



C=CC(=O)NC1=C(C=C2C(=C1)C(=NC=N2)NC3=CC(=C(C=C3)F)Cl)OCCCN4CCOCC4
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Nilotinib ニロチニブ;
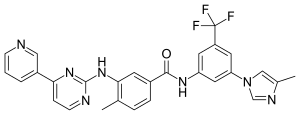
NILOTINIB
|
ニロチニブ;
|
- Molecular FormulaC28H22F3N7O
- Average mass529.516 Da
4-Methyl-N-(3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-(pyridin-3-yl)pyrimidin-2-yl)amino)benzamide
4-Methyl-N-(3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)benzamide
4-Methyl-N-(3-(4-methylimidazol-1-yl)-5-(trifluoromethyl)phenyl)-3-((4-pyridin-3-ylpyrimidin-2-yl)amino)benzamide
4-Methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluormethyl)phenyl]-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}benzolcarboxamid
4-Methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-{[4-(3-pyridinyl)-2-pyrimidinyl]amino}benzamide
4-Methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-{[4-(pyridin-3-yl)pyrimidin-2-yl]amino}benzamide
641571-10-0 [RN]
8654
Benzamide, 4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]-
Nilotinib (AMN107, trade name Tasigna[2]), in the form of the hydrochloride monohydrate salt, is a small-molecule tyrosine kinase inhibitor approved for the treatment of imatinib-resistant chronic myelogenous leukemia.[3] Structurally related to imatinib,[4] it was developed based on the structure of the Abl-imatinib complex to address imatinib intolerance and resistance.[5][6][7] Nilotinib is a selective Bcr-Abl tyrosine kinase inhibitor[5][6] that is 10–30 fold more potent than imatinib in inhibiting Bcr-Abl tyrosine kinase activity and proliferation of Bcr-Abl expressing cells.[4][6][7][8] Nilotinib was developed by Novartis and is sold under the trade name Tasigna.[9]
Medical uses

Crystal structure of Abl kinase domain(blue) in complex with nilotinib (red)
It is FDA– (29 October 2007),[10] EMA– (29 September 2009),[11] MHRA– (19 November 2007)[12] and TGA– (17 January 2008)[13] approved for use as a treatment for Philadelphia chromosome (Ph+)-positive chronic myelogenous leukaemia.[1]
The drug carries a black box warning for possible heart complications.[14][15]
Clinical trials
CML
In June 2006, a phase I clinical trial found nilotinib has a relatively favorable safety profile and shows activity in cases of CML resistant to treatment with imatinib, another tyrosine kinase inhibitor currently used as a first-line treatment.[16] In that study 92% of patients (already resistant or unresponsive to imatinib) achieved normal white blood cell counts after five months of treatment.[17]
Contraindications
Contraindications include long QT syndrome, hypokalaemia, hypomagnesaemia, pregnancy, planned pregnancy, lactation and galactose/lactose intolerance.[1][13]
Cautions include:[1]
- Myelosuppression
- Tumour lysis syndrome
- Liver impairment
- History of pancreatitis
- Check serum lipase periodically in order to detect pancreatitis
- Total gastrectomy
- Avoid pregnancy or impregnating women
Dose reduction of nilotinib has been recommended in hepatically impaired population which involves recommendation of lower starting dose and monitoring of any hepatic function abnormalities.[18]
Adverse effects
Nilotinib has a number of adverse effects typical of anti-cancer drugs. These include headache, fatigue, gastrointestinal problems such as nausea, vomiting, diarrhea and constipation, muscle and joint pain, rash and other skin conditions, flu-like symptoms, and reduced blood cell count. Less typical side effects are those of the cardiovascular system, such as hypertension (high blood pressure), various types of arrhythmia, and prolonged QT interval. Nilotinib can also affect the body’s electrolyte and glucosebalance.[10] Though pulmonary-related adverse effects are rare when compared with imatinib and dasatinib, there is a case report of acute respiratory failure from diffuse alveolar hemorrhage in a patient taking nilotinib.[19]
Interactions
Nilotinib has been reported as a substrate for OATP1B1 and OATP1B3. Interaction of nilotinib with OATP1B1 and OATP1B3 may alter its hepatic disposition and can lead to transporter mediated drug-drug interactions.[18] Nilotinib is an inhibitor of OATP-1B1 transporter but not for OATP-1B3.[20]
It is a substrate for CYP3A4 and hence grapefruit juice and other CYP3A4 inhibitors[21] will increase its action and inducers like St. John’s wort[22] will decrease it. Patients report that pomegranates and starfruit may also interfere.
Food should not be eaten two hours before or one hour afterwards because it unpredictably increases its bioavailability, approximately doubling it.
Pharmacology
Nilotinib inhibits the kinases BCR-ABL,[23] KIT, LCK, EPHA3, EPHA8, DDR1, DDR2, PDGFRB, MAPK11 and ZAK.[24]
Research
Parkinson’s disease
There is weak evidence that nilotinib may be beneficial with Parkinson’s Disease (PD), with a small clinical trial suggesting it might halt progression and improve symptoms.[25]However, there were significant side effects including infection, liver function tests abnormalities, hallucinations and heart attack, and the benefit in PD disappeared at follow up after drug discontinuation, raising question as to whether it was truly a disease modifying therapy. Nilotinib is currently undergoing phase II studies for treatment of Parkinson’s.[26]Scientists and medical professionals have advised caution with over-optimistic interpretation of its effects in Parkinson’s due to the significant media hype surrounding the small and early clinical trial.[27][28]
Other
Novartis announced on April 11, 2011 that it was discontinuing a phase III trial of Tasigna (nilotinib) for investigational use in the first-line treatment of gastrointestinal stromal tumor(GIST) based on the recommendation of an independent data monitoring committee. Interim results showed Tasigna is unlikely to demonstrate superiority compared to Novartis’s Gleevec (imatinib)*, the current standard of care in this setting.[29]
Low dose nilotinib is also being investigated for use for and Alzheimer’s disease, as well as for ALS, dementia and Huntington’s disease.[30]
Patent

WO 2016024289, NILOTINIB, New Patent by SUN
SUN PHARMACEUTICAL INDUSTRIES LTD [IN/IN]; 17/B, Mahal Industrial Estate, Off Mahakali Caves Road, Andheri (east), Mumbai 400093 (IN)
THENNATI, Rajamannar; (IN).
KILARU, Srinivasu; (IN).
VALANCE SURENDRAKUMAR, Macwan; (IN).
SHRIPRAKASH DHAR, Dwivedi; (IN)
The present invention provides novel salts of nilotinib and polymorphs thereof. The acid addition salts of nilotinib with benzenesulfonic acid, butanedisulfonic acid, 1-5- naphthalenedisulfonic acid, naphthalene-1-sulfonic acid and 1-hydroxynaphthoic acid; hydrates and anhydrates thereof.
Nilotinib, 4-methyl-N-[3-(4-methyl-lH-imidazol-l-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl] amino] -benzamide, having the following formula

is marketed under the name Tasigna® in US and Europe. Tasigna contains nilotinib monohydrate monohydrochloride salt and is available as capsules for the treatment of adult patients with newly diagnosed Philadelphia chromosome positive chronic myeloid leukemia (Ph+ CML) in chronic phase. Tasigna is also indicated for the treatment of chronic phase and accelerated phase Philadelphia chromosome positive chronic myelogenous leukemia (Ph+ CML) in adult patients resistant or intolerant to prior therapy that included imatinib.
Nilotinib is considered a low solubility/low permeability (class IV) compound in the Biopharmaceutics Classification System (BCS). Therefore, dissolution of nilotinib can potentially be rate limiting step for in-vivo absorption. It is soluble in acidic media; being practically insoluble in buffer solutions of pH 4.5 and higher.
WIPO publication 2014059518A1 discloses crystalline forms of nilotinib hydrochloride and methods of the preparation of various crystalline solvates of nilotinib hydrochloride including benzyl alcohol, acetic acid and propylene glycol.
WIPO publication 2011033307A1 discloses nilotinib dihydrochloride and its hydrates and method for their preparation.
WIPO publication 2011163222A1 discloses the preparation of nilotinib salts and crystalline forms thereof. The salts of nilotinib disclosed are hydrochloride, fumarate, 2-chloromandelate, succinate, adipate, L-tartrate, glutarate, p-toluenesulfonate, camphorsulfonate, glutamate, palmitate, quinate, citrate, maleate, acetate, L-malate, L-aspartate, formate, hydrobromide, oxalate and malonate.
WIPO publication number 2011086541A1 discloses a nilotinib monohydrochloride monohydrate salt and methods for preparing.
WIPO publication number 2010054056A2 describes several crystalline forms of nilotinib hydrochloride.
WIPO publication number 2007/015871A1 discloses the preparation of nilotinib salts and crystalline forms thereof. The salts are mixtures of nilotinib and one acid wherein the acids are selected from the group consisting of hydrochloric acid, phosphoric acid, sulfuric acid, sulfonic acid, methane sulfonic acid, ethane sulfonic acid, benzene sulfonic acid, p-toluene sul- fonic acid, citric acid, fumaric acid, gentisic acid, malonic acid, maleic acid, and tartaric acid.
WIPO publication number 2007015870A2 discloses several nilotinib salts including amorphous and crystalline forms of nilotinib free base, nilotinib HC1 and nilotinib sulfate along with their hydrate and solvates.
EXAMPLES:
Example 1: Preparation of nilotinib benzenesulfonate crystalline Form I
Nilotinib base (1 g) was suspended in water (20 ml). A solution of benzenesulfonic acid (0.4 g) in water (3ml) was added and the content was heated at 60 °C for 2-3 h. The mixture was cooled to 25-30 °C, filtered, washed with water (3 x 5 ml) and dried under vacuum for 2 h at 50-55 °C.
1H NMR (500 MHz, DMSO-d6) δ 2.40 (s,3H), 2.42 (s,3H), 7.35-7.37 (m,3H), 7.51-7.66 (m,5H),7.83 (d,lH), 7.96 (s,lH),8.08 (s,lH),8.30 (s,lH) 8.39 (s,lH),8.54 (d,lH), 8.61 (d,lH), 8.64 (s,lH), 8.75 (d,lH), 9.25 (s,lH), 9.34 (d,lH), 9.61 (s,lH), 10.84 (s,lH).
The salt provides an XRPD pattern substantially same as set forth in FIG. 1.
Example 2: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form II
Nilotinib base (100 g) was dissolved in 20 % water in THF solution (2000 ml) at 60-65 °C and insoluble matter was filtered. The filtrate was concentrated under vacuum below 60 °C. Filtered water (1000 ml) was added to the reaction mixture and it was heated at 50-55 °C, followed by addition of 1,4-butanedisulfonic acid -60% aqueous solution (28.6 ml) at same temperature. The content was stirred at 50-55 °C for 2-3h. Reaction mixture as cooled to 25-30 °C and product was filtered, washed with water (200 ml x 2) and dried in air oven at 50-55 °C (yield: 115 g).
Purity (by HPLC):99.76%
1H NMR (400 MHz,DMSO-d6) δ 1.63-1.66(m,2H), 2.40(d,3H),2.42(s,3H),2.43-2.47(m,2H), 7.51-7.62(m,3H),7.85(dd,lH),7.96(s,lH),8.08(s,lH),8.34(s,lH),8.38(d,lH),8.52-8.55(m,lH), 8.60-8.62 (m,2H), 8.75(d,lH), 9.25(S,1H),9.34(S,1H),9.59(S,1H),10.86(S,1H)
Water content: 7.95 %.
The salt has a XRPD pattern substantially same as set forth in FIG. 2.
Example 3: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form II
Nilotinib base (300 g) was suspended in methanol (3000 ml) and aqueous hydrochloric acid was added to get pH less than 2. Reaction contents were heated at reflux and was filtered and washed with methanol (100 ml). 5% (w/w) NaOH (1200 ml) solution was added at 40-45 °C within 15 min, reaction mixture was stirred for 2h. Product was filtered, washed with water
(300 ml x 3) and dried for lh. Wet material was suspended in water (3000 ml), heated at 50- 55 °C followed by addition of 1,4-butanedisulfonic acid -60% aqueous solution. The reaction mixture was stirred at 50-55°C for 2hrs. Product was filtered at room temperature, washed with water (500 ml x 2) and dried in air oven at 50-55 °C (yield: 293 g).
Purity (by HPLC): 99.88 %
1H NMR (400 MHz,DMSO-d6+TFA-dl) δ 1.75-1.78(m,2H), 2.36(d,3H),2.38(s,3H),2.69- 2.72(m,2H),7.45(d,lH),7.68(d,lH),7.83(s,lH),7.88(dd,lH),7.97(s,lH),8.16-8.19(m,lH), 8.35
(s,2H), 8.63(d,lH),8.68(d,lH),9.04(d,lH),9.21(d,lH),9.53(br s,lH),9.69(d,lH)10.80 (s,lH)
Water content: 6.44 %
Example 4: Preparation of nilotinib butanedisulfonate (2: 1) crystalline Form III
Nilotinib butanedisulfonate (210g) was dissolved in acetic acid water mixture (50:50) (2520 ml) at 75-80 °C and was filtered to remove insoluble matter and washed with acetic acid water mixture (50:50) (210 ml). Water (3150ml) was added to the filtrate and stirred first at room temperature and then at 0-5 °C. Product was filtered and washed with water. Material was dried in air oven at 70-75 °C. Dried material was leached with methanol (3438 ml) at reflux temperature, filtered and dried in air oven 70-75°C (yield: 152.6 g)
Purity (by HPLC): 99.89 %
1H NMR (400 MHz,DMSO-d6+TFA-dl) δ 1.73-1.77(m,2H), 2.40(s,6H),2.67-2.70(m,2H), 7.50 (d,lH), 7.70(d,lH), 7.88-7.92(m,2H), 8.07(s,lH),8.23 (dd,lH), 8.34(s,2H), 8.67 (d,lH), 8.72 (d,lH), 9.09(d,lH), 9.23 (s,lH), 9.54(d,lH), 9.74(d,lH), 10.86(s,lH).
Water content: 0.61 %
The salt provides an XRPD pattern substantially same as set forth in FIG. 3.
Example 5: Preparation of crystalline form of nilotinib butanedisulfonate (2: 1)
Crystalline Nilotinib butanedisulfonate (1 g) of Example 2 was suspended in methanol (20 ml) and was stirred at reflux for 60 min. The mixture was cooled to room temperature. Solid was filtered, washed with methanol (2 ml x 3) and dried in air oven at 70-75°C (yield: 0.8 g)
Example 6: Preparation of nilotinib butanedisulfonate (1: 1) crystalline Form IV
Nilotinib base (20 g) was suspended in methanol (800 ml) and 1,4-butanedisulfonic acid -60
% aqueous solution (6 ml) was added at 50-55 °C, and was filtered to remove insoluble matter. Filtrate was stirred at room temperature for 2-3 h. Product formed was filtered, washed with methanol (20 ml x 2) and dried the product in air oven at 70-75 °C (yield: 18.4 g).
Purity (by HPLC):99.86 %
1H NMR (400 MHz,DMSO-d6) δ 1.64-1.68(m,4H), 2.47-2.5 l(m,4H), 2.41(s,3H), 2.42(d,3H), 7.52(d,lH), 7.83-7.89(m,2H), 7.99(s,lH), 8.15(s,lH), 8.36 (d,lH), 8.39(s,lH), 8.65-8.66(m,2H), 8.79(d,lH), 8.89(br s,lH), 9.36(s,lH), 9.41(br s,lH), 9.74(d,lH), 10.91(s,lH).
The salt has XRPD pattern substantially same as set forth in FIG. 4.
Example 7: Preparation of nilotinib 1,5-napthalenedisulfonic acid salt (2: 1) crystalline Form V
Nilotinib base (1 g) was suspended in water (20 ml). A solution of 1,5-napthalenedisulfonic acid (0.4 g; 0.6 eq.) in water (5ml) was added and the content was heated at 50-55 °C for lh. The mixture was cooled to 25-30 °C, filtered and washed with water (10 ml). The product was dried in air oven at 50-55°C (yield: 1.2 g).
1H NMR (400 MHz,DMSO-d6) δ 2.39 (s,3H), 2.42 (s,3H), 7.45-7.61 (m,4H),7.84 (d,lH), 7.97(s,2H),8.08 (m,lH),8.31 (s,lH) 8.38 (s,lH),8.55 (d,lH), 8.63 (s,2H), 8.75 (s,lH), 8.92 (d,lH), 9.26 (s, 1H), 9.34 (s,lH),9.62 (s,lH), 10.85 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 5.
Example 8: Preparation of nilotinib 1,5-napthalenedisulfonic acid salt (1: 1) crystalline Form VI
Nilotinib base (1 g) was suspended in water (20 ml). A solution of 1,5-napthalenedisulfonic acid (0.8 g; 1.2eq) in water (5 ml) was added and the content was heated at 50-55 °C for 1 h. The mixture was cooled to 25-30 °C, filtered, washed with water (10 ml) and dried in air oven at 50-55 °C (yield: 1.4g).
1H NMR(400 MHz,DMSO-d6) δ 2.40 (s,3H),2.41 (s,3H), 7.43-7.52 (m,3H),7.61 (d,lH), 7.85-7.99(m,5H),8.11 (s,lH),8.34 (s,2H), 8.64-8.67 (m,2H), 8.89-8.92 (m,4H),9.40(d,2H), 9.72 (s,lH), 10.87 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 6.
Example 9: Preparation of nilotinib napthalene-1- sulfonic acid salt crystalline Form VII Nilotinib base (1 g) was suspended in water (10 ml) and heated to 50-55 °C. A solution of napthelene-1 -sulfonic acid and methanol (10 ml) was added to it and heated at 70-75 °C for 30 min. The mixture was cooled to 25-30 °C and stirred for 10 min. The product was filtered, washed with water (2 x 2 ml) and dried under vacuum for 1-2 h at 50-55 °C.
1H NMR (400 MHz,DMSO-d6) δ 2.41 (s,3H),2.42 (s,3H), 7.46-7.58 (m,5H), 7.70-8.00 (m,7H)8.11(s,lH)8.31(s,lH),8.37(s,lH),8.63-8.66 (m,3H), 8.81-8.89 (m,2H), 9.31 (s,lH), 9.37 (d,lH), 9.71 (d,lH), 10.86 (s,lH)
The salt has a XRPD pattern substantially same as set forth in FIG. 7.
Example 10: Preparation of nilotinib l-hydroxy-2-napthoic acid salt crystalline Form VIII Nilotinib base (1 g) was suspended in water (20 ml) and heated to 50-55 °C. l-Hydroxy-2-napthoic acid was added to it and the content was heated at 50-55 °C for 1 h. Methanol (5 ml) was added to the mixture and stirred for 30 min. The content was filtered, washed with water (2 x 2 ml) and dried under vacuum for 1 h at 50-55 °C.
1H NMR (400 MHz, DMSO-d6) δ 2.25 (s,3H), 2.41 (s,3H), 7.40-7.92 (m,l lH), 8.23-8.73 (m,8H), 9.24 (s,lH), 9.34(s,lH), 10.70 (s,lH).
The salt has a XRPD pattern substantially same as set forth in FIG. 8.
PATENT
https://patents.google.com/patent/WO2010009402A2/en
Nilotinib, 4-methyl-N-[3-(4-methyl-lH-imidazol-l-yl)-5-
(trifluoromethyl)phenyl] -3 – [ [4-(3 -pyridinyl)-2-pyrimidinyl] amino] -benzamide, having the following formula

is a tyrosine kinase inhibitor used for the treatment of drug-resistant chronic myelogenous leukemia (CML), and in particular, for the treatment of chronic phase and accelerated phase Philadelphia chromosome positive chronic myeloid leukemia (CML) in adult patients whose disease has progressed on or who cannot tolerate other therapies that included imatinib. Nilotinib is administered as a hydrochloride salt in forms of capsules that are marketed in the USA and the EU under the name Tasigna®.
[0004] US patent no. 7,169,791 (“US 791”) and its parallel PCT publication WO
2004/005281, the journal article in Synthesis, 2007, vol 14, pp 2121-2124, as well as PCT publication nos.: WO 2006/135640, WO 2006/135641 (“WO “641”), WO 2007/018325 and WO 2007/017734, report processes for preparing Nilotinib intermediate, 3-(trifluoromethyl)- 5-(4-methyl-lH-imidazole-l-yl)-benzeneamine of formula I

I by reacting 3-bromo-5-trifluoromethylaniline of formula II and 4-methylimidazole of formula III in the presence of a non-alkaline hydroxide inorganic base, such as potassium carbonate, cesium carbonate and sodium hydride, a copper (I) salt, such as copper iodide and a complexing amine ligand, such as ethylene diamine. The process can be illustrated by the following scheme:

Il ‘
Scheme 1
[0005] The journal article in Synthesis, 2007, VoI 14, pp 2121-2124, describes a purification process of 3-(trifluoromethyl-5-(4-methyl-lH-imidazole-l-yl)-benzeneamine of formula I.
[0006] US 791 describes processes for preparing Nilotinib and its different intermediates, using di-ethyl cyano phosphate, as described in the following scheme:

[0007] WO ‘641 further describes a process for preparing Nilotinib according to the following scheme:

Scheme 3
[0008] The present invention provides improved processes to prepare and/or purify 3-
(trifluoromethyl)-5-(4-methyl-lH-imidazole-l-yl)-benzeneamine of formula I without requiring the use of column chromatography, and thus can be easily applied to large scale manufacture, as well as new intermediates of Nilotinib, which result in higher yields in the preparation of Nilotinib.
[0009] PCT publications WO 2007/015870 (“WO ‘870”) and WO 2007/015871
(“WO ‘871”) describe several Nilotinib salts including crystalline forms of nilotinib free base, Nilotinib hydrochloride and Nilotinib Sulfate.
[0010] The present invention also relates to the solid state physical properties of
Nilotinib»3HCl, 4-methyl-N-[3-(4-methyl-lH-imidazol-l-yl)-5-(trifluoromethyl)phenyl]-3- [[4-(3-pyridinyl)-2-pyrimidinyl]amino]-benzamide trihydrochloride. These properties can be influenced by controlling the conditions under which Nilotinib-3HC1 is obtained in solid form. Solid state physical properties include, for example, the flowability of the milled solid. Flowability affects the ease with which the material is handled during processing into a pharmaceutical product. When particles of the powdered compound do not flow past each other easily, a formulation specialist must necessitate the use of glidants such as colloidal silicon dioxide, talc, starch, or tribasic calcium phosphate.
[0011 ] Another important solid state property of a pharmaceutical compound is its rate of dissolution in aqueous fluid. The rate of dissolution of an active ingredient in a patient’s stomach fluid can have therapeutic consequences since it imposes an upper limit on the rate at which an orally administered active ingredient can reach the patient’s bloodstream. The rate of dissolution is also a consideration in formulation syrups, elixirs, and other liquid medicaments. The solid state form of a compound can also affect its behavior on compaction and its storage stability. [0012] These practical physical characteristics are influenced by the conformation and orientation of molecules in the unit cell, which define a particular polymorphic form of a substance. The polymorphic form can give rise to thermal behavior different from that of the amorphous material or another polymorphic form. Thermal behavior is measured in the laboratory by such techniques as capillary melting point, thermogravimetric analysis (“TGA”), and differential scanning calorimetry (“DSC”) and can be used to distinguish some polymorphic forms from others. A particular polymorphic form can also give rise to distinct spectroscopic properties that can be detectable by powder x-ray crystallography, solid state 13C NMR spectroscopy, and infrared spectrometry.
[0013] Generally, a crystalline solid has improved chemical and physical stability over the amorphous form, and forms with low crystallinity. Crystalline forms may also exhibit improved solubility, hygroscopicity, bulk properties, and/or flowability.
[0014] The discovery of new polymorphic forms of a pharmaceutically useful compound provides a new opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for designing, for example, a pharmaceutical dosage form of a drug with a targeted release profile or other desired characteristic.
[0015] There is a need in the art for new intermediates of Nilotinib and processes for their preparation, new processes for preparing Nilotinib and new crystalline forms of Nilotinib»3HCl salt and processes for the preparation thereof.
xample 1: Preparation of 3-(ϊrifluoromethyl)-5-(4-methyl-lH-imidazole-l-yl)- benzeneamine of formula I
[00245] 200Og of 3-bromo-5- trifluoromethylaniline of formula II, 1368g of 4- methylimidazole of formula III , 181g of 8-hydroxyquinoline, 238g of CuI, 666.6g of NaOH, 933g of CaO and 7000ml of DMSO were loaded into a 1OL of 3-neck flask. The reaction mixture was protected with nitrogen and was then stirred at 12O0C for 69 hours while monitoring for the consumption of 3-bromo-5- trifluoromethy aniline by HPLC. Heating was stopped when 3-bromo-5- trifluoromethyaniline / 4-methylimidazole is not more than 5%. The reaction mixture was cooled down to 45-5O0C and poured into a 5OL reactor. 8.4L of 14% ammonia was added dropwise and then stirred for lhour at 45-5O0C. The mixture was cooled down to room temperature.16.8L of water and 1OL of ethyl acetate were added to the extract. The upper organic layer was separated and filtered through the filter aid. The lower aqueous layer was washed with 7.5L of ethyl acetate and combined with the above filtrate.
The combined organic layer was washed with 5L*3 of 5% of brine for three times. The upper organic layer was separated and dried over lkg of anhydrous Na2SO4overnight. The mixture was filtered and concentrated to obtain 2.3kg of solid. The residue was dissolved in 2L of ethyl acetate at 450C. To the solution was then added 8L of petroleum ether dropwise at 450C. The mixture was cooled down slowly to 0-150C and stirred for lhour. A large amount of precipitate was formed and filtered. The filtered cake was dissolved in 2L of ethyl acetate at 450C. The solution was then added 8L of petroleum ether dropwise at 450C. The mixture was cooled down slowly to 15-O0C and stirred for lhour. A large precipitate was formed and filtered. The filter cake was dried at 450C and 954g of 3-(trifluoromethyl)-5-(4-methyl-lH- imidazole-l-yl)-benzeneamine of formula I were obtained. (Yield: 47.5%). The obtained compound of formula I had purity of 99.7% on area by HPLC and contained 0.13% on area by HPLC, of the 5 methyl isomer impurity.
Example 2: Recrystallization of 3-ftrifluoromethyl)-5-f4-methyl-lH-imidazole-l-yl)- benzeneamine of formula I from IPA/water
[00246] A 5OmL flask was charged with Ig of the compound of Formula I crude
(purity of 82.5%) and 3.5mL of IPA. The mixture was heated to 45°C under stirring until the entire solid dissolved. At 45°C, 6mL of water was added drop-wise. The mixture was stirred for lOmin and cooled slowly to 0~10°C. The mixture was stirred at 0~10°C for 10 min and filtered to obtain the recrystallized compound of Formula I having a purity of 98%.
Example 3: Recrystallization of 3-ftrifluoromethyl)-5-f4-methyl-lH-imidazole-l-yl)- benzeneamine of formula I from Ethanol/water
[00247] A 5OmL flask was charged with 2g of the compound of Formula I crude
(purity of 83.1%) and 4mL of Ethanol. The mixture was heated to reflux under stirring until the entire solid dissolved. While refluxing, 1OmL of water was added drop-wise. The mixture was cooled slowly to 25±5°C. The mixture was filtered and washed with a mixture of ethanol/water to obtain the recrystallized compound of Formula I having a purity of 86.5%.
[00248] The purification factor can be seen in the following table:

Example 4: Preparation of compound of formula IV
[00249] The compound of formula X (31.Og, 0.1 Omol) was suspended in 310ml toluene, and SOCl2 (47.6g, 0.40mol) was added to the mixture under the protection of N2. The formed mixture was reacted at 5O0C for 2 h. The solvent was evaporated completely, and a compound of formula (X-Cl) was obtained as yellow solid. The compound of formula (X- Cl) was then added to a THF solution of the compound of formula II (27.Og, 0.1 lmol), DIPEA (15.Og, 0.12mol) and DMAP (0.5g, 4.0mmol). The reaction mixture was reacted at 3O0C for 12 h, and then quenched with 8% solution of sodium bicarbonate (620ml). The mixture was filtered, and washed with H2O, then dried in vacuum. The solid was re-slurried with MTBE, and dried in vacuum again. 49.5g of the compound of formula IV were obtained as light yellow powder. The yield is about 93.7% by weight. The purity of the isolated product is 98% (% on area by HPLC).
Example 5: Preparation of compound of formula IV
[00250] To a 50ml 3-neck flask was charged compound of formula X 3. Ig and 21ml of toluene. The suspension was charged 5.1g dichlorosulfoxide (SOCl2) under nitrogen protection. The reaction mixture was heated to 5O0C and reacted for 2hrs. The reaction was then concentrated to dry. To another 100ml 3-neck flask was charged 2.7g of compound of formula II, 1.5g of DIPEA, O.lg of DMAP and 30ml of THF. To the mixture was charged the above concentrated residue. The reaction mixture was stirred at 25±5°C overnight. The mixture was charged 45ml of ethyl acetate and 20ml of water. The mixture was then stirred at 25±5°C for lOmin, filtered and the filtrate was phase separated. The organic layer was washed by water 10ml twice. Then the organic layer was concentrated to dry. The residue was combined with the filter cake and slurried in MTBE. The mixture was filtered and dried under vacuum at 5O0C. The water layer was adjusted pH to 8 with NaHCO3solution. The second crop 0.5g was thus precipitated out. Total yield was 94%.
Example 6: Preparation of compound of formula IV
[00251] To a 50ml 3-neck flask was charged compound of formula X 3. Ig, 20 mL of toluene and 18ml of dichlorosulfoxide (SOCl2) under nitrogen protection. The reaction mixture was heated to 5O0C and reacted overnight. The reaction was then concentrated to dry and co-evaporated with 20ml of toluene of once. To another 100ml 3-neck flask was charged 2.7g of compound of formula II, 1.5g OfK2CO3, O.lg of DMAP and toluene. To the mixture was charged the above concentrated residue. The reaction mixture was stirred at 5O0C overnight. The mixture was charged 30ml of half saturated NaHCO3 solution, 15ml of MTBE and stirred for lOmin. Large amount of solid was precipitated out and filtered. The filter cake was washed with MTBE and fired under vacuum at 55 0C. The resulted product was of 81% of purity. There were about 9% of the compound of formula X.
Example 7: Preparation of compound of formula IV [00252] The compound of formula X (50 g), HOBt (26.5 g)/ EDCI (37.5 g) and DMF
(500 mL) were loaded into a reactor at 25±5°C. After being reacted for 3h, the compound of formula II (39 g) was added to the reactor. The reaction mixture was stirred at 800C for about 18 hours while monitoring for the consumption of active ester by HPLC. After being cooled to 25±5°C, the mixture was dropped to a solution of half-saturated aqueous solution of sodium hydrogen carbonate, and the product was precipitated as canary yellow solid. [00253] The yield of this step was about 29.0% by weight. The purity of the isolated product was 95% (% on area by HPLC method described in Appendix 1).
Example 8: Preparation of Nilotinib
[00254] The compound of formula IV (21.Og, 39.7mmol), NaI (12.Og, 79.8mmol), CuI
(1.3g, β.Ommol) and N,N-Dimethylethylenediamine (1.Ig, 12.0mmol) were dissolved in DMF (105ml) under the protection of N2. The formed solution was reacted at 12O0C for 24h. The temperature of the above solution was decreased to 6O0C.
[00255] 8-Hydroxyquinoline (1.8g, 1 l.βmmol), CuI (1.3g, β.Ommol), the compound of formula III (4.6g, 56.3mmol) and DBU (9.Og, 59.3mmol) were added to the above solution under the protection of N2. The formed solution was reacted at 12O0C for 48h. After the reaction was competed (detected by the consumption of the compound of formula IV, HPLC), the reaction solution was dropped to a mixture of saturated solution of NaHCO3 (15ml) and water (300ml) at 25±5°C. The mixture was then filtered, and the filter cake was washed with water. 26.9g crude product was obtained as pale brown powder with 69% purity after drying in vacuum.
[00256] The crude product was added to 3.8 vol. DMF, and heated to dissolution. The solution was filtered through Celite, and the filter cake was washed with 0.5 vol. DMF. 3.5 vol. of methanol/H2O (3:1) was added to the above solution at 6O0C. The formed solution was stirred at 25±5°C overnight and at ice bath for 2h. The mixture was filtered, and the filter cake was washed with methanol (0.05 volχ3). The first round re-crystallization solid was obtained after drying in vacuum. The above solid was added to 2.9 vol DMF, and heated to dissolution. Then filtered, and the filter cake was washed with 0.1vol. DMF. The resulting solution was stirred at 25±5°C for 0.5 h, and at ice bath for 2 h. The mixture was filtered, and the cake was washed with methanol (0.05volχ3). 9.1g solid was obtained with 99.1% purity after drying in vacuum. The total yield was about 43.5% by weight. The purity of the isolated product is 99.1% (% on area by HPLC). Example 9: Preparation of Nilotinib
[00257] The compound of formula IV, the compound of formula III, CS2CO3, CuI , 8- hydroxyquinoline and CaO were loaded into a reactor at 25±5°C under the protection of N2. The reaction mixture was then stirred at 1200C for about 24 hours while monitoring for the consumption of the compound of formula IV by HPLC. After cooled to 25±5°C, the mixture was treated with a half-saturated aqueous solution of sodium hydrogen carbonate and extracted three times with ethyl acetate, then dried by Na2SO4. After concentration, the crude product was obtained as yellow solid. Then the solid was dissolved by CH2CVMeOH (10 equ., 3:2), and the mixture was washed three times with water. After a period of time, the product would be crystallized from the organic solvent (purity: 95%, detected by HPLC). Few minutes later, the product would precipitate as yellow solid. Then the product was stirred in the solvent of CH2Cl2/Me0H (5 equ., 5:1) at 400C for 1 hour. After that, the mixture would be filtered. The solid we got was dried in vacuum, and the product with 98% purity was obtained by this means.
[00258] The yield of this step was about 31.1% by weight. The purity of the isolated product was 98% (% on area by HPLC method described in Appendix 1).
Example 10: Preparation of Nilotinib:
[00259] To 250 mL glass reactor was added the compound 4-methyl-3-{[4-(pyridin-3- yl)pyrimidin-2-yl] amino} benzoic acid of formula X (10.0 g, 0.032 mol), a compound of formula I (8.7 g, 0.036 mol), SOCl2 (7.5 mL, 0.103 mol) and N-Methyl-pyrrolidone (100 mL). The reaction mixture was stirred and heated to 900C for 5 h. The reaction was then cooled to 500C and an aqueous NaOH solution was added (12 g in 72 mL H2O) until pH 10- 11. Then, the suspension was cooled to room-temperature, stirred for 30 minutes at this temperature, filtered under reduced pressure and washed with 30 mL H2O to yield a beige solid. This material was dried under vacuum at 500C and 8.2 g of Nilotinib base was obtained. To the mother-liquor was added H2O (300 mL), and the mixture was stirred for 15 hours at room-temperature. A precipitate was formed and filtered under vacuum. The solid so-obtained was washed with H2O (20 mL), and dried in vacuum oven at 500C to yield additional 5.9 g of Nilotinib base. The total amount of Nilotinib base was 14.1 g in 81% yield. Example 11: Preparation of Nilotinib:
[00260] To 250 mL glass reactor was added the compound of formula 4-methyl-3- {[4-
(pyridin-3-yl)pyrimidin-2-yl]amino}benzoic acid of formula X (20.0 g, 0.065 mol), a compound of formula I (17.3 g, 0.072 mol), SOCl2 (15 mL, 0.206 mol) and N-Methyl- pyrrolidone (100 mL). The reaction mixture was stirred and heated to 900C for 3 h. The reaction was filtered under reduced pressure and washed with NMP (10 mL) and H2O (10 mL). The filtrate was then cooled to 700C and a 47% NaOH solution (30 mL) was added and stirred for 30 minutes until pH 11-12. Then, the suspension was cooled to 5°C during 3 hours, stirred at this temperature for 10 hours room-temperature, filtered under reduced pressure and washed with 100 mL H2O to yield a beige solid. This material was dried under vacuum at 500C and 27.1 g of Nilotinib base was obtained with 76% yield. (97.2% assay, 99.17% purity).
Example 12: Preparation of Nilotinib:
[00261] To IL glass reactor was added the compound of formula 4-methyl-3-{[4-
(pyridin-3-yl)pyrimidin-2-yl]amino}benzoic acid of formula X (80.0 g, 0.26 mol), and N- Methyl-pyrrolidone (400 mL). The mixture was heated to 600C, then SOCl2 (24 mL, 0.33 mol) was added during 15 minutes. The resulted mixture was stirred at 600C for 1 h. A compound of formula I (69.2 g, 0.29 mol) was added and the reaction mixture was stirred and heated to 900C for 3 h. Water (500 mL) was added and the solution was heated to 800C. NaOH 47% solution (65 mL) was added until pH 11-12. Then, the suspension was cooled to 400C and stirred at this temperature for 2 hours, filtered under reduced pressure at 400C, and washed with 500 mL H2O to yield a beige solid. This material was slurried in water (1 L) at 400C for 1 h, filtered, washed with water (500 mL), and dried under vacuum at 500C to obtain 135.25 g of Nilotinib base with 94% yield. (95.8% assay, 99.46% purity).
Example 13: Preparation of 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-4-methylbenzoyl chloride, dihydrochloride of the formula (X-C1)*2HC1:
[00262] Thionyl chloride (1400ML) was added to 3-(4-(pyridin-3-yl)pyrimidin-2- ylamino)-4-methylbenzoic acid of formula X (39 gms). This mixture was heated to 60-700C and stirred for 10-12 hours. The reaction mixture was then cooled to 30-270C. The obtained slurry was filtered and the solid was washed with dichloromethane. The wet product was dried at 55-600C under reduced pressure.
Dry wt: 140gm
Yield: 95.4
Purity: above 98% by HPLC
Hydrochloride content (by Argentometry titration): 27.48%
Example 14: Preparation of 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-4-methylbenzoyl chloride, dihydrochloride of the formula (X-ClWHCl:
[00263] Thionyl chloride (1000ML) was added to 3-(4-(pyridin-3-yl)pyrimidin-2- ylamino)-4-methylbenzoic acid of formula X (100 gms). This mixture was heated to 60-700C and stirred for 5-6 hours. The reaction mixture was then cooled to 30-350C. Dichloromethane
(1000ML) was then added to the recation mixture and stirred for 10-15 minutes. The obtained slurry was filtered and the solid was washed with dichloromethane. The wet product was dried at 55-600C under reduced pressure.
Dry wt: 100-106gm
Purity: above 98% by HPLC
Example 15: Preparation of Nilotinib*3HCl (crude):
[00264] 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-4-methylbenzoyl chloride dihydrochloride of formula (X-C1)-2HC1 (105 gms) was added to dichloromethane (1000ml) and 3-(trifluoromethyl)-5-(4-methyl-lH-imidazol-l-yl)benzenamine of formula I (71 gms) at
25-400C. The temperature was raised to reflux point and was stirred at this temperature for
10-12 Hours. The reaction mixture was then cooled to 30-200C. The obtained slurry was filtered and the solid was washed with dichloromethane (200ml). The wet product was dried at 40-60 0C under reduced pressure.
[00265] The X-ray powder diffraction of the obtained product is shown in Figure 3.
The X-ray powder diffraction of the obtained product after exposure to 100% humidity for
96% is shown in Figure 4.
Yield: 90-92%
Purity: 85-90%
Hydrochloride content (by Argentometry titration): 16.8%.
Example 16: Preparation of Nilotinib«3HCl: [00266] Methanol (50ml) was cooled to 0-50C and acetyl chloride (2.29gm) was slowly added to it. To this mixture, 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-N-(3- (trifluoromethyl)-5-(4-methyl-lH-imidazol-l-yl)phenyl)-4-methyl benzamide (Nilotinib free base) (5.00 gms) was added slowly and mixture was stirred for 2 hours. Acetone (50ml) was then added and mixture was stirred for 60 minutes. Reaction mass was filtered and washed with acetone (10ml). The obtained product was dried at 55-600C. Dry wt: 4.5gm Yield: 75% Purity: 95-98%
Example 17: Purification of Nilotinib«3HCl (Pure):
[00267] 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-N-(3-(trifluoromethyl)-5-(4-methyl- lH-imidazol-l-yl)phenyl)-4-methylbenzamide tri hydrochloride (5gm) and water (25ml) were added and the mass was heated to 60-700C. The mass was charcoalized (0.5gm carbon) and filtered through celite bed. Methanol (50ml) was added to the filtrate. The mixture was heated to 50-600C and acetone (100ml) was added. It was then cooled to 30-270C and stirred for 2hours. The obtained product was filtered and dried at 50-550C for 12 hours under vacuum. The X-ray powder diffraction of the obtained product is shown in Figure 5. Dry wt 3.5gm Yield 0.7w/w Purity: 95-98%
Example 18: Preparation of Nilotinib:
[00268] 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-N-(3-(trifluoromethyl)-5-(4-methyl- lH-imidazol-l-yl)phenyl)-4-methylbenzamide tri hydrochloride (185gms) was dissolved in 825ml water and heated to 45-55°C. A methanolic solution of sodium hydroxide (35.9gm Sodium hydroxide dissolve in 1800 ml methanol) was added to the reaction mixture over a period of 1-2 hours. The suspension was heated to 65-700C for 5-6 hours and the slurry was cooled to 35-300C. The solid was filtered and washed with equal amount of water: methanol mixture 200ml. The wet product was dried at 45-55°C under reduced pressure. Yield: 90% Purity: 99.5% Example 19: Purification of Nilotinib:
[00269] 3-(4-(pyridin-3-yl)pyrimidin-2-ylamino)-N-(3-(trifluoromethyl)-5-(4-methyl- lH-imidazol-l-yl)phenyl)-4-methylbenzamide (140gm) was taken into methanol (1.41it) and sodium hydroxide (14gm). The mixture was heated to reflux and stirred for 3-4 hours. The mixture was the cooled to 40-350C and filtered. The product was washed with methanol (2X50ml) and dried at 50-600C for 12 hours under vacuum. Dry wt. 120gm Yield: 0.85w/w

