
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HERE
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
.....FOR BLOG HOME CLICK HEREFlag and hits
ORGANIC SPECTROSCOPY

SUBSCRIBE

 Subscribe in a reader
Subscribe in a reader
Enter your email address:
Delivered by FeedBurner
Subscribe to New Drug Approvals by Email![]()

![]()



![]()
![]()





Recent Comments
| shivkr2 on SPSR Excellence Award 2025 – S… | |
| Bonkasaurus on Infigratinib phosphate | |
| PALOVAROTENE | ORGAN… on Palovarotene | |
| Pfizer just purchase… on Temanogrel | |
| Pfizer To Cash In On… on Temanogrel |
New FDA Draft Guidance ‘Data Integrity and Compliance with cGMP’ published
DRUG REGULATORY AFFAIRS INTERNATIONAL

In the last years, the topic “data integrity” has become a priority for the FDA. Recently, the Agency has published the draft of a Guidance for Industry on the topic which presents the comprehensive opinion of the FDA on data integrity. Read more about the draft of the Guidance for Industry “Data Integrity and Compliance with cGMP”.
In recent years, the topic “data integrity” has become a priority for European and American inspectors. At the beginning of 2015, the British authority MHRA published a first paper on that topic. Also in 2015, the World Health Organisation WHO issued another significant draft document on data integrity. Recently, the US American FDA has released the draft of a Guidance for Industry entitled “Data Integrity and Compliance with cGMP”. Although the FDA describes the Guidance as a non-binding recommendation, one may assume that the document presents the current thinking of the…
View original post 543 more words
Five new General Chapters in the European Pharmacopoeia on Genotoxic Impurities in Pharmaceutical APIs
DRUG REGULATORY AFFAIRS INTERNATIONAL
.jpg)
During the manufacture of APIs as sulfonate salts, esters of sulfonic acid may develop in undesired chemical side reactions. Recently, five new General Monographs have been included in the European Pharmacopoeia which describe how to cope with these impurities. Read more about these genotoxic impurities and the possibility to control them thanks to risk assessments.
Sulfonic acids are often used for the manufacture of pharmaceutical APIs. They serve as counterions in crystallisation processes, as protective groups or acid catalysts in API syntheses. Here, if short-chain alcohols such as methanol, ethanol or isopropanol are present, the formation of esters of these sulfonic acids can occur, which may have a genotoxic potential (alkylation of DNA).
The Mesilate Working Party which has been appointed in 2008 by the European Pharmacopoeia Commission has elaborated five General Chapters on different sulfonates which have been published in the European Pharmacopoeia Supplement 8.7 that came into force on 1 April 2016. The…
View original post 311 more words
FDA releases draft guidance on the use of comparability protocols for post approval changes
DRUG REGULATORY AFFAIRS INTERNATIONAL

The US FDA released a draft guidance for industry “Comparability Protocols for Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Information”. The guidance replaces the draft guidance published in February 2003. It provides recommendations on implementing postapproval changes through the use of comparability protocols (CPs). Read more about FDA´s draft guidance for industry “Comparability Protocols for Human Drugs and Biologics”.
On April 19, 2016, the US Food & Drug Administration (FDA) released a draft guidance for industry “Comparability Protocols for Human Drugs and Biologics: Chemistry, Manufacturing, and Controls Information”. Comments and suggestions regarding the draft guideline should be submitted within 60 days of publication.
The guidance replaces the draft guidance published in February 2003. It provides recommendations on implementing postapproval changes through the use of comparability protocols (CPs). A CP is a comprehensive, prospectively written plan for assessing the effect of proposed CMC postapproval changes on the identity, strength…
View original post 418 more words
энкломифен Enclomiphene citrate إينكلوميفان

Enclomiphene citrate
NDA FILED Hypogonadism, Repros Therapeutics
An estrogen receptor (ER) antagonist potentially for treatment of hypogonadotropic hypogonadism.
![]()
ICI-46476; RMI-16289
CAS No.15690-57-0(free)
7599-79-3(Enclomiphene citrate)
| Molecular Weight | 598.08 |
| Formula | C26H28ClNO▪C6H8O7 |
Ethanamine, 2-[4-[(1E)-2-chloro-1,2-diphenylethenyl]phenoxy]-N,N-diethyl-, 2-hydroxy-1,2,3-propanetricarboxylate (1:1)
- Ethanamine, 2-[4-(2-chloro-1,2-diphenylethenyl)phenoxy]-N,N-diethyl-, (E)-, 2-hydroxy-1,2,3-propanetricarboxylate (1:1)
- Triethylamine, 2-[p-(2-chloro-1,2-diphenylvinyl)phenoxy]-, citrate (1:1), (E)-
- (E)-Clomiphene citrate
- Androxal
- Clomiphene B citrate
- Enclomid
- Enclomiphene citrate
- trans-Clomiphene citrate

Clomifene is a mixture of two geometric isomers, enclomifene (E-clomifene) and zuclomifene (Z-clomifene). These two isomers have been found to contribute to the mixed estrogenic and anti-estrogenic properties of clomifene.
|
Enclomifene
|
Zuclomifene
|


EXAMPLE 1
Preparation of trans-clomiphene citrate from
1- {4- [2-(Oiethylamino)ethoxy| phenylj-1 ,2-diphenylethanol
Dehydration
[0023] l-{4-[2-(Diethylamino)ethoxy]phenyl}-l,2-diphenylethanol (6) dissolved in ethanol containing an excess of hydrogen chloride was refluxed 3 hours at 50 °C. The solvent and excess hydrogen chloride were removed under vacuum and the residue was dissolved in dichloromethane. 2-{4-[(Z)-l,2-diphenylvinyl]phenoxy}-N,N- diethylethanaminium hydrogen chloride (7) was obtained.
Chlorination
The hydrochloride salt (7) solution obtained above was treated with 1.05 equivalents of N-chlorosuccinimide and stirred at room temperature for about 20 hours. Completion of the reaction was confirmed by HPLC. The hydrochloride salt was converted to the free base by addition of saturated aqueous bicarbonate solution. The mixture was stirred at room temperature for 30 minutes after which the phases were separated and the organic phase was evaporated in vacuo. 2-{4-[2-chloro-l,2- diphenylvinyl]phenoxy}-N,N-diethylethanamine (clomiphene -1.8:1 E:Z mixture) (8) was obtained.
Separation of clomiphene isomers
Clomiphene (8) obtained above is dissolved in methanol and racemic binaphthyl- phosphoric acid (BPA) is added under stirring. When the precipitate begins separating from the solution, stirring is stopped and the mixture is allowed to settle at room temperature for 2 hours. The precipitate is filtered, washed with methanol and ether and dried. Trans-clomiphene-BPA salt (3) is obtained.
The enclomiphene-BPA salt (3) obtained above is extracted with ethyl acetate and NH3 solution. To the organic solution washed with water and dried, citric acid dissolved in ethanol is added. The solution is allowed to settle for about one hour at room temperature; the precipitate is then filtered and dried under vacuum. The obtained precipitate, trans-clomiphene citrate (1) is dissolved in 2-butanone for storage.
EXAMPLE 2
Synthesis of Clomiphene Using a Single Solvent
Step 1 – Dehydration of l-i4-r2-(Diethylamino)ethoxy1phenyl|-l,2- diphenylefhanol to form 2-{4-[(Z)-l,2-diphenylvinyllphenoxy}-N,N-diethylethanaminium hydrogen sulfate (7) [0030] The synthesis route described in Example 1 utilized HC1 for the dehydration step and utilized ethanol at 50 °C as the solvent. Sulfuric acid was investigated as an alternative to HC1 for the dehydration step (as described in Example 1) in part due to the more favorable corrosion profile of sulfuric acid. Dichloromethane (methylene chloride) was investigated as an alternative solvent for the dehydration step as this would render removal of the ethanol solvent prior to the chlorination step unnecessary.
A 100 mL 3-neck round bottom flask, fitted with a temperature probe and a stir bar, was charged with l- {4-[2-(Diethylamino)ethoxy]phenyl}-l,2-diphenylethanol (6) (6.60 g, 16.9 mmol) and 66 mL (lxlO3 mmol) of methylene chloride to give a yellow solution which was cooled in an ice bath to 0 °C. Concentrated sulfuric acid (H2S04, 0.96 mL, 18.1 mmol) was added at a rate such that the internal temperature did not exceed 5 °C. Upon completion of the addition, the mixture was allowed to stir one hour at ambient temperature. Completion of the reaction was confirmed by high performance liquid chromatography (HPLC). The reaction resulted in 7.96 grams of 2- (4-[(Z)- 1 ,2- diphenylvinyl]phenoxy}-N,N-diethylethanaminium hydrogen sulfate (7), a yield of 100%. Thus, sulfuric acid was demonstrated to be a suitable acid for the dehydration step.
[0042] Using these HPLC conditions, starting material has a retention time of 3.30 min and product has a retention time of 4.05 min.
It was determined that removal of water produced by the dehydration reaction was important before performing the chlorination step. When ethanol is used as the solvent for this reaction, as in Example 1, the water is removed azeotropically upon removal of the ethanol. Several methods of drying the dichloromethane solution were attempted. Drying with MgS04 had a deleterious effect on the subsequent chlorination step, rendering the chlorination process very messy with a number of new impurities observed following HPLC analysis which were determined to be the corresponding chlorohydrins. On the other hand, a wash with brine was sufficient to remove enough water and had no deleterious effect on the chlorination step. Accordingly, the solution was stirred vigorously with brine (66 ml) for 30 minutes and then the phases were separated prior to chlorination step.
Step 2- Synthesis of 2-|4-r2-chloro-L2-diphenylvinyl1phenoxyl-N,N- diethylethanamine 8
The solution of 2-{4-[(Z)-l,2-diphenylvinyl]phenoxy}-N,N-diethylethanaminium hydrogen sulfate (7.94 grams) in methylene chloride obtained in step 1 is stirred at room temperature and treated with N-chlorosuccinimide (2.37 g, 17.7 mmol, 1.05 equivalents) in a single portion and left to stir at room temperature for 12 hours. The yellow solution became orange and then went back to yellow. After 12 hours, a sample was removed, concentrated and assayed by HPLC to confirm the extent of reaction. HPLC analysis revealed that the reaction had proceeded but not to completion. Accordingly, an additional 0.09 equivalents of N-chlorosuccinimide (203 mg, 1.52 mmol) was added and the solution stirred at room temperature for an additional 4 hours. The reaction was again assayed by HPLC which revealed that the reaction was near completion. Accordingly, an additional 0.09 equivalents of N-chlorosuccinimide (203 mg, 1.52 mmol) was added and the solution stirred for an additional 12 hours at room temperature. The reaction was again assayed by HPLC and an additional 0.058 equivalents of N-chlorosuccinimide (131 mg, 0.98 mmol) was added and the solution stirred for an additional 4 hours. HPLC indicated that the reaction was complete at that point. The reaction was carefully quenched by slow addition of 66 mL (600 mmol) of saturated aqueous sodium bicarbonate solution and the quenched mixture was stirred for 30 minutes at room temperature – the reaction mixture pH should be about 8-9 after addition of saturated aqueous sodium bicarbonate solution. The reaction yielded 6.86 grams of 2-{4-[2-chloro-l,2-diphenylvinyl]phenoxy}-N,N- diethylethanamine (8). The phases were separated and the organic phase was evaporated in vacuo. The resulting light brown oil was transferred to a tared amber bottle using a small volume of dichloromethane.
[0055] Using these HPLC conditions, the retention time of product is 15 minutes.
Chromatographic Separation of Clomiphene Isomers
Clomiphene (mixture of isomers) in free base form obtained by steps 1 and 2 is loaded onto a chromatographic column (e.g. batch high pressure chromatography or moving bed chromatography) using the same solvent as used in steps 1 and 2 (here DCM) in order to separate the cis- and trans-clomiphene isomers. Trans-clomiphene is preferably eluted using a solvent suitable for recrystallization.
Journal of Chromatography (1984), 298, (1), 172-4.

