
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
MOLINDONE, молиндон موليندون 吗茚酮


MOLINDONE
C16H24N2O2,, 276.374
SPN 810, SPN 801M, AFX 2201
cas 15622-65-8 hcl
Molindone is used for the management of the manifestations of psychotic disorders.
Supernus Pharmaceuticals , under license from Afecta Pharmaceuticals , is developing molindone hydrochloride (SPN-810; SPN-801M; AFX-2201; presumed to be Zalvari), as a capsule formulation, for the potential oral treatment of conduct disorder in patients with attention deficit hyperactivity disorder. In 3Q15, the company initiated two phase III trials (CHIME 1 and CHIME 2) for compulsive aggression in ADHD. In November 2019, the trial was expected to complete in June 2020.
Molindone, sold under the brand name Moban, is an antipsychotic which is used in the United States in the treatment of schizophrenia.[1][2] It works by blocking the effects of dopamine in the brain, leading to diminished symptoms of psychosis. It is rapidly absorbed when taken orally.
It is sometimes described as a typical antipsychotic,[3] and sometimes described as an atypical antipsychotic.[4]
Molindone was discontinued by its original supplier, Endo Pharmaceuticals, on January 13, 2010.[5]
Availability and Marketing in the USA
After having been produced and subsequently discontinued by Core Pharma in 2015-2017, Molindone is available again from Epic Pharma effective December, 2018.[6]
Adverse effects
The side effect profile of molindone is similar to that of other typical antipsychotics. Unlike most antipsychotics, however, molindone use is associated with weight loss.[4][7]
Chemistry
Synthesis

Molindone synthesis: SCHOEN KARL, J PACHTER IRWIN; BE 670798 (1965 to Endo Lab).
Condensation of oximinoketone 2 (from nitrosation of 3-pentanone), with cyclohexane-1,3-dione (1) in the presence of zinc and acetic acid leads directly to the partly reduced indole derivative 6. The transformation may be rationalized by assuming as the first step, reduction of 2 to the corresponding α-aminoketone. Conjugate addition of the amine to 1 followed by elimination of hydroxide (as water) would give ene-aminoketone 3. This enamine may be assumed to be in tautomeric equilibrium with imine 4. Aldol condensation of the side chain carbonyl group with the doubly activated ring methylene group would then result in cyclization to pyrrole 5; simple tautomeric transformation would then give the observed product. Mannich reaction of 6 with formaldehyde and morpholine gives the tranquilizer molindone (7).
US-20200262788
Process for preparing molindone and its intermediates useful for treating schizophrenia..
|
Molindone is chemically known as 4H-Indol-4-one, 3-ethyl-1,5,6,7-tetrahydro-2-methyl-5-(4-morpholinylmethyl) and represented by formula I. Molindone is indicated for management of schizophrenia and is under clinical trial for alternate therapies.
|
EXAMPLES
Example 1: Preparation of methyl 2-chloro-2-ethyl-3-oxobutanoate
Example 2: Preparation of 3-chloropentan-2-one
Example 3: Preparation of 3-chloropentan-2-one
Example 4: Preparation of 2-(2-oxopentan-3-yl)cyclohexane-1,3-dione (4)
Example 5: Preparation of 2-methyl-3-ethyl-4-oxo-4,5,6,7-tetrahydroindole (5)
Example 6: Preparation of 2-methyl-3-ethyl-4-oxo-4,5,6,7-tetrahydroindole (5)
Example 7: Preparation of Molindone Hydrochloride
References
- ^ “molindone”. F.A. Davis Company.
- ^ “Molindone”.
- ^ Aparasu RR, Jano E, Johnson ML, Chen H (October 2008). “Hospitalization risk associated with typical and atypical antipsychotic use in community-dwelling elderly patients”. Am J Geriatr Pharmacother. 6 (4): 198–204. doi:10.1016/j.amjopharm.2008.10.003. PMID 19028375.
- ^ Jump up to:a b Bagnall A, Fenton M, Kleijnen J, Lewis R (2007). Bagnall A (ed.). “Molindone for schizophrenia and severe mental illness”. Cochrane Database Syst Rev (1): CD002083. doi:10.1002/14651858.CD002083.pub2. PMID 17253473.
- ^ https://www.fda.gov/Drugs/DrugSafety/DrugShortages/ucm050794.htm
- ^ “NEWS”. http://www.epic-pharma.com. Retrieved 2018-12-12.
