
Home » MAA
Category Archives: MAA
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
British drugmaker GlaxoSmithKline (GSK) has submitted a marketing authorisation application (MAA) to the European Medicines Agency (EMA) for its cervical cancer vaccine, Cervarix [Human papillomavirus bivalent (types 16 and 18) vaccine, recombinant].
The application in the EU is to allow administration of the vaccine in a two dosing schedule (0, 6 months) in 9-14 year old girls.
Intended to serve as an alternative dosing schedule, the two-dose schedule will help prevent premalignant genital (cervical, vulvar and vaginal) lesions and cervical cancer causally related to certain oncogenic Human Papillomavirus (HPV) types. It does not seek to replace the three-dose schedule.
read all at
Cervarix is a vaccine against certain types of cancer-causing human papillomavirus (HPV).
Cervarix is designed to prevent infection from HPV types 16 and 18, that cause about 70% of cervical cancer cases. These types also cause most HPV-induced genital and head and neck cancers. Additionally, some cross-reactive protection against virus strains 45 and 31 were shown in clinical trials. Cervarix also contains AS04, a proprietary adjuvant that has been found to boost the immune system response for a longer period of time.
Cervarix is manufactured by GlaxoSmithKline. An alternative product, from Merck & Co., is known as Gardasil.

…..
Ospemifene ….EMA accepts MAA submission of Shionogi’s ospemifene for the treatment of VVA

Ospemifene
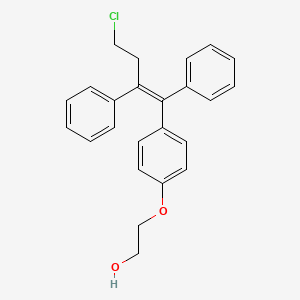
CAS Number: 128607-22-7
OSPHENA is indicated for the treatment of moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy, due to menopause
- CCRIS 9205
- Deamino-hydroxytoremifene
- Fc-1271
- FC-1271a
- Ospemifene
- Osphena
- UNII-B0P231ILBK
Molecular Formula: C24H23ClO2
Molecular Weight: 378.89 g.mol-1