US2914562https://www.google.co.in/patents/US2914562PATENTUS2914529
http://www.google.co.in/patents/US2914529
PAPER
J. Med. Chem.1967, 10, 84–86.

DOI: 10.1039/C5CC01968K

| CN103351304A * | Jul 1, 2013 | Oct 16, 2013 | 暨明医药科技(苏州)有限公司 | Synthesis method of clomiphene |
| US2914563 * | Aug 6, 1957 | Nov 24, 1959 | Wm S Merrell Co | Therapeutic composition |
| US3848030 * | Mar 10, 1972 | Nov 12, 1974 | Richardson Merrell Spa | Optical isomers of binaphthyl-phosphoric acids |
| US5681863 * | Dec 5, 1994 | Oct 28, 1997 | Merrell Pharmaceuticals Inc. | Non-metabolizable clomiphene analogs for treatment of tamoxifen-resistant tumors |
| Reference | ||
|---|---|---|
| 1 | * | RAO ET AL.: “Synthesis of carbon-14 labeled clomiphene.“, JOUMAL OF LABELLED COMPOUNDS AND RADIOPHARMACEUTICALS, vol. 22, no. 3, 1985, pages 245 – 255, XP055180053, Retrieved from the Internet <URL:http://onlinelibrary. wiley .com/doi/10.1002/jlcr.2580220306/abstract> [retrieved on 20150504] |
//////////энкломифен, Enclomiphene citrate, إينكلوميفان , ICI-46476, RMI-16289, nda filed, Hypogonadism, Repros Therapeutics
Cymipristone
Cymipristone
- Estra-4,9-dien-3-one, 11-[4-(cyclohexylmethylamino)phenyl]-17-hydroxy-17-(1-propynyl)-, (11β,17β)- (9CI)
- (11β,17β)-11-[4-(Cyclohexylmethylamino)phenyl]-17-hydroxy-17-(1-propyn-1-yl)estra-4,9-dien-3-one
- Saimisitong
NDA Filed china
Shanghai Siniwest Pharmaceutical Chemical Technology Co., Ltd., Shanghai Zhongxi Pharmaceutical Co. Ltd., Xianju Pharmaceutical Co., Ltd,
A progesterone receptor antagonist potentially for termination of intrauterine pregnancy.
![]()
CAS No.329971-40-6
- Molecular FormulaC34H43NO2
- Average mass497.711 Da
- Steroid Compounds, a Method for Preparation thereof, Pharmaceutical Compositions Containing the Same and Use thereof
-
This invention relates to steroid compounds and pharmaceutical acceptable salts thereof, a method for preparation thereof, pharmaceutical compositions containing the same as active component, and their use in the preparation of medicines for treating diseases associated with progestogen dependence and for fertility control, abortion or contraception and for anticancer use.
-
Mifepristone (11β-[4-(N,N-dimethylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one) is a steroid compound which is disclosed in French Patent No. 2,497,807 to Rousell-Uclaf, published May 31, 1983. It is the first progesterone receptor antagonist put into clinical application and is a new type of anti-progestin. It binds to progesterone receptor and glucocorticoid receptor, having an affinity with progesterone receptor in rabbit endometrium five-fold higher than that of progesterone and thereby having strong anti-progesterone effect. It causes degeneration of pregnant villus tissue and decidual tissue, endogenous prostaglandin (PG) release, luteinizing hormone decrease, corpus luteum dissolution, and necrosis of embryo sac whose development depends on corpus luteum, leading to abortion. Therefore, it can be used as a non-surgical medicine for stopping early pregnancy. It can also be used, inter alia, in contraception and as an antineoplastic. (The Antiprogestin Steroid Ru486 and Human Fertility Control, 1985, New York: Plenum Press) .
-
Onapristone (11β-[4-(N,N-diemthylamino)phenyl]-17α-hydroxy-17β-(3-hydroxypropyl)-13α-4,9-estradiene-3-one), is a steroid compound which is disclosed in German Patent No. 3,321,826 to Schering AG, published Dec. 20, 1984. It has a strong antiprogestin activity and can be used in abortion (American Journal of Obstetrics and Gyencology, 1987, 157:1065-1074), anticancer (Breast Cancer Research and Treatment, 1989, 14:275-288), etc. It was reported that onapristone had toxicity to human liver (European Journal of Cancer, 1999, 35(2):214-218).
-
Lilopristone (11β-[4-(N,N-dimethylamino) phenyl]-17α-[3-hydroxy-1(Z)-propenyl]-17β-hydroxy-4,9-estradiene-3-one) is a steroid compound which is disclosed in German Patent No. 3,347,126 to Schering AG, published July 11, 1985. It has a strong antiprogestin activity and can be used in abortion, contraception (American Journal of Obstetrics and Gyencology, 1987, 157:1065-1074), etc. It was reported that the clinical effect of lilopristone in stopping early pregnancy was only equivalent to that of mifepristone (Human Reproduction, 1994, 9(1):57-63).
-
ZK112993 (11β-(4-acetylphenyl)-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one) is as steroid compound which is disclosed in German Patent No. 3,504,421 to Schering AG, published Aug. 7, 1986. It has a potent antiprogestin activity and can be used in, inter alia, anticancer (Anticancer Res., 1990, 10:683-688).
-
In European Patent No. 321,010 to Akzo NV, The Netherland published June 21, 1989 are disclosed “11-arylsteroid compounds” having a strong antiprogestin activity.
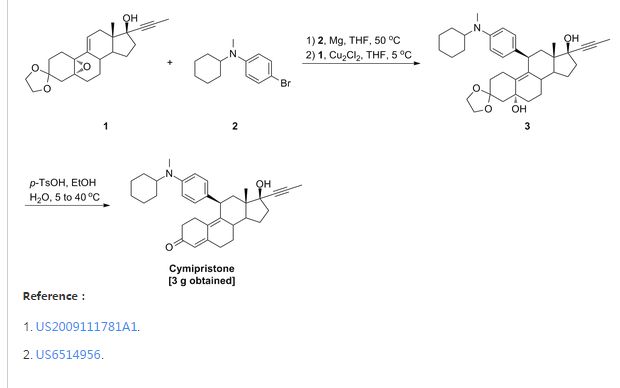
PATENT
WO 2001018026
http://www.google.com/patents/EP1219632A1?cl=en

The preparation method of the present invention includes the following single- or multi-step procedures:
1. Method for the preparation of 11β-[4-(N-methyl-N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (IV) which includes the following steps:
(1) Preparation of Grignard reagent (III)

4-bromo-N-methyl-N-cyclohexylaniline (II) is reacted with magnesium in tetrahydrofuran (THF) to obtain Grignard reagent of formula (III).
(2) C11 additive reaction

Compound of formula (IV) and the Grignard reagent of formula (III) prepared in step (1) are brought to an additive reaction to obtain compound of formula (V).
(3) Hydrolytic reaction

The compound of formula (V) prepared in step (2) is subjected to a hydrolytic reaction to obtain compound of form (VI).
2. Method for preparation of 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI) which includes the following steps:
(1) Preparation of Grignard reagent of formula (IX)

4-bromo-N-cyclohexylaniline (VII) is first protected by trimethylchlorosilane, then reacted with magnesium in THF to obtain Grignard reagent of formula (IX).
(2) C11 additive reaction

Compound of formula (IV) and the Grignard reagent of formula (IX) prepared in step (1) are brought to an additive reaction to obtain compound of formula (X).
(3) Hydrolytic reaction

The compound of formula (X) prepared in step (2) is subjects to a hydrolytic reaction to obtain compound of formula (XI).
Example 2:
-

-
9g 4-bromo-N-cyclohexylaniline (VII) (CA registration number [113388-04-8], see Synthetic Communications, 1986, 16(13): 1641-1645 for its preparation) was placed into a four-necked flask and 15 ml (1.5 mol/L) n-BuLi solution in n-hexane. The mixture was stirred for 30 min at room temperature. Then 8 g trimethylsilyl chloride (Me3SiCl) was added and the mixture was stirred for 1 hour. Solvent and excessive Me3SiCl was evaporated under reduced pressure to yield 4-bromo-(N-cyclohexyl-N-trimethylsilylaniline) (VIII) which was formulated into a solution with 7.5 ml anhydrous tetrahydrofuran for further use.
-
1.3 g magnesium was placed into a four-necked flask and a small amount of the above solution was added dropwise and slowly at 40°C. After completion of addition, the temperature was kept for 1 hour to yield a solution of 4-(N-cyclohexyl-N-trimethylsilylamino)phenylmagnesium bromide (IX) in tetrahydrofuran for further use.
- Preparation of 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI)(1) Preparation of 4-(N-cyclohexyl-N-trimethylsilylamino)phenyl magnesium bromide (IX)

(2) Preparation of 3,3-ethylenedioxy-5α,17β-dihydroxy-11β-[4-(N-cylohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene(X).

