

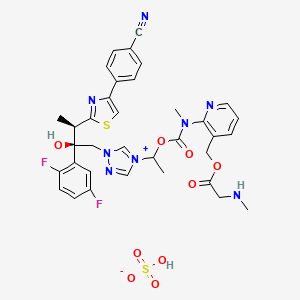
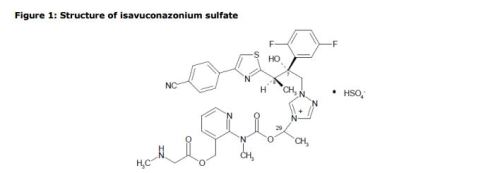
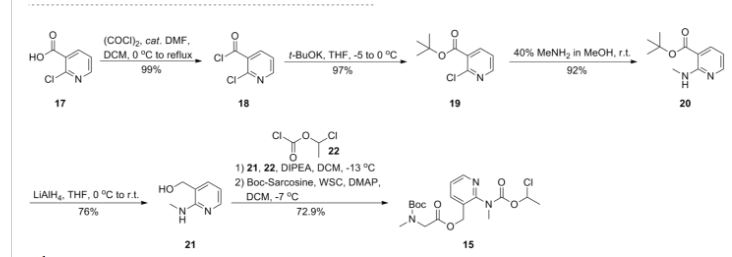
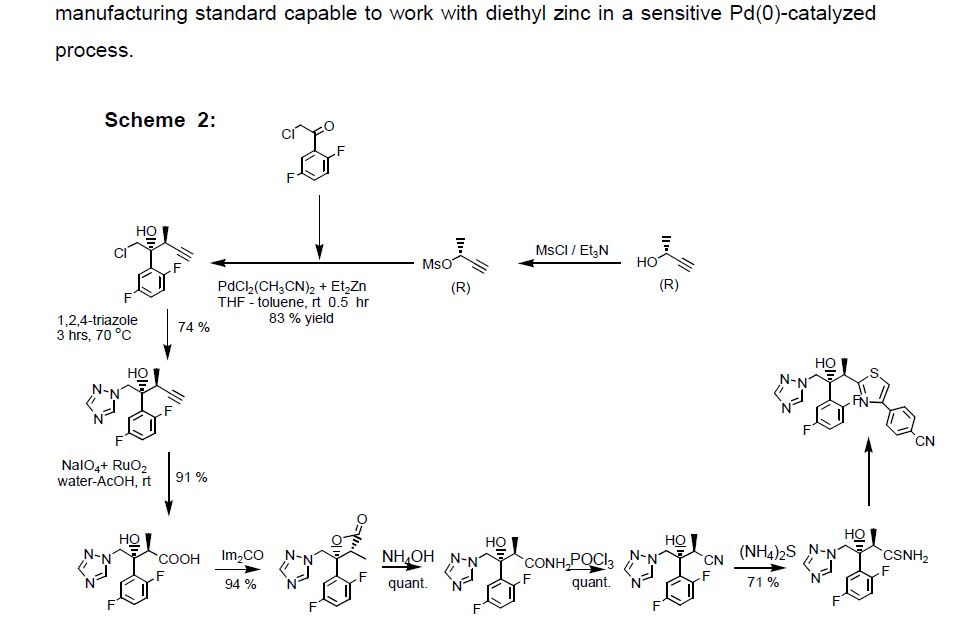
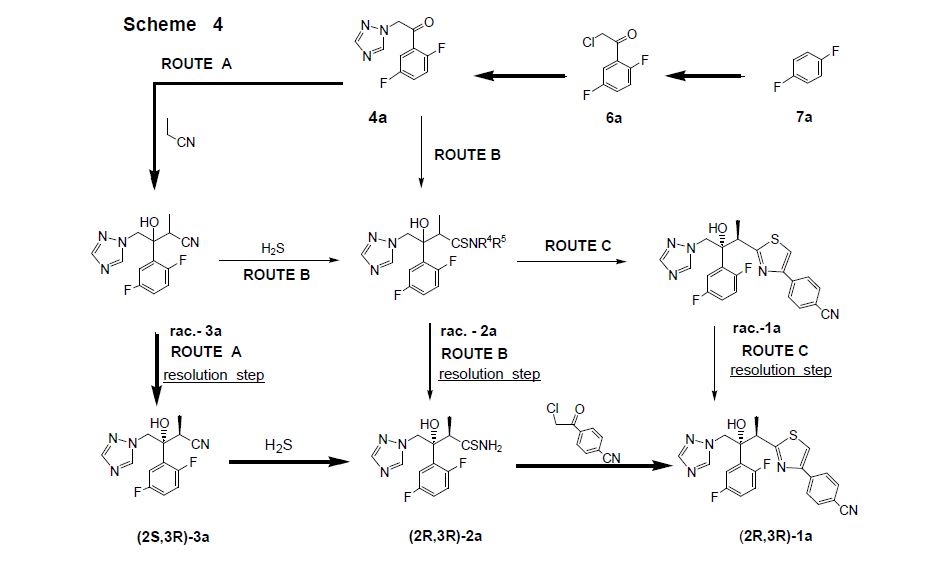
LYS228
BOS-228
LYS-228
Molecular Formula, C16-H18-N6-O10-S2
Molecular Weight, 518.4783
(3S,4R)-3-((Z)-2-(2-Ammoniothiazol-4-yl)-2-((1-carboxycyclopropoxy)imino)acetamido)-2-oxo-4-((2-oxooxazolidin-3-yl)methyl)azetidine-1-sulfonate
RN: 1810051-96-7
UNII: 29H7N9XI1B
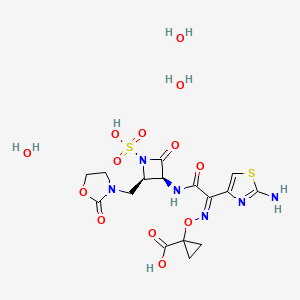
UNII-005B24W9YP
005B24W9YP
Lys-228 trihydrate
2091840-43-4
Yclopropanecarboxylic acid, 1-(((Z)-(1-(2-amino-4-thiazolyl)-2-oxo-2-(((3S,4R)-2-oxo-4-((2-oxo-3-oxazolidinyl)methyl)-1-sulfo-3-azetidinyl)amino)ethylidene)amino)oxy)-, hydrate (1:3)
1-[(Z)-[1-(2-amino-1,3-thiazol-4-yl)-2-oxo-2-[[(3S,4R)-2-oxo-4-[(2-oxo-1,3-oxazolidin-3-yl)methyl]-1-sulfoazetidin-3-yl]amino]ethylidene]amino]oxycyclopropane-1-carboxylic acid;trihydrate
BOS-228 (LYS-228) is a monobactam discovered at Novartis and currently in phase II clinical development at Boston Pharmaceuticals for the treatment of complicated urinary tract infection and complicated intraabdominal infections in adult patients.
The compound has been granted fast track and Qualified Infectious Disease Product (QIDP) designation from the FDA.
In October 2018, Novartis licensed to Boston Pharmaceuticals worldwide rights to the product.
Paper
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00330
Patent
US 20150266867
PATENT
WO 2017050218
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017050218&tab=FULLTEXT
Compound X: 1- ( ( (Z) – (1- (2-aminothiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) -1-sulfoazetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylic acid.
[0126]
Step 1: Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate. To a solution of (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetic acid (854 mg, 1.59 mmol) prepared according to published patent application US2011/0190254, Intermediate B (324 mg, 1.75 mmol) and HATU (785 mg, 2.07 mmol) in DMF (7.9 mL) , DIPEA was added (832 μL, 4.77 mmol) . After 1 h of stirring, it was poured into water and extracted with EtOAc. Brine was added to the aqueous layer, and it was further extracted with ethyl acetate (EtOAc) (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo. The crude residue was purified via silica gel chromatography (0-10%MeOH-DCM) to afford the title compound (1.09 g, 97%) as a beige foam. LCMS: R t = 0.97 min, m/z =705.3 (M+1) Method 2m_acidic.
[0127]
Instead of HATU, a variety of other coupling reagents can be used, such as any of the typical carbodiimides, or CDMT (2-chloro-4, 6-dimethoxy-1, 3, 5-triazine) and N-methylmorpholine to form the amide bond generated in Step 1.
[0128]
Step 2: (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid. Benzhydryl 1- ( ( (Z) – (1- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylate (1.00 g, 1.42 mmol) in DMF (7.0 mL) at 0 ℃ was treated with SO 3·DMF (448 mg, 2.84 mmol) . After 2 h of stirring at rt, the solution was poured into ice-cold brine and extracted with EtOAc (3x) . The combined organic layers were dried over Na 2SO 4 and concentrated in vacuo, affording the title compound (assumed quantitative) as a white solid. LCMS: Rt =0.90 min, m/z = 785.2 (M+1) Method 2m_acidic.
[0129]
Step 3: 1- ( ( (Z) – (1- (2-aminothiazol-4-yl) -2-oxo-2- ( ( (3S, 4R) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) -1-sulfoazetidin-3-yl) amino) ethylidene) amino) oxy) cyclopropanecarboxylic acid.
[0131]
