
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Lenacapavir sodium
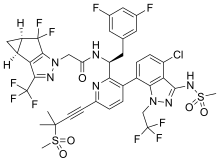
Lenacapavir sodium
レナカパビルナトリウム
| Formula |
C39H31ClF10N7O5S2. Na
C39H32ClF10N7O5S2 FREE FORM
|
|---|---|
| CAS |
2283356-12-5
2189684-44-2 FEE FORM
|
| Mol weight |
990.2641
968.28 FREE FORM
|
2022/8/17 EMA APPROVED, SUNLECA
N-[(1S)-1-[3-[4-chloro-3-(methanesulfonamido)-1-(2,2,2-trifluoroethyl)indazol-7-yl]-6-(3-methyl-3-methylsulfonylbut-1-ynyl)pyridin-2-yl]-2-(3,5-difluorophenyl)ethyl]-2-[(2S,4R)-5,5-difluoro-9-(trifluoromethyl)-7,8-diazatricyclo[4.3.0.02,4]nona-1(6),8-dien-7-yl]acetamide
|
Treatment of HIV-1 infection
|
PF-3540074, to GS-CA1, GS-6207, GS-HIV, GS-CA1, GS-CA2
Lenacapavir, sold under the brand name Sunlenca, is a medication used to treat HIV/AIDS.[1] It is taken by mouth or by subcutaneous injection.[1]
The most common side effects include reactions at the injection site and nausea.[1]
Lenacapavir was approved for medical use in the European Union in August 2022.[1]
HIV/AIDS remains an area of concern despite the introduction of numerous successful therapies, mainly due to the emergence of multidrug resistance and patient difficulty in adhering to treatment regimens.1,2 Lenacapavir is a first-in-class capsid inhibitor that demonstrates picomolar HIV-1 inhibition as a monotherapy in vitro, little to no cross-resistance with existing antiretroviral agents, and extended pharmacokinetics with subcutaneous dosing.1,2,3,5
Lenacapavir was first globally approved by the European Commission to treat adults with multi-drug resistant HIV infection.7 It is currently being investigated in clinical trials in the US.
U.S. Patent Application No. 15/680,041 discloses novel compounds useful for treating a Retroviridae viral infection, including an infection caused by the HIV virus. One specific compound identified therein is a compound of formula I:
PATENTS
- WO 2018/035359 A1
- Different formulations and salts: WO 2019/035904 A1; WO 2019/035973 A1
PATENT
WO 2019/161280 A1
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019161280
I. Synthesis of Starting Materials and Intermediates
Example la: Preparation of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan- 1-amine (VIII-02), or a co-crystal, solvate, salt, or combination thereof, and starting materials and/or intermediates therein
wherein R4 and R5 are each independently hydrogen, methyl, phenyl, benzyl, 4-nitrobenzyl, 4-chlorobenzyl, 4-brornobenzylamine, or 4-methoxybenzyl
Synthesis of 3,6-dibromopicolinaldehyde (1a)
[00553] A dry reaction flask with magnetic stir-bar was charged with 2,5-dibromopyridine (1.0 g). The flask was inerted under nitrogen, THF (4.2 mL) was added, and the thin slurry agitated. Separately, a dry glass reactor was charged with 2,2,6,6-tetramethylpiperidinylmagnesium chloride, lithium chloride complex (TMPMgCl●LiCl) (5.8 mL, 6.3 mmol). The TMPMgCl●LiCl solution was agitated and cooled to about -20 °C. The 2,5-dibromopyridine solution was added to the TMPMgCl●LiCl solution over about 30 min, maintaining a temperature below about -18 °C. Upon completing the addition, the flask was rinsed forward to the reactor with three additional portions of THF (1 mL x 2), and aged at about -20 for about 1 hour. A solution of N,N-dimethylformamide (1.6 mL, 20 mmol) in THF (1.6 mL) was added to the reactor over about 15 min. The reaction mixture was aged for a further 15 min. and quenched by the addition of a solution of acetic acid (1.9 mL, 34 mmol) in water (10 mL) over about 20 minutes, maintaining a temperature of no more than about 0 °C. To the reactor was added isopropyl acetate (10 mL) and the reaction mixture was warmed to about 20 °C. After aging for 30 min, the mixture was filtered through diatomaceous earth and the reactor rinsed with a mixture of isopropyl acetate (10 mL), saturated aqueous ammonium chloride (10 mL) and 0.2 M aqueous hydrochloric acid (10 mL). The reactor rinse was filtered and the pH of the combined reaction mixture was adjusted to about 8-9 by the addition of a 10% aqueous sodium hydroxide solution (about 6 mL). The mixture was filtered a second time to remove magnesium salts and transferred to a separatory funnel. The phases were separated and the aqueous phase was extracted with isopropyl acetate (3 x 10 mL). The combined organic extracts were washed with 50% saturated aqueous sodium chloride (20 mL), dried over anhydrous sodium sulfate, and filtered. The solution was concentrated to dryness by rotary evaporation and purified by chromatography (eluting with 0-100% ethyl acetate in heptane) to afford 3,6-dibromopicolinaldehyde (1a) as a solid. 1H NMR (400 MHz, DMSO-d6) δ 9.94 (q, J = 0.6 Hz, 1H), 8.19 (dq, J = 8.4, 0.6 Hz, 1H), 7.82 (dt, J = 8.4, 0.7 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 189.33, 148.59, 145.66, 140.17, 133.19, 120.27.
Synthesis of 3,6-dibromopicolinaldehyde (1a)
[00554] A solution of 2,5-dibromo-6-methylpyridine (8.03 g) in THF (81 mL) was cooled to about 0 °C. To this solution was charged tert-butyl nitrite (4.33 g), followed by a dropwise addition of potassium tert-butoxide (28 mL, 1.5 equiv, 20 wt% solution in THF). The reaction mixture was agitated at about 0 °C until the reaction was complete. The reaction mixture was diluted with THF (24 mL), and quenched with ammonium chloride (6.38 g, 119 mmol) in water (43 mL). The reaction mixture was distilled under vacuum to approximately 55 mL to afford a slurry, which was filtered and washed twice with water (2x 24 mL) to afford 1h. 1H NMR (400 MHz, DMSO-d6) δ 11.69 (s, 1H), 8.08 (d, J = 8.4 Hz, 1H), 7.67 (s, 1H), 7.61 (d, J = 8.5 Hz, 1H).
[00555] A solution of glyoxylic acid (407 L, 50 wt% in water) was heated to about 80 °C and in portions was charged with 1h (40.69 kg, 145.4 mol) . Reaction mixture was held at this temperature until the reaction was complete. The reaction mixture was cooled to about 20 °C, filtered, and the filter cake was washed with water until the filtrate had a pH ≥ 5, to afford 1a. 1H NMR (400 MHz, DMSO-d6) δ 9.95 (s, 1H), 8.22 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 8.4 Hz, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00556] Compound 1a (5.0 g, 18.0 mmol) in toluene (20 mL) was heated to about 50 °C and benzhydrylamine (3.47 g, 18.9 mmol) was charged in one portion and agitated at this temperature until the reaction was deemed complete. Methanol (61 mL) was charged and the reaction mixture was distilled to a volume of approximately 25 mL. Methanol (40 mL) was charged and the reaction mixture was distilled to a volume of approximately 30 mL. The resulting slurry was filtered and rinsed with two portions of methanol (15 mL each) and dried under vacuum to afford 1b-02.
[00557] Alternatively, compound 1a (10.0 g, 37.8 mmol) in 2-methyltetrahydrofuran (50 mL) was heated to about 50 °C and benzhydrylamine (7.28 g, 39.7 mmol) was charged dropwise. The reaction was agitated at this temperature until it was deemed complete. The reaction mixture was distilled to a volume of approximately 30 mL. To the reaction mixture was charged heptane (100 mL) and 1b-02 seed (59.3 mg, 0.138 mmol). The resulting slurry was filtered, rinsed with two portions of heptane (2x 20 mL), and dried under vacuum to afford 1b-02. 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.44 – 7.40 (m,
4H), 7.38 – 7.32 (m, 4H), 7.28 – 7.22 (m, 2H), 5.88 (s, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00558] 1a (2.00 g) was combined with isopropanol (7.6 mL) and agitated at ambient temperature. To this mixture was added potassium metabisulfite (0.96 g) in water (3.8 mL), dropwise. This mixture was agitated for at least 90 minutes and the resulting slurry was filtered. The filter cake was rinsed twice with isopropanol (6 mL then 12 mL) to afford 1i-1. 1H NMR (400 MHz, DMSO-d6) δ 7.92 (d, J = 8.3 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 5.48 – 5.38 (m, 2H).
[00559] li-1 (1.00 g) was combined with 2-methyltetrahydrofuran (3.5 mL) and agitated at ambient temperature. To this slurry was charged potassium hydroxide (443.8 mg, 7.91 mmol) in water (4 mL) and the biphasic mixture was agitated for 2 hours. The layers were separated and the aqueous layer was extracted with an additional portion of 2-methyltetrahydrofuran (3.5 mL). To the combined organics was charged benzhydrylamine (0.47 mL, 2.7 mmol). The reaction mixture was concentrated in vacuo (-300 mbar, 45 °C bath) to a volume of approximately 3 mL. Heptane (7 mL) was charged and the mixture was agitated. The resulting slurry was filtered to afford 1b-02 1H NMR (400 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.12 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.44 – 7.40 (m, 4H), 7.38 – 7.32 (m, 4H), 7.28 – 7.22 (m, 2H), 5.88 (s, 1H).
Synthesis of (E)-N-benzhydryl-1-(3,6-dibromopyridin-2-yl)methanimine (1b-02)
[00560] Compound 1a (1.0 g) was added to a reactor, and toluene (6.0 mL) was added to the reactor. The mixture was agitated. Aminodiphenylmethane (0.73 g, 1.05 equiv.) was added to the reaction mixture. The jacket was heated to about 60 °C, and the mixture was allowed to age for about 1 hour. After about one hour, the mixture was carried forward to the next step. 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.60 (d, J = 8.4 Hz, 4H), 7.40 – 7.34 (m, 7H), 7.29 (td, J = 6.9, 6.5, 1.7 Hz, 5H), 7.22 – 7.16 (m, 3H), 5.81 (s, 1H).
Synthesis of N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-1,1-diphenylmethanimine (1d-02)
[00561] A solution of1b-02 in toluene (1.0 g in 3.8 mL) was stirred in a reactor at about 60 °C. Tetrabutylammonium bromide (0. 08 g, 0.10 equiv.) was added, 3,5-difluorobenzylbromide (0.60 g, 1.20 equiv.) was added, and potassium hydroxide (50% in water, 1.3 g, 5 equiv.) was added. The mixture was aged for about 4 hours and sampled for conversion. When the reaction was complete, the aqueous phase was removed, and water (3.1 mL) was added to the reactor. Contents were agitated and phases were allowed to settle. The aqueous phase was removed, and the toluene solution of1d-02 was carried forward to the next step. 1H NMR (400 MHz, Chloroform-d) δ 7.78 (dd, J = 8.6, 1.0 Hz, 1H), 7.64 – 7.60 (m, 2H), 7.59 – 7.53 (m, 1H), 7.49 (d, J = 8.3 Hz, 1H), 7.47 (s, 0H), 7.45 (s, 0H), 7.43 (d, J = 0.7 Hz, 0H), 7.41 – 7.34 (m, 3H), 7.33 (t, J = 1.4 Hz, 1H), 7.28 (t, J = 7.3 Hz, 2H), 7.22 (s, 0H), 7.18 (d, J = 8.3 Hz, 1H), 6.87 (dd, J = 7.7, 1.7 Hz, 2H), 6.55 (dt, J = 9.0, 2.3 Hz, 1H), 6.50 (dd, J = 7.0, 4.9 Hz, 3H), 5.26 (s, 0H), 5.16 (t, J = 6.9 Hz, 1H), 3.32 (dd, J = 13.2, 6.6 Hz, 1H), 3.16 (dd, J = 13.1, 7.2 Hz, 1H).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) from N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-1,1-diphenylmethanimine (1d-02)
[00562] A solution of 1d-02 in toluene (1.0 g in 3.0 mL) was stirred in a reactor at about 60 °C. Sulfuric acid (0.93 g, 5 equiv.) was diluted into water (3.5 mL), and added to the reactor. The mixture was aged for about 4 hours. When the reaction was complete, the aqueous phase was removed. The aqueous phase was recharged to the reactor, and heptane (2.5 mL) was added. The mixture was agitated and agitation stopped and layers allowed to settle. The aqueous phase was removed, and heptane was discharged to waste. Toluene (5.0 mL) and potassium hydroxide (50% in water, 2.1 g, 10 equiv.) was added to the reactor. The aqueous acidic solution was added to the reactor. The mixture was agitated for about 10 minutes, and agitation stopped and phases allowed to settle. The aqueous phase was discharged to waste. Water (2.5 mL) was added to the reactor, and the mixture was agitated for about 5 minutes, and agitation was stopped and the phases were allowed to settle. The aqueous phase was discharged to waste. The toluene solution of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) was carried forward to the next step. 1H NMR (400 MHz, Chloroform-d) δ 7.60 (d, J = 8.3 Hz, 1H), 7.21 (d, J = 8.3 Hz, 1H), 6.74 – 6.67 (m, 2H), 6.66 – 6.58 (m, 1H), 4.57 – 4.45 (m, 1H), 3.02 (dd, J = 13.5, 5.2 Hz, 1H), 2.72 (dd, J = 13.5, 8.6 Hz, 1H), 1.77 (s, 3H).
Synthesis of (S)-1-(3.6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-2-hydroxy-2-phenyl acetate (VIII-03)
[00563] A solution of X in toluene (1.0 g in 7.1 mL) was stirred in a reactor at about 60 °C. The mixture was distilled to minimum volumes (2.9 mL), and methyl tert-butyl ether was added (7.1 mL). (R)-(-)-Mandelic acid (0.41 g, 1 equiv.) was added, and the mixture was cooled to about 0 °C. The newly formed slurry was filtered, providing (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-2-hydroxy-2-phenylacetate (VIII-03). 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 8.4 Hz, 1H), 7.49 (d, J = 8.4 Hz, 1H), 7.34 (d, J = 7.3 Hz, 2H), 7.28 – 7.14 (m, 4H), 7.01 (tt, J = 9.4, 2.3 Hz, 1H), 6.79 (d, J = 7.4 Hz, 3H), 4.77 (s, 1H), 4.55 (d, J = 6.6 Hz, 1H), 3.02 (s, 1H), 2.92 (d, J = 6.7 Hz, 2H), 1.05 (s, 2H).
Synthesis of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine N-acetyl-D- Leucine (VIII-04)
[00564] A reactor was charged with X (15.0 g), N-acetyl-D-leucine (8.28 g) and zinc oxide (0.311 g). Toluene (375 mL) was charged to the reactor followed by 2-pyridinecarboxaldehyde (183 μL). The mixture was aged at about 55 °C for about 6 hrs. and then held at about 35 °C for about 4 days. The mixture was cooled to about 0 °C and held for about 17 hrs. The product was isolated by filtration and the filter cake was washed with cold toluene (2 x 75 mL). The filter cake was re-charged to the reactor. Ethanol (150 mL) was added and the mixture distilled to remove residual toluene. Once the toluene was removed, the reactor volume was adjusted with ethanol to about 90 mL and the mixture was cooled to about 25 °C. Water (210 mL) was added over approximately 10 min. and the mixture aged for approximately 12 hrs. The slurry was filtered and the solids were dried to afford VIII-04. 1H NMR (400 MHz, DMSO-d6) δ 8.03 (d, J = 8.0 Hz, 1H). 7.95 (d, J = 8.3 Hz, 1H), 7.49 (d, 7 8.3 Hz, 1H), 7.03 (tt, J = 9.5, 2.4 Hz, 1H),
6.87 (dtd, J = 8.4, 6.2, 2.2 Hz, 2H), 5.49 (s, 3H), 4.42 (dd, J = 7.9, 5.9 Hz, 1H), 4.18 (q, J = 7.8 Hz, 1H), 2.93 (dd, J = 13.3, 5.9 Hz, 1H), 2.85 (dd, J = 13.2, 8.0 Hz, 1H), 1.83 (s, 3H), 1.71 -1.54 (m, 1H), 1.47 (dd, J = 8.4, 6.2 Hz, 2H), 0.88 (d, J = 6.6 Hz, 3H), 0.83 (d, J = 6.5 Hz, 3H).
13C NMR (101 MHz, DMSO-d6) δ 174.72, 169.03, 162.07 (dd, J = 245.5, 13.3 Hz), 161.79, 143.51, 142.82 (t, J = 9.4 Hz), 139.72, 128.39, 119.30, 113.36 – 111.39 (m), 101.73 (t, J = 25.7 Hz), 55.19, 50.69, 41.74 (d, J = 2.3 Hz), 40.51, 24.36, 22.91, 22.44, 21.46.
Example 1b: Preparation of alternative starting materials and intermediates for use in the formation of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difliiorophenyl)ethan-1-amine (VIII), or a co-crystal, solvate, salt, or combination thereof
Synthesis of (R)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-ol (XII)
[00565] A stainless steel autoclave equipped with a glass inner tube was charged with compound XI (1.00 g) and (A)-RuCY-XylBINAP (16 mg, 0.05 equiv.). EtOH (1.0 mL) and IPA (1.0 mL) followed by tert-BuOK (1.0 M solution in THE, 0.51 mL, 0.2 equiv.) were added to the autoclave. After being purged by H2, the autoclave was charged with 3 MPa
of H2. The mixture was stirred at about 20 °C for about 10 h. To the mixture, cone. HCl aqueous solution was added and pH was adjusted to 2. 1H NMR (400 MHz, CDCl3): δ 7.72 ( d, J = 8.2 Hz, 1H), 7.33 (d, J = 8.2 Hz, 1H), 6.80 -6.72 (m, 2H), 6.68 (tt, J = 9.2, 2.4 Hz, 1H), 5.16 (dd, J = 8.2, 3.4 Hz, 1H), 3.60 (br, 1H), 3.12 (dd, J = 13.8, 3.4 Hz, 1H), 2.81 (dd, J = 13.8, 8.2 Hz,
1H). 13C NMR (100 MHz, CDC13): d 162.8 (dd, J= 246.4, 12.9 Hz), 160.1, 143.0, 141.3 (t, j = 9.1 Hz), 139.8, 128.7 (t, J= 35.7 Hz), 117.9, 112.3 (m), 102.1 (t, J= 25.0 Hz), 72.0, 43.0. 19F NMR (376 MHz, CDCl3): δ -112.1 (m).
Synthesis of N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-15-chloranimine (X-02)
[00566] Compound XIII (.0 g) was dissolved in THF (4.2 mL) and was cooled over an ice bath. Diphenylphosphoryl azide (0.66 mL, 1.2 equiv.) was added followed by DBU (0.46 mL, 1.2 equiv.) over about 25 min at below about 4 °C. The dark mixture was aged about 1 hour, and the cooling bath was removed. After about 2.5 hours age at RT, some starting material was still present so more diphenylphosphoryl azide (0.15 equiv.) and DBU (0.15 equiv.) were added after cooling over an ice bath. After about 2 hours, more diphenylphosphoryl azide (0.08 equiv.) and DBU (0.08 equiv.) were added. The reaction mixture was allowed to age overnight for about 16 h to allow the conversion to azide intermediate complete. The reaction mixture was cooled over an ice bath and triphenylphosphine (1.0 g, 1.5 equiv.) was added over about 15 min at about 6 °C). The cooling bath was removed after about 10 min and the reaction mixture was agitated for additional about 2.5 hours. To this reaction mixture was added water (0.18 mL, 4 equivalents) and the resulting mixture was aged for about 15 hours at room temperature. The mixture was diluted with EtOAc (5.0 mL) and was washed with water (4.2 mL + 2.0 mL). The aqueous layer was back extracted with EtOAc (4.0 mL) and the EtOAc layer was washed with water (1.0 mL). The organic layers were combined, concentrated via rotary evaporation and evaporated with EtOAc (4 x 4.0 mL) to dry. The residue was dissolved to a 50 ml solution in EtOAc, and cooled over an ice bath to become slurry. To the cold slurry 4N HCl/dioxane (0.76 mL, 1.2 equiv.) was added and the slurry was aged about 2 hours at room temperature. The solid product was filtered and the filter cake was rinsed with EtOAc and dried at about 35 to 50 °C under vacuum to give X-02.
[00567] Recrystallization: A portion of the above obtained X-02 (1.0 g) was mixed with EtOAc (10 mL) and was heated to 65 °C to afford thick slurry. The slurry was aged at about 65 °C for about 2 hours, and overnight at room temperature. The solids were filtered with recycling the mother liquor to help transfer the solids. The filter cake was rinsed with EtOAc, and dried overnight at about 50 °C vacuum to afford X-02. 1H NMR (300 MHz, DMSO-d) δ 8.78 (br s, 3 H), 8.06-8.02 (m, 1 H), 7.64-7.61 (m, 1 H), 7.15-7.08 (m, 1 H), 6.83-6.78 (m, 2 H), 4.87-4.82 (m, 1 H), 3.35-3.25 (m, 1 H), 3.17-3.05 (m, 1 H). 19F NMR (282.2 MHz, Chloroform-d) δ – 109.9-110.1 (m).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl methanesulfonate (XIII-A)
[00568] Compound XIII (1.0 g) and DMAP (0.1 equiv.) were dissolved in THF (4.5 mL) and cooled over an ice bath. Triethylamine (Et3N) (0.39 mL, 1.1 equiv.) was added followed by methanesulfonyl chloride (218 μL, 1.1 equiv.). The cooling bath was removed, and the mixture was aged about 1.5 hours at room temperature. The reaction mixture was cooled over an ice bath and quenched with water (10 mL). The mixture was diluted with EtOAc and the phases were separated. The aqueous phase was extracted with EtOAc, and the combined organic phase was dried (Na2SO4) and was passed through silica gel with EtOAc. The filtrate was concentrated to afford the mesylate (XIII-A). 1H NMR (300 MHz, Chloroform-d) δ 7.72-7.66 (m, 1 H), 7.38-7.32 (m, 1 H), 6.78-6.63 (m, 3 H), 6.17-6.13 (m, 1 H), 3.40-3.25 (m, 2 H), 2.87 (s, 3 H). 19F NMR (282.2 MHz, Chloroform-d) δ -109.3—109.5 (m).
Synthesis of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (X) from 1-(3,6- dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl methanesulfonate (XIII-A)
[00569] A glass pressure bottle was charged with the mesylate (XIII-A) (1.0 g), 28-30% ammonium hydroxide (19 mL) and MeOH (4.7 mL). The mixture was sealed and heated at about 70 °C for about 16 hours, and extracted with 2-MeTHF/ EtOAc. The organic layer was dried (Na2SO4) and purified by silica gel chromatography (10-60% EtOAc/hexanes) to afford racemic amine X. 1H NMR (300 MHz, Chloroform-d) δ 7.70-7.60 (m, 1 H), 7.30-7.20 (m, 1 H), 6.78-6.60 (m, 3 H), 4.46-4.58 (m, 1 H), 3.00-3.16 (m, 1 H), 2.70-2.80 (m, 1 H). 19F NMR (282.2 MHz, Chloroform-d) δ -110.3 – 110.4 (m).
Synthesis of (Z)-N-(1-(3,6-dibrornopyridin-2-yl)-2-(3,5-difluorophenyl)vinyl)acetamide (1f)
[00570] A glass reactor was charged with XI (1.0 g). Ethanol (5.0 mL) was added, and the slurry was agitated while hydroxylamine hydrochloride (0.88 g) was charged. Pyridine (1.0 mL) was added and the mixture heated at about 55-65 °C for about two hours. The mixture was cooled to about 20 °C, transferred to a flask, and concentrated to approximately 75 mL by rotary evaporation. The concentrate was returned to the reactor, rinsing through with isopropyl acetate (5.0 mL). Residue remaining in the flask was carefully (gas evolution) rinsed into the reactor with saturated aqueous sodium bicarbonate (5.0 mL). The bi-phasic mixture was agitated, the phases separated, and the organic extract washed with water (3.2 mL) and saturated sodium chloride (3.2 mL). The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated to dryness by rotary evaporation to yield 1e which was used without further purification.
[00571] A glass reactor was charged with iron powder (<10 micron, 0.30 g mmol) followed by acetic acid (1.6 mL) and acetic anhydride (0.72 mL). The slurry was de-gassed by holding the reactor contents under vacuum until bubbling was observed, and back-filled with nitrogen (3 cycles). The mixture was heated at 115-120 °C for 2 hours and cooled to 40 °C. Compound le from the previous step in isopropyl acetate (2.0 mL) was added over 30 min. Upon completing the addition, the temperature was raised to 45-65 °C and the mixture aged for about 2 hours. A slurry of diatomaceous earth (1.0 g) in isopropyl acetate (2.0 mL) was added, followed by toluene (2.0 mL). The slurry was filtered, hot, through a Buchner funnel and the reactor and filter cake were washed with warm isopropyl acetate (3 x 1.8 mL). The filtrate was transferred to a reactor and the solution washed with 0.5% aqueous sodium chloride (4.2 mL). Water (3.1 mL) was added to the reactor and the mixture was cooled to about 5 °C. The pH was adjusted to 7-9 with the addition of 50 wt% aqueous sodium hydroxide; following separation, the organic extract was warmed to room temperature and washed with aqueous 1% (w/w) sodium chloride NaCl (3.6 mL). The organic extract was discharged to a flask and dried over anhydrous sodium sulfate (ca. 0.8 g), filtered through diatomaceous earth, and concentrated to approximately 4 mL at 100 mmHg and 45 °C water bath. The warm solution was returned to the reactor, rinsing forward with isopropyl acetate to a produce a total volume of approximately 5.2 mL. This solution was heated further to 50 °C with agitation, cooled to about 35 °C, and seeded with pure 1f (0.006 g). Heptane (9.6 mL) was added over a period of about 4 hours, the solution was cooled to about 10 °C, and the product was isolated by filtration. The filter cake was washed with 33.3% iPAc in heptane (4.0 mL) and dried in a vacuum oven at 40 °C with nitrogen sweep for approximately 24 hours. Compound 1f, a mixture of geometric isomers (approximately 94:6 ratio) was isolated. Major isomer: 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.04 (d, J = 8.4 Hz, 1H), 7.66 (d, J= 8.4 Hz, 1H), 7.05 (s, 1H), 6.97 (tt, J = 9.2, 2.2 Hz, 1H), 6.40 – 6.31 (m,
2H), 1.97 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 168.37, 162.04 (dd, J = 245.1, 13.9 Hz), 154.47, 143.63, 139.45, 139.40 – 139.18 (m), 135.99, 129.44, 120.66, 113.80, 111.23 – 109.68 (m), 101.77 (t, J = 26.0 Hz), 23.49.
Synthesis of (S)-N-(1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)acetamide (1g)
[00572] Preparation of catalyst solution: A flask was charged with [IrCl(cod)((S)-segphos)] (110 mg) and the internal atmosphere was replaced with N2. EtOAc (200 mL) was added to the flask and the mixture was stirred until the catalyst solid was dissolved.
[00573] A stainless steel autoclave was charged with compound 1f (1.0 mg). EtOAc (16 mL) and followed by the catalyst solution prepared above (4.0 mL, 0.001 equiv.) were added to the autoclave. After being purged by H2, the autoclave was charged with 3 MPa of H2.The mixture was stirred at about 130 °C for about 6 hours and cooled to room temperature and H2 was vented out. The reaction mixture was purified by silica gel column chromatography (EtOAc/Hexane = 1/4 to 1/1) to afford 1g. 1H NMR (400 MHz, CD2Cl2): d 7.70 ( d, J = 8.0 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 6.68 (tt, J = 9.2, 2.4 Hz, 1H), 6.64 -6.58 (m, 2H), 6.49 (brd, j = 8.0 Hz, 1H), 5.74 (ddt, J = 8.0, 7.2, 6.4 Hz, 1H), 3.10 (dd, J = 13.6, 6.4 Hz, 1H), 2.99 (dd, J = 13.6, 7.2 Hz), 1.95 (s, 3H). 13C NMR (100 MHz, CD2Cl2): δ 169.5, 163.3 (dd, J = 246.0, 12.9 Hz), 159.1, 143.6, 141.4 (t, J = 9.1 Hz), 140.7, 129.1, 119.9, 112.9 (m), 102.6 (t, J= 25.1 Hz), 53.0, 41.3, 23.6. 19F NMR (376 MHz, CD2Cl2): δ -111.3 (m).
Synthesis of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VIII) from 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-one (XI), Method 1
[00574] A glass-lined reactor was charged with isopropylamine (about 18 g) and triethanolamine (3.8 g). Water (231 mL) was added and the pH was adjusted to about 7.5 by the addition of concentrated hydrochloric acid. A portion of the buffer solution (23 mL) was removed. The transaminase enzyme (2.5 g) was added to the reactor as a suspension in buffer solution (12 mL), followed by addition of pyridoxal phosphate monohydrate (50 mg) as a solution in buffer solution (12 mL). A solution of XI (1.0 g) in dim ethyl sulfoxide (23 mL) was added to the reactor and the mixture was heated at about 35 °C for about 48 hours with constant nitrogen sparging of the solution. The reaction mixture was cooled to about 20 °C the unpurified amine was removed by filtration. The filter cake was washed with water (3 x 7.7 mL) and the product was dried at about 60 °C under vacuum with nitrogen sweep to afford VIII.
Synthesis of (S)-1-(3.6-dibromopyridin-2-yl)-2-(3.5-difluorophenyl)ethan-1-amine (VIII) from 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-one (XI), Method 2
[00575] A stainless steel reactor was charged with XI (1.0 g) and p-toluenesulfonic acid (0.49 g). Ammonia (7 M in methanol, 3.7 mL) was added and the vessel was sealed and heated at about 60 °C for about 18 hours. The mixture was cooled to about 20 °C and sparged for about 30 min to remove excess ammonia. A solution of diacetato[(R)-5,5′-bis(diphenylphosphino)-4,4′-bi-1,3-benzodioxole]ruthenium(II) (0.10 g) in methanol (0.5 mL) was added to the reactor, which was sealed and heated at about 60 °C under a hydrogen atmosphere (400 psi) for a further about 6-10 hours. Upon cooling to about 20 °C the mixture was filtered through a plug of silica, rinsing with additional methanol (5.0 mL). Concentration of the filtrate by rotary evaporation affords VIII.
Example 1c: Preparation of 1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyI)ethan-1-amine (X) by racemization of (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VIII)
[00576] A vial was charged with zinc acetate (25 mol %), enantioenriched VIII (1.0 g, 92:8 enantiomer ratio), toluene (10 mL), and 2-formylpyridine (5 mol %). The vial was wanned to about 60 °C and stirred for about 4 h.
Example 2: Preparation of (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI)
[00577] A glass-lined reactor was charged with (S)-1-(3,6-dibromopyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (R)-mandelic acid salt (VIII-03) (1.0 g), 3-methyl-3-(methylsulfonyl)but-1-yne (IX) (about 0.3 g), and dichlorobis(triphenylphosphine)palladium(II) (about 0.39 g). The reactor was evacuated and purged with nitrogen to inert. To this reactor was added 2-methyltetrahydrofuran (6.4 kg) and triethylamine (0.92 kg 5.0 equiv.). The reaction mixture was agitated at about 65-75 °C until the reaction was deemed complete by HPLC analysis. Upon cooling to about 30-40 °C the reaction mixture was discharged to another reactor and the parent reactor was rinsed with 2-methyltetrahydrofuran (4.6 g) and the resulting solution transferred to the receiving reactor. To the reactor was added water (5.0 g) and the biphasic mixture agitated at about 30-40 °C for about 30 min. Agitation was ceased and the mixture was allowed to layer for 30 min. The lower aqueous layer was discharged and the remaining organic solution held for about 15 hours. A solution of A-acetyl-L-cysteine (196 g) and sodium hydroxide (0.80 g) in water (11.8 g) was prepared. To the reactor was added approximately half of the N-acetyl-L-cysteine solution (6.7 g). The mixture was agitated at about 55-65 °C for about 30 min. The temperature was adjusted to about 30-40 °C and agitation was ceased. After about 30 min had elapsed, the lower aqueous phase was discharged. The remaining alkaline N-acetyl-L-cysteine solution (5.4 kg) was added and the mixture was heated, with agitation, to about 55-65 °C and held for about 30 min. The temperature was adjusted to about 30-40 °C and agitation was ceased. After about 30 min had elapsed, the lower aqueous phase was discharged. To the reactor was added a solution of sodium chloride (0.26 g) in water (4.9 g) and the mixture agitated at about 30-40 °C for about 30 min. Agitation was ceased and the biphasic mixture allowed to layer for about 30 min. The lower aqueous layer was discharged and the contents cooled to about 15-25 °C and held for about 16 hours. The mixture was concentrated at about 55-65 °C. The concentrated solution was cooled to about 30-40 °C and heptane (3.4 kg) was added over about 2 hours. The resulting slurry was cooled to about 20 °C and aged for about 20 h, and filtered. The filter cake was washed with 2-methyltetrahydrofuran/heptane (1:1 v/v,2 mL) and the solids dried in a vacuum oven at about 40 °C to yield (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI)). 1H NMR (400 MHz, DMSO-d6) δ 8.05 (d, J = 8.2 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 7.01 (tt, J = 9.5, 2.4 Hz, 1H), 6.97 – 6.84 (m, 2H), 4.41 (dd, J = 8.5, 5.2 Hz, 1H), 3.20 (s, 3H), 2.93 (dd, J = 13.3, 5.2 Hz, 1H), 2.79 (dd, J = 13.3, 8.5 Hz, 1H), 1.99 (s, 2H), 1.68 (s, 6H). 13C NMR (101 MHz, DMSO-d6) δ 162.25, 162.00 (dd, J = 245.2, 13.4 Hz), 143.88 (t, J= 9.4 Hz), 141.09, 139.72, 127.51, 120.08, 112.58 – 112.12 (m), 101.45 (t, J= 25.7 Hz), 87.94, 84.25, 57.24, 55.90, 42.57, 34.99, 22.19.
Example 2a: Preparation of 3-methyl-3-(methylsulfonyl)but-1-yne (IX)
[00578] Sodium methansulfmate (418.1 g), copper (II) acetate (26.6 g), N,N,N’,N’- Tetramethylethylenediamine (TMEDA, 34.0 g), and isopropyl acetate (2100 mL) were added to a reactor and the suspension was agitated at 20 – 25 °C. 3-Chloro-3-methylbut-1-yne (3-CMB,
300 g) was added slowly to maintain a constant temperature of about 20 – 25 °C. The reaction mixture was then heated to about 30 °C until the reaction was complete. The mixture was cooled to about 20 °C and washed twice with 5% aqueous sulfuric acid (600 mL). The combined
aqueous layers were then extracted with isopropyl acetate (600 mL). The combined organic layers were then washed with water (600 mL). The product was then isolated by crystallization from isopropyl acetate (900 mL) and n-heptane (1.8 kg) at about 0 °C. The wet cake was then washed with cold n-heptane to afford IX. 1H NMR (400 MHz, DMSO-d6) δ 3.61 (s, 1H), 3.07 (s, 3H), 1.55 (s, 6H); 13C NMR (10Q MHz, DMSO) d 82.59, 77.76, 56.95, 34.95, 22.77.
Example 3a: Preparation of (3bS,4aR)-3-(trifluoromethyI)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from lithium (Z)-2,2,2-trifluoro-1-(3-oxobicyclo[3.1.0]hexan-2-ylidene)ethan-1-olate (3a)
Synthesis of 3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazole (3b)
[00579] A reactor was charged with 3a (1.0 g) and AcOH (4.2 ml) and the resulting solution was adjusted to about 20 °C. Hydrazine hydrate (0.29 g, 1.4 equiv.) was added over about 60 min at about 17-25 °C and the reaction mixture was stirred for about 2 hours at about 20-25 °C, warmed up to about 45 to 50 °C over about 30 min, and aged at about 50 °C overnight. Water was slowly (5 mL) added at about 50 °C and product started to crystallize after addition of 5 mL of water. Another 5 mL of water was added at about 50 °C, and the slurry was cooled down to about 20 °C in about one hour and held overnight at about 20 °C. The solids were filtered, washed with water (4X 3 mL), and dried under vacuum at about 30 °C to yield 3b. 1H NMR (400 MHz, Chloroform-d) δ 2.99 (dd, J = 17.0, 6.1 Hz, 1H), 2.89 – 2.78 (m, 1H), 2.14 (dddd, J = 9.1, 7.9, 3.6, 2.5 Hz, 2H), 1.13 (td, J = 7.8, 5.1 Hz, 1H), 0.36 – 0.26 (m, 1H).
Isolation of (3bS,4aS)-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazole (3c)
[00580] Chiral purification of 3b (1.0 g) was achieved using a 8×50 mm simulated moving bed (SMB) chromatography system and Chiralpak IG (20 μ particle size) stationary phase using acetonitrile as a mobile phase to afford 3c. 1H NMR (400 MHz, Chloroform-d) δ 3.00 (dd, J = 17.0, 5.7 Hz, 1H), 2.90 – 2.77 (m, 1H), 2.21 – 2.05 (m, 2H), 1.13 (td, J = 7.8, 5.1 Hz, 1H), 0.35 – 0.27 (m, 1H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV)
[00581] A reactor was charged with water (7 mL) and CuCl2 ● 2H2O (0.09 g, 0.1 equiv). To the reactor was added pyridine (0.42 g, 1 equiv.) and 3c. tert-Butylhydroperoxide (70% in water, 5.5 g, 8 equiv.) was added over about 0.5 hour. The reaction mixture was stirred at about 20 °C for about 2.5 days and quenched with aqueous sodium metabisulfite solution (0.73 g in 2.5 mL water). The quenched reaction mixture was extracted with isopropyl acetate (20 mL), and the aqueous layer was back extracted with isopropyl acetate (2.0 ml). The organic layers were combined and washed with aqueous ethylenediaminetetraacetic acid (EDTA) solution 0.16 g EDTA 10 ml in water), the aqueous layer was dropped, and the organic layer was further washed with aqueous EDTA solution (0.015 g EDTA in 20 ml water). The washed organic layer was concentrated to dryness. To the residue was added isopropyl acetate (2.0 ml) and heptane (2.0 mL). The solution was seeded and stirred overnight at about 20 °C, further diluted with heptane (2.0 mL), and the mixture was concentrated to dryness. The residue was suspended in heptane (4.0 mL) at about 40 °C. The solid was filtered and the filter cake was washed with heptane (1.0 mL) and dried at about 40 °C to yield XV. 1H NMR (400 MHz, Chloroform-d) δ 2.84 (dt, J = 6.8, 4.2 Hz, 1H), 2.71 – 2.64 (m, 1H), 1.79 – 1.67 (m, 2H).
Example 3b: Preparation of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from lithium (Z)-1-((1S,5R)-4,4- dimethoxy-3-oxobicyclo[3.1.0]hexan-2-ylidene)-2,2,2-trifluoroethan-1-olate (3d-02)
[00582] Hydrazine sulfate (0.45 g, 0.95 equiv.) and ketal lithium salt 3d-02 (1.0 g) were dissolved in ethylene glycol (9.5 mL), and the solution was heated to about 40 °C for about 16 hours. Reaction was cooled to room temperature and water (9.0 mL) was added. Reaction was polish filtered andThe filtrate was collected and to this receiving flask was added water (10 mL, 2x). Slurry was cooled in ice water bath for about five hours, and filtered. Solids were washed with ice water (10 mL, 2x), deliquored, and dried to afford XV. 1H NMR (400 MHz, CDCl3) δ 11.83 (bs, 1H), 2.93 – 2.77 (m, 1H), 2.77 – 2.58 (m, 1H), 1.86 – 1.57 (m, 2H). 19F NMR (376 MHz, CDCl3) δ -61.69. 13C NMR (101 MHz, CDCl3) δ 188.56, 144.08, 142.92, 121.82, 119.15, 36.28, 31.87, 14.15.
Example 3c: Preparation of (3bS,4aR)-3-(trifiuoromethyl)-1,3b,4,4a-tetrahydro-5H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV) from (1S,2S)-2-iodo-N-methoxy-N- methylcyclopropane-1-carboxamide (3f) and 1-(4-methoxybenzyl)-4-(4,4,5,5-tetramethyl- 1,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)-1H-pyrazole (3i) and preparation of starting materials and/or intermediates therein
Synthesis of (1S,2S)-2-iodo-N-methoxy-N-methylcyclopropane-1-carboxamide (3f)
[00583] Starting material iodoacid 3e is a mixture of 3e and cyclopropane carboxylic acid (des-iodo 3e) with mole ratio of 3e to des-iodo 3e of 2:1 by NMR. A mixture of 3e (1.0 g),
N,O-dimethyl hydroxyl amine-HCl (0.46 g) and carbonyl diimidazole (1.72 g) in THF was stirred overnight at room temperature. The reaction mixture was diluted with water, extracted with CH2Cl2, and concentrated to afford unpurified 3f (1.8 g). The unpurified 3f was purified by column chromatography to afford 3f which was a mixture of Wei nr eb amide 3f and des-iodo-3f (about 80:20 by HPLC).
Synthesis of 1-(4-methoxybenzyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3- (trifluoromethyl)-1H-pyrazole (3i)
[00584] To a suspension of NaH (60%, 0.31 g, 1.1 equiv.) in DMF (7.5 mL), a solution of 3g (1.0 g) in DMF (7.5 mL) was added dropwise over about 15 min at about 3 to 7 °C. The reaction mixture was stirred at room temperature for about 1 h and a solution of PMBCl (1.2 g, 1.05 equiv.) in DMF (4.2 mL) was added dropwise in about 25 min at room temperature. The reaction mixture was stirred at room temperature overnight, poured into water (17 mL), and extracted with diethyl ether (3×17 mL). The ether layers were combined and washed with water (2 x 17 mL) and brine (17 mL), dried over Na2SO4, and concentrated in vacuo to give unpurified 3h. Unpurified 3h was absorbed in silica gel (4.3 g) and purified by silica gel chromatography (eluting with 5-25% EtOAc in hexanes) to give 3h (1.5 g).
[00585] To solution of iodopyrazole 3h (1.0 g) in THF (8 mL) i-PrMgCl (2M in ether, 1.8 mL, 1.1 equiv.) was added dropwise over about 10 min at below about 5 °C. The resulting solution was stirred at about 0 °C for about 70 min and 2-methoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (970 mg, 1.81 equiv.) was added at below about 6 °C. The reaction mixture was warmed up to room temperature, quenched by addition of saturated NH4Cl (20 mL), and
extracted with EtOAc (2 x 20 mL). The combined organic layer was washed with saturated NH4Cl (10 mL) and concentrated to unpurified oil, which was combined with the unpurified oil from a previous batch (prepared using 1.1 g of 3h), absorbed on silica gel (6 g), and purified via silica gel chromatography (eluting with 5-40% EtOAc/Hexanes,). Boronate 3i was obtained. 1H NMR (300 MHz, Chloroform-d) δ 7.60 (s, 1 H), 7.23-7.19 (m, 2 H), 6.90-6.85 (m, 2 H), 5.25
(s, 2 H), 3.81 (m, 3 H), 1.29 (s, 12 H).
Synthesis of (1R,2S)-N-methoxy-2-(1-(4-methoxybenzyl)-3-(trifluoromethyl)-1H-pyrazol-4-yl)-N-methylcyclopropane-1-carboxamide (3j)
[00586] A mixture of unpurified iodide 3f (1.0 g), boronate 3i (about 2.2 g), CsF (4.5 equiv.), Pd(OAc)2 (0.1 equiv.), and PPh3 (0.5 equiv.) in DMF (58 mL) was degassed by bubbling N2 and heated at about 87 °C for about 15 hours. The reaction mixture was diluted with water,
extracted with MTBE, concentrated and the unpurified product was purified by column chromatography to give 3j. 1H NMR (300 MHz, Chloroform-d) δ 7.18-7. 14 (m, 3 H), 6.86-6.82 (m, 2 H), 5.24-5.08 (m, 2 H), 3.77 (s, 3 H), 3.63 (s, 3 H), 3.05 (s, 3 H), 2.37-2.32 (m, 1 H), 1.50-1.42 (m, 1 H), 1.32-1.21 (m, 2 H).
Synthesis of (3bS,4aR)-1-(4-methoxybenzyl)-3-ftrifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta91,2-c]pyrazol-5-one (3k)
[00587] Compound 3j (1.0 g) was treated with freshly prepared LDA (3.3 eq then 0.7 equiv.) at about -67 °C for about 2.5 hours. The reaction mixture was quenched with saturated NH4Cl (12.5 mL) and diluted with MTBE (63 mL). The organic layer was washed with brine, concentrated, and purified by column chromatography to give 3k. 1H NMR (300 MHz, Chloroform-d) δ 7.36-7.33 (m, 2 H), 6.86-6.83 (m, 2 H), 5.28 (s, 2 H), 3.78 (s, 3 H), 2.73-2.65
(m, 1 H), 2.60-2.53 (1 H), 1.70-1.61 (m, 2 H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1.2-c]pyrazol-5-one (XV)
[00588] A mixture of 3k (1.0 g) and TFA (5 mL) was heated at about 75 °C for about 3 hours and concentrated. The residue was dissolved in DCM (50 mL), washed with saturated NaHCO3 and brine, concentrated, and purified by column chromatography to give XV. 1H NMR (300 MHz, Chloroform-d) δ 2.86-2.80 (m, 1 H), 2.68-2.63 (m, 1 H), 1.77-1.65 (m, 2 H).
Example 3d: Resolution of 2-(2,2,2-trifluoroacetyl)bicyclo[3.1.0]hexan-3-one (3I) with quinine
[00589] A flask was charged with 3I (1.0 g), acetone (2.5 ml), and quinine (1.7 g, 0.65 equiv). The mixture was stirred at about 15 to 25 °C for about 18 hours and the solids were isolated by filtration and washed with acetone to provide the quinine salt 3n.
Example 4a: Preparation of ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV) from (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydro-5H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-5-one (XV)
[00590] Acetonitrile (5 vol.) was added to a reactor containing XV (1.0 g). N,N-Diisopropylethylamine (0.80 g, 1.25equiv.) was added at about 0 °C. Ethyl bromoacetate (0.91 g, 1.1 equiv.) was added over about 1 hour at about 0 °C. The reaction was stirred at about 5 °C for about 30 minutes and warmed to about 10 °C. The reaction was stirred until complete as determined by HPLC, warmed to about 20 °C, and extracted with MTBE (2 vol.) and saturated NaCl (6 vol.). The aqueous layer was removed and the organic phase was concentrated and diluted with EtOH (3 vol.). The reaction was crystallized by the addition of H2O (7.8 vol.) at about 20 °C. The mixture was cooled to about 5 °C over about 2 hours and maintained at about 5 °C for about 0.5 hour. The mixture was filtered at about 5 °C and washed with cold water (4 vol). The product was dried at about 40 °C under vacuum to give XIV. 1H NMR (400 MHz, Chloroform-d) δ 4.97 (s, 2H), 4.31 – 4.17 (m, 2H), 2.77 (dddd, J= 6.4, 5.2, 2.9, 2.3Hz, 1H), 2.65 – 2.55 (m, 1H), 1.74 – 1.64 (m, 2H), 1.34 – 1.19 (m, 5H), 0.94 – 0.84 (m, 1H).
Example 4b: Preparation of ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV) from (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
Synthesis of (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-ol (4b-02)
[00591] Potassium hydroxide (KOH) (2.2 g, 3.50 equiv.) and anhydrous methanol (13 mL) were added to a reactor and the reaction mixture was warmed to about 55 °C and agitated until
KOH solids were dissolved completely. The mixture was adjusted to about 0 to 6 °C and compound 4a (1.0 g) was slowly added while maintaining the internal temperature at NMT 6 °C. The reaction mixture was agitated for about 45 min at about 0 to 6 °C. Diacetoxy iodobenzene (PhI(OAc)2, 5.0 g, 1.5 equiv.) was added over about 2 hours while maintaining the internal temperature at NMT 6 °C. The reaction mixture was agitated for NLT 1 hour at about 0 to 6 °C. Water (10 g) and heptane (10 mL) were added to the reaction mixture and the biphasic was agitated for NLT 30 min at about 19 to 25 °C The aqueous layer was separated and washed with heptane (10 mL). The combined organic layer was extracted twice with aqueous solution of methanol (MeOH, 10 mL) and water (5 g). The combined aqueous layer was concentrated under vacuum. The aqueous layer was extracted twice with DCM (15 mL and 5 mL). The combined organic layer was concentrated and dried under vacuum. The unpurified compound 4b-02 was obtained. 1H NMR (600 MHz, CDCl3): d 3.98 (d, 1H), 3.45 (s, 3H), 3.25 (s, 3H),
2.40 (s, 1H), 2.21 (m, 1H), 1.78 (d, 1H), 1.48 (m, 1H), 1.38 (m, 1H), 0.83 (q, 1H), 0.58 (m, 1H).
13C NMR (150 MHz, CDCl3): δ 110.91, 72.19, 51.18, 49.02, 34.08, 21.66, 14.75, 8.37.
Synthesis of (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-one (4c-02)
[00592] Oxalyl chloride (0.96 g, 1.20 equiv.) and dichloromethane (10 mL) were added to a reactor and the mixture was cooled to about -78 °C. Dimethyl sulfoxide (DMSO, 1.2 g, 2.4 equiv.) was added over about 30 min while maintaining the internal temperature below about -60 °C. After agitation for about 5 min, the solution of compound 4b-02 (1.0 g) in dichloromethane (6 mL) was added over about 30 min while maintaining the internal temperature below about -60 °C and the reaction mixture was agitated for about 20 min at about -60 °C. Triethylamine (TEA, 3.1 g, 4.8 equiv.) was added over about 40 min at about -60 °C, and the reaction mixture was warmed to about 10 to 20 °C. Water (15 g) was added and the biphasic was agitated about 30 min at about 10 to 20 °C. After phase separation, the aqueous layer was back-extracted with dichloromethane (10 mL). Combined organic layer was concentrated until no distillate was observed. To the residue was added MTBE (1 mL), filtered and evaporated to afford unpurified compound 4c-02. 1H NMR (600 MHz, CDCl3): d 3.45 (s,
3H), 3.27 (s, 3H), 2.79 (ddd, 1H), 2.30 (d, 1H), 1.73 (td, 1H), 1.63 (m, 1H), 0.96 (m, 1H), 0.25 (td, 1H). 13C NMR (150 MHz, CDCl3): δ 207.75, 102.13, 50.93, 50.50, 38.87, 19.15, 9.30, 8.56.
Synthesis of lithium (Z)-1-((1S,5R)-4,4-dimethoxy-3-oxobicyclo[3.1.0]hexan-2-ylidene)-2,2,2-trifluoroethan-1-olate (3d-02)
[00593] A reactor was charged with compound 4c-02 (1.0 g), ethyl trifluoroacetate (CF3COOEt, 0.91 g, 1.0 equiv.) and tetrahydrofuran (THF, 0.5 mL) and the reaction mixture was cooled to about -10 to 0 °C. The 1M solution of lithium bis(trimethylsilyl)amide (LiHMDS, 7.0 mL, 1.10 equiv.) was added over about 40 min while maintaining the internal temperature below about 0 °C. The reaction mixture was agitated for about 2 hours at about -10 to 0 °C until the reaction was complete. After then, the reaction mixture was wanned to about 20 °C followed by charging tert-butyl methyl ether (MTBE, 10 mL) and water (10 g). After agitating for about 30 min, the organic layer was separated and the aqueous layer was back-extracted twice with mixture of MTBE (6 mL) and THF (4 mL). The combi ned organic layer was concentrated until no distillate was observed. To the unpurified solids, THF (3 mL) and heptane (15 mL) were added at about 20 °C, and the reaction mixture was cooled to about 0 °C and agitated about 1 hour. The resulting slurry was filtered and wet cake was washed with heptane (7 g) and dried under vacuum at about 40 °C to afford compound 3d-02. 1H NMR (600
MHz, DMSO-d6): d 3.31 (s, 3H), 3.27 (s, 3H) 2.01 (m, 1H), 1.42 (td, 1H), 0.96 (m, 1H), 0.08 (q, 1H). (600 MHz, CDCl3 with THF) δ 3.44 (s, 3H), 3.24 (s, 3H), 2.26 (m, 1H), 1.48 (m, 1H), 1.04 (q, 1H), 0.25 (m, 1H). 13C NMR (150 MHz, DMSO-d6): 193.20, 120.78, 118.86, 105.53,
104.04, 50.66, 49.86, 17.34, 16.20, 13.78.
Synthesis of ethyl 2-((3bS.4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00594] Compound 3d-02 (1.0 g), ethyl hydrazinoacetate hydrochloride (EHA-HCl, 0.60 g,
1.0 equiv.) and absolute ethanol (EtOH, 15 mL) were added to a reactor and the reaction mixture was cooled to about 0 – 5 °C. Sulfuric acid (H2SO4, 0.19 g, 0.50 equiv.) was added while maintaining the internal temperature below about 5 °C. Triethyl orthoformate (0.86 g, 1.50 equiv.) was added and the reaction mixture was agitated at about 0 to 5 °C for about 15 hours. The reaction mixture was warmed to about 20 to 25 °C and water (30 g) was added over about 15 minutes. The content was cooled to about 0 to 5 °C and agitated for about 1 hour. The slurry was filtered and wet cake was washed with water (5 g) and dried under vacuum at about 45 °C to afford XIV 1H NMR (600 MHz, CDCl3): d 4.97 (s, 1H), 4.23 (qd, 2H), 2.77 (quint. 1H), 2.60 (quint, 1H), 1.69 (m, 2H), 1.28 (t, 3H). 13C NMR (150 MHz, CDCl3): d 187.14, 165.98, 143.35, 143.12, 121.37, 119.59, 62.34, 51.83, 35.35, 31.72, 14.00, 13.73.