- ^ Allison DB, Mentore JL, Heo M, et al. (1999). “Antipsychotic-induced weight gain: a comprehensive research synthesis”. Am J Psychiatry. 156 (11): 1686–96. doi:10.1176/ajp.156.11.1686 (inactive 2020-01-22). PMID 10553730. Free full text
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /moʊˈlɪndoʊn/ moh-LIN-dohn |
| Trade names | Moban |
| AHFS/Drugs.com | Consumer Drug Information |
| MedlinePlus | a682238 |
| Pregnancy category |
|
| Routes of administration |
By mouth (tablets) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Metabolism | Hepatic |
| Elimination half-life | 1.5 hours |
| Excretion | Minor, renal and fecal |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.254.109 |
| Chemical and physical data | |
| Formula | C16H24N2O2 |
| Molar mass | 276.380 g·mol−1 |
| 3D model (JSmol) | |
//////////MOLINDONE, SPN 810, SPN 801M, AFX 2201, молиндон, موليندون , 吗茚酮 ,
TILDACERFONT
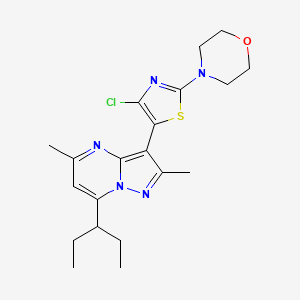
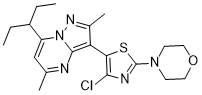
TILDACERFONT
| Synonyms: |
Tildacerfont 1014983-00-6 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(1-ethyl-propyl)-2,5-dimethyl-pyrazolo[1,5-a]pyrimidine 7-(1-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[1,5-a]pyrimidine |
|---|---|
| MW/ MF | 420 g/mol/ C20H26ClN5OS |
- Originator Spruce Biosciences
- Class2 ring heterocyclic compounds; Morpholines; Pyrazoles; Pyrimidines; Small molecules; Thiazoles
- Mechanism of Action Corticotropin receptor antagonists
- Orphan Drug Status Yes – Congenital adrenal hyperplasia
- New Molecular Entity Yes
- Phase II Congenital adrenal hyperplasia
- 09 Jul 2020 Spruce Biosciences initiates a phase II trial in Congenital adrenal hyperplasia in USA (PO) (NCT04457336)
- 24 Sep 2019 Spruce Biosciences completes a phase II trial in Congenital adrenal hyperplasia in USA (NCT03687242)
- 19 Sep 2019 Updated safety and efficacy data from a phase II trial in Congenital adrenal hyperplasia release by Spruce Biosciences
Deuterated pyrazolo[1,5-a]pyrimidine derivatives, particularly tildacerfont (SPR-001), useful as CRF antagonists for treating congenital adrenal hyperplasia. Spruce Bioscience is developing tildacerfont under license from Lilly as an oral capsule formulation for the treatment of congenital adrenal hyperplasia; in July 2017, a phase II trial for CAH was initiated.
Corticotropin releasing factor (CRF) is a 41 amino acid peptide that is the primary physiological regulator of proopiomelanocortin (POMC) derived peptide secretion from the anterior pituitary gland. In addition to its endocrine role at the pituitary gland, immunohistochemical localization of CRF has demonstrated that the hormone has a broad extrahypothalamic distribution in the central nervous system and produces a wide spectrum of autonomic, electrophysiological and behavioral effects consistent with a neurotransmitter or neuromodulator role in the brain. There is also evidence that CRF plays a significant role in integrating the response in the immune system to physiological, psychological, and immunological stressors.
PATENT
Product case, WO2008036579 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2008036579
Example 16
3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl- pyrazolo [ 1 ,5 -α]pyrimidine
Under a nitrogen atmosphere dissolve 3-(4-bromo-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine (116 mg, 0.25 mmol) in THF (1.5 mL) and chill to -78 0C. Add n-butyl lithium (0.1 mL. 2.5 M in hexane, 0.25 mmol) and stir at -78 0C for 30 min. Add N-chlorosuccinimide (33.4 mg, 0.25 mmol) and stir for another 30 min, slowly warming to room temperature. After stirring overnight, quench the reaction by adding a solution of saturated ammonia chloride and extract with ethyl acetate. Wash the organic layer with brine, dry over sodium sulfate, filter, and concentrate to a residue. Purify the crude material by flash chromatography, eluting with hexanes:dichloromethane: ethyl acetate (5:5:2) to provide the title compound (54 mg). MS (APCI) m/z (35Cl) 420.6 (M+l)+; 1H NMR (400 MHz, CDCl3): 6.44 (s, IH), 3.79 (t, 4H, J=4.8 Hz), 3.63-3.56 (m, IH), 3.47 (t, 4H, J=4.8 Hz), 2.55 (s, 3H), 2.45 (s, 3H), 1.88-1.75 (m, 4H), 0.87 (t, 6H, J=7.5 Hz).
Alternate Preparation from Preparation 6:
Combine 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine, (9 g,
26.2 mmol) and 4-chloro-2-morpholino-thiazole (7.5 g, 36.7 mmol) in
dimethylformamide (90 mL) previously degassed with nitrogen. Add cesium carbonate (17.8 g, 55 mmol), copper iodide (250 mg, 1.31 mmol), triphenylphosphine (550 mg, 2.09 mmol) and palladium acetate (117 mg, 0.52 mmol). Heat the mixture to 125 0C for 16 h and then cool to 22 0C. Add water (900 mL) and extract with methyl-?-butyl ether (3 x 200 mL). Combine the organic portions and evaporate the solvent. Purify by silica gel chromatography eluting with hexanes/ethyl acetate (4/1) to afford the title compound (6.4 g, 62%). ES/MS m/z (35Cl) 420 (M+l)+.
