
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
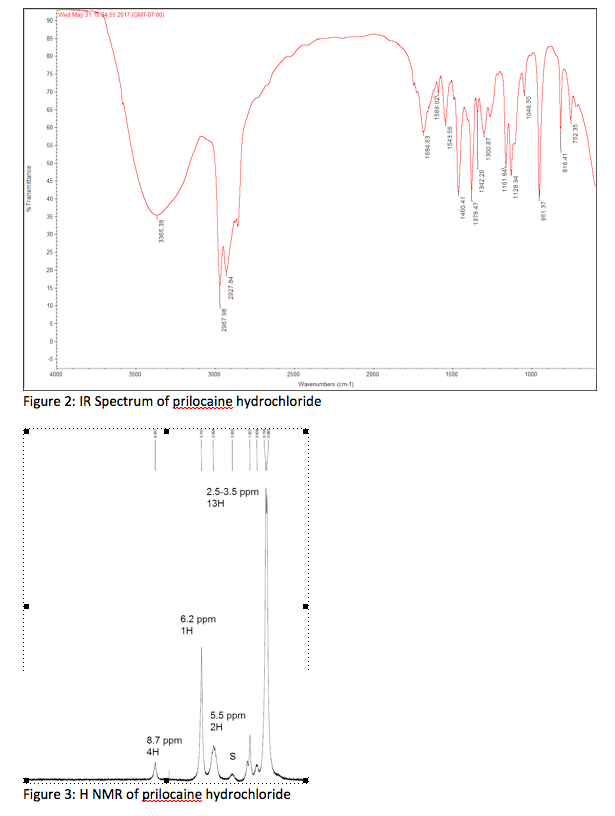
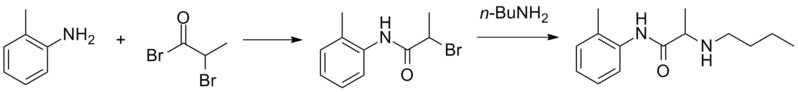
PRILOCAINE

PRILOCAINE
- Molecular FormulaC13H20N2O
- Average mass220.311 Da
Propanamide, N-(2-methylphenyl)-2-(propylamino)-
прилокаин [Russian] [INN]
بريلوكائين [Arabic] [INN]
丙胺卡因 [Chinese] [INN]
1641
211-957-0[EINECS]
721-50-6[RN]
N-(2-Methylphenyl)-2-(propylamino)propanamid
N-(2-méthylphényl)-2-(propylamino)propanamide
PrilocaineCAS Registry Number: 721-50-6
CAS Name:N-(2-Methylphenyl)-2-(propylamino)propanamide
Additional Names: 2-(propylamino)-o-propionotoluidide; N-(a-propylaminopropionyl)-o-toluidine; a-propylamino-2-methylpropionanilide; propitocaine
Molecular Formula: C13H20N2O
Molecular Weight: 220.31
Percent Composition: C 70.87%, H 9.15%, N 12.72%, O 7.26%
Literature References: Prepn: N. Löfgren, C. Tegner, Acta Chem. Scand.14, 486 (1960); GB839943; N. Löfgren, C. Tegner, US3160662 (1960, 1964 both to Astra).
Properties: Needles, mp 37-38°. bp0.1 159-162°. nD20 1.5298.
Melting point: mp 37-38°
Boiling point: bp0.1 159-162°
Index of refraction:nD20 1.5298 Derivative Type: Hydrochloride
CAS Registry Number: 1786-81-8
Manufacturers’ Codes: L-67
Trademarks: Citanest (AstraZeneca); Xylonest (AstraZeneca)
Molecular Formula: C13H20N2O.HCl
Molecular Weight: 256.77
Percent Composition: C 60.81%, H 8.24%, N 10.91%, O 6.23%, Cl 13.81%
Properties: Crystals from ethanol + isopropyl ether, mp 167-168°. Readily sol in water.
Melting point: mp 167-168° Therap-Cat: Anesthetic (local).Keywords: Anesthetic (Local).
- ASTRA 1512
- ASTRA 1515
- ASTRA-1512
- ASTRA-1515
- L 67
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Prilocaine hydrochloride | MJW015BAPH | 1786-81-8 | BJPJNTKRKALCPP-UHFFFAOYSA-N |
Agoneaze, Anodyne Lpt, Citanest, Citanest Forte, Dermacinrx Prikaan, Dermacinrx Prizopak, Emla, Fortacin, Lido Bdk, Lido-prilo Caine Pack, Lidopril, Oraqix, Prilolid, Prizotral, Relador
Prilocaine is a local anesthetic used in dental procedures.
A local anesthetic that is similar pharmacologically to lidocaine. Currently, it is used most often for infiltration anesthesia in dentistry. (From AMA Drug Evaluations Annual, 1992, p165)
Prilocaine (/ˈpraɪləˌkeɪn/[1]) is a local anesthetic of the amino amide type first prepared by Claes Tegner and Nils Löfgren. In its injectable form (trade name Citanest), it is often used in dentistry. It is also often combined with lidocaine as a topical preparation for dermal anesthesia (lidocaine/prilocaine or EMLA), for treatment of conditions like paresthesia. As it has low cardiac toxicity, it is commonly used for intravenous regional anaesthesia (IVRA).
Contraindications
In some patients, ortho-toluidine, a metabolite of prilocaine, may cause methemoglobinemia, which may be treated with methylene blue. Prilocaine may also be contraindicated in people with sickle cell anemia, anemia, or symptomatic hypoxia.[2]
Combinations
It is given as a combination with the vasoconstrictor epinephrine under the trade name Citanest Forte. It is used as an eutectic mixture with lidocaine, 50% w/w, as lidocaine/prilocaine. The mixture is an oil with a melting point of 18 °C (64 °F). A 5% emulsion preparation, containing 2.5% each of lidocaine/prilocaine, is marketed by APP Pharmaceuticals under the trade name EMLA (an abbreviation for eutectic mixture of local anesthetics).[3]
NMR
![1 H-nuclear magnetic resonance ( 1 H-NMR) spectra of prilocaine solution after sterilization with the assignment of the prilocaine hydrogens. [Prilocaine] = 5 mM, 20°C, 500 MHz.](https://www.researchgate.net/profile/Francisco-Groppo/publication/23458262/figure/fig3/AS:394557704425486@1471081295276/1-H-nuclear-magnetic-resonance-1-H-NMR-spectra-of-prilocaine-solution-after.png) 1 H-nuclear magnetic resonance ( 1 H-NMR) spectra of prilocaine solution after sterilization with the assignment of the prilocaine hydrogens. [Prilocaine] = 5 mM, 20°C, 500 MHz.
1 H-nuclear magnetic resonance ( 1 H-NMR) spectra of prilocaine solution after sterilization with the assignment of the prilocaine hydrogens. [Prilocaine] = 5 mM, 20°C, 500 MHz.
Compendial status

Table 1 The common types of local anesthetics
| COMPOUND | STRUCTURE | TIME TO MARKET | APPLICATION METHODS |
|---|---|---|---|
| Procaine | 1904 | Infiltration anesthesia, conduction anesthesia, subarachnoid anesthesia and epidural anesthesia | |
| Chloroprocaine | 1952 | Infiltration anesthesia, epidural anesthesia and conduction anesthesia | |
| Hydroxyprocaine | 1960 | Infiltration anesthesia | |
| Tetracaine | 1988 | Conduction anesthesia, subarachnoid anesthesia and epidural anesthesia | |
| Oxybuprocaine | 1975 | Topical anesthesia | |
| Tutocaine | 1976 | Topical anesthesia and infiltration anesthesia | |
| Butacaine | 1976 | Topical anesthesia and infiltration anesthesia | |
| Dimethocaine | 1938 | Topical anesthesia and infiltration anesthesia | |
| Thiocaine | Halt sales | Topical anesthesia and infiltration anesthesia | |
| Lidocaine | 1948 | Conduction anesthesia and epidural anesthesia | |
| Mepivacaine | 1986 | Infiltration anesthesia, conduction anesthesia, epidural anesthesia and topical anesthesia | |
| Bupivacaine | 2000 | Infiltration anesthesia, conduction anesthesia and epidural anesthesia | |
| Ropivacaine | 1996 | Infiltration anesthesia, conduction anesthesia and epidural anesthesia | |
| Trimecaine | 1965 | Infiltration anesthesia, surface anesthesia and epidural anesthesia | |
| Prilocaine | 1993 | Infiltration anesthesia, topical anesthesia and epidural anesthesia | |
| Etidocaine | 1976 | Epidural anesthesia | |
| Pyrrocaine | 1964 | Conduction anesthesia and epidural anesthesia | |
| Butanilicaine | 1982 | Infiltration anesthesia and conduction anesthesia | |
| Cinchocaine | 1985 | Topical anesthesia, subarachnoid anesthesia and epidural anesthesia | |
| Articaine | 2002 | Infiltration anesthesia and subarachnoid anesthesia | |
| Dyclonine | 1956 | Topical anesthesia | |
| Falicaine | 1957 | Topical anesthesia | |
| Quinisocaine | 1957 | Topical anesthesia | |
| Pramocaine | 1977 | Topical anesthesia | |
| Diperodon | 1980 | Topical anesthesia | |
| Heptacaine | 1984 | Infiltration anesthesia |

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Synthesis Reference
SYN
English: N. Lofgren and C. Tegner, Acta Chem. Scand., 14, 486 (1960). DOI number: 10.3891/acta.chem.scand.14-0486
SYN
SUN
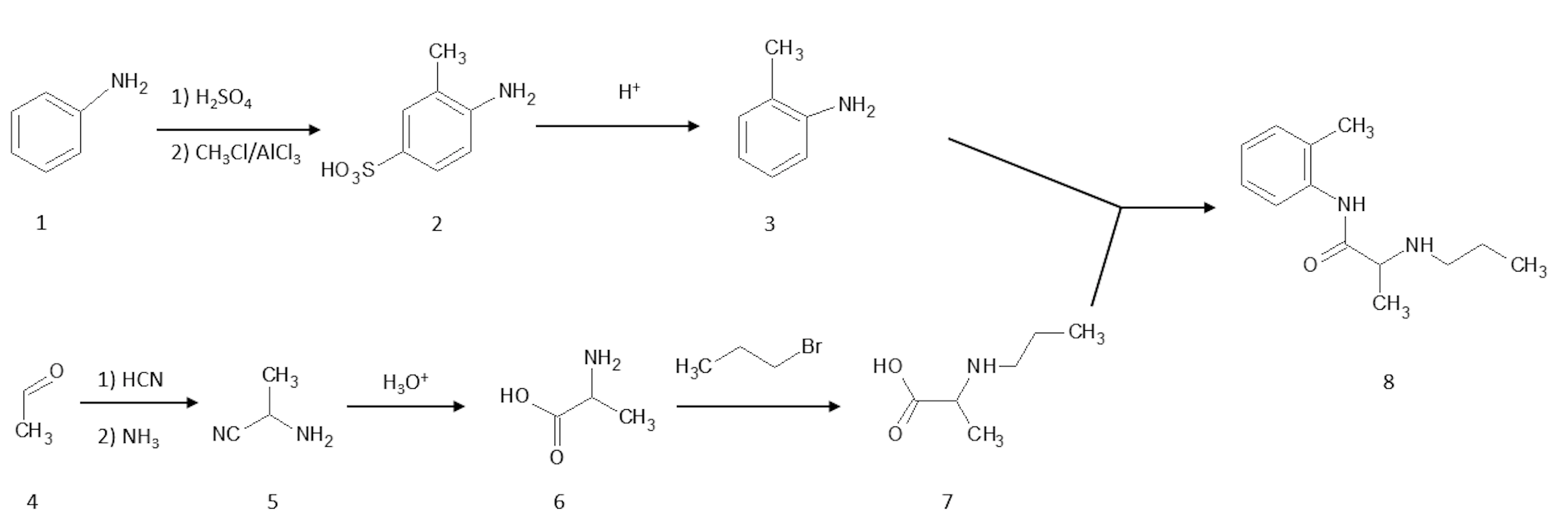
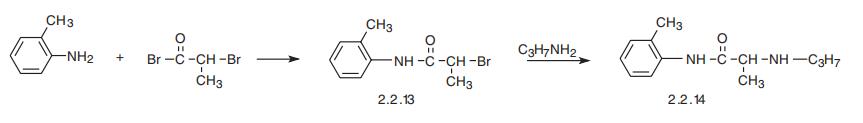
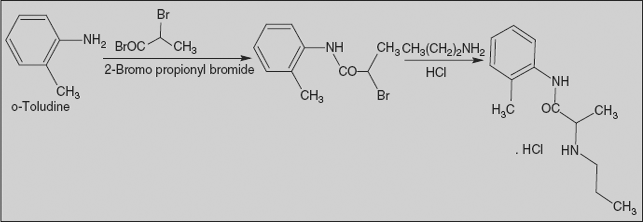
Prilocaine, 2-(propylamino)-o-propiontoluidine (2.2.14), is structurally related to the exact same group as ethidocaine, yet it differs structurally in that during synthesis, o-toluidine is used instead of 2,6-dimethylaniline, and instead of a butyric acid, a fragment of propionic acid, and a terminal propylethylamine group is replaced with a propylamine group. In order to synthesize prilocaine, o-toluidine is reacted with bromopropionyl bromide, and the resulting bromopropionyltoluidide (2.2.13) is then reacted with propylamine, which gives prilocaine [22,23].
SYN

SYN
| Clinical data | |
|---|---|
| Trade names | Citanest |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a603026 |
| License data | EU EMA: by INNUS DailyMed: Prilocaine |
| Pregnancy category | AU: A |
| Routes of administration | Subcutaneous |
| ATC code | N01BB04 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)US: ℞-only |
| Pharmacokinetic data | |
| Protein binding | 55% |
| Metabolism | Liver and kidney |
| Elimination half-life | 10-150 minutes, longer with impaired liver or kidney function |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 721-50-6 |
| PubChem CID | 4906 |
| IUPHAR/BPS | 7276 |
| DrugBank | DB00750 |
| ChemSpider | 4737 |
| UNII | 046O35D44R |
| KEGG | D00553 as HCl: D01243 |
| ChEBI | CHEBI:8404 |
| ChEMBL | ChEMBL1194 |
| CompTox Dashboard (EPA) | DTXSID7031955 |
| ECHA InfoCard | 100.010.871 |
| Chemical and physical data | |
| Formula | C13H20N2O |
| Molar mass | 220.316 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Chirality | Racemic mixture |
| Melting point | 37 to 38 °C (99 to 100 °F) |
| showSMILES | |
| showInChI | |
| (verify) |
References
- ^ “Prilocaine”. Merriam-Webster Dictionary. Retrieved 2016-01-21.
- ^ Patel V, Morrissey J (2011-09-15). Practical and Professional Clinical Skills. Oxford University Press. p. 267. ISBN 9780199585618.
- ^ “Topical Anesthesia Use in Children: Eutectic Mixture of Local Anesthetics”. Medscape.com. Retrieved 2014-01-07.
- ^ The United States Pharmacopeial Convention, Revision Bulletin: Lidocaine and Prilocaine Cream–Revision to Related Compounds Test, archived from the original on 5 July 2010, retrieved 10 July 2009
External links
- “Prilocaine”. Drug Information Portal. U.S. National Library of Medicine.
- “Prilocaine hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
//////////PRILOCAINE, Anesthetic, ASTRA 1512, ASTRA 1515, ASTRA-1512, ASTRA-1515, L 67,
CCCNC(C)C(=O)NC1=CC=CC=C1C

NEW DRUG APPROVALS
ONE TIME
$10.00
CEFOPERAZONE
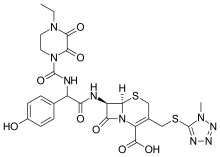
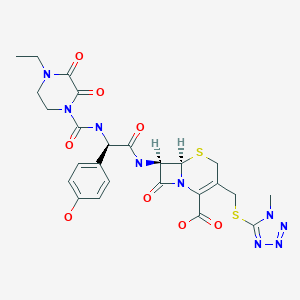
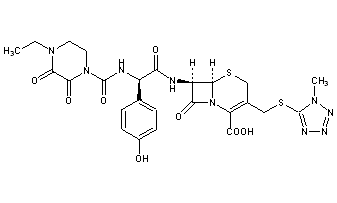
Cefoperazone
- Molecular FormulaC25H27N9O8S2
- Average mass645.667 Da
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Cefoperazone sodium | 5FQG9774WD | 62893-20-3 | NCFTXMQPRQZFMZ-WERGMSTESA-M |
(6R,7R)-7-{[(2R)-2-{[(4-ethyl-2,3-dioxopiperazin-1-yl)carbonyl]amino}-2-(4-hydroxyphenyl)acetyl]amino}-3-{[(1-methyl-1H-tetrazol-5-yl)thio]methyl}-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
(6R,7R)-7-[(2R)-2-[(4-ethyl-2,3-dioxopiperazine-1-carbonyl)amino]-2-(4-hydroxyphenyl)acetamido]-3-{[(1-methyl-1H-1,2,3,4-tetrazol-5-yl)sulfanyl]methyl}-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid
263-749-4[EINECS], 4742
5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[(2R)-2-[[(4-ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo- , (6R,7R)- [ACD/Index Name]
5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[(2R)-2-[[(4-ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-, (6R,7R)-
62893-19-0[RN]
7-[D-(-)-a-(4-Ethyl-2,3-dioxo-1-piperazinecarboxamido)-a-(4-hydroxyphenyl)acetamido]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-3-cephem-4-carboxylic Acid
7U75I1278D
Experimental Properties
| PROPERTY | VALUE | SOURCE |
|---|---|---|
| melting point (°C) | 188-190 | Saikawa, I., Takano, S., Yoshida, C., Takashima, 0..Momonoi, K., Kuroda, S., Komatsu, M., Yasuda, T.and Kodama, Y.; British Patent 1,508,071; April 19,1978; assigned to Toyama Chemical Co., Ltd. and U.S. Patent 4,110,327; August 29,1978; also assigned to Toyama Chemical Co., Ltd. |
| logP | -0.74 | HANSCH,C ET AL. (1995) |
Cefoperazone
CAS Registry Number: 62893-19-0
CAS Name: (6R,7R)-7-[[(2R)-[[(4-Ethyl-2,3-dioxo-1-piperazinyl)carbonyl]amino](4-hydroxyphenyl)acetyl]amino]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acidAdditional Names: 7-[D-(-)-a-(4-ethyl-2,3-dioxo-1-piperazinecarboxamido)-a-(4-hydroxyphenyl)acetamido]-3-[[(1-methyl-1H-tetrazol-5-yl)thio]methyl]-3-cephem-4-carboxylic acid
Molecular Formula: C25H27N9O8S2
Molecular Weight: 645.67
Percent Composition: C 46.50%, H 4.21%, N 19.52%, O 19.82%, S 9.93%
Literature References: Broad spectrum third generation cephalosporin antibiotic. Prepn: I. Saikawa et al.,BE837682; eidem,US4410522 (1976, 1983 both to Toyama); eidem,Yakugaku Zasshi99, 929 (1979). Stability in aq soln: eidem,ibid. 1207. In vitro activity: M. V. Borobio et al.,Antimicrob. Agents Chemother.17, 129 (1980). Kinetics in rats: J. Fabre et al.,Schweiz. Med. Wochenschr.110, 264 (1980); in humans: A. F. Allaz, ibid.109, 1999 (1979). Review of pharmacology and therapeutic efficacy: R. N. Brogden et al.,Drugs22, 423-460 (1981). Symposium on clinical studies: ibid. Suppl. 1, 1-124.
Properties: Crystals from acetonitrile/water, mp 169-171° (hydrated). Stable at pH 4.0-7.0; slightly unstable in acid; highly unstable in alkaline soln.
Melting point: mp 169-171° (hydrated)
Derivative Type: Sodium salt
CAS Registry Number: 62893-20-3
Manufacturers’ Codes: CP-52640-2; T-1551
Trademarks: Bioperazone (Biopharma); Cefazone (Firma); Cefobid (Pfizer); Cefobine (Pfizer); Cefobis (Pfizer); Cefogram (Metapharma); Cefoneg (Tosi); Cefosint (Proter); Dardum (Lisapharma); Farecef (Lafare); Kefazon (Esseti); Novobiocyl (Francia); Pathozone (Pfizer); Peracef (Pfizer); Perocef (Pulitzer); Tomabef (Aandersen)
Molecular Formula: C25H26N9NaO8S2
Molecular Weight: 667.65
Percent Composition: C 44.97%, H 3.93%, N 18.88%, Na 3.44%, O 19.17%, S 9.61%
Therap-Cat: Antibacterial., Therap-Cat-Vet: Antibacterial.
Keywords: Antibacterial (Antibiotics); ?Lactams; Cephalosporins.
Cefoperazone is a third-generation cephalosporin antibiotic, marketed by Pfizer under the name Cefobid. It is one of few cephalosporin antibiotics effective in treating Pseudomonas bacterial infections which are otherwise resistant to these antibiotics.
It was patented in 1974 and approved for medical use in 1981.[1] Cefoperazone/sulbactam (Sulperazon) is a co-formulation with sulbactam.
Cefoperazone is a broad-spectrum cephalosporin antibiotic used for the treatment of bacterial infections in various locations, including the respiratory tract, abdomen, skin, and female genital tracts.
Cefoperazone is a semisynthetic broad-spectrum cephalosporin proposed to be effective against Pseudomonas infections. It is a third-generation antiobiotic agent and it is used in the treatment of various bacterial infections caused by susceptible organisms in the body, including respiratory tract infections, peritonitis, skin infections, endometritis, and bacterial septicemia. While its clinical use has been discontinued in the U.S., cefoperazone is available in several European countries most commonly under the product name, Sulperazon.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

English: I. Saikawa, S. Takano, Y. Shuntaro, C. Yoshida, 0.
Takashima, K. Momonoi, S. Kuroda, M. Komatsu, T. Yasuda, and Y. Kodama, German Offen., DE 2,600,880 (1977); Chem.
Abstr., 87_, 184533b (1977).
SYN
Following is one of the synthesis routes:
alpha-(4-Ethyl-2,3-dioxo-1-piperazinocarbonylamino)-p-hydroxyphenylacetic acid (I) is condensed with 7-amino-3-[(1-methyl-1H-tetrazol-5-yl)thiomethyl]-3-cephem-4-carboxylic acid (II) in the presence of ethyl chlorocarbonate and N,O-bis(trimethylsilyl)acetamide in acetonitrile to produce Cefoperazone sodium.

SYN
Antibiotics
R.S. Vardanyan, V.J. Hruby, in Synthesis of Essential Drugs, 2006
Cefoperazone
Cefoperazone, (6R,7R)-7-[(R)-2-(4-ethyl-2,3-dioxo-1-piperazincarboxamido)-2-(p-hydroxyphenyl)acetamido]-3-[[(1-methyl-1 H-tetrazol-5-yl)thio]methyl]-8-oxo-5-thia-1-azabicyclo[4.2.0]oct-2-en-2-carboxylic acid (32.1.2.84), is synthesized by acylating 7-amino-3-(1-methyl-1,2,3,4-tetrazol-5-yl)-thiomethyl-3-cefem-4-carboxylic acid (32.1.2.24) with a mixed anhydride synthesized from ethyl chloroformate and α-(4-ethylpiperazin-2, 3-dion-1-carbonylamino)-4-hydroxyphenylacetic acid (32.1.2.83), which in turn is synthesized from 4-ethylpiperazin-2,3-dion-1-carboxylic acid (32.1.1.29) and the sodium salt of 4-hydroxyphenylglycine [163–168].


Cefoperazone also has a broad spectrum of antimicrobial action, including most clinically significant microorganisms: Gram-positive, Gram-negative, aerobic, and anaerobic. It is stable with respect to most beta-lactamases of Gram-positive and Gram-negative bacteria.
Cefoperazone is used for bacterial infections of the lower respiratory tract, urinary and sexual tracts, bones, joints, skin, soft tissues, abdominal, and gynecological infections. Synonyms of this drug are cefazon, cefobid, cefobis, and many others.
Spectrum of bacterial susceptibility
Cefoperazone has a broad spectrum of activity and has been used to target bacteria responsible for causing infections of the respiratory and urinary tract, skin, and the female genital tract. The following represents MIC susceptibility data for a few medically significant microorganisms.
- Haemophilus influenzae: 0.12 – 0.25 µg/ml
- Staphylococcus aureus: 0.125 – 32 µg/ml
- Streptococcus pneumoniae: ≤0.007 – 1 µg/ml[2]
Adverse effects
Cefoperazone contains an N-methylthiotetrazole (NMTT or 1-MTT) side chain. As the antibiotic is broken down in the body, it releases free NMTT, which can cause hypoprothrombinemia (likely due to inhibition of the enzyme vitamin K epoxide reductase) and a reaction with ethanol similar to that produced by disulfiram (Antabuse), due to inhibition of aldehyde dehydrogenase.[3]
Mechanism of action
Cefoperazone exerts its bactericidal effect by inhibiting the bacterial cell wall synthesis, and sulbactam acts as a beta-lactamase inhibitor, to increase the antibacterial activity of cefoperazone against beta-lactamase-producing organisms.
References
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 494. ISBN 9783527607495.
- ^ “Cefoperazone (Cefobid) – The Antimicrobial Index Knowledgebase – TOKU-E”. antibiotics.toku-e.com.
- ^ Stork CM (2006). “Antibiotics, antifungals, and antivirals”. In Nelson LH, Flomenbaum N, Goldfrank LR, Hoffman RL, Howland MD, Lewin NA (eds.). Goldfrank’s toxicologic emergencies. New York: McGraw-Hill. p. 847. ISBN 0-07-143763-0. Retrieved 2009-07-03.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a601206 |
| ATC code | J01DD12 (WHO) QJ51DD12 (WHO) |
| Pharmacokinetic data | |
| Excretion | Hepatic |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 62893-19-0 |
| PubChem CID | 44185 |
| DrugBank | DB01329 |
| ChemSpider | 40206 |
| UNII | 7U75I1278D |
| KEGG | D07645 |
| ChEMBL | ChEMBL507674 |
| CompTox Dashboard (EPA) | DTXSID2022759 |
| ECHA InfoCard | 100.057.936 |
| Chemical and physical data | |
| Formula | C25H27N9O8S2 |
| Molar mass | 645.67 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
//////////cefoperazone, Antibacterial, Antibiotics, Lactams, Cephalosporins, CP-52640-2, T-1551, CP 52640-2, T 1551
[H][C@]12SCC(CSC3=NN=NN3C)=C(N1C(=O)[C@@]2([H])NC(=O)[C@H](NC(=O)N1CCN(CC)C(=O)C1=O)C1=CC=C(O)C=C1)C(O)=O

NEWDRUG APPROVALS
one time
$10.00
CICLOPIROX
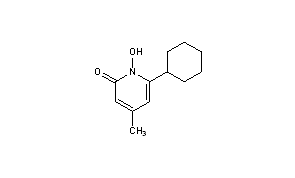

- Molecular FormulaC12H17NO2
- Average mass207.269 Da
CICLOPIROX(6-Cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridone)
2(1H)-Pyridinone, 6-cyclohexyl-1-hydroxy-4-methyl-
249-577-2[EINECS]
29342-05-0[RN]
KS-5085, циклопирокс , سيكلوبيروكس , 环吡酮 ,
Ciclopirox
CAS Registry Number: 29342-05-0
CAS Name: 6-Cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridinone
Molecular Formula: C12H17NO2
Molecular Weight: 207.27
Percent Composition: C 69.54%, H 8.27%, N 6.76%, O 15.44%
Literature References: Broad spectrum antimycotic agent with some antibacterial activity. Prepn: G. Lohaus, W. Dittmar, ZA6906039; eidem,US3883545 (1970, 1975 both to Hoechst). In vitro study: eidem,Arzneim.-Forsch.23, 670 (1973). Series of articles on pharmacokinetics, pharmacology, teratology, toxicity studies: Oyo Yakuri9, 57-115 (1975), C.A.83, 53159d, 53538b, 53539c, 71844c, 90833q (1975). Series of articles on chemistry, mechanism of action, toxicology, clinical trials: Arzneim.-Forsch.31, 1309-1386 (1981). Toxicity data: H. G. Alpermann, E. Schutz, ibid. 1328. Review: S. G. Jue et al.,Drugs29, 330-341 (1985). Review of clinical experience in seborrheic dermatitis: A. Starova, R. Aly, Expert Opin. Drug Saf.4, 235-239 (2005).
Properties: Solid, mp 144°.
Melting point: mp 144°
Derivative Type: Ethanolamine salt (1:1)
CAS Registry Number: 41621-49-2
Additional Names: Ciclopirox olamine
Manufacturers’ Codes: HOE-296
Trademarks: Batrafen (HMR); Brumixol (Bruschettini); Ciclochem (Novag); Dafnegin (Poli); Loprox (HMR); Micoxolamina (Domp?; Mycoster (Fabre)
Molecular Formula: C14H24N2O3
Molecular Weight: 268.35
Percent Composition: C 62.66%, H 9.01%, N 10.44%, O 17.89%
Properties: LD50 in mice, rats (mg/kg): 2898, 3290 orally (Alpermann, Schutz).
Toxicity data: LD50 in mice, rats (mg/kg): 2898, 3290 orally (Alpermann, Schutz)
- EINECS:255-464-9
- LD50:71 mg/kg (M, i.v.); 1740 mg/kg (M, p.o.);
72 mg/kg (R, i.v.); 2350 mg/kg (R, p.o.)
Therap-Cat: Antifungal.
Keywords: Antifungal (Synthetic).
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Ciclopirox (sometimes known by the abbreviation CPX[2]) is a synthetic antifungal agent for topical dermatologic treatment of superficial mycoses. It is most useful against Tinea versicolor. It is sold under many brand names worldwide.[1]
Medical uses
Ciclopirox is indicated for the treatment of tinea pedis and tinea corporis due to Trichophyton rubrum, Trichophyton mentagrophytes and Epidermophyton floccosum, as well as seborrheic dermatitis. It is not to be used in the eyes or vagina, and nursing women should consult their doctors before use, since it is not known whether ciclopirox passes into human milk. A burning sensation may be felt when first applying ciclopirox on the skin.[3]
Nail infections
In addition to other formulations, ciclopirox is used in lacquers for topical treatment of onychomycosis (fungal infections of the nails). A meta-analysis of the six trials of nail infections available in 2009 concluded that they provided evidence that topical ciclopirox had poor cure rates and that amorolfine might be substantially more effective, but more research was required.
“Combining data from 2 trials of ciclopiroxolamine versus placebo found treatments failure rates of 61% and 64% for ciclopiroxolamine. These outcomes followed long treatment times (48 weeks) and this makes ciclopiroxolamine a poor choice for nail infections. Better results were observed with the use of amorolfine lacquer; 6% treatment failure rates were found after 1 month of treatment but these data were collected on a very small sample of people and these high rates of success might be unreliable.”[4]
Pharmacology
Pharmacodynamics
In contrast to the azoles and other antimycotic drugs, the mechanism of action of ciclopirox is poorly understood.[5] However, loss of function of certain catalase and peroxidase enzymes has been implicated as the mechanism of action, as well as various other components of cellular metabolism. In a study conducted to further elucidate ciclopirox’s mechanism, several Saccharomyces cerevisiae mutants were screened and tested. Results from interpretation of the effects of both the drug treatment and mutation suggested that ciclopirox may exert its effect by disrupting DNA repair, cell division signals and structures (mitotic spindles) as well as some elements of intracellular transport.[6]
It is currently being investigated as an alternative treatment to ketoconazole for seborrhoeic dermatitis as it suppresses growth of the yeast Malassezia furfur. Initial results show similar efficacy to ketoconazole with a relative increase in subjective symptom relief due to its inherent anti-inflammatory properties.[7]
Chemistry
Ciclopirox is a considered a hydroxypyrimidine (sic) antifungal agent.[citation needed] Structurally, ciclopirox is the N-oxide of a 2-hydroxypyridine derivative and therefore ought to be termed a hydroxypyridine antifungal agent. Additionally, the structure as drawn above is the lactam tautomer and indicates the molecule being an N-Hydroxy-2-pyridone. Hence the classification of ciclopirox as a 2-pyridone antifungal agent.
Ciclopirox is used clinically as ciclopirox olamine, the olamine salt of ciclopirox.
Literatures:
Lohaus; Dittmar Arzneimittel-Forschung/Drug Research, 1981 , vol. 31, # 8 a p. 1311 – 1316
Literatures:
Hoechst Aktiengesellschaft Patent: US3972888 A1, 1976 ;
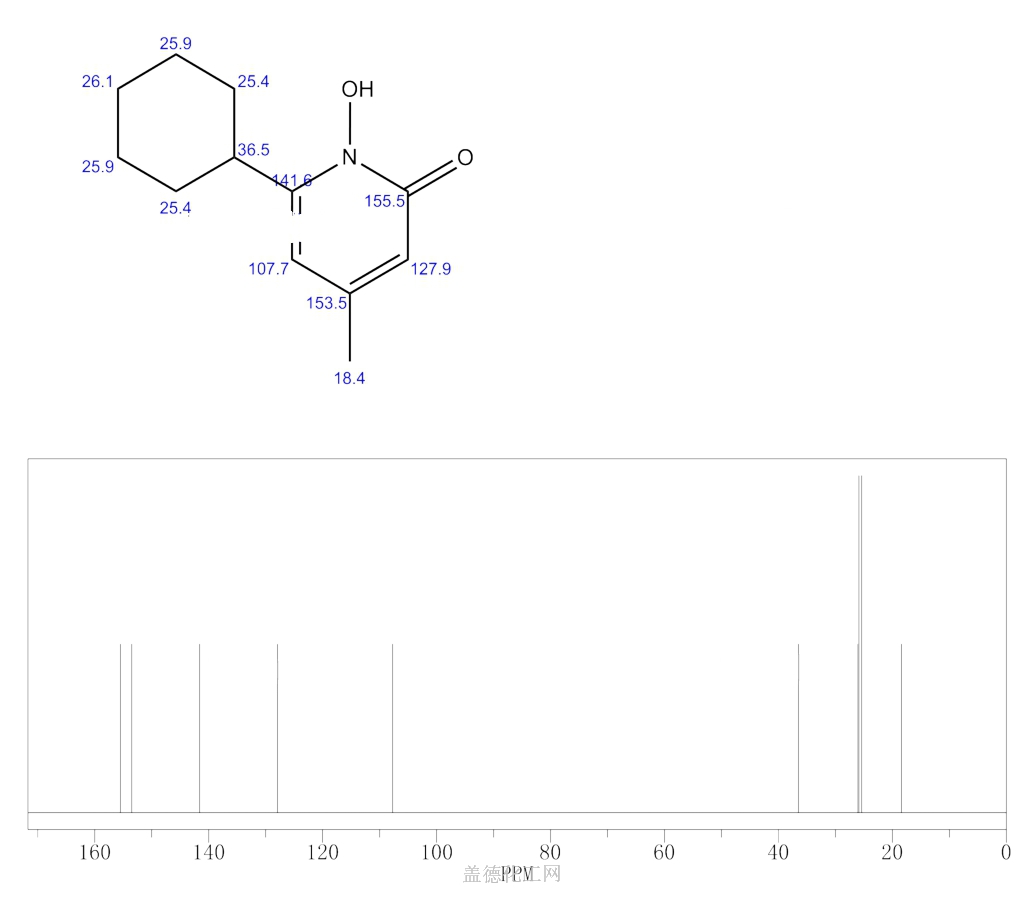
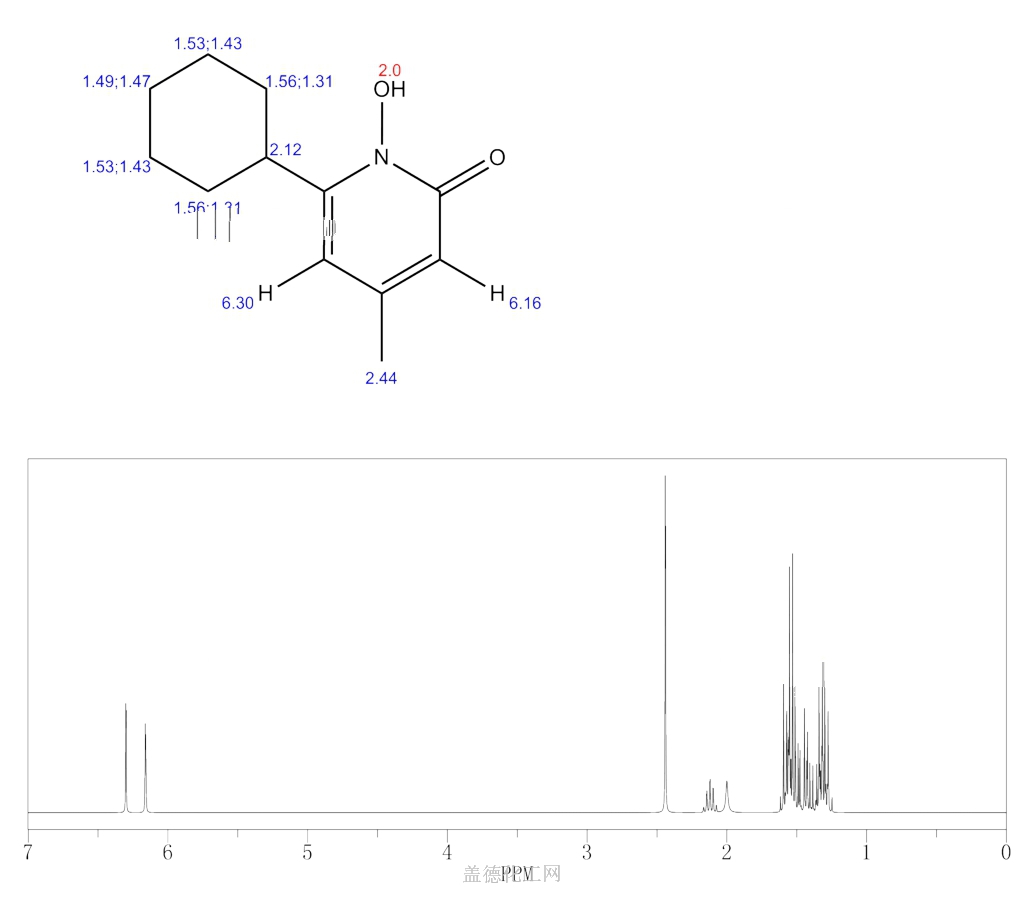
SYNTHESIS
SYN

SYN
W. Dittmar, E. Druckrey andBroad spectrum antimycotic agent with some antibacterial activity. Prepn: G. Lohaus, W. Dittmar, ZA 6906039; eidem, US 3883545 (1970, 1975 both to Hoechst). In vitro study: eidem, Arzneim.-Forsch. 23, 670 (1973).
H. Urbach, J. Med. Chem., 17, 753 (1974); W. Dittmar and G. Lohaus,
German Patent 2,214,608 (1973); Chem. Abstr., 79: 146419w (1973).

SYN
ethanolamine (CAS NO.: ), with other names as 6-Cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridinone 2-aminoethanol, could be produced through the following synthetic routes.
.jpg)
Compound can be prepared in two different ways: 1) The reaction of methyl 5-oxo-5-cyclohexyl-3-methylpentenoate (I) with NH2OH gives the corresponding oxime (II), which is then cyclized to 6-cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridone (III). Finally, this compound is salified with ethanolamine (IV). 2) Compound (III) can also be obtained by reaction of 6-cyclohexyl-4-methyl-2-pyron (V) with hydroxylamine hydrochloride in hot 2-aminopyridine.
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 14818-35-0 | C12H16O2 | 6-cyclohexyl-4-methyl-2-pyrone | 2H-Pyran-2-one, 6-cyclohexyl-4-methyl- |
| 141-43-5 | C2H7NO | ethanolamine | Ethanol, 2-amino- |
| 7803-49-8 | H3NO | hydroxylamine | Hydroxylamine |
PATENT
https://patents.google.com/patent/US9545413B2/en
The molecule 6-cyclohexyl-1-hydroxy-4-methylpyridin-2(1H)-one, also known as Ciclopirox, is a commercially available antifungal agent as an olamine salt. Ciclopirox olamine has been used to treat superficial mycoses and Tinea versicolor following topical application to the skin. Following enteral administration, ciclopirox undergoes significant first-pass effect resulting in low oral bioavailability. The oral route of administration is also associated with gastrointestinal toxicities observed in animals and humans limiting its benefit in animal and human health applications. Ciclopirox olamine has poor solubility, limiting opportunities to deliver the antifungal agent via parenteral administration of suitably potent solutions and suspensions. As such, it would be beneficial to configure ciclopirox for improved water solubility in order to deliver the drug by parenteral routes of administration.
References
- ^ Jump up to:a b Drugs.com International brand names for ciclopirox Page accessed January 201, 2016
- ^ Ciclopirox
- ^ “Ciclopirox Olamine Antifungal Shampoo”. Okdermo. Retrieved 2019-08-06.
- ^ Crawford F (2007). “Topical treatments for fungal infections of the skin and nails of the foot”. The Cochrane Database of Systematic Reviews. 2007 (3): CD001434. doi:10.1002/14651858.CD001434.pub2. PMC 7073424. PMID 17636672.
- ^ Niewerth M, Kunze D, Seibold M, Schaller M, Korting HC, Hube B (June 2003). “Ciclopirox Olamine Treatment Affects the Expression Pattern of Candida albicans Genes Encoding Virulence Factors, Iron Metabolism Proteins, and Drug Resistance Factors”. Antimicrobial Agents and Chemotherapy. 47 (6): 1805–1817. doi:10.1128/AAC.47.6.1805-1817.2003. PMC 155814. PMID 12760852.
- ^ Leem SH, Park JE, Kim IS, Chae JY, Sugino A, Sunwoo Y (2003). “The possible mechanism of action of ciclopirox olamine in the yeast Saccharomyces cerevisiae”. Mol. Cells. 15 (1): 55–61. PMID 12661761.
- ^ Ratnavel RC, Squire RA, Boorman GC (2007). “Clinical efficacies of shampoos containing ciclopirox olamine (1.5%) and ketoconazole (2.0%) in the treatment of seborrhoeic dermatitis”. J Dermatolog Treat. 18 (2): 88–96. doi:10.1080/16537150601092944. PMID 17520465. S2CID 34852507.
| Clinical data | |
|---|---|
| Trade names | Many brand names worldwide[1] |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a604021 |
| Pregnancy category | B |
| Routes of administration | Topical (applied as a nail lacquer, skin cream or shampoo) |
| ATC code | D01AE14 (WHO) G01AX12 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyRx-only (CA) |
| Pharmacokinetic data | |
| Bioavailability | <5% with prolonged use |
| Protein binding | 94 to 97% |
| Elimination half-life | 1.7 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 29342-05-0 |
| PubChem CID | 2749 |
| DrugBank | DB01188 |
| ChemSpider | 2647 |
| UNII | 19W019ZDRJ |
| KEGG | D03488 |
| ChEBI | CHEBI:453011 |
| ChEMBL | ChEMBL1413 |
| CompTox Dashboard (EPA) | DTXSID9048564 |
| ECHA InfoCard | 100.045.056 |
| Chemical and physical data | |
| Formula | C12H17NO2 |
| Molar mass | 207.269 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
////////CICLOPIROX OLAMINE

NEW DRUG APPROVALS
ONE TIME
$10.00
RITONAVIR
RITONAVIR
- Molecular FormulaC37H48N6O5S2
- Average mass720.944 Da
1,3-Thiazol-5-ylmethyl-[(1S,2S,4S)-1-benzyl-2-hydroxy-4-({(2S)-3-methyl-2-[(methyl{[2-(1-methylethyl)-1,3-thiazol-4-yl]methyl}carbamoyl)amino]butanoyl}amino)-5-phenylpentyl]carbamat
155213-67-5[RN]
7449Abbott 84538
UNII-O3J8G9O825
ритонавир
ريتونافير
利托那韦
(1E,2S)-N-[(2S,4S,5S)-4-Hydroxy-5-{(E)-[hydroxy(1,3-thiazol-5-ylmethoxy)methylene]amino}-1,6-diphenyl-2-hexanyl]-2-[(E)-(hydroxy{[(2-isopropyl-1,3-thiazol-4-yl)methyl](methyl)amino}methylene)amino]-3- methylbutanimidic acid
(2S,3S,5S)-5-[N-[N-[[N-methyl-N-[(2-isopropyl-4-thiazolyl)methyl]amino]carbonyl]valinyl]amino]-2-[N-[(5-thiazolyl)methoxycarbonyl]amino]-1,6-diphenyl-3-hydroxyhexane
- A-84538
- Abbott 84538
- ABBOTT-84538
- ABT 538
- ABT-538
- DRG-0244
- NSC-693184
- TMC 114r
Ritonavir
CAS Registry Number: 155213-67-5
CAS Name: (5S,8S,10S,11S)-10-Hydroxy-2-methyl-5-(1-methylethyl)-1-[2-(1-methylethyl)-4-thiazolyl]-3,6-dioxo-8,11-bis(phenylmethyl)-2,4,7,12-tetraazatridecan-13-oic acid 5-thiazolylmethyl ester
Additional Names: (2S,3S,5S)-5-[N-[N-[[N-methyl-N-[(2-isopropyl-4-thiazolyl)methyl]amino]carbonyl]valinyl]amino]-2-[N-[(5-thiazolyl)methoxycarbonyl]amino]-1,6-diphenyl-3-hydroxyhexane
Manufacturers’ Codes: A-84538; Abbott 84538; ABT-538
Trademarks: Norvir (Abbott)
Molecular Formula: C37H48N6O5S2
Molecular Weight: 720.94
Percent Composition: C 61.64%, H 6.71%, N 11.66%, O 11.10%, S 8.90%
Literature References: Peptidomimetic HIV-1 protease inhibitor. Prepn: D. J. Kempf et al.,WO9414436; eidem,US5541206 (1994, 1996 both to Abbott). Antiretroviral spectrum, pharmacokinetics: idemet al.,Proc. Natl. Acad. Sci. USA92, 2484 (1995). Structural model for drug resistance: M. Markowitz et al.,J. Virol.69, 701 (1995). HPLC determn in biological fluids: R. M. W. Hoetelmans et al.,J. Chromatogr. B705, 119 (1998). Review of clinical experience: A. P. Lea, D. Faulds, Drugs52, 541-546 (1996). Clinical trial with nucleoside analogs in HIV-infected children: S. A. Nachman et al.,J. Am. Med. Assoc.283, 492 (2000). Clinical trial with lopinavir, q.v., in HIV infection: S. Walmsley et al.,N. Engl. J. Med.346, 2039 (2002).
Therap-Cat: Antiviral.
Keywords: Antiviral; Peptidomimetics; HIV Protease Inhibitor.Company:Abbvie (Originator)Sales:$389 Million (Y2012); 
$419 Million (Y2011);ATC Code:J05AE03
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-02-10 | New dosage form | Norvir | HIV infection | Tablet | 100 mg | Abbvie | |
| 1999-06-29 | Additional approval | Norvir | HIV infection | Capsule | 100 mg | Abbvie | |
| 1996-03-01 | New dosage form | Norvir | HIV infection | Capsule | 100 mg | Abbvie | Priority |
| 1996-03-01 | First approval | Norvir | HIV infection | Solution | 80 mg/mL | Abbvie |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1996-08-26 | First approval | Norvir | HIV infection | Capsule | 100 mg | Abbvie | |
| 1996-08-26 | First approval | Norvir | HIV infection | Tablet, Film coated | 100 mg | Abbvie | |
| 1996-08-26 | First approval | Norvir | HIV infection | Solution | 80 mg/mL | Abbvie |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2011-02-28 | New dosage form | Norvir | HIV infection | Tablet | 100 mg | Abbvie | |
| 1998-09-25 | First approval | Norvir | HIV infection | Solution | 80 mg/mL | Abbvie |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-10-15 | Marketing approval | HIV infection | Tablet | 100 mg | Abbvie | ||
| 2010-07-29 | Marketing approval | 爱治威/Norvir | HIV infection | Capsule | 100 mg | Abbott | |
| 2010-07-13 | Marketing approval | 迈可欣 | HIV infection | Solution | 75 ml:6 g | 美吉斯制药(厦门) | 3.1类 |
Ritonavir was first approved by the U.S. Food and Drug Administration (FDA) on March 1, 1996, then approved by European Medicine Agency (EMA) on August 26, 1996, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on September 25, 1998. It was developed and marketed as Norvir® by Abbvie.
Ritonavir is an HIV protease inhibitor. It is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection.
Norvir® is available as solution for oral use, containing 80 mg of free Ritonavir per mL. The recommended dose is 600 mg twice-day with meals for adult patients.
Ritonavir is an HIV protease inhibitor used in combination with other antivirals in the treatment of HIV infection.
Ritonavir (RTV), sold under the brand name Norvir, is an antiretroviral medication used along with other medications to treat HIV/AIDS.[2] This combination treatment is known as highly active antiretroviral therapy (HAART).[2] Often a low dose is used with other protease inhibitors.[2] It may also be used in combination with other medications for hepatitis C.[3] It is taken by mouth.[2] The capsules of the medication do not work the same as the tablets.[2]
Common side effects include nausea, vomiting, loss of appetite, diarrhea, and numbness of the hands and feet.[2] Serious side effects include liver problems, pancreatitis, allergic reactions, and arrythmias.[2] Serious interactions may occur with a number of other medications including amiodarone and simvastatin.[2] At low doses it is considered to be acceptable for use during pregnancy.[4] Ritonavir is of the protease inhibitor class.[2] Typically, however, it is used to inhibit the enzyme that metabolizes other protease inhibitors.[5] This inhibition allows lower doses of these latter medication to be used.[5]
Ritonavir was patented in 1989 and came into medical use in 1996.[6][7] It is on the World Health Organization’s List of Essential Medicines.[8] Ritonavir capsules were approved as a generic medication in the United States in 2020.[9]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////

