
Home » JAPAN 2022
Category Archives: JAPAN 2022
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
Ozoralizumab
Ozoralizumab
| Formula | C1682H2608N472O538S12 |
|---|---|
| CAS | 1167985-17-2 |
| Mol weight | 38434.3245 |
PMDA JAPAN APPROVED 2022 2022/9/26 Nanozora
anti-TNFα Nanobody®; ATN-103; Nanozora; PF-5230896; TS-152
Ozoralizumab is a humanized monoclonal antibody designed for the treatment of inflammatory diseases.[1]
Ozoralizumab was developed by Pfizer Inc, and now belongs to Ablynx NV. Ablynx has licensed the rights to the antibody in China to Eddingpharm.
Ozoralizumab has been used in trials studying the treatment of Rheumatoid Arthritis and Active Rheumatoid Arthritis.
Ozoralizumab is a 38 kDa humanized trivalent bispecific construct consisting of two anti-TNFα NANOBODIES® and anti-HSA NANOBODY® that was generated at Ablynx by a previously described method (23). Llamas were immunized with human TNFα and human muscle extract, which is rich in HSA, to induce the formation of anti-TNFα VHH and anti-HSA VHH. Both the anti-TNFα VHH and anti-HSA VHH were humanized by a complementary determining regions (CDR) grafting approach in which the CDR of the gene encoding llama VHH was grafted onto the most homologous human VHH framework sequence. Since binding to serum albumin prolongs the half-life of VHH (23, 26, 27), an anti-HSA VHH which efficiently binds murine serum albumin as well was incorporated into the two anti-TNFα VHHs. The three components were fused using a flexible Gly-Ser linker.
////////

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
| Monoclonal antibody | |
|---|---|
| Type | Whole antibody |
| Source | Humanized |
| Clinical data | |
| ATC code | none |
| Identifiers | |
| CAS Number | 1167985-17-2 |
| ChemSpider | none |
| UNII | 05ZCK72TXZ |
| KEGG | D09944 |
| Chemical and physical data | |
| Formula | C1682H2608N472O538S12 |
| Molar mass | 38434.85 g·mol−1 |
- OriginatorAblynx
- DeveloperAblynx; Eddingpharm; Pfizer; Taisho Pharmaceutical
- ClassAnti-inflammatories; Antirheumatics; Monoclonal antibodies; Proteins
- Mechanism of ActionTumour necrosis factor alpha inhibitors
- Orphan Drug StatusNo
- New Molecular EntityYes
- RegisteredRheumatoid arthritis
- DiscontinuedAnkylosing spondylitis; Crohn’s disease; Psoriatic arthritis
- 05 Oct 2022Sanofi’s affiliate Ablynx has worldwide patent pending for Nanobodies® (Sanofi website, October 2022)
- 05 Oct 2022Sanofi’s affiliate Ablynx has worldwide patent protection for Nanobodies® (Sanofi website, October 2022)
- 26 Sep 2022First global approval – Registered for Rheumatoid arthritis in Japan (SC)
References
- ^ Kratz F, Elsadek B (July 2012). “Clinical impact of serum proteins on drug delivery”. J Control Release. 161 (2): 429–45. doi:10.1016/j.jconrel.2011.11.028. PMID 22155554.
////////Ozoralizumab, Nanozora, Monoclonal antibody, nanobody, Treatment inflammation, ATN 103, APPROVALS 2022, JAPAN 2022

NEW DRUG APPROVALS
ONE TIME
$10.00
Valemetostat tosilate
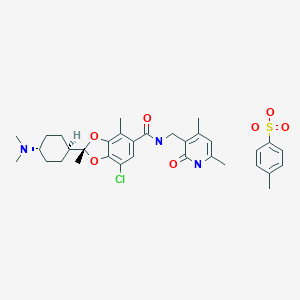
Valemetostat tosilate
バレメトスタットトシル酸塩
| Formula | C26H34ClN3O4. C7H8O3S |
|---|---|
| CAS | 1809336-93-3 |
| Mol weight | 660.2205 |
PMDA JAPAN approved 2022/9/26, Ezharmia
- 1,3-Benzodioxole-5-carboxamide, 7-chloro-N-((1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl)-2-(trans-4-(dimethylamino)cyclohexyl)-2,4-dimethyl-, (2R)-, compd. with 4-methylbenzenesulfonate (1:1)
Antineoplastic, histone methyltransferase inhibitor
1809336-39-7 (free base). 1809336-93-3 (tosylate) 1809336-92-2 (mesylate) 1809336-94-4 (fumarate) 1809336-95-5 (tarate)
Synonym: Valemetostat; DS-3201; DS 3201; DS3201; DS-3201b
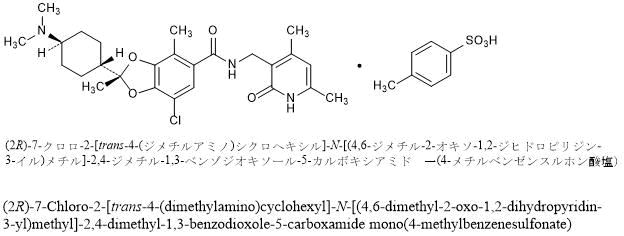
(2R)-7-Chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide mono(4-methylbenzenesulfonate)
C26H34ClN3O4▪C7H8O3S : 660.22
[1809336-93-3]
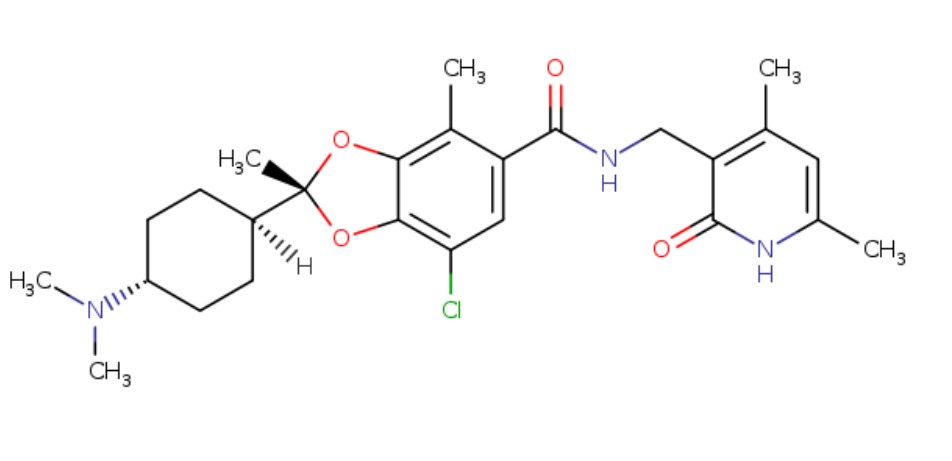
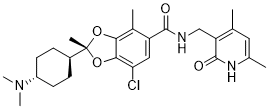
1809336-39-7 (free base)
Chemical Formula: C26H34ClN3O4
Exact Mass: 487.2238
Molecular Weight: 488.02
(2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide
Valemetostat, also known as DS-3201 is a potent, selective and orally active EZH1/2 inhibitor. DS-3201 selectively inhibits the activity of both wild-type and mutated forms of EZH1 and EZH2. Inhibition of EZH1/2 specifically prevents the methylation of lysine 27 on histone H3 (H3K27). This decrease in histone methylation alters gene expression patterns associated with cancer pathways, enhances transcription of certain target genes, and results in decreased proliferation of EZH1/2-expressing cancer cells.
- OriginatorDaiichi Sankyo Inc
- DeveloperCALYM Carnot Institute; Daiichi Sankyo Inc; Lymphoma Academic Research Organisation; Lymphoma Study Association; University of Texas M. D. Anderson Cancer Center
- ClassAmides; Amines; Antineoplastics; Benzodioxoles; Chlorinated hydrocarbons; Cyclohexanes; Pyridones; Small molecules
- Mechanism of ActionEnhancer of zeste homolog 1 protein inhibitors; Enhancer of zeste homolog 2 protein inhibitors
- Orphan Drug StatusYes – Adult T-cell leukaemia-lymphoma; Peripheral T-cell lymphoma
- New Molecular EntityYes
- RegisteredAdult T-cell leukaemia-lymphoma
- Phase IIB-cell lymphoma; Peripheral T-cell lymphoma
- Phase I/IISmall cell lung cancer
- Phase INon-Hodgkin’s lymphoma; Prostate cancer; Renal cell carcinoma; Urogenital cancer
- PreclinicalDiffuse large B cell lymphoma
- No development reportedAcute myeloid leukaemia; Precursor cell lymphoblastic leukaemia-lymphoma
- 26 Sep 2022First global approval – Registered for Adult T-cell leukaemia-lymphoma (Monotherapy, Second-line therapy or greater) in Japan (PO)
- 26 Sep 2022Updated efficacy and adverse events data from a phase II trial in Adult T-cell leukaemia-lymphoma released by Daiichi Sankyo
- 28 Dec 2021Preregistration for Adult T-cell leukaemia-lymphoma (Monotherapy, Second-line therapy or greater) in Japan (PO

