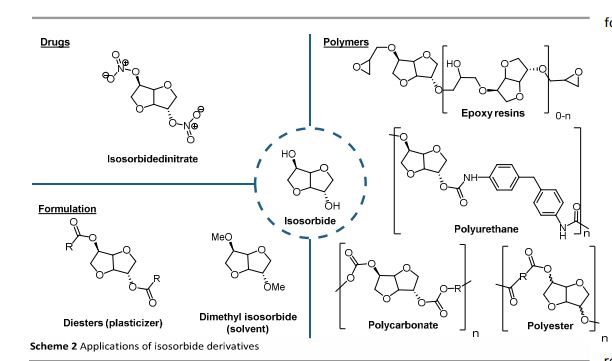
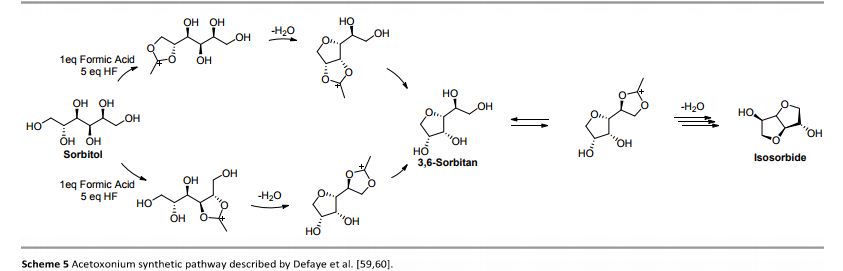
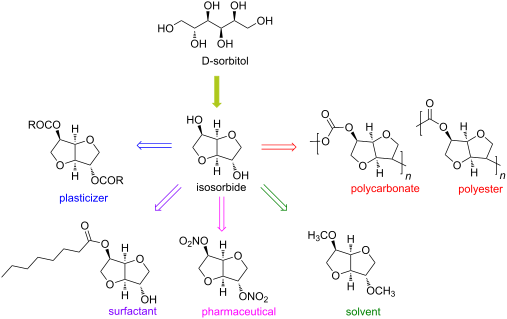
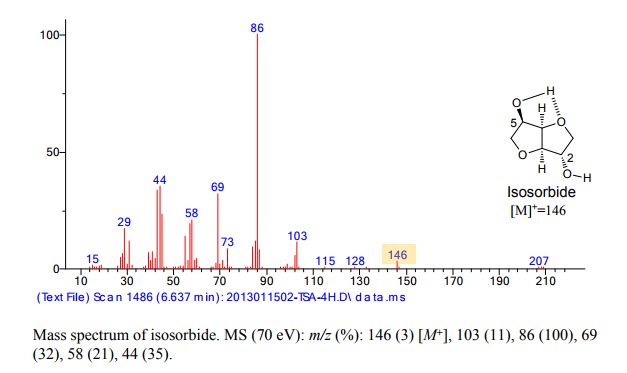
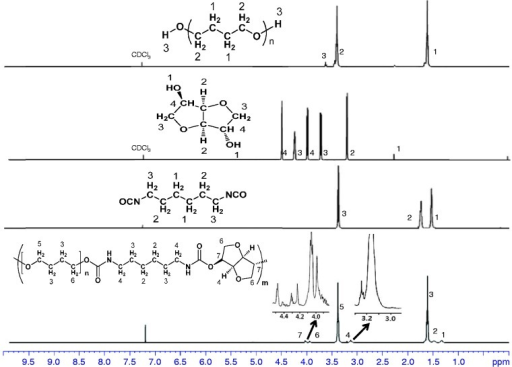
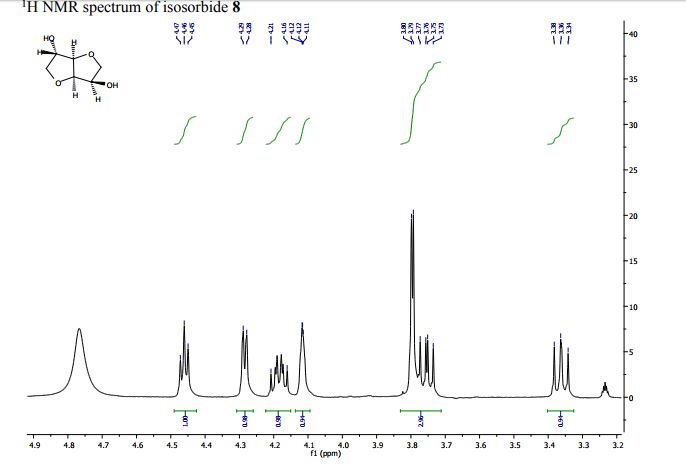
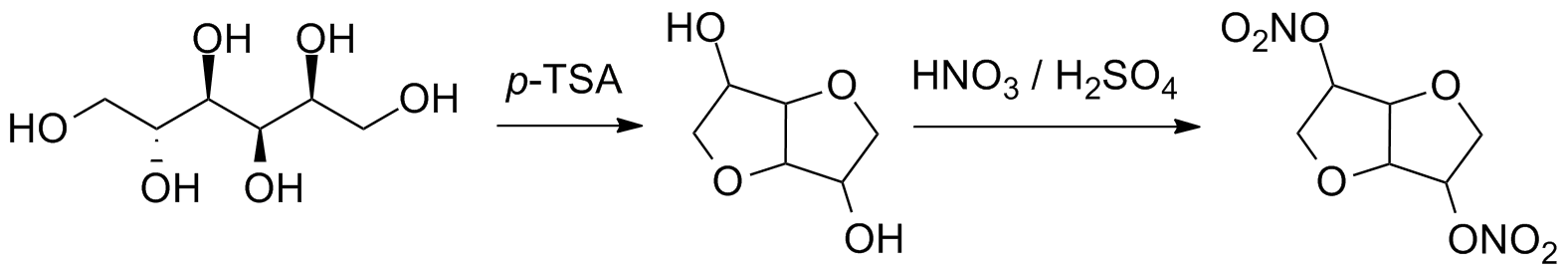
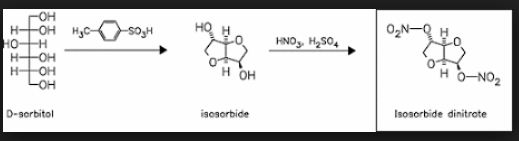
Continuous Processing
Continuous production is a flow production method used to manufacture, produce, or process materials without interruption. Continuous production is called a continuous process or a continuous flow process because the materials, either dry bulk or fluids that are being processed are continuously in motion, undergoing chemical reactions or subject to mechanical or heat treatment. Continuous processing is contrasted with batch production.
Continuous usually means operating 24 hours per day, seven days per week with infrequent maintenance shutdowns, such as semi-annual or annual. Some chemical plants can operate for more than one or two years without a shutdown. Blast furnaces can run four to ten years without stopping.[1]
Production workers in continuous production commonly work in rotating shifts.
Processes are operated continuously for practical as well as economic reasons. Most of these industries are very capital intensive and the management is therefore very concerned about lost operating time.
Shutting down and starting up many continuous processes typically results in off quality product that must be reprocessed or disposed of. Many tanks, vessels and pipes cannot be left full of materials because of unwanted chemical reactions, settling of suspended materials or crystallization or hardening of materials. Also, cycling temperatures and pressures from starting up and shutting down certain processes (line kilns, boilers, blast furnaces, pressure vessels, etc.) may cause metal fatigue or other wear from pressure or thermal cycling.

In the more complex operations there are sequential shut down and start up procedures that must be carefully followed in order to protect personnel and equipment. Typically a start up or shut down will take several hours.
Continuous processes use process control to automate and control operational variables such as flow rates, tank levels, pressures, temperatures and machine speeds.[2]
Semi-continuous processes
Many processes such as assembly lines and light manufacturing that can be easily shut down and restarted are today considered semi-continuous. These can be operated for one or two shifts if necessary.
