
Home » FDA 2019 (Page 2)
Category Archives: FDA 2019
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Afamelanotide, アファメラノチド , афамеланотид , أفاميلانوتيد , 阿法诺肽 ,

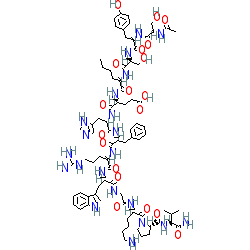
Afamelanotide
RN: 75921-69-6
Molecular Formula, C78-H111-N21-O19, Molecular Weight, 1646.8629
Synonyms
- 75921-69-6
- AFAMELANOTIDE [MI]
- AFAMELANOTIDE
- AFAMELANOTIDE [INN]
- .ALPHA.-MELANOTROPIN (SWINE), 4-L-NORLEUCINE-7-D-PHENYLALANINE-
- AC-SER-TYR-SER-NLE-GLU-HIS-D-PHE-ARG-TRP-GLY-LYS-PRO-VAL-NH2
- CUV1647
- AFAMELANOTIDE [WHO-DD]
- AFAMELANOTIDE [USAN]
- ACETYL(4-(2S)-2-AMINOHEXANOIC ACID,7-D-PHENYLALANINE)HUMAN MELANOTROPIN ALPHA
- CUV-1647
- MELANOTAN I
- MELANOTAN-1
alpha-Melanotropin, 4-L-norleucine-7-D-phenylalanine-
Prevention of Phototoxicity in Adults with Erythropoietic Protoporphyria (EPP)
| UNII: | QW68W3J66U | |
アファメラノチド;
Observations suggest that afamelanotide has beneficial effects in patients with erythropoietic protoporphyria, induces epidermal melanin formation.
SYN
Lensing, Cody J. et alFrom Journal of Medicinal Chemistry, 62(1), 144-158; 2019

FDA APPROVED
oct 2019
FDA approves first treatment to increase pain-free light exposure in patients with a rare disorder
The U.S. Food and Drug Administration today granted approval to Scenesse (afamelanotide) to increase pain-free light exposure in adult patients with a history of phototoxic reactions (damage to skin) from erythropoietic protoporphyria.
For patients who are suffering from erythropoietic protoporphyria, a rare disorder, exposure to light may be extremely painful. Prior to today’s approval, there were no FDA-approved treatments to help erythropoietic protoporphyria patients increase their light exposure,” said Julie Beitz, M.D., director of FDA’s Center for Drug Evaluation and Research Office of Drug Evaluation III. “Today’s approval is one example of the FDA’s ongoing commitment to encourage industry innovation of therapies to treat rare diseases, and work with drug developers to make promising new therapies available to patients as safely and efficiently as possible.”
Erythropoietic protoporphyria is a rare disorder caused by mutations leading to impaired activity of ferrochelatase, an enzyme involved in heme production. Heme is an important component in hemoglobin, the oxygen carrying molecule in red blood cells. The decrease in ferrochelatase activity leads to an accumulation of protoporphyrin IX (PPIX) in the body. Light reaching the skin can react with PPIX causing intense skin pain and skin changes, such as redness and thickening. Scenesse (afamelanotide), a melanocortin-1 receptor (MC1-R) agonist, increases the production of eumelanin in the skin independent of exposure to sunlight or artificial light sources. It is an implant that is administered subcutaneously (inserted under the skin).
The efficacy of Scenesse was established in two parallel group clinical trials with patients with erythropoietic protoporphyria who received Scenesse or placebo form of the implant subcutaneously every two months. The first clinical trial enrolled 93 subjects, of whom 48 received Scenesse, and were followed for 180 days. The primary endpoint was the total number of hours over 180 days spent in direct sunlight between 10 a.m. and 6 p.m. on days with no pain. The median total number of hours over 180 days spent in direct sunlight between 10 a.m. and 6 p.m. on days with no pain was 64 hours for patients receiving Scenesse and 41 hours for patients taking placebo.
The second clinical trial enrolled 74 patients, of whom 38 received Scenesse, and were followed for 270 days. The primary endpoint was the total number of hours over 270 days spent outdoors between 10 am and 3 pm on days with no pain for which “most of the day” was spent in direct sunlight. The analysis did not include sun exposure on days patients reported spending time in a combination of both direct sunlight and shade. The median total number of hours over 270 days spent outdoors between 10 am and 3 pm on days with no pain for which “most of the day” was spent in direct sunlight was six hours for patients receiving Scenesse and 0.75 hours for patients receiving placebo.
Scenesse’s most common side effects are implant site reaction, nausea, oropharyngeal (part of the throat just behind the mouth, where the oral cavity starts) pain, cough, fatigue, skin hyperpigmentation, dizziness, melanocytic nevus (moles), respiratory tract infection, somnolence (feeling drowsy), non-acute porphyria (build-up of normally occurring molecules created during heme production) and skin irritation. Scenesse should be administered by a health care professional who is proficient in the subcutaneous implantation procedure and has completed the applicant-provided training. Scenesse may induce skin darkening, and a full body skin examination is recommended for patients twice a year. In addition, patients are encouraged to maintain sun protection measures during treatment with Scenesse to prevent phototoxic reactions related to erythropoietic protoporphyria.
The FDA granted this application Priority Review designation. Scenesse also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases.The approval of Scenesse was granted to Clinuvel.
For more information:
- NIH: Erythropoietic protoporphyria
- FDA: Fast Track, Breakthrough Therapy, Accelerated Approval, and Priority Review
Afamelanotide (melanotan I, CUV1647; brand name Scenesse)[2] is a synthetic peptide and analogue of α-melanocyte stimulating hormone used to prevent skin damage from the sun in people with erythropoietic protoporphyria in Europe since January 2015. It is administered as an implant that is placed under the skin; the implant lasts for two months.
It is under development in other skin disorders in several jurisdictions. It causes skin to turn darker by causing the skin to make more melanin.
It was discovered at University of Arizona and initially developed there as a sunless tanning agent; the Australian company Clinuvel conducted further clinical trials in that and other indications, and brought the drug to market.
Unlicensed and untested powders sold as “melanotan” are found on the Internet marketed for tanning and other purposes, and multiple regulatory bodies have warned consumers that the peptides may be unsafe and ineffective.
Medical use
Afamelanotide is used in Europe to prevent phototoxicity in adults with erythropoietic protoporphyria (EPP).[1] It is an implant that is injected and placed under the skin; an implant lasts two months.[1]
People who have severe liver disease, liver impairment, or kidney impairment, should not use this drug. Pregnant women should not take it, and women who are active sexually should use contraception while they are taking it. It is not known if afamelanotide is secreted in breast milk.[1]
Adverse effects
Very common (up to 10% of people) adverse effects in people with EPP include headache and nausea. Common (between 1% and 10%) adverse effects include back pain, upper respiratory tract infections, decreased appetite, migraine, dizziness, weakness, fatigue, lethargy, sleepiness, feeling hot, stomach pain, diarrhea, vomiting, flushing and red skin, development of warts, spots, and freckles, itchy skin, and reactions at the injection site. There are many uncommon (less than 1%) adverse effects.[1]
Pharmacology
Afamelanotide is thought to cause skin to darken by binding to the melanocortin 1 receptor which in turn drives melanogenesis.[1]
Afamelanotide has a half-life of 30 minutes. After the implant is injected, most of the drug is released within the first 2 days, with 90% released by the fifth day. By the tenth day no drug is detectable in plasma.[1]
Its metabolites, distribution, metabolism and excretion were not understood as of 2017.[1]
Chemistry
The amino acid sequence is Ac-Ser-Tyr-Ser-Nle-Glu-His-D-Phe-Arg-Trp-Gly-Lys-Pro-Val-NH2, and it is additionally known as [Nle4,D-Phe7]-α-MSH, which is sometimes abbreviated as NDP-MSH or NDP-α-MSH. Afamelanotide is the International Nonproprietary Name.[3]
History
The role of α-MSH in promoting melanin diffusion has been known since the 1960s.[4] In the 1980s, scientists at University of Arizona began attempting to develop α-MSH and analogs as potential sunless tanning agents, and synthesized and tested several analogs, including melanotan-I.[5]
To pursue the tanning agent, melanotan-I was licensed by Competitive Technologies, a technology transfer company operating on behalf of University of Arizona, to an Australian startup called Epitan,[6][5] which changed its name to Clinuvel in 2006.[7]
Early clinical trials showed that the peptide had to be injected about ten times a day due to its short half-life, so the company collaborated with Southern Research in the US to develop a depot formulation that would be injected under the skin, and release the peptide slowly. This was done by 2004.[6]
As of 2010, afamelanotide was in Phase III trials for erythropoietic protoporphyria and polymorphous light eruption, and was in Phase II trials for actinic keratosis and squamous cell carcinoma, and had been trialled in phototoxicity associated with systemic photodynamic therapy and solar urticaria.[8] Clinuvel had also obtained orphan drug status for afamelanotide in the US and the EU by that time.[8]
In May 2010 the Italian Medicines Agency (AIFA, or Agenzia Italiana del Farmaco) approved afamelanotide as a treatment for erythropoietic protoporphyria.[9]
In January 2015 afamelanotide was approved by the EMA in Europe for the treatment of phototoxicity in people with EPP.[1]
Society and culture
Counterfeits
A number of products are sold online and in gyms and beauty salons as “melanotan” or “melanotan-1” which discuss afamelanotide in their marketing.[10][11] [12]
The products are not legal in any jurisdiction and are dangerous.[13][14][15][16]
Starting in 2007 health agencies in various counties began issuing warnings against their use.[17][18][19][20] [21][22]
PAPERS
1 Sawyer T K; Sanfilippo P J; Hruby V J; Engel M H; Heward C B; Burnett J B; Hadley M E
- From Proceedings of the National Academy of Sciences of the United States of America (1980), 77(10), 5754-8.
2 Journal of Medicinal Chemistry (1982), 25(9), 1022-7.
3 Journal of medicinal chemistry (1984), 27(11), 1406-10.
4 Journal of Medicinal Chemistry (2019), 62(1), 144-158
5 Journal of Medicinal Chemistry (2018), 61(17), 7729-7740.
6 Journal of medicinal chemistry (2017), 60(2), 805-813.
PATENT
US 4457864
References
- ^ Jump up to:a b c d e f g h i “Scenesse: Summary of Product Characteristics” (PDF). EMA. 27 January 2016. Retrieved 6 April 2017. For updates see EMA Index page
- ^ “Afamelanotide”. AdisInsight. Retrieved 6 April 2017.
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN)” (PDF). World Health Organization. 2009. Retrieved 2009-03-02.
- ^ Baker, BI (31 May 1993). “The role of melanin-concentrating hormone in color change”. Annals of the New York Academy of Sciences. 680: 279–89. doi:10.1111/j.1749-6632.1993.tb19690.x. PMID 8390154.
- ^ Jump up to:a b Hadley, ME; Dorr, RT (April 2006). “Melanocortin peptide therapeutics: historical milestones, clinical studies and commercialization”. Peptides. 27 (4): 921–30. doi:10.1016/j.peptides.2005.01.029. PMID 16412534.
- ^ Jump up to:a b “EpiTan focuses on Melanotan, a potential blockbuster”. The Pharma Letter. 1 November 2004.
- ^ “Epitan changes name to Clinuvel, announces new clinical program”. LabOnline. 27 February 2006.
- ^ Jump up to:a b Dean, Tim (3 May 2010). “Biotechnology profile: Bright future for Clinuvel (ASX:CUV)”. Australian Life Scientist. Archived from the original on 6 April 2017.
- ^ “GAZZETTA UFFICIALE: SOMMARIO”. Agenzia Nazionale Stampa Associata. 2010. Retrieved 2010-05-17.
- ^ “Believe It Or Not ‘Tanorexia’ A Very Real Problem”. WCBS-TV, CBS. 2009-05-20. Archived from the original on May 21, 2009. Retrieved 2009-07-23.
- ^ “Fools Gold”. Cosmopolitan (Australia). 2009-06-14. Retrieved 2009-07-25.
- ^ Madrigal, Alexis (2009-01-29). “Suntan Drug Greenlighted for Trials”. Wired. Archivedfrom the original on 5 May 2009. Retrieved 2009-04-11.
- ^ “Tanning drug a health risk”. Herald Sun. 2009-10-31. Retrieved 2009-10-31.
- ^ Ewan A Langan; Z. Nie; Lesley E Rhodes (June 2010). “Melanotropic peptides: More than just “Barbie drugs” and “sun tan jabs?““. British Journal of Dermatology. 163 (3): 451–5. doi:10.1111/j.1365-2133.2010.09891.x. PMID 20545686.
- ^ Ewan A Langan; Denise Ramlogan; Lynne A Jamieson; Lesley E Rhodes (January 2009). “Change in moles linked to use of unlicensed “sun tan jab““. BMJ. 338: b277. doi:10.1136/bmj.b277. PMID 19174439.
- ^ “Risky tan jab warnings ‘ignored‘“. BBC. 2009-02-18. Archived from the original on 21 February 2009. Retrieved 2009-03-04.
- ^ “Warning against the product Melanotan”. Danish Medicines Agency. 2008. Retrieved 2008-08-11.
- ^ ““Tan jab” is an unlicensed medicine and may not be safe”. MHRA. 2008. Archived from the original on 2014-12-05. Retrieved 2008-11-17.
- ^ “US Lab Research Inc Warning letter”. U.S. Food and Drug Administration. 2009-01-29. Archived from the original on 10 July 2009. Retrieved 2009-07-23.
- ^ “Melanotan Powder for Injection”. Notice Information: – Warning – 27 February 2009. Irish Medicines Board. 2009. Retrieved 2009-02-02.
- ^ “Legemiddelverket advarer mot bruk av Melanotan”. Norwegian Medicines Agency. 2007-12-13. Archived from the original on 17 April 2009. Retrieved 2009-03-11.
- ^ “Melanotan – farlig og ulovlig brunfarge”. Norwegian Medicines Agency. 2009-01-23. Archived from the original on 17 April 2009. Retrieved 2009-03-11.
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ˌæfəmɛˈlænoʊtaɪd/ ( |
| Trade names | Scenesse |
| Synonyms | Melanotan; Melanotan-1; Melanotan I; CUV1647; EPT1647; NDP-MSH; NDP-α-MSH; [Nle4,D-Phe7]α-MSH |
| AHFS/Drugs.com | UK Drug Information |
| License data | |
| Routes of administration |
S.C.; I.M.; I.V.; subcutaneous implant; intranasal |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Elimination half-life | 30 minutes[1] |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C78H111N21O19 |
| Molar mass | 1646.845 g/mol g·mol−1 |
| 3D model (JSmol) | |
|
|
/////////////fda 2019, Scenesse, afamelanotide, pain-free light exposure, erythropoietic protoporphyria, アファメラノチド , афамеланотид , أفاميلانوتيد , 阿法诺肽 ,
Afamelanotide acetate [USAN]
1566590-77-9
MW: 1706.9145
Edotreotide gallium Ga-68
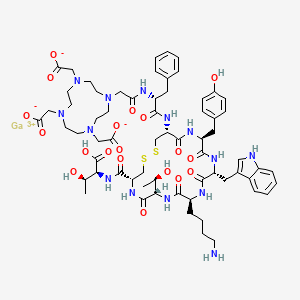