Marja Sodervall, Maire Eloranta, Arja Kalapudas, Brian Kearton, Michael McKenzie, “METHODS FOR THE PREPARATION OF FISPEMIFENE FROM OSPEMIFENE.” U.S. Patent US20080214860, issued September 04, 2008.
| Country | Patent Number | Approved | Expires (estimated) |
|---|---|---|---|
| United States | 6245819 | 2013-02-26 | 2020-07-21 |
| United States | 8236861 | 2013-02-26 | 2026-08-11 |
Data
| Patent No
US |
PatentExpireyDate | patent use code |
|---|---|---|
| 6245819 | Jul 21, 2020 | U-1369 |
| 8236861 | Aug 11, 2026 | U-1369 |
| 8236861 | Aug 11, 2026 | U-1370 |
| Exclusivity Code | ExclusivityDate |
|---|---|
| NCE | Feb 26, 2018 |
UPDATE……….ON OCT 2015
| Date of issue ofmarketing authorisation valid throughout the European Union | 15/01/2015 |
|---|
NMR……http://file.selleckchem.com/downloads/nmr/S428501-Ospemifene-HNMR-Selleck.pdf
HPLC….http://file.selleckchem.com/downloads/hplc/S428501-Ospemifene-HPLC-Selleck.pdf
Ospemifene appears as a white to almost white, non-hygroscopic crystalline powder. It is insoluble in water, soluble in ethanol and propanol, very slightly soluble in isopropanol. The partition coefficient was found 4.43 and the pKa was calculated 14.26. The molecule has two geometrical isomeric forms. The active substance ospemifene is the Z-isomer. Polymorphism was not observed.
The chemical name of the active substance ospemifene is Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl) phenoxy]ethanol, corresponding to the molecular formula C24H23O2Cl and has a relative molecular mass of 378.9.
Ospemifene is a new selective non-hormonal estrogen receptor modulator (SERM) that is used for the treatment of moderate to severe dyspareunia, a symptom of vulvar and vaginal atrophy, due to menopause. FDA approved on February 26, 2013.
Article
 OSPEMIFINE
OSPEMIFINEArticle 27 March 2013
Shionogi Limited, the London-based European subsidiary of Shionogi & Co., Ltd announced today that on 26th March 2013 the European Medicines Agency (EMA) accepted its Marketing Authorisation Application (MAA) submission for ospemifene for the treatment of vulvar and vaginal atrophy (VVA) in post-menopausal women.
this is already approved by FDA
“We are pleased to announce the MAA submission for ospemifene to the EMA following the US Food and Drug Administration (FDA) approval last month. The acceptance of the MAA submission for ospemifene not only represents an important step forward in expanding the treatment options for women living in Europe with this condition, but it is also an important milestone for Shionogi as it continues to build its business in Europe” said Takashi Takenoshita, CEO of Shionogi Limited.
Osphena (ospemifene) to treat women experiencing moderate to severe dyspareunia (pain during sexual intercourse), a symptom of vulvar and vaginal atrophy due to menopause.
Dyspareunia is a condition associated with declining levels of estrogen hormones during menopause. Less estrogen can make vaginal tissues thinner, drier and more fragile, resulting in pain during sexual intercourse.
Osphena, a pill taken with food once daily, acts like estrogen on vaginal tissues to make them thicker and less fragile, resulting in a reduction in the amount of pain women experience with sexual intercourse.
“Dyspareunia is among the problems most frequently reported by postmenopausal women,” said Victoria Kusiak, M.D., deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research. “Osphena provides an additional treatment option for women seeking relief.”
Osphena’s safety and effectiveness were established in three clinical studies of 1,889 postmenopausal women with symptoms of vulvar and vaginal atrophy. Women were randomly assigned to receive Osphena or a placebo. After 12 weeks of treatment, results from the first two trials showed a statistically significant improvement of dyspareunia in Osphena-treated women compared with women receiving placebo. Results from the third study support Osphena’s long-term safety in treating dyspareunia.
Common side effects reported during clinical trials included hot flush/flashes, vaginal discharge, muscle spasms, genital discharge and excessive sweating.
Osphena is marketed by Florham Park, N.J.-based Shionogi, Inc.
- Shionogi Files a New Drug Application for Ospemifene Oral Tablets 60mg for the Treatment of Vulvar and Vaginal Atrophy – May 9, 2012
- END OF ARTICLE
OSPHENA is an estrogen agonist/antagonist. The chemical structure of ospemifene is shown in Figure 1.
Figure 1: Chemical structure
 |
The chemical designation is Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethanol, and has the empirical formula C24H23ClO2, which corresponds to a molecular weight of 378.9. Ospemifene is a white to off-white crystalline powder that is insoluble in water and soluble in ethanol.
Each OSPHENA tablet contains 60 mg of ospemifene. Inactive ingredients include colloidal silicon dioxide, hypromellose, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, povidone, pregelatinized starch, sodium starch glycolate, titanium dioxide, and triacetin.
INTRODUCTION
Ospemifene (commercial name Osphena produced by Shionogi) is an oral medication indicated for the treatment of dyspareunia – pain during sexual intercourse – encountered by some women, more often in those who are post-menopausal. Ospemifene is aselective estrogen receptor modulator (SERM)[1] acting similarly to an estrogen on thevaginalepithelium, building vaginal wall thickness which in turn reduces the pain associated with dyspareunia. Dyspareunia is most commonly caused by “vulval and vaginal atrophy.”[2]
The medication was approved by the FDA in February 2013.[3]
Ospemifene is used to treat dyspareunia. It is available as a 60 mg tablet that is taken by mouth once a day. The fact that ospemifene can be taken orally is an advantage over other products that are used to treat dyspareunia, because these are generally in a topical dosage form and have to be applied locally.[2] The oral dosage form is much easier and more convenient for patients to administer.
It is “an agonist/antagonist that makes vaginal tissue thicker and less fragile resulting in a reduction in the amount of pain women experience with sexual intercourse.”[2] This drug should be used for the shortest amount of time possible due to associated adverse effects.[2]
Approval process
Hormos Medical Ltd., which is a part of QuatRx Pharmaceuticals, filed a patent on January 19, 2005 for a solid dosage form of ospemifene.[5] In March of 2010, QuatRX Pharmaceuticals licensed ospemifene to Shionogi & Co., Ltd. for them to develop it into a product and put it on the market.[6] A New Drug Application (NDA) was submitted to the FDA on April 26, 2012.[7] Amendments to the NDA were submitted in June, July, August, October, and November 2012, and January and February of 2013.[7] It was ultimately approved by the FDA on February 26, 2013.[6]
Preclinial and clinical trials
Preclinical trials were performed in ovariectomized rats to model menopause.[8] Oral ospemifene was compared with raloxifene (another SERM), its metabolites 4-hydroxy ospemifene and 4′-hydroxy ospemifene, estradiol, and ospemifene administered as an intravaginal suppository.[8] Estradiol was used as a positive control and raloxifene was used because it is in the same drug class as ospemifene.[8]Multiple doses of oral ospemifene were tested.[8] 10 mg/kg/day of Ospemifene was found to cause a greater increase in vaginal weight and vaginal epithelial height than 10 mg/kg/day of raloxifene.[8] Vaginal weight had a 1.46x increase after a two week treatment of 10mg/kg/day of ospemifene.[8] The number of progesterone receptors was increased in the vaginal stroma and epithelium, which indicates that ospemifene has “estrogenic activity.”[8]
A binding assay was also performed to measure the affinity of ospemifene for the estrogen receptor (ERα and ERβ).[8] The study showed that ospemifene bound ERα and ERβ with similar affinity.[8] Ospemifene bound the estrogen receptors with a lower affinity than estradiol.[8] Ospemifene was shown to be an antagonist of “ERE-mediated transactivation on MCF-7 cells,” which the authors concluded indicates “anti-estrogenic activity in breast cancer cells.”[8]
Two 12 week phase 3 clinical trials were performed for ospemifene.[9] To evaluate the efficacy of the drug, 4 signs and symptoms of dyspareunia were measured. These included the “change in percent parabasal cells,” “change in percent superficial cells,” “change in vaginal pH,” and “change in most bothersome symptom (vaginal dryness and vaginal pain associated with sexual activity.”[9]Ospemifene was more effective than placebo in all four of these categories.[9] A dose-response was also seen in the trial; ospemifene 60 mg had greater efficacy than ospemifene 30 mg.[9] Safety was also evaluated in these phase 3 trials. There was a 5.2% increase in the incidence of hot flushes, 1.6% increase in urinary tract infections, and 0.5% increase in the incidence of headache with ospemifene over placebo.[9] One of the phase 3 trials was double-blinded and randomized and involved 826 women who were post-menopausal.[10]The women in the study were required to have one or more vulvovaginal atrophy (VVA) symptom that was moderate or severe in nature, no more than 5% of cells that were superficial when given a vaginal smear, and have a vaginal pH of at least 5.0.[10] Another phase 3 trial involved 605 women who were between the ages of 40 and 80, were diagnosed with VVA, and whose worst symptom was dyspareunia.[11]
 OSPEMIFENE
OSPEMIFENE
In the first half of the 2013 fiscal year, Osphena® generated 0.1 B yen in revenue, which is probably roughly equivalent to $974, 944 U.S. dollars.[12] When Osphena® was put onto the market, it was predicted to earn $495 million in 2017.[13]
OSPHENA is an estrogen agonist/antagonist. The chemical structure of ospemifene is shown in Figure 1.
Figure 1: Chemical structure
 |
The chemical designation is Z-2-[4-(4-chloro-1,2-diphenylbut-1-enyl)phenoxy]ethanol, and has the empirical formula C24H23ClO2, which corresponds to a molecular weight of 378.9. Ospemifene is a white to off-white crystalline powder that is insoluble in water and soluble in ethanol.
Each OSPHENA tablet contains 60 mg of ospemifene. Inactive ingredients include colloidal silicon dioxide, hypromellose, lactose monohydrate, magnesium stearate, mannitol, microcrystalline cellulose, polyethylene glycol, povidone, pregelatinized starch, sodium starch glycolate, titanium dioxide, and triacetin.
“SERM”s (selective estrogen receptor modulators) have both estrogen-like and antiestrogenic properties (Kauffman & Bryant, 1995). The effects may be tissue-specific as in the case of tamoxifen and toremifene which have estrogen-like effects in the bone, partial estrogen-like effect in the uterus and liver, and pure antiestrogenic effect in breast cancer.
Raloxifene and droloxifen are similar to tamoxifen and toremifene, except that their antiestrogenic properties dominate. Based on the published information, many SERMs are more likely to cause menopausal symptoms than to prevent them. They have, however, other important benefits in elderly women: they decrease total and LDL cholesterol, thus deminishing the risk of cardiovascular diseases, and they may prevent osteoporosis and inhibit breast cancer growth in postmenopausal women.
Ospemifene is the Z-isomer of the compound of formula (I)