PAPER
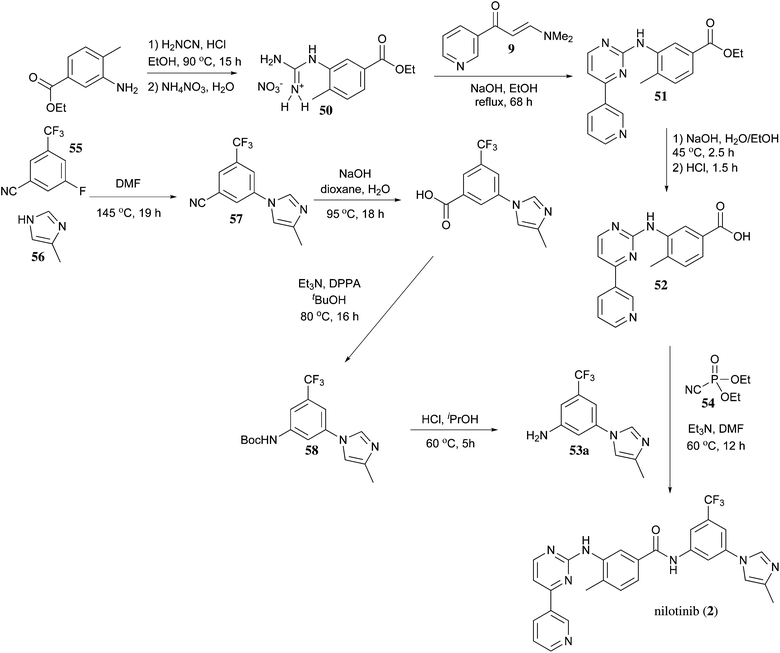
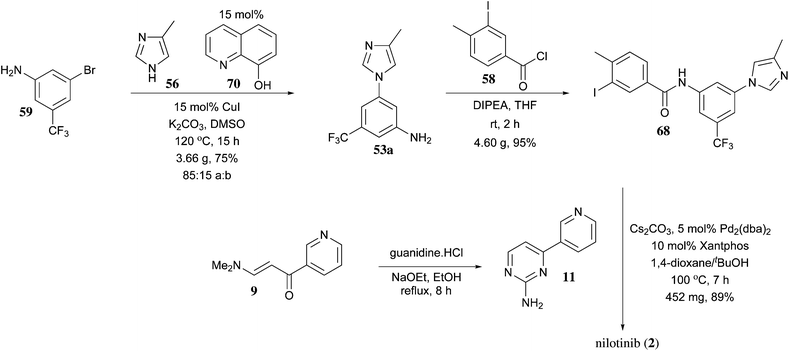
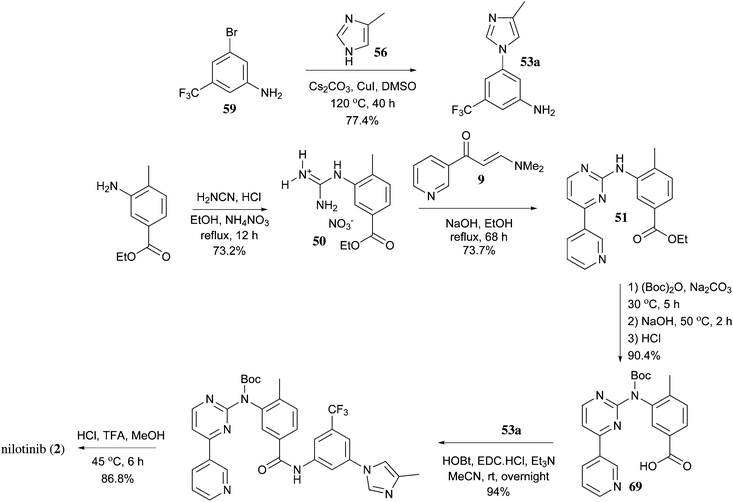
https://pubs.rsc.org/en/content/articlelanding/2013/ob/c2ob27003j/unauth#!divAbstract
References
- ^ Jump up to:a b c d e f g h “Tasigna (nilotinib) dosing, indications, interactions, adverse effects, and more”. Medscape Reference. WebMD. Retrieved 25 January 2014.
- ^ Official Manufacturer Website http://www.tasigna.com
- ^ “Cancer Drug Information: Nilotinib”.
- ^ Jump up to:a b Manley, P.; Cowan-Jacob, S.; Mestan, J. (2005). “Advances in the structural biology, design and clinical development of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid leukaemia”. Biochimica et Biophysica Acta. 1754 (1–2): 3–13. doi:10.1016/j.bbapap.2005.07.040. PMID 16172030.
- ^ Jump up to:a b Manley, P.; Stiefl, N.; Cowan-Jacob, S.; Kaufman, S.; Mestan, J.; Wartmann, M.; Wiesmann, M.; Woodman, R.; Gallagher, N. (2010). “Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib”. Bioorganic & Medicinal Chemistry. 18 (19): 6977–6986. doi:10.1016/j.bmc.2010.08.026. PMID 20817538.
- ^ Jump up to:a b c Jabbour, E.; Cortes, J.; Kantarjian, H. (2009). “Nilotinib for the treatment of chronic myeloid leukemia: An evidence-based review”. Core Evidence. 4: 207–213. doi:10.2147/CE.S6003. PMC 2899790.
- ^ Jump up to:a b Olivieri, A.; Manzione, L. (2007). “Dasatinib: a new step in molecular target therapy”. Annals of Oncology. 18 Suppl 6: vi42–vi46. doi:10.1093/annonc/mdm223. PMID 17591830.
- ^ Breccia, M.; Alimena, G. (2010). “Nilotinib: a second-generation tyrosine kinase inhibitor for chronic myeloid leukemia”. Leukemia Research. 34 (2): 129–134. doi:10.1016/j.leukres.2009.08.031. PMID 19783301.
- ^ https://www.cancer.gov/about-cancer/treatment/drugs/fda-nilotinib
- ^ Jump up to:a b “Complete Nilotinib information from Drugs.com”. Drugs.com. Retrieved 25 January2014.
- ^ “Tasigna : EPAR – Product Information” (PDF). European Medicines Agency. Novartis Europharm Ltd. 18 October 2013. Retrieved 25 January 2014.
- ^ “Tasigna 150mg Hard Capsules – Summary of Product Characteristics (SPC)”. electronic Medicines Compendium. Novartis Pharmaceuticals UK Ltd. 9 September 2013. Retrieved 25 January 2014.
- ^ Jump up to:a b “TASIGNA® nilotinib” (PDF). TGA eBusiness Services. 21 October 2013. Retrieved 25 January 2014.
- ^ “FDA Approves Tasigna for Treatment of Philadelphia Chromosome Positive Chronic Myeloid Leukemia”. U.S. Food and Drug Administration. 2007-10-30. Retrieved 2009-08-04.
- ^ “Prescribing information for Tasigna (nilotinib) Capsules” (PDF). NDA 022068. U.S. FDA. 2007-10-29. Retrieved 2009-08-04.
- ^ Kantarjian H; Giles, Francis; Wunderle, Lydia; Bhalla, Kapil; O’Brien, Susan; Wassmann, Barbara; Tanaka, Chiaki; Manley, Paul; Rae, Patricia; Mietlowski, William; Bochinski, Kathy; Hochhaus, Andreas; Griffin, James D.; Hoelzer, Dieter; Albitar, Maher; Dugan, Margaret; Cortes, Jorge; Alland, Leila; Ottmann, Oliver G.; et al. (2006). “Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL”. N Engl J Med. 354 (24): 2542–51. doi:10.1056/NEJMoa055104. PMID 16775235.
- ^ “Patients with treatment-resistant leukemia achieve high responses to Tasigna (nilotinib) in first published clinical trial results”. MediaReleases. Novartis. 2006-06-14. Retrieved 2009-08-04.
- ^ Jump up to:a b Khurana V, Minocha M, Pal D, Mitra AK (March 2014). “Role of OATP-1B1 and/or OATP-1B3 in hepatic disposition of tyrosine kinase inhibitors”. Drug Metabol Drug Interact. 29 (3): 179–90. doi:10.1515/dmdi-2013-0062. PMC 4407685. PMID 24643910.
- ^ Donatelli, Christopher; Chongnarungsin, Daych; Ashton, Rendell (2014). “Acute respiratory failure from nilotinib-associated diffuse alveolar hemorrhage”. Leukemia & Lymphoma. 55 (10): 1–6. doi:10.3109/10428194.2014.887714. PMID 24467220.
- ^ Khurana V, Minocha M, Pal D, Mitra AK (May 2014). “Inhibition of OATP-1B1 and OATP-1B3 by tyrosine kinase inhibitors”. Drug Metabol Drug Interact. 29 (4): 249–59. doi:10.1515/dmdi-2014-0014. PMC 4407688. PMID 24807167.
- ^ Bailey, David G; Malcolm, J; Arnold, O; David Spence, J (1998-08-01). “Grapefruit juice–drug interactions”. British Journal of Clinical Pharmacology. 46 (2): 101–110. doi:10.1046/j.1365-2125.1998.00764.x. ISSN 0306-5251. PMC 1873672. PMID 9723817.
- ^ Komoroski, Bernard J.; Zhang, Shimin; Cai, Hongbo; Hutzler, J. Matthew; Frye, Reginald; Tracy, Timothy S.; Strom, Stephen C.; Lehmann, Thomas; Ang, Catharina Y. W. (2004-05-01). “Induction and inhibition of cytochromes P450 by the St. John’s wort constituent hyperforin in human hepatocyte cultures”. Drug Metabolism and Disposition. 32 (5): 512–518. doi:10.1124/dmd.32.5.512. ISSN 0090-9556. PMID 15100173.
- ^ Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD (June 2006). “AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL”. Br. J. Cancer. 94 (12): 1765–9. doi:10.1038/sj.bjc.6603170. PMC 2361347. PMID 16721371.
- ^ Manley, PW; Drueckes, P; Fendrich, G; Furet, P; Liebetanz, J; Martiny-Baron, G; Mestan, J; Trappe, J; et al. (2010). “Extended kinase profile and properties of the protein kinase inhibitor nilotinib”. Biochimica et Biophysica Acta. 1804 (3): 445–53. doi:10.1016/j.bbapap.2009.11.008. PMID 19922818.
- ^ Pagan, F.; Hebron, M.; Valadez, E. H.; Torres-Yaghi, Y.; Huang, X.; Mills, R. R.; Wilmarth, B. M.; Howard, H.; Dunn, C.; Carlson, A.; Lawler, A.; Rogers, S. L.; Falconer, R. A.; Ahn, J.; Li, Z.; Moussa, C. (2016). “Nilotinib Effects in Parkinson’s disease and Dementia with Lewy bodies”. Journal of Parkinson’s Disease. 6 (3): 503–17. doi:10.3233/JPD-160867. PMC 5008228. PMID 27434297.
- ^ Dash, Deepa (2019). “Anticancer Drugs for Parkinson’s Disease: Is It a Ray of Hope or Only Hype?”. Annals of Indian Academy of Neurology. 22 (1): 13–16. doi:10.4103/aian.AIAN_177_18. PMC 6327695. PMID 30692753.
- ^ Robledo, I.; Jankovic, J. (2017). “Media hype: Patient and scientific perspectives on misleading medical news”. Movement Disorders. 32 (9): 1319–1323. doi:10.1002/mds.26993. PMID 28370445.
- ^ Wyse, R. K.; Brundin, P.; Sherer, T. B. (2016). “Nilotinib – Differentiating the Hope from the Hype”. Journal of Parkinson’s Disease. 6 (3): 519–22. doi:10.3233/JPD-160904. PMC 5044778. PMID 27434298.
- ^ “Global Novartis News Archive”.
- ^ “Cancer drug prevents build-up of toxic brain protein”. MedicalXpress.com. 10 May 2013. Retrieved 11 April 2017.
External links
- Discovery and development of Bcr-Abl tyrosine kinase inhibitors
- New drug information/Abbreviated Scientific Narrative
- Highlights of Prescription information Nilotinib (August 2007) Novartis Pharmaceuticals Corporation (USA)
- Summary of Product Characteristics Nilotinib (November 2007) Novartis AG (Europe)
 |
|
 |
|
| Clinical data | |
|---|---|
| Trade names | Tasigna |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a608002 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | 30%[1] |
| Protein binding | 98%[1] |
| Metabolism | Hepatic (mostly CYP3A4-mediated)[1] |
| Elimination half-life | 15-17 hours[1] |
| Excretion | Faeces (93%)[1] |
| Identifiers | |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| PDB ligand | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.166.395 |
| Chemical and physical data | |
| Formula | C28H22F3N7O |
| Molar mass | 529.5245 g/mol g·mol−1 |
| 3D model (JSmol) | |
Nilotinib
-
- Synonyms:AMN-107
- ATC:L01XE08
- Use:antineoplastic, kinase inhibitor
- Chemical name:4-methyl-N-[3-(4-methyl-1H-imidazol-1-yl)-5-(trifluoromethyl)phenyl]-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]benzamide
- Formula:C28H22F3N7O
- MW:529.53 g/mol
- CAS-RN:641571-10-0
- InChI Key:HHZIURLSWUIHRB-UHFFFAOYSA-N
- InChI:InChI=1S/C28H22F3N7O/c1-17-5-6-19(10-25(17)37-27-33-9-7-24(36-27)20-4-3-8-32-14-20)26(39)35-22-11-21(28(29,30)31)12-23(13-22)38-15-18(2)34-16-38/h3-16H,1-2H3,(H,35,39)(H,33,36,37)
Derivatives
Hydrochloride monohydrate
- Formula:C28H22F3N7O • HCl • H2O
- MW:586.01 g/mol
- CAS-RN:923288-90-8
Synthesis
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Tasigna | Novartis ,2008 | |
| F | Tasigna | Novartis | |
| GB | Tasigna | Novartis | |
| I | Tasigna | Novartis | |
| USA | Tasigna | Novartis ,2007 | |
| J | Tasigna | Novartis ,2010 |
Formulations
- cps. 150 and 200 mg as hydrochloride monohydrate
References
-
- a WO 2004 005281 (Novartis; 15.1.2004; GB-prior. 5.7.2002).
- US 7 169 791 (Novartis; 30.1.2007; appl. 4.7.2003; GB-prior. 5.7.2002).
- US 7 569 566 (Novartis; 4.8.2009; GB-prior. 5.7.2002, 20.12.2002).
- WO 2006 135641 (Novartis; 21.12.2006; USA-prior. 4.8.2005).
- US 7 956 053 (Novartis; 7.6.2011; appl. 22.6.2009; GB-prior. 5.7.2002).
-
Preparation of III:
- b Huang, W.-S., Shakesperare, W.C., Synthesis (SYNTBF) (2007) 14, 2121.
- c WO 2010 060074 (Teva Pharms.; 27.5.2010; appl. 24.11.2009; USA-prior. 24.11.2008).
- d Ueda, S. et al., J. Med. Chem. Soc., (2012) 134(1), 700-706.
- US 8 017 621 (Novartis; 13.9.2011; appl. 17.11.2007; USA-prior. 18.11.2003).
- WO 2006 135619 (Novartis; 21.12.2006; USA-prior. 6.9.2005).
- e EP 2 626 355 (Natco Pharma; 14.8.2013; appl. 9.2.2012).
-
Inhibitors of mutant form of KIT:
- US 8 017 621 (Novartis; 13.9.2011; appl. 17.11.2004; USA-prior. 18.11.2003).
-
Salts of Nilotinib:
- US 8 163 904 (Novartis; 24.4.2012; appl. 18.7.2006; USA-prior. 20.7.2005).
- US 8 389 537 (Novartis; 5.3.2013; appl. 13.3.2012; USA-prior. 20.7.2005).
-
Pharmaceutical compositions:
- US 8 293 756 (Novartis; 23.10.2012; appl. 25.9.2007; EP-prior. 27.9.2006).
- US 8 501 760 (Novartis; 6.8.2013; appl. 21.9.2012; EP-prior. 27.9.2006).
-
Crystalline forms:
- US 8 343 984 (Novartis; 1.1.2013; appl. 18.7.2006; USA-prior. 20.7.2005).
- US 8 415 363 (Novartis; 9.4.2013; appl. 3.8.2012; USA-prior. 20.7.2005).
//////////Nilotinib, AMN107, Tasigna, ニロチニブ,
LENALIDOMIDE, レナリドミド, леналидомид , ليناليدوميد , 来那度胺 ,
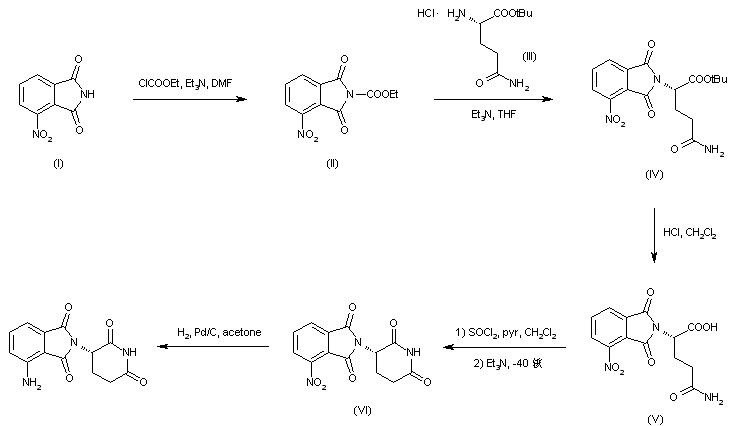
LENALIDOMIDE
- Molecular FormulaC13H13N3O3
- Average mass259.261 Da
|
レナリドミド;
|
леналидомид , ليناليدوميد , 来那度胺 ,
191732-72-6 [RN]
1-Oxo-4-amino-2-(2,6-dioxopiperidin-3-yl)isoindole
2,6-Piperidinedione, 3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-
3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-2,6-piperidinedione
3-(4-Amino-1-oxo-1,3-dihydro-2H-isoindol-2-yl)-2,6-piperidinedione
3-(4-Amino-1-oxo-1,3-dihydro-2H-isoindol-2-yl)piperidin-2,6-dion
3-(4-amino-1-oxo-1,3-dihydro-2H-isoindol-2-yl)piperidine-2,6-dione
3-(4-amino-1-oxoisoindolin-2-yl)piperidine-2,6-dione
3-(7-amino-3-oxo-1h-isoindol-2-yl)piperidine-2,6-dione
8505
E3 Ligase ligand
IMiD3
CAS Registry Number: 191732-72-6
CAS Name: 3-(4-Amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-2,6-piperidinedione
Additional Names: 1-oxo-2-(2,6-dioxopiperidin-3-yl)-4-aminoisoindoline
Manufacturers’ Codes: CC-5013
Trademarks: Revimid (Celgene); Revlimid (Celgene)
Molecular Formula: C13H13N3O3
Molecular Weight: 259.26
Percent Composition: C 60.22%, H 5.05%, N 16.21%, O 18.51%
Literature References: Immunomodulatory drug; analog of thalidomide, q.v. Prepn: G. W. Muller et al., US 5635517 (1997 to Celgene); and in vitro TNF-a inhibition: eidem, Bioorg. Med. Chem. Lett. 9, 1625 (1999). LC-MS determn in plasma: T. M. Tohnya et al., J. Chromatogr. B 811, 135 (2004). Clinical evaluation in multiple myeloma: P. G. Richardson et al., Blood 100, 3063 (2002); in myelodysplastic syndromes: A. List et al., N. Engl. J. Med. 352, 549 (2005). Review of development, pharmacology and therapeutic potential: J. B. Bartlett et al., Nature Rev. 4, 314-322 (2004); C. S. Mitsiades, N. Mitsiades, Curr. Opin. Invest. Drugs 5, 635-647 (2004).
Therap-Cat: Immunomodulator.
Keywords: Immunomodulator.
- 191732-72-6
- SYP-1512
- LENALIDOMIDE [VANDF]
- LENALIDOMIDE [WHO-DD]
- LENALIDOMIDE [EMA EPAR]
- LENALIDOMIDE [MI]
- LENALIDOMIDE [MART.]
- LENALIDOMIDE [ORANGE BOOK]
- LENALIDOMIDE [USAN]
- LENALIDOMIDE [INN]
- CDC-501
- REVLIMID
- LENALIDOMIDE
- 3-(4-AMINO-1-OXO-1,3-DIHYDRO-2H-ISOINDOL-2-YL)PIPERIDINE-2,6-DIONE
- 2,6-PIPERIDINEDIONE, 3-(4-AMINO-1,3-DIHYDRO-1-OXO-2H-ISOINDOL-2-YL)-
- CC-5013
Lenalidomide (trade name Revlimid) is a derivative of thalidomide approved in the United States in 2005.[1]
It was initially intended as a treatment for multiple myeloma, for which thalidomide is an accepted therapeutic treatment. Lenalidomide has also shown efficacy in the class of hematological disorders known as myelodysplastic syndromes (MDS). Along with several other drugs developed in recent years, lenalidomide has significantly improved overall survival in myeloma (which formerly carried a poor prognosis), although toxicity remains an issue for users.[2] It costs $163,381 per year for the average patient.[3]
It is on the World Health Organization’s List of Essential Medicines, the safest and most effective medicines needed in a health system.[4]
Medical uses
Multiple myeloma
Multiple myeloma is a cancer of the blood, characterized by accumulation of a plasma cell clone in the bone marrow.[5] Lenalidomide is one of the novel drug agents used to treat multiple myeloma. It is a more potent molecular analog of thalidomide, which inhibits tumor angiogenesis, tumor secreted cytokines and tumor proliferation through the induction of apoptosis.[6][7][8]
Compared to placebo, lenalidomide is effective at inducing a complete or “very good partial” response as well as improving progression-free survival. Adverse events more common in people receiving lenalidomide for myeloma were neutropenia (a decrease in the white blood cell count), deep vein thrombosis, infections, and an increased risk of other hematological malignancies.[9] The risk of second primary hematological malignancies does not outweigh the benefit of using lenalidomide in relapsed or refractory multiple myeloma.[10] It may be more difficult to mobilize stem cells for autograft in people who have received lenalidomide.[6]
On 29 June 2006, lenalidomide received U.S. Food and Drug Administration (FDA) clearance for use in combination with dexamethasone in patients with multiple myeloma who have received at least one prior therapy.[11] On 22 February 2017, the FDA approved lenalidomide as standalone maintenance therapy (without dexamethasone) for patients with multiple myeloma following autologous stem cell transplant.[12]
On 23 April 2009, The National Institute for Health and Clinical Excellence (NICE) issued a Final Appraisal Determination (FAD) approving lenalidomide, in combination with dexamethasone, as an option to treat patients with multiple myeloma who have received two or more prior therapies in England and Wales.[13]
On 5 June 2013, the FDA designated lenalidomide as a specialty drug requiring a specialty pharmacy distribution for “use in mantle cell lymphoma (MCL) in patients whose disease has relapsed or progressed after two prior therapies, one of which included bortezomib.” Revlimid is only available through a specialty pharmacy, “a restricted distribution program in conjunction with a risk evaluation and mitigation strategy (REMS) due to potential for embryo-fetal risk.”[14]
Myelodysplastic syndromes
With myelodysplastic syndromes (MDS), the best results of lenalidomide were obtained in patients with the Chromosome 5q deletion syndrome (5q- syndrome).[15] The syndrome results from deletions in human chromosome 5 that remove three adjacent genes, granulocyte-macrophage colony-stimulating factor, Platelet-derived growth factor receptor B, and Colony stimulating factor 1 receptor.[16][17]
It was approved by the FDA on 27 December 2005, for patients with low or intermediate-1 risk MDS with 5q- with or without additional cytogenetic abnormalities. A completed Phase II, multi-centre, single-arm, open-label study evaluated the efficacy and safety of Revlimid monotherapy treatment for achieving haematopoietic improvement in red blood cell (RBC) transfusion dependent subjects with low- or intermediate-1-risk MDS associated with a deletion 5q cytogenetic abnormality.
63.8% of subjects had achieved RBC-transfusion independence accompanied by a median increase of 5.8 g/dL in blood Hgb concentration from baseline to the maximum value during the response period. Major cytogenetic responses were observed in 44.2% and minor cytogenetic responses were observed in 24.2% of the evaluable subjects. Improvements in bone marrow morphology were also observed. The results of this study demonstrate the efficacy of Revlimid for the treatment of subjects with Low- or Intermediate-1-risk MDS and an associated del 5 cytogenetic abnormality.[15][18][19]
Lenalidomide was approved on 17 June 2013 by the European Medicines Agency for use in low- or intermediate-1-risk myelodysplastic syndromes (MDS) patients who have the deletion 5q cytogenetic abnormality and no other cytogenetic abnormalities, are dependent on red blood cell transfusions, and for whom other treatment options have been found to be insufficient or inadequate.[20]
Mantle cell lymphoma
Lenalidomide is approved by FDA for mantle cell lymphoma in patients whose disease has relapsed or progressed after at least two prior therapies.[1] One of these previous therapies must have included bortezomib.
Other cancers
Lenalidomide is undergoing clinical trial as a treatment for Hodgkin’s lymphoma,[21] as well as non-Hodgkin’s lymphoma, chronic lymphocytic leukemia and solid tumor cancers, such as carcinoma of the pancreas.[22] One Phase 3 clinical trial being conducted by Celgene in elderly patients with B-cell chronic lymphocytic leukemia was halted in July 2013, when a disproportionate number of cancer deaths were observed during treatment with lenalidomide versus patients treated with chlorambucil.[23]
Adverse effects
In addition to embryo-fetal toxicity, lenalidomide also carries Black Box Warnings for hematologic toxicity (including significant neutropenia and thrombocytopenia) and venous/arterial thromboembolisms.[1]
Serious potential side effects are thrombosis, pulmonary embolus, and hepatotoxicity, as well as bone marrow toxicity resulting in neutropenia and thrombocytopenia. Myelosuppression is the major dose-limiting toxicity, which is contrary to experience with thalidomide.[24] Lenalidomide may also be associated with adverse effects including second primary malignancy, severe cutaneous reactions, hypersensitivity reactions, tumor lysis syndrome, tumor flare reaction, hypothyroidism, and hyperthyroidism. [1]
Teratogenicity
Lenalidomide is related to thalidomide which is known to be teratogenic. Tests in monkeys have suggested lenalidomide is also teratogenic.[25] It therefore has the pregnancy category X and cannot be prescribed for women who are pregnant or who may become pregnant during therapy. For this reason, the drug is only available in the United States(under the brand name Revlimid) through a restricted distribution system called RevAssist. Females who may become pregnant must use at least two forms of reliable contraception during treatment and for at least four weeks after discontinuing treatment with lenalidomide.[1]
Venous thromboembolism
Lenalidomide, like its parent compound thalidomide, may cause venous thromboembolism (VTE), a potentially serious complication with their use. Bennett et al. have reviewed incidents of lenalidomide-associated VTE among patients with multiple myeloma.[26] They have found that there are high rates of VTE when patients with multiple myeloma received thalidomide or lenalidomide in conjunction with dexamethasone, melphalan, or doxorubicin. When lenalidomide and dexamethasone are used to treat multiple myeloma, a median of 14% of patients had VTE (range,3-75%). In patients who took prophylaxis to treat lenalidomide-associated VTE, such as aspirin, thromboembolism rates were found to be lower than without prophylaxis, frequently lower than 10%. Clearly, thromboembolism is a serious adverse drug reaction associated with lenalidomide, as well as thalidomide. In fact, a black box warning is included in the package insert for lenalidomide, indicating that lenalidomide-dexamethasone treatment for multiple myeloma is complicated by high rates of thromboembolism.
Currently,[when?] clinical trials are under way to further test the efficacy of lenalidomide to treat multiple myeloma, and to determine how to prevent lenalidomide-associated venous thromboembolism.[citation needed]
Stevens-Johnson syndrome
In March 2008, the U.S. Food and Drug Administration (FDA) included lenalidomide on a list of 20 prescription drugs under investigation for potential safety problems. The drug is being investigated for possibly increasing the risk of developing Stevens–Johnson syndrome, a life-threatening condition affecting the skin.[27]
FDA ongoing safety review
As of 2011, the FDA has initiated an ongoing review which will focus on clinical trials which found an increased risk of developing cancers such as acute myelogenous leukemia (AML) and B-cell lymphoma,[3] though the FDA is currently advising all people to continue their treatment.[28]
Mechanism of action
Lenalidomide has been used to successfully treat both inflammatory disorders and cancers in the past ten years.[when?] There are multiple mechanisms of action, and they can be simplified by organizing them as mechanisms of action in vitro and in vivo.[29] In vitro, lenalidomide has three main activities: direct anti-tumor effect, inhibition of angiogenesis, and immunomodulation. In vivo, lenalidomide induces tumor cell apoptosis directly and indirectly by inhibition of bone marrow stromal cell support, by anti-angiogenic and anti-osteoclastogenic effects, and by immunomodulatory activity. Lenalidomide has a broad range of activities that can be exploited to treat many hematologic and solid cancers.
On a molecular level, lenalidomide has been shown to interact with the ubiquitin E3 ligase cereblon[30] and target this enzyme to degrade the Ikaros transcription factors IKZF1 and IKZF3.[31] This mechanism was unexpected as it suggests that the major action of lenalidomide is to re-target the activity of an enzyme rather than block the activity of an enzyme or signaling process, and thereby represents a novel mode of drug action. A more specific implication of this mechanism is that the teratogenic and anti-neoplastic properties of lenalidomide, and perhaps other thalidomide derivatives, could be disassociated.
Research
The low level of research that continued on thalidomide, in spite of its scandalous history of teratogenicity, unexpectedly showed that the compound affected immune function. The drug was, for example, recently approved by the FDA for treatment of complications from leprosy; it has also been investigated as an adjunct for treating some malignancies. Recent research on related compounds has revealed a series of molecules which inhibit tumor necrosis factor (TNF-α).[citation needed]
Price
Lenalidomide costs $163,381 per year for the average person in the United States.[3] Lenalidomide made almost $9.7bn for Celgene in 2018.[32]
In 2013, the UK National Institute for Health and Care Excellence (NICE) rejected lenalidomide for “use in the treatment of people with a specific type of the bone marrow disorder myelodysplastic syndrome (MDS)” in England and Scotland, arguing that Celgene “did not provide enough evidence to justify the £3,780 per month (USD$5746.73) price-tag of lenalidomide for use in the treatment of people with a specific type of the bone marrow disorder myelodysplastic syndrome (MDS)”.[33]
SYN
https://link.springer.com/article/10.1007/s10593-015-1670-0

A new process for the synthesis of anticancer drug lenalidomide was developed, using platinum group metal-free and efficient reduction of nitro group with the iron powder and ammonium chloride. It was found that the bromination of the key raw material, methyl 2-methyl-3-nitrobenzoate, could be carried out in chlorine-free solvent methyl acetate without forming significant amounts of hazardous by-products. We also have compared the known synthetic methods for cyclization of methyl 2-(bromomethyl)-3-nitrobenzoate and 3-aminopiperidinedione to form lenalidomide nitro precursor.
SYN

SYN
EP 0925294; US 5635517; WO 9803502
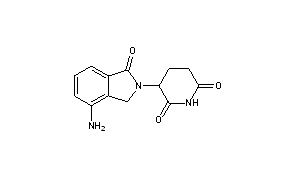
Cyclization of N-(benzyloxycarbonyl)glutamine (I) by means of CDI in refluxing THF gives 3-(benzyloxycarbonylamino)piperidine-2,6-dione (II), which is deprotected with H2 over Pd/C in ethyl acetate/4N HCl to yield 3-aminopiperidine-2,6-dione hydrochloride (III). Bromination of 2-methyl-3-nitrobenzoic acid methyl ester (IV) with NBS in CCl4 provides 2-(bromomethyl)-3-nitrobenzoic acid methyl ester (V), which is cyclized with the aminopiperidine (III) by means of triethylamine in hot DMF to afford 3-(4-nitro-1-oxoisoindolin-2-yl)piperidine-2,6-dione (VI). Finally, the nitro group of compound (VI) is reduced with H2 over Pd/C in methanol (1, 2).
SYN
Bioorg Med Chem Lett 1999,9(11),1625
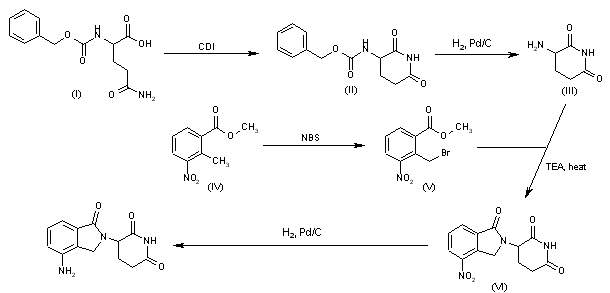
Treatment of 3-nitrophthalimide (I) with ethyl chloroformate and triethylamine produced 3-nitro-N-(ethoxycarbonyl)phthalimide (II), which was condensed with L-glutamine tert-butyl ester hydrochloride (III) to afford the phthaloyl glutamine derivative (IV). Acidic cleavage of the tert-butyl ester of (IV) provided the corresponding carboxylic acid (V). This was cyclized to the required glutarimide (VI) upon treatment with thionyl chloride and then with triethylamine. The nitro group of (VI) was finally reduced to amine by hydrogenation over Pd/C.
Lenalidomide
-
- Synonyms:CC-5013, CDC 501
- ATC:L04AX04
- Use:myelodysplastic syndrome (MDS)
- Chemical name:3-(4-amino-1,3-dihydro-1-oxo-2H-isoindol-2-yl)-2,6-piperidinedione
- Formula:C13H13N3O3
- MW:259.27 g/mol
- CAS-RN:191732-72-6
- InChI Key:GOTYRUGSSMKFNF-JTQLQIEISA-N
- InChI:InChI=1S/C13H13N3O3/c14-9-3-1-2-7-8(9)6-16(13(7)19)10-4-5-11(17)15-12(10)18/h1-3,10H,4-6,14H2,(H,15,17,18)/t10-/m0/s1
Synthesis
Trade Names
| Country | Trade Name | Vendor | Annotation |
|---|---|---|---|
| D | Revlimid | Celgene | |
| GB | Revlimid | Celgene | |
| USA | Revlimid | Celgene ,2005 |
Formulations
- cps. 5 mg, 10 mg
References
-
- WO 9 803 502 (Celgene; 29.1.1998; USA-prior. 24.7.1996).
- WO 2 006 028 964 (Celgene; 16.3.2006; USA-prior. 3.9.2004).
- US 5 635 517 (Celgene; 3.6.1997; USA-prior. 24.7.1996).
-
medical use for treatment of certain leukemias:
- US 2 006 030 594 (Celgene; 9.2.2006; USA-prior. 4.10.2005).
-
alternative preparation of III:
- WO 2 005 005 409 (Siegfried Ltd.; 20.1.2005; CH-prior. 9.7.2003).
References
- ^ Jump up to:a b c d e REVLIMID [package insert]. Summit, NJ: Celgene Corporation; 2017. Accessed at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021880s055lbl.pdf on 14 September 2018.
- ^ McCarthy PL, Owzar K, Hofmeister CC, et al. (2012). “Lenalidomide after stem-cell transplantation for multiple myeloma”. N. Engl. J. Med. 366 (19): 1770–81. doi:10.1056/NEJMoa1114083. PMC 3744390. PMID 22571201.
- ^ Jump up to:a b c Badros AZ (10 May 2012). “Lenalidomide in Myeloma — A High-Maintenance Friend”. N Engl J Med. 366 (19): 1836–1838. doi:10.1056/NEJMe1202819. PMID 22571206.
- ^ “World Health Organization model list of essential medicines: 21st list 2019”. 2019. hdl:10665/325771.
- ^ Armoiry X, Aulagner G, Facon T (June 2008). “Lenalidomide in the treatment of multiple myeloma: a review”. Journal of Clinical Pharmacy and Therapeutics. 33 (3): 219–26. doi:10.1111/j.1365-2710.2008.00920.x. PMID 18452408.
- ^ Jump up to:a b Li S, Gill N, Lentzsch S (November 2010). “Recent advances of IMiDs in cancer therapy”. Curr Opin Oncol. 22 (6): 579–85. doi:10.1097/CCO.0b013e32833d752c. PMID 20689431.
- ^ Tageja N (March 2011). “Lenalidomide – current understanding of mechanistic properties”. Anti-Cancer Agents Med. Chem. 11 (3): 315–26. doi:10.2174/187152011795347487. PMID 21426296.
- ^ Kotla V, Goel S, Nischal S, et al. (August 2009). “Mechanism of action of lenalidomide in hematological malignancies”. J Hematol Oncol. 2: 36. doi:10.1186/1756-8722-2-36. PMC 2736171. PMID 19674465.
- ^ Yang B, Yu RL, Chi XH, et al. (2013). “Lenalidomide treatment for multiple myeloma: systematic review and meta-analysis of randomized controlled trials”. PLoS ONE. 8 (5): e64354. doi:10.1371/journal.pone.0064354. PMC 3653900. PMID 23691202.
- ^ Dimopoulos MA, Richardson PG, Brandenburg N, et al. (22 March 2012). “A review of second primary malignancy in patients with relapsed or refractory multiple myeloma treated with lenalidomide”. Blood. 119 (12): 2764–7. doi:10.1182/blood-2011-08-373514. PMID 22323483.
- ^ “FDA approves lenalidomide oral capsules (Revlimid) for use in combination with dexamethasone in patients with multiple myeloma”. Food and Drug Administration (FDA). 29 June 2006. Retrieved 15 October 2015.
- ^ “Approved Drugs – Lenalidomide (Revlimid)”. Food and Drug Administration (FDA).
- ^ “REVLIMID Receives Positive Final Appraisal Determination from National Institute for Health and Clinical Excellence (NICE) for Use in the National Health Service (NHS) in England and Wales”. Reuters. 23 April 2009.
- ^ Ness, Stacey (13 March 2014). “New Specialty Drugs”. Pharmacy Times. Retrieved 5 November 2015.
- ^ Jump up to:a b List A, Kurtin S, Roe DJ, et al. (February 2005). “Efficacy of lenalidomide in myelodysplastic syndromes”. The New England Journal of Medicine. 352 (6): 549–57. doi:10.1056/NEJMoa041668. PMID 15703420.
- ^ “PDGFRB platelet derived growth factor receptor beta [Homo sapiens (human)] – Gene – NCBI”.
- ^ Nimer SD (2006). “Clinical management of myelodysplastic syndromes with interstitial deletion of chromosome 5q”. Journal of Clinical Oncology. 24 (16): 2576–82. doi:10.1200/JCO.2005.03.6715. PMID 16735711.
- ^ List AF (August 2005). “Emerging data on IMiDs in the treatment of myelodysplastic syndromes (MDS)”. Seminars in Oncology. 32 (4 Suppl 5): S31–5. doi:10.1053/j.seminoncol.2005.06.020. PMID 16085015.
- ^ List A, Dewald G, Bennett J, et al. (October 2006). “Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion”. The New England Journal of Medicine. 355 (14): 1456–65. doi:10.1056/NEJMoa061292. PMID 17021321.
- ^ “Revlimid Approved In Europe For Use In Myelodysplastic Syndromes”. The MDS Beacon. Retrieved 17 June 2013.
- ^ “Phase II Study of Lenalidomide for the Treatment of Relapsed or Refractory Hodgkin’s Lymphoma”. ClinicalTrials.gov. US National Institutes of Health. February 2009.
- ^ “276 current clinical trials world-wide, both recruiting and fully enrolled, as of 27 February 2009”. ClinicalTrials.gov. US National Institutes of Health. February 2009.
- ^ “Celgene Discontinues Phase 3 Revlimid Study after ‘Imbalance’ of Deaths”. Nasdaq. 18 July 2013.
- ^ Rao KV (September 2007). “Lenalidomide in the treatment of multiple myeloma”. American Journal of Health-System Pharmacy. 64 (17): 1799–807. doi:10.2146/ajhp070029. PMID 17724360.
- ^ “Revlimid Summary of Product Characteristics. Annex I” (PDF). European Medicines Agency. 2012. p. 6.
- ^ Bennett CL, Angelotta C, Yarnold PR, et al. (December 2006). “Thalidomide- and lenalidomide-associated thromboembolism among patients with cancer”. JAMA: The Journal of the American Medical Association. 296 (21): 2558–60. doi:10.1001/jama.296.21.2558-c. PMID 17148721.
- ^ “Potential Signals of Serious Risks/New Safety Information Identified from the Adverse Event Reporting System (AERS) between January – March 2008”. Food and Drug Administration (FDA). March 2008.
- ^ “FDA Drug Safety Communication: Ongoing safety review of Revlimid (lenalidomide) and possible increased risk of developing new malignancies”. Food and Drug Administration(FDA). April 2011.
- ^ Vallet S, Palumbo A, Raje N, et al. (July 2008). “Thalidomide and lenalidomide: Mechanism-based potential drug combinations”. Leukemia & Lymphoma. 49 (7): 1238–45. doi:10.1080/10428190802005191. PMID 18452080.
- ^ Zhu YX, Braggio E, Shi CX, et al. (2011). “Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide”. Blood. 118 (18): 4771–9. doi:10.1182/blood-2011-05-356063. PMC 3208291. PMID 21860026.
- ^ Stewart AK (2014). “Medicine. How thalidomide works against cancer”. Science. 343(6168): 256–7. doi:10.1126/science.1249543. PMC 4084783. PMID 24436409.
- ^ “Top 10 Best-Selling Cancer Drugs of 2018”. Genetic Engineering and Biotechnology News. 22 April 2019. Retrieved 25 April 2019.
- ^ “Revlimid faces NICE rejection for use in rare blood cancer Watchdog’s draft guidance does not recommend Celgene’s drug for NHS use in England and Wales”. Pharma News. 11 July 2013. Retrieved 5 November 2015.
Further reading
- Chang DH, Liu N, Klimek V, et al. (July 2006). “Enhancement of ligand-dependent activation of human natural killer T cells by lenalidomide: therapeutic implications”. Blood. 108 (2): 618–21. doi:10.1182/blood-2005-10-4184. PMC 1895497. PMID 16569772.
- Anderson KC (October 2005). “Lenalidomide and thalidomide: mechanisms of action–similarities and differences”. Seminars in Hematology. 42 (4 Suppl 4): S3–8. doi:10.1053/j.seminhematol.2005.10.001. PMID 16344099.
External links
- Official website Includes list of adverse reactions
- Prescribing Information
- International Myeloma Foundation article on Revlimid
- multiplemyeloma.org Revlimid April 2007 Summary
 |
|
| Clinical data | |
|---|---|
| Pronunciation | /ˌlɛnəˈlɪdoʊmaɪd/ |
| Trade names | Revlimid |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a608001 |
| License data | |
| Pregnancy category |
|
| Routes of administration |
Oral (capsules) |
| ATC code | |
| Legal status | |
| Legal status | |
| Pharmacokinetic data | |
| Bioavailability | Undetermined |
| Protein binding | 30% |
| Metabolism | Undetermined |
| Elimination half-life | 3 hours |
| Excretion | Renal (67% unchanged) |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| ECHA InfoCard | 100.218.924 |
| Chemical and physical data | |
| Formula | C13H13N3O3 |
| Molar mass | 259.261 g/mol g·mol−1 |
| 3D model (JSmol) | |
| Chirality | Racemic mixture |
//////////LENALIDOMIDE, レナリドミド ,REVLIMID, Celgene Corporation, леналидомид , ليناليدوميد , 来那度胺 ,
Benvitimod, Tapinarof, тапинароф , تابيناروف , 他匹那罗 ,
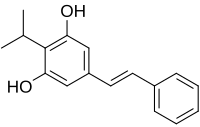
Benvitimod, Tapinarof
- Molecular FormulaC17H18O2
- Average mass254.324 Da
3,5-dihydroxy-4-isopropyl-trans-stilbene
Launched – 2019 CHINA, Psoriasis, Tianji Pharma
тапинароф [Russian] [INN]WBI-1001
تابيناروف [Arabic] [INN]
他匹那罗 [Chinese] [INN]
(E)-2-(1-Methylethyl)-5-(2-phenylethenyl)-1,3-benzenediol
1,3-Benzenediol, 2-(1-methylethyl)-5-(2-phenylethenyl)-, (E)-
1,3-Benzenediol, 2-(1-methylethyl)-5-[(E)-2-phenylethenyl]-
10253
2-Isopropyl-5-[(E)-2-phenylvinyl]-1,3-benzenediol
3,5-Dihydroxy-4-isopropyl-trans-stilbene
5-[(E)-2-phenylethenyl]-2-(propan-2-yl)benzene-1,3-diol
79338-84-4 [RN]
84HW7D0V04
Research Code:WB-1001; WBI-1001
Trade Name:MOA:NSAID
Indication:Atopic dermatitis; PsoriasisStatus:
Phase III (Active)
Company:GlaxoSmithKline (Originator), Welichem Biotech (Originator), 天济药业 (Originator)
2894512
DMVT-505
GSK-2894512
RVT-505
WB-1001
WBI-1001
84HW7D0V04 (UNII code)
DMVT-505
GSK-2894512
RVT-505
WB-1001
WBI-1001
84HW7D0V04 (UNII code)
In May 2019, the drug was appoved in China for the treatment of moderate stable psoriasis vulgaris in adults and, in July 2019, Tianji Pharma (subsidiary of Guanhao Biotech) launched the product in China for the treatment of moderate stable psoriasis vulgaris in adults.
Benvitimod is in phase III clinical trials, Dermavant Sciences for the treatment of atopic dermatitis and psoriasis.
The compound was co-developed by Welichem Biotech and Stiefel Laboratories (subsidiary of GSK). However, Shenzhen Celestial Pharmaceuticals acquired the developement rights in China, Taiwan, Macao and Hong Kong.
Benvitimod (also known as Tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes.It is a product of an alternative ketosynthase-directed stilbenoids biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters. It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis.
Benvitimod (also known as tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes. It is a product of an alternative ketosynthase-directed stilbenoids biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters .[1] It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis. Experiments with infected larvae of Galleria mellonella, the wax moth, support the hypothesis that the compound has antibiotic properties that help minimize competition from other microorganisms and prevents the putrefaction of the nematode-infected insect cadaver.[2]
Tapinarof is a non-steroidal anti-inflammatory drug originated by Welichem Biotech. Dermavant Sciences is developing the product outside China in phase III clinical trials for the treatment of plaque psoriasis. The company is also conducting phase II clinical trials for the treatment of atopic dermatitis. Phase II studies had also been conducted by Welichem Biotech and Stiefel (subsidiary of GlaxoSmithKline) for these indications.
Tapinarof was originated at Welichem Biotech, from which Tianji Pharma and Shenzen Celestial Pharmaceuticals obtained rights to the product in the Greater China region in 2005. In 2012, Welichem licensed development and commercialization rights in all other regions to Stiefel. In 2013, Welichem entered into an asset purchase agreement to regain Greater China rights to the product from Tianji Pharma and Celestial; however, this agreement was terminated in 2014. In 2018, Stiefel transferred its product license to Dermavant Sciences.
_Poinar,_1975.jpg)
Entomopathogenic nematodesemerging from a wax moth cadaver
//////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical research
Benvitimod is being studied in clinical trials for the treatment of plaque psoriasis.[3]
PATENTS
Route 1


