
Home » IND 2017
Category Archives: IND 2017
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Efonidipine, エホニジピン
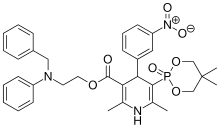
Efonidipine
- Molecular FormulaC34H38N3O7P
- Average mass631.655 Da
- エホニジピン
- CAS 111011-63-3; FREE FORM
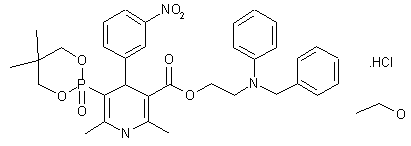
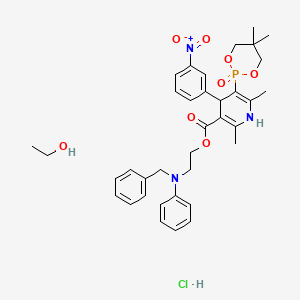
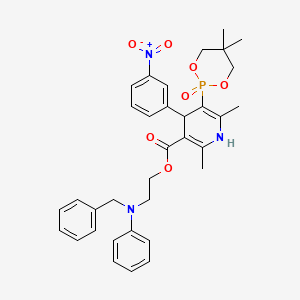
| Molecular Formula: | C36H45ClN3O8P |
|---|---|
| Molecular Weight: | 714.193 g/mol |
LD50:> 5 g/kg (R, p.o.)
- Synonyms:NZ-105
- ATC:C08CA
Efonidipine Hydrochloride Ethanolate Bulk & Tablets 10 mg/20mg/40mg, 
Efonidipine (INN) is a dihydropyridine calcium channel blocker marketed by Shionogi & Co. of Japan. It was launched in 1995, under the brand name Landel (ランデル). The drug blocks both T-type and L-type calcium channels.[1] Drug Controller General of India (DCGI) granted approval to M/s. Zuventus pharma Ltd for marketing efonidipine under brand name Efnocar in India .[2]
Structure Activity Relationship
Efonidipine is a dual Calcium Channel Blocker (L & T-type). It has a unique chemical structure. The phosphonate moiety (Figure 1) at the C5 position of the dihydropyridine ring is considered to be important for the characteristic pharmacological profile of the drug. (figure-1)