History
The oldest continuous flow processes is the blast furnace for producing pig iron. The blast furnace is intermittently charged with ore, fuel and flux and intermittently tapped for molten pig iron and slag; however, the chemical reaction of reducing the iron and silicon and later oxidizing the silicon is continuous.
Semi-continuous processes, such as machine manufacturing of cigarettes, were called “continuous” when they appeared.
Many truly continuous processes of today were originally batch operations.
The Fourdrinier paper machine, patented in 1799, was one of the earliest of the industrial revolution era continuous manufacturing processes. It produced a continuous web of paper that was formed, pressed, dried and reeled up in a roll. Previously paper had been made in individual sheets.
Another early continuous processes was Oliver Evans‘es flour mill (ca. 1785), which was fully automated.
Early chemical production and oil refining was done in batches until process control was sufficiently developed to allow remote control and automation for continuous processing. Processes began to operate continuously during the 19th century. By the early 20th century continuous processes were common.
Shut-downs
In addition to performing maintenance, shut downs are also when process modifications are performed. These include installing new equipment in the main process flow or tying-in or making provisions to tie-in sub-processes or equipment that can be installed while the process is operating.
Shut-downs of complicated processes may take weeks or months of planning. Typically a series of meetings takes place for co-ordination and planning. These typically involve the various departments such as maintenance, power, engineering, safety and operating units.
All work is done according to a carefully sequenced schedule that incorporates the various trades involved, such as pipe-fitters, millwrights, mechanics, laborers, etc., and the necessary equipment (cranes, mobile equipment, air compressors, welding machines, scaffolding, etc.) and all supplies (spare parts, steel, pipe, wiring, nuts and bolts) and provisions for power in case power will also be off as part of the outage. Often one or more outside contractors perform some of the work, especially if new equipment is installed.
Safety
Safety meetings are typically held before and during shutdowns. Other safety measures include providing adequate ventilation to hot areas or areas where oxygen may become depleted or toxic gases may be present and checking vessels and other enclosed areas for adequate levels of oxygen and insure absence of toxic or explosive gases. Any machines that are going to be worked on must be electrically disconnected, usually through the motor starter, so that it cannot operate. It is common practice to put a padlock on the motor starter, which can only be unlocked by the person or persons who is or are endangered by performing the work. Other disconnect means include removing couplings between the motor and the equipment or by using mechanical means to keep the equipment from moving. Valves on pipes connected to vessels that workers will enter are chained and locked closed, unless some other means is taken to insure that nothing will come through the pipes.
Continuous processor (equipment)
Continuous Production can be supplemented using a Continuous Processor. Continuous Processors are designed to mix viscous products on a continuous basis by utilizing a combination of mixing and conveying action. The Paddles within the mixing chamber (barrel) are mounted on two co-rotating shafts that are responsible for mixing the material. The barrels and paddles are contoured in such a way that the paddles create a self-wiping action between themselves minimizing buildup of product except for the normal operating clearances of the moving parts. Barrels may also be heated or cooled to optimize the mixing cycle. Unlike an extruder, the Continuous Processor void volume mixing area is consistent the entire length of the barrel ensuring better mixing and little to no pressure build up. The Continuous Processor works by metering powders, granules, liquids, etc. into the mixing chamber of the machine. Several variables allow the Continuous Processor to be versatile for a wide variety of mixing operations:[3]
- Barrel Temperature
- Agitator speed
- Fed rate, accuracy of feed
- Retention time (function of feed rate and volume of product within mixing chamber)
Continuous Processors are used in the following processes:
- Compounding
- Mixing
- Kneading
- Shearing
- Crystallizing
- Encapsulating
The Continuous Processor has an unlimited material mixing capabilities but, it has proven its ability to mix:
- Plastics
- Adhesives
- Pigments
- Composites
- Candy
- Gum
- Paste
- Toners
- Peanut Butter
- Waste Products
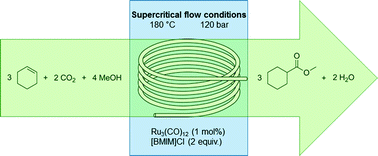
EXAMPLE…………….
