
Home » ANTI FUNGAL
Category Archives: ANTI FUNGAL
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Liranaftate
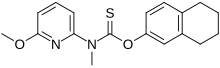
Liranaftate
リラナフタート
88678-31-3
(6-Methoxy-2-pyridinyl)methylcarbamothioic Acid O-(5,6,7,8-Tetrahydro-2-naphthalenyl) Ester
O-(5,6,7,8-Tetrahydronaphthalen-2-yl) (6-methoxypyridin-2-yl)methylcarbamothioate
Zefnart;Piritetrate;M-732
лиранафтат
ليرانافتات
利拉萘酯
| Formula | C18H20N2O2S |
|---|---|
| CAS | 88678-31-3 |
| Mol weight | 328.4286 |
| Efficacy | Antifungal, Ergosterol biosynthesis inhibitor |
|---|---|
| Comment | Thiocarbamate |
Liranaftate (trade name Zefnart) is a topical antifungal drug.[1] It is used as a 2% cream used to treat tinea pedis (athlete’s foot), tinea corporis (ringworm), and tinea cruris (jock itch).[2] It was approved for use in Japan in August 2000.[3][4]
Liranaftate works by inhibiting the fungal enzyme squalene epoxidase that is necessary for the fungus to synthesize sterols which are essential for cell membrane integrity.[5]
SYN
IN 2010MU02699
PAPER
Journal of Chemical and Pharmaceutical Research (2013), 5(11), 219-222,
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007010744
Conventionally, 0-aryl N- (6-alkoxy-2-pyridyl) -N-alkylthio-force rubamate has generally been produced by a method using thiophosgen. For example, in Patent Document 1, 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N- represented by the following reaction formula 0 or ii) A method for producing methylthiolbamate (4) is disclosed.
(Example 1)
1) Sodium 5, 6, 7, 8-Tetrahydro-2-naphthoside synthesis
[hua 6]
,She
To methanol (10 ml), 0.54 g (10.0 mmol) of sodium methoxide was added, and the mixture was stirred at room temperature. There, 1.50 g (10.0 mmol) of 5,6,7,8-tetrahydro-2-naphthol was added and he stirred for 1 hour at room temperature. The solvent was distilled off under reduced pressure to obtain 3.75 g ( q uant.) Of white powder. I left it overnight in a desiccator.
2) 2- [Ν- (1-imidazolithiocarbol) -Ν’-methyl] amino-6-methoxypyridin compound
[hua 7]
To ethyl acetate (30 ml), 2.07 g (15.0 mmol) of 6-methoxy-2-methylaminoviridin and 2.67 g (15.0 mmol) of 1,1, -thiocarboldiimidazole were added, and the mixture was heated under reflux for 2 hours. After allowing to cool, the solvent was distilled off under reduced pressure to obtain 3.70 g of brown oil. (Yield 99.3%). If necessary, further purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1) to obtain pale yellow crystals.
Melting point: 58.0~60.0°C
NMR(CDC1 3 ) δ ppm:3.86(3H,s), 3.87(3H,s), 6.38 (lH’dd, J=7.5Hz, 0.7Hz), 6.61 (1H
,dd, J=8.3Hz, 0.7Hz), 6.82 (lH,t, J=1.0Hz) , 7.03 (lH,t, J=1.0Hz) , 7.46 (lH’dd, J= 8.3Hz, 7.5Hz), 7.72 (lH,t, J=1.0Hz)
IR(KBr)cm_1: 1604, 1590, 1571, 1465, 1359, 1303, 1120, 1013, 986, 822, 798 MS m/z: 248(M+)
3) Synthesis of 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) -N- (6-methoxy-2-pyridyl) -N-methylthiocarbamate
Dissolve 2- [N- (1-imidazolithiocarbol) -N-methyl] amino-6-methoxypyridin 250 mg (1.0 mmol) in N, N-dimethylformamide (4 ml), and then dissolve. At room temperature, Natrium 5, 6, 7, 8-tetrahydro-2-naphthoside 360 mg (2.0 mmol) was added. -After stirring at room temperature, the reaction solution was extracted with ethyl acetate (10 mlx2), and the insoluble material was filtered off on the way. The organic layer was washed with saturated brine, dried over magnesium sulfate, filtered off magnesium sulfate, and the solvent was distilled off under reduced pressure. Purification by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) gave the title compound 266.6 mg (yield 81.3%).
Melting point: 99~100°C
NMR(CDCl 3) δ ppm:1.77(4H,bs), 2.75(4H,bs), 3.75(3H,s), 3.93(3H,s), 6.65(lH,d, J
=8.0Hz), 6.78-7.08(4H,m), 7.64(lH,t,J=8.0Hz)
IR(KBr) cm_1 : 1603, 1460, 1413, 1369, 1325, 1262, 1175, 1035, 808, 785
MS m/z: 328(M+)
(Example 2)
0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N-methylthio force Rubamate synthesis
[Chemical 9]
1.34 g (33.6 mmol) of 60% sodium hydride was added to N, N-dimethylformamide (20 ml), followed by the addition of 5, 6, 7, 8-tetrahydro-2-naphthol 4.65 g (30.5 mmol). After gas generation is complete, add 2- [N- (1-imidazolthiocarbonyl) -N-methyl] amino-6-methoxypyridin 7.45 g (30.0 mmol) and zinc chloride 2.05 g (15.0 mmol). rice field. After heating and stirring at 60 ° C for 3 hours and allowing to cool, the reaction solution was extracted with ethyl acetate (150 mlx2), and the insoluble material was filtered off on the way. The organic layer is washed with saturated brine, dried over magnesium sulfate, and filtered through magnesium sulfate.
Separately, the solvent was distilled off under reduced pressure. The obtained crystals were purified by one of the following methods.
[0028] A) Purification was performed by silica gel column chromatography (eco-gel C 200, hexane: ethyl silicate = 10: 1) to obtain 9.80 g of the indicated compound (yield 99.5%).
B) Suspended in hexane (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 9.65 g of crystals. Further, the mixture was suspended in methanol (10 ml), stirred for 30 minutes, and then the crystals were collected by filtration to obtain 8.62 g (yield 87.5%) of the indicated compound.
The physics and physics data of the obtained compound were consistent with the compounds obtained in the examples.
(Example 3)
1) Synthesis of 2- [N- [1-2 (1H) -pyridonylthiocarbol] -N-methyl] amino-6-methoxypyridine
[Chemical 10]
OMe
Add 6-methoxy-2-methylaminoviridin 690 mg (5.0 mmol) and 1, 1, -thiocarbol-di-2 (1H) -pyridone 1.16 g (5.0 mmol) to ethyl acetate (15 ml). Heated and refluxed for 1 hour. After allowing to cool, the solvent was distilled off under reduced pressure, and purification was performed by silica gel column chromatography (hexane: ethyl acetate = 10: 1)! ヽ, 297.4 mg of brown oil was obtained. (Yield 21.6%).
NMR(CDC1 3 ) δ ppm:3.77(3H,s), 3.93(3H,s), 6.66 (lH’dd, J=8.0Hz, 0.7Hz), 7.07 ( lH,d, J=8.0Hz), 7.14 (lH,d, J=7.5Hz) , 7.25 (lH’dd, J=8.0Hz, 4.0Hz) , 7.62 (lH’dd , J=8.0Hz, 7.5Hz), 7.78 (lH’dd, J=2.0Hz, 0.7Hz) , 8.43 (lH’dd, J=4.0Hz, 0.7Hz)
MS m/z: 275(M+)
[0031] 2) Synthesis of 0- (5, 6, 7, 8-tetrahydro-2-naphthyl) N- (6-methoxy-2-pyridyl) -N-methylthiocarbamate
[Chemical 11]
OMe
N, N-dimethylformamide (2 ml), 2- [N- [1-2 (1H) -pyridonylthiocarbol] –N-methyl] amino-6-methoxypyridin 297 mg (1.08 mmol) and sodium 5 , 6, 7, 8-Tetrahydro-2-naphthoside 390 mg (2.16 mmol) was added and stirred overnight at room temperature. The reaction mixture was extracted with ethyl acetate (50 mlx2), the organic layer was washed with saturated brine, dried over magnesium sulfate, magnesium sulfate was filtered off, and the solvent was distilled off under reduced pressure. The obtained crystals were purified by silica gel column chromatography (eco-gel C-200, hexane: ethyl acetate = 10: 1) to obtain the title compound 288.2 mg (81.4%).
SYN
CN 104725302
| Liranafate is a new-generation antifungal drug, a squalene cyclooxygenase inhibitor and a cell wall synthesis inhibitor, with the chemical name of 6-methoxy-2-N-methyl-pyridylamino-thio Formic acid-(5,6,7,8-tetrahydro)-β-naphthyl ester. A new type of antifungal drug jointly developed by Tosoh Corporation of Japan and Zenyaku Kogyo Corporation was first listed in Japan by Torii Corporation in August 2000. The antifungal drug exerts antifungal activity by inhibiting the squalene epoxidation reaction of fungal cells and inhibiting the synthesis of ergosterol, a component of cell membranes. effect is particularly evident. Today, with the increasing concern of the world about environmental pollution, the development of new green and effective drug synthesis methods is an important task faced by the research of drug synthesis. In recent years, room temperature ionic liquids have been widely used in various organic synthesis reactions as a new type of environmentally friendly reaction media. Compared with traditional organic solvents, ionic liquids have many advantages, such as extremely low vapor pressure, non-flammability, good thermal stability and recyclability. |
| At present, the main synthetic route of liranaftate is as follows: |
| |
| Among the four synthetic routes, the pyridine derivative intermediates of routes C and D need to be prepared through multi-step reactions, the routes are long, the steps are cumbersome, the actual operation is cumbersome, the cost is high, and they are not suitable for industrialized large-scale production. Although route A has simple steps, the yield of pyridine derivatives is low. Each intermediate structure in route B is relatively simple and easy to prepare, but this route uses 6-methoxy-2-methylaminopyridine and 5,6,7,8-tetrahydro-2-naphthoxysulfuryl chloride as raw materials to synthesize the In the process of lanaphthalate, isopropanol-water is used as the reaction medium, and the experiment shows that with the progress of the reaction, the reaction solution becomes viscous, and the reaction is difficult to complete. |
| Example 1 |
| (1) Ionic liquid [bmim]BF 4 Synthesis |
| |
| Add N-methylimidazole (14.8g, 0.18mol) and trichloroethane (80mL) to a dry 250mL three-neck flask, stir to make the mixture uniform, add 20.4mL of freshly distilled n-bromine to the dropping funnel Butane (26.03g, 0.19mol) was added dropwise for about 30min, and the reaction was refluxed for 4-5h (the reflux temperature was about 78±1℃). With the progress of the reaction, the reaction solution changed from colorless and transparent to white turbidity, light yellow turbidity, and the color gradually became darker until brownish red. After the reaction is completed, the liquids are separated into layers, the upper layer is lighter in color, which is the trichloroethane layer, and the lower layer is darker in color (brown red), which is the ionic liquid [bmim]Br layer. The prepared ionic liquid [bmim]Br and trichloroethane were separated, and the ionic liquid [bmim]Br was washed twice with trichloroethane, and then the trichloroethane in the ionic liquid [bmim]Br was washed with a water pump. The alkane was pumped away until the ionic liquid [bmim]Br liquid was no longer turbid, and then dried in a vacuum drying oven at 90 °C for 10-12 h to obtain relatively pure ionic liquid [bmim]Br. |
| |
| Then prepare 0.03mol NaBF 4 of aqueous solution. Add 6.58g (about 0.03mol) ionic liquid [bmim]Br and 5-10mL water to a 100mL round-bottomed single diameter flask, stir, ice-water bath, and dropwise add NaBF 4 The solution (completed dropwise addition in about 5min), continue to stir for 10-20min, the solution is yellow and transparent, pour it into a separatory funnel, extract twice with dichloromethane, combine the dichloromethane layers, and wash the dichloromethane layer 2 with 50 mL of water times, and then the dichloromethane layer was washed with anhydrous MgSO 4 Dry, filter, evaporate the dichloromethane under normal pressure in a water bath (50-52°C), and dry the remaining dark yellow viscous liquid in a vacuum drying oven at 90°C for 10-12h to obtain the ionic liquid [bmim]BF 4 。 |
| |
| (2) Synthesis of 6-methoxy-2-chloropyridine 2 |
| 2,6-dichloropyridine (10g, 0.068mol) and sodium methoxide (24.5g, 0.136mol) were put into the reaction flask, heated under reflux for 4-5h, and the reaction was completed by TLC (ethyl acetate: petroleum ether=1 : 15), concentrated to remove methanol, added 100 mL of water, extracted with ethyl acetate, combined the organic phases, washed with saturated brine, dried, filtered, and the filtrate was concentrated to obtain 9 g of a crude colorless oily product with a yield of 92.5%. used for the next reaction. |
| (3) Synthesis of 6-methoxy-2-methylaminopyridine 3 |
| Take 6-methoxy-2-chloropyridine 2 (9g, 0.127mol), cuprous chloride (1.72g, 0.0017mol) and methylamine aqueous solution (29mL, mass concentration is 25%-30%) and add it to the autoclave , sealed and heated to 120 °C for 7 h, the reaction was stopped, ethyl acetate was added for extraction, the organic phases were combined, washed with saturated brine, dried, and the filtrate was concentrated to obtain 6.18 g of brown oil, the yield was 71.2%, and the HPLC purity was 98% . |
| (4) Synthesis of 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride 4 |
| Mix 50 mL of ethyl acetate, thiophosgene (4.25 mL, 0.056 mol) and 5,6,7,8-tetrahydro-2-naphthol (6.3 g, 0.0425 mol), and cool it in an ice-salt bath to below 0 °C. Add 10 mL of potassium carbonate (3 g, 0.022 mol) solution, continue to stir the reaction after the dropwise addition, and check by TLC (developing solvent: petroleum ether) that the reaction is complete, add 100 mL of water, extract with ethyl acetate, wash the organic phase with saturated brine, Dry, filter, and concentrate the filtrate to obtain 8.7 g of yellow oil with a yield of 90.4%, which can be directly used in the next reaction without purification. |
| (5) Synthesis of Liranaftate 1 |
| The prepared ionic liquid [bmim]BF 4 (100mL), 6-methoxy-2-methylaminopyridine 3 (5.7g, 0.0413mol) and potassium carbonate (5.7g, 0.0413mol) were mixed, cooled with ice water, and slowly added dropwise 5,6,7,8 -Tetrahydro-2-naphthyloxysulfuryl chloride 4 (8.7g, 0.0385mol) was added dropwise for 4h, slowly added 150mL of water under full stirring, continued to stir for 20min, filtered, washed with deionized water to obtain 12.2g of crude product, collected The yield was 96.81%, and acetone was recrystallized to obtain 11 g of white crystalline powder, the yield was 90%, and the HPLC purity was 99.7%. mp: 98.8-99.5°C, IR (2973cm -1 , 2930cm -1 , 2852cm -1 , 1416cm -1 , 1264cm -1 , 1037cm -1 ), 1 HNMR: 1.8 (m, 4H); 6.68(d, 1H) ;6.86(dd,1H);3.78(s,3H);3.98(s,3H);6.68(d,1H);6.86(dd,1H);7.05(d,1H);7.10(d.1H); 7.65 (dd, 1H), MS (m/z: 328, 181, 165, 108). |
| Example 2 |
| Under the same conditions, the ionic liquid 1-n-butyl-3-methylimidazolium tetrafluoroborate ([bmim]BF 4 ), N-ethylpyridine tetrafluoroborate ([EPy]BF 4 ), 1-n-butyl-3-methylimidazolium hexafluorophosphate ([bmim]PF 6 ), 1-hydroxyethyl-2,3-dimethylimidazolium chloride (LOH), 1-cyanopropyl-3-methylimidazolium chloride (LCN), 1-carboxyethyl-3-methylimidazole Chloride salt (LOOH), [Hnmp]HSO 4 The effects of and [bmim]OH on the synthesis of liranaftate are shown in Table 1. The results show that different ionic liquids have little effect on the yield of the synthesis and the yields are relatively high. |
| Table 1 Effects of different ionic liquids on the reaction yield |
| ionic liquidYield/%[bmim] BF 496.81[EPy]BF 496.83[bmim]PF 696.82LOH96.75LCN96.67LOOH96.05[Hnmp]HSO 496.06[bmim]OH95.98 |
| Example 3 |
| Whether the reaction medium used can be recovered and reused is an important content of “green chemistry”. This example specifically examines the reuse of ionic liquid for synthesizing liranaftate. After 5 times of use of ionic liquid, the product yield It just started to decrease, which shows that the ionic liquid can be recovered and reused effectively, and the reuse performance is good. It is a recyclable green solvent. |
SYN
| Comparative Example 1: |
| Put 10 g of 2,6-dichloropyridine, 100 ml of methanol, and 15 g of sodium methoxide into a reaction flask, heat under reflux for about 4 to 5 hours, concentrate to remove methanol, add 150 ml of water, extract with ethyl acetate, and concentrate under reduced pressure to remove ethyl acetate. 6-Methoxy2-chloropyridine was obtained as a colorless oil. |
| 9 g of 6-methoxy 2-chloropyridine, 1.72 g of cuprous chloride, and 29 ml of 30% methylamine aqueous solution were put into the reaction flask, heated and added with a mass fraction of 11.6 g of cuprous chloride, and the temperature was kept at 120 ° C for the reaction 8h, extracted three times with 150 ml of ethyl acetate, washed with saturated brine, concentrated under reduced pressure to remove the ethyl acetate to obtain 6.18 g of 6-methoxy-2-methylaminopyridine as a brown oily product. The two-step yield was 71.2%. |
| 50ml of carbon tetrachloride, 4.25g of thiophosgene, 6.3g of 5,6,7,8-tetrahydro-2-naphthol were added to the reaction flask, the ice-salt bath was lowered to below 0°C, and 10ml of 3g potassium carbonate aqueous solution was added dropwise. , Continue the reaction at 0°C after the dropwise addition, and detect by TLC (developing solvent: petroleum ether) after the reaction is completed, separate the organic phase, wash three times with saturated brine, and concentrate under reduced pressure to obtain red oily products 5, 6, 7 , 8.7g of 8-tetrahydro-2-naphthyloxysulfuryl chloride was directly used in the next reaction. |
| 100ml of acetone, 5.7g of 6-methoxy-2-methylaminopyridine and 5.7g of potassium carbonate were added to the reaction flask, cooled with ice water, and 5,6,7,8-tetrahydro-2-naphthyloxysulfuryl chloride was added dropwise 8.7g, continue to stir and react for 4h after dropping, add 150ml of water, continue to stir for 30min, and filter to obtain the crude product. The crude product was recrystallized with acetone to obtain 11 g of off-white crystalline powder. The weight yield was 174.6% based on 5,6,7,8-tetrahydro-2-naphthol. The maximum single impurity content determined by HPLC was 1.5%, which did not meet the requirements of the Pharmacopoeia. |
SYN
CN 106632018
| Example 1 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours; |
| Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, and the consumption of purified water was 25.0 g each time; |
| Drying: put the wet product into a drying box, control the temperature to 45 ℃ and dry for 4 hours, to obtain 24 g of the crude product of lira naphthate; |
| The synthesis yield is 81%; |
| (2) preparation of Liranaftate fine product: |
| Impurity removal: put 23g of Liranaftate crude product and 115g of absolute ethanol into the reaction flask, add 1.38g of medicinal charcoal, decolorize at 55°C under temperature control, remove impurities for 30 minutes, filter, transfer the filtrate to the reaction flask, control the temperature Crystallize at 55°C, centrifuge, dry, pulverize, and pack to obtain 22g of Lira naphthate fine product. |
| The purification yield was 92%. |
| Example 2 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 500g of absolute ethanol was added to the reaction flask, 25g of 2-methoxy-6-methylaminopyridine, 17.6g of anhydrous sodium carbonate and 62.6g of purified water were added to the reaction flask in turn, stirred for 30 minutes, and slowly added 2-(5,6,7,8-tetrahydronaphthyloxy) chlorothioformate 37.6g, added in 2.5 hours; |
| Reaction: control the temperature at 25°C for 2.5 hours, add 250 g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 50 g each time; |
| Drying: put the wet product into a drying box, control the temperature to 55 ℃ and dry for 4 hours to obtain 49 g of the crude product of lira naphthate; |
| The synthesis yield is 82%; |
| (2) preparation of Liranaftate fine product: |
| Impurity removal: put 49g of Liranaftate crude product and 245g of absolute ethanol into the reaction flask, add 2.9g of medicinal charcoal, decolorize at 55~65 ℃ of temperature, remove impurities for 30 minutes, filter, and transfer the filtrate to the reaction flask, The temperature was controlled at 65°C for crystallization, centrifugation, drying, pulverization, and packaging to obtain 45g of fine lanaftate. |
| The purification yield was 92%. |
| Example 3 |
| A preparation method of liranaftate of the present invention comprises the following steps: |
| (1) preparation of Liranaftate crude product: |
| Feeding: 250g of absolute ethanol was added to the reaction flask, 12.5g of 2-methoxy-6-methylaminopyridine, 8.8g of anhydrous sodium carbonate and 31.3g of purified water were added to the reaction flask in turn, stirred for 30 minutes, slowly 18.8 g of 2-(5,6,7,8-tetrahydronaphthyloxy) thioformate chloride was added, and the addition was completed in 2 hours; |
| Reaction: control the temperature at 20°C for 2 hours, add 125.0g of purified water, and stir for 30 minutes; |
| Suction filtration: the reaction solution was suction filtered, and the filter cake was washed three times with purified water, 25.0 g each time; |
| Drying: put the wet product into a drying oven, control the temperature to 45~55 ℃ and dry for 4 hours, to obtain the crude product, 23.3 g of the crude liranaftate; |
| The synthesis yield is 82%; |
| (2) preparation of Liranaftate fine product: |
| Removal of impurities: 140g of absolute ethanol was added to the reaction flask, 23.3g of crude liranaftate was added, the temperature was controlled at 50°C and stirred for 30 minutes, 1.5g of medicinal charcoal was added, the temperature was controlled at 60°C for decolorization for 30 minutes, filtered, and the temperature was controlled Crystallize at 60°C, centrifuge, dry, pulverize, and package to obtain 23g of Lira naphthate fines. |
| The purification yield was 92%. |
SYN
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Koga H, Nanjoh Y, Makimura K, Tsuboi R (2009). “In vitro antifungal activities of luliconazole, a new topical imidazole”. Medical Mycology. 47 (6): 640–7. doi:10.1080/13693780802541518. PMID 19115136.
- ^ “Torii Pharmaceutical to Launch Antifungal Agent for External Use, “ZEFNART SOLUTION 2%”, in Japan” (Press release). Torii Pharmaceutical Co. Retrieved June 27, 2021.
- ^ “Liranaftate”. ncats.io. Retrieved June 27, 2021.
- ^ “Liranaftate”. Adis Insight. Retrieved June 27, 2021.
- ^ “Liranaftate”. targetmol.com. Retrieved June 27, 2021.
///////////////////Liranaftate , リラナフタート , Zefnart, Piritetrate, M-732, лиранафтат , ليرانافتات , 利拉萘酯 , ANTIFUNGAL, JAPAN 2000