To a solution of (3S, 4R) -3- ( (Z) -2- ( (1- ( (benzhydryloxy) carbonyl) cyclopropoxy) imino) -2- (2- ( (tert-butoxycarbonyl) amino) thiazol-4-yl) acetamido) -2-oxo-4- ( (2-oxooxazolidin-3-yl) methyl) azetidine-1-sulfonic acid (1.10 g, 1.40 mmol) in DCM (1.5 mL) at 0℃, TFA (5.39 mL, 70.0 mmol) was added, and after 10 minutes, the ice bath was removed. Additional TFA (3.24 mL, 42.0 mmol) was added after 1 hr at rt and the solution was diluted with DCM and concentrated in vacuo after an additional 30 min. Optionally, anisole may be added to the TFA reaction to help reduce by-product formation, which may increase the yield of desired product in this step. The crude residue was purified by reverse phase prep HPLC (XSelect CSH, 30 x 100 mm, 5 μm, C18 column; ACN-water with 0.1%formic acid modifier, 60 mL/min) , affording the title compound (178 mg, 23%) as a white powder. LCMS: R t = 0.30 min, m/z = 518.9 (M+1) Method 2m_acidic; 1H NMR (400 MHz, DMSO-d 6) δ 9.27 (d, J = 9.0 Hz, 1H) 6.92 (s, 1H) 5.23 (dd, J = 9.1, 5.7 Hz, 1H) 4.12-4.23 (m, 3H) 3.72-3.62 (m, 2H assumed; obscured by water) 3.61-3.52 (m, 1H assumed; obscured by water) 3.26 (dd, J = 14.5, 5.9 Hz, 1H) 1.36 (s, 4H) . 1H NMR (400 MHz, D 2O) δ 7.23 (s, 1H) , 5.48 (d, J = 5.8 Hz, 1H) , 4.71-4.65 (m, 1H) , 4.44 (t, J = 8.2 Hz, 2H) , 3.89-3.73 (m, 3H) , 3.54 (dd, J = 14.9, 4.9 Hz, 1H) , 1.65-1.56 (m, 2H) , 1.56-1.46 (m, 2H) . The product of this process is amorphous. Compound X can be crystallized from acetone, ethanol, citrate buffer at pH 3 (50 mM) , or acetate buffer at pH 4.5 (50 mM) , in addition to solvents discussed below.
PAPER
Bioorganic & Medicinal Chemistry Letters (2018), 28(4), 748-755.
https://www.sciencedirect.com/science/article/pii/S0960894X18300064
PATENT
WO 2019026004
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019026004&tab=PCTDESCRIPTION
Over the past several decades, the frequency of antimicrobial resistance and its association with serious infectious diseases have increased at alarming rates. The increasing prevalence of resistance among nosocomial pathogens is particularly disconcerting. Of the over 2 million (hospital-acquired) infections occurring each year in the United States, 50 to 60% are caused by antimicrobial-resistant strains of bacteria. The high rate of resistance to commonly used antibacterial agents increases the morbidity, mortality, and costs associated with nosocomial infections. In the United States, nosocomial infections are thought to contribute to or cause more than 77,000 deaths per year and cost approximately $5 to $10 billion annually.
Important causes of Gram-negative resistance include extended-spectrum 13- lactamases (ESBLs), serine carbapenemases (KPCs) and metallo-13-lactamases (for example NDM-1 ) in Klebsiella pneumoniae, Escherichia coli, and Proteus mirabilis, high-level third-generation cephalosporin (AmpC) 13-lactamase resistance among Enterobacter species and Citrobacter freundii, and multidrug-resistance genes observed in Pseudomonas, Acinetobacter, and Stenotrophomonas. The problem of antibacterial resistance is compounded by the existence of bacterial strains resistant to multiple antibacterials. For example, Klebsiella pneumonia harboring NDM-1 metallo-13- lactamase carries frequently additional serine-13-lactamases on the same plasmid that carries the NDM-1 .
Thus there is a need for new antibacterials, particularly antibacterial compounds that are effective against existing drug-resistant microbes, or are less susceptible to development of new bacterial resistance. Monobactam antibiotic, which is referred to herein as Compound X, is primarily effective against Gram-negative bacteria, including strains that show resistance to other monobactams.
The present invention relates to a process for the preparation of monobactam antibiotic Compound X and intermediates thereof.
More particularly, the present invention relates to a process for the preparation of Compound X

