

.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY
Read all about Organic Spectroscopy on
ORGANIC SPECTROSCOPY INTERNATIONAL

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()




Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Tripeptide Glycyl-L-Prolyl-L-Glutamate (Gly-Pro-Glu or GPE)

Gly-Pro-Glu
Synonym: GPE, Glycyl-prolyl-glutamic acid, (1-3)IGF-1
Pfizer (Originator)
Neuren Pharmaceuticals (Originator)
Glypromate; glycine-proline-glutamate (neuroprotectant), Neuren
- CAS Number 32302-76-4
- Empirical Formula C12H19N3O6
- Molecular Weight 301.30
- Psychiatric Disorders (Not Specified)
Neurologic Drugs (Miscellaneous)
Cognition Disorders, Treatment of
Antiepileptic Drugs
Antidepressants Biochem/physiol Actions
Gly-Pro-Glu is a neuroprotective compound and the N-terminal tripeptide of IGF-1. Gly-Pro-Glu is neuroprotective after central administration in animal models of neurodegenerative processes, such as Huntington’s, Parkinson’s, Alzheimer’s diseases, and varies acute brain injury animal models. The neuroprotective activity is not related to its affinity to glutamate receptor. Findings indicate that GPE mimics insulin-like growth factor I effects on the somatostatin system through a mechanism independent of β-amyloid clearance that involves modulation of calcium and glycogen synthase kinase 3β signaling.
GPE is a naturally occurring peptide fragment which had been in phase III clinical trials at Neuren Pharmaceuticals for use as prophylactic neuroprotection for patients undergoing coronary artery bypass graft (CABG) and valvuloplasty surgery. Although clinical evaluation in Australia continues, phase III trials evaluating the compound in the U.S. were discontinued based on negative results. The compound is found in normal brain tissue and, when injected intravenously, has been shown to act by multiple pathways to protect brain tissue from injury. The drug was originally developed by Pfizer, but rights were transferred to Neuren pursuant to a proprietary agreement between the companies.
When amino acids join together (forming short groups called polypeptides, or much longer chains called proteins) the amine group of one amino acid joins with the carboxyl group of the next, making a peptide bond. These bonds don’t ionise at different pHs, but can be hydrolised — broken — reforming the amino acids. GPE is formed from the amino acids glycine, proline and glutamic acid:

This tripeptide has 3 pH-sensitive groups, each with its own pKa. What the university chemists needed to do was work out what form GPE is in when it is active in the brain, what parts of the molecule are critical to its effectiveness, and how to ‘tweak’ the molecule (by changing the side chains) so that it will remain in the brain for longer than the naturally-occurring substance. They also needed to make sure the final compound passes through the blood-brain barrier (that prevents most substances in the blood from entering and affecting the brain). If possible, they also wanted a compound that could be taken in pill form without being broken down in the stomach. It was also essential that the compound was safe for people to take!
Neuren Pharmaceuticals
After initial work on GPE at the university, the research was passed to a spin-off research group called Neuren Pharmaceuticals Ltd, which takes compounds discovered by the University of Auckland and develops them into medicines. Neuren developed GPE intoGlypromate® and are working with researchers in the US (including the US Military, who have a keen interest in a medicine that will reduce brain damage after head injuries) to test the compound on patients. There is considerable interest in Glypromate® world-wide, because at present there is nothing that reduces cell death after brain injuries. The chances of winning a race are pretty high when you’re the only competitor!Glypromate® is being tested on heart-bypass patients because up to 70% of bypass patients are affected mentally after their surgery. It’s thought that tiny clots form after the heart is restarted, and that these travel to the brain and cause mini-strokes. Unlike naturally-occurring strokes, or the brain damage caused by accident or war, the bypass surgery is planned, so before and after tests can be done on the patients to see exactly what effect the treatment has. Early results look very promising.
Glypromate is just one of the compounds Neuren is working on. Others may develop into treatments for Multiple Sclerosis, Parkinson’s Disease or Alzheimer’s Disease as well as various kinds of cancer. The company’s links with overseas research groups mean that compounds developed in New Zealand are able to be tested in the US and gain the FDA approval which will allow them to be used in most countries in the world.
The tripeptide Glycyl-L-Prolyl-L-Glutamate (Gly-Pro-Glu or GPE) is a naturally occurring peptide, which is proteolytically cleaved from insulin-like growth factor-1 (IGF-1). IGF-1 is a potent neurotrophic factor produced endogenously in damaged regions of the brain. It has been postulated that some of the neuroprotective actions of IGF-1 are mediated by GPE although the precise mechanism of action remains unclear. GPE has a different mode of action to IGF-1 as GPE does not bind to the IGF-1 receptor. Rather, GPE has been shown to bind with low affinity to the N-methyl-D-aspartate (NMDA) receptor and also elicit a biological response via other mechanisms. GPE facilitates the release of dopamine through interaction with the NMDA receptor but GPE stimulated acetylcholine release is via an unknown, non-NMDA pathway.
It has been demonstrated that GPE can act as a neuronal rescue agent following brain injury or disease, including hypoxic-ischemic brain injury, NMDA challenge, chemical toxins and in animal models of Parkinson’s and Alzheimer’s disease. Analogs of GPE are thus of interest in the development of novel pharmaceutical agents for the treatment of central nervous system (CNS) injuries and neurodegenerative disorders among others.
CURRENT STATUS
Neuren Pharmaceuticals was developing Glypromate (glycine-proline glutamate), a naturally occurring small-molecule neuroprotectant derived from IGF-1 which inhibits caspase III dependent apoptosis, for the potential treatment of neurodegenerative diseases by iv infusion. By June 2008, a phase III trial had begun . However, in December 2008, the company discontinued further development of the drug after it failed to show an observable effect [972907]. In November 2005, the company was seeking to outlicense the drug [771417].
Neuren is also investigating the Glypromate analog, NNZ-2566 for similar indications.

In August 2006, Neuren expected Glypromate to be eligible for Orphan Drug status for neurodegenerative diseases and planned to apply for Fast Track status for the drug.
SYDNEY, Australia, Sept. 4 /PRNewswire-FirstCall/ — Neuren Pharmaceuticals today announced that physicians from Madigan Army Medical Center (Madigan) in Tacoma, Washington, will conduct an investigator- initiated Phase 2 trial to determine the safety and efficacy of Glypromate(R) in reducing brain injury caused by out of hospital cardiac arrest. The trial will start in mid-2007 and will be managed by The Henry M. Jackson Foundation for the Advancement of Military Medicine (Jackson Foundation) in consultation with the clinical investigators at Madigan.
The proposed study will be an investigator-initiated study which means that the Investigational New Drug (IND) application will be submitted to the FDA by the Army investigators rather than by Neuren. Neuren will provide the drug product as well as access to preclinical, clinical and regulatory documents related to Glypromate(R). The Company’s only financial commitment will be compensation to the Jackson Foundation for administrative costs incurred in coordinating the study. Neuren will retain all commercial rights to Glypromate(R) in these indications.
Cardiac arrest involves the sudden, complete cessation of heart function and circulation leading rapidly to neurological and other organ system damage. Among patients who survive, the consequences of neurological damage resulting from lack of blood flow and oxygen to the brain represent the primary adverse outcomes. This occurs in up to 80% of survivors and causes cognitive impairment such as occurs in patients undergoing major cardiac surgery, the focus for Neuren’s upcoming Phase 3 study with Glypromate(R). However recovery without residual neurological damage after cardiac arrest is rare.
There are no drugs approved to reduce the neurological damage caused by cardiac arrest. Neuren believes that Glypromate(R) for this indication will be eligible for Orphan Drug designation. Orphan Drug designation provides for a period of market exclusivity following approval as well as possible access to US government grants. In addition, because of the serious nature of neurological impairment resulting from cardiac arrest and the lack of available drug therapy, Neuren intends to apply for Fast Track designation which provides for accelerated clinical development and review.
While the Army’s investigator-initiated trial will focus on out of hospital cardiac arrest, if this trial is successful, Neuren, the Jackson Foundation and the Army investigators are considering additional trials of Glypromate(R) to reduce brain damage resulting from related conditions including in-hospital cardiac arrest and treatment of patients with ventricular fibrillation, the heart rhythm disturbance associated with more than 75% of cardiac arrests.
Under the agreement, the Jackson Foundation will provide support to the Army investigators in clinical trial preparations, protocol development, obtaining human subjects clearance, coordination of patient enrolment, data management and analysis, and preparation of study reports.
Mr David Clarke, CEO of Neuren said: “This is a very important development for Neuren in that it reflects a growing appreciation of the potential for Glypromate(R) to reduce neurological damage. It also, of course, reinforces the value and strength of Neuren’s relationship with the US Army physicians and scientists. Cardiac arrest is a devastating clinical event and one for which a drug to reduce the neurological consequences is clearly needed. The addition of this trial will now give Neuren a very strong and cost effective portfolio of clinical trials in 2007 — a Phase 3 and a Phase 2 for Glypromate(R) and the two Phase 2 trials with NNZ-2566.”
Approximately 300,000 deaths result from cardiac arrest in the US each year, making cardiac arrest one of the leading causes of death. According to the American Heart Association, each year approximately 160,000 people in the US experience sudden cardiac arrest outside of a hospital or in a hospital emergency department.
Neuren estimates that the number of patients in the US that could be treated for out of hospital cardiac arrest and related indications is approximately 400,000 which could represent a potential market of US$800 million.
About Madigan Army Medical Center
Madigan Army Medical Center, located in Tacoma, Washington, is one of the major US Army medical centers, providing clinical care to over 120,000 active, reserve and retired military personnel and dependents. The hospital has a medical staff of more than 1,000 with 200 physicians and nurses in training. Madigan’s Department of Clinical Investigations, which is dedicated to writing, performing, and regulating clinical research, is conducting approximately 200 clinical trials across a wide spectrum of indications from Phase I to IV.
About the Jackson Foundation
The Jackson Foundation is a private, not-for-profit organisation that supports the US military in conducting medical research and clinical trials and has established relationships with more than 160 military medical organisations worldwide. It was founded in 1983, in part, to foster cooperative relationships between the military medical community and the private sector, including pharmaceutical sponsors. The Jackson Foundation manages Phase I – IV clinical trials utilizing an established network of military medical centers across the United States.
About Glypromate(R)
Glypromate(R) is a peptide fragment of IGF-1 and is being developed by Neuren as a potential therapeutic candidate for diseases caused by some forms of chronic or acute brain injury. Glypromate(R) has been shown to act by multiple pathways to protect brain tissue from injury. Neuren has successfully completed a Phase I safety study and a Phase IIa safety and pharmacokinetics study and plans to initiate a Phase III study in late 2006.
About Neuren Pharmaceuticals
Neuren Pharmaceuticals is a biotechnology company developing novel therapeutics in the fields of brain injury and diseases and metabolic disorders. The Neuren portfolio consists of six product families, targeting markets with large unmet needs and limited competition. Neuren has three lead candidates, Glypromate(R) andNNZ-2566, presently in the clinic in development to treat a range of acute neurological conditions, and NNZ-2591, in preclinical development for Parkinson’s and other chronic conditions. Neuren has commercial and development partnerships with the US ArmyWalter Reed Army Institute of Research, Metabolic Pharmaceuticals,UCLA Medical Center and the National Trauma Research Institute in Melbourne.
For more information, please visit Neuren’s website at http://www.neurenpharma.com
Company David Clarke CEO of Neuren T: 1800 259 181 (Australia) T: +64 9 3 367 7167 ext 82308 (New Zealand) M: +64 21 988 052 Media and investor relations Rebecca Piercy Buchan Consulting T: +61 9827 2800 M: +61 422 916 422
CONTACT: David Clarke, CEO of Neuren, 1-800-259-181(Australia), or
+64-9-3-367-7167 ext 82308 (New Zealand), or +64-21-988-052 (mobile); or
Media and investor relations – Rebecca Piercy of Buchan Consulting,
+61-9827-2800, +61-422-916-422 (mobile)
Web site: http://www.neurenpharma.com/
REFERENCES
1 EP 0366638
2 WO 2005042000
3 WO 2008153929
4 WO 2009033805
5 WO 2009033806
Synthesis off isotopically labelled glycyl-L-prolyl-L-glutamic acid (Glypromate(R)) and derivatives
J Label Compd Radiopharm 2006, 49(6): 571
An efficient fmoc solid-phase synthesis of an amphiphile of the neuroprotective agent glycyl-prolyl-glutamic acid
Synlett (Stuttgart) 2014, 25(15): 2221
Intracellular pathways activated by Insulin-like growth factor 1 and its derivates
40th Annu Meet Soc Neurosci (November 13-17, San Diego) 2010, Abst 167.13
| EP2667715A1 * | Jan 27, 2012 | Dec 4, 2013 | Neuren Pharmaceuticals Limited | Treatment of autism spectrum disorderes using glycyl-l-2-methylprolyl-l-glutamic acid |
| EP2667715A4 * | Jan 27, 2012 | Jul 23, 2014 | Neuren Pharmaceuticals Ltd | Treatment of autism spectrum disorderes using glycyl-l-2-methylprolyl-l-glutamic acid |
| US8940732 | Jan 15, 2010 | Jan 27, 2015 | Massachusetts Institute Of Technology | Diagnosis of autism spectrum disorders and its treatment with an antagonist or inhibitor of the 5-HT2c receptor signaling pathway |
| US9212204 | Jan 26, 2015 | Dec 15, 2015 | Neuren Pharmaceuticals Limited |
| WO2005042000A1 * | 22 Oct 2004 | 12 May 2005 | David Charles Batchelor | Neuroprotective effects of gly-pro-glu following intravenous infusion |
| WO2005097161A2 * | 30 Mar 2005 | 20 Oct 2005 | Peter D Gluckman | Gpe and g-2mepe, caffeine and alkanol for treatment of cns injury |
| WO2006127702A2 * | 23 May 2006 | 30 Nov 2006 | Neuren Pharmaceuticals Ltd | Analogs of glycyl-prolyl-glutamate |
| EP0366638A2 * | 24 Oct 1989 | 2 May 1990 | KabiGen AB | Neuromodulatory peptide |
| US20020151522 * | 13 Mar 2002 | 17 Oct 2002 | Tajrena Alexi | Regulation of weight |
| Reference | ||
|---|---|---|
| 1 | * | ALONSO DE DIEGO, SERGIO A. ET AL: “New Gly-Pro-Glu (GPE) analogues: Expedite solid-phase synthesis and biological activity” BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 16, no. 5, 2006, – 1392 page 1396, XP002527092 |
| 2 | * | SARA V R ET AL: “IDENTIFICATION OF GLY-PRO-GLU (GPE), THE AMINOTERMINAL TRIPEPTIDE OF INSULIN-LIKE GROWTH FACTOR 1 WHICH IS TRUNCATED IN BRAIN, AS A NOVEL NEUROACTIVE PEPTIDE” BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ACADEMIC PRESS INC. ORLANDO, FL, US, vol. 165, no. 2, 15 December 1989 (1989-12-15), pages 766-771, XP000992688 ISSN: 0006-291X |
//////Gly-Pro-Glu, GPE, Glycyl-prolyl-glutamic acid, 32302-76-4, Tripeptide, Glycyl-L-Prolyl-L-Glutamate, Glypromate®, (1-3)IGF-1 , PHASE 3, Glypromate, glycine-proline-glutamate, neuroprotectant, Neuren
Neuren’s NNZ-2566 shows clinical benefit in Rett syndrome trial


Promising results in Phase 2 clinical trial
by Michael Tranfaglia, MDFRAXA Medical Director
This isn’t a Fragile X trial, but the Neuren compound, NNZ-2566, that is in trials now for Fragile X has shown significant positive effects in a Phase 2 trial for Rett syndrome.
The results of the trial are interesting, in that improvement was seen a Rett syndrome-specific rating scale compared to placebo, and there was also improvement noted on the CGI-I (Clinical Global Impression of Improvement) and Caregiver Top 3 Concerns. However, there was no effect seen on ABC scores (Aberrant Behavior Checklist) compared to placebo. Many in the Fragile X field have noted the inadequacies of the ABC; indeed, it was never designed or intended to be an outcome measure for clinical trials. In this case, a Rett-specific rating scale called the Motor-Behavior Assessment (MBA) showed a statistically significant and clinically meaningful treatment effect at the highest dose of the Neuren compound compared to placebo.
This is great news for those of us in the Fragile X community for several reasons:
- It shows that this compound really does something—it seems to have useful properties in actual patients, and that’s not trivial.
- It demonstrates that disease-specific symptoms can improve significantly on the drug, and that improvement can be measured in a relatively short clinical trial.
- It shows that a drug can have beneficial effects on core features of a genetically based developmental disorder, even if the more general rating scales (like the ABC) show no change.
–
This last point is strongly reminiscent of the experience of many families and clinicians in recent Fragile X clinical trials, where the drugs showed no advantage compared to placebo based on rating scales, but genuine improvement was noted in many subjects, with significant deterioration upon discontinuation of the drugs. Thus the calls for improved rating scales which can “capture” these core, disease-specific therapeutic effects. The NeurenFragile X trial is using some Fragile X-specific outcome measures which will hopefully lead to similar positive results.
The fact that this result is good news for Neuren also means that the company should remain financially viable for longer, so that they can continue the development of this compound for a number of indications—more “shots on goal”.
Of course, the usual caveats apply: this was a small study, and these results need to be replicated in a larger Phase 3 trial. Still, there’s a realistic possibility that we may see a similar result in Fragile X!
IACS -9571
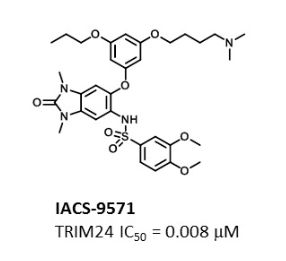

IACS-9571
TRIM24/BRPF1 bromodomain inhibitor
IACS-9571; IACS 9571; IACS9571.
| Molecular Formula: | C32H42N4O8S |
|---|---|
| Molecular Weight: | 642.76288 g/mol |
N-[6-[3-[4-(dimethylamino)butoxy]-5-propoxyphenoxy]-1,3-dimethyl-2-oxobenzimidazol-5-yl]-3,4-dimethoxybenzenesulfonamide
BOARD OF REGENTS, UNIVERSITY OF TEXAS SYSTEM
IACS-9571 is a potent and selective inhibitor TRIM24 and BRPF1. The bromodomain containing proteins TRIM24 (Tripartite motif containing protein 24) and BRPF1 (bromodomain and PHD finger containing protein 1) are involved in the epigenetic regulation of gene expression and have been implicated in human cancer. Overexpression of TRIM24 correlates with poor patient prognosis and BRPF1 is a scaffolding protein required for the assembly of histone acetyltransferase complexes, where the gene of MOZ (monocytic leukemia zinc finger protein) was first identified as a recurrent fusion partner in leukemia patients (8p11 chromosomal rearrangements). IACS-9571 has low nanomolar affinities for TRIM24 and BRPF1 (ITC Kd = 31 nM and 14 nM, respectively). With its excellent cellular potency (EC50 = 50 nM) and favorable pharmacokinetic properties (F = 29%), IACS-9571 is a high-quality chemical probe for the evaluation of TRIM24 and/or BRPF1 bromodomain function in vitro and in vivo. (J Med Chem. 2015 Jun 10. [Epub ahead of print] )

PAPER
http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.5b00405
Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor
†Institute for Applied Cancer Science, and ‡Core for Biomolecular Structure and Function, The University of Texas MD Anderson Cancer Center, 1881 East Road, Unit 1956, Houston, Texas 77054, United States
§ Department of Epigenetics and Molecular Carcinogenesis, The University of Texas MD Anderson Cancer Center,
1515 Holcombe Boulevard
, Houston, Texas 77030, United States
J. Med. Chem., 2016, 59 (4), pp 1440–1454
DOI: 10.1021/acs.jmedchem.5b00405
Publication Date (Web): June 10, 2015
Copyright © 2015 American Chemical Society