Example 16a
3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl- pyrazolo[l,5-α]pyrimidine, hydrochloride
Dissolve 3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-7-(l-ethyl-propyl)-2,5-dimethyl-pyrazolo[l,5-α]pyrimidine (1.40 g, 3.33 mmol) in acetone (10 mL) at 50 0C and cool to room temperature. Add hydrogen chloride (2 M in diethyl ether, 2.0 mL, 4.0 mmol) and stir well in a sonicator. Concentrate the solution a little and add a minimal amount of diethyl ether to crystallize the HCl salt. Cool the mixture in a refrigerator overnight. Add additional hydrogen chloride (2 M in diethyl ether, 2.0 mL, 4.0 mmol) and cool in a refrigerator. Filter the crystalline material and dry to obtain the title compound (1.15 g, 75%). ES/MS m/z (35Cl) 420 (M+l)+; 1H NMR(CDCO): 9.18 (br, IH), 6.86 (s, IH), 3.72 ( m, 4H), 3.49(m, IH), 3.39 (m, 4H), 2.48 (s, 3H), 2.38(s, 3H), 1.79 (m, 4H), 0.79 (m, 6H).
PATENT
US-20200255436
PATENT
WO2019210266
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019210266
claiming the use of CRF-1 antagonists (eg tildacerfont).
PATENT
WO 2010039678
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2010039678
EXAMPLES
Example 1 : 7-(l-ethyl-propyl)-3-(‘2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori ,5-alpyrimidine nthroline
Charge 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (1.03 g, 3.00 mmoles), K3PO4 (1.95 g, 9.00 mmoles), 2,4-dichlorothiazole (0.58 g, 3.75 mmoles), 1,10 phenanthroline (0.05 g, 0.30 mmoles) and anhydrous DMAC (5 mL) to a round bottom flask equipped with a magnetic stir bar, thermal couple and N2 inlet. Degas the yellow heterogeneous reaction mixture with N2 (gas) for 30 min. and then add CuI (0.06 g, 0.30 mmoles) in one portion followed by additional 30 min. degassing with N2 (gas). Stir the reaction mixture at 120 0C for about 6 hr. Cool the reaction mixture to room temperature overnight, add toluene (10 mL) and stir for 1 hr. Purify the mixture through silica gel eluting with toluene (10ml). Extract with 1 M HCl (10 mL), water (10 mL), brine (10 mL) and concentrate under reduced pressure to give a yellow solid. Recrystallize the solid from methanol (5ml) to yield the title compound as a yellow crystalline solid. (0.78 g, 70% yield, >99% pure by LC) MS(ES) = 369 (M+ 1). 1H NMR (CDCl3)= 6.5 (IH, s); 3.6 (IH, m); 2.6 (3H, s); 2.5 (3H, s); 1.9 (4H, m); 0.9 (6H, t).
Example 2: 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolol“! ,5-aipyrimidine
Charge 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (0.37 g, 1.00 mmoles), K2CO3 (0.28 g, 2.00 mmoles) and anhydrous morpholine (3 mL) to a round bottom flask equipped with a magnetic stir bar and N2 inlet. Stir the yellow mixture at 100 0C for about 4 hr., during which time the reaction becomes homogeneous. Cool the reaction mixture to room temperature, add H2O (10 mL) and stir the heterogeneous reaction mixture overnight at room temperature. Collected the yellow solid by filtration, wash with H2O and allowed to air dry overnight to give the crude title compound (391mg). Recrystallize from isopropyl alcohol (3 mL) to yield the title compound as a light yellow crystalline solid (380 mg, 90.6% yield, >99% by LC). MS(ES) = 420 (M+l). 1H NMR (CDCl3)= 6.45 (IH, s); 3.81 (m, 4H); 3.62 (IH, m); 3.50 (m, 4H); 2.6 (3H, s); 2.45 (3 H, s); 1.85 (4H, m); 0.9 (6H, t).
Example 3 :
The reactions of Example 1 are run with various other catalysts, ligands, bases and solvents, which are found to have the following effects on yield of 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine. (See Tables 1 – 4).
Table 1 : Evaluation of different li ands
(Reactions are carried out in parallel reactors with 1.2 mmol 2,4-dichlorothiazole, 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.5 mmol CuI, 0.5 mmol ligand and 2.1 mmol Cs2CO3 in 4 mL DMAC. The reactions are degassed under N2 for 30 min. and then heated at between 80 and
1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak. Longer reaction times are shown in parenthesis) Table 2: Evaluation of various solvents
(Reactions are carried out in parallel reactors with 1.2 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.25 mmol CuI, 0.25 mmol 1,10-phenanthroline and 2.1 mmol Cs2CO3 in 3 mL specified solvent. The reactions are degassed under N2 for 30 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Table 3 : Evaluation of different copper sources
(Reactions are carried out in in parallel reactors with 1 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.05 mmol CuX, 0.01 mmol 1,10-phenanthroline and 3 equivalents K3PO4 in 3 mL DMAC. The reactions are degassed under N2 for 30 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Table 4: Evaluation of various inorganic bases
(Reactions are carried out in in parallel reactors with 1 mmol 2,4-dichlorothiazole 1 mmol 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine, 0.1 mmol CuI, 0.1 mmol 1,10-phenanthroline and 2.1 mmol base and degassed for 30 minutes prior to the addition of 3 mL DMAC. The reactions are degassed under N2 for 10 minutes and then heated at 1000C overnight under N2. Percent product is measured as the percent of total area under the HPLC curve for the product peak.)