Example 4c: Kinetic resolution of ethyl 2-(5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XVII) to form ethyl 2- ((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00595] Compound XVII (1.0 g), (R)-2-methyl-CBS-oxazaborolidine (0.0.05 g, 0.05 equiv.), and tetrahydrofuran (11.9 g) were combined and cooled to about 0 to 5 °C. A solution of borane dimethyl sulfide complex (0.14 g, 0.55 equiv.) in tetrahydrofuran (0.67 g) was added to the mixture, and the mixture was agitated at about 0 to 5 °C until the reaction was deemed complete. Methanol (1 mL) was added to the mixture at about 0 to 5 °C over about 1 h, and the mixture was adjusted to about 15 to 25 °C. The mixture was concentrated under vacuum and combined with tetrahydrofuran (2.7 g). The mixture was combined with 4-dimethylaminopyridine (0.18, 0.44 equiv.) and succinic anhydride (0.30 g, 0.87 equiv.) and agitated at about 15 to 25 °C until the reaction was deemed complete. The mixture was combined with tert-butyl methyl ether (5.2 g) and washed with 1 M aqueous HCl (6.7 g), twice with 5 wt % aqueous potassium carbonate (6.7 g each), and 5 wt % aq. sodium chloride (6.7 g). The organics were concentrated under reduced pressure to an oil which was dissolved in dichloromethane (0.1 g) and purified by flash column chromatography (2.0 g silica gel, 20:80 to 80:20 gradient of ethyl acetate:hexanes). The combined fractions were concentrated under vacuum to give XIV.
Example 4d: Preparation of (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
[00596] 4-Tosyloxycyclohexanone (50 mg), (8α,9S)-6′-methoxycinchonan-9-amine trihydrochloride (16 mg), trifluoroacetic acid (28 μL), lithium acetate (49 mg), water (3.4 μL), and 2-methyltetrahydrofuran (0.75 mL) were combined in a vial. The mixture was agitated at about 20 °C until the reaction was complete. 4a was isolated by vacuum distillation. 1H NMR (400 MHz, CDCl3) δ2.05 (m, 5H), 1.74 (m, 1H), 1.18 (m, 1H), 0.91 (m, 1H).
Example 5: Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a- dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane]-1(3bH)- yl)acetate (5h) from (1R,5R)-2,2-dimethoxybicyclo[3.1.0]hexan-3-ol (4b-02)
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan1-3-ol (5d)
[00597] A mixture of ketal alcohol 4b-02 (1.0 g), ethanedi thiol (0.91 g), MeCN (7.5 ml) and BiCl3 (0.30 g) was agitated at r.t. overnight. The solids were removed by filtration and the filtrate was concentrated and the residue was further purified by flash column on silica gel to obtain the two isomers. Major product: 1H NMR (400 MHz, Chloroform-d) δ 3.82 (ddt, J = 6.1, 1.3, 0.6 Hz, 1H), 3.41 – 3.32 (m, 2H), 3.31 -3.23 (m, 1H), 3.14 – 3.06 (m, 1H), 2.71 (s, 1H),
2.33 (dddd, J = 14.0, 6.2, 4.8, 1.4 Hz, 1H), 2.00 (d, J = 13.9 Hz, 1H), 1.79 – 1.72 (m, 1H), 1.54 -1.46 (m, 1H), 1.04 (dt, J = 5.1, 3.9 Hz, 1H), 0.63 – 0.54 (m, 1H). Minor product: 1H NMR (400 MHz, Chloroform-d) δ 3.83 (q, J = 9.1 Hz, 1H), 3.43 – 3.34 (m, 2H), 3.33 – 3.25 (m, 2H), 2.35 (d, J= 11.2 Hz, 1H), 2.18 (ddd, J = 12.7, 6.7, 0.4 Hz, 1H), 1.84 (ddd, J= 8.1, 6.3, 3.7 Hz, 1H),
1.60 – 1.51 (m, 1H), 1.43 – 1.35 (m, 1H), 0.65 (tdt, J= 8.1, 5.9, 0.8 Hz, 1H), 0.57 (dddd, J= 5.9, 4.2, 3.7, 0.6 Hz, 1H).
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan1-3-one (5e)
[00598] To a dried flask was sequentially added dithiolane alcohol 5d (1.0 g), CH2Cl2 (25 ml), anhydrous DMSO (8.5 ml), and tri ethylamine (3.5 ml) and the resulting mixture was aged at room temperature for about 21 hours. The reaction mixture was transferred to a separatory funnel, diluted with CH2Cl2 (30 ml), washed with 1 M HCl (25 ml), and water (25 ml). The CH2Cl2 layer was concentrated to a solid and further purify by flash column chromatography on silica gel eluted with gradient EtOAc/n-heptane (0-20%) to obtain 5e. 1H NMR (400 MHz, Chloroform-d) δ 3.57 (dddd, J = 10.5, 5.6, 4.3, 0.5 Hz, 1H), 3.49 – 3.41 (m, 1H), 3.39 – 3.28 (m, 2H), 3.10 (ddd, J = 18.3, 5.6, 2.2 Hz, 1H), 2.29 (d, J = 18.3 Hz, 1H), 1.89 (ddd, J = 8.0, 7.0, 3.9
Hz, 1H), 1.63 (tdd, J= 7.3, 5.6, 4.1 Hz, 1H), 1.05 (tdd, J = 8.0, 6.3, 2.2 Hz, 1H), 0.21 (dt J = 6.4, 4.0 Hz, 1H).
Synthesis of lithium (Z)-2,2,2-trifluoro-1-((1R,5S)-3-oxospiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-4-ylidene)ethan-1-olate (5f)
[00599] To a flask with dithiolane ketone 5e (1.0 g) under N2 was added anhydrous THF (8.8 ml), and the mixture was cooled to about -78 °C and followed by addition of LiHMDS (1 M in THF, 7.4 ml) over about 5 min. The resulting mixture was agitated at about -78 °C for about 0.5 hours, and ethyl trifluoroacetate (0.88 ml) was added. The resulting mixture was agitated at about -78 °C for about 10 minutes, at about 0 °C for about 1 hour, and at room temperature overnight. THF was removed under reduced pressure and the residue was crystallized in n-heptane (about 18 ml). The solid product was isolated by filtration, and the filter cake was rinsed with n-heptane (4.1 ml), and dried at about 50 °C under vacuum to provide 5f. 1H NMR (400 MHz, Acetonitrile-d3) δ 6.98 (s, 0H), 5.20 (s, 0H), 3.60 – 3.50 (m, 2H), 3.46 – 3.36 (m, 2H), 2.28 – 2.20 (m, 1H), 1.80 (ddd, J = 8.3, 7.2, 4.1 Hz, 1H), 1.39 (s, 1H), 1.03 (ddd, J = 8.3, 6.7, 4.8 Hz, 1H), 0.17 (ddd, J = 4.7, 4.2, 3.6 Hz, 1H).
Synthesis of (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydrospiro[cvciopropa[3.4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane] (5g)
[00600] To flask containing the dithiolane lithium salt 5f (1.0 g) was added water (10 ml), hydrazine hydrate (0.88 ml) and acetic acid (10 ml). The reaction mixture was heated at about 35 °C for about 2 hours, and at about 55 °C for about 2 hours. Water was removed under reduced pressure and the residue was diluted with acetic acid (20 ml) and heated at about 55 °C for about 0.5 hour and held at room temperature overnight. The reaction mixture was further heated at about 65 °C for about 20 hours, and cooled down and concentrated to remove volatile components by rotavap. The residue was triturated with water (50 ml) at about 0 °C and the solid residue was isolated and further washed with ice-cold water (2×10 ml). The solids were further dried to afford unpurified 5g. 1H NMR (400 MHz, Chloroform-d) δ 3.65 – 3.46 (m, 4H), 2.60 (dddd, J = 8.3, 5.6, 4.2, 0.7 Hz, 1H), 2.47 – 2.38 (m, 1H), 1.33 (dddd, J= 8.2, 7.4, 5.7, 0.7 Hz, 1H), 0.66 (dddd, J = 5.7, 4.3, 3.6, 0.7 Hz, 1H)
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5.2′-[1,3]dithiolane]-1(3bH)-yl)acetate
(5h) from (3bS,4aR)-3-(trifluoromethyl)-1,3b,4,4a-tetrahydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane] (5g)
[00601] A reactor was charged with dithiolane pyrazole 5g (1.0 g) and THF (15 ml). The contents were adjusted to about 0 to -5 °C and followed by addition of ethyl bromoacetate (0.44 ml, 1.1 equiv.). To the resulting mixture NaHMDS (2 M, 2.0 ml, 1.1 equiv.) was added over about 10 min via syringe pump at about -2.5 to 0 °C and the mixture was held for about 3 hours, a second portion of ethyl bromoacetate (0.050 ml, 0.12 equiv.) was added, and the mixture was aged for about 1 hour. The reaction mixture was quenched by excess water (2 ml) to form 5h.
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolanel-1(3bH)-yl)acetate
(5h) from lithium (Z)-2,2,2-trifluoro-1-((1R,5S)-3-oxospiro[bicyclo[3.1.0]hexane-2.2′- [1,3]dithiolanl-4-ylidene)ethan-1-olate (5f)
[00602] A 100 ml flask was charged with ethanol (5 ml). The contents were cooled to about 0 °C and acetyl chloride (1.1 g, 4.0 equiv.) was added over about 10 min. The mixture was agitated at about 0 °C for about 20 minutes and at room temperature for about 20 minutes. To the freshly prepared HCl ethanol solution was added EHA.HCl (0.68 g, 1.2 equiv.) and dithiolane lithium salt 5f (1.0 g). The reaction mixture was heated at about 40 °C for about 22 hours. Ethanol was removed under reduced pressure, and the residue was partitioned between ethyl acetate (5 ml) and water (5 ml). The aqueous layer was discarded, and the organic layer was sequentially washed with aqueous NaHCO3 (5%, 5 ml) and brine (5%, 5 ml) and 5h was
obtained in the EtOAc layer. 1H NMR (400 MHz, DMSO-d6) d 5.14 – 4.97 (m, 2H), 4.14 (qd, J = 7.1, 1.0 Hz, 2H), 3.67 – 3.35 (m, 4H), 2.69 (ddd, J= 8.2, 5.6, 4.2 Hz, 1H), 2.44 (ddd, J= 7.2,
5.5, 3.5 Hz, 1H), 1.37 – 1.29 (m, 1H), 1.21 – 1.14 (m, 3H), 0.44 (ddd, J = 5.3, 4.2, 3.6 Hz, 1H).
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolanel-1 (3bH)-yl)acetate (5h) from (1R,5R)-spiro[bicyclo[3.1.0]hexane-2.2′-[1,3]dithiolanl-3-one (5e)
[00603] 5e (756 mg) was charged to a vessel and dissolved in 2-methyltetrahydrofuran (7.6 mL). To this solution was charged ethyl trifluoroacetate (0.57 g) and the resulting solution was cooled to about 0 °C. Lithium hexamethyldisilazide (1.0 M solution in THF, 4.5 g) was charged over about 60 minutes and reaction was agitated until complete. A solution of sulfuric acid (2.0 g) in water (5.6 mL) was charged, then the reaction was warmed to about 20 °C and agitated for about 20 minutes. Layers were separated and aqueous layer was extracted twice with 2-methyltetrahydrofuran (5.3 mL). Combined organic layer was concentrated to about 0.4 mL and N,N-diisopropylamine (0.5 g) was charged. The product was crystallized by the addition of heptane (11 ml). The slurry was filtered and the filter cake was washed with heptane, then deliquored thoroughly, and dried to afford 5f-01. 1H NMR (400 MHz, Acetonitrile-d3) δ 7.84 (m, 2H), 3.58 (d, J = 8.7 Hz, 2H), 3.47 – 3.27 (m, 4H), 2.20 (s, 1H), 1.81 – 1.68 (m, 1H), 1.24 (dd, J = 6.5, 0.6 Hz, 12H), 0.99 (q, J = 6.5 Hz, 1H), 0.13 (s, 1H).
[00604] Acetyl chloride (1.02 g) was charged to a cooled reaction vessel containing ethanol (5.0 mL) at about 0 °C, then warmed to about 20 °C and agitated for about 30 minutes. In a separate vessel, 5f-01 (1.00 g), ethyl hydrazinoacetate hydrochloride (0.48 g), and lithium chloride (0.39 g) were combined, and the acetyl chloride/ethanol solution was charged to this mixture, followed by tri ethyl orthoformate (1.16 g). The mixture was heated to about 45 °C and agitated until reaction was complete. The reaction was then concentrated to 2 volumes and dichlorom ethane (5.0 mL) was added followed by water (5.0 mL). Layers were separated and organic layer was washed with 5 wt % aqueous sodium bicarbonate followed by 10 wt % aqueous sodium chloride to afford a solution of 5h in dichloromethane that was carried forward into the subsequent step. 1H NMR (400 MHz, DMSO-d6) δ 5.27 – 4.79 (m, 2H), 4.14 (qd, J =
7.1, 1.1 Hz, 2H), 3.70 – 3.42 (m, 4H), 2.68 (dtd, J = 8.0, 6.4, 5.9, 4.4 Hz, 1H), 2.44 (ddd, J = 7.2, 5.5, 3.6 Hz, 1H), 1.32 (ddd, J = 8.2, 7.2, 5.4 Hz, 1H), 1.18 (t, J = 7.1 Hz, 3H), 0.44 (dt, J = 5.4, 3.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 167.14, 148.36, 133.80 (q, J = 38.3 Hz), 128.77 (m), 121.54 (q, J = 268.4 Hz), 65.33, 61.79, 51.14, 41.30, 40.98, 40.49, 23.57, 15.52, 14.33; 19F NMR (376 MHz, DMSO-d6) δ -60.31.
Synthesis of (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-3-one (5e) from (1R,5R)-spiro[bicyclo[3.1.0]hexane-2,2′-[1,3]dithiolan]-3-one (5e) from (1R,5S)-bicyclo[3.1.0]hexan-2-one (4a)
[00605] Tert-butyl nitrite (1.31 g) was charged to a vessel containing 4a (1.00 g, 1.0 equiv) and tetrahydrofuran (5.0 mL) at about 20 °C. Potassium tert-butoxide (6.1 g, 1.7M in tetrahydrofuran) was charged over not less than 30 minutes. The mixture was then agitated until the reaction was complete. The reaction was quenched with aqueous citric acid (2.00 g in 10.00 g water) and extracted with dichloromethane (10.0 mL, 3x). This solution was partially concentrated and the product was isolated by the addition of heptane (6.0 mL). The slurry was filtered and the filter cake was washed with heptane (2.0 mL), then deliquored thoroughly to afford 4d 1H NMR (400 MHz, DMSO-d6) δ 12.26 (s, 1H), 2.73 (d, J = 18.5 Hz, 1H), 2.63 (ddd, J = 18.6, 5.3, 2.0 Hz, 1H), 2.17 – 2.01 (m, 2H), 1.34 (dddd, J= 9.2, 7.1, 4.9, 2.0 Hz, 1H), 0.77 (td, J= 4.6, 3.4 Hz, 1H).
[00606] 1,2-Ethanedithiol (0.41 g) was charged to a vessel containing a solution of 4d (0.50 g, 4.0 mmol) in glacial acetic acid (2.5 mL) at about 20 °C. para-toluenesulfonic acid monohydrate (0.15 g) was added and the mixture was agitated until the reaction was complete. The product was isolated by the addition of water (2 mL). The slurry was filtered and the filter cake was washed with water, then deliquored thoroughly to afford 5i. 1H NMR (400 MHz,
DMSO-d6) δ 10.93 (s, 1H), 3.63 – 3.51 (m, 2H), 3.51 – 3.42 (m, 1H), 3.39 – 3.31 (m, 1H), 2.83 (d, J= 17.4 Hz, 1 H), 2.59 – 2.52 (m, 1H), 1.87 (ddd, J = 8.0, 6.2, 3.7 Hz, 1H), 1.65 (dddd, J=
7.7, 6.2, 5.2, 3.9 Hz, 1H), 0.93 (tdd, J = 7.6, 5.5, 1.7 Hz, 1H), 0.02 (dt, J= 5.5, 3.8 Hz, 1H).
[00607] Para-toluenesulfonic acid (0.90 g) was charged to a vessel containing a suspension of 5i (0.50 g, 2.5 mmol) in methyl ethyl ketone (2.5 mL) and water (2.5 mL). The mixture was agitated at about 85 °C until the reaction was complete. The product was isolated from the reaction mixture by cooling to about 20 °C, adding water (2.50 mL), and cooling to about 0 °C. The slurry was filtered and the filter cake was washed with water, then deliquored thoroughly to afford 5e. 1H NMR (400 MHz, DMSO-d6) δ 3.55 – 3.37 (m, 3H), 3.28 – 3.13 (m, 1H), 3.03 (ddd, J = 18.5, 5.6, 2.2 Hz, 1H), 2.20 (d, J = 18.5 Hz, 1H), 1.84 (ddd, J = 8.0, 7.0, 3.8 Hz, 1H), 1.66 (tdd, J = 7.2, 5.6, 4.1 Hz, 1H), 1.03 (tdd, J = 7.9, 5.9, 2.1 Hz, 1H), 0.06 (dt, J = 6.0, 4.0 Hz, 1H).
Example 6: Preparation of 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetic acid (VII) from ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
Synthesis of ethyl 2-((3bS,4aR)-3-(trifluoromethyl)-4,4a-dihydrospiro[cyclopropa[3,4]cyclopenta[1,2-c]pyrazole-5,2′-[1,3]dithiolane]-1(3bH)-yl)acetate (5h) from ethyl 2-((3bS,4aR)-5-oxo-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (XIV)
[00608] Dichloromethane (27 g) was added to a reactor containing XIV (1.0 g) and cooled to about 10 °C. To this was added 1,2-ethanedithiol (0.18 g, 1.2 equiv.). To this was added boron trifluoride acetic acid complex (3.3 g, 2.5 equivalents) over about 25 minutes, and the reaction mixture was agitated at about 20 °C until complete. A solution of calcium chloride dihydrate (0.80g, 0.78 equiv) in 0.10 N hydrochloric acid (16 g) was added over about 1 hour at about 10 °C, and the mixture was agitated for about 90 minutes at about 20 °C. The organic layer was washed successively with water (8 g) and sodium bicarbonate solution (5 wt/wt%). The organic layer was concentrated to afford 5h. 1H NMR (400 MHz, DMSO-d6) δ 5.27 – 4.79 (m, 2H),
4.14 (qd, J = 7.1, 1.1 Hz, 2H), 3.70 – 3.42 (m, 4H), 2.68 (dtd, J = 8.0, 6.4, 5.9, 4.4 Hz, 1H), 2.44 (ddd, J = 7.2, 5.5, 3.6 Hz, 1H), 1.32 (ddd, J = 8.2, 7.2, 5.4 Hz, 1H), 1.18 (t, J= 7.1 Hz, 3H), 0.44 (dt, J = 5.4, 3.9 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 167. 14, 148.36, 133.80 (q, J= 38.3 Hz), 128.77 (m), 121.54 (q, J= 268.4 Hz), 65.33, 61.79, 51.14, 41.30, 40.98, 40.49, 23.57,
15.52, 14.33. 19F NMR (376 MHz, DMSO-d6) δ -60.31.
Synthesis of ethyl 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetate (VII-A)
[00609] Dichloromethane (26 g) was added to a reactor containing 1,3-dibromo-5,5-dimethylhydantoin (DBDMH, 2.4 g, 3.1 equiv.) and cooled to about -10 °C. To this was added 70% hydrofluoric acid/pyridine complex (1.3 g, 17 equiv.), followed by a solution of 5h (1.0 g) in dichloromethane (3 g). The reaction was agitated at about 0 °C until complete. A solution of potassium hydroxide (3.7 g, 25 equivalents) and potassium sulfite (1 .9 g, 4 equiv.) in water (24 g) was added, maintaining an internal temperature of about 5 °C, and agitated for about 30 minutes at about 20 °C. Layers were separated and organic layer was washed with hydrochloric acid (1.1 g, 4 equiv.) in water (9.6 g). The organic layer was concentrated to afford VII-A. 1H NMR (400 MHz, DMSC-d6) δ 5.31 – 5.04 (m, 2H), 4.17 (q, J = 7.1 Hz, 2H), 2.78 – 2.57 (m,
2H), 1.47 (dddd, J = 8.5, 7.1, 5.5, 1.4 Hz, 1H), 1.19 (t, J = 7.1 Hz, 3H), 1.04 (tdt, J= 5.3, 4.0,
1.8 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 166.79, 143.15 (t, J= 29.4 Hz), 134.65 (q, J=
39.0 Hz), 132.99, 121.05 (q, J= 268.4 Hz), 120.52 (t, J= 243.3 Hz), 62.09, 52.49, 27.95 (dd, J = 34.7, 29.0 Hz), 23.82 (d, J = 2.6 Hz), 14.25, 12.14 (t, J = 3.1 Hz). 19F NMR (376 MHz, DMSO-d6) δ -60.47, -79.68 (dd, J= 253.5, 13.2 Hz), -103.09 (dd, J = 253.3, 9.8 Hz).
Synthesis of 2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetic acid (VII)
[00610] A reactor was charged with a solution of VII-A (1.0 g) in dichloromethane (18 g) and cooled to about 5 °C. To this was added ethanol (1.5 g), followed by potassium hydroxide (45 wt/wt%, 0.74 g, 2.0 equiv.). The reaction mixture was agitated at about 20 °C until complete. Water (3.7 g) was added and the reaction mixture was agitated for about 30 minutes. Organic layer was removed and reaction was cooled to about 10 °C. Dichloromethane (18 g) was added, followed by 2N hydrochloric acid (3.3 g, 2,2 equiv.). Reaction was warmed to about 20 °C and agitated for 10 minutes. Layers were separated and aqueous phase was washed with dichloromethane (18 g). Organic layers were combined and concentrated on rotary evaporator to afford VII. 1H NMR (400 MHz, DMSO-d6) δ 13.50 (s, 1H), 5.14 – 4.81 (m, 2H), 2.82 – 2.56 (m, 2H), 1.46 (dddd, J = 8.5, 7.1, 5.5, 1.4 Hz, 1H), 1.08 – 1.00 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 168.16, 143.05 (t, J = 29.4 Hz), 134.40 (q, J = 38.9 Hz), 132.80, 121.11 (q, J = 268.4 Hz), 120.55 (t, J = 243.3 Hz), 52.54, 27.97 (dd, J = 34.7, 29.0 Hz), 23.81 (d, J = 2.5 Hz), 12.13 (t, J = 3.1 Hz). 19F NMR (376 MHz, DMSO-d6) δ -60.39 (d, J = 1.4 Hz), -79.83 (dd, J = 253.2, 13.1 Hz), -102.97 (dd, J= 253.2, 9.8 Hz).
Example 7: Preparation of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1- (2,2,2-trifluoroethyl)-1H-indazol-3-amine (V-02) and its mesylated derivatives
Synthesis of 4-chloro-7-bromo-1-(2,2,2-trifluoroethyl)-1H-indazol-3-amine (V-A)
[00611] To a reactor was added tetrahydrofuran (THF, 275 kg) and diisopropyl amine (DIPA, 30 kg) and the mixture was cooled to about -35 °C. nButyl lithium (2.5 mol/L in hexanes, 74 kg) was charged slowly keeping the reaction temperature less than -30 °C. The mixture was agitated at-35 °C until the reaction was complete. 1-bromo-4-chloro-2-fluorobenzene (52 kg) was charged keeping reaction temperature less than 30 °C and the mixture was agitated at -35°C until reaction was complete. N,N-dimethylformamide (DMF, 36 kg) was charged keeping reaction temperature less than -30 °C and the mixture was agitated at about -35 °C until reaction was complete. Hydrochloric acid (HCl, 18 mass% in water, 147 kg) was charged keeping reaction temperature less than -5 °C. The reaction was warmed to about 0 °C, water (312 kg) was added, and the reaction was extracted with methyl tert-butyl ether (MTBE, 770 kg). The organic was warmed to about 20 °C and washed with brine (NaCl, 23.5 mass% in water, 1404 kg). The mixture was distilled to about 3-4 volumes and heptane was charged (354 kg). The product was isolated by distillation to 3-4 volumes. The slurry was filtered and washed with heptane (141 kg) and dried to afford 6a. 1H NMR (400 MHz, DMSO-d6) δ 10.23 (d, J = 1.2 Hz, 1H), 8.00 (dd, J = 8.7, 1.4 Hz, 1H), 7.44 (dd, J = 8.7, 1.4 Hz, 1H).
[00612] 6a (98.5 kg) was charged to a reactor containing acetic anhydride (105 kg) and acetic acid (621 kg) at 20 °C. The mixture was heated to about 45 °C and hydroxyl amine hydrochloride (31.5 kg) was charged. The reaction was heated to about 75 °C and agitated until the reaction was complete. The product was isolated from the reaction mixture by adding water (788 kg) at about 45 °C. The mixture was cooled to about 25 °C and then the slurry was filtered. The filtered cake was washed with water (197 kg,). The cake was dried to afford 6b. 1H NMR (400 MHz, DMSO-d6) δ 8.11 (dd, J= 8.8, 1.4 Hz, 1H), 7.58 (dd, J = 8.8, 1.4 Hz, 1H).
[00613] To a reactor was charged 6b (84 kg), isopropanol (318 kg), and water (285 kg).
Hydrazine hydrate (20 wt% in water, 178 kg) was charged and the mixture was heated to about 80 °C until the reaction was complete. The product was isolated by cooling the reaction to about 25 °C. The slurry was filtered and the filtered cake was washed with a mixture of isopropanol (127 kg) and water (168 kg). The wet solids were recharged to the reactor and water (838 g) was added. The mixture was agitated at about 25 °C and then filtered and washed with water
(168 g, 2 rel). The cake was dried to afford 6c 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 7.41 (d, J= 7.9 Hz, 1H), 6.84 (d, J= 7.9 Hz, 1H), 5.31 (s, 2H).
[00614] 6c (75 kg) was charged to a reactor containing N,N-dimethylformamide (75 kg). Potassium phosphate (99.8 kg) was charged to the reactor at about 25 °C and the mixture was agitated. 2,2,2-trifluoroethyl trifluoromethanesulfonate (74.3 kg) was charged at about 25 °C and the mixture was agitated until the reaction was complete. Water (375 kg) was charged and the mixture was agitated at about 20 °C. The slurry was filtered and washed with water (150 kg). N,N-dimethylformamide (424 kg) and the wet solid were charged to a reactor at about 20 °C.
The mixture was agitated at about 45 °C. 5 % hydrochloric acid (450 kg) was charged drop-wise to the mixture at about 45 °C. The mixture was cooled to about 25 °C. The slurry was filtered and washed with water (375 g). Water (375 kg) and the filtered solid were charged to a reactor at about 20 °C. The mixture was agitated for about 1 hour at about 20 °C. The slurry was filtered and washed with water (375 kg). The cake was dried to afford V-A. 1H NMR (400 MHz, DMSO-d6) δ 7.57 (d, J= 8.1 Hz, 1H), 6.98 (d, J = 8.1 Hz, 1H), 5.70 (s, 2H), 5.32 (q, J = 8.6 Hz,
2H).
Synthesis of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2-trifluoroethyl)- 1 H-indazol-3-amine (V-02)
[00615] A reactor containing tetrahydrofuran (27 g) and V-A (1.0 g) was cooled to about 0 °C. Chlorotrimethylsilane (7.6 g, 2.3 equiv) was added, followed by the slow addition of lithium bis(trimethylsilyl)amide (5.7 g, 1 M in THF, 2.1 equiv.). The mixture was stirred at about 0 °C until bistrimethylsilane protection was complete. The solution was washed with ammonium chloride in water (10 wt%, 52 g), toluene (44 g) was added, and the biphasic mixture was filtered through celite. The organic and aqueous phases were separated and the aqueous phase was washed with toluene (44 g). The organics were combined, washed with brine (58 g), and azeotropically distilled . The solution was cooled to about 0 °C, isopropylmagnesium chloride lithium chloride complex (2.7 g, 1.3 M in THF, 1.2 equiv.) was added and the reaction was stirred at about 0 °C until lithium halogen exchange was complete. Isopropoxyboronic acid pinacol ester (6.8 g, 1.2 equiv.) was added and the reaction was stirred at about 0°C until botylation was complete. At about 0 °C, The reaction was quenched with aqueous hydrochloric acid (52 g, 1 M), acetonitrile (16 g) was added, and the mixture was stirred until trimethylsilane deprotection was complete. The solution was extracted with ethyl acetate (45 g) and the organic was washed twice with brine (2 x 58 g). The solution was concentrated to low volumes (26 g), dim ethylformami de (47 g) was added, and the solution was concentrated again (51 g). The product was crystallized by the addition of water (50 g). The slurry was filtered and filter cake was washed with heptane (14 g). The solids were dried to afford V-02. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J = 7.6, 1.0 Hz, 1H), 7.07 (dd, J = 7.6, 1.0 Hz, 1H), 5.58 (s, 2H), 5.46 (q, J = 9.1Hz, 2H), 1.32 (s, 12H).
Synthesis of 4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2-trifiuoroethyl)- 1 H-indazol-3-amine (V-02)
[00616] To a reactor was charged V-A (30 kg), bis(pinacolato)diboron (27.9 kg), bis(triphenylphosphine)palladium (II) dichloride (0.9 kg, 1.5 mol%), N,N-dimethylformamide (56 kg, 2 rel. vol.) and toluene (157 kg, 6 rel vol.). The mixture was heated to about 105 °C until the reaction was complete. The mixture was cooled to about 25 °C, filtered through celite (15 kg, 0.5 rel. wt.) and rinsed forward with ethyl acetate (270 kg, 10 rel vol.). PSA-17 palladium scavenger (3 kg, 10 wt%) was added and the mixture was stirred at about 45 °C. The mixture was filtered and the cake was washed with ethyl acetate (54 kg, 2 rel. vol.). The mixture was washed twice with lithium chloride (180 kg, 6 rel. vol.) and once with brine (NaCl, 23.5 mass% in water, 180 kg, 6 rel. vol.). The mixture was then concentrated to about 5-6 rel. vol. under vacuum, heated to about 45 °C then cooled to about 25 °C. Heptane (102 kg, 5 rel. vol.) was charged and the mixture was concentrated to about 4-5 rel. vol. The product was isolated by charging heptane (41 kg, 2 rel. vol.) and cooling the mixture to about 0 °C. The slurry was filtered and washed with heptane (41 kg, 2 rel. vol.). The wet solids were recharged to the reactor with ethyl acetate (27 kg, 1 rel. vol.) and heptane (82 kg, 4 rel. vol.), heated to about 65 °C, and then cooled to about 5 °C. The slurry was filtered and washed with heptane (41 kg, 2 rel. vol.). The cake was dried to afford V-02. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (dd, J =
7.6, 1.0 Hz, 1H), 7.07 (dd, J = 7.6, 1.0 Hz, 1H), 5.58 (s, 2H), 5.46 (q, J = 9.1Hz, 2H), 1.32 (s, 12h).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)-N-(methylsulfonyl)methanesulfonamide (V-04)
[00617] To a 100 mL reactor was added V-02 (5.00 g), 2-methyltetrahydrofuran (50 mL), and triethylamine (11.1 mL). The mixture was cooled to about 10 °C and methanesulfonyl chloride (2.58 mL, 33.3 mmol) was added to the mixture. The mixture was agitated at about 10 °C until reaction was complete. The mixture was concentrated to dryness and the residue was purified by column chromatography to afford V-04. 1H NMR (400 MHz, DMSO-d6) δ 7.96 (d, J = 7.7 Hz, 1H), 7.50 (d, J = 7.6 Hz, 1H), 5.95 (q, J = 8.8 Hz, 2H), 3.66 (s, 6H), 1.37 (s, 12H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2)-1-(2,2,2,- trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-03)
[00618] To a 100 mL reactor was added V-02 (5.00 g), 2-methyltetrahydrofuran (50 mL), and triethylamine (11.1 mL, 79.6 mmol). The mixture was cooled to about 10 °C and methanesulfonyl chloride (2.58 mL) was added to the mixture. The mixture was agitated at about 10 °C until reaction was complete. To the mixture was added 2-methyltetrahydrofuran (21.5 g) and sodium hydroxide (0.43 g) and the mixture was agitated at about 25 °C until the reaction was complete. To the resulting solution was added 2-methyltetrahydrofuran (21.5 g), water (25 g) and acetic acid to achieve a pH of less than 7. The lower aqueous layer was then removed and the organic layer was washed with brine (5 wt%, 7.8g). The organic layer was then concentrated to dryness and the residue was purified by column chromatography to afford V-03. 1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 7.86 (d, J = 7.6 Hz, 1H), 7.34 (d, J = 7.6 Hz, 1H), 5.80 (q, J = 8.9 Hz, 2H), 3.22 (s, 3H), 1.36 (s, 12H).
Synthesis of N-(7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)-N- (methylsulfonyl)methanesulfonamide (V-06)
[00619] To a reactor was added V-A (3 g), 2-methyltetrahydrofuran (25.8 g), and triethylamine (7.6 mL). The mixture was cooled to about 10 °C, methanesulfonyl chloride (1.8 mL) was added, and the mixture was stirred until reaction was complete. The reaction mixture was washed with aqueous sodium chloride (30 mL) and the organic layer was evaporated to dryness. The residue was purified by column chromatography to afford V-06. 1H NMR (400 MHz, DMSO-d6) δ 7.83 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.1 Hz, 1H), 5.79 (q, J = 8.5 Hz, 2H), 3.62 (s, 6H).
Synthesis of N-(7-bromo-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-05)
[00620] To a reactor was added V-02 (3 g), 2-methyltetrahydrofuran (30 mL), and triethylamine (7.6 mL). The mixture was cooled to about 10 °C, methanesulfonyl chloride (1.8 mL) was added, and the mixture was stirred until reaction was complete. The reaction mixture was washed with aqueous sodium chloride (30 mL) and the organic portion was concentrated to dryness.
[00621] To the resulting mixture (2.7g) was added 2-methyltetrahydrofuran (15 mL) and sodium hydroxide (1M in water, 15 mL). The mixture was stirred at about 20 °C until the reaction was complete. The aqueous layer was removed and the organic was washed with acetic acid (0.7M in water, 10 mL) and sodium chloride (5 wt% in water, 10 mL).The organic layer was then concentrated to dryness and the residue was purified by column chromatography to afford V-05. 1H NMR (400 MHz, DMSO-D6) δ 10.03 (s, 1H), 7.71 (dd, J = 8.0, 1.6 Hz, 1H), 7.20 (dd, J = 8.1, 1.6 Hz, 1H), 5.64 (q, J = 8.7 Hz, 3H), 3.19 (2, 3H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2,-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)-N-(methylsulfonyl)methanesulfonamide (V-04)
[00622] To a reactor was charged V-06 (148 mg), bis(pinacolato)diboron (93 mg), potassium acetate (90 mg) and bis(triphenylphosphine)palladium (II) chloride (4.3 mg, 1.5 mol%). N,N- dimethylformamide (0.2 mL) and toluene (0.6 mL) were added and the reaction was heated to about 105 °C until completion. V-04 was formed. 1H NMR (400 MHz, DMSO-D6) δ 7.96 (d, J = 7.7 Hz, 1H), 7.50 (d, J= 7.6 Hz, 1H), 5.95 (q, J= 8.8 Hz, 2H), 3.66 (s, 6H), 1.37 (s, 12H).
Synthesis of N-(4-chloro-7-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)methanesulfonamide (V-03)
[00623] To a reactor was charged V-05 (124 mg), bis(pinacolato)diboron (93 mg), potassium acetate (90 mg) and bis(triphenylphosphine)palladium (II) chloride (4.3 mg, 1.5 mol%). N,N- dimethylform amide (0.2 mL.) and toluene (0.6 mL, 6 rel. vol.) were added and the reaction was heated to about 105 °C until completion. V-03 was formed. 1H NMR (400 MHz, DMSO-d6) δ
9.96 (s, 1 H), 7.86 (d, J= 7.6 Hz, 1H), 7.34 (d, J= 7.6 Hz, 1H), 5.80 (q, J = 8.9 Hz, 2H), 3.22 (s,
II. Synthesis of the Compound of Formula I
Example 8: Preparation of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1- yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
Synthesis of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2- (3,5-difluorophenyl)ethyl)-2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV) from (S)-1-(3-bromo-6-(3- methyl-3-(methylsulfbnyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3.5-difluorophenyl)ethan-1-amine (VI) Method 1
[00624] n-Propyl phosphonic anhydride (T3P, 3.1 g, 1.5 equiv.) was slowly added to a reactor containing amine VI (1.5 g), acid VII (1.0 g, 1.1 equiv.), triethylamine (Et3N, 0.5 g, 1.5 equiv.), and acetonitrile (MeCN, 8.0 g). The mixture was agitated at about 20 °C until the reaction was complete. The product was crystallized from the reaction mixture with DMF (0.63 g), and water (15 g). The slurry was filtered and the filter cake was washed with a mixture of acetonitrile and water (2 x 2.5 g). The cake was dried to afford IV. 1H NMR (400 MHz, DMSO-d6) δ9.19 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.07 (tt, J = 9.4, 2.4 Hz, 1H),
6.96 – 6.87 (m, 2H), 5.52 (td), J = 8.8, 5.3 Hz, 1 H), 4.93 – 4.73 (m, 2H), 3.22 (s, 3H), 3.11 -2.90 (m, 2H), 2.66 – 2.52 (m, 2H), 1.69 (s, 6H), 1.45 – 1.36 (m, 1H), 1.02 – 0.93 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.42, 163.62, 163.49, 161.17, 161.04, 158.19, 142.92, 142.20, 142.10, 142.01, 141.63, 140.23, 134.11, 133.73, 132.14, 128.66, 122.23, 120.49, 119.56, 112.49, 112.25, 104.75, 102.25, 88.62, 84.20, 57.44, 53.85, 53.03, 35.21, 23.41, 22.46, 22.40, 11.79.
Synthesis of N-((S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV) from (S)-1-(3-bromo-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethan-1-amine (VI) Method 2
[00625] N-methylmorpholine (NMM, 0.51 g, 2.3 equiv.) was added to a vessel containing amine VI (1.0 g), acid VII (1.0 g), 1-hydroxybenzotriazole hydrate (HOBt ● H2O, 0.17 g, 0.5 equiv.), N-(3-dimethylaminopropyi)-N’-ethylcarbodiimide (EDCI ● HCl, 0.52 g, 1.25 equiv.), and acetonitrile (MeCN, 7.8 g). The mixture was agitated at about 20 °C until the reaction was complete. The product was crystallized from the reaction mixture with DMF (2.8 g), and water (10 g). The slurry was filtered and the filter cake was washed with a mixture of acetonitrile and water. The cake was dried to afford IV. 1H NMR (400 MHz, DMSO-d6) δ9.19 (d, J = 8.3 Hz, 1H), 8.12 (d, J = 8.3 Hz, 1H), 7.50 (d, J = 8.3 Hz, 1H), 7.07 (tt, J = 9.4, 2.4 Hz, 1H), 6.96 – 6.87 (m, 2H), 5.52 (td), J = 8.8, 5.3 Hz, 1 H), 4.93 – 4.73 (m, 2H), 3.22 (s, 3H), 3.11 – 2.90 (m, 2H), 2.66 – 2.52 (m, 2H), 1.69 (s, 6H), 1.45 – 1.36 (m, 1H), 1.02 – 0.93 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 164.42, 163.62, 163.49, 161.17, 161.04, 158.19, 142.92, 142.20, 142.10, 142.01, 141.63, 140.23, 134.11, 133.73, 132.14, 128.66, 122.23, 120.49, 119.56, 112.49, 112.25, 104.75, 102.25, 88.62, 84.20, 57.44, 53.85, 53.03, 35.21, 23.41, 22.46, 22.40, 11.79.
Example 9: Preparation of N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H- indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5- difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro- 1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
Synthesis of compound III-03
[00626] To a reactor was added IV (1 .0 g), potassium bicarbonate (0.43 g, 1.3 equiv), dichlorobis(tricyclohexylphosphine)palladium(II) (28 mg, 2.5mol%), V-02 (0.67 g), butyl acetate (7.3 g) and water (2.1 g). The reactor was inerted and the mixture was agitated at about 85 °C (75-90 °C) until the reaction was complete. The mixture was cooled to about 40 °C and passed through celite (0.52 g). The celite cake was rinsed with butyl acetate (1.8 g). The filtrate and rinse were combined and this solution was washed twice with a mixture of N-acetyl-L-
cysteine (0.31 g) dissolved in water (5.2 g) and sodium hydroxide in water (5 wt%, 5.4 g). The organics were washed twice with sodium chloride in water (5 wt%, 11 g). The solution was azeotropically distilled into 1-propanol (3.3 g). To the propanol solution at about 50 °C was added methanesulfonic acid (0.31 g, 2.25 equiv.) and the product was crystallized using dibutyl ether (5.1 g). The slurry was cooled to about 10 °C, filtered, and the filter cake was washed with a 5:1 mixture of propanol in dibutyl ether (1.6 g). The solids were dried to afford III-03 1H NMR (400 MHz, DMSO-d6) δ 9.19 (d, J = 8.3 Hz, 2H), 7.84 – 7.69 (m, 4H), 7.11 (d, J = 7.7 Hz, 2H), 7.07 – 6.95 (m, 3H), 6.82 (d, J = 7.7 Hz, 2H), 6.54 – 6.40 (m, 4H), 4.90 (d, J = 16.4 Hz, 2H), 4.76 – 4.60 (m, 4H), 4.15 (dq, J = 16.6, 8.4 Hz, 2H), 3.75 (dt, J = 16.3, 8.7 Hz, 2H), 3.25 (s, 7H), 2.99 – 2.86 (m, 4H), 2.63 – 2.50 (m, 3H), 2.41 (s, 14H), 1.73 (d, J = 2.1 Hz, 13H), 0.93 (dd, J = 6.1, 3.9 Hz, 2H).
Synthesis of N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
[00627] Aqueous sodium hydroxide (0.2 M; 2.2 equivalents; 9.2 g) was added to a reactor containing III-03 (1.0 g) in MeTHF (8.3 g) at about 20 °C. The biphasic mixture was agitated for about 15 min, and the aqueous layer was removed. The organic layer was washed four times with 2.0 wt% aqueous sodium chloride (9.8 g) and was distilled. The solution containing III was used directly in the II process below. A sample was concentrated to dryness for analysis. 1H NMR (400 MHz, CDCl3): δ 7.44 ( m, 1H), 7.39 (br, 1H), 7.18 (m, 1H), 6.90 (m, 1H), 6.65 (m 1H), 4.10 (m, 2H), 3.72 (m, 4H), 2.78 (m 2H), 2.56 (br, 4H), 1.31 (s, 9H). 13C NMR (100 MHz, DMSO-d6): δ 176.88, 158.95, 141,06, 129.55, 112.79, 109.56, 106.83, 66.66, 65.73, 57.45,
Example 10: Preparation of N-((S)-1-(3-(4-chloro-3-(N- (methylsulfonyl)methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (II)
[00628] Methanesulfonyl chloride (0.32 g, 2.5 equivalents) was added to a reactor containing III (1.0 g), triethylamine (0.69 g, 6.0 equivalents), and MeTHF (11 g) at about 10 °C. The mixture was agitated at about 10 °C until the reaction was complete. The reaction mixture was washed with water (6.4 g) for about 15 minutes, and warmed to about 20 °C. The layers were separated and the organic layer was washed for about 15 minutes with 10 wt% aqueous sodium chloride (6.9 g). The layers were separated and the organic layer was used directly in the next step. An aliquot was concentrated to dryness for analysis. 1H NMR (400 MHz, δ6-DMSO; 9: 1 mixture of atropi somers): δ 9.20 (d, J = 7.9 Hz 1 H), 8.99* (d, J = 8.6 Hz, 1 H), 7.96* (d, J = 7.9 Hz, 1 H), 7.83 (d, J = 8.0 Hz, 1 H), 7.80* (d, J = 7,9 Hz, 1 H), 7.76 (d, J – 8.0 Hz, 1 H), 7.45 (d, J = 7.7 Hz, 1 H), 7.41* (d, J = 7.8 Hz, 1 H), 7.31* (d, J = 7.8 Hz, 1 H), 7.02 (tt, J = 9.4, 2.1 Hz,
1 H), 6.92* (s, 1 H), 6.91 (d, J = 7.7 Hz, 1 H), 6.48 (m, 2 H), 4.92* (s, 1 H), 4.88 (d, J = 16.4 Hz, 1 H), 4.79* (d, J = 16.8 Hz, 1 H), 4.73* (d, J = 16.4 Hz, 1 H), 4.71* (m, 1 H), 4.69 (m, 1 H), 4.62* (s, 1 H), 4.60 (m, 1 H), 4.38* (dq, J = 16.4, 8.2 Hz, 1 H), 4.12 (dq, J = 16.7, 8.4 Hz, 1 H), 3.68* (s, 3 H), 3.66* (s, 3 H), 3.63 (s, 3 H), 3.58 (s, 3 H), 3.26 (s, 3 H), 3.12* (dd, 7 = 13.8, 10.5 Hz, 1 H), 3.05 (dd, J = 13.5, 5.8 Hz, 1 H), 2.97 (dd, J = 13.5, 8.5 Hz, 1 H), 2.78* (dd, J = 13.7, 3.9 Hz, 1 H), 2.59 (m, 1 H), 2.53 (m, 1 H), 1.75 (s), 1.75 (s, 6 H), 1 .39 (m, 1 H), 0.98 (m, 1 H).
13C NMR (100 MHz, DMSO-d6, 9:1 mixture of atropi somers): δ 164.5, 163.6*, 162.1 (dd, ,7 = 246.3, 13.4 Hz), 162.0* (dd, J = 246.1, 13.3 Hz), 158.7, 158.4*, 142.7 (t, J = 29.3 Hz), 142.3, 142.0*, 141.8 (t, J= 9.4 Hz), 140.6*, 139.9, 139.7*, 139.3, 135.8*, 135.0, 133.8 (q, J = 39.0 Hz), 132.2*, 132.1 (m), 131.6, 129.6, 129.4*, 126.7, 125.3, 125.2*, 124.1*, 123.4, 122.8*, 122.7 (q, J= 280.9 Hz), 120.7 (q, J = 268.3 Hz), 119.9 (t, J = 243.7 Hz), 119.8, 119.5*, 119.0*, 118.9, 112.0, 102.2 (t, J= 225.7 Hz), 101.8*, 88.4, 84.5, 57.3, 52.93, 52.86, 52.7, 52.5*, 50.7 (q, J = 33.8 Hz), 50.3*, 42.6*, 42.4, 42.3*, 42.2, 39.51, 39.5, 38.9*, 35.1, 27.5 (dd, J = 35.0, 28.6 Hz), 23.1, 22.4, 22.3, 11.5. (* signals arising from minor atropisomer)
Example 11: Preparation of N-((S)-1-(3-(4-chIoro-3-(methylsuIfonamido)-1-(2,2,2- trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)- 2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5- tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I)
Synthesis of sodium (4-chloro-7-(2-((S)-1-(2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamido)-2-(3,5- difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2- trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide (1-02)
[00629] Sodium hydroxide (1 M, 2.9 g, 3.0 equiv.) was added to a reactor containing II (1.0 g) and 2-methyltetrahydrofuran (8.4 g) at about 35 °C. The mixture was agitated until the reaction was deemed complete. The reaction mixture was adjusted to between about 20 and 40 °C and the bottom layer was removed. The organic layer was washed with water (2.9 g) for about 15 minutes, and the bottom layer was removed. The organic solvent was swapped for ethanol and the solution was concentrated to about 5 volumes and the temperature was adjusted to about 35 °C. n-Heptane (3.4 g) was slowly added, and the mixture was aged for about 12 hours. The solids were collected by filtration, and the filter cake was washed with ethanol/n- heptane (1:1). The resultant wet cake was dried under vacuum to afford 1-02. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (d, J = 8.0 Hz, 1H), 8.93* (d, J = 8.5 Hz), 7.80 – 7.72* (m), 7.71 (s, 2H), 6.99 (tt, J = 9.5, 2.4 Hz, 1H), 6.94 (d, J = 7.6 Hz, 1H), 6.90* (d, J = 6.3 Hz), 6.69 (d, J = 7.6 Hz, 1H), 6.57 – 6.51* (m), 6.48 – 6.40 (m, 2H), 4.90 (d, J = 16.5 Hz, 1H), 4.77 (d, J = 16.4
Hz, 1H), 4.70 (td, J = 8.3, 5.2 Hz, 1H), 4.63* (d, J = 16.5 Hz), 4.22 (dq, J= 16.7, 8.4 Hz, 1H), 3.90 – 3.75 (m, 1H), 3.26 (s, 3H), 2.92 (td, J = 13.8, 8.5 Hz, 2H), 2.83* (s), 2.80 (s, 3H), 2.64 – 2.51 (m, 2H), 1.74 (d, J = 2,2 Hz, 6H), 1.44 – 1.34 (m, 1H), 0.94 (dq, J = 6.0, 3.7 Hz, 1H); 13C NMR (100 MHz, dmso) δ 164.39, 163.43, 163.39, 163.25, 160.94, 160.91, 160.81, 158.93,
158.22, 152.64, 151.94, 142.92, 142.72, 142.63, 142.43, 142.34, 142.19, 142.10, 142.00, 141.43,
141.14, 139.55, 139.36, 133.95, 133.56, 133.17, 132.12, 131.93, 131.68, 129.66, 129.56, 128.17,
127.91, 126.86, 126.76, 125.02, 122.35, 122.21, 122.08, 122.05, 119.93, 119.88, 119.38, 118.88,
118.18, 117.54, 117.21, 117.04, 112.18, 112.02, 111.95, 111.84, 111.78, 102.28, 102.03, 101.81,
88.14, 88.00, 84.69, 84.65, 57.33, 53.22, 52.96, 52.76, 52.44, 40.15, 39.94, 39.73, 39.52, 39.31, 39.10, 38.97, 38.89, 38.65, 35.10, 35.08, 27.86, 27.56, 27.52, 27.23, 23.19, 22.42, 22.41, 22.30, 22.28, 11.63. * Signals arising from minor atropisomer. 13C NMR data is reported for the mixture of atropisomers.
Synthesis of N-((S)-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I) from sodium (4-chioro-7-(2-((S)-1-(2-((3bS.4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-l-yl)acetamido)-2-(3.5-difluorophenyl)ethyl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-3-yl)-1-(2,2,2-trifluoroethyl)-1H-indazol-3-yl)(methylsulfonyl)amide (I-02)
[00630] Compound I-02 (1.0 g) and glacial acetic acid (2.1 g) were combined at about 20 °C and were agitated until dissolved. The resultant solution was transferred to a reactor containing water (15 g) over about 1 hour. The resultant slurry was further agitated for about one hour, and was filtered. The wet cake was washed with water (2 x 5 g), deliquored, and dried at about 60 °C under vacuum to provide I. 1H NMR (400 MHz, δ6-DMSO; 5:1 mixture of atropi somers) δ 10.11* (s), 10.00 (s, 1 H), 9.25 (d, J= 8.0 Hz, 1 H), 8.92* (d, J = 8.4 Hz), 7.90* (d, J = 7.6 Hz), 7.81 (d, J = 8.0 Hz, 1 H), 7.76 (d, J= 8.0 Hz, 1 H), 7.32 (d, J = 7.6 Hz, 1 H), 7.23* (d, J = 8.0 Hz), 7.19* (d, J = 8.0 Hz), 7.02 (tt, J = 9.4, 2,4 Hz, 1 H), 6.94* (m), 6.86 (d, J = 7.6 Hz, 1 H), 6.54* (m), 6.48 (m, 2 H), 4.92 (d, J = 16.4 Hz, 1 H), 4.77* (d, J = 16.4 Hz), 4.71 (d, J = 16.4 Hz, 1 H), 4.68* (m), 4.51 (dq, J = 16.4, 8.3 Hz, 1 H), 4.19* (dq, J = 16.4, 8.2 Hz), 3.96 (dq, J = 16.8,
8.4 Hz, 1 H), 3.27 (s, 3 H), 3.24* (s), 3.17 (s, 3 H), 3.11* (dd, J = 13.0, 3.4 Hz), 3.02 (dd, J = 13.6, 5.6 Hz, 1 H), 2.95 (dd, J = 13.8, 8.6 Hz, 1 H), 2.92* (m), 2.60 (m, 1 H), 2.55 (m, 1 H), 1.74 (s, 6 H), 1.40 (m, 1 H), 0.96 (m, 1 H); 13C NMR (100 MHz, δ6-DMSO; 5:1 mixture of atropisomers) δ 164.5, 163.4*, 162.1 (dd, 7 = 246.0, 13.4 Hz), 162.0* (dd, 7 = 246.1, 13.4 Hz), 158.8, 158.1 *, 142.7 (t, 7 = 29.3 Hz), 142.3, 142.1* (m), 141.9 (t, J= 9.5 Hz), 141.7*, 140.2*, 140.0*, 139.8*, 139.5, 139.3, 139.2, 133.8 (q, J= 38.7 Hz), 132.0 (m), 131.7*, 131.1, 130.3*, 130.0, 126.8, 126.4, 126.2*, 123.0* (m), 122.9 (q, J = 281.7 Hz), 122.7*, 122.1, 120.7 (q, J = 268.3 Hz), 119.9 (t, J= 243.4 Hz), 119.0, 118.7*, 117.5*, 117.4, H2.0 (m), 102.1 (t, J= 25.6 Hz), 101.9* (m), 88.5*, 88.4, 84.5, 57.3, 52.8, 52.7, 52.4*, 50.2 (q, J= 33.3 Hz), 50.0 (m),
41.4*, 41.2, 39.8, 38.7, 35.1, 27.5 (dd, J= 35.1, 29.0 Hz), 23.2, 22.4, 22.3, 22.2*, 11.6. * Signals arising from the minor atropisomer.
[00631] Alternatively, a premixed solution of acetic acid (1.5 g), ethanol (12 g), and water (0.3 g) were combined with Compound I-02 at 20 °C and were agitated until dissolved. The resultant solution was transferred to a reactor containing water (100 g) over about 30 minutes. The resultant slurry was further agitated for about one hour, and was filtered. The wet cake was washed with water (2 x 25 g), deliquored, and dried at about 60 °C under vacuum to provide I.
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,44a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide(I) from N-((S)-1-(3-(3-amino-4-chloro-1-(2,2,2-trifluoroethyl)-1H-indazol-7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2-((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)- 3b,4,4a,5-tetrahydro-1H-cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (III)
[00632] A reactor was charged with III (1.0 g) followed by cyclopentyl methyl ether (2.0 mL). The contents were adjusted to about 80 °C. In a separate reactor, methanesulfonic acid anhydride (0.3g, 1.5 equiv.) was dissolved in cyclopentyl methyl ether (6 mL). The solution was added to the first reactor via a syringe pump over 5 h. Following addition, the reaction mixture was aged for 16 h. The reaction mixture was quenched with water (10 mL). UPLC analysis of the organic phase showed I with 94.8% purity.
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (I) from N-((S)-1-(3-bromo-6-(3- methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
[00633] To a 40 mL vial was added IV (1 .00 g), potassium bicarbonate (420 mg), palladium(II) chloride (4.9 mg, 2.0 mol%), cyclohexyl diphenylphosphine (13.4 mg, 3.6 mol%), V-03 (849 mg), 2-methyltetrahydrofuran (8.0 mL) and water (2.0 mL). The vial was inerted and the mixture was agitated at about 68 °C (65-73 °C) until the reaction was complete. The mixture was cooled to about 40 °C and the aqueous layer was removed. The organic layer was washed with aqueous acetic acid (5% w/v, 5.1 g). The organic was then concentrated to dryness and the residue was purified by column chromatography to afford I. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 0.2H), 10.00 (s, 1H), 9.25 (d, J = 8.2 Hz, 1H), 8.92 (d, J = 8.6 Hz, 0H),
7.90 (d, J = 7.9 Hz, 0.1H), 7.85 – 7.71 (m, 2H), 7.52-7.50 (m, 0.1H), 7.32 (d, J = 7.7 Hz, 1H),
7.21 (q, J= 9.6 Hz, 0.4H), 7.11 – 6.97 (m, 1H), 6.94-6.89 (m, 0.2H), 6.86 (d, J = 7.7 Hz, 1H),
6.55 (d, J = 7.4 Hz, 0.4H), 6.52 – 6.43 (m, 2H), 4.92 (d, J = 16.4 Hz, 1H), 4.81-4.66 (m, 1.5H),
4.64-4.45 (m, 2.4H), 4.28-4.13 (m, 0.2H), 4.08-3.92 (m, 1.6H), 3.32 (s, 0.7H), 3.30-3.22 (m, 4.4H), 3.17 (s, 3H), 3.08-2.89 (m, 2.2H), 2.69 – 2.53 (m, 2.2H), 2.12 (s, 0.2H), 1.99 (s, 1H), 1.91 (s, 0.3H), 1.80 – 1.70 (m, 6H), 1.48-1.36 (m, 1.2H), 1.23 – 1.12 (m, 1.3H), 0.96 (s, 1.2H).
Synthesis of N-((S)-1-(3-(4-chloro-3-(methylsulfonamido)-1-(2,2,2-trifluoroethyl)-1H-indazol- 7-yl)-6-(3-methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3.5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cvclopenta[1,2-c]pyrazol-1-yl)acetamide(I) from N-((S)-1-(3-bromo-6-(3- methyl-3-(methylsulfonyl)but-1-yn-1-yl)pyridin-2-yl)-2-(3,5-difluorophenyl)ethyl)-2- ((3bS,4aR)-5,5-difluoro-3-(trifluoromethyl)-3b,4,4a,5-tetrahydro-1H- cyclopropa[3,4]cyclopenta[1,2-c]pyrazol-1-yl)acetamide (IV)
[00634] To a 40 mL vial was added IV (1.00 g), potassium bicarbonate (420 mg), palladium(II) chloride (4.9 mg, 2.0 mol%), cyclohexyl diphenylphosphine (13.4 mg, 3.6 mol%), V-04 (923 mg), 2-methyltetrahydrofuran (8.0 mL) and water (2.0 mL). The vial was inerted and the mixture was agitated at about 68 °C (65-73 °C) until the reaction was complete. The mixture was cooled to about 40 °C and the aqueous layer was removed. The organic was stirred with aqueous sodium hydroxide (5 % w/w, 6.3 g) at 40 °C until reaction was complete. The organic was washed with aqueous acetic acid (5% w/v, 5.1 g). The organic was then concentrated to dryness and the residue was purified by column chromatography to afford I. 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 0.2H), 10.00 (s, 1H), 9.25 (d, J = 8.2 Hz, 1H), 8.92 (d, J = 8.6 Hz, 0H), 7.90 (d, J = 7.9 Hz, 0.1H), 7.85 – 7.71 (m, 2H), 7.52-7.50 (m, 0.1H), 7.32 (d, J = 7.7 Hz, 1H), 7.21 (q, J = 9.6 Hz, 0.4H), 7.11 – 6.97 (m, 1H), 6.94-6.89 (m, 0.2H), 6.86 (d, J =
7.7 Hz, 1H), 6.55 (d, J = 7.4 Hz, 0.4H), 6.52 – 6.43 (m, 2H), 4.92 (d, J = 16.4 Hz, 1H), 4.81- 4.66 (m, 1.5H), 4.64-4.45 (m, 2.4H), 4.28-4.13 (m, 0.2H), 4.08-3.92 (m, 1.6H), 3.32 (s, 0.7H), 3.30-3.22 (m, 4.4H), 3.17 (s, 3H), 3.08-2.89 (m, 2.2H), 2.69 – 2.53 (m, 2.2H), 2.12 (s, 0.2H), 1.99 (s, 1H), 1.91 (s, 0.3H), 1.80 – 1.70 (m, 6H), 1.48-1.36 (m, 1.2H), 1.23 – 1.12 (m, 1.3H), 0.96 (s, 1.2H).
SYN
Luíza Cruz
https://drughunter.com/lenacapavir-synthesis-highlights/