The bromodomain containing proteins TRIM24 (tripartite motif containing protein 24) and BRPF1 (bromodomain and PHD finger containing protein 1) are involved in the epigenetic regulation of gene expression and have been implicated in human cancer. Overexpression of TRIM24 correlates with poor patient prognosis, and BRPF1 is a scaffolding protein required for the assembly of histone acetyltransferase complexes, where the gene of MOZ (monocytic leukemia zinc finger protein) was first identified as a recurrent fusion partner in leukemia patients (8p11 chromosomal rearrangements). Here, we present the structure guided development of a series of N,N-dimethylbenzimidazolone bromodomain inhibitors through the iterative use of X-ray cocrystal structures. A unique binding mode enabled the design of a potent and selective inhibitor 8i (IACS-9571) with low nanomolar affinities for TRIM24 and BRPF1 (ITC Kd = 31 nM and ITC Kd = 14 nM, respectively). With its excellent cellular potency (EC50 = 50 nM) and favorable pharmacokinetic properties (F = 29%), 8i is a high-quality chemical probe for the evaluation of TRIM24 and/or BRPF1 bromodomain function in vitro and in vivo.
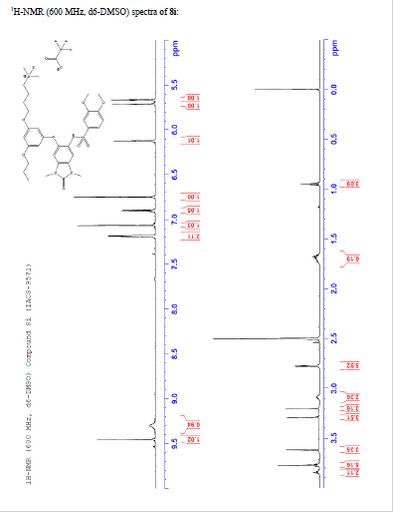
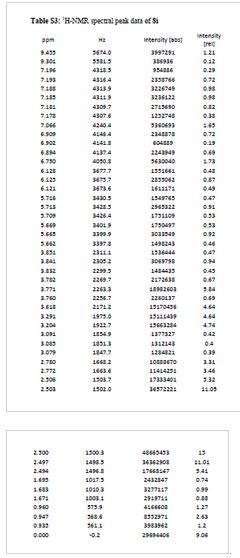
TFA salt of 8i (106 mg, 57%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 9.46 (s, 1H), 9.30 (br-s, 1H), 7.19 (m, 2H), 7.07 (s, 1H), 6.90 (d, J = 9.0 Hz, 1H), 6.75 (s, 1H), 6.13 (t, J = 2.2 Hz, 1H), 5.71 (t, J = 2.0 Hz, 1H), 5.67 (t, J = 2.0 Hz, 1H), 3.84 (t, J = 5.9 Hz, 2H), 3.77 (m, 5H), 3.62 (s, 3H), 3.29 (s, 3H), 3.20 (s, 3H), 3.12–3.05 (m, 2H), 2.78 (d, J = 4.7 Hz, 6H), 1.77–1.63 (m, 6H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (600 MHz, DMSO-d6) δ 160.3, 160.0, 159.3, 154.1, 152.0, 148.4, 143.9, 131.8, 128.2, 126.0, 121.9, 120.5, 110.4, 109.4, 106.4, 100.6, 95.9, 95.8, 95.2, 68.9, 66.7, 56.3, 55.6, 55.4, 42.1, 27.1, 27.0, 25.6, 21.9, 20.7, 10.4. MS (ESI) m/z 644 [M + H]+.
NMR
IACS -9571
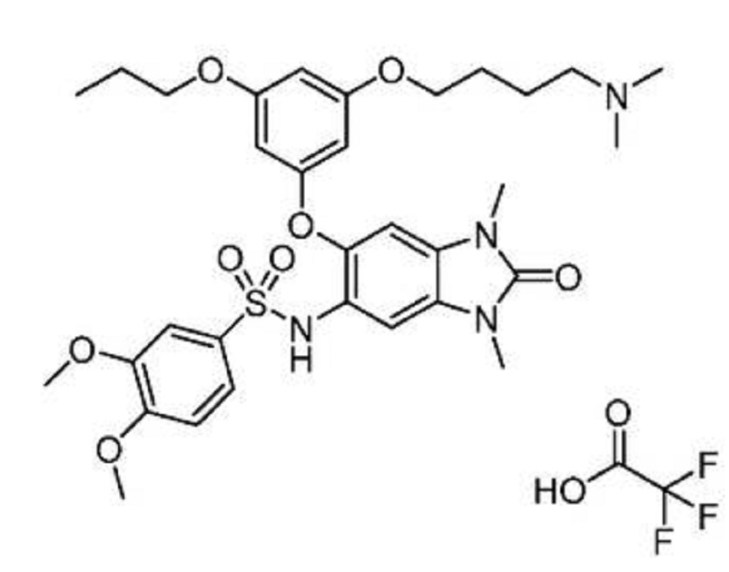
N-(6-(3-(4-(dimethylamino)butoxy)-5- propoxyphenoxy)-l,3-dimethyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-3,4- dimethoxybenzenesulfonamide 2,2,2-trifluoroacetate
 CLICK ON IMAGE
CLICK ON IMAGE
ABSTRACT
251st ACS National Meeting & Exposition
13–17 March 2016
San Diego, United States
MEDI 5
Discovery and development of a potent dual TRIM24/BRPF1 bromodomain inhibitor, IACS -9571, using structure- based drug design Wylie S. Palmer 1 , wpalmer@mdanderson.org, Guillaume Poncet -Montagne 1 , Gang Liu 1 , Alessia Petrocchi 1 , N aphtali Reyna 1 , Govindan Subramanian 1 , Jay Theroff 1 , Maria Kost -Alimova 1 , Jennifer Bardenhagen 1 , Elisabetta Leo 1 , Hannah Sheppard 1 , Trang Tieu 1 , Shi Xi 1 , Yanai Zhan 1 , Shuping Zhao 1 , Michelle Barton 2 , Giulio Draetta 1 , Carlo Toniatti 1 , Philip Jones 1 , Mary Ge ck Do 1 , Jannik Andersen 1 . (1) Institute for Applied Cancer Science, The University of Texas, MD Anderson Cancer Center, Houston, Texas, United States (2) Department of Epigenetics and Molecular Carcinogenesis, The University of Texas, MD Anderson Cancer Center, Houston, Texas, United States
Bromodomains are an important class of chromatin remodeling proteins that recognize acetylated lysine residues on histone tails. As epigenetic targets they regulate gene transcription and offer a new way to treat diseas es, particularly in inflammation and oncology. The bromodomain and extra- terminal (BET) family has emerged as an important and druggable example of this class of proteins with the successful entry of small- molecule inhibitors into the clinic. Other families of bromodomains are only starting to be explored, such as the Tripartite Motif -containing 24 protein (TRIM24) and bromodomain- PHD finger protein 1 (BRPF1). Both proteins contain a dual PHD -bromo motif which have a role in recognizing specific histone mar ks. TRIM24 recognizes the dual histone marks of unmodified H3K4 and acetylated- H3K23 within the same histone tail. TRIM24 is a potent co- activator of ER -alpha and overexpression of TRIM24 has been linked to poor survival rates in breast cancer patients.
This presentation will describe the structure guided development of a series of N,N- dimethyl -benzimidazolones through the iterative use of X -ray cocrystal structures. A unique binding mode enabled the design of a potent and selective inhibitor (IACS -9571) with low nanomolar affinities for TRIM24 and BRPF1 (ITC Kd = 31 nM and ITC Kd = 14 nM, respectively). With its excellent cellular potency (EC 50 = 50 nM) and favorable pharmacokinetic properties, IACS -9571 is a high- quality chemical probe for the evaluation of TRIM24 and/or BRPF1 bromodomain function in vitro and in vivo

PATENT
WO-2016033416-A1
Synthesis of Intermediates:
N-(6-bromo-l ,3-dimethyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-2,2,2- trifluoroacetamide (Intermediate 1):
Step 1 : 5-nitro-lH-benzo[d]imidazol-2(3H)-one:
To a 0 °C solution of 4-nitrobenzene- 1 ,2-diamine (44 g, 285 mmol) in 80 mL of DMF was added l, l’-carbonyldiimidazole (70 g, 428 mmol). The reaction mixture was stirred at RT for 4 h, then water (250 mL) was added. The resulting suspension was filtered, and the collected solids were washed with water (200 mL) and dried to give 5-nitro-lH- benzo[d]imidazol-2(3H)-one as a yellow solid (45 g, 88%). MS (ES+) C7H5N3O3 requires: 179, found: 180 [M+H]+.
Step 2: l,3-dimethyl-5-nitro-lH-benzo[d]imidazol-2(3H)-one:
To a solution of 5-nitro-lH-benzo[d]imidazol-2(3H)-one (55 g, 309 mmol) in 150 mL of DMF was added K2CO3 (85 g, 618 mmol), the reaction mixture was cooled to 0 °C, then iodomethane (109 g, 772 mmol) was slowly added. The reaction mixture was stirred at RT overnight, then water was added to the reaction mixture. The resulting suspension was filtered and the collected solids were washed with water (200 mL) and dried to give 1,3- dimethyl-5-nitro-lH-benzo[d]imidazol-2(3H)-one as a yellow solid (55 g, 86%). MS (ES+) C9H9N3O3 requires: 207, found: 208 [M+H] +.
Step 3: 5-amino-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one:
To a solution of l,3-dimethyl-5-nitro-lH-benzo[d]imidazol-2(3H)-one (50 g, 240 mmol) in 200 mL of EtOAc under an inert atmosphere was added 10% palladium on activated carbon (5 g, 24 mmol). The reaction mixture was then charged with hydrogen and stirred at RT under an ¾ atmosphere overnight. The reaction mixture was filtered through a pad of celite then concentrated to give 5-amino-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)- one as a yellow solid (32 g, 68%). MS (ES+) C9H11N3O requires: 177, found: 178 [M+H]+.
Step 4: 5-amino-6-bromo-l ,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one:
To a 0 °C solution of 5-amino-l ,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (4 g, 22.6 mmol) in 25 mL of CHCI3 and 25 mL of AcOH was slowly added drop wise bromine (3.5 g, 22.6mmol). The mixture was stirred at RT for 30 min, then concentrated and purified by silica gel chromatography (1 : 1 EtOAc/ hexanes) to afford 5-amino-6-bromo-l ,3-dimethyl- lH-benzo[d]imidazol-2(3H)-one as a yellow solid (3.2 g, 69%). MS (ES+) C9HioBrN30 requires: 256, found: 257 [M+H]+.
Step 5: N-(6-bromo-l ,3-dimethyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-2,2,2- trifluoroacetamide:
To a 0 °C solution of 5-amino-6-bromo-l ,3-dimethyl-lH-benzo[d]imidazol- 2(3H)-one (1.50 g, 5.9 mmol) in DCM (45 ml) was added DMAP (72 mg, 0.59 mmol), triethylamine (1.63 ml, 11.7 mmol) and trifluoroacetic anhydride (0.91 ml, 6.4 mmol). The reaction mixture was stirred for 2 h and warmed to RT. The reaction mixture was then quenched with water and the organic phase was washed with brine, dried over sodium sulfate, filtered and concentrated to give N-(6-bromo-l,3-dimethyl-2-oxo-2,3-dihydro-lH- benzo[d]imidazol-5-yl)-2,2,2-trifluoroacetamide (Intermediate 1) as a yellow solid (2.20 g, 100%). MS (ES+) CiiH9BrF3N302 requires: 352, found 353 [M+H]+.
5-amino-6-(3-hydroxyphenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (Intermediate 2, Route A):
To a mixture of 5-amino-6-(3-(benzyloxy)phenoxy)-l,3-dimethyl-lH- benzo[d]imidazol-2(3H)-one (400 mg, 1.07 mmol) in DCM (20 mL) at -78 °C was added tribromoborane (5.3 mL, 5.3 mmol). The mixture was warmed up to room temperature gradually, then quenched by methanol dropwise, concentrated, and purified by column chromatography (20-100% EtOAc/hexanes and then 0-40% methanol/EtOAc) to give 5- amino-6-(3-hydroxyphenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one as a solid (240 mg, 79%). MS (ES+) C15H15N3O3 requires: 285, found: 286 [M+H]+.
5-amino-6-(3-hydroxyphenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (Intermediate 2, Route B):
Step 2
Step 1: 3-[(ieri-butyldimethylsilyl)oxy]phenol:
A mixture of lH-imidazole (2.25 g, 33.1 mmol), ieri-butylchlorodimethylsilane (3.83 g, 25.4 mmol) and resorcinol (5.6 g, 51 mmol) in THF (30 ml) was stirred at 80 °C for 5 h. The resulting suspension of the cooled reaction mixture was filtered and the collected filtrate was concentrated and purified by silica-gel chromatography (20:80 to 0:100, EtOAc/hexanes) to give 3-((ieri-butyldimethylsilyl)oxy)phenol (2.78 g, 49%). MS (ES+) C12H20O2S1 requires: 224, found 225 [M+H]+.
Step 2: 5-amino-6-(3-((ier^butyldimethylsilyl)oxy)phenoxy)-l ,3-dimethyl-lH- benzo[d]imidazol-2(3H)-one:
A mixture of 3-((ieri-butyldimethylsilyl)oxy)phenol (1.39 g, 6.20 mmol), quinolin-8-ol (79 mg, 0.55 mmol), copper(I) chloride (20 mg, 0.21 mmol), potassium phosphate (526 mg, 2.48 mmol) and 5-amino-6-bromo-l ,3-dimethyl-lH-benzo[d]imidazol- 2(3H)-one (529 mg, 2.07 mmol) in diglyme (20 ml) in a 100 mL round-bottom flask was degassed under a nitrogen atmosphere and heated to 120 °C for 24 h. To the cooled reaction mixture was added silica gel, stirred for 2 min, then the mixture was filtered through a pad of silica gel. The collected filtrate was concentrated and purified by column chromatography (20:80 to 0: 100, EtOAc/hexanesthen 0: 100 to 40:60, MeOH/EtOAc) to give 5-amino-6-(3- ((ieri-butyldimethylsilyl)oxy)phenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (521 mg, 63%). MS (ES+) C21H29N3O3S1 requires: 399, found 400 [M+H]+.
Step 3: 5-amino-6-(3-hydroxyphenoxy)-l,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one:
To a 0 °C solution of 5-amino-6-(3-((ieri-butyldimethylsilyl)oxy)phenoxy)-l,3- dimethyl-lH-benzo[d]imidazol-2(3H)-one (623 mg, 1.56 mmol) in THF was added a solution of ieira-butylammonium fluoride (0.90 mL, 3.1 mmol) in THF, the reaction mixture was allowed to warm up to RT and then stirred for 1-2 h. The reaction mixture was quenched with 1 M hydrogen chloride (0.10 mL, 3.1 mmol) and then partitioned between EtOAc and water. The seperated organic layer was washed with water twice, then concentrated and purified by column chromatography (20-80% EtOAc/hexanes and 0-40% MeOH/DCM) to give 5-amino-6-(3-hydroxyphenoxy)-l ,3-dimethyl-lH-benzo[d]imidazol-2(3H)-one (120 mg, 27%) as a solid. MS (ES+) C15H15N3O3 requires: 285, found 286 [M+H]+.
EXAMPLE 10: N-(6-(3-(4-(dimethylamino)butoxy)-5-propoxyphenoxy)-l,3-dimethyl-2- oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-3,4-dimethoxybenzenesulfonamide 2,2,2-
To a solution of N-(6-(3-(4-aminobutoxy)-5-propoxyphenoxy)-l ,3-dimethyl-2- oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-3,4-dimethoxybenzenesulfonamide 2,2,2- trifluoroacetate (180 mg, 0.247 mmol) in methanol (3.0 ml) was added triethylamine (0.034 ml, 0.25 mmol), acetic acid (0.028 ml, 0.49 mmol), formaldehyde (0.054 ml, 2.0 mmol), and sodium triacetoxyborohydride (131 mg, 0.618 mmol). The reaction mixture was stirred at room temperature and checked by LCMS every 30 minutes. After 3 h the reaction was complete by LCMS. The reaction was quenched with a few drops of TFA and concentrated under reduced pressure. The residue was purified by prep-HPLC using a gradient of 20-60% ACN/water containing 0.1% TFA to afford N-(6-(3-(4-(dimethylamino)butoxy)-5- propoxyphenoxy)-l,3-dimethyl-2-oxo-2,3-dihydro-lH-benzo[d]imidazol-5-yl)-3,4- dimethoxybenzenesulfonamide 2,2,2-trifluoroacetate (106 mg, 57%) as a white solid. MS (ES+) C32H42N4O8S requires: 642, found 643 [M+H]+. ¾ NMR (600 MHz, DMSO-ifc) δ 9.46 (s, 1H), 9.30 (br-s, 1H), 7.19 (m, 2H), 7.07 (s, 1H), 6.90 (d, 7 = 9.0 Hz, 1H), 6.75 (s, 1H), 6.13 (t, 7 = 2.2 Hz, 1H), 5.71 (t, J = 2.0 Hz, 1H), 5.67 (t, J = 2.0 Hz, 1H), 3.84 (t, 7 = 5.9 Hz, 2H), 3.77 (m, 5H), 3.62 (s, 3H), 3.29 (s, 3H), 3.20 (s, 3H), 3.12-3.05 (m, 2H), 2.78 (d, 7 = 4.7 Hz, 6H), 1.77-1.63 (m, 6H), 0.95 (t, 7 = 7.3 Hz, 3H)
References
1: Palmer WS, Poncet-Montange G, Liu G, Petrocchi A, Reyna N, Subramanian G, Theroff J, Yau A, Kost-Alimova M, Bardenhagen JP, Leo E, Shepard HE, Tieu TN, Shi X, Zhan Y, Zhao S, Draetta G, Toniatti C, Jones P, Geck Do M, Andersen JN. Structure-Guided Design of IACS-9571, a Selective High-Affinity Dual TRIM24-BRPF1 Bromodomain Inhibitor. J Med Chem. 2015 Jun 10. [Epub ahead of print] PubMed PMID: 26061247.
US-20160060260-A1
Institute for Applied Cancer Science, The University of Texas, MD Anderson Cancer Center, Houston, Texas, United States

The University of Texas MD Anderson Cancer Center | University of Texas System

The new Institute for Applied Cancer Science will be located at the south campus of M.D.

Draetta arrived at MD Anderson in 2011 to direct the Institute for Applied Cancer Science. He oversees the moon shots platforms
Department of Epigenetics and Molecular Carcinogenesis, The University of Texas, MD Anderson Cancer Center, Houston, Texas, United States

///////IACS-9571, TRIM24, BRPF1 bromodomain inhibitor, IACS-9571, IACS 9571, IACS9571, BOARD OF REGENTS, UNIVERSITY OF TEXAS SYSTEM
CAS BASE 1800477-30-8
CAS OF 1:1 TRIFLUOROACETATE 1883598-69-3
c1(cc(cc(c1)OCCC)Oc3cc2N(C(N(c2cc3NS(=O)(=O)c4cc(c(cc4)OC)OC)C)=O)C)OCCCCN(C)C
CCCOC1=CC(=CC(=C1)OC2=C(C=C3C(=C2)N(C(=O)N3C)C)NS(=O)(=O)C4=CC(=C(C=C4)OC)OC)OCCCCN(C)C
TFA salt of 8i (106 mg, 57%) as a white solid. 1H NMR (600 MHz, DMSO-d6) δ 9.46 (s, 1H), 9.30 (br-s, 1H), 7.19 (m, 2H), 7.07 (s, 1H), 6.90 (d, J = 9.0 Hz, 1H), 6.75 (s, 1H), 6.13 (t, J = 2.2 Hz, 1H), 5.71 (t, J = 2.0 Hz, 1H), 5.67 (t, J = 2.0 Hz, 1H), 3.84 (t, J = 5.9 Hz, 2H), 3.77 (m, 5H), 3.62 (s, 3H), 3.29 (s, 3H), 3.20 (s, 3H), 3.12–3.05 (m, 2H), 2.78 (d, J = 4.7 Hz, 6H), 1.77–1.63 (m, 6H), 0.95 (t, J = 7.3 Hz, 3H). 13C NMR (600 MHz, DMSO-d6) δ 160.3, 160.0, 159.3, 154.1, 152.0, 148.4, 143.9, 131.8, 128.2, 126.0, 121.9, 120.5, 110.4, 109.4, 106.4, 100.6, 95.9, 95.8, 95.2, 68.9, 66.7, 56.3, 55.6, 55.4, 42.1, 27.1, 27.0, 25.6, 21.9, 20.7, 10.4. MS (ESI) m/z 644 [M + H]+.
Biocon’s Insulin Glargine gets approval in Japan

![]()
Avik Das | TNN | Mar 28, 2016, 02.52 PM IST
BENGALURU: Biopharmaceutical company Biocon said it got approval from Japan’s health ministry to sell its biosimilar Insulin Glargine in the country.
The product, which is a ready-to-use, prefilled disposable pen with 3 ml of 100IU Insulin Glargine, is expected to be launched in Japan in the first quarter of 2017 with its commercial partner FUJIFILM Pharma Co. Ltd, Biocon said on Monday.
The move will help Biocon capture a significant share of the Japanese Glargine market, which is about $144 million and second largest market outside of North America & Europe.
“The Insulin Glargine approval in the highly regulated market like Japan, marks a huge credibility milestone for Biocon. We see this as a significant achievement in our journey of making global impact in diabetes management through our affordable biosimilar insulins,” chairperson and managing director Kiran Mazumdar-Shaw said.