Example 4. Use of morpholine both as a reactant and base in 2-MeTHF as solvent.
solvent
7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-ajpyrimidine (15.2 g, 41.16 mmoles) is charged into a 250 mL 3-necked round bottomed flask, followed by addition of 2-MeTHF (61 mL, 4.0 volumes), the yellowish brown slurry is stirred at about 20 0C for 5 min. Then morpholine (19 g, 218.18 mmoles) is added over 2-5 minutes. Contents are heated to reflux and maintained at reflux for 12 hr. The slurry is cooled to 25 0C, followed by addition of 2-MeTHF (53 mL, 3.5 volumes) and water ( 38 mL 2.5 volumes). The reaction mixture is warmed to 40 0C, where upon a homogenous solution with two distinct layers formed. The layers are separated, the organic layer is filtered and concentrated to ~3 volumes at atmospheric pressure. Four volumes 2-propanol (61 mL) are added. The solution is concentrated to ~3 volumes followed by addition of 4 volumes 2-propanol (61 mL), re-concentrated to ~3 volumes, followed by addition of another 6 volumes 2-propanol (91 mL), and refluxed for 15 min. The clear solution is gradually cooled to 75 0C, seeded with 0.45 g 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine slurried in 2 mL 2-propanol, rinsed with an additional 2 mL 2-propanol and transferred to a crystallization flask. The slurry is cooled to between 0-5 0C, maintained for 1 hr, filtered and the product rinsed with 2-propanol (30 mL, 2 volumes). The solid is dried at 60 0C in a vacuum oven to afford 16.92 g 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine. Purity of product by HPLC assay is 100.00 %. XRPD and DSC data of product is consistant with reference sample. MS(ES) = 420 (M+ 1).
Example 5. Use of morpholine as both reactant and base in 2-propanol as solvent.
7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-ajpyrimidine (11.64 mmoles) is charged into a 100 mL 3 -necked round bottomed flask followed by addition of 2-propanol ( 16 mL, 3.72 volumes). The yellowish brown slurry is stirred at about 20 0C for 5 min. Then morpholine (3.3 g, 37.84 mmoles) is added over 2-5 minutes. Contents are refluxed for 6 hr. The slurry is cooled to 25 0C. 2-Propanol ( 32 mL, 7.44 volumes) and water ( 8.6 mL, 2.0 volumes) are added and the mixture warmed to 70-75 0C, filtered and concentrated to ~ 9 volumes at atmospheric pressure. The clear solution is gradually cooled to 55 0C, seeded with 0.06 g of crystalline 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine slurried in 0.5 mL 2-propanol, rinsed with additional 0.5 mL 2-propanol and added to crystallization flask. The slurry is cooled to 0-5 0C, maintained for 1 hr., filtered and the product rinsed with 2-propanol ( 9 mL, 2.1 volumes). Suctioned dried under vacuum at 60 0C to afford 4.6 g of dry 7-(l-ethyl-propyl)-3-(4-chloro-2-morpholin-4-yl-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (88.8 % yield, purity by HPLC assay is 99.88 % ). MS(ES) = 420 (M+ 1).
Example 6: 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori ,5-alpyrimidine
7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (10 g, 29.17 mmoles), 2, 4-dichlorothiazole (5.2 g , 33.76 mmoles), cesium carbonate(19.9g, 61.07 mmoles) and 1,10-phenanthroline (1 g, 5.5 mmoles) are charged into a 250 mL 3-necked round bottomed flask, followed by 2-MeTHF (36 mL, 3.6 volumes). The reaction mixture is degassed with nitrogen and then evacuated. Cuprous chloride (0.57 g, 5.7 mmoles), DMAC (10 mL, 1 volume) and 2-MeTHF (4 mL, 0.4 volumes) are added in succession. The reaction mixture is degassed with nitrogen and then evacuated. The contents are refluxed for 20 hr. The reaction mixture is cooled to -70 0C and 2-MeTHF (100 mL, 10 volumes) is added. The contents are filtered at ~70 0C and the residual cake is washed with 2-MeTHF (80 mL, 8 volumes) at about 65-72°C. The filtrate is transferred into a separatory funnel and extracted with water. The organic layer is separated and washed with dilute HCl. The resulting organic layer is treated with Darco G60, filtered hot (600C). The filtrate is concentrated at atmospheric pressure to -2.8 volumes. 25 mL 2-propanol is added, followed by re-concentration to -2.8 volumes. An additional 25 mL 2-propanol is added, followed again by re-concentration to -2.8 volumes. Finally, 48 mL 2-propanol is added. The contents are cooled to -7 0C, maintained at -7 0C for 1 hr., filtered and rinsed with 20 mL chilled 2-propanol. Product is suction dried and then vacuum dried at 60 0C to afford 9.41 g 7-(l-ethyl-propyl)-3-(2,4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (purity of product by HPLC assay is 95.88 %). MS(ES) = 369 (M+ 1).
Example 7. Synthesis of 7-(l-ethyl-propyl)-3-(2, 4-dichloro-thiazol-5-yl)-2,5-dimethyl-pyrazolori,5-a1pyrimidine using 1,4-Dioxane solvent and CuCl catalyst
Add dioxane (9.06X), Cs2CO3 (2.00X), 7-(l-ethyl-propyl)-3-iodo-2,5-dimethyl-pyrazolo[l,5-a]pyrimidine (1.0 equivalent), 2,4-dichlorothiazole (0.54 equivalent) to a reactor under N2. Purge the reactor with N2 three times, degas with N2 for 0.5-1 hr., and then add 1,10-phenanthroline (0.3 eq) and CuCl (0.3eq) under N2 , degassing with N2 for 0.5-1 hr. Heat the reactor to 1000C -1100C under N2 . Stir the mixture for 22-24 hr. at 100 0C -1100C. Cool to 10~20°C and add water (10V) and CH3OH (5V), stir the mixture for 1-1.5 hr. at 10~20°C. Filter the suspension, resuspend the wet cake in water, stirr for 1-1.5 hr. at 10~20°C, and filter the suspension again. Charge the wet cake to n-heptane (16V) and EtOAc (2V) under N2. Heat the reactor to 40 °C~500C under N2.