see below at end
/////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
 |
|
| Clinical data | |
|---|---|
| Trade names | Sunlenca |
| Other names | GS-CA1, GS-6207 |
| Routes of administration |
By mouth, subcutaneous |
| ATC code | |
| Legal status | |
| Legal status |
|
| Identifiers | |
| CAS Number | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| PDB ligand | |
| Chemical and physical data | |
| Formula | C39H32ClF10N7O5S2 |
| Molar mass | 968.28 g·mol−1 |
| 3D model (JSmol) | |
History
Lenacapavir is being developed by Gilead Sciences.[2]
As of 2021, it is in phase II/III clinical trials.[3] It is being investigated as a treatment for HIV patients infected with multidrug-resistant virus and as a twice-yearly injectable for pre-exposure prophylaxis (PrEP).[3][4]
Society and culture
Legal status
On 23 June 2022, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Sunlenca, intended for the treatment of adults with multidrug‑resistant human immunodeficiency virus type 1 (HIV‑1) infection.[5] The applicant for this medicinal product is Gilead Sciences Ireland UC.[5] Lenacapavir was approved for medical use in the European Union in August 2022.[1]
References
- ^ Jump up to:a b c d e f “Sunlenca EPAR”. European Medicines Agency (EMA). 22 June 2022. Archived from the original on 26 August 2022. Retrieved 25 August 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ Link JO, Rhee MS, Tse WC, Zheng J, Somoza JR, Rowe W, et al. (August 2020). “Clinical targeting of HIV capsid protein with a long-acting small molecule”. Nature. 584 (7822): 614–618. Bibcode:2020Natur.584..614L. doi:10.1038/s41586-020-2443-1. PMC 8188729. PMID 32612233. S2CID 220293679.
- ^ Jump up to:a b Boerner H (11 March 2021). “Lenacapavir Effective in Multidrug Resistant HIV”. Medscape. Archived from the original on 16 March 2021. Retrieved 15 March 2021.
- ^ Highleyman L (15 March 2021). “Lenacapavir Shows Promise for Long-Acting HIV Treatment and Prevention”. POZ. Archived from the original on 19 July 2021. Retrieved 15 March 2021.
- ^ Jump up to:a b “Sunlenca: Pending EC decision”. European Medicines Agency. 23 June 2022. Archived from the original on 26 June 2022. Retrieved 26 June 2022. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
External links
- “Lenacapavir”. Drug Information Portal. U.S. National Library of Medicine.
- “Lenacapavir sodium”. Drug Information Portal. U.S. National Library of Medicine.
- “Lenacapavir”. Clinical Info. National Institutes of Health.
////////////Lenacapavir sodium, approvals 2022, ema 2022, レナカパビルナトリウム , HIV, SUNLECA, GS-6207, GS-HIV, GS-CA1, GS-CA2, PF-3540074, GS-CA1, eu 2022
[H][C@]12C[C@@]1([H])C(F)(F)C1=C2C(=NN1CC(=O)N[C@@H](CC1=CC(F)=CC(F)=C1)C1=NC(=CC=C1C1=CC=C(Cl)C2=C1N(CC(F)(F)F)N=C2NS(C)(=O)=O)C#CC(C)(C)S(C)(=O)=O)C(F)(F)F
Syn
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
Lenacapavir (Sunlenca). Lenacapavir was first approved in the EU in 2022 for use in combination with
other antiretroviral(s) in adults with multidrug resistant HIV infection and for whom it is otherwise not possible to construct a suppressive antiviral regimen.1314Later in the year the drug also received approval in Canada and the US for similar indications.
Lenacapavir received first-in-class designation as 15,16 the first approved drug known to inhibit the capsid of HIV-1, aprotein shell that encompasses the genetic material of the virus and is known to be involved in multiple stages of the HIV life cycle.
Lenacapavir is available as both an oral and injectable therapy. As a slow-release, long lasting treatment
for HIV-1, dosing may be required as infrequently as every 6months when taken in combination with other antiretrovirals, with the goal of improving patient adherence and medication
compliance. Lenacapavir was developed by Gilead Sciences, Inc. and is currently being evaluated in a number of other clinical trials for both HIV-1 treatment and prevention. 13 The synthesis of lenacapavir is achieved by joining four advancedintermediates:17boronateester2.5,18 cyclopropane carboxylic acid 2.13a,17 chiral amino pyridine 2.19,17 and alkynyl sulfone2.20(Schemes2, 3, 4, and5, respectively).18 Thesynthesisof thesekeyintermediateshavebeendescribed inthepatent literatureemployingavarietyof routes17−22and is also exemplified on scales ranging fromsingle gram to multikilogramscale, albeit without reporting isolated yields. For thepurposeof this review, the largest reportedroute to each intermediate will be described. First, synthesis of boronateester2.5beganwitharylcyanofluorideintermediate 2.1 and reliedon a condensation/intramolecular cyclization processwithhydrazinehydrate in i-PrOH/water togenerate
aminopyrazole2.2onmultikilogramscaleafter coolingand precipitation of the product from the reaction mixture (Scheme2).18Next, nucleophilicdisplacementof triflate2.3, facilitated by potassium phosphate in DMF, generated ultimately providing 2.13a as a single isomer to carry forward to lenacapavir. Chiral amine intermediate 2.19 was accessed from 3,6 dibromopicolinaldehyde (2.14), using (S)-2-ethylpropane-2 sulfinamide (2.15) as a chiral auxiliary for installation of the chiral amine (Scheme 4). 17
Condensation of aldehyde 2.14 and sulfinamide 2.15 in the presence of Cs2CO3 and NMP
furnished chiral sulfinimine 2.16, which was precipitated from the reaction mixture by addition of water. The resulting solid was then treated with (3,5-difluorobenzyl)zinc bromide using a controlled addition protocol before HCl-mediated cleavage of the chiral auxiliary. Precipitation from the reaction mixture
yielded the HCl salt of 2.18 as a single isomer, which was treated with Boc2O under aqueous basic conditions to yield the Boc-protected chiral amine 2.19. 17 It is important to note that while this chiral auxiliary-based method constitutes the largest scale route to 2.13a published to date, patent literature
by Gilead has recently emerged, describing synthesis of similar analogs on a small scale using chiral salt-based resolution methods.
The final steps in the synthesis of lenacapavir began with the Sonogashira cross coupling of advanced pyridyl bromide intermediate 2.19 and 3-methyl-3-(methylsulfonyl)but-1-yne (2.20) (Scheme 5). 17
After precipitation from the reaction mixture, the resulting product, 2.21, was subjected to Suzuki
coupling with boronate ester 2.5, furnishing 2.22. Bis sulfonylation of amine 2.22 with MsCl in TEA and subsequent Boc group cleavage with TFA allowed for generation of 2.23 following a hexane wash and neutralization process. Finally, incorporation of the chiral cyclopropane fragment was made
possible by HATU-mediated coupling of amine 2.23 and cyclopropane-carboxylic acid intermediate 2.13a in DMF and DIPEA to give a bis-substituted methylsulfonamide. Selective cleavage of one methanesulfonyl group was performed by addition of 2 N NaOH to the crude reaction mixture,
completing the synthesis of lenacapavir (2).17
(13) Paik, J. Lenacapavir: First approval. Drugs 2022, 82, 1499−
1504.
(14) Tuan, J.; Ogbuagu, O. Lenacapavir: a twice-yearly treatment for
adults with multidrug-resistant HIV infection and limited treatment
options. Expert Rev. Anti Infect. Ther. 2023, 21, 565−570.
(15) Mullard, A. FDA approves first-in-class HIV capsid inhibitor.
Nat. Rev. Drug Discovery 2023, 22, 90.
(16) Dvory-Sobol, H.; Shaik, N.; Callebaut, C.; Rhee, M. S.
Lenacapavir: A first-in-class HIV-1 capsid inhibitor. Curr. Opin. HIV
AIDS 2022, 17, 15−21.
(17) Graupe, M.; Henry, S. J.; Link, J. O.; Rowe, C. W.; Saito, R. D.;
Schroeder, S. D.; Stefanidis, D.; Tse, W. C.; Zhang, J. R. Preparation
of cyclopropacyclopentapyrazolylacetamide compounds useful for the
prophylactic or therapeutic treatment of HIV virus infection. WO
2018035359, 2018.
(18) Allan, K. M.; Batten, A. L.; Brizgys, G.; Dhar, S.; Doxsee, I. J.;
Goldberg, A.; Heumann, L. V.; Huang, Z.; Kadunce, N. T.; Kazerani,
S.; et al. Methods and intermediates for preparation of antiretroviral
pyridine derivative useful for treatment of HIV-1 infections. WO
2019161280, 2019.
.
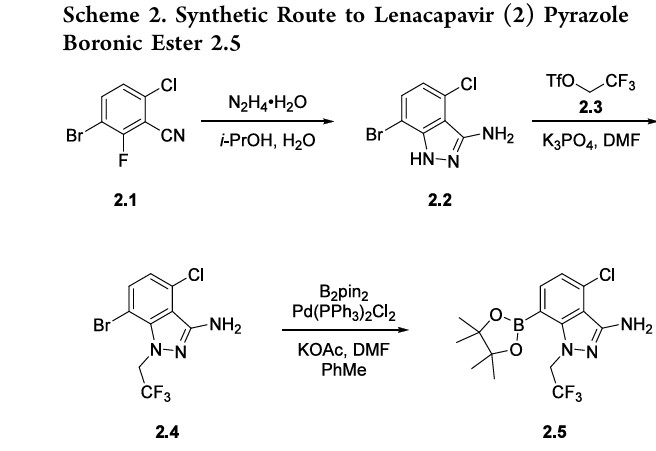