Edotreotide gallium Ga-68
エドトレオチドガリウム (68Ga);
- Edotreotide Gallium Ga-68
- Gallium (68Ga) edotreotide
- Gallium edotreotide Ga-68
- Gallium Ga 68-dotatoc
- Gallium Ga 68-edotreotide
- Gallium Ga-68 edotreotide
gallium;2-[4-[2-[[(2R)-1-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-4-[[(1S,2R)-1-carboxy-2-hydroxypropyl]carbamoyl]-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-2-oxoethyl]-7,10-bis(carboxylatomethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetate
L-threonine, N-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-19-[[(2R)-1-oxo-3-phenyl-2-[[2-[4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododec-1-yl]acetyl]amino]propyl]amino]-1,2-dithia-5,8,11,14,17-pentaazacycloeicos-4-yl]carbonyl]-, gallium salt (1:1)
| Formula |
C65H92N14O18S2. Ga
|
|---|---|
| CAS |
1027785-90-5
|
| Mol weight |
1491.362
|
FDA, 2019/8/21 APPROVED Ga-68-dotatoc
UNII Y68179SY2L
Indicated for use with positron emission tomography (PET) for the localization of somatostatin receptor positive neuroendocrine tumors (NETs)
|
Diagnostic aid (tumor), Radioactive agent
|
Edotreotide gallium Ga-68 is an 8 amino acid peptide bound to the chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA).8 Edotreotide gallium Ga-68 is indicated for localizing somatostatin receptor positive neuroendocrine tumors by positron emission tomography.7 Dotatate gallium Ga-68 is used for a similar indication.2 Dotatate gallium Ga-68 has lower tumor uptake but this data is highly variable between patients.2
Edotreotide gallium Ga-68 was granted FDA approval on 21 August 2019.7
Indication
Edotreotide gallium Ga-68 is a radioactive diagnostic compound used in positron emission tomography (PET) for diagnose somatostatin receptor positive neuroendocrine tumors in pediatrics and adults.7
Associated Conditions
Pharmacodynamics
Edotreotide gallium Ga-68 binds to somatostatin receptors where it emits beta particle radiation for detection by positron emission tomography.7 The duration of action is short as it has short radioactive and biological half lives.4,8 Patients should hydrate before and after the administration of this medication to encourage frequent urination and rapid clearance.7
Mechanism of action
Edotreotide gallium Ga-68 binds to somatostatin receptors, with higher affinity for somatostatin receptor type 2, where it emits beta particle radiation for detection by positron emission tomography (PET).7
Absorption
Edotreotide gallium Ga-68 reaches 80% activity in tumors within 30 minutes,4 and reaches its highest activity in tumors 70±20min post injection.8 Edotreotide is mostly taken up into the spleen, followed by kidneys, liver, pituitary, thyroid, and adrenal gland.3,4,8 Accumulation in non-tumor tissue reaches a maximum within 40 minutes.4
Volume of distribution
Data regarding the volume of distribution of this medication is not readily available.7
Protein binding
Data suggests edotreotide gallium-Ga 68 may bind to proteins in serum.6 The extent of serum protein binding and which proteins it binds to are not described in the literature.
Metabolism
Edotreotide gallium Ga-68 is largely unmetabolized.8 4 hours post injection there are no metabolites or degradation products detectable in serum.4
Route of elimination
16% of a Edotreotide gallium Ga-68 dose is eliminated in the urine within 2h.1,7 It is expected that Edotreotide gallium Ga-68 is exclusively eliminated in the urine.1,7 In animal studies, edotreotide Y-90 was >80% eliminated in the urine within 24h, with 95.6±3.4% being unmetabolized.3,8 <1% of a dose is detected in the feces.5
Half life
Edotreotide gallium Ga-68 has a radioactive half life of 68 minutes.4,8 Edotreotide gallium Ga-68 has two half lives, 2.0±0.3min and 48±7min for its removal from blood.4,8
Clearance
Data regarding the clearance of this medication is not readily available.7
Toxicity
The LD50 of this medication is not readily available.7
In the event of an overdose, give patients plenty of fluids and diuretics if necessary to encourage frequent urination.7 If possible, an estimation of radioactive dose should be performed.7
General References
- Hartmann H, Zophel K, Freudenberg R, Oehme L, Andreeff M, Wunderlich G, Eisenhofer G, Kotzerke J: [Radiation exposure of patients during 68Ga-DOTATOC PET/CT examinations]. Nuklearmedizin. 2009;48(5):201-7. doi: 10.3413/nukmed-0214. Epub 2009 Jul 28. [PubMed:19639164]
- Poeppel TD, Binse I, Petersenn S, Lahner H, Schott M, Antoch G, Brandau W, Bockisch A, Boy C: 68Ga-DOTATOC versus 68Ga-DOTATATE PET/CT in functional imaging of neuroendocrine tumors. J Nucl Med. 2011 Dec;52(12):1864-70. doi: 10.2967/jnumed.111.091165. Epub 2011 Nov 9. [PubMed:22072704]
- de Jong M, Bakker WH, Krenning EP, Breeman WA, van der Pluijm ME, Bernard BF, Visser TJ, Jermann E, Behe M, Powell P, Macke HR: Yttrium-90 and indium-111 labelling, receptor binding and biodistribution of [DOTA0,d-Phe1,Tyr3]octreotide, a promising somatostatin analogue for radionuclide therapy. Eur J Nucl Med. 1997 Apr;24(4):368-71. doi: 10.1007/bf00881807. [PubMed:9096086]
- Hofmann M, Maecke H, Borner R, Weckesser E, Schoffski P, Oei L, Schumacher J, Henze M, Heppeler A, Meyer J, Knapp H: Biokinetics and imaging with the somatostatin receptor PET radioligand (68)Ga-DOTATOC: preliminary data. Eur J Nucl Med. 2001 Dec;28(12):1751-7. doi: 10.1007/s002590100639. Epub 2001 Oct 31. [PubMed:11734911]
- Kwekkeboom DJ, Kooij PP, Bakker WH, Macke HR, Krenning EP: Comparison of 111In-DOTA-Tyr3-octreotide and 111In-DTPA-octreotide in the same patients: biodistribution, kinetics, organ and tumor uptake. J Nucl Med. 1999 May;40(5):762-7. [PubMed:10319747]
- Bangard M, Behe M, Guhlke S, Otte R, Bender H, Maecke HR, Biersack HJ: Detection of somatostatin receptor-positive tumours using the new 99mTc-tricine-HYNIC-D-Phe1-Tyr3-octreotide: first results in patients and comparison with 111In-DTPA-D-Phe1-octreotide. Eur J Nucl Med. 2000 Jun;27(6):628-37. doi: 10.1007/s002590050556. [PubMed:10901448]
- FDA Approved Drug Products: Gallium Dotatoc GA 68 [Link]
- EMA Assessment Report: SomaKit TOC [Link]
///////////Edotreotide gallium Ga-68, FDA 2019, エドトレオチドガリウム (68Ga),
 |
|
| Names | |
|---|---|
| IUPAC name
2-[4-[2-[[(2R)-1-[[(4R,7S,10S,13R,16S,19R)-10-(4-aminobutyl)-4-[[(2R,3R)-1,3-dihydroxybutan-2-yl]carbamoyl]-7-[(1R)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1H-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-2-oxoethyl]-7,10-bis(carboxymethyl)-1,4,7,10-tetrazacyclododec-1-yl]acetic acid
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C65H92N14O18S2 | |
| Molar mass | 1421.65 g·mol−1 |
| Pharmacology | |
| License data | |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
Edotreotide (USAN, codenamed SMT487, also known as (DOTA0–Phe1–Tyr3)octreotide, or DOTATOC) is a substance which, when bound to various radionuclides, is used in the treatment and diagnosis of certain types of cancer.[1]
Yttrium-90 labeled edoteotide has been the subject of a trial by the National Cancer Institute to determine its effects in young cancer patients (up to 25 years of age) for its ability to locate malignant cancer cells without harming normal cells. Specific cancers being included in the trial include neuroblastoma, childhood brain tumours and gastrointestinal cancer.[2][3]
-

Yttrium-90 labeled edotreotide

References
- ^ Martindale, The Extra Pharmacopoeia, 30th ed, p1161.
- ^ Bushnell, D. L.; O’Dorisio, T. M.; O’Dorisio, M. S.; Menda, Y.; Hicks, R. J.; Van Cutsem, E.; Baulieu, J. -L.; Borson-Chazot, F.; Anthony, L.; Benson, A. B.; Oberg, K.; Grossman, A. B.; Connolly, M.; Bouterfa, H.; Li, Y.; Kacena, K. A.; Lafrance, N.; Pauwels, S. A. (2010). “90Y-Edotreotide for Metastatic Carcinoid Refractory to Octreotide”. Journal of Clinical Oncology. 28 (10): 1652–1659. doi:10.1200/JCO.2009.22.8585. PMC 4872330. PMID 20194865.
- ^ Radiolabeled Octreotide in Treating Children With Advanced or Refractory Solid Tumors
Cilastatin, циластатин , سيلاستاتين , 西司他丁 ,
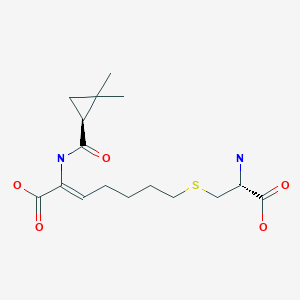
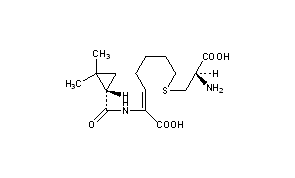

Cilastatin
シラスタチン
UNII141A6AMN38
CAS number 82009-34-5
WeightAverage: 358.453
Monoisotopic: 358.156242642
Chemical FormulaC16H26N2O5S
- (L)-7-(2-Amino-2-carboxy-ethylsulfanyl)-2-[(2,2-dimethyl-cyclopropanecarbonyl)-amino]-hept-2-enoic acid
- (Z)-(S)-6-carboxy-6-[(S)-2,2-dimethylcyclopropanecarboxamido]hex-5-enyl-L-cysteine
- (Z)-7-((R)-2-Amino-2-carboxy-ethylsulfanyl)-2-[((S)-2,2-dimethyl-cyclopropanecarbonyl)-amino]-hept-2-enoic acid
- (2Z)-7-{[(2R)-2-amino-2-carboxyethyl]sulfanyl}-2-{[(1S)-2,2-dimethylcyclopropyl]formamido}hept-2-enoic acid
FDA 2019 APPROVED 2019/7/16, Imipenem, cilastatin and relebactam, Recarbrio
|
Antibacterial
|
|
| Disease |
Uncomplicated urinary tract infection
|
|---|
Cilastatin inhibits the human enzyme dehydropeptidase.[1]
Yatendra Kumar, “Process for the preparation of amorphous cilastatin sodium.” U.S. Patent US20040152780, issued August 05, 2004.US20040152780
Cilastatin is an inhibitor of renal dehydropeptidase, an enzyme responsible for both the metabolism of thienamycin beta-lactam antibiotics as well as conversion of leukotriene D4 to leukotriene E4. Since the antibiotic, imipenem, is one such antibiotic that is hydrolyzed by dehydropeptidase, cilastatin is used in combination with imipenem to prevent its metabolism. The first combination product containing both drugs was approved by the FDA in November of 1985 under the trade name Primaxin, marketed by Merck & Co.9 A newer triple-drug product was approved in July 2019 under the trade name Recarbrio which also contains relebactam.8
Cilastatin is indicated, in combination with imipenem with or without relebactam, for the treatment of bacterial infections including respiratory, skin, bone, gynecologic, urinary tract, and intra-abdominal as well as septicemia and endocarditis.6,5