and it is one of the main metabolites of toremifene, is known to be an estrogen agonist and antagonist (Kangas, 1990; International patent publications WO 96/07402 and WO 97/32574 ). The compound is also called (deaminohydroxy)toremifene and it is also known under the code FC-1271a. Ospemifene has relatively weak estrogenic and antiestrogenic effects in the classical hormonal tests (Kangas, 1990). It has anti-osteoporosis actions and it decreases total and LDL cholesterol levels in both experimental models and in human volunteers (International patent publications WO 96/07402 and WO 97/32574 ). It also has antitumor activity in an early stage of breast cancer development in an animal breast cancer model.
Ospemifene is also the first SERM which has been shown to have beneficial effects in climacteric syndromes in healthy women. The use of ospemifene for the treatment of certain climacteric disorders in postmenopausal women, namely vaginal dryness and sexual dysfunction, is disclosed in WO 02/07718 . The published patent application WO 03/103649 describes the use of ospemifene for inhibition of atrophy and for the treatment or prevention of atrophyrelated diseases or disorders in women, especially in women during or after the menopause.
SYNTHESIS

SYNTHESIS

SYNTHESIS
Ospemifene simple structure, its point is to control the synthesis of the product cis-trans isomerization of the double bond. Chloride 1 and benzene ( 2 ) occurs pay – acylation reaction 3 . Ester4 aluminum trichloride under the action of Fries rearrangement of 5 , 5 on the propylene oxide under alkaline conditions to obtain 6 , 6 and 3 McMurry coupling occurs directly generated Ospemifene.
……………………………………………
SYNTHESIS
https://www.google.com/patents/EP2121553B1
-
Ospemifene is the Z-isomer of the compound of formula (Ib)

-
The common starting material in the syntheses of (Ib), namely compound (II), is previously known (Toivola, 1990; EP 0095875 ). According to a method disclosed in EP 095875 , this compound was prepared by dealkylation of a corresponding ether to give (II). The method may be used to produce a mixture of isomers of compounds (Ib), but most preferably is used to prepare the pure E- and Z-isomers of this compound.
-
Particularly in case the Z-isomer of the compound (Ib) is desired, a preferable method for the synthesis of compound (II) is a McMurry reaction of commercially available starting materials, 4-hydroxybenzophenone with 3-chloropropiophenone. The McMurry reaction is a well-known reductive coupling of ketones involving two steps: (1) a single electron transfer to the carbonyl groups from an alkali metal, followed by (2) deoxygenation of the 1,2-diol with low-valent titanium to yield the alkene. This reaction produces mainly the Z-isomer of compound (II)