.
ICENTICAFTOR
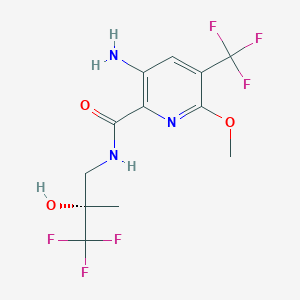
QBW 251, ICENTICAFTOR
- Molecular FormulaC12H13F6N3O3
- Average mass361.240 Da
Icenticaftor (development code QBW251) is a drug candidate for the treatment of chronic obstructive pulmonary disease (COPD)[1][2] and cystic fibrosis.[3][4] The drug is being developed by Novartis.[5]
Like ivacaftor (which is marketed as Kalydeco), icenticaftor functions by acting as a stimulator of the protein cystic fibrosis transmembrane conductance regulator (CFTR).[5]
Icenticaftor (QBW251) is an orally active CFTR channel potentiator, with EC50s of 79 nM and 497 nM for F508del and G551D CFTR, respectively. Icenticaftor can be used for chronic obstructive pulmonary disease (COPD) and cystic fibrosis research.
Cystic fibrosis (CF) is the most prevalent life-threatening Mendelian disorder in Caucasian populations. CF arises from mutations of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The CFTR ion channel orchestrates gating of chloride and bicarbonate ions across epithelial cell membranes in various tissues, including the lung, pancreas, intestine, reproductive tract, and sweat glands. While CF is a systemic disorder, the primary mortality derives from reduced CFTR activity in the airways. Subsequent acidification3 and dehydration leads to accumulation of a viscous mucus layer, occluding the airways and trapping bacteria, leading to infections, reduced lung function, and ultimately, respiratory failure. The most common CFTR mutation, F508del (Class II, found in 90% of CF patients), impairs folding of the CFTR protein (a Class II trafficking defect), resulting in a reduced amount of channel present at the plasma membrane. With the G551D mutation (class III), theamount of protein at the membrane is unaffected, but its open probability (Po) is reduced, also resulting in a reduced channel gating. Thus, to address the underlying causes of CF, two distinct CFTR modulators are required: correctors to increase CFTR levels at the plasma membrane and potentiators to enable effective opening of the channel
Chronic obstructive pulmonary disease (COPD) is anticipated to shortly become the third leading cause of death globally. COPD is characterized by persistent airflow obstruction with cigarette smoke exposure recognized as the primary risk factor. Airflow limitation is associated with all COPD patients; however, the disease is heterogeneous, with variable phenotypes ranging from chronic bronchitis (CB) to emphysema. Small airway disease exhibits increased numbers of goblet cells and mucus plugging with associated smooth muscle hyperplasia, airway fibrosis, and increased inflammation. Excess mucus secretion is believed to play an important role in COPD pathogenesis and is associated with progression of the disease.
Cystic fibrosis (CF) is a fatal genetic disease caused by mutations in the gene encoding the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), a protein kinase A activated epithelial anion channel involved in salt and fluid transport in multiple organs, including the lung. Most CF mutations either reduce the number of CFTR channels at the cell surface (e.g. synthesis or processing mutations) or impair channel function (e.g. gating or conductance mutations) or both.
PCT publication No. WO 2011/113894 describes compounds which restore or enhance the function of mutant and/or wild type CFTR for the treatment of cystic fibrosis, primary ciliary dyskinesia, chronic bronchitis, chronic obstructive pulmonary disease, asthma and other CFTR related diseases. The compounds described therein include (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (Example 5 of WO 2011/113894).
The synthesis described in WO 2011/113894 to make (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide is long, uses expensive starting materials and toxic reagents. Schemes 1 and 2 outline a synthesis from WO 2011/113894 used to make(S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
In Scheme 1, the intermediate ethyl 3-amino-5-(trifluoromethyl)picolinate (B4) is made via a Buchwald-Hartwig coupling reaction which requires the use of an expensive starting material (B1) and an expensive palladium catalyst which has to be controlled in the final product. Also, the conversion of B4 to B5 requires the use of NBS, a mutagenic reagent which has to be controlled in the API.
Moreover, the conversion of B5 to B8 is accomplished through the addition of 2,5-hexanedione, a well-known neurotoxin, as shown in Scheme 2. Transformation of the pyrrole in B8 to the amine B9 uses hydroxylamine which is a mutagenic and thermally unstable compound that is dangerous to use in large quantities. The overall process described in WO 2011/113894 requires many protecting group manipulations that lead to a low atom economy and afford a lot of waste. Thus there is a need for an improved synthetic process for making (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide.
PATENT
WO 2018116139,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2018116139&_cid=P21-L7D5PQ-39961-1
xample 1: 3-Bromo-6-methoxy-5-(trifluoromethyl)picolinic acid
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (1.4 kg, 5.47 mol), tetramethyl ethylene diamine (TMEDA) (1.75 kg, 15 mol) and tetrahydrofuran (THF) (10kg) were charged to a dry and inert reactor. At -25°C a solution of 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithiumchloride complex, 1 M in THF/toluene (TMPMgCl.LiCl)(14.5 kg, 15 mol) was slowly added. After stirring the reaction mixture for 30 min., CO2 gas was carefully bubbled into the reactor so that the temperature of the exothermic reaction did not exceed -20°C. The reaction mixture was then quenched onto a mixture of t-butyl methyl ether (TBME) and 5% aq. H2SO4 (50 kg). The biphasic mixture was separated and the organic phase was extracted with 2M NaOH solution. The aqueous phase was acidified to pH 1-2 with 5% aq. H2SO4 and extracted with TBME. After a distillative solvent change to cyclohexane the product was crystallized from cyclohexane to yield 1.1 kg 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (65% yield).
1H NMR (400 MHz, CDCl3): δ ppm 8.24 (d,J = 0.7Hz, 1 H), 4.12 (s, 3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 54.84, 106.37, 114 (m), 117.6/120.3/123.0/125.7 (m), 141.74, 152.43, 158.63, 165.63
HRMS: [M-H]- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9337
Example 2: Methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (5.0 g, 19.53 mmol) was added to a 100 ml reactor followed by toluene (20 ml) and dimethylcarbonate (17.59 g, 195.30 mmol). To the stirred solution at 20 °C was slowly added 2,2,6, 6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (27.34 ml, 27.34 mmol) within 45 minutes. A sample was taken and diluted in acetic acid for HPLC analysis in order to confirm full conversion of II to the methylester. Within the same vessel 5% aq. H2SO4 (36 ml) was slowly added to the reaction mixture until a pH below 2 was obtained (caution, exothermic). The biphasic mixture was separated and the lower aqueous phase back-extracted with toluene (10 ml).
In order to isolate the methylester the organic phases were combined and concentrated by rotary evaporation to yield a residue which was chromatographed on reverse-phase silica to yield the final product: methyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate as a yellow solid, 5.3 g, 86 % yield. The solid was optionally recrystallized from methanol and water to further increase purity.
1H NMR (400MHz, CDCl3): δ ppm 8.08 (br s, 1 H), 4.07 (s, 3H), 4.02 (s, 3H)
13C NMR (CDCl3): δ ppm 164.76, 159.22, 149.90, 141.49, 122.83, 120.12, 116.12, 108.05, 54.93, 53.09
HRMS: MH+ expected C9H8BrF3NO3, 313.9561 ; found C9H8BrF3NO3, 313.9634
HPLC Conditions:
HPLC: Column : Agilent Zorbax SB-C18 (150 mm x 3.0 mm, particle size 3.5 urn)
Eluent A : Water / TFA = 1000/1 (v/v)
Eluent B: Acetonitrile / TFA = 1000/1 (v/v)
Wavelength : 230 nm
Flow-rate : 0.8 ml/min
Gradient: eluent B: 45% to 90% over 9 mins
Retention time 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate: 5.80 min
Alternative synthesis for 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid:
Isolation of Example 1
In order to proceed to Example 1 without the isolation of VII, the work-up continues from the combined toluene phases post-H2SO4 quench as follows:
To the combined organic phases was slowly added 50% aq. sodium hydroxide (30 ml) until a pH of above 10 was obtained. The reaction mixture was heated to 35 °C and after 15 mins addition of water (30 ml) followed by 30 mins further stirring preceded sample-taking to ensure full hydrolysis of the methylester to Example 1 by HPLC. Water was added (130 ml), followed by TBME (60 ml) and the phases separated. To the aqueous phase was cautiously added concentrated H2SO4 (30 g) until a pH of below 2.5 was obtained (caution, exothermic and release of CO2 causes foaming). TBME (100 ml) was added and the phases separated. The organic phase contained the C2, and could be evaporated to dryness by rotary evaporation to confirm the yield, 5.4 g C2, 92 % yield.
1H NMR (400 MHz,CDCl3): δ ppm 8.24 (d,J=0.7Hz, 1 H), 4.12 (s, 3H)
13C NMR (101 MHz,DMSO-d6): δ ppm 54.84, 106.37, 114 (m), 117.6/120.3/123.0/125.7 (m), 141.74, 152.43, 158.63, 165.63
HRMS: M-H- expected C8H4BrF3NO3, 297.9405; found C8H4BrF3NO3, 297.9333
For HPLC method details see above. Retention time C2: 2.94 min
Alternative synthesis for ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate:
5-bromo-2-methoxy-3-(trifluoromethyl)pyridine (III) (0.5 g, 1.95 mmol) was added to a reactor followed by THF (2 ml) and the solution cooled to 0 °C. To the mixture was added 2,2,6,6-tetramethyl-piperidinylmagnesium chloride lithium chloride complex as a 1 M solution in THF/toluene (4.88 ml, 3.91 mmol), and the mixture was left to stir for 15 minutes at 0 °C. An aliquot of the solution (50 ul) was then added to a reactor containing diethylcarbonate (20 ul, 19.5 mmol). A second aliquot (50 ul) was taken of the metallated II and added to a reactor containing ethyl chloroformate (14 ul, 19.5 mmol). After 2 minutes both reactors were quenched with a 1 :1 mixture of acetonitrile/HCl (1 M). The reaction with diethylcarbonate gave 56 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate and the reaction with ethyl chloroformate gave 68 A% of ethyl 3-bromo-6-methoxy-5-(trifluoromethyl)picolinate product according to the HPLC method described above.
Example 3: Synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1: 3-bromo-6-methoxy-5-(trifluoromethyl)picolinic acid (1.3 kg, 4.33 mol) and
copper(II)sulfate pentahydrate (0.108 kg, 0.433 mol) were charged into an inert autoclave
followed by aqueous ammonia 25% (12 kg). The mixture was stirred and heated up to 100 °C, whereby a pressure of 7 bar resulted. The solution was stirred for 2 hr and then cooled down to
5 °C. Sulfuric acid (8 M) was dosed upon cooling, so that a temperature range of 5 °C to 30 °C was held until a pH of about 5 was reached. Isopropylacetate was added and the pH was
further adjusted to 1-2. The phases were separated and the organic phase was azeotropically dried by partial distillation. n-Heptane was added and the mixture stirred for 15 hr at 20 °C
during which the product crystallized out. After filtration and drying 3-amino-6-methoxy-5-(trifluoromethyl)picolinic acid was obtained as a yellow solid (0.92 kg, 90%).
1H NMR (400 MHz, DMSO-d6): δ ppm 7.70 (s, 1 H), 3.89 (s, 3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Step 2: 3-amino-6-methoxy-5-(trifluoromethyl) picolinic acid (20 g, 84.7 mmol) and HATU (38.6 g, 101.6 mmol) were charged to a reactor followed by a solution of (S)-3-amino-1 ,1 ,1-trifluoro-2- methylpropan-2-ol in isopropylacetate (7 %, 188 g, 93 mmol). The solution was stirred at room temperature, diisopropyl ethyl amine (21.9 g, 169 mmol) was added and stirring was continued for at least 16h at 25 °C. Water (250 ml) was then added dropwise within 15 min. keeping the temperature below 25 °C. The water phase was separated and the organic phase was extracted with 5% aqueous HCl , 5% potassium carbonate solution, and water. The organic layer was concentrated to about 60% solution. At 50 °C n-heptane (41 g) was added and the solution was cooled by a linear ramp to 5 °C while adding more n-heptane (131 g). The precipitate was filtered off and dried at 50 °C resulting in a yellow to beige product (S)-3-amino-6-methoxy-N- (3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (21.1 g, 69 % yield).
1H NMR (400 MHz, DMSO-d6): δ ppm 8.30 (m,1 H), 7.68 (s,1 H), 6.69 (s,2H), 6.29(s,1 H), 3.93(s,3H), 3.7-3.4(m,2H), 1.26(s,3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 18.92, 42.15, 53.52, 72.40, 115.5-116.5 m, 118-126 m, 122-130.7 m, 124.82, 128.3 m, 140.95, 148.49, 166.27
Example 4: Telescoped process for the synthesis of the HCl salt of 3-amino-6-methoxy- 5-(trifluoromethyl)picolinic acid (V)
1 Equivalent* of (III) and 6 equivalents of dimethyl carbonate (DMC) were dissolved in 3.5 parts** of toluene at room temperature. To this solution 1.5 equivalent of TMPMgCl.LiCl solution in THF was added at 15-25°C within ca. 1 h. Tert butyl methyl ether (MTBE, 5.9 parts) was added and the mixture was quenched in 7.3 parts of 10% sulfuric acid at 25-40°C. The water phase was discarded and to the organic phase 6.2 parts of 30% sodium hydroxide solution were added. The mixture was stirred well at 40°C for 1-2h. After the successful conversion of (VIII) to (IV), 2.5 parts of water were added to dissolve the partially precipitated sodium carbonate. The water phase was discarded and the organic phase was cooled to 20°C and extracted with 4.8 parts of 25% aqueous ammonia. The aqueous phase was transferred in an autoclave and 0.0979 parts (10mol%) of copper sulfate pentahydrate were added. The autoclave was well inertized by a pressure method and heated up to 100°C, while the pressure raises up to ca. 8 bar absolute pressure. After the successful conversion of (IV) to (V), the green solution was added to a mixture of 3.7 parts of MTBE and 6.8 parts of 50% sulfuric acid resulting in a biphasic solution of pH 1-2. The water phase was separated and the organic phase washed two times with 2.5 parts of water each. The organic phase was dried by distillation at JT 50°C/400mbar while 3.7 parts of MTBE were added/replaced. To the dried organic solution 0.41 parts of HCl gas was dosed at 0-5°C under or over solvent level. The suspension was stirred for ca.1 h, then filtered off and washed with 48 parts of TBME. The product was dried at 40°C/20 mbar for ca. 12h. (yield from (III): 72%, slightly beige solid).
*equivalents are based on the molar amount of the starting material (III) = 1 equivalent
**parts = weight/weight (III)
1H NMR (400 MHz, DMSO-d6): δ ppm 7.70 (s, 1 H), 3.89 (s, 3H)
13C NMR (101 MHz, DMSO-d6): δ ppm 53.59, 116.76 m, 123.27, 126.36-117.40 m, 128.04, 142.56, 148.65, 167.62
Example 5: Alternative synthesis of (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide
Step 1 : (VIII) (1.0 g), (S)-3-amino-1 ,1 ,1-trifluoro-2-methylpropan-2-ol as mandellic acid salt (1.128 g, 1.2 eq.) and 2,3,4,6, 7, 8-hexahydro-1H-pyrimido[1,2-a]pyrimidine (TBD, 0.588 g, 1.3 eq.) were added to a pre-dried flask as solids. To this was added the anhydrous THF (10 ml) and the cloudy solution heated to 55 °C. Sampling and analytical determination of purity at 2.5 hrs confirmed 88 A% product upon which water (10 ml) was added and the phases separated. The organic phase was distilled to a concentrated mixture upon which toluene (20 ml) was added. The organic layer was extracted with 10% aq. citric acid (10 ml) followed by three consecutive extractions with 1 M aq. NaOH. The organic phase was then dried with magnesium sulfate and evaporated to dryness to give 1.196 g of (S)-3-bromo-6-methoxy-N-(3,3,3-trifluoro- 2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (IX) as a white solid (95 A%, 88% yield).
1H NMR, CDCl3: δ ppm 8.08 (s, 1 H), 7.83 (br s, 1 H), 3.99 (s, 3H), 3.78-3.60 (m, 2H), 3.51 (br s, 1 H), 1.36 (s, 3H)
19F NMR, CDCl3: δ ppm -64.28, -81.44
13C DEPT135, CDCl3: δ ppm 144.20 (CH), 54.70 (CH3), 44.26 (CH2), 19.71 (CH3)
HRMS: MH+ expected C12H12BrF6N2O3, 424.9857; found C12H12BrF6N2O3, 424.9931
HPLC (method described above): retention time = 4.94 min
Step 2: IX (79 mg, 0.186 mmol) was combined with copper(II)sulfate pentahydrate (4.6 mg, 0.019 mmol), methanol (0.6 ml) and 23% aqueous ammonium hydroxide solution (559 ul) within a glass microwave vial. The headspace was inertized with nitrogen, then the vial sealed and placed in the microwave unit for heating to 105 °C for 7.5 hrs. Isopropylacetate (5 ml) was added to the deep green reaction mixture and a solvent-switch brought about by rotary evaporation. To the mixture now in water and isopropyl acetate was added 8M H2SO4 (5 ml), the phases mixed and then left to separate. The aqueous phase was further extracted with isopropylacetate and the combined organic phases washed with aq. NaCl (5 ml). The organic phase was dried over MgSO4 and evaporated to yield of a yellow residue, 66 mg.
A portion of the residue (16 mg) was re-dissolved in heptane / ethyl acetate and submitted for combiflash purification (n-heptane / ethyl acetate gradient, elution at 20% ethyl acetate) providing (S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2-methylpropyl)-5-(trifluoromethyl)picolinamide (VII) as a residue on evaporation in 91 A% purity containing trace residual solvents (17 mg, corrected to 13 mg by 1H NMR, 80 % yield back-calculated).
1H NMR, CDCl3: δ ppm 8.11 (br s, 1 H), 7.37 (s, 1 H), 3.97 (s, 3H), 3.76-3.72 (d, 2H, J=6.3Hz), 1.42 (s, 3H)
13C NMR, CDCl3: δ ppm 168.86, 150.55, 140.21 , 128.63, 127.26, 125.35, 124.42, 123.39, 120.68, 118.60, 74.16, 53.73, 44.39, 19.55
ESI-MS: expected mass 361.2. ELS detector, 100 A%, MH+ 362.1 , M- 360.1
HPLC (method described above): retention time = 4.39 min
PATENT
US20200383960
https://patentscope.wipo.int/search/en/detail.jsf?docId=US312969607&_cid=P21-L7D5H8-38258-1
Examples 4, 5 and 6: 3-Amino-6-methoxy-5-trifluoromethyl-pyridine-2-carboxylic acid (3,3,3-trifluoro-2-hydroxy-2-methyl-propyl)-amide and its enantiomers
was prepared according to the following procedure:
Examples 5 and 6 are Entantiomers
SYN
J. Med. Chem. 2021, 64, 11, 7241–7260
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) ion channel are established as the primary causative factor in the devastating lung disease cystic fibrosis (CF). More recently, cigarette smoke exposure has been shown to be associated with dysfunctional airway epithelial ion transport, suggesting a role for CFTR in the pathogenesis of chronic obstructive pulmonary disease (COPD). Here, the identification and characterization of a high throughput screening hit 6 as a potentiator of mutant human F508del and wild-type CFTR channels is reported. The design, synthesis, and biological evaluation of compounds 7–33 to establish structure–activity relationships of the scaffold are described, leading to the identification of clinical development compound icenticaftor (QBW251) 33, which has subsequently progressed to deliver two positive clinical proofs of concept in patients with CF and COPD and is now being further developed as a novel therapeutic approach for COPD patients.


