
Home » FDA 2019
Category Archives: FDA 2019
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Fam-trastuzumab deruxtecan-nxki

Fam-trastuzumab deruxtecan-nxki
| Formula | C6460H9972N1724O2014S44. (C52H57FN9O13)8 |
|---|---|
| CAS | 1826843-81-5 |
| Mol weight | 153701.9811 |
| Antineoplastic | |
| Disease | Breast cancer (HER2 positive) |
|---|
DS-8201a
Trastuzumab deruxtecan, sold under the brand name Enhertu, is an antibody-drug conjugate consisting of the humanized monoclonal antibody trastuzumab (Herceptin) covalently linked to the topoisomerase I inhibitor deruxtecan (a derivative of exatecan).[5][6] It is licensed for the treatment of breast cancer or gastric or gastroesophageal adenocarcinoma.[6][7] Trastuzumab binds to and blocks signaling through epidermal growth factor receptor 2 (HER2/neu) on cancers that rely on it for growth. Additionally, once bound to HER2 receptors, the antibody is internalized by the cell, carrying the bound deruxtecan along with it, where it interferes with the cell’s ability to make DNA structural changes and replicate its DNA during cell division, leading to DNA damage when the cell attempts to replicate itself, destroying the cell.[7]
It was approved for medical use in the United States in December 2019,[6] in Japan in March 2020,[8] in the European Union in January 2021,[3][4] and in Australia in October 2021.[1]

Trastuzumab deruxtecan (DS-8201a) is a HER2-targeting antibody-drug conjugate or ADC), structurally composed of a humanized anti-human HER2 (anti-hHER2) antibody, an enzymatically cleavable peptide-linker, and a proprietary topoisomerase I inhibitor payload (exatecan derivative or DX-8951 / DXd).

CLIP
Trastuzumab deruxtecan active substance, also referred to as DS-8201a, results from the conjugation of the following intermediates: – Trastuzumab monoclonal antibody (MAAL-9001); – A drug-linker (MAAA-1162a) comprised of a Topoisomerase I inhibitor derivative of exatecan (MAAA1181a) and a tetrapeptide based cleavable linker (MFAH). MAAL-9001 is covalently conjugated to approximately 8 molecules of MAAA-1162a. The linker is designed to be stable in plasma to reduce systemic exposure to the released MAAA-1181a drug. After cell internalisation, the released MAAA-1181a drug leads to apoptosis of the target tumour cells via the inhibition of topoisomerase I. The released MAAA-1181a drug is cell-membrane permeable, giving the ability to penetrate and act in surrounding cells. The effect of the ADC derives primarily from the released MAAA-1181a drug and to a lesser extent to the antibody-dependent cellular cytotoxic (ADCC) effector function of the conjugated antibody. The quality of MAAL-9001 antibody, MAAA-1162a drug-linker and the conjugated antibody is described in separate sections. The structures of DS-8201a, MAAA-1162a, MAAA-1181a, and MAAL-9001 are provided in Figure 1.

Full information for the active substance intermediate MAAA-1162a (C52H56FN9O13, MW 1034.05) was provided in the dossier. MAAA-1162a is composed of DX-8951·MsOH (drug intermediate) and MFAH (linker intermediate with maleimide functionality). The maleimide moiety reacts with the antibody (MAAL-9001) in the conjugation reaction to yield trastuzumab deruxtecan (DS-8201a). MAAA-1162a contains 3 stereogenic centres. General information was provided for solid state form, melting point, moisture sorption, UV-Vis absorption, optical rotation and solubility.
/////////////////////////////////////////////////////////////////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
Fam-trastuzumab deruxtecan-nxki is an ADC that is comprised of an anti-HER2 antibody and the potent topoisomerase inhibitor exatecan [206]. These two entities are connected via a liner consisting of a maleimide conjugation handle that includes a protease-cleavable Gly-Gly-Phe-Gly (GGFG) tetrapeptide linker. This conjugation handle could release deruxtecan after internalization of the conjugate by the cancer cells that recognize the antibody. Fam-trastuzumabderuxtecan-nxki was developed by Daiichi Sankyo and AstraZeneca, and granted approval by the FDA in December 2019 [207]. There are approximately eight payload molecules/antibodies. Fam-trastuzumab deruxtecan-nxki has been approved for the treatment of adult patients with HER2 positive breast cancer that is unresectable or metastatic [208]. The synthesis of GGFG linker is described in Scheme 38 [209,210]. First, commercial tert-butyl 2-aminoacetate 274 was treated with Fmoc-L-Phe-OH 275 in the presence of HOBt and DIC, giving the corresponding product 276. Further deprotection of Fmoc group and amide formation gave the intermediate 278, which then underwent removal of Fmoc group to give GGFG linker 279. Lastly, treatment of 279 with the activated ester 280 provided 281.
Preparation of the payload exatecan derivative is described in Scheme 39 [211]. Aluminum-catalyzed Friedel-Crafts acylation of o-fluorotoluene 282 with succinic andydride 283 gave intermediate 284 in 90% yield. Next, hydrogenation reduction ofthe carbonyl group of 284, followed by reaction with SOCl2 in MeOH, furnished 285, which then underwent nitration with H2SO4 and KNO3 to give compound 286 in 48% overall yield. Hydrolysis of 286 followed by treatment with polyphosphoric acid (PPA) gave the cyclization product 287 in only 27% yield. The transformation of 287 into 288 was realized following the four-step sequence: carbonyl reduction with NaBH4, acid-mediated elimination reaction, PtO2-catalyzed hydrogenation reduction, and acetylation with Ac2O. Regioselective benzylic oxidation of 288 in acetone with KMnO4 gave 289 in 65% yield, further functionalization with butyl nitrile and Zn-mediated acylation gave compound 290 in 66% yield over 2 steps. Treatment of 290 with aqueous HCl provided hydrolysis product 291 in 50% yield, which then coupled with ethyl trifluoroacetate to provide intermediate 292. Polycyclic compound 294 was prepared from 292 and 293 through a [4+2] cycloaddition reaction in refluxing toluene. The key intermediate 294 next underwent acidic hydrolysis and chiral resolution to provide the chiral product 295. Further condensation reaction with 296 in the presence of T3P and Et3N in DCM and TFA-promoted removal of the Boc group formed 297. The synthesis of fam-trastuzumab deruxtecan-nxki is described in Scheme 40 [212]. The linker 281 was coupled to 297 in the presence of T3P and Et3N to give the linker-payload 298. Through transformation of the disulfide bonds into free sulfhydryl groups for linkage (DTT in pH 8.0 buffer), followed by re-oxidation of the remaining disulfide bonds with cysteine, the linker-payload 298 was conjugated to the anti-HER2 mAb to give fam-trastuzumab deruxtecan-nxki (XXIX) based on the amount of protein with approximately eight linker/payloads per antibody.



[206] T.N. Iwata, K. Sugihara, T. Wada, T. Agatsuma, [Fam-] trastuzumab deruxtecan (DS-8201a)-induced antitumor immunity is facilitated by the anti-CTLA-4 antibody in a mouse model, PLoS One 14 (2019) 0222280.
[207] R. Voelker, Another targeted therapy for ERBB2-positive breast cancer, JAMA 323 (2020) 408.
[208] S. Modi, C. Saura, T. Yamashita, Y.H. Park, S.B. Kim, K. Tamura, F. Andre, H. Iwata, Y. Ito, J. Tsurutani, J. Sohn, N. Denduluri, C. Perrin, K. Aogi, E. Tokunaga, S.A. Im, K.S. Lee, S.A. Hurvitz, J. Cortes, C. Lee, S. Chen, L. Zhang, J. Shahidi, A. Yver, I. Krop, Trastuzumab deruxtecan in previously treated HER2-positive breast cancer, N. Engl. J. Med. 382 (2020) 610-621.
[209] C.L. Law, K. Klussman, A.F. Wahl, P. Senter, S. Doronina, B. Toki, Treatment of immunological disorders using anti-CD30 antibodies, 2003.WO2003043583.
[210] S. Doronina, P.D. Senter, B.E. Toki, Pentapeptide compounds and uses related thereto, 2002. WO2002088172. [211] H. Terasawa, A. Ejima, S. Ohsuki, K. Uoto, Hexa-cyclic compound, 1998. US5834476.
[212] G.M. Dubowchik, R.A. Firestone, L. Padilla, D. Willner, S.J. Hofstead, K. Mosure, J.O. Knipe, S.J. Lasch, P.A. Trail, Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: model studies of enzymatic drug release and antigen-specific in vitro anticancer activity, Bioconjugate. Chem 13 (2002) 855-869.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Target | HER2 |
| Clinical data | |
| Trade names | Enhertu |
| Other names | DS-8201a, fam-trastuzumab deruxtecan-nxki |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Trastuzumab_deruxtecanUS FDA: Enhertu |
| Pregnancy category | AU: D[1] |
| Routes of administration | Intravenous |
| ATC code | L01FD04 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1]US: ℞-only [2]EU: Rx-only [3]Rx-only[4] |
| Identifiers | |
| CAS Number | 1826843-81-5 |
| PubChem SID | 384585505 |
| DrugBank | DB14962 |
| UNII | 5384HK7574 |
| KEGG | D11529 |
| ChEMBL | ChEMBL4297844 |
| Chemical and physical data | |
| Formula | C6460H9972N1724O2014S44.(C52H57F1N9O13)8 |
Medical uses
Trastuzumab deruxtecan-nxki is indicated for the treatment of adults with unresectable (unable to be removed with surgery) or metastatic (when cancer cells spread to other parts of the body) HER2-positive breast cancer who have received two or more prior anti-HER2-based regimens in the metastatic setting and for adults with locally advanced or metastatic HER2-positive gastric or gastroesophageal junction adenocarcinoma who have received a prior trastuzumab-based regimen.[6][7]
Side effects and label warnings
The most common side effects are nausea, fatigue, vomiting, alopecia (hair loss), constipation, decreased appetite, anemia (hemoglobin in blood is below the reference range), decreased neutrophil count (white blood cells that help lead your body’s immune system response to fight infection), diarrhea, leukopenia (other white blood cells that help the immune system), cough and decreased platelet count (component of blood whose function is to react to bleeding from blood vessel injury by clumping, thereby initiating a blood clot).[6]
The prescribing information for fam-trastuzumab deruxtecan-nxki includes a boxed warning to advise health care professionals and patients about the risk of interstitial lung disease (a group of lung conditions that causes scarring of lung tissues) and embryo-fetal toxicity.[6] Interstitial lung disease and pneumonitis, including cases resulting in death, have been reported with fam-trastuzumab deruxtecan-nxki.[6]
History
The U.S. Food and Drug Administration (FDA) approved fam-trastuzumab deruxtecan-nxki in December 2019.[6][9] The application for fam-trastuzumab deruxtecan-nxki was granted accelerated approval, fast track designation, and breakthrough therapy designation.[6]
The FDA approved fam-trastuzumab deruxtecan-nxki based on the results of one clinical trial enrolling 184 female patients with HER2-positive, unresectable and/or metastatic breast cancer who had received two or more prior anti-HER2 therapies in the metastatic setting.[6] These patients were heavily pretreated in the metastatic setting, receiving between two and 17 therapies prior to receiving fam-trastuzumab deruxtecan-nxki.[6] Patients in the clinical trial received fam-trastuzumab deruxtecan-nxki every three weeks and tumor imaging was obtained every six weeks.[6] The overall response rate was 60.3%, which reflects the percentage of patients who had a certain amount of tumor shrinkage with a median duration of response of 14.8 months.[6]
The FDA granted the approval of Enhertu to Daiichi Sankyo.[6]
On 10 December 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a conditional marketing authorization for the medicinal product Enhertu, intended for the treatment of metastatic HER2-positive breast cancer.[10][11] Enhertu was reviewed under EMA’s accelerated assessment program. The applicant for this medicinal product is Daiichi Sankyo Europe GmbH. Trastuzumab deruxtecan was approved for medical use in the European Union in January 2021.[3][4]
In January 2021, the U.S. Food and Drug Administration (FDA) granted accelerated approval to fam-trastuzumab deruxtecan-nxki for the treatment of adults with locally advanced or metastatic HER2-positive gastric or gastroesophageal (GEJ) adenocarcinoma who have received a prior trastuzumab-based regimen.[7][12]
Efficacy was evaluated in a multicenter, open-label, randomized trial (DESTINY-Gastric01, NCT03329690) in participants with HER2-positive locally advanced or metastatic gastric or GEJ adenocarcinoma who had progressed on at least two prior regimens, including trastuzumab, a fluoropyrimidine- and a platinum-containing chemotherapy.[7] A total of 188 participants were randomized (2:1) to receive fam-trastuzumab deruxtecan-nxki 6.4 mg/kg intravenously every three weeks or physician’s choice of either irinotecan or paclitaxel monotherapy.[7]
References
- ^ Jump up to:a b c “Enhertu”. Therapeutic Goods Administration (TGA). 18 October 2021. Retrieved 22 October 2021.
- ^ “Enhertu- fam-trastuzumab deruxtecan-nxki injection, powder, lyophilized, for solution”. DailyMed. Retrieved 15 January 2021.
- ^ Jump up to:a b c “Enhertu EPAR”. European Medicines Agency (EMA). 9 December 2020. Retrieved 31 March 2021.
- ^ Jump up to:a b c “Enhertu approved in the EU for the treatment of HER2-positive metastatic breast cancer” (Press release). AstraZeneca. 20 January 2021. Retrieved 21 January 2021.
- ^ A HER2-Targeting Antibody–Drug Conjugate, Trastuzumab Deruxtecan (DS-8201a), Enhances Antitumor Immunity in a Mouse Model
- ^ Jump up to:a b c d e f g h i j k l m n “FDA approves new treatment option for patients with HER2-positive breast cancer who have progressed on available therapies”. U.S.Food and Drug Administration (FDA) (Press release). 20 December 2019. Archived from the original on 20 December 2019. Retrieved 20 December 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e f “FDA approves fam-trastuzumab deruxtecan-nxki for HER2-positive gastric adenocarcinomas”. U.S. Food and Drug Administration (FDA). 15 January 2021. Retrieved 15 January 2021. This article incorporates text from this source, which is in the public domain.
- ^ “Enhertu Approved in Japan for Treatment of Patients with HER2 Positive Unresectable or Metastatic Breast Cancer” (Press release). Daiichi Sankyo. 25 March 2020. Retrieved 21 January 2021 – via Business Wire.
- ^ “Drug Trials Snapshot: Enhertu”. U.S. Food and Drug Administration (FDA). 20 December 2019. Retrieved 24 January 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Enhertu: Pending EC decision”. European Medicines Agency (EMA). 10 December 2020. Retrieved 11 December 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Trastuzumab deruxtecan recommended for approval in the EU by CHMP for HER2-positive metastatic breast cancer” (Press release). AstraZeneca. 14 December 2020. Retrieved 21 January 2021.
- ^ “Enhertu approved in the US for the treatment of patients with previously treated HER2-positive advanced gastric cancer” (Press release). AstraZeneca. 18 January 2021. Retrieved 22 January 2021.
Further reading
- Iwata TN, Sugihara K, Wada T, et al. (October 2019). “[Fam-] trastuzumab deruxtecan (DS-8201a)-induced antitumor immunity is facilitated by the anti-CTLA-4 antibody in a mouse model”. PLOS ONE. 14 (10): e0222280. Bibcode:2019PLoSO..1422280I. doi:10.1371/journal.pone.0222280. PMC 6772042. PMID 31574081.
- Modi S, Saura C, Yamashita T, et al. (February 2020). “Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer”. N. Engl. J. Med. 382 (7): 610–621. doi:10.1056/NEJMoa1914510. PMC 7458671. PMID 31825192.
External links
- “Trastuzumab_deruxtecan”. Drug Information Portal. U.S. National Library of Medicine.
- Deruxtecan shows structure
- Clinical trial number NCT03329690 for “DS-8201a in Human Epidermal Growth Factor Receptor 2 (HER2)-Expressing Gastric Cancer [DESTINY-Gastric01]” at ClinicalTrials.gov
////////////Fam-trastuzumab deruxtecan-nxki ,, FDA 2019, APROVALS 2019, DS-8201a