- 5g 3,3-ethylenedioxy-5,10-epoxy-17α-(1-propinyl)-17β-hydroxy-9(11)-estrene (IV) was placed into a four-necked flask and 10 ml anhydrous tetrahydrofuran and a catalytic amount of cuprous chloride (Cu2Cl2) added. Then solution of 4-(N-cyclohexyl-N-trimethylsilylamino)phenyl magnesium bromide (IX) in tetrahydrofuran was added dropwise and slowly while controlling the temperature below 5°C. After completion of addition, the mixture was allowed to react for 2 hours at room temperature and to stand overnight. Saturated ammonium chloride aqueous solution was added and the tetrahydrofuran layer separated which was washed with saturated ammonium chloride solution. The solution in tetrahydrofuran was washed with saturated saline and dried over anhydrous sodium sulfate. Evaporation of tetrahydrofuran under reduced pressure yielded a residual which was chromatographed on silica gel column using cyclohexane: acetone (5:1) as developing agent to yield 3 g 3,3-ethylenedioxy-5α,17β-dihydroxy-11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene(X).
-
IR (KBr) cm-1: 3420 (C5, C17-OH), 1610, 1510 (benzene backbone), 840, 808 (ArH).
1H NMR (CDCl3) δ ppm: 0.52(3H, S, C13-CH3), 2.72(3H, S, N-CH3), 3.92(4H, m, -O-CH2CH2-O-), 4.24(1H, m, C11-H), 6.65-7.00 (4H, ArH).
(3) Preparation of 11β- [4- (N-cyclohexylamino)phenyl] -17α- (1-propinyl) -17β-hydroxy-4,9-estradiene-3-one (XI).

- 1.5g 3,3-ethylenedioxy-5,17β-dihydroxy-11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-9(10)-estrene (X) and 0.75 g para-toluenesulfonic acid (PTS) were dissolved in 15 ml 90 % ethanol (v/v). The mixture was stirred for 2 hours while controlling the temperature at 40°C-50°C. After completion of the reaction, the reactant was poured into diluted sodium hydroxide aqueous solution, extracted with dichloroethane, washed with water to neutrality, and dried over anhydrous sodium sulfate. Evaporation of the solvent and chromatography on silica gel column using cyclohexane: ethyl acetate (5:1) as developing agent yielded 0.9 g 11β-[4-(N-cyclohexylamino)phenyl]-17α-(1-propinyl)-17β-hydroxy-4,9-estradiene-3-one (XI).
-
IR (KBr) cm-1: 3400 (C17-OH), 1658 (unsaturated ketone), 1613, 1514 (benzene backbone), 865, 810 (ArH).
1H NMR (CDCl3) δ ppm: 0.50 (3H, S, C13-CH3), 1.76 (3H, S, C≡C-CH3), 4.32(1H, S, C11-H), 5.75(1H, S, C4-H), 6.9-7.10 (4H, ArH).
PATENT
PATENT
Example 1
Race meters mifepristone synthetic routes:

Epoxy adduct match rice mifepristone
(N- hexylamino methylcyclohexyl) phenyl magnesium bromide (1) 4-
In the four-necked flask, 1.4 g of magnesium into pieces (Mg) and 10 ml of anhydrous tetrahydrofuran (THF), no iodine or add a little change, at about 50 ° C, a solution of 10.86 g of 4-bromo-methyl -N- cyclohexyl aniline (dissolved in 24 ml of anhydrous tetrahydrofuran) dropwise Bi, incubation was continued for 1 hour with stirring to give 4- (N- methyl-cyclohexylamino) phenyl magnesium bromide tetrahydrofuran solution (to be used in the next step an addition reaction ).
(2) 3,3-ethylenedioxy -5 α, 17 β – dihydroxy -11 β – [4- (Ν- methyl -Ν- cyclohexylamino) phenyl] -17 α – (1- propyl block-yl) -9 (10) – Preparation of estra-ene (adduct) of
In the four-necked flask, into 5 g of 3,3-ethylenedioxy-5,10-epoxy -17 α – (1- propynyl) – 17 (3 – hydroxy – 9 (11) – estra-ene (epoxy), 29.1 ml anhydrous tetrahydrofuran (THF) and 0.1 g cuprous chloride (of Cu 2 of Cl 2 ), a solution of 4- (N- methyl -N-cyclohexylamino) phenyl magnesium bromide tetrahydrofuran
Nan solution, temperature control 5. C, the drop was completed, the incubation was continued for 5 hours, the reaction was completed, the reaction solution was poured into saturated aqueous ammonium chloride solution, points to the water layer, the organic layer was washed with saturated ammonium chloride solution, the aqueous layer extracted with ethyl acetate number times, the organic layers combined, washed with saturated aqueous sodium chloride, dried over anhydrous sodium sulfate, and concentrated under reduced pressure, a silica gel column, eluent cyclohexane: acetone = (5: 1) to give 3,3-ethylene dioxo -5 α, 17 β – dihydroxy -11 β – [4- (- methyl -Ν- cyclohexylamino) phenyl] -17 α – (1- propynyl) -9 (10) – female steroidal women (adduct) solid 6 grams.
IR. ‘KBi cm- ^ SlS OI ^ ^ -OH lS jSlS benzene backbone), 819 (aromatic hydrogen). NMR Ή: (CDC1 3 ) ppm by [delta]: 0.47 (3H, the S, the C IR CH 3 ), 1.88 (3H, the S, the C ≡ the C-CH 3 ), 2.72 (3H, the S, the N-CH 3 ), 6.65- 7.03 (4H, ArH) O
(3) 11 β – [4- (N- methyl -N- cyclohexylamino) phenyl] -17 α – (1- propynyl) -17 β – hydroxy-estra-4,9-diene – Preparation of 3-one (match rice mifepristone) of
‘2.5 g of p-toluenesulfonic acid (PTS) and 5 grams of 3,3-ethylenedioxythiophene -5 α, 17 β – dihydroxy -11 β – [4- (Ν- methyl cyclohexylamino) phenyl] -17 α – (1- propynyl) -9 (10) – estra-ene (adduct) was dissolved in 50 ml of ethanol 90% (V / V), and at 5 ° C – 40 ° C the reaction was stirred 3 hours, the reaction solution was poured into dilute aqueous sodium hydroxide solution, the precipitated solid was suction filtered, washed with water until neutral, the filter cake was dissolved in 50 ml of ethyl acetate, then with saturated aqueous sodium chloride solution to the water layer was evaporated part of the solvent, the precipitated solid was suction filtered, and dried to give a pale yellow solid 11 β – [4- (Ν- -N- methyl-cyclohexylamino)] -17 α – (1- propynyl) -17 β – hydroxy estra-4,9-dien-3-one (match rice mifepristone) 3 grams.
^ Cm & lt IRCKB 1 : 3447 (the C . 17 -OH), among 1655 (unsaturated ketone), 1607,1513 (benzene backbone), 865,819 (aromatic hydrogen).
NMR ¾: (CDC1 3 ) ppm by [delta]: 0.56 (3H, the S 5 the C 13 -CH 3 ), 1.89 (3H, the S 5 -C ≡ the C-the CH3), 2.74 (3H, the S, the N-the CH3), 4.34 ( lH, the S, the C N -H), 5.75 (lH, the S, the C 4 -H), 6.68-6.99 (4H, ArH).
PATENT
PATENT
PAPER
Volume 878, Issues 7–8, 1 March 2010, Pages 719–723
Determination of cymipristone in human plasma by liquid chromatography–electrospray ionization-tandem mass spectrometry
doi:10.1016/j.jchromb.2010.01.027
Abstract
A rapid, specific and sensitive liquid chromatography–electrospray ionization-tandem mass spectrometry method was developed and validated for determination of cymipristone in human plasma. Mifepristone was used as the internal standard (IS). Plasma samples were deproteinized using methanol. The compounds were separated on a ZORBAX SB C18 column (50 mm × 2.1 mm i.d., dp 1.8 μm) with gradient elution at a flow-rate of 0.3 ml/min. The mobile phase consisted of 10 mM ammonium acetate and acetonitrile. The detection was performed on a triple-quadruple tandem mass spectrometer by selective reaction monitoring (SRM) mode via electrospray ionization. Target ions were monitored at [M+H]+m/z 498 → 416 and 430 → 372 in positive electrospray ionization (ESI) mode for cymipristone and IS, respectively. Linearity was established for the range of concentrations 0.5–100 ng/ml with a coefficient correlation (r) of 0.9996. The lower limit of quantification (LLOQ) was identifiable and reproducible at 0.5 ng/ml. The validated method was successfully applied to study the pharmacokinetics of cymipristone in healthy Chinese female subjects.
CHEMICAL ABSTRACTS, vol. 115, no. 25, 23 December 1991 (1991-12-23) Columbus, Ohio, US; abstract no. 270851g, X. ZHAO ET AL.: “Synthesis and terminating early pregnancy effect of mifepristone derivatives” page 117; XP002219009 & ZHONGGUO YAOKE DAXUE XUEBAO, vol. 22, no. 3, 1991, pages 133-136,
//////////Cymipristone, Saimisitong, NDA Filed , china, Shanghai Siniwest Pharmaceutical Chemical Technology Co., Ltd., Shanghai Zhongxi Pharmaceutical Co. Ltd., Xianju Pharmaceutical Co., Ltd,
New Patent, Tedizolid phosphate, Suzhou MiracPharma Technology Co Ltd, Zheren Pharmaceutical Nanjing Co Ltd, WO 2016058467
Tedizolid phosphate
Suzhou MiracPharma Technology Co Ltd; Zheren Pharmaceutical Nanjing Co Ltd
WO-2016058467 click for patent
SUZHOU MIRACPHARMA TECHNOLOGY CO., LTD [CN/CN]; Room 1305, Building 1 Lianfeng Commercial Plaza, Industrial District Suzhou, Jiangsu 215000 (CN).
ZHEREN PHARMACEUTICAL NANJING CO., LTD [CN/CN]; Qiaolin Industry Park 32-71, Pukou District Nanjing, Jiangsu 211806 (CN)