PATENT
WO 2015141616
Watson, W. D. J. Org. Chem. 1985, 50, 2145.
Lengyel, I. ; Cesare, V. ; Stephani, R. Synth. Common. 1998, 28, 1891.
PATENT
WO2022009911
The equipment and measurement conditions for the powder X-ray diffraction measurement in the examples are as follows.
Model: Rigaku Rint TTR-III
Specimen: Appropriate
X-ray generation conditions: 50 kV, 300 mA
Wavelength: 1.54 Å (Copper Kα ray)
Measurement temperature: Room temperature
Scanning speed: 20°/min
Scanning range: 2 to 40°
Sampling width: 0.02°
[0043]
(Reference Example 1) Production of ethyl trans-4-[(tert-butoxycarbonyl)amino]cyclohexanecarboxylate
[0044]
[hua 6]
[0045]
Under a nitrogen atmosphere, ethanol (624 L) and ethyl trans-4-aminocyclohexanecarboxylate monohydrochloride (138.7 kg, 667.8 mol) were added to a reaction vessel and cooled. Triethylamine (151.2 kg, 1495 .5 mol) and di-tert-butyl dicarbonate (160.9 kg, 737.2 mol) were added dropwise while maintaining the temperature below 20°C. After stirring at 20-25°C for 4 hours, water (1526 kg) was added dropwise at 25°C or lower, and the mixture was further stirred for 2 hours. The precipitated solid was collected by filtration, washed with a mixture of ethanol:water 1:4 (500 L), and dried under reduced pressure at 40°C to obtain 169.2 kg of the title compound (yield 93.4%). .
1 H NMR (500 MHz, CDCl 3 ): δ 4.37 (br, 1H), 4.11 (q, J = 2.8 Hz, 2H), 3.41 (br, 1H), 2.20 (tt, J = 4.8, 1.4 Hz, 1H),2.07(m,2H),2.00(m,2H),1.52(dq,J=4.6,1.4Hz,2H),1.44(s,9H),1.24(t,J=2.8Hz,3H), 1.11(dq,J=4.6,1.4Hz,2H)
[0046]
(Reference Example 2) Production of tert-butyl = [trans-4-(hydroxymethyl)cyclohexyl]carbamate
[0047]
[hua 7]
[0048]
Under a nitrogen atmosphere, tetrahydrofuran (968 kg), ethyl = trans-4-[(tert-butoxycarbonyl)amino]cyclohexanecarboxylate (110 kg, 405.4 mol), lithium chloride (27.5 kg, 648 kg) were placed in a reaction vessel. .6 mol), potassium borohydride (32.8 kg, 608.1 mol), and water (2.9 L, 162.2 mol) were added, the temperature was slowly raised to 50°C, and the mixture was further stirred for 6 hours. Cooled to 0-5°C. Acetone (66 L) and 9 wt % ammonium chloride aqueous solution (1210 kg) were added dropwise while maintaining the temperature at 20° C. or lower, and the mixture was stirred at 20-25° C. for 1 hour. Additional ethyl acetate (550 L) was added, the aqueous layer was discarded and the organic layer was concentrated to 550 L. Ethyl acetate (1650 L) and 9 wt% aqueous ammonium chloride solution (605 kg) were added to the residue, and the aqueous layer was discarded after stirring. Washed sequentially with water (550 L). The organic layer was concentrated to 880 L, ethyl acetate (660 L) was added to the residue, and the mixture was concentrated to 880 L while maintaining the internal temperature at 40-50°C. The residue was cooled to 0-5° C. and stirred for 1 hour, petroleum ether (1760 L) was added dropwise over 30 minutes, and the mixture was stirred at the same temperature for 2 hours. The precipitated solid was collected by filtration, washed with a petroleum ether:ethyl acetate 3:1 mixture (220 L) cooled to 0-5°C, and dried at 40°C under reduced pressure to give 86.0 kg of the title compound (yield: obtained at a rate of 92.3%).
1 H NMR (500 MHz, CDCl 3 ): δ 4.37 (br, 1H), 3.45 (d, J = 2.2 Hz, 2H), 3.38 (br, 1H), 2.04 (m, 2H),
1.84(m,2H),1.44(m,10H),1.28-1.31(m,1H),1.00-1.13(m,4H)
[0049]
(Reference Example 3) Production of tert-butyl = [trans-4-(2,2-dibromoethenyl)cyclohexyl]carbamate
[0050]
[hua 8]
[0051]
(Step 1)
Under a nitrogen atmosphere, ethyl acetate (50 L), tert-butyl = [trans-4-(hydroxymethyl)cyclohexyl]carbamate (2.5 kg, 10.90 mol), potassium bromide ( 39.3 g, 0.33 mol), 2,2,6,6-tetramethylpiperidine 1-oxyl (51.1 g, 0.33 mol), 4.8% aqueous sodium hydrogen carbonate solution (26.25 kg ) was added and cooled to 0-5°C, 9.9% sodium hypochlorite (8.62 kg, 11.45 mol) was added at 5°C or lower, and the mixture was further stirred at 0°C for 4 hours. Sodium sulfite (250 g) was added to the mixture and stirred at 0-5°C for 30 minutes before warming to 20-25°C. Thereafter, the aqueous layer was discarded and washed with a 20% aqueous sodium chloride solution (12.5 kg), then the organic layer was dried over sodium sulfate and concentrated to 7.5 L. Ethyl acetate (12.5 L) was added to the residue, the mixture was concentrated again to 7.5 L, and used in the next reaction as a tert-butyl=(trans-4-formylcyclohexyl)carbamate solution.
[0052]
(Step 2)
Under a nitrogen atmosphere, tetrahydrofuran (30 L) and triphenylphosphine (5.72 kg, 21.8 mol) were added to a reaction vessel, heated to 40°C, and stirred for 5 minutes. Carbon tetrabromide (3.61 kg, 10.9 mol) was added over 30 minutes and stirred at 40-45° C. for another 30 minutes. A mixture of tert-butyl (trans-4-formylcyclohexyl)carbamate solution and triethylamine (2.54 kg, 25.1 mol) was added below 45°C over 20 minutes and stirred at 40°C for an additional 15 hours. After cooling the reaction solution to 0° C., water (0.2 L) was added at 10° C. or lower, and water (25 L) was added. After heating to 20-25° C., the aqueous layer was discarded, ethyl acetate (4.5 kg) and 10% aqueous sodium chloride solution (25 kg) were added, and after stirring, the aqueous layer was discarded again. After the obtained organic layer was concentrated to 15 L, 2-propanol (19.65 kg) was added and concentrated to 17.5 L. 2-Propanol (11.78 kg) and 5 mol/L hydrochloric acid (151.6 g) were added to the residue, and the mixture was stirred at 25-35°C for 2.5 hours. Water (16.8 L) was added dropwise to the resulting solution, and the mixture was stirred at 20-25°C for 30 minutes and then stirred at 0°C for 2 hours. The precipitated solid was collected by filtration, washed with a mixture (11 kg) of acetonitrile:water 60:40 cooled to 0-5°C, and dried at 40°C under reduced pressure to give 3.05 kg of the title compound (yield 73%). .0%).
1 H NMR (500 MHz, CDCl3):δ6.20(d,J=3.6Hz,1H),4.37(br,1H),3.38(br,1H),2.21(dtt,J=3.6,4.6,1.4Hz,1H),2.05-2.00(m,2H),1.80-1.83(m,2H),1.44(s,9H),1.23(ddd,J=9.9,5.3,1.2 Hz,2H), 1.13(ddt,J=4.6,1.4,5.2 Hz,2H)
[0053]
(Reference Example 4) Production of tert-butyl = (trans-4-ethynylcyclohexyl) carbamate
[0054]
[Chemical 9]
[0055]
Under a nitrogen atmosphere, toluene (1436 kg), tert-butyl = [trans-4-(2,2-dibromoethenyl)cyclohexyl]carbamate (110 kg, 287.1 mol), and N,N,N ‘,N’-Tetramethylethane-1,2-diamine (106.7 kg, 918.8 mol) was added and cooled to -10°C. An isopropylmagnesium chloride-tetrahydrofuran solution (2.0 mol/L, 418 kg, 863 mol) was added dropwise at -5°C or lower, and stirred at -10°C for 30 minutes. After the reaction, 5 mol/L hydrochloric acid (465 kg) was added at 5°C or lower, heated to 20-25°C, and further 5 mol/L hydrochloric acid (41.8 kg) was used to adjust the pH to 5.0-. adjusted to 6.0. After discarding the aqueous layer, the organic layer was washed twice with water (550 L) and concentrated to 550 L. 2-Propanol (1296 kg) was added to the concentrate and concentrated to 550 L again. Further, 2-propanol (1296 kg) was added to the residue, and after concentrating to 550 L, water (770 L) was added dropwise in 4 portions. At that time, it was stirred for 30 minutes after each addition. After the addition, the mixture was stirred for 1 hour and further stirred at 0° C. for 1 hour. The precipitated solid was collected by filtration, washed with a 5:7 mixture of 2-propanol:water (550 L) cooled to 0-5°C, and dried at 40°C under reduced pressure to yield 57.8 kg of the title compound. obtained at a rate of 90.2%).
1 H NMR (500 MHz, CDCl 3 ): δ 4.36 (br, 1H), 3.43 (br, 1H), 2.18-2.23 (m, 1H), 1.97-2.04 (m, 5H), 1.44-1.56 (m, 11H ),1.06-1.14(m,2H)
[0056]
(Reference Example 5) Production of 4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile
[0057]
[Chemical 10]
[0058]
Under a nitrogen atmosphere, water (300 L), 2-cyanoacetamide (20 kg, 238 mol), 1-pentane-2-4-dione (26.2 kg, 262 mol), potassium carbonate (3.29 mol) were added to a reaction vessel. kg, 23.8 mol) was added and stirred at room temperature for 6 hours or longer. After the reaction, the precipitated solid was collected by filtration, washed with water (60 L), further washed with a mixture of methanol (40 L) and water (40 L), and dried under reduced pressure at 40°C to give the title compound as 34 Obtained in .3 kg (97.3% yield).
1 H NMR (500 MHz, DMSO-d 6 ): δ 2.22 (s, 3H), 2.30 (s, 3H), 6.16 (s, 1H), 12.3 (brs, 1H)
[0059]
(Reference Example 6) Production of 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one monohydrochloride
[0060]
[Chemical 11]
[0061]
Under a nitrogen atmosphere, water (171 L), methanol (171 L), 4,6-dimethyl-2-oxo-1,2-dihydropyridine-3-carbonitrile (17.1 kg, 116 mol), concentrated After adding hydrochloric acid (15.8 kg, 152 mol) and 5% palladium carbon (55% wet) (3.82 kg), the inside of the reaction vessel was replaced with hydrogen. Then, the mixture was pressurized with hydrogen and stirred overnight at 30°C. After the reaction, the reaction vessel was purged with nitrogen, the palladium on carbon was removed by filtration, and the palladium on carbon was washed with a 70% aqueous solution of 2-propanol (51 L). Activated carbon (0.86 kg) was added to the filtrate and stirred for 30 minutes. Activated carbon was removed by filtration and washed with 70% aqueous 2-propanol solution (51 L). The filtrate was concentrated under reduced pressure until the liquid volume became 103 L, and 2-propanol (171 L) was added. The mixture was again concentrated under reduced pressure until the liquid volume reached 103 L, then 2-propanol (171 L) was added, and the mixture was stirred for 1 hour or longer. Precipitation of a solid was confirmed, and the solution was concentrated to a volume of 103 L. Further, 2-propanol (51 L) was added, and after concentration under reduced pressure until the liquid volume reached 103 L, the mixture was stirred at 50° C. for 30 minutes. After adding acetone (171 L) over 1 hour while keeping the internal temperature at 40° C. or higher, the mixture was stirred at 40 to 45° C. for 30 minutes. The solution was cooled to 25°C and stirred for 2 hours or longer, and the precipitated solid was collected by filtration, washed with acetone (86 L) and dried under reduced pressure at 40°C to give 19.7 kg of the title compound (yield 90.4%). ).
1 H NMR (500 MHz, methanol-d 4 ): δ 2.27 (s, 3H), 2.30 (s, 3H), 4.02 (s, 2H), 6.16 (s, 1H)
[0062]
(Example 1-1) Production of methyl 5-chloro-3,4-dihydroxy-2-methylbenzoate
[0063]
[Chemical 12]
[0064]
Under a nitrogen atmosphere, water (420 L), toluene (420 L), acetonitrile (420 L), and methyl 3,4-dihydroxy-2-methylbenzoate (1) (60 kg, 329 mol) were added to the reactor and cooled. After that, sulfuryl chloride (133.4 kg, 988 mol) was added dropwise while maintaining the temperature at 20°C or lower. After the reaction, the mixture was separated into an organic layer 1 and an aqueous layer, acetonitrile (60 L) and toluene (120 L) were added to the aqueous layer, and the mixture was stirred. Water (420 L) and acetonitrile (210 L) were added to the organic layer 1, and after cooling, sulfuryl chloride (88.9 kg, 659 mol) was added dropwise at 20°C or lower, and sulfuryl chloride (53.2 kg, 394 mol) was added. ) was added in portions. After the reaction, the mixture was separated into an organic layer 3 and an aqueous layer, and the organic layer 2 was added to the aqueous layer and stirred. Water (420 L), acetonitrile (210 L) were added to the combined organic layer, sulfuryl chloride (44.5 kg, 329 mol) was added dropwise below 20°C, and sulfuryl chloride (106.4 kg, 788 mol) was added. ) was added in portions. After the reaction, the organic layer 4 and the aqueous layer were separated, acetonitrile (60 L) and toluene (120 L) were added to the aqueous layer, and the mixture was stirred. The combined organic layers were washed three times with 20 wt % aqueous sodium chloride solution (300 L) and then concentrated under reduced pressure to 600 L. After repeating the operation of adding toluene (300 L) and concentrating under reduced pressure to 600 L again twice, the mixture was heated and stirred at 60° C. for 1 hour. After cooling to room temperature, the precipitated solid was collected by filtration, washed with toluene (120 L), and dried under reduced pressure at 40°C to give 52.1 kg of the crude title compound (2) (yield: 73.0%). ).
[0065]
Under a nitrogen atmosphere, toluene (782 L) and crude title compound (52.1 kg, 241 mol) were added to a reactor and heated to 80°C. After confirming that the crystals were completely dissolved, they were filtered and washed with heated toluene (261 L). The mixture was cooled to 60° C. and stirred for 0.5 hours after crystallization. After cooling to 10°C, the precipitated solid was collected by filtration, washed with toluene (156 L), and dried under reduced pressure at 40°C to give 47.9 kg of the title compound (2) (yield 91.9%). Acquired.
1 H NMR (500 MHz, methanol-d 4 ): δ 2.41 (s, 3H), 3.82 (s, 3H), 7.41 (s, 1H)
[0066]
(Example 1-2) Examination of chlorination conditions 1 Since
it is difficult to remove compound (1), which is the starting material, and compound (4), which is a by-product of the reaction, even in subsequent steps, need to control. Therefore, chlorination was investigated in the same manner as in Example 1-1 using compound (1) as a starting material. Table 1 shows the results.
[0067]
[Chemical 13]
[0068]
[Table 1]
[0069]
HPLC condition
detection: 220 nm
column: ACQUITY UPLC BEH C18 (2.1 mm ID x 50 mm, 1.7 μm, Waters)
column temperature: 40 ° C
mobile phase: A: 0.1 vol% trifluoroacetic acid aqueous solution, B: acetonitrile
Gradient conditions:
[0070]
[Table 2]
[0071]
Flow rate: 1.0 mL/min
Injection volume: 1 μL
Sample solution: acetonitrile/water (1:1)
wash solution: acetonitrile/water (1:1)
purge solution: acetonitrile/water (1:1)
seal wash solution : Acetonitrile/water (1:1)
Sample cooler temperature: None
Measurement time: 5 minutes
Area measurement time: about 0.5 minutes – 4.0 minutes
Comp. 1: 1.11 min, Comp. 2: 1.55 min,
Comp. 3: 1.44 min, Comp. 4: 1.70 min
[0072]
(Example 1-3) Examination of chlorination conditions 2
Compound (1) was used as a starting material, sulfuryl chloride was used as a chlorination reagent, and chlorination in various solvents was examined. Table 3 shows the results.
[0073]
[table 3]
[0074]
(Example 2) Methyl (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5- Manufacture of carboxylates
[0075]
[Chemical 14]
[0076]
Toluene (9.0 L), tert-butyl = (trans-4-ethynylcyclohexyl) carbamate (2.23 kg, 9.99 mol), methyl = 5-chloro-3,4- were added to a reaction vessel under a nitrogen atmosphere. Dihydroxy-2-methylbenzoate (1.80 kg, 8.31 mol), tri(o-tolyl)phosphine (76.0 g, 250 mmol), triruthenium dodecacarbonyl (53.0 g, 82.9 mmol) ) was added, and the mixture was heated and stirred at 80 to 90° C. for 7 hours under an oxygen-containing nitrogen stream. The reaction solution was cooled to room temperature to obtain a toluene solution of the title compound.
[0077]
(Example 3) (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carvone acid production
[0078]
[Chemical 15]
[0079]
Methyl = (2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole obtained in Example 2 -5-carboxylate toluene solution (13 L, equivalent to 7.83 mol), methanol (9.0 L), 1,2-dimethoxyethane (3.6 L), 5 mol / L sodium hydroxide aqueous solution ( 2.50 L, 12.5 mol) was added and stirred at 55-65° C. for 3 hours. After adding water (5.4 L), the mixture was allowed to stand and separated into an organic layer and an aqueous layer. After cooling to room temperature, 1,2-dimethoxyethane (16.2 L) was added to the aqueous layer, and after adjusting the pH to 4.0 to 4.5 with 3 mol/L hydrochloric acid, toluene (5.4 L) was added. added. After heating to 50-60° C., the organic layer and aqueous layer were separated, and the organic layer was washed with a 20 wt % sodium chloride aqueous solution (7.2 L). Then, 1,2-dimethoxyethane (21.6 L) was added to the organic layer, and after concentration under reduced pressure to 9 L, 1,2-dimethoxyethane (21.6 L) was added and heated to 50-60°C. After that, filtration was performed to remove inorganic substances. Then, after washing with 1,2-dimethoxyethane (1.8 L), the 1,2-dimethoxyethane solution of the title compound (quantitative value 89.6% (Example 2 total yield from ), corresponding to 7.45 mol).
[0080]
(Example 4) (1S)-1-phenylethanaminium (2R)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1, Preparation of 3-benzodioxole-5-carboxylate
[0081]
[Chemical 16]
[0082]
(2RS)-2-{trans-4-[(tert-butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5 obtained in Example 3 – A solution of carboxylic acid in dimethoxyethane (21.6 L, corresponding to 7.45 mol) was heated to 75-80°C, and then (1S)-1-phenylethanamine (1.02 kg, 8.42 mmol). was added and stirred for 4 hours. A mixture of 1,2-dimethoxyethane (9.2 L) and water (3.4 L) heated to 50-60° C. was added, stirred, and then cooled to room temperature. The precipitated solid was collected by filtration and washed with 1,2-dimethoxyethane (9 L) to give a crude title compound (1.75 kg (converted to dry matter), yield 38.5% (Example 2 total yield from ) and an optical purity of 93.8% ee).
[0083]
Under a nitrogen atmosphere, a 1,2-dimethoxyethane aqueous solution (13.6 L) was placed in a reaction vessel, and (1S)-1-phenylethanaminium obtained in step 1 (2R)-2-{trans-4-[(tert -Butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate crude (1.70 kg equivalent, 3.11 mol) was added. After that, 5 mol/L hydrochloric acid (0.56 L, 2.8 mol) was added dropwise. After stirring at room temperature for 10 minutes or longer, the mixture was heated to 75° C. or higher, and (1S)-1-phenylethanamine (360 g, 2.97 mmol) was dissolved in 1,2-dimethoxyethane (2.6 L). The solution was added dropwise over 1 hour. It was then washed with 1,2-dimethoxyethane (0.9 L), stirred for 2 hours and cooled to 0-5°C. The slurry was collected by filtration and washed with 1,2-dimethoxyethane (5.1 L) cooled to 0-5° C. to give the title compound (1.56 kg, yield 91.9%, obtained with an optical purity of 99.5% ee).
1 H NMR (500 MHz, methanol-d 4 ): δ 1.15-1.23(m,2H), 1.28-1.35(m,2H), 1.42(s,9H),
1.59(s,3H), 1.60-1.61(d ,3H,J=7.0Hz,3H),1.80-1.86(dt,J=12.0,3.0Hz,1H),1.95-1.96(m,4H),2.27(s,3H),3.24-3.28(m,1H ),4.39-4.43(q,J=7.0Hz,1H),7.07(s,1H),7.37-7.45(m,5H)
[0084]
(Example 5) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride Manufacturing A
[0085]
[Chemical 17]
[0086]
(Step 1)
Under a nitrogen atmosphere, 1,2-dimethoxyethane (200 L) and (1S)-1-phenylethanaminium (2R)-2-{trans-4-[(tert-butoxycarbonyl) were placed in a reaction vessel. Amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate (equivalent to 87.64 kg, 160 mol), 35% hydrochloric acid (16.7 kg, 160 mol) was added and heated to 45-55° C., 35% hydrochloric acid (36.7 kg, 352 mol) was added dropwise in 7 portions and stirred for 3 hours after dropping. After cooling to room temperature, the reaction solution was added to a mixture of water (982 L) and 5 mol/L sodium hydroxide (166.34 kg, 702 mol). 3 mol/L hydrochloric acid (22.4 kg) was added dropwise to the resulting solution at 30°C, crystal precipitation was confirmed, and the mixture was stirred for 30 minutes or more, cooled to 10°C, and further stirred for 2 hours. After stirring, 3 mol/L hydrochloric acid (95.1 kg) was added dropwise at 10°C to adjust the pH to 7.0. The slurry liquid was collected by filtration, washed with water (293 L) cooled to 10° C., and (2R)-2-(trans-4-aminocyclohexyl)-7-chloro-2,4-dimethyl-1,3- Benzodioxol-5-carboxylic acid trihydrate was obtained (57.63 kg (converted to dry matter), yield 94.7%).
1 H NMR (500 MHz, methanol- d4 + D2O): 1.32-1.44 ( m, 4H), 1.61 (s, 3H), 1.89-1.94 (m, 1H), 2.01-2.13 (m, 4H) ,2.27(s,3H),2.99-3.07(m,1H),7.06(s,3H)
[0087]
(Step 2)
Under nitrogen atmosphere, 1,2-dimethoxyethane (115 L), (2R)-2-(trans-4-aminocyclohexyl)-7-chloro-2,4-dimethyl-1,3 -benzodioxole-5-carboxylic acid trihydrate (57.63 kg equivalent, 152 mmol), formic acid (34.92 kg, 759 mol), 37% formaldehyde aqueous solution (93.59 kg, 1153 mol) was added and stirred at 55-65°C for 2 hours. Cool to room temperature, add 2-propanol (864 L) and concentrate to 576 L under reduced pressure. 2-Propanol (231 L) was added thereto and concentrated under reduced pressure to 576 L. Further, 2-propanol (231 L) was added and concentrated under reduced pressure to 576 L. After concentration, 35% hydrochloric acid (20.40 kg, 196 mol) was added dropwise over 2 hours and stirred at room temperature for 30 minutes. Ethyl acetate (576 L) was added to the resulting slurry over 30 minutes and concentrated to 692 L. Ethyl acetate (461 L) was added followed by further concentration to 519 L. Ethyl acetate (634 L) was added to the residue and the mixture was stirred at room temperature for 2 hours. The precipitated solid was collected by filtration, washed with ethyl acetate (491 L) and dried under reduced pressure at 40°C to give the title compound (51. 56 kg, 87.1% yield).
1 H NMR (500 MHz, methanol-d 4 ): δ 1.38-1.47 (m, 2H), 1.53-1.61 (m, 2H), 1.67 (s, 3H), 1.99-2.05 (m, 1H), 2.13 -2.18(m,4H),2.38(s,3H),2.84(s,6H),3.19-3.25(dt,J=12.5,3.5Hz,1H),
7.53(s,1H)
[0088]
(Example 6) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride Manufacturing B
[0089]
[Chemical 18]
[0090]
Under a nitrogen atmosphere, formic acid (20 mL), 37% formaldehyde aqueous solution (15 mL), dimethoxyethane (10 mL), (1S)-1-phenylethanaminium (2R)-2-{trans-4- [(tert-Butoxycarbonyl)amino]cyclohexyl}-7-chloro-2,4-dimethyl-1,3-benzodioxole-5-carboxylate (10 g, 18.3 mmol) was added and Stirred for 10 hours. After cooling to room temperature and filtering the insolubles, 2-propanol (100 mL) was added and the mixture was concentrated under reduced pressure until the liquid volume became 30 mL. While stirring at room temperature, ethyl acetate (120 mL) and concentrated hydrochloric acid (6.1 mL) were added to form a slurry. This was concentrated under reduced pressure to 30 mL, ethyl acetate (120 mL) was added, and then concentrated under reduced pressure to 30 mL again. After adding ethyl acetate (120 mL), the precipitated solid was collected by filtration, washed with ethyl acetate (50 mL) and dried under reduced pressure at 40°C to give 6.56 g of the title compound (yield 92.0%). Acquired.
[0091]
(Example 7) (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-N-[(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl ) Preparation of methyl]-2,4-dimethyl-1,3-benzodioxole-5-carboxamide p-toluenesulfonate
[0092]
[Chemical 19]
[0093]
Under nitrogen atmosphere, acetone (6.5 L), purified water (1.3 L), (2R)-7-chloro-2-[trans-4-(dimethylamino)cyclohexyl]-2,4- Dimethyl-1,3-benzodioxole-5-carboxylic acid monohydrochloride (650.4 g, 1.67 mol), 3-(aminomethyl)-4,6-dimethylpyridin-2(1H)-one Monohydrochloride (330.1 g, 1.75 mol) and triethylamine (337 g, 3.33 mol) were added and stirred at room temperature for 30 minutes. After that, 1-hydroxybenzotriazole monohydrate (255 g, 1.67 mol), 1-ethyl-3-(dimethylaminopropyl)carbodiimide hydrochloride (383 g, 2.00 mmol) were added, and the mixture was stirred overnight at room temperature. Stirred. After adjusting the pH to 11 with 5 mol/L sodium hydroxide, toluene (9.8 L) was added, and after stirring, the mixture was separated into an organic layer 1 and an aqueous layer. Toluene (3.3 L) was added to the aqueous layer, and after stirring, the aqueous layer was discarded, and the obtained organic layer was combined with the previous organic layer 1. The combined organic layers were concentrated under reduced pressure to 9.75 L, toluene (6.5 L) was added and washed twice with purified water (3.25 L). The resulting organic layer was concentrated under reduced pressure to 4.875 L and 2-propanol (1.625 L) was added. A solution of p-toluenesulfonic acid monohydrate (0.12 kg, 0.631 mol) dissolved in 4-methyl-2-pentanone (1.14 L) was added to the organic layer heated to 68°C. The mixture was added dropwise over 5 hours and stirred at 68°C for 30 minutes. Furthermore, a solution of p-toluenesulfonic acid monohydrate (0.215 kg, 1.13 mol) dissolved in 4-methyl-2-pentanone (2.11 L) was added dropwise over 3.5 hours, Stirred at 68° C. for 30 minutes. After that, 4-methyl-2-pentanone (6.5 L) was added dropwise over 1 hour. After cooling to room temperature, the precipitated solid was collected by filtration, washed with 4-methyl-2-pentanone (3.25 L) and dried under reduced pressure at 40°C to give 1.035 kg of the crude title compound (yield 94%). .2%).
[0094]
Under a nitrogen atmosphere, 2-propanol (6.65 L) and the obtained crude title compound (950 g) were added to the reactor and stirred. Purified water (0.23 L) was added to completely dissolve the solid at 68° C., filtered, and washed with warm 2-propanol (0.95 L). After confirming that the solid was completely dissolved at an internal temperature of 68°C, the solution was cooled to 50°C. After cooling, seed crystals* (9.5 g, 0.01 wt) were added and stirred at 50° C. overnight. tert-Butyl methyl ether (11.4 L) was added dropwise thereto in 4 portions over 30 minutes each. At that time, it was stirred for 30 minutes after each addition. After cooling to room temperature, the precipitated solid was collected by filtration, washed with a mixture of 2-propanol (0.38 L) and tert-butyl methyl ether (3.42 L), and further treated with tert-butyl methyl ether (4.75 L). ) and dried under reduced pressure at 40° C. to obtain the title compound (915.6 g, yield 96.4%).
1 H NMR (500 MHz, methanol-d 4 ): δ 1.35-1.43 (m, 2H), 1.49-1.57 (m, 2H), 1.62 (s, 3H),
1.94-2.00 (dt, J = 12.5, 3.0Hz ,1H),2.09-2.13(m,4H),2.17(s,3H),2.24(s,3H),2.35(s,3H),2.36(s,3H),2.82(s,6H),3.16- 3.22(dt,J=12.0,3.5Hz,1H),4.42(s,2H),
6.10(s,1H),6.89(s,1H),7.22-7.24(d,J=8.0Hz,2H),7.69 -7.71(dt,J=8.0,1.5 Hz,2H)
*Seed crystal preparation method
Under a nitrogen atmosphere, 2-propanol (79.0 L) and the obtained crude title compound (7.90 kg) were added to a reactor and stirred. Purified water (7.9 L) was added to completely dissolve the solid, and activated carbon (0.40 kg) was added and stirred. After filtering the activated carbon, it was washed with 2-propanol (79.0 L) and concentrated to 58 L. 2-Propanol (5 L) was added to the residue, and after heating to 64° C., tert-butyl methyl ether (19.8 L) was added, and after crystal precipitation was confirmed, tert-butyl methyl ether (75. 1 L) was added in three portions. At that time, it was stirred for 30 minutes after each addition. After cooling to room temperature, the precipitated solid was collected by filtration, washed with a mixture of 2-propanol (7.9 L) and tert-butyl methyl ether (15.8 L), and dried under reduced pressure at 40°C to obtain seed crystals. The title compound was obtained (7.08 kg, 89.6% yield).
/////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
///////Valemetostat tosilate, japan 2022, approvals 2022, Ezharmia, バレメトスタットトシル酸塩 , DS-3201, DS 3201, DS3201, DS-3201b, Orphan Drug
CN(C)[C@@H]1CC[C@H](CC1)[C@]2(C)Oc3c(C)c(cc(Cl)c3O2)C(=O)NCC4=C(C)C=C(C)NC4=O

NEW DRUG APPROVALS
ONE TIME
$10.00
Darinaparsin
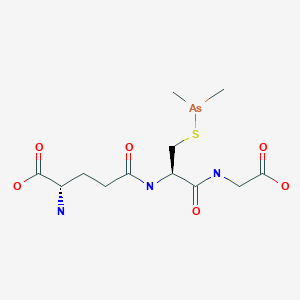
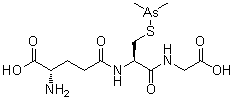
Darinaparsin
ダリナパルシン , Darvias
JAPAN 2022 APPROVED, PMDA 2022/6/20
(2S)-2-amino-5-[[(2R)-1-(carboxymethylamino)-3-dimethylarsanylsulfanyl-1-oxopropan-2-yl]amino]-5-oxopentanoic acid
Glycine, L-gamma-glutaMyl-S-(diMethylarsino)-L-cysteinyl-
| Formula | C12H22AsN3O6S |
|---|---|
| CAS | 69819-86-9 |
| Mol weight | 411.3062 |
| Efficacy | Antineoplastic |
|---|---|
| Comment | organic arsenical |
Zinapar, ZIO-101, DMAs(III)G, clarinaparsin, UNII-9XX54M675G, SP-02L
- OriginatorTexas A&M University; University of Texas M. D. Anderson Cancer Center
- DeveloperSolasia Pharma; ZIOPHARM Oncology
- ClassAmines; Antineoplastics; Arsenicals; Oligopeptides; Pentanoic acids; Small molecules; Sulfides
- Mechanism of ActionApoptosis stimulants; Cell cycle inhibitors; Reactive oxygen species stimulants
- Orphan Drug StatusYes – Peripheral T-cell lymphoma
- PreregistrationPeripheral T-cell lymphoma
- DiscontinuedLiver cancer; Lymphoma; Multiple myeloma; Non-Hodgkin’s lymphoma; Solid tumours
- 28 Mar 2022No recent reports of development identified for phase-I development in Peripheral-T-cell-lymphoma in China (IV, Injection)
- 26 Jan 2022ZIOPHARM Oncology is now called Alaunos Therapeutics
- 11 Dec 2021Safety and efficacy data from a phase II trial in Peripheral T-cell lymphoma presented at the 63rd American Society of Hematology Annual Meeting and Exposition (ASH-2021)
Darinaparsin is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects. Compared to inorganic arsenic compounds such as arsenic trioxide (As2O3), darinaparsin appears to exhibit a wide therapeutic window.
Darinaparsin, also know as ZIO-101 and SP-02, is a small-molecule organic arsenical with potential antineoplastic activity. Although the exact mechanism of action is unclear, darinaparsin, a highly toxic metabolic intermediate of inorganic arsenicals (iAs) that occurs in vivo, appears to generate volatile cytotoxic arsenic compounds when glutathione (GSH) concentrations are low. The arsenic compounds generated from darinaparsin disrupt mitochondrial bioenergetics, producing reactive oxygen species (ROS) and inducing ROS-mediated tumor cell apoptosis; in addition, this agent or its byproducts may initiate cell death by interrupting the G2/M phase of the cell cycle and may exhibit antiangiogenic effects.
Darinaparsin is an organic arsenical composed of dimethylated arsenic linked to glutathione, and is being investigated for antitumor properties in vitro and in vivo. While other arsenicals, including arsenic trioxide, have been used clinically, none have shown significant activity in malignancies outside of acute promyelocytic leukemia. Darinaparsin has significant activity in a broad spectrum of hematologic and solid tumors in preclinical models. Here, we review the literature describing the signaling pathways and mechanisms of action of darinaparsin and compare them to mechanisms of cell death induced by arsenic trioxide. Darinaparsin has overlapping, but distinct, signaling mechanisms. We also review the current results of clinical trials with darinaparsin (both intravenous and oral formulations) that demonstrate significant antitumor activity.
PAPER
Biochemical Pharmacology (Amsterdam, Netherlands), 126, 79-86; 2017