a Reagents and conditions: (i) aq NaOH, THF, RT, 97%; (ii) aq Me2NH or MeNH2, THF, RT, 56−92%; (iii) 41, HATU, Et3N, NMP, RT, 52− 78%; (iv) NH2OH·HCl, Et3N, EtOH−water, reflux, then chiral HPLC, 34−36%; (v) aq NaOH, MeOH, 60°C, 97%; (vi) cat H2SO4, MeOH, reflux, 75%; (vii) TMSCl, KI, MeCN, reflux, 54%; (viii) EtOH, DEAD, Ph3P, dioxane, RT, 61%; (ix) aq NaOH, THF, reflux, 26%; (x) (S)-41, HATU, DIPEA, DMF, RT, 89%; (xi) NH2OH·HCl, Et3N, EtOH−water, reflux, 37−53%; (xii) (S)-41, HATU, DIPEA, NMP, RT, 59%.
(S)-3-Amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2- methylpropyl)-5-(trifluoromethyl)picolinamide
(S)-3-amino-6-methoxy-N-(3,3,3-trifluoro-2-hydroxy-2- methylpropyl)-5-(trifluoromethyl)picolinamide 33 as a white solid (33.6 g, 59%). LRMS C12H13F6N3O3 requires M+ 361.08, found [MH]+ 362.2. Elemental analysis requires C, 39.90%; H, 3.63%; N, 11.63% found C, 40.22 ± 0.06%; H, 3.68 ± 0.11%; N, 11.76 ± 0.04%. 1 H NMR (DMSO-d6) 1.26 (3H s), 3.46 (1H dd J = 13.3, 5.6), 3.66 (1H dd J = 13.7, 7.3), 3.92 (3H s), 6.29 (1H s), 6.69 (2H br s), 7.68 (1H s), 8.30 (1H t J = 6.4). 13C NMR (DMSO-d6) 18.95 (q), 42.19 (t), 53.56 (q), 72.27 (s JF = 26.8), 116.07 (s JF = 32.3), 122.40 (s JF = 272.1), 124.85 (s), 126.43 (s JF = 287.1), 128.29 (d JF = 5.2), 141.0 (s), 148.51 (s), 166.3 (s). 19F NMR (DMSO-d6) −62.71 (s), −80.46 (s).
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Clinical data | |
|---|---|
| Other names | QBW251 |
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Chemical and physical data | |
| Formula | C12H13F6N3O3 |
| Molar mass | 361.244 g·mol−1 |
| 3D model (JSmol) | |
References
- ^ Rowe SM, Jones I, Dransfield MT, Haque N, Gleason S, Hayes KA, et al. (2020). “Efficacy and Safety of the CFTR Potentiator Icenticaftor (QBW251) in COPD: Results from a Phase 2 Randomized Trial”. International Journal of Chronic Obstructive Pulmonary Disease. 15: 2399–2409. doi:10.2147/COPD.S257474. PMC 7547289. PMID 33116455.
- ^ Grand DL, Gosling M, Baettig U, Bahra P, Bala K, Brocklehurst C, et al. (June 2021). “Discovery of Icenticaftor (QBW251), a Cystic Fibrosis Transmembrane Conductance Regulator Potentiator with Clinical Efficacy in Cystic Fibrosis and Chronic Obstructive Pulmonary Disease”. Journal of Medicinal Chemistry. 64 (11): 7241–7260. doi:10.1021/acs.jmedchem.1c00343. ISSN 0022-2623. PMID 34028270.
- ^ Kazani S, Rowlands DJ, Bottoli I, Milojevic J, Alcantara J, Jones I, et al. (March 2021). “Safety and efficacy of the cystic fibrosis transmembrane conductance regulator potentiator icenticaftor (QBW251)”. Journal of Cystic Fibrosis. 20 (2): 250–256. doi:10.1016/j.jcf.2020.11.002. PMID 33293212.
- ^ Ray F (December 9, 2020). “Icenticaftor Effective in CF Patients With Certain Mutations, Phase 1/2 Trial Shows”. cysticfibrosisnewstoday.com. BioNews Services.
- ^ Jump up to:a b “Icenticaftor – Novartis”. Adis Insight. Springer Nature Switzerland AG.
////////////QBW 251, ICENTICAFTOR, NOVARTIS, chronic obstructive pulmonary disease, COPD, cystic fibrosis,
C[C@](CNC(=O)C1=C(C=C(C(=N1)OC)C(F)(F)F)N)(C(F)(F)F)O
Chief Advisor Industry Advisory Board Amity University Noida, India. 26th Aug 2022