Figure-1:Efonidipine: Chemical Structure
Mechanism of action
Efonidipine, a new generation dihydropyridine (DHP) calcium channel blocker, inhibits both L-type and T-type calcium channels.[1]
Pharmacodynamics
- Efonidipine exhibits antihypertensive effect through vasodilatation by blocking L-type and T-type calcium channels.[1]
- Efonidipine has a negative chronotropic effect. Working on sino atrial node cells by inhibiting T-type calcium channel activation, Efonidipine prolongs the late phase-4 depolarization of the sino atrial node action potential and suppresses an elevated HR. The negative chronotropic effect of Efonidipine decreases heart rate, myocardial oxygen demand and increases coronary blood flow.[3]
- Efonidipine increases coronary blood flow by blocking L & T-type calcium channels and attenuates myocardial ischaemia.[4]
- By reducing synthesis and secretion of aldosterone, Efonidipine prevents hypertrophy and remodeling of cardiac myocytes.[5]
- Efonidipine increases glomerular filtration rate without increasing intra-glomerular pressure and filtration fraction. This prevents hypertension induced renal damage.[6]
- Efonidipine prevents Rho-kinase and NFB induced renal parenchymal fibrosis and provides long term renal protection.[7][8]
- Efonidipine suppresses renin secretion from the juxta glomerular apparatus in the kidneys.[9]
- Efonidipine enhances sodium excretion from the kidneys by suppressing aldosterone synthesis and secretion from the adrenal glands. Aldosterone induced renal parenchymal fibrosis is suppressed by Efonidipine.[5]
- Efonidipine prevents NFB induced hypertrophy and inflammation in the renal vasculature and protects the kidneys.[7]
- Efonidipine protects against endothelial dysfunction due to its anti-oxidant activity and by restoring NO bioavailability.[10][11]
- Efonidipine has anti-atherogenic activity and protects the blood vessels from atherosclerosis.[12]
- Efonidipine lowers blood pressure in cerebral resistance vessels and prevents hypertension induced brain damage.[4]
Pharmacokinetics
Absorption
Peak plasma concentration is achieved in about 1.5 to 3.67 hours after administration. Half life is approximately 4 hours. The pharmacokinetic parameters of Efonidipine are depicted in Table-1.
Table 1: PK Parameters in Adult Healthy Male Subjects
| Variable | Efonidipine | |
| Mean | Range | |
| Cmax(ng/ml) | 36.25 | 9.66-66.91 |
| Tmax (hour) | 2.59 | 1.50-3.67 |
| T1/2 (hour) | 4.18 | 2.15-6.85 |
*Data on file
Long Duration of Action
Efonidipine has a slow onset and a long duration of action. This unique characteristic of Efonidipine is because of the following reasons:[13]
- High lipophilicity of Efonidipine allows it to enter the phospholipid rich cell membrane and access the dihydropyridine binding site of the Ca2+ channels.
- Tight binding to the dihydropyridine receptors.
- The dissociation constant of Efonidipine from dihydropyridine receptors is very low (0.0042/min/nM), signifying very slow dissociation from the receptors. This explains the long duration of action of Efonidipine.
Metabolism
Efonidipine is primarily metabolized in the liver. The important metabolites are N-dephenylated Efonidipine (DPH), deaminated Efonidipine (AL) and N-debenzylated Efonidipine (DBZ). DBZ and DPH exhibit activity as calcium antagonists. The vasodilating properties of DBZ and DPH were about two-thirds and one-third respectively than that of the parent compound. Results suggest that the majority of the pharmacological effect after oral dosing of Efonidipine hydrochloride in man is due to unchanged compound and its metabolites make a small contribution to the pharmacological effect.[14]
Elimination
Biliary route is the main pathway of excretion. No significant amount of unchanged drug was excreted in urine. In the urine collected for 24 h after an oral dosing, 1.1 % of the dose was excreted as deaminated Efonidipine, and 0.5% as a pyridine analogue of deaminated Efonidipine.
Indications
- Essential hypertension and renal parenchymal hypertension
- Angina
Dosage and Administration
- Essential hypertension and renal parenchymal hypertension: 20-40 mg orally once daily. A dose of up to 80mg/day is seen to be safe and effective in clinical trials.[15][16]
- Angina: 40 mg/day.
Contraindications
- Contraindicated in patients hypersensitive to Efonidipine or any of the excipients
- It is also contraindicated in pregnancy and lactation.
Precautions
- Should be administered with caution in patients with hepatic impairment
- Dose adjustment may be required in elderly as hypotension can occur
- Efonidipine may worsen clinical condition in patients with sinus bradycardia, sinus arrest or sinus node dysfunction
- As dizziness can occur due to hypotensive action, one should be careful while operating machines, with aerial work platforms and driving of a motor vehicle
- Drug should not be stopped abruptly. Discontinuation should be gradual and under supervision of a qualified physician
Drug Interactions
- Other anti-hypertensive agents: Efonidipine enhances the antihypertensive action additively and may produce hypotension and shock. Blood pressure should be monitored regularly to adjust dose of concomitant drugs.
- Cimetidine: Cimetidine inhibits CYP450 enzymes involved in metabolism of CCBs. Blood concentration of calcium channel antagonists increase leading to higher incidence of side effects (hot flushes).
- Grape fruit juice: Grapefruit juice suppresses enzymes metabolizing calcium channel antagonists (cytochrome P450) and reduces the clearance. Thus, there is a possibility that blood concentration of the drug may increase and the anti-hypertensive effect is enhanced.
- Tacrolimus: Efonidipine inhibits metabolic enzymes involved in Tacrolimus metabolism and reduces its clearance. So, increase in blood concentration of Tacrolimus can occur.
Adverse Drug Reactions
The common side effects are hot flushes, facial flushing and headache. In addition, elevation in serum total cholesterol, ALT (SGPT), AST (SGOT) and BUN may occur. Frequent urination, pedal edema, increased triglycerides occurs in less than 0.1%.[17]
Lesser incidence of pedal edema (< 0.1%)
One common adverse effect of the L-type Ca2+ channel blockers like Amlodipine is vasodilatory Pedal edema. Combined L-/T-type Ca2+ channel blockers, such as Efonidipine, display antihypertensive efficacy similar to their predecessors (Amlodipine) with much less propensity of pedal edema formation. Efonidipine equalizes the hydrostatic pressure across the capillary bed through equal arteriolar and venular dilatation, thus reducing vasodilatory edema. These incremental microcirculatory benefits of efonidipine over the conventional L-type Ca2+ channel blockers (Amlodipine) are likely attributed to their additional T-type Ca2+ channel blocking properties and the increased presence of T-type Ca2+channels in the microvasculature (e.g. arterioles, capillaries, venules etc).[18]
Among the CCBs, Efonidipine (<0.1%)[17] has lowest incidence of pedal edema compared to amlodipine ( 5-16%)[19], cilnidipine (5%)[20], benidipine (5%)[21] and azelnidipine (15.5%).[22]
Use in Special Population
Administration to Elderly
The drug should be started at low dose (20 mg/day) in elderly. Patient should be carefully observed for development of hypo-tension. Dose may be halved if there is intolerance to the 20 mg/day dosage regimen.
Pregnancy and Lactation
The drug should not be administered to pregnant women and women suspected of being pregnant. Administration to lactating women should be avoided unless benefit significantly surpasses the risk to the child. Mothers on Efonidipine treatment should avoid breast feeding.
Pediatric Use
Safety of Efonidipine in low birth weight infants, newborns, infants and children has not been established.
Efonidipine-The Best in Class
Efonidipine is unique among clinically available CCBs. Its antihypertensive efficacy is superior or at par with other CCBs. But, in terms of pleiotropic effects leading to enhanced cerebral, cardiac and renal protection, Efonidipine scores over the other CCBs.
Advantages over Amlodipine
1. Better renoprotection by:
- Dual channel blockade [1]
- Prevention of Rho-kinase and NFkB induced tubulointerstitial fibrosis[23][24]
- Reduction of synthesis and secretion of aldosterone from the adrenal cortex[25]
2. Preferred in angina with hypertension due to negative chronotropic action[26]
3. Better control of reflex tachycardia[3]
4. Reduces cardiac remodelling, arterial stiffness and prevents atherogenesis[27]
5. More useful in patients with diabetes & nephropathy[28]
6. Better protection against cardiac hypertrophy by significant reduction in LVMI[29]
7. Less adverse effects compared to Amlodipine[30]
8. Reduces endothelial dysfunction and oxidative stress(anti-oxidant property)[10]
Advantages over Cilnidipine
1. Strong negative chronotropic effect (less tachycardia) compared to Cilnidipine[3]
2. Significant improvement in exercise tolerance.[31]Better choice in hypertensive patients with angina.
3. Better BP control by marked urinary Na+ excretion[32]
4. Better renoprotection by:
- a. Suppression of plasma renin release[33]
- b. Prevention of Rho-kinase and NFkB induced tubulointerstitial fibrosis[34][35]
- c. Reduction of synthesis and secretion of aldosterone from the adrenal cortex[5]
5. Better choice in diabetic hypertensives[36]
6. Prevents cardiac remodelling by suppression of aldosterone secretion[5]
7. Superior anti-oxidant activity[10]
8. Less adverse effects compared to Cilnidipine[30]
Advantages over Benidipine
L & T-type CCBs have invoked a lot of interest in the management of hypertension because of their unique pharmacological profile. Several novel agents have been developed including Azelnidipine, Barnidipine, Benidipine, Efonidipine, Manidipine and Nilvadipine. Among all the agents, Efonidipine has emerged as the best among its peers. The advantages of Efonidipine over Benidipine are summarized below.
1. More selective blockade of T-type calcium channels [37][38]
2. More balanced renal arteriolar dilatation than benidipine[37][38]
3. Superior anti-proteinuric effect [15]
4. Greater reduction of serum aldosterone [39]
5. Renoprotection by reducing plasma renin unlike Benidipine [39]
6. Greater negative chronotropic effect
7. Efonidipine has anti-platelet activity[12]
8. Efonidipine reduces Insulin Resistance [40]
9. Significantly lower incidence of pedal edema & constipation compared to Benidipine
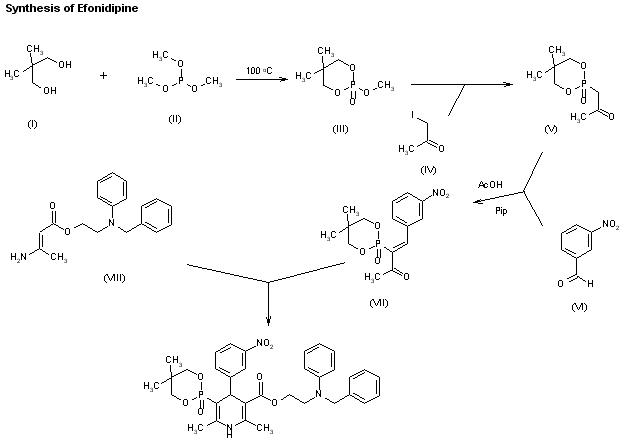
A new synthesis of efonidipine has been described: The cyclization of 2,2-dimethylbutane-1,4-diol (I) with triethyl phosphite (II) by heating at 100 C gives 2-methoxy-5,5-dimethyl-1,3,2-dioxaphosphorinan (III), which, by treatment with iodoacetone (IV) in refluxing ether, yields 2-acetonyl-5,5-dimethyl-1,3,2-dioxaphosphorinan-2-one (V). The condensation of (V) with 3-nitrobenzaldehyde (VI) by means of piperidine in acetic acid affords 3-(5,5-dimethyl-2-oxo-1,3,2-dioxaphosphorinan-2-yl)-4-(3-nitrophenyl)-3-buten-2-one (VII), which is finally cyclized with 3-amino-2-propenoic acid 2-(N-benzyl-N-phenylamino)ethyl ester (VIII) in refluxing toluene.ReferencesChem Pharm Bull 1992,40(9),2362
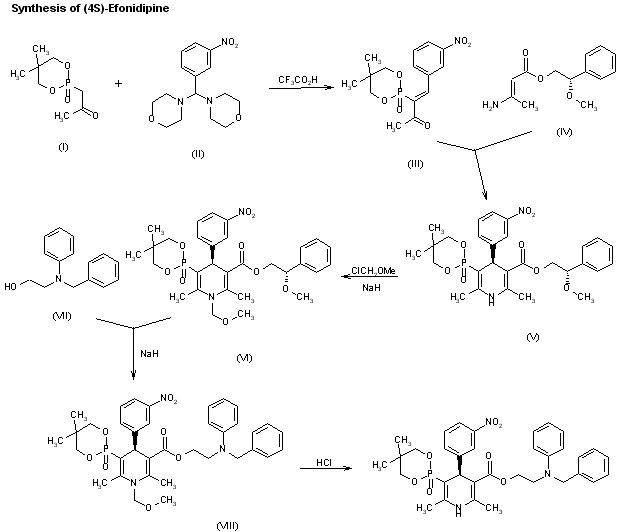