NEW DRUG APPROVALS
ONE TIME
$10.00
Zanubrutinib, ザヌブルチニブ , занубрутиниб , زانوبروتينيب ,
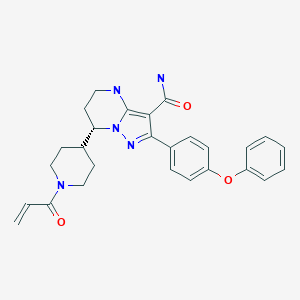
Zanubrutinib, BGB-3111
| Formula |
C27H29N5O3
|
|---|---|
| CAS |
1691249-45-2
|
| Mol weight |
471.5509
|
FDA , 2019/11/14, Brukinsa
ザヌブルチニブ ,
Antineoplastic, Bruton’s tyrosine kinase inhibitor, Mantle cell lymphoma
NEW PA
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2023062504&_gid=202316
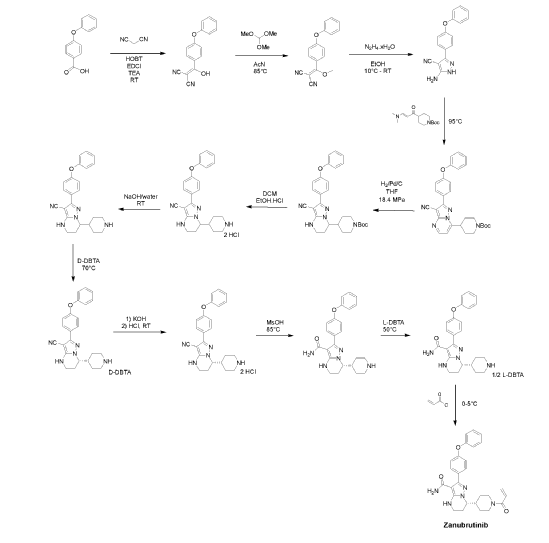
Zanubrutinib, sold under the brand name Brukinsa, is for the treatment of adult patients with mantle cell lymphoma (MCL) who have received at least one prior therapy.[3]
It was approved for medical use in the United States in November 2019.[4][3][5][6]
Zanubrutinib is classified as a Bruton’s tyrosine kinase (BTK) inhibitor. It is administered orally.
History
Efficacy was evaluated in BGB-3111-206 (NCT03206970), a phase II open-label, multicenter, single-arm trial of 86 patients with mantle cell lymphoma (MCL) who received at least one prior therapy.[5] Zanubrutinib was given orally at 160 mg twice daily until disease progression or unacceptable toxicity.[5] Efficacy was also assessed in BGB-3111-AU-003 (NCT 02343120), a phase I/II, open-label, dose-escalation, global, multicenter, single-arm trial of B‑cell malignancies, including 32 previously treated MCL patients treated with zanubrutinib administered orally at 160 mg twice daily or 320 mg once daily.[5][6]
The primary efficacy outcome measure in both trials was overall response rate (ORR), as assessed by an independent review committee.[5] In trial BGB-3111-206, FDG-PET scans were required and the ORR was 84% (95% CI: 74, 91), with a complete response rate of 59% (95% CI 48, 70) and a median response duration of 19.5 months (95% CI: 16.6, not estimable).[5] In trial BGB-3111-AU-003, FDG-PET scans were not required and the ORR was 84% (95% CI: 67, 95), with a complete response rate of 22% (95% CI: 9, 40) and a median response duration of 18.5 months (95% CI: 12.6, not estimable).[5] Trial 1 was conducted at 13 sites in China, and Trial 2 was conducted at 25 sites in the United States, United Kingdom, Australia, New Zealand, Italy, and South Korea.[6]
The U.S. Food and Drug Administration (FDA) granted zanubrutinib priority review, accelerated approval, breakthrough therapydesignation, and orphan drug designation.[3][5][7]
The FDA approved zanubrutinib in November 2019, and granted the application for Brukinsa to BeiGene USA Inc.[3][5][8]
PAPER
https://www.x-mol.com/paper/5799457
Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase Journal of Medicinal Chemistry ( IF 6.054 ) Pub Date: 2019-08-19 , DOI: 10.1021 / acs.jmedchem.9b00687
Yunhang Guo, Ye Liu, Nan Hu, Desheng Yu, Changyou Zhou, Gongyin Shi, Bo Zhang, Min Wei, Junhua Liu, Lusong Luo, Zhiyu Tang, Huipeng Song, Yin Guo, Xuesong Liu, Dan Su, Shuo Zhang, Xiaomin Song , Xing Zhou, Yuan Hong, Shuaishuai Chen, Zhenzhen Cheng, Steve Young, Qiang Wei, Haisheng Wang, Qiuwen Wang, Lei Lv, Fan Wang, Haipeng Xu, Hanzi Sun, Haimei Xing, Na Li, Wei Zhang, Zhongbo Wang, Guodong Liu, Zhijian Sun, Dongping Zhou, Wei Li, Libin Liu, Lai Wang, Zhiwei Wang
 |
Bruton’s tyrosine kinase (Btk) belongs to the Tec tyrosine kinase family (Vetrie et al., Nature 361: 226-233, 1993; Bradshaw, Cell Signal. 22: 1175-84, 2010). Btk is primarily expressed in most hematopoietic cells such as B cells, mast cells and macrophages (Smith et al., J. Immunol. 152: 557-565, 1994) and is localized in bone marrow, spleen and lymph node tissue. Btk plays important roles in B-cell receptor (BCR) and FcR signaling pathways, which involve in B-cell development, differentiation (Khan, Immunol. Res. 23: 147, 2001). Btk is activated by upstream Src-family kinases. Once activated, Btk in turn phosphorylates PLC gamma, leading to effects on B-cell function and survival (Humphries et al., J. Biol.Chem. 279: 37651, 2004).
[0003] These signaling pathways must be precisely regulated. Mutations in the gene encoding Btk cause an inherited B-cell specific immunodeficiency disease in humans, known as X-linked agammaglobulinemia (XLA) (Conley et al., Annu. Rev. Immunol. 27: 199-227, 2009). Aberrant BCR-mediated signaling may result in dysregulated B-cell activation leading to a number of autoimmune and inflammatory diseases. Preclinical studies show that Btk deficient mice are resistant to developing collagen- induced arthritis. Moreover, clinical studies of Rituxan, a CD20 antibody to deplete mature B-cells, reveal the key role of B-cells in a number of inflammatory diseases such as rheumatoid arthritis, systemic lupus erythematosus and multiple sclerosis (Gurcan et al, Int. Immunopharmacol. 9: 10-25, 2009). Therefore, Btk inhibitors can be used to treat autoimmune and/or inflammatory diseases.
[0004] In addition, aberrant activation of Btk plays an important role in pathogenesis of B-cell lymphomas indicating that inhibition of Btk is useful in the treatment of hematological malignancies (Davis et al, Nature 463: 88-92, 2010). Preliminary clinical trial results showed that the Btk inhibitor PCI-32765 was effective in treatment of several types of B-cell lymphoma (for example, 54thAmerican Society of Hematology (ASH) annual meeting abstract, Dec. 2012: 686 The Bruton’s Tyrosine Kinase (Btk) Inhibitor, Ibrutinib (PCI- 32765), Has Preferential Activity in the ABC Subtype of Relapsed/Refractory De Novo Diffuse Large B-Cell Lymphoma (DLBCL): Interim Results of a Multic enter, Open-Label, Phase I Study). Because Btk plays a central role as a mediator in multiple signal transduction pathways, inhibitors of Btk are of great interest as anti-inflammatory and/or anti-cancer agents {Mohamed et al., Immunol. Rev. 228: 58-73, 2009; Pan, Drug News perspect 21: 357-362, 200%; Rokosz et al., Expert Opin. Ther. Targets 12: 883-903, 2008; Uckun et al., Anti-cancer Agents Med. Chem. 7: 624-632, 2007; Lou et al, J. Med. Chem. 55(10): 4539-4550, 2012).
[0005] International application WO2014173289A disclosed a series of fused heterocyclic compounds as Btk inhibitors. In particular, WO2014173289A disclosed
(S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetra-hydropyrazolo[l,5-a]pyrimi dine-3-carboxamide (hereinafter C
Compound 1
[0006] Compound 1 is a potent, specific and irreversible BTK kinase inhibitor. The data generated in preclinical studies using biochemical, cell based and animal studies suggested that Compound 1 could offer significant benefit in inhibiting tumor growth in B-cell malignancies. As Compound 1 was shown to be more selective than ibrutinib for inhibition of BTK vs. EGFR, FGR, FRK, HER2, HER4, ITK, JAK3, LCK, and TEC, it is expected to give rise to less side-effects than ibrutinib in clinic. In addition, Compound 1 showed significantly less inhibition of rituximab-induced antigen-dependent cell-mediated cytotoxicity (ADCC) than ibrutinib due to weaker ITK inhibition, and therefore may provide better efficacy when combined with rituximab or other ADCC-dependent antibody in treating B-cell malignancies.
[0007] Preclinical safety evaluation has demonstrated that Compound 1 was safer than ibrutinib in terms of the overall tolerance and severe toxicities in both rat and dog single and repeat dose toxicity studies up to 28 days. Additionally, Compound 1 had better bioavailability without accumulation issues observed for ibrutinib. These unique characteristics warrant further evaluation of Compound 1 in clinical studies.
[0008] However, Compound 1 was found to be an amorphous form according to the preparation method for Compound 27 in WO 2014173289A, which was further confirmed by the X-Ray Powder Diffraction pattern of FIG. 7A. The amorphous form was shown to have a low glass transition temperature as shown in FIG. 7B, indicating some difficulties in the drug formulation with the amorphous form, such as low stability and hard to purify. Therefore, it’s necessary to develop a new form of Compound 1 which possesses characteristics such as high melting point and better stability, suitable for drug formulation.
Scheme 1: Preparation of Compound 1 and deuterium-labeled Compound 1
Deuterium-Labeled Compound 1
Step 15: Synthesis of
(S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetrahydropyrazolori,5-a1pyrimi dine-3-carboxamide (Compound 1
[0105] Under N2 atmosphere, ACN (12.0 v), water (12.5 v), BG-13 (8.0 Kg, 1.0 eq), and NaHC03 (2.5 eq.) were added to a reactor. The mixture was then cooled to -5-0 °C. To the mixture, the solution of acryloyl chloride (1.1 eq.) in MeCN (0.5 v) was added dropwise and
stirred until the reaction was completed. EA (6.0 v) was then added to the reactor, and stirred. The organic phase was collected. The aqueous layer was further extracted with EA (3.0 v). The organic phases were combined and washed with brine. The organic layer was collected and concentrated.
[0106] The residue was purified by silica gel (2 wt) column, eluted with 3% w/w methanol in DCM (21.0 v). The Compound 1 solution was collected and concentrated under vacuum. The residue was precipitated from EA/MTBE (2.0 v). The cake was collected by centrifugation as the product.
Step 15: Synthesis of (S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)
-4,5,6,7-tetrahydropyrazolori,5-a1pyrimidine-3-carboxamide (Compound 1, alternative method)
[0107] A mixture of CHsCN (10.0 v), purified water (5.0 v), NaOH (1.5 eq.) and BG-13 (1.0 eq.) was stirred to get a clear solution. EtOAc (6.0 v) was then charged to the reaction and separated. The organic phase was collected and washed with 15% brine (3.0 v) twice. The organic phase prepared above was concentrated and the solvent was swapped to CH3CN (residue volume: NMT 5.0 v). CH3CN (7.5 v) and purified water (12.5 v) were charged and cooled to 15-20°C. L-(+)-tartaric acid (0.5 eq) and NaHCCb (2.5 eq.) were charged to the reaction mixture. A solution of acryloyl chloride (1.1 eq.) in CH3CN (0.5 v) was charged drop-wise to the reaction mixture. After the reaction was completed, EtOAc (6.0 v) was charged to the reaction mixture and organic layer was collected. Aqueous phase was further extracted with EA (3.0 v). The organic layers were combined, washed with 15% brine (5.0 v) and concentrated. The solvent was swapped to DCM (volume of residue: 1.5-2.0 v) and purified by silica gel column (silica gel: 100-200 mush, 2.0 w/ w; eluent: 3%> w/ w MeOH in DCM (about 50 v). The collected solution was concentrated and swapped to EtOAc (4.0 v). MTBE (6.4 v) was charged drop-wise to residue at 50°C. The mixture was then cooled to 5°C and the cake was collected centrifugation.
Step 16: Preparation of Crystalline Form A of Compound 1
[0108] The above cake of Compound 1 was dissolved in 7.0 volumes of DCM, and then swapped to solvent EA. After recrystallization from EA/MTBE, the cakes was collected by centrifugation, and was dried under vacuum. This gave 4.44 Kg product (Yield: 70.2%).
[0109] The product was then characterized by X-ray powder diffraction (XRPD) pattern method, which was generated on a PANalytical Empyrean X-ray powder diffractometer with the XRPD parameters as follows: X-Ray wavelength (Cu, ka, Kal (A): 1.540598, Ka2(A): 1.544426; Ka2/Kal intensity ratio: 0.50); X-Ray tube setting (45 Kv, 40mA); divergence slit (automatic); scan mode (Continuous); scan range (°2TH) (3°-40); step size (°2TH) (0.0131); scan speed (°/min) (about 10). The XRPD result found the resultant product as a crystalline shown in FIG. 1.
[0110] The differential scanning calorimetry (DSC) curves shown as in FIG. 2 was generated on a TA Q2000 DSC from TA Instruments. The DSC parameters used includes: temperature (25°C-desired temperature); heating rate (10°C/min) ; method (ramp); sample pan (aluminum, crimped); purge gas (N2). DSC result showed a sharp melting point at 139.4°C (onset temperature).
[0111] The thermo-gravimetric analysis (TGA) curves shown as in FIG. 3 was generated on a TA Q5000 TGA from TA Instruments. The TGA parameters used includes: temperature
(RT-desired temperature); heating rate (10°C/min); method (ramp); sample pan (platinum, open); purge gas (N2). TGA result showed is anhydrous with no weight loss even up to 110 °C.
[0112] The proton nuclear magnetic resonance ^H-NMR) shown as in FIG. 4 was collected on a Bruker 400M NMR Spectrometer in DMSO-de. ¾-NMR (DMSO-de) δ 7.50 (d, J= 8.6 Hz, 2H), 7.46-7.38 (m, 2H), 7.17 (t, J = 7.6 Hz, 1H), 7.08 (d, J= 7.6 Hz, 2H), 7.05 (d, J= 8.8 Hz, 2H), 6.85-6.72 (m, 1H), 6.67 (s, 1H), 6.07 (dd, J= 16.8, 2.2 Hz, 1H), 5.64 (dd, J= 10.4 Hz, 2.2 Hz, 1H), 4.55-4.38 (m, 1H), 4.17-3.94 (m, 2H), 3.33-3.22 (m, 2H), 3.08-2.88 (m, 1H), 2.67-2.51 (m, 1H), 2.36-2.15 (m, 1H), 2.12-1.82 (m, 2H), 1.79-1.65 (m, 1H), 1.63-1.49 (m, 1H), 1.38-1.08 (m, 2H).
[0113] The carbon nuclear magnetic resonance (13C-NMR) shown as in FIG. 5 was collected on a Bruker 400M NMR Spectrometer in DMSO-de. 13C-NMR spectra for Crystalline Form A of Compound 1.
Step 15: Synthesis of (S)-7-(1-acrvlovlpiperidin-4-vl)-2-(4-phenoxvphenyl)-4.5.6.7-tetrahvdropvrazolo[1.5-a1pvrimidine-3-carboxamide (Compound 1)
[0119] Under N2 atmosphere, ACN (12.0 v), water (12.5 v), BG-13 (8.0 Kg, 1.0 eq), and NaHCO3 (2.5 eq.) were added to a reactor. The mixture was then cooled to -5-0 °C. To the mixture, the solution of acryloyl chloride (1.1 eq.) in MeCN (0.5 v) was added dropwise and stirred until the reaction was completed. EA (6.0 v) was then added to the reactor, and stirred. The organic phase was collected. The aqueous layer was further extracted with EA (3.0 v). The organic phases were combined and washed with brine. The organic layer was collected and concentrated.
[0120] The residue was purified by silica gel (2 wt) column, eluted with 3% w/w methanol in DCM (21.0 v). The Compound 1 solution was collected and concentrated under vacuum. The residue was precipitated from EA/MTBE (2.0 v). The cake was collected by centrifugation as the product.
Step 15: Synthesis of (S)-7-(l-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl) -4.5.6.7-tetrahvdropvrazolori.5-a1pvrimidine-3-carboxamide (Compound 1. alternative method)
[0121] A mixture of CH3CN (10.0 v), purified water (5.0 v), NaOH (1.5 eq.) and BG-13 (1.0 eq.) was stirred to get a clear solution. EtOAc (6.0 v) was then charged to the reaction and separated. The organic phase was collected and washed with 15% brine (3.0 v) twice. The organic phase prepared above was concentrated and the solvent was swapped to CH3CN (residue volume: NMT 5.0 v). CH3CN (7.5 v) and purified water (12.5 v) were charged and cooled to 15-20°C. L-(+)-tartaric acid (0.5 eq) and NaHCO3 (2.5 eq.) were charged to the reaction mixture. A solution of acryloyl chloride (1.1 eq.) in CH3CN (0.5 v) was charged drop-wise to the reaction mixture. After the reaction was completed, EtOAc (6.0 v) was charged to the reaction mixture and organic layer was collected. Aqueous phase was further extracted with EA (3.0 v). The organic layers were combined, washed with 15% brine (5.0 v) and concentrated. The solvent was swapped to DCM (volume of residue: 1.5-2.0 v) and purified by silica gel column (silica gel: 100-200 mush, 2.0 w/ w; eluent: 3% w/ w MeOH in DCM (about 50 v). The collected solution was concentrated and swapped to EtOAc (4.0 v). MTBE (6.4 v) was charged drop-wise to residue at 50°C. The mixture was then cooled to 5°C and the cake was collected centrifugation.
References
- ^ “Zanubrutinib (Brukinsa) Use During Pregnancy”. Drugs.com. 3 January 2020. Retrieved 26 January 2020.
- ^ “Zanubrutinib”. DrugBank. Retrieved 15 November 2019.
- ^ Jump up to:a b c d “FDA approves therapy to treat patients with relapsed and refractory mantle cell lymphoma supported by clinical trial results showing high response rate of tumor shrinkage”. U.S. Food and Drug Administration (FDA) (Press release). 14 November 2019. Retrieved 15 November 2019.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ “Brukinsa (zanubrutinib) FDA Approval History”. Drugs.com. 14 November 2019. Archived from the original on 15 November 2019. Retrieved 15 November 2019.
- ^ Jump up to:a b c d e f g h i “FDA grants accelerated approval to zanubrutinib for mantle cell lymphoma”. U.S. Food and Drug Administration (FDA)(Press release). 15 November 2019. Archived from the original on 28 November 2019. Retrieved 27 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c “Drug Trials Snapshots Brukinsa”. U.S. Food and Drug Administration (FDA). 14 November 2019. Retrieved 26 January 2020. This article incorporates text from this source, which is in the public domain.
- ^ “Zanubrutinib Orphan Drug Designation and Approval”. U.S. Food and Drug Administration (FDA). 28 November 2019. Archived from the original on 28 November 2019. Retrieved 27 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ “Drug Approval Package: Brukinsa”. U.S. Food and Drug Administration (FDA). 27 November 2019. Archived from the original on 28 November 2019. Retrieved 27 November 2019. This article incorporates text from this source, which is in the public domain.
External links
- “Zanubrutinib”. Drug Information Portal. U.S. National Library of Medicine.
 |
|
| Clinical data | |
|---|---|
| Trade names | Brukinsa |
| Other names | BGB-3111 |
| AHFS/Drugs.com | Monograph |
| License data |
|
| Pregnancy category |
|
| Routes of administration |
By mouth |
| Drug class | Bruton’s tyrosine kinase(BTK) inhibitor |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| PubChem SID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C27H29N5O3 |
| Molar mass | 471.5509 g·mol−1 |
| 3D model (JSmol) | |
/////////////////Zanubrutinib, FDA 2019, ザヌブルチニブ , занубрутиниб , زانوبروتينيب , BGB-3111
Brilliant blue G , ブリリアントブルーG ,
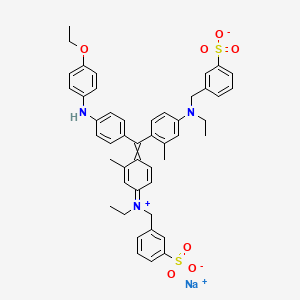
Brilliant blue G
FDA 2019, 12/20/2019, TISSUEBLUE, New Drug Application (NDA): 209569
Company: DUTCH OPHTHALMIC, PRIORITY; Orphan
OPQ recommends APPROVAL of NDA 209569 for commercialization of TissueBlue (Brilliant Blue G Ophthalmic Solution), 0.025%
Neuroprotectant
sodium;3-[[4-[[4-(4-ethoxyanilino)phenyl]-[4-[ethyl-[(3-sulfonatophenyl)methyl]azaniumylidene]-2-methylcyclohexa-2,5-dien-1-ylidene]methyl]-N-ethyl-3-methylanilino]methyl]benzenesulfonate
| Formula |
C47H48N3O7S2. Na
|
|---|---|
| CAS |
6104-58-1
|
| Mol weight |
854.0197
|
ブリリアントブルーG, C.I. Acid Blue 90
UNII-M1ZRX790SI
M1ZRX790SI
6104-58-1
Brilliant Blue G
Derma Cyanine G
SYN


