
Home » DIABETES
Category Archives: DIABETES
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
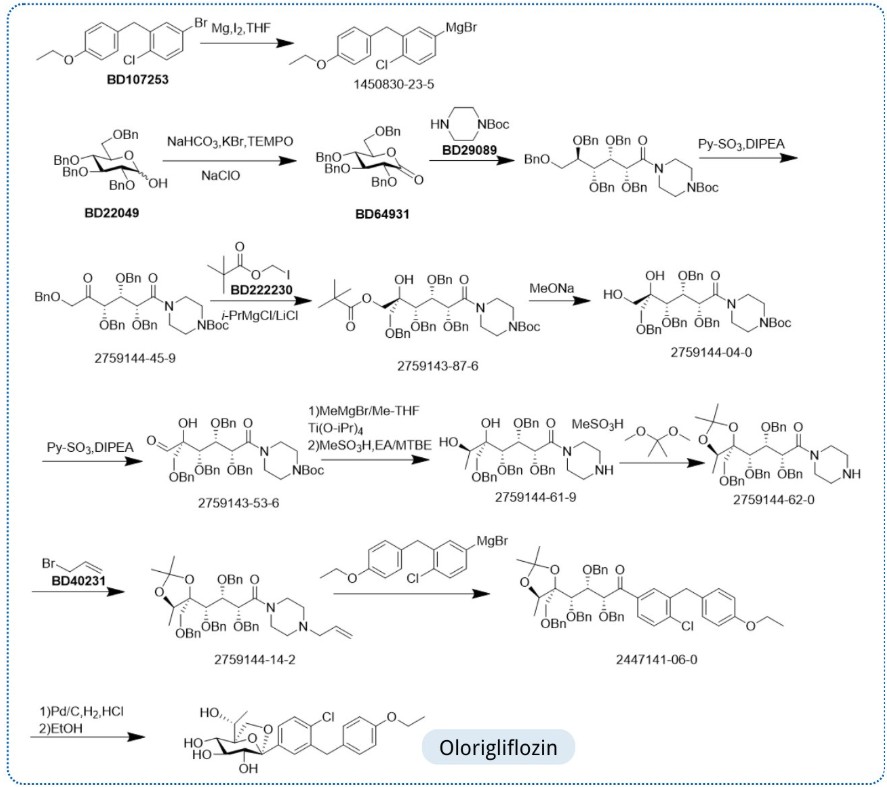
Olorigliflozin, Rongliflozin
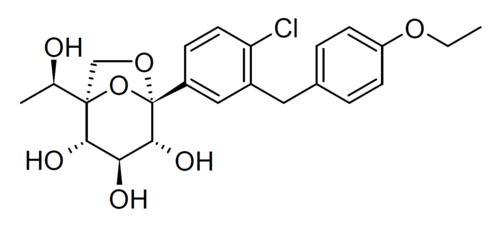
Rongliflozin
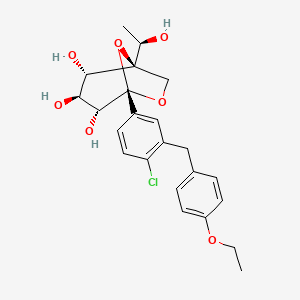
Olorigliflozin, 6FP3NST6ZQ, DJT1116PG
Cas 2035989-50-3
450.9 g/mol, C23H27ClO7
1-C-{4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl}-1-O,5-Cmethylene-D-glycero-α-D-gluco-heptopyranose
sodium glucose co-transporter inhibitor, antihyperglycaemic,
(1R,2S,3S,4R,5S)-5-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-1-[(1R)-1-hydroxyethyl]-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
- (1R,2S,3S,4R,5S)-5-(4-Chloro-3-(4-ethoxybenzyl)phenyl)-1-((R)-1-hydroxyethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
- 1,6-Anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-beta-L-idopyranose
- beta-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-
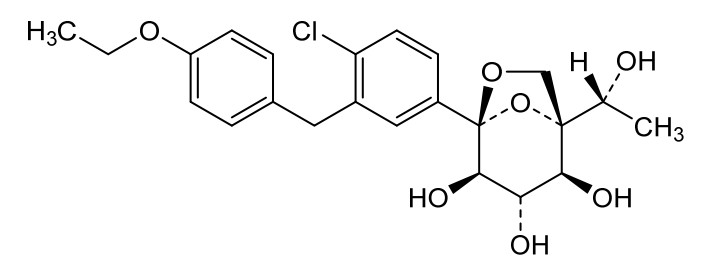
Rongliflozin 화학구조
CAS No. : 2648020-91-9
| MW | 602.55 |
|---|---|
| MF | C23H27ClO7.C5H7NO3.5/4H2O |
- OriginatorHEC Pharm
- DeveloperSunshine Lake Pharma
- ClassAntihyperglycaemics; Small molecules
- Mechanism of ActionSodium-glucose transporter 2 inhibitors
- PreregistrationType 2 diabetes mellitus
- 04 Sep 2025Chemical structure information added.
- 31 Dec 2023Preregistration for Type 2 diabetes mellitus in China (PO), in December 2023
- 31 Dec 2023Efficacy and adverse events data from a phase IIIa trial in Type 2 diabetes mellitus released by Sunshine Lake Pharma, before December 2023
HEC Pharma announced that its independently developed Olorigliflozin (previously known as rongliflozin pyroglutamate) capsules, has been approved for marketing by the China NMPA. It can be used as a monotherapy or in combination with metformin to improve glycemic control in adults with T2DM.
Olorigliflozin is a SGLT2 inhibitor that moderately inhibits SGLT1. It promotes urinary glucose excretion by potently inhibiting renal SGLT2 receptors and reduces glucose or galactose absorption by moderately inhibiting intestinal SGLT1 receptors, effectively reducing drastic postprandial blood glucose fluctuations.
Rongliflozin is an SGLT2 inhibitor developed as a potential treatment for diabetes.[1][2]
Rongliflozin (DJT1116PG) is a selective and orally active inhibitor of sodium-glucose co-transporter-2 (SGLT-2). Rongliflozin can be used for the research of type 2 diabetes mellitus (T2DM).
On January 16, 2026, Olorigliflozin, a small-molecule antidiabetic drug was approved by the National Medical Products Administration (NMPA) of China for improving glycemic control in adult patients with type 2 diabetes mellitus. Its synthetic route highlights a key and recurring feature of SGLT inhibitors
a glucose-derived core, as a sugar ring is present in the majority of approved SGLT2 inhibitors and serves as the structural foundation of these molecules.
PAT
- (1R,2S,3S,4R,5S)-5-(4-Chloro-3-(4-ethoxybenzyl)phenyl)-1-((R)-1-hydroxyethyl)-6,8-dioxabicyclo[3.2.1]octane-2,3,4-triol
- 1,6-Anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-beta-L-idopyranose
- beta-L-Idopyranose, 1,6-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-5-C-[(1R)-1-hydroxyethyl]-
- Complexes of glucopyranosyl derivatives and methods for their preparation and usePublication Number: JP-2018535237-APriority Date: 2015-11-27
- Complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: US-10555930-B2Priority Date: 2015-11-27Grant Date: 2020-02-11
- Complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: US-2018344689-A1Priority Date: 2015-11-27
- A complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: WO-2017088839-A1Priority Date: 2015-11-27
- Glucopyranosyl derivative complex and its preparation method and usePublication Number: JP-6916180-B2Priority Date: 2015-11-27Grant Date: 2021-08-11
- Preparation method and intermediate of glucopyranosyl derivativesPublication Number: CN-113195510-BPriority Date: 2019-01-08Grant Date: 2022-12-23
- Crystalline forms of glucopyranosyl derivativesPublication Number: CN-107778336-BPriority Date: 2016-08-24Grant Date: 2022-09-27
- Glucopyranosyl derivative compound, preparation method and applicationPublication Number: CN-106810582-APriority Date: 2015-11-27
- Glucopyranosyl derivative compound, preparation method and applicationPublication Number: CN-106810582-BPriority Date: 2015-11-27Grant Date: 2019-12-31
- A complex of a glucopyranosyl derivative and preparation method and use thereofPublication Number: EP-3371199-A1Priority Date: 2015-11-27
- Method for preparing glucopyranosyl derivatives and intermediates thereofPublication Number: WO-2022007838-A1Priority Date: 2020-07-08
- Method for preparing glucopyranosyl derivatives and intermediates thereofPublication Number: EP-4178970-A1Priority Date: 2020-07-08
- Method for preparing glucopyranosyl derivatives and intermediates thereofPublication Number: US-2023250121-A1Priority Date: 2020-07-08
- Preparation methods of glucopyranosyl derivatives and intermediates thereofPublication Number: CN-113912567-BPriority Date: 2020-07-08Grant Date: 2024-01-16
- Preparation method for glucopyranosyl derivative and intermediate thereofPublication Number: WO-2020143653-A1Priority Date: 2019-01-08
- Composition and use of sglt-2 inhibitor and angiotensin receptor blockersPublication Number: WO-2022036506-A1Priority Date: 2020-08-17
- Composition and use of sglt-2 inhibitor and angiotensin receptor blockersPublication Number: EP-4197543-A1Priority Date: 2020-08-17
- Compositions of SGLT-2 inhibitors and angiotensin receptor antagonists and uses thereofPublication Number: KR-20230057388-APriority Date: 2020-08-17
- Composition and application of SGLT-2 inhibitor and angiotensin receptor blockerPublication Number: CN-116490178-APriority Date: 2020-08-17
- Composition and use of sglt-2 inhibitor and angiotensin receptor blockersPublication Number: US-2023346817-A1Priority Date: 2020-08-17
- Nintedanib targeted combinationPublication Number: CN-118021812-APriority Date: 2023-12-30
- Preparation method of L-pyroglutamic acid co-crystal of glucopyranosyl derivativesPublication Number: CN-115141235-APriority Date: 2021-03-30
- Preparation method of L-pyroglutamic acid cocrystal of pyranose glucopyranose derivativePublication Number: CN-115141235-BPriority Date: 2021-03-30Grant Date: 2024-08-09
- Fixed-dose combination of sglt-2 inhibitor and angiotensin converting enzyme inhibitor, and use thereofPublication Number: WO-2022104621-A1Priority Date: 2020-11-19
- Compositions and uses of fixed-dose SGLT-2 inhibitors and angiotensin-converting enzyme inhibitorsPublication Number: CN-116234545-APriority Date: 2020-11-19
SYN
SYN
https://pubs.rsc.org/en/content/articlelanding/2021/ce/d1ce01305j/unauth
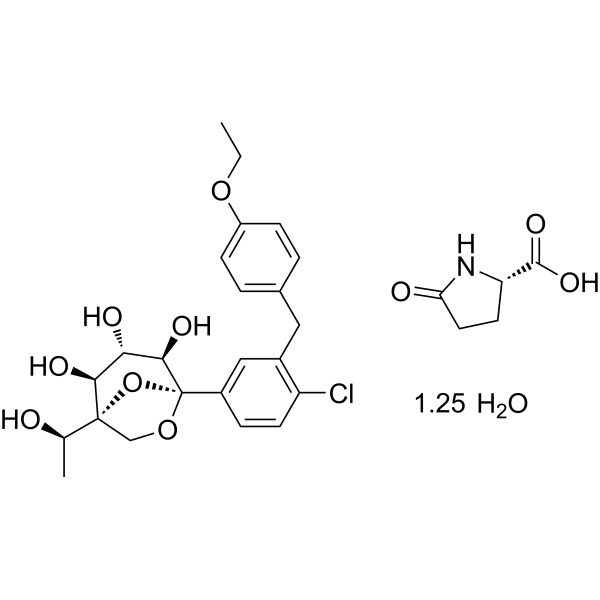
Rongliflozin L-pyroglutamic acid, a highly active SGLT-2 inhibitor cocrystal discovered and developed by our group, is currently undergoing clinical trials for the treatment of diabetes. Here, we report and design a simple and robust process to obtain a single and pure crystalline form I (1) of the cocrystal, containing Rongliflozin (2) with L-pyroglutamic acid (L-PA), based on coformer-induced purification (CoIP). Extensive experiments showed that the addition of L-pyroglutamic acid in the eluent was key to suppression of the dissociation equilibrium of the cocrystal during lessivation, with high efficiency. Importantly, based in this profile, this process exhibited strong robustness and margin of safety at multigram and multikilogram scales
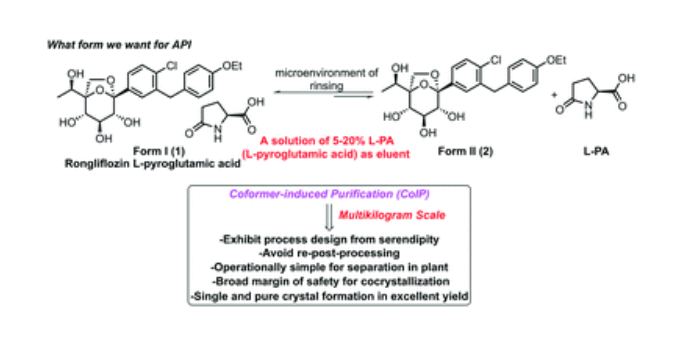
Kilogram scale Process of 1
A mixture of (1R,2S,3S,4R,5S)-5-(4-chloro-3-(4-ethoxybenzyl) phenyl)-1-((R)-1-
hydroxyethyl)-6,8-dioxabicyclo [3.2.1] octane-2,3,4-triol ethanolate form III (3) (23.45 kg, 47.3
mol), L-pyroglutamic acid (24.31 kg, 4.0 equiv.), EtOH (35.9 L) and H2O (70 L) was added into a
300 L reactor at room temperature. The slurry was heated to 65 °C and stirred until it is clear. The
clear solution was cooled to 35±5 °C typically. Seed crystal form I (1) (0.70 kg, 3% g/g) was added
when the solution was cooled to 34 °C and maintained for 1.5 h. Gradually, the slurry was cool to
30 °C and 25 °C in 3 hours, and finally stirred at 25 °C for 24 h. The slurry was collected on a
centrifuge filter. The filter cake was washed with a mixed solution of EtOH (31.3 L)/H2O (62.7 L)
with L-pyroglutamic acid (1.64 kg, 7% g/g) pre-cooled to -15°C. The wet cake was dried under
vacuum at 45 °C for 8 h. Pure cocrystal form I (1) was obtained as a white solid (24.91 kg, yield
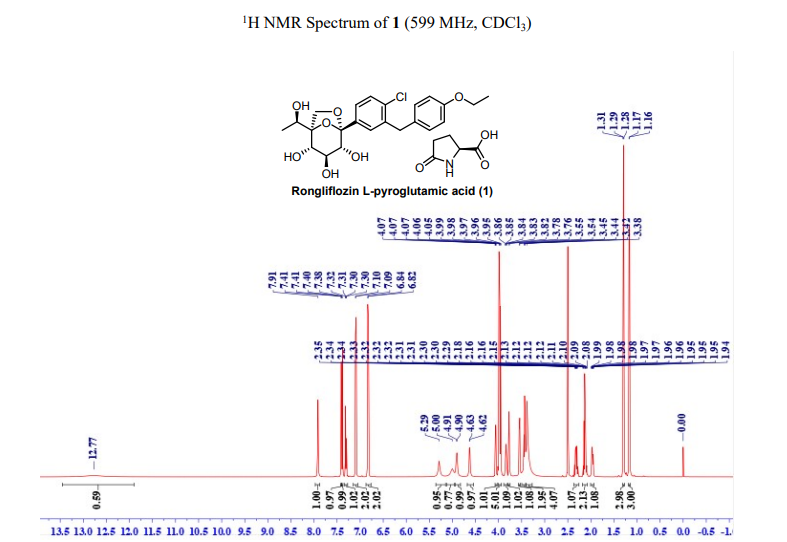
91%). MP (DSC onset) = 96.91 ℃. 1H NMR (599 MHz, DMSO-d6) δ 12.77 (br, 1H), 7.91 (s, 1H),
7.41 (d, J = 2.0 Hz, 1H), 7.39 (d, J = 12.0 Hz, 1H), 7.31 (dd, J = 12.0, 2.0 Hz, 1H), 7.10 (d, J = 2.0
Hz , 2H), 6.83 (d, J = 2.0 Hz, 2H), 5.29 (s, 1H), 5.00 (s, 1H), 4.91 (d, J = 6.7 Hz, 1H), 4.63 (d, J =
6.1 Hz, 1H), 4.06 (dd, J = 12.0, 6.0 Hz, 1H), 3.99– 3.95 (m, 5H), 3.84 (p, J = 6.0 Hz, 1H), 3.77 (d,
J = 12.0 Hz, 1H), 3.55 (d, J = 6.0 Hz, 1H), 3.44 (t, J = 12.0 Hz, 2H), 3.38 (s, 4H), 2.35-2.29 (m,
1H), 2.18-2.08 (m, 2), 1.99-1.94 (m, 1H), 1.29 (t, J = 12.0 Hz, 3H), 1.17 (d, J = 6.0 Hz, 3H). 13C
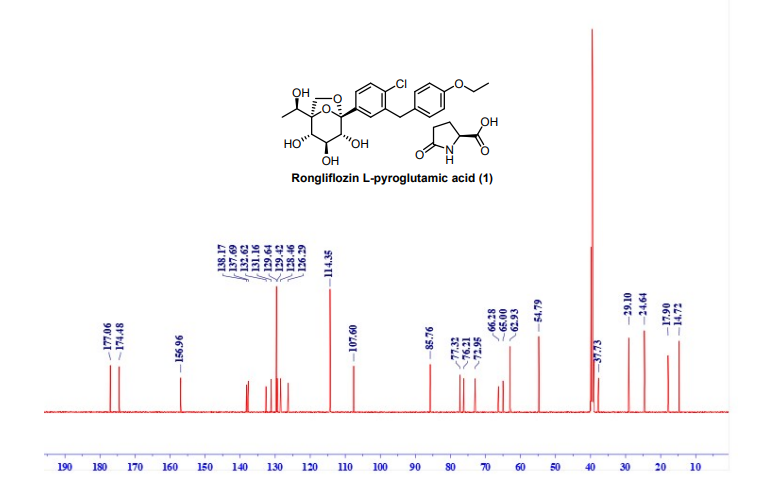
NMR (151 MHz, DMSO-d6) δ 177.06, 174.48, 156.96, 138.17, 137.69, 131.16, 129.64, 129.42,
128.46, 126.29, 114.35, 107.60, 85.76, 77.32, 76.21, 72.95, 66.28, 65.00, 62.93, 54.79, 37.73, 29.10,
24.64, 17.90, 14.72. HRMS: (ESI) Calcd for C23H27ClO7 [M+NH4]+: 468.1784, C5H7NO3 [M+H]+
:130.0499; Found: 468.1774, 130.0490 respectively. IR (KBr, cm-1): 3257, 2986, 2927, 1750, 1648,
1513, 1476, 1371, 1264, 1239, 1223, 1206, 1088, 1061, 821
13C NMR



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
References
- Zhang H, Liu J, Zhu X, Li X, Chen H, Wu M, et al. (May 2020). “A Phase I Study on the Pharmacokinetics and Pharmacodynamics of DJT1116PG, a Novel Selective Inhibitor of Sodium-glucose Cotransporter Type 2, in Healthy Individuals at Steady State”. Clinical Therapeutics. 42 (5): 892–905.e3. doi:10.1016/j.clinthera.2020.03.007. PMID 32265061.
- Zhang H, Zhu X, Li X, Chen H, Wu M, Li C, et al. (February 2020). “Pharmacokinetics and pharmacodynamics of rongliflozin, a novel selective inhibitor of sodium-glucose co-transporter-2, in people with type 2 diabetes mellitus”. Diabetes, Obesity & Metabolism. 22 (2): 191–202. doi:10.1111/dom.13887. PMID 31588657.
| Legal status | |
|---|---|
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 2035989-50-3 |
| PubChem CID | 122660464 |
| UNII | 6FP3NST6ZQ |
| ChEMBL | ChEMBL5314927 |
| Chemical and physical data | |
| Formula | C23H27ClO7 |
| Molar mass | 450.91 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////////Rongliflozin, diabetes, Olorigliflozin, 6FP3NST6ZQ, 2035989-50-3, DJT1116PG, DJT 1116PG,
ADVERTISEMENT
ANAX LABORATORIES
WEBSITE https://www.anaxlab.com/
Discovery Solutions, Supporting the chemistry needs of clients in the Medical, Analytical and Bio Sciences
Development Solutions, Developing from Lab scale to PR&D, Kilo Scale-ups and Commercial Scales
SEE MORE………Integrated Solutions, Manufacturing Solutions, Products,
Can’t Find? Let’s Connect

Phone : +91 897704 2010 / +91 9177075735, Email : info@anaxlab.com
#MedicinalChemistry, #DrugDiscovery, #OrganicSynthesis, #ChemicalLibrary, #BuildingBlocks, #SARStudies, #ChemistryInnovation, #medchem, #Drugdevelopment, #Biotech, #Biotechnology, #AnaxLaboratories, #Pharma

AS ON FEB2026 4.574 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
Ebselen
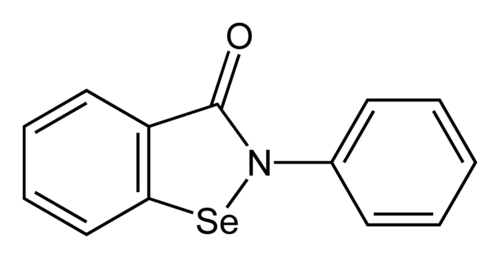
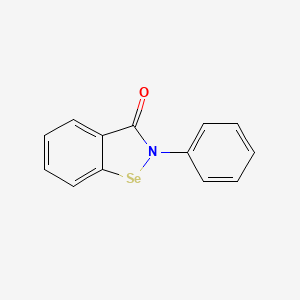
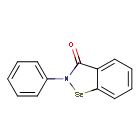
Ebselen
274.19 g/mol,
C13H9NOSe
2-phenyl-1,2-benzoselenazol-3-one
- CAS 60940-34-3
- 2-phenyl-1,2-benzoselenazol-3-one
- 2-Phenyl-1,2-benzisoselenazol-3(2H)-one
- Ebselene
- PZ 51, DR3305, and SPI-1005
- 40X2P7DPGH
Ebselen is a benzoselenazole that is 1,2-benzoselenazol-3-one carrying an additional phenyl substituent at position 2. Acts as a mimic of glutathione peroxidase. It has a role as a neuroprotective agent, an apoptosis inducer, an anti-inflammatory drug, an antioxidant, a hepatoprotective agent, a genotoxin, a radical scavenger, an enzyme mimic, an EC 1.3.1.8 [acyl-CoA dehydrogenase (NADP(+))] inhibitor, an EC 1.8.1.12 (trypanothione-disulfide reductase) inhibitor, an EC 1.13.11.33 (arachidonate 15-lipoxygenase) inhibitor, an EC 1.13.11.34 (arachidonate 5-lipoxygenase) inhibitor, an EC 2.5.1.7 (UDP-N-acetylglucosamine 1-carboxyvinyltransferase) inhibitor, an EC 2.7.10.1 (receptor protein-tyrosine kinase) inhibitor, an EC 3.5.4.1 (cytosine deaminase) inhibitor, an EC 5.1.3.2 (UDP-glucose 4-epimerase) inhibitor, a ferroptosis inhibitor, an antifungal agent, an EC 3.4.22.69 (SARS coronavirus main proteinase) inhibitor, an anticoronaviral agent, an antibacterial agent, an antineoplastic agent and an EC 3.1.3.25 (inositol–phosphate phosphatase) inhibitor.
Ebselen (also called PZ 51, DR3305, and SPI-1005), is a synthetic organoselenium molecule under preliminary investigation as a drug candidate.[1] It belongs to the class of compounds related to benzene and its derivatives.[1] It is being developed by the Seattle biotechnology company, Sound Pharmaceuticals, Inc.[1] It has also been reported to target tubulin, blocking its polymerization.[2]
Ebselen has been investigated for the treatment and basic science of Meniere’s Disease, Type 2 Diabetes Mellitus, and Type 1 Diabetes Mellitus.
Ebselen has been entered into clinical trials as a lead compound intended for the potential treatment of various diseases.[3] Its most advanced clinical trial is a Phase III study in people with Meniere’s disease, completed in July 2024.[4]
In vitro, ebselen is a mimic of glutathione peroxidase and reacts with peroxynitrite.[5] It is purported to have antioxidant and anti-inflammatory properties.[1][5]
Synthesis
Generally, synthesis of the characteristic scaffold of ebselen, the benzoisoselenazolone ring system, can be achieved either through reaction of primary amines (RNH2) with 2-(chloroseleno)benzoyl chloride (Route I),[6] by ortho-lithiation of benzanilides followed by oxidative cyclization (Route II) mediated by cupric bromide (CuBr2),[7] or through the efficient Cu-catalyzed selenation / heterocyclization of o-halobenzamides, a methodology developed by Kumar et al.[8] (Route III).

SYN
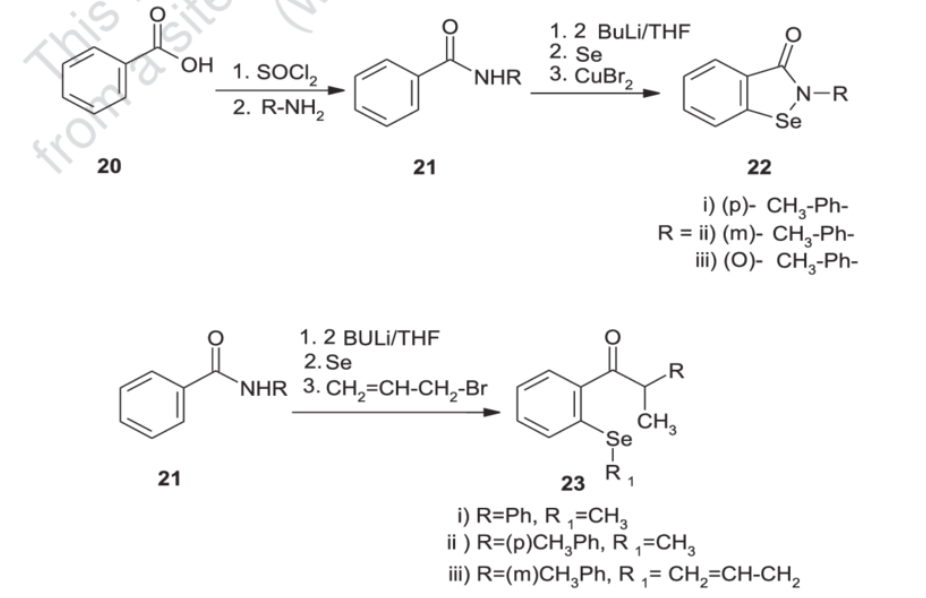
Synthesis of ebselen from benzoic acid by ortholithiation of benzanilide SOCl 2 =Thionyl chloride, R-NH 2 =Substituted aryl mine, BuLi/THF=n-butyllithium/ tetrahydrofuran, CuBr 2 =Cupper bromide, CH 2 =CH- CH 2 -Br = Allyl bromide.
SYN
New Chiral Ebselen Analogues with Antioxidant and Cytotoxic Potential
Molecules, March 2017, 22(3):492
SYN
https://pubs.acs.org/doi/10.1021/ol102027j

2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide (Typical
Procedure): Copper iodide (114 mg, 0.6 mmol) and 1,10-phenanthroline (108 mg, 0.6 mmol)
were added into DMF (3 mL) in a single neck flask. Resulted brownish solution was stirred for
15 min and then 2-iodo-N-phenylbenzamide1 (0.97 g, 3.0 mmol), selenium powder (0.29 g, 3.6
mmol), and potassium carbonate powder (0.65 g, 4.7 mmol) were added sequentially to same reaction flask. Brown colored reaction mixture was refluxed at 110oC using refluxing condenser
under nitrogen atmosphere. Progress of reaction was monitored by TLC. Reaction mixture was
refluxed for 8h. After this, reaction mixture poured over brine solution (60 mL) and stirred for 3
h. Product was precipitated as white solid which was collected by filtration over Buchner funnel,
product was washed with water (15 mL x 2), dried in air, dissolved in ethyl acetate, concentrated
over rotary evaporator, resulted brown solid which was purified by column chromatography
using hexane/ ethyl acetate (8:2) over silica gel. Yield 0.69 g (84%), mp 182-183 °C (180-181
°C).14,15 1H NMR (400 MHz, DMSO-d6) 8.09 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H),
7.71-7.62 (m, 3H), 7.51-7.43 (m, 3H), 7.28 (t, J = 8.0 Hz, 1H). 1H NMR (400 MHz, CDCl3)
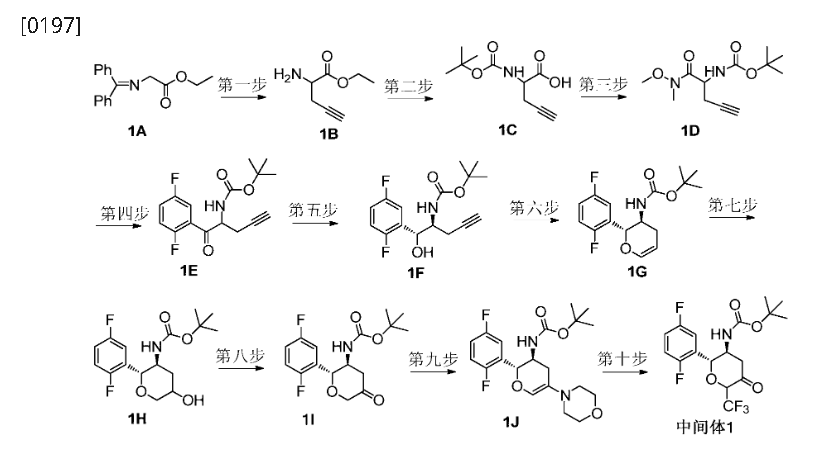
8.12 (d, 7.6 Hz, 1H), 7.68-7.62 (m, 4H), 7.52-7.41 (m, 3H), 7.29 (m, 1H). IR (plate): 3057, 2921,
1598, 1443, 1346, 1263, 1028 cm-1; ESMS m/z: 276 (M+H+).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Iodo-N-phenylbenzamide at 74 mmol
scale: Reaction was carried out at 74 mmol scale using 2-iodo-N-phenylbenzamide (24.00 g,
74.3 mmol), selenium powder (7.04 g, 89.1 mmol), CuI (2.83 g, 14.9 mmol), 1,10
phenanthroline (2.69 g, 14.9 mmol), and anhydrous potassium carbonate powder (15.40 g, 111.4
mmol) in DMF (50 mL) and procedure and workup followed are similar to 3.6 mmol scale
reaction. Yield 16.28 g (80%), Figure S1.
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Bromo-N-phenylbenzamide: Ebselen 1
was prepared from 2-bromo-N-phenylbenzamide2 (1.00 g, 3.6 mmol), selenium powder (0.34 g,
4.3 mmol), K2CO3 powder (0.74 g, 5.4 mmol), CuI (137 mg, 0.7 mmol), and 1,10-phenanthroline
(130 mg, 0.7 mmol) in DMF (3 mL). Reaction mixture was refluxed for 16 h at 110oC. Progress of reaction was monitored by TLC. After completion of reaction, mixture was poured into brine
solution (60 mL) and the resulted white precipitate was washed with water (20 mL x 2), and
dried in air. Purification by column chromatography on silica gel using CH2Cl2 provided white
crystalline solid (0.77 g, 78%).
2-Phenylbenzo[d][1,2]selenazol-3(2H)-one (1) from 2-Chloro-N-phenylbenzamide: Reaction
was carried out at 4 mmol scale using 2-chloro-N-phenylbenzamide3 (1.00 g, 4.3 mmol), CuI
(172 mg, 0.9 mmol), 1,10-phenanthroline (162 mg, 0.9 mmol), selenium powder (0.41 g, 5.2
mmol), K2CO3 (0.89 g, 6.4 mmol) in DMF (4 mL). Reaction mixture was refluxed for 24 h at
110oC. Workup procedure is similar as followed for bromo substrate. Yield 0.55 g (47%).
History
The first patent for 2-phenyl-1,2-benzoselenazol-3(2H)-one was filed in 1980 and granted in 1982.[9]
Research
Ebselen is in preliminary clinical development for the potential treatment of hearing loss and depression, among other medical indications.[3][10]
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- “Ebselen”. DrugBank. 29 January 2025. Retrieved 4 February 2025.
- Baksheeva VE, La Rocca R, Allegro D, Derviaux C, Pasquier E, Roche P, Morelli X, Devred F, Golovin AV, Tsvetkov PO (2025). “NanoDSF Screening for Anti-tubulin Agents Uncovers New Structure–Activity Insights”. Journal of Medicinal Chemistry. doi:10.1021/acs.jmedchem.5c01008.
- “Ebselen pipeline”. Sound Pharmaceuticals, Inc. 2025. Retrieved 4 February 2025.
- “SPI-1005 for the Treatment of Meniere’s Disease (STOPMD-3)”. ClinicalTrials.gov, US National Library of Medicine. 1 August 2024. Retrieved 4 February 2025.
- Schewe T (October 1995). “Molecular actions of ebselen – an antiinflammatory antioxidant”. General Pharmacology. 26 (6): 1153–69. doi:10.1016/0306-3623(95)00003-J. PMID 7590103.
- Kamigata N, Iizuka H, Izuoka A, Kobayashi M (July 1986). “Photochemical Reaction of 2-Aryl-1, 2-benzisoselenazol-3 (2 H)-ones”. Bulletin of the Chemical Society of Japan. 59 (7): 2179–83. doi:10.1246/bcsj.59.2179.
- Engman L, Hallberg A (1989-06-01). “Expedient synthesis of ebselen and related compounds”. The Journal of Organic Chemistry. 54 (12): 2964–2966. doi:10.1021/jo00273a035. ISSN 0022-3263.
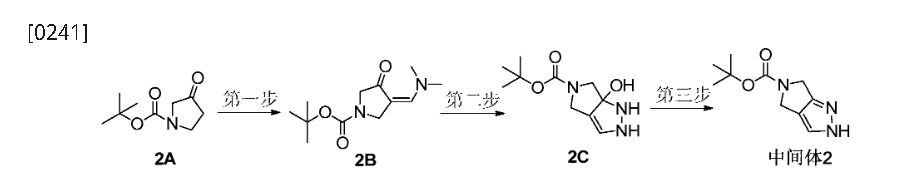
- Balkrishna SJ, Bhakuni BS, Chopra D, Kumar S (December 2010). “Cu-catalyzed efficient synthetic methodology for ebselen and related Se-N heterocycles”. Organic Letters. 12 (23): 5394–7. doi:10.1021/ol102027j. PMID 21053969.
- DE3027073A1, Etschenberg, Eugen Dr; Renson, Marcel Prof Dipl-Chem Jupille & Winkelmann, Johannes Dr 5000 Köln, “2-phenyl-1,2-benzisoselenazol-3(2h)-on enthaltende pharmazeutische praeparate und ihre verwendung”, issued 1982-02-18
- “Ebselen search: list of clinical trials sponsored by Sound Pharmaceuticals”. ClinicalTrials.gov, US National Library of Medicine. 2025. Retrieved 4 February 2025.
External links
| Names | |
|---|---|
| Preferred IUPAC name2-Phenyl-1,2-benzoselenazol-3(2H)-one | |
| Identifiers | |
| CAS Number | 60940-34-3 |
| 3D model (JSmol) | Interactive imageInteractive image |
| ChEBI | CHEBI:77543 |
| ChEMBL | ChEMBL51085 |
| ChemSpider | 3082 |
| ECHA InfoCard | 100.132.190 |
| PubChem CID | 3194 |
| UNII | 40X2P7DPGH |
| CompTox Dashboard (EPA) | DTXSID7045150 |
| InChI | |
| SMILES | |
| Properties | |
| Chemical formula | C13H9NOSe |
| Molar mass | 274.17666 |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
References
- Zhou Y, Zhang Y, Zhao D, Yu X, Shen X, Zhou Y, Wang S, Qiu Y, Chen Y, Zhu F: TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024 Jan 5;52(D1):D1465-D1477. doi: 10.1093/nar/gkad751. [Article]
////////Ebselen, Ebselene, PZ 51, DR 3305, SPI 1005, PHASE 3, 40X2P7DPGH, Meniere’s Disease, Type 2 Diabetes Mellitus, Type 1 Diabetes Mellitus
Velagliflozin
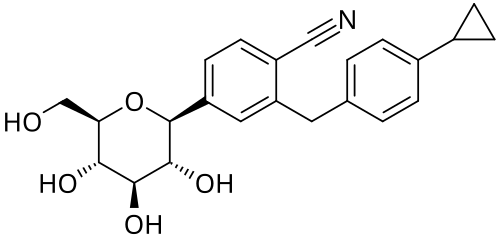
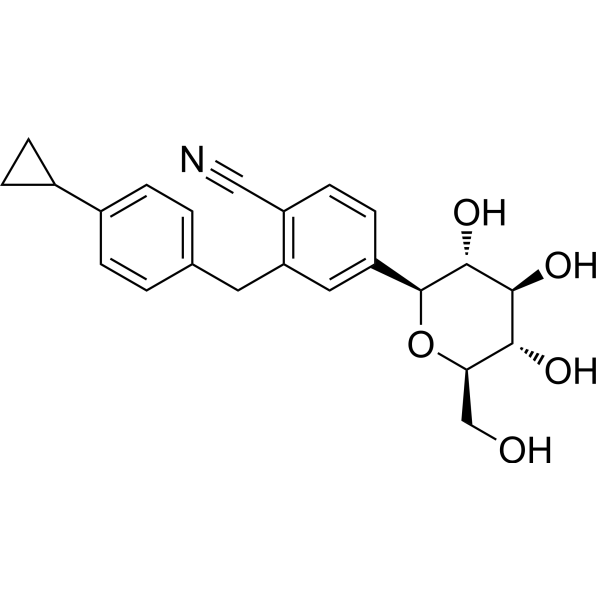
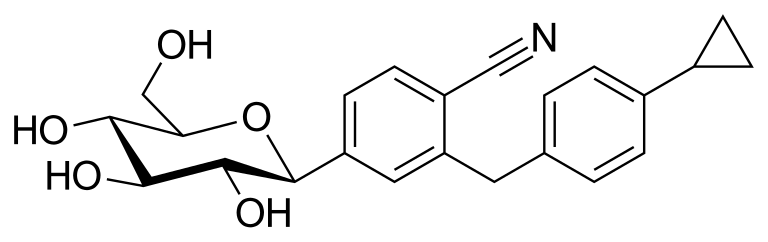
Velagliflozin
VETERINARY DRUG
- Cas 946525-65-1
- FV2YU8SL0P
- 2-((4-cyclopropylphenyl)methyl)-4-((2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl)benzonitrile
- 2-((4-Cyclopropylphenyl)methyl)-4-beta-D-glucopyranosylbenzonitrile
- 395.4 g/mol, C23H25NO5
2-[(4-cyclopropylphenyl)methyl]-4-[(2S,3R,4R,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]benzonitrile
- 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYLBENZONITRILE
- BENZONITRILE, 2-((4-CYCLOPROPYLPHENYL)METHYL)-4-.BETA.-D-GLUCOPYRANOSYL-
Velagliflozin L-proline H2O
Velagliflozin, sold under the brand name Senvelgo, is an antidiabetic medication used for the treatment of cats.[2][4][5] Velagliflozin is a sodium-glucose cotransporter 2 (SGLT2) inhibitor.[6] It is taken by mouth.[2]
Velagliflozin is the active ingredient of the first oral liquid medication approved by the Food and Drug Administration for the treatment of diabetes in cats. This compound belongs to the known class of sodium-glucose cotransporter 2 inhibitors approved to treat diabetes in human.
- Application: NADA 141-568Drug: Senvelgo®Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Patent(s): 7776830 (Exp: 05/01/2027); 8557782 (Exp: 05/01/2027); 9145434 (Exp: 09/07/2033); 10617666 (Exp: 06/06/2035); 11896574 (Exp: 12/17/2034); 10220017 (Exp: 09/29/2036); 10709683 (Exp: 08/24/2036); 11225500 (Exp: 12/17/2038)
- [Indication for Use] To improve glycemic control in otherwise healthy cats with diabetes mellitus not previously treated with insulin.Application: NADA 141-568Active Ingredient(s): VelagliflozinCompany: Boehringer lngelheim Animal Health USA Inc.Freedom of Information: FOIA Summary 14320Approval Date: August 10, 2023
APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO
Velagliflozin (brand name Senvelgo) is a veterinary medication approved for treating diabetes in cats, not humans.
Approved countries and years for velagliflozin:
- United States (US): Approved by the FDA in August 2023.
- European Union (EU): Received marketing authorization in November 2023.
- Switzerland: Approved in 2023.
- Great Britain: Approved in 2023.
SYN
https://patentscope.wipo.int/search/en/detail.jsf?docId=US310904480&_cid=P11-METCZG-99171-1
SYN
US7776830
https://patentscope.wipo.int/search/en/detail.jsf?docId=US41880220&_cid=P11-METD0X-00376-1
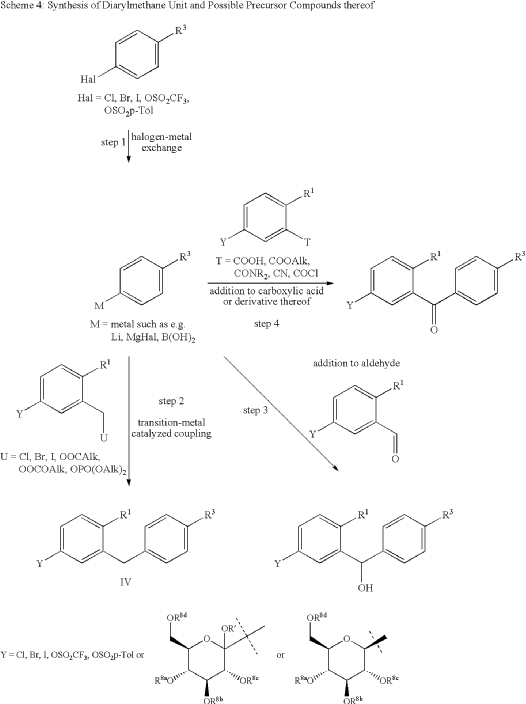
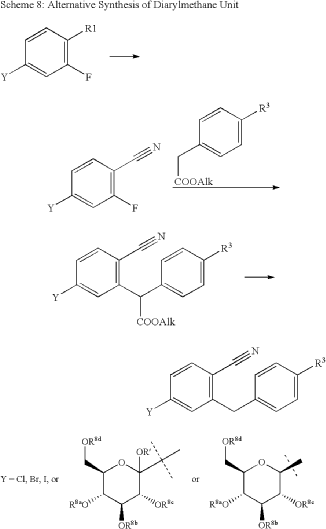
| The following compound is obtained analogously to Example XXIV: |
(1) 1-Cyano-2-(4-cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzene
EXAMPLE 17
2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile
| The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner. |
SYN
WO2007128749
https://patents.google.com/patent/WO2007128749A1/en
The following compound is obtained analogously to Example XXIV:
(1 ) 1 -Cvano-2-(4-cvclopropyl-benzyl)-4-(3-D-glucopyranos-1 -vD-benzene