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
AMOROLFINE
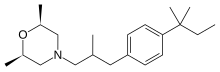
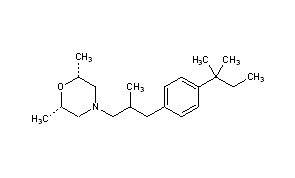
AMOROLFINE(2R,6S)-2,6-Dimethyl-4-{2-methyl-3-[4-(2-methyl-2-butanyl)phenyl]propyl}morpholine
(2R,6S)-2,6-Dimethyl-4-{2-methyl-3-[4-(2-methylbutan-2-yl)phenyl]propyl}morpholine
78613-35-1[RN]
(±)-cis-2,6-Dimethyl-4-(2-methyl-3-(p-tert-pentylphenyl)propyl)morpholine
Ro 14-4767-002
аморолфин , أمورولفين ,阿莫罗芬 ,
Title: Amorolfine
CAS Registry Number: 78613-35-1
CAS Name:cis-4-[3-[4-(1,1-Dimethylpropyl)phenyl]-2-methylpropyl]-2,6-dimethylmorpholine
Additional Names:cis-4-[3-(4-tert-amylphenyl)-2-methylpropyl]-2,6-dimethylmorpholine; (±)-cis-2,6-dimethyl-4-[2-methyl-3-(p-tert-pentylphenyl)propyl]morpholine
Manufacturers’ Codes: Ro-14-4767/000
Molecular Formula: C21H35NO
Molecular Weight: 317.51
Percent Composition: C 79.44%, H 11.11%, N 4.41%, O 5.04%
Literature References: Antimycotic morpholine derivative; inhibits fungal ergosterol biosynthesis. Prepn (unspec stereochem): A. Pfiffner, K. Bohnen, DE2752096; A. Pfiffner, US4202894 (1978, 1980 both to Hoffmann-La Roche); of cis-form: NL8004537 (1980 to Hoffmann-La Roche). In vitro comparative antifungal spectrum: S. Shadomy et al.,Sabouraudia22, 7 (1984). Mechanism of action: A. Polak-Wyss et al.,ibid.23, 433 (1985); A. Polak, Ann. N.Y. Acad. Sci.544, 221 (1988). LC determn in pharmaceutical formulations: M. A. Czech et al.,J. Pharm. Biomed. Anal.9, 1019 (1991). Series of articles on mode of action and clinical trials: Clin. Exp. Dermatol.17, Suppl. 1, 1-70 (1992). Review of pharmacology and clinical efficacy: M. Haria, H. M. Bryson, Drugs49, 103-120 (1995).
Properties: bp0.1 120°.
Boiling point: bp0.1 120°
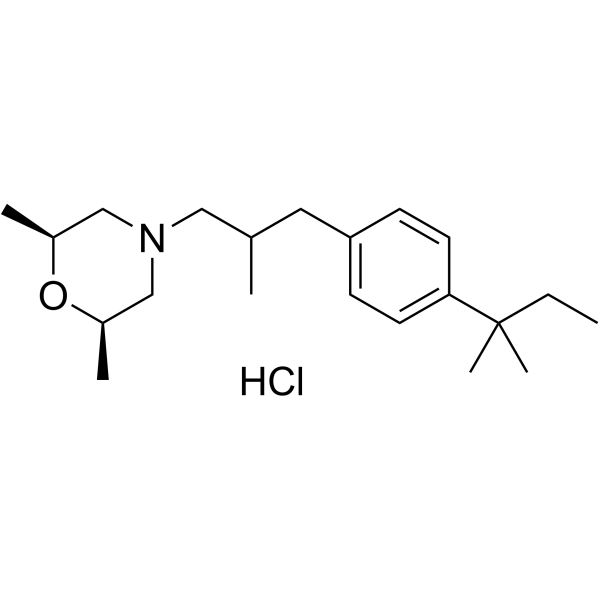
Amorolfine hydrochloride (Ro 14-4767/002) is a antifungal reagent.
Derivative Type: Hydrochloride
CAS Registry Number: 78613-38-4
Manufacturers’ Codes: Ro-14-4767/002
Trademarks: Loceryl (Roche)
Molecular Formula: C21H35NO.HCl
Molecular Weight: 353.97
Percent Composition: C 71.26%, H 10.25%, N 3.96%, O 4.52%, Cl 10.02%
Therap-Cat: Antifungal (topical).
Amorolfine hydrochloride (Ro 14-4767/002) is a antifungal reagent. Target: Antifungal Amorolfine is an antifungal showing activity against fungi pathogenic to plants, animals and humans. Amorolfine possesses a broad antifungal spectrum including dermatophytes, yeasts, dimorphic fungi and moulds and is not only fungistatic but fungicidal against most species [1]. At 0.2, 2 and 5 micrograms/ml amorolfine did not have any significant inhibitory or enhancing effect on phagocytosis whether following simultaneous addition of blastospores and drug to the neutrophils, prior treatment of neutrophils for 2 h before addition of blastospores or prior treatment of blastospores for 2 h. Simultaneous addition of amorolfine resulted in a significant increase in killing at all concentrations. This increase was not significantly enhanced by either preincubation of neutrophils or blastospores for 2 h with the drug [2].
Amorolfine (or amorolfin), is a morpholineantifungal drug that inhibits Δ14-sterol reductase and cholestenol Δ-isomerase, which depletes ergosterol and causes ignosterol to accumulate in the fungal cytoplasmiccell membranes. Marketed as Curanail, Loceryl, Locetar, and Odenil, amorolfine is commonly available in the form of a nail lacquer, containing 5% amorolfine hydrochloride as the active ingredient. It is used to treat onychomycosis (fungal infection of the toe- and fingernails). Amorolfine 5% nail lacquer in once-weekly or twice-weekly applications has been shown in two studies to be between 60% and 71% effective in treating toenail onychomycosis; complete cure rates three months after stopping treatment (after six months of treatment) were 38% and 46%. However, full experimental details of these trials were not available and since they were first reported in 1992 there have been no subsequent trials.[1]
It is a topical solution for the treatment of toenail infections.[2][3] Systemic treatments may be considered more effective.[1]
It is approved for sale over-the-counter in Australia, Brazil, Russia, Germany and the UK, and is approved for the treatment of toenail fungus by prescription in other countries. It is not approved for the treatment of onychomycosis in the United States or Canada, but can be ordered from there by mail from other countries.[4]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN
Indian Pat. Appl., 2010MU01980,







SYN
https://pubs.rsc.org/en/content/articlelanding/2017/ob/c6ob02765b/unauth
The acid-promoted crystallization-induced diastereoisomer transformation (CIDT) of naphthoxazines derived from racemic O-protected 2-substituted 4-hydroxybutyraldehydes and enantiopure Betti’s base allows the deracemization of the starting aldehydes with ee up to 96%. As an alternative, reduction with lithium aluminum hydride of the diastereoisomerically enriched naphthoxazines leads to enantioenriched primary amines. The utility of the latter strategy was demonstrated by applying it to the synthesis of enantioenriched fenpropimorph and to the first synthesis of enantiopure amorolfine, with ee up to 99.5%.
PATENT
https://patents.google.com/patent/WO2013097629A1/en
Amorolfine hydrochloride, chemical name is cis-4-[3-[4-(1,1-dimethyl-propyl)phenyl]-2-mercaptopropyl]-2 , 6-diamidino-morpholine hydrochloride, CAS registration number is 78613-38-4, the chemical knot is as follows:

Amoxifen hydrochloride is an antifungal drug developed by Roche and launched in 1991 under the trade name Leceryl. Regarding the synthesis process of amorolfine hydrochloride, the prior art has been described:


US7795425B2 synthetic route: (1) 2-nonyl cinnamaldehyde is condensed with cis-2,6-dimethylmorpholine to give cis-4-(3-phenyl-2-methylpropyl)-2,6- Dimercapto-morpholine hydrochloride, (2) cis-4-(3-phenyl-2-methylpropyl)-2,6-dimethyl-morpholine hydrochloride followed by 2-methyl – 2-chlorobutane, with acid Catalytic, Heck reaction occurs, and amorolfine is obtained. In step (1), palladium carbon catalytic hydrogenation is required, so the cost is high; in addition, there may be multiple rearrangement reactions in step (2), many by-products, difficult product purification, low quality of finished product and low yield. And it requires a low temperature reaction equipment of -40 ~ -65 °C, which consumes a lot of energy. International patent application WO2007113218A1 improves the synthesis method of amorolfine hydrochloride, the first step of Heck reaction, 4-iodo-t-amylbenzene and 2-methylallyl alcohol are reacted in the presence of a palladium catalyst and a base to obtain 3-un Butyl phenyl-2-methylpropanal, the reaction solvent is selected from N,N-dimercaptocarboxamide (abbreviated as DMF), polar protic solvent or non-polar solvent; second step reductive amination reaction, 3 – tert-amylphenyl-2-mercaptopropanal is reacted with cis-2,6-dimercaptomorpholine to give amorolfine, the reducing agent is selected from palladium

The WO2007113218A1 process still has defects: (1) The first step of the Heck reaction, the reaction solvent DMF is moderately toxic, and the International Agency for Research on Cancer (IARC) considers it to be a carcinogen. DMF is chemically stable and can exist for a long time in wastewater. It is highly polluted by water and difficult to biodegrade. Its BOD5/COD value is 0.065 ( BOD5/COD is an indicator of biodegradability of wastewater, and 0.3 is the lower limit of biodegradable degradation of wastewater). value). Wastewater treatment costs are high during large production. Although the boiling point of DMF is 154 ° C, it is unstable under alkaline conditions, especially at high temperatures, and decomposition starts at 100 ° C or higher. The polar protic solvent, such as the lower alcohol described in the patent, cannot meet the high temperature reaction requirements, and the high boiling polar protic solvent has poor solubility to the catalyst and is difficult to react. The non-polar solvent does not substantially dissolve the palladium catalyst, so the application value is not large. (2) The second step of reductive amination reaction, using expensive The cost of catalytic hydrogenation of heavy metal palladium is high, and the high pressure reaction equipment is unsafe; the reduction of metal borohydride is easy to generate a large amount of hydrogen, which poses a safety hazard, and also reduces 3-tert-pentylphenyl-2-methylpropanal to The corresponding alcohol increases the impurities; the reduction by-product of the metal cyanoborohydride is highly toxic. (3) The product yield was low, and the total yield of the product of the example was about 50%. None of the purity of the products and intermediates has been disclosed.The chemical reaction equation of the present invention is expressed as follows:

(la) (lb)In a 10L clean reaction kettle, add 2600 mL of acetic anhydride, 5200 mL of glacial acetic acid, 350 g of sodium periodate, break 1236 g, cool to 5 ° C, add 810 mL of sulfuric acid, control the dropwise addition within 1 hour, and then add 1130 g of t-amyl. The benzene was stirred at room temperature for more than 16 hours, and the reaction of the raw materials was confirmed by thin layer chromatography. The reaction mixture was poured into a mixture of 8 L of water and 4 L of dichloromethane, and the mixture was separated. The organic layer was washed with 4L of 25% aqueous sodium sulfite, and the organic layer was dried over anhydrous sodium sulfate. It was 4-iodo-t-amylbenzene 2013 g, yield: 96%, and the GC purity was 94.2%. NMR spectral data: (400 MHz, CDC1 3 ): 0.73 (3H, t, J = 7.4 Hz), 1.31 (6H, s), 1.67 (2H, q, J – 7.4 Hz), 7.13 (2H, d, J = 8.56 Hz), 7.66 (2H, d, J = 8.56 Hz) 0 Example 22 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 6 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel, and the mixture was stirred under nitrogen, stirring was carried out, and 300 g of palladium acetate and 1.7 kg of sodium hydrogencarbonate were added. Finally, 2.5 kg of 2-mercaptopropanol was added, the temperature was raised to 105 C, and the GC content of 4-iodo-t-amylbenzene was measured to monitor the progress of the reaction, and the reaction was completed for 2 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 12 L of ethyl acetate, wash with 20 L of water, rectify the organic phase, collect 125-128 ° C fraction (vacuum degree ≤ -0.099)\3⁄4^), and obtain 3- Tert-amylphenyl-2-mercaptopropanal L41 kg, yield: 88.6%, GC purity: 93.5%. NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1.11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J =7.43 Hz), 2.60 (13⁄4 dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9,75 (1H, s).The above 3-tert-pentylphenyl-2-methylpropanal lkg, 5 L of ethyl acetate was added to a 10 L reactor, protected with nitrogen, cooled to 10 ° C, and 600 g of 2,6-dimethylmorpholine was added dropwise. , add about 30 minutes. Then, 300 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 C, the addition was completed, and the temperature was raised to 18 ° C for 30 minutes. After cooling to 10 Torr, 1,3 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was maintained at 18 ° C, and the GC content of 3-tert-amylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. Ended in 2 hours. After cooling to 10 ° C or lower, the pH was adjusted to 10 with a sodium hydroxide solution, and the layers were allowed to stand, and the organic layer was washed with 4 L of water. The organic phase was added with concentrated hydrochloric acid, adjusted to pH 2, filtered, and the filter cake was dried under reduced pressure at 65 V for 14 hours to obtain 1.59 kg of amorolamine hydrochloride, yield: 85.6%, HPLC purity: 99.6%. R spectrum data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J=7, 2Hz), 1.03 (3H, d, J=6.8Hz), 1.15(6H, d, J=6 , 0 Hz), 1.25 (63⁄4 s), 1.64 (2H, m, J = 7.6 Hz), 2.34 (1H, d, J = 6.8 Hz), 2.48 (23⁄4 d, J = 6.8 Hz), 2.75 (2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz) 5 3.4(2H, d, J=11.2Hz), 3,9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 3 In a 10 L clean reaction kettle, 2 kg of 4-substituted tert-amylbenzene prepared according to the method of Example 1 and 6 L of N-mercaptopyrrolidone were protected by nitrogen, stirring was started, and 150 g of palladium acetate and 2.5 kg of dipotassium hydrogen phosphate were added. Finally, 1.8 kg of 2-methylallyl alcohol was added, and the temperature was raised to 130. C reaction, the GC content of 4-deuterated tert-amylbenzene was measured to control the progress of the reaction, and the reaction was completed for 10 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 12 L of ethyl acetate, dissolve 20 L of water, concentrate the organic phase, recover ethyl acetate, and add the residue to 10 L of saturated sodium hydrogen sulfite solution at room temperature to precipitate solid. The mixture was stirred for 6 hours, filtered, and filtered, washed with EtOAc EtOAc EtOAc EtOAc. The filtrate was concentrated to dry ethyl acetate to give 1. <RTI ID=0.0>#</RTI><RTIgt;</RTI><RTIgt;</RTI><RTIgt; -NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J-7.45 Hz), 1.11 (3H, d, J-6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (1H, dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz) ), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).Add 1 kg of the above 3-tert-pentylphenyl-2-methylpropanal, 5 L of ethyl acetate in a 10 L reactor, protect with nitrogen, cool to 10 C, and add 1.2 kg of 2,6-dimethylmorpholine dropwise. , 40 minutes added. Then, 780 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 20 ° C for 60 minutes. After cooling to 10 ° C, 2.3 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was checked at 25 ° C, and the GC content of 3-tert-amylpyridyl-2-methylpropanal was detected to monitor the progress of the reaction. The reaction was completed in 2 hours. Cool to below 10 ,, adjust the pH to 11 with sodium hydroxide solution, let stand for stratification, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 2, filter, filter cake at 70 ° C decompression After drying for 14 hours, 1.75 kg of amorolfine hydrochloride was obtained, yield: 84.6%, HPLC purity: 99.7%. R spectrum data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2Hz), 1.03 (3H, d, J = 6.8Hz), L15(6H, d, J=6.0Hz ), 1.25(6H, s), L64(2H 5 m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H 5 d, J=11.2Hz), 3·9(2Η, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H ; dd, J = 8.4 Hz). Example 4In a 10 L clean reaction kettle, 2 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1, 2 N of N-methylpyrrolidone, protected by nitrogen, stirring was started, and palladium nitrate 6 g, acetic acid was added. Sodium 627 g, and finally 592 g of 2-methylallyl alcohol was added thereto, and the temperature was raised to 140 ° C to carry out a reaction. The GC content of 4-deactivated t-amylbenzene was examined to monitor the progress of the reaction, and the reaction was terminated for 24 hours. Cool to room temperature, filter, concentrate the filtrate, add the residue to 8 L of ethyl acetate, dissolve in 16 L of water, rectify the organic phase, collect 125-128 C fraction (vacuum degree ≤ -0.0991 ^ & ) to give 3-tert-pentylphenyl 2-mercaptopropanal 1.37 kg, yield: 86%, GC purity: 93.0%. MR spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1 , 11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q , 3=1 A3 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (IH, s).The above 3-tert-pentylphenyl-2-mercaptopropanal lkg, 5 L of dichloromethane was added to a 10 L reactor, protected with nitrogen, cooled to 10 ° C, and 1.6 kg of 2,6-dimethyl was added dropwise. Morpholine, added in 45 minutes. Then, 300 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 23 Torr for 60 minutes. After cooling to 10 ° C, 1.6 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was checked at 23 ° C, and the GC content of 3-tert-pentylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. The end of the hour. Cool to below 10 °C, adjust the pH to 10 with sodium hydroxide solution, let stand for layering, wash the organic layer with 4L water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 1, filter, filter cake at 70 °C After drying under reduced pressure for 14 hours, 1.59 kg of amorolamine hydrochloride was obtained, yield: 83.6%, HPLC purity: 99.6%. iH-NMR spectral data: ! H NM (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J= 6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H , d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 52 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 4 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel, and the mixture was stirred under nitrogen, stirring was carried out, 30 g of palladium chloride and 750 g of sodium hydrogencarbonate were added. Finally, 1.3 kg of 2-methylallyl alcohol was added, and the mixture was heated to 120 ° C to measure the GC content of 4-iodo-t-amylbenzene to control the progress of the reaction, and the reaction was completed for 13 hours. It was cooled to room temperature, filtered, and the filtrate was concentrated. The residue was dissolved in 8 L of chloroform, washed with 16 L of water, and the organic phase was concentrated. The ethyl acetate was recovered. The residue was added dropwise to 10 L of saturated sodium hydrogensulfite solution at room temperature to precipitate a solid. Hour, filter, filter cake washed with 5 L of ethyl acetate, solid dispersed in 3 L 3 mol / liter The mixture was stirred at room temperature for 5 hours, and the reaction mixture was dried over EtOAcjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjjj Yield: 91,7%, GC purity: 98.8%. – Spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J=7.45 Hz), 1.11 (3H, d, J-6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz) ), 7.27 (2H, d, J = 8.27 Hz), 9.75 (IH, s).Add 1 kg of the above 3-tert-pentylphenyl-2-methylpropanal, 5 L of absolute ethanol in a 10 L reactor, protect with nitrogen, cool to 10 ° C, and add 600 g of 2,6-dimercaptomorpholine. , added in 30 minutes. Then, 500 mL of glacial acetic acid was added dropwise, the temperature was kept at 15 ° C, the addition was completed, and the temperature was raised to 23 ° C for 60 minutes. After cooling to 10 ° C, 1.2 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was monitored at 10 Torr, and the GC content of 3-tert-pentylphenyl-2-nonylpropionaldehyde was detected to monitor the progress of the reaction. The end of the hour. 10. Under C, adjust the pH value to 11 with sodium hydroxide solution, add 3 L of dichloromethane, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 2, filter, filter cake at 7 CTC minus After drying for 14 hours, 1.45% of amorolfine hydrochloride was obtained, yield: 87.0%, HPLC purity: 99.7% – NMR spectral data: J H NMR (400 MHz 5 CD 3 OD) 6: 0.64 (3H, t, J= 7,2Hz), 1.03(3H, d, J=6.8Hz), 1.15(6H, d, J=6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34( 1H ? d, J = 6.8 Hz), 2.48 (2H, d, J = 6.8 Hz), 2.75 (23⁄4 d, J = 6.0 Hz), 3.1 (2H, d, J = 8.8 Hz), 3.4 (2H, d , J = 11.2 Hz) 5 3.9 (2H, m), 7.16 (2H, dd, J = 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Example 62 kg of 4-iodo-t-amylbenzene prepared in accordance with the method of Example 1 and 4 L of N-methylpyrrolidone were added to a 10 L clean reaction vessel. The mixture was stirred under nitrogen, stirring was started, 10 g of palladium acetate was added, and 800 g of carbonic acid was added. 1.1 kg of 2-mercaptopropanol was heated to 80 ° C, and the GC content of 4-deactivated t-amylbenzene was measured to control the progress of the reaction, and the reaction was terminated for 24 hours. Cool to room temperature, filter, concentrate the filtrate, add 8 L of chloroform to dissolve, 16 L of water, rectify the organic phase, collect 125-128 ° C 真空 (vacuum degree ≤ -0.099 ^ ^ & ), to obtain 3-tert-amylbenzene Base-2-mercaptopropanal 1.42 kg, yield: 89.2%, GC purity: 92.5%. ^- MR Spectral Data: (400 MHz, CDC1 3 ): 0.69 (33⁄4 t, J=7.45 Hz), 1.11 (3H, d, J=6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2.60 (IH, dd, J=13.52 Hz), 2.69 (IH, J=7.06 Hz), 3.08 (IH, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).The above 3-tert-pentylphenyl-2-methylpropanal lkg, 5 L of decyl alcohol was added to a 10 L reactor, protected with nitrogen, cooled to 10 C, and 600 g of 2,6-dimethylmorpholine was added dropwise for 30 minutes. Plus finished. Then, 500 mL of water acetic acid was added dropwise, the temperature was kept at 10 ° C, the addition was completed, and the temperature was raised to 20 ° C for 60 minutes. After cooling to 10 C, 1.2 kg of sodium triacetoxyborohydride was added. After the addition, the temperature was maintained at 23 ° C, and the GC content of 3-tert-pentylphenyl-2-methylpropanal was detected to monitor the progress of the reaction. End of 2 hours. Cool to 10 ° C, adjust the pH to 10 with sodium hydroxide solution, add 3 L of dichloromethane, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust pH to 1.5, filter, filter The cake was dried under reduced pressure at 65 C for 15 hours to obtain 1.46 kg of amorolfine hydrochloride, yield: 90.1%, HPLC purity: 99,8%. ^-NMR spectral data: l R NMR (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), U5 (6H, d, J = 6.0Hz), 1.25(6H, s), 1.64(23⁄4 m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=l 1.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz ), 7.27 (2H, dd, J = 8.4 Hz). Example 72 kg of 4-iodo-t-amylbenzene prepared according to the method of Example 1 and 6 L of N-decylpyrrolidone were added to a 10 L clean reaction kettle, protected by nitrogen, stirring was started, and 75 g of palladium acetate and 2.0 kg of disodium hydrogen phosphate were added. Finally, 780 g of 2-methylallyl alcohol was added, and the temperature was raised to 125 Torr. The GC content of 4-iodo-t-amylbenzene was measured to control the progress of the reaction, and the reaction was terminated for 8 hours. The mixture was cooled to room temperature, filtered, and the filtrate was concentrated. The residue was evaporated, evaporated, evaporated, evaporated, evaporated. The solid was precipitated, stirred for 6 hours, filtered, and the filter cake was washed with 5 L of ethyl acetate. The solid was dispersed in 10 L 2 mol/L hydrochloric acid, stirred at room temperature for 5 hours, and the reaction mixture was extracted with 10 L of ethyl acetate. The mixture was dried, filtered, and the filtrate was evaporated to ethyl acetate to ethylamine (ethyldiethyldithioacetate). 3⁄4-NMR spectral data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz), 1.11 (3H, d, J = 6.87 Hz), 1.29 (6H, s), 1.65 (2H, q , J=7.43 Hz), 2.60 (1H, dd, J=13.52 Hz), 2.69 (1H, J=7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).Add the above 3-tert-pentylphenyl-2-mercaptopropanal lkg, 5L hydrazine, in a 10L reactor Under nitrogen atmosphere, cooled to 10 Torr, 700 g of 2,6-dimercaptomorpholine was added dropwise, then 280 mL of glacial acetic acid was added, the temperature was maintained at 15 C, and then the temperature was raised to 23 ° C for 60 minutes. After cooling to 10 ° C, 1.0 kg of sodium triacetoxyborohydride was added, and 20 was added. The temperature was maintained under C, and the GC content of 3-tert-amylphenyl-2-methylpropanal was examined to monitor the progress of the reaction, and the reaction was completed for 3 hours. Cool to below 10 ° C, adjust the pH to 11 with sodium hydroxide solution, let stand for layering, wash the organic layer with 4 L of water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 1, filter, filter cake at 70 ° C After drying under reduced pressure for 14 hours, 1.59 kg of amorolamine hydrochloride was obtained, yield: 83.8%, HPLC purity: 99.6%. ^-NMR spectral data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J- 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H ; d, J = 6.0 Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz) } 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz) , 7.27 (2H, dd, J = 8.4 Hz). Example 83-tert-pentylphenyl-2-mercaptopropanol lkg, 5 L of dichloromethane prepared by the method of Example 5 was added to a 10 L reactor, and was purged with nitrogen and cooled to 10. C, 1000 g of 2,6-dimethylmorpholine was added dropwise, then 400 mL of water acetic acid was added, the temperature was maintained at 15 ° C, and then the temperature was raised to 20 ° C for 60 minutes. After cooling to 0 C, 1.5 kg of sodium triacetoxyborohydride was added, and 6 C was added after the addition, and the GC content of 3-tert-pentylphenyl-2-mercaptopropanal was detected to monitor the progress of the reaction for 5 hours. End. Adjust the pH to 10 with sodium hydroxide solution at 6 °C, let stand for layering, wash the organic layer with 4L of water, add concentrated hydrochloric acid to the organic phase, adjust the pH to 2, filter, filter cake and dry at 65 Ό for 14 hours under reduced pressure. , Amofufen hydrochloride 1.48kg, yield: 91.2%, HPLC purity: 99.7%. ^- MR spectral data: NMR (400MHz, CD 3 OD) 5: 0·64 (3Η, ΐ, J=7, 2Hz), 1.03(3Η, d, J=6.8Hz), 1.15(6H, d, J =6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75( 2H, d, J=6.0Hz), 3,1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(23⁄4 m), 7.16(2H, dd, J- 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Example 9Add 3-tert-pentylphenyl-2-mercaptopropanol lkg prepared in the same manner as in Example 2, 4 L of tetrahydrofuran, protect with nitrogen, cool to 10 ° C, add 820 g of 2,6-two Mercaptomorpholine, Then, 380 mL of glacial acetic acid was added, the temperature was maintained at 15 ° C, and then kept at room temperature for 60 minutes. After cooling to 10 ° C, 1.8 kg of sodium triacetoxyborohydride was added, and after 10 liters of the addition, the GC content of 3-tert-amylphenyl-2-nonylpropionaldehyde was detected to monitor the progress of the reaction for 5 hours. End. The pH was adjusted to 10 with sodium hydroxide solution at 10 ° C, and the layers were allowed to stand. The organic layer was washed with 4 L of water, and the organic phase was added with concentrated hydrochloric acid, adjusted to pH 2, filtered, and the filter cake was dried under reduced pressure at 65 Torr for 14 hours. , amlofol hydrochloride 1.41 kg, yield: 87.1%, HPLC purity: 99.8%. NMR spectral data: J H NMR (400 MHz, CD 3 OD) 5: 0.64 (3H, t, J- 7.2 Hz), L03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J = 6.0 Hz) ), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J-6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d , J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz), 7.27 (2H, dd, J = 8.4 Hz). Comparative example 1In a 1000 mL four-necked flask, 137 g of 4-deuterated tert-amylbenzene prepared according to the method of Example 1, 1.12 g of palladium acetate, 50.4 g of sodium hydrogencarbonate, N,N-dimethylformamide 500 mL, nitrogen gas, added 54 g of 2-mercaptopropanol, warmed to 10 (TC for 10 hours, cooled to room temperature, filtered, filter cake washed with hydrazine, hydrazine-dimethylformamide 300 mL, combined filtrate, poured into 2000 mL of saturated brine and 1000 mL The mixture was extracted with ethyl acetate, and the organic phase was washed with water, dried over anhydrous magnesium sulfate, filtered, and concentrated, dried, and evaporated, and the residue was distilled in vacuo to collect fractions of 125-128 ° C (vacuum degree <-0.099 MPa) to obtain 3-un Amyl phenyl-2-mercaptopropanal 84 g, Yield: 77%, GC purity: 88.0% – R spectrum data: (400 MHz, CDC1 3 ): 0.69 (3H, t, J = 7.45 Hz) , 1.11 (3H : d, J=6.87 Hz), 1.29 (6H, s), 1.65 (2H, q, J=7.43 Hz), 2,60 (1H, dd, J=13.52 Hz), 2.69 (1H, J-7.06 Hz), 3.08 (1H, dd, J = 13.54 Hz), 7.12 (2H, d, J = 8.27 Hz), 7.27 (2H, d, J = 8.27 Hz), 9.75 (1H, s).109 g of the above 3-tert-amylphenyl-2-mercaptopropanal and 500 mL of ethanol were placed in a 1000 mL four-necked flask, cooled to 0 ° C, and 30 mL of glacial acetic acid and 69 g of 2,6-dimethylmorpholine were added. Stir at room temperature for 30 minutes, cool to -15 ° C, add 15.93 g of sodium borohydride in 1 hour. After the addition, warm to 0 C for 2 hours, adjust the pH to 12 with 25% sodium hydroxide solution. The mixture was extracted with 2000 mL of saturated brine and 1000 mL of ethyl acetate. The organic phase was washed with water and concentrated to dryness. The obtained residue was added to 500 mL of isopropyl ether, hydrogen chloride gas to pH 2, stirred at room temperature for 2 hours, filtered, and washed with isopropyl ether. , the filter cake is dried under reduced pressure at 70 ° C for 14 hours to obtain hydrochloric acid. Morofen 119 g, yield: 67%, HPLC purity: 97.1%. 3⁄4-NMR spectral data: ‘H NMR (400 MHz, CD 3 OD) 5: 0, 64 (3H, t, J = 7, 2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d , J=6.0Hz), 1.25(6H, s), 1.64(2H, m, J=7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz), 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J=11.2Hz), 3,9(2H, m), 7.16(2H, dd , J = 8.4 Hz), 7.27 (2H, dd, J = 8.4 Hz). Comparative example 2109 g of 3-tert-pentylphenyl-2-methylpropanal prepared according to the method of Comparative Example 1 and 500 mL of methanol were added to a 1000 mL four-necked flask, cooled to 0 ° C, and 30 mL of glacial acetic acid and 69 g of 2, 6 were added. – dimethylmorpholine, stirred at room temperature for 30 minutes, cooled to -15 ° C, replaced with nitrogen, added 5 g of 0% palladium on carbon, passed through hydrogen, reduced at 40 ° C, 4 atm, until the hydrogen pressure did not decrease, The reaction is complete. Cool to room temperature, replace with nitrogen, filter, adjust the pH of the filtrate with 25% sodium hydroxide solution, add 2000 mL of saturated brine and 1000 mL of ethyl acetate for extraction, wash the organic phase, concentrate and dry, add the residue to 500 mL Isopropyl ether, hydrogen chloride gas to pH 2, stirred at room temperature for 2 hours, filtered, washed with isopropyl ether, and the filter cake was dried under reduced pressure at 70 ° C for 14 hours to obtain amolofol hydrochloride 113 g, yield: 64%. HPLC purity: 97.8%. NMR spectral data: 3⁄4 NMR (400MHz, CD 3 OD) 5: 0.64 (3H, t, J = 7.2 Hz), 1.03 (3H, d, J = 6.8 Hz), 1.15 (6H, d, J = 6.0 Hz) , 1.25(6H, s), 1.64(2H, m, J-7.6Hz), 2.34(1H, d, J=6.8Hz), 2.48(2H, d, J=6.8Hz) ? 2.75(2H, d, J=6.0Hz), 3.1(2H, d, J=8.8Hz), 3.4(2H, d, J-11.2Hz), 3.9(2H, m), 7.16(2H, dd, J=8.4Hz), 7.27 (2H, dd, J=8, 4Hz).
Patent
Publication numberPriority datePublication dateAssigneeTitleEP0447947A1 *1990-03-231991-09-25BASF AktiengesellschaftN-(3-Phenyl-2-methylpropyl and -methyl-prop-2-enyl)-azaheterocyclesWO2007113218A1 *2006-04-032007-10-11Galderma S.A.Process for producing 3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propionaldehyde and cis-4-{3-[4-(1,1-dimethyl-propyl)-phenyl]-2-methyl-propyl}-2,6-dimethyl-morpholine (amorolfine)Family To Family CitationsEP1749826A1 *2005-07-282007-02-07Galderma S.A.Process of producing bepromolineCN101485625B *2009-02-192010-09-22中国药科大学Amoluofen emulsifiable paste
Publication numberPriority datePublication dateAssigneeTitle
CN105130808A *2015-08-132015-12-09上海瑞博化学有限公司High purity 2,5-dimethyl-3,4-dihydroxy methylbenzoate synthesis methodFamily To Family CitationsCN103288768B *2013-06-182015-02-18中国人民解放军第四军医大学Asymmetric synthetic method of optical pure amorolfine hydrochlorideCN104926629B *2015-05-302016-06-22江苏科本医药化学有限公司Domino reaction is utilized to prepare the green method of 3,3-diaryl acrylic aldehydeCN108997246B *2017-06-062021-08-31江苏礼华生物技术有限公司Preparation method of amorolfine hydrochlorideCN110498729A *2019-09-092019-11-26武汉诺安药业有限公司A kind of clean method for preparing of hydrochloric acid Amorolfine intermediate
Notes
- ^ Jump up to:a b Williams HC (2003). Evidence-Based Dermatology. Blackwell. ISBN 9781444300178.
- ^ Flagothier C, Piérard-Franchimont C, Piérard GE (March 2005). “New insights into the effect of amorolfine nail lacquer”. Mycoses. 48 (2): 91–4. doi:10.1111/j.1439-0507.2004.01090.x. PMID 15743424.
- ^ Feng X, Xiong X, Ran Y (May 2017). “Efficacy and tolerability of amorolfine 5% nail lacquer in combination with systemic antifungal agents for onychomycosis: A meta-analysis and systematic review”. Dermatologic Therapy. 30 (3): e12457. doi:10.1111/dth.12457. PMID 28097731.
- ^ It can readily be verified that Curanail is advertised on websites such as US Amazon.com, shipped from abroad.
| Clinical data | |
|---|---|
| AHFS/Drugs.com | International Drug Names |
| ATC code | D01AE16 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 78613-35-1 |
| PubChem CID | 54260 |
| ChemSpider | 49010 |
| UNII | AB0BHP2FH0 |
| KEGG | D02923 |
| ChEBI | CHEBI:599440 |
| ChEMBL | ChEMBL489411 |
| CompTox Dashboard (EPA) | DTXSID0046690 |
| Chemical and physical data | |
| Formula | C21H35NO |
| Molar mass | 317.517 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
/////////////AMOROLFINE, Ro 14-4767-002, аморолфин ,أمورولفين ,阿莫罗芬 , antifungal

NEW DRUG APPROVALS
one time
$10.00
SULCONAZOLE

SULCONAZOLEсульконазол , سولكونازول , 硫康唑
- Molecular FormulaC18H15Cl3N2S
- Average mass397.749 Da
1H-Imidazole, 1-[2-[[(4-chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]- [ACD/Index Name]
4332
5D9HAA5Q5S
61318-90-9[RN]
(±)-1-[2,4-Dichloro-b-[(p-chlorobenzyl)thio]phenethyl]imidazole
1-[2-[[(4-Chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole: SulconazoleCAS Registry Number: 61318-90-9
CAS Name: 1-[2-[[(4-Chlorophenyl)methyl]thio]-2-(2,4-dichlorophenyl)ethyl]-1H-imidazole
Additional Names: (±)-1-[2,4-dichloro-b-[(p-chlorobenzyl)thio]phenethyl]imidazole
Molecular Formula: C18H15Cl3N2S
Molecular Weight: 397.75
Percent Composition: C 54.35%, H 3.80%, Cl 26.74%, N 7.04%, S 8.06%
Literature References: Prepn: K. A. M. Walker, DE2541833; idem,US4055652 (1976, 1977 both to Syntex). HPLC determn in plasma: M. Fass et al.,J. Pharm. Sci.70, 1338 (1981). Mechanism of action study: W. H. Beggs, Biochem. Arch.10, 117 (1994). Clinical trial in tinea pedis: W. A. Akers et al.,J. Am. Acad. Dermatol.21, 686 (1989). Review of pharmacology and clinical efficacy: P. Benfield, S. P. Clissold, Drugs35, 143-153 (1988).
Derivative Type: Nitrate
CAS Registry Number: 61318-91-0
Manufacturers’ Codes: RS-44872
Trademarks: Exelderm (Syntex); Myk (Cassenne); Sulcosyn (Syntex)
Molecular Formula: C18H15Cl3N2S.HNO3
Molecular Weight: 460.76
Percent Composition: C 46.92%, H 3.50%, Cl 23.08%, N 9.12%, S 6.96%, O 10.42%
Properties: Colorless crystals from acetone, mp 130.5-132°.
Melting point: mp 130.5-132°
Therap-Cat: Antifungal.
Keywords: Antifungal (Synthetic); Imidazoles.
Sulconazole (trade name Exelderm) is an antifungal medication of the imidazole class. It is available as a cream or solution to treat skin infections such as athlete’s foot, ringworm, jock itch, and sun fungus.[1][2] Although not used commercially for insect control, sulconazole nitrate exhibits a strong anti-feeding effect on the keratin-digesting Australian carpet beetle larvae Anthrenocerus australis.[3]
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
SYN