Compound X
also referred to as 1 -(((Z)-(1 -(2-aminothiazol-4-yl)-2-oxo-2-(((3S,4R)-2-oxo-4-((2-oxooxazolidin-3-yl)methyl)-1 -sulfoazetidin-3-yl)amino)ethylidene)amino)oxy)cyclopropanecarboxylic acid, or a salt thereof, or a solvate including hydrate thereof.
Patent application number PCT/US2015/02201 1 describes certain monobactam antibiotics. Compound X may be prepared using the method disclosed in PCT/US2015/02201 1 , in particular example 22, and in PCT/CN2016/099482.
A drawback from these processes is that they exhibit a large number of process steps and intermediate nitrogen protection/deprotection steps, reducing the overall yield and efficiency. Furthermore, these processes require several chromatographic purification steps to be carried out in course of the processes. We have found that the preparation of Compound X, as previously prepared on a manufacturing scale, possesses a number of disadvantages, in particular poor handling characteristics.
It would thus be beneficial to develop alternative or improved processes for the production of Compound X that do not suffer from some or all of these disadvantages.

Compound x Compound x
Scheme 1
Preparation of Compound X from Intermediates 22 and 2A

Scheme 3
Examples
The Following examples are merely illustrative of the present disclosure and they should not be considered as limiting the scope of the disclosure in any way, as these examples and other equivalents thereof will become apparent to those skilled in the art in the light of the present disclosure, and the accompanying claims.
Synthesis of Compound 8 (R = benzyl)

1 .50kg oxazolidin-2-one (7b) was charged into the reactor. 7.50kg THF was charged and the stirring started. The mixture was cooled to 10~20°C. 2.18kg potassium fert-butoxide was charged intol 2.00kg THF and stirred to dissolve.
The potassium fert-butoxide solution was added dropwise into the reactor while maintaining the temperature at 10-20 °C. The reaction was stirred for 1 ~2hrs at 10-20 °C after the addition. The solution of 2.36kg methyl-2-chloroacetate (7a) in 3.00kg of THF was added to the reactor while maintaining the temperature at 10-20 °C. The reaction mixture was stirred for 16-18 h at 20-25 °C. The IPC (in process control) showed completion of the reaction. The mixture was centrifuged and the wet cake was washed with 7.50kg THF. The filtrate was concentrated and the crude 7 was provided as reddish brown liquid, which was used for the next step without further purification,
1H NMR (400 MHz, CHLOROFORM- /) δ ppm 3.65 – 3.71 (m, 2 H) 3.74 (s, 3 H) 4.02 (s, 2 H) 4.34 – 4.45 (m, 2 H).
The dried reactor was exchanged with N2 three times. 3.71 kg LiHMDS solution in THF/Hep (1 M) and 1 .30kg THF were charged under nitrogen protection. The stirring was started and the solution was cooled to -70—60 °C. The solution of 0.71 kg benzyl acetate (6) in 5.20 kg THF was added dropwisely at -70— 60 °C, and the resulted mixture was stirred for 1 -1 .5 h after the addition. The solution of 0.65kg 7 in 3.90kg THF was added dropwise while maintaining the temperature at -70—60 °C, then stirred for 30-40 minutes. The reaction mixture was warmed to 20-25 °C and stirring was continued for 0.5-1 .0 h. IPC showed 6 was less than1 .0% (Otherwise, continue the reaction till IPC passes). The reaction mixture was poured into 13.65 kg aqueous citric acid below 10 °C. The mixture was stirred for 15-20 minutes after the addition. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (6.50kg * 2). The organic layer was combined, washed by 6.50 kg 28% NaCI solution and dried with 0.65
kg anhydrous MgSC . The mixture was filtered and the wet cake was washed with 1 .30kg EA. The filtrate was concentrated under vacuum to provide crude 8. The crude 8 was stirred in 2.60 kg MTBE at 20-25 °C for 1 -1 .5 h. The mixture was cooled to 0-10 °C and stirred for 1 .5-2.0 h and filtered. The filter cake was washed with 0.65kg pre-cooled MTBE and dried under vacuum (<-0.096Mpa) at 20-25 °C for 12~16hrs till a constant weight to give 513 g of 8 as a white solid, Yield: 45%, HPLC purity 96.4%,1 H NMR (400 MHz, CHLOROFORM-c δ ppm 3.48 – 3.55 (m, 1 H) 3.56 – 3.63 (m, 2 H) 3.66 – 3.74 (m, 1 H) 4.17 – 4.26 (m, 2 H) 4.31 – 4.44 (m, 2H) 5.12 – 5.24 (m, 2 H) 7.30 – 7.44 (m, 5 H).
Synthesis of Compound 9 (R = benzyl)