Mass spectrum (ESI“): m/z = 413 [M+H] + Advantageously, the reduction of the anomeric carbon center of the appropriate intermediate obtained during the synthesis of this compound is conducted with the oxygen functionalities on the pyranose ring protected. Preferred protective groups are benzyl, p-methoxybenzyl, trimethylsilyl, triethylsilyl, terfbutyldimethylsilyl, triisopropylsilyl and allyl.
Example XXV

1-Cyano-2-(4-cyclopropyl-benzyl)-4-(tetra-O-acetyl-β-D-glucopyranos-1-yl)-benzene To a flask charged with a stir bar, 4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1-yl)-2-(4- trifluoromethylsulfonyloxy-benzyl)-benzonitrile (4.4 g), degassed toluene (12 ml.) and degassed water (8 ml.) and kept under argon atmosphere is added cyclopropylboronic acid (0.20 g), potassium phosphate (5.0 g), tricyclohexylphosphine (0.19 g) and at last palladium(ll)acetate (76 mg). The mixture is stirred at 1 10 °C for 6 h meanwhile cyclopropylboronic acid is added after each hour (5x 0.20 g). After cooling to room temperature, the mixture is diluted with aqueous sodium hydrogen carbonate solution and extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is removed under reduced pressure. The residue is chromatographed on silica gel (cyclohexane/ethyl acetate 20:1 -> 1 :1 ). Yield: 3.2 g (87% of theory ) Mass spectrum (ESI+): m/z = 581 [M+NH4] +
Example XXVI

4-(1 -Hvdroxy-cvclopropyD-phenylboronic acid A 3.0 M solution of ethylmagnesium bromide in diethylether (7.6 ml.) is added to a stirred solution of titanium(IV) isopropoxide (2.2 ml.) in diethylether (70 ml.) chilled to -78 °C. The resultant solution is stirred at -78 °C for 1.5 h, before 4-(4,4,5,5-tetramethyl-[1 ,3,2]dioxa borolan-2-yl)-benzoic acid methyl ester (2.0 g) is added. The reaction mixture is warmed to ambient temperature and stirred for an additional 12 h. Then, 1 M aqueous hydrochloric acid is added and the resulting mixture is extracted with ethyl acetate. The combined organic extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is dissolved in acetone (60 ml.) and 0.1 M aqueous NH4OAc solution (50 ml.) followed by NaIO4 (2.3 g) is added. The resulting reaction mixture is stirred at room temperature for 18 h. After removal of the acetone, the residue is extracted with ethyl acetate. The combined extracts are dried (sodium sulphate) and the solvent is evaporated. The residue is purified by chromatography on silicagel (cyclohexane/ethyl acetate). Yield: 0.45 g (33% of theory) Mass spectrum (ESI“): m/z = 223 [M+HCOO]“ Preparation of the end compounds:
Example 17: 2-(4-Cyclopropyl-benzyl)-4-(β-D-glucopyranos-1-yl)-benzonitrile

Mass spectrum (ESI+): m/z = 413 [M+NH4]+
The compound is obtained according to example 6 using 4-cyclopropyl-phenylboronic acid as the coupling partner.
Yield: 83% of theory
Alternatively this compound is obtained as described in Example XXIV(I ).
The compound of example 17 is also obtained by employing the following procedure:
A solution of 2-(4-cyclopropyl-benzyl)-4-(2,3,4,6-tetra-O-acetyl-D-glucopyranos-1 -yl)- benzonitrile (0.80 g) in methanol (5 ml.) and THF (5 ml.) is treated with aqueous potassium hydroxide solution (4 mol/l, 5 ml_). The reaction solution is stirred at ambient temperature for 1 h and then neutralized with 1 M hydrochloric acid. The organic solvents are evaporated and the residue is diluted with brine and extracted with ethyl acetate. The organic extracts are dried (sodium sulphate) and the solvent is removed. The residue is chromatographed on silica gel (dichloromethane/methanol 1 :0 -> 9:1 ). Yield: 0.54 g (96% of theory)
SYN
Synthesis 2024, 56, 906–943
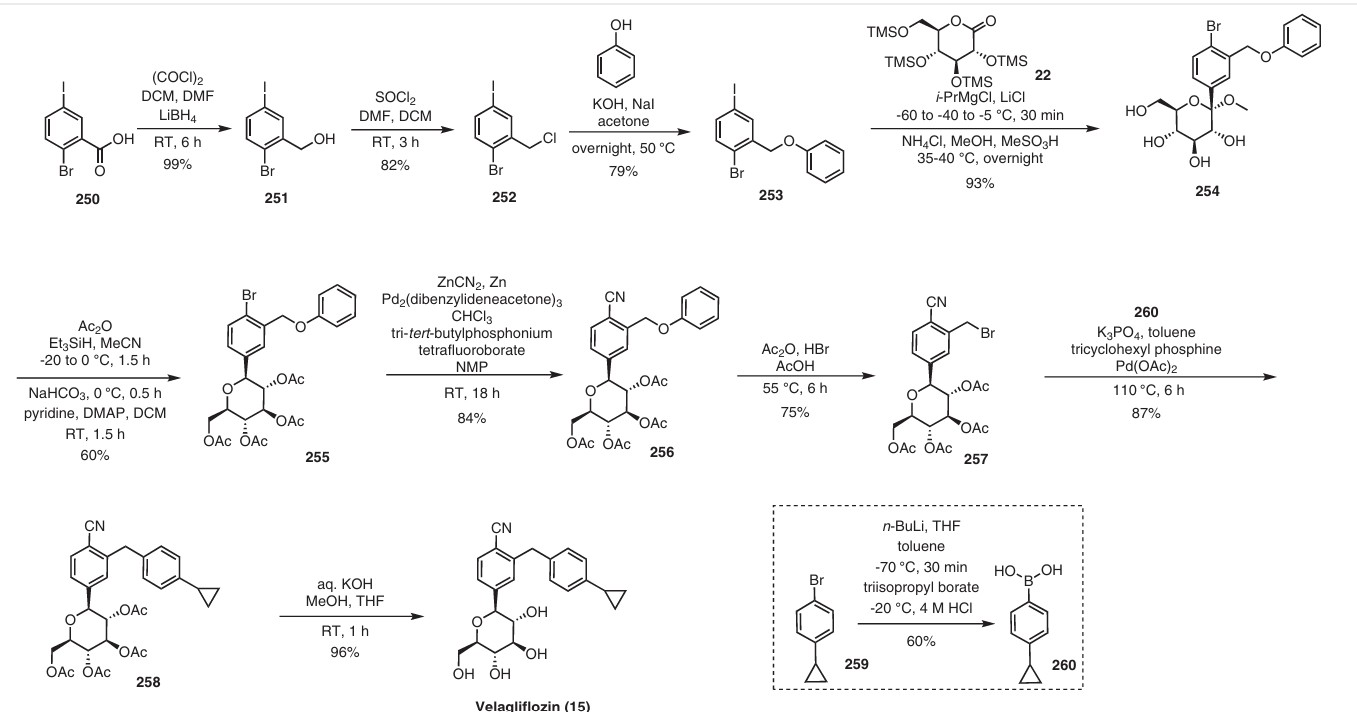
In 2007, Boehringer-Ingelheim Vetmedica GmbH pioneered the development of velagliflozin (15), subsequently submitting a patent application in the United States with the identification number US7776830B2.72a More recently, through clinical investigations, this compound has demonstrated its efficacy as an SGLT2 inhibitor, proving adept at curtailing glucose reabsorption, encouraging glucosuria,
and leading to reductions in both blood glucose and insulin levels.
The initial synthesis of velagliflozin (15) was also disclosed in the above patent,72a and in patent
WO2007128749A1.72b The synthesis, depicted in Scheme46, comprises of nine-steps starting with the readily available raw material 2-bromo-5-iodobenzoic acid (250), which undergoes reduction using LiBH4 to form the corresponding alcohol 251. Subsequently, chlorination is carried out using thionyl chloride, resulting in the formation of chloride 252. O-Alkylation of phenol with compound 252 is
then conducted in a basic medium, yielding intermediate 253.The C-glycosylation of 253 with 2,3,4,6-tetrakis-O(trimethylsilyl)-D-glucopyranone 22 in the presence of turbo Grignard reagent (isopropylmagnesium chloride and LiCl) and methanesulfonic acid in methanol gives compound
254 with an impressive 93% yield. The hydroxy group of in termediate 254 is protected using acetic anhydride, and themethoxy group is subsequently removed via Lewis acid (BF3·Et2O, Et3SiH) treatment, providing compound 255 in a yield of 60%. A metal-catalyzed cyano group installation is then performed on intermediate 255, leading to the formation of compound 256 in 84% yield. The subsequent steps involve benzylic bromination followed by coupling with cyclopropylphenyl boronic acid 260, resulting in the formation of intermediate 258. Finally, deacetylation of intermediate 258 using aqueous KOH produces the desired product
The overall yield obtained for velagliflozin (15) is calculated to be 11.3%, with this synthetic route providing a systematic and efficient approach. The highlight of the route is high-yielding chemical transformations. However, the drawback is the use of two palladium-mediated couplings
that increase the possibility of leaching of the toxic metal in scale-up batches. Additionally, the synthetic route requires a large number of chemical transformations and not best suited for commercial production.
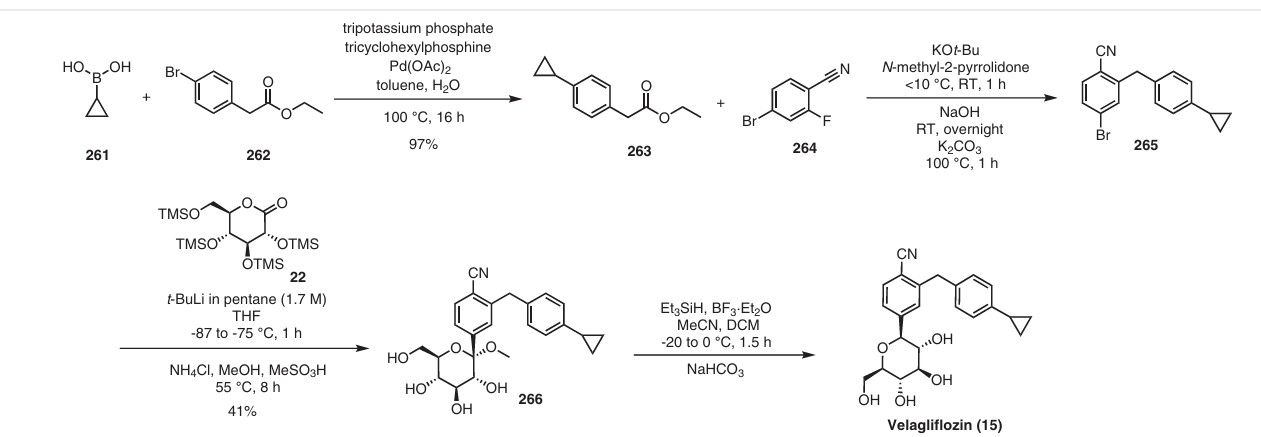
The same authors reported an alternative method (Scheme 47) for the synthesis of velagliflozin (15) in the product patent.72 The aglycone intermediate 265 is accessed in two steps starting from ethyl 2-(4-bromophenyl)acetate (262). O-Glycosylation takes place with the aglycone
4-bromo-2-(4-cyclopropylbenzyl)benzonitrile (265) using 2,3,4,6-tetrakis-O-(trimethylsilyl)-D-glucopyranone 22 in the presence of tert-butyllithium in pentane (1.7 M), resulting in the formation of compound 266. Reduction of compound 266 using boron trifluoride–diethyl etherate yields
the final API velagliflozin (15). This truncated synthetic route is well suited for scale-up due to the significantly low er number of transformations compared to the previous route. Unfortunately, the specific yields were not clearly in dicated for this process. This method presents an alternative approach to the synthesis of velagliflozin (15), providing a potential pathway for its preparation in 5 steps with
an overall yield of 40%.
(72) (a) Eckhardt, M.; Himmelsbach, F.; Eickelmann, P.; Sauer, A.;
Thomas, L. US7776830B2, 2010. (b) Eckhardt, M.; Himmelsbach,
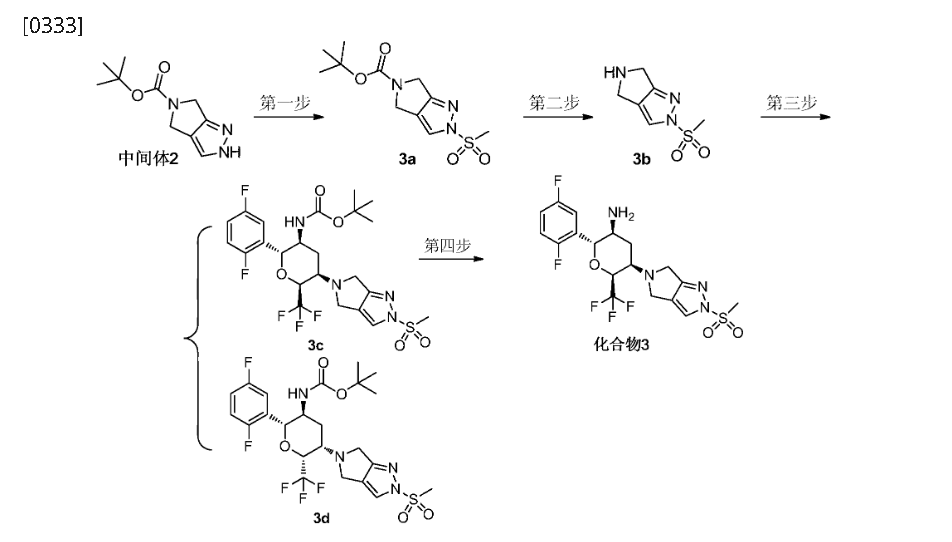
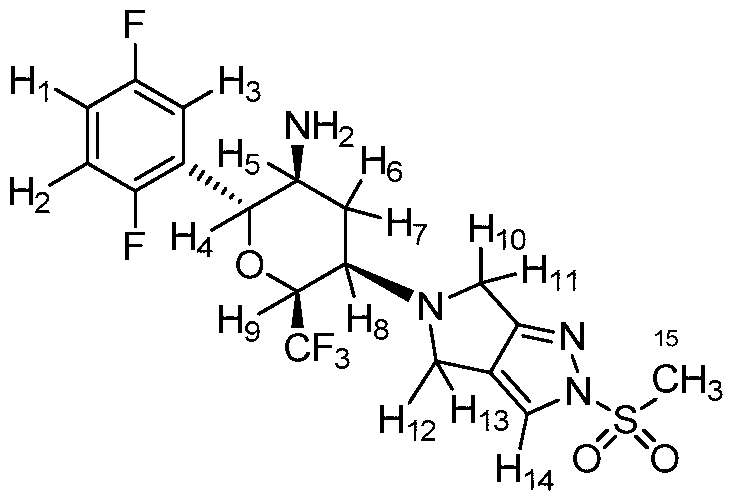
F.; Eickelmann, P.; Sauer, A.; Thomas, L. WO2007128749A1,
2007.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
Medical uses
Velagliflozin is indicated to improve glycemic control in otherwise healthy cats with diabetes not previously treated with insulin.[2][4][6]
References
- “Notice: Multiple additions to the Prescription Drug List (PDL) [2024-10-18]”. Health Canada. 18 October 2024. Retrieved 25 October 2024.
- “Senvelgo- velagliflozin solution”. DailyMed. 8 November 2023. Retrieved 13 December 2023.
- “Senvelgo Product information”. Union Register of veterinary medicinal products. 22 November 2023. Retrieved 29 August 2024.
- “NADA 141-568 Senvelgo (velagliflozin oral solution) Cats”.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - Cook AK, Behrend E (January 2025). “SGLT2 inhibitor use in the management of feline diabetes mellitus”. Journal of Veterinary Pharmacology and Therapeutics. 48 Suppl 1 (Suppl 1): 19–30. doi:10.1111/jvp.13466. PMC 11736986. PMID 38954371.
- “Dear Veterinarian Letter regarding important safety conditions associated with the use of Senvelgo (velagliflozin oral solution) for improving glycemic control in certain cats with diabetes mellitus”. U.S. Food and Drug Administration. 4 December 2023. Retrieved 13 December 2023. This article incorporates text from this source, which is in the public domain.
| Clinical data | |
|---|---|
| Trade names | Senvelgo |
| License data | US DailyMed: Velagliflozin |
| Routes of administration | By mouth |
| ATCvet code | QA10BK90 (WHO) |
| Legal status | |
| Legal status | CA: ℞-only[1]US: ℞-only[2]EU: Rx-only[3] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 946525-65-1 |
| PubChem CID | 24862817 |
| ChemSpider | 58827717 |
| UNII | FV2YU8SL0PEQE2P2T77I |
| Chemical and physical data | |
| Formula | C23H25NO5 |
| Molar mass | 395.455 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
- SGLT2 inhibitors: a novel therapy for cognitive impairment via multifaceted effects on the nervous systemPublication Name: Translational NeurodegenerationPublication Date: 2024-08-09PMCID: PMC11312905PMID: 39123214DOI: 10.1186/s40035-024-00431-y
- Demographic, morphologic, hormonal and metabolic factors associated with the rate of improvement from equine hyperinsulinaemia-associated laminitisPublication Name: BMC Veterinary ResearchPublication Date: 2022-01-18PMCID: PMC8764787PMID: 35042535DOI: 10.1186/s12917-022-03149-z
- The efficacy and safety of velagliflozin over 16 weeks as a treatment for insulin dysregulation in poniesPublication Name: BMC Veterinary ResearchPublication Date: 2019-02-26PMCID: PMC6390376PMID: 30808423DOI: 10.1186/s12917-019-1811-2
- The sodium-glucose co-transporter 2 inhibitor velagliflozin reduces hyperinsulinemia and prevents laminitis in insulin-dysregulated poniesPublication Name: PLOS ONEPublication Date: 2018-09-13PMCID: PMC6136744PMID: 30212530DOI: 10.1371/journal.pone.0203655
- Effects of the sodium‐glucose cotransporter 2 (<scp>SGLT</scp>2) inhibitor velagliflozin, a new drug with therapeutic potential to treat diabetes in catsPublication Name: Journal of Veterinary Pharmacology and TherapeuticsPublication Date: 2017-11-15PMID: 29139146DOI: 10.1111/jvp.12467
/////////Velagliflozin, APPROVALS 2023, GDA 2023, EU 2023, EMA 2023, SENVELGO, DIABETES, SENVELGO,
Cofrogliptin
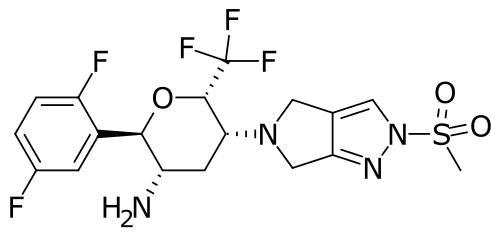
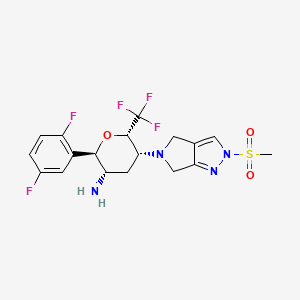
Cofrogliptin
HSK 7653
- Haisco HSK 7653
- CAS 1844874-26-5
- 466.4 g/mol
- C18H19F5N4O3S
(2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-(2-methylsulfonyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)-6-(trifluoromethyl)oxan-3-amine
- (2R,3S,5R,6S)-2-(2,5-Difluorophenyl)-5-(2- (methanesulfonyl)-2,6-dihydropyrrolo(3,4-C)pyrazol- 5(4H)-yl)-6-(trifluoromethyl)oxan-3-amine
- (2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-[2- (methanesulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol- 5(4H)-yl]-6-(trifluoromethyl)oxan-3-amine
- (6R)-5-Amino-2,6-anhydro-1,3,4,5-tetradeoxy-6-C-(2,5-difluorophenyl)-3-(2,6-dihydro-2-(methylsulfonyl)pyrrolo(3,4-C)pyrazol-5(4H)-yl)-1,1,1-trifluoro-D-arabino-hexitol
- (6R)-5-Amino-2,6-anhydro-1,3,4,5-tetradeoxy-6-C-(2,5-difluorophenyl)-3-[2,6-dihydro-2-(methylsulfonyl)pyrrolo[3,4-c]pyrazol-5(4H)-yl]-1,1,1-trifluoro-D-arabino-hexitol
- D-Arabino-hexitol, 5-amino-2,6-anhydro-1,3,4,5-tetradeoxy-6-C-(2,5-difluorophenyl)-3-(2,6-dihydro-2-(methylsulfonyl)pyrrolo(3,4-C)pyrazol-5(4H)-yl)-1,1,1-trifluoro-, (6R)-
- (2r,3s,5r,6s)-2-(2,5-difluorophenyl)-5-[2-(methylsulfonyl)-2,6-dihydropyrrolo[3,4-c]pyrazol-5(4h)-yl]-6-(trifluoromethyl)-tetrahydro-2h-pyran-3-amine
APPROVALS 2024, CHINA 2024, Haisco Pharmaceutical Group Co, Beichangping, DIABETES
Cofrogliptin (developmental name HSK7653) is a long-acting DPP4 inhibitor dosed once every two weeks.[1][2][3][4]
Cofrogliptin (HSK7653) (compound 2), a tetrahydropyran derivative, is a potent oral dipeptidyl aminopeptidase 4 (DPP-4) inhibitor with Long-acting antidiabetic efficacy. Cofrogliptin (compound 2) has a great potential for type 2 diabetes mellitus (T2DM) .
SYN
J Med Chem. 2020 Jul 9;63(13):7108-7126
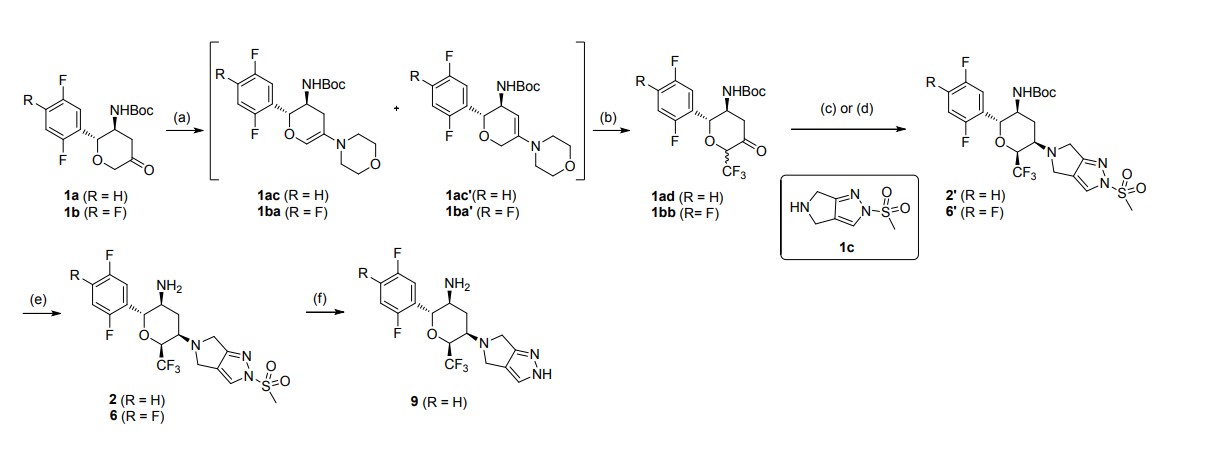
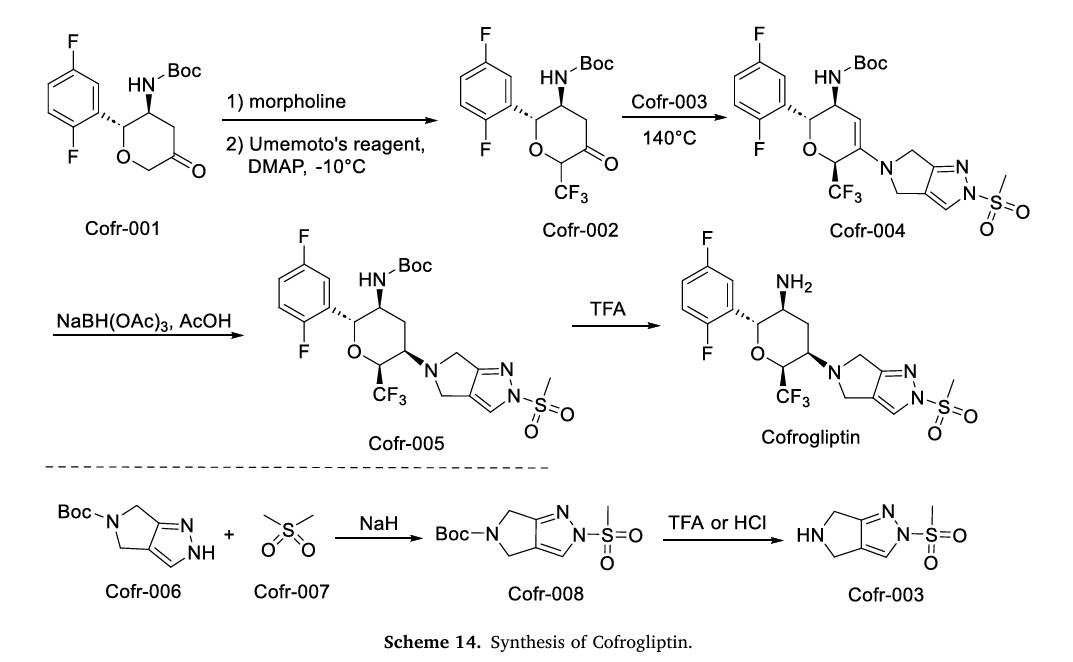
aReagents and conditions: (a) morpholine, toluene, reflux in Dean-Stark appartus; (b)
Umemoto’s reagent, DMAP, DMAc; (c) step 1: 1c, toluene, reflux; step 2: NaBH(OAc)3, CH3COOH, 1,2-DCE; (d) step 1: 1c, CHCl3, reflux in Dean-Stark apparatus; step 2:
NaBH(OAc)3, CH3COOH, 1,2-DCE; (e) TFA, DCM; (f) t-BuOK, THF
Step 2: To a stirred solution of tert-butyl N-[(2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-
(2-methylsulfonyl-4,6-dihydropyrrolo[3,4-c]pyrazol-5-yl)-6-
(trifluoromethyl)tetrahydropyran-3-yl]carbamate (2′) (407.5 mg, 0.72 mmol) in DCM (6
mL) was added CF3COOH (2 mL) under nitrogen at 0 ℃. After the addition, the reaction
mixture was allowed to warm to room temperature and stirred for 2 h. The reaction mixture
was quenched with a saturated solution of Na2CO3 (15 mL), and extracted with DCM (15
mL × 2). The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo.
The residue was purified by flash column chromatography (Eluent: DCM/MeOH = 80:1–
30:1) to afford the desired product 2 (301.9 mg, yield: 90%). White solid. Mp: 150.1–152.0
℃. [α]D20 = +17.6 (c = 2.000 in MeOH). Rf= 0.40 (1:15 MeOH/CH2Cl2, TLC).
1H NMR
(400 MHz, CDCl3) δ = 7.71 (s, 1H), 7.20 – 7.12 (m, 1H), 7.10 – 6.97 (m, 2H), 4.63 (d, J =
10.0 Hz, 1H), 4.49 – 4.38 (m, 1H), 4.07 – 3.97 (m, 2H), 3.93 – 3.81 (m, 2H), 3.53 – 3.42
(m, 1H), 3.29 (s, 3H), 3.01 – 2.91 (m, 1H), 2.45 – 2.35 (m, 1H), 2.07 – 1.93 (m, 1H), 1.19
(br. s, 2H). 13C NMR (100 MHz, CDCl3) δ = 163.6, 159.1 (dd, J = 2.3 Hz, 235.8 Hz), 156.6
SYN
https://www.sciencedirect.com/science/article/abs/pii/S0223523424003441
SYN
WO2015192701
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015192701&_cid=P20-MEQV3M-18104-1
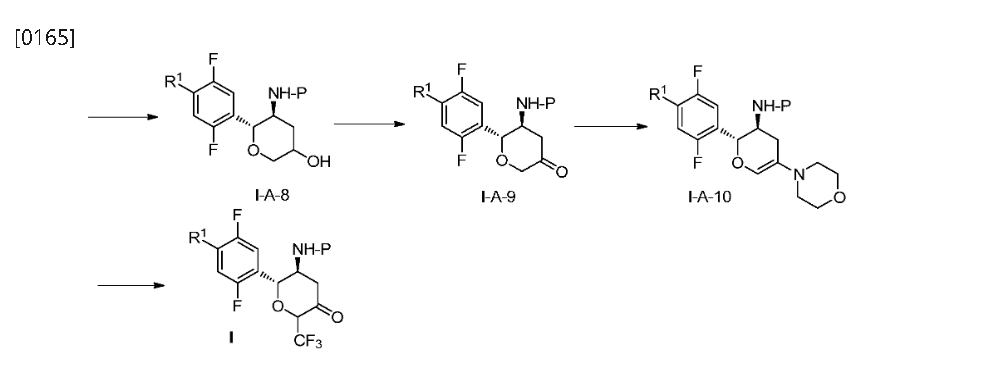
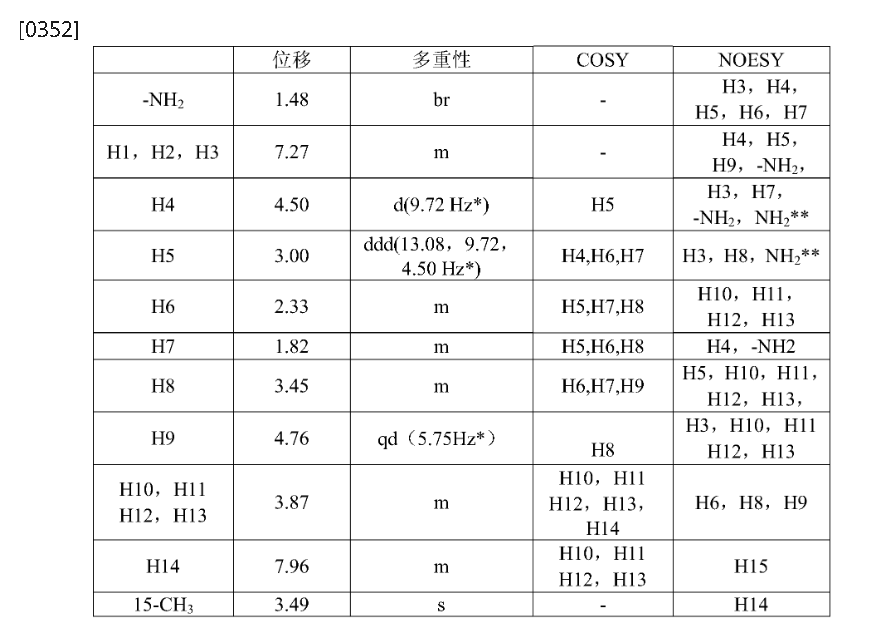
Step 4: (2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-(2-(methylsulfonyl)-pyrrolo[3,4]pyrazol-5(2H,4H,6H)-yl)-6-(trifluoromethyl)tetrahydro-2H-pyran-3-amine (Compound 3)
[0345]
(2R,3S,5R,6S)-2-(2,5-difluorophenyl)-5-(2-(methylsulfonyl)pyrrolo[3,4-c]pyrazol-5(2H,4H,6H)-yl)-6-(trifluoromethyl)tetrahydro-2H-pyran-3-amine
[0346]3c (410 mg, 0.72 mmol) was dissolved in 6 mL of dichloromethane and 2 mL of trifluoroacetic acid and stirred at room temperature for 1 hour. After completion, saturated aqueous sodium bicarbonate (30 mL) was added to quench the reaction. After separation, the aqueous phase was extracted with ethyl acetate (30 mL x 2). The combined organic phases were dried over anhydrous sodium sulfate, and concentrated. Purification by silica gel column chromatography (dichloromethane/methanol (v/v) = 30:1) afforded compound 3 (250 mg, 75% yield) as a white powder.
[0347]MS m/z(ESI): 467.1[M+1];
[0348]
1H NMR(400MHz,DMSO-d 6):δ7.96(m,1H),7.35–7.04(m,3H),4.86–4.63(qd,1H),4.50(d,1H),3.95(dd,2H),3.78(dd,2H),3.49(s,3H),3.45(m,1H),3.00(ddd,1H),2.33(m,1H),1.82(m,1H),1.48(br,2H)。
SYN
Cofrogliptin, developed by Haisco Pharmaceutical Group Co., Ltd., is a novel, ultra-long-acting dipeptidyl peptidase-4 (DPP-4) inhibitor designed for the treatment of T2DM. It is marketed under the brand name (Beichangping). In 2024, the NMPA approved Cofrogliptin for improving blood glucose control in adult patients with T2DM [59].Cofrogliptin acts pharmacologically by inhibiting DPP-4, an enzyme tasked with degrading incretin hormones like glucagon-like peptide-1(GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). By obstructing the degradation of these hormones, it amplifies their activity. This leads to a glucose-dependent rise in insulin secretion and a
corresponding decrease in glucagon release, which in turn improves glycemic control. The clinical efficacy of Cofrogliptin was demonstrated in Phase III, randomized, double-blind, non-inferiority trial
(NCT04556851), where its efficacy and safety were compared to those of daily linagliptin in patients with T2DM whose blood sugar was not well-controlled by metformin. The study reported that Cofrogliptin
administered once every two weeks achieved a reduction in HbA1c comparable to that of daily linagliptin, with a mean decrease of approximately 0.96 % over 24 weeks. Regarding toxicity, Cofrogliptin
was generally well-tolerated [60,61]. The incidence of hypoglycemia was low, and no severe hypoglycemic events directly attributed to the drug were reported.
The synthesis of Cofrogliptin, illustrated in Scheme 14, initiates with trifluoromethylation of Cofr-001 via oxidation, affording Cofr-002 [62]. Nucleophilic addition of Cofr-003 to Cofr-002 yields Cofr-004, followed by NaBH(OAc)3 reduction to Cofr-005. TFA-mediated deprotection of Cofr-005 ultimately delivers Cofrogliptin. Concurrently, Cofr-006 undergoes nucleophilic substitution with Cofr-007 to form Cofr-008, whose deprotection regenerates Cofr-003
[59] L. Gao, F. Bian, T. Pan, H. Jiang, B. Feng, C. Jiang, J. Sun, J. Xiao, P. Yan, L. Ji,
Efficacy and safety of cofrogliptin once every 2 weeks in Chinese patients with type
2 diabetes: a randomized, double-blind, placebo-controlled, phase 3 trial, Diabetes
Obes Metab 27 (2025) 280–290.
[60] C. Cui, F. Cao, I.I. Kong, Q. Wu, F. Li, H. Li, D. Liu, A model-informed approach to
accelerate the clinical development of cofrogliptin (HSK7653), a novel ultralong-
acting dipeptidyl peptidase-4 inhibitor, Diabetes Obes Metab 26 (2024) 592–601.
[61] Q. Ren, L. Li, X. Su, X. Hu, G. Qin, J. Han, Y. Liu, J. Wang, L. Ji, Cofrogliptin once
every 2 weeks as add-on therapy to metformin versus daily linagliptin in patients
with type 2 diabetes in China: a randomized, double-blind, non-inferiority trial,
Diabetes Obes Metab 26 (2024) 5013–5024.
[62] C. Zhang, J. Wang, C. Li, Y. Wei, Amino Pyranoid Ring Derivative as DPP-IV
Inhibitor and Its Preparation, 2015. WO2015192701A1.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Ji, Linong; Bian, Fang; Pan, Tianrong; Jiang, Hongwei; Jiang, Chengxia; Ren, Qian (20 June 2023). “55-OR: HSK7653, a Novel Ultralong-Acting DPP-4 Inhibitor, as Monotherapy in Patients With Type 2 Diabetes—A Randomized, Double-Blind, Placebo-Controlled Phase III Trial”. Diabetes. 72 (Supplement_1). doi:10.2337/db23-55-OR. S2CID 259433641.
- Zhang, Miao; Zhang, Shudong; Yu, Zhiheng; Yao, Xueting; Lei, Zihan; Yan, Pangke; Wu, Nan; Wang, Xu; Hu, Qin; Liu, Dongyang (October 2023). “Dose decision of HSK7653 oral immediate release tablets in specific populations clinical trials based on mechanistic physiologically-based pharmacokinetic model”. European Journal of Pharmaceutical Sciences. 189 106553. doi:10.1016/j.ejps.2023.106553. PMC 10485820. PMID 37532063.
- Liu, Yang; Yan, Shuai; Liu, Jie; Liu, Hongzhong; Song, Ling; Yao, Xueting; Jiang, Ji; Li, Fangqiong; Du, Ke; Liu, Dongyang; Hu, Pei (May 2023). “Development and validation of an HPLC coupled with tandem mass spectrometry method for the determination of HSK7653, a novel super long-acting dipeptidyl peptidase-4 inhibitor, in human plasma and urine and its application to a pharmacokinetic study”. Biomedical Chromatography. 37 (5): e5607. doi:10.1002/bmc.5607. PMID 36802077. S2CID 257048524.
- Bai, Nan; Wang, Jin; Liang, Wenxin; Gao, Leili; Cui, Wei; Wu, Qinghe; Li, Fangqiong; Ji, Linong; Cai, Yun (6 November 2023). “A Multicenter, Randomized, Double-Blind, Placebo-Controlled, and Dose-Increasing Study on the Safety, Tolerability and PK/PD of Multiple Doses of HSK7653 by Oral Administration in Patients with Type 2 Diabetes Mellitus in China”. Diabetes Therapy. 15 (1): 183–199. doi:10.1007/s13300-023-01496-0. PMC 10786778. PMID 37930584.
| Clinical data | |
|---|---|
| Other names | HSK7653 |
| Legal status | |
| Legal status | Investigational |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1844874-26-5 |
| PubChem CID | 118613788 |
| ChemSpider | 115037226 |
| UNII | LH4G6K6NKP |
| ChEMBL | ChEMBL4646510 |
| Chemical and physical data | |
| Formula | C18H19F5N4O3S |
| Molar mass | 466.43 g·mol−1 |
- [1]. International Nonproprietary Names for Pharmaceutical Substances (INN)[2]. Chen Zhang, et al. Design, Synthesis, and Evaluation of a Series of Novel Super Long-Acting DPP-4 Inhibitors for the Treatment of Type 2 Diabetes. J Med Chem. 2020 Jul 9;63(13):7108-7126. [Content Brief]
///////Cofrogliptin, APPROVALS 2024, CHINA 2024, Haisco Pharmaceutical Group Co, Beichangping, DIABETES, HSK 7653, Haisco HSK 7653, 1844874-26-5
Janagliflozin
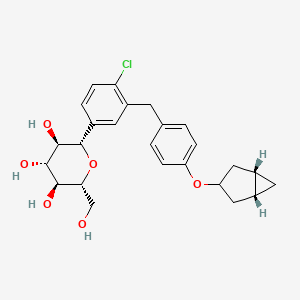
Janagliflozin
WeightAverage: 460.95
Monoisotopic: 460.1652664
Chemical FormulaC25H29ClO6
- WK4RT85HCA
- XZP-5695
- UNII-WK4RT85HCA
- 1800115-22-3
- (2S,3R,4R,5S,6R)-2-[3-[[4-[[(1S,5R)-3-bicyclo[3.1.0]hexanyl]oxy]phenyl]methyl]-4-chlorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
- D-Glucitol, 1,5-anhydro-1-C-(3-((4-((1alpha,3alpha,5alpha)-bicyclo(3.1.0)hex-3-yloxy)phenyl)methyl)-4-chlorophenyl)-, (1S)-
- (2S,3R,4R,5S,6R)-2-[3-[[4-[[(1S,5R)-3-bicyclo[3.1.0]hexanyl]oxy]phenyl]methyl]-4-chlorophenyl]-6-(hydroxymethyl)oxane-3,4,5-triol
- D-GLUCITOL, 1,5-ANHYDRO-1-C-(3-((4-((1.ALPHA.,3.ALPHA.,5.ALPHA.)-BICYCLO(3.1.0)HEX-3-YLOXY)PHENYL)METHYL)-4-CHLOROPHENYL)-, (1S)-
- (2S,3R,4R,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
China 2024, approvals 2024, Jilin Huisheng Biopharmaceutical Co, sihuan, SGLT2 inhibitors, Huiyoujing
Janagliflozin is an SGLT2 inhibitor developed by Sihuan Pharmaceutical.[1][2][3][4][5][6] It is approved in China for the treatment of type 2 diabetes.[7]
PAPER
https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-0042-1751524
(71) (a) Wu, F. US9315438B2, 2016. (b) Wu, F. EP2891654A1, 2014.
Initially, the two advanced intermediates were synthesized and then coupled under cryogenic conditions using nBuLi. The construction of 242 commences with the reaction of 5-bromo-2-chlorobenzoic acid (26c) with oxalyl chloride and a catalytic amount of DMF in DCM, yielding the acid chloride derivative 26c′. This intermediate is then subjected to Friedel–Crafts acylation with anisole to produce 240 in
71% yield. Subsequent reduction of 240 was carried out using boron trifluoride–diethyl etherate and triethylsilane in a DCM/acetonitrile mixture, leading to the formation of 241 in an excellent yield. Demethylation of compound 241 is accomplished using boron tribromide at low temperature, resulting in 242 with a yield of 97%. On the other hand, the synthesis of 245 involves two steps starting from commercially available cyclopent-3-en-1-ol (243). The Simmons Smith cyclopropanation of 243 is performed using a mixture of trifluoroacetic acid, diiodomethane, and diethylzinc in DCM, providing 244 with a yield of 48%. Compound 244 is then further treated with methanesulfonyl chloride to give the mesylated compound 245 in a yield of 68%. Subse quently, 4-(5-bromo-2-chlorobenzyl)phenol (242) is allowed to react with 245 in the presence of NMP, cesium carbonate, and BTEAC (benzyltriethylammonium chloride) to give 246. The next step involves a lithium–halogen exchange on
246 using n-butyllithium, with addition to 22 at –78 °C affording the hydroxy intermediate. Methylation of this hydroxy intermediate using methanesulfonic acid and methanol provides 247 in 98% yield. Reduction of 247 using borontrifluoride–diethyl etherate and triethylsilane at –78 °C furnishes 248. To achieve the desired isomer, all of the hydroxy groups of compound 248 were protected using acetic anhydride, DMAP, and pyridine in DCM at 0 °C to give the O-acylated compound 249. In the final step, 249 is hydrolyzed us ing lithium hydroxide monohydrate in a mixed solvent consisting of methanol, THF, and water to provide the desired compound janagliflozin (14) in a yield of 91%. This truncated synthetic route is protection-group-free, and is well suited for scale-up. The drawback of the synthetic route is
the late-stage enrichment of the desired isomer in the final product via acylated derivative 249. The poor isolated yield of 249 is not commercially favored due to low throughput and an increase in raw material and production costs.
PAPER
https://pubs.acs.org/doi/10.1021/acs.oprd.8b00017
SYN
https://www.sciencedirect.com/science/article/abs/pii/S022352342400223X
PAT
https://patentscope.wipo.int/search/en/detail.jsf?docId=US142552820&_cid=P11-MEPJES-88258-1
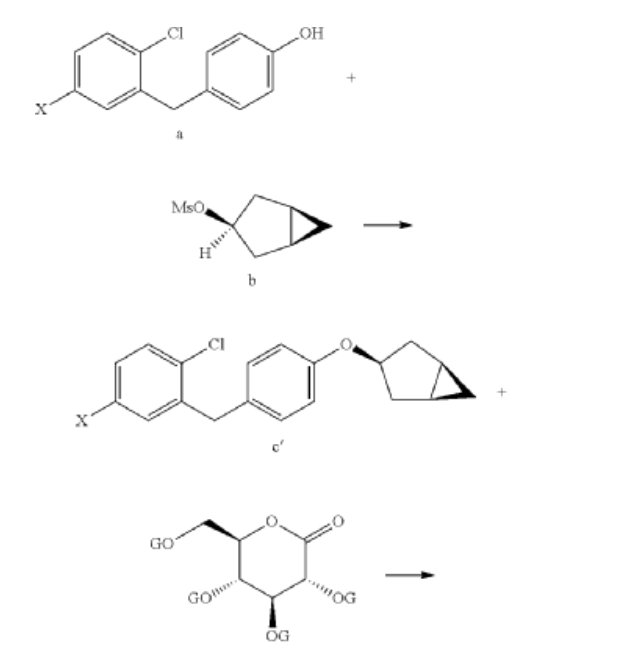
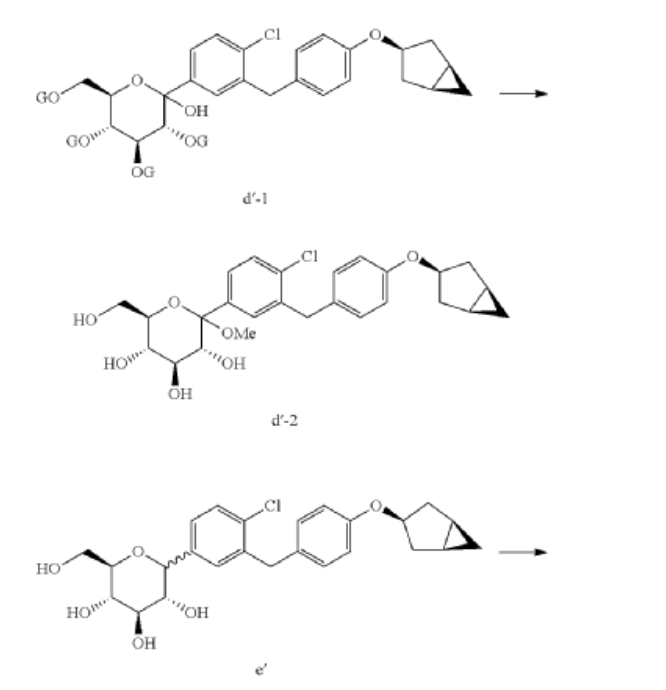
Example 1
Preparation of (2S,3R,4R,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Formula II)
(1) Preparation of 5-bromo-2-chlorobenzoyl chloride
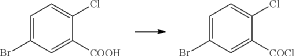
(2) Preparation of (5-bromo-2-chlorophenyl)(4-methoxyphenyl)methanone
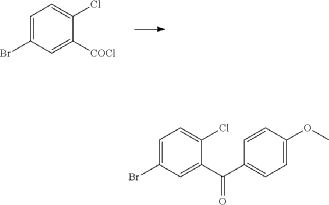
(3) Preparation of 4-bromo-1-chloro-2-(4-methoxybenzyl)benzene
(4) Preparation of 4-(5-bromo-2-chlorobenzyl)phenol
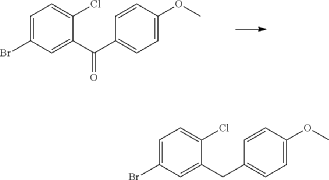
(5) Preparation of (1R,3r,5S)-bicyclo[3.1.0]hexan-3-ol
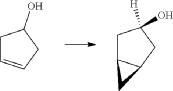
(6) Preparation of (1R,3r,5S)-bicyclo[3.1.0]hexan-3-yl methanesulfonate
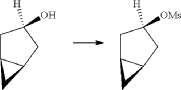
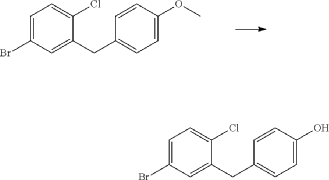
(7) Preparation of (1R,3s,5S)-3-(4-(5-bromo-2-chlorobenzyl)phenyloxy)bicyclo[3.1.0]hexane
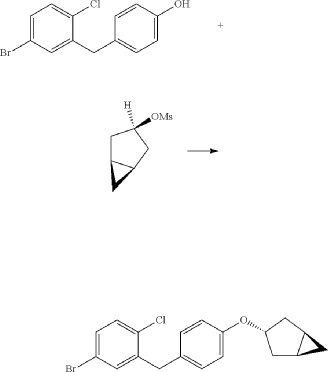
(8) Preparation of (3R,4S,5R,6R)-3,4,5-tri((trimethylsilyl)oxy)-6-(((trimethylsilyl)oxy)methyl)tetrahydro-2H-pyran-2-one
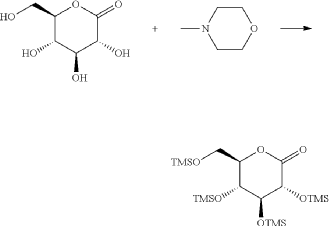
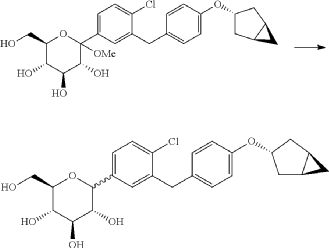
(9) Preparation of (3R,4S,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
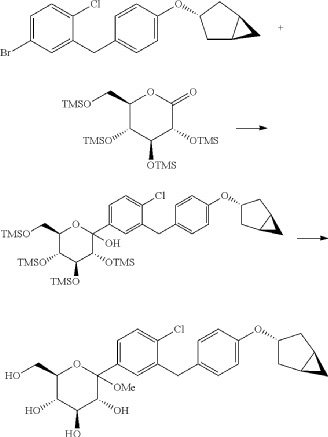
(10) Preparation of (3R,4R,5S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
(11) Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)tetrahydro-2H-pyran-3,4,5-triyl triacetate
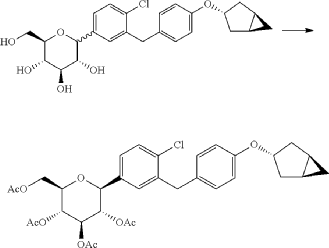
(12) Preparation of (2S,3R,4R,5 S,6R)-2-(3-(4-(((1R,3s,5S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
PAT
EP2891654
https://patentscope.wipo.int/search/en/detail.jsf?docId=EP142501978&_cid=P20-MEQIAN-96633-1
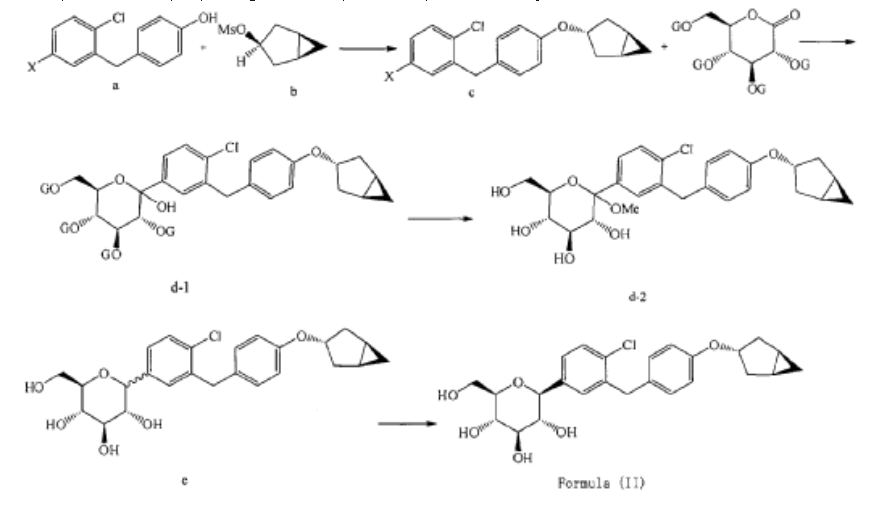
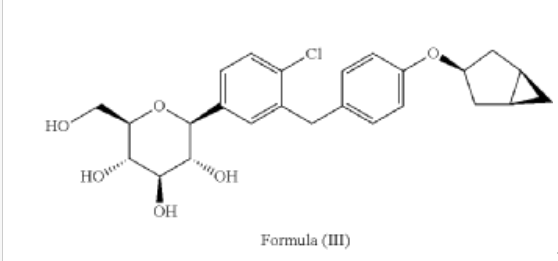
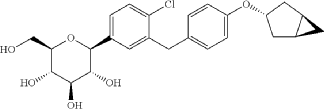
[0027] The compound represented by formula (II) as defined hereinbefore, lab-made, its chemical name and preparation process are described in the following Example 1.
Reference compound 1: Compound 4 as described in the PCT application WO2013/000275A1, lab-made (with reference to the PCT application WO2013/000275A1), its structure is as follows:
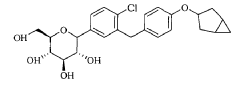
Compound 4, i.e. the compound represented by formula (I).
Reference compound 2: Compound 22 as described in the PCT application WO2013/000275A1, lab-made (with reference to the PCT application WO2013/000275A1), its structure is as follows:
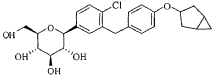
Compound 22.
(12) Preparation of
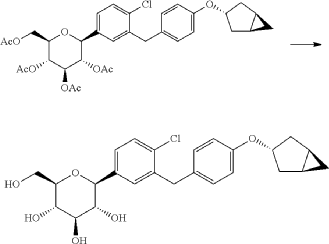
[0057] (2 S,3 R,4 R,5 S,6 R)-2-(3-(4-(((1 R,3 s,5 S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlorophenyl)-6-(hydr oxymethyl)tetrahydro-2 H-pyran-3,4,5-triol
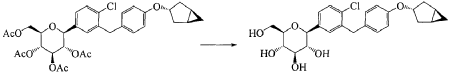
[0058] (2 R,3 R,4 R,5 S,6 S)-2-(acetoxymethyl)-6-(3-(4-(((1 R,3 s,5 S)-bicyclo[3.1.0]hexan-3-yl)oxy)benzyl)-4-chlo rophenyl)tetrahydro-2 H-pyran-3,4,5-triyl triacetate (81g, 0.129mol) was dissolved in a mixed solvent of tetrahydrofuran (313mL), methanol (470mL) and water (156mL). To the resulting mixture was added lithium hydroxide monohydrate (6.32g, 150mmol). The mixture was stirred at room temperature overnight. TLC indicated the completion of reaction. The solvent was removed from the reaction mixture by rotary evaporation. The residual reaction mixture was dissolved with ethyl acetate (400mL). The organic phase was washed with an aqueous saturated sodium chloride solution, with an aqueous KHSO 4 solution, and with water twice, dried over anhydrous sodium sulphate, and evaporated by rotation. The residue was purified with C18 reverse phase preparative chromatography to produce 54.2g of a final product in a yield of 91%.
Formula: C 25H 29ClO 6 Mw: 460.95 LC-MS( m/ z): 478.3 [M+NH 4] +
1H-NMR (400MHz, MeOD) δ: 7.35-7.26 (m, 3H), 7.08-7.06 (d, 2H), 6.76-6.74 (d, 2H), 4.45-4.41 (m, 1H), 4.10-4.00 (m, 3H), 3.89-3.88 (d, 1H), 3.71-3.69 (m, 1H), 3.45-3.38 (m, 3H), 3.31-3.26 (m, 1H), 2.34-2.29 (m, 2H), 1.87-1.81 (m, 2H), 1.37-1.33 (m, 2H), 0.43-0.42 (m, 1H), 0.11-0.10 (m, 1H).
SYN
European Journal of Medicinal Chemistry 291 (2025) 117643
Janagliflozin, engineered by Jilin Huisheng Biopharmaceutical Co., Ltd., a subsidiary under the umbrella of Sihuan Pharmaceutical Holdings Group, falls within the category of oral sodium-glucose co-transporter 2(SGLT2) inhibitors. This agent has been specifically designed with the aim of optimizing glycemic regulation in the adult population grappling with type 2 diabetes mellitus (T2DM) [54]. It is marketed under the brand name Huiyoujing. In 2024, the NMPA gave its approval for Janagliflozin, indicated for adult patients with T2DM, where it can be employed either as a standalone treatment (monotherapy) or in combination with metformin to optimize blood glucose regulation [55]. The clinical effectiveness of Janagliflozin was substantiated through a Phase III clinical trial (NCT03811548). This trial specifically assessed its application as a monotherapy in Chinese patients suffering from T2DM
whose blood glucose was not well – managed via diet and exercise alone. The findings of the study indicated notable decreases in glycated hemoglobin levels. Concurrently, improvements were observed in both body weight and blood pressure. Collectively, these outcomes serve as evidence of the drug’s ability to enhance glycemic regulation [56]. Regarding safety, Janagliflozin was generally well-tolerated. In line with the well-established safety characteristics of SGLT2 inhibitors, the frequently encountered adverse events associated with this treatment were urinary tract infections and genital mycotic infections. No serious adverse events were reported during the trial [57].
The synthesis of Janagliflozin, depicted in Scheme 13, commences with the acylation of 5-bromo-2-chlorobenzoic acid (Jana-001) using oxalyl chloride, yielding the acyl chloride intermediate Jana-002 [58]. Friedel-Crafts acylation of Jana-002 with anisole (Jana-003) affords ketone Jana-004. Subsequent reduction of the carbonyl group in Jana-004 produces Jana-005. Demethylation of Jana-005 with BBr3
generates phenol Jana-006, which undergoes substitution with intermediate Jana-007 to form ether Jana-008. Addition of gluconolactone (Jana-009) to Jana-008 affords Jana-010, where concurrent TMS
deprotection during etherification yields Jana-011. Reduction of Jana-011 using Et3SiH/BF3.ET2Oproduces Jana-012which is sequentially esterified with Ac2O , and hydrolyzed under LiOH conditions, ultimately yielding Janagliflozin
[54] L. Gao, Z. Cheng, B. Su, X. Su, W. Song, Y. Guo, L. Liao, X. Chen, J. Li, X. Tan, F. Xu,
S. Pang, K. Wang, J. Ye, Y. Wang, L. Chen, J. Sun, L. Ji, Efficacy and safety of
janagliflozin as add-on therapy to metformin in Chinese patients with type 2
diabetes inadequately controlled with metformin alone: a multicentre,
randomized, double-blind, placebo-controlled, phase 3 trial, Diabetes Obes Metab
25 (2023) 785–795.
[55] L. Ji, X. Jiang, Q. Hao, Z. Cheng, K. Wang, S. Pang, M. Liu, Y. Guo, X. Chen, X. Su,
T. Ning, J. Liu, F. Bian, Y. Li, Z. Zhang, W. Song, J. Sun, Efficacy and safety of
janagliflozin monotherapy in Chinese patients with type 2 diabetes mellitus
inadequately controlled on diet and exercise: a multicentre, randomized, double-
blind, placebo-controlled, phase 3 trial, Diabetes Obes Metab 25 (2023)
1229–1240.
[56] L. Song, X. Wang, J. Sun, X. Hu, H. Li, P. Hu, D. Liu, A model-informed approach to
accelerate the clinical development of janagliflozin, an innovative SGLT2 inhibitor,
Clin. Pharmacokinet. 62 (2023) 505–518.
[57] Canagliflozin, Drugs and Lactation Database (Lactmed®), National Institute of
Child Health and Human Development, Bethesda (MD), 2006.
[58] F. Wu, Optically Pure benzyl-4-chlorophenyl-C-glucoside Derivatives as SGLT
Inhibitors (Diabetes Mellitus), 2015. EP2891654.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Song, Ling; Yao, Xueting; Liu, Yang; Zhong, Wen; Jiang, Ji; Liu, Hongzhong; Zhou, Huimin; Shi, Chongtie; Zong, Kaiqi; Wang, Chong; Ma, Chuanxiang; Liu, Dongyang; Hu, Pei (April 2020). “Translational prediction of first-in-human pharmacokinetics and pharmacodynamics of janagliflozin, a selective SGLT2 inhibitor, using allometric scaling, dedrick and PK/PD modeling methods”. European Journal of Pharmaceutical Sciences. 147: 105281. doi:10.1016/j.ejps.2020.105281. S2CID 212405270.
- Liu, Dongyang; Song, Ling; Wang, Xiaoxu; Liu, Xu; Cao, Fangrui; Liu, Hongzhong; Ding, Yanhua; Xiao, Xinhua; Jiang, Ji; Hu, Pei (1 June 2019). “154-LB: Accelerating Clinical Development of Janagliflozin, a Novel Antidiabetic Drug, Using Model-Informed Drug Development Strategy”. Diabetes. 68 (Supplement_1). doi:10.2337/db19-154-LB. S2CID 195440798.
- Zhao, Hengli; Wei, Yilin; He, Kun; Zhao, Xiaoyu; Mu, Hongli; Wen, Qing (December 2022). “Prediction of janagliflozin pharmacokinetics in type 2 diabetes mellitus patients with liver cirrhosis or renal impairment using a physiologically based pharmacokinetic model”. European Journal of Pharmaceutical Sciences. 179: 106298. doi:10.1016/j.ejps.2022.106298. PMID 36162752. S2CID 252505056.
- Zhao, Hengli; Zhao, Zhirui; He, Kun; Mi, Nianrong; Lou, Kai; Dong, Xiaolin; Zhang, Wenyu; Sun, Jingfang; Hu, Xinyu; Pang, Shuguang; Cheng, Hong; Wen, Qing (August 2023). “Pharmacokinetics, Pharmacodynamics and Safety of Janagliflozin in Chinese Type 2 Diabetes Mellitus Patients with Renal Impairment”. Clinical Pharmacokinetics. 62 (8): 1093–1103. doi:10.1007/s40262-023-01256-0. PMID 37284974. S2CID 259097798.
- Gao, Leili; Cheng, Zhifeng; Su, Benli; Su, Xiuhai; Song, Weihong; Guo, Yushan; Liao, Lin; Chen, Xiaowen; Li, Jiarui; Tan, Xingrong; Xu, Fangjiang; Pang, Shuguang; Wang, Kun; Ye, Jun; Wang, Yuan; Chen, Lili; Sun, Jingfang; Ji, Linong (March 2023). “Efficacy and safety of janagliflozin as add‐on therapy to metformin in Chinese patients with type 2 diabetes inadequately controlled with metformin alone: A multicentre, randomized, double‐blind, placebo‐controlled, phase 3 trial”. Diabetes, Obesity and Metabolism. 25 (3): 785–795. doi:10.1111/dom.14926. PMID 36433709. S2CID 253967474.
- Ji, Linong; Jiang, Xiaozhen; Hao, Qingshun; Cheng, Zhifeng; Wang, Kun; Pang, Shuguang; Liu, Meiying; Guo, Yushan; Chen, Xiaowen; Su, Xiuhai; Ning, Tao; Liu, Jie; Bian, Fang; Li, Yulan; Zhang, Zhinong; Song, Weihong; Sun, Jingfang (May 2023). “Efficacy and safety of janagliflozin monotherapy in Chinese patients with type 2 diabetes mellitus inadequately controlled on diet and exercise: A multicentre, randomized, double‐blind, placebo‐controlled, Phase 3 trial”. Diabetes, Obesity and Metabolism. 25 (5): 1229–1240. doi:10.1111/dom.14971. PMID 36594724. S2CID 255474211.
- “NMPA approves China’s second homegrown SGLT2 inhibitor janagliflozin”. bioworld.com. January 23, 2024.
| Legal status | |
|---|---|
| Legal status | Rx in China; investigational elsewhere |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1800115-22-3 |
| PubChem CID | 91820686 |
| DrugBank | DB16209 |
| UNII | WK4RT85HCA |
| Chemical and physical data | |
| Formula | C25H29ClO6 |
| Molar mass | 460.95 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Janagliflozin, china 2024, approvals 2024, Jilin Huisheng Biopharmaceutical Co, sihuan, SGLT2 inhibitors, Huiyoujing, WK4RT85HCA, XZP 5695, UNII-WK4RT85HCA, 1800115-22-3
SYN
SYNTHESIS 2024, 56, 906–943
synthesis of janagliflozin (14) was achieved through an eleven-step process in an overall yield of 3% (Scheme 45).71 Initially, the two advanced intermediates were synthesized and then coupled under cryogenic conditions using nBuLi. The construction of 242 commences with the reaction of 5-bromo-2-chlorobenzoic acid (26c) with oxalyl chloride and a catalytic amount of DMF in DCM, yielding the acid
chloride derivative 26c′. This intermediate is then subjected to Friedel–Crafts acylation with anisole to produce 240 in 71% yield. Subsequent reduction of 240 was carried out using boron trifluoride–diethyl etherate and triethylsilane in a DCM/acetonitrile mixture, leading to the formation of 241 in an excellent yield. Demethylation of compound 241 is accomplished using boron tribromide at low temperature, re
sulting in 242 with a yield of 97%. On the other hand, the synthesis of 245 involves two steps starting from commercially available cyclopent-3-en-1-ol (243). The Simmons Smith cyclopropanation of 243 is performed using a mixture of trifluoroacetic acid, diiodomethane, and diethylzinc in DCM, providing 244 with a yield of 48%. Compound 244 is then further treated with methanesulfonyl chloride to
give the mesylated compound 245 in a yield of 68%. Subsequently, 4-(5-bromo-2-chlorobenzyl)phenol (242) is allowed to react with 245 in the presence of NMP, cesium carbonate, and BTEAC (benzyltriethylammonium chloride) to give 246. The next step involves a lithium–halogen exchange on
246 using n-butyllithium, with addition to 22 at –78 °C affording the hydroxy intermediate. Methylation of this hydroxy intermediate using methanesulfonic acid and methanol provides 247 in 98% yield. Reduction of 247 using boron trifluoride–diethyl etherate and triethylsilane at –78 °C furnishes 248. To achieve the desired isomer, all of the hydroxy groups of compound 248 were protected using acetic anhydride, DMAP, and pyridine in DCM at 0 °C to give the O-acylated compound 249. In the final step, 249 is hydrolyzed us ing lithium hydroxide monohydrate in a mixed solvent consisting of methanol, THF, and water to provide the desired compound janagliflozin (14) in a yield of 91%. This truncated synthetic route is protection-group-free, and is well suited for scale-up. The drawback of the synthetic route is
the late-stage enrichment of the desired isomer in the final product via acylated derivative 249. The poor isolated yield of 249 is not commercially favored due to low throughput and an increase in raw material and production costs