Uses
Dehydropeptidase is an enzyme found in the kidney and is responsible for degrading the antibiotic imipenem. Cilastatin can therefore be combined intravenously with imipenem in order to protect it from degradation, prolonging its antibacterial effect.
Imipenem alone is an effective antibiotic and can be given without cilastatin. Cilastatin itself does not have antibiotic activity, although it has been proved to be active against a zinc-dependent beta-lactamase that usually confers antibiotic resistance to certain bacteria, more precisely, the carbapenem family of antibiotics. This property is due to the physicochemical similarities between membrane dipeptidase (MDP), the compound it is usually set to target, and the bacterial metallo-beta-lactamase carried by the CphA gene.[1] The combination allows the antibiotic to be more effective by changing the pharmacokinetics involved. Thus imipenem/cilastatin, like amoxicillin/clavulanic acid, is a commonly used combination product.
PATENT
https://patents.google.com/patent/EP2402312A1
Cilastatin sodium is the sodium salt of a derivatized heptenoic acid. Its chemical name is [R-[R*,S*-(Z)]]-7-[(2-amino-2-carboxyethyl)thio]-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino]-2-heptenoic acid, monosodium salt. It is an off-white to yellowish-white, hygroscopic, amorphous compound. PRIMAXIN (Imipenem and Cilastatin) is a formulation of Imipenem (a thienamycin antibiotic) and Cilastatin sodium.
Imipenem with Cilastatin acts as an effective antibiotic for the treatment of infections of various body systems. PRIMAXIN is a potent broad-spectrum antibacterial agent for intramuscular administration. Imipenem can be further described as a semi-synthetic thienamycin that is administered intravenously or intramuscularly in combination with Cilastatin to reduce toxicity. Cilastatin, a renal dipeptidase inhibitor, inhibits the enzymatic breakdown of Imipenem and increases urinary excretion of the active drug.
Originally Cilastatin was disclosed in US patent number 5,147,868 . This patent also discloses various processes for the preparation of Cilastatin, particularly example 19 A of this patent disclose a process for the preparation of Cilastatin. According to this example the condensation of 7-chloro-2-oxoheptanoic acid ethyl ester (I) with (S)-2,2-dimethylcyclopropanecarboxamide (II) by means of p-toluene sulphonic acid in refluxing toluene gives (S)-7-chloro-2-(2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid ethyl ester (III), which is hydrolyzed in aq. NaOH to yield the corresponding carboxylic acid (IV). Finally, this compound is condensed with (R)-cysteine (V) by means of NaOH in water to afford the target Cilastatin, followed by isomerisation to at 3.0 pH. The process followed in this example is depicted as below:


Example 1Preparation of 7-chloro-2-[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid (II) (starting material):
- [0032]
To the solution of S-2, 2-dimethylcylopropyl carboxamide (100gm) in toluene (500) was added Ethyl-7-chloro-2-oxo-heptanoate (270gm) and p-toluene sulphonic acid (1.5gm). The resulted solution was refluxed for 20hrs azeotropically. The resulted mass was cooled to 5-10°C and added the solution of sodium hydroxide (140gm) in water 500 ml and the resulted two-layered solution was stirred for 8hrs at 25-30°C up to the complete disappearance of ester. The toluene layer was separated and the aqueous layer was washed with toluene. The pH of the aqueous layer was adjusted to 4.0 to 4.5 and extracted with toluene (1 lt). The toluene layer containing 7-chloro-2-[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid was washed with water and used as such for the next step. The ratio of Z and E isomer 90:10% was obtained.
Example 2Isomerisation of 7-chloro-2-[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid (II):
- [0033]
To the toluene layer, obtained from example -1, was added hydrochloric acid (11t) and stirred for 4hrs at 25-30°C till the disappearance of E isomer. The toluene layer was separated and washed with water and followed by brine. The toluene layer was distilled out under vacuum up to 50% of the original volume. To the reaction mass hexane/IPE was added at 50°C and cooled to 0-5°C. The precipitated mass was filtered and washed with hexane (200ml) and dried under vacuum to obtained 99% pure Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid (150gm) as white solid.
Example 3Preparation of Cilastatin Acid (I):
- [0034]
To the solution of sodium hydroxide (90gm) in water (11t) was added L-Cysteine hydrochloride monohydrate (96gm) and Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid and stirred at 25-30°C till the disappearance of Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid. After completion of reaction, the reaction mass was washed with dichloromethane (500ml). To the aqueous layer was added carbon (10 gm) and stirred and filtered. To the filtrate was added water (11t) and the pH of the solution was adjusted to 3.0 and stirred for 24 hrs. The precipitated mass was filtered, washed with water (200ml) and with acetone (500ml) and dried to obtain 110gm white solid with 97% purity. The solid was dissolved in water (700ml) and added MDC (700ml) and ethyl acetate (100ml) and stirred for 10hrs. The precipitated mass was filtered and washed with water (100ml) and acetone (200ml) and dried to obtain 100gm white Cilastatin acid with 99.5% purity.
Example 4Preparation of Cilastatin Sodium:
- [0035]
The Cilastatin acid (100gm, 99.5%) was dissolved in the mixture of ethanol (2.5lt) and triethylamine (30gm) at 25 to 30°C. To the resulted clear solution was added carbon (10gm) and stirred and filtered. The filtrated was filtered again through sterile micron (0.2 µ) filter. To the resulted clear solution was added solution of sodium ethyl hexanoate (70gm) in ethanol (70ml) and stirred for 3hrs at 25 to 30°C.The precipitated Cilastatin sodium was filtered and washed with ethanol (80ml) and followed by acetone (200ml) and dried under vacuum to obtained 95gm Cilastatin sodium as amorphous white solid with 99.5% purity.
Example 5Preparation of Cilastatin Acid:
- [0036]
To the solution of sodium hydroxide (88gm) in methanol (1500ml) was added Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid and stirred to dissolve. To the resulted clear solution was added L-Cysteine hydrochloride monohydrate (97gm) and stirred the resulted suspension at 60 to 65°C till the disappearance of Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid. After completion of reaction, the pH insoluble salts were filtered. The filtrate was distilled out under vacuum. The residue was dissolved in water (500ml) and washed with dichloromethane (500ml). The pH of aqueous layer was adjusted to 3 to 4 from the original pH in the range of 5.5, and with n-butanol (500ml). The butanol layer was washed with water and distilled. The residue was dissolved in water (100ml) and added acetonitrile (1500ml) at 50°C and further refluxed at 80°C for one hr. The precipitated cilastatin acid was filtered and washed with acetonitrile (100ml). The crude wet cake (60gm) was refluxed with acetonitrile water mixture (9:1,1500ml), and cooled to yield 60gm pure cilastatin acid with 99.5% purity.
Example 6Preparation of Cilastatin Acid:
- [0037]
To the solution of sodium hydroxide (88gm) in methanol (1500ml) was added Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid and stirred to dissolve. To the resulted clear solution was added L-Cysteine hydrochloride monohydrate (97gm) and stirred the resulted suspension at 60 to 65°C till the disappearance of Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid. The pH of the reaction mass was adjusted to 7.0 with conc.HCl and filterd the insoluble salts. The filtrated was distilled out under vacuum. The residue was dissolved in water (500ml) and washed with dichloromethane (500ml). The pH of aqueous layer was adjusted to 3 to 4 from the original pH in the range of 5.5, and with n-butanol (500ml). The butanol layer was washed with water and distilled up to 50% of original volume and stirred at 25°C. The precipitated cilastatin acid was filtered and washed with n-butanol (100ml) followed by acetone to yield 60gm pure cilastatin acid with 99.7% purity.
Example 7Preparation of Cilastatin, Sodium:
- [0038]
The Cilastatin acid (100gm, 99.5%) was dissolved in the mixture of n-butanol (2.5lt) and triethylamine (30gm) at 25 to 30°C. To the resulted clear solution was added carbon (10gm) and stirred and filtered. The filtrated was filtered again through sterile micron (0.2 µ) filter. To the resulted clear solution was added solution of sodium ethyl hexanoate (70gm) in n-butanol (70ml) and stirred for 3hrs at 25 to 30°C. The precipitated Cilastatin sodium was filtered and washed with n-butanol (80ml) and followed by acetone (200ml) and dried under vacuum to obtained 80gm Cilastatin sodium as amorphous white solid with 99.78% purity.
Abbreviations;
- [0039]
- DBU: diazabicyclo[5,4,0]undec-7-en
- DBN : 1,5-diazabicyclo[4,3,0]-non-5-ene
- TMG: 1,1,3,3-tetramethylguanidine
- DABCO: 1,4-diazabicyclo-[2,2,2]-octane
PATENT
https://patents.google.com/patent/WO2006022511A1/en
Cilasatin sodium salt i.e., [R-[R*, S*-(Z)]] –
7-[(2-amino-2-carboxyethylthio)-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino-2-hepa tenoic acid monosodium salt represented by following chemical formulae (1)1 has been used with imipenem in order to prevent its renal metabolism. Imipenem/cilastatin sodium is used as a potent broad spectrum antibacterial agent. [3] There have been several reports on the method for preparing a cilastatin sodium until now: for example, EP 48301 Bl discloses a method for the preparation of a cilastatin sodium salt using by Grignard reaction started from l-bromo-5-chloropentane (2′) explained by following Reaction Scheme 1; Donald W.
Graham et al discloses a preparation method using ethyl- 1, 3-dithian-2-carboxylate as a starting material (Donald W. Graham et al, J. Med. Chem., 30, pplO74, 1987) etc. [4] [Reaction Scheme 1]
[5]

[6]

[7] [8] As shown in the above Reaction Scheme 1, l-bromo-5-chloropentane (2′) is reacted with diethyl oxalate through Grignard reaction to afford ethyl 7-chloro-2-oxo-hepanoate (3′)at the 1st step; ethyl 7-chloro-2-oxo-heptanoate (3′) is reacted with (S)-2, 2-dimethylcyclopropanecarboxamide to obtain ethyl (Z )-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate (4′) at the 2n step.
[9] However, present inventors has confirmed that considerable amount (about 10 to 13%) of (E)-form isomer thereof (7′)was produced during the 2nd step as a reaction impurity by gas chromatography. The (E)-form isomer is further subjected to hydrolysis resulting in (E)-7-chloro-2-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (8′)as shown in following Reaction Scheme 2.
[10] [H] However, present inventors has confirmed that considerable amount (about 10 to 13%) of (E)-form isomer thereof (7′) was produced during the 2nd step as an reaction impurities by gas chromatography as shown in following Reaction Scheme 2. The (E )-form isomer is further subjected to hydrolysis resulting in (E)-7-chloro-2-((S)-2, 2-dimethylcyclopropylcarboxamide)-2-heptanoic acid (8′).
[12] [Reaction Scheme 2] [13]


[14] [15] There have been tried to solve the problems for example, the isomer impurity was removed by the acidification followed by recrystallization step or by adding cysteine to the reaction solution obtained in the 3r step at the above described 4 step, reacting with together to form (E)-7-(L-amino-2-carboxyethylthio)-2-((S )-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid and finally removing the reacted impurity by acidifying and heating step in the known preparation till now. However, the present inventors found that there remained unsolved problem such that the recrystallization yield of the product, i.e., (Z)-7-chloro-2-((S )-2,2-dimethylcyclopropylcarboxamide)-2-heptanoic acid was very poor because of the formed byproduct, i.e., (E
)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid in 3rdstep and further the unknown impurity (10′) and (S)-2,2-dimethylcyclopropanecarboxamide (H’) were produced by acidifying and heating reaction solution at the above described the 4 step as shown in following Reaction Scheme 3 confirmed by HPLC analysis, which give rise to another difficulty in the purification of final products. [16] [17] [Reaction Scheme 3] [18]

NH
(Z) and (E) m ix ture (91)