The dried reactor was charged with 3.75kg HOAc and 1 .50 kg 8. The stirring was started and the reaction mixture was cooled to 0-5 °C. 3.53kg aqueous NaN02 was added dropwise at 0-10 °C, and the reaction mixture was stirred for 15-30 minutes after the addition. IPC showed 8 was less than 0.2%. The reaction mixture was treated with 7.50kg EA and 7.50 kg water. Phases were separated and the organic layer was collected. The aqueous layer was extracted with EA (7.50kg * 2). The organic layers were combined, washed with 7.50 kg 28% NaCI solution, and concentrated under vacuum to provide crude 9. The crude 9 was slurried with 5.25 kg water at 10-20 °C for 3~4hrs, and filtered. The wet cake was washed with 1 .50kg water. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .44 Kg of 9, yield: 86.9%, HPLC purity 92.9%,1H NMR (400 MHz, CHLOROFORM- /) δ ppm 3.60 – 3.76 (m, 2 H) 4.44 (t, J=8.07 Hz, 2 H) 4.60 (s, 2 H) 5.25 – 5.41 (m, 2 H) 7.30 – 7.43 (m, 5 H) 1 1 .62 (br s, 1 H).
Synthesis of Compound 9a (R = benzyl)

9
The dried reactor was charged with 0.58 kg Zn, 4.72kg (Βο Ο, 6.00 kg water, 1 .20 kg NH4CI and 6.00kg THF. The reaction mixture was stirred and heated to 50-55 °C. The solution of 0.60 kg 9 in 4.20kg THF
was added dropwisely while maintaining the temperature at 50-55 °C. The reaction mixture was stirred for 0.5-1 .Ohrs after the addition. IPC showed 9 was less than 0.1 %. The reaction mixture was treated withl .50 kg ethyl acetate and stirred for 15-20 minutes. Phase was separated and the water layer was extracted by1 .50 kg ethyl acetate. The organic layers were combined, washed with 6.00 kg 28% NaCI solution and concentrated under vacuum to provide crude 9a. The crude 9a was stirred with 3.60kg*2 n-heptane to remove excess (Βο Ο. The residue was purified by silica gel chromatography column eluted with ethyl acetate: Heptane= 1 :1 to provide crude 9a solution. The solution was concentrated under reduced pressure to obtain crude 9a. The crude 9a was slurried with 1 .80 kg MTBE for 2.0-3. Ohrs, filtered, and the wet cake was washed with MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 50-55 °C for 16-18 h till a constant weight to give 392 g of 9a as a white solid, Yield: 51 %, HPLC purity 98.1 %,1H NMR (400 MHz, DMSO-cfe) δ ppm 1.17 – 1 .57 (m, 9 H) 3.39 – 3.61 (m, 2 H) 4.20 – 4.45 (m, 3 H) 5.10 – 5.32 (m, 3 H) 5.75 (s, 1 H) 7.38 (br s, 5 H) 7.75 – 7.99 (m, 1 H).
Synthesis of compound (VII) (R = benzyl, X = CI)

9a VII
The dried reactor was charged with 13.0kg HCI in IPA and the stirring was started. 1 .33 kg 9a was charged in portions at 20-25 °C. The mixture was stirred at 20-25 °C for 3-4 h. IPC showed 9a was less than 0.1 %. The reaction solution was concentrated under vacuum 40-45 °C. The residue was treated with 21 .58kg MTBE at 20-25 °C for 3-4 h. The mixture was filtered and the wet cake was washed with 2.60kg MTBE. The solid was dried under vacuum (<-0.096 Mpa) at 45-50 °C for 5-6 h till a constant weight to give 1 .045 Kg of compound VII (R = benzyl, X = CI) as a yellow solid, Yield: 93.7%, HPLC purity 99.2%,1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.16 – 3.74 (m, 3 H) 4.10 – 4.35 (m, 4 H) 5.09 – 5.39 (m, 2 H) 7.27 – 7.60 (m, 5 H) 8.72 (br s, 2 H).
Synthesis of compound (Vile) (R = benzyl)

VII Vile
To an autoclave (3L) were added VII (R = benzyl, X = CI) (100 g, 304.2 mmol, 1 .0 equiv.), DCM (2650 g, 26.5 equiv., w/w) and (S-BINAP)RuCl2 (2.4 g, 3.04 mmol, 0.01 equiv.), successively. Air in the autoclave was replaced with N2 5 times. N2 in the autoclave was was replaced with H2 5 times. The solution was stirred with 250-260 r/min and H2 (2.1 ±0.1 MPa) at 40±5°C for 24 h. The reaction mixture was filtered, and the filter cake was washed with DCM (400 g, 4.0 equiv., w/w). The filter cake was slurried with IPA (785 g, 7.85 equiv., w/w) and H2O (40 g, 0.4 equiv., w/w) overnight (18-20 h). The mixture was filtered. The filter cake was washed with IPA (200 g, 2.0 equiv., w/w) and dried at 45±5°C overnight (18-20 h). Vile (R = benzyl) was obtained as off-white solid, 80.4 g, 79.9% yield, 95.5% purity, 97.6% de, >99.5% ee. 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.34-3.38 (m, 2 H) 3.50-3.52 (m, 1 H) 3.60-3.62 (m, 1 H) 4.18-4.24 (m, 4 H) 5.23 (s, 2H) 6.16 (s, 1 H) 7.32 (m, 5H) 8.74 (s, 1 H).
Alternative synthesis of compound 9a (R = benzyl)