(71) (a) Wu, F. US9315438B2, 2016. (b) Wu, F. EP2891654A1, 2014.
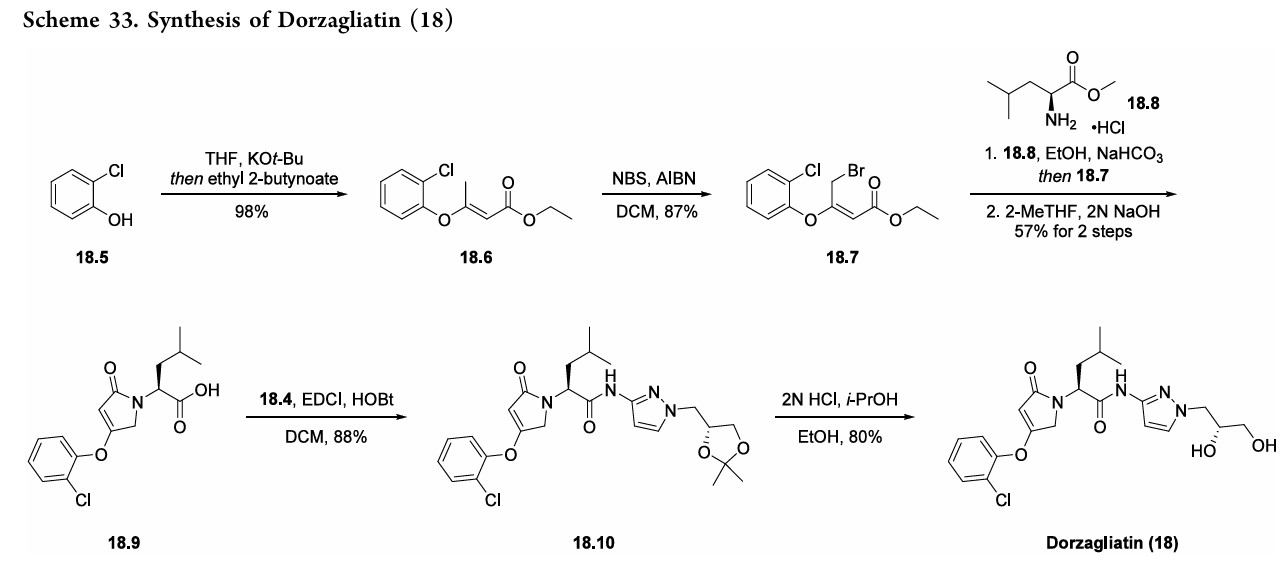
Dorzagliatin
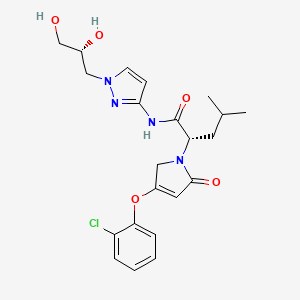
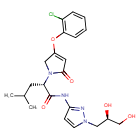
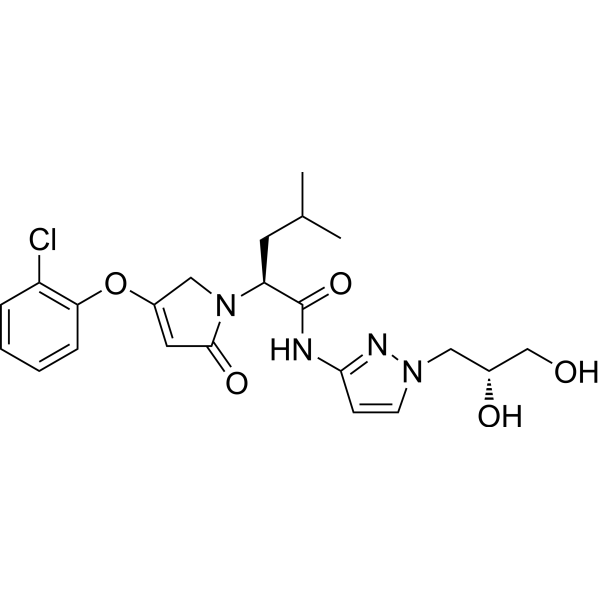
Dorzagliatin
- CAS 1191995-00-2
- HMS5552
- Sinogliatin
- HMS-5552
- MW 462.9 g/mol MF C22H27ClN4O5
- (2S)-2-[3-(2-chlorophenoxy)-5-oxo-2H-pyrrol-1-yl]-N-[1-[(2R)-2,3-dihydroxypropyl]pyrazol-3-yl]-4-methylpentanamide
- RO5305552
- RO-5305552
- X59W6980E8
- (2S)-2-[3-(2-chlorophenoxy)-5-oxo-2H-pyrrol-1-yl]-N-[1-[(2R)-2,3-dihydroxypropyl]pyrazol-3-yl]-4-methyl-pentanamide
- 1H-PYRROLE-1-ACETAMIDE, 4-(2-CHLOROPHENOXY)-N-(1-((2R)-2,3-DIHYDROXYPROPYL)-1H-PYRAZOL-3-YL)-2,5-DIHYDRO-.ALPHA.-(2-METHYLPROPYL)-2-OXO-, (.ALPHA.S)-
Dorzagliatin(18)was developed by Hua Medicine as a treatment for diabetic kidney disease(DKD), type1diabetes mellitus(T1DM), and type2 diabetes mellitus (T2DM). CHINA 2022
Dorzagliatin is a glucokinase activator that is being developed to treat diabetes.[1] Unlike other diabetes drugs, it is intended to increase insulin sensitivity.[2]
Dorzagliatin is under investigation in clinical trial NCT03173391 (Long-term Efficacy and Safety of HMS5552 in T2DM).
PATENT
https://patents.google.com/patent/CN112062754A/en
(R) -1- ((2, 2-dimethyl-1, 3-dioxolane-4-yl) methyl) -1H-pyrazole-3-ammonia (II) is a very important medical intermediate for synthesizing Dorzagliatin. Dorzagliatin is a novel medicine for treating type 2 diabetes mellitus, and (R) -1- ((2, 2-dimethyl-1, 3-dioxolane-4-yl) methyl) -1H-pyrazole-3-ammonia (II) is an essential intermediate in the synthetic process of the medicine, and along with the steady promotion of new Dorzagliatin medicines to the market, the demand of the chiral intermediate in the market is required to be rapidly increased.
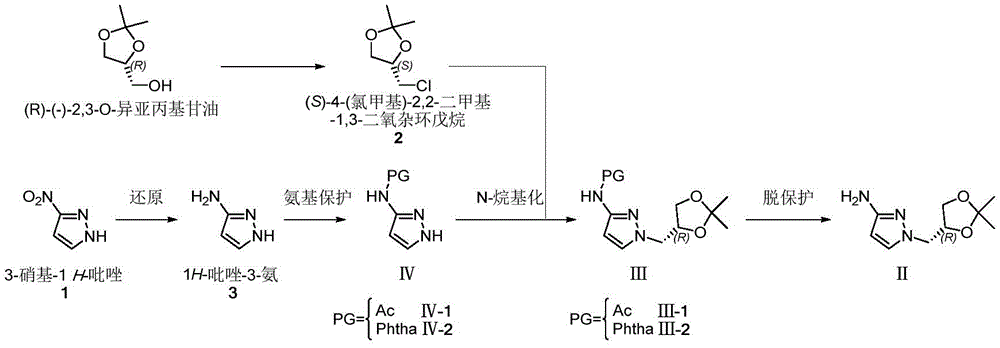
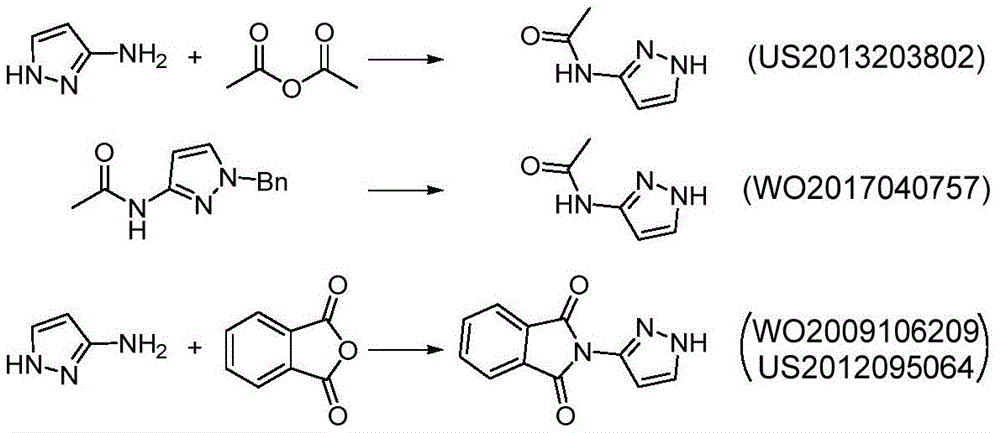
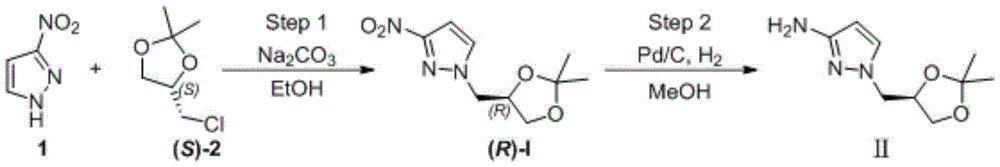
The main production method of the key chiral intermediate is shown as follows: reducing nitro in 3-nitro-1H-pyrazole substrate into amino, protecting free amino, carrying out N-alkylation reaction with (R) – (-) -2, 3-O-isopropylidene glycerol-OH derivative active intermediate, and deprotecting to obtain the final product. The synthetic route needs to be subjected to an N-protection process, so that route steps are added, and the cost is increased. The synthesis of N-protected substrate iv is reported: in the patent US2013203802, 1H-pyrazole-3-ammonia is protected by acetic anhydride, and in WO2017040757, N-acetyl-1H-pyrazole-3-ammonia is obtained by an N- (1-benzyl-1H-pyrazole-3-yl) acetamide debenzylation method; the protection of the N-benzoyl group of 1H-pyrazol-3-amine is reported in the patent US 6118008; in addition, WO2009106209, US2012095064, mention the phthalimide protection strategy of 1H-pyrazole-3-ammonia with phthalic anhydride.
Example 1
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine
The first step is as follows: intermediate (R) -I preparation:
under the protection of nitrogen, 3-nitro-1H-pyrazole (1) (100.00g,0.884mol), ethanol (1.0L) and sodium carbonate (133.90g, 1.26mol) are sequentially added into a 3L reaction bottle, and the system is stirred for 0.5H at room temperature; (S) – (-) -4-chloromethyl-2, 2-dimethyl-1, 3-dioxolane ((S) -2) (126.84g, 0.842mol) was dissolved and diluted with 634ml of ethanol and then added dropwise to the reaction flask. After the dropwise addition, the temperature is raised to 50 ℃ and the reaction is stirred for 5 hours. Ethanol was distilled off under reduced pressure, and the residue was diluted with (1.0L) of water and then extracted twice with dichloromethane (500ml × 2); the organic phase was washed with water and then with saturated sodium chloride brine. Concentrating under reduced pressure to remove dichloromethane to obtain crude oily substance; the crude product was purified by silica gel column chromatography (eluent: n-hexane/ethyl acetate mixed system) to give 166.5g of a pale yellow oily product, with a yield of 87% and an ee value of 98% or more.
The second step is that: reducing nitro to obtain target product
A2L autoclave was charged with (R) -I substrate (150g, 0.66mol), methanol (750mL), Pd/C (0.75g, 0.5% W/W), and the mixture was subjected to nitrogen substitution three times, then hydrogen substitution three times, under a hydrogen-charging pressure of 2.0MPa, at a temperature of 50 ℃ for reaction for 8 hours. Filtering, filtering to remove Pd/C catalyst, concentrating the filtrate to remove methanol to obtain 123.70g of light yellow oily matter, wherein the yield is 95%, and the ee value is more than or equal to 98%.
Example 2
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine by Raney-Ni reduction system
The first step is the same as in example 1.
The second step is that: reduction of nitro groups by Rany-Ni
The intermediate (R) -I (150g, 0.66mol) obtained in the first step was charged into a 2L reactor, and ethanol (1.2L) was added thereto and stirred, followed by adding Rany-Ni (75g) and stirring at room temperature for reaction for 15 hours. Filtering, filtering to remove the solid catalyst, and concentrating the filtrate to dryness to obtain 106.77g of light yellow oily substance with yield of 82% and ee value of more than or equal to 97%.
Example 3
Preparation of (R) -1- ((2, 2-dimethyl-1, 3-dioxolan-4-yl) methyl) -1H-pyrazol-3-amine by hydrazine hydrate system
The first step is the same as in example 1.
The second step is that: A2L reaction flask was charged with intermediate (R) -I (150g, 0.66mol), ferric trichloride (528mg, 3.3mmol), and ethanol (1.2L), stirred, charged with hydrazine hydrate (39.5g, 0.79mol), and heated to reflux for 6 h. Ethanol was removed by concentration under reduced pressure, the residue was diluted with 750ml of water and extracted twice with ethyl acetate (250 ml. times.2). The organic phase was washed with water and then with saturated brine. The ethyl acetate is removed by concentration to obtain 110.7g of crude light yellow oily substance, the yield is 85 percent, and the ee value is more than or equal to 97 percent.
SYN
https://doi.org/10.1021/acs.jmedchem.3c02374J.Med.Chem.2024,67,4376−4418
Dorzagliatin(HuaTangNing).
Dorzagliatin(18)was developed by Hua Medicine as a treatment for diabetic kidney disease(DKD), type1diabetes mellitus(T1DM), and type2 diabetes mellitus (T2DM).133 This first-in-class, small
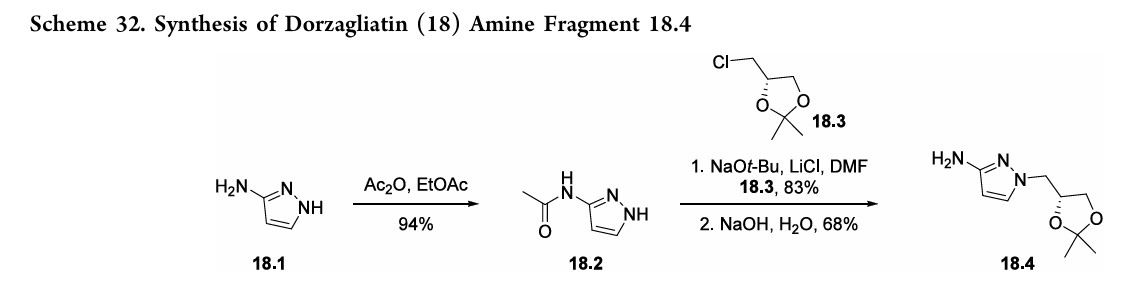
molecule,oral,glucokinaseactivator(GKA)wasfirst approved in ChinainSeptember2022foradultpatientswithT2DMasa monotherapy and in combination with metformin (an antidiabetic medication).134 Expression of glucokinase is reduced for individuals with T2DM, thus GKAs such as dorzagliatin serve as a novel class of antidiabetic treatment options.135,136 Theinitialpatent thatdisclosesthesynthesisofdorzagliatin (18)began fromreadily availablematerials 3-aminopyrazole
(18.1) and 2-chlorophenol (18.5). The synthetic strategy reliedonaconvergentamidecouplingofamine18.4(Scheme32) and carboxylic acid 18.9 (Scheme 33).137 A later disclosure provided an updated route toward amine 18.4 (Scheme 32), detailing the synthetic improvements with respect to yield and purity.138 This later disclosure also detailed the synthesis of dorzagliatinonmultikilogramscale fromtheamidationofacid18.9withamine18.4,yieldingover
10kgoftheactivepharmaceutical ingredient.Acetylationof3 aminopyrazole (18.1) with acetic anhydride provided the protectedpyrazole18.2(Scheme32). Subsequent alkylation with alkyl chloride 18.3 followed by base-mediated deprotectionyieldedamine18.4. The synthesis of acid 18.9 began with base-mediated
alkenylationof2-chlorophenol (18.5)withethyl 2-butynoate toprovideester18.6(Scheme33). Subsequentbromination withNBSandAIBNyieldsallylbromide18.7.Next,subjection
ofL-leucinemethylesterhydrochloride(18.8)tobaseresulted ina freeamine thatunderwent allylationwithbromide18.7. Acid 18.9was subsequently generated froma cyclization
condensation sequence and saponification reaction with NaOH. Final amidebondformationwas facilitatedbyEDCI andHOBt toprovideamide18.10, anddorzagliatin(18)was generatedonthemultikilogramscale followingacid-mediated acetonidedeprotectiontoreveal the1,2-diol.
(133) Syed, Y. Y. Dorzagliatin: First approval. Drugs 2022, 82,
1745−1750.
(134) Xu, H.; Sheng, L.; Chen, W.; Yuan, F.; Yang, M.; Li, H.; Li, X.;
Choi, J.; Zhao, G.; Hu, T.; et al. Safety, tolerability, pharmacokinetics,
and pharmacodynamics of novel glucokinase activator HMS5552:
results from a first-in-human single ascending dose study. Drug Des.
Devel. Ther. 2016, 10, 1619−26.
(135) Ren, Y.; Li, L.; Wan, L.; Huang, Y.; Cao, S. Glucokinase as an
emerging anti-diabetes target and recent progress in the development
of its agonists. J. Enzyme Inhib. Med. Chem. 2022, 37, 606−615.
(136) Toulis, K. A.; Nirantharakumar, K.; Pourzitaki, C.; Barnett, A.
H.; Tahrani, A. A. Glucokinase activators for type 2 diabetes:
Challenges and future developments. Drugs 2020, 80, 467−475.
(137) Berthel, S. J.; Brinkman, J. A.; Hayden, S.; Haynes, N.-E.;
Kester, R. F.; McDermott, L. A.; Qian, Y.; Sarabu, R.; Scott, N. R.;
Tilley, J. W. Pyrrolidinone as glucokinase activators and their
preparation, pharmaceutical compositions and use in the treatment
of metabolic disorders. WO 2009127546, 2009.
(138) Chen, J.; Ren, Y.; She, J.; Wang, L. Process for the preparation
of 1-([1,3]dioxolan-4-ylmethyl)-1h-pyrazol-3-ylamine. U.S. Patent US
20150315176, 2015.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Chow, Elaine; Wang, Ke; Lim, Cadmon K.P.; Tsoi, Sandra T.F.; Fan, Baoqi; Poon, Emily; Luk, Andrea O.Y.; Ma, Ronald C.W.; Ferrannini, Ele; Mari, Andrea; Chen, Li; Chan, Juliana C.N. (1 February 2023). “Dorzagliatin, a Dual-Acting Glucokinase Activator, Increases Insulin Secretion and Glucose Sensitivity in Glucokinase Maturity-Onset Diabetes of the Young and Recent-Onset Type 2 Diabetes”. Diabetes. 72 (2): 299–308. doi:10.2337/db22-0708. PMC 9871194.
- Zhu, Dalong; Li, Xiaoying; Ma, Jianhua; Zeng, Jiao’e; Gan, Shenglian; Dong, Xiaolin; Yang, Jing; Lin, Xiaohong; Cai, Hanqing; Song, Weihong; Li, Xuefeng; Zhang, Keqin; Zhang, Qiu; Lu, Yibing; Bu, Ruifang; Shao, Huige; Wang, Guixia; Yuan, Guoyue; Ran, Xingwu; Liao, Lin; Zhao, Wenjuan; Li, Ping; Sun, Li; Shi, Lixin; Jiang, Zhaoshun; Xue, Yaoming; Jiang, Hongwei; Li, Quanmin; Li, Zongbao; Fu, Maoxiong; Liang, Zerong; Guo, Lian; Liu, Ming; Xu, Chun; Li, Wenhui; Yu, Xuefeng; Qin, Guijun; Yang, Zhou; Su, Benli; Zeng, Longyi; Geng, Houfa; Shi, Yongquan; Zhao, Yu; Zhang, Yi; Yang, Wenying; Chen, Li (May 2022). “Dorzagliatin in drug-naïve patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial”. Nature Medicine. 28 (5): 965–973.
- [1]. Zhu XX, et al. Dorzagliatin (HMS5552), a novel dual-acting glucokinase activator, improves glycaemic control and pancreatic β-cell function in patients with type 2 diabetes: A 28-day treatment study using biomarker-guided patient selection. Diabetes Obes Metab. 2018 Sep;20(9):2113-2120. [Content Brief][2]. Wang P, et al. Effects of a Novel Glucokinase Activator, HMS5552, on Glucose Metabolism in a Rat Model of Type 2 Diabetes Mellitus. J Diabetes Res. 2017;2017:5812607. [Content Brief]
//////////Dorzagliatin, APPROVALS 22, CHINA 22, DIABETES, Hua Medicine, 1191995-00-2, HMS 5552, Sinogliatin, HMS-5552, RO 5305552, RO-5305552, X59W6980E8
Chiglitazar
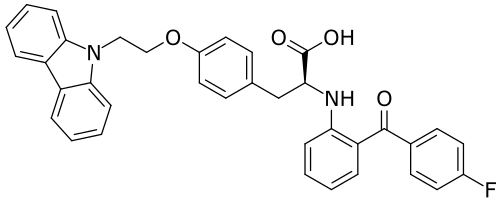
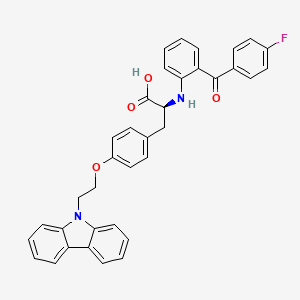
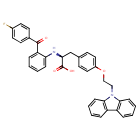
Chiglitazar
CAS 743438-45-1
| Ingredient | UNII | CAS | InChI Key |
|---|---|---|---|
| Chiglitazar sodium, (S)- | YN12H6OCV6 | 2390374-10-2 | RMVIEXHXRDCWBT-UCRKPPETSA-M |
- CS 038
- Carfloglitazar, (s)-
- E6EJV1J6Y0
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- C36H29FN2O4
- 572.6 g/mol
- (2S)-3-[4-(2-carbazol-9-ylethoxy)phenyl]-2-[2-(4-fluorobenzoyl)anilino]propanoic acid
- (2S)-3-(4-(2-CARBAZOL-9-YLETHOXY)PHENYL)-2-(2-(4-FLUOROBENZOYL)ANILINO)PROPANOIC ACID
- (2s)-3-(4-(2-carbazol-9-ylethoxy)phenyl)-2-(2-(4-fluorobenzoyl)anilino)propanoic acid
- Carfloglitazar, (s)-
- L-tyrosine, o-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-
- O-(2-(9h-carbazol-9-yl)ethyl)-n-(2-(4-fluorobenzoyl)phenyl)-l-tyrosine
Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
patients with type2 diabetes in October2021.
Chiglitazar (trade name Bilessglu) is a drug for the treatment of type 2 diabetes.[1] It is a peroxisome proliferator-activated receptor (PPAR) agonist.
In China, chiglitazar is approved for glycemic control in adult patients with type 2 diabetes when used in combination with diet and exercise.[2]
Chiglitazar is under investigation in clinical trial NCT06125587 (Chiglitazar/metformin in Non-obese Women With PCOS).
SYN
WO 2004048333
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2004048333&_cid=P12-MDMUOB-48741-1
Example 15
Preparation of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-carbazolylethoxy)-phenyl]
-propionic acid (compound CS038)
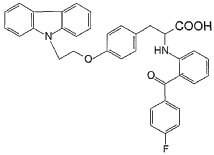
To a solution of 2-[(2-(4-fluorobenzoyl)phenyl)amino]-3-[4-(2-bromoethoxy)-phenyl] -propionic acid methyl ester (0.25 g, 0.49 mmol) and carbazole (0.082 g, 0.49 mmol) in benzene (10 ml) is added tetrabutyl ammonium bromide (0.08 g) and 50% NaOH aqueous solution (0.084 g, 1.08 mmol), then the mixture is heated to reflux for 10 h. After cooled, benzene (30ml) is added, and the mixture is washed with water (3×30 ml). Then the solvent is evaporated under a vacuum. The crude product is purified by silica gel chromatography using CHCl3/MeOH (4:1) as eluent to give the title compound (0.10 g, 36%). HRMS calcd for C36H29FN204: 572.6357. Found: 572.6354. MA calcd for C36H29FN204: C, 75.51%; H, 5.11%; N, 4.89%. Found: C, 75.83%; H, 5.10%; N, 4.90%.
PATENT
US 10640465
https://patentscope.wipo.int/search/en/detail.jsf?docId=US249083802&_cid=P12-MDMUQY-52500-1
The pharmacological activity of the compound is described in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157. 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is able to selectively activate PPAR-α, PPAR-γ and PPAR-6, and can be used to treat the diseases associated with metabolic syndrome such as diabetes, hypertension, obesity, insulin resistance, hypertriglyceridemia, hyperglycemia, high cholesterol, arteries atherosclerosis, coronary heart disease, etc. A preparation method of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic acid is disclosed in Chinese patent application No. CN03126974.5 and U.S. Pat. No. 7,268,157, and the synthetic route thereof is as follows:
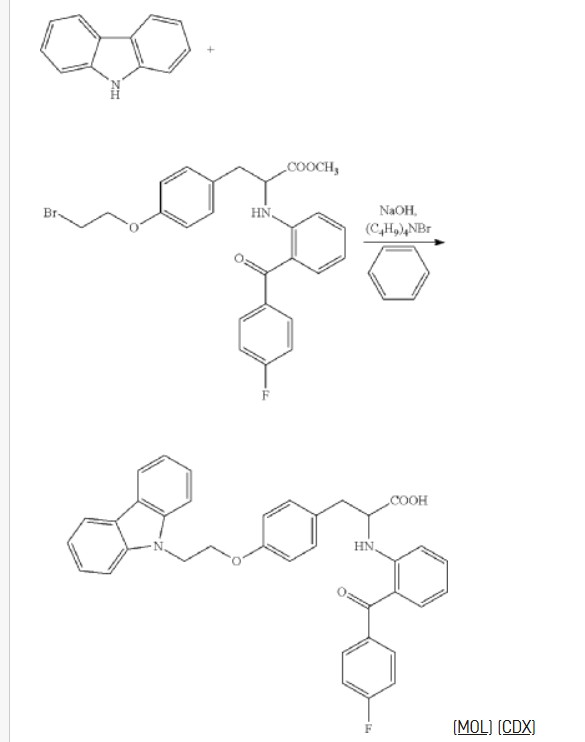
EXAMPLES
Example 1: Preparation of 2-(2-(4-fluorobenzoyl)phenylamino)-3-(4-(2-(9H-carbazol-9-yl)ethoxy)phenyl)propanoic Acid
SYN
J. Med. Chem. 2024, 67, 4376−4418
Chiglitazar (Bilessglu). Chiglitazar (17), a novel nonthiazolidinedione pan-agonist of α, δ, and γ peroxisome proliferator-activated receptors (PPARs), has shown promise for the treatment of type 2 diabetes. 126 Type 2 diabetes impacts over 374 million patients worldwide and continues to
rise in incidence and prevalence globally. 127 Chiglitazar preferentially regulates expression of ANGPTL4 and PDK4 genes, which are involved in glucose and lipid metabolism. 128 Chiglitazar was developed by Chipscreen Biosciences and was approved in China for improving glycemic control in adult
patients with type2 diabetes in October2021.129 Thesynthesisof17beganwithimineformationbetweenL
tyrosine methyl ester (17.1) and 2-(4-fluorobenzoyl) cyclohexanone(17.2)with tandemaromatizationunderPd/C catalysis to generate aniline derivative 17.3 (Scheme31).130,131 Alkylation of the phenol moiety of 17.3 with mesylate17.4furnishedphenyl alkyl etherderivative17.5.132
Hydrolysisof themethylester in17.5withlithiumhydroxide followedbyacidificationwithhydrochloricacidandrecrystal lization fromacetonitrile afforded chiglitazar (17) in 42% overall yield from17.3.Thisprocessdeliveredchiglitazar in 99.4%purityat24gscale.
(126) Ji, L.; Song, W.; Fang, H.; Li, W.; Geng, J.; Wang, Y.; Guo, L.;
Cai, H.; Yang, T.; Li, H.; et al. Efficacy and safety of chiglitazar, a
novel peroxisome proliferator-activated receptor pan-agonist, in
patients with type 2 diabetes: a randomized, double-blind, placebo
controlled, phase 3 trial (CMAP). Sci. Bull. 2021, 66, 1571−1580.
(127) Chatterjee, S.; Khunti, K.; Davies, M. J. Type 2 diabetes.
Lancet 2017, 389, 2239−2251.
(128) Pan, D.-S.; Wang, W.; Liu, N.-S.; Yang, Q.-J.; Zhang, K.; Zhu,
J.-Z.; Shan, S.; Li, Z.-B.; Ning, Z.-Q.; Huang, L.; Lu, X.-P. Chiglitazar
preferentially regulates gene expression via configuration-restricted
binding and phosphorylation inhibition of PPARγ. PPAR Research
2017 2017, 2017, 1−16.
(129) Deeks, E. D. Chiglitazar: First approval. Drugs 2022, 82, 87−
92.
(130) Li, Z.; Lu, X.-P.; Liao, C.; Shi, L.; Liu, Z.; Ma, B. Substituted
arylalcanoic acid derivatives as PPAR pan agonists with potent
antihyperglycemic and antihyperlipidemic activity. WO 2004048333
A1, 2004.
(131) Sutter, M.; Sotto, N.; Raoul, Y.; Métay, E.; Lemaire, M.
Straightforward heterogeneous palladium catalyzed synthesis of aryl
ethers and aryl amines via a solvent free aerobic and non-aerobic
dehydrogenative arylation. Green Chem. 2013, 15, 347−352.
(132) Lu, X.; Li, Z.; Wang, X. Method for preparing phenylalanine
compound. U.S. Patent US 10640465 B2, 2020.
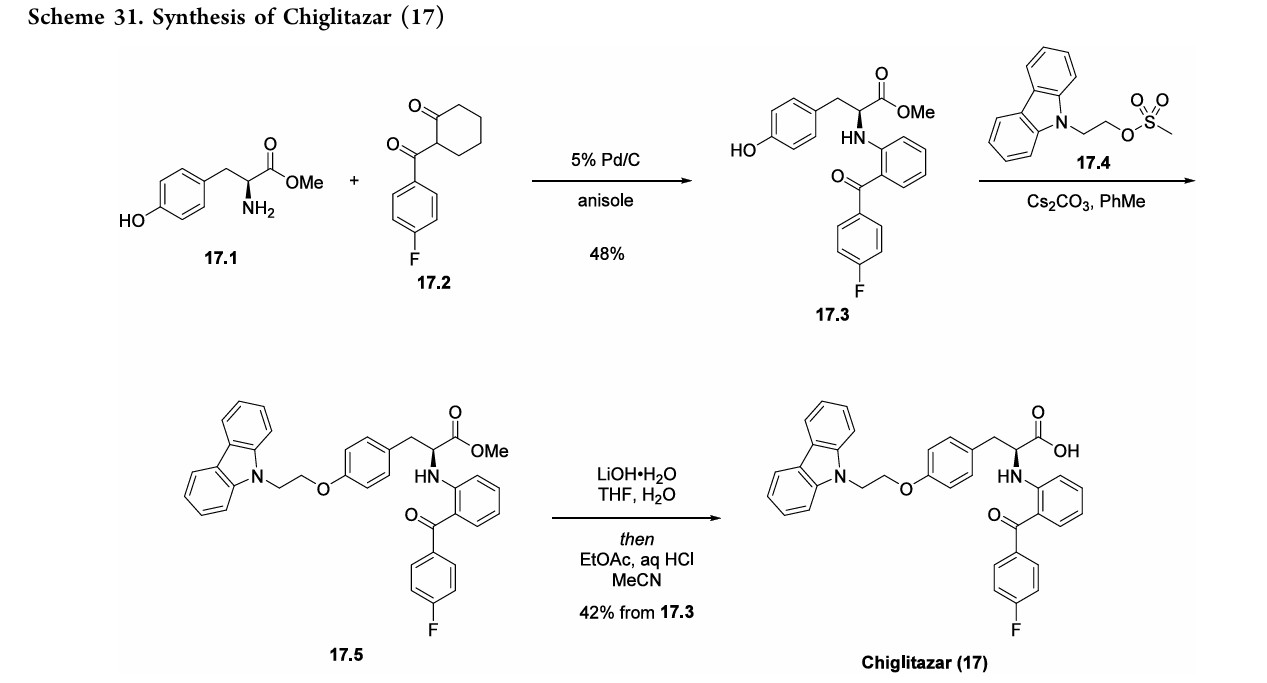
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
References
- Ji L, Song W, Fang H, Li W, Geng J, Wang Y, et al. (August 2021). “Efficacy and safety of chiglitazar, a novel peroxisome proliferator-activated receptor pan-agonist, in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled, phase 3 trial (CMAP)”. Science Bulletin. 66 (15): 1571–1580. Bibcode:2021SciBu..66.1571J. doi:10.1016/j.scib.2021.03.019. PMID 36654286. S2CID 233650336.
- Deeks ED (January 2022). “Chiglitazar: First Approval”. Drugs. 82 (1): 87–92. doi:10.1007/s40265-021-01648-1. PMID 34846697. S2CID 244716275.
| Clinical data | |
|---|---|
| Trade names | Bilessglu |
| Other names | Carfloglitazar |
| Legal status | |
| Legal status | Rx in China |
| Identifiers | |
| IUPAC name | |
| CAS Number | 743438-45-1 |
| PubChem CID | 71402018 |
| ChemSpider | 57523239 |
| UNII | E6EJV1J6Y0 |
| ChEMBL | ChEMBL4650349 |
| CompTox Dashboard (EPA) | DTXSID00225352 |
| Chemical and physical data | |
| Formula | C36H29FN2O4 |
| Molar mass | 572.636 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
///////////Chiglitazar, Chipscreen Biosciences, CHINA 2021, DIABETES, CS 038, Carfloglitazar, (s)-, E6EJV1J6Y0,
Tirzepatide
YXEGTFTSDY SIXLDKIAQK AFVQWLIAGG PSSGAPPPS