A new synthesis for (4S)-efonidipine has been described: The reaction of 5,5-dimethyl-2-(2-oxopropyl)-1,3,2-dioxaphosphorinan-2-one (I) with dimorpholino(3-nitrophenyl)methane (II) by means of trifluoroacetic acid in hot toluene gives 5,5-dimethyl-2-[1-acetyl-2-(3-nitrophenyl)vinyl]-1,3,2-dioxaphosphorina n-2-one (III), which is cyclized with 3-aminocrotonic acid 2(S)-methoxy-2-phenylethyl ester (IV) in refluxing toluene; the recrystallization of the resulting product affords 5-(5,5-dimethyl-2-oxo-1,3,2-dioxaphosphorinan-2-yl)-2,6-dimethyl-4(S)-(3-nitrophenyl)-1,4-dihydropyridine-3-carboxylic acid 2(S)-methoxy-2-phenylethyl ester (V). The protection of the NH group of (V) with chloromethyl methyl ether and NaH in THF yields the N-methoxymethyl derivative (VI), which is transesterified with 2-(N-benzyl-N-methylamino)ethanol (VII) and NaH in DMSO, giving the protected final product (VIII). Finally, this compound is deprotected with HCl in ethanol.
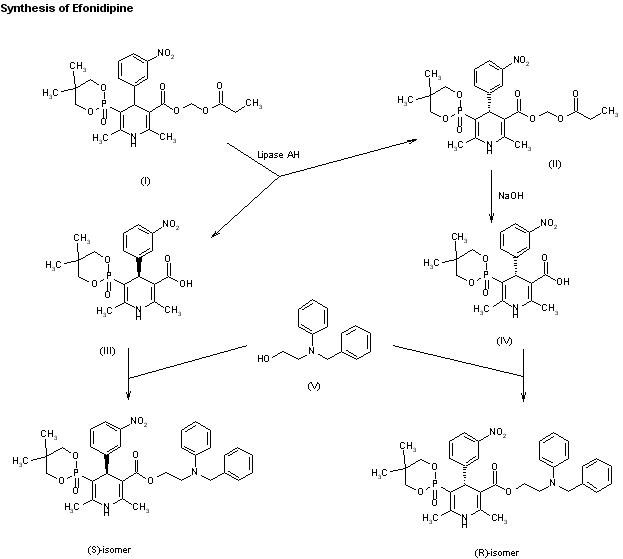
An enantioselective synthesis of efonidipine has been described: The enantioselective hydrolysis of 5-(5,5-dimethyl-2-oxo-1,3,2-dioxaphosphorinan-2-yl)-2,6-dimethyl-4-(3-n itrophenyl)-1,4-dihydropyridine-3-carboxylic acid propionyloxymethyl ester (I) with lipase AH in 2,5-dimethyltetrahydrofuran saturated with water gives the corresponding free acid of the (S)-isomer (III), while the propionyloxymethyl ester of the (R)-isomer (II) remains undisturbed. After chromatographic separation, the (R)-ester (II) is hydrolyzed with NaOH in methanol to the (R)-acid (IV). Finally, both enantiomerically pure acids (III) and (IV) are separately esterified with 2-(N-benzyl-N-phenylamino)ethanol in the usual way
CLIP
PAPER
Synthesis of 1,4-dihydropyridine-5-phosphonates and their calcium antagonistic and antihypertensive activities: Novel calcium-antagonist 2-[benzyl(phenyl)amino]ethyl 5-(5,5-dimethyl-2-oxo-1,3,2-dioxaphosphorinan-2-yl)-1,4-dihydro-2, 6-dimethyl-4-(3-nitrophenyl)-3-pyridinecarboxylate hydrochloride ethanol (NZ-105) and its crystal structure
Chem Pharm Bull 1992, 40(9): 2362
PATENT
IN 201501586
http://ipindiaservices.gov.in/PatentSearch/PatentSearch/ViewPDF
- ^ Jump up to:a b c d Tanaka H, Shigenobu K (2002). “Efonidipine hydrochloride: a dual blocker of L- and T-type Ca2+ channels”. Cardiovasc. Drug Rev. 20 (1): 81–92. PMID 12070536.
- Jump up^http://www.cdsco.nic.in/writereaddata/Minutes%20of%2034th%20SEC%20Cardiovascular%20&%20Renal%2008_11_2016.pdf
- ^ Jump up to:a b c Masumiya H, Shijuku T, Tanaka H, Shigenobu K. Inhibition of myo¬cardial L- and T-type Ca2+ currents by efonidipine: possible mecha¬nism for its chronotropic effect. Eur J Pharmacol. 1998; 349: 351-7.
- ^ Jump up to:a b Masuda Y, Tanaka S. Efonidipine Hydrochloride: A New Calcium Antagonist. Cardiovascular Drug Reviews. 1994; 12 ( 2): 123-135.
- ^ Jump up to:a b c d Ikeda K, Isaka T, Fujioka K, Manome Y, Tojo K. Suppression of Aldosterone Synthesis and Secretion by Ca2+ Channel Antagonists. International Journal of Endocrinology. 2012.
- Jump up^ Hayashi K, et al. T-Type Ca Channel Blockade as a Determinant of Kidney Protection. Keio J Med. 2010; 59(3): 84-95
- ^ Jump up to:a b Hayashi M, Yamaji Y, Nakazato Y, Saruta T. The effects of calcium channel blockers on nuclear factor kappa B activation in the mesangial cells. Hypertens Res. 2000;23:521–525.
- Jump up^ Sugano N, Sugano N, Wakino S, Tatematsu S, Homma K, Yoshioka K, Hasegawa K, Utsunomiya Y, Tokudome G, Hosoya T, Saruta T, Hayashi K. Role of T-type Ca2 channels (TCCs) as a determinant of Rho-kinase activation and epithelial-mesenchymal transition (EMT) in renal injury. J Hypertens. 2006;24(suppl 6):128
- Jump up^ Baylis C, Qiu C, Engels K. Comparison of L-type and mixed L- and T-type calcium channel blockers on kidney injury caused by deoxycorticosterone-salt hypertension in rats. Am J Kidney Dis. 2001;38: 1292–1297.
- ^ Jump up to:a b c Sasaki H, Saiki A, Endo K, Ban N, Yamaguchi T, Kawana H, Nagayama D, Ohhira M, Oyama T, Miyashita Y, Shirai K. Protective effects of efonidipine, a T- and L-type calcium channel blocker, on renal function and arterial stiffness in type 2 diabetic patients with hypertension and nephropathy. J Atheroscler Thromb. 2009 Oct; 16(5): 568-75.
- Jump up^ Oshima T, Ozono R, Yano Y, Higashi Y, Teragawa H, Miho N, Ishida T, Ishida M, Yoshizumi M, Kambe M. Beneficial effect of T-type calcium channel blockers on endothelial function in patients with essential hypertension. Hypertens Res. 2005 Nov;28(11):889-94.
- ^ Jump up to:a b Nomura S, Kanazawa S, Fukuhara S. Effects of efonidipine on platelet and monocyte activation markers in hypertensive patients with and without type 2 diabetes mellitus. J Hum Hypertens. 2002 Aug;16(8):539-47.
- Jump up^ Yamashita T, Masuda Y, et al. NZ-105, a New 1,4-Dihydropyridine Derivative: Correlation between Dihydropyridine Receptor Binding and Inhibition of Calcium Uptake in Rabbit Aorta. Japan J Pharmacol. 1991; 57: 337-348.
- Jump up^ Nakabeppu H, et al.Metabolism of Efonidipine in Man.Xenobiotica.1995 Aug;229-239.
- ^ Jump up to:a b Hayashi K, Kumagai H, Saruta T. Effects of efonidipine and ACE inhibitors on proteinuria in human hypertension with renal impairment. Am J Hypertens. 2003;16:116 –122
- Jump up^ Oh IY, Seo MK, Lee HY, Kim SG, Kim KS, Kim WH, Hyon MS, Han KR, Lim SJ, Kim CH. Beneficial Effect of Efonidipine, an L- and T-Type Dual Calcium Channel Blocker, on Heart Rate and Blood Pressure in Patients With Mild-to-Moderate Essential Hypertension. Korean Circ J. 2010 Oct;40(10):514-9.
- ^ Jump up to:a b http://www.kegg.jp/medicus-bin/japic_med?japic_code=00044638. Missing or empty
|title=(help) - Jump up^ Ge W, Ren J. Combined L-/T-type calcium channel blockers: ready for prime time. Hypertension. 2009 Apr;53(4):592-4. doi: 10.1161/HYPERTENSIONAHA.108.127548. Epub 2009 Feb 23. PubMed PMID 19237678.
- Jump up^ Osterloh IH. An update on the safety of amlodipine. J Cardiovasc Pharmacol.1991;17 Suppl 1:S65-8.
- Jump up^ http://www.kegg.jp/medicus-bin/japic_med?japic_code=00062065. Missing or empty
|title=(help) - Jump up^ http://www.kegg.jp/medicus-bin/japic_med?japic_code=00005939. Missing or empty
|title=(help) - Jump up^ Takihata M, Nakamura A, Kondo Y, Kawasaki S, Kimura M, Terauchi Y. Comparison of Azelnidipine and Trichlormethiazide in Japanese Type 2 Diabetic Patients with Hypertension: The COAT Randomized Controlled Trial. PLoS One. 2015 May 4;10(5):e0125519.
- Jump up^ Song I, KimD, Choi S, Sun M, Kim Y, Shin HS. Role of the α1g T-type calcium channel in spontaneous absence seizures in mutant mice. J Neurosci. 2004; 24: 5249–5257.
- Jump up^ Lory P, Bidaud I, Chemin J. T-Type calcium channels in differentiation and proliferation. Cell Calcium. 2006; 40: 135–146.
- Jump up^ Ikeda K, Isaka T, Fujioka K, Manome Y, Tojo K. Suppression of Aldosterone Synthesis and Secretion by Ca2+ Channel Antagonists. International Journal of Endocrinology. 2012.
- Jump up^ Oh IY, Seo MK, Lee HY, Kim SG, Kim KS, Kim WH, Hyon MS, Han KR, Lim SJ, Kim CH. Beneficial Effect of Efonidipine, an L- and T-Type Dual Calcium Channel Blocker, on Heart Rate and Blood Pressure in Patients With Mild-to-Moderate Essential Hypertension. Korean Circ J. 2010 Oct;40(10):514-9.
- Jump up^ Catena C, Colussi G, Marzano L, Sechi LA. Aldosterone and the heart: from basic research to clinical evidence. Horm Metab Res. 2012;44:181– 187.
- Jump up^ Sasaki H, Saiki A, Endo K, Ban N, Yamaguchi T, Kawana H, Nagayama D, Ohhira M, Oyama T, Miyashita Y, Shirai K. Protective effects of efonidipine, a T- and L-type calcium channel blocker, on renal function and arterial stiffness in type 2 diabetic patients with hypertension and nephropathy. J Atheroscler Thromb. 2009 Oct; 16(5): 568-75.
- Jump up^ Saito T, Fujii K, Takizawa T, Toyosaki T, Kuwabara Y, Kobayashi S, Ichikawa H, Karaki A, Yamazaki Y, Iwata J, Yamada K, Tomiya H, Takeda K, Inagaki Y. Effects of the new calcium antagonist efonidipine hydrochloride on resting and exercise hemodynamics in patients with stable effort angina. Arzneimittelforschung. 1996 Sep;46(9):861-7.
- ^ Jump up to:a b Saruta T. Current status of calcium antagonists in Japan. Am J Cardiol. 1998;82:32R-34R.
- Jump up^ Okayama S, Imagawa K, Naya N, Iwama H, Somekawa S, Kawata H, Horii M, Nakajima T, Uemura S, Saito Y. Blocking T-type Ca2+ channels with efonidipine decreased plasma aldosterone concentration in healthy volunteers. Hypertens Res. 2006 Jul;29(7):493-7.
- Jump up^ Honda M, Hayashi K, Matsuda H, Kubota E, Tokuyama H, Okubo K, Ozawa Y, Saruta T. Divergent natriuretic action of calcium channel antagonists in mongrel dogs: renal haemodynamics as a determinant of natriuresis. Clinical Science. 2001; 101: 421–427
- Jump up^ Wagner C, Kramer KB, Hinder M, Kieninger M, Kurtz A. T-type and L-type calcium channel blockers exert opposite effects on renin secretion and renin gene expression in conscious rats. Br J Pharmacol. 1998;124: 579 –585.
- Jump up^ Song I, KimD, Choi S, Sun M, Kim Y, Shin HS. Role of the α1g T-type calcium channel in spontaneous absence seizures in mutant mice. J Neurosci. 2004; 24: 5249–5257.
- Jump up^ Lory P, Bidaud I, Chemin J. T-Type calcium channels in differentiation and proliferation. Cell Calcium. 2006; 40: 135–146.
- Jump up^ Ando K, Ueshima K, Tanaka S, Kosugi S, Sato T, Matsuoka H, Nakao K, Fujita T. Comparison of the antialbuminuric effects of L-/N-type and L-type calcium channel blockers in hypertensive patients with diabetes and microalbuminuria: the study of assessment for kidney function by urinary microalbumin in randomized (SAKURA) trial. Int J Med Sci. 2013 Jul 30;10(9):1209-16.
- ^ Jump up to:a b Hayashi K, Wakino S, Sugano N, Ozawa Y, Homma K, Saruta T. Ca2+ Channel Subtypes and Pharmacology in the Kidney. Circ Res. 2007;100:342-353.
- ^ Jump up to:a b Hayashi K, Ozawa Y, Fujiwara K, Wakino S, Kumagai H, Saruta T. Role of actions of calcium antagonists on efferent arterioles with special references to glomerular hypertension. Am J Nephrol. 2003 Jul-Aug;23(4):229-44.
- ^ Jump up to:a b Tani S, Takahashi A, Nagao K, Hirayama A. Effects of the T/L-type calcium channel blocker benidipine on albuminuria and plasma aldosterone concentration. A pilot study involving switching from L-type calcium channel blockers to benidipine. Int Heart J. 2014;55(6):519-25
- Jump up^ Li M. Role of T-Type Ca2+ Channels in Basal Insulin Release. T-type Calcium Channels in Basic and Clinical Science. Springer Vienna. 2015; 137-150.
- (in Japanese) Landel ランデル (PDF) Shionogi & Co. April 2005.
 |
|
| Clinical data | |
|---|---|
| Trade names | Landel (ランデル) |
| AHFS/Drugs.com | International Drug Names |
| Routes of administration |
Oral |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C34H38N3O7P |
| Molar mass | 631.65 g/mol |
| 3D model (JSmol) | |
(R)-base
- Formula:C34H38N3O7P
- MW:631.67 g/mol
- CAS-RN:128194-13-8
(S)-base
- Formula:C34H38N3O7P
- MW:631.67 g/mol
- CAS-RN:128194-12-7
///////////Efonidipine, エホニジピン, IND 2017, Landel , NZ 105, Efonidipine Hydrochloride Ethanolate
CC1=C(C(C(=C(N1)C)P2(=O)OCC(CO2)(C)C)C3=CC(=CC=C3)[N+](=O)[O-])C(=O)OCCN(CC4=CC=CC=C4)C5=CC=CC=C5
Arterolane Maleate
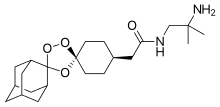
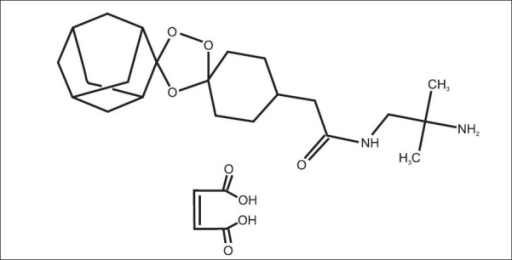