C ondition
(105 0 15
[19] In addition to above described problems, present inventors have found that the cilastatin isolated through the above described 4th step consisting of eluting the cation exchange resin with ammonia solution, concentrating the eluate and solidifying with ethanol and diethyl ether exists in the form of its ammonium salt not free acid form as disclosed in the patent. Using an acid such as hydrochloric acid in order to obtain free acid accompany with unwanted formation of inorganic ammonium salt such as ammonium chloride, which could not afford high purity of cilastatin sodium salt in the end.
[20] [21] Therefore, there have been tried to solve the above-described problems: for example, PCTAVO 0318544 (Al) discloses the isolation method using by neutral HP 20 resin column instead of cationic resin disclosed in EP 48301 Bl; PCTAVO 02094742 (Al) discloses the method for preparing cilastatin sodium salt (Ia) from cilastatin (6′), the disclosure of which cited documents are incorporated herein by reference.
[22]
[23] However, the above-described methods for preparing cilastatin using column chro¬ matographic process are not suitable for commercial mass production.
[24]
[25] The present inventors have made extensive researches to discover novel method for preparing cilastatin sodium salt with high yield and mass production and finally completed the invention by founding novel preparation for obtaining purposed cilastatin sodium salt; i.e., selectively hydrolyzing (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate, isolating (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt from the reaction mixture, adopting the cilastatin amine salt instead of free acid form disclosed in cited references and the use of sodium hydroxide and cationic exchange resin with pH control in order to obtain cilastatin sodium salt with high purity and high yield.
Example 1: Preparation of ethyl (Z)-7-chloro-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate (4)
[67]
[68] l-bromo-5-chloropentane (29 Ig, 1.57 mol) was reacted with diethyl oxalate
(206.5g) through Grignard reaction to obtain ethyl 7-chloro-2-oxo-heptanoate (3) and the compound (3) was reacted with (S)-2,2-dimethylcyclopropanecarboxamide to obtain ethyl (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptanoate (237g, 0.79 mol). The above-described step was performed by the procedure according to the procedure disclosed in EP 48301 (Bl).
[69]
[70] Example 1: Preparation of ethyl (Z)-7-chloro-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12)
[71]
[72] 1-1. ( Z V7-chloro-(YSV2. 2-dimethylcyclopropanecarboxamidoV2-heptanoic acid sodium salt
[73] The ethyl (Z)-7-chloro-2-((S
)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoate (237g, 0.79 mol) obtained in Comparative Example 1 was dissolved in 877ml of methanol and 1.8 L of sodium hydroxide solution (0.48 M) was added with stirring at room temperature. The reaction was finished when the area ratio of (Z) isomer and (E) isomer becomes 20: 1 by HPLC analysis and the un-reacted organic reagent was extracted with 490 ml of dichloromethane. The pH of the solution was adjusted to 7-8 with 3N HCl and the un- reacted organic reagent was extracted with 490 ml of dichloromethane again. The water layer was concentrated under reduced pressure and 650ml of ethanol was added and stirred until the solid had been dissolved at 50°C, for 30 minute to 1 hour. The un- dissolved solid was removed with filtration and the filtrate was concentrated under reduced pressure. 2.4 L of acetonitrile is added thereto and stirred to obtain 140.8g of ( Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12 ; 55% yield).
[74]
[75]
[76] m.p.: 219°C;
[77] 1H-NMR (D2O, 300MHz) δppm: 0.87 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.61 (m, 2H), 1.68 (dd, IH), 1.78 (m, 2H), 2.12 (m, 2H), 3.62 (t, 2H), 6.47 (t, IH);
[78]
13
[79] 13C ( -NMR (D2O, 300MHz) δppm: 19.47, 19.99, 22.55, 25.74, 26.75, 27.53, 29.44,
32.27, 46.11, 131.41, 136.52, 172.74, 174.62.
[80] [81] [82]
[83] 1-2. ( Z V7-chloro-(YSV2. 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid
(12-D
[84] 140.8g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12) obtained from Example 1-1 was dissolved in 422 ml of distilled water. The pH of the solution was adjusted to 2.0-3.0 with 3N HCl, extracted with 592 ml of isopropylether two times and 59.2g of anhydrous magnesium sulfate was added to isopropylether layer, stirred and subjected to filtration. The filtrate was concentrated to afford 127.7g of (Z)-7-chloro-2-((S )-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (12-1, 98% yield).
[85]
[86] 1H-NMR (CDCl3, 300MHz) δppm: 0.83 (dd, IH), 1.19 (s, 7H), 1.44 (dd, IH), 1.19
(s, 3H), 1.64 (m, 2H), 1.81 (m, 2H), 2.21 (m, 2H), 3.54 (t, 2H), 6.78 (t, IH), 7.04 (br, IH);
[87]
13
[88] 13C ( -NMR (CDCl3, 300MHz) δppm: 18.69, 20.82, 22.86, 25.36, 27.03, 28.53,
29.27, 32.17, 44.60, 124.88, 139.49, 168.96, 170.15.
[89]
[90]
[91] 1-3. ( Z V7-chloro-((SV2. 2-dimethylcyclopropanecarboxamidoV2-heptenoic acid ammonium salt (12-2)
[92] 127.7g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (12-1) obtained from Example 1-2 was dissolved in 422 ml of EtOH. 100 ml of 25% ammonia water solution was added thereto, stirred and concentrated to obtain 135.6g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid ammonium salt (12-2, 100% yield).
[93]
[94] Example 2: Preparation of cilastatin ammonium salt (13-1)
[95] 4Og of (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12, 0.14 mol) obtained in Example 1-1 was dissolved in 120 ml of 0.48 M sodium hydroxide solution and 240 ml of ethanol and the mixture of 1.4g of NaBr (0.013 mol) and 25.3g of L-cysteineDHClDH) was added thereto, stirred at 55°C, for 8 hours.
[96] The pH of the reaction solution was adjusted to 5.5-5.0 with 3N HCl, concentrated and 800ml of methanol was added, stirred at 55°C for 1 hour and un-dissolved salt was filtered out. The filtrate was concentrated to the extent that the volume of total solution was reduced to about 1/2. The concentrate was adsorbed with cationic exchange resin (PK208 model, Samyang Co.), washed with distilled water to the extent that the con¬ ductivity of the solution became less than lθμs(microsiemens), eluted with 2N ammonia water and the eluate was concentrated under the reduced pressure to give brown solid compound. The compound was dissolved in 40 ml of distilled water. 0.8 L of 2-propanol was added thereto and the solution was subjected to salting out method with reflux for 2 hours. The resulting solid was cooled and filtered to obtain 45.66g of cilastatin ammonium salt (13-1. 90% yield).
[97]
[98] m. p.: 161°C;
[99] Element Analysis: C16H29N3O5S (MW: 375.183): CaI. Q51.18; 7.78; N:11.19; Est.
C:51.01; H: 7.97; N: 11.04;
[100] MS m/z : 375 (M+, 49), 312(36), 97 (84.2), 69 (100);
[101] 1H-NMR (D2O, 300MHz) δppm: 0.87 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.62 (m, 5H), 2.1 l(q, 2H), 2.62 (t, 2H), 3.06 (m, 4H), 3.91 (dd, IH), 6.47 (t, IH);
13
[102] ” (C-NMR (D2O, 300MHz) δppm: 19.49, 19.97, 22.53, 26.74, 27.44, 27.86, 29.09,
29.43, 31.94, 32.85, 54.44, 131.23, 136.83, 172.70, 173.71, 174.64.
[103]
[104] Example 3: Preparation of cilastatin ethylamine salt (13-2)
[105] 4Og of (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12-1, 0.15 mol) obtained in Example 1-2 was dissolved in 165 ml of 0.66 M sodium hydroxide solution and 330 ml of ethanol and the mixture of 1.5g of NaBr (0.015 mol) and 27.6g of L-cysteineDHClDH) was added thereto, stirred at 55°C, for 8 hours.
[106] The pH of the reaction solution was adjusted to 5.5-5.0 with 3N HCl, concentrated and 800ml of methanol was added, stirred at 55°C for 1 hour and un-dissolved salt was filtered out.. The filtrate was concentrated to the extent that the volume of total solution was reduced to about 1/2. The concentrate was adsorbed with cationic exchange resin (PK208 model, Samyang Co.), washed with distilled water to the extent that the conductivity of the solution became less than 10μs(microsiemens), eluted with 2N ethylamine water and the eluate was concentrated under the reduced pressure to give brown solid compound. The compound was dissolved in 40 ml of distilled water. 0.8 L of 2-propanol was added thereto and the solution was subjected to salting out method with reflux for 2 hours. The resulting solid was cooled and purified with filtration to obtain 49.38g of cilastatin ethylamine salt (13-2. 90% yield).
[107]
[108] 1H-NMR (D2O, 300MHz) δppm: 0.86 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.27 (t, 3H), 1.60 (m, 5H), 2.1 l(q, 2H), 2.62 (t, 2H), 3.06 (m, 4H), 3.91 (dd, IH), 6.47 (t, IH); [109] 13C-NMR (D2O, 300MHz) δppm: 14.7, 21.57, 22.04, 24.63. 28.82, 29.52, 29.94,
31.16, 31.49, 34.00, 34.91, 37.78, 56.50, 133.27, 138.96, 174.75, 175.81, 176.74.
[HO]
[111] Example 4 : Purification of cilastatin ammonium salt
[112] 4-1. Purification using by water and ethanol
[113] 45.66g of cilastatin ammonium salt (13-1,0.12 mol) obtained in Example 2 was dissolved in 45.66 ml of distilled water and 1.3L of anhydrous ethanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 38.81g of cilastatin ammonium salt (Yield: 85%, Purity: 99.8%).
[114]
[115] 4-2. Purification using by ammonia water and propanol Q)
[116] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 50 ml of 25% ammonia water and 1.5L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 41.2g of cilastatin ammonium salt (Yield: 82.4%, Purity: 99.3%).
[117]
[118] 4-3. Purification using by ammonia water and propanol (1)
[119] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in
100 ml of 25% ammonia water and 2.0L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was purified with filtration to obtain 35.4g of cilastatin ammonium salt (Yield: 70.8%, Purity: 99.3%)
[120]
[121] 4-4. Purification using by ammonia water and ethanol
[122] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 50 ml of 25% ammonia water and 1.5 L of anhydrous ethanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 35.6g of cilastatin ammonium salt (Yield: 71.2%, Purity: 99.8%).
[123]
[124] 4-5. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (1)
[125] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 50ml of 4N ammonia water, and 2.0 L of 1 -propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 89.3g of cilastatin ammonium salt (Yield: 89.3%, Purity: 99.6%).
[126]
[127] 4-6. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (1) [128] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 50ml of 2N ammonia water, and 2.0 L of 1-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 95.0g of cilastatin ammonium salt (Yield: 95.0%, Purity: 99.5%).
[129]
[130] 4-7. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (3)
[131] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 100 ml of distilled water and 50ml of 25% ammonia water, and 2.0 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 89.7g of cilastatin ammonium salt (Yield: 89.7%, Purity: 99.8%).
[132]
[133] 4-8. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (4)
[134] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 100ml of 25% ammonia water, and 3.0 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 80.Og of cilastatin ammonium salt (Yield: 80.0%, Purity: 99.7%).
[135]
[136] 4-9. Purification using by water and propanol
[137] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 100 ml of distilled water and 1.5 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 87.2g of cilastatin ammonium salt (Yield: 87.2%, Purity: 99.6%).
[138]
[139] Example 5: Preparation of cilastatin sodium salt
[140] 4.28g of sodium hydroxide (0.107 mol) was dissolved in 38.3 ml of distilled water and 191.5 ml of ethanol. 38.81g of cilastatin ammonium salt (0.1 mol) obtained in Example 4-1 was added thereto and stirred for 30 minutes. The solution was con¬ centrated under reduced pressure at 60°C and 153 ml of distilled water was added to the concentrate. The solution was stirred to dissolve the concentrate and the pH of the solution was adjusted to 7.0 using by cationic exchange resin and filtered. The filtrate was lyophilized to obtain high purity (99.4%) of cilastatin sodium salt.
[141]
[142] Experimental Example 1: Purity Determination [143] The purity of cilastatin ammonium salt obtained in Example 4 was determined by
HPLC on condition as shown in Table 1 and the determined result was shown in Table 2.
[144] Table 1
[145] Table 2

[146]
Industrial Applicability
[147] The novel method of the present invention could prevent the formation of (E )-isomer from the preparation of novel intermediate for preparing cilastatin sodium, i.e., (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt and isolate the intermediate in situ providing simpler process with high yield and purity. Furthermore, it can provide with highly purified cilastatin sodium salt by isolating novel cilastatin amine salt and using sodium hydroxide and cationic exchange resin. Accordingly, the method can be very useful in preparing cilastatin sodium salt with high yield and high purity.