-
The alkylation in step a) is carried out in an organic solvent, preferably carried out in tetrahydrofuran. It is also preferable to add a base to the solvent, most preferably sodium hydride
-
Zinc (15.0 g, 0.23 mol) and tetrahydrofuran (THF) (180 ml) was added to the reaction vessel and cooled to -10 °C. Titan tetrachloride was added dropwise to the mixture (21.6 g, 0.114 mol) at about -10 °C. After the addition was completed the mixture was refluxed for two hours. Then the mixture was cooled to 40 °C and 4-hydroxybenzophenone (7.68 g, 0.039 mol) and 3-chloropropiophenone (6.48 g, 0.039 mol) dissolved in THF (75 ml) were added to the mixture. Refluxing was continued for additional 3.5 hours. The cooled reaction mixture was poured in aqueous potassium carbonate solution (21 g K2CO3 + 210 ml water) and allowed to stand overnight at the ambient temperature. The mixture was filtered and the precipitate was washed with THF. The filtrate was evaporated to dryness. The residue was dissolved in ethyl acetate and washed with water. Ethyl acetate phase was evaporated to dryness and the residue was crystallized first from methanol-water (8:2) and then from methanol-water (9:1). Yield 5.4 g.
-
Z-isomer:1H NMR (CDCl3): 2.92 (t, 2H, =CH2CH2Cl), 3.42 (t, 2H, =CH2CH2 Cl), 6.48 (d, 2H, aromatic proton ortho to hydroxy), 6.75 (d, 2H, aromatic proton meta to hydroxy), 7.1-7.4 (m, 10H, aromatic protons)
- EXAMPLE 14-(4-Chloro-1,2-diphenyl-but-1-enyl)phenol (Compound II)
EXAMPLE 2
2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethanol (Compound Ib)
-
4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenol (0.23 g, 0.689 mmol) was dissolved in tetrahydrofuran (3 ml) under nitrogen atmosphere. Sodium hydride (0.025 g, 1.03 mmol) was added to the solution and the mixture was stirred at room temperature for an hour. 2-(2-iodo-ethoxy)-tetrahydropyran (0.3 g, 1.17 mmol) was added and the mixture was refluxed for 2 hours. Additional portions of 2-(2-iodo-ethoxy)-tetrahydro-pyran (0.5 g, 2 mmol) were added to the mixture during seven hours. After cooling and adding water, THF was evaporated and the mixture was extracted three times with ethyl acetate. The organic phase was washed with 2 N aqueous sodium hydroxide and water, dried with sodium sulphate and evaporated to dryness. The residue (which is Compound (IV) where Pr is tetrahydropyranyl) was dissolved in ethanol and acidified with 2 N aqueous hydrogen chloride solution. The mixture was stirred at room temperature over night, evaporated and extracted with dichloromethane. After washing with water the organic phase was dried (Na2SO4) and evaporated. The residue was purified by flash chromatography with dichloromethane/methanol 9.5/0.5 as eluent. Yield 0.17 g, 59 %.
-
Z-isomer, 1H NMR (CDCl3): 2.92 (t, 2H, =CH2CH2Cl), 3.42 (t, 2H, =CH2CH2 Cl), 3.85-3.89 (m, 4H, OCH2CH2), 6.56 (d, 2H, aromatic proton ortho to hydroxy), 6.80 (d, 2H, aromatic proton meta to hydroxy), 7.1-7.43 (m, 10H, aromatic protons).
EXAMPLE 3
2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethanol (Compound Ib)
-
The compound was prepared by the same method as described in Example 2 using 2-(2-iodo-ethoxymethyl)-benzene as a reagent and removing the benzylic protecting group using the method described in Example (e) ofUS Patent No. 6,891,070 B2 . Briefly, the removal is carried out under a nitrogen atmosphere, in the presence of Zn powder and acetyl chloride.
- EXAMPLE 5
2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-ethanol (Compound Ib)
-
[4-(4-Chloro-1,2-diphenyl-but-1-enyl)-phenoxy]-acetic acid ethyl ester (Example 4) was dissolved in tetrahydrofuran at room temperature under nitrogen atmosphere. Lithium aluminium hydride was added to the solution in small portions until the reaction was complete. The reaction was quenched by adding saturated ammonium chloride solution to the mixture. The product was extracted into toluene, which was dried and evaporated in vacuo. The yield 100 mg, 43 %.
-
1H NMR (CDCl3): 2.92 (t, 2H, =CH2CH2Cl), 3.42 (t, 2H, =CH2CH2 Cl), 3.85-3.89 (m, 4H, OCH2CH2), 6.56 (d, 2H, aromatic proton ortho to hydroxy), 6.80 (d, 2H, aromatic proton meta to hydroxy), 7.1-7.43 (m, 10H, aromatic protons).
PATENT
https://www.google.com/patents/US6891070
e) 2-{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenoxy]ethoxy}ethanol:
Z-1-{4-[2-(2-Benzyloxy-ethoxy)ethoxy]phenyl}-4-chloro-1,2-diphenyl-but-1-ene (3.8 g, 7.4 mmol) is dissolved in ethyl acetate under nitrogen atmosphere, Zn powder (0.12 g, 1.85 mmol) and acetyl chloride (1.27 g, 16.3 mmol) are added and the mixture is stirred at 50° C. for 3 h (Bhar, 1995). The reaction mixture is cooled to room temperature, water (10 ml) is added and stirring is continued for additional 10 min. The aqueous layer is separated and the organic phase is washed with 1 M aqueous hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in methanol (16 ml) and water (4 ml). The acetate ester of the product is hydrolysed by making the mixture alkaline with sodium hydroxide (1 g) and stirring the mixture at room temperature for 1 h. Methanol is evaporated, water is added and the residue is extracted in ethyl acetate and washed with 1 M hydrogen chloride solution and with water. Ethyl acetate is evaporated and the residue is dissolved in toluene (25 ml), silica gel (0.25 g) is added and mixture is stirred for 15 min. Toluene is filtered and evaporated to dryness.
The residue is crystallised from heptane-ethyl acetate (2:1). The yield is 71%.
Z-isomer: 1H NMR (CDCl3): 2.92 (t, 2H), 3.41 (t, 2H), 3.58-3.63 (m, 2H), 3.69-3.80 (m, 4H), 3.96-4.01 (m, 2H), 6.56 (d, 2H), 6.78 (d, 2H), 7.10-7.40 (m, 10H).
E-2-{2-[4-(4-Chloro-1,2-diphenyl-but-1-enyl)phenoxy]ethoxy}ethanol is prepared analogously starting from E-1-{4-[2-(2-benzyloxy-ethoxy)ethoxy]phenyl}-4-chloro-1,2-diphenyl-but-1-ene. The product is purified by flash chromatography with toluene-methanol (10:0.5) as eluent.
E-isomer: 1H NMR (CDCl3): 2.97 (t, 2H), 3.43 (t, 2H), 3.65-3.79 (m, 4H), 3.85-3.90 (m, 2H), 4.13-4.17 (m, 2H), 6.85-7.25 (m, 2H).
Debenzylation of 1-{4-[2-(2-benzyloxy-ethoxy)ethoxy]phenyl}-4-chloro-1,2-diphenyl-but-1-ene is also carried out by hydrogenation with Pd on carbon as a catalyst in ethyl acetate-ethanol solution at room temperature.
PATENT
http://www.google.com/patents/WO2014060639A1?cl=en
EXAMPLE 5. Preparation of (Z)-2-[4-(4-chloro-l,2-diphenyl-but-l-enyl)- phenoxy]ethanol (ospemifene) by base hydrolysis of pivaloyl-groiip
; . (Z)-2-(4-(4-Chloro- l ,2-diphenylbut-l-en- l-yl)phenqxy)ethyl pivalate ( 1 g, 2.16 mmol) was dissolved in THF (8 ml) followed by addition of MeOH (1 ml) and water (1 ml). Sodium hydroxide (0.1 g, 2.5 mmol) was added in orie portion and the reaction was stirred at room temperature for 12 h. After completion of the reaction the mixture was partitioned between water (20 ml) and EtOAc (20 ml). Organic phase was washed with water (20 ml) and brine (20 ml); dried (Na2S04), filtered, and concentrated: The residue was crystallized from -PrOH yielding ospernifene (0:29 g, 35 %) as a white solid.
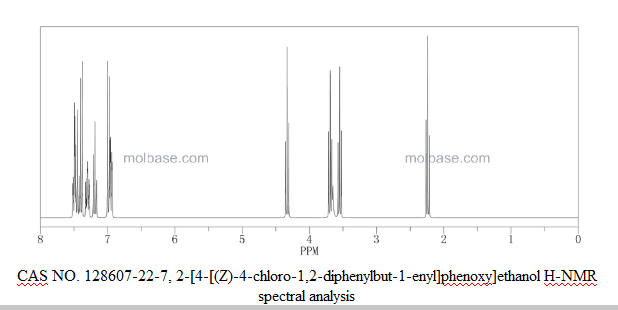
1H-NMR (400 MHz, CDC13) δ (ppm): 7.37 (2H, t, 7=8Hz, ArH), 7.29 (3Η, t, J=7.2Hz, ArH), 7.20 (2Η, t,7=7.6Hz, ArH), 7.16-7.13 (3Η, m, ArH), 6.80 (2Η, d, J=8.8Hz, ArH), 6.57 (2Η, d, 7=8.8Hz, ArH), 3.94 (2Η, t, y=4.4Hz, ArOCH2CH2OH), 3.87 (2H, m, ArOCH2CH OH), 3.42 (2H, t, J=7.2Hz, C1CH2CH2), 2.92 (2H, t, 7=7.2Hz, C1CH2CH2), 1.95 (1Η, t, 7=6.4Hz, OH).
13C- NMR (100 MHz, CDC13) δ (ppm): 157.2, 143.2, 142.1 , 141.3, 2 x 135.7, 132.2, 130.0, 129.8, 128.8, 128.7, 127.4, 127.0, 113.9, 69.3, 61.8, 43.3, 39.0.
EXAMPLE 6. Preparation of (Z)-2-[4-(4-chloro-l,2-diphenyl-but-l-enyl)- phenoxy]ethanol (ospernifene) by reductive cleavage of pivaloyl-grou
(Z)-2-(4-(4-Chloro- 1 ,2-diphenylbut- 1 -en- 1 -yl)phenoxy)ethyl pivalate (3.5 g, 7.56 mmol) was dissolved in toluene (35 ml) and stirred under nitrogen for 5 min at room temperature. Lithium aluminium hydride solution (1 M in THE) (7.56 ml, 7.56 n mbi) was added dropwise to the reaction and the mixture was stirred at room temperature for 30 min. After HPLC indicated completion, the reaction was quenched by addition of saturated NH4Cl-sblution (75 ml). Additional amount of toluene (30 ml) was added and the phases were separated. The organic phase was washed with water (50 ml), brine (50 ml), dried (Na2S04), filtered and concentrated in vacuo. The residue was crystallized from 90 % MeOH yielding ospernifene (1 ,75 g, 61 9c) as a white solid.
1H NMR PREDICT