////////////Brilliant blue G , ブリリアントブルーG , C.I. Acid Blue 90, FDA 2019, PRIORITY, Orphan
CCN(CC1=CC(=CC=C1)S(=O)(=O)[O-])C2=CC(=C(C=C2)C(=C3C=CC(=[N+](CC)CC4=CC(=CC=C4)S(=O)(=O)[O-])C=C3C)C5=CC=C(C=C5)NC6=CC=C(C=C6)OCC)C.[Na+]
- Benzenemethanaminium, N-[4-[[4-[(4-ethoxyphenyl)amino]phenyl][4-[ethyl[(3-sulfophenyl)methyl]amino]-2-methylphenyl]methylene]-3-methyl-2,5-cyclohexadien-1-ylidene]-N-ethyl-3-sulfo-, hydroxide, inner salt, monosodium salt
- Benzenemethanaminium, N-[4-[[4-[(4-ethoxyphenyl)amino]phenyl][4-[ethyl[(3-sulfophenyl)methyl]amino]-2-methylphenyl]methylene]-3-methyl-2,5-cyclohexadien-1-ylidene]-N-ethyl-3-sulfo-, inner salt, monosodium salt (9CI)
- Brilliant Indocyanine G (6CI)
- C.I. Acid Blue 90 (7CI)
- C.I. Acid Blue 90, monosodium salt (8CI)
- Acid Blue 90
- Acid Blue G 4061
- Acid Blue PG
- Acid Bright Blue G
- Acid Brilliant Blue G
- Acid Brilliant Cyanine G
- Acidine Sky Blue G
- Amacid Brilliant Cyanine G
- Anadurm Cyanine A-G
- BBG
- Benzyl Cyanine G
- Biosafe Coomassie Stain
- Boomassie blue silver
- Brilliant Acid Blue G
- Brilliant Acid Blue GI
- Brilliant Acid Blue J
- Brilliant Acid Cyanine G
- Brilliant Blue G
- Brilliant Blue G 250
- Brilliant Blue J
- Brilliant Indocyanine GA-CF
- Bucacid Brilliant Indocyanine G
- C.I. 42655
- CBB-G 250
- Colocid Brilliant Blue EG
- Coomassie Blue G
- Coomassie Blue G 250
- Coomassie Brilliant Blue G
- Coomassie Brilliant Blue G 250
- Coomassie G 250
- Cyanine G
- Daiwa Acid Blue 300
- Derma Cyanine G
- Derma Cyanine GN 360
- Dycosweak Acid Brilliant Blue G
- Eriosin Brilliant Cyanine G
- Fenazo Blue XXFG
- Impero Azure G
- Kayanol Cyanine G
- Lerui Acid Brilliant Blue G
- Milling Brilliant Blue 2J
- NSC 328382
- Optanol Cyanine G
- Orient Water Blue 105
- Orient Water Blue 105S
- Polar Blue G
- Polar Blue G 01
- Polycor Blue G
- Sandolan Cyanine N-G
- Sellaset Blue B
- Serva Blue G
- Serva Blue G 250
- Silk Fast Cyanine G
- Simacid Blue G 350
- Sumitomo Brilliant Indocyanine G
- Supranol Cyanin G
- Supranol Cyanine G
- TissueBlue
- Triacid Fast Cyanine G
- Water Blue 105
- Water Blue 105S
- Water Blue 150
- Xylene Brilliant Cyanine G
Enfortumab vedotin