References
- ^ Jump up to:a b Keynan S, Hooper NM, Felici A, Amicosante G, Turner AJ (1995). “The renal membrane dipeptidase (dehydropeptidase I) inhibitor, cilastatin, inhibits the bacterial metallo-beta-lactamase enzyme CphA”. Antimicrob. Agents Chemother. 39 (7): 1629–31. doi:10.1128/aac.39.7.1629. PMC 162797. PMID 7492120.
- Keynan S, Hooper NM, Felici A, Amicosante G, Turner AJ: The renal membrane dipeptidase (dehydropeptidase I) inhibitor, cilastatin, inhibits the bacterial metallo-beta-lactamase enzyme CphA. Antimicrob Agents Chemother. 1995 Jul;39(7):1629-31. [PubMed:7492120]
- Buckley MM, Brogden RN, Barradell LB, Goa KL: Imipenem/cilastatin. A reappraisal of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy. Drugs. 1992 Sep;44(3):408-44. [PubMed:1382937]
- Balfour JA, Bryson HM, Brogden RN: Imipenem/cilastatin: an update of its antibacterial activity, pharmacokinetics and therapeutic efficacy in the treatment of serious infections. Drugs. 1996 Jan;51(1):99-136. doi: 10.2165/00003495-199651010-00008. [PubMed:8741235]
- Koller M, Brom J, Raulf M, Konig W: Cilastatin (MK 0791) is a potent and specific inhibitor of the renal leukotriene D4-dipeptidase. Biochem Biophys Res Commun. 1985 Sep 16;131(2):974-9. doi: 10.1016/0006-291x(85)91335-x. [PubMed:3863619]
- FDA: Recarbrio Label [Link]
- FDA: Primaxin Label [Link]
- ChemSpider: Cilastatin [Link]
- FDA Label: Apadaz [Link]
- Drugs@FDA: Primaxin [Link]
Synthesis
By Panchapakesan, Ganapathy et alFrom Indian, 269299, 16 Oct 2015
IN 269299

SYN
Patent
Cilastatin
-
- ATC:J01DH51
- Use:dehydropeptidase inhibitor (for combination with imipenem)
- Chemical name:[R-[R*,S*-(Z)]]-7-[(2-amino-2-carboxyethyl)thio]-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino]-2-heptenoic acid
- Formula:C16H26N2O5S
- MW:358.46 g/mol
- CAS-RN:82009-34-5
- InChI Key:DHSUYTOATWAVLW-WFVMDLQDSA-N
- InChI:InChI=1S/C16H26N2O5S/c1-16(2)8-10(16)13(19)18-12(15(22)23)6-4-3-5-7-24-9-11(17)14(20)21/h6,10-11H,3-5,7-9,17H2,1-2H3,(H,18,19)(H,20,21)(H,22,23)/b12-6-/t10-,11+/m1/s1
- EINECS:279-875-8
- LD50:8 g/kg (M, route unreported);
8 g/kg (R, route unreported)
Derivatives
monosodium salt
- Formula:C16H25N2NaO5S
- MW:380.44 g/mol
- CAS-RN:81129-83-1
- EINECS:279-694-4
- LD50:6786 mg/kg (M, i.v.); >10 g/kg (M, p.o.);
5027 mg/kg (R, i.v.); >10 g/kg (R, p.o.)
 |
|
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686013 |
| Routes of administration |
IV |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.072.592 |
| Chemical and physical data | |
| Formula | C16H26N2O5S |
| Molar mass | 358.454 g/mol g·mol−1 |
| 3D model (JSmol) | |
/////////////cilastatin, シラスタチン , FDA 2019, циластатин , سيلاستاتين , 西司他丁 , MK-791, Recarbrio
CC1(C)C[C@@H]1C(=O)N\C(=C/CCCCSC[C@H](N)C(O)=O)C(O)=O
J-147

J-147
N-(2,4-Dimethylphenyl)-2,2,2-trifluoro-N’-[(E)-(3-methoxyphenyl)methylene]acetohydrazide
- Molecular FormulaC18H17F3N2O2
- Average mass350.335 Da
2,2,2-trifluoroacetic acid-1-(2,4-dimethylphenyl)-2-[(3-methoxyphenyl)methylene]hydrazide
Acetic acid, 2,2,2-trifluoro-, 1-(2,4-dimethylphenyl)-2-[(1E)-(3-methoxyphenyl)methylene]hydrazide
FDA UNII Z41H3C5BT9
Abrexa Pharmaceuticals, Dementia, Alzheimer’s type, PHASE1
Blanchette Rockefeller Neurosci Inst (Originator)
Salk Institute for Biological Studies (Originator)
Abrexa Pharmaceuticals is developing the oral curcumin derivative J-147 for the treatment of Alzheimer’s disease. A phase I clinical trial is under way in healthy young and older adults.
The Salk Institute for Biological Studies and Abrexa Pharmaceuticals are developing J-147, a curcumin derivative CNB-001 , and a 5-lipoxygenase inhibitor, for the oral treatment of Alzheimer’s disease (AD), aging and acute ischemic stroke; in January 2019, a phase I trial for AD was initiated.
J147 is an experimental drug with reported effects against both Alzheimer’s disease and ageing in mouse models of accelerated aging.[1][2][3][4]
The approach that lead to development of the J147 drug was to screen candidate molecules for anti-aging effects, instead of targeting the amyloid plaques. It is contrary to most other approaches to developing drugs against Alzheimer’s disease that target the plaque deposits in the brain.[5]
The J147 drug is also reported to address other biological aging factors, such as preventing the leakage of blood from microvessels in mice brains.[5] The development of J147 follows the chemical pharmacological way, contrary to biological ways that exploit e.g. use of bacteriophages.[6][7]
Enhanced neurogenic activity over J147 in human neural precursor cells has its derivative called CAD-31. CAD-31 is enhancing the use of free fatty acids for energy production by shifting of the metabolic profile of fatty acids toward the production of ketone bodies, a potent source of energy in the brain when glucose levels are low.[8]
The target molecule is a protein called ATP synthase, which is found in the mitochondria.[9]
PAPER
Organic & Biomolecular Chemistry (2015), 13(37), 9564-9569
https://pubs.rsc.org/en/content/articlelanding/2015/OB/C5OB01463H#!divAbstract
A series of novel J147 derivatives were synthesized, and their inhibitory activities against β-amyloid (Aβ) aggregation and toxicity were evaluated by using the oligomer-specific antibody assay, the thioflavin-T fluorescence assay, and a cell viability assay in the transformed SH-SY5Y cell culture. Among the synthesized J147 derivatives, 3j with a 2,2-dicyanovinyl substituent showed the most potent inhibitory activity against Aβ42oligomerization (IC50 = 17.3 μM) and Aβ42 fibrillization (IC50 = 10.5 μM), and disassembled the preformed Aβ42 fibrils with an EC50 of 10.2 μM. Finally, we confirmed that 3j is also effective at preventing neurotoxicity induced by Aβ42-oligomers as well as Aβ42-fibrils.



PAPER
https://www.sciencedirect.com/science/article/pii/S0960894X12014746

Figure 1. Chemical structures of previously developed [11C]PIB, [18F]Amyvid and [18F]-T808, and newly developed [11C]J147.

Scheme 1. Synthesis of the reference standard J147 (2).
PRODUCT PATENT
WO2009052116
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009052116&tab=PCTDESCRIPTION
PATENT
WO-2019164997
A process for preparing crystalline Form II of 2,2,2-trifluoroacetic acid-1-(2,4-dimethylphenyl)-2-[(3-methoxyphenyl)methylene]hydrazide (J-147; 98% of purity) comprising the steps of providing a slurry containing saturated amorphous or crystalline Form I of J-147 and mixing the slurry to obtain the crystalline Form II of J147. Also claimed are processes for preparing the crystalline Form I of 2,2,2-trifluoroacetic acid-1-(2,4-dimethylphenyl)-2-[(3-methoxyphenyl)methylene]hydrazide. Further claimed are isolation of the crystalline Form II and I of 2,2,2-trifluoroacetic acid-1-(2,4-dimethylphenyl)-2-[(3-methoxyphenyl)methylene]hydrazide. The compound is disclosed to be a neurotrophic agent and known to be a Trkb receptor agonist, useful for treating neurodegenerative disease, such as aging and motor neurone disease.
The present disclosure relates to polymorph forms of a pharmaceutical active agent. In particular, the present disclosure relates to polymorph forms of neuroprotective agent 2,2,2-trifluoroacetic acid l-(2,4-Dimethylphenyl)-2-[(3-methoxyphenyl)methylene] hydrazide (J147).
[0002] 2,2,2 -trifluoroacetic acid l-(2,4-Dimethylphenyl)-2-[(3-methoxyphenyl)methylene] hydrazide (J147) is a potent orally active neurotrophic agent discovered during screening for efficacy in cellular models of age-associated pathologies and has a structure given by Formula I:
[0003] J147 is broadly neuroprotective, and exhibited activity in assays indicating distinct neurotoxicity pathways related to aging and neurodegenerative diseases, with EC50 between 10 and 200 nM. It has been indicated to improve memory in normal rodents, and prevent the loss of synaptic proteins and cognitive decline in a transgenic AD mouse model.
Furthermore, it has displayed neuroprotective, neuroanti-inflammatory, and LTP-enhancing activity.
[0004] The neurotrophic and nootropic effects have been associated with increases in BDNF levels and BDNF responsive proteins. Interestingly, despite this mechanism of action, Jl47’s neuroprotective effects have been observed to be independent of TrkB receptor activation.
J147 has been indicated to reduce soluble Ab40 and Ab42 levels, and it is currently being researched for potential applications in treating ALS.
The Fourier transform infrared (FTIR) spectrum is shown in Figure 4. Based on visual inspection the spectrum is consistent with structure. The Raman spectrum is in agreement with the FTIR spectrum and is shown in Figure 5. The proton NMR data is consistent with the structure of J147 and is shown in Figure 6. The proton NMR data is also shown in tabulated form in Table B below.
Table B
EXAMPLE OF PREPARATION OF FORM II OF J 147
Batch Process: About 100 kg of crude J147 from its synthetic preparation was evaporated twice from about 80 kg of ethanol. The crude product was taken up in about 48 kg of ethanol and the batch temperature was adjusted to 28 °C. About 37 kg of water was added gradually to the batch. The batch was held at about 30 °C for about 1.7 hours. A sample of the batch was pulled from the reactor and solids precipitated by addition of 45 mL of water. The solids obtained were added back to the batch as seed crystals and the mixture stirred for 40 minutes at 30 °C. An additional about 34 kg of water was added. The batch was held at about 18 °C for about 58 hours and then cooled to about 10 °C for another about 5.5 hours. Analysis of the resultant solids indicated the presence of Form I. Form I was converted to Form II by heating the slurry to about 45 °C for about 16 hours and then cooling back to about 10 °C and holding the batch at this temperature for about 3 hours about 17.7 kg of solid Form II of J147 were recovered by filtration after washing and drying.

CLIP
https://cen.acs.org/articles/90/i31/Tumeric-Derived-Compound-Curcumin-Treat.html
Turmeric-Derived Compound Curcumin May Treat Alzheimer’s

More than 5 million people in the U.S. currently live with Alzheimer’s disease. And according to the Alzheimer’s Association, the situation is only going to get worse.
By 2050, the nonprofit estimates, up to 16 million Americans will have the memory-robbing disease. It will cost the U.S. $1.1 trillion annually to care for them unless a successful therapy is found.
Pharmaceutical companies have invested heavily in developing Alzheimer’s drugs, many of which target amyloid-β, a peptide that misfolds and clumps in the brains of patients. But so far, no amyloid-β-targeted medications have been successful. Expectation for the most advanced drugs—bapineuzumab from Pfizer and Johnson & Johnson and solanezumab from Eli Lilly & Co.—are low on the basis of lackluster data from midstage clinical trials. That sentiment was reinforced last week when bapineuzumab was reported to have failed the first of four Phase III studies.
Even if these late-stage hopefuls do somehow work, they won’t come cheap, says Gregory M. Cole, a neuroscientist at the University of California, Los Angeles. These drugs “would cost patients tens of thousands of dollars per year,” he estimates. That hefty price tag stems from bapineuzumab and solanezumab being costly-to-manufacture monoclonal antibodies against amyloid-β.
“There’s a great need for inexpensive Alzheimer’s treatments,” as well as a backup plan if pharma fails, says Larry W. Baum, a professor in the School of Pharmacy at the Chinese University of Hong Kong. As a result, he says, a great many researchers have turned their attention to less pricy alternatives, such as compounds from plants and other natural sources.
Curcumin, a spice compound derived from the rootstalk of the turmeric plant (Curcuma longa), has stood out among some of the more promising naturally derived candidates.
When administered to mice that develop Alzheimer’s symptoms, curcumin decreases inflammation and reactive oxygen species in the rodents’ brains, researchers have found. The compound also inhibits the aggregation of troublesome amyloid-β strands among the animals’ nerve cells. But the development of curcumin as an Alzheimer’s drug has been stymied, scientists say, both by its low uptake in the body and a lack of funds for effective clinical trials—obstacles researchers are now trying to overcome.
In addition to contributing to curry dishes’ yellow color and pungent flavor, curcumin has been a medicine in India for thousands of years. Doctors practicing traditional Hindu medicine admire turmeric’s active ingredient for its anti-inflammatory properties and have used it to treat patients for ailments including digestive disorders and joint pain.
Only in the 1970s did Western researchers catch up with Eastern practices and confirm curcumin’s anti-inflammatory properties in the laboratory. Scientists also eventually determined that the polyphenolic compound is an antioxidant and has chemotherapeutic activity.