Kiran Mazumdar–Shaw

Biosimilars are biologic products, made inside living cells and has no clinical differences in terms of safety and effectiveness from the main product. They are however not considered duplicates, like generics, by regulators as it is impossible to manufacture exact copies of biotech drugs.



| Public company | |
| Traded as | BSE: 532523 NSE: BIOCON |
| Industry | Biotechnology |
| Founded | 1978 |
| Founder | Kiran Mazumdar-Shaw |
| Headquarters | Bangalore, Karnataka, India |
|
Key people
|
Kiran Mazumdar-Shaw, (Chairman & MD) |
| Products | Pharmaceuticals Enzymes |
| Revenue | ₹22.41 billion (US$330 million) (2014–15)[1] |
|
Number of employees
|
5,585 (Mar 2011)[1] |
| Subsidiaries | Syngene Clinigene |
| Website | www.biocon.com |
//////Biocon, Insulin Glargine, approval, Japan
HELP, Need one time help to pay 10 year concessional subscription to this, your favorite blog to WordPress
Just One viewer please come forward
Dear Kind Viewer’s
WordPress is kind to me and negotiated a one time 10 year concessional subscription of 260 US dollars…….https://newdrugapprovals.org/
I need one time help to pay this one time 10 year concessional subscription to our favorite blog.
This is done to keep this blog running even after my death.
Currently I am paying 99 US Dollars per annum
email me
amcrasto@gmail.com
call +919323115463
Paypal will work for me via email request to you by me, Indian govt does not allow automatic transfer via paypal buttons on the blog.
email me at amcrasto@gmail.com and tell me amount, i will request you via paypal

DR ANTHONY CRASTO
LIONEL MY SON, MY MOTIVATION
.
He was only in first standard in school when I was hit by a deadly one in a million spine stroke called acute transverse mylitis, it made me 90% paralysed…
View original post 226 more words
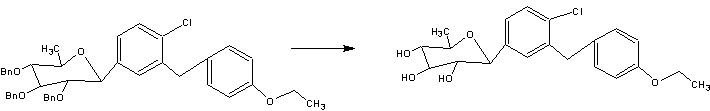
Tianagliflozin IND filed by Tianjin Institute of Pharmaceutical research


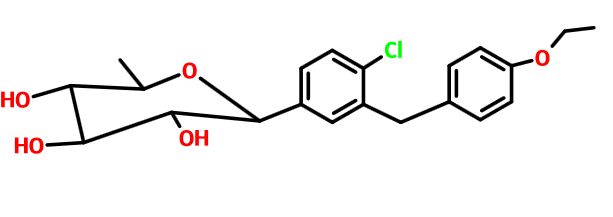
Tianagliflozin,
taigeliejing, 6-deoxydapagliflozin
| Molecular Formula: | C21H25ClO5 |
|---|---|
| Molecular Weight: | 392.8732 g/mol |
IND Filing…Tianjin Institute of Pharmaceutical research
Tianjin Institute Of Pharmaceutical Research,
(3R,4S,5S,6R)-2-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-methyloxane-3,4,5-triol
1-[4-Chloro-3-(4-ethoxybenzyl)phenyl]-1,6-dideoxy-b-D-glucopyranose
D-Glucitol, 1,5-anhydro-1-C-[4-chloro-3-[(4-ethoxyphenyl)methyl]phenyl]-6-deoxy-, (1S)-
1–[4–Chloro–3–(4–ethoxybenzyl)phenyl]–1,6–dideoxy–β–d–glucopyranose
6-deoxydapagliflozin
A SGLT-2 inhibitor potentially for the treatment of type 2 diabetes.
![]()
CAS N. 1461750-27-5
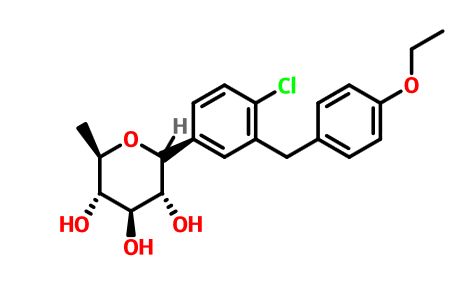

The structures of dapagliflozin and 6-deoxydapagliflozin (1)
,deletion of the 6-OH in the sugar moiety of dapagliflozin led to the discovery of a more potent SGLT2 inhibitor, 6-deoxydapagliflozin (1, ). In an in vitro assay, 1 was a more active SGLT2 inhibitor, with IC 50 = 0.67 nM against human SGLT2 (hSGLT2), as compared with 1.1 nM for dapagliflozin, leading to the identification of 1 as the most active SGLT2 inhibitor discovered so far in this field. Also in an in vivo assay, 1 also introduced more urinary glucose in a rat urinary glucose excretion test (UGE) and exhibited more potent blood glucose inhibitory activity in a rat oral glucose tolerance test (OGTT) than dapagliflozin.
Given the fact that 6-dexoydapagliflozin (1) is a very promising SGLT2 inhibitor that could be used to treat type 2 diabetes, led to preclinical trials
Tianjin Institute Of Pharmaceutical Research,天津药物研究院

SPECTRAL DATA of Tianagliflozin
1 as a white solid (3.65 g, 93 %). R f = 0.35 (EtOAc);
m.p.: 148–149 °C;
1H NMR (400 MHz, DMSO-d 6): δ = 7.35 (d, 1H, J = 8.4 Hz), 7.25 (s, 1H), 7.18 (d, 1H, J = 8.0 Hz), 7.08 (d, 2H, J = 8.4 Hz), 6.81 (d, 2H, J = 8.4 Hz), 4.95 (d, 1H, J = 5.2 Hz, OH), 4.90 (d, 1H, J = 4.4 Hz, OH), 4.79 (d, 1H, J = 5.6 Hz, OH), 3.92–4.01 (m, 5H), 3.24–3.29 (m, 1H), 3.18–3.22 (m, 1H), 3.09–3.15 (m, 1H), 2.89–2.95 (m, 1H), 1.29 (t, 3H, J = 7.0 Hz, CH2 CH 3 ), 1.15 (d, 3H, J = 6.0 Hz, CHCH 3 ) ppm;
13C NMR (100 MHz, DMSO-d 6): δ = 156.85, 139.65, 137.82, 131.83, 131.16, 130.58, 129.52, 128.65, 127.14, 114.26, 80.71, 77.98, 75.77, 75.51, 74.81, 62.84, 37.55, 18.19, 14.62 ppm;
IR (KBr): v¯¯¯ = 3,564 (w), 3,385 (s), 2,981 (s), 2,899 (s), 2,861 (s), 1,613 (m), 1,512 (s), 1,477 (m), 1,247 (s), 1,102 (s), 1,045 (s), 1,012 (s) cm−1;
HR–MS: calcd for C21H29ClNO5 ([M + NH4]+) 410.1729, found 410.1724.
PATENT
CN 103864737
http://www.google.com/patents/CN103864737A?cl=en
PATENT
WO 2014094544
http://www.google.com/patents/WO2014094544A1?cl=en


-27-


1 D1 -6 Optionally, the step (7 ‘) is the step (7’) in place:
LS l- [4 – D (I- Dl- 6)

A.

(DMSO-d 6, 400 MHz), δ 7.35 (d, 1H, J = 8.0 Hz), 7.28 (d, 1H, J ‘. 2.0 Hz), 7.17 (dd, IH, / = 2.0 Hz and 8.4 Hz), 7.05 (d, 2H, J: 8.8 Hz), 6.79 (d, 2H, 8.8 Hz): 4.924,95 (m, 2H), 4,81 (d, IH, 6,0 Hz), 3.93- 3.99 (m, 5H), 3,85 (d, 1H, J = 10,4 Hz), 3,66 (dd, IH, 5,2 Hz and 11,6 Hz), 3.17-3,28 (m, 3H), 3.02-3.08 (m: IH), 1.28 (t, 3H, J = 7,0 Hz), 0,80 (s, 9H), -0.05 (s, 3H), -0.09 (s, 3H) .
PATENT
[0066] The added 100mL dried over anhydrous methanol 0. 5g of sodium metal, nitrogen at room temperature with stirring, until the sodium metal disappeared. Followed by addition of 5. 2g (10mmol) of compound 6, stirring was continued at room temperature for 3 hours. To the reaction system was added 5g strong acid cation exchange resin, stirred at room temperature overnight, the reaction mixture until pH = 7. The resin was removed by suction, and the filtrate evaporated to dryness on a rotary evaporator, the residue was further dried on a vacuum pump to give the product I-D1-6, as a white foamy solid.
PATENT
WO 2014139447
PATENT related
http://www.google.com/patents/WO2013044608A1?cl=en
http://link.springer.com/article/10.1007%2Fs40242-014-4043-9#/page-1
Med Chem. 2015;11(4):317-28.
Design of SGLT2 Inhibitors for the Treatment of Type 2 Diabetes: A History Driven by Biology to Chemistry.
Abstract
A brief history of the design of sodium-dependent glucose cotransporter 2 (SGLT2) inhibitors is reviewed. The design of O-glucoside SGLT2 inhibitors by structural modification of phlorizin, a naturally occurring O-glucoside, in the early stage was a process mainly driven by biology with anticipation of improving SGLT2/SGLT1 selectivity and increasing metabolic stability. Discovery of dapagliflozin, a pioneering C-glucoside SGLT2 inhibitor developed by Bristol-Myers Squibb, represents an important milestone in this history. In the second stage, the design of C-glycoside SGLT2 inhibitors by modifications of the aglycone and glucose moiety of dapagliflozin, an original structural template for almost all C-glycoside SGLT2 inhibitors, was mainly driven by synthetic organic chemistry due to the challenge of designing dapagliflozin derivatives that are patentable, biologically active and synthetically accessible. Structure-activity relationships (SAR) of the SGLT2 inhibitors are also discussed.
http://www.ncbi.nlm.nih.gov/pubmed/25557661
Paper

Discovery of 6-Deoxydapagliflozin as a Highly Potent Sodium-dependent Glucose Cotransporter 2 (SGLT2) Inhibitor for the Treatment of Type 2 Diabetes
http://www.ingentaconnect.com/content/ben/mc/2014/00000010/00000003/art00009?crawler=true
CLIP

A facile synthesis of 6-deoxydapagliflozin
Keywords. Carbohydrates Drug research Hydrogenolysis Dapagliflozin SGLT2 inhibitor


The synthetic route to the target compound 1 is shown in Scheme 3. The starting material methyl 2,3,4-tri-O-benzyl-6-deoxy-6-iodo-α–d-glucopyranoside (3) was prepared from commercially available methyl α–d-glucopyranoside (2) according to a known method [5, 6].
Iodide 3 was reductively deiodinated to give 4 in 91 % yield under hydrogenolytic conditions using 10 % Pd/C as catalyst in the presence of Et3N as base in THF/MeOH at room temperature.
when the iodide 3 was treated with Barton–McCombie reagent (n-Bu3SnH/AIBN) [7] in toluene at room temperature no reaction occurred; however, when the reaction was carried out at elevated temperatures, such as reflux, a complex mixture formed with only a trace amount (3 %, entry 1) of the desired product 4.
When the iodide 3 was treated with LiAlH4 in THF at 0 °C to room temperature, another complex mixture was produced with only a trace amount (2 %, entry 2) of 4.
When Pd(OH)2 was used as the hydrogenolysis catalyst instead of 10 % Pd/C, the desired 4 was indeed formed (14 %, entry 4), but most of the starting material was converted to a few more polar byproducts, which were believed to result from the cleavage of at least one of the benzyl groups.
pdf available
Monatshefte für Chemie – Chemical Monthly
December 2013, Volume 144, Issue 12, pp 1903-1910
////////IND Filing, SGLT-2 inhibitor, type 2 diabetes, Tianagliflozin, taigeliejing, 6-deoxydapagliflozin, 1461750-27-5
Clc1c(cc(cc1)C2[C@@H]([C@H]([C@@H]([C@H](O2)C)O)O)O)Cc3ccc(cc3)OCC
CCOC1=CC=C(C=C1)CC2=C(C=CC(=C2)C3C(C(C(C(O3)C)O)O)O)Cl
c1(c(cc(cc1)C2OC(C(C(C2O)O)O)C)Cc3ccc(cc3)OCC)Cl
BMS 986120

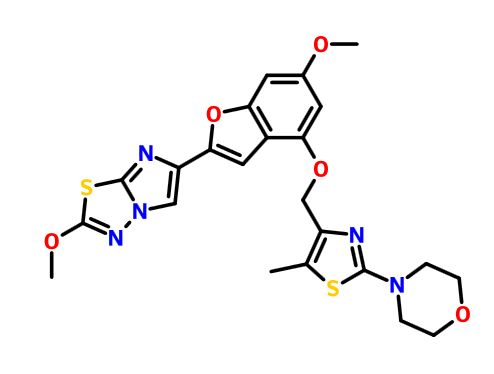
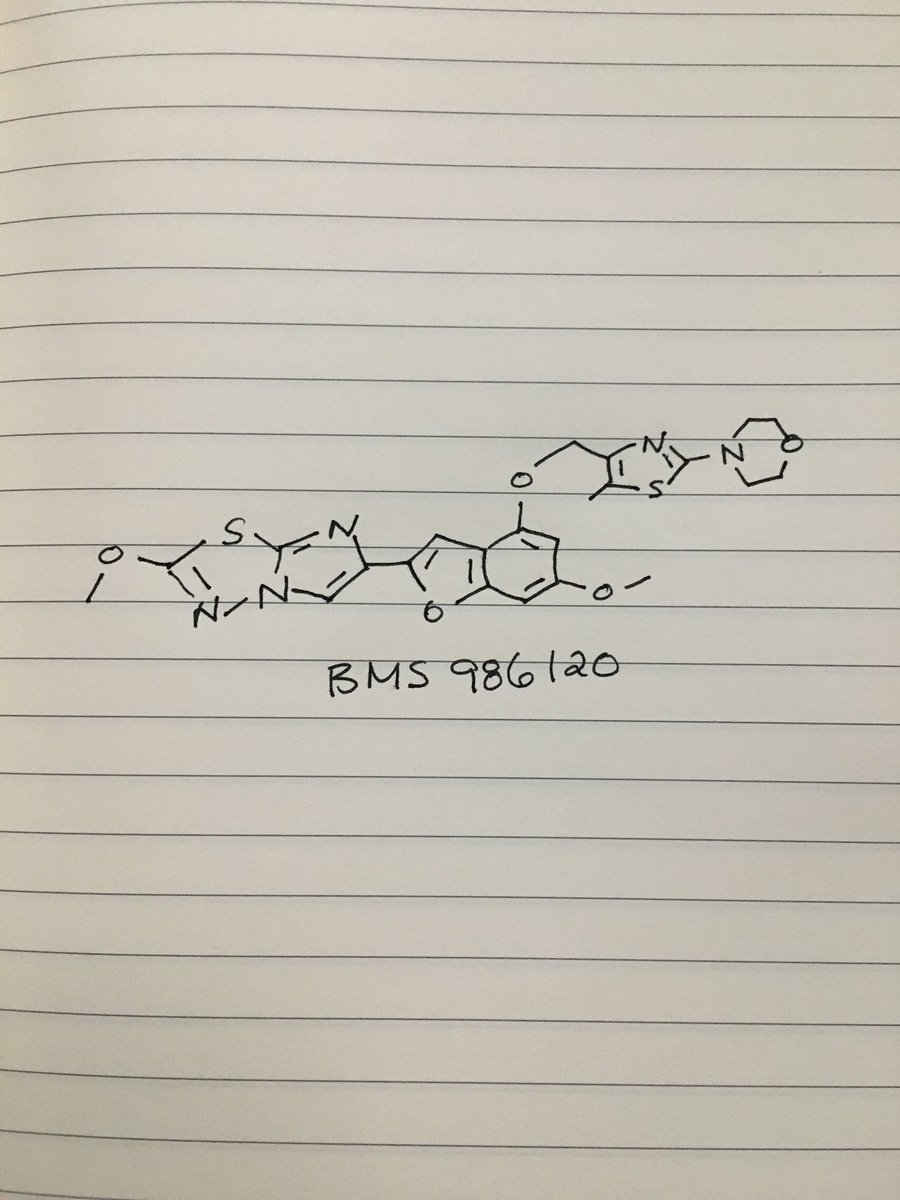 .
.
Picture credit….Bethany Halford
BMS 986120
Originator Bristol-Myers Squibb
Bristol-Myers Squibb Company, Université de Montréal
| Molecular Formula: | C23H23N5O5S2 |
|---|---|
| Molecular Weight: | 513.58922 g/mol |
4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]morpholine
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine
Imidazo[2,1-b] -1,3,4-thiadiazole, 2-methoxy-6-[6-methoxy-4-[[5-methyl-2-(4-morpholinyl)-4- thiazolyl]methoxy]-2-benzofuranyl]-
CAS 1478712-37-6
Phase I Thrombosis
- 02 Apr 2015 Bristol-Myers Squibb plans a phase I trial in Thrombosis (In volunteers) in United Kingdom (NCT02439190)
- 01 Aug 2014 Preclinical trials in Thrombosis in USA (PO)
https://clinicaltrials.gov/ct2/show/NCT02208882
https://clinicaltrials.gov/ct2/show/NCT02439190
Class Imidazoles; Small molecules; Thiadiazoles
$BMY antithrombic compound
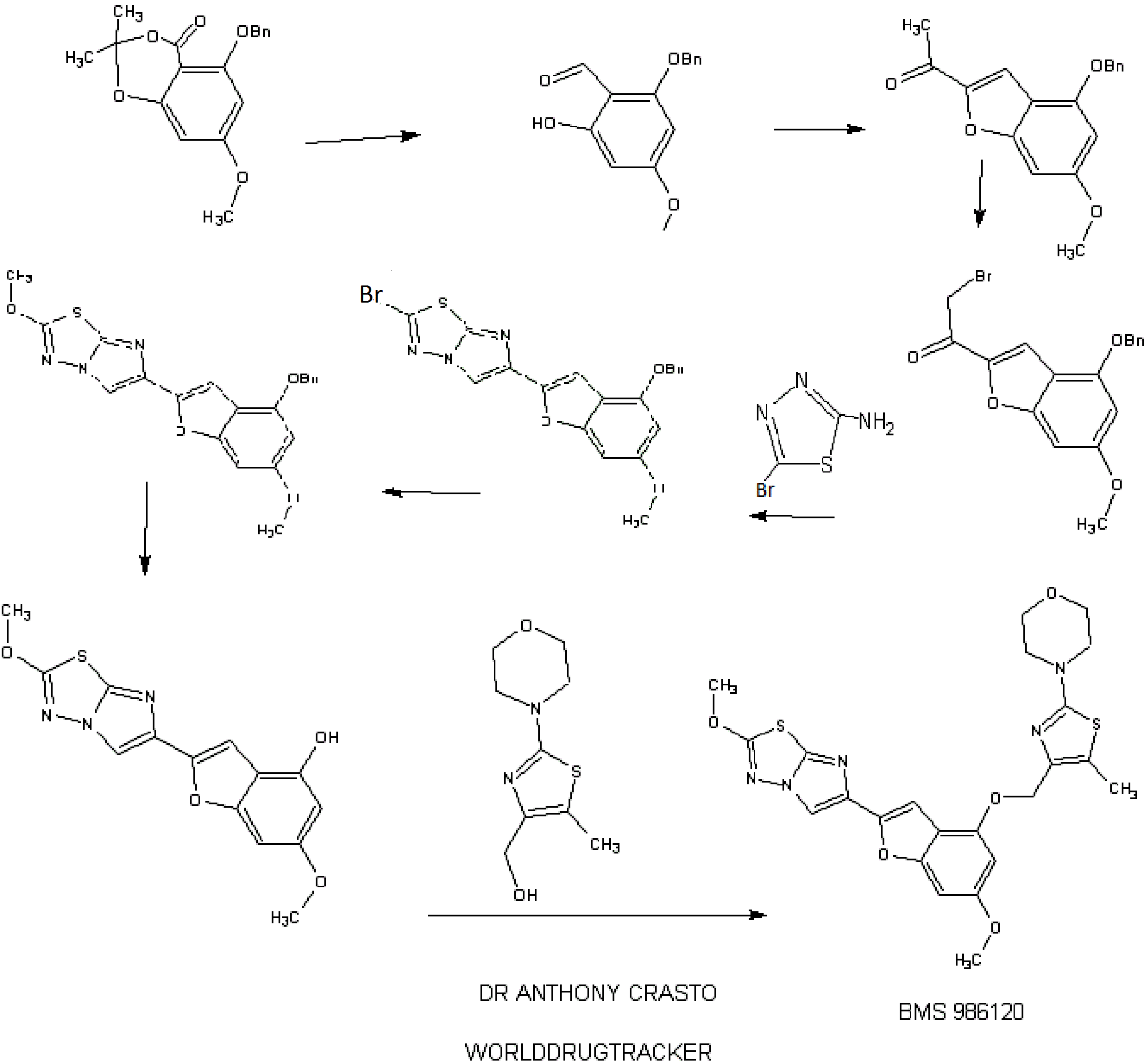
PATENT
http://www.google.com/patents/WO2013163279A1?cl=en
Thromboembolic diseases remain the leading cause of death in developed countries despite the availability of anticoagulants such as warfarin (COUMADIN®), heparin, low molecular weight heparins (LMWH), synthetic pentasaccharides, and antiplatelet agents such as aspirin and clopidogrel (PLAVIX®).
Current anti-platelet therapies have limitations including increased risk of bleeding as well as partial efficacy (relative cardiovascular risk reduction in the 20 to
30% range). Thus, discovering and developing safe and efficacious oral or parenteral antithrombotics for the prevention and treatment of a wide range of thromboembolic disorders remains an important goal.
Alpha-thrombin is the most potent known activator of platelet aggregation and degranulation. Activation of platelets is causally involved in atherothrombotic vascular occlusions. Thrombin activates platelets by cleaving G-protein coupled receptors termed protease activated receptors (PARs). PARs provide their own cryptic ligand present in the N-terminal extracellular domain that is unmasked by proteolytic cleavage, with subsequent intramolecular binding to the receptor to induce signaling (tethered ligand mechanism; Coughlin, S.R., Nature, 407:258-264 (2000)). Synthetic peptides that mimic the sequence of the newly formed N-terminus upon proteolytic activation can induce signaling independent of receptor cleavage. Platelets are a key player in atherothrombotic events. Human platelets express at least two thrombin receptors, commonly referred to as PARI and PAR4. Inhibitors of PARI have been investigated extensively, and several compounds, including vorapaxar and atopaxar have advanced into late stage clinical trials. Recently, in the TRACER phase III trial in ACS patients, vorapaxar did not significantly reduce cardiovascular events, but significantly increased the risk of major bleeding (Tricoci, P. et al, N. Eng. J. Med., 366(l):20-33 (2012). Thus, there remains a need to discover new antiplatelet agents with increased efficacy and reduced bleeding side effects.
There are several early reports of preclinical studies of PAR4 inhibitors. Lee, F-Y. et al., “Synthesis of l-Benzyl-3-(5′-hydroxymethyl-2′-furyl)indazole Analogues as Novel Antiplatelet Agents”, J. Med. Chem., 44(22):3746-3749 (2001) discloses in the abstract that the compound