5b
Mg(OtBu)2
To a flask was added 5a (1 .88 g, 12.93 mmol), THF (40 mL), and CDI (2.20 g, 13.58 mmol) at 25 °C. The mixture was stirred for 3 h. To the reaction mixture was added 5b (2.00 g, 6.47 mmol), and Mg(OfBu)2 (2.21 g, 12.93 mmol). The reaction mixture was stirred at 25 °C for 24 h. The reaction mixture was concentrated under vacuum to remove most of the THF solvent. To the concentrated solution was added MTBE (40 mL), followed by addition of an aqueous solution of HCI (1 M, 60mL) to adjust to pH = 2-3. Two phases were separated, and the water phase was extracted with MTBE (20 mL). The combined organic phase was washed with aqueous NaHCC (5%, 50 mL) and brine (20%, 40 mL). The organic phase was concentrated to a weight of -19 g, and a lot of white solid was obtained in the concentration process. The suspension was cooled to 0 °C, and filtered. The filter cake was washed with cold MTBE (5 mL) and dried under vacuum to obtain product 9a (1 .6g, 63% yield).
Synthesis of compound (Vile) (R = benzyl, PG = Cbz)

Vile Vile
To a flask (5 L) were added Vile (R = benzyl) (140 g, 423.2 mmol, LOequiv.), H20 (1273 g, 9.09 equiv., w/w) and toluene (2206 g, 15.76 equiv., w/w). The solution was stirred and cooled to 0-5 °C with ice bath. Then NaHCOa (78.4 g, 933 mmol, 2.22 equiv.) was added and CbzCI (89.6 g, 527 mmol, 1 .24 equiv.) was dropped into the stirring solution, respectively. The solution was stirred at 30±5 °C overnight (18-20 h). Heptane (3612 g, 25.8 equiv., w/w) was added dropwise to the stirring solution over 1 h at 20-30 °C. The mixture was filtered. The filter cake was washed with heptane (280 g, 2.00 equiv., w/w) and MTBE (377 g, 2.69 equiv., w/w), respectively. The filter cake was dried at 45±5°C overnight (18-20 h). Vile (R = benzyl, PG = Cbz) was obtained as an off-white solid, 169.4 g, 93% yield, 96.7% purity, 98% de, >99.5% ee, 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.23-3.24 (m, 1 H) 3.30 (m, 1 H) 3.51 -3.55 (m, 2 H) 3.99 (s, 1 H) 4.17-4.21 (m, 3 H) 5.02-5.03 (m, 2H) 5.12 (s, 2H) 5.46-5.48 (d, 1 H) 7.33-7.36 (m, 10H) 7.75-7.73 (d, 1 H).
Synthesis of compound (IV) (PG = Cbz)

Vile IV
Vile (R = benzyl) (220 g, 513.5 mmol, 1 .0 equiv.) was dissolved in THF (1464g, 6.65 equiv., w/w). The solution was filtered. The filter cake was washed with THF (488g, 2.22 equiv., w/w). The filtrate (Vile) was collected. To an autoclave (3L) were added the filtrate (Vile). The reactor was cooled down to -75 – -65 °C with dry-ice/EtOH bath, and bubbled with NH3 for not less than 4 h. Then the solution was stirred at 25±5 °C with NH3 (0.5-0.6 MPa) for 24 h. The autoclave was deflated to release NH3. The reaction solution was concentrated with a rotary evaporator to remove THF until the residue was around 440 g. The residue was slurried with EA (2200 g, 10 equiv., w/w) at 70±2 °C, then cooled to 25±5 °C and stirred for 16-18 h. The mixture was filtered. The filter cake was washed with EA (440 g). The filter cake was slurried with EA (1320 g, 6.00 equiv. w/w), and the temperature was raised to 70±2 °C, then cooled to 25±5 °C and stirred for 16-20 h. The mixture was filtered. The filter cake was washed with EA, and dried at 50±5 °C overnight (18-20 h). IV (PG = Cbz) was obtained as off-white solid, 141 g, 81 .5% yield, 99.1 % purity, >99.5% assay, 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.12 – 3.23 (m, 2 H) 3.31 (br s, 1 H) 3.56 (t, J=8.01 Hz, 2 H) 3.88 (quin, J=6.02 Hz, 1 H) 3.93 – 4.03 (m, 1 H) 4.20 (t, J=8.01 Hz, 2 H) 5.02 (s, 2 H) 5.27 (d, J=5.87 Hz, 1 H) 7.12 (s, 1 H) 7.22 – 7.45 (m, 5 H).
Synthesis of compound (III) (PG = Cbz, LG = S02CH3)