Arteolane Maleate
C26H40N2O8
Molecular Weight: 508.612
CAS 959520-73-1
664338-39-0 (free base) 959520-73-1 (maleate) 959520-79-7 (acetate) 664338-40-3 (tosylate) 959520-82-2 (tartrate) 959520-83-3 (citrate)
N-(2-amino-2-methylpropyl)-2-((1R,3R,4”S,5R,5’s,7R)-dispiro[adamantane-2,3′-[1,2,4]trioxolane-5′,1”-cyclohexan]-4”-yl)acetamide maleate
Dispiro[cyclohexane-1,3′-[1,2,4]trioxolane-5′,2”-tricyclo[3.3.1.13,7]decane]-4-acetamide, N-(2-amino-2-methylpropyl)-, cis-, (2Z)-2-butenedioate (1:1)
APPROVED 4.11.2017 CDSCO
Arteolane Maleate and Piperaquine phosphate Dispersible tablets (37.5 mg +187.5 mg

cas 664338-39-0
Arterolane
Arterolane, also known as OZ277 or RBx 11160, is a substance that was tested for antimalarial activity[1] by Ranbaxy Laboratories.[2] It was discovered by US and European scientists who were coordinated by the Medicines for Malaria Venture (MMV).[3] Its molecular structure is uncommon for pharmacological compounds in that it has both a ozonide (trioxolane) group and an adamantanesubstituent.[4]
Initial results were disappointing, and in 2007 MMV withdrew support, after having invested $20M in the research;[5] Ranbaxy said at the time that it intended to continue developing the drug combination on its own.[2] Ranbaxy started a Phase II clinical trial of arterolane, in combination with piperaquine in 2009 that published in 2015.[6][7]
In 2012, Ranbaxy obtained approval to market the arterolane/piperaquine combination drug in India, under the brand name Synriam,[5]and in 2014 received approval to market it in Nigeria, Uganda, Senegal, Cameroon, Guinea, Kenya and Ivory Coast; it had already received approval in Uganda.[8]

Ranbaxy launched India’s first new drug, SynriamTM, treating Plasmodium falciparummalaria in adults. The drug provides quick relief from most malaria-related symptoms, including fever, and has a high cure rate of over 95 %.
Just one tablet per day is required, for three days, instead of two to four tablets, twice daily, for three or more days with other medicines. The drug is independent of dietary restrictions for fatty foods or milk.
Ranbaxy developed Synriam as a fixed-dose combination of arterolane maleate and piperaquine phosphate, where arterolane is the new chemical entity (NCE) that was developed as an alternative to artemisinin. It is the first recently developed antimalarial not based on artemisinin, one of the most effective treatments for malaria, which has shown problems with resistance in recent years. Arterolane was discovered by a collaborative drug discovery project funded by the Medicines for Malaria Venture. Since SynriamTM has a synthetic source, unlike artemisinin-based drugs, production can be scaled up whenever required and a consistent supply can be maintained at a low cost.

The new drug, has been approved by the Drug Controller General of India (DCGI) for marketing in India and conforms to the recommendations of the World Health Organization (WHO) for using combination therapy in malaria. Ranbaxy is also working to make it available in African, Asian and South American markets where Malaria is rampant. SynriamTM trials are ongoing for Plasmodium vivax malaria and a paediatric formulation.
Derek Lowe of the famous In the Pipeline blog had written about arterolane in 2009. At the time it was in Phase III trial, which I assumed were the trials that Ranbaxy was conducting. But it turned out that arterolane was developed by a collaboration between researchers in the US, the UK, Switzerland and Australia who were funded by the World Health Organization and Medicines for Malaria Venture (a Swiss non-profit). They published this work in Nature in 2004 and further SAR (Structure Activity Relationship) studies in J Med Chem in 2010. So Ranbaxy did not develop the drug from scratch? But the press release quotes Arun Sawhney, CEO and Managing Director of Ranbaxy which misleads people to think so: “It is indeed gratifying to see that Ranbaxy’s scientists have been able to gift our great nation its first new drug, to treat malaria, a disease endemic to our part of the world. This is a historic day for science and technology in India as well as for the pharmaceutical industry in the country. Today, India joins the elite and exclusive club of nations of the world that have demonstrated the capability of developing a new drug”. So Ranbaxy mixes a known active compound (piperaquine) with a new compound that someone else found to be active (arterolane) and claims that they developed a new drug? In an interview in LiveMint, Sawhney says, “Ranbaxy spent around $30 million on Synriam and the contribution from DST [India’s Department of Science & Technology] was Rs.5 crore. The drug went through several phases of development since the project began in 2003. We did not look at this as a commercial development. Instead, this is a CSR [Corporate Social Responsibility] venture for us.” That’s a give away because developing a new drug from scratch has to cost more than $30 million + Rs.50 million.
- Ranbaxy Laboratories Limited (Ranbaxy), India
![]() now taken over by sun
now taken over by sun
SynriamTM
|
|
Description SynriamTM is a fixed dose combination of two antimalarial active ingredients arterolane maleate and piperaquine phosphate.
Arterolane maleate is a synthetic trioxolane compound. The chemical name of arterolane maleate is cis-adamantane-2-spiro-3’-8’-[[[(2’-amino-2’ methylpropyl) amino] carbonyl] methyl] 1’,2’,4’-trioxaspiro [4.5] decane hydrogen maleate. The molecular formula is C26H40N2O8 and molecular weight is 508.61. The structural formula is as follows:

| MALARIA |
| Malaria is one of the most prevalent and deadly parasitic diseases in the world. Up to 289 million cases of malaria may have occurred in 2010, causing between 660,000 and 1.25 million deaths, mainly in Africa and mostly of children younger than 5 years. |
| (WHO: http://www.who.int/malaria/publications/world_malaria_report_2012/en/index.html; Fidock, D. A. Eliminating Malaria. Science 2013, 340, 1531-1533.) |
|