Active carbon (0. IX) is added at 40 °C~500C. The reactor is heated to 55°C~650C under N2 and stirred at 55 °C~650C for 1-1.5 hr. The suspension is filtered at 40~55°C through diatomite (0.4 X). The cake is washed with n-heptane (2.5V). The filtrate is transferred to another reactor. EtOAc (10V) is added and the the organic layer washed with 2 N HCl (10V) three times, followed by washing two times with water (10X, 10V). The organic layer is concentrated to 3-4V below 500C. The mixture is heated to 80-90 0C. The mixture is stirred at this temperature for 40-60 min. The mixture is cooled to 0~5°C, stirred for 1-1.5 hr. at 0~5°C and filtered. The cake is washed with n-heptane (IV) and vacuum dried at 45-500C for 8-10 hr. The crude product is dissolved in 2-propanol (7.5V) under N2, and re-crystallized with 2-propanol. The cake is dried in a vacuum oven at 45°C~50°C for 10-12 hr. (55-80% yield). 1H NMR56.537 (s, IH) 3.591-3.659 (m, IH, J=6.8Hz), 2.593 (s, 3H), 2.512 (s, 3H), 1.793-1.921(m, 4H), 0.885-0.903 (m, 6H).
REFERENCES
1: Zorrilla EP, Logrip ML, Koob GF. Corticotropin releasing factor: a key role in the neurobiology of addiction. Front Neuroendocrinol. 2014 Apr;35(2):234-44. doi: 10.1016/j.yfrne.2014.01.001. Epub 2014 Jan 20. Review. PubMed PMID: 24456850; PubMed Central PMCID: PMC4213066.
/////////////tildacerfont, SPR 001, Orphan Drug Status, Congenital adrenal hyperplasia, SPRUCE BIOSCIENCES, PHASE 2
CCC(CC)C1=CC(=NC2=C(C(=NN12)C)C3=C(N=C(S3)N4CCOCC4)Cl)C
SULCARDINE SULPHATE
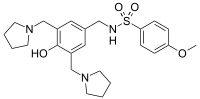
sulcardine, HBI-3000
B 87823
- Molecular FormulaC24H33N3O4S
- Average mass459.602 Da
N-[[4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)phenyl]methyl]-4-methoxybenzenesulfonamide
heart arrhythmia
.gif)
CAS No. : 343935-61-5 (Sulcardine sulfate)
| Synonyms: | B-87823; HBI-3000; B87823; HBI3000; B 87823; HBI 3000;N-(4-hydroxy-3,5-bis(pyrrolidin-1-ylmethyl)benzyl)-4-methoxybenzenesulfonamide sulfate |
| Molecular Formula: | C24H35N3O8S2 |
| Molecular Weight: | 557.67 |
- Originator Jiangsu Furui Pharmaceuticals; Shanghai Institute of Materia Medica
- Developer HUYA Bioscience International; Jiangsu Furui Pharmaceuticals
- Class Antiarrhythmics; Small molecules
- Mechanism of ActionIon channel antagonists
- Phase I Atrial fibrillation
- No development reported Arrhythmias
- 13 Mar 2020 Chemical structure information added
- 28 Feb 2020 No recent reports of development identified for preclinical development in Arrhythmias in USA (IV)
- 16 Dec 2019 Adverse events data from a phase I trial in Atrial fibrillation (In volunteers) presented at the American Heart Association Scientific Sessions 2019 (AHA-2019)
HUYA Bioscience , under license from Shanghai Institute of Materia Medica (SIMM), is developing sulcardine (HBI-3000, oral, i.v, heart arrhythmia), a myocardial ion channel inhibitory compound, for the treatment of arrhythmia; In September 2016, the drug was still in phase II development, as of August 2020, the company website states that a phase II trial was pending in China.