Enfortumab vedotin
| Formula |
C6642H10284N1742O2063S46
|
|---|---|
| Cas |
1346452-25-2
|
| Mol weight |
149022.148
|
AGS-22M6E, enfortumab vedotin-ejfv
Fda approved 2019/12/18, Padcev
Antineoplastic, Nectin-4 antibody, Tubulin polymerization inhibitor, Urothelial cancer
エンホルツマブベドチン (遺伝子組換え);
protein Based Therapies, Monoclonal antibody, mAb,
UNII DLE8519RWM
Other Names
- AGS 22CE
- AGS 22M6E
- AGS 22ME
- Enfortumab vedotin
- Enfortumab vedotin-ejfv
- Immunoglobulin G1 (human monoclonal AGS-22M6 γ1-chain), disulfide with human monoclonal AGS-22M6 κ-chain, dimer, tetrakis(thioether) with N-[[[4-[[N-[6-(3-mercapto-2,5-dioxo-1-pyrrolidinyl)-1-oxohexyl]-L-valyl-N5-(aminocarbonyl)-L-ornithyl]amino]phenyl]methoxy]carbonyl]-N-methyl-L-valyl-N-[(1S,2R)-4-[(2S)-2-[(1R,2R)-3-[[(1R,2S)-2-hydroxy-1-methyl-2-phenylethyl]amino]-1-methoxy-2-methyl-3-oxopropyl]-1-pyrrolidinyl]-2-methoxy-1-[(1S)-1-methylpropyl]-4-oxobutyl]-N-methyl-L-valinamide
- Padcev
Protein Sequence
Sequence Length: 1322, 447, 447, 214, 214multichain; modified (modifications unspecified)
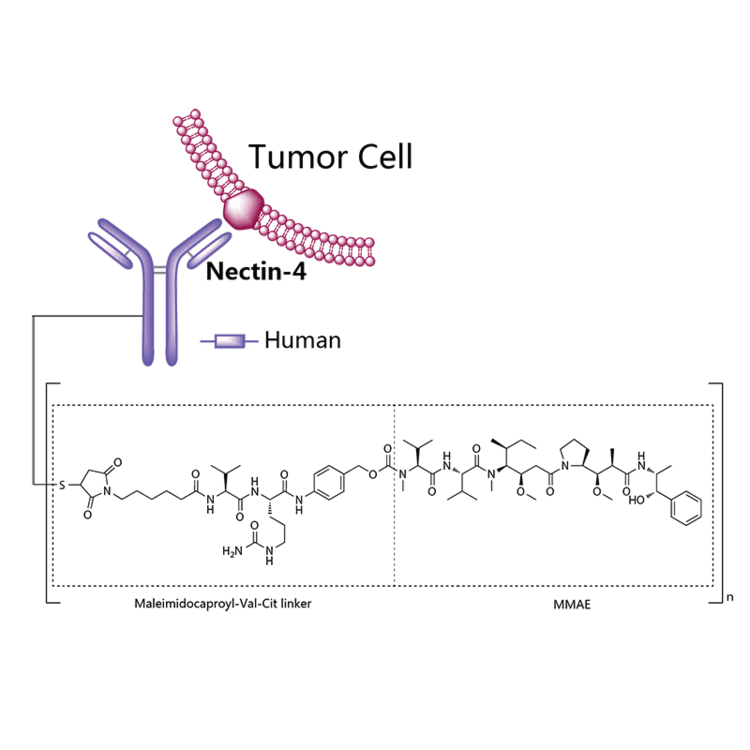
Enfortumab vedotin is an antibody-drug conjugate used in the treatment of patients with advanced, treatment-resistant urothelial cancers.3 It is comprised of a fully human monoclonal antibody targeted against Nectin-4 and a microtubule-disrupting chemotherapeutic agent, monomethyl auristatin E (MMAE), joined by a protease-cleavable link.3 It is similar to brentuximab vedotin, another antibody conjugated with MMAE that targets CD-30 instead of Nectin-4.
The clinical development of enfortumab vedotin was the result of a collaboration between Astellas Pharma and Seattle Genetics2 and it was first approved for use in the United States in December 2019 under the brand name PadcevTM.3
The most common side effects for patients taking enfortumab vedotin were fatigue, peripheral neuropathy (nerve damage resulting in tingling or numbness), decreased appetite, rash, alopecia (hair loss), nausea, altered taste, diarrhea, dry eye, pruritis (itching) and dry skin. [4]Enfortumab vedotin[1] (AGS-22M6E) is an antibody-drug conjugate[2] designed for the treatment of cancer expressing Nectin-4.[3]Enfortumab refers to the monoclonal antibody part, and vedotin refers to the payload drug (MMAE) and the linker.
The fully humanized antibody was created by scientists at Agensys (part of Astellas) using Xenomice from Amgen; the linker technology holding the antibody and the toxin together was provided by and licensed from Seattle Genetics.[5]
Results of a phase I clinical trial were reported in 2016.[2]
In December 2019, enfortumab vedotin-ejfv was approved in the United States for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death ligand 1 (PD-L1) inhibitor and a platinum-containing chemotherapy.[4]
Enfortumab vedotin was approved based on the results of a clinical trial that enrolled 125 patients with locally advanced or metastatic urothelial cancer who received prior treatment with a PD-1 or PD-L1 inhibitor and platinum-based chemotherapy.[4] The overall response rate, reflecting the percentage of patients who had a certain amount of tumor shrinkage, was 44%, with 12% having a complete response and 32% having a partial response.[4] The median duration of response was 7.6 months.[4]
The application for enfortumab vedotin-ejfv was granted accelerated approval, priority review designation, and breakthrough therapydesignation.[4] The U.S. Food and Drug Administration (FDA) granted the approval of Padcev to Astellas Pharma US Inc.[4]
Indication
Enfortumab vedotin is indicated for the treatment of adult patients with locally advanced or metastatic urothelial cancer who have previously received a programmed death receptor-1 (PD-1) or programmed death-ligand 1 (PD-L1) inhibitor, and a platinum-containing chemotherapy in the neoadjuvant/adjuvant, locally advanced, or metastatic setting.3
Associated Conditions
Pharmacodynamics
Enfortumab vedotin is an anti-cancer agent that destroys tumor cells by inhibiting their ability to replicate.3 Patients with moderate to severe hepatic impairment should not use enfortumab vedotin – though it has not been studied in this population, other MMAE-containing antibody-drug conjugates have demonstrated increased rates of adverse effects in patients with moderate-severe hepatic impairment.3 Enfortumab vedotin may also cause significant hyperglycemia leading, in some cases, to diabetic ketoacidosis, and should not be administered to patients with a blood glucose level >250 mg/dl.3
Mechanism of action
Enfortumab vedotin is an antibody-drug conjugate comprised of multiple components.3 It contains a fully human monoclonal antibody directed against Nectin-4, an extracellular adhesion protein which is highly expressed in urothelial cancers,1 attached to a chemotherapeutic microtubule-disrupting agent, monomethyl auristatin E (MMAE). These two components are joined via a protease-cleavable linker. Enfortumab vedotin binds to cells expressing Nectin-4 and the resulting enfortumab-Nectin-4 complex is internalized into the cell. Once inside the cell, MMAE is released from enfortumab vedotin via proteolytic cleavage and goes on to disrupt the microtubule network within the cell, arresting the cell cycle and ultimately inducing apoptosis.3
PATENT
WO 2016176089
WO 2016138034
WO 2017186928
WO 2017180587
WO 2017200492
US 20170056504
PAPER
Cancer Research (2016), 76(10), 3003-3013.
General References
- Hanna KS: Clinical Overview of Enfortumab Vedotin in the Management of Locally Advanced or Metastatic Urothelial Carcinoma. Drugs. 2019 Dec 10. pii: 10.1007/s40265-019-01241-7. doi: 10.1007/s40265-019-01241-7. [PubMed:31823332]
- McGregor BA, Sonpavde G: Enfortumab Vedotin, a fully human monoclonal antibody against Nectin 4 conjugated to monomethyl auristatin E for metastatic urothelial Carcinoma. Expert Opin Investig Drugs. 2019 Oct;28(10):821-826. doi: 10.1080/13543784.2019.1667332. Epub 2019 Sep 17. [PubMed:31526130]
- FDA Approved Drug Products: Padcev (enfortumab vedotin-ejfv) for IV injection [Link]
References
- ^ World Health Organization (2013). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 109”(PDF). WHO Drug Information. 27 (2).
- ^ Jump up to:a b Seattle Genetics and Agensys, an Affiliate of Astellas, Highlight Promising Enfortumab Vedotin (ASG-22ME) and ASG-15ME Phase 1 Data in Metastatic Urothelial Cancer at 2016 ESMO Congress. Oct 2016
- ^ Statement On A Nonproprietary Name Adopted By The USAN Council – Enfortumab Vedotin, American Medical Association.
- ^ Jump up to:a b c d e f g “FDA approves new type of therapy to treat advanced urothelial cancer”. U.S. Food and Drug Administration (FDA) (Press release). 18 December 2019. Archived from the original on 19 December 2019. Retrieved 18 December 2019. This article incorporates text from this source, which is in the public domain.
- ^ Challita-Eid PM, Satpayev D, Yang P, et al. (May 2016). “Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models”. Cancer Research. 76 (10): 3003–13. doi:10.1158/0008-5472.can-15-1313. PMID 27013195.
External links
- “Enfortumab vedotin”. Drug Information Portal. U.S. National Library of Medicine.
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Human |
| Target | Nectin-4 |
| Clinical data | |
| Trade names | Padcev |
| Other names | AGS-22M6E, AGS-22CE, enfortumab vedotin-ejfv |
| License data | |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChemSID | |
| DrugBank | |
| ChemSpider |
|
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C6642H10284N1742O2063S46 |
| Molar mass | 149.0 kg/mol g·mol−1 |
PADCEV™
(enfortumab vedotin-ejfv) for Injection, for Intravenous Use
DESCRIPTION
Enfortumab vedotin-ejfv is a Nectin-4 directed antibody-drug conjugate (ADC) comprised of a fully human anti-Nectin-4 IgG1 kappa monoclonal antibody (AGS-22C3) conjugated to the small molecule microtubule disrupting agent, monomethyl auristatin E (MMAE) via a protease-cleavable maleimidocaproyl valine-citrulline (vc) linker (SGD-1006). Conjugation takes place on cysteine residues that comprise the interchain disulfide bonds of the antibody to yield a product with a drug-to-antibody ratio of approximately 3.8:1. The molecular weight is approximately 152 kDa.
Figure 1: Structural Formula
|
Approximately 4 molecules of MMAE are attached to each antibody molecule. Enfortumab vedotin-ejfv is produced by chemical conjugation of the antibody and small molecule components. The antibody is produced by mammalian (Chinese hamster ovary) cells and the small molecule components are produced by chemical synthesis.
PADCEV (enfortumab vedotin-ejfv) for injection is provided as a sterile, preservative-free, white to off-white lyophilized powder in single-dose vials for intravenous use. PADCEV is supplied as a 20 mg per vial and a 30 mg per vial and requires reconstitution with Sterile Water for Injection, USP, (2.3 mL and 3.3 mL, respectively) resulting in a clear to slightly opalescent, colorless to slightly yellow solution with a final concentration of 10 mg/mL [see DOSAGE AND ADMINISTRATION]. After reconstitution, each vial allows the withdrawal of 2 mL (20 mg) and 3 mL (30 mg). Each mL of reconstituted solution contains 10 mg of enfortumab vedotin-ejfv, histidine (1.4 mg), histidine hydrochloride monohydrate (2.31 mg), polysorbate 20 (0.2 mg) and trehalose dihydrate (55 mg) with a pH of 6.0.
///////////////Enfortumab vedotin, AGS-22M6E, エンホルツマブベドチン (遺伝子組換え) , protein Based Therapies, Monoclonal antibody, mAb, FDA 2019
[*]SC1CC(=O)N(CCCCCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(=O)N)C(=O)Nc2ccc(COC(=O)N(C)[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@@H](CC(=O)N3CCC[C@H]3[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c4ccccc4)OC)cc2)C1=O
Cefiderocol, セフィデロコル , цефидерокол , سيفيديروكول , 头孢德罗 ,