DE 2541833 US 4038409
(Read example 5 and 9 in US patent.)
https://patents.google.com/patent/US4038409A/en
EXAMPLE 5Alternative Route to 1-[β-(R-carbonylthio)phenethyl]imidazolesA. Preparation of 1-[2,4-dichloro-β-(methylcarbonylthio)-phenethyl]imidazole, oxalate.1-(β,2,4-Trichlorophenethylimidazole (1.19g) in 5 ml of dry tetrahydrofuran was added to preformed sodium thioacetate, generated in situ from 720 mg thioacetic acid and sodium hydride (480 mg 57% dispersion in mineral oil) in 20 ml. tetrahydrofuran and the mixture stirred and refluxed under nitrogen for 18 hours. The solvent was removed under reduced pressure, water (20 ml) added and the product extracted with ether. The extracts were washed with water, dried (MgSO4), evaporated and the residue chromatographed on silica gel eluting with 10-20% acetone in dichloromethane. The pure product in ether was treated dropwise with ethereal oxalic acid until precipitation was complete, and the thus obtained oxalate salt of 1-[2,4-dichloro-β-(methylcarbonylthio)phenethyl]imidazole recrystallized from acetone/ethyl acetate with mpBy substituting other available sodium thioacids for sodium thioacetate, other compounds of this invention may be prepared.
EXAMPLE 9A. Preparation of 1-[2,4-dichloro-β-(4-chlorobenzylthio)-phenethyl]imidazoleTo a stirred solution of 330 mg sodium hydroxide in 30 ml methanol under nitrogen is added 810 mg of 1-[2,4-dichloro-β-(methylcarbonylthio)phenethyl]imidazole oxalate and the mixture is stirred at room temperature for ca. 30 minutes (until thin layer chromatography shows the disappearance of the ester). α,p-dichlorotoluene (350 mg) is then added, the solution stirred a further 15 minutes and the solvent removed under reduced pressure. Ether and water are then added to the residue and the ether extract washed with water, dried (MgSO4) and concentrated. Dropwise addition of nitric acid (d = 1.42) until precipitation is complete gives the nitrate salt of 1-[2,4-dichloro-β-(4-chlorobenzylthio)phenethyl]imidazole, recrystallized from acetone, mp 130.5°-132° C.B. By using other compounds of this invention exemplified by those set forth in Examples 2 and 4 and other suitable (substituted) hydrocarbyl halides (or mesylates, tosylates), other compounds may be prepared.
SYN
https://www.sciencedirect.com/science/article/pii/S2095177917301405
SYN
Synthesis Path
Substances Referenced in Synthesis Path
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 6258-66-8 | C7H7ClS | 4-chlorobenzyl mercaptan | Benzenemethanethiol, 4-chloro- |
| 24155-42-8 | C11H10Cl2N2O | 1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethanol | 1H-Imidazole-1-ethanol, α-(2,4-dichlorophenyl)- |
References
- ^ Drugs.com: sulconazole topical
- ^ Fromtling RA (April 1988). “Overview of medically important antifungal azole derivatives”. Clinical Microbiology Reviews. 1 (2): 187–217. doi:10.1128/CMR.1.2.187. PMC 358042. PMID 3069196.
- ^ Sunderland MR, Cruickshank RH, Leighs SJ (2014). “The efficacy of antifungal azole and antiprotozoal compounds in protection of wool from keratin-digesting insect larvae”. Textile Research Journal. 84 (9): 924–931. doi:10.1177/0040517513515312.
| Clinical data | |
|---|---|
| Trade names | Exelderm |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a698018 |
| Routes of administration | Topical |
| ATC code | D01AC09 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 61318-90-9 |
| PubChem CID | 5318 |
| ChemSpider | 5127 |
| UNII | 5D9HAA5Q5S |
| KEGG | D08535 |
| ChEBI | CHEBI:9325 |
| ChEMBL | ChEMBL1221 |
| CompTox Dashboard (EPA) | DTXSID8044129 |
| Chemical and physical data | |
| Formula | C18H15Cl3N2S |
| Molar mass | 397.74 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
/////////SULCONAZOLE, сульконазол , سولكونازول , 硫康唑 , Antifungal,

NEW DRUG APPROVALS
ONE TIME
$10.00
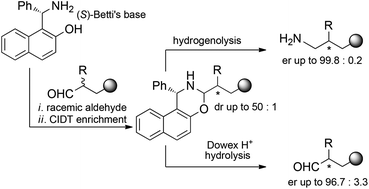
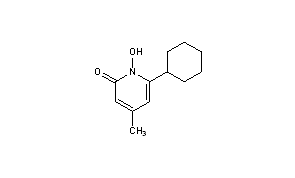
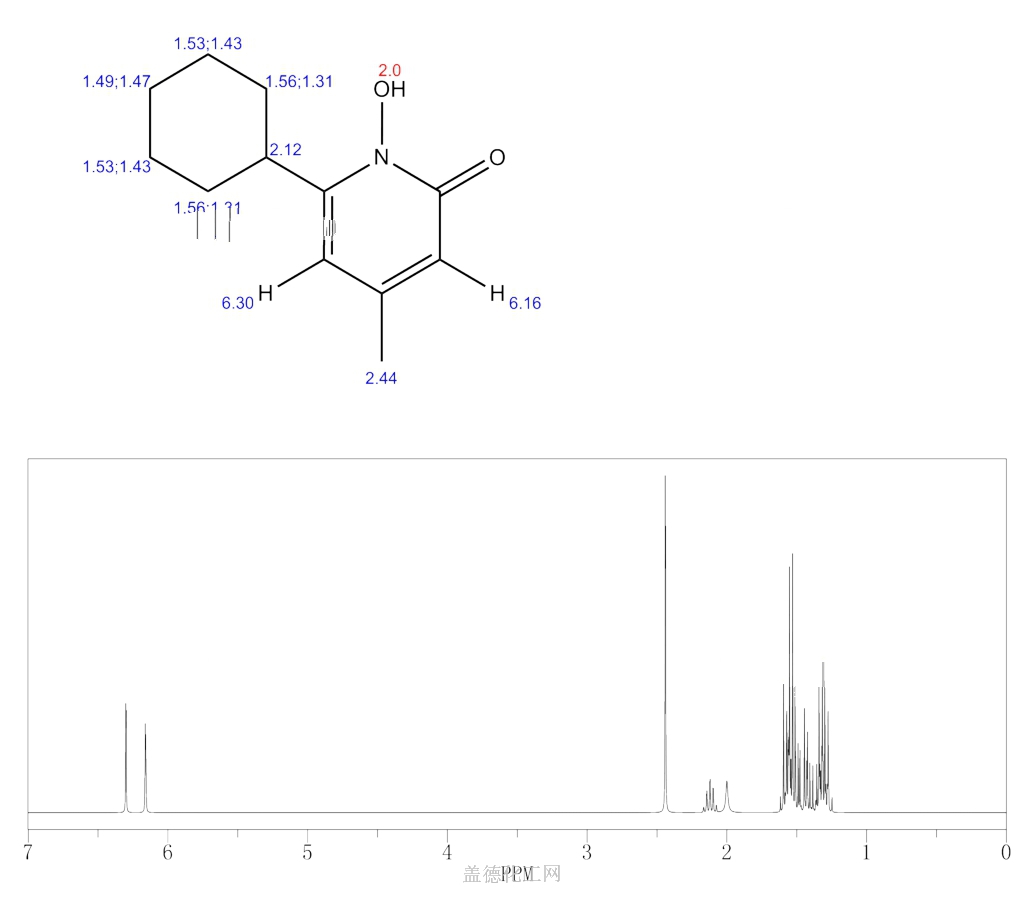
CICLOPIROX

- Molecular FormulaC12H17NO2
- Average mass207.269 Da
CICLOPIROX(6-Cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridone)
2(1H)-Pyridinone, 6-cyclohexyl-1-hydroxy-4-methyl-
249-577-2[EINECS]
29342-05-0[RN]
KS-5085, циклопирокс , سيكلوبيروكس , 环吡酮 ,
Ciclopirox
CAS Registry Number: 29342-05-0
CAS Name: 6-Cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridinone
Molecular Formula: C12H17NO2
Molecular Weight: 207.27
Percent Composition: C 69.54%, H 8.27%, N 6.76%, O 15.44%
Literature References: Broad spectrum antimycotic agent with some antibacterial activity. Prepn: G. Lohaus, W. Dittmar, ZA6906039; eidem,US3883545 (1970, 1975 both to Hoechst). In vitro study: eidem,Arzneim.-Forsch.23, 670 (1973). Series of articles on pharmacokinetics, pharmacology, teratology, toxicity studies: Oyo Yakuri9, 57-115 (1975), C.A.83, 53159d, 53538b, 53539c, 71844c, 90833q (1975). Series of articles on chemistry, mechanism of action, toxicology, clinical trials: Arzneim.-Forsch.31, 1309-1386 (1981). Toxicity data: H. G. Alpermann, E. Schutz, ibid. 1328. Review: S. G. Jue et al.,Drugs29, 330-341 (1985). Review of clinical experience in seborrheic dermatitis: A. Starova, R. Aly, Expert Opin. Drug Saf.4, 235-239 (2005).
Properties: Solid, mp 144°.
Melting point: mp 144°
Derivative Type: Ethanolamine salt (1:1)
CAS Registry Number: 41621-49-2
Additional Names: Ciclopirox olamine
Manufacturers’ Codes: HOE-296
Trademarks: Batrafen (HMR); Brumixol (Bruschettini); Ciclochem (Novag); Dafnegin (Poli); Loprox (HMR); Micoxolamina (Domp?; Mycoster (Fabre)
Molecular Formula: C14H24N2O3
Molecular Weight: 268.35
Percent Composition: C 62.66%, H 9.01%, N 10.44%, O 17.89%
Properties: LD50 in mice, rats (mg/kg): 2898, 3290 orally (Alpermann, Schutz).
Toxicity data: LD50 in mice, rats (mg/kg): 2898, 3290 orally (Alpermann, Schutz)
- EINECS:255-464-9
- LD50:71 mg/kg (M, i.v.); 1740 mg/kg (M, p.o.);
72 mg/kg (R, i.v.); 2350 mg/kg (R, p.o.)
Therap-Cat: Antifungal.
Keywords: Antifungal (Synthetic).
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////////////////////////////
Ciclopirox (sometimes known by the abbreviation CPX[2]) is a synthetic antifungal agent for topical dermatologic treatment of superficial mycoses. It is most useful against Tinea versicolor. It is sold under many brand names worldwide.[1]
Medical uses
Ciclopirox is indicated for the treatment of tinea pedis and tinea corporis due to Trichophyton rubrum, Trichophyton mentagrophytes and Epidermophyton floccosum, as well as seborrheic dermatitis. It is not to be used in the eyes or vagina, and nursing women should consult their doctors before use, since it is not known whether ciclopirox passes into human milk. A burning sensation may be felt when first applying ciclopirox on the skin.[3]
Nail infections
In addition to other formulations, ciclopirox is used in lacquers for topical treatment of onychomycosis (fungal infections of the nails). A meta-analysis of the six trials of nail infections available in 2009 concluded that they provided evidence that topical ciclopirox had poor cure rates and that amorolfine might be substantially more effective, but more research was required.
“Combining data from 2 trials of ciclopiroxolamine versus placebo found treatments failure rates of 61% and 64% for ciclopiroxolamine. These outcomes followed long treatment times (48 weeks) and this makes ciclopiroxolamine a poor choice for nail infections. Better results were observed with the use of amorolfine lacquer; 6% treatment failure rates were found after 1 month of treatment but these data were collected on a very small sample of people and these high rates of success might be unreliable.”[4]
Pharmacology
Pharmacodynamics
In contrast to the azoles and other antimycotic drugs, the mechanism of action of ciclopirox is poorly understood.[5] However, loss of function of certain catalase and peroxidase enzymes has been implicated as the mechanism of action, as well as various other components of cellular metabolism. In a study conducted to further elucidate ciclopirox’s mechanism, several Saccharomyces cerevisiae mutants were screened and tested. Results from interpretation of the effects of both the drug treatment and mutation suggested that ciclopirox may exert its effect by disrupting DNA repair, cell division signals and structures (mitotic spindles) as well as some elements of intracellular transport.[6]
It is currently being investigated as an alternative treatment to ketoconazole for seborrhoeic dermatitis as it suppresses growth of the yeast Malassezia furfur. Initial results show similar efficacy to ketoconazole with a relative increase in subjective symptom relief due to its inherent anti-inflammatory properties.[7]
Chemistry
Ciclopirox is a considered a hydroxypyrimidine (sic) antifungal agent.[citation needed] Structurally, ciclopirox is the N-oxide of a 2-hydroxypyridine derivative and therefore ought to be termed a hydroxypyridine antifungal agent. Additionally, the structure as drawn above is the lactam tautomer and indicates the molecule being an N-Hydroxy-2-pyridone. Hence the classification of ciclopirox as a 2-pyridone antifungal agent.
Ciclopirox is used clinically as ciclopirox olamine, the olamine salt of ciclopirox.
Literatures:
Lohaus; Dittmar Arzneimittel-Forschung/Drug Research, 1981 , vol. 31, # 8 a p. 1311 – 1316
Literatures:
Hoechst Aktiengesellschaft Patent: US3972888 A1, 1976 ;
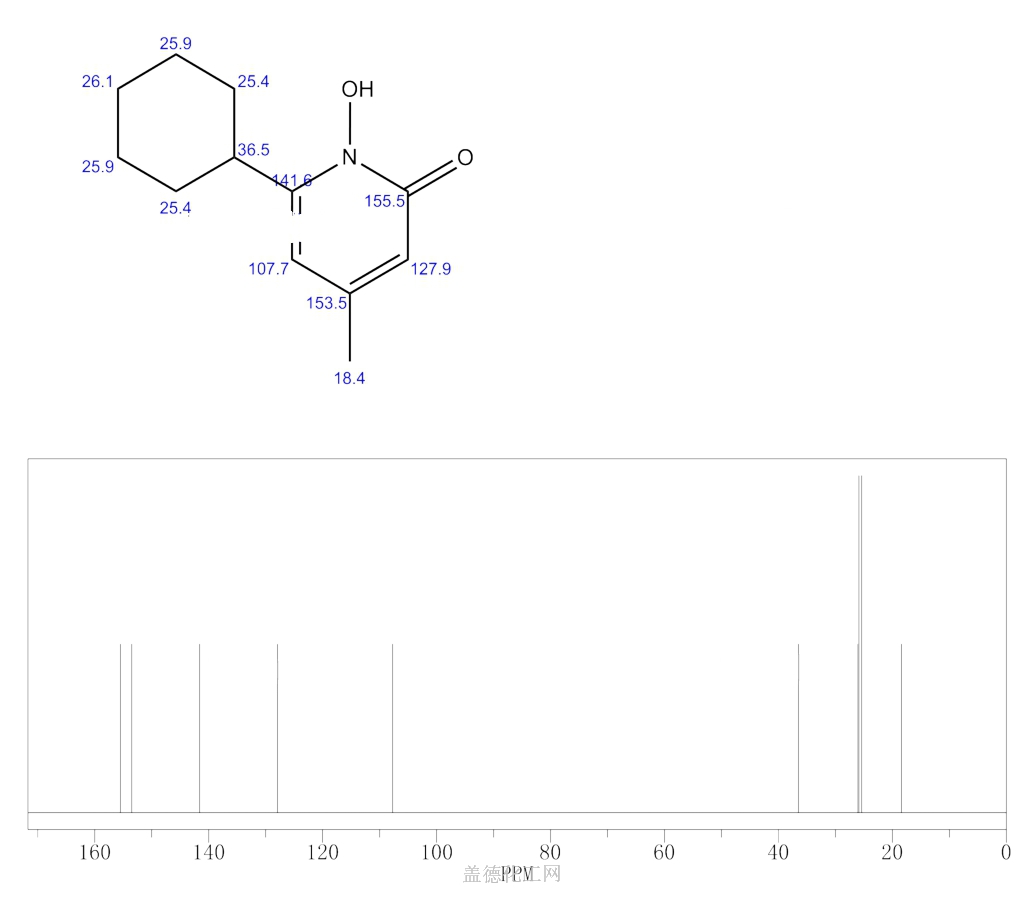
SYNTHESIS
SYN

SYN
W. Dittmar, E. Druckrey andBroad spectrum antimycotic agent with some antibacterial activity. Prepn: G. Lohaus, W. Dittmar, ZA 6906039; eidem, US 3883545 (1970, 1975 both to Hoechst). In vitro study: eidem, Arzneim.-Forsch. 23, 670 (1973).
H. Urbach, J. Med. Chem., 17, 753 (1974); W. Dittmar and G. Lohaus,
German Patent 2,214,608 (1973); Chem. Abstr., 79: 146419w (1973).

SYN
ethanolamine (CAS NO.: ), with other names as 6-Cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridinone 2-aminoethanol, could be produced through the following synthetic routes.
.jpg)
Compound can be prepared in two different ways: 1) The reaction of methyl 5-oxo-5-cyclohexyl-3-methylpentenoate (I) with NH2OH gives the corresponding oxime (II), which is then cyclized to 6-cyclohexyl-1-hydroxy-4-methyl-2(1H)-pyridone (III). Finally, this compound is salified with ethanolamine (IV). 2) Compound (III) can also be obtained by reaction of 6-cyclohexyl-4-methyl-2-pyron (V) with hydroxylamine hydrochloride in hot 2-aminopyridine.
SYN
| CAS-RN | Formula | Chemical Name | CAS Index Name |
|---|---|---|---|
| 14818-35-0 | C12H16O2 | 6-cyclohexyl-4-methyl-2-pyrone | 2H-Pyran-2-one, 6-cyclohexyl-4-methyl- |
| 141-43-5 | C2H7NO | ethanolamine | Ethanol, 2-amino- |
| 7803-49-8 | H3NO | hydroxylamine | Hydroxylamine |
PATENT
https://patents.google.com/patent/US9545413B2/en
The molecule 6-cyclohexyl-1-hydroxy-4-methylpyridin-2(1H)-one, also known as Ciclopirox, is a commercially available antifungal agent as an olamine salt. Ciclopirox olamine has been used to treat superficial mycoses and Tinea versicolor following topical application to the skin. Following enteral administration, ciclopirox undergoes significant first-pass effect resulting in low oral bioavailability. The oral route of administration is also associated with gastrointestinal toxicities observed in animals and humans limiting its benefit in animal and human health applications. Ciclopirox olamine has poor solubility, limiting opportunities to deliver the antifungal agent via parenteral administration of suitably potent solutions and suspensions. As such, it would be beneficial to configure ciclopirox for improved water solubility in order to deliver the drug by parenteral routes of administration.
References
- ^ Jump up to:a b Drugs.com International brand names for ciclopirox Page accessed January 201, 2016
- ^ Ciclopirox
- ^ “Ciclopirox Olamine Antifungal Shampoo”. Okdermo. Retrieved 2019-08-06.
- ^ Crawford F (2007). “Topical treatments for fungal infections of the skin and nails of the foot”. The Cochrane Database of Systematic Reviews. 2007 (3): CD001434. doi:10.1002/14651858.CD001434.pub2. PMC 7073424. PMID 17636672.
- ^ Niewerth M, Kunze D, Seibold M, Schaller M, Korting HC, Hube B (June 2003). “Ciclopirox Olamine Treatment Affects the Expression Pattern of Candida albicans Genes Encoding Virulence Factors, Iron Metabolism Proteins, and Drug Resistance Factors”. Antimicrobial Agents and Chemotherapy. 47 (6): 1805–1817. doi:10.1128/AAC.47.6.1805-1817.2003. PMC 155814. PMID 12760852.
- ^ Leem SH, Park JE, Kim IS, Chae JY, Sugino A, Sunwoo Y (2003). “The possible mechanism of action of ciclopirox olamine in the yeast Saccharomyces cerevisiae”. Mol. Cells. 15 (1): 55–61. PMID 12661761.
- ^ Ratnavel RC, Squire RA, Boorman GC (2007). “Clinical efficacies of shampoos containing ciclopirox olamine (1.5%) and ketoconazole (2.0%) in the treatment of seborrhoeic dermatitis”. J Dermatolog Treat. 18 (2): 88–96. doi:10.1080/16537150601092944. PMID 17520465. S2CID 34852507.
| Clinical data | |
|---|---|
| Trade names | Many brand names worldwide[1] |
| AHFS/Drugs.com | Micromedex Detailed Consumer Information |
| MedlinePlus | a604021 |
| Pregnancy category | B |
| Routes of administration | Topical (applied as a nail lacquer, skin cream or shampoo) |
| ATC code | D01AE14 (WHO) G01AX12 (WHO) |
| Legal status | |
| Legal status | US: ℞-onlyRx-only (CA) |
| Pharmacokinetic data | |
| Bioavailability | <5% with prolonged use |
| Protein binding | 94 to 97% |
| Elimination half-life | 1.7 hours |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 29342-05-0 |
| PubChem CID | 2749 |
| DrugBank | DB01188 |
| ChemSpider | 2647 |
| UNII | 19W019ZDRJ |
| KEGG | D03488 |
| ChEBI | CHEBI:453011 |
| ChEMBL | ChEMBL1413 |
| CompTox Dashboard (EPA) | DTXSID9048564 |
| ECHA InfoCard | 100.045.056 |
| Chemical and physical data | |
| Formula | C12H17NO2 |
| Molar mass | 207.269 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
////////CICLOPIROX OLAMINE