IV III
To a flask was added IV (PG = Cbz) (14.00 g, 41 .50 mmol, 1 .00 equiv), and dry 1 , 2-dimethoxyethane (300 mL) under N2. The mixture was stirred at -5°C ~ 0°C for 1 h to obtain a good suspension. MsCI (7.89 g, 68.89 mmol, 5.33 mL, 1 .66 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min, and Et3N (12.60 g, 124.50 mmol, 17.26 mL, 3.00 eq) in 1 , 2-dimethoxyethane (20.00 mL) was added dropwise during 30 min side to side. The reaction mixture was stirred for additional 5 min at -5°C ~ 0°C, and was quenched with water (6 mL). The reaction mixture was concentrated to remove DME. The solid was slurried in water (250 mL) and MTBE (125 mL) for 1 h. The solid was collected by filtration, and then slurried in water (250 mL) for 1 hr. The solid was collected by filtration, and washed with water (25 mL) to give white solid. The solid was slurried in EA (150 mL) and dried in vacuum at 60°C for 24 h to give III (PG = Cbz, LG = SO2CH3) (15.00 g, 36.1 1 mmol, 87.01 % yield), 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.17 (s, 3 H) 3.26 (br d, J=15.04 Hz, 1 H) 3.47 – 3.57 (m, 1 H) 3.64 (br d, J=6.36 Hz, 2 H) 4.22 (br dd, J=17.79, 8.50 Hz, 2 H) 4.50 (br s, 1 H) 4.95 – 5.17 (m, 3 H) 7.21 – 7.56 (m, 5H) 7.43 (s, 1 H) 7.63 – 7.89 (m, 2 H).
Synthesis of compound II (PG = Cbz, LG = SO2CH3, M+ = NBu4+)
O OMs o CISO3H, 2-picoline – ° O ?yO
HN Bu4NHS04< NHCbz
“Cbz
III II
To a flask was added 2-picoline (1 1 .50 g, 12.23 mL) and DMF (10 mL). The solution was cooled to 5 SC, followed by slow addition of chlorosulfonic acid (7.20 g, 4.14 mL). The temperature was increased to 20 SC. Ill (PG = Cbz, LG = SO2CH3) (5.13 g, 12.35 mmol) was added to the reaction mixture. The reaction mixture was heated to 42 SC for 18h. IPC (in process control) showed complete conversion of starting material. The reaction was cooled to 20 SC and dropwise added to a solution of tetrabutylammonium hydrogen sulfate (4.6 g, 13.6 mmol) in the mixed solvents of dichloromethane (100 mL) and water (100 mL) at 5SC. The phases were separated and the water phase was extracted with dichloromethane (2*50mL). The combined organic phase was washed with water (5*100mL). The organic phase was concentrated to dryness and purified by column chromatography (dichloromethane/methanol = 15/1 v/v) to afford II (PG = Cbz, LG = SO2CH3, M+ = NBii4+) (8.4 g, 92.30%), 1 H NMR (400 MHz, CHLOROFORM-c/) δ ppm 0.99 (t, J=7.34 Hz, 12 H) 1 .36 – 1 .50 (m, 8 H) 1 .54 – 1 .76 (m, 8 H) 3.15 (br d, J=8.31 Hz, 2 H) 3.21 – 3.35 (m, 8 H) 3.47 (br dd, J=14.73, 7.27 Hz, 1 H) 3.54 – 3.65 (m, 1 H) 3.67 – 3.81 (m, 2 H) 4.17 – 4.32 (m, 1 H) 4.39 – 4.62 (m, 1 H) 4.74 (br s, 1 H) 5.1 1 (s, 3 H) 5.32 – 5.50 (m, 1 H) 6.47 (br s, 1 H) 7.29 – 7.47 (m, 5 H) 8.69 – 8.94 (m, 1 H).
Synthesis of compound (IA)

A solution of II (PG = Cbz, LG = SO2CH3, M+ = NBu4+) (4.0 g) in dichloromethane (38 mL) was pumped to tube A at rate of 2.0844 mL/min, and a solution of KHCO3 (3.0 g) in water (100 mL) was pumped to tube B at a rate of 1 .4156 mL/min side to side. These two streams were mixed in a cross-mixer then flowed to a tube coil that was placed in an oil bath at 100 °C. The residence time of the mixed stream in the coil was 2 min. The reaction mixture flowed through a back-pressure regulator that was set at ~ 7 bars, and was collected to a beaker. After completion of the collection, two phases was separated. The organic phase was concentrated to dryness. The residue was slurried in ethyl acetate (5 mL). The solid was filtered and the filter cake was dried to give IA (2.6 g, 75%),
1H NMR (400 MHz, CHLOROFORM-c/) δ ppm 1.00 (t, J=7.27 Hz, 12 H) 1 .42 (sxt, J=7.31 Hz, 8 H) 1 .62 (quin, J=7.83 Hz, 8 H) 3.13 – 3.39 (m, 8 H) 3.54 – 3.69 (m, 2 H) 3.81 (dd, J=14.98, 2.51 Hz, 1 H) 3.96 – 4.13 (m, 1 H) 4.22 – 4.47 (m, 3 H) 4.99 – 5.23 (m, 3 H) 6.42 (br d, J=9.29 Hz, 1 H) 7.26 – 7.44 (m, 5 H).
Synthesis of compound 2A
Step 1

To a stirring solution of compound 16b (2 g, 10.14mmol, 1 .0 eq) in DMF (20 ml_) was added CS2CO3 (5.29g, 16.22 mmol, 1 .6 eq), then the resulting solution was stirred at room temperature for 10mins, then compound 16a (5.27g, 20.28mmol, 2eq) was added dropwise to the mixture for 2 minutes, then the resulting solution was stirred for another 2 hours. TLC showed the starting material was consumed completely. The mixture was added with water (60mL) and extracted with MTBE (20mL*3). The combined organic layers were dried over anhydrous sodium sulfate and concentrated. The crude was slurried in heptane to give 1 .65 g 16 as a white solid (Yield: 57%), 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.48-7.28 (m, 10 H), 5.00-4.96 (t, J=6.0 Hz, 1 H), 3.81 (s, 3H), 3.44-3.42 (m, 2H), 2.40-2.37 (m, 2H).