Tirzepatide
チルゼパチド
LY3298176,
| Formula | C225H348N48O68 |
|---|---|
| CAS | 2023788-19-2 |
| Mol weight | 4813.4514 |
FDA APPROVED 2022/5/13, Mounjaro
| Class | Antidiabetic agent GLP-1 receptor agonist |
|---|---|
| Efficacy | Antidiabetic, Gastric inhibitory polypeptide receptor agonist, Glucagon-like peptide 1 (GLP-1) receptor agonist |
| Disease | Type 2 diabetes mellitus |

Tirzepatide is an agonist of human glucose-dependent insulinotropic polypeptide (GIP) and human glucagon-like peptide-1 (GLP-1) receptors, whose amino acid residues at positions 2 and 13 are 2-methylAla, and the C-terminus is amidated Ser. A 1,20-icosanedioic acid is attached to Lys at position 20 via a linker which consists of a Glu and two 8-amino-3,6-dioxaoctanoic acids. Tirzepatide is a synthetic peptide consisting of 39 amino acid residues.
C225H348N48O68 : 4813.45
[2023788-19-2]
L-Serinamide, L-tyrosyl-2-methylalanyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-isoleucyl-2-methylalanyl-L-leucyl-L-α-aspartyl-L-lysyl-L-isoleucyl-L-alanyl-L-glutaminyl-N6-[(22S)-22,42-dicarboxy-1,10,19,24-tetraoxo-3,6,12,15-tetraoxa-9,18,23-triazadotetracont-1-yl]-L-lysyl-L-alanyl-L-phenylalanyl-L-valyl-L-glutaminyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-alanylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-prolyl-
Other Names
- L-Tyrosyl-2-methylalanyl-L-α-glutamylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-isoleucyl-2-methylalanyl-L-leucyl-L-α-aspartyl-L-lysyl-L-isoleucyl-L-alanyl-L-glutaminyl-N6-[(22S)-22,42-dicarboxy-1,10,19,24-tetraoxo-3,6,12,15-tetraoxa-9,18,23-triazadotetracont-1-yl]-L-lysyl-L-alanyl-L-phenylalanyl-L-valyl-L-glutaminyl-L-tryptophyl-L-leucyl-L-isoleucyl-L-alanylglycylglycyl-L-prolyl-L-seryl-L-serylglycyl-L-alanyl-L-prolyl-L-prolyl-L-prolyl-L-serinamide
Tirzepatide, sold under the brand name Mounjaro,[1] is a medication used for the treatment type 2 diabetes.[2][3][4] Tirzepatide is given by injection under the skin.[2] Common side effects may include nausea, vomiting, diarrhea, decreased appetite, constipation, upper abdominal discomfort and abdominal pain.[2]
Glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are hormones involved in blood sugar control.[2] Tirzepatide is a first-in-class medication that activates both the GLP-1 and GIP receptors, which leads to improved blood sugar control.[2] Tirzepatide was approved for medical use in the United States in May 2022.[2]
SYN
https://pubs.acs.org/doi/10.1021/acs.oprd.1c00108

The large-scale manufacture of complex synthetic peptides is challenging due to many factors such as manufacturing risk (including failed product specifications) as well as processes that are often low in both yield and overall purity. To overcome these liabilities, a hybrid solid-phase peptide synthesis/liquid-phase peptide synthesis (SPPS/LPPS) approach was developed for the synthesis of tirzepatide. Continuous manufacturing and real-time analytical monitoring ensured the production of high-quality material, while nanofiltration provided intermediate purification without difficult precipitations. Implementation of the strategy worked very well, resulting in a robust process with high yields and purity.
PATENT
- WO2016111971
- US2020023040
- WO2019245893
- US2020155487
- US2020155650
- WO2020159949CN112592387
- WO2021066600CN112661815
- WO2021154593
- US2021338769