1. US2003171429A1.
2. US2005059733A1.
Route 2


Reference:1. CN103265412A.

Patent
https://patents.google.com/patent/CN103992212A/en
phenalkenyl Maude (Benvitimod) is a new generation of anti-inflammatory drugs, are useful for treating a variety of major autoimmune diseases, such as psoriasis, eczema, hair and more concentrated colitis allergic diseases.Phenalkenyl Maud stilbene compound, comprising cis and trans isomers, the trans alkenyl benzene Maude has a strong physiological activity, stability and physical and chemical properties, and cis alkenyl benzene Modesto predominantly trans phenalkenyl Maud byproducts during synthesis, conventional methods such as benzene alkenyl Maude Wittig reaction of cis-isomer impurity is inevitable.

[0004] benzyl trans-alkenyl Maude as main impurities in the synthesis, whether a drug is detected, or monitored during the reaction, the synthesis and analysis methods established cis alkenyl benzene Maude has very important significance.Phenalkenyl Maud conventional synthetic methods the impurity content is very low, and the properties of the cis compound is extremely unstable, easily converted to trans-structure, the synthetic method according to the preceding, the cis compound difficult to separate. The synthesis method has not been reported before in the literature. Thus, to find a synthesis route of cis-alkenyl benzene Maude critical.
[0005] The synthesis of compounds of cis-stilbene, in the prior art, there have been many reports, however, the prior art method of synthesizing a reaction product of the cis starting materials and reagents difficult source, the catalyst used is expensive higher costs, operational difficulties, is not conducive to large-scale production, such as:
① Gaukroger K, John A.Hadfield.Novel syntheses of cis and trans isomers ofcombretastatin A-4 [J] .J.0rg.Chemj 2001, (66): 8135-8138, instead of styrene and substituted phenyl bromide boric acid as the raw material, the Suzuki coupling reaction is a palladium catalyst, to give the cis compound, the reaction follows the formula:

Yield and selectivity of the process the structure is good, but the reaction is difficult source of raw materials, catalyst more expensive, limiting the use of this method.
[0006] ② Felix N, Ngassaj Erick A, Lindsey, Brandon Ej Haines.The first Cu- and
amine-free Sonogashira-type cross-coupling in the C_6 -alkynylation of protected
2, -deoxyadenosine [J] .Tetrahedron Letters, 2009, (65): 4085-4091, with a substituted phenethyl m
Alkynyl easily catalyst Pd / CaC03, Fe2 (CO) 9, Pd (OAc) 2 and the like produce cis compound to catalytic reduction. The reaction follows the formula:

Advantage of this method is stereospecific reduction of alkynes in the catalyst, to overcome the phenomenon of cis-trans isomerization of the Wittig reaction, but the reaction requires at _78 ° C, is not conducive to the operation, and the reagent sources difficult, expensive than high cost increase is not conducive to mass production.
[0007] ③ Belluci G, Chiappe C, Moro G L0.Crown ether catalyzed stereospecificsynthesis of Z_and E-stilbenes by Wittig reaction in a solid-liquid two-phasessystem [J] .Tetrahedron Letters, 1996, (37): 4225-4228 using Pd (PPh3) 4 as catalyst, an organic zinc reagent with a halide compound of cis-coupling reaction formula as follows:

The advantage of this method is that selective, high yield to give cis; deficiency is difficult to handle, the catalyst is expensive.
[0008] ④ new Wang, Zhangxue Jing, Zhou Yue, Zouyong Shun, trans-3,4 ‘, 5-trihydroxy-stilbene China Pharmaceutical Synthesis, 2005, 14 (4);. 204-208, reported that the trans compound of formula was dissolved in DMSO solution at a concentration dubbed, ultraviolet irradiation was reacted at 365nm, converted into cis compounds, see the following reaction formula:

However, the concentration of the solution preparation method, the reaction time is more stringent requirements.

The synthesis of cis-alkenyl benzene Maude application embodiments Example 1 A synthesis of cis-alkenyl Maude benzene and benzene-cis-ene prepared Maude, the reaction was carried out according to the following scheme:

Specific preparation process steps performed in the following order:
(O methylation reaction
The 195.12g (Imol) of 3, 5-hydroxy-4-isopropyl benzoic acid, 414.57g (3mol) in DMF was added 5000ml anhydrous potassium carbonate, mixing, stirred at room temperature, then cooled in an ice-salt bath next, slowly added dropwise 425.85g (3mol) of iodomethane, warmed to room temperature after the addition was complete, the reaction 2h, after completion of the reaction was stirred with water, extracted with ethyl acetate, and concentrated to give 3,5-dimethoxy-4- isopropyl benzoate; yield 93%, purity of 99%.
[0033] (2) a reduction reaction
3000ml tetrahydrofuran and 240g (Imol) 3,5-dimethoxy-4-isopropyl benzoate, 151.40g (4mol) mixing at room temperature sodium borohydride was stirred and heated to reflux was slowly added dropwise 400ml methanol, reaction 4h, was added 3L of water was stirred, extracted with ethyl acetate, washed with water, the solvent was removed by rotary evaporation to give a white solid, to give 3,5-dimethoxy-4-isopropylbenzene methanol; 96% yield purity was 99%.
[0034] (3) the oxidation reaction
The 212g (ImoI) of 3,5-dimethoxy-4-isopropylbenzene methanol, DMSO 800ml and 500ml of acetic anhydride were mixed and stirred at rt After 2h, stirred with water, extracted with ethyl acetate, washed with water, dried , and concentrated to give 3,5-dimethoxy-4-isopropyl-benzaldehyde; 94% yield, 99% purity.
[0035] (4) a condensation reaction
The mixture was 209.18g (lmol) of 3,5-dimethoxy-4-isopropyl-benzoic awake and 136.15g (Imol) phenylacetic acid was added 5000ml of acetic anhydride, stirred to dissolve, sodium acetate was added 246.09g , heating to 135 ° C, the reaction after 6h, cooled to room temperature after adjusting the dilute acid 2 was added, extracted with ethyl acetate, the pH was concentrated, added saturated sodium bicarbonate solution adjusted to pH 7, stirred 2h, and extracted with dichloromethane , adding dilute aqueous hydrochloric acid pH 2, the yellow solid was filtered, to obtain 3,5-dimethoxy-4-isopropyl-stilbene acid; 96% yield, 80% purity.
[0036] (5) decarboxylation reaction
The 327g (Imol) of 3,5-dimethoxy-4-isopropyl-stilbene acid and 384g (6mol) of copper powder were added to 5000ml of quinoline, 180 ° C reaction 3h, cooled to room temperature ethyl acetate was added with stirring, filtered, and the filtrate was washed with dilute hydrochloric acid to the aqueous layer was colorless and the aqueous phase was extracted with ethyl acetate inverted, the organic layers were combined, washed with water and saturated brine until neutral, i.e., spin-dried to give 3,5 – dimethoxy-4-isopropyl-stilbene; 92% yield, 77% purity.
[0037] (6) Demethylation
The 282.32g (Imol) of 3,5-dimethoxy-4-isopropyl-stilbene 4000ml toluene was placed in an ice bath and stirring, was cooled to 0 ° C, and dissolved slowly added 605.9g (5mol after) in N, N- dimethylaniline, was added 666.7g (5mol) of anhydrous aluminum chloride. after stirring for 0.5h, warmed to room temperature, the reaction was heated to 100 ° C 2h, cooled to 60 ° C , hot toluene layer was separated, diluted hydrochloric acid was added to the aqueous phase with stirring to adjust the PH value of 2, extracted with ethyl acetate, washed with water, and concentrated to give the cis-alkenyl benzene Modesto; crude yield 95%, purity 74 %.After separation by column chromatography using 300-400 mesh silica gel, benzene-cis-ene was isolated Maude pure, 68% yield, 98.5% purity. The resulting cis-alkenyl benzene Maud NMR shown in Figure 1, NMR data are as follows:
1HNMR (CDCl3, 500 Hz, δ: ppm), 7.255 (m, 5H), 6.558 (d, 1H), 6.402 (d, 1H), 6.218 (s, 2H), 4.872 (s, 2H), 3.423 (m , 1H), 1.359 (q, 6H). Coupling constants / = 12.
[0038] trans-alkenyl benzene Maud NMR shown in Figure 2, the following NMR data:
1HNMR (CDCl3, 500 Hz, δ: ppm), 7.477 (d, 2H), 7.360 (t, 2H), 6.969 (q, 2H), 6.501 (s, 1H), 4.722 (s, 2H), 3.486 (m , 1H), 1.380 (t, 6H). Coupling constants / = 16.
[0039] HPLC conditions a cis alkenyl benzene Maude pure product: column was Nucleosil 5 C18; column temperature was 20 ° C; detection wavelength 318nm; mobile phase consisting of 50:50 by volume of acetonitrile and water; flow rate It was 0.6mL / min, injection volume of 5 μ L; cis phenalkenyl Maude 18.423min retention time of a peak in an amount of 96.39%, see Figure 3. Trans phenalkenyl Maude 17.630min retention time of a peak, the content was 99.8%, see Figure 4.After mixing the two, trans-alkenyl benzene Maude 17.664min retention time of the peak, cis-alkenyl benzene Maude 18.458min retention time of the peak, see Figure 5.
PATENT
https://patents.google.com/patent/CN103172497A/en

phenalkenyl Maude is a natural product, a metabolite as to be symbionts.Phenalkenyl Maud Escherichia coli, Staphylococcus aureus has a very significant inhibitory effect, in addition, there is a styrenic Maude suppression of inflammation and its reactive derivative with immunomodulating activity. Alkenyl benzene Modesto topical ointment as an active ingredient, as a class of drugs has been completed two clinical treatment of psoriasis and eczema, the results of ongoing clinical phase III clinical studies, it has been shown to be completed in both psoriasis and eczema clearly effect, together with a styrenic Maude is a non-hormonal natural small molecule compounds, can be prepared synthetically prepared, therefore, it exhibits good market prospect.
[0004] a styrenic Maude initial synthesis route is as follows:
[0005]

[0006] The reaction conditions for each step: 1) isopropanol, 80% sulfuric acid, 60 ° C, 65% .2) sodium borohydride, boron trifluoride, tetrahydrofuran, 0 ° C, 90% .3). of thionyl chloride, heated under reflux, 85% .4). triethyl phosphate, 120 ° C, 80% .5). benzaldehyde, sodium hydride, 85% .6) pyridine hydrochloride, 190 ° C, 60 %.
[0007] The chemical synthesis route, although ultimately obtained a styrenic Maude, but the overall yield is low, part of the reaction step is not suitable for industrial production, due to process conditions result in the synthesis of certain byproducts produced is difficult to remove impurities, difficult to achieve the quality standard APIs.
Preparation of 4-isopropyl-dimethoxy-benzoic acid [0011] 1,3,5_
[0012] 1000 l reactor 200 liters of 80% sulfuric acid formulation (V / V), the temperature was lowered to room temperature, put 80 kg 3,5_-dimethoxybenzoate ,, stirring gradually warmed to 60 ° C, in was added dropwise within 25 kg of isopropanol I hour, the reaction was complete after 5 hours, 500 liters of hot water, filtered, the filter cake was washed with a small amount of hot water I th, crushed cake was removed and dried. The dried powder was recrystallized from toluene, the product was filtered to give 78 kg `, yield 86%. Preparation 2,3,5_ dimethoxy-4-isopropylbenzene methanol
[0013] 1000 l reactor was added 50 kg 3,5_ _4_ isopropyl dimethoxy benzoic acid, 24 kg of potassium borohydride, 400 l of THF, at room temperature was slowly added dropwise 65 kg BF3.Et2O was stirred 12 hours, the reaction was complete, pure water was added dropwise to destroy excess BF3, filtered, concentrated to dryness, methanol – water to give an off-white recrystallized 40.3 kg, yield 90.1%.
[0014] Preparation of 3,3,5-_ ■ methoxy _4- isopropyl group gas section
[0015] 1000 l autoclave, 100 kg of 3,5-dimethoxy-4-isopropylbenzene methanol, 220 l of DMF, 0 ° C and added dropwise with stirring and 50 l of thionyl chloride, 24 hours after the reaction was complete, 300 liters of water and 300 liters of ethyl acetate, the aqueous phase was stirred layered discharged, and then washed with 200 liters of water was added 3 times, until complete removal of DMF, was added concentrated crystallized from petroleum ether to give 98 kg of white solid was filtered and dried a yield of 91%.
Preparation of methyl-dimethoxy-4-isopropylbenzene of diethyl [0016] 4,3,5_
[0017] 500 l autoclave, 98 kg 3,5_ _4_ isopropyl dimethoxy benzyl chloride and 120 l of triethyl phosphite, the reaction at 120 ° C 5h, fear distilled off under reduced pressure, the collection 145-155 ° C / 4mmHg fear minutes, cured at room temperature to give a colorless light solid was 118 kg, yield 81.6%.
, 3- [0018] 5, E-1 _ ■ methoxy-2-isopropyl-5- (2-phenylethyl lean-yl) – benzene
[0019] 500 l autoclave, 33 kg 3,5_-dimethoxy-4-isopropylbenzene acid diethyl ester, 10.8 kg of benzaldehyde, and 120 l of tetrahydrofuran, at 40 ° C, and nitrogen with stirring, was added dropwise a solution of 11.8 kg potassium tert-butoxide in 50 liters of tetrahydrofuran, the temperature dropping control not to exceed 50 ° C. after the dropwise addition stirring was continued for I h, the reaction was complete, 150 liters of ethyl acetate and extracted , washed twice with 150 liters of water, 100 l I washed with brine, and the organic phase was dried and concentrated, methanol – water (I: D as a white crystalline solid 25.3 kg, yield 91%.
[0020] 6> 1, 3 ~ _ ■ Light-2-isopropyl-5- (2-phenylethyl lean-yl) – benzene (I), (De Dae dilute benzene)
[0021] 100 l autoclave, 10 kg 1,3_-dimethoxy-2-isopropyl-5- (2-styryl) benzene _ pyridine hydrochloride and 25 kg nitrogen atmosphere was heated to 180 -190 ° C, stirred for 3 hours after the reaction was completed, 20 l HCl (2N) cooling to 100 ° C, and 20 liters of ethyl acetate the product was extracted, dried and concentrated to give the product 7.3 kg, 83% yield.
[0022] The method for purifying:
[0023] 100 l added to the reaction vessel 15.5 kg of crude product and 39 liters of toluene, heated to the solid all dissolved completely, filtered hot and left to crystallize, after crystallization, filtration, the crystals with cold toluene 10 washed liter at 60 ° C, protected from light vacuo dried for 24 hours, to obtain 14 kg of white needle crystals, yield 90%.
CLIP
https://www.eosmedchem.com/article/237.html
Design new synthesis of Route of Benvitimod
Nov 26, 2018
1.Benvitimod and intermediates
Benvitimod 79338-84-4 intermediate: 1999-10-5
Benvitimod 79338-84-4 intermediate: 2150-37-0
Benvitimod 79338-84-4 intermediate: 344396-17-4
Benvitimod 79338-84-4 intermediate: 344396-18-5
Benvitimod 79338-84-4 intermediate: 344396-19-6
Benvitimod 79338-84-4 intermediate: 1080-32-6
Benvitimod 79338-84-4 intermediate: 678986-73-7
Benvitimod 79338-84-4 intermediate: 55703-81-6
Benvitimod 79338-84-4 intermediate: 1190122-19-0
Benvitimod 79338-84-4 intermediate: 443982-76-1
Benvitimod 79338-84-4 intermediate: 100-52-72.ROS-Benvitimod
(1)

(2)
 3.
3.
Name: Benvitimod
CAS#: 79338-84-4
Chemical Formula: C17H18O2
Exact Mass: 254.1307
Molecular Weight: 254.329
Elemental Analysis: C, 80.28; H, 7.13; O, 12.58
Benvitimod 79338-84-4 intermediate: 1999-10-5
Benvitimod 79338-84-4 intermediate: 2150-37-0
Benvitimod 79338-84-4 intermediate: 344396-17-4
Benvitimod 79338-84-4 intermediate: 344396-18-5
Benvitimod 79338-84-4 intermediate: 344396-19-6
Benvitimod 79338-84-4 intermediate: 1080-32-6
Benvitimod 79338-84-4 intermediate: 678986-73-7
Benvitimod 79338-84-4 intermediate: 55703-81-6
Benvitimod 79338-84-4 intermediate: 1190122-19-0
Benvitimod 79338-84-4 intermediate: 443982-76-1
Benvitimod 79338-84-4 intermediate: 100-52-72.ROS-Benvitimod
(1)
(2)
Name: Benvitimod
CAS#: 79338-84-4
Chemical Formula: C17H18O2
Exact Mass: 254.1307
Molecular Weight: 254.329
Elemental Analysis: C, 80.28; H, 7.13; O, 12.58
References
- ^ Joyce SA; Brachmann AO; Glazer I; Lango L; Schwär G; Clarke DJ; Bode HB (2008). “Bacterial biosynthesis of a multipotent stilbene”. Angew Chem Int Ed Engl. 47 (10): 1942–5. doi:10.1002/anie.200705148. PMID 18236486.
- ^ Hu, K; Webster, JM (2000). “Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus–Heterorhabditis infected Galleria mellonella larvae”. FEMS Microbiology Letters. 189 (2): 219–23. doi:10.1111/j.1574-6968.2000.tb09234.x. PMID 10930742.
- ^ “New Topical for Mild to Moderate Psoriasis in the Works”. Medscape. March 5, 2017.
- https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fanie.201814016&file=anie201814016-sup-0001-misc_information.pdf
///Benvitimod, Tapinarof, WBI-1001, тапинароф , تابيناروف , 他匹那罗 , Welichem Biotech, Stiefel Laboratories, Shenzhen Celestial Pharmaceuticals,CHINA 2019 , Psoriasis, Tianji Pharma, Dermavant Sciences, PHASE 3, fda 2022, approvals 2022, vtama, tapinarof
update….
5/23/2022 fda approved, To treat plaque psoriasis, vtama, tapinarof
 |
|
| Names | |
|---|---|
| Preferred IUPAC name
5-[(E)-2-Phenylethen-1-yl]-2-(propan-2-yl)benzene-1,3-diol
|
|
| Other names
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
| Properties | |
| C17H18O2 | |
| Molar mass | 254.329 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
Benvitimod (also known as tapinarof or 3,5-dihydroxy-4-isopropyl-trans-stilbene) is a bacterial stilbenoid produced in Photorhabdus bacterial symbionts of Heterorhabditis nematodes. It is a product of an alternative ketosynthase-directed stilbenoid biosynthesis pathway. It is derived from the condensation of two β-ketoacyl thioesters.[1] It is produced by the Photorhabdus luminescens bacterial symbiont species of the entomopathogenic nematode, Heterorhabditis megidis. Experiments with infected larvae of Galleria mellonella, the wax moth, support the hypothesis that the compound has antibiotic properties that help minimize competition from other microorganisms and prevents the putrefaction of the nematode-infected insect cadaver.[2]
Medical research
Benvitimod is being studied in clinical trials for the treatment of plaque psoriasis.[3]
See also
- Pinosylvin, a molecule produced in pines that does not bear the isopropyl alkylation.
References
- ^ Joyce SA; Brachmann AO; Glazer I; Lango L; Schwär G; Clarke DJ; Bode HB (2008). “Bacterial biosynthesis of a multipotent stilbene”. Angew Chem Int Ed Engl. 47 (10): 1942–5. CiteSeerX 10.1.1.603.247. doi:10.1002/anie.200705148. PMID 18236486.
- ^ Hu, K; Webster, JM (2000). “Antibiotic production in relation to bacterial growth and nematode development in Photorhabdus–Heterorhabditis infected Galleria mellonella larvae”. FEMS Microbiology Letters. 189 (2): 219–23. doi:10.1111/j.1574-6968.2000.tb09234.x. PMID 10930742.
- ^ “New Topical for Mild to Moderate Psoriasis in the Works”. Medscape. March 5, 2017.
CANERTINIB
CANERTINIB
Canertinib
CAS Registry Number: 267243-28-7
CAS Name: N-[4-[(3-Chloro-4-fluorophenyl)amino]-7-[3-(4-morpholinyl)propoxy]-6-quinazolinyl]-2-propenamide
Additional Names: N-[4-(3-chloro-4-fluorophenylamino)-7-(3-morpholin-4-ylpropoxy)quinazolin-6-yl]acrylamide
Molecular Formula: C24H25ClFN5O3
Molecular Weight: 485.94
Percent Composition: C 59.32%, H 5.19%, Cl 7.30%, F 3.91%, N 14.41%, O 9.88%
Literature References: Irreversible pan-erbB tyrosine kinase inhibitor. Prepn: A. J. Bridges et al., WO 0031048; eidem, US 6344455 (2000, 2002 both to Warner-Lambert); J. B. Smaill et al., J. Med. Chem. 43, 1380 (2000). Clinical pharmacokinetics in patients with solid malignancies: E. Calvo et al., Clin. Cancer Res. 10, 7112 (2004); and tolerability in refractory cancer: J. Nemunaitis et al., ibid. 11, 3846 (2005). Review of pharmacology and mechanism of action: L. F. Allen et al., Semin. Oncol. 30, Suppl. 16, 65-78 (2003); of development and clinical experience: C. M. Galmarini, IDrugs 7, 58-63 (2004).
Properties: Crystals from methanol, mp 188-190°.
Melting point: mp 188-190°
Canertinib dihydrochloride, CI-1033, PD-183805(free base)
Derivative Type: Dihydrochloride
CAS Registry Number: 289499-45-2
Manufacturers’ Codes: CI-1033
Molecular Formula: C24H25ClFN5O3.2HCl
Molecular Weight: 558.86
Percent Composition: C 51.58%, H 4.87%, Cl 19.03%, F 3.40%, N 12.53%, O 8.59%
Properties: Sol in water.
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Tyrosine Kinase Inhibitors.
267243-28-7 [RN]
2-propenamide, N-[4-[(3-chloro-4-fluorophenyl)amino]-7-[3-(4-morpholinyl)propoxy]-6-quinazolinyl]- [ACD/Index Name]
8256
C78W1K5ASF
N-{4-[(3-Chloro-4-fluorophenyl)amino]-7-[3-(4-morpholinyl)propoxy]-6-quinazolinyl}acrylamide
Canertinib (CI-1033) is an experimental drug candidate for the treatment of cancer. It is an irreversible tyrosine-kinase inhibitor with activity against EGFR (IC50 0.8 nM), HER-2 (IC50 19 nM) and ErbB-4 (IC50 7 nM).[1][2] By 2015, Pfizer had discontinued development of the drug.[3]
Canertinib has been reported as a substrate for OATP1B3. Interaction of canertinib with OATP1B3 may alter its hepatic disposition and can lead to transporter mediated drug-drug interactions.[4] Also, canertinib is not an inhibitor of OATP-1B1 or OATP-1B3 transporter.[5]
SYN
J Med Chem 2000,43(7),1380
| EP 1131304; US 6344455; WO 0031048 |
4-Chloro-7-fluoro-6-nitroquinazoline (I) was condensed with 3-chloro-4-fluoroaniline (II) to afford the 4-anilino quinazoline (III). Displacement of the activated fluorine of (III) with the potassium alkoxide of morpholinopropanol (IV) gave the morpholinopropyl ether (V). Subsequent reduction of the nitro group of (V), either using iron dust and acetic acid or catalytic hydrogenation over Raney-Ni, furnished aminoquinazoline (VI). This was finally condensed with acrylic acid (VII), via activation as the mixed anhydride with isobutyl chloroformate or using EDC as the coupling reagent, to provide the title acrylamide.
PATENT
https://patents.google.com/patent/CN103242244A/en
canertinib (Canertinib, I), chemical name 4- (3-chloro-4-fluoroanilino) -7- [3- (4_-morpholinyl) propoxy] -6-propylene quinazoline amide group, and by the US Pfizer Warner Lambert developed jointly an irreversible epidermal growth factor receptor (pan-ErbB) selective inhibitor, which is capable of binding to the cell surface of all members of the ErbB family adenosine triphosphate binding site, thereby inhibiting the activation of these receptors and their downstream mitogenic signal transduction pathways. Clinical studies show that the product has good resistance, can be effective in treating metastatic breast cancer, ovarian cancer, cervical cancer and other tumors, and can be combined with a variety of antineoplastic agents exhibit a synergistic effect.
[0004]