Disclosed is a method for preparing tedizolid phosphate (I), and the preparation step thereof comprises producing the tedizolid phosphate (I) by means of a coupling reaction of a compound of formula II and a compound of formula III. The preparation method uses easily available raw materials and a simple process, is economical and environmentally friendly, and is suitable for industrial production.
Process for preparing tedizolid phosphate (TD-P), useful for treating bacterial infection. The present filing represents the first PCT and first filing to be seen from Suzhou Miracpharma and Zheren Pharmaceutical, respectively, that focuses on tedizolid; however this case was first seen as a Chinese national filing (assigned to Suzhou Miracpharma), published in February 2015. The drug was developed and launched by Dong-A ST and licensees Cubist Pharmaceuticals and Bayer, for treating acute bacterial skin and skin structure infections.
//////New Patent, Tedizolid phosphate, Suzhou MiracPharma Technology Co Ltd, Zheren Pharmaceutical Nanjing Co Ltd, WO 2016058467
Lurasidone hydrochloride, Jubilant Generics Ltd, WO 2016059649, New patent
 |
|
 |

Lurasidone hydrochloride, Jubilant Life Sciences Ltd, WO 2016059649, New patent
An improved process for the preparation of lurasidone hydrochloride
Jubilant Life Sciences Ltd
WO 2016059649
JUBILANT GENERICS LIMITED (FORMERLY JUBILANT LIFE SCIENCES DIVISION) [IN/IN]; Plot 1A, Sector 16 A, NOIDA Uttar Pradesh 201301 (IN)
MISHRA, Vaibhav; (IN).
DUBEY, Shailendr; (IN).
SINGH, Kumber; (IN).
CHOUDHARY, Alka Srivastava; (IN).
VIR, Dharam; (IN)

![]()
Disclosed herein is an improved process for the preparation of Lurasidone and its pharmaceutically acceptable salts via novel intermediate and use thereof for the preparation of an antipsychotic agent useful for the treatment of schizophrenia and bipolar disorder. Further, present invention provides a cost effective and eco-friendly process for producing Lurasidone hydrochloride of formula (I) substantially free of residual solvent(s) at industrial scale.
Improved process for preparing lurasidone or its hydrochloride, substantially free of residual solvent, useful for treating schizopherenia and bipolar disorder. Also claims novel intermediate of lurasidone eg ((R,R)-cyclohexane-1,2-diyl)bis((1H-imidazol-1-yl)methanone) and its preparation method.

In April 2016, Newport Premium™ reported that Jubilant Life Sciences was capable of producing commercial quantities of lurasidone and lists the drug as a molecule available under research and development on the company’s website.
This is the first patenting to be seen from Jubilant Life Sciences that focuses on lurasidone – it having been developed and launched by Sumitomo Dainippon Pharma and EU licensee Takeda, for treating schizophrenia.

May 2, 2014
Neeraj Agrawal: Took charge of API business for Jubilant Life Sciences at the age of 31
Position: CEO Generics, Jubilant Life Sciences
Education: IIIM-C, MBA, 1998; IIT, Bombay, Electrical Engg., 1995.
Previous Jobs: Associate-Business Strategy, Operations Improvement, McKinsey & Co.
Claim to Fame: Took charge of the API business for Jubilant when he was just 31-years-old
Management mantra: It revolves around trust, freedom and teams. I like my team to think and act like an entrepreneur – assess business risks and rewards suitably and then take decisions.
Lurasidone and its pharmaceutically acceptable salts like lurasidone hydrochloride is chemically, (3a ?,45,7 ?,7a5)-2-{ (1 ?,2 ?)-2-[4-(l,2-benzisothiazol-3-yl)piperazin-lyl-methyl] cyclohexylmethyl }hexahydro-4,7-methano-2H-isoindole- 1 ,3 -dione hydrochloride and has the structure represented by the Formula (I):

Formula-I
Lurasidone hydrochloride is marketed in the United States under the trade name Latuda®. Lurasidone and its pharmaceutically acceptable salts as well as process for their preparation was first disclosed in US patent no. 5,532,372. The patent discloses the preparation of lurasidone hydrochloride using racemic trans 1,2-cyclohexane dicarboxylic acid. Racemic trans 1,2-cyclohexane dicarboxylic acid on reduction with lithium aluminium hydride in THF at reflux temperature forms l,2-bis(hydroxymethyl)cyclohexane which is converted into racemic iran5-l,2-bis(methanesulfonyloxymethyl)cyclohexane by reaction with methane sulfonyl halide. l-(l,2-benzisothiazol-3-yl)piperazine on reaction with trans-l, 2-b (methanesulfonyloxymethyl)cyclohexane in the presence of sodium carbonate and acetonitrile forms iran5-3a,7a-octahydroisoindolium-2-spiro- -[4′-(l,2-benzisothiazol-3-yl)]piperazine methanesulfonate which on reaction with bicyclo[2.2.1]heptane-2-exo-3-exo-dicarboximide in the presence of potassium carbonate, dibenzo-18-crown-6-ether and xylene on refluxing forms racemic lurasidone free base. The compound is obtained by column chromatography and then treated the resulting lurasidone free base with IPA.HCl in acetone to obtain racemic lurasidone hydrochloride. Resolution of racemic lurasidone hydrochloride is carried out using tartaric acid as resolving agent. The process involves use of lithium aluminium hydride which is highly pyrophoric reagent and is not to utilize the same on commercial scale due to its handling problems associated with its reactivity. Also, the use of the column chromatography for purification is not viable on commercial scale. Further the process involves the usage of dibenzo-18-crown-6-ether as a phase transfer catalyst which is costly material and in turn increases the cost of production. Carrying out the resolution in the last stages is difficult due to the presence of six chiral centres in lurasidone and is also not suitable for an industrial scale preparation as it affects the overall yield and cost of the manufacturing process.

Chinese patent application no. CN102731512 discloses a process for preparation of lurasidone which comprises reaction of racemic irans-l,2-bis(methanesulfonyloxymethyl) cyclohexane and l-(l,2-benzisothiazol-3-yl)piperazine in toluene in the presence of sodium carbonate or potassium carbonate having particle size less than 200 micron and tetrabutyl ammonium bromide to give the intermediate /rans-3a,7a-octahydroisoindolium-2-spiro- -[4′-(l,2-benzisothiazol-3-yl)]piperazinemethanesulfonate which on reaction with bicyclo[2.2.1]heptane-2-exo-3-exo-dicarboximide in toluene using potassium carbonate having particle size less than 200 micron forms racemic lurasidone free base. The racemic free base is converted into racemic hydrochloride salt using acetone and cone, hydrochloric acid. Racemic lurasidone hydrochloride is resolved by following the method disclosed in US patent no. 5,532,372. The process involves resolution of product in the last stage which is not commercially viable as it affects the overall yield and cost of the manufacturing process.
Japanese patent no. JP4219696 discloses the resolution of trans 1,2-cycloheaxne dicarboxylic acid using (lS,2R)-(+)-norephedrine or (lR,2S)-(-)norephedrine to provide (R,R)-trans 1 ,2-cyclohexanedicarboxylic acid. The (R,R)-iran,sl,2-cyclohexane dicarboxylic acid obtained was esterified with ethanol and the obtained ester compound was reduced with vitride to provide (R,R)-l,2-bis(hydroxymethyl)cyclohexane followed by treatment with methane sulfonyl chloride to form (R,R)-1,2-bis(methanesulfonyloxymethyl)cyclohexane. The process requires large quantity of reducing agent viz., for reducing one lg of compound about 5g of reducing agent is required which is not conducive for industrial production.
Chinese patent application no. CN 102952001 discloses a process for the preparation of (lR,2R)cyclohexane-l,2-dimethanol by the reduction of (lR,2R)cyclohexane-l,2-
dicarboxylic acid using sodium borohydride or potassium borohydride and boron triflouoride diethyl ether in THF or diethyl ether as solvent. Boron triflouoride diethyl ether is used in large quantity and quite expensive which makes the process commercially unviable.
International publications no. WO 2012/131606 and WO 2014/037886 disclose a process for preparation of lurasidone which involves separating the racemic transl,2-cyclohexane dicarboxylic acid into its (R,R) trans and (S,S) trans isomers and then using the desired trans (R,R) isomer for the preparation of lurasidone hydrochloride using the chemistry disclosed in US patent no. 5,532,372 for preparation of racemic lurasidone hydrochloride. In these publications diisobutyl aluminium hydride (DIBAL) is used as the reducing agent for the preparation of (1R,2R) cyclohexane 1,2-dimethanol from (1R,2R) cyclohexane 1,2-dicarboxylic acid which is quite expensive. Further the process involves the usage of dibenzo-18-crown-6-ether as a phase transfer catalyst which is costly material and in turn increases the cost of production.
Some of the prior art processes disclose the process for the preparation of lurasidone hydrochloride from l,2-(lR,2R)-bis-(methanesulfonyloxymethyl)cyclohexane using different solvents and bases.
US patent no. 8,853,395 discloses a process for the preparation of lurasidone in which condensation of iran5-l,2-bis(methanesulfonyloxymethyl)cyclohexane with 1-(1,2-benz isothiazol-3-yl)piperazine and condensation of /rans-3a,7a-octahydroisoindolium-2-spiro- -[4′-(l,2-benzisothiazol-3-yl)]piperazine methanesulfonate with bicyclo[2.2.1] heptane-2-exo-3-exo-dicarboximide is carried out using organic bases with a ρ¾ higher than 10 such as l,4-diazabicycloundec-7-ene (DBU), l,5-diazabicyclo[4.3.0]non-5-ene (DBN), 1,4-diaza bicyclo[2.2.2] -octane (DABCO). These organic bases are comparatively expensive.
Indian patent application no. IN 2306/MUM/2014 and Chinese patent applications no. CN 102863437 and CN 103864774 disclose the use of dimethyl formamide (DMF), dimethyl sulphoxide (DMSO), dimethyl acetamide (DMA) and N-methyl pyrrolidine (NMP) for the condensation of iran5-3a,7a-octahydroisoindolium-2-spiro- -[4′-(l,2-benzisothiazol-3-yl)] piperazine methanesulfonate with bicyclo[2.2.1] heptane-2-exo-3-exo-dicarboximide to form lurasidone. These solvents have high boiling point so not preferred at commercial scale.
Some of the prior art processes are related to reduction of impurities or quality improvement of lurasidone hydrochloride.
International publication no. WO2011/136383 discloses a process for the preparation of lurasidone hydrochloride in which amount of by products are reduced by increasing the quantity of l-(l,2-benzisothiazol-3-yl)piperazine instead of sodium carbonate or potassium carbonate as base in the reaction mixture. Increasing the amount of l-(l,2-benzisothiazol-3-yl)piperazine causes an increase in cost of production and removal of excess compound makes the process less commercially viable.
International publication no. WO2011/136384 discloses a process for the preparation of lurasidone hydrochloride in which amount of by products are reduced by using dibasic potassium phosphate with a small amount of water as a base instead of sodium carbonate. Use of dibasic potassium phosphate as a base causes an increase in cost of production as dibasic potassium phosphate is expensive.
International publication no. WO2013/014665 discloses various processes for the preparation of lurasidone hydrochloride. In general the process is shown below:

Formula-(I)
In this process iran5-(lR,2R)-2-(aminomethyl)cyclohexyl)methanol of Formula (B) is first reacted with bicyclo[2.2.1]heptane-2-exo-3-exo-dicarboximide of Formula (A) to form (3aR,4S,7R,7aS)-2-(((lR,2R)-2-(hydroxymethyl)cyclohexyl)methyl)hexahydro-lH-4,7-methanoisoindole-l,3(2H)-dione of Formula (C) which on reaction with methane sulphonyl chloride followed by reaction with l-(l,2-benzisothiazol-3-yl)piperazine of Formula (D) forms lurasidone free base which was converted into lurasidone hydrochloride using acetone and cone, hydrochloric acid.
Some of the prior art processes disclose various combinations of hydrogen chloride and solvent for the preparation of lurasidone hydrochloride from lurasidone free base.
US 7,605,260 discloses use of acetone and aqueous HC1 having strength 1.8-14.4 % for preparing lurasidone hydrochloride. The yield of lurasidone hydrochloride is relatively low (85%) by this method. If the acid concentration during the salt formation is more than 5.0% then acetone quantity as the residual solvent in the reaction product is found to be greater than 0.5% in our hands which is above the ICH limits. If acid concentration during the salt formation is less than 1.8%, then yield is reduced drastically to 65%. Therefore, this method has limitations on the large-scale industrial production.
Chinese patent application no. CN102746289A discloses the process for the preparation of lurasidone hydrochloride by adding a mixture of acetone and aqueous HC1 to a solution of lurasidone free base in acetone. On reproducing this process in laboratory, it was observed that the XRPD of the product obtained does not match with XRPD of lurasidone hydrochloride.
Indian patent application IN 777/MUM/2013 discloses use of IPA, water and 35% Aqueous HC1 for the preparation of lurasidone hydrochloride. The IPA content in the product was found to be more than 5000ppm.
The methods described in the prior art are not suitable for large scale commercial production as the residual solvent is out of the ICH limits and thus the product obtained can’t be used as a drug. In order to keep the residual solvent(s) within ICH limits, repeated crystallization/purification are required which results in reduced yield and make the process quite expensive.
The prior art discloses various processes for the preparation of lurasidone hydrochloride and its intermediates. However, there still remains a need for alternative process for the preparation of lurasidone and its pharmaceutically acceptable salts substantially free of residual solvent(s) which can be used as a drug.
According to another embodiment of the present invention, novel process for the preparation of the compound of Formula (III), their isomers and pharmaceutically acceptable salts thereof, comprises condensing 1,2-cyclohexane dicarboxylic acid of Formula (II), their isomers with carbonyl diimidazole, optionally in a solvent.
(IV) 
Formula (III)
NaBH4 RT /H20
![]()
Formula (VII)
Scheme-1:

Example-1
Synthesis of trans(R,R)-l,2-cyclo exane dicarboxylic acid
A round bottom flask was charged with methanol (500 mL), IPA (500 mL) and trans (racemic)-l,2-cyclohexane dicarboxylic acid (100 g). In this reaction mass (R)-l-phenylethyl amine (74 mL) was added over a period of 30 minutes and stirred for 2-3 hrs at 30-40 °C. The solid obtained was filtered, washed with methanol and IPA solution (50+50 mL) and dried under reduced pressure to obtain crude salt of iran5(R,R)-l,2-cyclohexane dicarboxylic acid. The obtained salt was stirred in a solution of methanol (500 mL) and IPA (500 mL) at 65-70 °C for 2-3 hours, cooled to room temperature and filtered. The solid was washed with methanol and IPA solution (50+50 mL) and dried under reduced pressure. The solid thus obtained was dissolved in about 2N hydrochloric acid and extracted two times with ethyl acetate (1000 mL+200 mL). Organic layers were combined and washed with brine solution (100 mL). Ethyl acetate was distilled off under vacuum at 50-55 °C and cyclohexane was added to the residue. The solid separated out was filtered and washed with cyclohexane and dried under vacuum at 45-50 °C for 8-10 hours. Yield = 29.4 g
Example-2
Synthesis of ((R,R)-cyclohexane-L2-diyl)bis((lH-imidazol-l-yl)methanone)
To a solution of iran5(R,R)-l,2-cyclohexane dicarboxylic acid (25.0 g) in THF (250 mL), carbonyl diimidazole (60 g) is added and stirred for one hour at 25-30 °C . To the said solution of (R,R)2-(((lH-imidazole-lcarbonyl)oxy)carbonyl)cyclohexanecarboxylic acetic anhydride lH-imidazole (25.0 g) in THF (250 mL) is stirred for one hour at 45-50 °C. The compound obtained is isolated and is characterized by mass and NMR.
[m z = 272.75; 1H-NMR: 8.24 (s, 2H), 7.72 (d, 2H); 7.50 (d, 2H), 3.5 (m, 2H), 2.26-1.50 (m, 8H)]
Example-3
Synthesis of tra»,s(R,R)-l,2- bis(hydroxymethyl)cyclohexane
To a solution of ((R,R)-cyclohexane-l,2-diyl)bis((lH-imidazol-l-yl)methanone) (25 g) in THF (250 mL), sodium borohydride (22.0 g) followed by water (44.0 mL) are added and stirred for one hour. To this reaction mass, 10% solution of acetic acid (500 mL) and dichloromethane (500 mL) are added, stirred and layers separated. The organic layer is washed with 10% sodium bicarbonate solution followed by water. The dichloromethane is distilled off from organic layer under vacuum to give an oily mass. To the oily mass
dichloromethane (100 mL), water (100 mL) and 12.5mL cone, hydrochloric acid (35%) are added, stirred and layers obtained are separated. The dichloromethane is distilled off completely from organic layer at 40 °C to obtain oily mass (15.5 g).
Example-4
One pot process for synthesis of trans(R,R)-l,2- bis(hydroxymethyl)cyclohexane from trans(R,R)-l,2-cyclo exane dicarboxylic acid
To a solution of iran5(R,R)-l,2-cyclohexane dicarboxylic acid (25.0 g) in THF (250 mL), carbonyl diimidazole (60 g) was added and stirred for one hour at 25-30 °C. To the intermediate obtained sodium borohydride (22.0 g) and water (44.0 mL) were added and stirred for one hour. To this reaction mass, 10% solution of acetic acid (500 mL) and dichloromethane (500 mL) were added, stirred and layers separated. The aqueous layer was washed with dichloromethane (250 mL). The organic layer was washed with 10% sodium bicarbonate solution followed by water. The dichloromethane is distilled off from organic layer under vacuum to give an oily mass. To the oily mass dichloromethane (100 mL), water (100 mL) and 12.5mL cone, hydrochloric acid (35%) were added, stirred and layers obtained were separated. The dichloromethane was distilled off completely at 40 °C to obtain oily mass (15.5 g).
Example-5
Synthesis of m¾ns(R,R)- 2-bis(methanesulfonylmethyl) cyclohexane
To a suspension of irafts(R,R)-l,2-bis(hydroxymethyl)cyclohexane (15.0g) in dichloro methane (300 mL), triethyl amine (43.7 mL) followed by methane sulphonyl chloride (17.8 mL) were added over a period of 30-45 minutes. Reaction mass was stirred for 2-3 hrs. Reaction was monitored by HPLC (RI detector). After the completion of reaction, water was added, stirred and layers separated. The organic layer was washed with 10% sodium bicarbonate solution (150 mL) followed by water (150 mL). The dichloromethane was distilled off from organic layer under vacuum at 40-55 °C to give an oily mass. Methanol (30 mL) was added to the oily mass and strip off under vacuum at 40°C, added methanol (150 mL) and stirred for 1 h at 10-15°C and the solid obtained was filtered, washed with methanol (15 mL) and dried under vacuum to get the product (15.8g).
Example-6
Synthesis of ?ran (R,R)-3aJ(¾-octahvdroisoindolium-2-spiro- -r4-(L2-benzoisothiazole-3-yl)l piperazine methanesulfonate:
To a suspension of iran5(R,R)-l,2-bis(methanesulfonylmethyl)cyclohexane (15 g) in acetonitrile (150 mL) l-(l,2-benzisothiazol-3-yl)piperazine (10.95g) and sodium carbonate (7.8 g) were added, heated and stirred for 20 hrs at reflux temperature. Reaction was monitored by HPLC. After the completion of reaction, mass was cooled to 40-45 °C, filtered and washed with acetonitrile (20 mL). The acetonitrile was distilled off under vacuum at 45-50 °C. To the residue acetone (100 mL) was added, stirred for 1 hour, filtered, washed with acetone (10 mL), dried at 50-55°C for 6-8 hours to get the product (12.5 g).
Example-7
Synthesis of Lurasidone
To a suspension of iran5(R,R)-3<3,7(3-octahydroisoindolium-2-spiro- -[4-(l,2-benzo isothiazole-3-yl)]piperazinemethanesulfonate (10 g) in toluene (150 mL), bicycle[2.2.1] heptane-2-exo-3-exo-dicarboximide (5.9 g) and potassium carbonate (4.8 g) were added, heated to 110° C and stirred for 8-10 hours. Reaction was monitored by HPLC. After the completion of reaction, reaction mass was cooled to 20-30 °C, filtered and washed with toluene (10 mL). The toluene was distilled off at 55-60°C. To the residue IPA (100 mL) was added and stirred for 1-2 hours at room temperature. Lurasidone free base obtained was filtered and washed with IPA (10 mL). The solid was suck dried for 30 minutes to obtain lurasidone.
Example-8
Synthesis of Lurasidone hydrochloride
To lurasidone base (5g), acetone (75mL) and water (10 mL) were added. The mixture was heated to 55-60°C followed by the addition of IPA.HCl (10%) (lOmL) and stirred for 1-2 hours, reflux temperature. The clear solution obtained was stirred for 30 min and then 5ml IPA.HCl (10%) was added. The reaction mixture was stirred at reflux temperature for 30 min, cooled and stirred for 60 min. The solid obtained was filtered and washed with acetone (5ml) and dried under vacuum at 60°C for 8 hours.
Acetone: 542 ppm; IPA= 38ppm; Yield=93%
Example-9
Synthesis of Lurasidone hydrochloride
To lurasidone base (5g), acetone (75mL) and water (5 mL) were added. The mixture was heated to 55-60°C followed by the addition of IPA.HCl (10%) (5mL) and stirred for about 1-2 hours. The reaction mixture was stirred for 30 min. at 55-60°C, cooled and stirred for 60 min. The solid obtained was filtered and washed with acetone (5ml) and dried under vacuum at 70-80°C for 8 hours.
Jubilant Generics Limited