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG SUBSCRIPTION
$10.00
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
Tirzepatide in indicated to improve blood sugar control in adults with type 2 diabetes, as an addition to diet and exercise.[2]
Contraindications
Tirzepatide should not be used in people with a personal or family history of medullary thyroid cancer or in people with multiple endocrine neoplasia syndrome type 2.[2]
Adverse effects
Preclinical, phase I, and phase II trials have indicated that tirzepatide exhibits similar adverse effects to other established GLP-1 receptor agonists, such as GLP-1 receptor agonist dulaglutide. These effects occur largely within the gastrointestinal tract.[5] The most frequently observed adverse effects are nausea, diarrhoea and vomiting, which increased in incidence with the dosage amount (i.e. higher likelihood the higher the dose). The number of patients who discontinued taking tirzepatide also increased as dosage increased, with patients taking 15 mg having a 25% discontinuation rate vs 5.1% for 5 mg patients and 11.1% for dulaglutide.[6] To a slightly lesser extent, patients also reported reduced appetite.[5] Other side effects reported were dyspepsia, constipation, abdominal pain, dizziness and hypoglycaemia.[7][8]
Pharmacology
Tirzepatide is an analogue of gastric inhibitory polypeptide (GIP), a human hormone which stimulates the release of insulin from the pancreas. Tirzepatide is a linear polypeptide of 39 amino acids which has been chemically modified by lipidation to improve its uptake into cells and its stability to metabolism.[9] The compound is administered as a weekly subcutaneous injection.[10] It completed phase III trials globally in 2021.[11][12]
Mechanism of action
Tirzepatide has a greater affinity to GIP receptors than to GLP-1 receptors, and this dual agonist behaviour has been shown to produce greater reductions of hyperglycemia compared to a selective GLP-1 receptor agonist.[3] Signaling studies have shown that this is due to tirzepatide mimicking the actions of natural GIP at the GIP receptor.[13] However, at the GLP-1 receptor, tirzepatide shows bias towards cAMP (a messenger associated with regulation of glycogen, sugar and lipid metabolism) generation, rather than β-arrestin recruitment. This combination of preference towards GIP receptor and distinct signaling properties at GLP-1 suggest this biased agonism increases insulin secretion.[13] Tirzepatide has also been shown to increase levels of adiponectin, an adipokine involved in the regulation of both glucose and lipid metabolism, with a maximum increase of 26% from baseline after 26 weeks, at the 10 mg dosage.[3]
Chemistry
Structure
Tirzepatide is an analog of the human GIP hormone with a C20 fatty-diacid portion attached, used to optimise the uptake and metabolism of the compound.[9] The fatty-diacid section (eicosanedioic acid) is linked via a glutamic acid and two (2-(2-aminoethoxy)ethoxy)acetic acid units to the side chain of the lysine residue. This arrangement allows for a much longer half life, extending the time between doses, because of its high affinity to albumin.[14]
Synthesis
The synthesis of tirzepatide was first disclosed in patents filed by Eli Lilly and Company.[15] This uses standard solid phase peptide synthesis, with an allyloxycarbonyl protecting group on the lysine at position 20 of the linear chain of amino acids, allowing a final set of chemical transformations in which the sidechain amine of that lysine is derivatized with the lipid-containing fragment.
Large-scale manufacturing processes have been reported for this compound.[16]
History
Indiana-based pharmaceutical company Eli Lilly and Company first applied for a patent for a method of glycemic control using tirzepatide in early 2016.[15] The patent was published late that year. After passing phase 3 clinical trials, Lilly applied for FDA approval in October 2021 with a priority review voucher.[17]
Following the completion of the pivotal SURPASS-2 trial no. NCT03987919, the company announced on 28 April that tirzepatide had successfully met their endpoints in obese and overweight patients without diabetes.[18] Alongside results from the SURMOUNT-1 trial no. NCT04184622, they suggest that tirzepatide may potentially be a competitor for existing diabetic medication semaglutide, manufactured by Novo Nordisk.[19][20]
In industry-funded preliminary trials comparing tirzepatide to the existing diabetes medication semaglutide (an injected analogue of the hormone GLP-1), tirzepatide showed minor improvement of reductions (2.01%–2.30% depending on dosage) in glycated hemoglobin tests relative to semaglutide (1.86%).[21] A 10 mg dose has also been shown to be effective in reducing insulin resistance, with a reduction of around 8% from baseline, measured using HOMA2-IR (computed with fasting insulin).[3] Fasting levels of IGF binding proteins like IGFBP1 and IGFBP2 increased following tirzepatide treatment, increasing insulin sensitivity.[3] A meta-analysis published by Dutta et al. showed that over 1-year clinical use, tirzepatide was observed to be superior to dulaglutide, semaglutide, degludec, and insulin glargine with regards to glycemic efficacy and obesity reduction. Tirzepatide is perhaps the most potent agent developed to date to tackle the global problem of “diabesity“.[22]
Society and culture
Names
Tirzepatide is the international nonproprietary name (INN).[23]
References
- ^ Jump up to:a b “Highlights of prescribing information” (PDF). accessdata.fda.gov. FDA. May 2022. Retrieved 14 May 2022.
- ^ Jump up to:a b c d e f g h i “FDA Approves Novel, Dual-Targeted Treatment for Type 2 Diabetes”. U.S. Food and Drug Administration (FDA) (Press release). 13 May 2022. Retrieved 13 May 2022.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b c d e Thomas MK, Nikooienejad A, Bray R, Cui X, Wilson J, Duffin K, et al. (January 2021). “Dual GIP and GLP-1 Receptor Agonist Tirzepatide Improves Beta-cell Function and Insulin Sensitivity in Type 2 Diabetes”. The Journal of Clinical Endocrinology and Metabolism. 106 (2): 388–396. doi:10.1210/clinem/dgaa863. PMC 7823251. PMID 33236115.
- ^ Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. (December 2018). “LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept”. Molecular Metabolism. 18: 3–14. doi:10.1016/j.molmet.2018.09.009. PMC 6308032. PMID 30473097.
- ^ Jump up to:a b Min T, Bain SC (January 2021). “The Role of Tirzepatide, Dual GIP and GLP-1 Receptor Agonist, in the Management of Type 2 Diabetes: The SURPASS Clinical Trials”. Diabetes Therapy. 12 (1): 143–157. doi:10.1007/s13300-020-00981-0. PMC 7843845. PMID 33325008.
- ^ Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. (November 2018). “Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial”. The Lancet. 392 (10160): 2180–2193. doi:10.1016/S0140-6736(18)32260-8. PMID 30293770.
- ^ Frias JP, Nauck MA, Van J, Benson C, Bray R, Cui X, et al. (June 2020). “Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: A 12-week, randomized, double-blind, placebo-controlled study to evaluate different dose-escalation regimens”. Diabetes, Obesity & Metabolism. 22 (6): 938–946. doi:10.1111/dom.13979. PMC 7318331. PMID 31984598.
- ^ Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, Rodríguez Á (February 2022). “Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial”. JAMA. 327 (6): 534–545. doi:10.1001/jama.2022.0078. PMID 35133415.
- ^ Jump up to:a b Ahangarpour M, Kavianinia I, Harris PW, Brimble MA (January 2021). “Photo-induced radical thiol-ene chemistry: a versatile toolbox for peptide-based drug design”. Chemical Society Reviews. Royal Society of Chemistry. 50 (2): 898–944. doi:10.1039/d0cs00354a. PMID 33404559. S2CID 230783854.
- ^ Bastin M, Andreelli F (2019). “Dual GIP-GLP1-Receptor Agonists In The Treatment Of Type 2 Diabetes: A Short Review On Emerging Data And Therapeutic Potential”. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy. 12: 1973–1985. doi:10.2147/DMSO.S191438. PMC 6777434. PMID 31686879.
- ^ “Tirzepatide significantly reduced A1C and body weight in people with type 2 diabetes in two phase 3 trials from Lilly’s SURPASS program” (Press release). Eli Lilly and Company. 17 February 2021. Retrieved 28 October 2021 – via PR Newswire.
- ^ “Lilly : Phase 3 Tirzepatide Results Show Superior A1C And Body Weight Reductions In Type 2 Diabetes”. Business Insider. RTTNews. 19 October 2021. Retrieved 28 October 2021.
- ^ Jump up to:a b Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, et al. (September 2020). “Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist”. JCI Insight. 5 (17). doi:10.1172/jci.insight.140532. PMC 7526454. PMID 32730231.
- ^ Østergaard S, Paulsson JF, Kofoed J, Zosel F, Olsen J, Jeppesen CB, et al. (October 2021). “The effect of fatty diacid acylation of human PYY3-36 on Y2 receptor potency and half-life in minipigs”. Scientific Reports. 11 (1): 21179. Bibcode:2021NatSR..1121179O. doi:10.1038/s41598-021-00654-3. PMC 8551270. PMID 34707178.
- ^ Jump up to:a b US patent 9474780, Bokvist BK, Coskun T, Cummins RC, Alsina-Fernandez J, “GIP and GLP-1 co-agonist compounds”, issued 2016-10-25, assigned to Eli Lilly and Co
- ^ Frederick MO, Boyse RA, Braden TM, Calvin JR, Campbell BM, Changi SM, et al. (2021). “Kilogram-Scale GMP Manufacture of Tirzepatide Using a Hybrid SPPS/LPPS Approach with Continuous Manufacturing”. Organic Process Research & Development. 25 (7): 1628–1636. doi:10.1021/acs.oprd.1c00108. S2CID 237690232.
- ^ Sagonowsky, Eric (26 October 2021). “As Lilly gears up for key 2022 launches, Trulicity, Taltz and more drive solid growth”. Fierce Pharma. Retrieved 9 April 2022.
- ^ Kellaher, Colin (28 April 2022). “Eli Lilly’s Tirzepatide Meets Main Endpoints in Phase 3 Obesity Study >LLY”. Dow Jones Newswires. Retrieved 29 April 2022 – via MarketWatch.
- ^ Kahan, Scott; Garvey, W. Timothy (28 April 2022). “SURMOUNT-1: Adults achieve weight loss of 16% or more at 72 weeks with tirzepatide”. healio.com. Retrieved 29 April 2022.
- ^ Taylor, Nick Paul (28 April 2022). “SURMOUNT-able: Lilly’s tirzepatide clears high bar set by Novo’s Wegovy in obesity”. FierceBiotech. Retrieved 29 April 2022.
- ^ Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. (August 2021). “Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes”. The New England Journal of Medicine. 385 (6): 503–515. doi:10.1056/NEJMoa2107519. PMID 34170647. S2CID 235635529.
- ^ Dutta D, Surana V, Singla R, Aggarwal S, Sharma M (November–December 2021). “Efficacy and safety of novel twincretin tirzepatide a dual GIP and GLP-1 receptor agonist in the management of type-2 diabetes: A Cochrane meta-analysis”. Indian Journal of Endocrinology and Metabolism. 25 (6): 475–489. doi:10.4103/ijem.ijem_423_21.
- ^ World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1). hdl:10665/330896.
Further reading
- Bhagavathula AS, Vidyasagar K, Tesfaye W (September 2021). “Efficacy and Safety of Tirzepatide in Patients with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomized Phase II/III Trials”. Pharmaceuticals (Basel). 14 (10). doi:10.3390/ph14100991. PMC 8537322. PMID 34681215.
- Frías JP (November 2020). “Tirzepatide: a glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) dual agonist in development for the treatment of type 2 diabetes”. Expert Rev Endocrinol Metab. 15 (6): 379–394. doi:10.1080/17446651.2020.1830759. PMID 33030356.
- Ryan DH (September 2021). “Next Generation Antiobesity Medications: Setmelanotide, Semaglutide, Tirzepatide and Bimagrumab: What do They Mean for Clinical Practice?”. J Obes Metab Syndr. 30 (3): 196–208. doi:10.7570/jomes21033. PMC 8526285. PMID 34518444.
External links
- “Tirzepatide”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03954834 for “A Study of Tirzepatide (LY3298176) in Participants With Type 2 Diabetes Not Controlled With Diet and Exercise Alone (SURPASS-1)” at ClinicalTrials.gov
- Clinical trial number NCT03987919 for “A Study of Tirzepatide (LY3298176) Versus Semaglutide Once Weekly as Add-on Therapy to Metformin in Participants With Type 2 Diabetes (SURPASS-2)” at ClinicalTrials.gov
- Clinical trial number NCT03882970 for “A Study of Tirzepatide (LY3298176) Versus Insulin Degludec in Participants With Type 2 Diabetes (SURPASS-3)” at ClinicalTrials.gov
- Clinical trial number NCT03730662 for “A Study of Tirzepatide (LY3298176) Once a Week Versus Insulin Glargine Once a Day in Participants With Type 2 Diabetes and Increased Cardiovascular Risk (SURPASS-4)” at ClinicalTrials.gov
- Clinical trial number NCT04039503 for “A Study of Tirzepatide (LY3298176) Versus Placebo in Participants With Type 2 Diabetes Inadequately Controlled on Insulin Glargine With or Without Metformin (SURPASS-5)” at ClinicalTrials.gov
CLIP
FDA approves Lilly’s Mounjaro™ (tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes
May 13, 2022
Mounjaro delivered superior A1C reductions versus all comparators in phase 3 SURPASS clinical trials
While not indicated for weight loss, Mounjaro led to significantly greater weight reductions versus comparators in a key secondary endpoint
Mounjaro represents the first new class of diabetes medicines introduced in nearly a decade and is expected to be available in the U.S. in the coming weeks
INDIANAPOLIS, May 13, 2022 /PRNewswire/ — The U.S. Food and Drug Administration (FDA) approved Mounjaro™ (tirzepatide) injection, Eli Lilly and Company’s (NYSE: LLY) new once-weekly GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. Mounjaro has not been studied in patients with a history of pancreatitis and is not indicated for use in patients with type 1 diabetes mellitus.
As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP and GLP-1, which are natural incretin hormones.1
“Mounjaro delivered superior and consistent A1C reductions against all of the comparators throughout the SURPASS program, which was designed to assess Mounjaro’s efficacy and safety in a broad range of adults with type 2 diabetes who could be treated in clinical practice. The approval of Mounjaro is an exciting step forward for people living with type 2 diabetes given the results seen in these clinical trials,” said Juan Pablo Frías, M.D., Medical Director, National Research Institute and Investigator in the SURPASS program.
Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
The approval was based on results from the phase 3 SURPASS program, which included active comparators of injectable semaglutide 1 mg, insulin glargine and insulin degludec. Efficacy was evaluated for Mounjaro 5 mg, 10 mg and 15 mg used alone or in combination with commonly prescribed diabetes medications, including metformin, SGLT2 inhibitors, sulfonylureas and insulin glargine. Participants in the SURPASS program achieved average A1C reductions between 1.8% and 2.1% for Mounjaro 5 mg and between 1.7% and 2.4% for both Mounjaro 10 mg and Mounjaro 15 mg. While not indicated for weight loss, mean change in body weight was a key secondary endpoint in all SURPASS studies. Participants treated with Mounjaro lost between 12 lb. (5 mg) and 25 lb. (15 mg) on average.1
Side effects reported in at least 5% of patients treated with Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion (dyspepsia), and stomach (abdominal) pain. The labeling for Mounjaro contains a Boxed Warning regarding thyroid C-cell tumors. Mounjaro is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2.1
“Lilly has a nearly 100-year heritage of advancing care for people living with diabetes – never settling for current outcomes. We’re not satisfied knowing that half of the more than 30 million Americans living with type 2 diabetes are not reaching their target blood glucose levels,” said Mike Mason, president, Lilly Diabetes. “We are thrilled to introduce Mounjaro, which represents the first new class of type 2 diabetes medication introduced in almost a decade and embodies our mission to bring innovative new therapies to the diabetes community.”
Mounjaro is expected to be available in the United States in the coming weeks. Lilly is committed to helping people access the medicines they are prescribed and will work with insurers, health systems and providers to help enable patient access to Mounjaro. Lilly plans to offer a Mounjaro savings card for people who qualify. Patients or healthcare professionals with questions about Mounjaro can visit www.Mounjaro.com or call The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979).
Tirzepatide is also under regulatory review for the treatment of type 2 diabetes in Europe, Japan and several additional markets. A multimedia gallery is available on Lilly.com.
About the SURPASS clinical trial program
The SURPASS phase 3 global clinical development program for tirzepatide began in late 2018 and included five global registration trials and two regional trials in Japan. These studies ranged from 40 to 52 weeks and evaluated the efficacy and safety of Mounjaro 5 mg, 10 mg and 15 mg as a monotherapy and as an add-on to various standard-of-care medications for type 2 diabetes. The active comparators in the studies were injectable semaglutide 1 mg, insulin glargine and insulin degludec. Collectively, the five global registration trials consistently demonstrated A1C reductions for participants taking Mounjaro across multiple stages of their type 2 diabetes journeys, from an average around five to 13 years of having diabetes.2-8
- SURPASS-1 (NCT03954834) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=121), 10 mg (N=121) and 15 mg (N=120) as monotherapy to placebo (N=113) in adults with type 2 diabetes inadequately controlled with diet and exercise alone. From a baseline A1C of 7.9%, Mounjaro reduced participants’ A1C by a mean of 1.8%* (5 mg) and 1.7%* (10 mg and 15 mg) compared to 0.1% for placebo. In a key secondary endpoint, from a baseline weight of 189 lb., Mounjaro reduced participants’ weight by a mean of 14 lb.* (5 mg), 15 lb.* (10 mg) and 17 lb.* (15 mg) compared to 2 lb. for placebo.2,3
- SURPASS-2 (NCT03987919) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=470), 10 mg (N=469) and 15 mg (N=469) to injectable semaglutide 1 mg (N=468) in adults with type 2 diabetes inadequately controlled with ≥1500 mg/day metformin alone. From a baseline A1C of 8.3%, Mounjaro reduced participants’ A1C by a mean of 2.0%ꝉ (5 mg), 2.2%* (10 mg) and 2.3%* (15 mg) compared to 1.9% for semaglutide. In a key secondary endpoint, from a baseline weight of 207 lb., Mounjaro reduced participants’ weight by a mean of 17 lb.ꝉ (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to 13 lb. for semaglutide.4,5
- SURPASS-3 (NCT03882970) was a 52-week study comparing the efficacy of Mounjaro 5 mg (N=358), 10 mg (N=360) and 15 mg (N=358) to titrated insulin degludec (N=359) in adults with type 2 diabetes treated with metformin with or without an SGLT-2 inhibitor. From a baseline A1C of 8.2%, Mounjaro reduced participants’ A1C by a mean of 1.9%* (5 mg), 2.0%* (10 mg) and 2.1%* (15 mg) compared to 1.3% for insulin degludec. From a baseline weight of 208 lb., Mounjaro reduced participants’ weight by a mean of 15 lb.* (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to an increase of 4 lb. for insulin degludec.6
- SURPASS-4 (NCT03730662) was a 104-week study comparing the efficacy and safety of Mounjaro 5 mg (N=328), 10 mg (N=326) and 15 mg (N=337) to insulin glargine (N=998) in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The primary endpoint was measured at 52 weeks. From a baseline A1C of 8.5%, Mounjaro reduced participants’ A1C by a mean of 2.1%* (5 mg), 2.3%* (10 mg) and 2.4%* (15 mg) compared to 1.4% for insulin glargine. From a baseline weight of 199 lb., Mounjaro reduced weight by a mean of 14 lb.* (5 mg), 20 lb.* (10 mg) and 23 lb.* (15 mg) compared to an increase of 4 lb. for insulin glargine.7
- SURPASS-5 (NCT04039503) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=116), 10 mg (N=118) and 15 mg (N=118) to placebo (N=119) in adults with inadequately controlled type 2 diabetes already being treated with insulin glargine, with or without metformin. From a baseline A1C of 8.3%, Mounjaro reduced A1C by a mean of 2.1%* (5 mg), 2.4%* (10 mg) and 2.3%* (15 mg) compared to 0.9% for placebo. From a baseline weight of 210 lb., Mounjaro reduced participants’ weight by a mean of 12 lb.* (5 mg), 17 lb.* (10 mg) and 19 lb.* (15 mg) compared to an increase of 4 lb. for placebo.8
*p<0.001 for superiority vs. placebo or active comparator, adjusted for multiplicity
ꝉp<0.05 for superiority vs. semaglutide 1 mg, adjusted for multiplicity
About Mounjaro™ (tirzepatide) injection1
Mounjaro™ (tirzepatide) injection is FDA-approved as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1). Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
PURPOSE AND SAFETY SUMMARY WITH WARNINGS
Important Facts About MounjaroTM (mown-JAHR-OH). It is also known as tirzepatide.
- Mounjaro is an injectable prescription medicine for adults with type 2 diabetes used along with diet and exercise to improve blood sugar (glucose).
- It is not known if Mounjaro can be used in people who have had inflammation of the pancreas (pancreatitis). Mounjaro is not for use in people with type 1 diabetes. It is not known if Mounjaro is safe and effective for use in children under 18 years of age.
Warnings
Mounjaro may cause tumors in the thyroid, including thyroid cancer. Watch for possible symptoms, such as a lump or swelling in the neck, hoarseness, trouble swallowing, or shortness of breath. If you have a symptom, tell your healthcare provider.
- Do not use Mounjaro if you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC).
- Do not use Mounjaro if you have Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- Do not use Mounjaro if you are allergic to tirzepatide or any of the ingredients in Mounjaro.
Mounjaro may cause serious side effects, including:
Inflammation of the pancreas (pancreatitis). Stop using Mounjaro and call your healthcare provider right away if you have severe pain in your stomach area (abdomen) that will not go away, with or without vomiting. You may feel the pain from your abdomen to your back.
Low blood sugar (hypoglycemia). Your risk for getting low blood sugar may be higher if you use Mounjaro with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin. Signs and symptoms of low blood sugar may include dizziness or light-headedness, sweating, confusion or drowsiness, headache, blurred vision, slurred speech, shakiness, fast heartbeat, anxiety, irritability, or mood changes, hunger, weakness and feeling jittery.
Serious allergic reactions. Stop using Mounjaro and get medical help right away if you have any symptoms of a serious allergic reaction, including swelling of your face, lips, tongue or throat, problems breathing or swallowing, severe rash or itching, fainting or feeling dizzy, and very rapid heartbeat.
Kidney problems (kidney failure). In people who have kidney problems, diarrhea, nausea, and vomiting may cause a loss of fluids (dehydration), which may cause kidney problems to get worse. It is important for you to drink fluids to help reduce your chance of dehydration.
Severe stomach problems. Stomach problems, sometimes severe, have been reported in people who use Mounjaro. Tell your healthcare provider if you have stomach problems that are severe or will not go away.
Changes in vision. Tell your healthcare provider if you have changes in vision during treatment with Mounjaro.
Gallbladder problems. Gallbladder problems have happened in some people who use Mounjaro. Tell your healthcare provider right away if you get symptoms of gallbladder problems, which may include pain in your upper stomach (abdomen), fever, yellowing of skin or eyes (jaundice), and clay-colored stools.
Common side effects
The most common side effects of Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion, and stomach (abdominal) pain. These are not all the possible side effects of Mounjaro. Talk to your healthcare provider about any side effect that bothers you or doesn’t go away.
Tell your healthcare provider if you have any side effects. You can report side effects at 1-800-FDA-1088 or www.fda.gov/medwatch.
Before using
- Your healthcare provider should show you how to use Mounjaro before you use it for the first time.
- Before you use Mounjaro, talk to your healthcare provider about low blood sugar and how to manage it.
Review these questions with your healthcare provider:
- Do you have other medical conditions, including problems with your pancreas or kidneys, or severe problems with your stomach, such as slowed emptying of your stomach (gastroparesis) or problems digesting food?
- Do you take other diabetes medicines, such as insulin or sulfonylureas?
- Do you have a history of diabetic retinopathy?
- Are you pregnant or plan to become pregnant or breastfeeding or plan to breastfeed? It is not known if Mounjaro will harm your unborn baby.
- Do you take birth control pills by mouth? These may not work as well while using Mounjaro. Your healthcare provider may recommend another type of birth control when you start Mounjaro or when you increase your dose.
- Do you take any other prescription medicines or over-the-counter drugs, vitamins, or herbal supplements?
How to take
- Read the Instructions for Use that come with Mounjaro.
- Use Mounjaro exactly as your healthcare provider says.
- Mounjaro is injected under the skin (subcutaneously) of your stomach (abdomen), thigh, or upper arm.
- Use Mounjaro 1 time each week, at any time of the day.
- Do not mix insulin and Mounjaro together in the same injection.
- If you take too much Mounjaro, call your healthcare provider or seek medical advice promptly.
Learn more
For more information, call 1-800-LillyRx (1-800-545-5979) or go to www.mounjaro.com.
This information does not take the place of talking with your healthcare provider. Be sure to talk to your healthcare provider about Mounjaro and how to take it. Your healthcare provider is the best person to help you decide if Mounjaro is right for you.
MounjaroTM and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates.
Please click to access full Prescribing Information and Medication Guide.
TR CON CBS MAY2022
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
Lilly Cautionary Statement Regarding Forward-Looking Statements
This press release contains forward-looking statements (as that term is defined in the Private Securities Litigation Reform Act of 1995) about Mounjaro™ (tirzepatide 2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg and 15 mg) injection as a treatment to improve glycemic control in adults with type 2 diabetes, the timeline for supply of Mounjaro to become available, and certain other milestones and ongoing clinical trials of Mounjaro and reflects Lilly’s current beliefs and expectations. However, as with any pharmaceutical product or medical device, there are substantial risks and uncertainties in the process of research, development and commercialization. Among other things, there can be no guarantee that Mounjaro will be commercially successful, that future study results will be consistent with results to date, or that we will meet our anticipated timelines for the commercialization of Mounjaro. For further discussion of these and other risks and uncertainties, see Lilly’s most recent Form 10-K and Form 10-Q filings with the United States Securities and Exchange Commission. Except as required by law, Lilly undertakes no duty to update forward-looking statements to reflect events after the date of this release.
References
- Mounjaro. Prescribing Information. Lilly USA, LLC.
- Rosenstock, J, et. al. Efficacy and Safety of Once Weekly Tirzepatide, a Dual GIP/GLP-1 Receptor Agonist Versus Placebo as Monotherapy in People with Type 2 Diabetes (SURPASS-1). Abstract 100-OR. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
- Rosenstock, J, et. al. (2021). Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398(10295):143-155. doi: 10.1016/S0140-6736(21)01324-6.
- Frías JP, Davies MJ, Rosenstock J, et al; for the SURPASS-2 Investigators. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6)(suppl):503-515. doi: 10.1056/NEJMoa2107519
- Frias, J.P. Efficacy and Safety of Tirzepatide vs. Semaglutide Once Weekly as Add-On Therapy to Metformin in Patients with Type 2 Diabetes. Abstract 84-LB. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
- Ludvik B, Giorgino F, Jódar E, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. 2021;398(10300):583-598. doi: 10.1016/S0140-6736(21)01443-4
- Del Prato S, Kahn SE, Pavo I, et al; for the SURPASS-4 Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021;398(10313):1811-1824. doi: 10.1016/S0140-6736(21)02188-7
- Dahl D, Onishi Y, Norwood P, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327(6):534-545. doi:10.1001/jama.2022.0078
CLIP
Lilly’s tirzepatide delivered up to 22.5% weight loss in adults with obesity or overweight in SURMOUNT-1
April 28, 2022
Participants taking tirzepatide lost up to 52 lb. (24 kg) in this 72-week phase 3 study
63% of participants taking tirzepatide 15 mg achieved at least 20% body weight reductions as a key secondary endpoint
INDIANAPOLIS, April 28, 2022 /PRNewswire/ — Tirzepatide (5 mg, 10 mg, 15 mg) achieved superior weight loss compared to placebo at 72 weeks of treatment in topline results from Eli Lilly and Company’s (NYSE: LLY) SURMOUNT-1 clinical trial, with participants losing up to 22.5% (52 lb. or 24 kg) of their body weight for the efficacy estimandi. This study enrolled 2,539 participants and was the first phase 3 global registration trial evaluating the efficacy and safety of tirzepatide in adults with obesity, or overweight with at least one comorbidity, who do not have diabetes. Tirzepatide met both co-primary endpoints of superior mean percent change in body weight from baseline and greater percentage of participants achieving body weight reductions of at least 5% compared to placebo for both estimandsii. The study also achieved all key secondary endpoints at 72 weeks.
For the efficacy estimand, participants taking tirzepatide achieved average weight reductions of 16.0% (35 lb. or 16 kg on 5 mg), 21.4% (49 lb. or 22 kg on 10 mg) and 22.5% (52 lb. or 24 kg on 15 mg), compared to placebo (2.4%, 5 lb. or 2 kg). Additionally, 89% (5 mg) and 96% (10 mg and 15 mg) of people taking tirzepatide achieved at least 5% body weight reductions compared to 28% of those taking placebo.
In a key secondary endpoint, 55% (10 mg) and 63% (15 mg) of people taking tirzepatide achieved at least 20% body weight reductions compared to 1.3% of those taking placebo. In an additional secondary endpoint not controlled for type 1 error, 32% of participants taking tirzepatide 5 mg achieved at least 20% body weight reductions. The mean baseline body weight of participants was 231 lb. (105 kg).
“Obesity is a chronic disease that often does not receive the same standard of care as other conditions, despite its impact on physical, psychological and metabolic health, which can include increased risk of hypertension, heart disease, cancer and decreased survival,” said Louis J. Aronne, MD, FACP, DABOM, director of the Comprehensive Weight Control Center and the Sanford I. Weill Professor of Metabolic Research at Weill Cornell Medicine, obesity expert at NewYork-Presbyterian/Weill Cornell Medical Center and Investigator of SURMOUNT-1. “Tirzepatide delivered impressive body weight reductions in SURMOUNT-1, which could represent an important step forward for helping the patient and physician partnership treat this complex disease.”
For the treatment-regimen estimandiii, results showed:
- Average body weight reductions: 15.0% (5 mg), 19.5% (10 mg), 20.9% (15 mg), 3.1% (placebo)
- Percentage of participants achieving body weight reductions of ≥5%: 85% (5 mg), 89% (10 mg), 91% (15 mg), 35% (placebo)
- Percentage of participants achieving body weight reductions of ≥20%: 30% (5 mg, not controlled for type 1 error), 50% (10 mg), 57% (15 mg), 3.1% (placebo)
The overall safety and tolerability profile of tirzepatide was similar to other incretin-based therapies approved for the treatment of obesity. The most commonly reported adverse events were gastrointestinal-related and generally mild to moderate in severity, usually occurring during the dose escalation period. For those treated with tirzepatide (5 mg, 10 mg and 15 mg, respectively), nausea (24.6%, 33.3%, 31.0%), diarrhea (18.7%, 21.2%, 23.0%), vomiting (8.3%, 10.7%, 12.2%) and constipation (16.8%, 17.1%, 11.7%) were more frequently experienced compared to placebo (9.5% [nausea], 7.3% [diarrhea], 1.7% [vomiting], 5.8% [constipation]).
Treatment discontinuation rates due to adverse events were 4.3% (5 mg), 7.1% (10 mg), 6.2% (15 mg) and 2.6% (placebo). The overall treatment discontinuation rates were 14.3% (5 mg), 16.4% (10 mg), 15.1% (15 mg) and 26.4% (placebo).
Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and the potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
“Tirzepatide is the first investigational medicine to deliver more than 20 percent weight loss on average in a phase 3 study, reinforcing our confidence in its potential to help people living with obesity,” said Jeff Emmick, MD, Ph.D., vice president, product development, Lilly. “Obesity is a chronic disease that requires effective treatment options, and Lilly is working relentlessly to support people with obesity and modernize how this disease is approached. We’re proud to research and develop potentially innovative treatments like tirzepatide, which helped nearly two thirds of participants on the highest dose reduce their body weight by at least 20 percent in SURMOUNT-1.”
Tirzepatide is a novel investigational once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist, representing a new class of medicines being studied for the treatment of obesity. Tirzepatide is a single peptide that activates the body’s receptors for GIP and GLP-1, two natural incretin hormones. Obesity is a chronic, progressive disease caused by disruptions in the mechanisms that control body weight, often leading to an increase in food intake and/or a decrease in energy expenditure. These disruptions are multifactorial and can be related to genetic, developmental, behavioral, environmental and social factors. To learn more, visit Lilly.com/obesity.
Lilly will continue to evaluate the SURMOUNT-1 results, which will be presented at an upcoming medical meeting and submitted to a peer-reviewed journal. Additional studies are ongoing for tirzepatide as a potential treatment for obesity or overweight.
About tirzepatide
Tirzepatide is a once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1 receptor agonists. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with GLP-1 receptor agonism, may result in greater effects on markers of metabolic dysregulation such as body weight, glucose and lipids. Tirzepatide is in phase 3 development for adults with obesity or overweight with weight-related comorbidity and is currently under regulatory review as a treatment for adults with type 2 diabetes. It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH) and heart failure with preserved ejection fraction (HFpEF). Studies of tirzepatide in obstructive sleep apnea (OSA) and in morbidity/mortality in obesity are planned as well.
About SURMOUNT-1 and the SURMOUNT clinical trial program
SURMOUNT-1 (NCT04184622) is a multi-center, randomized, double-blind, parallel, placebo-controlled trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to placebo as an adjunct to a reduced-calorie diet and increased physical activity in adults without type 2 diabetes who have obesity, or overweight with at least one of the following comorbidities: hypertension, dyslipidemia, obstructive sleep apnea or cardiovascular disease. The trial randomized 2,539 participants across the U.S., Argentina, Brazil, China, India, Japan, Mexico, Russia and Taiwan in a 1:1:1:1 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or placebo. The co-primary objectives of the study were to demonstrate that tirzepatide 10 mg and/or 15 mg is superior in percentage of body weight reductions from baseline and percentage of participants achieving ≥5% body weight reduction at 72 weeks compared to placebo. Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg).
The SURMOUNT phase 3 global clinical development program for tirzepatide began in late 2019 and has enrolled more than 5,000 people with obesity or overweight across six clinical trials, four of which are global studies. Results from SURMOUNT-2, -3, and -4 are anticipated in 2023.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
CLIP
Tirzepatide results superior A1C and body weight reductions compared to insulin glargine in adults with type 2 diabetes
Newly published data show that participants maintained A1C and weight control up to two years in SURPASS-4, the largest and longest SURPASS trial completed to dateNo increased cardiovascular risk identified with tirzepatide; hazard ratio of 0.74 observed for MACE-4 events
SURPASS-4 is the largest and longest clinical trial completed to date of the phase 3 program studying tirzepatide as a potential treatment for type 2 diabetes. The primary endpoint was measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE) to assess CV risk. In newly published data from the treatment period after 52 weeks, participants taking tirzepatide maintained A1C and weight control for up to two years.
The overall safety profile of tirzepatide, assessed over the full study period, was consistent with the safety results measured at 52 weeks, with no new findings up to 104 weeks. Gastrointestinal side effects were the most commonly reported adverse events, usually occurring during the escalation period and then decreasing over time.
“We are encouraged by the continued A1C and weight control that participants experienced past the initial 52 week treatment period and up to two years as we continue to explore the potential impact of tirzepatide for the treatment of type 2 diabetes,” said John Doupis, M.D., Ph.D., Director, Diabetes Division and Clinical Research Center, Iatriko Paleou Falirou Medical Center, Athens, Greece and Senior Investigator for SURPASS-4.
Tirzepatide is a novel investigational once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single molecule, representing a new class of medicines being studied for the treatment of type 2 diabetes.
SURPASS-4 was an open-label global trial comparing the safety and efficacy of three tirzepatide doses (5 mg, 10 mg and 15 mg) to titrated insulin glargine in 2,002 adults with type 2 diabetes with increased CV risk who were treated with between one and three oral antihyperglycemic medicines (metformin, a sulfonylurea or an SGLT-2 inhibitor). Of the total participants randomized, 1,819 (91%) completed the primary 52-week visit and 1,706 (85%) completed the study on treatment. The median study duration was 85 weeks and 202 participants (10%) completed two years.
Study participants had a mean duration of diabetes of 11.8 years, a baseline A1C of 8.52 percent and a baseline weight of 90.3 kg. More than 85 percent of participants had a history of cardiovascular events. In the insulin glargine arm, the insulin dose was titrated following a treat-to-target algorithm with the goal of fasting blood glucose below 100 mg/dL. The starting dose of insulin glargine was 10 units per day, and the mean dose of insulin glargine at 52 weeks was 43.5 units per day.
About tirzepatide
Tirzepatide is a once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with a GLP-1 receptor agonist, may result in greater effects on glucose and body weight. Tirzepatide is in phase 3 development for blood glucose management in adults with type 2 diabetes, for chronic weight management and heart failure with preserved ejection fraction (HFpEF). It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH).
About SURPASS-4 and the SURPASS clinical trial program
SURPASS-4 (NCT03730662) is a randomized, parallel, open-label trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to insulin glargine in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The trial randomized 2,002 study participants in a 1:1:1:3 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or insulin glargine. Participants were located in the European Union, North America (Canada and the United States), Australia, Israel, Taiwan and Latin America (Brazil, Argentina and Mexico). The primary objective of the study was to demonstrate that tirzepatide (10 mg and/or 15 mg) is non-inferior to insulin glargine for change from baseline A1C at 52 weeks in people with type 2 diabetes and increased CV risk. The primary and key secondary endpoints were measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE). Study participants enrolled had to have a mean baseline A1C between 7.5 percent and 10.5 percent and a BMI greater than or equal to 25 kg/m2 at baseline. All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg). All participants in the titrated insulin glargine treatment arm started with a baseline dose of 10 units per day and titrated following a treat-to-target algorithm to reach a fasting blood glucose below 100 mg/dL.
The SURPASS phase 3 global clinical development program for tirzepatide has enrolled more than 20,000 people with type 2 diabetes across 10 clinical trials, five of which are global registration studies. The program began in late 2018, and all five global registration trials have been completed.
About Diabetes
Approximately 34 million Americans2 (just over 1 in 10) and an estimated 463 million adults worldwide3 have diabetes. Type 2 diabetes is the most common type internationally, accounting for an estimated 90 to 95 percent of all diabetes cases in the United States alone2. Diabetes is a chronic disease that occurs when the body does not properly produce or use the hormone insulin.
| Clinical data | |
|---|---|
| Trade names | Mounjaro |
| Other names | LY3298176, GIP/GLP-1 RA |
| License data | US DailyMed: Tirzepatide |
| Routes of administration | subcutaneous |
| Drug class | Antidiabetic, GLP-1 receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1][2] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 2023788-19-2 |
| PubChem CID | 156588324 |
| IUPHAR/BPS | 11429 |
| DrugBank | DB15171 |
| ChemSpider | 76714503 |
| UNII | OYN3CCI6QE |
| KEGG | D11360 |
| ChEMBL | ChEMBL4297839 |
| Chemical and physical data | |
| Formula | C225H348N48O68 |
| Molar mass | 4813.527 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Tirzepatide, FDA 2022, APPROVALS 2022, Mounjaro, PEPTIDE, チルゼパチド , LY3298176,
UNIIOYN3CCI6QE
chart 1 Structure of GLP-1 & TZP & Exenatide & Somalutide
DAPAGLIFLOZIN
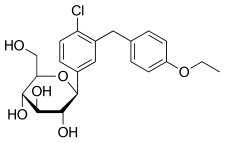

DAPAGLIFLOZIN, BMS-512148
ダパグリフロジン;
(2S,3R,4R,5S,6R)-2-[4-chloro-3-(4-ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol,
Cas 461432-26-8
| Molecular Formula: C21H25ClO6 |
| Molecular Weight: 408.87 |
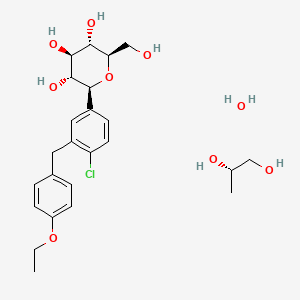
Dapagliflozin propandiol monohydrate; 960404-48-2
| Molecular Weight | 502.98 |
| Formula | C21H25ClO6•C3H8O2•H2O |
Bristol-Myers Squibb (Originator)
AstraZeneca
TYPE 2 DIABETES,SGLT-2 Inhibitors
launched 2012, as forxiga in EU, FDA 2014, JAPAN PMDA 2014
Dapagliflozin propanediol monohydrate was first approved by European Medicine Agency (EMA) on November 12, 2012, then approved by the U.S. Food and Drug Administration (FDA) on January 8, 2014, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on March 24, 2014. It was co-developed and co-marketed as Forxiga® by Bristol-Myers Squibb and AstraZeneca in EU.
Dapagliflozin propanediol monohydrate is a sodium-glucose co-transporter 2 (SGLT2) inhibitor indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Forxiga® is available as tablet for oral use, containing 5 mg or 10 mg of free Dapagliflozin. The recommended starting dose is 5 mg once daily in the morning.

Dapagliflozin propanediol is a solvate containing 1:1:1 ratio of the dapagliflozin, (S)-(+)-1,2-propanediol, and water.
US——-In 2011, the product was not recommended for approval by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee. In 2011, the FDA assigned a complete response letter to the application. A new application was resubmitted in 2013 by Bristol-Myers Squibb and AstraZeneca in the U.S
WILMINGTON, Del. & PRINCETON, N.J.--(BUSINESS WIRE)--December 12, 2013-- USFDA



Sales:$518.7 Million (Y2015); 
$235.8 Million (Y2014);
$33 Million (Y2013);ATC Code:A10BX09
Approved Countries or AreaUpdate Date:2015-07-29
- US
- EU
- JP
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-01-08 | Marketing approval | Farxiga | Type 2 diabetes | Tablet | 5 mg/10 mg | AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2012-11-12 | Marketing approval | Forxiga | Type 2 diabetes | Tablet, Film coated | Eq. 5 mg/10 mg Dapagliflozin | Bristol-Myers Squibb, AstraZeneca |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-03-24 | Marketing approval | Forxiga | Type 2 diabetes | Tablet, Film coated | 5 mg/10 mg | Bristol-Myers Squibb, AstraZeneca, Ono |
MoreChemical Structure
AstraZeneca (NYSE:AZN) and Bristol-Myers Squibb Company (NYSE:BMY) today announced the U.S. Food and Drug Administration’s (FDA) Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) voted 13-1 that the benefits of dapagliflozin use outweigh identified risks and support marketing of dapagliflozin as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. The Advisory Committee also voted 10-4 that the data provided sufficient evidence that dapagliflozin, relative to comparators, has an acceptable cardiovascular risk profile.
The FDA is not bound by the Advisory Committee’s recommendation but takes its advice into consideration when reviewing the application for an investigational agent. The Prescription Drug User Fee Act (PDUFA) goal date for dapagliflozin is Jan. 11, 2014.