NEW DRUG APPROVALS
ONE TIME
$10.00
OTESECONAZOLE
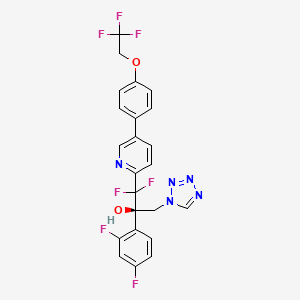

OTESECONAZOLE
VT 1161
オテセコナゾール;
(2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(tetrazol-1-yl)-1-[5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl]propan-2-ol
| C23H16F7N5O2 527.4 | |
| Synonyms | VT 1161 Oteseconazole CAS1340593-59-0 |
|---|
Other Names
- (αR)-α-(2,4-Difluorophenyl)-β,β-difluoro-α-(1H-tetrazol-1-ylmethyl)-5-[4-(2,2,2-trifluoroethoxy)phenyl]-2-pyridineethanol
- (2R)-2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-1,2,3,4-tetrazol-1-yl)- 1-{5-[4-(2,2,2-trifluoroethoxy)phenyl]pyridin-2-yl}propan-2-ol
UPDATE MAY 2022… FDA APPROVED 2022/4/26, Vivjoa
Oteseconazole, sold under the brand name Vivjoa, is a medication used for the treatment of vaginal yeast infections.[1]
It was approved for medical use in the United States in April 2022.[2][3] It was developed by Mycovia Pharmaceuticals.[3]
Names
Oteseconazole is the international nonproprietary name (INN).[4]
Oteseconazole is an azole antifungal used to prevent recurrent vulvovaginal candidiasis in females who are not of reproductive potential.
Oteseconazole, also known as VT-1161, is a tetrazole antifungal agent potentially for the treatment of candidal vaginal infection. VT-1161 Protects Immunosuppressed Mice from Rhizopus arrhizus var. arrhizus Infection. VT-1161 dosed once daily or once weekly exhibits potent efficacy in treatment of dermatophytosis in a guinea pig model.
Oteseconazole has been used in trials studying the treatment of Tinea Pedis, Onychomycosis, Candidiasis, Vulvovaginal, and Recurrent Vulvovaginal Candidiasis.
Mycovia Pharmaceuticals is developing oteseconazole, the lead from a program of metalloenzyme Cyp51 (lanosterol demethylase) inhibitors, developed using the company’s Metallophile technology, for treating fungal infections including onychomycosis and recurrent vulvovaginal candidiasis (RVVC). In July 2021, oteseconazole was reported to be in phase 3 clinical development. Licensee Jiangsu Hengrui Medicine is developing otesaconazole, as an oral capsule formulation, for treating fungal conditions, including RVVC, onychomycosis and invasive fungal infections, in Greater China and planned for a phase 3 trial in April 2021 for treating VVC.
- OriginatorViamet Pharmaceuticals
- DeveloperMycovia Pharmaceuticals; Viamet Pharmaceuticals
- ClassAntifungals; Foot disorder therapies; Pyridines; Small molecules; Tetrazoles
- Mechanism of Action14-alpha demethylase inhibitors
- PreregistrationVulvovaginal candidiasis
- Phase IIOnychomycosis
- No development reportedTinea pedis
- 01 Jun 2021Preregistration for Vulvovaginal candidiasis (In adolescents, In adults, In children, Recurrent) in USA (PO)
- 01 Jun 2021Mycovia intends to launch otesaconazole (Recurrent) for Vulvovaginal candidiasis in the US in early 2022
- 06 Jan 2021Interim efficacy and adverse events data from a phase III ultraVIOLET trial in Vulvovaginal candidiasis released by Mycovia Pharmaceuticals

Synthesis Reference
Hoekstra, WJ., et al. (2020). Antifungal compound process (U.S. Patent No. US 10,745,378 B2). U.S. Patent and Trademark Office. https://patentimages.storage.googleapis.com/f4/62/19/5ba525b1caad0e/US10745378.pdf
PATENT
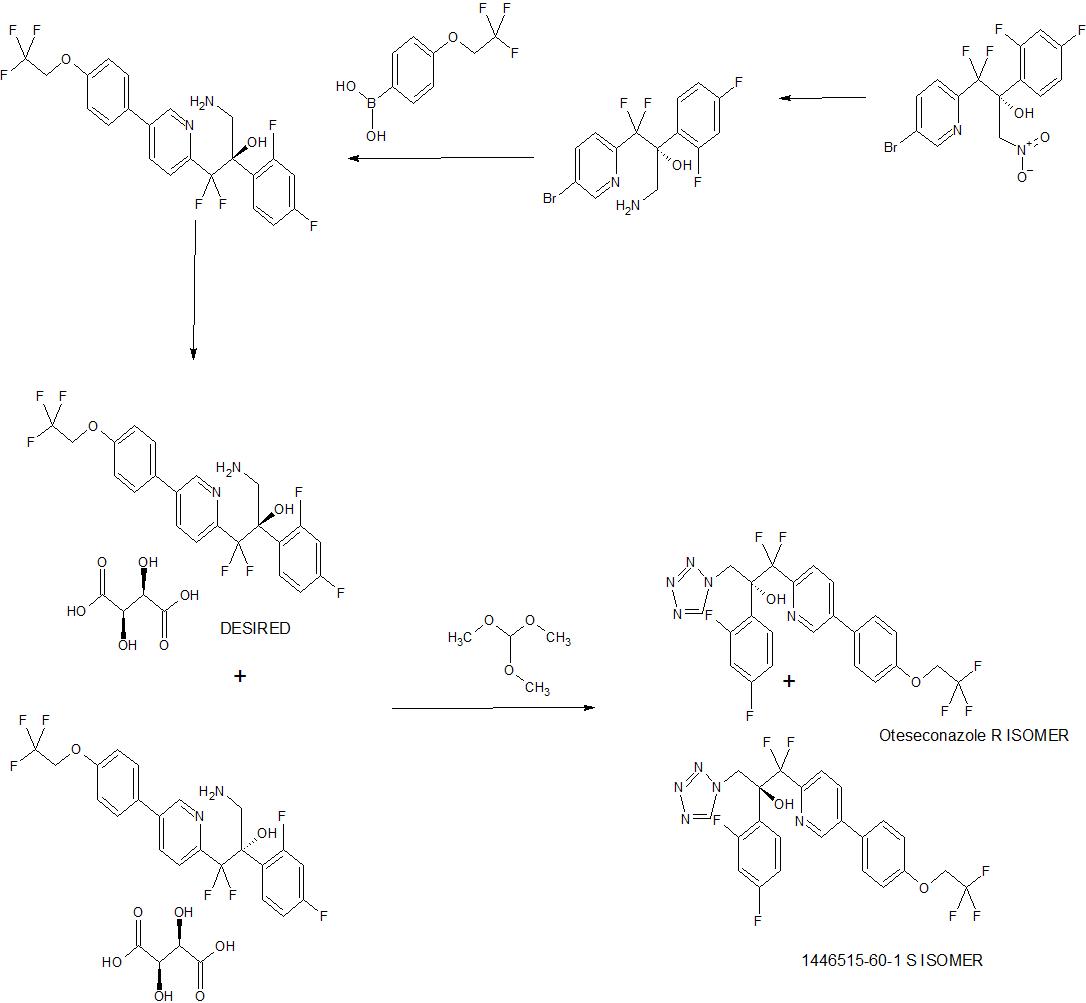
WO 2017049080
WO 2016149486
US 20150024938
WO 2015143172
WO 2015143184
WO 2015143180
WO 2015143142
WO 2013110002
WO 2013109998
WO 2011133875
PATENT
WO 2017049080,
Syn
J. Med. Chem. 2024, 67, 4376−4418
Oteseconazole was approved by the USFDA in April 2022 for the treatment of recurrent vulvovaginal candidiasis in women with a history of vulvovaginal candidiasis and who are not of reproductive
potential. Additional studies for other invasive and opportunistic infections and for onychomycosis are underway.40, The design and discovery of oteseconazole is published by a group from Viamet Pharmaceuticals, now part of Mycovia Pharmaceuticals. It details the racemic synthesis of the drug on
<1 g scale in which the metal-binding tetrazole is installed by treatment of ester 5.2 (Scheme 10) with diazomethane and tetrazole.42
A more scale-friendly asymmetric route that avoided the use of diazomethane was subsequently disclosed in patents and is detailed in Scheme 10 and Scheme 11.43
First, a mixture of ethyl bromodifluoroacetate, stoichiometric copper
powder, and 2,5-dibromopyridine (5.1) in DMSO provided ester 5.2 as an oil that was purified via distillation (Scheme10). Conversion to the aryl ketone 5.5 was achieved via direct addition of lithiated 5.3 or via a two-step process by first conversion to morpholine amide 5.4 followed by addition of
the Grignard generated from aryl bromide 5.3. The resulting ketone 5.5 was a liquid that was carried into the next step without purification.
The key step in the synthesis of 5 is an asymmetric Henry reaction using cinchona alkaloid catalyst 5.6. Addition of nitromethane to ketone 5.5 furnished alcohol 5.7 in 75% yield and ∼90:10 ratio of enantiomers. Next, reduction of the nitro group to the primary amine was accomplished using Pt
catalyzed hydrogenation. The chiral purity of the resulting amine was upgraded by classical resolution using di-p-toluoyl L-tartaric acid to provide 5.8·L-DTTA in 33% yield and >99% chiral purity.Conversion of amino alcohol 5.8 to oteseconazole (5) required two steps: cross coupling to introduce the aryltrifluoroethyl ether fragment and tetrazole formation. These steps were performed in either sequence in the patent. The route shown in Scheme 11 represents the largest scale demonstrated (>100 g input of 5.8). While the use of azide containing reagents presents significant safety risks, no information was provided on safe operation of the tetrazole forming step in the laboratory or on plant scale. Some of the
procedures for tetrazole formation described in the patent would likely require modification for safe scale-up.
To complete the synthesis of oteseconazole, resolved amino alcohol 5.8 first underwent a salt break followed by Suzuki coupling using boronic acid 5.9 to provide biaryl product 5.10 as the L-tartrate salt (Scheme 11). Conversion of 5.10 to 5 was accomplished using TMSN3 in acetic acid with sodium acetate and trimethoxy orthoformate. Treatment of the resulting solution with a Pd scavenger preceded crystallization of the product from EtOH and water after pH adjustment with potassium carbonate. The product was isolated in 85% yield as a hydrated form. Another patent described conversion of the oteseconazolehydrate totheanhydrous form byrecrystallizationfrom EtOHandn-heptanetofurnish5 in90%yield.45
(40) Hoy, S. M. Oteseconazole: First approval. Drugs 2022, 82,1017−1023.
(41) Sobel, J. D.; Nyirjesy, P. Oteseconazole: an advance in
treatment of recurrent vulvovaginal candidiasis. Future Microbiol 2021,
16, 1453−1461.
(42) Hoekstra, W. J.; Garvey, E. P.; Moore, W. R.; Rafferty, S. W.;
Yates, C. M.; Schotzinger, R. J. Design and optimization of highly
selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24,
3455−3458.
(43) Wirth, D. D.; Yates, C. M.; Hoekstra, W. J.; Bindl, M. F.;
Hartmann, E. Process for enantioselective preparation of tetrazolyl
pyridinyl diaryl propanols as antifungal drugs and their precursors.
WO 2017049080, 2017.
(44) González-Bobes, F.; Kopp, N.; Li, L.; Deerberg, J.; Sharma, P.;
Leung, S.; Davies, M.; Bush, J.; Hamm, J.; Hrytsak, M. Scale-up of
Azide Chemistry: A Case Study. Org. Process Res. Dev. 2012, 16,
2051−2057.
(45) Hoekstra, W. J.; Wirth, D. D.; Ehiwe, T.; Bonnaud, T.
Antifungal compounds and processes for making. WO 2016149486,
2016.


.
PATENT
WO-2021143811
Novel crystalline polymorphic form of VT-1161 (also known as oteseconazole) phosphate disodium salt, useful as a prodrug of oteseconazole, for treating systemic fungal infection (eg Candida albicans infection) or onychomycosis.The function of metalloenzymes is highly dependent on the presence of metal ions in the active site of the enzyme. It is recognized that reagents that bind to and inactivate metal ions at the active site greatly reduce the activity of the enzyme. Nature uses this same strategy to reduce the activity of certain metalloenzymes during periods when enzyme activity is not needed. For example, the protein TIMP (tissue inhibitor of metalloproteinases) binds to zinc ions in the active sites of various matrix metalloproteinases, thereby inhibiting enzyme activity. The pharmaceutical industry has used the same strategy in the design of therapeutic agents. For example, the azole antifungal agents fluconazole and voriconazole contain 1-(1,2,4-triazole) group, which exists in the active site of the target enzyme lanosterol demethylase The heme iron binds, thereby inactivating the enzyme. Another example includes zinc-bound hydroxamic acid groups, which have been introduced into most of the published inhibitors of matrix metalloproteinases and histone deacetylases. Another example is the zinc-binding carboxylic acid group, which has been introduced into most of the published angiotensin converting enzyme inhibitors.
VT-1161, the compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2, 2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol, is an antifungal drug developed by VIAMET, currently in the clinical research stage, its structure is as follows Shown:
This compound mainly acts on the CYP51 target of fungal cells. Compared with the previous triazole antifungal drugs, it has the advantages of wider antibacterial spectrum, low toxicity, high safety and good selectivity. However, this compound is not suitable for Liquid preparations (including or excluding the parenteral delivery carrier) are used to treat patients in need thereof.
2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-trifluoro Ethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate is a prodrug of VT-1161.
On the other hand, nearly half of the drug molecules are in the form of salts, and salt formation can improve certain undesirable physicochemical or biological properties of the drug. Relative to 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2- Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl dihydrogen phosphate, it is of great significance to develop salts with more excellent properties in terms of physical and chemical properties or pharmaceutical properties.To this end, the present disclosure provides a new pharmaceutically acceptable salt form of a metalloenzyme inhibitor.Example 1:[0161](R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2, 2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate disodium salt (Compound 1)[0162]
[0163](R)-2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2 ,2-Trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-yl phosphate (compound 1a, prepared according to the method of patent WO2013110002, 0.28g, 0.46mmol, 1.0eq) and ethanol (5mL ) Add to the reaction flask and stir evenly. A solution of NaOH (36.90 mg, 2.0 eq) dissolved in water (1 mL) was added dropwise into the above reaction flask, stirring was continued for 2 h, and concentrated to obtain compound 1, 300 mg of white solid.[0164]After X-ray powder diffraction detection, the XRPD spectrum has no sharp diffraction peaks, as shown in FIG. 10.[0165]Ms:608.10[M-2Na+3H] + .[0166]Ion chromatography detected that the sodium ion content was 6.23%.[0167]Example 2: (R)-((2-(2,4-Difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4 -(2,2,2-Trifluoroethoxy)phenyl)pyridin-2-yl)prop-2-yl)oxy)methyl phosphate disodium salt (compound 2)
[0169]Under ice-cooling, NaH (58mg, 0.87mmol) was added to the reaction flask, 1.5mL of N,N-dimethylformamide and 0.6mL of tetrahydrofuran were added, followed by iodine (38mg, 0.15mmol), and then Compound 2-(2,4-difluorophenyl)-1,1-difluoro-3-(1H-tetrazol-1-yl)-1-(5-(4-(2,2,2-tri Fluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (2b, prepared according to the method of patent WO2013110002, 158mg, 0.3mmol) tetrahydrofuran (1ml) solution was added to the reaction solution, stirred and reacted for 1-4h , And then add compound 2a (519mg, 2.01mmol) in tetrahydrofuran (1ml) solvent to the reaction, stir until the reaction is complete, 10% aqueous ammonium chloride solution to quench the reaction, extract, concentrate and drain, the crude product 2c is directly used for the next One-step reaction, Ms: 750.0[M+H] + .[0170]
[0171]Under ice-bath cooling, add trifluoroacetic acid (0.5mL) to the crude product 2c (300mg) in dichloromethane (2mL) solution, stir until the reaction is complete, and after concentration, the target compound 2d, 82mg, Ms was separated by high performance liquid phase separation. :638.0[M+H] + .[0172]
Add compound 2d (0.29g, 0.46mmol, 1.0eq) and ethanol (5mL) obtained in the previous step into the reaction flask, stir, and add NaOH (36.90mg, 2.0eq) water (1ml) solution dropwise to the aforementioned reaction solution , Stirred for 2-5 h, and concentrated to obtain 2,313 mg of the target compound.
Ms:638.10[M-2Na+3H] + .
PATENT
WO2011133875
https://patents.google.com/patent/WO2011133875A2/en
Product pat, WO2011133875 , protection in the EU states and the US April 2031.
PATENT
WO2015143184 ,
https://patents.google.com/patent/WO2015143184A1/en
Mycovia, claiming a process for preparing antifungal compounds, particularly oteseconazole.EXAMPLE 11

2-(2,4-Difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (11)Compound 11 was prepared using the conditions employed for 1: 0.33 g as a solid. The precursor l-bromo-4-(2,2,2-trifluoroethoxy)benzene was prepared as described below in one step.1H NMR (500 MHz, CDC13): δ 8.76 (s, 1 H), 8.70 (s, 1 H), 7.95 (d, / = 8.0 Hz, 1 H), 7.70 (s, 1 H), 7.64 (d, / = 8.5 Hz, 1 H), 7.54 (d, / = 8.5 Hz, 2 H), 7.42- 7.37 (m, 1 H), 7.08 (d, / = 8.5 Hz, 2 H), 6.79- 6.75 (m, 1 H), 6.69- 6.66 (m, 1 H), 5.58 (d, / = 14.0 Hz, 1 H), 5.14 (d, / = 14.0 Hz, 1 H), 4.44 – 4.39 (m, 2 H). HPLC: 99.1%. MS (ESI): m/z 528 [M++l].Chiral preparative HPLC Specifications for (+)-ll:Column: Chiralpak IA, 250 x 4.6mm, 5uMobile Phase: A) w-Hexane, B) IPAIsocratic: A: B (65:35)Flow Rte: l.OO mL/minOptical rotation [a]D: + 24° (C = 0.1 % in MeOH). 1 -Bromo-4-( 2,2,2-trifluoroethoxy )benzeneTo a stirred solution of trifluoroethyl tosylate (1.5 g, 5.8 mmol) in DMF (20 mL) was added K2CO3 (4 g, 29.4 mmol) followed by addition of p-bromo phenol (1.1 g, 6.46 mmol) at RT under inert atmosphere. The reaction mixture was stirred at 120 °C for 6 h. The volatiles were evaporated under reduced pressure; the residue was diluted with water (5 mL) and extracted with ethyl acetate (3 x 30 mL). The organic layer was washed with water, brine and dried over anhydrous Na2S04, filtered and concentrated in vacuo. The crude compound was purified by silica gel column chromatography eluting with 5% EtOAc/hexane to afford the desired product (0.8 g, 3.13 mmol, 53.3%) as semi solid. 1H NMR (200 MHz, CDC13): δ 7.44 – 7.38 (m, 2 H), 6.86-6.80 (m, 2 H), 4.38- 4.25 (m, 2 H).ExamplesThe present invention will now be demonstrated using specific examples that are not to be construed as limiting.General Experimental ProceduresDefinitions of variables in the structures in schemes herein are commensurate with those of corresponding positions in the formulae delineated herein.Synthesis of 1 or la

A process to prepare enantiopure compound 1 or la is disclosed. Syntheses of lor la may be accomplished using the example syntheses that are shown below (Schemes 1-4). The preparation of precursor ketone 3-Br is performed starting with reaction of 2,5-dibromo- pyridine with ethyl 2-bromo-difluoroacetate to produce ester 2-Br. This ester can be reacted with morpholine to furnish morpholine amide 2b-Br, followed by arylation to provide ketone 3-Br. Alternatively, ketone 3-Br can be afforded directly from ester 2-Br as shown in Scheme 1. Scheme 1. Synthesis of ketone 3-Br r

Ketone 3 may be prepared in an analogous fashion as described in Scheme 1 starting from corresponding substituted 2-bromo-pyridines, which can be prepared according to synthetic transformations known in the art and contained in the references cited herein (Scheme 2).Scheme 2. Synthesis of ketone 3