Bharat B. Aggarwal, a professor at the University of Texas M. D. Anderson Cancer Center, says curcumin is an example of a pleiotropic agent: It has a number of different effects and interacts with many targets and biochemical pathways in the body. He and his group have discovered that one important molecule targeted and subsequently suppressed by curcumin is NF-κB, a transcription factor that switches on the body’s inflammatory response when activated (J. Biol. Chem.,DOI: 10.1074/jbc.270.42.24995).
Aside from NF-κB, curcumin seems to interact with several other molecules in the inflammatory pathway, a biological activity that Aggarwal thinks is advantageous. “All chronic diseases are caused by dysregulation of multiple targets,” he says. “Chemists don’t yet know how to design a drug that hits multiple targets.” With curcumin, “Mother Nature has already provided a compound that does so.”
Curcumin’s pleiotropy also brought it to the attention of UCLA’s Cole during the early 1990s while he was searching for possible Alzheimer’s therapeutics. “That was before we knew about amyloid-β” and its full role in Alzheimer’s, he says. “We were working on the disease from an oxidative damage and inflammation point of view—two processes implicated in aging.”
When Cole and his wife, Sally A. Frautschy, also at UCLA, searched the literature for compounds that could tackle both of these age-related processes, curcumin jumped out at them. It also didn’t hurt that the incidence of Alzheimer’s in India, where large amounts of curcumin are consumed regularly, is lower than in other parts of the developing world (Lancet Neurol., DOI: 10.1016/s1474-4422(08)70169-8).
In 2001, Cole, Frautschy, and colleagues published the first papers that demonstrated curcumin’s potential to treat neurodegenerative disease (Neurobiol. Aging, DOI: 10.1016/s0197-4580(01)00300-1; J. Neurosci.2001, 8370). The researchers studied the effects of curcumin on rats that had amyloid-β injected into their brains, as well as mice engineered to develop amyloid brain plaques. In both cases, curcumin suppressed oxidative tissue damage and reduced amyloid-β deposits.
Those results, Cole says, “turned us into curcumin-ologists.”
Although the UCLA team observed that curcumin decreased amyloid plaques in animal models, at the time, the researchers weren’t sure of the molecular mechanism involved.
Soon after the team’s first results were published, Cole recalls, a colleague brought to his attention the structural similarity between curcumin and the dyes used to stain amyloid plaques in diseased brain tissue. When Cole and Frautschy tested the spice compound, they saw that it, too, could stick to aggregated amyloid-β. “We thought, ‘Wow, not only is curcumin an antioxidant and an anti-inflammatory, but it also might be an anti-amyloid drug,’ ” he says.
In 2004, a group in Japan demonstrated that submicromolar concentrations of curcumin in solution could inhibit aggregation of amyloid-β and break up preformed fibrils of the stuff (J. Neurosci. Res., DOI: 10.1002/jnr.20025). Shortly after that, the UCLA team demonstrated the same (J. Biol. Chem., DOI: 10.1074/jbc.m404751200).
As an Alzheimer’s drug, however, it’s unclear how important it is that the spice compound inhibits amyloid-β aggregation, Cole says. “When you have something that’s so pleiotropic,” he adds, “it’s hard to know” which of its modes of action is most effective.
Having multiple targets may be what helps curcumin have such beneficial, neuroprotective effects, says David R. Schubert, a neurobiologist at the Salk Institute for Biological Studies, in La Jolla, Calif. But its pleiotropy can also be a detriment, he contends.
The pharmaceutical world, Schubert says, focuses on designing drugs aimed at hitting single-target molecules with high affinity. “But we don’t really know what ‘the’ target for curcumin is,” he says, “and we get knocked for it on grant requests.”
Another problem with curcumin is poor bioavailability. When ingested, UCLA’s Cole says, the compound gets converted into other molecular forms, such as curcumin glucuronide or curcumin sulfate. It also gets hydrolyzed at the alkaline and neutral pHs present in many areas of the body. Not much of the curcumin gets into the bloodstream, let alone past the blood-brain barrier, in its pure, active form, he adds.
Unfortunately, neither Cole nor Baum at the Chinese University of Hong Kong realized the poor bioavailability until they had each launched a clinical trial of curcumin. So the studies showed no significant difference between Alzheimer’s patients taking the spice compound and those taking a placebo (J. Clin. Psychopharmacol., DOI: 10.1097/jcp.0b013e318160862c).
“But we did show curcumin was safe for patients,” Baum says, finding a silver lining to the blunder. “We didn’t see any adverse effects even at high doses.”
Some researchers, such as Salk’s Schubert, are tackling curcumin’s low bioavailability by modifying the compound to improve its properties. Schubert and his group have come up with a molecule, called J147, that’s a hybrid of curcumin and cyclohexyl-bisphenol A. Like Cole and coworkers, they also came upon the compound not by initially screening for the ability to interact with amyloid-β, but by screening for the ability to alleviate age-related symptoms.
The researchers hit upon J147 by exposing cultured Alzheimer’s nerve cells to a library of compounds and then measuring changes to levels of biomarkers for oxidative stress, inflammation, and nerve growth. J147 performed well in all categories. And when given to mice engineered to accumulate amyloid-β clumps in their brains, the hybrid molecule prevented memory loss and reduced formation of amyloid plaques over time (PLoS One, DOI: 10.1371/journal.pone.0027865).
Other researchers have tackled curcumin’s poor bioavailability by reformulating it. Both Baum and Cole have encapsulated curcumin in nanospheres coated with either polymers or lipids to protect the compound from modification after ingestion. Cole tells C&EN that by packaging the curcumin in this way, he and his group have gotten micromolar quantities of it into the bloodstream of humans. The researchers are now preparing for a small clinical trial to test the formulation on patients with mild cognitive impairment, who are at an increased risk of developing Alzheimer’s.
An early-intervention human study such as this one comes with its own set of challenges, Cole says. People with mild cognitive impairment “have good days and bad days,” he says. A large trial over a long period would be the best way to get any meaningful data, he adds.
Such a trial can cost up to $100 million, a budget big pharma might be able to scrape together but that is far out of reach for academics funded by grants, Cole says. “If you’re down at the level of what an individual investigator can do, you’re running a small trial,” he says, “and even if the result is positive, it might be inconclusive” because of its small size or short duration. That’s one of the reasons the curcumin work is slow-going, Cole contends.
The lack of hard clinical evidence isn’t stopping people from trying curcumin anyway. Various companies are selling the spice compound as a dietary supplement, both in its powdered form and in nanoformulations such as the ones Cole and Baum are working with. Indiana-based Verdure Sciences, for instance, licensed a curcumin nanoformulation from UCLA and sells it under the name Longvida (about $1.00 to $2.00 per capsule, depending on the distributor).
“There’s no proof that it works,” Cole says. “If you want to take it, you’re experimenting on yourself.” And he cautions that correct dosing for this more bioavailable form of curcumin hasn’t yet been established, so there could be safety concerns.
But on the basis of positive e-mails he’s received from caregivers and Alzheimer’s patients who are desperate for options and trying supplements, “I have some hope,” Cole says. “Maybe there’s something to curcumin after all.”
CLIP

Raw J 147 powder basic Characters
| Name: | J 147 powder |
| CAS: | 1146963-51-0 |
| Molecular Formula: | C18H17F3N2O2 |
| Molecular Weight: | 350.3349896 |
| Melt Point: | 177-178°C |
| Storage Temp: | 4°C |
| Color: | White or off white powder |
Raw J 147 powder in enhance brain function and an extra boost cycle
Names
J 147 powder
J 147 (1146963-51-0) Usage dosage
Using a drug discovery scheme for Alzheimer’s disease (AD) that is based upon multiple pathologies of old age, we identified a potent compound with efficacy in rodent memory and AD animal models. Since this compound, J-147 powder, is a phenyl hydrazide, there was concern that it can be metabolized to aromatic amines/hydrazines that are potentially carcinogenic. To explore this possibility, we examined the metabolites of J 147 powder in human and mouse microsomes and mouse plasma. It is shown that J-147(1146963-51-0) powder is not metabolized to aromatic amines or hydrazines, that the scaffold is exceptionally stable, and that the oxidative metabolites are also neuroprotective. It is concluded that the major metabolites of J 147(1146963-51-0) powder may contribute to its biological activity in animals.
J 147 , derived from the curry spice component curcumin, has low toxicity and actually reverses damage in neurons associated with Alzheimer’s.
J 147 (1146963-51-0) was the mitochondrial protein known as ATP synthase, specifically ATP5A, a subunit of that protein. ATP synthase is involved in the mitochondrial generation of ATP, which cells use for energy.
The researchers demonstrated that by reducing the activity of ATP synthase, they were able to protect neuronal cells from a number of toxicities associated with the aging of the brain. One reason for this neuroprotective effect is thought to be the role of excitotoxicity in neuronal cell damage.
Excitotoxicity is the pathological process by which neurons are damaged and killed by the overactivation of receptors for the excitatory neurotransmitter glutamate. Think of it being a bit like a light switch being turned on and off so rapidly that it ends up causing the light bulb to blow.
Recently, the role of ATP synthase inhibition for neuroprotection against excitotoxic damage was demonstrated in a mouse study[4]. The second study showed that mouse models expressing the human form of mutant ATPase inhibitory factor 1 (hIF1), which causes a sustained inhibition of ATP synthase, were more resilient to neuronal death after excitotoxic damage. This data is consistent with this new J 147 powder study, in which an increase in IF1 in the mice reduced the activity of ATP synthase (specifically ATP5A) and was neuroprotective.
Warning on Raw J 147 powder
Data presented here demonstrate that J-147 powder has the ability to rescue cognitive deficits when administered at a late stage in the disease. The ability of J-147 powder to improve memory in aged AD mice is correlated with its induction of the neurotrophic factors NGF (nerve growth factor) and BDNF (brain derived neurotrophic factor) as well as several BDNF-responsive proteins which are important for learning and memory. The comparison between J-147(1146963-51-0) powder and donepezil in the scopolamine model showed that while both compounds were comparable at rescuing short term memory, J-147 powder was superior at rescuing spatial memory and a combination of the two worked best for contextual and cued memory.
Further instructions
Alzheimer’s disease is a progressive brain disorder, recently ranked as the third leading cause of death in the United States and affecting more than five million Americans. It is also the most common cause of dementia in older adults, according to the National Institutes of Health. While most drugs developed in the past 20 years target the amyloid plaque deposits in the brain (which are a hallmark of the disease), few have proven effective in the clinic.
“While most drugs developed in the past 20 years target the amyloid plaque deposits in the brain (which are a hallmark of the disease), none have proven effective in the clinic,” says Schubert, senior author of the study.
Several years ago, Schubert and his colleagues began to approach the treatment of the disease from a new angle. Rather than target amyloid, the lab decided to zero in on the major risk factor for the disease–old age. Using cell-based screens against old age-associated brain toxicities, they synthesized J 147(1146963-51-0) powder.
Previously, the team found that J-147 powder could prevent and even reverse memory loss and Alzheimer’s pathology in mice that have a version of the inherited form of Alzheimer’s, the most commonly used mouse model. However, this form of the disease comprises only about 1 percent of Alzheimer’s cases. For everyone else, old age is the primary risk factor, says Schubert. The team wanted to explore the effects of the drug candidate on a breed of mice that age rapidly and experience a version of dementia that more closely resembles the age-related human disorder.