In the first step shown, an aldehyde derived from phenylalanine is treated with zinc dust in the presence of vanadium(III) chloride. This results in a pinacol coupling reaction which dimerises the material to provide an intermediate which is converted to its epoxide and then reduced to (2S,3S,5S)-2,5-diamino-1,6-diphenylhexan-3-ol. Importantly, this retains the absolute stereochemistry of the amino acid precursor. The diamine is then treated sequentially with two thiazole derivatives, each linked by an amide bond, to provide ritonavir.[23][24]
REF 23, 24 BELOW
- WO patent 1994014436, Kempf, Dale J.; Norbeck, Daniel W.; Sham, Hing Leung; Zhao, Chen; Sowin, Thomas J.; Reno, Daniel S.; Haight, Anthony R. and Cooper, Arthur J., “Retroviral protease inhibiting compounds”, published 1994-07-07, assigned to Abbott Laboratories
- ^ Vardanyan, Ruben; Hruby, Victor (2016). “34: Antiviral Drugs”. Synthesis of Best-Seller Drugs. pp. 698–701. doi:10.1016/B978-0-12-411492-0.00034-1. ISBN 9780124114920. S2CID 75449475.
SYN
EP 0674513; EP 0727419; JP 1996505844; JP 1997118679; JP 1998087639; WO 9414436 |
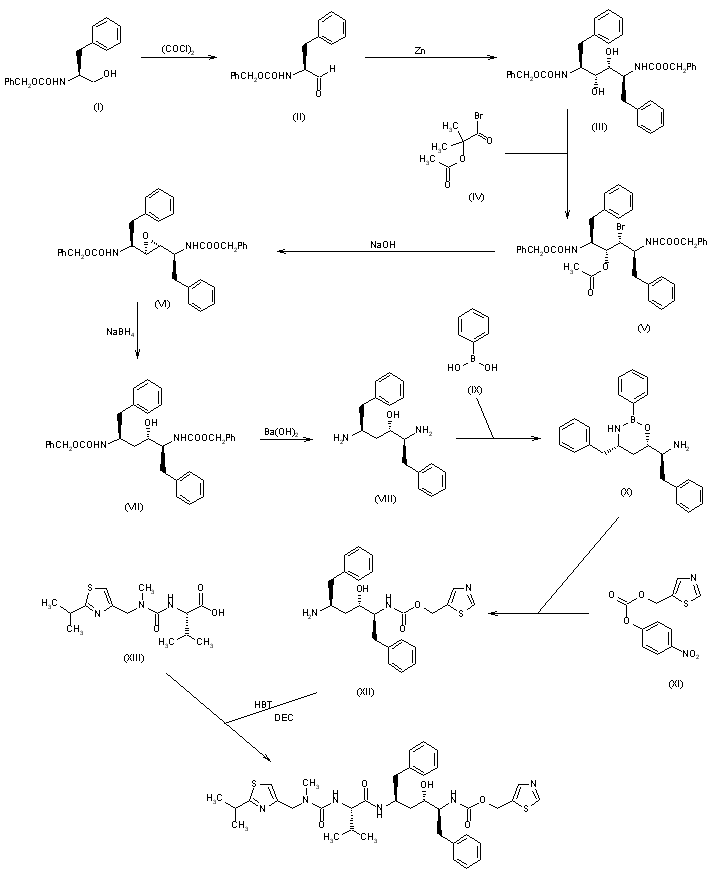
The oxidation of N-(benzyloxycarbonyl)-L-phenylalaninol (I) with oxalyl chloride in DMSO gives the corresponding aldehyde (II), which by dimerization with Zn dust in dichloromethane yields (2S,3R,4R,5S)-2,5-bis(benzyloxycarbonylamino)-1,6-diphenylhexane-3,4-diol (III) [along with the (2S,3S,4S,5S)-isomer]. The reaction of (III) with alpha-acetoxyisobutyryl bromide (IV) in hexane/dichloromethane affords (2S,3R,4R,5S)-2,5-bis(benzyloxycarbonylamino)-4-bromo-1,6-diphenylhexan-3-ol acetate ester (V), which is converted into the corresponding epoxide (VI) with NaOH in dioxane/water. The reduction of the epoxide (VI) with NaBH4 in THF affords (2S,3S,5S)-2,5-bis(benzyloxycarbonylamino)-1,6-diphenylhexan-3-ol (VII), which is deprotected with Ba(OH)2 in refluxing dioxane/water yielding the corresponding diamine (VIII). The cyclization of (VIII) with phenylboronic acid (IX) in refluxing toluene gives (4S,6S)-6-[1(S)-amino-2-phenylethyl]-4-benzyl-2-phenyl-1,3,2-oxaazaborinane (X), which is condensed with (5-thiazolylmethyl)(4-nitrophenyl)carbonate (XI) in THF to afford (2S,3S,5S)-5-amino-1,6-diphenyl-2-(5-thiazolylmethoxycarbonylamino) hexan-3-ol (XII). Finally, this compound is condensed with N-[N-(2-isopropylthiazol-4-ylmethyl)-N-methylaminocarbonyl]-L-valine (XIII) by means of 1-hydroxybenzotriazole (HBT) and N-[3-(dimethylamino)propyl]-N’-ethylcarbodiimide (DEC) in THF.
SYN
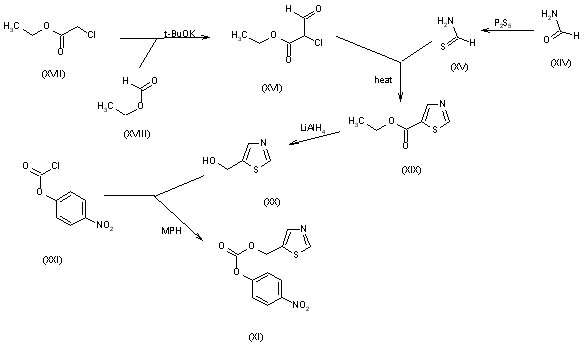
The thiazolyl carbonate (XI) has been synthesized as follows: The reaction of formamide (XIV) with P2S5 in ethyl ether gives thioformamide (XV), which is cyclized with 2-chloro-3-oxopropionic acid ethyl ester (XVI) [obtained by condensation of ethyl chloroacetate (XVII) with ethyl formate (XVIII) by means of t-BuOK in THF] yielding thiazol-5-carboxylic acid ethyl ester (XIX). The reduction of (XIX) with LiAlH4 in THF affords 5-thiazolylmethanol (XX), which is then esterified with 4-nitrophenyl chloroformate (XXI) by means of 4-methylmorpholine (MPH) in dichloromethane to give the desired product (XI).
SYN
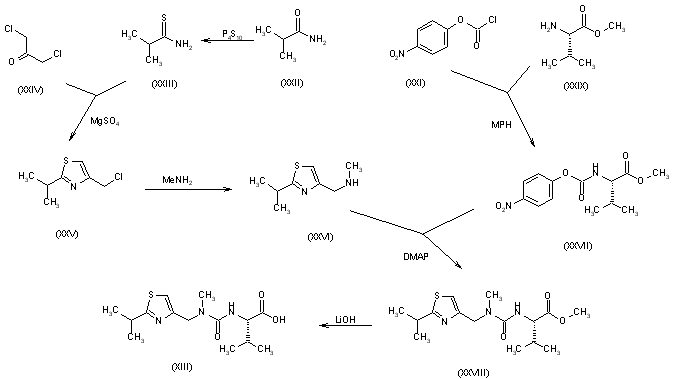
The N-substituted valine (XIII) has been synthesized as follows: The reaction of isobutyramide (XXII) with phosphorus pentasulfide (P4S10) in ethyl ether gives the corresponding thioamide (XXIII), which is cyclized with 1,3-dichloroacetone (XXIV) by means of MgSO4 in refluxing acetone yielding 4-(chloromethyl)-2-isopropylthiazole (XXV). The reaction of (XXV) with methylamine in water affords N-(2-isopropylthiazol-4-ylmethyl)-N-methylamine (XXVI), which is condensed with N-(4-nitrophenoxycarbonyl)-L-valine methyl ester (XXVII) by means of 4-(dimethylamino)pyridine (DMAP) and triethylamine in refluxing THF to give the thiazol-substituted L-valine ester (XXVIII). Finally, this compound is converted into the corresponding free acid with LiOH in dioxane/water. The N-(4-nitrophenoxycarbonyl)-L-valine methyl ester (XXVII) has been synthesized by reaction of chloroformate (XXI) with L-valine methyl ester (XXIX) by means of 4-methylmorpholine (MPH) in dichloromethane.
SYN
Org Process Res Dev 1999,3(2),94 |
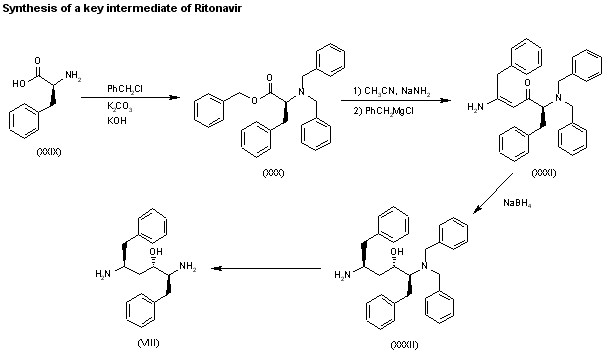
A new method for the preparation of a key diaminoalcohol intermediate in the synthesis of ritonavir has been described: The reaction of L-phenylalanine (XXIX) with benzyl chloride by means of K2CO3 and KOH in hot water gives N,N-dibenzyl-L-phenylalanine benzyl ester (XXX), which is condensed first with acetonitrile by means of NaNH2 and then with benzylmagnesium chloride, both reactions in methyl tert-butyl ether, yielding 5-amino-2-(dibenzylamino)-1,6-diphenylhex-4-en-3-one (XXXI). The reduction of (XXXI) with NaBH4 with an accurate control of the solvent and acidity, results in an enhanced enantioselectivity towards the desired (S,S,S)-enantiomer of 5-amino-2-(dibenzylamino)-1,6-diphenyl-3-hexanol (XXXII). Finally, this compound is deprotected by hydrogenation with ammonium formate over Pd/C in methanol/water to provide the target chiral diaminoalcohol (VIII).
SYN
Chem Commun (London) 2001,(2),203 |
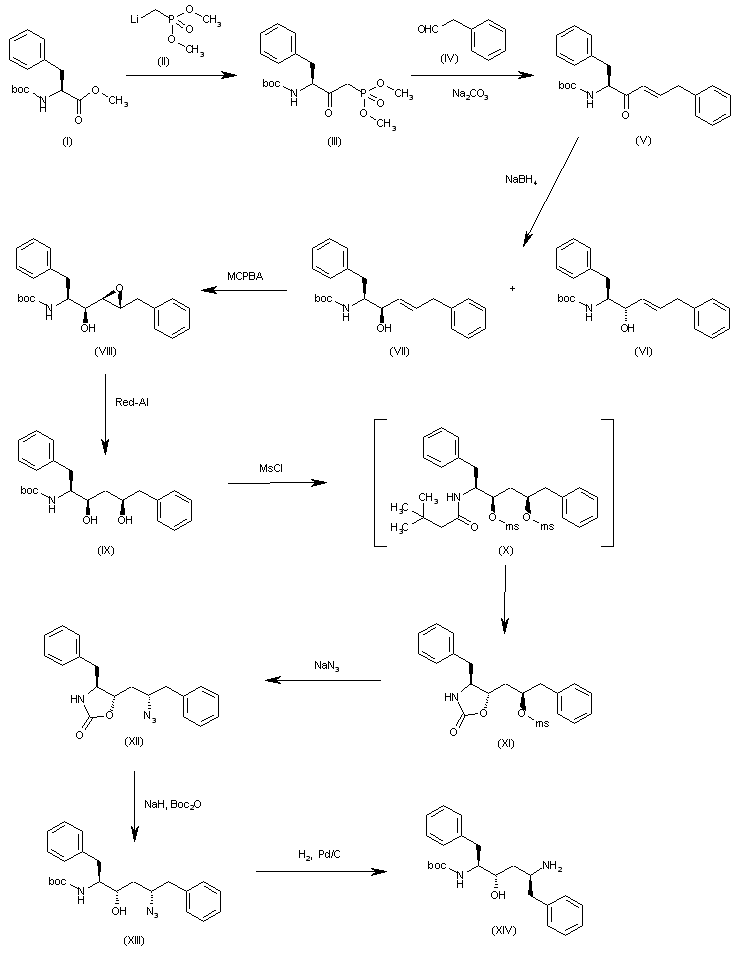
he reaction of N-(tert-butoxycarbonyl)-L-phenylalanine methyl ester (I) with trimethylphosphonate lithium salt (II) in THF gives the dimethyl L-phenylalanylmethylphosphonate (III), which is condensed with 2-phenylacetaldehyde (IV) by means of Na2CO3 in ethanol yielding the diphenylhexenone (V). The reduction of the carbonyl group of (V) with NaBH4 in methanol affords a diastereomeric mixture of alcohols (VI) and (VII) from which the desired major isomer (VII) is separated by column chromatography.. The epoxidation of the double bond of (VII) with MCPBA in dichloromethane gives the epoxide (VIII) (1), which is cleaved with Red-Al in THF providing the diol (IX). The reaction of (IX) with Ms-Cl and DIEA yields the intermediate dimesylate (X) which, without isolation, affords oxazolidinone (XI). The reaction of (XI) with sodium azide and 18-C-6 in DMSO gives the azide (XII), which is treated with NaH and Boc2O in THF in order to cleave the oxazolidine ring and furnish the protected aminoalcohol (XIII). Finally the azido group of (XIII) is hydrogenate with H2 over Pd/C in methanol to afford the monoprotected diaminoalcohol (XIV) the target compound.
SYN
https://www.scielo.br/j/mioc/a/FT4WXwhbCPCkm7W6XdX4GHm/?lang=en
Ritonavir (2) – The first disclosure of ritonavir (2) is presented in a patent from 1994, assigned to Abbott Laboratories. Even though the patent consists of a range of Markush structures, among which is found (2), its synthesis is not presented.249 The synthetic pathway to obtain (2) was disclosed for the first time in a second-generation patent in 1995, by the same company.250,251 Some drawbacks related to the synthesis presented by Abbott include the employment of expensive condensing agents, as 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and poor reaction yields on the first steps of the synthetic pathway, making this strategy too expensive and not suitable for scale-up production. Considering the above deficiencies, a recent Chinese patent concerning the synthesis of ritonavir (2) has been released.
The synthetic pathway described in the patent starts with a nucleophilic addition-elimination reaction between the starting material (2-isopropylthiazol-4-yl)- nitro-methylamine (71) and N-[(2,2,2-trichloroethoxy)carbonyl]-L-valine (72), generating intermediate (73). The intermediate is mixed with p-toluenesulfonyl chloride and triethylamine to activate the acid function, followed by the addition of reagent (74) in a one-pot procedure. The condensation allows the formation of intermediate (75), which is submitted to acidic conditions for a Boc deprotection, followed by another nucleophilic addition-elimination reaction with reagent (76), thus obtaining the final product ritonavir (2) (Fig. 16).252 When comparing both patents, it is possible to highlight important advantages present in the latest one, including the use of cheap and easily available reagents, for instance, the use of p-toluenesulfonyl chloride as an amide condensing agent, the synthetic pathway has a high yield (79% overall), low cost and it is ease to scale-up the production.

SYN
US5543551

Reference:1. US5541206A / EP0402646B1.
Reference:1. Org. Process Res. Dev. 1999, 3, 94-100.
https://pubs.acs.org/doi/abs/10.1021/op9802071
Patent
2. WO2006090264A1.
Reference:1. Tetrahedron Lett. 2011, 52, 6968-6970.
SYN
Synthesis ReferenceUS5484801
SYN
Method of synthesis
i. Valine condensed with bis-trichloromethyl carbonate to give an intermediate compound 4-isopropyloxazolidine-2,5-dione.
ii. The intermediate compound is reacted with compound (1) to give compound (2).
ii. (2) reacts with bis-trichloromethyl carbonate followed by reaction with (2-isopropylthiazol-4-yl)-N-methylmethanamine to give compound (3).
iv. Primary amine group of (3) is subjected to deprotection by the removal of two benzyl groups to give compound (4).
v. (4) reacts with (5) to give Ritonavir in good yield.
CLICK ON BELOW IMAGE
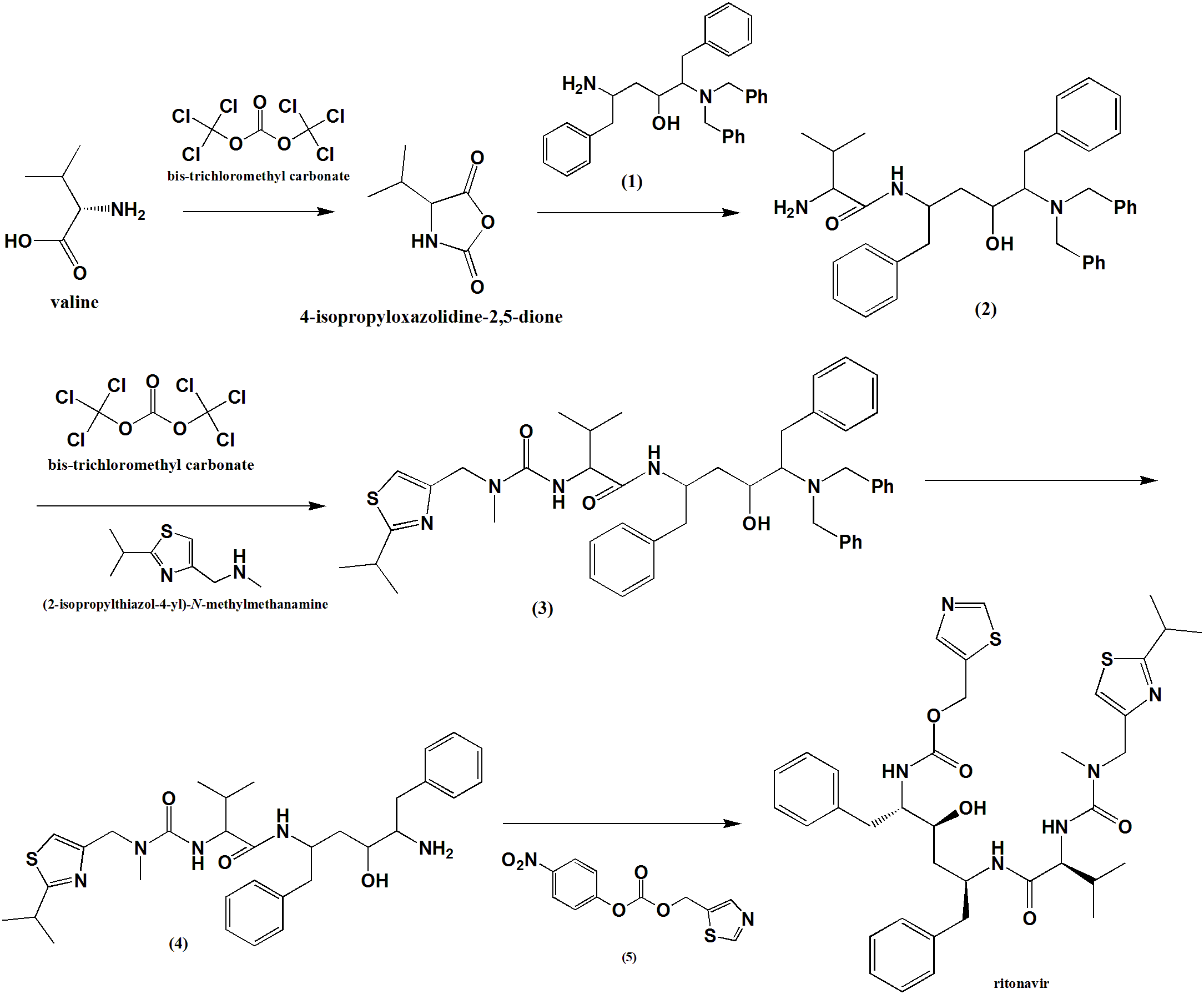
CLIP
SYN
US5543551
SYN
Ruben Vardanyan, Victor Hruby, in Synthesis of Best-Seller Drugs, 2016
Lopinavir–Kaletra
The first report of a new protease inhibitor candidate lopinavir (34.1.21) seems to be Abbott’s patent [134].
The core of lopinavir is identical to that of ritonavir. The 5-thiazolyl end group in ritonavir was replaced by the phenoxyacetyl group, and the 2-isopropylthiazolyl group in ritonavir was replaced by a modified valine in which the amino terminus had a six-membered cyclic urea attached.
Synthetic strategy employed for the synthesis of multikilogram quantities of lopinavir is very similar to that implemented for ritonavir (34.1.22), but using the protected version (34.1.79) of the “core” diamino alcohol (34.1.55), which was sequentially acylated with the acid chlorides of (S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoic acid (34.1.89) and 2-(2,6-dimethylphenoxy)acetic acid (34.1.94) [135,136].
The bulk synthesis of protected diamino alcohol (34.1.79) was proposed [137,138] by a method closely related to the method [122,123] for ritonavir. For that purpose L-phenylalanine (34.1.74) was sequentially trialkylated with benzyl chloride using a K2CO3/water system at reflux to produce a tribenzylated product (34.1.75). A solution with generated acetonitrile anion in THF was added to the benzylated product (34.1.75) at less than −40°C to yield the cyanomethylketone (34.1.76), which was exposed to Grignard reagent–benzyl magnesium chloride to produce an enaminone (34.1.77). No racemization was observed in these two steps. The addition of the obtained enaminone (34.1.77) in THF/PrOH to a solution of NaBH4 and MsOH in THF at 5°C produced an intermediate aminoketone (34.1.78). No further reduction of the keto group occurs under these conditions. Reduction of the keto group could proceed by addition of a preformed solution of sodium tris(trifluoroacetoxy)borohydride in tetrahydrofuran [NaBH3(OTFA)], which produces a mixture of amino alcohols composed of 93% of the desired (2S)-5-amino-2-(dibenzylamino)-1,6-diphenylhexan-3-ol (34.1.79) along with 7% of the three undesired diastereomers. The crude mixture was debenzylated (Pd-C, HCONH4), and the product was purified by precipitation from iPrOH/HCl (aq) to produce (34.1.55) in greater than 99% purity and in high yield (Scheme 34.8.).

An efficient synthesis of each of the side chain moieties and their coupling with the “core” diamino alcohol derivatives was developed as follows: (S)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanoic acid (34.1.85) was prepared starting from L-valine (34.1.80), which was first converted to N-phenoxycarbonyl-L-valine (34.1.82) with phenylchloroformate (34.1.81). Accurate pH monitoring (pH 9.5 to 10.2) was necessary and LiOH was found to be a superior base. Control of pH was essential as the valine dimer and its derivatives were formed as reaction byproducts outside of this pH margin. LiCl was added to provide a lower freezing point to the aqueous solution and neutral Al2O3 was added to prevent gumming and emulsion formation during the course of the reaction.
Treatment of N-phenoxycarbonyl-L-valine (34.1.82) with 3-chloropropylamine hydrochloride (34.1.3) and solid NaOH in THF produced the unisolated salt of chloropropylurea (34.1.84), which was then treated with t-BuOK, effecting cyclization to produce the desired acid (34.1.85) in 75 to 85% yield and in greater than 99% enantiomeric excess. Acylation of (34.1.79) with synthesized acid (34.1.85) was initially achieved by well-known peptide coupling methods. Optimization of this transformation allowed the discovery of a more cost-effective method for implementing acyl chloride (34.1.86), which was easily prepared using thionyl chloride in THF at room temperature.
The reaction of dibenzylamino alcohols (34.1.79) with acyl chloride (34.1.86) in the presence of 3.0 equivalents of imidazole in EtOAc and DMF produced acylated intermediate (34.1.87) as a mixture of diastereomers, which, without any further purification, was subjected to debenzylation with Pd/C and HCO2NH4 in MeOH at 50°C, which proceeded without significant complications to produce (34.1.88).
Exposure of crude (34.1.88) to L-pyroglutamic acid (34.1.89) in dioxane at 50°C followed by cooling, allowed for the isolation of (S)-N-((2S,4S,5S)-5-amino-4-hydroxy-1,6-diphenylhexan-2-yl)-3-methyl-2-(2-oxotetrahydropyrimidin-1(2H)-yl)butanamide (34.1.90) pyroglutamic salt as virtually a single diastereomer in high yield (Scheme 34.9.).

Acyl chloride (34.1.92) was prepared by the reaction of 2-(2,6-dimethylphenoxy)acetic acid (34.1.91) with thionyl chloride in EtOAc, at room temperature adding a single drop of DMF, and warming the slurry to 50°C, which produced a clear solution of (34.1.92) that was used in the subsequent acylation of amine (34.1.90). Reaction of pyroglutamate salt (34.1.90) with acyl chloride (34.1.95) in ethyl acetate under Schotten-Baumann reaction conditions (use of a two-phase solvent system) in the presence of a water solution of NaHCO3 for liberation of free amine, produced the desired lopinavir (34.1.21) in high yield and purity (Scheme 34.10.).

Lopinavir is a novel and strong protease inhibitor developed from ritonavir with high specificity for HIV-1 protease [139-141]. It is indicated, in combination with other antiretroviral agents, for the treatment of HIV-1 infection. Numerous clinical trials have shown that lopinavir/ritonavir (Kaletra) is highly effective as a component of highly active antiretroviral therapy [142,143].
Ritonavir is indicated in combination with other antiretroviral agents for the treatment of HIV-1-infected patients (adults and children of two years of age and older).[1][2]
PATENT
https://patents.google.com/patent/EP1133485B1/en
- Ritonavir (CAS Number [155213-67-5]), the structural formula of which is given below,is an inhibitor of human HIV protease which was described for the first time by Abbott in International Patent Application WO 94/14436, together with the process for the preparation thereof.
- [0002]
In the synthesis scheme given in WO 94/14436, Ritonavir is manufactured starting from valine and from compounds 1, 4 and 7, the structural formulae of which are also given below. - [0003]
The synthesis process in question was subsequently optimised in its various parts by Abbot, who then described and claimed the individual improvements in the patent documents listed below: US 5,354,866, US 5,541,206, US 5,491,253, WO98/54122, WO 98/00393 and WO 99/11636. - [0004]
The process for the synthesis of Ritonavir carried out on the basis of the above-mentioned patent documents requires, however, a particularly large number of intermediate stages; it is also unacceptable from the point of view of so-called “low environmental impact chemical synthesis” (B.M.Trost Angew. Chem. Int. Ed. Engl. (1995) 34, 259-281) owing to the increased use of activating groups and protective groups which necessitate not inconsiderable additional work in disposing of the by-products of the process, with a consequent increase in the overall manufacturing costs. - [0005]
The object of the present invention is therefore to find a process for the synthesis of Ritonavir which requires a smaller number of intermediate stages and satisfies the requirements of low environmental impact chemical synthesis, thus limiting “waste of material”. - [0006]
A process has now been found which, by using as starting materials the same compounds as those used in WO 94/14436, leads to the formation of Ritonavir in only five stages and with a minimum use of carbon atoms that are not incorporated in the final molecule.
EXAMPLESStage 1
- [0014]
ReagentMolecular weightAmountMmolEq.Amine 1464.6515 g32.2821Val-NCA 2142.136.9 g48.5471.5Triethylamine101.194.5 ml32.2831 (d = 0.726) Dichloromethane 108 ml Sol. 0.3 M - [0015]
Compound Val-NCA 2 was added to a solution of amine 1 (note 1, 2) in dichloromethane (60 ml) at -15°C under nitrogen followed by a solution of triethylamine in dichloromethane (48 ml) (note 3). The reaction mixture was maintained under agitation at from -15° to -13°C for two hours (note 4). The solution was used directly for the next stage without further purification.
- Note 1. Amine 1 was prepared using the procedure described in A.R. Haight et al. Org. Proc. Res. Develop. (1999) 3, 94-100.
- Note 2. Amine 1 was a mixture of stereoisomers with a ratio of 80 : 3.3 : 2.1 : 1.9.
- Note 3. This solution was added dropwise over a period of 25 minutes.
- Note 4. HPLC analysis after 2 hours: Amide 3 70.2 % – starting material 4.3 %
Stage 2
- [0016]
ReagentMolecular weightAmountMmolEq.Amine 3 Solution from stage 1563.78 32.2821Triethylamine101.19 (d = 0.726)2.6 ml18.6540.6Bis(trichloromethyl) carbonate (BTC)296.753.5 g11.7940.36dichloromethane 65 ml N-Methyl-4-aminomethyl-2-isopropylthiazole 4170.275.5 g32.2821triethylamine101.196.9 ml49.5041.5 (d = 0.726) Dichloromethane 52 ml - [0017]
The triethylamine was added slowly to the solution of amine 3 resulting from stage 1 and this mixture was in turn added slowly to a solution of BTC in dichloromethane (65 ml) at from -15° to -13°C and the reaction mixture was maintained under agitation at that temperature for 1.5 hours (note 1). A solution of amine 4 and triethylamine in dichloromethane (52 ml) was then added slowly to the reaction mixture (note 2) and maintained under agitation at that temperature for one hour (note 3). The reaction was stopped with water (97 ml) and the two phases were separated. The organic phase was washed with a 10% aqueous citric acid solution, filtered over Celite and evaporated with a yield of 25 g of crude urea 5 which was purified by flash chromatography on silica gel (eluant: toluene – ethyl acetate 6:4) to give 9.4 g of pure compound 5 (total yield of the two stages 38%) (note 4).
- Note 1. HPLC analysis after 1.5 hours of the reaction stopped in tert-butylamine: Amide 3 = 0.41 % – intermediate isocyanate = 61.3 %
- Note 2. The solution of amine 4 was added over a period of approximately 20 minutes.
- Note 3. HPLC analysis after 1 hour: Amide 3 = 7 % – Urea 5 = 54 %
- Note 4. 1H-NMR (CDCl3, 600 MHz) δ 7.31 (m, 4H), 7.28-7.24 (m, 4H), 7.21-7.14 (m, 8H), 7.11 (d, 2H), 7.05 (d, 2H), 6.96 (s, 1H), 6.75 (bd, 1H), 5.96 (bs, 1H), 4.48 (d, 1H), 4.39 ( d, 1H), 4.13 (dd, 1H), 4.09 (m, 1H), 3.93 (bd, 2H), 3.56 (bt, 1H), 3.39 (d, 2H), 3.28 (m, 1H), 3.04 (dd, 1H), 2.97 (s, 3H), 2.89 (m, 1H), 2.74 (q, 1H), 2.61 (m, 2H), 2.25 (m, 1H), 1.53 ( ddd, 1H), 1.38 (d, 6H), 1.28 (dt, 1H), 0.98 (d, 3H), 0.91 (d, 3H).
Stage 3
- [0018]
ReagentMolecular weightAmountMmolEq.Urea 5760.053.5 g4.6051Pd(OH)2/C 20% 5.25 g 30% w/wAcetic acid 31 ml Sol. 0.15 M - [0019]
The Pearlman catalyst (note 1) was added to a solution of urea 5 in acetic acid and the mixture was hydrogenated for 5 hours at from 4 to 4.5 bar and from 78 to 82°C (note 2). The catalyst was filtered and the solvent was removed under reduced pressure. The crude product was dissolved in water (35 ml), the pH was adjusted to a value of 8 with NaOH, and extracted with CH2Cl2 (2×15 ml). The organic phase was washed with water (10 ml) and the solvent was evaporated under reduced pressure with a yield of 1.8 g of amine 6 as a free base which was used for the next stage without further purification (note 3, 4).
- Note 1. Of the various reaction conditions tested, such as HCOONH4/Pd-C in MeOH, H2/Pd-C in methanol, H2Pd-C/CH3SO3H in methanol, H2/Pd(OH)2/C in methanol, etc., the best conditions we have found hitherto are those described in this Example.
- Note 2. HPLC analysis after 5 hours: amine 6 = 52 % – monobenzyl derivative = 9.3%.
- Note 3. HPLC analysis on the free base: amine 6 = 54 % – monobenzyl derivative = 6%.
- Note 4. 1H-NMR (CDCl3, 600 MHz) δ 7.29 (m, 4H), 7.22-7.14 (m, 6H), 7.00 (s, 1H), 6.83 (bd, 1H), 6.15 (bs, 1H), 4.49 (d, 1H), 4.41 ( d, 1H), 4.25 (m, 1H), 4.08 (m, 1H), 3.38 (m, 1H), 3.29 (m, 1H), 2.99 (s, 3H), 2.92 (dd 1H), 2.84 (m, 2H), 2.76 (m, 1H), 2.47 (m, 1H), 2.28 (m, 1H), 1.76 ( dt, 1H), 1.61 (dt, 1H), 1.38 (d, 6H), 0.95 (d, 3H), 0.89 (d, 3H).
Stage 4
- [0020]
ReagentMolecular weightAmountMmolEq.Amine 6579.801.8 g3.101Compound 7.HCl316.701.28 g4.031.3NaHCO384.00370 mg4.401.4Ethyl acetate 31 ml Sol. 0.1M - [0021]
A solution of the chloride of compound 7 in ethyl acetate was treated with an aqueous sodium bicarbonate solution. The phases were separated and the organic phase was added to a solution of amine 6 in ethyl acetate. The reaction mixture was heated at 60°C for 12 hours, then concentrated; ammonia was added and the solution was maintained under agitation for 1 hour. The organic phase was washed with a 10% aqueous potassium carbonate solution (3x5ml) and with a saturated sodium chloride solution (5 ml); the solvent was removed under reduced pressure. The crude product was purified by flash chromatography on silica gel (eluant ethyl acetate) to give 900 mg of pure Ritonavir (total yield of the two stages 27 %) (note 1).
- Note 1. 1H-NMR (DMSO, 600 MHz) δ 9.05(s, 1H), 7.86 (s, 1H), 7.69 (d, 1H), 7.22-7.10 (m,11H), 6.88 (d, 1H), 6.02 (d, 1H), 5.16 (d, 1H), 5.12 (d, 1H), 4.60 (bs, 1H), 4.48 (d, 1H), 4.42 (d, 1H), 4.15 (m, 1H), 3.94 (dd, 1H), 3.83 (m, 1H), 3.59 (bt, 1H), 3.23 (m, 1H), 2.87 (s, 3H), 2.69-2.63 (m, 3H), 2.60 (m, 1H), 1.88 (m, 1H), 1.45 (m, 2H), 1.30 (d, 6H), 0.74 (d, 6H).
Side effects
When administered at the initially tested higher doses effective for anti-HIV therapy, the side effects of ritonavir are those shown below.[10]
- asthenia, malaise
- diarrhea
- nausea and vomiting
- abdominal pain
- dizziness
- insomnia
- sweating
- taste abnormality
- metabolic effects, including
- hypercholesterolemia
- hypertriglyceridemia
- elevated transaminases
- elevated creatine kinase
One of ritonavir’s side effects is hyperglycemia, through inhibition of the GLUT4 insulin-regulated transporter, thus keeping glucose from entering fat and muscle cells.[medical citation needed] This can lead to insulin resistance and cause problems for people with type 2 diabetes.[medical citation needed]
Drug interactions
Ritonavir induces CYP 1A2 and inhibits the major P450 isoforms 3A4 and 2D6.[according to whom?][medical citation needed] Concomitant therapy of ritonavir with a variety of medications may result in serious and sometimes fatal drug interactions.[11] The list of clinically significant interactions of ritonavir includes the following drugs:
Mechanism of action
Ritonavir was originally developed as an inhibitor of HIV protease, one of a family of pseudo-C2-symmetric small molecule inhibitors.
Ritonavir (center) bound to the active site of HIV protease.[medical citation needed]
Ritonavir is now rarely used for its own antiviral activity but remains widely used as a booster of other protease inhibitors. More specifically, ritonavir is used to inhibit a particular enzyme, in intestines, liver, and elsewhere, that normally metabolizes protease inhibitors, cytochrome P450-3A4 (CYP3A4).[16] The drug binds to and inhibits CYP3A4, so a low dose can be used to enhance other protease inhibitors. This discovery drastically reduced the adverse effects and improved the efficacy of protease inhibitors and HAART. However, because of the general role of CYP3A4 in xenobiotic metabolism, dosing with ritonavir also affects the efficacy of numerous other medications, adding to the challenge of prescribing drugs concurrently.[medical citation needed][17][better source needed]
Pharmocodymanics and pharmacokinetics
The capsules of the medication do not have the same bioavailability as the tablets.[2]
History

New HIV infections and deaths, before and after the FDA approval of “highly active antiretroviral therapy”,[18] of which saquinavir and ritonavir were key as the first two protease inhibitors.[medical citation needed] As a result of the new therapies, HIV deaths in the United States fell dramatically within two years.[18]
Ritonavir is manufactured as Norvir by AbbVie, Inc..[citation needed] The US Food and Drug Administration (FDA) approved ritonavir on March 1, 1996,[19] making it the seventh U.S.-approved antiretroviral drug and the second U.S.-approved protease inhibitor (after saquinavir four months earlier).[citation needed] As a result of the introduction of new “highly active antiretroviral thearap[ies]”—of which the protease inhibitors ritonavir and saquinavir were critical[citation needed]—the annual U.S. HIV-associated death rate fell from over 50,000 to about 18,000 over a period of two years.[20][21]
In 2003, Abbott (now AbbVie, Inc.) raised the price of a Norvir course from US$1.71 per day to US$8.57 per day, leading to claims of price gouging by patients’ groups and some members of Congress. Consumer group Essential Inventions petitioned the NIH to override the Norvir patent, but the NIH announced on August 4, 2004, that it lacked the legal right to allow generic production of Norvir.[22]
In 2014, the FDA approved a combination of ombitasvir/paritaprevir/ritonavir for the treatment of hepatitis C virus (HCV) genotype 4,[3] where the presence of ritonavir again capitalizes on its inhibitory interaction with the human drug metabolic enzyme CYP3A4.
Polymorphism and temporary market withdrawal
Ritonavir was originally dispensed as an ordinary capsule that did not require refrigeration. This contained a crystal form of ritonavir that is now called form I.[23] However, like many drugs, crystalline ritonavir can exhibit polymorphism, i.e., the same molecule can crystallize into more than one crystal type, or polymorph, each of which contains the same repeating molecule but in different crystal packings/arrangements. The solubility and hence the bioavailability can vary in the different arrangements, and this was observed for forms I and II of ritonavir.[24]
During development—ritonavir was introduced in 1996—only the crystal form now called form I was found; however, in 1998, a lower free energy,[25] more stable polymorph, form II, was discovered. This more stable crystal form was less soluble, which resulted in significantly lower bioavailability. The compromised oral bioavailability of the drug led to temporary removal of the oral capsule formulation from the market.[24] As a consequence of the fact that even a trace amount of form II can result in the conversion of the more bioavailable form I into form II, the presence of form II threatened the ruin of existing supplies of the oral capsule formulation of ritonavir; and indeed, form II was found in production lines, effectively halting ritonavir production.[23] Abbott (now AbbVie) withdrew the capsules from the market, and prescribing physicians were encouraged to switch to a Norvir suspension.[citation needed]
The company’s research and development teams ultimately solved the problem by replacing the capsule formulation with a refrigerated gelcap.[when?][citation needed] In 2000, Abbott (now AbbVie) received FDA-approval for a tablet formulation of lopinavir/ritonavir (Kaletra) which contained a preparation of ritonavir that did not require refrigeration.[26]
Research
In 2020, the fixed-dose combination of lopinavir/ritonavir was found not to work in severe COVID-19.[27] In the trial the medication was started around thirteen days after the start of symptoms.[27] Virtual screening of the 1930 FDA-approved drugs followed by molecular dynamics analysis predicted ritonavir blocks the binding of the SARS-CoV-2 spike (S) protein to the human angiotensin-converting enzyme-2 (hACE2) receptor, which is critical for the virus entry into human cells.[28]
References
- ^ Jump up to:a b “Norvir EPAR”. European Medicines Agency (EMA). Retrieved 20 August 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Jump up to:a b c d e f g h i j k “Ritonavir”. The American Society of Health-System Pharmacists. Archived from the original on 2015-10-17. Retrieved Oct 23, 2015.
- ^ Jump up to:a b “FDA approves Viekira Pak to treat hepatitis C”. Food and Drug Administration. December 19, 2014. Archived from the original on October 31, 2015.
- ^ “Ritonavir Pregnancy and Breastfeeding Warnings”. drugs.com. Archived from the original on 7 September 2015. Retrieved 23 October 2015.
- ^ Jump up to:a b British National Formulary 69 (69 ed.). Pharmaceutical Pr. March 31, 2015. p. 426. ISBN 9780857111562.
- ^ Hacker, Miles (2009). Pharmacology principles and practice. Amsterdam: Academic Press/Elsevier. p. 550. ISBN 9780080919225.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 509. ISBN 9783527607495.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “First Generic Drug Approvals”. U.S. Food and Drug Administration (FDA). Retrieved 13 February 2021.
- ^ “Norvir side effects (Ritonavir) and drug interactions – prescription drugs and medications at RxList”. June 27, 2007. Archived from the original on 2007-06-27.
- ^ “Ritonavir: Drug Information Provided by Lexi-Comp: Merck Manual Professional”. April 30, 2008. Archived from the originalon 2008-04-30.
- ^ Henry, J. A.; Hill, I. R. (1998). “Fatal interaction between ritonavir and MDMA”. Lancet. 352 (9142): 1751–1752. doi:10.1016/s0140-6736(05)79824-x. PMID 9848354. S2CID 45334940.
- ^ Papaseit, E.; Vázquez, A.; Pérez-Mañá, C.; Pujadas, M.; De La Torre, R.; Farré, M.; Nolla, J. (2012). “Surviving life-threatening MDMA (3,4-methylenedioxymethamphetamine, ecstasy) toxicity caused by ritonavir (RTV)”. Intensive Care Medicine. 38 (7): 1239–1240. doi:10.1007/s00134-012-2537-9. PMID 22460853. S2CID 19375709.
- ^ Nieminen, Tuija H.; Hagelberg, Nora M.; Saari, Teijo I.; Neuvonen, Mikko; Neuvonen, Pertti J.; Laine, Kari; Olkkola, Klaus T. (2010). “Oxycodone concentrations are greatly increased by the concomitant use of ritonavir or lopinavir/ritonavir”. European Journal of Clinical Pharmacology. 66 (10): 977–985. doi:10.1007/s00228-010-0879-1. ISSN 0031-6970. PMID 20697700. S2CID 25770818.
- ^ Hsieh, Yi-Ling; Ilevbare, Grace A.; Van Eerdenbrugh, Bernard; Box, Karl J.; Sanchez-Felix, Manuel Vincente; Taylor, Lynne S. (2012-05-12). “pH-Induced Precipitation Behavior of Weakly Basic Compounds: Determination of Extent and Duration of Supersaturation Using Potentiometric Titration and Correlation to Solid State Properties”. Pharmaceutical Research. 29 (10): 2738–2753. doi:10.1007/s11095-012-0759-8. ISSN 0724-8741. PMID 22580905. S2CID 15502736.
- ^ Zeldin RK, Petruschke RA (2004). “Pharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patients”. Journal of Antimicrobial Chemotherapy. 53 (1): 4–9. doi:10.1093/jac/dkh029. PMID 14657084.
- ^ “Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers”. U.S. Food and Drug Administration(FDA). December 3, 2019.
- ^ Jump up to:a b (PDF)https://web.archive.org/web/20150924044850/http://www.cdc.gov/mmwr/PDF/wk/mm6021.pdf. Archived from the original (PDF)on 2015-09-24. Retrieved 2020-02-17. Missing or empty
|title=(help) - ^ “Ritonavir FDA approval package” (PDF). 1 March 1996.
- ^ “HIV Surveillance—United States, 1981-2008”. Archived from the original on 9 November 2013. Retrieved 8 November 2013.
- ^ The CDC, in its Morbidity and Mortality Weekly Report, ascribes this to “highly active antiretroviral therapy”, without mention of either of these drugs, see the preceding citation. A further citation is needed to make this accurate connection between this drop and the introduction of the protease inhibitors.
- ^ Ceci Connolly (2004-08-05). “NIH Declines to Enter AIDS Drug Price Battle”. The Washington Post. Archived from the original on 2008-08-20. Retrieved 2006-01-16.
- ^ Jump up to:a b Bauer J; et al. (2001). “Ritonavir: An Extraordinary Example of Conformational Polymorphism”. Pharmaceutical Research. 18 (6): 859–866. doi:10.1023/A:1011052932607. PMID 11474792. S2CID 20923508.
- ^ Jump up to:a b S. L. Morissette; S. Soukasene; D. Levinson; M. J. Cima; O. Almarsson (2003). “Elucidation of crystal form diversity of the HIV protease inhibitor ritonavir by high-throughput crystallization”. Proc. Natl. Acad. Sci. USA. 100 (5): 2180–84. doi:10.1073/pnas.0437744100. PMC 151315. PMID 12604798.
- ^ Lüttge, Andreas (February 1, 2006). “Crystal dissolution kinetics and Gibbs free energy”. Journal of Electron Spectroscopy and Related Phenomena. 150 (2): 248–259. doi:10.1016/j.elspec.2005.06.007.
- ^ “Kaletra FAQ”. AbbVie’s Kaletra product information. AbbVie. 2011. Archived from the original on 7 July 2014. Retrieved 5 July2014.
- ^ Jump up to:a b Cao, Bin; Wang, Yeming; Wen, Danning; Liu, Wen; Wang, Jingli; Fan, Guohui; et al. (18 March 2020). “A Trial of Lopinavir–Ritonavir in Adults Hospitalized with Severe Covid-19”. New England Journal of Medicine. 382 (19): 1787–1799. doi:10.1056/NEJMoa2001282. PMC 7121492. PMID 32187464.
- ^ Bagheri, Milad; Niavarani, Ahmadreza (2020-10-08). “Molecular dynamics analysis predicts ritonavir and naloxegol strongly block the SARS-CoV-2 spike protein-hACE2 binding”. Journal of Biomolecular Structure and Dynamics: 1–10. doi:10.1080/07391102.2020.1830854. ISSN 0739-1102. PMID 33030105. S2CID 222217607.
Further reading
- Chemburkar, Sanjay R.; Bauer, John; Deming, Kris; Spiwek, Harry; Patel, Ketan; Morris, John; Henry, Rodger; Spanton, Stephen; et al. (2000). “Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development”. Organic Process Research & Development. 4 (5): 413–417. doi:10.1021/op000023y.
External links
- “Ritonavir”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Norvir |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a696029 |
| License data | EU EMA: by INNUS DailyMed: Ritonavir |
| Pregnancy category | AU: B3 |
| Routes of administration | By mouth |
| ATC code | J05AE03 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)CA: ℞-onlyUK: POM (Prescription only)US: ℞-onlyEU: Rx-only [1] |
| Pharmacokinetic data | |
| Protein binding | 98-99% |
| Metabolism | Liver |
| Elimination half-life | 3-5 hours |
| Excretion | mostly fecal |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 155213-67-5 |
| PubChem CID | 392622 |
| DrugBank | DB00503 |
| ChemSpider | 347980 |
| UNII | O3J8G9O825 |
| KEGG | D00427 |
| ChEBI | CHEBI:45409 |
| ChEMBL | ChEMBL163 |
| NIAID ChemDB | 028478 |
| PDB ligand | RIT (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID1048627 |
| ECHA InfoCard | 100.125.710 |
| Chemical and physical data | |
| Formula | C37H48N6O5S2 |
| Molar mass | 720.95 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////////RITONAVIR, Antiviral, Peptidomimetics, HIV Protease Inhibitor, A-84538, Abbott 84538, ABBOTT-84538, ABT 538, ABT-538, DRG-0244, NSC-693184, TMC 114r
CC(C)[C@H](NC(=O)N(C)CC1=CSC(=N1)C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC1=CC=CC=C1)NC(=O)OCC1=CN=CS1)CC1=CC=CC=C1

NEW DRUG APPROVALS
ONE TIME
$10.00
BUPIVACAINE
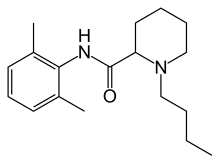
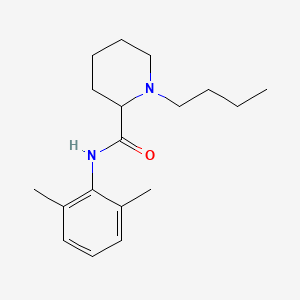
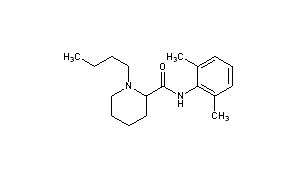
Bupivacaine
cas 38396-39-3, MF C18H28N2O, Average: 288.4277
1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide
- AH 250
- DUR-843
- LAC-43
- SKY 0402
- SKY-0402
- SKY0402
- Win 11318
2-Piperidinecarboxamide, 1-butyl-N-(2,6-dimethylphenyl)-, hydrochloride, hydrate (1:1:1), cas 73360-54-0
Molecular Formula, C18H28N2O.ClH.H2O
Bupivan (Sun) / Carbostesin (AstraZeneca) / Marcain (AstraZeneca) / Marcaina (AstraZeneca) / Posimir (Durect) / Sensorcaine-MPF (Astra Zeneca) / Xaracoll (Innocoll Holdings Limited)
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Bupivacaine hydrochloride | 7TQO7W3VT8 | 73360-54-0 | HUCIWBPMHXGLFM-UHFFFAOYSA-N |
| Bupivacaine hydrochloride anhydrous | AKA908P8J1 | 18010-40-7 | SIEYLFHKZGLBNX-UHFFFAOYSA-N |
BupivacaineCAS Registry Number: 2180-92-9
CAS Name: 1-Butyl-N-(2,6-dimethylphenyl)-2-piperidinecarboxamide
Additional Names:dl-1-butyl-2¢,6¢-pipecoloxylidide; 1-n-butyl-2¢,6¢-dimethyl-2-piperidinecarboxanilide; dl-N-n-butylpipecolic acid 2,6-xylidide; 1-butyl-2-(2,6-xylylcarbamoyl)piperidine; dl-1-n-butylpiperidine-2-carboxylic acid 2,6-dimethylanilide
Molecular Formula: C18H28N2O
Molecular Weight: 288.43
Percent Composition: C 74.95%, H 9.78%, N 9.71%, O 5.55%
Literature References: Prepn: B. Ekenstam et al.,Acta Chem. Scand.11, 1183 (1957); B. T. Ekenstam, B. G. Pettersson, US2955111 (1960 to AB Bofors). Resolution of isomers: B. F. Tullar, J. Med. Chem.14, 891 (1971). Stereospecific synthesis: B. Adger et al.,Tetrahedron Lett.37, 6399 (1996).Pharmacology of racemate: F. Henn, R. Brattsand, Acta Anaesthesiol. Scand. Suppl.21, 9 (1966), C.A.66, 17863u (1967); of isomers: F. P. Luduena et al.,Arch. Int. Pharmacodyn.200, 359 (1972). Clinical pharmacokinetics: D. W. Blake et al.,Anaesth. Intensive Care22, 522 (1994). Comprehensive description: T. D. Wilson, Anal. Profiles Drug Subs.19, 59-94 (1990). Review of use in spinal anesthesia: Acta Anaesthesiol. Scand.35, 1-10 (1991). Review of pharmacology and clinical efficacy of levobupivacaine: K. J. McClellan, C. M. Spencer, Drugs56, 355-362 (1998).Properties: mp 107.5-108°. pKa 8.09; also reported as 8.17. Partition coefficient: (oleyl alcohol/water) 1565; (n-heptane/pH 7.4 buffer) 27.5.
Melting point: mp 107.5-108°
pKa: pKa 8.09; also reported as 8.17
Log P: Partition coefficient: (oleyl alcohol/water) 1565; (n-heptane/pH 7.4 buffer) 27.5
Derivative Type: Hydrochloride monohydrate
CAS Registry Number: 14252-80-3
Manufacturers’ Codes: AH-2250; LAC-43
Trademarks: Carbostesin (AstraZeneca); Marcaine (AstraZeneca); Sensorcaine (AstraZeneca)
Molecular Formula: C18H28N2O.HCl.H2O
Molecular Weight: 342.90
Percent Composition: C 63.05%, H 9.11%, N 8.17%, O 9.33%, Cl 10.34%
Properties: White, odorless crystalline powder. mp 258.5°. Slightly sol in acetone, chloroform, ether. Soly (mg/ml): water 40; alcohol 125. LD50 in mice (mg/kg): 7.8 i.v., 82 s.c. (Henn, Brattsand).
Melting point: mp 258.5°
Toxicity data: LD50 in mice (mg/kg): 7.8 i.v., 82 s.c. (Henn, Brattsand)
Derivative Type: (-)-Form
CAS Registry Number: 27262-47-1
Additional Names: Levobupivacaine; (S)-bupivacaine
Properties: Crystals from isopropanol, mp 135-137°. [a]D25 -80.9° (c = 5 in methanol).
Melting point: mp 135-137°
Optical Rotation: [a]D25 -80.9° (c = 5 in methanol)
Derivative Type: (-)-Form hydrochloride
CAS Registry Number: 27262-48-2
Trademarks: Chirocaine (Abbott)
Molecular Formula: C18H28N2O.HCl
Molecular Weight: 324.89
Percent Composition: C 66.54%, H 9.00%, N 8.62%, O 4.92%, Cl 10.91%
Properties: mp 255-257°. [a]D25 -12.3° (c = 2 in water).
Melting point: mp 255-257°
Optical Rotation: [a]D25 -12.3° (c = 2 in water)
Therap-Cat: Anesthetic (local).
Keywords: Anesthetic (Local).
Other Names for this Substance
- 2-Piperidinecarboxamide, 1-butyl-N-(2,6-dimethylphenyl)-, hydrochloride, hydrate (1:1:1)
- 2-Piperidinecarboxamide, 1-butyl-N-(2,6-dimethylphenyl)-, monohydrochloride, monohydrate
- Bupivacaine hydrochloride monohydrate
- Marcain Heavy
- Marcain
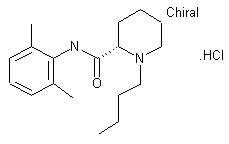
(-)-Bupivacaine hydrochloride, Levobupivacaine hydrochloride, Chirocaine
Synthesis Reference
Thuresson, B. and Egner, B.P.H.; U.S. Patent 2,792,399; May 14, 1957; assigned to AB Bofors, Sweden. Thuresson, B. and Pettersson, B.G.; US. Patent 2,955.1 11; October 4,1960; assigned to AB Bofors, Sweden., US2955111
SYN
British Patent 869,978 (1959).