58
“was found to be a selective and potent inhibitor or protease-activated receptor type 4 (PAR4)-dependent platelet activation. ”
Compound 58 is also referred to as YD-3 in Wu, C-C. et al, “Selective Inhibition of Protease-activated Receptor 4-dependent Platelet Activation by YD-3”, Thromb. Haemost., 87: 1026-1033 (2002). Also, see Chen, H.S. et al, “Synthesis and platelet activity”, J. Bioorg. Med. Chem., 16: 1262-1278 (2008).
EP1166785 Al and EP0667345 disclose various pyrazole derivatives which are useful as inhibitors of platelet aggregation.\
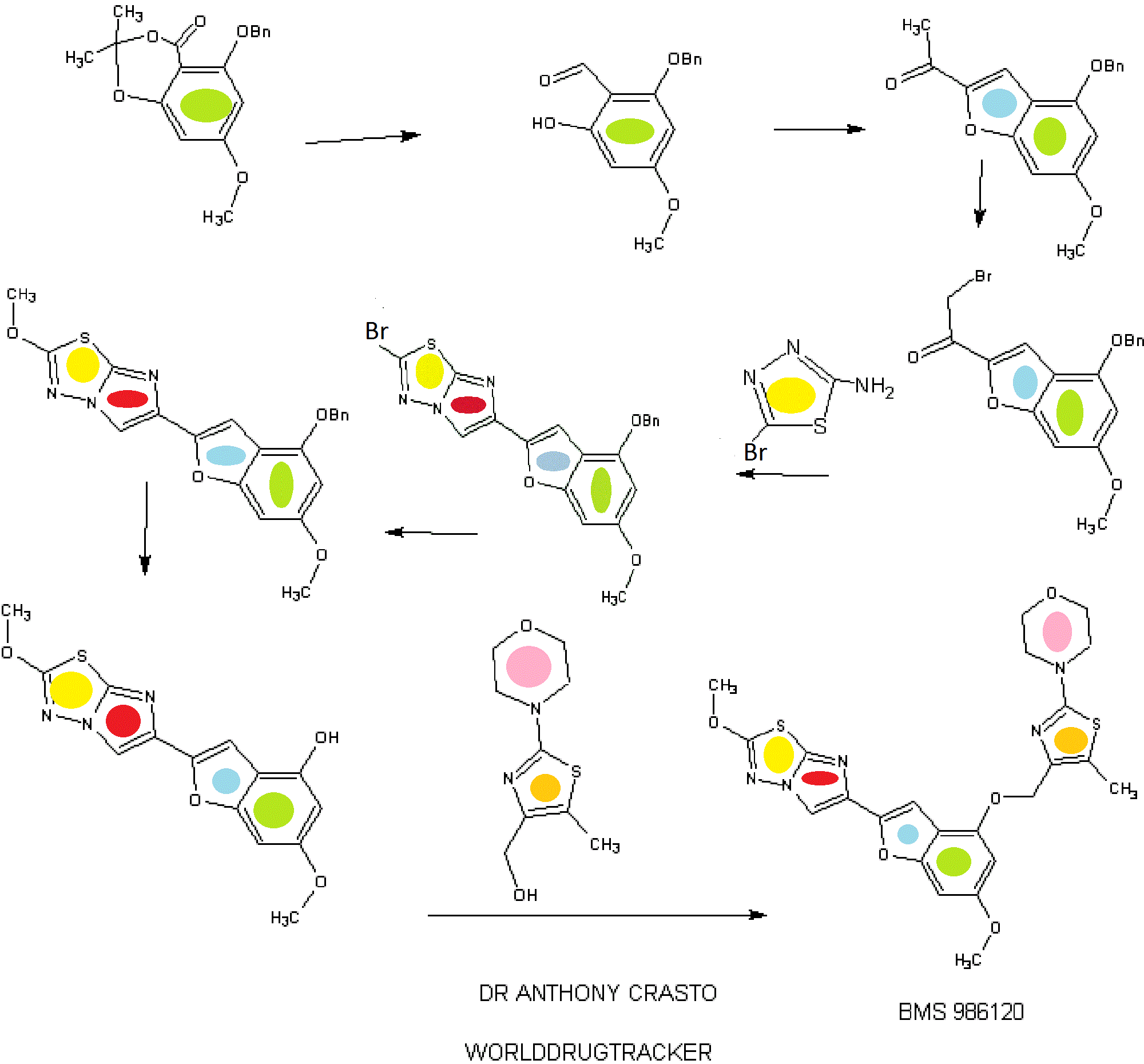
IB. 5-(Benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4-one

A solution of 5-hydroxy-7-methoxy-2,2-dimethyl-4H-benzo[d][l,3]dioxin-4- one (30.00 g, 0.134 mol, see Kamisuki, S. et al, Tetrahedron, 60:5695-5700 (2004) for preparation) in N,N-dimethylformamide (400 mL) was treated with powdered anhydrous potassium carbonate (19.41 g, 0.14 mol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with benzyl bromide (24.03 g, 0.14 mol) added dropwise over 15 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting material left by tic). The solid was filtered and washed with N,N- dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (500 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Crystallization form ethyl acetate (50 mL) and hexane (150 mL) gave 35.17 g of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H- benzo[d][l ,3]dioxin-4-one as large colorless prisms. Chromatography of the mother liquors on silica gel (4 x 13 cm, elution toluene – ethyl acetate 0-5%) gave 6.64 g of additional material to afford a total yield of 41.81 g (99%). HRMS(ESI) calcd for
Ci8Hi905 [M+H]+ m/z 315.1227, found 315.1386. 1H NMR (CDC13, 600 MHz) δ 1.68 (s, 6H), 3.77 (s, 3H), 5.19 (s, 2H), 5.19 (s, 2H), 6.04 (d, J = 2.03 Hz, 1H), 6.15 (d, J = 2.03 Hz, 1H), 7.27 (broad t, 1H), 7.36 (broad t, 2H), 7.52 (broad d, 2H).
1 C. 2-(Benzyloxy)-6-hydroxy-4-methoxybenzaldehyde

A solution of 5-(benzyloxy)-7-methoxy-2,2-dimethyl-4H-benzo[d][l ,3]dioxin- 4-one (Example IB, 6.76 g, 21.5 mmol) in dichloromethane (120 mL) was cooled to -78 °C and treated with 43 mL (64.5 mmol) of a 1.5 M solution of diisobutylaluminum hydride in toluene added dropwise over 20 min. The resulting mixture was then stirred at -78 °C for 3 h. The reaction mixture was quenched by the careful addition of methanol (5 mL) added dropwise over 15 min, followed by IN hydrochloric acid (50 mL) added dropwise over 15 min. The cooling bath was then removed and an additional 150 mL of IN hydrochloric acid was added over 20 min. The mixture was then stirred at 22 °C for 2 h and diluted with dichloromethane (400 mL). The organic phase was collected and the aqueous phase (pH ~1) was extracted with dichloromethane (3 x 50 mL). The combined organic extracts were washed with brine, dried over anhydrous magnesium sulfate and concentrated in vacuo. The residual oil was diluted with tetrahydrofuran (70 mL), treated with 10 mL of 0.1N hydrochloric acid and stirred at 20 °C for 2 h. The reaction mixture was diluted with ethyl acetate (300 mL), washed with brine, dried over anhydrous magnesium sulfate, evaporated in vacuo to give a clear oil. Chromatography on silica gel (4 x 13 cm, elution toluene) gave 4.08 g (73% yield) of the title aldehyde as a clear oil which solidified on standing. LC (Method C): 2.237 min. HRMS(ESI) calcd for Ci5Hi504 [M+H]+ m/z 259.0965, found 259.1153. 1H NMR (CDC13, 600 MHz) δ 3.80 (s, 3H), 5.07 (s, 2H), 5.97 (d, J= 2.1 Hz, 1H), 6.01 (d, J= 2.1 Hz, 1H), 7.3 – 7.4 (m, 5 H), 10.15 (s, 1H), 12.49 (s, 1H).
ID. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)ethanone

A solution of 2-(benzyloxy)-6-hydroxy-4-methoxybenzaldehyde (Example 1C, 3.46 g, 13.4 mmol) in N,N-dimethylformamide (50 mL) was treated with powdered anhydrous cesium carbonate (4.58 g, 14.05 mmol) added all at once. The resulting mixture was stirred in vacuo for 10 min. and then flushed with nitrogen. The reaction flask was placed in a water bath (22 °C) and treated with chloroacetone (1.74 g, 18.7 mmol) added dropwise over 5 min. The resulting mixture was then stirred at 22 °C for 18 h (no starting aldehyde left by tic and formation of the intermediate alkylated aldehyde). The solid was filtered and washed with N,N-dimethylformamide. The filtrate was evaporated in vacuo and the residual oil was diluted with ethyl acetate (300 mL), washed with cold 0.1 N hydrochloric acid, saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. This syrup was diluted with tetrahydrofuran (50 mL) and ethyl acetate (50 mL), treated p- toluenesulfonic acid monohydrate (0.2 g) and stirred at 20 °C for 1 h (tic indicated complete cyclization of the intermediate alkylated aldehyde to the benzofuran). The reaction mixture was diluted with ethyl acetate (300 mL), washed with saturated sodium bicarbonate and brine. After drying over anhydrous magnesium sulfate, evaporation of the solvent gave a thick syrup. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 2-4%) gave 3.51 g (88% yield) of the title benzofuran as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave the title material as large yellow prisms (3.15 g). LC (Method D): 2.148 min. HRMS(ESI) calcd for Ci8Hiv04 [M+H]+ m/z 297.1121, found 297.1092. 1H NMR (CDC13, 600 MHz) δ 2.51 (s, 3H), 3.82 (s, 3H), 5.13 (s, 2H), 6.37 (d, J= 1.77 Hz, 1H), 6.63 (broad s, 1H), 7.34 (broad t, 1H), 7.39 (broad t, 2H), 7.44 (broad d, 2H), 7.55 (d, J = 0.7 Ηζ,ΙΗ). IE. l-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone

A 250-mL, three-necked flask is equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with anhydrous tetrahydrofuran (25 mL) followed by 9.3 mL (9.3 mmol) of a 1M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran. The mixture was cooled to -78 °C and treated with a solution of l-(4- (benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 2.40 g, 8.1 mmole) in tetrahydrofuran (20 mL) added dropwise over 10 min. The resulting mixture was then stirred at -78 °C for 45 min. Then chlorotrimethylsilane (1.18 mL, 9.31 mmol) was added dropwise over 5 min and the resulting solution was stirred at -78 °C for another 20 min. The cooling bath was then removed and the mixture is allowed to warm to room temperature over 30 min. The reaction mixture was then quenched by addition to a cold solution of ethyl acetate (200 mL), saturated sodium bicarbonate (30 mL) and ice. The organic phase was rapidly dried over anhydrous magnesium sulfate (magnetic stirring) and evaporated in vacuo to give the silyl enol ether as an oil which is co-evaporated with toluene (20 mL). The silyl enol ether was then dissolved in dry tetrahydrofuran (40 mL), cooled to -20 °C and treated with solid sodium bicarbonate (0.10 g) followed by N- bromosuccinimide (1.44 g, 8.1 mmol) added in small portions over 15 min. The reaction mixture was allowed to warm to 0 °C over 2h and then quenched by addition of ethyl acetate (300 mL) and saturated sodium bicarbonate. The organic phase was washed with brine, dried over anhydrous magnesium sulfate and evaporated to give an orange oil. Chromatography on silica gel (4 x 12 cm, elution toluene – ethyl acetate 0-5%) gave 2.62 g (86% yield) of the title bromomethylketone as a yellow solid. Recrystallization from ethyl acetate (10 mL) and hexane (20 mL) gave yellow prisms (2.30 g). LC (Method E): 1.977 min. HRMS(ESI) calcd for Ci8Hi6Br04 [M+H]+ m/z 375.0226, found 375.0277. 1H NMR (CDCls, 600 MHz) δ 3.84 (s, 3H), 4.33 (s, 2H), 5.14 (s, 2H), 6.38 (d, J = 1.76 Hz, 1H), 6.64 (broad s, 1H), 7.35 (broad t, 1H), 7.40 (broad t, 2H), 7.44 (broad d, 2H), 7.70 (s, 1H). 1 EE. 1 -(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone

Benzyltrimethylammonium dichloroiodate (117 g, 169 mmol) was added to a solution of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)ethanone (Example ID, 50 g, 170 mmol) in THF (500 mL) in a 1 L multineck round bottom flask under nitrogen atmosphere. The reaction mixture was stirred at RT for 6 h, cooled to 0 °C and quenched with 10% NaHCC”3 solution. The organic layer was washed with 1 M sodium thiosulphate solution, water, and brine, dried over Na2S04, and concentrated in vacuo (bath temperature <45 °C). The residue was triturated with 5% EtOAc in pet. ether and dried to obtain the title chloromethylketone as a pale yellow solid (48 g, 130 mmol, 78%). 1H NMR (300 MHz, DMSO-d6) δ 3.84-3.82 (d, J =4.5Hz, 3H) 4.98 (s, 2H), 5.27(s, 2H), 6.62 -6.61 (d, J = 1.8Hz, 1H), 6.92-6.93 (m, 1H), 7.54-7.36 (m, 5H), 8.10-8.09 (d, J = 3Hz, 1H); MS m/z: [M+H]+ 331.0. IF. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoimidazo[2, 1 – b] [ 1 ,3 ,4]thiadiazole

A mixture of l-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-bromoethanone (Example IE, 3.00 g, 8.0 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (1.65 g, 9.16 mmol) in isopropanol (100 mL) was heated in a pressure flask equipped with a magnetic stirring bar at 78-80 °C for 18 h (homogeneous after 20 min and then formation of a precipitate after 2 h). The cooled mixture is then transferred into five 20 mL microwave vials and then heated in a microwave apparatus to 150 °C for 30 min. Each vial was then diluted with dichloromethane (250 mL) washed with saturated sodium bicarbonate (25 mL) and brine (25 mL), dried over anhydrous magnesium sulfate. The fractions were combined and concentrated in vacuo. Chromatography of the orange-brown residual solid on silica gel (4 x 10 cm, slow elution with dichloromethane due to poor solubility) gave 2.96 g of the title imidazothiadiazole contaminated with some l-(4-(benzyloxy)-6- methoxybenzofuran-2-yl)ethanone. The solid material was triturated with ethyl acetate (20 mL), filtered, washed with ethyl acetate (10 ml) and dried in vacuo to give 2.34 g (64% yield) of pure title imidazothiadiazole as an off white solid which is used as such for the next step. LC (Method E): 2.188 min. HRMS(ESI) calcd for C2oHi5BrN303S [M+H]+ m/z 456.00175, found 456.00397. 1H NMR (CDC13, 600 MHz) δ 3.82 (s, 3H), 5.16 (s, 2H), 6.38 (d, J= 1.67 Hz, 1H), 6.66 (broad s, 1H), 7.15 (s, 1H), 7.31 (broad t, 1H), 7.38 (broad t, 2H), 7.45 (broad d, 2H), 8.02 (s, 1H).
Alternatively, Example IF, 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole, was prepared as follows:
A 1000-mL, three-necked flask equipped with a magnetic stirring bar and purged with a nitrogen atmosphere was charged with dry NMP (200 mL) followed by 1- (4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2-chloroethanone (Example 1EE, 50 g, 150 mmol) and 5-bromo-l,3,4-thiadiazol-2-amine (27.2 g, 151 mmol). The resulting mixture was stirred at 80 °C for 8h. TLC (8:2 dichloromethane/pet. ether) and LC/MS showed intermediate uncyclized material (m/z 476) and the reaction mixture was stirred at 120 °C for 3h. The reaction mixture was cooled to RT, quenched with water and extracted with EtOAc (3X). The combined organic layers were washed with brine, dried over Na2S04, and concentrated in vacuo. The thick brown residue was purified by silica gel chromatography (0 to 100% dichloromethane in pet. ether) to give a brown solid. This material was triturated with EtOAc and dried to obtain the title imidazothiadiazole (24 g, 50 mmol, 33%>) as a light brown solid. (See the procedure set forth above for analytical data).
1 G. 6-(4-(Benzyloxy)-6-methoxybenzofuran-2-yl)-2-methoxyimidazo[2, 1 – b][l,3,4]thiadiazole

A solution of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- bromoimidazo[2,l-b][l,3,4]thiadiazole (Example IF, 2.30 g, 5.04 mmol) in a mixture of dichloromethane (180 mL) and methanol (45 mL) was treated at 22 °C with 4.2 mL of a 25 wt.% solution of sodium methoxide in methanol (0.2 mmol) added in one portion. More methanol (45 mL) was added and the mixture was stirred for 1 h. The reaction mixture was quenched by the addition of 25 mL of IN hydrochloric acid followed by 20 ml of saturated sodium bicarbonate. The solvent was evaporated under reduced pressure and the residue was diluted with dichloromethane (400 mL), washed with brine, dried over anhydrous magnesium sulfate and evaporated in vacuo. Chromatography of the residue on silica gel (3 x 10 cm, elution with dichloromethane – ethyl acetate 0-4%) gave 1.70 g (83% yield) of the title compound as a white solid. This material was recrystallized from ethyl acetate (30 mL per gram, 80% recovery) to give white needles. LC (Method
D): 2.293 min. HRMS(ESI) calcd for C21H18N3O4S [M+H]+ m/z 408.1013, found 408.1024. 1H NMR (CDC13, 600 MHz) δ 3.81 (s, 3H), 4.18 (s, 3H), 5.16 (s, 2H), 6.37 (d, J = 1.75 Hz, 1H), 6.67 (broad s, 1H), 7.07 (s, 1H), 7.31 (broad t, 1H), 7.37 (broad t, 2H), 7.45 (broad d, 2H), 7.81 (s, 1H).
1H. 6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-ol

A mixture of 6-(4-(benzyloxy)-6-methoxybenzofuran-2-yl)-2- methoxyimidazo[2,l-b][l,3,4]thiadiazole (Example 1G, 1.250 g, 3.06 mmol) and pentamethylbenzene (3.17 g, 21.4 mmol) in dichloromethane (200 mL) was cooled to -78 °C under a nitrogen atmosphere and then treated immediately (to avoid crystallization) with 8 mL (8 mmol) of a 1 M solution of boron trichloride in dichloromethane added dropwise over 3 min. The resulting mixture was stirred at -78 °C for 1 h. The reaction mixture was then quenched by the addition of a solution of sodium bicarbonate (6 g) in water (100 mL) added in one portion. The cooling bath was removed and the resulting mixture was stirred at room temperature for 1 h. The solid formed was filtered, washed successively with water (50 m) and dichloromethane (50 mL). The filter cake was allowed to soak with anhydrous ethanol (15 ml) and then sucked dry. The white solid obtained was then dried under vacuum for 24 h to give 0.788 g (80%> yield) of pure title material (> 95% by hplc). The combined filtrate and washings were diluted with dichloromethane (600 mL) and stirred in a warm water bath till the organic phase was clear with no apparent solid in suspension. The organic phase was collected, dried over anhydrous magnesium sulfate and rapidly filtered while still warm. The filtrate was evaporated and the residue (product and pentamethylbenzene) was triturated with toluene (20 mL), the solid collected and washed with toluene (20 mL) to give 0.186 g (19% yield, 99% combined yield) of title material as a tan solid (> 95% by hplc). LC (Method E): 1.444 min. HRMS(ESI) calcd for C14H12N3O4S [M+H]+ m/z 318.0543, found 318.0578. 1H NMR (DMSO-de, 600 MHz) 5 3.71 (s, 3H), 4.16 (s, 3H), 6.21 (d, J = 1.87 Hz, 1H), 6.61 (broad s, 1H), 6.95 (s, 1H), 8.29 (s, 1H), 9.96 (s, 1H).
Example 94
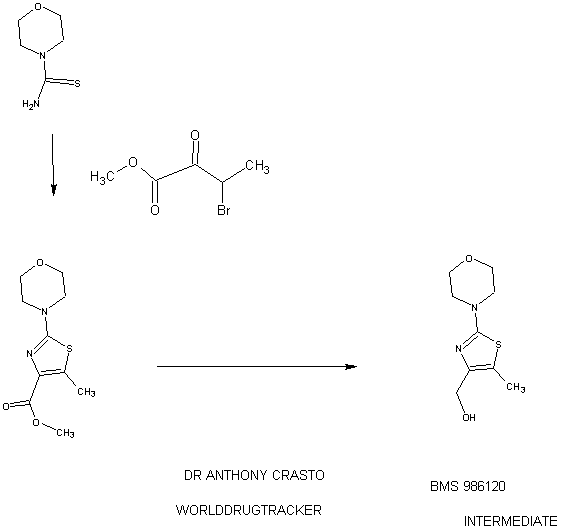
4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2,l-b][l,3,4]thiadiazol-6-yl)benzofuran-4-yl) oxy)methyl)-5-methylthiazol-2-yl)morpholine

94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate [00258] A solution of methyl 2-bromo-5-methylthiazole-4-carboxylate (2.80 g, 11.86 mmol) and morpholine (4.5 mL, 51.7 mmol) in THF (10 mL) was heated at reflux under nitrogen for 18 h. The volatiles were then removed under reduced pressure and the crude product was purified on the ISCO using a REDISEP® 40 g column (0 to 40% EtOAc- DCM), to give the title compound (2.20 g, 77%) as a yellow solid. LCMS (APCI): calcd for CioHisNzOsS [M+H]+ m/z 243.07, found 243.1. 1H NMR (CDC13, 400 MHz) δ ppm: 3.89 (s, 3H), 3.77-3.83 (m, 4H), 3.41-3.47 (m, 4H), 2.64 (s, 3H). [00259] Alternatively, Example 94A, methyl 5-methyl-2-morpholinothiazole-4- carboxylate, was prepared as follows:
94AA. Methyl 3-bromo-2-oxobutanoate

A 5L 4-neck round bottom flask equipped with a mechanical stirrer, temperature thermocouple, condenser and a 1L addition funnel, was charged copper(II) bromide (962 g, 4310 mmol) and ethyl acetate (2 L). A solution of methyl 2-ketobutyrate (250 g, 2150 mmol) in CHC13 (828 mL) was added dropwise. A scrubber (400 mL 1 N NaOH) was connected and the reaction mixture was heated to reflux (75 °C). The reaction started as a dark green color and as heating progressed, it became a light green with a white precipitate forming. NMR after one hour at reflux indicated that the reaction was complete. The reaction was cooled to RT and filtered through a pad of CELITE®. The filtrate was concentrated to an oil, dissolved in methylene chloride (500 mL) and filtered again through CELITE®. The filtrate was then passed through a pad of silica gel and eluted with ethyl acetate. Concentration of the filtrate provided the title bromoketoester (399 g, 2040 mmol, 95%) as a yellow oil. 1H NMR (400MHz, CDC13) δ 5.18 (q, J = 6.7 Hz, 1H), 3.94 (s, 3H), 1.83 (d, J = 6.8 Hz, 3H). 94AAA. Morpholine-4-carbothioamide

To a solution of morpholine (199 g, 2280 mmol) in CHC13 (1 L) was added isothiocyanatotrimethylsilane (150 g, 1140 mmol) dropwise. A white precipitate formed almost immediately, and the reaction was stirred for 1 h at RT. The reaction was then filtered and the resulting solid was washed with additional CHC13 and dried in vacuo to give the title thiourea as a white solid. (137 g, 937 mmol, 82%). 1H NMR (400MHz, DMSO-de) δ 3.81 – 3.71 (m, 2H), 3.17 – 3.08 (m, 2H).
94 A. Methyl 5-methyl-2-morpholinothiazole-4-carboxylate

To a solution of morpholine-4-carbothioamide (Example 94 AAA, 175 g, 1200 mmol) in methanol (500 mL) was charged methyl 3-bromo-2-oxobutanoate (Example 94AA, 233 g, 1200 mmol). The reaction was then heated to reflux for 1 hour, cooled to RT, and filtered. The filtrate was concentrated and the crude product was purified on by silica gel chromatography. The title thiazole (206g, 850 mmol, 71%) was isolated as a yellow oil. (See the procedure set forth above for analytical data).
(5-Methyl-2-morpholinothiaz l-4-yl)methanol