13C NMR PREDICT


References
- Rutanen EM, Heikkinen J, Halonen K, Komi J, Lammintausta R, Ylikorkala O (2003). “Effects of ospemifene, a novel SERM, on hormones, genital tract, climacteric symptoms, and quality of life in postmenopausal women: a double-blind, randomized trial”. Menopause10 (5): 433–9.doi:10.1097/01.GME.0000063609.62485.27.PMID14501605.
- Tanzi MG (April 2013). “Ospemifene: New treatment for postmenopausal women.”. Pharmacy Today. American Pharmacists Association.
- “FDA approves Osphena for postmenopausal women experiencing pain during sex”. FDA News Release (U.S. Food and Drug Administration). 2013-02-26.
- “Ospemifene: Indications, Side Effects, Warnings”. Drugs.com.
- EP application 2286806, Lehtola V-M, Halonen K, “Solid formulations of ospemifene”, published 2011-02-23, assigned to Hormos Medical Ltd.
- “Shionogi Files a New Drug Application for Ospemifene Oral Tablets 60mg for the Treatment of Vulvar and Vaginal Atrophy”. Drugs.com.
- Kusiak V (2013-02-13). “NDA Approval” (PDF). U.S. Food and Drug Administration.
- Unkila M, Kari S, Yatkin E, Lammintausta R (November 2013). “Vaginal effects of ospemifene in the ovariectomized rat preclinical model of menopause”. J. Steroid Biochem. Mol. Biol.138: 107–15.doi:10.1016/j.jsbmb.2013.04.004. PMID23665515.
- Center for Drug Evaluation and Research (2013-02-26). “Clinical Pharmacology and Biopharmaceutics Review Application Number 203505Orig1s000” (PDF). Office of Clinical Pharmacology Review. U.S. Food and Drug Administration.
- Bachmann GA, Komi JO (2010). “Ospemifene effectively treats vulvovaginal atrophy in postmenopausal women: results from a pivotal phase 3 study”. Menopause17 (3): 480–6.doi:10.1097/gme.0b013e3181c1ac01. PMID20032798.
- Portman DJ, Bachmann GA, Simon JA (June 2013). “Ospemifene, a novel selective estrogen receptor modulator for treating dyspareunia associated with postmenopausal vulvar and vaginal atrophy”. Menopause20 (6): 623–30.doi:10.1097/gme.0b013e318279ba64. PMID23361170.
- http://www.shionogi.co.jp/en/ir/pdf/e_p131101.pdf. First Half of Fiscal 2013 Financial Results. Nov. 1, 2013.
- http://www.thepharmaletter.com/article/fda-approves-shionogi-s-osphena-for-postmenopausal-women-experiencing-pain-during-sex. ThePharmaLetter
PATENTS
|
8-8-2012
|
Method for enhancing the bioavailablity of ospemifene
|
|
|
1-21-2011
|
METHOD FOR THE PREPARATION OF THERAPEUTICALLY VALUABLE TRIPHENYLBUTENE DERIVATIVES
|
|
|
11-5-2010
|
METHODS FOR THE INHIBITION OF ATROPHY OR FOR TREATMENT OR PREVENTION OF ATROPHY-RELATED SYMPTOMS IN WOMEN
|
|
|
10-13-2010
|
METHOD FOR THE PREPARATION OF THERAPEUTICALLY VALUABLE TRIPHENYLBUTENE DERIVATIVES
|
|
|
3-18-2009
|
METHODS FOR THE PREPARATION OF FISPEMIFENE FROM OSPEMIFENE
|
|
|
5-11-2007
|
Novel oral formulations of ospemifene
|
|
|
5-11-2007
|
Formulations of fispemifene
|
|
|
1-11-2006
|
Methods for the inhibition of atrophy or for treatment or prevention of atrophy-related symptoms in women
|
|
|
12-9-2005
|
Methods for the inhibition of atrophy or for treatment or prevention of atrophy-related symptoms in women
|
|
|
8-26-2005
|
Solid formulations of ospemifene
|
|
8-26-2005
|
Method for treatment or prevention of osteoporosis in individuals with high bone turnover
|
|
|
4-6-2005
|
Triphenylalkene derivatives and their use as selective estrogen receptor modulators
|
|
|
6-11-2003
|
Triphenylalkene derivatives and their use as selective estrogen receptor modulators
|
|
|
6-13-2001
|
Method for the treatment of vaginal dryness and sexual dysfunction in women during or after the menopause
|
Florbetaben (18F), FDA and EMA accept NDA and MAA for Piramal‘s Alzheimer’s imaging agent

Florbetaben (18F)
PHOTO CREDIT-KEGG
902143-01-5 cas no
(18F-AV-1/ZK; BAY-94-9172; 18F-BAY-94-9172; ZK-6013443)