Cefiderocol
セフィデロコル;
| Formula |
C30H34ClN7O10S2
|
|---|---|
| CAS |
1225208-94-5
|
| Mol weight |
752.2149
|
Antibacterial, Cell wall biosynthesis inhibitor, enicillin binding protein, Siderophore cephalosporin
Fetroja (TN)
FDA, Cefiderocol, APPROVED, 2019/11/14
(6R,7R)-7-{[(2Z)-2-(2-Amino-1,3-thiazol-4-yl)-2-{[(2-carboxy-2-propanyl)oxy]imino}acetyl]amino}-3-[(1-{2-[(2-chloro-3,4-dihydroxybenzoyl)amino]ethyl}-1-pyrrolidiniumyl)methyl]-8-oxo-5-thia-1-azabicycl o[4.2.0]oct-2-ene-2-carboxylate
S-649266, GSK 2696266D
Cefiderocol, sold under the brand name Fetroja, is an antibiotic used to treat complicated urinary tract infections when no other options are available.[2] It is indicated for the treatment of multi-drug-resistant Gram-negative bacteria including Pseudomonas aeruginosa.[3][4][5] It is given by injection into a vein.[6]
It is in the cephalosporin family of medications.[2][7] Cefiderocol was approved for medical use in the United States on November 14, 2019.[2][8]
Cefiderocol, also known as S-649266, is a potent siderophore cephalosporin antibiotic with a catechol moiety on the 3-position side chain. S-649266 shows potent in vitro activity against the non-fermenting Gram-negative bacteria Acinetobacter baumannii, Pseudomonas aeruginosa and Stenotrophomonas maltophilia, including MDR strains such as carbapenem-resistant A. baumannii and metallo-β-lactamase-producing P. aeruginosa. S-649266 showed potent in vitro activities against A. baumannii producing carbapenemases such as OXA-type β-lactamases, and P. aeruginosa producing metallo-β-lactamases such as IMP type and VIM type. FDA approved this drug in 11/14/2019 To treat patients with complicated urinary tract infections who have limited or no alternative treatment options
Medical uses
Cefiderocol is used to treat adults with complicated urinary tract infections, including kidney infections caused by susceptible Gram-negative microorganisms, who have limited or no alternative treatment options.[2][7]
Mechanism of action
Its mechanism of entry into bacterial cells is by binding to iron, which is actively transported into the bacterial cells along with the cefiderocol.[6][9][10][11][12] It is in a medication class known as siderophores,[6][7] and was the first siderophore antibiotic to be approved by the U.S. Food and Drug Administration (FDA).[13] It bypasses the bacterial porin channels by using the bacteria’s own iron-transport system for being transported in.[14]
History
In 2019, cefiderocol was approved in the United States as an antibacterial drug for treatment of adults 18 years of age or older with complicated urinary tract infections (cUTI), including kidney infections caused by susceptible Gram-negative microorganisms, who have limited or no alternative treatment options.[2][8]
The safety and effectiveness of cefiderocol was demonstrated in a study of 448 patients with cUTIs.[2] Of the patients who were administered cefiderocol, 72.6% had resolution of symptoms and eradication of the bacteria approximately seven days after completing treatment, compared with 54.6% in patients who received an alternative antibiotic.[2] The clinical response rates were similar between the two treatment groups.[2]
Labeling for cefiderocol includes a warning regarding the higher all-cause mortality rate observed in cefiderocol-treated patients compared to those treated with other antibiotics in a trial in critically ill patients with multidrug-resistant Gram-negative bacterial infections.[2] The cause of the increase in mortality has not been established.[2] Some of the deaths were a result of worsening or complications of infection, or underlying co-morbidities.[2] The higher all-cause mortality rate was observed in patients treated for hospital-acquired/ventilator-associated pneumonia (i.e.nosocomial pneumonia), bloodstream infections, or sepsis.[2] The safety and efficacy of cefiderocol has not been established for the treatment of these types of infections.[2]
Cefiderocol received a Qualified Infectious Disease Product designation from the U.S. Food and Drug Administration (FDA) and was granted priority review.[2] The FDA granted approval of Fetroja, on November 14, 2019, to Shionogi & Co., Ltd.[2]
PATENT
WO 2010050468
WO 2016035845
WO 2016035847
PATENT
WO 2017216765,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017216765&tab=PCTDESCRIPTION
Bacterial infections continue to remain one of the major causes contributing towards human diseases. One of the key challenges in treatment of bacterial infections is the ability of bacteria to develop resistance to one or more antibacterial agents over time. Examples of such bacteria that have developed resistance to typical antibacterial agents include: Penicillin-resistant Streptococcus pneumoniae, Vancomycin-resistant Enterococci, and Methicillin-resistant Staphylococcus aureus. The problem of emerging drug-resistance in bacteria is often tackled by switching to newer antibacterial agents, which can be more expensive and sometimes more toxic. Additionally, this may not be a permanent solution as the bacteria often develop resistance to the newer antibacterial agents as well in due course. In general, bacteria are particularly efficient in developing resistance, because of their ability to multiply very rapidly and pass on the resistance genes as they replicate. Therefore, there is a need for development of newer ways to treat infections that are becoming resistant to known therapies and methods.
Surprisingly, it has been found that the compositions comprising a compound of Formula (I) or a pharmaceutically acceptable salt thereof and at least one beta-lactamase inhibitor or a pharmaceutically acceptable salt thereof, exhibit synergistic antibacterial activity, even against resistant bacterial strains.
Formula (I)
Example 1
Synthesis of Compound of formula (I)
Step-1: Preparation of intermediate (1):
To the clear solution of (Z)-2[(2-tert-butoxycarbonyl amino-thiazol-4-yl)-carboxy-methyleneaminooxy]2-methyl-propionic acid tert-butyl ester (30 gm, 69.93 mmol) in N,N-dimethyl acetamide (300 ml) was charged triethylamine (17.68 ml, 125.87 mmol) under stirring. The reaction mixture was cooled to -15°C. Methane sulfonyl chloride (12.01 gm, 104. 89 mmol) was charged to this cooled reaction mixture via addition funnel while maintaining temperature at about -15°C. The reaction mixture was stirred for 30 minutes at -15°C after the addition. To the reaction mixture was charged (6 ?,75)-4-methoxybenzyl-7-amino-3-chloromethyl-8-oxo-5-thia-l-aza-bicyclo[4.2.0]oct-2-ene-2-carboxylate hydrochloride salt (28.25 gm, 69.93 mmol) along with N-methyl morpholine (15.5 ml, 139.86 mmol). The reaction mixture was stirred further for 1 hour at -15°C and the reaction progress was monitored using TLC. After completion of reaction, ethyl acetate (1.2 L) was charged followed by IN aqueous hydrochloric acid (1.2 L) under stirring and cooling was removed to warm up reaction mixture to room temperature. Layers were separated and organic layer was washed with saturated aqueous sodium bicarbonate solution (500 ml) followed by brine (500 ml). Organic layer was dried over sodium sulphate and was evaporated under vacuum to provide a crude mass. It was purified using silica gel column chromatography (60-120 mesh, 30% ethyl acetate in hexane) to provide 38 gm of intermediate (1).
Analysis:
1H NMR (CDCls) δ ppm: 8.29 (br s, 1H), 8.17 (d, 1H), 7.35 (d, 2H), 7.31 (s, 1H), 6.91 (d, 2H), 6.21 (dd, 1H), 5.23 (dd, 2H), 5.05 (d, 1H), 4.55 (d, 1H), 4.46 (d, 1H), 3.82 (s, 3H), 3.65 (d, 1H), 3.48 (d, 1H), 1.62 (s, 3H), 1.59 (s, 3H), 1.53 (s, 9H), 1.45 (s, 9H).
Step-2: Preparation of intermediate (2):
The solution of intermediate 1 (45 gm, 57.76 mmol) in dichloro methane (450 ml) was cooled to about -40°C and m-chloroperbenzoic acid (18 gm, 57.76 mmol) was added in three lots at -40°C under stirring. The mixture was stirred for 30 minutes and allowed to warm at -20°C. As TLC showed complete conversion, 5% aqueous sodium thiosulfate solution (1.2 L) was added at -15°C under stirring. The mixture was allowed to warm at room temperature and was charged with ethyl acetate (1.5 L) and stirred for 30 minutes and layers were separated. Organic layer was washed with saturated aqueous sodium bicarbonate solution (1 L) followed by brine (500 ml).
Organic layer was dried over sodium sulphate and evaporated under vacuum to provide 46 gm of intermediate (2).
Analysis:
1H NMR (CDC13) δ ppm: 8.48 (br s, 1H), 7.89 (d, 1H), 7.34 (d, 2H), 7.29 (s, 1H), 6.92 (d, 2H), 6.21 (dd, 1H), 5.27 (dd, 2H), 5.04 (br d, 1H), 4.58 (d, 1H), 4.23 (d, 1H), 3.83 (s, 3H), 3.82 (d, 1H), 3.43 (d, 1H), 1.60 (s, 3H), 1.58 (s, 3H), 1.53 (9H)1.42 (s, 9H).
Step-3: Preparation of intermediate (3):
Part-1: To the clear solution of intermediate 2 (35 gm, 44.02 mmol) in tetrahydrofuran (350 ml) was charged potassium iodide (14.61 gm, 88.05 mmol) under stirring at 25°C. The suspension was stirred for 5 hours at the same temperature and the reaction was monitored using mass spectroscopy. After completion of the reaction ethyl acetate (600 ml) was added to the reaction mixture followed by 5% aqueous sodium thiosulphate (600 ml). The reaction mixture was stirred for 15 minutes and layers were separated. Organic layer was washed with demineralised water (500 ml) followed by brine (500 ml). Organic layer was dried over sodium sulphate and evaporated to dryness under vacuum to provide 38 gm of corresponding iodo-methyl intermediate.
Part-2: To the iodo-methyl intermediate obtained (37.24 gm, 41.98 mmol) in N,N-dimethylformamide (35 ml) was added 2-chloro-3,4-di-(4-methoxybenzyloxy)-N-(pyrrolidin-l-ylethyl)-benzamide (22 gm, 42.98 mmol). The thick mass was stirred at 25°C for 15 hours and the reaction was monitored using mass spectroscopy. Potassium iodide (48.78 gm, 293.8 mmol) was charged to the reaction mass under stirring at 25 °C. The reaction mixture was cooled to -40°C and acetyl chloride (12 ml, 167.9 mmol) was added. After completion of the reaction ethyl acetate (1.2 L) followed by demineralised water (1.2 L) was added to the reaction mass at 0°C. Layers were separated and organic layer was washed with demineralised water (500 ml) followed by brine (500 ml). Organic layer was dried over sodium sulphate and was evaporated to dryness under vacuum to obtain quaternary intermediate (3) as iodide salt.
Step-4: Preparation compound of Formula (I):
Compound (3) (30 gm, 21.5 mmol) was dissolved in dichloro methane (300 ml) and anisole (30 gm, mmol) was added under stirring at 25°C. The mixture was cooled to -40° C and 2M aluminium chloride solution in nitromethane (150 ml) was added over 45 minutes at -40°C. As addition was completed reaction mixture was stirred for 1 hour at 0°C. To the reaction mixture 2M aqueous hydrochloric acid (750 ml) and acetonitrile (750 ml) were added and the stirring was
continued for 15 minutes. Di-isopropyl ether (1.5 L) was charged to the reaction mixture and the reaction mass was stirred for 15 minutes at 25°C, and the layers were separated. Aqueous layer was washed with additional di-isopropyl ether (500 ml). HP-21 resin (150 gm) was charged to the aqueous layer. The aqueous layer along with resin was loaded on a resin HP-21 column. The column was eluted with demineralised water till pH of eluent became neutral. Then the column was eluted with 10% acetonitrile in water mixture. Finally the column was eluted with 20% acetonitrile in water mixture. Evaporation of required fractions below 40°C under vacuum provided 5.5 gm of crude compound (I). The crude compound (I) was purified by dissolving in acetonitrile (200 ml) and demineralised water (200 ml) mixture followed by addition of HP-21 resin (200 gm).The slurry thus obtained was loaded on HP-21 resin column. The column was eluted first with demineralised water (3 L) followed by 10% acetonitrile in water mixture (2 L) then followed by 20% acetonitrile in water mixture till complete pure compound from the column is eluted. Pure fractions were collected and lyophilized under vacuum to provide titled compound (I) in pure form.
Analysis:
1H NMR (DMSO d6) δ ppm: 12.5 (br s, 2H), 9.42 (br s, 1H), 8.41 (br t, 1H), 7.28 (br s, 3H), 6.78 (s, 2H), 6.73 (s, 1H), 5.73 (dd, 1H), 5.15 (d, 1H), 5.08 (br d, 1H), 3.71-3.91 (m, 4H), 3.21-3.60 (m, 7H), 1.95-2.19 (m, 4H)1.76 (s, 3H), 1.44 (s, 3H).
HPLC purity: 90.80%
PATENT
WO 2019093450
Prior art documents
Non-patent literature
Non-patent Document 2: The Lancet Infrction diseases, 13 (9), 785-796,2013
Non-patent Document 3: Antimicrobial Agents and Chemotherapy, 61 (3), 1-11, 2017
PAPER
European Journal of Medicinal Chemistry (2018), 155, 847-868
References
- ^ Katsube, T.; Echols, R.; Arjona Ferreira, J. C.; et al. (2017). “Cefiderocol, a Siderophore Cephalosporin for Gram‐Negative Bacterial Infections: Pharmacokinetics and Safety in Subjects With Renal Impairment”. Journal of Clinical Pharmacology. 57 (5): 584–591. doi:10.1002/jcph.841. PMC 5412848. PMID 27874971.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves new antibacterial drug to treat complicated urinary tract infections as part of ongoing efforts to address antimicrobial resistance”. U.S. Food and Drug Administration (FDA) (Press release). 14 November 2019. Archived from the original on 16 November 2019. Retrieved 15 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Choi, Justin J; McCarthy, Matthew W. (24 January 2018). “Cefiderocol: a novel siderophore cephalosporin”. Expert Opinion on Investigational Drugs. 27 (2): 193–197. doi:10.1080/13543784.2018.1426745. PMID 29318906.
- ^ Aoki, Toshiaki; Yoshizawa, Hidenori; Yamawaki, Kenji; et al. (15 July 2018). “Cefiderocol (S-649266), A new siderophore cephalosporin exhibiting potent activities against Pseudomonas aeruginosa and other gram-negative pathogens including multi-drug resistant bacteria: Structure activity relationship”. European Journal of Medicinal Chemistry. 155: 847–868. doi:10.1016/j.ejmech.2018.06.014. ISSN 1768-3254. PMID 29960205.
- ^ Portsmouth, Simon; van Veenhuyzen, David; Echols, Roger; et al. (25 October 2018). “Cefiderocol versus imipenem-cilastatin for the treatment of complicated urinary tract infections caused by Gram-negative uropathogens: a phase 2, randomised, double-blind, non-inferiority trial”. The Lancet Infectious Diseases. 0 (12): 1319–1328. doi:10.1016/S1473-3099(18)30554-1. ISSN 1473-3099. PMID 30509675.
- ^ Jump up to:a b c “Fetroja (cefiderocol) for injection, for intravenous use full prescribing information”(PDF). November 2019. Retrieved 17 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b c Zhanel GG, Golden AR, Zelenitsky S, et al. (February 2019). “Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli”. Drugs. 79 (3): 271–289. doi:10.1007/s40265-019-1055-2. PMID 30712199.
- ^ Jump up to:a b “Cefiderocol New Drug Application”. U.S. Food and Drug Administration (FDA). Archived from the original on 4 December 2019. Retrieved 22 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ Sato T, Yamawaki K (November 2019). “Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin”. Clin. Infect. Dis. 69 (Supplement_7): S538–S543. doi:10.1093/cid/ciz826. PMC 6853759. PMID 31724047.
- ^ Matthews-King A (26 October 2018). “Antibiotic ‘Trojan horse’ could defeat superbugs causing global medical crisis, trial finds”. The Independent. Retrieved 26 October 2018.
- ^ Newey S (26 October 2018). “New ‘Trojan horse’ drug proves effective against antibiotic resistant bacteria”. The Telegraph. ISSN 0307-1235. Retrieved 26 October 2018.
- ^ Simpson DH, Scott P (2017). “Antimicrobial Metallodrugs”. In Lo K (ed.). Inorganic and Organometallic Transition Metal Complexes with Biological Molecules and Living Cells. Elsevier. ISBN 9780128038871.
- ^ Saisho, Yutaka; Katsube, Takayuki; White, Scott; et al. (March 2018). “Pharmacokinetics, Safety, and Tolerability of Cefiderocol, a Novel Siderophore Cephalosporin for Gram-Negative Bacteria, in Healthy Subjects” (PDF). Antimicrobial Agents and Chemotherapy. 62 (3): 1–12. doi:10.1128/AAC.02163-17. PMC 5826143. PMID 29311072. Retrieved 22 November 2019.
- ^ Ito A, Nishikawa T, Matsumoto S, et al. (December 2016). “Siderophore Cephalosporin Cefiderocol Utilizes Ferric Iron Transporter Systems for Antibacterial Activity against Pseudomonas aeruginosa”. Antimicrobial Agents and Chemotherapy. 60 (12): 7396–7401. doi:10.1128/AAC.01405-16. PMC 5119021. PMID 27736756.
External links
- “Cefiderocol”. Drug Information Portal. U.S. National Library of Medicine.
ADDITIONAL INFORMATION
S-649266 shows potent in vitro activity against the non-fermenting Gram-negative bacteria Acinetobacter baumannii, Pseudomonas aeruginosa and Stenotrophomonas maltophilia, including MDR strains such as carbapenem-resistant A. baumannii and metallo-β-lactamase-producing P. aeruginosa. MIC90s of S-649266 for A. baumannii, P. aeruginosa and S. maltophilia were 2, 1 and 0.5 mg/L, respectively, whereas MIC90s of meropenem were >16 mg/L. S-649266 showed potent in vitro activities against A. baumannii producing carbapenemases such as OXA-type β-lactamases, and P. aeruginosa producing metallo-β-lactamases such as IMP type and VIM type. MIC90 values for these A. baumannii strains and P. aeruginosa strains were 8 and 4 mg/L, respectively.
REFERENCES
1: Yamano Y. In Vitro Activity of Cefiderocol Against a Broad Range of Clinically Important Gram-negative Bacteria. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S544-S551. doi: 10.1093/cid/ciz827. PubMed PMID: 31724049; PubMed Central PMCID: PMC6853761.
2: Echols R, Ariyasu M, Nagata TD. Pathogen-focused Clinical Development to Address Unmet Medical Need: Cefiderocol Targeting Carbapenem Resistance. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S559-S564. doi: 10.1093/cid/ciz829. PubMed PMID: 31724048; PubMed Central PMCID: PMC6853756.
3: Sato T, Yamawaki K. Cefiderocol: Discovery, Chemistry, and In Vivo Profiles of a Novel Siderophore Cephalosporin. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S538-S543. doi: 10.1093/cid/ciz826. PubMed PMID: 31724047; PubMed Central PMCID: PMC6853759.
4: Bonomo RA. Cefiderocol: A Novel Siderophore Cephalosporin Defeating Carbapenem-resistant Pathogens. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S519-S520. doi: 10.1093/cid/ciz823. PubMed PMID: 31724046; PubMed Central PMCID: PMC6853757.
5: Katsube T, Echols R, Wajima T. Pharmacokinetic and Pharmacodynamic Profiles of Cefiderocol, a Novel Siderophore Cephalosporin. Clin Infect Dis. 2019 Nov 13;69(Supplement_7):S552-S558. doi: 10.1093/cid/ciz828. PubMed PMID: 31724042; PubMed Central PMCID: PMC6853762.
6: Kidd JM, Abdelraouf K, Nicolau DP. Efficacy of Humanized Cefiderocol Exposure is Unaltered by Host Iron Overload in the Thigh Infection Model. Antimicrob Agents Chemother. 2019 Oct 28. pii: AAC.01767-19. doi: 10.1128/AAC.01767-19. [Epub ahead of print] PubMed PMID: 31658966.
7: Chen IH, Kidd JM, Abdelraouf K, Nicolau DP. Comparative In Vivo Antibacterial Activity of Human-Simulated Exposures of Cefiderocol and Ceftazidime against Stenotrophomonas maltophilia in the Murine Thigh Model. Antimicrob Agents Chemother. 2019 Oct 7. pii: AAC.01558-19. doi: 10.1128/AAC.01558-19. [Epub ahead of print] PubMed PMID: 31591126.
8: Stevens RW, Clancy M. Compassionate Use of Cefiderocol in the Treatment of an Intraabdominal Infection Due to Multidrug-Resistant Pseudomonas aeruginosa: A Case Report. Pharmacotherapy. 2019 Nov;39(11):1113-1118. doi: 10.1002/phar.2334. Epub 2019 Oct 22. PubMed PMID: 31550054.
9: Sanabria C, Migoya E, Mason JW, Stanworth SH, Katsube T, Machida M, Narukawa Y, Den Nagata T. Effect of Cefiderocol, a Siderophore Cephalosporin, on QT/QTc Interval in Healthy Adult Subjects. Clin Ther. 2019 Sep;41(9):1724-1736.e4. doi: 10.1016/j.clinthera.2019.07.006. Epub 2019 Aug 1. PubMed PMID: 31378318.
10: Trecarichi EM, Quirino A, Scaglione V, Longhini F, Garofalo E, Bruni A, Biamonte E, Lionello R, Serapide F, Mazzitelli M, Marascio N, Matera G, Liberto MC, Navalesi P, Torti C; IMAGES Group . Successful treatment with cefiderocol for compassionate use in a critically ill patient with XDR Acinetobacter baumannii and KPC-producing Klebsiella pneumoniae: a case report. J Antimicrob Chemother. 2019 Nov 1;74(11):3399-3401. doi: 10.1093/jac/dkz318. PubMed PMID: 31369095.
11: Nakamura R, Ito-Horiyama T, Takemura M, Toba S, Matsumoto S, Ikehara T, Tsuji M, Sato T, Yamano Y. In Vivo Pharmacodynamic Study of Cefiderocol, a Novel Parenteral Siderophore Cephalosporin, in Murine Thigh and Lung Infection Models. Antimicrob Agents Chemother. 2019 Aug 23;63(9). pii: e02031-18. doi: 10.1128/AAC.02031-18. Print 2019 Sep. PubMed PMID: 31262762; PubMed Central PMCID: PMC6709502.
12: Katsube T, Saisho Y, Shimada J, Furuie H. Intrapulmonary pharmacokinetics of cefiderocol, a novel siderophore cephalosporin, in healthy adult subjects. J Antimicrob Chemother. 2019 Jul 1;74(7):1971-1974. doi: 10.1093/jac/dkz123. PubMed PMID: 31220260; PubMed Central PMCID: PMC6587409.
13: Jean SS, Hsueh SC, Lee WS, Hsueh PR. Cefiderocol: a promising antibiotic against multidrug-resistant Gram-negative bacteria. Expert Rev Anti Infect Ther. 2019 May;17(5):307-309. doi: 10.1080/14787210.2019.1612240. Epub 2019 May 6. PubMed PMID: 31055983.
14: Hackel MA, Tsuji M, Yamano Y, Echols R, Karlowsky JA, Sahm DF. Reproducibility of broth microdilution MICs for the novel siderophore cephalosporin, cefiderocol, determined using iron-depleted cation-adjusted Mueller-Hinton broth. Diagn Microbiol Infect Dis. 2019 Aug;94(4):321-325. doi: 10.1016/j.diagmicrobio.2019.03.003. Epub 2019 Mar 23. PubMed PMID: 31029489.
15: Miyazaki S, Katsube T, Shen H, Tomek C, Narukawa Y. Metabolism, Excretion, and Pharmacokinetics of [(14) C]-Cefiderocol (S-649266), a Siderophore Cephalosporin, in Healthy Subjects Following Intravenous Administration. J Clin Pharmacol. 2019 Jul;59(7):958-967. doi: 10.1002/jcph.1386. Epub 2019 Feb 7. PubMed PMID: 30730562; PubMed Central PMCID: PMC6593826.
16: Zhanel GG, Golden AR, Zelenitsky S, Wiebe K, Lawrence CK, Adam HJ, Idowu T, Domalaon R, Schweizer F, Zhanel MA, Lagacé-Wiens PRS, Walkty AJ, Noreddin A, Lynch Iii JP, Karlowsky JA. Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli. Drugs. 2019 Feb;79(3):271-289. doi: 10.1007/s40265-019-1055-2. Review. PubMed PMID: 30712199.
17: Huttner A. Cefiderocol for treatment of complicated urinary tract infections – Author’s reply. Lancet Infect Dis. 2019 Jan;19(1):24-25. doi: 10.1016/S1473-3099(18)30728-X. PubMed PMID: 30587291.
18: Portsmouth S, Echols R, Den Nagata T. Cefiderocol for treatment of complicated urinary tract infections. Lancet Infect Dis. 2019 Jan;19(1):23-24. doi: 10.1016/S1473-3099(18)30721-7. PubMed PMID: 30587290.
19: Wagenlehner FME, Naber KG. Cefiderocol for treatment of complicated urinary tract infections. Lancet Infect Dis. 2019 Jan;19(1):22-23. doi: 10.1016/S1473-3099(18)30722-9. PubMed PMID: 30587289.
20: Portsmouth S, van Veenhuyzen D, Echols R, Machida M, Ferreira JCA, Ariyasu M, Tenke P, Nagata TD. Cefiderocol versus imipenem-cilastatin for the treatment of complicated urinary tract infections caused by Gram-negative uropathogens: a phase 2, randomised, double-blind, non-inferiority trial. Lancet Infect Dis. 2018 Dec;18(12):1319-1328. doi: 10.1016/S1473-3099(18)30554-1. Epub 2018 Oct 25. PubMed PMID: 30509675.
 |
|
| Clinical data | |
|---|---|
| Trade names | Fetroja |
| Routes of administration |
Intravenous infusion |
| ATC code |
|
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Protein binding | 56–58%[1] |
| Elimination half-life | 2.8 hours |
| Excretion | mainly renal (60–70% unchanged) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| Chemical and physical data | |
| Formula | C30H34ClN7O10S2 |
| Molar mass | 752.21 g·mol−1 |
| 3D model (JSmol) | |
////////////Cefiderocol, セフィデロコル , FDA 2019, цефидерокол , سيفيديروكول , 头孢德罗 , S-649266, GSK 2696266D
Givosiran, ギボシラン ,