PATENT
WO 2015085208
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015085208
Preparation of Darinaparsin
[0071] Sterile water (15.5 L) and ethyl alcohol (200 proof, 15.5 L) were charged in a reaction flask prior to the addition of L-glutathione (3.10 kg). While being stirred, the reaction mixture was cooled to 0-5 °C prior to the addition of triethylamine (1.71 L). Stirring was continued until most of the solids were dissolved and the solution was filtered. After filtration, the reaction mixture was cooled to 0-5 °C prior to the addition of chlorodimethylarsine (1.89 kg) over 115 minutes while maintaining the temperature at 0-5 °C. Stirring continued at 0-5 °C for 4 hours before acetone (30.6 L) was added over 54 minutes while maintaining the temperature at 0-5 °C. The suspension was stored at 0-5°C overnight prior to filtration. The solid was collected in a filter funnel, washed successively with ethyl alcohol (200 proof, 13.5 L) and acetone (13.5 L) and dried in suction for 23 minutes. A second similar run was performed and the collected solids from both runs were combined. Ethyl alcohol (200 proof, 124 L) and the combined solids (11.08 kg) were charged in a vessel. The slurry was stirred at ambient temperature for 2 hours before filtration, washing successively with ethyl alcohol (200 proof, 27 L) and acetone (27 L) and dried in suction for 60 minutes. The resulting solid was transferred to drying trays and dried in a vacuum oven at ambient temperature for 66 hours to provide darinaparsin as a solid with the differential scanning calorimetry (DSC) thermogram of Figure 1, with an extrapolated onset temperature at about 191.36° C and a peak temperature at about 195.65° C.
PATENT
WO 2010021928
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109°C was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
SGLU-1: Glutathione (14.0 g, 45.6 mmol) was stirred rapidly in glyme while dimethylchoroarsine (6.5 g, 45.6 mmol) was added dropwise. Pyridine (6.9 g, 91.2 mmol) was then added to the slurry and the mixture was subsequently heated to reflux. The heat was removed immediately and the mixture stirred at room temperature for 4 h. Isolation of the resultant insoluble solid and recrystallization from ethanol afforded 4 as the pyridine hydrochloride complex (75% yield). mp 115-118°C; NMR (D20) δ1.35 (s, 6H), 1.9-4.1 (m’s, 10H), 7.8-9.0 (m, 5H); mass spectrum (m/e) 140, 125, 110, 105, 79, 52, 45, 36.
PATENT
WO 2009075870
Step 1
Example 1. Preparation of Dimethylchloroarsine (DMCA). A 3-neck round-bottom flask (500 mL) equipped with mechanical stirrer, inlet for nitrogen, thermometer, and an ice bath was charged with cacodylic acid (33 g, 0.23 mol) and cone. hydrochloric acid (67 mL). In a separate flask, a solution of SnCl2·2H2O (54 g, 0.239 mol) in cone. hydrochloric acid (10 mL) was prepared. The SnCl2·2 H2O solution was added to the cacodylic acid in HCl solution under nitrogen while maintaining the temperature between 5 °C and 10 °C. After the addition was complete, the ice bath was removed and the reaction mixture was stirred at ambient temperature for 1 h. The reaction mixture was transferred to a separatory funnel and the upper layer (organic) collected. The bottom layer was extracted with dichloromethane (DCM) (2 × 25 mL). The combined organic extract was washed with 1 N HCl (2 × 10 mL) and water (2 × 20 mL). The organic extract was dried over MgSO4 and DCM was removed by rotary evaporation (bath temperature 80 °C, under nitrogen, atmospheric pressure). The residue was further distilled under nitrogen. Two tractions of DMCA were collected. The first fraction contained some DCM and the second fraction was of suitable quality (8.5 g, 26% yield). The GC analysis confirmed the identity and purity of the product.
Step 2
Example 3. Preparation of S-Dimethylarsinoglutathione (SGLU-1). In a 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel and thermometer under an inert atmosphere was prepared a suspension of glutathione (114.5 g, 0.37 mol) in a 1:1 (v/v) mixture of water/ethanol (1140 mL) and cooled to below 5 °C. The mixture was treated slowly (over 15 min) with triethylamine (63.6 mL, 0.46 mol) while maintaining the temperature below 20 °C. The mixture was cooled to 4 °C and stirred for 15 min and then the traces of undissolved material removed by filtration. The filtrate was transferred in a clean 3 L three-neck flask equipped with a mechanic stirrer, dropping funnel, nitrogen inlet, and thermometer and DMCA (70 g, 0.49 mol) (lot # 543-07-01-44) was added slowly while maintaining the temperature at 3-4°C. The reaction mixture was stirred at 1-4°C for 4 h, and acetone (1.2 L) was added over a period of 1 h. The mixture was stirred for 90 min between 2 and 3°C and the resulting solid was isolated by filtration. The product was washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and the wet solids were suspended in ethanol 200 Proof (2000 mL). The product was isolated by filtration, washed with ethanol (2 × 250 mL) and acetone (2 × 250 mL) and dried in vacuum for 2 days at RT to give 115 g (75%) of SGLU-1, HPLC purity > 99.5% (in process testing).
PATENT
WO 2007027344
Example 2 Preparation of S-Dimethylarsinoglutathione A 5 L, three necked round bottom flask was equipped with a mechanical stirrer assembly, thermometer, addition funnel, nitrogen inlet, and a drying tube was placed in a cooling bath. A polyethylene crock was charged with glutathione-reduced (200 g) and deionized water (2 L) and stirred under a nitrogen atmosphere to dissolve all solids. The mixture was filtered to remove any insoluble material and the filtrate was transferred to the 5 L flask. While stirring, ethanol, 200 proof (2 L) was added and the clear solution was cooled to 0-5° C. using an ice/methanol bath. Pyridine (120 g) was added followed by a dropwise addition of Me2AsCl (120 g) over a minimum of 1 hour. The reaction mixture was stirred at 0-5° C. for a minimum of 2 hours prior to removal of the cooling bath and allowing the mixture to warm to room temperature under a nitrogen atmosphere with stirring. The reaction mixture was stirred overnight (>15 hrs) at room temperature under a nitrogen atmosphere at which time a white solid may precipitate. The reaction mixture was concentrated to a slurry (liquid and solid) at 35-45° C. using oil pump vacuum to provide a white solid residue. As much water as possible is removed, followed by two coevaporations with ethanol to azeotrope the last traces of water. The white solid residue was slurried in ethanol, 200 pf. (5 L) under a nitrogen atmosphere at room temperature overnight. The white solid was filtered and washed with ethanol, 200 pf. (2×500 mL) followed by acetone, ACS (2×500 mL). The resulting solid was transferred to drying trays and vacuum oven dried overnight at 25-35° C. using oil pump vacuum to provide pyridinium hydrochloride-free S-dimethylarsinoglutathione as a white solid. melting point of 189-190° C.
PATENT
WO 20060128682
Step 1
Dimethylchloroarsine. Dimethylarsinic acid, (CH3)2As(O)OH was supplied by the Luxembourg Chemical Co., Tel Aviv, Israel. The product was accompanied by a statement of its purity and was supplied as 99.7% pure. The dimethylarsinic acid was dissolved in water-hydrochloric acid to pH 3. A stream of sulfur dioxide was passed through this solution for about one hour. Dimethylchloroarsine separated as a heavy, colorless oil. The two liquid phases, water/(CH3)2AsCl were separated using a separatory funnel. The chlorodimethylarsine was extracted into diethylether and the ether solution was dried over anhydrous sodium sulfate. The dried solution was transferred to a distillation flask which was heated slowly to evaporate the ether. The remaining liquid, dimethylchloroarsine was purified by distillation. The fraction boiling at 106-109° C. was collected. The product, a colorless oil. 1H NMR resonance at 1.65 ppm.
Step 2
Pyridine Hydrochloride Free Synthesis of S-Dimethylarsinoglutathione (GLU) Dimethylarsinoglutathione is made using an adapted of Chen (Chen, G. C., et al. Carbohydrate Res. (1976) 50: 53-62) the contents of which are hereby incorporated by reference in their entirety. Briefly, dithiobis(dimethylarsinoglutamine) is dissolved in dichloromethane under nitrogen. Tetramethyldiarsine is added dropwise to the solution and the reaction is stirred overnight at room temperature under nitrogen and then exposed to air for 1 h. The mixture is then evaporated to dryness and the residue is washed with water and dried to give a crude solid that is recrystallized from methanol to give S-dimethylarsinoglutathione.
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

NEW DRUG APPROVALS
ONE TIME TO PAY BLOG SUBSCRIPTIONS
$10.00
Solasia Announces Submission of New Drug Application for Anti-cancer Drug DARINAPARSIN for Peripheral T-Cell Lymphoma in Japan
Solasia Pharma K.K. (TSE: 4597, Headquarters: Tokyo, Japan, President & CEO: Yoshihiro Arai, hereinafter “Solasia”) today announced submission of a New Drug Application (NDA) for its new anti-cancer drug darinaparsin (generic name, development code: SP-02) as a treatment for relapsed or refractory peripheral T-cell lymphoma to the Ministry of Health, Labour and Welfare (MHLW). Based on positive results of R&D on darinaparsin, centered primarily on the results of the Asian Multinational Phase 2 Study (study results released in June 2020), Solasia filed an NDA for the drug with the regulatory authority in Japan ahead of anywhere else in the world.
Solasia expects to obtain regulatory approval in 2022 and to also launch in the same year. If approved and launched, darinaparsin would be the third drug Solasia successfully developed and brought to market since its founding and is expected to contribute to the treatment of PTCL.
Mr. Yoshihiro Arai, President and CEO of Solasia, commented as follows:
“No standard treatment has been established for relapsed or refractory PTCL as of yet. I firmly believe that darinaparsin, with its novel mechanism of action that differs from those of already approved drugs, will contribute to patients and healthcare providers at clinical sites as a new treatment option for relapsed or refractory PTCL. Since founding, Solasia has conducted R&D on five pipeline drugs. Of the five, we have successfully developed and brought to market two drugs, i.e., began providing them to patients, and today, we submitted an NDA for our first anti-cancer drug. Under our mission to provide patients with ‘Better Medicine for a Brighter Tomorrow’, we will continue aiming to contribute to patients’ treatment and enhanced quality of life. ”
About darinaparsin (SP-02)
Darinaparsin, an organoarsenic compound with anticancer activity, is a novel mitochondrial-targeted agent being developed for the treatment of various hematologic and solid tumors. The proposed mechanism of action of the drug involves the disruption of mitochondrial function, increased production of reactive oxygen species, and modulation of intracellular signal transduction pathways. Darinaparsin is believed to exert anticancer effect by inducing cell cycle arrest and apoptosis. Darinaparsin has been granted orphan drug designation in the US and EU.
For more information, please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Asian Multinational Phase 2 Study
The Asian Multinational Phase 2 Study was a multinational, multicenter, single-arm, open-label, non-randomized study to evaluate the efficacy and safety of darinaparsin monotherapy in patients with relapsed or refractory PTCL conducted in Japan, Korea, Taiwan, and Hong Kong. (CT.gov Identifier: NCT02653976).
Solasia plans to present the results of the study at an international academic conference to be held in the near future.
About peripheral T-cell lymphoma (PTCL)
Please visit at https://solasia.co.jp/en/pipeline/sp-02.html
About Solasia
Please visit at https://solasia.co.jp/en/
/////////////Darinaparsin, Darvias, JAPAN 2022, APPROVALS 2022, PMDA, ダリナパルシン , Zinapar, ZIO-101, DMAs(III)G, clarinaparsin, UNII-9XX54M675G, SP-02L, Orphan Drug
C[As](C)SCC(C(=O)NCC(=O)O)NC(=O)CCC(C(=O)O)N
Pimitespib
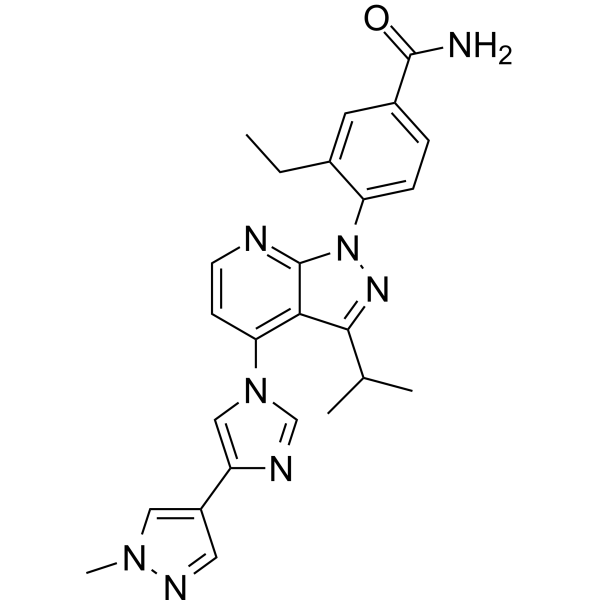
![Benzamide, 3-ethyl-4-[3-(1-methylethyl)-4-[4-(1-methyl-1H-pyrazol-4-yl)-1H-imidazol-1-yl]-1H-pyrazolo[3,4-b]pyridin-1-yl]-.png](https://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=67501411&t=l)
Pimitespib
TAS 116
CAS 1260533-36-5
Antineoplastic, Hsp 90 inhibitor
3-ethyl-4-[4-[4-(1-methylpyrazol-4-yl)imidazol-1-yl]-3-propan-2-ylpyrazolo[3,4-b]pyridin-1-yl]benzamide
Pimitespib (TAS-116) is an oral bioavailable, ATP-competitive, highly specific HSP90α/HSP90β inhibitor (Kis of 34.7 nM and 21.3 nM, respectively) without inhibiting other HSP90 family proteins such as GRP94. Pimitespib demonstrates less ocular toxicity.
| Formula | C25H26N8O |
|---|---|
| CAS | 1260533-36-5 |
| Mol weight | 454.5269 |
JAPAN APPROVED 2022/6/20, ピミテスピブ
| Jeselhy |
Taiho. originator
Pimitespib is a specific inhibitor of heat shock protein 90 (Hsp90) subtypes alpha and beta, with potential antineoplastic and chemo/radiosensitizing activities. Upon oral administration, pimitespib specifically binds to and inhibits the activity of Hsp90 alpha and beta; this results in the proteasomal degradation of oncogenic client proteins, which inhibits client protein dependent-signaling, induces apoptosis, and inhibits the proliferation of cells overexpressing HSP90alpha/beta. Hsp90, a family of molecular chaperone proteins that are upregulated in a variety of tumor cells, plays a key role in the conformational maturation, stability, and function of “client” proteins within the cell,; many of which are involved in signal transduction, cell cycle regulation and apoptosis, including kinases, cell-cycle regulators, transcription factors and hormone receptors. As TAS-116 selectively inhibits cytosolic HSP90alpha and beta only and does not inhibit HSP90 paralogs, such as endoplasmic reticulum GRP94 or mitochondrial TRAP1, this agent may have less off-target toxicity as compared to non-selective HSP90 inhibitors.
Patent
WO2011004610
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011004610
PATENT
CN108623496
| 3-Ethyl-4-fluorobenzonitrile is an important intermediate for the preparation of a variety of new drugs under development, such as TAS-116, a Phase II clinical drug of Taiho Pharmaceuticals for the treatment of gastrointestinal stromal tumors. |
| |
| Patent WO2005105760 discloses its preparation method. In the method, tetrakis(triphenylphosphine) palladium is used as a catalyst, and 3-bromo-4-fluorobenzonitrile is coupled with tetraethyl tin in a solvent hexamethylphosphoramide for a heating reaction for 15 hours to obtain 3 -Ethyl-4-fluorobenzonitrile. The method uses highly toxic tetraethyl tin, which brings great harm to operators and the environment, and is difficult to carry out industrial production. Meanwhile, the product 3-ethyl-4-fluorobenzonitrile obtained by the preparation method is an oily substance, which is purified by column chromatography with complicated operation, which is unfavorable for industrial production, and the specific purity of the product is not described. |
| |
| Therefore, looking for a new method for preparing 3-ethyl-4-fluorobenzonitrile with cheap and easy-to-obtain raw materials, safe and simple operation, high product purity and low cost suitable for industrial production, which will speed up the research process of related new drugs under development. , it is of great significance to reduce the production cost of related new drugs. |
| Example 1 3-Bromo-4-fluorobenzonitrile |
| |
| 3-Bromo-4-fluorobenzaldehyde (250g, 1.23mol) was dissolved in acetonitrile (1.5L), then hydroxylamine sulfonic acid (67g, 1.48mol) was added, and the reaction was refluxed for 4h. TLC showed that the conversion of the raw materials was complete, and the reaction solution was concentrated. To a small volume, add water (2L) and stir for 30min, cool to 5-10°C and continue stirring for 10min, filter, dissolve the filter cake with methyl tert-butyl ether (1.2L), wash twice with 500ml of water, saturated with 200ml Washed with sodium bicarbonate solution, dried over anhydrous sodium sulfate, filtered, the filtrate was adsorbed with activated carbon (10g), filtered, concentrated under reduced pressure to remove the solvent, added n-heptane (250ml), cooled and stirred in an ice-salt bath for 1h, filtered, reduced Press drying to give 3-bromo-4-fluorobenzonitrile (217 g, 88% yield). 1 H NMR (CDCl 3 ,400MHz):δ7.91(m,1H),7.63(m,1H),7.24(m,1H)。 |
| Example 2 3-Bromo-4-fluorobenzonitrile |
| |
| Add tetrahydrofuran (100ml) to a 250ml reaction flask, add 3-bromo-4-fluorobenzaldehyde (10g, 49.2mmol) and ammonia (40ml) under stirring, add elemental iodine (25g, 98.5mmol) in batches under cooling to 5°C ), then raised to ambient temperature and reacted for 2 to 3 hours, the reaction was completed, the reaction solution was poured into a 10% aqueous solution of sodium sulfite (200g), extracted twice with methyl tert-butyl ether (100ml), dried over anhydrous sodium sulfate , concentrated under reduced pressure to remove the solvent, added n-heptane (20 ml), cooled to 0-10 °C and stirred for 1 h, filtered, and dried under reduced pressure to obtain 3-bromo-4-fluorobenzonitrile (9.6 g, yield: 97.5 %). The NMR spectrum of this compound was determined and was identical to the product of Example 1. |
| Example 3 3-ethyl-4-fluorobenzonitrile |
| |
| 3-Bromo-4-fluorobenzonitrile (200 g, 1 mol) and [1,1-bis(diphenylphosphino)ferrocene]palladium(II) chloride dichloromethane complex (4.08 g, 5mmol) was dissolved in THF (1.2L), 1.0M/L diethylzinc n-hexane solution (600mL, 0.6mol) was added at 40-50°C, and the temperature was raised to 50-60°C for 4-5h. TLC showed The raw materials reacted completely. After the reaction solution was cooled to room temperature, it was added to 5% dilute hydrochloric acid (1 L), the layers were separated, the organic layer was washed twice with 500 ml of water, and then concentrated under reduced pressure to remove the solvent. Then n-hexane (600mL) and activated carbon (20g) were added, refluxed for 0.5h, cooled to room temperature, filtered, then added activated carbon (10g) to the filtrate, refluxed for 0.5h, cooled to room temperature, filtered, and cooled to -50°C to -60°C and filtered, and the filter cake was dried under reduced pressure at 10-20°C to obtain 3-ethyl-4-fluorobenzonitrile (112 g, yield: 75%) as an off-white solid, melting point 23.1-27.4°C. 1 H NMR (CDCl 3 , 400MHz): δ 7.50 (m, 2H), 7.09 (m, 1H), 2.69 (q, J=7.6Hz, 2H), 1.24 (t, 3H, J=7.6Hz), HPLC purity 99.6%. |
| HPLC assay conditions: |
| Chromatographic UV detector: DAD |
| Chromatography pump: 1100 quaternary pump |
| Chromatographic column: Agilent (USA) ZORBAX SB-C184.6×150mm, 5μm PN883975-902 Chromatographic conditions: |
| Mobile Phase A: Water |
| Mobile Phase B: Acetonitrile |
| |
| Injection volume: 5 μL, flow rate: 1.0 mL/min, column temperature: room temperature, detection wavelength: 210 nm. |


Acylation of 2-fluoro-4-iodopyridine with isobutyric anhydride in presence of BuLi and DIEA in THF at -78 °C gives 1-(2-fluoro-4-iodo-3-pyridinyl)-2-methylpropan-1-one ,
This upon cyclization using hydrazine hydrate at 65 °C gives 4-iodo-3-isopropylpyrazolo[3,4-b]pyridine.
N-Protection of intermediate with PMB-Cl in the presence of base NaH in solvent DMF at 0 °C affords 4-iodo-3-isopropyl-1-(4-methoxybenzyl)pyrazolo[3,4-b]pyridine,
This is coupled with 4-(4-imidazolyl)-1-methylpyrazole in the presence of Cu2O, 4,7-dimethoxy-1,10-phenanthroline, Cs2CO3 and PEG-diamine in solvent NMP or DMSO at 130 °C to furnish 4-[4-(4-pyrazolyl)-imidazol-1-yl]pyrazolo[3,4-b]pyridine derivative .
N-Deprotection of PMB-protected pyrazolo[3,4-b]pyridine derivative by using TFA and anisole gives free pyrazolo[3,4-b]pyridine ,
This on condensation with 3-ethyl-4-fluorobenzonitrile in the presence of Cs2CO3 in DMF at 95 °C yields 4-(pyrazolo[3,4-b]pyridin-1-yl)benzonitrile .
Finally, partial hydrolysis of nitrile by means of aqueous NaOH and H2O2 in DMSO/EtOH gives the Pimitespib TAS-116 .
CLIP
https://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.8b01085
J. Med. Chem.2019, 62, 2, 531–551
Publication Date:December 7, 2018
https://doi.org/10.1021/acs.jmedchem.8b0108