HBI-3000 (sulcardine sulfate) is an experimental drug candidate that is currently in phase II of human clinical trials as an antiarrhythmic agent.[1][needs update] Clinical investigation will test the safety and efficacy of HBI-3000 as a treatment for both atrial and ventricular arrhythmias.[2]
The molecular problem
Anti-arrhythmic medication is taken to treat irregular beating of the heart. This irregular beating results from a deregulation of the initiation or propagation of the electrical stimulus of the heart. The most common chronic arrhythmia is atrial fibrillation.[3] There is an increased incidence of atrial fibrillation in the elderly and some examples of complications include heart failure exacerbation, hypotension and thrombembolic events.[3]
Most anti-arrhythmic medications exert their effects by decreasing the permeability of potassium ion channels (IKr) in heart cells. These potassium channel blockers delay ventricular repolarization and prolong action potential duration (APD; the prolongation of the electrical stimulus within heart cells). These changes can lower heart rate, eliminate atrial fibrillation, and ultimately sudden cardiac death.[4][5]
Mechanism of action in ventricular myocytes
Ventricular myocytes are heart muscle cells found in the lower chambers of the heart. Heart rate is dependent on the movement of an electrical stimulus through the individual heart cells. This is mediated by the opening of ion channels on cell surfaces. HBI-3000 exerts its effects on the heart by inhibiting multiple ion channels (INa-F, INa-L, ICa-L and IKr), but predominantly the INa-L ion channel . By decreasing the ion permeability of these channels, HBI-3000 slightly prolongs APD (due to IKr); however, unlike pure IKr channel blockers, it is self-limited (due to the decreased permeability of INa-L and ICa-L). This is similar to the medications ranolazine and amiodarone.[5] HBI-3000 suppresses early afterdepolarizations (EADs; a change in the normal net flow of ions during repolarization), does not produce any electrical abnormalities, and displays minimally pronounced prolongation of APD during a slow heart rate (i.e. stimulated at a slower frequency). Pronounced prolongation of APD during a slow heart rate can lead to proarrythmias. Overall, HBI-3000 seems to have a low proarrhythmic risk. The effect of HBI-3000 on contractility and cardiac conduction requires further investigation.[5]
Studies
Animal model
In a canine model, the intravenous injection of HBI-3000 demonstrated to be an effective anti-arrhythmic and anti-fribrillatory agent.[6]
Cellular isolation
The administration of HBI-3000 to isolated heart muscle cells demonstrated the potential to improve arrhythmias while having low proarrhythmic risk.[5]
Human studies
Jiangsu Furui Pharmaceuticals Co., Ltd is currently recruiting participants in their study.[1][
PAPER
http://www.simm.cas.cn/wyp/wyp_lw/201804/W020180420480084769998.pdf

N-[3,5-bis(1-pyrrolidylmethyl)-4-hydroxybenzyl]-4-methoxybenzenesulfamide (sulcardine, 6f) and the sulfate (sulcardine sulfate) (1) To a suspension of 4-hydroxybenzylamine (133 g, 1.08 mol) in DMF (500 mL) was added dropwise 4-methoxybenzensul-fonyl chloride (206 g, 1.00 mol) in DMF (320 mL) over a period of 30 min at 0–10 °C with stirring, followed by the addition of triethylamine (158 mL, 1.12 mol) over 30 min at the same temperature. The stirring was continued for an additional 1.5 h at room temperature. The reaction mixture was poured into ice-water (5 L). After stirring for 10 min, the suspension was allowed to stand for 2 h. The solid was filtered, washed with water (300 mL×3), and dried in a desiccator over anhydrous calcium chloride, yielding N-(4-hydroxybenzyl)-4-methoxybenzenesulfamide (11) (248 g, 85%) as a white solid, mp 160–162 °C. The authentic sample was obtained by recrystallization from ethyl acetate, mp 161–162 °C. 1 H NMR (CD3OD) δ 3.70 (s, 3H), 3.76 (s, 2H), 6.48 (d, J=8.4 Hz, 2H), 6.82(d, J=8.4 Hz, 2H), 6.86 (d, J=8.7 Hz, 2H), 7.56 (d, J=8.7 Hz, 2H). EIMS (m/z): 293 (M+ ), 254, 195, 185, 171, 155, 149, 122 (100), 107, 99, 77, 65. Anal. (C14H15NO4S) C, H, N.
(2) A mixture of 11 (230 g, 0.78 mmol), pyrrolidine (200 mL, 2.44 mol) and 36% aqueous formaldehyde (250 mL, 3.30 mol) in ethanol (800 mL) was stirred under reflux for 8 h. The reaction mixture was concentrated under vacuum to dryness. The resulting oil residue was dissolved in chloroform (350 mL), and the solution was washed with water (300 mL×3). Under stirring, the organic layer was mixed with water (300 mL), and then concentrated hydrochloric acid (approximately 165 mL) was added portionwise at 0-10 °C to adjust the pH of the aqueous phase to ~2. The aqueous phase was washed with chloroform (200 mL) and then mixed with additional chloroform (300 mL). Under stirring, the two-phase mixture was treated portionwise with 25%–28% aqueous ammonia (~300 mL) to adjust the pH of the aqueous phase to 9–10. The organic layer was separated, and the aqueous layer was further extracted with chloroform (200 mL×2). The combined organic layer was dried over anhydrous sodium sulfate and concentrated under vacuum to dryness. The oily residue was treated with acetone (45 mL) and isopropyl ether (290 mL), and the mixture was heated under reflux until the suspension became a solution. The solution was cooled to room temperature, seeded with an authentic sample, and allowed to stand at 0°C overnight. The solid was filtered and dried under vacuum, yielding product 6f (290 g, 81%) as a yellowish solid, mp 96–98 °C. The authentic sample was obtained by preparative TLC or column chromatography (silica gel; CHCl3:MeOH:25% NH4OH=92:7:1). The compound could be recrystallized from ethanol-water, mp 101–102 °C. 1 H NMR (CDCl3) δ 1.77–1.86 (m, 8H), 2.53–2.63 (m, 8H), 3.68 (s, 4H), 3.86 (s, 3H), 3.97 (s, 2H), 6.86 (s, 2H), 6.95 (d, J=8.7 Hz, 2H), 7.78 (d, J=8.6 Hz 2H). EIMS (m/z): 459 (M+ ), 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S) C, H, N.