Givosiran
N-[1,3-bis[3-[3-[5-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxypentanoylamino]propylamino]-3-oxopropoxy]-2-[[3-[3-[5-[(2R,3R,4R,5R,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxypentanoylamino]propylamino]-3-oxopropoxy]methyl]propan-2-yl]-12-[(2R,4R)-4-hydroxy-2-methylpyrrolidin-1-yl]-12-oxododecanamide
Treatment of Acute Hepatic Porphyria (AHP)
| Formula |
C524H694F16N173O316P43S6
|
|---|---|
| CAS |
1639325-43-1
|
| Mol weight |
16300.3229
|
Treatment of acute hepatic porphyria, RNA interference (RNAi) drug
FDA APPROVED, Givlaari, 2019/11/20
ギボシラン;
Givosiran, sold under the brand name Givlaari, is for the treatment of adults with acute hepatic porphyria, a genetic disorder resulting in the buildup of toxic porphyrin molecules which are formed during the production of heme (which helps bind oxygen in the blood).[1][2]
History
The U.S. Food and Drug Administration (FDA) granted the application for givosiran breakthrough therapy designation, priority reviewdesignation, and orphan drug designation.[1] The FDA granted the approval of Givlaari to Alnylam Pharmaceuticals.[1]
The full ENVISION results demonstrated a 74 percent mean and 90 percent median reduction in the primary endpoint measure of annualized rate of composite attacks in patients on givosiran relative to placebo during the six-month double-blind period. In addition, givosiran achieved statistically significant positive results for five of nine secondary endpoints, with an overall safety and tolerability profile that the Company believes is encouraging, especially in this high unmet need disease. Adverse events (AEs) were reported in 89.6 percent of givosiran patients and 80.4 percent of placebo patients; serious adverse events (SAEs) were reported in 20.8 percent of givosiran patients and 8.7 percent of placebo patients. Ninety-three of 94 patients, or 99 percent, enrolled in the open-label extension (OLE) period of the study. Based on the ENVISION results, the Company plans to complete its rolling submission of a New Drug Application (NDA) and file a Marketing Authorisation Application (MAA) in mid-2019.
“Given the high unmet need in this disease setting, we are very pleased for the patients and families living with acute hepatic porphyria for whom these results signal hope for a potential new therapeutic option,” said Akshay Vaishnaw, M.D., Ph.D., President of R&D at Alnylam. “Givosiran substantially reduced the frequency of attacks, providing strong support for a treatment benefit, with a consistent effect across all components of the primary endpoint and all subgroups analyzed. In this disease with high burden and associated comorbidities, we’re encouraged by the overall tolerability profile. We firmly believe givosiran has the potential to be a transformative medicine for patients living with AHP.”
“Currently, there are no approved therapies aimed at preventing the painful, often incapacitating attacks and chronic symptoms associated with AHP,” said Manisha Balwani, M.D., M.S, Associate Professor of the Department of Genetics and Genomic Sciences and Department of Medicine at the Icahn School of Medicine at Mount Sinai and principal investigator of the ENVISION study. “The results from ENVISION are promising and demonstrate a strong treatment effect for givosiran, with reduction of attacks and improvement in patient-reported measures of overall health status and quality of life. Thus, givosiran represents a novel and targeted treatment approach that has the potential to make a significant impact on the lives of patients who are struggling with the disabling symptoms of this disease.”
Efficacy Results
Givosiran met the primary efficacy endpoint with a 74 percent mean reduction relative to placebo in the annualized rate of composite porphyria attacks, defined as those requiring hospitalization, urgent healthcare visit, or hemin administration, in patients with acute intermittent porphyria (AIP) over six months (p equal to 6.04×10-9). There was a corresponding 90 percent median reduction in composite annualized attack rate (AAR), with a median AAR of 1.0 in givosiran patients compared with a median AAR of 10.7 in placebo patients. Fifty percent of givosiran-treated patients were attack-free during the six-month treatment period as compared to 16.3 percent of placebo-treated patients. The reductions in attack rates were observed across all components of the primary endpoint. The treatment benefit for givosiran compared to placebo was maintained across all pre-specified patient subgroups, including age, race, geography, historical attack rates, prior hemin prophylaxis status, disease severity, and other baseline characteristics.
Givosiran also demonstrated statistically significant differences in five of nine hierarchically tested secondary endpoints relative to placebo. These included mean reductions of:
- 91 percent in urinary aminolevulinic acid (ALA) in patients with AIP at three months (p equal to 8.74×10-14).
- 83 percent in urinary ALA in patients with AIP at six months (p equal to 6.24×10-7).
- 73 percent in urinary levels of porphobilinogen (PBG) in patients with AIP at six months (p equal to 8.80×10-7).
- 77 percent in the number of annualized days on hemin in patients with AIP (p equal to 2.35×10-5).
- 73 percent in composite AAR for patients with any AHP (p equal to 1.35×10-8).
The remaining four secondary endpoints did not meet the prespecified criteria for statistical significance in hierarchical testing.

About Acute Hepatic Porphyria
Acute hepatic porphyria (AHP) refers to a family of rare, genetic diseases characterized by potentially life-threatening attacks and for some patients chronic debilitating symptoms that negatively impact daily functioning and quality of life. AHP is comprised of four subtypes, each resulting from a genetic defect leading to deficiency in one of the enzymes of the heme biosynthesis pathway in the liver: acute intermittent porphyria (AIP), hereditary coproporphyria (HCP), variegate porphyria (VP), and ALAD-deficiency porphyria (ADP). These defects cause the accumulation of neurotoxic heme intermediates aminolevulinic acid (ALA) and porphobilinogen (PBG), with ALA believed to be the primary neurotoxic intermediate responsible for causing both attacks and ongoing symptoms between attacks. Common symptoms of AHP include severe, diffuse abdominal pain, weakness, nausea, and fatigue. The nonspecific nature of AHP signs and symptoms can often lead to misdiagnoses of other more common conditions such as irritable bowel syndrome, appendicitis, fibromyalgia, and endometriosis, and consequently, patients afflicted by AHP often remain without a proper diagnosis for up to 15 years. In addition, long-term complications of AHP and its treatment can include chronic neuropathic pain, hypertension, chronic kidney disease and liver disease, including iron overload, fibrosis, cirrhosis and hepatocellular carcinoma. Currently, there are no treatments approved to prevent debilitating attacks or to treat the chronic manifestations of the disease.
About Givosiran
Givosiran is an investigational, subcutaneously administered RNAi therapeutic targeting aminolevulinic acid synthase 1 (ALAS1) in development for the treatment of acute hepatic porphyria (AHP). Monthly administration of givosiran has the potential to significantly lower induced liver ALAS1 levels in a sustained manner and thereby decrease neurotoxic heme intermediates, aminolevulinic acid (ALA) and porphobilinogen (PBG), to near normal levels. By reducing accumulation of these intermediates, givosiran has the potential to prevent or reduce the occurrence of severe and life-threatening attacks, control chronic symptoms, and decrease the burden of the disease. Givosiran utilizes Alnylam’s Enhanced Stabilization Chemistry ESC-GalNAc conjugate technology, which enables subcutaneous dosing with increased potency and durability and a wide therapeutic index. Givosiran has been granted Breakthrough Therapy Designation by the U.S. Food and Drug Administration (FDA) and PRIME Designation by the European Medicines Agency (EMA). Givosiran has also been granted Orphan Drug Designations in both the U.S. and the EU for the treatment of AHP. The safety and efficacy of givosiran were evaluated in the ENVISION Phase 3 trial with positive results; these results have not been evaluated by the FDA, the EMA or any other health authority.
About RNAi
RNAi (RNA interference) is a natural cellular process of gene silencing that represents one of the most promising and rapidly advancing frontiers in biology and drug development today. Its discovery has been heralded as “a major scientific breakthrough that happens once every decade or so,” and was recognized with the award of the 2006 Nobel Prize for Physiology or Medicine. By harnessing the natural biological process of RNAi occurring in our cells, a new class of medicines, known as RNAi therapeutics, is now a reality. Small interfering RNA (siRNA), the molecules that mediate RNAi and comprise Alnylam’s RNAi therapeutic platform, function upstream of today’s medicines by potently silencing messenger RNA (mRNA) – the genetic precursors – that encode for disease-causing proteins, thus preventing them from being made. This is a revolutionary approach with the potential to transform the care of patients with genetic and other diseases.
References
- ^ Jump up to:a b c “FDA approves first treatment for inherited rare disease”. U.S. Food and Drug Administration (FDA) (Press release). 20 November 2019. Archived from the original on 21 November 2019. Retrieved 20 November 2019. This article incorporates text from this source, which is in the public domain.
- ^ “FDA approves givosiran for acute hepatic porphyria”. U.S. Food and Drug Administration (FDA) (Press release). 20 November 2019. Archived from the original on 21 November 2019. Retrieved 20 November 2019. This article incorporates text from this source, which is in the public domain.
- The New England journal of medicine (2019), 380(6), 549-558.
- New England Journal of Medicine (2019), 380(6), 549-558.
- Toxicologic Pathology (2018), 46(7), 735-745.
External links
- “Givosiran”. Drug Information Portal. U.S. National Library of Medicine (NLM).
-
GIVLAARI
(givosiran) Injection, for Subcutaneous UseDESCRIPTION
GIVLAARI is an aminolevulinate synthase 1-directed small interfering RNA (siRNA), covalently linked to a ligand containing three N-acetylgalactosamine (GalNAc) residues to enable delivery of the siRNA to hepatocytes.
The structural formulas of the givosiran drug substance in its sodium form, and the ligand (L96), are presented below.
Abbreviations: Af = adenine 2′-F ribonucleoside; Cf = cytosine 2′-F ribonucleoside; Uf = uracil 2′-F ribonucleoside; Am = adenine 2′-OMe ribonucleoside; Cm = cytosine 2′-OMe ribonucleoside; Gf = guanine 2′-F ribonucleoside; Gm = guanine 2′-OMe ribonucleoside; Um = uracil 2′-OMe ribonucleoside; L96 = triantennary GalNAc (N-acetylgalactosamine)
GIVLAARI is supplied as a sterile, preservative-free, 1-mL colorless-to-yellow solution for subcutaneous injection containing 189 mg givosiran in a single-dose, 2-mL Type 1 glass vial with a TEFLON®-coated stopper and a flip-off aluminum seal. GIVLAARI is available in cartons containing one single-dose vial each. Water for injection is the only excipient used in the manufacture of GIVLAARI.
The molecular formula of givosiran sodium is C524 H651 F16 N173 Na43 O316 P43 S6 with a molecular weight of 17,245.56 Da.
The molecular formula of givosiran (free acid) is C524 H694 F16 N173 O316 P43 S6 with a molecular weight of 16,300.34 Da.
| Clinical data | |
|---|---|
| Trade names | Givlaari |
| Routes of administration |
Subcutaneous injection |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| UNII | |
| KEGG | |
| Chemical and physical data | |
| Formula | C524H694F16N173O316P43S6 |
| Molar mass | 16300.42 g·mol−1 |
| 3D model (JSmol) | |
/////////Givosiran, ギボシラン , FDA 2019, Acute Hepatic Porphyria,
Golodirsen, ゴロジルセン;