[0005] China Patent No. CN1160338C, CN1438994A and No. No. CN1745073A reported the preparation of canertinib: A nucleus 4- [(3-chloro-4-fluorophenyl) amino] -6-nitro 7-fluoro-quinazoline (VIII) as a starting material, under basic conditions with 3- (4-morpholinyl) -1-propanol 7-position substitution reaction occurs to give 4- [(3-chloro – 4-fluorophenyl) amino] -6-nitro-7- [3- (4-morpholinyl) -1-propoxy] quinazoline (IX); intermediate (IX) through the 6-position nitro reduction, to give the corresponding amino compound (X); amino compound (X) to give canertinib acylation reaction (I) with acrylic acid or acryloyl chloride occurs.
[0006] In addition, “Qilu Pharmaceutical Affairs” 30, 2011, Vol. 10, page 559, and “China Industrial Medicine” 2010 Volume 41, No. 6, pp. 404 also reported an improved method of the above-prepared and studied method from 7-fluoro-quinazolin-3-one (V) via nitration, chloro and condensation reaction of the preparation of intermediate (VIII) is.
[0007]

[0008] This shows that the current Kanai prepared for Nepal is mainly the 4-position through an intermediate (VII), respectively, a functional transformation of the 6-position and 7-position achieved. Since the intermediate (VII) a fluorine-containing compounds, materials are not readily available, many steps, and many steps are required to be isolated and purified by column chromatography, which is not required for industrialization.
Example a:
[0023] at room temperature, to a three-necked flask was added diisopropyl azodicarboxylate (3mL, 15mmol) and tetrahydrofuran 5mL, dropwise addition of triphenylphosphine (4.0g, 15mmol) in tetrahydrofuran 25mL solution at room temperature, kept at room temperature for 2 hours. Under nitrogen, 3- (4-morpholinyl) -1_-propanol (0.49g, 3.4mmol) in 5mL of tetrahydrofuran was added dropwise to the reaction system after the dropwise addition is complete, 6-amino – 7-hydroxy-3,4-dihydro-quinazolin-4-one (II) (0.53g,
3.0mmol), stirred at room temperature for 4 hours. Solution of 3- (4-morpholinyl) -1-propanol (0.38g, 2.6mmol) in 5mL of tetrahydrofuran was continued at room temperature for 2 hours, the end of the reaction was monitored TLC. Recovery of the solvent by distillation under reduced pressure, the residue was treated with dilute hydrochloric acid, pH = 5-6, extracted with ethyl acetate, the organic phase was washed with saturated sodium carbonate adjusted pH = 10-11. The aqueous phase was freeze-dried in vacuo to give an off-white solid 6-amino-7- [3- (4-morpholinyl) propoxy] _3,4- dihydroquinazolin-4-one (111) 0.80g yield 87.7%.
[0024] Example II:
[0025] to a three-neck flask was added 6-amino-7- [3- (4_ morpholino) propoxy] quinazolin-dihydro _3,4_ one _4_
(III) (0.76g, 2.5mmol), triethylamine (0.25g, 2.5mmol) and dichloromethane 20mL, warmed to 40-45 ° C, stirred until homogeneous dissolution system. Dropped below 10 ° C, was slowly added dropwise acryloyl chloride (0.25g, 2.8mmol) in dichloromethane IOmL solution dropwise at room temperature after continued for 6 h, TLC detection reaction was completed. The reaction solution was respectively 10% sodium bicarbonate solution and water, dried over anhydrous sodium sulfate. Recovery of the solvent under reduced pressure, the residue was recrystallized from ethyl acetate to give a white solid 7- [3- (4-morpholinyl) propoxy] -6-acrylamido-3,4-dihydro-quinazoline – 4-one (IV) 0.81g, 90.5% yield.
[0026] Example III:
Under [0027] nitrogen, to a three-necked flask was added 7- [3- (4_-morpholinyl) propoxy] -6-acrylamido-_3,4- dihydroquinazolin-4-one (IV ) (3.58g, IOmmol), benzotriazol-1-yloxytris (dimethylamino) phosphonium iron hexafluorophosphate (BOP) (6.63g, 15mmol) and acetonitrile 100mL. Under stirring, a solution of 1,8-diazabicyclo [5.4.0] ^ a-7-ene (DBU) (2.28g, 15mmol), dropwise, at room temperature for 12 hours. Warmed to 60 ° C, the reaction was continued for 12 hours. The solvent was removed by distillation under reduced pressure, ethyl acetate was added to dissolve IOOmL, washed with 2M sodium hydroxide and 20mL. The organic phase was separated, dried and concentrated under reduced pressure. The residue was dissolved in tetrahydrofuran IOOmL, 4-chloro-3-fluoroaniline (1.89g, 13mmol) and sodium hydride (0.32g, 13mmol), was heated to 50 ° C, reaction was stirred for 5 hours, the end of the reaction was monitored TLC. Quenched with saturated brine the reaction, the organic phase was separated, dried, evaporated under reduced pressure to recover the solvent to give an off-white solid. Recrystallized from ethanol to give an off-white solid canertinib (I) 4.05g, yield 83.5%.
[0028] Example IV:
Under [0029] nitrogen, to a three-necked flask was added 7- [3- (4_-morpholinyl) propoxy] -6-acrylamido-3,4-dihydro-quinazolin-4-one (IV ) (3.58g, IOmmol), benzotriazol-1-yloxytris (dimethylamino) phosphonium iron hexafluorophosphate (BOP) (6.63g, 15mmol) and acetonitrile lOOmL. Under stirring, dropwise power port I, 5- diazabicyclo [4.3.0] – non-5-ene (DBN) (1.86g, 15mmol), dropwise, at room temperature for 12 hours. Warmed to 60 ° C, the reaction was continued for 12 hours. The solvent was removed by distillation under reduced pressure, ethyl acetate was added to dissolve IOOmL, washed with 2M sodium hydroxide and 20mL. The organic phase was separated, dried and concentrated under reduced pressure. The residue was dissolved in tetrahydrofuran IOOmL, 4-chloro-3-fluoroaniline (1.89g, 13mmol) and sodium hydride (0.32g, 13mmol), was heated to 50 ° C, reaction was stirred for 5 hours, the end of the reaction was monitored TLC. Quenched with saturated brine the reaction, the organic phase was separated, dried, evaporated under reduced pressure to recover the solvent to give an off-white solid. Recrystallized from ethanol to give an off-white solid canertinib (I) 3.85g, yield 79.4%. ·
[0030] Example Five:
Under [0031] nitrogen, to a three-necked flask was added 7- [3- (4_-morpholinyl) propoxy] -6-acrylamido-3,4-dihydro-quinazolin-4-one (IV ) (3.58g, IOmmol), benzotriazol-1-yloxytris (dimethylamino) phosphonium hexafluorophosphate gun (BOP) (6.63g, 15mmol), 4_-chloro-3-fluoroaniline ( 1.89g, 13mmol) and N, N- dimethylformamide lOOmL. Under stirring, a solution of I, 8- diazabicyclo [5.4.0] – ^ a _7_ ene (DBU) (2.28g, 15mmol), dropwise, at room temperature for 12 hours. Warmed to 60 ° C, the reaction was continued for 12 hours. The solvent was removed by distillation under reduced pressure, ethyl acetate was added to dissolve IOOmL, washed with 2M sodium hydroxide and 20mL. The organic phase was separated, dried and concentrated under reduced pressure. The residue was recrystallized from ethanol to give an off-white solid canertinib (1) 2.32g, yield 47.8%.

References
GW; Loo, JA; Greis, KD; Chan, OH; Reyner, EL; Lipka, E; Showalter, HD; et al. (2000). “Tyrosine kinase inhibitors. 17. Irreversible inhibitors of the epidermal growth factor receptor: 4-(phenylamino)quinazoline- and 4-(phenylamino)pyrido3,2-dpyrimidine-6-acrylamides bearing additional solubilizing functions”. Journal of Medicinal Chemistry. 43 (7): 1380–97. doi:10.1021/jm990482t. PMID 10753475.
- ^ CI-1033 (Canertinib), Selleck Chemicals
- ^ http://adisinsight.springer.com/drugs/800012072
- ^ Khurana V, Minocha M, Pal D, Mitra AK (March 2014). “Role of OATP-1B1 and/or OATP-1B3 in hepatic disposition of tyrosine kinase inhibitors”. Drug Metabol Drug Interact. 29 (3): 1–11. doi:10.1515/dmdi-2013-0062. PMC 4407685. PMID 24643910.
- ^ Khurana V, Minocha M, Pal D, Mitra AK (May 2014). “Inhibition of OATP-1B1 and OATP-1B3 by tyrosine kinase inhibitors”. Drug Metabol Drug Interact. 29 (4): 1–11. doi:10.1515/dmdi-2014-0014. PMC 4407688. PMID 24807167.
 |
|
| Names | |
|---|---|
| IUPAC name
N-{4-[(3-Chloro-4-fluorophenyl)amino]-7-[3-(morpholin-4-yl)propoxy]quinazolin-6-yl}prop-2-enamide
|
|
| Other names
CI-1033; PD-183805
|
|
| Identifiers | |
|
3D model (JSmol)
|
|
| ChEBI | |
| ChEMBL | |
| ChemSpider | |
|
PubChem CID
|
|
| UNII | |
|
CompTox Dashboard (EPA)
|
|
| Properties | |
| C24H25ClFN5O3 | |
| Molar mass | 485.94 g·mol−1 |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
|
|
/////////////CANERTINIB
Cilastatin, циластатин , سيلاستاتين , 西司他丁 ,

Cilastatin
シラスタチン
циластатин [Russian] [INN]
سيلاستاتين [Arabic] [INN]
西司他丁 [Chinese] [INN]
UNII141A6AMN38
CAS number 82009-34-5
WeightAverage: 358.453
Monoisotopic: 358.156242642
Chemical FormulaC16H26N2O5S
- (L)-7-(2-Amino-2-carboxy-ethylsulfanyl)-2-[(2,2-dimethyl-cyclopropanecarbonyl)-amino]-hept-2-enoic acid
- (Z)-(S)-6-carboxy-6-[(S)-2,2-dimethylcyclopropanecarboxamido]hex-5-enyl-L-cysteine
- (Z)-7-((R)-2-Amino-2-carboxy-ethylsulfanyl)-2-[((S)-2,2-dimethyl-cyclopropanecarbonyl)-amino]-hept-2-enoic acid
- (2Z)-7-{[(2R)-2-amino-2-carboxyethyl]sulfanyl}-2-{[(1S)-2,2-dimethylcyclopropyl]formamido}hept-2-enoic acid
MK 0791|Primaxin®
Primaxin®
TL8005438
UNII:141A6AMN38
UNII-141A6AMN38
EINECS 279-875-8
Cilastatin
CAS Registry Number: 82009-34-5
CAS Name: (2Z)-7-[[(2R)-2-Amino-2-carboxyethyl]thio]-2-[[[(1S)-2,2-dimethylcyclopropyl]carbonyl]amino]-2-heptenoic acid
Manufacturers’ Codes: MK-791
Molecular Formula: C16H26N2O5S
Molecular Weight: 358.45
Percent Composition: C 53.61%, H 7.31%, N 7.82%, O 22.32%, S 8.95%
Literature References: Prevents renal metabolism of penem and carbapenem antibiotics by specific and reversible dehydropeptidase I inhibition.
Synthesis and combination with thienamycins: D. W. Graham et al., EP 48301; H. Kropp et al., EP48025 (both 1982 to Merck & Co.), C.A. 97, 145271b, 145270a (1982). Combination with penems: F. M. Kahan, H. Kropp, EP72014 (1983 to Merck & Co.), C.A. 99, 70272h (1983). The articles cited below discuss the activity of cilastatin alone and in combination with imipenem, q.v. Dipeptidase inhibition, pharmacokinetics: S. R. Norrby et al., Antimicrob. Agents Chemother. 23,300 (1983). Stimulation of granulocyte function: H. Gnarpe et al., ibid. 25, 179 (1984). HPLC determn in serum: C. M. Myers, J. L. Blumer, ibid. 26, 78 (1984). Enhances intrathecal and ocular penetration of imipenem: A. W. Chow et al., ibid. 23, 634 (1983) and K. R. Finlay et al., Invest. Ophthalmol. Visual Sci. 24, 1147 (1983), respectively. In experimental meningitis: D. E. Washburn et al.,J. Antimicrob. Chemother. 12, 39 (1983). Series of articles on pharmacokinetics, safety and tolerance and efficacy of cilastatin/imipenem: ibid. 12, Suppl. D, 1-155 (1983); Infection 14, Suppl. 2, S111-S180 (1986).
Derivative Type: Sodium salt
CAS Registry Number: 81129-83-1
Additional Names: Cilastatin sodium
Molecular Formula: C16H25N2NaO5S
Molecular Weight: 380.43
Percent Composition: C 50.51%, H 6.62%, N 7.36%, Na 6.04%, O 21.03%, S 8.43%
Properties: Off-white to yellowish-white hygroscopic, amorphous solid. pKa1 2.0; pKa2 4.4; pKa3 9.2. Very sol in water, methanol.
pKa: pKa1 2.0; pKa2 4.4; pKa3 9.2
Therap-Cat: Antibacterial adjunct (dipeptidase inhibitor).
Keywords: Antibacterial Adjuncts; Renal Dipeptidase Inhibitors.
FDA 2019 APPROVED 2019/7/16, Imipenem, cilastatin and relebactam, Recarbrio
|
Antibacterial
|
|
| Disease |
Uncomplicated urinary tract infection
|
|---|
Cilastatin inhibits the human enzyme dehydropeptidase.[1]
Yatendra Kumar, “Process for the preparation of amorphous cilastatin sodium.” U.S. Patent US20040152780, issued August 05, 2004.US20040152780
Cilastatin is an inhibitor of renal dehydropeptidase, an enzyme responsible for both the metabolism of thienamycin beta-lactam antibiotics as well as conversion of leukotriene D4 to leukotriene E4. Since the antibiotic, imipenem, is one such antibiotic that is hydrolyzed by dehydropeptidase, cilastatin is used in combination with imipenem to prevent its metabolism. The first combination product containing both drugs was approved by the FDA in November of 1985 under the trade name Primaxin, marketed by Merck & Co.9 A newer triple-drug product was approved in July 2019 under the trade name Recarbrio which also contains relebactam.8
Cilastatin is indicated, in combination with imipenem with or without relebactam, for the treatment of bacterial infections including respiratory, skin, bone, gynecologic, urinary tract, and intra-abdominal as well as septicemia and endocarditis.6,5

Uses
Dehydropeptidase is an enzyme found in the kidney and is responsible for degrading the antibiotic imipenem. Cilastatin can therefore be combined intravenously with imipenem in order to protect it from degradation, prolonging its antibacterial effect.
Imipenem alone is an effective antibiotic and can be given without cilastatin. Cilastatin itself does not have antibiotic activity, although it has been proved to be active against a zinc-dependent beta-lactamase that usually confers antibiotic resistance to certain bacteria, more precisely, the carbapenem family of antibiotics. This property is due to the physicochemical similarities between membrane dipeptidase (MDP), the compound it is usually set to target, and the bacterial metallo-beta-lactamase carried by the CphA gene.[1] The combination allows the antibiotic to be more effective by changing the pharmacokinetics involved. Thus imipenem/cilastatin, like amoxicillin/clavulanic acid, is a commonly used combination product.
PATENT
https://patents.google.com/patent/EP2402312A1
Cilastatin sodium is the sodium salt of a derivatized heptenoic acid. Its chemical name is [R-[R*,S*-(Z)]]-7-[(2-amino-2-carboxyethyl)thio]-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino]-2-heptenoic acid, monosodium salt. It is an off-white to yellowish-white, hygroscopic, amorphous compound. PRIMAXIN (Imipenem and Cilastatin) is a formulation of Imipenem (a thienamycin antibiotic) and Cilastatin sodium.
Imipenem with Cilastatin acts as an effective antibiotic for the treatment of infections of various body systems. PRIMAXIN is a potent broad-spectrum antibacterial agent for intramuscular administration. Imipenem can be further described as a semi-synthetic thienamycin that is administered intravenously or intramuscularly in combination with Cilastatin to reduce toxicity. Cilastatin, a renal dipeptidase inhibitor, inhibits the enzymatic breakdown of Imipenem and increases urinary excretion of the active drug.
Originally Cilastatin was disclosed in US patent number 5,147,868 . This patent also discloses various processes for the preparation of Cilastatin, particularly example 19 A of this patent disclose a process for the preparation of Cilastatin. According to this example the condensation of 7-chloro-2-oxoheptanoic acid ethyl ester (I) with (S)-2,2-dimethylcyclopropanecarboxamide (II) by means of p-toluene sulphonic acid in refluxing toluene gives (S)-7-chloro-2-(2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid ethyl ester (III), which is hydrolyzed in aq. NaOH to yield the corresponding carboxylic acid (IV). Finally, this compound is condensed with (R)-cysteine (V) by means of NaOH in water to afford the target Cilastatin, followed by isomerisation to at 3.0 pH. The process followed in this example is depicted as below:

WO 03/018544 claims a process for the purification of Cilastatin, which comprises contacting a solution of crude Cilastatin with a non-ionic adsorbent resin and recovering pure Cilastatin from a solution thereof. This publication also claims a process for the isomerisation of Cilastatin by heating a solution of Cilastatin containing the corresponding E isomer at a pH of about 0.5 to 1.5. This invention not suitable for plant point of view as it involves column chromatography.
US 2004/0152780 claims a process for the preparation of pure Cilastatin sodium in an amorphous form which comprises recovering Cilastatin sodium from a solution thereof which contains an organic solvent, homogeneous mixture of organic solvents, or homogeneous mixture of organic solvents and water, by solvent precipitation. According to this patent the pure Cilastatin sodium in amorphous form was recovered from the solution of Cilastatin sodium in a solvent (where Cilastatin sodium was soluble) by adding an anti-solvent (where Cilastatin sodium was insoluble).
WO 2006/022511 claims a process for preparing Cilastatin sodium via Cilastatin amine salt, also the said patent claims Cilastatin ammonium salt. However EP 0 048 301 page 2; line 33-37 & US 4,616,038 col 36; 40-44 anticipates the claim of the said publication. Also this patent utilizes the column chromatography for removing sodium chloride.
However taking the consideration the commercial importance of Cilastatin sodium and Imipenem, there remains a need of convenient process. Hence, we focused our research to find an alternative processes and succeeded with a process that eliminates the foregoing problems associated with earlier processes.

Example 1Preparation of 7-chloro-2-[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid (II) (starting material):
- [0032]
To the solution of S-2, 2-dimethylcylopropyl carboxamide (100gm) in toluene (500) was added Ethyl-7-chloro-2-oxo-heptanoate (270gm) and p-toluene sulphonic acid (1.5gm). The resulted solution was refluxed for 20hrs azeotropically. The resulted mass was cooled to 5-10°C and added the solution of sodium hydroxide (140gm) in water 500 ml and the resulted two-layered solution was stirred for 8hrs at 25-30°C up to the complete disappearance of ester. The toluene layer was separated and the aqueous layer was washed with toluene. The pH of the aqueous layer was adjusted to 4.0 to 4.5 and extracted with toluene (1 lt). The toluene layer containing 7-chloro-2-[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid was washed with water and used as such for the next step. The ratio of Z and E isomer 90:10% was obtained.
Example 2Isomerisation of 7-chloro-2-[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid (II):
- [0033]
To the toluene layer, obtained from example -1, was added hydrochloric acid (11t) and stirred for 4hrs at 25-30°C till the disappearance of E isomer. The toluene layer was separated and washed with water and followed by brine. The toluene layer was distilled out under vacuum up to 50% of the original volume. To the reaction mass hexane/IPE was added at 50°C and cooled to 0-5°C. The precipitated mass was filtered and washed with hexane (200ml) and dried under vacuum to obtained 99% pure Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid (150gm) as white solid.
Example 3Preparation of Cilastatin Acid (I):
- [0034]
To the solution of sodium hydroxide (90gm) in water (11t) was added L-Cysteine hydrochloride monohydrate (96gm) and Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid and stirred at 25-30°C till the disappearance of Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid. After completion of reaction, the reaction mass was washed with dichloromethane (500ml). To the aqueous layer was added carbon (10 gm) and stirred and filtered. To the filtrate was added water (11t) and the pH of the solution was adjusted to 3.0 and stirred for 24 hrs. The precipitated mass was filtered, washed with water (200ml) and with acetone (500ml) and dried to obtain 110gm white solid with 97% purity. The solid was dissolved in water (700ml) and added MDC (700ml) and ethyl acetate (100ml) and stirred for 10hrs. The precipitated mass was filtered and washed with water (100ml) and acetone (200ml) and dried to obtain 100gm white Cilastatin acid with 99.5% purity.
Example 4Preparation of Cilastatin Sodium:
- [0035]
The Cilastatin acid (100gm, 99.5%) was dissolved in the mixture of ethanol (2.5lt) and triethylamine (30gm) at 25 to 30°C. To the resulted clear solution was added carbon (10gm) and stirred and filtered. The filtrated was filtered again through sterile micron (0.2 µ) filter. To the resulted clear solution was added solution of sodium ethyl hexanoate (70gm) in ethanol (70ml) and stirred for 3hrs at 25 to 30°C.The precipitated Cilastatin sodium was filtered and washed with ethanol (80ml) and followed by acetone (200ml) and dried under vacuum to obtained 95gm Cilastatin sodium as amorphous white solid with 99.5% purity.
Example 5Preparation of Cilastatin Acid:
- [0036]
To the solution of sodium hydroxide (88gm) in methanol (1500ml) was added Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid and stirred to dissolve. To the resulted clear solution was added L-Cysteine hydrochloride monohydrate (97gm) and stirred the resulted suspension at 60 to 65°C till the disappearance of Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid. After completion of reaction, the pH insoluble salts were filtered. The filtrate was distilled out under vacuum. The residue was dissolved in water (500ml) and washed with dichloromethane (500ml). The pH of aqueous layer was adjusted to 3 to 4 from the original pH in the range of 5.5, and with n-butanol (500ml). The butanol layer was washed with water and distilled. The residue was dissolved in water (100ml) and added acetonitrile (1500ml) at 50°C and further refluxed at 80°C for one hr. The precipitated cilastatin acid was filtered and washed with acetonitrile (100ml). The crude wet cake (60gm) was refluxed with acetonitrile water mixture (9:1,1500ml), and cooled to yield 60gm pure cilastatin acid with 99.5% purity.
Example 6Preparation of Cilastatin Acid:
- [0037]
To the solution of sodium hydroxide (88gm) in methanol (1500ml) was added Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid and stirred to dissolve. To the resulted clear solution was added L-Cysteine hydrochloride monohydrate (97gm) and stirred the resulted suspension at 60 to 65°C till the disappearance of Z-7-chloro-2[[(1S)-2,2-dimethyl cyclopropane]carboxamide]-2-heptenoic acid. The pH of the reaction mass was adjusted to 7.0 with conc.HCl and filterd the insoluble salts. The filtrated was distilled out under vacuum. The residue was dissolved in water (500ml) and washed with dichloromethane (500ml). The pH of aqueous layer was adjusted to 3 to 4 from the original pH in the range of 5.5, and with n-butanol (500ml). The butanol layer was washed with water and distilled up to 50% of original volume and stirred at 25°C. The precipitated cilastatin acid was filtered and washed with n-butanol (100ml) followed by acetone to yield 60gm pure cilastatin acid with 99.7% purity.
Example 7Preparation of Cilastatin, Sodium:
- [0038]
The Cilastatin acid (100gm, 99.5%) was dissolved in the mixture of n-butanol (2.5lt) and triethylamine (30gm) at 25 to 30°C. To the resulted clear solution was added carbon (10gm) and stirred and filtered. The filtrated was filtered again through sterile micron (0.2 µ) filter. To the resulted clear solution was added solution of sodium ethyl hexanoate (70gm) in n-butanol (70ml) and stirred for 3hrs at 25 to 30°C. The precipitated Cilastatin sodium was filtered and washed with n-butanol (80ml) and followed by acetone (200ml) and dried under vacuum to obtained 80gm Cilastatin sodium as amorphous white solid with 99.78% purity.
Abbreviations;
- [0039]
- DBU: diazabicyclo[5,4,0]undec-7-en
- DBN : 1,5-diazabicyclo[4,3,0]-non-5-ene
- TMG: 1,1,3,3-tetramethylguanidine
- DABCO: 1,4-diazabicyclo-[2,2,2]-octane
PATENT
https://patents.google.com/patent/WO2006022511A1/en
Cilasatin sodium salt i.e., [R-[R*, S*-(Z)]] –
7-[(2-amino-2-carboxyethylthio)-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino-2-hepa tenoic acid monosodium salt represented by following chemical formulae (1)1 has been used with imipenem in order to prevent its renal metabolism. Imipenem/cilastatin sodium is used as a potent broad spectrum antibacterial agent. [3] There have been several reports on the method for preparing a cilastatin sodium until now: for example, EP 48301 Bl discloses a method for the preparation of a cilastatin sodium salt using by Grignard reaction started from l-bromo-5-chloropentane (2′) explained by following Reaction Scheme 1; Donald W.
Graham et al discloses a preparation method using ethyl- 1, 3-dithian-2-carboxylate as a starting material (Donald W. Graham et al, J. Med. Chem., 30, pplO74, 1987) etc. [4] [Reaction Scheme 1]
[5]