Compound 16 (1 g, 2.66mmol, 1 eq) was dissolved in THF (20mL) under Nitrogen, and cooled to -40 °C. NaHMDS (1 .6mL, 2.0M THF solution, 1 .2 eq) was added dropwise. The reaction was stirred for 1 h at -40 °C. HPLC indicated the reaction was finished. The reaction was quenched with 10% Citric acid, extracted with MTBE (25 ml_ x 2). The combined organic layers were washed with brine (30 ml_), dried with Na2S04, filtered and concentrated to give 17 as a yellow solid, which was used for the next step without purification (assay yield: 65%); 1H NMR (400 MHz, DMSO-cfe) δ ppm 7.27-7.13 (m, 10 H), 3.46 (s, 3H), 1 .21 -1 .17(dd, J=7.2, 10.4 Hz, 2H ); 1 .14-1 .1 1 (dd, J=7.2, 10.4 Hz, 2H).
Step 3

Compound 17 (100 mg) was dissolved in methanol (5 mL) and 2.0 M HCI IPAC solution (5 mL). The solution was heated at 45 °C for 3 days. HPLC indicated the reaction was finished. The reaction was cooled to room temperature and was diluted with 10 mL water. The reaction mixture was washed with MTBE (10 mL x 2), organic layer was discarded and the aqueous layer was concentrated to give compound 2A HCI (32 mg, 62% yield), 1 H NMR (400 MHz, DMSO-cfe) δ ppm 3.80-3.44 (br, 4H), 1 .56 (s, 2H), 1 .38 (s, 2H).
Step 4
To a solution of 2A HCI (0.70 g, 4.57 mmol) in methanol (5 mL) was added triethylamine (1 .26 mL, 9.14 mmol) at room temperature. The solution was stirred for 20 min, and the solvent was removed under vacuum. To the residue was added IPAC (10 mL) leading to precipitation. The solid was filtered, and the filtrate was concentrated to provide 2A (0.50g, 94% yield) containing ca. 6 wt% Et3N-HCI.
Synthesis of Compound X from compound of formula (I), (IA)

Compound x
To a flask was charged 21 (1 .00 g, 68.43 wt%, 2.50 mmol) and DMF (10 mL). The suspension was cooled to -20 °C, to which was added diphenylphosphinic chloride (0.52 mL, 2.75 mmol). The solution was stirred at -20 °C for 30 min, followed by addition of a mixed solution of (IA) (1 .52g, 3.00 mmol) and triethylamine (0.52 mL, 3.76 mmol) in DMF (2mL). The reaction mixture was stirred at 20 °C for 20 h, followed by addition of MTBE (20 mL). The reaction mixture was adjusted to pH = 2-3 using aqueous HCI solution (37%). To the mixture was added isopropanol (100 mL). The resulting mixture was stirred for 4 h to obtain a suspension. The suspension was filtered and the filter cake was dried under vacuum to afford crude 22 (1 .17 g). The crude 22 was slurried in a combined solvent of THF/H2O (= 12 mL / 3mL), and filtered to afford 22 (0.744 g, 75 wt% by Q-NMR, 53.3% yield). 1H NMR (400 MHz, DMSO-cfe) δ ppm 3.47 – 3.55 (m, 2 H) 3.59 – 3.63 (m, 2 H) 4.13 – 4.21 (m, 3 H ) 5.05 (dd, J=8.8, 5.6 Hz, 1 H) 8.22 (s, 1 H) 9.73 (d, J=8.7 Hz, 1 H).
To a suspension of 22 (580 mg, 75 wt%, 1 .037 mmol) in DMAC (1 .5 mL) was added 2A (214.3 mg, 85 wt%, 1 .556 mmol). The reaction was stirred at 25 °C for 3 days, and in process control showed 22, Compound X = 4/96, and Z/E = 91 /9. the mixture was slowly added into 15ml acetone to precipitate yellowish solid. The reaction mixture was filtered to afford Compound X (0.7 g, 34 wt% by QNMR, 44% yield).
Synthesis of compound 3 (R2 = CH(Ph)2)

R2 = CH(Ph)2
2-(2-aminothiazol-4-yl)-2-oxoacetic acid (Y) (10.00 g, 47.93 mmol) and compound W (R2 = CH(Ph)2) (13.31 g, 46.98 mmol) were suspended in DMAC (40 mL), followed by addition of triethylamine (5.01 mL, 35.95 mmol). The reaction mixture was stirred at 20 °C for 5 h. HPLC showed completion of the reaction, and Z/E
= 97/3. To the reaction mixture was added water (120 mL) with stirring. The mixture was stirred for 20 min to obtain a suspension. The suspension was filtered and the filter cake was washed with water (50 mL).
The filter cake was slurried in a combined solvent of THF/ethyl acetate (50 mL / 50 mL) at 60 °C and cooled to 20 °C. The solid was filtered and dried at 50 °C for 3 h to get 3 (R2 = CH(Ph)2) (19.5 g, 88% yield). 1H
NMR (400 MHz, DMSO-cfe) δ ppm 1.37 -1 .42 (m, 2 H) 1 .44 – 1 .49 (m, 2 H) 6.87 (s, 1 H) 6.94 (s, 1 H) 7.22
– 7.30 (m, 6 H) 7.45 – 7.49 (m, 4 H).
Alternative Synthesis of Compound X from compound of formula (I), (IA)