The compound was prepared according to the protocol described for Example 92B. The crude product was purified on the ISCO using a REDISEP® Gold 24 g column (0 to 50% EtOAc-DCM) to give the title compound as a white solid (0.086 g, 51%). LCMS (APCI): calcd for C9Hi5N202S [M+H]+ m/z 215.08, found 215.1. 1H NMR (CDCI3, 400 MHz) δ ppm: 4.48 (d, J= 4.7 Hz, 2H), 3.77-3.83 (m, 4H), 3.37-3.43 (m, 4H), 2.30 (t, J= 4.7 Hz, 1H), 2.28 (s, 3H).
Example 94. 4-(4-(((6-Methoxy-2-(2-methoxyimidazo[2, 1 -b] [ 1 ,3,4]thiadiazol-6-yl) benzofuran-4-yl)oxy)methyl)-5 -methylthiazol-2-yl)morpholine

The title compound was prepared according to the protocol described for Example 86. The crude product was purified on the ISCO using a REDISEP® 4 g column (0 to 40% EtOAc-DCM) and the obtained solid was suspended in MeOH, sonicated, filtered and dried to give the title compound as an off-white solid (0.094 g, 53%). LC (Method C): 2.314 min. HRMS(ESI): calcd for C23H24N505S2 [M+H]+ m/z 514.122, found 514.126. 1H NMR (CDC13, 400 MHz) δ ppm: 7.83 (s, 1H), 7.06 (d, J = 0.8 Hz, 1H), 6.69 (d, J= 0.8 Hz, 1H), 6.50 (d, J= 2.0 Hz, 1H), 5.05 (s, 2H), 4.21 (s, 3H), 3.85 (s, 3H), 3.78- 3.84 (m, 4H), 3.39- 3.46 (m, 4H), 2.37 (s, 3H).
ABSTRACT
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 263


| Patent ID | Date | Patent Title |
|---|---|---|
| US2015094297 | 2015-04-02 | IMIDAZOTHIADIAZOLE AND IMIDAZOPYRAZINE DERIVATIVES AS PROTEASE ACTIVATED RECEPTOR 4 (PAR4) INHIBITORS FOR TREATING PLATELET AGGREGATION |
////////BMS 986120, phase 1, Bristol-Myers Squibb , Imidazoles, Small molecules, Thiadiazoles, 1478712-37-6
c1(sc2nc(cn2n1)c3cc4c(cc(cc4o3)OC)OCc5nc(sc5C)N6CCOCC6)OC
CC1=C(N=C(S1)N2CCOCC2)COC3=C4C=C(OC4=CC(=C3)OC)C5=CN6C(=N5)SC(=N6)OC
PF 06650808
 .
.
Picture credit….Bethany Halford

PF 06650808
CAS 1822383-80-1
A biologic for cancer treatment (Pfizer Inc.)
- Originator Pfizer
- Class Antineoplastics
- Mechanism of Action Notch-3 receptor antagonists
- No development reported Solid tumours
- 24 Jun 2018 Biomarkers information updated
- 28 Apr 2018 No recent reports of development identified for phase-I development in Solid-tumours(Late-stage disease) in USA (IV)
- 01 Jul 2017 Pfizer completes a phase I trial in Solid tumours (Late-stage disease) in USA (IV) (NCT02129205)
Company: Pfizer
Target: Neurogenic locus notch homolog protein 3 (NOTCH3): Activation and mutation of the NOTCH signaling pathway can lead to cancer.
Disease: Cancer
Notes: PF06650808 is an antibody-drug conjugate that delivers a cytotoxic payload molecule directly to tumor cells, explained Andreas Maderna, an associate research fellow at Pfizer. The payload molecule in PF06650808 was inspired by the marine natural product dolostatin 10, which is produced by cyanobacteria consumed by a type of sea slug.
https://cen.acs.org/articles/94/i15/New-drug-candidates-shine-San-Diego.html
PATENT
WO 2015171907
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015171907
The present invention relates to stable isotopic identification of biologic products, methods of stable isotopic identification of such biologic products, and stable isotopic methods and systems for correlating biologic products to the processes by which they are made.
PATENT
WO 2018045058
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018045058&tab=PCTDESCRIPTION&maxRec=1000
CLIP
Rosen, L.S.; Wesolowski, R.; Gibson, B.; et al.
A Phase 1 dose escalation, safety, and pharmacokinetic study of PF-06650808, an anti-Notch3 antibody drug conjugate, in adult patients with advanced solid tumors
Eur Cancer Congr (September 25-29, Vienna) 2015, Abst 3OLBA
Maderna, A.
Therapeutic targeting the NOTCH3 receptor with antibody drug conjugates
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 262
Hurvitz, S.A.; von Euw, E.; O’Brien, N.; et al.
Preclinical evaluation of targeting Notch-3 in breast cancer
107th Annu Meet Am Assoc Cancer Res (AACR) (April 16-20, New Orleans) 2016, Abst 1206
Chen, J.; Geles, K.; Silva, M.; Waterhouse, R.; Ma, D.; Charati, M.; Sapra, P.; Mccarthy, T.
Evaluate the impact of conjugation on targeting capacity, pharmacokinetics and tissue distribution of antibody drug conjugate, PF-06650808, in tumor bearing mice
22nd Int Symp Radiopharm Sci (ISRS) (May 14-19, Dresden) 2017, Abst P 052
///////////
PF 06650808
| Phase 1 |
$PFE compound inspired by auristatins
https://clinicaltrials.gov/ct2/show/NCT02129205
http://www.pfizer.com/sites/default/files/product-pipeline/8_7_2014_Pipeline_Update.pdf
ALL DATA COMING………
Notch-3 receptor antagonists
Neoplasms
Breast
| Pfizer |
Cancer
PF-06650808, is currently being examined in a Ph1 clinical trial (Protocol B7501001).
Notch3
Researchers are also exploring the use of Notch3 targeting. “The Notch pathway plays an important role in the growth of several solid tumours, including breast and ovarian cancer and melanoma,” explained Joerger. “In particular, Notch3 alterations such as gene amplification and upregulation are associated with poor patient survival. Research using Notch3 targeting as an innovative approach to treat solid malignancies included 27 patients unselected for Notch3 who received increasing doses of the anti-Notch3 antibody-drug conjugate PF-06650808. Responses were seen in two breast cancer patients (LBA 30). While preliminary, targeting Notch3 may become a new treatment approach in patients with selected solid tumours.”
The anti-Notch3 antibody-drug conjugate PF-06650808 is being developed by Pfizer.
- 31 Jul 2014 Phase-I clinical trials in Solid tumours (Late-stage disease) in USA (Parenteral)
- 30 Apr 2014 Preclinical trials in Solid tumours in USA (Parenteral)
- 30 Apr 2014 Pfizer plans a phase I trial for Solid tumours (late-stage disease, second-line therapy or greater) in USA (NCT02129205)
251st Am Chem Soc (ACS) Natl Meet (March 13-17, San Diego) 2016, Abst MEDI 262


/////////PF 06650808, PF-06650808, PF-6650808, monoclonal antibody, pfizer, phase 1, Solid tumours , Notch-3 receptor antagonists
C1(C(N(C(C1)=O)CCCCCC(=O)NC([C@H](C)C)C(=O)NC(C(=O)Nc2ccc(cc2)COC(=O)NC(C)(C)C(=O)N[C@@H](C(C)C)C(=O)[N@](C)C(C(CC)C)[C@@H](OC)CC(=O)N3CCC[C@H]3C(OO)C(C)C(=O)N[C@H](c4nccs4)CC)CCCNC(=O)N)=O)SC
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE


Join me on twitter
Join me on google plus  Googleplus
Googleplus
Googleplus amcrasto@gmail.com
amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
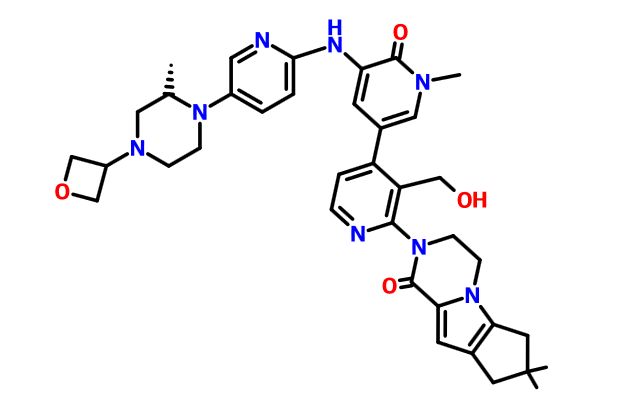
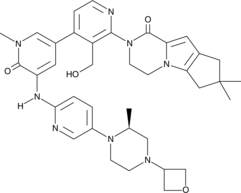
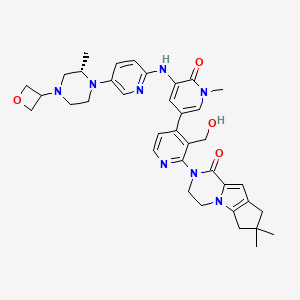
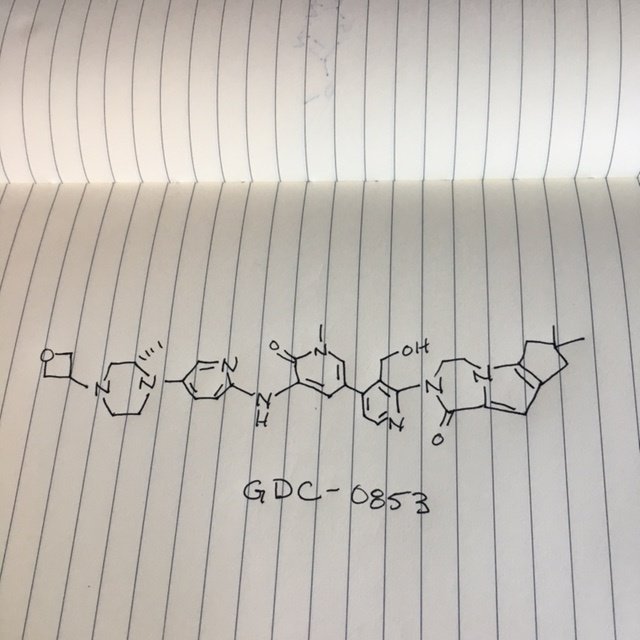
GDC 0853, Fenebrutinib
 .
.
Picture credit….Bethany Halford
GDC 0853, Fenebrutinib
GDC-0853; RG 7845
| Molecular Formula: | C37H44N8O4 |
|---|---|
| Molecular Weight: | 664.79646 g/mol |
2-[3-(hydroxymethyl)-4-[1-methyl-5-[(7-methyl-6,8-dihydro-5H-[1,2,4]triazolo[1,5-a]pyrazin-2-yl)amino]-6-oxo-3-pyridyl]-2-pyridyl]-3,4,6,7,8,9-hexahydropyrazino[1,2-a]indol-1-one
3-[3-(hydroxymethyl)-4-[1-methyl-5-[[5-[2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl]amino]-6-oxopyridin-3-yl]pyridin-2-yl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one
2H-Cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one, 2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S) -2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino] -6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7- dimethyl-
s ISoMER 1434048-34-6 desired
r iSoMER 1434048-57-3 undesired
Phase 1
Patients with Patients with Resistant B-Cell Lymphoma or Chronic Lymphocytic Leukemia..
@genentech‘s Btk inhibitor
https://clinicaltrials.gov/ct2/show/NCT01991184
Bruton tyrosine kinase inhibitor
- 01 Sep 2015 Phase-I clinical trials in Autoimmune disorders (In volunteers) in USA (PO, Capsule and Tablet) (NCT02699710)
- 16 Oct 2014 Discontinued – Phase-I for Non-Hodgkin’s lymphoma (Second-line therapy or greater) in USA (unspecified route)
- 16 Oct 2014 Discontinued – Phase-I for Chronic lymphocytic leukaemia (Second-line therapy or greater) in USA (unspecified route)

GDC-0853; RG 7845; RO 7010939
2-[1,6-dihydro-3′-(hydroxymethyl)-1-methyl-5-[[5-[(2S)-2-methyl-4-(3-oxetanyl)-1-piperazinyl]-2-pyridinyl]amino]-6-oxo[3,4′-bipyridin]-2′-yl]-3,4,7,8-tetrahydro-7,7-dimethyl-2H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1(6H)-one
GDC-0853 is an orally bioavailable, selective, and reversible Bruton’s tyrosine kinase (BTK) inhibitor with IC50s ranging from 2-9 nM for basophil activation, B cell receptor activation, and constitutive p-BTK activity in whole blood lysates.1,2 In rats, treatment for longer than 7 days leads to pancreatic toxicity but it does not occur in mice or dogs, even at higher doses.3 Formulations containing GDC-0853 were well-tolerated in Phase I clinical trials and are in additional clinical trials for rheumatoid arthritis, lupus erythematosus, lymphoma, and leukemia.
- Originator Genentech
- Class Antineoplastics; Antirheumatics; Piperazines; Pyrazines; Pyridines
- Mechanism of Action Agammaglobulinaemia tyrosine kinase inhibitors
Highest Development Phases
- Phase II Rheumatoid arthritis; Systemic lupus erythematosus; Urticaria
- Phase I Autoimmune disorders
- Discontinued Chronic lymphocytic leukaemia; Non-Hodgkin’s lymphoma
Most Recent Events
- 01 Jun 2018 Chemical structure information added
- 07 Nov 2017 Genentech initiates enrolment in a phase II extension trial for Systemic Lupus Erythematosus in Spain (EudraCT2017-001764-37)
- 13 Sep 2017 Genentech initiates enrolment in a phase I trial in Healthy volunteers in United Kingdom (NCT03290703)
BTK inhibitor GDC-0853 An orally available inhibitor of Bruton’s tyrosine kinase (BTK) with potential antineoplastic activity. Upon administration, GDC-0853 inhibits the activity of BTK and prevents the activation of the B-cell antigen receptor (BCR) signaling pathway. This prevents both B-cell activation and BTK-mediated activation of downstream survival pathways, which leads to the inhibition of the growth of malignant B-cells that overexpress BTK. BTK, a member of the Src-related BTK/Tec family of cytoplasmic tyrosine kinases, is overexpressed in B-cell malignancies; it plays an important role in B-lymphocyte development, activation, signaling, proliferation and survival.
SCHEME

MAIN

Patent
WO 2013067274
https://www.google.co.in/patents/WO2013067274A1?cl=en
part
Example 271a (S)-tert-Butyl 4-(6-(5-Chloro-2-methoxypyridin-3-ylamino)pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271a
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 1,4-dioxane (40 mL), (S)-tert-butyl 4-(6-amino pyridin-3-yl)-3-methylpiperazine-1-carboxylate 101h (2.04 g, 7.0 mmol), 3-bromo-5-chloro-2-methoxypyridine (2.8 g, 12.6 mmol), Pd2(dba)3 (640 mg, 0.70 mmol), XantPhos (404.6 mg, 0.70 mmol), and cesium carbonate (4.56 g, 14.0 mmol). After three cycles of vacuum/argon flush, the mixture was heated at 100 °C for 4 h. After this time the reaction was cooled to room temperature. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 1:3 ethyl acetate/petroleum ether to afford 271a (1.7 g, 57%) as a yellow solid. MS-ESI: [M+H]+ 434.2
Example 271btert-Butyl (3S)-4-(6-{[5-(2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}-3-(hydroxymethyl)pyridin-4-yl)-2-methoxypyridin-3-yl] amino}pyridin-3-yl)-3-methylpiperazine-1-carboxylate 271b
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 271a (650 mg, 1.50 mmol), {3-[(acetyloxy)methyl]-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridin-4-yl}boronic acid 199e (1.79 g, 4.5 mmol), Pd2(dba)3 (137.2 mg, 0.15 mmol), P(cy)3(167.4 mg, 0.60 mmol), Cs2CO3 (978 mg, 3.0 mmol), dioxane (20 mL), and water (0.5 mL). After three cycles of vacuum/argon flush, the mixture was heated at 110°C for 16 h. After this time the reaction was cooled to room temperature. Lithium hydroxide monohydrate (1.89 g, 45 mmol) and water (2.0 mL) were added. The resulting mixture was stirred at 45°C for 4 h. It was then filtered and the filtrate was evaporated under reduced pressure. The residue was purified by silica-gel column chromatography eluting with 3:1 ethyl acetate/petroleum ether to afford 271b (290 mg, 27%) as a yellow solid. MS-ESI: [M+H]+ 709.3
Example 271c 10-[3-(Hydroxymethyl)-4-[5-({5-[(2S)-2-methylpiperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridin-2-yl]-4,4-dimethyl-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-9-one 271c
A solution of 271b (286.6 mg, 0.40 mmol) in dioxane/HCl (30 mL) was stirred at 50 °C for 2 h. It was evaporated under reduced pressure to afford 271c (450 mg, crude) as a black solid. MS-ESI: [M+H]+ 595.3
Example 271 3-[3-(hydroxymethyl)-4-[5-[[5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]-2-pyridyl]amino]-6-oxo-1H-pyridin-3-yl]-2-pyridyl]-7,7-dimethyl-1,2,6,8-tetrahydrocyclopenta[3,4]pyrrolo[3,5-b]pyrazin-4-one 271
To a solution of 271c (450 mg, 0.75 mmol) in methanol (10 mL) was added oxetan-3-one (162 mg, 2.25 mmol), NaBH3CN (141.8 mg, 2.25 mmol), and ZnCl2 (306 mg, 2.25 mmol). The reaction was stirred at room temperature for 3 h. The mixture was evaporated under reduced pressure and the residue was diluted with water (5 mL). It was then extracted with dichloromethane (3 X 10 mL) and the combined dichloromethane extract was concentrated under reduced pressure. The residue was purified by reverse-phase prep-HPLC to afford 271 (23.0 mg, 8.8%, over two steps) as a yellow solid. MS-ESI: [M+H]+651.3. 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H), 8.74 (d, J = 2.0 Hz, 1H), 8.53 (d, J = 5.0 Hz, 1H), 7.99 (d, J = 3.0 Hz, 1H), 7.84 (s, 1H), 7.73 (s, 1H), 7.41 (d, J = 4.5 Hz, 1H), 7.35 (dd, J = 2.5 Hz, 8.5 Hz, 1H), 6.87 (s, 1H), 6.85 (d, J = 9.0 Hz, 1H), 5.16-5.13 (m, 1H), 4.72-4.69 (m, 5H), 4.54-4.53 (m, 1H), 4.36-4.35 (m, 1H), 4.19-4.17 (m, 2H), 3.89-3.87 (m, 1H), 3.56-3.49 (m, 2H), 3.11-3.09 (m, 2H), 2.60-2.48 (m, overlap, 7H), 2.24-2.21 (m, 1H), 1.29 (s, 6H), 1.02 (d, J = 6.0 Hz, 3H)
271
………………………..
syn of 191 j
is intermediatenot product, is acid
To a mixture of 4-chloro-2-{4,4-dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl}pyridine-3-carbaldehyde 108a (500 mg, 1.46 mmol), tert-butyl alcohol (20 mL), and dichloromethane (5 mL) was added 2-methyl-2-butene (3066 mg, 43.8 mmol). An aqueous solution (8 mL) of NaClO2 (263 mg, 2.92 mmol) and NaH2PO4·2water (683 mg, 4.38 mmol) was added dropwise at -10°C and the reaction mixture was stirred at -10 °C for overnight. It was concentrated under reduced pressure and the residue was extracted with ethyl acetate (4 × 20 mL). The combined organic extract was dried over MgSO4 and concentrated. The residue was purified with reverse-phase prep-HPLC to afford 210a (315 mg, 60%) as a pale yellow solid. MS-ESI: [M+H]+ 360.1
Example 210b 2-{4,4-Dimethyl-9-oxo-1,10-diazatricyclo[6.4.0.02,6]dodeca-2(6),7-dien-10-yl} -4-[1-methyl-5-({5-[(2S)-2-methyl-4-(oxetan-3-yl)piperazin-1-yl]pyridin-2-yl}amino)-6-oxo-1,6-dihydropyridin-3-yl]pyridine-3-carboxylic Acid 210b
A 25-mL round-bottomed flask equipped with a reflux condenser was charged with 210a (400 mg, 1.1 mmol), (S)-1-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-ylamino)-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2(1H)-one 191j (536 mg, 1.1 mmol), PdCl2(dppf) (81 mg, 0.11 mmol), K3PO4 (466 mg, 2.2 mmol), sodium acetate (216 mg, 2.2 mmol), acetonitrile (10 mL), and water (0.2 mL). After three cycles of vacuum/argon flush, the mixture was heated at 100°C for 3 h. It was then filtered and the filtrate was evaporated in vacuo. The residue was purified by silica-gel column chromatography eluting with 1:3 petroleum/ethyl acetate to afford 210b as a yellow solid (306 mg, 41%). MS-ESI: [M+H]+ 679.3
construction, use your discretion
Example 130a (3S)-tert- utyl 3-methyl-4-(6-nitropyridin-3-yl)piperazine-l-carboxylate 130a

130a
Following the procedures as described for compound lOlg, reaction of 5-bromo-2-nitropyridine (10.5 g, 50 mmol), and (JS)-tert-butyl-3 -methylpiperazine- 1 -carboxylate (10.0 g, 50 mmol) afforded 130a as a yellow solid (8.05 g, 50%). LCMS: [M+H]+ 323
Example 130b (3 S)-tert-butyl-4-(6-aminopyridin-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130b

130b
Following the procedures as described for compound lOlh, hydrogenation of 130a (5.8 g) afforded 130bas a brown solid (4.9 g, 96%). LCMS: [M+H]+ 293
Example 130c (3 S)-tert-Butyl-4-(6-(5 -bromo- 1 -methyl -2 -oxo- 1,2-dihydropyridin-3 -yl amino) pyridine-3 -yl)-3 -methylpiperazine- 1 -carboxylate 130c
N