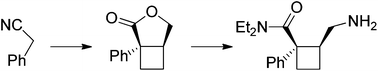
In the development of a new route to bendamustine hydrochloride, the API in Treanda, the key benzimidazole intermediate 5 was generated via catalytic heterogeneous hydrogenation of an aromatic nitro compound using a batch reactor. Because of safety concerns and a site limitation on hydrogenation at scale, a continuous flow hydrogenation for the reaction was investigated at lab scale using the commercially available H-Cube. The process was then scaled successfully, generating kilogram quantities on the H-Cube Midi. This flow process eliminated the safety concerns about the use of hydrogen gas and pyrophoric catalysts and also showed 1200-fold increase in space–time yield versus the batch processing.
Improved Continuous Flow Processing: Benzimidazole Ring Formation via Catalytic Hydrogenation of an Aromatic Nitro Compound
Org. Process Res. Dev., 2014, 18 (11), pp 1427–1433
EXAMPLE…………….
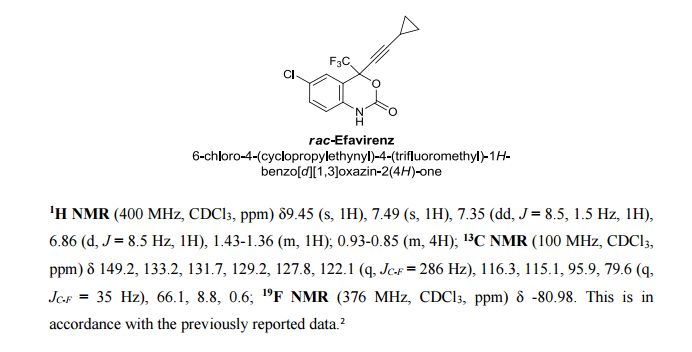
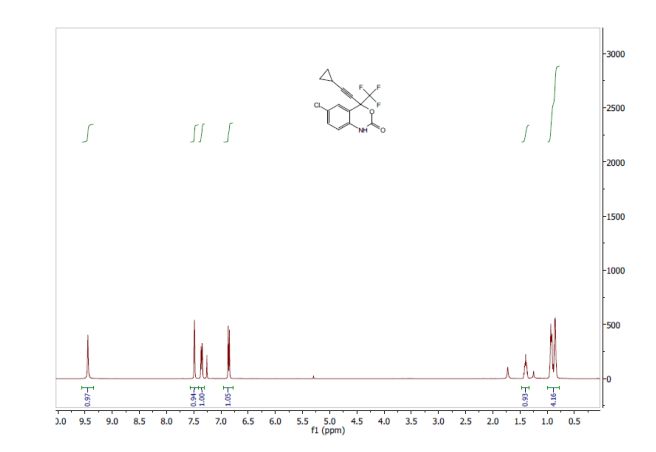
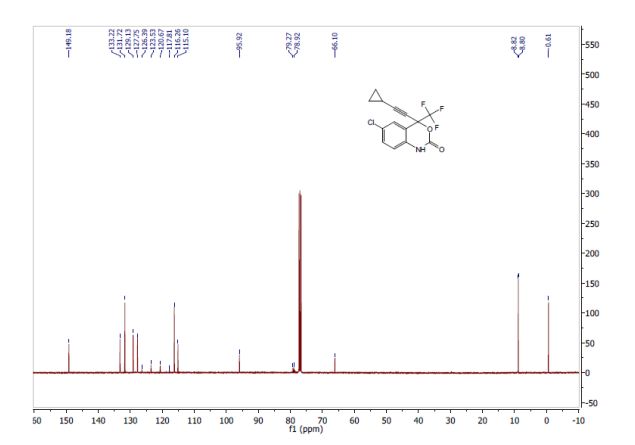
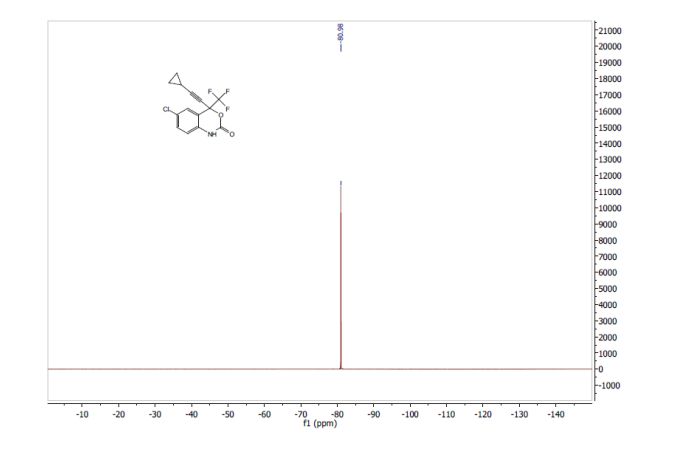
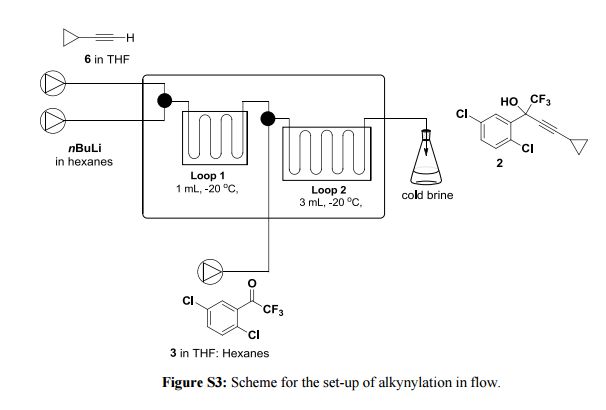
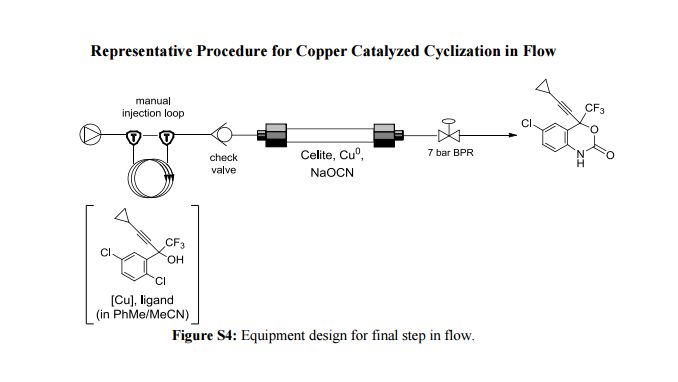
Correia et al. have published a three-step flow synthesis of rac-Effavirenz. This short synthetic route begins with cryogenic trifluoroacetylation of 1,4-dichlorobenzene. After quench and removal of morpholine using silica gel, this intermediate could either be isolated, or the product stream could be used directly in the next alkynylation step. Nucleophilic addition of lithium cyclopropylacetylide to the trifluoroacetate gave the propargyl alcohol intermediate in 90% yield in under 2 min residence time. This reaction was temperature-sensitive, and low temperatures were required to minimize decomposition. Again silica gel proved effective in the quench of the reaction. However, residual alkyne and other byproducts were difficult to remove. Thus, isolation of this intermediate was performed to minimize the impact of impurities on the