[6]

[7] [8] As shown in the above Reaction Scheme 1, l-bromo-5-chloropentane (2′) is reacted with diethyl oxalate through Grignard reaction to afford ethyl 7-chloro-2-oxo-hepanoate (3′)at the 1st step; ethyl 7-chloro-2-oxo-heptanoate (3′) is reacted with (S)-2, 2-dimethylcyclopropanecarboxamide to obtain ethyl (Z )-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate (4′) at the 2n step.
[9] However, present inventors has confirmed that considerable amount (about 10 to 13%) of (E)-form isomer thereof (7′)was produced during the 2nd step as a reaction impurity by gas chromatography. The (E)-form isomer is further subjected to hydrolysis resulting in (E)-7-chloro-2-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (8′)as shown in following Reaction Scheme 2.
[10] [H] However, present inventors has confirmed that considerable amount (about 10 to 13%) of (E)-form isomer thereof (7′) was produced during the 2nd step as an reaction impurities by gas chromatography as shown in following Reaction Scheme 2. The (E )-form isomer is further subjected to hydrolysis resulting in (E)-7-chloro-2-((S)-2, 2-dimethylcyclopropylcarboxamide)-2-heptanoic acid (8′).
[12] [Reaction Scheme 2] [13]


[14] [15] There have been tried to solve the problems for example, the isomer impurity was removed by the acidification followed by recrystallization step or by adding cysteine to the reaction solution obtained in the 3r step at the above described 4 step, reacting with together to form (E)-7-(L-amino-2-carboxyethylthio)-2-((S )-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid and finally removing the reacted impurity by acidifying and heating step in the known preparation till now. However, the present inventors found that there remained unsolved problem such that the recrystallization yield of the product, i.e., (Z)-7-chloro-2-((S )-2,2-dimethylcyclopropylcarboxamide)-2-heptanoic acid was very poor because of the formed byproduct, i.e., (E
)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid in 3rdstep and further the unknown impurity (10′) and (S)-2,2-dimethylcyclopropanecarboxamide (H’) were produced by acidifying and heating reaction solution at the above described the 4 step as shown in following Reaction Scheme 3 confirmed by HPLC analysis, which give rise to another difficulty in the purification of final products. [16] [17] [Reaction Scheme 3] [18]

NH
(Z) and (E) m ix ture (91)

C ondition
(105 0 15
[19] In addition to above described problems, present inventors have found that the cilastatin isolated through the above described 4th step consisting of eluting the cation exchange resin with ammonia solution, concentrating the eluate and solidifying with ethanol and diethyl ether exists in the form of its ammonium salt not free acid form as disclosed in the patent. Using an acid such as hydrochloric acid in order to obtain free acid accompany with unwanted formation of inorganic ammonium salt such as ammonium chloride, which could not afford high purity of cilastatin sodium salt in the end.
[20] [21] Therefore, there have been tried to solve the above-described problems: for example, PCTAVO 0318544 (Al) discloses the isolation method using by neutral HP 20 resin column instead of cationic resin disclosed in EP 48301 Bl; PCTAVO 02094742 (Al) discloses the method for preparing cilastatin sodium salt (Ia) from cilastatin (6′), the disclosure of which cited documents are incorporated herein by reference.
[22]
[23] However, the above-described methods for preparing cilastatin using column chro¬ matographic process are not suitable for commercial mass production.
[24]
[25] The present inventors have made extensive researches to discover novel method for preparing cilastatin sodium salt with high yield and mass production and finally completed the invention by founding novel preparation for obtaining purposed cilastatin sodium salt; i.e., selectively hydrolyzing (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate, isolating (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt from the reaction mixture, adopting the cilastatin amine salt instead of free acid form disclosed in cited references and the use of sodium hydroxide and cationic exchange resin with pH control in order to obtain cilastatin sodium salt with high purity and high yield.
Example 1: Preparation of ethyl (Z)-7-chloro-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoate (4)
[67]
[68] l-bromo-5-chloropentane (29 Ig, 1.57 mol) was reacted with diethyl oxalate
(206.5g) through Grignard reaction to obtain ethyl 7-chloro-2-oxo-heptanoate (3) and the compound (3) was reacted with (S)-2,2-dimethylcyclopropanecarboxamide to obtain ethyl (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptanoate (237g, 0.79 mol). The above-described step was performed by the procedure according to the procedure disclosed in EP 48301 (Bl).
[69]
[70] Example 1: Preparation of ethyl (Z)-7-chloro-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12)
[71]
[72] 1-1. ( Z V7-chloro-(YSV2. 2-dimethylcyclopropanecarboxamidoV2-heptanoic acid sodium salt
[73] The ethyl (Z)-7-chloro-2-((S
)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoate (237g, 0.79 mol) obtained in Comparative Example 1 was dissolved in 877ml of methanol and 1.8 L of sodium hydroxide solution (0.48 M) was added with stirring at room temperature. The reaction was finished when the area ratio of (Z) isomer and (E) isomer becomes 20: 1 by HPLC analysis and the un-reacted organic reagent was extracted with 490 ml of dichloromethane. The pH of the solution was adjusted to 7-8 with 3N HCl and the un- reacted organic reagent was extracted with 490 ml of dichloromethane again. The water layer was concentrated under reduced pressure and 650ml of ethanol was added and stirred until the solid had been dissolved at 50°C, for 30 minute to 1 hour. The un- dissolved solid was removed with filtration and the filtrate was concentrated under reduced pressure. 2.4 L of acetonitrile is added thereto and stirred to obtain 140.8g of ( Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12 ; 55% yield).
[74]
[75]
[76] m.p.: 219°C;
[77] 1H-NMR (D2O, 300MHz) δppm: 0.87 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.61 (m, 2H), 1.68 (dd, IH), 1.78 (m, 2H), 2.12 (m, 2H), 3.62 (t, 2H), 6.47 (t, IH);
[78]
13
[79] 13C ( -NMR (D2O, 300MHz) δppm: 19.47, 19.99, 22.55, 25.74, 26.75, 27.53, 29.44,
32.27, 46.11, 131.41, 136.52, 172.74, 174.62.
[80] [81] [82]
[83] 1-2. ( Z V7-chloro-(YSV2. 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid
(12-D
[84] 140.8g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12) obtained from Example 1-1 was dissolved in 422 ml of distilled water. The pH of the solution was adjusted to 2.0-3.0 with 3N HCl, extracted with 592 ml of isopropylether two times and 59.2g of anhydrous magnesium sulfate was added to isopropylether layer, stirred and subjected to filtration. The filtrate was concentrated to afford 127.7g of (Z)-7-chloro-2-((S )-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (12-1, 98% yield).
[85]
[86] 1H-NMR (CDCl3, 300MHz) δppm: 0.83 (dd, IH), 1.19 (s, 7H), 1.44 (dd, IH), 1.19
(s, 3H), 1.64 (m, 2H), 1.81 (m, 2H), 2.21 (m, 2H), 3.54 (t, 2H), 6.78 (t, IH), 7.04 (br, IH);
[87]
13
[88] 13C ( -NMR (CDCl3, 300MHz) δppm: 18.69, 20.82, 22.86, 25.36, 27.03, 28.53,
29.27, 32.17, 44.60, 124.88, 139.49, 168.96, 170.15.
[89]
[90]
[91] 1-3. ( Z V7-chloro-((SV2. 2-dimethylcyclopropanecarboxamidoV2-heptenoic acid ammonium salt (12-2)
[92] 127.7g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid (12-1) obtained from Example 1-2 was dissolved in 422 ml of EtOH. 100 ml of 25% ammonia water solution was added thereto, stirred and concentrated to obtain 135.6g of (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid ammonium salt (12-2, 100% yield).
[93]
[94] Example 2: Preparation of cilastatin ammonium salt (13-1)
[95] 4Og of (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12, 0.14 mol) obtained in Example 1-1 was dissolved in 120 ml of 0.48 M sodium hydroxide solution and 240 ml of ethanol and the mixture of 1.4g of NaBr (0.013 mol) and 25.3g of L-cysteineDHClDH) was added thereto, stirred at 55°C, for 8 hours.
[96] The pH of the reaction solution was adjusted to 5.5-5.0 with 3N HCl, concentrated and 800ml of methanol was added, stirred at 55°C for 1 hour and un-dissolved salt was filtered out. The filtrate was concentrated to the extent that the volume of total solution was reduced to about 1/2. The concentrate was adsorbed with cationic exchange resin (PK208 model, Samyang Co.), washed with distilled water to the extent that the con¬ ductivity of the solution became less than lθμs(microsiemens), eluted with 2N ammonia water and the eluate was concentrated under the reduced pressure to give brown solid compound. The compound was dissolved in 40 ml of distilled water. 0.8 L of 2-propanol was added thereto and the solution was subjected to salting out method with reflux for 2 hours. The resulting solid was cooled and filtered to obtain 45.66g of cilastatin ammonium salt (13-1. 90% yield).
[97]
[98] m. p.: 161°C;
[99] Element Analysis: C16H29N3O5S (MW: 375.183): CaI. Q51.18; 7.78; N:11.19; Est.
C:51.01; H: 7.97; N: 11.04;
[100] MS m/z : 375 (M+, 49), 312(36), 97 (84.2), 69 (100);
[101] 1H-NMR (D2O, 300MHz) δppm: 0.87 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.62 (m, 5H), 2.1 l(q, 2H), 2.62 (t, 2H), 3.06 (m, 4H), 3.91 (dd, IH), 6.47 (t, IH);
13
[102] ” (C-NMR (D2O, 300MHz) δppm: 19.49, 19.97, 22.53, 26.74, 27.44, 27.86, 29.09,
29.43, 31.94, 32.85, 54.44, 131.23, 136.83, 172.70, 173.71, 174.64.
[103]
[104] Example 3: Preparation of cilastatin ethylamine salt (13-2)
[105] 4Og of (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid sodium salt (12-1, 0.15 mol) obtained in Example 1-2 was dissolved in 165 ml of 0.66 M sodium hydroxide solution and 330 ml of ethanol and the mixture of 1.5g of NaBr (0.015 mol) and 27.6g of L-cysteineDHClDH) was added thereto, stirred at 55°C, for 8 hours.
[106] The pH of the reaction solution was adjusted to 5.5-5.0 with 3N HCl, concentrated and 800ml of methanol was added, stirred at 55°C for 1 hour and un-dissolved salt was filtered out.. The filtrate was concentrated to the extent that the volume of total solution was reduced to about 1/2. The concentrate was adsorbed with cationic exchange resin (PK208 model, Samyang Co.), washed with distilled water to the extent that the conductivity of the solution became less than 10μs(microsiemens), eluted with 2N ethylamine water and the eluate was concentrated under the reduced pressure to give brown solid compound. The compound was dissolved in 40 ml of distilled water. 0.8 L of 2-propanol was added thereto and the solution was subjected to salting out method with reflux for 2 hours. The resulting solid was cooled and purified with filtration to obtain 49.38g of cilastatin ethylamine salt (13-2. 90% yield).
[107]
[108] 1H-NMR (D2O, 300MHz) δppm: 0.86 (dd, IH), 1.00 (dd, IH), 1.14 (s, 3H), 1.19 (s,
3H), 1.27 (t, 3H), 1.60 (m, 5H), 2.1 l(q, 2H), 2.62 (t, 2H), 3.06 (m, 4H), 3.91 (dd, IH), 6.47 (t, IH); [109] 13C-NMR (D2O, 300MHz) δppm: 14.7, 21.57, 22.04, 24.63. 28.82, 29.52, 29.94,
31.16, 31.49, 34.00, 34.91, 37.78, 56.50, 133.27, 138.96, 174.75, 175.81, 176.74.
[HO]
[111] Example 4 : Purification of cilastatin ammonium salt
[112] 4-1. Purification using by water and ethanol
[113] 45.66g of cilastatin ammonium salt (13-1,0.12 mol) obtained in Example 2 was dissolved in 45.66 ml of distilled water and 1.3L of anhydrous ethanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 38.81g of cilastatin ammonium salt (Yield: 85%, Purity: 99.8%).
[114]
[115] 4-2. Purification using by ammonia water and propanol Q)
[116] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 50 ml of 25% ammonia water and 1.5L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 41.2g of cilastatin ammonium salt (Yield: 82.4%, Purity: 99.3%).
[117]
[118] 4-3. Purification using by ammonia water and propanol (1)
[119] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in
100 ml of 25% ammonia water and 2.0L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was purified with filtration to obtain 35.4g of cilastatin ammonium salt (Yield: 70.8%, Purity: 99.3%)
[120]
[121] 4-4. Purification using by ammonia water and ethanol
[122] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 50 ml of 25% ammonia water and 1.5 L of anhydrous ethanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 35.6g of cilastatin ammonium salt (Yield: 71.2%, Purity: 99.8%).
[123]
[124] 4-5. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (1)
[125] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 50ml of 4N ammonia water, and 2.0 L of 1 -propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 89.3g of cilastatin ammonium salt (Yield: 89.3%, Purity: 99.6%).
[126]
[127] 4-6. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (1) [128] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 50ml of 2N ammonia water, and 2.0 L of 1-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 95.0g of cilastatin ammonium salt (Yield: 95.0%, Purity: 99.5%).
[129]
[130] 4-7. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (3)
[131] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 100 ml of distilled water and 50ml of 25% ammonia water, and 2.0 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 89.7g of cilastatin ammonium salt (Yield: 89.7%, Purity: 99.8%).
[132]
[133] 4-8. Purification using by the mixture solvent mixed with water and ammonia water, and propanol (4)
[134] lOOg of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in the mixture solvent mixed with 50 ml of distilled water and 100ml of 25% ammonia water, and 3.0 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 80.Og of cilastatin ammonium salt (Yield: 80.0%, Purity: 99.7%).
[135]
[136] 4-9. Purification using by water and propanol
[137] 50g of cilastatin ammonium salt (13-1) obtained in Example 2 was dissolved in 100 ml of distilled water and 1.5 L of 2-propanol was added thereto in a dropwise manner. The resulting salted out solid was filtered to obtain 87.2g of cilastatin ammonium salt (Yield: 87.2%, Purity: 99.6%).
[138]
[139] Example 5: Preparation of cilastatin sodium salt
[140] 4.28g of sodium hydroxide (0.107 mol) was dissolved in 38.3 ml of distilled water and 191.5 ml of ethanol. 38.81g of cilastatin ammonium salt (0.1 mol) obtained in Example 4-1 was added thereto and stirred for 30 minutes. The solution was con¬ centrated under reduced pressure at 60°C and 153 ml of distilled water was added to the concentrate. The solution was stirred to dissolve the concentrate and the pH of the solution was adjusted to 7.0 using by cationic exchange resin and filtered. The filtrate was lyophilized to obtain high purity (99.4%) of cilastatin sodium salt.
[141]
[142] Experimental Example 1: Purity Determination [143] The purity of cilastatin ammonium salt obtained in Example 4 was determined by
HPLC on condition as shown in Table 1 and the determined result was shown in Table 2.
[144] Table 1
[145] Table 2

[146]
Industrial Applicability
[147] The novel method of the present invention could prevent the formation of (E )-isomer from the preparation of novel intermediate for preparing cilastatin sodium, i.e., (Z)-7-chloro-2-((S)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt and isolate the intermediate in situ providing simpler process with high yield and purity. Furthermore, it can provide with highly purified cilastatin sodium salt by isolating novel cilastatin amine salt and using sodium hydroxide and cationic exchange resin. Accordingly, the method can be very useful in preparing cilastatin sodium salt with high yield and high purity.
Claims
Hide Dependent
Claims
[1] A method for preparing (Z)-7-chloro-2-((S)-2,
2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt represented by general chemical formula (12) comprising the steps consisting of: selectively hy- drolyzing (Z)-7-chloro-2-((S
)-2,2-dimethylcyclopropanecarboxamido)-2-heptenoate represented by chemical formula (4) in reaction solvent under basic condition and removing un-reacted reactant remained in reaction solvent layer with washing organic solvent with controlling the pH of reaction solution with acid at the 1st step; concentrating remaining water layer, adding alcohol thereto with heating, stirring to the extent to dissolve the solid, filtering out un-dissolved salt, and concentrating the filtrate under reduced pressure at the 2n step; adding organic solvent thereto to solidify the concentrate, filtering the solution to isolate (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid metal salt represented by chemical formula (12) at the final step:

Wherein M+ is alkali metal salt.
[2] The method according to claim 1, said R group of general formula (12) is selected from lithium salt, sodium salt and potassium salt. [3] The method according to claim 1, said reaction solvent at the 1st step is selected from the mixture of water and methanol, water and ethanol or water and propanol.
[4] The method according to claim 1, said pH of the reaction solution at the 1ststep ranges from 6 to 8. [5] The method according to claim 1, said organic solvent for isolating final product from the salt thereof is acetonitrile, acetone or the mixture solvent mixed with water and alcohol.
[6] A method for preparing cilastatin sodium salt represented by chemical formula (1) comprising the steps consisting of: reacting (Z)-7-chloro-2-((S)-2, 2-dimethylcyclopropanecarboxamido)-2-heptenoic acid or the salt thereof represented by chemical formula (12) with cysteine in basic solution at the 1ststep; controlling the pH of the reaction solution obtained in step 1, concentrating, adsorbing the concentrate with cationic exchange resin, washing with water, eluting with amine solution to concentrate the eluant at the 2nd step; dissolving the concentrate in recrystallization solvent and subjecting to recrystallization process by adding alcohol in a dropwise manner to afford pure cilastatin amine salt (13) at the 3r step; reacting cilastatin amine salt (13) with sodium hydroxide and controlling the pH with cationic exchange resin at the 4thstep.

Wherein M+ is alkali metal salt.


Wherein R is a hydrogen atom or lower alkyl group.
[7] The method according to claim 6, said R group of general chemical formula (13) is a hydrogen atom or C1-C4 alkyl group. [8] The method according to claim 6, said recrystallization solvent at the 3 step is water, ammonia water, or the mixture thereof in the amount ranging from 1 :3 to
2:1 (w/v) of the weight of cilastatin amine salt.
[9] The method according to claim 6, said alcohol added for recrystallization at the 3 step is ethanol, 1-propanol, 2-propanol, n-butanol, or the mixture thereof. [10] The method according to claim 6, said recrystallization process at the 3rdstep is performed at the temperature ranging from 5 to 97°C.
[H] The method according to claim 6, said cationic exchange resin at the 4thstep is styrene strong acidic resin. [12] An intermediate represented by chemical formula (13)

Wherein R is a hydrogen atom or lower alkyl group.
References
- ^ Jump up to:a b Keynan S, Hooper NM, Felici A, Amicosante G, Turner AJ (1995). “The renal membrane dipeptidase (dehydropeptidase I) inhibitor, cilastatin, inhibits the bacterial metallo-beta-lactamase enzyme CphA”. Antimicrob. Agents Chemother. 39 (7): 1629–31. doi:10.1128/aac.39.7.1629. PMC 162797. PMID 7492120.
- Keynan S, Hooper NM, Felici A, Amicosante G, Turner AJ: The renal membrane dipeptidase (dehydropeptidase I) inhibitor, cilastatin, inhibits the bacterial metallo-beta-lactamase enzyme CphA. Antimicrob Agents Chemother. 1995 Jul;39(7):1629-31. [PubMed:7492120]
- Buckley MM, Brogden RN, Barradell LB, Goa KL: Imipenem/cilastatin. A reappraisal of its antibacterial activity, pharmacokinetic properties and therapeutic efficacy. Drugs. 1992 Sep;44(3):408-44. [PubMed:1382937]
- Balfour JA, Bryson HM, Brogden RN: Imipenem/cilastatin: an update of its antibacterial activity, pharmacokinetics and therapeutic efficacy in the treatment of serious infections. Drugs. 1996 Jan;51(1):99-136. doi: 10.2165/00003495-199651010-00008. [PubMed:8741235]
- Koller M, Brom J, Raulf M, Konig W: Cilastatin (MK 0791) is a potent and specific inhibitor of the renal leukotriene D4-dipeptidase. Biochem Biophys Res Commun. 1985 Sep 16;131(2):974-9. doi: 10.1016/0006-291x(85)91335-x. [PubMed:3863619]
- FDA: Recarbrio Label [Link]
- FDA: Primaxin Label [Link]
- ChemSpider: Cilastatin [Link]
- FDA Label: Apadaz [Link]
- Drugs@FDA: Primaxin [Link]
Synthesis
By Panchapakesan, Ganapathy et alFrom Indian, 269299, 16 Oct 2015
IN 269299

SYN
Patent
Publication numberPriority datePublication dateAssigneeTitle
EP0048301A11980-09-241982-03-31Merck & Co., Inc.2-(Cyclopropane-carboxamido)-2-alkenoic acids, their esters and salts, and antibacterial compositions comprising the same and a thienamycin-type compound
EP0072014A1 *1981-08-101983-02-16Merck & Co., Inc.Combination of 2-substituted penems with dipeptidase inhibitors
US4616038A1978-07-241986-10-07Merck & Co., Inc.Combination of thienamycin-type antibiotics with dipeptidase inhibitors
WO2003018544A12001-08-242003-03-06Ranbaxy Laboratories LimitedProcess for the preparation of cilastatin
US20040152780A12001-05-182004-08-05Yatendra KumarProcess for the preparation of amorphous cilastatin sodium
WO2006022511A12004-08-252006-03-02Dong Kook Pharm. Co., Ltd.Novel process for the preparation of cilastatin sodium salt
Publication numberPriority datePublication dateAssigneeTitle
Family To Family Citations
WO2011061609A22009-11-192011-05-26Ranbaxy Laboratories LimitedProcesses for the preparation of cilastatin
US20120253066A1 *2010-01-012012-10-04Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of cilastatin sodium
Cilastatin
-
- ATC:J01DH51
- Use:dehydropeptidase inhibitor (for combination with imipenem)
- Chemical name:[R-[R*,S*-(Z)]]-7-[(2-amino-2-carboxyethyl)thio]-2-[[(2,2-dimethylcyclopropyl)carbonyl]amino]-2-heptenoic acid
- Formula:C16H26N2O5S
- MW:358.46 g/mol
- CAS-RN:82009-34-5
- InChI Key:DHSUYTOATWAVLW-WFVMDLQDSA-N
- InChI:InChI=1S/C16H26N2O5S/c1-16(2)8-10(16)13(19)18-12(15(22)23)6-4-3-5-7-24-9-11(17)14(20)21/h6,10-11H,3-5,7-9,17H2,1-2H3,(H,18,19)(H,20,21)(H,22,23)/b12-6-/t10-,11+/m1/s1
- EINECS:279-875-8
- LD50:8 g/kg (M, route unreported);
8 g/kg (R, route unreported)
Derivatives
monosodium salt
- Formula:C16H25N2NaO5S
- MW:380.44 g/mol
- CAS-RN:81129-83-1
- EINECS:279-694-4
- LD50:6786 mg/kg (M, i.v.); >10 g/kg (M, p.o.);
5027 mg/kg (R, i.v.); >10 g/kg (R, p.o.)
 |
|
 |
|
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a686013 |
| Routes of administration |
IV |
| ATC code | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.072.592 |
| Chemical and physical data | |
| Formula | C16H26N2O5S |
| Molar mass | 358.454 g/mol g·mol−1 |
| 3D model (JSmol) | |
/////////////cilastatin, シラスタチン , FDA 2019, циластатин , سيلاستاتين , 西司他丁 , MK-791, Recarbrio
CC1(C)C[C@@H]1C(=O)N\C(=C/CCCCSC[C@H](N)C(O)=O)C(O)=O
J-147

J-147
N-(2,4-Dimethylphenyl)-2,2,2-trifluoro-N’-[(E)-(3-methoxyphenyl)methylene]acetohydrazide
- Molecular FormulaC18H17F3N2O2
- Average mass350.335 Da
2,2,2-trifluoroacetic acid-1-(2,4-dimethylphenyl)-2-[(3-methoxyphenyl)methylene]hydrazide
Acetic acid, 2,2,2-trifluoro-, 1-(2,4-dimethylphenyl)-2-[(1E)-(3-methoxyphenyl)methylene]hydrazide
N-(2,4-Dimethylphenyl)-2,2,2-trifluoro-N’-[(E)-(3-methoxyphenyl)methylene]acetohydrazide
[1146963-51-0]
1-(2,4-dimethylphenyl)-2-[(3-methoxyphenyl)methylene]hydrazide, 2,2,2-trifluoro-acetic acid
1146963-51-0 [RN] DOUBLE BOND GEOMETRY UNSPECIFIED
FDA UNII Z41H3C5BT9
Abrexa Pharmaceuticals, Dementia, Alzheimer’s type, PHASE1
Blanchette Rockefeller Neurosci Inst (Originator)
Salk Institute for Biological Studies (Originator)
Abrexa Pharmaceuticals is developing the oral curcumin derivative J-147 for the treatment of Alzheimer’s disease. A phase I clinical trial is under way in healthy young and older adults.
The Salk Institute for Biological Studies and Abrexa Pharmaceuticals are developing J-147, a curcumin derivative CNB-001 , and a 5-lipoxygenase inhibitor, for the oral treatment of Alzheimer’s disease (AD), aging and acute ischemic stroke; in January 2019, a phase I trial for AD was initiated.
J147 is an experimental drug with reported effects against both Alzheimer’s disease and ageing in mouse models of accelerated aging.[1][2][3][4]
The approach that lead to development of the J147 drug was to screen candidate molecules for anti-aging effects, instead of targeting the amyloid plaques. It is contrary to most other approaches to developing drugs against Alzheimer’s disease that target the plaque deposits in the brain.[5]
The J147 drug is also reported to address other biological aging factors, such as preventing the leakage of blood from microvessels in mice brains.[5] The development of J147 follows the chemical pharmacological way, contrary to biological ways that exploit e.g. use of bacteriophages.[6][7]
Enhanced neurogenic activity over J147 in human neural precursor cells has its derivative called CAD-31. CAD-31 is enhancing the use of free fatty acids for energy production by shifting of the metabolic profile of fatty acids toward the production of ketone bodies, a potent source of energy in the brain when glucose levels are low.[8]
The target molecule is a protein called ATP synthase, which is found in the mitochondria.[9]
PAPER
Organic & Biomolecular Chemistry (2015), 13(37), 9564-9569
https://pubs.rsc.org/en/content/articlelanding/2015/OB/C5OB01463H#!divAbstract
A series of novel J147 derivatives were synthesized, and their inhibitory activities against β-amyloid (Aβ) aggregation and toxicity were evaluated by using the oligomer-specific antibody assay, the thioflavin-T fluorescence assay, and a cell viability assay in the transformed SH-SY5Y cell culture. Among the synthesized J147 derivatives, 3j with a 2,2-dicyanovinyl substituent showed the most potent inhibitory activity against Aβ42oligomerization (IC50 = 17.3 μM) and Aβ42 fibrillization (IC50 = 10.5 μM), and disassembled the preformed Aβ42 fibrils with an EC50 of 10.2 μM. Finally, we confirmed that 3j is also effective at preventing neurotoxicity induced by Aβ42-oligomers as well as Aβ42-fibrils.