Chairman & Managing Director
Jubilant Bhartia Group Shyam, together with his brother Hari, is founder of Jubilant Bhartia Group (www.jubilantbhartia.com) headquartered in New Delhi, India. The Jubilant Bhartia Group, with 30,000 employees, has a strong presence in diverse sectors like Pharmaceuticals and Life Sciences, Oil and Gas (exploration and production), Agri products, Performance Polymers, Retail, Food and Consulting in Aerospace and Oilfield Services. Jubilant Bhartia Group has four flagships Companies- Jubilant Life Sciences Limited, Jubilant FoodWorks Limited and Jubilant Industries Limited, listed on Indian Stock Exchange and Jubilant Energy NV, listed at AIM market of London Exchange.Shyam, holds a bachelors’ degree in commerce from St. Xavier’s College, Calcutta University, and is a qualified cost and works accountant & a fellow member of the Institute of Cost and Works Accountants of India (ICWAI).Shyam has been associated with various institutions and has served as Member of Board of Governors, Indian Institute of Technology (IIT), Mumbai, and Indian Institute of Management (IIM), Ahmedabad. Shyam has also served as a Member of the Executive Committee of Federation of Indian Chamber of Commerce & Industry (FICCI) & Confederation of Indian Industry (CII) and was also a member of Task Force on Chemicals appointed by the Government of IndiaShyam’s immense contributions have been recognized by various awards. CHEMEXCIL has conferred Lifetime Achievement Award 2010-11 to him. He, along with his brother, was felicitated with the Entrepreneur of the Year Award at the prestigious AIMA Managing India Awards 2013, presented by the President of India. In 2010, the duo also shared the much-covetedErnst & Young Entrepreneur of the Year Award for Life Sciences & Consumer Products category.Shyam serves on the Board of several Public and Private and Foreign companies likes of Chambal Fertilizers and Chemicals Ltd, Putney Inc., CFCL Technologies Limited (Cayman Islands), Tower Promoters, BT Telecom India Pvt Ltd., American Orient Capital Partners India Pvt Ltd, IMACID, Morocco, Safe Food Corporation, etc. He was also a Director on the Board of Air India.Shyam is a regular participant at the World Economic Forum Annual Meeting in Davos and a member of the Chemical Governors Council of the World Economic Forum.Shyam is married to Shobhana, Former Member of Parliament & Chairperson, The Hindustan Times Media Ltd. They have two sons- Priyavrat and Shamit.
ISO Certification
ISO 9001:2008, 14001:2004 & OHSAS 18001:2007 certified
Code of Conduct
Code Of Conduct for Directors and Senior ManagementThis Code of Conduct highlights the standards of conduct expected from the Company’s Directors and Senior Management so as to align these with the Company’s Vision, Promise and Values.Jubilant Life Sciences Ltd. (Jubilant) has a well formulated Vision which drives the business and has the promise of Caring, Sharing, Growing to all the stakeholders–We will, with utmost care for the environment, continue to enhance value for our customers by providing innovative products and economically efficient solutions and for our shareholders through sales growth, cost effectiveness and wise investment of resources.
Director’s Desk

Co-Chairman & Managing Director
Jubilant Bhartia Group
Hari, together with his brother Shyam, is co-founder of Jubilant Bhartia Group (www.jubilantbhartia.com) headquartered in New Delhi, India.The Jubilant Bhartia Group, with 30,000 employees, has a strong presence in diverse sectors like Pharmaceuticals and Life Sciences, Oil and Gas (exploration and production), Agri products, Performance Polymers, Retail, Food and Consulting in Aerospace and Oilfield Services. Jubilant Bhartia Group has four flagships Companies- Jubilant Life Sciences Limited, Jubilant FoodWorksLimited and Jubilant Industries Limited, listed on Indian Stock Exchange and Jubilant Energy NV, listed at AIM market of London Exchange.A Chemical Engineering Graduate from the prestigious Indian Institute of Technology (IIT), Delhi, Hari was conferred the Distinguished Alumni award by his alma mater in 2000. He has been associated in various capacities with the IIT system and with the Ministry of Human Resource Development, Government of India.Hari is a past President of the Confederation of Indian Industry (CII) & a member of several educational, scientific and technological programmes of the Government of India. He is currently the Chairman of the Board of Governors of the Indian Institute of Management (IIM), Raipur and Member of the International Advisory Board of McGill University, Canada.Hari is the Co-Chairman of India-Canada CEO’s Forum appointed by the Prime Minister of India. He is also a member of CEO’s Forum for India-USA, India-France and India-Sri Lanka and Joint Task Force for India-Myanmar & India-UAE. He is a regular participant at the World Economic Forum Annual Meeting in Davos and is a member of the World Economic Forum’s International Business Council and the Health Governors.Hari’s immense contributions have been recognized by various awards. He, along with his brother, was felicitated with the Entrepreneur of the Year Award at the prestigious AIMA Managing India Awards 2013, presented by the President of India. In 2010, the duo also shared the much-coveted Ernst & Young Entrepreneur of the Year Award for Life Sciences & Consumer Products category.Hari serves on the board of several public and private companies like TV 18 Broadcast Ltd., Shriram Pistons & Rings Ltd., Export Credit Guarantee Corporation of India Ltd., BT Telecom India Pvt. Ltd & India Brand Equity Foundation.Hari is married to Kavita, a leading Fashion Designer and Retailer. They have a daughter, Aashti and a son, Arjun.

Executive Leadership Team

Shyam S Bhartia
Chairman

Hari S Bhartia
Co-Chairman & Managing Director

Shyamsundar Bang
Executive Director –Manufacturing & Supply Chain

R Sankaraiah
Executive Director – Finance

Pramod Yadav
Co-CEO
Life Science Ingredients
Rajesh Srivastava
Co-CEO
Life Science Ingredients
G. P. Singh
Fine Chemicals and CRAMS
CEO – Jubilant Pharma
Chandan Singh
President – Life Science Chemicals

Martyn Coombs
President – Jubilant DraxImage

Bryan Downey
President – Allergy Business

T. S. Parmar
President – India Branded Pharmaceuticals

Dr. Ashutosh Agarwal
Chief Scientific Officer –Chemicals and Life Science Ingredients