final copper catalyzed cyanate installation/cyclization step to afford Effavirenz. Optimization of this step in batch mode for both copper source and ligand identified Cu(NO3)2 and CyDMEDA in a 1:4 molar ratio (20 mol % and 80 mol %, respectively) produced the product in 60% yield. Adaptation of this procedure to flow conditions resulted in poor conversion due to slow in situ reduction of the Cu(II) to Cu(I). Thus, a packed bed reactor of NaOCN and Cu(0) was used. Under these conditions, the ligand and catalyst loading could be reduced without compromising yield. Due to solubility limitations of Cu(NO3)2, Cu(OTf)2 was used with CyDMEDA in 1:2 molar ratio (5 mol % and 10 mol % loading, respectively). Under these optimized conditions, rac-Effavirenz was obtained in 62% isolated yield in reaction time of 1 h. This three-step process provides 45% overall yield of rac-Effavirenz and represents the shortest synthesis of this HIV drug reported to date



1H NMR (400 MHz, CDCl3, ppm) δ9.45 (s, 1H), 7.49 (s, 1H), 7.35 (dd, J = 8.5, 1.5 Hz, 1H), 6.86 (d, J = 8.5 Hz, 1H), 1.43-1.36 (m, 1H); 0.93-0.85 (m, 4H);
13C NMR (100 MHz, CDCl3, ppm) δ 149.2, 133.2, 131.7, 129.2, 127.8, 122.1 (q, JC-F = 286 Hz), 116.3, 115.1, 95.9, 79.6 (q, JC-F = 35 Hz), 66.1, 8.8, 0.6;
19F NMR (376 MHz, CDCl3, ppm) δ -80.98.
1 T. J. Connolly; A. W.-Y Chan; Z. Ding; M. R. Ghosh; X. Shi; J. Ren, E. Hansen; R. Farr; M. MacEwan; A. Alimardanov; et al, PCT Int. Appl. WO 2009012201 A2 20090122, 2009.
2 (a) Z. Dai, X. Long, B. Luo, A. Kulesza, J. Reichwagen, Y. Guo, (Lonza Ltd), PCT Int. Appl. WO2012097510, 2012; (b) D. D. Christ; J. A. Markwalder; J. M. Fortunak; S. S. Ko; A. E. Mutlib; R. L. Parsons; M. Patel; S. P. Seitz, PCT Int. Appl. WO 9814436 A1 19980409, 1998 (c) C. A. Correia; D. T. McQuade; P. H. Seeberger, Adv. Synth. Catal. 2013, 355, 3517−3521.
A Concise Flow Synthesis of Efavirenz†
SUPP INFO
NEXT EXAMPLE…………….