Dapagliflozin is being reviewed by the FDA for use as monotherapy, and in combination with other antidiabetic agents, as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. It is a selective and reversible inhibitor of sodium-glucose cotransporter 2 (SGLT2) that works independently of insulin to help remove excess glucose from the body. Dapagliflozin, an investigational compound in the U.S., was the first SGLT2 inhibitor to be approved anywhere in the world. Dapagliflozin is currently approved under the trade name [Forxiga](TM) for the treatment of adults with type 2 diabetes, along with diet and exercise, in 38 countries, including the European Union and Australia.
http://online.wsj.com/article/PR-CO-20131212-910828.html?dsk=y
Reference:1. WO03099836A1 / US6515117B2.
2. WO2010048358.
3. J. Med. Chem. 2008, 51, 1145–1149.
4. WO2004063209A2 / US7375213B2.
5. WO2008002824A1 / US7919598B2.Route 2
Reference:1. WO2010022313 / US8283454B2.Route 3
Reference:1. WO2013068850.Route 4
Reference:1. Org. Lett. 2012, 14, 1480-1483.
PAPER
https://www.future-science.com/doi/10.4155/fmc-2020-0154
Patent
https://patents.google.com/patent/WO2016178148A1/en
1. A process for the preparation of dapagliflozin in amorphous form, the process comprising:
(a) reducing a compound of formula II to a compound of formula ΠΙ in the presence of a Lewis acid;

(b) silylating a compound of formula IV with hexamethyldisilazane to form a compound of formula V;

(c) reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;

(d) converting the compound of formula VII to dapagliflozin;
(e) acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;

(f) optionally, purifying the compound of formula VIII with a solvent selected from halogenated hydrocarbons, alcohols, ethers, or mixtures thereof; (g) hydrolyzing the compound of formula VIII obtained in step (f) to give dapagliflozin;
EXAMPLE 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
To a suspension of 5-bromo-2-chiorobenzoic acid (lOg) in methylene di chloride (40niL), dimethylformamide (0.2g) and thionyl chloride were added and the reaction mixture was refluxed for about 2h. After completion of reaction, the solvent was distilled out. The mass obtained was degassed under vacuum followed by stripping with cyclohexane to give crude 5-bromo-2-chlorobenzoyl chloride (10.8g).
[0189] EXAMPLE 2: Preparation of 5-bromo-2-chloro-4′-ethoxybenzophenone (compound of Formula II)
5-bromo-2-chlorobenzoyl chloride (10.7g) was dissolved in methylene dichloride (40mL) and the reaction mixture was cooled to about -8°C to about -12°C under inert atmosphere. Aluminum chloride (5.65g) was added to the reaction mixture followed by addition of a solution of ethoxybenzene in methylene dichloride. The reaction mixture was stirred for about lh at about -8°C to -12°C and then quenched in dilute hydrochloric acid followed by extraction with methylene dichloride. The organic layer was washed with sodium bicarbonate solution and concentrated. The residue obtained was crystallized from methanol to give 5-bromo-2-chloro-4′-ethoxybenzophenone (8.5g). HPLC purity: 99.34%
[0190] EXAMPLE 3: Preparation of 5-bromo-2-chloro-4′-ethoxydiphenylmethane (compound of formula III)
To a mixture of 5-bromo-2-chloro-4′-ethoxybenzophenone (lOg) and methylene dichloride (50mL), cooled to about 0°C to about 5°C, triethylsilane (11.98g) and titanium chloride (22.3g) were added. The reaction mixture was stirred for about 3h at about 10°C to about 15°C. The reaction mixture was quenched into chilled water. The organic layer was separated, washed with water and sodium bicarbonate solution and concentrated under vacuum followed by stripping with toluene. The residue obtained was stirred with methanol, filtered and dried to give 5-bromo-2-chloro-4′-ethoxydiphenylmethane (9g). HPLC purity: 99.4%
[0191] EXAMPLE 4: Preparation of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5- lactone (compound of Formula V)
To a mixture of D-glucono- 1,5 -lactone (lOg) and iodine (0.28g) in methylene dichloride (80mL), hexamethyldisilazane (36.1g) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed to give 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l,5-lactone as liquid (25g). HPLC purity: 95%
[0192] EXAMPLE 5: Preparation of D-glucopyranoside, methyl l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (compound of Formula VII wherein R is methyl)
To a mixture of 2,3,4,6-tetra-0-(trimethylsilyl)-D-glucono-l ,5-lactone (25g) and 5- bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) in tetrahydrofuran (174mL), cooled to about -75°C to about -88 °C under nitrogen atmosphere, n-butyl lithium in hexane (50mL) was slowly added. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. The residue was purified with a mixture of toluene and cyclohexane. Yield: 1 lg as thick mass with 80-85% HPLC purity.
reacting the compound of formula III with the compound of formula V in the presence of a strong base followed by treatment with an acid in the presence of an alcohol to prepare a compound of formula VII, wherein R is an alkyl group selected from C1-5 alkyl;
[0193] EXAMPLE 6: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl) methyl] phenyl]
To a mixture of D-glucopyranoside, methyl l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] in methylene di chloride (40mL) and acetonitrile (40mL), cooled to about -40°C to about -45°C, triethylsilane (8.74g) was added followed by addition of boron trifluoride etherate (10.67g) maintaining the temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum to give title compound (1 lg) as thick residue with 80-85% HPLC purity.
[0194] EXAMPLE 7: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (lS)-
To a cooled solution of D-glucitol, l,5-anhydro-l -C-[4-chloro-3-[(4-ethoxyphenyl) methyl] phenyl]- (l lg) in methylene dichloride (55mL) at about 0°C to about 5°C, diisopropylethylamme, dmiethylaminopyridine and acetic anhydride were added and the reaction mixture was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol. The residue was purified with methanol and charcoal, followed by diisopropyl ether and methanol crystallization. Yield: lOg; HPLC purity: 99.6%
acetylating dapagliflozin to give D-glucitol, l ,5-anhydro-l -C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, a compound of formula VIII;
[0195] EXAMPLE 8: Preparation of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl] (Dapagliflozin)
To a stirred solution of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]-, 2,3,4,6-tetraacetate, (IS)-, (lOg) in THF: methanol: water mixture (50mL: 50mL:30mL), sodium hydroxide was added and the reaction mixture was stirred. After completion of reaction, the solvents were distilled out under vacuum and the residue obtained was dissolved in methylene dichloride and washed with water and brine and dried over sodium sulfate. The reaction mixture was concentrated and degassed to give off- white to white solids of D-glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl] phenyl]- (dapagliflozin) Yield: 7g (XRD matches with amorphous form) HPLC purity: 99.8%
[0197] EXAMPLE 10: Preparation of D-Glucitol, l,5-anhydro-l-C-[4-chloro-3-[(4- ethoxyphenyl)methyl]phenyl]-, 2,3,4,6-tetraacetate, (IS)- from D-glucono-1,5- lactone (One-pot Synthesis)
To a mixture of D-glucono-l,5-lactone (lOg) in methylene dichloride (80mL), hexamethyldisilazane (36. lg) was added and the reaction mixture was refluxed. After completion of reaction, the reaction mixture was concentrated and degassed. The residue obtained was dissolved in tetrahydrofuran. 5-Bromo-2-chloro-4′-ethoxydiphenylmethane (8.7g) was added to the reaction mixture which was cooled to about -75°C to about-85°C under nitrogen atmosphere. n-Butyl lithium in hexane (50mL) was slowly added to the reaction mixture maintaining the temperature between -75°C to about -85°C. The reaction mixture was stirred at about the same temperature and then mixture of methanol and methanesulphonic acid was added to it. The reaction mixture was quenched into sodium bicarbonate solution and extracted with ethyl acetate. The organic layer was separated, washed with saturated sodium chloride solution and concentrated under vacuum to obtain a residue. This residue was purified by a mixture of toluene and cyclohexane. To the product obtained, methylene dichloride and acetonitrile were added and the reaction mixture was cooled to about -40°C to about -45°C. Triethylsilane (8.74g) was added to the reaction mixture followed by addition of boron trifluoride etherate (10.67g) maintaining temperature at about -40°C to about -45°C. The reaction mixture was quenched in sodium bicarbonate solution. The aqueous layer was separated and extracted with ethyl acetate. The organic layer was separated, concentrated and degassed under vacuum. The thick residue obtained was dissolved in methylene dichloride and cooled to about 0°C to about 5°C. Diisopropylethylamine, dimethylaminopyridine and acetic anhydride were added to the reaction mixture which was stirred. After completion of reaction, the reaction mixture was quenched by adding water. The aqueous layer was separated and extracted with methylene dichloride. The organic layer was separated, washed with sodium bicarbonate solution and concentrated under vacuum to obtain residue which was stripped out with methanol The residue obtained was recrystallized with methanol and charcoal to give title compound (iOg) with 99.7% HPLC purity.
PAPER
Bioorganic & Medicinal Chemistry, 26(14), 3947-3952; 2018
https://www.sciencedirect.com/science/article/abs/pii/S0968089618309386?
Abstract
The cardiovascular complications were highly prevalent in type 2 diabetes mellitus (T2DM), even at the early stage of T2DM or the state of intensive glycemic control. Therefore, there is an urgent need for the intervention of cardiovascular complications in T2DM. Herein, the new hybrids of NO donor and SGLT2 inhibitor were design to achieve dual effects of anti-hyperglycemic and anti-thrombosis. As expected, the preferred hybrid 2 exhibited moderate SGLT2 inhibitory effects and anti-platelet aggregation activities, and its anti-platelet effect mediated by NO was also confirmed in the presence of NO scavenger. Moreover, compound 2 revealed significantly hypoglycemic effects and excretion of urinary glucose during an oral glucose tolerance test in mice. Potent and multifunctional hybrid, such as compound 2, is expected as a potential candidate for the intervention of cardiovascular complications in T2DM.
Graphical abstract

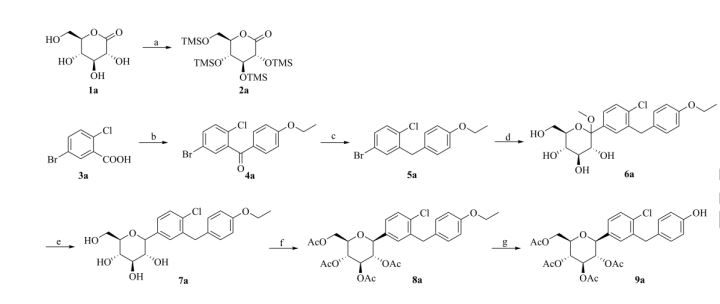
Scheme 1. Synthesis of target compounds 1-3. Reagents and conditions: (a) TMSCl, NMM, THF, 35 °C; (b) (COCl)2, CH2Cl2, DMF, then phenetole, AlCl3, 0 °C; (c) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, 25 °C; (d) n-BuLi, THF, toluene, -78 °C, then 2a followed by MeOH, CH3SO3H; (e) Et3SiH, BF3·OEt2, CH2Cl2, CH3CN, -10 °C; (f) Ac2O, pyridine, CH2Cl2, DMAP;
PATENT
Indian Pat. Appl., 2014MU03972,
PATENT
| Dapagliflozin, also known as SGLT2 inhibitor, chemical name is (2S,3R,4R,5S,6R)-2-[3-(4-ethoxybenzyl)-4-chlorophenyl]-6- Hydroxymethyltetrahydro-2H-pyran-3,4,5-triol, a sodium-glucose cotransporter 2 inhibitor, announced by the U.S. Food and Drug Administration (FDA) on January 8, 2014 , approved the use of dapagliflozin for the treatment of type 2 diabetes, the specific structural formula is as follows: |
| |
| Dapagliflozin works by inhibiting sodium-glucose transporter 2 (SGLT2), a protein in the kidney that allows glucose to be reabsorbed into the blood. This allows excess glucose to be excreted through the urine, thereby improving blood sugar control without increasing insulin secretion. |
| At present, there are two main methods for synthesizing dapagliflozin. One uses 5-bromo-2-chlorobenzoic acid as the starting material, which is chlorinated, Falk acylated, reduced, and then combined with 2,3,4 ,6-tetra-0-trimethylsilyl-D-glucopyranosic acid 1,5-lactone is condensed, methyl etherified, and demethoxylated to obtain dapagliflozin. The specific process route is as follows: |
| |
| The method has expensive starting materials and too many process steps, so it is not suitable for industrial production, and dangerous n-butyllithium needs to be used in the reaction process, so the requirements for experimental conditions are too high; |
| Another method is to use o-toluidine as the starting material, undergo bromination, diazotization, chlorination, and alkylation reactions, and then react with 2,3,4,6-tetra-0-trimethylsilyl -D-glucopyranosic acid 1,5-lactone is condensed, then methyl etherified and demethoxylated to obtain dapagliflozin. The specific process route is as follows: |
| |
| AIBN will be used in this reaction, which will produce highly toxic cyanide, which will seriously pollute the environment, and also requires the use of n-butyllithium, which requires high experimental conditions and is dangerous to operate, and is not suitable for large-scale production. |
| Example 1 |
| Weigh 16g (0.8mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodobenzene to it 2000mL of THF solution (total 2000mL: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsided, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene. |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 423.6 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 93%. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 2500mL of toluene, then add 5.6g of europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 259.49 g of solid with a yield of 85%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:3 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 147.1 g of white rice-like crystals with a yield of 80.2% and a purity of 99.2% (determined by high performance liquid chromatography, external standard method). |
| IR(cm-1):1689,1612,1588,1523,1455,1253,1089,820。1H NMR(500MHz,CDCl 3 ):δ:7.36(d,J=8.2Hz,1H),7.32(d,J=1.9Hz,1H),7.23(dd,J=8.3,2.0Hz,1H),7.09(d,J=8.6Hz,2H),6.82(d,J=8.6Hz,2H),4.85(s,1H),4.41(s,3H),3.93~4.02(m,5H),3.70(dd,J=11.7,1.3Hz,1H),3.44(dd,J=11.7,5.6Hz,1H),3.26~3.28(m,1H)。LC-MS,m/z:[M+Na]+=431。 |
| Example 2 |
| Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 272.75mL of 33% hydrogen bromide in acetic acid solution (1.1mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 381.5 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 83.8%. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , dried to obtain solid 281.2g, yield 92.1%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, dried the organic phase, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 150.9 g of white rice-like crystals with a yield of 88.2% and a purity of 99.5% (determined by high performance liquid chromatography, external standard method). |
| Example 3 |
| Weigh 37.4g (1.862mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 818.25mL of 33% hydrogen bromide in acetic acid solution (3.3mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 375.3 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose, and the yield was 82.4%. |
| Weigh 217.83g 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 7500mL toluene, then add 5.6g europium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, and control the temperature at -20~-15°C. After the dropwise addition, the temperature is raised to 5°C for reaction for 2h, vacuum concentrated to an oily substance, and then added to it. 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, and 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 273.25 g of solid with a yield of 89.5%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was finished, and then 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:5 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 152.7 g of white rice-like crystals with a yield of 82.9% and a purity of 99.4% (determined by high performance liquid chromatography, external standard method). |
| Example 4 |
| Weigh 26.7g (1.33mol) of activated magnesium, add two iodine pellets, heat to 35°C under nitrogen protection, then add 500mL of anhydrous THF to it, without magnesium, and then add 200mL of 10% iodine to it The THF solution of benzene (2000mL in total: 20g iodobenzene (0.53mol)+2000mL THF), after the color of the solution subsides, add the remaining iodobenzene solution dropwise, react for 5h, and filter to obtain the Grignard reagent THF solution of iodobenzene . |
| 433.4g (1.1mol) of peracetyl sugar and 1500mL of dichloromethane were added, cooled to 0°C in an ice-water bath, 545.5mL of 33% hydrogen bromide in acetic acid solution (2.2mol) was added dropwise to the reaction flask, and the mixture was gradually heated up The mixture was heated to 25° C. and stirred for 1.5 h. Saturated sodium bicarbonate solution was added to quench the reaction. The aqueous solution was extracted with dichloromethane, dried, and concentrated by rotary evaporation. 425.1 g of white solid were obtained, which was 2,3,4,6-tetraacetyl bromoglucose in 94% yield. |
| Weigh 217.83g of 2,3,4,6-tetraacetyl bromoglucose (0.53mol) and dissolve it in 5000mL of toluene, then add 5.76g of gadolinium oxide (3mol%) to it, under nitrogen protection, reduce the temperature to – 20°C, dropwise add the prepared iodobenzene Grignard reagent to it, control the temperature at -20~-15°C, after the dropwise addition, raise the temperature to 5°C for 2 hours, concentrate in vacuo to an oily substance, then add to it 2000mL of toluene was dissolved, extracted twice with saturated aqueous sodium chloride solution, the organic phase was dried, concentrated in vacuo to an oily substance, then 1000mL of 3:1 n-hexane/ethyl acetate solution was added to it, heated to dissolve, cooled, and a solid was precipitated out, filtered , and dried to obtain 278.6 g of solid with a yield of 91.2%. |
| The above-mentioned solid was dissolved in 1000mL 2:3:1 tetrahydrofuran/ethanol/water solution, then 40g sodium hydroxide solid (1mol) was added to it, stirred overnight, vacuum concentrated to obtain oil after the reaction was completed, and 1000mL of ethyl acetate was added thereto. The ester was dissolved, extracted twice with saturated aqueous sodium chloride solution and twice with saturated aqueous sodium thiosulfate solution, the organic phase was dried, concentrated in vacuo to an oil, to which was added 200 mL of 1:4 ethyl acetate/acetonitrile solution , heated to dissolve, cooled and recrystallized to obtain 155.62 g of white rice-like crystals with a yield of 84.5% and a purity of 99.7% (determined by high performance liquid chromatography, external standard method). |
Patent
Sodium-glucose co-transporter-2 (SGLT2) inhibitors are a group of oral medicines used for treating diabetes that have been approved since 2013. SGLT2 inhibitors prevent the kidneys from re-absorbing glucose back into the blood by passing into the bladder. Glucose is re-absorbed back into the blood via the renal proximal tubules. SGLT2 is a protein predominantly expressed in the renal proximal tubules and is likely to be major transporter responsible for this uptake. Glucose-lowering effect of SGLT-2 inhibitors occurs via an insulin-independent mechanism mostly through glucosuria by increasing the urinary excretion of glucose.
It has been shown that the treatment with SGLT2 inhibitors in patients with type II diabetes lowers HbAlc, reduces body weight, lowers systemic blood pressure (BP) and induces a small increase in LDL-C and HDL-C levels.
SGLT2 inhibitors inhibit the reabsorption of sodium and glucose from the tubule and hence, more sodium is delivered in the macula densa causing arteriole dilation, reduced intraglomerular pressure and decreased hyperfiltration. SGLT2 inhibitors cause natriuresis and volume depletion, and an increase in circulating levels of renin, angiotensin and aldosterone. They also reduce albuminuria and slow GFR loss through mechanisms that appear independent of glycemia.
Dapagliflozin is a highly potent and reversible SGLT2 inhibitor, which increases the amount of glucose excreted in the urine and improves both fasting and post-prandial plasma glucose levels in patients with type 2 diabetes. Dapagliflozin has also been shown to tend to reduce liver fat content in some studies in a diabetic population.
Dapagliflozin is available on the market in the form of dapagliflozin propanediol monohydrate and is sold under trade name Forxiga or Farxiga in the form of fdm-coated tablets. Further it is available on the market as a combination product with metformin hydrochloride which is sold under trade name Xigduo IR or Xigduo XR in the form of film-coated tablets. In addition, it is available on the market as a combination product with saxagliptin hydrochloride which is sold under trade name Qtem in the form of film-coated tablets. Moreover, it is available on the market as a combination product with saxagliptin hydrochloride and metformin hydrochloride which is sold under trade name Qtemmet XR in the form of film-coated tablets.
Dapagliflozin as a monotherapy and in a combination with other active substances has demonstrated its efficacy in improving glycaemic control and reducing body weight and blood pressure in a broad spectrum of patients with type II diabetes, including those with high baseline HbAlc and the elderly. A sustained reduction in serum uric acid concentration was also observed. Dapagliflozin provides significant improvement in HbAlc, reduction in insulin dose and reduction in body weight in patients with type 1 diabetes as adjunct therapy to adjustable insulin.
Dapagliflozin can be in its free form or any stereoisomer or any pharmaceutically acceptable salt or co crystal complex or a hydrate or a solvate thereof and in any polymorphic forms and any mixtures thereof.
Dapagliflozin as a substance was first disclosed in US 6,515,117. The process for the preparation of dapagliflozin involves the reaction of 4-bromo- 1 -chloro-2-(4-ethoxybenyl)benzene with 2,3,4,6-tetra-O-trimethyl silyl -D-gluconolactone, the obtained compound 3 on demethoxylation yields diastereomeric mixture of Dapagliflozin. Hie diastereomeric mixture of dapagliflozin is further acetylated with acetic anhydride in the presence of pyridine and dimethylaminopyridine yields, then recrystallized from absolute ethanol to yield the desired tetra-acetyJated b-C-glucoside as a white solid. Compound tetra-acetylated b-C-glucoside is treated with lithium hydroxide hydrate which undergoes deprotection to yield the compound dapagliflozin.
Several other documents, patents and applications disclose the process for the preparation of dapagliflozin such as for example WOO 127128, WO03099836, W02004063209, W02006034489,
W02010022313, WO2012019496, W02013064909, W02013068850, W02013079501, WO2014094544, WO2014159151, WO2014206299, W02015040571, WO2015044849, WO2015063726, WO2015132803, WO2015155739, W02016098016, WO2016128995, WO2016178148, WO2017042683, WO2017063617, W02018029611, WO2018029264, WO2018142422.
Prior art documents already provided some compositions of SGLT2 inhibitor dapagliflozin.
W02008116179 discloses immediate release formulation in the form of a stock granulation or in the form of a capsule or a tablet which comprises dapagliflozin propylene glycol hydrate, one or more bulking agent, one or more binder and one or more disintegrant.
WO2011060256 describes the bilayer tablet comprising dapagliflozin having sustained release profde in one layer and metformin in another layer while WO2011060290 describes immediate release formulation of dapagliflozin and metformin.
WO2012163546 discloses the pharmaceutical composition comprising cyclodextrin and dapagliflozin.
Co-crystals of dapagliflozin with lactose are described in WO2014178040.
Solid dispersion compositions comprising amorphous dapagliflozin and at least one polymer are disclosed in W02015011113 and in WO2015128853.
CN103721261 discloses the combination of SGLT2 inhibitor with vitamins such as vitamin B.
Pharmaceutical composition preparation comprising dapagliflozin L-proline and metformin and/or DPP-IV inhibitor is disclosed in WO2018124497.
EP2252289A1 provides a combination of SGLT inhibitor with DPP4 inhibitor showing synergistic effect in increasing plasma active GLP-1 level in a patient over that provided by administration of the SGLT inhibitor or the DPP4 inhibitor alone.
EP2395983A1 relates to a pharmaceutical composition comprising a SGLT2 inhibitor, a DPP4 inhibitor and a third antidiabetic agent which is suitable in the treatment or prevention of one or more conditions selected from type 1 diabetes mellitus, type 2 diabetes mellitus, impaired glucose tolerance and hyperglycemia.
Example A: HPLC method
The purity of Dapagliflozine in general may be determined with the following HPLC method: column: XBridge C18, 150×4.6mm, 3.5; flow-rate: 0.9ml/min; column temperature: 50°C, wavelength: UV 225 nm; mobile phase: eluent A: 0.1% H3PO4, Eluent B: methanol; gradient:
Sample preparation: Accurately weigh about 40mg of sample and dissolve in 50 ml of solvent. Calculation: Use area per cent method. Do not integrate solvent peaks.
Example 1: Preparation of 5-bromo-2-chlorobenzoyl chloride
5-bromo-2-chlorobenzoic acid (450 g) was suspended in dichloromethane (2.25 L) and dimethylformamide (0.74 ml). At 15 – 30°C oxalyl chloride (180.3 ml) was slowly added. During addition gas evolution of HC1 and CO2 occurred. The reaction was performed at 20-30°C. The reaction was considered to be complete if 2-chloro-5-bromobenzoic acid was below 1% (area percent purity). The mixture was concentrated at elevated temperature until oily residue was obtained.
Example 2: Preparation of (5-bromo-2-chlorophenyl)(4-ethoxyphenyl)methanone
Dichloromethane (900 ml) was charged into reactor and then aluminum chloride (267.6 g) was added. The reaction mixture was cooled below 5°C and ethoxybenzene (256.1 ml) was slowly added. After complete addition, the mixture was gradually cooled below -5°C. In a separate reactor, 5-bromo-2-chlorobenzoyl chloride (485g) was dissolved in dichloromethane (900 ml). This solution was slowly added to the mixture of aluminum chloride and ethoxybenzene with such rate that temperature was kept below -5°C. After complete addition the mixture was stirred below -5°C until reaction was finished. The reaction was considered to be complete if methyl ester was below 1 % (the reaction mixture is sampled in methanol). After reaction was completed the reaction mixture was slowly added into cooled 1M HC1 solution and flushed with of dichloromethane (450 ml). The organic phase was separated and water phase was washed again with dichloromethane. Organic phases were combined and washed with water and NaHC03 solution. So obtained organic phase was concentrated to oily residue and dissolved in methanol ethyl acetate mixture in 10 to 1 ratio at reflux temperature. The clear solution was gradually cooled down to 35-45 °C and seeded with pure 5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone. The reaction mass was gradually cooled down to 0-10°C and stirred at that temperature up to 4 hours. The precipitate was isolated and washed with precooled methanol. The product was dried to a final LOD (Loss on drying) content of less than 1.0% with a yield of 564g (87% mass yield).
Example 3: Preparation of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene
5-bromo-2-chlorophenyl(4-ethoxyphenyl)methanone (400g) was dissolved in 1.62L tetrahydrofuran. Into solution NaBTL (53.5g ) was added. After addition, the mixture was stirred at ambient temperature for 30-60 min followed by cooling of reaction mixture below -5°C. Aluminum chloride (314g) was added in portion and reaction mixture maintained below 5°C. After addition, the reaction mixture was gradually heated to reflux temperature and stirred until reaction was complete. Reaction mixture was cooled to ambient temperature and mixture of THF/water was slowly added into reaction mixture followed by addition of water and stirred at ambient temperature. Organic phase was collected and washed with saturated NaCl solution. Organic phase was concentrated to oily residue and dissolved in ethanol (800ml) at elevated temperature. Solution was cooled to 25-30°C and seeded with pure 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene. The reaction mass was gradually cooled to -2 to 10°C and stirred at that temperature. The product was isolated and washed with precooled ethanol and dried until final LOD (Loss on drying) content was less than 1.0%. Yield was 322 g (89%).
Example 4: Preparation of 3R,4S,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6- (hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol
4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (97.5g ) and toluene (1.46L) was charge into reactor. Solution was heated to reflux temperature and approximately half of the solvent was distilled out. Tetrahydrofuran (195 mL) was charged into the solution and mixture was cooled below -70°C. Solution of 15% «-Buli in hexane (227.5 ml) was slowly added and temperature was kept below -70°C. After complete addition solution was stirred at temperature below -70°C to complete reaction. Solution of 2,3,4,6-tetra-O-trimethylsilyl-D-gluconolactone (182 g) in toluene (243 mL) was added into reaction mixture at temperature below -70°C. After complete addition, the mixture was stirred below -70°C, warmed to approximately -65°C and then mixture of 57.6 g methanesulfonic acid in 488 ml methanol was added. After addition, the mixture was gradually warmed to ambient temperature and stirred until reaction was complete. After reaction was finished reaction mixture was slowly added into saturated NaHCCL solution (630ml) and stirred. Into quenched mixture 975 ml of heptane and 585 ml methanol was added. The mixture was stirred for additional 15 min. Organic phase was washed with water/methanol mixture several times. Water phases were combined and distilled to remove organic solvents. Into the residual water phase, toluene was added to perform extraction. Organic phases were combined and washed with water. Organic phase was distillated at elevated temperature until oily residue was obtained.
Example 5: Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl) tetrahydro2H-pyran-3,4,5-triol – Dapagliflozin
Dichloromethane (656 mL) was charged into oily residue from step 4 and stirred at ambient temperature until clear solution was obtained. Triethysilane (122 mL) was added into the so obtained solution. Reaction mixture was cooled below -30°C and 94.2 mL of boron trifluoride etherate was slowly added at temperatures below -30°C. After complete addition, the mixture was stirred below -30°C for one hour and gradually warmed to -5 to 0°C until reaction was completed. After reaction was finished saturated NaHCCL solution (468 mL) was slowly added. Reaction mixture was distilled to remove organic solvents followed by addition ethyl acetate into the residue. Organic phase was collected and washed again with saturated NaHC03 and water. So obtained organic phase was distillated at elevated temperature until oily residue is obtained.
Example 6: Preparation of (2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate.
Oily residual from example 5 was dissolved in dichloromethane (602 mL) at ambient temperature followed by addition of DMAP (6.22g). Reaction mixture was cooled to 0 – 10°C and 144.3 mL of acetic anhydride was added at temperatures below 10°C. Reaction mixture was gradually warmed to ambient temperature and stirred until reaction was completed. Reaction mass was washed with water with saturated NaHC03. Organic phase was collected and concentrated to oily residue to which ethanol (1.68L) was charged and approximately 300 ml ethanol was removed by distillation. The clear solution was gradually cooled to 60-65 °C and seeded. The reaction mass was gradually cooled to 20-25 °C and product was isolated. The product was dried at 50°C in vacuum until LOD (Loss on drying) below 1.0% 123g of product was obtained with yield 71%. HPLC purity: 99.97 %.
Formula 9 Formula 2
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (740 g) as prepared according to process described in examples 1 to 6 was charged into solution of methanol (2.27L), water (0.74L) and NaOH (23 lg) at 35-45°C and stirred at 35-45°C until reaction was completed. After reaction was finished 1M HC1 (1.63L) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. Combined organic phases were washed with water and concentrated at elevated temperature until oily residue was obtained. Content of impurity IMP A was below 0.02%.
Example 7a: Preparation of amorphous dapagliflozin.
Oily residue as prepared according to example 7 comprising approximately 262 g of dapagliflozin was dissolved in toluene (2.5L) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (5.3L) at temperature between 10 to 15°C and stirring rate with P/V at 4 W/m3. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was 14 · 104 m/s. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of impurity IMP A was below 0.02% and residual heptane and toluene were 1673 ppm and below 89 ppm.
(2R,3R,4R,5S,6S)-2-(acetoxymethyl)-6-(4-chloro-3-(4-ethoxybenzyl)phenyl) tetrahydro-2H-pyran-3,4,5-triyl triacetate (30 g) of 4 different qualities obtained by the process known in the prior art was charged into solution of methanol (90 mL), water (30 mL) and NaOH (9.36 g) and stirred at 35-45°C until reaction was completed and sampled for HPLC analysis (Sample 1).
After reaction was finished 1M HC1 (66 mL) was slowly added. Reaction mass was distilled to remove organic solvents and product was extracted by tert-butyl methyl ether. To the combined organic phases 68ml of 1M NaOH was added and pH was set to 12.5 to 13.5. Phases were separated and organic phase is washed again with 68ml of water without pH correction. So obtained organic phase was sampled for HPLC analysis (Sample 2) and concentrated at elevated temperature until oily residue was obtained.
Oily residue comprising approximately 21 g of dapagliflozin was dissolved in toluene (210 mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (420 mL) at temperature between 10 to 15°C and stirring rate with P/V as defined in Table 1. After complete addition, the suspension was cooled to 0°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Filtration rate was as defined in Table 1. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Amorphous dapagliflozin with content of impurity IMP A as shown in Table 1 and residual heptane and toluene as shown in Table 1 was obtained for each cases.
Table 1 : Process parameters used in the preparation of four different starting materials (cases).
As it is evident from Table 2 the final amorphous dapagliflozin prepared by the extraction process according to the present invention contains less than 0.02% of impurity IMP A irrespective of the level of impurity IMP A present in the starting material.
Table 2: Content of impurity IMP A in the final amorphous dapagliflozin obtained with and without extraction.
Example 9
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 25 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 10
Oily residue, as obtained by the procedure described in example 8 case 1, containing approximately 2 g of dapagliflozin was dissolved in 1.5ml of isopropyl acetate and 6ml of tert-butyl methyl ether at temperature 50-55 °C. So prepared solution was charged into 40 mL of heptane at 0°C. After complete addition the suspension was stirred at -10 to 0°C. Suspension was isolated and washed with precooled heptane at temperatures between 25°C to 50 °C. 1.5 g of dapagliflozin was obtained with content of impurity IMP A was below 0.02%.
Example 11
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -10°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1480 ppm and 732 ppm.
Example 12
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 20°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 5°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled hcptanc Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2873 ppm and 639 ppm.
Comparative Example 1
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at -5°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to -15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 1940 ppm and 1557 ppm.
Comparative Example 2
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 25°C and stirring rate with P V at 16 W/m3. After complete addition, the suspension was cooled to 20°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 3663 ppm and 2047 ppm.
Comparative Example 3
Dapagliflozin (30g) was dissolved in toluene (285mL) at temperature 60-70 °C. Solution of dapagliflozin in toluene was slowly added into heptane (600mL) at 30°C and stirring rate with P/V at 16 W/m3. After complete addition, the suspension was cooled to 15°C and stirred with unchanged stirring rate. Suspension was isolated and washed with precooled heptane. Isolated product was dried in vacuum dryer at temperatures between 25 °C to 50 °C. Content of residual heptane and toluene were 2425 ppm and 1812 ppm.
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
PATENT
https://patents.google.com/patent/WO2017206808A1/en
Daggliflozin (English name: Dapagliflozin) is a new Sodium glucose co-transporters 2 (SGLT-2) inhibitor developed by Bristol-Myers Squibb and AstraZeneca. Approved by the European Commission on November 14, 2012, and marketed in the United States on January 8, 2014, to improve glycemic control in adult patients with type 2 diabetes by combining diet and exercise; the trade name is Farxiga, currently offering 5 mg and 10 mg tablets. At the same time, a combination of dapagliflozin and metformin hydrochloride has also been marketed.The chemical name of dapagliflozin is (2S,3R,4R,5S,6R)-2-(3-(4-ethoxybenzyl)-4-chlorophenyl)-6-hydroxymethyltetrahydro-2H – pyran-3,4,5-triol, the chemical formula is C 21 H 25 ClO 6 , CAS No. 461432-26-8, the structural formula is shown as 2, clinically used as a pharmaceutical for dapagliflozin (S) -1,2-propanediol monohydrate, the structural formula is as shown in 1.

The synthesis of β-type C-aryl glycosidic bonds is a key point in the synthetic route during the preparation of dapagliflozin. At present, there are four synthetic methods for the synthesis of dapagliflozin reported in the literature and patents.Route 1: The synthetic route of dapagliflozin reported in patent WO03099836A1 is as follows:

The route uses 2-chloro-5-bromobenzoic acid (12) as raw material to react with phenethyl ether to form intermediate 11 and then triethylsilane to obtain intermediate 10; intermediate 10 and n-butyl The lithium is reacted at -78 ° C, and then subjected to a nucleophilic addition reaction with the intermediate 9, and then methoxylated to obtain the intermediate 8; the intermediate 8 is subjected to acylation reduction and deprotection to obtain the intermediate 2. The disadvantage of this method is that the β-type C-aryl glycosidic bond synthesis of the compound is carried out at a low temperature of -78 ° C, which is obviously difficult to meet the needs of industrial production; and, through nucleophilic addition, methoxylation, The five-step reaction of acetylation, reduction and hydrolysis can synthesize the β-type C-aryl glycosidic bond. The procedure is relatively long, and the purity of the intermediate 2 is only 94%.Route 2: The synthetic route of dapagliflozin reported in the literature OrgLett.2012, 14, 1480 is as follows:

The intermediate 14 of the route is reacted with di-n-butyl-n-hexylmagnesium for 48 hours at 0 ° C, and then reacted with zinc bromide to prepare an organozinc reagent by Br/Mg/Zn exchange reaction, and then with intermediate 4 Intermediate 3 was prepared by nucleophilic substitution reaction; finally, intermediate 2 was obtained by deprotection with sodium methoxide. The synthesis method is relatively novel, and the synthesis step is short. However, the research experiment is conducted only as a synthesis method, and the post treatment of the intermediate 3 is performed by column chromatography. The purity of the intermediate 2 produced was not reported. Moreover, the di-n-butyl-n-hexylmagnesium reagent used in the route is not a commonly used reagent, and is not commercially available in China. It can only be prepared by reacting dibutylmagnesium with n-hexyllithium reagent before the test, and the operation is cumbersome and difficult to mass. use.Route 3: The synthetic route of dapagliflozin reported in patent WO2013068850A2 is as follows:

The route uses 1,6-anhydroglucose (20) as a raw material, protects the 2,4-hydroxyl group by tert-butyldiphenylchlorosilane, and then protects the 3-position hydroxyl group with phenylmagnesium bromide. Intermediate 18. The intermediate 14 is subjected to an Br/Mg/Al exchange reaction to prepare an organoaluminum reagent 16, which is reacted with an intermediate 18 to form an intermediate 15, and finally, deprotected to obtain an intermediate 2. The synthesis method is very novel and is also used as a synthetic methodological study. The purification of the intermediates is carried out by column chromatography. The 1,6-anhydroglucose (20) used in the route is very expensive; and the multi-step reaction in the route uses a format reagent, a preparation format reagent or an organoaluminum reagent, which is cumbersome and cumbersome to perform, and is difficult to scale synthesis. The purity of the intermediate 2 produced was not reported.Route 4: The synthetic route of dapagliflozin reported in patent WO2013152476A1 is as follows:

The route uses 2-chloro-5-iodobenzoic acid (24) as raw material to form intermediate 22 by Friedel acylation and reduction reaction, and exchange with I-Mg at -5 ° C with isopropyl magnesium chloride lithium chloride. The intermediate 8 is obtained by nucleophilic addition and methoxylation with the intermediate 9, and then the intermediate 2 is obtained by reduction with triethylsilane, and the intermediate 2 is further purified by co-crystallizing with L-valine. Finally, The pure intermediate 2 was obtained by removing L-valine. This route is a modified route of Route 1, which replaces n-butyllithium with isopropylmagnesium chloride chloride to raise the reaction temperature of the reaction from -78 °C to -5 °C. However, the problem of a long step of synthesizing a β-type C-aryl glycosidic bond still exists. The obtained intermediate 2 is not optically pure, and needs to be purified by co-crystallizing with L-valine, and the work amount of post-treatment is increased, and finally the purity of the intermediate 2 is 99.3%.Among the four synthetic routes described above for dapagliflozin, route one and route four are commonly used synthetic methods for β-type C-aryl glycosidic bonds, and the route is long, and the optical purity of the obtained product is not high, and further purification is required. Post processing is cumbersome. Moreover, the reaction required at -78 °C in Route 1 requires high equipment and high energy consumption, which undoubtedly increases the cost. Although both Route 2 and Route 3 are new methods, most of the purification of intermediates used is column chromatography. Such a process is not suitable for scale production in factories; and some of the synthetic routes are used. Reagents are not commercially available or expensive, and there is no advantage in such route costs. Therefore, there is an urgent need to find a new method for the synthesis of dapagliflozin, and to enable industrial production, and the route has a cost advantage.Repeating the procedure reported in the literature in Equation 2, the yield of Intermediate 3 was only 46%. The organic zinc reagent is prepared by Br/Mg/Zn exchange reaction, and the exchange reaction yield is 78%; and the raw material is prepared by X/Li/Zn exchange reaction to prepare an organic zinc reagent, and the exchange reaction yield is 98.5%, which is also the two Different reaction pathways lead to the essential reason for the different yields of intermediate 3. Moreover, the price of commercially available 1.0 mol/L di-n-butyl magnesium n-heptane solution 500 mL is 1380 yuan, and the price of 1.6 mol/L n-hexyl lithium n-hexane solution 500 mL is 950 yuan, and 2.5 mol/L n-butyl lithium. The price of 500 mL of n-hexane solution is only 145 yuan. Therefore, the method for preparing dapagliflozin by preparing an organozinc reagent by X/Li/Zn and then synthesizing the β-type C-aryl glycosidic bond designed by the invention has the advantages of cost, ease of operation and industrialization. Very obvious advantage.In order to solve this problem, the original compound company uses a eutectic method in the production of dapagliflozin to make dapagliflozin together with a solvent or an amino acid compound, since the compound 2 sugar ring structure contains four hydroxyl groups and is easy to absorb moisture and deteriorate. The crystal is made into a relatively stable solid, easy to store, stable and controllable in quality, and easy to prepare. Among them, the marketed dapagliflozin forms a stable eutectic with (S)-1,2-propanediol and water (1). The original crystal form patent (CN101479287B, CN103145773B) reported that all 11 crystal forms are dapagliflozin solvate or dapagliflozin. Crystal. Among them, there are two preparation methods for the da forme (S)-1,2-propanediol monohydrate (1) having a crystal structure of type Ia:Method 1: The preparation method is as follows:

Compound 7 is deprotected with sodium hydroxide to obtain compound 2, then compound 2 is extracted with isopropyl acetate, (S)-1,2-propanediol ((S)-PG) is added, and seed crystal of compound 1 is added. Then, cyclohexane was added to crystallize and separated to obtain a eutectic of the compound (1) of the type Ia.Method 2: The preparation method is as follows:

Compound 8 is subjected to reduction of methoxy group by triethylsilane and boron trifluoride diethyl ether complex, and then the reaction solution is extracted with methyl tert-butyl ether (MTBE), and (S)-1,2-propanediol ( (S)-PG), a seed crystal of the compound 1 is added, and then cyclohexane is added to crystallize, and the mixture is separated and dried to obtain a eutectic of the compound (1) of the type Ia.The above two methods for preparing the eutectic are all used in the cyclohexane solvent, which is listed in the appendix of the 2015 edition of the Pharmacopoeia (four parts) as the second type of solvent that should be restricted, with a residual limit of 0.388%. The solvent residue of the final product obtained must reach the specified limit, and the post-treatment process is complicated, time-consuming and labor-intensive, and the production cost is correspondingly increased. The invention finds a suitable solvent on the basis of the synthetic route to prepare a medicinal crystal form, and has obvious advantages in both the method and the process operation steps.The synthetic route is as follows:

Comparative Example 1, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4- Preparation of chlorophenyl]glucosamine (Compound 3)Under nitrogen protection, 1.0 mol/L di-n-butylmagnesium-n-heptane solution (16 mL) was cooled to 0 ° C, and 1.6 mol/L n-hexane lithium n-hexane solution (10 mL) was slowly added dropwise. After the addition was completed, 0 ° C After stirring for 15 h, dry n-butyl ether (2.5 mL) was added to prepare a solution of di-n-butyl-n-hexylmagnesium lithium solution, which was calibrated with iodine and stored for use.Zinc bromide (2.7 g) and lithium bromide (1.04 g) were added with n-butyl ether (20 mL), heated to 50 ° C for 4 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (8 mL) and n-butyl ether (5 mL) under nitrogen, cooled to 0 ° C, and 0.61 mol/L was added dropwise. n-Butyl-n-hexylmagnesium lithium solution (13.1 mL), after the addition is completed, the reaction was kept at 0 ° C for 48 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution were added, and the reaction was kept at 0 ° C for 1 h, and added 2 , 3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (25 mL), heated to 100 ° C to stir the reaction, after TLC detection reaction, add 1 mol / L diluted hydrochloric acid (60 mL), taken after stirring extraction, the organic phase was washed with water (40 mL), then washed with saturated brine (40 mL), dried over anhydrous Na 2 SO 4, concentrated under reduced pressure, column chromatography (petroleum ether / Ethyl acetate = 20:1) 10.38 g of Compound 3 as a pale yellow oil. Yield: 46%. Purity: 99.02%. The organozinc reagent prepared by the method has an iodine calibration yield of 78%.The calibration method of the concentration of the prepared organic zinc reagent: accurately weighed iodine (1 mmol), placed in a three-necked flask, replaced nitrogen, and added anhydrous 0.5 mol/L LiCl tetrahydrofuran solution (5 mL), stirred and dissolved, and cooled to 0 ° C. The prepared organozinc reagent was slowly added dropwise until the color of the brownish yellow solution disappeared.Example 2 (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (2.25 g) and lithium bromide (0.87 g) were added with n-butyl ether (30 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and n-butyl ether (10 mL) under nitrogen, cooled to -20 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (14mL), control the internal temperature does not exceed -10 ° C, after the completion of the addition, the temperature is incubated at -20 ° C for 0.5 h, adding the above-mentioned spare zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at 20 ° C for 3 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (11.59g) toluene (50mL) solution, heat to 120 ° C and stir the reaction for 4h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (40 mL), water (20 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.8 g of Compound 3 as a white solid was obtained in a yield: 72.42%. Purity: 99.47%. Melting point: 99.5 to 101.6 °C. (The organic zinc reagent prepared by this method was iodine-calibrated in a yield of 98.5%.) ESI-MS (m/z): 767.30 [M+Na] + . 1 H-NMR (400 MHz, CDCl 3 ): δ 7.33 (1H, d), 7.14-7.17 (2H, m), 7.05 (2H, d), 6.79-6.81 (2H, dd), 5.39 (1H, t ), 5.21-5.31 (2H, m), 4.33 (1H, d), 4.17-4.20 (1H, dd), 3.94-4.11 (5H, m), 3.79-3.83 (1H, m), 1.39 (3H, t ), 1.20 (9H, s), 1.16 (9H, s), 1.11 (9H, s), 0.86 (9H, s).Example 3, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3) PrepareZinc bromide (3.38 g) and lithium bromide (1.3 g) were added with n-butyl ether (40 mL), heated to 50 ° C for 2 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (20 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -50 ° C, and slowly added dropwise 2.5 mol / L-butyllithium hexane solution (8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -50 ° C for 10 h, adding the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution, The reaction was stirred at -20 ° C for 10 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (34.77g) toluene (80mL) solution, heat to 100 ° C and stir the reaction for 24h, after TLC detection reaction, was added 1mol / L diluted hydrochloric acid (60 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, concentrated with n-heptane (15mL) and methanol (60 mL) and recrystallized 10.854 g of Compound 3 as a white solid. Yield: 72.81%. Purity: 99.53%.Example 4, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)N-butyl ether (50 mL) was added to zinc iodide (3.19 g) and lithium iodide (1.34 g), and the mixture was heated to 50 ° C for 1.5 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -60 ° C, and slowly added dropwise 1.6 mol / L-n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -60 ° C for 5 h, and the above-mentioned alternate zinc iodide and lithium iodide n-butyl ether solution is added. The reaction was stirred at 25 ° C for 1 h. Add 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (23.2g) toluene (50mL) solution, heat to 140 ° C reflux reaction for 0.5h, after TLC detection reaction was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous SO 4 Na 2, concentrated by weight of n-heptane (15mL) and methanol (60 mL) Crystallization gave 10.51 g of Compound 3 as a white solid, yield 70.5%. Purity: 99.41%.Example 5, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)To the zinc bromide (2.25 g) and lithium bromide (0.87 g), cyclopentyl methyl ether (30 mL) was added, and the mixture was heated to 50 ° C for 3 hours, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (10 mL) and cyclopentyl methyl ether (10 mL) under nitrogen, cooled to -5 ° C, and slowly added dropwise. Mol / L n-hexyl lithium n-hexane solution (12.5mL), control the internal temperature does not exceed 0 ° C, after the addition is completed, the reaction is kept at -5 ° C for 3 h, adding the above-mentioned spare zinc bromide and lithium bromide cyclopentyl methyl ether The solution was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (40 mL) was added and heated to 80 ℃ reaction was stirred 6h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (50 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous 2 SO 4 Na, and concentrated under reduced pressure, Recrystallization of n-heptane (15 mL) and methanol (60 mL) gave 8.15 g of Compound 3 as a white solid. Purity: 99.39%.Example 6, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Zinc bromide (4.5 g) and lithium bromide (1.74 g) were added with n-butyl ether (60 mL), heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-bromo-benzyl) phenyl ether (6.513 g) was added with toluene (15 mL) and n-butyl ether (5 mL) under nitrogen, cooled to -30 ° C, and slowly added dropwise 2.5 mol / L-butyllithium n-hexane solution (8.4mL), control the internal temperature does not exceed -20 ° C, after the addition is completed, the reaction is kept at -30 ° C for 3 h, and the above-mentioned alternate zinc bromide and lithium bromide n-butyl ether solution is added. The reaction was incubated at -5 ° C for 4 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (14.49 g) in toluene (50 mL) was added and heated to 120 ° C for stirring. the reaction 4h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (50 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure, n-heptyl Recrystallization of the alkane (15 mL) and methanol (60 mL) gave 10.38 g of Compound 3 as a white solid. Purity: 99.54%.Example 7, (1S)-2,3,4,6-tetra-O-pivaloyl-1,5-anhydro-1-[3-(4-ethoxyphenylmethyl)-4-chloro Preparation of phenyl]glucitol (compound 3)Methyl bromide (40 mL) was added to zinc bromide (2.25 g) and lithium bromide (0.87 g), and the mixture was heated to 50 ° C for 3 h, and cooled for use. 4-(2-Chloro-5-iodo-benzyl) phenyl ether (7.45 g) was added with toluene (15 mL), methyl tert-butyl ether (15 mL), cooled to -40 ° C, and slowly added dropwise. 1.6mol/L n-hexyl lithium n-hexane solution (13.8mL), control the internal temperature does not exceed -30 ° C, after the addition is completed, the reaction is kept at -40 ° C for 4 h, and the above-mentioned alternate zinc bromide and lithium bromide are added. The butyl ether solution was incubated at 5 ° C for 7 h, and a solution of 2,3,4,6-tetra-O-pivaloyl-α-D-bromoglucopyranose (17.39 g) in toluene (50 mL) was added and heated. to 90 deg.] C the reaction was stirred 8h, after completion of the reaction by TLC, was added 1mol / L diluted hydrochloric acid (40 mL), water (40 mL), and extracted, the organic phase was washed with water (40 mL), dried over anhydrous Na 2 SO 4, and concentrated under reduced pressure Recrystallization from n-heptane (15 mL) and methanol (60 mL) gave 9.41 g of Compound 3 as a white solid. Purity: 99.42%. Example 8. Preparation of dapagliflozin (S)-1,2-propanediol monohydrate eutectic (Compound 1)To the compound 3 (37.27 g), methanol (190 mL) was added, and sodium methoxide (10.8 g) was added thereto, and the mixture was heated under reflux for 3 hours. After the TLC reaction was completed, methanol was concentrated, and isopropyl acetate (100 mL) was added to the residue, and water was added. (60 mL), extracted with stirring and the organic phase washed with water (50 mL). (S)-1,2-propanediol (3.8g) and water (0.9g) were added to the organic phase, stirred until it was dissolved, and n-heptane (200 mL) was added, and the mixture was stirred for 2 hours under ice-cooling, suction filtration, filter cake Washing with n-heptane and drying at 30 ° C gave 23.89 g of Compound 1 as a white solid. Yield: 95%. Purity: 99.79%. Melting point: 69.1 to 75.6 °C. The product obtained was subjected to KF = 3.74% (theoretical value: 3.58%). ESI-MS (m/z): 431.22 [M+Na] + . 1 H-NMR (400 MHz, CD 3 OD): δ 7.33 – 7.37 (2H, m), 7.28-7.30 (1H, dd), 7.11 (2H, d), 6.80-6.83 (2H, dd), 4.1 ( 1H, d), 3.98-4.05 (4H, m), 3.88-3.91 (1H, dd), 3.74-3.82 (1H, m), 3.68-3.73 (1H, m), 3.37-3.49 (5H, m), 3.28-3.34 (1H, m), 1.37 (1H, t), 1.15 (3H, d).The crystal form of the obtained product was subjected to thermogravimetric analysis (TGA) by a Universal V4.7A TA instrument, and the TGA curve (Fig. 1) showed a weight loss of about 18.52% from about room temperature to about 240 ° C. The original form Ia crystal form The TGA plot shows a value of 18.7%.The crystal form of the obtained product was subjected to differential scanning calorimetry (DSC) by a Universal V4.7A TA instrument, and the DSC curve (Fig. 2) showed endotherm in the range of about 60 ° C to 85 ° C. The DSC plot shows a range of approximately 50 ° C to 78 ° C.
The crystal form of the obtained product was examined by a Bruker D8advance instrument for powder X-ray diffraction (PXRD), and the 2X value of the PXRD pattern (Fig. 3) (CuKα).

There are characteristic peaks at 3.749°, 7.52°, 7.995°, 8.664°, 15.134°, 15.708°, 17.069°, 18.946°, 20.049°, which are completely consistent with the characteristic peaks of the PXRD pattern of the Ia crystal form in the original patent.In combination with the nuclear magnetic data and melting point of the prepared crystal form, the crystal form of the product (Compound 1) obtained by the present invention is consistent with the pharmaceutically acceptable crystalline form Ia reported in the original patent.
Patent Citations
Publication numberPriority datePublication dateAssigneeTitleCN101479287A *2006-06-282009-07-08布里斯托尔-迈尔斯斯奎布公司Crystalline solvates and complexes of (is) -1, 5-anhydro-l-c- (3- ( (phenyl) methyl) phenyl) -d-glucitol derivatives with amino acids as sglt2 inhibitors for the treatment of diabetesCN104496952A *2014-11-282015-04-08深圳翰宇药业股份有限公司Synthesis method of dapagliflozinCN105153137A *2015-09-172015-12-16上海应用技术学院Preparation method of empagliflozinFamily To Family CitationsCN104829572B *2014-02-102019-01-04江苏豪森药业集团有限公司Dapagliflozin novel crystal forms and preparation method thereofCN105399735A *2015-12-292016-03-16上海应用技术学院Empagliflozin intermediate, and preparation method and application thereof* Cited by examiner, † Cited by third party
Non-Patent Citations
TitleCHEN DEJIN ET AL., CHINA MASTER’S THESES FULL-TEXT DATABASE, ENGINEERING TECHNOLOGY I, vol. B016-731, no. 3, 15 March 2016 (2016-03-15) *LEMAIRE S. ET AL.: “Stereoselective C-glycosylation of furanosyl halides with arylzinc reagents”, PURE APPL. CHEM., vol. 86, no. 3, 4 March 2014 (2014-03-04), pages 329 – 333 *LEMAIRE S. ET AL.: “Stereoselective C-Glycosylation Reactions with Arylzinc Reagents”, ORGANIC LETTERS, vol. 14, no. 6, 2 March 2012 (2012-03-02), pages 1480 – 1483, XP055069093 ** Cited by examiner, † Cited by third partyCLIP
Chemical Synthesis
Dapagliflozin propanediol hydrate, an orally active sodium glucose cotransporter type 2 (SGLT-2) inhibitor, was developed by Bristol-Myers Squibb (BMS) and AstraZeneca for the once-daily treatment of type 2 diabetes. As opposed to competitor SGLT-2 inhibitors, dapagliflozin was not associated with renal toxicity or long-term deterioration of renal function in phase III clinical trials. The drug exhibits excellent SGLT2 potency with more than 1200 fold selectivity over the SGLT1 enzyme.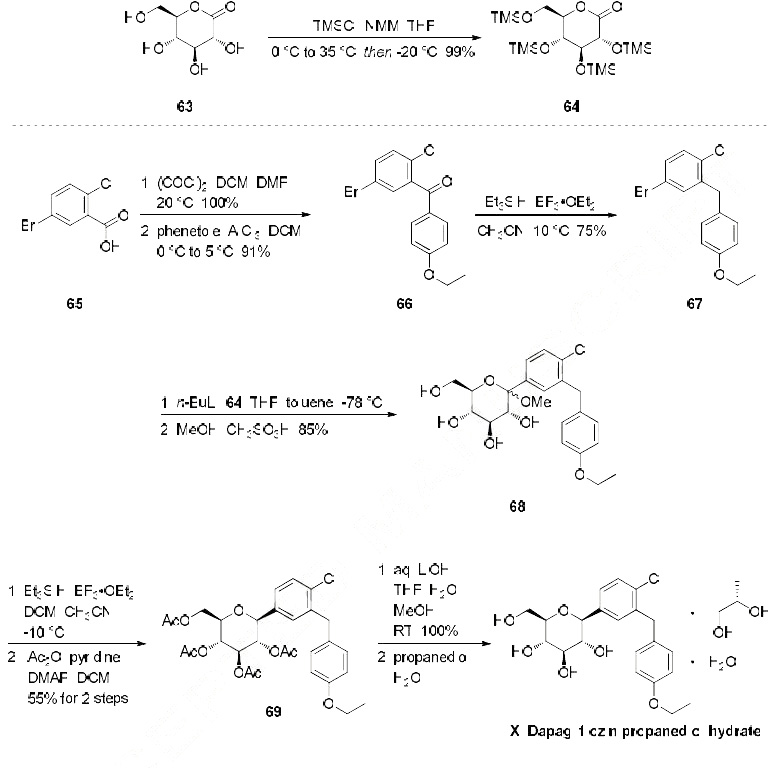
Dapagliflozin propanediol monohydrate
PAPER
https://link.springer.com/article/10.1007/s12039-020-1747-x
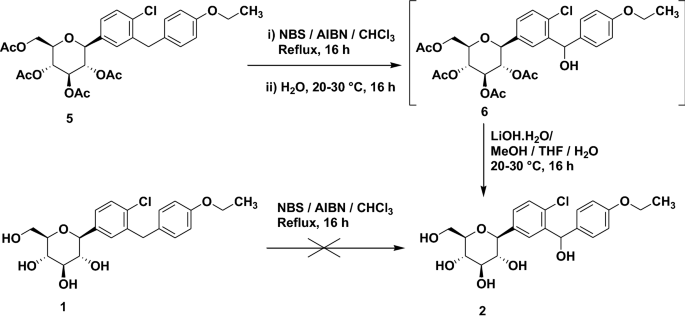
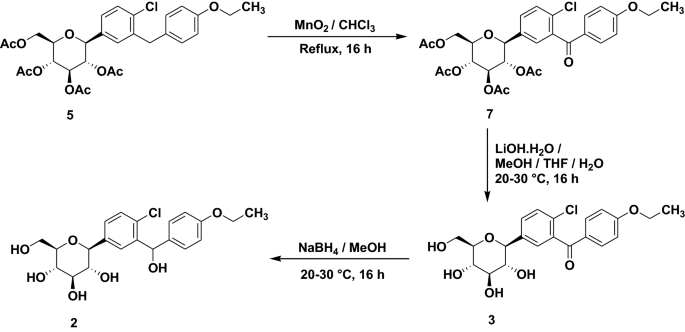

PATENTS
WO 2010138535
WO 2011060256
WO 2012041898
WO 2012163990
WO 2013068850
WO 2012163546
WO 2013068850
WO 2013079501

The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/l), so that the drug does not interfere with the intestinal glucose absorption.[7

dapagliflozin being an inhibitor of sodiumdependent glucose transporters found in the intestine and kidney (SGLT2) and to a method for treating diabetes, especially type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, Syndrome X, diabetic
complications, atherosclerosis and related diseases, employing such C-aryl glucosides alone or in combination with one, two or more other type antidiabetic agent and/or one, two or more other type therapeutic agents such as hypolipidemic agents.
Approximately 100 million people worldwide suffer from type II diabetes (NIDDM – non-insulin-dependent diabetes mellitus), which is characterized by hyperglycemia due to excessive hepatic glucose production and peripheral insulin resistance, the root causes for which are as yet unknown. Hyperglycemia is considered to be the major risk factor for the development of diabetic complications, and is likely to contribute directly to the impairment of insulin secretion seen in advanced NIDDM. Normalization of plasma glucose in NIDDM patients would be predicted to improve insulin action, and to offset the development of diabetic complications. An inhibitor of the sodium-dependent glucose transporter SGLT2 in the kidney would be expected to aid in the normalization of plasma glucose levels, and perhaps body weight, by enhancing glucose excretion.
Dapagliflozin can be prepared using similar procedures as described in U.S. Pat. No. 6,515,117 or international published applications no. WO 03/099836 and WO 2008/116179
WO 03/099836 A1 refers to dapagliflozin having the structure according to formula 1 .

formula 1
WO 03/099836 A1 discloses a route of synthesis on pages 8-10, whereby one major step is the purification of a compound of formula 2

formula 2
The compound of formula 2 provides a means of purification for providing a compound of formula 1 since it crystallizes. Subsequently the crystalline form of the compound of formula 2 can be deprotected and converted to dapagliflozin. Using this process, dapagliflozin is obtained as an amorphous glassy off-white solid containing 0.1 1 mol% of EtOAc. Crystallization of a pharmaceutical drug is usually advantageous as it provides means for purification also suitable for industrial scale preparation. However, for providing an active pharmaceutical drug a very high purity is required. In particular, organic impurities such as EtOAc either need to be avoided or further purification steps are needed to provide the drug in a
pharmaceutically acceptable form, i.e. substantially free of organic solvents. Thus, there is the need in the art to obtain pure and crystalline dapagliflozinwhich is substantially free of organic solvents.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Crystalline forms (in comparision to the amorphous form) often show desired different physical and/or biological characteristics which may assist in the manufacture or formulation of the active compound, to the purity levels and uniformity required for regulatory approval. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps.
PATENT
WO 2008/ 1 16179 Al seems to disclose an immediate release formulation comprising dapagliflozin and propylene glycol hydrate. WO 2008/ 116195 A2 refers to the use of an SLGT2 inhibitor in the treatment of obesity
http://www.google.com/patents/US20120282336
http://www.tga.gov.au/pdf/auspar/auspar-dapagliflozin-propanediol-monohydrate-130114.pdf
Example 2 Dapagliflozin (S) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (S)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (S) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in published applications WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
Example 3 Dapagliflozin (R) PGS—(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (R)-propane-1,2-diol hydrate (1:1:1)
Dapagliflozin (R) propylene glycol hydrate (1:1:1) can be prepared using similar procedures as described in WO 08/002824 and WO 2008/116179, the disclosures of which are herein incorporated by reference in their entirety for any purpose. SGLT2 EC50=1.1 nM.
WO 2008/002824 A1 discloses several alternative solid forms of dapagliflozin, such as e.g. solvates containing organic alcohols or co-crystals with amino acids such as proline and phenylalanine. For instance, the document discloses crystalline
dapagliflozin solvates which additionally contain water molecules (see e.g.
Examples 3-6), but is silent about solid forms of dapagliflozin which do not contain impurities such as organic alcohols. As described above, it is desirable to provide the pharmaceutical active drug in a substantially pure form, otherwise triggering further expensive and time-consuming purification steps. In contrast, the document relates to dapagliflozin solvates where an alcohol and water are both incorporated into the crystal lattice. Hence, there is the need in the art to obtain pure and crystalline dapagliflozin suitable for pharmaceutical production.
WO 2008/1 16179 A1 refers to an immediate release pharmaceutical composition comprising dapagliflozin and propylene glycol. Propylene glycol is a chiral
substance and (S)-propylene glycol used is very expensive. Consequently, also the immediate release pharmaceutical composition is more expensive.
Surprisingly, amorphous dapagliflozin can be purified with the process of the present invention. For instance amorphous dapagliflozin having a purity of 99,0% can be converted to crystalline dapagliflozin hydrate having a purity of 100% (see examples of the present application). Moreover, said crystalline dapagliflozin hydrate does not contain any additional solvent which is desirable. Thus, the process of purifying dapagliflozin according to the present invention is superior compared with the process of WO 03/099836 A1 .
Additionally, the dapagliflozin hydrate obtained is crystalline which is advantageous with respect to the formulation of a pharmaceutical composition. The use of expensive diols such as (S)-propanediol for obtaining an immediate release pharmaceutical composition as disclosed in WO 2008/1 16179 A1 can be avoided
PAPER
In Vitro Characterization and Pharmacokinetics of Dapagliflozin …
dmd.aspetjournals.org/content/…/DMD29165_supplemental_data_.doc
Dapagliflozin (BMS-512148), (2S,3R,4R,5S,6R)-2-(3-(4-Ethoxybenzyl)-4-chlorophenyl)
-6-hydroxymethyl-tetrahydro-2H-pyran-3,4,5-triol. 1H NMR (500 MHz, CD3OD) δ 7.33
(d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H),
6.78 (d, J = 8.8, 2H), 4.07-3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6,
1H), 3.42-3.25 (m, 4H), 1.34 (t, J = 7.0, 3H). 13C NMR (125 MHz, CD3OD) δ 158.8,
140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9,
64.5, 63.1, 39.2, 15.2.
HRMS calculated for C21H25ClNaO6 (M+Na)+
For C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
: 431.1237; found 431.1234. Anal. Calcd
SECOND SETJ. Med. Chem., 2008, 51 (5), pp 1145–1149DOI: 10.1021/jm701272q
1H NMR (500 MHz, CD3OD) δ 7.33 (d, J = 6.0, 1H), 7.31 (d, J = 2.2, 1H), 7.31 (dd, J = 2.2, 6.0, 1H), 7.07 (d, J = 8.8, 2H), 6.78 (d, J = 8.8, 2H), 4.07–3.90 (m, 7H), 3.85 (d, J = 10.6, 1H), 3.69 (dd, J = 5.3, 10.6, 1H), 3.42–3.25 (m, 4H), 1.34 (t, J = 7.0, 3H);
13C NMR (125 MHz, CD3OD) δ 158.8, 140.0, 139.9, 134.4, 132.9, 131.9, 130.8, 130.1, 128.2, 115.5, 82.9, 82.2, 79.7, 76.4, 71.9, 64.5, 63.1, 39.2, 15.2;
HRMS calcd for C21H25ClNaO6 (M + Na)+ 431.1237, found 431.1234. Anal. Calcd for C21H25ClO6: C, 61.68; H, 6.16. Found: C, 61.16; H, 6.58.
HPLC
- HPLC measurements were performed with an Agilent 1100 series instrument equipped with a UV-vis detector set to 240 nm according to the following method:
Column: Ascentis Express RP-Amide 4.6 x 150 mm, 2.7 mm;
Column temperature: 25 °C
– Eluent A: 0.1 % formic acid in water
– Eluent B: 0.1 % formic acid in acetonitrile
– Injection volume: 3 mL
– Flow: 0.7 mL/min
– Gradient:Time [min][%] B0.02525.06526.07029.07029.52535.025……………………..
PATENT
http://www.google.com/patents/WO2013068850A2?cl=en
EXAMPLE 24 – Synthesis of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside 2,4-di-6>-TBDPS-dapagliflozin; (IVj”))

[0229] l-(5-Bromo-2-chlorobenzyl)-4-ethoxybenzene (1.5 g, 4.6 mmol) and magnesium powder (0.54 g, 22.2 mmol) were placed in a suitable reactor, followed by THF (12 mL) and 1,2- dibromoethane (0.16 mL). The mixture was heated to reflux. After the reaction had initiated, a solution of l-(5-bromo-2-chlorobenzyl)-4-ethoxybenzene (4.5 g, 13.8 mmol) in THF (28 mL) was added dropwise. The mixture was allowed to stir for another hour under reflux, and was then cooled to ambient temperature, and then titrated to determine the concentration. The above prepared 4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl magnesium bromide (31 mL, 10 mmol, 0.32 M in THF) and A1C13 (0.5 M in THF, 8.0 mL, 4.0 mmol) were mixed at ambient temperature to give a black solution, which was stirred at ambient temperature for 1 hour. To a solution of
I, 6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (0.64 g, 1.0 mmol) in PhOMe (3.0 mL) at ambient temperature was added phenylmagnesium bromide (0.38 mL, 1.0 mmol, 2.6 M solution in Et20). After stirring for about 5 min the solution was then added into the above prepared aluminum mixture via syringe, followed by additional PhOMe (1.0 mL) to rinse the flask. The mixture was concentrated under reduced pressure (50 torr) at 60 °C (external bath temperature) to remove low-boiling point ethereal solvents and then PhOMe (6mL) was added. The reaction mixture was heated at 130 °C (external bath temperature) for 8 hours at which time HPLC assay analysis indicated a 51% yield of 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3- (4-ethoxybenzyl)phenyl)- -D-glucopyranoside. After cooling to ambient temperature, the reaction was treated with 10% aqueous NaOH (1 mL), THF (10 mL) and diatomaceous earth at ambient temperature, then the mixture was filtered and the filter cake was washed with THF. The combined filtrates were concentrated and the crude product was purified by silica gel column chromatography (eluting with 1:30 EtOAc/77-heptane) affording the product 2,4-di-6>- ieri-butyldiphenylsilyl- 1 – -(4-chloro-3 -(4-ethoxybenzyl)phenyl)- β-D-glucopyranoside (0.30 g, 34%) as a white powder.
1H NMR (400 MHz, CDC13) δ 7.56-7.54 (m, 2H), 7.43-7.31 (m, 13H), 7.29-7.22 (m, 6H), 7.07- 7.04 (m, 2H), 7.00 (d, J= 2.0 Hz, IH), 6.87 (dd, J= 8.4, 2.0 Hz, IH), 6.83-6.81 (m, 2H), 4.18 (d, J= 9.6 Hz, IH), 4.02 (q, J= 6.9 Hz, 2H), 3.96 (d, J= 10.8 Hz, 2H), 3.86 (ddd, J= 11.3, 7.7, 1.1 Hz, IH), 3.76 (ddd, J= 8.4, 8.4, 4.8 Hz, IH), 3.56 (ddd, J= 9.0, 6.4, 2.4 Hz, IH), 3.50 (dd, J=
I I.4, 5.4 Hz, IH), 3.44 (dd, J= 9.4, 8.6 Hz, IH), 3.38 (dd, J= 8.8, 8.8 Hz, IH), 1.70 (dd, J= 7.8, 5.4 Hz, IH, OH), 1.42 (t, J= 6.8 Hz, 3H), 1.21 (d, J= 5.2 Hz, IH, OH), 1.00 (s, 9H), 0.64 (s, 9H); 13C NMR (100 MHz, CDC13) δ 157.4 (C), 138.8 (C), 137.4 (C), 136.3 (CH x2), 136.1 (CH x2), 135.2 (CH x2), 135.0 (C), 134.9 (CH x2), 134.8 (C), 134.2 (C), 132.8 (C), 132.0 (C), 131.6 (CH), 131.1 (C), 129.9 (CH x2), 129.7 (CH), 129.6 (CH), 129.5 (CH), 129.4 (CH), 129.2 (CH), 127.58 (CH x2), 127.57 (CH x2), 127.54 (CH x2), 127.31 (CH), 127.28 (CH x2), 114.4 (CH x2), 82.2 (CH), 80.5 (CH), 79.3 (CH), 76.3 (CH), 72.7 (CH), 63.4 (CH2), 62.7 (CH2), 38.2 (CH2), 27.2 (CH3 x3), 26.6 (CH3 x3), 19.6 (C), 19.2 (C), 14.9 (CH3). EXAMPLE 25 -Synthesis of dapagliflozin ((25,3R,4R,55,6/?)-2-[4-chloro-3-(4- ethoxybenzyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol; (Ij))

IVj’ U
[0230] A solution of the 2,4-di-6>-ieri-butyldiphenylsilyl-l-C-(4-chloro-3-(4- ethoxybenzyl)phenyl)- -D-glucopyranoside (60 mg, 0.068 mmol) in THF (3.0 mL) and TBAF (3.0 mL, 3.0 mmol, 1.0 M in THF) was stirred at ambient temperature for 15 hours. CaC03 (0.62 g), Dowex^ 50WX8-400 ion exchange resin (1.86 g) and MeOH (5mL) were added to the product mixture and the suspension was stirred at ambient temperature for 1 hour and then the mixture was filtrated through a pad of diatomaceous earth. The filter cake was rinsed with MeOH and the combined filtrates was evaporated under vacuum and the resulting residue was purified by column chromatography (eluting with 1 : 10 MeOH/DCM) affording dapagliflozin (30 mg).
1H NMR (400 MHz, CD3OD) δ 7.37-7.34 (m, 2H), 7.29 (dd, J= 8.2, 2.2 Hz, 1H), 7.12-7.10 (m, 2H), 6.82-6.80 (m, 2H), 4.10 (d, J= 9.6 Hz, 2H), 4.04 (d, J= 9.2 Hz, 2H), 4.00 (q, J= 7.1 Hz, 2H), 3.91-3.87 (m, 1H), 3.73-3.67(m, 1H), 3.47-3.40 (m, 3H), 3.31-3.23 (m, 2H), 1.37 (t, J= 7.0 Hz, 3H);
13C NMR (100 MHz, CD3OD) δ 157.4 (C), 138.6 (C), 138.5 (C), 133.1 (C), 131.5 (C), 130.5 (CH), 129.4 (CH x2), 128.7 (CH), 126.8 (CH), 114.0 (CH x2), 80.5 (CH), 80.8 (CH), 78.3 (CH), 75.0 (CH), 70.4 (CH), 63.0 (CH2), 61.7 (CH2), 37.8 (CH2), 13.8 (CH3);
LCMS (ESI) m/z 426 (100, [M+NH4]+), 428 (36, [M+NH4+2]+), 447 (33, [M+K]+).
Example 1 – Synthesis of l,6-anhydro-2,4-di-6>-ieri-butyldiphenylsilyl- -D-glucopyranose (II”)

III II”
[0206] To a suspension solution of l,6-anhydro- -D-glucopyranose (1.83 g, 11.3 mmol) and imidazole (3.07 g, 45.2 mmol) in THF (10 mL) at 0 °C was added dropwise a solution of TBDPSC1 (11.6 mL, 45.2 mmol) in THF (10 mL). After the l,6-anhydro-P-D-gJucopyranose was consumed, water (10 mL) was added and the mixture was extracted twice with EtOAc (20 mL each), washed with brine (10 mL), dried (Na2S04) and concentrated. Column
chromatography (eluting with 1 :20 EtOAc/rc-heptane) afforded 2,4-di-6>-ieri-butyldiphenylsilyl- l,6-anhydro- “D-glucopyranose (5.89 g, 81%).
1H NMR (400 MHz, CDC13) δ 7.82-7.70 (m, 8H), 7.49-7.36 (m, 12H), 5.17 (s, IH), 4.22 (d, J= 4.8 Hz, IH), 3.88-3.85 (m, IH), 3.583-3.579 (m, IH), 3.492-3.486 (m, IH), 3.47-3.45 (m, IH), 3.30 (dd, J= 7.4, 5.4 Hz, IH), 1.71 (d, J= 6.0 Hz, IH), 1.142 (s, 9H), 1.139 (s, 9H); 13C NMR (100 MHz, CDCI3) δ 135.89 (CH x2), 135.87 (CH x2), 135.85 (CH x2), 135.83 (CH x2), 133.8 (C), 133.5 (C), 133.3 (C), 133.2 (C), 129.94 (CH), 129.92 (CH), 129.90 (CH), 129.88 (CH), 127.84 (CH2 x2), 127.82 (CH2 x2), 127.77 (CH2 x4), 102.4 (CH), 76.9 (CH), 75.3 (CH), 73.9 (CH), 73.5 (CH), 65.4 (CH2), 27.0 (CH3 x6), 19.3 (C x2).
PATENT
WO 2016147197, DAPAGLIFLOZIN, NEW PATENT, HARMAN FINOCHEM LIMITED
LINK>>> (WO2016147197) A NOVEL PROCESS FOR PREPARING (2S,3R,4R,5S,6R)-2-[4-CHLORO-3-(4-ETHOXYBENZYL)PHENY 1] -6-(HY DROXY METHYL)TETRAHYDRO-2H-PY RAN-3,4,5-TRIOL AND ITS AMORPHOUS FORM
PATENT
PATENT
WO2016018024, CRYSTALLINE COMPOSITE COMPRISING DAPAGLIFLOZIN AND METHOD FOR PREPARING SAME
HANMI FINE CHEMICAL CO., LTD. [KR/KR]; 59, Gyeongje-ro, Siheung-si, Gyeonggi-do 429-848 (KR)
Dapagliflozin, sold under the brand name Farxiga among others, is a medication used to treat type 2 diabetes and, with certain restrictions, type 1 diabetes.[2] It is also used to treat adults with certain kinds of heart failure.[3][4][5]
Common side effects include hypoglycaemia (low blood sugar), urinary tract infections, genital infections, and volume depletion (reduced amount of water in the body).[6] Diabetic ketoacidosis is a common side effect in type 1 diabetic patients.[7] Serious but rare side effects include Fournier gangrene.[8] Dapagliflozin is a sodium-glucose co-transporter-2 (SGLT-2) inhibitor and works by removing sugar from the body with the urine.[9]
It was developed by Bristol-Myers Squibb in partnership with AstraZeneca. In 2018, it was the 227th most commonly prescribed medication in the United States, with more than 2 million prescriptions.[10][11]
Medical uses
Dapagliflozin is used along with diet and exercise to improve glycemic control in adults with type 2 diabetes and to reduce the risk of hospitalization for heart failure among adults with type 2 diabetes and known cardiovascular disease or other risk factors.[12][3] It appears more useful than empagliflozin.[13][verification needed]
In addition, dapagliflozin is indicated for the treatment of adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3][4][5] It is also indicated to reduce the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14]
In the European Union it is indicated in adults:
- for the treatment of insufficiently controlled type 2 diabetes mellitus as an adjunct to diet and exercise:
- as monotherapy when metformin is considered inappropriate due to intolerance;
- in addition to other medicinal products for the treatment of type 2 diabetes;
- for the treatment of insufficiently controlled type 1 diabetes mellitus as an adjunct to insulin in patients with BMI ≥ 27 kg/m2, when insulin alone does not provide adequate glycaemic control despite optimal insulin therapy; and
- for the treatment of heart failure with reduced ejection fraction.[5]
Adverse effects
Since dapagliflozin leads to heavy glycosuria (sometimes up to about 70 grams per day) it can lead to rapid weight loss and tiredness. The glucose acts as an osmotic diuretic (this effect is the cause of polyuria in diabetes) which can lead to dehydration. The increased amount of glucose in the urine can also worsen the infections already associated with diabetes, particularly urinary tract infections and thrush (candidiasis). Rarely, use of an SGLT2 drug, including dapagliflozin, is associated with necrotizing fasciitis of the perineum, also called Fournier gangrene.[15]
Dapagliflozin is also associated with hypotensive reactions. There are concerns it may increase the risk of diabetic ketoacidosis.[16]
Dapagliflozin can cause dehydration, serious urinary tract infections and genital yeast infections.[3] Elderly people, people with kidney problems, those with low blood pressure, and people on diuretics should be assessed for their volume status and kidney function.[3] People with signs and symptoms of metabolic acidosis or ketoacidosis (acid buildup in the blood) should also be assessed.[3] Dapagliflozin can cause serious cases of necrotizing fasciitis of the perineum (Fournier gangrene) in people with diabetes and low blood sugar when combined with insulin.[3]
To lessen the risk of developing ketoacidosis (a serious condition in which the body produces high levels of blood acids called ketones) after surgery, the FDA has approved changes to the prescribing information for SGLT2 inhibitor diabetes medicines to recommend they be stopped temporarily before scheduled surgery. Canagliflozin, dapagliflozin, and empagliflozin should each be stopped at least three days before, and ertugliflozin should be stopped at least four days before scheduled surgery.[17]
Symptoms of ketoacidosis include nausea, vomiting, abdominal pain, tiredness, and trouble breathing.[17]
Use is not recommended in patients with eGFR < 45ml/min/1.73m2, though data from 2021 shows the reduction in the kidney failure risks in people with chronic kidney disease using dapagliflozin.[18]
Mechanism of action
Dapagliflozin inhibits subtype 2 of the sodium-glucose transport proteins (SGLT2) which are responsible for at least 90% of the glucose reabsorption in the kidney. Blocking this transporter mechanism causes blood glucose to be eliminated through the urine.[19] In clinical trials, dapagliflozin lowered HbA1c by 0.6 versus placebo percentage points when added to metformin.[20]
Regarding its protective effects in heart failure, this is attributed primarily to haemodynamic effects, where SGLT2 inhibitors potently reduce intravascular volume through osmotic diuresis and natriuresis. This consequently may lead to a reduction in preload and afterload, thereby alleviating cardiac workload and improving left ventricular function.[21]
Selectivity
The IC50 for SGLT2 is less than one thousandth of the IC50 for SGLT1 (1.1 versus 1390 nmol/L), so that the drug does not interfere with intestinal glucose absorption.[22]
Names
Dapagliflozin is the International nonproprietary name (INN),[23] and the United States Adopted Name (USAN).[24]
There is a fixed-dose combination product dapagliflozin/metformin extended-release, called Xigduo XR.[25][26][27]
In July 2016, the fixed-dose combination of saxagliptin and dapagliflozin was approved for medical use in the European Union and is sold under the brand name Qtern.[28] The combination drug was approved for medical use in the United States in February 2017, where it is sold under the brand name Qtern.[29][30]
In May 2019, the fixed-dose combination of dapagliflozin, saxagliptin, and metformin hydrochloride as extended-release tablets was approved in the United States to improve glycemic control in adults with type 2 diabetes when used in combination with diet and exercise. The FDA granted the approval of Qternmet XR to AstraZeneca.[31] The combination drug was approved for use in the European Union in November 2019, and is sold under the brand name Qtrilmet.[32]
History
In 2012, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) issued a positive opinion on the drug.[5]
Dapagliflozin was found effective in several studies in participants with type 2 and type 1 diabetes.[5] The main measure of effectiveness was the level of glycosylated haemoglobin (HbA1c), which gives an indication of how well blood glucose is controlled.[5]
In two studies involving 840 participants with type 2 diabetes, dapagliflozin when used alone decreased HbA1c levels by 0.66 percentage points more than placebo (a dummy treatment) after 24 weeks.[5] In four other studies involving 2,370 participants, adding dapagliflozin to other diabetes medicines decreased HbA1c levels by 0.54-0.68 percentage points more than adding placebo after 24 weeks.[5]
In a study involving 814 participants with type 2 diabetes, dapagliflozin used in combination with metformin was at least as effective as a sulphonylurea (another type of diabetes medicines) used with metformin.[5] Both combinations reduced HbA1c levels by 0.52 percentage points after 52 weeks.[5]
A long-term study, involving over 17,000 participants with type 2 diabetes, looked at the effects of dapagliflozin on cardiovascular (heart and circulation) disease.[5] The study indicated that dapagliflozin’s effects were in line with those of other diabetes medicines that also work by blocking SGLT2.[5]
In two studies involving 1,648 participants with type 1 diabetes whose blood sugar was not controlled well enough on insulin alone, adding dapagliflozin 5 mg decreased HbA1c levels after 24 hours by 0.37% and by 0.42% more than adding placebo.[5]
Dapagliflozin was approved for medical use in the European Union in November 2012.[5] It is marketed in a number of European countries.[33]
Dapagliflozin was approved for medical use in the United States in January 2014.[34][14]
In 2020, the U.S. Food and Drug Administration (FDA) expanded the indications for dapagliflozin to include treatment for adults with heart failure with reduced ejection fraction to reduce the risk of cardiovascular death and hospitalization for heart failure.[3] It is the first in this particular drug class, sodium-glucose co-transporter 2 (SGLT2) inhibitors, to be approved to treat adults with New York Heart Association’s functional class II-IV heart failure with reduced ejection fraction.[3]
Dapagliflozin was shown in a clinical trial to improve survival and reduce the need for hospitalization in adults with heart failure with reduced ejection fraction.[3] The safety and effectiveness of dapagliflozin were evaluated in a randomized, double-blind, placebo-controlled study of 4,744 participants.[3] The average age of participants was 66 years and more participants were male (77%) than female.[3] To determine the drug’s effectiveness, investigators examined the occurrence of cardiovascular death, hospitalization for heart failure, and urgent heart failure visits.[3] Participants were randomly assigned to receive a once-daily dose of either 10 milligrams of dapagliflozin or a placebo (inactive treatment).[3] After about 18 months, people who received dapagliflozin had fewer cardiovascular deaths, hospitalizations for heart failure, and urgent heart failure visits than those receiving the placebo.[3]
In July 2020, the FDA granted AstraZeneca a Fast Track Designation in the US for the development of dapagliflozin to reduce the risk of hospitalisation for heart failure or cardiovascular death in adults following a heart attack.[35]
In August 2020, it was reported that detailed results from the Phase III DAPA-CKD trial showed that AstraZeneca’s FARXIGA® (dapagliflozin) on top of standard of care reduced the composite measure of worsening of renal function or risk of cardiovascular (CV) or renal death by 39% compared to placebo (p<0.0001) in patients with chronic kidney disease (CKD) Stages 2-4 and elevated urinary albumin excretion. The results were consistent in patients both with and without type 2 diabetes (T2D)[36]
In April 2021, the FDA expanded the indications for dapagliflozin (Farxiga) to include reducing the risk of kidney function decline, kidney failure, cardiovascular death and hospitalization for heart failure in adults with chronic kidney disease who are at risk of disease progression.[14] The efficacy of dapagliflozin to improve kidney outcomes and reduce cardiovascular death in people with chronic kidney disease was evaluated in a multicenter, double-blind study of 4,304 participants.[14]
///////////////////////////////////////////
POST YOUR INTERMEDIATES LIST HERE FOR WORLDWIDE REACH
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYN
https://pharmacia.pensoft.net/article/70626/
Synthesis
Dapagliflozin is an approved drug by U.S. Food and Drug Administration (FDA). Dapagliflozin is a representative of SGLT-2 inhibitors, actively considered to cure diabetes type 2. Thus, methodology of dapagliflozin synthesis has rarely published (Ellsworth et al. 2002; Meng 2008). Scheme 1 have shown the general synthetic route for the synthesis of dapagliflozin. Gluconolactone 3 which was protected by trimethylsilyl TMS was treated with aryl lithium. Aryl lithium was obtained by reacting aryl bromide 2 (exchange of Li/Br) with n-BuLi. Methyl C-aryl glucoside 4 was produced by treatment of resulting mixture with methane sulfonic acid in the presence of methanol. Compound 4 was subjected to acetylation in the presence of Ac2O, resulted in the formation of 5 followed by reduction of 5 to 6 with the help of Et3SiH and BF3.OEt2. Finally, dapagliflozin 1 was produced via hydrolysis of 6 by LiOH (Deshpande et al. 2008; Meng 2008).
Ellsworth B, Washburn WN, Sher PM, Wu G, Meng W (2002) C-Aryl glucoside SGLT2 inhibitors and method, Google Patents. https://patents.google.com/patent/US6515117B2/en
Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN (2008) “Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. ” Journal of Medicinal Chemistry 51(5): 1145–1149. https://doi.org/10.1021/jm701272q
Deshpande PP, Ellsworth BA, Singh J, Denzel TW, Lai C, Crispino G, Randazzo ME, Gougoutas JZ (2008) Methods of producing C-aryl glucoside SGLT2 inhibitors, Google Patents. https://patents.google.com/patent/US7375213B2/en