Following the procedures as described for compound lOli, reaction of 130b (4.0 g) and 3,5-dibromo-l-methylpyridin-2(lH)-one (5.5 g) afforded 130c as a yellow solid (5.4 g, 83%). LCMS: [M+H]+ 478
Example 130d (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- 2-ylamino)pyridine-2(lH)-one 130d

Following the procedures as described for compound lOlj, acidic hydrolysis of the Boc group of 130c (3.1 g) afforded 130d as a yellow solid (2.3 g, 95%). LCMS: [M+H]+ 380.
Example 130e (3 S)-5 -Bromo- 1 -methyl-3 -(5 -(2 -methyl-4-(ox etan-3-yl)piperazin-l-yl) pyridine -2-ylamino)pyridin-2(lH)-one 130e

Following the procedures as described for compound 101k, reductive amination of 130d (2.35 g) with oxetan-3-one (0.4 mL) afforded 130e as a yellow solid (2.6 g, 98%). LCMS: [M+H]+ 434.
Example 13 Of (3S)-l-methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)pyridin-2-ylamino) -5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 130f
 check pyridine ring position
check pyridine ring position
A 100 mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with 130e (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), AcOK (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90 °C for 4 hrs, then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3: 1 PE/EA (80 mL) to afford 130f as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
check pyridine ring position, use your discretion
Example 191h ( 3S)-5 -Bromo- 1 -methyl-3 -(5 -(2-methylpiperazin- 1 -yl)pyridin- -ylamino)pyridine-2(lH)-one 191h

Following the procedure described for compound lOlj and starting with (3S)-tert-butyl 4-(6-(5 -bromo- 1 -methyl-2-oxo- 1 ,2-dihydropyridin-3 -ylamino)pyridine-3 -yl)-3 -methyl-piperazine-l-carboxylate 191g (3.1 g, 6.5 mmol) afforded 191h as a yellow solid (2.3 g, 94%). MS-ESI: [M+H]+ 378.
Example 1 1 i (S)-5 -Bromo- 1 -methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin- 1 -yl)pyridin-2-ylamino)pyridin-2(lH)-one 191i
A mixture of (5)-5-bromo-l-methyl-3-(5-(2-methylpiperazin-l-yl)pyridin-2-ylamino)pyridin-2(lH)-one 191h (40.0 g, 106 mmol), oxetan-3-one (1 1.4 g, 159 mmol), NaBH3CN (10.0 g, 159 mmol), and zinc chloride (21.3 g, 159 mmol) in methanol (700 mL) was stirred at 50°C for 5 hours. The mixture was added to water (100 mL) and concentrated under reduced pressure. The residue was extracted with dichloromethane (200 mL x 3). The combined organic layer was concentrated under reduced pressure and the residue was purified by silica-gel column chromatography eluting with 40: 1 dichloromethane /methanol to afford 191i (35 g, 73%). MS: [M+H]+ 434.
Example 191j (J5)-l-Methyl-3-(5-(2-methyl-4-(oxetan-3-yl)piperazin-l-yl)-pyridin- -ylamino) -5-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)pyridin-2(lH)-one 191j

191 i 191j
A 100-mL single-neck round-bottomed flask equipped with a magnetic stirrer and a reflux condenser was charged with (5)-tert-butyl-4-(6-(5-bromo-l-methyl-2-oxo-l ,2-dihydropyridin-3-ylamino)pyridine-3-yl)-3-methylpiperazine-l-carboxylate 191i (1.0 g, 1.0 eq., 2.3 mmol), Pin2B2 (1.46 g, 2.50 eq., 5.75 mmol), Pd2(dba)3 (105 mg, 0.05 eq., 0.125 mmol), X-Phos (93 mg, 0.1 eq., 0.23 mmol), potassium acetate (676 mg, 3.0 eq., 6.9 mmol), and dioxane (50 mL). After three cycles of vacuum/argon flush, the mixture was heated at 90°C for 4 h. It was then cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and the resulting residue was washed with 3 : 1 petroleum ether/ethyl acetate (80 mL) to afford 191j as yellow solid (1.0 g, 90%). MS: [M+H]+ 482.
pipeline
http://www.gene.com/medical-professionals/pipeline

Pictrelisib, GDC-0941, RG7321 and GNE0941
| Patent ID | Date | Patent Title |
|---|---|---|
| US8921353 | 2014-12-30 | Heteroaryl pyridone and aza-pyridone compounds |
| US2014378432 | 2014-12-25 | HETEROARYL PYRIDONE AND AZA-PYRIDONE COMPOUNDS |
| US8716274 | 2014-05-06 | Heteroaryl pyridone and aza-pyridone compounds |
Development of an Efficient Manufacturing Process for Reversible Bruton’s Tyrosine Kinase Inhibitor GDC-0853
Haiming Zhang*†  , Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin†
, Theresa Cravillion†, Ngiap-Kie Lim†, Qingping Tian†, Danial Beaudry†, Jessica L. Defreese†, Alec Fettes‡, Philippe James¶, David Linder¶, Sushant Malhotra†, Chong Han† , Remy Angelaud†, and Francis Gosselin† †Department of Small Molecule Process Chemistry, Genentech, Inc., 1 DNA Way, South San Francisco, California 94080, United States
‡Department of Process Chemistry and Catalysis and ¶Department of Drug Substance Scale-up and Supply, F. Hoffmann-La Roche AG, Grenzacherstrasse 124, 4070 Basel, Switzerland
ACS Editors’ Choice – This is an open access article published under an ACS AuthorChoice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. https://pubs.acs.org/doi/10.1021/acs.oprd.8b00134

Efforts toward the process development of reversible Bruton’s tyrosine kinase (BTK) inhibitor GDC-0853 (1) are described. A practical synthesis of GDC-0853 was accomplished via a key highly regioselective Pd-catalyzed C–N coupling of tricyclic lactam 5 with 2,4-dichloronicotinaldehyde (6) to afford the C–N coupling product 3, a Suzuki–Miyaura cross-coupling of intermediate 3 with boronic ester 4 derived from a Pd-catalyzed borylation of tetracyclic bromide 7, to generate penultimate aldehyde intermediate 2 and subsequent aldehyde reduction and recrystallization. Process development of starting materials 5, 6, and 7 is also discussed.
(S)-2-(3′-(Hydroxymethyl)-1-methyl-5-((5-(2-methyl-4-(oxetan-3-yl)piperazin-1-yl)pyridin-2-yl)amino)-6-oxo-1,6-dihydro-[3,4′-bipyridin]-2′-yl)-7,7-dimethyl-2,3,4,6,7,8-hexahydro-1H-cyclopenta[4,5]pyrrolo[1,2-a]pyrazin-1-one (crude GDC-0853, 1)
GDC-0853 (1, 196 kg, 81% yield, >99 A%, Pd < 10 ppm): mp 271 °C (DSC);
FTIR (cm–1, neat) 3430, 3313, 2945, 2865, 1606, 1573;
1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 2.2 Hz, 1H), 8.48 (d, J = 5.1 Hz, 1H), 7.96 (d, J = 2.7 Hz, 1H), 7.83 (d, J = 2.3 Hz, 2H), 7.36 (d, J = 5.1 Hz, 1H), 7.31 (dd, J = 8.9, 2.8 Hz, 1H), 6.87–6.76 (m, 2H), 5.18–4.98 (m, 1H), 4.77–4.58 (m, 5H), 4.50 (m, 1H), 4.33 (m, 1H), 4.16 (m, 2H), 3.86 (m, 1H), 3.71 (s, 3H), 3.61–3.38 (m, 2H), 3.07 (m, 2H), 2.67–2.39 (m, 7H), 2.20 (dd, J = 10.8, 6.3 Hz, 1H), 1.27 (s, 6H), 0.98 (d, J = 6.3 Hz, 3H);
13C NMR (101 MHz, CDCl3) δ 161.7, 157.6, 154.3, 150.3, 148.4, 141.9, 140.0, 131.4, 131.1, 129.7, 128.8, 127.7, 125.8, 123.9, 117.2, 116.3, 112.4, 111.3, 75.5, 75.5, 59.4, 59.1, 56.3, 52.9, 50.0, 49.2, 48.2, 45.9, 42.7, 40.9, 39.6, 38.5, 30.3, 15.3.
HRMS (ESI+) calcd for C37H45N8O4 ([M + H]+), 665.3564; found, 665.3588.
https://pubs.acs.org/doi/suppl/10.1021/acs.oprd.8b00134/suppl_file/op8b00134_si_001.pdf


/////////////
O=C1N(C)C=C(C2=CC=NC(N3CCN4C(C3=O)=CC5=C4CC(C)(C)C5)=C2CO)C=C1NC(N=C6)=CC=C6N7CCN(C8COC8)C[C@@H]7C
//////GDC 0853, genentech, Btk inhibitor, phase 1, Patients with Resistant B-Cell Lymphoma, Chronic Lymphocytic Leukemia, Bruton tyrosine kinase inhibitor, GDC-0853, RG 7845, 1434048-34-6, Fenebrutinib
N1(CCN(CC1C)C2COC2)c3cnc(cc3)NC=4C(N(\C=C(/C=4)c5c(c(ncc5)N6CCn7c(C6=O)cc8CC(Cc78)(C)C)CO)C)=O
CC1CN(CCN1C2=CN=C(C=C2)NC3=CC(=CN(C3=O)C)C4=C(C(=NC=C4)N5CCN6C7=C(CC(C7)(C)C)C=C6C5=O)CO)C8COC8
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus Googleplus
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
SIDE CHAIN
MAIN
GLPG 1690
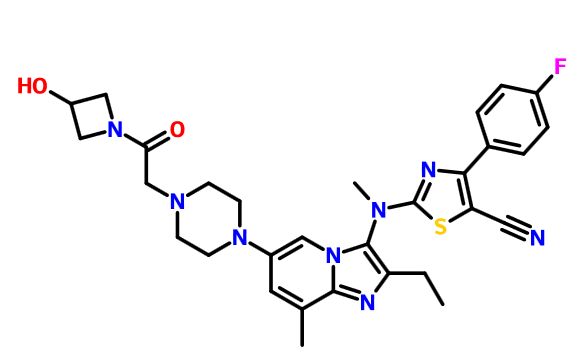

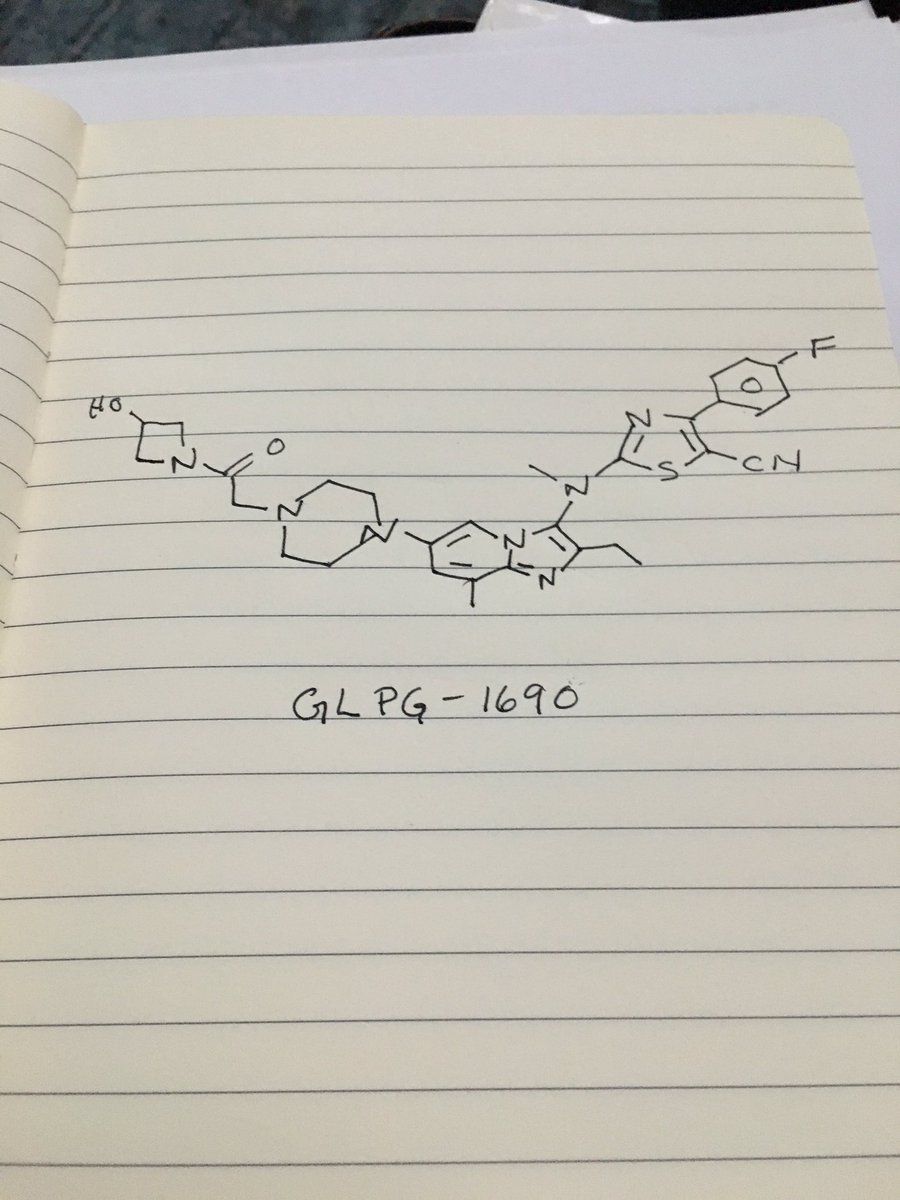
Picture credit….Bethany Halford
GLPG 1690
2-[[2-ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methylimidazo[1,2-a]pyridin-3-yl]-methylamino]-4-(4-fluorophenyl)-1,3-thiazole-5-carbonitrile
5- Thiazolecarbonitrile, 2-[[2-ethyl-6-[4-[2-(3-hydroxy-1-azetidinyl)-2-oxoethyl]- 1-piperazinyl]-8-methylimidazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)-
CAS 1628260-79-6
![]()
$GLPG compound for treating idiopathic pulmonary fibrosis
| Molecular Formula: | C30H33FN8O2S |
|---|---|
| Molecular Weight: | 588.698823 g/mol |
| Galapagos Nv |
http://files.glpg.com/docs/website_1/Poster_ERS_2015_final.pdf
http://www.glpg.com/docs/view/56b360a81f6b2-en
Phase I Idiopathic pulmonary fibrosis
| Description | Selective autotaxin (ENPP2; ATX) inhibitor |
| Molecular Target | Autotaxin (ENPP2) (ATX) |
- Originator Galapagos NV
- Class Anti-inflammatories; Small molecules
- Mechanism of Action ENPP2 protein inhibitors
- 23 Sep 2015 Pharmacodynamics data from a preclinical trial in Indiopathic pulmonary fibrosis released by Galapagos
- 22 Sep 2015 Pharmacokinetics data from a phase I trial in healthy volunteers released by Galapagos
- 22 Sep 2015 Updated adverse events data from a phase I trial in healthy volunteers released by Galapagos
GLPG1690
GLPG1690 is a selective autotaxin inhibitor discovered by Galapagos, with potential application in idiopathic pulmonary disease (IPF). In a Phase 1 study in healthy human volunteers, GLPG1690 demonstrated favorable safety and tolerability, as well as a strong pharmacodynamic signal implying target engagement. Galapagos is currently preparing a Phase 2 study in IPF, to be filed for approval before the end of 2015. GLPG1690 is fully proprietary to Galapagos.
| Source: Galapagos NV
- Fully owned and proprietary clinical asset for pulmonary fibrosis
- GLPG1690 acts on autotaxin target
- Novel mode of action, originating from Galapagos target discovery engine
- Filing for Phase 2 clinical trial in 2015
MECHELEN, Belgium, March 16, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext: GLPG) announced that Janssen Pharmaceutica NV and Galapagos have mutually agreed to terminate the inflammation alliance and option agreements between the companies. Galapagos views the molecules emerging from the alliance as strong additions to its growing proprietary pipeline. Among others, all rights to candidate drug GLPG1690, a selective autotaxin inhibitor, return to Galapagos. Galapagos has successfully completed a First-in-Human Phase 1 trial for GLPG1690 and is preparing a Phase 2 clinical trial in idiopathic pulmonary fibrosis (IPF).
“We are pleased to regain the rights to GLPG1690 to pursue the most suitable clinical application of autotaxin inhibition. There is a large unmet medical need in IPF, and our pre-clinical data with GLPG1690 supports its potential as a competitive and novel approach in this disease area,” said Dr Piet Wigerinck, Chief Scientific Officer of Galapagos. “The alliance with Janssen has been underway since October 2007 and has generated three clinical molecules, two of which are now proprietary Phase 2 assets of Galapagos: GLPG1205 and GLPG1690. This program is a valuable component of our development portfolio, and regaining the rights is a next step in our transformation into a mature biotech company with a proprietary product pipeline.”
Galapagos identified autotaxin as playing a key role in inflammation, using an inflammation assay in its unique target discovery platform. Pharmacology and translational studies published by other parties in the literature since then suggest autotaxin may play a key role in metabolic disease, arthritic pain, oncology, and lung disease.
GLPG1690 is a potent and selective inhibitor of autotaxin. In a Phase 1 study in healthy human volunteers, GLPG1690 demonstrated favorable safety and tolerability, as well as a strong pharmacodynamic signal implying target engagement. Galapagos is currently preparing a Phase 2 study in IPF, to be filed for approval before the end of 2015.
About IPF
Idiopathic pulmonary fibrosis (IPF) is a chronic and ultimately fatal disease characterized by a progressive decline in lung function. Pulmonary fibrosis involves scarring of lung tissue and is the cause of shortness of breath. Fibrosis is usually associated with a poor prognosis. The term “idiopathic” is used because the cause of pulmonary fibrosis is still unknown. Estimated incidence of IPF is up to 16.3 per 100,000 persons in the US and 7.4 per 100,000 persons in Europe, with approximately 30,000-35,000 new patients diagnosed with IPF worldwide each year. The goals of treatment in IPF are essentially to reduce the symptoms, slow down disease progression, reduce acute exacerbations, and prolong survival. Approved treatments thus far have improved the overall survival of IPF patients, but unwanted side effects with these treatments are common, presenting an unmet need for effective treatments with safer side effect profiles.
| Source: Galapagos NV
MECHELEN, Belgium, Sept. 22, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext & NASDAQ: GLPG) presents pre-clinical and Phase 1 results for autotaxin inhibitor GLPG1690 at the European Respiratory Society Annual Meeting in Amsterdam, Netherlands. Galapagos expects to file an exploratory Phase 2 study in idiopathic pulmonary fibrosis before year end. GLPG1690 has potential application in other pulmonary diseases such as chronic obstructive pulmonary disease (COPD), as supported by the presentation on pre-clinical findings at ERS this year:
“Pharmacological profile and efficacy of GLPG1690, a novel ATX inhibitor for COPD treatment,” poster PA2129 in Poster Discussion Session: “New targets and modalities for the treatment of asthma and COPD” (September 28, 2015; Room D201-202, 10:45 AM – 12:45 PM)
Galapagos is the first to show efficacy of an autotaxin inhibitor in pre-clinical models for COPD and IPF, pointing to novel therapeutic areas for autotaxin inhibition. The poster shows how GLPG1690 acts as a potent inhibitor of mouse and human autotaxin (IC50: 100 -500 nM range). Furthermore, GLPG1690 reduces inflammation in a mouse steroid-resistant tobacco smoke model to a similar extent as a standard therapy for COPD.
Galapagos also presents the topline results with GLPG1690 in Phase 1 in healthy human volunteers: “Favorable human safety, pharmacokinetics and pharmacodynamics of the autotaxin inhibitor GLPG1690, a potential new treatment in COPD,” oral presentation OA484 in session “Advances in the future treatment of COPD” (September 27, 2015; Room 2.1, 10:45 AM – 12:45 PM)
GLPG1690 was safe and well tolerated up to a single oral dose of 1500 mg and up to 1000 mg twice daily for 14 days, with no significant adverse effects on ECGs, vital signs or laboratory parameters. The compound also showed good oral bioavailability with a half-life of 5 hours and a dose-proportional increase in exposure. GLPG1690 showed concentration-dependent reduction of a relevant biomarker (plasma LPA18:2 levels) with a maximum of approximately 90%. At steady state, continuous reduction of this biomarker levels of >60% was observed from 0 to 24 hours. The presentation will also include relevant pre-clinical model data for COPD and IPF with GLPG1690.
Both the presentation and the posters will be made available on the Galapagos website after the conference.
About Galapagos
Galapagos (Euronext & NASDAQ: GLPG) is a clinical-stage biotechnology company specialized in the discovery and development of small molecule medicines with novel modes of action, with a pipeline comprising three Phase 2 programs, two Phase 1 trials, five pre-clinical studies, and 20 discovery small-molecule and antibody programs in cystic fibrosis, inflammation, and other indications. In the field of inflammation, AbbVie and Galapagos signed a collaboration agreement for the development and commercialization of filgotinib. Filgotinib is an orally-available, selective inhibitor of JAK1 for the treatment of rheumatoid arthritis and potentially other inflammatory diseases, currently in Phase 2B studies in RA and in Phase 2 in Crohn’s disease. Galapagos reported good activity and a favorable safety profile in both the DARWIN 1 and 2 trials in RA. AbbVie and Galapagos also signed a collaboration agreement in cystic fibrosis to develop and commercialize molecules that address mutations in the CFTR gene. Potentiator GLPG1837 is currently in a Phase 1 trial, and corrector GLPG2222 is at the pre-clinical candidate stage. GLPG1205, a first-in-class inhibitor of GPR84 and fully-owned by Galapagos, is currently being tested in a Phase 2 proof-of-concept trial in ulcerative colitis patients. GLPG1690, a fully proprietary, first-in-class inhibitor of autotaxin, has shown favorable safety in a Phase 1 trial and is expected to enter Phase 2 in idiopathic pulmonary fibrosis. The Galapagos Group, including fee-for-service subsidiary Fidelta, has approximately 400 employees, operating from its Mechelen, Belgium headquarters and facilities in The Netherlands, France, and Croatia. More info at www.glpg.com
CONTACT
Galapagos NV
Elizabeth Goodwin, Head of Corporate Communications & IR
Tel: +31 6 2291 6240
ir@glpg.com
MECHELEN, Belgium, Feb. 16, 2015 (GLOBE NEWSWIRE) — Galapagos NV (Euronext: GLPG) announced today that GLPG1690, a first-in-class molecule for pulmonary disease, has demonstrated target engagement, a good safety profile, and favorable drug properties in a Phase 1 study. Galapagos is developing GLPG1690 within its alliance with Janssen Pharmaceutica NV.
The aim of the Phase 1 study was to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of oral single and multiple ascending doses of GLPG1690. The randomized, double-blind, placebo-controlled, single center study was conducted in 40 healthy volunteers in Belgium. In the first part of the study, single ascending doses were evaluated. In the second part, the new compound was administered daily for 14 days.
GLPG1690 proved to be safe and well-tolerated over a wide dose range in healthy volunteers. Engagement of the thus far undisclosed novel target was confirmed using a relevant biomarker. GLPG1690 displayed a favorable pharmacokinetic and pharmacodynamic profile. The data shown in Phase 1 encourage Galapagos to explore a Phase 2 study design in pulmonary disease.
“GLPG1690 is the first molecule against this target ever to be evaluated clinically, and we are pleased with the outcome of the Phase 1 study,” said Dr Piet Wigerinck, CSO of Galapagos. “Galapagos continues to deliver novel therapeutics from its unique target and drug discovery engine.”
In 2007, Galapagos announced an alliance agreement with Janssen Pharmaceutica NV providing the option to worldwide, commercial licenses to certain Galapagos internal inflammatory disease programs. These programs are based on novel targets for inflammatory disorders that were identified and validated by Galapagos using its proprietary target discovery engine. Subsequent Galapagos research led to the discovery of GLPG1690, a first-in-class molecule that entered the clinic for inflammatory disorders. Galapagos is responsible for execution of Phase 1 and Phase 2A studies with GLPG1690.
SYNTHESIS
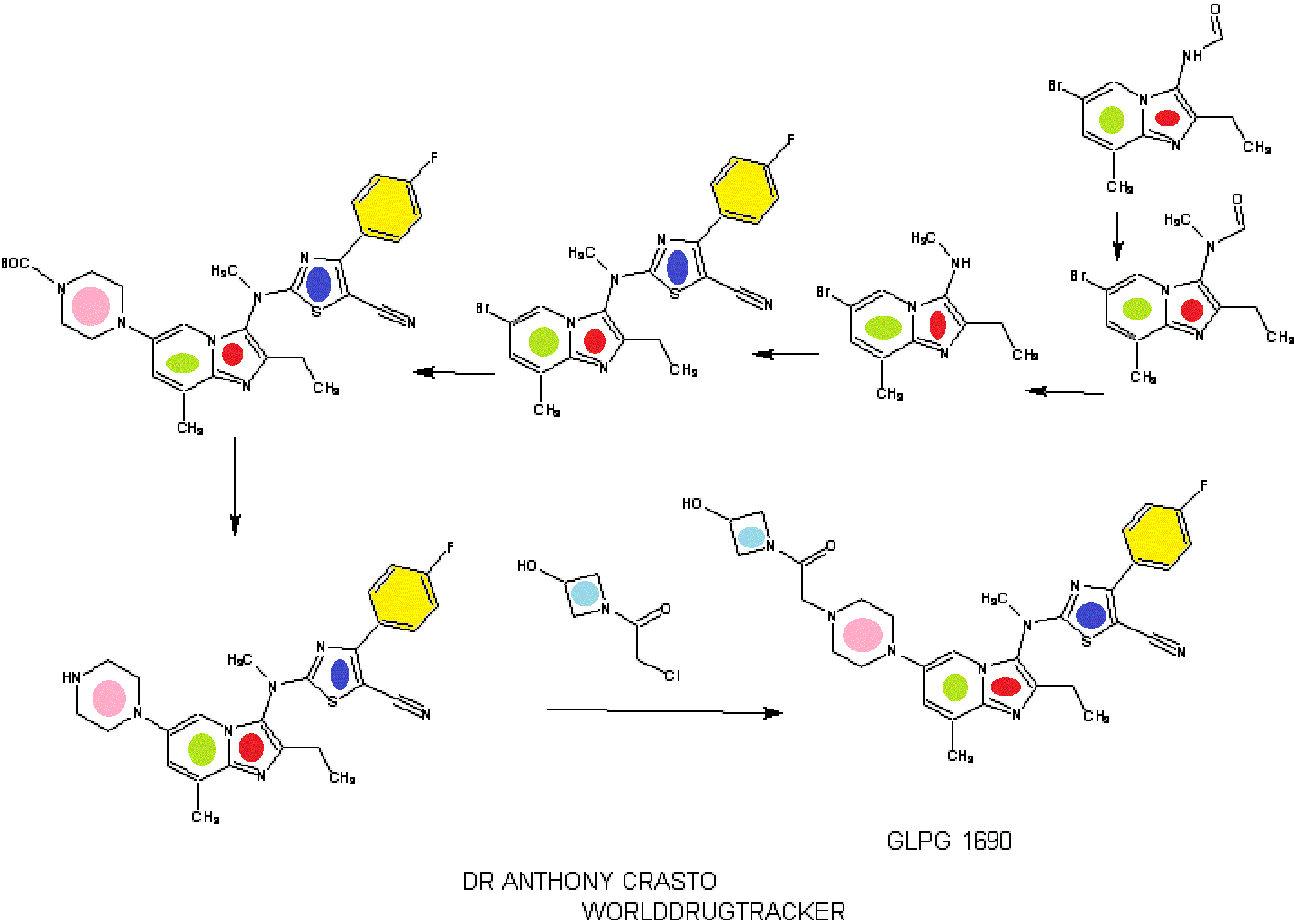
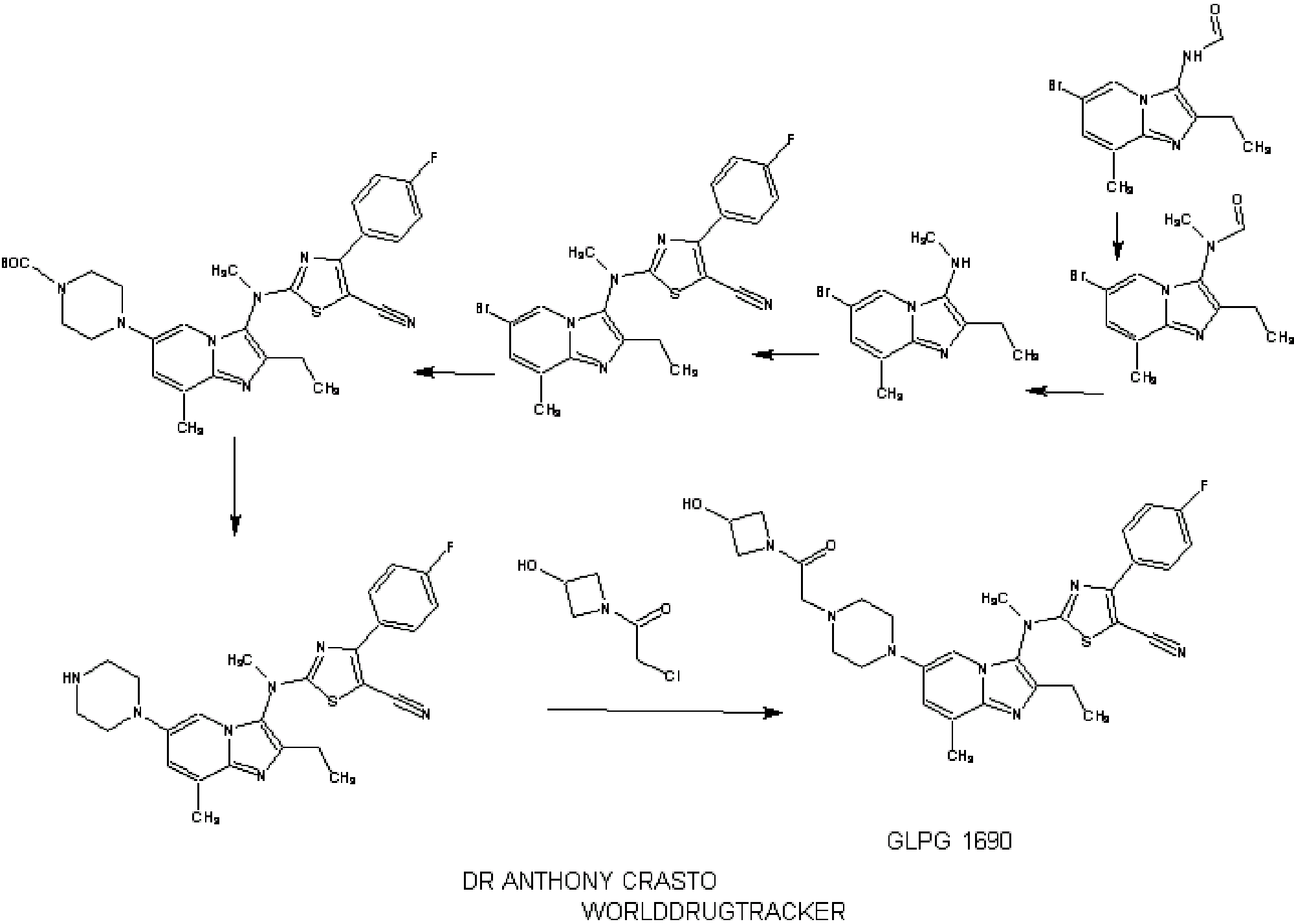
INTRODUCTION
relates to compounds that are inhibitors of autotaxin, also known as ectonucleotide pyrophosphatase/phosphodiesterase 2 (NPP2 or ENPP2), that is involved in fibrotic diseases, proliferative diseases, inflammatory diseases, autoimmune diseases, respiratory diseases, cardiovascular diseases, neurodegenerative diseases, dermatological disorders, and/or abnormal angiogenesis associated diseases. The present invention also provides methods for the production of a compound of the invention, pharmaceutical compositions comprising a compound of the invention, methods for the prophylaxis and/or treatment of diseases involving fibrotic diseases, proliferative diseases, inflammatory diseases, autoimmune diseases, respiratory diseases, cardiovascular diseases, neurodegenerative diseases, dermatological disorders, and/or abnormal angiogenesis associated diseases by administering a compound
STAGE 1