SYN
Bupivacaine, N-2,6-(dimethyl)1-butyl-2-piperidincarboxamide (2.2.7), is chemically similar to mepivacaine and only differs in the replacement of the N-methyl substituent on the piperidine ring with an N-butyl substituent. There are also two suggested methods of synthesis. The first comes from α-picolin-2,6-xylidide (2.2.4). The alkylation of the last with butyl bromide gives the corresponding pyridine salt (2.2.6). Finally, it is reduced by hydrogen using platinum oxide as a catalyst into a piperidine derivative—bupivacaine [13,16].

The other method results directly from the piperidine-2-carboxylic acid chloride, which is reacted with 2,6-dimethylaniline. The resulting amide (2.2.8) is further alkylated with butyl bromide to bupivacaine [17–19].

Like lidocaine and mepivacaine, bupivacaine is used in infiltration, spinal, and epidural anesthesia in blocking nerve transmission. Its most distinctive property is its long-lasting action. It is used for surgical intervention in urology and in lower thoracic surgery from 3 to 5 h in length, and in abdominal surgery lasting from 45 to 60 min. It is used to block the trifacial nerve, the sacral and brachial plexuses, in resetting dislocations, in epidural anesthesia, and during Cesarian sections. The most common synonym for bupivacaine is marcaine.
SYN
3.7 Bupivacaine (21293) and Levobupivacaine (1976)
Bupivacaine (3.1.41) (Marcaine) is a local anesthetic of great potency and long duration that has been widely used for years, but it has cardio and CNS toxic sideeffects. For many years it was nearly the only local anesthetic applicable to almost all kinds of loco-regional anesthetic techniques, and nowadays, in many occasions, it is still the only alternative available [61–64].
Bupivacaine is currently used in racemic form. At high doses, however, the racemate is potentially hazardous due to toxicity problems.
Currently, racemic bupivacaine (3.1.41) is produced from picolinic acid (3.1.38) either by reduction to pipecolic acid (3.1.39) and then, after conversion to corresponding acid chloride (3.1.40) coupling with 2,6-xylidine to give pipecolic acid-2,6-xylidide (3.1.33), or by reducing the pyridyl amide (3.1.43) prepared from picolinic acid chloride (3.1.42) over platinum oxide. The amide intermediate (3.1.33), which can also be used to prepare the anesthetics ropivacaine (3.1.37) and mepivacaine (3.1.31), was transformed to desired bupivacaine (3.1.41) either by direct alkylation using butyl bromide and potassium carbonate or by reductive amination using butyraldehyde [45,59,65–69] (Scheme 3.7).

Enantiomers of bupivacaine can be prepared via diastereomeric salt resolution with tartaric acid or by resolution of the amide (3.1.33) with O,O-dibenzoyl tartaric acid followed by alkylation [47,70].
One of enantiomers, S(–) isomer of the racemic bupivacaine (levobupivacaine), has equal potency but less cardiotoxic and CNS effects in comparison with both R(+) bupivacaine and bupivacaine racemate. The reduced toxicity of levobupivacaine (3.1.48) gives a wider safety margin in clinical practice [71,72].
Stereospecific synthesis of levobupivacaine from (S)-lysine have been proposed (Scheme 3.8).

Treatment of N-CBZ (S)-lysine (3.1.44) with sodium nitrite in acetic acid yields the acetate (3.1.45). The prepared acetate (3.1.45) was then coupled with dimethyl aniline using N,N′-dicyclohexylcarbodiimide to give the amide (3.1.46) in good yield. The acetate group was then converted into the tosylate (3.1.47), which was deprotected and cyclized stereospecifically in one-pot reaction to give the amide (3.1.33) in high yield. Alkylation is easily achieved using an alkyl bromide and K2CO3 without any racemization. Alkylation can also be carried out using butyraldehyde/formic acid although the former is a much simpler process [73] (Scheme 3.8).
SYN
WO 9611181
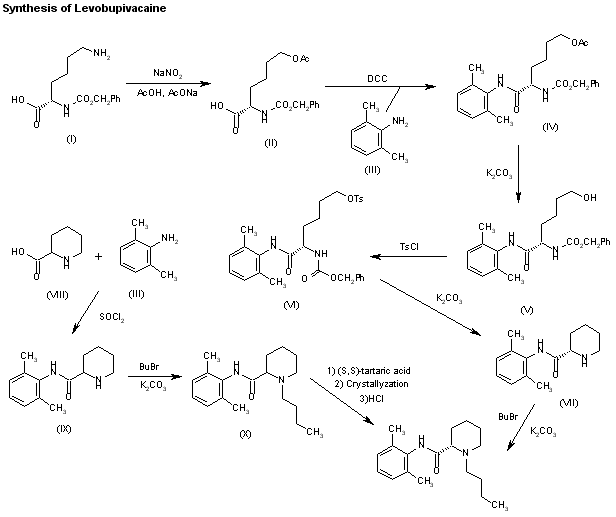
Levobupivacaine has been obtained by two different ways: 1) The deamination of N-benzoyloxycarbonyl-L-lysine (I) with NaNO2/acetic acid gives 6-acetoxy-2(S)-(benzyl-oxycarbonylamino)hexanoic acid (II), which is amidated with 2,6-dimethylaniline (III) and dicyclohexylcarbodiimide (DCC) to the expected amide (IV). The deacetylation of (IV) with K2CO3 in methanol affords compound (V), which is tosylated as usual with tosyl chloride giving intermediate (VI), which is stereospecifically cyclized by means of K2CO3 in ethanol yielding N-(2,6-dimethyl-phenyl)piperidine-2 (S)-carboxamide (VII). Finally, this compound is alkylated with butyl bromide and K2CO3 or by reductoalkylation with butyraldehyde. 2) The amidation of piperidine-2-carboxylic acid (VIII) with 2,6-dimethylaniline (III) by means of SOCl2 in toluene gives the corresponding amide (IX), which is alkylated with butyl bromide as before yielding racemic bupivacaine (X) (3). This compound is then submitted to optical resolution by treatment with (S,S)-(?-tartaric acid followed by crystallization of the resulting tartrate and acidification with HCl in isopropanol.
SYN
| Org Process Res Dev 2000,4(6),530 |
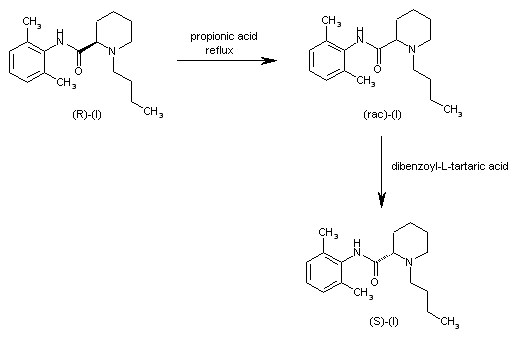
Improved yield in the synthesis of levobupivacaine. An improved yield in the synthesis of levobupivacaine can be obtained by recovering the unwanted (R)-enantiomer side product in the optical resolution of the racemic bupivacaine. The treatment of (R)-(I) with refluxing propionic acid causes its racemization, yielding racemic-(I) (bupivacaine), which is then submitted to a new optical resolution process using dibenzoyl-L-tartaric acid.
Literatures:
Acta Chemica Scandinavica (1947-1973), , vol. 11, p. 1183,1184
Literatures:
BRIDGE PHARMA, INC. Patent: WO2008/88756 A1, 2008 ; Location in patent: Page/Page column 30-31 ;
Yield: ~94%
nmr
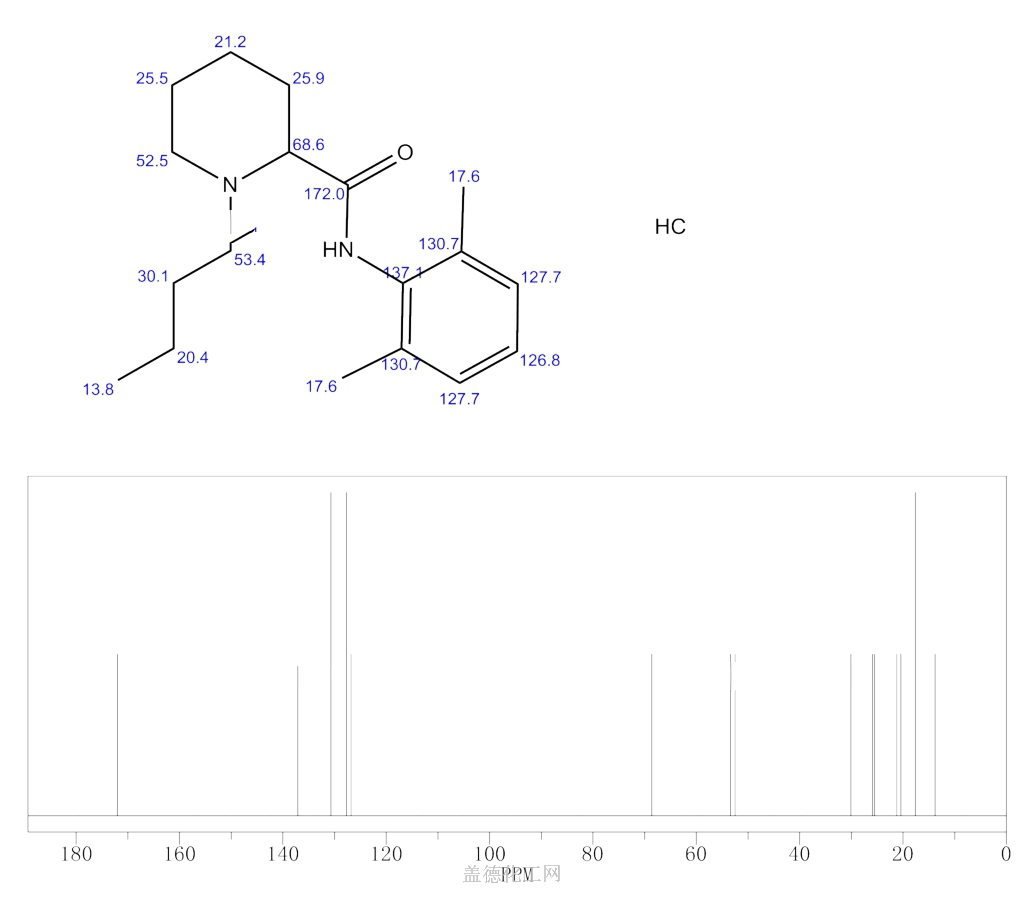
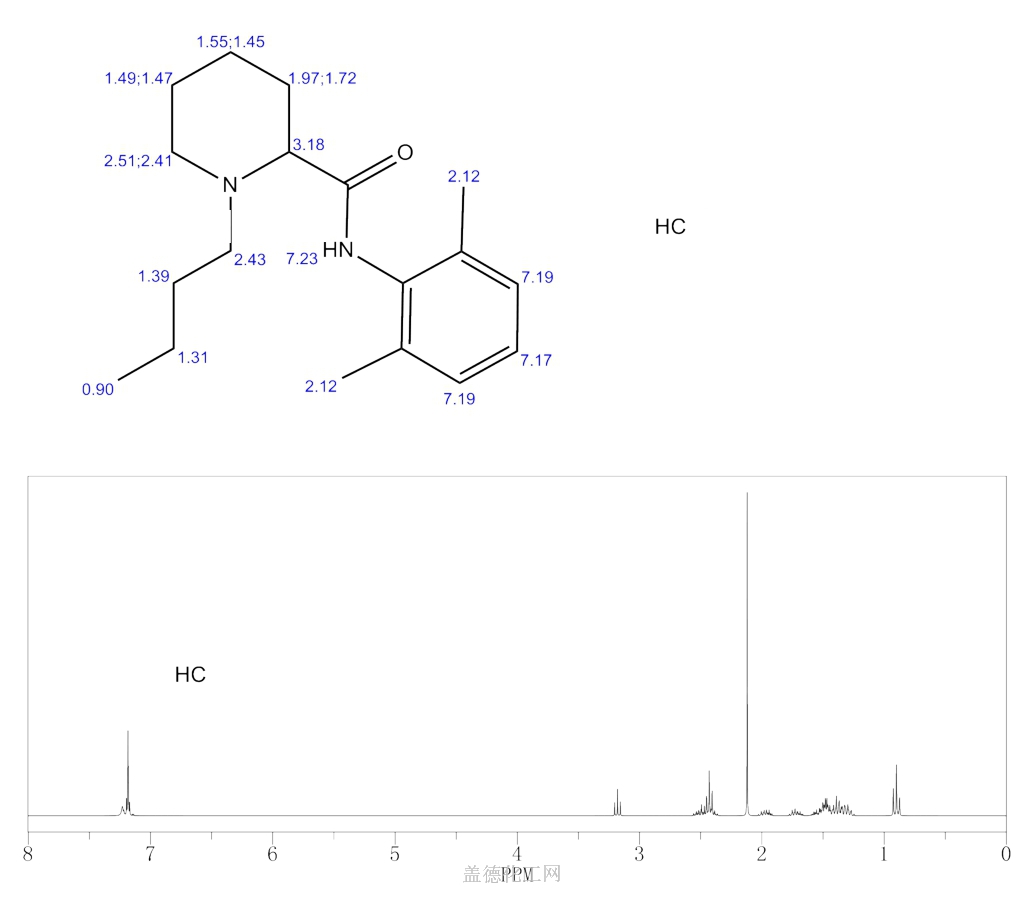
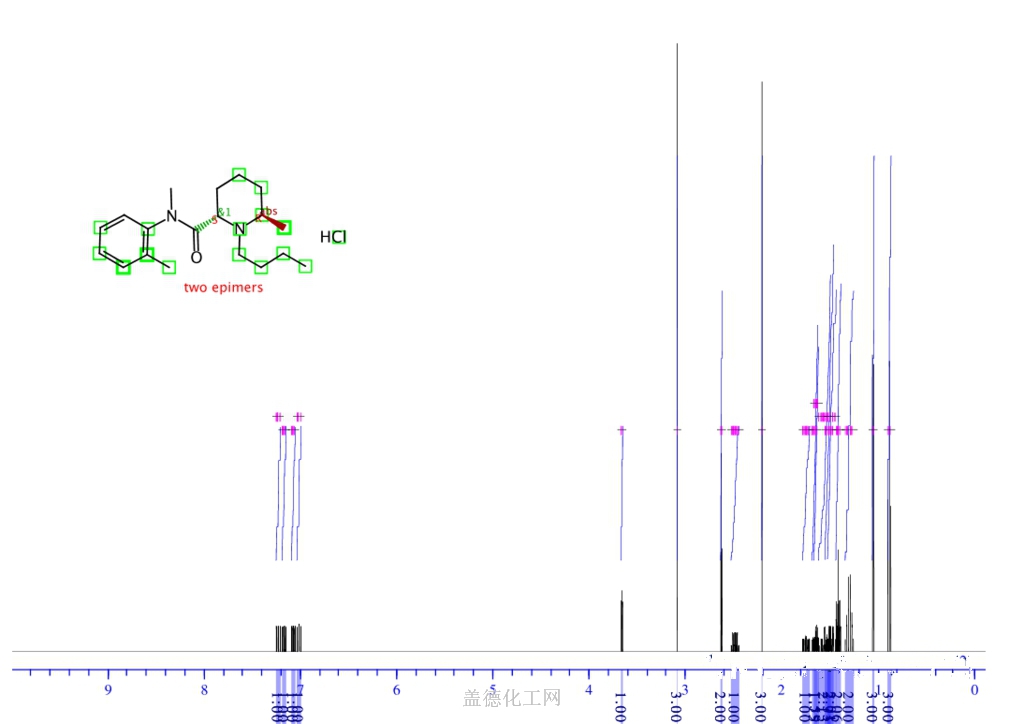
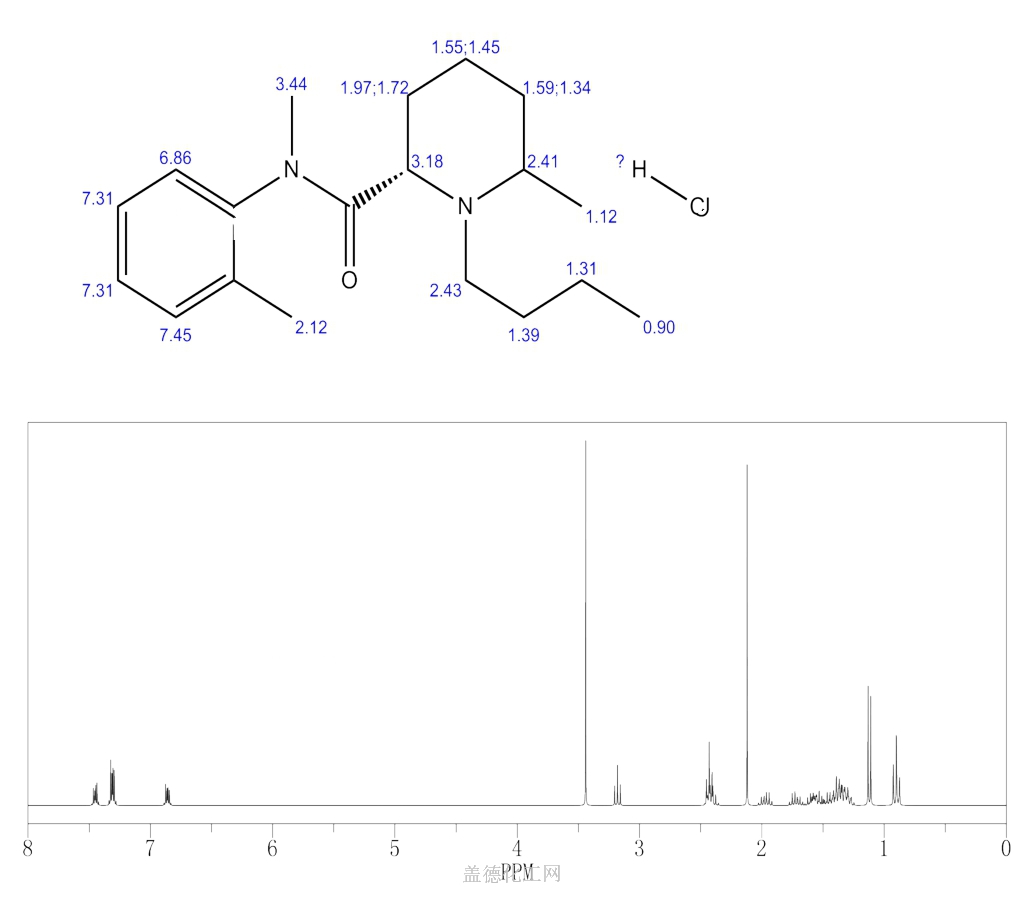
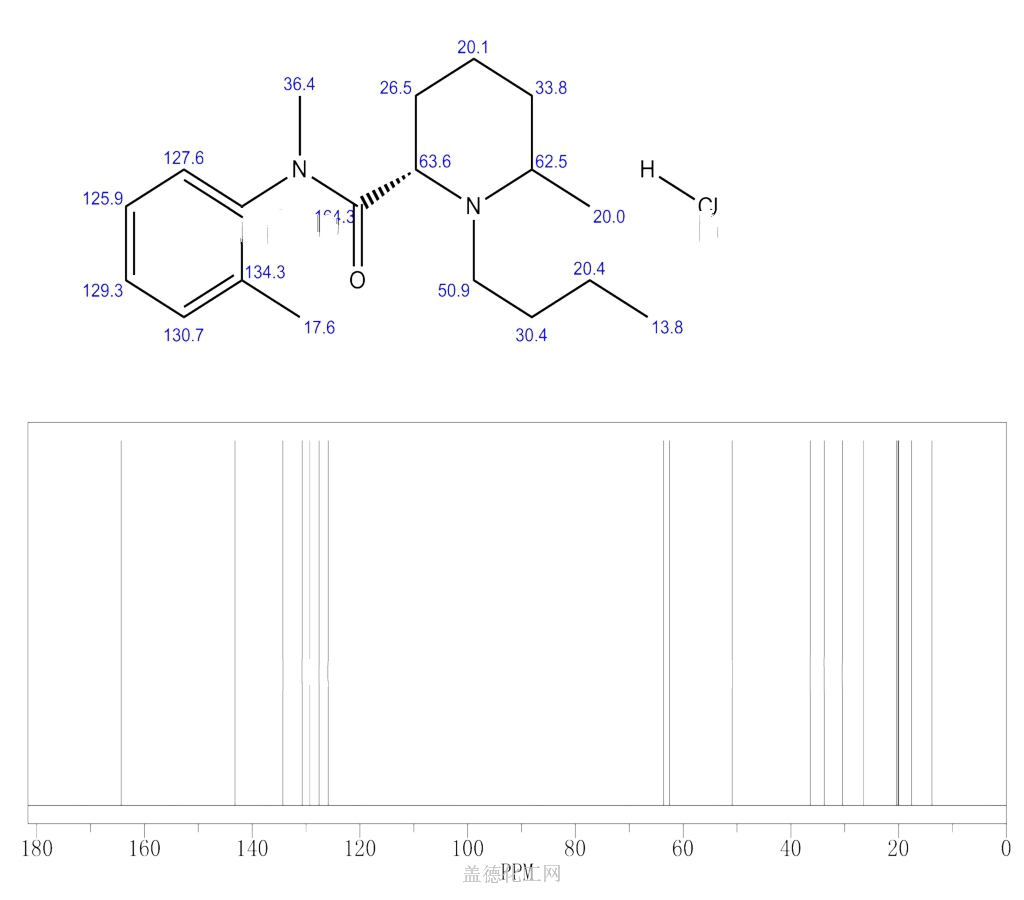
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Bupivacaine is a local anesthetic used in a wide variety of superficial and invasive procedures.
Bupivacaine, marketed under the brand name Marcaine among others, is a medication used to decrease feeling in a specific area.[4] In nerve blocks, it is injected around a nerve that supplies the area, or into the spinal canal’s epidural space.[4] It is available mixed with a small amount of epinephrine to increase the duration of its action.[4] It typically begins working within 15 minutes and lasts for 2 to 8 hours.[4][5]
Possible side effects include sleepiness, muscle twitching, ringing in the ears, changes in vision, low blood pressure, and an irregular heart rate.[4] Concerns exist that injecting it into a joint can cause problems with the cartilage.[4] Concentrated bupivacaine is not recommended for epidural freezing.[4] Epidural freezing may also increase the length of labor.[4] It is a local anaesthetic of the amide group.[4]
Bupivacaine was discovered in 1957.[6] It is on the World Health Organization’s List of Essential Medicines.[7] Bupivacaine is available as a generic medication.[4][8] An implantable formulation of bupivacaine (Xaracoll) was approved for medical use in the United States in August 2020.[9][10][11]
Medical uses
Bupivacaine is indicated for local infiltration, peripheral nerve block, sympathetic nerve block, and epidural and caudal blocks. It is sometimes used in combination with epinephrine to prevent systemic absorption and extend the duration of action. The 0.75% (most concentrated) formulation is used in retrobulbar block.[12] It is the most commonly used local anesthetic in epidural anesthesia during labor, as well as in postoperative pain management.[13] Liposomal formulations of bupivacaine (brand name EXPAREL) have shown to be more effective in providing pain relief than plain solutions of bupivacaine.[14][15]
The fixed-dose combination of bupivacaine with Type I collagen (brand name Xaracoll) is indicated for acute postsurgical analgesia (pain relief) for up to 24 hours in adults following open inguinal hernia repair.[10][11]
Bupivacaine (Posimir) is indicated in adults for administration into the subacromial space under direct arthroscopic visualization to produce post-surgical analgesia for up to 72 hours following arthroscopic subacromial decompression.[16][17]
Contraindications
Bupivacaine is contraindicated in patients with known hypersensitivity reactions to bupivacaine or amino-amide anesthetics. It is also contraindicated in obstetrical paracervical blocks and intravenous regional anaesthesia (Bier block) because of potential risk of tourniquet failure and systemic absorption of the drug and subsequent cardiac arrest. The 0.75% formulation is contraindicated in epidural anesthesia during labor because of the association with refractory cardiac arrest.[18]
Adverse effects
Compared to other local anaesthetics, bupivacaine is markedly cardiotoxic.[19] However, adverse drug reactions (ADRs) are rare when it is administered correctly. Most ADRs are caused by accelerated absorption from the injection site, unintentional intravascular injection, or slow metabolic degradation. However, allergic reactions can rarely occur.[18]
Clinically significant adverse events result from systemic absorption of bupivacaine and primarily involve the central nervous system (CNS) and cardiovascular system. CNS effects typically occur at lower blood plasma concentrations. Initially, cortical inhibitory pathways are selectively inhibited, causing symptoms of neuronal excitation. At higher plasma concentrations, both inhibitory and excitatory pathways are inhibited, causing CNS depression and potentially coma. Higher plasma concentrations also lead to cardiovascular effects, though cardiovascular collapse may also occur with low concentrations.[20] Adverse CNS effects may indicate impending cardiotoxicity and should be carefully monitored.[18]
- CNS: circumoral numbness, facial tingling, vertigo, tinnitus, restlessness, anxiety, dizziness, seizure, coma
- Cardiovascular: hypotension, arrhythmia, bradycardia, heart block, cardiac arrest[13][18]
Toxicity can also occur in the setting of subarachnoid injection during high spinal anesthesia. These effects include: paresthesia, paralysis, apnea, hypoventilation, fecal incontinence, and urinary incontinence. Additionally, bupivacaine can cause chondrolysis after continuous infusion into a joint space.[18]
Bupivacaine has caused several deaths when the epidural anaesthetic has been administered intravenously accidentally.[21]
Treatment of overdose
Further information: Lipid rescue
Animal evidence[22][23] indicates intralipid, a commonly available intravenous lipid emulsion, can be effective in treating severe cardiotoxicity secondary to local anaesthetic overdose, and human case reports of successful use in this way.[24][25] Plans to publicize this treatment more widely have been published.[26]
Pregnancy and lactation
Bupivacaine crosses the placenta and is a pregnancy category C drug. However, it is approved for use at term in obstetrical anesthesia. Bupivacaine is excreted in breast milk. Risks of discontinuing breast feeding versus discontinuing bupivacaine should be discussed with the patient.[18]
Postarthroscopic glenohumeral chondrolysis
Bupivacaine is toxic to cartilage and its intra-articular infusions may lead to postarthroscopic glenohumeral chondrolysis.[27]
Pharmacology
Pharmacodynamics
Bupivacaine binds to the intracellular portion of voltage-gated sodium channels and blocks sodium influx into nerve cells, which prevents depolarization. Without depolarization, no initiation or conduction of a pain signal can occur.
Pharmacokinetics
The rate of systemic absorption of bupivacaine and other local anesthetics is dependent upon the dose and concentration of drug administered, the route of administration, the vascularity of the administration site, and the presence or absence of epinephrine in the preparation.[28]
- Onset of action (route and dose-dependent): 1-17 min
- Duration of action (route and dose-dependent): 2-9 hr
- Half life: neonates, 8.1 hr, adults: 2.7 hr
- Time to peak plasma concentration (for peripheral, epidural, or caudal block): 30-45 min
- Protein binding: about 95%
- Metabolism: hepatic
- Excretion: renal (6% unchanged)[18]
Chemical structure
Like lidocaine, bupivacaine is an amino-amide anesthetic; the aromatic head and the hydrocarbon chain are linked by an amide bond rather than an ester as in earlier local anesthetics. As a result, the amino-amide anesthetics are more stable and less likely to cause allergic reactions. Unlike lidocaine, the terminal amino portion of bupivacaine (as well as mepivacaine, ropivacaine, and levobupivacaine) is contained within a piperidine ring; these agents are known as pipecholyl xylidines.[13]
Society and culture
Legal status
On 17 September 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Exparel, intended for the treatment of post-operative pain.[29] The applicant for this medicinal product is Pacira Ireland Limited.[29] Exparel liposomal was approved for medical use in the European Union in November 2020.[30]
Economics
Bupivacaine is available as a generic medication.[4][8]
Research
Levobupivacaine is the (S)-(–)-enantiomer of bupivacaine, with a longer duration of action, producing less vasodilation. Durect Corporation is developing a biodegradable, controlled-release drug delivery system for after surgery. It has currently[when?] completed a phase-III clinical trial.[31]
References
- ^ “Bupivacaine Use During Pregnancy”. Drugs.com. 13 April 2020. Retrieved 21 September 2020.
- ^ “Marcaine- bupivacaine hydrochloride injection, solution Marcaine with epinephrine- bupivacaine hydrochloride and epinephrine bitartrate injection, solution”. DailyMed. Retrieved 13 February2021.
- ^ “Sensorcaine MPF- bupivacaine hydrochloride injection, solution”. DailyMed. Retrieved 13 February 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “Bupivacaine Hydrochloride”. The American Society of Health-System Pharmacists. Archived from the original on 2015-06-30. Retrieved August 1, 2015.
- ^ Jump up to:a b Whimster, David Skinner (1997). Cambridge textbook of accident and emergency medicine. Cambridge: Cambridge University Press. p. 194. ISBN 9780521433792. Archived from the original on 2015-10-05.
- ^ Egan, Talmage D. (2013). Pharmacology and physiology for anesthesia : foundations and clinical application. Philadelphia, PA: Elsevier/Saunders. p. 291. ISBN 9781437716795. Archivedfrom the original on 2016-05-12.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
- ^ Jump up to:a b Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 22. ISBN 9781284057560.
- ^ “Xaracoll: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 2 September 2020.
- ^ Jump up to:a b “FDA approval letter” (PDF). U.S. Food and Drug Administration (FDA). 28 August 2020. Retrieved 2 September2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b “FDA Approves Xaracoll (bupivacaine HCl) Implant, a Non-opioid, Drug-device Treatment Option for Acute Postsurgical Pain Relief for up to 24 Hours Following Open Inguinal Hernia Repair in Adults” (Press release). Innocoll Pharmaceuticals. 31 August 2020. Retrieved 2 September 2020 – via PR Newswire.
- ^ Lexicomp. “Bupivacaine (Lexi-Drugs)”. Archived from the original on 2014-04-10. Retrieved 20 April 2014.
- ^ Jump up to:a b c Miller, Ronald D. (November 2, 2006). Basics of Anesthesia. Churchill Livingstone.
- ^ Ma, Ting-Ting, et al. (2017). “Liposomal bupivacaine versus traditional bupivacaine for pain control after total hip arthroplasty: A meta-analysis”. Medicinevol. 96 (96, 25 (2017): e7190): e7190. doi:10.1097/MD.0000000000007190. PMC 5484209. PMID 28640101.
- ^ Mont, M. A., Beaver, W. B., Dysart, S. H., Barrington, J. W., & Gaizo, D. J. (2018). “Local Infiltration Analgesia With Liposomal Bupivacaine Improves Pain Scores and Reduces Opioid Use After Total Knee Arthroplasty: Results of a Randomized Controlled Trial”. The Journal of Arthroplasty. 33 (1): 33(1), 90–96. doi:10.1016/j.arth.2017.07.024. PMID 28802777.
- ^https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/204803Orig1s000ltr.pdf
- ^ “Durect Corporation Announces U.S. FDA Approval of Posimir For Post-Surgical Pain Reduction for up to 72 Hours Following Arthroscopic Subacromial Decompression” (Press release). Durect Corporation. 2 February 2021. Retrieved 13 February 2021 – via PR Newswire.
- ^ Jump up to:a b c d e f g “Bupivacaine (Lexi-Drugs)”. Archived from the original on 2014-04-10. Retrieved 20 April 2014.
- ^ de La Coussaye, J. E.; Eledjam, J. J.; Brugada, J.; Sassine, A. (1993). “[Cardiotoxicity of local anesthetics]”. Cahiers d’Anesthésiologie. 41 (6): 589–598. ISSN 0007-9685. PMID 8287299.
- ^ Australian Medicines Handbook. Adelaide. 2006. ISBN 978-0-9757919-2-9.
- ^ ABS-CBN Interactive: Filipino nurse dies in UK due to wrong use of anaesthetic
- ^ Weinberg, GL; VadeBoncouer, T; Ramaraju, GA; Garcia-Amaro, MF; Cwik, MJ. (1998). “Pretreatment or resuscitation with a lipid infusion shifts the dose-response to bupivacaine-induced asystole in rats”. Anesthesiology. 88 (4): 1071–5. doi:10.1097/00000542-199804000-00028. PMID 9579517. S2CID 1661916.
- ^ Weinberg, G; Ripper, R; Feinstein, DL; Hoffman, W. (2003). “Lipid emulsion infusion rescues dogs from bupivacaine-induced cardiac toxicity”. Regional Anesthesia and Pain Medicine. 28 (3): 198–202. doi:10.1053/rapm.2003.50041. PMID 12772136. S2CID 6247454.
- ^ Rosenblatt, MA; Abel, M; Fischer, GW; Itzkovich, CJ; Eisenkraft, JB (July 2006). “Successful use of a 20% lipid emulsion to resuscitate a patient after a presumed bupivacaine-related cardiac arrest”. Anesthesiology. 105 (1): 217–8. doi:10.1097/00000542-200607000-00033. PMID 16810015.
- ^ Litz, RJ; Popp, M; Stehr, S N; Koch, T. (2006). “Successful resuscitation of a patient with ropivacaine-induced asystole after axillary plexus block using lipid infusion”. Anaesthesia. 61 (8): 800–1. doi:10.1111/j.1365-2044.2006.04740.x. PMID 16867094. S2CID 43125067.
- ^ Picard, J; Meek, T (February 2006). “Lipid emulsion to treat overdose of local anaesthetic: the gift of the glob”. Anaesthesia. 61(2): 107–9. doi:10.1111/j.1365-2044.2005.04494.x. PMID 16430560. S2CID 29843241.
- ^ Gulihar, Abhinav; Robati, Shibby; Twaij, Haider; Salih, Alan; Taylor, Grahame J.S. (December 2015). “Articular cartilage and local anaesthetic: A systematic review of the current literature”. Journal of Orthopaedics. 12 (Suppl 2): S200–S210. doi:10.1016/j.jor.2015.10.005. PMC 4796530. PMID 27047224.
- ^ “bupivacaine hydrochloride (Bupivacaine Hydrochloride) injection, solution”. FDA. Archived from the original on 21 April 2014. Retrieved 20 April 2014.
- ^ Jump up to:a b “Exparel: Pending EC decision”. European Medicines Agency (EMA). 17 September 2020. Retrieved 21 September 2020.Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “Exparel liposomal EPAR”. European Medicines Agency (EMA). 15 September 2020. Retrieved 11 December 2020.
- ^ “Bupivacaine Effectiveness and Safety in SABER Trial (BESST)”. ClinicalTrials.gov. 20 January 2010. Archived from the original on 2011-12-27. Retrieved 2012-03-01.
External links
- “Bupivacaine”. Drug Information Portal. U.S. National Library of Medicine.
///////////Bupivacaine, AH 250, DUR-843, LAC-43, SKY 0402, SKY-0402, SKY0402, Win 11318, ANAESTHETIC
- CCCCN1CCCCC1C(=O)NC1=C(C)C=CC=C1C

NEW DRUG APPROVALS
ONE TIME
$10.00
ROPIVACAINE

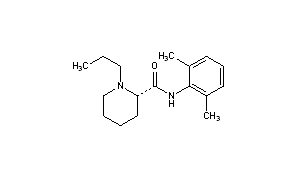
Ropivacaine
CAS No.84057-95-4 (Ropivacaine);
- Molecular FormulaC17H26N2O
- Average mass274.401 Da
HCL SALTCAS Registry Number: 98717-15-8
HCL MONOHYDRATE
Molecular Weight328.88, FormulaC17H26N2O • HCl • H2O
132112-35-7 (Ropivacaine HCl Monohydrate);
Chemical Name S-(-)-1-propyl-2′,6′-pipecoloxylidide hydrochloride monohydrate(S)-(-)-1-Propyl-2′,6′-pipecoloxylidide
(S)-N-(2,6-dimethylphenyl)-1-propyl-2-piperidinecarboxamide
2-Piperidinecarboxamide, N-(2,6-dimethylphenyl)-1-propyl-, (2S)-
5376
5421606[Beilstein]
7IO5LYA57N
84057-95-4[RN]
854056-07-8[RN]
(S)-ropivacaine
(2S)-N-(2,6-Dimethylphenyl)-1-propyl-2-piperidinecarboxamide
ропивакаин [Russian] [INN]
روبيفاكائين [Arabic] [INN]
罗哌卡因 [Chinese] [INN]
Drug Name:Ropivacaine Hydrochloride Hydrate
Research Code:LEA-103; NA-001; (-)-LEA-103;
Trade Name:Naropin® / Anapeine®
MOA:Sodium channels blockers
Indication:Anaesthetic
Company:AstraZeneca (Originator) , Fresenius Kabi
ATC Code:N01BB09APPROVED
- US
- JP
- CN
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1996-09-26 | First approval | Naropin | Anaesthetic | Injection | 2 mg/ml; 5 mg/ml; 7.5 mg/ml; 10 mg/ml | APP Pharmaceuticals |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2001-04-04 | First approval | Anapeine | Anaesthetic | Injection | 2 mg/ml; 7.5 mg/ml; 10 mg/ml | AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2010-02-11 | Marketing approval | 耐乐品/Naropin | Anaesthetic | Injection | 20 mg/10 ml;100 mg/10 ml; 75 mg/10 ml; 50 mg/10 ml | AstraZeneca | |
| 2010-02-03 | Marketing approval | 耐乐品/Naropin | Anaesthetic | Injection | 2 mg/mL | AstraZeneca |
| No. | NDA No. | Major Technical Classification | Patent No. | Estimated Expiry Date | Drug Substance Claim | Drug Product Claim | Patent Use Code (All list) |
| 1 | N020533 | Uses(Indication) | 5670524 | 2014-09-23 | Y | Y | U – 833 |
| 2 | N020533 | Device | 7828787 | 2025-10-18 | Y | ||
| 3 | N020533 | Device | 7857802 | 2026-11-28 | Y | ||
| 4 | N020533 | Device | 8118802 | 2023-05-18 | Y | ||
| 5 | N020533 | Device | 8162915 | 2024-05-23 | Y |
Ropivacaine
CAS Registry Number: 84057-95-4
CAS Name: (2S)-N-(2,6-dimethylphenyl)-1-propyl-2-piperidinecarboxamide
Additional Names: (S)-(-)-1-propyl-2¢,6¢-pipecoloxylidide; l-N-n-propylpipecolic acid-2,6-xylidide
Manufacturers’ Codes: LEA-103
Molecular Formula: C17H26N2O, Molecular Weight: 274.40
Percent Composition: C 74.41%, H 9.55%, N 10.21%, O 5.83%
Literature References: Prepn: A. F. Thuresson, C. Bovin, WO8500599 (1985 to Apothekernes); H.-J. Federsel et al.,Acta Chem. Scand.B41, 757 (1987).Physicochemical properties: G. R. Strichartz et al.,Anesth. Analg.71, 158 (1990).HPLC determn in human plasma: Z. Yu et al.,J. Chromatogr. B654, 221 (1994). In vitro metabolism: Y. Oda et al.,Anesthesiology82, 214 (1995). Clinical pharmacokinetics: D. J. Kopacz et al.,ibid.81, 1139 (1994). Toxicity study in sheep: A. C. Santos et al.,ibid.82, 734 (1995). Clinical evaluation in relief of surgical pain: I. Cederholm et al.,Reg. Anesth.19, 18 (1994); B. Johansson et al.,Anesth. Analg.78, 210 (1994); labor pain: R. Stienstra et al.,ibid.80, 285 (1995).
Properties: Crystals from toluene, mp 144-146°. [a]D25 -82.0° (c = 2 in methanol). pKa 8.16. Distribution coefficient (1-octanol/aq buffer, pH 7.4): 115.0.
Melting point: mp 144-146°
pKa: pKa 8.16
Optical Rotation: [a]D25 -82.0° (c = 2 in methanol)
Derivative Type: Hydrochloride
CAS Registry Number: 98717-15-8
Trademarks: Naropin (AstraZeneca)
Molecular Formula: C17H26N2O.HCl, Molecular Weight: 310.86
Percent Composition: C 65.68%, H 8.75%, N 9.01%, O 5.15%, Cl 11.40%
Properties: Crystals from isopropyl alcohol, mp 260-262°. [a]D25 -6.6° (c = 2 in water).
Melting point: mp 260-262°
Optical Rotation: [a]D25 -6.6° (c = 2 in water)
Derivative Type: Hydrochloride monohydrate
CAS Registry Number: 132112-35-7
Properties: Crystals from acetone + water, mp 269.5-270.6°. [a]D20 -7.28° (c = 2 in water).
Melting point: mp 269.5-270.6°
Optical Rotation: [a]D20 -7.28° (c = 2 in water)
Therap-Cat: Anesthetic (local).
Keywords: Anesthetic (Local).Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Ropivacaine hydrochloride | V910P86109 | 132112-35-7 | VSHFRHVKMYGBJL-CKUXDGONSA-N |
| Ropivacaine hydrochloride anhydrous | 35504LBE2T | 98717-15-8 | NDNSIBYYUOEUSV-RSAXXLAASA-N |
Ropivacaine is an analgesic drug used for local or regional anesthesia for surgery and short-term management of pain.Ropivacaine is an aminoamide local anaesthetic drug commonly marketed by AstraZeneca under the trade name Naropin. It is present as a racemic mixture of the enantiomers containing equal proportions of the “S” and “R” forms. The marketed form contains the single S-enantiomer as the active ingredient.
Ropivacaine hydrochloride hydrate was first approved by the U.S. Food and Drug Administration (FDA) on September 26, 1996, then approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) in April 4, 2001. It was developed by AstraZeneca, then marketed as Naropin® by APP Pharmaceuticals, LLC. in the US and as Anapeine® by AstraZeneca in JP.
Ropivacaine is a local anaesthetic drug belonging to the amino amide group. It is indicated for the production of local or regional anesthesia for surgery and for acute pain management.
Naropin® is available as injection solution for intravenous use, containing 2, 5, 7.5 or 10 mg of Ropivacaine hydrochloride one mL. Common concentration is 7.5 mg/mL, and the maximum single dose is 200 mg.
Ropivacaine (rINN) /roʊˈpɪvəkeɪn/ is a local anaesthetic drug belonging to the amino amide group. The name ropivacaine refers to both the racemate and the marketed S–enantiomer. Ropivacaine hydrochloride is commonly marketed by AstraZeneca under the brand name Naropin.
Table 1 The common types of local anesthetics
Syn
Synthesis Reference
Peter Jaksch, “Process for the preparation of ropivacaine hydrochloride monohydrate.” U.S. Patent US5959112, issued February, 1970.
US5959112Route 1
Reference:1. US2799679A.
Reference:1. WO8500599A1.
https://patents.google.com/patent/WO1985000599A1/enA large variety of N-alkyl-pipecolic acid amides have been synthesized. A number of these compounds have found use as local anesthetics, such as Mepivacaine, namely the racemate of N-methylpipecolic–acid-2,6-xylidide:

and Bupivacaine, namely the racemate of N-butylpipecolic- acid-2,6-xylidide:

References disclosing homologs of this series of compounds include U.S. Patent 2,799,679; British Patent 775,749; British Patent 775,750; British Patent 800,565; British Patent 824,542; British Patent 869,978; British Patent 949,729; U.S. Patent 4,110,331; and U.S. Patent 4,302,465.There is a summary paper dealing with these types of anesthetics, and related compounds in a paper in Acta Che ica Scandinavica 11, (1957) No. 7 pp. 1183-1190 by Bo Thuresson af Ekenstam et al.There is a discussion of the“ effect of optical isomers in related compounds in J. Med. Chem., 14 (1971) pp. 891-892 entitled “Optical Isomers Of Mepivacaine And Bupivacaine” by Benjamin F. Tullar; Acta Pha m. Suecica, 8 (1971) pp. 361- 364 entitled “Some Physicochemical Properties Of The Racemates And The Optically Active Isomers Of Two Local Anaesthetic Compounds”, by . Friberger et al -.; Acta Pharmacol et Toxicol, 31 (1972). pp. 273-286 entitled “Toxicological And Local Anaesthetic Effects Of Optically Active Isomers Of Two Local Anaesthetic Compounds”, by G. Aberg; Annual Review Of Pharmacology, 9 (1969) pp. 5Q3-520 entitled “Duration Of Local Anaesthesia”, by F.P. Luduena and Acta Pharmacol, et Toxicol, 41 (1977). pp. 432-443 entitled “Studies On The Duration Of Local Anaesthesia: Structure/Activity Relationships In A Series Of Homologous Local Anaesthetics”, by G. Aberg et al.