(3) Under stirring, the Mannich base 6f (150.5 g, 0.327 mol) was mixed with 2 mol/L H2SO4 (172 mL, 0.344 mol), and the mixture was heated at 80 °C until the solid dissolved. The solution was cooled to room temperature, seeded with an authentic sample, and the sulfate of 6f was formed as crystals. To the stirred mixture was added anhydrous ethanol (520 mL), and the mixture was allowed to stand at 0°C for 24 h. The solid was filtered, washed with ethanol, and recrystallized with 80% ethanol (250 mL). The sulfate was dried over concentrated sulfuric acid in a desiccator, giving the sulfate of 6f (143 g, 71%) as a trihydrate, mp 125–140°C. 1 H NMR (D2O) δ 2.00–2.13 (m, 4H), 2.14–2.25 (m, 4H), 3.12–3.22 (m, 4H), 3.45– 3.55 (m, 4H), 3.90 (s, 3H), 4.20 (s, 2H), 4.33 (s, 4H), 7.06 (d, J=8.7 Hz, 2H), 7.28 (s, 2H), 7.66 (d, J=8.9 Hz, 2H). 13C NMR (D2O) δ 24.7, 47.6, 55.7, 56.1, 58.1, 116.6, 122.5, 131.3, 132.3, 133.3, 136.0, 155.8, 164.8. EIMS (m/z): 459, 390, 388, 202, 171, 148, 107, 84, 70 (100). Anal. (C24H33N3O4S∙H2SO4∙3H2O) C, H, N, S.
PATENT
Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
https://patents.google.com/patent/US6605635
Synthesis and antiarrhythmic activities of changrolin (1) have been reported (Liangquan Li, et al., Scientia Sinica, 1979, 7, 723; Weizhou Chen, et al., Acta Pharmaceutica Sinica, 1979, 14, 710). Thereafter, investigations of the chemical structural modifications and the physiological activities have successively been carried out by domestic and foreign scientists (Cunji Sun, et al., Acta Pharmaceutica Sinica, 1981, 16, 564; 1986, 21, 692; Mulan Lin, et al., ibid., 1982, 17, 212; D. M. Stout, et al. J. Med. Chem., 1983, 26, 808; 1984, 27, 1347; 1985, 28, 295; 1989, 32, 1910; R. J. Chorvat, et al., ibid., 1993, 36, 2494).

Changrolin is an effective antiarrhythmic agent. Ventricular premature beats disappear 2-3 days after oral administration of changrolin to patients suffering from arrhythmia; I.v. injection or instillaton may result in significant reduction or even disappearence of ventricular premature beats and ventricular tachycardia. However, oral administration of changrolin for a period of over one month may cause a reversible pigmentation on the skin of patients, which gradually retrogresses after ceasing the administration. This pigmentation is associated to the subcutaneous oxidation of certain structural moieties in changrolin molecule or to its instability in solution.
EXAMPLE 1N-[3,5-bis(1-Piperidinomethyl)-4-hydroxy]phenyl-1-naphthalenesulfonamide (B-87836)
(1) To a solution of 4-aminophenol (4.5 g) in dioxane (20 ml) was added dropwise a solution of 1-naphthalenesulfonyl chloride (4.4 g) in dioxane (20 ml). The mixture was further stirred at room temperatue for 4.5 hours and poured into water. The precipitate was collected by filtration, recrystallized from ethanol and decolored with activated carbon to give N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (4.2 g), mp 195-196° C.
(2) A mixture of N-(ρ-hydroxyphenyl)-1-naphthalenesulfonamide (2.0 g), 37% aqueous formaldehyde (4.5 g) and piperidine (5.6 g) in ethanol (100 ml) was heated to reflux for 50 hours. The ethanol was removed by evaporation in vacuo and chloroform was added to the residue. The organic layer was washed with water then dried over anhydrous Na2SO4. Then the chloroform was removed in vacuo and the residue was triturated in water to give a solid, which was then recrystallized from ethanol to give the titled product (1.4 g), mp 197-198° C.
1HNMR(CDCl3): 1.30-1.50(m, 12H), 2.10-2.21(m, 8H), 3.28(s, 4H), 6.45(s, 2H), 7.24-8.04(m, 6H), 8.56(m, 1H). Elemental analysis (C28H35N3O3S ): Calcd. (%): C, 68.12; H, 7.15; N, 8.51. Found (%): C, 67.96; H, 7.16; N, 8.56.
PATENT
WO-2020159959
Novel crystalline forms of acid salts of sulcardine useful for treating arrhythmia and atrial fibrillation.
4-Methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)-4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), also known as sulcardine, and its salts, such as sulcardine sulfate, constitute a group of compounds with potent anti -arrhythmic activity. Sulcardine is a multi-ion channel blocker that specifically inhibits iNa-Peak, iNa-Late, Ica,L, and Ixrwith similar in vitro potencies (and Ito and IKUT to a lesser degree) in human atrial cardiomyocytes and represents what may be the sole example of a substituted sulfonamide class of anti-arrhythmic. Sulcardine salts can be used as an intravenous injectable or as oral doses for the treatment of arrhythmias, including supraventricular tachyarrhythmia, premature ventricular contractions, ventricular tachycardia, ventricular fibrillation, and atrial fibrillation. See, e.g ., U.S. Patent Nos. 8,541,464 and 8,637,566. Preparation of sulcardine sulfate salt has been reported in U.S. Patent No. 6,605,635.