Malaria, the most common parasitic disease of humans, remains a major health and economic burden in most tropical countries. Large areas of Central and South America, Hispaniola (Haiti and the Dominican Republic), Africa, the Middle East, the Indian subcontinent, Southeast Asia, and Oceania are considered as malaria-risk areas. It leads to a heavy toll of illness and death, especially amongst children and pregnant women.
According to the World Health Organization, it is estimated that the disease infects about 400 million people each year, and around two to three million people die from malaria every year. There are four kinds of malaria parasites that infect human: Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale and Plasmodium malariae.
Malaria spreads from one person to another by the bite of mosquito, Anopheles gambiae, which serves as vector. When a mosquito sucks the blood of human, sporozoites are transfused into the human body together with saliva of the mosquito. The sporozoites enter into the hepatocytes, reproduce asexually and finally enter into the blood stream. The parasites continue to multiply inside the red blood cells, until they burst and release large number of merozoites. This process continues, destroying a significant number of blood cells and causing the characteristic paroxysm (“chills and fever”) associated with the disease. In the red blood cells, some of the merozoites become male or female gametocytes. These gametocytes are ingested by the mosquito when it feeds on blood. The gametocytes fuse in the vector’s gut; sporozoites are produced and are migrated to the vector’s salivary glands.
The clinical symptoms of malaria are generally associated with the bursting of red blood cells causing an intense fever associated with chills that can leave the infected individual exhausted and bedridden. More severe symptoms associated with repeat infections and/or infection by Plasmodium falciparum include anaemia, severe headaches, convulsions, delirium and, in some instances, death.
Quinine, an antimalarial compound that is extracted from the bark of cinchona tree, is one of the oldest and most effective drugs in existence. Chloroquine and mefloquine are the synthetic analogs of quinine developed in 1940’s, which due to their effectiveness, ease of manufacture, and general lack of side effects, became the drugs of choice. The downside to quinine and its derivatives is that they are short-acting and have bitter taste. Further, they fail to prevent disease relapses and are also associated with side effects commonly known as “Chinchonism syndrome” characterized by nausea, vomiting, dizziness, vertigo and deafness. However, in recent years, with the emergence of drug- resistant strains of parasite and insecticide-resistant strains of vector, the treatment and/or control of malaria is becoming difficult with these conventional drugs.
Malarial treatment further progressed with the discovery of Artemisinin
(qinghaosu), a naturally occurring endoperoxide sesquiterpene lactone isolated from the plant Artemisia annua (Meshnick et al., Microbiol. Rev. 1996, 60, p. 301-315; Vroman et al., Curr. Pharm. Design, 1999, 5, p. 101-138; Dhingra et al., 2000, 66, p. 279-300), and a number of its precursors, metabolites and semi-synthetic derivatives which have shown to possess antimalarial properties. The antimalarial action of artemisinin is due to its reaction with iron in free heme molecules of the malaria parasite, with the generation of free radicals leading to cellular destruction. This initiated a substantial effort to elucidate its molecular mechanism of action (Jefford, dv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297) and to identify novel antimalarial peroxides (Dong and Vennerstrom, Expert Opin. Ther. Patents 2001, 1 1, p. 1753-1760).
Although the clinically useful artemisinin derivatives are rapid acting and potent antimalarial drugs, they have several disadvantages including recrudescence,
neurotoxicity, (Wesche et al., Antimicrob. Agents. Chemother. 1994, 38, p. 1813-1819) and metabolic instability (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43). A fair number of these compounds are quite active in vitro, but most suffer from low oral activity (White, Trans. R. Soc. Trop. Med. Hyg., 1994, 88, p. 41-43 and van Agtmael et al., Trends Pharmacol. Sci., 1999, 20, p. 199-205). Further all these artemisinin derivatives are conventionally obtained from plant source and are therefore expensive. As the cultivation of the plant material is dependent on many factors including the weather conditions, the supply source thus becomes finite and there are chances of varying yield and potency. This leads to quality inconsistencies and supply constraints. As malaria is more prevalent in developing countries, a switch to cheaper and effective medicine is highly desirable.
Thus there exists a need in the art to identify new peroxide antimalarial agents, especially those which are not dependent on plant source and can be easily synthesized, are devoid of neurotoxicity, and which possess improved solubility, stability and pharmacokinetic properties.
Following that, many synthetic antimalarial 1 ,2,4-trioxanes (Jefford, Adv. Drug Res. 1997, 29, p. 271-325; Cumming et al., Adv. Pharmacol. 1997, 37, p. 254-297), 1,2,4,5-tetraoxanes (Vennerstrom et al., J. Med. Chem., 2000, 43, p. 2753-2758), and other endoperoxides have been prepared. Various patents/applications disclose means and method for treating malaria using Spiro or dispiro 1,2,4-trioxolanes for example, U.S.
Patent Application No. 2004/0186168 and U.S. Patent Nos. 6,486, 199 and 6,825,230. The present invention relates to solid dosage forms of the various spiro or dispiro 1 ,2,4- trioxolanes antimalarial compounds disclosed in these patents/applications and are incorporated herein by reference.
Active compounds representing various Spiro and dispiro 1 ,2,4-trioxolane derivatives possess excellent potency, efficacy against Plasmodium parasites, and a lower degree of neurotoxicity, in addition to their structural simplicity and ease of synthesis. Furthermore, these compounds have half-lives which are believed to permit short-term treatment regimens comparing favorably to other artemisinin-like drugs. In general, the therapeutic dose of trioxolane derivative may range between about 0.1-1000 mg/kg/day, in particular between about 1-100 mg/kg/day. The foregoing dose may be administered as a single dose or may be divided into multiple doses. For malaria prevention, a typical dosing schedule could be, for example, 2.0-1000 mg/kg weekly beginning 1-2 weeks prior to malaria exposure, continued up to 1-2 weeks post-exposure.
Monotherapy with artemisinin (natural or synthetic) class of drugs might cure the patients within 3 days, however perceiving the potential threat of the malarial parasite developing resistance towards otherwise very potent artemisinin class of drugs, WHO had strictly called for an immediate halt to the provision of single-drug artemisinin malaria pills. Combination therapy in case of malaria retards the development of resistance, improve efficacy by lowering recrudescence rate, provides synergistic effect, and increase exposure of the parasite to the drugs.
Artemsinin based combinations are available in the market for a long time.
Artemether-lumafentrine (Co-artem®) was the first fixed dose antimalarial combination containing an artemisinin derivative and has been known since 1999. This combination has passed extensive safety and efficacy trials and has been approved by more than 70 regulatory agencies. Co-artem® is recommended by WHO as the first line treatment for uncomplicated malaria.
Other artemisinin based combinations include artesunate and amodiaquine (Coarsucam®), and dihydroartemisin and piperaquine (Eurartesim®). Unfortunately, all the available artemisinin based combinations have complicated dosage regimens making it difficult and inconvenient for a patient to comply completely with the total prescribed duration. For example, the dosage regimen of Co-artem® for an adult having body weight of more than 35 kg includes 6 doses over three days. The first dose comprises four tablets initially, the second dose comprises four tablets after eight hours, the third to sixth doses comprise four tablets twice for another two days; making it a total of 24 tablets. The dosage regimen of Coarsucam® for an adult having body weight of more than 36 kg or age above 14 years includes three doses over three days; each dose comprises two tablets; making it a total of six tablets. The dosage regimen of Eurartesim® for an adult having body weight between 36 kg – 75 kg includes 3 doses over three days, each dose comprises of three tablets, making it a total of nine tablets.
It is evident that the available artemisinin-based combinations have a high pill burden on patients as they need to consume too many tablets. As noted above, this may increase the possibility of missing a few doses, and, consequently, could result in reduced efficacy due to non-compliance and may even lead to development of resistance for the drug. Therefore, there is an urgent and unmet need for anti-malarial combinations with a simplified daily dosing regimen that reduces the pill burden and would increase patient compliance.
Apart from simplifying the regimen, there are certain limitations for formulators developing formulations with trioxolones, the first being their susceptibility to degradation in presence of moisture that results in reduced shelf lives. Another is their bitter taste, which can result in poor compliance of the regimen or selection of another, possibly less effective, therapeutic agent.
……………………..
http://www.google.st/patents/US6906205

……………………
http://www.google.st/patents/WO2013008218A1?cl=en
structural Formula II.