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, – 0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, – 0(S02)-aryl, or -0(S02)-substituted aryl.Alternatively, compound 1 can be prepared according to Scheme 3 utilizing diols 2-6b (or 2- 6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof). Olefins 2-5a and 2-5 can be prepared by reacting ketones 3 and 1-4 under Wittig olefination conditions (e.g., Ph3PCH3Br and BuLi). Also, as indicated in Scheme 5, any of pyridine compounds, 3, 2-5a, 2-6b, 2-7b, 4*, 4b, or 6 can be converted to the corresponding 4-CF3CH2O-PI1 analogs (e.g., 1-4, 2-5, 2-6a, 2-7a, 5*, 1-6*, or 1 or the corresponding enantiomers, or mixtures thereof) by cross-coupling with 4,4,5, 5-tetramethyl-2- (4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (or the corresponding alkyl boronates or boronic acid or the like), in a suitable solvent system (e.g., an organic-aqueous solvent mixture), in the presence of a transition metal catalyst (e.g., (dppf)PdCl2), and in the presence of a base (e.g., KHCO3, K2C03, Cs2C03, or Na2C03, or the like). Olefins 2-5a and 2-5 can be transformed to the corresponding chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), through exposure to Sharpless asymmetric dihydroxylation conditions: 1) commercially available AD- mix alpha or AD-mix beta with or without additional osmium oxidant and methanesulfonamide, 2) combination of a catalytic osmium oxidant (e.g., Os04 or K20sC>2(OH)4), a stoichiometric iron oxidant (e.g., K3Fe(CN)6), a base (e.g., KHCO3, K2CO3, Cs2C03, or Na2C03, or the like), and a chiral ligand (e.g., (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, (DHQ)2AQN, (DHQD)2PYR, or (DHQ)2PYR; preferably (DHQ)2PHAL, (DHQD)2PHAL, (DHQD)2AQN, and (DHQD)2PYR), or 3) option 2) with methanesulfonamide. The primary alcohol of the resultant chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), can then be activated to afford compounds 2-7b (or 2-7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof). For example, the mesylates can be prepared by exposing chiral diols, 2-6b (or 2-6d, the enantiomer of 2-6b, or mixtures thereof) or 2-6a (or 2-6c, the enantiomer of 2-6a, or mixtures thereof), to methanesulfonyl chloride and a base. Epoxide formation can be affected by the base-mediated (e.g., KHCO3, K2CO3, CS2CO3, or Na2CC>3, or the like) ring closure of compounds 2-7b (or 2- 7d, the enantiomer of 2-7b, or mixtures thereof) or 2-7a (or 2-7c, the enantiomer of 2-7a, or mixtures thereof) to provide epoxides 4* (or 4c*, the enantiomer of 4*, or mixtures thereof) and 5* (or 5-b*, the enantiomer of 5*, or mixtures thereof). The epoxides can then be converted into amino-alcohols 4b (or 4c, the enantiomer of 4b, or mixtures thereof) and 1-6* (or 1-7*, the enantiomer of 1-6*, or mixtures thereof) through ammonia-mediated epoxide opening using ammonia in a suitable solvent (e.g., MeOH, EtOH, or water). Subsequent treatment with TMS-azide in the presence of trimethylorthoformate and sodium acetate in acetic acid would yield compounds 6 (or 6a, the enantiomer of 6, or mixtures thereof) or 1 (or la, the enantiomer of 1, or mixtures thereof) (US 4,426,531).Scheme 3. Synthesis of 1 via Asymmetric Dihydroxylation Method


Y is -OS02-alkyl, -OS02-substituted alkyl, -OS02-aryl, -OS02- substituted aryl, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, – 0(C=0)-aryl, -0(C=0)-substituted aryl, or halogen

R-i = halo, -0(C=0)-alkyl, -0(C=0)-substituted alkyl, -0(C=0)-aryl, -0(C=0)-substituted aryl, -0(C=0)-0-alkyl, -0(C=0)-0-substituted alkyl, -0(C=0)-0-aryl, -0(C=0)-0-substituted aryl, -0(S02)-alkyl, -0(S02)-substituted alkyl, -0(S02)-aryl, or -0(S02)-substituted aryl.Compound 1 (or la, the enantiomer of 1, or mixtures thereof) prepared by any of the methods presented herein can be converted to a sulfonic salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof), as shown in Scheme 4. This can be accomplished by a) combining compound 1 (or la, the enantiomer of 1, or mixtures thereof), a crystallization solvent or crystallization solvent mixture (e.g., EtOAc, i‘PrOAc, EtOH, MeOH, or acetonitrile, or oZ-S-OHcombinations thereof), and a sulfonic acid o (e.g., Z = Ph, p-tolyl, Me, or Et), b) diluting the mixture with an appropriate crystallization co-solvent or crystallization co-solvent mixture (e.g., pentane, methyl i-butylether, hexane, heptane, or toluene, or combinations thereof), and c) filtering the mixture to obtain a sulfonic acid salt of formula IX (or IXa, the enantiomer of IX, or mixtures thereof). cheme 4. Synthesis of a Sulfonic Acid Salt of Compound 1 or la

The following describes the HPLC method used in assessing HPLC purity of the examples and intermediates presented below:Column: Waters XBridge Shield RP18, 4.6 x 150 mm, 3.5 μιηMobile Phase: A = 0.05% TFA/H20, B = 0.05% TFA/ACNAutosampler flush: 1 : 1 ACN/H20Diluent: 1:1 ACN/H20Flow Rate: 1.0 ml/minTemperature: 45 °CDetector: UV 275 nmPump Parameters:

EXAMPLE 1Preparation of ethyl 2-(5-bromopyridin-2-yl)-2,2-difluoroacetate (2-Br)

2-Br Dialkylated impurity In a clean multi-neck round bottom flask, copper powder (274.7 g, 2.05 eq) was suspended in dimethyl sulfoxide (3.5 L, 7 vol) at 20 – 35 °C. Ethyl bromodifluoroacetate (449 g, 1.05 eq) was slowly added to the reaction mixture at 20 – 25 °C and stirred for 1 – 2 h. 2, 5- dibromopyridine (500 g, 1 eq) was added to the reaction mixture and the temperature was increased to 35 – 40 °C. The reaction mixture was maintained at this temperature for 18 – 24 h and the reaction progress was monitored by GC.After the completion of the reaction, ethyl acetate (7 L, 14 vol) was added to the reaction mixture and stirring was continued for 60 – 90 min at 20 – 35 °C. The reaction mixture was filtered through a Celite bed (100 g; 0.2 times w/w Celite and 1L; 2 vol ethyl acetate). The reactor was washed with ethyl acetate (6 L, 12 vol) and the washings were filtered through a Celite bed. The Celite bed was finally washed with ethyl acetate (1 L, 2 vol) and all the filtered mother liquors were combined. The pooled ethyl acetate solution was cooled to 8 – 10 °C, washed with the buffer solution (5 L, 10 vol) below 15 °C (Note: The addition of buffer solution was exothermic in nature. Controlled addition of buffer was required to maintain the reaction mixture temperature below 15 °C). The ethyl acetate layer was washed again with the buffer solution until (7.5 L; 3 x 5 vol) the aqueous layer remained colorless. The organic layer was washed with a 1: 1 solution of 10 % w/w aqueous sodium chloride and the buffer solution (2.5 L; 5 vol). The organic layer was then transferred into a dry reactor and the ethyl acetate was distilled under reduced pressure to get crude 2-Br.The crude 2-Br was purified by high vacuum fractional distillation and the distilled fractions having 2-Br purity greater than 93 % (with the dialkylated not more than 2 % and starting material less than 0.5 %) were pooled together to afford 2-Br.Yield after distillation: 47.7 % with > 93 % purity by GC (pale yellow liquid). Another 10 % yield was obtained by re-distillation of impure fractions resulting in overall yield of ~ 55 – 60 %.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz): 8.85 (1H, d, 1.6 Hz), 8.34 (1H, dd, J = 2.0 Hz, 6.8 Hz), 7.83 (1H, d, J = 6.8 Hz), 4.33 (2H, q, J = 6.0 Hz), 1.22 (3H, t, J = 6.0 Hz). 13C NMR: 162.22 (i, -C=0), 150.40 (Ar-C-), 149.35 (t, Ar-C), 140.52 (Ar-C), 123.01 (Ar-C), 122.07 (Ar-C), 111.80 (t, -CF2), 63.23 (-OCH2-), 13.45 (-CH2CH3).EXAMPLE 2
Preparation of2-( 5-bromopyridin-2-yl )-l -(2,4-difluorophenyl )-2, 2-difluoroethanone ( 3-Br ) A. One-step Method

l-Bromo-2,4-difluorobenzene (268.7 g; 1.3 eq) was dissolved in methyl tert butyl ether (MTBE, 3.78 L, 12.6 vol) at 20 – 35 °C and the reaction mixture was cooled to -70 to -65 °C using acetone/dry ice bath. n-Butyl lithium (689 rriL, 1.3 eq; 2.5 M) was then added to the reaction mixture maintaining the reaction temperature below -65 °C (Note: Controlled addition of the n-Butyl Lithium to the reaction mixture was needed to maintain the reaction mixture temperature below – 65 °C). After maintaining the reaction mixture at this temperature for 30 – 45 min, 2-Br (300 g, 1 eq) dissolved in MTBE (900 rriL, 3 vol) was added to the reaction mixture below – 65 °C. The reaction mixture was continued to stir at this temperature for 60 – 90 min and the reaction progress was monitored by GC.The reaction was quenched by slow addition of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) below -65 °C. The reaction mixture was gradually warmed to 20 – 35 °C and an additional amount of 20 % w/w ammonium chloride solution (750 mL, 2.5 vol) was added. The aqueous layer was separated, the organic layer was washed with a 10 % w/w sodium bicarbonate solution (600 mL, 2 vol) followed by a 5 % sodium chloride wash (600 mL, 2 vol). The organic layer was dried over sodium sulfate (60 g; 0.2 times w/w), filtered and the sodium sulfate was washed with MTBE (300 mL, 1 vol). The organic layer along with washings was distilled below 45 °C under reduced pressure until no more solvent was collected in the receiver. The distillation temperature was increased to 55 – 60 °C, maintained under vacuum for 3 – 4 h and cooled to 20 – 35 °C to afford 275 g (73.6 % yield, 72.71 % purity by HPLC) of 3-Br as a pale yellow liquid.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.63 (1H, d, 1.6 Hz, Ar-H), 8.07 – 8.01 (2H, m, 2 x Ar-H), 7.72 (1H, d, J = 6.8 Hz, Ar-H), 7.07 – 6.82 (1H, m, Ar-H), 6.81 – 6.80 (1H, m, Ar-H). 13C NMR: 185.60 (t, -C=0), 166.42 (dd, Ar-C-), 162.24 (dd, Ar-C),150.80 (Ar-C), 150.35 (Ar-C), 140.02 (Ar-C), 133.82 (Ar-C), 123.06 (Ar-C), 1122.33 (Ar-C), 118.44 (Ar-C), 114.07 (-CF2-), 122.07 (Ar-C), 105.09 (Ar-C).
B. Two-step Method via 2b-Br

2-Br (147.0 g) was dissolved in n-heptane (1.21 L) and transferred to a 5-L reactor equipped with overhead stirrer, thermocouple, condenser and addition funnel. Morpholine (202 ml) was added. The solution was heated to 60 °C and stirred overnight. The reaction was complete by HPLC analysis (0.2% 2-Br; 94.7% 2b-Br). The reaction was cooled to room temperature and 1.21 L of MTBE was added. The solution was cooled to ~4 °C and quenched by slow addition of 30% citric acid (563 ml) to maintain the internal temperature <15 °C. After stirring for one hour the layers were allowed to settle and were separated (Aq. pH=5). The organic layer was washed with 30% citric acid (322 ml) and 9% NaHC03 (322 ml, aq. pH 7+ after separation). The organic layer was concentrated on the rotary evaporator (Note 1) to 454 g (some precipitation started immediately and increased during concentration). After stirring at room temperature the suspension was filtered and the product cake was washed with n-heptane (200 ml). The solid was dried in a vacuum oven at room temperature to provide 129.2 g (77%) dense powder. The purity was 96.5% by HPLC analysis.To a 1-L flask equipped with overhead stirring, thermocouple, condenser and addition funnel was added magnesium turnings (14.65 g), THF (580 ml) and l-bromo-2,4-difluorobenzene (30.2 g, 0.39 equiv). The mixture was stirred until the reaction initiated and self-heating brought the reaction temperature to 44 °C. The temperature was controlled with a cooling bath as the remaining l-bromo-2,4-difluorobenzene (86.1 g, 1.11 equiv) was added over about 30 min. at an internal temperature of 35-40 °C. The reaction was stirred for 2 hours while gradually cooling to room temperature. The dark yellow solution was further cooled to 12 °C.During the Grignard formation, a jacketed 2-L flask equipped with overhead stirring, thermocouple, and addition funnel was charged with morpholine amide 2b-Br (129.0 g) and THF (645 ml). The mixture was stirred at room temperature until the solid dissolved, and then the solution was cooled to -8.7 °C. The Grignard solution was added via addition funnel over about 30 min. at a temperature of -5 to 0 °C. The reaction was stirred at 0 °C for 1 hour and endpointed by HPLC analysis. The reaction mixture was cooled to -5 °C and quenched by slow addition of 2N HC1 over 1 hour at <10 °C. The mixture was stirred for 0.5 h then the layers were allowed to settle and were separated. The aqueous layer was extracted with MTBE (280 ml). The combined organic layers were washed with 9% NaHCC>3 (263 g) and 20% NaCl (258 ml). The organic layer was concentrated on the rotary evaporator with THF rinses to transfer all the solution to the distillation flask. Additional THF (100 ml) and toluene (3 x 100 ml) were added and distilled to remove residual water from the product. After drying under vacuum, the residue was 159.8 g of a dark brown waxy solid (>theory). The purity was approximately 93% by HPLC analysis.EXAMPLE 3Preparation of 3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (±ib-Br)

4-Br (200g, 1 eq) was added into methanolic ammonia (8.0 L; 40 vol; ammonia content: 15 – 20 % w/v) in an autoclave at 10 – 20 °C. The reaction mixture was gradually heated to 60 – 65 °C and at 3 – 4 kg/cm2 under sealed conditions for 10 – 12 h. The reaction progress was monitored by GC. After completion of the reaction, the reaction mixture was cooled to 20 – 30 °C and released the pressure gradually. The solvent was distilled under reduced pressure below 50 °C and the crude obtained was azeotroped with methanol (2 x 600 mL, 6 vol) followed by with isopropanol (600 mL, 2 vol) to afford 203 g (96.98 % yield, purity by HPLC: 94.04 %) of +4b-Br. EXAMPLE 4Preparation of3-amino-l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoropropan- -ol (4b-Br or 2c-Br)

Amino alcohol ±4b-Br (150 g, 1 eq) was dissolved in an isopropanol /acetonitrile mixture (1.5L, 8:2 ratio, 10 vol) and Di-p-toluoyl-L-tartaric acid (L-DPTTA) (84.05 g, 0.55 eq) was added into the reactor at 20 – 30 °C. The reaction mixture was heated to 45 – 50 °C for 1 – 1.5 h (Note: The reaction mixture becomes clear and then became heterogeneous). The reaction mixture was gradually cooled to 20 – 30 °C and stirred for 16 – 18 h. The progress of the resolution was monitored by chiral HPLC analysis.After the completion of the resolution, the reaction mixture was gradually cooled to 20 – 35 °C. The reaction mixture was filtered and the filtered solid was washed with a mixture of acetonitrile and isopropanol (8:2 mixture, 300 mL, 2 vol) and dried to afford 75 g of the L- DPTTA salt (95.37 % ee). The L-DPTTA salt obtained was chirally enriched by suspending the salt in isopropanol /acetonitrile (8:2 mixture; 750 mL, 5 vol) at 45 – 50 °C for 24 – 48 h. The chiral enhancement was monitored by chiral HPLC; the solution was gradually cooled to 20 – 25 °C, filtered and washed with an isoporpanol /acetonitrile mixture (8:2 mixture; 1 vol). The purification process was repeated and after filtration, the salt resulted in chiral purity greater than 96 % ee. The filtered compound was dried under reduced pressure at 35 – 40 °C to afford 62 g of the enantio-enriched L-DPPTA salt with 97.12% ee as an off-white solid. The enantio-enriched L-DPTTA salt (50 g, 1 eq) was dissolved in methanol (150 mL, 3 vol) at 20 – 30 °C and a potassium carbonate solution (18.05 g K2CO3 in 150 mL water) was slowly added at 20 – 30 °C under stirring. The reaction mixture was maintained at this temperature for 2 – 3 h (pH of the solution at was maintained at 9). Water (600 mL, 12 vol) was added into the reaction mixture through an additional funnel and the reaction mixture was stirred for 2 – 3 h at 20 – 30 °C. The solids were filtered; washed with water (150 mL, 3 vol) and dried under vacuum at 40 – 45 °C to afford 26.5 g of amino alcohol 4b-Br or 4c-Br with 99.54 % chemical purity, 99.28 % ee as an off-white solid. (Water content of the chiral amino alcohol is below 0.10 % w/w).1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):8.68 (1H, d, J = 2.0 Hz, Ar- H), 8.16 (1H, dd, J = 8.0 Hz, 2.0 Hz, Ar-H), 7.49 – 7.43 (1H, m, Ar-H), 7.40 (1H, d, J = 8 Hz, Ar-H), 7.16 – 7.11 (1H, m, Ar-H), 7.11 – 6.99 (1H, m, Ar-H), 3.39 – 3.36 (1H, m, -OCHAHB– ), 3.25 – 3.22 (1H, m, -OCHAHB-).13C NMR: 163.87 -158.52 (dd, 2 x Ar-C-), 150.88 (Ar-C), 149.16 (Ar-C), 139.21 (Ar-C), 132.39 (Ar-C), 124.49 (Ar-C), 122.17 (Ar-C), 121.87 (d, Ar- C), 119.91 (t, -CF2-), 110.68 (Ar-C), 103.97 (i, Ar-C), 77.41 (i,-C-OH), 44.17 (-CH2-NH2).EXAMPLE 5
Preparation of l-(5-bromopyridin-2-yl)-2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l- yl)propan-2-ol (l-6*-Br or l-7*-Br)

4b-Br or 4c-Br (20.0 g, 1 eq.) was added to acetic acid (50 mL, 2.5 vol) at 25 – 35 °C followed by the addition of anhydrous sodium acetate (4.32 g, 1 eq), trimethyl orthoformate (15.08 g, 2.7 eq). The reaction mixture was stirred for 15 – 20 min at this temperature and trimethylsilyl azide (12.74 g, 2.1 eq) was added to the reaction mixture (Chilled water was circulated through the condenser to minimize the loss of trimethylsilyl azide from the reaction mixture by evaporation). The reaction mixture was then heated to 70 – 75 °C and maintained at this temperature for 2 -3 h. The reaction progress was monitored by HPLC. Once the reaction was complete, the reaction mixture was cooled to 25 – 35 °C and water (200 mL, 10 vol) was added. The reaction mixture was extracted with ethyl acetate (400 mL, 20 vol) and the aqueous layer was back extracted with ethyl acetate (100 mL, 5 vol). The combined organic layers were washed with 10 % potassium carbonate solution (3 x 200 mL; 3 x 10 vol) followed by a 10 % NaCl wash (1 x 200 mL, 10 vol). The organic layer was distilled under reduced pressure below 45 °C. The crude obtained was azeotroped with heptanes (3 x 200 mL) to get 21.5g (94 % yield, 99.26 5 purity) of tetrazole 1-6* or 1-7* compound as pale brown solid (low melting solid).1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz NMR instrument): 9.13 (1H, Ar-H), 8.74 (1H, Ar-H), 8.22 – 8.20 (1H, m, Ar-H), 7.44 (1H, d, J = 7.2 Hz, Ar-H), 7.29 (1H„Ar-H), 7.23 – 7.17 (1H, m, Ar-H), 6.92 – 6.88 (1H, Ar-H), 5.61 (1H, d, J = 1 1.2 Hz, – OCHAHB-), 5.08 (1H, d, J = 5.6 Hz, -OCHAHB-).13C NMR: 163.67 -161.59 (dd, Ar-C-), 160.60 – 158.50 (dd, Ar-C-), 149.65 (Ar-C), 144.99 (Ar-C), 139.75 (Ar-C), 131.65 (Ar-C), 124.26 (Ar-C), 122.32 (d, Ar-C), 119.16 (t, -CF2-), 118.70 (d, Ar-C), 1 11.05 (d, Ar-C) 104.29 (t, Ar-C), 76.79 (i,-C-OH), 59.72 (Ar-C), 50.23 (-OCH2N-). EXAMPLE 6Preparation of 2-(2,4-difluorophenyl)-l , 1 -difluoro-3-( 1 H-tetrazol-1 -yl)-l -(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)A. Preparation of 1 or la via l-6*-Br or l-7*-Br