Wang et al. developed a flow process that uses metal catalyzed hydrogenation of NAB (2-nitro-2′-hydroxy-5′-methylazobenzene) to BTA (2-(2′-hydroxy-5′-methylphenyl)benzotriazole), a commonly used ultraviolet absorber. The major challenge in this process was to optimize the reduction of the diazo functionality over the nitro group and control formation of over reduction side products. The initial screen of metals adsorbed onto a γ-Al2O3 support indicated Pd to be superior to the other metals and also confirmed that catalyst preparation plays an important role in selectivity. To better understand the characteristics of the supported metal catalyst systems, the best performing were analyzed by TEM, XRD, H2-TPR, and N2 adsorption–desorption. Finally, solvents and bases were screened ultimately arriving at the optimized conditions using toluene, 2 equiv n-butylamine over 1% Pd/Al2O3, which provided 90% yield BTA in process with 98% conversion. The process can run over 200 h without a decrease in performance
( ACS Sustainable Chem. Eng. 2015, 3,1890−1896)
.


The synthesis of 2-(2′-hydroxy-5′-methylphenyl)benzotriazole from 2-nitro-2′-hydroxy-5′-methylazobenzene over Pd/γ-Al2O3 in a fixed-bed reactor was investigated. Pd/γ-Al2O3 catalysts were prepared by two methods and characterized by XRD, TEM, H2-TPR, and N2 adsorption–desorption. Employed in the above reaction, the palladium catalyst impregnated in hydrochloric acid exhibited much better catalytic performance than that impregnated in ammonia–water, which was possibly attributed to the better dispersion of palladium crystals on γ-Al2O3. This result demonstrated that the preparation process of the catalyst was very important. Furthermore, the reaction parameters were optimized. Under the optimized conditions (toluene, NAB/triethylamine molar ratio 1:2, 60 °C, 2.5 MPa hydrogen pressure, 0.23 h–1 liquid hourly space velocity), about 90% yield of 2-(2′-hydroxy-5′-methylphenyl)benzotriazole was obtained. Finally, the time on stream performance of the catalyst was evaluated, and the reaction could proceed effectively over 200 h without deactivation of the catalyst.