Golodirsen
- RNA, [P-deoxy-P-(dimethylamino)](2′,3′-dideoxy-2′,3′-imino-2′,3′-seco)(2’a→5′)(G-m5U-m5U-G-C-C-m5U-C-C-G-G-m5U-m5U-C-m5U-G-A-A-G-G-m5U-G-m5U-m5U-C), 5′-[P-[4-[[2-[2-(2-hydroxyethoxy)ethoxy]ethoxy]carbonyl]-1-piperazinyl]-N,N-dimethylphosphonamidate]
- Nucleic Acid Sequence
- Sequence Length: 25
| Formula |
C305H481N138O112P25
|
|---|---|
| CAS |
1422959-91-8
|
| Mol weight |
8647.2841
|
- Exon 53: NG-12-0163
- Golodirsen
- SRP 4053
Nucleic Acid Sequence
Sequence Length: 252 a 6 c 8 g 9 umodified
FDA APPROVED, Vyondys 53, 019/12/12
Antisense oligonucleotide
|
ゴロジルセン;
|
Duchenne muscular dystrophy (DMD variant amenable to exon 53 skipping)
VYONDYS 53 (golodirsen) injection is a sterile, aqueous, preservative-free, concentrated solution for dilution prior to intravenous administration. VYONDYS 53 is a clear to slightly opalescent, colorless liquid. VYONDYS 53 is supplied in single-dose vials containing 100 mg golodirsen (50 mg/mL). VYONDYS 53 is formulated as an isotonic phosphate buffered saline solution with an osmolality of 260 to 320 mOSM and a pH of 7.5. Each milliliter of VYONDYS 53 contains: 50 mg golodirsen; 0.2 mg potassium chloride; 0.2 mg potassium phosphate monobasic; 8 mg sodium chloride; and 1.14 mg sodium phosphate dibasic, anhydrous, in water for injection. The product may contain hydrochloric acid or sodium hydroxide to adjust pH.
Golodirsen is an antisense oligonucleotide of the phosphorodiamidate morpholino oligomer (PMO) subclass. PMOs are synthetic molecules in which the five-membered ribofuranosyl rings found in natural DNA and RNA are replaced by a six-membered morpholino ring. Each morpholino ring is linked through an uncharged phosphorodiamidate moiety rather than the negatively charged phosphate linkage that is present in natural DNA and RNA. Each phosphorodiamidate morpholino subunit contains one of the heterocyclic bases found in DNA (adenine, cytosine, guanine, or thymine). Golodirsen contains 25 linked subunits. The sequence of bases from the 5′ end to 3′ end is GTTGCCTCCGGTTCTGAAGGTGTTC. The molecular formula of golodirsen is C305H481N138O112P25 and the molecular weight is 8647.28 daltons. The structure of golodirsen is:
|
SIDE EFFECTS
- Hypersensitivity Reactions [see WARNINGS AND PRECAUTIONS]
Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In the VYONDYS 53 clinical development program, 58 patients received at least one intravenous dose of VYONDYS 53, ranging between 4 mg/kg (0.13 times the recommended dosage) and 30 mg/kg (the recommended dosage). All patients were male and had genetically confirmed Duchenne muscular dystrophy. Age at study entry was 6 to 13 years. Most (86%) patients were Caucasian.
VYONDYS 53 was studied in 2 double-blind, placebo-controlled studies.
In Study 1 Part 1, patients were randomized to receive once-weekly intravenous infusions of VYONDYS 53 (n=8) in four increasing dose levels from 4 mg/kg to 30 mg/kg or placebo (n=4), for at least 2 weeks at each level. All patients who participated in Study 1 Part 1 (n=12) were continued into Study 1 Part 2, an open-label extension, during which they received VYONDYS 53 at a dose of 30 mg/kg IV once weekly [see Clinical Studies].
In Study 2, patients received VYONDYS 53 (n=33) 30 mg/kg or placebo (n=17) IV once weekly for up to 96 weeks, after which all patients received VYONDYS 53 at a dose of 30 mg/kg.
Adverse reactions observed in at least 20% of treated patients in the placebo-controlled sections of Studies 1 and 2 are shown in Table 1.
Table 1: Adverse Reactions That Occurred in At Least 20% of VYONDYS 53-Treated Patients and at a Rate Greaterthan Placebo in Studies 1 and 2
| Adverse Reaction | VYONDYS 53 (N = 41) % |
Placebo (N = 21) % |
| Headache | 41 | 10 |
| Pyrexia | 41 | 14 |
| Fall | 29 | 19 |
| Abdominal pain | 27 | 10 |
| Nasopharyngitis | 27 | 14 |
| Cough | 27 | 19 |
| Vomiting | 27 | 19 |
| Nausea | 20 | 10 |
Other adverse reactions that occurred at a frequency greater than 5% of VYONDYS 53-treated patients and at a greater frequency than placebo were: administration site pain, back pain, pain, diarrhea, dizziness, ligament sprain, contusion, influenza, oropharyngeal pain, rhinitis, skin abrasion, ear infection, seasonal allergy, tachycardia, catheter site related reaction, constipation, and fracture.
Hypersensitivity reactions have occurred in patients treated with VYONDYS 53 [see WARNINGS AND PRECAUTIONS].
Antisense technology provides a means for modulating the expression of one or more specific gene products, including alternative splice products, and is uniquely useful in a number of therapeutic, diagnostic, and research applications. The principle behind antisense technology is that an antisense compound, e.g., an oligonucleotide, which hybridizes to a target nucleic acid, modulates gene expression activities such as transcription, splicing or translation through any one of a number of antisense mechanisms. The sequence specificity of antisense compounds makes them attractive as tools for target validation and gene functionalization, as well as therapeutics to selectively modulate the expression of genes involved in disease.
Duchenne muscular dystrophy (DMD) is caused by a defect in the expression of the protein dystrophin. The gene encoding the protein contains 79 exons spread out over more than 2 million nucleotides of DNA. Any exonic mutation that changes the reading frame of the exon, or introduces a stop codon, or is characterized by removal of an entire out of frame exon or exons, or duplications of one or more exons, has the potential to disrupt production of functional dystrophin, resulting in DMD.
Recent clinical trials testing the safety and efficacy of splice switching
oligonucleotides (SSOs) for the treatment of DMD are based on SSO technology to induce alternative splicing of pre-mRNAs by steric blockade of the spliceosome (Cirak et al., 2Q\ \; Goemans et al., 2011; Kinali et al., 2009; van Deutekom et al., 2007). However, despite these successes, the pharmacological options available for treating DMD are limited. Golodirsen is a phosphorodiamidate morpholino oligomer (PMO) designed to skip exon 53 of the human dystrophin gene in patients with DMD who are amendable to exon 53 skipping to restore the read frame and produce a functional shorter form of the dystrophin protein.
Although significant progress has been made in the field of antisense technology, there remains a need in the art for methods of preparing phosphorodiamidate morpholino oligomers with improved antisense or antigene performance.
PATENT
https://patents.google.com/patent/WO2017205880A1/en
Provided herein are processes for preparing phosphorodiamidate morpholino oligomers (PMOs). The synthetic processes described herein allow for a scaled-up PMO synthesis while maintaining overall yield and purity of a synthesized PMO.
Accordingly, in one aspect, provided herein is a process for preparing an oligomeric compound of Formula A):

(A).
In certain embodiments, provided herein is a process for preparing an oligomeric compound of Formula (G):

In yet another embodiment, the oligomeric compound of the disclosure including, for example, some embodiments of an oligomeric compound of Formula (G), is an oligomeric compound of Formula (XII):

(XII).
For clarity, the structural formulas including, for example, oligomeric compound of Formula (C) and Golodirsen depicted by Formula (XII), are a continuous structural formula from 5′ to 3′, and, for the convenience of depicting the entire formula in a compact form in the above structural formulas, Applicants have included various illustration breaks labeled “BREAK A” and “BREAK B.” As would be understood by the skilled artisan, for example, each indication of “BREAK A” shows a continuation of the illustration of the structural formula at these points. The skilled artisan understands that the same is true for each instance of “BREAK B” in the structural formulas above including Golodirsen. None of the illustration breaks, however, are intended to indicate, nor would the skilled artisan understand them to mean, an actual discontinuation of the structural formulas above including
Example 1: NCP2 Anchor Synthesis
1. Preparation of Meth l 4-Fluoro-3-Nitrobenzoate (1)

To a 100L flask was charged 12.7kg of 4-fluoro-3-nitrobenzoic acid was added 40kg of methanol and 2.82kg concentrated sulfuric acid. The mixture was stirred at reflux (65° C) for 36 hours. The reaction mixture was cooled to 0° C. Crystals formed at 38° C. The mixture was held at 0° C for 4 hrs then filtered under nitrogen. The 100L flask was washed and filter cake was washed with 10kg of methanol that had been cooled to 0° C. The solid filter cake was dried on the funnel for 1 hour, transferred to trays, and dried in a vacuum oven at room temperature to a constant weight of 13.695kg methyl 4-fluoro-3-nitrobenzoate (100% yield; HPLC 99%).
2. Preparation of 3-Nitro-4-(2-oxopropyl)benzoic Acid
A. (Z)-Methyl 4-(3 -Hydroxy- l-Methoxy-l-Oxobut-2-en-2-yl)-3-Nitrobenzoate (2)

To a 100L flask was charged 3.98kg of methyl 4-fluoro-3-nitrobenzoate (1) from the previous step 9.8kg DMF, 2.81kg methyl acetoacetate. The mixture was stirred and cooled to 0° C. To this was added 3.66kg DBU over about 4 hours while the temperature was maintained at or below 5° C. The mixture was stirred an additional 1 hour. To the reaction flask was added a solution of 8.15kg of citric acid in 37.5kg of purified water while the reaction temperature was maintained at or below 15° C. After the addition, the reaction mixture was stirred an addition 30 minutes then filtered under nitrogen. The wet filter cake was returned to the 100L flask along with 14.8kg of purified water. The slurry was stirred for 10 minutes then filtered. The wet cake was again returned to the 100L flask, slurried with 14.8kg of purified water for 10 minutes, and filtered to crude (Z)-methyl 4-(3 -hydroxy- 1 – methoxy-l-oxobut-2-en-2-yl)-3-nitrobenzoate.
B. 3-Nitro-4-(2-oxopropyl)benzoic Acid

2 3
The crude (Z)-m ethyl 4-(3 -hydroxy- 1-methoxy-l -ox obut-2-en-2-yl)-3-nitrobenzoate was charged to a 100L reaction flask under nitrogen. To this was added 14.2kg 1,4-dioxane and the stirred. To the mixture was added a solution of 16.655kg concentrated HC1 and 13.33kg purified water (6M HC1) over 2 hours while the temperature of the reaction mixture was maintained below 15° C. When the addition was complete, the reaction mixture was heated at reflux (80° C) for 24 hours, cooled to room temperature, and filtered under nitrogen. The solid filter cake was triturated with 14.8kg of purified water, filtered, triturated again with 14.8kg of purified water, and filtered. The solid was returned to the 100L flask with 39.9kg of DCM and refluxed with stirring for 1 hour. 1.5kg of purified water was added to dissolve the remaining solids. The bottom organic layer was split to a pre-warmed 72L flask, then returned to a clean dry 100L flask. The solution was cooled to 0° C, held for 1 hour, then filtered. The solid filter cake was washed twice each with a solution of 9.8kg DCM and 5kg heptane, then dried on the funnel. The solid was transferred to trays and dried to a constant weight of 1.855kg 3-Nitro-4-(2-oxopropyl)benzoic Acid. Overall yield 42% from compound 1. HPLC 99.45%.
3. Preparation of N-Tritylpiperazine Succinate (NTP)

To a 72L jacketed flask was charged under nitrogen 1.805kg triphenylmethyl chloride and 8.3kg of toluene (TPC solution). The mixture was stirred until the solids dissolved. To a 100L jacketed reaction flask was added under nitrogen 5.61kg piperazine, 19.9kg toluene, and 3.72kg methanol. The mixture was stirred and cooled to 0° C. To this was slowly added in portions the TPC solution over 4 hours while the reaction temperature was maintained at or below 10° C. The mixture was stirred for 1.5 hours at 10° C, then allowed to warm to 14° C. 32.6kg of purified water was charged to the 72L flask, then transferred to the 100L flask while the internal batch temperature was maintained at 20+/-50 C. The layers were allowed to split and the bottom aqueous layer was separated and stored. The organic layer was extracted three times with 32kg of purified water each, and the aqueous layers were separated and combined with the stored aqueous solution.
The remaining organic layer was cooled to 18° C and a solution of 847g of succinic acid in 10.87kg of purified water was added slowly in portions to the organic layer. The mixture was stirred for 1.75 hours at 20+/-50 C. The mixture was filtered, and the solids were washed with 2kg TBME and 2kg of acetone then dried on the funnel. The filter cake was triturated twice with 5.7kg each of acetone and filtered and washed with 1kg of acetone between triturations. The solid was dried on the funnel, then transferred to trays and dried in a vacuum oven at room temperature to a constant weight of 2.32kg of NTP. Yield 80%. 4. Preparation of (4-(2-Hydroxypropyl)-3-NitrophenyI)(4-Tritylpiperazin-l-yl)Methanone A. Preparation of l-(2-Nitro-4(4-Tritylpiperazine-l-Carbonyl)Phenyl)Propan-2-one

3 4
To a 100L jacketed flask was charged under nitrogen 2kg of 3-Nitro-4-(2- oxopropyl)benzoic Acid (3), 18.3 kg DCM, 1.845kg N-(3-dimethylaminopropyl)-N’- ethylcarbodiimide hydrochloride (EDC.HC1). The solution was stirred until a homogenous mixture was formed. 3.048kg of NTP was added over 30 minutes at room temperature and stirred for 8 hours. 5.44kg of purified water was added to the reaction mixture and stirred for 30 minutes. The layers were allowed to separate and the bottom organic layer containing the product was drained and stored. The aqueous layer was extracted twice with 5.65kg of DCM. The combined organic layers were washed with a solution of 1.08kg sodium chloride in 4.08kg purified water. The organic layers were dried over 1.068kg of sodium sulfate and filtered. The sodium sulfate was washed with 1.3kg of DCM. The combined organic layers were slurried with 252g of silica gel and filtered through a filter funnel containing a bed of 252g of silica gel. The silica gel bed was washed with 2kg of DCM. The combined organic layers were evaporated on a rotovap. 4.8kg of THF was added to the residue and then evaporated on the rotovap until 2.5 volumes of the crude l-(2-nitro-4(4-tritylpiperazine-l- carbonyl)phenyl)propan-2-one in THF was reached.
B. Preparation of (4-(2-Hydroxypropyl)-3-NitrophenyI)(4-Tritylpiperazin-l- yl)Methano

To a 100L jacketed flask was charged under nitrogen 3600g of 4 from the previous step and 9800g THF. The stirred solution was cooled to <5° C. The solution was diluted with 11525g ethanol and 194g of sodium borohydride was added over about 2 hours at <5° C. The reaction mixture was stirred an additional 2 hours at <5° C. The reaction was quenched with a solution of about 1.1kg ammonium chloride in about 3kg of water by slow addition to maintain the temperature at <10° C. The reaction mixture was stirred an additional 30 minutes, filtered to remove inorganics, and recharged to a 100L jacketed flask and extracted with 23kg of DCM. The organic layer was separated and the aqueous was twice more extracted with 4.7kg of DCM each. The combined organic layers were washed with a solution of about 800g of sodium chloride in about 3kg of water, then dried over 2.7kg of sodium sulfate. The suspension was filtered and the filter cake was washed with 2kg of DCM. The combined filtrates were concentrated to 2.0 volumes, diluted with about 360g of ethyl acetate, and evaporated. The crude product was loaded onto a silica gel column of 4kg of silica packed with DCM under nitrogen and eluted with 2.3kg ethyl acetate in 7.2kg of DCM. The combined fractions were evaporated and the residue was taken up in 11.7kg of toluene. The toluene solution was filtered and the filter cake was washed twice with 2kg of toluene each. The filter cake was dried to a constant weight of 2.275kg of compound 5 (46% yield from compound 3) HPLC 96.99%. 5. Preparation of 2,5-Dioxopyrrolidin-l-yl(l-(2-Nitro-4-(4-triphenylmethylpiperazine-l Carbon l)Phenyl)Propan-2-yl) Carbonate (NCP2 Anchor)