The molecular chaperone heat shock protein 90 (HSP90) is a promising target for cancer therapy, as it assists in the stabilization of cancer-related proteins, promoting cancer cell growth, and survival. A novel series of HSP90 inhibitors were discovered by structure–activity relationship (SAR)-based optimization of an initial hit compound 11a having a 4-(4-(quinolin-3-yl)-1H-indol-1-yl)benzamide structure. The pyrazolo[3,4-b]pyridine derivative, 16e (TAS-116), is a selective inhibitor of HSP90α and HSP90β among the HSP90 family proteins and exhibits oral availability in mice. The X-ray cocrystal structure of the 16e analogue 16d demonstrated a unique binding mode at the N-terminal ATP binding site. Oral administration of 16e demonstrated potent antitumor effects in an NCI-H1975 xenograft mouse model without significant body weight loss.
3-Ethyl-4-(3-Isopropyl-4-(4-(1-methyl-1H-Pyrazol-4-yl)-1H-Imidazol-1-yl)-1H-Pyrazolo[3,4-b]pyridin-1-yl)benzamide (16e). Yield 64% (2 steps), white powder. UPLC−MS (ESI) m/z: 454.8 [M + H]+ , tR = 1.19 min. UPLC purity 99.65%. 1 H NMR (400 MHz, CDCl3): δ 1.14 (t, J = 7.5 Hz, 3H), 1.25 (d, J = 7.0 Hz, 6H), 2.62 (q, J = 7.3 Hz, 2H), 3.18 (spt, J = 6.8 Hz, 1H), 3.98 (s, 3H), 5.88 (br s,1H), 6.22 (br s, 1H), 7.13 (d, J = 5.1 Hz, 1H), 7.39 (d, J = 1.1 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 7.78−7.81 (m, 3H), 7.86 (d, J = 1.5 Hz, 1H), 7.96 (d, J = 1.8 Hz, 1H), 8.59 (d, J = 4.7 Hz, 1H). HRMS: calcd for C25H26N8O, 455.2308 [M + H]+ ; found, 455.2311.
PAPER
Journal of Medicinal Chemistry (2021), 64(5), 2669-2677.
PATENT
WO 2016181990
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016181990
Compound 1 in the present invention is 3-ethyl-4- {3-isopropyl-4- (4- (1-methyl-1H-pyrazol-4-yl) -1H-imidazole-1-yl) -1H-. Pyrazolo [3,4-b] pyridin-1-yl} benzamide (formula below). Compound 1 is known to have HSP90 inhibitory activity and exhibit excellent antitumor activity. Compound 1 can be synthesized based on the production methods described in Patent Documents 1 and 2.
[0013]
[hua 1]
Patent Document 1: International Publication No. 2012/093708
Patent Document 2: International Publication No. 2011/004610
Comparative Example 1 3-Ethyl-4- {3-isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazole-1-yl) -1H-pyrazolo [3, 4-b] Pyridine-1-yl} Synthesis of type I crystals of benzamide
3-Ethyl-4 obtained according to the production method described in International Publication No. 2012/093708 and International Publication No. 2011/004610. -{3-Isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazole-1-yl) -1H-pyrazolo [3,4-b] pyridin-1- A white solid (3.58 g) of yl} benzamide was added to ethanol (7.84 mL) and stirred at room temperature for 2 hours. After sampling, it was washed with ethanol (7.84 mL) and dried under reduced pressure at 70 to 80 ° C. for 20 hours to obtain type I crystals (yield: 2.40 g, yield: 61.2%, purity: 98.21%). rice field.
Further, as shown in FIG. 1, the type I crystal has a diffraction angle (2θ) of 8.1 °, 10.9 °, 12.1 °, 14.0 °, and 14.9 in the powder X-ray diffraction spectrum. °, 16.2 °, 17.7 °, 20.2 °, 21.0 °, 21.5 °, 22.6 °, 24.3 °, 25.4 ° 26.4 °, 27.0 ° , 28.3 °, 30.2 °, 30.9 °, 31.5 °, 32.7 °, 34.7 °, 35.4 ° and 36.6 ° showed characteristic peaks.
[0032]
1H-NMR (DMSO-d 6):δppm 9.35 (1H,d,J=4.88Hz), 8.93 (1H,d,J=1.22Hz), 8.84 (1H,brs), 8.72 (1H,d,J=1.95Hz), 8.70 (1H,s) ,8.63 (1H,d,J=1.22Hz), 8.60 (1H,dd,J=8.29,1.95Hz), 8.46 (1H,s) ,8.25 (1H,d,J=8.29Hz), 8.22 (1H,brs), 8.12 (1H,d,J=4.88Hz), 4.59 (3H,s) ,3.95 (1H,tt,J=6.83,6.83Hz), 3.21 (2H,q,J=7.56Hz), 1.83(6H,d,J=6.83Hz), 1.75 (3H,t,J=7.56Hz):LRMS(ESI)m/z 455[M+H]
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2015050235
Synthesis of Test Compound The
following synthesis example compounds (Synthesis Examples 1 to 3) were synthesized according to the method described in WO2011 / 004610.
[0361]
Synthesis Example 1: 4- {3-Isopropyl-4- (4- (1-methyl-1H-pyrazole-4-yl) -1H-imidazol-1-yl) -1H-pyrazolo [3,4-b] pyridine -1-yl} -3-methylbenzamide
[0362]
[Changing 22]
PATENT
WO 2011004610
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2011004610
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

NEW DRUG APPROVALS
ONE TIME TO PAY BLOG SUBSCRIPTIONS
$10.00
Pimitespib
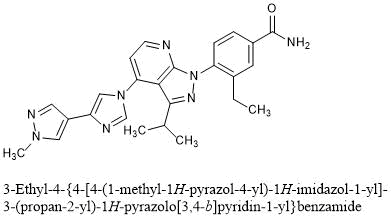
3-Ethyl-4-{4-[4-(1-methyl-1H-pyrazol-4-yl)-1H-imidazol-1-yl]-3-(propan-2-yl)-1H-pyrazolo[3,4-b]pyridin-1-yl}benzamide
C25H26N8O : 454.53
[1260533-36-5]
//////////Pimitespib, ピミテスピブ, JAPAN 2022, APPROVALS 2022, TAS 116, Jeselhy
O=C(N)C1=CC=C(N2N=C(C(C)C)C3=C(N4C=C(C5=CN(C)N=C5)N=C4)C=CN=C32)C(CC)=C1
Olipudase alfa
| HPLSPQGHPA RLHRIVPRLR DVFGWGNLTC PICKGLFTAI NLGLKKEPNV ARVGSVAIKL CNLLKIAPPA VCQSIVHLFE DDMVEVWRRS VLSPSEACGL LLGSTCGHWD IFSSWNISLP TVPKPPPKPP SPPAPGAPVS RILFLTDLHW DHDYLEGTDP DCADPLCCRR GSGLPPASRP GAGYWGEYSK CDLPLRTLES LLSGLGPAGP FDMVYWTGDI PAHDVWHQTR QDQLRALTTV TALVRKFLGP VPVYPAVGNH ESTPVNSFPP PFIEGNHSSR WLYEAMAKAW EPWLPAEALR TLRIGGFYAL SPYPGLRLIS LNMNFCSREN FWLLINSTDP AGQLQWLVGE LQAAEDRGDK VHIIGHIPPG HCLKSWSWNY YRIVARYENT LAAQFFGHTH VDEFEVFYDE ETLSRPLAVA FLAPSATTYI GLNPGYRVYQ IDGNYSGSSH VVLDHETYIL NLTQANIPGA IPHWQLLYRA RETYGLPNTL PTAWHNLVYR MRGDMQLFQT FWFLYHKGHP PSEPCGTPCR LATLCAQLSA RADSPALCRH LMPDGSLPEA QSLWPRPLFC (Disulfide bridge: 43-119, 46-111, 74-85, 175-180, 181-204, 339-385, 538-542, 548-561) |
Olipudase alfa
Xenpozyme, Japan 2022, APPROVALS 2022, 2022/3/28
PEPTIDE, オリプダーゼアルファ (遺伝子組換え)
Alternative Names: Acid sphingomyelinase Niemann Pick disease type B – Sanofi; Acid-sphingomyelinase – Sanofi; GZ-402665; Recombinant human acid sphingomyelinase – Sanofi; rhASM – Sanofi; Sphingomyelinase-C (synthetic human) – Sanofi; Synthetic human sphingomyelinase-C – Sanofi; Xenpozyme
| Formula | C2900H4373N783O791S24 |
|---|---|
| CAS | 927883-84-9 |
| Mol weight | 63631.0831 |
| Efficacy | Lysosomal storage disease treatment, Enzyme replacement (acid sphingomyelinase) |
|---|---|
| Comment | Enzyme replacement therapy product Treatment of Niemann-Pick disease type A/B |
- OriginatorGenzyme Corporation
- DeveloperSanofi
- ClassRecombinant proteins; Sphingomyelin phosphodiesterases
- Mechanism of ActionSphingomyelin-phosphodiesterase replacements
- Orphan Drug StatusYes – Niemann-Pick diseases
- RegisteredNiemann-Pick diseases
- 28 Mar 2022Registered for Niemann-Pick diseases (In adolescents, In children, In adults) in Japan (IV) – First global approval
- 09 Feb 2022FDA assigns PDUFA action date of (03/07/2022) for Olipudase alfa (In children, In adults) for Niemann-Pick diseases
- 09 Feb 2022Adverse e
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
CLIP
Olipudase Alfa Improves Lung Function, Spleen Volume in ASMD
Olipudase Alfa Improves Lung Function, Spleen Volume in ASMD
Olipudase alfa was associated with significant improvements in clinically relevant disease end points among patients with chronic visceral acid sphingomyelinase (ASM) deficiency (ASMD), according to results from the phase 2/3 ASCEND trial presented at the 17th Annual WORLDSymposium.
ASMD is a rare, debilitating lysosomal storage disease characterized by a deficiency of the enzyme acid sphingomyelinase, which results in the accumulation of sphingomyelin in various tissues of the body. Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM.
The multicenter, randomized, double-blind, placebo-controlled ASCEND trial evaluated the efficacy and safety of olipudase alfa in 36 adults with chronic visceral ASMD. Patients were randomly assigned 1:1 to receive olipudase alfa 3mg/kg intravenously every 2 weeks or placebo for 52 weeks. The coprimary end points were the percent change in spleen volume and percent-predicted diffusing capacity of the lung for carbon monoxide (DLCO).
At week 52, treatment with olipudase alfa resulted in a 39.45% reduction in spleen volume, compared with a 0.5% increase for placebo (P <.0001). A decrease in spleen volume of at least 30% was observed in 17 patients (94%) treated with olipudase afla compared with no patients treated with placebo. Additionally, olipudase alfa significantly improved lung function by 22% from baseline compared with 3% for patients receiving placebo (P =.0004), as measured by percent predicted DLCO.
Olipudase alfa also met key secondary end points including a 31.7% reduction in liver volume (vs a 1.4% reduction for placebo; P <.0001) and a 16.8% improvement in mean platelet counts (vs 2.5% with placebo; P =.019) at week 52. Significant improvements in HDL, LDL, AST, ALT, chitotriosidase (54% vs 12% with placebo; P =.0003), and lyso-sphingomyelin (78% vs 6% with placebo) were also observed in the olipudase alfa group at week 52.
With regard to Splenomegaly Related Score, a patient-reported outcome measurement that evaluates patient symptoms associated with an enlarged spleen, findings showed no meaningful difference between olipudase alfa and placebo (-8 point vs -9.3 points, respectively).
As for safety, olipudase alfa was well tolerated with most adverse events being mild to moderate in severity. There were no treatment-related serious adverse events and no adverse event-related discontinuations.
Disclosure: Some authors have declared affiliations with or received funding from the pharmaceutical industry. Please refer to the original study for a full list of disclosures.
Reference
Wasserstein M, Arash-Kaps L, Barbato A, et al. Adults with chronic acid sphingomyelinase deficiency show significant visceral, pulmonary, and hematologic improvements after enzyme replacement therapy with olipudase-alfa: 1-year results of the ASCEND placebo-controlled trial. Presented at: 17th Annual WORLDSymposium; February 8-12, 2021. Abstract 265.
CLIP
https://www.sanofi.com/en/media-room/press-releases/2021/2021-12-06-14-00-00-2346501
EMA accepts regulatory submission for olipudase alfa, the first potential therapy for ASMD
- Olipudase alfa has been granted PRIority MEdicines (PRIME) designation in Europe, Breakthrough Therapy designation in the United States, and SAKIGAKE designation in Japan
- European regulatory decision anticipated second half of 2022
DECEMBER 6, 2021
The European Medicines Agency (EMA) has accepted for review under an accelerated assessment procedure the Marketing Authorization Application (MAA) for olipudase alfa, Sanofi’s investigational enzyme replacement therapy which is being evaluated for the treatment of acid sphingomyelinase deficiency (ASMD). Historically referred to as Niemann-Pick disease (NPD) type A and type B, ASMD is a rare, progressive, and potentially life-threatening disease for which no treatments are currently approved. The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan. If approved, olipudase alfa will become the first and only therapy for the treatment of ASMD.
“Today’s milestone has been decades in the making and our gratitude goes to the ASMD community who has stood by us with endless patience while olipudase alfa advanced through clinical development,” said Alaa Hamed, MD, MPH, MBA, Global Head of Medical Affairs, Rare Diseases, Sanofi. “Olipudase alfa represents the kind of potentially life-changing innovation that is possible when industry, medical professionals and the patient community work together toward a common goal.”
The MAA is based on positive results from two separate clinical trials (ASCEND and ASCEND-Peds) evaluating olipudase alfa in adult and pediatric patients with non-central nervous system (CNS) manifestations of ASMD type A/B and ASMD type B.
Olipudase alfa has received special designations from regulatory agencies worldwide, recognizing the innovation potential of the investigational therapy.
“Scientific innovation is the greatest source of hope for people living with diseases like ASMD where there are no approved treatments and is a critical component for ensuring a viable healthcare ecosystem,” said Bill Sibold, Executive Vice President of Sanofi Genzyme. “At Sanofi, we have a long history of pioneering scientific innovation, and we remain committed to finding solutions to address unmet medical needs, including those of the rare disease community.”
The EMA awarded olipudase alfa the PRIority MEdicines designation, also known as PRIME, intended to aid and expedite the regulatory process for investigational medicines that may offer a major therapeutic advantage over existing treatments, or benefit patients without treatment options.
The U.S. Food and Drug Administration (FDA) has granted Breakthrough Therapy designation to olipudase alfa. This designation is intended to expedite the development and review of drugs intended to treat serious or life-threatening diseases and conditions. The criteria for granting Breakthrough Therapy designation include preliminary clinical evidence indicating that the molecule may demonstrate substantial improvement on a clinically significant endpoint over available therapies.
In Japan, olipudase alfa was awarded the SAKIGAKE designation, which is intended to promote research and development in Japan for innovative new medical products that satisfy certain criteria, such as the severity of the intended indication. In September, Sanofi filed the J-NDA submission for olipudase alfa.
About ASMD
ASMD results from a deficient activity of the enzyme acid sphingomyelinase (ASM), which is found in special compartments within cells called lysosomes and is required to breakdown lipids called sphingomyelin. If ASM is absent or not functioning as it should, sphingomyelin cannot be metabolized properly and accumulates within cells, eventually causing cell death and the malfunction of major organ systems. The deficiency of the lysosomal enzyme ASM is due to disease-causing variants in the sphingomyelin phosphodiesterase 1 gene (SMPD1). The estimated prevalence of ASMD is approximately 2,000 patients in the U.S., Europe (EU5 Countries) and Japan.
ASMD represents a spectrum of disease caused by the same enzymatic deficiency, with two types that may represent opposite ends of a continuum sometimes referred to as ASMD type A and ASMD type B. ASMD type A is a rapidly progressive neurological form of the disease resulting in death in early childhood due to central nervous system complications. ASMD type B is a serious and potentially life-threatening disease that predominantly impacts the lungs, liver, and spleen, as well as other organs. ASMD type A/B represents an intermediate form that includes varying degrees of neurologic involvement. Patients with ASMD type A/B or ASMD type B were studied in the ASCEND trial program. Another type of NPD is NPD type C, which is unrelated to ASMD.
About olipudase alfa
Olipudase alfa is an investigational enzyme replacement therapy designed to replace deficient or defective ASM, allowing for the breakdown of sphingomyelin. Olipudase alfa is currently being investigated to treat non-CNS manifestations of ASMD. Olipudase alfa has not been studied in ASMD type A patients. Olipudase alfa is an investigational agent and the safety and efficacy have not been evaluated by the FDA, EMA, or any other regulatory authority worldwide.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions.
With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe.
///////Olipudase alfa, japan 2022, APPROVALS 2022, Xenpozyme, PEPTIDE, オリプダーゼアルファ (遺伝子組換え) , ORPHAN DRUG, GZ-402665 , GZ 402665

NEW DRUG APPROVALS
ONE TIME TO MAINTAIN THIS BLOG
$10.00
Andexanet alfa
(heavy chain)
IVGGQECKDG ECPWQALLIN EENEGFCGGT ILSEFYILTA AHCLYQAKRF KVRVGDRNTE
QEEGGEAVHE VEVVIKHNRF TKETYDFDIA VLRLKTPITF RMNVAPACLP ERDWAESTLM
TQKTGIVSGF GRTHEKGRQS TRLKMLEVPY VDRNSCKLSS SFIITQNMFC AGYDTKQEDA
CQGDAGGPHV TRFKDTYFVT GIVSWGEGCA RKGKYGIYTK VTAFLKWIDR SMKTRGLPKA
KSHAPEVITS SPLK
(light chan)
ANSFLFWNKY KDGDQCETSP CQNQGKCKDG LGEYTCTCLE GFEGKNCELF TRKLCSLDNG
DCDQFCHEEQ NSVVCSCARG YTLADNGKAC IPTGPYPCGK QTLER
(Disulfide bridge: H7-H12, H27-H43, H108-L98, H156-H170, H181-H209, L16-L27, L21-L36, L38-L47, L55-L66, L62-L75, L77-L90)
Andexanet alfa
JAPAN 2022, PEPTIDE
| Ondexxya |
| 2022/3/28 |
| Anticoagulant reversal (factor Xa inhibitors) |
CAS: 1262449-58-0
アンデキサネットアルファ (遺伝子組換え)
- Andexanet alfa
- r-Antidote
- rfXa Inhibitor Antidote
- PRT-4445
- PRT064445
Andexanet alfa, sold under the trade name Andexxa among others, is an antidote for the medications rivaroxaban and apixaban, when reversal of anticoagulation is needed due to uncontrolled bleeding.[1] It has not been found to be useful for other factor Xa inhibitors.[2] It is given by injection into a vein.[2]
Common side effects include pneumonia and urinary tract infections.[2] Severe side effects may include blood clots, heart attacks, strokes, or cardiac arrest.[2] It works by binding to rivaroxaban and apixaban.[2]
It was approved for medical use in the United States in May 2018.[1] It was developed by Portola Pharmaceuticals.[3]
ndexanet alfa is a recombinant human coagulation Factor Xa that promotes blood coagulation. It was developed by Portola Pharmaceuticals and was approved in in May 2018. It is marketed as Andexxa for intravenous injection or infusion and is indicated for the reversal of anticoagulation in combination with rivaroxaban and apixaban in cases of life-threatening or uncontrolled bleeding. Rivaroxaban and apixaban are Factor Xa inhibitors that promote anticoagulation in situations where blood clotting is unfavourable, such as in deep vein thrombosis and pulmonary embolism. However, the use of these agents is associated with a risk for uncontrollable bleeding episodes that can lead to can cause serious or fatal bleeding. Andexanet alfa is currently under regulatory review by the European Union and is undergoing clinical development in Japan 1.
Andexanet alfa works by binding to Factor Xa inhibitors and prevent them from interacting with endogenous Factor Xa. It displayed high affinity (0.53–1.53 nmol/L) to apixaban, betrixaban, edoxaban and rivaroxaban 1. However, the effectiveness of andexanet alfa on treating bleeding related to any FXa inhibitors other than apixaban and rivaroxaban was not demonstrated, thus such use is limited 7. Its pharmacokinetic properties are not reported to be affected by factor Xa inhibitors 1. Andexanet alfa retains the structural similarity to that of endogenous human factor Xa, but exists in its mature functional form without the need for activation via the intrinsic or extrinsic coagulation pathways 5 and remains catalytically inactive due to structural modification 1. The procoagulation potential of andexanet alfa is eliminated through the removal of a 34-residue fragment containing Gla: via this truncation, andexanet alfa is unable to bind to membrane surfaces and assemble the prothrombinase complex 5. It also prevents andexanet alfa from taking up space on phospholipid surface membranes, so that native FXa may bind and assemble the prothrominase complex 5. The amino acid residue modification from serine to alanine in the binding site of the catalytic domain allows more effective binding to FXa inhibitors and deters the andexanet alfa from converting prothrombin to thrombin 5.
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////

Structure of andexant alfa. Andexanet alfa is a modified activated human factor Xa (FXa) that binds FXa with high affinity and a 1:1 stoichiometric ratio but does not have intrinsic catalytic activity (the amino acid serine at position 419 is replaced by alanine) and lacks the membrane-binding-carboxyglutamic acid domain (Gla domain) of native FX. The Gla domains are responsible for the binding of FXa to phospholipids
Medical uses
Andexanet alfa is used to stop life threatening or uncontrollable bleeding in people who are taking rivaroxaban or apixaban.[1]
There are no randomised clinical trials as of 2019. Studies in healthy volunteers show that the molecule binds factor Xa inhibitors and counters their anti-Xa-activity.[4] The only published clinical trial is a prospective, open label, single group study.[5] This study reports results on 352 people and demonstrates a reduction of anti-Xa-activity while also showing an excellent or good hemostatic efficacy in 82%. While people who were expected to die in 30 days were excluded from the study, 14% of participants died. There was no relationship between hemostatic efficacy and reduced anti-Xa-activity.[6] The FDA has demanded a randomised clinical trial: the first results are not expected before 2023.[7]
Adverse effects
Common side effects include pneumonia and urinary tract infections.[2] Severe side effects may include blood clots or cardiac arrest.[2]
Andexanet alfa has a boxed warning that it is associated with arterial and venous blood clots, ischemic events, cardiac arrest, and sudden deaths.[1]
Pharmacology
Mechanism of action
Andexanet alfa is a biologic agent, a recombinant modified version of human activated factor X (FXa).[8] Andexanet alfa differs from native FXa due to the removal of a 34 residue fragment that contains the Gla domain. This modification reduces andexanet alfa’s procoagulant potential. Additionally, a serine to alanine (S419A) mutation in the active site eliminates its activity as a prothrombin to thrombin catalyst, but still allows the molecule to bind to FXa inhibitors.[9] FXa inhibitors bind to andexanet alfa with the same affinity as to natural FXa. As a consequence in the presence of andexanet alfa natural FXa is partially freed, which can lead to effective hemostasis.[3][10] In other words, it acts as a decoy receptor. Andexanet alfa reverses effect of all anticoagulants that act directly through FXa or by binding antithrombin III. The drug is not effective against factor IIa inhibitor dabigatran.[11]
History[edit]
It was approved in the United States in 2018 based on data from two phase III studies on reversing the anticoagulant activity of FXa inhibitors rivaroxaban and apixaban in healthy volunteers.[4] As a condition of its accelerated approval there is a study being conducted comparing it to other currently used reversal agents (“usual care”).[5][12]
Society and culture
Economics
Initial pricing (AWP) is $58,000 per reversal (800 mg bolus + 960 mg infusion, $3,300 per 100 mg vial) which is higher than reversal agents for other DOAC agents (idarucizumab for use in dabigatran reversal is $4,200 per reversal).[13]
References
- ^ Jump up to:a b c d e “Andexxa- andexanet alfa injection, powder, lyophilized, for solution”. DailyMed. 21 September 2020. Retrieved 12 November 2020.
- ^ Jump up to:a b c d e f g “Andexxa Monograph for Professionals”. Drugs.com. Retrieved 19 December 2018.
- ^ Jump up to:a b Dolgin E (March 2013). “Antidotes edge closer to reversing effects of new blood thinners”. Nature Medicine. 19 (3): 251. doi:10.1038/nm0313-251. PMID 23467222. S2CID 13340319.
- ^ Jump up to:a b Siegal DM, Curnutte JT, Connolly SJ, Lu G, Conley PB, Wiens BL, Mathur VS, Castillo J, Bronson MD, Leeds JM, Mar FA, Gold A, Crowther MA (December 2015). “Andexanet Alfa for the Reversal of Factor Xa Inhibitor Activity”. New England Journal of Medicine. 373 (25): 2413–24. doi:10.1056/NEJMoa1510991. PMID 26559317.
- ^ Jump up to:a b Connolly SJ, Crowther M, Eikelboom JW, Gibson CM, Curnutte JT, Lawrence JH, et al. (April 2019). “Full Study Report of Andexanet Alfa for Bleeding Associated with Factor Xa Inhibitors”. New England Journal of Medicine. 380 (14): 1326–1335. doi:10.1056/NEJMoa1814051. PMC 6699827. PMID 30730782.
- ^ Justin Morgenstern, “Andexanet Alfa: More garbage science in the New England Journal of Medicine”, First10EM blog, February 11, 2019. Available at: https://first10em.com/andexanet-alfa/.
- ^ “A Randomized Clinical Trial of Andexanet Alfa in Acute Intracranial Hemorrhage in Patients Receiving an Oral Factor Xa Inhibitor”. 11 January 2022.
- ^ Lu, Genmin; DeGuzman, Francis R.; Lakhotia, Sanjay; Hollenbach, Stanley J.; Phillips, David R.; Sinha, Uma (2008-11-16). “Recombinant Antidote for Reversal of Anticoagulation by Factor Xa Inhibitors”. Blood. 112 (11): 983. doi:10.1182/blood.V112.11.983.983. ISSN 0006-4971.
- ^ Kaatz, Scott; Bhansali, Hardik; Gibbs, Joseph; Lavender, Robert; Mahan, Charles E.; Paje, David G. (2017-09-13). “Reversing factor Xa inhibitors – clinical utility of andexanet alfa”. Journal of Blood Medicine. 8: 141–149. doi:10.2147/JBM.S121550. PMC 5602457. PMID 28979172.
- ^ Lu G, Deguzman FR, Hollenbach SJ, et al. (March 2013). “A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa”. Nature Medicine. 19 (4): 446–51. doi:10.1038/nm.3102. PMID 23455714. S2CID 11235887.
- ^ H. Spreitzer (23 December 2013). “Neue Wirkstoffe – Andexanet Alfa”. Österreichische Apothekerzeitung (in German) (26/2013): 40.
- ^ “Trial of Andexanet in ICH Patients Receiving an Oral FXa Inhibitor”. ClinicalTrials.gov. 11 January 2022.
- ^ “Lexi Comp Drug Information Online”. 24 May 2018.
Further reading
- Connolly SJ, Milling TJ, Eikelboom JW, Gibson CM, Curnutte JT, Gold A, et al. (September 2016). “Andexanet Alfa for Acute Major Bleeding Associated with Factor Xa Inhibitors”. New England Journal of Medicine. 375 (12): 1131–41. doi:10.1056/NEJMoa1607887. PMC 5568772. PMID 27573206.
External links
- “Andexanet alfa”. Drug Information Portal. U.S. National Library of Medicine.
- “Ondexxya (andexanet alfa): Avoid use of andexanet prior to heparinization”. European
| Clinical data | |
|---|---|
| Trade names | Andexxa, Ondexxya, others |
| Other names | Coagulation factor Xa (recombinant), inactivated-zhzo, PRT06445, r-Antidote, PRT4445 |
| AHFS/Drugs.com | Monograph |
| License data | US DailyMed: Andexanet_alfa |
| Routes of administration | Intravenous injection |
| ATC code | V03AB38 (WHO) |
| Legal status | |
| Legal status | UK: POM (Prescription only)US: ℞-only [1]EU: Rx-only |
| Pharmacokinetic data | |
| Metabolism | Not studied |
| Elimination half-life | 5 h to 7 h |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1262449-58-0 |
| IUPHAR/BPS | 7576 |
| DrugBank | DB14562 |
| ChemSpider | none |
| UNII | BI009E452R |
| KEGG | D11029 |
| ChEMBL | ChEMBL3301583 |
//////////Andexanet alfa, JAPAN 2022, APPROVALS 2022, アンデキサネットアルファ (遺伝子組換え) , Ondexxya , PRT-4445, PRT064445