Construction of 2-(2′-Hydroxy-5′-methylphenyl)benzotriazole over Pd/γ-Al2O3 by a Continuous Process
ACS Sustainable Chem. Eng., 2015, 3 (8), pp 1890–1896
DOI: 10.1021/acssuschemeng.5b00507
Publication Date (Web): July 06, 2015
NEXT EXAMPLE…………….
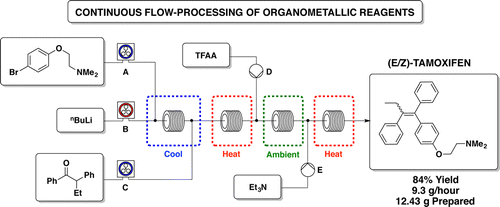
Continuous Flow-Processing of Organometallic Reagents Using an Advanced Peristaltic Pumping System and the Telescoped Flow Synthesis of (E/Z)-Tamoxifen
A new enabling technology for the pumping of organometallic reagents such as n-butyllithium, Grignard reagents, and DIBAL-H is reported, which utilises a newly developed, chemically resistant, peristaltic pumping system. Several representative examples of its use in common transformations using these reagents, including metal–halogen exchange, addition, addition–elimination, conjugate addition, and partial reduction, are reported along with examples of telescoping of the anionic reaction products. This platform allows for truly continuous pumping of these highly reactive substances (and examples are demonstrated over periods of several hours) to generate multigram quantities of products. This work culminates in an approach to the telescoped synthesis of (E/Z)-tamoxifen using continuous-flow organometallic reagent-mediated transformations.
https://www.vapourtec.com/flow-chemistry-resource-centre/publications-citing-vapourtec/continuous-flow-processing-of-organometallic-reagents-using-an-advanced-peristaltic-pumping-system-and-the-telescoped-flow-synthesis-of-ez-tamoxifen/
NEXT EXAMPLE…………….
Multi-step Continuous Flow Pyrazole Synthesis via a Metal-free Amine-redox Process

A versatile multi-step continuous flow synthesis for the preparation of substituted pyrazoles is presented.
The automated synthesis utilises a metal-free ascorbic acid mediated reduction of diazonium salts prepared from aniline starting materials followed by hydrolysis of the intermediate hydazide and cyclo-condensation with various 1,3-dicarbonyl equivalents to afford good yields of isolated functionalised pyrazole products.
The synthesis of the COX-2 selective NSAID was demonstrated using this approach.
NEXT EXAMPLE…………….
Synthesis of a Precursor to Sacubitril Using Enabling Technologies

Continuous flow methodologyhas been used to enhance several steps in the synthesis of a precursor to Sacubitril.
In particular, a key carboethoxyallylation benefited from a reducedprocessing time and improved reproducibility, the latter attributable toavoiding the use of a slurry as in the batch procedure. Moreover, in batchexothermic formation of the organozinc species resulted in the formation ofside products, whereas this could be avoided in flow because heat dissipationfrom a narrow packed column of zinc was more efficient
NEXT EXAMPLE…………….