Reference:1. J. Labelled Compd. Rad. 1987, 24, 521-528.Route 4
Reference:1. CN104003930A.
https://patents.google.com/patent/CN104003930A/enRopivacaine (Ropivacaine) is the long-acting local anesthetics of amide derivatives of Novel pure levo form of Astra drugmaker of Sweden listing in 1996, there is analgesia and anesthesia dual function, be widely used in nerve block anesthesia, local infiltration anesthesia and epidural anesthesia , be particularly useful for Postoperative Analgesia After and obstetrical analgesia.On piperidine ring in ropivacaine structure, having a chiral carbon atom, is chipal compounds, and levoisomer is low compared with dextrorotatory isomer toxicity, and action effect is good.Ropivacaine HCL is the hydrochloride of ropivacaine, and chemistry is by name: (-)-(S)-N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-carboxamide hydrochloride, molecular formula is C 17 h 26 n 2 oHCl, structural formula:At present, in prior art, the synthetic method of ropivacaine mainly contains:Taking L-2-piperidine formyl chlorine as starting raw material, through phosphorus pentachloride or sulfur oxychloride acidylate, then with the condensation of 2,6-xylidine, and then react and obtain ropivacaine with n-propyl bromide.Although this method production technique is simple, reactions steps is also shorter, but commercially available L-2-piperidine carboxylic acid average price is 4~5 times of racemization Pipecolic Acid, raw materials cost is too high, and may there is racemization phenomenon in subsequent reactions process, affect optical purity of products, for example US Patent No. 4695576 and “Chinese Medicine magazine” o. 11th in 2012 “Ropivacaine HCL a synthetic” literary composition and Chinese patent CN201310041390.2 all adopt this kind of method.”synthetic chemistry” the 14th the 4th phase of volume “Synthesis of Ropivacaine Hydrochloride by Triphosgene” in 2006 and Hunan University’s Master’s thesis “synthesising process research of Ropivacaine HCL and” disclose the synthetic method of another ropivacaine, the Pipecolic Acid that adopts inexpensive racemization is raw material, prepare Ropivacaine HCL through reactions such as amidation, alkylations, use triphosgene or thionyl chloride to prepare acyl chlorides, but triphosgene danger in the time of storage and aftertreatment is larger, is not suitable for suitability for industrialized production; And partial condition in the latter’s method (reagent that the pH separating as intermediate (I) and reagent, catalyzer and recrystallization are used etc.) haves much room for improvement,under its test conditions, be difficult to take into account high purity and high yield simultaneously, according to prior art, the separation of ropivacaine raceme is also not ideal.The preparation method of embodiment 1, a kind of Ropivacaine HCL, comprises the steps:(1) preparation of intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea10.0g2-piperidine carboxylic acid, 160ml toluene are added in 500ml reaction flask.Pass into HCl gas, to pH2, be warming up to 48 ± 2 DEG C, add 1.5mlDMF, drip 11.2g (1.2 equivalent) sulfur oxychloride and 20ml toluene mixture liquid, drip and finish, be incubated 48 ± 2 DEG C of reaction 3h.Drip 2 of 4.0 equivalents, 6-xylidine and 20ml toluene mixture liquid, be incubated 58 ± 2 DEG C of reaction 3h.Filter, obtain yellow-green colour wet product 65g, dry to obtain gray solid 56g, solid is added in 280ml purified water, stir the molten reaction solution that obtains; 10%NaOH solution is slowly dropped in reaction solution, adjust pH to 4.5-5.0, use 100ml toluene wash , layering, retains water layer, continues to adjust pH to 9-10 with 10%NaOH solution, adds 100ml methylene dichloride.Layering, gets organic layer,and water layer continues to use 50ml dichloromethane extraction, merges organic layer, adds anhydrous sodium sulfate dehydration, 40 DEG C of concentrating under reduced pressure.Obtain pale yellow oily liquid body 15.5g, yield 86.2%, is intermediate (I) N-( 2,6-dimethyl benzene)-2-piperidyl urea.(2) preparation of intermediate (II) N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amideIntermediate 15.5 g of (the I) Dissolved in 60mlDMF IS, ADDS 8.9gK 2 cO . 3 , 8.2 g of drip (1.0 equivalent)-n-propyl bromide, and drip BE Finishing After Warming up to 78 ± 2 of DEG C, Insulation Reaction 2H; Ice Bath is down to room temperature, filters, and filtrate is added in 150ml frozen water, separates out a large amount of white solids, filter, dry, obtain white solid 17.4g, yield 95.0%, is intermediate (II) N-(2, 6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amide.(3) preparation of left-handed ropivacaine tartrate17.4g intermediate (II) is dissolved in 100ml Virahol, heats up 40 DEG C and stir molten; Treat entirely moltenly, add successively 1.80g (0.1 equivalent) titanium isopropylate, 1.91g (0.2 equivalent) D-tartrate, be warming up to backflow, after solution clarification, continue reaction 2h; Be cooled to 30 DEG C of crystallizatioies, filter, 75 DEG C of oven dry, obtain white solid 8.7g, and yield 39.2% is left-handed ropivacaine tartrate; After testing, ropivacaine purity 99.02%, dextrorotatory isomer per-cent 0.98%.(4) preparation of Ropivacaine HCL crude productLeft-handed 8.7g ropivacaine tartrate is joined in 50ml Virahol, be warming up to 50 DEG C, drip concentrated hydrochloric acid, surveying pH is 1~2, insulation reaction 2h.Be cooled to 0 DEG C of crystallization, separate out a large amount of white solids, filter, dry, obtain white solid 6.6g, yield 85.5%, is Ropivacaine HCL crude product.After testing, ropivacaine purity 99.11%, dextrorotatory isomer per-cent 0.89%.(5) refining6.6g crude product and 40ml dehydrated alcohol-concentrated hydrochloric acid mixed solution (20:1) are added in reaction flask, be heated to 50 DEG C and make to dissolve; Complete molten after, naturally cool to room temperature, ice-water bath is cooled to 0 DEG C, crystallization 2h; Filter, 5ml mixed solution washing for filter cake, obtains wet product, dries, and obtains white solid 6.0g, and yield 91.7%, is Ropivacaine HCL fine work.After testing, ropivacaine purity 99.91 %, dextrorotatory isomer per-cent 0.09%.The preparation method of embodiment 2, a kind of Ropivacaine HCLStep is as follows:(1) preparation of intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea100.0g2-piperidine carboxylic acid, 1600ml toluene are added in 3000ml reaction flask.Pass into HCl gas, to pH2 left and right, be warming up to 45~50 DEG C, add 15mlDMF, drip 111.5g (1.2 equivalent) sulfur oxychloride and 200ml toluene mixture liquid, drip and finish, be incubated 50-55 DEG C of reaction 3h.Drip 2 of 4.0 equivalents, 6-xylidine and 200ml toluene mixture liquid, be incubated 55~60 DEG C of reaction 2h.Filter, obtain the about 660g of yellow-green colour wet product, dry to obtain gray solid 545g, solid is added in 3000ml purified water, stir the molten reaction solution that obtains; 10%NaOH solution is slowly dropped in reaction solution, adjust pH to 4.5~5.0 , use 1000ml toluene wash, layering, retains water layer, continues to adjust pH to 9~10 with 10%NaOH solution, adds 1000ml methylene dichloride.Layering, gets organic layer,and water layer continues to use 750ml dichloromethane extraction, merges organic layer, adds anhydrous sodium sulfate dehydration, 40 DEG C of concentrating under reduced pressure.Obtain the about 151.8g of pale yellow oily liquid body, yield 84.5%, is intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea.(2) preparation of intermediate (II) N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amideIntermediate 150.0 g (the I) Dissolved in 600mlDMF IS, ADDS 86.5gK 2 cO . 3 , drip 95.4 g (1.2 equivalent)-n-propyl bromide, and drip BE Finishing After Warming up to 85 ~ 90 of DEG C, Insulation Reaction 2H; of Be Down to room temperature, filter, filtrate is added in 1500ml frozen water, separate out a large amount of white solids, filter, dry, obtain the about 167.6g of white solid, yield 94.6%, is intermediate (II) N-(2, 6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amide.(3) preparation of left-handed ropivacaine tartrate160.0g intermediate (II) is dissolved in 1000ml Virahol, heats up 50 DEG C and stir molten; Treat entirely moltenly, add successively 16.58g (0.1 equivalent) titanium isopropylate, 43.8g (0.5 equivalent) D-tartrate, be warming up to backflow, after solution clarification, continue reaction 3h; Cooling, is down to 30-35 DEG C of crystallization, filters, and 75 DEG C of oven dry, obtain white solid 84.2g, and yield 41.3% is left-handed ropivacaine tartrate; After testing, ropivacaine purity 98.97%, dextrorotatory isomer per-cent 1.03%.(4) preparation of Ropivacaine HCL crude productLeft-handed 80.0g ropivacaine tartrate is joined in 500ml Virahol, be warming up to 50 DEG C, drip concentrated hydrochloric acid, surveying pH is 1~2, insulation reaction 4h.Be cooled to 0~5 DEG C of crystallization, separate out a large amount of white solids, filter, dry, obtain the about 61.6g of white solid, yield 86.5%, is Ropivacaine HCL crude product.After testing, ropivacaine purity 99.07%, dextrorotatory isomer per-cent 0.93%.(5) refining60.0g crude product and 500ml dehydrated alcohol-concentrated hydrochloric acid mixed solution (20:1) are added in reaction flask, be heated to 50 DEG C and make to dissolve; Complete molten after, cooling crystallization, ice-water bath is cooled to 0-5 DEG C, crystallization 4h; Filter, a small amount of cold mixed solution washing for filter cake, obtains wet product, dries, and obtains white solid 55.6g, and yield 92.7%, is Ropivacaine HCL fine work.After testing, ropivacaine purity 99.87%, dextrorotatory isomer per-cent 0.13%.The preparation method of embodiment 3, a kind of Ropivacaine HCLStep is as follows:(1) preparation of intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea10.0g2-piperidine carboxylic acid, 160ml toluene are added in 500ml reaction flask.Pass into HCl gas, to pH3 left and right, be warming up to 48 ± 2 DEG C, add 1.5mlDMF, drip 9.3g (1.0 equivalent) sulfur oxychloride and 20ml toluene mixture liquid, drip and finish, be incubated 48 ± 2 DEG C of reaction 2h.Drip 2 of 4.0 equivalents, 6-xylidine and 20ml toluene mixture liquid, be incubated 58 ± 2 DEG C of reaction 3h.Filter, obtain yellow-green colour wet product 63.6g, dry to obtain gray solid 55g, solid is added in 280ml purified water, stir the molten reaction solution that obtains; 10%NaOH solution is slowly dropped in reaction solution, adjust pH to 4.5-5.0, use 100ml toluene wash, layering, retains water layer, continues to adjust pH to 9-10 with 10%NaOH solution, adds 100ml methylene dichloride.Layering, gets organic layer,and water layer continues to use 50ml dichloromethane extraction, merges organic layer, adds anhydrous sodium sulfate dehydration, 40 DEG C of concentrating under reduced pressure.Obtain the about 14.8g of pale yellow oily liquid body, yield 82.4%, is intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea.(2) preparation of intermediate (II) N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amideIntermediate 14.8 g of (the I) Dissolved in 60mlDMF IS, ADDS 8.5gK 2 cO . 3 , 7.8 g of drip (1.0 equivalent)-n-propyl bromide, and drip After Finishing of DEG BE Warming up to 75 C, Reaction Insulation 2H; IS Down Ice Bath to room temperature, filters, and filtrate is added in 150ml frozen water, separates out a large amount of white solids, filter, dry, obtain the about 16.0g of white solid, yield 91.5%, is intermediate (II) N-(2 ,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amide.(3) preparation of left-handed ropivacaine tartrate15g intermediate (II) is dissolved in 100ml Virahol, heats up 40 DEG C and stir molten; Treat entirely moltenly, add successively 1.72g (0.1 equivalent) titanium isopropylate, 1.82g (0.2 equivalent) D-tartrate, be warming up to backflow , after solution clarification, continue reaction 1h; Be cooled to 32 DEG C of crystallizatioies, filter, 75 DEG C of oven dry, obtain white solid 7.5g, and yield 39.2% is left-handed ropivacaine tartrate; After testing, ropivacaine purity 98.92 %, dextrorotatory isomer per-cent 0.99%.(4) preparation of Ropivacaine HCL crude productLeft-handed 7.5g ropivacaine tartrate is joined in 50ml Virahol, be warming up to 40 DEG C, drip concentrated hydrochloric acid, surveying pH is 1~2, insulation reaction 1h.Be cooled to 0 DEG C of crystallization, separate out a large amount of white solids, filter, dry, obtain the about 5.7g of white solid, yield 85.3%, is Ropivacaine HCL crude product.After testing, ropivacaine purity 99.12%, dextrorotatory isomer per-cent 0.96%.(5) refining5.7g crude product and 40ml dehydrated alcohol-concentrated hydrochloric acid mixed solution (20:1) are added in reaction flask, be heated to 50 DEG C and make to dissolve; Complete molten after, naturally cool to room temperature, ice-water bath is cooled to 0 DEG C, crystallization 2h; Filter, 10ml mixed solution washing for filter cake, obtains wet product, dries, and obtains white solid 5.5g, and yield 92.1%, is Ropivacaine HCL fine work.After testing, ropivacaine purity 99.92 %, dextrorotatory isomer per-cent 0.15%.The preparation method of embodiment 4, a kind of Ropivacaine HCLStep is as follows:(1) preparation of intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea10.0g2-piperidine carboxylic acid, 160ml toluene are added in 500ml reaction flask.Pass into HCl gas, to pH3 left and right, be warming up to 48 ± 2 DEG C, add 1.5mlDMF, drip 10.2g (1.1 equivalent) sulfur oxychloride and 20ml toluene mixture liquid, drip and finish, be incubated 48 ± 2 DEG C of reaction 6h.Drip 2 of 4.0 equivalents, 6-xylidine and 20ml toluene mixture liquid, be incubated 58 ± 2 DEG C of reaction 8h.Filter, obtain yellow-green colour wet product 64.2g, dry to obtain gray solid 55.6g, solid is added in 280ml purified water, stir the molten reaction solution that obtains; 10%NaOH solution is slowly dropped in reaction solution, adjust pH to 4.5-5.0 , use 100ml toluene wash, layering, retains water layer, continues to adjust pH to 9-10 with 10%NaOH solution, adds 100ml methylene dichloride.Layering, gets organic layer,and water layer continues to use 50ml dichloromethane extraction, merges organic layer, adds anhydrous sodium sulfate dehydration, 40 DEG C of concentrating under reduced pressure.Obtain the about 14.9g of pale yellow oily liquid body, yield 82.9%, is intermediate (I) N-(2,6-dimethyl benzene)-2-piperidyl urea.(2) preparation of intermediate (II) N-(2,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amideIntermediate 14.9 g of (the I) Dissolved in 60mlDMF IS, ADDS 8.5gK 2 cO . 3 , 7.8 g of drip (1.0 equivalent)-n-propyl bromide, and drip After Finishing of DEG BE Warming up to 75 C, Reaction Insulation 2H; IS Down Ice Bath to room temperature, filters, and filtrate is added in 150ml frozen water, separates out a large amount of white solids, filter, dry, obtain the about 16.1g of white solid, yield 92.0%, is intermediate (II) N-(2 ,6-3,5-dimethylphenyl)-1-n-propyl piperidines-2-methane amide.(3) preparation of left-handed ropivacaine tartrate15g intermediate (II) is dissolved in 100ml Virahol, heats up 60 DEG C and stir molten; Treat entirely moltenly, add successively 1.72g (0.1 equivalent) titanium isopropylate, 1.82g (0.2 equivalent) D-tartrate, be warming up to backflow , after solution clarification, continue reaction 4h; Be cooled to 30 DEG C of crystallizatioies, filter, 75 DEG C of oven dry, obtain white solid 7.6g, and yield 39.7% is left-handed ropivacaine tartrate; After testing, ropivacaine purity 99.01 %, dextrorotatory isomer per-cent 1.05%.(4) preparation of Ropivacaine HCL crude productLeft-handed 7.6g ropivacaine tartrate is joined in 50ml Virahol, be warming up to 40 DEG C, drip concentrated hydrochloric acid, surveying pH is 1~2, insulation reaction 4h.Be cooled to 5 DEG C of crystallizatioies, separate out a large amount of white solids, filter, dry, obtain the about 5.7g of white solid, yield 85.3%, is Ropivacaine HCL crude product.After testing, ropivacaine purity 99.06%, dextrorotatory isomer per-cent 0.95%.(5) refining5.7g crude product and 40ml dehydrated alcohol-concentrated hydrochloric acid mixed solution (volume ratio 20:1) are added in reaction flask, be heated to 80 DEG C and make to dissolve; Complete molten after, naturally cool to room temperature, ice- water bath is cooled to 5 DEG C, crystallization 2h; Filter, 10ml mixed solution washing for filter cake, obtains wet product, dries, and obtains white solid 5.2g, and yield 91.2%, is Ropivacaine HCL fine work.After testing, ropivacaine purity 99.81%, dextrorotatory isomer per-cent 0.11%.The optical isomer method for detecting purity of left-handed ropivacaine tartrate, Ropivacaine HCL crude product and the Ropivacaine HCL purified product obtaining in above-described 1-4 is: measure according to high performance liquid chromatography (annex VD), with alpha- acid glycoprotein post (AGP, 100mm × 4.0mm, 5 μ m are suitable for); Agilent-1260 type high performance liquid chromatograph; (get potassium primary phosphate 2.72g with Virahol-phosphate buffered saline buffer, the 800ml that adds water dissolves, regulating pH value with 0.1mol/L sodium hydroxide solution is 7.1, be diluted with water to 1000ml) be (10:90) moving phase, detection wavelength is: 210nm, column temperature: 30 DEG C, flow velocity 1.0ml/min, limit is: dextrorotatory isomer must not be greater than 0.5%.
PATENThttps://patents.google.com/patent/CN109503465A/enThe embodiment of 1 intermediate (-) of-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamideL- piperidinecarboxylic acid hydrochloride (30.00g, 0.18mol), toluene are sequentially added in three mouthfuls of reaction flasks of 500ml cleaning N,N-Dimethylformamide (1ml), thionyl chloride (25.85g, 0.2 2mol) is added in (300ml) , stirring.It finishes, is warming up to 50~55 DEG C insulation reaction 3 hours.Snubber device is added to vacuumize 1 hour.The toluene solution of 2,6- dimethylaniline is added dropwise (2,6- dimethylanilines (109.75g , 0.91mol) are mixed with toluene (60ml)).It finishes, 60 DEG C of insulation reaction 2.0h.Cooling It to 20~30 DEG C, is added purified water (300ml), water phase is collected in layering; Fresh toluene (300ml), 10% hydrogen-oxygen is added in water phase Change sodium regulation system pH=6- 7, water phase is collected in layering; Water phase 10% sodium hydroxide regulation system pH=11~12,room temperature Stirring 4 hours, filter, purified water (150ml) elute filter cake, filter cake in 60 DEG C of air dry ovens it is dry 35.88g (yield 85%, HPLC purity 94.023% is calculated by areas of peak normalization method) .The purification of 2 intermediate (-) of-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide1 gained intermediate (-) of embodiment-(2S)-N- (2,6- diformazan is sequentially added in three mouthfuls of reaction flasks of 100ml cleaning Base phenyl) piperidines -2- formamide (5.00g, 21.52mmol), ether (50ml), stir and are warming up to reflux, flow back insulated and stirred 1 Hour, it is cooled to room temperature, insulated and stirred 1 hour, is filtered, ether (10ml) elutes filter cake, and filter cake is dry in 50 DEG C of air dry ovens Dry 2 hours 2.66g (yield 53.2% calculates HPLC purity 99.837% by areas of peak normalization method), map is shown in attached drawing 1.The purification of 3 intermediate (-) of-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide1 gained intermediate (-) of embodiment-(2S)-N- (2,6- diformazan is sequentially added in three mouthfuls of reaction flasks of 100ml cleaning Base phenyl) piperidines -2- formamide (5.00g, 21.52mmol), isopropyl ether (50ml), stir and are warming up to reflux, reflux heat preservation is stirred It mixes 1 hour, is cooled to room temperature, insulated and stirred 1 hour, filters, isopropyl ether (10m l) elutes filter cake, and filter cake is dry in 50 DEG C of air blast Dry 2 hours 3.45g of dry case (yield 69.0% calculates HPLC purity 99.332% by areas of peak normalization method).Map is shown in attached Fig. 2.The purification of 4 intermediate (-) of-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide1 gained intermediate (-) of embodiment-(2S)-N- (2,6- diformazan is sequentially added in three mouthfuls of reaction flasks of 100ml cleaning Base phenyl) piperidines -2- formamide (5.00g, 21.52mmol), methyl tertiary butyl ether(MTBE) (50ml), stir and are warming up to reflux, flow back Insulated and stirred 1 hour, be cooled to room temperature, insulated and stirred 1 hour, filter, methyl tertiary butyl ether(MTBE) (10ml) elutes filter cake, filter cake in Dry 2 hours 4.75g (yield 95% calculates HPLC purity 99.709% by areas of peak normalization method) of 50 DEG C of air dry ovens, Map is shown in attached drawing 3.5 intermediate (-) of embodiment-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide preparation and purificationA) the preparation of intermediate (-)-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamideL- piperidinecarboxylic acid hydrochloride (3kg, 18.1mol), toluene (30L) are added in 50L reaction kettle, N, N- is added in stirring Dimethylformamide (1L), thionyl chloride (2.59kg, 21.8mol). It finishes, is warming up to 50~55 DEG C of insulation reactions 3 hours. Snubber device is added to vacuumize 6 hours.Be added dropwise 2,6- dimethylaniline toluene solution (2,6- dimethylanilines (11kg, It 90.8mol) is mixed with toluene (6L)).It finishes, 60 DEG C of insulation reaction 2.0h. 20~30 DEG C are cooled to, is added purified water (30L), Water phase is collected in layering; Water phase is added fresh toluene (30L) , 10% sodium hydroxide regulation system pH=6-7, and water is collected in layering Phase; 10% sodium hydroxide regulation system pH=11~12 of water phase, are stirred at room temperature 4 hours, filter,purified water (15L) elution filter Cake, filter cake in 60 DEG C of air dry ovens it is dry 3.52kg (yield 84%, by areas of peak normalization method calculate HPLC purity 98.092%), map is shown in attached drawing 4.B) the purification of intermediate (-)-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamideIntermediate (-)-(2S)-N- (2,6- 3,5-dimethylphenyl) piperidines -2- formamide (3. is sequentially added in 50L reaction kettle 5kg, 15.1mol), methyl tertiary butyl ether(MTBE) (35L), stirring is warming up to reflux, flows back insulated and stirred 1 hour, be cooled to room temperature, guarantor Temperature stirring 1 hour, filters, and methyl tertiary butyl ether(MTBE) (7L) elutes filter cake, and filter cake is in the dry 8 hours 3.3kg of 50 DEG C of air dry ovens (yield 94% calculates HPLC purity 99.889% by areas of peak normalization method), map is shown in attached drawing 5.
Literatures:
Navinta LLC Patent: US2006/276654 A1, 2006 ; Location in patent: Page/Page column 5 ;
Yield: ~82%
Literatures:
US2006/276654 A1, ; Page/Page column 5 ;
Yield: null
SYN
https://pubs.rsc.org/en/content/articlehtml/2019/ra/c9ra09287k
Ropivacaine is the S-enantiomer of an N-alkyl pipecoloxylidine derivative, which is the first local anesthetic with chiral activity, and is widely used in clinical infiltration anesthesia, conduction anesthesia and epidural anesthesia. It has a long of local anesthesia and analgesic effect. However, ropivacaine also has serious safety risks in clinical practice. When the concentration of ropivacaine in human blood is too high, it may cause toxicity to the cardiovascular and central nervous system, and even cause allergic reactions in some patients. Thus far, the mechanism of the effect of ropivacaine on local anesthesia is not clear. Ropivacaine is a multitarget drug that acts on the gamma-aminobutyric acid a receptor (GABAA-R) and N-methyl-D-aspartate acid receptor (NDMA-R). Sodium (Na+) channels are a key target of local anesthetics and these two receptors regulate sodium channels. Previous studies on the structural modification of ropivacaine mainly focused on the substitution of –CH3 on the phenyl group or the substitution of –CH2CH2CH3 on piperidine with different alkyl groups. In 2017, Wen L. et al.69 reported the design and synthesis of ropivacaine analogues for local anesthesia. In the process of structural design, they used ropivacaine as the lead compound to design two series of compounds, 4a–4q (17 new substituted imines). In the first series of compounds, 4a–4i, different substituents were selected to replace –CH2CH2CH3 on piperidine. In the second series of compounds, 4j–4q, the methyl groups were replaced by –CF3 at the o-positions, m-positions and p-positions. Meanwhile, the –CH2CH2CH3 on piperidine ring was also substituted and modified. The process for the synthesis of the target compounds is shown in Scheme 8. The synthetic route takes piperic acid (compound 1) as the starting material, hydrochloric acid and sulfoxide chloride as additives, and toluene as the reaction solvent to convert compound 1 into acyl chloride salt (compound 2). Compound 2 was then treated with substituted aniline and reacted at 58 °C for 5 h to form compounds 3a–3i and 3j–3q. Finally, bromoalkyl and hydrochloric acid were used to treat compounds 3a–3i and 3j–3q. Potassium carbonate (K2CO3) was used as an acid dressing agent and dimethylformamide (DMF) as the reaction solvent in N-alkylation reaction. The N-alkylation reaction lasted 10 h at 80 °C, and the salt reaction lasted 5 min at room temperature to obtain the final target compounds 4a–4q. The total yield of the target compounds ranged from 17.5% to 87.7%. The synthetic route has the advantages of mild reaction conditions, cheap reagents and simple operation. However, using this synthetic route, the total yield of some products is too low, and too low yield will bring great problems to the synthesis cost, which needs to be further optimized in follow-up work. In the evaluation of the local anesthesia effect, sciatic nerve block activity, infiltration anesthesia activity, corneal anesthesia activity and spinal cord anesthesia activity were used as evaluation indexes. Ropivacaine was used as a positive control substance to test the local anesthesia activity in vitro. Firstly, the local anesthesia effect of all the target compounds 4a–4q was screened by a sciatic nerve block test in toads in vitro (Table 6). The preliminary screening results in vitro showed that these compounds increased the blocking effect of the sciatic nerve on electrical stimulation, with ED50 values ranging from 0.012 to 0.64 (positive control for ropivacaine was 0.013), with the highest activity shown by compound 4b. In terms of latent period, that of target compounds 4a–4q ranged from 27.7 to 59.4 min. Based on the results of the preliminary in vitro screening, compounds 4a, 4b, 4c, 4j and 4l were selected to test the efficacy of invasive anesthesia in guinea pigs. The results of the infiltration anesthesia test showed that the local anesthetic effect of compounds 4c and 4l was similar to that of the positive control ropivacaine, and the local anesthetic activity of other compounds was lower than that of the positive control. Furthermore, compounds 4a, 4b, 4c, 4j and 4l were used to test the local surface anesthesia effect of these compounds (Table 7). The results of the surface anesthesia test showed that compound 4l had a similar local anesthetic effect as the positive control ropivacaine, while the effect of the other compounds was poor in comparison with the positive control. Finally, compounds 4a, 4b, 4c, 4j and 4l were tested for spinal anesthesia in order to further study their local surface anesthesia effect. The experimental results showed that the ED50 produced by compounds 4l and 4b was 5.02 and 7.87, respectively, while the effects of compounds 4a, 4c and 4j were poor. The evaluation of local anesthesia in vitro found that compound 4l had the best activity, and thus molecular docking of compound 4l and ropivacaine was conducted to further study its local anesthesia mechanism. The molecular docking results showed that compound 4l interacts with receptor proteins of VGSC, GABAA-R and NDMA-R, which may help optimize and predict the activity of these ropivacaine analogues as potential local anesthetics.
| Scheme 8 Reagents and conditions: (a) (i) HCl,PhCH3, r.t., 1 h; (ii) PhCH3, SOCl2, 55 °C, 1 h; (b) substituted anilines, 58 °C, 5 h; and (c) (i) RBr, K2CO3, DMF, 80 °C, 10 h; (ii) HCl, r.t., 5 min. |
SYN
Prepn: A. F. Thuresson, C. Bovin, WO 8500599 (1985 to Apothekernes); H.-J. Federsel et al., Acta Chem. Scand. B41, 757 (1987).

CLIP
Ropivacaine hydrochloride was synthesized from L-2-pipecolic acid by successive reaction with SOCl2 and 2,6-dimethylaniline at 40 °C under ultrasonic irradiation to yield L-N-(2,6-dimethylphenyl)piperidin-2-carboxamide (4), and 4 was reacted with 1-bromopropane at 50 °C for 1 h under ultrasonic irradiation. The effects of reaction solvent, temperature and time under ultrasonic irradiation were investigated. Compared with conventional methods, present procedures have the advantages in milder conditions, shorter reaction time and higher yields. The total yield was 67.5%, [α]25 D= – 6.6°(c = 2, H2O).

SYN
Ropivacaine (3.1.37) (Naropin) is the pure S(–)-enantiomer of propivacaine released for clinical use in 1996. It is a long-acting, well tolerated local anesthetic agent and first produced as a pure enantiomer. Its effects and mechanism of action are similar to other local anesthetics working via reversible inhibition of sodium ion influx in nerve fibers. It may be a preferred option among other drugs among this class of compounds because of its reduced CNS and cardiotoxic potential and its lower propensity for motor block in the management of postoperative pain and labor pain [48–58].
The synthesis of ropivacaine (3.1.37) was carried out starting with l-pipecolic acid (3.1.34), prepared by a resolution of (±)-pipecolic acid with (+)-tartaric acid, which was dissolved in acetyl chloride and converted to acid chloride (3.1.35) with phosphorus pentachloride. The obtained compound (3.1.35) dissolved in toluene a solution of 2,6-xylidine (3.1.28) dissolved in the mixture of equal volumes of acetone, and N-methyl-2-pyrrolidone was added at 70°C to give (+)-l-pipecolic acid-2,6-xylidide (3.1.36). Reaction of this compound with propyl bromide in presence of potassium carbonate in i-PrOH/H2O gave the desired ropivacaine (3.1.37) [59] (Scheme 3.6).

Another approach for the synthesis of ropivacaine (3.1.37) was proposed via a resolution of enantiomers of chiral pipecolic acid-2,6-xylidide [60].
SYN

Scheme 21. Generation of ‘cation pool’ and its applications.
Reproduced from Yoshida, J.; Suga, S.; Suzuki, S.; et al. J. Am. Chem. Soc. 1999, 121, 9546–9549, and Shankaraiah, N.; Pilli, R. A.; Santos, L. S. Tetrahedron Lett. 2008, 49, 5098–5100.
CLIP
Process R&D under the magnifying glass: Organization, business model, challenges, and scientific context
Hans-Jürgen Federsel, in Bioorganic & Medicinal Chemistry, 2010
The synthesis of ropivacaine is achieved in only three steps, as in the previous example, comprised of a resolution of a racemic, commercially available starting material (pipecoloxylidide) followed by an N-alkylation and the final precipitation of the product as its HCl salt.14,24 Focusing on the middle step—the attachment of a propyl moiety onto the piperidine nitrogen—this reaction when developed in the laboratory and scaled up to maximum pilot plant volume (1000 L) behaved very well (Scheme 3). Thus, boiling the reaction mixture (reactants in a H2O/organic solvent mixture in the presence of a solid inorganic base) for an extended period of time (6 h) at high temperature (100 °C), the transformation was considered complete once a sample of the process solution showed <1% of remaining starting material. In preparation for launch, the method that had been thoroughly investigated and tested over a number of years and proven reliable on scale up had to be validated in the authentic 4000 L production equipment. Much to our surprise (and shock) we, however, found that the reaction came to a complete stand still long before reaching the expected end point. With a large amount of un-reacted starting material (30–40%) we were facing a situation that had never occurred during the lengthy development phase and this put the whole project in a very critical state as we were not able to reproduce the manufacturing method.

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
History
Ropivacaine was developed after bupivacaine was noted to be associated with cardiac arrest, particularly in pregnant women. Ropivacaine was found to have less cardiotoxicity than bupivacaine in animal models.
Clinical use
Contraindications
Ropivacaine is contraindicated for intravenous regional anaesthesia (IVRA). However, new data suggested both ropivacaine (1.2-1.8 mg/kg in 40ml) and levobupivacaine (40 ml of 0.125% solution) be used, because they have less cardiovascular and central nervous system toxicity than racemic bupivacaine.[1]
Adverse effects
Adverse drug reactions (ADRs) are rare when it is administered correctly. Most ADRs relate to administration technique (resulting in systemic exposure) or pharmacological effects of anesthesia, however allergic reactions can rarely occur.
Systemic exposure to excessive quantities of ropivacaine mainly result in central nervous system (CNS) and cardiovascular effects – CNS effects usually occur at lower blood plasma concentrations and additional cardiovascular effects present at higher concentrations, though cardiovascular collapse may also occur with low concentrations. CNS effects may include CNS excitation (nervousness, tingling around the mouth, tinnitus, tremor, dizziness, blurred vision, seizures followed by depression (drowsiness, loss of consciousness), respiratory depression and apnea). Cardiovascular effects include hypotension, bradycardia, arrhythmias, and/or cardiac arrest – some of which may be due to hypoxemia secondary to respiratory depression.[2]
Postarthroscopic glenohumeral chondrolysis
Ropivacaine is toxic to cartilage and their intra-articular infusions can lead to Postarthroscopic glenohumeral chondrolysis.[3]
Treatment of overdose
As for bupivacaine, Celepid, a commonly available intravenous lipid emulsion, can be effective in treating severe cardiotoxicity secondary to local anaesthetic overdose in animal experiments[4] and in humans in a process called lipid rescue.[5][6][7]
References
- ^ (Basic of Anesthesia, Robert Stoelting, page 289)
- ^ Rossi S, editor. Australian Medicines Handbook 2006. Adelaide: Australian Medicines Handbook; 2006. ISBN 0-9757919-2-3
- ^ Gulihar A, Robati S, Twaij H, Salih A, Taylor GJ (December 2015). “Articular cartilage and local anaesthetic: A systematic review of the current literature”. Journal of Orthopaedics. 12 (Suppl 2): S200-10. doi:10.1016/j.jor.2015.10.005. PMC 4796530. PMID 27047224.
- ^ Weinberg G, Ripper R, Feinstein DL, Hoffman W (2003). “Lipid emulsion infusion rescues dogs from bupivacaine-induced cardiac toxicity”. Regional Anesthesia and Pain Medicine. 28 (3): 198–202. doi:10.1053/rapm.2003.50041. PMID 12772136. S2CID 6247454.
- ^ Picard J, Meek T (February 2006). “Lipid emulsion to treat overdose of local anaesthetic: the gift of the glob”. Anaesthesia. 61 (2): 107–9. doi:10.1111/j.1365-2044.2005.04494.x. PMID 16430560. S2CID 29843241.
- ^ Rosenblatt MA, Abel M, Fischer GW, Itzkovich CJ, Eisenkraft JB (July 2006). “Successful use of a 20% lipid emulsion to resuscitate a patient after a presumed bupivacaine-related cardiac arrest”. Anesthesiology. 105 (1): 217–8. doi:10.1097/00000542-200607000-00033. PMID 16810015.
- ^ Litz RJ, Popp M, Stehr SN, Koch T (August 2006). “Successful resuscitation of a patient with ropivacaine-induced asystole after axillary plexus block using lipid infusion”. Anaesthesia. 61 (8): 800–1. doi:10.1111/j.1365-2044.2006.04740.x. PMID 16867094. S2CID 43125067.
External links
- “Ropivacaine”. Drug Information Portal. U.S. National Library of Medicine.
- “Ropivacaine hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Trade names | Naropin |
| AHFS/Drugs.com | Monograph |
| Pregnancy category | AU: B1 |
| Routes of administration | Parenteral |
| ATC code | N01BB09 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 87%–98% (epidural) |
| Metabolism | Liver (CYP1A2-mediated) |
| Elimination half-life | 1.6–6 hours (varies with administration route) |
| Excretion | Kidney 86% |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 84057-95-4 |
| PubChem CID | 175805 |
| IUPHAR/BPS | 7602 |
| DrugBank | DB00296 |
| ChemSpider | 153165 |
| UNII | 7IO5LYA57N |
| KEGG | D08490 as HCl: D04048 |
| ChEBI | CHEBI:8890 |
| ChEMBL | ChEMBL1077896 |
| CompTox Dashboard (EPA) | DTXSID4040187 |
| ECHA InfoCard | 100.128.244 |
| Chemical and physical data | |
| Formula | C17H26N2O |
| Molar mass | 274.408 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 144 to 146 °C (291 to 295 °F) |
| showSMILES | |
| showInChI | |
| (verify) |
Patent
Publication numberPriority datePublication dateAssigneeTitleUS4695576A *1984-07-091987-09-22Astra Lake Medel AktiebolagLNn-propylpipecolic acid-2,6-xylidideUS20050065345A1 *2001-09-102005-03-24Toshio TsuchidaMethod for producing pipecolamide derivativeCN103086954A *2013-02-042013-05-08Shandong Pharmaceutical Industry Research InstituteMethod for preparing ropivacaineCN104003930A *2014-06-132014-08-27Shandong Alura Pharmaceutical Research and Development Co., Ltd.Method for preparing hydrochloric acid ropivacaineCN107325041A *2017-06-202017-11-07Guangzhou Tonghui Pharmaceutical Co., Ltd.A kind of preparation method of Ropivacaine HCL
Non-Patent
TitleNAGULA SHANKARAIAH, etc.: “Enantioselective total syntheses of ropivacaine and its analogues”, “TETRAHEDRON LETTERS” *Liu Yi, et al.: “Synthesis of Ropivacaine Hydrochloride”, “Chinese Journal of Pharmaceutical Industry” *Ye Jiao, et al.: “Synthesis of Ropivacaine Hydrochloride by Triphosgene Method”, “Synthetic Chemistry” *Jiang Yao: “Study on the Synthetic Process of Ropivacaine Hydrochloride and Bupivacaine Hydrochloride”, “Engineering Science and Technology Series Ⅰ” *
/////////////Ropivacaine, Anesthetic, ропивакаин , روبيفاكائين , 罗哌卡因 , DRopivacaine Hydrochloride Hydrate, LEA-103, NA-001, (-)-LEA-103
CCCN1CCCC[C@H]1C(=O)NC1=C(C)C=CC=C1C

NEW DRUG APPROVALS
ONE TIME
$10.00
LIDOCAINE
LIDOCAINE
CAS Registry Number: 137-58-6
CAS Name: 2-(Diethylamino)-N-(2,6-dimethylphenyl)acetamide
Additional Names: 2-diethylamino-2¢,6¢-acetoxylidide; w-diethylamino-2,6-dimethylacetanilide; lignocaine
Trademarks: Cuivasil (IDC); Lidoderm (Hind); LidoPosterine (Kade); Vagisil (Combe)
Molecular Formula: C14H22N2O
Molecular Weight: 234.34
Percent Composition: C 71.75%, H 9.46%, N 11.95%, O 6.83%
Literature References: Long-acting, membrane stabilizing agent against ventricular arrhythmia. Originally developed as a local anesthetic. Prepn: N. M. Löfgren, B. J. Lundqvist, US2441498 (1948 to Astra); A. D. H. Self, A. P. T. Easson, GB706409 (1954 to May & Baker); I. P. S. Hardie, E. S. Stern, GB758224 (1956 to J. F. Macfarlane & Co.); Zhuravlev, Nikolaev, Zh. Obshch. Khim.30, 1155 (1960). Toxicity studies: E. R. Smith, B. R. Duce, J. Pharmacol. Exp. Ther.179, 580 (1971); G. H. Kronberg et al.,J. Med. Chem.16, 739 (1973). Review of pharmacokinetics: N. L. Benowitz, W. Meister, Clin. Pharmacokinet.3, 177 (1978). Review of action as local anesthetic: Löfgren, Studies on Local Anesthetics: Xylocaine, A New Synthetic Drug (Hoeggstroms, Stockholm, 1948); Cooper, Pharm. J.171, 68 (1953). Reviews of anti-arrhythmic agents: J. L. Anderson et al.,Drugs15, 271 (1978); L. H. Opie, Lancet1, 861 (1980); E. Carmeliet, Ann. N.Y. Acad. Sci.427, 1 (1984). Comprehensive description: K. Groningsson et al.,Anal. Profiles Drug Subs.14, 207-243 (1985); M. F. Powell, ibid.15, 761-779 (1986). Review of use in treatment of postherpetic neuralgia: P. S. Davies, B. S. Galer, Drugs64, 937-947 (2004).Properties: Needles from benzene or alcohol, mp 68-69°. bp4 180-182°; bp2 159-160°. Insol in water. Sol in alcohol, ether, benzene, chloroform, oils. Partition coefficient (octanol/water, pH 7.4): 43.
Melting point: mp 68-69°
Boiling point: bp4 180-182°; bp2 159-160°
Log P: Partition coefficient (octanol/water, pH 7.4): 43
Derivative Type: Hydrochloride
CAS Registry Number: 73-78-9; 6108-05-0 (monohydrate)
Trademarks: Basicaina (Galenica); Batixim (So.Se.); Dynexan (Kreussler); Heweneural (Hevert); Licain (DeltaSelect); Lidesthesin (Ritsert); Lidofast (Angelini); Lidoject (Hexal); Lidrian (Baxter); Odontalg (Giovanardi); Sedagul (Wild); Xylocaine (AstraZeneca); Xylocard (AstraZeneca); Xylocitin (Jenapharm); Xyloneural (Strathmann)
Molecular Formula: C14H22N2O.HCl
Molecular Weight: 270.80
Percent Composition: C 62.09%, H 8.56%, N 10.34%, O 5.91%, Cl 13.09%
Properties: Crystals, mp 127-129°; monohydrate, mp 77-78°. Very sol in water, alcohol; sol in chloroform. Insol in ether. pH of 0.5% aq soln: 4.0-5.5. LD50 in mice (mg/kg): 292 orally (Smith, Duce); 105 i.p.; 19.5 i.v. (Kronberg).
Melting point: mp 127-129°; mp 77-78°
Toxicity data: LD50 in mice (mg/kg): 292 orally (Smith, Duce); 105 i.p.; 19.5 i.v. (Kronberg)
Therap-Cat: Anesthetic (local); antiarrhythmic (class IB).
Therap-Cat-Vet: Anesthetic (local).
Keywords: Anesthetic (Local); Antiarrhythmic.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
CLIP
https://pubs.acs.org/doi/10.1021/ed076p1557
http://www.asianjournalofchemistry.co.in/User/ViewFreeArticle.aspx?ArticleID=19_7_12
PATENT
https://patents.google.com/patent/CN102070483B/en#:~:text=The%20method%20comprises%20the%20following,as%20solvent%20and%20carbonate%20isPreparation method of the present invention, it can be two-step approach, comprises the steps:1) 2,6-xylidine is dissolved in the acetone, adds carbonate then, the back that stirs drips chloroacetyl chloride, and 20~35 ℃ (room temperature) be stirring reaction 3h down; After-filtration is finished in reaction, and after filter cake was washed with water to filtrate and is neutrality, drying made intermediate chloracetyl-2, the 6-xylidine, and yield is about about 94%;
2) intermediate that step 1) is made is dissolved in the acetone, adds carbonate then, and the back that stirs drips diethylamine, back flow reaction 8h; After-filtration is finished in reaction, and filtrate is recrystallization, drying after removing solvent under reduced pressure, makes lignocaine.Wherein, in the step 1) 2, the mol ratio of 6-xylidine, chloroacetyl chloride and carbonate is 1: 1.2~1.7: 1.3~2.0, is preferably 1: 1.5: 1.6.
Step 2) intermediate chloracetyl-2 in, the mol ratio of 6-xylidine, diethylamine and carbonate is 1: 1.5~2.5: 1.2~2.0, is preferably 1: 2: 1.5.In addition, preparation method of the present invention owing to all be that solvent, carbonate are catalyzer with acetone in the two-step reaction, therefore can further optimize reaction process on the basis of two-step approach, namely the intermediate of Sheng Chenging needn’t pass through aftertreatment, prepares lignocaine by one kettle way.Described one kettle way comprises the steps: 2,6-xylidine is dissolved in the acetone, adds carbonate then, after stirring, drips chloroacetyl chloride, and 20~35 ℃ (room temperature) be reaction 3h down; After reaction is finished, without processing, directly drip diethylamine, back flow reaction 8h, after-filtration is finished in reaction, and filtrate is recrystallization, drying after removing solvent under reduced pressure, makes lignocaine.Wherein, described 2, the mol ratio of 6-xylidine, chloroacetyl chloride, diethylamine and carbonate is 1: 1.2~1.7: 1.5~2.5: 2.5~3.5, is preferably 1: 1.5: 2: 2.5.
In addition, preparation method of the present invention adopts TLC monitoring reaction progress, and the developping agent of TLC is sherwood oil: ethyl acetate (V/V)=3: 1.The invention has the advantages that, the method synthesis technique for preparing lignocaine of the present invention is simple, do not need in the intermediate aftertreatment first pickling numerous and diverse step of alkali cleaning again, avoided unnecessary loss, therefore the yield of the intermediate that makes of the inventive method and lignocaine is all higher, and the lignocaine purity that makes is good, reaches more than 99%, has favorable industrial application prospect; In addition, the inventive method uses acetone to make solvent, and this solvent is nontoxic substantially non-stimulated, and can recycle, and is environmentally friendly.
EmbodimentBelow further specify the present invention by specific embodiment, but be not used for limiting the scope of the invention.
Embodiment 1 two-step approach prepares lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineAdd 102g 2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, adds 200g salt of wormwood again, and mechanical stirring evenly back drips 100mL chloroacetyl chloride (1.5h drips off), (20 ℃) stirring reaction 3h under the room temperature; Reaction finishes the back suction filtration, and filter cake is washed with water to filtrate and is neutral, and under 100 ℃ of temperature dry 1 hour then, make the 156g white powder, be intermediate chloracetyl-2,6-xylidine, yield are 94%, fusing point is 145.0~147.0 ℃.2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, and the dissolving back adds 112g salt of wormwood, drips the 60g diethylamine fast, back flow reaction 8h; Reaction finishes the back suction filtration, and filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, uses 150mL sherwood oil recrystallization then, suction filtration, vacuum-drying 6h under 40 ℃ of temperature makes the 90g white powder, is lignocaine, yield is 95%, and fusing point is 67.0~68.0 ℃, and content is 99.05%.
Embodiment 2 two-step approachs prepare lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineAdd 102g 2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, adds 163g salt of wormwood again, and mechanical stirring evenly back drips 80mL chloroacetyl chloride (1.5h drips off), (20 ℃) stirring reaction 3h under the room temperature; Reaction finishes the back suction filtration, and filter cake is washed with water to filtrate and is neutral, and under 100 ℃ of temperature dry 1 hour then, make the 136g white powder, be intermediate chloracetyl-2,6-xylidine, yield are 82%, fusing point is 145~146 ℃.
2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, and the dissolving back adds 90g salt of wormwood, drips the 45g diethylamine fast, back flow reaction 8h; Reaction finishes the back suction filtration, and filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, uses 150mL sherwood oil recrystallization then, suction filtration, vacuum-drying 6h under 40 ℃ of temperature makes the 84g white powder, is lignocaine, yield is 89%, and fusing point is 66~67 ℃, and content is 99.15%.
Embodiment 3 two-step approachs prepare lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineAdd 102g 2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, adds 250g salt of wormwood again, and mechanical stirring evenly back drips 113mL chloroacetyl chloride (1.5h drips off), (20 ℃) stirring reaction 3h under the room temperature; Reaction finishes the back suction filtration, and filter cake is washed with water to filtrate and is neutral, and under 100 ℃ of temperature dry 1 hour then, make the 150g white powder, be intermediate chloracetyl-2,6-xylidine, yield are 90%, fusing point is 147~148 ℃.
2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, and the dissolving back adds 150g salt of wormwood, drips the 75g diethylamine fast, back flow reaction 8h; Reaction finishes the back suction filtration, and filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, uses 150mL sherwood oil recrystallization then, suction filtration, vacuum-drying 6h under 40 ℃ of temperature makes the 88g white powder, is lignocaine, yield is 93%, and fusing point is 68~69 ℃, and content is 98.75%.
Embodiment 4 one kettle ways prepare lignocaineIn the 5000mL there-necked flask, add 305g 2, the 6-xylidine, make solvent with 2000mL acetone, add 700g salt of wormwood again, mechanical stirring evenly back slowly drips 230mL chloroacetyl chloride (1.5h drips off), room temperature (35 ℃) is stirring reaction 3h down, and TLC point plate (use sherwood oil: ethyl acetate (V/V)=3: 1 is made developping agent) demonstration reacts completely; Dropwise 5 50g diethylamine then, the back back flow reaction 8h that stirs, TLC monitoring (developping agent is the same) shows and reacts completely; The reaction solution suction filtration, filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, gets light yellow solid, uses sherwood oil recrystallization secondary then, makes 482g white lignocaine crystal, and total recovery is 82%, and fusing point is 68.0~69.0 ℃, and content is 99.75%.
Comparative example 1 existing method prepares lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineIn the 1000mL there-necked flask, with 102g 2, the 6-xylidine is dissolved in the 400mL glacial acetic acid, stirs slowly to add the 100mL chloroacetyl chloride down, is heated to 45 ℃, adds 200g solid sodium acetate (containing crystal water) then, reaction 2h; After reaction finished, ice bath was cooled to below 10 ℃, suction filtration, filter cake is washed with water to filtrate and is neutral, and drying is 1 hour under 100 ℃ of temperature, makes the 111g white powder, be intermediate chloracetyl-2,6-xylidine, yield are that 67% fusing point is 145.0~148.0 ℃.
2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL toluene, and the dissolving back drips 60g diethylamine, back flow reaction 3.5h fast; After reaction finished, ice bath was cooled to 5 ℃, suction filtration, filtrate is used the 3mol/L hcl as extraction agent, and the acid solution that extraction is obtained is cooled to 10 ℃ then, stirs slowly to add 6mol/L KOH solution down, be alkalescence (pH8~9) to solution, with pentane extraction, organic layer was after washing, Anhydrous potassium carbonate drying after ice bath was cooled to 20 ℃, vapor bath is steamed and is desolventized, make the 74g white powder, be lignocaine, yield is 78%, fusing point is 66.0~67.0 ℃, and content is 97.15%.Embodiment 1 compares with comparative example 1, its intermediate chloracetyl-2, and the yield of 6-xylidine obviously improves, and reaches 94%, and the total recovery of final product lignocaine also is significantly improved, and the content of lignocaine is brought up to about 99%.Embodiment 4 compares with comparative example 1, and the total recovery of lignocaine is significantly improved, and reach 82%, and content is brought up to more than 99%.
Though above with a general description of the specific embodiments, the present invention is described in detail, on basis of the present invention, can make some modifications or improvements it, this will be apparent to those skilled in the art.Therefore, these modifications or improvements all belong to the scope of protection of present invention without departing from theon the basis of the spirit of the present invention.
CLIPhttps://www.cerritos.edu/chemistry/chem_212/Documents/Lab/10_lidocaine.pdf
Procedure: (1st week)A: Synthesis of 2,6-Dimethylaniline via Reduction of 2,6-Dimethylnitrobenzene 1. Dissolve1.0 g of 2,6-dimethylnitrobenzene in 10 mL of glacial acetic acid in a 50 mL Erlenmeyer flask. 2. In a 25 mL flask, dissolve 4.6 grams of SnCl2 · 2H2O in 8 mL of concentrated HCl, inside the fume hood. 3. Add the SnCl2 solution in one portion to the nitroxylene solution, magnetically swirl and mix, and let the mixture stand for 15 minutes. 4. Cool the mixture and collect the crystalline salt (dimethylaniline in the salt form: C6H5NH3 +Cl- ) in a Buchner funnel. 5. Transfer the moist crystals to an Erlenmeyer flask, add 5-10 mL of water, and make the solution strongly basic (to remove the acid and change C6H5NH3 +Clback intoC6H5NH2) by adding 30% KOH solution (12 to 17 mL required). 6. After cooling extract with three 10 mL portions of ether, rinse the ether extracts twice with 10 mL of water, and dry over K2CO3. 7. Evaporate the dried and filtered solution to an oil, transfer and rinse into a 50 mL Erlenmeyer flask, complete evaporation, weigh, and calculate the %yield of 2,6-dimethylaniline.
B: Synthesis of α-Chloro-2,6-dimethylacetanilide (prepare for a steam bath ahead of time) 1. For every 7 grams (from this step on, you need to calculate proportionally how much you need to add according to the actual weight that you got) of dimethylaniline from the previous step, add 50 mL of glacial acetic acid, and 7.2 g (or 5.2 mL) of chloroacetyl chloride, in that order. 2. Warm the solution on a steam bath to (40–50)ºC, remove, and add a solution of 1 gram of sodium acetate in 100 mL of water. 3. Cool the mixture and collect the product in a Buchner funnel. 4. Transfer the product to a disk of medium–sized filter paper, finely divide it with a spatula, and let air dry until the next laboratory period. 5. Upon drying, measure the mass and the melting point. Also, calculate the % yield.
B: Synthesis of α-Chloro-2,6-dimethylacetanilide (prepare for a steam bath ahead of time) 1. For every 7 grams (from this step on, you need to calculate proportionally how much you need to add according to the actual weight that you got) of dimethylaniline from the previous step, add 50 mL of glacial acetic acid, and 7.2 g (or 5.2 mL) of chloroacetyl chloride, in that order. 2. Warm the solution on a steam bath to (40–50)ºC, remove, and add a solution of 1 gram of sodium acetate in 100 mL of water. 3. Cool the mixture and collect the product in a Buchner funnel. 4. Transfer the product to a disk of medium–sized filter paper, finely divide it with a spatula, and let air dry until the next laboratory period. 5. Upon drying, measure the mass and the melting point. Also, calculate the % yield.
D. Synthesis of the bisulfate salt of lidocaine 1. Dissolve the lidocaine in ether (10 mL per gram of lidocaine) and add 2 mL of 2.2 M sulfuric acid in ethanol per gram of lidocaine. 2. Stir and scratch with a glass rod to mix and induce crystallization. 3. Dilute the mixture with an equal volume of acetone to aid filtration and collect the salt in a small Buchner funnel. 4. Rinse the solid on the funnel with a few milliliters of acetone and air dry and weigh the product. 5. Calculate the % yield of this step. *** Overall % Yield The overall % YCLIPhttp://home.sandiego.edu/~khuong/chem302L/Handouts/Lidocaine_handout_Su07.pdfSynthetic Strategy Lidocaine will be prepared via a three-step linear synthesis starting from 2,6-dimethylnitrobenzene. The reduction of 2,6-dimethylnitrobenzene 1 with three equivalents of stannous chloride (SnCl2) yields the ammonium salt 2. It is very important that the reaction mixture is strongly acidic during this reaction because the reduction of nitrobenzene using different reducing reagents and conditions can afford a variety of functional groups: nitroso, hydroxylamine (zinc dust, pH 4), azoxy (sodium arsenite), azo (zinc, weakly basic), or hydrazo (zinc, strongly basic). In industrial settings, often iron or tin with hydrochloric acid is used instead of stannous chloride because iron and tin are cheaper, but the reduction takes much longer. In the workup portion of the reaction, the ammonium salt 2 is reacted with an aqueous potassium hydroxide solution, liberating the free 2,6-dimethylaniline 3 in an acid-base reaction.
The reaction of 3 with the bifunctional α-chloroacetyl chloride leads to α-chloro-2,6-dimethylacetanilide 4. A slight excess of the acid chloride is used to ensure the complete conversion of the amine to the amide. The formation of the amide is a result of the significantly higher reactivity (~106 times) of the acyl chloride over the alkyl chloride. The addition of sodium acetate solution avoids the formation of HCl which would protonate unreacted 3 causing it to co-precipitate with the desired product 4.
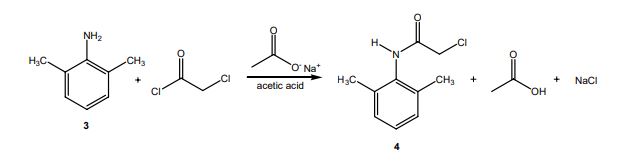
In the last step, diethylamine performs a nucleophilic substitution (SN2) on the remaining alkyl chloride. Diethylamine serves both as a nucleophile to form lidocaine 5, and as acid scavenger, leading to formation of NH2Et2 + Cl- in this reaction. Since diethylamine is not a very strong nucleophile, it is used in excess here to improve the yield and speed up the reaction. The unreacted amine is later removed by extraction with water. The aqueous extraction of lidocaine with acid separates the unreacted chloroanilide 4 and the lidocaine. After addition of a strong base like aqueous potassium hydroxide, crude lidocaine is obtained.
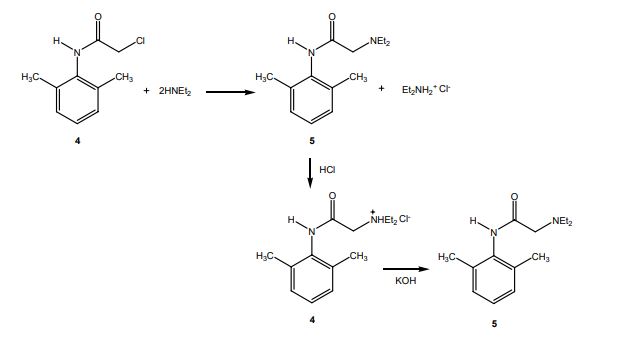
Procedure Synthesis of 2,6-dimethylaniline (3) Dissolve 15 g of SnCl2•2H2O in 27 mL of concentrated hydrochloric acid. If necessary, heat the mixture gently. Add this solution in one portion to a solution of 3 mL of 2,6-dimethylnitrobenzene in 34 mL of glacial acetic acid. Swirl the resulting mixture and then allow it to stand for 15 minutes before placing the mixture in an ice bath. Collect the formed precipitate by vacuum filtration. Place the wet precipitate obtained above in a beaker and add 20 mL of water. Neutralize the acidic mixture by carefully adding an 8 M aqueous potassium hydroxide with continuous stirring until basic to litmus. Place the mixture in an ice bath. Upon cooling to room temperature, extract the mixture three times with diethyl ether. Combine the organic layers and wash them twice with water and once with brine. Dry the organic layer over anhydrous potassium carbonate. Decant away from the drying agent and evaporate the diethyl ether from a dry, preweighed flask using a rotary evaporator. The oily residue will be your crude product 3. Obtain and record the following information: 1. crude product description (co2. crude weight/percent yieldSynthesis of α-chloro-2,6-dimethylacetanilide (4) Dissolve 3 in 17 mL of glacial acetic acid. Add 1.1 equivalents (based on the moles of 3) of α-chloroacetyl chloride to this solution. Heat the solution to 40-50 o C for ten minutes to complete the reaction. Upon cooling, add a solution of ~3.3 g sodium acetate trihydrate in 67 mL water and then place the resulting mixture in an ice bath. Collect the precipitate by vacuum filtration. Rinse the filter cake with copious amounts of water in order to remove the acetic acid. It is important that the product be completely free of acetic acid after this step (why?). The pH of the individual water rinses can be checked with litmus paper to determine if the product is acid free. Allow for the product to air-dry on a watch glass until the next meeting. There is a reasonable chance that you will not obtain a precipitate as described above. If this is the case, you can try “seeding” using a small sample of authentic product from a classmate. If this does not work, check the TLC to be sure that you have formed product and devise an extractive workup that will separate the unreacted aniline 3 from the desired product 4. (Make sure you understand how to do this even if you obtain a precipitate in the first place). After the aqueous workup and following removal of solvent, you should obtain a solid. If not, check the TLC, using a sample of authentic product from a classmate as a standard. If the product appears relatively pure, you can continue even though the material is not a solid. Obtain and record the following information: 1. crude product description (color, physical state, etc.) 2. crude weight/percent yield 3. mp (if a solid)
4. TLC analysis 5. IR (check for presence of amide functional group) Synthesis of lidocaine; α-(N,N-diethylamino)-2,6-dimethylacetanilide (5). In a round bottom flask, dissolve α-chloro-2,6-dimethylacetanilide 4 in 17 mL of toluene. Before continuing, spot several (4 to 5) TLC plates in advance with this solution of 4. Provide three lanes and spot the 4 on the “SM” and “CO-SPOT” lanes. You will use these plates to monitor the progress of this reaction. Add three equivalents of diethylamine to the round bottom flask, and reflux the mixture vigorously until the reaction is complete. The amount of time required for complete reaction depends on many factors but it will likely take anywhere from more than a few minutes up to several hours. If the reaction is not complete when your lab period ends, you can stopper the reaction and reflux it for additional time at the next period. Usually a white precipitate forms during the reflux. Upon cooling, transfer the reaction mixture to a separatory funnel and extract the mixture three times with water. Next, extract the organic layer with two portions of 3 M hydrochloric acid. Cool the combined acidic aqueous extracts in an ice bath and then add 8 M aqueous potassium hydroxide slowly until the mixture is strongly basic again. The formation of a thin, dark yellow oily layer on top or a white solid is observed at this point. Place the mixture in an ice bath. Once the mixture is chilled, try to initiate the crystallization of the final product if no solid has formed at this point. Collect the obtained precipitate by filtration using a Büchner funnel. Wash it with twice with water and then press it as dry as possible. Obtain and record the following information: 1. crude product description (color, physical state, etc.) 2. crude weight/percent yield 3. TLC analysis Recrystallize the crude product from hexanes. Regardless of the final physical state of your product (solid or oil), obtain and record the following: 1. pure product description (color, physical state, etc.) 2. pure product weight/percent yield 3. overall (three-step) percent yield (from starting material 1) 4. TLC analysis 5. melting point (if a solid) 6. IR 7. 1 H and 13C NMR spectra of lidocaine will be given to you. Turn in a sample of your final product.
1H NMR
13C NMR
MS
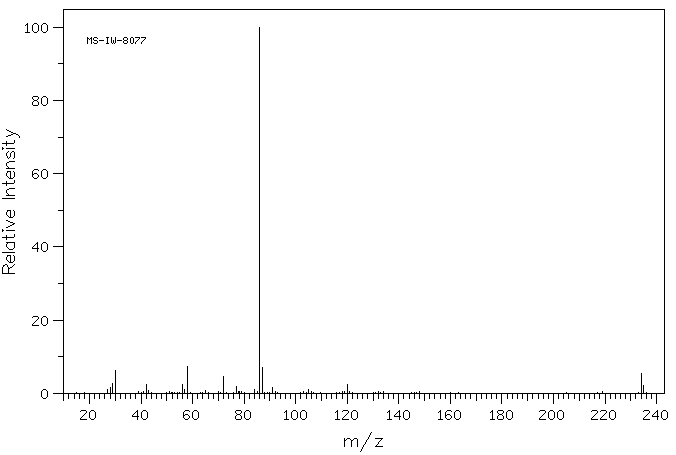
IR KBR
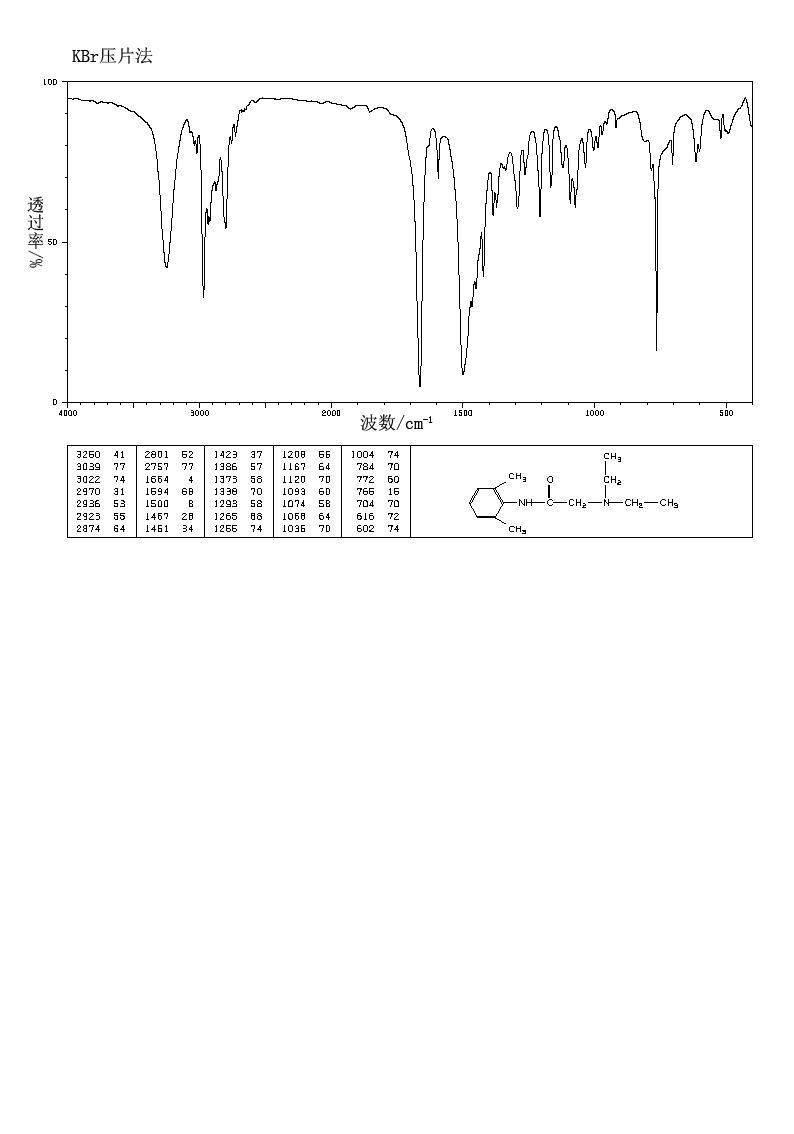
Lidocaine is an antiarrhythmic medicine and also serves as a local anaesthetic drug. It is utilized in topical application to relieve pain, burning and itching sensation caused from skin inflammations. This drug is mainly used for minor surgeries. Figure 1 shows the 1H NMR spectrum of 200 mM lidocaine in CDCl3.
.jpg)
Figure 1. Proton NMR spectrum of 200 mM lidocaine in CDCl3.
1H NMR Relaxation
Figures 2, 3 and 4 show the relaxation time measurements. It can be seen that the relaxation times are shortest for the CH2 protons and longest for the CH protons. The first data point amplitude increases with the number of protons for the related peak.
.jpg)
Figure 2. Proton T1 relaxation time measurement of 200 mM lidocaine in CDCl3.
.jpg)
Figure 3. Proton T2 relaxation time measurement of 200 mM lidocaine in CDCl3.
.jpg)
Figure 4. COSY spectrum of 200 mM lidocaine in CDCl3. The cross-peaks and corresponding exchanging protons are labeled by colour-coded arrows and ellipses.
2D COSY
Figure 4 shows the 2D COSY spectrum where two spin systems (6,7,8) to (10,11) can be clearly seen. For instance, the methyl groups at 10 and 11 positions bond to aromatic protons at 6 and 8 positions, while the methyl groups at 16 and 17 positions bond to the ethylene groups at 14 and 15 positions. No coupling occurs at positions (6,7,8) to (16,17) or (14,15).
2D Homonuclear J-Resolved Spectroscopy
The chemical shift in the 2D homonuclear j-resolved spectrum appears along the direct (f2) direction and the effects of coupling between protons appear along the indirect (f1) dimension. This enables the assignment of chemical shifts of multiplets and may help in measuring unresolved couplings. Also, a decoupled 1D proton spectrum is produced by the projection along the f1 dimension. The 2D homonuclear j-resolved spectrum of lidocaine, plus the 1D proton spectrum (blue line) are shown in Figure 5.
.jpg)
Figure 5. Homonuclear j-resolved spectrum of 200 mM lidocaine in CDCl3. The multiplet splitting frequencies for different couplings are colour- coded.
The projection which is vertical reveals how the multiplets disintegrate into a single peak, which makes the 1D spectrum more simplified. Peak multiplicities are produced by vertical traces from peaks in the 2D spectrum and help in determining the frequencies of proton-proton coupling. When coupling frequencies are compared between different peaks, information can be obtained regarding which peaks are bonded to each other. Also, Information regarding the coupling strength can be obtained from the size of the coupling frequencies. These couplings substantiate the results of the COSY experiment.
However, in this experiment, the effects of second order coupling appear in the f1 direction as additional peaks which are equidistant from the coupling partners detached from the zero frequency in the f1 dimension. These peaks provide proof of second order coupling partners, but are generally considered as artifacts. Figure 6 shows these coupling partners and additional peaks marked by colour-coded arrows and ellipses.
.jpg)
Figure 6. Homonuclear j-resolved spectrum of 200 mM lidocaine in CDCl3 showing the extra peaks due to strong couplings.
1D 13C Spectra
Figure 7 shows the 13C NMR spectra of 1 M lidocaine in CDCl3. Since the 1D Carbon experiment is highly susceptible to the 13C nuclei in the specimen, it easily and clearly resolves 9 resonances. In this experiment, only carbons coupled to protons are seen.
.jpg)
Figure 7. Carbon spectra of 1 M lidocaine in CDCl3.
Given the fact that the DEPT spectra do not display the peaks at 170 and 135ppm, they must be part of quaternary carbons. The DEPT-135 and the DEPT-45 experiments provide signals of CH3, CH2 and CH groups, while the DEPT-90 experiment provides only the signal of CH groups. However, in DEPT-135 the CH2 groups occur as negative peaks. It can thus be summed up that the peaks between 45 and 60ppm belong to ethylene groups; the peaks between 10 and 20ppm are part of the methyl groups; and the peaks between 125 and 130ppm belong to methyne groups. A similar study can be carried out on the C and CH peaks.
Heteronuclear Correlation
The Heteronuclear Correlation (HETCOR) experiment identifies the proton signal that appears along the indirect dimension and the carbon signal along the direct dimension. Figure 8 shows the HETCOR spectrum of 1 M lidocaine in CDCl3. in the 2D spectrum, the peaks reveal which proton is attached to which carbon. This experiment helps in resolving assignment uncertainty from the ID carbon spectra.
.jpg)
Figure 8. HETCOR spectrum of 1 M lidocaine in CDCl3.
Heteronuclear Multiple Quantum Coherence
Heteronuclear Multiple Quantum Coherence (HMQC) is similar to the HETCOR experiment and is utilized to associate proton resonances to the carbons that are coupled directly to those protons. But in the HMQC experiment, the proton signal appears along the direct dimension and the carbon signal along the indirect dimension. Figure 9 shows the HMQC spectrum of 1 M lidocaine in CDCl3. In the 2D spectrum, the peaks show which proton is attached to which carbon. For conclusive peak assignment, a similar study with the HETCOR spectrum can be carried out.
.jpg)
Figure 9. HMQC spectrum of 1 M lidocaine in CDCl3.
Heteronuclear Multiple Bond Correlation
The Heteronuclear Multiple Bond Correlation (HMBC) experiment can be employed to achieve long-range correlations of proton and carbon via two or three bond couplings. Similar to the HMQC experiment, the proton signal appears along the direct dimension and the carbon signal along the indirect dimension. Figure 10 shows the HMBC spectrum of 1 M lidocaine in CDCl3.
.jpg)
Figure 10. HMBC spectrum of 1 M lidocaine in CDCl3, with some of the long-range couplings marked.
The couplings amid the molecular positions appear analogous to the couplings seen in the COSY spectrum; however, the HMBC also displays couplings to quaternary carbons, which are not seen either in HMQC or COSY experiments. In addition, there is a correlation between protons and carbons. This is attributed to three-bond bonding from 14 and 15 and vice versa, as shown in light green in Figure 1.
SYN
Synthesis of lidocaine T. J. Reilly (1999). “The Preparation of Lidocaine”. J. Chem. Ed. 76 (11): 1557.