Compound x
IA (40.14 g, 62.63 mmol) was dissolved in methanol (200 ml_), followed by addition of Pd/C (10%, 1 .1 g). The reaction mixture was maintained under hydrogen atmosphere (1 -2 bar) at 20 °C for 24 h. In process control showed completion of the reaction. The reaction mixture was filtered. The filtrate was concentrated to give an oil of IB (M+ = NBu4+) (58.20 g, 55 wt% by Q-NMR, 100% yield). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 0.93 (t, J=7.3 Hz, 12 H) 1 .23 – 1 .36 (m, 8 H) 1 .57 (m, 8 H) 2.99 – 3.28 (m, 8 H) 3.37 (dd, J=14.3, 7.5 Hz, 1 H) 3.65 – 3.70 (m, 3 H) 3.84 – 3.88 (m, 1 H) 4.08 (d, J=5.6 Hz, 1 H) 4.18 – 4.22 (m, 2 H).
3 (R2 = CH(Ph)2) (0.95 g, 2.17 mmol) was dissolved in THF (20 ml_). To the solution was added /V-methyl morpholine (0.77 g, 7.60 mmol) and 2-chloro-4,6-dimethoxy-1 ,3,5-triazine (0.57 g, 3.26 mmol). The reaction mixture was stirred at 20 °C for 1 h followed by addition of IB (M+ = NBu +) (2.70 g, 48.98 wt%, 2.61 mmol). The reaction was stirred at 20 °C for 5 h. In process control showed completion of the reaction. To the reaction mixture was added ethyl acetate (20 ml_). The organic phase was washed with brine (10 ml_). Solvent was removed. Acetone (40ml) was added to dissolve residue. TFA (1 .24 g, 10.86 mmol) dissolved in acetone (3 ml) was added slowly. The white solid was filtered and washed by acetone (10 ml) two times. Dried at 40 °C for 5h to get compound 4 (R2 = CH(Ph)2). 1 H NMR (400 MHz, DMSO-cfe) δ ppm 1 .49 – 1 .55 (m, 4 H) 3.27 (dd, J=14.4, 6.2 Hz, 1 H) 3.49 – 3.65 (m, 2 H) 3.71 (dd, J=14.4, 6.2 Hz, 1 H) 4.04 – 4.10 (m, 1 H) 4.07 (dd, J=16.0, 8.6 Hz, 1 H) 4.17 (dd, J=1 1 .8, 6.0 Hz, 1 H) 5.28 (dd, J=9.0, 5.7 Hz, 1 H) 6.88 (s, 1 H) 7.03 (s, 1 H) 7.18 – 7.32 (m, 6 H) 7.43 (m, 4 H) 9.45 (d, J=9.0 Hz, 1 H).
Crude 4 (R2 = CH(Ph)2) (2.13 g) was dissolved in dichloromethane (20 ml_). The solution was cooled to 0 °C. To the solution was added anisole (0.68 ml_, 6.24 mmol) and trifluoroacetic acid (2.16 ml_, 28.08 mmol). The reaction was warmed to 20 °C, and stirred for 15 h. In process control showed completion of the
reaction. The aqueous phase was separated and added to acetone (40 mL) to obtain a suspension. The suspension was filtered to afford Compound X (0.98 g, 54.5% yield over two steps). 1 H NMR (400 MHz, DMSO-c/e) δ ppm 1.40 (m, 4 H) 3.26 (dd, J=14.4, 6.0 Hz, 1 H) 3.54 – 3.69 (m, 3 H) 4.14 – 4.21 (m, 3 H) 5.25 (dd, J= 8.9, 5.7 Hz, 1 H) 7.02 (s, 1 H) 9.38 (d, J=9.0 Hz, 1 H).
REF
Synthesis and optimization of novel monobactams with activity against carbapenem-resistant Enterobacteriaceae – Identification of LYS228
57th Intersci Conf Antimicrob Agents Chemother (ICAAC) (June 1-5, New Orleans) 2017, Abst SATURDAY-297
//////////////LYS228, LYS 228, BOS-228, LYS-228, monobactam, Novartis, phase II, Boston Pharmaceuticals, complicated urinary tract infection, complicated intraabdominal infections, fast track, Qualified Infectious Disease Product, QIDP,
Nc1nc(cs1)\C(=N\OC2(CC2)C(=O)O)\C(=O)N[C@H]3[C@@H](CN4CCOC4=O)N(C3=O)S(=O)(=O)O

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....















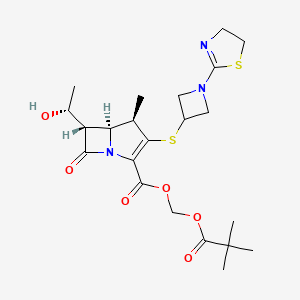




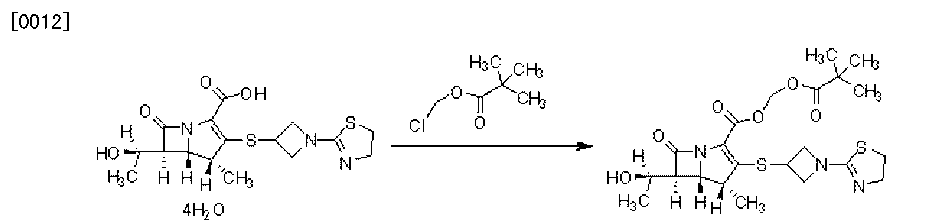
 EP 0632039; EP 0717042; JP 1996053453; US 5534510; US 5659043; US 5783703
EP 0632039; EP 0717042; JP 1996053453; US 5534510; US 5659043; US 5783703 1-azabicyclobutane (III) was opened with thioacetic acid with concomitant N-acetylation yielding (XII). Further acid hydrolysis of (XII) gave 3-mercaptoazetidine (XIII). Condensation of (XIII) with either 2-(methylthio)thiazoline (V) or 2-chloroethyl isothiocyanate (VII) then produced thiazolinylazetidine (XI).
1-azabicyclobutane (III) was opened with thioacetic acid with concomitant N-acetylation yielding (XII). Further acid hydrolysis of (XII) gave 3-mercaptoazetidine (XIII). Condensation of (XIII) with either 2-(methylthio)thiazoline (V) or 2-chloroethyl isothiocyanate (VII) then produced thiazolinylazetidine (XI).





























































{kind=link}