Synthesis of 4,4,5, 5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane Potassium carbonate (59.7 g, 2.2 eq.) was added to a slurry of DMF (190 mL, 3.8 Vol.), 4- Bromo phenol (37.4g, 1.1 eq.) and 2,2,2-trifluroethyl tosylate (50.0 g, 1.0 eq.) at 20 – 35 °C under an inert atmosphere. The reaction mixture was heated to 115 – 120 °C and maintained at this temperature for 15 – 18 h. The reaction progress was monitored by GC. The reaction mixture was then cooled to 20 – 35 °C, toluene (200 mL, 4.0 vol.) and water (365 mL, 7. 3 vol.) were added at the same temperature, stirred for 10 – 15 minutes and separated the layers. The aqueous layer was extracted with toluene (200 mL, 4.0 vol.). The organic layers were combined and washed with a 2M sodium hydroxide solution (175 mL, 3.5 vol.) followed by a 20 % sodium chloride solution (175 mL, 3.5 vol.). The organic layer was then dried over anhydrous sodium sulfate and filtered. The toluene layer was transferred into clean reactor, spurged with argon gas for not less than 1 h. Bis(Pinacolato) diborane (47 g, 1.1 eq.), potassium acetate (49.6 g, 3.0 eq.) and 1,4-dioxane (430 mL, 10 vol.) were added at 20 -35 °C, and spurged the reaction mixture with argon gas for at least 1 h. Pd(dppf)Cl2 (6.88 g, 0.05eq) was added to the reaction mixture and continued the argon spurging for 10 – 15 minutes. The reaction mixture temperature was increased to 70 – 75 °C, maintained the temperature under argon atmosphere for 15 – 35 h and monitored the reaction progress by GC. The reaction mixture was cooled to 20 – 35 °C, filtered the reaction mixture through a Celite pad, and washed with ethyl acetate (86 mL, 2 vol.). The filtrate was washed with water (430 mL, 10 vol.). The aqueous layer was extracted with ethyl acetate (258 mL, 6 vol.) and washed the combined organic layers with a 10 % sodium chloride solution (215 mL, 5 vol.). The organic layer was dried over anhydrous sodium sulfate (43g, 1 time w/w), filtered and concentrated under reduced pressure below 45 °C to afford crude 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g; 71 % yield with the purity of 85.18 % by GC). The crude 4,4,5,5-tetramethyl-2-(4-(2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (65 g, 1 eq.) was dissolved in 10 % ethyl acetate – n-Heptane (455 mL, 7 vol.) and stirred for 30 – 50 minutes at 20 – 35 °C. The solution was filtered through a Celite bed and washed with 10 % ethyl acetate in n-Heptane (195 mL, 3 vol.). The filtrate and washings were pooled together, concentrated under vacuum below 45 °C to afford 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a thick syrup (45.5 g; 70 % recovery). This was then dissolved in 3 % ethyl acetate-n-heptane (4 vol.) and adsorbed on 100 – 200 M silica gel (2 times), eluted through silica (4 times) using 3 % ethyl acetate – n- heptane. The product rich fractions were pooled together and concentrated under vacuum. The column purified fractions (> 85 % pure) were transferred into a round bottom flask equipped with a distillation set-up. The compound was distilled under high vacuum below 180 °C and collected into multiple fractions. The purity of fractions was analyzed by GC (should be > 98 % with single max impurity < 1.0 %). The less pure fractions (> 85 % and < 98 % pure fraction) were pooled together and the distillation was repeated to get 19g (32% yield) of 4,4,5, 5-tetramethyl-2-(4- (2,2,2-trifluoroethoxy)phenyl)-l,3,2-dioxaborolane as a pale yellow liquid.*H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):7.64 (2H, d, 6.8 Hz), 7.06 (2H, d, J = 6.4 Hz), 4.79 (2H, q, J = 6.8 Hz), 1.28 (12H, s).13C NMR: 159.46 (Ar-C-O-), 136.24 (2 x Ar-C-), 127.77 – 120.9 (q, -CF3), 122.0 (Ar-C-B), 114.22 (2 x Ar-C-), 64.75 (q, J = 27.5 Hz).Synthesis of 2-(2.4-difluorophenyl)-l.l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)l-6*-Br or l-7*-Br (14 g, 0.03 mol, 1 eq) was added to tetrahydrofuran (168 mL, 12 vol) at 25 – 35 °C and the resulting solution was heated to 40 – 45 °C. The reaction mixture was maintained at this temperature for 20 – 30 min under argon bubbling. Sodium carbonate (8.59 g, 0.08 mol, 2.5 eq) and water (21 mL, 1.5 vol) were added into the reaction mixture and the bubbling of argon was continued for another 20 – 30 min. 4,4,5, 5-tetramethyl-2-(4-(2,2,2- trifluoroethoxy)phenyl)-l,3,2-dioxaborolane (10.76 g, 1.1 eq) dissolved in tetrahydrofuran (42 mL, 3 vol) was added into the reaction mixture and argon bubbling was continued for 20 – 30 min. Pd(dppf)Cl2 (2.65 g, 0.1 eq) was added to the reaction mixture under argon bubbling and stirred for 20 – 30 min (Reaction mixture turned into dark red color). The reaction mixture was heated to 65 – 70 °C and maintained at this temperature for 3 – 4 h. The reaction progress was monitored by HPLC. The reaction mixture was cooled to 40 – 45 °C and the solvent was distilled under reduced pressure. Toluene (350 mL, 25 vol.) was added to the reaction mixture and stirred for 10 – 15 min followed by the addition of water (140 mL, 10 vol). The reaction mixture was filtered through Hyflo (42 g, 3 times), the layers were separated and the organic layer was washed with water (70 mL, 5 vol) and a 20 % w/w sodium chloride solution (140 mL, 10 vol). The organic layer was treated with charcoal (5.6 g, 0.4 times, neutral chalrcoal), filtered through Hyflo. (lS)-lO-Camphor sulfonic acid (7.2 g, 1 eq.) was added to the toluene layer and the resulting mixture was heated to 70 – 75 °C for 2 – 3 h. The reaction mixture was gradually cooled to 25 – 35 °C and stirred for 1 – 2 h. The solids were filtered, washed with toluene (2 x 5 vol.) and then dried under vacuum below 45 °C to afford 18.0 g of an off white solid. The solids (13.5 g, 1 eq.) were suspended in toluene (135 mL, 10 vol) and neutralized by adding 1M NaOH solution (1.48 vol, 1.1 eq) at 25 – 35 °C and stirred for 20 – 30 min. Water (67.5 mL, 5 vol) was added to the reaction mixture and stirred for 10 – 15 min, and then the layers were separated. The organic layer was washed with water (67.5 mL, 5 vol) to remove the traces of CSA. The toluene was removed under reduced pressure below 45 °C to afford crude 1 or la. Traces of toluene were removed by azeotroping with ethanol (3 x 10 vol), after which light brown solid of crude 1 or la (7.5 g, 80% yield) was obtained.The crude 1 or la (5 g) was dissolved in ethanol (90 mL, 18 vol.) at 20 – 35 °C, and heated to 40 – 45 °C. Water (14 vol) was added to the solution at 40 – 45 °C, the solution was maintained at this temperature for 30 – 45 min and then gradually cooled to 20 – 35 °C. The resulting suspension was continued to stir for 16 – 18 h at 20 – 35 °C, an additional amount of water (4 vol.) was added and the stirring continued for 3 – 4 h. The solids were filtered to afford 4.0 g (80% recovery) of 1 or la (HPLC purity >98%) as an off-white solid.1H NMR: δ values with respect to TMS (DMSO-d6; 400 MHz):9.15 (1H, s, Ar-H), 8.93 (1H, d, J = 0.8 Hz, Ar-H), .8.22 – 8.20 (1H, m, Ar-H), 7.80 (2H, d, J = 6.8 Hz, Ar-H), 7.52 (1H, d, J = 6.8 Hz, Ar-H), 7.29 (1H, d,J = 3.2Hz, Ar-H), 7.27 – 7.21 (1H, m, Ar-H), 7.23 – 7.21 (2H, d, J = 6.8 Hz, Ar-H), 7.19 (1H, d, J = 6.8 Hz, Ar-H), 6.93 – 6.89 (1H, m, Ar-H), 5.68 (1H, / = 12 Hz, -CHAHB), 5.12 (2H, d, J = 11.6 Hz, -CHAHB), 4.85 (2H, q, J = 1.6 Hz).13C NMR: 163.93 – 158.33 (m, 2 x Ar-C), 157.56 (Ar-C), 149.32 (i, Ar-C), 146.40 (Ar-C), 145.02 (Ar-C), 136.20 (Ar-C), 134.26 (2 x Ar-C), 131.88 – 131.74 (m, AR-C), 129.72 (Ar-C), 128.47 (2 x Ar-C), 123.97 (q, -CF2-), 122.41 (Ar-C), 119.30 (-CF3), 118.99 (Ar-C), 115.65 (2 x Ar-C), 110.99 (d, Ar-C), 104.22 (i, Ar-C), 77.41 – 76.80 (m, Ar-C), 64.72 (q, -OCH2-CF3), 50.54 (-CH2-N-).B. Preparation of 1 or la via 4b-Br or 4c-Br


Synthesis of 3-amino-2-(2.4-difluorophenyl)-l.l-difluoro-l-(5-(4-(2.2.2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (8a or 8b)Potassium carbonate (30.4 g) and water (53.3 g) were charged to a 1-L flask equipped with overhead stirring, thermocouple, and nitrogen/vacuum inlet valve, and stirred until dissolved. The boronic acid (19.37 g), a solution of 4b-Br or 4c-Br in 2-butanol (103.5 g, 27.8 g theoretical 4b-Br or 4c-Br)) and 2-BuOH (147.1 g) were added and stirred to form a clear mixture. The flask was evacuated and refilled with nitrogen 3 times. Pd(d f)2Cl2 (0.30 g) was added and stirred to form a light orange solution. The flask was evacuated and refilled with nitrogen 4 times. The mixture was heated to 85 °C and stirred overnight and endpointed by HPLC analysis. The reaction mixture was cooled to 60 °C and the layers were allowed to settle. The aqueous layer was separated. The organic layer was washed with 5% NaCl solution (5 x 100 ml) at 30-40 °C. The organic layer was filtered and transferred to a clean flask with rinses of 2-BuOH. The combined solution was 309.7 g, water content 13.6 wt% by KF analysis. The solution was diluted with 2-BuOH (189 g) and water (10 g). Theoretically the solution contained 34.8 g product, 522 ml (15 volumes) of 2-BuOH, and 52.2 ml (1.5 volumes) of water. L-Tartaric acid (13.25 g) was added and the mixture was heated to a target temperature of 70-75 °C. During the heat-up, a thick suspension formed. After about 15 minutes at 70-72 °C the suspension became fluid and easily stirred. The suspension was cooled at a rate of 10 °C/hour to 25 °C then stirred at 25 °C for about 10 hours. The product was collected on a vacuum filter and washed with 10:1 (v/v) 2-BuOH/water (50 ml) and 2- butanol (40 ml). The salt was dried in a vacuum oven at 60 °C with a nitrogen purge for 2 days. The yield was 40.08 g of 8a or 8b as a fluffy, grayish-white solid. The water content was 0.13 wt% by KF analysis. The yield was 87.3% with an HPLC purity of 99.48%. Synthesis of 2-(2,4-difluorophenyl)-l,l-difluoro-3-(lH-tetrazol-l-yl)-l-(5-(4-(2,2,2- trifluoroethoxy)phenyl)pyridin-2-yl)propan-2-ol (1 or la)To a 350 ml pressure bottle were charged acetic acid (73 ml), 8a or 8b (34.8 g), sodium acetate (4.58 g) and trimethylorthoformate (16.0 g). The mixture was stirred for 18 min. at room temperature until a uniform suspension was obtained. Azidotrimethylsilane (8.88 g) was added and the bottle was sealed. The bottle was immersed in an oil bath and magnetically stirred. The oil bath was at 52 °C initially, and was warmed to 62-64 °C over about ½ hour. The suspension was stirred at 62-64 °C overnight. After 20.5 hours the suspension was cooled to room temperature and sampled. The reaction was complete by HPLC analysis. The reaction was combined with three other reactions that used the same raw material lots and general procedure (total of 3.0 g additional starting material). The combined reactions were diluted with ethyl acetate (370 ml) and water (368 ml) and stirred for about ½ hour at room temperature. The layers were settled and separated. The organic layer was washed with 10% K2C03 solution (370 ml/ 397 g) and 20% NaCl solution (370 ml/ 424 g). The organic layer (319 g) was concentrated, diluted with ethanol (202 g) and filtered, rinsed with ethanol (83 g). The combined filtrate was concentrated to 74 g of amber solution.The crude 1 or la solution in ethanol (74 g solution, containing theoretically 31.9 g 1 or la) was transferred to a 2-L flask equipped with overhead stirring, thermocouple, and addition funnel. Ethanol (335 g) was added including that used to complete the transfer of the 1 or la solution. The solution was heated to nominally 50 °C and water (392 g) was added over 12 minutes. The resulting hazy solution was seeded with 1 or la crystals and stirred at 50 °C. After about ½ hour the mixture was allowed to cool to 40 °C over about ½ hour during which time crystallization started. Some darker colored chunky solid separated out from the main suspension. The pH of the crystallizing mixture was adjusted from 4.5 to 6 using 41% KOH (1.7 g). After about 1 hour a good suspension had formed. Additional water (191 g) was added slowly over ½ hour. The suspension was heated to 50 °C and cooled at 5 °C/min to room temperature. After stirring overnight the suspension was cooled in a water bath to 16 °C and filtered after 1 hour. The wet cake was washed with 55:45 (v/v) water/ethanol (2 x 50 ml) and air-dried on the vacuum filter funnel overnight. Further drying at 40 °C in a vacuum oven with a nitrogen bleed resulted in no additional weight loss. The yield was 30.2 g of off-white fine powder plus some darker granular material. By in-process HPLC analysis there was no difference in the chemical purity of the darker and lighter materials. The purity was 99.4%. The water content was 2.16 wt% by KF analysis. The residual ethanol was 1.7 wt% estimated by ‘Ft NMR analysis. The corrected yield was 29.0 g, 91.0% overall yield for tetrazole formation and crystallization. The melting point was 65 °C by DSC analysis.
FDA Approves Mycovia Pharmaceuticals’ VIVJOA™ (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)
– Approval of VIVJOA™ marks a significant therapeutic advancement for reducing the incidence of RVVC, a condition with substantial unmet need, in permanently infertile and postmenopausal women
– VIVJOA™ is the first FDA approval in Mycovia’s pipeline of novel treatments for fungal infections
– U.S. commercial launch of VIVJOA™ expected in Q2
April 28, 2022 07:55 AM Eastern Daylight Time
DURHAM, N.C.–(BUSINESS WIRE)–The U.S. Food and Drug Administration (FDA) approved VIVJOA™ (oteseconazole capsules), an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication for this condition and provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. VIVJOA is the first FDA-approved product for Mycovia Pharmaceuticals, Inc. (Mycovia), an emerging biopharmaceutical company dedicated to recognizing and empowering those living with unmet medical needs by developing novel therapies.
“We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans”Tweet this
RVVC, also known as chronic yeast infection, is defined by the Centers for Disease Control and Prevention (CDC) as three or more symptomatic acute episodes of yeast infection in 12 months. RVVC is a distinct condition from vulvovaginal candidiasis (VVC), and until now, there have been no FDA-approved medications specifically indicated for it. Nearly 75% of all adult women will have at least one yeast infection in their lifetime, with approximately half experiencing a recurrence. Of those women, up to 9% develop RVVC.
“After nearly two decades of living with chronic yeast infection and feeling like there was no hope from the itchiness, irritation and constant dread of when the next yeast infection would return, I was overjoyed to even be a part of this clinical trial,” said Leslie Ivey, RVVC patient and clinical trial participant. “It is gratifying to see RVVC finally get the attention it deserves.”
Symptoms of RVVC include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain.
VIVJOA’s FDA approval is based upon the positive results from three Phase 3 trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries. In the two global VIOLET studies, 93.3% and 96.1% of women with RVVC who received VIVJOA did not have a recurrence for the 48-week maintenance period compared to 57.2% and 60.6% of patients who received placebo (p <0.001). In the ultraVIOLET study, 89.7% of women with RVVC who received VIVJOA cleared their initial yeast infection and did not have a recurrence for the 50-week maintenance period compared to 57.1% of those who received fluconazole followed by placebo (p <0.001). The most common side effects reported in Phase 3 clinical studies were headache (7.4%) and nausea (3.6%). VIVJOA is contraindicated in those with a hypersensitivity to oteseconazole, and based on data from rat studies, also in females who are of reproductive potential, pregnant, or lactating. Please see additional Important Safety Information below.
Patrick Jordan, CEO of Mycovia Pharmaceuticals and Partner at NovaQuest Capital Management, stated, “We celebrate this important milestone for Mycovia, as VIVJOA is the first antifungal in our pipeline to obtain FDA approval and achieves our goal to fulfill a previously unmet medical need among women suffering from RVVC. We are honored to lead this advancement in women’s health.”
“We believe the market need for VIVJOA is strong, and we are eager to execute our commercial plans,” Jordan continued. “As we enter a new chapter of our history as a commercial biopharmaceutical company, we will continue driving our mission forward to develop novel therapies for overlooked conditions.”
Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA granted oteseconazole Qualified Infectious Disease Product and Fast Track designations.
“A medicine with VIVJOA’s sustained efficacy combined with the clinical safety profile has been long needed, as until now, physicians and their patients have had no FDA-approved medications for RVVC,” stated Stephen Brand, Ph.D., Chief Development Officer of Mycovia. “We are excited to be the first to offer a medication designed specifically for RVVC, a challenging and chronic condition that is expected to increase in prevalence over the next decade.”
Mycovia is planning its commercial launch of VIVJOA™ in the second quarter of 2022.
About Recurrent Vulvovaginal Candidiasis
RVVC is a debilitating, chronic infectious condition that affects 138 million women worldwide each year. RVVC, also known as chronic yeast infection, is a distinct condition from vulvovaginal candidiasis (VVC) and defined as three or more symptomatic acute episodes of yeast infection in 12 months. Primary symptoms include vaginal itching, burning, irritation and inflammation. Some women may experience abnormal vaginal discharge and painful sexual intercourse or urination, causing variable but often severe discomfort and pain.
About VIVJOA™
VIVJOA™ (oteseconazole) is an azole antifungal indicated to reduce the incidence of recurrent vulvovaginal candidiasis (RVVC) in females with a history of RVVC who are NOT of reproductive potential. VIVJOA is the first and only FDA-approved medication that provides sustained efficacy demonstrated by significant long-term reduction of RVVC recurrence through 50 weeks versus comparators. Oteseconazole is designed to inhibit fungal CYP51, which is required for fungal cell wall integrity, and this selective interaction is also toxic to fungi, resulting in the inhibition of fungal growth. Due to its chemical structure, oteseconazole has a lower affinity for human CYP enzymes as compared to fungal CYP enzymes. The FDA approved VIVJOA based upon the positive results from three Phase 3 clinical trials of oteseconazole – two global, pivotal VIOLET studies and one U.S.-focused ultraVIOLET study, including 875 patients at 232 sites across 11 countries.
References
- ^ Jump up to:a b https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/215888s000lbl.pdf
- ^ “Vivjoa: FDA-Approved Drugs”. U.S. Food and Drug Administration (FDA). Retrieved 27 April 2022.
- ^ Jump up to:a b “FDA Approves Mycovia Pharmaceuticals’ VIVJOA (oteseconazole), the First and Only FDA-Approved Medication for Recurrent Vulvovaginal Candidiasis (Chronic Yeast Infection)” (Press release). Mycovia Pharmaceuticals. 28 April 2022. Retrieved 28 April 2022 – via Business Wire.
- ^ World Health Organization (2016). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 76”. WHO Drug Information. 30 (3). hdl:10665/331020.
Further reading
- Sobel JD, Nyirjesy P (December 2021). “Oteseconazole: an advance in treatment of recurrent vulvovaginal candidiasis”. Future Microbiology. 16: 1453–1461. doi:10.2217/fmb-2021-0173. PMID 34783586.
External links
- “Oteseconazole”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03562156 for “A Study of Oral Oteseconazole for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03561701 for “A Study of Oral Oteseconazole (VT-1161) for the Treatment of Patients With Recurrent Vaginal Candidiasis (Yeast Infection) (VIOLET)” at ClinicalTrials.gov
- Clinical trial number NCT03840616 for “Study of Oral Oteseconazole (VT-1161) for Acute Yeast Infections in Patients With Recurrent Yeast Infections (ultraVIOLET)” at ClinicalTrials.gov
| Clinical data | |
|---|---|
| Trade names | Vivjoa |
| Other names | VT-1161 |
| License data | US DailyMed: Oteseconazole |
| Routes of administration | By mouth |
| Drug class | Antifungal |
| ATC code | J02AC06 (WHO) |
| Legal status | |
| Legal status | US: ℞-only [1] |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1340593-59-0 |
| PubChem CID | 77050711 |
| DrugBank | DB13055 |
| ChemSpider | 52083215 |
| UNII | VHH774W97N |
| KEGG | D11785 |
| ChEBI | CHEBI:188153 |
| ChEMBL | ChEMBL3311228 |
| ECHA InfoCard | 100.277.989 |
| Chemical and physical data | |
| Formula | C23H16F7N5O2 |
| Molar mass | 527.403 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
/////////OTESECONAZOLE, vt 1161, fungal infection, Candida albicans infection, onychomycosis, PHASE 3,



AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
C1=CC(=CC=C1C2=CN=C(C=C2)C(C(CN3C=NN=N3)(C4=C(C=C(C=C4)F)F)O)(F)F)OCC(F)(F)F
NEW DRUG APPROVALS
ONE TIME
$10.00
Ibrexafungerp citrate, Brexafemme
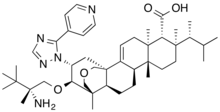
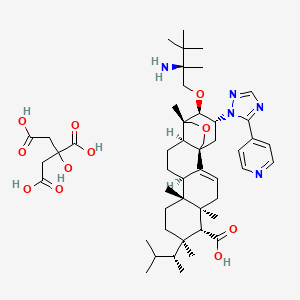

Ibrexafungerp citrate
| アイブレキサフンジェルプクエン酸塩; |
| Formula | C44H67N5O4. C6H8O7 |
|---|---|
| cas | Citrate1965291-08-0 free 1207753-03-4 |
| Mol weight | 922.1574 |
Brexafemme, fda approved 2021, 2021/6/1
Antifungal, Cell wall biosynthesis inhibitor, Treatment of invasive fungal infections due to Candida spp. or Aspergillus spp., vulvovaginal candidiasis
SCY-078 citrate, MK-3118; SCY-078,
- WHO 10597
(1R,5S,6R,7R,10R,11R,14R,15S,20R,21R)-21-[(2R)-2-amino-2,3,3-trimethylbutoxy]-5,7,10,15-tetramethyl-7-[(2R)-3-methylbutan-2-yl]-20-(5-pyridin-4-yl-1,2,4-triazol-1-yl)-17-oxapentacyclo[13.3.3.01,14.02,11.05,10]henicos-2-ene-6-carboxylic acid;2-hydroxypropane-1,2,3-tricarboxylic acid
- Originator Merck & Co; SCYNEXIS
- Class Antifungals; Glycosides; Triterpenes
- Mechanism of ActionBeta-1,3-D glucan synthetase inhibitors
- Orphan Drug StatusYes – Invasive bronchopulmonary aspergillosis; Candidiasis
- RegisteredVulvovaginal candidiasis
- Phase IIICandidiasis
- Phase IIInvasive bronchopulmonary aspergillosis
- Phase IUnspecified
- PreclinicalPneumocystis pneumonia
- 01 Jun 2021Registered for Vulvovaginal candidiasis (In adolescents, In children, In the elderly, In adults) in USA (PO)
- 01 May 2021Ibrexafungerp – SCYNEXIS receives Qualified Infectious Disease Product status for Vulvovaginal candidiasis (Recurrent, Prevention) in USA
- 30 Apr 2021Efficacy data from phase III VANISH-303 and VANISH-306 trials in Vulvovaginal Candidiasis presented at the 2021 American College of Obstetricians and Gynecologists Annual Meeting (ACOG-2021)
Ibrexafungerp, sold under the brand name Brexafemme, is an antifungal medication used to treat vulvovaginal candidiasis (VVC) (vaginal yeast infection).[1] It is taken by mouth.[1]
Ibrexafungerp is a triterpenoid antifungal.[1]
Ibrexafungerp was approved for medical use in the United States in June 2021.[1][2] It is the first approved drug in a novel antifungal class.[2]
Medical uses
Ibrexafungerp is indicated for the treatment of adult and postmenarchal pediatric females with vulvovaginal candidiasis (VVC).[1][2]
Syn
https://www.sciencedirect.com/science/article/abs/pii/S0960894X20307721

Abstract
We previously reported medicinal chemistry efforts that identified MK-5204, an orally efficacious β-1,3-glucan synthesis inhibitor derived from the natural product enfumafungin. Further extensive optimization of the C2 triazole substituent identified 4-pyridyl as the preferred replacement for the carboxamide of MK-5204, leading to improvements in antifungal activity in the presence of serum, and increased oral exposure. Reoptimizing the aminoether at C3 in the presence of this newly discovered C2 substituent, confirmed that the (R) t-butyl, methyl aminoether of MK-5204 provided the best balance of these two key parameters, culminating in the discovery of ibrexafungerp, which is currently in phase III clinical trials. Ibrexafungerp displayed significantly improved oral efficacy in murine infection models, making it a superior candidate for clinical development as an oral treatment for Candida and Aspergillus infections.
AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com
……
SYN
Bioorg. Med. Chem. Lett. 2021, 32, 127661.
PATENT
WO 2010019204
https://patents.google.com/patent/WO2010019204A1/en
SYN
https://doi.org/10.1021/acs.jmedchem.3c00501
Ibrexafungerp (Brexafemme). Ibrexafungerp (1), formerly SCY-078 or MK-3118 and developed by Scynexis Inc., is a first-in-class triterpenoid antifungal that inhibits the biosynthesis of β-(1,3)
D-glucan in the fungal cell wall. This mechanism of action provides an opportunity for the treatment
of fungal infections that are azole- or echinocandrin-resistant strains. In June 2021, ibrexafungerp received its first approval by the United States Food and Drug Administration (USFDA) for
the treatment of vulvovaginal candidiasis in adult and postmenarchal pediatric females.
24,25 Ibrexafungerp is a semisynthetic derivative of the natural product enfumafungin that
incorporates a pyridine triazole moiety on the core phenanthropyran ring system as well as a pendant 2-amino-2,3,3trimethyl-butyl ether. The drug demonstrates potent, broad spectrum activity against Candida sp. and is orally bioavailable.
As shown in Scheme 1, the synthesis of ibrexafungerp started with the natural product enfumafungin (1.1). The lactol of enfumafungin was reduced using triethylsilane and trifluoroacetic acid to give pyran 1.2. Treatmentwith H2SO4 in methanol resulted in cleavage of the glucose moiety to generate 1.3 in 87%
yield over 2 steps. Carboxylic acid 1.3 was converted to the corresponding benzyl ester upon treatment with benzyl bromide to give compound 1.4in an89%yield. Reaction of 1.4 with (R)
N-sulfonyl aziridine 1.5 (prepared as shown in Scheme 2) in the presence of potassium t-pentylate and the cation complexing agent 18-crown-6 provided ether 1.6 in 78% yield. Metal reduction with sodiumin liquid ammoniaconcurrently removed the N-sulfonyl benzyl groups to generate compound 1.7, which
was converted to hydrazine intermediate 1.8 with anhydrous hydrazine and BF32·OEt 28-30 in 1,2-dichloroethane (DCE). Cyclocondensation of 1.8 with acyl amidine derivative 1.9 upon heating in acetic acid then provided ibrexafungerp (1) in 66% yield.
Thepreparationof(R)-N-sulfonylaziridine1.5 isdescribedin Scheme 2. Condensation of 3,3-dimethylbutan-2-one (1.10)with (R)-p-toluenesulfinamide (1.11) gave an 84% yield of compound 1.12, which cyclized upon treatment with trimethylsulfoxonium chloride and n-butyllithium to give chiral toluenesulfinyl aziridine 1.13 in 64% yield. Oxidation of 1.13 with meta-chloroperoxybenzoic acid afforded the tosyl-pro
tected (R)-alpha-disubstituted aziridine 1.5..
(24) Lee, A. Ibrexafungerp: First approval. Drugs 2021, 81, 1445−
1450.
(25) Jallow, S.; Govender, N. P. Ibrexafungerp: A first-in-class oral
triterpenoid glucan synthase inhibitor. J. Fungi 2021, 7, 163.
(26) Lamoth, F.; Alexander, B. D. Antifungal activities of SCY-078
(MK-3118) and standard antifungal agents against clinical non
aspergillus mold isolates. Antimicrob. Agents Chemother. 2015, 59,
4308−4311
(27) Scorneaux, B.; Angulo, D.; Borroto-Esoda, K.; Ghannoum, M.;
Peel, M.; Wring, S. SCY-078 is fungicidal against Candida species in
time-kill studies. Antimicrob. Agents Chemother. 2017, 61, e01961-16.
(28) Apgar, J. M.; Wilkening, R. R.; Parker, D. L.; Meng, D.;
Wildonger, K. J.; Sperbeck, D.; Greenlee, M. L.; Balkovec, J. M.;
Flattery, A. M.; Abruzzo, G. K.; Galgoci, A. M.; Giacobbe, R. A.; Gill, C.
J.; Hsu, M.-J.; Liberator, P.; Misura, A. S.; Motyl, M.; Nielsen Kahn, J.;
Powles, M.; Racine, F.; Dragovic, J.; Fan, W.; Kirwan, R.; Lee, S.; Liu,
H.; Mamai, A.; Nelson, K.; Peel, M. Ibrexafungerp: an orally active β
1,3-glucan synthesis inhibitor. Bioorg. Med. Chem. Lett. 2021, 32,
127661.
(29) Greenlee, M. L.; Wilkening, R.; Apgar, J.; Sperbeck, D.;
Wildonger, K. J.; Meng, D.; Parker, D. L.; Pacofsky, G. J.; Heasley, B.
H.; Mamai, A.; Nelson, K. Antifungal Agents. WO 2010019204, 2010.
(30) Greenlee, M. L.; Wilkening, R.; Apgar, J.; Wildonger, K. J.; Meng,
D.; Parker, D. L. Antifungal Agents. WO 2010019203A1, 2010.
(31) Imran, M.; Khan, S. A.; Alshammari, M. K.; Alqahtani, A. M.;
Alanazi, T. A.; Kamal, M.; Jawaid, T.; Ghoneim, M. M.; Alshehri, S.;
Shakeel, F. Discovery, development, inventions and patent review of
fexinidazole: The first all-oral therapy for human African trypanoso
miasis. Pharmaceuticals 2022, 15, 128.


SYN
European Journal of Medicinal Chemistry 245 (2023) 114898
The gram-scale synthesis of this drug is demonstrated in Scheme 3 [50]. Starting with triterpene glycoside enfumafungin 14, a reduction of the bridging hemiacetal with triethylsilane provided the intermediate 15, followed by hydrolysis, etherification and benzyl protection, gave compound 16 in 76% yield over 2 steps. Subsequent ring-opening alkylation reaction of 16 with tosyl protected aziridine 17 gave com pound 18, which then underwent Borch reduction to provide the in termediate 19. Treatment of 19 with biaryl 20 in the presence of boron trifluoride diethyl etherate gave rise to the substitution product ibrexafungerp. In this synthetic method, the pyridine-triazolium biaryl and chiral benzene sulfonamide were elegantly introduced into the triterpene enfumafungin through ring-opening and substitution reactions to give the triterpene derivative. These elegant and practical synthetic
methods could be employed as the versatile tools for the synthesis of other drug molecules.
[50] J.M. Apgar, R.R. Wilkening, D.L. Parker, J.D. Meng, K.J. Wildonger, D. Sperbeck,
M.L. Greenlee, J.M. Balkovec, A.M. Flattery, G.K. Abruzzo, A.M. Galgoci, R.
A. Giacobbe, C.J. Gill, M.J. Hsu, P. Liberator, A.S. Misura, M. Motyl, J.N. Kahn,
M. Powles, F. Racine, J. Dragovic, W. Fan, R. Kirwan, S. Lee, H. Liu, A. Mamai,
K. Nelson, M. Peel, Ibrexafungerp: an orally active β-1, 3-glucan synthesis
inhibitor, Bioorg, Med. Chem. Lett. 32 (2021), 127661.

.
References
- ^ Jump up to:a b c d e f g https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/214900s000lbl.pdf
- ^ Jump up to:a b c “Scynexis Announces FDA Approval of Brexafemme (ibrexafungerp tablets) as the First and Only Oral Non-Azole Treatment for Vaginal Yeast Infections”. Scynexis, Inc. (Press release). 2 June 2021. Retrieved 2 June 2021.
Further reading
- Azie N, Angulo D, Dehn B, Sobel JD (September 2020). “Oral Ibrexafungerp: an investigational agent for the treatment of vulvovaginal candidiasis”. Expert Opin Investig Drugs. 29 (9): 893–900. doi:10.1080/13543784.2020.1791820. PMID 32746636.
- Davis MR, Donnelley MA, Thompson GR (July 2020). “Ibrexafungerp: A novel oral glucan synthase inhibitor”. Med Mycol. 58 (5): 579–592. doi:10.1093/mmy/myz083. PMID 31342066.
- Petraitis V, Petraitiene R, Katragkou A, Maung BB, Naing E, Kavaliauskas P, et al. (May 2020). “Combination Therapy with Ibrexafungerp (Formerly SCY-078), a First-in-Class Triterpenoid Inhibitor of (1→3)-β-d-Glucan Synthesis, and Isavuconazole for Treatment of Experimental Invasive Pulmonary Aspergillosis”. Antimicrob Agents Chemother. 64 (6). doi:10.1128/AAC.02429-19. PMC 7269506. PMID 32179521.
External links
- “Ibrexafungerp”. Drug Information Portal. U.S. National Library of Medicine.
- Clinical trial number NCT03734991 for “Efficacy and Safety of Oral Ibrexafungerp (SCY-078) vs. Placebo in Subjects With Acute Vulvovaginal Candidiasis (VANISH 303)” at ClinicalTrials.gov
- Clinical trial number NCT03987620 for “Efficacy and Safety of Oral Ibrexafungerp (SCY-078) vs. Placebo in Subjects With Acute Vulvovaginal Candidiasis (Vanish 306)” at ClinicalTrials.gov
Ibrexafungerp, also known as SCY-078 or MK-3118, is a novel enfumafungin derivative oral triterpene antifungal approved for the treatment of vulvovaginal candidiasis (VVC), also known as a vaginal yeast infection.1,9 It was developed out of a need to treat fungal infections that may have become resistant to echinocandins or azole antifungals.1 Ibrexafungerp is orally bioavailable compared to the echinocandins caspofungin, micafungin, and anidulafungin; which can only be administered parenterally.1,2 Similar to echinocandins, ibrexafungerp targets the fungal β-1,3-glucan synthase, which is not present in humans, limiting the chance of renal or hepatic toxicity.6,9
Ibrexafungerp was granted FDA approval on 1 June 2021.9
β-1,3-glucan synthase is composed of a catalytic subunit, FKS1 or FKS2, and a GTP-binding regulatory subunit, Rho1.5,6 This synthase is involved in the synthesis of β-1,3-glucan, a fungal cell wall component.6
Ibrexafungerp acts similarly to the echinocandin antifungals, by inhibiting the synthesis of β-1,3-glucan synthase.1,9 While echinocandins bind to the FKS1 domain of β-1,3-glucan synthase, enfumafungin and its derivatives bind at an alternate site which allows them to maintain their activity against fungal infections that are resistant to echinocandins.3,4
Ibrexafungerp has been shown in animal studies to distribute well to vaginal tissue, making it a favourable treatment for vulvovaginal candidiasis.4
- Wring SA, Randolph R, Park S, Abruzzo G, Chen Q, Flattery A, Garrett G, Peel M, Outcalt R, Powell K, Trucksis M, Angulo D, Borroto-Esoda K: Preclinical Pharmacokinetics and Pharmacodynamic Target of SCY-078, a First-in-Class Orally Active Antifungal Glucan Synthesis Inhibitor, in Murine Models of Disseminated Candidiasis. Antimicrob Agents Chemother. 2017 Mar 24;61(4). pii: AAC.02068-16. doi: 10.1128/AAC.02068-16. Print 2017 Apr. [Article]
- Hector RF, Bierer DE: New beta-glucan inhibitors as antifungal drugs. Expert Opin Ther Pat. 2011 Oct;21(10):1597-610. doi: 10.1517/13543776.2011.603899. Epub 2011 Jul 25. [Article]
- Kuhnert E, Li Y, Lan N, Yue Q, Chen L, Cox RJ, An Z, Yokoyama K, Bills GF: Enfumafungin synthase represents a novel lineage of fungal triterpene cyclases. Environ Microbiol. 2018 Sep;20(9):3325-3342. doi: 10.1111/1462-2920.14333. Epub 2018 Sep 13. [Article]
- Larkin EL, Long L, Isham N, Borroto-Esoda K, Barat S, Angulo D, Wring S, Ghannoum M: A Novel 1,3-Beta-d-Glucan Inhibitor, Ibrexafungerp (Formerly SCY-078), Shows Potent Activity in the Lower pH Environment of Vulvovaginitis. Antimicrob Agents Chemother. 2019 Apr 25;63(5). pii: AAC.02611-18. doi: 10.1128/AAC.02611-18. Print 2019 May. [Article]
- Ha YS, Covert SF, Momany M: FsFKS1, the 1,3-beta-glucan synthase from the caspofungin-resistant fungus Fusarium solani. Eukaryot Cell. 2006 Jul;5(7):1036-42. doi: 10.1128/EC.00030-06. [Article]
- Perlin DS: Mechanisms of echinocandin antifungal drug resistance. Ann N Y Acad Sci. 2015 Sep;1354:1-11. doi: 10.1111/nyas.12831. Epub 2015 Jul 17. [Article]
- Wring S, Murphy G, Atiee G, Corr C, Hyman M, Willett M, Angulo D: Clinical Pharmacokinetics and Drug-Drug Interaction Potential for Coadministered SCY-078, an Oral Fungicidal Glucan Synthase Inhibitor, and Tacrolimus. Clin Pharmacol Drug Dev. 2019 Jan;8(1):60-69. doi: 10.1002/cpdd.588. Epub 2018 Jun 27. [Article]
- Ghannoum M, Arendrup MC, Chaturvedi VP, Lockhart SR, McCormick TS, Chaturvedi S, Berkow EL, Juneja D, Tarai B, Azie N, Angulo D, Walsh TJ: Ibrexafungerp: A Novel Oral Triterpenoid Antifungal in Development for the Treatment of Candida auris Infections. Antibiotics (Basel). 2020 Aug 25;9(9). pii: antibiotics9090539. doi: 10.3390/antibiotics9090539. [Article]
- FDA Approved Drug Products: Brexafemme (Ibrexafungerp) Oral Tablet [Link]
| Clinical data | |
|---|---|
| Pronunciation | /aɪˌbrɛksəˈfʌndʒɜːrp/ eye-BREKS-ə-FUN-jurp |
| Trade names | Brexafemme |
| Other names | SCY-078 |
| License data | US DailyMed: Ibrexafungerp |
| Pregnancy category | Contraindicated[1] |
| Routes of administration | oral, intravenous |
| Drug class | Antifungal |
| ATC code | J02AX07 (WHO) |
| Legal status | |
| Legal status | US: ℞-only[1] |
| Pharmacokinetic data | |
| Protein binding | >99%[1] |
| Metabolism | Hydroxylation (CYP3A4) then conjugation (glucuronidation, sulfation)[1] |
| Elimination half-life | 20 hours[1] |
| Identifiers | |
| IUPAC name | |
| CAS Number | 1207753-03-4as citrate: 1965291-08-0 |
| PubChem CID | 46871657as citrate: 137552087 |
| DrugBank | DB12471as citrate: DBSALT003185 |
| UNII | A92JFM5XNUas citrate: M4NU2SDX3E |
| KEGG | D11544as citrate: D11545 |
| ChEMBL | ChEMBL4297513as citrate: ChEMBL4298168 |
| CompTox Dashboard (EPA) | DTXSID901336871 |
| Chemical and physical data | |
| Formula | C44H67N5O4 |
| Molar mass | 730.051 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| SMILES | |
| InChI | |
/////////Ibrexafungerp citrate, Brexafemme, アイブレキサフンジェルプクエン酸塩 , SCY-078 citrate, UNII-M4NU2SDX3E, M4NU2SDX3E, MK-3118; SCY-078, Orphan Drug, Merck, SCYNEXIS, WHO 10597, ANTI FUNGAL
CC(C)C(C)C1(CCC2(C3CCC4C5(COCC4(C3=CCC2(C1C(=O)O)C)CC(C5OCC(C)(C(C)(C)C)N)N6C(=NC=N6)C7=CC=NC=C7)C)C)C.C(C(=O)O)C(CC(=O)O)(C(=O)O)O
NEW DRUG APPROVALS
ONE TIME
$10.00

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}