CLIP
The Present Synthesis Of Lidocaine Begins With 2,6-Dimethylnitrobenzene (1). This Compound Can Be Made From 1,3-Dimethylbenzene, Also Known As M-Xylene, Which Is More Difficult To Make. Luckily,
This problem has been solved!
See the answer
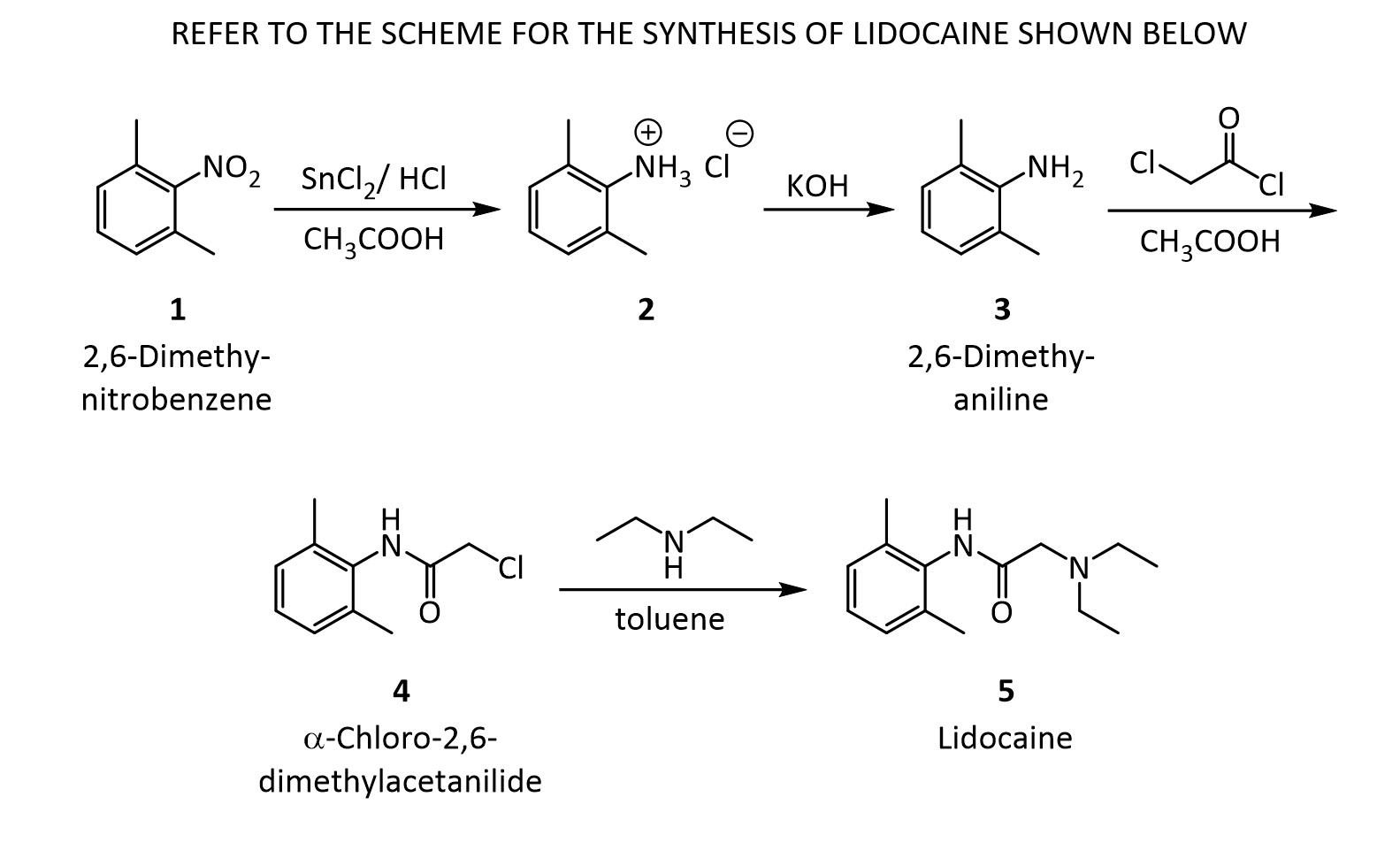
- The present synthesis of lidocaine begins with 2,6-dimethylnitrobenzene (1). This compound can be made from 1,3-dimethylbenzene, also known as m-xylene, which is more difficult to make. Luckily, m-xylene is commercially available, so a synthesis of 1 from m-xylene is a practical alternative if one wants to begin the synthesis of lidocaine with m-xylene. Suppose you want to prepare 1 from m-xylene. Show with chemical equations the reagents that you would use, and the possible isomers that would result.
2. The practical transformation of 1 into 3 is carried out by the following scheme:
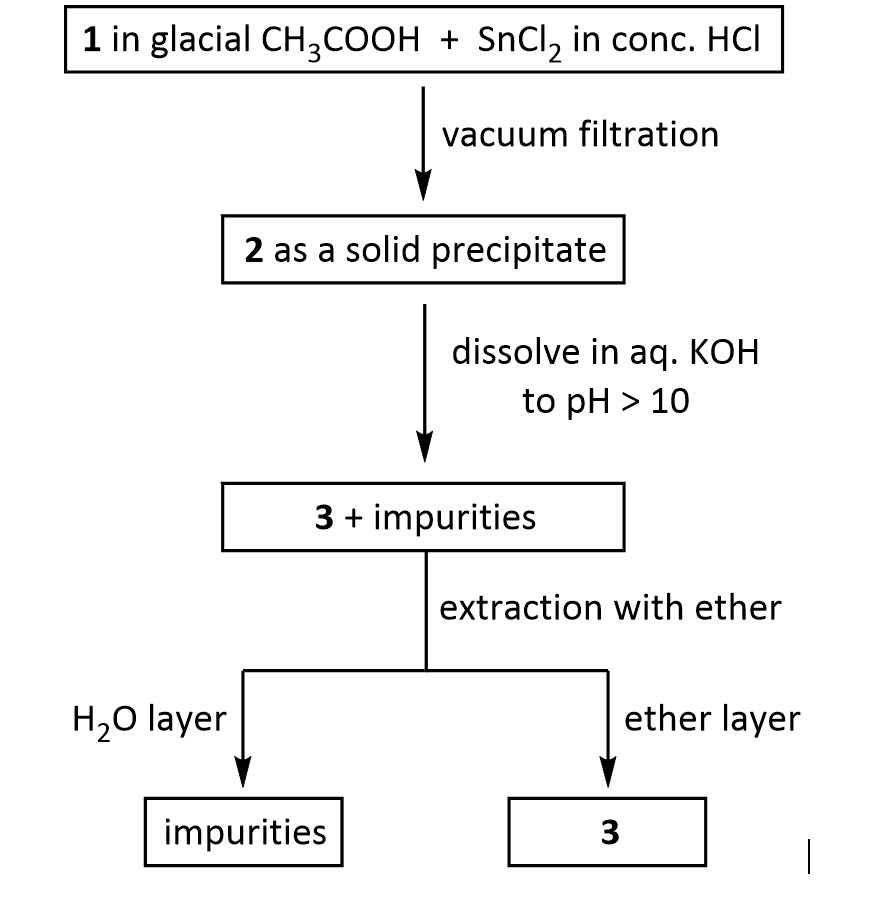
Suppose you dissolve the solid precipitate of 2 in water, but forget to include the KOH in the second step above. What would happen after the extraction with ether? Give your answer in terms of what would be found in the ether layer, and in the aqueous layer.
3. Suppose you’re out of acetic acid (CH3COOH) and decide to use ethanol (CH3 CH2OH) as the solvent in the transformation of 3 into 4. Would this be a wise choice, and why?
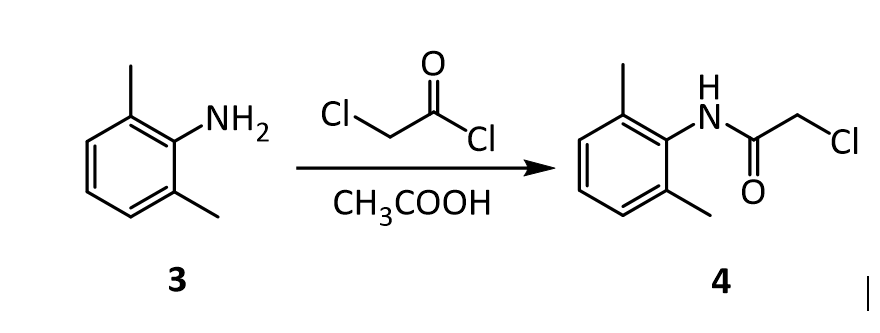
4.The amide 4 has a nitrogen attached to the benzene ring, and a chlorine attached to a primary carbon. Yet, it doesn’t react with itself in a nucleophilic displacement. Why is the nitrogen in the amide not nucleophilic? Give your answer in terms of the resonance forms of amides in general:
5. In the reaction below, what factors come into play to favor attack of the aniline 3 on the carbonyl carbon of the acid chloride (carbon 1 in red), rather than at the a-carbon (carbon 2 in red)?
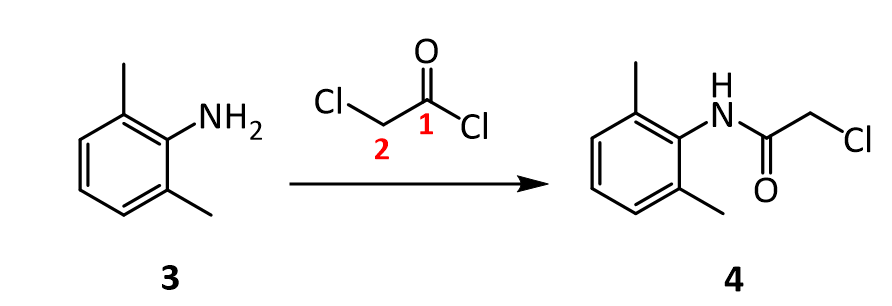
6. Before carrying out the transformation below, compound 4 and the glassware used must be oven-dried. What would happen if the reaction was attempted using wet 4?
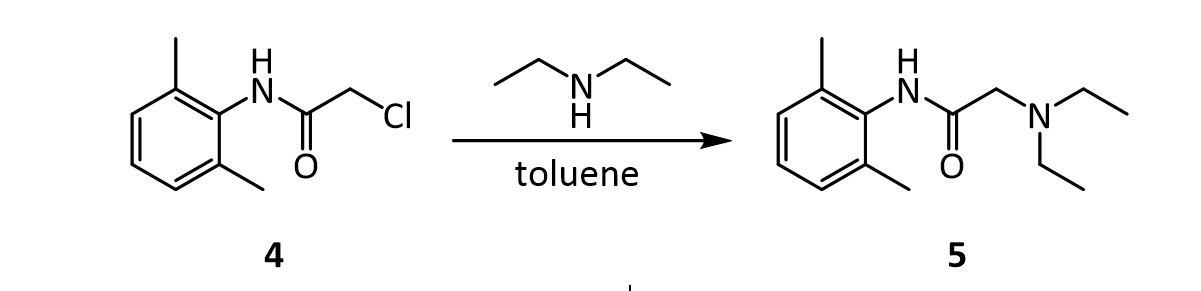
7.In the reaction below, what factors come into play to favor attack of diethylamine on the a-carbon (carbon 1 in red), rather than on the amide C=O carbon (carbon 2 in red)?
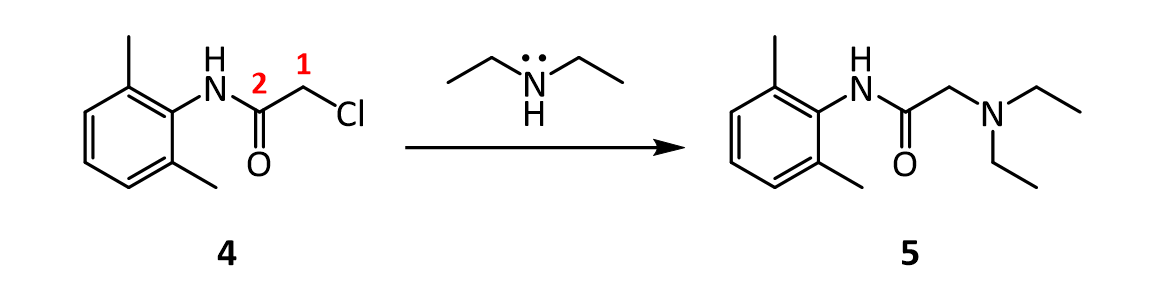
8. In the reaction below, why does the amine nitrogen (#1 in red) undergo protonation with H2SO4 preferentially over the amide nitrogen (#2 in red)? In other words, why is nitrogen 1 basic, but nitrogen 2 is not?
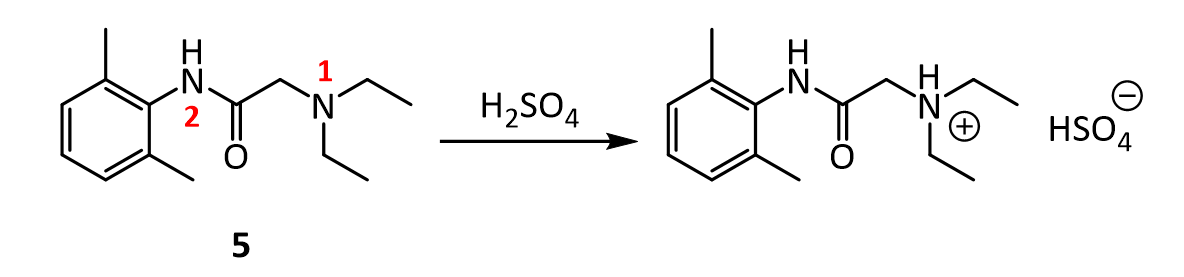
9.Lidocaine and other drugs containing amino groups are usually marketed as their hydrochloride or hydrogen sulfate salts, rather than as “free amines.” Provide two reasons why this practice makes sense.
10.Although lidocaine is marketed as its hydrochloride salt, it doesn’t exhibit the same level of physiological activity as the free amine. The free amine is more lipophilic and diffuses across a neuron cell membrane more rapidly than the ionic salt, resulting in a more rapid onset of anesthesia. Therefore, sodium bicarbonate (NaHCO3) is added to a solution of lidocaine prior to injection. How does the addition of sodium bicarbonate promote a faster anesthetic effect?
CLIP
CLIP
Lidocaine, also known as lignocaine and sold under the brand name Xylocaine among others, is a local anesthetic of the amino amide type. It is also used to treat ventricular tachycardia.[7][8] When used for local anaesthesia or in nerve blocks, lidocaine typically begins working within several minutes and lasts for half an hour to three hours.[8][9] Lidocaine mixtures may also be applied directly to the skin or mucous membranes to numb the area.[8] It is often used mixed with a small amount of adrenaline (epinephrine) to prolong its local effects and to decrease bleeding.[8]
If injected intravenously, it may cause cerebral effects such as confusion, changes in vision, numbness, tingling, and vomiting.[7] It can cause low blood pressure and an irregular heart rate.[7] There are concerns that injecting it into a joint can cause problems with the cartilage.[8] It appears to be generally safe for use in pregnancy.[7] A lower dose may be required in those with liver problems.[7] It is generally safe to use in those allergic to tetracaine or benzocaine.[8] Lidocaine is an antiarrhythmic medication of the class Ib type.[7] This means it works by blocking sodium channels and thus decreasing the rate of contractions of the heart.[7] When injected near nerves, the nerves cannot conduct signals to or from the brain.[8]
Lidocaine was discovered in 1946 and went on sale in 1948.[10] It is on the World Health Organization’s List of Essential Medicines.[11] It is available as a generic medication.[8][12] In 2018, it was the 233rd most commonly prescribed medication in the United States, with more than 2 million prescriptions.[13][14]
Medical uses
Local numbing agent
The efficacy profile of lidocaine as a local anaesthetic is characterized by a rapid onset of action and intermediate duration of efficacy. Therefore, lidocaine is suitable for infiltration, block, and surface anaesthesia. Longer-acting substances such as bupivacaine are sometimes given preference for spinal and epidural anaesthesias; lidocaine, though, has the advantage of a rapid onset of action. Adrenaline vasoconstricts arteries, reducing bleeding and also delaying the resorption of lidocaine, almost doubling the duration of anaesthesia.
Lidocaine is one of the most commonly used local anaesthetics in dentistry. It can be administered in multiple ways, most often as a nerve block or infiltration, depending on the type of treatment carried out and the area of the mouth worked on.[15]
For surface anaesthesia, several formulations can be used for endoscopies, before intubations, etc. Buffering the pH of lidocaine makes local numbing less painful.[16] Lidocaine drops can be used on the eyes for short ophthalmic procedures. There is tentative evidence for topical lidocaine for neuropathic pain and skin graft donor site pain.[17][18] As a local numbing agent, it is used for the treatment of premature ejaculation.[19]
An adhesive transdermal patch containing a 5% concentration of lidocaine in a hydrogel bandage, is approved by the US FDA for reducing nerve pain caused by shingles.[20] The transdermal patch is also used for pain from other causes, such as compressed nerves and persistent nerve pain after some surgeries.
Heart arrhythmia
Lidocaine is also the most important class-1b antiarrhythmic drug; it is used intravenously for the treatment of ventricular arrhythmias (for acute myocardial infarction, digoxin poisoning, cardioversion, or cardiac catheterization) if amiodarone is not available or contraindicated. Lidocaine should be given for this indication after defibrillation, CPR, and vasopressors have been initiated. A routine preventive dose is no longer recommended after a myocardial infarction as the overall benefit is not convincing.[21]
Epilepsy
A 2013 review on treatment for neonatal seizures recommended intravenous lidocaine as a second-line treatment, if phenobarbital fails to stop seizures.[22]
Other
Intravenous lidocaine infusions are also used to treat chronic pain and acute surgical pain as an opiate sparing technique. The quality of evidence for this use is poor so it is difficult to compare it to placebo or an epidural.[23]
Inhaled lidocaine can be used as a cough suppressor acting peripherally to reduce the cough reflex. This application can be implemented as a safety and comfort measure for patients who have to be intubated, as it reduces the incidence of coughing and any tracheal damage it might cause when emerging from anaesthesia.[24]
Lidocaine, along with ethanol, ammonia, and acetic acid, may also help in treating jellyfish stings, both numbing the affected area and preventing further nematocyst discharge.[25][26]
For gastritis, drinking a viscous lidocaine formulation may help with the pain.[27]
Adverse effects
Adverse drug reactions (ADRs) are rare when lidocaine is used as a local anesthetic and is administered correctly. Most ADRs associated with lidocaine for anesthesia relate to administration technique (resulting in systemic exposure) or pharmacological effects of anesthesia, and allergic reactions only rarely occur.[28] Systemic exposure to excessive quantities of lidocaine mainly result in central nervous system (CNS) and cardiovascular effects – CNS effects usually occur at lower blood plasma concentrations and additional cardiovascular effects present at higher concentrations, though cardiovascular collapse may also occur with low concentrations. ADRs by system are:
- CNS excitation: nervousness, agitation, anxiety, apprehension, tingling around the mouth (circumoral paraesthesia), headache, hyperesthesia, tremor, dizziness, pupillary changes, psychosis, euphoria, hallucinations, and seizures
- CNS depression with increasingly heavier exposure: drowsiness, lethargy, slurred speech, hypoesthesia, confusion, disorientation, loss of consciousness, respiratory depression and apnoea.
- Cardiovascular: hypotension, bradycardia, arrhythmias, flushing, venous insufficiency, increased defibrillator threshold, edema, and/or cardiac arrest – some of which may be due to hypoxemia secondary to respiratory depression.[29]
- Respiratory: bronchospasm, dyspnea, respiratory depression or arrest
- Gastrointestinal: metallic taste, nausea, vomiting
- Ears: tinnitus
- Eyes: local burning, conjunctival hyperemia, corneal epithelial changes/ulceration, diplopia, visual changes (opacification)
- Skin: itching, depigmentation, rash, urticaria, edema, angioedema, bruising, inflammation of the vein at the injection site, irritation of the skin when applied topically
- Blood: methemoglobinemia
- Allergy
ADRs associated with the use of intravenous lidocaine are similar to toxic effects from systemic exposure above. These are dose-related and more frequent at high infusion rates (≥3 mg/min). Common ADRs include: headache, dizziness, drowsiness, confusion, visual disturbances, tinnitus, tremor, and/or paraesthesia. Infrequent ADRs associated with the use of lidocaine include: hypotension, bradycardia, arrhythmias, cardiac arrest, muscle twitching, seizures, coma, and/or respiratory depression.[29]
It is generally safe to use lidocaine with vasoconstrictor such as adrenaline, including in regions such as the nose, ears, fingers, and toes.[30] While concerns of tissue death if used in these areas have been raised, evidence does not support these concerns.[30]
Interactions
Any drugs that are also ligands of CYP3A4 and CYP1A2 can potentially increase serum levels and potential for toxicity or decrease serum levels and the efficacy, depending on whether they induce or inhibit the enzymes, respectively. Drugs that may increase the chance of methemoglobinemia should also be considered carefully. Dronedarone and liposomal morphine are both absolutely a contraindication, as they may increase the serum levels, but hundreds of other drugs require monitoring for interaction.[31]
Contraindications
Absolute contraindications for the use of lidocaine include:
- Heart block, second or third degree (without pacemaker)
- Severe sinoatrial block (without pacemaker)
- Serious adverse drug reaction to lidocaine or amide local anesthetics
- Hypersensitivity to corn and corn-related products (corn-derived dextrose is used in the mixed injections)
- Concurrent treatment with quinidine, flecainide, disopyramide, procainamide (class I antiarrhythmic agents)
- Prior use of amiodarone hydrochloride
- Adams-Stokes syndrome[32]
- Wolff-Parkinson-White syndrome[32]
- Lidocaine viscous is not recommended by the FDA to treat teething pain in children and infants.[33]
Exercise caution in patients with any of these:
- Hypotension not due to arrhythmia
- Bradycardia
- Accelerated idioventricular rhythm
- Elderly patients
- Ehlers-Danlos Syndrome
- Pseudocholinesterase deficiency
- Intra-articular infusion (this is not an approved indication and can cause chondrolysis)
- Porphyria, especially acute intermittent porphyria; lidocaine has been classified as porphyrogenic because of the hepatic enzymes it induces,[34] although clinical evidence suggests it is not.[35] Bupivacaine is a safe alternative in this case.
- Impaired liver function – people with lowered hepatic function may have an adverse reaction with repeated administration of lidocaine because the drug is metabolized by the liver. Adverse reactions may include neurological symptoms (e.g. dizziness, nausea, muscle twitches, vomiting, or seizures).[36]
Overdosage
Overdoses of lidocaine may result from excessive administration by topical or parenteral routes, accidental oral ingestion of topical preparations by children (who are more susceptible to overdose), accidental intravenous (rather than subcutaneous, intrathecal, or paracervical) injection, or from prolonged use of subcutaneous infiltration anesthesia during cosmetic surgery.
Such overdoses have often led to severe toxicity or death in both children and adults. Lidocaine and its two major metabolites may be quantified in blood, plasma, or serum to confirm the diagnosis in potential poisoning victims or to assist forensic investigation in a case of fatal overdose.
Lidocaine is often given intravenously as an antiarrhythmic agent in critical cardiac-care situations.[37] Treatment with intravenous lipid emulsions (used for parenteral feeding) to reverse the effects of local anaesthetic toxicity is becoming more common.[38]
Postarthroscopic glenohumeral chondrolysis
Lidocaine in large amounts may be toxic to cartilage and intra-articular infusions can lead to postarthroscopic glenohumeral chondrolysis.[39]
Pharmacology
Mechanism of action
Lidocaine alters signal conduction in neurons by prolonging the inactivation of the fast voltage-gated Na+ channels in the neuronal cell membrane responsible for action potential propagation.[40] With sufficient blockage, the voltage-gated sodium channels will not open and an action potential will not be generated. Careful titration allows for a high degree of selectivity in the blockage of sensory neurons, whereas higher concentrations also affect other types of neurons.
The same principle applies for this drug’s actions in the heart. Blocking sodium channels in the conduction system, as well as the muscle cells of the heart, raises the depolarization threshold, making the heart less likely to initiate or conduct early action potentials that may cause an arrhythmia.[41]
Pharmacokinetics
When used as an injectable it typically begins working within four minutes and lasts for half an hour to three hours.[8][9] Lidocaine is about 95% metabolized (dealkylated) in the liver mainly by CYP3A4 to the pharmacologically active metabolites monoethylglycinexylidide (MEGX) and then subsequently to the inactive glycine xylidide. MEGX has a longer half-life than lidocaine, but also is a less potent sodium channel blocker.[42] The volume of distribution is 1.1 L/kg to 2.1 L/kg, but congestive heart failure can decrease it. About 60% to 80% circulates bound to the protein alpha1 acid glycoprotein. The oral bioavailability is 35% and the topical bioavailability is 3%.
The elimination half-life of lidocaine is biphasic and around 90 min to 120 min in most patients. This may be prolonged in patients with hepatic impairment (average 343 min) or congestive heart failure (average 136 min).[43] Lidocaine is excreted in the urine (90% as metabolites and 10% as unchanged drug).[44]
History
Lidocaine, the first amino amide–type local anesthetic, was first synthesized under the name ‘xylocaine’ by Swedish chemist Nils Löfgren in 1943.[45][46][47] His colleague Bengt Lundqvist performed the first injection anesthesia experiments on himself.[45] It was first marketed in 1949.
Society and culture
Dosage forms
Lidocaine, usually in the form of its hydrochloride salt, is available in various forms including many topical formulations and solutions for injection or infusion.[48] It is also available as a transdermal patch, which is applied directly to the skin.
- Lidocaine hydrochloride 2% epinephrine 1:80,000 solution for injection in a cartridge
- Lidocaine hydrochloride 1% solution for injection
- Topical lidocaine spray
- 2% viscous lidocaine
Names
Lidocaine is the International Nonproprietary Name (INN), British Approved Name (BAN), and Australian Approved Name (AAN),[49] while lignocaine is the former BAN[citation needed] and AAN. Both the old and new names will be displayed on the product label in Australia until at least 2023.[50]
Xylocaine is a brand name.
Recreational use
As of 2021, lidocaine is not listed by the World Anti-Doping Agency as a substance whose use is banned in sport.[51] It is used as an adjuvant, adulterant, and diluent to street drugs such as cocaine and heroin.[52] It is one of the three common ingredients in site enhancement oil used by bodybuilders.[53]
Adulterant in cocaine
Lidocaine is often added to cocaine as a diluent.[54][55] Cocaine and lidocaine both numb the gums when applied. This gives the user the impression of high-quality cocaine, when in actuality the user is receiving a diluted product.[56]
Compendial status
Veterinary use
It is a component of the veterinary drug Tributame along with embutramide and chloroquine used to carry out euthanasia on horses and dogs.[58][59]
References
- ^ “Lidocaine”. Merriam-Webster Dictionary.
- ^ “Lidocaine”. Dictionary.com Unabridged. Random House.
- ^ “Poisons Standard February 2021”. Federal Register of Legislation. 1 January 2021. Retrieved 11 April 2021.
- ^ “Lidocaine Hydrochloride Injection BP 1% w/v – Summary of Product Characteristics (SmPC)”. (emc). 29 June 2020. Retrieved 11 April 2021.
- ^ “Xylocaine MPF- lidocaine hydrochloride injection, solution Xylocaine- lidocaine hydrochloride injection, solution Xylocaine- lidocaine hydrochloride,epinephrine bitartrate injection, solution”. DailyMed. Retrieved 11 April 2021.
- ^ “Ztlido- lidocaine patch”. DailyMed. Retrieved 11 April 2021.
- ^ Jump up to:a b c d e f g h i j k “Lidocaine Hydrochloride (Antiarrhythmic)”. The American Society of Health-System Pharmacists. Archivedfrom the original on 2015-08-10. Retrieved Aug 26, 2015.
- ^ Jump up to:a b c d e f g h i j “Lidocaine Hydrochloride (Local)”. The American Society of Health-System Pharmacists. Archived from the original on 2015-09-06. Retrieved Aug 26, 2015.
- ^ Jump up to:a b c J. P. Nolan; P. J. F. Baskett (1997). “Analgesia and anaesthesia”. In David Skinner; Andrew Swain; Rodney Peyton; Colin Robertson (eds.). Cambridge Textbook of Accident and Emergency Medicine. Project co-ordinator, Fiona Whinster. Cambridge, UK: Cambridge University Press. p. 194. ISBN 9780521433792. Archived from the original on 2017-09-08.
- ^ Scriabine, Alexander (1999). “Discovery and development of major drugs currently in use”. In Ralph Landau; Basil Achilladelis; Alexander Scriabine (eds.). Pharmaceutical Innovation: Revolutionizing Human Health. Philadelphia: Chemical Heritage Press. p. 211. ISBN 9780941901215. Archived from the original on 2017-09-08.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
- ^ Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 22. ISBN 9781284057560.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Lidocaine – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ “Local anaesthetic drugs”.
- ^ Cepeda MS, Tzortzopoulou A, Thackrey M, Hudcova J, Arora Gandhi P, Schumann R (December 2010). Tzortzopoulou A (ed.). “Adjusting the pH of lidocaine for reducing pain on injection”. The Cochrane Database of Systematic Reviews (12): CD006581. doi:10.1002/14651858.CD006581.pub2. PMID 21154371.(Retracted, see doi:10.1002/14651858.cd006581.pub3. If this is an intentional citation to a retracted paper, please replace
{{Retracted}}with{{Retracted|intentional=yes}}.) - ^ Derry S, Wiffen PJ, Moore RA, Quinlan J (July 2014). Derry S (ed.). “Topical lidocaine for neuropathic pain in adults”. The Cochrane Database of Systematic Reviews. 7 (7): CD010958. doi:10.1002/14651858.CD010958.pub2. PMC 6540846. PMID 25058164.
- ^ Sinha S, Schreiner AJ, Biernaskie J, Nickerson D, Gabriel VA (November 2017). “Treating pain on skin graft donor sites: Review and clinical recommendations”. The Journal of Trauma and Acute Care Surgery. 83 (5): 954–964. doi:10.1097/TA.0000000000001615. PMID 28598907. S2CID 44520644.
- ^ “Lidocaine/prilocaine spray for premature ejaculation”. Drug and Therapeutics Bulletin. 55 (4): 45–48. April 2017. doi:10.1136/dtb.2017.4.0469. PMID 28408390. S2CID 19110955.
- ^ Kumar M, Chawla R, Goyal M (2015). “Topical anesthesia”. Journal of Anaesthesiology Clinical Pharmacology. 31 (4): 450–6. doi:10.4103/0970-9185.169049. PMC 4676230. PMID 26702198.
- ^ Martí-Carvajal AJ, Simancas-Racines D, Anand V, Bangdiwala S (August 2015). “Prophylactic lidocaine for myocardial infarction”. The Cochrane Database of Systematic Reviews. 8 (8): CD008553. doi:10.1002/14651858.CD008553.pub2. PMC 8454263. PMID 26295202.
- ^ Slaughter LA, Patel AD, Slaughter JL (March 2013). “Pharmacological treatment of neonatal seizures: a systematic review”. Journal of Child Neurology. 28 (3): 351–64. doi:10.1177/0883073812470734. PMC 3805825. PMID 23318696.
- ^ Weibel S, Jelting Y, Pace NL, Helf A, Eberhart LH, Hahnenkamp K, et al. (June 2018). “Continuous intravenous perioperative lidocaine infusion for postoperative pain and recovery in adults”. The Cochrane Database of Systematic Reviews. 2018 (6): CD009642. doi:10.1002/14651858.cd009642.pub3. PMC 6513586. PMID 29864216.
- ^ Biller JA (2007). “Airway obstruction, bronchospasm, and cough”. In Berger AM, Shuster JL, Von Roenn JH (eds.). Principles and practice of palliative care and supportive oncology. Hagerstwon, MD: Lippincott Williams & Wilkins. pp. 297–307. ISBN 978-0-7817-9595-1.
Inhaled lidocaine is used to suppress cough during bronchoscopy. Animal studies and a few human studies suggest that lidocaine has an antitussive effect…
- ^ Birsa LM, Verity PG, Lee RF (May 2010). “Evaluation of the effects of various chemicals on discharge of and pain caused by jellyfish nematocysts”. Comp. Biochem. Physiol. C. 151 (4): 426–30. doi:10.1016/j.cbpc.2010.01.007. PMID 20116454.
- ^ Morabito R, Marino A, Dossena S, La Spada G (Jun 2014). “Nematocyst discharge in Pelagia noctiluca (Cnidaria, Scyphozoa) oral arms can be affected by lidocaine, ethanol, ammonia and acetic acid”. Toxicon. 83: 52–8. doi:10.1016/j.toxicon.2014.03.002. PMID 24637105.
- ^ James G. Adams (2012). “32”. Emergency Medicine: Clinical Essentials. Elsevier Health Sciences. ISBN 9781455733941. Archived from the original on 2017-09-08.
- ^ Jackson D, Chen AH, Bennett CR (October 1994). “Identifying true lidocaine allergy”. J Am Dent Assoc. 125 (10): 1362–6. doi:10.14219/jada.archive.1994.0180. PMID 7844301.
- ^ Jump up to:a b Australian Medicines Handbook. Adelaide, S. Aust: Australian Medicines Handbook Pty Ltd. 2006. ISBN 978-0-9757919-2-9.[page needed]
- ^ Jump up to:a b Nielsen LJ, Lumholt P, Hölmich LR (October 2014). “[Local anaesthesia with vasoconstrictor is safe to use in areas with end-arteries in fingers, toes, noses and ears]”. Ugeskrift for Laeger. 176(44). PMID 25354008.
- ^ “Lidocaine”. Epocrates. Archived from the original on 2014-04-22.
- ^ Jump up to:a b “Lidocaine Hydrochloride and 5% Dextrose Injection”. Safety Labeling Changes. FDA Center for Drug Evaluation and Research (CDER). January 2014. Archived from the original on 2013-04-03.
- ^ “Lidocaine Viscous: Drug Safety Communication – Boxed Warning Required – Should Not Be Used to Treat Teething Pain”. FDA Center for Drug Evaluation and Research (CDER). June 2014. Archived from the original on 2014-07-14.
- ^ “Table 96–4. Drugs and Porphyria” (PDF). Merck Manual. Merck & Company, Inc. 2011. Archived from the original on 2014-04-20.
- ^ “Lidocaine – N01BB02”. Drug porphyrinogenicity monograph. The Norwegian Porphyria Centre and the Swedish Porphyria Centre. Archived from the original on 2014-04-20.
strong clinical evidence points to lidocaine as probably not porphyrinogenic
- ^ Khan, M. Gabriel (2007). Cardiac Drug Therapy (7th ed.). Totowa, NJ: Humana Press. ISBN 9781597452380.
- ^ Baselt R (2008). Disposition of Toxic Drugs and Chemicals in Man(8th ed.). Foster City, CA: Biomedical Publications. pp. 840–4. ISBN 978-0-9626523-7-0.
- ^ Picard J, Ward SC, Zumpe R, Meek T, Barlow J, Harrop-Griffiths W (February 2009). “Guidelines and the adoption of ‘lipid rescue’ therapy for local anaesthetic toxicity”. Anaesthesia. 64 (2): 122–5. doi:10.1111/j.1365-2044.2008.05816.x. PMID 19143686. S2CID 25581037.
- ^ Gulihar A, Robati S, Twaij H, Salih A, Taylor GJ (December 2015). “Articular cartilage and local anaesthetic: A systematic review of the current literature”. Journal of Orthopaedics. 12 (Suppl 2): S200-10. doi:10.1016/j.jor.2015.10.005. PMC 4796530. PMID 27047224.
- ^ Carterall, William A. (2001). “Molecular mechanisms of gating and drug block of sodium channels”. Sodium Channels and Neuronal Hyperexcitability. Novartis Foundation Symposia. 241. pp. 206–225. doi:10.1002/0470846682.ch14. ISBN 9780470846681.
- ^ Sheu SS, Lederer WJ (Oct 1985). “Lidocaine’s negative inotropic and antiarrhythmic actions. Dependence on shortening of action potential duration and reduction of intracellular sodium activity”. Circulation Research. 57 (4): 578–90. doi:10.1161/01.res.57.4.578. PMID 2412723.
- ^ Lewin NA, Nelson LH (2006). “Chapter 61: Antidysrhythmics”. In Flomenbaum N, Goldfrank LR, Hoffman RL, Howland MD, Lewin NA, Nelson LH (eds.). Goldfrank’s Toxicologic Emergencies(8th ed.). New York: McGraw-Hill. pp. 963–4. ISBN 978-0-07-143763-9.
- ^ Thomson PD, Melmon KL, Richardson JA, Cohn K, Steinbrunn W, Cudihee R, Rowland M (April 1973). “Lidocaine pharmacokinetics in advanced heart failure, liver disease, and renal failure in humans”. Ann. Intern. Med. 78 (4): 499–508. doi:10.7326/0003-4819-78-4-499. PMID 4694036.
- ^ Collinsworth KA, Kalman SM, Harrison DC (1974). “The clinical pharmacology of lidocaine as an antiarrhythymic drug”. Circulation. 50 (6): 1217–30. doi:10.1161/01.CIR.50.6.1217. PMID 4609637.
- ^ Jump up to:a b Löfgren N (1948). Studies on local anesthetics: Xylocaine: a new synthetic drug (Inaugural dissertation). Stockholm, Sweden: Ivar Heggstroms. OCLC 646046738.[page needed]
- ^ Löfgren N, Lundqvist B (1946). “Studies on local anaesthetics II”. Svensk Kemisk Tidskrift. 58: 206–17.
- ^ Wildsmith JAW (2011). “Lidocaine: A more complex story than ‘simple’ chemistry suggests” (PDF). The Proceedings of the History of Anaesthesia Society. 43: 9–16. Archived (PDF) from the original on 2014-04-22.
- ^ “Lidocaine international forms and names”. Drugs.com. Retrieved 29 October 2017.
- ^ “Lidocaine Ingredient Summary”. Therapeutic Goods Administration. Retrieved 20 September 2018.
- ^ “Updating medicine ingredient names – list of affected ingredients”. Therapeutic Goods Administration. 24 June 2019. Retrieved 16 February 2020.
- ^ “The 2021 Prohibited List International Standard” (PDF). The World Anti-Doping Code. World Anti-Doping Agency (WADA). 1 January 2021. Archived from the original (PDF) on 13 May 2021. Retrieved 18 May 2021.
- ^ “New York Drug Threat Assessment”. National Drug Intelligence Center. November 2002. Archived from the original on 2012-08-12.
- ^ Pupka A, Sikora J, Mauricz J, Cios D, Płonek T (2009). “[The usage of synthol in the body building]”. Polimery W Medycynie. 39(1): 63–5. PMID 19580174.
- ^ Bernardo NP; Siqueira MEPB; De Paiva MJN; Maia PP (2003). “Caffeine and other adulterants in seizures of street cocaine in Brazil”. International Journal of Drug Policy. 14 (4): 331–4. doi:10.1016/S0955-3959(03)00083-5.
- ^ “UNITED STATES of America, Plaintiff-Appellee, v. Luis A. CUELLO, Alvaro Bastides-Benitez, John Doe, a/k/a Hugo Hurtado, and Alvaro Carvajal, Defendants-Appellants”. Docket No. 78-5314. United States Court of Appeals, Fifth Circuit. 1979-07-25. Archived from the original on 2012-05-24.
- ^ Winterman, Denise (2010-09-07). “How cutting drugs became big business”. BBC News Online. BBC News Magazine. Archivedfrom the original on 2 February 2017. Retrieved 20 January 2017.
- ^ “Revision Bulletin: Lidocaine and Prilocaine Cream–Revision to Related Compounds Test”. The United States Pharmacopeial Convention. November 30, 2007. Archived from the original on May 1, 2013.
- ^ Peterson, Michael E.; Talcott, Patricia A. (2013-08-07). Small Animal Toxicology. Elsevier Health Sciences. ISBN 978-0323241984. Archived from the original on 2017-09-08.
- ^ “FDA Freedom of Information Summary – Tributame” (PDF). Archived from the original (PDF) on 2015-05-18.
External links
- “Lidocaine”. Drug Information Portal. U.S. National Library of Medicine.
- “Lidocaine Transdermal Patch”. MedlinePlus.
- US patent 2441498, Nils Magnus Loefgren & Bengt Josef Lundqvist, “Alkyl glycinanilides”, published 1948-05-11, issued 1948-05-11, assigned to ASTRA APOTEKARNES KEM FAB
| IUPAC name2-(diethylamino)- N-(2,6-dimethylphenyl)acetamide | |
| CAS Number | 137-58-6 as HCl: 73-78-9 |
|---|---|
| PubChem CID | 3676as HCl: 6314 |
| IUPHAR/BPS | 2623 |
| DrugBank | DB00281 as HCl: DBSALT001508 |
| ChemSpider | 3548 as HCl: 6075 |
| UNII | 98PI200987as HCl: EC2CNF7XFP |
| KEGG | D00358 as HCl: D02086 |
| ChEBI | CHEBI:6456 as HCl: CHEBI:50512 |
| ChEMBL | ChEMBL79 as HCl: ChEMBL541521 |
| PDB ligand | LQZ (PDBe, RCSB PDB) |
| CompTox Dashboard (EPA) | DTXSID1045166 |
| ECHA InfoCard | 100.004.821 |
| Chemical and physical data | |
| Formula | C14H22N2O |
| Molar mass | 234.343 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| Melting point | 68 °C (154 °F) |
| showSMILES | |
| showInChI |
////////LIDOCAINE, lignocaine