Mr Ajay Piramal, Chairman, Piramal Healthcare
Imaging with amyloid-β PET can potentially aid the early and accurate diagnosis of Alzheimer’s disease. Florbetaben (¹⁸F) is a promising ¹⁸F-labelled amyloid-β-targeted PET tracer in clinical development. We aimed to assess the sensitivity and specificity of florbetaben (¹⁸F) PET in discriminating between patients with probable Alzheimer’s disease and elderly healthy controls.
METHODS:
We did a multicentre, open-label, non-randomised phase 2 study in 18 centres in Australia, Germany, Switzerland, and the USA. Imaging with florbetaben (¹⁸F) PET was done on patients with probable Alzheimer’s disease (age 55 years or older, mini-mental state examination [MMSE] score=18-26, clinical dementia rating [CDR]=0·5-2·0) and age-matched healthy controls (MMSE ≥ 28, CDR=0). Our primary objective was to establish the diagnostic efficacy of the scans in differentiating between patients with probable disease and age-matched healthy controls on the basis of neocortical tracer uptake pattern 90-110 min post-injection. PET images were assessed visually by three readers masked to the clinical diagnosis and all other clinical findings, and quantitatively by use of pre-established brain volumes of interest to obtain standard uptake value ratios (SUVRs), taking the cerebellar cortex as the reference region. This study is registered with ClinicalTrials.gov, number NCT00750282.
FINDINGS:
81 participants with probable Alzheimer’s disease and 69 healthy controls were assessed. Independent visual assessment of the PET scans showed a sensitivity of 80% (95% CI 71-89) and a specificity of 91% (84-98) for discriminating participants with Alzheimer’s disease from healthy controls. The SUVRs in all neocortical grey-matter regions in participants with Alzheimer’s disease were significantly higher (p < 0·0001) compared with the healthy controls, with the posterior cingulate being the best discriminator. Linear discriminant analysis of regional SUVRs yielded a sensitivity of 85% and a specificity of 91%. Regional SUVRs also correlated well with scores of cognitive impairment such as the MMSE and the word-list memory and word-list recall scores (r -0·27 to -0·33, p ≤ 0·021). APOE ɛ4 was more common in participants with positive PET images compared with those with negative scans (65%vs 22% [p=0·027
MAR 21 2013
Piramal Imaging SA, a division of Piramal Enterprises, today announced that the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have accepted its applications for review of the investigational PET amyloid imaging agent [18F] florbetaben. A New Drug Application (NDA) was submitted to the U.S. Food and Drug Administration (FDA) and a Marketing Authorization Application to the EMA for [18F] florbetabenuse in the visual detection of beta-amyloid in the brains of adultswith cognitive impairment who are being evaluated for Alzheimer’s disease and other causes of cognitive decline.[18F] florbetaben binds to beta-amyloid plaques in the human brain, a hallmark characteristic in Alzheimer’s disease.
Today, Alzheimer’s disease is usually diagnosed after a person with a cognitive impairment undergoes an extensive clinical examination which typically includes family and medical history, physical and neurological examinations, laboratory tests, and imaging procedures such as computed tomography (CT) and magnetic resonance imaging (MRI) scans. Still, a definitive diagnosis of Alzheimer’s disease can only be made after death where an autopsy can reveal the presence of beta-amyloid plaques and neurofibrillary tangles in the brain. However, post-mortem studies looking for accumulations of beta-amyloid in the brain have shown that 10 to 30 percent of diagnoses based on clinical examinations are incorrect. [18F] florbetaben is being studied to determine its potential ability to detect beta-amyloid plaquesin living subjects with cognitive impairment.
FLORBETABEN F18
Diagnostic radiopharmaceutical
1. Benzenamine, 4-[(1E)-2-[4-[2-[2-[2-(fluoro-18F)ethoxy]ethoxy]ethoxy]phenyl]
ethenyl]-N-methyl-
2. 4-{(1E)-2-(4-{2-[2-(2-[18F]fluoroethoxy)ethoxy]ethoxy}phenyl)eth- 1-en-1-yl}-N-methylaniline
C21H26[18F]NO3
358.5
Bayer Healthcare
UNII-TLA7312TOI
CAS REGISTRY NUMBER 902143-01-5
https://www.ama-assn.org/resources/doc/usan/florbetaben-f18.pdf

4-[(E)-2-(4-{2-[2-(2-fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline has been labeled with [F-18]fluoride and is claimed by patent application WO2006066104 and members of the corresponding patent family.

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
The usefulness of this radiotracer for the detection of Αβ plaques have been reported in the literature (W. Zhang et al., Nuclear Medicine and Biology 32 (2005) 799-809; C. Rowe et al., Lancet Neurology 7 (2008) 1 -7).
The synthesis of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)- vinyl]-N-methylaniline has been described before:
a) W. Zhang et al., Nuclear Medicine and Biology 32 (2005) 799-809.