3 NCP2 Anchor
To a 100L jacketed flask was charged under nitrogen 4.3kg of compound 5 (weight adjusted based on residual toluene by 1H MR; all reagents here after were scaled accordingly) and 12.7kg pyridine. To this was charged 3.160 kg of DSC (78.91 weight % by 1H NMR) while the internal temperature was maintained at <35° C. The reaction mixture was aged for about 22 hours at ambience then filtered. The filter cake was washed with 200g of pyridine. In two batches each comprising ½ the filtrate volume, filtrate wash charged slowly to a 100L jacketed flask containing a solution of about 11kg of citric acid in about 50 kg of water and stirred for 30 minutes to allow for solid precipitation. The solid was collected with a filter funnel, washed twice with 4.3kg of water per wash, and dried on the filter funnel under vacuum.
The combined solids were charged to a 100L jacketed flask and dissolved in 28kg of DCM and washed with a solution of 900g of potassium carbonate in 4.3kg of water. After 1 hour, the layers were allowed to separate and the aqueous layer was removed. The organic layer was washed with 10kg of water, separated, and dried over 3.5kg of sodium sulfate. The DCM was filtered, evaporated, and dried under vacuum to 6.16kg of NCP2 Anchor (114% yield).
Example 2: Anchor Loaded Resin Synthesis
To a 75L solid phase synthesis reactor was charged about 52L of NMP and 2600g of aminomethyl polystyrene resin. The resin was stirred in the NMP to swell for about 2 hours then drained. The resin was washed twice with about 39L DCM per wash, then twice with 39L Neutralization Solution per wash, then twice with 39L of DCM per wash. The NCP2 Anchor Solution was slowly added to the stirring resin solution, stirred for 24 hours at room temperature, and drained. The resin was washed four times with 39L of NMP per wash, and six times with 39L of DCM per wash. The resin was treated and stirred with ½ the DEDC Capping Solution for 30 minutes, drained, and was treated and stirred with the 2nd ½ of the DEDC Capping Solution for 30 minutes and drained. The resin was washed six times with 39L of DCM per wash then dried in an oven to constant weight of 3573.71g of Anchor Loaded Resin.
Example 3: Preparation of Activated EG3 Tail
1. Preparation of Trityl Piperazine Phenyl Carbamate 35

To a cooled suspension of NTP in dichloromethane (6 mL/g NTP) was added a solution of potassium carbonate (3.2 eq) in water (4 mL/g potassium carbonate). To this two- phase mixture was slowly added a solution of phenyl chloroformate (1.03 eq) in
dichloromethane (2 g/g phenyl chloroformate). The reaction mixture was warmed to 20° C. Upon reaction completion (1-2 hr), the layers were separated. The organic layer was washed with water, and dried over anhydrous potassium carbonate. The product 35 was isolated by crystallization from acetonitrile. Yield=80%
2. Preparation of Carbamate Alcohol (36)

Sodium hydride (1.2 eq) was suspended in l-methyl-2-pyrrolidinone (32 mL/g sodium hydride). To this suspension were added triethylene glycol (10.0 eq) and compound 35 (1.0 eq). The resulting slurry was heated to 95° C. Upon reaction completion (1-2 hr), the mixture was cooled to 20° C. To this mixture was added 30% dichloromethane/methyl tert- butyl ether (v:v) and water. The product-containing organic layer was washed successively with aqueous NaOH, aqueous succinic acid, and saturated aqueous sodium chloride. The product 36 was isolated by crystallization from dichloromethane/methyl tert-butyl ether/heptane. Yield=90%.
3. Preparation of EG3 Tail Acid (37)

To a solution of compound 36 in tetrahydrofuran (7 mL/g 36) was added succinic anhydride (2.0 eq) and DMAP (0.5 eq). The mixture was heated to 50° C. Upon reaction completion (5 hr), the mixture was cooled to 20° C and adjusted to pH 8.5 with aqueous NaHC03. Methyl tert-butyl ether was added, and the product was extracted into the aqueous layer. Dichloromethane was added, and the mixture was adjusted to pH 3 with aqueous citric acid. The product-containing organic layer was washed with a mixture of pH=3 citrate buffer and saturated aqueous sodium chloride. This dichloromethane solution of 37 was used without isolation in the preparation of compound 38. 4. Preparation of Activated EG3 Tail (38)

To the solution of compound 37 was added N-hydroxy-5-norbornene-2,3-dicarboxylic acid imide (HONB) (1.02 eq), 4-dimethylaminopyridine (DMAP) (0.34 eq), and then l-(3- dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) (1.1 eq). The mixture was heated to 55° C. Upon reaction completion (4-5 hr), the mixture was cooled to 20° C and washed successively with 1 : 1 0.2 M citric acid/brine and brine. The dichloromethane solution underwent solvent exchange to acetone and then to Ν,Ν-dimethylformamide, and the product was isolated by precipitation from acetone/N,N-dimethylformamide into saturated aqueous sodium chloride. The crude product was reslurried several times in water to remove residual Ν,Ν-dimethylformamide and salts. Yield=70% of Activated EG3 Tail 38 from compound 36.
Example 4: 50L Solid-phase Synthesis of
Golodirsen [Oligomeric Compound (XII)] Crude Drug Substance
1. Materials
Table 2: Starting Materials

Activated Phosphoramidochloridic acid, 1155373-31-1 C37H37CIN5O5P 698.2 C Subunit N,N-dimethyl-,[6-[4-
(benzoylamino)-2-oxo-l(2H)- pyrimidinyl]-4-
(triphenylmethyl)-2- morpholinyljmethyl ester
Activated Propanoic Acid, 2,2-dimethyl- 1155309-89-9 C5iH53ClN707P 942.2
DPG ,4-[[[9-[6-
Subunit [[[chloro(dimethylamino)phosp
hinyl]oxy]methyl]-4-
(triphenylmethyl)-2- morpholinyl]-2-[(2- phenylacetyl)amino]-9H-purin-
6-yl]oxy]methyl]phenyl ester
Activated Phosphoramidochloridic acid, 1155373-34-4 C3iH34ClN405P 609.1 T Subunit N,N-dimethyl-,[6-(3,4-dihydro- 5-methyl-2,4-dioxo- 1 (2H)- pyrimidinyl)]-4- (triphenylmethyl)-2- morpholinyljmethyl ester
Activated Butanedioic acid, 1- 1380600-06-5 C43H47N3Oio 765.9 EG3 Tail [3aR,4S,7R,7aS)-l,3,3a,4,7,7a- hexahydro- 1 ,3 -dioxo-4,7- methano-2H-isoindol-2-yl] 4- [2-[2-[2-[[[4-(triphenylmethyl)- 1- piperazinyl ] carb onyl ] oxy] ethox
y]ethoxy] ethyl] ester
Golodirsen.
Example 5: Purification of Golodirsen Crude Drug Substance
The deprotection solution from Example 4, part E, containing the Golodirsen crude drug substance was loaded onto a column of ToyoPearl Super-Q 650S anion exchange resin (Tosoh Bioscience) and eluted with a gradient of 0-35% B over 17 column volume (Buffer A: 10 mM sodium hydroxide; Buffer B: 1 M sodium chloride in 10 mM sodium hydroxide) and fractions of acceptable purity (CI 8 and SCX HPLC) were pooled to a purified drug product solution. HPLC: 93.571% (C18; Fig. 3) 88.270% (SCX; Fig. 4).
The purified drug substance solution was desalted and lyophilized to 1450.72 g purified Golodirsen drug substance. Yield 54.56 %; HPLC: 93.531% (Fig. 5; C18) 88.354% (Fig. 6; SCX).
PATENT
WO 2019067979
Duchenne Muscular Dystrophy (DMD) is a serious, progressively debilitating, and ultimately fatal inherited X-linked neuromuscular disease. DMD is caused by mutations in the dystrophin gene characterized by the absence, or near absence, of functional dystrophin protein that disrupt the mRNA reading frame, resulting in a lack of dystrophin, a critically important part of the protein complex that connects the cytoskeletal actin of a muscle fiber to the extracellular matrix. In the absence of dystrophin, patients with DMD follow a predictable disease course. Affected patients, typically boys, develop muscle weakness in the first few years of life, lose the ability to walk during childhood, and usually require respiratory support by their late teens. Loss of functional abilities leads to loss of independence and increasing caregiver burden. Once lost, these abilities cannot be recovered. Despite improvements in the standard of care, such as the use of glucocorticoids, DMD remains an ultimately fatal disease, with patients usually dying of respiratory or cardiac failure in their mid to late 20s.
Progressive loss of muscle tissue and function in DMD is caused by the absence or near absence of functional dystrophin; a protein that plays a vital role in the structure and function of muscle cells. A potential therapeutic approach to the treatment of DMD is suggested by Becker muscular dystrophy (BMD), a milder dystrophinopathy. Both dystrophinopathies are caused by mutations in the DMD gene. In DMD, mutations that disrupt the pre-mRNA reading frame,
referred to as “out-of-frame” mutations, prevent the production of functional dystrophin. In BMD, “in-frame” mutations do not disrupt the reading frame and result in the production of internally shortened, functional dystrophin protein.
An important approach for restoring these “out-of-frame” mutations is to utilize an antisense oligonucleotide to exclude or skip the molecular mutation of the DMD gene
(dystrophin gene). The DMD or dystrophin gene is one of the largest genes in the human body and consists of 79 exons. Antisense oligonucleotides (AONs) have been specifically designed to target specific regions of the pre-mRNA, typically exons to induce the skipping of a mutation of the DMD gene thereby restoring these out-of-frame mutations in-frame to enable the production of internally shortened, yet functional dystrophin protein.
The skipping of exon 53 in the dystrophin gene has been an area of interest for certain research groups due to it being the most prevalent set of mutations in this disease area, representing 8% of all DMD mutations. A prominent AON being developed by Sarepta
Therapeutics, Inc., for DMD patients that are amenable to exon 53 skipping is golodirsen.
Golodirsen is a phosphorodiamidate morpholino oligomer, or PMO. Another AON being developed by Nippon Shinyaku CO., LTD., for DMD patients that are amenable to exon 53 skipping is viltolarsen (NS-065 which is a PMO.
Exondys 51 ® (eteplirsen), is another PMO that was approved in 2016 by the United States Food and Drug Administration (FDA) for the treatment of Duchenne muscular dystrophy (DMD) in patients who have a confirmed mutation of the DMD gene that is amenable to exon 51 skipping. However, the current standard of care guidelines for the treatment of DMD in patients that are not amenable to exon 51 skipping include the administration of glucocorticoids in conjunction with palliative interventions. While glucocorticoids may delay the loss of ambulation, they do not sufficiently ameliorate symptoms, modify the underlying genetic defect or address the absence of functional dystrophin characteristic of DMD.
Previous studies have tested the efficacy of an antisense oligonucleotides (AON) for exon skipping to generate at least partially functional dystrophin in combination with a steroid for reducing inflammation in a DMD patient (see WO 2009/054725 and van Deutekom, et al., N. Engl. J. Med. 2007; 357:2677-86, the contents of which are hereby incorporated herein by reference for all purposes). However, treatment with steroids can result in serious complications, including compromise of the immune system, reduction in bone strength, and growth
suppression. Notably, none of the previous studies suggest administering an antisense
oligonucleotide for exon skipping with a non-steroidal anti-inflammatory compound to a patient for the treatment of DMD.
Thus, there remains a need for improved methods for treating muscular dystrophy, such as DMD and BMD in patients.
EXAMPLE 1
CAT- 1004 in Combination with M23D PMO Reduces Inflammation and Fibrosis in Mdx Mice.
To assess the effectiveness of a combination treatment of an exon skipping antisense oligonucleotide and an F-Kb inhibitor in Duchenne muscular dystrophy, M23D PMO and
CAT-1004 were utilized in the Mdx mouse model. The effect on inflammation and fibrosis was determined by analyzing samples of muscle taken from the quadriceps, of (1) wild-type mice treated with saline, (2) mdx mice treated with saline, (3) mdx mice treated with CAT-1004, (4) mdx mice treated with the M23D PMO, and (5) mdx mice treated with the M23D PMO in combination with CAT-1004. The tissue sections were analyzed for fibrosis by picrosirius red staining and for inflammation and fibrosis by Hematoxylin and Eosin (H&E) staining, as described in the Materials and Methods section above.
Treatment of Mdx mice with either M23D PMO or CAT-1004 as monotherapies resulted in a reduction of inflammation and fibrosis as compared to Mdx mice treated with saline.
Surprisingly, treatment of Mdx mice with the M23D PMO in combination with CAT-1004 resulted in reduced inflammation and fibrosis as compared with mice treated with CAT-1004
alone or M23D alone (Fig. 9). These results indicate the combination treatment enhances muscle fiber integrity.
EXAMPLE 2
Exon Skipping and Dystrophin Production in Mdx Mice Treated with the M23D
PMO and the M23D PMO in Combination with CAT- 1004
To analyze the extent of exon skipping and dystrophin production in mice treated with the M23D PMO in combination with CAT- 1004, samples of muscle were taken from the quadriceps, diaphragm, and heart of (1) wild-type mice treated with saline, (2) mdx mice treated with saline, (3) mdx mice treated with CAT- 1004, (4) mdx mice treated with the M23D PMO, and (5) mdx mice treated with the M23D PMO in combination with CAT- 1004. RT-PCR analysis for exon 23 skipping was performed as well as Western blot analysis to determine dystrophin protein levels.
Exon skipping was observed in the muscle of the quadriceps, diaphragm, and heart of the Mdx mice treated with the M23D PMO as well as mice treated with the M23D PMO in combination with CAT-1004 (Fig. 10). Surprisingly, enhanced dystrophin production was observed in the muscle of the quadriceps, diaphragm, and heart of the mice treated with the M23D PMO in combination with CAT-1004 as compared to treatment with M23D PMO monotherapy (Fig. 11). These results indicated the increase in dystrophin levels extended to the heart, a tissue known to have low efficiency of dystrophin upregulation by these agents when used alone. Notably, neither exon skipping nor dystrophin production were observed in mdx mice treated with CAT-1004 monotherapy (Figs. 10 and 11).
PATENT
WO 2019046755
PAPER
Methods in Molecular Biology (New York, NY, United States) (2018), 1828(Exon Skipping and Inclusion Therapies), 31-55.
PAPER
Human Molecular Genetics (2018), 27(R2), R163-R172.
///////////Golodirsen, ゴロジルセン , FDA 2019, ANTISENSE, Exon 53: NG-12-0163, SRP 4053, OLIGONUCLEOTIDE, Duchenne Muscular Dystrophy
FDA approves novel treatment Oxbryta (voxelotor) to target abnormality in sickle cell disease
Sickle cell disease is a lifelong, inherited blood disorder in which red blood cells are abnormally shaped (in a crescent, or “sickle” shape), which restricts the flow in blood vessels and limits oxygen delivery to the body’s tissues, leading to severe pain and organ damage. It is also characterized by severe and chronic inflammation that worsens vaso-occlusive crises during which patients experience episodes of extreme pain and organ damage. Nonclinical studies have demonstrated that Oxbryta inhibits red blood cell sickling, improves red blood cell deformability (ability of a red blood cell to change shape) and improves the blood’s ability to flow.
“Oxbryta is an inhibitor of deoxygenated sickle hemoglobin polymerization, which is the central abnormality in sickle cell disease,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research. “With Oxbryta, sickle cells are less likely to bind together and form the sickle shape, which can cause low hemoglobin levels due to red blood cell destruction. This therapy provides a new treatment option for patients with this serious and life-threatening condition.”
Oxbryta’s approval was based on the results of a clinical trial with 274 patients with sickle cell disease. In the study, 90 patients received 1500 mg of Oxbryta, 92 patients received 900 mg of Oxbryta and 92 patients received a placebo. Effectiveness was based on an increase in hemoglobin response rate in patients who received 1500 mg of Oxbryta, which was 51.1% for these patients compared to 6.5% in the placebo group.
/////////fda 2019, Fast Track designation, Oxbryta, Orphan Drug designation, voxelotor, Global Blood Therapeutics, sickle cell disease
FDA approves new treatment XCOPRI (cenobamate tablets) for adults with partial-onset seizures
FDA approves first treatment Givlaari (givosiran) for inherited rare disease
///////////Givlaari, givosiran, fda 2019, Breakthrough Therapy designation, Priority Review, Orphan Drug

{kind=link}