[0004] In addition, the evidence to date suggests that one advantage of sulcardine and its salts is that they lack significant pro-arrhythmic activity, as demonstrated in rigorous preclinical safety models, including a post-MI sudden-death conscious canine model and the validated rabbit ventricular wedge model. Additionally, it has been shown that they do not significantly increase defibrillation threshold, nor increase defibrillation failure risk in a post-MI canine model as was seen with flecainide. On the basis of these data, sulcardine and salts, with their very low apparent pro-arrhythmic potential, could potentially be used to treat acute and recurrent atrial fibrillation in the presence of organic heart disease, prolonged QR syndrome, and ventricular arrhythmias, including premature ventricular contractions (PVCs), ventricular tachycardia (VT), and ventricular fibrillation (VF), in either acute- or chronic-administration settings owing to their ability to be formulated into intravenous and oral dosing formulations.
Sulcardine has a chemical name of 4-methoxy-N-(3,5-bis-(l-pyrrolidinylmethyl)- 4-hydroxybenzyl)benzene sulfonamide (or N-(4-hydroxy-3,5-bis(pyrrolidin-l-ylmethyl)benzyl)-4-methoxybenzenesulfonamide), and has the following structure:
[0062] Sulcardine sulfate has the following structure:
[0063] Sulcardine sulfate can exist in a hydrated form. One such form is a trihydrate.
HPLC analysis was performed on a Dionex Ultimate 3000 instrument with the following parameters:
Column: Phenomenex Luna C18, 150×4.6mm, 5pm
Column Temperature: 30°C
Mobile Phase A: 0.2% Phosphoric Acid
Mobile Phase B: Methanol
Diluent: 50:50 MeOH:H20
Runtime: 12 minutes
Flow Rate: l.OmL/min
Injection Volume: 5pL
Detection: 237 nm
Gradient:
EXAMPLE 2 – PREPARATION OF FREE BASE AND SCREENING
[00348] Sulcardine sulfate trihydrate was dissolved in ethyl acetate (16 vol.) and saturated sodium bicarbonate solution (16 vol.). The biphasic solution was transferred to a separating funnel and the layers separated. The organic layer was dried over sodium sulfate and then the solvent was removed by rotary evaporation and the resulting oil dried under vacuum at ambient temperature for ca. 3 hr. FIG. 4 is an XRPD pattern of the resulted amorphous sulcardine free base. In all cases, the initial screening work detailed below was performed on 10 mg of sulcardine free base. All XRPD diffractograms were compared with sulcardine sulfate trihydrate, sulcardine free base and relevant counterions and found to be distinct.
Patent
WO2020123824
claiming treatment of atrial fibrillation (AF) by intravenously administering sulcardine sulfate .
PATENT
References
- ^ Jump up to:a b Jiangsu Furui Pharmaceuticals (November 5, 2010). “Efficacy and safety of sulcardine sulfate tablets in patients with premature ventricular contractions”. ClinicalTrials.gov. U.S. National Library of Medicine. Retrieved 2019-12-20.
- ^ “HUYA Bioscience Int’l announces clinical trial milestones in China for promising new anti-arrhythmic compound; Data supports desirable safety profile” (Press release). San Francisco, California: HUYA Bioscience International. Retrieved 2019-12-20.
- ^ Jump up to:a b Mashal, Abdallah; Katz, Amos; Shvartzman, Pesach (2011). “Atrial fibrillation: A primary care cross-sectional study”. Israel Medical Association Journal. 13 (11): 666–671. PMID 22279699.
- ^ Farkas, András; Leprán, István; Papp, Julius Gy. (1998). “Comparison of the antiarrhythmic and the proarrhythmic effect of almokalant in anaesthetised rabbits”. European Journal of Pharmacology. 346 (2–3): 245–253. doi:10.1016/S0014-2999(98)00067-3. PMID 9652366.
- ^ Jump up to:a b c d Guo, Donglin; Liu, Que; Liu, Tengxian; Elliott, Gary; Gingras, Mireille; Kowey, Peter R.; Yan, Gan-Xin (2011). “Electrophysiological properties of HBI-3000: A new antiarrhythmic agent with multiple-channel blocking properties in human ventricular myocytes”. Journal of Cardiovascular Pharmacology. 57 (1): 79–85. doi:10.1097/FJC.0b013e3181ffe8b3. PMID 20980921.
- ^ Lee, Julia Y.; Gingras, Mireille; Lucchesi, Benedict R. (2010). “HBI-3000 prevents sudden cardiac death in a conscious canine model”. Heart Rhythm. 7 (11): 1712. doi:10.1016/j.hrthm.2010.09.028.
 |
|
| Names | |
|---|---|
| IUPAC name
N-({4-Hydroxy-3,5-bis[(pyrrolidin-1-yl)methyl]phenyl}methyl)-4-methoxybenzene-1-sulfonamide
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C24H33N3O4S | |
| Molar mass | 459.61 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
| Infobox references | |
////////////////sulcardine sulfate, phase 2, china, HBI 3000, atrial fibrillation, B 87823,
COC1=CC=C(C=C1)S(=O)(=O)NCC2=CC(=C(C(=C2)CN3CCCC3)O)CN4CCCC4