DR ANTHONY MELVIN CRASTO is Chief Advisor Industry Advisory Board Amity Univ. Noida, INDIA
Journey begins with online meet. 26th aug 2022
Service to education is service to humanity
Brochure for First Industry Advisory Board meeting at Amity Institute of Pharmacy is attached. Date 26th Aug 2022
it as hybrid-Online/offline.


Thnx
Dr Sandeep Arora
#amity
@amity #education #pharmacy
Amity University
Amity University, Greater Noida Campus


Camizestrant, AZD 9833


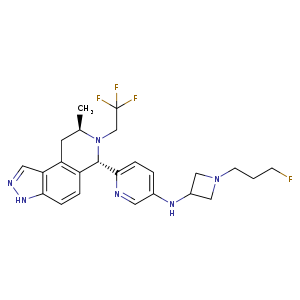
Camizestrant, AZD 9833
AZ 14066724
PHASE 2
CAS: 2222844-89-3
Chemical Formula: C24H28F4N6
Exact Mass: 476.2312
Molecular Weight: 476.5236
Elemental Analysis: C, 60.49; H, 5.92; F, 15.95; N, 17.64
N-(1-(3-fluoropropyl)azetidin-3-yl)-6-((6S,8R)-8-methyl-7-(2,2,2-trifluoroethyl)-6,7,8,9-tetrahydro-3H-pyrazolo[4,3-f]isoquinolin-6-yl)pyridin-3-amine
- AZ14066724
- AZD-9833
- AZD9833
- Camizestrant
- UNII-JUP57A8EPZ
- WHO 11592
- OriginatorAstraZeneca
- ClassAmines; Antineoplastics; Azetidines; Fluorinated hydrocarbons; Isoquinolines; Pyrazolones; Pyridines; Small molecules
- Mechanism of ActionSelective estrogen receptor degraders
- Phase IIIBreast cancer
- 13 Jun 2022AstraZeneca initiates a phase I drug-drug interaction trial of AZD 9833 Healthy postmenopausal female volunteers, in USA (NCT05438303)
- 10 Jun 2022AstraZeneca and Quotient Sciences complete the phase I QSC205863 trial in Breast cancer (In volunteers) in United Kingdom (PO, Liquid) (NCT05364255)
- 03 Jun 2022Safety, efficacy and pharmacokinetics data from the phase I SERENA 1 trial for Breast cancer presented at the 58th Annual Meeting of the American Society of Clinical Oncology (ASCO-2022)
- Mechanism:selective estrogen receptor degrader
- Area under investigation:estrogen receptor +ve breast cancer
- Date commenced phase:Q1 2019
- Estimated Filing Acceptance:
- CountryDateUS: EU: Japan: China:
AZD9833 is an orally available selective estrogen receptor degrader (SERD), with potential antineoplastic activity. Upon administration, SERD AZD9833 binds to the estrogen receptor (ER) and induces a conformational change that results in the degradation of the receptor. This prevents ER-mediated signaling and inhibits the growth and survival of ER-expressing cancer cells
Camizestrant is an orally available selective estrogen receptor degrader (SERD), with potential antineoplastic activity. Upon administration, camizestrant binds to the estrogen receptor (ER) and induces a conformational change that results in the degradation of the receptor. This prevents ER-mediated signaling and inhibits the growth and survival of ER-expressing cancer cells
SYN
https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-0040-1719368
Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist J. Med. Chem. 2020, 63, 14530–14559, DOI: 10.1021/acs.jmedchem.0c01163.

SYN
doi: 10.1021/acs.jmedchem.0c01163.

aReagents and Conditions: (a) n-BuLi, THF, −78 oC to 0 oC, 1 h, then 4 N HCl/dioxane, RT, 1 h, 60%; (b) alkyl triflate, DIPEA, 1,4-dioxane, 90 oC, 63-74% or isobutyrylaldehyde, Na(OAc)3BH, THF, 0 oC, 56%; (c) benzophenone imine, Pd2dba3, Rac-BINAP, NaOtBu, toluene, 90 oC, then 1 N aq. HCl, 71-85%; (d) nBuLi, THF, −78 oC to 0 oC, 1 h, then 4 N HCl/dioxane, RT, 4 h; e) NH2OH, NH2OH.HCl, EtOH, reflux. 84% over 2 steps; (f) alkyl triflate, DIPEA, 1,4-dioxane, 90 oC, 44-100% or 1-fluorocyclopropane-1- carboxylic acid, HATU, Et3N, DMF, RT, 61%, then BH3.THF, THF, 65 oC, 82%.

[α]26 D -147 (c 2.3, MeOH); 1H NMR (500 MHz, DMSO-d6, 27 °C) 1.08 (d, J = 6.6 Hz, 3H), 1.64 (dp, J = 25.0, 6.3 Hz, 2H), 2.45 (t, J = 6.9 Hz, 2H), 2.73(t, J = 6.8 Hz, 2H), 2.84 (dd, J = 17.1, 8.2 Hz, 1H), 2.96 (dt, J = 19.6, 9.8 Hz, 1H), 3.07 (dd, J = 17.2, 4.6 Hz, 1H), 3.49 (m, 1H), 3.50 – 3.58 (m, 1H), 3.58 – 3.66 (m, 2H), 3.92 (h, J = 6.5 Hz, 1H), 4.44 (dtd, J = 47.4, 6.1, 1.3 Hz, 2H), 4.93 (s, 1H), 6.23 (d, J = 6.9 Hz, 1H), 6.80 (d, J = 8.6 Hz, 1H), 6.83 (dt, J = 8.8, 2.0 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 7.22 (d, J = 8.6 Hz, 1H), 7.73 (d, J = 2.8 Hz, 1H), 8.05 (d, J = 1.3 Hz, 1H), 12.97 (s, 1H); 13C NMR (125 MHz, DMSO-d6, 27 °C) 16.2, 28.2 (d, J = 19.4 Hz), 30.1, 43.0, 47.3, 48.7 (q, J = 30.1 Hz), 54.8 (d, J = 5.6 Hz), 61.3 (2C), 67.1, 82.0 (d, J = 161.3 Hz), 107.5, 119.0, 122.4, 123.7, 126.1, 126.2 (q, J = 278.5 Hz), 126.4, 127.5, 131.7, 132.9, 138.5, 142.3, 150.0; 19F NMR (376 MHz, DMSO-d6, 27 °C) -218.1 (1F), -69.7 (3F); m/z (ES+), [M+H]+ = 477, HRMS (ESI) (MH+ ); calcd, 477.2408; found, 477.2390




/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
AZD9833 is selective oestrogen receptor degrader (SERD). It works by breaking down the site where oestrogen attaches to the cancer cell. This can help stop or slow the growth of hormone receptor breast cancer. Researchers think that AZD9833 with palbociclib might work better than anastrozole and palbociclib.
AZD9833 + palbociclib
The patients will receive AZD9833 (75 mg, PO, once daily) + palbociclib (PO, once daily, 125 mg for 21 consecutive days followed by 7 days off treatment) + anastrozole placebo (1 mg, PO, once daily)
SERENA-1: Study of AZD9833 Alone or in Combination in Women With Advanced Breast Cancer. (clinicaltrials.gov)…..https://veri.larvol.com/news/azd9833/drug
P1, N=305, Recruiting, AstraZeneca | Trial primary completion date: Dec 2022 –> Oct 2023
2 months ago
Trial primary completion date
|
HER-2 (Human epidermal growth factor receptor 2) • ER (Estrogen receptor) • PGR (Progesterone receptor)
|
HER-2 negative
Ibrance (palbociclib) • everolimus • Verzenio (abemaciclib) • capivasertib (AZD5363) • camizestrant (AZD9833)
| Description | Camizestrant (AZD-9833) is a potent and orally active estrogen receptor (ER) antagonist. Camizestrant is used for the study of ER+ HER2-advanced breast cancer[1]. |
|---|---|
| IC50 & Target | IC50: estrogen receptor (ER)[1] |
| In Vitro | Camizestrant is extracted from patent US20180111931A1, example 17[1].MCE has not independently confirmed the accuracy of these methods. They are for reference only. |
| In Vivo | Camizestrant (oral administration; 0.2-50 mg/kg; 20 days) exhibits anti-tumour efficacy as a dose-dependent manner in human parental MCF7 mice xenograft[1]. Camizestrant (oral administration; 0.8-40 mg/kg; 30 days) decreases tumor growth as a dose-dependent manner. It gives almost complete tumour growth inhibition at the doses >10 mg/kg in mice[1]. MCE has not independently confirmed the accuracy of these methods. They are for reference only.Animal Model:Human ESR1 mutant breast cancer patient derived xenograft with CTC174 cells in female NSG mice[1]Dosage:0.8 mg/kg, 3 mg/kg, 10 mg/kg, 20 mg/kg, 40 mg/kgAdministration:Oral administration; 30 days; once dailyResult:Inhibited tumor growth in a dose-dependent manner. |
| Clinical Trial | NCT NumberSponsorConditionStart DatePhaseNCT04711252AstraZenecaER-Positive HER2-Negative Breast CancerJanuary 28, 2021Phase 3NCT04964934AstraZenecaER-Positive HER2-Negative Breast CancerJune 30, 2021Phase 3NCT04214288AstraZenecaAdvanced ER-Positive HER2-Negative Breast CancerApril 22, 2020Phase 2NCT04588298AstraZenecaHER2-negative Breast CancerNovember 2, 2020Phase 2NCT04541433AstraZenecaER&addition; HER2- Advanced Breast CancerSeptember 29, 2020Phase 1NCT03616587AstraZenecaER&addition; HER2- Advanced Breast CancerOctober 11, 2018Phase 1NCT04546347AstraZeneca|Quotient SciencesHealthy VolunteersSeptember 17, 2020Phase 1NCT04818632AstraZenecaER&addition;, HER2-, Metastatic Breast CancerOctober 11, 2021Phase 1 |
////////////Camizestrant, AZD 9833, AZ 14066724, UNII-JUP57A8EPZ, WHO 11592, PHASE 2, ASTRA ZENECA, CANCER
C[C@@H]1CC2=C3C(NN=C3)=CC=C2[C@@H](C4=NC=C(NC5CN(CCCF)C5)C=C4)N1CC(F)(F)F

NEW DRUG APPROVALS
ONE TIME
$10.00
GEMCITABINE

GEMCITABINE
95058-81-4
WeightAverage: 263.1981
Monoisotopic: 263.071762265
Chemical FormulaC9H11F2N3O4
4-amino-1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2-dihydropyrimidin-2-one
Product Ingredients
| INGREDIENT | UNII | CAS | INCHI KEY |
|---|---|---|---|
| Gemcitabine hydrochloride | U347PV74IL | 122111-03-9 | OKKDEIYWILRZIA-OSZBKLCCSA-N |
- LY-188011
- LY188011
Gemcitabine
CAS Registry Number: 95058-81-4
CAS Name: 2¢-Deoxy-2¢,2¢-difluorocytidine
Additional Names: 1-(2-oxo-4-amino-1,2-dihydropyrimidin-1-yl)-2-deoxy-2,2-difluororibose; dFdC; dFdCyd
Manufacturers’ Codes: LY-188011
Trademarks: Gemzar (Lilly)
Molecular Formula: C9H11F2N3O4
Molecular Weight: 263.20
Percent Composition: C 41.07%, H 4.21%, F 14.44%, N 15.97%, O 24.32%
Literature References: Prepn: L. W. Hertel, GB2136425; idem,US4808614 (1984, 1989 both to Lilly); L. W. Hertel et al.,J. Org. Chem.53, 2406 (1988); T. S. Chou et al.,Synthesis1992, 565. Antitumor activity: L. W. Hertel et al.,Cancer Res.50, 4417 (1990). Mode of action study: V. W. T. Ruiz et al.,Biochem. Pharmacol.46, 762 (1993). Clinical pharmacokinetics and toxicity: J. L. Abbruzzese et al.,J. Clin. Oncol.9, 491 (1991). Review of clinical studies: B. Lund et al.,Cancer Treat. Rev.19, 45-55 (1993).
Properties: Crystals from water, pH 8.5. [a]365 +425.36°; [a]D +71.51° (c = 0.96 in methanol). uv max (ethanol): 234, 268 (e 7810, 8560). LD10 i.v. in rats: 200 mg/m2 (Abbruzzese).
Optical Rotation: [a]365 +425.36°; [a]D +71.51°
Absorption maximum: uv max (ethanol): 234, 268 (e 7810, 8560)
Toxicity data: LD10 i.v. in rats: 200 mg/m2 (Abbruzzese)
Derivative Type: Hydrochloride
CAS Registry Number: 122111-03-9
Molecular Formula: C9H11F2N3O4.HCl
Molecular Weight: 299.66
Percent Composition: C 36.07%, H 4.04%, F 12.68%, N 14.02%, O 21.36%, Cl 11.83%
Properties: Crystals from water-acetone, mp 287-292° (dec). [a]D +48°; [a]365 +257.9° (c = 1.0 in deuterated water). uv max (water): 232, 268 nm (e 7960, 9360).
Melting point: mp 287-292° (dec)
Optical Rotation: [a]D +48°; [a]365 +257.9° (c = 1.0 in deuterated water)
Absorption maximum: uv max (water): 232, 268 nm (e 7960, 9360)
Therap-Cat: Antineoplastic.
Keywords: Antineoplastic; Antimetabolites; Pyrimidine Analogs.
Gemcitabine is a nucleoside metabolic inhibitor used as adjunct therapy in the treatment of certain types of ovarian cancer, non-small cell lung carcinoma, metastatic breast cancer, and as a single agent for pancreatic cancer.
Gemcitabine hydrochloride was first approved in ZA on Jan 10, 1995, then approved by the U.S. Food and Drug Administration (FDA) on May 15, 1996, and approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on Aug 31, 2001. It was developed and marketed as Gemzar® by Eli Lilly.
Gemcitabine hydrochloride is a nucleoside metabolic inhibitor. It kills cells undergoing DNA synthesis and blocks the progression of cells through the G1/S-phase boundary. It is indicated for the treatment of advanced ovarian cancer that has relapsed at least 6 months after completion of platinum-based therapy, in combination with paclitaxel, for first-line treatment of metastatic breast cancer after failure of prior anthracycline-containing adjuvant chemotherapy, unless anthracyclines were clinically contraindicated, and it is also indicated in combination with cisplatin for the treatment of non-small cell lung cancer, and treated as a single agent for the treatment of pancreatic cancer.
Gemzar® is available as injection of lyophilized powder for intravenous use, containing 200 mg or 1000 mg of free Gemcitabine per vial. The recommended initial dosage is 1000 mg/m2 over 30 minutes on days 1 and 8 of each 21 day cycle for ovarian cancer, 1250 mg/m2 over 30 minutes on days 1 and 8 of each 21 day cycle for breast cancer, 1000 mg/m2 over 30 minutes on days 1, 8, and 15 of each 28 day cycle or 1250 mg/m2 over 30 minutes on days 1 and 8 of each 21 day cycle for non-small cell lung cancer, and 1000 mg/m2 over 30 minutes once weekly for the first 7 weeks, then one week rest, then once weekly for 3 weeks of each 28 day cycle for pancreatic cancer.
Approved Countries or AreaUpdate US, JP, CN, ZA
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1996-05-15 | First approval | Gemzar | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection, Lyophilized powder, For solution | Eq. 200 mg/1000 mg Gemcitabine/vial | Lilly | Priority |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2013-02-01 | New indication | Gemzar | Relapsed or refractory malignant lymphoma | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2011-02-23 | New indication | Gemzar | Advanced ovarian cancer | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2010-02-05 | New indication | Gemzar | Advanced breast cancer | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2008-11-25 | New indication | Gemzar | Urothelial cancer | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2006-06-15 | New indication | Gemzar | Biliary cancer | Injection, Lyophilized powder, For solution | 200 mg; 1 g | Lilly | |
| 2001-08-31 | First approval | Gemzar | Pancreatic cancer,Non small cell lung cancer (NSCLC) | Injection, Lyophilized powder, For suspension | 200 mg; 1 g | Lilly |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 2014-04-15 | Marketing approval | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | Eq. 1000 mg Gemcitabine per vial | 湖北一半天制药 | ||
| 2014-04-15 | Marketing approval | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | Eq. 200 mg Gemcitabine per vial | 湖北一半天制药 | 6类 | |
| 2014-04-08 | Marketing approval | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | Eq.1000 mg Gemcitabine per vial | 南京正大天晴制药 | 6类 | |
| 2011-12-02 | Marketing approval | 健择/Gemzar | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | Eq. 200 mg/1000 mg Gemcitabine per vial | Lilly | |
| 2010-08-31 | Marketing approval | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection | 1000 mg/200 mg | 北京协和药厂 | 6类 |
| Approval Date | Approval Type | Trade Name | Indication | Dosage Form | Strength | Company | Review Classification |
|---|---|---|---|---|---|---|---|
| 1995-01-10 | First approval | Gemzar | Ovarian cancer,Breast cancer,Non small cell lung cancer (NSCLC),Pancreatic cancer | Injection, Lyophilized powder, For solution | Eq. 200 mg/1000 mg Gemcitabine per vial | Lilly |
Gemcitabine, with brand names including Gemzar,[1] is a chemotherapy medication.[2] It treats cancers including testicular cancer,[3]breast cancer, ovarian cancer, non-small cell lung cancer, pancreatic cancer, and bladder cancer.[2][4] It is administered by intravenous infusion.[2] It acts against neoplastic growth, and it inhibits the replication of Orthohepevirus A, the causative agent of Hepatitis E, through upregulation of interferon signaling.[5]
Common side effects include bone marrow suppression, liver and kidney problems, nausea, fever, rash, shortness of breath, mouth sores, diarrhea, neuropathy, and hair loss.[2] Use during pregnancy will likely result in fetal harm.[2] Gemcitabine is in the nucleoside analog family of medication.[2] It works by blocking the creation of new DNA, which results in cell death.[2]
Gemcitabine was patented in 1983 and was approved for medical use in 1995.[6] Generic versions were introduced in Europe in 2009 and in the US in 2010.[7][8] It is on the WHO Model List of Essential Medicines.[9]
Medical uses
Gemcitabine treats various carcinomas. It is used as a first-line treatment alone for pancreatic cancer, and in combination with cisplatin for advanced or metastatic bladder cancer and advanced or metastatic non-small cell lung cancer. It is used as a second-line treatment in combination with carboplatin for ovarian cancer and in combination with paclitaxel for breast cancer that is metastatic or cannot be surgically removed.[10][11][12]
It is commonly used off-label to treat cholangiocarcinoma[13] and other biliary tract cancers.[14]
It is given by intravenous infusion at a chemotherapy clinic.[2]
Contraindications and interactions
Taking gemcitabine can also affect fertility in men and women, sex life, and menstruation. Women taking gemcitabine should not become pregnant, and pregnant and breastfeeding women should not take it.[15]
As of 2014, drug interactions had not been studied.[11][10]
SYN
. Hertel, L. W.; Kroin, J. S.; Misner, J. W.; Tustin, J. M. J. Org. Chem. 1988, 53, 2406– 2409.