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
4 mg precursor 2a (2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate) in 0.2 mL
DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediate was deprotected with HCI and neutralized with
NaOH. The mixture was extracted with ethyl acetate. The solvent was dried and evaporated, the residue was dissolved in acetonitrile and purified by semi-preparative HPLC. 20% (decay corrected), 1 1 % (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N- methylaniline were obtained in 90 min.
WO2006066104
4 mg precursor 2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate in 0.2 mL DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediates was deprotected with HCI and neutralized with NaOH. The mixture was extracted with ethyl acetate. The solvent was dried and evaporated, the residue was dissolved in acetonitrile and purified by semi- preparative HPLC. 30% (decay corrected), 17% (not corrected for decay) 4- [(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N- methylaniline were obtained in 90 min. to yield N-Boc protected 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline. The unreacted perfluorinated precursor was removed using a fluorous phase cartridge.
Deprotection, final purification and formulation to obtain a product, suitable for injection into human is not disclosed. Furthermore, the usefulness (e.g. regarding unwanted F-19/F-18 exchange) of this approach at a higher radioactivity level is not demonstrated. Finally, this method would demand a two-pot setup (first reaction vessel: fluorination, followed by solid-phase- extraction, and deprotection in the second reaction vessel).
However, the focus of the present invention are compounds and methods for an improved “one-pot process” for the manufacturing of 4-[(E)-2-(4-{2-[2-(2- [F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline.
Very recently, further methods have been described:
d) US201001 13763
The mesylate precursor 2a was reacted with [F-18]fluoride species in a solvent mixture consisting of 100 μΙ_ acetonitrile and 500 μΙ_ tertiary alcohol. After fluorination for 10 min at 100-150 °C, the solvent was evaporated. After deprotection (1 N HCI, 5 min, 100-120 °C), the crude product was purified by HPLC (C18 silica, acetonitrile / 0.1 M ammonium formate).
e) H. Wang et al., Nuclear Medicine and Biology 38 (201 1 ) 121 -127
5 mg precursor 2a (2-[2-(2-{4-[(E)-2-{4-[(tert-butoxycarbonyl)(methyl)amino]- phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl methanesulfonate) in 0.5 ml_
DMSO were reacted with [F-18]fluoride/kryptofix/potassium carbonate complex. The intermediate was deprotected with HCI and neutralized with NaOH. The crude product was diluted with acetonitrile / 0.1 M ammonium dformate (6/4) and purified by semi-preparative HPLC. The product fraction was collected, diluted with water, passed through a C18 cartridge and eluted with ethanol, yielding 17% (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline within 50 min. In the paper, the conversion of an unprotected mesylate precursor (is described:
5 mg unprotected mesylate precursor (2-{2-[2-(4-{(E)-2-[4- (methylamino)phenyl]vinyl}phenoxy)ethoxy]-ethoxy}ethyl 4- methanesulfonate) in 0.5 ml_ DMSO were reacted with [F- 18]fluoride/kryptofix/potassium carbonate complex. The crude product was diluted with acetonitrile / 0.1 M ammonium formate (6/4) and purified by semi- preparative HPLC. The product fraction was collected, diluted with water, passed through a C18 cartridge and eluted with ethanol, yielding 23% (not corrected for decay) 4-[(E)-2-(4-{2-[2-(2-[F-
18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline within 30 min. Beside the purification by HPLC, a process based on solid-phase-extraction was investigated, but the purity was inferior to that with HPLC purification. So far, one-pot radiolabelings have been performed using a mesylate precursor. It is know, that for F-18 labeling of stilbenes, mesylates have advantages over corresponding tosylates by providing more clean reactions with less amount of by-products (W. Zhang et al. Journal of Medicinal Chemistry 48 (2005) 5980- 5988), whereas the purification starting from the tosylate precursor was tedious and time consuming resulting in a low yield.
In contrast to this teaching of the prior art, we found advantages of tosylate and further arylsulfonate precursors for 4-[(E)-2-(4-{2-[2-(2-[F- 18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline compared to the corresponding mesylate. Less non-radioactive by-products that eluted close to the retention time of 4-[(E)-2-(4-{2-[2-(2-[F-
18]fluoroethoxy)ethoxy]ethoxy}phenyl)vinyl]-N-methylaniline were found in the crude products if arylsulfonate precursors were used (Example 2 – Example 6) compared to the crude mixture that was obtained after conversion of the mesylate precursor (Example 1 ).
The favorable by-product profile after radiolabeling of tosylate precursor 2b (Figure 10) compared to the radiolabeling of mesylate precursor 2a (Figure 7) supported an improved cartridge based purification (Example 8, Example 9).
…………………
The term “F-18” means fluorine isotope 18F. The term”F-19″ means fluorine isotope 19F. EXAMPLES
Example 1 Radiolabeling of mesylate precursor 2a

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2a (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 1 ). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 21 % (corrected for decay).
Example 2 Synthesis and radiolabeling of tosylate precursor 2b


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
4-Dimethylaminopyridine (26.7 mg) and triethylamine (225 μΙ_) were added to a solution of 1 .0 g terf-butyl {4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate (4) in dichloromethane (12 mL) at 0 °C. A solution of p- toluenesulfonyl chloride (417 mg) in dichloromethane (13.5 mL) was added at 0 °C. The resulting mixture was stirred at room temperature over night. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography (silica, 0- 80% ethyl acetate in hexane). 850 mg 2b were obtained as colorless solid.
1 H NMR (300 MHz, CDCI3) δ ppm 1 .46 (s, 9 H), 2.43 (s, 3 H), 3.27 (s, 3 H), 3.59-3.73 (m, 6 H), 3.80- 3.86 (m, 2 H), 4.05-4.19 (m, 2 H), 6.88-7.05 (m, 4 H), 7.21 (d, J = 8.3 Hz, 2 H), 7.32 (d, J = 8.3 Hz, 2 H), 7.39-7-47 (m, 4 H), 7.80 (d, J = 8.3 Hz, 2 H). MS (ESIpos): m/z = 612 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2b (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 2). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 25% (corrected for decay).
Example 3 Synthesis and radiolabeling of 2c (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-bromobenzenesulfonate)

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline To a stirred solution of 100 mg (0,219 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104) in 2 mL THF was added a solution of 140 mg (0.548 mmol) 4-brombenzene sulfonylchlorid in 3 mL THF drop by drop. The reaction mixture was cooled to 0°C. 156.8 mg (1 .1 mmol) potassium trimethylsilanolat was added. The milky suspension was stirred at 0°C for 2 hours and at 80°C for another 2 hours. The reaction mixture was poured onto ice-cooled water. The aqueous solution was extracted with dichloromethane several times. The combined organic phases were dried with sodium sulphate and concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2c was obtained with 77 mg (0.1 14 mmol, 52.0 % yield).
1 H NMR (300 MHz, CDCI3) δ ppm 1 .39 (s, 10 H) 3.20 (s, 3 H) 3.50 – 3.57 (m, 2 H) 3.57 – 3.61 (m, 2 H) 3.61 – 3.66 (m, 2 H) 3.72 – 3.80 (m, 2 H) 4.02 – 4.10 (m, 2 H) 4.10 – 4.17 (m, 2 H) 6.79 – 6.85 (m, 2 H) 6.91 (d, J=8.10 Hz, 2 H) 7.10 – 7.17 (m, 2 H) 7.32 – 7.41 (m, 5 H) 7.57 – 7.65 (m, 2 H) 7.67 – 7.74 (m, 2 H)
MS (ESIpos): m/z = 676/678 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2c (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 3). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 43% (corrected for decay). Example 4 Synthesis and radiolabeling of 2d (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-(adamantan-1 -yl)benzenesulfonate)