STAGE2

STAGE 3

STAGE4

STAGE 5

FINAL

PATENT
US2014303140
http://www.google.com/patents/US20140303140
1.2.4.4. Illustrative Synthesis of Intermediate Gen-3-e: N-(6-bromo-2-ethyl-8-methylimidazo[1,2-a]pyridin-3-yl)-N-methylformamide
-

-
To a suspension of formamide Gen-2-d (720 g, 2.55 mol, 1 eq.) in 5 L of acetone were added potassium carbonate (1 kg, 7.66 mol, 3 eq.) and methyl iodide (700 g, 4.93 mol, 1.9 eq.). The reaction mixture was heated to 40° C. overnight. Additional methyl iodide (25 g, 0.18 mol, 0.07 eq.) was then introduced and stirring continued for 1 h at 40° C. The reaction mixture was filtered and washed with acetone (2×300 mL) and DCM (2×300 mL). The filtrate was concentrated in vacuo and the residue was partitioned between DCM (3 L) and water (1 L). The aqueous layer was further extracted with DCM. The combined organic layers were then washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The solid was triturated with Et2O (1 L) at r.t. for 1 h, filtered off and dried to afford Intermediate Gen-3-e.
-
Rotamer A (Major): 1H NMR δ (ppm) (400 MHz, CDCl3): 8.19 (1H, s), 7.78 (1H, s), 7.15 (1H, s), 3.24 (3H, s), 2.72 (2H, q), 2.59 (3H, s), 1.31 (3H, t)
-
Rotamer B (Minor): 1H NMR δ (ppm) (400 MHz, CDCl3): 8.49 (1H, s), 7.65 (1H, s), 7.08 (1H, s), 3.36 (3H, s), 2.72 (2H, q), 2.59 (3H, s), 1.31 (3H, t)
-
LC-MS: MW (calcd): 295 (79Br), 297 (81Br); m/z MW (obsd): 296 (79Br M+1), 298 (81Br M+1)
1.2.5.2. Illustrative Synthesis of Intermediate Gen-4-d: (6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amine
-

-
Intermediate Gen-3-e (80 g, 270 mmol, 1 eq.) was dissolved in a 1.25 M HCl solution in MeOH (540 mL, 2.5 eq.) and the resulting mixture was refluxed overnight. 270 mL of 1.25 M HCl solution in MeOH were added and heating continued overnight. After 48 h, additional 70 mL of the 1.25 M HCl solution in MeOH were introduced in the reaction mixture. Heating was maintained overnight until conversion was complete. The crude mixture was then concentrated in vacuo and the residue was partitioned between EtOAc (300 mL) and water (700 mL). A saturated NaHCO3 solution was added until pH reached 8-9. The aqueous layer was extracted twice with EtOAc (2×300 mL). The combined organic layers were then washed with brine (200 mL), dried over Na2SO4, filtered and concentrated in vacuo to give Intermediate Gen-4-d (6-bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amine) as a free base.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.05 (1H, s), 7.04 (1H, s), 2.84-2.78 (5H, m), 2.60 (3H, s), 1.35 (3H, t)
-
LC-MS: MW (calcd): 267 (79Br), 269 (81Br); m/z MW (obsd): 268 (79Br M+1), 270 (81Br M+1)
1.2.6.4. Illustrative Synthesis of Intermediate Gen-5-t: 2-[(6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile
-
-
To a solution of amine Gen-4-d (4.4 g, 16.6 mmol, 1 eq.) in THF (44 mL) under argon was slowly added NaH (60% in oil suspension, 2.0 g, 50.0 mmol, 3 eq.). The reaction mixture was heated at 90° C. for 30 min then cooled to 40° C. before adding the chlorothiazole Gen-12-a (4.74 g, 19.9 mmol, 1.2 eq.). The reaction mixture was stirred at 90° C. overnight. After cooling to r.t. the mixture was slowly quenched by addition of water and then diluted with EtOAc. The organic layer was separated and the aqueous layer extracted with EtOAc. The combined organic layers were then washed with water and brine, dried over Na2SO4, filtered and concentrated in vacuo. The residue was triturated in Et2O, filtered and washed with Et2O and MeCN. Recrystallization was performed in MeCN (180 mL) to afford Intermediate Gen-5-t (2-[(6-Bromo-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile).
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.15 (2H, dd), 7.80 (1H, s), 7.22-7.14 (3H, m), 3.62 (3H, s), 2.77 (2H, q), 2.64 (3H, s), 1.35 (3H, t)
-
LC-MS: MW (calcd): 469 (79Br), 471 (81Br); m/z MW (obsd): 470 (79Br M+1), 472 (81Br M+1)

1.2.7.1.4. Illustrative Synthesis of 4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester
-

-
To a solution of Intermediate Gen-5-t (24.2 g, 51.5 mmol, 1 eq.) in toluene under argon were successively added N-Boc piperazine (14.4 g, 77.3 mmol, 1.5 eq.), sodium tert-butoxide (9.9 g, 103 mmol, 2 eq.), JohnPhos (1.54 g, 5.15 mmol, 0.1 eq.) and Pd2(dba)3 (2.36 g, 2.58 mmol, 0.05 eq.). The reaction mixture was heated at 115° C. for 1 h. After cooling to r.t., the crude product was filtered on Celpure® P65 and the residue dissolved in EtOAc and washed with water. The organic layer was further washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The crude product was purified by chromatography on silica gel (elution with heptane/EtOAc:90/10 to 20/80) to afford the expected product.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.16 (2H, dd), 7.17 (2H, app t), 6.99 (2H, bs), 3.62-3.53 (4H, m), 3.60 (3H, s), 3.04-2.93 (4H, m), 2.74 (2H, q), 2.62 (3H, s), 1.47 (9H, s), 1.33 (3H, t).
-
LC-MS: MW (calcd): 575; m/z MW (obsd): 576 (M+1)
1.2.7.8.4. Illustrative Synthesis of Compound 1: 2-[(2-Ethyl-8-methyl-6-piperazin-1-yl-imidazo[1,2-a]pyridin-3-yl)-methyl-amino]-4-(4-fluoro-phenyl)-thiazole-5-carbonitrile
-

-
4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester was prepared from intermediate Gen-5-t using Boc-piperazine and method Flb.
-
To a solution of 4-(3-{[5-Cyano-4-(4-fluoro-phenyl)-thiazol-2-yl]-methyl-amino}-2-ethyl-8-methyl-imidazo[1,2-a]pyridin-6-yl)-piperazine-1-carboxylic acid tert-butyl ester (24.4 g, 42 mmol, 1 eq.) in MeOH (100 mL) was added a 2 M HCl solution in Et2O (127 mL, 254 mmol, 6 eq.). The reaction mixture was stirred at r.t. for 3.5 h then concentrated in vacuo. The residue was partitioned between EtOAc and water. The aqueous layer was extracted twice with EtOAc. A 2 M NaOH solution was added to the aqueous layer until pH reached 8-9 and further extraction with EtOAc was performed. The combined organic layers were then washed with brine, dried over Na2SO4, filtered and concentrated in vacuo. The solid was triturated with heptane (100 mL) at r.t. overnight, filtered off, washed with heptane and Et2O, and dried to afford the expected compound.
-
1H NMR δ (ppm) (400 MHz, CDCl3): 8.17 (2H, dd), 7.18 (2H, app t), 6.99 (2H, bs), 3.61 (3H, s), 3.09-2.98 (8H, m), 2.75 (2H, q), 2.61 (3H, s), 1.34 (3H, t).
-
LC-MS: MW (calcd): 475; m/z MW (obsd): 476 (M+1)
1.2.7.14. Illustrative Synthesis of Compound 2: 2-((2-ethyl-6-(4-(2-(3-hydroxyazetidin-1-yl)-2-oxoethyl)piperazin-1-yl)-8-methylimidazo[1,2-a]pyridin-3-yl)(methyl)amino)-4-(4-fluorophenyl)thiazole-5-carbonitrile
-

-
To a solution of amine compound 1 (12.6 g, 27 mmol, 1 eq.) in 100 mL of MeCN were added potassium carbonate (7.3 g, 53 mmol, 2 eq.) and Gen13-a (5.2 g, 34 mmol, 1.3 eq.). The reaction mixture was refluxed for 5.5 h then cooled to r.t. and stirred for 40 h. The crude product was filtered and washed with MeCN. The collected precipitate was then suspended in 300 mL of water, stirred for 1 h, filtered, and finally washed with water and MeCN. The solid obtained was dried in vacuo for 48 h to afford Compound 2.
-
1H NMR (400 MHz, CDCl3) δ ppm 8.20-8.12 (2H, m), 7.22-7.13 (2H, m), 6.99 (2H, s), 4.68 (1H, m), 4.43 (1H, dd), 4.26 (1H, dd), 4.14-4.05 (1H, m), 3.88 (1H, dd), 3.61 (3H, s), 3.58-3.52 (1H, m), 3.14-3.02 (6H, m), 2.74 (2H, q), 2.70-2.62 (4H, m), 2.59 (3H, s), 1.33 (3H, t)
-
LC-MS: MW (calcd): 588; m/z MW (obsd): 589 (M+1)
| US9249141 | Dec 17, 2014 | Feb 2, 2016 | Galapagos Nv | Compounds and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
1 to 2 of 2
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015111872 | 2015-04-23 | NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS |
| US2014303140 | 2014-10-09 | NOVEL COMPOUNDS AND PHARMACEUTICAL COMPOSITIONS THEREOF FOR THE TREATMENT OF INFLAMMATORY DISORDERS |
////////////GLPG 1690, idiopathic pulmonary fibrosis, PHASE 1, GALAPAGOS, 1628260-79-6
n12c(c(nc1c(cc(c2)N3CCN(CC3)CC(=O)N4CC(C4)O)C)CC)N(C)c5nc(c(s5)C#N)c6ccc(cc6)F
CCC1=C(N2C=C(C=C(C2=N1)C)N3CCN(CC3)CC(=O)N4CC(C4)O)N(C)C5=NC(=C(S5)C#N)C6=CC=C(C=C6)F
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus Googleplus
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
DISCLAIMER
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
AZD 7594
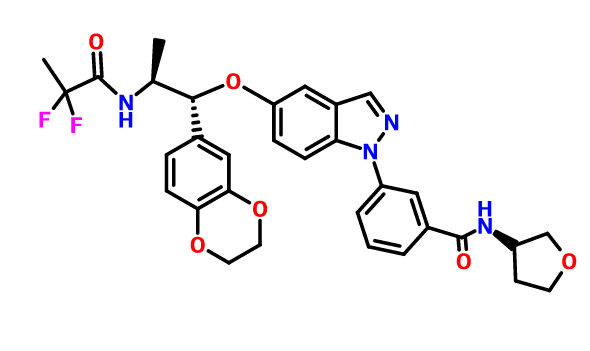
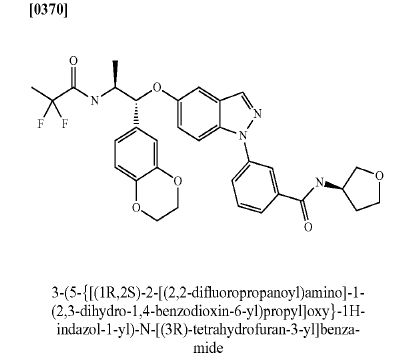
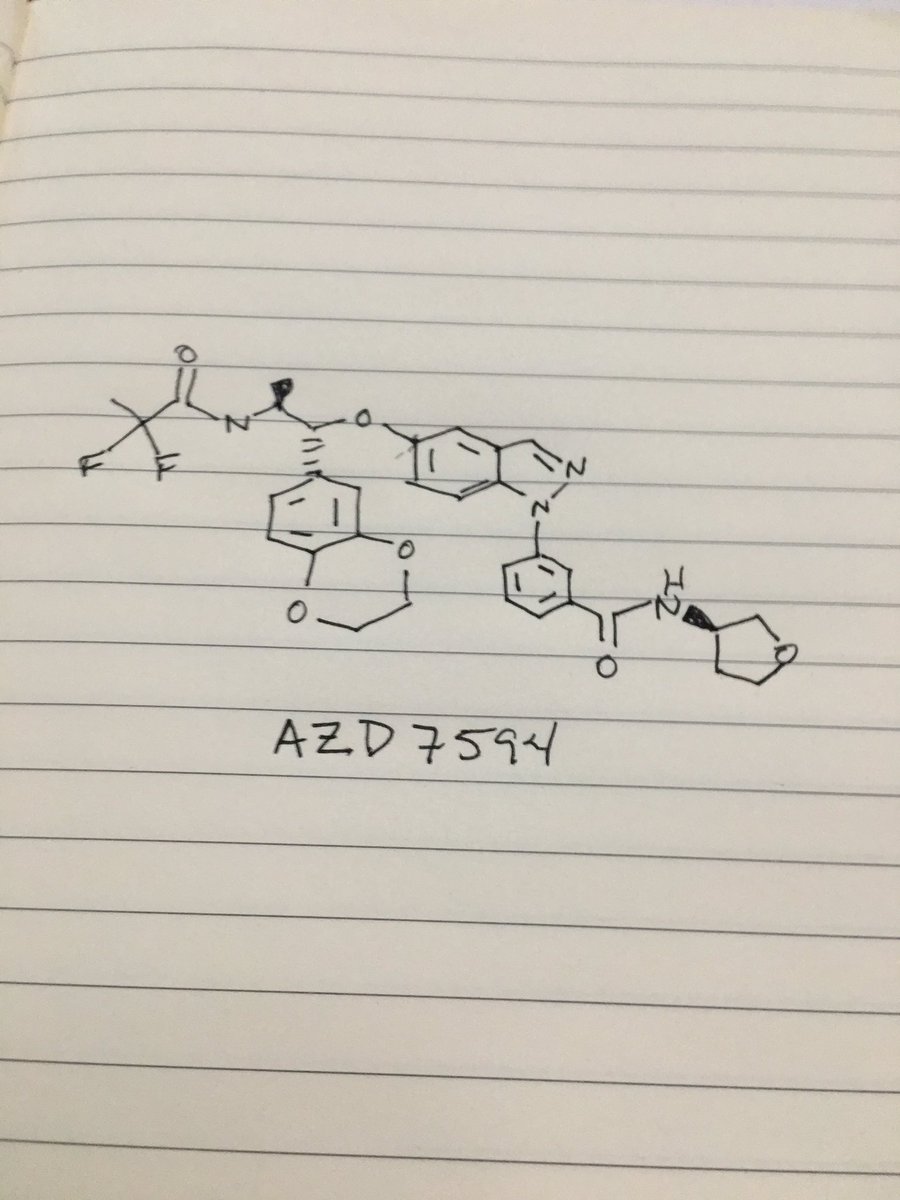 .
.
Picture credit….Bethany Halford