NEW DRUG APPROVALS
$10.00
Carotegrast methyl
Carotegrast methyl
| Formula | C28H26Cl2N4O5 |
|---|---|
| CAS | 401905-67-7 |
| Mol weight | 569.4358 |
PMDA APROVED, CAROGRA, カロテグラストメチル
| ON 2022/3/28 |
Antiasthmatic, Integrin alpha 4 inhibitor
- An alpha4 integrin antagonist.
401905-67-7[RN]
L-Phenylalanine, N-(2,6-dichlorobenzoyl)-4-[6-(dimethylamino)-1,4-dihydro-1-methyl-2,4-dioxo-3(2H)-quinazolinyl]-, methyl ester
methyl (2S)-2-[(2,6-dichlorophenyl)formamido]-3-{4-[6-(dimethylamino)-1-methyl-2,4-dioxo-1,2,3,4-tetrahydroquinazolin-3-yl]phenyl}propanoate
Methyl N-(2,6-dichlorobenzoyl)-4-[6-(dimethylamino)-1-methyl-2,4-dioxo-1,4-dihydro-3(2H)-quinazolinyl]-L-phenylalaninate
Carotegrast Methyl
Methyl (2S)-2-(2,6-dichlorobenzamido)-3-{4-[6-(dimethylamino)-1-methyl-2,4-dioxo-1,4-dihydroquinazolin-3(2H)-yl]phenyl}propanoate
C28H26Cl2N4O5 : 569.44
[401905-67-7]
PATENT
WO 2008062859
https://patents.google.com/patent/WO2008062859A1/en

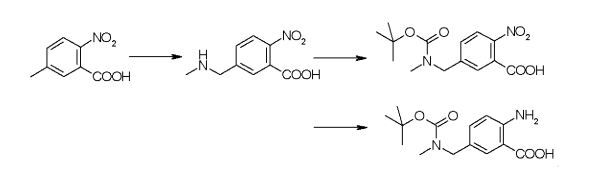


Step 1
(Method 2): The title compound was prepared starting from 2-amino-5-dimethylamino- benzoic acid methyl ester dihydrochloride through the hydrolysis under basic condition To 5.0 g of 2-amino-5-dimethylamino-benzoic acid methyl ester di-hydrochloride, there were added 15 mL of water and 15.6 mL of a 6M aqueous solution of sodium hydroxide and the resulting mixture was heated to 40°C for 2 hours. After the confirmation of the progress of the reaction according to HPLC, the reaction system was cooled to room temperature, a 6M hydrochloric acid aqueous solution was dropwise added to the reaction system to thus neutralize the same and to separate out crystals (pH 4.9) and then the reaction system was stirred at 10°C for 2 hours. The solid thus obtained was isolated through the filtration under reduced pressure, washed with 30 mL of water and then dried under reduced pressure at 60°C for 14 hours. Title compound 3.14 g was obtained as gray-colored solid. The physical properties determined were almost identical to those observed for the same compound prepared in the above-mentioned synthesis example. H-NMR (400MHz, DMSO-d6): δ 8.21 (bs, 3H), 7.10 (d, 1H, J=2.8Hz), 6.97 (dd, 1H, J=9.1, 2.8Hz), 6.70 (d, 1H, J=9.1 Hz), 2.72 (s, 6H); 13C-NMR (100MHz, DMSO-d6): δ168.89, 144.55, 141.61, 123.29, 117.90, 114.78, 110.11,41.95; MS (ESI+): m/z 181.3 (MH+), (ESI-): m/z 179.2 (M-H–).
Step 2
Step 1: Synthesis of Nα-(2,6-dichlorobenzoyl) -4-{2-ethoxycarbonylamino-5-dimethyl- amino-benzoylamino}-L-phenylalanine methyl ester To 1.96 g of 2-amino-5-dimethylaminobenzoic acid, there were added 12 mL of acetonitrile and 5.29 mL of pyridine to form a suspension and then the resulting suspension was cooled to 4°C. To this suspension there was dropwise added 4.17 mL of ethyl chloroformate over 5 minutes and then the mixture was stirred at 25°C for one hour. After confirming the disappearance of the starting material by HPLC, 0.7 mL of ethanol was added to the mixture to thus decompose the excess ethyl chloroformate and the mixture was further stirred for additional one hour. To this reaction solution there were added 4.0 g of 4-amino-Nα-(2,6-dichlorobenzoyl)-L-phenylalanine methyl ester and 12 mL of N,Ndimethylformamide, and the resulting mixture was stirred overnight. Subsequently, 48 mL of methanol was drop-wise added, the resulting mixture was stirred at 10°C overnight and then the solid separated from the mixture was isolated through filtration under reduced pressure. The solid was then washed with 8 mL of methanol and dried at 70°C for 5 hours under reduced pressure. Title compound 5.50 g was obtained as pale yellow solid. 1H-NMR (400MHz, DMSO-d6): δ 10.29 (s, 1H), 9.42 (bs, 1H), 9.24 (d, 1H, J=7.9Hz), 7.73 (bs, 1H), 7.62 (d, 2H, J=8.4Hz), 7.48-7.44 (m, 2H), 7.41 (dd, 1H, J=9.5, 6.2Hz), 7.27 (d, 2H,J=8.4Hz), 7.01 (d, 1H, J=2.7Hz), 6.93 (dd, 1H, J=9.1, 2.9Hz), 4.71 (ddd, 1H, J=9.2, 8.1, 5.7Hz), 4.05 (q, 2H, J=7.0Hz), 3.66 (s, 3H), 3.10 (dd, 1H, J=14.0, 5.6Hz), 2.96 (dd, 1H, J=14.0, 9.2Hz), 2.93 (s, 6H), 1.18 (t, 3H, J=7.2Hz); MS (ESI+): m/z 601.2 (MH+) and 623.2 (M+Na), (ESI–): m/z 599.1 (M-H–).
Step 3
Step2: Synthesis of Na-(2,6-dichlorobenzoyl)-4-{6-dimethylamino-1-methylquinazoline-2,4[1H,3H]-dion-3-yl}-L-phenylalanine methyl ester To 2.0 g of Na-(2,6-dichlorobenzoyl)-4-{2-ethoxycarbonylamino -5-dimethyl- amino-benzoylamino}-L-phenylalanine methyl ester prepared in above-mentioned step 1, were added 16 mL of N,N-dimethylfbrmamide, 0.8 mL of methanol and 0.91 g of potassium carbonate, followed by the stirring of the resulting mixture at 25°C overnight. To this reaction solution, there was added 0.75 mL of methyl p-toluenesulfonate for subjecting the methyl ester to alkylation at 25~40°C. After confirming the disappearance of the starting material by HPLC, 0.75 mL of acetic acid was added to quench the reaction, 16 mL of water was dropped and the solid was separated. Further, 8 mLof N,N-dimethylformamide/water = 1/1 mixed liquid was added to the resulting mixture, followed by the stirring of the mixture at 25°C. Then the solid thus separated was isolated through filtration under reduced pressure and then washed with 8 mL of water. Thereafter, the isolated solid was dried at 70°C for 4 hours under reduced pressure. Desired compound 1.77 g was obtained as pale yellow solid. 1H-NMR (400MHz, DMSO-d6): δ 9.28 (d, 1H, J=8.1 Hz), 7.48-7.36 (m, 6H), 7.31 (dd, 1H, J=3.0, 9.0Hz), 7.24 (d, 1H, J=3.0Hz), 7.20-7.15 (m, 2H), 4.18 (ddd, 1H, J=10.2, 8.1,4.8Hz), 3.69 (s, 3H), 3.49 (s, 3H), 3.22 (dd, 1H, J=14.1, 4.8Hz), 3.02 (dd, 1H, J=14.2, 10.5Hz), 2.94 (s, 6H); MS (ESI+): m/z 569.2 (MH+) and 591.1 (M+Na), (ESI-): m/z 567.2 (M-H–).
PATENT
https://patents.google.com/patent/WO2004074264A1/en
PATENT’ WO 2003070709
https://patents.google.com/patent/WO2003070709A1/en
PATENT
WO 2002016329
///////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
/////////////Carotegrast methyl, CAROGRA, カロテグラストメチル , JAPAN 2022, APPROVALS 2022,
COC(=O)[C@H](Cc1ccc(cc1)N2C(=O)N(C)c3ccc(cc3C2=O)N(C)C)NC(=O)c4c(Cl)cccc4Cl

NEW DRUG APPROVALS
ONE TIME
$10.00
ENSITRELVIR
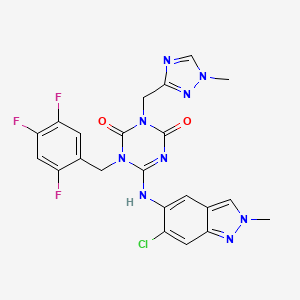
Ensitrelvir
S-217622, S 217622, Xocova, SHIONOGI,
6-[(6-chloro-2-methylindazol-5-yl)amino]-3-[(1-methyl-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-1,3,5-triazine-2,4-dione
CAS 2647530-73-0
| C22H17ClF3N9O2531.9 | |
| Synonyms | BDBM513874bioRxiv20220126.477782, S-217622 |
|---|
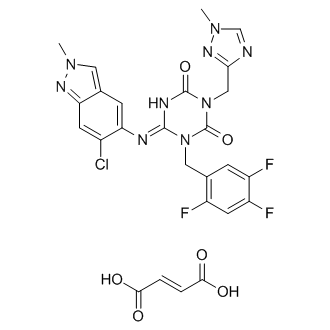
Ensitrelvir fumarate
CAS No. : 2757470-18-9
C22 H17 Cl F3 N9 O2 . C4 H4 O4
1,3,5-Triazine-2,4(1H,3H)-dione, 6-[(6-chloro-2-methyl-2H-indazol-5-yl)imino]dihydro-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-[(2,4,5-trifluorophenyl)methyl]-, (6E)-, (2E)-2-butenedioate (1:1)
| Formula: | C26H21ClF3N9O6 |
|---|---|
| M. Wt. : | 647.95 |
FDA 2026, APPROVALS 2026, 5/29/2026, Xocova
To use as post-exposure prophylaxis of coronavirus disease 2019 (COVID-19) following contact with an individual who has COVID-19
A Phase 1 study of S-217622 in healthy adult participants (jRCT2031210202)
Japan Registry of Clinical Trials Web Site 2021, July 16
PMDA APPROVED 2022/11/22, Xocova
Ensitrelvir[1] (code name S-217622, brand name Xocova)[2] is an antiviral drug developed by Shionogi in partnership with Hokkaido University, which acts as an orally active 3C-like protease inhibitor for the treatment of COVID-19 infection.[3][4] It is taken by mouth, and has been successfully tested against the recently emerged Omicron variant.[5]
About S-217622
S-217622, a therapeutic drug for COVID-19, is a 3CL protease inhibitor created through joint research between Hokkaido University and Shionogi. SARS-CoV-2 has an enzyme called 3CL protease, which is essential for the replication of the virus. S-217622 suppresses the replication of SARS-CoV-2 by selectively inhibiting 3CL protease. Shionogi has already been submitting the non-clinical, manufacturing/CMC data, and clinical trial data obtained so far to the PMDA. Currently the Phase 3 part of a Phase 2/3 clinical trial in patients with mild/moderate symptoms and the Phase 2b/3 part in patients with asymptomatic/only mild symptoms are in progress.
SYN
J.Med.Chem.2024,67,4376−4418
Ensitrelvir fumaric acid (3), also referred to as S-217622, is an oral noncovalent SARS-CoV-2 main protease (Mpro) inhibitordeveloped by Shionogi & Co. that was approved by the japan Pharmaceuticals and Medical Devices Agency (PMDA)for the treatment of disease caused by SARS-CoV-2 (COVID
19) infection. Dosed once daily for 5 days, ensitrelvirsuppresses the replication of SARS-CoV-2 in infected patients as a result of its inhibition of the viral mpro.25,26
Ensitrelvir retains potent inhibitory activity against many of the most common M mutants and exhibits antiviral activity against a wide variety of circulating SARS-CoV-2 variants. 27is the second Mpro
Ensitrelvir inhibitor approved for the treatment of 28 disease caused by COVID-19. Unlike the first approved treatment, Paxlovid, ensitrelvir does not require coadministration with a CYP3A4 inhibitor to attenuate metabolism in vivo.
Furthermore, crystal structures of ensitrelvir in complex with the main proteases of three other human-infecting coronaviruses (MERS-CoV, SARS-CoV, and HCoV-NL63)
A convergent, kilogram-scale synthesis of ensitrelvir suitable for manufacturing has been described in the literature by researchers at Shionogi. 30 The synthetic approach involved the union of two key building blocks indazole 3.7 and 1,3,5triazinone 3.14, each necessitating development of a scale worthy route. The preparation of triazinone 3.14 necessitated construction of a triazolyl methylene chloride subunit which
began with the reduction of triazole ester 3.1 with aluminum hydride 3.2 (a less pyrophoric alternative to LAH yet still required aqueous Rochelle salt quench to chelate excess aluminum) 31to provide alcohol 3.3, which was then convertedto the corresponding chloride and isolated as the triazole HCl
salt 3.4 (Scheme 6). Assembly of indazole intermediate 3.7began with regioselective nitration of benzaldehyde 3.5followed by treatment with hydrazine hydrate in aqueous EtOHtoprovide indazole3.6(Scheme7).Fascinatingly, the Shionogi team isolated a variety of byproducts during the
conversionof3.5to3.6whichsupportedtheirhypothesisforareaction mechanism that likely equilibrated through a dibenzylidenehydrazine intermediateenroute tothedesired
indazole3.6.Ascreenofelectrophilicmethyl sourcesrevealed thatMeerwein’s salt facilitatedthebest conversionof 3.6to the correspondingN2-monomethylated indazole; subsequenthydrogenative nitro reduction furnished the key indazole intermediate 3.7.Construction of the ensitrelvir core started with reaction of carboximidamide 3.8 with t-butyl isocyanate followed by N,N′carbonyldiimidazole (CDI) to secure 1,3,5-triazinone 3.10(Scheme 8). Subsequent N-alkylation with bromide 3.11provided benzyl triazinone 3.12. Substitution of the pyrazolewith m-cresol was accomplished under acidic conditions. The
authors report that m-cresol was identified as a leaving group that facilitated introduction of indazole 3.7 with a minimal number of byproducts in a later step of the synthesis. The TFA-mediated reaction concomitantly removed the N-tertbutyl group providing compound 3.13 in 91% yield. Nalkylation with chloride 3.4 in the presence of a base resulted in intermediate 3.14 which was then treated with building
block 3.7 in the presence of anhydrous acetic acid. Isolation of ensitrelvir fumaric acid was achieved by exposure to fumaric acid in aqueous acetone.
(25) Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura, T.;
Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Tsuge, Y.; Uehara, T.;
Mukae, H. A phase 2/3 study of S-217622 in participants with SARS
CoV-2 infection (Phase 3 part). Medicine 2023, 102, No. e33024.
(26) Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Imamura,
T.; Sonoyama, T.; Fukuhara, T.; Ichihashi, G.; Sanaki, T.; Baba, K.;
Takeda, Y.; Tsuge, Y.; Uehara, T. A randomized phase 2/3 study of
ensitrelvir, a novel oral SARS-CoV-2 3C-like protease inhibitor, in
Japanese patients with mild-to-moderate COVID-19 or asymptomatic
SARS-CoV-2 infection: results of the phase 2a part. Antimicrob. Agents
Chemother. 2022, 66, No. 00697.
(27) Kawashima, S.; Matsui, Y.; Adachi, T.; Morikawa, Y.; Inoue, K.;
Takebayashi, S.; Nobori, H.; Rokushima, M.; Tachibana, Y.; Kato, T.
Ensitrelvir is effective against SARS-CoV-2 3CL protease mutants
circulating globally. Biochem. Biophys. Res. Commun. 2023, 645, 132−
136.
(28) Unoh, Y.; Uehara, S.; Nakahara, K.; Nobori, H.; Yamatsu, Y.;
Yamamoto, S.; Maruyama, Y.; Taoda, Y.; Kasamatsu, K.; Suto, T.;
et al. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL
protease inhibitor clinical candidate for treating COVID-19. J. Med.
Chem. 2022, 65, 6499−6512
(29) Lin, C.; Jiang, H.; Li, W.; Zeng, P.; Zhou, X.; Zhang, J.; Li, J.
Structural basis for the inhibition of coronaviral main proteases by
ensitrelvir. Structure 2023, 31, 1016.
(30) Kawajiri, T.; Kijima, A.; Iimuro, A.; Ohashi, E.; Yamakawa, K.;
Agura, K.; Masuda, K.; Kouki, K.; Kasamatsu, K.; Yanagisawa, S.; et al.
Development of a manufacturing process toward the convergent
synthesis of the COVID-19 antiviral Ensitrelvir. ACS Cent. Sci. 2023,
9, 836−843.
(31) Gugelchuk, M.; Silva, III, L. F.; Vasconcelos, R. S.; Quintiliano,
S. A. P. Sodium bis(2-methoxyethoxy)aluminum hydride. In
Encyclopedia of Reagents for Organic Synthesis; Charette, A., Bode, J.,
Rovis, T., Shenvi, R., Eds.; John Wiley & Sons, Ltd., 2007.


Syn
Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19
View ORCID ProfileYuto Unoh, View ORCID ProfileShota Uehara, View ORCID ProfileKenji Nakahara, View ORCID ProfileHaruaki Nobori, Yukiko Yamatsu, View ORCID ProfileShiho Yamamoto, View ORCID ProfileYuki Maruyama, View ORCID ProfileYoshiyuki Taoda, View ORCID ProfileKoji Kasamatsu, View ORCID ProfileTakahiro Suto, Kensuke Kouki, View ORCID ProfileAtsufumi Nakahashi, Sho Kawashima, View ORCID ProfileTakao Sanaki, Shinsuke Toba, Kentaro Uemura, Tohru Mizutare, View ORCID ProfileShigeru Ando, View ORCID ProfileMichihito Sasaki, View ORCID ProfileYasuko Orba, View ORCID ProfileHirofumi Sawa, View ORCID ProfileAkihiko Sato, View ORCID ProfileTakafumi Sato, View ORCID ProfileTeruhisa Kato, View ORCID ProfileYuki Tachibana
doi: https://doi.org/10.1101/2022.01.26.477782
https://www.biorxiv.org/content/10.1101/2022.01.26.477782v1.full
The coronavirus disease 2019 (COVID-19) pandemic, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has resulted in millions of deaths and threatens public health and safety. Despite the rapid global spread of COVID-19 vaccines, effective oral antiviral drugs are urgently needed. Here, we describe the discovery of S-217622, the first oral non-covalent, non-peptidic SARS-CoV-2 3CL protease inhibitor clinical candidate. S-217622 was discovered via virtual screening followed by biological screening of an in-house compound library, and optimization of the hit compound using a structure-based drug-design strategy. S-217622 exhibited antiviral activity in vitro against current outbreaking SARS-CoV-2 variants and showed favorable pharmacokinetic profiles in vivo for once-daily oral dosing. Furthermore, S-217622 dose-dependently inhibited intrapulmonary replication of SARS-CoV-2 in mice, indicating that this novel non-covalent inhibitor could be a potential oral agent for treating COVID-19.
Chemistry

The synthetic scheme for compound 1 is described in Scheme 1. Starting from the pyrazole derivative 4, cyclization with Ethyl isocyanatoacetate and CDI was conducted, giving 5 in 90% yield. Then, an alkylation with 5-bromomethyl-1,2,3-trifluorobenzene followed by introduction of a 4-difluoromethoxy-2-methylaniline unit, to give 7 (40% in 2 steps). The ester group in 7 was hydrolyzed and then amidated with methylamine, yielding 1 (58% in 2 steps). Compound 2 was synthesized similarly as shown in Scheme 2.
S-217622 (3) was synthesized as described in Scheme 3. Starting from known compound 9,21 an alkylation with 1-(bromomethyl)-2,4,5-trifluorobenzene gave 10 in 93% yield. Then, the 3-tert-Bu group was removed and the triazole unit was introduced, and the substitution of the SEt moiety with the indazole unit finally gave S-217622 (3).
21 Kai, H.; Kameyama, T.; Horiguchi, T.; Asahi, K.; Endoh, T.; Fujii, Y.; Shintani, T.; Nakamura, K.; Matsumoto, S.; Hasegawa, T.; Oohara, M.; Tada, Y.; Maki, T.; Iida, A. Preparation of triazine derivatives and pharmaceutical compound that contains same and exhibits analgesic activity. WO 2012020749 A1, Feb 16, 2012

Scheme 1.
Reagents and Conditions: (a) ethyl isocyanato-acetate, DBU, CDI, DMA, –10 °C to rt, 90%; (b) 5-bromomethyl-1,2,3-trifluorobenzene, N,N-diisopropylethylamine, DMA, 60 °C; (c) 4-difluoromethoxy-2-methylaniline, tert-butanol, 100 °C, 40% in 2 steps; (d) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 58% in 2 steps.