References
- ^ “Experimental drug targeting Alzheimer’s disease shows anti-aging effects” (Press release). Salk Institute. 12 November 2015. Retrieved November 13, 2015.
- ^ Chen Q, Prior M, Dargusch R, Roberts A, Riek R, Eichmann C, Chiruta C, Akaishi T, Abe K, Maher P, Schubert D (14 December 2011). “A novel neurotrophic drug for cognitive enhancement and Alzheimer’s disease”. PLoS One. 6 (12): e27865. doi:10.1371/journal.pone.0027865. PMC 3237323. PMID 22194796.
- ^ Currais A, Goldberg J, Farrokhi C, Chang M, Prior M, Dargusch R, Daugherty D, Armando A, Quehenberger O, Maher P, Schubert D (11 November 2015). “A comprehensive multiomics approach toward understanding the relationship between aging and dementia” (PDF). Aging. 7 (11): 937–55. doi:10.18632/aging.100838. PMC 4694064. PMID 26564964.
- ^ Prior M, Dargusch R, Ehren JL, Chiruta C, Schubert D (May 2013). “The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer’s disease mice”. Alzheimer’s Research & Therapy. 5 (3): 25. doi:10.1186/alzrt179. PMC 3706879. PMID 23673233.
- ^ Jump up to:a b Brian L. Wang (13 November 2015). “Experimental drug targeting Alzheimer’s disease shows anti-aging effects in animal tests”. nextbigfuture.com. Retrieved November 16, 2015.
- ^ Krishnan R, Tsubery H, Proschitsky MY, Asp E, Lulu M, Gilead S, Gartner M, Waltho JP, Davis PJ, Hounslow AM, Kirschner DA, Inouye H, Myszka DG, Wright J, Solomon B, Fisher RA (2014). “A bacteriophage capsid protein provides a general amyloid interaction motif (GAIM) that binds and remodels misfolded protein assemblies”. Journal of Molecular Biology. 426: 2500–19. doi:10.1016/j.jmb.2014.04.015. PMID 24768993.
- ^ Solomon B (October 2008). “Filamentous bacteriophage as a novel therapeutic tool for Alzheimer’s disease treatment”. Journal of Alzheimer’s Disease. 15 (2): 193–8. PMID 18953108.
- ^ Daugherty, D., Goldberg, J., Fischer, W., Dargusch, R., Maher, P., & Schubert, D. (2017). A novel Alzheimer’s disease drug candidate targeting inflammation and fatty acid metabolism. Alzheimer’s research & therapy, 9(1), 50. https://doi.org/10.1186/s13195-017-0277-3
- ^ “Researchers identify the molecular target of J147, which is nearing clinical trials to treat Alzheimer’s disease”. Retrieved 2018-01-30.
 |
|
| Legal status | |
|---|---|
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| Chemical and physical data | |
| Formula | C18H17F3N2O2 |
| Molar mass | 350.341 g·mol−1 |
| 3D model (JSmol) | |
////////////J-147, J 147, J147, Alzheimer’s disease, neurotrophic agent, The Salk Institute for Biological Studies, Abrexa Pharmaceuticals, PHASE 1, CURCUMIN

CAS 1417911-00-2
- Acetic acid, 2,2,2-trifluoro-, 1-(2,4-dimethylphenyl)-2-[[3-(methoxy-11C)phenyl]methylene]hydrazide
Pretomanid, プレトマニド;
Pretomanid
プレトマニド;
| Formula |
C14H12F3N3O5
|
|---|---|
| CAS |
187235-37-6
|
| Mol weight |
359.2574
|
- (S)-PA 824
2019/8/14 FDA 2109 APPROVED
Antibacterial (tuberculostatic),
MP 149-150 °C, Li, Xiaojin; Bioorganic & Medicinal Chemistry Letters 2008, Vol 18(7), Pg 2256-2262 and Orita, Akihiro; Advanced Synthesis & Catalysis 2007, Vol 349(13), Pg 2136-2144
150-151 °C Marsini, Maurice A.; Journal of Organic Chemistry 2010, Vol 75(21), Pg 7479-7482
Pretomanid is an antibiotic used for the treatment of multi-drug-resistant tuberculosis affecting the lungs.[1] It is generally used together with bedaquiline and linezolid.[1] It is taken by mouth.[1]
The most common side effects include nerve damage, acne, vomiting, headache, low blood sugar, diarrhea, and liver inflammation.[1] It is in the nitroimidazole class of medications.[2]
Pretomanid was approved for medical use in the United States in 2019.[3][1] Pretomanid was developed by TB Alliance,[4] a not-for-profitproduct development partnership dedicated to the discovery and development of new, faster-acting and affordable medicines for tuberculosis (TB).[5]
Global Alliance for the treatment of tuberculosis (TB).
The compound was originally developed by PathoGenesis (acquired by Chiron in 2000). In 2002, a co-development agreement took place between Chiron (acquired by Novartis in 2005) and the TB Alliance for the development of the compound. The compound was licensed to Fosunpharma by TB Alliance in China.
History
Pretomanid is the generic, nonproprietary name for the novel anti-bacterial drug compound formerly called PA-824.[6] Pretomanid is referred to as “Pa” in regimen abbreviations, such as BPaL. The “preto” prefix of the compound’s name honors Pretoria, South Africa, the home of a TB Alliance clinical development office where much of the drug’s development took place. The “manid” suffix is used to group compounds with similar chemical structures. This class of drug is variously referred to as nitroimidazoles, nitroimidazooxazines or nitroimidazopyrans. Development of this compound was initiated because of the urgent need for new antibacterial drugs effective against resistant strains of tuberculosis. Also, current anti-TB drugs are mainly effective against replicating and metabolically active bacteria, creating a need for drugs effective against persisting or latent bacterial infections as often occur in patients with tuberculosis.[7]
Discovery and pre-clinical development
Pretomanid was first identified in a series of 100 nitroimidazopyran derivatives synthesized and tested for antitubercular activity. Importantly, pretomanid has activity against static M. tuberculosis isolates that survive under anaerobic conditions, with bactericidal activity comparable to that of the existing drug metronidazole. Pretomanid requires metabolic activation by Mycobacterium for antibacterial activity. Pretomanid was not the most potent compound in the series against cultures of M. tuberculosis, but it was the most active in infected mice after oral administration. Oral pretomanid was active against tuberculosis in mice and guinea pigs at safely tolerated dosages for up to 28 days.[7]
Limited FDA approval
FDA approved pretomanid only in combination with bedaquiline and linezolid for treatment of a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug resistant pulmonary tuberculosis. Pretomanid was approved under the Limited Population Pathway (LPAD pathway) for antibacterial and antifungal drugs. The LPAD Pathway was established by Congress under the 21st Century Cures Act to expedite development and approval of antibacterial and antifungal drugs to treat serious or life-threatening infections in a limited population of patients with unmet need. Pretomanid is only the third tuberculosis drug to receive FDA approval in more than 40 years.[3][8]
PATENT
IN 201641030408
HETERO RESEARCH FOUNDATION
http://ipindiaservices.gov.in/PatentSearch/PatentSearch/ViewPDF
- By Reddy, Bandi Parthasaradhi; Reddy, Kura Rathnakar; Reddy, Adulla Venkat Narsimha; Krishna, Bandi Vamsi
- From Indian Pat. Appl. (2018), IN 201641030408
The nitroimidazooxazine Formula I (PA-824) is a new class of bioreductive drug for tuberculosis. The recent introduction of the nitroimidazooxazine Formula I (PA-824) to clinical trial by the Global Alliance for TB Drug Development is thus of potential significance, since this compound shows good in vitro and in vivo activity against Mycobacterium tuberculosis in both its active and persistent forms. Tuberculosis (TBa) remains a leading infectious cause of death worldwide, but very few new drugs have been approved for TB treatment ifi the past 35 years, the current drug therapy for TB is long and complex, involving multidrug combinations.
The mechanism of actiém of Pretomanid is thoughrto involve reduction of the nitro group, in a‘ process dependent on the Bacterial ‘ m E Nfilw‘fieéFPEOEPEa‘e fillyeifiaasnfi (F8189); $943“; 20mm; “q Mcyarecent Swiss on mutant strains showed that a 151-amino acid (17.37 kDa) protein of unknown function, Rv3547, also, appears to be critical for this activation. Equivalent genes are present in M. boVis and MaVium.
Pretomanid and its pharmace’utically acceptable salts Were generically disclosed in US 5,668,127 A and Specifically disclosed in US 6,087,358 A. US ‘358 patent discloses a process for the preparation of Pretomanid, which is as shown below in scheme 1:

CN 104177372 A discloses a process for the preparation of Pretomanid, which is as shown below in scheme II: 
Bioorganic & Medicinal Chemistry Letters 2008, Volume: 18, Issue: 7, Pages: 2256-2262 discloses a process for the preparation of Pretomanid, which is as shown below in scheme Ill: 
US 7,!15,736 B2-discloses_a process fdr the preparation of 3S-Hydroxy-6-nitrQ-2H-3, 4— dihydro-[2-1b]-imidazopyran which is a key intermediate of Pretomanid, which is as shown below in scheme IV:

Journal Medicinal Chemistry 2009, Volume: 52, Pages: 637 — 645 discloses a process for the preparation of ‘Pretomanid, which is as shown below in scheme V:

Joumal Organic Chemistry 2010; Volume: 75 (2]), Pages: 7479—82 discloses a process for. the preparation of Pretomanid, which is as shown below in scheme VI:


Example 3: Preparation of Pretomanid (S) 1- -(3 (tert- -Butyldomethylsilyloxy)- -2- -(-4 -(trifluoromethoxy)-71benzyloxy2‘- propyl)- 2- -methylP AT E N4Tnitro- fi-Eimigazole (Efgm Awlas (3315;501:1691 gin! %etra%1y7drofuraen (18(150 ml) at room temperature and stirred for 5 to 10 minutes then TBAF (9516 ml) was added to the reaction mixture and stirred for 2 hours, at room temperature, afler completion of the reaction removed solvent through vacuum to obtained residue, dissolved the residue in MDC (1800 ml) and water (1800 ml) stirred, separated the layers and the organic layer washed with 10% ‘ sodium bicarbonate the obtained organic solution was concentrated under atmospheric pressure to obtained residue added MeOH (1730 ml) at room temperature and the reaction mixture was cooled to 0°C to 5°C, added KOH (24.5 gm) at the same temperaturé then cooled to room temperature and stirred for 24 hours. After completion of reaction DM Water added drop wise over 30 minutes at 10°C to 15° C and stirred for 1 hour to 1 hour 30 minutes at room’lemperature, filtrated the compound and washed with DM wa‘er (133 ml) and dried under vacuum for 10 hours at 50° C. Yield: 53 gm , Chromatographic purity: 97.69% (by HPLC):
Example 4: Purification of Pretomanid Pretomanid (53 gm) was dissolved in MDC (795 ml) at room temperatur’e and stirred for 10 to 15 minutes, added charcoal (10 gm) and stirred for 30-35 minutes, remove the charcoal and concentrated to obtained residue: Dissolved the obtained residue in IPA (795 ml) and the reaction mixture was heated to 80°C maintained for 10-15 minutes, added cyclohexane (1600ml) for 30 minutes at 80° C, then cooled to room temperature and stirred the reaction mass for overnight, filtered the solid and washed with cyclohexane (265 ml), and dried under vacuum for 10 hours at 50° C. Yield: 48 gm (Percentage of Yield: 90%) Chromatographic purity: 99.97% by HPLC).
CLIP

ReferencE
CN104177372A.
WO9701562A1.
IN 201641030408
IN 201621026053
CN 107915747
CN 106632393
CN 106565744
CN 104177372
WO 9701562
US 6087358
PAPER
Science (Washington, DC, United States) (2008), 322(5906), 1392-1395.
Paper
PAPER
Huagong Shikan (2010), 24(4), 32-34, 51.
Xiaojin; Bioorganic & Medicinal Chemistry Letters 2008, Vol 18(7), Pg 2256-2262
PAPER
Orita, Akihiro; Advanced Synthesis & Catalysis 2007, Vol 349(13), Pg 2136-2144
https://onlinelibrary.wiley.com/doi/abs/10.1002/adsc.200700119
https://application.wiley-vch.de/contents/jc_2258/2007/f700119_s.pdf



Marsini, Maurice A.; Journal of Organic Chemistry 2010, Vol 75(21), Pg 7479-7482

Scheme 1

aDHP = 3,4-dihydropyran; p-TsOH = p-toluenesulfonic acid; MsOH = methanesulfonic acid.
Scheme 3

aCl3CCN = trichloroacetonitrile; TBME = tert-butylmethyl ether; TfOH = trifluoromethanesulfonic acid.



PAPER
Journal of Medicinal Chemistry (2010), 53(1), 282-294.
Journal of Medicinal Chemistry (2009), 52(3), 637-645.
PATENT