Synthesis of (E)-N-(2,4-dimethylphenyl)-2,2,2-trifluoro-N’-(3-methoxybenzylidene)- 32 acetohydrazide (3a). To a solution of 3-methoxybenzaldehyde (1a) (0.10 g, 0.7 mmol) in EtOH (10 33 mL) was added (2,4-dimethylphenyl)hydrazine hydrochloride (0.13 g, 0.7 mmol), and the resulting 34 mixture was stirred for 1 h at room temperature (RT). After the reaction, the mixture was concentrated 35 under reduced pressure to yield the corresponding benzylidenehydrazine, which was used for the next 36 step without further purification. The intermediate benzylidenehydrazine was dissolved in CH2Cl2, 37 and the resulting solution was treated with Et3N (0.3 mL, 2.2 mmol). Trifluoroacetic anhydride (0.1 38 mL, 1.1 mmol) was added to this solution in drops at 0 °C. After stirring for 1 h, the mixture was 39 concentrated under reduced pressure, and the residue was purified by column chromatography on 40 silica gel (8:1 = hexanes:ether) to yield 3a (0.12 g, 0.3 mmol, 47% yield) as a yellow solid:
1H NMR 41 (400 MHz, CDCl3) δ 7.29-7.24 (m, 4H), 7.20 (d, J = 7.9 Hz, 1H), 7.12 (d, J = 7.6 Hz, 1H), 7.04 (d, J 42 = 7.9 Hz, 1H), 6.94 (ddd, J = 8.1, 2.2,0.8 Hz, 1H), 3.81 (s, 1H), 2.41 (s, 3H), 2.08 (s, 3H);
13C NMR 43 (100 MHz, CDCl3) δ 160.7, 158.9 (q, J = 36.4 Hz), 155.0, 143.4, 143.1, 142.3, 137.7, 134.4, 130.9, 44 130.8, 130.6, 129.9, 123.5, 123.0, 118.4 (q, J = 287.3 Hz), 113.8, 57.4, 23.5, 19.1;
LC-MS (ESI) m/z found 373.2 [M + Na]+ , calcd for C18H17F3N2O2Na 373.1.


PAPER
https://www.sciencedirect.com/science/article/pii/S0960894X12014746

Figure 1. Chemical structures of previously developed [11C]PIB, [18F]Amyvid and [18F]-T808, and newly developed [11C]J147.

Scheme 1. Synthesis of the reference standard J147 (2).
PRODUCT PATENT
WO2009052116
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2009052116&tab=PCTDESCRIPTION
PATENT
WO-2019164997
A process for preparing crystalline Form II of 2,2,2-trifluoroacetic acid-1-(2,4-dimethylphenyl)-2-[(3-methoxyphenyl)methylene]hydrazide (J-147; 98% of purity) comprising the steps of providing a slurry containing saturated amorphous or crystalline Form I of J-147 and mixing the slurry to obtain the crystalline Form II of J147. Also claimed are processes for preparing the crystalline Form I of 2,2,2-trifluoroacetic acid-1-(2,4-dimethylphenyl)-2-[(3-methoxyphenyl)methylene]hydrazide. Further claimed are isolation of the crystalline Form II and I of 2,2,2-trifluoroacetic acid-1-(2,4-dimethylphenyl)-2-[(3-methoxyphenyl)methylene]hydrazide. The compound is disclosed to be a neurotrophic agent and known to be a Trkb receptor agonist, useful for treating neurodegenerative disease, such as aging and motor neurone disease.
The present disclosure relates to polymorph forms of a pharmaceutical active agent. In particular, the present disclosure relates to polymorph forms of neuroprotective agent 2,2,2-trifluoroacetic acid l-(2,4-Dimethylphenyl)-2-[(3-methoxyphenyl)methylene] hydrazide (J147).
[0002] 2,2,2 -trifluoroacetic acid l-(2,4-Dimethylphenyl)-2-[(3-methoxyphenyl)methylene] hydrazide (J147) is a potent orally active neurotrophic agent discovered during screening for efficacy in cellular models of age-associated pathologies and has a structure given by Formula I:
[0003] J147 is broadly neuroprotective, and exhibited activity in assays indicating distinct neurotoxicity pathways related to aging and neurodegenerative diseases, with EC50 between 10 and 200 nM. It has been indicated to improve memory in normal rodents, and prevent the loss of synaptic proteins and cognitive decline in a transgenic AD mouse model.
Furthermore, it has displayed neuroprotective, neuroanti-inflammatory, and LTP-enhancing activity.
[0004] The neurotrophic and nootropic effects have been associated with increases in BDNF levels and BDNF responsive proteins. Interestingly, despite this mechanism of action, Jl47’s neuroprotective effects have been observed to be independent of TrkB receptor activation.
J147 has been indicated to reduce soluble Ab40 and Ab42 levels, and it is currently being researched for potential applications in treating ALS.
The Fourier transform infrared (FTIR) spectrum is shown in Figure 4. Based on visual inspection the spectrum is consistent with structure. The Raman spectrum is in agreement with the FTIR spectrum and is shown in Figure 5. The proton NMR data is consistent with the structure of J147 and is shown in Figure 6. The proton NMR data is also shown in tabulated form in Table B below.
Table B
EXAMPLE OF PREPARATION OF FORM II OF J 147
Batch Process: About 100 kg of crude J147 from its synthetic preparation was evaporated twice from about 80 kg of ethanol. The crude product was taken up in about 48 kg of ethanol and the batch temperature was adjusted to 28 °C. About 37 kg of water was added gradually to the batch. The batch was held at about 30 °C for about 1.7 hours. A sample of the batch was pulled from the reactor and solids precipitated by addition of 45 mL of water. The solids obtained were added back to the batch as seed crystals and the mixture stirred for 40 minutes at 30 °C. An additional about 34 kg of water was added. The batch was held at about 18 °C for about 58 hours and then cooled to about 10 °C for another about 5.5 hours. Analysis of the resultant solids indicated the presence of Form I. Form I was converted to Form II by heating the slurry to about 45 °C for about 16 hours and then cooling back to about 10 °C and holding the batch at this temperature for about 3 hours about 17.7 kg of solid Form II of J147 were recovered by filtration after washing and drying.

CLIP
https://cen.acs.org/articles/90/i31/Tumeric-Derived-Compound-Curcumin-Treat.html
Turmeric-Derived Compound Curcumin May Treat Alzheimer’s
Curry chemical shows promise for treating the memory-robbing disease

CURRY WONDER
Curcumin, derived from the rootstalk of the turmeric plant, not only gives Indian dishes their color but might treat Alzheimer’s.
Credit: Shutterstock
More than 5 million people in the U.S. currently live with Alzheimer’s disease. And according to the Alzheimer’s Association, the situation is only going to get worse.
By 2050, the nonprofit estimates, up to 16 million Americans will have the memory-robbing disease. It will cost the U.S. $1.1 trillion annually to care for them unless a successful therapy is found.
Pharmaceutical companies have invested heavily in developing Alzheimer’s drugs, many of which target amyloid-β, a peptide that misfolds and clumps in the brains of patients. But so far, no amyloid-β-targeted medications have been successful. Expectation for the most advanced drugs—bapineuzumab from Pfizer and Johnson & Johnson and solanezumab from Eli Lilly & Co.—are low on the basis of lackluster data from midstage clinical trials. That sentiment was reinforced last week when bapineuzumab was reported to have failed the first of four Phase III studies.
Even if these late-stage hopefuls do somehow work, they won’t come cheap, says Gregory M. Cole, a neuroscientist at the University of California, Los Angeles. These drugs “would cost patients tens of thousands of dollars per year,” he estimates. That hefty price tag stems from bapineuzumab and solanezumab being costly-to-manufacture monoclonal antibodies against amyloid-β.
“There’s a great need for inexpensive Alzheimer’s treatments,” as well as a backup plan if pharma fails, says Larry W. Baum, a professor in the School of Pharmacy at the Chinese University of Hong Kong. As a result, he says, a great many researchers have turned their attention to less pricy alternatives, such as compounds from plants and other natural sources.
Curcumin, a spice compound derived from the rootstalk of the turmeric plant (Curcuma longa), has stood out among some of the more promising naturally derived candidates.
When administered to mice that develop Alzheimer’s symptoms, curcumin decreases inflammation and reactive oxygen species in the rodents’ brains, researchers have found. The compound also inhibits the aggregation of troublesome amyloid-β strands among the animals’ nerve cells. But the development of curcumin as an Alzheimer’s drug has been stymied, scientists say, both by its low uptake in the body and a lack of funds for effective clinical trials—obstacles researchers are now trying to overcome.
In addition to contributing to curry dishes’ yellow color and pungent flavor, curcumin has been a medicine in India for thousands of years. Doctors practicing traditional Hindu medicine admire turmeric’s active ingredient for its anti-inflammatory properties and have used it to treat patients for ailments including digestive disorders and joint pain.
Only in the 1970s did Western researchers catch up with Eastern practices and confirm curcumin’s anti-inflammatory properties in the laboratory. Scientists also eventually determined that the polyphenolic compound is an antioxidant and has chemotherapeutic activity.

Bharat B. Aggarwal, a professor at the University of Texas M. D. Anderson Cancer Center, says curcumin is an example of a pleiotropic agent: It has a number of different effects and interacts with many targets and biochemical pathways in the body. He and his group have discovered that one important molecule targeted and subsequently suppressed by curcumin is NF-κB, a transcription factor that switches on the body’s inflammatory response when activated (J. Biol. Chem.,DOI: 10.1074/jbc.270.42.24995).
Aside from NF-κB, curcumin seems to interact with several other molecules in the inflammatory pathway, a biological activity that Aggarwal thinks is advantageous. “All chronic diseases are caused by dysregulation of multiple targets,” he says. “Chemists don’t yet know how to design a drug that hits multiple targets.” With curcumin, “Mother Nature has already provided a compound that does so.”
Curcumin’s pleiotropy also brought it to the attention of UCLA’s Cole during the early 1990s while he was searching for possible Alzheimer’s therapeutics. “That was before we knew about amyloid-β” and its full role in Alzheimer’s, he says. “We were working on the disease from an oxidative damage and inflammation point of view—two processes implicated in aging.”
When Cole and his wife, Sally A. Frautschy, also at UCLA, searched the literature for compounds that could tackle both of these age-related processes, curcumin jumped out at them. It also didn’t hurt that the incidence of Alzheimer’s in India, where large amounts of curcumin are consumed regularly, is lower than in other parts of the developing world (Lancet Neurol., DOI: 10.1016/s1474-4422(08)70169-8).
In 2001, Cole, Frautschy, and colleagues published the first papers that demonstrated curcumin’s potential to treat neurodegenerative disease (Neurobiol. Aging, DOI: 10.1016/s0197-4580(01)00300-1; J. Neurosci.2001, 8370). The researchers studied the effects of curcumin on rats that had amyloid-β injected into their brains, as well as mice engineered to develop amyloid brain plaques. In both cases, curcumin suppressed oxidative tissue damage and reduced amyloid-β deposits.
Those results, Cole says, “turned us into curcumin-ologists.”
Although the UCLA team observed that curcumin decreased amyloid plaques in animal models, at the time, the researchers weren’t sure of the molecular mechanism involved.
Soon after the team’s first results were published, Cole recalls, a colleague brought to his attention the structural similarity between curcumin and the dyes used to stain amyloid plaques in diseased brain tissue. When Cole and Frautschy tested the spice compound, they saw that it, too, could stick to aggregated amyloid-β. “We thought, ‘Wow, not only is curcumin an antioxidant and an anti-inflammatory, but it also might be an anti-amyloid drug,’ ” he says.
In 2004, a group in Japan demonstrated that submicromolar concentrations of curcumin in solution could inhibit aggregation of amyloid-β and break up preformed fibrils of the stuff (J. Neurosci. Res., DOI: 10.1002/jnr.20025). Shortly after that, the UCLA team demonstrated the same (J. Biol. Chem., DOI: 10.1074/jbc.m404751200).
As an Alzheimer’s drug, however, it’s unclear how important it is that the spice compound inhibits amyloid-β aggregation, Cole says. “When you have something that’s so pleiotropic,” he adds, “it’s hard to know” which of its modes of action is most effective.
Having multiple targets may be what helps curcumin have such beneficial, neuroprotective effects, says David R. Schubert, a neurobiologist at the Salk Institute for Biological Studies, in La Jolla, Calif. But its pleiotropy can also be a detriment, he contends.
The pharmaceutical world, Schubert says, focuses on designing drugs aimed at hitting single-target molecules with high affinity. “But we don’t really know what ‘the’ target for curcumin is,” he says, “and we get knocked for it on grant requests.”
Another problem with curcumin is poor bioavailability. When ingested, UCLA’s Cole says, the compound gets converted into other molecular forms, such as curcumin glucuronide or curcumin sulfate. It also gets hydrolyzed at the alkaline and neutral pHs present in many areas of the body. Not much of the curcumin gets into the bloodstream, let alone past the blood-brain barrier, in its pure, active form, he adds.
Unfortunately, neither Cole nor Baum at the Chinese University of Hong Kong realized the poor bioavailability until they had each launched a clinical trial of curcumin. So the studies showed no significant difference between Alzheimer’s patients taking the spice compound and those taking a placebo (J. Clin. Psychopharmacol., DOI: 10.1097/jcp.0b013e318160862c).
“But we did show curcumin was safe for patients,” Baum says, finding a silver lining to the blunder. “We didn’t see any adverse effects even at high doses.”
Some researchers, such as Salk’s Schubert, are tackling curcumin’s low bioavailability by modifying the compound to improve its properties. Schubert and his group have come up with a molecule, called J147, that’s a hybrid of curcumin and cyclohexyl-bisphenol A. Like Cole and coworkers, they also came upon the compound not by initially screening for the ability to interact with amyloid-β, but by screening for the ability to alleviate age-related symptoms.
The researchers hit upon J147 by exposing cultured Alzheimer’s nerve cells to a library of compounds and then measuring changes to levels of biomarkers for oxidative stress, inflammation, and nerve growth. J147 performed well in all categories. And when given to mice engineered to accumulate amyloid-β clumps in their brains, the hybrid molecule prevented memory loss and reduced formation of amyloid plaques over time (PLoS One, DOI: 10.1371/journal.pone.0027865).
Other researchers have tackled curcumin’s poor bioavailability by reformulating it. Both Baum and Cole have encapsulated curcumin in nanospheres coated with either polymers or lipids to protect the compound from modification after ingestion. Cole tells C&EN that by packaging the curcumin in this way, he and his group have gotten micromolar quantities of it into the bloodstream of humans. The researchers are now preparing for a small clinical trial to test the formulation on patients with mild cognitive impairment, who are at an increased risk of developing Alzheimer’s.
An early-intervention human study such as this one comes with its own set of challenges, Cole says. People with mild cognitive impairment “have good days and bad days,” he says. A large trial over a long period would be the best way to get any meaningful data, he adds.
Such a trial can cost up to $100 million, a budget big pharma might be able to scrape together but that is far out of reach for academics funded by grants, Cole says. “If you’re down at the level of what an individual investigator can do, you’re running a small trial,” he says, “and even if the result is positive, it might be inconclusive” because of its small size or short duration. That’s one of the reasons the curcumin work is slow-going, Cole contends.
The lack of hard clinical evidence isn’t stopping people from trying curcumin anyway. Various companies are selling the spice compound as a dietary supplement, both in its powdered form and in nanoformulations such as the ones Cole and Baum are working with. Indiana-based Verdure Sciences, for instance, licensed a curcumin nanoformulation from UCLA and sells it under the name Longvida (about $1.00 to $2.00 per capsule, depending on the distributor).
“There’s no proof that it works,” Cole says. “If you want to take it, you’re experimenting on yourself.” And he cautions that correct dosing for this more bioavailable form of curcumin hasn’t yet been established, so there could be safety concerns.
But on the basis of positive e-mails he’s received from caregivers and Alzheimer’s patients who are desperate for options and trying supplements, “I have some hope,” Cole says. “Maybe there’s something to curcumin after all.”
CLIP

Raw J 147 powder basic Characters
| Name: | J 147 powder |
| CAS: | 1146963-51-0 |
| Molecular Formula: | C18H17F3N2O2 |
| Molecular Weight: | 350.3349896 |
| Melt Point: | 177-178°C |
| Storage Temp: | 4°C |
| Color: | White or off white powder |
Raw J 147 powder in enhance brain function and an extra boost cycle
Names
J 147 powder
J 147 (1146963-51-0) Usage dosage
Using a drug discovery scheme for Alzheimer’s disease (AD) that is based upon multiple pathologies of old age, we identified a potent compound with efficacy in rodent memory and AD animal models. Since this compound, J-147 powder, is a phenyl hydrazide, there was concern that it can be metabolized to aromatic amines/hydrazines that are potentially carcinogenic. To explore this possibility, we examined the metabolites of J 147 powder in human and mouse microsomes and mouse plasma. It is shown that J-147(1146963-51-0) powder is not metabolized to aromatic amines or hydrazines, that the scaffold is exceptionally stable, and that the oxidative metabolites are also neuroprotective. It is concluded that the major metabolites of J 147(1146963-51-0) powder may contribute to its biological activity in animals.
J 147 , derived from the curry spice component curcumin, has low toxicity and actually reverses damage in neurons associated with Alzheimer’s.
J 147 (1146963-51-0) was the mitochondrial protein known as ATP synthase, specifically ATP5A, a subunit of that protein. ATP synthase is involved in the mitochondrial generation of ATP, which cells use for energy.
The researchers demonstrated that by reducing the activity of ATP synthase, they were able to protect neuronal cells from a number of toxicities associated with the aging of the brain. One reason for this neuroprotective effect is thought to be the role of excitotoxicity in neuronal cell damage.
Excitotoxicity is the pathological process by which neurons are damaged and killed by the overactivation of receptors for the excitatory neurotransmitter glutamate. Think of it being a bit like a light switch being turned on and off so rapidly that it ends up causing the light bulb to blow.
Recently, the role of ATP synthase inhibition for neuroprotection against excitotoxic damage was demonstrated in a mouse study[4]. The second study showed that mouse models expressing the human form of mutant ATPase inhibitory factor 1 (hIF1), which causes a sustained inhibition of ATP synthase, were more resilient to neuronal death after excitotoxic damage. This data is consistent with this new J 147 powder study, in which an increase in IF1 in the mice reduced the activity of ATP synthase (specifically ATP5A) and was neuroprotective.
Warning on Raw J 147 powder
Data presented here demonstrate that J-147 powder has the ability to rescue cognitive deficits when administered at a late stage in the disease. The ability of J-147 powder to improve memory in aged AD mice is correlated with its induction of the neurotrophic factors NGF (nerve growth factor) and BDNF (brain derived neurotrophic factor) as well as several BDNF-responsive proteins which are important for learning and memory. The comparison between J-147(1146963-51-0) powder and donepezil in the scopolamine model showed that while both compounds were comparable at rescuing short term memory, J-147 powder was superior at rescuing spatial memory and a combination of the two worked best for contextual and cued memory.
Further instructions
Alzheimer’s disease is a progressive brain disorder, recently ranked as the third leading cause of death in the United States and affecting more than five million Americans. It is also the most common cause of dementia in older adults, according to the National Institutes of Health. While most drugs developed in the past 20 years target the amyloid plaque deposits in the brain (which are a hallmark of the disease), few have proven effective in the clinic.
“While most drugs developed in the past 20 years target the amyloid plaque deposits in the brain (which are a hallmark of the disease), none have proven effective in the clinic,” says Schubert, senior author of the study.
Several years ago, Schubert and his colleagues began to approach the treatment of the disease from a new angle. Rather than target amyloid, the lab decided to zero in on the major risk factor for the disease–old age. Using cell-based screens against old age-associated brain toxicities, they synthesized J 147(1146963-51-0) powder.
Previously, the team found that J-147 powder could prevent and even reverse memory loss and Alzheimer’s pathology in mice that have a version of the inherited form of Alzheimer’s, the most commonly used mouse model. However, this form of the disease comprises only about 1 percent of Alzheimer’s cases. For everyone else, old age is the primary risk factor, says Schubert. The team wanted to explore the effects of the drug candidate on a breed of mice that age rapidly and experience a version of dementia that more closely resembles the age-related human disorder.

References
- ^ “Experimental drug targeting Alzheimer’s disease shows anti-aging effects” (Press release). Salk Institute. 12 November 2015. Retrieved November 13, 2015.
- ^ Chen Q, Prior M, Dargusch R, Roberts A, Riek R, Eichmann C, Chiruta C, Akaishi T, Abe K, Maher P, Schubert D (14 December 2011). “A novel neurotrophic drug for cognitive enhancement and Alzheimer’s disease”. PLoS One. 6 (12): e27865. doi:10.1371/journal.pone.0027865. PMC 3237323. PMID 22194796.
- ^ Currais A, Goldberg J, Farrokhi C, Chang M, Prior M, Dargusch R, Daugherty D, Armando A, Quehenberger O, Maher P, Schubert D (11 November 2015). “A comprehensive multiomics approach toward understanding the relationship between aging and dementia” (PDF). Aging. 7 (11): 937–55. doi:10.18632/aging.100838. PMC 4694064. PMID 26564964.
- ^ Prior M, Dargusch R, Ehren JL, Chiruta C, Schubert D (May 2013). “The neurotrophic compound J147 reverses cognitive impairment in aged Alzheimer’s disease mice”. Alzheimer’s Research & Therapy. 5 (3): 25. doi:10.1186/alzrt179. PMC 3706879. PMID 23673233.
- ^ Jump up to:a b Brian L. Wang (13 November 2015). “Experimental drug targeting Alzheimer’s disease shows anti-aging effects in animal tests”. nextbigfuture.com. Retrieved November 16, 2015.
- ^ Krishnan R, Tsubery H, Proschitsky MY, Asp E, Lulu M, Gilead S, Gartner M, Waltho JP, Davis PJ, Hounslow AM, Kirschner DA, Inouye H, Myszka DG, Wright J, Solomon B, Fisher RA (2014). “A bacteriophage capsid protein provides a general amyloid interaction motif (GAIM) that binds and remodels misfolded protein assemblies”. Journal of Molecular Biology. 426: 2500–19. doi:10.1016/j.jmb.2014.04.015. PMID 24768993.
- ^ Solomon B (October 2008). “Filamentous bacteriophage as a novel therapeutic tool for Alzheimer’s disease treatment”. Journal of Alzheimer’s Disease. 15 (2): 193–8. PMID 18953108.
- ^ Daugherty, D., Goldberg, J., Fischer, W., Dargusch, R., Maher, P., & Schubert, D. (2017). A novel Alzheimer’s disease drug candidate targeting inflammation and fatty acid metabolism. Alzheimer’s research & therapy, 9(1), 50. https://doi.org/10.1186/s13195-017-0277-3
- ^ “Researchers identify the molecular target of J147, which is nearing clinical trials to treat Alzheimer’s disease”. Retrieved 2018-01-30.
 |
|
| Legal status | |
|---|---|
| Legal status | |
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| Chemical and physical data | |
| Formula | C18H17F3N2O2 |
| Molar mass | 350.341 g·mol−1 |
| 3D model (JSmol) | |
////////////J-147, J 147, J147, Alzheimer’s disease, neurotrophic agent, The Salk Institute for Biological Studies, Abrexa Pharmaceuticals, PHASE 1, CURCUMIN

CAS 1417911-00-2
- Acetic acid, 2,2,2-trifluoro-, 1-(2,4-dimethylphenyl)-2-[[3-(methoxy-11C)phenyl]methylene]hydrazide
DICYCLOPLATIN

Dicycloplatin
Platinum(2+) 1-carboxycyclobutanecarboxylate ammoniate (1:2:2)
- Molecular FormulaC12H20N2O8Pt
- Average mass515.380 Da
- 287402-09-9
Has antineoplastic activity; a supramolecular complex of 1,1-cyclobutane dicarboxylic acid and cis-diammine(1,1-cyclobutane dicarboxylate)platinum (II).
1,1-Cyclobutanedicarboxylic acid, ammonium platinum(2+) salt (2:2:1) [ACD/Index Name]
Platinum(2+) 1-carboxycyclobutanecarboxylate ammoniate (1:2:2)
287402-09-9 [RN]
DICYCLOPLATIN
UNII:0KC57I4UNB
Dicycloplatin is a chemotherapy medication used to treat a number of cancers which includes the Non-small-cell lung carcinoma and prostate cancer.[1]
Some side effects which are observed from the treatment by dicycloplatin are nausea, vomiting, thrombocytopenia, neutropenia, anemia, fatigue, loss of appetite, liver enzyme elevation and alopecia. The drugs is a form of Platinum-based antineoplastic and it works by causing the mitochondrial dysfunction which leads to the cell death.[2]
Dicycloplatin was developed in China and it was used for phase I human trial clinical in 2006. The drug was approved for chemotherapy by the Chinese FDA in 2012.[3]