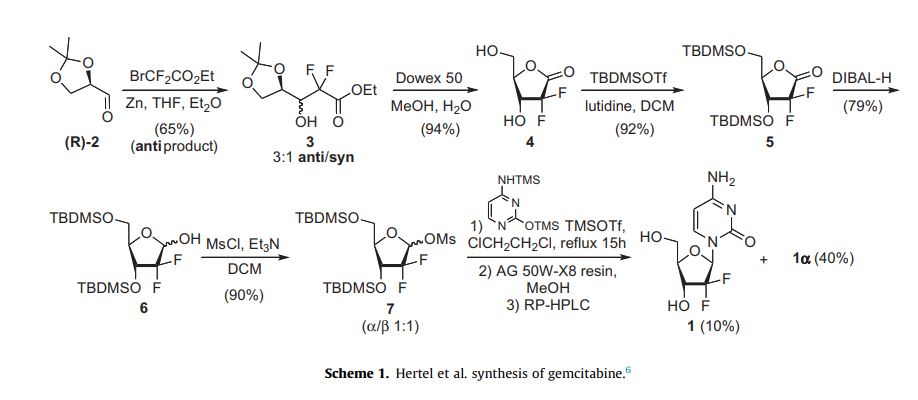
NEXT
a) Noe, C. R.; Jasic, M.; Kollmann, H.; Saadat, K. WO009147, 2007.; b) Noe, C. R.; Jasic, M.; Kollmann, H.; Saadat, K. US0249119, 2008. Note: no stereochemistry was indica


NExT
15. Hanzawa, Y.; Inazawa, K.; Kon, A.; Aoki, H.; Kobayashi, Y. Tetrahedron Lett. 1987, 28, 659–662. 16. Wirth, D. D. EP0727432, 1996


Synthesis Reference
John A. Weigel, “Process for making gemcitabine hydrochloride.” U.S. Patent US6001994, issued May, 1995.US6001994Route 1
Reference:1. J. Org. Chem. 1988, 53, 2406-2409.
2. US4808614A.Route 2
Reference:1. CN102417533A.Route 3
Reference:1. Nucleosides, Nucleotides and Nucleic Acids 2010, 29, 113-122.Route 4
Reference:1. CN102617677A.Route 5
Reference:1. CN103012527A.
SYN
U.S. Patent No. 4,808,614 (the ‘614 patent) describes a process for synthetically producing gemcitabine, which process is generally illustrated in Scheme Scheme 1

5

SYN
U.S. Patent No. 4,965,374 (the ‘374 patent) describes a process for producing gemcitabine from an intermediate 3,5-dibenzoyl ribo protected lactone of the formula:

11 where the desired erythro isomer can be isolated in a crystalline form from a mixture of erythro and threo isomers. The process described in the ‘374 patent is generally outlined in Scheme 2.
Scheme 2

mixture of α and β anomers
SYN
U.S. Patent No. 5,521,294 (the ‘294 patent) describes l-alkylsulfonyl-2,2- difluoro-3 -carbamoyl ribose intermediates and intermediate nucleosides derived therefrom. The compounds are reportedly useful in the preparation of 2′-deoxy-2′,2’- difluoro-β-cytidine and other β-anomer nucleosides. The ‘294 patent teaches, inter alia, that the 3-hydroxy carbamoyl group on the difluororibose intermediate may enhance formation of the desired β-anomer nucleoside derivative. The ‘294 patent describes converting the lactone 4 to the dibenzoyl mesylate 13, followed by deprotection at the 3 position to obtain the 5-monobenzoyl mesylate intermediate 15, which is reacted with various isocyanates to obtain the compounds of formula 16. The next steps involve coupling and deprotection using methods similar to those described in previous patents. The process and the intermediates 15 and 16 are illustrated by scheme 3 below: Scheme 3

13 15
PhCOCK
PhNCO/TEA -o. -~- j*«0Ms
PhNHCOO -r F
16
1 coupling 2 deprotection


16 gemcitabine
CLIP
https://www.sciencedirect.com/science/article/abs/pii/S0008621514000500
PATENT
https://patents.google.com/patent/WO2008129530A1/en
Scheme 4

e3
13A deprotection isomer separation

deprotection


EXAMPLE 1
[0045] This example demonstrates the preparation of 2-deoxy-2,2-difluoro-D- ribofuranose-3,5-dicinnamate-l-p-toluenesulfonate.
[0046] Crude 2-deoxy-2,2-difluoro-D-riboufuranose-3,5-dicinnamate (2.5g, 6 mmol) was dissolved in dichloromethane (20 ml) in a round flask, and diethylamine (0.7g, 9.6 mmol) was added followed by p-toluenesulfonyl chloride (1.32 g, 6.92 mmol), which was added drop wise while cooling to 0-50C. The mixture was stirred for 1 hour, and washed with IN HCl (15 ml), concentrated solution OfNaHCO3 (15 ml), and dried over MgSO4. The solvent was distilled off under reduced pressure to obtain crude 2-deoxy-2,2-difluoro-D-ribofuranose-3,5-dicinnamate-l-p- toluenesulfonate as light oil. Yield: 3.22 g, (5.6 mmol), 93%.
EXAMPLE 2
[0047] This example demonstrates the preparation of 3′,5′-dicinnamoyl-2′-deoxy- 2′,2′-difluorocytidine.
[0048] Dry 1 ,2-dichloroethane (800 ml) was added to N,O-bis(trimethylsilyl)- cytosine (136 g, 487 mmol) under nitrogen blanket to produce a clear solution, followed by adding trimethylsilyl triflate (Me3SiOTf), (100 ml, 122.8 g, 520 mmol) and stirred for 30 minutes. A solution of 2-deoxy-2,2-difluoro-D-ribofuranose-3,5- dicinnamate-1-p-toluenesulfonate (128 g, 224 mmol) in 1 ,2-dichloroethane (400 ml) was added drop wise, and the mixture was refluxed overnight. After cooling, the solvent was distilled off to obtain crude 3,5-dicinnamoyl-N4-trimethylsilyl-2′-deoxy- 2′,2′-difluorocytidine as a light yellow solid. The residue was dissolved in ethyl acetate (1600 ml) and washed 3 times with water (3X400 ml). The ethyl acetate phase was mixed with concentrated solution OfNaHCO3 (800 ml) for about 5 minutes, and then the mixture was set aside for about 20 minutes without stirring. The thus formed solid, which was precipitated in the inter-phase of the two layers, was filtered off and washed with 60 ml of ethyl acetate. The solid was dried under reduced pressure to obtain 116.7 g (223 mmol, 99.5%) of the crude 3′,5′-dicinnamoyl- 2′-deoxy-2′,2′- difluorocytidine containing 73.3 % of the β-anomer and 11.8 % of the α-anomer.
EXAMPLE 3
[0049] This example demonstrates the preparation of 3′,5′-dicinnamoyl-2′-deoxy- 2′,2′-difluorocytidine.
[0050] Dry 1,2-dichloroethane (1.5 L) was added to bis(trimethylsilyl)cytosine (417 g, 1.49 mol) under nitrogen blanket to produce a clear solution followed by adding trimethylsilyl triflate (Me3SiOTf), (300 ml, 368.4 g, 1.56 mol) and stirred for 30 minutes. A solution of 2-deoxy-2,2-difluoro-D-ribofuranose-3,5-dicinnamate-l-p- toluenesulfonate (384 g, 673 mmol) in 1,2-dichloroethane (1.2 L) was added drop wise, and the mixture was refluxed overnight. After cooling, the solvent was distilled off to obtain crude 3,5-dicinnamoyl-N4-trimethylsilyl-2l-deoxy-2′,2′-difluorocytidine as a light yellow solid. The residue was dissolved in ethyl acetate (2.4 L) and washed 3 times with water (3X1.2 L). The ethyl acetate phase was mixed with concentrated solution OfNaHCO3 (1.34 L) for about 20 minutes. The thus formed solid, which was precipitated in the inter-phase of the two layers, was filtered off and washed with 180 ml of ethyl acetate. The solid was dried under reduced pressure to obtain 346.5 g (0.66 mol, 99.9% yield) of the crude 3l,5l-dicinnamoyl-2′-deoxy-2′,2′-difluorocytidine containing 43 % of the β-anomer and 52 % of the α-anomer.
EXAMPLE 4
[0051] This example demonstrates the preparation of gemcitabine hydrochloride. [0052] To a solution of ammonia-methanol (15.8 %, 4.57 L), the crude 3,5- dicirmamoyl-2′-deoxy-2′,2′-difluorocytidine of example 3 was added (346.5 g, 0.66 mol), and stirred at ambient temperature for 6 hours. The mixture was concentrated to afford a light yellow solid (306 g). Purified water (3 L) was added to the solid, followed by addition of ethyl acetate (1.8 L), and stirring was maintained for about 10 minutes. The aqueous layer was separated and the organic layer was extracted with water (1.05 L). The aqueous layers were combined and water was removed by evaporation under reduced pressure to obtain an oil (154.7 g). Water was added (660 ml) and the mixture was heated to 50-550C to dissolve the solid. The mixture was cooled to 5-1O0C during about one hour and mixed for about 16 hours at that temperature. The thus formed solid was filtered and dried to afford 46.75 g (0.177 mol), containing 98 % of the β-anomer and 1.3 % of the α-anomer. 0.5N HCl (936 ml) was added followed by addition of dichloromethane (300 ml) with stirring. The water phase was separated and the aqueous phase was washed with dichloromethane (300 ml). After filtration, the aqueous phase was concentrated to dryness under reduced pressure to obtain gemcitabine hydrochloride as a solid (46.9 g). The solid was dissolved in water (187 ml) at ambient temperature and the mixture was heated to 500C to afford a clear solution and cooled to ambient temperature. Acetone (1.4 L) was added and stirring was maintained for about one hour. Then, the precipitate was collected by filtration and washed twice with acetone (2X30 ml) and dried at 450C under vacuum to obtain 39.2 g of gemcitabine hydrochloride, containing 99.9% of the β-anomer
EXAMPLE 5
[0053] This example demonstrates the preparation of gemcitabine hydrochloride. [0054] To a solution of ammonia-methanol (about 15.8 %, 1.35 L), the crude 3′,5′- dicinnamoyl-2′-deoxy-2′,2′-difluorocytidine prepared as described in example 2 was added (96 g, 183.4 mmol), and stirred at ambient temperature for 4 hours. The mixture was concentrated to afford a light yellow solid (80.5 g). Purified water (1 L) was added to the solid, followed by addition of ethyl acetate (600 ml), and stirring was maintained for about 10 minutes. The aqueous layer was separated and the organic layer was extracted with water (350 ml). The aqueous layers were combined and water was removed by evaporation under reduced pressure to obtain an oil (46.4 g). Water was added (220 ml) and the mixture was heated to 50-550C to dissolve the solid. The mixture was cooled to 0-50C during about one hour and mixed for about 16 hours at that temperature. The thus formed solid was filtered and dried to afford 11.1 g of gemcitabine free base. 0.5N HCl (240 ml) was added followed by addition of dichloromethane (100 ml) with stirring. The water phase was separated and the aqueous phase was washed with dichloromethane (300 ml). After filtration, the aqueous phase was concentrated to dryness under reduced pressure to obtain gemcitabine hydrochloride as a solid (12.0 g). The solid was dissolved in water (48 ml) at ambient temperature and the mixture was heated to 5O0C to afford a clear solution and cooled to ambient temperature. Acetone (360 ml) was added and stirring was maintained for about one hour. Then, the precipitate was collected by filtration and washed twice with acetone (2X30 ml) and dried at 450C under vacuum to obtain 9.9 g of gemcitabine hydrochloride, containing 99.6% of the β-anomer.
EXAMPLE 6
[0055] This example demonstrates the slurrying procedure of the 3 ‘,5′- dicinnamoyl-2′-deoxy-2’,2l-difluorocytidine in different solvents. [0056] 1 g of the crude 3′,5′-dicinnamoyl-2′-deoxy-2l,2′-difluorocytidine, containing 73.7 % of the β-anomer and 17.5 % of the α-anomer, was placed in flask and 10 ml of a solvent was added and the mixture was mixed at ambient temperature for one hour. Then, the solid was obtained by filtration, washed with 5 ml of the solvent and dried. The liquid obtained after filtering the solid and the liquid obtained after washing the solid were combined (hereinafter the mother liquor). The ratio between the β-anomer and the α-anomer in the solid and in the mother liquor was determined by HPLC and the results are summarized in Table 1.
Table 1

/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Adverse effects
Gemcitabine is a chemotherapy drug that works by killing any cells that are dividing.[10] Cancer cells divide rapidly and so are targeted at higher rates by gemcitabine, but many essential cells also divide rapidly, including cells in skin, the scalp, the stomach lining, and bone marrow, resulting in adverse effects.[16]: 265
The gemcitabine label carries warnings that it can suppress bone marrow function and cause loss of white blood cells, loss of platelets, and loss of red blood cells, and that it should be used carefully in people with liver, kidney, or cardiovascular disorders. People taking it should not take live vaccines. The warning label also states it may cause posterior reversible encephalopathy syndrome, that it may cause capillary leak syndrome, that it may cause severe lung conditions like pulmonary edema, pneumonia, and adult respiratory distress syndrome, and that it may harm sperm.[10][17]
More than 10% of users develop adverse effects, including difficulty breathing, low white and red blood cells counts, low platelet counts, vomiting and nausea, elevated transaminases, rashes and itchy skin, hair loss, blood and protein in urine, flu-like symptoms, and edema.[10][15]
Common adverse effects (occurring in 1–10% of users) include fever, loss of appetite, headache, difficulty sleeping, tiredness, cough, runny nose, diarrhea, mouth and lip sores, sweating, back pain, and muscle pain.[10]
Thrombotic thrombocytopenic purpura (TTP) is a rare but serious side effect that been associated with particular chemotherapy medications including gemcitabine. TTP is a blood disorder and can lead to microangipathic hemolytic anemia (MAHA), neurologic abnormalities, fever, and renal disease.[18]
Pharmacology
Gemcitabine is hydrophilic and must be transported into cells via molecular transporters for nucleosides (the most common transporters for gemcitabine are SLC29A1 SLC28A1, and SLC28A3).[19][20] After entering the cell, gemcitabine is first modified by attaching a phosphate to it, and so it becomes gemcitabine monophosphate (dFdCMP).[19][20] This is the rate-determining step that is catalyzed by the enzyme deoxycytidine kinase (DCK).[19][20] Two more phosphates are added by other enzymes. After the attachment of the three phosphates gemcitabine is finally pharmacologically active as gemcitabine triphosphate (dFdCTP).[19] [21]
After being thrice phosphorylated, gemcitabine can masquerade as deoxycytidine triphosphate and is incorporated into new DNA strands being synthesized as the cell replicates.[2][19][20]
When gemcitabine is incorporated into DNA it allows a native, or normal, nucleoside base to be added next to it. This leads to “masked chain termination” because gemcitabine is a “faulty” base, but due to its neighboring native nucleoside it eludes the cell’s normal repair system (base-excision repair). Thus, incorporation of gemcitabine into the cell’s DNA creates an irreparable error that leads to inhibition of further DNA synthesis, and thereby leading to cell death.[2][19][20]
The form of gemcitabine with two phosphates attached (dFdCDP) also has activity; it inhibits the enzyme ribonucleotide reductase (RNR), which is needed to create new DNA nucleotides. The lack of nucleotides drives the cell to uptake more of the components it needs to make nucleotides from outside the cell, which also increases uptake of gemcitabine.[2][19][20][22]
Chemistry
Gemcitabine is a synthetic pyrimidine nucleoside prodrug—a nucleoside analog in which the hydrogen atoms on the 2′ carbon of deoxycytidine are replaced by fluorine atoms.[2][23][24]
The synthesis described and pictured below is the original synthesis done in the Eli Lilly Company labs. Synthesis begins with enantiopure D-glyceraldehyde (R)-2 as the starting material which can made from D-mannitol in 2–7 steps. Then fluorine is introduced by a “building block” approach using ethyl bromodifluroacetate. Then, Reformatsky reaction under standard conditions will yield a 3:1 anti/syn diastereomeric mixture, with one major product. Separation of the diastereomers is carried out via HPLC, thus yielding the anti-3 gemcitabine in a 65% yield.[23][24] At least two other full synthesis methods have also been developed by different groups.[24]