AZD 7594
$AZN‘s asthma candidate
AZ13189620; AZD-7594
Bayer Pharma Aktiengesellschaft, Astrazeneca Ab
| Molecular Formula: | C32H32F2N4O6 |
|---|---|
| Molecular Weight: | 606.616486 g/mol |
3-[5-[(1R,2S)-2-(2,2-difluoropropanoylamino)-1-(2,3-dihydro-1,4-benzodioxin-6-yl)propoxy]indazol-1-yl]-N-(oxolan-3-yl)benzamide
Benzamide, 3-[5-[(1R,2S)-2-[(2,2-difluoro-1-oxopropyl)amino]-1-(2,3-dihydro-1,4-benzodioxin-6-yl)propoxy]-1H-indazol-1-yl]-N-[(3R)-tetrahydro-3-furanyl]-
- Cas 1196509-60-0
AZD-7594 is in phase II clinical trials by AstraZeneca for the treatment of mild to moderate asthma.
It is also in phase I clinical trials for the treatment of chronic obstructive pulmonary disorder (COPD).
https://clinicaltrials.gov/ct2/show/NCT02479412
| Company | AstraZeneca plc |
| Description | Inhaled selective glucocorticoid receptor (GCCR) modulator |
| Molecular Target | Glucocorticoid receptor (GCCR) |
- Phase II Asthma
- Phase I Chronic obstructive pulmonary disease
- 01 Feb 2016 AstraZeneca completes a phase II trial in Asthma in Bulgaria and Germany (Inhalation) (NCT02479412)
- 09 Jan 2016 AstraZeneca plans to initiate a phase I trial in Healthy volunteers in USA (IV and PO) (NCT02648438)
- 01 Jan 2016 Phase-I clinical trials in Chronic obstructive pulmonary disease (In volunteers) in USA (PO, IV, Inhalation) (NCT02648438)
PATENT
http://www.google.com/patents/WO2009142569A1
PATENT
US20100804345
UNWANTED ISOMER

WANTED COMPD


PATENT
Example 6
WANTED ISOMER

3-(5- { TC 1 R,2SV2-r(2,2-difluoropropanoyl)aminol- 1 -(2,3-dihydro-l ,4-benzodioxin-6-5 yDpropylioxy) – 1 H-indazol- 1 -ylVN-[(3R)-tetrahydrofuran-3-vnbenzamide. APCI-MS: m/z 607 [MH+] 1H NMR ^OO MHz, DMSOd6) δ 8.71 (IH, d), 8.65 (IH, d), 8.24 (IH, s), 8.18 (IH, s), 7.90 – 7.84 (2H, m), 7.77 (IH, d), 7.65 (IH, t), 7.21 (IH, dd), 7.13 (IH, d), 6.89 – 6.78 (3H, m), 5.17 (IH, d), 4.48 (IH, m), 4.23 – 4.10 (5H, m), 3.89 – 3.82 (2H, m), 3.72 (IH, td), 3.61 (IH, dd), 2.16 (IH, m), 1.94 (IH, m), 1.55 (3H, t), 1.29 (3H, d). LC (method A) rt = 12.03 min LC (method B) rt = 11.13 min Chiral SFC (method B) rt = 4.71 min M.p. = 177 °C
UNWANTED

o 3-(5- { IY 1 R,2S V2-r(2,2-difluoropropanoyl‘)amino‘|- 1 -(2,3-dihydro- 1 ,4-benzodioxin-6- yl)propyl]oxy } – 1 H-indazol- 1 -yP-N-IO S)-tetrahydrofuran-3 -yl“|benzamide
APCI-MS: m/z 607 [MH+]
1H NMR (400 MHz, DMSO-J6) δ 8.71 (IH, d), 8.65 (IH, d), 8.24 (IH, s), 8.18 (IH, s),
7.90 – 7.84 (2H, m), 7.77 (IH, d), 7.65 (IH, t), 7.21 (IH, dd), 7.13 (IH, d), 6.89 – 6.78 (3H,s m), 5.17 (IH, d), 4.48 (IH, m), 4.24 – 4.11 (5H, m), 3.90 – 3.81 (2H, m), 3.72 (IH, td), 3.61
(IH, dd), 2.16 (IH, m), 1.94 (IH, m), 1.55 (3H, t), 1.29 (3H, d).
LC (Method A) rt = 12.02 min
LC (Method B) rt = 11.12 min
Chiral SFC (method B) rt = 5.10 min o M.p. = 175 0C
PATENT
WO 2011061527
http://www.google.com/patents/WO2011061527A1?cl=en
Intermediate 12
( 1 R,2S)-2-amino- 1 -(2,3 -dihydrobenzo b [ 1 ,41dioxin-6-yl)propan- 1 -ol hydrochloride. (12)

5-6 N HC1 in 2-propanol (8 mL, 40-48 mmol) was added to tert-butyl (lR,2S)-l-(2,3- dihydrobenzo[b][l,4]dioxin-6-yl)-l-hydroxypropan-2-ylcarbamate (I2a) (3.1 g, 10.02 mmol) in ethyl acetate (40 mL) at 40°C and stirred for 3 hours. The reaction mixture was allowed to reach r.t. and was concentrated by evaporation. Ether was added and the salt was filtered off and washed with ether. The salt was found to be hygroscopic. Yield 2.10 g (85%)
APCI-MS: m/z 210 [MH+-HC1]
1H-NMR (300 MHz, DMSO-^): δ 8.01 (brs, 3H), 6.87-6.76 (m, 3H), 5.93 (brd, 1H), 4.79 (brt, 1H), 4.22 (s, 4H), 3.32 (brm, 1H), 0.94 (d, 3H).
tert-butyl (1R,2S)- 1 -(2,3-dihvdrobenzorbl Γ 1 ,41dioxin-6-yl)- 1 -hvdroxypropan-2-ylcarbamate.

The diastereoselective catalytic Meerwein-Ponndorf-Verley reduction was made by the method described by Jingjun Yin et. al. J. Org. Chem. 2006, 71, 840-843.
(S)-tert-butyl 1 -(2,3-dihydrobenzo[b] [ 1 ,4]dioxin-6-yl)- 1 -oxopropan-2-ylcarbamate (I2b) (3.76 g, 12.23 mmol), aluminium isopropoxide (0.5 g, 2.45 mmol) and 2-propanol (12 mL, 157.75 mmol) in toluene (22 mL) were stirred at 50°C under argon for 16 hours. The reaction mixture was poured into 1M HC1 (150 mL) and the mixture was extracted with ethyl acetate (250 mL). The organic phase was washed with water (2×50 mL) and brine (100 mL), dried over Na2SC”4, filtered and concentrated. The crude product was purified by flash- chromatography on silica using ethyl acetate/hexane (1/2) as eluent. Fractions containing product were combined. Solvent was removed by evaporation to give the desired product as a colourless solid. Yield 3.19 g (84%) APCI-MS: m/z 236, 210, 192 [MH -tBu-18, MH -BOC, MH -BOC- 18]
1H NMR (300 MHz, DMSO-^): δ 6.80-6.70 (m, 3H), 6.51 (d, IH), 5.17 (d, IH), 4.36 (t, IH),
4.19 (s, 4H), 3.49 (m, IH), 1.31 (s, 9H), 0.93 (d, 3H).
(S)-tert-butyl 1 -(2,3-dihydrobenzo[bl [ 1 ,41dioxin-6-yD- 1 -oxopropan-2-ylcarbamate. (I2b)

A suspension of (S)-tert-butyl l-(methoxy(methyl)amino)-l-oxopropan-2-ylcarbamate (3 g, 12.92 mmol) in THF (30 mL) was placed under a protective atmosphere of argon and cooled down to -15 to -20°C. Isopropylmagnesium chloride, 2M in THF (6.5 mL, 13.00 mmol), was added keeping the temperature below -10°C. The temperature was allowed to reach 0°C. A freshly prepared solution of (2,3-dihydrobenzo[b][l,4]dioxin-6-yl)magnesium bromide, 0.7M in THF (20 mL, 14.00 mmol) was added. The temperature was allowed to reach r.t. overnight. The reaction mixture was poured into ice cooled IN HC1 (300 mL). TBME (300 mL) was added and the mixture was transferred to a separation funnel. The water phase was back extracted with TBME (200 mL). The ether phases were washed with water, brine and dried (Na2S04). The crude product was purified by flash chromatography using TBME /Heptane 1/2 as eluent. Fractions containing the product were combined and solvents were removed by evaporation to give the subtitle compound as a slightly yellow sticky oil/gum. Yield 3.76g
(95%)
APCI-MS: m/z 208 [MH+ – BOC]
1H NMR (300 MHz, DMSO-^): δ 7.50 (dd, IH), 7.46 (d, IH), 7.24 (d, IH), 6.97 (d, IH), 4.97 (m, IH), 4.30 (m, 4H), 1.36 (s, 9H), 1.19 (d, 3H).
Intermediate 13
(lR,2S)-2-amino-l-(4H-benzo[dl[l,31dioxin-7- l)propan-l-ol hydrochloride (13)

Tert-butyl ( 1 R,2S)- 1 -(4H-benzo[d] [ 1 ,3]dioxin-7-yl)- 1 -hydroxypropan-2-ylcarbamate (I3b) (403 mg, 1.30 mmol) was dissolved in ethyl acetate (5 mL) and 5-6 N HC1 solution in 2- propanol (1.5 mL, 7.5-9 mmol) was added. The mixture was stirred at 50 °C for 1.5 hours. The solvents was removed by evaporation. The residual sticky gum was treated with ethyl acetate and evaporated again to give a solid material that was suspended in acetonitrile and stirred for a few minutes. The solid colourless salt was collected by filtration and was found to be somewhat hygroscopic. The salt was quickly transferred to a dessicator and dried under reduced pressure. Yield 293 mg (92%)
APCI-MS: m/z 210 [MH+ -HC1]
1H NMR (300 MHz, DMSO-^) δ 8.07 (3H, s), 7.05 (IH, d), 6.92 (IH, dd), 6.85 (IH, d), 6.03 (IH, d), 5.25 (2H, s), 4.87 (3H, m), 3.42 – 3.29 (IH, m), 0.94 (3H, d).
(4S.5R -5-(4H-benzordiri.31dioxin-7-vn- -methyloxazolidin-2-one (I3a

A mixture of (lR,2S)-2-amino-l-(4H-benzo[d][l,3]dioxin-7-yl)propan-l-ol hydrochloride (I3b) (120 mg, 0.49 mmol), DIEA (0.100 mL, 0.59 mmol) and CDI (90 mg, 0.56 mmol) in THF (2 mL) was stirred at r.t. for 2 hours. The reaction mixture was concentrated by evaporation and the residual material was partitioned between ethyl acetate and water. The organic phase was washed with 10% NaHS04, dried over MgS04, filtered and evaporated. The crude product was analysed by LC/MS and was considered pure enough for further analysis by NMR. Yield 66 mg (57%)
The relative cis conformation of the product was confirmed by comparing the observed 1H- NMR with the literature values reported for similar cyclised norephedrine (Org. Lett. 2005 (07), 13, 2755-2758 and Terahedron Assym. 1993, (4), 12, 2513-2516). In a 2D NOESY experiment a strong NOE cross-peak was observed for the doublet at 5.64 with the multiplet at 4.19 ppm. This also confirmed the relative czs-conformation.
APCI-MS: m/z 236 [MH+]
1H NMR (400 MHz, CDC13) δ 6.99 (d, J= 8.0 Hz, IH), 6.88 (dd, J= 8.0, 1.4 Hz, IH), 6.83 (s, IH), 5.81 (brs,lH), 5.64 (d, J= 8.0 Hz, IH), 5.26 (s, 2H), 4.91 (s, 2H), 4.19 (m, IH), 0.85 (d, J = 6.4 Hz, 3H). Tert-butyl ( 1 R,2S)- 1 -(4H-benzord1 Γ 1 ,31dioxin-7-yl)- 1 -hvdroxypropan-2-ylcarbamate (I3b)

A mixture (S)-tert-butyl l-(4H-benzo[d][l,3]dioxin-7-yl)-l-oxopropan-2-ylcarbamate (I3c) (680 mg, 2.21 mmol), triisopropoxyaluminum (140 mg, 0.69 mmol) and propan-2-ol (3 mL, 38.9 mmol) in toluene (3 mL) was stirred at 65 °C for 15 hours. The reaction mixture was allowed to cool down, poured into 1M HC1 (50 mL) and extracted with ethyl acetate (2×50 mL). The organic phase was washed with water, brine, dried over MgS04, filtered and solvents were removed by evaporation to afford a colourless solid. The crude product was purified by flash chromatography, (solvent A = Heptane, solvent B = EtOAc + 10% MeOH. A gradient of 10%B to 50%B in A was used). The obtained product was crystallised from DCM / heptane to afford the subtitle compound as colourless needles. Yield 414 mg (60%)
APCI-MS: m/z 210 [MH+ -BOC]
1H NMR (400 MHz, DMSO- ¾ δ 6.97 (1H, d), 6.88 (1H, d), 6.77 (1H, s), 6.56 (1H, d), 5.27 (1H, d), 5.22 (2H, s), 4.83 (2H, s), 4.44 (1H, t), 3.53 (1H, m), 1.32 (9H, s), 0.93 (3H, d). (S)-Tert-butyl 1 -(4H-benzord1 Γ 1 ,31dioxin-7-vD- 1 -oxopropan-2-ylcarbamate (I3c)

7-Bromo-4H-benzo[d][l,3]dioxine (1 g, 4.65 mmol) was dissolved in THF (5 mL) and added to magnesium (0.113 g, 4.65 mmol) under a protective atmosphere of argon. One small iodine crystal was added. The coloured solution was heated with an heat gun in short periods to initiate the Grignard formation. When the iodine colour vanished the reaction was allowed to proceed at r.t. for 1.5 hours.
In a separate reaction tube (S)-tert-butyl l-(methoxy(methyl)amino)-l-oxopropan-2- ylcarbamate (1 g, 4.31 mmol) was suspended in THF (5 mL) and cooled in an ice/acetone bath to below -5 °C. Isopropylmagnesium chloride, 2M solution in THF (2.5 mL, 5.00 mmol) was slowly added to form a solution. To this solution was added the above freshly prepared Grignard reagent. The mixture was allowed to reach r.t. and stirred for 4 hours. The reaction mixture was slowly poured into ice-cold 150 mL 1M HC1. Ethyl acetate (150 mL) was added and the mixture was stirred for a few minutes and transferred to a separation funnel. The organic phase was washed with water and brine, dried over MgS04, filtered and concentrated. The obtained crude product was further purified by flash chromatography using a prepacked 70g silica column with a gradient of 10% TBME to 40% TBME in heptane as eluent. The subtitle compound was obtained as a colourless solid. Yield 790 mg (59%>)
APCI-MS: m/z 208 [MH+ -BOC]
1H NMR (400 MHz, DMSO-^) δ 7.53 (IH, dd), 7.39 (IH, s), 7.30 (IH, d), 7.22 (IH, d), 5.30 (2H, s), 4.98 (IH, m), 4.95 (2H, s), 1.35 (9H, s), 1.20 (3H, d).
Preparation 4
3-(5-([(lR,2S)-2-[(2,2-difluoropropanoyl)aminol-l-(2,3-dihydro-l,4-benzodioxin-6- yl)propyl]oxy| – 1 H-indazol- 1 -yl)-N-[(3R)-tetrahydrofuran-3-yllbenzamide

TEA (2.0 g, 20.65 mmol) was added to a mixture of 3-(5-((lR,2S)-2-(2,2- difluoropropanamido)- 1 -(2,3-dihydrobenzo[b] [ 1 ,4]dioxin-6-yl)propoxy)-l H-indazol-1 – yl)benzoic acid (14) (3.6 g, 6.70 mmol), (R)-tetrahydrofuran-3 -amine hydrochloride (0.99 g, 8.0 mmol) and HBTU (2.65 g, 6.99 mmol) in DCM (15 mL). The reaction was stirred at r.t. for 3h, then quenched by addition of a mixture of water and ethyl acetate. The mixture was shaken and the organic layer was collected. The water phase was extracted twice with ethyl acetate. The combined organic layers were washed with a small portion of water and dried over magnesium sulphate. The product was purified by flash chromatography (silica, eluent: a gradient of ethyl acetate in heptane). The residue was crystallized by dissolving in refluxing acetonitrile (50 mL) and then allowing to cool to r.t. over night. The solid was collected by filtration, washed with a small volume of acetonitrile and dried at 40°C in vaccum to give the title compound (2.5 g, 61%).
APCI-MS: m/z 607 [MH+]
1H NMR (400 MHz, DMSO-d6) δ 8.71 (IH, d), 8.65 (IH, d), 8.24 (IH, s), 8.18 (IH, s), 7.90 – 7.84 (2H, m), 7.77 (IH, d), 7.65 (IH, t), 7.21 (IH, dd), 7.13 (IH, d), 6.89 – 6.78 (3H, m), 5.17 (IH, d), 4.48 (IH, m), 4.23 – 4.10 (5H, m), 3.89 – 3.82 (2H, m), 3.72 (IH, td), 3.61 (IH, dd), 2.16 (IH, m), 1.94 (IH, m), 1.55 (3H, t), 1.29 (3H, d).
LC (method A) rt = 12.03 min
LC (method B) rt = 11.13 min
Chiral SFC (method B) rt = 4.71 min
M.p. = 177 °C
| Patent ID | Date | Patent Title |
|---|---|---|
| US2015080434 | 2015-03-19 | PHENYL AND BENZODIOXINYL SUBSTITUTED INDAZOLES DERIVATIVES |
| US8916600 | 2014-12-23 | Phenyl and benzodioxinyl substituted indazoles derivatives |
| US8211930 | 2012-07-03 | Phenyl and Benzodioxinyl Substituted Indazoles Derivatives |
REFERENCES
https://www.astrazeneca.com/content/dam/az/press-releases/2014/Q2/Pipeline-table.pdf
////////AZD 7594, AZ13189620, AZD-7594 , phase 2, astrazeneca, 1196509-60-0
c21cc(ccc1n(nc2)c3cc(ccc3)C(=O)NC4COCC4)O[C@H](c5cc6c(cc5)OCCO6)[C@@H](NC(=O)C(F)(F)C)C
CC(C(C1=CC2=C(C=C1)OCCO2)OC3=CC4=C(C=C3)N(N=C4)C5=CC=CC(=C5)C(=O)NC6CCOC6)NC(=O)C(C)(F)F
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO …..FOR BLOG HOME CLICK HERE
Join me on twitter
Join me on google plus Googleplus
Googleplus amcrasto@gmail.com
P.S
THE VIEWS EXPRESSED ARE MY PERSONAL AND IN NO-WAY SUGGEST THE VIEWS OF THE PROFESSIONAL BODY OR THE COMPANY THAT I REPRESENT, amcrasto@gmail.com, +91 9323115463 India.
I , Dr A.M.Crasto is writing this blog to share the knowledge/views, after reading Scientific Journals/Articles/News Articles/Wikipedia. My views/comments are based on the results /conclusions by the authors(researchers). I do mention either the link or reference of the article(s) in my blog and hope those interested can read for details. I am briefly summarising the remarks or conclusions of the authors (researchers). If one believe that their intellectual property right /copyright is infringed by any content on this blog, please contact or leave message at below email address amcrasto@gmail.com. It will be removed ASAP