Medical uses
Dicycloplatin can inhibit the proliferation of tumor cells via the induction of apoptosis . It is used to treat a number types of cancer which are Non-small-cell lung carcinoma and prostate cancer.[4]
Side effects
Similar to cisplatin and carboplatin, dicycloplatin also contains some side effects, which are nausea, vomiting, thrombocytopenia, neutropenia, anemia, fatigue, anorexia, liver enzyme elevation, and alopecia. However, with doses up to 350 mg/m(2), there is no significant toxicity; these effects are observed only at higher doses. Furthermore, the nephrotoxicity of dicycloplatin is reported to be less than that of cisplatin, and its myelosuppressive potency is similar to that of carboplatin.[5]
Chemical structure
Dicycloplatin consists of carboplatin and cyclobutane-1,1-dicarboxylic acid (CBDC) linked by the hydrogen bond. In the structure of dicycloplatin, there are two types of bond: O-H…O is the bond between the hydroxyl group of CBDC with carboxyl oxygen atom. It creates the one-dimensional polymer chain of carboplatin and CBDC. The second one is N-H…O which links between the ammoniagroup of carboplatin and oxygen of CBDC. It forms the two-dimensional polymer chain of carboplatin and CBDC. In aqueous solution, the 2D-hydrogen bonded polymeric structure of dicycloplatin is destroyed. Firstly, the bond between ammonia group of carboplatin and oxygen of CBDC breaks, thus inducing the formation of one-dimensional dicycloplatin. After that, the strong hydrogen bond breaks and creates an intermediate state of dicycloplatin. Finally, the rearrangement of different orientation of carboplatin and CBDC leads to the formation of intramolecular hydrogen bond and a supramolecule of dicycloplatin with two O-H…O and N-H…O is created.[6]
Mechanism of action
Similar to carboplatin, dicycloplatin inhibits the proliferation of cancer cells by inducing cell apoptosis. When treated with dicycloplatin, some changes in the properties of Hep G2 cells are observed: the declination of Mitochondria Membrane Potential, the release of cytochrome c from mitocondria to cytosol, the activation of caspase-9, caspase-3 and the decrease of Bcl-2.[4] Those phenomena indicate the role of mitochondrial in the apoptosis by intrisic way.[7] Furthermore, the increase in caspase-8 activation is also observed. This can stimulate the apoptosis by activating downstream caspase-3 [8] or by cleaving Bid.[9] As a result, the cleavage of Bid (tBid) transfers to the mitochondria and induce mitochondrial dysfunction which promotes the release of cytochrome c from mitochondria to cytosol.[10] From the dicycloplatin-treated Hep G2 cell, an excessive amount of reactive oxygen species was detected,[4] which plays an important role in the release of cytochrome c. In the mitochondria, the release of hemoprotein happens through 2-step process: Firstly, the dissociation of cytochrome c from its binding to cardiolipin happens. Due to the reactive oxygen species, the cardiolipin is oxidized, thus reducing the cytochrome c binding and increase the concentration of free cytochrome c [11]
PATENT
WO2018171371
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018171371
Since the FDA approved cisplatin as an anticancer drug in 1978, the mortality rate of testicular cancer patients has been reduced from 100% to less than 10%. For patients with early detection, the cure rate can reach 100%, making cisplatin An outstanding representative of anticancer drugs. In 1986, the FDA approved the second-generation platinum anticancer drug carboplatin. Its anticancer spectrum is similar to that of cisplatin, but it has good water solubility and light toxicity. In 2002, the FDA approved the third-generation platinum anticancer drug oxaliplatin to enter clinical treatment of colorectal cancer. Its anticancer spectrum is different from cisplatin, and it does not produce cross-resistance with cisplatin.
In addition to the above three products, four products, including Nida Platinum, Shuplatin, Lobaplatin and Miplatin, have been listed in different countries and are the first in other countries.
In CN1311183A, Yang Xuqing et al. designed and prepared a new class of platinum antitumor drugs, diammonium platinum dichloride (II) derivatives, based on the abnormal changes in the spatial configuration of cancer cells DNA and RNA. A typical representative drug is bicycloplatinum. Bicycloplatinum in English is called Dicycloplatin, which is called bis(1,1-cyclobutanedicarboxylic acid) diammine platinum (II) (English name [Bis-(1,1-cyclobutane dicarboxylic acid)]diammine platinum(II) ), the structural formula is:
It is a supramolecular compound composed of carboplatin and 1,1-cyclobutanedicarboxylic acid through four hydrogen bonds. It is the first self-developed platinum antitumor drug in China with broad spectrum, low toxicity and high efficiency. It does not produce cross-resistance and good penetrability.
Bicycloplatinum is usually obtained by reacting carboplatin with 1,1-cyclobutanedicarboxylic acid. The prior art discloses various preparation methods, but both have the problems of complicated preparation process and low product purity.
CN1311183A As the earliest publication of bicycloplatin and its preparation method, it is disclosed that bicycloplatinum is prepared by the following method: carboplatin is dissolved in pure water at normal temperature, and then an equimolar amount of 1,1-cyclobutanedicarboxylic acid is added. After the reaction was completed, it was evaporated to dryness, washed with ethanol, and then recrystallized from distilled water. This method is cumbersome in operation due to the need for evaporation and recrystallization steps, and the yield of bicycloplatinum is low.
CN104693245A discloses a preparation method of bicyclo platinum, which is prepared by using carboplatin as a raw material in a ratio of 1:11 to 1,1-cyclobutanedicarboxylic acid in a molar ratio of 1:1, and is protected from light at 0-60 ° C. After -9 days, the excess water is removed by concentration under reduced pressure or freeze-drying to obtain a bicyclic platinum product. Although according to reports, the HPLC purity of the product is more than 99%, it requires a long standing process, is inefficient, and greatly increases the risk of carboplatin decomposition, especially for the process of amplification; The heating and concentration in the final process makes the bicyclic platinum product exist in the higher temperature aqueous solution for a long time, and the product has a high risk of degradation, and the quality stability is inevitably affected. In fact, bicycloplatinum with the reported yield and purity was not obtained according to this method.
CN106132408A discloses a process for the preparation of another bicyclic platinum in which carboplatin is mixed with a corresponding ratio of 1,1-cyclobutanedicarboxylic acid and a solvent to form a suspension, and the precipitated solid formed is separated from the suspension. Although the report states that the obtained product does not contain XRPD detectable amount of carboplatin, the suspension method uses a small amount of solvent, so that the product formed during the reaction is also precipitated as a solid, which is mixed with the unreacted raw material solid. This prevents the reaction from proceeding and makes the purification of the product more difficult. Especially in the case where the product is coated with carboplatin, the carboplatin can hardly be removed by purification. Therefore, the suspension method has the disadvantages of difficulty in control, poor operability, and incapability of industrial scale-up production. In fact, bicycloplatinum with the reported yield and purity cannot be obtained according to this method as well.
1 is a nuclear magnetic resonance-hydrogen spectrum of the bicyclic platinum product of Example 1.
2 is a nuclear magnetic resonance-carbon spectrum of the bicyclic platinum product of Example 1.
Drawing
[ figure 1] 
[ figure 2] 
Preparation Example 1:
Take 20.0 g of cis-diiododiammine platinum (II), add 600 ml of purified water, stir well and heat to 80 ° C in water bath, then add 14.1 g of silver 1,1-cyclobutanedicarboxylate, after reacting for 30 minutes. The AgI slag was filtered off, and the filtrate was concentrated under reduced pressure to a residue of about 50 ml, cooled to room temperature, and the precipitated product was filtered. After recrystallization, the mixture was dried at 60 ° C to obtain 11.26 g of carboplatin, and the yield was 69.88%.
Example 1
32.0 g (222.2 mmol) of 1,1-cyclobutanedicarboxylic acid was taken, and 260 ml of water was added thereto, and the mixture was heated to 80 ° C in a water bath. Add 10.0 g (26.95 mmol) of carboplatin, stir for 40 minutes, cool at 10 ° C for 8 hours, filter the precipitated solid, wash the filter cake with appropriate amount of purified water, drain the washing water, and dry at 40 ° C under reduced pressure to obtain bicyclo platinum 9.32 g. The yield is 67.15% and the content is 99.78%. The obtained products were characterized by elemental analysis, negative ion electrospray mass spectrometry, nuclear magnetic resonance-hydrogen spectroscopy, nuclear magnetic resonance-carbon spectroscopy and X-ray diffraction. The content of bicycloplatin was measured by high performance liquid chromatography.
The test results are shown in Figure 1. The attribution of each peak is as follows:
The peak of chemical shift 1.7159-1.7793ppm is H a , the actual number of hydrogen nuclei is 2, and it is divided into 5 heavy peaks by 4 H b on both sides ; the peak of chemical shift 1.8281-1.8928ppm is H c , actual hydrogen the number of cores 2, a total of four sides by H D impact crack 5 doublet; 2.3965-2.4288ppm peak chemical shift of H B , the actual number of hydrogen nuclei to 4, were subjected to unilateral 2 H a of Effect split into three doublet; 2.7140-2.7457ppm peak chemical shift of H D , the actual number of hydrogen nuclei is 4, were subjected to unilateral 2 H Caffected divided into three split doublet; chemical shifts of the peaks 4.0497ppm is H E , the actual number of hydrogen nuclei 6 as broad singlet; due to D 2 exchange interaction of O, carboxy FIG active hydrogen protons H does not appear f peaks. 4. Nuclear Magnetic Resonance – Carbon Spectrum (D 2 O, 500MHz)
The test results are shown in Figure 2, where the peaks are as follows:
The peak of chemical shift 15.25ppm is C a ; the peak of chemical shift 15.39ppm is C h ; the peak of chemical shift 28.60ppm is C b ; the peak of chemical shift 31.02ppm is C g ; the peak of chemical shift 52.93ppm is C c ; The peak of chemical shift 56.19 ppm is C f ; the peak of chemical shift 176.11 ppm is C d ; the peak of chemical shift 181.85 ppm is C e .
PATENT
WO-2019161526




One-pot method for preparing twin dicarboxylic acid diamine complex platinum (II) derivatives ( dicycloplatin ) comprising the separation of intermediate carboplatin or carboplatin analogue.
For the preparation of bicycloplatin, CN1311183A, as the earliest publication of bicycloplatin and its preparation method, discloses the preparation of bicycloplatinum by the following method: carboplatin is dissolved in pure water at normal temperature, and then an equimolar amount of 1,1-ring is added. Butane dicarboxylic acid was evaporated to dryness after completion of the reaction, washed with ethanol, and recrystallized from distilled water. The method needs to completely evaporate the solvent water, which increases the risk of degradation of the bicyclic platinum, and also introduces more impurities into the crude bicycloplatinum. Therefore, ethanol washing and recrystallization are required, and the operation is cumbersome, and the yield of the bicyclic platinum is low.
[0015]
CN104693245A discloses a preparation method of bicyclo platinum, which is prepared by using carboplatin as a raw material in a ratio of 1:11 to 1,1-cyclobutanedicarboxylic acid in a molar ratio of 1:1, and is protected from light at 0-60 ° C. After -9 days, the excess water is removed by concentration under reduced pressure or freeze-drying to obtain a bicyclic platinum product. Although according to reports, the HPLC purity of the product is more than 99%, it requires a long standing process, is inefficient, and greatly increases the risk of carboplatin decomposition, especially for the process of amplification; In the final process, the solvent water is completely evaporated to make the bicyclic platinum product exist in a relatively high temperature aqueous solution for a long time, and the product has a high risk of degradation, and the quality stability is inevitably affected. In fact, bicycloplatinum with the reported yield and purity was not obtained according to this method.
[0016]
CN106132408A also discloses a process for the preparation of another bicyclic platinum in which carboplatin is mixed with a corresponding ratio of 1,1-cyclobutanedicarboxylic acid and a solvent to form a suspension, and the precipitated solid formed is separated from the suspension. Although the report states that the obtained product does not contain XRPD detectable amount of carboplatin, the suspension method uses a small amount of solvent, so that the product formed during the reaction is also precipitated as a solid, which is mixed with the unreacted raw material solid. This prevents the reaction from proceeding and makes the purification of the product more difficult. Especially in the case where the product is coated with carboplatin, the carboplatin can hardly be removed by purification. Therefore, the suspension method has the disadvantages of difficulty in control, poor operability, and incapability of industrial scale-up production. In fact, bicycloplatinum with the reported yield and purity cannot be obtained according to this method as well.
Notes
- ^ D., Zhao; Y., Zhang; C., Xu; C., Dong; H., Lin; L., Zhang; C., Li; S., Ren; X., Wang; S., Yang; D., Han; X., Chen (February 2012). “Pharmacokinetics, Tissue Distribution, and Plasma Protein Binding Study of Platinum Originating from Dicycloplatin, a Novel Antitumor Supramolecule, in Rats and Dogs by ICP-MS”. Biological Trace Element Research. 148 (2): 203–8. doi:10.1007/s12011-012-9364-2. PMID 22367705.
- ^ G.Q., Li; X.G., Chen; X.P., Wu; J.D., Xie; Y.J., Liang; X.Q., Zhao; W.Q, Chen; L.W., Fu (November 2012). “Effect of Dicycloplatin, a Novel Platinum Chemotherapeutical Drug, on Inhibiting Cell Growth and Inducing Cell Apoptosis”. PLOS ONE. 7 (11): e48994. Bibcode:2012PLoSO…748994L. doi:10.1371/journal.pone.0048994. PMC 3495782. PMID 23152837.
- ^ J.J, Yu; X.Q, Yang; Q.H, Song; M. D., Mueller; S. C., Remick (2014). “Dicycloplatin, a Novel Platinum Analog in Chemotherapy: Synthesis of Chinese Pre-clinical and Clinical Profile and Emerging Mechanistic Studies”. Anticancer Research. 34: 455–464.
- ^ Jump up to:a b c Guang-quan, Li; Xing-gui, Chen; Xing-ping, Wu; Jing-dun, Xie; Yong-ju, Liang; Xiao-qin, Zhao; Wei-qiang, Chen; Li-wu, Fu (November 2012). “Effect of Dicycloplatin, a Novel Platinum Chemotherapeutical Drug, on Inhibiting Cell Growth and Inducing Cell Apoptosis”. PLOS ONE. 7 (11): e48994. Bibcode:2012PLoSO…748994L. doi:10.1371/journal.pone.0048994. PMC 3495782. PMID 23152837.
- ^ Li.S; Huang H; Liao H; Zhan J; Guo Y; Zou BY; Jiang WQ; Guan ZZ; Yang XQ (2015). “Phase I clinical trial of the novel platin complex dicycloplatin: clinical and pharmacokinetic results”. International Journal of Clinical Pharmacology and Therapeutics. 51 (2): 96–105. doi:10.5414/CP201761. PMID 23127487.
- ^ Y., Xu Qing; J., Xiang Lin; S., Q.; TANG, Ka Luo; Y., Zhen Yun; Z., Xiao Feng; T., You Qi (June 2010). “Structural studies of dicycloplatin, an antitumor supramolecule”. Science China Chemistry. 53 (6): 1346–1351. doi:10.1007/s11426-010-3184-z.
- ^ R., Kumar; P.E., Herbert; A.N., Warrens (September 2005). “An introduction to death receptors in apoptosis”. International Journal of Surgery. 3 (4): 268–77. doi:10.1016/j.ijsu.2005.05.002. PMID 17462297.
- ^ Yang, BF; Xiao, C; Li, H; Yang, SJ (2007). “Resistance to Fas-mediated apoptosis in malignant tumours is rescued by KN-93 and cisplatin via downregulation of cFLIP expression and phosphorylation”. Clinical and Experimental Pharmacology and Physiology. 34 (12): 1245–51. doi:10.1111/j.1440-1681.2007.04711.x. PMID 17973862.
- ^ Blomgran, R; Zheng, L; Stendahl, O (2007). “Cathepsin-cleaved Bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization”. Journal of Leukocyte Biology. 81 (5): 1213–23. doi:10.1189/jlb.0506359. PMID 17264306.
- ^ Yin, XM (2006). “Bid, a BH3-only multi-functional molecule, is at the cross road of life and death”. Gene. 369: 7–19. doi:10.1016/j.gene.2005.10.038. PMID 16446060.
- ^ Ott, M; Gogvadze, V; Orrenius, S; Zhivotovsky, B (May 2007). “Mitochondria, oxidative stress and cell death”. Apoptosis. 12 (5): 913–22. doi:10.1007/s10495-007-0756-2. PMID 17453160.

Chemical structure of Dicycloplatin
|
|
| Clinical data | |
|---|---|
| Trade names | Dicycloplatin |
| Synonyms | Platinum(2+) 1-carboxycyclobutanecarboxylate ammoniate (1:2:2), 1,1-Cyclobutanedicarboxylic acid, compd. with (sp-4-2)-diammine(1,1-cyclobutanedi(carboxylato-kappaO)(2-))platinum (1:1) |
| Routes of administration |
Intravenous |
| Pharmacokinetic data | |
| Bioavailability | 100% (IV) |
| Protein binding | < 88.7% |
| Elimination half-life | 24.49 – 108.93 hours |
| Excretion | Renal |
| Identifiers | |
| CAS Number | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C12H20N2O8Pt |
| Molar mass | 515.382 g/mol |
| 3D model (JSmol) |
|
/////////////Dicycloplatin
C1CC(C1)(C(=O)O)C(=O)O.C1CC(C1)(C(=O)[O-])C(=O)[O-].N.N.[Pt+2]
Pretomanid, プレトマニド;
Pretomanid
プレトマニド;
| Formula |
C14H12F3N3O5
|
|---|---|
| CAS |
187235-37-6
|
| Mol weight |
359.2574
|
(6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine
187235-37-6 [RN]
2XOI31YC4N
5H-Imidazo(2,1-b)(1,3)oxazine, 6,7-dihydro-2-nitro-6-((4-(trifluoromethoxy)phenyl)methoxy)-, (6S)-
5H-Imidazo[2,1-b][1,3]oxazine, 6,7-dihydro-2-nitro-6-[[4-(trifluoromethoxy)phenyl]methoxy]-, (6S)-
9871
PA824
PA-824; Pretomanid
- (S)-PA 824
2019/8/14 FDA 2109 APPROVED
Antibacterial (tuberculostatic),
MP 149-150 °C, Li, Xiaojin; Bioorganic & Medicinal Chemistry Letters 2008, Vol 18(7), Pg 2256-2262 and Orita, Akihiro; Advanced Synthesis & Catalysis 2007, Vol 349(13), Pg 2136-2144
150-151 °C Marsini, Maurice A.; Journal of Organic Chemistry 2010, Vol 75(21), Pg 7479-7482
Pretomanid is an antibiotic used for the treatment of multi-drug-resistant tuberculosis affecting the lungs.[1] It is generally used together with bedaquiline and linezolid.[1] It is taken by mouth.[1]
The most common side effects include nerve damage, acne, vomiting, headache, low blood sugar, diarrhea, and liver inflammation.[1] It is in the nitroimidazole class of medications.[2]
Pretomanid was approved for medical use in the United States in 2019.[3][1] Pretomanid was developed by TB Alliance,[4] a not-for-profitproduct development partnership dedicated to the discovery and development of new, faster-acting and affordable medicines for tuberculosis (TB).[5]
Global Alliance for the treatment of tuberculosis (TB).
The compound was originally developed by PathoGenesis (acquired by Chiron in 2000). In 2002, a co-development agreement took place between Chiron (acquired by Novartis in 2005) and the TB Alliance for the development of the compound. The compound was licensed to Fosunpharma by TB Alliance in China.
History
Pretomanid is the generic, nonproprietary name for the novel anti-bacterial drug compound formerly called PA-824.[6] Pretomanid is referred to as “Pa” in regimen abbreviations, such as BPaL. The “preto” prefix of the compound’s name honors Pretoria, South Africa, the home of a TB Alliance clinical development office where much of the drug’s development took place. The “manid” suffix is used to group compounds with similar chemical structures. This class of drug is variously referred to as nitroimidazoles, nitroimidazooxazines or nitroimidazopyrans. Development of this compound was initiated because of the urgent need for new antibacterial drugs effective against resistant strains of tuberculosis. Also, current anti-TB drugs are mainly effective against replicating and metabolically active bacteria, creating a need for drugs effective against persisting or latent bacterial infections as often occur in patients with tuberculosis.[7]
Discovery and pre-clinical development
Pretomanid was first identified in a series of 100 nitroimidazopyran derivatives synthesized and tested for antitubercular activity. Importantly, pretomanid has activity against static M. tuberculosis isolates that survive under anaerobic conditions, with bactericidal activity comparable to that of the existing drug metronidazole. Pretomanid requires metabolic activation by Mycobacterium for antibacterial activity. Pretomanid was not the most potent compound in the series against cultures of M. tuberculosis, but it was the most active in infected mice after oral administration. Oral pretomanid was active against tuberculosis in mice and guinea pigs at safely tolerated dosages for up to 28 days.[7]
Limited FDA approval
FDA approved pretomanid only in combination with bedaquiline and linezolid for treatment of a limited and specific population of adult patients with extensively drug resistant, treatment-intolerant or nonresponsive multidrug resistant pulmonary tuberculosis. Pretomanid was approved under the Limited Population Pathway (LPAD pathway) for antibacterial and antifungal drugs. The LPAD Pathway was established by Congress under the 21st Century Cures Act to expedite development and approval of antibacterial and antifungal drugs to treat serious or life-threatening infections in a limited population of patients with unmet need. Pretomanid is only the third tuberculosis drug to receive FDA approval in more than 40 years.[3][8]
PATENT
IN 201641030408
HETERO RESEARCH FOUNDATION
http://ipindiaservices.gov.in/PatentSearch/PatentSearch/ViewPDF
- By Reddy, Bandi Parthasaradhi; Reddy, Kura Rathnakar; Reddy, Adulla Venkat Narsimha; Krishna, Bandi Vamsi
- From Indian Pat. Appl. (2018), IN 201641030408
The nitroimidazooxazine Formula I (PA-824) is a new class of bioreductive drug for tuberculosis. The recent introduction of the nitroimidazooxazine Formula I (PA-824) to clinical trial by the Global Alliance for TB Drug Development is thus of potential significance, since this compound shows good in vitro and in vivo activity against Mycobacterium tuberculosis in both its active and persistent forms. Tuberculosis (TBa) remains a leading infectious cause of death worldwide, but very few new drugs have been approved for TB treatment ifi the past 35 years, the current drug therapy for TB is long and complex, involving multidrug combinations.
The mechanism of actiém of Pretomanid is thoughrto involve reduction of the nitro group, in a‘ process dependent on the Bacterial ‘ m E Nfilw‘fieéFPEOEPEa‘e fillyeifiaasnfi (F8189); $943“; 20mm; “q Mcyarecent Swiss on mutant strains showed that a 151-amino acid (17.37 kDa) protein of unknown function, Rv3547, also, appears to be critical for this activation. Equivalent genes are present in M. boVis and MaVium.
Pretomanid and its pharmace’utically acceptable salts Were generically disclosed in US 5,668,127 A and Specifically disclosed in US 6,087,358 A. US ‘358 patent discloses a process for the preparation of Pretomanid, which is as shown below in scheme 1:

CN 104177372 A discloses a process for the preparation of Pretomanid, which is as shown below in scheme II: 
Bioorganic & Medicinal Chemistry Letters 2008, Volume: 18, Issue: 7, Pages: 2256-2262 discloses a process for the preparation of Pretomanid, which is as shown below in scheme Ill: 
US 7,!15,736 B2-discloses_a process fdr the preparation of 3S-Hydroxy-6-nitrQ-2H-3, 4— dihydro-[2-1b]-imidazopyran which is a key intermediate of Pretomanid, which is as shown below in scheme IV:

Journal Medicinal Chemistry 2009, Volume: 52, Pages: 637 — 645 discloses a process for the preparation of ‘Pretomanid, which is as shown below in scheme V:

Joumal Organic Chemistry 2010; Volume: 75 (2]), Pages: 7479—82 discloses a process for. the preparation of Pretomanid, which is as shown below in scheme VI:


Example 3: Preparation of Pretomanid (S) 1- -(3 (tert- -Butyldomethylsilyloxy)- -2- -(-4 -(trifluoromethoxy)-71benzyloxy2‘- propyl)- 2- -methylP AT E N4Tnitro- fi-Eimigazole (Efgm Awlas (3315;501:1691 gin! %etra%1y7drofuraen (18(150 ml) at room temperature and stirred for 5 to 10 minutes then TBAF (9516 ml) was added to the reaction mixture and stirred for 2 hours, at room temperature, afler completion of the reaction removed solvent through vacuum to obtained residue, dissolved the residue in MDC (1800 ml) and water (1800 ml) stirred, separated the layers and the organic layer washed with 10% ‘ sodium bicarbonate the obtained organic solution was concentrated under atmospheric pressure to obtained residue added MeOH (1730 ml) at room temperature and the reaction mixture was cooled to 0°C to 5°C, added KOH (24.5 gm) at the same temperaturé then cooled to room temperature and stirred for 24 hours. After completion of reaction DM Water added drop wise over 30 minutes at 10°C to 15° C and stirred for 1 hour to 1 hour 30 minutes at room’lemperature, filtrated the compound and washed with DM wa‘er (133 ml) and dried under vacuum for 10 hours at 50° C. Yield: 53 gm , Chromatographic purity: 97.69% (by HPLC):
Example 4: Purification of Pretomanid Pretomanid (53 gm) was dissolved in MDC (795 ml) at room temperatur’e and stirred for 10 to 15 minutes, added charcoal (10 gm) and stirred for 30-35 minutes, remove the charcoal and concentrated to obtained residue: Dissolved the obtained residue in IPA (795 ml) and the reaction mixture was heated to 80°C maintained for 10-15 minutes, added cyclohexane (1600ml) for 30 minutes at 80° C, then cooled to room temperature and stirred the reaction mass for overnight, filtered the solid and washed with cyclohexane (265 ml), and dried under vacuum for 10 hours at 50° C. Yield: 48 gm (Percentage of Yield: 90%) Chromatographic purity: 99.97% by HPLC).
CLIP

ReferencE
CN104177372A.
WO9701562A1.
IN 201641030408
IN 201621026053
CN 107915747
CN 106632393
CN 106565744
CN 104177372
WO 9701562
US 6087358
PAPER
Science (Washington, DC, United States) (2008), 322(5906), 1392-1395.
Paper
PAPER
Huagong Shikan (2010), 24(4), 32-34, 51.
Xiaojin; Bioorganic & Medicinal Chemistry Letters 2008, Vol 18(7), Pg 2256-2262
PAPER
Orita, Akihiro; Advanced Synthesis & Catalysis 2007, Vol 349(13), Pg 2136-2144
https://onlinelibrary.wiley.com/doi/abs/10.1002/adsc.200700119
https://application.wiley-vch.de/contents/jc_2258/2007/f700119_s.pdf



Marsini, Maurice A.; Journal of Organic Chemistry 2010, Vol 75(21), Pg 7479-7482

Scheme 2. General Synthetic Strategy
Scheme 1

Scheme 1. Original Production Process for PA-824a
aDHP = 3,4-dihydropyran; p-TsOH = p-toluenesulfonic acid; MsOH = methanesulfonic acid.
Scheme 3

Scheme 3. Synthesis of a Functionalized Glycidol Derivativea
aCl3CCN = trichloroacetonitrile; TBME = tert-butylmethyl ether; TfOH = trifluoromethanesulfonic acid.

Scheme 4. Synthesis of PA-824
The combined organic extracts were washed with brine, dried (Na2SO4), filtered, and concentrated. Chromatography (75% EtOAc/hexanes) followed by recrystallization (i-PrOH/hexanes) affords PA-824 (1) (2.41 g, 62%) as a crystalline solid. Mp 150−151 °C (lit.(11a) mp 149−150); Rf 0.2 (75% EtOAc/hexanes); ee >99.9% as determined by chiral SFC (see the Supporting Information);
1H NMR (500 MHz, d6-DMSO) δ 8.09 (s, 1H), 7.48 (d, J = 8.6 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 4.81−4.62 (m, 3H), 4.51 (d, J = 11.9 Hz, 1H), 4.39−4.19 (m, 3H);
13C NMR (126 MHz, d6-DMSO) δ 148.7, 148.1, 143.0, 138.3, 130.4, 122.0, 120.0, 119.8, 69.7, 68.8, 67.51, 47.73;
IR [CH2Cl2 solution] νmax (cm−1) 2877, 1580, 1543, 1509, 1475, 1404, 1380, 1342, 1281, 1221, 1162, 1116, 1053, 991, 904, 831, 740;
HRMS (ESI-TOF) calcd for C14H12F3N3O5 359.0729, found 359.0728.


PAPER
Journal of Medicinal Chemistry (2010), 53(1), 282-294.
Journal of Medicinal Chemistry (2009), 52(3), 637-645.
PATENT

References
- ^ Jump up to:a b c d e “FDA approves new drug for treatment-resistant forms of tuberculosis that affects the lungs”. FDA. 14 August 2019. Retrieved 28 August 2019.
- ^ “Compounds | TB Alliance”. http://www.tballiance.org. Retrieved 2019-04-18.
- ^ Jump up to:a b Abutaleb Y (14 August 2019). “New antibiotic approved for drug-resistant tuberculosis”. Washington Post.
- ^ “TB Medicine Pretomanid Enters Regulatory Review Process in the United States | TB Alliance”. http://www.tballiance.org. Retrieved 2019-04-18.
- ^ “About TB Alliance”. TB Alliance. Retrieved 2019-04-18.
- ^ “PA-824 has a New Generic Name: Pretomanid”. TB Alliance. Retrieved 2019-04-18.
- ^ Jump up to:a b Lenaerts AJ, Gruppo V, Marietta KS, Johnson CM, Driscoll DK, Tompkins NM, Rose JD, Reynolds RC, Orme IM (June 2005). “Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models”. Antimicrobial Agents and Chemotherapy. 49 (6): 2294–301. doi:10.1128/AAC.49.6.2294-2301.2005. PMC 1140539. PMID 15917524.
- ^ FDA News Release. FDA approves new drug for treatment-resistant forms of tuberculosis that affects the lungs.
 |
|
| Legal status | |
|---|---|
| Legal status |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard(EPA) | |
| Chemical and physical data | |
| Formula | C14H12F3N3O5 |
| Molar mass | 359.261 g·mol−1 |
| 3D model (JSmol) | |
//////////////Pretomanid, FDA 2109, プレトマニド , Antibacterial, tuberculostatic, PA-824, ANTI tuberculostatic