Scheme 2.
Reagents and Conditions: (a) 6-chloro-2-methyl-2H-indazol-5-amine, tert-amyl alcohol, 100 °C, 44% in 2 steps from 5; (b) (i) NaOH aq., THF/MeOH, rt; (ii) methylamine, HATU, N,N-diisopropylethylamine, THF, rt., 29% in 2 steps.

Scheme 3.
Reagents and Conditions: (a) 1-(bromomethyl)-2,4,5-trifluorobenzene, K2CO3, MeCN, 80 °C, 93%; (b) TFA, rt, 97%; (c) 3-(chloromethyl)-1-methyl-1H-1,2,4-triazole hydrochloride, K2CO3, DMF, 60 °C, 45%; (d) 6-chloro-2-methyl-2H-indazol-5-amine, LHMDS, THF, 0 °C to rt., 25%.
(6E)-6-[(6-Chloro-2-methyl-2H-indazol-5-yl)imino]-3-[(1-methyl-1H-1,2,4-triazol-3-yl)methyl]-1-(2,4,5-trifluorobenzyl)-1,3,5-triazinane-2,4-dione (3, S-217622)
To a solution of 12 (300 mg, 0.727 mmol) and 6-chloro-2-methyl-2H-indazol-5-amine (172 mg, 0.946 mmol) in THF (6 mL) was added LHMDS (1M in THF; 1.46 mL, 1.46 mmol) dropwisely at 0 °C. The reaction mixture was stirred at 0 °C for 2.5 h and then at rt for 40 min. The reaction was quenched with aqueous NH4Cl solution, and the aqueous layer was extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3/MeOH gradient, 0-20% MeOH). The solid was recrystallized from acetone/H2O to afford 3 (S-217622) (95.3 mg, 25%) as a pale brown solid. 1H NMR (400 MHz, DMSO-d6, DCl in D2O) δ 3.90 (3H, s), 4.15 (3H, s), 5.04 (2H, s), 5.26 (2H, s), 7.44 (1H, m), 7.52-7.65 (2H, m), 7.73 (1H, s), 8.40 (1H, s), 9.31 (1H, s). 13C NMR (100 MHz, DMSO-d6, DCl in D2O) δ 37.34, 38.04, 40.06, 40.29, 106.16 (dd, J = 28.2, 21.6 Hz), 116.46-116.70, 116.70, 120.54-120.76, 120.76, 125.93, 129.10, 132.35, 143.84, 145.98, 146.38 (ddd, J = 241.4, 12.5, 3.7 Hz), 146.60, 148.52 (td, J = 247.7, 13.6 Hz), 150.43, 150.50, 155.22 (ddd, J = 244.3, 10.3, 2.2 Hz), 155.58. HRMS-ESI (m/z): [M + H]+ calcd for [C22H18 F3ClN9O2]+ 532.1219; found 532.1221.
Preparation of Compound 3 (S-217622) fumaric acid co-crystal
A mixture of 3 (S-217622) (1.17 g, 2.2 mmol) and fumaric acid (278 mg, 2.4 mmol) in EtOAc (5.9 mL) was stirred at room temperature for 45 min. The suspension was filtrated to afford 3 (S-217622) fumaric acid co-crystal (1.37 g, 95 %) as a white solid. 1H NMR (400 MHz, pyridine-d5) δ 3.64 (s, 3H), 3.99 (s, 3H), 5.56 (s, 2H), 5.61 (s, 2H), 7.16-7.25 (m, 2H), 7.44 (s, 2H), 7.81 (s, 1H), 7.89 (s, 1H), 7.89-7.97 (m, 1H), 8.32 (s, 1H).
Notes
SHIONOGI has applied for a patent covering 1, 2, and 3 (S-217622). Y.U., S.U., K.N., H.N., Y.Y., S.Y., Y.M., Y.T., K.K., T.S., K.K., A.N., S.K., T.S., S.T., K.U., T.M., S.A., A.S., T.S., T.K., and Y.T. are employees of SHIONOGI & Co., Ltd. S.U., K.N., H.N., Y.M., Y.T., K.K., T.S., K.K., S.K., TS, S.T., K.U., T.S., and T.K. are shareholders in SHIONOGI & Co., Ltd. M.S., Y.O., and H.S. are financially supported by the joint research fund from SHIONOGI & Co., Ltd.
- Supporting information[supplements/477782_file02.pdf]
see spectrum at end of page
///////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE
join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Oral antiviral medications, in addition to vaccines, are expected to play an important role in treating coronavirus disease 2019 (COVID-19), which is caused by infection with the severe acute respiratory disease coronavirus-2 (SARS-CoV-2).
These drugs must have significant antiviral activity, as well as target specificity, oral bioavailability, and metabolic stability. Although several antiviral compounds have been reported as possible SARS-CoV-2 inhibitors in vitro, only a few of these drugs have been shown to be effective in vivo.
Ensitrelvir, a novel SARS-CoV-2 antiviral
Ensitrelvir (code name S-217622, brand name Xocova), is a new inhibitor of the SARS-CoV-2 major protease (Mpro), also known as 3C-like protease, has been shown to reduce the viral load and help alleviate the severity of SARS-CoV-2 in infected hamsters. In cells, low nanomolar to sub-micromolar doses of S-217622 suppress viral growth. In hamsters, oral treatment of S-217622 showed excellent pharmacokinetic qualities and hastened recovery from acute SARS-CoV-2 infection.
S-217622 also demonstrated antiviral effectiveness against SARS-CoV-2 variants of concern (VOCs), such as the highly pathogenic Delta variant and the newly discovered Omicron variant. Overall, these findings show that S-217622, which is an antiviral drug that is currently being tested in Phase II/III clinical trials, has impressive antiviral efficiency and effectiveness against SARS-CoV-2 and could be a viable oral treatment option for COVID-19.
History
It has reached Phase III clinical trials.[3] The Japanese government is reportedly considering allowing Shionogi permission to apply for approval for medical use before the final steps of trials are completed, potentially speeding up the release for sale. This conditional early approval system has previously been used in Japan to accelerate the progression to market of other antiviral drugs targeting COVID-19, including remdesivir and molnupiravir.[6] In a study of 428 patients, viral load was reduced, but symptoms were not significantly reduced. [7]
It became the first Japanese domestic pill to treat COVID-19, third to be regulatorally approved in Japan; in February 2022.[8]

NEW DRUG APPROVALS
ONE TIME
$10.00
References
- ^ World Health Organization (2021). “International Nonproprietary Names for Pharmaceutical Substances. Proposed INN: List 126” (PDF). WHO Drug Information. 35 (4): 1135.
- ^ Xocova: Powerful New Japanese Pill for Coronavirus Treatment. BioPharma Media, February 2022
- ^ Jump up to:a b Unoh Y, Uehara S, Nakahara K, Nobori H, Yamatsu Y, Yamamoto S, et al. (January 2022). “Discovery of S-217622, a Non-Covalent Oral SARS-CoV-2 3CL Protease Inhibitor Clinical Candidate for Treating COVID-19”. bioRxiv. doi:10.1101/2022.01.26.477782. S2CID 246367525.
- ^ “Shionogi presents positive Ph II/III results for COVID-19 antiviral S-217622”. thepharmaletter.com. 31 January 2022.
- ^ Shionogi’s new COVID pill appears to ease omicron symptoms. Nikkei Asia, 21 December 2021
- ^ Japan to consider early approval for Shionogi COVID-19 pill. Japan Times, 8 February 2022
- ^ https://www.reuters.com/business/healthcare-pharmaceuticals/japans-shionogi-seeks-approval-oral-covid-19-drug-2022-02-25/[bare URL]
- ^ “Japan’s Shionogi seeks approval for COVID-19 pill”. Reuters. Reuters. 25 February 2022.
| Clinical data | |
|---|---|
| Other names | S-217622 |
| Identifiers | |
| showIUPAC name | |
| PubChem CID | 162533924 |
| Chemical and physical data | |
| Formula | C22H17ClF3N9O2 |
| Molar mass | 531.88 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
Journal reference:
- Sasaki, M., Tabata, K., Kishimoto, M., et al. (2022). Oral administration of S-217622, a SARS-CoV-2 main protease inhibitor, decreases the viral load and accelerates recovery from clinical aspects of COVID-19. bioRxiv. doi:10.1101/2022.02.14.480338. https://www.biorxiv.org/content/10.1101/2022.02.14.480338v1.full.
///////////Ensitrelvir, S-217622, S 217622, Xocova, SHIONOGI, CORONA VIRUS, covid 19


Somatrogon

>Somatrogon amino acid sequence SSSSKAPPPSLPSPSRLPGPSDTPILPQFPTIPLSRLFDNAMLRAHRLHQLAFDTYQEFE EAYIPKEQKYSFLQNPQTSLCFSESIPTPSNREETQQKSNLELLRISLLLIQSWLEPVQF LRSVFANSLVYGASDSNVYDLLKDLEEGIQTLMGRLEDGSPRTGQIFKQTYSKFDTNSHN DDALLKNYGLLYCFRKDMDKVETFLRIVQCRSVEGSCGFSSSSKAPPPSLPSPSRLPGPS DTPILPQSSSSKAPPPSLPSPSRLPGPSDTPILPQ
Somatrogon
CAS: 1663481-09-1
Protein Chemical FormulaC1359H2125N361O420S7
Protein Average Weight30465.1 Da (Aglycosylated)
NGENLA, JAPAN PMDA APPROVED 2022/1/20
ソマトロゴン;
- MOD-4023
Replenisher (somatotoropin)
- OriginatorModigene
- DeveloperOPKO Health; Pfizer
- ClassBiological proteins; Growth hormones; Hormonal replacements; Recombinant proteins
- Mechanism of ActionHuman growth hormone replacements
- Orphan Drug StatusYes – Somatotropin deficiency
- RegisteredSomatotropin deficiency
- 21 Jan 2022Pfizer and OPKO health receives complete response letter from the US FDA for somatrogon in Somatotropin deficiency (In children)
- 20 Jan 2022Registered for Somatotropin deficiency (In children) in Japan (SC)
- 01 Dec 2021CHMP issues a positive opinion and recommends approval of somatrogon for Somatotropin deficiency in the European Union
Somatrogon, sold under the brand name Ngenla, is a medication for the treatment of growth hormone deficiency.[1][2] Somatrogon is a glycosylated protein constructed from human growth hormone and a small part of human chorionic gonadotropin which is appended to both the N-terminal and C-terminal.[2]
Somatrogon is a long-acting recombinant human growth hormone used as the long-term treatment of pediatric patients who have growth failure due to growth hormone deficiency.
omatrogon is a long-acting recombinant human growth hormone. Growth hormone is a peptide hormone secreted by the pituitary gland that plays a crucial role in promoting longitudinal growth during childhood and adolescence and regulating metabolic function in adulthood.2 Recombinant growth hormone therapy for growth hormone deficiency and other conditions has been available since 1985, with daily administration being the standard treatment for many years. More recently, longer-acting forms of growth hormone were developed to improve patient adherence and thus, improve the therapeutic efficacy of treatment.1 Somatrogon was produced in Chinese Hamster Ovary (CHO) cells using recombinant DNA technology. It is a chimeric product generated by fusing three copies of the C-terminal peptide (CTP), or 28 carboxy-terminal residues, from the beta chain of human chorionic gonadotropin (hCG) to the N-terminus and C-terminus of human growth hormone.2,6 The glycosylation and the presence of CTPs in the protein sequence prolongs the half-life of somatrogon and allows its once-weekly dosing.6
In October 2021, Health Canada approved somatrogon under the market name NGENLA as the long-term treatment of pediatric patients who have growth failure due to an inadequate secretion of endogenous growth hormone caused by growth hormone deficiency, marking Canada as the first country to approve this drug.4 It is available as a once-weekly subcutaneous injection.5
////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
About Somatrogon©
Somatrogon©, a long-acting human growth hormone (hGH) molecule, is a once-weekly injectable, created using recombinant technology, for the treatment of pediatric and adult growth hormone deficiency (GHD). The molecule consists of the natural peptide sequence of native growth hormone and the 28 amino acids of the C-Terminus Peptide (CTP) of the human chorionic gonadotropin hormone. This molecule, as compared to current GH replacement therapies, is intended to reduce the injection frequency from a daily to once a week in adults and children with GHD.
| Clinical data | |
|---|---|
| Trade names | Ngenla |
| Other names | MOD-4023 |
| Pregnancy category | AU: B1[1] |
| Routes of administration | Subcutaneous injection |
| ATC code | H01AC08 (WHO) |
| Legal status | |
| Legal status | AU: S4 (Prescription only) [1] |
| Identifiers | |
| CAS Number | 1663481-09-1 |
| DrugBank | DB14960 |
| UNII | 6D848RA61B |
Somatrogon© COMPETITIVE ADVANTAGES
In 2014, Pfizer and OPKO entered into a worldwide agreement for the development and commercialization of Somatrogon©. Under the agreement, OPKO is responsible for conducting the clinical program and Pfizer is responsible for registering and commercializing the product.
- New molecular entity (NME) that maintains natural native sequence of growth hormone
- Once weekly injection vs. current products requiring daily injections
- Human growth hormone is used for:
- Growth hormone deficient children and adults
- SGA, PWS, ISS
- Final presentation:
- Refrigerated, liquid, non-viscous formulation
- Disposable easy to handle pen injection device with thin needle and small injection volume
- Orphan drug designation in the U.S. and the EU for children and adults
Somatrogon© PROGRAM STATUS
Phase 3 Pediatric Somatrogon©
- Phase 3 study in naive growth hormone deficiency pediatric population was completed.
The study was conducted in over 20 countries. This study enrolled and treated 224 pre-pubertal, treatment-naive children with growth hormone deficiency.
- OPKO and Pfizer Announce Positive Phase 3 Top-Line Results for Somatrogon© during Oct 2019.
- Achieved Primary Endpoint
- Somatrogon© was proven non-inferior to daily Genotropin® (somatropin) with respect to height velocity after 12 months
- Height velocity at 12 months of treatment was higher in the Somatrogon© group (10.12 cm/year) than in the somatropin group (9.78 cm/year)
- Secondary Endpoints Achieved
- Change in height standard deviation scores at six and 12 months were higher with Somatrogon© in comparison to somatropin
- At six months, change in height velocity was higher with Somatrogon© in comparison to somatropin
- Somatrogon© was generally well tolerated in the study and comparable to that of somatropin dosed once-daily with respect to the types, numbers and severity of the adverse events observed between the treatment arms
- Children completing this study had the opportunity to enroll in a global, open-label, multicenter, long-term extension study, in which they were able to either continue receiving or switch to Somatrogon© Approximately 95% of the patients switched into the open-label extension study and received Somatrogon© treatment
Phase 3 adults Somatrogon© completed
- Primary endpoint of change in trunk fat mass from baseline to 26 weeks did not demonstrate a statistical significance between the Somatrogon© treated group and placebo
- Completed post hoc outlier analysis in June 2017 to assess the influence of outliers on the primary endpoint results
- Analyses which excluded outliers showed a statistically significant difference between Somatrogon© and placebo on the change in trunk fat mass: additional analyses that did not exclude outliers showed mixed results
- No safety concerns
- OPKO and Pfizer have agreed that OPKO may proceed with a pre-BLA meeting with FDA to discuss a submission plan
- OPKO plans to carry out an additional study in adults using a pen device
Pediatric Somatrogon© registration study in Japan- expected to be completed in Q1 2020
- 44 patients, comparison of weekly Somatrogon to daily growth hormone.
- Same pen device, dosage and formulation used in global study.
Somatrogon© Path to Approval
- BLA submission in US anticipated second half of 2020
- Completion of analysis of immunogenicity and safety data from pivotal Phase 3 study and open label extension study
- Two abstracts accepted for oral presentation of data set at the Endo Society’s Annual Meeting in March 2020
- “Somatrogon© Growth Hormone in the Treatment of Pediatric Growth Hormone Deficiency: Results of the Pivotal Phase 3”
- “Interpretation of Insulin-like Growth Factor (IGF-1) Levels Following Administration of Somatrogon© (a long acting Growth Hormone-hGH-CTP)”
- MAA submission in Europe to follow upon completion of open label study demonstrating benefit and compliance with reduced treatment burden
- Study expected to be completed in Q3 2020
References
Hershkovitz O, Bar-Ilan A, Guy R, et al. In vitro and in vivo characterization of MOD-4023, a long-acting carboxy-terminal peptide (CTP)-modified human growth hormone. Mol Pharm. 2016; 13:631–639 [PDF]
Strasburger CJ, Vanuga P, Payer J, et al. MOD-4023, a long-acting carboxy-terminal peptide-modified human growth hormone: results of a Phase 2 study in growth hormone-deficient adults. Eur J Endocrinol. 2017;176:283–294 [PDF]
Zelinska N, Iotova V, Skorodok J, et al. Long-acting CTP-modified hGH (MOD-4023): results of a safety and dose-finding study in GHD children. J Clin Endocrinol Metab. 2017;102:1578–1587 [PDF]
Fisher DM, Rosenfeld RG, Jaron-Mendelson M, et al. Pharmacokinetic and pharmacodynamic modeling of MOD-4023, a long-acting human growth hormone, in GHD Children. Horm Res Paediatr. 2017;87:324–332 [PDF]
Kramer W, Jaron-Mendelson M, Koren R, et al. Pharmacokinetics, Pharmacodynamics and Safety of a Long-Acting Human Growth Hormone (MOD-4023) in Healthy Japanese and Caucasian Adults. Clin Pharmacol Drug Dev. 2017 [in press]
Society and culture
Legal status
On 16 December 2021, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Ngenla, intended for the treatment of growth hormone deficiency (GHD) in children and adolescents from 3 years of age.[3] The applicant for this medicinal product is Pfizer Europe MA EEIG.[3]
Somatrogon was approved for medical use in Australia in November 2021.[1]
References
- ^ Jump up to:a b c d “Ngenla”. Therapeutic Goods Administration (TGA). 13 December 2021. Retrieved 28 December 2021.
- ^ Jump up to:a b “Pfizer and OPKO Announce Extension of U.S. FDA Review of Biologics License Application of Somatrogon for Pediatric Growth Hormone Deficiency” (Press release). Opko Health. 24 September 2021. Retrieved 18 December 2021 – via GlobeNewswire.
- ^ Jump up to:a b “Ngenla: Pending EC decision”. European Medicines Agency (EMA). 16 December 2021. Retrieved 18 December 2021. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
Further reading
- Fisher DM, Rosenfeld RG, Jaron-Mendelson M, Amitzi L, Koren R, Hart G (2017). “Pharmacokinetic and Pharmacodynamic Modeling of MOD-4023, a Long-Acting Human Growth Hormone, in Growth Hormone Deficiency Children”. Horm Res Paediatr. 87 (5): 324–32. doi:10.1159/000470842. PMC 5637306. PMID 28399519.
External links
- “Somatrogon”. Drug Information Portal. U.S. National Library of Medicine.
///////////Somatrogon, NGENLA, APPROVALS 2022, JAPAN 2022, ソマトロゴン , MOD-4023, Modigene, OPKO Health, Pfizer

NEW DRUG APPROVALS
one time
$10.00
Gefapixant citrate

Gefapixant
- Molecular FormulaC14H19N5O4S
- Average mass353.397 Da
1015787-98-0[RN]
10642
AF 217
5-[(2,4-Diamino-5-pyrimidinyl)oxy]-4-isopropyl-2-methoxybenzenesulfonamide
5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzene- sulfonamide
Gefapixant Citrate
| Formula | C14H19N5O4S. C6H8O7 |
|---|---|
| CAS | 2310299-91-1 |
| Mol weight | 545.5203 |
APPROVED JAPAN PMDA 2022/1/20, Lyfnua
ゲーファピキサントクエン酸塩
吉法匹生
| Efficacy | Analgesic, Anti-inflammatory, Antitussive, P2X3 receptor antagonist |
|---|---|
| Comment | Treatment of disorders associated with purinergic receptor activation |
Gefapixant (MK-7264) is a drug which acts as an antagonist of the P2RX3 receptor, and may be useful in the treatment of chronic cough.[1][2][3] It was named in honour of Geoff Burnstock.[4]
Gefapixant is under investigation in clinical trial NCT02397460 (Effect of Gefapixant (AF-219/MK-7264) on Cough Reflex Sensitivity).
PAPER
Organic Process Research & Development (2020), 24(11), 2445-2452.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00248
A robust, green, and sustainable manufacturing process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. The newly developed commercial process features low process mass intensity (PMI), short synthetic sequence, high overall yield, minimal environmental impact, and significantly reduced API costs. The key innovations are the implementation of a highly efficient two-step methoxyphenol synthesis, an innovative pyrimidine synthesis in flow, a simplified sulfonamide synthesis, and a novel salt metathesis approach to consistently deliver the correct active pharmaceutical ingredient (API) salt form in high purity.