References
- ^ Jump up to:a b c d e “FDA approves new drug for treatment-resistant forms of tuberculosis that affects the lungs”. FDA. 14 August 2019. Retrieved 28 August 2019.
- ^ “Compounds | TB Alliance”. http://www.tballiance.org. Retrieved 2019-04-18.
- ^ Jump up to:a b Abutaleb Y (14 August 2019). “New antibiotic approved for drug-resistant tuberculosis”. Washington Post.
- ^ “TB Medicine Pretomanid Enters Regulatory Review Process in the United States | TB Alliance”. http://www.tballiance.org. Retrieved 2019-04-18.
- ^ “About TB Alliance”. TB Alliance. Retrieved 2019-04-18.
- ^ “PA-824 has a New Generic Name: Pretomanid”. TB Alliance. Retrieved 2019-04-18.
- ^ Jump up to:a b Lenaerts AJ, Gruppo V, Marietta KS, Johnson CM, Driscoll DK, Tompkins NM, Rose JD, Reynolds RC, Orme IM (June 2005). “Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models”. Antimicrobial Agents and Chemotherapy. 49 (6): 2294–301. doi:10.1128/AAC.49.6.2294-2301.2005. PMC 1140539. PMID 15917524.
- ^ FDA News Release. FDA approves new drug for treatment-resistant forms of tuberculosis that affects the lungs.
 |
|
| Legal status | |
|---|---|
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| Chemical and physical data | |
| Formula | C14H12F3N3O5 |
| Molar mass | 359.261 g·mol−1 |
| 3D model (JSmol) | |
//////////////Pretomanid, FDA 2109, プレトマニド , Antibacterial, tuberculostatic, PA-824, ANTI tuberculostatic
FDA approves first treatment Dupixent (Dupilumab) for chronic rhinosinusitis with nasal polyps
The U.S. Food and Drug Administration today approved Dupixent (dupilumab) to treat adults with nasal polyps (growths on the inner lining of the sinuses) accompanied by chronic rhinosinusitis (prolonged inflammation of the sinuses and nasal cavity). This is the first treatment approved for inadequately controlled chronic rhinosinusis with nasal polyps.
“Nasal polyps can lead to loss of smell and often patients require surgery to remove the polyps,” said Sally Seymour, M.D., Director of the Division of Pulmonary, Allergy and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research. “Dupixent provides an important treatment option for patients whose nasal polyps are not …
- June 26, 2019
The U.S. Food and Drug Administration today approved Dupixent (dupilumab) to treat adults with nasal polyps (growths on the inner lining of the sinuses) accompanied by chronic rhinosinusitis (prolonged inflammation of the sinuses and nasal cavity). This is the first treatment approved for inadequately controlled chronic rhinosinusis with nasal polyps.
“Nasal polyps can lead to loss of smell and often patients require surgery to remove the polyps,” said Sally Seymour, M.D., Director of the Division of Pulmonary, Allergy and Rheumatology Products in the FDA’s Center for Drug Evaluation and Research. “Dupixent provides an important treatment option for patients whose nasal polyps are not adequately controlled with intranasal steroids. It also reduces the need for nasal polyp surgery and oral steroids.”
Dupixent is given by injection. The efficacy and safety of Dupixent were established in two studies with 724 patients, 18 years and older with chronic rhinosinusitis with nasal polyps who were symptomatic despite taking intranasal corticosteroids. Patients who received Dupixent had statistically significant reductions in their nasal polyp size and nasal congestion compared to the placebo group. Patients taking Dupixent also reported an increased ability to smell and required less nasal polyp surgery and oral steroids.
Dupixent may cause serious allergic reactions and eye problems, such as inflammation of the eye (conjunctivitis) and inflammation of the cornea (keratitis). If patients experience new or worsening eye symptoms, such as redness, itching, pain or visual changes, they should consult their health care professional. The most common side effects reported include injection site reactions as well as eye and eyelid inflammation, which included redness, swelling and itching. Patients receiving Dupixent should avoid receiving live vaccines.
Dupixent was originally approved in 2017 for patients 12 and older with eczema that is not controlled adequately by topical therapies or when those therapies are not advisable. In 2018, Dupixent was approved as an add-on maintenance treatment for patients 12 years and older with moderate-to-severe eosinophilic asthma or with oral corticosteroid-dependent asthma.
The FDA granted this application Priority Review. The approval of Dupixent was granted to Regeneron Pharmaceuticals.
///////////Dupixent, dupilumab, fda 2019, nasal polyps, chronic rhinosinusitis, Priority Review, Regeneron Pharmaceuticals,
FDA approves new add-on drug Nourianz (istradefylline) to treat off episodes in adults with Parkinson’s disease
READ AT https://newdrugapprovals.org/2016/04/25/istradefylline/
FDA approves new add-on drug Nourianz (istradefylline) to treat off episodes in adults with Parkinson’s disease
The U.S. Food and Drug Administration today approved Nourianz (istradefylline) tablets as an add-on treatment to levodopa/carbidopa in adult patients with Parkinson’s disease (PD) experiencing “off” episodes. An “off” episode is a time when a patient’s medications are not working well, causing an increase in PD symptoms, such as tremor and difficulty walking.
“Parkinson’s disease is a debilitating condition that profoundly impacts patients’ lives,” said Eric Bastings, M.D., acting director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research. “We are committed to helping make additional treatments for Parkinson’s disease available to patients.”
According to the National Institutes of Health, PD is the second-most common neurodegenerative disorder in the U.S. after Alzheimer’s disease. An estimated 50,000 Americans are diagnosed with PD each year, and about one million Americans have the condition. The neurological disorder typically occurs in people over age 60, although it can occur earlier. It happens when cells in the brain, which produce a chemical called dopamine, become impaired or die. Dopamine helps transmit signals between the areas of the brain that produce smooth, purposeful movements – such as eating, writing, and shaving. Early symptoms of the disease are subtle and typically worsen gradually; however, the disease progresses more quickly in some people than in others.
The effectiveness of Nourianz in treating “off” episodes in patients with PD who are already being treated with levodopa/carbidopa was shown in four 12-week placebo-controlled clinical studies that included a total of 1,143 participants. In all four studies, patients treated with Nourianz experienced a statistically significant decrease from baseline in daily “off” time compared to patients receiving a placebo.
The most common adverse reactions observed in patients taking Nourianz were involuntary muscle movement (dyskinesia), dizziness, constipation, nausea, hallucination and sleeplessness (insomnia). Patients should be monitored for development of dyskinesia or exacerbation of existing dyskinesia. If hallucinations, psychotic behavior, or impulsive/compulsive behavior occurs, a dosage reduction or stoppage of Nourianz should be considered. Use of Nourianz during pregnancy is not recommended. Women of childbearing potential should be advised to use contraception during treatment.
The FDA granted approval of Nourianz to Kyowa Kirin, Inc.
////// Nourianz, istradefylline, Kyowa Kirin, FDA 2019, Parkinson’s disease
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
FDA approves treatment Inrebic (fedratinib) for patients with rare bone marrow disorder
Today, the U.S. Food and Drug Administration approved Inrebic (fedratinib) capsules to treat adult patients with certain types of myelofibrosis.
“Prior to today, there was one FDA-approved drug to treat patients with myelofibrosis, a rare bone marrow disorder. Our approval today provides another option for patients,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “The FDA is committed to encouraging the development of treatments for patients with rare diseases and providing alternative options, as not all patients respond in the same way.”
Myelofibrosis is a chronic disorder where scar tissue forms in the bone marrow and the production of the blood cells moves from the bone marrow to the spleen and liver, causing organ enlargement. It can cause extreme fatigue, shortness of breath, pain below the ribs, fever, night sweats, itching and bone pain. When myelofibrosis occurs on its own, it is called primary myelofibrosis. Secondary myelofibrosis occurs when there is excessive red blood cell production (polycythemia vera) or excessive platelet production (essential thrombocythemia) that evolves into myelofibrosis.
Jakafi (ruxolitinib) was approved by the FDA in 2011. The approval of Inrebic for intermediate-2 or high-risk primary or secondary (post-polycythemia vera or post-essential thrombocythemia) myelofibrosis was based on the results of a clinical trial where 289 patients with myelofibrosis were randomized to receive two different doses (400 mg or 500 mg daily by mouth) of fedratinib or placebo. The clinical trial showed that 35 of 96 patients treated with the fedratinib 400 mg daily dose (the dose recommended in the approved label) experienced a significant therapeutic effect (measured by greater than or equal to a 35% reduction from baseline in spleen volume at the end of cycle 6 (week 24) as measured by an MRI or CT scan with a follow-up scan four weeks later). As a result of treatment with Inrebic, 36 patients experienced greater than or equal to a 50% reduction in myelofibrosis-related symptoms, such as night sweats, itching, abdominal discomfort, feeling full sooner than normal, pain under ribs on left side, and bone or muscle pain.
The prescribing information for Inrebic includes a Boxed Warning to advise health care professionals and patients about the risk of serious and fatal encephalopathy (brain damage or malfunction), including Wernicke’s, which is a neurologic emergency related to a deficiency in thiamine. Health care professionals are advised to assess thiamine levels in all patients prior to starting Inrebic, during treatment and as clinically indicated. If encephalopathy is suspected, Inrebic should be immediately discontinued.
Common side effects for patients taking Inrebic are diarrhea, nausea, vomiting, fatigue and muscle spasms. Health care professionals are cautioned that patients may experience severe anemia (low iron levels) and thrombocytopenia (low level of platelets in the blood). Patients should be monitored for gastrointestinal toxicity and for hepatic toxicity (liver damage). The dose should be reduced or stopped if a patient develops severe diarrhea, nausea or vomiting. Treatment with anti-diarrhea medications may be recommended. Patients may develop high levels of amylase and lipase in their blood and should be managed by dose reduction or stopping the mediation. Inrebic must be dispensed with a patient Medication Guide that describes important information about the drug’s uses and risks.
The FDA granted this application Priority Review designation. Inrebic also received Orphan Drug designation, which provides incentives to assist and encourage the development of drugs for rare diseases. The FDA granted the approval of Inrebic to Impact Biomedicines, Inc., a wholly-owned subsidiary of Celgene Corporation.
LINK
///////Inrebic , fedratinib, FDA 2019, Priority Review , Orphan Drug, Biomedicines, Celgene , bone marrow disorder
FDA approves third oncology drug Rozlytrek (entrectinib) that targets a key genetic driver of cancer, rather than a specific type of tumor
FDA also approves drug for second indication in a type of lung cancer
The U.S. Food and Drug Administration today granted accelerated approval to Rozlytrek (entrectinib), a treatment for adult and adolescent patients whose cancers have the specific genetic defect, NTRK (neurotrophic tyrosine receptor kinase) gene fusion and for whom there are no effective treatments.
“We are in an exciting era of innovation in cancer treatment as we continue to see development in tissue agnostic therapies, which have the potential to transform cancer treatment. We’re seeing continued advances in the use of biomarkers to guide drug development and the more targeted delivery of medicine,” said FDA Acting Commissioner Ned Sharpless, M.D. “Using the FDA’s expedited review pathways, including breakthrough therapy designation and accelerated approval process, we’re supporting this innovation in precision oncology drug development and the evolution of more targeted and effective treatments for cancer patients. We remain committed to encouraging the advancement of more targeted innovations in oncology treatment and across disease types based on our growing understanding of the underlying biology of diseases.”
This is the third time the agency has approved a cancer treatment based on a common biomarker across different types of tumors rather than the location in the body where the tumor originated. The approval marks a new paradigm in the development of cancer drugs that are “tissue agnostic.” It follows the policies that the FDA developed in a guidance document released in 2018. The previous tissue agnostic indications approved by the FDA were pembrolizumab for tumors with microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) tumors in 2017 and larotrectinib for NTRK gene fusion tumors in 2018.
“Today’s approval includes an indication for pediatric patients, 12 years of age and older, who have NTRK-fusion-positive tumors by relying on efficacy information obtained primarily in adults. The FDA continues to encourage the inclusion of adolescents in clinical trials. Traditionally, clinical development of new cancer drugs in pediatric populations is not started until development is well underway in adults, and often not until after approval of an adult indication,” said Richard Pazdur, M.D., director of the FDA’s Oncology Center of Excellence and acting director of the Office of Hematology and Oncology Products in the FDA’s Center for Drug Evaluation and Research. “Efficacy in adolescents was derived from adult data and safety was demonstrated in 30 pediatric patients.”
The ability of Rozlytrek to shrink tumors was evaluated in four clinical trials studying 54 adults with NTRK fusion-positive tumors. The proportion of patients with substantial tumor shrinkage (overall response rate) was 57%, with 7.4% of patients having complete disappearance of the tumor. Among the 31 patients with tumor shrinkage, 61% had tumor shrinkage persist for nine months or longer. The most common cancer locations were the lung, salivary gland, breast, thyroid and colon/rectum.
Rozlytrek was also approved today for the treatment of adults with non-small cell lung cancer whose tumors are ROS1-positive (mutation of the ROS1 gene) and has spread to other parts of the body (metastatic). Clinical studies evaluated 51 adults with ROS1-positive lung cancer. The overall response rate was 78%, with 5.9% of patients having complete disappearance of their cancer. Among the 40 patients with tumor shrinkage, 55% had tumor shrinkage persist for 12 months or longer.
Rozlytrek’s common side effects are fatigue, constipation, dysgeusia (distorted sense of taste), edema (swelling), dizziness, diarrhea, nausea, dysesthesia (distorted sense of touch), dyspnea (shortness of breath), myalgia (painful or aching muscles), cognitive impairment (confusion, problems with memory or attention, difficulty speaking, or hallucinations), weight gain, cough, vomiting, fever, arthralgia and vision disorders (blurred vision, sensitivity to light, double vision, worsening of vision, cataracts, or floaters). The most serious side effects of Rozlytrek are congestive heart failure (weakening or damage to the heart muscle), central nervous system effects (cognitive impairment, anxiety, depression including suicidal thinking, dizziness or loss of balance, and change in sleep pattern, including insomnia and excessive sleepiness), skeletal fractures, hepatotoxicity (damage to the liver), hyperuricemia (elevated uric acid), QT prolongation (abnormal heart rhythm) and vision disorders. Health care professionals should inform females of reproductive age and males with a female partner of reproductive potential to use effective contraception during treatment with Rozlytrek. Women who are pregnant or breastfeeding should not take Rozlytrek because it may cause harm to a developing fetus or newborn baby.
Rozlytrek was granted accelerated approval. This approval commits the sponsor to provide additional data to the FDA. Rozlytrek also received Priority Review, Breakthrough Therapy and Orphan Drug designation. The approval of Rozlytrek was granted to Genentech, Inc.
///////////////Rozlytrek, entrectinib, accelerated approval, priority Review, Breakthrough Therapy, Orphan Drug designation, fda 2019, Genentech, cancer