Ajay Khanna
Chief – Strategic & Public Affairs
///////Lurasidone hydrochloride, Jubilant Life Sciences Ltd, WO 2016059649, New patent
Albutrepenonacog alfa
Albutrepenonacog alfa
recombinant factor IX
(Idelvion®)Approved, 2016-03-04 USFDA
A recombinant albumin-human coagulation factor IX (FIX) fusion protein indicated for the treatment and prevention of bleeding in patients with hemophilia B.
![]()
Research Code CSL-654
| Type | Recombinant coagulation factor |  |
| Source | Human | |
| Molecular Formula | C5077H7846N1367O1588S67 | |
| Molecular Weight | ~125000 |
Other Names
- Albutrepenonacog alfa
Protein Sequence
Sequence Length: 1018modified (modifications unspecified)
- Originator CSL Behring
- Class Albumins; Antihaemorrhagics; Blood coagulation factors; Recombinant fusion proteins
- Mechanism of Action Blood coagulation factor replacements; Factor X stimulants
- Orphan Drug Status Yes – Haemophilia B
- Marketed Haemophilia B
Most Recent Events
- 21 Mar 2016 Launched for Haemophilia B (In adolescents, In children, In adults) in USA (IV) – First global launch
- 07 Mar 2016 Preregistration for Haemophilia B in Australia (IV) before March 2016
- 04 Mar 2016 Registered for Haemophilia B (In children, In adolescents, In adults) in USA (IV)
| Latest Stage of Development | Approved |
| Standard Indication | Hemophilia |
| Indication Details | Treat and prevent bleeding episodes in hemophilia B patients; Treat hemophilia B |
| Regulatory Designation | U.S. – Orphan Drug (Treat and prevent bleeding episodes in hemophilia B patients); EU – Orphan Drug (Treat and prevent bleeding episodes in hemophilia B patients); Switzerland – Orphan Drug (Treat and prevent bleeding episodes in hemophilia B patients) |
-
BNF Category:Antifibrinolytic drugs and haemostatics (02.11)
Pharmacology: Albutrepenonacog alfa is a recombinant factor IX (rIX-FP) albumin fusion protein, designed to exhibit an extended half-life. Factor IX has a short half-life which necessitates multiple injections. Epidemiology: Haemophilia B is a genetic disorder caused by missing or defective factor IX, a clotting protein. It has a prevalence of around 1 in 50,000 live births in the UK and is more common in males. In 2012-13, there were 476 hospital admissions in England due to haemophilia B, accounting for 508 finished consultant episodes and 125 bed days. Indication: Haemophilia B

Albutrepenonacog alfa was approved by the U.S. Food and Drug Administration (FDA) on March 4, 2016. It was developed and marketed as Idelvion® by CSL Behring.
Albutrepenonacog alfa is a recombinant albumin-human coagulation factor IX (FIX) fusion protein, which replaces the missing FIX needed for effective hemostasis. It is indicated for the treatment and prevention of bleeding in children and adults with hemophilia B.
Idelvion® is available as injection (lyophilized powder) for intravenous use, containing 250 IU, 500 IU, 1000 IU or 2000 IU of albutrepenonacog alfa in single-use vials. In control and prevention of bleeding episodes and perioperative management, the required dosage is determined using the following formulas: Required Dose (IU) = Body Weight (kg) x Desired Factor IX rise (% of normal or IU/dL) x (reciprocal of recovery (IU/kg per IU/dL)). In routine prophylaxis, the recommended dose is 25-40 IU/kg (for patients ≥12 years of age) or 40-55 IU/kg (for patients <12 years of age) every 7 days.
On 25 February 2016, the Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion, recommending the granting of a marketing authorisation for the medicinal product IDELVION, intended for treatment and prophylaxis of bleeding in patients with Haemophilia B. IDELVION was designated as an orphan medicinal producton 04 February 2010. The applicant for this medicinal product is CSL Behring GmbH.
IDELVION will be available as 250 IU, 500 IU, 1000 IU and 2000 IU Powder and solvent for solution for injection. The active substance of IDELVION is albutrepenonacog alfa, an antihaemorrhagic, blood coagulation factor IX, (ATC code: B02BD04). It works as replacement therapy and temporarily increases plasma levels of factor IX, helping to prevent and control bleeding.
The benefits with IDELVION are its ability to stop the bleeding when given on demand and prevent bleeding when used as routine prophylaxis or for surgical procedures. The most common side effects are injection site reaction and headache.
The full indication is: “the treatment and prophylaxis of bleeding in patients with Haemophilia B (congenital factor IX deficiency)”. Idelvion can be used in all age groups. It is proposed that IDELVION be prescribed by physicians experienced in the treatment of haemophilia B.
Detailed recommendations for the use of this product will be described in the summary of product characteristics (SmPC), which will be published in the European public assessment report (EPAR) and made available in all official European Union languages after the marketing authorisation has been granted by the European Commission.
| Name | Idelvion |
|---|---|
| INN or common name | albutrepenonacog alfa |
| Therapeutic area | Hemophilia B |
| Active substance | albutrepenonacog alfa |
| Date opinion adopted | 25/02/2016 |
| Company name | CSL Behring GmbH |
| Status | Positive |
| Application type | Initial authorisation |
//////Albutrepenonacog alfa, CSL-654, Idelvion; Recombinant factor IX – CSL Behring, Recombinant factor IX fusion protein linked with human albumin, rFIX-FP – CSL Behring; rIX-FP, Orphan Drug Status, Haemophilia B, recombinant factor IX , FDA 2016
update
| Human medicines European public assessment report (EPAR): Idelvion, albutrepenonacog alfa, Hemophilia B, 11/05/2016, |
Asfotase alfa
![]()

> Asfotase Alfa Sequence LVPEKEKDPKYWRDQAQETLKYALELQKLNTNVAKNVIMFLGDGMGVSTVTAARILKGQL HHNPGEETRLEMDKFPFVALSKTYNTNAQVPDSAGTATAYLCGVKANEGTVGVSAATERS RCNTTQGNEVTSILRWAKDAGKSVGIVTTTRVNHATPSAAYAHSADRDWYSDNEMPPEAL SQGCKDIAYQLMHNIRDIDVIMGGGRKYMYPKNKTDVEYESDEKARGTRLDGLDLVDTWK SFKPRYKHSHFIWNRTELLTLDPHNVDYLLGLFEPGDMQYELNRNNVTDPSLSEMVVVAI QILRKNPKGFFLLVEGGRIDHGHHEGKAKQALHEAVEMDRAIGQAGSLTSSEDTLTVVTA DHSHVFTFGGYTPRGNSIFGLAPMLSDTDKKPFTAILYGNGPGYKVVGGERENVSMVDYA HNNYQAQSAVPLRHETHGGEDVAVFSKGPMAHLLHGVHEQNYVPHVMAYAACIGANLGHC APASSLKDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEV KFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIE KTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKT TPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGKDIDDDD DDDDDD
Asfotase alfa
Indicated for the treatment of patients with perinatal/infantile and juvenile onset hypophosphatasia (HPP).
(Strensiq®)Approved
A mineralized tissue targeted fusion protein used to treat hypophosphatasia.
![]()
Research Code ALXN-1215; ENB-0040; sALP-FcD-10
CAS No.1174277-80-5
| 180000.0
C7108H11008N1968O2206S56 |

| Company | Alexion Pharmaceuticals Inc. |
| Description | Fusion protein incorporating the catalytic domain of human tissue non-specific alkaline phosphatase (TNSALP; ALPL) and a bone-targeting peptide |
| Molecular Target | |
| Mechanism of Action | Enzyme replacement therapy |
| Therapeutic Modality | Biologic: Fusion protein |
| Latest Stage of Development | Approved |
| Standard Indication | Metabolic (unspecified) |
| Indication Details | Treat hypophosphatasia (HPP); Treat hypophosphatasia (HPP) in children; Treat hypophosphatasia (HPP) in patients whose first signs or symptoms occurred prior to 18 years of age; Treat perinatal, infantile and juvenile-onset hypophosphatasia (HPP) |
| Regulatory Designation | U.S. – Breakthrough Therapy (Treat hypophosphatasia (HPP) in children); U.S. – Breakthrough Therapy (Treat hypophosphatasia (HPP) in patients whose first signs or symptoms occurred prior to 18 years of age); U.S. – Fast Track (Treat hypophosphatasia (HPP)); U.S. – Orphan Drug (Treat hypophosphatasia (HPP)); U.S. – Priority Review (Treat hypophosphatasia (HPP) in children); EU – Accelerated Assessment (Treat hypophosphatasia (HPP)); EU – Accelerated Assessment (Treat hypophosphatasia (HPP) in children); EU – Orphan Drug (Treat hypophosphatasia (HPP)); Japan – Orphan Drug (Treat hypophosphatasia (HPP)); Australia – Orphan Drug (Treat hypophosphatasia (HPP) |
Asfotase Alfa is a first-in-class bone-targeted enzyme replacement therapy designed to address the underlying cause of hypophosphatasia (HPP)—deficient alkaline phosphatase (ALP). Hypophosphatasia is almost always fatal when severe skeletal disease is obvious at birth. By replacing deficient ALP, treatment with Asfotase Alfa aims to improve the elevated enzyme substrate levels and improve the body’s ability to mineralize bone, thereby preventing serious skeletal and systemic patient morbidity and premature death. Asfotase alfa was first approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on July 3, 2015, then approved by the European Medicine Agency (EMA) on August 28, 2015, and was approved by the U.S. Food and Drug Administration (FDA) on October 23, 2015. Asfotase Alfa is marketed under the brand name Strensiq® by Alexion Pharmaceuticals, Inc. The annual average price of Asfotase Alfa treatment is $285,000.
Hypophosphatasia (HPP) is a rare inheritable disease that results from loss-of-function mutations in the ALPL gene encoding tissue-nonspecific alkaline phosphatase (TNSALP). Therapeutic options for treating the underlying pathophysiology of the disease have been lacking, with the mainstay of treatment being management of symptoms and supportive care. HPP is associated with significant morbidity and mortality in paediatric patients, with mortality rates as high as 100 % in perinatal-onset HPP and 50 % in infantile-onset HPP. Subcutaneous asfotase alfa (Strensiq(®)), a first-in-class bone-targeted human recombinant TNSALP replacement therapy, is approved in the EU for long-term therapy in patients with paediatric-onset HPP to treat bone manifestations of the disease. In noncomparative clinical trials in infants and children with paediatric-onset HPP, asfotase alfa rapidly improved radiographically-assessed rickets severity scores at 24 weeks (primary timepoint) as reflected in improvements in bone mineralization, with these benefits sustained after more than 3 years of treatment. Furthermore, patients typically experienced improvements in respiratory function, gross motor function, fine motor function, cognitive development, muscle strength (normalization) and ability to perform activities of daily living, and catch-up height-gain. In life-threatening perinatal and infantile HPP, asfotase alfa also improved overall survival. Asfotase alfa was generally well tolerated in clinical trials, with relatively few patients discontinuing treatment and most treatment-related adverse events being of mild to moderate intensity. Thus, subcutaneous asfotase alfa is a valuable emerging therapy for the treatment of bone manifestations in patients with paediatric-onset HPP.
October 23, 2015
Release
Today, the U.S. Food and Drug Administration approved Strensiq (asfotase alfa) as the first approved treatment for perinatal, infantile and juvenile-onset hypophosphatasia (HPP).
HPP is a rare, genetic, progressive, metabolic disease in which patients experience devastating effects on multiple systems of the body, leading to severe disability and life-threatening complications. It is characterized by defective bone mineralization that can lead to rickets and softening of the bones that result in skeletal abnormalities. It can also cause complications such as profound muscle weakness with loss of mobility, seizures, pain, respiratory failure and premature death. Severe forms of HPP affect an estimated one in 100,000 newborns, but milder cases, such as those that appear in childhood or adulthood, may occur more frequently.
“For the first time, the HPP community will have access to an approved therapy for this rare disease,” said Amy G. Egan, M.D., M.P.H., deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research (CDER). “Strensiq’s approval is an example of how the Breakthrough Therapy Designation program can bring new and needed treatments to people with rare diseases.”
Strensiq received a breakthrough therapy designation as it is the first and only treatment for perinatal, infantile and juvenile-onset HPP. The Breakthrough Therapy Designation program encourages the FDA to work collaboratively with sponsors, by providing timely advice and interactive communications, to help expedite the development and review of important new drugs for serious or life-threatening conditions. In addition to designation as a breakthrough therapy, the FDA granted Strensiq orphan drug designation because it treats a disease affecting fewer than 200,000 patients in the United States.
Orphan drug designation provides financial incentives, like clinical trial tax credits, user fee waivers, and eligibility for market exclusivity to promote rare disease drug development. Strensiq was also granted priority review, which is granted to drug applications that show a significant improvement in safety or effectiveness in the treatment of a serious condition. In addition, the manufacturer of Strensiq was granted a rare pediatric disease priority review voucher – a provision intended to encourage development of new drugs and biologics for the prevention and treatment of rare pediatric diseases. Development of this drug was also in part supported by the FDA Orphan Products Grants Program, which provides grants for clinical studies on safety and/or effectiveness of products for use in rare diseases or conditions.
Strensiq is administered via injection three or six times per week. Strensiq works by replacing the enzyme (known as tissue-nonspecific alkaline phosphatase) responsible for formation of an essential mineral in normal bone, which has been shown to improve patient outcomes.
The safety and efficacy of Strensiq were established in 99 patients with perinatal (disease occurs in utero and is evident at birth), infantile- or juvenile-onset HPP who received treatment for up to 6.5 years during four prospective, open-label studies. Study results showed that patients with perinatal- and infantile-onset HPP treated with Strensiq had improved overall survival and survival without the need for a ventilator (ventilator-free survival). Ninety-seven percent of treated patients were alive at one year of age compared to 42 percent of control patients selected from a natural history study group. Similarly, the ventilator-free survival rate at one year of age was 85 percent for treated patients compared to less than 50 percent for the natural history control patients.
Patients with juvenile-onset HPP treated with Strensiq showed improvements in growth and bone health compared to control patients selected from a natural history database. All treated patients had improvement in low weight or short stature or maintained normal height and weight. In comparison, approximately 20 percent of control patients had growth delays over time, with shifts in height or weight from the normal range for children their age to heights and weights well below normal for age. Juvenile-onset patients also showed improvements in bone mineralization, as measured on a scale that evaluates the severity of rickets and other HPP-related skeletal abnormalities based on x-ray images. All treated patients demonstrated substantial healing of rickets on x-rays while some natural history control patients showed increasing signs of rickets over time.
The most common side effects in patients treated with Strensiq include injection site reactions, hypersensitivity reactions (such as difficulty breathing, nausea, dizziness and fever), lipodystrophy (a loss of fat tissue resulting in an indentation in the skin or a thickening of fat tissue resulting in a lump under the skin) at the injection site, and ectopic calcifications of the eyes and kidney.
Strensiq is manufactured by Alexion Pharmaceuticals Inc., based in Cheshire, Connecticut.