Illustration of the original synthesis process used and published by Hertel et al. in 1988 of Lilly laboratories.
History[
Gemcitabine was first synthesized in Larry Hertel’s lab at Eli Lilly and Company during the early 1980s. It was intended as an antiviral drug, but preclinical testing showed that it killed leukemia cells in vitro.[25]
During the early 1990s gemcitabine was studied in clinical trials. The pancreatic cancer trials found that gemcitabine increased one-year survival time significantly, and it was approved in the UK in 1995[10] and approved by the FDA in 1996 for pancreatic cancers.[4] In 1998, gemcitabine received FDA approval for treating non-small cell lung cancer and in 2004, it was approved for metastatic breast cancer.[4]
European labels were harmonized by the EMA in 2008.[26]
By 2008, Lilly’s worldwide sales of gemcitabine were about $1.7 billion; at that time its US patents were set to expire in 2013 and its European patents in 2009.[27] The first generic launched in Europe in 2009,[7] and patent challenges were mounted in the US which led to invalidation of a key Lilly patent on its method to make the drug.[28][29] Generic companies started selling the drug in the US in 2010 when the patent on the chemical itself expired.[29][8] Patent litigation in China made headlines there and was resolved in 2010.[30]
Society and culture
As of 2017, gemcitabine was marketed under many brand names worldwide: Abine, Accogem, Acytabin, Antoril, axigem, Bendacitabin, Biogem, Boligem, Celzar, Citegin, Cytigem, Cytogem, Daplax, DBL, Demozar, Dercin, Emcitab, Enekamub, Eriogem, Fotinex, Gebina, Gemalata, Gembin, Gembine, Gembio, Gemcel, Gemcetin, Gemcibine, Gemcikal, Gemcipen, Gemcired, Gemcirena, Gemcit, Gemcitabin, Gemcitabina, Gemcitabine, Gemcitabinum, Gemcitan, Gemedac, Gemflor, Gemful, Gemita, Gemko, Gemliquid, Gemmis, Gemnil, Gempower, Gemsol, Gemstad, Gemstada, Gemtabine, Gemtavis, Gemtaz, Gemtero, Gemtra, Gemtro, Gemvic, Gemxit, Gemzar, Gentabim, Genuten, Genvir, Geroam, Gestredos, Getanosan, Getmisi, Gezt, Gitrabin, Gramagen, Haxanit, Jemta, Kalbezar, Medigem, Meditabine, Nabigem, Nallian, Oncogem, Oncoril, Pamigeno, Ribozar, Santabin, Sitagem, Symtabin, Yu Jie, Ze Fei, and Zefei.[1]
Research
Because it is clinically valuable and is only useful when delivered intravenously, methods to reformulate it so that it can be given by mouth have been a subject of research.[31][32][33]
Research into pharmacogenomics and pharmacogenetics has been ongoing. As of 2014, it was not clear whether or not genetic tests could be useful in guiding dosing and which people respond best to gemcitabine.[19] However, it appears that variation in the expression of proteins (SLC29A1, SLC29A2, SLC28A1, and SLC28A3) used for transport of gemcitabine into the cell lead to variations in its potency. Similarly, the genes that express proteins that lead to its inactivation (deoxycytidine deaminase, cytidine deaminase, and NT5C) and that express its other intracellular targets (RRM1, RRM2, and RRM2B) lead to variations in response to the drug.[19] Research has also been ongoing to understand how mutations in pancreatic cancers themselves determine response to gemcitabine.[34]
It has been studied as a treatment for Kaposi sarcoma, a common cancer in people with AIDS which is uncommon in the developed world but not uncommon in the developing world.[35]
References
- ^ Jump up to:a b c “Gemcitabine International Brands”. Drugs.com. Archived from the original on 25 May 2014. Retrieved 6 May 2017.
- ^ Jump up to:a b c d e f g h i j k l “Gemcitabine Hydrochloride”. The American Society of Health-System Pharmacists. Archived from the original on 2 February 2017. Retrieved 8 December 2016.
- ^ “Drug Formulary/Drugs/ gemcitabine – Provider Monograph”. Cancer Care Ontario. Retrieved 6 December 2020.
- ^ Jump up to:a b c National Cancer Institute (2006-10-05). “FDA Approval for Gemcitabine Hydrochloride”. National Cancer Institute. Archived from the original on 5 April 2017. Retrieved 22 April 2017.
- ^ Li Y, Li P, Li Y, Zhang R, Yu P, Ma Z, Kainov DE, de Man RA, Peppelenbosch MP, Pan Q (December 2020). “Drug screening identified gemcitabine inhibiting hepatitis E virus by inducing interferon-like response via activation of STAT1 phosphorylation”. Antiviral Research. 184: 104967. doi:10.1016/j.antiviral.2020.104967. PMID 33137361.
- ^ Fischer J, Ganellin CR (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 511. ISBN 9783527607495.
- ^ Jump up to:a b Myers, Calisha (13 March 2009). “Gemcitabine from Actavis launched on patent expiry in EU markets”. FierceBiotech. Archived from the original on 11 September 2017.
- ^ Jump up to:a b “Press release: Hospira launches two-gram vial of gemcitabine hydrochloride for injection”. Hospira via News-Medical.Net. 16 November 2010. Archived from the original on 2 October 2015.
- ^ World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
- ^ Jump up to:a b c d e f g “UK label”. UK Electronic Medicines Compendium. 5 June 2014. Archived from the original on 10 July 2017. Retrieved 6 May 2017.
- ^ Jump up to:a b “US formLabel” (PDF). FDA. June 2014. Archived (PDF) from the original on 16 February 2017. Retrieved 6 May 2017. For label updates see FDA index page for NDA 020509 Archived 2017-04-29 at the Wayback Machine
- ^ Zhang XW, Ma YX, Sun Y, Cao YB, Li Q, Xu CA (June 2017). “Gemcitabine in Combination with a Second Cytotoxic Agent in the First-Line Treatment of Locally Advanced or Metastatic Pancreatic Cancer: a Systematic Review and Meta-Analysis”. Targeted Oncology. 12 (3): 309–321. doi:10.1007/s11523-017-0486-5. PMID 28353074. S2CID 3833614.
- ^ Plentz RR, Malek NP (December 2016). “Systemic Therapy of Cholangiocarcinoma”. Visceral Medicine. 32 (6): 427–430. doi:10.1159/000453084. PMC 5290432. PMID 28229078.
- ^ Jain A, Kwong LN, Javle M (November 2016). “Genomic Profiling of Biliary Tract Cancers and Implications for Clinical Practice”. Current Treatment Options in Oncology. 17 (11): 58. doi:10.1007/s11864-016-0432-2. PMID 27658789. S2CID 25477593.
- ^ Jump up to:a b Macmillan Cancer Support. “Gemcitabine”. Macmillan Cancer Support. Archived from the original on 25 March 2017. Retrieved 6 May 2017.
- ^ Rachel Airley (2009). Cancer Chemotherapy. Wiley-Blackwell. ISBN 978-0-470-09254-5.
- ^ Siddall E, Khatri M, Radhakrishnan J (July 2017). “Capillary leak syndrome: etiologies, pathophysiology, and management”. Kidney International. 92 (1): 37–46. doi:10.1016/j.kint.2016.11.029. PMID 28318633.
- ^ Kasi PM (January 2011). “Thrombotic thrombocytopenic purpura and gemcitabine”. Case Reports in Oncology. 4 (1): 143–8. doi:10.1159/000326801. PMC 3114619. PMID 21691573.
- ^ Jump up to:a b c d e f g h i Alvarellos ML, Lamba J, Sangkuhl K, Thorn CF, Wang L, Klein DJ, Altman RB, Klein TE (November 2014). “PharmGKB summary: gemcitabine pathway”. Pharmacogenetics and Genomics. 24 (11): 564–74. doi:10.1097/fpc.0000000000000086. PMC 4189987. PMID 25162786.
- ^ Jump up to:a b c d e f Mini E, Nobili S, Caciagli B, Landini I, Mazzei T (May 2006). “Cellular pharmacology of gemcitabine”. Annals of Oncology. 17 Suppl 5: v7-12. doi:10.1093/annonc/mdj941. PMID 16807468.
- ^ Fatima, M., Iqbal Ahmed, M. M., Batool, F., Riaz, A., Ali, M., Munch-Petersen, B., & Mutahir, Z. (2019). Recombinant deoxyribonucleoside kinase from Drosophila melanogaster can improve gemcitabine based combined gene/chemotherapy for targeting cancer cells. Bosnian Journal of Basic Medical Sciences, 19(4), 342-349. https://doi.org/10.17305/bjbms.2019.4136
- ^ Cerqueira NM, Fernandes PA, Ramos MJ (2007). “Understanding ribonucleotide reductase inactivation by gemcitabine”. Chemistry. 13 (30): 8507–15. doi:10.1002/chem.200700260. PMID 17636467.
- ^ Jump up to:a b Brown K, Weymouth-Wilson A, Linclau B (April 2015). “A linear synthesis of gemcitabine”. Carbohydrate Research. 406: 71–5. doi:10.1016/j.carres.2015.01.001. PMID 25681996.
- ^ Jump up to:a b c Brown K, Dixey M, Weymouth-Wilson A, Linclau B (March 2014). “The synthesis of gemcitabine”. Carbohydrate Research. 387: 59–73. doi:10.1016/j.carres.2014.01.024. PMID 24636495.
- ^ Sneader, Walter (2005). Drug discovery: a history. New York: Wiley. p. 259. ISBN 978-0-471-89979-2.
- ^ “Gemzar”. European Medicines Agency. 24 September 2008. Archived from the original on 11 September 2017.
- ^ Myers, Calisha (18 August 2009). “Patent for Lilly’s cancer drug Gemzar invalidated”. FiercePharma. Archived from the original on 11 September 2017.
- ^ Holman, Christopher M. (Summer 2011). “Unpredictability in Patent Law and Its Effect on Pharmaceutical Innovation” (PDF). Missouri Law Review. 76 (3): 645–693. Archived from the original (PDF) on 2017-09-11. Retrieved 2017-05-06.
- ^ Jump up to:a b Ravicher, Daniel B. (28 July 2010). “On the Generic Gemzar Patent Fight”. Seeking Alpha. Archived from the original on 9 December 2012.
- ^ Wang M, Alexandre D (2015). “Analysis of Cases on Pharmaceutical Patent Infringement in Great China”. In Rader RR, et al. (eds.). Law, Politics and Revenue Extraction on Intellectual Property. Cambridge Scholars Publishing. p. 119. ISBN 9781443879262. Archived from the original on 2017-09-11.
- ^ Dyawanapelly S, Kumar A, Chourasia MK (2017). “Lessons Learned from Gemcitabine: Impact of Therapeutic Carrier Systems and Gemcitabine’s Drug Conjugates on Cancer Therapy”. Critical Reviews in Therapeutic Drug Carrier Systems. 34 (1): 63–96. doi:10.1615/CritRevTherDrugCarrierSyst.2017017912. PMID 28322141.
- ^ Birhanu G, Javar HA, Seyedjafari E, Zandi-Karimi A (April 2017). “Nanotechnology for delivery of gemcitabine to treat pancreatic cancer”. Biomedicine & Pharmacotherapy. 88: 635–643. doi:10.1016/j.biopha.2017.01.071. PMID 28142120.
- ^ Dubey RD, Saneja A, Gupta PK, Gupta PN (October 2016). “Recent advances in drug delivery strategies for improved therapeutic efficacy of gemcitabine”. European Journal of Pharmaceutical Sciences. 93: 147–62. doi:10.1016/j.ejps.2016.08.021. PMID 27531553.
- ^ Pishvaian MJ, Brody JR (March 2017). “Therapeutic Implications of Molecular Subtyping for Pancreatic Cancer”. Oncology. 31 (3): 159–66, 168. PMID 28299752. Archived from the original on 3 July 2017.
- ^ Krown SE (September 2011). “Treatment strategies for Kaposi sarcoma in sub-Saharan Africa: challenges and opportunities”. Current Opinion in Oncology. 23 (5): 463–8. doi:10.1097/cco.0b013e328349428d. PMC 3465839. PMID 21681092.
External links
- “Gemcitabine”. Drug Information Portal. U.S. National Library of Medicine.
| Clinical data | |
|---|---|
| Pronunciation | /dʒɛmˈsaɪtəbiːn/ |
| Trade names | Gemzar, others[1] |
| Other names | 2′, 2′-difluoro 2’deoxycytidine, dFdC |
| AHFS/Drugs.com | Monograph |
| Pregnancy category | AU: D |
| Routes of administration | Intravenous |
| ATC code | L01BC05 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only)UK: POM (Prescription only)US: ℞-onlyIn general: ℞ (Prescription only) |
| Pharmacokinetic data | |
| Protein binding | <10% |
| Elimination half-life | Short infusions: 32–94 minutes Long infusions: 245–638 minutes |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 95058-81-4 |
| PubChem CID | 60750 |
| IUPHAR/BPS | 4793 |
| DrugBank | DB00441 |
| ChemSpider | 54753 |
| UNII | B76N6SBZ8R |
| KEGG | D02368 |
| ChEBI | CHEBI:175901 |
| ChEMBL | ChEMBL888 |
| CompTox Dashboard (EPA) | DTXSID3040487 |
| ECHA InfoCard | 100.124.343 |
| Chemical and physical data | |
| Formula | C9H11F2N3O4 |
| Molar mass | 263.201 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (verify) |
/////////////GEMCITABINE, LY 188011, LY188011, CANCER
NC1=NC(=O)N(C=C1)[C@@H]1O[C@H](CO)[C@@H](O)C1(F)F
Patent
Publication numberPriority datePublication dateAssigneeTitle
EP0577303A1 *1992-06-221994-01-05Eli Lilly And CompanyStereoselective glycosylation process
WO2006070985A1 *2004-12-302006-07-06Hanmi Pharm. Co., Ltd.METHOD FOR THE PREPARATION OF 2#-DEOXY-2#,2#-DIFLUOROCYTIDINE
WO2007027564A2 *2005-08-292007-03-08Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
WO2007069838A1 *2005-12-142007-06-21Dong-A Pharm.Co., Ltd.A manufacturing process of 2′,2′-difluoronucleoside and intermediate
Family To Family Citations
JPS541315B2 *1974-11-221979-01-23
US4526988A *1983-03-101985-07-02Eli Lilly And CompanyDifluoro antivirals and intermediate therefor
US4751221A *1985-10-181988-06-14Sloan-Kettering Institute For Cancer Research2-fluoro-arabinofuranosyl purine nucleosides
US5223608A *1987-08-281993-06-29Eli Lilly And CompanyProcess for and intermediates of 2′,2′-difluoronucleosides
US4965374A *1987-08-281990-10-23Eli Lilly And CompanyProcess for and intermediates of 2′,2′-difluoronucleosides
US5256798A *1992-06-221993-10-26Eli Lilly And CompanyProcess for preparing alpha-anomer enriched 2-deoxy-2,2-difluoro-D-ribofuranosyl sulfonates
US5371210A *1992-06-221994-12-06Eli Lilly And CompanyStereoselective fusion glycosylation process for preparing 2′-deoxy-2′,2′-difluoronucleosides and 2′-deoxy-2′-fluoronucleosides
US5256797A *1992-06-221993-10-26Eli Lilly And CompanyProcess for separating 2-deoxy-2,2-difluoro-D-ribofuranosyl alkylsulfonate anomers
US5480992A *1993-09-161996-01-02Eli Lilly And CompanyAnomeric fluororibosyl amines
US5521294A *1995-01-181996-05-28Eli Lilly And Company2,2-difluoro-3-carbamoyl ribose sulfonate compounds and process for the preparation of beta nucleosides
US5559222A *1995-02-031996-09-24Eli Lilly And CompanyPreparation of 1-(2′-deoxy-2′,2′-difluoro-D-ribo-pentofuranosyl)-cytosine from 2-deoxy-2,2-difluoro-β-D-ribo-pentopyranose
US5602262A *1995-02-031997-02-11Eli Lilly And CompanyProcess for the preparation of 2-deoxy-2,2-difluoro-β-D-ribo-pentopyranose
US5633367A *1995-03-241997-05-27Eli Lilly And CompanyProcess for the preparation of a 2-substituted 3,3-difluorofuran
GB9514268D0 *1995-07-131995-09-13Hoffmann La RochePyrimidine nucleoside
US5756775A *1995-12-131998-05-26Eli Lilly And CompanyProcess to make α,α-difluoro-β-hydroxyl thiol esters
CA2641719A1 *2006-02-072007-08-16Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
US20070249823A1 *2006-04-202007-10-25Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
Publication numberPriority datePublication dateAssigneeTitle
CN102617483A *2011-06-302012-08-01江苏豪森药业股份有限公司Process for recycling cytosine during preparing process of gemcitabine hydrochloride
WO2013164798A12012-05-042013-11-07Tpresso AgPackaging of dry leaves in sealed capsules
CN105566418A *2014-10-092016-05-11江苏笃诚医药科技股份有限公司2′,3′-di-O-acetyl-5′-deoxy-5-fluorocytidine synthesis method
EP3817732A4 *2018-08-032022-06-08Cellix Bio Private LimitedCompositions and methods for the treatment of cancer
Family To Family Citations
CA2641719A1 *2006-02-072007-08-16Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
US20070249823A1 *2006-04-202007-10-25Chemagis Ltd.Process for preparing gemcitabine and associated intermediates
IT1393062B1 *2008-10-232012-04-11Prime Europ TherapeuticalsPROCEDURE FOR THE PREPARATION OF GEMCITABINE CHLORIDRATE
PublicationPublication DateTitle
CA2509687C2012-08-14Process for the production of 2'-branched nucleosides
WO2008129530A12008-10-30Gemcitabine production process
AU2005328519B22012-03-01Intermediate and process for preparing of beta- anomer enriched 21deoxy, 21 ,21-difluoro-D-ribofuranosyl nucleosides
EP1931693A22008-06-18Process for preparing gemcitabine and associated intermediates
EP2164856A12010-03-24Processes related to making capecitabine
US8193354B22012-06-05Process for preparation of Gemcitabine hydrochloride
EP3638685A12020-04-22Synthesis of 3'-deoxyadenosine-5'-0-[phenyl(benzyloxy-l-alaninyl)]phosphate (nuc-7738)
EP2456778A12012-05-30Process for producing flurocytidine derivatives
US20090221811A12009-09-03Process for preparing gemcitabine and associated intermediates
JP5114556B22013-01-09A novel highly stereoselective synthetic process and intermediate for gemcitabine
EP0350292B11994-05-04Process for preparing 2'-deoxy-beta-adenosine
KR100908363B12009-07-20Stereoselective preparation method of tri-O-acetyl-5-deoxy-β-D-ribofuranose and separation method thereof
US20070249823A12007-10-25Process for preparing gemcitabine and associated intermediates
WO2007070804A22007-06-21Process for preparing gemcitabine and associated intermediates
KR101259648B12013-05-09A manufacturing process of 2′,2′-difluoronucloside and intermediate
US8338586B22012-12-25Process of making cladribine
WO2010029574A22010-03-18An improved process for the preparation of gemcitabine and its intermediates using novel protecting groups and ion exchange resins
KR20080090950A2008-10-09Process for preparing gemcitabine and associated intermediates
WO2012115578A12012-08-30Synthesis of flg

NEW DRUG APPROVALS
ONE TIME
$9.00
OTERACIL POTTASIUM

OTERACIL
UNII4R7FFA00RX, CAS Number2207-75-2, WeightAverage: 195.175, Monoisotopic: 194.96823705, Chemical FormulaC4H2KN3O4
[K+].OC1=NC(=NC(=O)N1)C([O-])=O
1,3,5-Triazine-2-carboxylic acid, 1,4,5,6-tetrahydro-4,6-dioxo-, potassium salt (1:1)
218-627-5[EINECS]
2207-75-2[RN]
4,6-Dihydroxy-1,3,5-triazine-2-carboxylic acid potassium salt
- KOX
- NSC 28841
- Oxonate
- Oxonate, potassium
CDSCO APPROVED,01.02.2022

Gimeracil bulk & Oteracil potassium bulk and Tegafur 15mg/20mg, Gimeracil 4.35mg/5.8mg and Oteracil 11.8mg/15.8mg capsules
indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.
Oteracil Potassium is the potassium salt of oxonate, an enzyme inhibitor that modulates 5- fluorouracil (5-FU) toxicity. Potassium oxonate inhibits orotate phosphoribosyltransferase, which catalyzes the conversion of 5-FU to its active or phosphorylated form, FUMP. Upon oral administration, Oxonate is selectively distributed to the intracellular sites of tissues lining the small intestines, producing localized inhibitory effects within the gastrointestinal tract. As a result, 5-FU associated gastrointestinal toxic effects are reduced and the incidence of diarrhea or mucositis is decreased in 5-FU related therapy.
Oteracil is an adjunct to antineoplastic therapy, used to reduce the toxic side effects associated with chemotherapy. Approved by the European Medicines Agency (EMA) in March 2011, Oteracil is available in combination with Gimeracil and Tegafur within the commercially available product “Teysuno”. The main active ingredient in Teysuno is Tegafur, a pro-drug of Fluorouracil (5-FU), which is a cytotoxic anti-metabolite drug that acts on rapidly dividing cancer cells. By mimicking a class of compounds called “pyrimidines” that are essential components of RNA and DNA, 5-FU is able to insert itself into strands of DNA and RNA, thereby halting the replication process necessary for continued cancer growth.
Oteracil’s main role within Teysuno is to reduce the activity of 5-FU within normal gastrointestinal mucosa, and therefore reduce’s gastrointestinal toxicity 1. It functions by blocking the enzyme orotate phosphoribosyltransferase (OPRT), which is involved in the production of 5-FU.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
SYNTHESIS
https://patents.google.com/patent/CN103435566A/zh


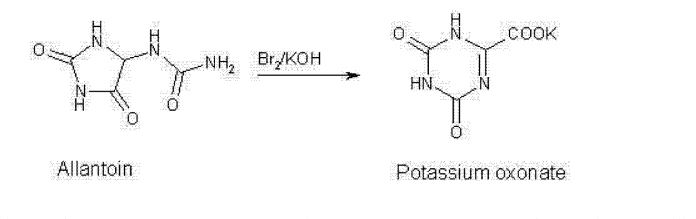
SYN
https://europepmc.org/article/pmc/pmc7717319
Poje et al. reported a two-step, gram-scale preparation of the TS-1 additive oteracil 21 (Scheme 16).226 Iodine-mediated-oxidation of uric acid 116 produced dehydroallantoin 117 as the major product, and subsequent treatment with potassium hydroxide resulted in the rearranged product oteracil 21.227

Synthesis of Oteracil 21a
aReagents and conditions: (a) LiOH, I2, H2O, 5 °C, 5 min, then AcOH, 75%; (b) aq KOH, 20 min, rt, 82%.
(226) Poje M; Sokolić-Maravić L The mechanism for the conversion of uric acid into allantoin and dehydro-allantoin: A new look at an old problem. Tetrahedron 1986, 42 (2), 747–751. [Google Scholar]
(227) Sugi M; Igi M EP Patent 0957096, 1999.
EP0957096A1 *1998-05-111999-11-17SUMIKA FINE CHEMICALS Co., Ltd.Method for producing potassium oxonate
CN101475539A *2009-02-112009-07-08鲁南制药集团股份有限公司Refining method for preparing high-purity oteracil potassium
CN102250025A *2011-05-182011-11-23深圳万乐药业有限公司Preparation method suitable for industrially producing oteracil potassium
CN102746244A *2012-07-272012-10-24南京正大天晴制药有限公司Refining method of oteracil potassium
//////////OTERACIL POTTASIUM, KOX, NSC 28841, Oxonate, Oxonate potassium, INDIA 2022, APPROVALS 2022, CANCER
[K+].OC1=NC(=NC(=O)N1)C([O-])=O

NEW DRUG APPROVALS
ONE TIME
$10.00
GIMERACIL
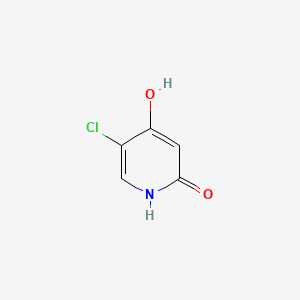

GIMERACIL
C5H4ClNO2, 145.54
5-chloro-4-hydroxy-1H-pyridin-2-one
5-Chloro-2,4-dihydroxypyridine
5-Chloro-4-hydroxy-2(1H)-pyridone
CDSCO APPROVED,01.02.2022
Gimeracil bulk & Oteracil potassium bulk and Tegafur 15mg/20mg, Gimeracil 4.35mg/5.8mg and Oteracil 11.8mg/15.8mg capsules
indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.
| Combination of | |
|---|---|
| Tegafur | Antineoplastic drug |
| Gimeracil | Enzyme inhibitor |
| Oteracil | Enzyme inhibitor |
| Clinical data | |
| Trade names | Teysuno, TS-1 |
| Other names | S-1[1] |
| AHFS/Drugs.com | UK Drug Information |
| License data | EU EMA: by Tegafur |
| Pregnancy category | Contraindicated |
| Routes of administration | By mouth |
| ATC code | L01BC53 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only) [2]EU: Rx-only [3]In general: ℞ (Prescription only) |
| Identifiers | |
| CAS Number | 150863-82-4 |
| PubChem CID | 54715158 |
Tegafur/gimeracil/oteracil, sold under the brand names Teysuno and TS-1,[3][4] is a fixed-dose combination medication used for the treatment of advanced gastric cancer when used in combination with cisplatin,[3] and also for the treatment of head and neck cancer, colorectal cancer, non–small-cell lung, breast, pancreatic, and biliary tract cancers.[5]: 213
The most common severe side effects when used in combination with cisplatin include neutropenia (low levels of neutrophils, a type of white blood cell), anaemia (low red blood cell counts) and fatigue (tiredness).[3]
Tegafur/gimeracil/oteracil (Teysuno) was approved for medical use in the European Union in March 2011.[3] It has not been approved by the U.S. Food and Drug Administration (FDA).[5]: 213
Medical uses
In the European Union tegafur/gimeracil/oteracil is indicated in adults for the treatment of advanced gastric cancer when given in combination with cisplatin.[3]
Contraindications
In the European Union, tegafur/gimeracil/oteracil must not be used in the following groups:
- people receiving another fluoropyrimidine (a group of anticancer medicines that includes tegafur/gimeracil/oteracil) or who have had severe and unexpected reactions to fluoropyrimidine therapy;[3]
- people known to have no DPD enzyme activity, as well as people who, within the previous four weeks, have been treated with a medicine that blocks this enzyme;[3]
- pregnant or breastfeeding women;[3]
- people with severe leucopenia, neutropenia, or thrombocytopenia (low levels of white cells or platelets in the blood);[3]
- people with severe kidney problems requiring dialysis;[3]
- people who should not be receiving cisplatin.[3]
Mechanism of action
Tegafur is the actual chemotherapeutic agent. It is a prodrug of the active substance fluorouracil (5-FU).[3] Tegafur, is a cytotoxic medicine (a medicine that kills rapidly dividing cells, such as cancer cells) that belongs to the ‘anti-metabolites’ group. Tegafur is converted to the medicine fluorouracil in the body, but more is converted in tumor cells than in normal tissues.[3] Fluorouracil is very similar to pyrimidine.[3] Pyrimidine is part of the genetic material of cells (DNA and RNA).[3] In the body, fluorouracil takes the place of pyrimidine and interferes with the enzymes involved in making new DNA.[3] As a result, it prevents the growth of tumor cells and eventually kills them.[3]
Gimeracil inhibits the degradation of fluorouracil by reversibly blocking the dehydrogenase enzyme dihydropyrimidine dehydrogenase (DPD). This results in higher 5-FU levels and a prolonged half-life of the substance.[6]
Oteracil mainly stays in the gut because of its low permeability, where it reduces the production of 5-FU by blocking the enzyme orotate phosphoribosyltransferase. Lower 5-FU levels in the gut result in a lower gastrointestinal toxicity.[6]
Within the medication, the molar ratio of the three components (tegafur:gimeracil:oteracil) is 1:1:0.4.[7]
The maximum tolerated dose differed between Asian and Caucasian populations (80 mg/m2 and 25 mg/m2 respectively), perhaps due to differences in CYP2A6 genotype.[5]: 213
Research
It is being developed for the treatment of hepatocellular carcinoma.[8] and has activity in esophageal,(Perry Chapter 33) breast,[citation needed] cervical,[citation needed] and colorectal cancer.[9]
References
- ^ Liu TW, Chen LT (201). “S-1 with leucovorin for gastric cancer: how far can it go?”. Lancet Oncol. 17 (1): 12–4. doi:10.1016/S1470-2045(15)00478-7. PMID 26640038.
- ^ “Teysuno 20mg/5.8mg/15.8mg hard capsules – Summary of Product Characteristics (SmPC)”. (emc). Retrieved 30 July 2020.
- ^ Jump up to:a b c d e f g h i j k l m n o p q r “Teysuno EPAR”. European Medicines Agency (EMA). Retrieved 30 July 2020. Text was copied from this source which is © European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
- ^ “ティーエスワン 患者さん・ご家族向け総合情報サイト | 大鵬薬品工業株式会社”.
- ^ Jump up to:a b c DeVita, DeVita; Lawrence, TS; Rosenberg, SA (2015). DeVita, Hellman, and Rosenberg’s Cancer: Principles and Practice of Oncology (10th ed.). LWW. ISBN 978-1451192940.
- ^ Jump up to:a b A. Klement (22 July 2013). “Dreier-Kombination gegen Magenkrebs: Teysuno”. Österreichische Apothekerzeitung (in German) (15/2013): 23.
- ^ Peters GJ, Noordhuis P, Van Kuilenburg AB et al. (2003). “Pharmacokinetics of S-1, an oral formulation of ftorafur, oxonic acid and 5-chloro-2,4-dihydroxypyridine (molar ratio 1:0.4:1) in patients with solid tumors”. Cancer Chemother. Pharmacol. 52 (1): 1–12. doi:10.1007/s00280-003-0617-9. PMID 12739060. S2CID 10858817.
- ^ “BCIQ”.
- ^ Miyamoto Y, Sakamoto Y, Yoshida N, Baba H (2014). “Efficacy of S-1 in colorectal cancer”. Expert Opin Pharmacother. 15 (12): 1761–70. doi:10.1517/14656566.2014.937706. PMID 25032886. S2CID 23637808.
External links
- “Tegafur”. Drug Information Portal. U.S. National Library of Medicine.
- “Gimeracil”. Drug Information Portal. U.S. National Library of Medicine.
- “Oteracil”. Drug Information Portal. U.S. National Library of Medicine.
Gimeracil is an adjunct to antineoplastic therapy, used to increase the concentration and effect of the main active componets within chemotherapy regimens. Approved by the European Medicines Agency (EMA) in March 2011, Gimeracil is available in combination with Oteracil and Tegafur within the commercially available product “Teysuno”. The main active ingredient in Teysuno is Tegafur, a pro-drug of Fluorouracil (5-FU), which is a cytotoxic anti-metabolite drug that acts on rapidly dividing cancer cells. By mimicking a class of compounds called “pyrimidines” that are essential components of RNA and DNA, 5-FU is able to insert itself into strands of DNA and RNA, thereby halting the replication process necessary for continued cancer growth.
Gimeracil’s main role within Teysuno is to prevent the breakdown of Fluorouracil (5-FU), which helps to maintin high enough concentrations for sustained effect against cancer cells 2. It functions by reversibly and selectively blocking the enzyme dihydropyrimidine dehydrogenase (DPD), which is involved in the degradation of 5-FU 1. This allows higher concentrations of 5-FU to be achieved with a lower dose of tegafur, thereby also reducing toxic side effects.
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////


SYNTHESIS

SYN
https://europepmc.org/article/pmc/pmc7717319
Synthesis of Gimeracil 20a
aReagents and conditions: (a) CH3C(OCH3)3, MeOH, then (CH3)2NHCH(OCH3)2, reflux, 92%; (b) aq AcOH, 130 °C, 2 h, 95%; (c) SO2Cl2, HOAc, 50 °C, 0.5 h, 91%; (d) 40% H2SO4, 130 °C, 4 h, 91%; (e) SO2Cl2, HOAc, 50 °C, 45 min, 86%; (f) 75% H2 SO4, 140 °C, 3 h, then NaOH, then pH 4–4.5, 89%


In 1953, Kolder and Hertog reported a synthesis of the TS-1 additive gimeracil 20, which was completed in seven steps using 4-nitropyridine N-oxide as starting material.222 Later, Yano et al. reported an alternative gram-scale synthesis (Scheme 15).223 The one-pot, three component condensation of malononitrile 111, 1,1,1-trimethoxyethane, and 1,1-dimethyoxytrimethylamine generated the dicyano intermediate 112, which was into 2(1H)-pyridinone 113.224 Selective chlorination of 113 was followed by acid-mediated demethylation, hydrolysis, and decarboxylation, to afford gimeracil 20. Interestingly, Xu et al. found that treatment of intermediate 113 with sulfuryl chloride resulted in dichloro 115 formation, which could still be converted to gimeracil 20 by treatment with sulfuric acid.225
(222) Kolder CR; den Hertog HJ Synthesis and reactivity of 5-chloro-2,4-dihydroxypyridine. Rec. Trav. Chim 1953, 72, 285–295. [Google Scholar]
(223) Yano S; Ohno T; Ogawa K Convenient and practical synthesis of 5-chloro-4-hydroxy-2(1H)-pyridinone. Heterocycles 1993, 36, 145–148. [Google Scholar]
(224) Mittelbach M; Kastner G; Junek H Synthesen mit Nitrilen, 71. Mitt. Zur Synthese von 4-Hydroxynicotinsaure aus Butadiendicarbonitrilen. Arch. Pharm 1985, 318 (6), 481–486. [Google Scholar]
(225) Xu Y; Mao D; Zhang F CN Patent 1915976, 2007.

NEW DRUG APPROVALS
THIS MAY NOT RUN WITHOUT SUBSCRIPTION HELP. AVOID CLOSURE OF THIS BLOG
$10.00
//////////GIMERACIL, APPROVALS 2022, INDIA 2022
OC1=CC(=O)NC=C1Cl

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}