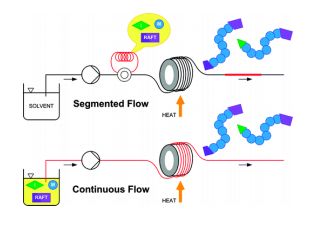
RAFT RAFT (Reversible Addition Fragmentation chain Transfer), a type of controlled radical polymerization, was invented by CSIRO in 1998 but developed in partnership with DuPont over a long term collaboration. Conventional polymerisation is fast but gives a wide distribution of polymer chain lengths. (known as a high polydispersity index ). RAFT is more versatile than other living polymerization techniques, such as atom transfer radical polymerization (ATRP) or nitroxide-mediated polymerization (NMP), it not only leads to polymers with a low polydispersity index and a predetermined molecular weight, but it permits the creation of complex architectures, such as linear block copolymers, comblike, star, brush polymers and dendrimers. Monomers capable of polymerizing by RAFT include styrenes, acrylates, acrylamides, and many vinyl monomers. CSIRO is the owner of the RAFT patents and is actively commercialising the technology. There are 12 licences in force and CSIRO is pursuing interest in a number of fields including human health, agriculture, animal health and personal care. RAFT is the dominant polymerization technique for the creation of polymer-protein or polymer-drug conjugates, permitting (for example) the combination of a polymer exhibiting high solubility with a drug molecule with poor solubility.. Though RAFT can be carried out in batch, it also lends itself to continuous flow processing, as this processing method offers an easy and reproducible scale-up route of the oxygen sensitive RAFT process. The possibility to effectively exclude oxygen using continuous flow reactors in combination with inline degassing methods offers advantages over batch processing at scales beyond the laboratory environment. Challenges associated with the high viscosity of the polymer product solution can be controlled using pressuriseable continuous flow reactor systems.
http://www.csiro.au/products/RAFT.html
Examples………..
Cyclohexaneperoxycarboxylic acid (6, has been developed as a safe, inexpensive oxidant, with demonstrated utility in a Baeyer−Villiger rearrangement.34 Solutions of cyclohexanecarboxylic acid in hexane and 50% aqueous H2O2 were continuously added to 45% H2SO4 at 50−70 °C and slightly reduced pressure. The byproduct H2O was removed azeotropically, and the residence time in the reactor was 3 h. Processing was adjusted to maintain a concentration of 6 at 17−19%, below the detonable level, and the product was kept as a stable solution in hexane. These operations enhanced the safety margin in preparing 6.
Examples………..
The conversion of a batch process to continuous (flow) operation has been investigated. The manufacture of 4,d-erythronolactone at kilogram scale was used as an example. Fully continuousprocessing was found to be impracticable with the available plant because of the difficulty in carrying out a multiphase isolation step continuously, so hybrid batch–continuous options were explored. It was found that very little additional laboratory or process safety work other than that required for the batch process was required to develop the hybrid process. A hybrid process was chosen because of the difficulty caused by the precipitation of solid byproduct during the isolation stage. While the project was a technical success, the performance benefits of the hybrid process over the batch were not seen as commercially significant for this system.
Multikilogram Synthesis of 4-d-Erythronolactone via Batch andContinuous Processing
Org. Process Res. Dev., 2012, 16 (5), pp 1003–1012
Examples………..

Continuous Biocatalytic Processes
Org. Process Res. Dev., 2009, 13 (3), pp 607–616
Scheme . Biotransformation of sodium l-glutamate to γ-aminobutyric acid (GABA) by single-step α-decarboxylation with glutamate decarboxylase
PICS…………..
References
- American Iron and Steel Institute
- Benett, Stuart (1986). A History of Control Engineering 1800-1930. Institution of Engineering and Technology. ISBN 978-0-86341-047-5.
- Ziegler, Gregory R.; Aguilar, Carlos A. (2003). “Residence Time Distribution in a Co-rotating, Twin-screw Continuous Mixer by the Step Change Method”. Journal of Food Engineering(Elsevier) 59 (2-3): 1–7.
Sources and further reading
- R H Perry, C H Chilton, C W Green (Ed), Perry’s Chemical Engineers’ Handbook (7th Ed), McGraw-Hill (1997), ISBN 978-0-07-049841-9
- Major industries typically each have one or more trade magazines that constantly feature articles about plant operations, new equipment and processes and operating and maintenance tips. Trade magazines are one of the best ways to keep informed of state of the art developments.

 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....