Jun et al. has reported a few improvements to the scheme 1. In the improved methodology scheme 2, trimethylsilyl chloride was added to gluconolactone 7 in the presence of N-methylmorpholine and tetrahydrofuran THF (Horton et al. 1981), followed by the formation of persilyated lactone 3. After completing the reaction of aryl bromide 2 with n-BuLi, added to persilyated lactone 3. Intermediate lactol 8 was produced by treating resulting reaction mixture with trifluoroacetic acid in aqueous form. Then ethyl C-aryl glycoside 9 yielded when subsequently compound 8 was subjected to methane-sulfonic acid in ethyl alcohol. Crude product 9 in the form of oil was secured after the screening of solvents. Jun et al. proposed that more than 98% pure 9 was collected as crystalline solvate after the crystallization of crude oil from n-propanol and n-heptane mixture (Yu et al. 2019). Moreover, Wang et al. proposed that a high extent of diastereoselectivity obtained after the reduction of tetra-O-unprotected methyl C-aryl glucoside by utilizing Et3SiH and BF3.Et2O (Wang et al. 2014). The nature of active pharmaceutical ingredient is amorphous foam which is isolated after the reduction of 9. Production of cocrystalline complex facilitate the isolation and purification of API (Deng et al. 2017). It is concluded that more than 99.7% pure dapagliflozin produced in overall 79% yield, after the crystallization of a mixture consists of n-heptane and ethyl acetate (Yu et al. 2019).
Zheng et al. designed the production methodology of dapagliflozin by introducing NO donor group at the last steps of general route of dapagliflozin synthesis (scheme 1). Novel hybrids achieved by the combination of dapagliflozin and NO donor, having excellent dual characteristics of anti-hyperglycemic and anti-thrombosis. The figure 2 represent the modifiable site (4-position) of dapagliflozin.
Horton D, Priebe W (1981) “Synthetic routes to higher-carbon sugars. Reaction of lactones with 2-lithio-, 3-dithiane. ” Carbohydrate Research 94(1): 27–41. https://doi.org/10.1016/S0008-6215(00)85593-7
Yu J, Cao Y, Yu H, Wang JJ (2019) “A Concise and Efficient Synthesis of Dapagliflozin. ” Organic Process Research & Development 23(7): 1458–1461. https://doi.org/10.1021/acs.oprd.9b00141
Wang X-j, Zhang L, Byrne D, Nummy L, Weber D, Krishnamurthy D, Yee N, Senanayake CH (2014) “Efficient synthesis of empagliflozin, an inhibitor of SGLT-2, utilizing an AlCl3-promoted silane reduction of a β-glycopyranoside. ” Organic Letters 16(16): 4090–4093. https://doi.org/10.1021/ol501755h
Deng J-H, Lu T-B, Sun CC, J-M Chen (2017) “Dapagliflozin-citric acid cocrystal showing better solid state properties than dapagliflozin. ” European Journal of Pharmaceutical Sciences 104: 255–261. https://doi.org/10.1016/j.ejps.2017.04.008

Scheme 3 has shown the formation strategy of new hybrids of nitric oxide with dapagliflozin. During the synthesis process, the compound 6 was treated with BBr3 that result in the formation of phenol 10, which was further subjected to condensation with bromoalkane and then undergo hydrolysis and produce 11 intermediates. Target compound was obtained by the reacting 11 with silver nitrate in acetonitrile (Li et al. 2018).
Li Z, Xu X, Deng L, Liao R, Liang R, Zhang B, Zhang LJB (2018) Design, synthesis and biological evaluation of nitric oxide releasing derivatives of dapagliflozin as potential anti-diabetic and anti-thrombotic agents. Bioorganic & Medicinal Chemistry 26(14): 3947–3952. https://doi.org/10.1016/j.bmc.2018.06.017

Lin et al. fabricated green route (scheme 4) for the production of dapagliflozin. 5-bromo-2-chlorobenzoic acid 12 and gluconolactone were utilized to initiate the synthesis. By taking BF3.Et2O in catalytic amount to produce 13, overall yield of 76% was obtained in one-pot way via considering the Friedel-Craft acylation and ketallization. There was no need to do work-up operations to separate the diaryl ketal 13 as it was easily crystallized from the mixture. Compound 14 was produce as a result of condensation between 13 and 3. Overall yield of 68% of compound 15 was produced by the deprotection of silyl group in ethyl alcohol media. In THF presence, single crystals of 15 was achieved and characterized by XRD-analysis. High yield of dapagliflozin was obtained after the reduction of 15 that was carried out by triethylsilane in the presence of boron trifluoride diethyl etherate in dichloromethane. Upon crystallization from the mixture having heptane and ethyl acetate, greater than 98% pure dapagliflozin was produced by green synthetic pathway (Hu et al. 2019).
Hu L, Zou P, Wei W, Yuan X-M, Qiu X-L, Gou S-H (2019) “Facile and green synthesis of dapagliflozin. ” Synthetic Communications 49(23): 3373–3379. https://doi.org/10.1080/00397911.2019.1666283
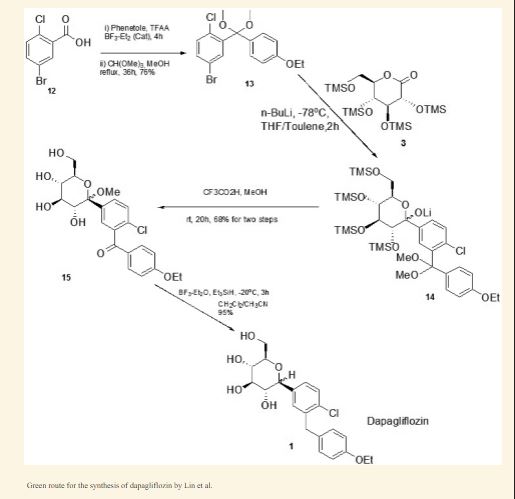
PAPER
A Concise and Efficient Synthesis of Dapagliflozin
Cite this: Org. Process Res. Dev.2019, 23, 7, 1458–1461
Publication Date:June 27, 2019
https://doi.org/10.1021/acs.oprd.9b00141
https://pubs.acs.org/doi/10.1021/acs.oprd.9b00141
file:///C:/Users/Inspiron/Downloads/op9b00141_si_001.pdf SUPP
Abstract

A concise and efficient synthesis of the SGLT-2 inhibitor dapagliflozin (1) has been developed. This route involves ethyl C-aryl glycoside 9 as the key intermediate, which is easily crystallized and purified as the crystalline n-propanol solvate with high purity (>98.5%). The tetra-O-unprotected compound 9 could be directly reduced to crude dapagliflozin with high diastereoselectivity. The final pure API product 1 was isolated and purified with high purity (>99.7%). The process has been implemented on a multikilogram scale.
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetra-hydro-2Hpyran-3,4,5-triol (1)
Compound 1 has a melting point of 88.3oC.
The MsOH solution should be used immediately to avoid sulfonate formation and we have no data on the presence of sulfonates in the API.
1HNMR (400 MHz, DMSO-d6) δ (ppm) 7.32-7.37 (m, 2H), 7.22-7.24 (m, 1H), 7.08-7.10 (m, 2H), 6.80-6.84 (m, 2H), 4.94-4.96 (m, 2H), 4.82-4.83 (d, J=5.6 Hz, 1H), 4.43-4.46 (m, 1H), 3.93-4.02 (m, 5H), 3.68-3.73 (m, 1H), 3.42-3.48 (m, 1H), 3.10-3.28 (m, 4H), 1.27-1.30 (m, 3H).
13C NMR (100 MHz, DMSOd6) δ(ppm) 157.38, 140.14, 138.27, 132.40, 131.69, 131.27, 130.03, 129.13, 127.81, 114.78, 81.67, 81.18, 78.80, 75.19, 70.80, 63.37, 61.86, 38.14, 15.15.
IR(KBr): 3415, 2979, 2918, 1617, 1512, 1475, 1391, 1242, 1103, 1045, 913, 825 cm-1.
MS(m/z):431.12[M + Na]+ .
1H NMR and 13C NMR spectra for Compound
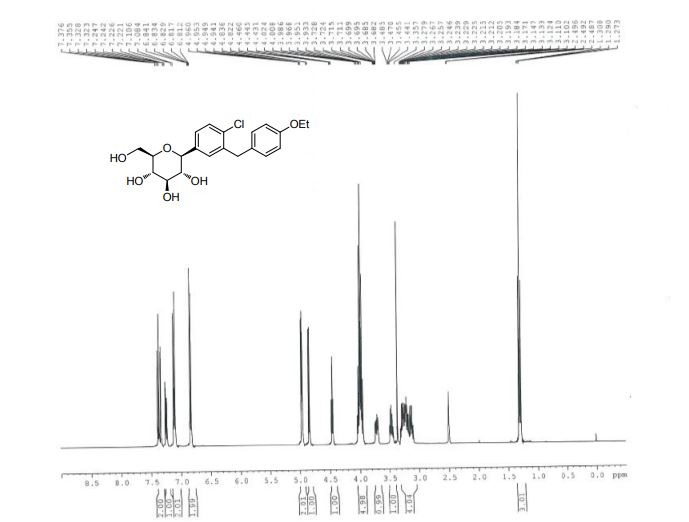
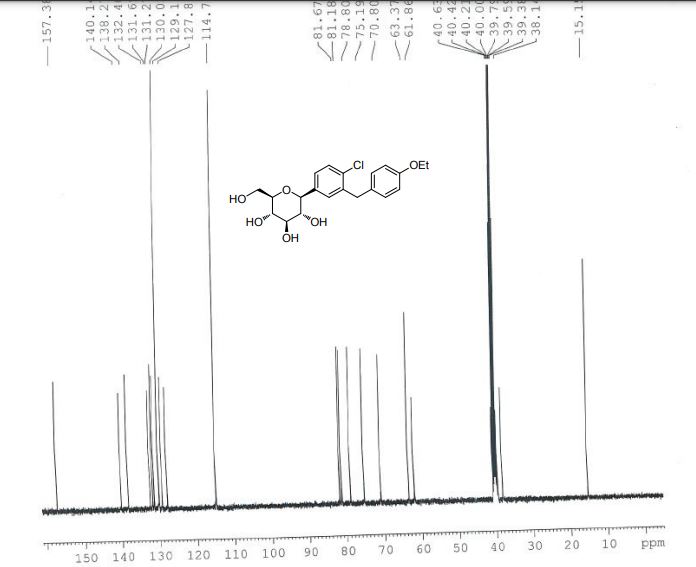
IR and Mass spectra for Compound
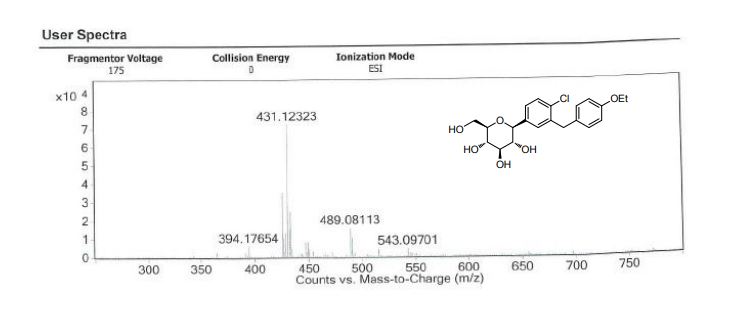
DSC spectra for Compound
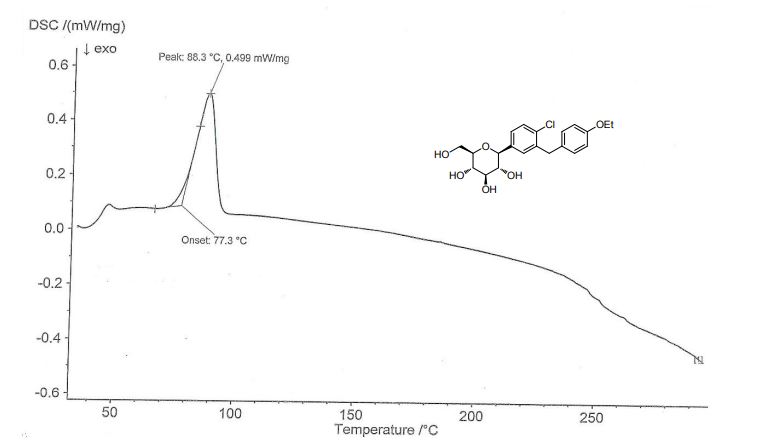
PAPER
https://pubs.acs.org/doi/10.1021/ol300220p
rg. Lett.2012, 14, 6, 1480–1483
Publication Date:March 2, 2012
https://doi.org/10.1021/ol300220p
Abstract

A general, transition-metal-free, highly stereoselective cross-coupling reaction between glycosyl bromides and various arylzinc reagents leading to β-arylated glycosides is reported. The stereoselectivity of the reaction is explained by invoking anchimeric assistance via a bicyclic intermediate. Stereochemical probes confirm the participation of the 2-pivaloyloxy group. Finally, this new method was applied to a short and efficient stereoselective synthesis of Dapagliflozin and Canagliflozin.
CROSS REF J. Med. Chem 2008, 51, 1145
H NMR (360 MHz, MeOD): δ 7.35‐7.28 (m, 3H); 7.09 (d, J=8.4Hz, 2H), 6.80 (d, J=8.8Hz, 2H), 4.09 (d, J=9.5Hz, 1H); 4.03‐3.96 (m, 4H); 3.89‐3.85 (m, 1H); 3.71‐3.66 (m, 1H); 3.45‐3.36 (m, 4H), 3.28‐3.26 (m, 2H); 1.36 (t, J=6.9Hz, 3H).
13CNMR (90 MHz, MeOD): δ 14.80, 38.31, 61.80, 63.38, 69.88, 74.67, 77.93, 79.35, 81.08, 114.48, 126.35, 128.20, 129.01, 129.72, 130.62, 131.14, 134.15, 137.04, 139.01, 157.31.
PAPER
https://pubs.acs.org/doi/10.1021/ja00199a028
Research
One study found that it had no benefit on heart disease risk or overall risk of death in people with diabetes.[37] Another study found that in heart failure with a reduced ejection fraction, dapagliflozin reduced the risk of worsening of heart failure or progression to death from cardiovascular causes, irrespective of diabetic status.[38]
References
- ^ Jump up to:a b “Dapagliflozin (Farxiga) Use During Pregnancy”. Drugs.com. 30 August 2018. Retrieved 5 May 2020.
- ^ Jump up to:a b “Farxiga- dapagliflozin tablet, film coated”. DailyMed. 3 February 2020. Retrieved 5 May 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o “FDA approves new treatment for a type of heart failure”. U.S. Food and Drug Administration (FDA) (Press release). 5 May 2020. Retrieved 5 May 2020.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain. - ^ Jump up to:a b National Institute for Health and Care Excellence (24 February 2021). “Dapagliflozin for treating chronic heart failure with reduced ejection fraction”. NICE Technology Appraisal Auidance [TA679]. NICE. Retrieved 9 May 2021.
- ^ Jump up to:a b c d e f g h i j k l m n “Forxiga EPAR”. European Medicines Agency (EMA). Retrieved 17 February 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Ptaszynska, Agata; Johnsson, Kristina M.; Parikh, Shamik J.; De Bruin, Tjerk W. A.; Apanovitch, Anne Marie; List, James F. (2014). “Safety Profile of Dapagliflozin for Type 2 Diabetes: Pooled Analysis of Clinical Studies for Overall Safety and Rare Events”. Drug Safety. 37 (10): 815–829. doi:10.1007/s40264-014-0213-4. PMID 25096959. S2CID 24064402.
- ^ Dandona, Paresh; Mathieu, Chantal; Phillip, Moshe; Hansen, Lars; Tschöpe, Diethelm; Thorén, Fredrik; Xu, John; Langkilde, Anna Maria; DEPICT-1 Investigators (2018). “Efficacy and Safety of Dapagliflozin in Patients with Inadequately Controlled Type 1 Diabetes: The DEPICT-1 52-Week Study”. Diabetes Care. 41(12): 2552–2559. doi:10.2337/dc18-1087. PMID 30352894. S2CID 53027785.
- ^ Hu, Yang; Bai, Ziyu; Tang, Yan; Liu, Rongji; Zhao, Bin; Gong, Jian; Mei, Dan (2020). “Fournier Gangrene Associated with Sodium-Glucose Cotransporter-2 Inhibitors: A Pharmacovigilance Study with Data from the U.S. FDA Adverse Event Reporting System”. Journal of Diabetes Research. 2020: 1–8. doi:10.1155/2020/3695101. PMC 7368210. PMID 32695827.
- ^ FARXIGA- dapagliflozin tablet, film coated. DailyMed. Retrieved 6 May 2021.
- ^ “The Top 300 of 2021”. ClinCalc. Retrieved 18 February 2021.
- ^ “Dapagliflozin – Drug Usage Statistics”. ClinCalc. Retrieved 18 February 2021.
- ^ “FDA Approves Farxiga to Treat Type 2 Diabetes” (Press release). U.S. Food and Drug Administration (FDA). 8 January 2014. Archived from the original on 9 January 2014. Retrieved 15 November 2016. This article incorporates text from this source, which is in the public domain.
- ^ Zelniker TA, Wiviott SD, Raz I, et al. (January 2019). “SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials”. Lancet. 393(10166): 31–9. doi:10.1016/S0140-6736(18)32590-X. PMID 30424892. S2CID 53277899.
However, in patients with atherosclerotic cardiovascular disease, the effect of empagliflozin on cardiovascular death was more pro-nounced than that of canagliflozin or dapagliflozin
- ^ Jump up to:a b c d “FDA Approves Treatment for Chronic Kidney Disease”. U.S. Food and Drug Administration (FDA) (Press release). 30 April 2021. Retrieved 30 April 2021. This article incorporates text from this source, which is in the public domain.
- ^ “FDA warns about rare occurrences of a serious infection of the genital area with SGLT2 inhibitors for diabetes”. U.S. Food and Drug Administration (FDA). 9 February 2019. This article incorporates text from this source, which is in the public domain.
- ^ “SGLT2 inhibitors: Drug Safety Communication – FDA Warns Medicines May Result in a Serious Condition of Too Much Acid in the Blood”. U.S. Food and Drug Administration (FDA). 15 May 2015. Archived from the original on 27 October 2016. Retrieved 15 November 2016. This article incorporates text from this source, which is in the public domain.
- ^ Jump up to:a b “FDA revises labels of SGLT2 inhibitors for diabetes to include warning”. U.S. Food and Drug Administration. 19 March 2020. Retrieved 6 June 2020. This article incorporates text from this source, which is in the public domain.
- ^ McMurray, John J.V.; Wheeler, David C.; Stefánsson, Bergur V.; Jongs, Niels; Postmus, Douwe; Correa-Rotter, Ricardo; Chertow, Glenn M.; Greene, Tom; Held, Claes; Hou, Fan-Fan; Mann, Johannes F.E.; Rossing, Peter; Sjöström, C. David; Toto, Roberto D.; Langkilde, Anna Maria; Heerspink, Hiddo J.L.; DAPA-CKD Trial Committees Investigators (2021). “Effect of Dapagliflozin on Clinical Outcomes in Patients with Chronic Kidney Disease, with and Without Cardiovascular Disease” (PDF). Circulation. 143 (5): 438–448. doi:10.1161/CIRCULATIONAHA.120.051675. PMID 33186054. S2CID 226948086.
- ^ “Life Sciences – Clarivate”. Clarivate. Archived from the original on 5 November 2007.
- ^ “UEndocrine: Internet Endocrinology Community”. uendocrine.com. Archived from the original on 5 February 2013.
- ^ Lan NS, Fegan PG, Yeap BB, Dwivedi G (October 2019). “The effects of sodium-glucose cotransporter 2 inhibitors on left ventricular function: current evidence and future directions”. ESC Heart Fail. 6 (5): 927–935. doi:10.1002/ehf2.12505. PMC 6816235. PMID 31400090.
- ^ Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- ^ “International Nonproprietary Names for Pharmaceutical Substances (INN). Recommended International Nonproprietary Names: List 59” (PDF). World Health Organization. 2008. p. 50. Retrieved 15 November 2016.
- ^ “Statement on a Nonproprietary Name Adopted by the USAN Council” (PDF). American Medical Association. Archived from the original (PDF) on 7 February 2012. Retrieved 15 November2016.
- ^ “US FDA Approves Once-Daily Xigduo XR Tablets for Adults with Type 2 Diabetes”. AstraZeneca. 30 October 2014.
- ^ “Drug Approval Package: Xigduo XR (dapagliflozin and metformin HCl) Extended-Release Tablets”. U.S. Food and Drug Administration (FDA). 7 April 2015. Retrieved 5 May 2020.
- ^ “Xigduo XR- dapagliflozin and metformin hydrochloride tablet, film coated, extended release”. DailyMed. 3 February 2020. Retrieved 5 May 2020.
- ^ “Qtern EPAR”. European Medicines Agency (EMA). Retrieved 7 May 2020.
- ^ “Drug Approval Package: Qtern (dapagliflozin and saxagliptin)”. U.S. Food and Drug Administration (FDA). 10 October 2018. Retrieved 8 May 2020.
- ^ “Qtern- dapagliflozin and saxagliptin tablet, film coated”. DailyMed. 24 January 2020. Retrieved 17 February 2020.
- ^ “Drug Approval Package: Qternmet XR”. U.S. Food and Drug Administration (FDA). 27 January 2020. Retrieved 17 February2020.
- ^ “Qtrilmet EPAR”. European Medicines Agency (EMA). Retrieved 30 March 2020.
- ^ “Forxiga”. Drugs.com. 4 May 2020. Retrieved 5 May 2020.
- ^ “Drug Approval Package: Farxiga (dapagliflozin) Tablets NDA #202293”. U.S. Food and Drug Administration (FDA). 24 December 1999. Retrieved 5 May 2020.
- ^ “FARXIGA Granted Fast Track Designation in the US for Heart Failure Following Acute Myocardial Infarction Leveraging an Innovative Registry-Based Trial Design”. http://www.businesswire.com. 16 July 2020. Retrieved 20 July 2020.
- ^https://www.businesswire.com/news/home/20200830005009/en/FARXIGA-Demonstrated-Unprecedented-Reduction-Risk-Kidney-Failure
- ^ “Type 2 diabetes. Cardiovascular assessment of dapagliflozin: no advance”. Prescrire International. 29 (211): 23. January 2020. Retrieved 2 February 2020.
- ^ McMurray JJ, Solomon SD, Inzucchi SE, et al. (November 2019). “Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction”. New England Journal of Medicine. 381 (21): 1995–2008. doi:10.1056/NEJMoa1911303. PMID 31535829.
External links
- “Dapagliflozin”. Drug Information Portal. U.S. National Library of Medicine.
- “Dapagliflozin mixture with metformin hydrochloride”. Drug Information Portal. U.S. National Library of Medicine.
- “Dapagliflozin mixture with saxagliptin”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trials
- Clinical trial number NCT00528372 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled With Diet and Exercise” at ClinicalTrials.gov
- Clinical trial number NCT00643851 for “An Efficacy & Safety Study of BMS-512148 in Combination With Metformin Extended Release Tablets” at ClinicalTrials.gov
- Clinical trial number NCT00859898 for “Study of Dapagliflozin in Combination With Metformin XR to Initiate the Treatment of Type 2 Diabetes” at ClinicalTrials.gov
- Clinical trial number NCT00528879 for “A Phase III Study of BMS-512148 (Dapagliflozin) in Patients With Type 2 Diabetes Who Are Not Well Controlled on Metformin Alone” at ClinicalTrials.gov
- Clinical trial number NCT00660907 for “Efficacy and Safety of Dapagliflozin in Combination With Metformin in Type 2 Diabetes Patients” at ClinicalTrials.gov
- Clinical trial number NCT00680745 for “Efficacy and Safety of Dapagliflozin in Combination With Glimepiride (a Sulphonylurea) in Type 2 Diabetes Patients” at ClinicalTrials.gov
- Clinical trial number NCT01392677 for “Evaluation of Safety and Efficacy of Dapagliflozin in Subjects With Type 2 Diabetes Who Have Inadequate Glycaemic Control on Background Combination of Metformin and Sulfonylurea” at ClinicalTrials.gov
- Clinical trial number NCT00683878 for “Add-on to Thiazolidinedione (TZD) Failures” at ClinicalTrials.gov
- Clinical trial number NCT00984867 for “Dapagliflozin DPPIV Inhibitor add-on Study” at ClinicalTrials.gov
- Clinical trial number NCT00673231 for “Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin” at ClinicalTrials.gov
- Clinical trial number NCT02229396 for “Phase 3 28-Week Study With 24-Week and 52-week Extension Phases to Evaluate Efficacy and Safety of Exenatide Once Weekly and Dapagliflozin Versus Exenatide and Dapagliflozin Matching Placebo” at ClinicalTrials.gov
- Clinical trial number NCT02413398 for “A Study to Evaluate the Effect of Dapagliflozin on Blood Glucose Level and Renal Safety in Patients With Type 2 Diabetes (DERIVE)” at ClinicalTrials.gov
- Clinical trial number NCT01730534 for “Multicenter Trial to Evaluate the Effect of Dapagliflozin on the Incidence of Cardiovascular Events (DECLARE-TIMI58)” at ClinicalTrials.gov
- Clinical trial number NCT03036124 for “Study to Evaluate the Effect of Dapagliflozin on the Incidence of Worsening Heart Failure or Cardiovascular Death in Patients With Chronic Heart Failure (DAPA-HF)” at ClinicalTrials.gov
///////////DAPAGLIFLOZIN, ダパグリフロジン, BMS 512148, TYPE 2 DIABETES, SGLT-2 Inhibitors, EU 2012, forxiga, FDA 2014, JAPAN 2014, DIABETES
- Statement on a nonproprietory name adopted by the USAN council
- Efficacy and Safety of Dapagliflozin, Added to Therapy of Patients With Type 2 Diabetes With Inadequate Glycemic Control on Insulin, ClinicalTrials.gov, April 2009
- Trial Details for Trial MB102-020, Bristol-Myers Squibb, May 2009
- “FDA panel advises against approval of dapagliflozin”. 19 July 2011.
- Prous Science: Molecule of the Month November 2007
- UEndocrine: Internet Endocrinology Community
- Schubert-Zsilavecz, M, Wurglics, M, Neue Arzneimittel 2008/2009
- more1) Pal, Manojit et al; Improved Process for the preparation of SGLT2 inhibitor dapagliflozin via glycosylation of 5-bromo-2-Chloro-4′-ethoxydiphenylmethane with Gluconolactone ;. Indian Pat Appl,. 2010CH03942 , 19 Oct 20122) Lemaire, Sebastien et al; Stereoselective C-Glycosylation Reactions with Arylzinc Reagents ;
- Organic Letters , 2012, 14 (6), 1480-1483;3) Zhuo, Biqin and Xing, Xijuan; Process for preparation of Dapagliflozin amino acid cocrystals ;
- Faming Zhuanli Shenqing , 102 167 715, 31 Aug 20114) Shao, Hua et al; Total synthesis of SGLT2 inhibitor Dapagliflozin ;
- Hecheng Huaxue , 18 (3), 389-392; 20105) Liou, Jason et al; Processes for the preparation of C-Aryl glycoside amino acid complexes as potential SGLT2 Inhibitors ;. PCT Int Appl,.
- WO20100223136) Seed, Brian et al; Preparation of Deuterated benzyl-benzene glycosides having an inhibitory Effect on sodium-dependent glucose co-transporter; . PCT Int Appl,.
- WO20100092437) Song, Yanli et al; Preparation of benzylbenzene glycoside Derivatives as antidiabetic Agents ;. PCT Int Appl,.
- WO20090265378) Meng, Wei et al; D iscovery of Dapagliflozin: A Potent, Selective Renal Sodium-Dependent Glucose cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes ;
- Journal of Medicinal chemistr y, 2008, 51 (5), 1145 -1149;9) Gougoutas, Jack Z. et al; Solvates Crystalline complexes of amino acid with (1S)-1 ,5-anhydro-LC (3 – ((phenyl) methyl) phenyl)-D-glucitol were prepared as for SGLT2 Inhibitors the treatment of Diabetes ;. PCT Int Appl,.
- WO200800282410) Deshpande, Prashant P. et al; Methods of producing C-Aryl glucoside SGLT2 Inhibitors ;..
- U.S. Pat Appl Publ,. 20,040,138,439
NEW DRUG APPROVALS
one time
$10.00
Dasiglucagon




Dasiglucagon
Treatment of Hypoglycemia in Type 1 and Type 2 Diabetes Patients
| Formula | C152H222N38O50 |
|---|---|
| CAS | 1544300-84-6 |
| Mol weight | 3381.6137 |
FDA APPROVED, 2021/3/22, Zegalogue
Zealand Pharma A/S
UNIIAD4J2O47FQ
HypoPal rescue pen
(4S)-4-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-6-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S,3R)-2-[[2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-amino-3-(1H-imidazol-4-yl)propanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]acetyl]amino]-3-hydroxybutanoyl]amino]-3-phenylpropanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-3-hydroxypropanoyl]amino]hexanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-methylpentanoyl]amino]-3-carboxypropanoyl]amino]-2-methylpropanoyl]amino]propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]amino]-5-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S)-6-amino-1-[[(2S)-1-[[(2S)-1-[[(2S)-4-carboxy-1-[[(2S)-1-[[(1S,2R)-1-carboxy-2-hydroxypropyl]amino]-3-hydroxy-1-oxopropan-2-yl]amino]-1-oxobutan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-(1H-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxohexan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-4-carboxy-1-oxobutan-2-yl]amino]-5-oxopentanoic acid
. [16-(2-methylalanine)(S>X),17-L-alanine(R>A),20-L-α-glutamyl(Q>E),21-L-αglutamyl(D>E),24-L-lysyl(Q>K),27-L-α-glutamyl(M>E),28-L-serine(N>S)]human glucagon
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L- phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L- lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L- arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L- valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl
L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-alpha-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-alpha-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-alpha-glutamyl-L-alphaC152 H222 N38 O50L-Threonine, L-histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-Molecular Weight3381.61
Other Names
- L-Histidyl-L-seryl-L-glutaminylglycyl-L-threonyl-L-phenylalanyl-L-threonyl-L-seryl-L-α-aspartyl-L-tyrosyl-L-seryl-L-lysyl-L-tyrosyl-L-leucyl-L-α-aspartyl-2-methylalanyl-L-alanyl-L-arginyl-L-alanyl-L-α-glutamyl-L-α-glutamyl-L-phenylalanyl-L-valyl-L-lysyl-L-tryptophyl-L-leucyl-L-α-glutamyl-L-seryl-L-threonine
- Developer Beta Bionics; Zealand Pharma
- ClassAntihyperglycaemics; Antihypoglycaemics; Peptides
- Mechanism of ActionGlucagon receptor agonists
- Orphan Drug StatusYes – Hypoglycaemia; Congenital hyperinsulinism
- RegisteredHypoglycaemia
- Phase IIICongenital hyperinsulinism
- Phase II/IIIType 1 diabetes mellitus
- 22 Mar 2021Registered for Hypoglycaemia (In children, In adolescents, In adults, In the elderly) in USA (SC) – First global approval
- 22 Mar 2021Zealand Pharma anticipates the launch of dasiglucagon in USA (SC, Injection) in June 2021
- 22 Mar 2021Pooled efficacy and safety data from three phase III trials in Hypoglycaemia released by Zealand Pharma
NEW DRUG APPROVALS
one time
$10.00
PATENTS
WO 2014016300
US 20150210744
PAPER
Pharmaceutical Research (2018), 35(12), 1-13
Dasiglucagon, sold under the brand name Zegalogue, is a medication used to treat severe hypoglycemia in people with diabetes.[1]
The most common side effects include nausea, vomiting, headache, diarrhea, and injection site pain.[1]
Dasiglucagon was approved for medical use in the United States in March 2021.[1][2][3] It was designated an orphan drug in August 2017.[4]
Dasiglucagon is under investigation in clinical trial NCT03735225 (Evaluation of the Safety, Tolerability and Bioavailability of Dasiglucagon Following Subcutaneous (SC) Compared to IV Administration).
Medical uses
Dasiglucagon is indicated for the treatment of severe hypoglycemia in people aged six years of age and older with diabetes.[1][2]
Contraindications
Dasiglucagon is contraindicated in people with pheochromocytoma or insulinoma.[1]
References
- ^ Jump up to:a b c d e f https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214231s000lbl.pdf
- ^ Jump up to:a b “Dasiglucagon: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 22 March 2021.
- ^ “Zealand Pharma Announces FDA Approval of Zegalogue (dasiglucagon) injection, for the Treatment of Severe Hypoglycemia in People with Diabetes” (Press release). Zealand Pharma. 22 March 2021. Retrieved 22 March 2021 – via GlobeNewswire.
- ^ “Dasiglucagon Orphan Drug Designations and Approvals”. U.S. Food and Drug Administration (FDA). 10 August 2017. Retrieved 22 March 2021.
External links
- “Dasiglucagon”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03378635 for “A Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Type 1 Diabetes Subjects” at ClinicalTrials.gov
- Clinical trial number NCT03688711 for “Trial to Confirm the Clinical Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in Subjects With T1DM” at ClinicalTrials.gov
- Clinical trial number NCT03667053 for “Trial to Confirm the Efficacy and Safety of Dasiglucagon in the Treatment of Hypoglycemia in T1DM Children” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Zegalogue |
| AHFS/Drugs.com | Zegalogue |
| License data | US DailyMed: Dasiglucagon |
| Routes of administration | Subcutaneous |
| Drug class | Glucagon receptor agonist |
| ATC code | None |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1544300-84-6 |
| PubChem CID | 126961379 |
| DrugBank | DB15226 |
| UNII | AD4J2O47FQ |
| KEGG | D11359 |
| Chemical and physical data | |
| Formula | C152H222N38O50 |
| Molar mass | 3381.664 g·mol−1 |
| 3D model (JSmol) | Interactive image |
///////////Dasiglucagon, FDA 2021, APPROVALS 2021, Zegalogue, ダシグルカゴン, ZP 4207, ZP-GA-1, Hypoglycemia, Type 1, Type 2 , Diabetes Patients, Zealand Pharma A/S, Orphan Drug Status, Hypoglycaemia, Congenital hyperinsulinism, HypoPal rescue pen, DIABETES
#Dasiglucagon, #FDA 2021, #APPROVALS 2021, #Zegalogue, #ダシグルカゴン, #ZP 4207, ZP-GA-1, #Hypoglycemia, #Type 1, #Type 2 , #Diabetes Patients, #Zealand Pharma A/S, #Orphan Drug Status, #Hypoglycaemia, #Congenital hyperinsulinism, #HypoPal rescue pen, #DIABETESSMILES
- C[C@H]([C@@H](C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CC2=CC=C(C=C2)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC3=CC=C(C=C3)O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(=O)O)C(=O)NC(C)(C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(=N)N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CC4=CC=CC=C4)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC5=CNC6=CC=CC=C65)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)O)NC(=O)CNC(=O)[C@H](CCC(=O)N)NC(=O)[C@H](CO)NC(=O)[C@H](CC7=CNC=N7)N)O

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}