Formula II
Active compound includes one or more of the various spiro and dispiro trioxolane derivatives disclosed in U.S. Application No. 2004/0186168 and U.S. Patent Nos.
6,486,199 and 6,825,230, which are incorporated herein by reference. These trioxolanes are relatively sterically hindered on at least one side of the trioxolane heterocycle which provides better in vivo activity, especially with respect to oral administration. Particularly, spiro and dispiro 1,2,4-trioxolanes derivatives possess excellent potency and efficacy against Plasmodium parasites, and a lower degree of neurotoxicity.
The term “Active compound I” herein means cis-adamantane-2-spiro-3′-8′-[[[(2′- amino-2′-methylpropyl)amino]carbonyl]-methyl]- 1 ‘,2′,4’-trioxaspiro[4.5]decane hydrogen maleate. The Active compound I may be present in an amount of from about 5% to about 25%, w/w based on the total dosage form.

………………
http://www.google.st/patents/WO2007138435A2?cl=en
A synthetic procedure for preparing compounds of Formula I, salts of the free base c«-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]- 1 ‘, 2′, 4’-trioxaspiro [4.5] decane has been disclosed in U.S. 6,906,205.

The process for the preparation of compounds of Formula I wherein a compound of Formula II (wherein R is lower alkyl) is reacted with a compound of Formula III (wherein R is lower alkyl) to obtain compound of Formula IV;


Formula Formula IV
followed by hydrolysis of the compounds of Formula IV to give a compound of Formula V;

Formula V followed by the reaction of the compound of Formula V with an activating agent, for example, methyl chloroformate, ethyl chloroformate, propyl chloro formate, n-butyl chloro formate, isobutyl chloroformate or pivaloyl chloride leads to the formation of mixed anhydride, which is reacted in situ reaction with 1 ,2-diamino-2-methyl propane to give a compound of Formula VI; and

Formula Vl reacting the compound of Formula VI with an acid of Formula HX (wherein X can be the same as defined earlier) to give compounds of Formula I.
Example 1 : Preparation of O-methyl-2-adamantanone oxime
To a solution of 2-adamantanone (50 g, 0.3328 mol, 1 equiv.) in methanol (0.25 lit), sodium hydroxide solution (15 g, 0.3761mol, 1.13 equiv, in 50 mL water) was added followed by methoxylamine hydrochloride (37.5 g x 81.59% Purity= 30.596 g, 0.366 mol, 1.1 equiv) at room temperature under stirring. The reaction mixture was stirred at room temperature for 1 to 2 h. The reaction was monitored by HPLC. The reaction mixture was concentrated at 40- 45°C under vacuum to get a thick residue. Water (250 mL) was added at room temperature and the reaction mixture was stirred for half an hour. The white solid was filtered, washed with water (50 mL), and dried at 40 to 45°C under reduced pressure. O-methyl 2- adamantanone oxime (57 g, 95 % yield) was obtained as a white solid.
(M++l) 180, 1HNMR (400 MHz, CDCl3 ): δ 1.98 – 1.79 (m, 12H), 2.53 (s, IH), 3.46 ( s, IH), 3.81 (s, 3H).
Example 2: Preparation of 4-(methoxycarbonvmethvPcvclohexanone A high pressure autoclave was charged with a mixture of methyl (4- hydroxyphenyl)acetate (50 g, 0.30 mol), palladium ( 5g) (10 %) on carbon (50 % wet) and O- xylene (250 mL). The reaction mixture was stirred under 110 to 115 psi of hydrogen pressure for 7 to 8 h at 1400C. The reaction was monitored by HPLC. The reaction mixture was then cooled to room temperature, and the catalyst was filtered off. Filtrate was concentrated under reduced pressure to get 4-(methoxycarbonylmethyl)cyclohexanone as light yellow to colorless oily liquid (48.7 g, 97.4 %).
(M++!) 171, ‘ HNMR (400 MHz, CDCl 3): δ 1.48 – 1.51 ( m, 2H), 2.1 1-2.07 (m, 2H), 2.4- 2.23 (m, 7H), 3.7 (s, 3H).
Example 3: Preparation of methyl (Is, 4s)-dispiro [cyclohexane-l, 3′-f 1,2,4] trioxolane-5′, 2″-tricvclor3.3.1.13–71decan1-4-ylacetate
A solution of O-methyl-2-adamantanone oxime (example 1) (11.06 g, 61.7 mmol, 1.5 equiv.) and 4-(methoxycarbonymethyl)cyclohexanone (example 2) (7.0 g, 41.1 mmol, 1 equiv.) in cyclohexane ( 200ml) and dichloromethane (40 mL) was treated with ozone (ozone was produced with an OREC ozone generator [0.6 L/min. O2, 60 V] passed through an empty gas washing bottle that was cooled to -780C). The solvent was removed after the reaction was complete. After removal of solvents, the crude product was purified by crystallization from 80% aqueous ethanol (200 mL) to afford the title compound as a colorless solid. Yield: 10.83 g, 78%, mp: 96-980C; 1HNMR (500 Hz3CDCl3): δ 1.20-1.33 (m, 2H), 1.61-2.09 (m, 5 21H), 2.22 (d, J = 6.8Hz, 2H), 3.67(s,3H).
Example 4: Preparation of (Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″- tricvclo [3.3.1.13‘7] decanl-4-ylacetic acid
Sodium hydroxide (3.86 g, 96.57 mmol, 3 equiv.) in water (80 mL) was added to a solution of methyl (\s, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo
10 [3.3.1.I3‘7] decan]-4-ylacetate (example 3) (10.83 g, 32.19 mmol, 1 equiv.) in 95% ethanol (150 mL). The mixture was stirred at 500C for about 4 h, cooled to O0C, and treated with IM hydrochloric acid (129ml, 4 equiv). The precipitate was collected by filtration, washed with 50 % aqueous ethanol (150 mL) and dried in vacuum at 40 0C to give the title compound as colorless solid. Yield: 9.952 g, 96%, mp: 146-1480C ( 95% ethanol), 1HNMR (500 Hz,
15 CDCl3): δ 1.19-1.41 (m,2H), 1.60-2.05 (m,21H), 2.27 (d, J=6.8 Hz,2H).
Example 5: Preparation of c?s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2′-methyl propyl) amino] carbonyl] methyl]-! ‘, T , 4’-trioxaspiro [4.5] decane
Method A:
(Is, 4s)-dispiro[cyclohexane- 1 ,3 ‘-[ 1 ,2,4]trioxolane-5 ‘,2 ‘ ‘-tricyclo[3.3.1.13‘7]decan]-4-
.0 ylacetic acid (example 4) (5 g ,15.5mmol, 1 equiv) was mixed with triethylamine (2.5 g , 24.8 mmol, 1.6 equiv) in 100ml of dichloromethane. The reaction mixture was cooled to – 1O0C to 00C. Ethyl chloro formate (1.68 g, 17 mmol, 1.0 equiv) in 15 mL dichloromethane was charged to the above reaction mixture at – 100C to 00C. The reaction mixture was stirred at the same temperature for 10 to 30 minutes. The resulting mixed anhydride reaction mixture
15 was added dropwise to a previously prepared solution of l,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv), in 100 mL dichloromethane at -100C to O0C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the same temperature till the reaction was complete. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete
>0 within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (50 mL) was charged, organic layer was separated and washed with 10% sodium bicarbonate solution (50 mL) and water (50 mL) at room temperature. The organic layer was dried over sodium sulphate and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (50ml) was added to obtain residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. The solid was dried under reduced pressure at room 5 temperature.
Yield: 5.2 g (85.4 %), (M++l) 393, 1HNMR (400 MHz, DMSO-J6 ): δ 0.929 ( s, 6H), 1.105 – 1.079 (m, 2H), 1.887-1.641 (m, 21H), 2.030-2.017 (d, 2H), 2.928 (d, 2H).
Method B:
(Is, 4s)-dispiro [cyclohexane-1, 3′-[l,2,4] trioxolane-5′, 2″-tricyclo [3.3.1.I3‘7]
10 decan]-4-ylacetic acid (example 4) (10 g, 31mmol, 1 equiv) was treated with isobutyl chloroformate (4.5 g, 33mmol, 1.1 equiv) in presence of organic base like triethyl amine (5 g, 49.6mmol, 1.6 equiv) at 00C to 7°C in 250ml of dichloromethane. The solution was stirred at O0C to 7°C for aboutlO to 30 minutes. To the above reaction mixture, previously prepared solution of l,2-diamino-2-methylpropane (3.27 g, 37 mmol, 1.2 equiv), in 50 mL of
15 dichloromethane was added at O0C to 7°C in one lot. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. Reaction was complete within 2 h. The reaction nitrogen atmosphere was maintained throughout the reaction. Water (250 mL) was charged, organic
20 layer was separated and washed with 10% sodium bicarbonate solution (200 mL) and water (100 mL) at room temperature and the solvent was removed at 25 to 4O0C under reduced pressure. Hexane (100ml) was added to the residue, under stirring, at room temperature. The mixture was filtered and washed with chilled hexane (10 mL). The resultant solid was dried under reduced pressure at room temperature. Yield: 10.63 g (87%), (M++l) 393, 1HNMR
>5 (400 MHz, DMSO-J6 ) :δ 0.928 ( s, 6H), 1.102 – 1.074 (m, 2H), 1.859-1.616 (m, 21H), 2.031- 2.013 (d, 2H), 2.94-2.925 (d, 2H). Method C:
(\s, 4s)-dispiro[cyclohexane-l,3′-[l,2,4]trioxolane-5′,2″-tricyclo[3.3.1.13>7]decan]-4- ylacetic acid (example 4) (5 g, 15.5mmol, 1 equiv) was treated with pivaloyl chloride (1.87 g, 15.5 mmol, 1 equiv) and triethylamine (2.5gm, 24.8mmol, 1.6 equiv) at -15°C to -100C in dichloromethane (125 mL). The solution was stirred at -150C to -100C for aboutlO to 30 minutes. It resulted in the formation of mixed anydride. To the above reaction mixture, previously prepared solution of 1 ,2-diamino-2-methylpropane (1.64 g, 18.6 mmol, 1.2 equiv) in 25 mL dichloromethane was added at -15°C to -100C. The temperature of reaction mixture was raised to room temperature. The reaction mixture was stirred at the room temperature till reaction was over. Reaction monitoring was done by thin layer chromatography using 5 to 10% methanol in dichloromethane. The reaction was complete within 2 h. Nitrogen atmosphere was maintained throughout the reaction. Water (125 mL) was charged, organic layer was separated and washed with 50 mL of 10% sodium bicarbonate solution and 125 mL of water, respectively at room temperature. Finally solvent was removed at 25 to 4O0C under reduced pressure. 50 mL of 5% Ethyl acetate – hexane solvent mixture was added to the residue under stirring at room temperature. The mixture was filtered and washed with 5 mL of chilled hexane. Solid was dried under reduced pressure at room temperature. Yield: 5.03 g (83 %), (M++l) 393, 1JINMR (400 MHz, OMSO-d6 ):δ 0.93 ( s, 6H), 1.113 – 1.069 (m, 2H), 1.861-1.644 (m, 21H), 2.033-2.015 (d, 2H), 2.948-2.933 (d, 2H).
Example 6: Preparation of c/s-adamantane-2-spiro-3′ -8 ‘-πT(2′-amino-2’ -methyl propyl) amino! carbonyl] methyli-l ‘, 2\ 4′-U-JoXaSpJrQ [4.51 decane maleate To a solution of c/s-adamantane-2-spiro-3′-8′-[[[(2′-amino-2’-methyl propyl) amino] carbonyl] methyl]-! ‘, 2′, 4’-trioxaspiro [4.5] decane (example 5) (60 g, 0.153 moles) in ethanol (150 mL) was added a solution of maleic acid (17.3 g, 0.15 moles, 0.98 equiv. in ethanol 90 mL) and the reaction mixture was stirred for about 1 h. To this clear solution, n- heptane (720 mL) was added at room temperature in 1 h and the reaction mixture was stirred for 3 h. It was then cooled to 0 to 100C and filtered. The cake was washed with n-heptane (60 mL) and dried under vacuum at 40-450C.
Yield: 67 g, 77.4%, mp: 1490C (decomp), (M++l) 393.5, 1HNMR (300 MHz, DMSO-^ ): δ 1.05-1.11 (2H,m), 1.18 (6H,s), 1.64-1.89 (21H,m), 2.07(2H,d), 3.21 (2H,d), 6.06 (2H,d), 7.797 (2H, bs), 8.07 (IH, t).