| Patent Number | Pediatric Extension | Approved | Expires (estimated) | |
|---|---|---|---|---|
| US7763712 | No | 2004-04-21 | 2026-07-15 |
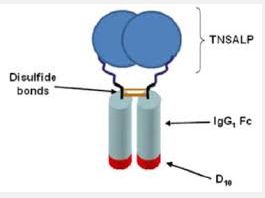
STRENSIQ is a formulation of asfotase alfa, which is a soluble glycoproteincomposed of two identical polypeptide chains. Each chain contains 726amino acids with a theoretical mass of 161 kDa. Each chain consists of the catalytic domain of human tissue non-specific alkaline phosphatase (TNSALP), the human immunoglobulin G1 Fc domain and a deca-aspartatepeptide used as a bone targeting domain. The two polypeptide chains are covalently linked by two disulfide bonds.
STRENSIQ is a tissue nonspecific alkaline phosphatase produced byrecombinant DNA technology in a Chinese hamster ovary cell line. TNSALP is a metallo-enzyme that catalyzes the hydrolysis of phosphomonoesters with release of inorganic phosphate and alcohol. Asfotase alfa has a specific activity of 620 to 1250 units/mg. One activity unit is defined as the amount of asfotase alfa required to form 1 μmol of p-nitrophenol from pNPP per minute at 37°C.
STRENSIQ (asfotase alfa) is a sterile, preservative-free, nonpyrogenic, clear, slightly opalescent or opalescent, colorless to slightly yellow, with few small translucent or white particles, aqueous solution for subcutaneous administration. STRENSIQ is supplied in glass single-use vials containing asfotase alfa; dibasic sodium phosphate, heptahydrate; monobasic sodium phosphate, monohydrate; and sodium chloride at a pH between 7.2 and 7.6. Table 5 describes the content of STRENSIQ vial presentations.
Table 5: Content of STRENSIQ Vial Presentations
| INGREDIENT | QUANTITY PER VIAL | |||
| ASFOTASE ALFA | 18 MG/0.45 ML | 28 MG/0.7 ML | 40 MG/ML | 80 MG/0.8 ML |
| Dibasic sodium phosphate, heptahydrate | 2.48 mg | 3.85 mg | 5.5 mg | 4.4 mg |
| Monobasic sodium phosphate, monohydrate | 0.28 mg | 0.43 mg | 0.62 mg | 0.5 mg |
| Sodium chloride | 3.94 mg | 6.13 mg | 8.76 mg | 7.01 mg |
REFERNCES
- Whyte MP: Hypophosphatasia – aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016 Apr;12(4):233-46. doi: 10.1038/nrendo.2016.14. Epub 2016 Feb 19. [PubMed:26893260 ]
- Whyte MP, Rockman-Greenberg C, Ozono K, Riese R, Moseley S, Melian A, Thompson DD, Bishop N, Hofmann C: Asfotase Alfa Treatment Improves Survival for Perinatal and Infantile Hypophosphatasia. J Clin Endocrinol Metab. 2016 Jan;101(1):334-42. doi: 10.1210/jc.2015-3462. Epub 2015 Nov 3. [PubMed:26529632 ]
- Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, Van Sickle BJ, Simmons JH, Edgar TS, Bauer ML, Hamdan MA, Bishop N, Lutz RE, McGinn M, Craig S, Moore JN, Taylor JW, Cleveland RH, Cranley WR, Lim R, Thacher TD, Mayhew JE, Downs M, Millan JL, Skrinar AM, Crine P, Landy H: Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012 Mar 8;366(10):904-13. doi: 10.1056/NEJMoa1106173. [PubMed:22397652 ]
//////Asfotase alfa, Strensiq, treat hypophosphatasia, ALXN-1215, ENB-0040, sALP-FcD-10, FDA 2015
Idarucizumab

![]()
Idarucizumab
(Praxbind®) Approved
An antidote for rapid reversal of dabigatran-induced anticoagulation indicated for emergency surgery (urgent procedures) and life-threatening or uncontrolled bleeding in patients treated with dabigatran.
![]()
BI-655075
CAS No.1362509-93-0
Other Names
- BI 655075
- Idarucizumab
- Praxbind
Protein Sequence
Sequence Length: 444, 225, 219multichain; modified (modifications unspecified)
Idarucizumab, sold under the brand name Praxbind, is a monoclonal antibody designed for the reversal of anticoagulant effects ofdabigatran.[1][2]
This drug was developed by Boehringer Ingelheim Pharmaceuticals. A large study sponsored by the manufacturer found that idarucizumab effectively reversed anticoagulation by dabigatran within minutes.[3] It was FDA approved in October 2015.[4] In the United States the wholesale cost is $3500 US.[5]
References
- Statement On A Nonproprietary Name Adopted By The USAN Council – Idarucizumab, American Medical Association.
- World Health Organization (2013). “International Nonproprietary Names for Pharmaceutical Substances (INN). Proposed INN: List 109” (PDF). WHO Drug Information 27 (2).
- Pollack, Charles V.; Reilly, Paul A.; Eikelboom, John; Glund, Stephan; Verhamme, Peter; Bernstein, Richard A.; Dubiel, Robert; Huisman, Menno V.; Hylek, Elaine M. (2015-08-06).“Idarucizumab for Dabigatran Reversal”. The New England Journal of Medicine 373 (6): 511–520. doi:10.1056/NEJMoa1502000. ISSN 1533-4406. PMID 26095746.
- “Press Announcements – FDA approves Praxbind, the first reversal agent for the anticoagulant Pradaxa”. http://www.fda.gov. Retrieved 2015-10-17.
- Elia, Joe. “Dabigatran-Reversal Agent Price Set”. Retrieved 20 October 2015.
| Monoclonal antibody | |
|---|---|
| Type | Fab fragment |
| Source | Humanized (from mouse) |
| Target | Dabigatran |
| Clinical data | |
| Trade names | Praxbind |
| Identifiers | |
| CAS Number | 1362509-93-0 |
| ATC code | V03AB37 (WHO) |
| IUPHAR/BPS | 8298 |
| ChemSpider | none |
| Chemical data | |
| Formula | C2131H3299N555O671S11 |
| Molar mass | 47.8 kg/mol |
/////Idarucizumab

{kind=link}