4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
To a stirred solution of 151 mg (0,330 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 4.03 mg (0,033 mmol) DMAP und 36.7 mg (363 mmol) triethylamine in 4 mL dichlormethane was added a solution of 97,4 mg (0,313 mmol) 4-(adamantan-1 -yl)benzene sulfonylchloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred at 0°C for 1 hour and over night at room temperature. 7.3 mg (0,072 mmol) triethylamin und 19.5 mg (0.062 mmol) 4- (adamantan-l -yl)benzenesulfonyl chloride were added to the reaction mixture. The reaction mixture was stirred at room temperature for 3 days. It was concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2d was obtained with 104 mg (0.142 mmol, 43.4% yield).
1 H NMR (300 MHz, CDCI3) δ ppm 1 .51 (s, 9 H), 1 .62 (s, 1 H), 1 .74 – 1 .91 (m, 6 H), 1 .94 (d, J=3.20 Hz, 6 H), 2.16 (br. s., 3 H), 3.31 (s, 3 H), 3.63 – 3.69 (m, 2 H), 3.69 – 3.73 (m, 2 H), 3.76 (dd, J=5.27, 4.52 Hz, 2 H), 3.89 (d, J=4.90 Hz, 2 H), 4.13 – 4.26 (m, 4 H), 6.95 (d, J=8.85 Hz, 2 H), 7.02 (d, J=8.29 Hz, 2 H), 7.25 (d, J=8.48 Hz, 2 H), 7.40 – 7.52 (m, 4 H), 7.55 (m, J=8.67 Hz, 2 H), 7.89 (m, J=8.67 Hz, 2 H)
MS (ESIpos): m/z = 732 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2d (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 4). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 27% (corrected for decay).
Example 5 Synthesis and radiolabeling of 2e (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
4-cyanobenzenesulfonate)


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- ethoxy}phenyl)vinyl]-N-methylaniline
To a stirred solution of 151 mg (0.330 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 4.03 mg (0.033 mmol) DMAP und 36.7 mg (0.363 mmol) triethylamine in 4 mL dichlormethane was added a solution of 63.2 mg (0.313 mmol) 4-cyanobenzenesulfonyl chloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred over night and concentrated in vacuum. The crude product was purified using silica gel with ethyl acetate/hexane-gradient as mobile phase. The desired product 2e was obtained with 118 mg (0.190 mmol, 57.6 % yield).
1 H NMR (400 MHz, CDCI3) δ ppm 1 .47 (s, 9 H) 3.28 (s, 3 H) 3.58 – 3.63 (m, 2 H) 3.63 – 3.68 (m, 2 H) 3.70 – 3.75 (m, 2 H) 3.81 – 3.87 (m, 2 H) 4.1 1 – 4.18 (m, 2 H) 4.24 – 4.30 (m, 2 H) 6.91 (d, J=8.59 Hz, 2 H) 6.99 (dt, 2 H) 7.22 (d, J=8.34 Hz, 2 H) 7.39 – 7.50 (m, 4 H) 7.83 (m, J=8.59 Hz, 2 H) 7.98 – 8.1 1 (m, 2 H)
MS (ESIpos): m/z = 623 (M+H)+
Radiolabeling was performed on a remote controlled synthesis module (Tracerlab FXN). Precursor 2e (2 mg) in 0.5 mL DMSO + 0.5 mL acetonitrile was treated with dried potassium carbonate/kryptofix/[F-18]fluoride complex for 6 min at 100 °C. 1 M HCI (1 mL) + 10 mg ascorbic acid was added and the mixture was heated for 4 min at 100 °C. 2M NaOH (0.5 mL), water (6 mL) and ethanol (1 mL) were added and the crude mixture was trapped on a C18 cartridge. The crude product mixture was eluted with acetonitrile and diluted with 0.1 M ammonium formiat buffer (1 mL) + 5 mg ascorbic acid. A sample of the crude product was taken and analyzed by analytical HPLC (Figure 5). After purification by semi- preparative HPLC, the product was diluted with water + 5 mg ascorbic acid, trapped on a C18 cartridge and eluted with 1 mL ethanol.
Yield of 4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]ethoxy}phenyl)-vinyl]-N- methylaniline: 31 % (corrected for decay).
Example 6 Synthesis and radiolabeling of 2f (2-[2-(2-{4-[(E)-2-{4-[(tert- butoxycarbonyl)(methyl)amino]phenyl}vinyl]phenoxy}ethoxy)ethoxy]ethyl
2-nitrobenzenesulfonate)


4-[(E)-2-(4-{2-[2-(2-[F-18]fluoroethoxy)ethoxy]- eth oxy} phe nyl )vi ny I] -N -methyla n i I i ne
To a stirred solution of 200 mg (0.437 mmol) tert-butyl-{4-[(E)-2-(4-{2-[2-(2- hydroxyethoxy)ethoxy]ethoxy}phenyl)vinyl]phenyl}methylcarbamate
(WO2006/066104), 5.34 mg (0.044 mmol) DMAP und 47.5 mg (0.470 mmol) triethylamine in 4 mL dichlormethane was added a solution of 92 mg (0,415 mmol) 2-nitrobenzenesulfonyl chloride in 1 mL dichlormethane at 0°C. The reaction mixture was stirred over night and concentrated in vacuum. The crude product was purified with ethyl acetate/hexane-gradient as mobile phase using silica gel. The desired product 2f was obtained with 77 mg (0.1 19 mmol, 59.5 % yield). 1 H NMR (400 MHz, CDCI3) δ ppm 1 .39 (s, 9 H) 3.20 (s, 3 H) 3.55 – 3.63 (m, 4 H) 3.59 (m, 4 H) 3.69 – 3.74 (m, 2 H) 3.75 – 3.80 (m, 2 H) 4.06 (dd, J=5.68, 3.92 Hz,
2 H) 4.32 – 4.37 (m, 2 H) 6.80 – 6.84 (m, 2 H) 6.84 – 6.98 (dt, 2 H) 7.14 (d, J=8.59 Hz, 2 H) 7.35 (d, J=3.03 Hz, 2 H) 7.37 (d, J=2.78 Hz, 2 H) 7.62 – 7.74 (m,
3 H) 8.06 – 8.1 1 (m, 1 H)