SYN
Organic Process Research & Development (2020), 24(11), 2478-2490.
https://pubs.acs.org/doi/10.1021/acs.oprd.0c00252
Gefapixant citrate (MK-7264) is a P2X3 antagonist for the treatment of chronic cough. The second generation manufacturing route developed for the Step 3A/3B formylation–cyclization reaction to generate the key intermediate diaminopyrimidine (1) (AF-072) required a significant excess of ethyl formate (EF), potassium tert-butoxide (KOt-Bu), and guanidine•HCl (G•HCl) when both steps were run as batch processes. It was imperative to develop an alternative process that required less of each reagent and generated less carbon monoxide byproducts, as the annual production of the final active pharmaceutical ingredient (API) is expected to be over 50 MT. In addition, the second generation process was misaligned with our company’s strategy of having the best science in place at the first regulatory filing. The final flow–batch process described herein, which features a flow-based formylation combined with a batch cyclization, has been performed on a 500 kg scale and now requires 35% less EF (leading to a 70% reduction in waste carbon monoxide), 38% less KOt-Bu, and 50% less G•HCl. These improvements, along with a twofold increase in concentration, have resulted in a 54% reduction in the step process mass intensity (step-PMI) from the second generation two-step batch–batch process (PMI of 17.16) to the flow–batch process (PMI of 7.86), without sacrificing reaction performance.

SYN
H. REN*, K. M. MALONEY* ET AL. (MERCK & CO., INC., RAHWAY USA) Development of a Green and Sustainable Manufacturing Process for Gefapixant Citrate (MK-7264) Part 1: Introduction and Process Overview Org. Process Res. Dev. 2020, 24, 2445–2452, DOI: 10.1021/acs.oprd.0c00248.

Syn
https://doi.org/10.1021/acs.jmedchem.3c02374
J. Med. Chem. 2024, 67, 4376−4418
Gefapixant (Lyfnua). Gefapixant (34), also known as MK-7264, prior to that AF-219 and RO-4926219, is a P2 × 3antagonist for the treatment of chronic cough that was recently approved by the Japan Ministry of Health.243 Chronic cough is one of the most frequent reasons for patients to request medical consultation and is defined as cough ≥8 weeks in the past 12 months for those aged 18 years or older.244 The prevalence of chronic cough among US adults is 5% and can be associated with a deterioration of quality of life.244 The commercial manufacturing process of gefapixant has been described by Merck & Co., Inc., Rahway, NJ, USA, and is outlined in Scheme 59. Synthesis of 34 began with the regioselective bromination of isopropyl phenol 34.1. 245−247 The choice of polar MeCN solvent was found to play a critical role in the bromination regioselectivity providing the parabromophenol 34.2 in high yield. Interestingly, when toluene was used as the solvent the undesired ortho-substituted brominated phenol was the major product. In trial experiments it was discovered that a small amount of dibrominated product was formed which was alleviated using 1 mol % of methanesulfonic acid. Copper-mediated C−O bond formation proceeded with the use of NaOMe and CuBr in DMF to provide 34.3 in 92% yield. The authors describe in detail the screening conditions employed and the dimerization biproducts initially observed when obtaining 34.3. Ultimately, the use of DABCO in the first step allowed for the crystallization of the brominated phenol 34.2 as a DABCO
adduct. This enabled the Cu-catalyzed methoxylation to proceed without the need for phenol protection as well as the suppression of undesired dimerization products.247 Alkylation of phenol 34.3 with chloroacetonitrile in the presence of aqueous sodium hydroxide provided cyanomethyl intermediate 34.4.248 The diaminopyrimidine heterocycle was formed by formylation using ethyl formate and KOtBu
followed by reaction with guanidine HCl to complete the cyclization and obtain 34.5 in 81% yield.249 This was performed in a hybrid flow-batch telescoped process. Treat ment of 34.5 with chlorosulfonic acid in MeCN followed by ammonium hydroxide provided sulfonamide 34.6 in high yield.250,251 The final step in the manufacturing process was the isolation of gefapixant as a mono citrate salt.252,253 The free base of gefapixant was converted to a highly soluble glycolate salt which enabled complete dissolution in MeOH. Citric acid was added to crystallize final API as a mono citrate salt in 93%
yield.
(243) Merck & Co. Inc. Merck provides U.S. and Japan regulatory
update for gefapixant. https://www.merck.com/news/merck-providesu-s-and-japan-regulatory-update-for-gefapixant/ (accessed 2023-06).
(244) Yang, X.; Chung, K. F.; Huang, K. Worldwide prevalence, risk
factors and burden of chronic cough in the general population: a
narrative review. J. Thorac. Dis. 2023, 15, 2300−2313.
(245) Kocienski, P. Synthesis of gefapixant. Synfacts 2021, 17,
No. 0123.
(246) Ren, H.; Maloney, K. M.; Basu, K.; Di Maso, M. J.;
Humphrey, G. R.; Peng, F.; Desmond, R.; Otte, D. A. L.; Alwedi, E.;
Liu, W. J.; et al. Development of a green and sustainable
manufacturing process for gefapixant citrate (MK-7264). Part 1:
Introduction and process overview. Org. Process Res. Dev. 2020, 24,
2445−2452.
(247) Peng, F.; Humphrey, G. R.; Maloney, K. M.; Lehnherr, D.;
Weisel, M.; Levesque, F.; Naber, J. R.; Brunskill, A. P. J.; Larpent, P.;
Zhang, S. W.; et al. Development of a green and sustainable
manufacturing process for gefapixant citrate (MK-7264). Part 2:
Development of a robust process for phenol synthesis. Org. Process
Res. Dev. 2020, 24, 2453−2461.
(248) Basu, K.; Lehnherr, D.; Martin, G. E.; Desmond, R. A.; Lam,
Y.-h.; Peng, F.; Chung, J. Y. L.; Arvary, R. A.; Zompa, M. A.; Zhang,
S.-W.; et al. Development of a green and sustainable manufacturing
process for gefapixant citrate (MK-7264). Part 3: development of a
one-pot formylation−cyclization sequence to the diaminopyrimidine
core. Org. Process Res. Dev. 2020, 24, 2462−2477.
(249) Otte, D. A. L.; Basu, K.; Jellett, L.; Whittington, M.; Spencer,
G.; Burris, M.; Corcoran, E. B.; Stone, K.; Nappi, J.; Arvary, R. A.;
et al. Development of a green and sustainable manufacturing process
for gefapixant citrate (MK-7264). Part 4: Formylation−cyclization as
a flow−batch process leads to significant improvements in process
mass intensity (PMI) and CO generated versus the batch−batch
process. Org. Process Res. Dev. 2020, 24, 2478−2490.
(250) Di Maso, M. J.; Ren, H.; Zhang, S.-W.; Liu, W.; Desmond, R.;
Alwedi, E.; Narsimhan, K.; Kalinin, A.; Larpent, P.; Lee, A. Y.; et al.
Development of a green and sustainable manufacturing process for
gefapixant citrate (MK-7264). Part 5: Completion of the API free
base via a direct chlorosulfonylation process. Org. Process Res. Dev.
2020, 24, 2491−2497.
(251) Rivera, N. R.; Cohen, R. D.; Zhang, S.-W.; Dance, Z. E. X.;
Halsey, H. M.; Song, S.; Bu, X.; Reibarkh, M.; Ren, H.; Lee, A. Y.;
et al. Gefapixant citrate (MK-7264) sulfonamide step speciation
study: Investigation into precipitation−dissolution events during
addition of chlorosulfonic acid. Org. Process Res. Dev. 2023, 27,
286−294.
(252) Maloney, K. M.; Zhang, S.-W.; Mohan, A. E.; Lee, A. Y.;
Larpent, P.; Ren, H.; Humphrey, G. R.; Desmond, R.; DiBenedetto,
M.; Liu, W.; et al. Development of a green and sustainable
manufacturing process for gefapixant citrate (MK-7264). Part 6:
Development of an improved commercial salt formation process. Org.
Process Res. Dev. 2020, 24, 2498−2504.
(253) Mohan, A. E.; DiBenedetto, M.; Alwedi, E.; Ang, Y. S.; Asi
Sihombing, M. S. B.; Chang, H. Y. D.; Cote, A.; Desmond, R.; DiazSantana, A.; Khong, E.; et al. Development and Demonstration of a
Co-feed Process to Address Form and Physical Attribute Control of
the Gefapixant (MK-7264) Citrate Active Pharmaceutical Ingredient.
Org. Process Res. Dev. 2021, 25, 541−551.


AS ON JUNE2025 4.45 LAKHS VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@gmail.com

……
SYN
https://pubs.acs.org/doi/abs/10.1021/acs.oprd.0c00247

A scalable two-pot sulfonamidation through the process has been developed for the synthesis of gefapixant citrate, a P2X3 receptor antagonist that is under investigation for the treatment of refractory and unexplained chronic cough. Direct conversion of the diaryl ether precursor to a sulfonyl chloride intermediate using chlorosulfonic acid, followed by treatment with aqueous ammonia hydroxide, provided the desired sulfonamide in high yield. A pH-swing crystallization allowed for the formation of a transient acetonitrile solvate that enables the rejection of two impurities. After drying, the desired anhydrous free base form was isolated in high yield and purity.
SYN
https://www.sciencedirect.com/science/article/abs/pii/S1566070221000898
Gefapixant is the approved generic name for a compound also known as MK-7264, and prior to that AF-219 and RO-4926219. It is the first-in-class clinically developed antagonist for the P2X3 subtype of trimeric ionotropic purinergic receptors, a family of ATP-gated excitatory ion channels, showing nanomolar potency for the human P2X3 homotrimeric channel and essentially no activity at related channels devoid of P2X3 subunits. As the first P2X3 antagonist to have progressed into clinical studies it has now progressed to the point of successful completion of Phase 3 investigations for the treatment of cough, and the NDA application is under review with US FDA for treatment of refractory chronic cough or unexplained chronic cough. The molecule was discovered in the laboratories of Roche Pharmaceuticals in Palo Alto, California, but clinical development then continued with the formation of Afferent Pharmaceuticals for the purpose of identifying the optimal therapeutic indication for this novel mechanism and establishing a clinical plan for development in the optimal patient populations selected. Geoff Burnstock was a close collaborator and advisor to the P2X3 program for close to two decades of discovery and development. Progression of gefapixant through later stage clinical studies has been conducted by the research laboratories of Merck & Co., Inc., Kenilworth, NJ, USA (MRL; following acquisition of Afferent in 2016), who may commercialize the product once authorization has been granted by regulatory authorities.
PATENT
WO 2008040652
https://patents.google.com/patent/WO2008040652A1/en

SCHEME AExample 1: 5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamideThe synthetic procedure used in this Example is outlined in Scheme B.


not isolated


SCHEME BStep 1 2-Isopropyl-4-methoxy-phenolTo a cooled solution of l-(2-hydroxy-5-methoxy-phenyl)-ethanone (10.0 kg) in 79.0 kg of THF was gradually added 46.4 kg of 3M solution of MeMgCl in THF at a rate such that the reaction mixture temperature did not exceed 25°C. Following addition of the MeMgCl solution, the reaction mixture was stirred at ambient temperature for 18 hours, at which point HPLC (high pressure liquid chromatography) analysis showed more than 98% conversion of l-(2-hydroxy-5-methoxy-phenyl)-ethanone to 2- (1 -hydroxy- 1- methyl-ethyl)-4-methoxy-phenol (not shown in Scheme D). To the stirred solution was then added 10% palladium on carbon (1.02 kg, 50% water wet) suspended in 3.5 kg of THF. The reaction mixture was cooled and placed under a hydrogen atmosphere at 0.34 atmosphere pressure, and concentrated HCl (19.5 kg) was added while maintaining the reaction temperature at 25°C. The resultant mixture was stirred at ambient temperature for 18 hours, then treated with 44.4 kg water and filtered through a bed of Celite to remove suspended catalyst. The filter cake was rinsed with EtOAc and the combined filtrate was separated. The organic phase was washed with water, then concentrated by distillation to provide an oil. This oil was dissolved in 2-butanone (20.4 kg) and the crude solution was employed directly in the next step. A 161.8 g aliquot of the solution was concentrated under vacuum to provide 49.5 g of 2-isopropyl-4-methoxyphenol as an oil, projecting to 10.4 kg crude contained product in the bulk 2-butanone solution. 1H NMR (DMSO) delta: 1.14 (d, 6H, J = 6.9 Hz), 3.18 (septet, IH, J = 6.9 Hz), 3.65 (s, 3H), 6.56, (dd, IH, J = 8.6 Hz, 3.1 Hz), 6.67 (d, IH, J = 3.1 Hz), 6.69 (d, IH, 8.6 Hz).Step 2 (2-Isopropyl-4-methoxy-phenoxy)-acetonitrileA stirred slurry of toluene-4-sulfonic acid cyanomethyl ester (13.0 kg), potassium carbonate (13.0 kg) and 2-isopropyl-4-methoxyphenol (9.57 kg) in 79.7 kg of 2-butanone was heated to 55-600C for 4 days, then heated to reflux for 18 hours. The resultant slurry was cooled and filtered to remove solids. The filtrate was concentrated under reduced pressure and the residue was redissolved in toluene. The toluene solution was extracted with IN KOH, and the organic phase was concentrated by distillation to give 20.6 g of a 1:1 (by weight) solution of (2-isopropyl-4-methoxy-phenoxy)-acetonitrile in toluene, which was used directly in the next step. A aliquot (96.7 g) of this solution was concentrated to dryness to give 50.9 g of crude (2-isopropyl-4-methoxy-phenoxy)- acetonitrile, projecting to a yield of 10.9 kg in the bulk solution: MS (M+H) = 206; 1H NMR (CDCl3) delta: 1.25 (d, J = 6.9 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 4.76 (s, 2H), 6.73 (dd. IH, J = 8.8 Hz, 3.1 Hz), 6.87 (d, IH, J = 3.1 Hz), 6.91 (d, IH, J = 8.8 Hz).Step 3 5-(2-Isopropyl-4-methoxy-phenoxy)-pyrimidine-2,4-diamine An approximately 1:1 (by weight) solution of 10.6 kg of (2-isopropyl-4-methoxy-phen- oxy) -acetonitrile in toluene was concentrated under reduced pressure and the residue was treated with 10.8 kg of tert-butoxybis(dimethylamino)methane (Brederick’s Reagent). The resulting mixture was dissolved in 20.2 kg of DMF and the solution was heated to 1100C for 2 hours, at which point HPLC analysis showed essentially complete conversion to 3,3-bis-dimethylamino-2-(2-isopropyl-4-methoxy-phenoxy)-propionitrile (not isolated, 1H NMR (CDCl3) delta: 1.21 (d, 3H, J = 7.2 Hz), 1.23 (d, 3H, J = 7.1 Hz), 2.46 (s, 6H), 2.48 (s, 6H), 3.43 (d, IH, J = 5.0 Hz), 3.31 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.93 (d, IH, J = 5.0 Hz), 6.70 (dd, IH, J = 8.8 Hz, 3.0 Hz), 6.82 (d, IH, J = 3.0 Hz), 6.98 (d, IH, J = 8.8 Hz). The DMF solution was cooled and transferred onto 14.7 kg of aniline hydrochloride. The resulting mixture was heated to 1200C for 22 hours, at which point HPLC analysis showed greater than 97% conversion to 2-(2-isopropyl-4-methoxy-phenoxy)-3- phenylamino-acrylonitrile (not isolated, 1H nmr (CDCl3) delta: 1.31 (d, 6H, J = 6.9 Hz), 3.39 (septet, IH, J = 6.9 Hz), 3.82 (s, 3H), 6.61 (d (br), IH, J = 12.7 Hz), 6.73 (dd, IH, J = 8.9 Hz, 3.1 Hz), 6.88 (d, IH, J = 3.0 Hz), 6.93 (m, 2H), 6.97 (d, IH, J = 8.9 Hz), 7.05 (m, IH), 7.17 (d, IH, J = 12.6 Hz), 7.35 (m. 2H)).The mixture was cooled, diluted with 21.5 kg toluene, then with 72.2 L of water. The organic layer was separated, washed with water, and concentrated by distillation. The concentrate was transferred into 23.8 kg DMF, and the DMF solution was transferred onto 6.01 kg of guanidine carbonate. The resulting mixture was heated to 1200C for 3 days, at which point HPLC analysis showed greater than 95% conversion of 2-(2- isopropyl-4-methoxy-phenoxy)-3-phenylamino-acrylonitrile into 5-(2-Isopropyl-4- methoxy-phenoxy)-pyrimidine-2,4-diamine. The reaction mixture was cooled, diluted with 7.8 kg of EtOAc, then reheated to 600C. Water (75.1 L) was added and the resultant mixture was allowed to cool to ambient temperature. The precipitated solid was collected by filtration, rinsed with isopropanol and dried under vacuum at 50 degrees to give 9.62 kg of 5-(2-isopropyl-4-methoxy- phenoxy)-pyrimidine-2,4-diamine: m.p. 170-171 degrees C; MS (M+H) = 275; H nmr (chloroform) delta: 1.25 (d, 6H, J = 6.9 Hz), 3.30 (septet, IH, J = 6.9 Hz), 3.79 (s, 3H), 4.68 (br, 2H), 4.96 (br, 2H), 6.64 (dd, IH, J = 8.9 Hz, 3.0 Hz), 6.73, d, J = 8.9 Hz), 6.85 (d, IH, J = 3 Hz), 7.47 (s, IH).Step 4 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide, sulfolane solvate Chlorosulfonic acid (13.82 kg) was added to a slurry of 5-(2-isopropyl-4-methoxy-phen- oxy)-pyrimidine-2,4-diamine (10.07 kg) in sulfolane (50.0 kg) at a rate to maintain an internal pot temperature below 65°C. The reaction mixture was aged at 60-650C for 12 hours, at which point HPCL showed that all 5-(2-isopropyl-4-methoxy-phenoxy)- pyrimidine-2,4-diamine starting material had been converted to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonic acid. MS (M+H) = 355. Phosphorus oxychloride (3.41 kg) was then added to the reaction mixture at 600C. The reaction mixture was heated to 75°C and aged for 12 hours, at which point HPLC showed that approximately 99% of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonic acid had been converted to 5-(2,4-diamino-pyrimidin-5-yloxy)-4-iso- propyl-2-methoxy-benzenesulfonyl chloride. MS (M+H) = 373. The solution of 5-(2,4- diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride was then cooled to around 2°C).To a cooled (ca. 2°C) solution of ammonia (7N) in MeOH (74.1 kg) was added the cooled sulfolane solution of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonyl chloride (a homogeneous syrup) at a rate such that the internal temperature did not exceed 23°C. The resultant slurry was stirred for 18 hours at ambient temperature, then filtered on a coarse porosity frit filter. The collected solids were rinsed with MeOH (15.9 kg), then dried under reduced pressure at 700C to a constant weight of 23.90 kg. HPLC showed 97.5% conversion of 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride to 5-(2,4-diamino- pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide sulfolane solvate. H nmr (DMSOd6) delta: 1.26 (d, 6H, J = 6.9 Hz), 2.07 (sym. m, 8H), 2.99 (sym. m, 8H), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 6.03 (s (br), 2H), 6.58 (s (br), 2H), 7.00 (s, IH), 7.04 (s (br), 2H), 7.08 (s, IH), 7.35 (s, IH).
Step 5 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzene- sulfonamideA slurry of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfon- amide sulfolane solvate (23.86 kg) in a mixture of ethanol (74.3 kg) and 0.44 N HCl (109.4 kg) was heated to reflux to provide a homogeneous solution of the monohydrochloride salt of 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonamide. This solution was filterd while hot, then treated with concentrated ammonium hydroxide (3.4 L) to liberate the free base of 5-(2,4-diamino-pyrimidin-5- yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide. The resultant mixture was cooled slowly to 200C and the crystalline product isolated by filtration. The filter cake was washed with water (20.1 kg) and dried under reduced pressure at 700C to a constant weight of 8.17 kg (57.7% yield based on di-solvate of sulfolane).MP = 281-282 0C.1H nmr (DMSOd6) delta: 1.27 (d, 6H, J = 6.9 Hz), 3.41 (septet, IH, J = 6.9 Hz), 3.89 (s, 3H), 5.87 (s (br), 2H), 6.40 (s (br), 2H), 6.98 (s, IH), 7.01 (s (br), 2H), 7.07 (s, IH), 7.36 (s, IH).
PATENT
US 20080207655https://patents.google.com/patent/US20080207655
PATENThttps://patentscope.wipo.int/search/en/detail.jsf?docId=WO2016004358
xample 20
5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-N-methyl-benzenemethylsulfonamide Step 1. 5-(2,4-Diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy- benzenesulfonyl chloride
[211] A mixture of pyrimidine (0.400 g, 1.5 mmol) in 2 ml chlorosulfonic acid was allowed to stir 20 min. The mixture was poured over ice. The precipitate was filtered, washed by cold H2O and dried under vacuum to afford 5-(2,4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonyl chloride (0.515 g, 95%) as a white solid; [MH]+= 373.
PATENT
WO 2017058645
https://patents.google.com/patent/WO2017058645A1/en
PATENTDisclosed herein is a novel process for preparing Compound A, a phenoxy diaminopyrimidine compound of the following formula, or a pharmaceutically acceptable salt thereof:

Compound A.Also disclosed herein are various salts and solvates of Compound A.
Scheme 1


Step 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalStep 1. Preparation of 4-Bromo-2-isopropylphenol DABCO Co-crystalThe following 4-bromo-2-isopropylphenol hemi-DABCO co-crystal is obtained in greater than 99% purity and at about 85-92% yield by the following process:

To a solution of 2-isopropyl phenol (75.0 g, 550 mmol) in acetonitrile (225 mL) was added MSA (0.520 g, 5.41 mmol). The mixture was cooled to -10 °C and NBS (98.01 g, 550 mmol) was added in portions while maintaining the internal temperature below 10 °C. The reaction was aged for 30 min to 1 h and then warmed to 20 °C, diluted with water (450 mL), and extracted with toluene (225 mL). The organic layer was sequentially washed with 9 wt% phosphoric acid (150 mL) and 5 wt% NaCl (150 mL). The organic layers were concentrated to roughly 150 mL and filtered into a clean reactor. The mixture was heated to 30-40 °C and n- heptane (28.5 mL) was added followed by DABCO (30.89 g, 275 mmol). The mixture was seeded (a seed can be synthesized from a previous batch of this procedure preformed without seeding) with 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (75 mg, 0.277 mmol), diluted with 52.5 mL of n-heptane, and stirred for 1 h. The slurry was cooled to 20 °C over 1 h and 370 mL of n-heptane is added over 2 h. The slurry was cooled to 5 °C over 2 h, aged for 2 h, filtered, and washed with n-heptane (2 x 75 mL). The solid was dried at 20-25 °C under vacuum to yield 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (134.8 g, 90 %) as a solid. 1H NMR (400 MHz, DMSO-76) d 7.20 (d, J= 2.5 Hz, 1H), 7.13 (dd, J= 8.5, 2.6 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 3.16 (hept, J= 6.9 Hz, 2H), 2.60 (s, 12H), 1.14 (d, J= 6.9 Hz, 12H).The crystallization of step 1 generates 4-bromo-2-isopropylphenol hemi-DABCO co-crystal, bromophenol mono-DABCO co-crystal, or a mixture of bromophenol hemi-DABCO co-crystal and bromophenol mono-DABCO co-crystal. An XRPD pattern of bromophenol hemi- DABCO co-crystal is shown in Figure 1.
The bromo-phenol mono-DABCO co-crystal can be generated in the following procedure:

bromophenol DABCO co-crystalTo a vial with a stir bar was charged DABCO (1.7 g, 15 mmol), phenol (2.5 g, 15 mmol), and 2 mL of n-heptane. The resulting slurry was stirred at 23 °C overnight. The slurry was then filtered and the resulting wet cake was washed with 2 mL of 5 °C n-heptane. The cake was dried under vacuum with nitrogen sweep to afford 4-bromo-2-isopropylphenol mono- DABCO co-crystal (2.9 g, 70% yield) as a solid. 1H NMR (500 MHz, DMSO-76) d 9.65 (s, 1H), 7.20 (s, 1H), 7.14 (d, J= 8.5 Hz, 1H), 6.74 (d, J= 8.5 Hz, 1H), 3.17 (hept, J= 6.8 Hz, 1H), 2.61(s, 12H), 1.15 (d, 7 = 6.9 Hz, 6H).An XRPD pattern of bromophenol mono-DABCO co-crystal is shown in Figure 2.Step 2a. Preparation of 2-Isopropyl-4-Methoxyphenol
The 2-isopropyl-4-Methoxyphenol shown below is obtained at about 92% yield by the following process:

bromophenol DABCO co-crystal methoxy phenolTo a solution of 4-bromo-2-isopropylphenol hemi-DABCO co-crystal (120 g, 442 mmol) in 25 wt% sodium methoxide in methanol (430 g) was added 60 mL of DMF. The solution was pressure purged with nitrogen, copper (I) bromide (3.23 g, 22.5 mmol) was added to the mixture, and the reaction was heated to reflux for 12-16 h. The reaction is cooled to 0-5 °C and quenched with 6M HC1 until the pH of the solution is less than 5. The slurry is diluted with 492 mL of toluene and 720 mL of water to provide a homogeneous solution with a rag between the layers. The aqueous layer is cut to waste. The organic layer is filtered to remove the rag and washed with 240 mL of water to provide 2-isopropyl-4-methoxylphenol (491 g, 13.3 wt%, 89% assay yield) as a solution in toluene. 1H NMR (500 MHz, DMSO-76) d 8.73 (s, 1H), 6.68 (d, J = 8.6 Hz, 1H), 6.66 (d, 7= 3.0 Hz, 1H), 6.55 (dd, 7= 8.6, 3.1 Hz, 1H), 3.65 (s, 3H), 3.17 (hept, j = 6.9 Hz, 1H), 1.14 (d, 7= 6.9 Hz, 6H).Step 2b. Preparation of 2-Isopropyl-4-Methoxyphenol
Alternatively, the methoxy phenol is obtained by the following process:

To a high-pressure vessel were charged 400 mL of anhydrous toluene, Re2(CO)io (3.16 g, 4.84 mmol) and mequinol (100 g, 806 mmol) at RT. The vessel was then degassed with propylene, and charged with propylene (85.0 g, 2.02 mol). The vessel was sealed and heated to 170 °C. Internal pressure was measured near 250 psi. The reaction was stirred at this condition for 72 h. The vessel was then allowed to cool down to 23 °C. The internal pressure was carefully released to 1 atmospheric pressure, and the toluene solution was assayed as 91% and used directly in the next step or isolated as a solid.Step 2a/2b results in anhydrous 2-isopropyl-4-methoxyphenol form 1. An XRPD pattern of the methoxy phenol form 1 is shown in Figure 3.In another embodiment, the product is isolated as a DMAP co-crystal:

To a vial with a stir bar was charged DMAP (3.67 g, 30.1 mmol), 2.5 ml of toluene, and 2-isopropyl-4-methoxylphenol (5.00 g, 30.1 mmol). The reaction mixture was stirred at RT for 5 min, and a homogeneous solution was formed. The reaction mixture was then cooled to 5 °C. Ten mL of n-heptane was slowly charged over 20 min. The resulting slurry was stirred at 5 °C overnight. The slurry was filtered and the resulting wet cake was washed with 3 mL of 5 °C n-heptane. The cake was dried under vacuum with a nitrogen sweep to provide 2- isopropyl-4-methoxylphenol DMAP co-crystal (7.01 g, 81%) as a solid. 1H NMR (500 MHz, DMSO-76) d 8.78 (s, 1H), 8.10 (d, J= 6.1 Hz, 2H), 6.71 – 6.65 (m, 2H), 6.57 (dd, J= 11.3, 6.0 Hz, 3H), 3.66 (s, 3H), 3.17 (hept, J= 6.8 Hz, 1H), 2.95 (s, 6H), 1.14 (d, J= 6.9 Hz, 6H).The crystallization generates anhydrous 2-isopropyl -4-methoxyphenol DMAP co crystal. An XRPD pattern of the 2-isopropyl-4-methoxyphenol DMAP co-crystal is shown in Figure 4.Step 3a. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
The cyanoether is obtained at about 95 % yield by the following process:

A 12-15 wt% solution of 2-isopropyl-4-methoxylphenol (314.3 g, 12 wt%, 226.8 mmol) was concentrated to greater than 50 wt% 2-isopropyl-4-methoxyphenol in toluene under vacuum at 40-50°C. To the solution was added 189 mL of NMP, and the mixture was cooled to 5 °C. Sodium hydroxide (27.2 g, 50 wt% in water, 340 mmol) and chloroacetonitrile (36 g, 340 mmol) were added sequentially to the mixture while maintaining the internal temperature below 10 °C. The reaction was aged for 2 h and then diluted with 150 mL of toluene and 226 mL of water while maintaining the temperature below 10 °C. The mixture was warmed to 20-25 °C, the layers were separated, and the organic layer was washed with 75 mL of 20 wt% NaCl (aq.). The organic layer was and filtered to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (56.8 g, 74.6 wt%) as a solution in toluene. The filter was washed with NMP to provide additional 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile (27.1 g, 5.0 wt%) as a solution in NMP. The combined yield was about 94 %. 1H NMR (500 MHz, DMSO-i¾) d 7.05 (d, J= 8.8 Hz, 1H), 6.81 (d, 7= 3.0 Hz, 1H), 6.78 (dd, j= 8.8, 3.1 Hz, 1H), 5.11 (s, 2H), 3.73 (s, 3H), 3.20 (hept, j = 6.9 Hz, 1H), 1.17 (d, 7= 6.9 Hz, 6H).Step 3b. Preparation of the Cvanoether. 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile
Alternatively, the cyanoether shown below is obtained at about 92% yield by the following process:

A solution of 2-isopropyl-4-methoxyphenol in toluene (491 g, 13.3 wt%, 393 mmol) was concentrated and solvent switched to acetonitrile under vacuum at 40-50 °C.Potassium carbonate (164.5 g, 1190 mmol) and tetrabutylammonium hydrogensulfate (1.5 g, 4.42 mmol) were added to a separate vessel, and the vessel was pressure purged with nitrogen gas.The solution of phenol in acetonitrile and chloroacetonitrile was added sequentially to the reaction vessel. The vessel was heated to 40 °C and aged for 4 h. The mixture was allowed to cool to 25 °C, and was diluted with 326 mL water. The layers were separated, and the organic layer was washed with 130 mL of 10 wt% NaCl. A solvent switch to toluene was performed under vacuum, and the organic layer was filtered through two 16D Cuno #5 cartridges. The organic layer was concentrated to provide 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile in toluene (128.2 g, 58 wt%, 92% yield).Step 4 Preparation of the Dia inopyrimidine 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2.4-di amineThe diaminopyrimidine is obtained at about 90 % yield by the following process:

A solution of potassium tert-butoxide (44.8 g, 0399 mmol) in NMP (180 mL) was cooled to -10 °C. A solution of 2-(2-isopropyl-4-methoxyphenoxy)acetonitrile, the cyanoether, (59.3 g, 61.4 wt%, 177 mmol) in toluene and ethyl formate (26.3 g, 355 mmol) was charged to the base solution while maintaining the internal temperature between -12 °C and -8 °C. After a 3 h age, guanidine hydrochloride (136 g, 1420 mmol) was added to the mixture and the reaction was heated to 115 °C for 6 h. The mixture was allowed to cool to 90 °C, diluted with 200 mL of water, and aged until the reaction mixture was homogeneous (about 30-45 min). After all solids dissolved, vacuum (400 mm Hg) was applied to the reactor to remove toluene. Vacuum was disconnected and the solution was allowed to cool to 85°C. 5-(2-Isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine seed (49.8 mg) (a seed can be synthesized by a route described in U.S. Patent 7,741,484) was charged, the solution was aged for 2 h, 200 mL of water was added, and the batch was allowed to cool to 20 °C over 6 h. The slurry was aged for 10 h at 20 °C, filtered, washed with 2: 1 water :NMP (3 x 100 mL) and water (3 x 100 mL), and dried under vacuum at 50 °C to provide the title compound (42.2 g, 88%) as a solid. 1H NMR (500 MHz, DMSO-r¾) d 7.23 (s, 1H), 6.83 (d, J= 3.0 Hz, 1H), 6.70 (dd, J= 8.9, 3.0 Hz, 1H), 6.63 (d, j= 8.8 Hz, 1H), 6.32 (s, 2H), 5.75 (s, 2H), 3.71 (s, 3H), 3.28 (hept, j= 6.9 Hz, 1H), 1.20 (d, j = 6.9 Hz, 6H); 13C NMR (126 MHz, DMSO-r¾) d 159.7, 157.2, 155.1, 148.4, 144.2, 139.0, 130.4,116.9, 112.5, 111.3, 55.4, 26.57, 22.83.The crystallization of step 4 generates an anhydrous 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1. An XRPD pattern of the 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 is shown in Figure 5.In one embodiment, 5-(2-isopropyl-4-methoxyphenoxy)pyrimidine-2, 4-diamineNMP solvate 1 is obtained by adding excess amount of 5-(2-isopropyl-4- methoxyphenoxy)pyrimidine-2, 4-diamine form 1 into NMP in a closed vessel to form a suspension. The suspension is stirred at RT until the completion of form transition. The crystals of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of the 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine NMP solvate 1 is shown in Figure 6.Step 5. Preparation of Compound A Free BaseCompound A free base is obtained at about 91% yield by a process comprising the steps:

To a suspension of 5-(2 -isopropyl -4-methoxyphenoxy)pyrimidine-2, 4-diamine, the diaminopyrimidine, (47.0 g, 171 mmol) in 141 mL of acetonitrile at -10 °C was added chlorosulfonic acid (63.1 mL, 942 mmol) while maintaining the internal temperature below 25 °C. The solution was aged for 1 h at 25 °C and then heated to 45 °C for 12 h. The solution was allowed to cool to 20 °C and added to a solution of 235 mL ammonium hydroxide and 71 mL of acetonitrile at -10 °C while maintaining the internal temperature below 15 °C. The slurry was aged at l0°C for 1 h, heated to 25 °C, and aged for 1 h. The slurry was diluted with 564 mL of water and 188 mL of 50 wt% sodium hydroxide to provide a homogeneous solution that was heated to 35 °C for 2 h. The solution was allowed to cool to 22 °C and the pH of the solution was adjusted to 12.9 with a 2M solution of citric acid. The solution was seeded with Compound A free base (470 mg, 1.19 mmol) (a seed can be synthesized by a route described in U.S. Patent 7,741,484), aged for 2 h, acidified to pH 10.5-11.3 with a 2M solution of citric acid over 5-10 h, and then aged for 2 h. The slurry was filtered, the resulting cake was washed with 90: 10 water: acetonitrile (2 x 118 mL) and water (2 x 235 mL), and dried at 55 °C under vacuum to provide Compound A free base (50.9 g, 91%) as a solid. 1H NMR (500 MHz, DMSO-i¾) d 7.36 (s, 1H), 7.07 (s, 1H), 7.05 – 6.89 (m, 3H), 6.37 (s, 2H), 5.85 (s, 2H), 3.89 (s, 3H), 3.41 (hept, J = 6.6 Hz, 1H), 1.27 (d, J= 6.8 Hz, 6H).The crystallization of step 5 generates anhydrous Compound A free base form 1. In one embodiment, Compound A free base acetonitrile solvate 1 can be prepared by adding excess amount of Compound A free base form 1 into acetonitrile in a closed vessel to form a suspension. The suspension is stirred at 50 °C until the completion of form transition.The crystals of Compound A free base acetonitrile solvate 1 can be collected by filtration and measured immediately by XRPD to prevent desolvation. An XRPD pattern of Compound A free base acetonitrile solvate 1 is shown in Figure 7.Step 6a. Preparation of Compound A Citrate SaltCompound A citrate salt is obtained by a process comprising the steps:

Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) were added to methanol (360 mL). The solution was heated to 60 °C, aged for 1 h, and filtered through a 0.6 pm filter into a clean vessel. A solution of citric acid (32.6 g, 170 mmol) in 2- propanol (180 mL) at RT was filtered through a 0.6 pm filter into the methanol solution over 30 min while the temperature of the methanol solution was maintained between 58-62 °C. The solution was seeded with Compound A citrate salt (450 mg, 0.825 mmol) (a seed can be synthesized by a route described in patent application number PCT/US17/66562), aged for 1 h, and diluted with 180 mL of 2-propanol over 3 h while the temperature was maintained between 58-62 °C. The slurry was cooled to 50 °C over 3 h. The slurry was filtered at 50 °C, washed with 1 : 1 methanol :2-propanol (120 mL) and 2-propanol (120 mL) at 50 °C, and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 97%) as a solid. 1H NMR (400 MHz, DMSO-76) d 10.89 (s, 3H), 7.33 (s, 1H), 7.10 (s, 1H), 7.07 (s, 3H), 7.04 (s, 2H), 6.44 (s, 2H), 3.91 (s, 3H), 3.34 (hept, J= 6.7 Hz, 1H), 2.69 (d, 7= 15.3 Hz, 2H), 2.60 (d, 7= 15.3 Hz, 2H), 1.26 (d, 7= 6.9 Hz, 6H). Step 6b. Alternative preparation of Compound A Citrate SaltAlternatively, Compound A citrate salt is obtained by a process comprising the steps:

To a suspension of Compound A citrate salt (4.5 g, 8.25 mmol) in methanol (72 mL) and 2-propanol (36 mL) at 50 °C were added simultaneously through separate 0.6 pm filters a solution of Compound A free base (30.0 g, 84.9 mmol) and glycolic acid (22.6 g, 297 mmol) in 360 mL of methanol at 50 °C and a solution of citric acid (19.5 g, 101 mmol) in 180 mL of 2- propanol at 25 °C over 8 h while maintaining the seed solution temperature of 60 °C. After the simultaneous addition is complete, citric acid (13.2 g, 68.7 mmol) in 180 mL of 2-propanol was added to the slurry over 8 h while the temperature was maintained at 60 °C. The slurry was allowed to cool to 50 °C and aged for 1 h, filtered at 50 °C, washed with 1 : 1 methanol :2- propanol (2 x 120 mL) and 2-propanol (120 mL), and dried under vacuum at 35 °C to provide Compound A citrate salt (45.1 g, 88%) as a solid.The crystallization of step 6a/6b generates anhydrous Compound A citrate form 1. In another embodiment, Compound A citrate methanol solvate 1 can be prepared via a saturated solution of Compound A citrate form 1 in methanol at 50C. The solution is naturally cooled to ambient temperature or evaporated at ambient temperature until the crystals of Compound A citrate methanol solvate 1 can be acquired. An XRPD pattern of Compound A citrate methanol solvate 1 is shown in Figure 8.
PATENT
https://patents.google.com/patent/CN111635368B/enPreparation of the Compound Gefapixant of example 11Adding compound 7(16g) and dichloromethane (64mL) into a 250mL three-necked bottle, stirring for dissolving, cooling to below 5 ℃ in an ice bath, dropwise adding a mixed solution of chlorosulfonic acid (21.1g) and dichloromethane (16mL) into the reaction solution, and stirring for 1 hour at the temperature of not higher than 5 ℃; then heating to room temperature and continuing stirring for 10 hours, after the reaction is finished, pouring the reaction liquid into ice water, and quickly separating a water layer; the organic layer was washed once with ice water, dried over anhydrous magnesium sulfate and concentrated under reduced pressure to give a crude product. Dissolving the crude product with 30ml of acetonitrile, and cooling to below 5 ℃; 16ml of ammonia water (25-28%) is dripped into the solution, and after the dripping is finished, the solution is heated to room temperature and stirred for 20 hours. After the reaction is completed, concentrating the reaction solution under reduced pressure to remove acetonitrile, and separating out a white solid; and filtering again, and drying the filter cake at 70 ℃ under reduced pressure for 24h to obtain Gefapixant: white powder (19.50g), yield 94.6%, purity: 97.2 percent.Example 12 purification of the Compound GefapixantAdding a compound Gefapixant (20.77g) into a 500mL reaction bottle, adding 0.44N hydrochloric acid (95.4mL), absolute ethyl alcohol (64.4g) and nitrogen protection, heating to 75 ℃, stirring for dissolving, then carrying out heat preservation and reflux for 1 hour, filtering while hot, after filtering, heating the filtrate again to 60 ℃, dropwise adding ammonia water (25-28 percent and 2.96mL), closing and heating after dropwise adding, slowly cooling to room temperature, and gradually precipitating white solids. And continuously cooling the reaction solution to 20 ℃, keeping the temperature and stirring for 4h, filtering, washing a filter cake with 15ml of water, and performing vacuum drying on the obtained wet product at 60 ℃ for 24h to obtain Gefapixant: white powder (6.58g), yield 53.2%, purity: 99.5 percent.1H NMR(400MHz,DMSO)δ7.37(s,1H),7.08(s,1H),7.02(s,2H),7.00(s,1H),6.43(brs,2H),5.89(s,2H),3.90(s,3H),3.42(m,1H),1.28(d,J=8.0Hz,6H);LC-MS:m/z=354.1[M+H]+。
//////////////////////////////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
References
- ^ Muccino D, Green S (June 2019). “Update on the clinical development of gefapixant, a P2X3 receptor antagonist for the treatment of refractory chronic cough”. Pulmonary Pharmacology & Therapeutics. 56: 75–78. doi:10.1016/j.pupt.2019.03.006. PMID 30880151.
- ^ Richards D, Gever JR, Ford AP, Fountain SJ (July 2019). “Action of MK-7264 (gefapixant) at human P2X3 and P2X2/3 receptors and in vivo efficacy in models of sensitisation”. British Journal of Pharmacology. 176 (13): 2279–2291. doi:10.1111/bph.14677. PMC 6555852. PMID 30927255.
- ^ Marucci G, Dal Ben D, Buccioni M, Martí Navia A, Spinaci A, Volpini R, Lambertucci C (December 2019). “Update on novel purinergic P2X3 and P2X2/3 receptor antagonists and their potential therapeutic applications”. Expert Opinion on Therapeutic Patents. 29 (12): 943–963. doi:10.1080/13543776.2019.1693542. hdl:11581/435751. PMID 31726893. S2CID 208037373.
- ^ Ford, Anthony P.; Dillon, Michael P.; Kitt, Michael M.; Gever, Joel R. (November 2021). “The discovery and development of gefapixant”. Autonomic Neuroscience. 235: 102859. doi:10.1016/j.autneu.2021.102859.
| Clinical data | |
|---|---|
| ATC code | R05DB29 (WHO) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 1015787-98-0 |
| PubChem CID | 24764487 |
| DrugBank | DB15097 |
| ChemSpider | 58828660 |
| UNII | 6K6L7E3F1L |
| KEGG | D11349 |
| ChEMBL | ChEMBL3716057 |
| Chemical and physical data | |
| Formula | C14H19N5O4S |
| Molar mass | 353.40 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI |
////////////Gefapixant, Lyfnua, JAPAN 2022, APPROVALS 2022, ゲーファピキサントクエン酸塩 , MK 7264, 吉法匹生 , AF 217

NEW DRUG APPROVALS
ONE TIME
$10.00
UPDATE
.WO/2022/060945SOLID STATE FORMS OF GEFAPIXANT AND PROCESS FOR PREPARATION THEREOF
TEVA
Gefapixant, 5-(2, 4-diamino-pyrimidin-5-yloxy)-4-isopropyl-2-methoxy-benzenesulfonamide, has the following chemical structure:
[0003] Gefapixant is a purinergic P2X3 receptor antagonist, and it is developed for the treatment of chronic cough. Gefapixant is also under clinical investigation as a treatment for asthma, interstitial cystitis, musculoskeletal pain, pelvic pain, and sleep apnea syndrome.
[0004] The compound is described in International Publication No. WO 2005/95359.
International Publication No. WO 2008/040652 disclosed a sulfonate solvate of Gefapixant. International Publication Nos. WO 2018/118668 and WO 2019/209607 disclose crystalline forms of Gefapixant as well as Gefapixant salts.
[0005] Polymorphism, the occurrence of different crystalline forms, is a property of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, thermal behaviors (e.g., measured by thermogravimetric analysis (“TGA”), or differential scanning calorimetry (“DSC”)), X-ray diffraction (XRD) pattern, infrared absorption fingerprint, and solid state (13C) NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
[0006] Different salts and solid state forms (including solvated forms) of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms and solvates may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the dissolution profile in a favorable direction, or improving stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also offer improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts and solid state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
[0007] Discovering new solid state forms and solvates of a pharmaceutical product may yield materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other polymorphic forms. New solid state forms of a pharmaceutically useful compound can also provide an opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, including a different crystal habit, higher crystallinity, or polymorphic stability, which may offer better processing or handling characteristics, improved dissolution profile, or improved shelf-life (chemi cal/phy si cal stability). For at least these reasons, there is a need for additional solid state forms (including solvated forms) of Gefapixant or salts or co-crystals thereof.

{kind=link}
{kind=link}
{kind=link}