References
- Dong, Yuxiang; Wittlin, Sergio; Sriraghavan, Kamaraj; Chollet, Jacques; Charman, Susan A.; Charman, William N.; Scheurer, Christian; Urwyler, Heinrich et al. (2010). “The Structure−Activity Relationship of the Antimalarial Ozonide Arterolane (OZ277)”. Journal of Medicinal Chemistry 53 (1): 481–91. doi:10.1021/jm901473s. PMID 19924861.
- Blow to Ranbaxy drug research plans at LiveMint.com, Sep 21 2007
- Vennerstrom, Jonathan L.; Arbe-Barnes, Sarah; Brun, Reto; Charman, Susan A.; Chiu, Francis C. K.; Chollet, Jacques; Dong, Yuxiang; Dorn, Arnulf et al. (2004). “Identification of an antimalarial synthetic trioxolane drug development candidate”. Nature 430 (7002): 900–4.doi:10.1038/nature02779. PMID 15318224.
- In the Pipeline: “Ozonides As Drugs: What Will They Think Of Next?”, by Derek Lowe, November 23, 2009, at Corante.com
- Indian company starts Phase III trials of synthetic artemisinin, May 4 2009, at the WorldWide Antimalarial Resistance Network
- http://www.nature.com/nature/journal/v430/n7002/full/nature02779.html
|
5-27-2011
|
PROCESS FOR THE PREPARATION OF DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS (OZ277)
|
|
|
2-13-2009
|
STABLE DOSAGE FORMS OF SPIRO AND DISPIRO 1,2,4-TRIOXOLANE ANTIMALARIALS
|
|
|
6-15-2005
|
Spiro and dispiro 1,2,4-trioxolane antimalarials
|
|
|
11-31-2004
|
Spiro and dispiro 1,2,4-trixolane antimalarials
|
ANTIMALARIALS




http://www.rsc.org/chemistryworld/2013/03/new-antimalarial-drug-class-resistance-elq-300-quinolone