NEW DRUG APPROVALS
ONE TIME
$10.00
Divalproex sodium
Divalproex
- 44089
WeightAverage: 144.2114
Chemical FormulaC8H16O2
UNII614OI1Z5WI, CAS number99-66-1, 76584-70-8
2-propylpentanoic acid, DIVALPROEX SODIUM, 76584-70-8, Valproate semisodium, Epival, Depakote, Sodium divalproate, Semisodium Valproate, Abbott 50711, ValdisovalValproic Acid
CAS Registry Number: 99-66-1
CAS Name: 2-Propylpentanoic acid
Additional Names: 2-propylvaleric acid; di-n-propylacetic acid
Trademarks: Convulex (Pharmacia); Depakene (Abbott)
Molecular Formula: C8H16O2
Molecular Weight: 144.21
Percent Composition: C 66.63%, H 11.18%, O 22.19%
Literature References: Antiepileptic; increases levels of g-aminobutyric acid (GABA) in the brain. Prepn: B. S. Burton, Am. Chem. J.3, 385 (1882); E. Oberreit, Ber.29, 1998 (1896); M. Tiffeneau, Y. Deux, Compte Rend.212, 105 (1941). Anticonvulsant activity: H. Meunier et al.,Therapie18, 435 (1963). Toxicity data: Jenner et al.,Food Cosmet. Toxicol.2, 327 (1964). Comprehensive description: Z. L. Chang, Anal. Profiles Drug Subs.8, 529-556 (1979). Review of teratogenicity studies: H. Nau et al.,Pharmacol. Toxicol.69, 310-321 (1991); R. Alsdorf, D. F. Wyszynski, Expert Opin. Drug Safety4, 345-353 (2005). Review of pharmacology and clinical experience in epilepsy: E. M. Rimmer, A. Richens, Pharmacotherapy5, 171-184 (1985); in psychiatric disease: D. R. P. Guay, ibid.15, 631-647 (1995); in migraine prophylaxis: C. E. Shelton, J. F. Connelly, Ann. Pharmacother.30, 865-866 (1996). Review of pharmacodynamics and mechanisms of action: W. Löscher, Prog. Neurobiol.58, 31-59 (1999).
Properties: Colorless liquid with characteristic odor. bp 219.5°. nD24.5 1.425. d40 0.9215. pKa 4.6. Very sol in organic solvents. Soly in water: 1.3 mg/ml. LD50 orally in rats: 670 mg/kg (Jenner).
Boiling point: bp 219.5°
pKa: pKa 4.6
Index of refraction:nD24.5 1.425
Density: d40 0.9215
Toxicity data: LD50 orally in rats: 670 mg/kg (Jenner)
Derivative Type: Sodium salt (1:1)
CAS Registry Number: 1069-66-5
Additional Names: Sodium valproate
Trademarks: Depacon (Abbott); Depakin (Sanofi-Synthelabo); Dépakine (Sanofi-Aventis); Epilim (Sanofi-Aventis); Ergenyl (Sanofi-Synthelabo); Leptilan (Dolorgiet); Orfiril (Desitin)
Molecular Formula: C8H15NaO2
Molecular Weight: 166.19
Percent Composition: C 57.82%, H 9.10%, Na 13.83%, O 19.25%
Properties: White, odorless, crystalline, deliquescent powder. pKa 4.8. Hygroscopic. One gram is sol in 0.4 ml water; 1.5 ml ethanol; 5 ml methanol. Practically insol in common organic solvents. LD50 orally in mice: 1700 mg/kg (Meunier).
pKa: pKa 4.8
Toxicity data: LD50 orally in mice: 1700 mg/kg (Meunier)
Derivative Type: Sodium salt (2:1)
CAS Registry Number: 76584-70-8
Additional Names: Sodium hydrogen bis(2-propylpentanoate); divalproex sodium; valproate semisodium
Manufacturers’ Codes: Abbott 50711
Trademarks: Depakote (Abbott); Valcote (Abbott)
Molecular Formula: C16H31NaO4
Molecular Weight: 310.40
Percent Composition: C 61.91%, H 10.07%, Na 7.41%, O 20.62%
Derivative Type: Magnesium salt
Trademarks: Depamag (Sigma-Tau)
Molecular Formula: C16H30MgO4
Molecular Weight: 310.71
Percent Composition: C 61.85%, H 9.73%, Mg 7.82%, O 20.60%
Therap-Cat: Anticonvulsant; antimanic; antimigraine.Keywords: Anticonvulsant; Antimigraine; Antimanic.
Synthesis Reference
Daniel Aubert, Francis Blanc, Henri Desmolin, Michel Morre, Lucette Sindely, “Valproic acid preparations.” U.S. Patent US5017613, issued January, 1965.
Patent
https://patents.google.com/patent/WO2007004238A2/enDivalproex sodium is one of the most widely used epileptic agents presently available in the market. Both the constituents, valproic acid and sodium valproate themselves have also been used for the treatment of epileptic seizures and convulsions. But their utility has remained restricted since valproic acid is a liquid and is difficult to formulate for an oral dosage form whereas sodium valproate is a hygroscopic solid with poor stability characteristics. Divalproex sodium is an oligomer having 1:1 molar ratio of valproic acid and sodium valproate containing 4 to 6 units. The relevant prior art includes US 4,988,731 (’73I) relates to a non-hygroscopic stable sodium hydrogen divalproate oligomer. Its probable structure is shown in Fig 1

Fig 1 – Divalproex sodiumWhere M is a about 2.As can be seen from the displayed structure, one mole each of the valproic acid forms coordinate bonds with the sodium of the sodium valproate molecule, and the valproate ion is ionically bonded to the sodium atom. The structure is thus consistent with the unique characteristic of the compound. However the preferred mode of representing Divalproex sodium is by reference to single compound of the formula{(CH3CH2CH2)2CHCO2} {(CH3CH2CH2)2CHCO2}Na, HThe said patent also describes two alternative processes for the preparation of the oligomer. According to one aspect, the oligomer is produced by dissolving sodium valproate and valproic acid in equimolar amount in acetone and crystallizing from chilled acetone at around O0C. Alternatively Divalproex sodium can be isolated from a two component liquid medium, which includes acetone where in half equivalent of NaOH to the valproic acid present, preferable as a solution in an acetone miscible solvent eg. water. The new compound can be recovered from the liquid phase by evaporating the solvent(s) and, if desired, the new compound can be recrystallized, for instance from acetonitrile or others or the material may be spay-dried, lyophilized or purified by chromatography.US ‘731 claims yield of 90% of theory.Drawbacks of the above mentioned reported methods for the preparation of Divalproex sodium described in US 4988731 are difficult to reproduce on a large scale and provides inconsistent yields and the material obtained is not always free flowing in nature. The process involves the crystallization of a 1:1 mixture of valproic acid and sodium valproate from a chilled solution of acetone, followed by washing with chilled acetone. Divalproex sodium is as such fairly soluble in acetone at temperatures above 1O0C and extreme care has to be. taken while performing washing with chilled acetone as any rise in temperature would lead to the loss of yield. This problem actually comes to the fore while scaling up the process during commercialization since during centrifugation of the large volume the temperature of the mixture rises and acetone has to be cooled below O0C, which require large amount of liquid nitrogen or dry ice. Moreover it was also observed that due to the cooled nature of the solvent, the isolated Divalproex sodium absorbs considerable amount of moisture and therefore requires longer time to dry eventually leading to longer time cycle for the otherwise simple single step process. Also the high moisture content in the recovered acetone makes it unsuitable for reuse. Alternatively, to avoid absorption of water, the centrifugation had to be carried out under a blanket of dry nitrogen gas. These additional infrastructural loads add to input costs eventually making the otherwise single step low cost process becoming uncompetitive and economically unviable.Similarly the other process involves the addition of half molar equivalent of sodium hydroxide dissolved in water to valproic acid and the solvent has to be evaporated to obtain crude product, which has to be recrystallized to get Divalprox of the desired specification. The process is operationally tedious and requires the reduction in the level of water in the reaction mass via evaporation of the solvent followed by re- crystallization from acetonitrile making the process lengthy and economically unviable. There is therefore a need for operationally making this single step process more efficient and high yieldingExample I:To lOOg of Valproic acid with stirring at 20-300C, powdered NaOH ( 13g; half molar) is added & the resulting reaction mixture is stirred at 40-500C for 1 hr. Then acetonitrile(600ml) is added to obtain clear solution at 40-500C and the solution is charcoalized at 40-500C followed by filtration at 40-500C through hyflo-bed. The resultant reaction mixture was stirred at 10-200C for 2-3 hr. The solid , thus obtained, was filtered and product was dried at 40-450C for 10-12 hr. (102.25g, 95%)Example II;To lOOg of Valproic acid with stirring at 20-300C, powdered NaOH (13g; half molar) is added & the resulting reaction mixture is stirred at 30-400C for 1 hr. Then acetone (600ml) is added to obtain clear solution at 30-400C and the material is charcoalized at 30-400C followed by filtration through hyflo-bed. The resultant reaction solution was stirred at -5°C to -1O0C for 2-3 hr. The solid , thus obtained, was filtered and product was dried at 40-450C for 10-12 hr. ( 55g, 51.11%) Example III:To a solution of Valproic acid (10Og) in dichloromethane (200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 30- 400C for 1 hr to get clear solution. Then acetonitrile (600ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (10Og; 93%)Example IV:To a solution of Valproic acid (10Og) in diisopropyl ether(200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (104g; 96.65%)Example V:To a solution of Valproic acid (10Og) in methyl tertiary butyl ether(200ml) at 20- 300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (102g;94.79%)Example VI:To a solution of Valproic acid (10Og) in toluene (200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (101g; 93.87%)Example VII: A mixture of sodium valproate (6Og) and valproic acid (52.04g) was taken in acetonitrile (800ml) and heated at reflux to obtain a clear solution, which was filtered through hyflo-bed to remove suspended particles. Then the solution was stirred at 10- 200C for 2-3 hr. The solid, thus obtained, was filtered and washed with acetonitrile (100ml). The product was dried at 40-450C for 10-12 hr. (105g ; 93.75%)Example VIII;To a solution of valproic acid (10Og) in methanol (200ml) at 20-300C5 caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the methanol was recovered at reduced pressure and acetonitrile (600ml) is added to it with stirring. The reaction mixture was further stirred at 0-50C for 2-3 hr. The solid, thus obtained, is filtered, washed with acetonitrile (100ml) and product was dried at 40-45°C for 10-12 hr.(102g; ~ 95%) Example IX:To a solution of valproic acid (10Og) in methanol (200ml) at 20-300C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the methanol was recovered at reduced pressure and acetone (600ml) is added to it with stirring. The reaction mixture was further stirred at -5°C to -1O0C for 2-3 hr. The solid, thus obtained, is filtered, washed with chilled acetone (100ml) and product was dried at 40-450C for 10-12 hr.(54g; ~ 50.11%)Example X:To a solution of valproic acid (10Og) in ethanol (200ml) at 20-300C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the ethanol was recovered at reduced pressure and acetonitrile (600ml) is added to it with stirring.The reaction mixture was further stirred at 0-50C for 2-3 hr. The solid, thus obtained, is filtered, washed with acetonitrile (100ml) and product was dried at 40-450C for 10-12 hr.(101g; ~ 93.87%)Example XI: To a solution of valproic acid (10Og) in ethanol (200ml) at 20-30°C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the ethanol was recovered at reduced pressure and acetone (600ml) is added to it with stirring. The reaction mixture was further stirred at -5°C to -100C for 2-3 hr. The solid, thus obtained, is filtered, washed with chilled acetone (100ml) and product was dried at 40-450C for 10-12 hr.(55g; ~ 51%)ADVANTAGES:> The process is high yielding. > The process produces Divalproex sodium with improved flowability.> The process produces Divalproex sodium that is non-hygroscopic and more stable.> The process is industrially feasible, precise, reproducible and does not require sophisticated infrastructure.
Divalproex Sodium is a stable coordination compound comprised of sodium valproate and valproic acid with anticonvulsant and antiepileptic activities. Divalproex dissociates to the valproate ion in the gastrointestinal tract. This agent binds to and inhibits gamma-aminobutyric acid (GABA) transaminase and its anticonvulsant activity may be exerted by increasing brain concentration of GABA and by inhibiting enzymes that catabolize GABA or block the reuptake of GABA into glia and nerve endings. Divalproex may also work by suppressing repetitive neuronal firing through inhibition of voltage-sensitive sodium channels.
Valproate semisodium is a mixture of valproic acid and its sodium salt in a 1:1 molar ratio. It is used for the management and treatment of seizure disorders, mania, and prophylactic treatment of migraine headache. It has a role as an antimanic drug, an anticonvulsant and a GABA agent. It contains a valproic acid and a sodium valproate.
Divalproex sodium, valproate sodium, and valproic acid, are all similar medications that are used by the body as valproic acid. Therefore, the term valproic acid will be used to represent all of these medications in this discussion.
Valproate (VPA) and its valproic acid, sodium valproate, and valproate semisodium forms are medications primarily used to treat epilepsy and bipolar disorder and prevent migraine headaches.[2] They are useful for the prevention of seizures in those with absence seizures, partial seizures, and generalized seizures.[2] They can be given intravenously or by mouth, and the tablet forms exist in both long- and short-acting formulations.[2]
Common side effects of valproate include nausea, vomiting, sleepiness, and dry mouth.[2] Serious side effects can include liver failure, and regular monitoring of liver function tests is therefore recommended.[2] Other serious risks include pancreatitis and an increased suicide risk.[2] Valproate is known to cause serious abnormalities in babies if taken during pregnancy,[2][3] and as such it is not typically recommended for women of childbearing age who have migraines.[2]
Valproate’s precise mechanism of action is unclear.[2][4] Proposed mechanisms include affecting GABA levels, blocking voltage-gated sodium channels, and inhibiting histone deacetylases.[5][6] Valproic acid is a branched short-chain fatty acid (SCFA) made from valeric acid.[5]
Valproate was first made in 1881 and came into medical use in 1962.[7] It is on the World Health Organization’s List of Essential Medicines[8] and is available as a generic medication.[2] It is marketed under the brand names Depakote, among others.[2] In 2018, it was the 131st most commonly prescribed medication in the United States, with more than 5 million prescriptions.[9][10]
Terminology
Valproic acid (VPA) is an organic weak acid. The conjugate base is valproate. The sodium salt of the acid is sodium valproate and a coordination complex of the two is known as valproate semisodium.[11]
Medical uses
It is used primarily to treat epilepsy and bipolar disorder. It is also used to prevent migraine headaches.[12]
Epilepsy
Valproate has a broad spectrum of anticonvulsant activity, although it is primarily used as a first-line treatment for tonic–clonic seizures, absence seizures and myoclonic seizures and as a second-line treatment for partial seizures and infantile spasms.[12][13] It has also been successfully given intravenously to treat status epilepticus.[14][15]
Mental illness
Bipolar disorder
Valproate products are also used to treat manic or mixed episodes of bipolar disorder.[16][17]
Schizophrenia
A 2016 systematic review compared the efficacy of valproate as an add-on for people with schizophrenia:[18]
| There is limited evidence that adding valproate to antipsychotics may be effective for overall response and also for specific symptoms, especially in terms of excitement and aggression. Valproate was associated with a number of adverse events among which sedation and dizziness appeared more frequently than in the control groups.[18] |
| showOutcomeFindings in wordsFindings in numbersQuality of evidence |
Dopamine dysregulation syndrome
Based upon five case reports, valproic acid may have efficacy in controlling the symptoms of the dopamine dysregulation syndrome that arise from the treatment of Parkinson’s disease with levodopa.[19][20][21]
Migraines
Valproate is also used to prevent migraine headaches. Because this medication can be potentially harmful to the fetus, valproate should be considered for those able to become pregnant only after the risks have been discussed.[22]
Other
The medication has been tested in the treatment of AIDS and cancer, owing to its histone-deacetylase-inhibiting effects.[23]
Contraindications
Contraindications include:
- Pre-existing acute or chronic liver dysfunction or family history of severe liver inflammation (hepatitis), particularly medicine related.[24]
- Known hypersensitivity to valproate or any of the ingredients used in the preparation[24]
- Urea cycle disorders[24]
- Hepatic porphyria[24]
- Hepatotoxicity[24]
- Mitochondrial disease[24]
- Pancreatitis[24]
- Porphyria[25]
Adverse effects
See also: List of adverse effects of valproic acid and List of adverse effects of valproate semisodium
Most common adverse effects include:[22]
- Nausea (22%)
- Drowsiness (19%)
- Dizziness (12%)
- Vomiting (12%)
- Weakness (10%)
Serious adverse effects include:[22]
- Bleeding
- Low blood platelets
- Encephalopathy
- Suicidal behavior and thoughts
- Low body temperature
Valproic acid has a black box warning for hepatotoxicity, pancreatitis, and fetal abnormalities.[22]
There is evidence that valproic acid may cause premature growth plate ossification in children and adolescents, resulting in decreased height.[26][27][28][29] Valproic acid can also cause mydriasis, a dilation of the pupils.[30] There is evidence that shows valproic acid may increase the chance of polycystic ovary syndrome (PCOS) in women with epilepsy or bipolar disorder. Studies have shown this risk of PCOS is higher in women with epilepsy compared to those with bipolar disorder.[31] Weight gain is also possible.[32]
Pregnancy
Valproate causes birth defects;[33] exposure during pregnancy is associated with about three times as many major abnormalities as usual, mainly spina bifida with the risks being related to the strength of medication used and use of more than one drug.[34][35] More rarely, with several other defects, including a “valproate syndrome”.[36] Characteristics of this valproate syndrome include facial features that tend to evolve with age, including a triangle-shaped forehead, tall forehead with bifrontal narrowing, epicanthic folds, medial deficiency of eyebrows, flat nasal bridge, broad nasal root, anteverted nares, shallow philtrum, long upper lip and thin vermillion borders, thick lower lip and small downturned mouth.[37] While developmental delay is usually associated with altered physical characteristics (dysmorphic features), this is not always the case.[38]
Children of mothers taking valproate during pregnancy are at risk for lower IQs.[39][40][41] Maternal valproate use during pregnancy increased the probability of autism in the offspring compared to mothers not taking valproate from 1.5% to 4.4%.[42] A 2005 study found rates of autism among children exposed to sodium valproate before birth in the cohort studied were 8.9%.[43] The normal incidence for autism in the general population is estimated at less than one percent.[44] A 2009 study found that the 3-year-old children of pregnant women taking valproate had an IQ nine points lower than that of a well-matched control group. However, further research in older children and adults is needed.[45][46][47]
Sodium valproate has been associated with paroxysmal tonic upgaze of childhood, also known as Ouvrier–Billson syndrome, from childhood or fetal exposure. This condition resolved after discontinuing valproate therapy.[48][49]
Women who intend to become pregnant should switch to a different medication if possible or decrease their dose of valproate.[50] Women who become pregnant while taking valproate should be warned that it causes birth defects and cognitive impairment in the newborn, especially at high doses (although valproate is sometimes the only drug that can control seizures, and seizures in pregnancy could have worse outcomes for the fetus than exposure to valproate). Studies have shown that taking folic acid supplements can reduce the risk of congenital neural tube defects.[22] The use of valproate for migraine or bipolar disorder during pregnancy is contraindicated in the European Union, and the medicines are not recommended for epilepsy during pregnancy unless there is no other effective treatment available.[51]
Elderly
Valproate in elderly people with dementia caused increased sleepiness. More people stopped the medication for this reason. Additional side effects of weight loss and decreased food intake were also associated with one-half of people who become sleepy.[22]
Overdose and toxicity
| Form | Lower limit | Upper limit | Unit |
| Total (including protein bound) | 50[52] | 125[52] | µg/mL or mg/l |
| 350[53] | 700[53] | μmol/L | |
| Free | 6[52] | 22[52] | µg/mL or mg/l |
| 35[53] | 70[53] | μmol/L |
Excessive amounts of valproic acid can result in sleepiness, tremor, stupor, respiratory depression, coma, metabolic acidosis, and death.[54] In general, serum or plasma valproic acid concentrations are in a range of 20–100 mg/l during controlled therapy, but may reach 150–1500 mg/l following acute poisoning. Monitoring of the serum level is often accomplished using commercial immunoassay techniques, although some laboratories employ gas or liquid chromatography.[55] In contrast to other antiepileptic drugs, at present there is little favorable evidence for salivary therapeutic drug monitoring. Salivary levels of valproic acid correlate poorly with serum levels, partly due to valproate’s weak acid property (pKa of 4.9).[56]
In severe intoxication, hemoperfusion or hemofiltration can be an effective means of hastening elimination of the drug from the body.[57][58] Supportive therapy should be given to all patients experiencing an overdose and urine output should be monitored.[22] Supplemental L-carnitine is indicated in patients having an acute overdose[59][60] and also prophylactically[59] in high risk patients. Acetyl-L-carnitine lowers hyperammonemia less markedly[61] than L-carnitine.
Interactions
Valproate inhibits CYP2C9, glucuronyl transferase, and epoxide hydrolase and is highly protein bound and hence may interact with drugs that are substrates for any of these enzymes or are highly protein bound themselves.[24] It may also potentiate the CNS depressant effects of alcohol.[24] It should not be given in conjunction with other antiepileptics due to the potential for reduced clearance of other antiepileptics (including carbamazepine, lamotrigine, phenytoin and phenobarbitone) and itself.[24] It may also interact with:[22][24][62]
- Aspirin: may increase valproate concentrations. May also interfere with valproate’s metabolism.
- Benzodiazepines: may cause CNS depression and there are possible pharmacokinetic interactions.
- Carbapenem antibiotics: reduce valproate levels, potentially leading to seizures.
- Cimetidine: inhibits valproate’s metabolism in the liver, leading to increased valproate concentrations.
- Erythromycin: inhibits valproate’s metabolism in the liver, leading to increased valproate concentrations.
- Ethosuximide: valproate may increase ethosuximide concentrations and lead to toxicity.
- Felbamate: may increase plasma concentrations of valproate.
- Mefloquine: may increase valproate metabolism combined with the direct epileptogenic effects of mefloquine.
- Oral contraceptives: may reduce plasma concentrations of valproate.
- Primidone: may accelerate metabolism of valproate, leading to a decline of serum levels and potential breakthrough seizure.
- Rifampicin: increases the clearance of valproate, leading to decreased valproate concentrations
- Warfarin: valproate may increase free warfarin concentration and prolong bleeding time.
- Zidovudine: valproate may increase zidovudine serum concentration and lead to toxicity.
Pharmacology
Pharmacodynamics
Although the mechanism of action of valproate is not fully understood,[24] traditionally, its anticonvulsant effect has been attributed to the blockade of voltage-gated sodium channels and increased brain levels of gamma-aminobutyric acid (GABA).[24] The GABAergic effect is also believed to contribute towards the anti-manic properties of valproate.[24] In animals, sodium valproate raises cerebral and cerebellar levels of the inhibitory synaptic neurotransmitter, GABA, possibly by inhibiting GABA degradative enzymes, such as GABA transaminase, succinate-semialdehyde dehydrogenase and by inhibiting the re-uptake of GABA by neuronal cells.[24]
Prevention of neurotransmitter-induced hyperexcitability of nerve cells, via Kv7.2 channel and AKAP5, may also contribute to its mechanism.[63] Also, it has been shown to protect against a seizure-induced reduction in phosphatidylinositol (3,4,5)-trisphosphate (PIP3) as a potential therapeutic mechanism.[64]
It also has histone-deacetylase-inhibiting effects. The inhibition of histone deacetylase, by promoting more transcriptionally active chromatin structures, likely presents the epigenetic mechanism for regulation of many of the neuroprotective effects attributed to valproic acid. Intermediate molecules mediating these effects include VEGF, BDNF, and GDNF.[65][66]
Endocrine actions
Valproic acid has been found to be an antagonist of the androgen and progesterone receptors, and hence as a nonsteroidal antiandrogen and antiprogestogen, at concentrations much lower than therapeutic serum levels.[67] In addition, the drug has been identified as a potent aromatase inhibitor, and suppresses estrogen concentrations.[68] These actions are likely to be involved in the reproductive endocrine disturbances seen with valproic acid treatment.[67][68]
Valproic acid has been found to directly stimulate androgen biosynthesis in the gonads via inhibition of histone deacetylases and has been associated with hyperandrogenism in women and increased 4-androstenedione levels in men.[69][70] High rates of polycystic ovary syndrome and menstrual disorders have also been observed in women treated with valproic acid.[70]
Pharmacokinetics

