: 27791347.
5: Walker G, Croasdell G. The European League Against Rheumatism (EULAR) – 17th Annual European Congress of Rheumatology (June 8-11, 2016 – London, UK). Drugs Today (Barc). 2016 Jun;52(6):355-60. doi: 10.1358/dot.2016.52.6.2516435. PubMed PMID: 27458612.
References
- ^ Jump up to:a b “Litfulo (ritlecitinib)”. Therapeutic Goods Administration (TGA). 30 July 2024. Retrieved 12 October 2024.
- ^ https://www.tga.gov.au/resources/prescription-medicines-registrations/litfulo-pfizer-australia-pty-ltd
- ^ “Litfulo Product information”. Health Canada. 13 February 2024. Retrieved 3 March 2024.
- ^ Jump up to:a b c “Summary Basis of Decision for Litfulo”. Health Canada. 18 July 2024. Retrieved 28 August 2024.
- ^ “Regulatory Decision Summary for Litfulo”. Health Canada. 29 November 2023. Retrieved 2 April 2024.
- ^ Jump up to:a b c d e f “Litfulo- ritlecitinib capsule”. DailyMed. U.S. National Library of Medicine. 23 June 2023. Archived from the original on 29 August 2023. Retrieved 28 August 2023.
- ^ Jump up to:a b c d “Litfulo EPAR”. European Medicines Agency. 18 September 2023. Archived from the original on 19 September 2023. Retrieved 20 September 2023.
- ^ “Litfulo Product information”. Union Register of medicinal products. 18 September 2023. Archived from the original on 1 October 2023. Retrieved 1 October 2023.
- ^ “Ritlecitinib”. Inxight Drugs. Archived from the original on 25 June 2023. Retrieved 24 June 2023.
- ^ Ramírez-Marín HA, Tosti A (February 2022). “Evaluating the Therapeutic Potential of Ritlecitinib for the Treatment of Alopecia Areata”. Drug Design, Development and Therapy. 16: 363–374. doi:10.2147/DDDT.S334727. PMC 8860347. PMID 35210753.
- ^ Jump up to:a b c d e f g h “Drug Trials Snapshots: Litfulo”. U.S. Food and Drug Administration. 23 June 2023. Retrieved 29 April 2024.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “FDA Approves Pfizer’s Litfulo (Ritlecitinib) for Adults and Adolescents With Severe Alopecia Areata” (Press release). Pfizer. 23 June 2023. Archived from the original on 25 June 2023. Retrieved 24 June 2023 – via Business Wire.
- ^ Kansteiner F (26 June 2023). “Pfizer’s Litfulo enters the scene in alopecia with adolescent nod to rival Lilly’s Olumiant”. Fierce Pharma. Archived from the original on 8 July 2023. Retrieved 18 September 2023.
Further reading
- Guttman-Yassky E, Pavel AB, Diaz A, Zhang N, Del Duca E, Estrada Y, et al. (April 2022). “Ritlecitinib and brepocitinib demonstrate significant improvement in scalp alopecia areata biomarkers”. The Journal of Allergy and Clinical Immunology. 149 (4): 1318–1328. doi:10.1016/j.jaci.2021.10.036. PMID 34863853. S2CID 244824663.
- King B, Zhang X, Harcha WG, Szepietowski JC, Shapiro J, Lynde C, et al. (May 2023). “Efficacy and safety of ritlecitinib in adults and adolescents with alopecia areata: a randomised, double-blind, multicentre, phase 2b-3 trial”. Lancet. 401 (10387). London, England: 1518–1529. doi:10.1016/S0140-6736(23)00222-2. PMID 37062298. S2CID 258114404.
External links
- Clinical trial number NCT03732807 for “PF-06651600 for the Treatment of Alopecia Areata (ALLEGRO-2b/3)” at ClinicalTrials.gov
 |
|
| Clinical data | |
|---|---|
| Trade names | Litfulo |
| Other names | PF-06651600 |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a624015 |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| ATC code | |
| Legal status | |
| Legal status | |
| Identifiers | |
|
show
|
|
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank |
|
| ChemSpider | |
| UNII |
|
| KEGG |
|
| ChEBI | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C15H19N5O |
| Molar mass | 285.351 g·mol−1 |
| 3D model (JSmol) | |
|
show
|
|
|
show
|
|
////////////PF-06651600, PF 06651600, PF06651600, Breakthrough Therapy designation, PHASE 2, alopecia areata, rheumatoid arthritis, Crohn’s disease, ulcerative colitis, Ritlecitinib, Litfulo
C=CC(N1[C@@H](C)CC[C@@H](NC2=C3C(NC=C3)=NC=N2)C1)=O