http://www.nature.com/nrd/journal/v9/n11/full/nrd3301.html
Structure of NITD609; the 1R,3Sconfiguration is fundamental for its antimalarial activity
References
- Jump up^ Dong, Yuxiang; Wittlin, Sergio; Sriraghavan, Kamaraj; Chollet, Jacques; Charman, Susan A.; Charman, William N.; Scheurer, Christian; Urwyler, Heinrich; et al. (2010). “The Structure−Activity Relationship of the Antimalarial Ozonide Arterolane (OZ277)”. Journal of Medicinal Chemistry. 53 (1): 481–91. doi:10.1021/jm901473s. PMID 19924861.
- ^ Jump up to:a b Blow to Ranbaxy drug research plans at LiveMint.com, Sep 21 2007
- Jump up^ Vennerstrom, Jonathan L.; Arbe-Barnes, Sarah; Brun, Reto; Charman, Susan A.; Chiu, Francis C. K.; Chollet, Jacques; Dong, Yuxiang; Dorn, Arnulf; et al. (2004). “Identification of an antimalarial synthetic trioxolane drug development candidate”. Nature. 430 (7002): 900–4. doi:10.1038/nature02779. PMID 15318224.
- Jump up^ In the Pipeline: “Ozonides As Drugs: What Will They Think Of Next?”, by Derek Lowe; published November 23, 2009; retrieved November 17, 2015; at Sciencemag.org
- ^ Jump up to:a b Akshat Rathi for Chemistry World. 3 May 2012 Ranbaxy launches new anti-malarial Synriam
- Jump up^ India Clinical trials registry CTRI/2009/091/000531
- Jump up^ Toure OA et al. Efficacy and safety of fixed dose combination of arterolane maleate and piperaquine phosphate dispersible tablets in paediatric patients with acute uncomplicated Plasmodium falciparum malaria: a phase II, multicentric, open-label study. Malar J. 2015 Nov 25;14(1):469. Clinical Trial Registry India: CTRI/2009/091/000531. PMID 26608469 PMC4660726
- Jump up^ Staff, Business Standard. December 16, 2014 Ranbaxy receives approval for malaria drug Synriam from 7 African countries
REFERENCES
1: Valecha N, Savargaonkar D, Srivastava B, Rao BH, Tripathi SK, Gogtay N, Kochar SK, Kumar NB, Rajadhyaksha GC, Lakhani JD, Solanki BB, Jalali RK, Arora S, Roy A, Saha N, Iyer SS, Sharma P, Anvikar AR. Comparison of the safety and efficacy of fixed-dose combination of arterolane maleate and piperaquine phosphate with chloroquine in acute, uncomplicated Plasmodium vivax malaria: a phase III, multicentric, open-label study. Malar J. 2016 Jan 27;15:42. doi: 10.1186/s12936-016-1084-1. PubMed PMID: 26818020; PubMed Central PMCID: PMC4728808.
2: Saha N, Moehrle JJ, Zutshi A, Sharma P, Kaur P, Iyer SS. Safety, tolerability and pharmacokinetic profile of single and multiple oral doses of arterolane (RBx11160) maleate in healthy subjects. J Clin Pharmacol. 2014 Apr;54(4):386-93. doi: 10.1002/jcph.232. PubMed PMID: 24242999.
3: Toure OA, Rulisa S, Anvikar AR, Rao BS, Mishra P, Jalali RK, Arora S, Roy A, Saha N, Iyer SS, Sharma P, Valecha N. Efficacy and safety of fixed dose combination of arterolane maleate and piperaquine phosphate dispersible tablets in paediatric patients with acute uncomplicated Plasmodium falciparum malaria: a phase II, multicentric, open-label study. Malar J. 2015 Nov 25;14:469. doi: 10.1186/s12936-015-0982-y. PubMed PMID: 26608469; PubMed Central PMCID: PMC4660726.
4: Toure OA, Valecha N, Tshefu AK, Thompson R, Krudsood S, Gaye O, Rao BH, Sagara I, Bose TK, Mohanty S, Rao BS, Anvikar AR, Mwapasa V, Noedl H, Arora S, Roy A, Iyer SS, Sharma P, Saha N, Jalali RK; AM–PQP Study Team.. A Phase 3, Double-Blind, Randomized Study of Arterolane Maleate-Piperaquine Phosphate vs Artemether-Lumefantrine for Falciparum Malaria in Adolescent and Adult Patients in Asia and Africa. Clin Infect Dis. 2016 Apr 15;62(8):964-71. doi: 10.1093/cid/ciw029. PubMed PMID: 26908796; PubMed Central PMCID: PMC4803108.
5: Fontaine SD, Spangler B, Gut J, Lauterwasser EM, Rosenthal PJ, Renslo AR. Drug delivery to the malaria parasite using an arterolane-like scaffold. ChemMedChem. 2015 Jan;10(1):47-51. doi: 10.1002/cmdc.201402362. PubMed PMID: 25314098; PubMed Central PMCID: PMC4420023.
6: Gupta A, Singh Y, Srinivas KS, Jain G, Sreekumar VB, Semwal VP. Development and validation of a headspace gas chromatographic method for the determination of residual solvents in arterolane (RBx11160) maleate bulk drug. J Pharm Bioallied Sci. 2010 Jan;2(1):32-7. doi: 10.4103/0975-7406.62706. PubMed PMID: 21814428; PubMed Central PMCID: PMC3146089.
7: Gautam A, Ahmed T, Sharma P, Varshney B, Kothari M, Saha N, Roy A, Moehrle JJ, Paliwal J. Pharmacokinetics and pharmacodynamics of arterolane maleate following multiple oral doses in adult patients with P. falciparum malaria. J Clin Pharmacol. 2011 Nov;51(11):1519-28. doi: 10.1177/0091270010385578. PubMed PMID: 21148048.
8: Patil C, Katare S, Baig M, Doifode S. Fixed dose combination of arterolane and piperaquine: a newer prospect in antimalarial therapy. Ann Med Health Sci Res. 2014 Jul;4(4):466-71. doi: 10.4103/2141-9248.139270. Review. PubMed PMID: 25221689; PubMed Central PMCID: PMC4160665.
9: Valecha N, Looareesuwan S, Martensson A, Abdulla SM, Krudsood S, Tangpukdee N, Mohanty S, Mishra SK, Tyagi PK, Sharma SK, Moehrle J, Gautam A, Roy A, Paliwal JK, Kothari M, Saha N, Dash AP, Björkman A. Arterolane, a new synthetic trioxolane for treatment of uncomplicated Plasmodium falciparum malaria: a phase II, multicenter, randomized, dose-finding clinical trial. Clin Infect Dis. 2010 Sep 15;51(6):684-91. doi: 10.1086/655831. PubMed PMID: 20687837.
10: Lanteri CA, Chaorattanakawee S, Lon C, Saunders DL, Rutvisuttinunt W, Yingyuen K, Bathurst I, Ding XC, Tyner SD. Ex vivo activity of endoperoxide antimalarials, including artemisone and arterolane, against multidrug-resistant Plasmodium falciparum isolates from Cambodia. Antimicrob Agents Chemother. 2014 Oct;58(10):5831-40. doi: 10.1128/AAC.02462-14. PubMed PMID: 25049252; PubMed Central PMCID: PMC4187925.
11: Valecha N, Krudsood S, Tangpukdee N, Mohanty S, Sharma SK, Tyagi PK, Anvikar A, Mohanty R, Rao BS, Jha AC, Shahi B, Singh JP, Roy A, Kaur P, Kothari M, Mehta S, Gautam A, Paliwal JK, Arora S, Saha N. Arterolane maleate plus piperaquine phosphate for treatment of uncomplicated Plasmodium falciparum malaria: a comparative, multicenter, randomized clinical trial. Clin Infect Dis. 2012 Sep;55(5):663-71. doi: 10.1093/cid/cis475. PubMed PMID: 22586253.
12: Dong Y, Wittlin S, Sriraghavan K, Chollet J, Charman SA, Charman WN, Scheurer C, Urwyler H, Santo Tomas J, Snyder C, Creek DJ, Morizzi J, Koltun M, Matile H, Wang X, Padmanilayam M, Tang Y, Dorn A, Brun R, Vennerstrom JL. The structure-activity relationship of the antimalarial ozonide arterolane (OZ277). J Med Chem. 2010 Jan 14;53(1):481-91. doi: 10.1021/jm901473s. PubMed PMID: 19924861.
13: Jourdan J, Matile H, Reift E, Biehlmaier O, Dong Y, Wang X, Mäser P, Vennerstrom JL, Wittlin S. Monoclonal Antibodies That Recognize the Alkylation Signature of Antimalarial Ozonides OZ277 (Arterolane) and OZ439 (Artefenomel). ACS Infect Dis. 2016 Jan 8;2(1):54-61. PubMed PMID: 26819968; PubMed Central PMCID: PMC4718528.
14: Fügi MA, Wittlin S, Dong Y, Vennerstrom JL. Probing the antimalarial mechanism of artemisinin and OZ277 (arterolane) with nonperoxidic isosteres and nitroxyl radicals. Antimicrob Agents Chemother. 2010 Mar;54(3):1042-6. doi: 10.1128/AAC.01305-09. PubMed PMID: 20028825; PubMed Central PMCID: PMC2825978.
15: Mossallam SF, Amer EI, El-Faham MH. Efficacy of Synriam™, a new antimalarial combination of OZ277 and piperaquine, against different developmental stages of Schistosoma mansoni. Acta Trop. 2015 Mar;143:36-46. doi: 10.1016/j.actatropica.2014.12.005. PubMed PMID: 25530543.
16: Longo M, Zanoncelli S, Brughera M, Colombo P, Wittlin S, Vennerstrom JL, Moehrle J, Craft JC. Comparative embryotoxicity of different antimalarial peroxides: in vitro study using the rat whole embryo culture model (WEC). Reprod Toxicol. 2010 Dec;30(4):583-90. doi: 10.1016/j.reprotox.2010.07.011. PubMed PMID: 20708075.
17: Abiodun OO, Brun R, Wittlin S. In vitro interaction of artemisinin derivatives or the fully synthetic peroxidic anti-malarial OZ277 with thapsigargin in Plasmodium falciparum strains. Malar J. 2013 Jan 31;12:43. doi: 10.1186/1475-2875-12-43. PubMed PMID: 23368889; PubMed Central PMCID: PMC3566918.
18: White NJ. Can new treatment developments combat resistance in malaria? Expert Opin Pharmacother. 2016 Jul;17(10):1303-7. doi: 10.1080/14656566.2016.1187134. PubMed PMID: 27191998.
19: Wang X, Dong Y, Wittlin S, Charman SA, Chiu FC, Chollet J, Katneni K, Mannila J, Morizzi J, Ryan E, Scheurer C, Steuten J, Santo Tomas J, Snyder C, Vennerstrom JL. Comparative antimalarial activities and ADME profiles of ozonides (1,2,4-trioxolanes) OZ277, OZ439, and their 1,2-dioxolane, 1,2,4-trioxane, and 1,2,4,5-tetraoxane isosteres. J Med Chem. 2013 Mar 28;56(6):2547-55. doi: 10.1021/jm400004u. PubMed PMID: 23489135.
20: Marfurt J, Chalfein F, Prayoga P, Wabiser F, Wirjanata G, Sebayang B, Piera KA, Wittlin S, Haynes RK, Möhrle JJ, Anstey NM, Kenangalem E, Price RN. Comparative ex vivo activity of novel endoperoxides in multidrug-resistant plasmodium falciparum and P. vivax. Antimicrob Agents Chemother. 2012 Oct;56(10):5258-63. doi: 10.1128/AAC.00283-12. PubMed PMID: 22850522; PubMed Central PMCID: PMC3457353.
 |
|
| Clinical data | |
|---|---|
| Routes of administration |
Oral |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C22H36N2O4 |
| Molar mass | 392.531 g/mol |
| 3D model (JSmol) | |
////////////Arteolane Maleate, OZ-277, RBx-11160, OZ 277, RBx 11160, OZ277, RBx11160, IND 2017
O=C(NCC(C)(N)C)C[C@H](CC1)CC[C@@]21OC3(OO2)[C@H]4C[C@H]5C[C@@H]3C[C@@H](C4)C5.O=C(O)/C=C\C(O)=O