Some metabolites of valproic acid. Glucuronidation and β-oxidation are the main metabolic pathways; ω-oxidation metabolites are considered hepatotoxic.[71][72] Details see text.
Taken by mouth, valproate is rapidly and virtually completely absorbed from the gut.[71] When in the bloodstream, 80–90% of the substance are bound to plasma proteins, mainly albumin. Protein binding is saturable: it decreases with increasing valproate concentration, low albumin concentrations, the patient’s age, additional use of other drugs such as aspirin, as well as liver and kidney impairment.[73][74] Concentrations in the cerebrospinal fluid and in breast milk are 1 to 10% of blood plasma concentrations.[71]
The vast majority of valproate metabolism occurs in the liver.[75] Valproate is known to be metabolized by the cytochrome P450 enzymes CYP2A6, CYP2B6, CYP2C9, and CYP3A5.[75] It is also known to be metabolized by the UDP-glucuronosyltransferase enzymes UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7, and UGT2B15.[75] Some of the known metabolites of valproate by these enzymes and uncharacterized enzymes include (see image):[75]
- via glucuronidation (30–50%): valproic acid β-O-glucuronide
- via beta oxidation (>40%): 2E-ene-valproic acid, 2Z-ene-valproic acid, 3-hydroxyvalproic acid, 3-oxovalproic acid
- via omega oxidation: 5-hydroxyvalproic acid, 2-propyl-glutaric acid
- some others: 3E-ene-valproic acid, 3Z-ene-valproic acid, 4-ene-valproic acid, 4-hydroxyvalproic acid
All in all, over 20 metabolites are known.[71]
In adult patients taking valproate alone, 30–50% of an administered dose is excreted in urine as the glucuronide conjugate.[75] The other major pathway in the metabolism of valproate is mitochondrial beta oxidation, which typically accounts for over 40% of an administered dose.[75] Typically, less than 20% of an administered dose is eliminated by other oxidative mechanisms.[75] Less than 3% of an administered dose of valproate is excreted unchanged (i.e., as valproate) in urine.[75] Only a small amount is excreted via the faeces.[71] Elimination half-life is 16±3 hours and can decrease to 4–9 hours when combined with enzyme inducers.[71][74]
Chemistry
Valproic acid is a branched short-chain fatty acid and the 2-n–propyl derivative of valeric acid.[5]
History
Valproic acid was first synthesized in 1882 by Beverly S. Burton as an analogue of valeric acid, found naturally in valerian.[76] Valproic acid is a carboxylic acid, a clear liquid at room temperature. For many decades, its only use was in laboratories as a “metabolically inert” solvent for organic compounds. In 1962, the French researcher Pierre Eymard serendipitously discovered the anticonvulsant properties of valproic acid while using it as a vehicle for a number of other compounds that were being screened for antiseizure activity. He found it prevented pentylenetetrazol-induced convulsions in laboratory rats.[77] It was approved as an antiepileptic drug in 1967 in France and has become the most widely prescribed antiepileptic drug worldwide.[78] Valproic acid has also been used for migraine prophylaxis and bipolar disorder.[79]
Society and culture
Valproate is available as a generic medication.[2]
Approval status
| Indications | FDA-labelled indication?[1] | TGA-labelled indication?[12] | MHRA-labelled indication?[80] | Literature support |
|---|---|---|---|---|
| Epilepsy | Yes | Yes | Yes | Limited (depends on the seizure type; it can help with certain kinds of seizures: drug-resistant epilepsy, partial and absence seizures, can be used against glioblastoma and other tumors both to improve survival and treat seizures, and against tonic–clonic seizures and status epilepticus).[81][82][83][84] |
| Bipolar mania | Yes | Yes | Yes | Limited.[85] |
| Bipolar depression | No | No | No | Moderate.[86] |
| Bipolar maintenance | No | No | No | Limited.[87] |
| Migraine prophylaxis | Yes | Yes (accepted) | No | Limited. |
| Acute migraine management | No | No | No | Only negative results.[88] |
| Schizophrenia | No | No | No | Weak evidence.[89] |
| Agitation in dementia | No | No | No | Weak evidence. Not recommended for agitation in people with dementia.[90] Increased rate of adverse effects, including a risk of serious adverse effects.[90] |
| Fragile X syndrome | Yes (orphan) | No | No | Limited.[66] |
| Familial adenomatous polyposis | Yes (orphan) | No | No | Limited. |
| Chronic pain & fibromyalgia | No | No | No | Limited.[91] |
| Alcohol hallucinosis | No | No | No | One randomised double-blind placebo-controlled trial.[92] |
| Intractable hiccups | No | No | No | Limited, five case reports support its efficacy, however.[93] |
| Non-epileptic myoclonus | No | No | No | Limited, three case reports support its efficacy, however.[94] |
| Cluster headaches | No | No | No | Limited, two case reports support its efficacy.[95] |
| West syndrome | No | No | No | A prospective clinical trial supported its efficacy in treating infantile spasms.[96] |
| HIV infection eradication | No | No | No | Double-blind placebo-controlled trials have been negative.[97][98][99] |
| Myelodysplastic syndrome | No | No | No | Several clinical trials have confirmed its efficacy as a monotherapy,[100] as an adjunct to tretinoin[100] and as an adjunct to hydralazine.[101] |
| Acute myeloid leukaemia | No | No | No | Two clinical trials have confirmed its efficacy in this indication as both a monotherapy and as an adjunct to tretinoin.[102][103][104] |
| Cervical cancer | No | No | No | One clinical trial supports its use here.[105] |
| Malignant melanoma | No | No | No | One phase II study has seemed to discount its efficacy.[106] |
| Breast cancer | No | No | No | A phase II study has supported its efficacy.[107] |
| Impulse control disorder | No | No | No | Limited.[108][109] |
Off-label uses
In 2012, pharmaceutical company Abbott paid $1.6 billion in fines to US federal and state governments for illegal promotion of off-label uses for Depakote, including the sedation of elderly nursing home residents.[110][111]
Some studies have suggested that valproate may reopen the critical period for learning absolute pitch and possibly other skills such as language.[112][113]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Formulations
| Clinical data | |
|---|---|
| Other names | valproate sodium (USAN US) |
| License data | US DailyMed: Valproate_sodium |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1069-66-5 |
| PubChem CID | 16760703 |
| DrugBank | DBSALT001257 |
| ChemSpider | 13428 |
| UNII | 5VOM6GYJ0D |
| KEGG | D00710 |
| ChEBI | CHEBI:9925 |
| ChEMBL | ChEMBL433 |
| CompTox Dashboard (EPA) | DTXSID6023733 |
| ECHA InfoCard | 100.002.525 |
| Chemical and physical data | |
| Formula | C8H15NaO2 |
| Molar mass | 166.196 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
| Clinical data | |
|---|---|
| Trade names | Depakote, others |
| Other names | semisodium valproate, divalproex sodium (USAN US) |
| License data | US DailyMed: Divalproex_sodium |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 76584-70-8 |
| PubChem CID | 23663956 |
| DrugBank | DBSALT000185 |
| ChemSpider | 48337 |
| UNII | 644VL95AO6 |
| KEGG | D00304 |
| ChEBI | CHEBI:4667 |
| ChEMBL | ChEMBL2105613 |
| CompTox Dashboard (EPA) | DTXSID6023733 |
| ECHA InfoCard | 100.002.525 |
| Chemical and physical data | |
| Formula | C16H31NaO4 |
| Molar mass | 310.410 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Valproate exists in two main molecular variants: sodium valproate and valproic acid without sodium (often implied by simply valproate). A mixture between these two is termed semisodium valproate. It is unclear whether there is any difference in efficacy between these variants, except from the fact that about 10% more mass of sodium valproate is needed than valproic acid without sodium to compensate for the sodium itself.[114]
Brand names of valproic acid
Branded products include:
- Absenor (Orion Corporation Finland)
- Convulex (G.L. Pharma GmbH Austria)
- Depakene (Abbott Laboratories in US and Canada)[115]
- Depakine (Sanofi Aventis France)
- Depakine (Sanofi Synthelabo Romania)
- Depalept (Sanofi Aventis Israel)
- Deprakine (Sanofi Aventis Finland)
- Encorate (Sun Pharmaceuticals India)
- Epival (Abbott Laboratories US and Canada)
- Epilim (Sanofi Synthelabo Australia and South Africa)
- Stavzor (Noven Pharmaceuticals Inc.)
- Valcote (Abbott Laboratories Argentina)
- Valpakine (Sanofi Aventis Brazil)
- Orfiril (Desitin Arzneimittel GmbH Norway)
Brand names of sodium valproate
Portugal
- Tablets – Diplexil-R by Bial.
United States
- Intravenous injection – Depacon by Abbott Laboratories.
- Syrup – Depakene by Abbott Laboratories. (Note Depakene capsules are valproic acid).
- Depakote tablets are a mixture of sodium valproate and valproic acid.
- Tablets – Eliaxim by Bial.
Australia
- Epilim Crushable Tablets Sanofi[116]
- Epilim Sugar Free Liquid Sanofi[116]
- Epilim Syrup Sanofi[116]
- Epilim Tablets Sanofi[116]
- Sodium Valproate Sandoz Tablets Sanofi
- Valpro Tablets Alphapharm
- Valproate Winthrop Tablets Sanofi
- Valprease tablets Sigma
New Zealand
- Epilim by Sanofi-Aventis
All the above formulations are Pharmac-subsidised.[117]
UK
- Depakote Tablets (as in USA)
- Tablets – Orlept by Wockhardt and Epilim by Sanofi
- Oral solution – Orlept Sugar Free by Wockhardt and Epilim by Sanofi
- Syrup – Epilim by Sanofi-Aventis
- Intravenous injection – Epilim Intravenous by Sanofi
- Extended release tablets – Epilim Chrono by Sanofi is a combination of sodium valproate and valproic acid in a 2.3:1 ratio.
- Enteric-coated tablets – Epilim EC200 by Sanofi is a 200-mg sodium valproate enteric-coated tablet.
UK only
- Capsules – Episenta prolonged release by Beacon
- Sachets – Episenta prolonged release by Beacon
- Intravenous solution for injection – Episenta solution for injection by Beacon
Germany, Switzerland, Norway, Finland, Sweden
- Tablets – Orfiril by Desitin Pharmaceuticals
- Intravenous injection – Orfiril IV by Desitin Pharmaceuticals
South Africa
- Syrup – Convulex by Byk Madaus[118]
- Tablets – Epilim by Sanofi-synthelabo
Malaysia
- Tablets – Epilim by Sanofi-Aventis
Romania
- Companies are SANOFI-AVENTIS FRANCE, GEROT PHARMAZEUTIKA GMBH and DESITIN ARZNEIMITTEL GMBH
- Types are Syrup, Extended release mini tablets, Gastric resistant coated tablets, Gastric resistant soft capsules, Extended release capsules, Extended release tablets and Extended release coated tablets
Canada
- Intravenous injection – Epival or Epiject by Abbott Laboratories.
- Syrup – Depakene by Abbott Laboratories its generic formulations include Apo-Valproic and ratio-Valproic.
Japan
- Tablets – Depakene by Kyowa Hakko Kirin
- Extended release tablets – Depakene-R by Kyowa Hakko Kogyo and Selenica-R by Kowa
- Syrup – Depakene by Kyowa Hakko Kogyo
Europe
In much of Europe, Dépakine and Depakine Chrono (tablets) are equivalent to Epilim and Epilim Chrono above.
Taiwan
- Tablets (white round tablet) – Depakine (Chinese: 帝拔癲; pinyin: di-ba-dian) by Sanofi Winthrop Industrie (France)
Iran
- Tablets – Epival 200 (enteric coated tablet) and Epival 500 (extended release tablet) by Iran Najo
- Slow release tablets – Depakine Chrono by Sanofi Winthrop Industrie (France)
Israel
Depalept and Depalept Chrono (extended release tablets) are equivalent to Epilim and Epilim Chrono above. Manufactured and distributed by Sanofi-Aventis.
India, Russia and CIS countries
- Valparin Chrono by Torrent Pharmaceuticals India
- Valprol CR by Intas Pharmaceutical (India)
- Encorate Chrono by Sun Pharmaceutical (India)
- Serven Chrono by Leeven APL Biotech (India)
Brand names of valproate semisodium
- Brazil – Depakote by Abbott Laboratories and Torval CR by Torrent do Brasil
- Canada – Epival by Abbott Laboratories
- Mexico – Epival and Epival ER (extended release) by Abbott Laboratories
- United Kingdom – Depakote (for psychiatric conditions) and Epilim (for epilepsy) by Sanofi-Aventis and generics
- United States – Depakote and Depakote ER (extended release) by Abbott Laboratories and generics[22]
- India – Valance and Valance OD by Abbott Healthcare Pvt Ltd, Divalid ER by Linux laboratories Pvt Ltd, Valex ER by Sigmund Promedica, Dicorate by Sun Pharma
- Germany – Ergenyl Chrono by Sanofi-Aventis and generics
- Chile – Valcote and Valcote ER by Abbott Laboratories
- France and other European countries — Depakote
- Peru – Divalprax by AC Farma Laboratories
- China – Diprate OD
References
- ^ Jump up to:a b c d “Depakene, Stavzor (valproic acid) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Archived from the original on 21 February 2014. Retrieved 13 February 2014.
- ^ Jump up to:a b c d e f g h i j k l “Valproic Acid”. The American Society of Health-System Pharmacists. Archived from the original on 2017-07-31. Retrieved Oct 23, 2015.
- ^ “Valproate banned without the pregnancy prevention programme”. GOV.UK. Retrieved 26 April 2018.
- ^ Owens MJ, Nemeroff CB (2003). “Pharmacology of valproate”. Psychopharmacol Bull. 37 Suppl 2: 17–24. PMID 14624230.
- ^ Jump up to:a b c Ghodke-Puranik Y, Thorn CF, Lamba JK, Leeder JS, Song W, Birnbaum AK, Altman RB, Klein TE (April 2013). “Valproic acid pathway: pharmacokinetics and pharmacodynamics”. Pharmacogenet. Genomics. 23 (4): 236–241. doi:10.1097/FPC.0b013e32835ea0b2. PMC 3696515. PMID 23407051.
- ^ “Valproic acid”. DrugBank. University of Alberta. 29 July 2017. Archived from the original on 31 July 2017. Retrieved 30 July2017.
- ^ Scott, D.F. (1993). The history of epileptic therapy : an account of how medication was developed (1. publ. ed.). Carnforth u.a.: Parthenon Publ. Group. p. 131. ISBN 9781850703914.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Divalproex Sodium – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ Brayfield, Alison (ed.). Martindale: The Complete Drug Reference. London: Pharmaceutical Press. Retrieved March 3,2018.
- ^ Jump up to:a b c Rossi, S, ed. (2013). Australian Medicines Handbook(2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN 978-0-9805790-9-3.
- ^ Löscher W (2002). “Basic pharmacology of valproate: a review after 35 years of clinical use for the treatment of epilepsy”. CNS Drugs. 16 (10): 669–694. doi:10.2165/00023210-200216100-00003. PMID 12269861. S2CID 67999301.
- ^ Olsen KB, Taubøll E, Gjerstad L (2007). “Valproate is an effective, well-tolerated drug for treatment of status epilepticus/serial attacks in adults”. Acta Neurol. Scand. Suppl. 187: 51–4. doi:10.1111/j.1600-0404.2007.00847.x. PMID 17419829. S2CID 11159645.
- ^ Kwan SY (2010). “The role of intravenous valproate in convulsive status epilepticus in the future” (PDF). Acta Neurol Taiwan. 19(2): 78–81. PMID 20830628.[permanent dead link]
- ^ “Valproate Information”. Fda.gov. Archived from the original on 2015-05-03. Retrieved 2015-04-24.
- ^ Jochim, Janina; Rifkin-Zybutz, Raphael; Geddes, John; Cipriani, Andrea (7 October 2019). “Valproate for acute mania”. Cochrane Database of Systematic Reviews. 10: CD004052. doi:10.1002/14651858.CD004052.pub2. PMC 6797024. PMID 31621892.
- ^ Jump up to:a b Wang, Y; Xia, J; Helfer, B (2016). “Valproate for schizophrenia”. Cochrane Database of Systematic Reviews. 11: CD004028.pub4. doi:10.1002/14651858.CD004028.pub4. PMC 6734130. PMID 27884042. Archived from the originalon 2017-07-29. Retrieved 2017-07-27.
- ^ Pirritano D, Plastino M, Bosco D, Gallelli L, Siniscalchi A, De Sarro G (2014). “Gambling disorder during dopamine replacement treatment in Parkinson’s disease: a comprehensive review”. Biomed Res Int. 2014: 1–9. doi:10.1155/2014/728038. PMC 4119624. PMID 25114917.
- ^ Connolly B, Fox SH (2014). “Treatment of cognitive, psychiatric, and affective disorders associated with Parkinson’s disease”. Neurotherapeutics. 11 (1): 78–91. doi:10.1007/s13311-013-0238-x. PMC 3899484. PMID 24288035.
- ^ Averbeck BB, O’Sullivan SS, Djamshidian A (2014). “Impulsive and compulsive behaviors in Parkinson’s disease”. Annu Rev Clin Psychol. 10: 553–80. doi:10.1146/annurev-clinpsy-032813-153705. PMC 4197852. PMID 24313567.
- ^ Jump up to:a b c d e f g h i “Depakote- divalproex sodium tablet, delayed release”. Archived from the original on 5 March 2016. Retrieved 10 November 2015.
- ^ Činčárová L, Zdráhal Z, Fajkus J (2013). “New perspectives of valproic acid in clinical practice”. Expert Opin Investig Drugs. 22(12): 1535–1547. doi:10.1517/13543784.2013.853037. PMID 24160174. S2CID 11855893.
- ^ Jump up to:a b c d e f g h i j k l m n o “Valpro sodium valproate” (PDF). TGA eBusiness Services. Alphapharm Pty Limited. 16 December 2013. Retrieved 14 February 2014.
- ^ “Depakote 250mg Tablets – Summary of Product Characteristics”. electronic Medicines Compendium. Sanofi. 28 November 2013. Archived from the original on 1 February 2014. Retrieved 18 January 2014.
- ^ Wu S, Legido A, De Luca F (2004). “Effects of valproic acid on longitudinal bone growth”. J Child Neurol. 19 (1): 26–30. doi:10.1177/088307380401900105011. PMID 15032379. S2CID 19827846.
- ^ Robinson PB, Harvey W, Belal MS (1988). “Inhibition of cartilage growth by the anticonvulsant drugs diphenylhydantoin and sodium valproate”. Br J Exp Pathol. 69 (1): 17–22. PMC 2013195. PMID 3126792.
- ^ Guo CY, Ronen GM, Atkinson SA (2002). “Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy”. Epilepsia. 42 (9): 1141–7. doi:10.1046/j.1528-1157.2001.416800.x. PMID 11580761. S2CID 25499280.
- ^ Guo CY, Ronen GM, Atkinson SA (2002). “Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy”. Epilepsia. 42 (9): 1141–7. doi:10.1046/j.1528-1157.2001.416800.x. PMID 11580761. S2CID 25499280.
- ^ “Could Depakote cause Mydriasis”. eHealthMe.com. 2014-11-18. Archived from the original on 2014-12-05. Retrieved 2015-04-24.
- ^ Bilo, Leonilda; Meo, Roberta (October 2008). “Polycystic ovary syndrome in women using valproate: a review”. Gynecological Endocrinology. 24 (10): 562–70. doi:10.1080/09513590802288259. PMID 19012099. S2CID 36426338.
- ^ Chukwu, J; Delanty, N; Webb, D; Cavalleri, GL (January 2014). “Weight change, genetics and antiepileptic drugs”. Expert Review of Clinical Pharmacology. 7 (1): 43–51. doi:10.1586/17512433.2014.857599. PMID 24308788. S2CID 33444886.
- ^ New evidence in France of harm from epilepsy drug valproateArchived 2017-04-21 at the Wayback Machine BBC, 2017
- ^ Koch S, Göpfert-Geyer I, Jäger-Roman E, et al. (February 1983). “[Anti-epileptic agents during pregnancy. A prospective study on the course of pregnancy, malformations and child development]”. Dtsch. Med. Wochenschr. (in German). 108 (7): 250–7. doi:10.1055/s-2008-1069536. PMID 6402356.
- ^ Moore SJ, Turnpenny P, Quinn A, et al. (July 2000). “A clinical study of 57 children with fetal anticonvulsant syndromes”. J. Med. Genet. 37 (7): 489–97. doi:10.1136/jmg.37.7.489. PMC 1734633. PMID 10882750.
- ^ Ornoy A (2009). “Valproic acid in pregnancy: how much are we endangering the embryo and fetus?”. Reprod. Toxicol. 28 (1): 1–10. doi:10.1016/j.reprotox.2009.02.014. PMID 19490988.
- ^ Kulkarni ML, Zaheeruddin M, Shenoy N, Vani HN (2006). “Fetal valproate syndrome”. Indian J Pediatr. 73 (10): 937–939. doi:10.1007/bf02859291. PMID 17090909. S2CID 38371596.
- ^ Adab N, Kini U, Vinten J, et al. (November 2004). “The longer term outcome of children born to mothers with epilepsy”. J. Neurol. Neurosurg. Psychiatry. 75 (11): 1575–83. doi:10.1136/jnnp.2003.029132. PMC 1738809. PMID 15491979.
This argues that the fetal valproate syndrome constitutes a real clinical entity that includes developmental delay and cognitive impairments, but that some children might exhibit some developmental delay without marked dysmorphism.
- ^ Umur AS, Selcuki M, Bursali A, Umur N, Kara B, Vatansever HS, Duransoy YK (2012). “Simultaneous folate intake may prevent adverse effect of valproic acid on neurulating nervous system”. Childs Nerv Syst. 28 (5): 729–737. doi:10.1007/s00381-011-1673-9. PMID 22246336. S2CID 20344828.
- ^ Cassels, Caroline (December 8, 2006). “NEAD: In Utero Exposure To Valproate Linked to Poor Cognitive Outcomes in Kids”. Medscape. Archived from the original on July 31, 2011. Retrieved 2007-05-23.
- ^ Meador KJ, Baker GA, Finnell RH, Kalayjian LA, Liporace JD, Loring DW, Mawer G, Pennell PB, Smith JC, Wolff MC (2006). “In utero antiepileptic drug exposure: fetal death and malformations”. Neurology. 67 (3): 407–412. doi:10.1212/01.wnl.0000227919.81208.b2. PMC 1986655. PMID 16894099.
- ^ Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, Vestergaard M (2013). “Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism”. JAMA. 309 (16): 1696–1703. doi:10.1001/jama.2013.2270. PMC 4511955. PMID 23613074.
- ^ Rasalam AD, Hailey H, Williams JH, et al. (August 2005). “Characteristics of fetal anticonvulsant syndrome associated autistic disorder”. Dev Med Child Neurol. 47 (8): 551–5. doi:10.1017/S0012162205001076. PMID 16108456.
- ^ Autism Society of America: About Autism Archived 2011-01-10 at the Wayback Machine
- ^ I.Q. Harmed by Epilepsy Drug in Utero Archived 2015-12-29 at the Wayback Machine By RONI CARYN RABIN, New York Times, April 15, 2009
- ^ Meador KJ, Baker GA, Browning N, Clayton-Smith J, Combs-Cantrell DT, Cohen M, Kalayjian LA, Kanner A, Liporace JD, Pennell PB, Privitera M, Loring DW (2009). “Cognitive function at 3 years of age after fetal exposure to antiepileptic drugs”. N. Engl. J. Med. 360 (16): 1597–1605. doi:10.1056/NEJMoa0803531. PMC 2737185. PMID 19369666.
- ^ Valproate Products: Drug Safety Communication – Risk of Impaired Cognitive Development in Children Exposed In Utero (During Pregnancy) Archived 2011-09-02 at the Wayback Machine. FDA. June 2011
- ^ Luat, AF (20 September 2007). “Paroxysmal tonic upgaze of childhood with co-existent absence epilepsy”. Epileptic Disorders. 9(3): 332–6. doi:10.1684/epd.2007.0119 (inactive 31 May 2021). PMID 17884759.
- ^ Ouvrier, RA (July 1988). “Benign paroxysmal tonic upgaze of childhood”. Journal of Child Neurology. 3 (3): 177–80. doi:10.1177/088307388800300305. PMID 3209843. S2CID 38127378.
- ^ Valproate Not To Be Used for Migraine During Pregnancy, FDA Warns Archived 2013-07-09 at the Wayback Machine
- ^ “New measures to avoid valproate exposure in pregnancy endorsed”. European Medicines Agency. 31 May 2018.
- ^ Jump up to:a b c d Bentley, Suzanne (Dec 11, 2013). “Valproic Acid Level”. Medscape. Archived from the original on 2015-05-04. Retrieved 2015-06-05.
- ^ Jump up to:a b c d “Free Valproic Acid Assay (Reference – 2013.03.006) Notice of Assessment” (PDF). Canadian Agency for Drugs and Technologies in Health (CADTH) with INESSS’s permission. April 2014. Archived from the original (PDF) on 2016-03-03. Retrieved 2015-06-05.
- ^ Rissardo, Jamir Pitton; Caprara, Ana Letícia Fornari; Durante, Ícaro (2021). “Valproate-associated Movement Disorder: A Literature Review”. Prague Medical Report. 122 (3): 140–180. doi:10.14712/23362936.2021.14. ISSN 1214-6994.
- ^ Sztajnkrycer MD (2002). “Valproic acid toxicity: overview and management”. J. Toxicol. Clin. Toxicol. 40 (6): 789–801. doi:10.1081/CLT-120014645. PMID 12475192. S2CID 23031095.
- ^ Patsalos PN, Berry DJ (2013). “Therapeutic drug monitoring of antiepileptic drugs by use of saliva”. Ther Drug Monit. 35 (1): 4–29. doi:10.1097/FTD.0b013e31827c11e7. PMID 23288091. S2CID 15338188.
- ^ Thanacoody RH (2009). “Extracorporeal elimination in acute valproic acid poisoning”. Clin Toxicol. 47 (7): 609–616. doi:10.1080/15563650903167772. PMID 19656009. S2CID 13592043.
- ^ R. Baselt, Disposition of Toxic Drugs and Chemicals in Man, 8th edition, Biomedical Publications, Foster City, CA, 2008, pp. 1622–1626.
- ^ Jump up to:a b Lheureux PE, Penaloza A, Zahir S, Gris M (2005). “Science review: carnitine in the treatment of valproic acid-induced toxicity – what is the evidence?”. Crit Care. 9 (5): 431–440. doi:10.1186/cc3742. PMC 1297603. PMID 16277730.
- ^ Mock CM, Schwetschenau KH (2012). “Levocarnitine for valproic-acid-induced hyperammonemic encephalopathy”. Am J Health Syst Pharm. 69 (1): 35–39. doi:10.2146/ajhp110049. PMID 22180549.
- ^ Matsuoka M, Igisu H (1993). “Comparison of the effects of L-carnitine, D-carnitine and acetyl-L-carnitine on the neurotoxicity of ammonia”. Biochem. Pharmacol. 46 (1): 159–164. doi:10.1016/0006-2952(93)90360-9. PMID 8347126.
- ^ Herzog, Andrew; Farina, Erin (June 9, 2005). “Serum Valproate Levels with Oral Contraceptive Use”. Epilepsia. 46 (6): 970–971. doi:10.1111/j.1528-1167.2005.00605.x. PMID 15946343. S2CID 7696039.
- ^ Kay, Hee Yeon; Greene, Derek L.; Kang, Seungwoo; Kosenko, Anastasia; Hoshi, Naoto (2015-10-01). “M-current preservation contributes to anticonvulsant effects of valproic acid”. The Journal of Clinical Investigation. 125 (10): 3904–3914. doi:10.1172/JCI79727. ISSN 0021-9738. PMC 4607138. PMID 26348896.
- ^ Chang P, Walker MC, Williams RS (2014). “Seizure-induced reduction in PIP3 levels contributes to seizure-activity and is rescued by valproic acid”. Neurobiol. Dis. 62: 296–306. doi:10.1016/j.nbd.2013.10.017. PMC 3898270. PMID 24148856.
- ^ Kostrouchová M, Kostrouch Z, Kostrouchová M (2007). “Valproic acid, a molecular lead to multiple regulatory pathways” (PDF). Folia Biol. (Praha). 53 (2): 37–49. PMID 17448293. Archived from the original (PDF) on 2014-02-21. Retrieved 2014-02-13.
- ^ Jump up to:a b Chiu CT, Wang Z, Hunsberger JG, Chuang DM (2013). “Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder”. Pharmacol. Rev. 65 (1): 105–142. doi:10.1124/pr.111.005512. PMC 3565922. PMID 23300133.
- ^ Jump up to:a b Death AK, McGrath KC, Handelsman DJ (2005). “Valproate is an anti-androgen and anti-progestin” (PDF). Steroids. 70 (14): 946–53. doi:10.1016/j.steroids.2005.07.003. hdl:10453/16875. PMID 16165177. S2CID 25958985.
- ^ Jump up to:a b Wyllie E, Cascino GD, Gidal BE, Goodkin HP (17 February 2012). Wyllie’s Treatment of Epilepsy: Principles and Practice. Lippincott Williams & Wilkins. pp. 288–. ISBN 978-1-4511-5348-4. Archived from the original on 6 June 2014.
- ^ Uchida, Hiroshi; Maruyama, Tetsuo; Arase, Toru; Ono, Masanori; Nagashima, Takashi; Masuda, Hirotaka; Asada, Hironori; Yoshimura, Yasunori (2005). “Histone acetylation in reproductive organs: Significance of histone deacetylase inhibitors in gene transcription”. Reproductive Medicine and Biology. 4 (2): 115–122. doi:10.1111/j.1447-0578.2005.00101.x. ISSN 1445-5781. PMC 5891791. PMID 29662388.
- ^ Jump up to:a b Isojärvi, Jouko I T; Taubøll, Erik; Herzog, Andrew G (2005). “Effect of Antiepileptic Drugs on Reproductive Endocrine Function in Individuals with Epilepsy”. CNS Drugs. 19 (3): 207–223. doi:10.2165/00023210-200519030-00003. ISSN 1172-7047. PMID 15740176. S2CID 9893959.
- ^ Jump up to:a b c d e f Haberfeld H, ed. (2021). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Depakine chrono retard 300 mg Filmtabletten.
- ^ Kumar, S.; Wong, H.; Yeung, S. A.; Riggs, K. W.; Abbott, F. S.; Rurak, D. W. (2000). “Disposition of valproic acid in maternal, fetal, and newborn sheep. II: Metabolism and renal elimination”. Drug Metabolism and Disposition: The Biological Fate of Chemicals. 28(7): 857–864. PMID 10859160.
- ^ Schneemann; Young, Lloyd; Koda-Kimble, Mary Anne, eds. (2001). Angewandte Arzneimitteltherapie (in German). Springer. pp. 28–29. ISBN 3540413561.
- ^ Jump up to:a b Valproate FDA Professional Drug Information. Accessed 2021-08-06.
- ^ Jump up to:a b c d e f g h “Pharmacology”. Valproic Acid. DrugBank. University of Alberta. 31 August 2017.
- ^ Burton BS (1882). “On the propyl derivatives and decomposition products of ethylacetoacetate”. Am. Chem. J. 3: 385–395.
- ^ Meunier H, Carraz G, Neunier Y, Eymard P, Aimard M (1963). “[Pharmacodynamic properties of N-dipropylacetic acid]” [Pharmacodynamic properties of N-dipropylacetic acid]. Thérapie (in French). 18: 435–438. PMID 13935231.
- ^ Perucca E (2002). “Pharmacological and therapeutic properties of valproate: a summary after 35 years of clinical experience”. CNS Drugs. 16 (10): 695–714. doi:10.2165/00023210-200216100-00004. PMID 12269862. S2CID 803106.
- ^ Henry TR (2003). “The history of valproate in clinical neuroscience”. Psychopharmacol Bull. 37 Suppl 2: 5–16. PMID 14624229.
- ^ Joint Formulary Committee (2013). British National Formulary (BNF) (65 ed.). London, UK: Pharmaceutical Press. ISBN 978-0-85711-084-8.
- ^ Rimmer EM, Richens A (May–June 1985). “An update on sodium valproate”. Pharmacotherapy. 5 (3): 171–84. doi:10.1002/j.1875-9114.1985.tb03413.x. PMID 3927267. S2CID 7700266.
- ^ Glauser, Tracy A; Cnaan, Avital; Shinnar, Shlomo; Hirtz, Deborah G; Dlugos, Dennis; Masur, David; Clark, Peggy O; Capparelli, Edmund V; Adamson, Peter C (2010). “Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy”. New England Journal of Medicine. 362 (9): 790–9. doi:10.1056/NEJMoa0902014. PMC 2924476. PMID 20200383.
- ^ Jiang, Mei (6 April 2015). “Co-Administration of Valproic Acid and Lamotrigine in the Treatment of Refractory Epilepsy (P1.238)”. Neurology. 84 (14 Supplement): P1.238 – via http://www.neurology.org.
- ^ Berendsen, S.; Kroonen, J.; Seute, T.; Snijders, T.; Broekman, M. L. D.; Spliet, W. G. M.; Willems, M.; Artesi, M.; Bours, V.; Robe, P. A. (1 September 2014). “O9.06 * Prognostic Relevance and Oncogenic Correlates of Epilepsy in Glioblastoma Patients”. Neuro-Oncology. 16 (suppl_2): ii21. doi:10.1093/neuonc/nou174.77. PMC 4185847.
- ^ Vasudev K, Mead A, Macritchie K, Young AH (2012). “Valproate in acute mania: is our practice evidence based?”. Int J Health Care Qual Assur. 25 (1): 41–52. doi:10.1108/09526861211192395. PMID 22455007.
- ^ Bond DJ, Lam RW, Yatham LN (2010). “Divalproex sodium versus placebo in the treatment of acute bipolar depression: a systematic review and meta-analysis”. J Affect Disord. 124 (3): 228–334. doi:10.1016/j.jad.2009.11.008. PMID 20044142.
- ^ Haddad PM, Das A, Ashfaq M, Wieck A (2009). “A review of valproate in psychiatric practice”. Expert Opin Drug Metab Toxicol. 5(5): 539–51. doi:10.1517/17425250902911455. PMID 19409030. S2CID 74028228.
- ^ Frazee LA, Foraker KC (2008). “Use of intravenous valproic acid for acute migraine”. Ann Pharmacother. 42 (3): 403–7. doi:10.1345/aph.1K531. PMID 18303140. S2CID 207263036.
- ^ Wang, Yijun; Xia, Jun; Helfer, Bartosz; Li, Chunbo; Leucht, Stefan (2016). “Valproate for schizophrenia”. The Cochrane Database of Systematic Reviews. 11: CD004028. doi:10.1002/14651858.CD004028.pub4. ISSN 1469-493X. PMC 6734130. PMID 27884042.
- ^ Jump up to:a b Baillon, Sarah F.; Narayana, Usha; Luxenberg, Jay S.; Clifton, Andrew V. (2018-10-05). “Valproate preparations for agitation in dementia”. The Cochrane Database of Systematic Reviews. 2018(10): CD003945. doi:10.1002/14651858.CD003945.pub4. ISSN 1469-493X. PMC 6516950. PMID 30293233.
- ^ Gill D, Derry S, Wiffen PJ, Moore RA (2011). “Valproic acid and sodium valproate for neuropathic pain and fibromyalgia in adults”. Cochrane Database Syst Rev (10): CD009183. doi:10.1002/14651858.CD009183.pub2. PMC 6540387. PMID 21975791.
- ^ Aliyev ZN, Aliyev NA (July–August 2008). “Valproate treatment of acute alcohol hallucinosis: a double-blind, placebo-controlled study”. Alcohol Alcohol. 43 (4): 456–459. doi:10.1093/alcalc/agn043. PMID 18495806.
- ^ Jacobson PL, Messenheimer JA, Farmer TW (1981). “Treatment of intractable hiccups with valproic acid”. Neurology. 31 (11): 1458–60. doi:10.1212/WNL.31.11.1458. PMID 6796902. S2CID 1578958.
- ^ Sotaniemi K (1982). “Valproic acid in the treatment of nonepileptic myoclonus”. Arch. Neurol. 39 (7): 448–9. doi:10.1001/archneur.1982.00510190066025. PMID 6808975.
- ^ Wheeler SD (July–August 1998). “Significance of migrainous features in cluster headache: divalproex responsiveness”. Headache. 38 (7): 547–51. doi:10.1046/j.1526-4610.1998.3807547.x. PMID 15613172. S2CID 27948702.
- ^ Siemes H, Spohr HL, Michael T, Nau H (September–October 1988). “Therapy of infantile spasms with valproate: results of a prospective study”. Epilepsia. 29 (5): 553–60. doi:10.1111/j.1528-1157.1988.tb03760.x. PMID 2842127. S2CID 23789333.
- ^ Smith SM (2005). “Valproic acid and HIV-1 latency: beyond the sound bite” (PDF). Retrovirology. 2 (1): 56. doi:10.1186/1742-4690-2-56. PMC 1242254. PMID 16168066. Archived from the original (PDF) on 2015-09-24. Retrieved 2014-02-13.
- ^ Routy JP, Tremblay CL, Angel JB, Trottier B, Rouleau D, Baril JG, Harris M, Trottier S, Singer J, Chomont N, Sékaly RP, Boulassel MR (2012). “Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: results from a multicentre randomized clinical study”. HIV Med. 13 (5): 291–6. doi:10.1111/j.1468-1293.2011.00975.x. PMID 22276680. S2CID 27571864.
- ^ Archin NM, Cheema M, Parker D, Wiegand A, Bosch RJ, Coffin JM, Eron J, Cohen M, Margolis DM (2010). “Antiretroviral intensification and valproic acid lack sustained effect on residual HIV-1 viremia or resting CD4+ cell infection”. PLOS ONE. 5 (2): e9390. Bibcode:2010PLoSO…5.9390A. doi:10.1371/journal.pone.0009390. PMC 2826423. PMID 20186346.
- ^ Jump up to:a b Hardy JR, Rees EA, Gwilliam B, Ling J, Broadley K, A’Hern R (2001). “A phase II study to establish the efficacy and toxicity of sodium valproate in patients with cancer-related neuropathic pain” (PDF). J Pain Symptom Manage. 21 (3): 204–9. doi:10.1016/S0885-3924(00)00266-9. PMID 11239739.[permanent dead link]
- ^ Candelaria M, Herrera A, Labardini J, González-Fierro A, Trejo-Becerril C, Taja-Chayeb L, Pérez-Cárdenas E, de la Cruz-Hernández E, Arias-Bofill D, Vidal S, Cervera E, Dueñas-Gonzalez A (2011). “Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial”. Ann. Hematol. 90 (4): 379–387. doi:10.1007/s00277-010-1090-2. PMID 20922525. S2CID 13437134.
- ^ Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, Romanski A, Kramer OH, Kampfmann M, Hoelzer D, Neubauer A, Ruthardt M, Ottmann OG (2005). “Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia”. Cancer. 104 (12): 2717–2725. doi:10.1002/cncr.21589. PMID 16294345. S2CID 1802132.
- ^ Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, Germing U, Haas R, Dohner H, Gattermann N (2006). “The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia”. Cancer. 106 (1): 112–119. doi:10.1002/cncr.21552. PMID 16323176. S2CID 43747497.
- ^ Fredly H, Gjertsen BT, Bruserud O (2013). “Histone deacetylase inhibition in the treatment of acute myeloid leukemia: the effects of valproic acid on leukemic cells, and the clinical and experimental evidence for combining valproic acid with other antileukemic agents” (PDF). Clin Epigenetics. 5 (1): 12. doi:10.1186/1868-7083-5-12. PMC 3733883. PMID 23898968. Archived from the original (PDF) on 2014-02-21. Retrieved 2014-02-13.
- ^ Coronel J, Cetina L, Pacheco I, Trejo-Becerril C, González-Fierro A, de la Cruz-Hernandez E, Perez-Cardenas E, Taja-Chayeb L, Arias-Bofill D, Candelaria M, Vidal S, Dueñas-González A (2011). “A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results”. Med. Oncol. 28 Suppl 1: S540–6. doi:10.1007/s12032-010-9700-3. PMID 20931299. S2CID 207372333.
- ^ Rocca A, Minucci S, Tosti G, Croci D, Contegno F, Ballarini M, Nolè F, Munzone E, Salmaggi A, Goldhirsch A, Pelicci PG, Testori A (2009). “A phase I-II study of the histone deacetylase inhibitor valproic acid plus chemoimmunotherapy in patients with advanced melanoma”. Br. J. Cancer. 100 (1): 28–36. doi:10.1038/sj.bjc.6604817. PMC 2634690. PMID 19127265.
- ^ Munster P, Marchion D, Bicaku E, Lacevic M, Kim J, Centeno B, Daud A, Neuger A, Minton S, Sullivan D (2009). “Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC” (PDF). Clin. Cancer Res. 15 (7): 2488–96. doi:10.1158/1078-0432.CCR-08-1930. PMID 19318486. S2CID 3230087.
- ^ Hicks CW, Pandya MM, Itin I, Fernandez HH (2011). “Valproate for the treatment of medication-induced impulse-control disorders in three patients with Parkinson’s disease”. Parkinsonism Relat. Disord. 17 (5): 379–81. doi:10.1016/j.parkreldis.2011.03.003. PMID 21459656.
- ^ Sriram A, Ward HE, Hassan A, Iyer S, Foote KD, Rodriguez RL, McFarland NR, Okun MS (2013). “Valproate as a treatment for dopamine dysregulation syndrome (DDS) in Parkinson’s disease”. J. Neurol. 260 (2): 521–7. doi:10.1007/s00415-012-6669-1. PMID 23007193. S2CID 21544457.
- ^ Aizenman, N. C. (7 May 2012). “Abbott Laboratories to pay $1.6 billion over illegal marketing of Depakote”. Washington Post. Retrieved 27 June 2018.
- ^ Schmidt, Michael; Thomas, Katie (8 May 2012). “Abbott settles marketing lawsuit”. New York Times. Retrieved 27 June 2018.
- ^ Gervain J, Vines BW, Chen LM, Seo RJ, Hensch TK, Werker JF, Young AH (2013). “Valproate reopens critical-period learning of absolute pitch”. Frontiers in Systems Neuroscience. 7: 102. doi:10.3389/fnsys.2013.00102. PMC 3848041. PMID 24348349.
- ^ Thomson, Helen. “Learning drugs reawaken grown-up brain’s inner child”. New Scientist. New Scientist Ltd. Retrieved 8 May2021.
- ^ David Taylor; Carol Paton; Shitij Kapur (2009). The Maudsley Prescribing Guidelines, Tenth Edition (10, revised ed.). CRC Press. p. 124. ISBN 9780203092835.
- ^ “Depakene- valproic acid capsule, liquid filled”. DailyMed. 19 September 2019. Retrieved 14 April 2020.
- ^ Jump up to:a b c d “Australian product information epilim (sodium valproate) crushable tablets, enteric-coated tablets, syrup, liquid” (PDF). TGA eBS. 15 April 2020. Retrieved 15 April 2020.
- ^ “Sodium valproate — Pharmaceutical Schedule”. Pharmaceutical Management Agency. Archived from the originalon 4 March 2016. Retrieved 22 June 2014.
- ^ South African Electronic Package Inserts: Convulex
External links
- “Valproic acid”. Drug Information Portal. U.S. National Library of Medicine.
- “Valproate sodium”. Drug Information Portal. U.S. National Library of Medicine.
- “Divalproex sodium”. Drug Information Portal. U.S. National Library of Medicine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCA1144558A *1979-10-221983-04-12Francis E. FischerProcess for making sodium hydrogen divalproateUS4988731A *1979-08-201991-01-29Abbott LaboratoriesSodium hydrogen divalproate oligomerUS5212326A *1979-08-201993-05-18Abbott LaboratoriesSodium hydrogen divalproate oligomerWO2001032595A1 *1999-11-022001-05-10Cilag AgMethod for producing compounds of the valproinic acidUS20030018215A1 *2001-06-292003-01-23Procos S.P.A.Process for the preparation of sodium divalproatePublication numberPriority datePublication dateAssigneeTitleUS20110040122A1 *2009-08-112011-02-17Sci Pharmtech, Inc.Method for preparing metal salt of valproic acidCN102942467A *2012-10-172013-02-27山东方明药业集团股份有限公司Preparation method of divalproex sodiumCN103183600A *2011-12-302013-07-03北大方正集团有限公司Method for preparing divalproex sodium
////// divalproex, Anticonvulsant, Antimigraine, Antimanic, valproic acid, sodium valproate
CCCC(CCC)C(O)=O

NEW DRUG APPROVALS
ONE TIME
$10.00
LORNOXICAM
LORNOXICAM
chlortenoxicam
- Molecular FormulaC13H10ClN3O4S2
- Average mass371.819 Da
70374-39-9[RN], Chlortenoxicam, CTX, ER09126G7A
2H-thieno[2,3-e]-1,2-thiazine-3-carboxamide, 6-chloro-4-hydroxy-2-methyl-N-2-pyridinyl-, 1,1-dioxide
6233
6-Chlor-4-hydroxy-2-methyl-N-(pyridin-2-yl)-2H-thieno[2,3-e][1,2]thiazin-3-carboxamid-1,1-dioxid
6-Chloro-4-hydroxy-2-methyl-N-(2-pyridinyl)-2H-thieno[2,3-e][1,2]thiazine-3-carboxamide 1,1-dioxide
- Chlortenoxicam, Ro-13-9297
- ATC:M01AC05
- CCRIS 8589
- Ro 13-9297
Lorcam (Taisho Pharmaceutical Co.) / Xafon (Nycomed)LornoxicamCAS Registry Number: 70374-39-9
CAS Name: 6-Chloro-4-hydroxy-2-methyl-N-2-pyridinyl-2H-thieno[2,3-e]-1,2-thiazine-3-carboxamide 1,1-dioxide
Additional Names: 6-chloro-4-hydroxy-2-methyl-3-(2-pyridylcarbamoyl)-2H-thieno[2,3-e]-1,2-thiazine-1,1-dioxide; chlortenoxicam
Manufacturers’ Codes: Ro-13-9297; TS-110
Trademarks: Xefo (Nycomed)
Molecular Formula: C13H10ClN3O4S2
Molecular Weight: 371.82
Percent Composition: C 41.99%, H 2.71%, Cl 9.53%, N 11.30%, O 17.21%, S 17.25%
Literature References: Cyclooxygenase inhibitor; structurally similar to tenoxicam, q.v.
Prepn: R. Pfister et al.,DE2838851; eidem,US4180662 (both 1979 to Hoffmann-La Roche).Clinical pharmacokinetics: S. I. Ankier et al.,Postgrad. Med. J.64, 752 (1988). Symposium on pharmacology and clinical experience: ibid.66, Suppl. 4, S1-S50 (1990). Overview of pharmacology and safety assessment: T. P. Pruss et al.,ibid. S18.
Properties: Orange to yellow crystals, mp 225-230° (dec). pKa2 4.7. uv max: 371 nm. Partition coefficient (n-octanol/pH 7.4 buffer): 1.8. LD50 orally in mice, rats, rabbits, dogs, monkeys: >10 mg/kg (Pruss).
Melting point: mp 225-230° (dec)
pKa: pKa2 4.7
Log P: Partition coefficient (n-octanol/pH 7.4 buffer): 1.8
Absorption maximum: uv max: 371 nm
Toxicity data: LD50 orally in mice, rats, rabbits, dogs, monkeys: >10 mg/kg (Pruss)
Therap-Cat: Anti-inflammatory; analgesic.
Keywords: Analgesic (Non-Narcotic); Anti-inflammatory (Nonsteroidal); Thiazinecarboxamides.
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 504-29-0 | C5H6N2 | 2-aminopyridine | 2-Pyridinamine |
| 7790-94-5 | ClHO3S | chlorosulfonic acid | Chlorosulfuric acid |
| 56946-84-0 | C5H5Cl2NO2S2 | 2,5-dichloro-N-methyl-3-thiophenesulfonamide | 3-Thiophenesulfonamide, 2,5-dichloro-N-methyl- |
| 3172-52-9 | C4H2Cl2S | 2,5-dichlorothiophene | Thiophene, 2,5-dichloro- |
SYN
Synthesis of lornoxicam (DE2838851)

The sulfonation of 2,5-dichlorothiophene (I) with ClSO3H -SOCl2 gives 2,5-dichlorothiophene-3-sulfonic acid chloride (II), which by reaction with methylamine in CHCl3 yields the corresponding methylamide (III). The carboxylation of (III) with butyllithium and CO2 in ether affords 5-chloro-3-(N-methylsulfamoyl)thiophene-2-carboxylic acid (IV), which is esterified with PCl5 and methanol to the methyl ester (V). The condensation of (V) with methyl iodoacetate (VI) by means of NaH in DMF gives 5-chloro-3-[N-(methoxycarbonylmethyl)-N-methylsulfamoyl]thiophene-2-carboxylic acid methyl ester (VII), which is cyclized with sodium methoxide in methanol yielding 6-chloro-4-hydroxy-2-methyl-2H-thieno[2,3-e]-1,2-thiazine-3-carboxylic acid methyl ester 1,1-dioxide (VIII). Finally, this compound is treated with 2-aminopyridine (IX) in refluxing xylene.
Lornoxicam is an NSAID indicated in the treatment of mild to moderate pain, as well as rheumatoid arthritis and osteoarthritis.
Lornoxicam, also known as chlortenoxicam, is a nonsteroidal anti-inflammatory drug (NSAID) of the oxicam class with analgesic (pain relieving), anti-inflammatory and antipyretic (fever reducing) properties. It is available in oral and parenteral formulations.
It was patented in 1977 and approved for medical use in 1997.[1] Brand names include Xefo and Xefocam among others.
Lornoxicam (chlortenoxicam) is a new nonsteroidal anti-inflammatory drug (NSAID) of the oxicam class with analgesic, anti-inflammatory and antipyretic properties. Lornoxicam differs from other oxicam compounds in its potent inhibition of prostaglandin biosynthesis, a property that explains the particularly pronounced efficacy of the drug. Lornoxicam is approved for use in Japan.
Medical uses
Lornoxicam is used for the treatment of various types of pain, especially resulting from inflammatory diseases of the joints, osteoarthritis, surgery, sciatica, and other inflammations.[2]

Contraindications
The drug is contraindicated in patients who must not take other NSAIDs, possible reasons including salicylate sensitivity, gastrointestinal bleeding and bleeding disorders, and severe impairment of heart, liver or kidney function. Lornoxicam is not recommended during pregnancy and breastfeeding and is contraindicated during the last third of pregnancy.[2]
Adverse effects
Lornoxicam has side effects similar to other NSAIDs, most commonly mild ones like gastrointestinal disorders (nausea and diarrhea) and headache. Severe but seldom side effects include bleeding, bronchospasms and the extremely rare Stevens–Johnson syndrome.[2]
Interactions
Interactions with other drugs are typical of NSAIDs. Combination with vitamin K antagonists like warfarin increases the risk of bleeding. Combination with ciclosporin can lead to reduced kidney function, and to acute kidney injury in rare cases. Lornoxicam can also increase the adverse effects of lithium, methotrexate and digoxin and its derivatives. The effect of diuretics, ACE inhibitors and angiotensin II receptor antagonists can be reduced, but this is only relevant in patients with special risks like heart failure. As with piroxicam, cimetidine can increase plasma levels but is unlikely to cause relevant interactions.[3]
PAPER
https://www.mdpi.com/2218-0532/71/4/303

PATENT
CN 113480561
The present invention relates to the prepn. of high purity loroxicam. In particular, the prepn. method comprises a step of taking 6-chloro-4-hydroxy-2-methyl-2H-thieno[2,3-e]-1,2-Me thiazinecarboxylate-1,1-dioxide and 2-amino pyridine is used as the raw material and xylene is used as the solvent undergoes distn. reaction with solid acid catalyst, mixed gas obtained by the distn. reaction is condensed to obtain a condensate and solid acid catalyst is used to adsorb methanol in the condensate and the adsorbed condensate is recycled, filtering and refining to obtain loroxicam. The present inventive method distills out the methanol produced by the reaction to promote the pos. progress of the reaction and then catalyzes the absorption of methanol by H2SO4/MxOy solid super acid, so that the xylene returned to the reaction system does not contain methanol, which reduces the coking of the reaction, thereby improving product quality and yield. The prepd. lornoxicam has high purity, which can reach more than 99.9%, reduces the amt. of solvent and also suitable for industrial prodn.
PATENT
CN 112592356
The present invention relates to the prepn. of lornoxicam. In particular, the prepn. method comprises a step of taking 6-chloro-4-hydroxy-2-methyl-2-H-thieno[2,3-e]-1,2-thiazidecarboxylic acid Me ester-1,1-dioxide and 2-aminopyridine as raw materials, xylene is used as solvent, adding stabilizer, and carrying out aminolysis reaction, the solvent was removed by concn. under reduced pressure, adding org. solvent to make the slurry, filtering and refining to obtain lornoxicam. The inventive method uses p-toluene sulfonic acid as a stabilizer, while lowering the reaction temp., it promotes the reaction to proceed forward, and improve the product quality and yield; at the same time reduce the amt. of industrial solvents, the post-treatment process is optimized and the cost of the three wastes treatment is reduced.
PATENT
IN 2014CH02116
Example: 1Preparation of 6-chloro-4-hydroxy-l,l-dioxo-l,2-dihydro-lX6-thieno [2,3-e][l,2] thiazine-3-carboxylic acid methyl ester To the mixture of methanol ( 1000 ml) and 5-chloro-3-(methoxy carbonyl methyl sulfamoyl)-thiophene-2-carboxylicacid methyl ester ( 100 g ,0.305 moles), added sodium methoxide solution (200 ml ) at 25-30°C over a period of 30-45 min. The resulting mixture was stirred for 60 min at same temperature; allowed to heat at 65-75°C and stirred for 10-12 hrs. After completion of reaction, methanol was distilled out under reduced pressure to obtained titled residual product which is directly used to next step
(Example-2). Example: – 2:Preparation of 6-chloro-4-hydroxy-2-methyl-l,l-dioxo-l,2-dihydro-U6- thieno[2,3-e][l,2] thiazine-3-carboxylic acid methyl ester 6-chloro-4-hydroxy-1,1 -dioxo-1,2-dihydro-1 X,6-thieno [2,3-e][ 1,2] thiazine-3-carboxylic acid methyl ester was suspended in DM water (500 ml) and cooled to 10-15° C, dimethyl sulphate ( 70 g) was slowly added to the mixture at 10-15°C in 30 min. The reaction mixture was raised to 25-30°C and maintained for 2-3 hours at same temperature. After completion of reaction, mixture was cooled to 10-15°C, methylene dichloride (1600 ml) was added, reaction mixture pH was adjust to 1.0 -2.0 with hydrochloric acid at 10-15° C, stir reaction mixture to separate the layers. The methylene dichloride layer was distilled out completely at below 30°C to get an residue, followed by addition of methanol (60 ml) and distilled out methanol completely under vacuum at below 50°C to get an residue; further it was crystallized by addition of methanol 190 ml and stirred for 30 min at 50-55°C; cooled the reaction mixture at 25-30°C and stirred for 60 min at same temperature. The resultant solid was filtered, washed with methanol (40 ml) and dried at 50-55°C for 4 – 6 hrs to obtain the titled product
Example: 3Preparation of 6-Chloro-4-hydroxy-2-methyl-N-2-pyridinyl-2H-thieno[2,3-e]-l,2-thiazine-3-carboxamide 1,1-dioxide (Lornoxicam) 6-chloro-4-hydroxy-2-methyl-l, 1 -dioxo-1,2-dihydro-l X.6-thieno[2,3-e][l ,2] thiazine-3-carboxylic acid methyl ester ( 50 g 0.161 moles) was suspended in O-xylene (500 ml) and allow to stirred at 70-75°C to obtained clear solution. To this clear solution slowly added the mixture of THF ( 50 ml) solution of 2-Amino pyridine ( 14 g ) and ethyl magnesium bromide 2 molar solution (100 ml) at 70-75°C and allow to stirred for 3-4 hrs at same temperature. After completion of reaction, the dilute hydrochloric acid was added to the mixture at 10-15°C and stirred for 60 min. The resultant solid was filtered, washed with water (100 ml) to obtain crude Lornoxicam.
Example: 4Preparation of 6-Chloro-4-hydroxy-2-methyl-N-2-pyridinyl-2H-thieno[2,3-e)-l,2-thiazine-3-carboxamide 1,1-dioxide (Lornoxicam) 6-chloro-4-hydroxy-2-methyl-l,l-dioxo-l,2-dihydro-R6-thieno[2,3-e][l,2] thiazine-3-carboxylic acid methyl ester ( 50 g 0.161 moles) was suspended in O-xylene (500 ml) and allow to stirred at 70-75°C to obtained clear solution. To this clear solution slowly added the mixture of THF ( 50 ml) solution of 2-Amino pyridine ( 14 g ) and isopropyl magnesium bromide 2 molar solution (100 ml) at 70-75°C and allow to stirred for 3-4 hrs at same temperature. After completion of reaction, the dilute hydrochloric acid was added to the mixture at 10-15°C and stirred for 60 min. The resultant solid was filtered, washed with water (100 ml) to obtain crude Lornoxicam.
Example: 5Purification of Lornoxicam.The crude Lornoxicam was suspended in methanol (500 ml) and cooled to 5-10°C, resulting suspension was basified to pH 11-13 by using sodium hydroxide solution to get clear solution; followed by filtration through hyflo bed; the obtain filtrate was acidified to pH 4.5 – 5.0 with dil. HC1 (1:1) at 5-10°C; stirred the slurry for 30 min. at 5-10°C. The resultant solid was filtered, washed with DM water (100 ml) and dried at 50-55°C to obtained pure Lornoxicam.
PATENT
.EXAMPLES:Preparation of Lornoxicam crudeExample ITo 1200ml o-xylene, 20gm Methyl-6-chloro-4-hydroxy-2-methyl-2//-thieno [2, 3-e] [1, 2] thiazine-3- carboxyate 1,1-dioxide and 6.44gm 2-aminopyridine was added. The reaction mass was stirred under nitrogen atmosphere. Temperature was raised to 140-145°C and maintained for 6hrs. The reaction mass was cooled to 30-35°C and nitrogen was removed. Reaction mass was further stirred for 3hrs- Filtered and washed twice with 50ml of o-xylene. 19.8gm of crude Lornoxicam was obtained. Purification of Lornoxicam crude
Example 219.8gm of crude Lornoxicam was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and methanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around Ihr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C, til! the reaction mass reached pH of 2-3, and then stirred for around I hi*. The reaction mass was cooled to room temperature, filtered, and then washed with 1:1 mixture of methanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 19.1 gm of pure Lornoxicam was obtained. (HPLC purity- 99.95%)
Example 3!7.9gm of crude Lornoxicam (prepared as per example 1) was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and methanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution, and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around Ihr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C till the reaction mass reached pH of 2-3, and then stirred for around Ihr. The reaction mass was cooled to room temperature, filtered and then washed with 1:1 mixture of methanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 17.2 gm of pure Lornoxicam was obtained. (HPLC purity- 99.9%) clear solution and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around lhr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C, till the reaction mass reached pH of 2-3, and then stirred for around lhr. The reaction mass was cooled to 30-35°C, filtered and then washed with 1:1 mixture of isopropyl alcohol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 4.85 gm of pure Lornoxicam was obtained. (HPLC purity- 99.8%)
Example 55 gm of crude Lornoxicam (prepared as per example 1) was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and ethanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution, and 5% activated charcoal was further added. The reaction mass was heated to 50-55°C and stirred for around lhr followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added at 50-55° C, til! the reaction mass reached pH of 2-3 and then stirred for around lhr. The reaction mass was cooled to 30-35°C and filtered, washed with 1:1 mixture of ethanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 4.8 gm of pure Lornoxicam was obtained. (HPLC purity- 99.8%)
Example 619.4 gm of crude Lornoxicam (prepared as per example I) was added to the solvent mixture of water (5 vol with respect to Lornoxicam) and methanol (10 vol with respect to Lornoxicam) under stirring. Subsequently 48% sodium hydroxide was added to form a clear solution, and 20% activated charcoal was further added. The reaction mass was stirred for around lhr at room temperature followed by filtration through Hyflo. To the filtrate, mixture of hydrochloric acid and water in the ratio of 1:1 was added till the reaction mass reached pH of 2-3 and then stirred for around 1 hr. The reaction mass was * filtered and washed with 1:1 mixture of methanol and water. Purified wet Lornoxicam was dried at 60-65°C for 6-8hrs. 18.9 gm of pure Lornoxicam was obtained. (HPLC purity- 99.3%).
PATENT
https://www.sciencedirect.com/science/article/abs/pii/S0968089603007624?via%
PATENT
https://patents.google.com/patent/WO2002000167A2/en
References
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 519. ISBN 9783527607495.
- ^ Jump up to:a b c Haberfeld H, ed. (2009). Austria-Codex (in German) (2009/2010 ed.). Vienna: Österreichischer Apothekerverlag. Xefo Filmtabletten. ISBN 978-3-85200-196-8.
- ^ Klopp T, ed. (2010). Arzneimittel-Interaktionen (in German) (2010/2011 ed.). Arbeitsgemeinschaft für Pharmazeutische Information. ISBN 978-3-85200-207-1.
| Clinical data | |
|---|---|
| Trade names | Xefo, Xefocam others |
| AHFS/Drugs.com | International Drug Names |
| Pregnancy category | Not recommended; contraindicated in months 7–9 |
| Routes of administration | By mouth, parenteral |
| ATC code | M01AC05 (WHO) |
| Legal status | |
| Legal status | In general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Bioavailability | 90–100% |
| Protein binding | 99% |
| Metabolism | CYP2C9 |
| Elimination half-life | 3–4 hours |
| Excretion | 2/3 liver, 1/3 kidney |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 70374-39-9 |
| PubChem CID | 5282204 |
| DrugBank | DB06725 |
| ChemSpider | 10442760 |
| UNII | ER09126G7A |
| KEGG | D01866 |
| ChEBI | CHEBI:31783 |
| CompTox Dashboard (EPA) | DTXSID6046133 |
| ECHA InfoCard | 100.158.646 |
| Chemical and physical data | |
| Formula | C13H10ClN3O4S2 |
| Molar mass | 371.81 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
//////////LORNOXICAM, Ro-13-9297, TS-110, Anti-inflammatory, analgesic, chlortenoxicam, CCRIS 8589
CN1C(C(=O)NC2=CC=CC=N2)=C(O)C2=C(C=C(Cl)S2)S1(=O)=O
General References
- Balfour JA, Fitton A, Barradell LB: Lornoxicam. A review of its pharmacology and therapeutic potential in the management of painful and inflammatory conditions. Drugs. 1996 Apr;51(4):639-57. [Article]
- Vane JR: Introduction: mechanism of action of NSAIDs. Br J Rheumatol. 1996 Apr;35 Suppl 1:1-3. [Article]
- Radhofer-Welte S, Rabasseda X: Lornoxicam, a new potent NSAID with an improved tolerability profile. Drugs Today (Barc). 2000 Jan;36(1):55-76. [Article]
- Skjodt NM, Davies NM: Clinical pharmacokinetics of lornoxicam. A short half-life oxicam. Clin Pharmacokinet. 1998 Jun;34(6):421-8. [Article]
- Olkkola KT, Brunetto AV, Mattila MJ: Pharmacokinetics of oxicam nonsteroidal anti-inflammatory agents. Clin Pharmacokinet. 1994 Feb;26(2):107-20. [Article]
- Hitzenberger G, Radhofer-Welte S, Takacs F, Rosenow D: Pharmacokinetics of lornoxicam in man. Postgrad Med J. 1990;66 Suppl 4:S22-7. [Article]
- Pruss TP, Stroissnig H, Radhofer-Welte S, Wendtlandt W, Mehdi N, Takacs F, Fellier H: Overview of the pharmacological properties, pharmacokinetics and animal safety assessment of lornoxicam. Postgrad Med J. 1990;66 Suppl 4:S18-21. [Article]
- Bonnabry P, Leemann T, Dayer P: Role of human liver microsomal CYP2C9 in the biotransformation of lornoxicam. Eur J Clin Pharmacol. 1996;49(4):305-8. [Article]

NEW DRUG APPROVALS
ONE TIME
$10.00
FAROPENEM
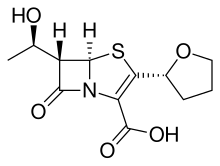
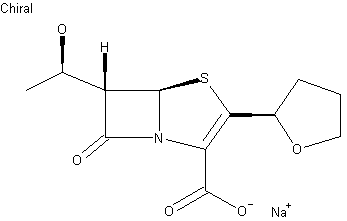
- Molecular FormulaC12H15NO5S
- Average mass285.316 Da
Faropenem
7086
(+)-(5R,6S)-6-((1R)-1-Hydroxyethyl)-7-oxo-3-((2R)-tetrahydro-2-furyl)-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic Acid
(5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
(5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydrofuran-2-yl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
106560-14-9[RN]
4-Thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid, 6-[(1R)-1-hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-, (5R,6S)-
6α-[(R)-1-hydroxyethyl]-2-[(R)-tetrahydrofuran-2-yl]pen-2-em-3-carboxylic acid
4-Oxofenretinide
4-Oxo-N-(4-hydroxyphenyl)retinamide
6α-[(1R)-1-hydroxyethyl]-2-[(2R)-tetrahydrofuran-2-yl]-2,3-didehydropenam-3-carboxylic acid
7305146 [Beilstein]
FaropenemCAS Registry Number: 106560-14-9
CAS Name: (5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid
Additional Names: fropenem; (5R,6S,8R,2¢R)-2-(2¢-tetrahydrofuryl)-6-hydroxyethylpenem-3-carboxylate
Molecular Formula: C12H15NO5S
Molecular Weight: 285.32
Percent Composition: C 50.51%, H 5.30%, N 4.91%, O 28.04%, S 11.24%
Literature References: Orally active, b-lactamase stable, penem antibiotic.Prepn: M. Ishiguro et al.,EP199446; eidem,US4997829 (1986, 1991 both to Suntory); eidem,J. Antibiot.41, 1685 (1988).Pharmacokinetics: A. Tsuji et al.,Drug Metab. Dispos.18, 245 (1990). In vitro antimicrobial spectrum: J. M. Woodcock et al.,J. Antimicrob. Chemother.39, 35 (1997). b-Lactamase stability: A. Dalhoff et al., Chemotherapy (Basel)49, 229 (2003).HPLC determn in plasma: R. V. S. Nirogi et al., Arzneim.-Forsch.55, 762 (2005). Clinical trial in urinary tract infections: S. Arakawa et al.,Nishinihon J. Urol.56, 300 (1994); in bacterial sinusitis: R. Siegert et al., Eur. Arch. Otorhinolaryngol.260, 186 (2003).
Derivative Type: Sodium salt
CAS Registry Number: 122547-49-3
Additional Names: Furopenem
Manufacturers’ Codes: ALP-201; SUN-5555; SY-5555; WY-49605
Trademarks: Farom (Daiichi)
Molecular Formula: C12H15NNaO5S
Molecular Weight: 308.31
Percent Composition: C 46.75%, H 4.90%, N 4.54%, Na 7.46%, O 25.95%, S 10.40%
Properties: [a]D22 +60° (c = 0.10).
Optical Rotation: [a]D22 +60° (c = 0.10)
Derivative Type: Daloxate
CAS Registry Number: 141702-36-5
CAS Name: (5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl ester
Additional Names: faropenem medoxomil
Manufacturers’ Codes: Bay-56-6854; SUN-208
Trademarks: Orapem (Replidyne)
Molecular Formula: C17H19NO8S
Molecular Weight: 397.40
Percent Composition: C 51.38%, H 4.82%, N 3.52%, O 32.21%, S 8.07%
Literature References: Prepn: H. Iwata et al., WO9203442; eidem, US5830889 (1992, 1998 both to Suntory).
Properties: Pale yellow crystals.
Therap-Cat: Antibacterial (antibiotics).
Keywords: Antibacterial (Antibiotics); ?Lactams; Penems.
Faropenem is an orally active beta-lactam antibiotic belonging to the penem group.[1] It is resistant to some forms of extended-spectrum beta-lactamase.[2] It is available for oral use.[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Forms
Faropenem was developed by Daiichi Asubio Pharma, which markets it in two forms.
- The sodium salt faropenem sodium, available under the trade name Farom, has been marketed in Japan since 1997. (CID 636379 from PubChem)
- The prodrug form faropenem medoxomil[4] (also known as faropenem daloxate) has been licensed from Daiichi Asubio Pharma by Replidyne, which plans to market it in conjunction with Forest Pharmaceuticals. The trade name proposed for the product was Orapem, but company officials recently announced this name was rejected by the FDA.[5]
Clinical use
As of 8 September 2015, Faropenem has yet to receive marketing approval in the United States, and was submitted for consideration by the United States Food and Drug Administration (FDA) on 20 December 2005. The new drug application dossier submitted included these proposed indications:
- acute bacterial sinusitis
- community-acquired pneumonia
- acute exacerbations of chronic bronchitis
- uncomplicated skin and skin structure infections
- urinary tract infections
History
The FDA refused to approve faropenem, an antibiotic manufactured by Louisville-based Replidyne. The FDA said the drug was “nonapprovable”, but did not refer to specific safety concerns about the product. The company will have to conduct new studies and clinical trials, lasting an estimated two more years, to prove the drug treats community-acquired pneumonia, bacterial sinusitis, chronic bronchitis, and skin infections.[citation needed]
In India it is available as Farobact 200/300ER CIPLA.
PATENT
https://patents.google.com/patent/WO2008035153A2/enFaropenem is an orally active β-lactam antibiotic belonging to the penem group. Faropenem is chemically known as 6-(l-hydroxyethyl)-7-oxo-3-(oxolan-2-yl)-4-thia-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid. The known forms of Faropenem are Faropenem sodium and the prodrug form, FaropenemMedoxomil (also known as Faropenem Daloxate). In view of the importance of the compound of the formula (I), several synthetic procedures to prepare the compound have been reported.US 4,997,829 provides process for the preparation of faropenem according to the following scheme. The process is exemplified with the allyl protected carboxyl group. One of the process involves the reaction of A- acetoxyazetidinone with tetrahydrothiofuroic acid, condensation with allyl glyoxalate in refluxing benzene, chlorination with thionyl chloride, reaction of triphenylphosphine with lutidine in hot THF, cyclization in refluxing toluene, deprotection of silyl protecting group with tetrabutylammonium fluoride, treating with triphenylphosphine and, treating with sodium 2-ethylhexanoate and (PP^)4Pd to result faropenem sodium. The process exemplified utilizes benzene as solvent, which is not environmentally acceptable. Tetrabutylammonium fluoride was used as desilylating agent that is expensive. Even though the description teaches that optically active compounds can be employed, the examples utilized the dl-compound of tetrahydrothiofuroic acid further requiring resolution.

Methods are provided for the synthesis of series of penem compounds in J Antibiotics 1988, 41(11), 1685-1693. The provided methods utilize sulfonylazetidinone as the starting materials. As one of the procedures gives lesser yield, another procedure was adopted which uses silver salts.Japanese patent, JP2949363 describes a process for deallylation and salt formation with an alkali metal salt of carboxylic acid in the presence of a catalytic amount of palladium complex for the preparation of faropenem.EP410727 describes a process for removing allyl group from a penem compound using cyclic 1,3-diketone such as dimedone.The yield and quality of the final product is always less in the above prior art methods. With the continued research, the present inventors have undertaken extensive studies for developing a process for the preparation of compound of formula (I), which is commercially viable, involves simple techniques such as crystallizations, with improved yields and quality of the product, and with lesser reaction time. None of the prior art suggests or teaches the techniques provided herein.The process is shown in Scheme-I as given below:

One-pot process for the preparation of Faropenem sodium:Sodium salt of R(+)-tetrahydrofuran-2-thiocarboxylic acid (67 g) in aqueous acetone was added slowly to a solution of AOSA (100 g) in acetone (200 mL) and stirred for 3 h at pH 8.0 to 8.5 using sodium bicarbonate solution.After completion of the reaction, the product was extracted with toluene. The combined toluene layer was washed with saturated sodium bicarbonate solution and brine solution. Toluene was removed under vacuum completely and the mass obtained, 3-(l’-tert-butyldimethylsilyloxyethyl)-4-(2′- tetrahydrofuranoylthio)-2-azetidinone was directly taken for next step.3-(r-tert-Butyldimethylsilyloxyethyl)-4-(2′-tetrahydrofuranoylthio)-2- azetidinone obtained was dissolved in toluene (1000 mL) and cooled to -10 to -5 °C under nitrogen. Triethylamine (124 mL) was added to it followed by allyl oxalyl chloride (82 g) at -10 to- 5 0C for 2 h. After completion of the reaction, cold water was added to the mass and washed with dilute hydrochloric acid and sodium bicarbonate solution. Toluene layer was separated and washed with purified water. The toluene layer containing compound of formula (VI) was concentrated under vacuum at 50 to 60 °C and taken for next step as such.Compound of formula (VI) (150 g) was dissolved in triethyl phosphite (150 mL), heated to 60 0C and stirred under nitrogen atmosphere. Toluene (3000 mL) was added, heated to 100 to 110 °C and stirred for 20- 24 h. Toluene was distilled under vacuum completely. Product obtained, allyl (1 ‘R,2″R,5R,6S)-6-(l 5-tert-butyldimethylsilyloxyethyl)-2-(2″-tetrahydrofuranyl) penem-3-carboxylate (VII) was directly taken for next step.Compound (VII) obtained was dissolved in DMF (700 mL) at 30 °C.Ammonium hydrogen difluoride (80 g) and NMP (210 mL) were added and stirred at room temperature for 25 to 35 h. The reaction mass was quenched into a mixture of water-ethyl acetate and stirred at room temperature. The ethyl acetate layer was separated and the aqueous layer extracted with ethyl acetate. ■ The combined ethyl acetate layer was washed with water followed by saturated sodium bicarbonate solution. The ethyl acetate layer was charcoal treated. The ethyl acetate layer containing allyl (l’R,2″R,5R,6S)-6-(l’-hydroxyethyl)-2-(2″- tetrahydrofuranyl)penem-3-carboxylate (XII) was partially distilled and taken for the next step.The ethyl acetate layer containing compound of formula (XII), Pd/C, sodium bicarbonate and purified water (1000 mL) were taken in an autoclave and maintained 5 to 10 kg pressure of hydrogen gas for 2-5 h. After completion of the reaction the Pd/C was filtered off and ethyl acetate layer separated. The pH of the mass was adjusted to 1.5 and extracted with ethyl acetate. The aqueous layer was extracted again with ethyl acetate twice. The combined ethyl acetate layer was carbon treated. Sodium-2-ethylhexanoate in ethyl acetate was added slowly and stirred. The precipitated title compound was filtered under vacuum, washed with acetone and dried. Dry weight of the product: 65-75 g.Example 9Purification of Faropenem sodiumCrude Faropenem sodium (50 g) was dissolved in purified water (200 mL) at 25-30 0C. The solution was charcoalised. Acetone (1500 mL) was added. The reaction mass was stirred further for 10 min. The precipitated solid was cooled to 0 —2 °C then filtered, washed with acetone and dried at room temperature. Weight of pure Faropenem sodium is 43 to 46 g (Purity 99.95%).Example 9aPurification of Faropenem sodiumCrude Faropenem sodium (50 g) was dissolved in purified water (200 mL) at 25-30 °C. Acetone (150O mL) was added. The reaction mass was stirred further for 10 min. The precipitated solid was cooled to 0-2 °C then filtered, washed with acetone and dried at room temperature. Weight of pure Faropenem sodium is 43 to 46 g (Purity 99.95%).
PATENT
https://patents.google.com/patent/CN103880864B/enFaropenem sodium is developed by Japanese Suntory companies, and first penemss antibiosis in listing in 1997 Element, it are similar to the several carbapenem antibiotics for listing, strong with has a broad antifungal spectrum, antibacterial activity, to beta-lactamase Stably, the features such as also having good action to extended spectrumβ-lactamase producing strains, citrobacter, enterococcus and anaerobe etc.. It is first orally active, penems antibiotics stable to beta-lactamase in the world so far.Its structural formula As follows:
Report about Faropenem sodium preparation method is a lot, mainly has several as follows:1st, J. Antibiotics 1988, the method that reports in 41,1685, see below row reaction equation:
Acyl group substitution reaction is carried out in the basic conditions with 4-AA and three beneze methane thiols and obtains thio trityl as protecting group Aza cyclo-butanone, then when 2-TETRAHYDROFUROYL chlorine is connected with lactams, using silver nitrate as condensing agent, but nitric acid Silver is expensive, and cost is too high, while the silver chloride for generating is difficult to filter, is not suitable for large-scale production.2nd, the classical preparation method of United States Patent (USP) US4997829 report:There is acyl with (R) tetrahydrofuran -2- thiocarboxylic acids Base substitution reaction generates thioesters, then through condensation, chlorine replacement, intramolecular Witting cyclization, slough hydroxyl protecting group and carboxylic Base protection group obtains product, and this synthetic route yield is very low, while side chain is thio-compoundss, abnormal smells from the patient is extremely smelly, and prepares complexity, There is-fixed harm to human body and environment.It is also required in chloro building-up process using pungent thionyl chloride, these factors are all It is unfavorable for industrialized production
3rd, the method that reports in Chinese patent CN1314691 is as follows:
Said method route is shorter, is produced using one kettle way, more convenient.But said method is related to some other salt such as acetate using heavy metal palladium in last operation The deprotecting regent of compound and triphenyl phosphorus together as pi-allyl, metal palladium reagent is expensive, while triphenyl phosphorus are most More difficult removing in step afterwards, increases operation difficulty, affects product quality.Allyloxy is used easily to produce as protection group simultaneously A kind of double bond olefinic polymerization species impurity of life, affects product quality, reduces yield.Embodiment one(R) tetrahydrofuran -2- thiocarboxylic acids (198g, 1.5 mol) are put in 3L reaction bulbs, plus 1 mol/L hydrogen-oxygens Change sodium body lotion (I.5 L) to be adjusted at 5 DEG C of pH 9- 10,0-, Deca 4AA(287g, 1. 0mo l) acetone (1 L) Solution, drop are finished, and are adjusted to pH 8 or so, 2 h of room temperature reaction with 1 mol/L sodium hydroxide. and add water (500 ml) dilution, second Acetoacetic ester (600 ml x3) is extracted, and merges organic layer, successively with 5 % sodium bicarbonate solutions (300 ml x 2) and water (300 m1 x 2) is washed, and anhydrous sodium sulphate is dried, and is filtered, and filtrate concentrates, and obtains pale yellow oil (about 360 g), directly Input the next step.Embodiment twoThe mixing of concentrated solution as obtained above, triethylamine (l70g, 1.7 mol) and dichloromethane (1.5 L), 0-5 DEG C Deca chlorine oxalic acid is finished to p-Nitrobenzyl (414.1 g, 1 .7 mo l), drop, and equality of temperature reacts 2 h, and add water (1 L) dilution, Extracted with dichloromethane (500 ml x 4), merge organic layer, molten with water (300m1 x 2) and 5 % sodium bicarbonate successively Liquid (300 m1 x 2) is washed, anhydrous sodium sulfate drying, is filtered, and concentration obtains pale yellow oil (about 530g), direct plunges into The next step..Embodiment threeAbove-mentioned gained grease, dimethylbenzene (4L) and NSC 5284 (500ml) are mixed, heating reflux reaction 5h , reduce pressure and boil off dimethylbenzene and NSC 5284, residue ethyl acetate-hexane (1:5,1 L) recrystallization, obtain yellowish Color solid (334.3g, 61%, in terms of 4AA).Example IVAbove-mentioned solid (0.60 mol of 330g.) is dissolved in methanol (2 L), adds 1.0M hydrochloric acid (0.4 L), adds palladium carbon (15.0 g), hydrogen is passed through, 40 DEG C of stirrings, response time are 16 h, and the pressure of system is 4atm, after reaction terminates, crosses and filters Catalyst is removed, is concentrated.Embodiment fiveThe product obtained after above-mentioned concentration is dissolved in tetrahydrofuran 600ml, the 2 ethyl hexanoic acid sodium of 100.0g is added Tetrahydrofuran(200ml)And water(200 ml)Mixed solution, 2 h are stirred at room temperature, have faint yellow solid generate, filter, be method Faropenem crude product 147.0g.Embodiment sixBy above-mentioned solid deionized water(2200ml)Acetone is slowly added under dissolved solution, stirring to start to become to solution Muddiness, when about adding acetone 750ml, solution starts to become cloudy, and stops adding, and continues stirring and allows its crystallize overnight, sucking filtration, acetone Washing, dries, and obtains the Faropenem sodium fine work 125.0g of white.
Syn
AU 8654460; EP 0199446; JP 1994128267; US 4997829
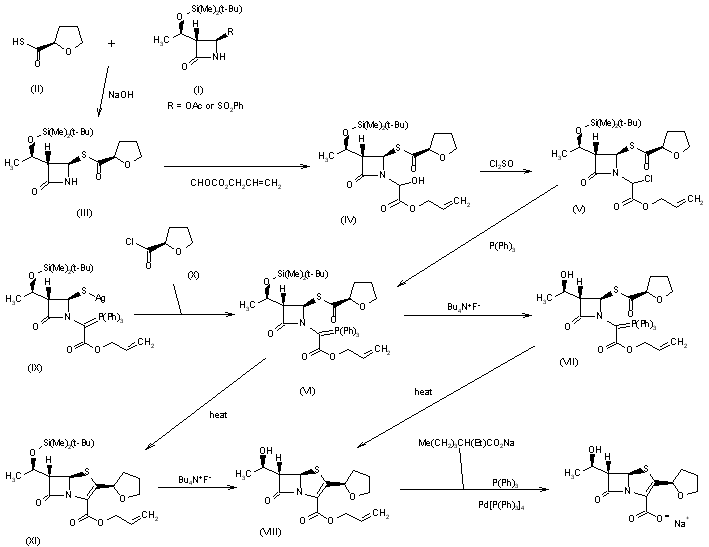
This compound is prepared by several related ways: 1) The reaction of silylated azetidinone (I) with tetrahydrofuran-2-thiocarboxylic acid (II) by means of NaOH in THF – water gives the azetidinone thioester (III), which is condensed with allyl glyoxylate in refluxing benzene yielding the hydroxyester (IV). The reaction of (IV) with SOCl2 affords the chloroester (V), which by reaction with triphenylphosphine by means of lutidine in hot THF is converted into the phosphoranylidene derivative (VI). The elimination of the silyl protecting group of (VI) with tetrabutylammonium fluoride gives the azetidinone (VII), which is cyclized in refluxing toluene yielding the (5R,6S)-6-[1(R)-hydroxyethyl]-2-[2(R)-tetrahydrofuryl]penem-3-carboxyli c acid allyl ester (VIII). Finally, this compound is hydrolyzed with triphenylphosphine, sodium 2-ethylhexanoate and Pd-tetrakis(triphenylphosphine). 2) The condensation of the silver salt of protected azetidinone (IX) with tetrahydrofuran-2(R)-carbonyl chloride (X) also yields the phosphoranylidene salt (VI). 3) Phosphoranylidene ester (VI) can also be cyclized first in refluxing benzene yielding the silylated penem ester (XI), which is deprotected with tetrabutylammonium fluoride to (VIII). 4) The hydrolysis of allyl ester (VIII) to the final product can also be performed with paladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)-5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethylacetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene, or ethylene glycol dimethyl ether. 5) The preceding hydrolysis can also be performed with triphenylphosphine and paladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone.
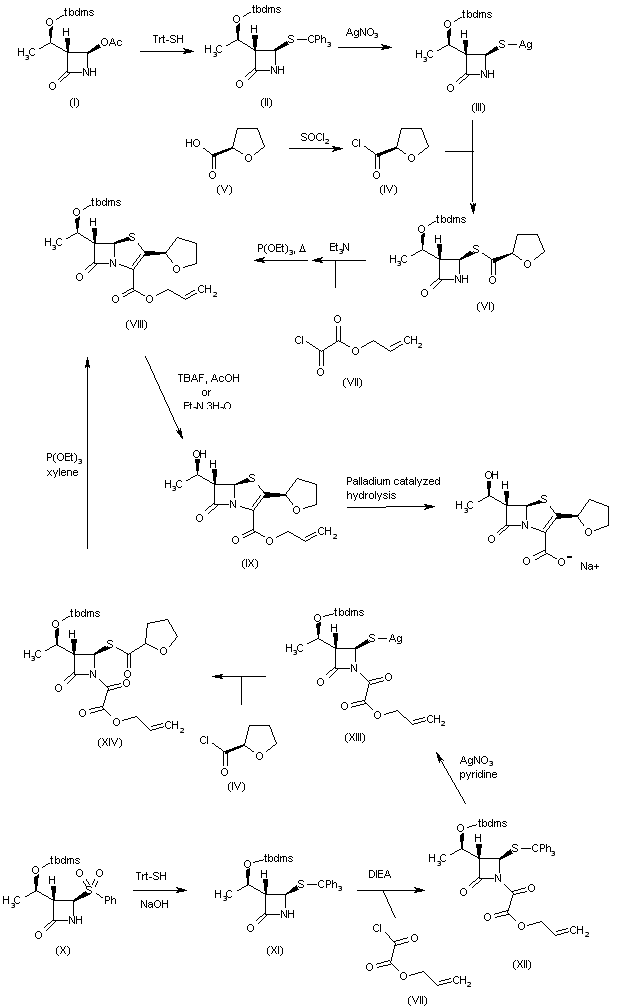
Treatment of the silylated azetidinone (I) with tritylmercaptan affords the tritylsulfanyl-azetidinone (II), which is converted into the silver salt (III) by reaction with AgNO3. Compound (III) is coupled with tetrahydrofuran-2(R)-carbonyl chloride (IV) — obtained by treatment of carboxylic acid (V) with thionyl chloride — providing the azetidinone thioester (VI). Coupling of azetidinone (VI) with allyl oxalyl chloride (VII) in CH2Cl2 by means of Et3N, followed by intramolecular Wittig cyclization by means of triethyl phosphite in refluxing xylene, affords penem (VIII). Alternatively, compound (VIII) can also be obtained as follows: Substitution of phenyl sulfonyl group of azetidinone (X) by tritylmercaptan by means of NaOH in acetone/water provides tritylsulfanyl-azetidinone (XI), which is condensed with allyl oxalyl chloride (VII) by means of DIEA in CH2Cl2 to give the oxalyl amide (XII). Compound (XII) is then treated with AgNO3 and pyridine in acetonitrile, providing the silver mercaptide (XIII), which is acylated with tetrahydrofuran-2(R)-carbonyl chloride (IV) in acetonitrile to afford the penem precursor (XIV). Penem (VIII) is obtained by intramolecular Wittig cyclization of (XIV) with P(OEt)3 in refluxing xylene. Finally, faropenem sodium can be obtained by removal of the tbdms protecting group of (VIII) by means of either Et3N tris(hydrogen fluoride) in ethyl acetate or tetrabutylammonium fluoride (TBAF) and HOAc in THF to give compound (IX). This is followed by allyl ester group removal of (IX), which can be performed under several different conditions: i) triphenylphosphine, sodium 2-ethylhexanoate and palladium tetrakis(triphenylphosphine); ii) palladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)-5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethyl acetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene or ethylene glycol dimethyl ether; iii) triphenylphosphine and palladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone; or iv) palladium acetate in the presence of P(OBu)3 and sodium propionate in THF.
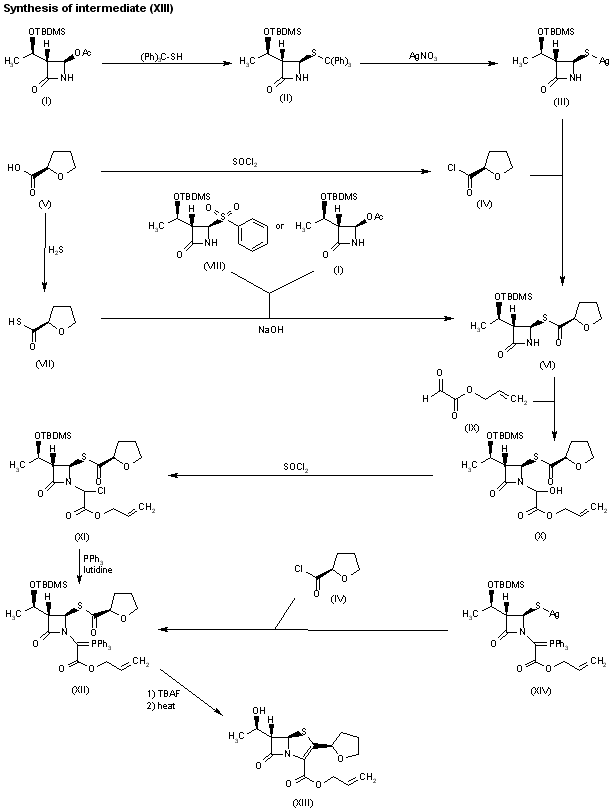
Treatment of the silylated azetidinone (I) with tritylmercaptan affords the tritylsulfanylazetidinone (II), which by reaction with AgNO3 is converted into the silver salt (III). Compound (III) is coupled with tetrahydrofuran-2(R)-carbonyl chloride (IV) ?obtained by treatment of carboxylic acid (V) with thionyl chloride ?to provide the azetidinone thioester (VI). Alternatively, compound (VI) can be obtained by condensation of tetrahydrofuran-2(R)-thiocarboxylic S-acid (VII) ?obtained by treatment of carboxylic acid (V) with hydrogen sulfide ?with silylated azetidinones (I) or (VIII) by means of NaOH in THF/water. Condensation of azetidinone thioester (VI) with allyl glyoxylate (IX) in refluxing benzene gives the hydroxy ester (X), which is treated with SOCl2 to yield the chloro ester (XI). Reaction of compound (XI) with triphenylphosphine and lutidine in hot THF provides the phosphoranylidene derivative (XII), which is converted into (5R,6S)-6-[1(R)-hydroxyethyl]-2-[2(R)-tetrahydrofuryl]penem-3-carboxylic acid allyl ester, faropenem allyl ester (XIII) by removal of the silyl protecting group with tetrabutylammonium fluoride, followed by cyclization in refluxing toluene. Compound (XII) can also be obtained by condensation of the silver salt of protected azetidinone (XIV) with tetrahydrofuran-2(R)-carbonyl chloride (V).
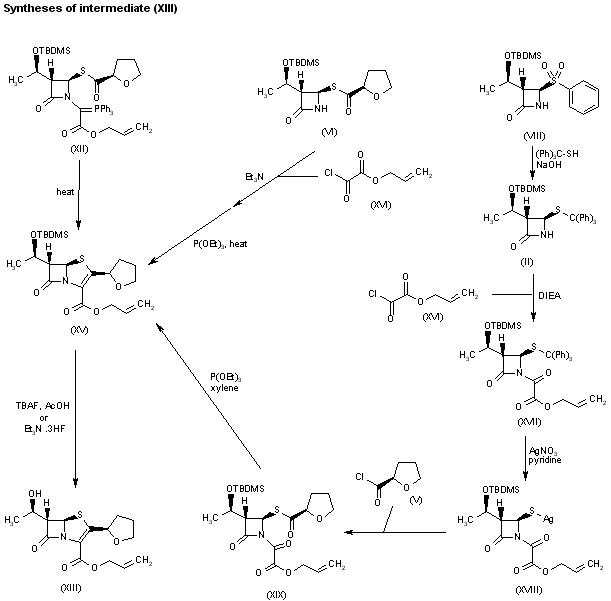
Alternatively, faropenem allyl ester (XIII) can also be prepared by cyclization of compound (XII) in refluxing benzene to yield silylated penem allyl ester (XV), which is then deprotected with either tetrabutylammonium fluoride in AcOH or triethylamine tris(hydrogen fluoride) in methyl isobutyl ketone or toluene. Penem (XV) can also be synthesized by several related ways: a) By coupling of azetidinone (VI) with allyl oxalyl chloride (XVI) in CH2Cl2 by means of Et3N, followed by intramolecular Wittig cyclization by means of triethyl phosphite in refluxing xylene. b) Substitution of phenyl sulfonyl group of azetidinone (VIII) by tritylmercaptan by means of NaOH in acetone/water provides tritylsulfanyl-azetidinone (II), which is condensed with allyl oxalyl chloride (XVI) by means of DIEA in CH2Cl2 to give the oxalyl amide (XVII). Compound (XVII) is then treated with AgNO3 and pyridine in acetonitrile to provide the silver mercaptide (XVIII), which is acylated with tetrahydrofuran-2(R)-carbonyl chloride (IV) in acetonitrile to afford the penem precursor (XIX). Finally, compound (XV) is obtained by intramolecular Wittig cyclization of (XX) with P(OEt)3 in refluxing xylene.
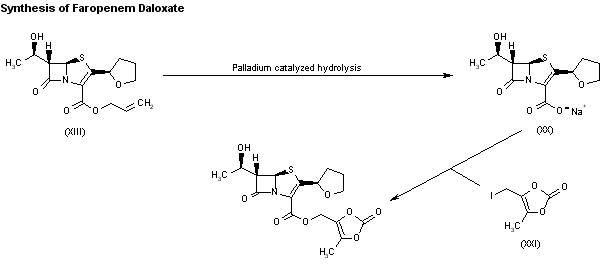
Hydrolysis of faropenem allyl ester (XIII) to faropenem sodium (XX) can be performed under several different conditions: i) triphenylphosphine, sodium 2-ethylhexanoate and palladium tetrakis(triphenylphosphine); ii) palladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)- 5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethyl acetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene, or ethylene glycol dimethyl ether; iii) triphenylphosphine and palladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone; and iv) palladium acetate in the presence of P(OBu)3 and sodium propionate in THF. Finally, faropenem daloxate can be directly obtained from faropenem sodium (XX) by esterification with 4-(iodomethyl)-5-methyl-1,3-dioxol-2-one (XXI) in DMF.
PATENT
https://patents.google.com/patent/CN103059046A/enFaropenem (Faropenem), chemistry (5R, 6S)-6-[(1R)-hydroxyethyl by name]-2-[(2R)-and tetrahydrofuran (THF)] penem-3-carboxylic acid list sodium salt, by the first exploitation listing in 1997 years of Japanese Suntory company.This medicine is a kind of atypical beta-lactam penems antibiotics, has very strong anti-microbial activity, especially to the anti-microbial activities of the anerobes such as the gram positive organisms such as golden Portugal bacterium, penicillin-fast streptococcus pneumoniae, streptococcus faecium and bacteroides fragilis apparently higher than existing cynnematin, anti-gram-negative bacteria is active similar to oral cephalosporin, and is stable to various β-lactamases.Various clinical studyes show that this medical instrument has clinical effectiveness good, safe, the advantage that renal toxicity and neurotoxicity are little.Its structural formula is as follows:
For synthesizing of Faropenem, existing many reports in the prior art, for example CN101125857A has reported following synthetic route:
Take (3R, 4R)-3-[(R)-1-tert-butyl dimethyl silica ethyl]-4-[(R)-and acetoxyl group] nitrogen heterocyclic din-2-ketone is as starting raw material, and warp gets intermediate compound I with R-(+)-sulfo-tetrahydrofuran (THF)-2-formic acid condensation; Intermediate compound I is carried out acylation reaction with monoene propoxy-oxalyl chloride under the catalysis of alkali, get intermediate II; Intermediate II cyclization under the effect of triethyl-phosphite gets intermediate III; Intermediate III is sloughed hydroxyl protecting group through the effect of tetrabutylammonium, gets intermediate compound IV; Intermediate compound IV decarboxylize protecting group under [four (triphenylphosphine)] palladium and triphenylphosphine effect gets Faropenem.Find that after deliberation the method for the present synthetic Faropenem of reporting is all similar with the disclosed method of above-mentioned CN101125857A, all need remove in two steps the protecting group of hydroxyl and carboxyl, reaction scheme is longer.When removing above-mentioned protecting group, need to use a large amount of tetrabutylammonium and [four (triphenylphosphine)] palladium and triphenylphosphine; these reagent costs are high, toxicity is large; be unfavorable for large industrial production; and can introduce the heavy metal palladium; so that the heavy metal remnants in the Faropenem exceed standard, be not suitable for the production of bulk drug.And when adopting aforesaid method deprotection base, the yield in per step only can reach 60%-75%, has further increased production cost.Embodiment 6The preparation of FaropenemWith intermediate 3(364.5g, 0.8mol) use the 700mL acetic acid ethyl dissolution, to open and stir, 0 ℃ of lower dropping with the 36g trifluoroacetic acid after the dilution of 100mL ethyl acetate dripped off in 1 hour, 0 ℃ of lower reaction 2h that continues.Stopped reaction stirs the sodium bicarbonate aqueous solution of lower dropping 5%, until reaction solution pH is neutral.Emit water layer from the reactor lower end, discard.In reactor, add gradually the ethanolic soln of sodium bicarbonate, until till no longer including solid and separating out.Suction filtration, filter cake gets white solid powder 230g(productive rate 93.7% with acetone-water (10:3, v/v) recrystallization), M.P. 163-164 ℃, detect through HPLC, purity is 99.8%Reference examples 1(5R, 6S)-6-[(R)-1-hydroxyethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] preparation of penem-3-carboxylic acid propyleneWith (5R, 6S)-6-[(R)-the 1-tert-butyl dimethyl silica ethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] penem-3-carboxylic acid propylene (150g, 0.342mol) and ammonium bifluoride (59.5g, 1.025mmol) add successively among the 400mL DMF, 55~60 ℃ were reacted 5 hours, stopped reaction, suction filtration, filtrate adds water 800ml, uses ethyl acetate extraction, and organic phase is washed with 5% sodium hydrogen carbonate solution, anhydrous sodium sulfate drying, concentrated, gained incarnadine oily matter gets yellow solid 73g through the petrol ether/ethyl acetate recrystallization, yield 66%.Reference examples 2The preparation of Faropenem(the 5R that reference examples 1 is prepared, 6S)-6-[(R)-the 1-hydroxyethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] penem-3-carboxylic acid propylene (73g, 0.224mol), 6.5g triphenylphosphine, 6.5g [four (triphenylphosphine)] palladium adds among the 500mL methylene dichloride l successively, the ethyl acetate solution that adds the 2 ethyl hexanoic acid sodium preparation of 500mL 0.5M, stirring at room 1 hour, stopped reaction adds 15mL water in reaction solution, stir 30min, suction filtration, this solid is dissolved in the 100mL water again, adds decolorizing with activated carbon 30min, filter, filtrate adds in the 500mL acetone, place crystallization, get Faropenem 66g, yield 96%.Find that by contrast the total recovery that two steps of reference examples remove hydroxyl and carboxyl-protecting group only has about 63.4%, and single stage method of the present invention removes the yield of hydroxyl and carboxyl-protecting group and can reach more than 90%.Preparation method of the present invention can the one-step removal hydroxyl and carboxyl on protecting group, shortened the production cycle, the deprotecting regent cost is low, toxicity is little, can not cause heavy metal remaining, and have higher reaction yield, is fit to very much the industrial production of raw material medicine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCN1939924A *2006-09-082007-04-04鲁南制药集团股份有限公司Industrial production of Fallopeinan sodiumWO2008035153A2 *2006-08-022008-03-27Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of beta-lactam antibioticCN103059046A *2013-01-282013-04-24苏州二叶制药有限公司Preparation method of faropenemFamily To Family CitationsCN100522975C *2007-08-232009-08-05东北制药集团公司沈阳第一制药厂Method for preparing faropenemPublication numberPriority datePublication dateAssigneeTitleCN1884284A *2005-06-212006-12-27浙江金华康恩贝生物制药有限公司Process for the preparation of sodium faropenemCN1939924A *2006-09-082007-04-04鲁南制药集团股份有限公司Industrial production of Fallopeinan sodiumCN101125857A *2007-08-232008-02-20东北制药集团公司沈阳第一制药厂Method for preparing faropenemWO2008035153A2 *2006-08-022008-03-27Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of beta-lactam antibiotic
Publication numberPriority datePublication dateAssigneeTitle
EP0410727A1 *1989-07-261991-01-30Suntory LimitedProcesses for removing allyl groupsUS4997829A *1985-03-091991-03-05Suntory LimitedPenem compounds, and use thereofEP0574940A1 *1992-06-181993-12-22Tanabe Seiyaku Co., Ltd.Method for removing the protecting group for carboxyl groupWO2007039885A1 *2005-10-052007-04-12Ranbaxy Laboratories LimitedA process for the preparation of faropenemFamily To Family Citations
Publication numberPriority datePublication dateAssigneeTitleCN102964357A *2012-11-112013-03-13苏州二叶制药有限公司Faropenem sodium and tablet thereofCN103059046A *2013-01-282013-04-24苏州二叶制药有限公司Preparation method of faropenemCN103880864A *2014-03-252014-06-25江苏正大清江制药有限公司Method for synthesizing faropenem sodiumCN104086516A *2014-07-182014-10-08成都樵枫科技发展有限公司Synthetic method of R-(+)-sulfotetrahydrofuran-2-formic acidCN101941981B *2009-07-032015-01-21湖南华纳大药厂有限公司Catalyst composition and method for preparing faropenem sodiumCN106860405A *2015-12-142017-06-20山东新时代药业有限公司A kind of faropenem sodium granules and preparation method thereofCN108840877A *2018-06-122018-11-20赤峰迪生药业有限责任公司A kind of preparation method of oxygen cephalosporin intermediate
References
- ^ Critchley IA, Brown SD, Traczewski MM, Tillotson GS, Janjic N (December 2007). “National and regional assessment of antimicrobial resistance among community-acquired respiratory tract pathogens identified in a 2005-2006 U.S. Faropenem surveillance study”. Antimicrob. Agents Chemother. 51 (12): 4382–9. doi:10.1128/AAC.00971-07. PMC 2168020. PMID 17908940.
- ^ Mushtaq S, Hope R, Warner M, Livermore DM (May 2007). “Activity of faropenem against cephalosporin-resistant Enterobacteriaceae”. J. Antimicrob. Chemother. 59 (5): 1025–30. doi:10.1093/jac/dkm063. PMID 17353220.
- ^ Milazzo I, Blandino G, Caccamo F, Musumeci R, Nicoletti G, Speciale A (March 2003). “Faropenem, a new oral penem: antibacterial activity against selected anaerobic and fastidious periodontal isolates”. J. Antimicrob. Chemother. 51 (3): 721–5. doi:10.1093/jac/dkg120. PMID 12615878.
- ^ Gettig JP, Crank CW, Philbrick AH (January 2008). “Faropenem medoxomil”. Ann Pharmacother. 42 (1): 80–90. doi:10.1345/aph.1G232. PMID 18094341. Archived from the original on 2013-02-03.
- ^ (Q1 06 Investor Conf Call)(CID 6918218 from PubChem)
External links
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| Routes of administration | Oral |
| ATC code | J01DI03 (WHO) |
| Identifiers | |
| CAS Number | 106560-14-9 |
| PubChem CID | 65894 |
| ChemSpider | 59303 |
| UNII | F52Y83BGH3 |
| ChEBI | CHEBI:51257 |
| ChEMBL | ChEMBL556262 |
| CompTox Dashboard (EPA) | DTXSID0046430 |
| Chemical and physical data | |
| Formula | C12H15NO5S |
| Molar mass | 285.31 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
///////////Faropenem, ALP-201, SUN-5555, SY-5555, WY-49605, ANTIBACTERIALS, DIICHI, Daiichi Asubio Pharma

NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
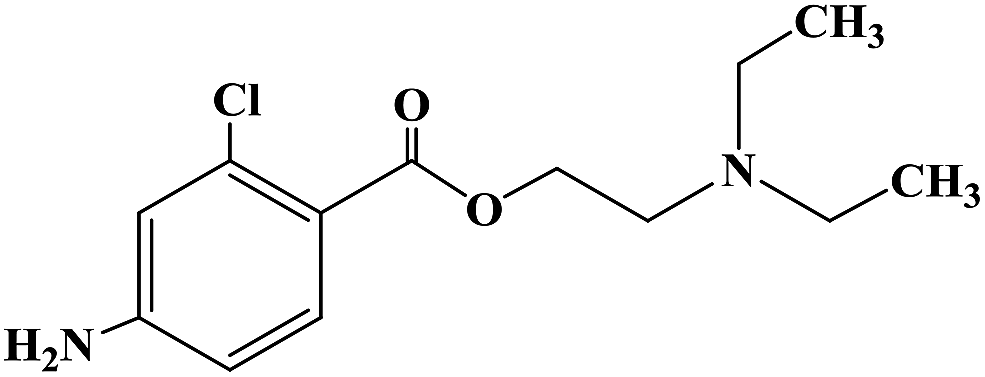{kind=link}
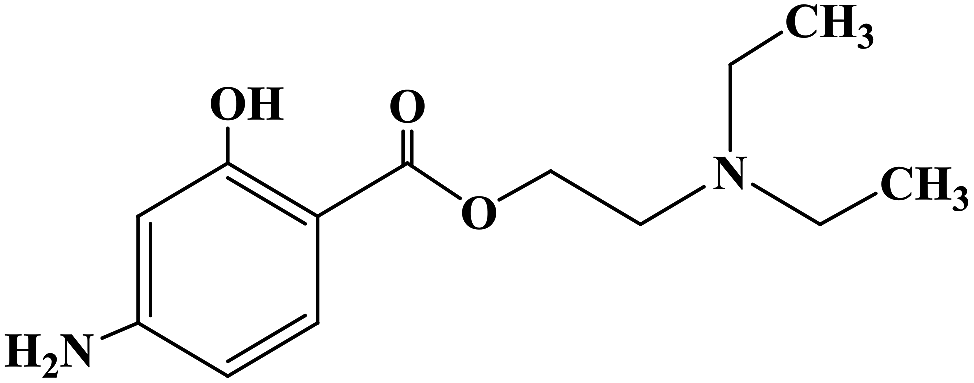{kind=link}
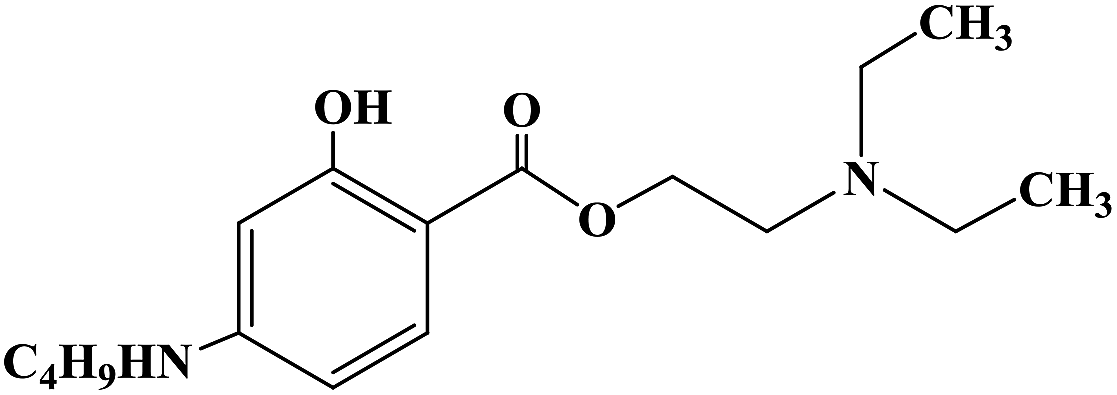{kind=link}
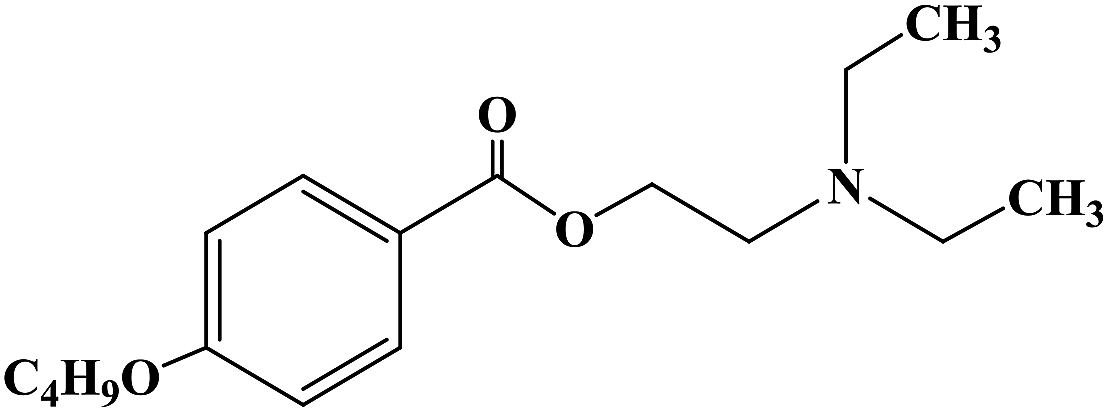{kind=link}
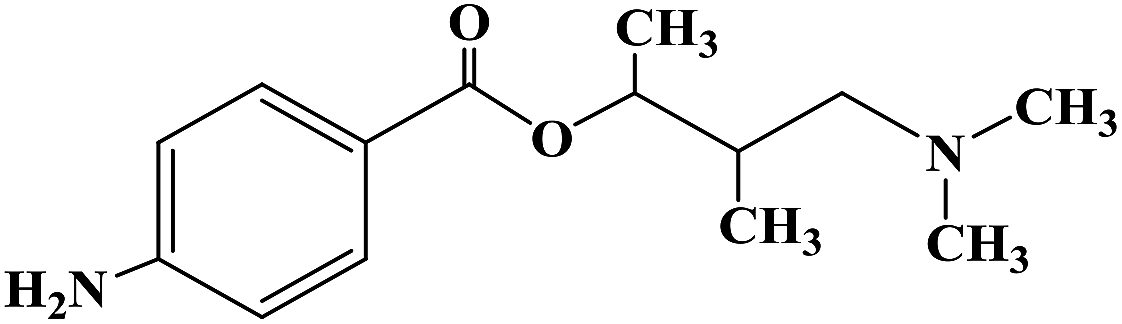{kind=link}
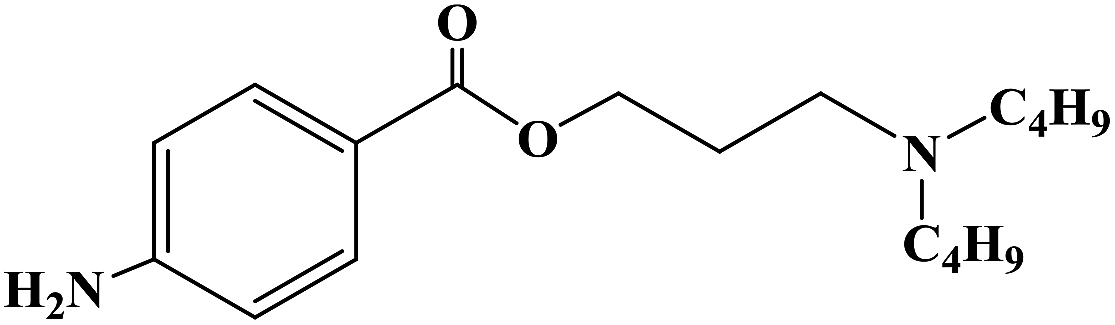{kind=link}
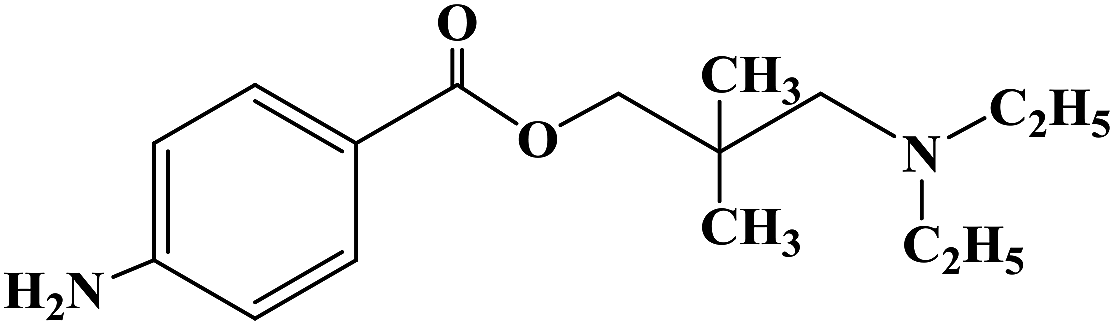{kind=link}
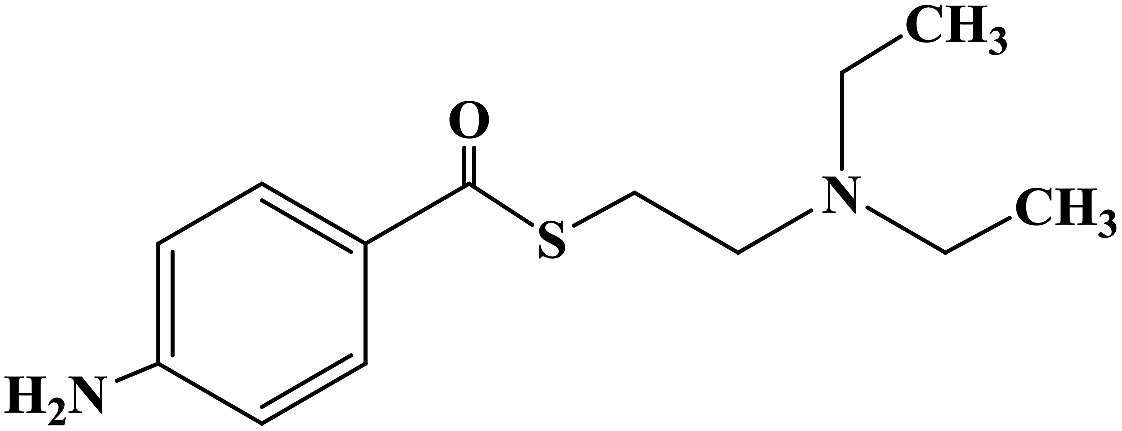{kind=link}
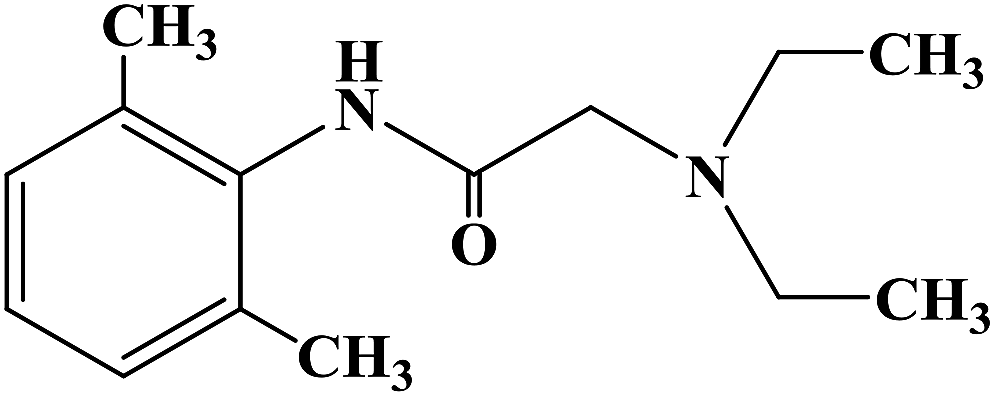{kind=link}
{kind=link}
{kind=link}
{kind=link}
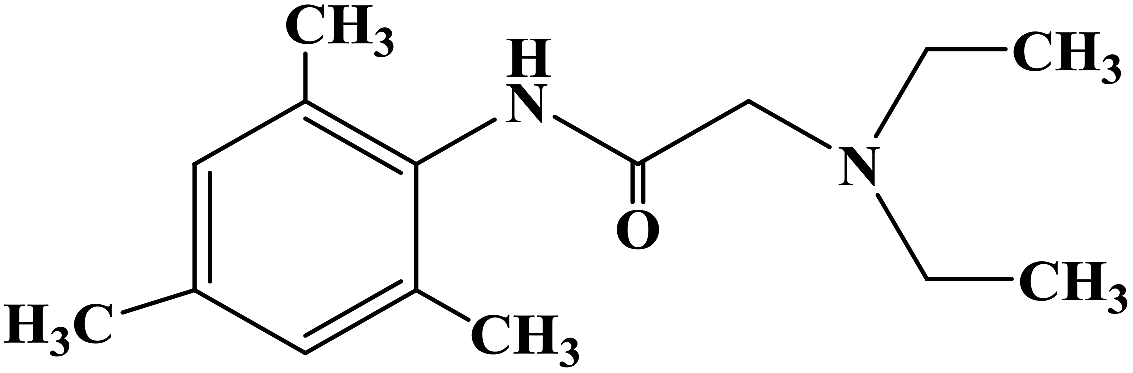{kind=link}
{kind=link}
{kind=link}
{kind=link}
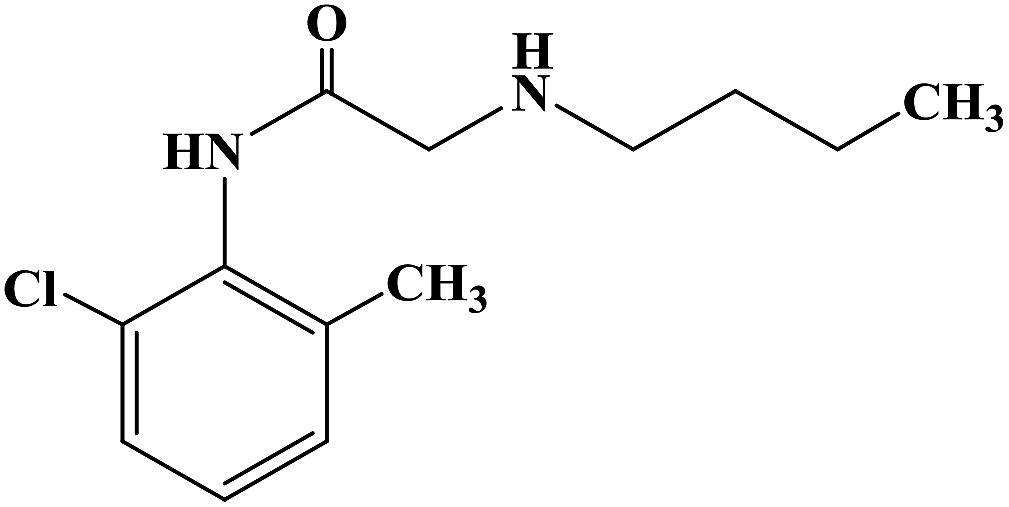{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
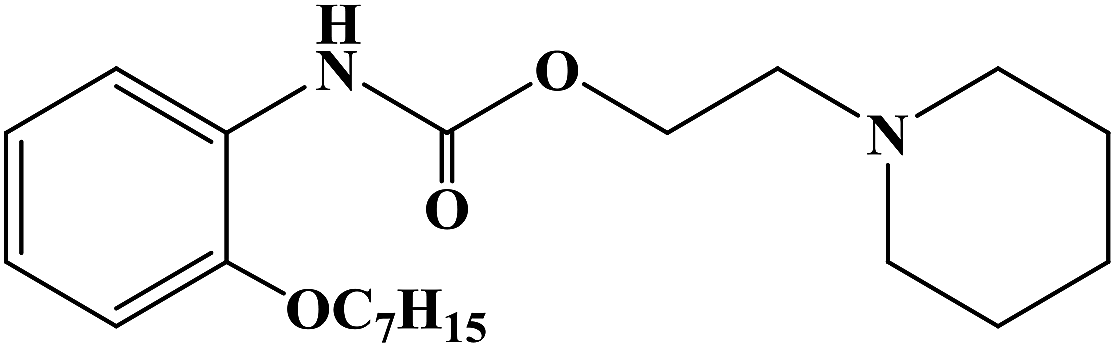{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
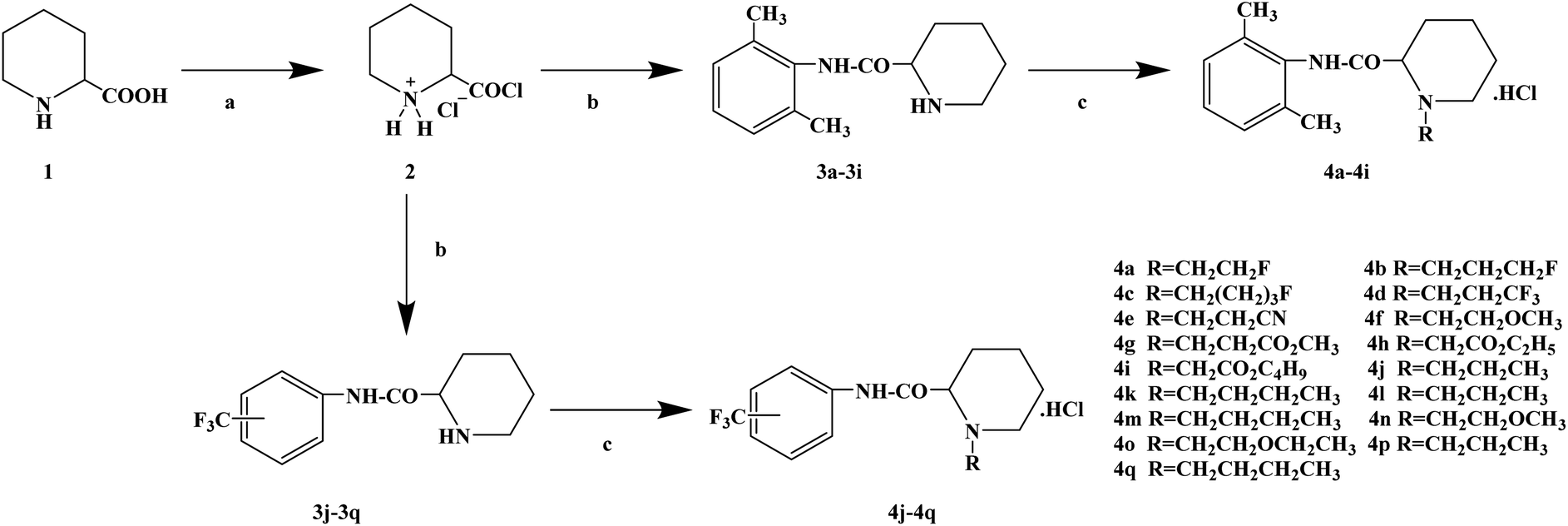{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}